Способ изомеризации ксилола и этилбензола с использовнием uzm-35 - RU2514423C1
Код документа: RU2514423C1
Чертежи
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к использованию цеолитной композиции UZM-35 в способе изомеризации ксилолов и этилбензола. Цеолитная композиция UZM-35 может присутствовать в катализаторе в виде немодифицированной цеолитной композиции UZM-35 или в виде модифицированной цеолитной композиции UZM-35. Содержащий UZM-35 катализатор может принимать одну из нескольких форм, включая, например, катализатор в виде сфер, полученный способом прикапывания в масло, или экструдированный катализатор.
Уровень техники
Цеолиты представляют собой кристаллические алюмосиликатные композиции, являющиеся микропористыми и имеющими трехмерный оксидный каркас, образованный имеющими общую вершину тетраэдрами AlO2 и SiO2. В различных производственных процессах используется множество различных цеолитов, как естественного происхождения, так и изготовленных синтетическим путем. Синтетические цеолиты готовятся гидротермальным синтезом, использующим подходящие источники Si, Al и структурообразующие агенты, такие как щелочные металлы, щелочноземельные металлы, амины или аммонийорганические катионы. Структурообразующие агенты локализуются в порах цеолита и в значительной степени отвечают за конкретную, образующуюся в конечном счете структуру. Эти соединения уравновешивают сообщаемый каркасу алюминием заряд и могут также служить в качестве заполнителей пустот. Цеолиты отличаются наличием отверстий пор однородных размеров, наличием существенной ионообменной способности и способности к обратимой десорбции поглощенной фазы, которая распределяется по внутренним пустотам кристалла без значительных изменений положения каких-либо атомов, составляющих постоянную структуру кристалла цеолита. Топологические структуры цеолитов описаны в Atlas of Zeolite Framework Types (Атлас типов каркасов цеолитов), который поддерживается Структурной комиссией Международной цеолитной ассоциации (International Zeolite Association Structure Commission) по адресу http://www.iza-structure.org/databases/. Цеолиты могут использоваться в качестве катализаторов для реакций конверсии углеводородов, которые могут происходить на внешних поверхностях, а также на внутренних поверхностях в порах.
Катализаторы для изомеризации ароматических соединений C8 обычно классифицируются по способу переработки этилбензола, связанного с изомерами ксилола. Этилбензол не очень легко изомеризуется в ксилолы, но обычно подвергается конверсии на установках изомеризации, поскольку его отделение от ксилолов сверхчеткой ректификацией или адсорбцией слишком дорого. Широко применяемый подход состоит в деалкилировании этилбензола с преимущественным образованием бензола при изомеризации ксилолов до почти равновесной смеси. Альтернативный подход заключается в проведении реакции этилбензола с образованием смеси ксилолов через конверсию в нафтены и обратное преобразование из нафтенов в присутствии твердого кислотного катализатора с функцией гидрогенизации-дегидрогенизации.
Первый подход обычно приводит к более высокой степени конверсии этилбензола, уменьшающей тем самым количество рециклов в установку извлечения параксилола и снижающей сопутствующие затраты на переработку, но последний подход увеличивает выход ксилола посредством образования ксилолов из этилбензола.
Каталитическая композиция и способ, которые увеличивают конверсию согласно последнему подходу, то есть обеспечивают изомеризацию этилбензола в ксилолы с высокой конверсией, способны приводить к значительному улучшению экономики производства ксилола.
Особенно предпочтительным был бы пригодный для промышленного применения катализатор, содержащий 12-членные кольца и 10-членные кольца в одной и той же трехмерной структуре. Применимость для промышленных целей обычно наблюдается в алюмосиликатных структурах, которые синтезируются в гидроксидных средах с легкодоступными структурообразующими агентами. Цеолиты, которые содержат и 12-членные, и 10-членные кольца в трехмерных структурах, принадлежат к структурным типам CON, DFO, IWR, IWW и MSE. Синтез CIT-1, цеолита со структурой типа CON, описан в патенте США 5512267 и в J. Chem. Soc. 1995, 117, 3766-79 в виде боросиликатной формы. После завершения синтеза может быть осуществлена следующая стадия, обеспечивающая замену Al на В. Цеолиты SSZ-26 и SSZ-33, также со структурным типом CON, описаны в патенте США 4910006 и патенте США 4963337 соответственно. SSZ-33 также описан в виде боросиликата. Все 3 члена структурного типа CON используют очень сложные, трудно синтезируемые структурообразующие агенты, что осложняет их коммерческое использование. Известным представителем структурного типа DFO является DAF-1, который описан как алюмофосфат в Chem. Commun. 1993, 633-35 и в Chem. Mater. 1999, 11, 158-63. Цеолиты со структурными типами IWR и IWW синтезируются только способами, подразумевающими использование фтористоводородной кислоты, что делает сложным их коммерческое применение.
Один специфический цеолит со структурным типом MSE, названный МСМ-68, был раскрыт Calabro и др. в 1999 г. (патент США 6049018). Этот патент описывает синтез МСМ-68 из дикатионных структурных агентов N,N,N',N'-тетрааликилбицикло[2.2.2]окт-7-ен-2R,3S:5R,6S-дипирролидиний-дикатиона и N,N,N',N'-тетрааликилбицикло[2.2.2]октан-2R,3S:5R.,6S-дипирролидиний-дикатиона. Найдено, что МСМ-68 имеет по меньшей мере одну систему каналов, в которой каждый канал ограничен 12-членным кольцом из тетраэдрически координированных атомов и по меньшей мере две дополнительные независимые системы каналов, в которых каждый канал ограничен 10-членным кольцом тетраэдрически координированных атомов, при этом число уникальных каналов из 10-членных колец вдвое больше числа каналов из 12-членных колец, см. патент США 2009/318696.
Заявители успешно приготовили новое семейство материалов, обозначенное UZM-35. Топология основного компонента этих материалов подобна наблюдаемой для МСМ-68. Данные материалы были получены с использованием простых, коммерчески доступных структурообразующих агентов, таких как гидроксид диметилдипропиламмония, во взаимодействии с совместно применяемыми в небольших количествах К+ и Na+ и применяя к синтезу цеолитов метод несогласованности плотности заряда (Charge Density Mismatch Approach), как показано в патенте США 7578993.
Семейство материалов UZM-35 способно обеспечивать и поддерживать высокую степень конверсии при реакциях изомеризации этилбензола и ксилола и минимизировать потери ароматических соединений. Это, как полагают, происходит из-за его специфической геометрии пор и мольного соотношения Si/Al в каркасе. Цеолитная композиция UZM-35 содержит значительные количества Al в тетраэдрическом каркасе, с мольным соотношением Si/Al в пределах от 2 до 12. Содержание Al в каркасе, как известно, обеспечивает кислотные центры, необходимые для обеспечения высокой активности в процессах изомеризации.
Благодаря уникальной структуре UZM-35, изготовленные из UZM-35 катализаторы при испытании и подтверждении принципа их действия способны демонстрировать 30-40% преимущество в сохранении ароматических соединений по сравнению с катализатором на основе цеолита MTW с каналами, образованными 12-членными кольцами. Кроме того, экструдат, содержащий UZM-35, также показывает различное распределение побочных ароматических продуктов и уникальный характер в течение начального периода по сравнению с экструдатами, содержащими цеолиты MTW. В частности, образование побочных продуктов снижается без какого-либо уменьшения активности в отношении изомеризации ксилолов, предполагая деактивацию участков, специфических для протекания нежелательных реакций.
Раскрытие изобретения
Настоящее изобретение относится к способу изомеризации ксилолов и этилбензола с использованием катализатора, содержащего композицию UZM-35. Данный способ содержит приведение ксилолов и этилбензола в контакт с цеолитной композицией UZM-35 в условиях изомеризации с целью получения каталитически изомеризованного продукта. Условия изомеризации обычно включают температуру от 100 до 500°С, абсолютное давление от 10 кПа до 5 МПа, часовую объемную скорость жидкости от 0,5 до 10 час-1 и мольное соотношение водорода к углеводороду от 0,5:1 до 10:1.
Композиция UZM-35 представляет собой композицию микропористого кристаллического цеолита, имеющего трехмерный каркас из по меньшей мере AlO2 и SiO2 тетраэдрических звеньев и эмпирический состав в состоянии «как синтезировано», отображаемый в безводном представлении эмпирической формулой
где "М" представляет комбинацию способных к обмену катионов калия и натрия, "m" - мольное отношение М к (Al+E), варьирующее от 0,05 до 3, "R" - однозарядный аммонийорганический катион, выбранный из группы, состоящей из диметилдипропиламмония (DMDPA+), диметилдиизопропиламмония (DMDIP+), холина, этилтриметиламмония (ЕТМА+), диэтилдиметиламмония (DEDMA+), триметилпропиламмония, триметилбутиламмония, диметилдиэтаноламмония, тетраэтиламмония (TEA+), тетрапропиламмония (ТРА+), метилтрипропиламмония и их смесей, "r" - мольное отношение R к (Al+E), имеющее величину от 0,25 до 2,0, "Е" является элементом, выбранным из группы, состоящей из галлия, железа, бора и их смесей, "х" - мольная доля Е, имеющая величину от 0 до 1,0, "у" является мольным отношением Si к (Al+E), варьирующим от более 2 до 12, и "z" - мольное отношение О к (Al+E), величина которого определяется уравнением
z=(m+r+3+4-y)/2
и характеризуется демонстрируемыми на рентгеновской дифрактограмме величинами межплоскостных расстояний d и интенсивностей, представленными в таблице А.
и в одном воплощении является термически устойчивой вплоть до температур, превышающих 400°С, а в другом воплощении - 600°С. При использовании описанного в J.Appl. Cryst. (1969) 2, 65-71) уточнения по Ритвельду показатели межплоскостных расстояний d и интенсивностей на рентгеновской дифрактограмме имеют по меньшей мере величины, представленные в таблице А'
Описанная выше композиция кристаллического микропористого цеолита может синтезироваться посредством приготовления реакционной смеси, содержащей реакционно-способные источники М, R, Al, Si и, при необходимости, Е, и нагреванием реакционной смеси при температуре от 150°С до 200°С или от 165°С до 185°С в течение времени, достаточного для образования композиции цеолита, при этом реакционная смесь имеет следующий состав в выражении мольных соотношений оксидов:
аМ2О:bR2/pO:1-cAl2O3:сЕ2О3:dSiO2:eH2O
где "а" имеет величину от 0,05 до 1,25, "b" имеет величину от 1,5 до 40, "р" - средневзвешенная валентность R, варьирующая от 1 до 2, "с - имеет величину от 0 до 1,0, "d" имеет величину от 4 до 40, "е" имеет величину от 25 до 4000.
Краткое описание чертежей
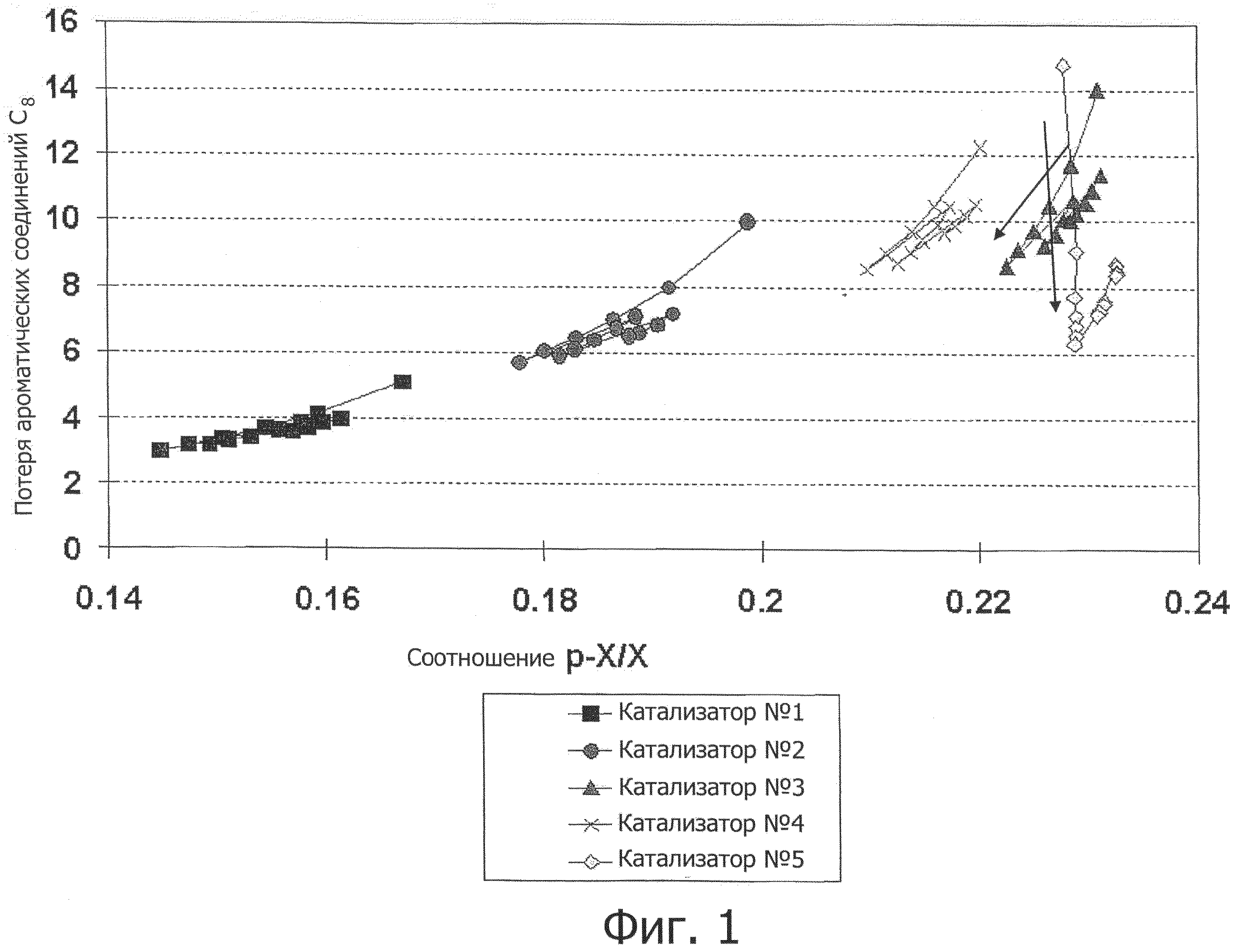
Фиг.1 - график результатов каждого эксперимента, где на оси Y отложены показатели потерь ароматических соединений С8, а по оси Х - отношение количества параксилола к общему количеству ксилола (РХ:Х). «Потери ароматических соединений C8» представлены в единицах мол.%, определенных как (1 - (нафтены и ароматические соединения C8 на выходе)/нафтены и ароматические соединения C8 на входе))·100.
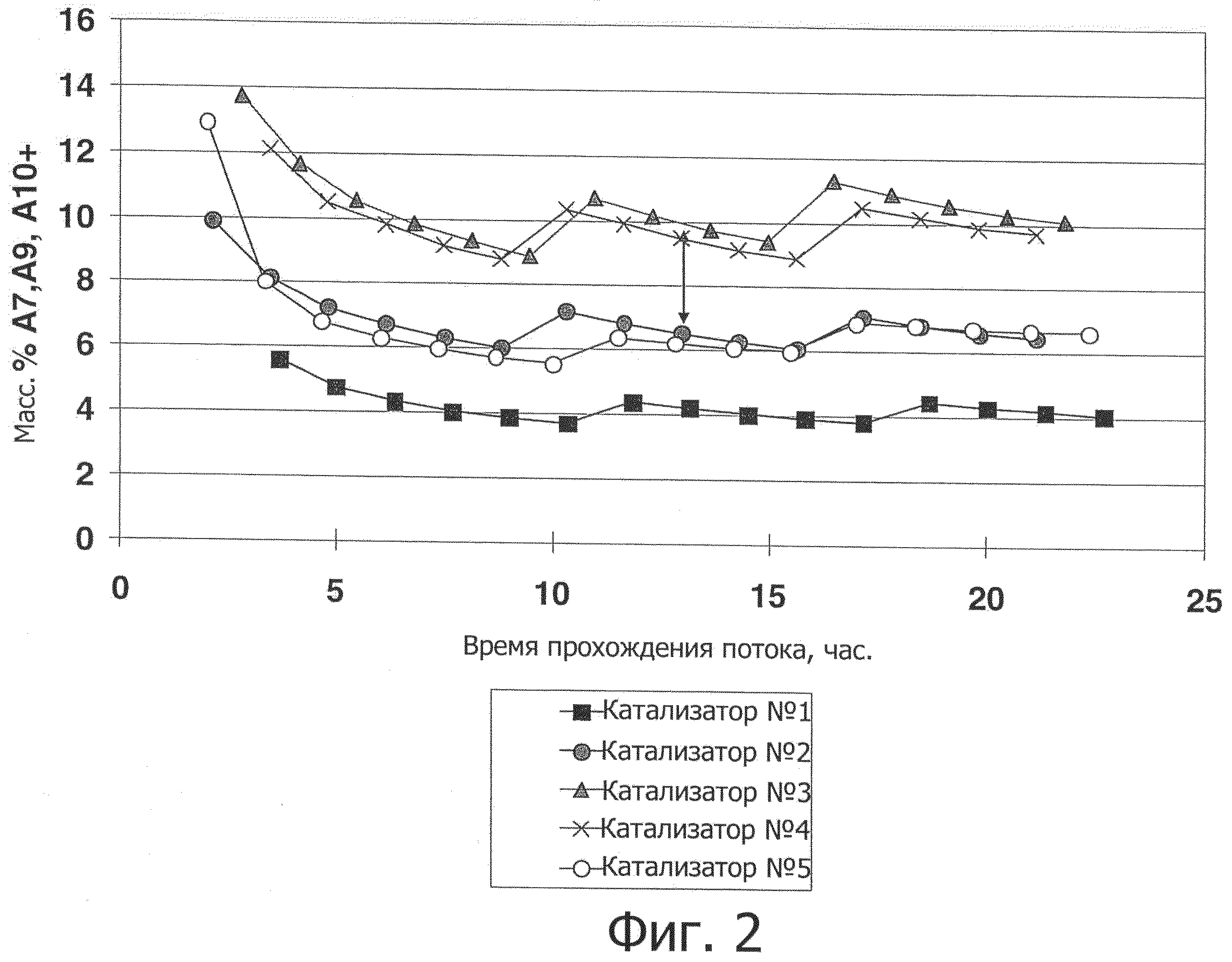
Фиг.2 представляет график масс.% А7, А9 и А10+ в зависимости от времени действия (в часах) для каждого из экспериментов, демонстрирующий, что наблюдающаяся потеря ароматических соединений главным образом обусловлена переалкилированием.
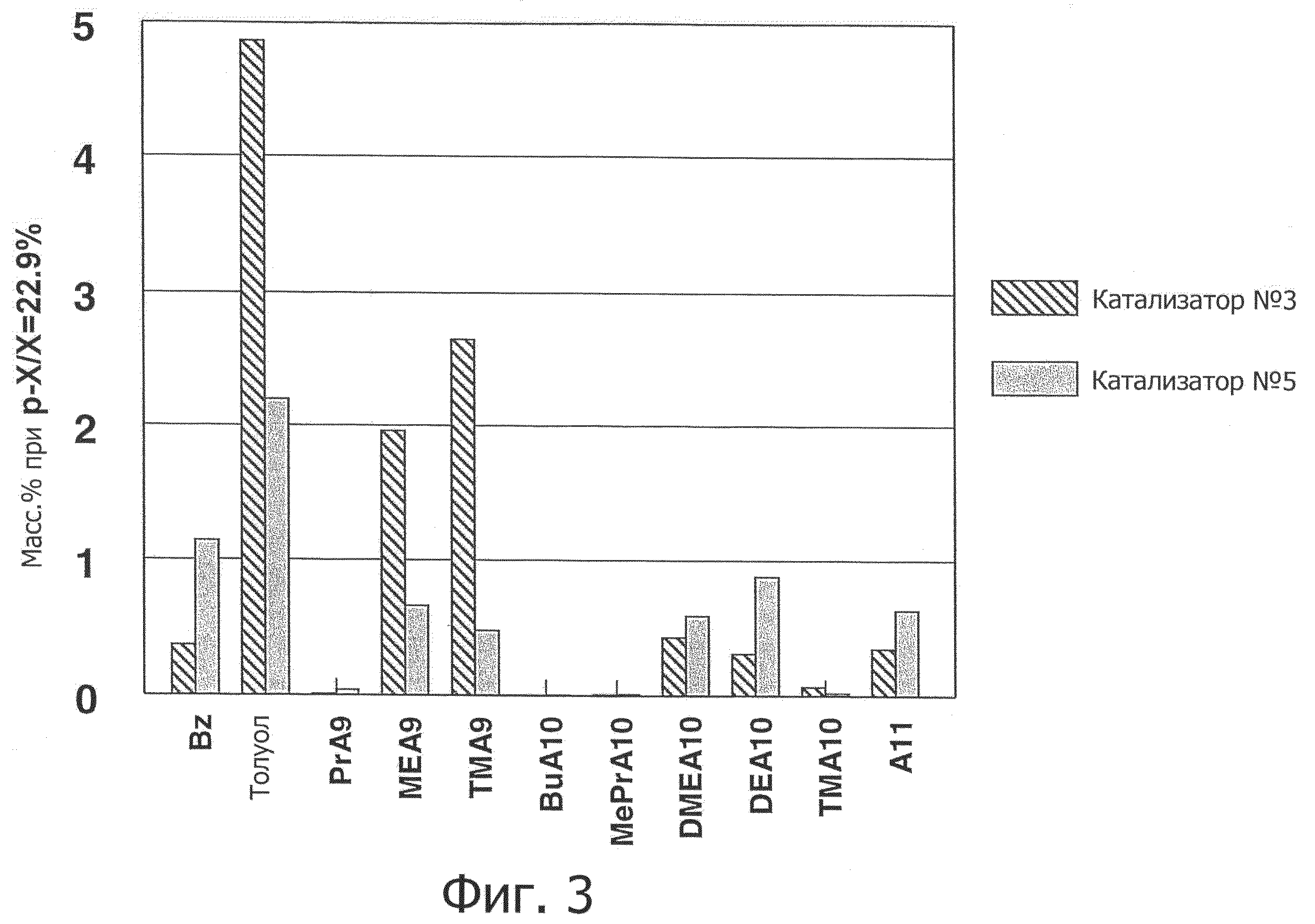
Фиг.3 показывает распределение продуктов, образующихся в результате использования представленных в примерах катализатора №3 и катализатора №5.
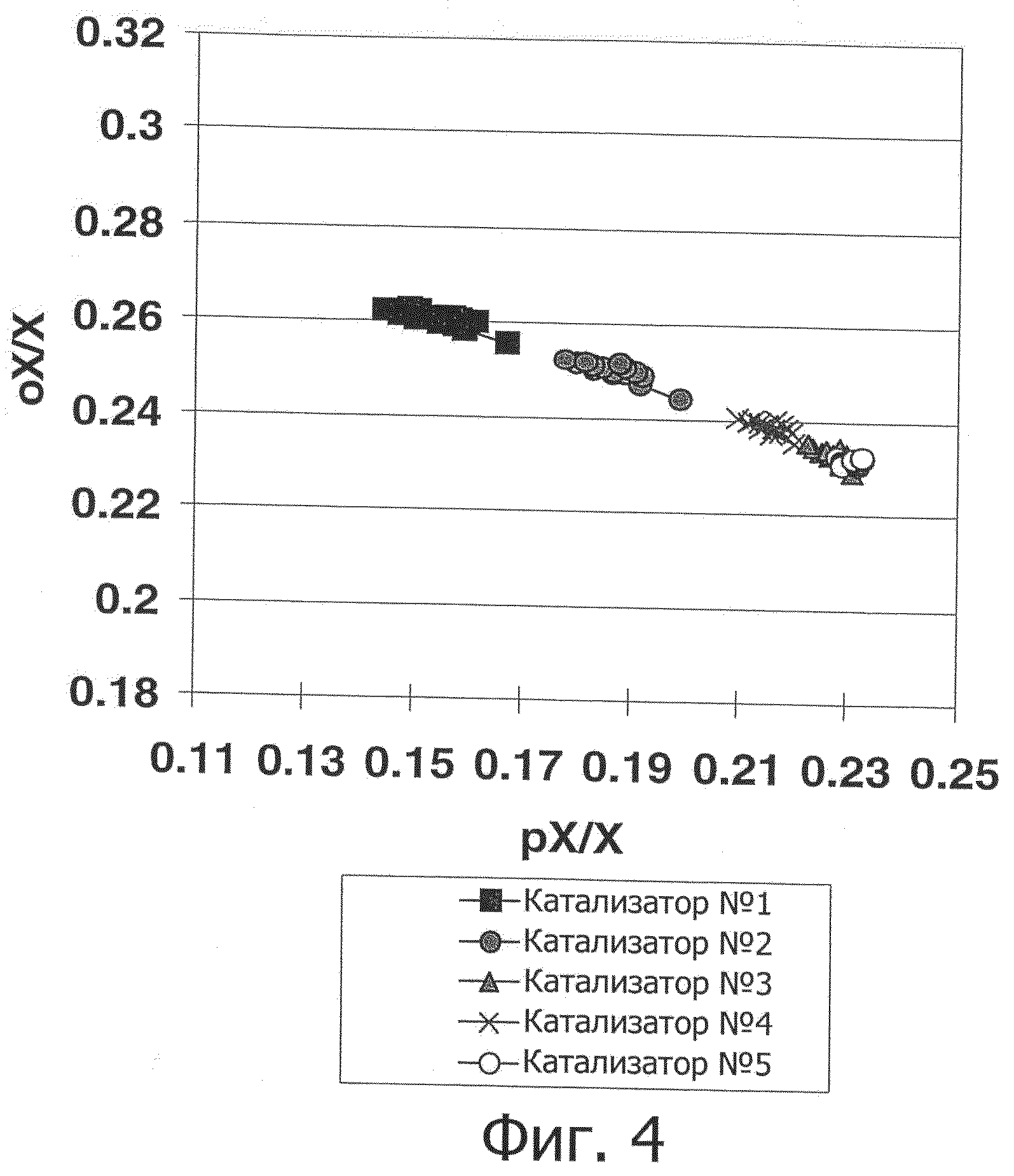
Фиг.4 показывает, что катализатор изобретения обеспечивает получение ксилолов с такой же кривой соотношений орто-ксилола и параксилола, как и катализаторы на основе цеолита MTW.
Осуществление изобретения
Заявители получили композицию алюмосиликатного цеолита UZM-35, которая имеет главный компонент, топологическая структура которого согласно описанию в Atlas of Zeolite Framework Types, поддерживаемом Структурной комиссией Международной цеолитной ассоциации (http://www.iza-structure.org/databases) относится к типу MSE. Как подробно показано в USAN 12/241,302, композиция UZM-35 отличается от МСМ-68 по целому ряду своих особенностей. Настоящая композиция микропористого кристаллического цеолита UZM-35 имеет эмпирический состав в состоянии «как синтезировано», отображаемый в безводном представлении эмпирической формулой
где "М" представляет комбинацию способных к обмену катионов калия и натрия. "R" является однозарядным аммонийорганическим катионом, примеры которого включают, но не ограничиваются катионом диметилдипропиламмония (DMDPA+), диметилдиизопропиламмонием (DMDIP+), холином [(CH3)3N(CH2)2OH]+, ЕТМА+, DEDMA+, триметилпропиламмонием, триметилбутиламмонием, диметилдиэтаноламмонием, метилтрипропиламмонием, ТЕА+, TPA+ и их смесями, "r" - мольное отношение R к (Al+Е), варьирующее от 0,25 до 2,0, в то время как "m" - мольное отношение М к (Al+Е), варьирующее от 0,05 до 3. Мольное отношение кремния к (Al+Е) представлено параметром "у", который изменяется от 2 до 30. Е является тетраэдрически координированным элементом, который присутствует в каркасе и выбран из группы, состоящей из галлия, железа и бора. Мольная доля Е представлена показателем "х" и имеет величину от 0 до 1,0, в то время как "z" - мольное отношение О к (Al+Е), определяемое по уравнению
z=(m·n+r+3+4·у)/2
В случае когда М является только одним металлом, средневзвешенный показатель валентности представляется валентностью этого одного металла, то есть +1 или +2. Однако, когда присутствует более одного металла М, общее количество составляет
и средневзвешенная величина валентности "n" задается уравнением
Композиция микропористого кристаллического цеолита UZM-35 готовится гидротермальной кристаллизацией реакционной смеси, полученной объединением реакционно-способных источников М, R, алюминия, кремния и, при необходимости, Е. Источники алюминия включают, но не ограничиваются алкоксидами алюминия, осажденными оксидами алюминия, металлическим алюминием, солями алюминия и золями оксида алюминия. Конкретные примеры алкоксидов алюминия включают, но не ограничиваются орто-втор-бутоксидом алюминия и орто-изопропилатом алюминия. Источники диоксида кремния включают, но не ограничиваются тетраэтилортосиликатом, коллоидным диоксидом кремния, осажденным диоксидом кремния и силикатами щелочных металлов. Источники элементов Е включают, но не ограничиваются боратами щелочных металлов, борной кислотой, осажденным метагидроксидом галлия, сульфатом галлия, сульфатом железа и хлоридом железа. Источники М металлов, калия и натрия, включают галоидные соли, нитратные соли, ацетатные соли и гидроксиды соответствующих щелочных металлов. R является аммонийорганическим катионом, выбираемым из группы, состоящей из диметилдипропиламмония, холина, ЕТМА, DEDMA, TEA, ТРА, триметилпропиламмония, триметилбутиламмония, диметилдиэтаноламмония и их смесей, и данные источники включают гидроксидные, хлоридные, бромидные, йодидные и фторидные соединения. Конкретные примеры включают без ограничения гидроксид диметилдипропиламмония, хлорид диметилдипропиламмония, бромид диметилдипропиламмония, гидроксид диметилдиизопропиламмония, хлорид диметилдиизопропиламмония, бромид диметилдиизопропиламмония, гидроксид этилтриметиламмония, гидроксид диэтилдиметиламмония, гидроксид тетраэтиламмония, гидроксид тетрапропиламмония и хлорид тетрапропиламмония.
Следует заметить, что в ходе синтеза металл М, в частности калий и натрий, имеет валентность +1. Однако в одном альтернативном воплощении композиция может подвергнуться дополнительным постсинтетическим стадиям ионного обмена с целью снабжения материала одним или несколькими металлами М, имеющими валентность +2.
Реакционная смесь, содержащая реакционно-способные источники требуемых компонентов, в представлении мольных соотношений оксидов может быть описана формулой
аМ2О:bR2/pO:1-cAl2O3:сЕ2О3:dSiO2:eH2O
где "а" варьирует от 0,05 до 1,25, "b" варьирует от 1,5 до 40, "с - варьирует от 0 до 1,0, "d" варьирует от 4 до 40, "е" варьирует от 25 до 4000 и "р" - средневзвешенная валентность R, варьирующая от 1 до 2. В случае использования алкоксидов предпочтительно включение стадии дистилляции или выпаривания с целью удаления продуктов гидролиза спирта. Реакционная смесь вступает в реакцию при температуре от 150°С до 200°С, от 165°С до 185°С или от 170°С до 180°С в течение времени от 1 дня до 3 недель и предпочтительно в течение времени от 5 дней до 12 дней в герметизированном реакторе под автогенным давлением. После завершения кристаллизации твердый продукт выделяется из гетерогенной смеси с помощью таких способов, как фильтрация или центрифугирование, а затем промывается деминерализованной водой и высушивается на воздухе при температуре окружающей среды вплоть до 100°С. Следует отметить, что для ускорения образования цеолита к реакционной смеси при необходимости могут быть добавлены затравки кристаллизации UZM-35.
Предпочтительный путь синтеза для получения композиции UZM-35 использует концепцию несогласованности плотности заряда, которая раскрывается в патенте США 7578993 и в публикации Studies in Surface Science and Catalysis, (2004), том 154А, 364-372. Раскрываемый в патенте США 7578993 способ использует четверичные гидроксиды аммония для солюбилизации алюмосиликатных соединений, тогда как индуцирующие кристаллизацию агенты, такие как щелочные и щелочноземельные металлы и более высокозаряженные аммонийорганические катионы, часто вносятся на отдельной стадии. Как только в ходе применения этого подхода образуется некоторое количество затравок кристаллизации UZM-35, данные затравки могут быть использованы на отдельной стадии синтеза UZM-35 с использованием, например, комбинации гидроксида диметилдипропиламмония и катионов щелочных металлов. Использование для приготовления UZM-35 коммерчески доступного гидроксида диметилдипропиламмония предполагает значительные экономические преимущества перед структурообразующими агентами, используемыми ранее для приготовления алюмосиликатов с топологией MSE (N,N,N',N'-тетраалкилбицикло[2.2.2]окт-7-ен-2R,38:5R,68-дипирролидиниум-дикатионом, N,N,N',N'-тетраалкилбицикло[2.2.2]октан-2R,38:5R,68-дипирролидиниум-дикатионом и 1,1-диметил-4-циклогексилпиперазиниум-катионом). Помимо этого, опираясь на концепцию несогласованности плотности заряда, в целях дальнейшего снижения затрат гидроксид диметилдипропиламмония может использоваться в виде гидроксида или хлорида совместно с другими недорогими аммонийорганическими гидроксидами.
Полученная с помощью вышеописанного способа алюмосиликатная цеолитная композиция UZM-35 характеризуется рентгеновской дифрактограммой с величинами межплоскостных расстояний d и интенсивностей, представленными в таблице А.
Как будет подробно показано в примерах, материал UZM-35 является термически и каталитически устойчивым вплоть до температур по меньшей мере в 400°С и в другом воплощении вплоть до 600°С. Композиция UZM-35 в состоянии «как синтезировано» содержит цеолит топологии MSE, цеолит топологии MFI и цеолит топологии ERI. Как правило, количество цеолита MSE в композиции варьирует от около 55 масс.% до около 75 масс.% или от около 55 масс.% до около 90 масс.%. Количество цеолита MFI варьирует от около 20 масс.% до около 35 масс.% от массы композиции или от около 10 масс.% до около 35 масс.%, а количество цеолита ERI варьирует от около 3 масс.% до около 9 масс.% от массы композиции или от около 3 масс.% до около 10 масс.%. Разумеется, суммарное количество этих трех цеолитов при отсутствии каких-либо других примесей в целом составляет 100 масс.% композиции. При использовании описанного в J.Appl. Cryst. (1969) 2, 65-71) уточнения по Ритвельду межплоскостные расстояния d и интенсивности на рентгеновской дифрактограмме имеют по меньшей мере величины, представленные в таблице А'.
Одно преимущество материала UZM-35 состоит в том, что он может использоваться в качестве катализатора изомеризации ксилола и этилбензола без необходимости удаления из синтезированного материала калия. Другими словами, калий для обеспечения активности катализатора изомеризации удаляться не должен. Катализатор в его каталитически активном состоянии может содержать калий и алюминий в мольном соотношении менее 0,90.
Синтезированный материал UZM-35 будет содержать в своих порах некоторые из способных к обмену или уравновешивающих заряд катионов. Эти способные к обмену катионы могут замещаться другими катионами или в случае органических катионов они могут быть удалены нагреванием при контролируемых условиях. Поскольку UZM-35 содержит цеолит с большими порами, также оказывается возможным удаление некоторых органических катионов непосредственно с помощью ионного обмена. Цеолитная композиция UZM-35 может модифицироваться различными способами для адаптации к использованию в конкретных применениях. Модификации включают прокаливание, ионный обмен, обработку паром, экстрагирование различными кислотами, обработку гексафторосиликатом аммония или любые их комбинации, как это изложено для случая UZM-4M в патенте США 6776975 В1, который является полностью включенным в настоящее описание посредством ссылки. Модифицируемые свойства включают пористость, адсорбцию, отношение Si/Al, кислотность, термическую стабильность и т.п.
Для удобства получения частиц катализатора композиция UZM-35 предпочтительно смешивается со связующим веществом в пропорции от 1 до 100 масс.% цеолита и от 0 до 99 масс.% связующего вещества, при этом композиция предпочтительно содержит от 2 до 90 масс.% композита. Связующее вещество должно предпочтительно быть пористым, иметь площадь поверхности от 5 до 800 м2/г и быть относительно тугоплавким в условиях, применяемых в процессе конверсии углеводородов. Неограничивающими примерами связующих веществ являются оксид алюминия, диоксид титана, диоксид циркония, оксид цинка, оксид магния, оксид бора, алюмосиликаты, магнийсиликаты, хромсиликаты, борсиликаты, цирконийсиликаты, диоксид кремния, силикагель и глины. Предпочтительными связующими веществами являются аморфный диоксид кремния и оксид алюминия, включая гамма-, эта- и тета-оксид алюминия, при этом гамма- и эта-оксид алюминия являются особенно предпочтительными.
Композиция UZM-35 с или без связующего вещества может формоваться в виде различных форм, таких как таблетки, драже, экструдаты, сферы и т.д. Предпочтительные формы представлены экструдатами и сферами. Экструдаты готовятся обычным способом, который включает смешивание цеолита либо до, либо после добавления металлических компонентов со связующим веществом и подходящим химическим пластификатором для образования гомогенного теста или густой пасты, имеющей влагосодержание, подходящее для обеспечения возможности образования экструдатов с целостностью, приемлемой для противостояния прямому прокаливанию. Затем тесто экструдируется через формующую головку для получения отформованного экструдата. Возможно множество различных форм экструдата, включая, но не ограничиваясь, цилиндрами, в виде трилистника, гантели и симметричными и асимметричными многолопастными формами. Также в рамках данного изобретения экструдатам может быть далее с помощью любых известных в данной области средств придана любая требуемая форма, например сферическая.
Сферы могут быть приготовлены известным способом прикапывания в масло, который описан в патенте США 2620314, включенном в настоящее описание посредством ссылки. Данный способ включает прикапывание смеси цеолита и, например, золя оксида алюминия и гелеобразующего агента в масляную баню, поддерживаемую при повышенных температурах. Капельки смеси остаются в масляной бане до затвердевания и образования сфер из гидрогеля. После этого сферы в непрерывном режиме извлекаются из масляной бани и обычно подвергаются специальной обработке по старению в масле и аммиачном растворе для дальнейшего улучшения их физических свойств. Получающиеся состаренные гелеобразные частицы после этого промываются, высушиваются при относительно низкой температуре 50-200°С и подвергаются процедуре обжига при температуре 450-700°С в течение от 1 до 20 часов. Такая обработка приводит к преобразованию гидрогеля в соответствующую матрицу из оксида алюминия.
Катализаторы изобретения содержат компонент гидрирующего катализатора, который является металлом платиновой группы, включая один или более из платины, палладия, родия, рутения, осмия и иридия. Предпочтительным металлом платиновой группы является платина. Компонент металла платиновой группы может существовать в готовой каталитической композиции в виде соединения, такого как оксид, сульфид, галоидное соединение, оксисульфид и т.д., или же в виде элементарного металла, либо в комбинации с одним или более другими ингредиентами каталитической композиции. Предполагается, что наилучшие результаты достигаются, когда по существу весь компонент металла платиновой группы присутствует в восстановленном состоянии. Компонент металла платиновой группы в целом в пересчете на элемент содержится в количестве от 0,01 до 5 масс.% и предпочтительно от 0,1 до 2 масс.% от массы готовой каталитической композиции.
Компонент металла платиновой группы может быть включен в каталитическую композицию любым подходящим образом. Один способ приготовления катализатора включает использование растворимого в воде, разложимого соединения металла платиновой группы для пропитки прокаленного композита молекулярное сито/связующее вещество. В качестве варианта соединение металла платиновой группы может быть добавлено во время объединения цеолита и связующего вещества. Еще один способ обеспечения подходящего распределения металла состоит в объединении металлического компонента со связующим веществом перед совместным экструдированием цеолита и связующего вещества. Комплексные соединения металлов платиновой группы, которые могут использоваться согласно вышеупомянутым или другим известным способам, включают платинохлористоводородную кислоту, палладийхлористоводородную кислоту, хлороплатинат аммония, платинабромистоводородную кислоту, трихлорид платины, гидрат тетрахлорида платины, дихлориддихлоркарбонил платины, хлороплатинат тетрамин, динитродиаминоплатину, тетранитроплатинат (II) натрия, хлорид палладия, нитрат палладия, сульфат палладия, гидроксид диаминохлорида палладия (II), хлорид тетраминпалладия (II) и т.п.
В объеме настоящего изобретения каталитический композит может содержать другие металлические компоненты, известные способностью модифицировать действие компонента, представленного металлом платиновой группы. Такие металлы-модификаторы могут включать рений, олово, германий, свинец, кобальт, никель, индий, галлий, цинк, уран, диспрозий, таллий и их смеси. Каталитически эффективные количества таких металлов-модификаторов могут быть включены в катализатор любым известным в данной области способом, обеспечивающим их гомогенное или послойное распределение.
Каталитический композит настоящего изобретения может содержать галоидный компонент. Галоидный компонент может быть либо фтором, хлором, бромом, либо йодом, или их смесью, предпочтительным при этом является хлор. Галоидный компонент обычно присутствует в виде химического соединения с неорганическим оксидом-носителем. Используемый при необходимости галоидный компонент предпочтительно является хорошо диспергированным по всему катализатору и в пересчете на элементы может составлять от более 0,2 до 5 масс.% конечного катализатора. Галоидный компонент может вводиться в каталитическую композицию любым подходящим образом либо в ходе приготовления неорганического оксида-носителя, либо до, во время или после внесения других каталитических компонентов.
Каталитический композит высушивается при температуре от 100 до 320°С в течение времени от 2 до 24 или более часов и обычно прокаливается при температуре от 400 до 650°С в течение времени от 1 до 10 часов до тех пор, пока присутствующие металлические соединения не преобразуются по существу в оксидную форму. Если желательно, используемый при необходимости галоидный компонент может быть отрегулирован введением галогена или содержащего галоген соединения в воздушной атмосфере.
Получающийся в результате обожженный композит возможно подвергается стадии восстановления в по существу безводном состоянии для обеспечения однородного высокодисперсного распределения используемых при необходимости металлических компонентов. Восстановление может при необходимости осуществляться in situ (на месте). В качестве восстановителя на этой стадии используется по существу чистый и сухой водород (то есть содержащий по объему менее 20 ч./млн. Н2О). Восстановитель вступает в контакт с катализатором в условиях, подходящих для восстановления по существу всех компонентов-металлов VIII группы в металлическое состояние и включающих температуру от 200 до 650°С и продолжительность от 0,5 до 10 часов. В некоторых случаях также может быть полезным подвергнуть полученный восстановленный каталитический композит пресульфидированию известным в данной области способом для введения в каталитический композит от 0,05 до 1,0 масс.% серы в пересчете на элемент.
Исходное сырье для изомеризации ароматических соединений содержит изомеризуемые алкилароматические углеводороды общей формулы C6H(6-n)Rn, где n является целым числом от 1 до 5 и R представлен CH3, C2H5, C3H7 или C4H9, в любой комбинации и включая все их изомеры в целях получения более ценных алкилароматических изомеров. Подходящие алкилароматические углеводороды включают без ограничения ортоксилол, метаксилол, параксилол, этилбензол, этилтолуолы, триметилбензолы, диэтилбензолы, триэтилбензолы, метилпропилбензолы, этилпропилбензолы, диизопропилбензолы и их смеси.
Изомеризация смеси ароматических соединений C8, содержащей этилбензол и ксилолы, является особенно предпочтительным применением для цеолитов изобретения. В целом такая смесь будет иметь содержание этилбензола в приблизительном диапазоне от 5 до 50 масс.%, содержание орто-ксилола в приблизительном диапазоне от 0 до 35 масс.%, содержание метаксилола в приблизительном диапазоне от 20 до 95 масс.% и содержание параксилола в приблизительном диапазоне от 0 до 15 масс.%. Предпочтительно, чтобы упомянутые выше ароматические соединения С8 содержали неравновесную смесь, то есть чтобы по меньшей мере один изомер ароматического соединения C8 присутствовал в концентрации, которая отличалась по существу (определяется здесь как различие по меньшей мере в 5 масс.% от общего количества ароматических соединений C8) от термодинамически равновесной концентрации такого изомера в условиях изомеризации. Обычно неравновесная смесь готовится удалением пара- и/или орто-ксилола из свежей ароматической смеси C8, приготовленной способом получения ароматических соединений, и предпочтительно неравновесная смесь содержит менее 5 масс.% параксилола.
В одном из вариантов осуществления изобретения, в исходном потоке от 1 до 60 масс.% ароматических соединений C8 представляет собой этилбензол.
В настоящем изобретении могут использоваться алкилароматические углеводороды, обнаруживаемые в соответствующих фракциях различных потоков нефтеперерабатывающих заводов, например, в виде индивидуальных компонентов или в виде фракций с определенным интервалом кипения, полученных селективным фракционированием и дистилляцией углеводородов, подвергнутых каталитическому крекингу или риформингу. Изомеризуемые ароматические углеводороды не обязательно должны быть сконцентрированы; способ данного изобретения допускает изомеризацию содержащих алкилароматические соединения потоков, таких как продукты каталитического риформинга, с или без последующей экстракции ароматических соединений с целью получения определенных изомеров ксилола и, в частности, параксилола. Исходные для настоящего способа ароматические соединения C8 могут содержать неароматические углеводороды, то есть нафтены и парафины, в количестве вплоть до 30 масс.%. Однако предпочтительно изомеризуемые углеводороды состоят по существу из ароматических соединений для гарантирования чистоты продуктов, получаемых после следующих далее операций извлечения.
Согласно способу настоящего изобретения исходная смесь алкилароматических углеводородов, предпочтительно в смеси с водородом, вступает в контакт с описанным здесь содержащим UZM-35 катализатором в зоне изомеризации алкилароматических углеводородов. Контактирование может осуществляться с использованием катализатора в системе с неподвижным слоем, системе с подвижным слоем, системе с псевдоожиженным слоем или в режиме периодического действия. В системе с неподвижным слоем может быть минимизирована опасность потерь ценного катализатора вследствие истирания и упрощена эксплуатация. В этой системе богатый водородом газ и исходная смесь подогреваются подходящим нагревательным устройством до требуемой температуры реакции и затем подаются в зону изомеризации, содержащую неподвижный слой катализатора. Зона конверсии может быть представлена одним или более отдельными реакторами с соответствующими устройствами между ними, предназначенными для обеспечения того, чтобы на входе в каждую зону поддерживалась требуемая температура изомеризации. Реагенты могут вступать в контакт со слоем катализатора в режиме потока, направленного вверх, вниз или в радиальном направлении, и реагенты при вступлении в контакт с катализатором могут находиться в жидкой фазе, в смешанной фазе пар-жидкость или же в паровой фазе.
Исходная смесь алкилароматических соединений, предпочтительно неравновесная смесь ароматических соединений C8, входит в контакт с катализатором изомеризации в подходящих для алкилароматической изомеризации условиях. Такие условия включают температуру в пределах от 0 до 600°C или более, с одним конкретным воплощением в диапазоне от 100 до 500°C. Абсолютное давление в целом находится в диапазоне от 10 кПа до 5 МПа, с одним воплощением, абсолютное давление в котором составляет менее 5 МПа. В зоне изомеризации содержится достаточно катализатора, чтобы обеспечивать часовую объемную скорость жидкости относительно исходной смеси углеводородов от 0,1 до 30 час-1, с одним конкретным воплощением, в котором она составляет от 0,5 до 10 час-1. Исходная смесь углеводородов оптимально вводится в реакцию в смеси с водородом при мольном соотношении водород/углеводород от 0,5:1 до 10:1 или более. Могут присутствовать другие инертные разбавители, такие как азот, аргон и легкие углеводороды.
Реакция проходит по механизму изомеризации ксилолов, при котором из этилбензола образуется смесь ксилолов через конверсию в нафтены и обратное преобразование из нафтенов. Таким образом, посредством образования ксилолов из этилбензола увеличивается выход ксилолов в продукте. При этом обеспечивается желаемый невысокий уровень потерь ароматических соединений C8 в ходе реакции.
Какая-либо конкретная схема извлечения изомеризованного продукта из потока, исходящего из реакторов зоны изомеризации, в качестве критической для настоящего изобретения не рассматривается, и использоваться может любая известная в данной области эффективная схема извлечения. Как правило, продукты реакции конденсируются, а водород и компоненты, представленные легкими углеводородами, удаляются контактным дегазированием. Сконденсированный жидкий продукт затем фракционируется для удаления легких и/или тяжелых побочных продуктов и получения изомеризованного продукта. В некоторых случаях определенные соединения продукта, такие как орто-ксилол, могут быть извлечены из изомеризованного продукта селективным фракционированием. Продукт изомеризации ароматических соединений C8 обычно подвергается обработке с целью селективного извлечения параксилольного изомера, возможно кристаллизацией. Предпочтительной является селективная адсорбция с использованием кристаллических алюмосиликатов согласно патенту США 3201491. Усовершенствования и возможные варианты в рамках предпочтительного способа адсорбционного извлечения описаны в патентах США 3626020, 3696107, 4039599, 4184943, 4381419 и 4402832, каждый из которых включен в настоящее описание посредством ссылки.
В комбинации процесса разделения/изомеризации, касающейся обработки смеси этилбензол/ксилол, исходный поток свежих ароматических соединений C8 объединяется с изомеризованным продуктом, содержащим ароматические соединения C8 и нафтены из зоны реакции изомеризации, и подается в зону отделения параксилола; обедненный по параксилолу поток, содержащий неравновесную смесь ароматических соединений C8, направляется к зоне реакции изомеризации, где изомеры ароматических соединений C8изомеризуются до близких к равновесным уровням с получением изомеризованного продукта. При такой схеме способа остающиеся неизвлеченными ароматические изомеры C8 предпочтительно рециклируются исчерпывающим образом - до тех пор, пока они или преобразуются в параксилол, или утрачиваются вследствие побочных реакций. Отделение орто-ксилола, предпочтительно выполняемое фракционированием, также может производиться из исходного потока свежих ароматических соединений C8, или из изомеризованного продукта, или из комбинации обоих, до отделения параксилола.
Следующие далее примеры представлены для иллюстрирования данного изобретения и не имеют цели наложения неоправданных ограничений на общий объем изобретения, определяемый в прилагаемой формуле изобретения.
Структура цеолитной композиции UZM-35 данного изобретения была определена с помощью рентгенографического анализа. Рентгенограммы, представленные в следующих примерах, были получены с использованием стандартных методик рентгеновской дифракции на порошке. Источником излучения была рентгеновская трубка высокой интенсивности, действовавшая на 45 кВ и 35 мА. Основываясь на соответствующих компьютерных методах была получена дифракционная картина от K-альфа-линий меди. Образцы в виде таблеток из спрессованных порошков в непрерывном режиме сканировались на углах от 2° до 56° (2θ). Межплоскостные расстояния (d) в единицах ангстрем были получены от положения дифракционных пиков, выраженных в виде θ, где θ является брэгговским углом, наблюдаемым из оцифрованных данных. Величины интенсивностей определялись по интегрированной площади дифракционных пиков за вычетом фона, при этом "IO" представляет интенсивность самой сильной линии или пика, а "I" является интенсивностью каждого из других пиков.
Специалистами в данной области ясно, что определение параметра 2θ подвержено влиянию как человеческой ошибки, так и ошибки прибора, которые в комбинации могут привносить в каждую приводимую величину 2θ погрешность ±0,4°.
Эта погрешность, естественно, также проявляется и в приводимых показателях d-расстояний, которые вычисляются из величин 2θ. Такая погрешность является общей для всей данной области и поэтому не мешает различать кристаллические материалы настоящего изобретения один от другого и от композиций предшествующего уровня техники. В некоторых из приводимых рентгенограмм относительные интенсивности d-расстояний отмечены примечаниями «оч.сильн.», «сильн.», «ср.» и «сл.», которые, соответственно, означают «очень сильный», «сильный», «средний» и «слабый». В выражении 100×I/IO вышеуказанные обозначения определяются как:
сл.=0-15; ср.=15-60; сильн.=60-80 и оч.сильн.=80-100.
В некоторых случаях по дифракционной картине рентгеновских лучей на порошке синтезированного продукта может быть оценена его чистота. Таким образом, если, например, образец объявляется чистым, это подразумевает только то, что рентгенограмма образца не содержит линий, относящихся к кристаллическим примесям, и не означает, что в нем нет никаких аморфных материалов.
Для более полного иллюстрирования данного изобретения далее приводятся следующие примеры. Следует понимать, что эти примеры приводятся только в иллюстративных целях и не предполагают наложения неоправданных ограничений на общий объем изобретения, определяемый прилагаемой формулой изобретения.
Пример 1
Был приготовлен реакционный алюмосиликатный раствор смешиванием вначале 27,17 г гидроксида алюминия (27,78 масс.% Al) и 1053,58 г гидроксида диметилдипропиламмония (18,8 масс.% раствор) при интенсивном перемешивании. После полного смешивания было добавлено 505,6 г Ludox™ AS-40 (40 масс.% SiO2). Реакционная смесь гомогенизировалась еще в течение часа с помощью высокоскоростной механической мешалки, закупоривалась в тефлоновом флаконе и помещалась на ночь в нагретую до 100°C печь. Анализ показал, что раствор алюмосиликата содержал 6,16 масс.% Si и 0,67 масс.% Al (мольное отношение Si/Al 8,83).
1200 г порция указанного выше алюмосиликатного раствора непрерывно перемешивалась. К раствору алюмосиликата по каплям был добавлен составной водный раствор, содержащий 28,56 г КОН и 3,6 г NaOH, растворенных в 150 г дистиллированной воды. После завершения добавления полученная реакционная смесь была гомогенизирована в течение 1 часа, перенесена в автоклав Парра из нержавеющей стали емкостью 2000 мл, который был нагрет до 175°C и выдерживался при этой температуре в течение 216 часов.
Твердый продукт отделялся центрифугированием, промывался деминерализованной водой и высушивался при температуре от 95°C до 100°C. Дифракционным рентгеновским анализом продукт был идентифицирован как UZM-35. Наблюдаемые для данного продукта представительные дифракционные линии показаны в таблице 1. Композиция продукта, определенная по данным элементного анализа, представлена следующими мольными соотношениями: Si/Al=7,92, Na/Al=0,1, K/Al=0,48.
Пример 2
UZM-35 из примера 1 был прокален в течение 7 часов при 570°C под азотом, а затем в воздушной атмосфере. После чего UZM-35 был подвергнут ионному обмену для замещения катионов Na+ или K+ на NH4+. Ионный обмен в UZM-35 на аммоний был осуществлен контактированием 40 г UZM-35 с 500 мл 1 М раствора NH4NO3 при 80°С и перемешивании в течение 1 часа, фильтрацией и промывкой. Эта процедура была повторена три раза. После чего подвергнутый ионному обмену UZM-35 прокаливался в течение 2 час при 550°C на воздухе для преобразования в H+ с потерей аммиака.
Пример 3
В качестве варианта, выполненный вначале обмен на аммоний затем сопровождался прокаливанием для удаления матрицы и обмена катионов Na+ или K+ на UZM-35 из примера 1 был подвергнут ионному обмену на аммоний контактированием 100 г UZM-35 со 1000 мл 1 М раствора NH4NO3 при 80°С и перемешивании в течение 1 часа. Подвернутый ионному обмену UZM-35 был после этого прокален при 560°C в течение 7 часов под азотом и затем воздухом. Была выполнена вторая операция ионного обмена контактированием 95 г UZM-35 со 1000 мл 1 М раствора NH4NO3 при 80°С и перемешивании в течение 1 часа. Подвергнутый ионному обмену UZM-35 отфильтровывался и высушивался.
Пример 4
UZM-35 из примера 3 далее обрабатывался в течение 2 час водяным паром при 600°C в вертикальной паровой камере, пропусканием над UZM-35 воздушного потока, содержащего 50 об.% пара. Обработанный паром UZM-35 был вновь подвергнут ионному обмену с использованием раствора NH4NO3. Получающийся обработанный паром и аммонием UZM-35 экструдировался с оксидом алюминия в пропорции 70/30. Экструдаты обжигались при 550°C на воздухе в течение 2 часов для преобразования в H+ с потерей аммиака.
Сравнительный пример 5
Три различных катализатора были подвергнуты сравнению с одним воплощением заявляемого катализатора в отношении их производительности при изомеризации ксилола. Первый и второй катализаторы являлись экструдатами, изготовленными с использованием порошка цеолита MTW, имеющего мольное соотношение Si/Al, равное 20, приготовленного согласно описанному в патенте США 7525008 способу, и оксида алюминия V-251 (предлагается UOP, LLC). Первый катализатор имел концентрацию цеолита 20 масс.%, в то время как второй катализатор имел концентрацию цеолита 50 масс.% по отношению к массе экструдата.
Третий и четвертый катализаторы были изготовлены с цеолитом MTW, имевшим мольное соотношение Si/Al, равное 20, при этом концентрация цеолита в расчете на массу каталитического композита составляла в обоих 80 масс.%, а в качестве связующего использовался оксид алюминия. Третий катализатор был представлен в виде полученных прикапыванием в масло сфер, а четвертый катализатор был экструдатом, подвергнутым ионному обмену с нитратом аммония, промытым и прокаленным.
Воплощение изобретения, исследованное в качестве пятого катализатора, было представлено цеолитной композицией, содержащей 70 масс.% UZM-35 и имевшей форму экструдата с использованным в качестве связующего оксидом алюминия.
В каждом эксперименте по 2 грамма катализатора загружалось в реактор с неподвижным слоем. В реактор подавались исходная смесь и водород для обеспечения контакта с катализатором. Исходная смесь состояла из 56 масс.% метаксилола, 22 масс.% орто-ксилола, 1 масс.% параксилола, 1 масс.% толуола и 6 масс.% нафтенов C8 с остальной до 100% частью, представленной этилбензолом. Исходная смесь подавалась с объемной скоростью в 10 единиц смеси на единицу массы катализатора в час, а отношение Н2/НС равнялось 4. Реактор функционировал под абсолютным давлением 786 кПа и каждый катализатор был проверен при температурах 365°C, 375°C и 385°C. Исходящий из реактора продукт контролировался с использованием газовой хроматографии. Результаты каждого эксперимента отображались в виде графика, как показано на фиг.1, где на оси Y отложены показатели потерь ароматических соединений C8, а по оси X - отношение количества параксилола к общему количеству ксилола (PX:X). «Потери ароматических соединений C8» представлены в единицах мол.%, определенных как (1 - (нафтены и ароматические соединения C8 на выходе) / нафтены и ароматические соединения C8 на входе)) ·100.
Как показано на фиг.1, данное воплощение изобретения продемонстрировало интересную тенденцию изменения потока в зависимости от времени. Селективность катализатора изобретения улучшается на ранних стадиях без существенного снижения активности в отношении изомеризации ксилола, таким образом обеспечивая исходное сокращение количества побочных продуктов. Кроме того, катализатор изобретения обеспечил преимущество более низкой потери ароматических соединений по сравнению с полученными прикапыванием в масло сферами, содержащими 80 масс.% MTW с используемым в качестве связующего оксидом алюминия. Имеющаяся потеря ароматических соединений в основном является следствием переалкилирования, как видно из фиг.2, где сравнение Катализатора №5 с Катализатором №3 демонстрирует его более низкий показатель потерь ароматических соединений при эквивалентном P-X/X. Катализатор изобретения имеет иное по сравнению с цеолитом MTW распределение продуктов, приводящее к различиям в их соотношении, см. фиг.3, на которой показано распределение продуктов в массовых процентах при P-X/X, равном 22,9 масс.%. Сопоставимо с цеолитом MTW, нет никаких ограничений по ортоксилолу, см. фиг.4, которая показывает, что катализатор изобретения обеспечивает ксилолы с такой же кривой соотношения орто-ксилола и параксилола, как и цеолит MTW.
Сравнительный пример 6
Три различных катализатора были подвергнуты сравнению с одним воплощением заявляемого катализатора в отношении производительности при переалкилировании ксилола. Первый катализатор представлял собой экструдат порошка композиции UZM-35 и оксида алюминия V-251, где первый катализатор имел концентрацию цеолита в 70 масс.% по отношению к массе готового экструдата. Второй катализатор являлся экструдатом 2,2 масс.% Ga и 0,6 масс.% Al на цеолите MFI со связующим оксидом алюминия V-251 в соответствии с указаниями патента США 4957891. Второй катализатор имел концентрацию цеолита по отношению к массе экструдата в 50 масс.%. Третий катализатор представлял собой подвернутые ионообменной обработке нитратом аммония, пропаренные сферы, полученные прикапыванием в масло и содержащие 65 масс.% MFI и алюмофосфат в качестве связующего вещества, приготовленные с использованием способа примера 1 из патента США 6143941.
В каждом эксперименте в реактор с неподвижным слоем загружалось по 1 грамму катализатора. В реактор подавались исходная смесь и водород для обеспечения контакта с катализатором. Исходная смесь состояла из 60 масс.% метаксилола, 25 масс.% орто-ксилола, 15 масс.% этилбензола. Исходная смесь подавалась с объемной скоростью в 10 единиц смеси на единицу массы цеолита в час, а отношение H2/HC равнялось 4. Реактор функционировал под абсолютным давлением 786 кПа и каждый катализатор был проверен при температурах 375°C, 385°C и 395°C. Катализаторы были представлены в виде физической смеси 0,4 г катализатора с дисперсностью 14/20 меш, содержащего 0,3 масс.% платины на оксиде алюминия, модифицированного 0,6 масс.% индия и 0,3 масс.% олова в соответствии с примером III из патента США 6048449.
Исходящий из реактора продукт контролировался с помощью газовой хроматографии. Результаты каждого эксперимента показаны в таблице 2. Из таблицы 2 видно, что по сравнению с цеолитом структуры MFI конверсия этилбензола относительно невелика. Большая часть преобразований представляет перенос этила в другие ароматические соединения. Активность изомеризации ксилола находилась на высоком уровне, что отображено соотношением параксилола к общему ксилолу, и близка к равновесной.
Пример 7
Был приготовлен реакционный алюмосиликатный раствор смешиванием вначале 86,33 г гидроксида алюминия (26,97 масс.% Al) и 1437,67 г гидроксида диметилдипропиламмония (40,66 масс.% раствор) при интенсивном перемешивании. После полного смешивания было добавлено 1366,88 г Ludox™ AS-40 (40 масс.% SiO2). Реакционная смесь гомогенизировалась в течение 20 минут высокоскоростной механической мешалкой, коллоидный раствор алюмосиликата непрерывно перемешивался и к раствору алюмосиликата по каплям был добавлен водный раствор, содержащий 83,04 г КОН и 17,38 г NaOH в 808,7 г H2O. После завершения добавления полученная реакционная смесь была гомогенизирована в течение 1/2 часа, перенесена в 3 автоклава Парра из нержавеющей стали емкостью 2000 мл, которые были нагреты до 175°C и выдерживались при этой температуре в течение 9 дней. Твердые продукты отделялись фильтрацией, промывались деминерализованной водой и высушивались при 100°C.
Образующийся в результате этой реакции продукт был идентифицирован рентгеновской дифракцией (метод уточнения по Ритвельду, описанный в J.Appl. Cryst. (1969) 2, 65-71), как являющийся композицией UZM-35 из 72,1 масс.% цеолита типа MSE с параметром решетки в 18,372 ангстрем для «a» и 20,285 ангстрем для «c»; 24,1 масс.% цеолита типа MFI с параметром решетки 20,101 ангстрем для «a», 19,862 ангстрем для «b» и 13,402 для «c», и 3,7 масс.% цеолита типа ERI с параметром решетки 13,222 ангстрем для «а» и 14,900 ангстрем для «с». По данным химического анализа мольное соотношение Si/Al в композиции продукта составляла 8,9. Удельная поверхность по методу BET была определена как равная 408 м2/г, а объем микропор составлял 0,197 см3/г. Наблюдаемые для данного продукта представительные дифракционные линии показаны в таблице 3.
Данный образец прокаливался при 600°C в течение 5 час под азотом и затем в атмосфере воздуха. Образующийся в результате прокаливания продукт был идентифицирован рентгеновской дифракцией (метод уточнения по Ритвельду, описанный в J.Appl. Cryst. (1969) 2, 65-71), как являющийся смесью из 64,4 масс.% цеолита типа MSE с параметром решетки в 18,371 ангстрем для «a» и 20,235 ангстрем для «c»; 30,7 масс.% цеолита типа MFI с параметром решетки 20,048 ангстрем для «a», 19,880 ангстрем для «b» и 13,403 для «c»; и 4,8 масс.% цеолита типа ERI с параметром решетки 13,071 ангстрем для «a» и 15,238 ангстрем для «c». Порция в 160 г прокаленного образца UZM-35 (мольное соотношение Si/Al=8,9) была подвергнута ионному обмену на NH4. Был приготовлен раствор растворением 160 г NH4NO3 в 1800 г деминерализованной воды. Перед добавлением прокаленного UZM-35 раствор нагревался до 75°C. Суспензия перемешивалась в течение 1 часа при 75°C. Продукт отделялся фильтрацией и промывался деминерализованной водой. Эта операция обмена NH4 была повторена трижды, а затем была выполнена сушка при 100°C в течение 12 час.
Элементный анализ этих образцов показал величины мольных соотношений Si/Al=9,07, Na/Al=0,01, K/Al - 0,11.
Наблюдаемые для данного продукта представительные дифракционные линии показаны в таблице 4.
Таблица 4
Пример 8
Был приготовлен реакционный алюмосиликатный раствор смешиванием вначале 29,01 г гидроксида алюминия (26,97 масс.% Al) и 483,08 г гидроксида диметилдипропиламмония (40,66 масс.% раствор) при интенсивном перемешивании. После полного смешивания было добавлено 461,58 г Ludox™ AS-40 (40 масс.% SiO2). Реакционная смесь гомогенизировалась в течение 20 минут высокоскоростной механической мешалкой, коллоидный раствор алюмосиликата непрерывно перемешивался и к раствору алюмосиликата по каплям был добавлен водный раствор, содержащий 27,90 г КОН и 3,46 г NaOH в 269,98 г H2O. После завершения добавления полученная реакционная смесь была гомогенизирована в течение 1/2 часа, перенесена в автоклав Парра из нержавеющей стали емкостью 2000 мл, который был нагрет до 175°C и выдерживался при этой температуре в течение 10 часов. Твердые продукты отделялись фильтрацией, промывались деминерализованной водой и высушивались при 100°C.
Образующийся в результате этой реакции продукт был идентифицирован рентгеновской дифракцией (метод уточнения по Ритвельду, описанный в J.Appl. Cryst. (1969) 2,65, 65-71), как являющийся композицией UZM-35 из 66,3 масс.% цеолита типа MSE с параметром решетки в 18,369 ангстрем для «a» и 20,284 ангстрем для «c»; 25,5 масс.% цеолита типа MFI с параметром решетки 20,136 ангстрем для «a», 19,976 ангстрем для «b» и 13,443 для «c», и 8,2 масс.% цеолита типа ERI с параметром решетки 13,152 ангстрем для «a» и 15,107 ангстрем для «c». Данные химического анализа представили мольные соотношения композиции продукта как Si/Al=7,65, N/Al=0,38, K/Al=0,68, Na/Al=0,03. Удельная поверхность по методу BET была определена равной 404 м2/г, а объем микропор составлял 0,188 см3/г. Наблюдаемые для данного продукта представительные дифракционные линии показаны в таблице 5.
Таблица 5
Данный образец прокаливался при 600°C в течение 5 час под азотом и затем в атмосфере воздуха. Образующийся в результате прокаливания продукт был идентифицирован рентгеновской дифракцией (метод уточнения по Ритвельду, описанный в J.Appl. Cryst. (1969) 2,65, 65-71), как являющийся композицией UZM-35 из 61,9 масс.% цеолита типа MSE с параметром решетки в 18,401 ангстрем для «a» и 20,280 ангстрем для «c»; 30,8 масс.% цеолита типа MFI с параметром решетки 20,114 ангстрем для «a», 19,919 ангстрем для «b» и 13,432 для «c», и 7,3 масс.% цеолита типа ERI с параметром решетки 13,189 ангстрем для «a» и 15,174 ангстрем для «c». Порция в 100 г прокаленного образца UZM-35 (мольное соотношение Si/Al=7,65) была подвергнута ионному обмену на NH4. Был приготовлен раствор растворением 160 г NH4NO3 в 1800 г деминерализованной воды. Перед добавлением прокаленного UZM-35 раствор нагревался до 75°C. Суспензия перемешивалась в течение 1 часа при 75°C. Продукт отделялся фильтрацией и промывался деминерализованной водой. Эта операция обмена NH4 была повторена трижды, а затем была выполнена сушка при 100°C в течение 12 час.
Элементный анализ этих образцов показал величины мольных соотношений Si/Al=9,20, Na/Al=0,01, K/Al=0,10.
Наблюдаемые для данного продукта представительные дифракционные линии показаны в таблице 6.
Реферат
Изобретение относится к области катализа. Описан способ изомеризации ксилола и этилбензола, катализируемый семейством UZM-35 кристаллических алюмосиликатных цеолитных композиций. Технический результат - увеличение селективности и активности изомеризации ксилолов и этилбензола. 9 з.п. ф-лы, 4 ил., 6 табл., 8 пр.
Формула
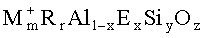
где "M" представляет комбинацию способных к обмену катионов калия и натрия, "m" - мольное отношение M к (Al+E), варьирующее от 0,05 до 3, "R" - по меньшей мере один однозарядный катион диметилдипропиламмония, "r" - мольное отношение R к (Al+E), имеющее величину от 0,25 до 2,0, "E" является по меньшей мере одним элементом, выбираемым из группы, состоящей из галлия, железа, бора и их смесей, "x" - мольная доля E, имеющая величину от 0 до 1,0, "y" является мольным отношением Si к (Al+E), варьирующим от более 2 до 12, и "z" - мольное отношение O к (Al+E), величина которого определяется уравнением
z=(m+r+3+4·y)/2
и характеризуется тем, что на рентгеновской дифрактограмме имеет величины межплоскостных расстояний d и интенсивностей, представленные в таблице А':
и обладает термической устойчивостью вплоть до температур по меньшей мере 400°C.
Документы, цитированные в отчёте о поиске
Способ каталитической изомеризации неравновесной смеси c8- ароматических углеводородов
Комментарии