Цисплатиновый комплекс и способ его получения - RU2245340C2
Код документа: RU2245340C2
Чертежи








Описание
Область техники
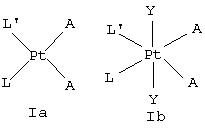
Настоящее изобретение относится к области лекарственных препаратов, содержащих платину. В частности, оно относится к усовершенствованному способу получения платиновых комплексов, имеющих общую формулу (Iа) или (Ib):


где:
L и L’ могут быть одинаковыми или различными, при условии, что L’ может представлять NН3, а L не может представлять NН3; и
L и L’, каждый, представляет амин или замещенный амин, скоординированный с атомом Pt через атом азота, и означает гетероциклический или гетероароматический амин, или представлен NRR’R’’, где R, R’ или R’’ независимо выбраны из группы, состоящей из водорода, замещенных или незамещенных прямых, разветвленных или циклических алифатических, арильных, неароматических или ароматических гетероциклических групп; при этом предпочтительно L представляет замещенный амин, в котором заместитель пространственно затрудняет доступ атома Pt к цепи ДНК клетки, предпочтительно опухолевой клетки; и
А могут быть одинаковыми или различными и представляют галоген или уходящую группу, такую как гидрокси, алкоксид, карбоксилат, и могут быть одинаковыми или различными или образовывать би-дентат карбоксилат, фосфонкарбоксилат, дифосфонат или сульфат; и
Y представляет галоген, гидрокси, сложный эфир карбоксилата, карбамата или карбоната.
Предпосылки изобретения
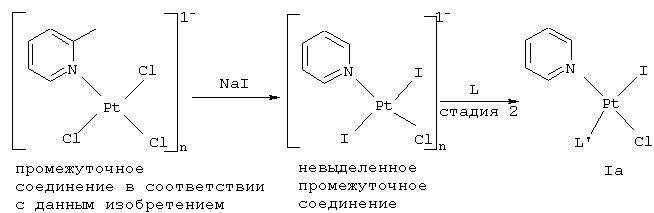
Патенты США №№4329299 и 5665771 описывают платиновые соединения и их применимость в качестве противоопухолевых лекарственных препаратов. Указанные два патента описывают платиновые соединения, которые охватывают комплексы, имеющие формулы cis-[PtA2(L’)(L)] и с,t,с-[PtA2Y2(L’)(L)], где А представляет уходящую группу, такую как галоген, гидрокси или карбоксилат, L представляет амин, скоординированный через атом азота, и L’ представляет аммоний или замещенный амин. Способы получения таких комплексов, описанных в указанных патентах, известны в данной области (Hydes P.С. Патент США 4329299 (1982); Murrer В.А. Патент США 5665771 (1997); Braddock P.D.; Connors T.A.; Jones M., Khokhar A.R.; Melzack D.H.; Tobe M.L. Chem.-Biol. Interactions. 1975, 11, 145-161; and Giandomenico C.M.; Abrams M.J.; Murrer B.A.; Vollano J.F.; Rheiheimer M.I., Wyer S.B.; Bossard G.E.; Higgins (III) J.D. Inorg. Chem. 1995, 34, 1015-1021). Данный процесс проиллюстрирован на Фиг.1, где примерами служат синтез cis-[PtCl2(NH3)(L)] и c,t,c-[PtCl2(OH2(NH3)(L)]. Синтез cis-[PtCl2(NH3)(L)] из легкодоступного и обычно применяемого исходного материала K2[PtCl4] включает четыре стадии, а синтез с,t,c-[PtCl2Y2(NН3)(L)] требует пяти стадий. Синтез указанных комплексов в соответствии со способом, известным в данной области, обеспечивает низкий общий выход. В патенте США №4329299 указано, что общий выход при применении K2[PtCl4] составляет менее 8%, в то время как общий выход, указанный в патенте США №5665771 и в литературе (Khokhar et al. и Giandomenico et al.), составляет 20-30%. Низкий общий выход обусловлен большим количеством стадий, составляющих данный способ, а также трудной и малоэффективной конверсией [PtCl2(NH3)2] в [РtСl3(NН3 )]-, требующей применения дорогого Pt катализатора. Синтез K[PtCl3(NH3)] из [PtCl2(NН3)2] не особенно надежен и трудно достичь крупномасштабного синтеза К[РtСl3(NН3)] постоянного качества. Вышеописанный способ требует также применения ионов серебра и иодида и приводит к получению загрязненных серебром и иодидом отходов производства.
Патент США №4533502 и патент Соединенного Королевства GB 2137198А описывают синтетический способ получения [PtX2(L)(L’)], где L и L’ представляют лиганды, связанные через азот амина, при этом L≠L’ (Rochon F.D.; Kong P.-С. UК Patent GB2137198A (1984) and Rochon F.D.; Kong P.-C. US Patent 4533502 (1985)). Указанный способ известен в данной области, и подробности такого синтетического способа были опубликованы (Courtot P.; Rumin R.; Peron A.; Girault J.P.J. Organometallic Chem. 1978, 145, 343-357, and Rochon F.D.; Kong P.-С. Can. J. Chem. 1986, 64, 1894-1896). Фиг.2 иллюстрирует способ с применением [PtCl2(L)(L’)] в качестве примера. Способ с применением K2[PtCl4], описанный в патенте США №4533502 и патенте Соединенного Королевства GB 2137198A, включает 4 стадии и выделение 3 промежуточных продуктов. Промежуточный олигомер представлен [PtLI2]x, где х=2-4; возможно также получение различных видов олигомера. Общий выход в результате применения K2[PtCl4] в патенте не указан. Данный способ включает применение ионов серебра и иодида с получением соответствующих отходов, загрязненных серебром и иодидом.
[PtCl3L]-, где L представляет амин, отличный от NН3, является промежуточным соединением в данном изобретении. Было описано получение [PtCl3L]-из разбавленного раствора K2[PtCl4] в диметилформамиде (ДМФ), где L представляет пиридин и его производные (Rochon F.D.; Kong P.-С. Can. J. Chem. 1978, 56, 441-445 and Rochon F.D.; Beauchamp A.L.; Bensimon C. Can. J. Chem. 1996, 74, 2121-2130). Получение [PtCl3L]-в растворителях, отличных от ДМФ или Н2O, или с применением амина, отличного от пиридина и его производных, не было описано. Синтез [PtCl3L] в ДМФ, как отмечается в литературе, проводят при 65-80°С, при этом выход выделенного продукта составляет от 40 до 90%, в зависимости от производного пиридина. Синтез [PtCl3L]-в ДМФ может приводить к получению реакционноспособных или неустойчивых комплексов Pt-ДМФ, которые могут воздействовать на последующие реакции или разлагаться с образованием нерастворимых черных примесей Pt. Например, Can. J. Chem. 1978, 56, 441 (также см. Chemical Abstracts, V. 89 (July 1978), Abstract No. 35686) Rochon et al. описывают осаждение нерастворимого черного материала при растворении К[РtСl3(2, 6-диметилпиридина)] в водном растворе. Также было описано получение во время выделения К[РtСl3(4-метилпиридина)] и К [PtCl3 (пиридина)] маслянистой пасты, содержащей [РtСl2(ДМФ) (производное пиридина)] и другие примеси. Были описаны примеры комплексов [PtCl2 (ДМФ) L] (Kong P.-С.; Rochon. F.D.; Can. J. Chem., 1979, 57, 682-684; Rochon F.D.; Kong Р.-С.; Melanson R. Can. J. Chem., 1980, 58, 97-101; and Rochon F.D.; Melanson R.; Doyon M.; Butler I.S. Inorg. Chem., 1994, 33, 4485-4493).
Chemical Abstracts, V.126 (April 1997), Abstract No. 194433 and Inorg. Chem. (1997), 36:854-861 описывают применение [Pt (с-С6H11NН2)I2]2 в качестве исходного материала для получения Pt(NH3) (с-С6Н11NH2) Cl2 (Фиг.2). Однако данная реакция приводит к разрыву связи Pt-A-Pt группой NH3 после образования промежуточного соединения, имеющего такие же заместители.
Chemical Abstracts, V. 108 (June 1988), Absract No.215224 and Inorg. Chim. Acta (1988), 143:81-7 описывают превращение [Pt(Cl)4]2-в промежуточное соединение [Pt (Cl)3NН3]1-, но не описывают добавление к данному промежуточному соединению замещенного циклического амина.
Цитирование вышеприведенных документов не предполагает, что какой-либо из них является подходящим уровнем техники. Все указания в отношении дат или ссылок на содержание данных документов основаны на информации, доступной заявителям, и не предполагают сомнения в их правильности. Кроме того, все документы, упоминаемые в данной заявке, приведены здесь во всей своей полноте в качестве ссылок. Конкретно, данная заявка испрашивает дату приоритета на основании предварительной заявки на патент США, серийный №60/128939, поданной 13 апреля 1999 г. и приводимой здесь во всей своей полноте в качестве ссылки.
Описание изобретения
Данное изобретение предлагает способ получения цис-платинового комплекса общей формулы Iа или Ib


включающий стадии:
a) взаимодействие [PtA4]2-или его соли с L в растворителе с получением [РtА3(L)]-;
b) взаимодействие [РtA3 (L)]-с L’ во втором растворителе с получением cis-[PtA2 (L’) (L)];
c) в случае, когда цисплатиновый комплекс имеет общую формулу Ib, и Y представляет гидрокси или галоген, взаимодействие cis-[PtA2 (L’) (L)], полученного на стадии b), с H2O2, когда Y представляет гидрокси, или галогеном, когда Y представляет галоген, с получением с,t,с-[PtA2Y2(L’)(L)]; и
d) в случае, когда цисплатиновый комплекс имеет общую формулу Ib, и Y представляет сложный эфир карбоксилата, карбамата или карбоната, вначале получение [PtA2OH2(L’)(L)] из [PtA2(L’)(L)] взаимодействием cis-[PtA2(L’)(L)], полученного на стадии b), с H2O2 в соответствии со стадией с), а затем взаимодействие [PtA2OH2(L’)(L)] с ацилирующим агентом с получением [PtA2Y2(L’)(L)]; и
где L и L’ имеют различные значения и каждый представляет амин или замещенный амин, скоординированный с атомом Pt через атом азота, и означает гетероциклический или гетероароматический амин, либо представлен NRR’R’’, где R, R’ и R’’ независимо выбраны из группы, состоящей из водорода, замещенных или незамещенных прямых, разветвленных или циклических алифатических, арильных, неароматических или ароматических групп; при условии, что только L’ может означать NН3 и что, по меньшей мере, один из L и L’ представляет замещенный гетероциклический или гетероароматический амин; и
где А могут быть одинаковыми или различными и представляют галоидную или не галоидную уходящую группу.
Данное изобретение также предлагает способ получения цисплатинового комплекса общей формулы Iа или Ib


включающий стадии:
a) взаимодействие [PtA4]2-или его соли с L в растворителе с получением [РtА3(L)]-;
b) взаимодействие [РtА3(L)]-с L’ во втором растворителе с получением cis-[PtA2(L’)(L)];
c) в случае, когда цисплатиновый комплекс имеет общую формулу Ib, и Y представляет гидрокси или галоген, взаимодействие cis-[PtA2(L’)(L)], полученного на стадии b), с H2O2, когда Y представляет гидрокси, или галогеном, когда Y представляет галоген, с получением с,t,c-[PtA2Y2(L’)(L)]; и
d) в случае, когда цисплатиновый комплекс имеет общую формулу Ib и Y представляет сложный эфир карбоксилата, карбамата или карбоната, вначале получение [PtA2OH2(L’)(L)] из [PtA2(L’)(L)] взаимодействием cis-[PtA2(L’)(L)], полученного на стадии b), с H2O2, в соответствии со стадией с), а затем взаимодействие [PtA2OH2(L’)(L)] с ацилирующим агентом с получением [PtA2Y2(L’)(L)]; и
e) превращение А в А’, где А’ представляет иную галоидную или не галоидную уходящую группу, чем А;
где L и L’ имеют различные значения и каждый представляет амин или замещенный амин, скоординированный с атомом Pt через атом азота, и означает гетероциклический или гетероароматический амин, либо представлен NRR’R’’, где R, R’ и R’’ независимо выбраны из группы, состоящей из водорода, замещенных или незамещенных прямых, разветвленных или циклических алифатических, арильных, неароматических или ароматических гетероциклических групп; при условии, что только L’ может означать NН3, и что, по меньшей мере, один из L и L’ представляет замещенный гетероциклический или гетероароматический амин; и
где каждый А может быть одинаковым или различным и представляет галоидную или не галоидную уходящую группу.
Данное изобретение также предлагает цисплатиновый комплекс формулы Ib

где L представляет

L’ представляет NH3, А представляет Сl и ОН, и Y представляет ОН.
Настоящее изобретение раскрывает более эффективный и экономичный способ получения Pt-комплексов вида cis-[PtA2(L’)(L)] (формула Ia) и c,t,c-[PtA2Y2(L’)(L)] (формула Ib) непосредственно из недорогого и легкодоступного платинового исходного материала, предпочтительно тетрагалоплатинита, подобного [PtCl4]2-или [PtBr4]2-,
где L и L’ могут быть одинаковыми или различными, при условии, что L’ может представлять NН3, но L не может представлять NН3; и
L и L’, каждый, представляет амин или замещенный амин, скоординированный с атомом Pt через атом азота, и означает гетероциклический или гетероароматический амин, либо представлен NRR’R’’, где R, R’ или R’’ независимо выбраны из группы, состоящей из водорода, замещенных или незамещенных прямых, разветвленных или циклических алифатических, арильных, неароматических или ароматических гетероциклических групп; при этом предпочтительно L представляет замещенный амин, в котором заместитель пространственно затрудняет доступ атома Pt к цепи ДНК-клетки, предпочтительно опухолевой клетки; и
А могут быть одинаковыми или различными и представляют галоген или уходящую группу, такую как гидрокси, алкоксид, карбоксилат, а также могут быть одинаковыми или различными или образовывать би-дентат карбоксилат, фосфонкарбоксилат, дифосфонат или сульфат; и
Y представляет галоген, гидрокси, сложный эфир карбоксилата, карбамата или карбоната.
В соответствии с одним из вариантов его осуществления способ по данному изобретению является предпочтительным для получения соединения формулы Iа.
Применяемые в данном описании термины основаны на их значениях, признанных в данной области, и должны быть без сомнения понятны рядовому специалисту в данной области техники. Для ясности, термины могут также иметь конкретное значение, понятное из их контекста. Например, лиганд представляет собой ион или молекулу, связанную с и считающуюся связанной с атомом или ионом металла. "Моно-дентат" означает наличие одной позиции, через которую могут быть образованы ковалентные или координационные связи с металлом. "Би-дентат" означает наличие двух позиций, через которые могут быть образованы ковалентные или координационные связи с металлом. Настоящее изобретение предпочтительно включает моно-дентатную координацию амина L и L’ через атом азота с Pt. Далее, термин "пространственно затрудненный" применяется в соответствии со своим обычным употреблением в данной области. Следовательно, "пространственно затрудненный амин" относится к компоненту амина, который из-за своего размера или массы затрудняет или влияет на вращение или другую функцию или свойство любого другого компонента описываемых здесь Pt-комплексов. Способы в соответствии с настоящим изобретением предпочтительно применяют для получения соединений, описанных в патенте США №5665771 (особенно пространственно затрудненных аминов, представленных формулой Iа в патенте ‘771), который включен в описание во всей своей полноте, в частности, определения замещающих групп приведены в данном описании в качестве ссылки. Термин "замещенный" по отношению как к L, так и L’, когда представляет соединенный с азотом гетероциклический или гетероароматический амин (амины), означает, что замещающую группу независимо выбирают из группы, состоящей из водорода, замещенных или незамещенных прямых, разветвленных или циклических алифатических, арильных, неароматических или ароматических гетероциклических групп; и предпочтительно L представляет замещенный амин, а заместитель таким образом пространственно затрудняет доступ атома Pt к цепи ДНК клетки, предпочтительно опухолевой клетки. Примеры такого замещенного L или L’ включают, но не ограничиваются ими, алкиламины, которые могут включать метиламин; диметиламин; трибутиламин; диизопропиламин; ариламины, которые могут включать анилин, толуидин, аминонафталин и амино-антрацен; гетероциклические амины, которые могут включать пиперидин, пиперазин и пирролидин; и гетероароматические амины, которые могут включать пиридин, пиразолы, имидазолы, оксазолы, изооксазолы; пиримидин и пиразин. Специалисту в данной области известны другие заместители, которые могут быть использованы в настоящем изобретении в соответствии с данным описанием.
Например, более конкретно, в случае замещенных циклических аминов заместителем может быть низший алкил или алкокси, имеющий от 1 до 4 атомов углерода (особенно метил или метокси), галоген (особенно хлор или бром), или арил (особенно бензил). Заместитель может быть сам замещен низшим алкилом или галогеном. Термин "низший алкил" обозначает алкильную группу, имеющую от 1 до 6 атомов углерода. Циклический амин может нести другие заместители, либо смежные с координационным атомом азота, либо находящиеся в другом месте на кольце. Другие заместители включают электрон-оттягивающие или электрон-донорные заместители, такие как нитро и алкокси, например метокси. Если циклический амин представляет собой конденсированную кольцевую систему, в которой конденсированным кольцом является ароматическое кольцо в позициях 2 и 3 циклического амина, то другого заместителя не требуется, несмотря на то, что он может присутствовать. Следует также отметить, что данное изобретение может быть использовано для получения транс-изомеров. Предпочтительный вариант данного изобретения предусматривает получение цис-изомеров.
Для иллюстрации данного изобретения в качестве примеров используют синтез cis-[PtCl2(NН3)(L)] и c,t,c-[PtCl2(ОН)2(NН3)(L)] из [PtCl4]2-. Аналоги cis-[PtBr2(NН3)(L)] и c,t,c-[PtBr2(ОН)2(NH3)(L)] также могут быть получены подобным способом из [PtBr4]2-.
Усовершенствованный способ получения cis-[PtCl2(NH3)(L)] включает две стадии, при этом первая стадия включает превращение суспензии или концентрированного раствора [PtCl4]2-в [PtCl3L]-в апротонных растворителях. Вторая стадия включает превращение суспензии или концентрированного раствора [РtСl3L]-в cis-[PtCl2(NH3)(L)] в растворе гидроокиси аммония. По сравнению с синтетическими способами, используемыми в настоящее время в данной области, усовершенствованный способ включает меньше синтетических стадий, меньше выделяемых продуктов, требует меньших объемов экологически вредных растворителей, производит меньше загрязненных металлами отходов и обеспечивает более высокий общий выход cis-[PtCl2(NH3)(L)]. Он также не требует применения ионов серебра и иодида и не приводит к получению отходов, загрязненных серебром и иодидом. Все стадии способа надежны, воспроизводимы и всегда обеспечивают одинаковое качество продуктов.
Первая стадия усовершенствованного способа включает взаимодействие [PtCl4]2 -с амином L в соответствующих условиях в первом растворителе с получением [PtCl3L]-. Обычно применяют наиболее легкодоступную калиевую соль [PtCl4]2-. Однако могут быть использованы также другие соли [PtCl4]2-. Соответствующие условия в данном описании включают реакционные условия, способствующие и облегчающие описываемую и заявленную здесь химическую реакцию. Конкретно, настоящее изобретение предусматривает, чтобы такие подходящие условия включали, но не ограничивались ими, температуру, рН, концентрацию реагентов, степень перемешивания, ситовой размер реактивов и другие подобные условия, облегчающие описываемые химические реакции. Однако специалисту в данной области известны и другие подходящие условия, обеспечивающие проведение стадий описываемых химических реакций. Для облегчения растворения K2[PtCl4] предпочтительно применение тонкоизмельченного порошка K2[PtCl4]. Предпочтительно, чтобы размер K2[PtCl4] составлял примерно 240 мкм или менее. Более предпочтительно, размер K2[PtCl4] составляет примерно 100 мкм или менее. Во время реакции 1-1,3 экв. амина подвергают взаимодействию с 1 экв. K2[PtCl4]. Более предпочтительно применяют 1-1,2 экв. амина. Наиболее предпочтительно 1,05-1,15 экв. амина подвергают взаимодействию с 1 экв. K2[PtCl4]. Применение высоких эквивалентов амина повышает скорость реакции, но также может повысить образование побочных продуктов и снизить выход реакционного продукта. Кроме того, амин, L, добавляют к реакционной смеси небольшими порциями в течение периода времени. Предпочтительно амин добавляют в двух или более эквивалентных количествах или более предпочтительно в 4 или более количествах.
Реакция может быть осуществлена при температуре примерно 30-100°С, но более предпочтительно проведение реакции при примерно 40-70°С. Наиболее предпочтительно применяют температурный интервал примерно 50-65°С. Как правило, чем выше температура реакции, тем выше скорость реакции между [PtCl4]2-и амином. Однако высокая температура реакции может усилить образование побочных продуктов или вызвать образование реакционноспособных и нестабильных Pt-примесей. В растворителях, способных координироваться с атомами металла, таких как ДМФ, температура реакции выше чем или равная примерно 60°С может способствовать образованию комплексов Pt-растворитель, которые будут разлагаться или влиять на следующую стадию способа.
Реакцию осуществляют в апротонных растворителях. Предпочтительно, чтобы растворитель содержал менее чем примерно 25% воды, однако предпочтительным является содержание воды менее чем примерно 10%. Наиболее предпочтительно, требуется содержание воды менее чем примерно 3%. Реакция может быть осуществлена в апротонных растворителях, таких как ацетон, хлороформ, дихлорметан, диметилацетамид, диметил-формамид, N-метилпирролидинон и тетрагидрофуран. Наиболее предпочтительным растворителем является N-метилпирролидинон.
Первую реакционную стадию осуществляют при соотношении, составляющем менее примерно (15 мл растворителя)/(1 ммол. платины). Предпочтительный вариант осуществления данного изобретения предполагает отношение растворителя (мл) к Pt (ммол.), составляющее примерно 3-6:1. Однако в более предпочтительном варианте данного способа первую реакционную стадию осуществляют при отношении растворителя к Pt, составляющем примерно 1-2:1.
Синтез [PtCl3L]-в диметилформамиде (ДМФ), где L представляет пиридин или его производные, известен из литературы (Rochon F.D.; Kong Р.-С. Can. J. Chem., 1978, 56, 441-445, Rochon F.D.; Beauchamp A.L.; Bensimon С. Can. J. Chem., 1996, 74, 2121-2130). При синтезе K[PtCl3(L)] в качестве иллюстративного примера используют К[РtСl3(2-пиколин)] для сравнения опубликованного способа со способом, раскрываемым в данном изобретении. В соответствии с опубликованным способом выделение К[РtСl3(2-пиколина)] требует проведения двух отдельных стадий, во время каждой из которых растворители выпаривают при пониженном давлении. При выпаривании ДМФ при пониженном давлении требуется нагревание при 40°С. При крупномасштабном промышленном синтезе выпаривание растворителей при пониженном давлении, особенно с нагреванием, является дорогостоящей и длительной процедурой. В соответствии с предпочтительной методикой заявителей синтез и выделение К[РtСl3(2-пиколина)] не требуют выпаривания растворителей и переноса материала из одного растворителя в другой. Способ, раскрытый в данном изобретении, более эффективен и больше подходит для крупномасштабного промышленного получения соединения. Осуществление обоих способов приводит к получению сопоставимых выхода и качества K[PtCl3(2-пиколина)]. Результаты инфракрасной (ИК) и ЯМР-спектроскопии К [РtСl3(2-пиколина)], полученные с применением способа, известного в данной области, и способа, раскрытого в данном изобретении, представлены на Фиг.4 и 5. Данное описание также демонстрирует синтез [PtCl3L]-в других апротонных растворителях, таких как ацетон, хлороформ, дихлорметан и N-метилпирролидинон.
Координирование молекул растворителя с Pt, вызывающее образование реакционноспособных или нестойких соединений Pt, является проблемой для способа, раскрытого в данном изобретении. В опубликованном способе (Rochon F.D.; Kong P.-С. Can. J. Chem., 1978, 56, 441-445) описано образование [PtCl2 (ДМФ)(производное пиридина)] и других примесей во время синтеза [РtСl3(производное пиридина)]. В соответствии с данным изобретением авторы применяют температурный интервал, сводящий к минимуму образование нежелательных соединений, таких как [PtCl2 (ДМФ) (производное пиридина)]. Образование черного осадка во время выделения продукта является показателем присутствия реакционноспособных Pt-примесей. При осуществлении синтеза К[РtСl3 (2-пиколина)] в диметилформамиде в качестве примера образование нерастворимых черных осадков не наблюдается, если синтез проходит при температуре ниже примерно 60°С. В соответствии с наиболее предпочтительным вариантом осуществления данного изобретения, на первой стадии взаимодействия применяют реакционную температуру примерно 50-65°С. Однако данный способ предусматривает любую температуру, при которой образование нежелательных соединений или примесей продукта реакции, таких как [PtCl2 (ДМФ)(производное пиридина)], сведено к минимуму (<10%) или устранено.
Превращение [PtCl3L]-в [PtCl2(NH3)L] в водной гидроокиси аммония используется для иллюстрации стадии 2 данного изобретения. Стадия 2 включает взаимодействие суспензии или концентрированного раствора [PtCl3L]-с NН3, во втором растворителе с получением [PtCl2(NH3)L]. Синтез [PtCl2 (NH3)L] осуществляют при температуре примерно 30-60°С в растворе гидроокиси аммония. Более предпочтительно осуществлять взаимодействие при примерно 35-55°С, в то время как наиболее предпочтительной является температура взаимодействия примерно 40-50°С. Как правило, более высокая температура реакции сокращает ее продолжительность, но также может способствовать образованию Pt-мультиаминовых/аминовых побочных продуктов. Более высокий уровень образования побочных продуктов снижает выход продуктов реакции.
Взаимодействие осуществляют при рН примерно 7-14. Более предпочтительным является рН примерно 7-12, в то время как наиболее предпочтительным является рН 8-10. Осуществление взаимодействия при рН>10 может вновь привести к снижению выхода из-за повышенного образования Pt-мультиаминовых побочных продуктов.
Взаимодействие осуществляют при концентрации, составляющей 1 г К[РtСl3L] на 3-10 мл растворителя. Концентрация 1 г К[РtСl3L] на 4-8 мл растворителя является более предпочтительной, в то время как наиболее предпочтительной является концентрация 1 г К[РtСl3L] на 5-7 мл растворителя. Неожиданно было обнаружено, что осуществление взаимодействия при высокой концентрации будет эффективно обеспечивать получение продукта с высоким выходом. Взаимодействие может быть осуществлено при гораздо более разбавленной концентрации, однако выход реакции был низким из-за образования побочных продуктов. Большие объемы растворителей и более разбавленная концентрация также требуют утилизации больших объемов экологически вредных растворителей и отходов. Предпочтительно осуществление данного взаимодействия обязательно в водных растворах. Однако может быть также использовано сочетание органических и водных растворителей. Второй растворитель может содержать примерно от 0,1 до 6 N хлорид. В частности, данный способ предусматривает проведение второй стадии взаимодействия 1b) при соотношении растворителя к платине ниже чем или равном примерно 5:1 (мл растворителя)/(ммол. платины). Вторую стадию способа осуществляют при интервале соотношения NН3/Pt примерно 3-7. Предпочтительным является соотношение NН3/Pt примерно 4-6, в то время как наиболее предпочтительным является соотношение NH3/Pt, составляющее примерно 4,5-5,5. Данный способ предусматривает осуществление второй стадии взаимодействия 1b) при молярном соотношении свободного основания L’ и платины примерно от 3:1 до 1:1. Большой избыток NН3 снижает период взаимодействия, но может также увеличить образование Pt-мультиаминовых/аминовых побочных продуктов.
с,t, c-[PtA2(ОН)2(NH3)(L)] может быть получен из cis-[PtA2(NH3)(L)] в результате взаимодействия суспензии cis-[PtA2(NH3)L] с перекисью водорода. Из с, t, с-[PtA2(ОН)2(NH3)(L)] могут быть получены другие Pt(IV) комплексы формулы c,t,c-[PtA2Y2(NH3)(L)] с применением способов, известных в данной области, где Y представляет галоген, гидрокси, сложный эфир карбоксилата, карбамата или карбоната, другой, чем, когда оба Y представляют гидроокись.
Примеры, используемые для иллюстрации получения cis-[PtCl2(NH3)(L)] и c,t,c-[PtCl2(OH)2(NH3)(L)], могут также быть использованы для получения соединений общей формулы cis-[PtA2(L)(L’)] и c,t,c-[PtA2Y2(L)(L’)], где L и L’ могут быть одинаковыми или различными, при условии, что L’ может представлять NН3, а L не может представлять NН3; и L и L’, каждый, представляет амин или замещенный амин, скоординированный с атомом Pt через атом азота, и означает гетероциклический или гетероароматический амин, либо представлен NRR’R’’, где R, R’ или R’’ независимо выбраны из группы, состоящей из водорода, замещенных или незамещенных прямых, разветвленных или циклических алифатических, арильных, неароматических или ароматических гетероциклических групп, L предпочтительно представляет замещенный амин, в котором заместитель пространственно затрудняет доступ атома Pt к цепи ДНК клетки, предпочтительно опухолевой клетки. А могут быть одинаковыми или различными и могут представлять галоген или уходящую группу, такую как гидрокси, алкоксид, карбоксилат, или образовывать би-дентат карбоксилат, фосфонкарбоксилат, дифосфонат или сульфат; и Y представляет галоген, гидрокси, сложный эфир карбоксилата, карбамата или карбоната.
Что касается комплексов Iа или Ib, то в данной области известны способы превращения лиганда А в различные уходящие группы, такие как галоид, гидрокси, алкоксид, или моно-дентат карбоксилат, или би-дентат карбоксилат, или би-дентат фосфонкарбоксилат, или би-дентат фосфонат, или би-дентат сульфат. Примеры таких превращений представляют уравнения 1 и 2. Может быть задумано много других перестановок и сочетаний превращений уходящей группы, приводящих к получению желательных комплексов. Способ получения описываемых промежуточных соединений подходит для получения всех подобных соединений.
Уравнение 1. Способ получения комплекса формулы Iа, в которой две уходящие группы А представляют галоиды и имеют различные значения
Уравнение 2. Превращение обоих лигандов А (где А=галоид) для получения нового соединения, в котором оба А имеют одинаковые значения и образуют би-дентат карбоксилат

После общего описания данного изобретения то же самое будет объяснено более подробно со ссылкой на нижеприведенные примеры, представленные лишь с целью иллюстрации, а не ограничения данного изобретения, если не указано иначе.
Краткое описание чертежей
Фиг.1. Фигура иллюстрирует синтез cis-[PtCl2(NH3)(L)] и c,t,c-[PtX2Y2(NH3)(L)] через К[РtСl3(NH3)].
Фиг.2. Фигура иллюстрирует синтез [PtCl2(L)L’] через [PtI2(L)]x олигомер.
Фиг.3. Фигура иллюстрирует синтез [PtA2 (L’)L] с применением способа в соответствии с данным изобретением.
Фиг.4. Фигура иллюстрирует данные инфракрасной и ЯМР-спектрометрии для [PtCl2(NН3) (2-пиколина)], полученного с применением способа, раскрытого в данном изобретении.
Фиг.4А. Инфракрасный спектр [PtCl2(NH3) (2-пиколина)], полученного с применением способа, раскрытого в данном изобретении.
Фиг.4В.195Pt ЯМР-спектр [PtCl2 (NН3) (2-пиколина)], полученного с применением способа, раскрытого в данном изобретении.
Фиг.4С.1H ЯМР-спектр [PtCl2(NН3) (2-пиколина)], полученного с применением способа, раскрытого в данном изобретении.
Фиг.5. Фигура иллюстрирует данные инфракрасной и ЯМР-спектрометрии для [PtCl2(NН3)(2-пиколина)], полученного с применением известного в данной области способа, представленного на Фиг.1.
Фиг.5А. Инфракрасный спектр [PtCl2(NH3)(2-пиколина)], полученного с применением известного в данной области способа, представленного на Фиг.1.
Фиг.5В.195Pt ЯМР-спектр [PtCl2(NН3)(2-пиколина)], полученного с применением известного в данной области способа, представленного на Фиг.1.
Фиг.5С.1Н ЯМР-спектр [PtCl2(NН3)(2-пиколина)], полученного с применением известного в данной области способа, представленного на Фиг.1.
ПРИМЕРЫ
В соответствии с приведенными ниже примерами соединения анализируют с применением1H и195Pt ЯМР-спектроскопии, элементного анализа и ВЭЖХ. ЯМР-спектры записывают на спектрометре Bruker Avance 300 (1Н и195Pt ЯМР) в ДМФ-d7 и сравнивают со спектрами сравнительных соединений, синтезированных с применением известных в данной области способов. Элементный анализ (%С,%Н,%N) осуществляют с применением анализатора Perkin Elmer 2400 или Carlo Erba 1108. Содержание %Сl определяют путем титрования нитратом серебра. Для анализа соединений, иллюстрируемых в нижеприведенных примерах, применяют два способа ВЭЖХ (анионный и катионный). При использовании анионного метода ВЭЖХ время удерживания K[PtCl3 (2-пиколина)] и [PtCl2(NН3)(2-пиколина)] составляет 21,9 и 4,2 мин соответственно. При использовании катионного метода время удерживания [РtСl2(NН3)(2-пиколина)] составляет 3 мин. Время удерживания ВЭЖХ синтезированных соединений сравнивают со временем удерживания сравнительных соединений, полученных с применением известного в данной области способа. Применяют следующие рабочие условия анионного и катионного методов ВЭЖХ:
Катионный метод ВЭЖХ:
Общее время процесса: 35,01 мин
Скорость потока: 1,0 мл/мин
Температура: 25°С
Детектор: DAD @ 267 нм
Инъекция: 10 мкл
Анионный метод ВЭЖХ:
Общее время процесса: 40 мин
Скорость потока: 1,0 мл/мин
Температура: 35°С
Детектор: DAD @ 230 нм
Инъекция: 15 мкл
Примеры 1-9 иллюстрируют стадию 1 данного способа.
Пример 1
Синтез K[PtCl3(2-пиколина)] в N-метилпирролидиноне
K2[PtCl4] измельчают до тонкодисперсного порошка пестиком в ступке. 3,5047 г (8,443 ммол.) K2[PtCl4] помещают в 25-мл круглодонную колбу и добавляют 6-7 мл сухого NMP. 0,8648 г (9,286 ммол.) 2-пиколина помещают в 3-4 мл NMP и делят на 5 равных порций. Первую порцию 2-пиколина добавляют к смеси Pt. Смесь полностью погружают в 60°С масляную баню и перемешивают при 1200 об/мин. Следующие порции 2-пиколина добавляют через 30-35-минутные интервалы. Скорость добавления 2-пиколина составляет 20% каждые 30-35 минут. После добавления последней порции реакции дают возможность протекать в течение еще 50-60 минут. В конце реакции реакционный раствор имеет оранжевый цвет. Реакционному раствору дают возможность охладиться до температуры окружающей среды. К реакционному раствору при температуре окружающей среды добавляют 100 мл метиленхлорида. Добавление метиленхлорида вызывает осаждение К[РtСl3(2-пиколина)] и КСl. Осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают метиленхлоридом (3×5 мл), а затем диэтиловым эфиром (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. Выход: 3,8440 г (86,8%). Анализ. Вычислено (найдено) для С6Н7N1Сl3КРt·1,2 K1Cl1: С 13,74 (13,54); Н 1,35 (1,39); N 2,67 (2,59); Cl 28,51 (28,32).1H ЯМР (300 МГц, ДМФ-d6): 9,12 (д, 1 пиридин Н); 7,61 (д, 1 пиридин Н); 7,40 (т, 1 пиридин Н); 3,40 (с, 3 метил Н).
195Pt ЯМР (300 МГц, ДМФ-d6): соответствует195Pt ЯМР-спектру К [РtСl3(2-пиколина)], полученного способом, известным в данной области. ВЭЖХ (анионная): время удерживания соответствует времени удерживания сравнительного соединения.
Пример 2
Синтез К[РtСl3(2,6-лутидина)] в N-метилпирролидиноне
K2[PtCl4] измельчают до тонкодисперсного порошка пестиком в ступке. 1,9427 г (4,68 ммол.) K2[PtCl4] помещают в 15-мл круглодонную колбу и добавляют 4 мл сухого NMP. 0,5501 г (5,13 ммол.) 2,6-лутидина помещают в 3-4 мл NMP и делят на 5 равных порций. Первую порцию 2-пиколина добавляют к смеси Pt. Смесь полностью погружают в 60°С масляную баню и перемешивают при 1200 об/мин. Следующие порции 2-пиколина добавляют через 30-35-минутные интервалы. Скорость добавления 2-пиколина составляет 20% каждые 30-35 минут. Общая продолжительность реакции составляет 24 часа. В конце реакции реакционный раствор имеет оранжевый цвет. Реакционному раствору дают возможность охладиться до температуры окружающей среды. К реакционной смеси при температуре окружающей среды добавляют 200 мл метиленхлорида. Добавление метиленхлорида вызывает осаждение К[РtСl3(2-пиколина)] и КСl. Осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают метиленхлоридом (3×5 мл), а затем диэтиловым эфиром (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. Выход: 2,1415 г (84,7%). Анализ. Вычислено (найдено) для C7H9N1Cl3KPt·1,24K1Cl1: С 15,57 (15,40); Н 1,68 (1,72); N 2,59 (2,60); Cl 27,83 (27,70);1H ЯМР (300 МГц, ДМФ-d6): 7,6 (т, 1 пиридин Н); 7,28 (д, 2 пиридин Н); 3,51 (с, 3 метил Н); 3,43 (с, 3 метил Н).
Пример 3
Синтез К[РtCl3(2-пиколина)] в диметилформамиде при 50°С K2[PtCl4] измельчают до тонкодисперсного порошка пестиком в ступке. 2,6461 г (6,375 ммол.) K2[PtCl4] помещают в 25-мд круглодонную колбу и добавляют 6 мл сухого ДМФ. 0,6233 г (6,693 ммол.) 2-пиколина добавляют к раствору Pt. Реакционную смесь погружают в 50°С масляную баню и позволяют реакции продолжаться в течение приблизительно 120 минут. В конце реакции реакционный раствор имеет оранжевый цвет. Данному раствору дают возможность охладиться до температуры окружающей среды. К реакционной смеси при температуре окружающей среды добавляют 100 мл хлороформа. Добавление хлороформа вызывает осаждение K[PtCl3 (2,6-лутидина)] и КСl. Осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают метиленхлоридом (3×5 мл), а затем диэтиловым эфиром (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. При растворении продукта в водном растворе черного осадка не наблюдается. Выход: 2,8565 г (84%). Анализ: Вычислено (найдено) для C6H7N1Cl3KPt·1,3K1C1: С 13,58 (13,65); Н 1,33 (1,31); N 2,67 (2,64); Cl 28,73 (28,78).1H ЯМР (300 МГц, ДМФ-d6): 9,12 (д, 1 пиридин Н); 7,90 (т, 1 пиридин Н); 7,61 (д, 1 пиридин Н); 7,40 (т, 1 пиридин Н); 3,40 (с, 3 метил Н).195Pt ЯМР (300 МГц, ДМФ-d6): соответствует195Pt ЯМР-спектру К[РtСl3(2-пиколина)], полученного с применением известного в данной области способа. ВЭЖХ (анионная): время удерживания соответствует времени удерживания сравнительного соединения.
Пример 4
Синтез К[РtСl3(2, 6-лутидина)] в диметилформамиде при 50°С K2[PtCl4] измельчают до тонкодисперсного порошка пестиком в ступке. 1,0900 г (2,62 ммол.) K2[PtCl4] помещают в 15-мл круглодонную колбу и добавляют 2-3 мл сухого ДМФ. 0,3078 г (2,87 ммол.) 2,6-лутидина помещают в 1-2 мл ДМФ и делят на 5 равных порций. Первую порцию 2-пиколина добавляют к смеси Pt. Смесь полностью погружают в 50°С масляную баню и перемешивают при 1200 об/мин. Следующие порции 2-пиколина добавляют через 30-35-минутные интервалы. Скорость добавления 2-пиколина составляет 20% каждые 30-35 мин. Общая продолжительность реакции составляет 72 часа. В конце реакции реакционный раствор имеет оранжевый цвет. Данному раствору дают возможность охладиться до температуры окружающей среды и фильтруют. К реакционной смеси при температуре окружающей среды добавляют 100 мл метиленхлорида. Добавление метиленхлорида вызывает осаждение К[РtСl3(2,6-лутидина)]. Осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают метиленхлоридом (3×5 мл), а затем диэтиловым эфиром (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. При растворении продукта в водном растворе черного осадка не наблюдается. Выход: 0,6815 г (53,1%). Анализ: Вычислено (найдено) для C7H9N1Cl3KPt·0,1 K2[PtCl4]: С 17,19 (17,20); Н 1,85 (1,90); N 2,86 (2,935); Сl 24,64 (24,61);1H ЯМР (300 МГц, ДМФ-d6): 7,6 (т, 1 пиридин Н); 7,28 (д, 2 пиридин Н); 3,51 (с, 3 метил Н); 3,43 (с, 3 метил Н).
Пример 5
Синтез [РtСl3(2-пиколина)] в ацетоне, дихлорметане или хлороформе.
1,0040 г (2,419 ммол.) K2[PtCl4] помещают в 25-мл круглодонную колбу и добавляют 1 мл ацетона. 0,67 г хлорида тетрабутиламмония растворяют в 2 мл ацетона и добавляют к раствору K2[PtCl4]. 0,2783 г (2,988 ммол.) 2-пиколина растворяют в 2 мл ацетона и добавляют к раствору Pt. Реакционную смесь нагревают при 60°С. K2[PtCl4] постепенно растворяется в течение часа и превращается в более растворимую соль тетрабутиламмония [PtCl4]2-. Реакционный раствор перемешивают при 50°С в течение 16 часов. Реакционный раствор фильтруют, удаляя КСl, а ацетон удаляют при пониженном давлении, получая масло оранжевого цвета, соответствующее продукту [РtСl3(2-пиколин)]1-.1H ЯМР (300 МГц, ДМФ-d6): 9,0 (д, 1 пиридин Н); 7,8 (т, 1 пиридин Н); 7,45 (д, 1 пиридин Н); 7,25 (т, 1 пиридин Н), 3,20 (с, 3 метил Н).195Pt ЯМР (300 МГц, ДМФ-d6): соответствует сравнительному соединению.
Подобную процедуру применяют для получения [РtСl3(2-пиколина)]1-с хлороформом или дихлорметаном в качестве растворителя.1H ЯМР: соответствует сравнительному соединению.
Чтобы выделить [PtCl3 (2-пиколин)]1-в виде соли калия, масло оранжевого цвета растворяют в 2 мл метанола. Добавляют ацетат калия, растворенный в метаноле, вызывая осаждение К[РtСl3(2-пиколина)]. Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. Выход: 0,5762 г (58%).
Пример 6
Синтез [PtCl3(трибутиламина)]-из тетрахлорплатината тетрабутиламмония в ацетоне.
0, 2715 г (0,33 мол.) тетрахлорплатината тетрабутиламмония растворяют в ацетоне. К раствору Pt добавляют 0,1323 г (0,7135 ммол.) трибутиламина. Реакционный раствор нагревают при 60°С в течение ночи. Реакционный раствор фильтруют, удаляя КСl, а ацетон удаляют при пониженном давлении, получая оранжевое масло, соответствующее продукту [PtCl3 (трибутиламин)]-.195Pt ЯМР (300 МГц, ДМФ-d6): соответствует сравнительному соединению. Чтобы выделить [PtCl3 (трибутиламин)]2-в виде соли калия, масло оранжевого цвета растворяют в 2 мл метанола. Добавляют ацетат калия, растворенный в метаноле, вызывая осаждение К[РtСl3(трибутиламина)]. Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. Выход: 0,1577 г (64%).
Пример 7
Синтез К[РtCl3(2,5-диметилпиразина)] в N-метилпирролидиноне (NMP)
K2PtCl4 измельчают до тонкодисперсного порошка пестиком в ступке. 1,0724 г (2,58 ммол.) K2PtCl4 помещают в 10-мл круглодонную колбу и добавляют ~5 мл NMP. Содержимое колбы перемешивают при ~700 об/мин и погружают в масляную баню при 65°С. 0,3196 г (2,96 ммол.) 2,5-диметилпиразина смешивают с ~1 мл NMP. К реакционной смеси с 30-минутными интервалами добавляют приблизительно четыре равные порции раствора 2,5-диметилпиразина. После последнего добавления реакции дают возможность протекать в течение 60 мин, а затем охлаждают до температуры окружающей среды. К реакционной смеси добавляют 150 мл метиленхлорида. Добавление метиленхлорида вызывает осаждение продукта. Осадок собирают вакуумной фильтрацией, применяя стеклянную фритту, и промывают метиленхлоридом (3×30 мл) и диэтиловым эфиром (3×10 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16 часов и взвешивают. Выход: 1,0507 г (66,3%). Анализ: Вычислено (найдено) для С6 Н8N2Сl3КРt·2,2КСl: С 11,73 (11,50); Н 1,31 (1,50); N 4,56 (4,27); Cl 30,14 (29,86).1H ЯМР (300 МГц, ДМФ-d7) 9,11 (с, 1 пиразин Н); 8,68 (с, 1 пиразин Н); 3,31 (с, 3 метил Н), 2,68 (с, 3 метил Н).
Пример 8
Синтез K[PtCl3] (4, 6-диметилпиримидина)] в NMP
K2PtCl4 измельчают до тонкодисперсного порошка пестиком в ступке. 0,5277 г (1,27 ммол.) K2PtCl4 помещают в 15-мл круглодонную колбу и добавляют ~3 мл NMP. Содержимое колбы энергично перемешивают и погружают в масляную баню при 65°С. 0,1549 г (1,43 ммол.) 4,6-диметилпиримидина смешивают с ~1 мл NMP. Раствор 4,6-диметилпиримидина добавляют к реакционной смеси приблизительно четырьмя равными порциями через 30-минутные интервалы. После последнего добавления реакции дают возможность протекать в течение 60 мин, а затем смесь охлаждают до температуры окружающей среды. Реакционную смесь гасят ~80 мл метиленхлорида, вызывающего осаждение твердой фазы. Осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают метиленхлоридом (3×30 мл) и диэтиловым эфиром (3×10 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16 часов и взвешивают. Выход: 0,4353 г (76,3%).1H ЯМР (300 МГц, ДМФ-d7 ) 9,58 (с, 1 пиримидин Н); 7,65 (с, 1 пиримидин Н); 3,32 (с, 3 метил Н), 2,65 (с, 3 метил Н).
Пример 9
Синтез [РtСl3(диизопропиламина)]-из тетрахлорплатината тетрабутиламмония в ацетоне.
0,7961 г (0,9687 ммол.) тетрахлорплатината тетрабутиламмония помещают в 25-мл круглодонную колбу и добавляют 8 мл ацетона. 0,1699 г (1, 679 ммол.) диизопропиламина растворяют в 2 мл ацетона и добавляют к раствору Pt. Реакционный раствор погружают в масляную баню при 60°С и перемешивают в течение 60 часов. Красный [PtCl4 ]-превращают в оранжевый [РtСl3(диизопропиламин]-, что подтверждается195Pt ЯМР-спектроскопией. [PtCl3(диизопропиламин)]-может быть использован для получения [PtCl2 (NН3) (диизопропиламина)] непосредственно без выделения в виде соли калия или тетрабутиламмония.195Pt ЯМР (300 МГц, ДМФ-d7 ): соответствует195Pt ЯМР-спектру [РtСl3(диизопропиламина)], полученного с применением известного в данной области способа.
Примеры 10-18 иллюстрируют стадию 2 данного способа.
Пример 10
Синтез [PtCl2 (NН3) (2-пиколина)] в водном растворе.
6,819 г (12,50 ммол.) К[PtCl3 (2-пиколина)]·1,5 КСl помещают в 25-мл круглодонную колбу и добавляют 10 мл раствора 2,5 N КСl. 8,2688 г (63,12 ммол.) тригидрата ацетата аммония растворяют в 25 мл 2,5 N раствора гидроокиси аммония и добавляют к перемешиваемой смеси Pt. Общий объем реакционной смеси составляет ~35 мл. Смесь оранжевого цвета погружают в 45°С масляную баню и перемешивают в течение 1 часа в темноте при >1000 об/мин. Оранжевая смесь постепенно превращается в желтую. Желтый осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают водой (2×5 мл) и ацетоном (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. Выход: 3,8996 г (83%). Анализ: Вычислено (найдено) для C6H10N2Cl2Pt: С 19,16 (19,25); Н 2,68 (2,72); N 7,45 (7,43), Cl 18,85 (18,81).1H ЯМР (300 МГц/ ДМФ-d6): 9,19 (д, 1 пиридин Н); 8,03 (т, 1 пиридин Н); 7,15 (д, 1 пиридин Н); 7,51 (т, 1 пиридин Н); 4,39 (бс, 3 NН3 Н); 3,34 (с, 3 метил Н).195Pt ЯМР (300 МГц, ДМФ-d6): соответствует195Pt ЯМР спектру [PtCl2 (NН3) (2-пиколина)], полученного с применением известного в данной области способа. ВЭЖХ (катионная): время удерживания соответствует времени удерживания сравнительного соединения.
Пример 11
Синтез [PtCl2(NH3) (2,6-лутидина)3 в водном растворе.
1,7412 г (3,224 ммол.) К[PtCl3 (2, 6-лутидина)]·1,24 КСl помещают в 25-мл круглодонную колбу и добавляют 3 мл 2,5 N раствора КСl. 1,3478 г (17,48 ммол.) ацетата аммония растворяют в 6,4 мл 2,5 N раствора гидроокиси аммония и добавляют к перемешиваемой смеси Pt. Общий объем реакционной смеси составляет ~9,5 мл. Оранжевую смесь погружают в 45°С масляную баню и перемешивают в течение 40 часов в темноте при ~1000 об/мин. Оранжевая смесь постепенно превращается в желтую. Желтый осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают водой (2×5 мл) и ацетоном (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. Выход: 0,9791 г (78%).1H ЯМР (300 МГц, ДМФ-d6): 7,87 (т, 1 лутидин Н); 7,59 (д, 2 лутидин Н); 4,28 (бс, 3 NН3 Н); 3,49 (с, 6 метил Н).195ЯМР (300 МГц, ДМФ-d6): соответствует сравнительному соединению. Анализ: Вычислено (найдено) для C7H12N2Cl2Pt: С 21,55 (21,70); Н 3,10 (3,13); N 7,18 (7,07); Cl 18,17 (18,28).
Пример 12
Синтез [РtСl2(NН3)(2,5-диметилпиразина)] в водном растворе.
0,5325 г (0,8665 ммол.) К[РtСl3 (2,5-диметилпиразина)]·2,2 КСl помещают в 15-мл круглодонную колбу и добавляют 1,0 мл 2,8 М раствора КСl. 0,335 г (4,35 ммол.) ацетата аммония растворяют в 1,75 мл 2,5 М (4,38 ммол.) раствора гидроокиси аммония и добавляют к перемешиваемой реакционной смеси. Реакционную смесь погружают в 45°С масляную баню. Через 15 мин смесь приобретает желтый цвет. Через 1 час смесь охлаждают до температуры окружающей среды и желтый осадок собирают вакуумной фильтрацией с применением стеклянной фритты. Осадок промывают водой (2×10 мл) и ацетоном (1×10 мл) и сушат в вакууме при температуре окружающей среды.1H ЯМР (300 МГц, ДМФ-d7): 9,16 (с, 1 пиразин Н); 8,80 (с, 1 пиразин Н); 4,70 (бс, 3 NН3 Н); 3,26 (с, 3 метил Н); 2,69 (2, 3 метил Н).
Пример 13
Синтез [PtCl2(NH3) (2-пиколина)] в N-метилпирролидиноне/водном растворе.
1,84 г (14,0 ммол.) тригидрата ацетата аммония растворяют в 4,63 мл 2,9 N гидроокиси аммония. Водный раствор добавляют к 2,68 ммол. [РtСl3(2-пиколина)]-в 2,5 мл N-метилпирролидинона. Реакционный раствор перемешивают при 45°С в течение 80 мин. Образуется желтый осадок. Осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают водой (2×5 мл) и ацетоном (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов и взвешивают. Выход: 0,3391 г (34%). Анализ: Вычислено (найдено) для C6H10N2Cl2Pt: С 19,16 (19,22); Н 2,68 (2,69); N 7,45 (7,23); Cl 18,85 (18,83).1H ЯМР (300 МГц, ДМФ-d6): 9,2 (д, 1 пиридин Н); 8,0 (т, 1 пиридин Н); 7,2 (д, 1 пиридин, Н); 7,5 (т, 1 пиридин Н); 3,4 (с, 3 метил Н).195Pt ЯМР (300 МГц, ДМФ-d6): соответствует195Pt ЯМР-спектру [PtCl2(NH3) (2-пиколина)], полученного известным в данной области способом. ВЭЖХ (катионная): время удерживания соответствует времени удерживания сравнительного соединения.
Пример 14
Синтез [PtCl2(NH3)(2-пиколина)] в диметилформамиде/водном растворе.
[PtCl2(NН3)(2-пиколин)] получают в диметилформамиде/водном растворе в соответствии с примером 13. Анализ: Вычислено (найдено) для C6H10N2Cl2Pt: С 19,16 (19,30), Н 2,68 (2,62); N 7,45 (7,18); Cl 18,85 (18,59).1H ЯМР (300 МГц, ДМФ-d6): 9,1 (д, 1 пиридин Н); 8,1 (т, 1 пиридин Н); 7,3 (д, 1 пиридин Н); 7,4 (т, 1 пиридин Н); 3,4 (с, 3 метил Н).195Pt ЯМР (300 МГц, ДМФ-d6 ): соответствует195Pt ЯМР-спектру [PtCl2(NH3)(2-пиколина)], полученного известным в данной области способом.
Пример 15
Синтез [PtCl2(NH3)(диизопропиламина)] в ацетоне/водном растворе
6 мл 2,5 N гидроокиси аммония добавляют к [PtCl2(NH3) (диизопропиламину)]-(~2,69 ммол.) в 2,5 мл ацетона. рН раствора составляет 12. Реакционный раствор перемешивают при 45°С в течение 48 часов. Образуется желтый осадок, который собирают вакуумной фильтрацией с применением стеклянной фритты и промывают водой (2×5 мл) и диэтиловым эфиром (3×5 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16-24 часов. Анализ: Вычислено (найдено) для C6H18N2Cl2Pt·0,095C6H30N2Cl2Pt: С 20,00 (19,98); Н 4,90 (4,89); N 7,16 (7,12); Cl 18,11 (17, 93).1H ЯМР (300 МГц, ДМФ-d-): 4,5 (бс, 1 диизопропиламин Н), 3,9 (бс, 3 NН3 Н); 3,3 (м, 2 метил Н в диизопропиламине); 1,7 (д, 6 метил Н в диизопропиламине); 1,5 (д, 6 метил Н в диизопропиламине).195Pt ЯМР (300 МГц, ДМФ-d6): соответствует195Pt ЯМР-спектру [PtCl2(NH3) (диизопропиламина)], полученного известным в данной области способом.
Пример 16
Синтез [PtCl2 (2-пиколина) (NН2СН3)] в водном растворе.
0,5055 г (1,17 ммол.) К[PtCl3 (2-пиколина)] помещают в 15 мл круглодонную колбу и добавляют 1 мл 2/5 М раствора КСl. Суспензию погружают в 45°С масляную баню и перемешивают при ~1000 об/мин. Через пять минут к реакционной смеси добавляют раствор, состоящий из 0,1704 г 40% метиламина (2,19 ммол.) и 1 мл воды. рН раствора составляет 12. Нагревание прекращают после того, как общая продолжительность взаимодействия составит 1 час. Затем реакционную смесь охлаждают до температуры окружающей среды. Бледно-желтый осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают водой (2×20 мл) и ацетоном (3×20 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16 часов. Анализ: Вычислено (найдено) для C7H12N2Cl2Pt: С 21,55 (21,73); Н 3,10 (3,09); N 7,18 (7,14); Cl 18,17 (18,20).1H ЯМР (300 МГц, ДМФ-d7): 9,24 (д, 1 пиридин Н); 8,06 (т, 1 пиридин Н); 7,75 (д, 1 пиридин Н); 5,22 (бс, 2 метиламин Н); 3,35 (с, 3 метил Н 2-пиколина); 2,45 (т, 3 метил Н метиламина).
Пример 17
Синтез [PtCl2(2-пиколина)(NН(СН3)2)] в водном растворе.
0,5459 г (1,26 ммол.) К[РtСl3(2-пиколина)] помещают в 15-мл круглодонную колбу и добавляют 1,5 мл 2,5 М раствора КСl. Суспензию погружают в 45°С масляную баню и энергично перемешивают. Через 5 мин раствор, состоящий из 0,1426 г 40% диметиламина (1,27 ммол.) и ~1 мл воды, добавляют к реакционной смеси. рН раствора составляет 12. После часового взаимодействия нагревание прекращают и реакционную смесь охлаждают до температуры окружающей среды. Желтый осадок собирают вакуумной фильтрацией с применением стеклянной фритты и промывают водой (2×20 мл) и ацетоном (2×10 мл). Осадок сушат в вакууме при температуре окружающей среды в течение 16 часов. Анализ: Вычислено (найдено) для C8H14Cl2Pt: С 23,77 (24,00); Н 3,48 (3,49); N 6,93 (6,80); Cl 17,54 (17,63);1H ЯМР (300 МГц, ДМФ-d7): 9,31 (д, 1 пиридин Н); 7,78 (д, 1 пиридин Н); 7,58 (т, 1 пиридин Н); 6,06 (бс, 1 NH Н); 3, 37 (с, 3 метил Н пиколина); 2,76 (д, 3 метил Н диметиламина); 2,70 (д, 3 метил Н диметиламина).
Пример 18
Синтез [РtСl2(2-пиколина) (NВu3)] в водном растворе.
0,6289 г (1,45 ммол.) К[РtСl3 (2-пиколина)] помещают в 1,5-мл круглодонную колбу и добавляют 1,0 мл 2,5 М раствора КСl. Реакционную смесь погружают в 45°С масляную баню и энергично перемешивают в течение 5 мин. 0,2735 г (1,47 ммол.) трибутиламина растворяют в 1,0 мл воды и добавляют к оранжевой реакционной смеси. рН раствора составляет 12. Через час нагревание прекращают. После охлаждения до температуры окружающей среды осадок собирают вакуумной фильтрацией с применением стеклянной фритты. Собранную твердую фазу сушат в вакууме при температуре окружающей среды.1H ЯМР (300 МГц, ДМФ-d7): 9,14 (1 пиридин Н); 7,90 (т, 1 пиридин Н); 7,60 (д, 1 пиридин Н); 7,42 (т, 1 пиридин Н); 3,41 (с, 3 метил Н 2-пиколина); 3,28 (д, 2 метилен Н трибутиламина); 1,88 (тт, 2 метилен Н трибутиламина); 1,56 (м, 2 метилен Н трибутиламина); 1,10 (т, 3 метил Н трибутиламина).
Примеры 19-33 иллюстрируют дополнительные стадии данного способа.
Пример 19
Синтез c,t,c-[PtCl2(OH)2(NH3) (2-пиколина)].
5,0 мл воды и 5,0 мл 30% H2O2 добавляют к суспензии 3,142 г ZD0473 в 15-20 мл гептана. Данную смесь перемешивают и нагревают до ~80°С в течение 2 часов. Смесь охлаждают до комнатной температуры, а затем перемешивают в течение 1 часа на ледяной бане. Ярко-желтую твердую фазу собирают вакуумной фильтрацией и промывают водой и метанолом. Продукт сушат в вакууме при температуре окружающей среды в течение ночи. Выход: 2, 975 г (87%). Анализ: Вычислено (найдено) для C6H12N2Cl2O2Pt: С 17,57 (17,67); Н 2,95 (2,93); N 6,83 (6,79); Cl 17,29 (17,38).
Пример 20
Синтез с, t, с-[PtCl2(ОН)2(NH3)(2,3-диметилпиразина)].
2,5 мл воды и 3,5 мл 30% H2O2 добавляют к суспензии 1,6731 г cis-[(PtCl2(NH3)(2,3-диметилпиразина)] в 10 мл гептана. Данную смесь перемешивают и нагревают до ~80°С в течение 2 часов. Смесь охлаждают до комнатной температуры, а затем перемешивают в течение 1 часа на ледяной бане. Ярко-желтую твердую фазу собирают вакуумной фильтрацией и промывают водой и метанолом. Продукт сушат в вакууме при температуре окружающей среды в течение ночи. Выход: 1,1341 г (62%). Анализ: Вычислено (найдено) для С6Н13N3Сl2О2Рt: С 16,95 (16,81); Н 3,08 (3,12); Н 9,88 (9, 66); Cl 16,68 (16,44).
Пример 21
Синтез [PtCl(ОН)3(NH3)(2-пиколина)].
0,246 г LiOH·H2O растворяют в 5 мл воды. 2, 402 г c,t,c-[PtCl2(ОН)2 (NH3) (2-пиколина)] суспендируют в данном растворе. Смесь перемешивают в течение ночи при температуре окружающей среды. Желтая твердая фаза постепенно растворяется в течение ночи. рН раствора доводят до 7. Растворитель удаляют при пониженном давлении, получая твердое желтое вещество. Чтобы смыть образующийся LiCl, твердое вещество перемешивают в 10 мл этанола в течение 30 мин. Смесь центрифугируют и надосадочную жидкость сливают. Данный процесс промывания повторяют до удаления хлорида лития. Продукт сушат в вакууме при температуре окружающей среды в течение ночи. Выход: 1,209 (50%). Анализ: Вычислено (найдено) для С6Н13N2СlO3Р·2Н2O·0,12LiCl: С 16, 65 (16,45); Н 3,96 (4,04); N 6,47 (6,75); Cl 9,17 (9,47).
Пример 22
Синтез [PtCl(OAc)3(NH3)(2-пиколина)].
0,352 г [PtCl(ОН)3(NН3)(2-пиколина)] добавляют небольшими порциями к 1,1 мл уксусного ангидрида при 0°С. Данную смесь энергично перемешивают при температуре окружающей среды. Через 3 дня твердую фазу растворяют, получая раствор. Растворитель удаляют при пониженном давлении, получая желтое твердое вещество. Продукт сушат в вакууме при температуре окружающей среды в течение ночи. Выход: 0,314 г (70%). Анализ: Вычислено (найдено) для C12H19N2ClO6Pt: С 27,83 (27,93); Н 3,70 (3,66); N 5,41 (5,34); Cl 6,85 (7,00).
Пример 23
Синтез PtCl2(OAc)2(NH3)(2-пиколина).
1,367 г с,t,с-[PtCl2(ОН)2(NH3)(2-пиколина)] добавляют небольшими порциями к 3,1 мл уксусного ангидрида при 0°С. Данную смесь энергично перемешивают при комнатной температуре. Через 4 дня твердую фракцию собирают вакуумной фильтрацией и промывают диэтиловым эфиром. Продукт сушат в вакууме при температуре окружающей среды в течение ночи. Выход: 1,318 г (96%). Анализ: Вычислено (найдено) для C10H16N2Cl2O4Pt: С 24,30 (24,32); Н 3,26 (3,15); N 5,67 (5,66); Сl 14,35 (14,29).



















Данное изобретение представлено непосредственно описанием и примерами. Как указано выше, примеры предназначены только для иллюстрации и никоим образом не для ограничения данного изобретения. Кроме того, специалист в данной области, к которой относится данное изобретение, понимает, изучая описание и прилагаемую к нему формулу изобретения, что существуют эквивалентные решения заявленным аспектам данного изобретения. Авторы считают, что такие эквивалентные решения входят в разумный объем заявленного изобретения.
Реферат
Изобретение относится к усовершенствованному способу получения платиновых комплексов, имеющих формулу (Ia) или (Ib), включающему: 1а) первую стадию, на которой [PtA4]2- взаимодействует с L в соответствующих условиях в первом растворителе, образуя [PtA3(L)]-; 1b) вторую стадию, на которой [PtA3(L)]- взаимодействует с L' в соответствующих условиях во втором растворителе, образуя cis-[PtA2 (L')(L)]; 1с) в случае, когда Y представляет галоген или гидрокси, третью стадию, на которой cis-[PtA2(L')(L)] взаимодействует с Н2O2, Y2 или галогеном, содержащим оксидант, образуя с,t,с-[PtA2Y2(L')(L)]; в случае, когда Y представляет сложный эфир карбоксилата, карбамата или карбоната, четвертую стадию, на которой промежуточное соединение, в котором Y представляет гидрокси, полученный на стадии 1с), функционализируют соответствующим ацилирующим агентом; и 1d) в случае, когда А не представляет галоид или отличен от первоначального галоида, дополнительную стадию (стадии), на которой первоначальный галоид А промежуточного соединения, полученного на стадии 1а или 1b, 1с или 1d, превращают в другой галоид или новую уходящую группу (группы) А, такую как монодентат гидрокси, алкокси, карбоксилат или бидентат карбоксилат, фосфонокарбоксилат, дифосфонат или сульфат; где L, L' и Y имеют значения, приведенные в описании. Технический результат состоит в расширении ассортимента лекарственных препаратов, содержащих платину. 3 н. и 30 з.п. ф-лы, 3 табл. 9 ил.

Формула






Комментарии