Водорастворимое соединение бензоазепина и содержащая его фармацевтическая композиция - RU2418804C2
Код документа: RU2418804C2
Чертежи


Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к новому соединению бензоазепина и содержащей его фармацевтической композиции.
Уровень техники
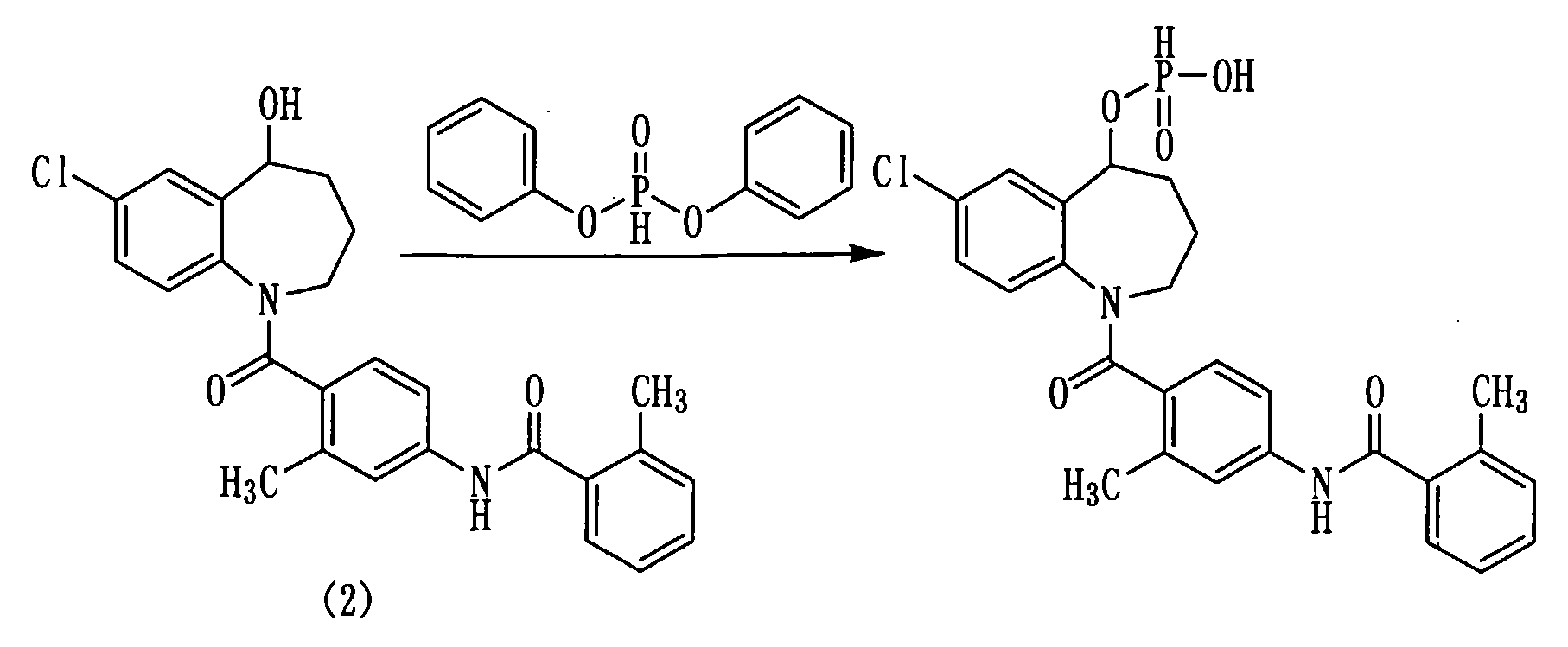
Толваптан, представленный следующей формулой (2), является известным соединением, и описан, например, в описании патента США №5258510 (пример 1199).

Известно, что толваптан применяется в качестве антагониста вазопрессина, обладающего акваретической активностью (Circulation, 107, pp.2690-2696 (2003)). Однако вследствие его низкой растворимости в воде проблема, связанная с толваптаном, заключается в том, что он плохо всасывается в кишечном канале, его дозировки и способы введения ограничены, и так далее. Несмотря на то, что предпринимались попытки решить данные проблемы, так что, например, толваптан можно вводить в виде препарата с композицией на основе аморфного твердого вещества (нерассмотренная японская патентная публикация №1999-21241), при использовании толваптана его дозировка и способ введения все еще остаются ограниченными.
Описание изобретения
Задачей настоящего изобретения является создание нового соединения бензоазепина для улучшения растворимости толваптана в воде.
Для решения упомянутой выше проблемы авторами настоящего изобретения проведено всестороннее исследование, в результате которого найдено, что когда толваптан находится в виде соединения эфира фосфорной кислоты, его растворимость в воде можно существенно повысить.
Настоящее изобретение основано на данном открытии.
Более конкретно, в настоящем изобретении предложены следующие соединения бензоазепина и содержащие их композиции, описанные в приведенных далее пунктах с 1 по 13.
Пункт 1. Соединение бензоазепина, представленное общей формулой (1)

или его соль,
в котором R представляет собой атом водорода, гидрокси-группу, необязательно защищенную защитной группой, меркапто-группу, необязательно защищенную защитной группой, или амино-группу, необязательно защищенную одной или двумя защитными группами, R1 представляет собой атом водорода, или гидрокси-защитную группу, а Х представляет собой атом кислорода, или атом серы.
Пункт 2. Соединение бензоазепина по пункту 1, или его соль, в котором Х представляет собой атом кислорода.
Пункт 3. Соединение бензоазепина по пункту 1, или 2, или его соль, в котором R представляет собой гидрокси-группу, необязательно защищенную защитной группой.
Пункт 4. Соединение бензоазепина по пункту 1, или 2, или его соль, в котором R представляет собой атом водорода, меркапто-группу, необязательно защищенную защитной группой, или амино-группу, необязательно защищенную одной или двумя защитными группами.
Пункт 5. Соединение бензоазепина по любому из пунктов 1, 2, 3 и 4, или его соль, в котором R1 представляет собой гидрокси-защитную группу.
Пункт 6. Соединение бензоазепина по любому из пунктов 1, 2, 3 и 4, или его соль, в котором R1 представляет собой атом водорода.
Пункт 7. Соединение бензоазепина по пункту 1, или его соль, в котором Х представляет собой атом серы.
Пункт 8. Соединение бензоазепина по пункту 1, или его соль, в котором Х представляет собой атом кислорода, R представляет собой гидрокси-группу, а R1 представляет собой атом водорода.
Пункт 9. Фармацевтическая композиция, содержащая соединение бензоазепина по пункту 1, или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым разбавителем и/или носителем.
Пункт 10. Фармацевтическая композиция по пункту 9 для использования в качестве сосудорасширяющего, гипотензивного, акваретического агента, PKD, или ингибитора агрегации тромбоцитов.
Пункт 11. Композиция в виде водного раствора, содержащего соединение бензоазепина по пункту 1, или его фармацевтически приемлемую соль.
Пункт 12. Композиция в виде водного раствора по пункту 11, содержащего соединение бензоазепина по пункту 1, или его фармацевтически приемлемую соль, вместе с буфером, агентом для придания изотоничности и растворителем для инъекции, и которая имеет форму препарата для инъекций.
Пункт 13. Композиция в виде водного раствора по пункту 12, дополнительно содержащего регулятор рН.
Использованный в настоящем описании термин «низший» означает С1-6, если не обозначено иначе.
Примеры защитных групп для «гидрокси-группы, необязательно защищенной защитной группой», «меркапто-группы, необязательно защищенной защитной группой» и «гидрокси-защитной группы» включают в себя низшие алкильные группы, фенил(низшие)алкильные группы, циано низшие алкильные группы и низшие алкилоксикарбонил низшие алкильные группы.
Примеры защитных групп для «амино-группы, необязательно защищенной одной или двумя защитными группами», включают в себя низшие алкильные группы, необязательно содержащие гидрокси-группу(ы).
Примеры низших алкильных групп и низших алкильных групп в фенил(низших)алкильных группах, циано низших алкильных группах и низших алкилоксикарбонил низших алкильных группах и низших алкильных групп, необязательно содержащих гидрокси-группу(ы), включают в себя С1-6 линейные или разветвленные алкильные группы, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, третбутил, втор-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, 3-метилпентил и так далее.
Предпочтительными фенил(низшими)алкильными группами являются, например, бензил, фенетил, 3-фенилпропил, тритил и так далее.
Предпочтительными циано низшими алкильными группами являются С1-6 линейные или разветвленные алкильные группы, содержащие от одной до трех циано-групп, например, цианометил, 2-цианоэтил, 1-, 2-, или 3-циано-н-пропил, 1-, 2-, или 3-цианоизопропил, 1-, 2-, 3- или 4-циано-н-бутил, 1-, 2-, 3- или 4-цианоизобутил, 1-, 2-, 3- или 4-цианотретбутил, 1-, 2-, 3- или 4-циано-втор-бутил, 1-, 2-, 3-, 4- или 5-циано-н-пентил, 1-, 2-, 3-, 4- или 5-цианоизопентил, 1-, 2-, 3-, 4- или 5-цианонеопентил, 1-, 2-, 3-, 4-, 5- или 6-циано-н-гексил, 1-, 2-, 3-, 4-, 5- или 6-циано-изогексил, 1-, 2-, 3-, 4-, 5- или 6-циано-3-метилпентил, и так далее.
Предпочтительными низшими алкилоксикарбонил низшими алкильными группами являются алкилоксикарбонилалкильные группы, в которых алкилокси-фрагмент представляет собой С1-6 линейную, или разветвленную алкилокси-группу, а алкильный фрагмент представляет собой С1-6 линейную, или разветвленную алкильную группу, например, метоксикарбонилметил, этоксикарбонилметил, н-пропоксикарбонилметил, изопропоксикарбонилметил, н-бутоксикарбонилметил, изобутоксикарбонилметил, н-пентоксикарбонилметил, н-гексилоксикарбонилметил, 2-метоксикарбонилэтил, 3-метоксикарбонилпропил, 4-метоксикарбонилбутил, 5-метоксикарбонилпентил, 6-метоксикарбонилгексил и так далее.
Предпочтительными низшими алкильными группами, необязательно замещенными гидрокси-группой(группами), являются С1-6 линейные, или разветвленные алкильные группы, необязательно содержащие от одной до трех гидрокси-групп, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, третбутил, втор-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, 3-метилпентил, гидроксиметил, 2-гидроксиэтил, 1-, 2-, или 3-гидрокси-н-пропил, 1-, 2-, или 3-гидроксиизопропил, 1-, 2-, 3-, или 4-гидрокси-н-бутил, 1-, 2-, 3-, или 4-гидрокси-изобутил, 1-, 2-, 3-, или 4-гидрокси-третбутил, 1-, 2-, 3-, или 4-гидрокси-вторбутил, 1-, 2-, 3-, 4-, или 5-гидрокси-н-пентил, 1-, 2-, 3-, 4-, или 5-гидрокси-изопентил, 1-, 2-, 3-, 4-, или 5-гидрокси-неопентил, 1-, 2-, 3-, 4-, 5-, или 6-гидрокси-н-гексил, 1-, 2-, 3-, 4-, 5-, или 6-гидрокси-изогексил, 1-, 2-, 3-, 4-, 5-, или 6-гидрокси-3-метилпентил и так далее.
Предпочтительными амино-группами, необязательно замещенными одной или двумя защитными группой(группами), являются амино-группы, необязательно содержащие одну или две С1-6 линейных, или разветвленных алкильных группы, необязательно содержащих от одной до трех гидрокси-групп, например, амино, метиламино, диметиламино, этиламино, диэтиламино, н-пропиламино, ди-н-пропиламино, изопропиламино, диизопропиламино, н-бутиламино, ди-н-бутиламино, изобутиламино, диизобутиламино, третбутиламино, дитретбутиламино, н-пентиламино, ди-н-пентиламино, н-гексиламино, ди-н-гексиламино, гидроксиметиламино, 2-гидроксиэтиламино, диэтиламино, ди-(2-гидроксиэтил)амино, 3-гидроксипропиламино, 4-гидроксибутиламино и так далее.
В ряду соединений бензоазепина, представленных приведенной выше общей формулой (1), предпочтительными являются следующие соединения и их соли:
в случае, когда Х является атомом кислорода,
(1) соединения, в которых R представляет собой гидрокси-группу, а R1 представляет собой атом водорода,
(2) соединения, в которых R представляет собой гидрокси-группу, а R1 представляет собой гидрокси-защитную группу,
(3) соединения, в которых R представляет собой меркапто-группу, а R1 представляет собой гидрокси-защитную группу, и
(4) соединения, в которых R представляет собой амино-группу, а R1 представляет собой гидрокси-защитную группу, и
в случае, когда Х является атомом серы,
(1) соединения, в которых R представляет собой гидрокси-группу, а R1 представляет собой атом водорода, или гидрокси-защитную группу.
Особенно предпочтительным из них является соединение, в котором Х является атомом кислорода, R представляет собой гидрокси-группу, а R1 представляет собой атом водорода; или его соль.
Соединения бензоазепина, представленные приведенной выше общей формулой (1), можно получить различными способами, и их примером является способ, представленный на следующих схемах реакций с 1 по 7:
Схема реакции-1

в которой R3 и R4независимо представляют собой низшую алкильную группу, или необязательно замещенную фенильную группу, или вместо этого R3 и R4могут быть связаны друг с другом через один, или более дополнительных гетероатомов, или без них, с образованием, вместе с атомом азота, с которым они связаны, 5-8-членного насыщенного, или ненасыщенного цикла, а R1а и R2амогут быть одинаковыми, или различными, и каждый из них представляет собой гидрокси-защитную группу.
Примерами низших алкильных групп являются упомянутые выше группы, включая С1-6 линейные, или разветвленные алкильные группы, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, третбутил, втор-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, 3-метилпентил и так далее.
Примеры заместителей для необязательно замещенных фенильных групп включают в себя упомянутые выше низшие алкильные группы, С1-6 линейные, или разветвленные алкокси-группы, например, метокси, этокси, пропокси, изопропокси, бутокси, третбутокси, пентилокси, гексилокси и так далее, и атомы галогена, например, фтор, хлор, бром, йод и так далее.
Предпочтительные примеры необязательно замещенных фенильных групп включают в себя фенил, 2-, 3-, или 4-метилфенил, 2-, 3-, или 4-хлорфенил, 2-, 3-, или 4-метоксифенил и так далее.
Примеры 5-8-членных насыщенных, или ненасыщенных циклов, образованных связанными друг с другом R3 и R4, включают в себя морфолиновый цикл и так далее.
Соединение (4) можно получить, вводя соединение (2) во взаимодействие с соединением (3) в подходящем растворителе в присутствии кислоты.
Примеры растворителей включают в себя галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, ацетонитрил и так далее.
Примеры кислот включают в себя мягкие кислоты, например, 1Н-тетразол, 5-метилтетразол, гидробромид пиридиния и так далее.
Количество кислоты обычно составляет, по меньшей мере, около 1 моль, а предпочтительно, от около 1 до около 10 моль на моль соединения (2).
Количество соединения (3) обычно составляет от 0,5 до 2 моль, а предпочтительно, от 0,7 до 1,5 моль на моль соединения (2).
Температура реакции обычно составляет от -20 до 50°С, предпочтительно, от 0 до 50°С, а более предпочтительно, от 0°С до комнатной температуры. Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 30 минут до 6 часов, а более предпочтительно, от 1 до 3 часов.
Соединение (1а) можно получить, вводя соединение (4) во взаимодействие с окисляющим агентом в подходящем растворителе.
Примеры растворителей включают в себя галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, ацетонитрил и так далее.
Примеры окисляющих агентов включают в себя надкислоты, например, перекись водорода и метахлорпербензойную кислоту, надуксусную ксилоту, надмалеиновую кислоту и так далее.
Количество окисляющего агента обычно составляет, по меньшей мере, около 1 моль, а предпочтительно, от около 1 до около 3 моль на моль соединения (4).
Температура реакции обычно составляет от -100 до 50°С, предпочтительно, от -40°С до комнатной температуры, а более предпочтительно, от -40 до 0°С. Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 30 минут до 6 часов, а более предпочтительно, от 30 минут до 2 часов.
Соединение (1b) можно получить путем снятия защиты с защищенных гидрокси-групп соединения (1а) стандартными методами.
Например, в том случае, когда гидрокси-защитные группы представляют собой низшие алкильные группы, снятие защиты можно осуществить в стандартных условиях гидролиза.
Подобный гидролиз предпочтительно проводят в присутствии основания или кислоты (включая кислоту Льюиса).
В качестве подобного основания можно использовать широкий ряд известных неорганических и органических оснований. Предпочтительными неорганическими основаниями являются, например, щелочные металлы (например, натрий, калий и так далее), щелочноземельные металлы (например, магний, кальций и так далее) и их гидроксиды, карбонаты и гидрокарбонаты. Предпочтительными органическими основаниями являются, например, триалкиламины (например, триметиламин, триэтиламин и так далее), пиколин и 1,5-диазабицикло[4,3,0]нон-5-ен.
В качестве подобной кислоты можно использовать широкий ряд известных неорганических и органических кислот. Предпочтительными органическими кислотами являются жирные кислоты, например, муравьиная кислота, уксусная кислота, пропионовая кислота и так далее, и тригалогенуксусные кислоты, например, трихлоруксусная кислота, трифторуксусная кислота и так далее. Предпочтительными неорганическими кислотами являются, например, хлористоводородная кислота, бромистоводородная кислота, серная кислота, хлористый водород, бромистый водород и так далее. Примеры кислот Льюиса включают в себя эфирный комплекс трехфтористого бора, трехбромистый бор, хлорид алюминия, хлорид железа и так далее.
При использовании тригалогенуксусной кислоты, или кислоты Льюиса, гидролиз предпочтительно проводят в присутствии ловушки для катионов (например, анизола, фенола и так далее).
Количество основания или кислоты не ограничено, пока оно удовлетворяет требованиям гидролиза.
Температура реакции обычно составляет от -20 до 100°С, предпочтительно, от 0 до 50°С, а более предпочтительно, от 0°С до комнатной температуры. Продолжительность реакции обычно составляет от 5 минут до 24 часов, предпочтительно, от 15 минут до 6 часов, а более предпочтительно, от 15 минут до 3 часов.
В том случае, когда, например, гидрокси-защитные группы представляют собой фенил(низшие)алкильные группы, снятие защиты можно осуществить стандартным методом каталитического восстановления.
Катализаторы, подходящие для подобного каталитического восстановления, представляют собой платиновые катализаторы (например, платиновую пластину, губчатую платину, платиновую чернь, коллоидную платину, оксид платины, платиновую проволоку и так далее), палладиевые катализаторы (например, губчатый палладий, палладиевую чернь, оксид палладия, палладий на угле, палладий/сульфат бария, палладий/карбонат бария и так далее), никелевые катализаторы (например, восстановленный никель, оксид никеля, никель Ренея и так далее), кобальтовые катализаторы (например, восстановленный кобальт, кобальт Ренея и так далее), железные катализаторы (например, восстановленное железо и так далее) и так далее. При использовании катализатора палладия на угле, каталитическое восстановление предпочтительно проводят в присутствии бромида цинка.
Количество катализатора, используемого при каталитическом восстановлении, не ограничено и может представлять собой стандартное количество.
Температура реакции обычно составляет от 0 до 100°С, предпочтительно, от 0 до 50°С, а более предпочтительно, от комнатной температуры до 50°С. Продолжительность реакции обычно составляет от 5 минут до 24 часов, предпочтительно, от 5 минут до 3 часов, а более предпочтительно, от 5 минут до 1 часа.
Схема реакции-2

Соединение (2) вводят во взаимодействие с хлорокисью фосфора, а затем гидролизуют, получая соединение (1b).
Количество хлорокиси фосфора обычно составляет от 1 моль до большого избытка, а предпочтительно, от 1 до 5 моль на моль соединения (2).
Упомянутую выше реакцию проводят в присутствии основного соединения в подходящем растворителе. Примеры растворителей для реакции с хлорокисью фосфора включают в себя простые эфиры, например, диэтиловый эфир, диоксан, тетрагидрофуран, моноглим, диглим и так далее, галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, ацетонитрил и так далее.
Примеры основных соединений включают в себя карбонаты, например, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат цезия и так далее, гидроксиды щелочных металлов, например, гидроксид натрия, гидроксид калия и так далее, гидроксиды щелочноземельных металлов, например, гидроксид кальция и так далее, фосфаты, например, фосфат натрия, фосфат калия и так далее; органические основания, например, пиридин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триэтиламин, триметиламин, диметиланилин, N-метилморфолин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), 1,4-диазабицикло[2.2.2]октан (DABCO) и так далее, и их смеси.
Количество основного соединения обычно составляет, по меньшей мере, около 3 моль, а предпочтительно, от около 3 до около 10 моль на моль соединения (2). Температура реакции обычно составляет от -100 до 50°С, предпочтительно, от -50°С до комнатной температуры, а более предпочтительно, от -30°С до комнатной температуры. Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 30 минут до 6 часов, а более предпочтительно, от 1 до 3 часов.
Гидролиз можно осуществить, добавляя воду к упомянутой выше реакционной смеси или прибавляя реакционную смесь к воде.
Поскольку это обычно сопровождается избыточным разложением реагентов и, в результате этого, выделяется теплота, гидролиз предпочтительно проводят при охлаждении. Для завершения реакции нагревание предпочтительно осуществляют после замедления первоначальной реакции.
Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 30 минут до 6 часов, а более предпочтительно, от 1 до 3 часов.
Схема реакции-3

в которой R1 аналогичен указанному выше.
Соединение (2) вводят во взаимодействие с дифенилфосфитом, а затем вводят во взаимодействие со спиртом (R1ОН), получая соединение (1с).
Количество дифенилфосфита составляет обычно от 1 моль до большого избытка, а предпочтительно, от 1 до 5 моль на моль соединения (2). Количество спирта (R1ОН) составляет обычно от 1 моль до большого избытка, а предпочтительно, от 1 до 10 моль на моль соединения (2).
Упомянутую выше реакцию проводят в присутствии основного соединения в подходящем растворителе.
Примеры растворителей включают в себя простые эфиры, например, диэтиловый эфир, диоксан, тетрагидрофуран, моноглим, диглим и так далее, галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, и ацетонитрил.
Примеры основных соединений включают в себя карбонаты, например, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат цезия и так далее, гидроксиды щелочных металлов, например, гидроксид натрия, гидроксид калия и так далее, гидроксиды щелочноземельных металлов, например, гидроксид кальция и так далее, фосфаты, например, фосфат натрия, фосфат калия и так далее; органические основания, например, пиридин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триэтиламин, триметиламин, диметиланилин, N-метилморфолин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), 1,4-диазабицикло[2.2.2]октан (DABCO) и так далее, и их смеси.
Количество основного соединения обычно составляет, по меньшей мере, около 1 моль, а предпочтительно, от около 1 до около 10 моль на моль соединения (2). В качестве растворителя можно также использовать органические растворители.
Температура реакции обычно составляет от -100 до 50°С, предпочтительно, от -50°С до комнатной температуры, а более предпочтительно, от -30°С до комнатной температуры. Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 30 минут до 6 часов, а более предпочтительно, от 1 до 3 часов.
Схема реакции-4

в которой R1 аналогичен указанному выше.
Окисление фосфита можно осуществить при использовании от около 1 до около 3 эквивалентов агента, окисляющего фосфористую кислоту, при температуре в интервале от около 0°С до 50°С. Предпочтительно реакцию проводят при использовании от около 5 до около 15% избытка агента, окисляющего фосфористую кислоту, в интервале от 0°С до комнатной температуры.
Агент, окисляющий фосфористую кислоту, представляет собой реагент, окисляющий фосфит до фосфата. Его примеры включают в себя перекиси, например, перекись водорода, метахлорпербензойную кислоту и так далее, йод в воде, бром, четырехокись азота и так далее. Предпочтительным является йод в воде.
Указанную выше реакцию проводят в подходящем растворителе.
Примеры растворителей включают в себя простые эфиры, например, диэтиловый эфир, диоксан, тетрагидрофуран, моноглим, диглим и так далее, галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, ацетонитрил и пиридин.
Температура реакции обычно составляет от -100 до 50°С, предпочтительно, от -50°С до комнатной температуры, а более предпочтительно, от -30°С до комнатной температуры. Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 15 минут до 6 часов, а более предпочтительно, от 15 минут до 3 часов.
Схема реакции-5

в которой R1 аналогичен указанному выше, а R11и R12 могут быть одинаковыми или различными и каждый из них представляет собой атом водорода, или низшую алкильную группу, необязательно содержащую гидрокси-группу(группы).
Амин (R11R12NH) и четыреххлористый углерод вводят во взаимодействие с диэфиром фосфористой кислоты (1с), получая амидофосфат (1е).
Вместо четыреххлористого углерода можно также использовать гипохлорит натрия.
Количество четыреххлористого углерода обычно составляет от 1 моль до большого избытка, а предпочтительно, от 1 до 5 моль на моль соединения (1с). Количество амина (R11R12NH) обычно составляет от 1 моль до большого избытка, а предпочтительно, от 1 до 10 моль на моль соединения (1с).
Указанную выше реакцию проводят в присутствии основного соединения в подходящем растворителе.
Примеры растворителей включают в себя простые эфиры, например, диэтиловый эфир, диоксан, тетрагидрофуран, моноглим, диглим и так далее, галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, ацетонитрил и так далее.
Примеры основных соединений включают в себя карбонаты, например, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат цезия и так далее, гидроксиды щелочных металлов, например, гидроксид натрия, гидроксид калия и так далее, гидроксиды щелочноземельных металлов, например, гидроксид кальция и так далее, фосфаты, например, фосфат натрия, фосфат калия и так далее; органические основания, например, пиридин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триэтиламин, триметиламин, диметиланилин, N-метилморфолин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), 1,4-диазабицикло[2.2.2]октан (DABCO) и так далее, и их смеси. Количество основного соединения обычно составляет, по меньшей мере, около 1 моль, а предпочтительно, от около 1 до около 10 моль на моль соединения (2). В качестве растворителя можно также использовать органические растворители.
Температура реакции обычно составляет от -100 до 50°С, предпочтительно, от -50°С до комнатной температуры, а более предпочтительно, от -30°С до комнатной температуры. Продолжительность реакции обычно составляет от 1 минуты до 24 часов, предпочтительно, от 1 минуты до 6 часов, а более предпочтительно, от 1 минуты до 3 часов.
Схема реакции-6

в которой R1 аналогичен указанному выше.
Диэфир фосфористой кислоты (1с) вводят во взаимодействие с серой, получая диэфир тиофосфорной кислоты (1f).
Количество серы обычно составляет от 1 моль до большого избытка, а предпочтительно, от 1 до 5 моль на моль соединения (1с).
Указанную выше реакцию проводят в присутствии основного соединения в подходящем растворителе.
Примеры растворителей включают в себя простые эфиры, например, диэтиловый эфир, диоксан, тетрагидрофуран, моноглим, диглим и так далее, галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, ацетонитрил и пиридин.
Примеры основных соединений включают в себя карбонаты, например, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат цезия и так далее, гидроксиды щелочных металлов, например, гидроксид натрия, гидроксид калия и так далее, гидроксиды щелочноземельных металлов, например, гидроксид кальция и так далее, фосфаты, например, фосфат калия, фосфат натрия и так далее, гидриды щелочных металлов, например, гидрид натрия, гидрид калия и так далее, щелочные металлы, например, калий, натрий и так далее, амид натрия, алкоголяты металлов, например, метилат натрия, этилат натрия, н-бутилат натрия, третбутилат натрия, третбутилат калия и так далее; органические основания, например, пиридин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триэтиламин, триметиламин, диметиланилин, N-метилморфолин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), 1,4-диазабицикло[2.2.2]октан (DABCO) и так далее, и их смеси. Количество основного соединения обычно составляет, по меньшей мере, около 1 моль, а предпочтительно, от около 1 до около 10 моль на моль соединения (2). В качестве растворителя можно также использовать органические растворители.
Температура реакции обычно составляет от -100 до 50°С, предпочтительно, от -50°С до комнатной температуры, а более предпочтительно, от -30°С до комнатной температуры. Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 30 минут до 6 часов, а более предпочтительно, от 1 до 3 часов.
Схема реакции-7

в которой R1' представляет собой гидрокси-защитную группу.
Защитную группу в соединении (1g), которое представляет собой соединение (1f), полученной по схеме реакции 6, в которой R1 представляет собой гидрокси-защитную группу, снимают, получая соединение (1h).
Если R1 представляет собой цианоэтильную группу, данную защитную группу можно удалить действием основного соединения.
Указанную выше реакцию проводят в присутствии основного соединения в подходящем растворителе.
Примеры растворителей включают в себя воду, спирты, например, метанол, этанол, изопропиловый спирт и так далее, простые эфиры, например, диэтиловый эфир, диоксан, тетрагидрофуран, моноглим, диглим и так далее, галогенсодержащие углеводородные растворители, например, хлористый метилен, хлороформ, 1,2-дихлорэтан, четыреххлористый углерод и так далее, сложные эфиры, например, этилацетат и так далее, ароматические углеводороды, например, бензол, толуол, ксилол и так далее, апротонные полярные растворители, например, диметилформамид (ДМФА), диметилсульфоксид (ДМСО) и так далее, кетоны, например, ацетон, метилэтилкетон, метилизобутилкетон и так далее, ацетонитрил, и их смеси.
Примеры основных соединений включают в себя карбонаты, например, карбонат натрия, карбонат калия, гидрокарбонат натрия, гидрокарбонат калия, карбонат цезия и так далее, гидроксиды щелочных металлов, например, гидроксид натрия, гидроксид калия и так далее, гидроксиды щелочноземельных металлов, например, гидроксид кальция и так далее, фосфаты, например, фосфат калия, фосфат натрия и так далее, гидриды щелочных металлов, например, гидрид натрия, гидрид калия и так далее, щелочные металлы, например, калий, натрий и так далее, амид натрия, алкоголяты металлов, например, метилат натрия, этилат натрия, н-бутилат натрия, третбутилат натрия, третбутилат калия и так далее; органические основания, например, пиридин, имидазол, N-этилдиизопропиламин, диметиламинопиридин, триэтиламин, триметиламин, диметиланилин, N-метилморфолин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), 1,4-диазабицикло[2.2.2]октан (DABCO) и так далее, и их смеси. Количество основного соединения обычно составляет, по меньшей мере, около 1 моль, а предпочтительно, от около 1 до около 10 моль на моль соединения (2). В качестве растворителя можно также использовать органические растворители.
Температура реакции обычно составляет от -100 до 50°С, предпочтительно, от -50°С до комнатной температуры, а более предпочтительно, от -30°С до комнатной температуры. Продолжительность реакции обычно составляет от 15 минут до 24 часов, предпочтительно, от 30 минут до 6 часов, а более предпочтительно, от 1 до 3 часов.
Соединения (2), (3), (4), (1а), (1b), (1c), (1d), (1e), (1f), (1g) и (1h) в приведенных выше схемах реакций могут представлять собой свои подходящие соли. Примеры подобных подходящих солей включают в себя те же типы солей, что и для соединения (1).
Соединения, полученные по приведенным выше схемам реакций, можно выделить и очистить из реакционной смеси обычными способами, например, после охлаждения реакционной смеси, выделения сырого продукта фильтрованием, концентрированием, экстракцией, или подобным способом выделения, а затем очисткой полученного продукта колоночной хроматографией, перекристаллизацией, или подобным стандартным способом очистки.
Соединения, представленные общей формулой (1) настоящего изобретения, включают в себя стереоизомеры, оптические изомеры и их сольваты (гидраты, этаноляты и так далее).
Примеры солей соединений, представленных общей формулой (1) настоящего изобретения, включают в себя фармацевтически приемлемые соли, например, соли металлов, например, соли щелочных металлов (например, соли натрия, соли калия и так далее), соли щелочноземельных металлов (например, соли кальция, соли магния и так далее), аммониевые соли, соли органических оснований (например, соли триметиламина, соли триэтиламина, соли пиридина, соли пиколина, соли дициклогексиламина, соли этилендиамина, соли N,N'-дибензилэтилендиамина, соли трис(гидроксиметил)аминометана, соли этаноламина и так далее) и так далее. В их ряду предпочтительными являются соли щелочных металлов, а более предпочтительными являются соли натрия.
Подобные соли можно легко получить, действуя на соединение настоящего изобретения соответствующим фармацевтически приемлемым основным соединением. Примеры применимых основных соединений включают в себя гидроксид натрия, гидроксид калия, гидроксид кальция, карбонат натрия, гидрокарбонат калия и так далее.
Соединение настоящего изобретения обладает, например, антагонизмом по отношению к вазопрессину, сосудорасширяющей активностью, гипотензивной активностью, активностью ингибирования выделения сахаридов в печени, активностью ингибирования роста мезангиальных клеток, акваретической активностью, и ингибирующей активностью к агрегации тромбоцитов. Соединение применимо в качестве сосудорасширяющего агента, гипотензивного агента, акваретического агента и ингибитора агрегации тромбоцитов и эффективно для предотвращения и лечения гипертензии, отека (например, сердечного отека, отека печени, почечного отека, церебрального отека), асцита, сердечной недостаточности (например, тяжелой сердечной недостаточности), почечной дисфункции, синдрома несоответствующей секреции вазопрессина (SIADH), цирроза печени, гипонатриемии, гипокалиемии, диабета, циркуляторной недостаточности, болезни поликистоза почек (PKD), инфаркта мозга, инфаркта миокарда и так далее.
При введении в организм человека в форме лекарственного средства соединение настоящего изобретения можно использовать одновременно, или отдельно с другими антагонистами вазопрессина, ингибиторами АСЕ, β-блокирующими агентами, акваретическими агентами, антагонистами ангиотензина II (ARB) и/или подобными фармацевтическими лекарственными средствами.
Соединение настоящего изобретения обычно используют в виде общей фармацевтической композиции. Такую фармацевтическую композицию можно получить стандартным способом при использовании обычно используемых разбавителей и/или эксципиентов, например, наполнителей, расширителей, связывающих веществ, увлажняющих агентов, разрыхлителей, поверхностно-активных веществ, смазывающих веществ и так далее.
Форму фармацевтической композиции, содержащей соединение настоящего изобретения, можно выбрать подходящим образом в зависимости от цели лечения. Например, она может быть в форме таблетки, пилюли, порошка, раствора, суспензии, эмульсии, капсулы, суппозитория, мази или гранул. В особенности предпочтительна композиция в виде водного раствора, например, препарат для инъекции, вливания и так далее.
Например, при получении препарата для инъекций с использованием соединения настоящего изобретения, подобный препарат для инъекций предпочтительно получают в виде раствора, эмульсии или суспензии, которую стерилизуют и которая изотонична крови. Для получения подобного раствора, эмульсии или суспензии с использованием соединения настоящего изобретения можно использовать любые обычно применяемые в данной области разбавители. Примеры подобных разбавителей включают в себя воду, водные растворы молочной кислоты, этиловый спирт, пропиленгликоль, этоксилированный изостеариловый спирт, полиоксиэтилированный изостеариловый спирт и полиоксиэтиленовые сложные эфиры сорбита и жирных кислот. Кроме того, в данном случае к фармацевтической композиции можно добавить хлорид натрия, глюкозу, маннит, глицерин и подобные агенты, придающие изотоничность, в количествах, достаточных для получения изотонического раствора. Можно также добавить обычные регуляторы рН, солюбилизаторы, буферные агенты, успокаивающие средства и так далее.
Препарат для инъекций, содержащий соединение настоящего изобретения, можно получить стандартным способом при использовании соединения, представленного общей формулой (1), или его фармацевтически приемлемой соли, вместе с буферным агентом, агентом для придания изотоничности, растворителем для инъекции и, при необходимости, регулятором рН.
Примеры буферных агентом включают в себя карбонаты, бораты, фосфаты, цитраты, трис(гидроксиметил)аминометан, малаты и тартраты. Можно также отдельно использовать кислоту или основание, образующие подобный буфер.
Примеры регуляторов рН включают в себя основные соединение, например, гидроксид натрия и так далее, кислоты, например, хлористоводородную кислоту и так далее.
Кроме того, при необходимости в фармацевтическую композицию можно добавлять красители, консерванты, ароматизирующие вещества, подсластители и так далее, а также другие лекарственные вещества.
Содержание соединения, представленного общей формулой (1) настоящего изобретения, или его соли, в фармацевтической композиции не ограничено и может быть подходящим образом выбрано из широкого интервала. Содержание обычно составляет от 0,01 до 70 мас.% фармацевтической композиции.
Способ введения подобной фармацевтической композиции не ограничен и ее можно вводить подходящим способом в зависимости от формы данной фармацевтической композиции, возраста пациента, пола и так далее, степени симптомов пациента и так далее. Например, таблетки, пилюли, растворы, суспензии, эмульсии, гранулы и капсулы можно вводить перорально. Препараты для инъекций можно вводить путем внутривенной инъекции сами по себе или в виде смеси с глюкозой, аминокислотой и/или подобными стандартными добавками. Кроме того, при необходимости инъекции можно вводить сами по себе путем внутримышечного, внутрикожного, подкожного или внутрибрюшинного введения.
Дозировку фармацевтической композиции настоящего изобретения можно выбрать в зависимости от применения, возраста пациента, пола и так далее, степени заболевания и так далее. Доза обычно такова, что соединение, представленное общей формулой (1), которое является активным ингредиентом, вводят в количестве от 0,001 до 100 мг, а предпочтительно, от 0,001 до 50 мг на 1 кг массы тела в день за одно или более введений.
Доза изменяется при различных условиях. Может быть достаточно дозы, которая меньше указанного выше интервала, в то время как может потребоваться доза, превышающая указанный выше интервал.
Эффект изобретения
Соединение (1) настоящего изобретения или его соль обладают превосходной растворимостью в воде, превосходной абсорбируемостью и так далее.
В частности, соединение (1b) или его соль обладают превосходной растворимостью в воде, превосходной абсорбируемостью и так далее.
При введении в организм человека соединение (1) настоящего изобретения, или его соль, в частности, соединение (1b) или его соль дает возможность легко получить активный ингредиент толваптан.
Кроме того, соединение (1) настоящего изобретения или его соль легко кристаллизуется и чрезвычайно просто в обращении. Кроме того, соединение (1) настоящего изобретения или его соль обладают превосходной химической стабильностью.
Соединение (1) настоящего изобретения или его соль можно подходящим образом использовать в качестве исходного вещества для получения соединения (1b).
Использование соединения (1) настоящего изобретения или его соли дает возможность предоставления композиций в различных формах, которые проявляют эффективность лекарственного средства, равную толваптану, являющемуся эффективным лекарственным средством.
Краткое описание чертежей
Фигура 1 представляет собой график, на котором показано изменение концентрации толваптана в сыворотке крови самок крыс после быстрого введения раствора соединения (1b) в хвостовую вену в такой дозе, чтобы на кг массы тела приходился 1 мг толваптана.
Фигура 2 представляет собой график, на котором показано изменение в концентрации толваптана в сыворотке крови самок крыс после перорального введения раствора соединения (1b) в такой дозе, чтобы на кг массы тела приходился 1 мг толваптана.
Наилучший способ осуществления изобретения
Примеры, экспериментальные примеры и примеры получения приведены ниже для иллюстрации настоящего изобретения в дальнейших подробностях, но рамки изобретения не ограничены данными примерами.
Пример 1

Толваптан (соединение (2)) в количестве 1,0 г и 460 мг 1Н-тетразола растворяли в 30 мл хлористого метилена и прибавляли к данному раствору по каплям 1,2 г дибензилдиизопропиламидофосфита при комнатной температуре при перемешивании. После этого смесь перемешивали в течение 2 часов при той же температуре.
Полученную реакционную смесь охлаждали до -40°С и прибавляли к ней по каплям 6 мл раствора 920 мг метахлорпербензойной кислоты в хлористом метилене. После этого смесь перемешивали при той же температуре в течение 30 минут и при 0°С в течение 30 минут. Реакционную смесь промывали водным раствором тиосульфата натрия и насыщенным водным раствором гидрокарбоната натрия, а затем сушили над безводным сульфатом натрия. Полученную реакционную смесь фильтровали и концентрировали, а остаток очищали колоночной хроматографией на силикагеле (элюент: н-гексан:этилацетат=1:1), получая 1,5 г аморфного соединения (1а-1) (выход 97,2%).
ЯМР (ДМСО-d6, 100°C) δ м.д.: 9,86 (1H, ушир.с), 7,56 (1H, с), 7,50-7,10 (17H, м), 7,00-6,80 (2H, м), 5,60-5,50 (1H, м), 5,15-5,00 (4H, m), 5,00-2,75 (2H, м), 2,36 (3H, с), 2,34 (3H, с), 2,10-1,70 (4H, м).
Пример 2

Толваптан (соединение (2)) в количестве 4,5 г и 2,2 г 1Н-тетразола растворяли в 120 мл хлористого метилена и прибавляли по каплям к данному раствору раствор 4,0 г дитретбутилдиизопропиламидофосфита в 10 мл хлористого метилена при охлаждении льдом и перемешивании. После этого смесь перемешивали в течение 2 часов при комнатной температуре.
Полученную реакционную смесь охлаждали до -40°С и прибавляли к ней по каплям 20 мл раствора 4,0 г метахлорпербензойной кислоты в хлористом метилене. После этого смесь перемешивали при той же температуре в течение 30 минут и при 0°С в течение 40 минут. Реакционную смесь промывали водным раствором тиосульфата натрия и насыщенным водным раствором гидрокарбоната натрия, а затем сушили над безводным сульфатом натрия. Полученную реакционную смесь фильтровали и концентрировали, а остаток очищали колоночной хроматографией на силикагеле (элюент: гексан:этилацетат=1:1), получая 3,0 г аморфного соединения (1а-2) (выход 46,7%).
ЯМР (ДМСО-d6) δ м.д.: 10,50-10,20 (1H, м), 8,00-6,50 (10H, м), 5,55-5,20 (1H, м), 4,90-4,50 (1H, м), 2,85-2,60 (1H, м), 2,40-2,20 (6H, м), 2,20-1,60 (4H, м), 1,60-1,30 (18H, м).
Пример 3

Соединение (1а-1) в количестве 5,3 г растворяли в 100 мл этанола и подвергали данный раствор каталитическому восстановлению при комнатной температуре и атмосферном давлении в течение 10 минут при использовании 2 г 5% палладия на угле в качестве катализатора. Катализатор удаляли из раствора фильтрованием, а полученный фильтрат концентрировали (4,2 г). Полученный остаток кристаллизовали из смеси метанол/вода. Кристаллы выделяли фильтрованием, а затем сушили при пониженном давлении (пятиокись фосфора), получая 3,5 г белого порошкообразного соединения (1b) (выход 88,5%).
Температура плавления: от 150 до 152°С
ЯМР (ДМСО-d6) δ м.д.: 7,50-6,70 (10Н, м), 5,50-5,40 (1Н, м); 5,00-2,50 (2Н, м), 2,37 (6Н, с), 2,40-1,50 (4Н, м).
Пример 4

Соединение (1а-2) в количестве 3,0 г растворяли в 100 мл хлористого метилена и прибавляли по каплям к данному раствору раствор 10 мл трифторуксусной кислоты в 5 мл хлористого метилена при охлаждении льдом и перемешивании. После этого смесь перемешивали при той же температуре в течение 2 часов. Из данного раствора удаляли растворитель. Полученный остаток повторно растворяли в хлористом метилене, а затем концентрировали. Полученный остаток кристаллизовали из смеси метанол/вода. Кристаллы выделяли фильтрованием, а затем сушили при пониженном давлении (пятиокись фосфора), получая 1,9 г белого порошкообразного соединения (1b) (выход 76,8%).
Пример 5

1,2-диметоксиэтан (DME) в количестве 240 мл и 84 мл триэтиламина (0,60 моль, 9 эквивалентов) прибавляли к 30 г (66 ммоль) толваптана (соединение (2)) и охлаждали смесь в потоке азота до -15°С. К полученной смеси прибавляли по каплям хлорокись фосфора (POCl3) в количестве 19 мл (0,20 моль, 3 эквивалента) при внутренней температуре не более -12°С и осуществляли перемешивание при -12°С в течение 2 часов. К 1 кг измельченного льда прибавляли 5N водный раствор гидроксида натрия в количестве 200 мл и прибавляли к нему небольшими порциями при перемешивании указанную выше реакционную смесь. К полученной смеси добавляли 500 мл толуола. Смесь нагревали до 50°С, а затем разделяли на водный слой и толуольный слой. К водному слою снова прибавляли толуол в количестве 500 мл, перемешивали при 50°С и после этого разделяли смесь на водный слой и толуольный слой. Водный слой охлаждали до 10°С, прибавляли к нему 80 мл 6N хлористоводородной кислоты и дважды экстрагировали 500 мл этилацетата. Экстракт сушили над сульфатом натрия и фильтровали, а фильтрат концентрировали. Концентрат сушили при пониженном давлении при комнатной температуре, получая 34 г аморфного соединения (1b).
Выход: 97%
Пример 6
Получение кальциевой соли соединения (1b).

(1) Соединение (1b) в количестве 2,6 г (5,0 ммоль) растворяли в 25 мл изопропилового спирта и прибавляли к нему 2,2 мл 5N водного раствора гидроксида натрия при комнатной температуре. Полученную смесь концентрировали при пониженном давлении. К остатку прибавляли 30 мл воды для растворения твердого содержимого, а после этого добавляли к нему водный раствор 0,61 г (5,5 ммоль) хлорида кальция. Выпавший осадок выделяли фильтрованием, промывали водой и сушили горячим воздухом при 60°С, получая 2,2 г белой порошкообразной кальциевой соли соединения (1b).
Выход:78%
1Н-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,3-2,4(10H, м), 2,8-4,5 (2Н, м), 5,2-5,8 (1Н, м), 6,4-8,1 (10Н, м), 9,0-10,2 (1Н, м).
(2) Соединение (1b) в количестве 280 мг (0,53 ммоль) растворяли в смешанном растворе 2 мл метанола и 1 мл воды, а затем прибавляли туда 43 мг (0,58 ммоль) гидроксида кальция. Смесь перемешивали при комнатной температуре в течение 1 часа. Выпавший осадок выделяли фильтрованием. Отфильтрованное вещество суспендировали в метаноле, перемешивали при нагревании, а затем фильтровали в горячем состоянии. Фильтрат концентрировали, а остаток перекристаллизовывали из метанола, получая 75,4 мг белой порошкообразной кальциевой соли соединения (1b).
Выход: 25%
Температура плавления: от 263 до 265°С
Пример 7
Получение магниевой соли соединения (1b).

(1) Соединение (1b) в количестве 1,0 г (1,9 ммоль) растворяли в 15 мл метанола и прибавляли к нему 0,76 мл 5N водного раствора гидроксида натрия. Смесь концентрировали при пониженном давлении. Остаток повторно растворяли в 10 мл метанола и прибавляли к полученному раствору раствор 0,18 г хлорида магния в 3 мл метанола при комнатной температуре. Выпавшее нерастворимое вещество (NaCl) удаляли фильтрованием, а фильтрат концентрировали. К остатку добавляли 10 мл воды и перемешивали при нагревании. После перемешивания смесь оставляли охлаждаться до комнатной температуры. После этого нерастворимое вещество выделяли фильтрованием, промывали водой и сушили при пониженном давлении при 60°С, получая 400 мг белой порошкообразной магниевой соли соединения (1b).
Выход: 38%
1H-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,4-2,4 (10H, м), 2,8-4,5 (2H, м), 5,3-5,5 (1H, м), 6,4-7,8 (10H, м), 9,7 (1H, ушир.).
(2) Соединение (1b) в количестве 282 мг (0,53 ммоль) растворяли в 2 мл метанола и прибавляли к нему 41 мг (0,70 ммоль) этилата магния при охлаждении льдом. К полученной смеси прибавляли еще 2 мл этанола и водную суспензию 36 мг (0,58 ммоль) гидроксида магния и перемешивали при комнатной температуре в течение 1 часа. Нерастворимое вещество удаляли фильтрованием, а фильтрат оставляли стоять на ночь. Выпавшее нерастворимое вещество выделяли фильтрованием, а затем сушили при пониженном давлении, получая 24,9 мг белой порошкообразной магниевой соли соединения (1b).
Выход: 11%
Температура плавления: от 250 до 252°С
Пример 8
Получение мононатриевой соли соединения (1b).

К раствору 266 мг (0,5 ммоль) соединения (1b) в метаноле (2 мл) прибавляли 1N водный раствор гидроксида натрия в количестве 0,5 мл и 1 мл воды при охлаждении льдом и перемешивали полученный раствор при комнатной температуре в течение 1 часа. Полученную реакционную смесь концентрировали при пониженном давлении, а остаток перекристаллизовывали из смеси метанол-вода, получая 45,2 мг белой порошкообразной мононатриевой соли соединения (1b).
Выход: 16%
Температура плавления: от 235 до 238°С
Пример 9
Получение динатриевой соли соединения (1b).

К раствору 276 мг (0,52 ммоль) соединения (1b) в метаноле (2 мл) прибавляли 1N водный раствор гидроксида натрия в количестве 1,0 мл при охлаждении льдом и перемешивали полученный раствор при комнатной температуре в течение 5 минут. Полученную реакционную смесь концентрировали при пониженном давлении, а остаток перекристаллизовывали из смеси ацетон-вода, получая 221 мг белой порошкообразной динатриевой соли соединения (1b).
Выход: 73%
Температура плавления: от 250 до 252°С
Пример 10
Получение диаммониевой соли соединения (1b).

К раствору 271 мг (0,51 ммоль) соединения (1b) в метаноле (2 мл) прибавляли 25%-ный водный раствор аммиака в количестве 1,0 мл при охлаждении льдом и перемешивали полученную смесь в течение 10 минут. Реакционную смесь концентрировали при пониженном давлении, а остаток перекристаллизовывали из смеси метанол-вода, получая 104 мг белой порошкообразной диаммониевой соли соединения (1b).
Выход: 36%
Температура плавления: от 195 до 198°С
Пример 11
Получение монокалиевой соли соединения (1b).
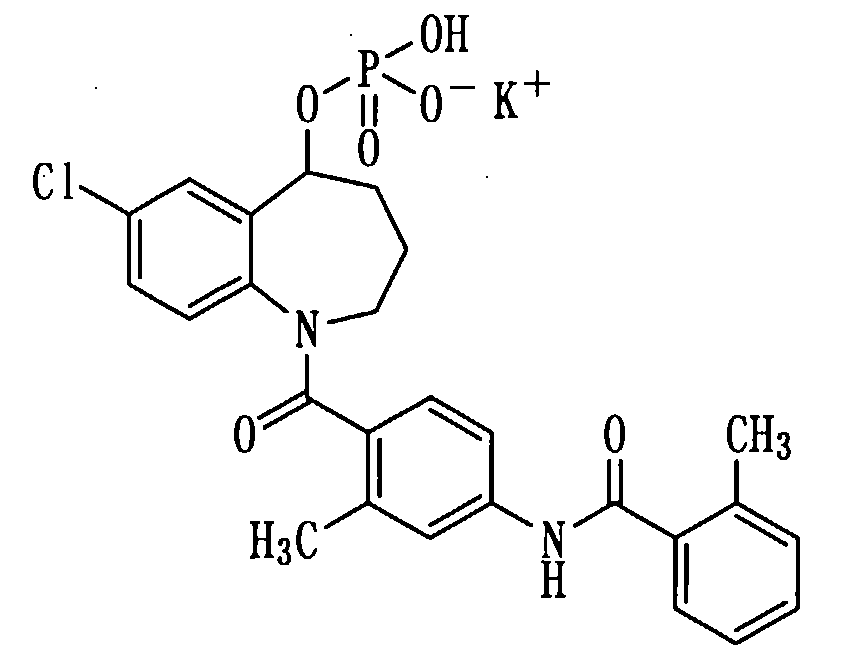
К раствору 276 мг (0,52 ммоль) соединения (1b) в метаноле (2 мл) прибавляли 1N водный раствор гидроксида калия в количестве 0,5 мл при охлаждении льдом и перемешивали полученную смесь в течение 10 минут. Реакционную смесь концентрировали при пониженном давлении, а остаток перекристаллизовывали из изопропилового спирта, получая 110,6 мг белой порошкообразной монокалиевой соли соединения (1b).
Выход: 37%
Температура плавления: от 200 до 203°С
Пример 12
Получение дикалиевой соли соединения (1b).

К раствору 276 мг (0,52 ммоль) соединения (1b) в метаноле (2 мл) прибавляли 1N водный раствор гидроксида калия в количестве 1,0 мл при охлаждении льдом и перемешивали полученную смесь в течение 5 минут. Реакционную смесь концентрировали при пониженном давлении и добавляли к остатку диэтиловый эфир. Нерастворимое вещество выделяли фильтрованием, а затем сушили, получая 273,9 мг белой порошкообразной дикалиевой соли соединения (1b).
Выход: 86%
Температура плавления: от 255 до 265°С (разложение)
Пример 13
Получение цинковой соли соединения (1b).

Соединение (1b) в количестве 1,0 г (1,9 ммоль) растворяли в 15 мл метанола и прибавляли к данному раствору 0,76 мл 5N водного раствора гидроксида натрия. Смесь концентрировали при пониженном давлении. Полученный остаток растворяли в 10 мл метанола и прибавляли к нему раствор 259 мг хлорида цинка в 3 мл метанола при комнатной температуре. Выпавшее нерастворимое вещество (NaCl) выделяли фильтрованием, а фильтрат концентрировали. К полученному остатку добавляли 10 мл воды и перемешивали при нагревании. Затем смеси давали остыть до комнатной температуры. Нерастворимое вещество выделяли фильтрованием, промывали водой и сушили при пониженном давлении при 60°С, получая 900 мг белой порошкообразной цинковой соли соединения (1b).
Выход: 80%
Температура плавления: от 235 до 239°С (разложение)
1Н-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,3-2,4 (10H, м), 2,8-4,5 (2Н, м), 5,3-5,7 (1Н, м), 6,6-7,7 (10Н, м), 9,7 (1Н, ушир.).
Пример 14
Получение соли соединения (1b) с этилендиамином.

Этилендиамин в количестве 0,074 мл (1,1 ммоль) прибавляли к раствору 600 мг (1,1 ммоль) соединения (1b) в этаноле (10 мл). Полученную реакционную смесь концентрировали при пониженном давлении, а остаток перекристаллизовывали из изопропилового спирта, получая 250 мг белой порошкообразной соли соединения (1b) с этилендиамином.
1H-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,5-2,0 (3H, м), 2,1-2,4 (7H, м), 2,77 (4H, с), 2,8-4,3 (2H, м), 5,3-5,5 (1H, м), 6,6-6,9 (1Н, м), 6,9-7,2 (5Н, м), 7,2-7,5 (5Н, м), 7,58 (2Н, д, J=7,6 Гц), 9,80 (1Н, ушир.).
Пример 15
Получение соли соединения (1b) с диэтаноламином.

Этаноламин в количестве 0,14 мл (2,3 ммоль) прибавляли к раствору 600 мг (1,1 ммоль) соединения (1b) в изопропиловом спирте (6 мл). К полученной смеси добавляли изопропиловый спирт в количестве 6 мл, растворяли при нагревании и в результате перекристаллизации из изопропилового спирта получали 280 мг белой порошкообразной соли соединения (1b) с диэтаноламином.
1H-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,4-2,0 (3H, м), 2,2-2,5 (7H, м), 2,75 (4H, т, J=5,5 Гц), 3,52 (4H, т, J=5,5 Гц), 2,8-4,3 (2H, м), 5,3-5,5 (1H, м), 6,7-6,9 (1H, м), 6,9-7,2 (2H, м), 7,2-7,4 (4H, м), 7,42 (1H, д, J=7,7 Гц), 7,57 (2H, d, J=6,5 Гц), 7,58 (2H, д, J=7,6 Гц), 9,80 (1H, ушир.).
Пример 16

Дифенилфосфит в количестве 1,3 мл (6,6 ммоль) прибавляли к раствору 1,0 г (2,2 ммоль) толваптана (соединение (2)) в пиридине (10 мл) при охлаждении льдом. Полученную смесь перемешивали при 0°С в течение 30 минут, а затем при комнатной температуре в течение 30 минут. К реакционной смеси добавляли 0,58 мл этанола и перемешивали при комнатной температуре в течение 30 минут. К данной смеси добавляли 1N хлористоводородную кислоту и экстрагировали этилацетатом. Этилацетатный слой промывали насыщенным водным раствором гидрокарбоната натрия, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=27:73→0:100). Очищенный продукт концентрировали при пониженном давлении, а остаток растворяли в смеси растворителей из 10 мл ацетонитрила и 10 мл воды, а затем подвергали сублимационной сушке, получая 450 мг белого аморфного твердого желаемого соединения.
Выход: 38%
1H-ЯМР (толуол-d8, 100°C) δ м.д.: 1,0-1,1 (3H, м), 1,4-1,9 (4H, м), 2,31 (3H, с), 2,42 (3H, с), 2,0-4,0 (2H, м), 3,7-4,1 (2H, м), 5,5 (0,5H, д, J=4,8 Гц), 6,4-7,5 (10H, м), 7,8 (0,5H, д, J =8,6 Гц).
Пример 17
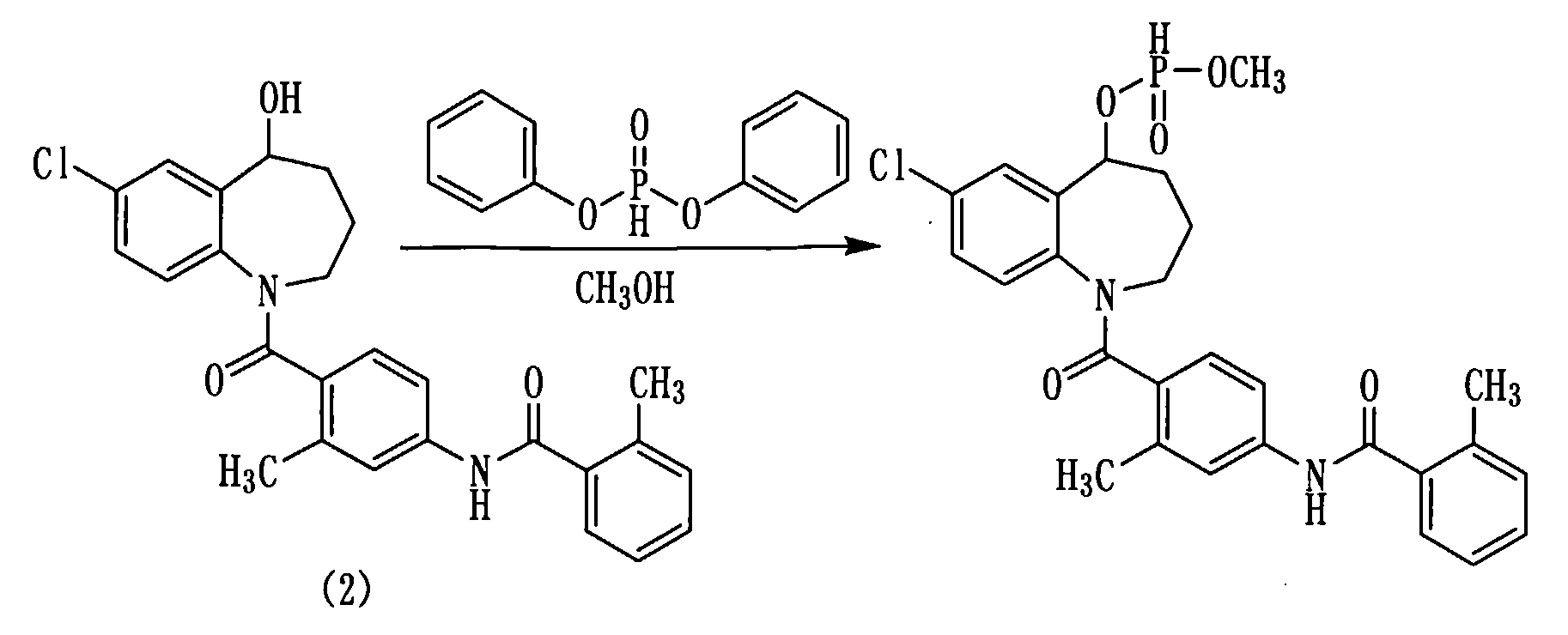
Раствор 10,0 г (22 ммоль) толваптана (соединение (2)) в пиридине (50 мл) охлаждали льдом и медленно прибавляли к нему 13 мл (66 ммоль) дифенилфосфита в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 30 минут. К данной смеси добавляли 4,5 мл метанола и перемешивали при комнатной температуре в течение 30 минут. Полученную реакционную смесь прибавляли к 325 мл 2N хлористоводородной кислоты при охлаждении льдом и экстрагировали этилацетатом. Этилацетатный слой промывали насыщенным раствором соли, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=100:0→93:7). Очищенный продукт концентрировали при пониженном давлении, получая 10,5 мг белого аморфного твердого желаемого соединения.
Выход: 91%
1H-ЯМР (толуол-d8, 100°C) δ м.д.: 1,5-2,0 (4H, м), 2,41 (3H, с), 2,49 (3H, с), 3,02-4,2 (2H, м), 5,5 (0,5H, д, J=4,8 Гц), 5,5-5,8 (1H, м), 6,6 (1H, д, J=8,3 Гц), 6,7-6,9 (1H, м), 6,9-7,2 (6H, м), 7,3-7,5 (2H, м), 7,81, 7,84 (0,5H, д, J=8,1 Гц).
Пример 18
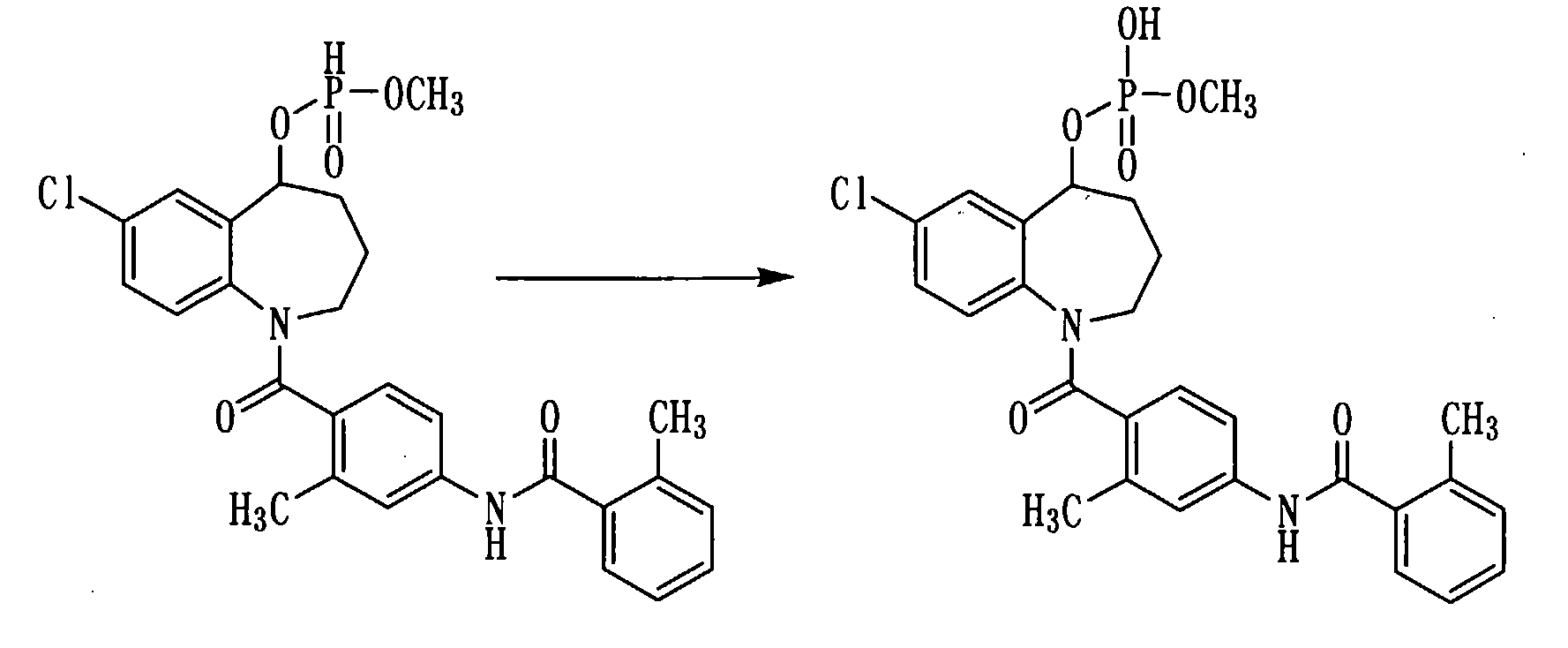
К раствору 500 мг (0,95 ммоль) соединения примера 17 в пиридине (5 мл) прибавляли воду в количестве 0,1 мл и 254 мг (1,0 ммоль) йода и перемешивали полученную смесь при комнатной температуре в течение 30 минут. К данной смеси добавляли 2 мл триэтиламина и концентрировали при пониженном давлении. К остатку прибавляли толуол в количестве 20 мл и концентрировали при пониженном давлении. К остатку добавляли воду и промывали смесью растворителей из этилацетата и диэтилового эфира. К водному слою добавляли 1N хлористоводородную кислоту и экстрагировали этилацетатом. Этилацетатный слой сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (хлористый метилен:метанол=90:10→50:50). Очищенный продукт концентрировали при пониженном давлении, а остаток растворяли в 30 мл воды. Полученный раствор фильтровали через целит, а фильтрат подвергали сублимационной сушке, получая 140 мг белого аморфного твердого желаемого соединения.
1H-ЯМР (толуол-d8, 100°C) δ м.д.: 1,4-2,0 (4H, м), 2,33 (3H, с), 2,34 (3H, с), 2,5-4,5 (5H, м), 5,4-5,7 (2H, м), 6,5 (2H, д, J=7,9 Гц), 6,7 (2H, д, J=7,9 Гц), 6,8-7,2 (5H, м), 7,2-7,4 (2H, м), 7,55 (1H, с).
Пример 19
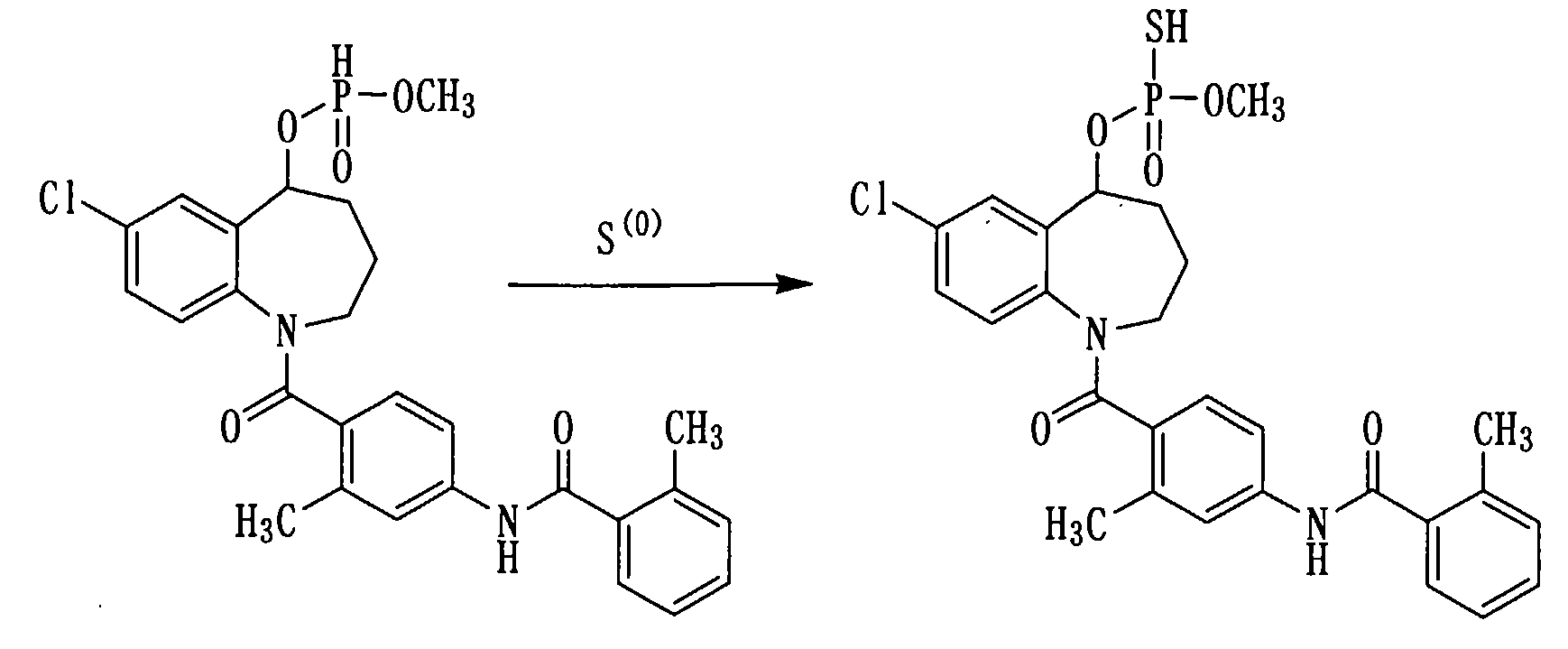
К раствору 500 мг (0,9 ммоль) соединения примера 17 в пиридине (5 мл) прибавляли серу в количестве 64 мг и перемешивали полученную смесь при комнатной температуре в течение 2 часов. К данной смеси добавляли 1 мл триэтиламина и концентрировали при пониженном давлении. К полученному остатку прибавляли толуол в количестве 10 мл и концентрировали при пониженном давлении. К остатку добавляли воду для растворения и фильтровали с использованием целита. К фильтрату прибавляли 1N хлористоводородную кислоту и проводили экстракцию этилацетатом. Этилацетатный слой сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. К полученному остатку добавляли воду, а нерастворимое вещество выделяли фильтрованием, а затем сушили, получая 300 мг белого порошкообразного твердого желаемого соединения.
1H-ЯМР (толуол-d8, 100°C) δ м.д.: 1,1-2,0 (4H, м), 2,2-2,5 (6H, м), 3,5 (3H, дд, J=13,9, 14,9 Гц), 2,5-5,0 (2H, м), 3,5-5,7 (1H, м), 6,4-7,5 (10Н, м).
Пример 20

К раствору 500 мг (0,95 ммоль) соединения примера 17 в ацетонитриле (5 мл) прибавляли воду в количестве 0,5 мл, 0,5 мл четыреххлористого углерода, 0,5 мл триэтиламина и 0,072 мл (1,2 ммоль) этаноламина и перемешивали полученную смесь при комнатной температуре в течение 10 минут. К данной смеси добавляли воду и проводили экстракцию этилацетатом. Этилацетатный слой промывали насыщенным раствором соли, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=100:0→80:20). Очищенный продукт концентрировали при пониженном давлении, получая 540 мг белого аморфного твердого желаемого соединения.
1Н ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,6-2,3 (4H,м), 2,36 (6Н, с), 2,7-3,1 (2Н, м), 2,5-4,5 (2Н, м), 3,3-3,5 (2Н, м), 3,65 (3Н, дд, J=9,6, 11,2 Гц), 4,0-4,3 (1Н, м), 4,4-4,8 (1Н, м), 5,3-5,7 (1Н, м), 6,7-7,1 (2Н, м), 7,1-7,5 (5Н, м), 7,57 (1Н, с), 9,76 (1Н, с).
Пример 21

К раствору 500 мг (0,95 ммоль) соединения примера 17 в ацетонитриле (5 мл) прибавляли воду в количестве 0,5 мл, 0,5 мл четыреххлористого углерода, 0,5 мл триэтиламина и 0,119 мл (1,2 ммоль) метиламина (40%-ный раствор в метаноле) и перемешивали полученную смесь при комнатной температуре в течение 10 минут. К данной смеси добавляли воду и проводили экстракцию этилацетатом. Этилацетатный слой промывали насыщенным раствором соли, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=94:6→85:15). Очищенный продукт концентрировали при пониженном давлении, получая 250 мг белого аморфного твердого желаемого соединения.
1Н ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,7-2,3 (4H, м), 2,37 (6Н, с), 2,4-2,6 (3Н, м), 2,8-4,3 (2Н, м), 3,63 (3Н, т, J=10,7 Гц), 4,4-4,8 (1Н, м), 5,3-5,6 (1Н, м), 6,6-7,1 (2Н, м), 7,1-7,5 (5Н, м), 7,58 (1Н, с), 9,81 (1Н, с).
Пример 22

К раствору 500 мг (0,95 ммоль) соединения примера 17 в ацетонитриле (5 мл) прибавляли воду в количестве 0,5 мл, 0,5 мл четыреххлористого углерода, 0,5 мл триэтиламина и 0,115 мл (1,2 ммоль) диэтаноламина и перемешивали полученную смесь при комнатной температуре в течение 10 минут. К данной смеси добавляли воду и осуществляли экстракцию этилацетатом. Этилацетатный слой промывали насыщенным раствором соли, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=88:12→70:30). Очищенный продукт концентрировали при пониженном давлении, а остаток перекристаллизовывали из водного метанола, получая 250 мг белого порошкообразного желаемого соединения.
1Н ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,6-2,2 (4H,м), 2,37 (6Н, с), 3,0-3,2 (4Н, м), 3,5-3,7 (7Н, м), 2,8-4,3 (2Н, м), 4,1-4,4 (1Н, м), 5,3-5,7 (1Н, м), 6,7-7,1 (2Н, м), 7,1-7,5 (7Н, м), 7,5-7,7 (1Н, м), 9,80 (1Н, ушир).
Пример 23
Дифенилфосфит в количестве 3,8 мл (20 ммоль) прибавляли к раствору 3,0 г (6,7 ммоль) толваптана (соединение (2)) в пиридине (10 мл) и перемешивали полученную смесь при комнатной температуре в течение 1 часа. К данной смеси добавляли 2 мл воды и осуществляли перемешивание при комнатной температуре в течение 30 минут. Полученную реакционную смесь концентрировали при пониженном давлении, к остатку добавляли 1N хлористоводородную кислоту и проводили экстракцию этилацетатом. Этилацетатный слой дважды промывали насыщенным раствором соли, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=100:0→50:50). Очищенный продукт концентрировали при пониженном давлении. Остаток растворяли в воде, а нерастворимое вещество, выпавшее при добавлении 1N хлористоводородной кислоты, выделяли фильтрованием, а затем сушили, получая 0,83 г белого порошкообразного желаемого соединения.
Выход: 24%
1Н-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,7-2,2 (4H,м), 2,35 (3Н, с), 2,36 (3Н, с), 2,8-4,3 (2Н, м), 5,4-5,6 (1Н, м), 5,8 (0,5Н, ушир.), 6,7-7,4 (8Н, м), 7,47 (1Н, д, J=2,3 Гц)), 7,55 (1Н, с), 9,79 (1Н, ушир.).
Пример 24

Треххлористый фосфор в количестве 2,9 мл прибавляли к тетрагидрофурану (ТГФ) (29 мл) в токе азота. Полученную смесь охлаждали льдом и добавляли к ней 6,1 мл (44 ммоль) триэтиламина. Данную смесь охлаждали на бане лед-метанол. После этого к ней прибавляли по каплям раствор 10,0 г (22 ммоль) толваптана (соединение (2)) в ТГФ (120 мл) при внутренней температуре не выше -10°С и осуществляли перемешивание при той же температуре в течение 2 часов. К полученной реакционной смеси прибавляли по каплям 1N водный раствор гидроксида натрия в количестве 130 мл при внутренней температуре не выше 0°С, добавляли туда еще воды в количестве 200 мл и дважды промывали толуолом. Полученный водный раствор охлаждали на бане лед-метанол, добавляли к нему по каплям 1N HCl при внутренней температуре не выше 0°С и осуществляли экстракцию этилацетатом. Этилацетатный слой сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали, получая 6,8 г белого аморфного твердого желаемого соединения.
Выход: 60%
Пример 25

Дифенилфосфит в количестве 3,8 мл (20 ммоль) прибавляли к раствору 3,0 г (6,7 ммоль) толваптана (соединение (2)) в пиридине (10 мл) и перемешивали полученную смесь при комнатной температуре в течение 1 часа. К данной смеси добавляли 5,2 мл (66,6 ммоль) метилгликолята и осуществляли перемешивание при комнатной температуре в течение 12 часов. К реакционной смеси добавляли 50 мл воды и осуществляли экстракцию этилацетатом. Этилацетатный слой дважды промывали 1N хлористоводородной кислотой, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (н-гексан:этилацетат=50:50→0:100).
Очищенный продукт концентрировали при пониженном давлении, получая 0,79 г белого аморфного желаемого соединения.
Выход: 20%
1H-ЯМР (толуол-d8, 100°C) δ м.д.: 1,6-2,2 (4H, м), 2,51 (3H, с), 2,60 (3H, с), 3,2-4,4 (2H, м), 3,53 (3H, с), 4,43 (1H, с), 4,47 (1H, с), 5,87 (0,5H, с), 5,9-6,1 (1H, м), 6,6-6,8 (1H, м), 6,8-7,0 (2H, м), 7,0-7,4 (5H, м), 7,48 (1Н, с), 7,63 (1Н, с), 8,27 (0,5H, с).
Пример 26

К раствору 0,79 г (1,35 ммоль) соединения примера 25 в пиридине (7,9 мл) прибавляли воду в количестве 0,8 мл. К полученной смеси прибавляли 0,34 г (2,7 ммоль) йода при охлаждении льдом и осуществляли перемешивание при комнатной температуре в течение 1 часа. К реакционной смеси добавляли 1N хлористоводородную кислоту и проводили экстракцию этилацетатом. Этилацетатный слой промывали насыщенным раствором соли, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток растворяли в воде, а затем подвергали сублимационной сушке, получая 80 мг белого аморфного твердого желаемого соединения.
Выход: 9,9%
1Н ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,7-2,3 (4H,м), 2,35 (3Н, с), 2,36 (3Н, с), 2,8-4,3 (2Н, м), 4,49 (2Н, дд, J=1,7, 10,1 Гц), 5,4-5,6 (1Н, м), 6,7-7,1 (2Н, м), 7,1-7,5 (7Н, м), 7,54 (1Н, с), 9,79 (1Н, ушир.).
Пример 27

Толваптан (соединение (2)) в количестве 3,0 г (6,7 ммоль) прибавляли небольшими порциями к раствору 3,8 мл (20 ммоль) дифенилфосфита в пиридине (15 мл) и перемешивали полученную смесь при комнатной температуре в течение 0,5 часа. К данной смеси добавляли 2,8 мл (40 ммоль) 3-гидроксипропионитрила и перемешивали полученную смесь при комнатной температуре в течение 0,5 часа. К полученной реакционной смеси добавляли 1N хлористоводородную кислоту и проводили экстракцию этилацетатом. Этилацетатный слой промывали водой, сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=100:0→10:1). Очищенный продукт концентрировали при пониженном давлении, получая 2,8 г белого аморфного твердого желаемого соединения.
Выход: 75%
1H-ЯМР (толуол-d8, 100°C) δ м.д.: 1,4-2,0 (6H, м), 2,33 (3H, с), 2,40 (3H, с), 3,1-3,8 (4H, м), 5,40 (0,5H, д, J=3,1 Гц), 5,3-5,4 (1H, м), 6,5-6,7 (1H, м), 6,7-6,9 (1H, м), 6,9-7,2 (6H, м), 7,2-7,5 (2Н, м), 7,76 (0,5H, д, J=8,5 Гц).
Пример 28

К раствору 1,0 г (1,8 ммоль) соединения примера 27 в пиридине (10 мл) прибавляли серу в количестве 0,115 г (3,6 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 2 часов. К данной смеси добавляли 1N хлористоводородную кислоту и проводили экстракцию этилацетатом. Этилацетатный слой сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=100:0→85:15). Очищенный продукт концентрировали при пониженном давлении, получая 0,91 г белого аморфного твердого желаемого соединения.
Выход: 85%
1Н-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,6-1,9 (3H,м), 2,0-2,3 (1Н, м), 2,10 (3Н, м), 2,36 (6Н, с), 2,3-4,2 (2Н, м), 2,7-2,8 (2Н, м), 3,9-4,2 (2Н, м), 5,5-5,8 (1Н, м), 6,7-6,9 (1Н, м), 7,0-7,4 (7Н, м), 7,4-7,5 (1Н, м), 7,56 (1Н, с), 7,7-7,8 (0,3Н, м), 8,5-8,6 (м, 0,7Н), 9,76 (1Н, ушир.).
Пример 29

Соединение примера 28 в количестве 300 мг (0,5 ммоль) прибавляли к 5 мл 28%-ного водного аммиака и перемешивали полученную смесь при комнатной температуре в течение 3 дней. К данной смеси добавляли 1N хлористоводородную кислоту. Выпавший осадок выделяли фильтрованием, а затем сушили, получая 100 мг белого порошкообразного желаемого соединения.
Выход: 37%
1Н-ЯМР (пиридин-d5-D2O, 90°C) δ м.д.: 1,6-2,4 (4H, м), 2,43 (3Н, с), 2,53 (3Н, с), 2,8-4,3 (2Н, м), 5,1-5,4 (1Н, м), 6,8-7,3 (6Н, м), 7,4-7,7 (2Н, м), 7,7-8,1 (2Н, м).
Пример 30

К тетрагидрофурану (5 мл) прибавляли хлорокись фосфора в количестве 0,62 мл (6,6 ммоль) и 0,92 мл (6,6 ммоль) триэтиламина в токе азота. Полученную смесь охлаждали на бане лед-метанол. После этого к ней прибавляли по каплям раствор 1,0 г (2,2 ммоль) толваптана (соединение (2)) в ТГФ (10 мл) и осуществляли перемешивание при той же температуре в течение 30 минут. К данной смеси добавляли 2,8 мл (20 ммоль) триэтиламина и 1,1 мл (26,4 ммоль) метанола и осуществляли перемешивание в течение 30 минут. К полученной реакционной смеси добавляли воду и проводили экстракцию этилацетатом. Этилацетатный слой сушили над сульфатом натрия, а затем фильтровали, а фильтрат концентрировали. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат:метанол=100:0→80:20). Очищенный продукт концентрировали при пониженном давлении, а остаток перекристаллизовывали из водного метанола, получая 400 мг белого порошкообразного желаемого соединения.
Выход: 33%
1Н-ЯМР (ДМСО-d6, 100°C) δ м.д.: 1,7-2,2 (4H, м), 2,36 (6Н, с), 2,3-4,3 (2Н, м), 3,71 (2Н, дд, J=10,2, 11,1 Гц), 5,5-5,6 (1Н, м), 6,8-7,1 (2Н, м), 7,1-7,5 (7Н, м), 7,58 (1Н, с), 9,80 (1Н, ушир.).
Экспериментальный пример 1
Растворимость соединения (1b)
Избыток соединения (1b), полученного в примере 3, или 4, прибавляли к 0,1N буферу фосфата натрия (рН 5, рН 6, рН 7, рН 8, рН 9, или рН 10), 0,1 N Tris/HCl буферу (рН 8, или рН 9), 0,1N буферу гидрокарбоната натрия/HCl (рН 8) или 0,1 N буферу цитрата натрия (рН 8), а затем встряхивали при комнатной температуре в течение 16 дней. Если тестируемое соединение растворялось даже после того, как туда было добавлено от около 6 до около 8 мас./об.%, тестируемое соединение больше не добавляли.
Каждый раствор фильтровали через 0,45 мкм фильтр, а затем определяли растворимость соединения (1b) путем абсолютной калибровки в следующих условиях ВЭЖХ.
Условия ВЭЖХ
Детектирование: ультрафиолетовый абсорбционный фотометр (длина волны измерения: 254 нм)
Колонка: YMC (ODS) AM-302 (4,6 × 150 нм)
Температура колонки: постоянная температура приблизительно 25°С
Элюат: ацетонитрил/вода/фосфорная кислота=450/550/1
Скорость потока: 1 мл/мин
Объем инъекции: 10 мкл

Экспериментальный пример 2
Растворимость соли соединения (1)
В пробирку прибавляют подходящее количество тестируемого соединения и прибавляют к нему 2,5 мл воды. После встряхивания при 37°С в течение 30 минут смесь фильтруют через 0,45-мкм мембранный фильтр и аккуратно отвешивают 0,5 мл фильтрата. К нему добавляют подвижную фазу для получения ровно 50 мл, приготавливая тестовый раствор (коэффициент разбавления: 100-кратный). Аккуратно взвешивают приблизительно 5 мг достоверного образца в виде свободной формы и прибавляют к нему ацетонитрил для получения ровно 50 мл. Аккуратно взвешивают 2 мл данной жидкости и добавляют к ней подвижную фазу, чтобы получить ровно 20 мл, получая стандартный раствор (эквивалентный 10 мкг/мл). Тестируют 20 мкл тестового раствора и стандартного раствора методом жидкостной хроматографии в следующих условиях, получая площади пиков At и As для тестового раствора и стандартного раствора.
Концентрация (мкг/мл)=Ws/5 × 10 × At/As × 100 = Ws × At/As × 200
Ws: взвешенное количество достоверного образца (мг)
Условия эксперимента
Детектирование: ультрафиолетовый абсорбционный фотометр (длина волны измерения: 254 нм)
Колонка: TOSOH TSKgel ODS-80Ts (0,46 см × 15 см)
Температура колонки: постоянная температура приблизительно 40°С
Подвижная фаза: вода/ацетонитрил/трифторуксусная кислота=500/500/1
Скорость потока: 1 мл/мин

Экспериментальный пример 3
Растворимость толваптана
К буферу Бриттона-Робинсона (рН 2, рН 7 или рН 12) или очищенной воде прибавляли избыток толваптана, а затем встряхивали при 25°С±1°С в течение 4 часов. Каждый раствор фильтровали через фильтр, а затем методом ВЭЖХ проводили количественную оценку растворимости толваптана при абсолютной калибровке.
Экспериментальный пример 4
Концентрация толваптана в сыворотке крови у самок крыс после введения раствора соединения (1b) в хвостовую вену
Способ эксперимента
Получали раствор соединения (1b) (эквивалентный 1 мг толваптана на мл раствора).
Способ получения
Дигидрат дигидрофосфата натрия в количестве 79 мг и 5 г маннита растворяли примерно в 90 мл воды для инъекций. В полученный раствор добавляли раствор гидроксида натрия и получали раствор с рН 7. В данном растворе растворяли соединение (1b), эквивалентное 100 мг толваптана. Добавляли туда раствор гидроксида натрия и доводили рН до 7. К полученному раствору прибавляли растворитель для инъекций для доведения объема раствора до 100 мл и осуществляли стерильное фильтрование на 0,2-мкм фильтре, получая раствор соединения (1b) (эквивалентный 1 мг толваптана на мл раствора).
Данный раствор быстро вводили самкам крыс через хвостовую вену в такой дозе, чтобы на кг массы тела приходился 1 мг толваптана. Время от времени отбирали кровь через яремную вену при легкой анастезии диэтиловым эфиром и определяли концентрацию толваптана в сыворотке крови методом высокоскоростной жидкостной хроматографии (ВЭЖХ).
Результаты представлены на фигуре 1.
Толваптан первоначально детектировали через пять минут после внутривенного введения раствора соединения (1b) самкам крыс. Этот факт указывает на то, что соединение (1b) быстро гидролизуется в толваптан у крыс.
Экспериментальный пример 5
Концентрация толваптана в сыворотке крови у самок крыс после перорального введения раствора соединения (1b)
Способ эксперимента
Получали раствор соединения (1b) (эквивалентный 0,4 мг толваптана на мл раствора).
Способ получения
Гидрокарбонат натрия в количестве 1 г растворяли примерно в 400 мл воды для инъекций. К нему прибавляли раствор гидроксида натрия, чтобы довести рН до 9,0 и к полученному раствору добавляли воды, получая 500 мл 0,2%-ного раствора гидрокарбоната натрия. Примерно к 40 мл данного 0,2%-ного раствора гидрокарбоната натрия прибавляли и растворяли 1N раствор гидроксида натрия в количестве 89 мкл и соединение (1b), эквивалентное 20 мг толваптана. Добавляли в полученный раствор еще 0,2%-ного раствора гидрокарбоната натрия для доведения объема до 50 мл, получая таким образом раствор соединения (1b) (эквивалентный 0,4 мг толваптана на мл раствора). рН данного раствора составлял 9,1. В дальнейшем данный раствор называют «раствор А».
Высушенный методом распылительной сушки порошок толваптана, эквивалентный 60 мг толваптана, который получали по аналогии с примером 3 JP1999-21241-А, суспендировали в 50 мл воды для инъекций в фарфоровой ступке. Данную суспензию разбавляли трехкратным объемом воды для инъекций, получая суспензию высушенного методом распылительной сушки порошка, эквивалентную 0,4 мг толваптана на мл суспензии. В дальнейшем данную суспензию называют «суспензия В».
С целью изучения характеристик пероральной абсорбции раствора А и суспензии В осуществляли следующие эксперименты. В качестве подопытных животных использовали самок крыс Wistar (масса тела около 160 г), которых не кормили в течение 18 часов. Раствор А и суспензию В вводили методом принудительного перорального введения при помощи зонда для перорального введения при дозировке 2,5 мл/кг массы тела, получая 1 мг толваптана на кг массы тела. После введения дозы периодически отбирали пробы крови через яремную вену при легкой анастезии диэтиловым эфиром и определяли концентрацию толваптана в сыворотке крови методом UPLC-МС/МС (Waters).
Результаты приведены на фигуре 2 и в таблице 6. На фигуре 2 показаны профили зависимости концентрация в сыворотке-время для толваптана после перорального введения раствора А и суспензии В (n=4). В таблице 6 приведены средние значения фармакокинетических параметров (n=4). Данные параметры в таблице 6 имеют следующие значение.
AUC8ч: площадь под кривой концентрация в сыворотке-время до 8 часов после введения (нг·ч/мл)
AUC∞: площадь под кривой концентрация в сыворотке-время до бесконечного времени после введения (нг·ч/мл)
Смакс: максимальная концентрация в сыворотке (нг/мл)
Тмакс: время для достижения максимальной концентрации в сыворотке (ч)
В результате было доказано, что в случае раствора соединения (1b) (раствор А) для достижения максимальной концентрации в сыворотке требуется меньше времени, чем в случае суспензии высушенного методом распылительной сушки толваптана (суспензия В), и, кроме того, это приводит к большему максимуму концентрации в сыворотке (Смакс) и большим площадям под кривой концентрация в сыворотке-время (AUC8ч, AUC∞).
Из данных результатов видно, что при введении in vivo соединения настоящего изобретения, в частности, соединения (1b), абсорбция увеличивается даже больше, чем в случае стандартного улучшения абсорбции методом аморфизации, и, следовательно, повышается биодоступность толваптана.
Пример получения 1
Дигидрат дигидрофосфата натрия в количестве 79 мг и 5 г маннита растворяли примерно в 90 мл растворителя для инъекций. Добавляли в полученный раствор раствор гидроксида натрия до рН 7. К данному раствору прибавляли соединение (1b), эквивалентное 100 мг толваптана. Добавляли к нему раствор гидроксида натрия, доводя рН до 7. К полученному раствору добавляли растворитель для инъекций для получения 100 мл и осуществляли стерильное фильтрование на 0,2-мкм фильтре, получая препарат для инъекций настоящего изобретения, содержащий соединение (1b) (эквивалентное 1 мг толваптана на мл препарата для инъекций).
Пример получения 2
79 мг дигидрата дигидрофосфата натрия и 5 г маннита растворяли примерно в 90 мл растворителя для инъекций. Добавляли в полученный раствор раствор гидроксида натрия, получая раствор с рН 7,5. В данном растворе растворяли соединение (1b), эквивалентное 10 мг толваптана. К полученному раствору добавляли растворитель для инъекций для получения объема 100 мл и проводили стерильное фильтрование на 0,2-мкм фильтре, получая препарат для инъекций настоящего изобретения, содержащий соединение (1b) (эквивалентное 0,1 мг толваптана на мл препарата для инъекций).
Пример получения 3
Додекагидрат тринатрийфосфата в количестве 380 мг и 4 г маннита растворяли примерно в 90 мл растворителя для инъекций. В полученном растворе растворяли соединение (1b), эквивалентное 100 мг, 300 мг или 1000 мг толваптана. В случае растворения соединения (1b), эквивалентного 1000 мг толваптана, для улучшения растворимости добавляли раствор гидроксида натрия. рН каждого из полученных растворов доводили до 8-9 при помощи гидроксида натрия, или хлористоводородной кислоты и к полученному раствору добавляли растворитель для инъекций для получения объема 100 мл. Полученный раствор подвергали стерильному фильтрованию на 0,2-мкм фильтре, получая препараты для инъекций настоящего изобретения, содержащие соединение (1b) (эквивалентное 1 мг, 3 мг или 10 мг толваптана на мл препарата для инъекций).
Реферат
Изобретение относится к соединению бензоазепина, представленному следующей общей формулой (I): ! ! или к его соли, где R представляет собой атом водорода, гидрокси-группу, необязательно защищенную защитной группой, выбранной из низшей алкильной группы, фенил(низшей)алкильной группы, меркапто-группу или амино-группу, необязательно защищенную одной или двумя защитными группами, выбранными из низшей алкильной группы, необязательно содержащей гидрокси-группу, R1 представляет собой атом водорода или гидрокси-защитную группу, где защитная группа включает низшую алкильную группу, фенил(низшую)алкильную группу, циано низшую алкильную группу и низшую алкилоксикарбонил низшую алкильную группу, а Х представляет собой атом кислорода или атом серы. Изобретение также относится к фармацевтической композиции, обладающей антагонистической активностью к вазопрессину, на основе указанного соединения. Данное соединение бензоазепина обладают высокой растворимостью в воде и могут быть использованы в качестве сосудорасширяющего, гипотензивного, акваретического агента, ингибитора агрегации тромбоцитов или для лечения болезни поликистоза почек (PKD). 3 н. и 10 з.п. ф-лы, 2 ил., 6 табл.
Формула

или его соль,
в которой R представляет собой атом водорода, гидроксигруппу, необязательно защищенную защитной группой, выбранной из низшей алкильной группы, фенил(низшей)алкильной группы, меркаптогруппу или аминогруппу, необязательно защищенную одной или двумя защитными группами, выбранными из низшей алкильной группы, необязательно содержащей гидроксигруппу,
R1 представляет собой атом водорода или гидроксизащитную группу, где защитная группа включает низшую алкильную группу, фенил(низшую)алкильную группу, цианонизшую алкильную группу и низшую алкилоксикарбонил низшую алкильную группу, а
Х представляет собой атом кислорода, или атом серы.
Документы, цитированные в отчёте о поиске
Производные бензазепинон-n-уксусной кислоты, замещенные фосфоновой кислотой, способ их получения и лекарственные средства, содержащие эти соединения
Комментарии