Способ получения хлорида или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия - RU2762826C2
Код документа: RU2762826C2
Описание
Предшествующий уровень техники
1. Область техники
Настоящее изобретение относится к способу получения бромида или хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия; способу превращения бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия в хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия; и очистке хлорида 7-бис-(диметиламино)-фенотиазин-5-ия посредством кристаллизации из водного раствора соляной кислоты, приводящим к получению фармацевтически приемлемого хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (метилтиониния хлорид, метиленовый синий, МТС) представленной ниже формулы I

Хлорид 3,7-бис(диметиламино)-фенотиазин-5-ия
Формула I
2. Родственная область
Хлорид 3,7-Бис-(диметиламино)-фенотиазин-5-ия (также известный как метилтиониния хлорид, метиленовый синий, МТС, C. I. Основной синий, хлорид 9-метилтиониния, Швейцарский синий, C. I. 52015, C. I. Растворитель синий 8, Уролен синий) представляет собой хорошо известный фенотиазиновый краситель для волос, кожи и целлюлозных волокон, окислительно-восстановительный индикатор, фотосенсибилизатор для генерации синглетного кислорода, антиоксидант и антисептический краситель для фиксированной и живой ткани, диагностическое средство в исследованиях функции почек.
МТС также используют в качестве лекарственного средства, например, в качестве ингибитора синтетазы оксида азота и гуанилат циклазы. Обнаружено, что он устраняет гипотензию, ассоциированную с разными клиническими состояниями; он устраняет гипоксию и гипердинамическую циркуляцию при циррозе печени и тяжелом гепатопульмональном синдроме. Также он действует во временном и воспроизводимом улучшении кровяного давления и сердечной функции при септическом шоке. Раньше МТС использовался в лечении малярии (P. Guttmann and P. Ehrlich, 1981); в качестве лекарственного средства для метгемоглобинемии и в последнее время проводят исследования в фотодинамическом лечением рака. МТС оценивается на поздней фазе клинической разработки в отношении лечения болезни Альцгеймера (Wischik et al. US 7737138 B2) и недавно был описан в качестве диагностического маркера в форме визуализирующего красителя во множестве процедур, включая биопсию сторожевого лимфоузла у больных раком (например, пациенты с раком молочной железы), эндоскопическую оценку очаговых поражений у пациентов с ГЭРБ (Гастроэзофагеальная рефлюксная болезнь) или с пищеводом Барретта, урологическую оценку у пациентов с повреждением мочеиспускательного канала или почечной лоханки и торакоскопические процедуры у пациентов с легочными узелками.
Метиленовый синий, в комбинации со светом, использовался для лечения резистентного бляшечного псориаза, СПИД (Синдром приобретенного иммунного дефицита)-ассоциированной саркомы Капоши, вируса Западного Нила и для инактивации золотистого стафилококка, ВИЧ (вирус иммунодефицита человека)-1, вируса гепатита уток B, аденовирусных векторов и гепатита С. Фенотиазиновые красители и свет обладают вирулицидными свойствами, как известно на протяжении более 70 лет. Однако в некоторых обстоятельствах комбинация фенотиазиновых красителей и света может вызывать повреждение ДНК, что может проводить к раку.
МТС использовался в форме таблетки или в форме инъекции. Широкое применение МТС в качестве фармацевтического средства требует способов синтеза, способных обеспечить синтез продукта с высокой химической чистотой.
Синтез, очистка и биологическая активность МТС описаны во многих патентах и публикациях.
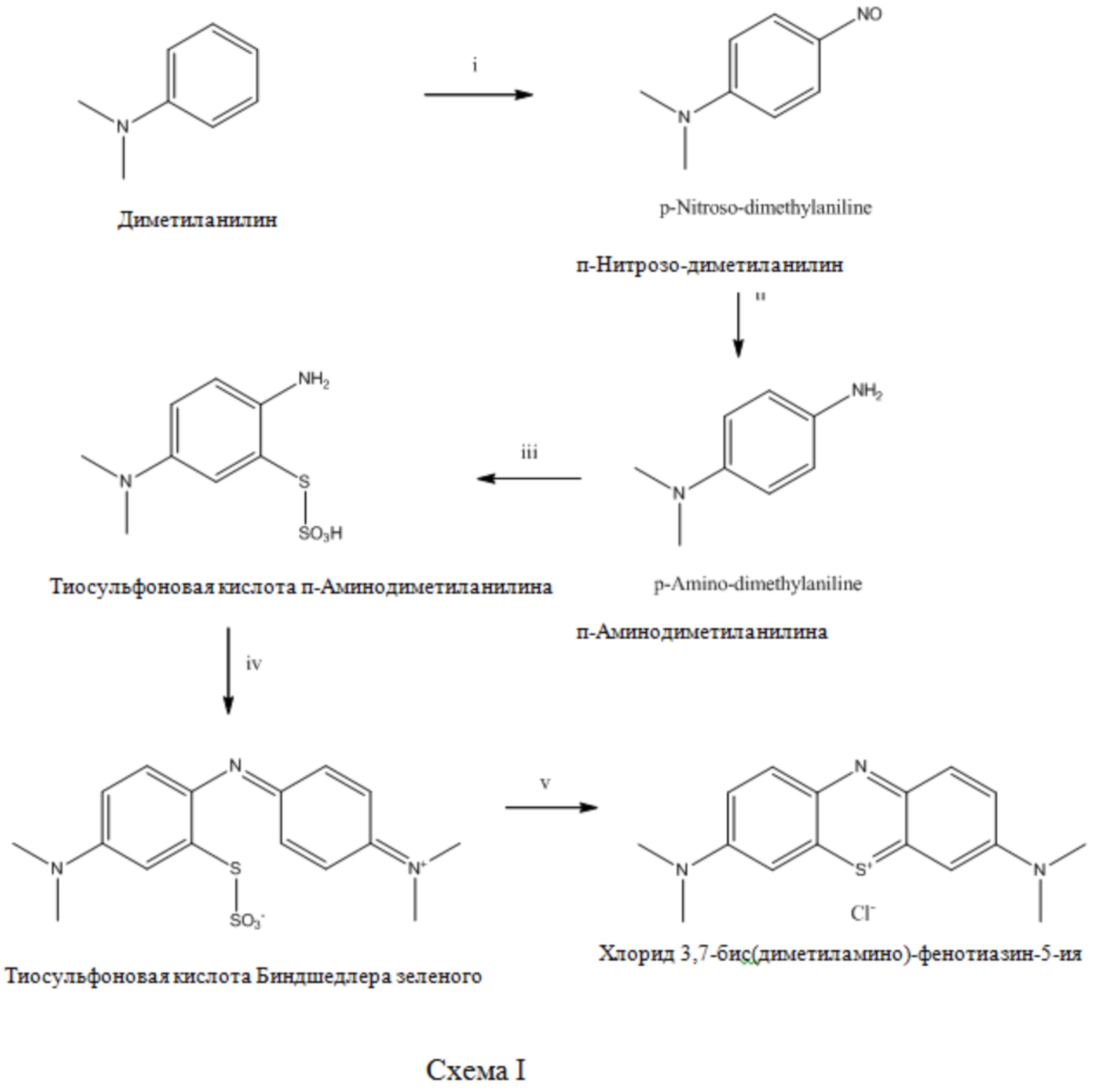
Хлорид 3,7-Бис(диметиламино)-фенотиазин-5-ия (MTC) исходно был синтезирован немецким химиком H. Caro в 1876, и имеется в продаже с тех пор, как он был впервые произведен BASF в 1876 (немецкий патент № DE-1886, Badishe Anilin-und Soda Fabrik, 1877). Согласно патенту BASF MTC синтезировали посредством нитросилации диметиланилина, последующего восстановления с образованием N,N-диметил-1,4-диаминобензола и последующего окислительного сочетания с использованием сульфида водорода и хлорида железа (III). Позже многие авторы описывали похожие способы синтеза MTC. В принципе способы являются одинаковыми, и разными являются только используемые реагенты. A. Bernthsen (1885a, 1885b и 1889), H. E. Fierz-David и L. Blangley (1949) опубликовали получение MTC из диметиланилина, как проиллюстрировано на Схеме 1. Аналогичный способ описан в Colour Index (Vol. 43rd Edition, 1971).
Согласно Схеме 1 исходное вещество диметиланилин на стадии i нитрозилируют посредством реакции с нитритом натрия в водной соляной кислоте. На стадии ii нитрозосоединение восстанавливают до диметиламиноанилина с использованием цинковой пыли и дополнительного количества соляной кислоты. Диметиламиноанилин окисляют (стадия iii) под действием тиосульфоновой кислоты в серной кислоте в присутствии хлорида цинка с получением производного тиосульфоновой кислоты п-аминодиметиламина, на стадии iv тиосульфоновую кислоту п-аминодиметиламина окисляют до тиосульфоновой кислоты Биндшедлера зеленого (химическое название N-[4-[[4-(диметиламино)-2-(сульфотио)фенил]имино]-2,5-циклогексадиен-1-илиден]-N-метил-Метанаминий). Замыкание кольца с получением MTC (стадия v) проводят в присутствии диоксида марганца или сульфата меди.
Лаборатории Wista (WO 2010130977) пытались исключить тяжелые металлы из путей синтеза посредством использования персульфата щелочного металла и персульфата аммония с бихроматом натрия в качестве окислителя. Используя те же стадии, как на Схеме 1, с некоторыми улучшениями, они описали получение в 3-реакторном способе (три реактора), где главную часть получения методом синтеза проводили в реакторе 1 (первый реактор) с последующей циклизацией в реакторе 2 (второй реактор) и перекристаллизацией МТС в реакторе 3 (третий реактор). Кроме того, они описали 2-реакторный способ, где перекристаллизацию проводили во втором реакторе.
Очистка MTC описана, например, P. N. Marshall and S. M. Lewis Stain Technol. 1975a, 1975b; 50(6), 375-81 and 143-147 посредством экстракции растворителем с последующей кристаллизацией, W. Lőhr etal. (The azure dyes: their purification and physicochemical properties. I. Purification of Azure B, Stain Technologies (1974) 49(6), 359-366 и H. E. Fierz-David (Fundamental Processes of Dye Chemistry (1949), Interscience deel, Oxazine and Thiazine Dyes) через образование двойной соли хлорид цинка. MTC, полученный согласно приведенному выше способу, изображенному на Схеме 1, содержит большое количество примесей металлов, таких как Cu, Fe, Cr, Mn, Al, Zn, превышая пределы безопасности, установленные европейскими организациями здравоохранения.
Другой подход был опубликован C. M. Wischik et al., где имеющийся в продаже MTC использовали технической чистоты с чистотой меньше чем 95% (WO2008007074). Сначала его восстанавливали борогидридом натрия (или, в качестве альтернативы, гидразином, метилгидразином) до белой (бесцветной) восстановленной формы (лейкоформа) и затем осуществляли ацилирование уксусным ангидридом. Кристаллизация производного ацетила обеспечивала получение очищенного продукта без металлов. Производное ацетила гидролизовали обратно до белой восстановленной формы MTC и на последней стадии окисляли до MTC (синяя окраска), который, по сравнению с MTC технической чистоты, используемым в качестве исходного вещества, имел гораздо более высокую чистоту. Для реакции окисления использовали хлорид железа (III), изоамилнитрит, трет-бутиламилнитрит и амберлит (см. Схема 2). В этой же международной заявке на патент также описано получение промежуточного соединения 1-ацетил-3,7-диметиламинофенотиазин путем проведения последовательности реакций, которые включают нитрование фенотиазина нитритом натрия, N ацетилирование с использованием уксусного ангидрида с получением 3,7-динитро-10-ацетилфенотиазина, который восстанавливали в атмосфере водорода с использованием палладия на активированном угле в качестве катализатора и, наконец, осуществляли реакцию с п-формальдегидом и цианоборогидридом натрия.

M. Feraud et al. обнаружили новый путь синтеза и разработали его химический способ, приводящий к изготовлению метиленового синего фармацевтической степени чистоты (WO2008006979). В данном патенте описана очистка неочищенного MTC посредством последовательности стадий, включая бензоилирование неочищенного MCT, которая ведет к образованию соответствующей бензоильной восстановленной формы с последующей кристаллизацией, окислением 2,3-дихлор-5,6-дициано-1,4-бензохиноном (DDQ - 2,3-dichloro-5,6-dicyano-1,4-benzoquinone) и конечной очисткой, проводимой последовательно посредством ионообменной хроматографии и кристаллизации продукта из водной среды (см. Схему 3).

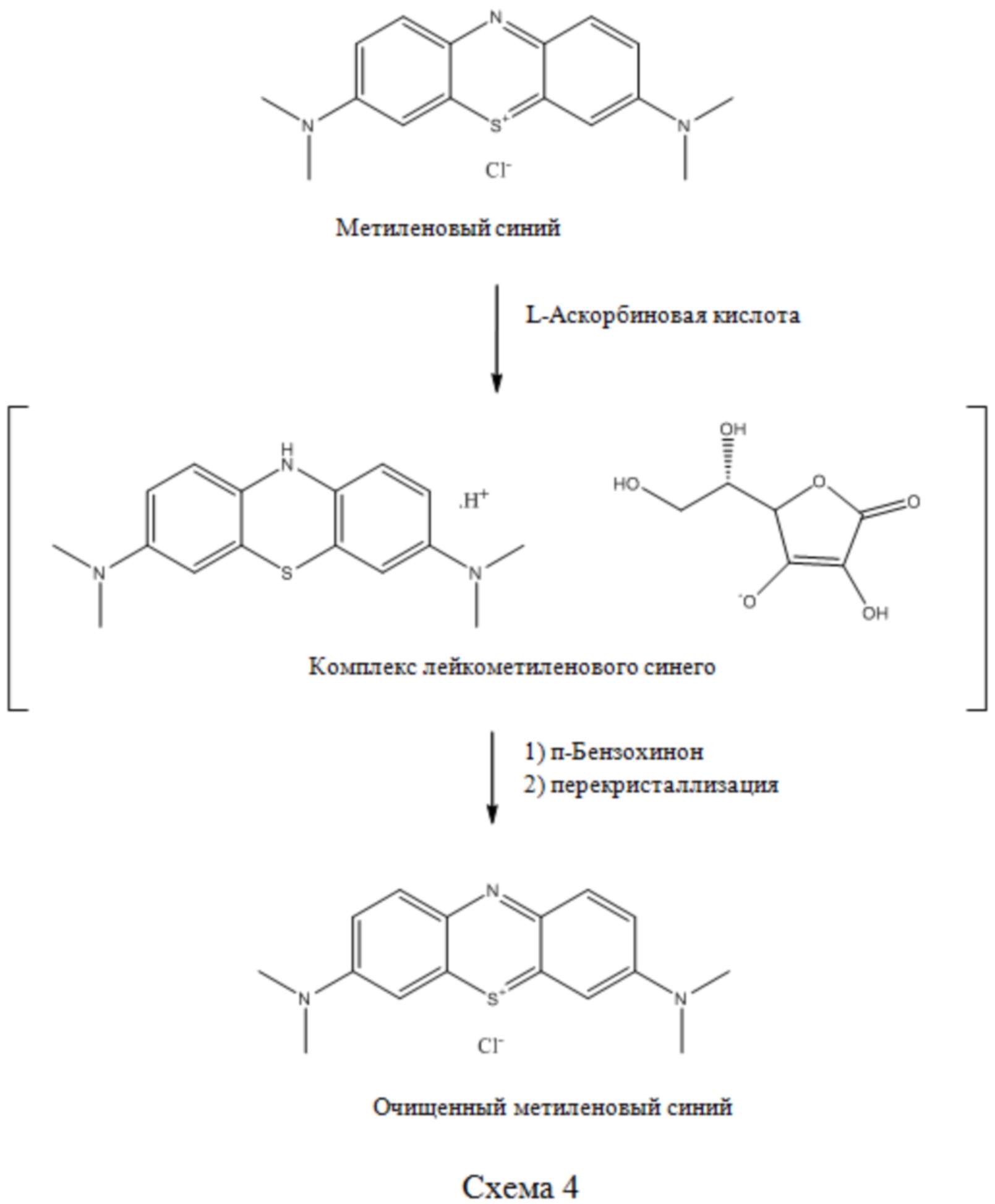
E. Malvin в WO 2015021500 описывает очистку МТС технической степени чистоты с использованием аскорбиновой кислоты с получением лейкометиленового синего и последующего окисленияп-бензохиноном (см. Схему 4).
Схема 4
Очищенный метиленовый синий
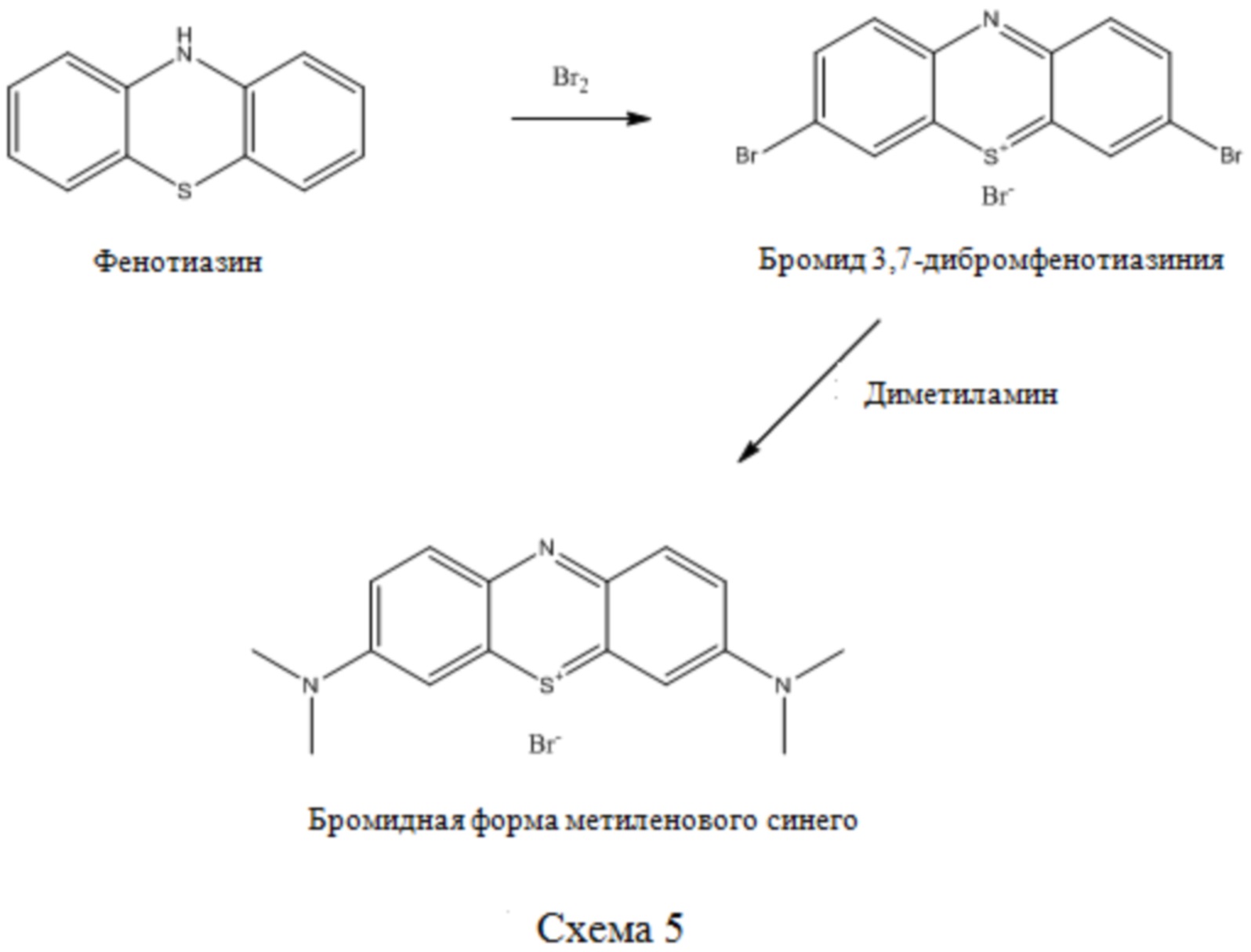
MTC можно также получать из фенотиазина, используя реакцию нитрирования. В незначительном изменении данного способа на первой стадии используется бром для бромирования фенотиазина в положениях 3 и 7 вместе с одновременным получением бромидов 3,7-дибромфенотиазин-5-ия с последующей реакцией с диметиламином с получением бромидной формы MTC (см. Схему 5). В литературе данный способ получения MTC известен как синтез по Керманну (Kehrmann F. Ber., 1916, 49, 53; Kehrmann F.: Diserens L. Ber., 1915, 48, 318) и в настоящее время используется для изготовления MTC.
Незначительное изменение способа синтеза по Керманну использовали N. Leventis et al. Tetrahedron Vol. 53 N 29, p. 10083-10092 (1997) для получения аналогов MTC, начиная с фенотиазина. Leventis et al. описывали 2-стадийный способ, начиная с превращения фенотиазина в бромид 3,7-дибромфенотиазин-5-ия посредством реакции с бромом в уксусной кислоте с последующей реакцией ароматического нуклеофильного замещения бромида 3,7-дибромфенотиазин-5-ия двухзамещенным амином в этаноле или хлороформе в качестве растворителя с образованием бромидной соли аналогов МТС. Указанный выше способ имел низкие выходы (27-63%), при получении дивинилового аналога метиленового синего, но имел хороший выход (70-85%) при получении метиленового синего (бромидная соль). Однако, данный способ имел некоторые недостатки: он требовал больших количеств брома на стадии 1 (20-молярный избыток брома) для достижения полного превращения фенотиазина в бромид 3,7-дибромфенотиазин-5-ия, так как применение меньших количеств приводило к получению смеси бромида 3,7-дибромфенотиазин-5-ия (окисленная форма) и 3,7-дибромфенотиазина (восстановленная форма, побочный продукт); бромид 3,7-дибромфенотиазин-5-ия нужно было выделить и очистить, поскольку данное вещество не очень стабильно, особенно в растворе; кроме того, на второй стадии, выбранный растворитель (этанол) конкурировал с двухзамещенным амином за нуклеофильное замещение с образованием побочных продуктов.
Обычными недостатками приведенных способов синтеза являются низкие выходы продукта из-за низкой региоселективности реакции, необходимость выделять бромид 3,7-дибромфенотиазин-5-ия перед следующей стадией реакции; длительное время протекания реакций; применение огромного количества растворителей для реакции и разделения промежуточных соединений; и необходимость применять большой молярный избыток брома.
Более того, широкое применение MTC в качестве фармацевтического средства требует очень чистого вещества, содержащего только небольшие количества металлов.
Краткое описание изобретения
Одной целью настоящего изобретения является предложение улучшенного способа получения бромида или хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия в пределах стандартов качества, установленных американскими и европейскими организациями здравоохранения, подходящего для применения в качестве фармацевтического средства. В одном воплощении настоящее изобретение представляет собой способ получения бромида или хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия, имеющего характеристики качества, удовлетворяющие требованиям американских и европейских организаций здравоохранения, который можно осуществлять быстрым и экономически выгодным путем. В частности, способ по данному изобретению позволяет выполнять 2 химические стадии способа, а именно превращение фенотиазина в соответствующий галогенид 3,7-дигалогенфенотиазин-5-ия и последующая трансформация данного галогенида 3,7-дигалогенфенотиазин-5-ия в бромид или хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия в виде однореакторной реакции, которую проводят в одном и том же реакционном сосуде и с использованием одной системы растворителей, общей для обеих стадий реакции.
Преимущественно, в конкретном способе по изобретению не используется выделение и/или очистка какого-либо промежуточного соединения реакции после осуществления реакции фенотиазина с подходящим галогеном, перед тем, как перейти к последующим стадиям способа. Способ по изобретению позволяет исключить время, необходимое для выделения и очистки промежуточных соединений, уменьшить производственные отходы, исключить риск того, что нестабильные промежуточные соединения могут деградировать во время стадий выделения/очистки, и применять нетоксичные растворители. Таким образом, способ по изобретению представляет собой значительное улучшение, по сравнению с предшествующим уровнем техники.
Подробное описание изобретения
Одной целью настоящего изобретения является предложение улучшенного способа получения бромида или хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия в пределах стандартов, установленных американскими или европейскими организациями здравоохранения, подходящего для фармацевтического применения.
Согласно настоящему изобретению предложен способ получения бромида или хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (Схема 6 и Схема 7), включающий следующие стадии:
(а) осуществление реакции хлора или брома с фенотиазином, необязательно в присутствии подходящего катализатора на основе металла, с получением хлорида 3,7-дихлорфенотиазин-5-ия или бромида 3,7-дибромфенотиазин-5-ия, соответственно;
(б) добавление диметиламина к реакционной смеси со стадии (а) с получением хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия, соответственно;
(в) необязательно, очистка хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия, полученного на стадии (б), предпочтительно посредством сливания данных соединений в подходящем растворителе или смеси растворителей;
(г) необязательно, элюирование веществ (бромида) со стадии (б) или (в) в ионообменной колонке с обменом бромида на хлорид;
(д) необязательно, растворение хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия со стадии (б) или (в) в водном растворе соляной кислоты и очистка продуктов; и
(е) необязательно, кристаллизация хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромида 3,7-дибром-фенотиазин-5-ия со стадии б), в) или г);
где указанную первую (а) и вторую (б) стадии проводят без выделения и/или очистки промежуточных соединений, образованных на стадии (а) (хлорид 3,7-дихлорфенотиазин-5-ия или бромид 3,7-дибромфенотиазин-5-ия).
Согласно настоящему изобретению стадии (а) и (б) способа представляют собой однореакторную реакцию, которую проводят в одном и том же реакционном сосуде и с использованием одной системы растворителей, общей для обеих стадий реакции. Стадия (г), которая представляет собой превращение из формы бромида в форму хлорида, является необязательной и ее проводят, когда вещество, полученное на стадии (в), представляет собой бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия.
Для получения хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) стадия (г) элюирования вещества со стадии (в) в ионообменной колонке с обменом бромида на хлорид является необходимой, когда вещество, полученное на стадии (в), представляет собой 3,7-бис-(диметиламино)-фенотиазин-5-ия бромид.

Преимущественно, согласно способу по данному изобретению получают MTC за две стадии реакции, (а) и (б), которые проводят в одном и том же реакционном сосуде без необходимости разделения и очистки промежуточных соединений между данными стадиями, он позволяет исключить время, необходимое для выделения и очистки промежуточных соединений, и позволяет повысить выходы продукта за один проход в единицу времени на единицу объема реакторов. Кроме того, поскольку на обеих стадиях реакции (а) и (б) используют одну и ту же систему растворителей, способ по изобретению не требует процесса смены растворителя после стадии (а) с заменой его другим растворителем для проведения стадии (б). Более того, способ по изобретению делает возможным уменьшение молярного избытка хлора или брома на стадии реакции (а) относительно фенотиазина, по сравнению с тем, о чем сообщается в предшествующем уровне техники. Уменьшение молярных эквивалентов бромида или хлора на стадии (а) и одна единственная система растворителей для стадий (а) и (б) обеспечивает значительное уменьшение отходов растворителей, что является огромным уменьшением затрат при работе в промышленном масштабе,. Дополнительным преимуществом способа по изобретению является то, что исключение необходимости в выделении и очистке бромида 3,7-дибромфенотиазин-5-ия или хлорида 3,7-дихлорфенотиазин-5-ия после стадии (а) обеспечивает уменьшение риска того, что те вещества, которые, как известно, являются довольно нестабильными, особенно в растворе, могут деградировать во время стадии выделения и/или очистки. Дополнительным преимуществом способа по изобретению является то, что он позволяет применять растворители с низкой токсичностью, например, этилацетат. Данный последний аспект настоящего изобретения является особенно благоприятным, когда продукт химического способа является активным фармацевтическим ингредиентом, для которого применение растворителей с низкой токсичностью настоятельно рекомендуется руководствами основных организаций здравоохранения, таких как EMEA (European Medicines Agency - Европейское агентство лекарственных средств) и FDA (Food and Drug Administration - Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) (см. ICH руководство Q3(R5)). В способе используются недорогие способы очистки конечного продукта, обеспечивая получение MTC с очень низким содержанием побочных продуктов и металлов, вследствие этого, продукт подходит для фармацевтического применения.
В одном воплощении настоящее изобретение представляет собой «невыделенное» промежуточное соединение, полученное со стадии (а) Схемы 6, а именно хлорид 3,7-дихлорфенотиазин-5-ия. Данное соединение было выделено и охарактеризовано авторами настоящего изобретения лишь с аналитической целью.
Одно воплощение настоящего изобретения представляет собой хлорид 3,7-дихлорфенотиазин-5-ия.
Одно воплощение настоящего изобретения представляет собой хлорид 3,7-дихлорфенотиазин-5-ия для применения в качестве промежуточного соединения.
В одном воплощении изобретение представляет собой способ получения хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия, включающий следующие стадии:
а) осуществление реакции хлора или брома с фенотиазином с получением хлорида 3,7-дихлорфенотиазин-5-ия или бромида 3,7-дибромфенотиазин-5-ия, соответственно;
б) добавление диметиламина к реакционной смеси стадии а) с получением хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия, соответственно;
где стадии а) и б) проводят без выделения и/или очистки промежуточного соединения, образованного на стадии а). В одном воплощении на стадии (а) способа по изобретению фенотиазин растворяют в системе растворителей, которая контактирует с хлором или бромом с получением промежуточного соединения хлорида 3,7-дихлорфенотиазина или бромида 3,7-дибромфенотиазина. Все молярные отношения хлор или бром:фенотиазин выше чем 1:1 подходят для стадии (а) способа по данному изобретению, при условии, что в качестве промежуточных соединений получают хлорид 3,7-дихлорфенотиазина или бромид 3,7-дибромфенотиазина. Подходящие молярные отношения хлора или брома к фенотиазину на стадии (а) обычно находятся в интервале от примерно 1:1 до примерно 4:1 бром или хлор:фенотиазин, предпочтительно от примерно 2,2:1 до примерно 2,8:1 бром или хлор:фенотиазин. В некоторых воплощениях молярное отношение хлора или брома к фенотиазину на стадии (а) составляет примерно 2:1 бром или хлор:фенотиазин. В некоторых воплощениях молярное отношение хлора или брома к фенотиазину на стадии (а) составляет примерно 2,5:1 бром или хлор:фенотиазин. В некоторых воплощениях молярное отношение хлора или брома к фенотиазину на стадии (а) составляет примерно 3:1 бром или хлор:фенотиазин. В предпочтительном воплощении молярное отношение хлора или брома к фенотиазину на стадии (а) составляет примерно 2,5:1 бром или хлор:фенотиазин. В дополнительном предпочтительном воплощении молярное отношение хлора или брома к фенотиазину на стадии (а) составляет примерно 3:1 бром или хлор:фенотиазин.
Стадию (а) способа по изобретению можно в некоторых воплощениях проводить в присутствии инертного растворителя.
Термин «инертный» означает, что растворитель является нереакционноспособным в отношении реагентов и продуктов и не препятствует способу по данному изобретению. Любой инертный носитель подходит при условии, что он обеспечивает задуманную солюбилизацию, и при условии, что в качестве промежуточных соединений получают хлорид 3,7-дихлорфенотиазина или бромид 3,7-дибромфенотиазина.
Подходящие инертные растворители для стадии (а) включают полярные протонные и апротонные органические растворители, такие как дихлорметан, уксусная кислота, метилацетат, этилацетат, бутилацетат, хлороформ или их смеси. В одном воплощении растворитель представляет собой дихлорметан. В одном воплощении растворитель представляет собой метилацетат. В одном воплощении растворитель представляет собой бутилацетат. В предпочтительном воплощении растворитель представляет собой этилацетат. В некоторых воплощениях бром или хлор добавляют в реакционную смесь стадии (а) в форме растворов в указанном инертном растворителе. В других воплощениях бром или хлор добавляют в реакционную смесь стадии (а) непосредственно в виде чистых веществ, то есть в жидкой или в газовой форме, соответственно. Как общее правило, объемное/массовое отношение растворителя к фенотиазину составляет от примерно 1 до примерно 40, и предпочтительно составляет от примерно 2,5 до примерно 25. В одном воплощении реакцию фенотиазина с бромом или хлором на стадии (а) проводят при отсутствии катализатора. В одном воплощении реакция фенотиазина с бромом или хлором на стадии (а) протекает в присутствии катализатора на основе металла. Катализатор на основе металла может быть использован в своей безводной форме или во всех своих состояниях гидратации. Катализатор на основе металла, который используют на стадии (а) реакции, может представлять собой любой, который приводит к получению желательных промежуточных соединений. В некоторых воплощениях указанный катализатор на основе металла выбран из металлов группы VIII Периодической таблицы химических элементов Менделеева, например, железа, кобальта, никеля. В других воплощениях указанный катализатор на основе металла выбран из металлов группы IB Периодической таблицы химических элементов Менделеева, например, меди и/или серебра. В других воплощениях указанный катализатор на основе металла выбран из металлов группы IIIB Периодической таблицы химических элементов Менделеева, таких как, в качестве примера, алюминий. В некоторых воплощениях катализатор ан основе металла добавляют к реакционной смеси на стадии (а) в элементной форме. В других воплощениях катализатор на основе металла добавляют к реакционной смеси на стадии (а) в форме соли. Согласно последним воплощениям подходящие соли указанных выше металлов могут быть выбраны из группы, состоящей из хлоридов, бромидов, йодидов, фторидов, карбонатов, нитратов, сульфатов, фосфатов, цитратов, ацетатов, малеатов, но не ограничивающейся ими. В других воплощениях катализатор на основе металла находится в форме оксида. В одном воплощении катализатор на основе металла стадии (а) представляет собой железо. В предпочтительном воплощении катализатор на основе металла стадии (а) представляет собой соль железа. Согласно последнему воплощению указанная соль железа может представлять собой соль железа (II) или соль железа (III), предпочтительно соль железа (III). Соль железа (III) можно использовать в ее безводной форме или во всех ее состояниях гидратированных форм. В более предпочтительном воплощении хлорид железа (III) используют в его форме гексагидрата. В некоторых воплощениях указанная соль железа (II) может быть выбрана из группы, состоящей из: хлорида железа (II), бромида железа (II), йодида железа (II), фторида железа (II), карбоната железа (II), нитрата железа (II), сульфата железа (II), фосфата железа (II), ацетата железа (II), малеата железа (II), но не ограничивающейся ими. В некоторых воплощениях указанная соль железа (III) может быть выбрана из группы, состоящей из: хлорида железа (III), бромида железа (III), йодида железа (III), фторида железа (III), карбоната железа (III), нитрата железа (III), сульфата железа (III), фосфата железа (III), ацетата железа (III), малеата железа (III), но не ограничивающейся ими. В предпочтительном воплощении указанная соль железа (II) представляет собой сульфат железа (II). В предпочтительном воплощении указанная соль железа (III) представляет собой хлорид железа (III).
Несмотря на имеющиеся знания в данной области, авторами настоящего изобретения по существу неожиданно обнаружено, что описанные выше катализаторы на основе металла могут быть полезным образом получены также с использованием соли железа (II), предпочтительно сульфата железа (II).
Согласно изобретению можно использовать любую гидратную форму солей железа.
Любые рабочие условия способа могут быть использованы в способе по данному изобретению, при условии, что образуются желаемы промежуточные продукты. Согласно способу по изобретению интервалы температур способа для стадии (а) составляют от примерно минус 35°C до примерно 45°C, предпочтительно от примерно минус 30°C до примерно 35°C, более предпочтительно от примерно -20°C до примерно 15°C, гораздо более предпочтительно от примерно минус 20°C до примерно минус 10°C. В одном предпочтительном воплощении температура для стадии (а) составляет примерно минус 15°C. В одном воплощении температуру стадии (а) поддерживают в интервале от примерно минус 25°C до примерно 0°C. В другом воплощении температуру стадии (а) поддерживают в интервале от примерно минус 20°C до примерно минус 5°C. В предпочтительном воплощении температуру стадии (а) поддерживают в интервале от примерно минус 20°C до примерно минус 10°C. В одном предпочтительном воплощении температуру стадии (а) поддерживают примерно при минус 15°C. В одном воплощении стадию (а) проводят при температуре меньше чем примерно 45°C, 35°C, 25°C, 15°C, 10°C, 5°C, 0°C, минус 5°C, минус 10°C, минус 15°C, минус 20°C или минус 25°C. В одном воплощении стадию (а) проводят при температуре меньше чем примерно 25°C. В одном воплощении стадию (а) проводят при температуре меньше чем примерно 0°C. В одном воплощении стадию (а) проводят при температуре меньше чем примерно минус 10°C. В одном предпочтительном воплощении стадию (а) проводят при температуре от примерно минус 20°C до примерно минус 10°C. В еще одном более предпочтительном воплощении стадию (а) проводят при температуре примерно минус 15°C.
Время реакции стадии (а) зависит от температуры реакции, скорости смешивания реагентов и концентрации реагентов в реакционной зоне. В одном воплощении, необязательно в реакторе периодического действия, время реакции стадии (а) составляет примерно 6 часов или менее. В одном воплощении время реакции стадии (а) составляет примерно 5 часов. В одном воплощении время реакции стадии (а) составляет примерно 4 часа. В одном воплощении время реакции стадии (а) составляет примерно 3 часа. В одном воплощении время реакции стадии (а) составляет примерно 2 часа. В некоторых воплощениях время реакции стадии (а) составляет примерно 3 часа, предпочтительно примерно 2 часа, более предпочтительно примерно 1 час. В предпочтительном воплощении время реакции стадии (а) составляет примерно 1 час. Время реакции может быть должным образом адаптировано, исходя из реакционных сосудов и масштаба способа (например, переходя от полупромышленного к промышленному масштабу).
В одном воплощении настоящего изобретения стадию (б) способа по настоящему способу можно проводить посредством добавления диметиламина в реакционную смесь, богатую хлоридом 3,7-дихлорфенотиазина или бромидом 3,7-дибромфенотиазина, образованным на стадии (а), без какой-либо промежуточной стадии очистки или выделения. В одном воплощении указанный диметиламин добавляют в реакционную смесь стадии (б) в виде чистого диметиламина. Согласно такому воплощению указанный чистый диметиламин может находиться в газообразной или жидкой форме. Согласно другому воплощению указанный диметиламин добавляют в реакционную смесь стадии (б) в форме раствора в подходящем растворителе. Согласно такому воплощению указанный подходящий растворитель должен быть инертным и может быть выбран из группы, состоящей из метанола, воды, бутилацетата и этилацетата и их смесей, но не ограничивающейся ими. Согласно предпочтительному воплощению указанный диметиламин добавляют в реакционную смесь стадии (б) в форме раствора в этилацетате. Согласно предпочтительному воплощению указанный диметиламин добавляют в реакционную смесь стадии (б) в форме раствора в метаноле. Подходящее молярное отношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), находится в интервале от примерно 1:1 до примерно 1:5, предпочтительно в интервале от примерно 1:1,5 до примерно 1:4,5, более предпочтительно в интервале от примерно 1:2,0 до примерно 1:4,0 бром или хлор:диметиламин. В одном воплощении молярное отношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), составляет примерно 1:2. В другом воплощении молярное соотношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), составляет примерно 1:2,6. В другом воплощении молярное соотношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), составляет примерно 1:3,0. В другом воплощении молярное соотношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), составляет примерно 1:3,4. В другом воплощении молярное соотношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), составляет примерно 1:4,0. В предпочтительном воплощении молярное соотношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), составляет примерно 1:3,0. В предпочтительном воплощении молярное соотношение хлора или брома, используемого на стадии (а), к диметиламину, используемому на стадии (б), составляет примерно 1:3,1.
Согласно способу по изобретению реакцию стадии (б) можно проводить при температуре от примерно минус 35°C до примерно 45°C, предпочтительно при температуре от примерно минус 30°C до примерно 35°C, более предпочтительно при температуре от примерно минус 25°C до примерно 20°C, гораздо более предпочтительно при температуре от примерно минус 20°C до примерно 10°C. В одном предпочтительном воплощении стадию (б) проводят при температуре примерно минус 15°C.
В одном воплощении температуру стадии (б) поддерживают в интервале от примерно минус 25°C до примерно 25°C. В другом воплощении температуру стадии (б) поддерживают в интервале от примерно минус 20°C до примерно 20°C. В другом воплощении температуру стадии (б) поддерживают в интервале от примерно минус 20°C до примерно 15°C. В предпочтительном воплощении температуру стадии (б) поддерживают в интервале от примерно минус 20°C до примерно 10°C. В одном предпочтительном воплощении температуру стадии (б) поддерживают при примерно минус 15°C.
В одном воплощении стадию (б) проводят при температуре меньше чем примерно 45°C, 40°C, 35°C, 30°C, 25°C, 20°C, 15°C или 10°C. В одном воплощении стадию (б) проводят при температуре меньше чем примерно 25°C. В одном воплощении стадию (б) проводят при температуре меньше чем примерно 20°C. В одном воплощении стадию (б) проводят при температуре меньше чем примерно 10°C. В одном воплощении стадию (б) проводят при температуре от примерно минус 15°C до примерно 10°C.
В одном воплощении стадию (а) проводят при температуре меньше чем примерно 0°C, предпочтительно меньше чем примерно минус 10°C, более предпочтительно от примерно минус 20°C до примерно минус 10°C, и стадию (б) проводят при температуре меньше чем примерно 20°C , предпочтительно меньше чем примерно 10°C, более предпочтительно от примерно минус 20°C до примерно 10°C. В одном предпочтительном воплощении стадию (а) проводят при температуре примерно минус 15°C и стадию (б) проводят при температуре примерно минус 15°C.
Время реакции стадии (б) зависит от температуры реакции, скорости смешивания реагентов и концентрации реагентов в реакционной зоне. В одном воплощении возможно в реакторе периодического действия время реакции стадии (б) равно или меньше чем примерно 6 часов. В одном воплощении время реакции стадии (б) составляет примерно 5 часов. В одном воплощении время реакции стадии (б) составляет примерено 4 часа. В одном воплощении время реакции стадии (б) составляет примерно 3 часа. В одном воплощении время реакции стадии (б) составляет примерено 2 часа. В некоторых воплощениях время реакции стадии (б) составляет примерено 3 часа, предпочтительно примерно 2 часа, более предпочтительно примерно 1 час. В предпочтительном воплощении время реакции стадии (б) составляет примерно 2 часа.
Время реакции может быть должным образом адаптировано, исходя из реакционных сосудов и масштаба способа (например, переходя от полупромышленного к промышленному масштабу).
В одном воплощении настоящего изобретения стадии (а) и (б) проводят в одном и том же реакционном сосуде и в одной и той же системе растворителей. Поскольку региоселективность реакции на стадии (а) способа является высокой, разделение и очистка основных промежуточных соединений, образованных на стадии (а), не нужны. Не желая быть связанными какой-либо теорией, полагают, что высокая региоселективность стадии (а) обусловлена, по меньшей мере частично, выбранными температурными интервалами или комбинацией температурных интервалов и выбранного растворителя. Реакционную смесь со стадии (а) непосредственно используют для реакции с диметиламином.
В способе по изобретению конечные продукты реакции со стадии (б) хлорид или бромид 3,7-бис-(диметиламин)-фенотиазин-5-ия представляют собой твердые вещества, которые только слабо растворимы в инертном растворителе, используемом для обеих реакций стадий (а) и (б). Указанные продукты реакции могут быть отделены от реакционной смеси посредством использования обычных методик разделения твердой фазы и жидкости, хорошо известных в данной области техники, таких как, например, фильтрование или осаждение или центрифугирование.
В одном воплощении после отделения неочищенного продукта, полученного после стадии (а) и (б), от реакционной смеси посредством использования обычных методик разделения твердой фазы и жидкости, твердый неочищенный продукт промывают используемой системой инертных растворителей и затем разбавленным водным раствором соляной кислоты. Концентрация соляной кислоты находится в интервале от примерно 0,05 моль/л до примерно 2,0 моль/л, предпочтительно от примерно 0,1 моль/л до примерно 1,0 моль/л, более предпочтительно от примерно 0,1 моль/л до примерно 0,4 моль/л. После промывки гидробромидом или гидрохлоридом диметиламина, образованных в способе, их удаляют из желаемых продуктов.
Согласно изобретению дополнительную очистку продуктов, полученных на стадии (а) и (б), можно проводить для получения очень низкой концентрации побочных продуктов и металлов (в пределах спецификаций американских и европейских фармакопейных статей). Такая очистка (стадия (в)) может включать разные стадии сливания хлорида или бромида 3,7-бис-(диметиламин)-фенотиазин-5-ия при перемешивании при разных температурах и с подходящим растворителем. В одном воплощении указанная очистка включает следующие стадии:
1) сливание указанных продуктов со стадий (а) и (б) с растворителем или смесями растворителей при температуре в интервале от примерно 2°C до примерно 35°C, предпочтительно от примерно 5°C до примерно 30°C, более предпочтительно от примерно 20°C до примерно 25°C;
сливание указанных продуктов со стадий (а) и (б) с растворителем или смесями растворителей при температуре в интервале от примерно 25°C до примерно 75°C, предпочтительно от примерно 30°C до примерно 65°C, более предпочтительно от примерно 40°C до примерно 60°C; и, возможно,
2) вновь сливание указанных продуктов со стадий (а) и (б) с растворителем или смесями растворителей при температуре в интервале от примерно -5°C до примерно 20°C, предпочтительно от примерно 0°C до примерно 15°C, более предпочтительно от примерно 5°C до примерно 10°C.
Любые из указанных выше стадий можно повторять несколько раз для получения продукта желаемого качества, имеющего очень низкую концентрацию побочных продуктов и металлов (в пределах спецификаций американских и европейских фармакопейных статей). После каждой стадии твердое вещество можно отделять от промывочного растворителя посредством обычной методики разделения твердой фазы и жидкости, хорошо известной в данной области техники, такой как, например, фильтрование или осаждение или центрифугирование. Преимущественно, перед любой стадией отделения твердой фазы от жидкой фазы смесь, подлежащая отфильтровыванию, должна находиться при температуре, составляющей примерно 30°C или менее, для максимизации выхода твердого вещества. Стадию отделения проводят при температуре, составляющей примерно 30°C или менее, предпочтительно в интервале от примерно минус 5°C до примерно 25°C, более предпочтительно от примерно 0°C до примерно 15°C, гораздо более предпочтительно от примерно 5°C до примерно 10°C.
Подходящие растворители или смеси растворителей для очистки включают полярные апротонные и/или протонные растворители. Указанные полярные протонные и апротонные растворители включают дихлорметан, метилацетат, этилацетат, бутилацетат, метанол, этанол, 2-пропанол, соляную кислоту, воду или их смеси, но не ограничиваются ими. Предпочтительно, в качестве растворителей используют чистые спирты или смеси водных спиртов с относительным. соотношением объемов, находящимся в интервале от 1/4 до 5/1. В некоторых воплощениях растворитель для промывки представляет собой спирт, предпочтительно метанол, этанол или 2-пропанол. В некоторых воплощениях указанный спирт является чистым. В других воплощениях указанный спирт содержит процент воды, предпочтительно в интервале от примерно 5% до примерно 80%, более предпочтительно от примерно 10% до примерно 40%, гораздо более предпочтительно от примерно 10% до примерно 30%. В некоторых воплощениях растворитель представляет собой раствор соляной кислоты в воде. Согласно таким воплощениям концентрация соляной кислоты в воде предпочтительно находится в интервале от примерно 0,05 моль/л до примерно 2,0 моль/л, более предпочтительно от примерно 0,1 моль/л до примерно 0,4 моль/л. В некоторых воплощениях очистку проводят с использованием смеси вышеупомянутых растворителей.
Стадии очистки можно повторять несколько раз и можно использовать один единственный растворитель или системы на основе смеси растворителей.
В одном воплощении очистку хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромида 3,7-дибром-фенотиазин-5-ия, полученного на стадии (б), (в) или (г), можно проводить посредством кристаллизации.
Подходящие растворители или смеси растворителей для кристаллизации включают метанол, этанол, 2-пропанол, соляную кислоту, воду или их смеси.
Кристаллизацию можно повторять несколько раз и можно использовать один единственный растворитель или системы на основе смеси растворителей.
В одном воплощении, когда продукт, полученный после стадий (а), (б) и возможно (в), представляет собой бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия, для получения хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) указанный продукт нужно элюировать в ионообменной колонке с обменом бромид-иона на хлорид-ионом. Согласно такому воплощению после необязательной стадии очистки (в) продукт, полученный со стадий (а) - (б), растворяют в желательном растворителе и элюируют через колонку, содержащую макропористую анионообменную смолу в активированной форме хлорида, с осуществлением анионного обмена на продукт (стадия (г)).
Подходящие растворители для растворения продукта могут представлять собой метанол, этанол, 2-пропанол, воду или их смеси. В некоторых воплощениях используют смесь этанола и воды, причем смесь имеет процентное сооотношение объемов воды, находящееся в интервале от примерно 1% до примерно 40%, более предпочтительно от примерно 5% до примерно 35%, более предпочтительно от примерно 8% до примерно 25%, гораздо более предпочтительно от примерно 9% до примерно 20%. В некоторых воплощениях используют смесь метанола и воды, причем смесь имеет процентное соотношение объемов воды, находящееся в интервале от примерно 1% до примерно 40%, более предпочтительно от примерно 5% до примерно 35%, более предпочтительно от примерно 8% до примерно 25%, гораздо более предпочтительно от примерно 9% до примерно 20%. В некоторых воплощениях используют смесь 2-пропанола и воды, причем смесь имеет об./об. процентное содержание воды, находящееся в интервале от примерно 1% до примерно 40%, более предпочтительно от примерно 5% до примерно 35%, более предпочтительно от примерно 8% до примерно 25%, гораздо более предпочтительно от примерно 9% до примерно 20%. Затем данный раствор элюируют на колонке, наполненной анионообменной смолой в активированной форме хлорида.
В одном воплощении макропористая анионообменная смола представляет собой макропористый полистирол, сшитый с дивинилбензолом, содержащий группы четвертичного аммония.
В одном воплощении макропористая анионообменная смола представляет собой Purolite A500® в форме хлорида.
В одном воплощении стадию конечной кристаллизации хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия можно проводить в водном растворе соляной кислоты, предпочтительно в интервале концентраций от примерно 0,05 моль/л до примерно 2,0 моль/л, более предпочтительно от примерно 0,1 моль/л до примерно 0,4 моль/л (стадии (д) и (е)).
В одном воплощении чистота полученных продуктов бромида (3,7-бис-(диметиламино)-фенотиазин-5-ия и хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC)) попадает в текущую предварительную версию и следующие уточнения в отношении стандартов, изложенных в американских и европейских фармакопейных статьях хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC).
В одном воплощении продукт по настоящему изобретению имеет очень низкое содержание металлов, например, меньше чем примерно 1 млн-1 мышьяка, меньше чем примерно 2 млн-1 меди и/или меньше чем примерно 2 млн-1 цинка.
В одном воплощении, таким образом, согласно настоящему изобретению также предложен хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия, имеющий чистоту, выше чем примерено 97%, и меньше чем примерно 1 млн-1 мышьяка.
В другом воплощении согласно настоящему изобретению также предложен хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия, имеющий чистоту выше чем примерно 97% и ниже чем примерно 2 млн-1 меди и меньше чем примерно 2 млн-1 цинка.
В другом воплощении согласно настоящему изобретению также предложен хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия, имеющий чистоту, выше чем примерно 97%, меньше чем примерно 1 млн-1 мышьяка, меньше чем примерно 2 млн-1 меди и меньше чем примерно 2 млн-1 цинка.
В одном дополнительном воплощении согласно настоящему изобретению также предложен хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия, полученный или который можно получить способом получения хлорида или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия, как раскрыто выше.
В одном дополнительном воплощении согласно настоящему изобретению предложено соединение, полученное на стадии (а) Схемы 6, а именно собственно хлорид 3,7-дихлорфенотиазин-5-ия.
В одном дополнительном воплощении согласно настоящему изобретению предложено соединение, полученное на стадии (а) Схемы 6, а именно хлорид 3,7-дихлорфенотиазин-5-ия, в качестве промежуточного соединения способа поучения хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия.
В одном дополнительном воплощении согласно настоящему изобретению предложена фармацевтическая композиция, содержащая хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC) или бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия, имеющий характеристики, как раскрыто выше, в отношении чистоты/металлов, и/или полученный или которыйможно получить способом получения хлорида или бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия, как раскрыто выше.
В одном дополнительном воплощении согласно настоящему изобретению предложен хлорид или бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия (MTC), как раскрыто выше, или фармацевтическая композиция, как раскрыто выше, для применения в области диагностики, предпочтительно в оценке на основе эндоскопии желудочно-кишечного тракта воспалительных, язвенных, предраковых, неопластических, диспластических патологий и/или очагового поражения желудочно-кишечного тракта. Согласно такому воплощению изобретение способно обеспечивать усиление выявления очаговых поражений слизистой кишечника, как, например, предраковых форм, интервальных видов рака, аденом, карцином, пилоообразных очаговых поражений, внутриэпителиальной неоплазии, дисплазии, полипов, псевдополипов, предполипов или разных воспалительных патологий, очаговых повреждений на широком основании, плоских или на ножке.
Примеры
Пример 1. Неочищенный бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия
В охлажденный реакционный сосуд (при 10°C) добавляют фенотиазин (10,86 кг), этилацетат (216 литров) и катализатор, хлорид железа (III) (30 г). Реакционную смесь охлаждают (при 10°C) и затем добавляют бром (21,84 кг, 7,0 литров) в этилацетате (21,0 литр). Температуру реакции поддерживают в интервале от минус 10°C до минус 15°C. После добавления всего брома реакционную смесь перемешивают еще в течение одного часа при температуре от минус 10°C до минус 15°C.
Затем добавляли диметиламин (19,0 кг) в этилацетате (77 литров) и температуру реакции поддерживают в интервале от минус 15°C до 10°C. После добавления всего диметиламина реакционную смесь перемешивают еще в течение двух часов при температуре от минус 15°C до 10°C.
Неочищенный продукт отфильтровывали, промывали охлажденным этилацетатом и соляной кислотой концентрации 0,1 моль/л. Выделенный продукт затем промывали с использованием описанных ниже способов.
Пример 1. Копия. Неочищенный бромид 3,7-бис-(диметиламино)-фенотиазин-5-ия
В охлажденный реакционный сосуд (при 10°C) добавляют фенотиазин (10,86 кг), этилацетат (216 литров) и катализатор, хлорид железа (III) (30 г). Реакционную смесь охлаждают (при 10°C) и затем добавляют бром (21,84 кг, 7,0 литров) в этилацетате (21,0 литр). Температуру реакции поддерживают в интервале от минус 10°C до минус 15°C. После добавления всего брома реакционную смесь перемешивают еще в течение одного часа при температуре от минус 10°C до минус 15°C.
Затем добавляют диметиламин (19,0 кг) в метаноле (77 литров) и температуру реакции поддерживают в интервале от минус 15°C до 10°C. После добавления всего диметиламина реакционную смесь перемешивали еще в течение двух часов при температуре от минус 15°C до 10°C.
Неочищенный продукт отфильтровывают, промывают охлажденным этилацетатом и соляной кислотой концентрации 0,1 моль/л. Затем выделенный продукт промывают с использованием описанных ниже способов
Пример 2. Неочищенный хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия
В охлажденный реакционный сосуд (при 10°C) добавляют фенотиазин (10,86 г), этилацетат (250 мл) и катализатор, хлорид железа (III) (30 мг). Реакционную смесь охлаждают (при 10°C) и добавляют газообразный хлор (9,66 г, сжиженный под действием минус 30°C). Температуру реакции поддерживают в интервале от минус 10°C до минус 15°C. После добавления всего хлора реакционную смесь перемешивают еще в течение одного часа при температуре в интервале от минус 10°C до минус 15°C.
Затем добавляют диметиламин (19,0 г) в этилацетате (77 мл) и температуру реакции поддерживают в интервале от минус 15°C до 10°C. После добавления всего диметиламина реакционную смесь перемешивают еще в течение двух часов при температуре от минус 15°C до 10°C.
Неочищенный продукт отфильтровывали, промывают охлажденным этилацетатом и соляной кислотой концентрации 0,1 моль/л. Затем выделенный продукт очищают с использованием описанных ниже способов аликвотными объемами растворителей.
Пример 3. Очищенный хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия из неочищенного бромида 3,7-бис(диметиламино)-фенотиазин-5-ия
Выделенный продукт из Примера 1 (бромид 3,7-бис(диметиламино)-фенотиазин-5-ия) промывают при перемешивании в 2-пропаноле (60 литров) при комнатной температуре в течение одного часа и отфильтровывают. Добавляют 2-пропанол и повторяют промывку при перемешивании при температуре примерно 50°C, смесь охлаждают до комнатной температуры и отфильтровывают.
В конце добавляют смесь 2-пропанола (12 литров) и соляной кислоты концентрации 0,5 моль/л (48 литров), и процедуру получения суспензии повторяют при температуре примерно 50°C. Смесь охлаждают до комнатной температуры и отфильтровывают.
Продукт растворяют в смеси 2-пропанола (100 литров) и воды (20 литров) при 60°C и элюируют через колонку с анионообменной смолой (45 литров Purolite A500, форма хлорида); затем элюаты концентрируют посредством выпаривания. Сухой продукт растворяют в соляной кислоте концентрации 0,4 моль/л (100 литров) при 60°C и затем охлаждают при 10 °C, раствор перемешивают при данной температуре в течение двух часов и продукт отфильтровывают и сушат. Выход 9,6 кг.
Спектры13C и1H ЯМР (ядерный магнитный резонанс) ((CD3)2SO; 600 МГц) данного продукта подтверждают структуру соединения. Спектр1H ЯМР (значения в млн-1): 3.34 (s, 12H, 2xN(CH3)2, 7.43 (m, 4H, ароматические соединения; 2, 4, 6 и 8 CH), 7,83 (d, 2H, ароматические соединения; 1 и 9 CH). Спектр13C ЯМР (значения в млн-1): 41.1 (N(CH3)2), 106,8 (4 и 6 CH), 119,0 (2 и 8 CH), 133,5 (12 и 13 CS+), 134,9 (11 и 14 CN), 137,8 (1 и 9 CH) и 153,8 (3 и 7 C-N(CH3)2).
Пример 4. Очищенный хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия из неочищенного бромида 3,7-бис(диметиламино)-фенотиазин-5-ия
Выделенный продукт из Примера 1 (бромид 3,7-бис(диметиламино)-фенотиазин-5-ия) суспендируют в этаноле (60 литров) при комнатной температуре в течение одного часа и отфильтровывают. Добавляют этанол и процедуру получения суспензии повторяют при температуре примерно 50°C. Смесь охлаждают до комнатной температуры и отфильтровывают.
В конце добавляют смесь этанола (12 литров) и соляной кислоты концентрации 0,5 моль/л (48 литров), и процедуру получения суспензии повторяют при температуре примерно 50 °C. Смесь охлаждают до комнатной температуры и отфильтровывают.
Продукт растворяют в смеси этанола (100 литров) и воды (20 литров) при 60°C и элюируют через колонку, наполненную анионообменной смолой (45 литров Purolite A500, форма хлорида); затем элюированные фракции выпаривают досуха под действием вакуума. Сухой продукт растворяют в соляной кислоте концентрации 0,4 моль/л (100 литров) при 60°C и охлаждают при 10°C, раствор перемешивают при данной температуре в течение двух часов и продукт отфильтровывают и сушат. Выход 10,8 кг.
Зарегистрированные спектры13C и1H ЯМР ((CD3)2SO; 600 МГц) подтверждают структуру исследуемого соединения: сигналы ЯМР практически совместимы с сигналами, полученными посредством анализа образца, выделенного в соответствии с примером 3.
Пример 5. Очищенный хлорид 3,7-бис-(диметиламино)-фенотиазин-5-ия из неочищенного бромида 3,7-бис(диметиламино)-фенотиазин-5-ия
Выделенный продукт из Примера 1 (бромид 3,7-бис(диметиламино)-фенотиазин-5-ия) суспендируют в метаноле (60 литров) при комнатной температуре в течение одного часа и отфильтровывают. Добавляют метанол, и процедуру получения суспензии повторяют при температуре примерно 50°C. Смесь охлаждают до комнатной температуры и отфильтровывают.
В конце добавляют смесь метанола (12 литров) и соляной кислоты концентрации 0,5 моль/л (48 литров), и способ получения суспензии повторяют при температуре примерно 50°C. Смесь охлаждают до комнатной температуры и отфильтровывают.
Продукт растворяют в смеси метанола (100 литров) и воды (20 литров) при 60°C и элюируют через колонку, наполненную анионообменной смолой (45 литров Purolite A500, форма хлорида); затем собранные элюаты концентрируют посредством выпаривания. Сухой продукт растворяют в соляной кислоте концентрации 0,2 моль/л (100 литров) при 60°C и охлаждают при 10°C. Раствор перемешивают при данной температуре в течение двух часов, и продукт отфильтровывают и сушат. Выход 12,4 кг.
Спектры13C и1H ЯМР ((CD3)2SO; 600 МГц), зарегистрированные на данном продукте, подтверждают структуру соединения: сигналы ЯМР практически совместимы с сигналами, полученными посредством анализа образцов, выделенных в соответствии с примерами 3 и 4.
Пример 6. Сравнительный пример: получение очищенного бромида 3,7-бис-(диметиламино)-фенотиазин-5-ия без применения FeCl3
В охлажденный реакционный сосуд (при 10°C) добавляют фенотиазин (10,86 кг) и этилацетат (216 мл). Реакционную смесь охлаждают (при 10°C) и добавляют бром (21,84 кг, 7,0 литров) в этилацетате (21,0 литр). Температуру реакции поддерживают в интервале от минус 10°C до минус 15°C. После добавления всего брома реакционную смесь перемешивают еще в течение двух часов при температуре от минус 10°C до минус 15°C.
Затем добавляют диметиламин (19,0 кг) в этилацетате (77 литров) и температуру реакции поддерживают в интервале от минус 15°C до 10°C. После добавления всего диметиламина реакционную смесь перемешивали еще в течение двух часов при температуре от минус 15°C до 10°C.
Неочищенный продукт отфильтровывают, промывают охлажденным этилацетатом и соляной кислотой концентрации 0,1 моль/л.
Выделенный неочищенный продукт (бромид 3,7-бис(диметиламино)-фенотиазин-5-ия) суспендируют в метаноле (60 литров) при комнатной температуре в течение одного часа и отфильтровывают. Добавляют метанол, и процедуру получения суспензии повторяют при температуре примерно 50°C. Смесь охлаждают до комнатной температуры и отфильтровывают.
В конце добавляют смесь метанола (12 литров) и соляной кислоты концентрации 0,5 моль/л (48 литров), и процедуру получения суспензии повторяют при температуре примерно 50°C. Смесь охлаждают до комнатной температуры и отфильтровывают.
Продукт растворяют в смеси метанола (100 литров) и воды (20 литров) при 60°C и элюируют через колонку, наполненную анионообменной смолой (45 литров Purolite A500, форма хлорида); затем собранные элюаты концентрируют посредством выпаривания. Сухой продукт растворяют в соляной кислоте концентрации 0,2 моль/л (100 литров) при 60°C и охлаждают при 10°C. Раствор перемешивают при данной температуре в течение двух часов, и продукт отфильтровывают и сушат. Выход 11,1 кг.
Спектры13C и1H ЯМР ((CD3)2SO; 600 МГц), зарегистрированные на данном продукте, подтверждают структуру соединения: сигналы ЯМР практически совместимы с сигналами, полученными посредством анализа образцов, выделенных в соответствии с примерами 3, 4 и 5.
Таблица 1. Соответствующие аналитические результаты образцов, полученных согласно приведенным выше Примерам 3-5
*ВЭЖХ способы и спецификации в соответствии с PhEur (European Pharmacopoeia - Европейская фармакопея)
Реферат
Изобретение относится к новому способу получения бромида или хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия, а также к применению хлорида 3,7-дихлорфенотиазин-5-ия в качестве промежуточного соединения. Технический результат: предложен улучшенный способ получения бромида или хлорида 3,7-бис-(диметиламино)-фенотиазин-5-ия, где не используется выделение и/или очистка какого-либо промежуточного соединения реакции после осуществления реакции фенотиазина с подходящим галогеном. Способ по изобретению позволяет исключить время, необходимое для выделения и очистки промежуточных соединений, уменьшить производственные отходы. 2 н. и 9 з.п. ф-лы, 1 табл., 6 пр.
Формула
Документы, цитированные в отчёте о поиске
Небелковые конъюгатные соединения бороновой кислоты и способ их получения
Комментарии