Палладиево-медные катализаторы гомогенного селективного окисления тиольных групп, комбинация и композиция на их основе и способ терапевтического воздействия - RU2451010C1
Код документа: RU2451010C1
Чертежи



Описание
Область техники, к которой относится изобретение
Данное изобретение относится к бионеорганической химии, медицинской химии и медицине, а именно к области получения лекарственных препаратов, и может быть использовано в бионеорганической химии, фармакологии, медицине и ветеринарии.
Предшествующий уровень техники
Повышение терапевтической эффективности фармакологических молекул посредством оптимизации их фармакокинетики и/или фармакодинамики и/или снижения токсичности за счет химической модификации молекулы лекарственного средства и/или ее сочетанным использованием с другим химическим соединением или соединениями является одним из направлений создания лекарственных препаратов нового поколения, проявляющих свою активность в физиологически более оптимальных дозах.
Так, известен ряд комбинированных средств, в том числе Амоксиклав, содержащий в своем составе амоксициллин и клавулановую кислоту, Тиенам, содержащий имипенем в сочетании со специфическим ингибитором фермента дигидропептидазы почек циластатином. Клавулановая кислота препятствует разложению бактериальными ферментами амоксициллина, а циластатин тормозит метаболизм имипенема в почках, что значительно повышает концентрацию неизмененного антибиотика в почках и мочевыводящих путях.
Существует, однако, масса других лекарственных средств, потенциально полезных для лечения заболеваний, однако не обеспечивающих желаемого эффекта в силу развития к ним того или иного вида устойчивости.
В настоящее время известно вещество дисульфид N-глутамил-L-цистеинил-глицина - окисленный глутатион (GSSG), которое само по себе обладает разнообразной фармакологической активностью. В частности, выявлена способность окисленного глутатиона инициировать процессы, осуществляющие различные виды химической модификации: фосфорилирование, глутатионелирование, окисление и др., - которые предшествуют формированию определенной структурной конформации с высоким сродством к лиганду и способностью к выполнению физиологической функции. Показана способность окисленного глутатиона усиливать продукцию широкого спектра цитокинов, контролирующих комплекс защитных реакций организма, включая противовирусное, антибактериальное, противоопухолевое, антифибротическое действие. Предложен спектр фармакологических решений по созданию композитов, включающих комплексное соединение окисленного глутатиона и цисплатина в сочетании с фармакологически активными молекулами для лечения различных заболеваний, включая сахарный диабет, ишемическую болезнь сердца, вирусные гепатиты, злокачественные опухоли, гнойные инфекции и ряд других. В частности, для лечения лекарственно резистентных форм вирусных гепатитов В и С предложено фармакологическое решение по усилению противовирусной активности инозина, применяемого в форме органической соли с окисленным глутатионом (RU 2153350, RU 2153351).
Однако в состав препарата окисленного глутатиона, раскрытого в RU 2153350, входит цисплатин, представляющий собой комплексное соединение платины (Pt), применение которой сопряжено с опасностью токсического и мутагенного действия. При этом именно платина проявляет каталитический эффект при использовании ее в минимальном количестве.
Опасность токсического действия платины в составе препарата окисленного глутатиона при многократном введении доказана в эксперименте на клетках, где ежедневное введение комплексного соединения платины на протяжении пяти дней привело к подавлению пролиферативной активности культуры и ее гибели. Использование окисленного глутатиона также накладывает определенные ограничения на возможные фармакологические решения, так как позволяет создавать лекарственные формы преимущественно парентерального введения.
Поэтому существует необходимость в разработке новых препаратов, обладающих способностью повышать терапевтическую эффективность фармакологических молекул посредством оптимизации их фармакокинетики и/или фармакодинамики, пригодных для создания лекарственные формы энтерального, парентерального и иных возможных способов введения.
Сущность изобретения
Данная задача решена тем, что предложен палладиево-медный катализатор гомогенного селективного окисления тиолов, сочетающий в себе функциональное биядерное тиолатмостиковое координационное соединение палладия(II) и модифицирующий тиолатный комплекс меди(I), имеющий общую формулу

где
SR представляет собой остаток тиолатного лиганда, выбранный из группы, включающей в себя остаток глутатиона и ацетилцистеина,
k=2÷14,
m≥3k.
Предпочтительно окисление представляет собой гомогенное селективное окисление тиолов с образованием дисульфидных связей между тиольными остатками, причем тиол, в отношении окисления которого осуществляется каталитическая функция, представляет собой N-ацетил-цистеин или N-глутамил-L-цистеинил-глицин.
Предпочтительно катализатор получают путем взаимодействия моноядерных аминатных комплексов палладия(II) и соответствующих тиолов с комплексами, образующимися из солей меди(II) и соответствующих тиолов.
Целесообразно в катализаторе по изобретению мольное соотношение Pd:Cu лежит в интервале от 1:0,1 до 1:2, более предпочтительно в интервале от 1:0,2 до 1:1.
Катализатор по изобретению можно применять в терапии.
Предложена также каталитическая комбинация, образованная тиолом, выбранным из ацетилцистеина, глутатиона, их сольватов и солей, и катализатором по изобретению.
Предпочтительно в комбинации катализатор присутствует в количестве от 1·10-2 до 1·10-7 г на моль тиола. Причем предложенная комбинация может по существу состоять только из дисульфида ацетилцистеина и/или глутатиона, их сольватов и солей и катализатора по изобретению.
Комбинацию по изобретению можно применять в терапии.
Предложена также фармакологическая комбинация, включающая в себя указанную каталитическую комбинацию и фармакологически активное соединение, способное к вступлению в реакцию присоединения с компонентами комбинации.
Эту комбинацию целесообразно применять для повышения терапевтической активности фармакологически активного соединения.
Указанное фармакологически активное соединение может представлять собой лекарственную или биологически активную молекулу, выбранную из пуриновых или пиримидиновых оснований или их производных.
Эту комбинацию можно применять в терапии инфекционных и неинфекционных заболеваний.
Фармакологически активное соединение может представлять собой инозин или рибавирин.
Предложена также фармацевтическая композиция, включающая в себя описанные катализатор или комбинацию и фармацевтически приемлемый эксципиент. Такая фармацевтическая композиция повышает терапевтическую активность фармакологически активных соединений.
Предложен также способ терапевтического воздействия на организм пациента для повышения терапевтической активности фармакологически активного соединения, при котором нуждающемуся в этом пациенту вводят эффективное количество катализатора, комбинации или композиции по изобретению.
Описание графических материалов
На Фиг.1 представлена зависимость накопления дисульфида N-глутамил-L-цистеинил-глицина как функция соотношения числа молей Pd:Cu в системе: «

Фиг.2 демонстрирует относительную каталитическую эффективность комплексов

На Фиг.3 представлены кривые окисления N-глутамил-L-цистеинил-глицина комплексами палладия, меди и бинарным катализатором Pd-Cu (25±0.1°C, CGSH 2 мг/мл, рН 6.0, Cм 6.3е-6 моль/литр).
Подробное описание изобретения
Попытки авторов изобретения использовать препарат дисульфида N-глутамил-L-цистеинил-глицина, получаемый без использования цисплатина или с использованием другого металла, привели к снижению терапевтических результатов.
Авторами данного изобретения было обнаружено, что множество проявлений фармакологической активности дисульфида N-глутамил-L-цистеинил-глицина, получаемого по способу из RU 2153350, сопряжено со способностью препарата осуществлять каталитическое окисление сульфгидрильных групп до дисульфидов в составе молекул пептидной природы.
Авторы изобретения выявили необходимость осуществления управляемого катализа, что было достигнуто с помощью предложенного катализатора по изобретению.
Тиолатные комплексы меди(I), будучи добавленными к биядерным тиолатмостиковым координационным соединениям палладия(II), способны значительно изменять скорость, но не глубину окисления тиола. На Фиг.1 в качестве примера представлены зависимости накопления дисульфида N-глутамил-L-цистеинил-глицина (GSSG) как функции соотношения Pd:Cu в системе «

Как можно видеть из результатов, приведенных на Фиг.1, каталитическая эффективность палладиевого катализатора, модифицированного ионами меди(I), возрастает при изменении соотношения Pd:Cu от 1:0 до 1:2.
Хорошо известно, что ионы Cu2+ сами способны эффективно катализировать окисление тиоаминокислот до образования в них связей -S-S-. Поэтому для установления области изменения соотношения Pd:Cu в предлагаемых бинарных каталитических системах Pd-Cu авторами были отдельно рассмотрены каталитические эффективности биядерных тиолатмостиковых комплексов палладия(II), тиолатных комплексов меди(I) и их совместная работа.
Окисление тиолов биядерными комплексами палладия
При каталитическом окислении тиолов RSH одной из наиболее важных функций палладия является образование координационных соединений - продуктов присоединения к иону палладия тиолат-ионов RS-, с последующим их окислением и разрушением координационного полиэдра.
Пространственная близость тиолатных ионов, входящих как лиганды в координационную сферу металла, легко может быть достигнута в биядерных соединениях палладия(II), в которых тиолатные ионы RS- занимают бидентатномостиковую координацию, приводящую к формированию остова

Для окисления тиолат-ионов был рассмотрен каталитический цикл с участием биядерного гидроксомостикового аммиачного комплекса палладия(II) - [Pd2(µ-OH)2(NH3)4]2+, для которого в качестве доминирующих были рассмотрены следующие реакции:

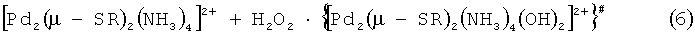

Уравнения (5) и (7) представляют ступени, в ходе которых катализатор [Pd2(µ-ОН)2(NH3)4]2+ расходуется и вновь регенерируется. Реакция (6) является основной ступенью, по которой возможно образование промежуточного неустойчивого комплекса палладия {[Pd2(µ-SR)2(NH3)4(OH)2]2+}.
Квантово-химические расчеты показали, что при окислении биядерного металлического остова

Квантово-химические расчеты координационных соединений проводилась методом DFT B3LYP в 6-31G** базисе по программе Jaguar 7.5. Для атомов Pd использовался эффективный псевдопотенциал остова HW с соответствующим валентным базисом. Анализ частот нормальных колебаний показал, что все полученные в результате оптимизации геометрии структуры соединений в газовой фазе соответствуют минимумам на поверхности потенциальной энергии. Энергии сольватации соединений рассчитывались в модели поляризуемого континуума. Для сокращения числа базисных функций молекулы глутатиона, ацетилцистеина или тиогликолевой кислоты RSH были моделированы простейшим тиолом CH3SH.
Причина нестабильности промежуточного координационного соединения PdIIPdIV связана с наличием в его внутренней сфере окислителя (иона PdIV) и восстановителей (лигандов µ-SR), что приводит к внутрисферному окислительно-восстановительному процессу. Этот процесс включает синхронный перенос двух электронов с пары координированных тиольных мостиков на ион PdIV, приводящий к разрыву мостиковых связей Pd-SR и объединению двух тиольных радикалов RS · в дисульфид R2S2. Далее в координационной сфере восстановленного палладиевого димера происходит внутримолекулярная перегруппировка лигандов ОН-, координированных ранее к PdIV, в мостиковое положение. В результате образуется соединение


Следует отметить, что в отсутствие пероксида водорода (O2 или каких-либо других окислителей) водные растворы биядерных тиолатмостиковых комплексов палладия(II) не катализируют окисление тиоаминокислоты.
Оценка эффективности каталитического действия соединения [Pd2(µ-SG)2(NH3)4]2+ на процесс окисления глутатиона (GSH) пероксидом водорода, проводимая с помощью метода высокоэффективной жидкостной хроматографии (ВЭЖХ), показала их большую (~30%) каталитическую эффективность, чем используемый в настоящее время цис-[Pt(NH3)2Cl2].
Окисление тиолов комплексами меди(I)
При внесении в водный раствор простых солей меди(II), таких как CuCl2 или CuBr2, происходит их аквотация, сопровождающаяся последующим гидролизом и образованием в растворе олигоядерных аквагидроксокомплексов меди(II).
Эти аквагидроксокомплексы меди(II) в водных растворах, содержащих тиолы, почти мгновенно восстанавливаются, образуя тиолатные комплексы меди(I) -


На Фиг.2 в качестве примера приведены результаты исследования относительной каталитической эффективности комплексов

Как можно видеть из результатов, приведенных на Фиг.2, комплексы



Окисление тиолов с использованием Cu-Pd катализаторов
Экспериментальные исследования каталитических систем с применением медно-палладиевых катализаторов



Исследования показывают, что в Pd-Cu катализаторах соотношения Pd:Cu лежат в пределах от 1:0.2 до 1:2, в зависимости от необходимости варьирования активностью работы катализатора.
Суммируя, для процессов мягкого селективного окисления тиолов GSH до GSSG, можно сделать вывод о том, что биядерные тиолатмостиковые комплексы палладия(II) выполняют основную функцию катализаторов окисления, а тиолатные комплексы меди(I) следует отнести к химическим сайтам, меняющим их каталитическую активность или, говоря иначе, управляющим их каталитической активностью.
Таким образом, сочетание функциональных биядерных палладиевых координационных соединений



Повышение каталитической эффективности палладий-медных катализаторов общей формулы



Управляющий химический сайт, образующийся

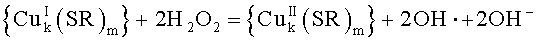
выступает в роли окислителя функционального сайта

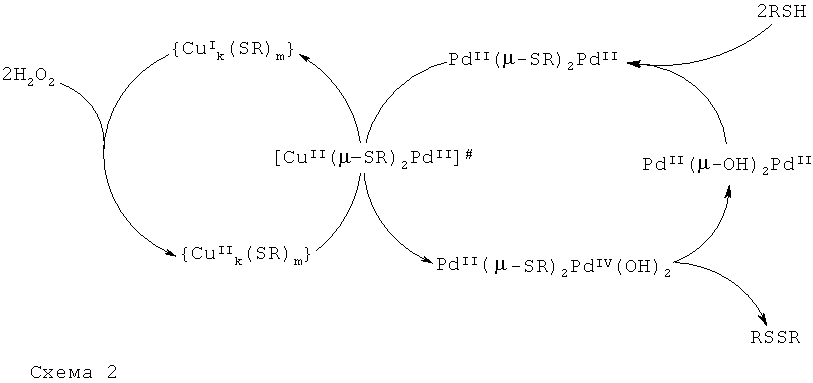
Для окислительно-восстановительных реакций координационных соединений d-элементов существуют два общих типа переходных состояний, так называемые внешнесферный и внутрисферный типы. Для внешнесферного типа внутренние координационные оболочки ионов обоих металлов не затрагиваются. Для внутрисферного типа ионы обоих металлов связываются мостиковым лигандом, общим для обеих координационных оболочек.
Результаты квантово-химических расчетов показали, что в каталитических системах на основе биядерного металлического остова


Анализ структуры граничных молекулярных орбиталей (МО) объясняет экспериментально выявленную более высокую реакционную способность каталитической системы на основе смешанных Cu-Pd координационных соединений, чем на основе биядерных комплексов, содержащих только ионы PdII.
Участие атомных d-орбиталей (d-AO) металлов в каталитических процессах приводит к снятию запретов на протекание стадии реакции по симметрии молекулярных орбиталей (МО), часто определяющих высокую энергию активации процессов. Основная идея для объяснения катализа ионами металлов для реакций, запрещенных по симметрии, состоит в том, что ионы переходных металлов имеют доступные и близколежащие по энергии d-AO. Это дает возможность частицам, координирующимся к иону металла, отдавать пару своих электронов на одни d-орбитали металла, а получать их с других d-орбиталей металла.
Анализ характера граничных МО в координационном соединении с центром CuII(µ-SR)2PdII показал, что его высшая занятая молекулярная орбиталь (ВЗМО) состоит в большой степени из d-AO меди и не пригодна для образования окисленного продукта, содержащего ион PdIV. Для осуществления процесса необходим перенос пары электронов на низшую свободную молекулярную орбиталь (НСМО) с большой долей 4d-орбиталей PdII. Чем меньше разница в энергиях этих МО, тем меньшие затраты требуются для достижения переходного состояния. В рассматриваемом координационном соединении со смешанным биметаллическим центром CuII(µ-SR)2PdII энергетическая разность между этими орбиталями составляет 2.61 эВ. В координационном соединении с центром PdII(µ-SR)2PdII разница в энергиях соответствующих занятых и свободных орбиталей значительно больше: 4.18 эВ (для PtII(µ-SR)2PtII 4.79 эВ).
Таким образом, каталитическая система для селективного окисления тиолов на основе смешанных промежуточно образуемых комплексов CuI и PdII должна обладать большей активностью, чем система на основе аналогичных комплексов PdII. Причина этого заложена на уровне энергетики d-орбиталей меди.
Образование промежуточного биметаллического центра [CuII(µ-SR)2PdII]# в цикле объясняет причину повышения каталитической эффективности катализаторов общей формулы

Предложенные катализаторы по изобретению могут сочетаться с дисульфидами N-глутамил-L-цистеинил-глицина и/или N-ацетил-L-цистеина, формируя комбинацию, обладающую как естественной биологической, так и каталитической активностью. Избыток тиола обеспечивает возможность изготовления и использования малых и ультрамалых доз непосредственно катализатора, обеспечивая его к тому же необходимым для каталитического цикла субстратом.
Свободные молекулы дисульфидов N-ацетил-цистеина и/или N-глутамил-L-цистеинил-глицина в составе препарата могут находиться как в катионной, так и в анионной форме или же в форме нейтральных частиц.
В качестве противоиона могут быть использованы неорганические ионы, например катионы натрия, лития, калия, кальция, магния, селена, марганца, цинка, ванадия и других химических элементов, или ионы органических соединений, например аминокислот, алифатические и ароматические ионы органических молекул из различных химических групп, обладающие биологической активностью (пример 12).
Комбинации по изобретению могут содержать также другие фармакологически активные соединения, в частности пуриновые и/или пиримидиновые основания, их производные или соединения на их основе (примеры 10 и 11).
Под фармакологически активным соединением подразумевается любое вещество, которое используется с терапевтическими целями, представляющее собой молекулы лекарственных или биологически активных веществ, в частности пуриновых и/или пиримидиновых оснований и соединений на их основе, например: Аденозин (9-β-D-рибофуранозиладенин), Гуанозин (9-β-D-рибофуранозилгуанидин), Дезоксиаденозин (9-β-D-дезоксирибофуранозиладенин), Дезоксигуанозин (9-β-D-дезоксирибофуранозилгуанидин), 9-β-D-рибофуранозиладенина моно-, ди-, трифосфат, 9-β-D-рибофуранозилгуанидин моно-, ди-, трифосфат, 9-β-D-дезоксирибофуранозиладенина моно-, ди-, трифосфат (кордицепин), 9-β-D-дезоксирибофуранозилгуанидин моно-, ди-, трифосфат, Цитидин (4-амино-1-[3,4-дигидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]пиримидин-2-он), 1-[(2R,4S,5R)-4-гидрокси-5-(гидрокси)тетрагидрофуран-2-ил]-5-пиридин-2,4-дион, Дезоксицитидин (4-амино-1-[4-гидрокси-5-(гидроксиметил)тетрагидрофуран-2-ил]пиримидин-2-он), Тимидин или дезокситимидин 1-[(2R,4S,5R)-4-гидрокси-5-(гидрокси)тетрагидрофуран-2-ил]-5-метилпиридин-2,4-дион, Инозин 9-[(2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-6,9-дигидро-3Н-пурин-6-он, Рибаверин: 1-(3,4-дигидрокси-5-гидроксиметил-тетрагидрофуран-2-ил)-1Н-[1,2,4]триазол-3-карбоновой кислоты амид, Занамивир (2R,3R,4S)-4-[(диаминометилиден)амино]-3-ацетамидо-2-[(1R,2R)-1,2,3-тригидроксипропил]-3,4-дигидро-2H-пиран-6-карбоновая кислота, Фармцикловир: 2-[2-(2-амино-9Н-пурин-9-ил)этил]-1,3-пропандиол диацетат(эфир), Ганцикловир (2-амино-1,9-дигидро-9-2-гидрокси-1-(гидроксиметил)этоксиметил-6Н-пурин-6-он), Зидовудин (3′-азидо-3′-дезокситимидин), Ацикловир: 2-амино-9-((2-гидроксиэтокси)метил)-1Н-пурин-6(9Н)-он, Фторурацил: 5-фтор-1Н-пиримидин-2,4-дион, 1-(2,3-Дидезокси-бета-D-глицеропент-2-енофуранозил)Тимин, Фтортиоурацил: 5-фтор-1Н-пиримидин-2-тион-4-он, Тиоурацил: 1Н-пиримидин-2-тион-4-он, S-аденозилгомоцистеин: 4[5-(6-амино-пурин-9-ил)-3,4-дигидрокси-тетрагидро-фуран-2-илметилсульфанил]-2-меркапто-бутановая кислота, S-Аденозилметионин: (2S)-2-амино-4-[[(2S,3S,4R,5R)-5-(6-аминопурин-9-ил)-3,4-дигидрокмиоксолан-2-ил]метил-метилсульфонио]бутаноат, циклический аденозинмонофосфат (цАМФ) 6-(6-амино-пурин-9-ил)-2-оксо-тетрагидро-2λ-5-фуро[3,2-d][1,2,3]d-изооксофосфинин-2,7-диол, 8-хлор-цАМФ: 6-(6-амино-8-хлор-пурин-9-ил)-2-оксо-тетрагидро-2λ-5-фуро[3,2-d][1,2,3]d-изооксофосфинин-2,7-диол, N-6,2′-O-дибутирил-8-SH-цАМФ, N-6,2′-O-дибутирил-8-SH-цАМФ, циклический гуанозинмонофосфат, cGMP, N6-монобутирил-8-S-метил-цАМФ, Формицин: 2-(7-амино-1Р-пиразоло[4,3-d]пиримидин-3-ил)-5-гидроксиметил-тетрагидрофуран-3,4-диол, Метилизогуанозин: 6-амино-1-метил-1,9-дигидро-пурин-2-он.
Фармакологически активное соединение может быть связанно с избытком дисульфидов N-ацетил-цистеина и/или N-глутамил-L-цистеинил-глицина ван-дер-ваальсовыми силами (ионными, водородными и иными нековалентными связями).
Комбинации по изобретению могут быть изготовлены согласно известным из уровня техники методам с учетом особенностей химических свойств палладиево-медных катализаторов по изобретению, дисульфидов (N-ацетил-цистеина и/или N-глутамил-L-цистеинил-глицина) и фармакологически активных веществ. Предпочтительно, когда доля катализатора по изобретению в препарате составляет от 1·10-2 до 1·10-7 г на моль дисульфида алифатического тиола - N-ацетил-цистеина и/или N-глутамил-L-цистеинил-глицина.
Согласно изобретению предложенный палладиево-медный катализатор или каталитическая комбинация по изобретению могут быть использованы для усиления терапевтической активности пуринового и/или пиримидинового основания или производного на их основе. В контексте данного описания повышение терапевтической эффективности фармакологически активного соединения представляет собой снижение разовой или курсовой дозы или снижение общей токсичности и достижение более выраженного терапевтического эффекта при принятой терапевтической дозе или меньшей для этого фармакологически активного соединения.
Палладиево-медный катализатор по изобретению, каталитическая комбинация и фармакологическая комбинация по изобретению могут быть использованы в форме фармацевтических композиций.
Для получения фармацевтических композиций по изобретению используют фармацевтически приемлемые эксципиенты. В частности, это неорганические или органические носители. Лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и т.п. могут быть использованы, например, в качестве таких носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т.п. Подходящими носителями для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар, глюкоза и т.п. Подходящими носителями для суппозиториев являются, например, природные или отвержденные масла, воски, жиры, полужидкие или жидкие полиолы и т.п.
Кроме того, фармацевтические композиции могут содержать консерванты, солюбилизаторы, стабилизаторы, смачивающие вещества, эмульгаторы, подсластители, красители, корригенты, соли для регулирования осмотического давления, буферы, маскирующие агенты или антиоксиданты и другие необходимые компоненты.
Палладиево-медный катализатор или каталитическая комбинация по изобретению и фармакологически активные вещества, эффективность которых они усиливают, могут находиться как в одной лекарственной форме, так и в отдельных лекарственных формах. Введение в отдельных лекарственных формах может быть осуществлено одновременно (одновременный прием двух твердых дозированных лекарственных форм, например таблеток, одновременная инъекция, в частности в одном шприце) либо последовательно, когда пациенту дают или вводят сначала первую лекарственную форму, а затем вторую лекарственную форму. Интервал между введением предпочтительно составляет не более 1 часа, однако может быть увеличен до тех пор, пока наблюдается синергический эффект. Оптимальный порядок введения зависит от фармакокинетики и фармакодинамики фармакологически активного вещества, эффективность которого должна быть усилена (скорость всасывания, распределение, скорость выведения, особенности клеточной, органной тропности или системной тропности) и может быть подобрана для каждого конкретного вещества индивидуально.
Количество вводимого палладиево-медного катализатора определяется массовой долей Pd и Cu в составе катализатора, которое может соответствовать или быть ниже суточной потребности в каждом металле. В противном случае количество вводимого d-металла в составе координационного соединения определяется необходимостью достижения результата лечения.
Терапевтический результат может быть достигнут при введении катализатора в количестве от 1·10-3 до 1·10-8 г на кг массы тела пациента, что в пересчете на количество дисульфида в комбинации составит от 1×10-2 до 1×10-5моль дисульфида на кг массы тела.
ПРИМЕРЫ
Далее изобретение поясняется конкретными примерами.
Пример 1. Получение катализатора Pd-Cu на основе аммиачного комплекса палладия(II) с N-глутамил-L-цистеинил-глицином (GSH)
50 мг (237 мкмоль) цис-[Pd(NH3)2Cl2] вносят в 10 мл раствора, содержащего 75 мг (244 мкмоль) GSH, и гомогенизируют в ультразвуковой ванне до образования светло-желтого раствора. К полученной системе приливают 5 мл раствора, содержащего 20 мг (117 мкмоль) CuCl2·2H2O, и корректируют рН полученного желто-зеленого раствора до 5.0-5.2 раствором гидроксида натрия (~0.01 М).
Полученный раствор катализатора может быть использован для проведения окисления водорастворимых тиолов (например: GSH или ацетилцистеина).
В полученном растворе катализатора мольное соотношение палладий-медь составляет 2:1.
Пример 2. Получение катализатора Pd-Cu на основе аммиачного комплекса палладия(II) с глутатионом и его использование для синтеза дисульфида N-глутамил-L-цистеинил-глицина (GSSG).
Стадия 1. Получение аммиачного комплекса палладия(II) с глицилцистеинилглутаматом
10 мг (47.3 мкмоль) цис-[Pd(NH3)2Cl2] на холоде диспергируют в 10-15 мл дистиллированной воды, в полученную суспензию вносят 145 мг (0.473 ммоль) GSH и перемешивают на магнитной мешалке до образования светло-желтого гомогенного раствора.
Стадия 2. Получение Pd-Cu катализатора
В ранее полученную реакционную смесь приливают 5 мл раствора, содержащего 8.06 мг (47.3 мкмоль) дигидрата хлорида меди(II). рН полученного желто-зеленого раствора катализатора доводят до значения 5.5-5.8 0.01 М раствором гидроксида натрия.
Стадия 3. Использование каталитической системы Pd-Cu для синтеза GSSG
В стеклянный стакан отвешивают 29.09 г (0.946 ммоль) GSH, приливают при перемешивании 150-200 мл дистиллированной воды, охлаждают до 10-15°C. Отдельно растворяют в 50-60 мл дистиллированной воды 3.78 г NaOH (0.946 ммоль) и полученный раствор приливают к суспензии GSH при интенсивном перемешивании и не позволяя реакционной массе разогреваться выше 15-20°C и перемешивают до образования прозрачного гомогенного раствора. рН реакционной смеси корректируют до 5.5-5.8 0.1 М раствором гидроксида натрия. В полученную реакционную систему приливают ранее приготовленный раствор катализатора и небольшими порциями приливают 50 мл 1 М свежеприготовленного раствора пероксида водорода при интенсивном перемешивании, не допуская нагревания реакционной смеси свыше 15°C. Полноту протекания реакции контролируют методом ВЭЖХ.
После окончания реакции и стерилизующей фильтрации раствор замораживают и подвергают вакуумно-сублимационной (лиофильной) сушке.
В полученном препарате мольное соотношение «натриевая соль дисульфида N-глутамил-L-цистеинил-глицина - палладий - медь» составляет 1000-1-1.
Пример 3. Получение катализатора Pd-Cu на основе аммиачного комплекса палладия(II) с N-глутамил-L-цистеинил-глицином (GSH) и его использование для получения глицилцистеинилглутамат рибофуранозилгипоксантина-динатрия
Стадия 1. Получение аммиачного комплекса палладия(II) с GSH
20 мг (94.6 мкмоль) цис-[Pd(NH3)2Cl2] на холоде диспергируют в 20 мл дистиллированной воды, в полученную суспензию вносят 30 мг GSH (97.6 мкмоль) и перемешивают до образования светло-желтого гомогенного раствора.
Стадия 2. Получение Pd-Cu катализатора
В ранее полученную реакционную смесь приливают 5 мл раствора, содержащего 14.52 мг (85.2 мкмоль) дигидрата хлорида меди(II). рН полученного желто-зеленого раствора катализатора доводят до значения 5.5-6.0 0.01 М раствором гидроксида натрия.
Стадия 3. Использование каталитической системы Pd-Cu для получения N-глутамил-L-цистеинил-глицина рибофуранозилгипоксантина-динатрия
В стеклянный стакан отвешивают 58.19 г (0.189 моль) GSH, приливают при перемешивании 200 мл дистиллированной воды, охлаждают до 10-15°C. Отдельно растворяют в 50-60 мл дистиллированной воды 7.57 г NaOH (0.189 моль) и полученный раствор приливают к суспензии GSH при интенсивном перемешивании и не позволяя реакционной массе разогреваться выше 15-20°C и перемешивают до образования прозрачного гомогенного раствора. Если необходимо, рН реакционной смеси корректируют до 5.5-6.0 0.01 М раствором гидроксида натрия. В полученную реакционную систему приливают ранее приготовленный раствор катализатора, стакан переносят на ледяную баню (5-10°С) и небольшими порциями, в течение 45-60 мин приливают 100-102 мл (~0.1 моля) 1 М свежеприготовленного раствора пероксида водорода при интенсивном перемешивании. Полноту протекания реакции контролируют методом ВЭЖХ.
Отдельно в 150 мл горячей дистиллированной воды (60-70°C) растворяют 25.38 г (0.095 моль) рибофуранозилгипоксантина (инозин). После полного растворения раствор охлаждают до комнатной температуры и приливают к реакционной смеси. После стерилизующей фильтрации раствор замораживают и подвергают вакуумно-сублимационной (лиофильной) сушке.
В полученном препарате мольное соотношение GSSG-инозин-палладий-медь составляет 1000-1000-1-0.9.
Пример 4. Получение катализатора Pd-Cu на основе аммиачного производного биядерного координационного соединения Pd(II) с N-ацетил-L-цистеином
120 мг (568 мкмоль) цис-[Pd(NH3)2Cl2] диспергируют в ультразвуковой ванне в 20 мл водного раствора N-ацетил-L-цистеина (648.5 мг, 3.97 ммоль) до образования желтого гомогенного раствора. В полученную систему приливают 5 мл раствора, содержащего 48.4 мг (284 мкмоль) CuCl2·2H2O. Выпавший белый осадок растворяется при подщелачивании реакционной системы до рН 4.5-5.0 0.1 М раствором гидроксида натрия.
Полученный желто-зеленый раствор катализатора может быть использован для проведения окисления водорастворимых тиолов (например: восстановленного глутатиона или ацетилцистеина) или лиофилизирован для дальнейшего использования.
В полученном растворе катализатора мольное соотношение количеств палладий-медь составляет 2:1.
Пример 5. Получение катализатора Pd-Cu на основе аммиачного производного биядерного координационного соединения Pd(II) с N-ацетил-L-цистеином и его использование для синтеза натриевой соли дисульфида N-ацетил-L-цистеина
Стадия 1. Получение аммиачного комплекса палладия(II) с N-ацетил-L-цистеином
16 мг (75.7 мкмоль) цис-[Pd(NH3)2Cl2] диспергируют в ультразвуковой ванне в 20 мл водного раствора N-ацетил-L-цистеина (123.5 мг, 757 мкмоль) до образования желтого гомогенного раствора.
Стадия 2. Получение Pd-Cu катализатора
К полученному раствору приливают 5 мл раствора, содержащего 6.5 мг (37.8 мкмоля) CuCl2·2H2O. Полученный раствор катализатора подщелачивают до рН 4.5-5.0 насыщенным раствором гидроксида лития.
Стадия 3. Использование каталитической системы Pd-Cu для получения натриевой соли N-ацетил-L-цистеина
В 250 мл дистиллированной воды растворяют 24.7 г (0.151 моль) N-ацетил-L-цистеина, приливают раствор катализатора и в полученный раствор при интенсивном перемешивании вносят 6.35 г (0.151 моль) гидроксида натрия. После полного растворения NaOH в H2O рН раствора корректируют до 5.5-6.0 насыщенным раствором гидроксида натрия и охлаждают до 10-15°C.
В полученный раствор на холоде небольшими порциями и при интенсивном перемешивании вносят 1 М раствор пероксида водорода (~80 мл), не допуская повышения температуры реакционной смеси свыше 15-20°C. Полноту протекания реакции контролируют методом ВЭЖХ.
После окончания реакции и стерилизующей фильтрации раствор замораживают и подвергают вакуумно-сублимационной (лиофильной) сушке.
В полученном препарате мольное соотношение «натриевая соль дисульфида N-ацетил-L-цистеина - палладий - медь» составляет 1000-1-0.5.
Пример 6. Получение катализатора Pd-Cu на основе аммиачного производного биядерного координационного соединения Pd(II) с N-ацетил-L-цистеином и его использование для синтеза литиевой соли дисульфида N-ацетил-L-цистеина
Стадия 1. Получение аммиачного комплекса палладия(II) с N-ацетил-L-цистеином
16 мг (75.7 мкмоль) цис-[Pd(NH3)2Cl2] диспергируют в ультразвуковой ванне в 20 мл водного раствора N-ацетил-L-цистеина (123.5 мг, 757 мкмоль) до образования желтого гомогенного раствора.
Стадия 2. Получение Pd-Cu катализатора
К полученному раствору приливают 5 мл раствора, содержащего 6.5 мг (37.8 мкмоля) CuCl2·2H2O. Полученный раствор катализатора подщелачивают до рН 4.5-5.0 насыщенным раствором гидроксида лития.
Стадия 3. Использование каталитической системы Pd-Cu для получения литиевой соли N-ацетил-L-цистеина
В 250 мл дистиллированной воды растворяют 24.7 г (0.151 моль) N-ацетил-L-цистеина, приливают раствор катализатора и в полученный раствор при интенсивном перемешивании вносят 6.35 г (0.151 моль) моногидрата гидроксида лития. После полного растворения LiOH·H2O pH раствора корректируют до 5.5-6.0 насыщенным раствором гидроксида лития и охлаждают до 10-15°C.
В полученный раствор на холоде небольшими порциями и при интенсивном перемешивании вносят 1 М раствор пероксида водорода (~80 мл), не допуская повышения температуры реакционной смеси свыше 15-20°C. Полноту протекания реакции контролируют методом ВЭЖХ.
После окончания реакции и стерилизующей фильтрации раствор замораживают и подвергают вакуумно-сублимационной (лиофильной) сушке.
В полученном препарате мольное соотношение «литиевая соль дисульфида N-ацетил-L-цистеина - палладий - медь» составляет 1000-1-0.5.
Пример 7. Получение катализатора Pd-Cu на основе аммиачного производного биядерного координационного соединения Pd(II) с N-глутамил-L-цистеинил-глицином (GSH) и его использование для синтеза литиевой соли дисульфида N-глутамил-L-цистеинил-глицина
Стадия 1. Получение аммиачного комплекса палладия(II) с GSH
21.1 мг (100 мкмоль) цис-[Pd(NH3)2Cl2] диспергируют в ультразвуковой ванне в 20 мл водного раствора GSH (307.5 мг, 1 ммоль) до образования желтого гомогенного раствора.
Стадия 2. Получение Pd-Cu катализатора
К полученному раствору приливают 5 мл раствора, содержащего 34.1 мг (200 мкмоль) CuCl2·2H2O. Полученный раствор катализатора подщелачивают до pH 5.0-5.5 насыщенным раствором гидроксида лития.
Стадия 3. Использование каталитической системы Pd-Cu для получения литиевой соли дисульфида N-глутамил-L-цистеинил-глицина
В 400-500 мл дистиллированной воды растворяют 61.5 г (0.2 моль) GSH, приливают раствор катализатора и в полученный раствор при интенсивном перемешивании вносят 8.39 г (0.2 моль) моногидрата гидроксида лития. После полного растворения LiOH·H2O pH раствора корректируют до 5.5-5.8 насыщенным раствором гидроксида лития и охлаждают до 10-15°C.
В полученный раствор на холоде небольшими порциями и при интенсивном перемешивании вносят 1 М раствор пероксида водорода (~110 мл), не допуская повышения температуры реакционной смеси свыше 15°C. Полноту протекания реакции контролируют методом ВЭЖХ.
После окончания реакции и стерилизующей фильтрации раствор замораживают и подвергают вакуумно-сублимационной (лиофильной) сушке.
В полученном препарате мольное соотношение литиевая соль дисульфида N-глутамил-L-цистеинил-глицина - палладий - медь составляет 1000-1-2.
Пример 8. Получение катализатора на основе аммиачного производного биядерного координационного соединения Pd(II) с N-глутамил-L-цистеинил-глицином (GSH) и его использование для синтеза дисульфида глицилцистеинилглутамата 1-[(2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-1Н-1,2,4-триазол-3-карбоксамида (GSSG-рибавирин)
Стадия 1. Получение аммиачного комплекса палладия(II) с GSH
20 мг (94.6 мкмоль) цис-[Pd(NH3)2Cl2] диспергируют в ультразвуковой ванне в 20 мл водного раствора GSH (291 мг, 947 мкмоль) до образования желтого гомогенного раствора.
Стадия 2. Получение Pd-Cu катализатора
К полученному раствору приливают 5 мл раствора, содержащего 24.2 мг (142 мкмоль) CuCl2·2H2O. Полученный раствор катализатора подщелачивают до рН 5.5-5.8 0.01 М раствором гидроксида натрия.
Стадия 3. Использование каталитической системы Pd-Cu для получения GSSG-рибаверина
В 400-500 мл дистиллированной воды растворяют 87.3 г (0.284 моль) GSH, при интенсивном перемешивании приливают раствор катализатора и раствор 11.35 гр (0.284 моль) гидроксида натрия в 50 мл воды. рН раствора корректируют до 5.5-5.8 0.1 М раствором гидроксида натрия и охлаждают до 10-15°C.
В полученный раствор на холоде небольшими порциями и при интенсивном перемешивании вносят 1 М раствор пероксида водорода (~150 мл), не допуская повышения температуры реакционной смеси свыше 15°C. Полноту протекания реакции контролируют методом ВЭЖХ.
Отдельно в 100-150 мл горячей (60-70°C) дистиллированной воды растворяют 34.65 г (0.142 моль) рибавирина. После полного растворения раствор охлаждают до комнатной температуры и приливают к реакционной смеси.
После окончания реакции и стерилизующей фильтрации раствор замораживают и подвергают вакуумно-сублимационной (лиофильной) сушке.
В полученном препарате мольное соотношение натриевая соль GSSG-рибавирина - палладий - медь составляет 1000-1000-1-1.5.
Пример 9. Сравнительная оценка биологической активности и токсичности координационного соединения платины и палладиево-медного катализатора при воздействии на рост клеток
Соединения платины имеют фармакологическую активность, обусловленную каталитическим действием в реакциях окислительной модификации сульфгидрильных групп молекул пептидной природы, что лежит в основе стимулирующего действия на продукцию цитокинов клеточными эффекторами иммунной системы, избирательного ингибирования реакций множественной лекарственной устойчивости к антибиотикам, способности подавлять развитие аутоиммунных реакций, лежащих в основе многих хронических социально значимых заболеваний - псориаз, нейродегенеративные и вирусные заболевания.
Таким образом, соединениям платины присущ ряд свойств, востребованных в фармакологических решениях и обусловленных каталитическим действием в реакциях окислительной модификации сульфгидрильных групп молекул пептидной природы. Однако соединения платины отличает высокая токсичность, механизм которой не связан с каталитической активностью. Токсичность химических соединений платины носит острый характер, т.е. проявляется относительно быстро после введения или попадания содержащего платину вещества в организм, если превышена предельно допустимая концентрация платины. При введении в организм платины в составе веществ ниже предельно допустимой концентрации может происходить постепенное накопление платины в тканях различных органов. В этом случае токсическое действие соединений платины проявляется позже или с характерной картиной отравления не проявляется вообще, однако мутагенное действие платины может служить причиной развития злокачественных новообразований.
Синтезированные катализаторы на основе координационных соединений алифатических тиолов (N-глутамил-L-цистеинил-глицином - GSH, N-ацетил-L-цистеином) и d-металлов палладия и меди (примеры 1, 4) также характеризует каталитическая активность в химической реакции окисления тиолов в составе молекул пептидной природы и фармакологически востребованных тиолсодержащих молекул, свойственная координационным соединениям платины. Соединения палладия и меди не токсичны в сравнении с соединениями платины, им не присущи мутагенное и тератогенное действие. Учитывая, что каталитическая активность соединений платины связана с востребованными фармакологической эффектами (исключая химиотерапию онкологических заболеваний, где востребовано токсическое действие соединений платины), необходимо было сопоставить биологические эффекты координационных соединений алифатических тиолов GSH, N-ацетил-L-цистеина и d-металлов палладия и меди с биологической активностью координационных соединений платины. В качестве объекта воздействия координационных соединений были выбраны клетки А431, на которых относительно полно исследованы биохимические аспекты действия координационных соединений платины с алифатическими тиолами. Характер клеточного ответа позволил оценить схожесть и/или различия в действии на клетки катализатора на основе координационных соединений GSH и d-металлов палладия и меди, N-ацетил-L-цистеина и d-металлов палладия и меди, окисленного глутатиона и платины.
Цель работы - изучение влияния катализатора на основе координационных соединений алифатических тиолов GSH, N-ацетил-L-цистеина и d-металлов палладия и меди, координационного соединения окисленного глутатиона и платины на рост культуры клеток.
В качестве исследуемых соединений использовались катализатор на основе координационного соединения алифатического тиола с N-глутамил-L-цистеинил-глицином и d-металлов палладия и меди (соединение №1, синтезировано в соответствии с описанием в примерах 1 и 2), катализатор на основе координационного соединения N-ацетил-L-цистеина и d-металлов палладия и меди (соединение №2, синтезировано в соответствии с описанием в примерах 4, 5), координационное соединение окисленного глутатиона и цисплатина (соединение №3, синтезировано в соответствии с методикой, описанной в тексте патента 2153350).
Приготовление препаратов для проведения исследования
Исследуемые соединения хранили при +4°С; непосредственно перед началом эксперимента вещества растворяли в деионизированной воде (super Q). Концентрация исходного раствора в 1000 и более раз превышает концентрации, используемые в эксперименте. Приготовленный концентрированный раствор хранится не более 5 часов при +4°С.
Путь введения
Соединения добавляли в среду культивирования клеток до исследуемой конечной концентрации.
Число доз
В одной из серий экспериментов препараты добавляются к клеткам однократно и клетки инкубировали в течение 48 часов.
В другой серии на указанный период времени экспериментов препараты добавляются к клеткам раз в сутки, через 24 часа пятикратно клетки инкубировали в течение 120 часов. В контрольных сериях вносили соответствующий объем физраствора.
Концентрации
Все соединения вносили до конечной концентрации 0.15 мкмоль/мл (концентрация рассчитана по содержанию избытка дисульфида алифатического тиола).
Критерии оценки действия
Определяли количество живых и погибших клеток в культуре через 24 и 48 часов в первой серии экспериментов и через 24, 48, 72, 96, 120 часов во второй серии экспериментов.
Использованная линия клеток
Клетки эпидермоидной карциномы человека линии А431, полученные из Всероссийской коллекции клеточных культур (Институт цитологии РАН, С.-Петербург).
Условия культивирования клеток
Клетки культивируются в CO2-инкубаторе (New Brunswick Scientific) при +37°С и при содержании CO2 5%. В этих условиях клетки выращиваются до монослойной культуры и подвергаются действию исследуемых соединений.
Среда для культивирования клеток
Для культивирования клеток используется среда DMEM (ООО "ПанЭко", Москва) с добавлением гентамицина К (80 мг/л), L-глутамина (300 мг/л) и фетальной сыворотки (РАА, Австрия) до конечной концентрации 10%. За сутки до начала эксперимента равные количества клеток (1000/2.5 мкл-400×10-3/мл) переводят в среду с пониженным содержанием сыворотки - 0,5%.
Динамика роста клеток
Клетки А431 высевали на пластиковые чашки Петри (Nunc) в концентрации 10000/см2, через сутки при достижении ими 10-15% монослоя подвергали действию исследуемых препаратов.
Подготовка клеток для окраски
Среду культивирования клеток А431 собирали в пробирки (Falkon) для полного анализа открепившихся мертвых клеток, а клетки на чашках Петри промывали PBS (который объединяли с собранной средой) и обрабатывали раствором трипсина 0.25% в Версене (ПанЭко) около 10 мин при комнатной температуре до открепления клеток. Далее клетки суспендировали пипетированием автоматической пипеткой и объединяли с собранной ранее средой. Пробы центрифугировали 5 мин 400 g при комнатной температуре, супернатант удаляли, а осадок ресуспендировали в фосфатно-солевом буфере PBS рН 7.4.
Окраска клеток иодидом пропидия
Иодид пропидия добавляли к суспензии клеток до концентрации 50 мкг/мл за 5-10 мин до измерения на проточном цитофлуориметре Bruker ACR 1000. Этот краситель способен проникать через поврежденную клеточную мембрану, и окрашенные клетки являются мертвыми.
Методы статистической обработки результатов
Статистическую обработку полученных результатов проводили на персональном компьютере с помощью пакета прикладных программ «STATISTICA 6,0».
Результаты влияния исследуемых соединений на пролиферативную активность и гибель клеток
Согласно полученным данным все исследуемые соединения при однократном введении в среду культивирования клеток стимулируют пролиферативную активность (табл.1).
Полученные данные свидетельствуют о том, что при однократном воздействии исследуемых соединений на культуру клеток имеет место стимуляция пролиферативной активности в сравнении с контрольными сериями. Выявленный эффект сопоставим у всех исследуемых веществ. По критерию количества погибших клеток отмечено уменьшение их количества в два и более раза при воздействии соединений №1 и №2 в сравнении с контролем и несколько меньшее при воздействии соединения №3 (50-60%). Схожесть результатов ответа культуры клеток при воздействии различных координационных соединений является одним из свидетельств общности механизма воздействия и клеточного ответа на него. Вносимые однократно количества металла относительно малы. Если учесть, что цисплатин токсичен для клеток, то, очевидно, недостаточно однократного введения, чтобы заметить эффект комплекса алифатического тиола и цисплатина и собственно цисплатина, который может освобождаться при распаде координационного соединения. В этой связи был поставлен эксперимент 120-часовой инкубации клеток с пятикратным воздействием на клетки каждого исследуемого соединения раз в 24 часа. Результаты приведены в таблице 2.
Результаты экспериментов показывают практически одинаковую пролиферативную активность клеток через сутки как в контроле, так и при воздействии всех исследуемых соединений. Сохраняется и выявленная ранее закономерность меньшей гибели клеток через сутки при воздействии исследуемых соединений в сравнении с контролем. Однако воздействие на клетки координационным соединением дисульфида глутатиона с цисплатином на вторые и последующие сутки приводит к падению пролиферативной активности культуры и усилению гибели клеток, практически двукратно превосходя значения аналогичных показателей в контроле. Напротив, воздействие на культуры клеток соединения №1 и соединения №2 предопределило практически линейный ежесуточный прирост количества клеток и относительно постоянную величину их гибели.
Совокупность полученных результатов указывает на определенные закономерности, которые, с одной стороны, определяются активностью исследуемого соединения в целом, с другой, отражают особенности воздействия на биологический объект металлов, образующих координационные соединения. Так, схожая динамика роста и гибели клеток через 24 часа при действии всех исследуемых координационных соединений указывает на схожесть механизма их воздействия независимо от металла и алифатического тиола. Однако из культуральной среды координационные соединения не удаляются, а это означает, что всякое последующее введение увеличивает их концентрацию. Если учесть, что лиганд является активно метаболизируемой структурой, то логичным является предположение об освобождении комплекса металла. Цисплатин представляет собой стойкую молекулу. Будучи нетоксичным в составе комплексного соединения, цисплатин, при деградации лиганда, проявляет свое биологическое цитотоксическое действие. В этой связи мы наблюдаем активный прирост погибших клеток и низкую пролиферативную активность. Не исключено, что помимо цисплатина в замкнутой системе цитотоксическое воздействие оказывают модифицированные цисплатином нуклеотиды при их участии в реакциях синтеза нуклеиновых кислот.
Биологическая активность d-металлов палладия и меди отлична от цисплатина. Палладий и его соединения представляют собой биологически относительно нейтральную молекулу. Медь является биоэлементом и активно используется клетками в составе различных ферментов, участвующих в реакциях освобождения энергии, детоксикации, физиологически упорядоченного синтеза и распада биомолекул. В этой связи в условиях эксперимента выявлены биологически позитивные эффекты катализатора на основе координационных соединений алифатических тиолов d-металлов палладия и меди.
Заключение. Таким образом, катализаторы на основе координационных соединений алифатических тиолов N-глутамил-L-цистеинил-глицина и N-ацетил-L-цистеина с d-металлами палладием и медью характеризует схожий механизм воздействия на клетки, присущий координационному соединению окисленного глутатиона и цисплатина, однако у них отсутствует токсичность, присущая соединениям платины. В этой связи катализаторы на основе комплексных соединений алифатических тиолов координационных соединений алифатических тиолов N-глутамил-L-цистеинил-глицина и N-ацетил-L-цистеина с d-металлами палладием и медью могут быть использованы в фармакологических решениях с лекарственными средствами для терапии различной длительности, при которой востребована каталитическая активность в реакциях окислительной модификации тиолов до дисульфидов в составе молекул пептидной природы.
Пример 10. Сравнительная оценка способности координационного соединения окисленного глутатиона и цисплатина и способности катализатора на основе координационного соединения N-глутамил-L-цистеинил-глицина палладия и меди потенцировать противовирусную активность инозина
В патентах RU 2153350, RU 2153351 рассматривается фармакологическое решение по потенцированию противовирусной активности инозина предпочтительно с использованием координационного соединения цисплатина и окисленного глутатиона. Эффект потенцирования достигается предположительно за счет способности соединения цисплатина и окисленного глутатиона катализировать комплекс реакций окислительной модификации мишени, повышая ее сродство к воздействию инозина. Если механизм потенцирования противовирусного эффекта инозина связан с каталитической активностью координационных соединений, то средство по изобретению должно оказывать подобное более выраженное действие, так как их отличает более высокая каталитическая активность в сравнении с координационным соединением цисплатина и окисленного глутатиона.
Цель исследования: сравнительная оценка противовирусной активности инозина в сочетании с координационным соединением окисленного глутатиона и цисплатина и катализатором на основе координационного соединения N-глутамил-L-цистеинил-глицина палладия и меди.
Исследуемые соединения
Фармакологическая композиция (ФК) №1 - N-глутамил-L-цистеинил-глицина рибофуранозилгипоксантина-динатрия, содержащий средство по изобретению (синтез приведен в примере 3)
Фармакологическая композиция (ФК) №2 - N-глутамил-L-цистеинил-глицина рибофуранозилгипоксантина-динатрия, содержащий координационное соединение цисплатина и окисленного глутатиона (синтез осуществлен в соответствии с методикой, приведенной в патентах 2153350, 2153351)
Экспериментальная модель
- вирус венесуэльского энцефалита лошадей (ВЭЛ) - патогенный штамм Тринидад. Накопление вируссодержащего материала для последующего заражения лабораторных животных осуществляли с использованием 9-11-дневных развивающихся куриных эмбрионов - 30-50 шт. Первоначально готовили пять последовательных десятикратных разведений вируссодержащей суспензии. По 0.2 мл каждого разведения вируссодержащей суспензии вносили в аллантоисную полость развивающихся куриных эмбрионов. Место инъекции вируссодержащей суспензии покрывали расплавленным парафином. Затем развивающиеся куриные эмбрионы помещают в термостат при температуре (37±0.5)°C на 18 ч, периодически оценивая их жизнеспособность с помощью овоскопа. По истечении времени инкубации в термостате оценивали жизнеспособность развивающихся куриных эмбрионов и из «тушек» живых эмбрионов готовили 10% суспензию вируссодержащего материала с использованием физиологического раствора с добавлением антибиотиков (пенициллин из расчета 100 ЕД на 1 мл, стрептомицин - 200 ЕД на 1 мл). Полученную суспензию центрифугировали в течение 10 мин при 1.5-2.0 тыс. об/мин и температуре плюс (3±0.5)°C. Надосадочную жидкость разливали во флаконы объемом 1.0 мл и использовали для дальнейшего заражения экспериментальных животных мышей. Исходный титр вируса 107-108ЛД50/мл;
- вирус лихорадки долины Рифт (ЛДР) - патогенный штамм 8-87. Накопление вируссодержащего материала для заражения лабораторных животных осуществляют с использование 3-5-дневных мышей-сосунков - 10-15 гол. Первоначально готовили пять последовательных десятикратных разведений вируссодержащего материала. По 0.02 мл каждого разведения вводили в мозг мышам-сосункам, за которыми устанавливали наблюдение в течение 24-48 ч, по окончании которого животных забивали эфиром, извлекали головной мозг и депонировали его по три образца в пенициллиновые флаконы, которые хранили в морозильной камере при температуре минус (20±0.5)°С. В дальнейшем 10% суспензию головного мозга использовали в качестве вируссодержащего материала. Исходный титр вируса 105-106 ЛД50/мл;
- вирус клещевого энцефалита (КЭ) - патогенный штамм Абсетаров. Накопление вируссодержащего материала для заражения лабораторных животных проводили на мышах-сосунках. Первоначально готовили пять последовательных десятикратных разведений вируссодержащего материала, которым служил центрифугат 10% суспензии мозга зараженных ранее мышей или регидратированный из лиофильного состояния вируссодержащий материал. По 0.02 мл каждого разведения вводили в мозг мышам-сосункам, за которыми устанавливали наблюдение в течение 24-48 ч, по окончании которого животных забивали эфиром, извлекали головной мозг и депонировали его по три образца в пенициллиновые флаконы, которые хранили в морозильной камере при температуре минус (20±0.5)°С. В дальнейшем 10% суспензию головного мозга использовали в качестве вируссодержащего материала. Исходный титр вируса 102-103 ЛД50/мл.
Исследования по оценке эффективности фармакологических композиций №1 и №2 выполнены на белых неинбредных мышах-самцах массой 16-18 г (360 голов).
В исследования брали животных, в обязательном порядке выдержавших карантин в течение 1 нед в клинике экспериментальных биологических моделей НИИЦ (МБЗ) ФГУ «ГосНИИИ ВМ Минобороны России».
Применительно к каждому возбудителю величину ЛД50 определяли на белых неинбредных мышах с расчетом этого критерия по методу Кербера в модификации И.П.Ашмарина и А.А.Воробьева.
Эффективность изучаемых препаратов определяли по сопоставлению величин показателей выживаемости животных в подопытных (получавших соответствующие препараты) и контрольных группах. Процент выживших животных в подопытных и контрольных группах определяли по таблицам Генеса B.C. При этом наблюдение за инфицированными животными проводили в течение 21 сут, ежедневно регистрируя число живых и павших в подопытных и контрольных группах.
Методы статистической обработки результатов
Статистическую обработку результатов экспериментов проводили на персональном компьютере, используя специальные программы, реализующие традиционные статистические методы.
Результаты изучения противовирусной активности фармакологических композиций №1 и №2
На всех экспериментальных моделях ООВИ эффективность фармакологических композиций №1 и №2 оценивали при их применении по двум схемам:
- за 24 ч до заражения, одновременно с заражением, через 24 ч, 48 ч и 72 ч после заражения (схема 1 - экстренной профилактики);
- одновременно с заражением, через 24 ч, 48 ч и 72 ч после заражения (схема 2 - раннего этиотропного лечения).
Фармакологические композиции №1 и №2 вводили подкожно в объеме 0.5 мл в разовой дозе 30 мг/кг массы тела (10 мкг/мышь).
Эффективность фармакологических композиций №1 и №2 при экспериментальной инфекции ВЭЛ
Результаты проведенных исследований приведены в таблице 3.
Представленные в таблице 3 данные свидетельствуют о том, что оцениваемые препараты оказывали протективное действие в отношении экспериментального ВЭЛ. Наиболее эффективной в данных условиях оказалась ФМ №1. При этом если препарат применяли по схеме 1 (за 24 ч до заражения, одновременно с заражением, через 24 ч, 48 ч и 72 ч после заражения), то вне зависимости от заражающей дозы вируса ВЭЛ выживаемость инфицированных мышей находилась на уровне 100% на фоне 100% летальности в контроле. Если же препарат применяли по схеме 2 (одновременно с заражением, через 24 ч, 48 ч и 72 ч после заражения), то в этих условиях в зависимости от заражающей дозы возбудителя протективный эффект находился на уровне 100% (при заражении возбудителем в дозе 12 ЛД50) и 100% (при заражении возбудителем в дозе 2 ЛД50). При применении ФК №2 показатели защиты в зависимости от заражающей дозы возбудителя были на 40-60% ниже, чем при применении ФК №1.
Таким образом, на основании проведенных исследований можно заключить, что среди оцененных препаратов наиболее эффективной в отношении экспериментальной инфекции ВЭЛ оказалась ФК №1, включающая средство по изобретению. ФК №1, примененная по схеме экстренной профилактики, обеспечивала защиту 100% инфицированных животных на фоне 100% летальности в контроле.
Эффективность фармакологических композиций №1 и №2 пои экспериментальной вирусной инфекции ЛДР
Результаты проведенных исследований приведены в таблице 4.
Представленные в таблице 4 данные свидетельствуют о том, что наилучший протективный эффект в отношении ЛДР был получен при применении ФК №1. Вне зависимости от схемы применения препарат обеспечивал 100% защиту инфицированных мышей на фоне 100% летальности в контроле. Фармакологическая композиция №2 в данных условиях была практически неэффективна.
Эффективность фармакологических композиций №1 и №2 при экспериментальной вирусной инфекции - экспериментальном клещевом энцефалите
Результаты исследований приведены в таблице 5.
Как следует из представленных данных, все оцененные препараты при применении у животных, инфицированных возбудителем в дозе 2 ЛД50, проявляли практически одинаковую защитную эффективность, обеспечивая выживаемость 80-100% инфицированных мышей на фоне их 100% летальности в контроле. В то же время если препараты применяли у животных, инфицированных возбудителем в дозе 12 ЛД50, то в этих условиях ФК №1 оказалась более эффективной, чем ФК №2.
Заключение
Совокупность результатов проведенных исследований позволяет сделать заключение о том, что фармакологические композиции №1 и №2 обладают противовирусной активностью. Однако ФК №1, включающая средство по изобретению, обладала более выраженной противовирусной активностью в сравнении с фармакологической композицией №2, включающей координационное соединение окисленного глутатиона и цисплатины.
Пример 11. Определение способности средства по изобретению потенцировать противовирусную активность рибавирина
Рибавирин является специфическим противовирусным препаратом. Его воздействие на вирусинфицированные клетки подобно инозину. Средство по изобретению, способное стимулировать процессы окислительной модификации белков, потенцировало противовирусный эффект инозина. Подобие противовирусного действия инозина и рибавирина позволяет предполагать возможность потенцирования противовирусного эффекта 1-[(2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-1H-1,2,4-триазол-3-карбоксамида (действующее начало фармакопейного препарата Рибавирин®) средством по изобретению.
Цель исследования: оценить способность средства по изобретению потенцировать противовирусную активность 1-[(2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-1Н-1,2,4-триазол-3-карбоксамида.
Исследуемые соединения
Фармакологическая композиция 1-[(2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-1H-1,2,4-триазол-3-карбоксамид в сочетании со средством по изобретению (синтез приведен в примере 8) - препарат №1.
Фармакопейный противовирусный препарат Рибавирин® производства «Канонфарма Продакшн» (Россия) - препарат №2.
Экспериментальная модель
Исследования выполнены на экспериментальных моделях, описанных в примере 10. Заражающая доза возбудителя в каждой модели соответствовала 10 ЛД50.
Результаты изучения противовирусной активности препаратов №1 и №2
На всех экспериментальных моделях ООВИ эффективность препаратов №1 и №2 оценивали при их применении по двум схемам:
- за 24 ч до заражения, одновременно с заражением, через 24 ч, 48 ч и 72 ч после заражения (схема 1 - экстренной профилактики);
- одновременно с заражением, через 24 ч, 48 ч и 72 ч после заражения (схема 2 - раннего этиотропного лечения).
Препараты №1 и №2 вводили подкожно в объеме 0.5 мл.
Препарат №1 в разовой дозе 30 мг/кг массы тела (10 мкг/мышь).
Препарат №2 в разовой дозе 30 мг/кг массы тела (10 мкг/мышь).
Эффективность препаратов №1 и №2 при экспериментальной вирусной инфекции - Венесуэльский энцефалит лошадей (ВЭЛ)
Результаты проведенных исследований приведены в таблице 6.
Представленные в таблице 6 данные свидетельствуют о том, что оцениваемые препараты по формируемому при их введении в организм животных протективному эффекту в отношении экспериментальной инфекции ВЭЛ отличались друг от друга.
Препарат №1 обеспечивал выживаемость инфицированных мышей на уровне 80% на фоне 100% летальности в контроле (р<0.05) при обеих схемах введения, тогда как аналогичные эффекты препарата №2 (рибавирина) не превышали 50%.
Таким образом, на основании проведенных исследований можно заключить, что препарат №1, представляющий собой действующее начало фармакопейного препарата Рибавирин®, в сочетании со средством по изобретению по протективной и терапевтической эффективности в отношении экспериментальной инфекции ВЭЛ в 1.5-2.0 раза превосходил действие фармакопейного препарата Рибавирин®.
Эффективность препаратов №1 и №2 при экспериментальной вирусной инфекции - Лихорадка долины Рифт (ЛДР)
Результаты проведенных исследований приведены в таблице 7.
Представленные в таблице 7 данные свидетельствуют о том, что в отношении экспериментальной инфекции ЛДР протективное и терапевтическое действие препарата №1 проявлялось в большей степени, чем препарата №2. В частности, защитный эффект препарата №1 составил 70% на фоне 30% у препарата №2 и 100% летальности в контроле.
Таким образом, на основании проведенных исследований можно заключить, что препарат №1, представляющий собой действующее начало фармакопейного препарата Рибавирин®, в сочетании со средством по изобретению по протективной и терапевтической эффективности в отношении экспериментальной вирусной инфекции более чем в 2 раза превосходил действие фармакопейного препарата Рибавирин®.
Эффективность препаратов №1 и №2 при экспериментальной вирусной инфекции - Клещевой энцефалит (КЭ)
Результаты проведенных исследований приведены в таблице 8.
Как следует из представленных данных, препарат №1 при применении у животных, инфицированных возбудителем в дозе 10 ЛД50, вне зависимости от использованной схемы проявлял и практически одинаковую защитную эффективность, обеспечивая выживаемость 70-80% инфицированных мышей на фоне их 100% летальности в контроле. В то же время если у инфицированных животных применяли препарат №2, то его эффективность оказалась менее выраженной. Препарат обеспечивал уровень защиты 50% вне зависимости от использованной схемы его введения, что было на 10-30% ниже, чем у препарата №1.
Таким образом, на основании проведенных исследований можно заключить, что препарат №1, представляющий собой действующее начало фармакопейного препарата Рибавирин®, в сочетании со средством по изобретению по протективной и терапевтической эффективности в отношении экспериментальной вирусной инфекции в 1.5-1.8 раза превосходил действие фармакопейного препарата Рибавирин®.
Заключение
Совокупность результатов проведенных исследований позволяет сделать заключение о том, что средство по изобретению потенцировало противовирусное действие 1-[(2R,3R,4S,5R)-3,4-дигидрокси-5-(гидроксиметил)оксолан-2-ил]-1Н-1,2,4-триазол-3-карбоксамида, действующего начала фармакопейного препарата Рибавирин®. В этой связи использование средства по изобретению может быть перспективным в фармакологических решения по созданию лекарственных препаратов нового поколения для лечения и профилактики вирусных заболеваний человека и животных.
Пример 12. Влияние средства по изобретению на гемостимулирующую активность ионов лития, вводимых в форме литиевой соли дисульфида N-ацетил-L-цистеина
В настоящее время отсутствует эффективное гемостимулирующие средство длительного хранения. В медицинской практике известна гемостимулирующая активность лития в концентрациях, которые сопряжены с негативным влиянием лития на функции центральной нервной системы.
Цель исследования - определение способности средства по изобретению потенцировать гемостимулирующее действие ионов лития.
Приготовление растворов исследуемых соединений для проведения исследования
Кристаллические вещества хранили при 4°С; непосредственно перед началом эксперимента вещество растворяли в физиологическом растворе. Раствор стерилизовали пропусканием через фильтры 0.22 мкм Millex-GS (Millipore) в стерильном ламинарном боксе.
Модель исследования
Исследование было проведено на белых рандомбредных крысах-самцах массой 140-160 г, которым однократно вводили циклофосфан в дозе 120 мг/кг подкожно в физиологическом растворе.
Было сформировано 4 группы экспериментальных животных.
№1 - интактные животные, получавшие инъекции растворителя изучаемых соединений (физиологический раствор) (контроль растворителя);
№2 - животные, получившие инъекцию ЦФ, которым в дальнейшем в качестве лечебного средства вводили физиологический раствор (контроль);
опытные группы:
№3 - животные, получившие инъекцию циклофосфана, которым в дальнейшем в качестве лечебного средства вводили фармакологическую композицию №1 (ФК №1 синтезирована в соответствии с описанием в примере 6), содержащую ионы лития, дисульфид N-ацетил-L-цистеина и средство по изобретению в физиологическом растворе в дозе 10 мг/кг (количество координационного соединения 7.8×10-8 М/кг);
№4 - животные, получившие инъекцию циклофосфана, которым в дальнейшем в качестве лечебного средства вводили aC-Li (препарат №2) в физиологическом растворе в дозе 10 мг/кг;
№5 - животные, получившие инъекцию циклофосфана, которым в дальнейшем в качестве лечебного средства вводили карбонат лития (препарат №3) в физиологическом растворе в дозе 3 мг/кг (количество карбоната лития рассчитывали исходя из массовой доли иона лития (0.55) в 10 мг литиевой соли цистеина, аналогичное количество лития содержится в 3 мг карбоната лития);
№6 - животные, получившие инъекцию циклофосфана, которым в дальнейшем в качестве лечебного средства вводили дисульфид ацетилцистеина (препарат №4) в физиологическом растворе в дозе 9.5 мг/кг;
Исследуемые препараты вводили на третий день после введения циклофосфана.
Исследуемый материал
Забор крови для гематологических исследований проводили из хвостовой вены через 3, 7, 14 суток после введения ЦФ. В конце каждой серии (3, 7 и 14 сут) экспериментальных животных подвергали эвтаназии передозировкой эфира и получали кровь из хвостовой вены и костный мозг из бедренной кости.
Анализируемые показатели
В данном исследовании оценивали клеточность крови (число эритроцитов, тромбоцитов, лейкоцитов, лимфоцитов и нейтрофилов, а также СОЭ) и костного мозга при 7-дневном введении исследуемых соединений гемодепрессированным животным.
Результаты исследования
Результаты проведенных исследований клеточности крови представлены в таблицах 9-11.



- Циклофосфан в дозе 120 мг/кг вызывает достаточно выраженную цитопению по всем форменным элементам крови с абсолютной и относительной лимфопенией, максимально выраженную на 14-й день (1-3).
- Литий, вводимый в составе литиевой соли дисульфида N-ацетил-L-цистеина, в сочетании со средством по изобретению оказывает выраженный гемостимулирующий эффект, что проявилось в практически полном восстановлении клеточности крови. Действие дисульфида N-ацетил-L-цистеина и литиевой соли дисульфида N-ацетил-L-цистеина, карбоната лития, в отличие от фармакологической композиции №1, проявляется в виде позитивных тенденций, которые в целом можно оценить как гемостимулирующий эффект (табл.1-3).
Заключение
Совокупность результатов проведенных исследований позволяет сделать заключение о том, что литий в составе фармакологической композиции №1, представляющей собой литиевую соль дисульфида N-ацетил-L-цистеина, в сочетании со средством по изобретению обладает гемостимулирующим действием, тогда как без средства по изобретению литий, вводимый в составе карбоната или дисульфида N-ацетил-L-цистеина, не оказывал гемостимулирующего действия.
Таким образом, средство по изобретению характеризует способность потенцировать специфическую гемостимулирующую активность ионов лития.
Реферат
Изобретение относится к бионеорганической химии, медицинской химии и медицине. Палладиево-медный катализатор гомогенного селективного окисления тиолов сочетает в себе функциональное биядерное тиолатмостиковое координационное соединение палладия(II) и модифицирующий тиолатный комплекс меди(I), имеющий общую формулу SR представляет собой остаток тиолатного лиганда глутатиона или ацетилцистеина, k=2÷14, m≥3k. Также предложены каталитическая комбинация, стимулирующая пролиферативную активность фармакологическая комбинация для усиления терапевтической активности пуринового и (или) пиримидинового основания или ионов лития, фармацевтические композиции, стимулирующие пролиферативную активность и для усиления терапевтической активности указанных оснований или ионов лития, и способы терапевтического воздействия на организм пациента на основе указанного катализатора. Катализатор обладает сниженной токсичностью и может быть использован в фармакологических решениях с лекарственными средствами для терапии различной длительности, при которой востребована каталитическая активность в реакциях окислительной модификации тиолов до дисульфидов в составе молекул пептидной природы. 7 н. и 12 з.п. ф-лы, 3 ил., 11 табл., 12 пр.
Формула

где
SR представляет собой остаток тиолатного лиганда, выбранный из группы, включающей в себя остаток глутатиона и ацетилцистеина,
k=2÷14,
m≥3k.
Документы, цитированные в отчёте о поиске
Композит гексапептида со стабилизированной дисульфидной связью с веществом металлом, фармацевтические композиции на его основе, способы их получения и применения для лечения заболеваний на основе регуляции метаболизма, пролиферации, дифференцировки и меха
Цисплатиновый комплекс и способ его получения
Лекарственные средства на основе олигоядерных координационных соединений d-металлов, способ терапевтического воздействия на организм пациента и способ повышения терапевтической эффективности фармакологически активного вещества
Комментарии