Способ получения синтетического пентапептида - RU2664545C2
Код документа: RU2664545C2
Описание
Область техники, к которой относится изобретение
[0001] Настоящее изобретение относится к способу получения синтетического пентапептида и к его промежуточному продукту.
Предпосылки создания изобретения
[0002] Известно, что агонисты опиоидного κ рецептора полезны в качестве терапевтических средств против различных типов боли. Из них, агонист опиоидного κ рецептора, обладающий высокой селективностью в отношении периферийного опиоидного κ рецептора, прогнозируется в качестве лекарственного средства, не вызывающего побочных эффектов на центральную нервную систему. Таким периферически-селективным агонистом опиоидного κ рецептора являются синтетические пентапептиды, о которых имеются сообщения в литературе (патентная литература 1 и 2).
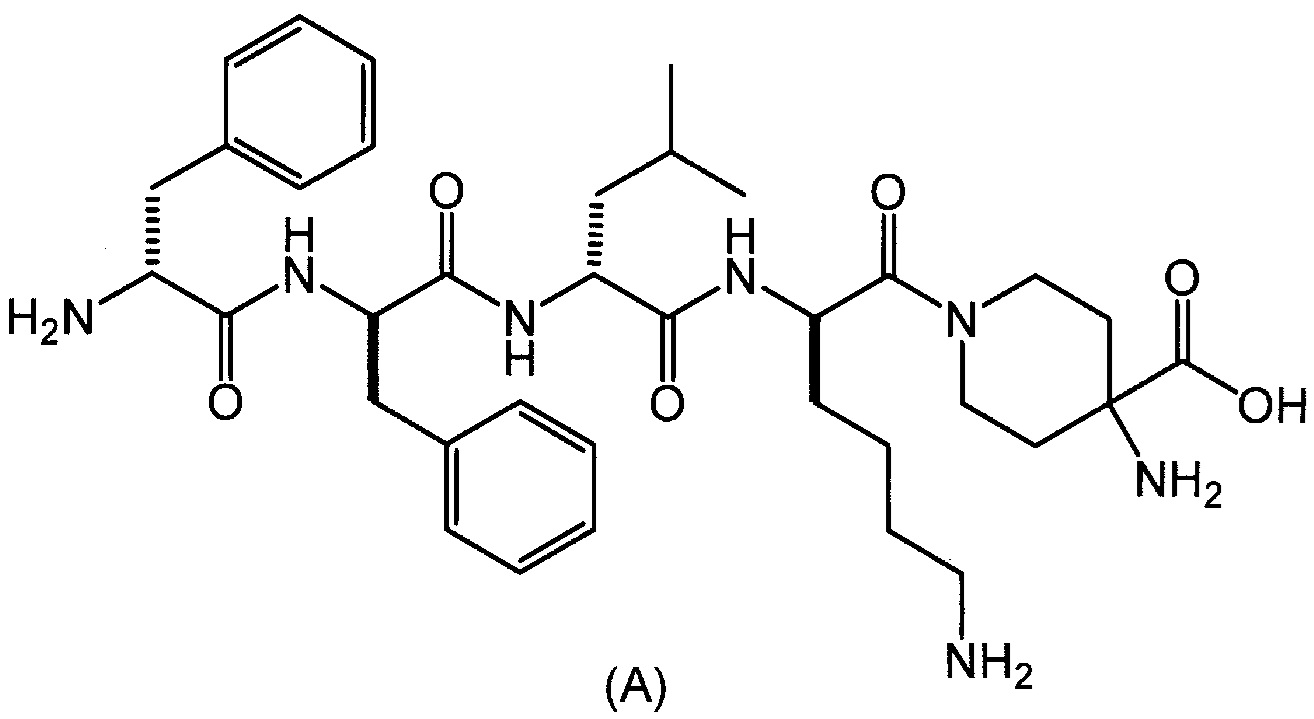
[0003] Соединение синтетических пентапептидов, представленное следующей формулой (A):
[0004]

[0005] полезно в качестве терапевтического средства против боли. В качестве способа получения данного соединения в патентной литературе 1 и 2, приведенной выше, описан твердофазный способ синтеза пептидов.
Перечень ссылок
[Патентная литература]
[0006] [Патентная литература 1] JP-A-2010-510966
[Патентная литература 2] JP-A-2013-241447
Сущность изобретения
Техническая задача
[0007] Несмотря на это, при твердофазном способе синтеза пептидов синтезируется пентапептид с защитными группами, и затем осуществляется удаление из смолы и удаление всех защитных групп, с последующей очисткой препаративной ВЭЖХ. Очистка требует крупномасштабного оборудования для препаративной ВЭЖХ и занимает много времени. Напротив, пептид может быть синтезирован жидкофазным методом. Однако, когда пентапептид, имеющий защитные группы на стадии промежуточного соединения, подвергался удалению этих групп и последующей очистке с получением соединения (A), чистота полученного в результате соединения (A) составляла менее чем 80%. Исходя из этого, было обнаружено, что невозможно получить очень чистое соединение (A).
[0008] Соответственно, технической проблемой, решаемой настоящим изобретением, является предоставление способа промышленно выгодного получения соединения (A) высокой чистоты.
Решение задач
[0009] Авторы настоящего изобретения провели исследования, относительно способа получения соединения (А) высокой чистоты. Когда они выделили следующее соединение (1) путем удаления только N-защитной группы из соединения (A), имеющего N- и O-защитные группы, к общему удивлению, было обнаружено, что очень чистое соединение (1) можно получить суспензионным методом очистки и методом перекристаллизации; и что, если соединение (1) подвергнуть гидролизу, можно промышленно выгодно получить соединение (A), имеющее чистоту выше, чем 90% или более. На основе приведенных выводов, было выполнено настоящее изобретение.
[0010] Настоящее изобретение далее предоставляет следующие объекты [1]-[4].
[1] Соединение, представленное следующей формулой (1), или его соль:
[0011]

[0012] где R1 представляет собой алкильную группу или аралкильную группу.
[2] Соединение или его соль по пункту [1], где R1 представляет собой алкильную группу.
[3] Соединение или его соль по пункту [1] или [2], где соединение или его соль представляет собой кислотно-аддитивную соль данного соединения.
[4] Способ получения соединения, представленного формулой (A), или его соли:
[0013]
[0014] включающий гидролиз соединения или его соли по любому из пунктов [1]-[3].
Эффект изобретения
[0015] Соединение (1) может быть очищено простой операцией, и если соединение (1) подвергнуть гидролизу, можно промышленно выгодно получить соединения (А) высокой чистоты.
Описание вариантов осуществления
[0016] Соединение (1) настоящего изобретения или его соль полезно в качестве промежуточного соединения для синтеза соединения (A).
[0017] В формуле (1) R1 представляет собой алкильную группу или аралкильную группу. Примеры алкильной группы включают линейную или разветвленную алкильную группу, имеющую 1-12 атомов углерода. Из них, линейная или разветвленная алкильная группа, имеющая 1-8 атомов углерода, является предпочтительной, и линейная или разветвленная алкильная группа, имеющая 1-4 атома углерода, является наиболее предпочтительной. Примеры алкильной группы включают метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу и трет-бутильную группу. Метильная группа является наиболее предпочтительной.
[0018] В качестве аралкильной группы, аралкильная группа, имеющая 7-18 атомов углерода, является предпочтительной; C6-14арил-C1-4алкильная группа является наиболее предпочтительной; фенил-C1-4алкильная группа является даже более предпочтительной; и бензильная группа является наиболее предпочтительной.
[0019] Из групп, представленных R1, алкильная группа является предпочтительной; C1-6алкильная группа является наиболее предпочтительной; C1-4алкильная группа является даже более предпочтительной; и метильная группа является наиболее предпочтительной.
[0020] В качестве соли соединения (1), представлена кислотно-аддитивная соль. Конкретные примеры кислотно-аддитивной соли включают соли неорганических кислот, такие как гидрохлорид, сульфат и нитрат; и соли органических кислот, такие как ацетат и трифторацетат. Гидрохлорид является предпочтительным в качестве соли с неорганической кислотой; и трифторацетат является предпочтительным в качестве соли с органической кислотой. Из них, гидрохлорид является наиболее предпочтительным.
[0021] Из соединений (1) и их солей, соль соединения (1), где R1 представляет собой C1-6алкильную группу, является предпочтительной, поскольку данную соль можно легко выделить в виде кристаллического вещества и легко очистить; кислотно-аддитивная соль соединения (1), где R1 представляет собой C1-6алкильную группу, является наиболее предпочтительной; кислотно-аддитивная соль соединения (1), где R1 представляет собой C1-4алкильную группу, является даже более предпочтительной; и кислотно-аддитивная соль соединения (1), где R1 представляет собой метильную группу, является наиболее предпочтительной.
[0022] Соединение (1) или его соль, и соединение (A) могут быть получены методом жидкофазного пептидного синтеза, в котором 4-аминопиперидин-4-карбоновую кислоту, D-лизин (D-Lys), D-лейцин (D-Leu), D-фенилаланин (D-Phe) и D-фенилаланин (D-Phe) подвергают последовательной конденсации, как показано, например, на следующей реакционной схеме.
[0023]

[0024]

[0025] где P1 и P2, каждый представляет собой N-защитную группу, и R1 является таким, как определено выше.
[0026] Как указано выше на реакционной схеме, предпочтительным является, чтобы N-защитные группы, представленные P1 и P2, являлись защитными группами, которые могут быть удалены раздельно различными средствами удаления. Примеры таких защитных групп включают (1) защитные группы, которые могут быть удалены при помощи кислоты (например, трет-бутоксикарбонильная группа (Boc), п-метоксибензилоксикарбонильная группа (Moz), формильная группа (CHO), 2-(триметилсилил)этоксикарбонильная группа (Teoc), 1-адамантилоксикарбонильная группа (Adoc), 2-(п-бифенил)изопропилоксикарбонильная группа (Bpoc), трифенилметильная группа (Tr), метоксиметильная группа (MOM)); (2) защитные группы, которые могут быть удалены путем восстановления (например, бензилоксикарбонильная группа (Cbz), аллильная группа (Allyl), N-бензилоксиметильная группа (BOM)); (3) защитные группы, которые могут быть удалены вторичным аминированием (например, 9-флуоренилметилоксикарбонильная группа (Fmoc), 2-(4-нитрофенил)этоксикарбонильная группа (Npeoc)); (4) защитные группы, которые могут быть удалены при помощи, например, цинкового порошка-уксусной кислоты (например, 2,2,2-трихлорэтоксикарбонильная группа (Troc), N-дитиасукциноильная группа (Dts), бензотиозол-2-сульфонильная группа (Betsyl), 1,1-диметил-2,2,2-трихлорэтоксикарбонильная группа (TcBoc), N-(дифенил-4-пиридил)метильная группа (Dppm)); и (5) защитные группы, которые могут быть удалены, например, при помощи амина в присутствии палладиевого катализатора (например, аллилоксикарбонильная группа (Alloc)). В качестве P1 и P2 защитных групп, которые могут быть удалены в различных условиях, можно использовать и выбрать из перечисленных выше защитных групп. Например, Boc является предпочтительной в качестве P1, и Cbz является предпочтительной в качестве P2.
[0027] Реакции конденсации между соединением (2) и защищенным D-Leu; между соединением (4) и защищенным D-Leu; между соединением (6) и защищенным D-Phe; и между соединением (8) и защищенным D-Phe могут быть осуществлены в присутствии конденсирующего агента, такого как молекулярные сита, 1-гидроксибензотриазол (HOBt), гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC⋅HCl) и N,N-дициклогексилкарбодиимид (DCC); и более конкретно, в растворителе, таком как галогеновый, сложноэфирный или простой эфирный растворитель, в присутствии конденсирующего агента, при температуре от 0 до 40°C в течение 1-48 часов.
[0028] Реакция удаления защиты может быть осуществлена путем выбора подходящего метода в зависимости от типа защитной группы. Для получения соединения (1) в форме кислотно-аддитивной соли, такой как гидрохлорид, предпочтительно защитную группу (например, Boc), которая может быть удалена при помощи кислоты, выбирают в виде P1, и защитную группу (например, (Cbz)), которая может быть удалена путем восстановления, выбирают в виде P2. При использовании Boc в виде P1, если P1 удаляют, например, путем гидролиза с использованием кислоты, может быть получена кислотно-аддитивная соль соединения (1). Удаление защиты путем восстановления может быть осуществлено, например, путем взаимодействия в растворителе, таком как сложноэфирный растворитель или простой эфирный растворитель, в присутствии металлического катализатора при 0-40°C в течение 1-48 часов. Удаление защиты при помощи кислоты может быть осуществлено в растворителе, таком как сложноэфирный растворитель или простой эфирный растворитель, в присутствии, например, неорганической кислоты или трифторуксусной кислоты, при 0-40°C в течение 1-48 часов.
[0029] Соль соединения (1) может быть легко очищена, например, перекристаллизацией. Соль соединения (1) имеет по существу высокую чистоту, даже если она не подвергалась очистке. Соответственно, соединения (А) высокой чистоты могут быть получены гидролизом соли соединения (1). Реакцию гидролиза, в данном случае, предпочтительно осуществляют в присутствии, например, основания; и более конкретно, может быть осуществлена в присутствии, например, гидроксида натрия, например, в воде или спирто-основном растворителе, при 0-40°C в течение 1-48 часов.
[0030] В способе получения соединения (A) согласно методу (жидкофазный метод) настоящего изобретения, трудно удаляемые примеси, такие как диастереомеры и дефектные пептиды, редко образуются, в отличие от твердофазного метода синтеза, но даже если примеси образуются, они могут быть удалены на стадии промежуточного продукта. Кроме того, поскольку промежуточное соединение может быть выделено, в отличие от твердофазного метода синтеза, можно использовать метод очистки фильтрованием, который является относительно простой операцией, не требующей затрат на дорогостоящее оборудование, таких как при суспензионном методе и методе перекристаллизации, с легкостью получая в результате желаемый продукт, имеющий высокую чистоту 99% (значение, измеренное, например, ВЭЖХ).
В отличие от твердофазного метода синтеза, при котором растворитель для использования ограничен, например, метиленхлоридом и диметилформамидом; в жидкофазном синтезе можно использовать подходящий для промышленного получения безопасный и недорогой растворитель. В отличие от твердофазного метода синтеза, легко синтезируется большое количество продукта. В твердофазном методе синтеза для очистки конечного соединения часто используется «препаративная ВЭЖХ» (как правило, требующая дорогостоящего оборудования и большого количества органических растворителей); однако эти проблемы можно избежать при жидкофазном синтезе.
Примеры
[0031] Настоящее изобретение будет более конкретно описано ниже при помощи примеров.
[0032] Пример 1
(1) Синтез Cbz-D-Lys(Boc)-α-Boc-Pic-OMe (3)
Четырех-горлую колбу (2L) загружали α-Boc-Pic-OMe⋅HCl [гидрохлорид метил α-Boc-4-аминопиперидин-4-карбоксилата] (2) (43,7 г (148 ммоль)), который суспендировали в EtOAc (656 мл (15 об./масс.)). К полученному раствору суспензии добавляли 1-гидроксибензотриазол (HOBt) (27,2 г (178 ммоль)) и Cbz-D-Lys(Boc)-OH (59,2 г (156 ммоль)). При охлаждении колбы на бане со льдом добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид⋅HCl (EDC⋅HCl) (34,1 г (178 ммоль)). Спустя двадцать минут, температуру полученной смеси поднимали до комнатной температуры, и смесь перемешивали в течение 12 часов. После завершения реакции добавляли 1Н HCl (218 мл (5,0 об./масс.)) к отделенному органическому слою. К полученному органическому слою, добавляли NaHCO3 водн. (218 мл (5,0 об./масс.)) и Et3N (33,0 г (326 ммоль)). Смесь перемешивали в течение 30 минут для разделения слоев. Органический слой последовательно промывали 1Н HCl (218 мл (5,0 об./масс.)), NaHCO3 водн. (218 мл (5,0 об./масс.)) и NaCl водн. (218 мл (5,0 об./масс.)), сушили над добавленным туда Na2SO4 (8,74 г (0,2 масс./масс.)) и фильтровали при пониженном давлении. Полученный в результате фильтрат концентрировали выпариванием при пониженном давлении и откачивали при помощи вакуумного насоса, с получением Cbz-D-Lys(Boc)-α-Boc-Pic-OMe (3) (88,9 г) в виде белого твердого вещества (выход 96,5%, чистота по данным ВЭЖХ 96,5%).
[0033] (2) Синтез D-Lys(Boc)-α-Boc-Pic-OMe (4)
2 л колбу со сборником дистиллята загружали Cbz-D-Lys(Boc)-α-Boc-Pic-OMe (3) (88,3 г (142 ммоль)), который растворяли добавлением EtOAc (441 мл (5,0 об./масс.)). К полученному реакционному раствору добавляли 5% Pd/C (17,7 г (0,2 масс./масс.)), и замену воздуха в колбе газообразным азотом осуществляли три раза при пониженном давлении атмосферы, с последующей трех-разовой заменой газообразным водородом. Реакционный раствор энергично перемешивали при комнатной температуре в течение 18 часов. После завершения реакции реакционный раствор фильтровали при пониженном давлении для удаления Pd/C. К полученному в результате фильтрату добавляли NaHCO3 водн. (441 мл (5,0 об./масс.)) для разделения слоев. К водному слою добавляли EtOAc (200 мл (2,3 об./масс.)), и органический слой экстрагировали. Экстрагированные таким же образом органические слои объединяли, и туда добавляли NaHCO3 водн. (441 мл (5,0 об./масс.)) и разделяли слои. К полученному водному слою добавляли EtOAc (200 мл (2,3 об./масс.)), и органический слой экстрагировали. Полученные таким же образом органические слои объединяли и туда добавляли NaCl водн. (441 мл (5,0 об./масс.)) для разделения слоев. К полученному водному слою добавляли EtOAc (200 мл (2,3 об./масс.)) и проводили экстракцию. Полученные таким же образом органические слои объединяли, сушили над добавленным туда Na2SO4 (17,7 г (0,2 масс./масс.)) и фильтровали при пониженном давлении. Полученный фильтрат концентрировали выпариванием при пониженном давлении и откачивали при помощи вакуумного насоса, с получением D-Lys(Boc)-α-Boc-Pic-OMe (4) (62,7 г) (выход 90,5%, чистота по данным ВЭЖХ 93,6%).
[0034] (3) Синтез Cbz-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (5)
Четырех-горлую колбу (2 л) загружали D-Lys(Boc)-α-Boc-Pic-OMe (4) (57,7 г (120 ммоль)), который суспендировали в EtOAc (576 мл (10 об./масс.)). К полученному раствору суспензии добавляли HOBt (19,3 г (126 ммоль)) и Cbz-D-Leu-OH (33,4 г (126 ммоль)). При охлаждении колбы на бане со льдом добавляли EDC⋅HCl (24,2 г (126 ммоль)). Спустя двадцать минут, температуру полученной смеси поднимали до комнатной температуры, и смесь перемешивали в течение 5 часов. К полученной смеси дополнительно добавляли EDC⋅HCl (1,15 г (6,00 ммоль)) и перемешивали в течение 16 часов. После завершения реакции добавляли 1Н HCl (576 мл (10 об./масс.)) для разделения слоев. К полученному органическому слою добавляли NaHCO3 водн. (576 мл (10 об./масс.)) и Et3N (24,3 г (240 ммоль)) и перемешивали в течение 30 минут для разделения слоев. Органический слой последовательно промывали 1Н HCl (576 мл (10 об./масс.)), NaHCO3 водн. (576 мл (10 об./масс.)) и NaCl водн. (576 мл (10 об./масс.)), сушили над добавленным туда Na2SO4 (11,5 г (0,2 масс./масс.)) и фильтровали при пониженном давлении. Полученный в результате фильтрат концентрировали выпариванием при пониженном давлении и откачивали при помощи вакуумного насоса, с получением Cbz-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (5) (85,8 г) в виде белого твердого вещества (выход 98,7%, чистота по данным ВЭЖХ 96,9%).
[0035] (4) Синтез D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (6)
1 л колбу со сборником дистиллята загружали Cbz-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (5) (91,9 г (125 ммоль)), который растворяли добавлением EtOAc (459 мл (5,0 об./масс.)). К полученному реакционному раствору добавляли 5% Pd/C (18,4 г (0,2 масс./масс.)), и замену воздуха в колбе газообразным азотом осуществляли три раза при пониженном давлении атмосферы, с последующей трех-разовой заменой газообразным водородом. Реакционный раствор энергично перемешивали при комнатной температуре в течение 8 часов. После завершения реакции реакционный раствор фильтровали при пониженном давлении для удаления Pd/C. К полученному в результате фильтрату добавляли NaHCO3 водн. (200 мл (2,2 об./масс.)) для разделения слоев. К органическому слою последовательно добавляли NaHCO3 водн. (200 мл (2,2 об./масс.)) и NaCl водн. (200 мл (2,2 об./масс.)) для промывки органического слоя. Полученный органический слой сушили над добавленным туда Na2SO4 (18,4 г (0,2 масс./масс.)) и фильтровали при пониженном давлении. Полученный в результате фильтрат концентрировали выпариванием при пониженном давлении и откачивали при помощи вакуумного насоса. Полученное аморфное твердое вещество растворяли добавлением EtOAc (200 мл (2,2 об./масс.)) и кристаллизовали из добавленного гептана (50 мл (1,8 об./масс.)). Выпавшее в осадок кристаллическое вещество отделяли фильтрованием при пониженном давлении и промывали смешанным растворителем из EtOAc (120 мл (1,3 об./масс.)) и гептана (50 мл (0,3 об./масс.)). Полученное кристаллическое вещество (46,1 г) растворяли добавлением EtOAc (480 мл (5,2 об./масс.)) и кристаллизовали из добавленного циклогексана 660 мл (7,2 об./масс.). Выпавшее в осадок кристаллическое вещество отделяли фильтрованием при пониженном давлении, промывали смешанным растворителем из циклогексана (120 мл (1,3 об./масс.)) и EtOAc (20 мл (0,2 об./масс.)) и сушили при 30°C при пониженном давлении, с получением D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (6) (36,6 г) в виде белого твердого вещества (выход 48,7%, чистота по данным ВЭЖХ 99,9%).
[0036] (5) Синтез Cbz-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (7)
Четырех-горлую колбу (1 л) загружали D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (6) (35,8 г (59,6 ммоль)), который суспендировали в EtOAc (358 мл (10 об./масс.)). К полученному раствору суспензии добавляли HOBt (9,59 г (62,6 ммоль)) и Cbz-D-Phe-OH (18,7 г (62,6 ммоль)). При охлаждении колбы на бане со льдом добавляли EDC⋅HCl (12,0 г (62,6 ммоль)). Спустя двадцать минут, температуру полученной смеси поднимали до комнатной температуры, и смесь перемешивали в течение 16 часов, и дополнительно добавляли EDC⋅HCl (3,09 г (16,1 ммоль)). После завершения реакции добавляли 1Н HCl (358 мл (10 об./масс.)), и органический слой отделяли. К полученному органическому слою добавляли NaHCO3 водн. (358 мл (10 об./масс.)) и Et3N (12,1 г (119 ммоль)), и смесь перемешивали в течение 30 минут для разделения слоев. Органический слой последовательно промывали 1Н HCl (358 мл (10 об./масс.)), NaHCO3 водн. (358 мл (10 об./масс.)) и NaCl водн. (358 мл (10 об./масс.)), сушили над добавленным туда Na2SO4 (7,16 г (0,2 масс./масс.)) и фильтровали при пониженном давлении. Полученный в результате фильтрат концентрировали выпариванием при пониженном давлении и откачивали при помощи вакуумного насоса, с получением Cbz-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (7) (52,5 г) в виде белого твердого вещества (выход количественный, чистота по данным ВЭЖХ 97,6%).
[0037] (6) Синтез D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (8)
2 л колбу со сборником дистиллята загружали Cbz-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (7) (46,9 г (53,3 ммоль)), который растворяли добавлением EtOAc (840 мл (18 об./масс.)) и H2O (93,8 мл (2,0 об./масс.)). К полученному реакционному раствору добавляли 5% Pd/C (9,38 г (0,2 масс./масс.)), замену воздуха в колбе газообразным азотом осуществляли три раза при пониженном давлении атмосферы, с последующей трех-разовой заменой газообразным водородом. Реакционный раствор энергично перемешивали при комнатной температуре в течение 10 часов. После завершения реакции реакционный раствор фильтровали при пониженном давлении для удаления Pd/C. К полученному в результате фильтрату добавляли NaHCO3 водн. (235 мл (5,0 об./масс.)) для разделения слоев. К органическому слою последовательно добавляли NaHCO3 водн. (235 мл (5,0 об./масс.)) и NaCl водн. (235 мл (5,0 об./масс.)) для промывки органического слоя. Полученный органический слой сушили над добавленным туда Na2SO4 (9,38 г (0,2 масс./масс.)) и фильтровали при пониженном давлении. Полученный в результате фильтрат концентрировали выпариванием при пониженном давлении и откачивали при помощи вакуумного насоса, с получением D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (8) (39,7 г) (выход количественный, чистота по данным ВЭЖХ 97,3%).
[0038] (7) Синтез Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (9)
Четырех-горлую колбу (1 л) загружали D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (8) (35,1 г), который суспендировали при добавлении EtOAc (351 мл (10 об./масс.)). К полученному раствору суспензии добавляли HOBt (7,92 г (51,7 ммоль)) и Boc-D-Phe-OH (13,1 г (49,4 ммоль)). При охлаждении колбы на бане со льдом добавляли EDC⋅HCl (9,91 г (51,7 ммоль)). Спустя двадцать минут, температуру полученной смеси поднимали до комнатной температуры, и смесь перемешивали в течение 8 часов, и дополнительно добавляли EDC⋅HCl (2,25 г (11,7 ммоль)). После завершения реакции добавляли 1Н HCl (351 мл (10 об./масс.)) к отделенному органическому слою. К полученному органическому слою добавляли NaHCO3 водн. (351 мл (10 об./масс.)) и Et3N (9,51g (94,0 ммоль)). Смесь перемешивали в течение 30 минут для разделения слоев. Органический слой последовательно промывали 1Н HCl (351 мл (10 об./масс.)), NaHCO3 водн. (351 мл (10 об./масс.)) и NaCl водн. (351 мл (10 об./масс.)), сушили над добавленным туда Na2SO4 (7,02 г (0,2 масс./масс.)) и фильтровали при пониженном давлении. Полученный в результате фильтрат концентрировали выпариванием при пониженном давлении и откачивали при помощи вакуумного насоса, с получением Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (9) (46,7 г) в виде белого твердого вещества (выход количественный, чистота по данным ВЭЖХ 98,6%).
[0039] (8) Синтез гидрохлорида D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1)
20 мл колбу со сборником дистиллята загружали Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (9) (2,00 г), который суспендировали при добавлении IPA (3,3 мл (1,65 об./масс.)) и PhMe (10 мл (5 об./масс.)). К полученной смеси добавляли 6Н HCl/IPA (6,7 мл (3,35 об./масс.)) и перемешивали при комнатной температуре в течение 19 часов. Выпавшее в осадок твердое вещество отделяли фильтрованием при пониженном давлении и сушили при пониженном давлении, с получением гидрохлорида D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (1,59 г) в виде белого твердого вещества (выход 99,0%, чистота по данным ВЭЖХ 98,2%).
[0040] (9) Очистка гидрохлорида D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1)
20 мл колбу со сборником дистиллята загружали неочищенное кристаллическое вещество, гидрохлорид D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (200 мг), к которому добавляли смешанный растворитель (4,0 мл (20 об./масс.)), содержащий EtOH:MeCN в соотношении 1:5. Полученную смесь нагревали до 40°C и перемешивали в течение одного часа, и дополнительно перемешивали при комнатной температуре в течение двух часов, с получением суспензии. Полученную смесь отделяли фильтрованием при пониженном давлении, и полученное в результате твердое вещество сушили при пониженном давлении, с получением белого твердого вещества ((1) очищенное кристаллическое вещество) (161 мг) (выход 80%, чистота по данным ВЭЖХ 99,2%).
[0041] (10) Синтез D-Phe-D-Phe-D-Leu-D-Lys-Pic (A) (использовали очищенное кристаллическое вещество (1))
10 мл колбу со сборником дистиллята загружали очищенное кристаллическое вещество, гидрохлорид D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (38,5 мг (0,0488 ммоль)), который растворяли добавлением H2O (0,2 мл (5,2 об./масс.)). К полученной смеси по каплям добавляли 1Н NaOH (197 мкл (0,197 ммоль)) при комнатной температуре и перемешивали в течение 1,5 часов. После завершения реакции добавляли 1Н HCl (48,8 мкл (0,0488 ммоль)) и полученную в результате смесь концентрировали выпариванием при пониженном давлении, с получением D-Phe-D-Phe-D-Leu-D-Lys-Pic (A) (выход количественный, чистота по данным ВЭЖХ 99,7%).
[0042] Физические свойства D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1)
1H ЯМР (400 МГц, 1M DCl) δ м.д.: 0,85-1,02 (м, 6H), 1,34-1,63 (м, 5H), 1,65-2,12 (м, 5H), 2,23-2,45 (м, 2H), 2,96-3,12 (м, 4H), 3,19 (ддт, J=5,0 и 5,0 и 10,0 Гц), 3,33-3,62 (м, 1H), 3,68-3,82 (м, 1H), 3,82-3,95 (м, 4H), 3,95-4,18 (м, 1H), 4,25-4,37 (м, 2H), 4,61-4,77 (м, 2H), 7,21-7,44 (м, 10H);
13C ЯМР (400 МГц, 1M DCl) δ м.д.: 21,8, 22,5, 24,8, 27,0, 30,5, 30,8, 31,0, 31,2, 31,7, 37,2, 37,8, 38,4, 39,0, 39,8, 40,4, 40,6, 41,8, 42,3, 49,8, 50,2, 52,2, 52,6, 54,6, 55,2, 57,7, 57,9, 127,6, 128,4, 129,2, 129,6, 129,7, 129,8.
Т. разл. 209,5°C
[0043] Пример 2
(Использование трифторуксусной кислоты (ТФУК))
(1) Синтез соли ТФУК D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1)
В 50 мл колбу со сборником дистиллята последовательно добавляли ТФУК (18 мл (18 об./масс.)), 1-додекантиол (1,6 мл (1,6 об./масс.)), триизопропилсилан (0,2 мл (0,2 об./масс.)) и H2O (0,2 мл (0,2 об./масс.)) и перемешивали. К полученному раствору при помощи шпателя понемногу добавляли Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (9) (1,00 г (1,01 ммоль)). После завершения реакции полученную смесь концентрировали выпариванием при пониженном давлении, и полученный в результате остаток по каплям добавляли в IPE (20 мл (20 об./масс.)). Выпавшее в осадок твердое вещество отделяли фильтрованием, и полученное в результате твердое вещество сушили при пониженном давлении, с получением соли ТФУК D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (выход 93,0%, чистота по данным ВЭЖХ 95,2%) в виде белого твердого вещества.
[0044] (2) Синтез D-Phe-D-Phe-D-Leu-D-Lys-Pic (A)
10 мл колбу со сборником дистиллята загружали соль ТФУК D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (83 мг (0,0843 ммоль)), которую растворяли добавлением H2O (431 мкл (5,2 об./масс.)). К полученному раствору по каплям добавляли 1Н NaOH (345 мкл (0,345 ммоль)) при комнатной температуре и перемешивали в течение 12 часов. После завершения реакции добавляли 1Н HCl (84,3 мкл (0,0843 ммоль)) и концентрировали выпариванием при пониженном давлении, с получением D-Phe-D-Phe-D-Leu-D-Lys-Pic (A) (выход количественный, чистота по данным ВЭЖХ 95,4%).
[0045] Пример 3
(Использование HCl/EtOAc)
(1) 30 мл колбу со сборником дистиллята загружали Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (9) (1,00 г (1,01 ммоль)), который растворяли добавлением EtOAc (7,0 мл (7,0 об./масс.)). К полученному раствору добавляли 4Н HCl/EtOAc (5,0 мл (5,0 об./масс.)) и перемешивали при комнатной температуре в течение 24 часов, и выпавшее в осадок твердое вещество отделяли фильтрованием при пониженном давлении и промывали EtOAc (2 мл (2,0 об./масс.)). Полученное в результате твердое вещество сушили при пониженном давлении, с получением гидрохлорида D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (781 мг) в виде белого твердого вещества (выход 96,7%, чистота по данным ВЭЖХ 95,4%).
[0046] (2) Синтез D-Phe-D-Phe-D-Leu-D-Lys-Pic (A)
10 мл колбу со сборником дистиллята загружали гидрохлорид D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (90 мг (0,112 ммоль)), который растворяли добавлением H2O (0,47 мл (5,2 об./масс.)). К полученному раствору по каплям добавляли 1Н NaOH (459 мкл (0,459 ммоль)) при комнатной температуре и перемешивали в течение 12 часов. После завершения реакции добавляли 1Н HCl (0,112 мкл (0,112 ммоль)), и полученную смесь концентрировали выпариванием при пониженном давлении, с получением D-Phe-D-Phe-D-Leu-D-Lys-Pic (A) (выход количественный, чистота по данным ВЭЖХ 93,1%).
[0047] Пример 4
Синтез соединения (A) гидролизом соединения (1) (без очистки соединения (1))
10 мл колбу со сборником дистиллята загружали гидрохлорид D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1) (114,5 мг (0,142 ммоль)) (без очистки на предыдущей стадии), который растворяли добавлением H2O (595 мкл (5,2 об./масс.)). К полученному раствору по каплям добавляли 1Н NaOH (586 мкл (0,586 ммоль)) при комнатной температуре и перемешивали в течение 14 часов. После завершения реакции добавляли 1Н HCl (0,15 мкл (0,150 ммоль)) и концентрировали выпариванием при пониженном давлении, с получением D-Phe-D-Phe-D-Leu-D-Lys-Pic (A) (выход количественный, чистота по данным ВЭЖХ 95,2%).
[0048] Сравнительный пример 1
Последовательность реакций не через посредство соединения (1) (использование Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (9), полностью защищенный)
(1) Синтез Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OH
30 мл колбу со сборником дистиллята загружали Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OMe (9) (1,00 г (1,00 ммоль)), который растворяли добавлением MeOH (5,0 мл (5,0 об./масс.)). К полученному раствору добавляли 1Н NaOH (1,1 мл (1,10 ммоль)) и перемешивали при комнатной температуре в течение 4 дней, и дополнительно добавляли MeOH (5,0 мл (5,0 об./масс.)) и 1Н NaOH (2,0 мл (2,0 ммоль)) и перемешивали при 35°C в течение 3 часов. После завершения реакции добавляли 1Н HCl (6,1 мл), и полученную смесь концентрировали при пониженном давлении для отгонки растворителя. После этого добавляли EtOAc (5,0 мл (5,0 об./масс.)), и органический слой отделяли. Органический слой промывали добавлением NaCl водного (5,0 мл (5,0 об./масс.)) и концентрировали при пониженном давлении, с получением Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OH (975,1 мг) (выход 99,3%, чистота по данным ВЭЖХ 80,8%) в виде белого твердого вещества.
[0049] (2) Синтез D-Phe-D-Phe-D-Leu-D-Lys-Pic (A)
20 мл колбу со сборником дистиллята загружали Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-α-Boc-Pic-OH (959 мг (0,978 ммоль)), который растворяли добавлением EtOAc (4,9 мл (5,0 об./масс.)). К полученному раствору по каплям добавляли 4Н HCl/EtOAc (4,9 мл (5,0 мл)) при комнатной температуре и перемешивали при комнатной температуре в течение 4 часов. После завершения реакции проводили фильтрование при пониженном давлении, с получением D-Phe-D-Phe-D-Leu-D-Lys-Pic (A) (выход 96,4%, чистота по данным ВЭЖХ 79,2%) в виде белого твердого вещества.
[0050] В случае последовательности реакций не через посредство соединения (1) настоящего изобретения, чистота полученного соединения (A) составляла менее чем 80%.
[0051] Пример 5
(Синтез гидрохлорида D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1))
Реакцию осуществляли по методике, аналогично примеру 1 (8), заменяя реакционный растворитель IPA (1,65 об./масс.), как показано в таблице 1 (растворитель и количество). Результаты показаны в таблице 1,
[0052]
[0053] Пример 6
(Очистка гидрохлорида D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe (1))
Реакцию осуществляли по методике, аналогично примеру 1 (9), заменяя очищающий растворитель EtOH/MeCN (16,7:83,5, 20 об./масс.), как показано в таблице 2 (растворитель и количество). Результаты показаны в таблице 2.
[0054]
Реферат
Изобретение относится к способу получения синтетического пентапептида, являющегося агонистом опиоидногорецептора, полезного в качестве терапевтического средства против боли, представленного формулой (А):или его соли, а также к промежуточному соединению для его получения, представленному формулой (1):(1) или его соли, где Rпредставляет собой алкильную группу или аралкильную группу. Технический результат – осуществление промышленно выгодного получения пентапептида формулы (А) высокой чистоты. 2 н. и 2 з.п. ф-лы, 2 табл., 5 пр.
Формула

Документы, цитированные в отчёте о поиске
Пептиды-агонисты k-опиоидных рецепторов
Комментарии