Производные бензазепинон-n-уксусной кислоты, замещенные фосфоновой кислотой, способ их получения и лекарственные средства, содержащие эти соединения - RU2211219C2
Код документа: RU2211219C2
Чертежи



Описание
Настоящее изобретение касается новых производных бензазепинон-N-уксусной кислоты, которые замещены в положении 3 циколопентилкарбониламинным остатком, несущим в положении 1 остаток метилфосфоновой кислоты, их солей и биолабильных сложных эфиров, а также содержащих указанные соединения фармацевтических препаратов и способов получения этих соединений.
Из европейской патентной заявки, номер публикации 0733642, известны производные бензазепин-, бензоксазепин- и бензодиазепин-N-уксусной кислоты, оказывающие тормозящее действие на нейтральную эндопептидазу (NEP).
Задачей изобретения является создание новых NEP-ингибирующего действия, фармацевтических активных веществ с благоприятным профилем действия для лечения сердечной недостаточности и высокого кровяного давления.
Было установлено, что новые производные бензазепинон-N-уксусной кислоты, замещенные в положении 3 каркаса бензазепинона циклопентилкарбониламинным остатком, несущим в положении 1 остаток метилфосфоновой кислоты, согласно изобретению, обладают ценными эффективными для сердца фармакологическими свойствами и отличаются благоприятным профилем действия для лечения сердечно-сосудистых заболеваний, в частности сердечной недостаточности, отличающейся комбинацией ясно выраженного тормозящего действия на нейтральную эндо-пептидазу с тормозящим действием на фермент преобразования эндотелина (ЕСЕ) и хорошей совместимостью.
Предметом изобретения
являются новые
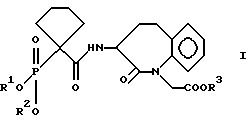
соединения общей формулы I
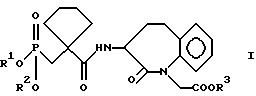
где R1 означает водород или группу, образующую биолабильный сложный эфир фосфоновой кислоты,
R2 означает водород или группу, образующую биолабильный сложный эфир фосфоновой кислоты,
R3 означает водород или группу, образующую биолабильный сложный эфир карбоновой кислоты,
а также физиологически переносимые соли кислот формулы I, способы получения этих соединений и содержащие указанные соединения лекарственные средства.
Соединения формулы I представляют собой, в случае необходимости, кислотные производные, содержащие группы карбоновой и фосфоновой кислот, этерифицированные образующими биолабильные сложные эфиры группами. Биолабильные сложные эфиры формулы I являются пролекарствами со свободными кислотами. В зависимости от формы применения предпочтительными являются биолабильные сложные эфиры или кислоты, причем последние пригодны, в частности, для внутривенного введения.
В качестве групп R1 и R2, образующих биолабильные сложные эфиры фосфоновой кислоты, пригодны группы, способные отщепляться в физиологических условиях in vivo с выделением соответствующих функций фосфоновой кислоты. Так, например, для этого пригодны низшие алкильные группы, в случае необходимости, С2-С6-алканоилоксиметильные группы или фенильные, или фенил-низший-алкилгруппы, фенильное кольцо которых, при необходимости, замещено одно- или многократно низшим алкилом, низшим алкоксильным радикалом или связанной через два соседних атома углерода низшей алкиленовой цепочкой. Если образующая биолабильный сложный эфир группа R1 и/или R2 означает или содержит низший алкил, то последний может быть разветвленным или неразветвленным и содержать от 1 до 4 атомов углерода. В том случае, когда R1 и/или R2 представляет собой, в случае необходимости, замещенную алканоилоксиметиловую группу, то последняя может содержать в себе предпочтительно разветвленную алканоилоксигруппу с 2-6, предпочтительно 3-5, атомами углерода и может означать, например, пивалоилоксиметиловый остаток (трет-бутилкарбонилоксиметиловый остаток). Если R1 и/или R2 представляют собой, при необходимости, замещенную фенил-низший-алкилгруппу, то эта группа может содержать алкиленовую цепь с 1-3, предпочтительно 1, атомами углерода. Если фенильное кольцо замещено цепью низших алкилов, то последняя может содержать 3-4, предпочтительно 3, атома углерода, замещенное фенильное кольцо является, в частности, инданилом.
В качестве групп R3, образующих биолабильные сложные эфиры карбоновой кислоты, пригодны группы, способные отщепляться в физиологических условиях in vivo с выделением карбоновой кислоты. Так, например, для этого пригодны низшие алкильные группы, при необходимости, фенильные или фенил-низший-алкилгруппы, в случае необходимости, замещенные в фенильном кольце одно- или многократно низшим алкилом или низшим алкоксильным радикалом или связанной через два соседних углеродных атома цепью низших алкиленов, замещенные фенил- или фенил-низший-алкилгруппы, замещенные в диоксолановом кольце низшим алкилом диоксоланилметиловые группы или, при необходимости, замещенные в оксиметиловой группе низшим алкилом C2-C6-алканоилоксиметиловые группы. Если образующая биолабильный сложный эфир группа R3 означает или содержит низший алкил, то последний может быть разветвленным или неразветвленным и содержать от 1 до 4 атомов углерода. Если образующая биолабильный сложный эфир группа представляют собой, при необходимости, замещенную фенил-низший-алкилгруппу, то эта группа может содержать алкиленовую цепь с 1-3, предпочтительно 1, атомами углерода и означает преимущественно бензил.
Если фенильное кольцо замещено цепью низших алкиленов, то последняя может содержать 3-4, предпочтительно 3, атома углерода. Если R3 представляет собой замещенную, при необходимости, алканоилоксиметильную группу, то последняя может содержать преимущественно разветвленную алканоилоксигруппу с 2-6, предпочтительно 3-5, атомами углерода и может являться, например, пивалоилоксиметиловым остатком.
Согласно изобретению новые соединения формулы I и их соли получают известным способом, при котором
а) для
получения соединений общей формулы
IV

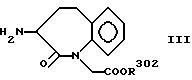
где R101 и R201, независимо друг от друга, означают водород или защитную группу фосфоновой кислоты, R302 означает защитную группу карбоновой кислоты,
соединения общей формулы II

где R101 и R201 имеют приведенные выше значения,
вводят во взаимодействие с соединениями общей формулы III

где R302 имеет указанное выше значение,
и в случае, если R101 и/или R201 означают водород, переводят свободную функцию (свободные функции) фосфоновой кислоты, при необходимости, путем этерификации с соединением общей формулы Va и/или Vb
R110-Y (Va), R210-Y (Vb),
где R110 и R210 обозначают соответственно группу, образующую биолабильный сложный эфир фосфоновой кислоты, Y означает гидроксильный радикал или отщепляемую летучую группу,
в биолабильные эфирные группы фосфоновой кислоты,
б) если в соединениях формулы IV защитные группы R101, R201 и/или R302 не являются желаемыми группами, образующими биолабильный сложный эфир, то их отщепляют одновременно или в отдельности друг за другом в любой последовательности и, при желании, переводят соответствующие высвободившиеся кислотные функции в биолабильные эфирные группы, этерифицируя при этом свободные функции фосфоновой кислоты соединением формулы Va или Vb и/или свободные функции карбоновой кислоты с соединением общей формулы Vc:
R310-Y (Vc),
где R310 означает группу, образующую биолабильный сложный эфир карбоновой кислоты, Y имеет указанное выше значение,
и, при необходимости, кислоты формулы I переводят в их физиологически переносимые соли или соли кислот формулы I - в свободные соединения.
В качестве физиологически переносимых солей кислот формулы I могут применяться соответственно их соли щелочных металлов, щелочноземельных металлов и аммония, например соли натрия, калия или кальция, или соли с физиологически совместимыми фармакологически нейтральными органическими аминами, такими, как, например, диэтиламин, трет-бутиламин или фенил-низший-алкиламины, такие, как α -метилбензиламин.
В качестве защитных групп R101 и R201 фосфоновой кислоты можно выбирать для защиты функций фосфоновой кислоты обычные защитные группы, которые затем снова отщепляются известными само по себе способами. В качестве защитных групп карбоновой кислоты R302 могут выбираться для защиты функций карбоновой кислоты обычные защитные группы, которые затем могут отщепляться известными само по себе способами. Приемлемые защитные группы для карбоновых кислот известны, например, из McOmie, "Protective Groups in Organic Chemistry" (Защитные группы в органической химии), Plenum Press und Green, Wuts, "Protective Groups in Organic Syntesis" (Защитные группы в органическом синтезе), Wiley Interscience Publication. Приемлемые защитные группы для фосфоновых кислот известны, например, из Houben, Weyl "Methoden der Organischen Chemie" (Методы органической химии), изд-во G. Thieme Verlag, г. Штуттгарт, Нью-Йорк, 1982 г. , стр. 313-341, а также из М. Kluba, A. Zwierak "Synthesis", 1978 г., стр. 134-137, и из McOmie, "Protective Groups in Organic Chemistry", Plenum Press. В качестве кислотных защитных групп могут также применяться группы, образующие биолабильный сложный эфир. Соединения формулы IV, полученные в результате взаимодействия между соединениями формул II и III, представляют собой уже сложные эфиры формулы I согласно изобретению.
Согласно изобретению в качестве защитных групп R101 и R201 фосфоновой кислоты пригодными являются такие группы, которые соответствующими методами, независимо друг от друга и независимо от вероятного присутствия защитной группы R302 карбоновой кислоты в молекуле, могут быть отщеплены или селективно введены. Защитные группы фосфоновой кислоты могут легко отщепляться посредством триметилсилилбромида селективно в присутствии защитных групп карбоновой кислоты. В качестве примеров отщепляемых в различных условиях защитных групп фосфоновой кислоты, которые могут выступать и в качестве групп, образующих биолабильные сложные эфиры фосфоновой кислоты, следует назвать следующие: неразветвленные низшие алкилгруппы, такие, как этил, которые могут легко отщепляться, например, кислотами, такими, как трифторуксусная кислота, причем в случае, если обе функции фосфоновой кислоты этерифицированы группами низших неразветвленных алкилов, то в условиях применения основания может отщепляться только одна из этих алкильных групп; разветвленные низшие алкилы, такие как трет-бутил, которые могут быть легко отщеплены в условиях применения кислоты, например трифторуксусной кислоты; замещенные, при необходимости, в фенильном кольце фенилметильные группы, такие, как бензил, способные легко отщепляться при деструктивной гидрогенизации; алканоилоксиметиловые группы, такие, как пивалоилоксиметил, которые легко могут отщепляться, например, кислотами, такими, как трифторуксусная кислота; фенилметиловые группы, такие, как п-метоксибензил, которые замещены в фенильном кольце одно- или многократно низшим алкоксильным радикалом и которые в условиях окисления, например, под действием 2,3-дихлор-5,6-дициан-1, 4-бензохинона (DDQ) или нитрита аммония-церия (CAN) могут отщепляться относительно легко.
В качестве защитных групп R302 карбоновой кислоты пригодными являются такие группы, которые независимо от вероятного присутствия защитных групп фосфоновой кислоты в молекуле могут быть отщеплены или селективно введены. В качестве примеров отщепляемых в различных условиях защитных групп карбоновой кислоты, которые могут выступать и в качестве групп, образующих биолабильные сложные эфиры карбоновой кислоты, следует назвать следующие: неразветвленные низшие алкилы, такие, как этил, которые могут относительно легко отщепляться в условиях применения основания; разветвленные низшие алкилы, такие, как трет-бутил, которые могут быть легко отщеплены кислотами, такими, как трифторуксусная кислота; замещенные, при необходимости, в фенильном кольце фенилметильные группы, такие, как бензил, способные легко отщепляться при деструктивной гидрогенизации или в условиях применения основания; фенилметильные группы, такие, как п-метоксибензил, которые замещены в фенильном кольце одно- или многократно низшим алкоксильным радикалом и которые в условиях окисления, например, под воздействием DDQ или CAN могут отщепляться относительно легко.
Соединения формулы I содержат хиральный атом углерода, а именно углеродный атом в положении 3 структуры бензазепина, несущий амидную боковую цепь. Следовательно, соединения могут присутствовать в двух оптически активных стереоизомерных формах или в виде рацемической смеси. Настоящее изобретение включает в себя как рацемические смеси, так и чистоизомерные соединения формулы I. Если в соединениях формулы I R1 и R2 не означают водород и соответственно имеют разные значения, то и атом фосфора в группе фосфоновой кислоты может быть хиральным. Образуемые хиральными атомами фосфора изомерные смеси и чистоизомерные соединения формулы I также являются предметом данного изобретения.
Взаимодействие кислот формулы II с аминами формулы III с образованием амидов формулы IV может проводиться методами, обычными при образовании амидных группировок путем аминоацилирования. В качестве средств ацилирования могут применяться карбоновые кислоты формулы II или их реакционноспособные производные. Реакционноспособными производными могут служить, в частности, смешанные ангидриды кислоты и галогениды кислоты. Так, например, могут применяться хлорангидриды или бромангидриды кислот формулы II или смешанные сложные эфиры кислот формулы II вместе с органическими сульфокислотами, например низкоалкановыми сульфокислотами, замещенными, при необходимости, галогенами, такими, как метансульфокислота или трифторметансульфокислота, или вместе с ароматическими сульфокислотами, как, например, бензолсульфокислотами, или вместе с замещенными низшим алкилом или галогенами бензолсульфокислотами, например толуолсульфокислотами или бромбензолсульфокислотами. Ацилирование может протекать в инертном в реакционных условиях органическом растворителе при температуре от -20oС до комнатной. В качестве растворителей пригодны галогенированные углеводороды, такие, как дихлорметан, или ароматические углеводороды, такие, как бензол или толуол, или циклические эфиры, такие, как тетрагидрофуран (THF) или диоксан, или смеси этих растворителей.
Целесообразно проводить ацилирование, в частности, если в качестве ацилирующего средства применяется смешанный ангидрид кислот формулы II вместе с одной из сульфокислот, в присутствии реактива, способного связывать кислоту. В качестве связывающих кислоту веществ пригодны, например, растворимые в реакционной смеси органические основания, такие, как третичные азотные основания, например трет-низкоалкильные амины и пиридины, такие, как, например, триэтиламин, трипропиламин, N-метилморфолин, пиридин, 4-диметиламинопиридин, 4-диэтиламинопиридин или 4-пирролидинопиридин. Применяемые в избытке органические основания одновременно могут служить и растворителями.
В случае, когда в качестве средства ацилирования используются сами кислоты формулы II, то взаимодействие аминных соединений формулы III с карбоновыми кислотами формулы II целесообразно проводить в присутствии связывающего реагента, известного из химии пептидов в качестве пригодного для образования амидов средства. В качестве примера связующих реагентов, способствующих образованию амидов со свободными кислотами в результате того, что они реагируют с кислотой в естественных условиях с образованием реакционноспособного производного кислоты, следует, в частности, назвать алкилкарбодиимиды, например циклоалкилкарбодиимиды, такие, как дициклогексилкарбодиимиды или N-(3-диметиламинопропил)-N'-этилкарбодиимид, карбонилдиимидазол и соли N-низкоалкильного-2-галогенпиридиния, в частности галогениды или толуолсульфонаты. Взаимодействие в присутствии связующего реагента целесообразно проводить при температуре от -30 до +50oС в растворителях, таких, как галогенированные углеводороды и/или ароматические растворители и, при необходимости, в присутствии описанного выше, связывающего кислоту амина.
Из соединений формулы IV, полученных взаимодействием между соединениями формулы II и соединениями формулы III, можно отщепить известным способом защитные группы R101, R201 и R302 при условии, что они не являются желаемыми группами, образующими биолабильный сложный эфир.
В том случае, когда требуется получить соединения формулы I, в которых R1, R2 и R3 означают идентичные группы, образующие биолабильный сложный эфир, целесообразно выбирать идентичные защитные группы в исходных соединениях формулы II и в исходных соединениях формулы III. При этом целесообарэно выбирать защитные группы, которые являются одновременно группами, образующими биолабильный сложный эфир. Если требуется получить свободные кислоты формулы I, в которых R1, R2 и R3 означают соответственно водород, то в качестве защитных групп R101, R201 и R302 могут выбираться, при прочих равных условиях, преимущественно в условиях деструктивной гидрогенизации, отщепляемые группы. Например, для R101, R201 и R302 могут выбираться бензиловые группы, которые в условиях каталитической гидрогенизации могут одновременно расщепляться с образованием свободных кислотных групп. В качестве катализаторов при каталитическом гидрировании могут использоваться, например, драгоценные металлы, такие, как палладий на носителе из активированного угля. Реакция может проводиться в среде инертного в условиях реакции растворителя, например низшего спирта, такого, как этанол, или низшего алкильного сложного эфира, такого, как сложный этиловый эфир уксусной кислоты, или в смесях этих растворителей. Целесообразно проводить каталитическое гидрирование при давлении водорода от 2 до 6 бар и при комнатной температуре.
В случае, когда требуется этерификация свободных групп фосфоновой кислоты и/или свободных групп карбоновой кислоты в соединениях формулы I, то для этого свободные группы фосфоновой кислоты соединений формулы I вводят во взаимодействие с соединениями формулы Va или Vb известным само по себе способом. Свободные группы карбоновой кислоты в соединениях формулы I могут известным способом вводиться во взаимодействие с соединениями формулы Vc. В качестве летучих групп Y в соединениях формул Va, Vb и Vc пригодны, например, галогены, в частности хлор или бром, или остатки низших алкановых сульфокислот, как, например, трифторметансульфонилоксильный остаток, или ароматических сульфокислот, таких, как бензолсульфокислоты, или бензолсульфоки слот, замещенных низшим алкилом или галогеном, таких, как толуолсульфокислоты.
В том случае, когда требуется получить соединения формулы I, в которых R1и R2 имеют одинаковое значение, но отличающееся от R3, целесообразно использовать исходные соединения формулы II, в которых R101 и R201 имеют идентичные значения, а также исходные соединения формулы III, в которых R302 имеет значение, отличающееся от R101 и R201. Так, например, при гидрогенолитических условиях могут выбираться устойчивые защитные группы R101 и R201 фосфоновой кислоты, такие, как низший алкил, предпочтительно этил. Одновременно в качестве защитной группы карбоновой кислоты R302 может применяться группа, отщепляемая при гидрогенолитических условиях, такая, как бензиловая группа. В этом случае в условиях каталитического гидрирования будет отщепляться от полученных соединений формулы IV с получением свободной карбоновой кислоты только бензиловая группа R302, тогда, как этиловые группы R101 и R201 останутся сохранными. При желании в заключение можно этерифицировать свободную карбоновую кислоту с соединением формулы Vc. Точно также в соединениях формулы I, где защитные группы R101 и R201 фосфоновой кислоты при гидрогенолитических условиях означают стабильные группы, такие, как группы низших алкилов, преимущественно этил, и R302 означает группу, отщепляемую гидрогенолитически, такую, как бензиловая группа, могут отщепляться в кислых условиях сначала этиловые группы R101, R201, при этом сохраняется бензиловая группа R302. При желании в заключение можно этерифицировать свободные группы фосфоновой кислоты соединениями формулы Va и Vb, например пивалоилоксиметилхлоридом. После этого бензиловую группу R302, отщепляемую при гидрогенолитических условиях, можно отщепить каталитическим восстановлением с помощью водорода в известных условиях с целью получения соединений формулы I, в которой R3 означает водород.
Если требуется получить соединения формулы I, где R1 и R2 имеют разные значения, то целесообразно использовать исходные соединения формулы II, где R101 и R201 имеют разные значения. Например, исходными соединениями могут быть выбраны соединения формулы II, где R101 означает водород, R201 - стабильную защитную группу фосфоновой кислоты в гидрогенолитических условиях. Например, R201 может означать низший алкил, предпочтительно этил. При желании полученные соединения формулы I, в которой R101 означает водород, могут быть затем введены во взаимодействие с соответствующими соединениями формулы Va с целью получения соединений формулы I, где R1 и R2 означают различные группы, образующие биолабильный сложный эфир. Исходные соединения формулы II, в которой R101 означает водород, могут быть получены, например, из соединений формулы II, в которой R101 означает группу, отщепляемую при гидрогенолитических условиях, такую, как бензил, путем каталитического гидрирования в известных условиях.
При описанных выше реакциях взаимодействия хиральные углеродные атомы не претерпевают изменений в исходных соединениях формулы III, так что в зависимости от вида исходных соединений возможно получать чисто изомерные соединения формулы I или изомерные смеси. Для получения стереохимически единых соединений формулы I целесообразно вводить во взаимодействие стереохимически единые соединения формулы II со стереохимически едиными соединениями формулы III. В случае, если соединения формулы II не содержат хирального атома фосфора, вступают во взаимодействие с рацемическим соединением формулы III, то получают смесь, состоящую из двух энантиомеров соединения формулы I. При желании смесь энатиомеров можно разделить известным способом, например, хроматографией и на хиральных разделительных материалах или взаимодействием свободной карбоновой кислоты формулы I с соответствующими оптически активными основаниями, например (-)-α-метилбензиламином, и при последующем разделении оптических антиподов фракционированной кристаллизацией полученных солей.
Исходные соединения формулы II можно получать известными способами.
Таким образом можно
получать, например,
соединения формулы II, для чего соединения общей формулы VI

где R102 и R202 означают соответственно защитные группы фосфоновой кислоты, Y имеет указанное выше значение,
вводят во взаимодействие с циклопентанкарбоновой кислотой формулы VII
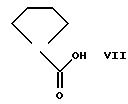
после чего при желании снова отщепляют защитные группы R102 и/или R202, пользуясь известным способом. Например, могут быть использованы соединения формулы VI, в которой Y означает остаток низшей алкансульфокислоты, предпочтительно трифторме-тансульфонилоксильный остаток.
Реакция может проводиться известным образом в условиях нуклеофильного замещения в среде инертного в условиях реакции органического растворителя в результате взаимодействия циклопентанкарбоновой кислоты с сильным основанием, способным образовать дианион циклопентанкарбоновой кислоты, с последующим взаимодействием с производным эфира фосфоновой кислоты формулы VI. В качестве растворителей пригодны, например, диалкиловые эфиры с открытой цепью, такие, как диэтиловый эфир или циклические эфиры, такие, как тетрагидрофуран (THF). В качестве сильных оснований пригодны, например, ненуклеофильные органические амиды щелочных металлов, такие, как литийдиизопропиламид (LDA). Целесообразно подвергать взаимодействию циклопентанкарбоновую кислоту в THF с двумя эквивалентными количествами LDA, затем реакционную смесь дополнительно вводить во взаимодействие с соединением формулы VI. Температура реакции может составлять от -70 до 0oС.
Соединения формулы VI можно получать известным само по себе способом, например взаимодействием диэфиров фосфоновой кислоты общей формулы VIII

где R102 и R202 имеют указанные выше значения,
с источником формальдегида, например с параформальдегидом. Целесообразно проводить реакцию без растворителя, но при участии растворимых в реакционной смеси оснований. В качестве оснований могут применяться ненуклеофильные основания, которые были описаны выше и предназначены для взаимодействия соединений формулы II с соединениями формулы III. Целесообразно, чтобы реакция проводилась при температуре от 50 до 130oС, предпочтительно от 80 до 120oС. Полученные соединения формулы VI, в которой Y означает гидроксильный радикал, при желании можно перевести известным способом в соединения формулы VI, в которой Y будет означать летучую отщепляемую группу.
Соединения формулы VIII известны или могут быть получены известными способами. Так, например, можно получить производные фосфоновой кислоты формулы VIII, этерифицированныe двумя разными биолабильными группами, для чего от диэфиров фосфоновой кислоты общей формулы VIII, в которой R101 и R201 означают одинаковую группу, например низший алкил, отщепляют под действием основания, такого, как гидроокись щелочного металла, например гидроокись натрия, сначала одну из двух групп сложного эфира, и полученный сложный моноэфир или его соль вводят затем во взаимодействие с соответствующим соединением формулы Va или Vb. Для ускорения хода реакции можно вводить соответствующие катализаторы, такие, как соли тетра-низший-алкиламмония, например, гидроокись тетрабутиламмония. Целесообразно добавлять в реакционную смесь соответствующие галогениды щелочных металлов, такие, как йодиды щелочных металлов, например йодид натрия, для ускорения процесса реакции. Реакция может проводиться в диполярноапротическом растворителе, таком, как низший алкильный цианид, например ацетонитрил, в низшем алифатическом эфире, таком, как диэтилэфир, THF или диоксан, в диметилформамиде (DMF), в диметилсульфоокиси (DMSO) или в смесях этих растворителей. Необходимые для этого температуры лежат в диапазоне от 0 до 80oС, предпочтительно от 5 до 40oС.
Соединения формулы III известны из европейской патентной заявки, номер публикации 0733642, и могут быть получены раскрытыми в ней методами.
Соединения формулы I и их фармакологически приемлемые соли характеризуются интересными фармакологическими свойствами. В частности, вещества замедляют действие фермента преобразования эндотелина (ЕСЕ) и нейтральной эндопептидазы (NEP) и обладают, следовательно, особо благоприятным профилем действия для лечения сердечной недостаточности.
При сердечной недостаточности вызванное болезнью снижение его выбрасывающей способности приводит к рефлекторному увеличению сопротивления периферийных сосудов. В результате миокарду приходится преодолевать дополнительную повышенную нагрузку. Это создает замкнутый круг, приводит к повышенной нагрузке на сердце и дополнительно ухудшает состояние. Увеличение сопротивления периферийных сосудов вызывается в числе прочего вазоактивным пептидом эндотелином. Эндотелин является сильнейшим аутогенным, известным в настоящее время сосудосужающим веществом и образуется из предстадии биг-эндотелина под действием фермента преобразования эндотелина (ЕСЕ).
При заболеваниях, связанных с сердечной недостаточностью, вследствие пониженной выбрасывающей способности сердца и повышения сопротивления периферийных сосудов происходят явления ретроградного застоя крови в малом круге кровообращения и в самом сердце. В результате происходит увеличение напряжения стенки миокарда в зоне предсердия и камер. При такой ситуации сердце функционирует как эндокринный орган и выделяет в числе прочего также пептид ANP (атриальный пептид натрийуреза) в кровеносное русло. Благодаря его выраженной активности в отношении расширения сосудов, натрийуреза и диуреза ANP вызывают уменьшение как сопротивления периферийных сосудов, так и циркулирующего объема крови. В результате происходит ярко выраженное снижение начальной и дополнительной нагрузок. Это представляет собой эндогенный механизм защиты сердца. Такой положительный эндогенный механизм ограничен тем, что ANP имеет лишь очень короткий период полураспада внутри плазмы. Причиной этого является очень быстрое разложение гормона нейтральной эндопептидазой (NEP).
Соединения согласно изобретению препятствуют благодаря замедлению активности ЕСЕ возникновению эндотелина и противодействуют таким образом увеличению сопротивления периферийных сосудов, что имеет своим следствием разгрузку сердечной мышцы. Вещества согласно изобретению вызывают, кроме того, в результате замедления активности NEP повышение уровня ANP и увеличивают длительность его действия. Это приводит к усилению действия, обеспечивающего ANP, эндогенного кардиозащитного механизма. В частности, вещества обладают высокой эффективностью относительно усиления диуретически/натрийуретической ANP-индуцирующей активности.
Нейтральная эндопептидаза (NEP) участвует не только в разложении атриального пептида натрийурезы (ANP), но также и в разрушении эндотелина. Отсюда следует, что одно только подавление нейтральной эндопептидазы (NEP) привело бы наряду с требуемым повышением уровня атриального пептида натрийуреза (ANP) и к нежелательному повышению уровня эндотелина. По этой причине следует считать особенно оптимальным смешанный профиль, заключающийся в подавлении фермента преобразования эндотелина (ЕСЕ) и нейтральной эндопептидазы, так как препятствуется разрушению атриального пептида натрийуреза/диуреза (блокада нейтральной пептидазы), а также одновременно замедляется образование эндотелина (подавление фермента преобразования эндотелина). Тем самым более уже не проявляется отрицательный сопутствующий эффект чистых NEP-ингибиторов нейтральной эндопептидазы (повышение уровня эндотелина).
1.Определение минимальной токсической дозы.
Группам, каждая из 10 крыс, с весом тела 250 г (в возрасте от 5 до 6 недель) внутривенно вводили тестируемые вещества в максимальной дозе 250 мг/кг (растворены в 0,1 н. водном растворе NaOH, pH 7,1). За животными внимательно наблюдали с момента введения веществ в течение 5 часов на проявление клинических признаков токсичности. Кроме того, на протяжении одной недели за ними наблюдали дважды в день. По истечении недели производили полное вскрытие каждого животного в отдельности и макроскопически исследовали все органы. Если отмечались гибель или сильные токсические симптомы, то последующим крысам вводили существенно меньшие дозы до тех пор, пока не исчезали симптомы токсичности. Наименьшая доза, при которой происходила гибель или проявлялись сильные симптомы токсичности, определяли как минимальную токсическую дозу. Испытуемое вещество, полученное в примере 2 и введенное внутривенно в количестве 215 мг/кг, не обнаружило значительных признаков токсичности.
2. Исследование веществ in vitro на тормозящее действие NEP - нейтральной эндопептидазы.
Для подтверждения тормозящего действия веществ согласно изобретению на нейтральную эндопептидазу (NEP) исследовали путем стандартного теста in vitro тормозящее действие веществ на гидролитическое разложение метионин-энкефалина (мет-энкефалин) путем воздействия ферментативной активности нейтральной эндопептидазы. В качестве единицы тормозящей эффективности веществ при этом была определена их величина IC50. Величиной IC50 тестируемого вещества с эффектом торможения фермента является концентрация тестируемого вещества, при которой блокируются 50% ферментативной активности нейтральной эндопептидазы (NEP).
Проведение теста
Для проведения теста были приготовлены пробы по 100 мкл разных
инкубационных растворов каждая с содержанием 10 нано-грамм очищенной
нейтральной эндопептидазы (Е.С. 3.4.24.11) и соответственно различные количества тестируемого вещества, а также 20 мкМ субстрата
(мет-энкефалина) и 50 мМ трис-буфера
(трис(гидроксиметил)аминометан/HCl, рН 7,4).
По каждому тестируемому веществу было приготовлено 6 разных инкубационных растворов с 3 разными концентрациями этого вещества для двукратного определения.
При каждом тестировании соответственно обрабатывали двукратно и контрольные инкубационные растворы, во-первых, проводился контроль за ферментом без содержания тестируемого вещества, и, во-вторых, контроль за субстратом, в котором не содержались ни фермент, ни тестируемое вещество.
Инкубационные растворы инкубировали в течение 30 минут при 37oС на встряхиваемой водяной бане. Ферментную реакцию запускали через 15 минут добавкой субстрата (мет-энкефалина) и прекращали в конце инкубационного периода нагревом в течение 5 минут при 95oС. Затем инкубационный раствор центрифугировали в течение 3 минут при 12000 х g и в надосадочной жидкости определяли концентрацию непрореагировавшего субстрата и гидролизных продуктов, образовавшихся в результате ферментной реакции. Для этого производили разделение проб надосадочной жидкости посредством жидкостной хроматографии высокого давления (HPLC) на гидрофобном силикагеле и продукты ферментативной реакции и непрореагировавший субстрат определяли фотометрически при длине волны 205 нм. При разделении жидкостной хроматографией высокого давления применялась разделительная колонка (4,6 х 125 мм), содержавшая в себе разделительный материал с обращенной фазой Nucleosin® С 18,5 мкм. Поток растворителя составил 1,0 мл/мин, колонку нагревали до 40oС. Текучим средством А служили 5 мМ Н3РO4, рН 2,5, текучим средством В - ацетонитрил + 1% 5 мМ Н3 РO4, рН 2,5.
Исходя из замеренной концентрации гидролизных продуктов и непрореагировавшего субстрата в разных пробах, определяли известным способом показатель IC50 для тестируемых веществ. Тестируемое вещество, полученное в примере 2, при данном тесте имело показатель IC50 подавления нейтральной эндопептидазы 1,7 нМ и, следовательно, зарекомендавало себя как высокоэффективный ингибитор нейтральной эндопептидазы.
3. Определение in vivo влияния веществ на диурез/натрийурез в крысах с объемной нагрузкой.
Активность in vivo исследовали на крысе с объемной нагрузкой. При этом эксперименте применением изотонического раствора хлорида натрия вызывали высокое давление наполнения сердца, в результате чего выделялся атриальный пептид натрийуреза и происходил диурез/натрийуреза.
Проведение теста
Опыты проводились на крысах-самцах Wistar весом от 200 до 400 г. При неврологической анальгезии
(Fentanyl; Hypnorm®, изготовитель фирма Janssen) катетер вводили в правую бедренную вену для фонового вливания и объемной нагрузки изотоническим раствором хлорида натрия. После
вскрытия брюшной полости вводили
второй катетер в пузырь и перевязывали уретру, что позволяло замерить объем мочи, натрийурез и калийурез.
Брюшную полость снова закрывали и животным вводили раствор хлорида натрия (0, 5 мл/100 г веса тела) на протяжении всего эксперимента, длившегося 2 часа. По истечении 30 минут, необходимых для приведения в равновесное состояние, на стадии, предшествующей вводу тестируемого вещества, проводили трехкратный сбор мочи через каждые 10 минут. Эти предварительные данные (данные о "предлекарстве") определяли с целью проверки того, что у подопытных животных происходит непрерывный отток мочи.
Затем растворы с содержанием тестируемых веществ внутривенно (инъекция в бедренную вену) или орально (с помощью желудочного зонда) вводили группам из 10 крыс каждая. При обоих видах применения одна контрольная группа животных получала только ложные растворы, не содержавшие в себе активного начала. Через 5 минут после внутривенного применения или через 120 минут после орального введения веществ, крыс нагружали повышенным объемом раствора хлорида натрия путем внутривенного введения (2 мл/100 г веса тела через 2 мин) и собирали мочу через 60 минут. Определяли количество образовавшейся за этот период мочи и замеряли содержание в ней натрия и калия. Образовавшееся количество мочи при объемной нагрузке свидетельствовало об увеличении выделения против предварительных данных.
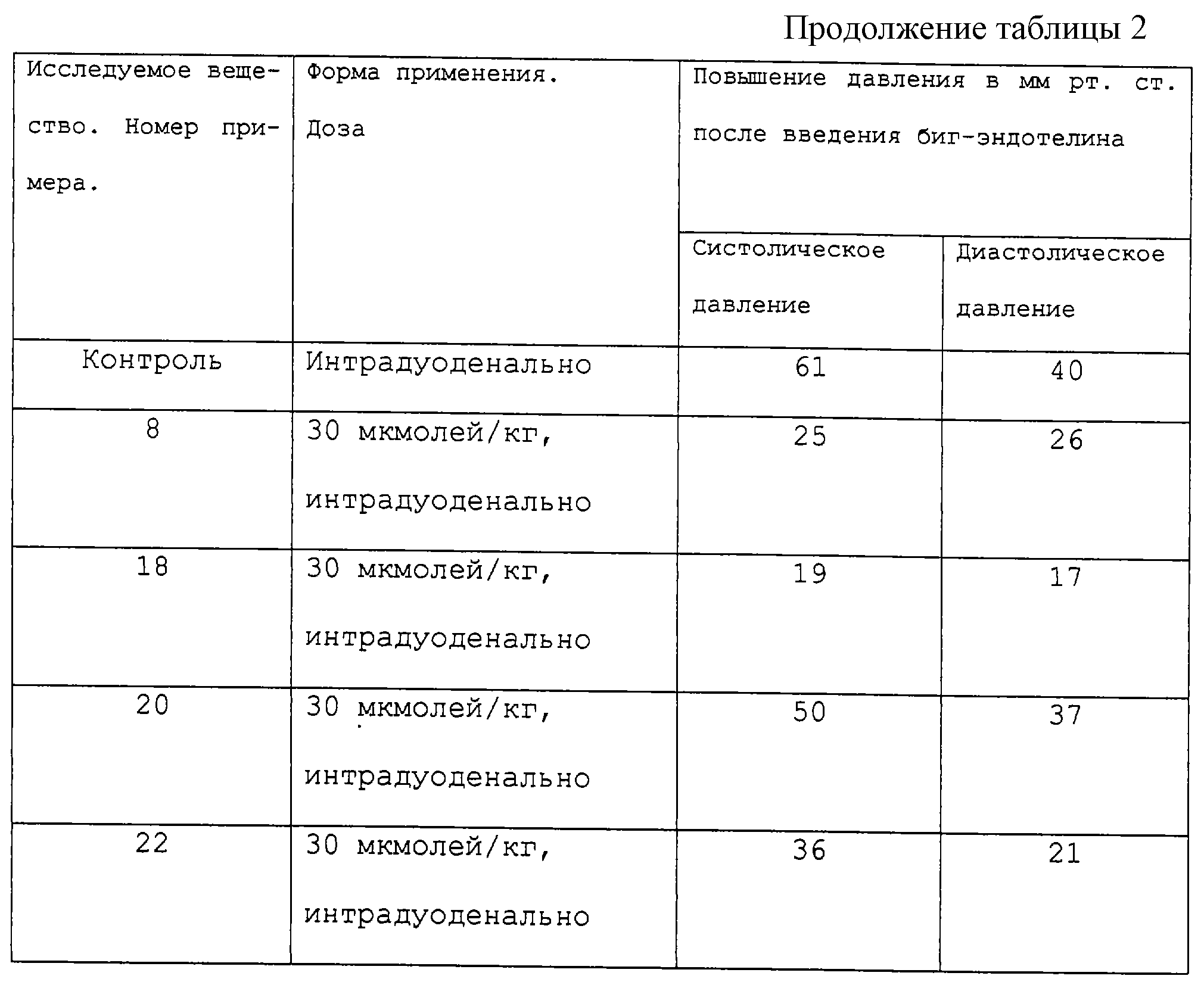
В приводимой табл. 1 указаны значения, на которые возросло выделение мочи при объемной нагрузке, а также после введения тестируемого вещества в % от количества выделившейся мочи при объемной нагрузке и после введения плацебо. Приводятся также количества выделившихся натрия и калия, при объемной нагрузке и после введения тестируемого вещества, в % от количества натрия и калия, выделившихся при объемной нагрузке после дачи плацебо. Номера примеров в табл. 1 и 2 соответствуют приводимым ниже примерам на получение.
4. Исследования in vivo ЕСЕ-тормозящего действия веществами на крысах.
Для подтверждения тормозящего действия, оказываемого веществами согласно изобретению, на фермент преобразования эндотелина (ЕСЕ), исследовали в виде стандартного теста in vivo замедляющее воздействие веществ на гидролитическое разложение биг-эндотелина (BIG-ET), происходящее в результате ферментативной активности ЕСЕ, с образованием эндотелина (ЕТ). Эндотелин представляет собой сильнодействующее сосудосужающее аутогенное вещество. Увеличение уровня эндотелина приводит к повышению давления крови. При вводе BIG-ET давление крови повышается по мере образования эндотелина в результате каталитического расщепления ЕСЕ. В качестве степени подавления ЕСЕ веществами определяли эффект замедления увеличения давления крови, вызываемого введением биг-эндотелина.
Проведение теста
Опыты проводили на крысах-самцах CD® фирмы
Charles River Wiga с весом тела от 220 до 280 г.
Под наркозом, вызванным кетамином/ксилацином, животным вводили один катетер в левую яремную вену для введения вещества и другой - в левую шейную
артерию для измерения кровяного давления. Через 30
минут отдыха животным вводили испытуемое вещество в виде раствора внутривенно или интрадуоденально. После введения испытуемых веществ животные
получали внутривенно соответственно биг-эндотелин в
количестве 0,5 нмоля/кг. Период между применением испытуемого вещества и вводом биг-эндотелина составил: при внутривенном введении 5 минут, при
интрадуоденальном введении испытуемых веществ,
полученных в примерах 18 и 22, 15 минут, испытуемых веществ, полученных в примерах 8 и 20, 30 минут. В течение последующих 30 минут измеряли каждые 5
минут систолическое и диастолическое давление
крови. У животных, не прошедших лечение, введение 0,5 нмоля/кг биг-эндотелина приводило к резкому повышению кровяного давления, которое было
воспроизводимым и продолжалось около 30 минут.
Максимальное повышение кровяного давления происходило через приблизительно 5 минут.
В табл. 2 приводится максимальное повышение кровяного давления после введения биг-эндотелина у контрольных животных, обработанных ложным раствором, и у животных, которых предварительно обработали растворами тестируемых веществ с применением разной дозировки.
Приведенные выше результаты тестирования показывают, что соединения формулы I обладают большим сродством к ферменту преобразования эндотелина и к нейтральной эндопептидазе и что в зависимости от дозы путем замедления активности фермента преобразования эндотелина ЕСЕ противодействуют образованию эндотелина и вызываемому им повышению сопротивления периферийных сосудов и кровяного давления. Результаты тестирования говорят и о том, что вещества способствуют также благодаря подавлению фермента (NEP), разрушающего атриальный пептид натрийурезы, повышению уровня последнего в крови и тем самым повышают эффекты диуреза/натрийуреза, вызываемые атриальным пептидом натрийуреза, без существенной потери калия.
Благодаря описанному выше воздействию соединений формулы I последние пригодны в качестве лекарственных средств и для более крупных млекопитающих, в частности для людей, при терапии сердечной недостаточности и для повышения диуреза/натрийурезы, особенно у пациентов, страдающих сердечной недостаточностью. В этом случае соединения формулы I и их соли, а также биолабильные сложные эфиры целесообразно применять в лекарственных формах, предназначенных для орального введения. Применяемые дозы могут назначаться индивидуально и естественно варьироваться в зависимости от состояния больного, применяемого вещества и лекарственной формы. Однако в целом при назначении для крупных млекопитающих, в частности людей, пригодны лекарственные формы с содержанием активного вещества от 1 до 200 мг в одной дозе.
В качестве лечебного средства соединения формулы I могут содержаться вместе со вспомогательными веществами в галеновых препаратах, таких, например, как таблетки, капсулы, свечи или растворы. Такие галеновые препараты можно получать известными методами с использованием обычных твердых или жидких наполнителей, как, например, лактоза, крахмал или тальк или жидкие парафины, и/или с использованием традиционных фармацевтических вспомогательных веществ, например веществ для разрушения таблетки, агентов растворения или консервантов.
Приводимые ниже примеры поясняют более подробно изобретение, не ограничивая при этом ни в коем случае его объема.
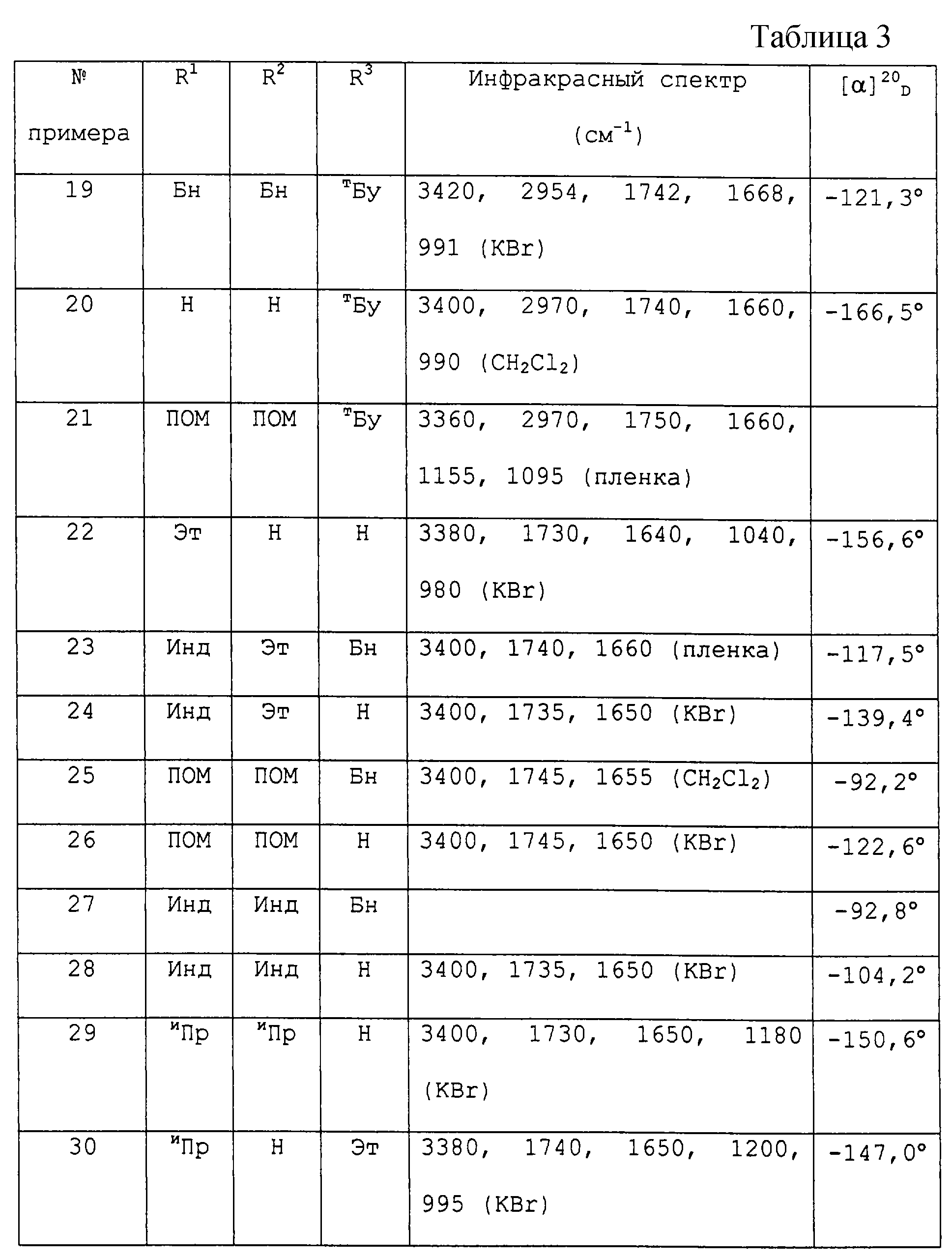
Структуры новых соединений были подтверждены спектроскопическими исследованиями, в частности анализом инфракрасных спектров, и, при необходимости, определением оптических значений вращения.
ПРИМЕР 1
Сложный бензиловый эфир
(3S)-3-(1-дибензилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной
кислоты.
А. При перемешивании объединяли 100 мл дибензилфосфита, 12,5 г параформальдегида и 6,2 мл триэтиламина. При медленном нагревании до 55oС температуру повышали до 120oС. Прозрачный раствор охлаждали до 90oС и перемешивали при этой температуре в течение 30 минут. После охлаждения до комнатной температуры проводили хроматографию на 1 кг силикагеля при повышенном давлении (растворитель: н-гексан/сложный этиловый эфир уксусной кислоты 1:4). После концентрирования фракций и 12-часовой сушки остатка в вакууме при 60oС получали 96,1 г чистого маслянистого дибензилгидроксиметилфосфоната, который использовали в реакции без дополнительной очистки.
Б. 17,8 г дибензилгидроксиметилфосфоната растворяли в 120 мл сухого дихлорметана. После охлаждения до -50oС вводили без доступа влаги капельно и последовательно друг за другом сначала 7,3 г 2,6-лутидина, затем 10,6 мл ангидрида трифторметанфосфоновой кислоты. Реакционную смесь перемешивали в течение одного часа сначала при -50oС, затем в течение еще одного часа при 0oС. Для разделения смеси ее выливали в ледяную воду, органическую фазу промывали сначала разбавленной ледяной соляной кислотой, затем ледяной водой. После высушивания органической фазы с помощью сульфата натрия и фильтрации производили выпаривание в вакууме. Полученный сырой продукт хроматографировали на 200 г силикагеля (разбавитель: н-гексан/сложный этиловый эфир уксусной кислоты 3:2). После сгущения и высушивания фракций продукта получали 17,0 г маслянистого дибензилфосфонометилтрифторметилсульфоната.
В. В атмосфере азота растворяли без доступа влаги 16,5 мл диизопропиламина в 100 мл сухого тетрагидрофурана (THF) и охлаждали до -70oС. В эту смесь добавляли капельно 65,5 мл 1,6-молярного раствора н-бутиллития в н-гексане. Затем перемешивали в течение 30 минут при 0o С, охлаждали до -20oС и при этой температуре каплями добавляли 5,3 мл раствора циклопентанкарбоновой кислоты в 20 мл тетрагидрофурана. Эту реакционную смесь перемешивали в течение 30 минут сначала при -20oС, затем в течение 2 часов при 0oС, после чего охлаждали до -60oС. В смесь каплями медленно добавляли 20,0 г раствора продукта, полученного, как описано в п. Б, в 20 мл тетрагидрофурана. После этой добавки в течение 1 часа перемешивали при -30oС, затем в течение 1 часа при -20oС. Затем реакционную смесь выливали в ледяной водный раствор гидросульфата калия и экстрагировали метил-трет-бутиловым эфиром (МТВЕ). Отделяли органическую фазу, промывали ее насыщенным раствором поваренной соли, высушивали сульфатом натрия и после фильтрации в вакууме сгущали. Полученный сырой продукт очищали хроматографией на 300 г силикагеля с помощью чистого МТВЕ, в который примешивался метанол, содержание которого постоянно повышали с 5 до 10%. Полученный таким образом продукт снова хроматографировали на 200 г силикагеля с целью дополнительной очистки, в результате получили 6,7 г чистой 1-дибензилфосфонометил-1-циклопентакарбоновой кислоты, точка плавления 89-92oС.
Г. В 24,5 г нагретого до 65oС раствора рацемического сложного третичного бутилового эфира 3-амино-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты в 54 мл этанола добавляли 12,65 г L-(+)-винной кислоты в 54 мл нагретого до 65oС этанола. Реакционную смесь перемешивали в течение часа при комнатной температуре. Затем капельно вводили 1,72 мл раствора бензальдегида в 1,3 мл этанола. Полученную суспензию кипятили в течение 14 часов при 80o С с обратным холодильником и охлаждали до комнатной температуры. Образовавшийся кристаллический осадок отсасывали, помещали в 80 мл этанола и снова кипятили в течение 8 часов с обратным холодильником. Затем охлаждали до комнатной температуры, отсасывали кристаллы и сушили при понижем давлении и 50oС. Было получено 23,6 г соли винной кислоты с точкой плавления 195-196oС; [α] = -152,0°(с=0,5 в метаноле).
Д. Для выделения основания 23,6 г соли винной кислоты в смеси из 250 мл воды и 108 мл дихлорметана и при размешивании охлаждали до 0oС, добавкой водного аммиачного раствора доводили до рН 9,6. Отделяли органическую фазу, повторно экстрагировали водную фазу с использованием 30 мл дихлорметана и сгущали органическую фазу, сушили сульфатом натрия и сгущали при пониженном давлении. Оставшийся осадок выделили кристаллизацией из МТВЕ и сушили при пониженном давлении. Было получено 12,2 г сложного трет-бутилового эфира (3S)-3-амино-2,3, 4, 5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты, точка плавления 113-115oС; [α] = -276,2°(с=0,5 в метаноле).
Е. Смешивали 3,6 г полученного выше, чистого энатиомерного сложного трет-бутилового эфира с 2,8 г п-толуолсульфоновой кислоты и 6,9 мл бензилового спирта в 60 мл толуола. Затем кипятили эту смесь в течение 3 часов на водоотделителе, отделяли толуол в вакууме и перемешивали образовавшийся осадок со сложным метил-трет-бутиловым эфиром (МТВЕ). После декантирования растворителя осадок помещали в дихлорметан и при встряхивании смешивали с ледяным разбавленным водным раствором карбоната натрия. Водную фазу экстрагировали дихлорметаном, объединенные органические фазы промывали водой. Затем сушили органическую фазу сульфатом натрия и выпаривали в вакууме. Остаток выделяли кристаллизацией из МТВЕ и высушивали. Получали 3, 2 г сложного бензилового эфира (3S)-3-амино-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты, точка плавления 113-115oС; [α] = -236,8°(с=0,5 в метаноле).
Ж. 5,8 г кислоты, полученной выше в п. В, помещали в 148 мл сухого дихлорметана. В полученный раствор последовательно добавляли при охлаждении до температуры льда 4,8 г полученного выше продукта, 3,7 мл N-метилморфолина, 1,84 г 1-гидроксибензатриазола и 5,8 г N-(3-диметиламинопропил)-N'-этилкарбодиимид-гидрохлорида. Затем реакционную смесь перемешивали без доступа влаги в течение часа при комнатной температуре. Для разделения реакционной смеси ее разбавляли дихлорметаном и последовательно промывали водой, водным раствором гидросульфата калия, водой, водным раствором карбоната натрия и снова водой. Высушиванием органической фазы сульфатом натрия и упариванием в вакууме получали 10,5 г сырого продукта, который хроматографически очищали на 200 г силикагеля (растворитель: н-гексан/сложный этиловый эфир уксусной кислоты 3: 7). После выпаривания фракций продукта и сушки в вакууме выделяли 6,5 г чистого соединения, указанного в названии примера, в виде твердой пены, инфракрасный спектр: 3400, 3310, 2940, 1740, 1650 см-1 (пленка); [α] = -104,6°(с=0,754 в метаноле).
ПРИМЕР 2
(3S)-3-(1-Фосфонометилциклопентан-1-карбониламино)-2,3,4,
5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусная кислота.
А. 1,9 г сложного бензилового эфира (3S)-3-(1-дибензилфосфонометилциклопентан-1-карбониламино)-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 1Ж) растворяли в 100 мл этанола. Добавляли 1,2 г 5%-ного палладиевого катализатора на активированном угле и гидрировали в течение 3 часов при давлении водорода 5,5 бара. Для разделения катализатор отфильтровывали, упаривали в вакууме и сушили. Получали 0,9 г указанного в названии примера соединения в виде пенистого продукта, инфракрасный спектр: 3400, 1720, 1630 см-1 (KBr); [α] = -140,8°(с=0,5 в метаноле).
Б. 701 мг полученной выше свободной кислоты и 238 мг карбоната натрия растворяли в 60 мл воды, раствор упаривали в вакууме. Образовавшийся осадок смешивали с небольшим количеством МТВЕ и снова упаривали в вакууме. Полученную твердую пену выделяли из изопропанола кристаллизацией, кристаллы отделяли от растворителя и высушивали в течение двух суток при 60oС в высоком вакууме. Получали 700 мг натриевой соли указанного в названии примера соединения, точка плавления >270oС; [α] = -159,7°(с=0,149 в метаноле).
ПРИМЕР 3
Сложный бензиловый эфир
(3S)-3-(1-бензилэтилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
А. При ледяном охлаждении 8,0 г раствора NaOH в 30 мл воды и 30 мл этанола капельно добавляли в 27,6 г диэтилфосфита и перемешивали в течение 2 часов при комнатной температуре. Затем упаривали в вакууме и экстрагировали водный остаток четыре раза с помощью МТВЕ. После упаривания водной фазы в вакууме получали 25,0 г метилового гидрофосфита натрия в виде порошка белого цвета, который использовали в реакции без дополнительной очистки.
Б. В 33,9 г раствора гидросульфата тетра бутиламмония в 20 мл воды капельно добавляли при ледяном охлаждении 4,0 г раствора NaOH в 22 мл воды, причем температуру поддерживали ниже 25oС. Затем добавляли капельно при комнатной температуре 12,5 г полученного выше продукта, растворенного в 15 мл воды. После 15-минутного перемешивания производили отсос выпавшего в осадок сульфата натрия и экстрагировали фильтрат 4 раза с помощью 50 мл дихлорметана. Объединенные орагнические фазы сушили сульфатом натрия и упаривали в вакууме. Остаток сушили в течение часа при 40oС в вакууме, растворяли в 120 мл безводного ацетонитрила и примешивали 7,07 мл бензилбромида и 0,4 г йодида натрия. Перемешивали в течение 12 часов при 50oС, отделяли растворитель в вакууме и помещали остаток в н-гексан. Отсасывали твердый остаток, дополнительно промывали смесью из н-гексана и МТВЕ и высушивали. Полученный раствор упаривали в вакууме, остаток хроматографировали на 200 г силикагеля (растворитель: н-гексан/сложный этиловый эфир уксусной кислоты 2:3). Получали 6,7 г бензилэтилфосфита в виде масла, инфракрасный спектр: 2420, 1255, 970 см-1 (пленка).
В. 18,0 г указанного выше продукта вводили во взаимодействие с 2,5 г параформальдегида и 1,2 мл триэтиламина способом, описанным в примере 1А. При хроматографии на 200 г силикагеля (растворитель: сложный этиловый эфир уксусной кислоты) было получено 16,5 г бензилэтилгидроксиметилфосфоната в виде масла, инфракрасный спектр: 3300, 1230, 1030 см-1 (пленка).
Г. 12,0 г полученного выше продукта приводили во взаимодействие с 6,2 г 2,6-лутидина и 9,0 мл ангидрида трифторметансульфокислоты способом, описанным в примере 1В. Хроматографией на 200 г силикагеля (растворитель: н-гексан/сложный этиловый эфир уксусной кислоты 2:3) получали 16,3 г маслянистого бензилэтилфосфонометилтрифторметилсульфоната, инфракрасный спектр: 1410, 1245, 1210, 1010 cм-1 (пленка).
Д. Из 16,08 мл диизопропиламина, 63,8 мл 1,6-молярного раствора н-бутиллития в н-гексане и 5,3 мл циклопентанкарбоновой кислоты получали способом, описанным в примере 1В, дианион циклопентанкарбоновой кислоты и приводили во взаимодействие описанным там же способом с 16,0 г продукта, получаемого, как описано в п. Г. Хроматографией сырого продукта на 300 г силикагеля (растворитель: сначала н-гексан/сложный этиловый эфир уксусной кислоты 1:1, который затем постепенно заменяли чистым сложным этиловым эфиром уксусной кислоты) получали 7,1 г чистой маслянистой 1-(бензилэтилфосфонометил)-1-циклопентанкарбоновой кислоты, инфракрасный спектр: 2950, 1720, 1210, 1175, 1010 см-1 (пленка).
Е. 3,1 г полученной выше кислоты вводили во взаимодействие с 3,2 г сложного бензилового эфира (3S)-3-амино-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 1Е), 3,3 мл N-метилморфолина, 1,35 г гидроксибензотриазола и 3,8 г N-(3-диметиламинопропил)-N'-этилкарбодиимид-гидрохлорида способом, описанным в примере 1Ж. Хроматографией на 200 г силикагеля (растворитель: сложный этиловый эфир уксусной кислоты) получали 2,3 г заглавного соединения в виде вязкого масла, инфракрасный спектр: 3410, 2940, 1735, 1660, 1230, 1020 см-1 (KBr); [α] = -121,6° (с= 0,495 в метаноле).
ПРИМЕР 4
Сложный этиловый эфир
(3S)-3-(1-бензилэтилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
А. 5,0 г сложного трет-бутилового эфира (3S)-3-амино-2,3,4, 5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты (получение см. в примере 1Д) и 3,75 г п-толуолсульфокислоты кипятили в 80 мл толуола в течение 2,5 часов на водоотделителе. Затем добавляли отдельными порциями этанол в суммарном количестве 200 мл, образующуюся реакционную смесь кипятили в течение 3,5 часов с обратным холодильником. После этого сгущали смесь в вакууме, остаток помещали в дихлорметан. Перемешивали при встряхивании с ледяным раствором карбоната натрия и нейтрально промывали органическую фазу водой. Органическую фазу высушивали сульфатом натрия, упаривали в вакууме и образовавшийся осадок высушивали. Получали 3,6 г сложного этилового эфира (3S)-3-амино-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты, точка плавления: 106,5-108oС; инфракрасный спектр: 3350, 3300, 2930, 1735, 1660 см-1 (пленка); [α] = -288, 4°(с=0,5 в метаноле).
Б. 3,1 г 1-(бензилэтилфосфонометил)-1-циклопентанкарбоновой кислоты (получение см. в примере 3Д) вводили во взаимодействие с 2,6 г полученного выше продукта, 3,3 мл N-метилморфолина, 1,35 г гидроксибензотриазола и 3,8 г N-(3-диметиламинопропил)-N'-этилкарбодиимид-гидрохлорида способом, описанным в п. 1Ж. Хроматографией на 200 г силикагеля (растворитель: сначала н-гексан/сложный этиловый эфир уксусной кислоты 1:1, который затем непрерывно изменяли до получения состава 3:7) получали 3,7 г указанного в названии примера соединения в виде масла, инфракрасный спектр: 3410, 2950, 1735, 1660 см-1 (пленка); [α] = -113, 6°(с=0,639 в метаноле).
ПРИМЕР 5
Сложный этиловый эфир (3S)-3-(1-этилфосфонометилциклопентан-1-карбониламино)-2,3,4,
5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
В 3,2 г сложного этилового эфира (3S)-3-(1-бензилэтилфосфонометилциклопентан-1-карбониламино)-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 4) вводили 1,0 г 5%-ного палладиевого катализатора на активированном угле и гидрировали при давлении водорода 2,2 бара способом, описанным в примере 2. После разделения получали 2,4 г указанного в названии примера соединения в виде вспененной смолы, инфракрасный спектр: 3400, 2950, 1740, 1650 см-1 (KBr); [α] = -162,0°(с=0,324 в метаноле).
ПРИМЕР 6
Сложный этиловый эфир (3S)-3-[1-(пивалоилоксиметилэтилфосфонометил)циклопентан-1-карбониламино] -2,3,4,5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты.
0,6 г сложного этилового эфира (3S)-3-(1-этилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 5) растворяли без доступа влаги в 20 мл диметилформамида (DMF), затем добавляли 1,86 мл триэтиламина, 0,88 мл пивалоилоксиметилхлорида и 0,1 г диметиламинопиридина. Реакционную смесь перемешивали в течение ночи, выпаривали растворитель при пониженном давлении и помещали остаток в дихлорметан. Органическую фазу отмывали водой и затем сушили сульфатом натрия. После сгущения в вакууме получали сырой продукт, который хроматографировали для его очистки на 50 г силикагеля (растворитель: сначала н-гексан/сложный этиловый эфир уксусной кислоты 3:7, затем содержание эфира постепенно доводили до 100%). Получали 188 мг указанного в названии примера соединения в виде масла, инфракрасный спектр: 1740, 1650 см-1 (СН2Сl2); [α] = -124,1°(с=0,228 в метаноле).
ПРИМЕР 7
Сложный этиловый эфир
(3S)-3-[1-(5-инданилэтилфосфонометил)циклопентан-1-карбониламино] -2,3,4,5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты.
480 мг сложного этилового эфира (3S)-3-(1-этилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 5) растворяли в 10 мл сухого дихлорметана и примешивали 0, 28 мл триэтиламина. Полученный раствор охлаждали до -50oС, после чего добавляли в него 0,09 мл оксалилхлорида. После этого к реакционной смеси примешивали при -50oС 200 мг 5-инданола, нагревали до 0oС и перемешивали при комнатной температуре в течение 5 часов. Органическую фазу отмывали водой, отделяли, сушили сульфатом натрия и упаривали при пониженном давлении. После хроматографии на 80 г силикагеля (растворитель: н-гексан/сложный этиловый эфир уксусной кислоты 1:1, причем соотношение растворителей непрерывно изменяли до соотношения 1:4) после высушивания в высоком вакууме получали 220 мг указанного в названии примера соединения в виде вязкой смолы, инфракрасный спектр: 1740, 1655 см-1 (СН2Сl); [α] = -135,1°(с=0,205 в метаноле).
ПРИМЕР 8
Сложный
трет-бутиловый эфир (3S)-3-(1-бензилэтилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты.
5,0 г 1-(бензилэтилфосфонометил)-1-циклопентанкарбоновой кислоты (получение см. в примере 3Д) вводят во взаимодействие с 5,15 г сложного трет-бутилового эфира (3S)-3-амино-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 1Д), 4,1 мл N-метилморфолина, 2,0 г гидроксибензотриазола и 6,3 г N-(3-диметиламинопропил)-N'-этилкарбодиимид-гидрохлорида способом, описанным в примере 1Ж. Полученный сырой продукт хроматографировали на 200 г силикагеля (растворитель: сначала н-гексан/сложный этиловый эфир уксусной кислоты 1:1, затем чистый сложный эфир). Получали 2,6 г указанного в названии примера соединения в виде вспененной смолы, инфракрасный спектр: 3410, 3350, 1735, 1655 см-1; [α] = -118,1°(с= 0,609 в метаноле).
ПРИМЕР
9
Сложный бензиловый эфир (3S)-3-(1-этилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты.
А. 3,5 г 1-(бензилэтилфосфонометил)-1-циклопентанкарбоновой кислоты (получение см. в примере 3Д) растворяли в 150 мл этанола и примешивали 1,0 г 5%-ного палладиевого катализатора на активированном угле. Затем гидрировали в течение 4 часов при давлении водорода 2,1 бара. Дважды отфильтровывали катализатор, упаривали в вакууме и сушили в высоком вакууме. Получали 2,60 г маслянистой 1-этилфосфонометил-1-циклопентанкарбоновой кислоты, которую использовали в реакции без дополнительной очистки.
Б. 2,6 г полученного выше продукта растворяли без доступа влаги в 100 мл сухого дихлорметана, примешивали 3,5 г карбонилдиимидазола и 3,56 г сложного бензилового эфира (3S)-3-амино-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 1Е) и перемешивали в течение ночи. Затем сливали в насыщенный водный раствор гидросульфата калия, нейтрально промывали водой органическую фазу и сушили сульфатом натрия. Полученный сырой продукт хроматографировали на 150 г силикагеля (растворитель: сначала сложный этиловый эфир уксусной кислоты, к которому затем постепенно примешивали дихлорметан, пока соотношение растворителей не составило 1:1).
После высушивания фракций продукта в вакууме получали 1,4 г указанного в названии примера соединения в виде пены, инфракрасный спектр: 3410, 1740, 1645 см-1 (KBr); [α] = -130,7° (с=0,339 в метаноле).
ПРИМЕР 10
Сложный бензиловый эфир (3S)-3-(1-диэтилфосфонометил-1-циклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты.
A. 69,05 г диэтилфосфита вводили во взаимодействие с 14,5 г параформальдегида и с 6,69 мл триэтиламина способом, аналогичным описанному в примере 1А. Получали 66,02 г диэтилгидроксиметилфосфоната, который после сушки в высоком вакууме без дополнительной очистки применяли при последующей реакции.
Б. 21,02 г полученного выше фосфоната, 15,0 г 2,6-лутидина и 21,8 мл ангидрида трифторметансульфокислоты вводили во взаимодействие способом, описанным в примере 1Б. Получали 32,5 г маслянистого диэтилфосфонометилтрифторметилсульфоната.
B. 30,0 г полученного выше трифторметилсульфоната вводили во взаимодействие с 133 мл 1,6-молярного раствора н-бутиллития в н-гексане и с 10,8 мл циклопентанкарбоновой кислоты способом, описанным в примере 1В. Получали 11,1 г диэтилфосфонометил-1-циклопентанкарбоновой кислоты, инфракрасный спектр: 2970, 1730, 1240, 1030 см-1 (пленка).
Г. 5,74 г полученного выше производного карбоновой кислоты вводили во взаимодействие с 7,05 г сложного бензилового эфира (3S)-3-амино-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 1Е) способом, описанным в примере 1Ж. Полученный сырой продукт очищали хроматографией на силикагеле (растворитель: сложный этиловый эфир уксусной кислоты). Получали 7,95 г указанного в названии примера соединения, инфракрасный спектр: 3400, 1745, 1650 см-1 (пленка); [α] = -130,3° (с=0,538 в метаноле).
ПРИМЕР 11
(3S)-3-(1-Диэтилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусная кислота.
5,3 г сложного бензилового эфира (3S)-3-(1-диэтилфосфонометил-1-циклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты (получение см. в примере 10) растворяли в 250 г этанола, смешивали с 1,5 г 5%-ного палладиевого катализатора на активированном угле и гидрировали способом, описанным в примере 2. Получали 4,3 г указанного в названии примера соединения, инфракрасный спектр: 3390, 1730, 1650 см-1 (KBr); [α] = -156,6° (с=0,514 в метаноле).
ПРИМЕР 12
Сложный этиловый эфир (3S)-3-(1-диэтилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
2,34 г (3S)-3-(1-диэтилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 11) растворяли без доступа влаги в дихлорметане, смешивали с 1,6 мл N-метилморфолина, 0,63 г 1-гидроксибензотриазола, 2,0 г N-(3-диметиламинопропил)-N'-этилкарбодиимид-гидрохлорида и 0,6 мл этанола и перемешивали в течение 4 часов при комнатной температуре. Затем реакционную смесь последовательно промывали водой, раствором гидросульфата калия, водой, раствором гидрокарбоната натрия и снова водой. После этого отделяли органическую фазу, сушили сульфатом натрия и выпаривали в вакууме. Полученный продукт хроматографировали на 200 г силикагеля (растворитель: вначале этиловый сложный эфир уксусной кислоты, позже в него добавляли 5%-ный метанол), фракции продукта обогащали и сушили в вакууме. Получали 1,6 г указанного в названии примера соединения, инфракрасный спектр: 3410, 1740, 1650, 1200, 1030 см-1 (пленка); [α] = -126,1° (с=0,584 в метаноле).
ПРИМЕР 13
Сложный этиловый эфир (3S)-3-(1-фосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
1,3 г сложного этилового эфира (3S)-3-(1-диэтилфосфонометилциклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 12) растворяли в атмосфере азота в 13 мл дихлорметана. При охлаждении льдом примешивали 0,5 мл бромтриметилсилана и 0,4 мл триэтиламина и перемешивали в течение ночи. Избытки растворителя удаляли в вакууме, остаток размешивали в течение 15 минут в водном ацетоне. Оставшийся после выпаривания растворителя осадок был помещен в МТВЕ, куда добавили небольшое количество дихлорметана, и смешивали с 0,53 г (S)-(-)-α-метилбензиламина. Выпавшее в осадок твердое вещество отделили от этанола перекристаллизацией, причем указанное в названии примера соединение получали в виде соли α-метилбензиламмония с точкой плавления 210-213o С. Инфракрасный спектр: 2940, 1750, 1650, 1200, 1045 см-1 (КВr); [α] = -141, 0° (с=0,2 в метаноле).
ПРИМЕР 14
Сложный бензиловый эфир (3S)-3-1-фосфонометилциклопентан-1-карбониламино)-2,3,4,
5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной
кислоты.
3,8 г сложного бензилового эфира (3S)-3-(1-диэтилфосфонометил-1-циклопентан-1-карбониламино)-2,3,4, 5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты (получение см. в примере 10) растворяли в 10 мл дихлорметана, при охлаждении льдом смешивали с 10,3 мл трифторуксусной кислоты и перемешивали в течение 18 часов при комнатной температуре. Растворитель отделяли в вакууме, оставшийся осадок несколько раз обрабатывали толуолом и снова выпаривали. Полученный сырой продукт растворяли в дихлорметане, трижды промывали водой, затем отделяли органическую фазу, сушили сульфатом натрия и выпаривали растворитель в вакууме. После сушки в высоком вакууме получали 3,0 г указанного в названии примера соединения в виде масла, инфракрасный спектр: 3400, 2950, 1745, 1640 см-1 (КВr); [α] = -146,5° (с=0,2 в метаноле).
ПРИМЕР 15
Сложный бензиловый эфир (3S)-3-(1-диизопропилфосфонометилциклопентан-1-карбониламино)-2,3,4,
5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
А. 50,0 г диизопропилфосфата, 8,5 г параформальдегида и 4,0 мл триэтиламина вводили во взаимодействие методом, описанным в примере 1А. После хроматографии сырого продукта на силикагеле (растворитель: н-гексан/сложный этиловый эфир уксусной кислоты 1:4) получали 37,5 г диизопропилметилфосфоната в виде масла, который в дальнейшем использовали без очистки.
Б. 19,6 г полученного выше соединения вводили во взаимодействие с 17,4 мл ангидрида трифторметансульфокислоты и 11,96 г 2,6-лутидина способом, описанным в примере 1Б. После хроматографии сырого продукта на силикагеле (растворитель: н-гексан/сложный этиловый эфир уксусной кислоты 3:7) получали 27,4 г диизопропилфосфонометилтрифторметилсульфоната в виде масла, инфракрасный спектр: 2980, 1410, 1205, 1000 см-1 (пленка).
В. 27,4 г полученного выше соединения, 10,05 мл циклопентанкарбоновой кислоты и 120 мл 1,6-молярного раствора н-бутиллития в н-гексане вводили во взаимодействие способом, описанным в примере 1В. После хроматографии сырого продукта на силикагеле (растворитель: сначала н-гексан/сложный этиловый эфир уксусной кислоты 3: 7, в который постепенно примешивали в возрастающем количестве сложный эфир до 100%) получали 10,6 г диизопропилфосфонометил-1-циклопентанкарбоновой кислоты с точкой плавления 53-57oС.
Г. 2,05 г полученного выше соединения вводили во взаимодействие с 2,24 г сложного бензилового эфира (3S)-3-амино-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 1Е) способом, описанным в примере 1Ж. Получали 3,5 г указанного в названии примера соединения в виде масла, инфракрасный спектр: 3410, 1735, 1650, 1240, 1180 см-1 (пленка); [α] = -127,5° (с=0,287 в метаноле).
ПРИМЕР 16
Сложный этиловый эфир
(3S)-3-(1-бензилизопропилфосфонометилциклопентан-1-карбониламино)-2,3,4,
5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
A. 92,0 мл диизопропилфосфита и 22,2 г NaOH вводили во взаимодействие способом, описанным в примере 3А. Получали 88,0 г изопропилгидрофосфита натрия, который использовали в реакции без дополнительной очистки.
Б. 88,0 г полученного выше соединения и 34 мл бензилбромида вводили во взаимодействие способом, аналогичным описанному в примере 3Б. Получали 46,3 г бензилизопропилфосфита в виде масла, который использовали в реакции без дополнительной очистки.
B. 46,3 г полученного выше соединения вводили во взаимодействие с 6,1 г параформальдегида и 2,87 мл триэтиламина способом, описанным в примере 1А. Получали 24, 0 г бензилизопропилгидроксиметилфосфоната в виде масла, инфракрасный спектр: 3300, 1230, 995 см-1 (пленка).
Г. 24,0 г полученного выше соединения вводили во взаимодействие с 18,01 мл ангидрида трифторметансульфокислоты и 13,57 мл 2,6-лутидина способом, описанным в примере 1Б. Получали 32,5 г бензилизопропилфосфонометилтрифторметилсульфоната в виде масла, инфракрасный спектр: 2980, 1410, 1245, 1000 см-1 (пленка).
Д. 32,5 г полученного выше соединения, 9,65 мл циклопентанкарбоновой кислоты и 13,4 мл 1,6-молярного раствора н-бутиллития в н-гексане вводили во взаимодействие способом, описанным в примере 1В. После хроматографии сырого продукта на силикагеле (растворитель: сначала н-гексан/сложный этиловый эфир уксусной кислоты 1:1, затем чистый сложный эфир, затем сложный этиловый эфир уксусной кислоты с содержанием 5 об.% изопропанола) получали 7,0 г 1-бензилизопропилфосфонометил-1-циклопентанкарбоновой кислоты, которую использовали в реакции без дополнительной очистки.
Е. 1,25 г полученного выше соединения и 1,06 г сложного этилового эфира (3S)-3-амино-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 4А) вводили во взаимодействие способом, описанным в примере 1Ж. Получали 0,68 г указанного в названии примера соединения, инфракрасный спектр: 2400, 1735, 1655, 1200, 985 см-1 (пленка); [α] = -123, 0° (с=0,1 в изопропаноле).
ПРИМЕР 17
Сложный
трет-бутиловый эфир (3S)-3-(1-этилфосфонометилциклопентан-1-карбониламино)-2,3,4,
5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
2,2 г сложного трет-бутилового эфира (3S)-3-(1-бензилэтилфосфонометилциклопентан-1-карбониламино)-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 8) гидрировали с применением 1,0 г 5%-ного палладиевого катализатора на активированном угле способом, описанным в примере 2, при давлении водорода 2,5 бара. Получали 1,7 г указанного в названии примера соединения, инфракрасный спектр: 3470, 1735, 1650 см-1 (пленка); [α] = -158,2° (с=0,515 в метаноле).
Пример 18
Сложный трет-бутиловый эфир
(3S)-3-[1-(пивалоилоксиметил-этилфосфонометил)циклопентан-1-карбониламино] -2,3,4,
5- тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты.
0,6 г сложного трет-бутилового эфира (3S)-3-(1-этилфосфонометилциклопентан-1-карбониламино)-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты (получение см. в примере 17) вводили во взаимодействие с 1,73 мл триэтиламина, 0,86 мл пивалоилоксиметилхлорида и 0,1 г диметиламинопиридина способом, описанным в примере 6. После хроматографии на силикагеле (растворитель: сложный этиловый эфир уксусной кислоты) получали 392 мг указанного в названии примера соединения в виде вязкой смолы, инфракрасный спектр: 1740, 1650 см-1 (СН2Сl2); [α] = -122, 9° (с=0,257 в метаноле).
К примеру 20.
Форма соли - сложный трет-бутиловый эфир (3S)-3-(1-фосфонометилциклопентан-1-карбониламино)-2,3,4, 5-тетрагидро-2-оксо-1Н-1-бензазепин-1-уксусной кислоты.
А. 961 мг приведенной выше, свободной фосфоновой кислоты смешивали с 212 мг карбоната натрия и 20 мл воды. Образовавшуюся смесь фильтровали, полученный фильтрат выпаривали в вакууме. Образовавшийся остаток отделяли от этанола кристаллизацией, кристаллы сушили в течение суток в вакууме при 60oС. Получали 750 мг натриевой соли заглавного соединения, точка плавления >270oС; [α] = -141,5° (с=0,25 в метаноле).
Б. 961 мг указанной выше свободной фосфоновой кислоты растворяли в 20 мл МТВЕ и смешивали с 0,42 мл трет-бутиламина. Полученный раствор выпаривали в вакууме, образовавшийся остаток помещали в смесь МТВЕ/н-гексана. Образовавшиеся в этой смеси растворителей кристаллы отделяли и высушивали в вакууме при 60oС. Получали 950 мг соли аммония указанного в названии примера соединения, точка плавления 215-220oС; [α] = -149,8° (с=0,26 в метаноле).
Способами, описанными в приведенных выше примерах, могут быть также получены соединения формулы I, указанные в табл. 3.
Пример 1. Лекарственное средство.
Капсулы, содержащие сложный трет-бутиловый эфир (3S)-3-[1-(пивалоилоксиметилэтилфосфонометил)циклопентан-1-карбониламино] -2,3,4, 5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты.
Готовят капсулы, каждая из которых имеет
следующий состав,мг:
Сложный трет-бутиловый эфир
(3S)-3-[1-(пивалоилоксиметилэтилфосфонометил)циклопентан-1-карбониламино] -2,3,4,5-тетрагидро-2-оксо-1Н-бензазепин-1-уксусной кислоты - 20
Кукурузный крахмал - 60
Лактоза - 301
Сложный этиловый эфир уксусной кислоты - Достаточное - количество
Из активного вещества, кукурузного крахмала и лактозы с помощью
сложного этилового эфира уксусной кислоты была
приготовлена однородная пастообразная смесь. Пасту измельчали, полученный гранулят помещали на соответствующий противень и сушили при 45oС для
удаления растворителя. Высушенный гранулят
пропускали через измельчающую машину и в миксере смешивали со следующими вспомогательными веществами, мг:
Тальк - 5
Стеарат магния - 5
Кукурузный крахмал - 9
Затем
капсулы вместимостью по 400 мг (размер капсулы=0) заполняли.
Реферат
Изобретение относится к новым производным бензазепинон-N-уксусной
кислоты, замещенным фосфоновой кислотой, которые
являются фармацевтически активными соединениями. Описываются производные бензазепинон-N-уксусной кислоты, замещенные фосфоновой кислотой общей формулы
I
где R1, R2, R3 означают водород или группу, образующую биoлабильный сложный эфир фосфоновой кислоты, и физиологически приемлемые соли кислот формулы I. Также описываются лекарственное средство, обладающее тормозящим нейтральную эндопептидазу действием, в состав которого входит соединение формулы (I) и способ получения соединений формулы (I). Технический результат - создание новых фармацевтически активных веществ. 3 c. и 1 з.п.ф-лы, 3 табл.
Формула
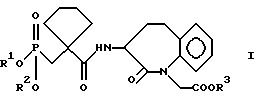
где R1 означает водород или группу, образующую биолабильный сложный эфир фосфоновой кислоты;
R2 означает водород или группу, образующую биолабильный сложный эфир фосфоновой кислоты;
R3 означает водород или группу, образующую биолабильный сложный эфир карбоновой кислоты,
и физиологически приемлемые соли кислот формулы I.
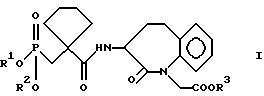
где R1 означает водород или группу, образующую биолабильный сложный эфир фосфоновой кислоты,
R2 означает водород иди группу, образующую биолабильный сложный эфир фосфоновой кислоты,
R3 означает водород или группу, образующую биолабильный сложный эфир карбоновой кислоты,
физиологически приемлемых солей кислот формулы I, отличающийся тем, что для получения соединений общей формулы IV

где R101 и R201, независимо друг от друга, означают водород или защитную группу фосфоновой кислоты;
R302 означает защитную группу карбоновой кислоты,
соединения общей формулы II

где R101 и R201 имеют приведенные выше значения,
вводят во взаимодействие с соединениями общей формулы III
где R302 имеет указанное выше значение,
и в случае, если R101 и/или R201 означают водород, свободную функцию (свободные функции) фосфоновой кислоты, при необходимости, путем этерификации с соединением общей формулы Va и/или Vb,
R110-Y (Va), R210-Y (Vb),
где R110 и R210 обозначают соответственно группу, образующую биолабильный сложный эфир фосфоновой кислоты;
Y означает гидроксильный радикал или отщепляемую летучую группу,
переводят в биолабильные эфирные группы фосфоновой кислоты, и в случае, если в соединениях формулы IV защитные группы R101, R201 и/или R302 не являются желаемыми группами, образующими биолабильный сложный эфир, то их отщепляют одновременно или в отдельности друг за другом в любой последовательности и, при желании, переводят соответствующие высвободившиеся кислотные функции в биолабильные эфирные группы, этерифицируя при этом свободные функции фосфоновой кислоты с соединением формулы Va или Vb и/или свободные функции карбоновой кислоты с соединением общей формулы Vc
R310-Y, (Vc)
где R310 означает группу, образующую биолабильный сложный эфир карбоновой кислоты;
Y имеет указанное выше значение,
и, при необходимости, кислоты формулы I переводят в их физиологически приемлемые соли или соли кислот формулы I - в свободные соединения.
Комментарии