Фосфатные производные фтороксиндолов и способ лечения с их использованием - RU2312857C2
Код документа: RU2312857C2
Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым фосфатным производным, фтороксиндольным соединениям, которые являются модуляторами активируемых кальцием калиевых каналов (ВК) высокой проводимости и поэтому полезны для защиты нейронных клеток и лечения заболеваний, являющихся результатом дисфункции поляризации клеточной мембраны и проводимости. Настоящее изобретение также обеспечивает способ лечения новыми замещенными производными фтороксиндола, а также их фармацевтические композиции.
УРОВЕНЬ ТЕХНИКИ
В настоящее время инсульт поставлен на третье место при определении причины недееспособности и смертности взрослого населения в США и Европе. За прошедшее десятилетие разрабатывалось несколько терапевтических подходов для снижения связанного с инсультом мозгового повреждения, включая ингибиторы АМРА/кинат, N-метил-D-аспартат (NMDA) и ингибиторы перезахвата аденозина. Целью настоящего изобретения является обеспечение новыми соединениями, которые модулируют калиевые каналы, в частности активируемые кальцием каналы калия (ВК) большой проводимости, которые применимы для ослабления нейронного повреждения при возникновении ишемических состояний во время инсульта.
Калиевые каналы играют ключевую роль в регулировании мембранного потенциала клетки и в модуляции возбудимости клетки. Калиевые каналы непосредственно регулируются напряжением, метаболизмом клетки, ионом кальция и процессами, опосредованными рецептором [Cook, N.S., Trends in Pharmacol. Sciences. - 9, pp.21-28 (1988); and Quast, U. и Cook, N.S., Trends in Pharmacol. Sciences., 10, pp.431-435 (1989)]. Активируемые кальцием каналы (КCa) представляют собой разнообразную группу ионных каналов, которые для активности распределяют зависимость на внутриклеточных ионах кальция. Активность каналов КCa регулируется внутриклеточным [Са2+], мембранным потенциалом и фосфорилированием. На основе проводимости их одиночного канала в симметрических К+растворах КСа каналы разделяют на три подкласса: большой проводимости (ВК)>150 pS; промежуточной проводимости 50-150 pS; малой проводимости<50 pS (pS обозначает пикосименс, единицу электрической проводимости). Активируемые кальцием каналы калия с большой проводимостью присутствуют во многих легковозбудимых клетках, включая нейроны, кардиальные клетки и различные типы клеток гладкой мышцы [Singer, J.J. and Walsh, J.V., Pflugers Archiv., 408. pp.98-111 (1987); Baro, L, and Escande, D., Pflugers Archiv., 414 (Suppl. 1), pp.S168-S170 (1989); and Ahmed, F. et al, Br. J. Pharmacol, 83, pp.227-233 (1984)].
Ионы калия играют доминирующую роль в контролировании постоянного мембранного потенциала в большинстве легковозбудимых клеток и в поддержании трансмембранного напряжения около K+ потенциала равновесия (Ek), приблизительно равного 90 мВ. Показано, что открытие калиевых каналов изменяет мембранный потенциал клетки к мембранному потенциалу равновесия (Ek) калия, приводя к гиперполяризации клетки. [Cook, N.S., Trends in Phannacol Sciences. 9, pp.21-28 (1988)]. Гиперполяризованные клетки демонстрируют пониженную реакцию на потенциальное повреждение стимулов деполяризации. Каналы BK, которые регулируются и напряжением и внутриклеточным Са2+, действуют так, что ограничивают деполяризацию и проникновение кальция и могут быть особенно эффективны для блокирования разрушительных стимулов. Поэтому гиперполяризация клетки через вскрытие каналов BK может привести к защите нейронных клеток при состоянии ишемии.
Роль калиевых каналов на действие гладкой мышцы человеческого мочевого пузыря описана у S.Trivedi et al. in Biochemical and Biophysical Research Communications (1995), 213. No.2, pp.404-409.
Было сообщено о ряде синтетических и встречающихся в природе соединений с BK открывающей активностью. Avena пирон, извлеченный из avena sativa обыкновенного овса, был идентифицирован как активирующий канал BK, использующий технику двойного слоя липида [РСТ WO 93/08800, опубликованная 13 мая 1993]. Было обнаружено, что плоретин, Флаванойд воздействуют на открытие Са2+-активируемых калиевых каналов в миелинизированных волокнах нерва Xenopus laevis (гладкой шпорцевой лягушки), используемого вне участка тела [Koh, D-S., et ai., Neuroscience Lett., 165. pp.167-170(1994)].
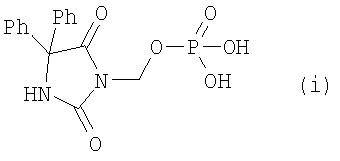
Varia, SA, et al., раскрыл использование фосфонометоксипроизводных (i) в качестве пролекарств гидантоина Фенитоина [J. Pharm. Sci. 73, pp.1068-1073 (1984)].
В РСТ WO 99/33846, опубликованной 8 июля 1999, раскрыты четвертичные аминофосфаты (ii) в качестве пролекарств для аминосодержащих лекарств.

В патенте США 5187173, опубликованном 16 февраля 1993, показано, что фосфонометилсахариновые производные (iii) являются полезными в качестве протеолитических ферментных ингибиторов.

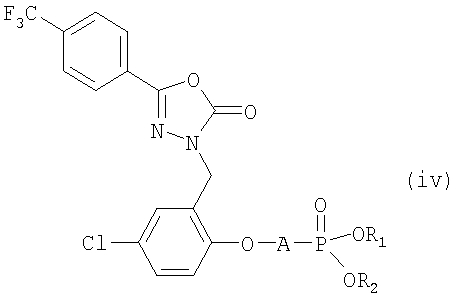
В патенте США 5939405, опубликованном 17 августа 1999, раскрыты фосфатные производные (iv), как полезные пролекарства диарил-1,3,4-оксадиазолонов, которые являются модуляторами активируемых кальцием калиевых каналов (ВК) большой проводимости.
В патенте США 5602169, опубликованном 11 февраля 1997, продемонстрировано, что (3S)-(+)-(5-хлор-2-метоксифенил)-1, 3-дигидро-3-фтор-6-(трифторметил)-2Н-индол-2-он (соединение формулы (S)-II) представляет собой модулятор активируемых кальцием калиевых каналов большой проводимости и является полезным для лечения ишемии.
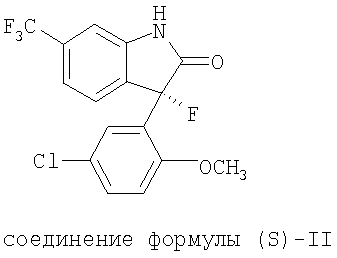
Синтез соединения формулы (S)-II и ее применимость для лечения нарушений, чувствительных к раскрытию калиевого канала, включая мозговую ишемию и травматическое мозговое повреждение, описан в патенте США 5602169. Вследствие низкой растворимости в воде соединения формулы (S)-II, добавки, например, такие как диметилсульфоксид и пропиленгликоль, должны быть применены, чтобы получить растворы соединения формулы (S)-II, пригодные для внутривенной инъекции (Gribkoff, et al., Nature Medicine. 2001, 7, 471-477).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Объектом настоящего изобретения являются новые фосфатные производные 3-фтороксиндолов, имеющие общую формулу

где волнообразная связь

Данные фосфаты повышают растворимость в воде 3-фтороксиндолов и, таким образом, уменьшают количество добавок, которые должны быть применены для доставки внутривенной дозы оксиндола. При систематическом введении производные оксиндола преобразуются в свободные системные уровни фтороксиндола. Настоящее изобретение также охватывает фармацевтические композиции, содержащие указанные фосфатные производные и способ лечения нарушений, чувствительных к активности при раскрытии калиевого канала, таких как ишемия, инсульт, конвульсии, астма, эпилепсия, синдром раздражительного кишечника, мигрень, травматическое повреждение мозга, повышенное внутричерепное давление, повреждение спинного мозга, половая дисфункция, отравление монооксидом углерода и недержание мочи.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение охватывает новые фосфатные производные в виде рацемата, (R)-энантиомера и (S)-энантиомера 3-(5-хлор-2-метоксифенил)-1,3-дигидро-3-фтор-6-(трифторметил)-2Н-индол-2-она (соединение формулы II), которые являются мощным вскрывателем активируемых кальцием K+-каналов (BK каналы) большой проводимости и новые соединения, имеющие общую формулу I

где волнообразная связь
А представляет собой прямую связь или (С=O);
В представляет собой прямую связь, кислород или азот;
m имеет значение 0 или 1;
n имеет значение 1, 2 или 3;
R1 и R2 каждый независимо являются водородом или C1-6 алкилом, и когда R1представляет собой водород, R2 может также быть P(O)OR5OR6;
R3 и R4 каждый независимо является водородом или C1-4 алкилом;
R5 и R6 каждый независимо является водородом,
или нетоксичную фармацевтически приемлемую соль или сольват.
Настоящее изобретение также раскрывает способ лечения или профилактики заболеваний, которые опосредуются активируемых кальцием K+ каналов (ВК каналов) большой проводимости у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или его нетоксичной фармацевтически приемлемой соли.
Предпочтительно соединения формулы I являются полезными при лечении ишемии, инсульта, травматического мозгового повреждения и повышенного внутричерепного давления.
Термины "C1-4 алкил" и " C1-6 алкил", как использовано в описании настоящего изобретения и в его формуле (если контекст не указывает иначе), означает прямые или разветвленные цепи алкильных групп, таких как метильная, этильная, пропильная, изопропильная, бутильная, пентильная, гексильная. Предпочтительно эти группы содержат от 1 до 2 атомов углерода. Термин "гетероарил" предназначен, чтобы включать пиридинил, тиофенил, пиримидинил, тиазоил, оксазоил, изоксазоил и им подобные.
Если иначе не определено, термин " гидролизуемой группой сложного эфира" предназначен, чтобы включать группу сложного эфира, которая является физиологически приемлемой и гидролизуемой, такие как C1-6 алкил, бензил, 4-метоксибензил, (низший)-алканоилокси(низший)алкил, например, ацетоксиметил, пропионилоксиметил или пивалоилоксиметил, (низший) алкоксикарбонилокси(низший)алкил, например метоксикарбонилоксиметил или этоксикарбонилоксиметил, (низший)-алкоксикарбонил(низший)алкил, например метоксикарбонилметил или трет-бутоксикарбонилметил, 2-метоксикарбонилоксиэтил, (5-метил-2-оксо-1,3-диоксол-4-ил)метил, дигидроксипропил и им подобные.
Термин "нетоксичная фармацевтически приемлемая соль" предназначен, чтобы включать нетоксичные аддитивно-основные соли с неорганическими и органическими основаниями. Соль соединения I, которая может быть представлена в настоящем изобретении с помощью

Вообще, фармацевтически приемлемыми солями по изобретению являются те, в которых противоположный ион не вносит значительный вклад в токсичность или фармакологическую активность соли. В некоторых образцах они имеют физические свойства, которые делают их более желательными для фармацевтических композиций, такие как растворимость, отсутствие гигроскопичности, возможность прессования при формировании таблетки и совместимость с другими ингредиентами, с которыми вещество может использоваться для фармацевтических целей. Соли обычно готовят путем смешивания соединения формулы I, где R1 и R2 представляют собой водород с подобранным основанием, предпочтительно с помощью контакта в растворе, используя избыток обычно применяемых инертных растворителей, таких как вода, эфир, ацетонитрил, диоксан, метиленхлорид, изопропиловый спирт, метанол, этиловый спирт, этилацетат и ацетонитрил. Они могут также быть получены путем обмена на ионообменной смоле в условиях, при которых соответствующий ион соли вещества формулы I заменяется другим ионом в условиях, которые позволяют провести разделение соединений, таких как осаждение из раствора или экстракция в растворитель или элюирование или удержание на ионообменной смоле.
Конкретные соединения по данному изобретению, включая их фармацевтически приемлемые соли, могут существовать как сольватные формы, включая гидратные формы, такие как моногидрат, дигидрат, полугидрат, тригидрат, тетрагидрат и т.п. Продукты могут быть настоящими сольватами, в то время как в других случаях продукты могут просто удерживать дополнительное количество растворителя или быть смесью сольвата плюс некоторого количества добавочного растворителя. Среднему специалисту должно быть понятно, что сольватные формы эквивалентны формам несольватным и предназначены, чтобы быть включенными в границы настоящего изобретения.
Термин "терапевтически эффективное количество" означает общую сумму каждого активного ингредиента композиции, которая является достаточной, чтобы оказать значимое воздействие на пациента, то есть произвести исцеление острых состояний, характеризующихся раскрытием активируемых кальцием K+ каналов большой проводимости или повышением степени исцеления таких состояний. Когда определение применяют к индивидуальному активному ингредиенту, вводимому самому по себе, термин относится к указанному одному ингредиенту. Когда определение применяют к комбинации, термин относится к объединенным количествам активных ингредиентов, которые приводят к терапевтическому эффекту, вводимому ли в комбинации, последовательно или одновременно. Термины "лечат, лечение, обработка" означают профилактику или уменьшение симптомов болезней, повреждения ткани и/или симптомов, связанных с дисфункцией поляризации клеточной мембраны и проводимости. В другом аспекте настоящее изобретение обеспечивает получение растворимого в воде пролекарства соединения, которое находится в виде рацемата, (R)-энантиомера и (S)-энантиомера 3-(5-хлор-2-метоксифенил)-1,3-дигидро-3-фтор-6-(трифторметил)-2Н-индол-2-она, который описан в патенте США 5602169.
Термин пролекарство означает производное активного лекарства, которое превращается после введения в активное лекарственное средство. В особенности, это относится к фосфатным производным лекарственных средств на основе 3-фтороксиндола, которые являются способными к осуществлению гидролиза эфирного остатка или к окислительному расщеплению сложного эфира, так, чтобы перевести лекарственное средство в свободное состояние. Например, фосфат может быть гидролизован ферментами фосфатазы в клетке-хозяине, чтобы получить более активную форму желательного 3-фтороксиндола. Физиологически гидролизуемые группы также служат пролекарствами, которые могут подвергаться гидролизу в организме, чтобы получить на выходе по существу исходное лекарственное средство и, таким образом, растворимые в воде пролекарства предпочтельны для введения исходного лекарственного средства.
В еще одном аспекте настоящее изобретение включает в себя способ лечения или профилактики заболеваний, которые опосредованы раскрытием активируемых кальцием K+ каналов (каналы ВК) большой проводимости у млекопитающего, нуждающегося в этом, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или его нетоксичной фармацевтически приемлемой соли, сольвата или гидрата. Предпочтительно соединения формулы I являются полезными при лечении ишемии, инсульта, травматического мозгового повреждения и повышенного внутричерепного давления.
Соединения формулы I могут быть приготовлены в соответствии с различными методиками, такими как иллюстрированные в настоящем описании примерами, схемами реакций и их вариациями, которые очевиды среднему специалисту. Различные пролекарственные соединения формулы I могут быть успешно приготовлены из активного вещества лекарственного средства формулы II, которое в свою очередь получают в соответствии с общей методикой, описанной в патенте США 5602169 и используют как исходный материал в способах, иллюстрированных на реакционных схемах 1-9.
РЕАКЦИОННАЯ СХЕМА 1
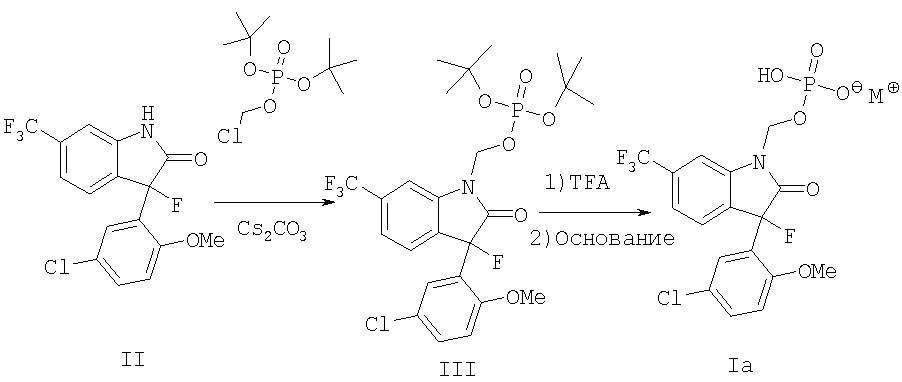
Получение 3-фтороксидолов формулы Ia иллюстрируется на реакционной схеме 1, где
РЕАКЦИОННАЯ СХЕМА 2

Альтернативный путь синтеза соединений формулы Ia, в котором противоположный ион может быть заменен простым способом, изображен на реакционной схеме 2, где R представляет собой C1-4 алкил и
РЕАКЦИОННАЯ СХЕМА 3

Когда имеется необходимость получить соединения формулы Ib, как иллюстрируется на реакционной схеме 3, соединение формулы II ацилируют агентом, таким как фосген, чтобы обеспечить получение хлорформиата формулы VI. Замена уходящей группы в виде хлора гидроксиалкилфосфатом, дает в результате соединения формулы VII, где n имеет значение, как определено в настоящем изобретении. Удаление трет-бутильных групп обработкой кислотой, такой как трифторуксусная кислота, дает на выходе соединения формулы Ib.
РЕАКЦИОННАЯ СХЕМА 4

Получение соединений формулы Ic приведено на реакционной схеме 4, где n имеет значение, как определено в настоящем изобретении. Ацилирование соединения формулы IV хлорформиата обеспечивает получение карбоната формулы VIII. Замена уходящей группы в виде нитрофенола аминоалкильным спиртом дает карбаматы формулы IX, которые могут быть фосфорилированны фосфорамидированием, с последующим окислением фосфора, чтобы обеспечить получение фосфатов формулы X. Удаление трет-бутильных групп обработкой кислотой, такой как трифторуксусная кислота, дает на выходе соединения формулы Ic.
РЕАКЦИОННАЯ СХЕМА 5
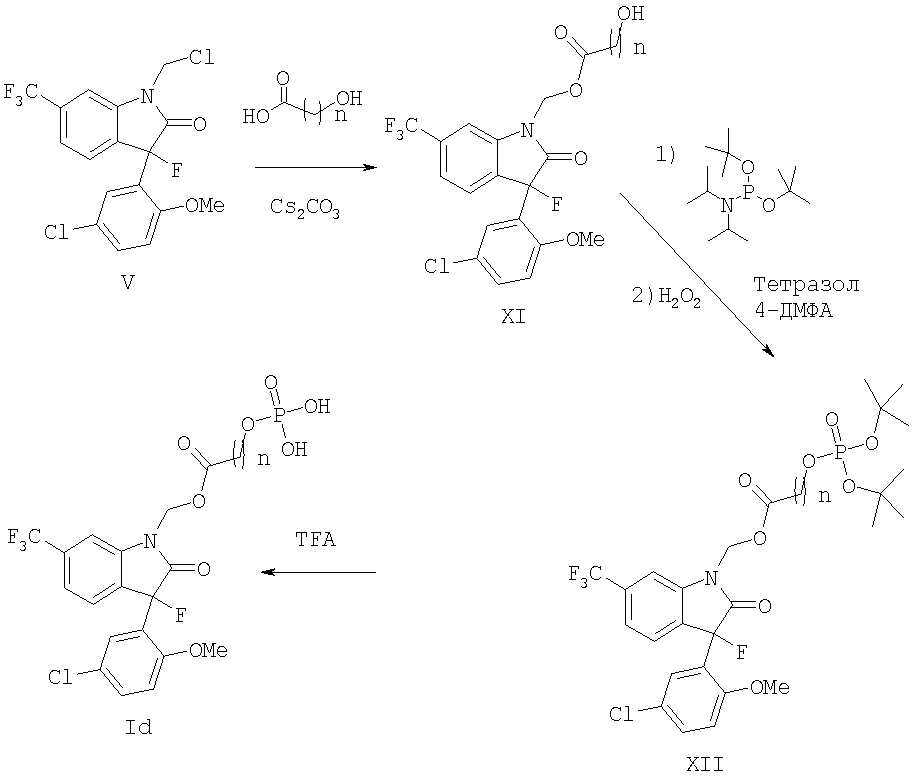
Сложные эфиры формулы Id могут быть получены, как показано на реакционной схеме 5, где n имеет значение, как определено в настоящем изобретении. Замена уходящей группы в виде хлора в соединениях формулы V гидроксиалкильными кислотами, обеспечивает получение сложных эфиров формулы XI, которые могут быть фосфорилированы фосфорамидированием с последующим окислением фосфора, что дает фосфаты формулы XII. Фосфаты формулы Id могут быть получены путем снятия защиты соединений формулы XII обработкой кислотой, такой как трифторуксусная кислота.
РЕАКЦИОННАЯ СХЕМА 6

Получение карбаматов формулы Ie иллюстрируется на реакционной схеме 6. Ацилирование соединений формулы II агентом ацилирования, таким как хлорметилхлорформиат, обеспечивает получение галогенметилкарбаматов формулы XIII. Замена хлора с помощью иодида натрия дает карбаматы иодметила формулы XIV. Замена иода фосфатами дает защищенные фосфаты формулы XV, с которых может быть снята защита обработкой кислотой, такой как трифторуксусная кислота, что дает карбаматы формулы Ie.
РЕАКЦИОННАЯ СХЕМА 7
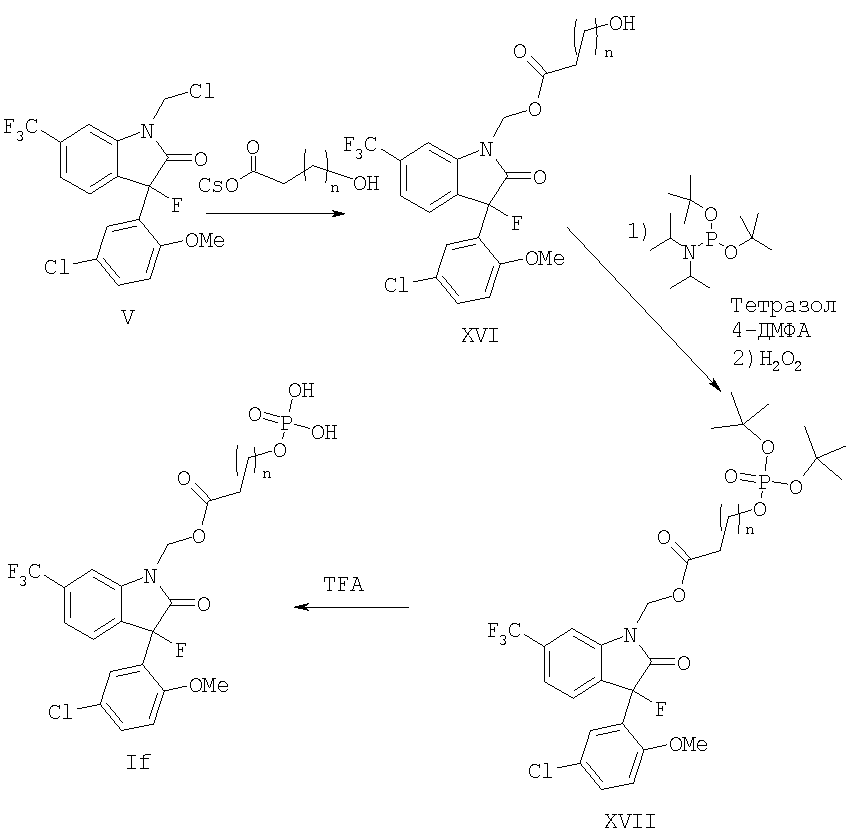
Фосфоноалкильные эфиры формулы If могут быть получены как иллюстрируется на реакционной схеме 7, где n имеет значения, определенные в настоящем изобретении. Замена уходящей группы в виде хлора в соединении формулы V гидроксиалкилом кислоты обеспечивает получение гидроксиалкильных сложных эфиров формулы XVI. Фосфорилирование спирта фосфорамидированием с последующем окислением фосфора дает соединения формулы XVII, с которых может быть снята защита обработкой кислотой, такой как трифторуксусная кислота, что дает на выходе фосфаты формулы If.
РЕАКЦИОННАЯ СХЕМА 8

Реакционная схема 8 демонстрирует получение пирофосфатов формулы Ig, где R и
РЕАКЦИОННАЯ СХЕМА 9

Замена группы хлора в соединениях формулы V фосфатами обеспечивает получение фосфатов алкиламмония формулы Ih1, как изображено на реакционной схеме 9, где R, R1 и М+ имеют значения, как определено в настоящем изобретении. Обработка фосфатов аммония ионообменной смолой дает алкилфосфаты формулы Ih2.
В предпочтительном воплощении изобретения соединения имеют формулу I'

где волнообразная связь
А представляет собой прямую связь или (С=O);
В представляет собой прямую связь или кислород;
m имеет значение 0 или 1;
n имеет значение 1, 2 или 3;
R1 и R2 каждый независимо является водородом или гидролизуемой группой сложного эфира и когда R1 представляет собой водород, R2 может также быть -P(O)OR5OR6;
R3 и R4 каждый независимо является водородом или C1-4 алкилом и
R5 и R6 каждый независимо является водородом или гидролизуемой группой сложного эфира;
или его нетоксичная фармацевтически приемлемая соль или его сольват.
В более предпочтительном воплощении изобретения волнообразная связь
В другом предпочтительный воплощении изобретения соединения формулы I выбраны из группы, содержащей:
(S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый] эфир;
(R)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый]эфир;
(S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-фосфоноксипропиловый эфир;
(S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-фосфоноксиэтиловый эфир;
(S)-(2-фосфонооксиэтил)карбаминовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир;
(S)-фосфоноксиуксусной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2, 3-дигидроиндол-1-илметиловый эфир;
(S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислотаы фосфоноксиметиловый эфир;
(S)-3-фосфоноксипропионовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир;
(S)-пирофосфорной кислоты [3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир;
(S)-фосфорной кислоты [3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2, 3-дигидроиндол-1-илметиловый эфир]метиловый эфир и
(S)-фосфорной кислоты-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир]этиловый эфир или
его нетоксичная фармацевтически приемлемая соль или его сольват.
В другом воплощении настоящее изобретение включает фармацевтические композиции, содержащие, по крайней мере, одно соединение формулы I в комбинации с адъювантом, носителем или разбавителем.
В еще другом воплощении настоящее изобретение относится к способу лечения или профилактики заболеваний, чувствительных к раскрытию калиевых каналов у млекопитающего, нуждающегося в этом, включающему введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или его нетоксичной фармацевтически приемлемой соли, сольвата или гидрата.
В еще другом воплощении настоящее изобретение относится к способу лечения ишемии, конвульсии, эпилепсии, астмы, раздражительного синдрома кишечника, мигрени, травматического мозгового повреждения, повышенного внутричерепного давления, травмы спинного мозга, отравления монооксидом углерода, мужской и женской половой дисфункции, недержания мочи и особенно инсульта у млекопитающего, нуждающегося в этом, включающему введение указанному млекопитающему терапевтически эффективного количества соединения Формулы I или его нетоксичной фармацевтически приемлемой соли, сольвата или гидрата.
Биологическая активность
Каналы калия (K+) являются структурно и функционально различными семействами K+-селективных канальных белков, которые присутствуют в клетках, что определяет их центральную роль для регулирования множества ключевых функций клетки [Rudy, В., Neuroscience. 25, pp.729-749 (1988)]. Хотя они широко распространены как класс, K+ каналы по-разному распределены как индивидуальные члены этого класса или как члены семейства [Gehlert, D.R. и др., Neuroscience. 52, pp.191-205 (1993)]. Вообще, активация K+ каналов в клетках, и особенно в легковозбудимых клетках, таких как нейроны и мышечные клетки, приводит к гиперполяризации клеточной мембраны или в случае деполяризованных клеток - к реполяризации. В дополнение к действию в качестве эндогенного мембранного зажима напряжения, K+ каналы могут обусловливать важные клеточные явления, такие как изменения внутриклеточной концентрации АТФ или внутриклеточной концентрации кальция (Са2+). Центральная роль K+ каналов в регулировании многочисленных функций клетки делает их особенно важными мишенями для терапевтических разработок [Cook, N.S., Potassium channels: Structure, classification, function and therapeutic potential. Ellis Horwood, Chinchester (1990)]. Один класс K+ каналов Са2+-активируемых K+ каналов (ВК или ВК каналы) с большой проводимостью регулируется трансмембранным напряжением, внутриклеточным Са+и другими разнообразными факторами, такими как состояние фосфорилирования канального белка [Latorre, R. и др., Ann. Rev. Physiol. 51, pp.385-399 (1989)]. Большая отдельная канальная проводимость (обычно >150 pS) и высокая степень специфичности для K+ BK каналов показывает, что небольшое число каналов может глубоко затрагивать мембранную проводимость и клеточную возбудимость. Дополнительно, увеличение открытой вероятности с увеличением внутриклеточного Са2+ указывает на причастность BK каналов к модуляции Са2+-зависимого явления, такого как секреция и мускульное сокращение [Asano, М. и др., J. Pharmacol. Exp.Ther. 267. pp.1277-1285 (1993)].
Вещества, раскрывающие ВК каналы, оказывают их клеточное воздействие путем увеличения вероятности раскрытия этих каналов [МсКау, М.С. и др., J. Neurophvsiol. 71. рр.1873-1882 (1994); Olesen, S.-P, exp.Opin.Invest. Drugs 3, pp.1181-1188 (1994)]. Это увеличение раскрытия всех индивидуальных ВК каналов приводит к гиперполяризации клеточных мембран, особенно в деполяризованных клетках, вызванное существенным увеличением BK-опосредованной проводимости в целой клетке.
Способность соединения формулы II открывать BK каналы и увеличивать направленные наружу (K+) BK-опосредованные потоки целой клетки оценивали в условиях зажима напряжения, определяя их способность увеличивать (mSlo или hSlo) направленного наружу ВК-медиированного потока в клонированном млекопитающем, гетерологически экспрессированного в овоцитах Xenopus [Butler, А. и др., Science, 261. pp.221-224 (1993); Dworetzky, S.I. и др., Mol. Brain Res., 27, pp.189-193 (1994)]. Две используемые ВК конструкции представляют почти структурно почти идентичные гомологичные белки и, как оказалось, являются фармакологически идентичными в испытаниях. Для выделения BK потока из нативного (фон, не-BK) потока использовали специфический и мощный блокирующий ВК канал, блокирующий токсин ибериотоксин (IBTX) [Galvez, А. и др., J. Biol. Chem. 265. pp.11083-11090 (1990)] при супрамаксимальной концентрации (50 нМ). Относительный вклад потока ВК каналов в общий, направленный наружу поток определяли вычитанием потока, остающегося в присутствии IBTX (не-BK поток) из профилей потока, полученных во всех других экспериментальных условиях (контроль, лекарственное средство и промывка). Было определено, что при тестируемой концентрации соединение не производит нативных не-BK потоков в ооцитах. Показано, что соединение Формулы II по крайней мере в 5 ооцитах в концентрации 10 мкМ увеличивает ВК на 170% от контроля IBTX-чувствительного потока. Запись проводили с помощью стандартных способов зажима напряжения с двумя электродами [Stuhmer, W. и др., Methods in Enzymology. 207. pp.319-339 (1992)]; протоколы зажима напряжения состояли из стадий деполяризации продолжительностью 500-750 мсек от держащегося потенциала от -60 до +140 мВ с шагом 20 мВ. Экспериментальная среда (модифицированный раствор Барта) состояла из (в мМ): NaCl (88), NaHCO3 (2.4), KCl (1.0), HEPES (10), MgSO4 (0.82), Ca(NO3)2 (0.33), CaCl2 (0.41); pH 7.5.
Соединение формулы (S)-II или пролекарство Формулы I вводили внутривенно болюсами самцам крыс Sprague-Dawley (n=3 крысы/время) в эквиваленте дозы-мишени 1 мг/кг соединения Формулы (S)-II. В Т=0.25, 1 или 2 часа после дозы собирали образцы общей крови и экстрагировали ацетонитрилом. Экстракты крови проанализировали с помощью LC/MS/MS на уровни соединения Формулы (S)-II. Таблица показывает сравнительные оценки стволовых 0.25-2 часа AUC соединения Формулы (S)-Второй после введения соединения Формулы II или пролекарства Формулы I. Например, как показано в таблице, после введения пролекарств Формулы I, соединение Формулы (S)-II было обнаружено в крови этой модели крыс.
Для определения способности соединений настоящего изобретения уменьшать гибель клеток, обусловленную нейронной ишемией, вызывали стандартную центральную мозговую ишемию путем постоянного перекрывания левой средней мозговой артерии (МСА) и общей сонной артерии (ССА) с одночасовым перекрыванием правой ССА в крысах Wistar. Операции выполняли, используя подвременный подход A. Tamura и др., J. Cereb. Blood Flow Metab., 1, pp.53-60, (1981), и его модификации [K.Osbome и др., J. Neurol Neurosurg. Psychiatry, 50, pp.402-410 (1987) и S. Menzies и др., Neurosurgery. 31., pp.100-107, (1992)].
Соединение Формулы II оценивали в модели центрального паралича, включающей постоянное перекрывание левого МСА (МСАО) и ССА (ССАО) и временного перекрывания правой ССА в крысах Wistar [Gribkoff и др., Nature Med. 7, pp.471-477 (2001)]. Эта процедура приводит к устойчиво большим объемам неокортикального инфаркта, которые измеряют посредством исключения жизненного красителя в последовательных пластинах в мозге через 24 часа после МСАО. В настоящем испытании соединения вводились внутривенным или внутрибрюшинным способом через два часа после перекрывания. Например, в этой модели соединение формулы II значительно уменьшило объем коркового инфаркта, примерно до 28%, при введении внутривенно (0.3 мг / кг) в виде отдельного болюса через два часа после перекрывания средней мозговой артерии по сравнению с контролем в виде инертного вещества.
Для определения способности соединений настоящего изобретения уменьшать количество поврежденных нейронов после травмы головного мозга использовали стандартную модель травмы головного мозга. Модель травмы головного мозга крыс (TBI) использовали для оценки соединения на эффективность изменения или предотвращения вредных эффектов подобной травмы сотрясения мозга. Вообще, крыс в этой модели обезболивали, осуществляли краниотомию (хирургическое вскрытие черепа) и затем в разрез вводили соляной раствор для произведения точного потока увеличенного интракраниального давления (обычно называемое жидкой перкуссионной травмой). Животным вводили соединение в указанных дозах через 15 минут после травмы. Животных умерщвляли через 48 часов после TBI.
Умеренную диффузионную мозговую травму (определенная Mclntosh и др. Neuroscience. 28:233-44 (1989)) вызывали устройством для жидкой перкуссии. Аппарат производит контузию быстрым введением потока соляного раствора [давление ˜от 2.1 до 2.7 атмосфер (атм)] в постоянной продолжительности (21-23 миллисекунды) в закрытую черепную полость. Поток соляного раствора приводит к краткому смещению и деформации основной коры головного мозга. Эта модель, как полагают, имитирует клиническую ситуацию, в которой пациент испытывает подобное сотрясение мозга, характеризующееся краткими неврологическими и системными физиологическими изменениями без серьезного структурного повреждения. Устройство для жидкого вливания производит мозговую травму без непосредственно воздействия на мозг. Распространение мозговой травмы достигается выпуском взвешенного (4.8 кг) металлического маятника с предопределенной высоты (Mclntosh и др., 1989), который ударяет закрытый в конце пробкой поршень Plexiglass цилиндрической емкости, заполненной изотоническим соляным раствором. Различные объемы соляного раствора вводят в закрытую черепную полость, производя поток увеличенного интракраниального давления (ICP). Изменение высоты маятника влияет на величину травмы.
В этом эксперименте пульсацию давления измеряли экстракраниально преобразователем, расположенным в травмирующем устройстве. После анестезии, травмирующий винт сильно связывали с устройством жидкого вливания и вызывали травму умеренной серьезности [от -2.1 до 2.7 атм], основываясь на шкале, установленной Mclntosh и др., 1989. Пульсацию регистрировали на осциллографе, запускаемом фотоэлектрически пуском маятника. После жидкого вливания крышка, созданная винтом травмы, стальной винт и краниопластический цемент были удалены и рану закрывали непроницаемым швом (3-0). Животные, остающиеся под анестезией в течение более 60 секунд после травмы, были немедленно умерщвлены. Крыс оставляли на водной рециркулирующей электрогрелке до нормализации дыхания и они двигались. Животных также умерщвляли и мозг удаляли для оценки отека через 48 часов измерением содержания воды, как описано ранее (Mclntosh и др., 1989).
Ранее было показано, что соединение формулы (S)-II производит существенные сокращения отека в нескольких областях, смежных с зоной воздействия [Cheney и др. J. Cer. Blood Flow & Metab.21:396-403 (2001)]. Соединения Формулы Ia согласно настоящему изобретению производят существенные сокращения отека в смежной коре головного мозга по сравнению с сокращением соединением Формулы (S)-II, когда обоих вводят в эквивалентных молярных дозах в той же самой животной модели травмы головного мозга.
В альтернативном способе серьезность повреждения головного мозга оценивали измерением интракраниального давления (ICP) в различных интервалах после жидкого вливания через 24 часа. Кратко, до вливания жидкости было сделано буровое отверстие 1 мм от стреловидного шва, с центром у правой теменной коры мозга в 5 мм от лямбды, 5 мм от брегмы. После травмы жидким вливанием крышку поверх травмы и краниопластический цемент удаляли. Для достижения адекватных значений ICP, находясь под анастезией, крышку краниотомии ставят в ее первоначальное местоположение после жидкого вливания. Предварительно разрезанную стерильную фольгу также помещают поверх крышки краниотомии. Краниопластический цемент помещали поверх фольги для запечатывания мозговой полости. Для получения непрерывных измерений ICP пробу вставляли в буровое отверстие и фиксировали краниопластическим цементом, животным затем накладывали швы (использование 3-0) и крыс помещали в клетку Hopper, чтобы учесть свободное движение в течение эксперимента. Затем ICP преобразователь связывали с Codman ICP монитором для непрерывных измерений ICP через 24 часа после TBI или после травмы. В это время животное имело регулярный доступ к воде и продовольствию. Через 24 часа после TBI эксперимент заканчивали и животных умерщвляли передозировкой пентабарбитала натрия (130 мг/кг; внутрибрюшинно).
Данные эксперименты состоят из группы инертного переносчика (SV), группы TBI-переносчика (TV), групп, обрабатываемых инертным лекарственным средством (SD; 0.08 мг/кг соединений примера 14 [эквивалент дозы 0.05 соединений мг/кг Формулы (S)-II]) и групп, обрабатываемых TBI-лекарственным средством (TD; 0.08 мг/кг соединений Примера 14). Каждая инертная группа действовала как контроль для соответствующей TBI группы. Всех животных анестезировали и оперировали. В TBI группах вызывали мозговую травму, используя боковую модель жидкого потока. Лечение лекарственным средством начинали через 15 минут после TBI или инертной травмы. Соединение рассматривалось эффективным, если оно значительно уменьшало ICP по сравнению с его соответствующей инертной поврежденной группой в любой данной точке времени (определено ANOVA, по сравнению с соответствующей инертной группой; определено t-испытанием). Через пять минут после травмы абсолютный ICP (необработанные данные) увеличили на 6.8 мм Hg единиц в TV животных, по сравнению с SV животными (SV-5.55 по сравнению с TV-12.35). В течение следующих 30 минут спустя значения ТВПСР уменьшали и наконец стабилизировали в 8.55 мм Hg (TV) и 5 мм Hg (SV) соответственно. TD значительно отличался от TV через 45 минут, 60 минут, 2 часа, 3 часа, 4 часа и 24 часа после TBI с наиболее существенным уменьшением (3.1 мм Hg единицы) через 60 минут после травмы (или 45 минут после введения препарата (TV-8.15 по сравнению с TD-5.05 мм Hg)). Внутренняя оценка животного подразумевает ICP сокращение, TD значительно уменьшено по сравнению с TV через 30 минут (-2.85 по сравнению с -1.15 мм Hg), 45 минут (-3.95 по сравнению с -1.35), 60 мин (-4.5 по сравнению с -1.55), 2 часа (-4.3 по сравнению с -1.95), 3 часа (-4.3 по сравнению с 1.8), 4 часа (-4.15 по сравнению с -1.3) и 24 часа (-3.05 по сравнению с -0.9) после травмы с максимальным сокращением, наблюдаемым через 60 минут после травмы (или 45 минут после введения препарата). SD значения значительно не отличались от SV до 4 часов после инертной травмы (-1.5 [SV] по сравнению с -2.1 [SD] мм Hg), однако через 24 часа после введения препарата наблюдаемое ICP сокращение между группами уменьшалось.
Результаты вышеупомянутых испытаний показывают, что новые соединения оксиндола настоящего изобретения полезны для лечения заболеваний человека, являющихся результатом дисфункции клеточной мембранной поляризации и проводимости и предпочтительно предназначены для лечения ишемии, паралича, конвульсий, эпилепсии, астмы, синдрома раздраженного кишечника, мигрени, травмы головного мозга, повышенного интракраниального давления, травмы спинного мозга, отравления монооксидом углерода, сексуальной дисфункции и недержания мочи и других заболеваний, чувствительных к активизирующей активности ВК канала. Наиболее предпочтительно соединения формулы I полезны для лечения мозговой ишемии/паралича, травмы головного мозга и повышенного интракраниального давления.
Соединения Формулы I или их фармацевтические композиции полезны для лечения, облегчения или устранения заболеваний или других болезней, связанных с ВК каналами. Такие заболевания включают ишемию, паралич, конвульсии, эпилепсию, астму, синдром раздраженного кишечника, мигрень, травму головного мозга, повышенное интракраниальное давление, травму спинного мозга, отравление монооксидом углерода, сексуальную дисфункцию, недержание мочи и другие заболевания, чувствительные к веществам, раскрывающим калиевый канал.
Для терапевтического применения фармакологически активные соединения Формулы I будут обычно вводиться в виде фармацевтической композиции, содержащей в качестве существенного активного компонента, по крайней мере, одно такое соединение совместно с твердым или жидким фармацевтически приемлемым носителем и необязательно с фармацевтически приемлемыми адъювантами и эксципиентами, используя стандартные и обычные способы.
Фармацевтические композиции включают подходящие формы дозировки для орального, парентерального (включая подкожное, внутримышечное, интрадермальное и внутривенное) бронхиальное или назальное введение. Таким образом, если используется твердый носитель, состав может быть таблетирован, помещен в твердую желатиновую капсулу в порошке или форме шарика или находиться в форме пастилки или лепешки. Твердый носитель может содержать обычные эксципиенты, такие как связующие агенты, наполнители, таблетирующие смазки, разрыхлители, увлажняющие агенты и т.п. Таблетка, если желательно, может быть покрыта пленкой обычными способами. Если используется жидкий носитель, состав может находиться в форме сиропа, эмульсии, мягкой желатиновой капсулы, стерильного транспортного средства для инъекции, водной или неводной жидкой суспензии или может представлять собой сухой продукт для соединения с водой или другим подходящим транспортным средством перед использованием. Жидкие составы могут содержать обычные добавки, такие как суспендирующие агенты, эмульсифицирующие агенты, увлажняющие агенты, неводные транспортные средства (включая съедобные масла), консерванты, а также отдушки и/или красители. Для парентерального введения транспортное средство обычно будет включать стерильную воду, по крайней мере в значительной степени, хотя могут использоваться растворы соляного раствора, растворы глюкозы и им подобные. Вводимые суспензии также могут использоваться, когда могут использоваться обычные суспендирующие агенты. Обычные консерванты, буферные агенты и т.п. также могут быть добавлены к парентеральным формам дозировки. Особенно полезным является введение соединения Формулы I непосредственно в парентеральных составах. Фармацевтические композиции получают обычными способами, соответствующими желаемому составу, содержащему соответствующие количества активного компонента, то есть соединения Формулы I согласно изобретению. См., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, 17th edition, 1985.
Дозировка соединений Формулы I для достижения терапевтического эффекта будет зависеть не только от таких факторов, как возраст, вес и пол пациента и способ введения, но также и от желаемой степени активизирующей активности калиевого канала и активности конкретного соединения, используемого для лечения конкретной болезни. Предполагается, что лечение и дозировка конкретного соединения могут вводиться в форме дозированной единицы и что форма дозированной единицы регулируется соответственно, как известно среднему специалисту, для отражения относительного уровня активности. Решение относительно конкретной используемой дозировки (и количества раз, которые нужно вводить в день), принимает врач по усмотрению, и может изменяться в зависимости от конкретных обстоятельств данного изобретения для произведения желаемого терапевтического эффекта.
Подходящая доза соединения Формулы I или его фармацевтической композиции для млекопитающего, включая человека, страдающего или подверженного любому описанному здесь состоянию, составляет количество активного компонента приблизительно от 0.1 нг/кг до 10 мг/кг веса тела. Для парентерального введения доза может составлять в диапазоне от 0.1 нг/кг веса тела до 10 мг/кг для внутривенного введения. Активный компонент будет предпочтительно вводиться болюсной инъекцией или болюсной инъекцией непрерывного вливания; непрерывно или в равных дозах от одного до четырех раз в день.
Однако следует понимать, что количество фактически вводимого соединения будет определено врачом в свете конкретных обстоятельств, включая состояние болезни, выбор вводимого соединения, выбранный способ введения, возраст, вес и индивидуальный ответ пациента и серьезность симптомов пациента.
Следующие примеры приводятся для иллюстрации и никаким образом не должны ограничивать изобретение, поскольку в пределах осуществления изобретения возможно множество вариантов изобретения.
ОПИСАНИЕ КОНКРЕТНЫХ ВОПЛОЩЕНИЙ
В следующих примерах все значения температур даются в градусах по шкале Цельсия. Температуру плавления регистрируют на некорректируемом приборе Gallenkamp определения температур плавления в капиллярах. Магнитный протоновый резонанс (1Н ЯМР) регистрируют на спектрометре Bruker AC 300. Все спектры снимают в обозначенных растворителях при химических сдвигах, переданных в δ единицах нижней области внутреннего стандарта тетраметилсилана (TMS) и межпротонных константах взаимодействия, переданных в герцах (Гц). Разделение структуры определяется следующим образом: с, синглет; д, диполь; т, триплет; кв., квартет; м, мультиплет; уширенный, широкий пик; дд, диполь диполя; бд, широкий диполь; дт, диполь триплета; бс, широкий синглет; dq, диполь квартета. Инфракрасные (IR) спектры, используя бромистый калий (KBr), определяют на Perkin Elmer 781 спектрометре в пределах от 4000 до 400 см-1, калиброванном до 1601 см-1 поглощения пленкой из полистирола и выраженного в обратных сантиметрах (см-1). Нижнее разрешение масс-спектра (MS) и молекулярная масса (МН+) или (М-Н)+ определяют на приборе Finnigen TSQ 7000. Верхнее разрешение масс-спектра определяют Kratos MS50 в FAB способом, используя иодит цезия /глицерин в качестве внутренней ссылки. Элементарный анализ выражают как процент от веса.
Следующие примеры приготовления иллюстрируют методики получения промежуточных соединений и способов получения продуктов в соответствии с настоящим изобретением. Среднему специалисту очевидно, что подходящее замещение и продуктов, и способов, раскрытых в настоящем изобретении, представленных примерами, иллюстрируемыми ниже, находится в границах настоящего изобретения.
ПРИМЕР 1
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, бис-трет-бутиламмонийная соль ((S)Ia)
Стадия А. Фосфорной кислоты ди-трет-бутиловый эфир 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-III)
В колбу объемом 250 мл, содержащую (3S)-3-(5-хлор-2-метоксифенил)-3-фтор-6-трифторметил-1,3-дигидроиндол-2-он ((S)-II) (полученный в соответствии с US 5808095) (2.00 г, 5.57 ммоль) добавляют 20 мл безводного ацетонитрила, карбонат цезия (2,35 г, 7.24 ммоль) и хлорметил-ди-трет-бутилфосфат (2.16 г, 8.36 ммоль). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре, затем растворитель выпаривают в вакууме. Полученный сырой остаток очищают с помощью флеш-хроматографии (силикагель; 4:1 гексан: EtOAc), получая названное соединение (2.12 г, 65%) в виде твердого вещества белого цвета.1H ЯМР (CDCl3, 400 МГц): δ=7.78 (с, 1Н), 7.47 (с, 1Н), 7.34-7.31 (м, 2Н), 7.25 (д, 1Н, J=7 Гц), 6.74 (д, 1Н, J=7 Гц), 5.71 (дд, 1Н, J=12.0, 6.8 Гц), 5.59 (дд, 1Н, J=12.0, 6.8 Гц), 3.53 (с, 3Н), 1.52 (с, 9Н), 1.51 (с, 9Н).
Стадия В. (S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, бис-трет-бутиламмонийная соль ((S)-Ia)
В колбу объемом 100 мл, содержащую фосфорной кислоты ди-трет-бутиловый эфир 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2, 3-дигидроиндол-1-ил метилового эфира ((S)-III) (0.500 г, 0.860 ммоль) добавляют 10 мл безводного дихлорметана и трифторуксусную кислоту (0.196 г, 1.72 ммоль). Реакционной смеси дают возможность перемешиваться при комнатной температуре в течение 24 часов. Дихлорметан выпаривают в вакууме и сырую пену очищают с помощью хроматографии на реверсивной фазе (C18, 2:1 СН3 CN:Н2O), затем выделенный свободный фосфат растворяют в этилацетате и добавляют трет-бутиловый амин (0.251 г, 3.44 ммоль) и растворитель выпаривают в вакууме, получая названное соединение (0.100 г, 19%) в виде порошка белого цвета.1H ЯМР (D2O 500 МГц): δ=7.89 (с, 1Н), 7.78 (с, 1Н), 7.50 (м, 2Н), 7.41 (д, 1Н, J=6.4 Гц), 7.03 (д, 1Н, J=6.4 Гц), 5.63 (дд, 1Н, J=12.0, 6.8 Гц), 5.56 (дд, 1Н, J=12.0, 6.8 Гц), 3.56 (с, 3Н), 1.36 (с, 18Н).31P ЯМР (D2О, 202 МГц) δ=1.68; LRMS [М-H]-467.9.
ПРИМЕР 2
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, монотетрабутиламмонийная соль ((S)-Ia1)
Стадия А. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-1-гидроксиметил-6-трифторметил-1.3-дигидроиндол-2-он ((S)-IV)
К смеси (S)-3-(5-хлор-2-метоксифенил)-3-фтор-6-трифторметил-1, 3-дигидроиндол-2-она ((S)-II) (60.0 г, 0.167 моль) и K2СО3 (27.7 г, 0.20 моль) в ТГФ (600 мл) добавляют формальдегид (37% раствор в Н2O, 240 мл, 3.2 моль), затем Н2O (300 мл). Слегка мутную смесь перемешивают при комнатной температуре в течение 3 часов и разбавляют диэтиловым эфиром (1000 мл). Органический слой отделяют. Водный слой промывают эфиром (200 мл х 2). Объединенный органический слой промывают рассолом, сушат над Na2SO4. Выпаривание растворителей обеспечивает получение названного соединения в виде сухой пены белого цвета (64.5 г, 99% выход). LC/MS м/е: 390 (МН+), 96% чистота.1H ЯМР (CDCl3): δ 3.50 (с, 3Н), 5.28 (м, 1Н), 5.45 (м, 1Н), 6.75 (дд, J=1.5, 6.5 Гц, 1Н), 7.24 (м, 1Н), 7.34 (м, (м, 3Н), 7.79 (дд. J=1.0, 3.0 Гц, 1Н).
Стадия В. (S)-3-(5-Хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-он ((S)-V)
К раствору (S)-3-(5-хлор-2-метоксифенил)-3-фтор-1-гидроксиметил-6-трифторметил-1,3-дигидроиндол-2-она ((S)-IV) (64.5 г, 0.166 моль) в СН2Cl2 (700 мл) добавляют по каплям PCl3 (2.0 М в СН2Cl2, 255.0 мл, 0.510 моль) при температуре 0°С в атмосфере N2 в течение 50 минут. Полученную смесь нагревают до комнатной температуры и перемешивание продолжают в течение ночи (16 часов). Смесь гасят льдом при температуре 0°С и полученную смесь перемешивают энергично в течение 30 минут. Органический слой отделяют и водный слой промывают СН2Cl2. Объединенный органический слой промывают рассолом и сушат над Na2SO4. Хроматография (силикагель, EtOAc/гексан) обеспечивает получение названного соединения в виде сухой пены белого цвета (42.0 г, 62% выход).1H ЯМР (CDCl3): δ 3.55 (с, 3Н), 5.39 (д, J=10.5 Гц, 1Н), 5.95 (д, J=11.0 Гц, 1Н), 6.76 (дд, J=1.5, 9.0 Гц, 1Н), 7.28 (м, 2Н), 7.35 (дд, J=1.5, 9.0 Гц, 1Н), 7.40 (д, J=8.0 Гц, 1Н), 7.78 (дд, J=1.0,2.5 Гц, 1Н).
Стадия С. (S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, монотетрабутиламмонийная соль ((S)-Ia1)
Удаляют Н2О из коммерчески доступного тетрабутиламмонийн дигидрофосфата (Aldrich, 1.0 М, 1000 мл, 1.0 моль), удаляют путем выпаривания при температуре 28°С и сушат в течение ночи в высоком вакууме. Остаток повторно растворяют в сухом ацетонитриле (4Å, 2500 мл). В полученный раствор добавляют по каплям раствор (S)-3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-она ((S)-V) (42.0 г, 0.103 моль) в СН3CN (200 мл) при температуре 0°С в атмосфере N2 в течение 30 минут. Реакционную смесь перемешивают при температуре 0°С в течение еще 30 минут. Перемешивание продолжают при комнатной температуре в течение 2.5 часов и растворитель удаляют. Остаток растворяют в СН2Cl2 (1200 мл), промывают Н2O (4 х 200 мл) и сушат над Na2SO4. Хроматография (силикагель, MeOH/CH2Cl2) обеспечивает получение названного соединения в виде твердого вещества белого цвета (28.0 г, 38% выход). LC/MS м/е: 470 (МН+), 99% чистота.1H ЯМР (D2O): δ 0.89 (т, J=7.4 Гц, 12Н), 1.29 (м, 8Н), 1.58 (м, 8Н), 3.11 (т, J=7.3 Гц, 8Н), 3.44 (с, 3Н), 5.48 (м, 1Н), 5.59 (м, 1Н), 6.80 (д, J=9.0 Гц, 1Н), 6.98 (д, J=7.0 Гц, 1Н), 7.16 (д, J=7.5 Гц, 1Н), 7.26 (д, J=8.5 Гц, 1Н), 7.66 (с, 1Н), 7.71 (с, 1Н).
Пример 3
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, мононатриевая соль ((S)Ia)
Смолу Dowex-50wx8-100 (1350 г) промывают Н2О, МеОН, Н2O и затем подщелачивают до значения рН>10 с помощью раствора гидроксида натрия (1.0М). Затем ее промывают Н2О до значения рН ˜7 и полученную смолу разделяют на три порции (˜450 г каждая). К раствору (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1 -ил метилового] эфира, монотетрабутиламмонийной соли ((S)-Ia1) (27 г, 0.038 моль) в 500 мл H2О добавляют одну порцию смолы (˜450 г). Смесь перемешивают в течение 10 минут, отфильтровывают и промывают Н2О. В объединенный фильтрат добавляют еще порцию смолы. Смесь перемешивают в течение 10 минут, отфильтровывают и промывают Н2О. В этот фильтрат добавляют последнюю порцию смолы. Смесь перемешивают в течение 10 минут, отфильтровывают и промывают Н2O. Объединенные фильтраты отфильтровывают через небольшой слой (1 см) С-18 силикагеля с обращенной фазой и промывают Н2О. Растворители объединенных фильтратов выпаривают. Остаток растворяют в ацетонитриле и отфильтровывают. Ацетонитрил выпаривают, остаток повторно растворяют в СН2 Cl2 и гексан добавляют к раствору. Выпаривание растворителей обеспечивает получение названного соединения в виде порошка ярко-белого цвета (15.4 г, 87% выход). MS м/е: 468.0 (М-Н- ). LC/MS м/е: 470 (МН+), 98% чистота. Аналитически вычислено для C17H12ClF4NO6PN·1.05Na·0.18H2O: С 41.15; Н 2.51; N 2.82, Na 4.89. Найдено: С 41.54; Н 2.50; N 2.70; Na 4.97.1H ЯМР (D2O): δ 3.52 (с, 3Н), 5.56 (м, 1Н), 5.65 (м, 1Н), 6.94 (д, J=9.0 Гц, 1Н), 7.21 (д, J=7.5 Гц, 1Н), 7.36 (д, J=7.5 Гц, 1Н), 7.43 (дд, J=1.5, 9.0 Гц, 1Н), 7.72 (с, 1Н), 7.79 (с, 1Н).13С ЯМР (D2O): δ 58.79, 67.99 94.23 110.52 116.42 124.01 126.38, 128.04, 128.35, 128.49, 128.74, 131.48, 133.52, 135.98, 145.96, 156.38, 175.98.19F ЯМР (D2O): δ - 63.2, -164.31P ЯМР (D2O): δ 0.12. 0.12. ИК (KBr, см-1): 3433, 1756, 1319, 1131, 1026. [α]20 (H2O) +131.95. Анализ хирального капиллярного электрофореза (СЕ) показывает 99+% (S)-изомер.
ПРИМЕР 4
(R) Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, мононатриевая соль ((R)-Ia2)
Исходя из (R)-3-(5-хлор-2-метоксифенил)-3-фтор-6-трифторметил-1,3-дигидроиндол-2-она ((R)-H) (полученного в соответствии с US 5602169), названное соединение получают, используя общие методики, описанные в Примерах 2 и 3. [α]20(H2O) -104.42. Анализ хирального капиллярного электрофореза (СЕ) показывает 89% (R)-изомер и 10% (S)-изомер вместе с 1% примеси неизвестного строения.
ПРИМЕР 5
(S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновая кислота 2-фосфонооксипропиловый эфир ((S)Ib. n=2)
Стадия А. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-(ди-трет-бутоксифосфорилокси)пропиловый эфир (YS)-VII. n=2)
К раствору (S)-3-(5-хлор-2-метоксифенил)-3-фтор-6-трифторметил-1,3-дигидроиндол-2-она ((S)-II) (720 мг, 2 ммоль) в дихлорметане добавляют по каплям фосген (2.2 мл, 20% раствор в толуоле, 4 ммоль) и пиридин (0.6 мл). Полученный хлорформиат (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбонилхлорида ((S)-VI) перемешивают при комнатной температуре в атмосфере N2 в течение ночи. В эту реакционную смесь добавляют по каплям другую порцию 0.6 мл пиридина, затем добавляют фосфорную кислоту ди-трет-бутиловый эфир 3-гидроксипропилового эфира (1.55 г, 5.78 ммоль), получение которой приведено ниже. Полученную смесь перемешивают при комнатной температуре еще в течение часа, разбавляют дихлорметаном и промывают водой. Органический слой сушат над Na2SO4 и конденсируют путем выпаривания. Остаток хроматографируют на флеш-колонке из силикагеля, заполненной смесью этилацетат : гексан (объем/объем, 4:1) и элюируют этилацетат : гексан (объем/объем, 4:1˜2:1), что дает названное соединение (818 мг, 63%).1H ЯМР (CDCl3, 500 МГц): δ 10.06 (с, 1Н), 8.31 (с, 1Н), 7.78 (т, 1Н), 7.43 (д, 1Н), 7.35 (дд, 1Н), 7.27 (д, 1Н), 6.76 (дд, 1Н), 4.59 (м, 2Н), 4.16 (м, 2Н), 3.52 (с, 3Н), 2.18 (м, 2Н), 1.48 (с, 18Н). LC-MS: 654.1 (MH+).
Получение фосфорной кислоты ди-трет-бутиловый эфир 3-гидроксипропилового эфира
В колбу объемом 100 мл добавляют тетра-н-бутиламмонийнной соли фосфорной кислоты ди-трет-бутилового эфира (3.08 г, 6.8 ммоль), диметилэтиленгликоль (10 мл) и 3-бром-1-пропанол (свежеперегнанный из карбоната калия). Реакционную смесь нагревают до кипения в течение 2.5 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют этиловым эфиром (60 мл) и охлаждают в холодильнике, пока осаждают твердое вещество белого цвета. Твердое вещество отфильтровывают через целит и фильтрат выпаривают в вакууме, получая названное соединение в виде прозрачного масла (1.85 г, 100%).1H ЯМР (CDCl3, 500 МГц): δ 4.10 (м, 2Н), 3.73 (м, 2Н), 1.84 (м, 2Н), 1.46 (с, 18Н).
Стадия В. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-фосфоноксипропиловый эфир ((S)-Ib. n=2)
Раствор (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1 -карбоновой кислоты 2-(ди-трет-бутоксифосфорилокси)пропилового эфира ((S)-VII, n=2) (540 мг, 0.82 ммоль) в дихлорметане (30 мл) перемешивают с трифторуксусной кислотой (0.6 мл) при комнатной температуре в течение 3 часов. LC-MS показывает завершение реакции. После удаления растворителя остаток растворяют в метаноле и сырой продукт очищают с помощью препаративной HPLC, получая названное соединение 255 мг (57%) в виде твердого вещества белого цвета после лиофилизации.1H ЯМР (CDCl3, 500 МГц): δ 8.27 (с, 1Н), 7.73 (д, 1Н), 7.42 (д, 1Н), 7.32 (д, 1Н), 7.27 (д, 1Н), 6.75 (д, 1Н), 4.50 (б, 6Н, содержащий некоторое количество Н2О в виде сольвата), 4.54 (м, 2Н), 4.17 (б, 2Н), 3.49 (с, 3Н), 2.1 (м, 2Н); MS (м/е) 539.92 (M+-1): LC-MS, 542 (MH+, 98.5%);31P ЯМР (CDCl3, 500 МГц): δ 2.03.19F ЯМР (CDCl3, 500 МГц): δ -63.4,153.8. Вычислено для C20H17ClF4NO8P: С 44.34%, Н 3.16%, N 2.58%. Найдено: С 44.01%, Н 3.11%, N 2.45%.
ПРИМЕР 6
(S)-3-(5-Хлор-2-метоксифенил-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-фосфоноксиэтиловый эфир ((S)-Ib. n=1)
Стадия А. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-(ди-трет-бутоксифосфорилокси)этиловый эфир ((S)-VII. n=1)
Названное соединение получают аналогично как (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-(ди-трет-бутоксифосфорилокси)пропиловый эфир ((S)-VII, n=2) с 52% выходом, используя фосфорной кислоты ди-трет-бутиловый эфир 2-гидроксиэтилового эфира.1Н ЯМР (CDCl3, 500 МГц): δ 8.29 (с, 1Н), 7.77 (д, 1Н), 7.43 (д, 1Н), 7.35 (д, 1Н), 7.27 (д, 1Н), 6.75 (д, 1Н), 4.67 (м, 2Н), 4.31 (м, 2Н), 3.53 (с, 3Н), 1.47 (с, 18Н); LC-MS, 640.15 (МН+, 96).31P ЯМР (CDCl3, 500 МГц) 8 -8.71;19F ЯМР (CDCl3, 500 МГц): δ -63.3,153.6.
Стадия В. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-фосфоноксиэтиловый эфир ((S)-Ib. n=1)
Используя (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-(ди-трет-бутоксифосфорилокси)этиловый эфир ((S)-VII, n=1) в виде исходного продукта, названное соединение получают аналогично как (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты 2-фосфоноксипропиловый эфир ((S)-Ib, n=2) с 83%.1H ЯМР (CDCl3, 500 МГц): δ 8.23 (с, 1Н), 7.73 (д, 1Н), 7.45 (д, 1Н), 7.30 (д, 1Н), 7.28 (д, 1Н), 6.75 (д, 1Н), 4.59 (м, 2Н), 4.33 (м, 2Н), 3.51 (с, 3Н). MS (м/е) 525.95 (М+-1);31P ЯМР (CDCl3, 500 МГц) δ 1.87;19F ЯМР (CDCl3, 500 МГц) δ - 63.25, 153.8. Вычислено для С19Н15ClF4NO8Р·1.159Н2О: С 41.60%, Н 3.18%, N 2.55%. Найдено: С 41.57%, Н 2.92%, N 2.51%.
ПРИМЕР 7
(S)-(2-Фосфонооксиэтил)карбаминовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир ((S)-Ic. n=1)
Стадия А. (S)-(2-Гидроксиэтилкарбаминовой кислоты 3-(5-хлор-2-метоксифенил-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир ((S)-IX. n=1)
В круглодонную колбу, объемом 25 мл, содержащую (S)-3-(5-хлор-2-метоксифенил)-3-фтор-1-гидроксиметил-6-трифторметил-1,3-дигидроиндол-2-он ((S)-IV) (0.397 г, 1.02 ммоль) добавляют 5 мл безводного дихлорметана, триэтиламин (0.204 г, 2.04 ммоль) и 4-нитрофенилхлорформиат (0.246 г, 1.22 ммоль). Реакционной смеси дают возможность перемешиваться в течение ночи при комнатной температуре, получая угольной кислоты 3-(5-хлор-2- метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир 4-нитрофенилового эфира (VIII), после чего добавляют 2-аминоэтанол (0.186 г, 3.05 ммоль) и реакционной смеси дают возможность перемешиваться в течение еще 24 часов. Растворитель удаляют в вакууме и сырой остаток очищают с помощью хроматография на колонке (силикагель; от 1:1 гексан : EtOAc до 4:1 EtOAc : гексан), получая 0.250 г (51%) названного соединения.1H ЯМР (CDCl3 400 МГц): δ 7.78 (с, 1Н), 7.51 (с, 1Н), 7.33 (д, 2Н, J=7 Гц), 7.22 (д, 1Н, J=7 Гц), 6.74 (д. 1Н, J=7 Гц), 5.88 (д, 1Н, J=12 Гц), 5.83 (д, 1Н, J=12 Гц), 3.75 (м, 2Н), 3.50 (с, 3Н), 3.39 (м, 2Н, J=7 Гц).
Стадия В. (S)-[2-(Ди-трет-бутоксифосфорилокси)этилкарбаминовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир ((S)-X. n=1)
В грушевидную колбу объемом 25 мл, содержащую (S)-(2-гидроксиэтил)карбаминовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-IX, n=1) (0.250 г, 0.526 ммоль) добавляют 3 мл безводного дихлорметана, 1H-тетразол (0.110 г 1.58 ммоль), 4-ДМФА (0.026 г, 0.21 ммоль) и ди-трет-бутилдиизопропилфосфорамидит (0.291 г, 1.05 ммоль). Реакционную смесь перемешивают в течение 2 часов при комнатной температуре, затем охлаждают до температуры 0°С и добавляют 30% водный раствор пероксида водорода (0.7 мл) и реакционную смесь перемешивают еще в течение часа. Органический слой отделяют, сушат (Na2SO4) и выпаривают в вакууме. Сырое масло очищают с помощью флеш-хроматографии (силикагель; от 1:1 EtOAc : гексан до 2:1 EtOAc : гексан), получая 0.280 г (80%) названного соединения.1H ЯМР (CDCl3 400 МГц): δ 7.78 (с, 1Н), 7.49 (с, 1Н), 7.32 (д, 2Н, J=7 Гц), 7.25 (д, 1Н, J=7 Гц), 6.74 (д, 1Н, J=7 Гц), 5.84 (дд, 2Н, 12 Гц), 4.05 (м, 2Н), 3.51 (с, 3Н), 3.48 (м, 2Н), 1.46 (с, 9Н), 1.45 (с, 9Н).
Стадия С. (S)-[2-Фосфонооксиэтил)карбаминовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир ((S)-Ic. n=1)
В грушевидную колбу объемом 25 мл, содержащую эфир (S)-[2-(ди-трет-бутокси-фосфорилокси)этил]карбаминовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2, 3-дигидроиндол-1-ил метиловый эфир ((S)-X, n=1) (0.280 г, 0.419 ммоль) добавляют 5 мл безводного дихлорметана и трифторуксусную кислоту (0.143 г, 1.26 ммоль). Реакционную смесь перемешивают в течение 3 часов при комнатной температуре, органический слой выпаривают в вакууме и сырую пену помещают в высокий вакуум в течение ночи. Полученный порошок белого цвета очищают с помощью хроматографии на реверсивной фазе (C18; 1:1 СН3CN:Н2O), получая названное соединение (0.134 г, 58%) в виде порошка белого цвета.1Н ЯМР (D2O=400 МГц): δ 7.88 (S1 1Н), 7.74 (с, 1Н), 7.50 (м, 2Н), 7.40 (д, 1Н, J=7 Гц), 7.00 (д, 1Н, J=7 Гц), 5.99 (д, 1Н, J=12 Гц), 5.77 (д, 1Н, J=12 Гц), 3.90 (м, 2Н), 3.48 (с, 3Н), 3.39 (м, 2Н); LRMS [M-HT554.91, [M+NH4]+574.0.
ПРИМЕР 8
(S)-Фосфоноксиуксусной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-Id. n=1)
Стадия А. (S)-Гидроксиуксусной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-XI)
В колбу объемом 10 мл, содержащую (8)-3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-он ((S)-V) (0.200 г, 0.489 ммоль) добавляют 3 мл безводного ацетонитрила, гликоливую кислоту (0.045 г, 0.588 ммоль) и карбонат цезия (0.079 г, 0.245 ммоль). Реакционную смесь перемешивают в течение 36 часов при комнатной температуре, затем органический слой разбавляют 5 мл диэтилового эфира и органический раствор отделяют декантированием от твердого хлорида цезия. Растворитель выпаривают в вакууме и полученную белого цвета пену очищают с помощью флеш-хроматографии (силикагель; 4:1 гексан : ацетон), получая названное соединение (0.126 г, 58%) в виде порошка белого цвета.1Н ЯМР (CDCl3 400 МГц): δ 7.79 (с, 1Н), 7.38-7.33 (м, 3Н), 7.26 (д, 1Н, J=7 Гц), 6.75 (д, 1Н, J=7 Гц), 5.99 (д, 1Н, J=12 Гц), 5.91 (д, 1Н, J=12 Гц), 4.26 (с, 2Н), 3.49 (с, 3Н).
Стадия В. (Ди-трет-бутоксифосфорилокси)уксусной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-XII)
В колбу, объемом 25 мл, содержащую (S)-гидроксиуксусную кислоту 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-XI) (0.126 г, 0.282 ммоль) добавляют 3 мл безводного дихлорметана, 1H-тетразол (0.059 г, 0.846 ммоль), 4-диметиламинопиридин (0.0138 г, 0.113 ммоль) и ди-трет-бутил-диизопропилфосфорамидит (0.156 г, 0.560 ммоль). Реакционной смеси дают возможность перемешиваться при комнатной температуре в течение 2 часов, затем реакционную смесь охлаждают до температуры 0°С и добавляют 30% пероксида водорода (0.8 мл) и реакционную смесь энергично перемешивают еще в течение 2 часов. Органический слой отделяют, промывают дистиллированной водой, сушат (Na2SO4) и выпаривают в вакууме. Сырую пену очищают с помощью флеш-хроматографии (силикагель; 4:1 гексан : EtOAc), получая названное соединение (0.144 г, 80%) в виде порошка белого цвета.1H ЯМР (CDCl3 400 МГц): δ 7.78 (с, 1Н), 7.36-7.33 (м, 3Н), 7.26 (д, 1Н, J=7 Гц), 6.75 (д, 1Н, J=7 Гц), 5.99 (д, 1Н, J=12 Гц), 5.89 (д, 1Н, J=12 Гц), 4.58 (д, 2Н, J=8.6 Гц), 3.52 (с, 3Н), 1.47 (с, 18Н).
Стадия С. (S)-Фосфонооксиуксусной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-Id. n=1)
В колбу объемом 25 мл, содержащую (S)-гидроксиуксусной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-XI) (0.144 г, 0.225 ммоль) добавляют 4 мл безводного дихлорметана и трифторуксусную кислоту (0.077 г, 0.675 ммоль). Реакционной смеси дают возможность перемешиваться при комнатной температуре в течение 3 часов. Дихлорметан выпаривают в вакууме и сырую пену очищают с помощью хроматографии на реверсивной фазе (C18; 1:1 СН3CN:H2О), получая названное соединение (0.075 г, 63%) в виде порошка белого цвета.1H ЯМР (CD3OD, 400 МГц): δ 7.73 (с, 1Н), 7.74 (с, 1Н), 7.62 (с, 1Н), 7.43-7.40 (м, 2Н), 7.33 (д, 1Н, J=7 Гц), 6.95 (д, 1Н, J=7 Гц), 6.05 (д, 1Н, J=11.2 Гц), 5.96 (д, 1Н, J=11.2 Гц), 4.62 (д, 2Н, J=9.6 Гц), 3.52 (с, 3Н);31P ЯМР (CD3OD, 162 МГц) δ=0.89;19F ЯМР (CD3OD, 376 МГц) δ-64.7, -160.9; LRMS [M-KT525.84, [М+NH4]+ 545.0.
ПРИМЕР 9
(S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты фосфоноксиметиловый эфир ((S)-Ie)
Стадия А. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты хлорметиловый эфир ((S)-XII)
В колбу объемом 10 мл, содержащую (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-1,3-дигидроиндол-2-он (S)-II (0.400 г, 1.1 ммоль) добавляют 6 мл безводного дихлорметана, триэтиламин (0.167 г, 1.67 ммоль) и хлорметилхлорформиат (0.171 г, 1.33 ммоль). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре, затем органический слой разбавляют 75 мл диэтилового эфира и твердый триэтиламингидрохлорид отфильтровывают через целит. Растворитель выпаривают в вакууме и названное соединение (0.414 г, 83%) используют в последующих реакциях без очистки.1H ЯМР (CDCl3, 400 МГц): δ 8.36 (с, 1Н), 7.79 (с, 1Н), 7.49 (д, 1Н, J=7 Гц), 7.36 (д, 1Н, J=7 Гц), 7.34 (д, 1Н, J=7 Гц), 6.77 (д, 1Н, J=7 Гц), 6.10 (д, 1Н, J=6.4 Гц), 5.96 (д, 1Н, J=6.4 Гц), 3.56 (с, 3Н).
Стадия В. 3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторэтил-2,3-дигидроиндол-1-карбоновой кислоты диметиловый эфир ((S)-XIV)
В колбу объемом 25 мл, содержащую (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты хлорметиловый эфир ((S)-XII) (0.633 г, 1.40 ммоль) добавляют 6 мл безводного ацетона и иодида натрия (0.314 г, 2.10 ммоль). Реакционную смесь нагревают до температуры кипения и перемешивают в течение 4 часов, затем реакцию охлаждают до комнатной температуры и растворитель выпаривают в вакууме. Сырое масло растворяют в дихлорметане и промывают 1% водный раствором тиосульфата натрия. Органический слой отделяют, сушат (Na2SO4) и выпаривают в вакууме, получая названное соединение (0.640 г, 84%), которое используют в последующих реакциях без очистки.1Н ЯМР (CDCl3, 400 МГц): δ 8.38 (с, 1Н), 7.78 (с, 1Н), 7.48 (д, 1Н, J=7 Гц), 7.36 (д, 1Н, J=7 Гц), 7.31 (д, 1Н, J=7 Гц), 6.77 (д, 1Н, J=7 Гц), 6.28 (д, 1Н, J=6.4 Гц), 6.18 (д, 1Н, J=6.4 Гц), 3.56 (с, 3Н).
Стадия С. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты ди-трет-бутоксифосфорилоксиметиловый эфир ((S)-XV)
В колбу объемом 25 мл, содержащую 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты иодметиловый эфир ((S)-XIV) (0.200 г, 0.368 ммоль) добавляют 5 мл безводного тетрагидрофурана и тетрабутиламмонийн ди-трет-бутилфосфат (0.199 г, 0.441 ммоль). Реакционную смесь нагревают до температуры кипения и дают возможность перемешиваться в течение часа. Реакционную смесь затем охлаждают до комнатной температуры и растворитель выпаривают в вакууме. Сырую пену очищают с помощью флеш-хроматографии (силикагель, 4:1 гексан: EtOAc), получая названное соединение (0.110 г, 48%) в виде прозрачного бесцветного масла.1H ЯМР (CDCl3, 400 МГц): δ 8.35 (с, 1Н), 7.78 (с, 1Н), 7.48 (д, 1Н, J=7 Гц), 7.34 (д, 1Н, J=7 Гц), 7.30 (д, 1Н, J=7 Гц), 6.76 (д, 1Н, J=7 Гц), 5.90 (д, 1Н, J=12.4 Гц), 3.53 (с, 3Н), 1.50 (с, 18Н).
Стадия D. (S)-3-(5-Хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-карбоновой кислоты фосфоноксиметиловый эфир ((S)-Ie)
В колбу, объемом 25 мл, содержащую (S)-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1 -карбоновой кислоты ди-трет-бутоксифосфорилоксиметиловый эфир ((S)-XV) (0.110 г, 0.176 ммоль) добавляют 2 мл безводного дихлорметана и трифторуксусную кислоту (0.060 г, 0.527 ммоль). Реакционной смеси дают возможность перемешиваться при комнатной температуре в течение 4 часов. Дихлорметан выпаривают в вакууме и сырую пену очищают с помощью хроматографии с обращенной фазой (C18, 2:1 СН3CN:Н2O), получая названное соединение (0.070 г, 78%) в виде порошка белого цвета.1H ЯМР (CDCl3, 400 МГц): δ 8.26 (с, 1Н), 7.68 (с, 1Н), 7.45 (д, 1Н, J=7 Гц), 7.28-7.24 (м, 2Н), 6.72 (д, 1Н, J=7 Гц), 5.90-5.81 (м, 2Н), 3.48 (с, 3Н);31P ЯМР (CDCl3, 162 МГц) δ -0.233;19F ЯМР (CDCl3, 376 МГц) δ -63.3, -153.8; LRMS [М-Н]- 511.4, [M+NH4]+530.9.
ПРИМЕР 10
(S)-3-Фосфоноксипропионовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-If. n=1)
Стадия А. (S)-3-Гидроксипропионовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-XVI. n=1)
В колбу объемом 10 мл, содержащую (S)-3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-он ((S)-V) (0.223 г, 0.546 ммоль) добавляют 3 мл безводного ацетонитрила и пропаноат цезия (0.182 г, 0.819 ммоль). Реакционную смесь перемешивают в течение 24 часов при комнатной температуре. Растворитель выпаривают в вакууме и полученную белого цвета пену очищают с помощью флеш-хроматографии (силикагель, 3:1 гексан: ацетон), получая названное соединение (0.115 г, 46%) в виде порошка белого цвета.1Н ЯМР (CDCl3, 400 МГц): δ 7.79 (с, 1Н), 7.37-7.32 (м, 3Н), 7.25 (д, 1Н, J=7 Гц), 6.75 (д, 1Н, J=7 Гц), 5.93 (д, 1Н, J=12 Гц), 5.85 (д, 1Н, J=12 Гц), 3.92 (т, 2Н, J=7 Гц), 3.51 (с, 3Н), 2.66 (т, 2Н, J=7 Гц).
Стадия В. (S)-3-(Ди-трет-бутоксифосфорилокси)пропионовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир (TS)-XVII. n=1)
В колбу объемом 25 мл, содержащую (S)-S-гидроксипропионовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-XVI, n=1) (0.115 г, 0.249 ммоль) добавляют 4 мл безводного дихлорметана, 1Н-тетразол (0.052 г, 0.747 ммоль), 4-диметиламинопиридин (0.012 г, 0.099 ммоль) и ди-трет-бутилдиизопропилфосфорамидит (0.138 г, 0.498 ммоль). Реакционной смеси дают возможность перемешиваться при комнатной температуре в течение 2 часов, затем реакционную смесь охлаждают до температуры 0°С и добавляют 30% пероксида водорода (0.8 мл) и реакционную смесь энергично перемешивают еще в течение 2 часов. Органический слой отделяют, промывают дистиллированной водой, сушат (Na2SO4) и выпаривают в вакууме. Сырую пену очищают с помощью флеш-хроматографии (силикагель, 4:1 гексан: EtOAc), получая названное соединение (0.144 г, 88%) в виде порошка белого цвета.1H ЯМР (CDCl3, 400 МГц): δ 7.78 (с, 1Н), 7.36-7.32 (м, 3Н), 7.26 (д, 1Н, J=7 Гц), 6.75 (д, 1Н, J=7 Гц), 5.90 (д, 1H, J=12 Гц), 5.84 (д, 1Н, J=12 Гц), 4.25 (кв, 2Н, J=7.6, 6.4 Гц), 3.51 (с, 3Н), 2.77 (т, 2Н, J=6.4 Гц), 1.46 (с, 18Н).
Стадия С. (S)-3-Фосфонооксипропионовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-If. n=1)
В колбу объемом 25 мл, содержащую (S)-3-(ди-трет-бутоксифосфорилокси)пропионовой кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир ((S)-XVII, n=1) (0.144 г, 0.22 ммоль) добавляют 2 мл безводного дихлорметана и трифторуксусную кислоту (0.075 г, 0.66 ммоль). Реакционной смеси дают возможность перемешиваться при комнатной температуре в течение 4 часов. Дихлорметан выпаривают в вакууме и сырую пену очищают с помощью хроматографии на реверсивной фазе (C18, 2:1 СН3CN:Н2O), получая названное соединение (0.070 г, 59%) в виде порошка белого цвета.1H ЯМР (CDCl3, 400 МГц): δ 7.70 (с, 1Н), 7.36 (с, 1Н), 7.32-7.27 (м, 3Н), 7.20 (д, 1Н, J=6 Гц), 6.73 (д, 1Н, J=8 Гц), 5.91 (д, 1Н, J=11.2 Гц), 5.73 (д, 1Н, J=11.2 Гц), 4.27 (м, 2Н), 3.44 (с, 3Н), 2.75 (м, 2Н);31P ЯМР (CDCl3, 162 МГц): δ 1.15;19F ЯМР (CDCl3, 376 МГц): δ -63.2, -159.4; LRMS [M-H]- 539.89, [M+NH4]+ 564.1.
ПРИМЕР 11
(S)-Пирофосфорной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, тринатривая соль ((S)-Ig2. M+=Na+)
Стадия А. (3S)-Пирофосфорной кислоты 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, трис(тетрабутиламмония) соль ((S)-Ig1. R=n-бутил)
Раствор (S)-3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-она ((S)-V) (0.45 г, 1.15 ммоль) и трис-(тетрабутиламмонийн)-гидропирофосфат (1.04 г, 1.15 ммоль) смешивают в CH3CN (25 мл) и перемешивают при комнатной температуре в течение около 45 минут. Растворитель удаляют при пониженном давлении и маслянистый остаток отфильтровывают через подушку С-18 из силикагеля с обращенной фазой, элюируя СН3CN/Н2О (1:1). Растворители удаляют в вакууме и остаток очищают с помощью флеш-хроматографии (SiO2), элюируя iPrOH/NH4OH (70:30), чтобы обеспечить получение названного соединения в виде стекловидного масла и использовать его непосредственно в последующей ионобменной реакции.
Стадия В. (S)-Пирофосфорной кислоты 3-(5-хлор-2-метоксифенил-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, тринатривая соль ((S)-Ia2. М+=Na+)
Восстановленное стекловидное масло, полученное выше, представляющее собой ((S)-пирофосфорную кислоту 3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, трис(тетрабутиламмонийная) соль ((S)-Ig2, R=н-бутил) растворяют в деионизированной Н2 О и наносят на колонку с ионообменной смолой (DOWEX 50WX8-200, Na+ форма). Фракции, содержащие названное соединение, лиофилизуют, получая пушистое твердое вещество белого цвета (0.34 г, 48% после двух стадий) с вкраплением длинных тонких игл.1H ЯМР (500 МГц, D2O): δ 3.58 (с, 3Н), 5.67 (дд, J=6.41, 10.68 Гц, 1Н), 5.78 (дд, 7=7.02, 10.68 Гц, 1Н), 7.01 (д, J=8.55 Гц, 1Н), 7.41 (м, 1Н), 7.51 (м, 2Н), 7.83 (с, 1Н), 789 (с, 1Н).31P ЯМР (200 МГц, D2O): -6.86 (д, J=22.05), -11.85 (д, J=22.05).19F ЯМР (470 МГц, D2 O): -63.0 (с), -161.7 (c). LRMS: 548 (М-3Н).
ПРИМЕР 12
(S)-Фосфорной кислоты-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир] метиловый эфир, мононатриевая соль ((S)-Ih2. М+=Na+. R1=СН3)
Стадия А. (S)-Фосфорной кислоты-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир]метиловый эфир, монотетра-бутиламмонийнная соль ((S)-Ih1. R=nBu. R1=CH3)
Раствор (S)-3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-она ((S)-V) (1.10 г, 2.81 ммоль) и гексагидрата метил-бис(тетрабутиламмонийн)фосфата (1.97 г, 2.81 ммоль, полученного в соответствии с Julia, М; Mestdagh, Н.; Rolando, G. Tettrahedron. 1986, 42, 3841-3849) смешивают в СН3CN (60 мл) и перемешивают при комнатной температуре в течение приблизительно 30 минут. Растворитель удаляют при пониженном давлении и маслянистый остаток очищают с помощью флеш-хроматографии (SiO2), элюируя iPrOH/NH4OH (95:05), что дает названное соединение в виде стекловидного масла, которое применяют непосредственно в последующей ионообменной реакции.
Стадия В. (S)-Фосфорной кислоты-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир]метиловый эфир, мононатриевая соль ((S)-Ih2. M+=Na+. R1 =СН3)
Соль аммония, описанную выше, ((S)-фосфорной кислоты-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового эфира метилового эфира монотетрабутиламмонийную соль ((S)-Ih1, R=nBu, R1=СН3)) растворяют в деионизированной Н2O и наносят на колонку с ионообменной смолой (DOWEX 50WX8-200, Na+ форма). Фракции, содержащие названное соединение, лиофилизуют, получая твердое вещество белого цвета (1.02 г, 72% после двух стадий). Найдено, протоновый ЯМР зависит от концентрации.1Н ЯМР (500 МГц, D2O, <<10 мг/мл): δ 3.56 (с, 3Н), 3.57 (д, J=10.99 Гц, 3Н), 5.51 (дд, J=7.32, 10.68 Гц, 1Н), 5.58 (дд, J=7.32, 10.68 Гц, 1Н), 7.02 (д, J=8.85 Гц, 1Н), 7.44 (м, 1Н), 7.51 (дд, J=2.44, 8.85 Гц, 1Н), 7.54 (д, J=7.33 Гц, 1Н), 7.72 (с, 1Н) 7.90 (с, 1Н).1Н ЯМР (500 МГц, D2O, >10 мг/мл): δ 3.39 (с, 3Н), 3.46 (д, J=10.38 Гц, 3Н), 5.51 (дд, J=7.02, 10.38 Гц, 1Н), 5.58 (дд, J=7.02, 10.38 Гц, 1Н), 6.75 (д, J=8.55 Гц, 1Н), 6.92 (уширенный, 1Н), 7.05 (уширенный, 1Н), 7.24 (д, J=1.83, 8.55 Гц, 1Н), 7.60 (м, 2Н).31P ЯМР (200 МГц, D2O): 0.95 (мультиплет).19F ЯМР (470 МГц, D2O): -63.3 (с). -160.7 (с). LRMS: 482 (M-H).
ПРИМЕР 13
(S)-Фосфорной кислоты-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир] этиловый эфир, мононатриевая соль ((S)-Ih2. М+=Na+. R1=СН2СН3)
Стадия А. (S)-Фосфорной кислоты-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир]этиловый эфир, монотетрабутиламмонийная соль ((S)-Ih1. R=nBu. R1=CH2CH3)
Раствор (S)-3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1, 3-дигидроиндол-2-она ((S)-V) (1.0 г, 2.57 ммоль) и гексагидрата этил-бис(тетрабутиламмонийн) гексафосфата (1.84 г, 2.57 ммоль (полученного в соответствии с Julia, М; Mestdagh, Н.; Rolando, G. Tettrahedron, 1986, 42, 3841-3849) смешивают в СН3CN (60 мл) и перемешивают при комнатной температуре в течение приблизительно 30 минут.
Растворитель удаляют при пониженном давлении и маслянистый остаток очищают с помощью флеш-хроматографии (SiO2), элюируя IPrOH/NH4OH (98:02), что дает названное соединение в виде стекловидного масла, которое применяют непосредственно в последующей ионообменной реакции.
Стадия В. (S)-Фосфорной кислоты-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир]этиловый эфир, мононатриевая соль ((S)-Ih2. М+=Na+. R1=СН2СН3)
Соль аммония, описанную выше, ((S)-фосфорной кислоты-3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир этиловый эфир монотетрабутиламмонийную соль ((S)-Ihl, R=nBu, R1=СН2 СН3)) растворяют в деионизированной Н2О и применяют к колонке с ионообменной смолой (DOWEX 50WX8-200, Na+ форма). Фракции, содержащие названное соединение, лиофилизуют, получая твердое вещество белого цвета (0.89 г, 65%).1H ЯМР (500 МГц, D2O): δ 1.20 (т, J=7.02 Гц, 3Н), 3.56 (с, 3Н), 3.93 (м, 2Н), 5.65 (м, 2Н), 7.02 (д, J=8.85 Гц, 1Н), 7.40 (м, 1Н), 7.51 (bd, J=7.33 Гц, 2Н), 7.72 (с, 1Н) 7.89 (с, 1Н).31P ЯМР (200 МГц, D2O): 0.03 (мультиплет).19F ЯМР (470 МГц, D2O): -63.1 (с), -161.1 (с). LRMS: 496 (M-H).
ПРИМЕРЫ 14-24. Общая методика получения солей
Смолу Dowex-50wx8-100 промывают Н2О, МеОН, снова Н2О, как описано у производителя (Aldrich), подщелачивают ее до значения рН>10 с помощью раствора гидроксида натрия (или тетраметиламмония) (или обрабатывают аминокислотой или органическими аминами), а затем промывают Н2O. Смолу, которая готова для применения, вначале разделяют на три равные порции. В раствор (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2, 3-дигидроиндол-1-ил метилового] эфира, монотетрабутиламмонийной соли в Н2О добавляют одну порцию смолы. Смесь перемешивают в течение 10 минут, отфильтровывают и промывают Н2О. В объединенный фильтрат добавляют еще порцию смолы. Смесь перемешивают в течение 10 минут, отфильтровывают и промывают Н2O. В полученный фильтрат добавляют последнюю порцию смолы. Смесь перемешивают в течение 10 минут, отфильтровывают и промывают Н2О. Растворители объединенных фильтратов упаривают. Остаток растворяют в ацетонитриле и отфильтровывают, чтобы удалить следы нерастворимого продукта. Ацетонитрил упаривают. Остаток повторно растворяют в СН2Cl2. Затем добавляют гексан. Упаривание растворителей обеспечивает получение желаемого продукта в виде порошка ярко-белого цвета.
ПРИМЕР 14
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, калийная соль ((S)-Ia2)
Следуя общей методике, описанной выше, используют 40 г смолы Dowex-50wx8-100, чтобы получить 0.27 г калийной соли (порошок белого цвета) (95% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового] эфира, монотетрабутиламмонийной соли (0.40 г, 0.56 ммоль). MS м/е: 468.0 (М-Н-). LC/MS м/е: 469.9 (МН+), 1.52 мин >95% чистота. Вычислено для C17H12ClF4NO6PN·1.10K·0.45H2O: С 39.27; Н 2.50; N 2.69, K 8.31. Найдено: С 39.41; Н 2.62; N 2.51; K 8.43.1H ЯМР (D2О): δ 3.56 (с, 3Н), 5.55 (м, 1Н), 5.61 (м, 1Н), 7.00 (д, J=8.5 Гц, 1Н), 7.40 (д, J=7.0 Гц, 1Н), 7.49 (м, 2Н), 7.77 (с, 1Н), 7.88 (с, 1Н).19F ЯМР (D2О): δ -63.1, -161.7.31P ЯМР (D2O): δ 1.27.
ПРИМЕР 15
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир, тетраметиламмонийная соль ((S)-Ia2)
Следуя общей методике, описанной выше, используют 20 г смолы Dowex-50wx8-100, чтобы получить 0.153 г тетраметиламмонийной соли (порошок белого цвета) (93% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового]эфира, монотетрабутиламмонийной соли (0.20 г, 0.28 ммоль). MS м/е: 468.1 (М-Н-). LC/MS м/е: 470.0 (МН+), 1.35 мин >95% чистота. Вычислено для С17Н12ClF4NO6PN·1.6C4H12N·0.8H2О: С 46.7; Н 5.50; N 6.05. Найдено: С 47.03; Н 5.84; N 5.74.1Н ЯМР (D2O): δ 3.14 (с, 19.2Н), 3.52 (с, 3Н), 5.50 (м, 1Н), 5.53 (м, 1Н), 6.96 (д, J=9.0 Гц, 1Н), 7.40 (м, 1Н), 7.45 (м, 2Н), 7.75 (с, 1Н), 7.84 (с, 1Н). IR (KBr, см-1): 3429, 1752, 1319, 1129.
ПРИМЕР 16
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир, глициновая соль ((S)-Ia2)
Следуя общей методике, описанной выше, используют 20 г смолы Dowex-50wx8-100 и глицина (7.5 г), чтобы получить 0.156 г глициновой соли (порошок белого цвета) (94% выход) из (S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового]эфира, монотетрабутиламмонийной соли (0.20 г, 0.28 ммоль). MS м/е: 468.0 (М-H-). Вычислено для C17H12ClF4NO6PN·1.42C2H6NO2·0.80H2O: С 40.31; Н 3.77; N 5.73. Найдено: С 40.57; Н 3.59; N 5.41.1Н ЯМР (D2O): δ 3.58 (с, 3Н), 3.65 (с, 2.8 Н), 5.63 (м, 1Н), 5.71 (м, 1Н), 7.03 (д, J=9.0 Гц, 1Н), 7.42 (д, J=7.0 Гц, 1Н), 7.52 (м, 2Н), 7.75 (с, 1Н), 7.92 (с, 1Н). IR (KBr, см-1): 3431, 1759, 1319, 1131, 1023.
ПРИМЕР 17
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир], пролиновая соль ((S)-Ia2)
Следуя общей методике, описанной выше, используют 20 г смолы Dowex-50wx8-100 и проллина (11.3 г), чтобы получить 0.160 г пролиновой соли (порошок белого цвета) (97% выход) из (З)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового] эфира, монотетрабутиламмонийной соли (0.20 г, 0.28 ммоль). MS м/е: 468.1 (М-Н-). LC/MS м/е: 470.0 (МН+), 1.35 мин >95% чистота. Вычислено для C17H12ClF4NO6PN·1.0C5H10NO2: C 45.10; Н 3.95; N 4.78. Найдено: С 45.19; Н 3.96; N 4.56.1H ЯМР (D2O): δ 1.95 (м, 2Н), 2.03 (м, 1Н), 2,35 (м, 1Н), 3.28 (м, 1Н), 3.35 (м, 1Н), 3.58 (с, 3Н), 4.11 (м, 1Н), 5.55 (м, 1Н), 5.61 (м, 1Н), 6.94 (д, J=9.0 Гц, 1Н), 7.34 (дд, J=2.0, 8.0 Гц, 1Н), 7.44 (м, 2Н), 7.66 (с, 1Н), 7.82 (с, 1Н). IR (KBr, см-1): 3431, 1757, 1319, 1131, 1026.
ПРИМЕР 18
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2, 3-дигидроиндол-1-ил метиловый эфир], магнивая соль ((S)-Ia2)
Магнивую соль получают простой заменой Na+/Mg++. Продукт минимально растворимый в воде, но более растворимый в органических растворителях. К раствору Mg2SO4 (0.130 г, 1.1 ммоль) в Н2O (5 мл) добавляют раствор (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового] эфира, мононатриевой соли (натривая соль) (0.050 г, 0.070 ммоль) в Н2O (2 мл). Смесь перемешивают при комнатной температуре в течение 10 минут. Осадок отфильтровывают, промывают H2O и затем растворяют в EtOAc. Остаток из упаренного растворителя повторно растворяют в СН2Cl2 и гексане. Упаривание растворителя обеспечивает получение желаемого продукта в виде порошка белого цвета (0.035 г, 72% выход). MS м/е: 468.0 (М-Н-). LC/MS м/е: 469.9 (МН+), 1.52 мин >95% чистота.
Вычислено для C17H12ClF4NO6PN·0.49Mg·1.0H2O: С 40.95; Н 2.83; N 2.81; Mg 2,39. Найдено: С 40.87; Н 3.04; N 2.44; Mg 2.42.1H ЯМР (D2O): δ 3.54 (с, 3Н), 5.50 (д, J=5.0 Гц, 1Н), 5.52 (д, J=5.0 Гц, 1Н), 6.99 (д, J=7.5 Гц, 1Н), 7.34 (д, J=7.0 Гц, 1Н), 7.47 (м, 2Н), 7.78 (с, 1Н), 7.86 (с, 1Н). IR (KBr, см-1): 3433, 1757, 1319, 1132, 1025.
ПРИМЕР 19
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, соль аммония ((S)-Ia2)
Следуя общей методике, описанной выше, используют 50 г смолы Dowex-50wx8-100, чтобы получить 0.6 г соли аммония (порошок белого цвета) (87.8% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2, 3-дигидроиндол-1-ил метилового] эфира, монотетрабутиламмонийной соли (1.0 г, 1.4 ммоль). MS м/е: 468.1 (М-Н)". LC/MS м/е: 486.8 (МН+), 4.54 мин >95% чистота. Вычислено для C17H12ClF4NO6PN·1.0NH4·0.35H2O: С 41.43; Н 3.39; N 5.69. Найдено: С 41.41; Н 3.32; N 5.71.1Н ЯМР (CD3 CN): 3.47 (с, 3Н), 5.61 (д, J=6.5 Гц, 2Н), 5.95 (б, 2Н), 6.83 (д, J=8.8 Гц, 1Н), 7.31 (д, J=7.8 Гц, 1Н), 7.36 (м, 2Н), 7.64 (с, 1Н), 7.72 (с, 1Н), 8.1 (б, 2Н).
ПРИМЕР 20
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, литиевая соль ((S)Ia2)
Следуя общей методике, описанной выше, используют 50 г смолы Dowex-50wx8-100, чтобы получить 0.2 г литиевой соли (порошок белого цвета) (78.8% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового]эфира, монотетрабутиламмонийной соли (0.3 г, 0.42 ммоль). MS м/е: 468.1 (М-Н)-. LC/MS м/е: 486.8 для [(M-)+NH4]+, 4.48 мин >95% чистота. Вычислено для C17H12ClF4NO6PN·1.1Li·1.2H2O: С 40.93; Н 2.93; N 2.81; Li 1.40. Найдено: С 41.01; Н 2.94; N 2.81; Li 1.54.1H ЯМР (D2O): 3.52 (с, 3Н), 5.55 (м, 1Н), 5.62 (м, 1Н), 6.96 (д, J=9.0 Гц, 1Н), 7.30 (д, J=7.5 Гц, 1Н), 7.44 (м, 2Н), 7.73 (с, 1Н), 7.82 (с, 1Н).
ПРИМЕР 21
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, метанаммонийнная соль ((S)-Ia2)
Следуя общей методике, описанной выше, используют смолу (15 г) Dowex-50wx8-100, чтобы получить 0.15 г метанаммонийной соли (порошок белого цвета) (71.1% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового]эфира, монотетрабутиламмонийной соли (0.3 г, 0.42 ммоль). MS м/е: 468.11 (М-Н)-. LC/MS м/е: 468.11 (М-Н)-, 4.55 мин >95% чистота. Вычислено для С17Н12ClF4NO6 PN·1.0СН6N·0.29Н2О: С 42.74; Н 3.68; N 5.54. Найдено: С 42, 38; Н 3.65; N 5.49.1H ЯМР (D2O): 2.57 (с, 3Н), 3.47 (с, 3Н), 5.53 (м, 1Н), 5.56 (м, 1Н), 6.88 (д, J=8.8 Гц, 1Н), 7.17 (д, J=6.4 Гц, 1Н), 7.31 (д, J=7.2 Гц, 1Н), 7.37 (д, J=8.0 Гц, 1Н), 7.67 (с, 1Н), 7.74 (с, 1Н).
ПРИМЕР 22
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, циклогексиламмонийная соль ((S)-Ia2)
Следуя общей методике, описанной выше, используют смолу (25 г) Dowex-50wx8-100, чтобы получить 0.36 г циклогексиламмонийной соли (порошок белого цвета) (91.6% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового] эфира, монотетрабутиламмонийной соли (0.5 г, 0.70 ммоль). MS м/е: 487.1 для [(M-)+NH4]+. LC/MS м/е: 467.99 (М-Н)-, 3.82 мин >95%. Вычислено для С17Н12ClF4NO6PN·1.0С6Н14N: С 48.56; Н 4.60; N 4.92. Найдено: С 48.80; Н 4.80; N 4.90.1Н ЯМР (D2O): 1.14 (м, 1Н), 1.31 (м, 4Н), 1.62 (м, 1Н), 1.77 (м, 2Н), 1.96 (м, 2Н), 3.12 (м, 1Н), 3.54 (с, 3Н), 5.52 (м, 1Н), 5.57 (м, 1Н), 6.95 (д, J=9.0 Гц, 1Н), 7.36 (м, 1Н), 7.47 (м, 2Н), 7.75 (с, 1Н), 7.85 (с, 1Н).
ПРИМЕР 23
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метиловый эфир, соль пиперидиния ((S)-Ia2)
Следуя общей методике, описанной выше, используют смолу (20 г) Dowex-50wx8-100 смолы, чтобы получить 0.25 г соли пиперидиния (порошок белого цвета) (78.2% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового]эфира, монотетрабутиламмонийной соли (0.4 г, 0.56 ммоль). MS м/е: 487.1 для [(M-)+NH4 ]+. LC/MS м/е: 467.99 (М-Н)-, 3.65 мин >95% чистота. Вычислено для C17H12ClF4NO6PN·1.2C5H12 N·0.55CH3Cl·0.45H2O: С 43.16; Н 3.93; N 4.46. Найдено: С 42.99; Н 4.18; N 4.53.1H ЯМР (D2O): 1.64 (м, 2Н), 1.76 (м, 4Н), 3.13 (м, 2Н), 3.53 (с, 3Н), 5.55 (м, 1Н), 5.61 (м, 1Н), 6.98 (д, J=9.0 Гц, 1Н), 7.35 (д, J=7.5 Гц, 1Н), 7.47 (д, J=7.5 Гц, 2Н), 7.73 (с, 1Н), 7.85 (с, 1Н).
ПРИМЕР 24
(S)-Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2.3-дигидроиндол-1-ил метиловый эфир, диметиламмонийная соль ((S)-Ia2)
Следуя общей методике, описанной выше, используют смолу (15 г) Dowex-50wx8-100, чтобы получить 0.20 г диметиламмонийной соли (порошок белого цвета) (78.9% выход) из (S)-фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-ил метилового] эфира, монотетрабутиламмонийной соли (0.3 г, 0.42 ммоль). MS м/е: 487.1 для [(М-)+NH4]+. LC/MS м/е: 468.01 (М-Н)-, 3.66 мин >95% чистота. Вычислено для С17Н12Cl F4NO6PN·1.2С2Н8 N·0.52СН3Cl·0.26Н2O: С 40.58; Н 3.67; N 5.23. Найдено: С 40.30; Н 3.75; N 5.50.1H ЯМР (D2O): 2.69 (с, 6Н), 3.49 (с, 3Н), 5.52 (м, 1Н), 5.60 (м, 1Н), 6.91 (д, J=8.5 Гц, 1Н), 7.25 (м, 1Н), 7.36 (м, 2Н), 7.70 (с, 1Н), 7.76 (с, 1Н).
ПРИМЕР 25
Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый] эфир, бис[трис(гидроксиметил)аммонийметановая]соль
Стадия А. 3-(5-Хлор-2-метоксифенил)-3-фтор-1-метилсульфанилметил-6-трифторметил-1,3-дигидроиндол-2-он
В 3-горлую круглодонную колбу объемом 1 л, оборудованную делительной воронкой, добавляют 3-(5-хлор-2-метоксифенил)-3-фтор-6-трифторметил-1,3-дигидроиндол-2-он (25 г, 69.8 ммоль) и безводный ТГФ (150 мл). Раствор охлаждают до температуры 0°С и добавляют бис(триметил)амид калия (0.5М в толуоле, 153.6 мл, 76.8 ммоль) в течение около 25 минут для того, чтобы поддержать температуру реакции между 0 и 5°С. Реакционная смесь сразу меняет цвет с черного на оранжевый. Раствору дают возможность нагреться до комнатной температуры, после чего добавляют хлорметилметилсульфид (6.39 мл, 76.8 ммоль). Темно-коричневого цвета реакционной смеси затем дают возможность перемешиваться при температуре кипения. После 3 часов реакционная смесь содержит 84% (АР) желаемого продукта. Реакционную смесь поддерживают при кипении в течение ночи. Затем реакционную смесь оставляют стоять в течение этого периода (82% АР продукта после ночи перемешивания). Реакционной смеси затем дают возможность остыть и медленно добавляют 1 н. HCl (100 мл) для того, чтобы поддержать температуру реакции ниже 30°С. После перемешивания в течение 5 минут добавляют гептан (200 мл) и слои отделяют. Органический слой затем промывают насыщенным раствором NaCl (200 мл), сушат над безводным сульфатом натрия и концентрируют, что дает на выходе сырой продукт (29.3 г). Сырой продукт затем перекристаллизовывают из гептана : ТГФ (15:1), чтобы обеспечить получение 3-(5-хлор-2-метоксифенил)-3-фтор-1-метилсульфанилметил-6-трифторметил-1,3-дигидроиндол-2-она в виде порошка светло-желтого цвета (19.5 г, 67%).1H ЯМР (CDCl3, 400 МГц): δ 7.78 (дд, 1Н, J=2.7, 0.9 Гц), 7.32 (м, 2Н), 7.21 (м, 2Н), 6.73 (дд, 1Н, J=8.9, 1.2 Гц), 4.95 (д, 1Н, J=14.6 Гц), 4.79 (д, 1Н, J=14.6 Гц), 3.48 (с, 3Н), 2.23 (с, 3Н).
Стадия В. 3-(5-Хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1.3-дигидроиндол-2-он
В круглодонную колбу, оборудованную мешалкой и патрубком для ввода азота, загружают 82 г (0.195 моль) 3-(5-хлор-2-метоксифенил)-3-фтор-1-метилсульфанилметил-6-трифторметил-1,3-дигидроиндол-2-она со стадии А и дихлорметан (500 мл). Раствор охлаждают до температуры 0°С и в раствор подают газообразный хлор при температуре, при которой скорость потока составляет около 50 мл/мин, поддерживая температуру реакционной смеси в интервале между 0-3°С. Хлор пробулькивают в течение порядка 90 минут (около 1 экв. хлора). Ход реакции отслеживают с помощью линейной комбинационной спектроскопии, HPLC и через массовый расходомер газа. После завершения реакции смесь нагревают до комнатной температуры и используют на стадии С.1H ЯМР (300 МГц, ДМСО-d6): δ 7.88 (с, 1Н), 7.69 (д, J=2,3 Гц, 1Н), 7.49 (дд, J=2.6, 8.8 Гц,1H), 7.41 (с, 1Н), 7.04 ((дд, J=1.2, 8.9 Гц, 1Н), 5.40 (д, 3-5.9, 2Н), 5.25 (уширенный, 12Н), 3.52 (с, 3Н), 3.35 (с, 12Н), 2.15 (с, 4.4Н, пик ацетона).
Альтернативный способ. Сульфонилхлорид (135 мг, 80.2 мкл, 1 ммоль) добавляют при комнатной температуре к раствору в дихлорметане (10 мл) 3-(5-хлор-2-метоксифенил)-3-фтор-1-метилсульфанилметил-6-трифторметил-1,3-дигидроиндол-2-она (420 мг, 1 ммоль) в круглодонной колбе, оборудованной патрубком для ввода азота и магнитной мешалкой. После перемешивания реакционной смеси при комнатной температуре в течение 20 минут сырой раствор концентрируют в вакууме, что дает 3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-он (425 мг, 100% выход сырого продукта) в виде раствора светло-желтого цвета.
Стадия С. Фосфорной кислоты моно-[3-(5-хлор-2-метоксифенил)-3-фтор-2-]оксо-6-трифторметил-2,3-дигидроиндол-1-илметиловый эфир, бис-[трис(гидроксиметил)аммонийметановая]соль
Раствор со стадии В, содержащий 3-(5-хлор-2-метоксифенил)-1-хлорметил-3-фтор-6-трифторметил-1,3-дигидроиндол-2-он, добавляют к перемешиваемому раствору в дихлорметане (1500 мл) тетра-н-бутиламмония дигидрофосфату (801 г) при температуре 40°С в течение около 90 минут. Полученный прозрачный раствор перемешивают при температуре 40°С в течение 10 минут, чтобы завершить реакцию.
Порцию указанного выше раствора (160 мл, 10.1 ммоль, 0.063 М, как определяют с помощью HPLC калибрирования) загружают в круглодонную колбу объемом 250 мл, оснащенную в верхней части обратным холодильником. Раствор нагревают на масляной бане (70°С) и затем удаляют дихлорметан путем отгонки, пока внутренняя температура не достигнет значения 60°С (температура дистиллята составляет 34-40°С). После охлаждения до комнатной температуры добавляют HCl (1 н. 140 мл, 140 ммоль) и трет-бутилметиливой эфир (80 мл) и полученную смесь перемешивают в течение 10 минут, пока все твердые вещества не растворятся. Фазы отделяют и водный слой снова экстрагируют трет-бутилметиловым эфиром (40 мл). Объединенный слой трет-бутилметилового эфира промывают HCl (1 н., 2×140 мл) и водой (5×100 мл, рН водяных промывок 0, 2, 4, 5, 6 соответственно). Слой трет-бутилметилового эфира затем экстрагируют раствором трис(гидроксиметил)аминометана (2.58 г, 21.3 ммоль) в воде (20 мл). Добавляют ацетон к водному слою, обогащенному продуктом (70 мл), пока раствор не станет мутным, и затем его перемешивают при комнатной температуре в течение 1.75 часов. Вновь добавляют ацетон (20 мл) и перемешивают при комнатной температуре в течение 16.5 часов, затем еще раз добавляют ацетон (100 мл) в течение 30 минут. Полученную суспензию охлаждают до температуры 0°С и перемешивают в течение 2.5 часов. Твердое вещество собирают путем фильтрования и промывают ацетоном (2×20 мл). Мокрую лепешку сушат в вакууме (25 мм рт.ст.) при комнатной температуре в течение 21 часов, что дает кристаллический продукт, бис[трис(гидроксиметил)аммонийметановую] соль (5.08 г) в виде сольвата с ацетоном (0.7 экв.).1H ЯМР (300 МГц, ДМСО-d6): δ 7.88 (с, 1Н), 7.69 (д, J=2, 3 Гц, 1Н), 7.49 (дд, J=2.6, 8.8 Гц, 1Н), 7.41 (с, 1Н), 7.04 (дд, J=1.2, 8.9 Гц, 1Н), 5.40 (д, J=5.9 Гц, 2H), 5.25 (уширенный, 12Н), 3.52 (с, 3Н), 3.35 (с, 12Н), 2.15 (с, 4.4Н, пик ацетона).31Р ЯМР (300 МГц, ДМСО-d6): S 0.11.19F ЯМР (500 МГц, ДМСО-d6): δ -67.9, -166.0.
Реферат
Изобретение относится к группе новых соединений формулы (I)


Изобретение также относится к способу лечения заболеваний, чувствительных к раскрытию активируемых кальцием калиевых каналов. Технический результат - получение новых биологически активных соединений и способ лечения заболеваний, чувствительных к раскрытию активируемых кальцием калиевых каналов большой проводимости у млекопитающего. 2 н. и 7 з.п. ф-лы, 1 табл.
Формула
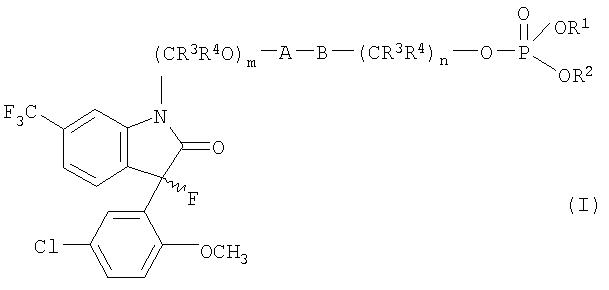

Документы, цитированные в отчёте о поиске
3-замещенные оксиндольные производные в качестве модуляторов калийных каналов, фармацевтическая композиция, способ лечения
Комментарии