Внутрижелудочковая доставка ферментов при лизосомных болезнях накопления - RU2665381C2
Код документа: RU2665381C2
Чертежи












Описание
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Изобретение относится к области лизосомных болезней накопления. В частности, изобретение относится к лечению и/или профилактике указанных заболеваний с помощью ферментной заместительной терапии.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Группа метаболических расстройств, известных как лизосомные болезни накопления (ЛБН), включает около сорока генетических расстройств, многие из которых связаны с генетическими дефектами в различных лизосомных гидролазах. Типичные лизосомные болезни накопления и связанные с ними дефектные ферменты перечислены в таблице 1.
Основным признаком ЛБН является патологическое накопление метаболитов в лизосомах, которое приводит к формированию в перикарионе больших количеств растянутых лизосом. Главной трудностью лечения ЛБН (в противоположность лечению органоспецифичной энзимопатии, например, специфичной для печени энзимопатии) является необходимость обратить лизосомную патологию накопления во множестве отдельных тканей. При некоторых ЛБН эффективна внутривенная инфузия недостающего фермента, известная как ферментная заместительная терапия (ФЗТ). Например, у пациентов с болезнью Гоше типа 1 поражены только висцеральные органы, поэтому такие пациенты хорошо отвечают на ФЗТ рекомбинантной глюкоцереброзидазой (Cerezyme™, Genzyme Corp.). Однако пациенты с метаболическим заболеванием, которое поражает ЦНС (например, болезнь Гоше типа 2 или 3), отвечают на внутривенную ФЗТ только частично, потому что поступлению заместительного фермента в головной мозг препятствует гематоэнцефалический барьер. Кроме того, попытки ввести заместительный фермент в головной мозг непосредственно путем инъекции ограничены, частично из-за цитотоксичности ферментов при высоких местных концентрациях и ограниченной скорости паренхиматозной диффузии в головном мозге (Partridge, Peptide Drug Delivery to the Brain, Raven Press, 1991).
Одной из типичных ЛБН является болезнь Нимана-Пика типа А (NPA). Согласно статье P17405 UniProtKB/Swiss-Prot, причиной болезни Нимана-Пика типа А, также носящей название классической инфантильной формы заболевания, являются дефекты гена SMPD1, расположенного на 11-й хромосоме (11p15.4-p15.1). Болезнь Нимана-Пика - клинически и наследственно гетерогенное рецессивное заболевание. Указанное заболевание вызывается накоплением в лизосомах сфингомиелина и других метаболически связанных липидов, что приводит к дегенерации нервной системы, начинающейся с молодости. У пациентов могут возникать ксантомы, пигментация, гепатоспленомегалия, лимфаденопатия и олигофрения. Болезнь Нимана-Пика более часто возникает у евреев-ашкенази, чем среди населения в целом. NPA характеризуется ранним началом в грудном возрасте и быстро прогрессирующим течением, приводящем к смерти к возрасту 3 года. Фермент кислая сфингомиелиназа (ASM), которая дефектна при NPA, превращает сфингомиелин в церамид. ASM также имеет активность фосфолипазы C по отношению к 1,2-диацилглицеринфосфохолину и 1,2-диацилглицеринфосфоглицерину. Указанный фермент преобразует
Сфингомиелин + H2O → N-ацилсфингозин + холина фосфат.
В соответствии с настоящим изобретением лечение и/или профилактику лизосомных болезней накопления, например, любого из заболеваний, указанных выше в таблице 1, например, болезнь Нимана-Пика типа A или B, проводят, используя внутрижелудочковую доставку в головной мозг фермента, который этиологически дефицитен при указанном заболевании. Введение можно осуществлять медленно, чтобы достичь максимального эффекта. Эффекты отмечаются с обеих сторон гематоэнцефалического барьера, что делает его эффективным путем введения при лизосомных болезнях накопления, которые поражают головной мозг и/или висцеральные органы. Таким образом, первый аспект изобретения относится к способу лечения или профилактики лизосомной болезни накопления у пациента, болезни, вызванной дефицитом фермента, причем способ предусматривает введение фермента пациенту посредством внутрижелудочковой доставки в головной мозг. Связанный с ним аспект изобретения относится к применению фермента для получения лекарственного средства для лечения или профилактики лизосомной болезни накопления у пациента, болезни, вызванной дефицитом фермента у пациента, где лечение или профилактика включают внутрижелудочковое введение фермента в головной мозг. Дефицит фермента может быть вызван, например, дефектом экспрессии фермента или мутацией фермента, которая снижает уровень его активности (например, фермент является неактивным) или повышает скорость клиренса/распада фермента in vivo. Дефицит может привести к накоплению субстрата фермента, и введение фермента может привести к снижению уровня субстрата в головном мозге. Лизосомная болезнь накопления может быть любым из заболеваний, перечисленных выше в таблице 1. Фермент может быть лизосомной гидролазой.
Согласно одному из вариантов осуществления изобретения лечение проводят у пациента с болезнью Нимана-Пика типа A или B. Кислую сфингомиелиназу вводят пациенту посредством внутрижелудочковой доставки в головной мозг в количестве, достаточном для уменьшения уровня сфингомиелина в указанном головном мозге.
Другой аспект изобретения относится к набору для лечения или профилактики лизосомной болезни накопления у пациента, болезни, вызванной ферментным дефицитом. Набор содержит фермент, имеющийся в дефиците, и катетер и/или насос для доставки фермента в один желудочек головного мозга и более. Катетер и/или насос может быть специально предназначен и/или приспособлен для внутрижелудочковой доставки. Согласно одному из вариантов осуществления изобретение относится к набору для лечения пациента с болезнью Нимана-Пика типа A или В. Набор содержит кислую сфингомиелиназу и катетер для доставки указанной кислой сфингомиелиназы в желудочки головного мозга пациента.
Еще один аспект изобретения относится к набору для лечения пациента с болезнью Нимана-Пика типа A или В. Набор содержит кислую сфингомиелиназу и насос для доставки указанной кислой сфингомиелиназы в желудочки головного мозга пациента.
В соответствии с изобретением можно лечить пациента, имеющего лизосомную болезнь накопления, вызванную дефицитом фермента, что приводит к накоплению субстрата фермента. Такие заболевания включают, среди прочего, болезнь Гоше, MPS I и II, болезнь Помпе и болезнь Баттена (CLN2). Фермент, дефектный при конкретном заболевании, вводят пациенту в головной мозг посредством внутрижелудочковой доставки. Фермент можно вводить в боковые желудочки и/или четвертый желудочек. Скорость введения фермента по настоящему изобретение такова, что введение одной дозы может быть струйным или может занимать приблизительно 1-5 минут, приблизительно 5-10 минут, приблизительно 10-30 минут, приблизительно 30-60 минут, приблизительно 1-4 часа или занимать более 4, 5, 6, 7 или 8 часов. Тем самым уровень субстрата в указанном головном мозге может снижаться. Введение одной дозы может занять более 1 минуты, более 2 минут, более 5 минут, более 10 минут, более 20 минут, более 30 минут, более 1 часа, более 2 часов или более 3 часов.
Указанные и другие варианты осуществления, которые будут очевидны специалистам в данной области техники после ознакомления с описанием изобретения, относящегося к способам и наборам для лечения и/или профилактики лизосомных болезней накопления, в особенности тех, которые вызывают патологию и ЦНС, и висцеральных органов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
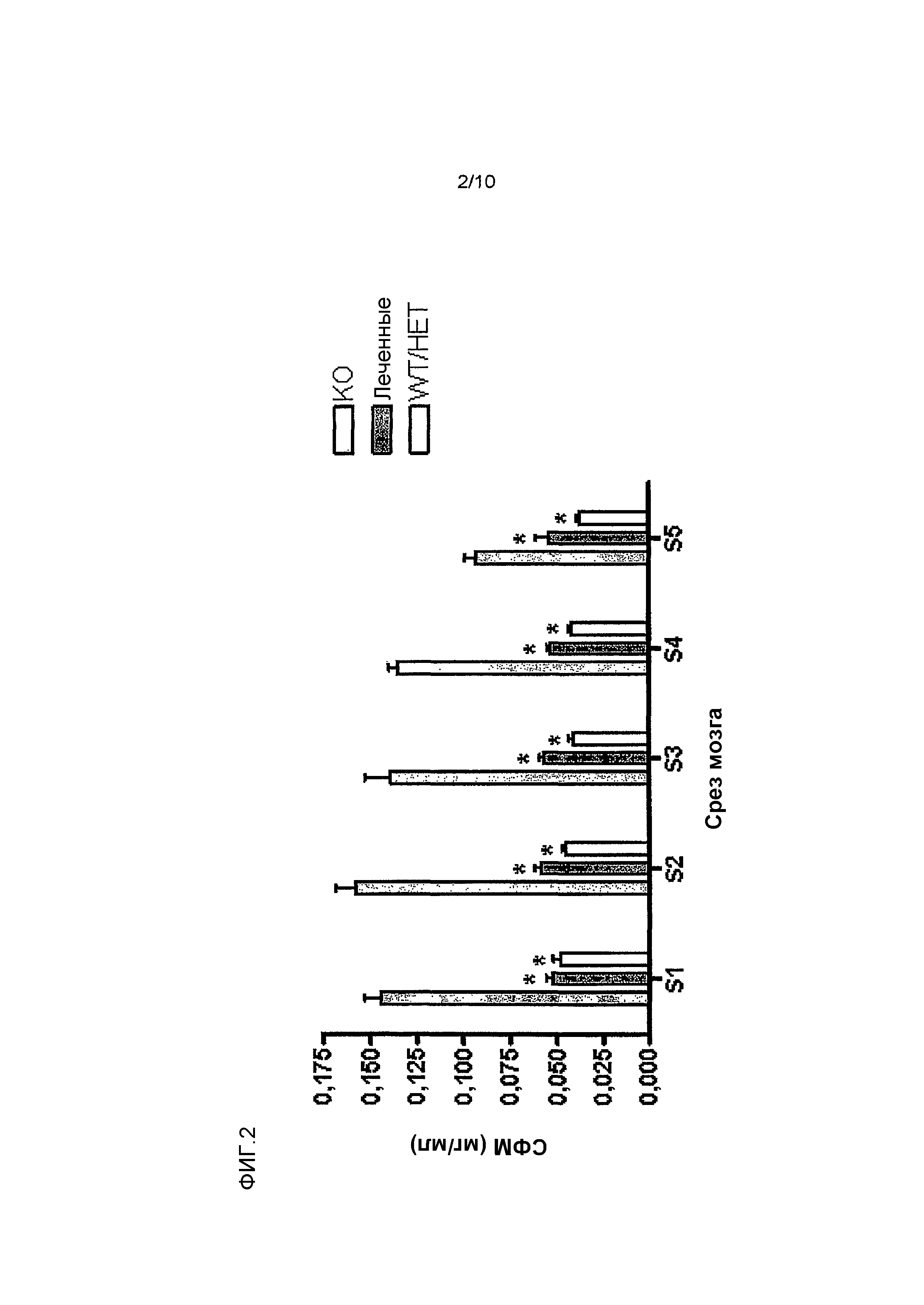
На фиг. 1 изображена схема срезов головного мозга, которые анализировались на сфингомиелин. S1 находится спереди головного мозга, а S5 - сзади.
Фиг. 2 показывает, что внутрижелудочковое введение rhASM снижает уровень сфингомиелина (СФМ) в головном мозге мыши линии ASMKO.
Фиг. 3 показывает, что внутрижелудочковое введение rhASM снижает уровень СФМ в печени, селезенке и легком мыши линии ASMKO.
Фиг. 4 показывает окрашивание головного мозга hASM после внутрижелудочковой инфузии.
Фиг. 5 показывает, что внутрижелудочковая инфузия rhASM длительностью 6 часов снижает уровень СФМ в головном мозге мыши линии ASMKO.
Фиг. 6 показывает, что внутрижелудочковая инфузия rhASM длительностью 6 часов снижает уровень СФМ в печени, сыворотке и легких мыши линии ASMKO.
На фиг. 7 изображены документально зафиксированные варианты hASM и их отношение к заболеванию или ферментной активности.
На фиг. 8 изображен поперечный срез головного мозга, на котором показаны желудочки.
На фиг. 9A и 9B представлен вид желудочков соответственно сбоку и сверху.
На фиг. 10 изображена инъекция в боковые желудочки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В практическом осуществлении настоящего изобретения будут использоваться, если не указано иного, обычные способы иммунологии, молекулярной биологии, микробиологии, цитобиологии и рекомбинантной ДНК, которые находятся в пределах компетентности в соответствующей области техники. См., например, Sambrook, Fritsch and Maniatis, Molecular Cloning: A Laboratory Manual, 2nd edition (1989); Current Protocols In Molecular Biology (F. M. Ausubel, et al. eds., (1987)); the series Methods in Enzymology (Academic Press, Inc.): PCR 2: A Practical Approach (M.J. MacPherson, B.D. Hames and G.R. Taylor eds. (1995)), Harlow and Lane, eds. (1988) Antibodies, a Laboratory Manual, and Animal Cell Culture (R.I. Freshney, ed. (1987)).
Как используется в описании и формуле изобретения, формы единственного числа включают ссылку на множественное число, если из контекста ясно не следует иного. Например, термин «клетка» включает множество клеток, включая их смеси.
Как используется в настоящем документе, под термином «содержащий» понимают композиции и способы, включающие перечисленные элементы, не исключая другие. «Состоящий по существу из», если используется для определения композиций и способов, должен означать исключение других элементов любой существенной значимости для комбинации. Таким образом, композиция, состоящая по существу из элементов, как определено в настоящем документе, не исключала бы следовые примеси метода выделения и очистки, а также фармацевтически приемлемые носители, например, физиологического раствора с фосфатным буфером, консерванты и т.п. «Состоящий из» должен означать исключение более чем следовых элементов других ингредиентов и исключение существенных стадий способа введения композиции или лекарственных средств по изобретению. Варианты осуществления, определенные каждым из этих переходных терминов, находятся в пределах объема настоящего изобретения.
Все числовые обозначения, например, pH, температура, время, концентрация и молекулярная масса, включая диапазоны, являются приближениями, которые меняются (+) или (-) с шагом 0,1. Следует понимать, хотя это не всегда явно указано, что числовым обозначениям предшествует термин «около». Также следует понимать, хотя это не всегда явно указано, что описанные в настоящем документе реагенты являются только примерами, и что также могут использоваться их эквиваленты, которые известны в данной области техники.
Термины «терапевтический», «терапевтически эффективное количество» и аналогичные термины относятся к тому количеству вещества, например, фермента или белка, которое приводит к профилактике или задержке начала, или ослаблению одного симптома заболевания и более у индивида или приводит к достижению желаемого биологического результата, такого как коррекция неврологической патологии. Термин «терапевтическая коррекция» относится к той степени коррекции, которая приводит к профилактике или задержке начала, или ослаблению одного симптома заболевания и более у индивида. Эффективное количество может быть определено известными эмпирическими способами.
Предполагается, что «композиция» или «лекарственное средство» включает комбинацию активного средства, например, фермента, с носителем или другим материалом, например, соединением или композицией, которые инертны (например, определимое вещество или метка) или активны, например, комбинацию со вспомогательным веществом, разбавителем, связующим, стабилизатором, буфером, солью, липофильным растворителем, консервантом, вспомогательным веществом и тому подобным, или со смесью двух таких веществ и более. Предпочтительно носители фармацевтически приемлемые. Они могут включать фармацевтические наполнители и добавки, белки, пептиды, аминокислоты, липиды и углеводы (например, сахара, включая моносахариды, ди-, три-, тетра- и олигосахариды; продукты замещения сахаров, например, альдитолы, альдоновые кислоты, этерифицированные сахара и тому подобное; и полисахариды или полимеры сахаров), которые могут присутствовать по отдельности или в комбинации, составляя по отдельности или в комбинации 1-99,99% по массе или объему. Примеры белковых наполнителей включают сывороточный альбумин, например, сывороточный альбумин человека (HSA), рекомбинантный альбумин человека (rHA), желатин, казеин и тому подобное. Характерные компоненты-аминокислоты/антитела, которые также могут функционировать как буфер, включают аланин, глицин, аргинин, бетаин, гистидин, глутаминовую кислоту, аспарагиновую кислоту, цистеин, лизин, лейцин, изолейцин, валин, метионин, фенилаланин, аспартам и тому подобное. Углеводные наполнители тоже входят в объем настоящего изобретения, примеры их включают, но не ограничены этим, моносахариды, например, фруктозу, мальтозу, галактозу, глюкозу, D-маннозу, сорбозу и тому подобное; дисахариды, такие как лактоза, сахароза, трегалоза, целлобиоза и тому подобное; полисахариды, такие как раффиноза, мелецитоза, мальтодекстрины, декстраны, крахмалы и тому подобное; и альдитолы, такие как маннит, ксилит, мальтит, лактит, ксилит, сорбит (глюцит) и миоинозит.
Термин «носитель» также включает буфер или средство для регулировки pH, или композицию, содержащую то же самое; обычно буфер является солью, полученной из органической кислоты или основания. Характерные буферы включают соли органических кислот, такие как соли лимонной кислоты, аскорбиновой кислоты, глюконовой кислоты, угольной кислоты, винной кислоты, янтарной кислоты, уксусной кислоты или фталевой кислоты, Трис-буфер, трометамина гидрохлорид или фосфатный буфер. Дополнительные носители включают полимерные наполнители/добавки, такие как поливинилпирролидоны, фиколлы (полимерный сахар), декстраты (например, циклодекстрины, такие как 2-гидроксипропил-.квадратура.-циклодекстрин), полиэтиленгликоли, ароматические средства, антимикробные средства, подслащивающие вещества, антиоксиданты, антистатические средства, поверхностно-активные вещества (например, полисорбаты, такие как «Твин-20» и «Твин-80»), липиды (например, фосфолипиды, жирные кислоты), стероиды (например, холестерин) и хелатирующие средства (например, ЭДТА).
Как используется в настоящем документе, термин «фармацевтически приемлемый носитель» включает любой стандартный фармацевтический носитель, такой как физиологический раствор с фосфатным буфером, воду и эмульсии, например, эмульсию масло/вода или вода/масло, и различные типы увлажнителей. Композиции и лекарственные средства, которые получают и/или используют по настоящему изобретению и включают конкретный фермент, дефицит которого должен быть скорректирован, могут содержать стабилизаторы и консерванты и любой из вышеупомянутых носителей с дополнительным условием, что они могут быть приемлемыми для применения in vivo. Примеры носителей, стабилизаторов и вспомогательных веществ см. в Martin REMINGTON'S PHARM. SCI., 15th Ed. (Mack Publ. Co., Easton (1975) and Williams & Williams, (1995), и «PHYSICIAN'S DESK REFERENCE», 52nd ed., Medical Economics, Montvale, N.J. (1998).
«Индивид», «человек» или «пациент» в настоящем документе используются взаимозаменяемо и относятся к позвоночному, предпочтительно млекопитающему, более предпочтительно человеку. Млекопитающие включают, но ими не ограничены, мышей, крыс, обезьян, людей, сельскохозяйственных животных, спортивных животных и домашних животных.
Как используется в настоящем документе, термин «модулировать» означает изменять количество или интенсивность эффекта или результата, например, усилить, увеличить, уменьшить или снизить.
Как используется в настоящем документе, термин «ослаблять» является синонимом термину «облегчать» и означает уменьшать или делать более легким. Например, можно ослабить симптомы заболевания или расстройства, сделав их более терпимыми.
Для идентификации структур в мозге человека, см., например, The Human Brain: Surface, Three-Dimensional Sectional Anatomy With MRI, and Blood Supply, 2nd ed., eds. Deuteron et al., Springer Vela, 1999; Atlas of the Human Brain, eds. Mai et al., Academic Press; 1997; и Co-Planar Stereotaxic Atlas of the Human Brain: 3-Dimensional Proportional System: An Approach to Cerebral Imaging, eds. Tamarack et al., Thyme Medical Pub., 1988. Для идентификации структур в головном мозге мыши, см., например, The Mouse Brain in Stereotaxic Coordinates, 2nd ed., Academic Press, 2000.
Авторы обнаружили, что внутрижелудочковая доставка лизосомных гидролитических ферментов в головной мозг пациентам с дефицитом ферментов, улучшает метаболический статус, как головного мозга, так и пораженных висцеральных (не ЦНС) органов. Это особенно верно, если скорость введения медленна по сравнению со струйным введением. Лизосомные болезни накопления, вызванные дефицитом конкретного фермента, такие как заболевания, перечисленные выше в таблице 1, поэтому можно лечить или предотвратить внутрижелудочковым введением соответствующего фермента. Один особенно используемый фермент для лечения Нимана-Пика A или B представляет собой кислую сфингомиелиназу (ASM), такую как показана в SEQ ID NO: 1 (Остатки 1-46 составляют сигнальную последовательность, которая при секретировании отщепляется.) Один особо используемый фермент для лечения болезни Гоше представляет собой глюкоцереброзидазу. Один особо используемый фермент для лечения MPS I представляет собой альфа-L-идуронидазу. Один особо используемый фермент для лечения MPS II представляет собой идуронат-2-сульфатазу. Один особо используемый фермент для лечения болезни Помпе или гликогеноза типа II (GSDII), также называемой дефицитом кислой мальтазы (AMD), представляет собой кислую альфа-глюкозидазу. Один особо используемый фермент для лечения классической поздней инфантильной болезни Баттена (CLN2) представляет собой трипептидилпептидазу. Ферменты, которые применяются и/или вводятся по настоящему изобретению, могут быть рекомбинантными формами ферментов, полученными с использованием способов, которые известны в данной области техники. В одном из вариантов осуществления фермент является рекомбинантным ферментом человека.
Введение лизосомных ферментов и конкретнее лизосомных гидролитических ферментов пациентам с дефицитом ферментов может производиться в любой один и более желудочек головного мозга, которые заполнены цереброспинальной жидкостью (ЦСЖ). ЦСЖ - прозрачная жидкость, которая заполняет желудочки, присутствует в субарахноидальном пространстве и окружает головной и спинной мозг. ЦСЖ вырабатывается сосудистыми сплетениями за счет просачивания или перехода тканевой жидкости мозга в желудочки. Сосудистое сплетение представляет собой структуру, выстилающую дно бокового желудочка и крышу третьего и четвертого желудочков. Определенные исследования показали, что указанные структуры способны вырабатывать 400-600 ccs жидкости в сутки - указанного количества достаточно, чтобы заполнить пространства центральной нервной системы 4 раза в сутки. Подсчитано, что у взрослых объем этой жидкости равен от 125 до 150 мл (4-5 унций). ЦСЖ непрерывно образуется, циркулирует и всасывается. Определенные исследования показали, что каждые сутки может образовываться приблизительно 430-450 мл (почти 2 стакана) ЦСЖ. Согласно определенным вычислениям ее образование равно приблизительно 0,35 мл в минуту у взрослых и 0,15 в минуту у грудных детей. Основная часть ЦСЖ вырабатывается сосудистыми сплетениями боковых желудочков. Через отверстие Монро ЦСЖ поступает в третий желудочек, где к ней примешивается ЦСЖ, произведенная в третьем желудочке, и продолжает движение вниз, через сильвиев водопровод к четвертому желудочку. В четвертом желудочке добавляется еще ЦСЖ, затем жидкость поступает в субарахноидальное пространство через отверстия Мажанди и Лушки. После этого ЦСЖ циркулирует вокруг основания головного мозга, направляется вниз вокруг спинного мозга и вверх вокруг полушарий головного мозга. ЦСЖ всасывается в кровь через ворсины паутинной оболочки и внутричерепные сосудистые синусы, тем самым доставляя ферменты, введенные в желудочки, не только к головному мозгу, но и к висцеральным органам, которые, как известно, поражаются при ЛБН.
Хотя конкретная аминокислотная последовательность показана в SEQ ID NO: 1, варианты указанной последовательности, которые сохраняют активность, например, нормальные варианты в популяции людей, тоже могут использоваться. Как правило, такие нормальные варианты отличаются от последовательности, показанной в SEQ ID NO: 1, только одним или двумя остатками. Варианты SEQ ID NO: 1, которые должны применяться по настоящему изобретению, природные или нет, должны быть по меньшей мере на 95%, 96%, 97%, 98% или 99% идентичны SEQ ID NO: 1. Варианты других ферментов могут применяться по настоящему изобретению. Однако независимо от фермента, который применяется, не должны использоваться варианты, которые связаны с заболеванием или сниженной активностью. Как правило, будет доставляться зрелая форма фермента. В случае SEQ ID NO: 1, зрелая форма будет начинаться с остатка 47, как показано в SEQ ID NO: 1. Варианты, которые связаны с заболеванием, изображены на фиг.7. Сходным образом, также могут использоваться присутствующие в популяции людей нормальные варианты таких ферментов ЛБН, как глюкоцереброзидаза, альфа-L-идуронидаза, идуронат-2-сульфатаза, кислая альфа-глюкозидаза и трипептидилпептидаза, которые сохраняют ферментативную активность.
Наборы по настоящему изобретению представляют собой совокупности отдельных компонентов. Хотя они могут быть упакованы в один контейнер, внутри упаковки они могут быть дополнительно упакованы по отдельности. Даже один контейнер может быть разделен на отделения. Как правило, набор будет сопровождаться комплектом инструкций и содержать инструкции по доставке ферментов, например, лизосомных гидролитических ферментов, в желудочки. Инструкции могут находиться в печатной форме, в электронной форме, в виде учебного видео или DVD, на компакт-диске, на гибком диске, в Интернете с адресом, вложенном в упаковку, или в виде комбинации этих средств. В дополнение к ферменту, одной и более канюле или катетеру могут предоставляться другие компоненты, такие как разбавители, буферы, растворители, лента, винты и эксплуатационные инструменты.
Популяции, для которых лечение способами по изобретению может быть эффективно, включают, но не ограничены этим, пациентов, страдающих или подвергающихся риску развития нейрометаболического расстройства, например, ЛБН, такого как заболевания, перечисленные в таблице 1, особенно если такое заболевание затрагивает ЦНС и висцеральные органы. В характерном варианте осуществления заболевание представляет собой болезнь Нимана-Пика типа А.
ASM или другой лизосомный фермент-гидролаза может быть включен в фармацевтическую композицию, применимую для лечения, например, ингибирования, ослабления, профилактики или облегчения состояния, которое характеризуется недостаточным уровнем активности лизосомных гидролаз. Фармацевтическую композицию будут вводить индивиду, страдающему дефицитом лизосомной гидролазы, или тому, кто подвергается риску развития указанного дефицита. Композиции должны содержать терапевтическое или профилактическое количество ASM или другого лизосомного гидролитического фермента, в фармацевтически приемлемом носителе. Фармацевтический носитель может быть любым совместимым, нетоксичным веществом, подходящим для того, чтобы доставить полипептиды пациенту. В качестве носителя могут использоваться стерилизованная вода, спирт, жиры и воски. В фармацевтические композиции также могут включаться фармацевтически приемлемые вспомогательные вещества, буферные вещества, диспергаторы и тому подобное. Носитель может быть объединен с ASM или другим лизосомным гидролитическим ферментом в любой форме, подходящей для введения посредством внутрижелудочковой инъекции или инфузии (возможно, указанная форма также подходит для внутривенного или интратекального введения) или иного способа. Подходящие носители включают, например, физиологический раствор, бактериостатическую воду, Кремофор EL.TM. (BASF, Parsippany, шт. Нью-Джерси) или физиологический раствор с фосфатным буфером (PBS), другие солевые растворы, растворы декстрозы, растворы глицерина, воду и эмульсии масел, например, полученные из нефтяных масел, масел животного, растительного или синтетического происхождения (масло арахиса, масло сои, минеральное масло или кунжутное масло). В качестве носителя может использоваться искусственная ЦСЖ. Предпочтительно носитель стерилен и свободен от пирогенов. Концентрация ASM или другого лизосомного гидролитического фермента в фармацевтической композиции может меняться в широких пределах, то есть по меньшей мере от приблизительно 0,01 масс.% до 0,1 масс.%, до приблизительно 1 масс.%, до 20 масс.% или более в расчете на общую массу композиции.
Для внутрижелудочкового введения ASM или другого лизосомного гидролитического фермента композиция должна быть стерильной и представлять собой жидкость. Композиция должна быть стабильной при условиях изготовления и хранения, и ее следует предохранять от воздействия загрязняющих микроорганизмов, таких как бактерии и грибы. Действие микроорганизмов можно предотвратить с помощью различных антибактериальных и противогрибковых средств, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимеросала и тому подобного. Во многих случаях в композиции предпочтительно включают изотонические средства, например, сахар, многоатомные спирты, например, маннит, сорбит, хлорид натрия.
Доза ASM или другого лизосомного гидролитического фермента может в некоторой мере изменяться от человека к человеку, в зависимости от конкретного фермента и его специфичной активности in vivo, путь введения, медицинского состояния, возраста, массы или пола пациента, чувствительности пациента к ASM или другому лизосомному гидролитическому ферменту или компонентам растворителя и других факторов, которые лечащий врач сможет легко принять во внимание. Хотя дозы могут изменяться в зависимости от заболевания и пациента, фермент, как правило, вводится пациенту в количестве от приблизительно 0,1 до приблизительно 1000 мг на 50 кг массы пациента в месяц. В одном из вариантов осуществления фермент вводят пациенту в количестве от приблизительно 1 до приблизительно 500 миллиграммов на 50 кг массы пациента на месяц. В других вариантах осуществления фермент вводят пациенту в количестве от приблизительно 5 до приблизительно 300 миллиграммов на 50 кг массы пациента в месяц или от приблизительно 10 до приблизительно 200 миллиграммов на 50 кг массы пациента в месяц.
Скорость введения такова, что введение одной дозы может происходить струйно. Кроме того, инфузия одной дозы может продолжаться в течение приблизительно 1-5 минут, приблизительно 5-10 минут, приблизительно 10-30 минут, приблизительно 30-60 минут, приблизительно 1-4 часов или занимать более 4, 5, 6, 7 или 8 часов. Введение может потребовать более 1 минуты, более 2 минут, более 5 минут, более 10 минут, более 20 минут, более 30 минут, более 1 часа, более 2 часов или более 3 часов. Авторы заметили, что хотя струйное внутрижелудочковое введение может быть эффективным, медленная инфузия высокоэффективна. Хотя авторы не желают быть связанными любой конкретной теорией операции, считается, что медленная инфузия более эффективна из-за обновления цереброспинальной жидкости (ЦСЖ). Хотя приведенные в литературе оценки и вычисления изменяются, считается, что у человека цереброспинальная жидкость обновляется в пределах приблизительно 4, 5, 6, 7 или 8 часов. В одном из вариантов осуществления изобретения длительность медленной инфузии должна быть отмерена такой, чтобы она была равной времени обновления ЦСЖ или превышало его. Время обновления может зависеть от биологического вида, размера и возраста индивида, но его можно определить, используя способы, известные в данной области техники. Инфузия также может быть непрерывной в течение одних суток и более. Лечение пациента можно проводить один, два или три и более раз в месяц, например, еженедельно, например, каждые две недели. Инфузии можно повторять в течение жизни индивида, что связано с повторным накоплением субстрата заболевания в головном мозге или висцеральных органах. Повторное накопление можно определить любым из способов, известных в данной области техники для идентификации и определения количества соответствующего субстрата, причем эти способы могут быть выполнены на одной пробе и более, взятой из головного мозга и/или одного висцерального органа и более. Такие способы включают ферментативные и/или иммунологические исследования, например, радиоиммунологическое исследование или ELISA.
ЦСЖ поступает в кровь через ворсины паутинной оболочки и внутричерепные сосудистые синусы, тем самым доставляя введенные посредством инфузии ферменты в висцеральные органы, которые, как известно, поражаются при ЛБН. Висцеральные органы, часто поражаемые при болезни Нимана-Пика, - это легкие, селезенка, почки и печень. Медленная внутрижелудочковая инфузия снижает количество субстрата для введенного фермента по меньшей мере в головном мозге и потенциально в висцеральных органах. Снижение количества субстрата, накопленного в головном мозге, легких, селезенке, почках и/или печени, может быть резким. Может быть достигнуто снижение более чем на 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%. Достигнутое снижение не обязательно одинаковое у разных пациентов и даже в разных органах одного пациента. Снижение можно определить любым способом, который известен в данной области техники, например, методами ферментного и/или иммунологического исследования, которые рассмотрены в другом месте данного документа.
Если желательно, структура мозга человека может быть скоррелирована с подобными структурами в головном мозге другого млекопитающего. Например, у большинства млекопитающих, включая людей и грызунов, отмечается похожая топографическая организация проекций энторинальной коры-гиппокампа, с нейронами в латеральной части и латеральной, и медиальной энторинальной коры, проецирующимися на дорсальную часть или септальный полюс гиппокампа, в то время как проекции на вентральный гиппокамп берут начало преимущественно от нейронов медиальных частей энторинальной коры (Principles of Neural Science, 4th ed., eds Kandel et al., McGraw-Hill, 1991; The Rat Nervous System, 2nd ed., ed. Paxinos, Academic Press, 1995). Кроме того, слой II клеток энторинальной коры проецируется на зубчатую извилину, и они оканчиваются в наружных двух третях молекулярного слоя зубчатой извилины. Аксоны клеток слоя III проецируются билатерально на поля СА1 и СА3 гиппокампа, оканчиваясь в губчатом слое молекулярного слоя.
В характерном варианте осуществления введение осуществляется путем инфузии фермента ЛБН в один или оба боковых желудочка индивида или пациента. Посредством инфузии в боковые желудочки фермент доставляют к месту в головном мозге, в котором образуется самое большое количество ЦСЖ. Инфузию фермента также можно произвести более чем в один желудочек головного мозга. Лечение может состоять из одной инфузии в намеченное место, или инфузия может повторяться. Могут использоваться множественные места инфузии/инъекции. Например, желудочки, в которые вводится фермент, могут включать боковые желудочки и четвертый желудочек. В некоторых вариантах осуществления, в дополнение к первому месту введения, композицию, содержащую фермент ЛБН, вводят в другое место, которое может быть контралатеральным или ипсилатеральным по отношению к первому месту введения. Инъекции/инфузии могут быть единичными или множественными, односторонними или двусторонними.
Для доставки раствора или другой композиции, содержащей фермент, именно к конкретной области центральной нервной системы, такую как конкретный желудочек, например, в боковые желудочки в четвертый желудочек головного мозга, введение может осуществляться в виде стереотаксической микроинъекции. Например, в день операции у пациентов будет закреплена стереотаксическая рамка (прикреплена винтами к черепу). Будет получено изображение мозга с основанием стереотаксической рамки (совместимой с МРТ с фидуциарной разметкой) с помощью МРТ высокого разрешения. Затем магнитно-резонансные томограммы будут перенесены в компьютер, на котором запущено стереотаксическое программное обеспечение. Будет использоваться ряд фронтальных, сагиттальных и горизонтальных изображений для определения намеченного места векторной инъекции и траекторию. Программное обеспечение непосредственно переводит траекторию в 3-мерные координаты, подходящие для стереотаксической рамки. Над местом проникновения сверлят фрезевые отверстия, и размещают стереотаксический аппарат с иглой, введенной на заданную глубину. После этого вводят раствор фермента в фармацевтически приемлемом растворителе. Могут использоваться дополнительные пути введения, например, поверхностное кортикальное нанесение под визуальным контролем или другое нестереотаксическое нанесение.
Один из способов медленной инфузии состоит в использовании насоса. Такие насосы поступают в продажу, например, от Alzet (Cupertino, Калифорния) или Medtronic (Миннеаполис, Миннесота). Насос может быть имплантируемым. Другой удобный способ введения ферментов состоит в том, чтобы использовать канюлю или катетер. Канюля или катетер могут использоваться для множественных введений, разнесенных во времени. Полые иглы и катетеры могут вводиться стереотаксически. Предполагается, что для лечения пациента с лизосомной болезнью накопления будут использоваться множественные введения. Катетеры и насосы могут использоваться по отдельности или в комбинации.
Лизосомные болезни накопления (ЛБН) включают свыше сорока генетических нарушений, при многих из которых имеются генетические дефекты в различных лизосомных гидролазах. Характерные лизосомные болезни накопления и связанные дефектные ферменты перечислены в таблице 1.
Болезнь Гоше является следствием наследственного дефицита лизосомной гидролазы глюкоцереброзидазы (GC), который приводит к накоплению ее субстрата - гликозилцерамида (GL-1) в лизосомах гистиоцитов. Прогрессирующее накопление GL-1 в тканевых макрофагах (клетках Гоше) происходит в различных тканях. Пределы накопления частично определяются генотипом. Клинически выделяют три различных фенотипа Гоше: ненейропатический тип 1, наиболее распространенный с началом в пределах от раннего детства до взрослой жизни, и нейропатическими типами 2 и 3, которые проявляются в грудном возрасте и раннем детстве соответственно. По настоящему изобретению можно проводить лечение любого из этих фенотипов. Первичные клинические проявления, общие для всех форм болезни Гоше, включают гепатоспленомегалию, цитопению, патологические переломы костей и, изредка, легочную недостаточность. Подробное обсуждение болезни Гоше можно найти в Online Metabolic & Molecular Bases of Inherited Diseases, Part 16, Chapter 146 и 146.1 (2007). У пациентов с болезнью Гоше типа 2 и 3, у которых имеется существенное поражение ЦНС, внутрижелудочковая доставка дефектного фермента ЛБН улучшает метаболический статус головного мозга и потенциально пораженных висцеральных (не ЦНС) органов. Внутрижелудочковая доставка дефектного фермента ЛБН индивидам с болезнью Гоше типа 1 улучшает метаболический статус пораженных висцеральных (не ЦНС) органов. Существуют животные модели болезни Гоше, полученные из мышиных моделей с прицельным разрушением соответствующего гена мыши. Например, существует мышиная модель болезни Гоше, несущая мутацию D409V в локусе GC (Xu, Y-H et al. (2003). Am. J. Pathol. 163: 2093-2101). У гетерозиготной мыши gbaD409V/null активность GC в висцеральных тканях составляет ~5% нормальной, и у мыши появляются наполненные липидами макрофаги (клетки Гоше) в печени, селезенке, легких и костном мозге к 4-месячному возрасту. Указанная модель является подходящей системой для оценки преимуществ и определения условий внутрижелудочковой доставки дефектного фермента ЛБН индивидам с болезнью Гоше.
Болезнь Нимана-Пика (NPD) является лизосомной болезнью накопления и представляет собой наследственное нейрометаболическое заболевание, характеризующееся генетической недостаточностью кислой сфингомиелиназы (ASM; сфингомиелинхолинфосфогидролаза, EC 3.1.3.12). Отсутствие функционального белка ASM приводит к накоплению субстрата сфингомиелина внутри лизосом нейронов и глии по всему головному мозгу. Это приводит к появлению в перикарионе больших количеств растянутых лизосом, что характерно и является первичным клеточным фенотипом NPD типа А. Наличие растянутых лизосом коррелирует с утратой нормальной клеточной функции и прогрессированием нейродегенеративных нарушений, которое приводит к смерти заболевшего в раннем детстве (The Metabolic and Molecular Bases of Inherited Diseases, eds. Scriver et al., McGraw-Hill, New York, 2001, pp. 3589-3610). Вторичные клеточные фенотипы (например, дополнительные метаболические расстройства), также связаны с этим заболеванием, особенно высокий уровень накопления холестерина в лизосомном компартменте. Сфингомиелин обладает высокой аффинностью к холестерину, что приводит к секвестрации большого количества холестерина в лизосомах мышей ASMKO и пациентов-людей (Leventhal et al. (2001) J. Biol. Chem., 276: 44976-44983; Slotte (1997) Subcell. Biochem., 28: 277-293; и Viana et al. (1990) J. Med. Genet, 27:499-504). Подробное обсуждение болезни NPD приведено Online Metabolic & Molecular Bases of Inherited Diseases, Part 16, Chapter 144 (2007). Существуют животные модели NPD. Так, мыши ASMKO - принятая модель болезни Нимана-Пика типов А и В (Horinouchi et al. (1995) Nat. Genetics, 10: 288-293; Jin et al. (2002) J. Clin. Invest., 109: 1183-1191; и Otterbach (1995) Cell, 81: 1053-1061). Внутрижелудочковая доставка дефектного фермента ЛБН улучшает метаболический статус головного мозга и поврежденных висцеральных (не ЦНС) органов.
Мукополисахаридозы (MPS) являются группой лизосомных расстройств накопления, вызванных дефицитом ферментов, катализирующих разложение гликозаминогликанов (мукополисахаридов). Существует 11 известных дефицитов ферментов, которые дают начало 7 различным MPS, включают MPS I (синдромы Гурлер, Шейе и Гурлер-Шейе) и MPS II (синдром Хантера). Любой MPS можно лечить по настоящему изобретению. Подробное обсуждение MPS можно обнаружить в Online Metabolic & Molecular Bases of Inherited Diseases, Part 16, Chapter 136 (2007). Существует множество животных моделей MPS, которые получены из природных мутаций собак, кошек, крыс, мышей и коз, а также мышиные модели, созданные нацеленным разрушением соответствующего гена. Биохимические и метаболические особенности указанной животной модель, как правило, очень близки обнаруженным у людей; однако клинические проявления могут быть более слабыми. Например, принятые модели MPS I включаю мышиную модель [Clark, LA et al., Hum. Mol. Genet. (1997), 6: 503] и собачью модель [Menon, KP et al., Genomics (1992), 14: 763]. Например, принятые модели для MPS II включают мышиную модель [Muenzer, J. et al., (2002), Acta Paediatr. Suppl.; 91(439): 98-9]. При MPS с поражением центральной нервной системы, таким как обнаруживаемое у больных с MPS I и MPS II, внутрижелудочковая доставка дефектного фермента ЛБН улучшает метаболический статус головного мозга и потенциально поврежденных висцеральных (не ЦНС) органов.
Болезнь Помпе или болезнь накопления гликогена типа II (GSD II), также называемая дефицитом кислой мальтазы (AMD), представляет собой наследственное расстройство метаболизма гликогена, возникающее из-за дефектов активности лизосомной гидролазы, кислой альфа-глюкозидазы во всех тканях пораженных людей. Дефицит фермента приводит к накоплению в лизосомах многих тканей гликогена нормального строения. Накопление наиболее выражено в мышце сердца, скелетных мышцах и в печеночных тканях грудных детей с генерализованным расстройством. При поздно проявляющемся GSD II накопление гликогена в лизосомах фактически ограничено скелетными мышцами и менее выражено. Электромиографические аномалии, указывающие на диагноз, включают псевдомиотонические разряды и раздражимость, но у пациентов с началом болезни в юношеском или взрослом возрасте аномалии могут быть неодинаково выражены в различных мышцах. Компьютерные аксиальные томограммы часто выявляют место (места) поражения мышц. У большинства пациентов, особенно при начале болезни во взрослом возрасте, повышены уровни в плазме креатинкиназы и ферменты печени. Существует несколько природных животных моделей болезни с началом в грудном или более позднем возрасте. Существует модель мыши с нокаутированным геном [Bijyoet AG et al., Hum. Mol. Genet. (1998); 7: 53-62]. Описано улучшение под действием ферментной терапии у мышей с нокаутированным геном [Raben, N et al., Mol. Genet. Metab. (2003); 80: 159-69] и у перепелов. Внутрижелудочковая доставка дефектного фермента ЛБН улучшает метаболический статус головного мозга и потенциально поврежденных висцеральных (не ЦНС) органов.
Нейрональные цероид-липофусцинозы (NCL) представляет собой группу нейродегенеративных расстройств, которые отличаются от других нейродегенеративных заболеваний накоплением в головном мозге и других тканях аутофлюоресцирующего вещества («пигмента старения»). Основные клинические особенности включают судороги, психомоторную деградацию, слепоту и преждевременную смерть. Выделены различные подгруппы NCL, которые отличаются по возрасту появления симптомов и внешнему виду накапливающегося вещества при электронной микроскопии. Три главные группы представляют собой инфантильную (INCL), классическую позднюю инфантильную (LINCL) и ювенильную (JNCL), также называемую болезнью Баттена) - вызываются аутосомно-рецессивными мутациями в генах CLN1, CLN2 и CLN3 соответственно. Белковые продукты генов CLN1 (пальмитоилпротеинтиоэстераза) и CLN2 (трипептидилпептидаза или пепиназа) представляет собой растворимые ферменты лизосом, в то время как белок CLN3 (баттенин) является мембранным белком лизосом, как и (предварительно) белок CLN5. Идентификация мутаций в генах, кодирующих белки лизосом при нескольких формах NCL, привела к распознаванию липофусцинозов как истинных лизосомных болезней накопления. Любую подгруппу NCL можно лечить по настоящему изобретению. Подробное обсуждение NCL можно обнаружить в Online Metabolic & Molecular Bases of Inherited Diseases, Part 16, Chapter 154 (2007). Природные NCL описаны у овец, собак, получены мышиные модели с помощью прицельного разрушения соответствующего гена мыши [см. например, Katz, ML et al., J. Neurosci. Res. (1999); 57: 551-6; Cho, SK et al., Glycobiology (2005); 15: 637-48]. Внутрижелудочковая доставка дефектного фермента ЛБН улучшает метаболический статус головного мозга и, возможно, поврежденных висцеральных (не ЦНС) органов.
Подробное обсуждение дополнительных лизосомных болезней накопления, раскрытых в таблице 1, с внутрижелудочковой доставкой дефектного фермента ЛБН при заболевании, имеется в Online Metabolic & Molecular Bases of Inherited Diseases, Part 16 (2007).
Вышеуказанное описание описывает настоящее изобретение в общих чертах. Все ссылки, указанные в настоящем документе, включены посредством ссылки. Более полного понимания можно достичь путем ссылки на следующие конкретные примеры, которые приведены исключительно с иллюстративными целями и не предназначены для ограничения объема изобретения.
ПРИМЕР 1
Животная модель
Мыши ASMKO представляют собой принятую модель болезни Нимана-Пика типов A и B (Horinouchi et al. (1995) Nat. Genetics, 10: 288-293; Jin et al. (2002) J. Clin. Invest., 109: 1183-1191; и Otterbach (1995) Cell, 81: 1053-1061). Болезнь Нимана-Пика (NPD) относится к лизосомным болезням накопления и является наследственным нейрометаболическим расстройством, которое характеризуется генетическим дефицитом кислой сфингомиелиназы (ASM; сфингомиелинхолинфосфогидролаза, EC 3.1.3.12). Отсутствие функционального белка ASM приводит к накоплению субстрата сфингомиелина внутри лизосом нейронов и глии по всему головному мозгу. Это приводит к формированию в перикарионе больших количеств растянутых лизосом, которые являются отличительной особенностью и первичным клеточным фенотипом NPD типа А. Наличие растянутых лизосом коррелирует с потерей нормальной клеточной функции и прогрессирующим нейродегенеративным течением, которое приводит к смерти пораженного в раннем детстве (The Metabolic and Molecular Bases of Inherited Diseases, eds. Scriver et al., McGraw-Hill, New York, 2001, pp. 3589-3610). Вторичные клеточные фенотипы (например, дополнительные метаболические расстройства) тоже связаны с указанным заболеванием, особенно высокий уровень накопления холестерина в лизосомном компартменте. Сфингомиелин имеет высокую аффинность к холестерину, что приводит к секвестрации большого количества холестерина в лизосомах мышей ASMKO и пациентов-людей (Leventhal et al. (2001) J. Biol. Chem., 276: 44976-44983; Slotte (1997) Subcell. Biochem., 28: 277-293; and Viana el al. (1990) J. Med. Genet., 27: 499-504.)
ПРИМЕР 2
Непрерывная внутрижелудочковая инфузия rhASM у мыши линии ASMKO
Цель: определить, какое влияние внутрижелудочковая инфузия рекомбинантного ASM человека (rhASM) оказывает на болезнь накопления (то есть накопление сфингомиелина и холестерина) в головном мозге мыши ASMKO.
Методы: мышам линии ASMKO в возрасте 12-13 недель стереотаксически имплантировали постоянную канюлю. В возрасте 14 недель мышам проводили инфузию 0,250 мг hASM (n=5) длительностью 24 ч (~0,01 мг/ч) на протяжении 4 дней подряд (в общей сложности ввели 1 мг), используя зонд для инфузий (соответствующий внутреннему диаметру направляющей канюли), соединенный с насосом. Перед инфузией лиофилизированный hASM растворяли в искусственной цереброспинальной жидкости (иЦСЖ). Мышей умерщвляли через 3 дня после инфузии. Для умерщвления мышам вводили чрезмерную дозу эутазола (>150 мг/кг), а затем перфузировали PBS или 4% параформальдегида. Извлекали головной мозг, печень, легкие и селезенку, и определяли в них уровень сфингомиелина (СФМ). Перед анализом на СФМ ткань мозга разделяли на 5 срезов (S1 = передняя часть мозга, S5 = задняя часть мозга; см. фиг.1).
Резюме результатов: внутрижелудочковая инфузия hASM в дозе 0,250 мг/24 ч в течение 4 дней подряд (общее количество 1 мг) привела к окрашиванию hASM и исчезновению филипина (то есть скоплений холестерина) по всему головному мозгу ASMKO. Биохимический анализ показал, что внутрижелудочковая инфузия hASM также привела к общему снижению уровней СФМ по всему головному мозгу. Уровень СФМ снизился по сравнению с уровнем дикого типа (WT). Существенное снижение СФМ также наблюдали в печени и селезенке (в легких отмечена тенденция к снижению).
ПРИМЕР 3
Внутрижелудочковая доставка hASM мышам ASMKO II
Цель: определить самую низкую эффективную дозу в течение 6-часового периода инфузии.
Методы: мышам линии ASMKO в возрасте 12-13 недель стереотаксически имплантировали постоянную канюлю. В возрасте 14 недель мышам проводили 6-часовую инфузию одной из следующих доз hASM: 10 мг/кг (0,250 мг; n=12), 3 мг/кг (0,075 мг; n=7), 1 мг/кг (0,025 мг; n=7), 0,3 мг/кг (0,0075 мг; n=7) или иЦСЖ (искусственная цереброспинальная жидкость; n=7). От мышей, получивших каждую дозу, отобрали по 2 и провели им перфузию 4% раствора параформальдегида сразу после 6-часовой инфузии, чтобы оценить распределение фермента в головном мозге (у них также взяли кровь, чтобы определить сывороточный уровень hASM). Остальных мышей из каждой группы умерщвляли через 1 неделю после инфузии. В тканях головного мозга, печени и легких этих мышей определяли уровень СФМ, как в исследовании 05-0208.
Резюме результатов: внутрижелудочковая 6-часовая инфузия hASM привела к существенному снижению уровней СФМ по всему головному мозгу независимо от дозы. Содержание СФМ в головном мозге мышей, получивших >0,025 мг, снизились до уровня WT. Содержание СФМ в висцеральных органах также значительно и дозозависимо уменьшилось (но не до уровня WT). В поддержку этих данных белок hASM также обнаружен в сыворотке мышей ASMKO, получивших инфузию белка hASM. Гистологический анализ показал, что после внутрижелудочкового введения hASM белок hASM широко распределился по всему головному мозгу (от S1 до S5).
ПРИМЕР 4
Внутрижелудочковая инфузия rhASM у мышей линии ASMKO III
Цель: 1) определить время, необходимое для повторного накопления СФМ в головном (и спинном) мозге после 6-часовой инфузии hASM (доза 0,025 мг); 2) выяснить, имеются ли половые различия в ответе на внутрижелудочковое введение hASM (предыдущие эксперименты показали, что есть половые различия в накоплении субстрата в печени; происходит ли это в головном мозге, неизвестно).
Методы: мышам линии ASMKO в возрасте 12-13 недель стереотаксически имплантировали постоянную канюлю. В возрасте 14 недель мышам проводили 6-часовую инфузию 0,025 мг hASM. После внутрижелудочковой доставки hASM мышей умерщвляли через 1 неделю после инфузии (n=7 самцов, 7 самок) или через 2 недели после инфузии (n=7 самцов, 7 самок) или через 3 недели после инфузии (n=7 самцов, 7 самок). После умерщвления извлекали головной мозг, спинной мозг, печень и легкие для анализа на СФМ.
Пробы тканей подготовили к анализу СФМ.
ПРИМЕР 5
Влияние внутрижелудочковой инфузии rhASM на когнитивные функции мышей линии ASMKO
Цель: определить, ослабляет ли внутрижелудочковая инфузия rhASM когнитивный дефицит, вызванный заболеванием, у мышей линии ASMKO.
Методы: мышам линии ASMKO в возрасте 9-10 недель стереотаксически имплантировали постоянную направляющую канюлю. В возрасте 13 недель мышам проводили 6-часовую инфузию 0,025 мг hASM. В возрасте 14 и 16 недель мыши подверглись когнитивному тестированию с использованием лабиринта Barnes.
ПРИМЕР 6
Распределение белка hASM в пределах ЦНС мышей линии ASMKO после внутрижелудочковой инфузии
Цель: определить распределение белка hASM (как функцию времени) в головном и спинном мозге мышей ASMKO после внутрижелудочковой инфузии.
Методы: мышам линии ASMKO в возрасте 12-13 недель стереотаксически имплантировали постоянную направляющую канюлю. В возрасте 14 недель мышам проводили 6-часовую инфузию 0,025 мг hASM. После инфузии мышей умерщвляли немедленно либо через 1, 2 или 3 недели.
Таблица 5. Указывает длительность инфузии, которая может использоваться с конкретным ферментом для лечения заболевания, при котором имеется дефицит указанного фермента, как указано в таблице 1.
Литература
Раскрытие каждой процитированной ссылки включено в настоящий документ.
1. Belichenko PV, Dickson PI, Passage M, Jungles S, Mobley WC, Kakkis ED. Penetration, diffusion, and uptake of recombinant human alpha-1-iduronidase after intraventricular injection into the rat brain. Mol Genet Metab. 2005; 86(1-2): 141-9.
2. Kakkis E, McEntee M, Vogler C, Le S, Levy B, Belichenko P, Mobley W, Dickson P, Hanson S, Passage M. Intrathecal enzyme replacement therapy reduces lysosomal storage in the brain and meninges of the canine model of MPS I. Mol Genet Metab. 2004; S3(1-2): 163-74.
3. Bembi B, Ciana G, Zanatta M, et al. Cerebrospinal-fluid infusion of alglucerase in the treatment for acute neuronopathic Gaucher's disease. Pediatr Res 1995; 38: A425.
4. Lonser RR, Walbridge S, Murray GJ, Aizenberg MR, Vortmeyer AO, Aerts JM, Brady RO, Oldfield EH. Convection perfusion of glucocerebrosidase for neuronopathic Gaucher's disease. Ann Neurol. 2005 Apr; 57(4): 542-8.
Реферат
Изобретение относится к медицине, а именно к терапии, и касается лечения лизосомных болезней накопления. Для этого осуществляют внутрижелудочковое введение фермента, этиологическая недостаточность которого имеется при данном заболевании. Введение производят в течение времени более трех часов. Такое медленное введение обеспечивает максимальный терапевтический эффект, заключающийся в снижении патологического уровня метаболитов в лизосомах как тканей мозга, так и во внутренних органах. 12 з.п. ф-лы, 11 ил., 5 табл.
Формула
Документы, цитированные в отчёте о поиске
Лечение в случае дефицита α-галактозидазы а
Комментарии