Синтетические пептидные амиды - RU2500685C2
Код документа: RU2500685C2
Чертежи






Описание
СВЯЗАННЫЕ ЗАЯВКИ
[0001] Данная заявка претендует на приоритет временной заявки США, серийный номер 60/858,109, поданной 10 ноября 2006 г., и временной заявки США, серийный номер 60/928,550, поданной 10 мая, 2007 г., каждая из которых явно включена в данное описание путем ссылки во всей ее полноте.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[0002] Изобретение относится к синтетическим пептидным амидам, в пептидную цепь которых инкорпорированы D-аминокислоты и, более конкретно, к таким синтетическим пептидным амидам, которые являются агонистами каппа-опиатного рецептора и к способам их применения в качестве профилактических и терапевтических агентов.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0003] Каппа-опиатные рецепторы предложены в качестве мишеней для вмешательства с целью лечения или предотвращения широкого спектра заболеваний и состояний введением агонистов каппа-опиатного рецептора. См., например, Jolivalt et al., Diabetologia, 49(11):2775-85; Epub Aug. 19, 2006), где описана эффективность азимадолина, агониста рецептора каппа при диабетической невропатии у грызунов; и Bileviciute-Ljungar et al., Eur. J. Pharm. 494:139-46 (2004), где описана эффективность агониста каппа U-50,488 на модели невропатической боли в результате хронического ограничения у крыс (CCI) и блокирование его воздействия опиатным антагонистом налоксоном. Эти наблюдения поддерживают применение агонистов каппа-опиатного рецептора для лечения диабетической, вирусной и индуцированной химиотерапией невропатической боли. Также рассмотрено применение агонистов рецепторов каппа для лечения или предотвращения висцеральной боли, включая гинекологические нарушения, например, спазмы в связи с дисменореей и эндометриоз. См., например, Riviere, Br. J. Pharmacol. 141:1331-4 (2004).
[0004] Агонисты каппа-опиатного рецептора также предложены для лечения боли, в том числе гипералгезии. Считается, что гипералгезию вызывают изменения в среде периферического сенсорного окончания, вторичные к местному повреждению ткани. Повреждение ткани (например, ссадины, ожоги) и воспаление может значительно повышать возбудимость полимодальных ноцицепторов (волокна С) и механических рецепторов с высоким порогом (Handworker et al. (1991) Proceeding of the VIth World Congress on Pain, Bond et al., eds., Elsevier Science Publishers BV, pp.59-70; Schaible et al. (1993) Pain 55:5-54). Считается, что такое повышение возбудимости и чрезмерно усиленные реакции чувствительных афферентных окончаний лежит в основе гипералгезии, где реакция боли является результатом чрезмерно усиленной реакции на стимул. Значение гипералгезического состояния в состоянии боли после повреждения повторно продемонстрировано и, по-видимому, ответственно за основную часть состояния боли после повреждения/при воспалении. Woold et al. (1993) Anesthesia and Analgesia 77:362-79; Dubner et al. (1994) в Textbook of Pain, Melzack et al., eds., Churchill-Livingstone, London, pp.225-242.
[0005] Каппа-опиатные рецепторы предложены в качестве мишеней для предотвращения и лечения сердечнососудистых заболеваний. См., например, Wu et al. "Cardioprotection of Preconditioning by Metabolic Inhibition in the Rat Ventricular Myocyte - Involvement of kappa Opioid Receptor" (1999) Circulation Res vol.84: pp.1388-1395. См. также Yu et al. «Anti-Arrythmic Effect of kappa Opioid Receptor Stimulation in the Perfused Rat Heart: Involvement of cAMP-Dependent Pathway" (1999) J Mol Cell Cardiol. vol. 31(10): pp.1809-1819.
[0006] Также было обнаружено, что развитие или прогрессирование таких заболеваний и состояний, в которые вовлечены нейродегенерация или гибель клеток нейронов, может быть предупреждено или как минимум замедлено лечением агонистами каппа-опиатного рецептора. Считается, что улучшенный результат - это следствие нейропротекции агонистами каппа-опиатного рецептора. См., например, Kaushik et al. "Neuroprotection in Glaucoma" (2003) J. Postgraduate Medicine vol. 49 (I): pp.90-95.
[0007] Присутствие каппа-опиатных рецепторов на иммунных клетках (Bidlak et al., (2000) CIjin. Diag. Lab. Immunol. 7 (5):719-723) вовлечено в ингибирующее воздействии агониста каппа-опиатного рецептора, который доказанно подавляет экспрессию ВИЧ-1. См. Peterson РК et al., Biochem Pharmacol. 2001, 61(19):1145-51.
[0008] Walker, Adv. Exp.Med. Biol. 521:148-60 (2003) оценивал противовоспалительные свойства агонистов каппа для лечения остеоартрита, ревматоидного артрита, воспалительного заболевания кишечника и экземы. Bileviciute-Ljungar et al., Rheumatology 45:295-302 (2006) описывают уменьшение боли и дегенерации при индуцированном адъювантом Фрейнда с помощью агониста каппа U-50,488.
[0009] Wikstrom et al., J. Am. Soc. Nephrol. 16:3742-7 (2005) описывают применение агониста каппа, TRK-820 для лечения уремического и индуцированного опиоидами зуда и Ко et al., J. Pharmacol. Exp.Ther. 305:173-9 (2003) описывают эффективность U-50,488 при индуцированном морфином зуде у обезьян.
[0010] Также внимательно рассматривалось применение опиоидов периферического действия, включая применение агонистов каппа, для лечения желудочно-кишечных заболеваний. См., например, Lembo, Diges. Dis. 24:91-8 (2006) для обсуждения применения опиоидов в лечении расстройств пищеварительного тракта, в том числе синдром раздраженного кишечника (IBS), непроходимость кишечника и функциональная диспепсия.
[0011] Также показана возможность применения каппа-опиоидов при расстройствах глаз, в том числе воспалении глаз и глаукоме. См. Potter efc al., J. Pharmacol. Exp.Ther. 309:548-53 (2004), где описана роль высоко активного агониста каппа-опиатного рецептора, бремазоцина в уменьшении внутриглазного давления и блокирование данного эффекта норбиналторфимином (norBNI), составляющим прототип антагонистом каппа-опиатного рецептора; и Dortch-Carnes et al., CNS Drug Rev. 11(2) - .195-212 (2005). В патенте США 6,191,126, выданном Gamache, раскрыто применение каппа-опиатных агонистов для лечения глазной боли. Также показано, что боль в ушах поддается лечению введением каппа-опиатных агонистов. См. патент США 6,174,878, также выданный Gamache.
[0012] Каппа-опиатные агонисты увеличивают экскрецию воды почками и уменьшают выделение натрия с мочой (т.е., вызывают селективный диурез воды, также известный как акварез). Многие, но не все исследователи относят данный эффект на счет подавлению секреции вазопрессина гипофизом. Исследования, в которых сравнивали опиоиды центрального действия и предположительно периферически селективные каппа-опиоиды, привели к выводу о том, что каппа-опиатные рецепторы в пределах гематоэнцефалического барьера ответственны за опосредование такого эффекта. Другие исследователи предложили лечить гипонатриемию ноцицептиновыми пептидами или заряженными конъюгатами пептидов, которые воздействуют периферически на рецепторы ноцицептина, что представляет собой родственный, но отличный от каппа-опиатных рецепторов механизм. (D.R.Kapusta, Life Sci., 60:15-21, 1997; патент США 5,840,696, патентная заявка США 20060052284.)
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0013] В настоящем изобретении предлагаются синтетические пептидные амиды формулы I ниже и стереоизомеры, смеси стереоизомеров, пролекарства, фармацевтически приемлемые соли, гидраты, сольваты, гидраты солей кислот, N-оксиды и изоморфные кристаллические формы синтетических пептидных амидов формулы I:

[0014] В формуле I Xaa1 представляет N-концевую аминокислоту, которая может представлять собой любой из (A)(A')D-Phe, (A) (A') (α-Me)D-Phe, D-Tyr, D-1,2,3,4-тетрагидроизохинолин-3-карбоновой кислоты, D-грет-лейцина, D-неопентилглицина, D-фенилглицина, D-гомофенилаланина или β(E)D-Ala, где каждый (А) и каждый (А') представляет собой заместитель в фенильном кольце, выбранный из -Н, -F, -Cl, -NO2, -СН3, -CF3, -CN и -CONH2, и где каждый (Е) независимо выбран из следующих заместителей: циклобутил, циклопентил, циклогексил, пиридил, тиенил и тиазолил.
[0015] Хаа2 представляет собой вторую аминокислоту, которая может быть любой из (А)(А')D-Phe, 3,4-дихлор-0-Phe, (A)(A')(α-Ме)D-Phe, D-1-нафтилаланина, D-2-нафтилаланина, D-Tyr, (E)D-Ala или D-Trp; где каждый из (А), (А') и (Е) независимо выбран из заместителей, перечисленных выше для каждого из (А), (А') и (Е).
[0016] Хаа3 представляет собой третью аминокислоту, которая может быть любой из D-норлейцина, D-Phe, (E)D-Ala, D-Leu, (α-Ме)D-Leu, D-гомолейцина, D-Val или D-Met, где (Е) независимо выбран из заместителей, перечисленных выше (Е).
[0017] Хаа4 представляет собой четвертую аминокислоту, которая может быть любой из (B)2D-arginine, (В)2D-нораргинина, (В)2D-гомоarginine, ζ-(B)D-гомолизина, D-2,3-диаминопропионовой кислота, ε-(B)D-Lys, ε-(В)2-D-Lys, D-аминометилфенилаланина, амидино-D-аминометил-фенилаланина, γ-(В)2D-γ-Диаминомасляной кислоты, δ-(В)2α-(В')D-Orn, D-2-амино-3(4-пиперидил)пропионовой кислоты, D-2-амино-3(2-аминопирролидил)пропионовой кислоты, D-α-амино-β-амидинопропионовой кислоты, α-амино-4-пиперидинуксусной кислоты, цис-α,4-диаминоциклогексануксусной кислоты, транс-α,4-диаминоциклогексануксусной кислоты, цис-α-амино-4-метиламиноциклогексануксусной кислоты, транс-α-амино-4-метиламиноциклогексануксусной кислоты, α-амино-1-амидино-4-пиперидинуксусной кислоты, цис-α-амино-4-гуанидиноциклогексануксусной кислоты или транс-α-амино-4-гуанидино-циклогексануксусной кислоты, где каждый (В) независимо представляет собой -Н или C1-C4 алкил, и (В') представляет собой - Н или (α-Me).
[0018] Линкерный фрагмент W может представлять собой любую из следующих трех альтернатив: (i) нуль, при условии, что если W равен 0, Y представляет собой N и присоединен к С-концу Хаа4, чтобы образовать амид; (ii) -NH-(CH2)b-, где b равно 0, 1, 2, 3, 4, 5 или 6; или (iii) -NH-(CH2)c-O-, где с равно 2 или 3, при условии, что Y представляет собой С. В каждой из вышеизложенных альтернатив, (ii) и (iii) атом азота W присоединен к С-концу Хаа4 с образованием амида.
[0019] Фрагмент

в формуле I представляет собой необязательно замещенный 4-8-членный гетероциклический кольцевой фрагмент, где все гетероатомы кольца в кольцевом фрагменте представляют собой N, и где каждый из Y и Z независимо представляет собой С или N. Однако, если данный гетероциклический кольцевой фрагмент представляет собой 6-, 7- или 8-членное кольцо, Y и Z должны быть разделены как минимум двумя атомами кольца. В дальнейшем, если данный гетероциклический кольцевой фрагмент содержит один гетероатом в кольце, который представляет собой N, то кольцевой фрагмент является неароматическим.
[0020] Фрагмент V в формуле I представляет собой -C1-С6 алкил. Оператор е равен 0 или 1, таким образом, что если е равно 0, то V равен 0, и R1 и R2 непосредственно присоединены к одному и тому же или различным атомам кольца.
[0021] В первом из четырех альтернативных вариантов, фрагмент R1 в формуле I может быть любой из следующих групп: -Н, -ОН, галоген, -CF3, -NH2, -СООН, C1-C6 алкил, амидино, C1-С6 алкилзамещенный амидино, арил, необязательно замещенный гетероциклил, Pro-амид, Pro, Gly, Ala, Val, Leu, He, Lys, Arg, Orn, Ser, Thr, CN, CONH2, COR', SO2R', CONR'R'', NHCOR', OR' или SO2NR'R''; где необязательно замещенный гетероциклил необязательно содержит 1 или 2 заместителя, независимо выбранные из C1-С6 алкила, C1-С6 алкокси, оксо, -ОН, -Cl, -F, -NH2, -NO2, -CN, -СООН и амидино. Каждый из фрагментов R' и R'' независимо представляет собой Н, C1-C8 алкил, арил или гетероциклил. Альтернативно, R' и R'' могут вместе образовывать 4-8-членное кольцо, которое необязательно содержит 1 или 2 заместителя, независимо выбранные из C1-C6 алкила, C1-C6 алкокси, -ОН, -Cl, -F, -NH2, -NO2, -CN, -СООН и амидино. Фрагмент R2 может быть любым из -Н, амидино, амидино, замещенного одным или двумя C1-С6 алкилом, -CN, -CONH2, -CONR'R'', -NHCOR', -SO2NR'R'' или -СООН.
[0022] Во втором альтернативном варианте, фрагменты R1 и R2 вместе могут образовывать необязательно замещенный 4-9-членный гетероциклический моноциклический или бициклический кольцевой фрагмент, который присоединен к одному атому кольца Y- и Z-содержащего кольцевого фрагмента.
[0023] В третьем альтернативном варианте, фрагменты R1 и R2 вместе с одним атомом кольца Y- и Z-содержащего кольцевого фрагмента могут образовывать необязательно замещенный 4-8-членный гетероциклический кольцевой фрагмент с образованием спироструктуры.
[0024] В четвертом альтернативном варианте, фрагменты R1 и R2 вместе с двумя или больше соседными атомами кольца Y- и Z-содержащего кольцевого фрагмента могут образовывать необязательно замещенный 4-9-членный гетероциклический моноциклический или бициклический кольцевой фрагмент, конденсированный с Y- и Z-содержащим кольцевым фрагментом.
[0025] В формуле I, каждый из необязательно замещенных 4-, 5-, 6-, 7-, 8- и 9-членных гетероциклических кольцевых фрагментов, содержащих R1 и R2, может содержать 1 или 2 заместителя, независимо выбранные из C1-C6 алкила, C1-C6 алкокси, необязательно замещенного фенила, оксо, -ОН, -Cl, -F, -NH2, -NO2, -CN, -СООН и амидино. В дальнейшем, если Y- и Z-содержащий кольцевой фрагмент формулы I представляет собой 6- или 7-членное кольцо, которое содержит один гетероатом в кольце, и е равно 0, то R1 не может быть -ОН, и R1 и R2 оба не являются -Н.
[0026] Если Y- и Z-содержащий кольцевой фрагмент в формуле I представляет собой 6-членное кольцо, в котором присутствует два гетероатома в кольце, где как Y, так и Z представляет собой N, и W равен 0, то фрагмент -(V)eR1R2 присоединен к атому кольца, отличному от Z. Кроме того, при вышеуказанных условиях, если е равно 0, то R1 и R2 оба не могут быть - Н. Наконец, если Хааз представляет собой D-Nle, то Хаа4 не может быть (B)2D-Arg; и если Хаа3 представляет собой D-Leu или (αMe)D-Leu, то Хаа4 не может быть δ-(В)2α-(В')D-Orn.
[0027] Изобретение также предлагает селективный агонист каппа-опиатного рецептора (в данном описании также называется агонистом или просто агонистом каппа), который является синтетическим пептидным амидом по изобретению, как изложено выше.
[0028] Изобретение также предлагает фармацевтическую композицию, которая содержит синтетические пептидные амиды по изобретению и фармацевтически приемлемый разбавитель, вспомогательное вещество или носитель.
[0029] Также предлагается способ лечения или предупреждения связанного с каппа-опиатными рецепторами заболевания или состояния у млекопитающего. Способ включает введение млекопитающему композиции, которая содержит эффективное количество синтетического пептидного амида по изобретению. Изобретение также предлагает применение синтетических пептидных амидов по изобретению для приготовления лекарственных средств и фармацевтических композиций, пригодных для лечения связанного с каппа-опиатными рецепторами заболевания или состояния у млекопитающего.
[0030] Изобретение предлагает также способ лечения или предупреждения связанного с каппа-опиатными рецепторами заболевания или состояния у млекопитающего, где синтетические пептидные амиды по изобретению вводят совместно с уменьшенной дозой анальгетика мю-опиатного агониста, для достижения терапевтического анальгетического эффекта, причем болеутоляющее соединение мю-опиатный агонист вызывает сопутствующий побочный эффект (особенно, угнетения дыхания, седацию, эйфорию, антидиуретический эффект, тошноту, рвоту, запор и физическое привыкание, зависимость и пристрастие). Уменьшенная доза анальгетика мю-опиатного агониста, которую вводят в соответствии с данным способом, вызывает менее выраженные сопутствующие побочные эффекты, чем побочные эффекты, связанные с дозой анальгетического соединения опиатного агониста мю, необходимой, чтобы достичь такого же терапевтического болеутоляющего эффекта при монотерапии.
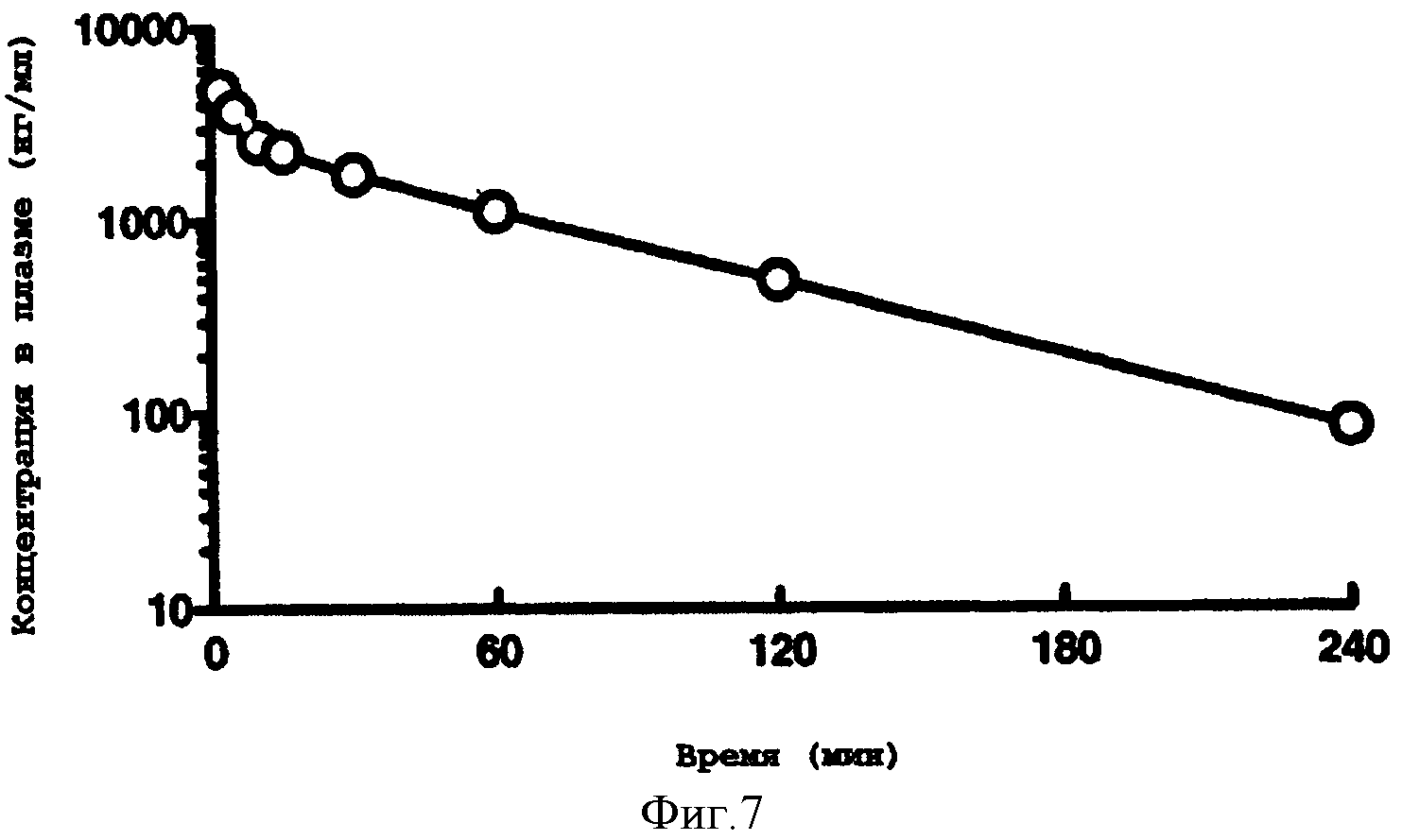
[0031] Изобретение также предлагает способ лечения или предупреждения периферической гипералгезии, где способ включает местное нанесение или введение млекопитающему, нуждающемуся в лечении, эффективного количества композиции, которая содержит эффективное против гипералгезии количество синтетических пептидных амидов по изобретению в носителе, предназначенном для местного нанесения или введения.
[0032] Изобретение также предлагает способ лечения или предупреждения гипонатриемии или гипокалиемии и, таким образом, лечения или предупреждения заболевания или состояния, связанного с гипонатриемией или гипокалиемией, такого как застойная сердечная недостаточность, цирроз печени, нефротический синдром, гипертензия или отек и, предпочтительно, где повышенная секреция вазопрессина связана с указанным заболеванием или расстройством, причем способ включает введение млекопитающему акваретически эффективного количества синтетических пептидных амидов по изобретению в фармацевтически приемлемом разбавителе, вспомогательном веществе или носителе.
КРАТКОЕ ОПИСАНИЕ ФИГУР
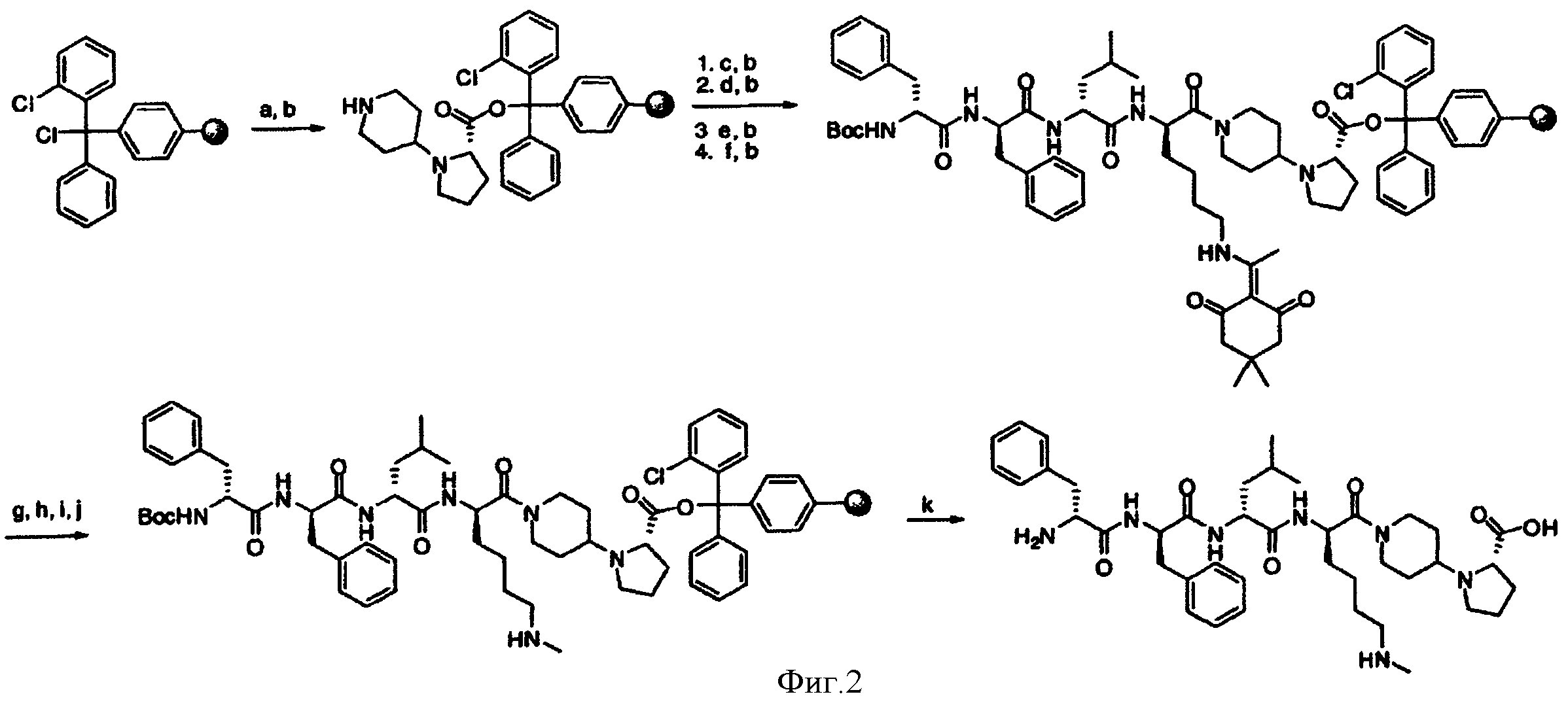
[0033] Фигура 1 показывает общую схему, используемую в синтезе соединений (1), (6), (7), (10) и (11). Стадии a-s осуществляют с применением следующих реагентов или условий: а) гомопиперазин, ДХМ; b) Fmoc-D-Dap(ivDde)-OH или Fmoc-D-Dab(ivDde)-ОН или Fmoc-D-Orn(Aloc)-OH или Fmoc-D-Orn(Cbz)-ОН или Fmoc-D-Lys(Dde)-OH или Fmoc-D-Arg(Pbf)-ОН, DIC, HOBt, ДМФА; с) 25% пиперидина в ДМФА; d) Fmoc-D-Leu-OH или Fmoc-D-Nle-ОН, DIC, HOBt, ДМФА; е) Fmoc-D-Phe-OH, DIC, HOBt, ДМФА; f) Cbz-D-Phe-OH, DIC, HOBt, ДМФА; g) 4% гидразина в ДМФА; h) Pd(PPh3)4, CHCl3/AcOH/NMM; i) O-NBS-Cl, коллидин, NMP; 3) диметилсульфат, DBU, NMP; k) меркаптоэтанол, DBU, NMP; 1) Cbz-OSu, ДМФА; m) ацетон, AcOH, NaBH(OAc)3, TMOF; n) 1Н-пиразол-1-карбоксамидин, ДИЭА, ДМФА; о) 50% ТФУ/ДХМ; р) S-метил-N-метилизотио-мочевины гидройодид, ДИЭА, ДМФА; q) 2-метилтио-2-имидазолина гидройодид, ДИЭА, ДМФА; r) йодэтан, ДИЭА, ДМФА; с) TMCOTf/ТФУ/м-крезол.
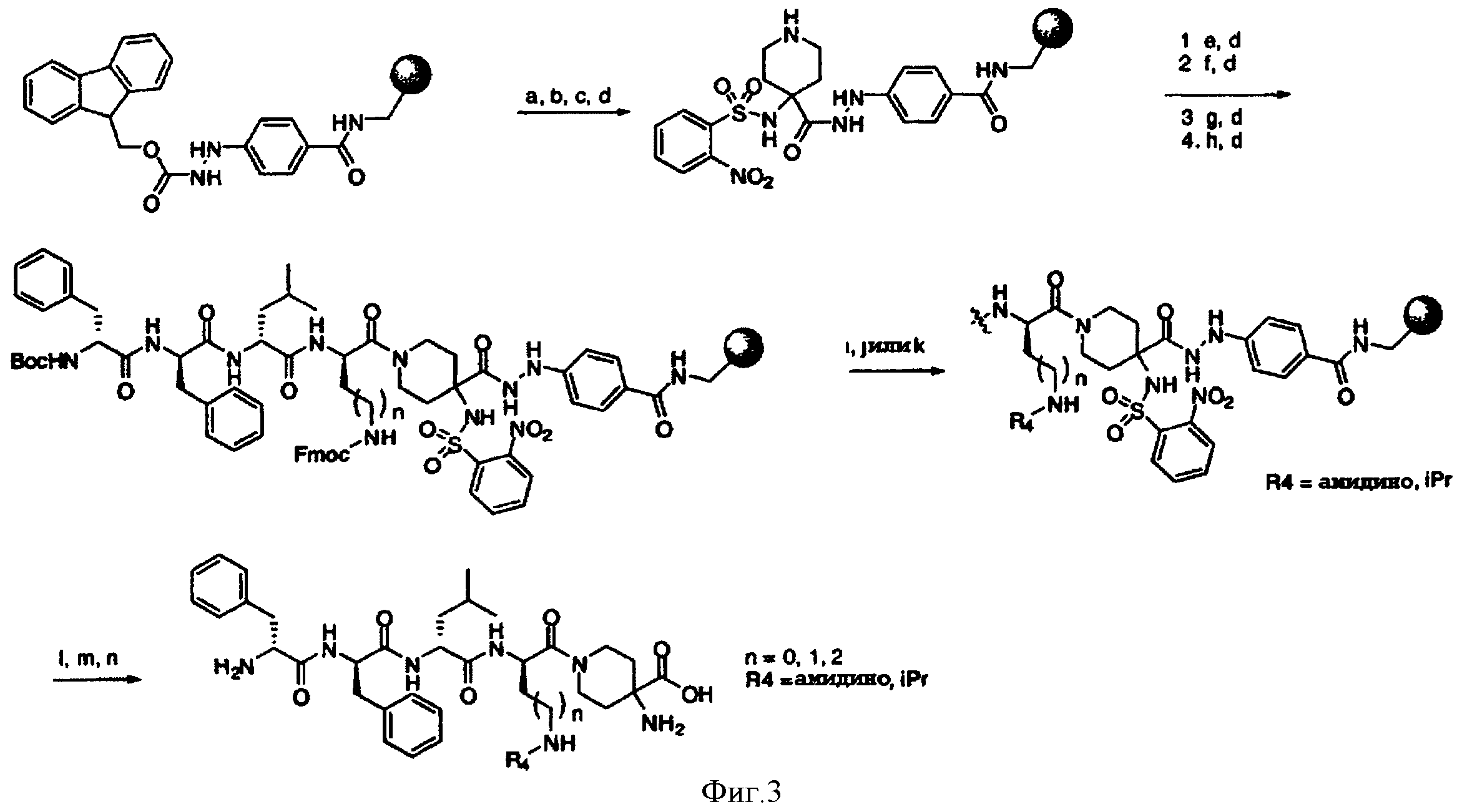
[0034] Фигура 2 - общая схема, используемая в синтезе соединений (2)-(5), (8), (9) и (12)-(14). Стадии a-k осуществляют с применением следующих реагентов или условий: а) N-(1-Fmoc-пиперидин-4-ил)-L-пролин, ДИЭА, ДХМ; b) 25% пиперидина/ДМФА; с) Fmoc-D-Lys(Dde)-ОН, DIC, HOBt, ДМФА; d) Fmoc-D-Leu-OH, DIC, HOBt, ДМФА; е) Fmoc-D-Phe-OH, DIC, HOBt, ДМФА; f) Boc-D-Phe-OH, DIC, HOBt, ДМФА; g) 4% гидразина в ДМФА; h) o-NBS-Cl, коллидин, NMP; i) диметилсульфат, DBU, NMP;]) меркаптоэтанол, DBU, NMP; k) ТФУ/TIS/Н2О.
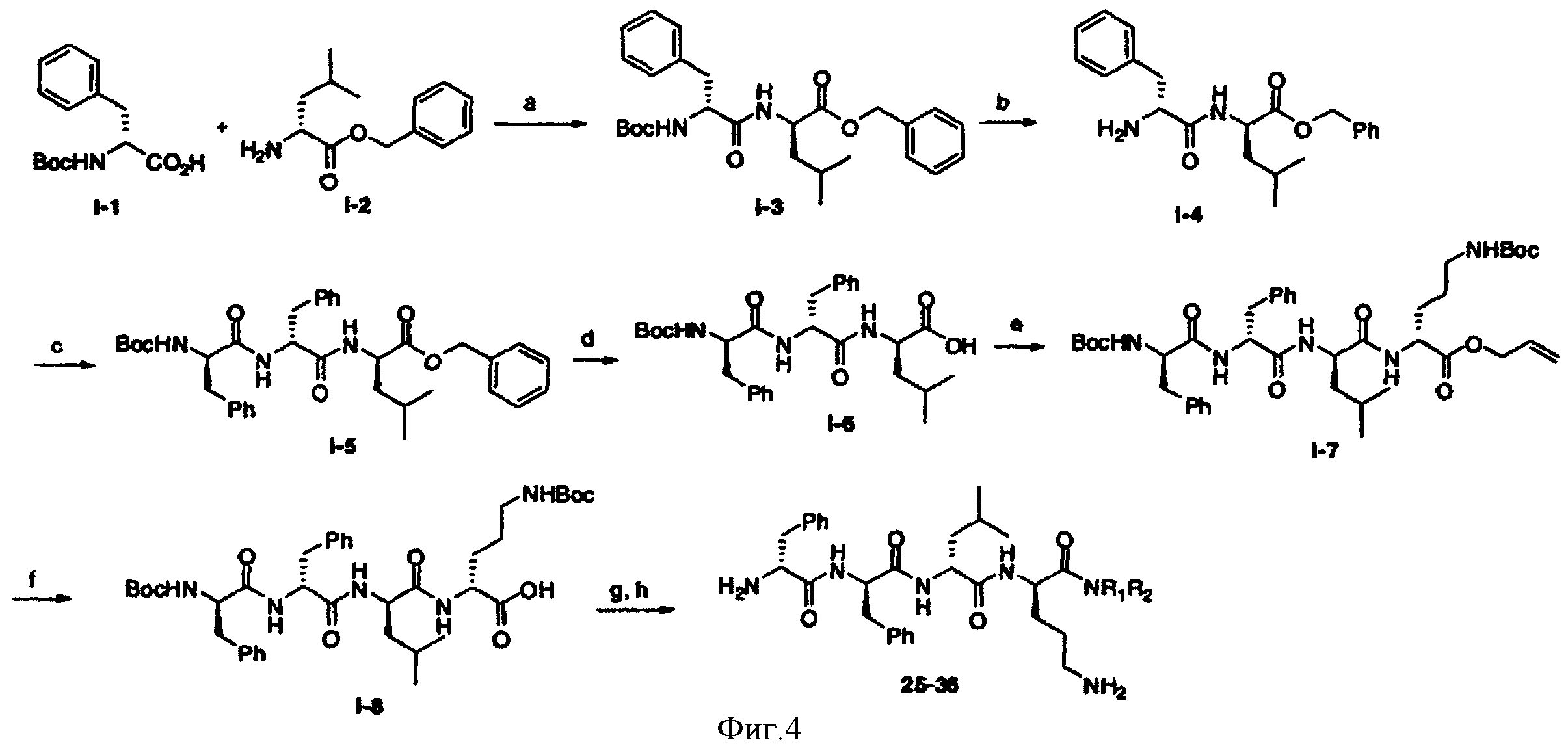
[0035] Фигура 3 - общая схема, используемая в синтезе соединений (15)-(24). Стадии а-n осуществляют с применением следующих реагентов или условий: а) 35% пиперидина, ДМФА; b) 1-Вос-4-N-Fmoc-амино-пиперидин-4-карбоновая кислота, РуВОР, ДИЭА, ДМФА; с) (i) 35% пиперидина, ДМФА; (ii) O-NBS-Cl, коллидин, NMP; d) 30% ТФУ в ДХМ; е) Boc-D-Dap(Fmoc)-OH или Boc-D-Dab(Fmoc)-ОН или Boc-D-Orn(Fmoc)-OH, РуВОР, ДИЭА, ДМФА; f) Boc-D-Leu-OH, РуВОР, ДИЭА, ДМФА; g) Boc-D-Phe-OH, РуВОР, ДИЭА, ДМФА; h) Boc-D-PHE-OH, РуВОР, ДИЭА, ДМФА; i) 2% DBU/ДМФА;]) 1Н-пиразол-1-карбоксамидин, ДИЭА, ДМФА; k) (i) ацетон, TMOF, (ii) NaBH(ОАс)3, ДМФА; 1) меркаптоэтанол, DBU, NMP; m) Cu(OAc)2, пиридин, DBU, ДМФА/H2O; n) 95% ТФУ/H2O.
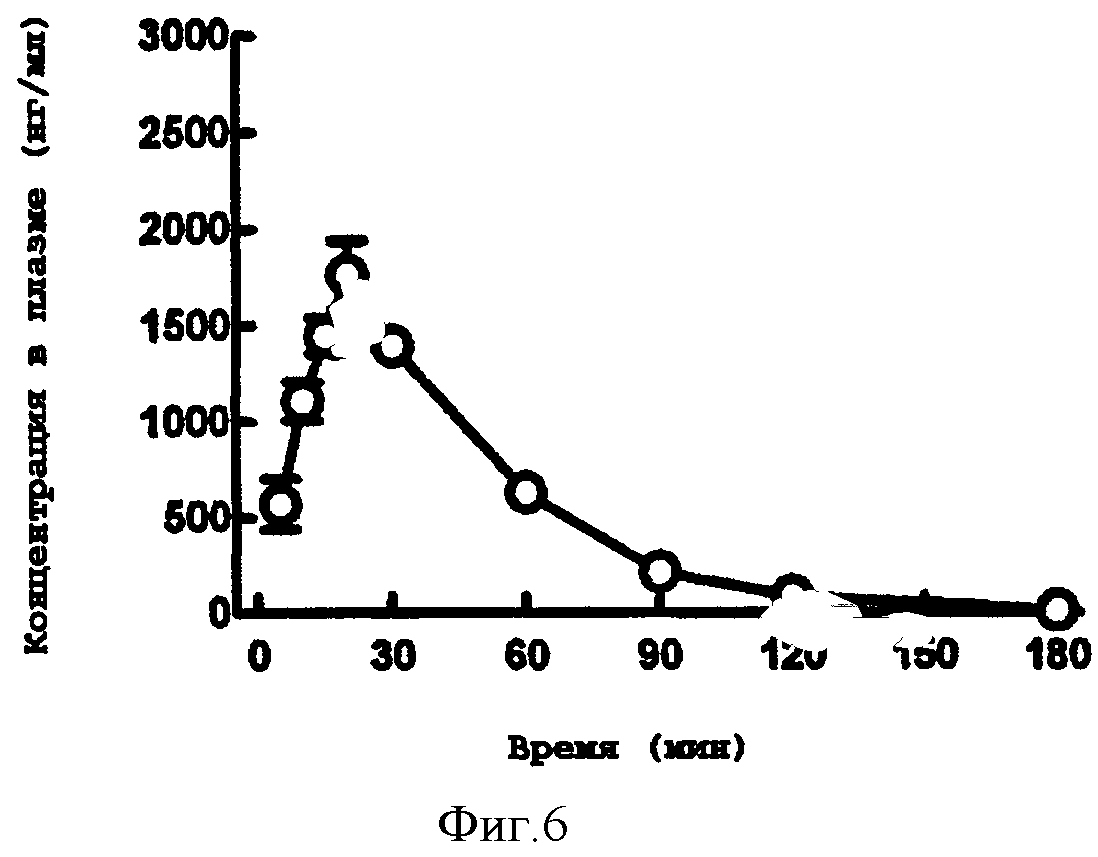
[0036] Фигура 4 - общая схема используемая в синтезе соединений (25)-(37). Стадии а-h осуществляют с применением следующих реагентов или условий: a) EDCI, HOBt, ДИЭА, ТГФ; b) ТФУ, ДХМ; с) Boc-D-Phe-OH, EDCI, HOBt, ДИЭА; d) H2, Pd/C; e) D-Lys(Вое)-ОА11, TBTU, ДИЭА, ДМФА; f) Pd(PPh3)4, пирролидин; g) HNRaRb, HBTU; h) HCl, диоксан.
[0037] Фигура 5 - концентрация, обнаруженная в плазме и мозге крыс после введения 3 мг/кг соединения (2) в виде инфузии длительностью 5 минут через катетер шейной вены. Концентрация соединения (2) в нг/мл: открытые круги: плазма, закрашенные круги: мозг.
[0038] Фигура 6 - концентрация в плазме соединения (6) после подкожного введения одного болюса 1 мг/кг соединения мышам ICR. ОБразцы плазмы отбирали в точках 5, 10, 15, 20, 30 60, 90 120 и 180 минут после инъекции.
[0039] Фигура 7 - концентрация в плазме соединения (3) после внутривенного введения одного болюса 0,56 мг/кг соединения яванским макакам. Образцы плазмы отбирали в точках 2, 5, 10, 15, 30, 60, 120 и 240 минут после инъекции.
[0040] Фигура 8 - кривые «доза-реакция» для соединения (3) у мышей ICR на модели индуцированных уксусной кислотой корчей (закрашенные круги) и в испытании перемещения (закрашенные квадраты).
[0041] Фигура 9 - зависимость «доза-реакция» для опосредованного соединением (2) угнетения индуцированных уксусной кислотой корчей у мышей при внутривенном введении.
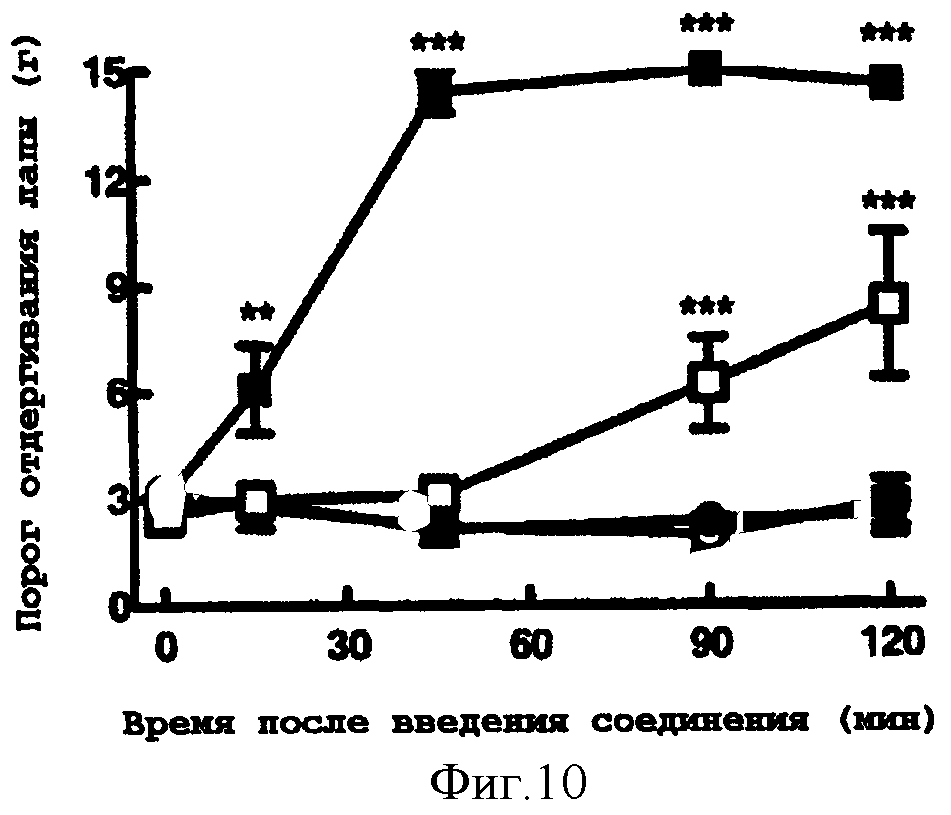
[0042] Фигура 10 - влияние соединения (2) на механическую гиперчувствительность, индуцированную перевязкой спинного нервна L5/L6 у крыс. Открытые круги - только растворитель; закрашенные круги - соединение (2) в дозе 0,1 мг/кг; открытые квадраты - соединение (2) в 0,3 мг/кг; закрашенные квадраты - соединение (2) в дозе 1,0 мг/кг. ** обозначает p<0,01; *** обозначает p<0,001 против растворителя (2-сторонний ANOVA, Бонферрони).
[0043] Фигура 11 - влияние соединения (2) в различных концентрациях на индуцированную панкреатитом чрезмерную чувствительность брюшины у крыс.Дибутилина дихлорид или только растворитель вводили внутривенно, и гиперчувствительность оценивали зондированием брюшины с помощью нити вон Фрея с интервалом 30 минут. Гиперчувствительность выражали как количество отдергиваний от десяти попыток зондирования. Открытые круги - только растворитель; закрашенные круги - соединение (2) в дозе 0,1 мг/кг; открытые квадраты - соединение (2) в дозе 0,3 мг/кг; закрашенные квадраты - соединение (2) в дозе 1,0 мг/кг. ** обозначает р<0,01; *** обозначает p<0,001 против растворителя (2-сторонний ANOVA, Бонферрони).
[0044] Фигура 12 - блокирование влияния соединения (2) на индуцированную панкреатитом гиперчувствительность брюшины nor-BNI и налоксона метйодидом у крыс.Открытый столбик - только растворитель, закрашенный столбик - соединение (2) в дозе 1 мг/кг с налоксона метйодидом или norBNI, как показано. *** обозначает p<0,001 против растворителя + растворитель (2-сторонний ANOVA, Бонферрони).
ПОДРОБНОЕ ОПИСАНИЕ
[0045] В данном описании термин "синтетические пептидные амиды" обозначает соединение по изобретению, соответствующее формуле I или его стереоизомер, смесь стереоизомеров, пролекарство, фармацевтически приемлемую соль, гидрат, сольват, гидрат соли кислоты, N-оксид или изоморфную кристаллическую форму. Если Xaa1, Хаа2, Хаа3 и Хаа4 представляют D-аминоки слоты в соединениях по изобретению, стереоизомеры соединений по изобретению, соответствующие формуле I, ограничиваются соединениями, содержащими аминокислоты в D-конфигурации, где это указано в Формуле I. Стереоизомеры соединений по изобретению включают соединения, с D- или L-конфигурацией хиральных центров, кроме альфа-атомов углерода четырех аминокислот в положениях Xaa1, Хаа2, Хаа3 и Хаа4. Термин «смеси стереоизомеров» обозначает смеси таких стереоизомеров по изобретению. В данном описании термин «рацематы» обозначает смеси стереоизомеров, содержащие равные пропорции соединений с D- и L-конфигурацией одного или больше хиральных центров, кроме альфа-атомов углерода Xaa1, Xaa2, Хаа3 и Хаа4, без изменения хиральности альфа-атомов углерода Xaa1, Хаа2, Хаа3 и Хаа4.
[0046] Номенклатура, используемая для определения пептидов в данном описании, описана Schroder & Lubke, The Peptides, Academic Press, 1965, где, в соответствии с обычным представлением, N-конец размещается слева, и С-конец справа. Если остаток аминокислоты присутствует в изомерных формах, L-изомерная форма, так и D-изомерная форма аминокислоты включены, если не указано иное. Аминокислоты в данном описании обычно идентифицированы стандартным трехбуквенным кодом. D-изомер аминокислоты указан приставкой "D-", например, в "D-Phe", что обозначает D-фенилаланин, D-изомер фенилаланина. Также, L-изомер указан приставкой "L-", например, в "L-Phe". Пептиды в данном описании представлены согласно традиционной договоренности, как последовательности аминокислот слева направо: от N-конца к С-концу, если не указано иное.
[0047] В данном описании, D-Arg представляет D-аргинин, D-Har представляет D-гомоаргинин, который содержит боковую цепь на одну метиленовую группу длиннее, чем D-Arg, и D-Nar представляет D-нораргинин, который содержит боковую цепь на одну метиленовую группу короче, чем D-Arg. Подобным образом, D-Leu обозначает D-лейцин, D-NIe обозначает D-норлейцин, и D-Н1е представляет D-гомолейцин. D-Ala обозначает D-аланин, D-Tyr обозначает D-тирозин, D-Trp обозначает D-триптофан, и d-tic обозначает D-1,2,3,4-тетрагидроизохинолин-3-карбоновую кислоту. D-Val кислоту D-валин, и D-Met кислоту D-метионин. D-Pro обозначает D-пролин, Pro-амид обозначает D- или L-форму амида пролина. D-Pro-амид представляет D-пролин с амидной группой, образованной на карбоксильном фрагменте, где амидный азот может представлять собой замещенный алкил, как в случае -NRaRb, где каждый из Ra и Rb независимо представляет собой C1-С6 алкильную группу, или один из Ra и Rb представляет собой -Н. Gly обозначает глицин, D-Ile обозначает D-изолейцин, D-Ser обозначает D-серин, и D-Thr обозначает D-треонин. (E)D-Ala обозначает D-изомер аланина, содержащий заместитель (Е) при β-углероде. Примеры таких групп заместителя (Е) включают циклобутил, циклопентил, циклогексил, пиридил, тиенил и тиазолил. Таким образом, циклопентил-D-Ala обозначает D-изомер аланина, замещенный циклопентилом при β-атоме углерода. Подобным образом, D-Ala(2-тиенил) и (2-тиенил)D-Ala взаимозаменяемы, и оба обозначают D-изомер аланина, замещенный при β-атоме углерода тиенилом, который присоединен в положении 2 кольца.
[0048] В данном описании D-Nal обозначает D-изомер аланина, замещенный нафтилом при β-атоме углерода. D-2Nal обозначает замещенный нафтилом D-аланин, где нафтален присоединен в положении 2 кольцевой структуры, и D-lNal обозначает замещенный нафтилом D-аланин, где нафтален присоединен в положении 1 кольцевой структуры.
(A)(A')D-Phe обозначает D-фенилаланин, замещенный на фенильном кольце 1 или 2 заместителями, независимо выбранными из галогена, нитро, метила, галогенметила (например, такого как трифторметил), пергалогенметила, циано и карбоксамида. D-(4-F)Phe обозначает D-фенилаланин, замещенный фтором в положении 4 фенильного кольца. D-(2-F)Phe обозначает D-фенилаланин, замещенный фтором в положении 2 фенильного кольца. D-(4-Cl)Phe обозначает D-фенилаланин, замещенный хлором в положении 4 фенильного кольца. (α-Me)D-Phe обозначает D-фенилаланин, замещенный метилом при альфа-атоме углерода. (αMe)D-Leu обозначает D-лейцин, замещенный метилом при альфа-атоме углерода.
[0049] Обозначения (B)2D-Arg, (B)2D-Nar и (B)2D-Har представляют D-аргинин и D-нораргинин, D-гомоаргинин, соответственно, каждый из которых содержит две группы заместителя (В) в боковой цепи. D-Lys обозначает D-лизин, и D-Hlys обозначает D-гомолизин. ζ-(В)D-Hlys, ε-(B)D-Lys и ε-(B)2-D-Lys представляют D-гомолизин и D-лизин, каждый из которых содержит в боковой цепи аминогруппу, замещенную 1 или 2 группами заместителя (В), как показано. D-Orn обозначает D-орнитин, и δ-(В)α-(В')D-Orn обозначает D-орнитин, замещенный (В') при альфа-атоме углерода, и замещенный (В) в боковой цепи δ-аминогруппы.
[0050] D-Dap обозначает D-2,3-диаминопропионовую кислоту. D-Dbu представляет D-изомер альфа,гамма-диаминомасляной кислоты, и (B)2D-Dbu представляет альфа,гамма-диаминомасляную кислоту, замещенную двумя группами заместителя (В) в гамма-аминогруппе. Если не указано иное, каждая из (В) групп таких дважды замещенных остатков независимо выбрана из Н- и C1-C4-алкила. В данном описании, D-Amf обозначает D-(NH2CH2)Phe, т.е. D-изомер фенилаланина, замещенный аминометилом в фенильном кольце, и D-4Amf представляет конкретный D-Amf, в котором аминометил присоединен в положении 4 кольца. D-Gmf обозначает D-Amf(амидино), который представляет D-Phe, где фенильное кольцо замещено -CH2NHC(NH)NH2. Amd представляет амидино, C(NH)NH2, и обозначения (Amd) D-Amf и D-Amf(Amd) также равнозначно применяются для D-Gmf. Обозначения Ilу и lor, соответственно, применяются для обозначения Lys изопропила и Оrn изопропила, где аминогруппа боковой цепи алкилирована изопропильной группой.
[0051] Алкил обозначает радикал алкана, который может представлять собой разветвленную, неразветвленную и циклическую алкильную группу, такую как, не ограничиваясь ими, метил, этил, пропил, изопропил, циклопропил, бутил, грег-бутил, вгор-бутил, пентил, циклопентил, гексил, циклогексил, циклогексилэтил. C1-C8 алкил обозначает алкильные группы, содержащие от 1 до 8 атомов углерода. Подобным образом, C1-С6 алкил обозначает алкильные группы, содержащие от 1 до 6 атомов углерода. Также, C1-C4 алкил обозначает алкильные группы, содержащие от 1 до 4 атомов углерода. Низший алкил обозначает C1-C6 алкил. Me, Et, Pr, Ipr, Bu и Pn равнозначно используются для представления распространенных алкильных групп: метил, этил, пропил, изопропил, бутил и пентил, соответственно. Хотя соединение для алкильной группы находится обычно на одном конце алкильной цепи, соединение может быть расположено где-либо в другом месте цепи, например 3-пентил, который может также быть обозначен как этилпропил или 1-этилпроп-1-ил. Алкилзамещенный, например, C1-C6 алкилзамещенный амидино, показывает, что соответствующий фрагмент замещен 1 или больше алкильных групп.
[0052] Если указанный фрагмент равен 0, такой фрагмент отсутствует, и если указано, что такой фрагмент присоединен к двум другим фрагментам, такие два других фрагмента соединены одной ковалентной связью. Если соединительный фрагмент показан в данном описании как присоединенный к кольцу в каком-либо положении кольца и присоединен к двум другим фрагментам, таким как R1 и R2, в случае, когда указано, что соединительный фрагмент равен 0, каждый из фрагментов R1 и R2 может быть независимо присоединен в каком-либо положении на кольце.
[0053] Термины "гетероцикл", "гетероциклическое кольцо" и "гетероциклил" применяются равнозначно в данном описании и обозначают кольцо или кольцевой фрагмент, содержащий как минимум один не углеродный атом в кольце, который также называют гетероатомом, и который может представлять собой атом азота, серы или кислорода. Если указано, что в кольце присутствует определенное количество членов, количество определяет число атомов в кольце без обозначения каких-либо заместителей или атомов водорода, присоединенных к атомам кольца. Гетероциклы, гетероциклические кольца и гетероциклильные фрагменты могут содержать несколько гетероатомов, независимо выбранных из атома азота, серы или кислорода в кольце. Кольца могут содержать заместители в каком-либо доступном положении. Например, не ограничиваясь ими, 6- и 7-членные кольца часто содержат заместители в положении 4 кольца, и 5-членные кольца обычно содержат заместители в положении 3, где кольцо присоединено к цепи пептидного амида в положении 1 кольца.
[0054] Термин «насыщенный» обозначает отсутствие двойных или тройных связей, и применение термина в связи с кольцами обозначает кольца, не содержащие двойных или тройных связей в пределах кольца, но не обозначает отсутствия двойных или тройных связей в заместителях, присоединенных к кольцу. Термин "неароматический" употребляется в контексте конкретного кольца для обозначения отсутствия ароматичности в таком кольце, но не обозначает отсутствия двойных связей в пределах кольца, в том числе двойных связей, которые являются частью ароматического кольца, конденсированного с указанным кольцом. Также не запрещается присутствие двойной связи между атомом кольца насыщенного гетероциклического кольцевого фрагмента и атомом за пределами кольца, например, атом серы в кольце связан двойной связью с атомом кислорода заместителя. В данном описании, гетероциклы, гетероциклические кольца и гетероциклильные фрагменты также включают насыщенные, частично ненасыщенные и гетероароматические кольца, а также конденсированные бициклические структуры колец, если не определено иное. Гетероцикл, гетероциклическое кольцо или гетероциклильный фрагмент может быть конденсирован со вторым кольцом, которое может быть насыщенным, частично ненасыщенным или ароматическим кольцом, которое может представлять собой гетероцикл или углеродный цикл. Если указано, два заместителя могут необязательно вместе образовывать дополнительное кольцо. Кольца могут быть замещены в каком-либо доступном положении. Гетероцикл, гетероциклическое кольцо и гетероциклильный фрагмент может, если это указано, быть необязательно замещенным в одном или больше положений кольца одним или больше независимо выбранных заместителей, таких как, например, C1-С6 алкил, С3-С8 циклоалкил, C1-C6 алкокси, галоген C1-C6 алкил, необязательно замещенный фенил, арил, гетероциклил, оксо, -ОН, -Cl, -F, -NH2, -NO2, -CN, -СООН и амидино. Подходящие необязательные заместители фенила включают, например, не ограничиваясь ими, одну или больше групп, выбранных из C1-С3 алкила, C1-С3 алкокси, галогена C1-С3 алкила, оксо, -ОН, -Cl, -F, -NH2, -NO2, -CN, -СООН и амидино.
[0055] D-Phe и замещенный D-Phe представляют собой примеры подходящей аминокислоты для остатка Xaa1 в Формуле I. Фенильное кольцо может быть замещено в любом из положений 2, 3 и/или 4. Конкретные примеры разрешенных замен включают, например, хлор или фтор в положении 2 или 4. Также альфа-углеродный атом может быть метилирован. Другие равноценные остатки, которые представляют консервативные замены для D-Phe, также могут использоваться. Они включают D-Ala(циклопентил), D-Ala(тиенил), D-Tyr и D-Tic. Остаток в положении 2, Xaa2 может также представлять собой D-Phe или замещенный D-Phe с такими заменами, в том числе заместителями при атоме углерода в положении 4 фенильного кольца или как положении 3, так и положении 4. Альтернативно, Хааг может представлять собой D-Trp, D-Tyr или D-аланин, замещенный нафтилом. Остаток в положении 3, Хаа3 может представлять собой каким-либо неполярный остаток аминокислоты, такой как D-Nle, D-Leu, (α-Me)D-Leu, D-Hie, D-Met или D-Val. Однако, D-Ala(циклопропил, циклобутил, циклопентил или циклогексил) или D-Phe может также использоваться в качестве Хаа3. Остаток в положении 4, Хаа4 может представлять собой какой-либо положительно заряженный остаток аминокислоты, например, D-Arg и D-Har, который может быть необязательно замещен низшими алкильными группами, такими как одна или две этильные группы. Альтернативно, могут использоваться D-Nar и любые другие равноценные остатки, такие как D-Lys или D-Orn (любой из которых может быть алкилирован ω-аминогруппой, например, метильными или изопропильными группами, или метилирован при α-атоме углерода). Кроме того, D-Dbu, D-4-Amf (который может быть необязательно замещен амидино) и D-Hlys также являются подходящими аминокислотами для данного положения.
[0056] Соединения по изобретению содержат один или больше хиральных центров, каждый из которых имеет два возможных трехмерных пространственных расположения (конфигурации) четырех заместителей вокруг центрального атома углерода. Они известны как стереоизомеры и, более специфично, как энантиомеры (все хиральные центры в обратной конфигурации) или диастереоизомеры (два или больше хиральных центров, как минимум один хиральный центр в такой же конфигурации). В конкретном варианте изобретения, указано, что аминокислоты, которые составляют основу тетрапептида, Хаа1Хаа2Хаа3Хаа4, представляют собой D-аминоаминокислоты, т.е. их конфигурация противоположна обычно присутствующей у млекопитающих. Ссылка на стереоизомеры синтетических пептидных амидов по изобретению касается хиральных центров, кроме альфа-атомов углерода D-аминокислот, которые составляют Xaa1-Xaa4. Таким образом, стереоизомеры синтетических пептидных амидов, которые являются вариантами изобретения, где указано, что каждый из Xaa1-Xaa4 представляет собой D-аминокислоту, не включают L-аминокислоты или рацемические смеси аминокислот в данных положениях. Подобным образом, ссылка на рацематы в данном описании касается центров, кроме альфа-атомов углерода D-аминокислот, которые составляют Xaa1-Xaa4. Хиральные центры в синтетических пептидных амидах по изобретению, для которых стереоизомер может находиться в R или S конфигурация, включают хиральные центры во фрагменте, присоединенном к С-концу Хаа4, а также хиральным центрам в каких-либо заместителях боковой цепи аминокислот Xaa1-Xaa4.
[0057] Синтетические пептидные амиды по изобретению, описанные в данном описании (также равнозначно обозначаются как соединения синтетических пептидных амидов, соединения по изобретению, соединение (номер) или просто "соединения"), могут применяться или быть получены в альтернативных формах. Например, многие аминосодержащие соединения могут применяться или быть получены в виде соли с кислотой. Часто такие соли улучшают выделение и свойства соединения в процессе обращения с ним. Например, в зависимости от реактивов, условий реакции и т.п., соединения, например, синтетические пептидные амиды, описанные в данном описании, могут применяться или быть получены, например, как гидрохлоридные или тозилатные соли. Изоморфные кристаллические формы, все хиральные и рацемические формы, N-оксиды, гидраты, соль ваты и гидраты солей кислот, также находятся в пределах контекста настоящего изобретения.
[0058] Определенные кислотные или основные синтетические пептидные амиды по настоящему изобретению могут существовать как цвиттер-ионы. Все формы таких синтетических соединений пептидных амидов, в том числе свободная кислота, свободное основание и цвиттер-ионы находятся в пределах контекста настоящего изобретения. В данной области хорошо известно, что соединения, содержащие как амино- так и карбоксильные группы, часто существуют в равновесии с их циттер-ионными формами. Таким образом, должно быть понятно, что для любого соединения, описанного в данном описании, которое содержит, например, как амино-, так и карбоксильные группы включен соответствующий цвиттер-ион.
[0059] В некоторых вариантах синтетические пептидные амиды по изобретению описываются Формулой I:
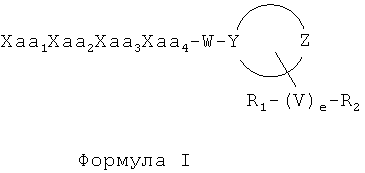
[0060] В таких вариантах Хаа1-Хаа2-Хаа3-Хаа4 представляет собой тетрапептидный фрагмент, где Хаа1 является аминокислотой на амино-конце, которая может представлять собой (А) (А')D-Phe, (А) (А')(α-Ме)D-Phe, D-Tyr, d-Tic, D-трег-лейцин, D-неопентилглицин, D-фенилглицин, D-гомофенилаланин, или β-(E)D-Ala, где каждый (А) и каждый (А') представляет собой заместитель в фенильном кольце, независимо выбранный из -Н, -F, -Cl, -NO2, -СН3, -CF3, -CN и -CONH2, и (Е) выбран как любой из циклобутила, циклопентила, циклогексила, тиенила, пиридила или тиазолила. Вторая аминокислота в цепи тетрапептида, Xaa2, может представлять собой любую из (А)(А')D-Phe, 3,4-дихлор-D-Phe, (А) (А') (α-Ме)D-Phe, D-lNal, D-2Nal, D-Tyr, (E)D-Ala или D-Trp. Третья аминокислота в цепи тетрапептида, Хаа3, может представлять собой D-NIe, D-Phe, (E)D-Ala, D-Leu, (α-Me)D-Leu, D-Hle, D-Val или D-Met. Четвертая аминокислота в цепи тетрапептида, Хаа4, может представлять собой любую из следующего: (B)2D-Arg, (B)2D-Nar, (B)2D-Har, ζ-(B)D-Hlys, D-Dap, ε-(B)D-Lys, ε-(B)2-D-Lys, D-Amf, амидино-D-Amf, γ-(B)2D-Dbu, δ-(B)2α-(B')D-Orn, D-2-амино-3(4-пиперидил)пропионовую кислоту, D-2-амино-3(2-амино-пирролидил)-пропионовую кислоту, D-α-амино-β-амидинопропионовую кислоту, α-амино-4-пиперидинуксусную кислоту, цис-α,4-диаминоциклогексануксусную кислоту, транс-α,4-диаминоциклогексануксусную кислоту, цис-α-амино-4-метиламино-циклогексануксусную кислоту, транс-α-амино-4-метиламиноциклогексануксусную кислоту, α-амино-1-амидино-4-пиперидинуксусную кислоту, цис-α-амино-4-гуанидиноциклогексануксусную кислоту или транс-α-амино-4-гуанидиноциклогексануксусную кислоту, где каждый (В) может быть отдельно выбран из -Н и C1-C4 алкила, и (В') может представлять собой -Н или группу (α-Ме).
[0061] В определенных вариантах изобретения, необязательная линкерная группа W отсутствует (т.е., равна нулю), при условии, что в таком случае Y представляет собой N. В других вариантах, W представляет собой -N-(CH2)b, где b равно 0, 1, 2, 3, 4, 5 или 6. В других вариантах, W представляет собой -N-(CH2)с-O-, где с равно 2 или 3, при условии, что в таких вариантах Y представляет собой С.
[0062] В конкретных вариантах изобретения, фрагмент
в формуле I представляет собой необязательно замещенный 4-8-членный гетероциклический кольцевой фрагмент, где все гетероатомы кольца в кольцевом фрагменте представляют собой N, и где каждый из Y и Z независимо представляет собой С или N, и они не являются соседными атомами кольца. В таких вариантах, если данный гетероциклический кольцевой фрагмент представляет собой 6-, 7- или 8-членное кольцо, атомы Y и Z в кольце разделены как минимум двумя другими атомами кольца. В таких вариантах, если данный гетероциклический кольцевой фрагмент содержит единственный гетероатом в кольце, который представляет собой N, кольцевой фрагмент является неароматическим.
[0063] В определенных конкретных вариантах изобретения, соединительный фрагмент V непосредственно присоединен к Y- и Z-содержащему кольцу. V представляет собой C1-С6 алкил, который может быть замещен группами R1 и R2. Заместитель R1 может быть выбран из -Н, -ОН, галогена, -CF3, -NH2, -СООН, C1-C6 алкила, амидино, C1-С6алкилзамещенного амидино, арила, необязательно замещенного гетероциклила, Pro-амида, Pro, Gly, Ala, Val, Leu, Ile, Lys, Arg, Orn, Ser, Thr, -CN, -CONH2, -COR', -SO2R', -CONR'R'', -NHCOR', OR' или -SO2NR'R''. Необязательно замещенный гетероциклил может содержать 1 или 2 заместителя, независимо выбранные из C1-C6 алкила, C1-C6 алкокси, оксо, -ОН, -Cl, -F, -NH2, -NO2, - CN, -СООН и амидино. Каждый из фрагментов R' и R'' независимо представляют собой Н, C1-C6 алкил, арил или гетероциклил. Альтернативно, R' и R'' вместе могут образовывать 4-8-членное кольцо, которое необязательно содержит 1 или 2 заместителя, независимо выбранные из C1-С6 алкила, C1-С6 алкокси, -ОН, -Cl, -F, -NH2, -NO2, -CN, -СООН и амидино. Заместитель R1 может быть любым из -Н, амидино, амидино, замещенного одным или двумя C1-С6 алкилом, -CN, -CONH2, -CONR'R'', -NHCOR', -SO2NR'R'' или -СООН.
[0064] В других конкретных вариантах, V отсутствует и группы заместителей R1 и R2 непосредственно присоединены к одному и тому же или различным атомам кольца Y- и Z-содержащего гетероциклического кольца.
[0065] В одном альтернативном аспекте определенных вариантов, фрагменты R1 и R2, взятые вместе, могут образовывать необязательно замещенный 4-9-членный гетероциклический моноциклический или бициклический кольцевой фрагмент, который присоединен к одному атому кольца Y- и Z-содержащего кольцевого фрагмента. В одном конкретном варианте, фрагменты R1 и R2 образуют необязательно замещенный 4-9-членный гетероциклический моноциклический или бициклический кольцевой фрагмент, который непосредственно присоединен к одному атому кольца Y- и Z-содержащего кольцевого фрагмента.
[0066] Во втором альтернативном аспекте определенных вариантов, фрагменты R1 и R2 вместе с одним атомом кольца Y- и Z-содержащего кольцевого фрагмента могут образовывать необязательно замещенный 4-8-членный гетероциклический кольцевой фрагмент с образованием спироструктуры.
[0067] В третьем альтернативном аспекте определенных вариантов, фрагменты R1 и R2 вместе с двумя или больше соседными атомами кольца Y- и Z-содержащего кольцевого фрагмента, могут образовывать необязательно замещенный 4-9-членный гетероциклический моноциклический или бициклический кольцевой фрагмент, конденсированный с Y-и Z-содержащим кольцевым фрагментом.
[0068] В конкретных вариантах, каждый из необязательно замещенных 4-, 5-, 6-, 7-, 8- и 9-членные гетероциклических кольцевых фрагментов, содержащих R1 и R2, может содержать 1 или 2 заместителя, независимо выбранные из С1-С6 алкила, C1-C6 алкокси, необязательно замещенного фенила, оксо, -ОН, -Cl, -F, -NH2, -NH2, -CN, -СООН и амидино.
[0069] В определенных конкретных вариантах, если Y- и Z-содержащий кольцевой фрагмент представляет собой 6- или 7-членное кольцо, которое содержит единственный гетероатом в кольце, и если один из Y и Z представляет собой С, а другой из Y и Z представляет собой N, и е равно 0, то R1 не может быть -ОН, и R1 и R2 оба не являются -Н. В дальнейшем, если Y- и Z-содержащий кольцевой фрагмент представляет собой 6-членное кольцо, которое содержит два гетероатома в кольце, где как Y, так и Z представляет собой N, и W равен 0, то фрагмент -(V)eR1R2 присоединен к атому кольца, отличному от Z. Кроме того, если е равно 0, то R1 и R2 оба не могут быть -Н. Наконец, если Хаа3 представляет собой D-Nle, то Хаа4 не может быть (B)2D-Arg; и если Хаа3 представляет собой D-Leu или (αМе) D-Leu, то Хаа4 не может быть δ-(B)2α-(B')D-Orn.
[0070] В определенных вариантах настоящего изобретения также предлагаются синтетические пептидные амиды формулы:

или их стереоизомер, рацемат, пролекарство, фармацевтически приемлемая соль, гидрат, сольват, гидрат соли кислоты, N-оксид или изоморфная кристаллическая форма; где Xaa1 выбран из группы, состоящей из (А) (А')D-Phe, (α-Ме)D-Phe, D-Tyr, D-Tic, D-фенилглицина, D-гомофенилаланина и β-(E)D-Ala, где каждый из (А) и (А') представляет собой заместитель в фенильном кольце, независимо выбранный из группы, состоящей из -Н, -F, -Cl, -NO2, -СН3, -CF3, -CN и -CONH2, и где (Е) выбран из группы, состоящей из циклобутила, циклопентила, циклогексила, пиридила, тиенила и тиазолила; Xaaz выбран из группы, состоящей из (А) (А')D-Phe, (α-Ме)D-Phe, D-1Nal, D- 2Nal, D-Tyr, (E)D-Ala и D-Trp; Хаа3 выбран из группы, состоящей из D-Nle, D-Phe, (E)D-Ala, D-Leu, (α-Me)D-Leu, D-Hle, D-Val и D-Met; Xaa4 выбран из группы, состоящей из (B)2D-Arg, (B)2D-nArg, (B)2D-Har, ζ-(B)D-Hlys, D-Dap, ε-(B)D-Lys, ε-(В)2-D-Lys, D-Amf, амидино-D-Amf, γ-(B)2D-Dbu, δ-(В)2α-(В')D-Orn, D-2-амино-3(4-пиперидил)пропионовой кислоты, D-2-амино-3(2-аминопирролидил)пропионовой кислоты, D-α-амино-β-амидинопропионовой кислота, (R)-α-амино-4-пиперидинуксусной кислоты, цис-α,4-диаминоциклогексануксусной кислоты, транс-α,4-диаминоциклогексануксусной кислоты, цис-α-амино-4-метиламиноциклогексануксусной кислоты, транс-α-амино-4-метиламиноциклогексануксусной кислоты, α-амино-1-амидино-4-пиперидинуксусной кислоты, цис-α-амино-4-гуанидиноциклогексануксусной кислоты и транс-α-амино-4-гуанидиноциклогексануксусной кислоты, где каждый (В) независимо выбран из группы, состоящей из Н и C1-C4 алкила, и (В') представляет собой Н или (α-Me). Группа W выбрана из группы, состоящей из:
0, при условии, что если W равен 0, Y представляет собой N;
-N-(CH2)b, где b равно 0, 1, 2, 3, 4, 5 или 6; и
-N-(CH2)c-O-, где с равно 2 или 3, при условии, что Y представляет собой С;
и фрагмент
представляет собой необязательно замещенный 4-8-членный насыщенный гетероциклический кольцевой фрагмент, содержащий 1 или 2 атома азота, где ни один из атомов кольца, кроме Y и Z, не является гетероатомом, Y представляет собой С или N, Z представляют собой С или N, и как минимум один из Y и Z представляет собой N, и при условии, что в случае 4- или 5-членных гетероциклических колец или Y, или Z представляет собой С, ив случае гетероцикла, содержащего 2 атома азота, Y и Z разделены двумя или больше атомами углерода кольца; V представляет собой C1-C6 алкил, и е равно 0 или 1, где если е равно 0, то V равен 0, и R1 и R2 непосредственно присоединены к одному и тому же или различным атомам кольца;
R1 представляет собой Н, ОН, -NH2, -СООН, C1-C6 алкил, амидино, C1-С6 алкилзамещенный амидино, дигидроимидазол, рго-амиды. Pro, Gly, Ala, Val, Leu, Ile, Lys, Arg, Orn, Ser, Thr, -CN, -CONH2, -CONR'R'', -NHCOR', -OR' или -SO2NR'R'', где каждый из R' и R'' независимо представляет собой Н или C1-C8 алкил, или R' и R" образуют 4-8-членное кольцо, которое необязательно замещено 1 или 2 заместителями, независимо выбранными из группы, состоящей из C1-С6 алкила, C1-C6 алкокси, -ОН, -Cl, -F, -NH2, -NO2, -CN и -СООН, амидино; и R2 представляет собой Н, амидино, амидино, замещенный одным или двумя C1-С6 алкилом, -CN, -CONH2, -CONR'R'', -NHCOR', -SO2NR'R'' или -СООН;
при условии, что если Y- и Z-содержащий кольцевой фрагмент представляет собой 6- или 7-членное кольцо, и если один из Y и Z представляет собой С, и е равно 0, то R1 не является -ОН, и R1 и R2 оба не являются Н; при условии, что если Y- и Z-содержащий кольцевой фрагмент представляет собой 6-членное кольцо, как Y, так и Z представляет собой N, и W равен 0, то -(V)eR1R2 присоединен к атому кольца, отличному от Z; и если е равно 0, то R1 и R2 оба не являются -Н; и, наконец, при условии, что если Хаа3 представляет собой D-Nle, то Хаа4 не может быть (B)2D-Arg, и если Хаа3 представляет собой D-Leu или (αМе)D-Leu, то Хаа4 не является δ-(В)2α-(В')D-Orn.
[0071] В одном варианте настоящего изобретения предлагаются синтетические пептидные амиды формулы I, где Xaa1Xaa2 представляет собой D-Phe-D-Phe, Хаа3 представляет собой D-Leu или D-Nle, и Хаа4 выбран из (B)2D-Arg, D-Lys, (B)2D-Har, ζ-(В)D-Hlys, D-Dap, ε-(B)D-Lys, ε-(B)2-D-Lys, D-Amf, амидино-D-Amf, γ-(B)2D-Dbu и 5-(В)2α-(В')D-Orn. В конкретном аспекте каждого варианта, Хаа4 выбран из D-Lys, (B)2D-Har, ε-(В)D-Lys и ε-(В)2-D-Lys.
[0072] В другом варианте изобретения предлагаются синтетические пептидные амиды формулы I, где W равен 0, Y представляет собой N, и Z представляет собой С. В конкретном аспекте упомянутого выше варианта, Y- и Z-содержащий кольцевой фрагмент представляет собой 6-членное насыщенное кольцо, содержащее один гетероатом в кольце.
[0073] В другом варианте изобретения предлагаются синтетические пептидные амиды формулы I, где Y и Z как представляют собой N, так и являются единственными гетероатомами в кольце Y- и Z-содержащего кольцевого фрагмента.
[0074] В другом варианте, если Y- и Z-содержащий кольцевой фрагмент представляет собой насыщенное 6-членное кольцо, которое содержит два гетероатома в кольце, и W равен 0, то Z представляет собой атом углерода.
[0075] В другом варианте изобретения предлагаются синтетические пептидные амиды формулы I, где R1 и R2 вместе с 0, 1 или 2 атомами кольца Y- и Z-содержащего кольцевого фрагмента образуют моноциклический или бициклический 4-9-членный гетероциклический кольцевой фрагмент. В конкретном аспекте упомянутого выше варианта, R1 и R2 вместе с одним атомом кольца Y- и Z- содержащкго кольцевого фрагмента образуют 4-8-членный гетероциклический кольцевой фрагмент, который вместе с Y- и Z-содержащим кольцевым фрагментом образует спироструктуру, и W равен 0.
[0076] В другом варианте изобретения предлагаются синтетические пептидные амиды формулы I, где е равно 0, и R1 и R2 присоединены к одному и тому же атому углерода.
[0077] В дальнейшем варианте изобретения предлагаются синтетические пептидные амиды формулы I, где R1 представляет собой Н, ОН, -NH2, -COOH, -CH2COOH, C1-С3 алкил, амидино, C1-С3 алкилзамещенный амидино, дигидроимидазол, D-Pro, D-Pro амид или -CONH2, и где R2 представляет собой Н, -COOH или C1-С3 алкил.
[0078] В другом варианте изобретения предлагаются синтетические пептидные амиды формулы I, где фрагмент:
выбран из группы, состоящей из:


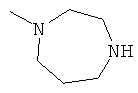
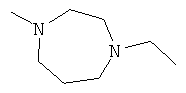
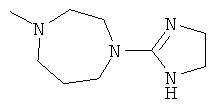

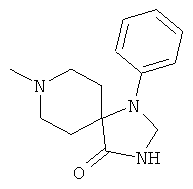
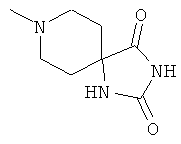

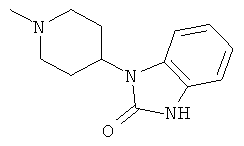

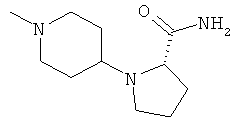



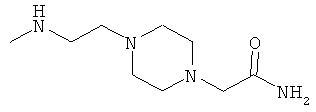
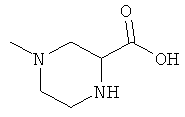

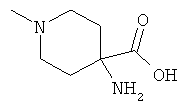
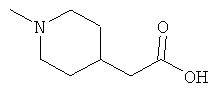








[0079] В другом варианте изобретения предлагаются синтетические пептидные амиды формулы 1, где фрагмент:
не является пролиновым фрагментом, замещенным пролиновым фрагментом, а также не является пролиновым фрагментом, если R1 или R2 содержат амидный фрагмент.
[0080] В другом варианте изобретения предлагаются синтетические пептидные амиды формулы I, при условии, что если W равен 0, Y- и Z-содержащий кольцевой фрагмент представляет собой насыщенное 5-членное кольцом с единственным гетероатомом в кольце, е равно 0, и R1 или R2 присоединен к атому углерода кольца, соседному с Y, то R1 выбран из группы, состоящей из -Н, -ОН, галогена, -CF3, -NH2, C1-С6-алкила, амидино, C1-C6-алкилзамещенного амидино, арила. Pro, Gly, Ala, Val, Leu, He, Lys, Arg, Orn, Ser, Thr, CN, -SO2R', -NHCOR', -OR' и -SO2NR'R'' и R2 выбран из группы, состоящей из -Н, амидино, амидино, замещенного одным или двумя C1-С6 алкилом, -CN, -NHCOR' и -SO2NR'R''.
[0081] В другом варианте изобретения предлагается синтетический пептидный амид формулы I, демонстрирующий EC50 меньше приблизительно 500 нМ для каппа-опиатного рецептора. В конкретном аспекте, синтетический пептидный амид может демонстрировать EC50 меньше приблизительно 100 нМ для каппа-опиатного рецептора. В более конкретном аспекте, синтетический пептидный амид может демонстрировать EC50 меньше приблизительно 20 нМ для каппа-опиатного рецептора. В наиболее конкретном аспекте, синтетический пептидный амид может демонстрировать EC50 меньше приблизительно 1 нМ для каппа-опиатного рецептора. Соединения в соответствии с вышеуказанным вариантом могут демонстрировать ЕС50 как минимум в 10 раз больше для мю- и дельта-опиатного рецептора, чем для каппа-опиатного рецептора, предпочтительно как минимум в 100 раз больше, и наиболее предпочтительно как минимум в 1000 раз больше (например, EC50 меньше приблизительно 1 нМ для каппа-опиатного рецептора, и EC50 больше приблизительно 1000 нМ для мю-опиатного рецептора и дельта-опиатного рецептора).
[0082] В другом варианте изобретение предлагается синтетический пептидный амид формулы 1, который в эффективной концентрации демонстрирует не более чем приблизительно 50% ингибирования любого из P4so CYP1A2, СУР2С9, CYP2C19 или CYP 2D6 синтетическим пептидным амидом в концентрации 10 мкМ после инкубации в течение 60 минут с микросомами печени человека.
[0083] Еще в одном варианте изобретения предлагается синтетический пептидный амид формулы I, который в дозе приблизительно 3 мг/кг у крыс обеспечивает пиковую концентрацию в плазме синтетического пептидного амида как минимум приблизительно в 5 раз выше, чем пиковая концентрация в мозге. В одном конкретном аспекте вышеуказанного варианта, синтетический пептидный амид демонстрирует ED50 для седативного эффекта в анализе на модели уменьшения перемещений у мышей как минимум приблизительно в 10 раз больше ED50 синтетических пептидного амида для анальгетического эффекта на модели корчей у мышей.
[0084] Еще в одном варианте изобретения предлагается синтетический пептидный амид формулы I, демонстрирующий как минимум приблизительно 50% максимальной эффективности приблизительно через 3 часа после введения дозы приблизительно 3 мг/кг синтетического пептидного амида у крыс.
[0085] В одном варианте изобретения предлагается фармацевтическая композиция, которая содержит синтетический пептидный амид формулы I и фармацевтически приемлемое вспомогательное вещество или носитель.
[0086] В другом варианте изобретения предлагается способ лечения или предупреждения связанного с каппа-опиатным рецептором заболевания или состояния у млекопитающего; где способ включает введение млекопитающему композиции, содержащей эффективное количество синтетических пептидного амида в соответствии с формулой I, достаточное для лечения или предупреждения связанного с каппа-опиатным рецептором заболевания или состояния.
[0087] Различные анализы могут применяться для проверки наличия у синтетических пептидных амидов по изобретению высокого сродства и селективности в отношении каппа-опиатного рецептора, продолжительность биологической активности in vivo и отсутствия побочного действия на ЦНС. Анализы с использованием рецепторов хорошо известны из уровня техники, и клонированы каппа-опиатные рецепторы нескольких видов, например, мю- и дельта-опиатные рецепторы. Каппа-опиатные рецепторы, также как мю- и дельта-опиатные рецепторы, являются классическими, семь трансмембранно-охватывающих, связанных с протеином G рецепторов. Хотя такие клонированные рецепторы позволяют легко провести скрининг конкретного соединения-кандидата, например, пептида или производного пептида, природные источники опиатных рецепторов млекопитающих также пригодны для скрининга, как хорошо известно из уровня техники (Dooley CT et al. Selective ligands for the mu, delta, and kappa opioid receptors identified from a single mixture based tetrapeptide positional scanning combinatorial library. J. Biol. Chem. 273:18848-56, 1998). Таким образом, скрининг как в отношении каппа-, так и мю-опиатных рецепторов, рекомбинантного или природного происхождения, может быть осуществлен для определения селективности синтетических пептидных амидов по изобретению в отношении каппа-опиатных рецепторов по сравнению с mu.
[0088] В конкретном варианте, синтетические пептидные амиды по изобретению представляют собой селективные агонисты каппа-опиатного рецептора. Активность синтетических пептидных амидов по изобретению в качестве агонистов конкретного рецептора может быть измерена как концентрация, в которой достигается половина максимального эффекта, выраженная как значение EC50. Активность синтетических пептидных амидов по изобретению в качестве агонистов каппа-опиатного рецептора, выраженная как процент максимального эффекта, который поддается наблюдению, может быть определена разнообразными способами, хорошо известными из уровня техники. См., например, Endoh T et а1., 1999, Potent Antinociceptive Effects of TRK-820, a Novel к-Opioid Receptor Agonist, Life Sci. 65 (16) 1685-94; и Kumar V et а1.. Synthesis and Evaluation of Novel peripherally Restricted к-Opioid Receptor Agonists, 2005 Bioorg Med Chem Letts 15: 1091-1095.
[0089] Примеры таких методов анализа для определения значений EC50 предложены ниже. Многие стандартные способы анализа для характеристики опиатных лигандов хорошо известны специалисту в данной области. См., например, Waldhoer et а1., (2004) Ann. Rev. Biochem. 73:953-990, и Satoh & Minami (1995) Pharmac. Ther. 68(3):343-364 и процитированные в данном описании ссылки.
[0090] В определенных конкретных вариантах, синтетические пептидные амиды по изобретению представляют собой агонисты каппа-опиатного рецептора с ЕС50 меньше приблизительно 500 нМ. В других вариантах, синтетические пептидные амиды как агонисты каппа-опиатного рецептора демонстрируют ЕС50 меньше приблизительно 100 нМ. В других вариантах, синтетические пептидные амиды как агонисты каппа-опиатного рецептора демонстрируют EC50 меньше приблизительно 10 нМ. В конкретных вариантах синтетические пептидные амиды по изобретению как агонисты каппа-опиатного рецептора демонстрируют EC50 меньше приблизительно 1,0 нМ, меньше приблизительно 0,1 нМ или меньше приблизительно 0,1 нМ или даже меньше приблизительно 0,01 нМ.
[0091] В конкретных вариантах, синтетические пептидные амиды по изобретению высокоселективны в отношении каппа по сравнению с мю-опиатными рецепторами. В определенных вариантах синтетические пептидные амиды по изобретению демонстрируют значения EC50 для мю-опиатного рецептора как минимум в 100 раз выше, чем соответствующие значения EC50 Для каппа-опиатного рецептора. В конкретных вариантах, синтетические пептидные амиды по изобретению демонстрируют значения EC50 для мю-опиатного рецептора как минимум в 1000 раз выше, чем соответствующие значения EC50 Для каппа-опиатного рецептора. Альтернативно, селективность синтетических пептидных амидов по изобретению может быть выражена, как более высокое значение EC50 для мю-опиатного рецептора по сравнению с каппа-опиатным рецептором. Таким образом, в конкретных вариантах, синтетические пептидные амиды по изобретению демонстрируют значения EC50 больше приблизительно 10 мкМ для мю-опиатного рецептора, и значения EC50 меньше приблизительно 10 нМ, и в других вариантах - меньше приблизительно 1,0 нМ или даже меньше приблизительно 0,01 нМ для каппа-опиатного рецептора. В другом варианте, конкретные синтетические пептидные амиды могут демонстрировать EC50 меньше приблизительно 1 нМ для каппа-опиатного рецептора, и EC50 больше приблизительно 1000 нМ для мю-опиатного рецептора или для дельта-опиатного рецептора.
[0092] Другое свойство синтетических пептидных амидов по изобретению состоит в их характерном свойстве низкой степени ингибирования изоферментов цитохром Р450 Изоферменты цитохром Р450 составляют большое суперсемейство гем-тиолятных белков, ответственных за метаболическую окислительную инактивацию многих терапевтических и других биологически активных соединений. Обычно, они действуют, как терминальные оксидазы в многокомпонентных цепях переноса электронов, которые также называют цитохром Р450содержащими монооксигеназными системами.
[0093] Свыше 50 различных изоферментов цитохром Р450 идентифицированы и классифицированы в семейства, сгруппированные в соответствии с генетическим родством, которое оценивают по гомологии последовательностей нуклеиновых кислот. Из изоферментов цитохром P450 в клетках человека наиболее распространены изоферменты 1А2 и 3А4, хотя изоферменты 2В6, 2С9, 2С19, 2D6 и 2Е1 также вносят значительный вклад в окислительную инактивацию введенных терапевтических средств. Хотя ингибирование изоферментов цитохром P450 может быть полезным с точки зрения удлинения времени после введения in vivo, в ходе которого поддерживается эффективная концентрация синтетических пептидных амидов по изобретению, такой эффект также удлиняет присутствие любого одновременно введенного терапевтического соединения, которое подвергается окислению под действием цитохром Р450. Такое увеличение периода присутствия может вызывать присутствие одновременно введенного терапевтического средства после окончания оптимального для лечения периода или может вызывать превышение желательных уровней или безопасных уровней концентрации in vivo. Такое удлинение присутствия и/или повышение концентрации трудно точно определить количественно и предпочтительно их избегают. Терапевтические средства, которые демонстрируют незначительное или отсутствие ингибирования активности изоферментов цитохром P450, не несут в себе потенциала данной проблемы и могут более безопасно вводиться одновременно с другими видами терапии без риска воздействия на скорость инактивации терапевтического соединения, которое вводится одновременно, под действием изоферментов цитохром P450.
[0094] Конкретные варианты синтетических пептидных амидов по изобретению демонстрируют низкую степень ингибирования изоферментов цитохром Р450 в терапевтических концентрациях синтетических пептидных амидов, в то время как другие в значительное мере не демонстрируют ингибирования изоферментов цитохром Р450 в терапевтических концентрациях. В некоторых вариантах, синтетические пептидные амиды в концентрации 10 мкМ демонстрируют ингибирование изоферментов цитохром P450 CYP1A2, CYP2C9, CYP2C19 или CYP2D6 меньше приблизительно 50%. В конкретных вариантах, синтетические пептидные амиды в концентрации 10 мкМ демонстрируют ингибирование любого из указанных изоферментов цитохром Р450 меньше приблизительно 20%. В отдельных вариантах, синтетические пептидные амиды в концентрации 10 мкМ демонстрируют ингибирование любого из указанных изоферментов цитохром P450 меньше приблизительно 10%.
[0095] В другом варианте, синтетические пептидные амиды по изобретению в эффективной концентрации демонстрируют не более чем приблизительно 50% ингибирование любого из P450 CYP1A2, CYP2C9, CYP2C19 или CYP2D6 синтетическими пептидными амидами в концентрации 10 мкМ после инкубации в течение 60 минут с микросомами печени человека.
[0096] Синтетические пептидные амиды по изобретению при введении, млекопитающему или пациенту-человеку в терапевтически эффективном концентрации демонстрируют низкую степень или по существу отсутствие проникновения через гематоэнцефалический барьер. Каппа-опиатные рецепторы (в дальнейшем равнозначно называются рецепторами каппа) распространены в периферических тканях, в том числе коже и соматических тканях, а также во внутренних органах человека и других млекопитающих. Рецепторы каппа также найдены в мозге. Активация рецепторов каппа в периферических тканях вызывает подавление боли и воспалительных реакций, в то время как активация рецепторов каппа в мозге вызывает седативный эффект, и, возможно, приводит также к тяжелой дисфории и галлюцинациям. В определенных вариантах, синтетический пептидный амид по изобретению, при введении в терапевтически эффективных концентрациях демонстрирует по существу отсутствие проникновения через гематоэнцефалический барьер и, таким образом, минимизируют или даже полностью свободны от седативного, галлюциногенного эффекта многих другие агонистов каппа, которые демонстрирую некоторую степень проникновения через гематоэнцефалический барьер.
[0097] Один из пригодных показателей степени, в которой синтетические пептидные амиды по изобретению проникают сквозь гематоэнцефалический барьер, представляет собой соотношение пиковой концентрации в плазме и концентрации в ткани мозга. В конкретных вариантах, синтетические пептидные амиды по изобретению, при введении в дозе приблизительно 3 мг/кг, демонстрируют как минимум 5-кратное снижение концентрации синтетических пептидных амидов в мозге по сравнению с плазмой в момент времени, когда достигается пиковая концентрация в плазме.
[0098] Другой пригодный показатель степени, в которой синтетические пептидные амиды по изобретению проникают сквозь гематоэнцефалический барьер, представляет собой соотношение дозы, необходимой для достижения седативного эффекта, и дозы, необходимой для достижения анальгетического эффекта. Анальгетический и седативный эффекты стимулирования рецептора каппа агонистами рецептора каппа могут быть измерены в помощью стандартных анализов, хорошо известных специалисту в данной области.
[0099] В конкретных вариантах, синтетические пептидные амиды по изобретению демонстрируют ED50 для седативного эффекта как минимум приблизительно в 10 раз больше ED50 для анальгетического эффекта. В конкретных вариантах, синтетические пептидные амиды по изобретению демонстрируют ED50 для седативного эффекта как минимум приблизительно в 30 раз больше ED50 для анальгетического эффекта. В других вариантах, синтетические пептидные амиды по изобретению демонстрируют ED50 для седативного эффекта как минимум приблизительно в 50 раз больше ED50 для анальгетического эффекта.
[00100] В другом варианте, синтетические пептидные амиды по изобретению демонстрируют ED50 для седативного эффекта на модели снижения перемещений у мышей как минимум приблизительно в 10 раз больше ED50 синтетических пептидных амидов для анальгетического эффекта на модели корчей у мышей.
[00101] Другой пригодный прогностический фактор ожидаемой степени проникновения синтетических пептидных амидов по изобретению сквозь гематоэнцефалический барьер, обеспечивается значениями проницаемости мембраны синтетических пептидных амидов в клетку человека или клетку другого млекопитающего, при введении терапевтически адекватной концентрации. В определенных вариантах, синтетические пептидные амиды по изобретению демонстрируют в терапевтически адекватных концентрациях низкую степень или по существу отсутствие способности проникать сквозь монослой подходящим образом культивированных клеток человека или других млекопитающих. Данный параметр проницаемости может быть выражен, как очевидная проницаемость, Рарр, которая представляет проницаемость конкретного монослоя клеток для целевого соединения. Любой пригодный для культивирования монослой клеток млекопитающих может использоваться для определения его проницаемости для конкретного целевого соединения, хотя для данной цели часто используются определенные линии клеток. Например, линия клеток Сасо-2 представляет собой аденокарциному ободочной и прямой кишки человека, которая может использоваться в качестве модельной системы культуры монослоя для определения проницаемости мембран для соединений по изобретению. В определенных вариантах, синтетические пептидные амиды по изобретению демонстрируют Рарр меньше приблизительно 10-6 см/сек. В некоторых других вариантах, синтетические пептидные амиды по изобретению демонстрируют Рарр меньше приблизительно 10-7 см/сек.
[00102] В одном варианте, синтетические пептидные амиды по изобретению в дозе приблизительно 3 мг/кг у крыс достигают пиковой концентрации в плазме синтетического пептидного амида, и демонстрирует как минимум в 5 раз более низкие концентрации в мозге, чем аналогичная пиковая концентрация в плазме.
[00103] В другом варианте, синтетические пептидные амиды по изобретению демонстрируют как минимум приблизительно 50% максимальной эффективности приблизительно после 3 часа после введения дозы приблизительно 3 мг/кг синтетического пептидного амида у крыс.
[00104] В одном из вариантов, синтетические пептидные амиды по изобретению демонстрируют продолжительное действие у млекопитающего, например, человека. В одном аспекте, синтетические пептидные амиды демонстрируют продолжительность действия на уровне как минимум приблизительно 50% максимальной эффективности через 3 часа после введения 0,1 мг/кг синтетического пептидного амида. В другом аспекте синтетические пептидные амиды демонстрируют продолжительность действия на уровне как минимум приблизительно 75% максимальной эффективности через 3 часа после введения 0,1 мг/кг синтетического пептидного амида. В конкретном аспекте синтетические пептидные амиды демонстрируют продолжительность действия на уровне как минимум приблизительно 90% максимальной эффективности через 3 часа после введения 0,1 мг/кг синтетического пептидного амида. В конкретном аспекте, синтетические пептидные амиды демонстрируют продолжительность действия как минимум приблизительно 95% максимальной эффективности через 3 часа после введения 0,1 мг/кг синтетического пептидного амида.
[00105] В другом варианте изобретения предлагается фармацевтическая композиция, которая содержит синтетические пептидные амиды в соответствии с любым из приведенных выше вариантов и фармацевтически приемлемое вспомогательное вещество или носитель. В изобретении предлагаются способы, композиции или лекарственные формы, в которых применяются и/или содержатся синтетические пептидные амиды по изобретению, селективные в отношении каппа-опиатных рецепторов. В конкретных вариантах, синтетические пептидные амиды по изобретению демонстрируют выраженное сродство к каппа-опиатным рецепторам и обладают высокой активностью в качестве агонистов каппа-опиатного рецептора.
[00106] Пролекарство соединения, такого как синтетические пептидные амиды по изобретению, включает фармацевтически приемлемые производные, которые при введении могут превращаться, посредством метаболизма или другого процесса, в биологически активную форму соединения. Пролекарства особенно желательны, если Пролекарство обладает более благоприятными свойствами, чем активное соединение, с точки зрения биодоступности, стабильности или пригодности для конкретной рецептуры.
[00107] В данном описании, связанное с каппа-опиатным рецептором заболевание, состояние или расстройство представляет собой любое заболевание, состояние или расстройство, поддающееся профилактике или поддающееся лечению активацией каппа-опиатного рецептора. В одном аспекте, синтетические пептидные амиды по изобретению представляют собой агонисты каппа-опиатного рецептора, которые активируют каппа-опиатный рецептор. В некоторых вариантах, конкретная доза и способ введения синтетических пептидных амидов по изобретению может быть выбрана клиницистом таким образом, чтобы полностью предупредить или излечить заболевание, состояние или расстройство. В других вариантах конкретная доза и способ введения синтетических пептидных амидов по изобретению, выбранные клиницистом, облегчают или уменьшают выраженность одного или больше симптомов заболевания, состояния или расстройства.
[00108] В данном описании, "эффективное количество" или "достаточное количество" синтетических пептидных амидов по изобретению обозначает количество соединения, как описано в данном описании, которое может быть терапевтически эффективным с точки зрения подавления, предупреждать или лечения симптома конкретного заболевания, расстройства, состояния или побочного эффекта. В данном описании "сниженная доза" анальгетика мю-опиатного агониста обозначает дозу, которая, при введении в комбинации с каппа-опиатным агонистом, таким как синтетические пептидные амиды по изобретению, будет ниже, чем обычно предлагаемая конкретному больному, с целью уменьшения выраженности одного или больше побочных эффектов соединения. Снижение дозы может быть выбрано таким образом, чтобы уменьшение анальготического или другого терапевтического эффекта соединения представляло собой приемлемый компромисс с учетом уменьшения побочного эффекта(ов), где указанное уменьшение выраженности указанного анальготического или другого терапевтического эффекта анальгетика мю-опиатного агониста как минимум частично и, более предпочтительно, полностью компенсировалось анальготическим или другим терапевтическим эффектом синтетического пептидного амида по изобретению. Совместное введение анальгетика мю-опиатного агониста с синтетическим пептидным амидом по изобретению, который действует как каппа-опиатный агонист, также позволяет введение сниженной дозы синтетического пептидного амида и/или анальгетика мю-опиатного агониста для достижения такого же терапевтического эффекта, как и при введении более высокой дозы синтетического пептидного амида или анальгетика мю-опиатного агониста, при монотерапии.
[00109] В данном описании, "фармацевтически приемлемый" обозначает соединения, материалы, композиции и/или лекарственные формы, которые являются, в пределах медицинского суждения, пригодными для контакта с тканями человеческих существ и животных без выраженной, раздражения, аллергической реакции или других осложнений, пропорционально соотношению выгода/риск, которое является разумным для медицинского состояния, подлежащего лечению.
[00110] В данном описании "единица лекарственной формы" обозначает физически дискретную единицу, пригодную в качестве единичной дозы для конкретного индивидуума или состояния, подлежащего лечению. Каждая единица может содержать предварительно определенное количество соединения(ий) активного синтетического пептидного амида, рассчитанных таким образом, чтобы обеспечить желательный терапевтический эффект(ы), необязательно в сочетании с фармацевтическим носителем. Спецификация для дозированных лекарственных форм может быть продиктована (а) уникальными характеристиками активного соединения или соединений и конкретным терапевтическим эффектом, который должен быть достигнут, и (b) ограничениями, свойственными в данной области рецептур такого активного соединения или соединений. Единицу дозы часто выражают как массу соединения на единицу массы тела, например, в миллиграммах соединения на килограмм массы тела субъекта или больного (мг/кг). Альтернативно, дозы могут быть выражены, как количество соединения на единицу массы тела в единицу времени (мг/кг/день) в конкретной схеме лечения. В альтернативных вариантах, дозы могут быть выражены, как количество соединения на единицу площади поверхности тела (мг/м2) или на единицу площади поверхности тела в единицу времени (мг/м2/сутки). Для местных препаратов, дозы могут быть выражены способом, обычным для таких препаратов, например, лента мази длиной1/2 дюйма, введенная в глаз, где концентрация соединения в препарате выражается как процент рецептуры.
[00111] В данном описании выражение "фармацевтически приемлемые соли" обозначает производные соединений, где исходное соединение модифицируют путем образования его соли с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, не ограничиваясь ими, соли основных остатков, таких как амины с минеральными или органическими кислотами; соли кислотных остатков, таких как карбоновые кислоты и т.п., с щелочами или органическими соединениями. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные соли аммония и исходного соединения, образованные, например, нетоксичными неорганическими или органическими кислотами. Например, такие традиционные нетоксичные соли включают полученные из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная кислоты и т.п.; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изэтиновая кислота и т.п. Такие физиологически приемлемые соли получают способами, известными из уровня техники, например, растворением свободных оснований амина с избытком кислоты в водном спирте или нейтрализацией свободной карбоновой кислоты основанием щелочного металла, таким как гидроксид, или амином. Таким образом, фармацевтически приемлемая соль синтетических пептидных амидов может быть образована любым таким пептидным амидом, содержащим кислотную, основную или обе функциональные группы. Например, пептидные амиды, содержащие группу карбоновой кислоты, могут в присутствие фармацевтически подходящей основы образовывать анион карбоксилата, соединяющийся с катионом, таким как катион натрия или калия. Подобным образом, пептидные амиды, содержащие функциональную аминогруппу, могут в присутствие фармацевтически подходящей кислоты, такой как HCl, образовывать соль.
[00112] Один из примеров фармацевтически приемлемого соль вата синтетических пептидных амидов представляет собой комбинацию пептидных амидов с молекулами растворителя, что дает комплекс таких молекул растворителя в сочетании с пептидными амидами. Особенно подходящими гидратами соединений являются такие гидраты, которые обладают сравнимой активностью, или гидраты, которые превращаются назад в активные соединения после введения. Фармацевтически приемлемый N-оксид синтетического пептидного амида, который содержит амин, представляет собой такое соединение, где атом азота амина присоединен к атому кислорода.
[00113] Фармацевтически приемлемая кристаллическая, изоморфная кристаллическая или аморфная форма синтетического пептидного амида по изобретению может представлять собой любую кристаллическую или некристаллическую форму фармацевтически приемлемой кислотной, основной, циттер-ионной, солевой, гидратной или любой другой соответствующим образом стойкой, физиологически совместимой формы синтетического пептидного амида по изобретению.
[00114] Синтетические пептидные амиды по изобретению могут быть введены в фармацевтические композиции. Композиции могут содержать эффективное количество синтетического пептидного амида в фармацевтически приемлемом разбавителе, вспомогательном веществе или носителе. Обычные вспомогательные вещества, носители и/или разбавители для применения в фармацевтических композициях в целом инертны и составляют большую часть препарата.
[00115] В конкретном варианте, синтетический пептидный амид представляет собой агонист каппа-опиатного рецептора. В другом варианте, синтетический пептидный амид представляет собой селективный агонист каппа-опиатного рецептора. Сайт-мишень может быть рецептором каппа у больного или субъекта, нуждающегося в таком лечении или профилактике. Определенные агонисты каппа-опиатного рецептора - синтетические пептидные амиды по изобретению - действуют периферически и демонстрируют слабо выраженное или отсутствия влияние на ЦНС в терапевтически эффективных дозах.
[00116] Фармацевтическое вспомогательное вещество или носитель может быть любой подходящей совместимой, нетоксичной субстанцией, такой как носитель для доставки синтетического пептидного амида по изобретению. Подходящие вспомогательные вещества или носители включают, не ограничиваясь ими, стерильную воду (предпочтительно, апирогенную), солевой раствор, буферизованный фосфатом солевой раствор (фосфатный буферный раствор), воду/этанол, воду/глицерин, воду/сорбитол, воду/полиэтиленгликоль, пропиленгликоль, цетилстеариловый спирт, карбоксиметилцеллюлозу, кукурузный крахмал, лактозу, глюкозу, микрокристаллическую целлюлозу, магния стеарат, поливинилпирролидон (ПВП), лимонную кислоту, винную кислоту, масла, липидные субстанции, воски или подходящие смеси любых вышеназванных компонентов.
[00117] Фармацевтическая композиция по изобретению может представлять собой жидкую, полутвердую или твердую лекарственную форму. Например, фармацевтическая композиция может иметь форму раствора для инъекций, капель, сиропа, спрея, суспензии, таблетки, пластыря, капсулы, повязки, суппозитория, мази, крема, лосьона, геля, эмульсии, аэрозоля или сыпучую форму, например, пилюли или гранулы, необязательно, спрессованные в таблетки или пастилки, помещенные в капсулы или суспендированные в жидкости. Таблетки могут содержать связующие вещества, смазывающие вещества, разбавители, красящие вещества, ароматизаторы, увлажняющие вещества, и могут быть покрыты кишечно-растворимой оболочкой, чтобы проходить неповрежденными через кислую среду желудка и растворяться в более щелочных условиях просвета кишечника. Альтернативно, таблетки могут быть покрыты сахарной оболочкой или пленочной оболочкой из водорастворимой пленки. Фармацевтически приемлемые адъюванты, буферные агенты, диспергирующие агенты и т.п., также могут быть включены в фармацевтические композиции.
[00118] Связующие вещества включают, например, крахмал, слизь, желатин и сахарозу. Смазывающие вещества включают тальк, ликоподий, магний и кальция стеарат/стеариновую кислоту. Разбавители включают лактозу, сахарозу, маннитол, соль, крахмал и каолин.
Увлажняющие агенты включают пропиленгликоль и сорбитана.
[00119] В данном описании, местное нанесение или введение обозначает введение фармацевтической композиции по изобретению в/на участок, такой воспаленный сустав, который демонстрирует болезненное и/или воспаленное состояние. Такое местное введение включает внутрисуставное, например, внутрисуставное нанесение, путем инъекции, введение через катетер или доставку в составе биосовместимого устройства. Таким образом, местное введение обозначает введение в дискретную внутреннюю область организма, такую как сустав, участок мягких тканей (например, мышца, сухожилие, связка, внутриглазные или другие внутренние области мягких тканей) или другой внутренний участок тела. В частности, в данном описании местное введение обозначает введение, которое не приводит в существенной мере к системному введению и/или системной доставке активных агентов в настоящих композициях. Также, в данном описании местное введение предназначено обозначать введение в дискретные участки тела, кроме различных крупных полостей тела (например, брюшная и/или плевральная полость).
[00120] В данном описании, местное нанесение обозначает нанесение на поверхность тела, такую как кожа, глаза, слизистая оболочка и губы, которая может располагаться в или на какой-либо части тела, в том числе, не ограничиваясь ими, эпидерма, любой другой слой кожи или любая другая ткань организма. Местное введение или нанесение подразумевает прямой контакт фармацевтической композиции по изобретению с тканью, такой как кожа или мембрана, особенно роговая оболочка или слизистая оболочка рта, влагалища или аноректальной области. Таким образом, для целей данного описания местное нанесение обозначает нанесение на ткани доступной поверхности тела, такой как кожа (внешняя оболочка или поверхность) и слизистая оболочка (вырабатывающие, выделяющие и/или содержащие слизь поверхности). В частности, местное нанесение обозначает нанесение, которое не предполагает или в существенной мере не предполагает системной доставки активных соединений в настоящих композициях. Примеры поверхностей слизистой оболочки включают поверхность слизистой оболочки глаз, рта (например, губы, язык, десны, щеки, подъязычная область и небо), гортань, пищевод, бронхи, трахея, носовые ходы, влагалище и прямая кишка/задний проход.
[00121] В случае перорального введения, активный ингредиент может вводиться в твердых дозированных лекарственных формах, таких как капсулы, таблетки и порошки, или в жидких дозированных лекарственных формах, таких как эликсиры, сиропы и суспензии. Активный компонент(ы) может быть инкапсулирован в желатиновые капсулы вместе с неактивными ингредиентами и порошкообразными носителями, такими как глюкоза, лактоза, сахароза, маннитол, крахмал, целлюлоза или производные целлюлозы, магния стеарат, стеариновая кислота, натрия сахаринат, тальк, магния карбонат и т.п. Примеры дополнительных неактивных ингредиентов, которые могут быть добавлены для обеспечения желательного цвета, вкуса, стабильности, буферной емкости, дисперсии или других известных желательных признаков, - железа оксид красный, силикагель, натрий лаурилсульфат, титана диоксид, съедобные белые чернила и т.п. Подобные наполнители могут применяться для получения прессованных таблеток. Как таблетки, так и капсулы могут быть произведены в виде продуктов с пролонгированным высвобождением, чтобы обеспечить непрерывное высвобождение терапевтического средства на протяжении часов. Прессованные таблетки могут быть покрыты сахарной или пленочной оболочкой, чтобы замаскировать неприятный вкус и защитить таблетку от воздействия атмосферы, или кишечно-растворимой оболочкой для селективного распада в желудочно-кишечном тракте. Жидкие лекарственные формы для перорального введения могут содержать красители и ароматизаторы, чтобы улучшить переносимость пациентом. Чтобы улучшить стабильность и абсорбцию лекарственного средства, пептиды по изобретению могут высвобождаться из капсулы после прохождения через жесткую протеолитическую среду желудка. Способы повышения стабильности и абсорбции пептидов после перорального введения хорошо известны из уровня техники (например, Mahato RI. Emerging trends in oral delivery of peptide and protein drugs. Critical Reviews in Therapeutic Drug Carrier Systems. 20:153-214, 2003).
[00122] Такие дозированные лекарственные формы, как пастилки, жевательные таблетки и жевательная резинка, обеспечивают быстрый терапевтический эффект по сравнению с дозированными лекарственными формами соединений синтетических пептидных амидов по изобретению для перорального введения, демонстрирующими значительную буккальную абсорбцию. Препараты в форме жевательной резинки представляют собой твердые, однодозовые препараты с основой, состоящей в основном из резины, которые предназначены для жевания, но не предназначены для глотания и содержат одно или больше соединений по изобретению, которые высвобождаются при жевании и предназначены для местного лечения боли и воспаления в ротовой полости или системной доставки после абсорбции через буккальную слизистую оболочку. См., например, патент США 6,322,828, выданный Athanikar и Gubler под названием «Способ производства фармацевтической жевательной резинки».
[00123] Для назального введения, селективные агонисты каппа-опиатного рецептора периферического действия могут быть введены в аэрозоли. Термин "аэрозоль" включает любую суспендированную в газообразной среде фазу соединений по настоящему изобретению, которую можно вдыхать в бронхиолы или носовые ходы. Конкретно, аэрозоль включает суспензию в газообразной фазе капелек соединений по настоящему изобретению, которая может быть произведена дозирующим ингалятором или небулайзером или мелкокапельным распылителем. Аэрозоль также включает сухую порошковую композицию соединения по настоящему изобретению, суспендированную в воздухе или другом газе-носителе, которая может быть введена вдуванием из ингаляторного устройства, например, см. Ganderton & Jones, Drug Delivery to the Respiratory Tract, Ellis Horwood (1987); Gonda (1990) Cr.it.ica2 Reviews in Therapeutic Drug Carrier Systems 6:273-313; и Raeburn et al. (1992) J. Pharmacol. Toxicol. Methods 27:143-159.
[00124] Фармацевтические композиции изобретения могут быть приготовлены в форме, подходящей для системного введения, например, внутривенного, подкожного, внутримышечного, внутрибрюшинного, интраназального, трансдермального, интравагинального, ректального, внутрилегочного или перорального введения. Альтернативно, фармацевтические композиции по изобретению могут быть подходящими для местного введения, например, для местного или ионофоретического введения или для трансдермального введения с помощью пластыря, покрытого или пропитанного препаратом, и местного введения в суставы, например, внутрисуставной инъекции.
[00125] Препараты для парентерального введения включают стерильные растворы, готовые для инъекций, стерильные сухие растворимые продукты, готовые для разбавления растворителем непосредственно перед применением, в том числе таблетки для подкожного введения, стерильные суспензии, готовые для инъекций, стерильные, сухие нерастворимые продукты, готовые для разбавления растворителем непосредственно перед применением, и стерильные эмульсии. Растворы могут быть водными или неводными и, таким образом, предназначены для введения инъекцией, инфузией или с применением имплантируемых насосов. Для внутривенного, подкожного и внутримышечного введения пригодные препараты по изобретению включают препараты микрокапсул со свойствами контролируемого высвобождения (R. Pwar et al. Protein and peptide parenteral controlled delivery. Expert Opin Biol Ther. 4(8):1203-12, 2004) или инкапсулированием в липосомах, например, в покрытых полиэтиленом липосомах, которые известны из уровня техники как обеспечивающие удлиненный период нахождения в кровотоке (например, Koppal, Т. "Drug delivery technologies are right on target". Drug Discov. Dev. 6, 49-50, 2003).
[00126] Для введения в глаза, настоящее изобретение предлагает способ лечения глаукомы или боли и воспаления глаз, включающий введение в глаз больного, нуждающемся в таком лечении, терапевтически эффективного количества синтетических пептидных амидов по изобретению. Синтетические пептидные амиды можно применять местно с приемлемым для глаз фармацевтическим носителем или в форме не системного применения в виде контактной линзы или внутриглазного имплантанта, который может необязательно содержать полимеры, которые обеспечивают пролонгированное высвобождение синтетических пептидных амидов. Такие приемлемые для глаз фармацевтические носители могут включать адъюванты, противомикробные консерванты, поверхностно-активные вещества, агенты для повышения вязкости и т.п. Из уровня техники известно, что высокие концентрации многих соединений раздражают глаз, и низкие концентрации оказывают меньшее раздражающее действие; поэтому препарат часто разрабатывают таким образом, чтобы включить самые низкие эффективные концентрации активного соединения, консерванта, поверхностно-активного вещества и/или агента для повышения вязкости, где указанный агент для повышения вязкости предпочтительно обладает высоким поверхностным напряжением, чтобы уменьшить раздражение глаз при увеличении удержания глазных растворов на поверхности глаз. Такое контролируемое высвобождение синтетических пептидных амидов по изобретению может продолжаться для имплантантов от 6 месяцев до года, или в течение более коротких периодов (3-14 дней) для контактных линз. Такие имплантанты могут представлять собой осмотические насосы, поддающиеся биологическому разложению матрицы или внутриглазные устройства пролонгированного высвобождения. Такие композиции для местного применения могут включать буферизованный солевой раствор, содержащий или не содержащий липосомы.
[00127] Водные растворы полимеров, водные суспензии, мази и гели могут использоваться для препаратов синтетических пептидных амидов по изобретению, предназначенных для местного применения в глаза. Водные препараты также могут содержать липосомы для создания резервуара синтетических пептидных амидов. Некоторые из таких местных препаратов представляют собой гели, которые увеличивают удержание на роговице без неудобств и нарушений зрения, связанного с мазями. Совместимый с глазом фармацевтический носитель может также включать поддающийся биологическому разложению синтетический полимер. Поддающиеся биологическому разложению композиции микросфер, зарегистрированные для применения у человека, включают полилактиды: поли (молочную кислоту), поли(гликолевую кислоту) и поли(молочную-согликолевую) кислоту. Дополнительные поддающиеся биологическому разложению препараты включают, не ограничиваясь ими: поли (ангидрид-со-имид), поли(молочную гликолевую кислоту), полиэтил-2-цианоакрилат, поликапролактон, полигидроксибутирата валерат, полиортоэфир и полиэтиленоксид/полибутилен терефталат.Внутриглазной имплантат или инъекция композиций с пролонгированным высвобождением, которые содержат синтетические пептидные амиды по изобретению, может обеспечить длительный контроль (варьирует от месяцев до лет) внутриглазного давления и, таким образом, устранить или уменьшить необходимость в местных препаратах. Пригодные способы введения в препараты и отпуска глазных лекарственных форм раскрыты в патенте США 7,122,579, выданном Schwartz и др., и в патенте США 7,105,512, выданном Monzono и др. Способы введения глазных лекарственных форм в контактные линзы раскрыты в Gulsen and Chauhan, Ophthalmic drug delivery through contact lenses. Investigative Ophthalmology and Visual Science, (2004) 45:2342-2347.
[00128] Препараты для трансдермального введения вводят в устройство, подходящее для такого введения, где в указанном устройстве используют, например, ионофорез (Kalia YN et al. lontophoretic Drug Delivery. Adv Drug Deliv Rev. 56:619-58, 2004) или поверхность, проникающую сквозь дерму (Prausnitz MR. Microneedles for Transdermal Drug Delivery. Adv Drug Deliv Rev. 56:581-7, 2004), такие как известны из уровня техники, которые пригодны для улучшения трансдермальной доставки лекарственных средств. Устройство электропереноса и способы его применения раскрыты в патенте США 6,718,201. Способы применения ионофореза для содействия трансдермальной доставке пептидов раскрыты в патенте США 6,313,092 и патенте США 6,743,432.
[00129] В данном описании термины "электроперенос", "ионофорез" и "ионофоретический" обозначают введение (доставку) через поверхность тела (например, кожи или слизистой оболочки) одного или больше фармацевтически активных соединений с помощью приложения электродвижущей силы к резервуару, содержащему агент. Соединение может быть доставлено электромиграцией, электропорацией, электроосмосом или любой их комбинацией. Электроосмос также носит название электрогидрокинеза, электроконвекции и электрически индуцированного осмоса. В целом, электроосмос соединения в ткани является результатом миграции растворителя, в котором содержится соединение, в результате приложения электродвижущей силы к резервуару терапевтического соединения, например, потока растворителя, индуцированного электромиграцией других ионов. В процессе электропереноса, могут происходить определенные модификации или изменения кожи, такие как образование временных пор в коже, что также называется "электропорацией". Любой носитель веществ с помощью электричества, усиленный модификациями или изменениями поверхности тела (например, образование пор в коже), также охватывается термином "электроперенос" в данном описании. Таким образом, в данном описании, применительно к соединениям по настоящему изобретению, термины "электроперенос", "ионофорез" и "ионофоретический" обозначают (1) доставку заряженных агентов электромиграцией, (2) доставку незаряженных агентов в процессе электроосмоса, (3) доставку заряженных или незаряженных агентов электропорацией, (4) доставку заряженных агентов объединенными процессами электромиграции и электроосмоса и/или (5) доставку смеси заряженных и незаряженных агентов объединенными процессами электромиграции и электроосмоса. В устройствах электропереноса в целом используются два электрода, оба из которых размещены в тесном электрическом контакте с некоторой частью кожи. Один электрод, который называется активным или донорным электродом, представляет собой электрод, из которого терапевтический агент доставляется в тело. Другой электрод, которые называется счетчиком или возвратным электродом, служит для замыкания электрического контура через тело. В соединении с кожей пациента, контур замыкается подсоединением электродов к источнику электрической энергии, например, аккумулятору, и обычно к контуру, способному управлять текущим прохождением через устройство.
[00130] В зависимости от электрического заряда соединения для трансдермальной доставки, анод или катод может представлять собой активный или донорский электрод. Таким образом, если соединение, которое подлежит переносу, заряжено положительно, например, соединение, приведенное в качестве примера в Примере 1 данного описания, то положительный электрод (анод) будет активным электродом, и отрицательный электрод (катод) будет служить возвратным электродом, замыкая контур. Однако, если соединение для доставки заряжено отрицательно, то катодный электрод будет активным электродом, и анодный электрод будет возвратным электродом. Устройства электропереноса дополнительно требуют резервуара или источника терапевтического агента, который подлежит доставке в организм. Такие резервуары лекарственного средства соединяют с анодом или катодом устройства электропереноса, чтобы обеспечить фиксированный или возобновляемый источник одного или больше желательных веществ или агентов. Каждый электродный узел состоит из электрически проводящего электрода, связанного проведением ионом с проводящим ионы жидкостным резервуаром, который в ходе применения размещен таким образом, чтобы контактировать с кожей больного. Резервуары геля, такие как описаны Webster (патент США 4,383,529) представляют собой одну из форма резервуара, поскольку гидратированные гели удобнее в обращении и производстве, чем заполненные жидкостью контейнеры. Вода представляет собой один из жидких растворителей, который может применяться в таких резервуарах, отчасти потому что соли пептидных соединений по изобретению водорастворимы, и отчасти, потому что вода не раздражает кожу, таким образом, позволяя длительный контакт между резервуаром гидрогеля и кожей. Для электропереноса, синтетические пептиды по изобретению могут быть введены в препарат с усиливающими текучесть веществами, такими как ионные поверхностно-активные вещества или сорастворители, кроме воды (см., например, патент США 4,722,726 и Европейскую патентную заявку 278,473, соответственно). Альтернативно, внешний слой кожи (т.е. stratum corneinn) может быть механически разрушен до введения электропереносом через кожу, например, как описано в патенте США 5,250,023.
[00131] Синтетические пептидные амиды периферического действия, которые хорошо подходят для электропереноса, могут быть выбраны измерением их тока в процессе электропереноса через поверхность тела (например, кожу или слизистую оболочку), например, по сравнению со стандартизированным тестовым пептидом с известными характеристиками тока электропереноса, например, тиреотропин-высвобождающий гормон (R.Burnette et al. J. Pharm. Sci. (1986) 75:738) или вазопрессин (Nair et al. Pharmacol Res. 48:175-82, 2003). Трансдермальный ток электропереноса может быть определен с использованием целого ряда способов in vivo или in vitro, хорошо известных из уровня техники. Способы in vitro включают зажимание куска кожи подходящего млекопитающего (например, кожи человеческого трупа) между донорным и рецепторным отсеком кюветы потока электропереноса, где сторона stratum corneum среза кожи обращена к донорному отсеку. Жидкий раствор или гель, содержащий лекарственное средство, которое подлежит доставке, размещают в контакте со stratum corneum, и электрический ток подают на электроды, один электрод в каждом отсеке. Трансдермальный ток вычисляют, отбирая образцы для количественного определения лекарственного средства в отсеке рецептора. Две успешные модели используют, чтобы оптимизировать трансдермальную доставку лекарственного средства электропереносом, - модель изолированного образца кожи свиньи (Heit МС et al. Transdermal iontophoretic peptide delivery: in vitro and in vivo studies with luteinizing hormone releasing hormone. J. Pharm. Sci. 82:240-243, 1993), и использование изолированной безволосой кожи безволосых грызунов или морских свинок, например, см. Hadzija BW et al. Effect of freezing on iontophoretic transport through hairless rat skin. J. Pharm. Pharmacol. 44, 387-390, 1992. Соединения по изобретению для трансдермальной ионофоретической доставки могут содержать один или обычно, два заряженных атома азота, чтобы облегчить доставку.
[00132] В других пригодных для трансдермальной доставки устройствах используют доставку с высокой скоростью под давлением, чтобы достичь проникновения кожи без применения иглы. Трансдермальная доставка может быть улучшена, как известно из уровня техники, применением химических усилителей, который в данной области иногда называют "усилителями проникания", т.е. соединений, которые вводят наряду с лекарственным средством (или в некоторых случаях используют, чтобы предварительно обработать кожу, до введения лекарственного средства) с целью увеличения проницаемости stratum corneum и, таким образом, обеспечивают повышенное проникновение лекарственного средства через кожу. Химические усилители проникания представляет собой соединения, которые нетоксичны и служат только для облегчения диффузии лекарственного средства через stratum corneuia, пассивной диффузией или энергетически управляемым процессом, таким как электроперенос. См., например, Meidan VM et al. Enhanced lontophoretic delivery of buspirone hydrochlonde across human skin using chemical enhancers. Int. J. Pharm. 264:73-83, 2003.
[00133] Фармацевтические лекарственные формы для ректального введения включают ректальные суппозитории, капсулы и таблетки для системного эффекта. Ректальные суппозитории в данном описании обозначают твердые тела для вставки в прямую кишку, которые тают или размягчаются при температуре тела, высвобождая один или больше фармакологически или терапевтически активных ингредиентов. Фармацевтически приемлемые субстанции, применяемые в ректальных суппозиториях, включают основы или носители и агенты, которые повышают температуру плавления суппозиториев. Примеры основ включают масло какао, глицерин-желатин, карбовоск (полиоксиэтиленгликоль) и подходящие смеси моно-, ди- и триглицеридов жирных кислот. Комбинации различных основ также могут использоваться. Агенты, которые повышают температуру плавления суппозиториев, включают спермацет и воск. Ректальные суппозитории могут быть приготовлены методом прессования или формованием. Масса ректальных суппозиториев обычно составляет приблизительно от 2 г до приблизительно 3 г. Таблетки и капсулы для ректального введения производят, используя такую же фармацевтически приемлемую субстанцию(и) и такие же способы, как и в случае препаратов для перорального введения.
[00134] Фармацевтически приемлемые носители, используемые в парентеральных препаратах, включают водный носитель, неводный носитель, противомикробные агенты, изотонические агенты, буферы, антиоксиданты, местно-анестезирующие средства, суспендирующие и диспергирующие агенты, эмульгаторы, секвестранты или хелатные агенты и другие фармацевтически приемлемые субстанции.
[00135] Примеры водного носителя включают раствор натрия хлорида для инъекций, раствор Рингера для инъекций, изотонической раствор глюкозы для инъекций, стерильную воду для инъекций, раствор глюкозы и раствор Рингера с лактатом для инъекций. Неводный парентеральный носитель включает нелетучие масла растительного происхождения, хлопковое масло, кукурузное масло, кунжутное масло и арахисовое масло. Противомикробные агенты в бактериостатических или фунгистатических концентрациях должны быть добавлены в парентеральные препараты, упакованные в многодозовые контейнеры, что включает фенолы или крезолы, соединения ртути, бензиловый спирт, хлорбутанол, метиловый и пропиловый эфиры п-гидроксибензойной кислоты, тимеросал, бензалкония хлорид и бензэтония хлорид. Изотонические агенты включают натрия хлорид и глюкозу. Буферные вещества включают фосфат и цитрат. Антиоксиданты включают натрия бисульфит. Местноанестезирующие средства включают прокаина гидрохлорид. Суспендирующие и диспергирующие агенты включают натрий карбоксиметилцеллюлозу, гидроксипропилметилцеллюлозу и поливинилпирролидон. Эмульгаторы включают полисорбат 80 (Твин 80). Секвестранты или хелатные агенты для ионов металла, такие как ЭДТА, также могут быть введены. Фармацевтические носители также включают этиловый спирт, полиэтиленгликоль и пропиленгликоль для смешиваемых с водой носителей, и значение рН может быть доведено до физиологически совместимых значений добавлением натрия гидроксида, кислоты хлористоводородной, лимонной кислоты или молочной кислоты.
[00136] Активный ингредиент может вводиться в виде одной дозы или может быть разделен на целый ряд меньших доз, которые вводят с промежутками времени или в форме препарата контролируемого высвобождения. Термин «препарат контролируемого высвобождения» включает препараты, которые позволяют непрерывную доставку синтетических пептидных амидов по изобретению субъекту за период времени, например, от нескольких дней до недель. Такие препараты могут вводиться подкожно или внутримышечно и позволяют непрерывную беспрерывное постоянное высвобождение предварительно определенного количества соединения в организме субъекта на протяжении периода времени. Препарат синтетического пептидного амида с контролируемым высвобождением может представлять собой, например, препарат лекарственного средства, содержащий полимерные микрокапсулы, такие как описаны в патентах США №№4,677,191 и 4,728,721, включенных в данное описание путем ссылки. Осуществляют коррекцию концентрации фармацевтически активного соединения таким образом, что введение обеспечивает эффективное количество для обеспечения желательного эффекта. Точная доза зависит от возраста, массы тела и состояния больного или животного, как известно из уровня техники. Для любого конкретного субъекта, специфические схемы лечения могут быть скорректированы через какое-то время согласно индивидуальной потребности и профессиональному суждению лица, которое вводит или наблюдает за введением препаратов. Таким образом, интервалы концентрации, определенные в данном описании, приведены только для примера и не предназначены ограничивать контекст или практику заявленного изобретения.
[00137] Единица дозы парентерального препарата включают упаковку в ампулу или расфасовку в шприц с иглой или без нее, для введения. Все препараты для парентерального введения обычно стерильны, как практикуется в данной области. Для иллюстрации, внутривенная инфузия стерильного водного буферизованного раствора, содержащего активное соединение, представляет собой эффективный способ введения. В другом варианте, стерильный водный или масляный раствор или суспензия, содержащая активный материал, может быть введена инъекцией по мере необходимости, чтобы обеспечить желательный фармакологический эффект.
[00138] Фармацевтические композиции по изобретению могут быть доставлены или введены внутривенно, трансдермально, через слизистую оболочку, интраназально, подкожно, внутримышечно, перорально или местно (например, в глаз). Композиции можно вводить для профилактического лечения индивидуумов, страдающих от или подвергающихся риску заболевания или расстройства. Для терапевтического применения, фармацевтическую композицию обычно вводят субъекту, страдающему от заболевания или расстройства, в количестве, достаточном для подавления, предупреждения или облегчения заболевания или расстройства. Количество, адекватное для обеспечения такого воздействия, определено как "терапевтически эффективная доза."
[00139] Фармацевтические композиции по изобретению можно вводить млекопитающему для профилактических или терапевтических целей в виде любого из вышеописанных препаратов и способов введения. Млекопитающее может представлять собой любое млекопитающее, такое как домашнее или сельскохозяйственное млекопитающее или даже дикое млекопитающее. Млекопитающее может быть любым приматом, копытным животным, из семейства собачьих или кошачьих. Например, не ограничиваясь ими, млекопитающее может представлять собой домашнее животное, такое как собака или кот; ценное млекопитающее, такое как чистокровный конь или выставочное животное; сельскохозяйственное животное, такое как корова, коза, овца или свинья; или примат, такой как обезьяна, горилла, орангутанг, лемур, обезьяна или шимпанзе. Подходящим млекопитающим для профилактики или лечения с применением фармацевтических композиций по изобретению, является человек.
[00140] Фармацевтические композиции по изобретению можно вводить млекопитающему, у которого присутствует заболевание или состояние, поддающееся лечению активацией каппа-опиатного рецептора. Альтернативно, фармацевтические композиции можно вводить в составе профилактических способов млекопитающему, подвергающемуся риску приобретения или развития заболевания или состояния, поддающегося профилактике активацией каппа-опиатного рецептора. Заболевания или состояния, которые поддаются лечению или профилактике введением фармацевтических композиций по изобретению, включают, не ограничиваясь ими, любое состояние, которое может быть облегчено активацией каппа-опиатного рецептора, в том числе такие состояния как боль, воспаление, зуд, гипонатриемия, гипокалиемия, застойная сердечная недостаточность, цирроз печени, нефротический синдром, артериальная гипертензия, отек, непроходимость кишечника, кашель и глаукома.
[00141] В конкретном варианте, фармацевтические композиции по изобретению могут вводиться совместно или могут включать один или больше других терапевтических соединений или адъювантов, такие как, не ограничиваясь ими, другие опиоиды, каннабиноиды, антидепрессанты, противосудорожные препараты, нейролептики, антигистаминные средства, ацетаминофен, кортикостероиды, блокаторы ионных каналов, нестероидные противовоспалительные лекарственные препараты (НПВП) и диуретики, многие из которых действуют синергетически с синтетическими пептидными амидами по изобретению.
[00142] Подходящие опиаты включают, не ограничиваясь ими, алфентанил, альфародин, анилеридин, бремазоцин, бупренорфин, буторфанол, кодеин, конорфон, декстроморамид, декстропропоксифен, дезоцин, диаморфин, дигидрокодеин, дигидроморфин, дифеноксилат, дипипанон, докспикомин, этогептазин, этилкетазоцин, этилморфин, эторфин, фентанил, гидрокодон, гидроморфон, кетобемидон, левометадил, леворфанол, лофентанил, лоперамид, меперидин (петидин), мептазинол, метадон, морфин, морфин-6-глюкуронид, налбуфин, налорфин, никоморфин, оксикодон, оксиморфон, пентазоцин, феназоцин, феноперидин, пиритрамид, пропирам, пропоксифен, ремифентанил, суфентанил, тилидат, тоназоцин и трамадол.
[00143] Один из вариантов изобретения представляет собой комбинированный препарат и/или совместное введение опиатного с выраженной активностью агониста по отношению к мю-опиатному рецептору, такого как морфин, фентанил, гидроморфон или оксикодон, вместе с синтетическими пептидными амидами по изобретению, с целью снижения дозы мю-опиоида, где дозу мю-опиоида снижают для минимизации общих побочных эффектов мю-опиоидов, особенно у больных, не получавших ранее опиоиды. Такие побочные эффекты включают запор, тошноту, рвоту, седацию, угнетение дыхания, зуд, спутанность мыслей, дезориентацию и нарушение когнитивной функции, задержку мочи, спазм желчных путей, бред, миоклонические рывки и припадки. Выбор сниженной дозы мю-опиоида требует суждения клинического эксперта и зависит от уникальных характеристик различных мю-опиоидов, а также от характеристик пациента, таких как интенсивность боли, возраст пациента, сопутствующее заболевание, текущая схема лечения и потенциальное межмедикаментозное взаимодействие, результаты предшествующего лечения и предпочтения пациента (McCaffery, M. and Pasero, С., Pain Clinical Manual, Second Edition, Mosby, 1999).
[00144] Каннабиноиды, подходящие для совместного введения или введения в фармацевтические композиции по изобретению, включают любой природный каннабиноид, например, тетрагидроканнабинол (ТГК) или производное ТГК, или синтетический каннабиноид, например, левонантрадол, маринол, набилон, римонабант или савитекс.
[00145] Антидепрессанты, подходящие для совместного введения или введения в фармацевтические композиции по изобретению, включают, например, трициклические антидепрессанты, такие как имипрамин, дезипрамин, тримипрамин, протриптилин, нортриптилин, амитриптилин, доксепин и кломипрамин; атипичные антидепрессанты, такие как амоксапин, мапротилин, тразодон, бупропион и венлафаксин; специфические ингибиторы обратного захвата серотонина, такие как флуоксетин, сертралин, пароксетин, циталопрам и флувоксамин; специфические ингибиторы обратного захвата норадреналина, такие как ребоксетин; или антидепрессанты двойного действия, такие как нефазодон и миртазапин.
[00146] Нейролептики, пригодные для совместного введения или введения в фармацевтические композиции по изобретению: включают любой нейролептик, например, соединение с активностью антагониста рецептора дофамина D2, такое как домперидон, метоклопрамид, левосульпирид, сульпирид, триэтилперазин, зипрасидон, зотепин, клозапин, хлорпромазин, ацетофеназин, карфеназин, хлорпротиксен, флуфеназин, локсапин, мезоридазин, молиндон, перфеназин, пимозид, пиперацетазин, перхлорперазин, тиоридазин, тиотиксен, трифлуоперазин, трифлупромазин, пипамперон, амперозид, кветиапин, мелперон, ремоксиприд, галоперидол, рисперидон, оланзапин, сертиндол, зипрасидон, амисульприд, прохлорперазин и тиотиксен.
[00147] Противосудорожные препараты, такие как фенобарбитал, фенитоин, примидон, карбамазепин, этосукцимид, ламотриджин, вальпроевая кислота, вигабатрин, фелбамат, габапентин, леветирацетам, окскарбазепин, ремацемид, тиагабин и топирамат могут также подходящим образом быть введены в фармацевтические композиции по изобретению.
[00148] Миорелаксанты, такие как метокарбамол, орфенадрин, каризопродол, мепробамат, хлорфенезин карбамат, диазепам, хлордиазепоксид и хлорзоксазон; противомигренозные агенты, такие как суматриптан, аналептики, такие как кофеин, метилфенидат, амфетамин и модафинил; антигистаминные средства, такие как хлорфенирамин, ципрогептадин, прометазин и пириламин, в также кортикостероиды, такие как метилпреднизолон, бетаметазон, гидрокортизон, преднизолон, кортизон, дексаметазон, преднизон, алклометазон, клобетазол, клокортолон, десонид, дезоксиметазон, дифлоразон, флуоцинолон, флуоцинонид, флурандренолид, флутиказон, фторметалон, галцинонид, галобетазол, лотепреднол, мометазон, предникарбат и триамцинолон, также могут быть введены в фармацевтические композиции по изобретению.
[00149] Блокаторы ионных каналов, такие как, блокатор натриевых каналов, карбамазепин, которые обычно применяют для лечения звона в ушах, аритмии, ишемического инсульта и эпилепсии, можно вводить совместно или вводить в фармацевтические композиции по изобретению. Альтернативно или дополнительно, блокаторы кальциевых каналов, такие как зиконотид, также могут применяться, как и антагонисты ионных каналов, связанных с рецептором NMDA, такие как кетамин. Существуют доказательства того, что как минимум некоторые из таких блокаторов ионных каналов могут потенцировать анальгетический эффект агониста каппа и, таким образом, уменьшать дозу, необходимую для эффективного облегчения боли. См., например, Wang et al., 2000, Pain 84: 271-81.
[00150] Подходящие НПВП, или другие неопиатные соединения с противовоспалительной и/или болеутоляющей активностью, которые можно вводить совместно или вводить в фармацевтические композиции по изобретению, включают, не ограничиваясь ими, одно или больше из следующего перечня: производные аминоарилкарбоновой кислоты, такие как этофенамат, меклофенамовая кислота, мефенамовая кислота, нифлумовая кислота; производные арилуксусной кислоты, такие как ацеметацин, амфенак, цинметацин, клопирак, диклофенак, фенклофенак, фенклорак, фенклозик, фентиазак, глюкаметацин, изоксепак, лоназолак, метиазиновая кислота, напроксин, оксаметацин, проглуметацин, сулиндак, тиарамид и толметин; производные арилмасляной кислоты, такие как бутибуфен и фенбуфен; арилкарбоновой кислоты, такие как клиданак, кеторолак и тиноридин. производные арилпропионовой кислоты, такие как, карпрофен, фенопрофен, флуноксапрофен ибупрофен, ибупроксам, оксапрозин, производные фенилалкановой кислоты, такие как флурбипрофен, пикетопрофен, пирпрофен, пранопрофен, протизинова кислота и тиапрофеновая кислота; пиранкарбоновой кислоты, такие как этодолак; производные пиразола, такие как мепиризол; пиразолоны, такие как клофезон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон, фенилпиразолидиноны, суксибузон и тиазолинобутазон; производные салициловой кислоты, такие как аспирин, бромсалигенин, дифлунизал, фендосал, гликольсалицилат, месаламин, 1-нафтилсалицилат, магния салицилат, олсалазин и салициламид, сальсалат и сульфасалазин; тиазинкарбоксамиды, такие как дроксикам, изоксикам и пироксикам, другие, такие как 8-ацетамидокапроновая кислота, ацетаминофен, s-аденозилметионин, 3-амино-4-гидроксимасляная кислота, амиксетрин, бендазак, буколом, карбазоны, кромолин, дифенпирамид, дитазол, гидроксихлорохин, индометацин, кетопрофен и его активный метаболита 6-метокси-2-нафтилуксусная кислота; гуайазулен, гетероциклические аминоалкильные эфиры микофеноловой кислоты и ее производных, набуметон, нимесулид, орготеин, оксацепрол, производные оксазола, паранилин, пифоксим, 2-замещенные 4,6-ди-трег-бутил-s-гидрокси-1,3-пиримидины, проквазон и тенидап, а также ингибиторы ЦОГ-2 (циклооксигеназа II), такие как целекоксиб или рофекоксиб.
[00151] Подходящие диуретики, которые можно вводить совместно или вводить в фармацевтические препараты по изобретению, включают, например ингибиторы карбоангидразы, такие как ацетазоламид, дихлорфенамид и метазоламид; осмотические диуретики, такие как глицерин, изосорбид, маннитол и мочевина; ингибиторы Na+-K+-2Cl--симпорта (петлевые диуретики или диуретики, действующие в верхних отделах), такие как фуросемид, буметанид, этакриновая кислота, торсемид, аксосемид, пиретанид и трипамид; ингибиторы Na+-Cl--симпорта (тиазидные и тиазидоподобные диуретики), такие как бендрофлуметазид, хлортиазид, гидрохлортиазид, гидрофлуметазид, метиклотиазид, политиазид, трихлорметиазид, хлорталидон, индапамид, метолазон и хинетазон; и, кроме того, ингибиторы почечных эпителиальных каналов Na+, такие как амилорид и триамтерен, и антагонисты минералокортикоидных рецепторов (антагонисты альдостерона), такие как спиронолактон, канренон, калия канреноат и эплеренон, которые вместе также классифицируются как диуретики К+-сберегающие. Один из вариантов представляет собой комбинированный препарат и/или совместное введение петлевого и тиазидного диуретика вместе с синтетическими пептидными амидами по изобретению с целью снижения дозы петлевого или тиазидного диуретика, где доза петлевого или тиазидного диуретика снижена для минимизации нежелательной задержки воды и предупреждения или уменьшения выраженности гипонатриемии, особенно в контексте застойной сердечной недостаточности, а также других медицинских состояний, при которых уменьшение задержки жидкости в организме и нормализация натриевого баланса могло бы быть полезно больному, нуждающемуся в таком лечении. См. R М Reynolds et al. Disorders of sodium balance Brit. Med. J. 2006; 332:702-705.
[00152] Связанная с каппа-опиатным рецептором гипонатриемия может представлять собой какое-либо заболевание или состояние, при котором присутствует гипонатриемия (низкое содержание натрия), например, у человека, если концентрация натрия в плазме снижается ниже 135 ммоль/л, нарушение, которое может развиваться отдельно, или, чаще, как осложнение других медицинских состояний или как следствие применения терапии, которая может вызвать истощение натрия.
[00153] Другой вариант представляет собой комбинированный препарат и/или совместное введение калий-сберегающего диуретика, например, антагониста рецептора минералокортикоида, такого как спиронолактон или эплеренон, вместе с синтетическими пептидными амидами по изобретению, с целью снижения дозы указанного калий-сберегающего диуретика, где доза указанного диуретика снижена с целью минимизации гиперкалиемии или метаболического ацидоза, например, у больных циррозом печени.
[00154] В конкретных вариантах, синтетические пептидные амиды по изобретению демонстрируют длительную продолжительность действия при введении в терапевтически адекватных дозах in vivo. Например, в некоторых вариантах, синтетические пептидные амиды по изобретению при введении млекопитающему в дозе 3 мг/кг синтетического пептидного амида сохраняют как минимум приблизительно 50% максимальной эффективности в анализе, зависимом от каппа-опиатного рецептора через 3 часа после введения. В некоторых других вариантах, синтетические пептидные амиды по изобретению при введении млекопитающему в дозе 0,1 мг/кг синтетического пептидного амида сохраняют как минимум приблизительно 50% максимальной эффективности в анализе, зависимом от каппа-опиатного рецептора через 3 часа после введения. Максимальная эффективность оперативно определяется как самый высокий уровень эффективности, определенный для конкретного анализа, зависимого от каппа-опиатного рецептора для всех исследуемых агонистов.
[00155] В определенных вариантах, синтетические пептидные амиды по изобретению при введении млекопитающему в дозе 0,1 мг/кг сохраняют как минимум приблизительно 75% максимальной эффективности через 3 часа после введения. В других вариантах, синтетические пептидные амиды по изобретению при введении млекопитающему в дозе 0,1 мг/кг сохраняют как минимум приблизительно 90% максимальной эффективности через 3 часа после введения. В других вариантах, синтетические пептидные амиды по изобретению при введении млекопитающему в дозе 0,1 мг/кг сохраняют как минимум приблизительно 95% максимальной эффективности в почтовых введениях трех часов.
[00156] В изобретении дополнительно предлагается способ лечения или предупреждения связанного с каппа-опиатными рецепторами заболевания или состояния у млекопитающего, где способ включает введение млекопитающему композиции, содержащей эффективное количество синтетического пептидного амида по изобретению. Млекопитающее может быть любым млекопитающим, таким как домашнее или сельскохозяйственное млекопитающее, или даже дикое млекопитающее. Альтернативно, млекопитающее может быть приматом, копытным животным, собачьим или кошачьим. Например, не ограничиваясь ими, млекопитающее может представлять собой домашнее животное, ценное млекопитающее, такое как чистокровное животное или выставочное животное; сельскохозяйственное животное, такое как корова, коза, овца или свинья; или примат, такой как обезьяна или мартышка. В одном конкретном аспекте, млекопитающее представляет собой человека.
[00157] Эффективное количество может быть определено средним специалистом в данной области согласно шаблонным способам. Например, эффективное количество может быть определено как единица дозы, достаточная для предупреждения или лечения связанного с рецептором каппа заболевания или состояния у млекопитающего. Альтернативно, эффективное количество может быть определено как количество, достаточное, чтобы приблизиться к концентрации EC50, или количество, достаточное, чтобы приблизиться к величине, в 2 или 3 или приблизительно до 5 или даже приблизительно до 10 раз превышающей концентрацию EC50 в терапевтически адекватной жидкости биологического происхождения млекопитающего, например, если жидкость биологического происхождения находится в прямом контакте с тканью-мишенью, например, синовиальная жидкость воспаленного сустава у пациента, страдающего от ревматоидного артрита.
[00158] В одном варианте синтетический пептидный амид по изобретению представляет собой фармацевтическую композицию, которая содержит эффективное количество синтетического пептидного амида по изобретению и фармацевтически приемлемое вспомогательное вещество или носитель. В одном аспекте, фармацевтическая композиция содержит синтетические пептидные амиды по изобретению в количестве, эффективном для лечения или предупреждения связанного с каппа-опиатным рецептором состояние у млекопитающего, такого как человек. В другом аспекте связанное с каппа-опиатным рецептором состояние представляет собой боль, воспаление, зуд, отек, непроходимость кишечника, кашель или глаукому.
[00159] В одном варианте фармацевтическая композиция по изобретению дополнительно содержит одно или больше следующих соединений: опиоид, каннабиноид, антидепрессант, антиконвульсант, нейролептик, кортикостероид, блокатор ионных каналов или нестероидный противовоспалительный лекарственный препарат (НПВП).
[00160] Фармацевтические композиции, которые содержат синтетические пептидные амиды по изобретению и фармацевтически приемлемый растворитель или носитель могут применяться, для лечения или предупреждения одного или больше разнообразных связанных с каппа-опиатным рецептором заболеваний, расстройств или состояний.
[00161] Связанное с каппа-опиатным рецептором заболевание, расстройство или состояние, поддающееся профилактике или поддающееся лечению синтетическими пептидными амидами по изобретению, может быть любым связанным с каппа-опиатным рецептором состоянием, в том числе, не ограничиваясь ими, острой или хронической болью, воспалением, зудом, гипонатриемией, отеком, непроходимостью кишечника, кашлем и глаукомой. Например, связанная с каппа-опиатным рецептором боль может быть невропатической болью, соматической болью, висцеральной болью или кожной болью. Некоторые заболевания, расстройства или состояния связаны более чем с одной формой боли, например послеоперационная боль может включать любой или все из невропатического, соматического, висцерального и кожного компонентов боли, в зависимости от вида и протяженности хирургической процедуры.
[00162] Связанное с каппа-опиатным рецептором воспаление может быть любым воспалительным заболеванием или состоянием, в том числе, не ограничиваясь ими, синусит, ревматоидный артрит, теносиновиит, бурсит, тендонит, латеральный эпикондилит, адгезивный капсулит, остеомиелит, остеоартритное воспаление, воспалительное заболевание кишечника (IBD), синдромом раздраженного кишечника (IBS), воспаление глаз, воспаление уха или воспаление аутоиммунного генеза.
[00163] Связанный с каппа-опиатным рецептором зуд может представлять собой любое заболевание или состояние, связанное с зудом, например, зуд глаз, например, связанный с конъюнктивитом, зуд ушей, зуд: связанный с терминальной стадией почечного заболевания, при котором многие больные получают почечный диализ, и другие формы холестаза, в том числе первичный билиарный цирроз, внутрипеченочный холестаз при беременности, хроническое холестатическое заболевание печени, уремия, злокачественный холестаз, желтуха, а также дерматологическое состояние, такое как экзема (дерматит), в том числе атопический или контактный дерматит, псориаз, истинная полицитемия, красный плоский лишай, простой хронический лишай, педикулез (вши), тиреотоксикоз, дермофития стопы, крапивница, чесотка, вагинит, анальный зуд, связанный с геморроем, зуд в результате укусов насекомых и индуцированный лекарственными средствами зуд, а также индуцированный мю-опиоидом зуд.
[00164] Связанный с каппа-опиатным рецептором отек может представлять собой любое связанное с отеком заболевание или состояние, такое как, отек в результате застойной сердечной недостаточности или синдрома ненадлежащей секреции антидиуретического гормона (АДГ).
[00165] Связанная с каппа-опиатным рецептором непроходимость кишечника может представлять собой любое связанное с непроходимостью кишечника заболевание или состояния, в том числе, не ограничиваясь ими, послеоперационная непроходимость кишечника или индуцированная опиоидами дисфункция кишечника.
[00166] Связанная с каппа-опиатным рецептором невропатическая боль может представлять собою любую невропатическую боль, например, невралгия тройничного нерва, боль диабетического происхождения, боль вирусного происхождения, например, боль, связанная с опоясывающим лишаем, индуцированная химиотерапией боль, боль при раковых заболеваниях, связанная с вторжением метастазов в нервную ткань, невропатическая боль, связанная с травматическим повреждением и хирургическими процедурами, а также виды головной боли, для которых предполагается невропатический компонент, например, мигрень.
[00167] Связанная с каппа-опиатным рецептором боль также включает боль в глазах, например, после фото-рефрактивной кератэктомии (PRK), разрыва глаз, перелома орбитального дна, химических ожогов, царапин или раздражения роговицы, или связанная с конъюнктивитом, язвами роговицы, склеритом вследствие опоясывающего лишая, эписклеритом, склерокератитом, офтальмическим герпесом, интерстициальным кератитом, острым иритом (воспаление радужной оболочки глаз), сухим кератоконъюнктивитом, орбитальным целлюлитом, орбитальной псевдоопухолью, пузырчаткой, трахомой или увеитом.
[00168] Связанная с каппа-опиатным рецептором боль также включает боль в горле, особенно связанную с воспалительными состояниями, такими как аллергический ринит, острый бронхит, обычная простуда, контактные язвы, поражение вирусом простого герпеса, инфекционный мононуклеоз, грипп, рак гортани, острый ларингит, острый некротизирующий язвенный гингивит, перитонзиллярный абсцесс, ожоги глотки, фарингит, грипп, ларингофарингит, острый синусит и тонзиллит.
[00169] Кроме того, связанная с каппа-опиатным рецептором боль может быть болью при артрите, почечнокаменной болезни, камнях в мочевых путях, камнях в желчных путях и болью при проходе камней по желчным путям, болью в результате спазмов матки, дисменореи, эндометриоза, мастита, диспепсии, послеоперационной болью (например, после аппендэктомии, открытого хирургического вмешательства в области ободочной и прямой кишки, лечения грыжи, простатэктомии, резекции ободочной кишки, гастрэктомии, спленэктомии, колестомии, колостомии, тазовой лапароскопии, перевязки труб, гистерэктомии, вазэктомии или холецистэктомии), болью после медицинских процедур (например, после колоноскопии, цистоскопии, гистероскопии или затылочного или биопсии шейки матки или эндометрия), ушной болью, прорывной болью при раковых заболеваниях и болью, связанной с расстройством ЖКТ, таким как воспалительное заболевание кишечника или синдром раздраженного кишечника или другие воспалительные состояния, особенно внутренних органов (например, гастроэзофагеальная рефлюксная болезнь, панкреатит, острый полинефрит, язвенный колит, острый пиелонефрит, холецистит, цирроз, абсцесс печени, воспаление печени, дуоденальная язва или язва желудка, эзофагит, гастрит, гастроэнтерит, колит, дивертикулит, непроходимость кишечника, киста яичника, воспалительное заболевание органов таза, перфорированная язва, перитонит, простатит, интерстициальный цистит) или контакт с токсичными агентами, такими как токсины насекомого, или лекарственными средствами, такими как салицилаты или НПВП.
[00170] В настоящем изобретении предлагается способ лечения или предупреждения связанного с каппа-опиатными рецепторами заболевания или состояния у млекопитающего, такого как человек, где способ включает введение млекопитающему композиции, содержащей эффективное количество синтетических пептидных амидов по изобретению. В другом варианте связанное с каппа-опиатным рецептором состояние представляет собой боль, воспаление (такое как воспаление при ревматоидном артрите, воспаление при остеоартрите, воспаление при воспалительном заболевании кишечника, воспаление при синдроме раздраженного кишечника, воспаление глаз, воспаление уха или воспаление аутоиммунного генеза), зуд (такой как атопический дерматит, связанный с почечным диализом зуд, зуд глаз, зуд ушей, зудом в результате укусов насекомых или индуцированным опиоидами зуд), отек, непроходимость кишечника, кашель или глаукома. В одном аспекте, боль представляет собой невропатическую боль (такую как невралгия тройничного нерва, мигрень, боль диабетического происхождения, боль вирусного происхождения, индуцированная химиотерапией боль или боль у больных метастатическим раком), соматическую боль, висцеральную боль или кожную боль. В другом аспекте боль представляет собой боль при артрите, боль при почечно-каменной болезни, боль в результате спазмов матки, дисменореи, эндометриоза, диспепсии, послеоперационную боль, боль после медицинских процедур, боль в глазах, боль в ушах, прорывную боль при раковых заболеваниях или боль, связанную с расстройством ЖКТ, таким как воспалительное заболевание кишечника или синдром раздраженного кишечника. В другом аспекте боль представляет собой боль, связанную с хирургическим вмешательством, где хирургическое вмешательство представляет собой лапароскопию тазовой области, перевязку труб, гистерэктомию и холецистэктомию. Альтернативно, боль может быть болью, связанной с медицинской процедурой, такой как колоноскопия, цистоскопия, гистероскопия или биопсия эндометрия. В конкретном аспекте, атопический дерматит может быть псориазом, экземой или контактным дерматитом. В другом специфическом аспекте, непроходимость кишечника представляет собой послеоперационную непроходимость кишечника или индуцированную опиоидами дисфункцию кишечника.
[00171] Связанная с каппа-опиатным рецептором боль включает гипералгезию, которая, как предполагается, вызвана изменениями в микроокружении периферического сенсорного окончания и развивается вторично к местному повреждению ткани. Повреждение (например, ссадины, ожоги) и воспаление ткани может существенно увеличивать возбудимость полимодальных ноцицепторов (волокна С) и механических рецепторов высокого порога (Handworker et al. (1991) Proceeding of the VIth World Congress on Pain, Bond et al., eds., Elsevier Science Publishers BV, pp.59-70; Schaible et al. (1993) Pain 55:5-54). Считается, что такое повышение возбудимости и чрезмерно усиленные реакции чувствительных афферентных окончаний лежит в основе гипералгезии, где реакция боли является результатом чрезмерно усиленной реакции на стимул. Значение гипералгезического состояния в состоянии боли после повреждения повторно продемонстрировано и, по-видимому, ответственно за основную часть состояния боли после повреждения/при воспалении. Woold et al. (1993) Anesthesia and Analgesia 77:362-79; Dubner et al. (1994) в Textbook of Pain, Melzack et al., eds., Churchill-Livingstone, London, pp.225-242.
[00172] В другом варианте связанное с каппа-опиатным рецептором состояние представляет собой боль, воспаление (такое как воспаление при ревматоидном артрите, воспаление при остеоартрите, воспаление при воспалительном заболевании кишечника, воспаление при синдроме раздраженного кишечника, воспаление глаз, воспаление уха или воспаление аутоиммунного генеза), зуд (такой как атопический дерматит, связанный с почечным диализом зуд, зуд глаз, зуд ушей, зуд в результате укусов насекомых или индуцированный опиоидами зуд), отек, непроходимость кишечника, кашель или глаукому. В одном аспекте, боль представляет собой невропатическую боль (такую как невралгия тройничного нерва, мигрень, боль диабетического происхождения, боль вирусного происхождения, индуцированная химиотерапией боль или боль у больных метастатическим раком), соматическую боль, висцеральную боль или кожную боль. В другом аспекте боль представляет собой боль при артрите, боль при почечно-каменной болезни, боль в результате спазмов матки, дисменореи, эндометриоза, диспепсии, послеоперационную боль, боль после медицинских процедур, боль в глазах, боль в ушах, прорывную боль при раковых заболеваниях или боль, связанную с расстройством ЖКТ, таким как воспалительное заболевание кишечника или синдром раздраженного кишечника. В другом аспекте боль представляет собой боль, связанную с хирургическим вмешательством, где хирургическое вмешательство представляет собой лапароскопию тазовой области, перевязку труб, гистерэктомию и холецистэктомию. Альтернативно, боль может быть болью, связанной с медицинской процедурой, такой как колоноскопия, цистоскопия, гистероскопия или биопсия эндометрия. В конкретном аспекте, атопический дерматит может быть псориазом, экземой или контактным дерматитом. В другом специфическом аспекте, непроходимость кишечника представляет собой послеоперационную непроходимость кишечника или индуцированную опиатами дисфункцию кишечника.
[00173] В другом варианте связанное с каппа-опиатным рецептором состояние представляет собой связанное с каппа-опиатным рецептором состояние, поддающееся профилактике или поддающееся лечению натрий- и калий-сберегающим диурезом, также известным как акварез. Пример таких связанных с каппа-опиатным рецептором состояний, поддающихся профилактике или поддающихся лечению введением синтетических пептидных амидов по изобретению, включает отек. Отек может быть результатом любого из многочисленных заболеваний или состояний, таких как застойная сердечная недостаточность или синдром ненадлежащей секреции антидиуретического гормона.
[00174] В другом варианте связанное с каппа-опиатным рецептором состояние представляет собой гипонатриемию или другое связанное с отеком заболевание. Связанная с каппа-опиатным рецептором гипонатриемия или отек может представлять собой любое заболевание или состояние, связанное с гипонатриемией или отеком, такое как гипонатриемия и отек, связанный с застойной сердечной недостаточностью или с синдромом ненадлежащей секреции антидиуретического гормона (АДГ), или гипонатриемию, связанную с интенсивной терапией мочегонными средствами с применением тиазидных и/или петлевых диуретиков. Синтетические пептидные амиды по изобретению демонстрируют выраженный натрий- и калий-сберегающий акваретический эффект, который благоприятен в лечении патологических состояний отечности, связанной с гипонатриемией и/или гипокалиемией. Соответственно, синтетические пептидные амиды по изобретению также пригодны для способов лечения или предупреждения связанных с гипонатриемией состояний, примеры которых приведены ниже. Связанные с гипонатриемией состояния могут быть распределены по категориям в соответствии с волемическим статусом как гиперволемические, эуволемические или гиповолемические.
[00175] Гиперволемическая гипонатриемия обычно вызвана увеличением общего содержания воды в организме, поскольку может наблюдаться в случаях застойной сердечной недостаточности, нефротического синдрома и цирроза печени.
[00176] Эуволемическая гипонатриемия часто обнаруживается при синдроме ненадлежащей секреции антидиуретического гормона (АДГ) и может также быть связана с пневмонией, мелкоклеточным раком легкого, полидипсией, случаями травм головы и органическими причинами (например, применение некоторых лекарственных средств, такие как галоперидол) или психогенной причиной.
[00177] Гиповолемическая гипонатриемия - результат относительного уменьшения общего содержания натрия в организме, и может быть связана с применением диуретиков, случаями интерстициального нефрита или чрезмерного потоотделения.
[00178] Такие формы гипонатриемии могут быть дополнительно того классифицированы в соответствии с концентрацией натрия в моче (т.е. концентрация больше или меньше 30 ммоль/л. См., R M Reynolds et al. Disorders of sodium balance, Brit. Med. J. 2006;332:702-705.
[00179] Связанная с каппа-опиатным рецептором гипонатриемия может представлять собой любое заболевание или состояние, где присутствует гипонатриемия (низкое содержание натрия), например, у человека, если концентрация натрия в плазме снижается ниже 135 ммоль/л, аномалия, которая может развиваться отдельно или, более часто, как осложнение других медицинских состояний или как последствие применения терапии, которая может вызывать истощение натрия.
[00180] В дополнение к перечисленным состояниям, различные другие состояния связаны с гипонатриемией, в том числе, не ограничиваясь ими: неопластические причины чрезмерной секреции АДГ, в том числе злокачественные новообразования легкого, двенадцатиперстной кишки, поджелудочной железы, яичника, мочевого пузыря и мочеточника, тимомы, мезотелиомы, бронхиальные аденомы, карциноид, ганглиоцитома и саркома Юинга; инфекции, такие как: пневмония (бактериальная или вирусная), абсцессы (легкого или мозга), образование каверн (аспергиллез), туберкулез (легкого или мозга), менингит (бактериальный или вирусный), энцефалит и СПИД; сосудистые причины, такие как: цереброваскулярная окклюзия или кровоизлияние и тромбоз кавернозного синуса; неврологические причины, такие как: синдром Гийена-Барре, рассеянный склероз, белая горячка, амиотрофный латеральный склероз, водянка головного мозга, психоз, периферическая неврастения, травма головы (закрытая и открытая), опухоли или инфекция ЦНС и инсульты ЦНС, затрагивающие гипоталамические осморецепторы; врожденные уродства, в том числе: агенез corpus callosum, волчья пасть/заячья губа и другие дефекты средней линии; метаболические причины, такие как: острая перемежающаяся порфирия, астма, пневмоторакс и дыхание с положительным давлением; применение лекарственных средств, таких как: тиазидные диуретики, ацетаминофен, барбитураты, холинергические агенты, эстроген, пероральные гипогликемические средства, вазопрессин или десмопрессин, высокая доза окситоцина, хлорпропамид, винкристин, карбамазепин, никотин, фенотиазины, циклофосфамид, трициклические антидепрессанты, ингибиторы моноаминооксидазы и ингибиторы обратного захвата серотонина; дополнительное введение гипотонических жидкостей, например, в ходе госпитализации, хирургического вмешательства или в ходе или после атлетической работы (т.е. связанная с физической нагрузкой гипонатриемия), а также применение пищевых добавок с низким содержанием натрия пожилыми людьми. См., например, Harnson's Principles of Internal Medicine, 16th Ed. (2005), p.2102.
[00181] Другие состояния, связанные с почечной недостаточностью, включают гипонатриемию, нефротический синдром (мембранозная нефропатия и заболевание минимальных изменений), кахексию, недоедание, рабдомиолиза, хирургические процедуры, плановую катетеризацию сердца, потерю крови, а также гиперкальциемию, гипокалиемию и гипергликемию с последующей гликозурией, ведущей к осмотическому диурезу.
[00182] В изобретении также предлагается способ лечения или предупреждения нейродегенеративного заболевания или состояния у млекопитающего, такого как человек, где способ включает введение млекопитающему композиции, которая содержит эффективное количество синтетических пептидных амидов, как изложено выше. Нейродегенеративное заболевание или состояние может представлять собой любое Нейродегенеративное заболевание или состояние, например, ишемию, аноксию, инсульт, травму мозга, травму спинного мозга или реперфузионное повреждение. Альтернативно, нейродегенеративное заболевание или состояние может быть нейродегенеративным заболеванием глаз. Конкретные нейродегенеративные заболевания глаз, поддающиеся лечению или поддающиеся профилактике способом по изобретению, включают глаукому, дегенерацию желтого пятна, ишемическое заболевание сетчатки и диабетическую неврастению.
[00183] В определенных вариантах изобретения предлагаются способы предотвращения или лечения определенных заболеваний и состояний, нейронов таких как заболевания и состояния, включающие нейродегенеративный компонент. Синтетические пептидные амиды по изобретению можно вводить в количестве, эффективном для защиты клетки нейронов против воздействия патологии или повреждения, которое приводило бы к нейродегенерации и/или гибели клетки нейрона в отсутствие лечения. Например, несколько заболеваний или состояния глаз, которые включают нейродегенеративный компонент, можно предупреждать или лечить введением эффективного количества синтетических пептидных амидов по изобретению. Такие заболевания и состояния глаз включают глаукому, дегенерацию желтого пятна, ишемическое заболевание сетчатки и диабетическую неврастению. Считается, что в прогресс этих заболеваний и состояний вовлечена нейродегенерация или гибель клеток нейронов, например, запрограммированная гибель клеток (апоптоз), при которой клетки нейронов направляются на путь, который в отсутствие вмешательства приведет к гибели клетки. Было обнаружено, что развитие или прогрессирование таких заболеваний и состояний может быть предупреждено или как минимум замедлено, лечением агонистами каппа-опиатного рецептора. Считается, что такой улучшенный результат возникает благодаря нейропротекции агонистами каппа-опиатного рецептора. См., например, Kaushik et al. "Neuroprotection in Glaucoma" (2003) J. Postgraduate Medicine vol. 49 (1): pp.90-95.
[00184] В случае глаукомы считается, что профилактика и лечение введением агонистов каппа-опиатного рецептора опосредованы как минимум двумя различными видами активности, индуцированными активацией каппа-опиатного рецептора: нейропротекцией и уменьшением внутриглазного давления (ЮР). Не привязываясь к теории, считается, что нейропротекция, как минимум частично, является результатом индукции атриального натрийуретического пептида (ANP) в глазу, что ведет к защите против окислительного повреждения и других видов повреждения.
[00185] Также считается, что аномально высокое внутриглазное давление является фактором, ведущим к развитию глаукомы. Повышенное внутриглазное давление также можно предупреждать или лечить введением агонистов каппа-опиатного рецептора с тремя различными видами активности, которые запускаются активацией рецептора: уменьшение секреции водного хумора, увеличение истечения водного хумора и акварез (натрия- и калий-сберегающий диурез, что ведет к выведению воды).
[00186] В изобретении также предлагается способ лечения или предупреждения связанного с рецептором каппа заболевания или состояния глаз млекопитающего, такого как повышенное внутриглазное давление (ЮР). Способ включает введение млекопитающему композиции, которая содержит эффективное количество синтетических пептидных амидов, как изложено выше. В одном аспекте изобретения, синтетические пептидные амиды применяют местно. В другом аспекте, синтетические пептидные амиды вводят в виде имплантанта.
[00187] В других вариантах изобретения предлагаются способы предупреждения или лечения некоторых сердечно-сосудистых заболеваний и состояний, включающих компонент дегенерации клеток. Синтетические пептидные амиды по изобретению может вводить в количестве, эффективном для защиты клеток миокарда против воздействия патологии или повреждения, которое приводило бы к дегенерации и/или гибели клеток в отсутствие лечения. Например, несколько сердечнососудистых заболеваний или состояний можно предупреждать или лечить введением эффективного количества синтетических пептидных амидов по изобретению. Такие сердечно-сосудистые заболевания и состояния включают, не ограничиваясь ими, заболевание коронарных сосудов сердца, ишемию, инфаркт миокарда, реперфузионное повреждение и аритмии. См., например, Wu et al. "Cardioprotection of Preconditioning by Metabolic Inhibition in the Rat Ventncular Myocyte-Involvement of kappa Opioid Receptor" (1999) Circulation Res vol.84: pp.1388-1395. См. также Yu et al. "Anti-Arrythmic Effect of kappa Opioid Receptor Stimulation in the Perfused Rat Heart: Involvement of a cAMP-Dependent Pathway" (1999) J Mol Cell Cardiol. vol. 31(10): pp.1809-1819.
[00188] Заболевания и состояния других тканей и органов, которые можно предупреждать или лечить введением эффективного количества синтетических пептидных амидов по изобретению включают, не ограничиваясь ими, ишемию, аноксию, инсульт, повреждение мозга или спинного мозга и реперфузионное повреждение.
[00189] Другая форма связанной с каппа-опиатным рецептором боли, поддающейся лечению или поддающейся профилактике синтетическими пептидными амидами по изобретению, представляет собой гипералгезию. В одном варианте, способ включает введение эффективного количества синтетического пептидного амида по изобретению млекопитающему, страдающему от или подвергающемуся риску развития гипералгезии для предупреждения, облегчения или полного устранения гипералгезии.
[00190] Синтетические пептидные амиды по изобретению можно вводить способами, раскрытыми в данном описании, для лечения или предотвращения какого-либо гипералгезического состояния, например, не ограничиваясь ими, гипералгезического состояния, связанного с аллергическим дерматитом, контактным дерматитом, язвами кожи, воспалением, сыпью, раздражением грибковой природы, и гипералгезических состояний, связанных с инфекционными агентами, ожогами, царапинами, синяками, ушибами, обморожением, сыпью, угрями, укусами/жалом насекомого, язвами кожи, мукозитом, гингивитом, бронхитом, ларингитом, болью в горле, опоясывающим лишаем, раздражением грибковой природы, лихорадочными волдырями, фурункулами, подошвенными бородавками, хирургическими процедурами или поражением влагалища. Например, синтетические пептидные амиды по изобретению можно применять местно на поверхности слизистой оболочки, такой как ротовая полость, пищевод или гортань, или в бронхиальные или носовые ходы. Альтернативно, синтетические пептидные амиды по изобретению можно применять местно по влагалище или прямую кишку/задний проход.
[00191] Кроме того, синтетические пептидные амиды по изобретению можно вводить способами, раскрытыми в данном описании, для лечения или предотвращения какого-либо гипералгезического состояния, связанного с ожогами, ссадинами, синяками, царапинами (например, царапины роговицы), ушибами, обморожением, сыпью, угрями, укусами/жалом насекомого, язвами кожи (например, диабетические язвы или пролежни), мукозитом, воспалением, гингивитом, бронхитом, ларингитом, болью в горле, опоясывающим лишаем, раздражением грибковой природы (такие как стопа атлета или зуд жокея), лихорадочными волдырями, фурункулами, подошвенными бородавками или поражением влагалища (например, поражение влагалища, связанное с микозом или заболеваниями, передаваемыми половым путем). Способы, предусмотренные для введения синтетических пептидных амидов по изобретению с целью лечения или предотвращения гипералгезии, включают способы, где соединение применяется местно на поверхности глаз, ротовой полости, гортани, пищевода, бронхиальных ходов, носовых ходов, влагалища или прямой кишки/заднего прохода.
[00192] Гипералгезическими состояниями, связанными с восстановлением после хирургических вмешательств, можно также управлять введением синтетических пептидных амидов по изобретению. Гипералгезические состояния, связанные с восстановлением после хирургического вмешательства, могут быть любыми Гипералгезическими состояниями, связанными с восстановлением после хирургического вмешательства, например, радиальной кератэктомии, удаления зубов, лампэктомии, эпизиотомии, лапароскопии и артроскопии.
[00193] Гипералгезическими состояниями, связанными с воспалением, также можно управлять введением синтетических пептидных амидов по изобретению. Воспаление периодонта, воспаление ортодонта, воспалительный конъюнктивит, геморрой и воспаление, связанное с заболеваниями, передающимися половым путем, можно лечить или предупреждать местным введением или нанесением синтетических пептидных амидов по изобретению.
[00194] В изобретении также предлагается способ индукции диуреза у млекопитающего, нуждающегося в этом. Способ включает введение млекопитающему композиции, содержащей эффективное количество синтетических пептидных амидов по изобретению, как изложено выше.
[00195] В изобретении также предлагается способ индукции секреции пролактина у млекопитающего. Способ включает введение млекопитающему композиции, содержащей эффективное количество синтетических пептидных амидов по изобретению, как изложено выше. Способ индукции секреции пролактина пригоден для лечения млекопитающего, такого как человечек, страдающего от недостаточной лактации, неадекватной лактации, субоптимальной лактации, пониженной подвижности сперматозоидов, возрастного расстройства, диабета I типа, бессонницы или неадекватного сна с быстрым движением глаз (REM сна). В конкретном аспекте, способ включает комбинированное введение синтетических пептидных амидов со сниженной дозой анальгетика мю-опиатного агониста для достижения терапевтического анальготического эффекта, где соединение вызывает сопутствующий побочный эффект, причем сниженная доза соединения вызывает менее выраженный сопутствующий побочный эффект, чем побочный эффект, вызываемый дозой анальгетика мю-опиатного агониста, необходимой для достижения терапевтического анальгетического эффекта при монотерапии.
[00196] В настоящем изобретении также предлагается способ связывания каппа-опиатного рецептора у млекопитающего, где способ включает стадию введения млекопитающему композиции, содержащей эффективное количество синтетического пептидного амида по настоящему изобретению. Эффективное количество может быть определено средним специалистом в данной области в соответствии с шаблонными способами. Например, эффективное количество может быть определено как единица дозирования, достаточная, чтобы связаться с каппа-опиатными рецепторами у млекопитающего и вызвать антиноцицептивный эффект, противовоспалительный эффект, акваретический эффект или повышение уровней пролактина в сыворотке или любой другой связанный с каппа-опиатным рецептором эффект. Альтернативно, эффективное количество может быть определено как количество, достаточное, чтобы достичь уровней, близких к EC50, в биологической жидкости организма млекопитающего, или количество,, достаточное, чтобы достичь уровней, в 2 или 3 или до приблизительно 5 раз или даже приблизительно до 10 раз превышающих EC50 в подходящей для терапии биологической жидкости в организме млекопитающего.
СИНТЕЗ ПЕПТИДНЫХ АМИДОВ ПО ИЗОБРЕТЕНИЮ
[00197] В данном описании химическое обозначение "тетрапептида-[ω(4-амино-пиперидин-4-карбоновая кислота)]" применяется для указания на аминоацильный фрагмент синтетических пептидных амидов по изобретению, полученный из 4-аминопиперидин-4-карбоновой кислоты, где атом азота пиперидинового кольца присоединен к С-концевому карбонильному атому углерода тетрапептидного фрагмента, если не указано иное.
[00198] На фиг.1 показана общая схема синтеза, используема для получения соединений (1) (6), (7), (10) и (11); на фиг.2 показана общая схема синтеза, используемая для получения соединений (2) to (5), (8), (9) и (12)-(14); на фиг.3 показана общая схема синтеза, используемая для получения синтетических пептидных амидов (15)-(24); и на фиг.4 показана схема, используемая для получения синтетических пептидных амидов (25)-(37):
[00199] Соединение (1): D-Phe-D-Phe-D-Leu-(e-Me)D-Lys-[4-Амидиногомопиперазина амид] (SEQ ID NO: 1):

[00200] Соединение (2): D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 2):
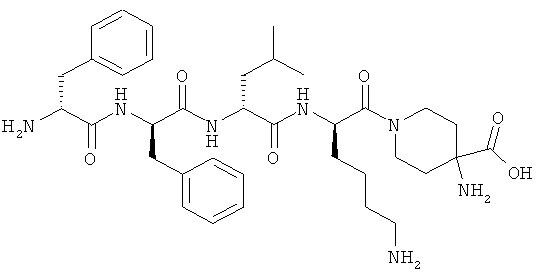
[00201] Соединение (3): D-Phe-D-Phe-D-Leu-(ε-Me) D-Lys-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 1):
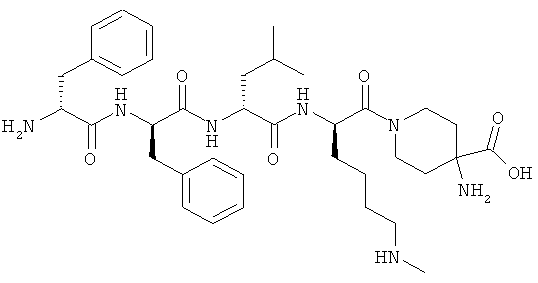
[00202] Соединение (4): D-Phe-D-Phe-D-Leu-D-Lys-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 2):

[00203] Соединение (5): D-Phe-D-Phe-D-Leu-D-Har-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 3):

[00204] Соединение (6): D-Phe-D-Phe-D-Leu-(ε-Me)D-Lys-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 1):

[00205] Соединение (7): D-Phe-D-Phe-D-Leu-D-Arg-[гомопиперазина амид] (SEQ ID NO: 4):
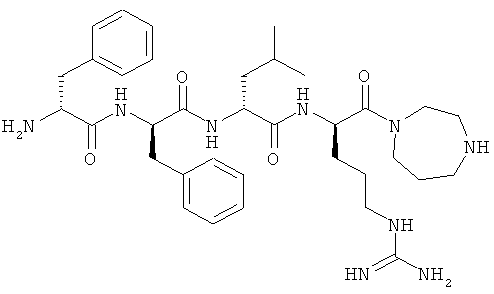
[00206] Соединение (8): D-Phe-D-Phe-D-Leu-D-Har-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 3):

[00207] Соединение (9): D-Phe-D-Phe-D-Leu-(ε-iPr)D-Lys-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 5):

Соединение (10): D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 6):
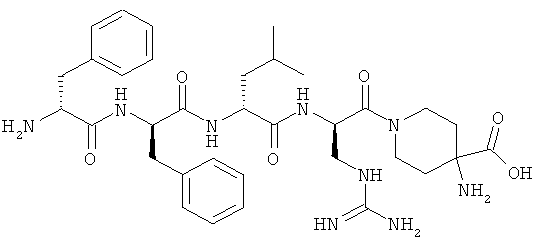
[00208] Соединение (11): D-Phe-D-Phe-D-Leu-D-Nar-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 7):

[00209] Соединение (12): D-Phe-D-Phe-D-Leu-D-Dbu-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 8):
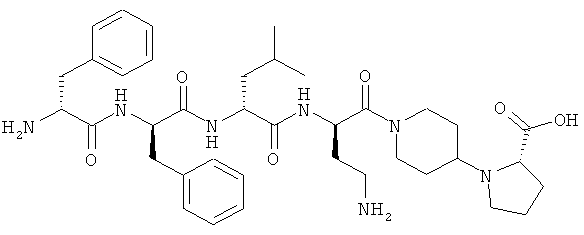
[00210] Соединение (13): D-Phe-D-Phe-D-Leu-D-Nar-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 7):

[00211] Соединение (14): D-Phe-D-Phe-D-Leu-D-Dap(амидино)-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID N0:6):

[00212] Соединение (15): D-Phe-D-Phe-D-Leu-D-Lys-[4-амидиногомопиперазина амид] (SEQ ID NO: 2):

[00213] Соединение (16): D-Phe-D-Phe-D-Leu-D-Har-[4-амидиногомопиперазина амид] (SEQ ID NO: 3):

[00214] Соединение (17): D-Phe-D-Phe-D-Leu-(ε-iPr)D-Lys-[4-амидиногомопиперазина амид] (SEQ ID NO: 5):

[00215] Соединение (18): D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[4-амидиногомопиперазина амид] (SEQ ID NO: 6):

[00216] Соединение (19): D-Phe-D-Phe-D-Nle-(β-амидино)D-Dap-[4-амидиногомопиперазина амид] (SEQ ID 20 NO: 6):
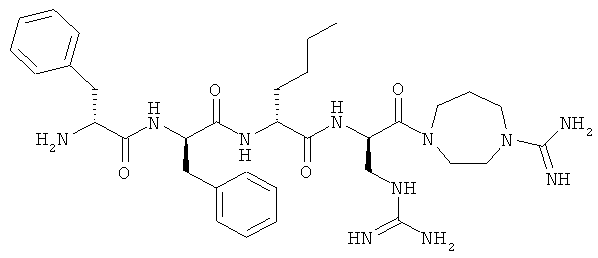
[00217] Соединение (20): D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[гомопиперазина амид] (SEQ ID NO: 6):

[00218] Соединение (21): D-Phe-D-Phe-D-Nle-(β-амидино)D-Dap-[гомопиперазина амид] (SEQ ID NO: 6):
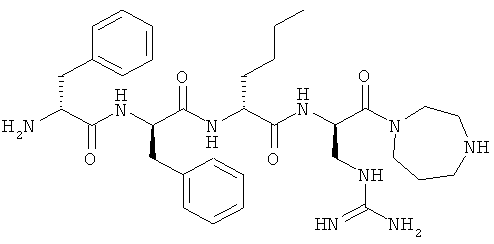
[00219] Соединение (22): D-Phe-D-Phe-D-Leu-D-Dbu-[4-амидиногомопиперазина амид] (SEQ ID NO: 8):

[00220] Соединение (23): D-Phe-D-Phe-D-Leu-D-Nar-[4-амидиногомопиперазина амид] (SEQ ID NO: 7):
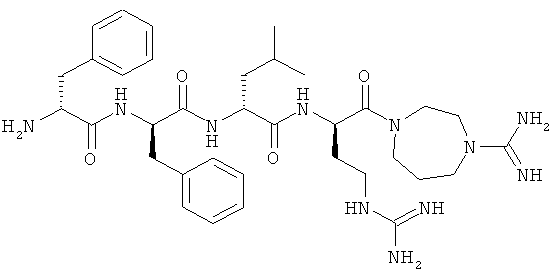
[00221] Соединение (24): D-Phe-D-Phe-D-Leu-D-Arg-[4-амидиногомопиперазина амид] (SEQ ID NO: 4):
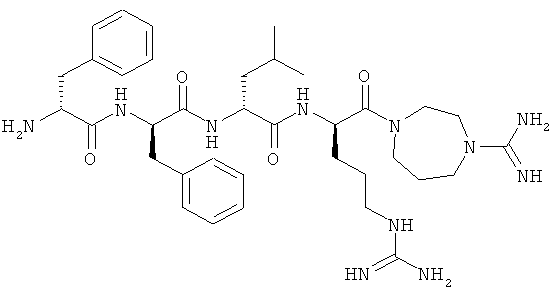
[00222] Соединение (25): D-Phe-D-Phe-D-Leu-D-Lys-[2,8-диазаспиро[4,5]декан-1-он амид] (SEQ ID NO: 2):

[00223] Соединение (26): D-Phe-D-Phe-D-Leu-D-Lys-[2-метил-2,8-диазаспиро[4,5]декан-1-он амид] (SEQ ID NO: 2):

[00224] Соединение (27): D-Phe-D-Phe-D-Leu-D-Lys-[1,3,8-триазаспиро[4,5]декан-2,4-дион амид] (SEQ ID NO: 2):
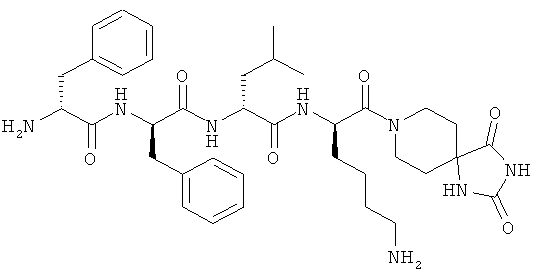
[00225] Соединение (28): D-Phe-D-Phe-D-Leu-D-Lys-[5-хлор-1-(пиперидин-4-ил)-1H-бензо[d]имидазол-2(3)Н-он амид] (SEQ ID NO: 2):
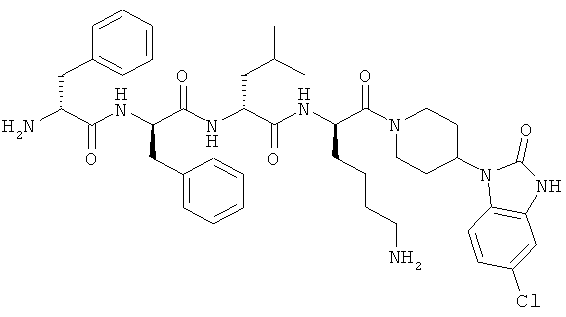
[00226] Соединение (29): D-Phe-D-Phe-D-Leu-D-Lys-[морфолино(пиперидин-4-ил)метанона амид] (SEQ ID NO: 2):
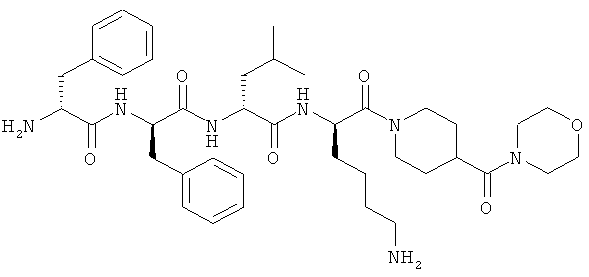
[00227] Соединение (30): D-Phe-D-Phe-D-Leu-D-Lys-[4-фенил-1-(пиперидин-ил-1H-имидазол-2(3H)-она амид] (SEQ ID NO: 2):
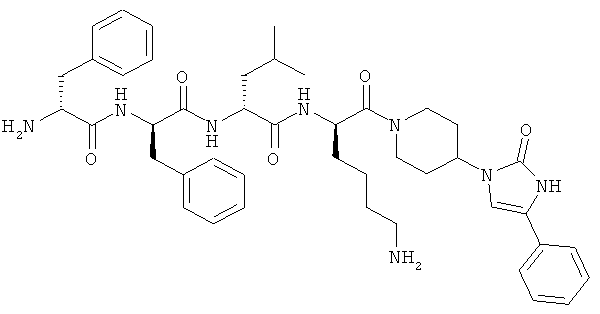
[00228] Соединение (31): D-Phe-D-Phe-D-Leu-D-Lys-[4-(3,5-диметил-4H-1,2,4-триазол-4-ил)пиперидина амид] (SEQ ID NO: 2):
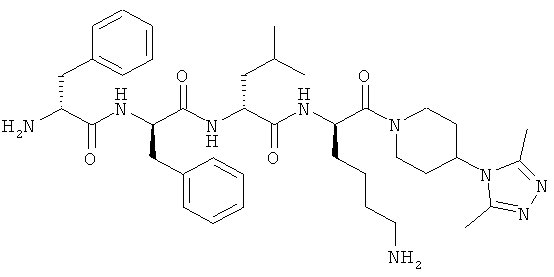
[00229] Соединение (32): D-Phe-D-Phe-D-Leu-D-Lys-[1-(пиперидин-4-ил)индолин-2-она амид] (SEQ ID NO: 10 2):
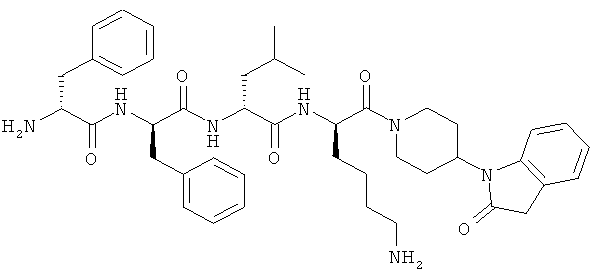
[00230] Соединение (33): D-Phe-D-Phe-D-Leu-D-Lys-[1-фенил-1,3,8-триазаспиро[4,5]декан-4-она амид] (SEQ ID NO: 2):
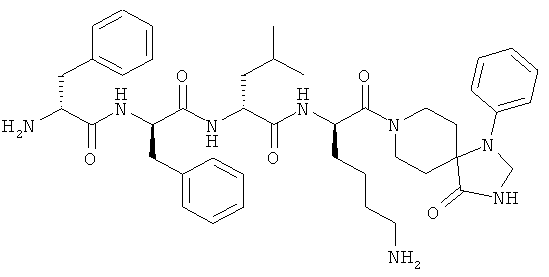
[00231] Соединение (34): D-Phe-D-Phe-D-Leu-D-30 Lys-[имидазо[1,2-а]пиридин-2-илметила амид] (SEQ ID NO; 2):
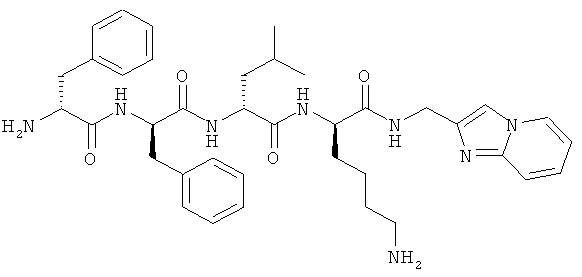
[00232] Соединение (35)D-Phe-D-Phe-D-Leu-D-Lys-[(5-метилпиразин-2-ил)метил амид] (SEQ ID NO: 2):
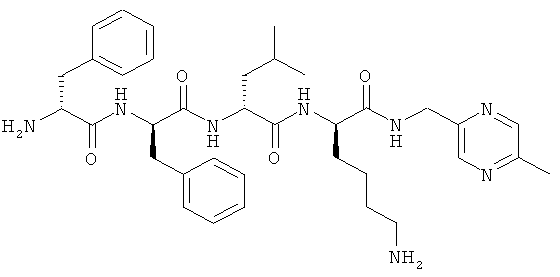
[00233] Соединение (36): D-Phe-D-Phe-D-Leu-D-Lys- [1-(пиперидин-4-ил)-1H-бензо[d]имидазол-2(3Н)-она амид] (SEQ ID NO: 2):

[00234] Соединение (37): D-Phe-D-Phe-D-Leu-D-Lys- [4,5,6,7-тетрагидро-1H-пиразоло[4,3-е]пиридина амид] (SEQ ID NO: 2):
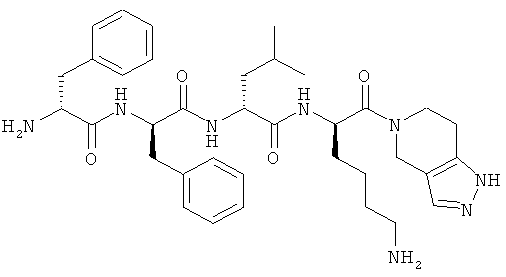
ПРИМЕРЫ
[00235] Общие экспериментальные методы синтеза:
[00236] Производные аминокислот и смолы приобретали у коммерческих поставщиков (Novabiochem, Bachem, Peptide International и PepTech Corporation). Другие реагенты и растворители приобретали у Sigma-Aldrich, Fisher Scientific и VWR. Соединения в данном описании синтезировали стандартными методами твердофазной химии пептидов с использованием как Fmoc, так и Boc методологии. Если не указано иное, все реакции проводят при комнатной температуре.
[00237] В следующих стандартных ссылках содержится общее экспериментальное руководство и описана доступность необходимых исходных материалов и реагентов: Kates, S.A., Albencio, F., Eds., Solid Phase Synthesis, A Practical Guide, Marcel Dekker, New York, Basel, (2000); Bodanszky, M., Bodanszky, A., Eds., The Practice of Peptide Synthesis, Second Edition, Spnnger-Verlag, (1994); Atherton, E., Sheppard, R.C., Eds., Solid Phase Peptide Synthesis, A Practical Approach, IRL Press at Oxford University Press, (1989); Stewart, J.M., Young, J.D., Solid Phase Synthesis, Pierce Chemical Company, (1984); Bisello, et al., J. Biol. Chem. 273, 22498-22505 (1998); and Mernfield, R.В., J. Am. Chem. Soc. 85, 2149-2154 (1963).
[00238] Дополнительные сокращения, используемые в данном описании:
[00239] ACN: ацетонитрил
[00240] Aloe: аллилоксикарбонил
[00241] Вое: трет-бутоксикарбонил
[00242] ВОР: бензотриазол-1-ил-окси-трис(диметиламино)-фосфония гексафторфосфат
[00243] Cbz: бензилоксикарбонил
[00244] Cbz-OSu: No-(бензилоксикарбонилокси)сукцинимид
[00245] DBU: 1,8-диазабицикло[5,4,0]ундец-7-ен
[00246] ДХМ: дихлорметан
[00247] Dde: 1-(4,4-диметил-2,6-диоксоциклогекс-1-илиден)этил
[00248] DIC: N,N'-диизопропилкарбодиимид
[00249] DIEA: N,N-диизопропилэтиламин
[00250] ДМФА: N,N-диметилформамид
[00251] Fmoc: 9-фторенилметоксикарбонил
[00252] HATU: 2-(1Н-9-азабензотриазол-1-ил)-1,1,3,3-тетраметиламиния гексафторфосфат
[00253] HBTU: 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламиния гексафторфосфат
[00254] HOBt: 1-гидроксибензотриазол
[00255] ВЭЖХ: высокоэффективная жидкостная хроматография
[00256] i: изо
[00257] ivDde: 1-(4,4-диметил-2,б-диоксоциклогекс-1-илиден)-3-метилбутил
[00258] NMM: 4-метилморфолино
[00259] NMP: N-метилпирролидинон
[00260] All: аллил
[00261] o-NBS-Cl: o-нитробензолсульфонилхлорид [00262] Pbf: 2,2,4,б,7-пентаметилдигидро-бензофуран-5-сульфонил
[00263] РуВОР: бензотриазол-1-илокси-трис-пирролидино-фосфоният гексафторфосфат
[00264] RP: обращенно-фазовая [00265] TBTU: 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония тетрафторборат
[00266] ТЕАР: триэтиламмония фосфат
[00267] ТФУ: трифторуксусная кислота
[00268] TIS: триизопропилсилан
[00269] TMOF: триметил ортоформиат
[00270] TMSOTf: триметилсилил трифторметансульфонат
[00271] Trt: тритил
[00272] Пептиды, синтезированные по методологии Fmoc, расщепляют смесью ТФУ/TIS/H2O (об./об./об.=95:2,5:2,5). Стадию расщепления в методологии Вое выполняют с использованием смеси HF/анизол (об./об.=9:1) или смеси TMSOTf/ТФУ/м-крезол (об./об./об. 2:7:1).
[00273] Реакции сочетания с удлинением цепи пептида осуществляют вручную на синтезаторе пептидов, реакции опосредованы реагентами сочетания с 2-4-кратным избытком производных аминокислот. Реагенты сочетания, применяемые в синтезе различных соединений по изобретению, выбирают из следующих комбинация: DIC/HOBt, HATU/DIEA, HBTU/DIEA, TBTU/DIEA, PyBOP/DIEA и BOP/DIEA.
[00274] Снятие защиты с боковой цепи аминокислот в положении 4 (обозначено Хаа4 в конечном продукте - синтетическом пептидном амиде) связанных со смолой пептидов обеспечивают следующим образом: пептиды собирают, используя в качестве исходных материалов Хаа4 и постепенно добавляя Хаа3, затем Хаа2 и, в конце, Xaa1. Защитные группы боковой цепи диаминокислоты, введенные в положении Хаа4, избирательно удаляют следующим образом: (i) группы N-Dde или N-ivDde удаляют с помощью 2-4% гидразина в ДМФА. См. Chabra, S.R., et al., Tetrahedron Lett. 35:1603-1606 (1998) и Rohwedder, В., et al., Tetrahedron Lett., 39: 1175 (1998); (ii) N-Aloc: удаляют с помощью 3 экв. (Ph3P)4Pd в CHCl3/AcOH/NMM (об./об./об.=37:2:1). См. Kates, S.A., et al. в "Peptides Chemistry, Structure and Biology, Proc. 13th American Peptide Symposium", Hodges, R.S. и Smith, J.A. (Eds), ESCOM, Leiden, 113-115 (1994).
[00275] Если пептиды собирают по методологии защиты с помощью Boc, защитную группу боковой цепи диаминокислоты, введенную в положении Хаа4, т.е. N-Fmoc, удаляют с помощью 20-30% пиперидина в ДМФА.
[00276] Изопропилирование концевого азота в боковой цепи аминокислоты в положении Xaa4 связанных со смолой пептидов осуществляют следующим образом: После снятия защиты осуществляют реакцию связанного со смолой пептида со свободной ω-аминогруппой в положении Xaa4 со смесью ацетона и NaBH(OAc)3 в TMOF с образованием связанного со смолой Nω-изопропилпептида.
[00277] Монометилирование концевого азота в боковой цепи аминокислоты в положении Хаа4 связанных со смолой пептиды: В синтезированных, связанных со смолой Nω-метилпептидах свободную ω-аминогруппу сначала дериватизируют с помощью о-нитробензолсульфонилхлорида (o-NBS-Cl; Biron, E.; Chatterjee, J.; Kessler, H. Optimized selective N-methylation of peptides on solid support. J. Pep.Sci. 12:213-219 (2006). Полученный сульфонамид далее метилируют смесью диметилсульфата и 1,8-диаза-бицикло[5,4,0]ундец-7-ена в NMP. Защитную группу o-NBS далее удаляют с помощью смеси меркаптоэтанола и 1,8-диазабицикло[5,4,0]ундец-7-ена в NMP.
[00278] Гуанилирование концевого азот в боковой цепи аминокислоты в положении Хаа4 связанных со смолой пептидов: После снятия защиты, осуществляют реакцию связанного со смолой пептида со свободной ω-аминогруппой в положении 4 со смесью 1Н-пиразол-1-карбоксамидина гидрохлорида (Bernatowicz, M.S., et al., J. Org. Chem. 57, 2497-2502 (1992) и DIEA в ДМФА с образованием связанного со смолой Nω-гуанидинопептида.
[00279] Пептиды очищают препаративной ВЭЖХ в буферных растворах триэтиламмония фосфата (ТЕАР) или трифторуксусной кислоты (ТФУ). При необходимости, соединения в конце превращают в трифторацетатные или ацетатные соли с использованием традиционной методологии ВЭЖХ. Фракции с чистотой свыше 97% объединяют и лиофилизируют.Чистоту синтезированных пептидов определяют аналитической обращенно-фазовой ВЭЖХ.
[00280] Аналитическую обращенно-фазовую ВЭЖХ осуществляли на системе доставки нескольких растворителей Waters 600 с адаптируемым детектором оптической плотности в УФ-области Waters 486 и модулем данных Waters 746. Анализ пептидов методом ВЭЖХ осуществляли с использованием колонки Vydac C18 (0,46×25 см, размер частиц 5 мкм, размер пор 300 Å) со скоростью потока 2,0 мл/мин. Растворители А и В: 0,1% ТФУ в H2O и 0,1% ТФУ в смеси 80% ACN/20% H2O, соответственно. Значения времени удерживания (tn) приведены в минутах. Препаративную RP-ВЭЖХ осуществляли с использованием препаративного картриджа Vydac C18 (5×30 см, размер частиц 15-20 мкм, размер пор 300 Å) со скоростью потока 100 мл/мин, на системе для препаративной хроматографии Waters Prep LC 2000 с адаптируемым детектором оптической плотности в УФ-области Waters 486 и ленточным графопостроителем Servogor 120. Буферные растворы А и В: 0,1% ТФУ в H2O и 0,1% ТФУ в 60% ACN/40% H2O, соответственно. Анализ методом ВЭЖХ конечного соединения осуществляли на жидкостном хроматографе Hewlett Packard 1090 с использованием колонки Phenomenex Synergi MAX-RP C12 (2,0×150 мм, размер частиц 4 мкм, размер пор 80 Å) со скоростью потока 0,3 мл/мин при температуре 40°C. Буферные растворы А и В: 0,01% ТФУ в H2O и 0,01% ТФУ в 70% ACN/30% H2O, соответственно. Идентичность синтетических пептидных амидов подтверждена масс-спектрометрией с электрораспылением. Масс-спектры регистрировали на масс-спектрометре Finnigan LCQ с источником ионизации электрораспылением (ESI).
ПРИМЕР 1: Синтез соединения (1)
[00281] D-Phe-D-Phe-D-Leu-(e-Me)D-Lys-[4-амидиногомопиперазина амид] (SEQ ID NO: 1).
[00282] См. схему, показанную на фиг.1. Использовали следующие производные аминокислот: Bcz-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu-OH и Fmoc-D-Lys(Dde)-ОН. Полностью защищенный, связанный со смолой пептид синтезируют вручную, используя в качестве исходных материалов л-нитрофенилкарбонатную смолу Wang (5,0 г, 4,4 ммоль; Novabiochem). Присоединение гомопиперазина к смоле обеспечивают, перемешивая ее с раствором гомопиперазина (8,7 г, 87 ммоль; Acros Organics) в ДХМ (100 мл) в течение ночи при комнатной температуре. Смолу промывают ДМФА и ДХМ и сушат под вакуумом. Полученный гомопиперазин-карбаматную смолу Wang (5,1 г; гомопиперазин-[карбаматная смола Wang]) делят на несколько порций, и порцию 1,5 г (1,3 ммоль) используют для продолжения синтеза пептида. Опосредованные DIC/HOBt одинарные сочетания проводят с 3-кратным избытком производных аминокислот. Группу Fmoc удаляют с помощью 25% пиперидин в ДМФА. По окончании удлинения пептидной цепи смолу обрабатывают 4% гидразина в ДМФА 3×3 мин для удаления Dde. Смолу промывают ДМФА и ДХМ, сушат под вакуумом. Полученную пептидную смолу (2,4 г; Bcz-D-Phe-D-Phe-DLeu-DLys-гомопиперазин-[карбаматная смола Wang]) делят снова, и порцию 0,6 г (0,3 ммоль) используют для последующей дериватизации (N-метилирование).
[00283] Метилирование ω-аминогруппы D-Lys в положении Хаа4 осуществляют в 2 стадии: (i) [защита о-NBS]: Связанный со смолой пептид (0,3 ммоль) сначала обрабатывают раствором o-NBS-Cl (0,4 г, 2 ммоль) и коллидином (0,7 мл, 5 ммоль) в NMP (7 мл) при комнатной температуре в течение 30 мин. Смолу затем промывают NMP. (ii) [N-Метилирование]: Далее осуществляют реакцию связанного со смолой o-NBS защищенного пептида с раствором 1,8-диазабицикло[5,4,0]ундек-7-ена (0,5 мл, 3 ммоль) и диметилсульфата (1,0 мл, 10 ммоль; Aldnch) в NMP (7 мл) при комнатной температуре в течение 5 мин. Далее смолу промывают NMP, и процесс промывания повторяют еще раз. (iii) [снятие защиты o-NBS]: Пептидную смолу обрабатывают раствором меркаптоэтанола (0,7 мл, 10 ммоль) и 1,8-диазабицикло[5,4,0]ундек-7-еном (0,8 мл, 5 ммоль) в NMP (7 мл) при комнатной температуре в течение 5 мин. Далее смолу промывают NMP и процесс промывания повторяют еще раз.
[00284] Для защиты полученного вторичного N-метиламина D-Lys в положении Хаа4, осуществляют реакцию связанного со смолой метилированного пептида с раствором Bcz-OSu (6 ммоль) в ДМФА (7 мл). Смолу промывают ДМФА и ДХМ, сушат под вакуумом. Далее пептид отщепляют от смолы обработкой раствором ТФУ/ДХМ (15 мл, об./об.=1:1) при комнатной температуре в течение 2 час. Далее смолу фильтруют и промывают ТФУ. Фильтрат упаривают под вакуумом, и сырой пептид (0,3 ммоль; Bcz-D-Phe-D-Phe-D-Leu-D-Lys(Me,Bcz)-[гомопиперазина амид]) осаждают из диэтилового эфира.
[00285] Для гуанилирования гомопиперазина на С-конце, описанный выше пептид (0,3 ммоль) обрабатывают раствором 1Н-пиразол-1-карбоксамидина гидрохлорида (0,4 г, 3,0 ммоль) и DIEA (0,5 мл, 6 ммоль) в ДМФА (3 мл) в течение ночи при комнатной температуре. Уксусную кислоту и HzO добавляют для гашения реакции, раствор замораживают и сушат на лиофилизаторе с получением целевого защищенного пептида, Bcz-D-Phe-D-Phe-D-Leu-D-Lys(Me,Z)-[4-амидиногомопиперазина амид] (0,6 г).
[00286] Для окончательного снятия защиты/гидролиза, описанный выше пептид (0,6 г) обрабатывают смесью TMSOTf/ТФУ/м-крезол (10 мл, об./об./об.=2:7:1) при комнатной температуре в течение 2 час. Смесь упаривают, и сырой пептид (0,6 г) осаждают из диэтилового эфира.
[00287] Для очистки описанный выше сырой пептид (0,6 г) растворяют в 0,1% ТФУ в Н20 (50 мл), раствор загружают на колонку ВЭЖХ и очищают с использованием буферной системы ТФУ (буферные растворы: А=0,1% ТФУ в H2O и В=0,1% ТФУ в 60% ACN/40% H2O). Соединение элюируют с линейным градиентом буфера В, от 25% В до 75% В на протяжении 30 мин, tR=37% В. Фракции с чистотой свыше 97% объединяют, замораживают и сушат на лиофилизаторе с получением очищенного пептида в виде белого аморфного порошка (153 мг). Анализ методом ВЭЖХ: tR=14,41 мин, чистота 99,8%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 692,5, зарегистрировано 692,5.
ПРИМЕР 2: Синтез соединения (2)
[00288] D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 2):
[00289] См. схему на фиг.2 и Biron et al., Optimized selective N-methylation of peptides on solid support. J. Peptide Science 12: 213-219 (2006). Использовали следующие производные аминокислот: Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Dde)-ОН и N-Boc-амино-(4-Н-Fmoc-пиперидинил)карбоновая кислота. Анализ методом ВЭЖХ и МС проводят, как описано в синтезе соединения (1) выше.
[00290] Полностью защищенный, связанный со смолой пептид синтезируют вручную, используя в качестве исходных материалов 2-хлортритилхлоридную смолу (1,8 г, 0,9 ммоль; Peptide International). Присоединение N-Boc-амино-(4-N-Fmoc-пиперидинил) карбоновой кислоты с последующим удлинением цепи пептида и снятием защиты Dde в D-Lys(Dde) при Хаа4 осуществляют в соответствии с методикой, описанной в синтезе соединения (1). См. выше. Полученную пептидную смолу (0,9 ммоль; Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N-Вос-амино-4-пиперидинилкарбоновая кислота)-[2-Cl-Trt смола]) делят на порции, и порцию 0,3 ммоль используют для последующего расщепления. Пептидную смолу (0,3 ммоль) затем обрабатывают смесью ТФУ/Т13/H2O (15 мл, об./об./об.=95:2,5:2,5) при комнатной температуре в течение 90 мин. Далее смолу отфильтровывают и промывают ТФУ. Фильтрат упаривают под вакуумом, и сырой пептид (0,3 ммоль; D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН) осаждают из диэтилового эфира.
[00291] Для очистки сырой пептид (0,3 ммоль) растворяют в 2% уксусной кислоте в H2O (50 мл), раствор загружают на колонку ВЭЖХ и очищают с использованием буферной системы ТЕАР с pH 5,2 (буферные растворы: А=ТЕАР 5,2 и В=20% ТЕАР 5,2 в 80% ACN). Соединение элюируют с линейным градиентом от буфера В, 7% В до 37% В на протяжении 60 мин. Фракции с чистотой свыше 95% объединяют, и полученный раствор разбавляют 2 объемами воды. Разбавленный раствор далее загружают на колонку ВЭЖХ для солевого обмена и дополнительно очищают с использованием буферной системы ТФУ (буферные растворы: А=0,1% ТФУ в H2O и В=0,1% ТФУ в 80% ACN/20% H2O) и линейный градиента от буфера В, 2% В до 75% В на протяжении 25 мин. Фракции с чистотой свыше 97%, объединяют, замораживают и сушат на лиофилизаторе с получением очищенного пептида в виде белого аморфного порошка (93 мг). Анализ методом ВЭЖХ: tR=16,43 мин, чистота 99,2%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 680,4, зарегистрировано 680,3.
[00292] Соединение (2) также получают с использованием схемы реакции, аналогичной показанной на фиг.2 с использованием следующих производных аминокислот: Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Boc)-OH и Boc-4-амино-1-Fmoc-(пиперидин)-4-карбоновая кислота.
[00293] Полностью защищенный, связанный со смолой пептид синтезируют вручную, используя в качестве исходных материалов 2-хлортритилхлоридную смолу (PS 1% DVB, 500 г, 1 мэкв. /г). Смолу обрабатывают Вос-4-амино-1-Fmoc-4-(пиперидин)-4-карбоновой кислотой (280 г, 600 ммоль) в смеси ДМФА, добавляют ДХМ и DIEA (по 260 мл каждого). Смесь перемешивают в течение 4 час, после чего смолу покрывают в течение 1 час добавлением МеОН (258 мл) и DIEA (258 мл).
[00294] Смолу отделяют и промывают ДМФА (3×3 л). Смолу, содержащую первую аминокислоту, обрабатывают пиперидином в ДМФА (3×3 л of 35%), промывают ДМФА (9×3 л), и осуществляют сочетание с Fmoc-D-Lys(Вое)-ОН (472 г) с использованием PyBOP (519 г) в присутствии HOBt (153 г) и DIEA (516 мл) и в ДХМ/ДМФА (500 мл/500 мл) при перемешивании в течение 2,25 час. Содержащую дипептид смолу отделяют и промывают ДМФА (3×3,6 л). Группу Fmoc удаляют обработкой пиперидином в ДМФА
[00295] (3×3,6 л, 35%), смолу промывают ДМФА (9×3,6 л) и обрабатывают Fmoc-D-Leu-OH (354 г), DIC (157 мл) и HOBt (154 г) в ДХМ/ДМФА (500 мл/500 мл) и перемешивают в течение 1 час. Последующее промывание ДМФА (3×4,1 л) с последующим отщеплением группы Fmoc с помощью пиперидина в ДМФА (3×4,2 л, 35%) и промыванием смолы ДМФА (9×4,2 л) дает связанный со смолой трипептид. Полученный материал обрабатывают Fmoc-D-Phe-OH (387 г), DIC (157 мл) и HOBt (153 г) в ДХМ/ДМФА (500 мл/500 мл) и перемешивают в течение ночи. Смолу отделяют, промывают ДМФА (3×4,7 л), и затем обрабатывают пиперидином в ДМФА (3×4,7 л 35%) для отщепления группы Fmoc и затем снова промывают ДМФА (9×4,7 л). Тетрапептид загружают на смолу, обрабатывают Fmoc-D-Phe-OH (389 г), DIC (157 мл) и HOBt (154 г) в ДХМ/ДМФА (500 мл/500 мл) и перемешивают в течение 2,25 час. Смолу отделяют, промывают ДМФА (3×5,2 л) и затем обрабатывают пиперидином (3×5,2 л, 35%) в ДМФА. Смолу отделяют и последовательно промывают ДМФА (9×5,2 л), затем ДХМ (5×5,2 л). Сушат с получением выхода 90,4% защищенного пептида, связанного со смолой. Пептид отщепляют от смолы с использованием смеси ТФУ/вода (4,5 L, 95/5), которая также служит для удаления защитной группы Boc. Смесь отфильтровывают, упаривают (1/3) и затем осаждают добавлением МТВЕ (42 л). Твердое вещество отделяют фильтрацией и сушат при сниженном давлении с получением сырого пептида.
[00296] Для очистки сырой пептид растворяют в 0,1% ТФУ в H2O и очищают препаративной обращенно-фазовой ВЭЖХ (С18) с использованием 0,1% градиента ТФУ/вода - ACN как подвижной фазы. Фракции с чистотой свыше 95% объединяют, упаривают и лиофилизируют с получением чистого пептида (чистота >95,5%). Ионный обмен проводят с использованием ионно-обменной смолы Dowex, элюируя водой. Водную фракцию фильтруют (фильтровальная капсула с размером пор 0,22 мкм) и сушат вымораживанием с получением ацетатной соли пептида (общий выход, 71,3%, чистота >99%).
ПРИМЕР 3: Синтез соединения (3)
[00297] D-Phe-D-Phe-D-Leu-(ε-Me)D-Lys-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 1):
[00298] Синтез начинают с 0,3 ммоль пептидной смолы: Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N-Вос-амино-4-пиперидинилкарбоновая кислота)-[2-Cl-Trt смола], которую получают в ходе синтеза соединения (2), как описано ниже. Анализ методом ВЭЖХ и МС также проводят, как описано в синтезе соединения (2) выше.
[00299] Для метилирования ω-аминогруппы D-Lys в положении Хаа4, следуют 3-хстадийной методике, как описано в синтезе соединения (1). См. описание выше. Связанный со смолой метилированный пептид (Boc-D-Phe-D-Phe-D-Leu-(ε-Me)D-Lys-(N-Boc-амино-4-пиперидинилкарбоновая кислота)-[2-Cl-Trt смола]) далее обрабатывают смесью ТФУ/TIS/H2O (15 мл, об./об./об.=95:2,5:2,5) при комнатной температуре в течение 90 мин. Затем смолу отфильтровывают и промывают ТФУ. Фильтрат упаривают под вакуумом, и сырой пептид (0,3 ммоль; D-Phe-D-Phe-D-Leu-(ε-Me)D-Lys-[ω(4-амино-пиперидин-4-карбоновая кислота)]-ОН) осаждают из диэтилового эфира.
[00300] Сырой пептид (0,3 ммоль) очищают препаративной ВЭЖХ в соответствии с протоколом, описанным в синтезе соединения (2). См. выше. Фракции с чистотой свыше 97% объединяют, замораживают и сушат на лиофилизаторе с получением очищенного пептида в виде белого аморфного порошка (185 мг). Анализ методом ВЭЖХ: tR=16,93 мин, чистота 99,2%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 694,4, зарегистрировано 694,4.
ПРИМЕР 4: Синтез соединения (4)
[00301] D-Phe-D-Phe-D-Leu-D-Lys-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 2):
[00302] Использовали следующие производные аминокислот: Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Dde)-ОН и N-(1-Fmoc-пиперидин-4-ил)-L-пролин. Анализ методом ВЭЖХ и МС проводят, как описано в синтезе соединения (1). См. Подробное описание выше. В значительной степени следуют схеме, показанной на фиг.2, за исключением того, что сочетание опосредовано скорее HATU/DIEA, чем DIC.
[00303] Полностью защищенный, связанный со смолой пептид синтезируют вручную, используя в качестве исходных материалов 2-хлортритилхлоридную смолу (3,2 г, 2,4 ммоль; NeoMPS). Присоединение первой аминокислоты к смоле обеспечивают обработкой смесью N-(1-Fmoc-пиперидин-4-ил)-L-пролин (2,0 г, 4,8 ммоль) и DIEA (3,3 мл, 19,2 ммоль) в ДХМ (40 мл) и ДМФА (10 мл) при комнатной температуре в течение 4 час. Смолу промывают 3×ДХМ/MeOH/DIEA (об./об./об.=17:2:1) и 3×ДХМ, сушат под вакуумом. Полученную смолу (3,7 г; N-(4-пиперидинил)-L-пролин-[2-Cl-Trt смола]) делят на несколько порций, и порцию 1,9 г (1,2 ммоль) используют для продолжения синтеза пептида. Опосредованные HATU/DIEA одинарные сочетания проводят с 3-хкратным избытком производных аминокислот. Группу Fmoc удаляют с помощью 25% пиперидина в ДМФА. По окончании удлинения пептидной цепи смолу обрабатывают 4% гидразина в ДМФА трижды на протяжении 3 мин для удаления Dde. Смолу промывают ДМФА и ДХМ и сушат под вакуумом. Полученную пептидную смолу (2,1 г; Boc-D-Phe-D-Phe-D-Leu-D-Lys-N-(4-пиперидинил)-L-пролин-[2-Cl-Trt смола]) делят снова, и часть 0,7 г (0,4 ммоль) используют для последующего расщепления. Пептидную смолу обрабатывают смесью ТФУ/TIS/H2O (15 мл, об./об./об.=95:2,5:2,5) при комнатной температуре в течение 90 мин. Смолу отфильтровывают и промывают ТФУ. Фильтрат упаривают под вакуумом, и сырой пептид (220 мг, D-Phe-D-Phe-D-Leu-D-Lys-[N-(4-пиперидинил)-L-пролин]-ОН) осаждают из диэтилового эфира.
[00304] Для очистки описанный выше сырой пептид (220 мг) растворяют в 0,1% ТФУ в H2O (50 мл), раствор загружают на колонку ВЭЖХ и очищают с использованием буферной системы ТФУ (буферные растворы: А=0,1% ТФУ в H2O и В=0,1% ТФУ в 60% ACN/40% H2O). Соединение элюируют с линейным градиентом от буфера В, 25% В до 75% В на протяжении 25 мин, tR=43% В. Фракции с чистотой свыше 97% объединяют, замораживают и сушат на лиофилизаторе с получением очищенного пептида в виде белого аморфного порошка (89 мг). Анализ методом ВЭЖХ: tR=18,22 мин, чистота 99,5%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 734,5, зарегистрировано 734,4.
ПРИМЕР 5: Синтез соединения (5)
[00305] D-Phe-D-Phe-D-Leu-D-Har-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 3):
[00306] Пептид-смолу: Вос-D-Phe-D-Phe-D-Leu-D-Lys-N-(4-пиперидинил)-L-пролин-[2-Cl-Trt смола], которую получают в ходе синтеза соединения (4), описанного выше, используют в качестве исходного материала. Анализ методом ВЭЖХ и МС проводят, как описано в синтезе соединения (1) выше.
[00307] Для гуанилирования ω-аминогруппы D-Lys в положении Хаа4, пептидную смолу (0,7 г, 0,4 ммоль) обрабатывают смесью 1Н-пиразол-1-карбоксамидина гидрохлорида (0,6 г, 4,0 ммоль) и DIEA (0,7 мл, 4,0 ммоль) в ДМФА (15 мл) в течение ночи при комнатной температуре. Смолу промывают ДМФА и ДХМ и сушат под вакуумом. Далее пептид отщепляют от смолы обработкой смесью ТФУ/TIS/H2O (15 мл, об./об./об.=95:2,5:2,5) при комнатной температуре в течение 90 мин. Затем смолу фильтруют и промывают ТФУ. фильтрат упаривают под вакуумом, и сырой пептид (170 мг; D-Phe-D-Phe-D-Leu-D-Har-[N-(4-пиперидинил)-L-пролин]-ОН) осаждают из диэтилового эфира.
[00308] Для очистки описанный выше сырой пептид (170 мг) растворяют в 0,1% ТФУ в Н20 (50 мл), раствор загружают на колонку ВЭЖХ и очищают с использованием буферной системы ТФУ (буферные растворы: А=0,1% ТФУ в H2O и В=0,1% ТФУ в 60% ACN/40% H2O). Соединение элюируют с линейным градиентом от буфера В, 25% В до 75% В на протяжении 25 мин, tR=46% В. Фракции с чистотой свыше 97% объединяют, замораживают и лиофилизируют с получением очищенного пептида в виде белого аморфного порошка (81 мг). Анализ методом ВЭЖХ: tR=19,42 мин, чистота 100%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+H+): ожидаемая масса молекулярного иона 776,5, зарегистрировано 776,5.
ПРИМЕР 6: Синтез соединения (6)
[00309] D-Phe-D-Phe-D-Leu-(ε-Me)D-Lys-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 1):
[00310] Синтез инициируют с помощью 0,7 г (0,4 ммоль) пептидной смолы, Boc-D-Phe-D-Phe-D-Leu-D-Lys-N-(4-пиперидинил)-L-пролин-[2-Cl-Trt смола], которую получают в ходе синтеза соединения (4), как описано выше. Анализ методом ВЭЖХ и МС проводят, как описано в синтезе соединения (1) выше. В этом случае, пептид Xaa1-Хаа4 предварительно синтезируют и сочетают, в противоположность постадийной сборке пептида, показанной на фиг.2.
[00311] Для метилирования ω-аминогруппы D-Lys в положении Xaa4 следуют 3-хстадийной методике, как описано в синтезе соединения (1) выше. Связанный со смолой метилированный пептид (Boc-D-Phe-D-Phe-D-Leu-(ε-Ме)D-Lys-N-(4-пиперидинил)-L-пролин-[2-Cl-Trt смола]) далее обрабатывают смесью ТФУ/TIS/H2O (15 мл, об./об./об.=95:2,5:2,5) при комнатной температуре в течение 90 минут. Смолу отфильтровывают и промывают ТФУ. Фильтрат упаривают под вакуумом, и сырой пептид (200 мг; D-Phe-D-Phe-D-Leu-(ε-Me)D-Lys-[N-(4-пиперидинил)-L-пролин]-ОН) осаждают из диэтилового эфира.
[00312] Для очистки описанный выше сырой пептид (200 мг) растворяют в 0,1% ТФУ в H2O (50 мл), раствор загружают на колонку ВЭЖХ и очищают с использованием буферной системы ТФУ (буферные растворы: А=0,1% ТФУ в H2O и В=0,1% ТФУ в 60% ACN/40% H2O). Соединение элюируют с линейным градиентом от 25% до 75% буферного раствора В на протяжении 30 мин, tR=42% В. Фракции с чистотой свыше 97% объединяют, замораживают и сушат на лиофилизаторе с получением очищенного пептида в виде белого аморфного порошка (41 мг). Анализ методом ВЭЖХ: tR=18,66 мин, чистота 98,1%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 748,5, зарегистрировано 748,5.
ПРИМЕР 7: Синтез соединения (7)
[00313] D-Phe-D-Phe-D-Leu-D-Arg-[гомопиперазина амид] (SEQ ID NO: 4):
[00314] Использовали следующие производные аминокислот: Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu-OH и Fmoc-D-Arg(Pbf)-ОН. Анализ методом ВЭЖХ и МС проводят, как описано в синтезе соединения (1), описанного выше. Полностью защищенный, связанный со смолой пептид синтезируют на синтезаторе SYMPHONY Multiple Synthesizer (Protein Technology Inc.), используя в качестве исходных материалов гомопиперазин-карбаматную смолу Wang (0,35 ммоль; гомопиперазин-[карбаматная смола Wang]), полученную в ходе синтеза соединения (1). Опосредованные HBTU/DIEA одинарные сочетания проводят с 4-хкратным избытком производных аминокислот.Группу Fmoc удаляют с помощью 25% пиперидина в ДМФА. После окончания автоматизированного синтеза, пептидную смолу (Boc-D-Phe-D-Phe-D-Leu-D-Arg(Pbf)-[гомопиперазина амид]) переносят в емкость для синтеза пептида вручную и обрабатывают смесью ТФУ/TIS/H2O (15 мл, об./об./об.=95:2,5:2,5) при комнатной температуре в течение 90 мин. Смолу отфильтровывают и промывают ТФУ. Фильтрат упаривают под вакуумом, и сырой пептид (380 мг; D-Phe-D-Phe-D-Leu-D-Arg-[гомопиперазина амид]) осаждают из диэтилового эфира.
[00315] Для очистки описанный выше сырой пептид (380 мг) растворяют в 0,1% ТФУ в H2O (50 мл), раствор загружают на колонку ВЭЖХ и очищают с использованием буферной системы ТФУ (буферные растворы: А=0,1% ТФУ в H2O и В=0,1% ТФУ в 60% ACN/40% H2O). Соединение элюируют с линейным градиентом от буфера В, от 25% В до 75% В на протяжении 25 мин, tR=36% В. Фракции с чистотой свыше 97% объединяют, замораживают и лиофилизируют с получением очищенного пептида в виде белого аморфного порошка (222 мг). Анализ методом ВЭЖХ: tR=16,75 мин, чистота 100%, градиент 2% В до 22% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 664,4, зарегистрировано 664,5.
ПРИМЕР 8: Синтез соединения (8)
[00316] D-Phe-D-Phe-D-Leu-D-Har-[ω(4-аминопиперидин-4-карбоновая кислота]-ОН (SEQ ID NO: 3):
[00317] Указанное соединение получают в существенной мере в соответствии с методикой, описанной выше для синтеза соединения (5), за исключением того, что N-Boc-амино-(4-N-Fmoc-пиперидинил)карбоновая кислота заменена на N-(1-Fmoc-пиперидин-4-ил)-L-пролин в процессе присоединения к смоле 2-Cl-Trt. Конечный очищенный пептид: аморфный порошок, выход 85 мг в масштабах синтеза 1 ммоль. Анализ методом ВЭЖХ: tR=17,87 мин, чистота 100%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 722,4, зарегистрировано 722,5.
ПРИМЕР 9: Синтез соединения (9)
[00318] D-Phe-D-Phe-D-Leu-(ε-iPr)D-Lys-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 5):
[00319] Синтез инициируют из 0,15 ммоль пептидной смолы, Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N-Boc-амино-4-пиперидинилкарбоновая кислота)-[2-Cl-Trt смола]), которую получают в ходе синтеза соединения (2) выше. Для изопропилирования ω-аминогруппы D-Lys в положении Xaa4, пептидную смолу обрабатывают смесью натрия триацетоксиборгидрида (3 ммоль) и ацетона (6 ммоль) в TMOF (10 мл) в течение 4 час при комнатной температуре. Последующие стадии отщепления и очистки осуществляют в соответствии с методикой, описанной в синтезе соединения (2). Конечный очищенный пептид: аморфный порошок, выход 67 мг. Анализ методом ВЭЖХ: tR=19,29 мин, чистота 98,4%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 722,5, зарегистрировано 722,5.
ПРИМЕР 10: Синтез соединения (10):
[00320] D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 6):
[00321] См. схему на фиг.3. Использовали следующие производные аминокислот: Boc-D-Phe-OH, Boc-D-Phe-OH, Boc-D-Leu-OH, Boc-D-Dap(Fmoc)-ОН и N-Fmoc-амино-(4-N-Вос-пиперидинил) карбоновая кислота. Анализ методом ВЭЖХ и МС проводят, как описано в синтезе соединения (1). Полностью защищенный, связанный со смолой пептид синтезируют вручную, начиная с 4-Fmoc-гидразинобензоильной смолы AM NovaGel (3 ммоль; Novabiochem). Защитную группу Fmoc на исходной смоле сначала удаляют обработкой 25% пиперидина в ДМФА, и смолу затем обрабатывают смесью N-Fmoc-амино-(4-N-Boc-пиперидинил)карбоновой кислоты (7,5 ммоль), PyBOP (7,5 ммоль) и DIEA (15 ммоль) в ДМФА в течение ночи при комнатной температуре. Группу Fmoc на присоединенной аминокислоте заменяют на o-NBS в 2 стадии: (i) удаление Fmoc обработкой 25% пиперидина в ДМФА. (ii) защита о-NBS в соответствии с методикой, описанной в синтезе соединения (1). Полученную пептидную смолу, N-o-NBS-амино-(4-N-Вос-пиперидинил)карбоновая кислота-[гидразинобензоильная смола AM NovaGel], делят на несколько порций, и порцию 1 ммоль используют для продолжения синтеза пептида. Опосредованные PyBOP/DIEA одинарные сочетания проводят с 3-хкратным избытком производных аминокислот. Группу Вое удаляют с помощью 30% ТФУ в ДХМ. По окончании удлинения пептидной цепи смолу обрабатывают 2% DBU в ДМФА на протяжении 2х8 мин для удаления Fmoc, с последующим гуанилированием ω-аминогруппы D-Dap в положении Xaa4 в соответствии с методикой, описанной в синтезе соединения (5), выше. Окончательное снятие защиты o-NBS осуществляют в соответствии с методикой, описанной в синтезе соединения (1).
[00322] Для окислительного расщепления, высушенную пептидную смолу смешивают со смесью Cu(OAc)2 (1 ммоль), пиридина (4 ммоль) и DBU (2 ммоль) в 5% Н2О в ДМФА и пропускают воздух сквозь смолу на протяжении 6 час при комнатной температуре. Смолу отфильтровывают, промывают ДМФА, и фильтрат выпаривают под вакуумом. Остаток, Boc-D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН, обрабатывают 95% ТФУ в H2O для удаления Boc. Раствор выпаривают под вакуумом, и сырой пептид (1 ммоль; D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН) осаждают из диэтилового эфира.
[00323] Очистку описанного выше сырого пептида обеспечивают в соответствии с протоколом, описанным в синтезе соединения (2). Очищенный пептид представляет собой аморфный порошок (16 мг). Анализ методом ВЭЖХ: tR=16,97 мин, чистота 99,9%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 680,4, зарегистрировано 680,4.
ПРИМЕР 11: Синтез соединения (11)
[00324] D-Phe-D-Phe-D-Leu-D-Nar-[ω(4-аминопиперидин-4-карбоновая кислота)]-ОН (SEQ ID NO: 7):
[00325] Указанное соединение получают в соответствии с методикой, описанной в синтезе соединения (10), за исключением того, что Boc-D-Dbu(Fmoc)-ОН заменяют на Boc-D-Dap(Fmoc)-ОН в сочетании производных аминокислоты в положении Хаа4. Конечный очищенный пептид: аморфный порошок, выход 23 мг в масштабах синтеза 1 ммоль. Анализ методом ВЭЖХ: tp=17,12 мин, чистота 99,2%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 694,4, зарегистрировано 694,5.
ПРИМЕР 12: Синтез соединения (12)
[00326] D-Phe-D-Phe-D-Leu-D-Dbu-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 8):
[00327] Указанное соединение получают в соответствии с методикой, описанной в синтезе соединения (4), как описано выше. Отличие состоит в замене Fmoc-D-Dbu(ivDde)-ОН для Fmoc-D-Lys(Dde)-ОН в сочетании производных аминокислот в положении Xaa4. Конечный очищенный пептид: аморфный порошок, выход 7 мг в масштабах синтеза 0,4 ммоль. Анализ методом ВЭЖХ: tR=18,15 мин, чистота 98,9%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 706,4, зарегистрировано 706,4.
ПРИМЕР 13: Синтез соединения (13)
[00328] D-Phe-D-Phe-D-Leu-D-Nar-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 7):
[00329] Синтез инициируют из 0,4 ммоль пептидной смолы, Boc-D-Phe-D-Phe-D-Leu-D-Dbu-N-(4-пиперидинил)-L-пролин-[2-Cl-Trt смола], которую получают в ходе синтеза соединения (12). Гуанилирование ω-аминогруппы D-Dbu в положении Xaa4 обеспечивают в соответствии с методикой, описанной в синтезе соединения (5), выше. Последующие стадии расщепления и очистки осуществляют в соответствии с методикой, описанной в синтезе соединения (1). Конечный очищенный пептид: аморфный порошок, выход 7 мг. Анализ методом ВЭЖХ: tR=18,68 мин, чистота 97,3%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 748,5, зарегистрировано 748,5.
ПРИМЕР 14: Синтез соединения (14)
[00330] D-Phe-D-Phe-D-Phe-D-Dap(амидино)-[N-(4-пиперидинил)-L-пролин]-ОН (SEQ ID NO: 6):
[00331] Соединение получают в соответствии с методикой, описанной в синтезе соединения (13), за исключением того, что Fmoc-D-Dap(ivDde)-ОН заменяют на Fmoc-D-Dbu(ivDde)-ОН при сочетании производных аминокислот в положении Xaa4. Конечный очищенный пептид: аморфный порошок, выход 12 мг в масштабах синтеза 0,4 ммоль. Анализ методом ВЭЖХ: tR=18,55 мин, чистота 98,0%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 734,4, зарегистрировано 734,4.
ПРИМЕР 15: Синтез соединения (15)
[00332] D-Phe-D-Phe-D-Leu-D-Lys-[4-амидиногомопиперазина амид] (SEQ ID NO: 2):
[00333] Соединение получают в соответствии с методикой, описанной в синтезе соединения (1), за исключением того, что отсутствует стадия метилирования ω-аминогруппы D-Lys в положении Xaa4. Конечный очищенный пептид: аморфный порошок, выход 140 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=14,02 мин, чистота 99,3%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 678,4, зарегистрировано 678,5.
ПРИМЕР 16: Синтез соединения (16)
[00334] D-Phe-D-Phe-D-Leu-D-Har-[4-амидиногомопиперазина амид] (SEQ ID NO: 3):
[00335] Соединение получают в соответствии с методикой, описанной в синтезе соединения (1), за исключением того, что стадия гуанилирования заменена на метилирование ω-аминогруппы D-Lys в положении Xaa4. Гуанилирование проводят в соответствии с методикой, описанной в синтезе соединения (5) выше. Конечный очищенный пептид: аморфный порошок, выход 173 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=15,05 мин, чистота 98,6%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 720,5, зарегистрировано 720,5.
ПРИМЕР 17: Синтез соединения (17)
[00336] D-Phe-D-Phe-D-Leu-(ε-iPr)D-Lys-[4-амидиногомопиперазина амид] (SEQ ID NO: 5):
[00337] Соединение получают в соответствии с методикой, описанной в синтезе соединения (1), за исключением того, что стадия изопропилирования заменена на метилирование ω-аминогруппы D-Lys в положении Xaa4. Изопропилирование осуществляют в соответствии с методикой, описанной в синтезе соединения (9). Конечный очищенный пептид: аморфный порошок, выход 233 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=16,16 мин, чистота 94,5%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 720,5, зарегистрировано 720,5.
ПРИМЕР 18: Синтез соединения (18)
[00338] D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[4-Амидиногомопиперазина амид] (SEQ ID NO: 6):
[00339] Соединение получают в соответствии с методикой, описанной в синтезе соединения (16), за исключением замены Fmoc-D-Dap(ivDde)-ОН на Fmoc-D-Lys(Dde)-OH в сочетании производных аминокислот в положении Хаа4. Конечный очищенный пептид: аморфный порошок, выход 155 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=14,44 мин, чистота 99,1%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 678,4, зарегистрировано 678,5.
ПРИМЕР 19: Синтез соединения (19)
[00340] D-Phe-D-Phe-D-Nle-(β-амидино)D-Dap-[4-Амидиногомопиперазина амид] (SEQ ID NO: 6):
[00341] Соединение получают в соответствии с методикой, описанной в синтезе соединения (18) выше, за исключением замены Fmoc-D-Nle-OH на Fmoc-D-Leu-OH при сочетании производных аминокислот в положении Xaa3. Конечный очищенный пептид: аморфный порошок, выход 190 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=14,69 мин, чистота 98,9%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+); ожидаемая масса молекулярного иона 678,2, зарегистрировано 678,5.
ПРИМЕР 20: Синтез соединения (20)
[00342] D-Phe-D-Phe-D-Leu-(β-амидино)D-Dap-[гомопиперазина амид] (SEQ ID NO: 6):
[00343] Соединение получают в соответствии с методикой, описанной в синтезе соединения (18) выше, за исключением того, что отсутствует гуанилирование гомопиперазина на С-конце. Конечный очищенный пептид: аморфный порошок, выход 172 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=13,84 мин, чистота 99,1%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 636,4, зарегистрировано 636,5.
ПРИМЕР 21: Синтез соединения (21)
[00344] D-Phe-D-Phe-D-Nle-(β-амидино)D-Dap-[гомопиперазина амид] (SEQ ID NO: 6):
[00345] Соединение получают в соответствии с методикой, описанной в синтезе соединения (19), за исключением того, что отсутствует гуанилирование гомопиперазина на С-конце. Конечный очищенный пептид: аморфный порошок, выход 149 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=14,06 мин, чистота 98,5%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 636,4, зарегистрировано 636,5.
ПРИМЕР 22: Синтез соединения (22)
[00346] D-Phe-D-Phe-D-Leu-D-Dbu-[4-амидиногомопиперазина амид] (SEQ ID NO: 8):
[00347] Соединение получают в соответствии с методикой, описанной в синтезе соединения (15) за исключением замены Fmoc-D-Dbu(ivDde)-ОН на Fmoc-D-Lys(Dde)-OH при сочетании производных аминокислот в положении Хаа4. Конечный очищенный пептид: аморфный порошок, выход 152 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=14,03 мин, чистота 98,1%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 650,4, зарегистрировано 650,5.
ПРИМЕР 23: Синтез соединения (23)
[00348] D-Phe-D-Phe-D-Leu-D-Nar-[4-Амидиногомопиперазина амид] (SEQ ID NO: 7):
[00349] Соединение получают в соответствии с методикой, описанной в синтезе соединения (16), за исключением замены Fmoc-D-Dbu(ivDde)-ОН на Fmoc-D-Lys(Dde)-OH при сочетании производных аминокислот в положении Xaa4. Конечный очищенный пептид: аморфный порошок, выход 227 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=14,37 мин, чистота 99,3%, градиент от 5% В до 25% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 664,4, зарегистрировано 664,5.
ПРИМЕР 24; Синтез соединения (24)
[00350] D-Phe-D-Phe-D-Leu-D-Arg-[4-амидиногомопиперазина амид] (SEQ ID NO: 4):
[00351] Соединение получают гуанилированием гомопиперазина на С-конце Bcz-D-Phe-D-Phe-D-Leu-D-Arg-(гомопиперазина амид], который синтезируют в соответствии с методикой, описанной в синтезе соединения (7) выше. Последующее расщепление и чистку осуществляют в соответствии с методикой, описанной в синтезе соединения (1), выше. Конечный очищенный пептид: аморфный порошок, выход 102 мг в масштабах синтеза 0,3 ммоль. Анализ методом ВЭЖХ: tR=17,34 мин, чистота 98,4%, градиент 2% В до 22% В на протяжении 20 мин; МС (М+Н+): ожидаемая масса молекулярного иона 706,5, зарегистрировано 706,5.
ПРИМЕР 25: Синтез соединения (25):
[00352] D-Phe-D-Phe-D-Leu-D-Lys-[2,8-диазаспиро[4,5]декан-1-он амид] (SEQ ID NO: 2):
[00353] Указанный синтез осуществляют в соответствии со схемой, показанной на фиг.4. Описанные ниже полупродукты соответствуют показанным на фиг.4. К суспензии Boc-D-Phe-OH промежуточного соединения 1-1 (7,96 г, 30,0 ммоль), D-Leu-OBn п-TsOH промежуточного соединения 1-2 (11,80 г, 30,0 ммоль), HOBt моногидрата (4,46 г, 33,0 ммоль) и DIEA (8,53 г, 66,0 ммоль) в безводном ТГФ (250 мл), охлажденной на бане, содержащей воду со льдом, добавляют EDCI (6,33 г, 33,0 ммоль) в виде 4-х порций на протяжении 20 минут с интервалом 5 минут между порциями. Суспензию перемешивают в течение ночи от исходной температуры 0°С до комнатной температуры. После выпаривания ТГФ, остаток растворяют в этилацетате и последовательно промывают 10% раствором лимонной кислоты, насыщенным раствором NaHCO3 и водой. Органическую фракцию сушат над натрия сульфатом и выпаривают при сниженном давлении. Остаток растворяют в ДХМ, пропускают сквозь слой силикагеля и элюируют 20% этилацетатом в гексане. Елюат упаривают с получением чистого продукта, Boc-D-Phe-D-Leu-OBn, промежуточного соединения 1-3 (12,40 г, 88%) в виде прозрачного масла. ЖХ-МС: соотношение массы к заряду=469 (М+Н).
[00354] Промежуточное соединение 1-3 (12,40 г, 26,5 ммоль) растворяют в ДХМ (50 мл). Добавляют ТФУ (25 мл), и раствор перемешивают при комнатной температуре в течение 2 час. После выпаривания ДХМ и ТФУ, дважды осуществляют азеотропную перегонку остатка с толуолом с получением соли ТФУ D-Phe-Leu-ОВп, промежуточного соединения 1-4. Полученный сырой дипептид суспендируют в ТГФ, и добавляют Boc-D-Phe-OH (6,36 г, 24 ммоль), HOBt моногидрат (4,04 г, 26,4 ммоль) и DIEA (8,7 мл, 50,0 ммоль) при 0°C. Добавляют EDCI (6,33 г, 6,4 ммоль) в виде 4-х порций на протяжении 20 минут с интервалом 5 минут между порциями. Суспензию перемешивают при температуре от 0°C до комнатной температуры в течение ночи. После выпаривания ТГФ, остаток растворяют в этилацетате и промывают последовательно 10% раствором лимонной кислоты, насыщенным раствором NaHCO3 и водой. Органическую фракцию сушат над натрия сульфатом и выпаривают при сниженном давлении. Остаток перекристаллизуют из 400 мл смеси ацетон/гексан (1:3) с получением 9,1 г чистого продукта. Маточник упаривают и снова перекристаллизуют из смеси ацетон/гексан (1:3) с получением 2,0 г продукта. Общий выход составляет 11,1 г (68% для 2 стадий). ЖХ-МС: соотношение массы к заряду=616 (М+Н).
[00355] В колбу, продутую азотом, помещают влажный палладий на угле (1,8 г) и раствор Boc-D-Phe-D-Phe-D-Leu-OBn, промежуточного соединения 1-5 (11,1 г, 18,05 ммоль) в метаноле (50 мл). Смесь перемешивают в атмосфере водорода (баллон) в течение ночи. После фильтрации сквозь броунмиллерит, метанол выпаривают при сниженном давлении. Остаток растворяют в ацетоне (20 мл) и медленно добавляют к 500 мл воды с 25 мл 1 н НС1 при энергичном перемешивании. Чистый продукт Boc-D-Phe-D-Phe-D-Leu-OH, промежуточное соединение 1-6, получают фильтрацией 9,4 г (99%). ЖХ-МС: соотношение массы к заряду=526 (М+Н).
[00356] К раствору промежуточное соединение 1-6 (2,06 г, 3,90 ммоль), D-Lys(Вое)-ОА11 гидрохлорида (1,26 г, 3,90 ммоль) и DIEA (1,7 мл, 9,8 ммоль) в ДМФА добавляют TBTU (1,56 г, 4,88 ммоль) в виде 3-х порций на протяжении 15 мин при 0°C. После перемешивания в течение ночи при температуре от 0°C до комнатной температуры, ДМФА выпаривают под глубоким вакуумом. Сырую реакционную смесь осаждают в 400 мл смеси воды со льдом и фильтруют для отделения осадка, Boc-D-Phe-D-Phe-D-Leu-D-Lys(Boc)-OAll, промежуточное соединение I-7 (2,60 г), который использовали без дополнительной очистки на следующей стадии.
[00357] К раствору промежуточного соединения 1-7 (2,60 г, 3,3 ммоль) в MeCN (75 мл) добавляют пирролидин (1,1 мл, 13,3 ммоль) и тетракис(трифенилфосфин)палладия (400 мг, 0,35 ммоль) при 0°C. Реакционную смесь перемешивают при комнатной температуре в течение 3 час и выпаривают до сухого состояния. Остаток очищают обращенно-фазовой колоночной хроматографией с использованием смеси от 30% MeCN/вода до 90% MeCN/вода с получением чистой кислоты, промежуточного соединения 1-8 (2,0 г, 80%) после выпаривания смеси ацетонитрил/вода. ЖХ-МС: соотношение массы к заряду=754 (М+Н).
[00358] К раствору кислоты, промежуточного соединения I-8 (150 мг, 0,20 ммоль), амина HNRaRb, 2,8-диазаспиро-[4,5]декан-1-она (57 мг, 0,30 ммоль) и DIEA (175 мкл, 1,0 ммоль) в ДМФА (5 мл) добавляют HBTU (113 мг, 3,0 ммоль) при 0°C. После перемешивания в течение ночи при температуре от 0°C до комнатной температуры, ДМФА выпаривают при сниженном давлении. Остаток перемешивают с 4 н HCl в 1,4-диоксане (2,0 мл) при комнатной температуре в течение 1 час. После удаления диоксана, остаток растворяют в воде и очищают обращенно-фазовой колоночной хроматографией с градиентом от смеси 10% MeCN/вода до смеси 60% MeCN/вода на протяжении 30 минут с получением чистого продукта, соединение (25) (108 мг, выход 78% для 2 стадий) после выпаривания растворителя. ЖХ-МС: соотношение массы к заряду=690 (М+Н).
ПРИМЕР 26: Синтез соединения (26):
[00359] D-Phe-D-Phe-D-Leu-D-Lys-[2-метил-2,8-диазаспиро[4,5]декан-1-он амид] (SEQ ID NO: 2):
[00360] Соединение (26) получают в существенной мере таким же способом, как описано для соединения 25, выше, за исключением того, что амин (HNRaRb на схема фиг.4) на стадии концевого сочетания с амидом представляет собой 2-метил-2,8-диазаспиро[4,5]декан-1-он вместо 2,8-диазаспиро [4,5]декан-1-она. ЖХ-МС: соотношение массы к заряду=704 (М+Н).
ПРИМЕР 27: Синтез соединения (27):
[00361] D-Phe-D-Phe-D-Leu-D-Lys-[1,3,8-триазаспиро[4,5]декан-2,4-дион амид] (SEQ ID NO: 2):
[00362] Соединение (27) получают в существенной мере таким же способом, как описано для соединения 25 выше, за исключением того, что на последней стадии используют амин 1,3,8-триазаспиро[4,5]декан-2,4-дион. ЖХ-МС: соотношение массы к заряду=705 (М+Н).
ПРИМЕР 28: Синтез соединения (28):
[00363] D-Phe-D-Phe-D-Leu-D-Lys-[5-хлор-1-(пиперидин-4-ил)-1H-бензо[d]имидазол-2(3)Н-он амид] (SEQ ID NO: 2):
[00364] Соединение (28) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что амин 5-хлор-1-(пиперидин-4-ил)-1H-бензо[d]имидазол-2(3)Н-он используют. ЖХ-МС: соотношение массы к заряду=394.
ПРИМЕР 29: Синтез соединения (29):
[00365] D-Phe-D-Phe-D-Leu-D-Lys-[морфолино(пиперидин-4-ил)метанона амид] (SEQ ID NO: 2):
[00366] Соединение (29) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин морфолино(пиперидин-4-ил)метанон. ЖХ-МС: соотношение массы к заряду=366.
ПРИМЕР 30: Синтез соединения (30):
[00367] D-Phe-D-Phe-D-Leu-D-Lys-[4-фенил-1-(пиперидин-ил-1Н-имидазол-2(3Н)-он амид] (SEQ ID NO: 2):
[00368] Соединение (30) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин 4-фенил-1-(пиперидин-ил-1Н-имидазол-2(3Н)-он. ЖХ-МС: соотношение массы к заряду=779 (М+Н).
ПРИМЕР 31: Синтез соединения (31):
[00369] D-Phe-D-Phe-D-Leu-D-Lys-[4-(3,5-диметил-4H-1,2,4-триазол-4-ил)пиперидин амид] (SEQ ID NO: 2):
[00370] Соединение (31) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин 4-(3,5-диметил-4H-1,2,4-триазол-4-ил)пиперидин. ЖХ-МС: соотношение массы к заряду=716 (М+Н).
ПРИМЕР 32: Синтез соединения (32):
[00371] D-Phe-D-Phe-D-Leu-D-Lys-[1-(пиперидин-4-ил)индолин-2-он амид] (SEQ ID NO: 2):
[00372] Соединение (32) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин 1-(пиперидин-4-ил)индолин-2-он.
[00373] ЖХ-МС: соотношение массы к заряду=752 (М+Н).
ПРИМЕР 33: Синтез соединения (33):
[00374] D-Phe-D-Phe-D-Leu-D-Lys-[1-фенил-1,3,8-триазаспиро[4,5]декан-4-он амид] (SEQ ID NO: 2):
[00375] Соединение (33) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин 1-фенил-1,3,8-триазаспиро[4,5]декан-4-он. ЖХ-МС: соотношение массы к заряду=767 (М+Н).
ПРИМЕР 34: Синтез соединения (34):
[00376] D-Phe-D-Phe-D-Leu-D-Lys-[имидазо[1,2-а]пиридин-2-илметанамин амид] (SEQ ID NO: 2):
[00377] Соединение (34) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин имидазо[1,2-а]пиридин-2-илметанамин. ЖХ-МС: соотношение массы к заряду=683 (М+Н).
ПРИМЕР 35: Синтез соединения (35):
[00378] D-Phe-D-Phe-D-Leu-D-Lys-[(5-метилпиразин-2-ил)метиламин амид] (SEQ ID NO: 2):
[00379] Соединение (35) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что на последней стадии используют амин (5-метил пиразин-2-ил)метанамин. ЖХ-МС: соотношение массы к заряду=659 (М+Н).
ПРИМЕР 36: Синтез соединения (36):
[00380] D-Phe-D-Phe-D-Leu-D-Lys-[1-(пиперидин-4-ил)-1H-бензо[d]имидазол-2(3Н)-он амид] (SEQ ID NO: 2):
[00381] Соединение (36) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин 1-(пиперидин-4-ил)-1H-бензо[d]имидазол-2(3Н)-он. ЖХ-МС: соотношение массы к заряду=753 (М+Н).
ПРИМЕР 37: Синтез соединения (37):
[00382] D-Phe-D-Phe-D-Leu-D-Lys-[4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридин амид] (SEQ ID NO: 2):
[00383] Соединение (37) получают в существенной мере таким же способом, как описано выше для соединения 25, за исключением того, что используют амин 4,5,6,7-тетрагидро-3-Я-пиразоло [4, 3-c] пиридин. ЖХ-МС: соотношение массы к заряду=659 (М+Н).
Притер 38: Подтверждение структуры синтетических пептидных амидов 1-24.
[00384] В табл.I приведена рассчитанная молекулярная масса молекулярного иона, NH+ для каждого соединения, и фактическая молекулярная масса, зарегистрированная методом масс-спектрометрии. Также показан тип фазы синтеза, использованной в синтезе каждого соединения: твердой фазы или смешанной; и тип смолы, которую используют в синтезе, где смола представляет собой "2-хлортритил 2-Cl-Trt" смолу, гидразинобензоил "гидразиновую" смолу Rink AM или п-нитрофенил-карбонатную (Wang) "карбонатную" смолу. Номер фигуры, где показана соответствующая схема синтеза для синтеза каждого соединения, приведен в последней колонке.
ПРИМЕР 39: Ингибирование выработки цАМФ стимулированием эндогенного каппа-опиатного рецептора мышей в R1.G1 клетках.
[00385] Активность синтетических пептидных амидов как агонистов каппа-опиатного рецептора определяют измерением ингибирования стимулированной форсколином активности аденилатциклазы. Клетки R1.G1 (линия клетки тимомы мыши, которая экспрессирует только каппа-опиатный рецептор и не экспрессирует других подтипов опиатного рецептора) сначала подвергают обработке форсколином (для индукции цАМФ) плюс синтетическим пептидным амидом в исследуемой концентрации. После инкубации, уровень цАМФ в обработанных клетках R1.G1 определяют, используя иммуноанализ цАМФ на основе переноса энергии флуоресцентного резонанса с разрешением во времени (TR-FRET, LANCE™, Perkin Elmer). Ниже способ описан подробно.
[00386] Мышиные клетки R1.G1 (АТСС, Manassas, VA) культивируют в суспензии в среде DMEM с высоким содержанием глюкозы (модифицированная Дульбекко среда Игла, Cellgro, Herndon, VA), содержащей 10% лошадиной сыворотки и 2% glutaMax (Invitrogen, Carlsbad. CA) без добавления антибиотиков. В день эксперимента, клетки центрифугируют со скоростью 1000 об./мин в течение 5 минут при комнатной температуре, а затем промывают 1 раз HBSS (буферизованный солевой раствор HEPES, Invitrogen, Carlsbad, CA). Далее клетки снова центрифугируют и ресуспендируют в буферном растворе для стимуляции (HBSS с добавлением 0,05% альбумина телячьей сыворотки-FAF [свободный от жирных кислот альбумин телячьей сыворотки, Roche Applied Science, Indianapolis, IN], 5 мМ HEPES) в концентрации 2 млн клеток/мл. Антитело, поставляемое в наборе для иммуноанализа цАМФ LANCE™, далее добавляют к клеткам согласно инструкции производителя, и 12 ООО клеток/лунку помещают в лунки, содержащие форсколин в предварительно определенной конечной концентрации (обычно приблизительно 2,5 мкМ) и предварительно определенное количество исследуемого синтетического пептидного амида. Синтетические пептидные амиды исследовали в интервале концентраций, чтобы определить их активность. Клетки инкубировали с синтетическими пептидными амидами плюс форсколином в течение приблизительно 20 минут при комнатной температуре. После инкубации, осуществляли лизис клеток, добавляя 12 мкл проявляющей смеси, поставляемой в наборе LANCE™, с последующей инкубацией в течение 1 час при комнатной температуре. Флуоресценцию с разрешением во времени измеряли, используя фильтр возбуждения 330-380 нм, фильтр эмиссии 665 нм, дихроическое зеркало 380 и Z=1 мм. Стандартную кривую концентрации цАМФ в данном испытании позволяет определить количество цАМФ, присутствующее в каждой лунке. Кривую строили, нанося на график концентрации синтетического пептидного амида против уровня цАМФ в исследуемых клетках, и обрабатывают нелинейной регрессией с использованием алгоритма аппроксимации 4-параметрической кривой для вычисления EC50, концентрации синтетического пептидного амида, необходимой для достижения 50% максимального подавления выработки цАМФ синтетическими пептидными амидами. В табл.II показаны значения ЕС50, полученные в данном анализе для соединений синтетических пептидных амидов (1)-(36).
ПРИМЕР 40: Активность синтетических пептидных амидов в отношении каппа-опиатного рецептора человека
[00387] Эмбриональные почечные клетки человека (клетки НЕК-293, АТСС, Manassas, VA) в чашках диаметром 100 мм трансфицируют реактивом для трансфекции, Fugene6 (Roche Molecular Biochemicals) и конструктами ДНК в соотношении 3,3:1. Конструкты ДНК, применяемые для трансфекции, были такими, как указано ниже: (i) вектор экспрессии для человеческого каппа-опиатного рецептора, (ii) вектор экспрессии для человеческого химерного G-протеина и (in) конструкт репортера люциферазы, в котором экспрессию люциферазы индуцируют чувствительным к кальцию фактором транскрипции NFAT.
[00388] Вектор экспрессии, содержащий человеческую каппа-опиатный рецептор, конструируют, как указано ниже: Человеческий ген OPRK1 клонируют из общей РНК спинного корневого ганглия человека методом ПЦР, и ген вставляют в вектор экспрессии pcDNA3 (Invitrogen, Carlsbad, CA), чтобы сконструировать вектор экспрессии млекопитающих pcDNA3-hOPRKl человеческого OPRK1.
[00389] Для конструирования вектора экспрессии химерного G-протеина человека, сначала конструируют химерный G-протеин Gaqi5 путем замены последних 5 аминокислот человеческого Gaq последовательностью последних 5 аминокислот Gai методом PCR. Вторую мутацию вводят в данный человеческий ген Gaqi5 в положении аминокислоты 66 для замены глицина (G) на аспарагиновую кислоту (D) методом сайт-направленного мутагенеза. Полученный ген далее субклонируют в вектор экспрессии млекопитающих pcDNA5/FRT (Invitrogen) с получением вектора экспрессии человеческого химерного G-протеина, pcDNA5/FRT-hGNAq-G66D-i5.
[00390] Для получения конструкта репортерного гена люциферазы, синтетические элементы реакции, в том числе 3 копии TRE (элементы 12-O-тетрадеканоилфорбол-13-ацетат-реагирующий) и 3 копии NFAT (ядерный фактор активированных Т-клеток) инкорпорируют в направлении 3'-5' по отношению к минимальному промотору c-fos.
Данный реагирующий элемент и кассету промотора затем вставляют в рСЬ3-основной вектор репортерного гена люциферазы (Promega), чтобы конструировать конструкт плазмиды репортерного гена люциферазы pGL3b-3TRE-3NFAT-cfos-Luc.
[00391] Смесь для трансфекции для каждого планшета клеток включала 6 мкм pcDNA3-hOPRKl, 6 мкг pcDNA5/FRT-hGNAq-G66D-i5 и 0,6 мкг pGL3b-3TRE-3NFAT-cfos-Luc. Клетки инкубируют в течение одного дня при 37°С во влажной атмосфере, содержащей 5% CO2, с последующей трансфекцией и нанесением на непрозрачные 96-луночные планшеты в концентрации 45000 клеток на лунку в 100 мкл средства. На следующий день, исследуемые соединения и соединения сравнения добавляют к клеткам в индивидуальных лунках. Интервал концентраций исследуемых соединений добавляют к одному набору лунок, и подобный интервал концентраций соединений сравнения добавляют к набору контрольных лунок. Клетки далее инкубируют в течение 5 часов при 37°С. В конце инкубации осуществляют лизис клеток, добавляя 100 мкл проявляющей смеси, содержащей субстрат люциферазы (AMP [22 мкг/мл], АТР [1,1 мг/мл], дитиотрейтол [3,85 мг/мл], HEPES [конечная концентрация 50 мМ], ЭДТА [0,2 мг/мл]. Тритон N-101 [4 мкл/мл], фенилуксусную кислоту [45 мкг/мл], щавелевую кислоту [8,5 мкг/мл], люциферин [28 мкг/мл], рН 7,8). Планшеты запечатывают и измеряют флуоресценцию в пределах 30 минут. Данные наносят на график концентрации каждого из соединений против импульсов флуоресценции в секунду (cps) и полученные кривые «доза-реакция» аппроксимируют нелинейной регрессией с использованием алгоритма 4-параметрической кривой для вычисления EC50 (концентрация соединения, необходимая для достижения 50% максимального увеличения активности люциферазы) и эффективности (максимальная активация в процентах по сравнению с полной индукцией любым из известных агонистов каппа-опиатного рецептора, таким как азимадолин (EMD-61753: см. Joshi et al., 2000, J. Neuroscl. 20(15):5874-9), или U-69593: cm. Heidbreder et al., 1999, Brain Res. 616(1-2):335-8).
[00392] В табл.II показаны значения EC50, полученные в результате анализа ингибирования цАМФ с примерами соединений, синтезированных в соответствии с настоящим изобретением и исследованных на модели каппа-опиатного рецептора (mKOR) мышей с подтверждением результатов на модели человеческого каппа-опиатного рецептора (hKOR) вышеописанными способами.
[00393] Синтетические пептидные амиды по изобретению были исследованы в подобном анализе активности на модели человеческого мю-опиатного рецептора. Каждое из исследованных соединений продемонстрировало EC50 для человеческого мю-опиатного рецептора ≥1 мкМ.
ПРИМЕР 41 Влияние синтетических пептидных амидов на проницаемость мембраны
[00394] Линия клеток Сасо-2 представляет собой линию клеток аденокарциномы ободочной и прямой кишки человека, которая дифференцируется в культуре и используется для моделирования эпителиальной выстилки тонкого кишечника человека. Соединения по настоящему изобретению были исследованы в испытании на проницаемость мембран с использованием субклона ТС7 Сасо-2 в стандартном анализе (Сегер, Сиэтл, WA). Если коротко, коэффициент очевидной проницаемости (Рарр) был определен в апикально-базолатеральном направлении (А-В) через монослои клеток, культивирование на 96-луночных поликарбонатных мембранных фильтрах.
[00395] Соединения по изобретению были исследованы в такой концентрации, как 10 мкМ при рН 6,5 в 1% ДМСО, с реципиентной стороной, удерживаемой при рН 7,4. Планшет для анализа инкубировали в течение 60 минут при 37°C с осторожным встряхиванием. Образцы отбирали в нулевой точке времени с донорской стороны и в конце периода инкубации, как с донорской, так и с реципиентной сторон. Образцы предпочтительно анализировали методом ВЭЖХ-МС/МС. Значение (выраженное, как 10-6 см/сек.) Рарр далее вычисляют на основании скорости появления соединения на реципиентной стороне. Рарр вычисляли с использованием уравнения:
где Рарр - очевидная проницаемость; S - площадь поверхности мембраны, С0 - донорная концентрация в момент времени 0, и dQ/dt - количество лекарственного средства, перенесенного за период времени. Четыре соединения сравнения (лабеталол, пропранолол, ранитидин и винбластин) были исследованы параллельно, чтобы гарантировать валидность испытания, а также азимадолин, который доказанно является каппа-опиоидом периферического действия. Результаты показаны в табл.III.
Считается, что соединения, которые демонстрируют низкую проницаемость в данном анализе, обладают низким потенциалом пересечения гематоэнцефалического барьера in vivo, поскольку высокая пассивная проницаемость, по-видимому, является ключевой особенностью действующих на ЦНС лекарственных средств (Mahar Doan et al. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002; 303:1029-37).
ПРИМЕР 42 Ингибирование оксидаз цитохром Р450
[00396] Ингибирование изоферментов оксидазы цитохром Р450 CYP1A, CYP2C9, CYP2C19, CYP2D6 и CYP3A4 синтетическими соединениями пептидными амидами по изобретения определяли в соответствии со следующими методами, выполненными Сегер (Сиэтл, WA):
[00397] В анализе цитохром P450 CYP1A, микросомы печени человека (0,2 мг/мл белка) инкубируют в течение 15 минут при 37°C с 10 мкМ исследуемого соединения, 1 мкМ этоксирезоруфина, 1,3 мМ НАДФ, 3,3 мМ глюкозо-6-фосфата и 0,4 Ед/мл глюкозо-6-фосфатдегидрогеназы. В отсутствии исследуемого соединения, этоксирезоруфин, добавленный как субстрат, окисляется до резоруфина, и в присутствии ингибитора изофермента CYP, количество образовавшегося резоруфина снижается. Фурафиллин использовали в качестве ингибитора сравнения.
[00398] Реакционную смесь анализа цитохром Р450 СУР2С9, содержащую микросомы печени человека (белок 0,2 мг/мл), инкубируют в течение 15 минут при 37°C с 10 мкМ исследуемого соединения, 10 мкМ толбутамида, 1,3 мМ НАДФ, 3,3 мМ глюкозо-6-фосфата и 0,4 Ед/мл глюкозо-6-фосфатдегидрогеназы. В отсутствие исследуемого соединения, толбутамид окисляется до 4-гидрокситолбутамида, и в присутствии ингибитора изофермента CYP, количество образовавшегося 4-гидрокситолбутамида снижается. Сульфафеназол (IC50: 0,35 мкМ) служит ингибитором сравнения.
[00399] Для анализа цитохром Р450 CYP2C19, микросомы печени человека (0,2 мг/мл белка) инкубируют в течение 15 минут при 37°C с 10 мкМ исследуемого соединения, 10 мкМ омепразола, 1,3 мМ НАДФ, 3,3 мМ глюкозо-6-фосфата и 0,4 Ед/мл глюкозо-6-фосфат дегидрогеназы. В отсутствие исследуемого соединения, омепразол окисляется до 5-омепразола, и в присутствии ингибитора изофермента CYP, количество образовавшегося 5-гидроксиомепразола снижается. Оксибутинин (ICso; 7,1 мкМ) служит ингибитором сравнения.
[00400] Реакционную смесь анализа цитохром Р450 CYP2D6, содержащую микросомы печени человека (0,2 мг/мл белка) инкубируют в течение 15 минут при 37°C с 10 мкМ исследуемого соединения, 5 мкМ декстрометорфана, 1,3 мМ НАДФ, 3,3 мМ глюкозо-6-фосфата и 0,4 Ед/мл глюкозы-6-фосфатдегидрогеназы. В отсутствие исследуемого соединения, декстрометорфана окисляется, и в присутствии ингибитора изофермента CYP, количество продукта окисления снижается. Хинидин (IC50: 0,093 мкМ) служит ингибитором сравнения.
[00401] Рекомбинантный человеческий цитохром P450 CYP3A4 (20 пмоль/мл) инкубируют в течение 20 минут при 37°C с 10 мкМ исследуемого соединения, 5 мкМ мидазолама, 1,3 мМ НАДФ, 3,3 мМ глюкозо-6-фосфата и 0,4 Ед/мл глюкозо-6-фосфатдегидрогеназы. В отсутствии исследуемого соединения, мидазолам окисляется, и в присутствии ингибитора рекомбинантного изофермента, количество продукта окисления снижается. Содержание продукта окисления определяют на основе площади под кривой после разделения методом ВЭЖХ-МС/МС.Кетоконазол (IC50: 0,55 мкМ) служит ингибитором сравнения.
[00402] В каждом испытании, процентное ингибирование изофермента цитохром P450 CYP P450 было определено как 100-кратное соотношение (1 - количество продукта в образце в присутствии исследуемого соединения), разделенное на количество продукта в образце, содержащем необработанный изофермент. Результаты анализов в двойном повторении (выраженные, как процент сохраненной активности CYP) показаны в табл.IV.
ПРИМЕР 43: Стабильность соединения (2) в контакте с микросомами печени человека
[00403] Микросомы печени человека (конечная концентрация белка 0,3 мг/мл) в 0,1 М фосфатном буфере (рН 7,4) и системе восстановления НАДФН (1 мМ НАДФ, 5 мМ глюкозо-6-фосфата и 1 Ед/мл глюкозо-6-фосфат-дегидрогеназы) предварительно инкубируют при 37°C до добавления субстрата, соединения (2), до конечной концентрации субстрата 1 мкМ; и конечная концентрация метанола 0,6%. Аликвоты отбирают через 0 и 60 минут. Добавляют равный объем смеси ацетонитрил/метанол (50:50), образцы центрифугируют и количество соединения, оставшегося в супернатанте, измеряют методом ВЭЖХ в сочетании с тандемной масс-спектрометрией, сравнивая значения площади пика, генерируемые образцами, отобранными в точках 0 и 60 минут. Образцы в двойном повторении показали 96% и 118% соединения (2) после инкубации на протяжении 60 минут с микросомами печени человека.
ПРИМЕР 44: Фармакокинетика соединения (2) у крыс
[00404] Для определения соотношения концентрации соединения (2) в мозге к концентрации в плазме, группе 6 находящихся в сознании крыс с катетером шейной вены вводили 3 мг/кг пептида в виде инфузии длительностью 5 минут через катетер шейной вены. Образцы крови отбирали через 30, 60 и 180 минут после начала инфузии у 2 животных в каждой точке времени терминальной пункцией сердца, и быстро извлекали мозг целиком. Плазму отделяли центрифугированием. Тандем жидкостной хроматографии/масс-спектрометрии (ЖХ-МС/МС) применяли для определения концентрации лекарственного средства в плазме и мозге крыс. Результаты показаны на фиг.5.
ПРИМЕР 45: Фармакокинетика соединения (6) у мышей и яванских макак
[00405] Однократный болюс соединения синтетического пептидного амида вводили подкожной инъекцией мышам ICR (n=6 самцов, масса тела 23-37 г, Charles River, Wilmington, MA), и отбирали образцы плазмы через 5, 10, 15, 20, 30, 60, 90, 120 и 180 минут после инъекции. На фиг.6 показаны результаты, полученные после подкожной инъекции дозы 1 мг/кг соединения (6) мышам ICR. Период полувыведения в данном исследовании определяли как время, необходимое для снижения концентрации в плазме на 50% после достижения максимальной концентрации в плазме; ожидается, что рассчитанный элиминационный период полувыведения на базе константы скорости элиминации самой медленной фазы элиминации будет длиннее. См. табл.V ниже.
ПРИМЕР 46: Фармакокинетика соединений синтетических пептидных амидов у обезьян
[00406] Образцы вводят самцам обезьян Масаса fasciculans (SNBL USA, Ltd., Everett, WA, специально выведенные яванские макаки. Близко родственны человеку, как филогенетически, так и физиологически), в возрасте 3-7 лет, с массой тела 3-5 кг. Образцы вводят в поверхностную вену руки или ноги (например, плечевую или подкожную вену ноги) в 0,9% растворе натрия хлорида для инъекций, Фарм. США (Baxter Healthcare, Deerfield, Ill.), как указано ниже. Образец, содержащий 4 мг соединения (6) по настоящему изобретению, подлежащего изучению, готовят в 2 мл 0,9% раствора натрия хлорида для инъекций. Дозу 2 мл вводят в виде внутривенного болюса экспериментальному животному, что составляет дозу приблизительно 0,4-0,65 мг/кг, в зависимости от массы тела животного. Образцы крови объемом 0,6 мл отбирают венепункцией периферической вены через 2, 5, 10, 15 и 30 минут после введения дозы, а затем через 1, 2 и 4 часа. Каждый образец помещают в предварительно охлажденную стеклянную пробирку, содержащую гепарин лития, и немедленно охлаждают на льду. Плазму отбирают после центрифугирования со скоростью 2000 g в течение 15 минут при 2-8°С. Слои плазмы каждого образца перемещают в полипропиленовую пробирку и хранят в замороженном состоянии при температуре -60°C или ниже до проведения анализа.
[00407] Аликвоты по 100 мкл размороженной плазмы фиксируют 5 мкл раствора подходящего внутреннего стандарта 400 мкг/мл (в данном случае известное стандартное соединение синтетического пептидного амида) в 0,1% ТФУ, и белки осаждают 100 мкл 0,1% ТФУ в ацетонитриле. Образцы центрифугируют при 1000 х д в течение 5 минут, и супернатант анализируют методом ЖХ-МС.Анализ методом ЖХ-МС выполняют на масс-спектрометре Finnigan LCQ Deca, соединенном с системой ВЭЖХ Surveyor (Thermo Electron Corporation, Waltham, Massachusetts, США). Анализ методом ВЭЖХ проводят на обращенно-фазовых колонках С18 2,1×150 мм с градиентом от 0,01% ТФУ в ацетонитриле до 0,01% ТФУ в воде. Определение массы выполняли в режиме избирательного мониторинга реакции (SRM).
[00408] Количественную оценку выполняли против калибровочной кривой анализируемого вещества в холостой плазме яванской макаки с использованием такого внутреннего стандарта. Анализ данных и вычисление фармакокинетических параметров выполняли с помощью программного обеспечения РК Solutions 2.0 (Summit Research Services, Ashland, Ohio, США). В табл.V ниже показан период полувыведения у мышей ICR после подкожной инъекции соединения (6), и период полувыведения для данного соединения после внутривенного введения болюса яванским макакам.
[00409] Присутствие соединения (3) в плазме яванских макак после внутривенного введения болюса 0,56 мг/кг показано на фиг.7.
ПРИМЕР 47: Индуцированные уксусной кислотой корчи у мышей
[00410] В данном анализе идентифицируют соединения, которые демонстрируют анальгетическую активность против висцеральной боли или боли, связанной с активацией чувствительных к низким значениям рН ноцицепторов [см. Barber and Gottschlich (1986) Med. Res. Rev. 12: 525-562; Ramabadran and Bansinath (1986) Pharm. Res. 3: 263-270]. Внутрибрюшинное введение разбавленного раствора уксусной кислоты вызывает поведение корчей у мышей. Корчи определяют как сокращение мышц живота, сопровождаемое вытягиванием передних конечностей и удлинением тела. Количество корчей, наблюдаемых в присутствии и отсутствие исследуемых соединений, подсчитывают, чтобы определить анальгетическую активность соединений.
[00411] В каждый день проведения испытания на модели корчей включали группу контрольных мышей (n=6-8), получавших растворитель, которых обрабатывали так же, как и исследуемую группу (за исключением того, что исследуемое соединение отсутствовало в инъекционной дозе), и среднее общее количество корчей в данной группе использовали в качестве абсолютного ориентира, определяющего 0% уменьшения восприятия боли для всех других мышей, получавших исследуемое соединение в указанный день. Конкретно, общее количество корчей для каждой мыши, получавшей исследуемое соединение, переводили в % уменьшения восприятия боли в соответствии со следующим уравнением:
,
где Wv представляют собой среднее количество корчей в получавшей растворитель группе, и Wс представляют собой количество корчей у мышей, получавших исследуемое соединение. Данные были проанализированы с использованием 2-параметрического уравнения Хилла (также известно как модель Emax), где Emax принимается за 100% антиноциперцепции (т.е. отсутствие корчей на протяжении 15 минут после введения уксусной кислоты).
[00412] Мышей-самцов ICR массой 23-37 г взвешивали и размещали в индивидуальных камерах (обычно, стеклянный стакан объемом 4000 мл) для наблюдения с тонким слоем подстилки для грызунов SANI-CHIPS на дне камеры. Для определения активности и мощности исследуемых соединений, различные дозы раствора соединения или растворителя вводили подкожной инъекцией в заднюю область шеи через 15 или 180 минут до введения раствора уксусной кислоты. После введения соединения или контрольного растворителя, мышей возвращали в индивидуальные камеры для наблюдения ожидать внутрибрюшинного введения раствора уксусной кислоты. Через 15 минут или 3 часа, в соответствии с интервалом времени, определенном в каждом эксперименте от введения соединения до инъекции уксусной кислоты, дозу, соответствующую 10 мл/кг 0,6% (об./об.) раствора уксусной кислоты, вводили инъекционно в нижний правый квадрант живота. Сразу после инъекции мышь возвращали в камеру для наблюдения и немедленно начинали регистрировать количество корчей. Количество корчей подсчитывали за период 15 минут, начинающийся с момента инъекции уксусной кислоты, причем данные регистрировали для 3-х отдельных периодов времени длительностью 5 минут каждый (0-5 минут, 5-10 минут и 10-15 минут).
[00413] Данные регистрировали как ED50 и коэффициент Хилла. ED50 выражают как среднее значение±стандартная ошибка среднего (sem) (ED50+/- sem) или как среднее геометрическое значение с 95% доверительными интервалами (95% CI) с использованием t-шкалы. Коэффициент Хилла выражают, как арифметическое среднее значение ± sem, вычисленное на основе значений, полученных в результате наблюдения за животными. Результаты для соединения (2) показаны на фиг.8 (закрашенные круги).
[00414] Для анализа зависимости «доза-реакция» первичные данные переводили в % максимальное возможного эффекта (% МРЕ) с помощью формулы: %МРЕ=((балл исследования - балл в группе растворителя) / (0 - балл в группе растворителя)) * 100. Первичные данные были проанализированы с помощью одностороннего ANOVA с последующим тестом Дуннетта. Доза, которая продемонстрировала 50% снижение гиперчувствительности (ED50), была определена с использованием анализа методом линейной регрессии. Соединения вводили внутривенно. В табл.VI приведены результаты описанных экспериментов.
[00415] Зависимость «доза-реакция» для соединения (2) на модели индуцированных уксусной кислотой корчей у мышей генерировали с использованием доз 0,01, 0,03, 0,1 и 0,3 мг/кг, которые вводили внутривенно, как изложено выше. С использованием описанного выше метода, линейная зависимость «доза-реакция» была определена для соединения (2) для доз в интервале от 0,01 мг/кг до 0,3 мгам/кг, как показано на фиг.9.
ПРИМЕР 48: Ингибирование перемещения у мышей для измерения седативного эффекта соединений после подкожной инъекции (модель уменьшения перемещений)
[00416] Соединения, которые демонстрируют седативную активность, подавляют самопроизвольное перемещение мышей в камере исследования. Для определения потенциального седативного эффекта исследуемых соединений, степень уменьшения перемещения после введения исследуемого соединения или контрольного растворителя может быть определена и сравнена с помощью специализированного устройства, разработанного для данной цели (измеритель активности Opto-Vanmex). В начале каждого эксперимента каждую мышь взвешивают и рассматривают, чтобы определить надлежащее состояние здоровья. Для определения активности и мощности соединений, различные дозы раствора соединения или растворителя вводят подкожной инъекцией через 15 или 180 минут до начала регистрации данных. Подкожную инъекцию выполняют в заднюю область шеи мыши, защипнутую в "тент", чтобы позволить доступ иголки шприца. После инъекции каждое животное размещают в индивидуальную коробку Plexiglas (43×43 см) внутри устройства для измерения активности Opto-Vanmex. Перед тем, как животное помещают в устройство, тонкий слой подстилки для грызунов SANI-CHIPS помещают на дно коробки Plexiglas, чтобы создать удобное окружение. Каждое устройство для измерения активности Opto-Vanmex затем включают в режим регистрации данных, которая начинается системой автослежения АТМ3. Данные обрабатывают, и результаты выражают таким же образом, как описано для данных анализа на модели корчей в Примере 47.
ПРИМЕР 49 Анальготический эффект против седативного эффекта синтетического пептидного амида (3)
[00417] Ингибирование соединением индуцированных уксусной кислотой корчей является показателем анальготического эффекта (также называется антиноцицептивным эффектом). Подобным образом, уменьшение перемещения, вызванное введением соединения, может использоваться в качестве показателя общего седативного эффекта.
[00418] Значение ED50, определенное на модели индуцированных уксусной кислотой корчей у мышей ICR, составило 52 мкг/кг при подкожном введении синтетического пептидного амида (3) как описано в Примере 34 и показано на фиг.8 (закрашенные круги). Значение ED50, определенное на модели подавления перемещения, как описано в Примере 35, составило 2685 мкг/кг для того же синтетических пептидного амида при подкожном введении (см. фиг.8, закрашенные квадраты). Терапевтическое соотношение анальгетического эффекта и седативного эффекта представляет собой кратность значения ED50, необходимого для достижения седативного эффекта, по сравнению со значением ED50, необходимым для достижения анальгетического эффекта. Таким образом, соединение (3) демонстрирует соотношение (2685/52) раз, т.е. 51,6 раз. Таким образом, терапевтическое соотношение для соединения (3) составляет приблизительно 52 раза.
ПРИМЕР 50: Модель перевязывания спинальных нервов (SNL)
[00419] Модель SNL (Kim и Chung, 1992) применяли для индукции хронической невропатической боли. Крыс подвергали анестезии изофлураном, левый поперечный процесс L5 удаляли, и спинальные нервы L5 и L6 туго перевязывали накладыванием шва с помощью шелковой хирургической нити 6-0. Рану зашивали внутренними швами и внешними скобами. Через 14 дней после SNL, начальное значение, значение после повреждения и после лечения для нетоксичной механической чувствительности были оценены с использованием 8 нитей Semmes-Weinstein (Stoelting, Wood Dale, IL, США) с варьирующей жесткостью (0,4, 0,7, 1,2, 2,0, 3,6, 5,5, 8,5 и 15 г) согласно способу "вниз-вверх" (Chaplan et al., 1994). Животных размещали на перфорированной металлической платформе и позволяли акклиматизироваться к окружению в течение минимум 30 минут перед проверкой. Среднее значение и стандартную ошибку среднего (SEM) определяли для каждой лапы в каждой группе лечения. Поскольку данный стимул обычно не считается болезненным, существенное повышение чувствительности, вызванное повреждением, интерпретировали как меру механической аллодинии. Дозу, которая вызывала 50% снижение механической гиперчувствительности (ED50), определяли с использованием линейного регрессионного анализа.
Соединение (2) вводили внутривенно. На фиг.10 приведены результаты экспериментов. Вычисленное значение ED50 для соединения (2) в данной модели составило 0,38 мг/кг (0,31-0,45; 95% доверительный интервал).
ПРИМЕР 51: Глазная анальгезия, вызванная соединениями (2), (3) и (4)
[00420] Глазную анальгезию оценивали путем инсталляции 5 аликвот исследуемого соединения, по 50 мкл каждая, в физиологическом растворе натрия хлорида, в исследуемой концентрации, в правый глаз новозеландских кроликов-альбиносов, ранее не принимавших участия в экспериментах, на протяжении 20 минут. Через 15 минут после последней инстилляции исследуемого соединения, каждому животному проводят однократную инстилляцию 30 мкл раствора 10 мг/мл капсаицина (33 мМ) в обработанный глаз. Известно, что капсаицин вызывает боль в роговице. Боль в роговице оценивают измерением открывания века в миллиметрах с использованием прозрачной линейки в обработанном и необработанном глазах. На данной животной модели, уменьшение размера открывания века после инстилляции капсаицина является показателем степени глазной боли. Таким образом, любое зарегистрированное восстановление (увеличение) размера открывания века после лечения исследуемым соединением рассматривается как мера облегчения индуцированной капсаицином глазной боли.
[00421] Такие оценки выполняют перед лечением исследуемым соединением (перед испытанием), непосредственно перед инстилляцией капсаицина, а затем через 1, 5, 10, 15, 20, 25, 30, 40, 50 и 60 минут после инстилляции капсаицина. В табл.VII показано среднее значение результатов измерения открывания века (относительно не получавшего лечение глаза, выраженное, как процент от контроля), выведенное за период 10-30 минут после инстилляции капсаицина кроликам, которые предварительно получали инстилляцию каппа-опиатного агониста по изобретению, а также после предварительной инстилляции стандартной концентрации дилтиазема, бензотиазепинового блокатора кальциевых каналов с местноанестезирующими свойствами. См. Gonzalez et al., (1993) Invest. Ophthalmol. Vis. Sci. 34: 3329-3335.
Среднее значения для 5 животных.
ПРИМЕР 52: Зависимость «доза-реакция» для соединения (2) при индуцированной капсаицином глазной боли
[00422] Глазную анальгезию, индуцированную соединением (2), исследуют при инстилляции в нескольких концентрациях в правый глаз новозеландских кроликов-альбиносов, ранее не принимавших участия в экспериментах, и оценивают, как описано выше. Результаты сравнивают с анальгезией, индуцированной 10 мг/мл морфина (неселективный опиатный агонист) в качестве контроля с системной активностью, и 10 мМ дилтиазема в качестве контрольного соединения местного действия в таком же эксперименте и при таких же условиях. Полученные результаты показаны в табл.VIII ниже.
5 Среднее значение для 10 животных.
ПРИМЕР 53: Влияние Соединения (2) на модели панкреатита у крыс
[00423] Хроническое воспаление поджелудочной железы вызывают у крыс внутривенным введением дибутилина дихлорида (DBTC, Aldrich Milwaukee, WI) растворенного в 100% этаноле, в дозе 8 мг/кг под изофлурановой анестезией (2-3 л/мин, 4%/об, до наступления анестезии, затем 2,5%/об. до окончания процедуры. Контрольные животные получали только такой же объем растворителя (100% этанол). Боль вследствие панкреатита оценивали определением чувствительности брюшины к зондированию живота крыс откалиброванной нитью von Frey (4 г). Крысам позволяли акклиматизироваться в подвешенных проволочно-петлевых клетках в течение 30 минут перед проверкой. На реакцию указывало острое отдергивание живота, облизывания области живота или отдергивание всего тела. Одно испытание состояло из 10 попыток нити von Frey, по одной каждые 10 сек, чтобы позволить животному прекратить любую реакцию и вернуться к относительно неактивному положению. Среднее количество отдергиваний в каждом испытании выражают, как количество реакций из 10 попыток. Крысы без воспаления поджелудочной железы обычно показывают частоту отдергивания в ответ на зондирование нитью von Frey 0-1. Животным позволяли восстановиться в течение 6 дней после введения DBTC до любых фармакологических манипуляций. Животных, не демонстрирующих достаточную гиперчувствительность брюшины (т.е. крыс с менее чем 5 позитивными реакциями из возможных 10), были исключены из исследования.
[00424] Количество позитивных реакций после зондирования брюшины (из возможных 10) регистрировали в каждой точке времени. Данные представлены как среднее число отдергиваний (± стандартная ошибка среднего) для каждой группы в каждой соответствующей точке времени. Для анализа зависимости «доза-реакция» первичные данные переводили в % максимально возможного эффекта (% МРЕ) с помощью формулы: % МРЕ = ((балл исследования - балл после введения DBTC) / (балл перед введением DBTC - балл после введения DBTC)) * 100. Первичные данные были проанализированы с использованием двухстороннего повторного ANOVA, с последующим тестом Бонферрони. Доза, которая обеспечивала 50% снижение гиперчувствительности (ED50), была определена с использованием линейного регрессионного анализа. Соединения вводили внутрибрюшинным способом. На фиг.11 приведены результаты экспериментов. Вычисленное значение ED50 для соединения (2) в данной модели составило 0,03 мг/кг (0,006-0,14; доверительный интервал 95%).
[00425] Чтобы определить, опосредована ли эффективность соединения (2) (1 мг/кг) активацией периферических каппа-опиатных рецепторов, группам из 8 крыс предварительно вводили селективный антагонист каппа-опиатного рецептора nor-BNI (1 мг/кг) или неселективный антагонист опиатных рецепторов, налоксона метйодид (10 мг/кг), который не пересекает гематоэнцефалический барьер, до лечения соединением (2). На фиг.12 представлены результаты экспериментов.
ПРИМЕР 54: Модель зуда у мышей
[00426] Использовали группы по 10 (и в одном случае, 11) самцов мышей Swiss Webster (25-30 г). Каждое животное взвешивали и позволяли акклиматизироваться как минимум в течение 1 часа в индивидуальных, прямоугольных коробках для наблюдения. Хвосты мышей погружали на 30 секунд в теплую воду, чтобы расширить хвостовые вены, после чего животные получали внутривенную инъекцию растворителя (солевой раствор) или соединения (2) (0,01, 0,03, 0,10 и 0,30 мг свободного основания/кг). Через 15 минут каждой мыши вводили GNTI дигидрохлорид (Tocris) (0,30 мг/кг; 0,25 мл/25 г) или соединение 48/80 (Sigma) (50 мкг в 0,10 мл солевого раствора) подкожно позади шеи. Затем за животными вели наблюдение попарно (иногда в тройках), количество царапающих движений задней лапы, направленных на шею, регистрировали в течение 30 минут.Средний процент ингибирования царапанья, вызванного соединением (2) наносили на график, и определяли дозу, связанную с 50% ингибированием, линейным регрессионным анализом (PharmProTools). В табл.IX приведены результаты экспериментов.
[00427] Описание каждого из патентов США и опубликованных патентных заявок, а также тексты литературных ссылок, процитированные в данном описании, включены путем ссылки во всей их полноте. В случае если какое-либо определение или описание, найденное в одной или больше их указанных ссылок, приходит в противоречие с соответствующим определением или описанием в данном описании, то применяется определение или описание, раскрытое в данном описании.
[00428] Примеры, приведенные в данном описании, предназначены только, в данном описании и не предназначены для ограничения изобретения, полный контекст которого будет легко понятен специалисту в данной области.
Реферат
Изобретение относится к синтетическим пептидным амидам формулы I, которые являются агонистами каппа-опиатного рецептора и демонстрируют низкую степень ингибирования PCYP и низкую степень проникновения в мозг. Также изобретение относится к фармацевтическим композициям, содержащим указанные соединения, и их применению для профилактики и лечения боли и воспаления, связанных с различными заболеваниями и состояниями. 3 н. и 14 з.п. ф-лы, 9 табл., 12 ил., 39 пр.
Формула
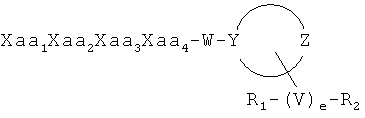
или стереоизомер, смесь стереоизомеров, фармацевтически приемлемая соль, гидрат, сольват, гидрат соли кислоты, N-оксид синтетического пептидного амина, который содержит амин, атом азота которого связан с атомом кислорода, или изоморфная кристаллическая форма указанного соединения;
где Xaa1 выбран из группы, состоящей из (A)(A')D-Phe, (A)(A')(α-Me)D-Phe, D-Tyr, D-Tic, D-трет-лейцина, D-неопентилглицина, D-фенилглицина, D-гомофенилаланина и β-(E)D-Ala, где каждый (А) и каждый (А') представляют собой заместители в фенильном кольце, независимо выбранные из группы, состоящей из -Н, -F, -Cl, -NO2, -CH3, -CF3, -CN и -CONH2, и где каждый (Е) независимо выбран из группы, состоящей из циклобутила, циклопентила, циклогексила, пиридила, тиенила и тиазолила;
Хаа2 выбран из группы, состоящей из (A)(A')D-Phe, 3,4-дихлор-D-Phe, (A)(A')(α-Me)D-Phe, D-1Nal, D-2Nal, D-Tyr, (E)D-Ala и D-Trp;
Хаа3 выбран из группы, состоящей из D-Nle, D-Phe, (E)D-Ala, D-Leu, (α-Me)D-Leu, D-Hle, D-Val и D-Met;
Xaa4 выбран из группы, состоящей из (B)2D-Arg, (В)2D-Nar, (B)2D-Har, ζ-(B)D-Hlys, D-Dap, ε-(B)D-Lys, ε-(B)2-D-Lys, D-Amf, амидино-D-Amf, γ-(B)2D-Dbu, δ-(B)2α-(B')D-Orn, D-2-амино-3(4-пиперидил)пропионовой кислоты, D-2-амино-3(2-аминопирролидил)пропионовой кислоты, D-α-амино-β-амидинопропионовой кислоты, α-амино-4-пиперидинуксусной кислоты, цис-α,4-диаминоциклогексануксусной кислоты, транс-α,4-диаминоциклогексануксусной кислоты, цис-α-амино-4-метиламиноциклогексануксусной кислоты, транс-α-амино-4-метиламиноциклогексануксусной кислоты, α-амино-1-амидино-4-пиперидинуксусной кислоты, цис-α-амино-4-гуанидиноциклогексануксусной кислоты и транс-α-амино-4-гуанидиноциклогексануксусной кислоты, где каждый (В) независимо выбран из группы, состоящей из Н и C1-C4 алкила, и (В') представляет собой Н или (α-Me);
W отсутствует, при условии, что, если W отсутствует, Y представляет собой N;
или выбран из группы, состоящей из:
-NH-(СН2)b-, где b равно 0, 1, 2, 3, 4, 5 или 6; и
-NH-(CH2)c-O-, где с равно 2 или 3, при условии, что Y представляет собой С;
фрагмент
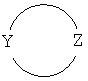
представляет собой необязательно замещенный 4-8-членный гетероциклический кольцевой фрагмент, где все гетероатомы кольца в указанном кольцевом фрагменте представляют собой N; где каждый из Y и Z независимо представляет собой С или N; при условии, что, если такой кольцевой фрагмент представляет собой 6-, 7- или 8-членное кольцо, Y и Z разделены как минимум двумя атомами кольца; и при условии, что, если такой кольцевой фрагмент содержит в кольце один гетероатом, который представляет собой N, то такой кольцевой фрагмент является неароматическим;
V представляет собой C1-C6 алкил, и е равно 0 или 1, где, если е равно 0, то V отсутствует, и R1 и R2 непосредственно присоединены к одному и тому же или различным атомам кольца;
где
(i) R1 выбран из группы, состоящей из -Н, -OH, галогена, -CF3, -NH2, -COOH, C1-С6 алкила, C1-С6 алкокси, амидино, C1-C6 алкилзамещенного амидино, арила, необязательно замещенного гетероциклила, Pro-амида, Pro, Gly, Ala, Val, Leu, Ile, Lys, Arg, Orn, Ser, Thr, -CN, -CONH2, -COR', -SO2R', -CONR'R'', -NHCOR', OR' и SO2NR'R''; где указанный необязательно замещенный гетероциклил необязательно содержит 1 или 2 заместителя, независимо выбранные из группы, состоящей из C1-С6 алкила, C1-C6 алкокси, оксо, -ОН, -Cl, -F, -NH2, -NO2, -CN, -COOH и амидино; где каждый из R' и R'' независимо представляет собой -Н, C1-C8 алкил, арил или гетероциклил, или R' и R'' образуют 4-8-членное кольцо, которое необязательно содержит 1 или 2 заместителя, независимо выбранные из группы, состоящей из C1-С6 алкила, -C1-С6 алкокси, -OH, -Cl, -F, -NH2, -NO2, -CN, -COOH и амидино; и R2 выбран из группы, состоящей из: -H; амидино; амидино, замещенного одним или двумя C1-C6 алкилами; -CN; -CONH2; -CONR'R''; -NHCOR'; -SO2NR'R'' и -COOH; или
(ii) R1 и R2 вместе могут образовывать необязательно замещенный 4-9-членный гетероциклический моноциклический или бициклический кольцевой фрагмент, который присоединен к одному атому кольца Y- и Z-содержащего кольцевого фрагмента; или
(iii) R1 и R2 вместе с одним атомом кольца Y- и Z-содержащего кольцевого фрагмента могут образовывать необязательно замещенный 4-8-членный гетероциклический кольцевой фрагмент с образованием спироструктуры; или
(iv) R1 и R2 вместе с двумя или больше соседними атомами кольца Y- и Z-содержащего кольцевого фрагмента могут образовывать необязательно замещенный 4-9-членный гетероциклический моноциклический или бициклический кольцевой фрагмент, конденсированный с Y- и Z-содержащим кольцевым фрагментом;
где каждый из указанных необязательно замещенных 4-, 5-, 6-, 7-, 8- и 9-членных гетероциклических кольцевых фрагментов, содержащих R1 и R2, необязательно содержит 1 или 2 заместителя, независимо выбранные из группы, состоящей из C1-C6 алкила, C1-C6 алкокси, необязательно замещенного фенила, оксо, -OH, -Cl, -F, -NH2, -NO2, -CN, -COOH и амидино;
при условии, что, если Y- и Z-содержащий кольцевой фрагмент представляет собой 6-или 7-членное кольцо, содержащее один гетероатом в кольце, и е равно 0, то R1 не является -OH, и R1 и R2 оба одновременно не являются -Н;
при условии, что, если Y- и Z-содержащий кольцевой фрагмент представляет собой 6-членное кольцо, содержащее два гетероатома в кольце, Y и Z представляют собой N, и W равен 0, то -(V)eR1R2 присоединен к атому кольца, отличному от Z; и если е равно 0, то R1 и R2 оба одновременно не являются -Н; и
наконец, при условии, что, если Xaa3 представляет собой D-Nle, то Хаа4 не является (B)2D-Arg, и если Хаа3 представляет собой D-Leu или (α-Me)D-Leu, то Хаа4 не является δ-(B)2α-(B')D-Orn.
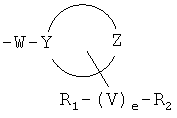
выбран из группы, состоящей из:

















D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-аминопиперидин-4-карбоновой кислоты)]-ОН (SEQ ID NO:2).
Документы, цитированные в отчёте о поиске
Пептиды-агонисты k-опиоидных рецепторов
Комментарии