Способ получения 1-метилциклопропена - RU2667511C1
Код документа: RU2667511C1
Описание
Изобретение относится к технологии получения 1-метилциклопропена, применяемого в сельском хозяйстве для послеуборочной обработки урожая плодоовощной продукции с целью сохранения ее потребительских свойств и увеличения сроков ее хранения.
Известен способ получения циклопропенов, в т.ч. 1-метилциклопропена, взаимодействием амидов щелочных металлов с галогенсодержащими олефинами в присутствии инертного растворителя /1, 2/. Процесс осуществляют путем дозировки галогенсодержащего олефина в суспензию амида (или алкиламида) щелочного металла в минеральном масле при температуре 0-75°C (предпочтительно при 20-40°C) с последующим добавлением воды. Полученные газообразные продукты реакции пропускают через холодильник-конденсатор, в котором конденсируют высококипящие продукты (главным образом, исходные непрореагировавшие галогенсодержащие олефины), а очищенный газообразный поток, содержащий в основном циклопропены, поглощают суспензией циклодекстрина в буферном растворе. Образующийся в процессе пропускания газообразного потока через суспензию циклодекстрина в буферном растворе комплекс - 1-метилциклопропен/циклодестрин - отделяют на фильтре, сушат на воздухе 24-48 часов, перемалывают и хранят до использования в виде сухого комплекса 1-метилциклопропен/циклодекстрин.
Однако в описании патентов не приводится выход циклопропенов на взятые исходные реагенты и, как следствие, отсутствие возможности оценки эффективности данных способов.
Известен способ получения циклопропенов взаимодействием амида натрия с аллилхлоридами /3/.
Основным недостатком данного способа является низкий выход целевого продукта, трудности, связанные с использованием дополнительных стадий очистки конечного продукта, а также необходимость использования большого избытка амида натрия.
Известен способ получения циклопропенов взаимодействием фениллития с аллилхлоридами /4/. Этот метод позволяет получать метилциклопропен с выходом до 60%.
Основным недостатком данного метода является наличие в целевом продукте больших примесей метиленциклопропана, который очень трудно отделить из-за близости температур их кипения.
Известен способ получения метилциклопропена взаимодействием 3-хлор-2-метилпропена с амидом лития в среде кипящего диоксана в присутствии добавок воды. Выход продукта на взятый 3-хлор-2-метилпропен достигает 40% мол. /5/.
Основным недостатком данного способа является наличие в целевом продукте изобутена, который очень трудно отделить из-за близости температур их кипения.
Известен способ получения циклопропенов взаимодействием галогенсодержащих аллильных соединений с сильными основаниями в среде инертного растворителя в присутствии каталитических количеств слабых оснований. Предпочтительно в качестве сильных оснований используют амиды щелочных металлов или металлоорганические основания, в качестве слабых оснований - силиламин или дисилазан, а процесс осуществляют в среде алифатических или ароматических углеводородов, простых эфиров, галогенсодержащих углеводородов, жидкого аммиака, метиламина или диметиламина /6/.
Несмотря на то что данный способ позволяет снизить образование метиленциклопропана, основным недостатком данного способа является относительно низкий выход метилциклопропена, не превышающий величины 24% мол. на взятый в реакцию 3-хлор-2-метилпропен.
Известен способ получения 1-метилциклопропена взаимодействием аллильных соединений (галогенированных карбенов) с основанием или смесью оснований в растворителе в присутствии ненуклеофильных слабых оснований /7/.
Недостатком данного способа является низкий выход целевого продукта и существенное присутствие примеси метиленциклопропана. Так, выход 1-метилциклопропена не превышает 29,4% мол. (по взятому 3-хлор-2-метилпропену), а содержание метиленциклопропана составляет 1,5% масс.
Наиболее близким к заявленному способу является способ получения 1-метилциклопропена, включающий взаимодействие 3-хлор-2-метилпропена или 3-бром-2-метилпропена с основанием в среде растворителя в присутствии каликсарена /8/.
Недостатком способа-прототипа является низкий выход целевого продукта, который не превышает 34,9% мол. (по взятому 3-хлор-2-метилпропену), но главным недостатком данного способа является образование большого количества метиленциклопропена, который, являясь по сути аналогом этилена, вредным образом действует на потребительские качества конечного продукта при использовании его в виде коммерческого препарата в сельском хозяйстве для послеуборочной обработки урожая плодоовощной продукции. Так, по данным заявителя, «Собранный в ловушке конденсат разделяют низкотемпературной ректификацией и получают жидкий 1-метилциклопропен с содержанием метиленциклопропана не более 0,1 масс. % (по данным ГЖХ анализа). Дело в том, что целевой продукт - 1-метилциклопропен - и его вредный изомер - метиленциклопропан - имеют настолько близкие температуры кипения (разница температур всего несколько градусов), что разделить их достаточно тщательно низкотемпературной ректификацией не представляется возможным без огромных потерь целевого продукта, поэтому на практике этого никогда не делают. В реакционных газах же (после осуществления синтеза по прототипу) и очистки их от аммиака после конденсации в низкотемпературной ловушке содержание метиленциклопропана достигает 15% масс. и более (см. сравнительный пример №1).
Задачей предлагаемого способа является увеличение выхода метилциклопропена и снижение образования вредного изомера - метиленциклопропана.
Поставленная задача достигается заявленным способом получения 1-метилциклопропена взаимодействием металлил-хлорида (3-хлор-2-метилпропена) или металлил-бромида (3-бром-2-метилпропена) с основаниями в среде полярного апротонного органического растворителя, отличающимся тем, что процесс осуществляют в присутствии четвертичных аммониевых солей в количестве от 0,01 до 0,5 мольных долей от суммарного содержания оснований в присутствии олефина этиленового ряда или их смесей в количестве от 0,01 до 0,50 мольных долей от содержания металлил-хлорида или металлил-бромида.
В предпочтительном варианте способа в качестве четвертичных аммониевых солей используют тетраэтиламмоний-хлорид или бромид, тетрапропиламмоний-хлорид или бромид, тетрабутиламмоний-хлорид или бромид, бензилтри-этиламмоний-хлорид или бромид, а в качестве олефина этиленового ряда используют этилен.
Температура кипения этилена и 1-метилциклопропена отличаются практически на 80°C, поэтому даже в низкотемпературной ловушке без какой-либо ректификации они легко отделяются друг от друга.
Способ иллюстрируется следующими примерами.
Пример 1 (по прототипу)
В реактор, представляющий собой круглодонную колбу объемом 0,5 литра, снабженную рубашкой, мешалкой, капельной воронкой, термометром и обратным холодильником, предварительно продутый сухим азотом (инертный газ), последовательно загружают 120 мл парафина с14-с17 (растворитель), 2,72 г (1 масс. %) смеси краун-эфира Бенз-15-краун-5 + и криптан марки Kryptofix® фирмы Merck Kryptofix® 22 DD. Затем включают мешалку и при перемешивании загружают 50 г амида натрия и 2 г гексаметидисилоза. После добавления всего количества смеси оснований включают подачу теплоносителя (30°C) в рубашку реактора и хладагента (10°C) в рубашку обратного холодильника. После достижения в реакторе заданной температуры (30°C) в реактор подают металлилхлорид в количестве 100 г (1,1 моль) в течение 70 минут. После прибавления всего количества металлилхлорида реакционную массу выдерживают при перемешивании в течение еще 70 минут. Образующийся в процессе реакции газ через обратный холодильник и поглотительную склянку, содержащую 5 масс. % водного раствора серной кислоты, направляют в ловушку, охлаждаемую сухим льдом. Таким образом, способ полностью копирует условия синтеза прототипа, указанные в таблицах 1 и 2 прототипа.
Собранный в ловушке конденсат пытаются делить низкотемпературной ректификацией, но более или менее успешно удается отделить исходный непрореагироваший металлилхлорид, поскольку отделить метиленциклопропан - невозможно. В результате получают 10,4 г жидкого 1-метилциклопропена (выход 17,8%) с содержанием метиленциклопропана 15,4 мас. % (по данным ГЖХ анализа).
Пример 2 (по заявленному способу)
В реактор, представляющий собой круглодонную колбу объемом 0,5 литра, предварительно продутую сухим азотом, снабженную рубашкой, мешалкой, перистальтическим насосом, термометром, устройством для ввода этилена (включающим отградуированный дроссельный кран и барбатер) и обратным холодильником-дефлегматором, последовательно загружают 120 мл тетрагидрофурана (растворитель), 10,0 г тетрабутиламмоний-хлорида (0,034 мольн. долей от суммарного количества амида натрия и гексаметилдисилозана). Затем включают мешалку, включают подачу этилена со скоростью 22,4 мл в минуту и при перемешивании загружают 40 г амида натрия и 7,5 г гексаметидисилоза. После добавления всего количества смеси оснований включают подачу теплоносителя (60°C) в рубашку реактора и хладагента (-15°C) в рубашку обратного холодильника-дефлегматора. После достижения в реакторе заданной температуры (60°C) в реактор подают металлилхлорид в количестве 100 г (1,1 моль) в 150 мл тетрагидрофурана в течение 70 минут. После прибавления всего количества металлилхлорида реакционную массу выдерживают при перемешивании в течение еще 70 минут.
Общее количество поданного этилена составляет 22,4×140=3,136 литра (0,14 моля), что составляет около 0,13 мольн. долей от загруженного металлилхлорида. Образующийся в процессе реакции газ через обратный холодильник и поглотительную склянку, содержащую 5 масс. % водного раствора уксусной кислоты, направляют в ловушку, охлаждаемую сухим льдом.
Отделившийся в ловушке этилен в виде газа пропускают через поглотительную склянку, содержащую 10%раствор пермангатата калия в 5% серной кислоте, где он полностью поглощается, а азотную сдувку сбрасывают в тягу лаборатории.
Собранный в ловушке конденсат без какой-либо дополнительной ректификации взвешивают и анализируют методом ГЖХ на содержание целевого 1-метилциклопропена, исходного металлилхлорида, вредного метиленциклопропана и инертного в отношении конечных свойств коммерческого препарата метилциклопропана. В результате получают 27,0 г жидкого 1-метилциклопропена (выход 46,2%) с содержанием метиленциклопропана 2,5 масс. %, 0,16 масс. % метилциклопропана и 0,038 масс. % исходного металлилхлорида (по данным ГЖХ анализа).
Полученный целевой продукт без какой-либо дальнейшей очистки направляют на получение коммерческого препарата «Фреш-Форма».
Результаты примеров выполнения способа №№2-15 сведены в таблицы 1 и 2.
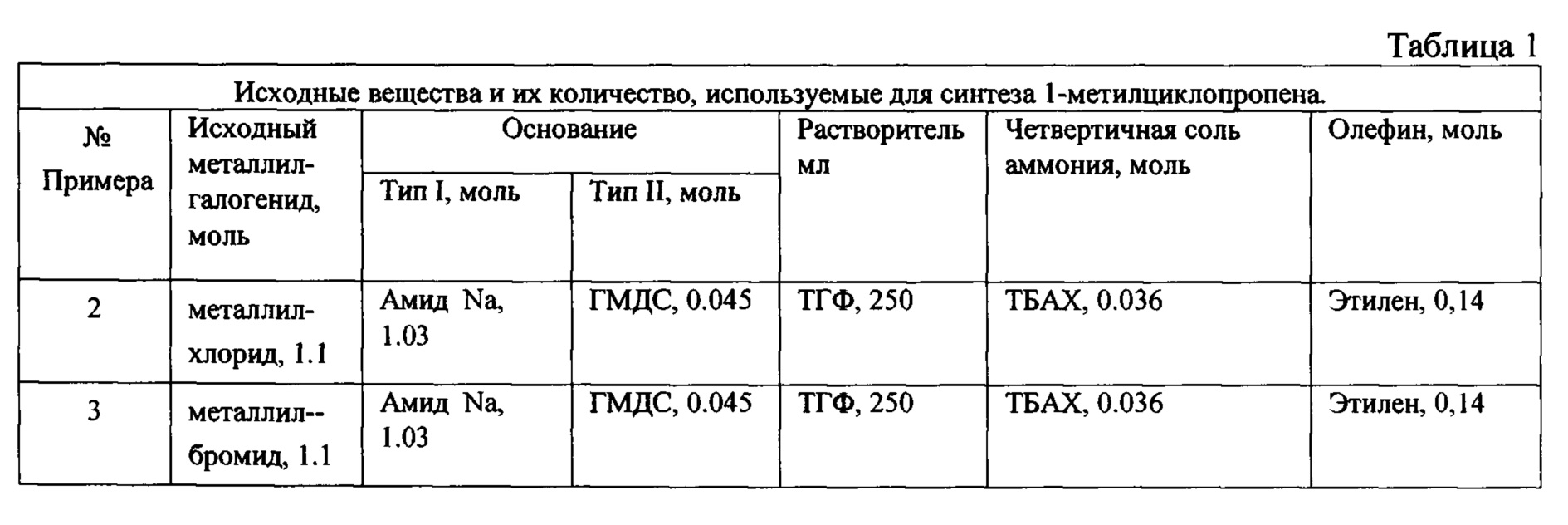
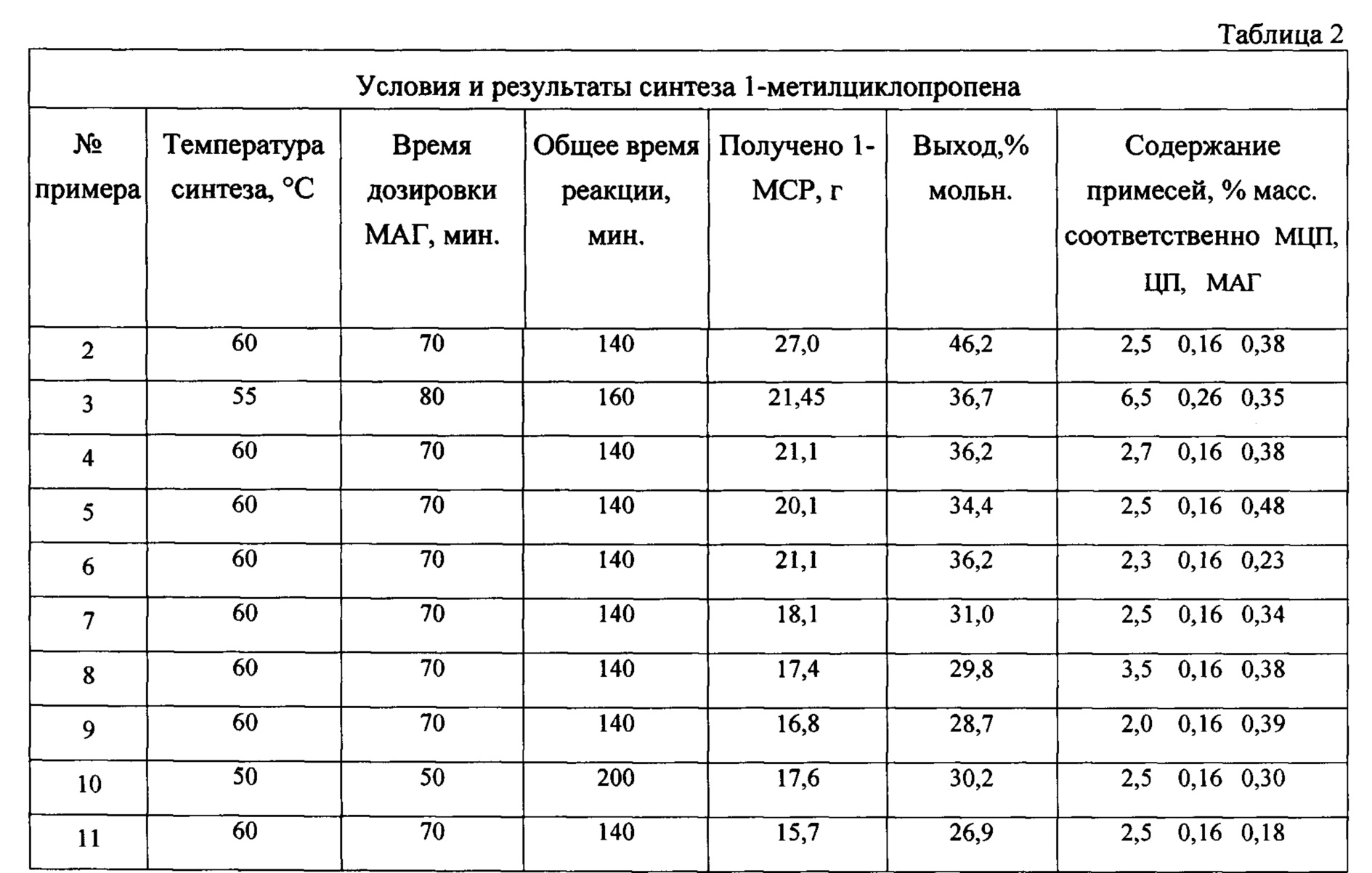
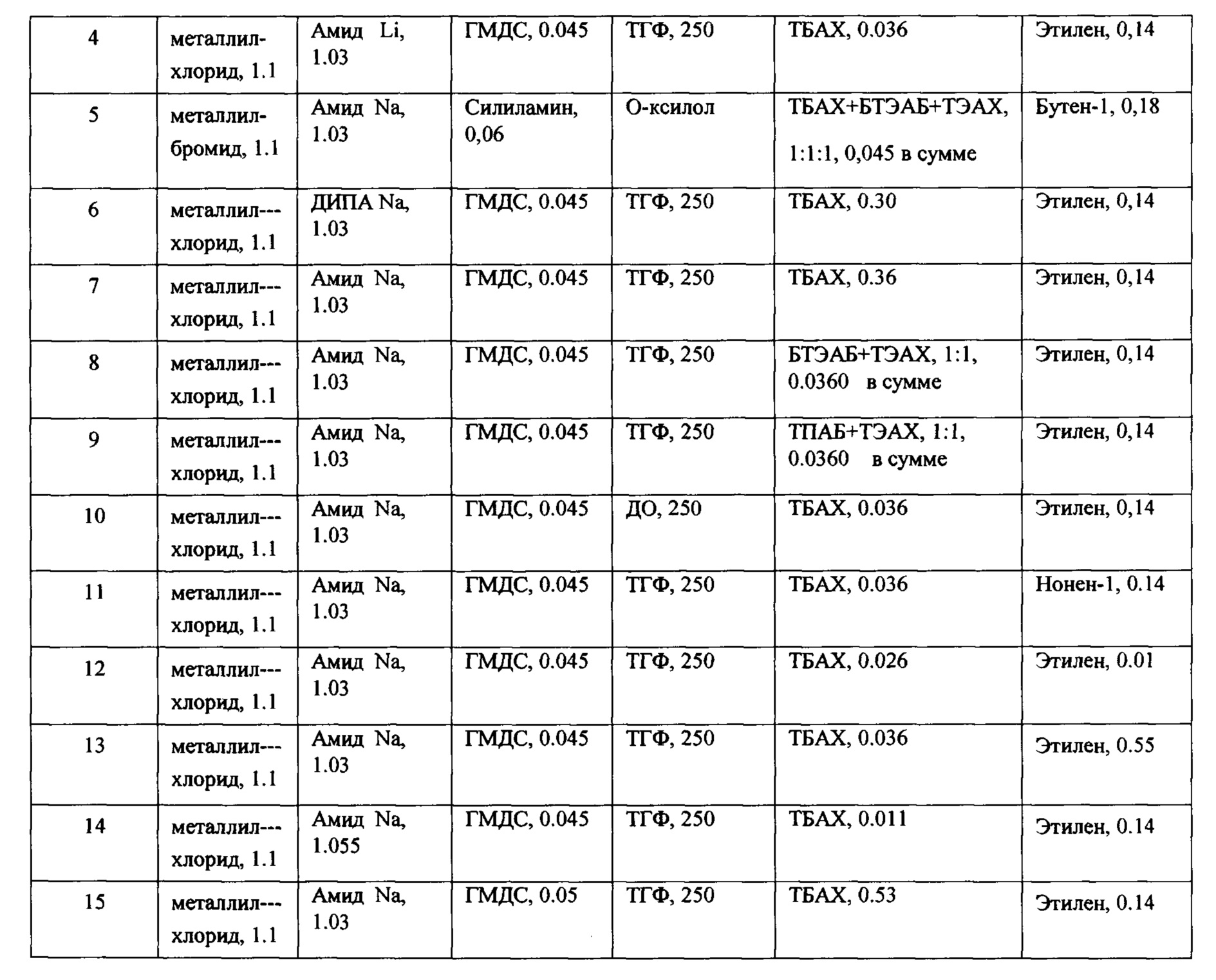
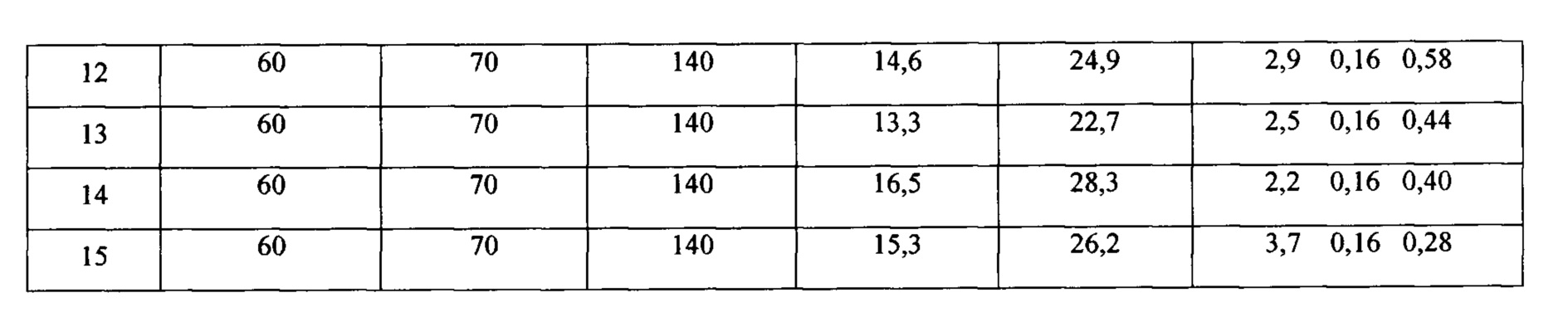
Приняты сокращения
МАГ - металлил-галогенид;
МЦП - метиленциклопропен;
ЦП - метилциклопропан;
ГМДС - гексаметилдисилозан;
ТГФ - тетрагидрофуран;
ДО - 1,4-диоксан;
ТБАХ - тетрабутиламмоний-хлорид;
ТЭАХ - тетраэтиламмоний-хлорид;
ТПАБ - тетрапропилам моний-бромидид;
БТЭАХ - бензилтри-этиламмоний-хлорид.
Источники информации, принятые во внимание:
1. Патент US 2004082480.
2. Патент US 6313068.
3. F. Fisher and D. Applequist, J. Org. Chem., 30, 2089, 1965.
4. R. Magid, и. ал., J. Org. Chem., 36, 1320, 1971.
5. Koster, et. al., Liebigs Ann. Chem., 1219, 1973.
6. Европейский патент EP 1146028.
7. Патент US 6452060.
8. Патент на изобретение РФ №2267477 (прототип).
Реферат
Изобретение относится к способу получения 1-метилциклопропена взаимодействием металлил-хлорида (3-хлор-2-метилпропена) или металлил-бромида (3-бром-2-метилпропена) с основаниями в среде полярного апротонного органического растворителя. Способ характеризуется тем, что процесс осуществляют в присутствии четвертичных аммониевых солей в количестве от 0,01 до 0,5 мольных долей от содержания оснований в присутствии олефина этиленового ряда или их смесей в количестве от 0,01 до 0,50 мольных долей от содержания металлил-хлорида или металлил-бромида. Способ позволяет увеличить выход целевого продукта до 46,2% и снизить содержание вредного для дальнейшей переработки в коммерческий препарат метилен-циклопропана без стадии низкотемпературной ректификации до 0,025%. 1 з.п. ф-лы, 2 табл., 2 пр.
Формула
Документы, цитированные в отчёте о поиске
Композиция для обработки растений и плодов и способы повышения урожая плодоовощной и растениеводческой продукции и увеличение срока его хранения
Комментарии