Способы получения (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений - RU2712224C2
Код документа: RU2712224C2
Описание
ПРИОРИТЕТ ИЗОБРЕТЕНИЯ
По настоящей заявке испрашивается приоритет на основании предварительной заявки США № 62/055893, поданной 26 сентября 2014 года. Содержание этой предварительной заявки в полном объеме включено в настоящее описание в качестве ссылки.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
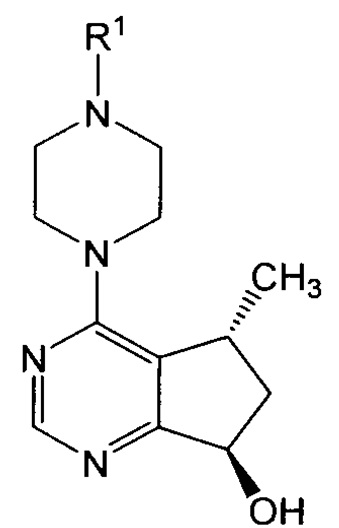
Настоящее изобретение относится к способам получения (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений, и, более конкретно, относится к способам получения (R)-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазина и его N-защищенным производным, которые могут быть использованы в качестве промежуточных соединений в синтезе ипатасертиба (то есть, (S)-2-(4-хлорфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)-пропан-1-она). Настоящее изобретение, кроме того, относится к различным соединениям, которые являются промежуточными соединениями, используемыми в этих способах.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
AKT (также известна как протеинкиназа B) представляет собой серин/треониновую протеинкиназу, которая сверхэкспрессируется в некоторых опухолях человека. Ипатасертиб является ингибитором АКТ, который в настоящее время исследуется в клинических испытаниях при лечении солидных опухолей, рака желудка и рака предстательной железы. Ипатасертиб описан, например, в патенте США № 8063050 (см., например, пример 14), а также в публикации международной патентной заявки WO 2008/006040.
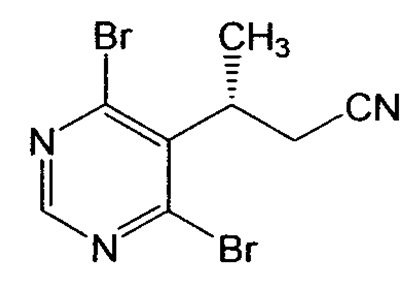
(R)-4-(5-Метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин или его N-защищенное производное можно использовать в качестве промежуточного продукта в синтезе ипатасертиба. Способы получения этого промежуточного продукта приведены, например, в публикации международной патентной заявки WO 2013/173736 и в публикации международной патентной заявки WO 2013/173768. Схема 1 из WO 2013/173768 показана ниже:
Схема 1


В настоящем изобретении предложены улучшенные способы масштабного производства (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений и, более конкретно, (R)-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазина, а также его N-защищенных производных. По сравнению с известными в настоящее время в способах по настоящему изобретению успешно улучшены, например, условия способа, выбор реагентов, уменьшена сложность необходимых устройств и тому подобное.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
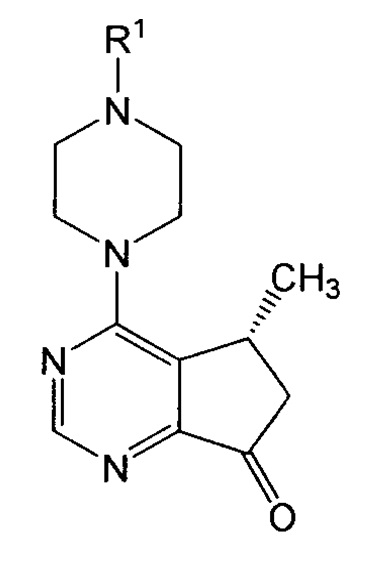
В настоящем изобретении предложены улучшенные способы получения (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений и, более конкретно, (R)-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазина, а также его N-защищенного производного, такого как, например, трет-бутил-(R)-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата. В настоящем изобретении предложены, кроме того, способы получения ингибиторов AKT и, в частности, ипатасертиба, с использованием таких улучшенных способов получения указанных (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений и их N-защищенных производных.
В одном варианте осуществления настоящее изобретение относится к способу получения соединения формулы I:

или его соли, где способ включает взаимодействие соединения формулы III:

или его соли с металлирующим агентом с образованием соединения формулы I или его соли, где R1 представляет собой водород или аминозащитную группу.
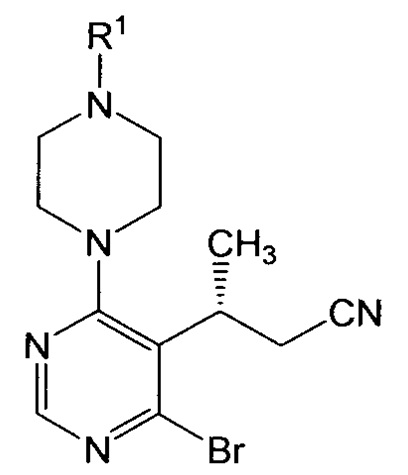
В этом или ином варианте осуществления настоящее изобретение относится далее к такому способу, где соединение формулы III или его соль получают путем взаимодействия соединения формулы IV:

или его соли, где Y выбран из хлора и брома, с пиперазиновым соединением, обладающим структурой:

или его солью, где Lv представляет собой удаляемую группу, и R1 представляет собой аминозащитную группу.
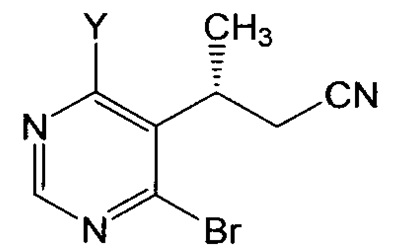
В этом или еще другом варианте осуществления настоящее изобретение, кроме того, относится к такому способу, где соединение формулы IV или его соль получают путем бромирования соединения формулы V:
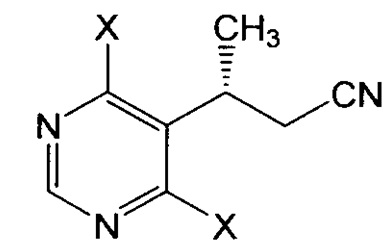
или его соли; где каждый X независимо выбран из хлора и гидроксила. В одном конкретном варианте осуществления настоящее изобретение относится к такому способу, где соединение или соль формулы IV не выделяют после осуществления реакции бромирования соединения или соли формулы V и перед взаимодействием с пиперазиновым соединением, как указано выше.
В этом или еще другом варианте осуществления настоящее изобретение, кроме того, относится к такому способу, где соединение или соль формулы IV (где Y представляет собой Br) получают путем бромирования соединения формулы Vb:
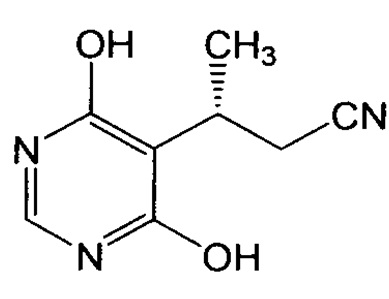
или его соли с получением соединения или соли формулы IV. Альтернативно, соединение или соль формулы IV (где Y представляет собой Cl или, в особенности, Br) получают путем хлорирования соединения формулы Vb или его соли с получением соединения формулы Vc:

или его соли; и бромирования соединения или соли формулы Vc с получением соединения формулы IV или его соли. В одном конкретном варианте осуществления настоящее изобретение относится к такому способу, где соединение или соль формулы Vc не выделяют после осуществления реакции хлорирования соединения или соли формулы Vb и перед осуществлением реакции бромирования.
В этом или еще другом варианте осуществления настоящее изобретение, кроме того, относится к такому способу, где соединение или соль формулы Vc не выделяют после осуществления реакции хлорирования соединения или соли формулы Vb и перед осуществлением реакции бромирования с получением соединения или соли формулы IV, и, кроме того, где соединение или соль формулы IV не выделяют после осуществления реакции бромирования соединения или соли формулы Vc и перед взаимодействием с пиперазиновым соединением с образованием соединения или соли формулы III.
В этом или еще другом варианте осуществления соединение или соль формулы Vb получают путем циклизации соединения формулы VIb:

или его соли.
В этом или еще другом варианте осуществления соединение формулы IVb или его соль получают путем (i) взаимодействия кротононитрила с малонатом с образованием изомерной смеси, содержащей соединение формулы VIa и соединение формулы VIb:

или их соли, и затем (ii) отделения соединения или соли формулы VIb от соединения или соли формулы VIa в изомерной смеси. В одном конкретном варианте осуществления соединение формулы VIb или его соль, выделяют из изомерной смеси путем ферментативного разделения. В этом или другом конкретном варианте осуществления изомерную смесь не выделяют из смеси продуктов реакции, образующейся при взаимодействии кротононитрила с малонатом, до выделения соединения формулы VIb или его соль; то есть, соединение формулы VIb или его соль, выделяют непосредственно из смеси продуктов реакции.
В еще одном варианте осуществления настоящее изобретение относится также к способу получения соединения формулы IX:

или его соли, где R2 представляет собой водород или аминозащитную группу, где способ включает: (i) взаимодействие соединения формулы III,

или его соли, где R1 представляет собой водород или аминозащитную группу, с металлирующим агентом с получением соединения формулы I:

или его соли; (ii) восстановление соединение формулы I или его соли с получением соединения формулы VIIa:
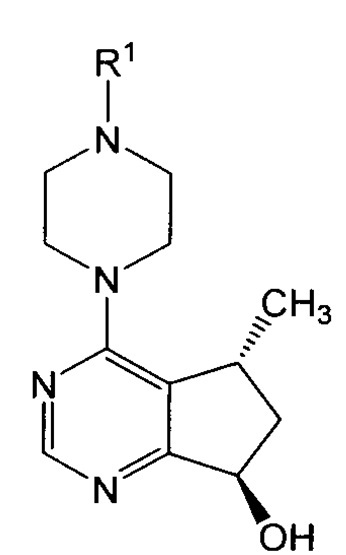
или его соли; (iii) необязательное удаление защитных групп у соединения формулы VIIa или его соли с получением соединения формулы VIIb:

или его соли; и (iv) взаимодействие соединения формулы VIIb или его соли с соединением формулы VIII:
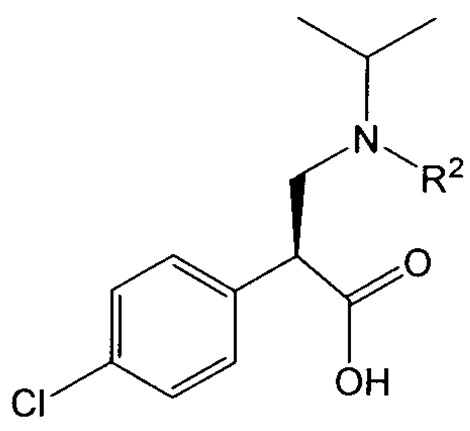
или его соли с получением соединения формулы IX или его соли.
В еще одном варианте осуществления настоящее изобретение относится также, кроме того, к соединению формулы Vb:

или его соли.
В еще одном варианте осуществления настоящее изобретение относится также, кроме того, к соединению формулы Vc:

или его соли.
В еще одном варианте осуществления настоящее изобретение относится также, кроме того, к соединению формулы IVb:
или его соли.
В еще одном варианте осуществления настоящее изобретение относится также, кроме того, к соединению формулы IVc:

или его солям.
В еще одном варианте осуществления настоящее изобретение относится также, кроме того, к соединению формулы III:

или его соли, где R1 представляет собой водород или аминозащитную группу.
Возможные модификации одного или более из вышеприведенных вариантов осуществления, а также дополнительные подробности, связанные с ними, представлены в настоящем документе далее.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Как более подробно в настоящем документе далее, настоящее изобретение главным образом относится к улучшенному способу получения (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений и, более конкретно, относится к улучшенному способу получения (R)-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазина, а также его N-защищенных производных, таких как, например, трет-бутил-(R)-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат, как в целом показано на схеме 2 далее:
Схема 2

Что касается схемы 2, то необходимо отметить, что дигидрокси-нитрил пиримидин может быть бромирован напрямую, или, альтернативно, может быть сначала хлорирован, и затем бромирован полученный продукт реакции хлорирования.
Далее, в отношении схемы 2 следует отметить, что одно или несколько соединений, показанных на ней, могут быть получены и/или использованы в конкретной изомерной или стереохимической конфигурации или, альтернативно, могут быть получены и/или использованы в виде рацемата или смеси стереоизомеров. В одном из конкретных вариантов осуществления, однако, R-изомер одного или нескольких продуктов реакции получают или выделяют, используя средства, обычно известные в данной области и/или описанные подробно ниже, и, необязательно, далее использованы на любой последующей стадии реакции. Например, для преимущественного получения R-изомера продукта реакции добавления конъюгата (соединение формулы VIb) может быть использовано ферментативное разложение, и R-изомер затем использован на последующих стадиях реакции, как более детально показано на схеме 3 ниже:
Схема 3

Преимущество состоит в том, что способ по настоящему изобретению исключает необходимость стадии йодирования и/или использования содержащего йодид реагента, таким образом, является более экономически выгодным и экологически чистым, чем другие способы, в которых эти реагенты используются. В частности, способ по настоящему изобретению включает стадию реакции циклизации или замыкания кольца с образованием циклопентильного кольца соединения формулы I, в которой используется бромнитрилзамещенное соединение формулы III, а не, например, йод-эфир, йод-кислота или йод-амид-замещенное аналогичное соединение. Как дополнительно показано в сравнительных результатах, приведенных ниже (см., например, пример 6), опыт, накопленный к настоящему времени, указывает на то, что реакция циклизации или реакция замыкания кольца менее эффективна, когда используется хлор-нитрил-замещенное аналогичное соединение.
Благодаря наличию нитрильной группы, например, в соединениях формулы Vb и формулы Vс, при осуществлении реакции бромирования способ по настоящему изобретению позволяет, кроме того, использовать (i) более реакционноспособный или более сильный бромирующий агент, и/или (ii) более жесткие или в более широком диапазоне условия реакции галогенирования (например, более высокие температуры реакции). Напротив, применение такого бромирующего агента и/или таких жестких условий реакции при получении аналогичного соединения с бромированным сложноэфирным заместителем приводит к отщеплению сложноэфирной группы и к сопутствующему образованию лактона.
Способ по настоящему изобретению имеет, кроме того, еще такое преимущество, что правильный выбор бромирующего агента, используемого на стадии реакции бромирования, приводит к образованию легколетучих побочных продуктов, которые могут быть удалены путем дистилляции. В этой связи, обычно считается, что удаление этих побочных продуктов из смеси в процессе проведения реакции позволяет лучше контролировать равновесие реакции, таким образом, что реакция смещается в сторону обмена брома по обоим местам; то есть, например, на схеме 3 выше оба атома хлора в соединении формулы Vс или гидроксильные группы в соединении формулы Vb, заменяются на бром. Такой способ позволяет осуществить более эффективное преобразование в желаемый продукт реакции (то есть, в соединение формулы IVb) и уменьшает количество примесей, которые в противном случае присутствовали бы в реакционной смеси.
Способ по настоящему изобретению имеет, кроме того, еще такое преимущество, что бромирование нитрил-замещенного соединения формулы Vс позволяет использовать меньшее количество эквивалентов бромирующего агента по сравнению, например, с эквивалентами йодирующего агента в реакции йодирования сложноэфирного замещенного аналогичного соединения, как показано на схеме 1 выше (например, йодирование соединения 1.1 до соединения 1.2).
Способ по настоящему изобретению имеет, кроме того, еще такое преимущество, что взаимодействие бром-нитрил-замещенных соединений формулы IVb и формулы IVс с пиперазиновым соединением может быть осуществлено при более низких температурах и, в частности, при комнатной температуре, по сравнению, например, с взаимодействием йодированных сложноэфирных замещенных аналогичных соединений с пиперазиновым соединением, как показано на схеме 1 выше (см., например, из соединения 1.2 в соединение 1.3), которое обычно проводят при 60°С. Более низкие температуры реакции позволяют успешно экономить энергетические расходы и/или уменьшать вероятность образования нежелательных побочных продуктов.
Способ по настоящему изобретению имеет, кроме того, еще такое преимущество, что позволяет проводить одну или несколько стадий реакции непрерывным способом, тем самым устраняя необходимость выделения промежуточного продукта реакции перед тем, как будут осуществлены одна или несколько последующих стадий реакции. В частности, (i) продукт реакции со стадии бромирования (то есть, соединения формул IVb и IVс) не нужно выделять перед осуществлением взаимодействия с пиперазиновым соединением, и/или (ii) продукт реакции со стадии хлорирования (то есть, соединение формулы Vс) не нужно выделять перед стадией изомеризации, и/или (iii) продукты реакции стадии добавления конъюгата (то есть, соединения формул VIa и VIb) не нужно выделять из содержащей их смеси продуктов реакции перед разделением (например, путем ферментативного разделения).
В одном конкретном варианте осуществления способа по настоящему изобретению используются все вышеописанные преимущества непрерывного способа для сокращения продолжительность общего производственного цикла (как показано, например, на схеме 3) примерно на 20%, примерно на 30%, примерно на 40%, примерно на 50%, примерно на 60% или более, по сравнению, например, с таким способом, в котором при получении сложноэфирного замещенного аналогичного соединения не используются эти преимущества непрерывного способа (как показано, например, на схеме 1). Кроме того, в данном конкретном варианте осуществления соединение формулы Vс не выделяют перед осуществлением реакции бромирования, реакцию бромирования проводят с использованием более реакционноспособного бромирующего агента и в более жестких условиях реакции бромирования, как подробно описано далее. Кроме того, реакцию бромирования проводят с дистилляцией легколетучих побочных продуктов реакции. Кроме того, изомерную смесь, содержащую соединения формул VIa и VIb не выделяют из смеси продуктов реакции перед тем, как подвергнуть ферментативному разделению. Кроме того, бром-нитрил-замещенные соединения формул IVb и IVс подвергают взаимодействию с пиперазиновым соединением при около комнатной температуре.
Настоящее изобретение относится также к одному или нескольким промежуточным продуктам реакции или соединениям или к их солям, получаемым в результате этого способа.
A. (Циклопента[d]пиримидин-4-ил)пиперазиновые соединения
1. Стадия циклизации
В одном из вариантов осуществления настоящее изобретение относится к способу получения соединения формулы I:
или его соли, где R1 представляет собой водород или аминозащитную группу. Способ включает осуществление взаимодействия соединения формулы III:
или его соли с металлирующим агентом с образованием соединения формулы I или его соли, где R1 представляет собой водород или аминозащитную группу. Более конкретно, способ включает осуществление взаимодействия соединения формулы III или его соли с металлирующим агентом с образованием соединения формулы II:

или его соли, где R1 имеет значения, описанные выше, и М представляет собой металл или переходный металл (такой как литий или магний), как подробно описано далее, и затем циклизацию соединения формулы II или его соли с образованием соединения формулы I или его соли.
В некоторых вариантах осуществления R1 представляет собой аминозащитную группу, как определено далее. В одном или нескольких конкретных вариантах осуществления R1 может быть выбран из фталимидила, бензила, трифенилметила, бензилиденила, п-толуолсульфонила и п-метоксибензила. R1 также может быть выбран из -C(O)-Rd или -C(O)ORd, где Rd независимо выбран из водорода, замещенного или незамещенного алкила, замещенного или незамещенного алкенила, замещенного или незамещенного алкинила, замещенного или незамещенного циклоалкила, замещенного или незамещенного фенила или замещенного или незамещенного гетероциклила. Примеры вариантов осуществления включают случаи, где R1 представляет собой (i) -C(O)ORd и, более того, где Rd представляет собой трет-бутил, бензил или флуоренилметил (то есть R1 представляет собой трет-бутоксикарбонил (ВОС), бензилоксикарбонил или флуоренилметилоксикарбонил (FMOC); или (ii) -C(O)Rd и, более того, где Rd представляет собой метил или трифторметил (то есть, R1 представляет собой ацетил или трифторацетил). В альтернативных вариантах осуществления R1 представляет собой -C(O)ORd или -C(O)Rd, где Rd выбран из водорода и C1-C10 алкила и, кроме того, где указанный алкил необязательно замещен оксогруппой, галогеном или фенильной группой. В некоторых предпочтительных вариантах осуществления R1 выбран из ацетила, трифторацетила, фталимидила, бензила, трифенилметила, бензилиденила, п-толуолсульфонила, п-метоксибензила, трет-бутилоксикарбонила, 9-флуоренилметилоксикарбонила и карбобензилокси.
Металлирующий агент, который, как следует понимать, включает металлы и переходные металлы, обычно может быть выбран из любого металлирующего агента, который облегчает циклизацию или замыкание кольца с образованием циклопентильного кольца. Обычно металлирующим агентом является металлорганическое соединение, которое может содержать, например, один или несколько из атомов лития и магния и/или галогена. Более конкретно, металлирующим агентом может быть литийорганическое соединение или реагент (например, RxLi), магнийорганическое соединением или реагент (например, RxMgZ) или магний- или литийорганическое соединение или реагент (например, (Rx)3MgLi), где: (i) каждый Rx независимо выбран из необязательно замещенного C1-10 алкила, необязательно замещенного C3-7 циклоалкила, необязательно замещенного арила, необязательно замещенного гетероарила или необязательно замещенного гетероциклила, или две группы Rx, взятые вместе с атомом, к которому они присоединены, образуют 5-7-членное необязательно замещенное кольцо; и (ii) Z представляет собой галоген, и, более конкретно, представляет собой Cl, Br или I. В некоторых вариантах осуществления каждый Rx независимо выбран из необязательно замещенного C1-10 алкила и необязательно замещенного C3-7циклоалкила, но, более конкретно, выбран из изопропила (изо-Pr) или бутила (н-бутила, втор-бутила или трет-бутила). Необязательно, также может быть использована добавка, которая действует для модуляции реакционной способности и/или стабильности металлирующего агента (например, добавка или модификатор на основе амина и, более конкретно, диамина).
Примеры магнийорганических соединений или реагентов включают реактивы Гриньяра, такие как C1-C6 алкилмагний галогениды и, более конкретно, включают изо-PrMgCl или втор-бутилMgCl, которые могут быть использованы по отдельности или в составе комплекса с хлоридом лития (например, изо-PrMgCl·LiCl). (см., например, Organomet. Chem., 2011, 37, 1-26, pp. 7-13; и A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 333.) Примеры литийорганических соединений или реагентов включают C1-C6 алкиллитий и, более конкретно, включают н-бутиллитий, втор-бутиллитий и трет-бутиллитий. Примеры магний-литийорганических соединений или реагентов (то есть, (Rx)3MgLi), включают случаи, где Rx представляет собой C1-C6 алкил и, более конкретно, например, изопропил или бутил (например, н-бутил, втор-бутил или трет-бутил), например, такие соединения включают литий три-н-бутилмагний, литий триизопропилмагний и литий (изопропил)(ди-н-бутил)магний.
Хотя конкретные условия осуществления способа, включая одно или несколько из следующих: время реакции, температура, растворитель, реагент, количество реагента (реагентов), порядок добавления реагентов, рН и тому подобное, могут быть подобраны для оптимизации чистоты и/или выхода реакционного продукта, в частности, соединение формулы III может быть подвергнуто взаимодействию с примерно от 1 до 1,5 молярными эквивалентами металлирующего агента и, более конкретно, с от примерно 1 до примерно 1,4 или от примерно 1, 05 до примерно 1,2 молярными эквивалентами металлирующего агента. Кроме того, в одном из конкретных вариантов осуществления осуществление объединения или взаимодействия соединения формулы III с металлирующим агентом может происходить в процессе или поэтапно в течение этой стадии реакции, количество и/или время каждого добавления определяется с целью оптимизации выхода, и/или чистоты, и/или для того, чтобы обеспечить последнее добавление ближе к концу времени реакции. Например, металлирующий агент может быть добавлен к реакционной смеси, содержащей соединение формулы III, приблизительно равными порциями в течение периода времени (например, приблизительно 4 часов), его последняя часть добавляется ближе к концу желаемого времени реакции.
В различных вариантах осуществления способ получения соединения формулы I или его соли может быть осуществлен в эфирном или углеводородном растворителе или в смеси этих растворителей (например, тетрагидрофуран (ТГФ), 2-метилтетрагидрофуран (MeТГФ), метил-трет-бутиловый эфир (MTBE), циклопентилметиловый эфир (CPME), диэтиловый эфир, диизопропиловый эфир, дифениловый эфир, толуол, этилбензол, ксилол, кумол, пентан или гептан). Примеры реакционных условий включают: (i) температуру реакции около 20°С, около 15°С, около приблизительно 10°С, около 5°С, около 0°С или менее (например, около -10°С, около -25°С, около -50°С или около -75°С); и/или (ii) проведение реакции в практически безводных условиях (например, приблизительно 100 частей на миллион, приблизительно 50 частей на миллион, приблизительно 25 частей на миллион или приблизительно 10 частей на миллион воды или меньше); и/или (iii) проведение реакции в инертной атмосфере (например, в атмосфере гелия, неона, аргона или азота). В конкретном варианте осуществления способ получения соединения формулы I или его соли из соединения формулы III или его соли проводят в MeТГФ, отдельно или в сочетании с толуолом, при температуре от около -15°С до около 15°С, от около -10°С до около 10°С или от около 0°С до около 5°С, необязательно, в безводных условиях и/или, необязательно, в инертной (например, азот) атмосфере.
Кроме того, должно быть понятно, что для получения желаемого конечного продукта (то есть, соединения формулы I) может быть проведена дополнительная обработка или обработка одного или нескольких продуктов, полученных в результате вышеуказанной циклизации или реакции замыкания кольца, с использованием известных в данной области методов, таких как, например, гидролиз енаминового промежуточного продукта реакции с получением конечного кетонового продукта. См., например, WO 2013/173784, содержание которого включено в качестве ссылки для всех соответственных и соответствующих целей.
В различных вариантах осуществления преобразование или циклизация соединения формулы III до соединения формулы I проходит примерно на 90%, примерно на 95%, примерно на 99% или более, и/или выход соединения формулы I составляет примерно 75%, предпочтительно, примерно 80%, примерно 85%, примерно 90% или больше.
2. Стадия добавления пиперазина
В одном из вариантов осуществления соединение формулы III или его соль получают путем осуществления взаимодействия соединения формулы IV:
или его соли, где Y выбран из хлора и брома, с пиперазиновым соединением, имеющим структуру:
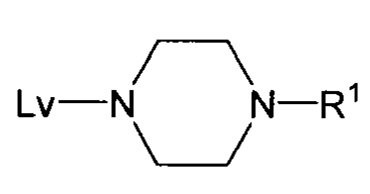
или его соли, где Lv представляет собой удаляемую группу, и R1 представляет собой аминозащитную группу, как определено далее. В примере варианта осуществления Y представляет собой бром. В этом или другом примере варианта осуществления R1 может быть, например, алкоксикарбонилом (таким как трет-бутоксикарбонил) или арилоксикарбонилом (таким как бензилоксикарбонил). В этих или других примерах вариантов осуществления Lv может быть, например, водородом или галогеном. Альтернативно, однако, один или оба R1 и Lv могут быть выбраны из других вариантов, данных в определениях, приведенных в настоящем документе ниже, или, альтернативно, из аминозащитных групп и удаляемых групп, известных специалистам в данной области, не отклоняясь от предполагаемого объема настоящего раскрытия.
Хотя для оптимизации чистоты и/или выхода продукта реакции могут быть выбраны определенные условия осуществления способа, включая время реакции, температуру, растворитель, реагент, количество реагента(ов), порядок добавления реагентов, рН и тому подобное, в частности, соединение формулы IV может быть подвергнуто взаимодействию с примерно от 1 до 1,5 молярными эквивалентами пиперазинового соединения, и, более обычно, будет подвергаться взаимодействию с примерно от 1,05 до примерно 1,4, или от примерно 1,1 до примерно 1,2, молярных эквивалентов пиперазинового соединения, причем в одном конкретном варианте осуществления используется примерно 1,15 эквивалента пиперазинового соединения.
В этой связи следует отметить, что температуру реакции и/или количество добавляемого пиперазинового соединения, помимо других соображений (например, вида и/или количества добавленного основания или используемого растворителя), как правило, контролируют или оптимизируют для того, чтобы ограничить количество образующегося побочного продукта реакции замещения дипиперазина (то есть, образование соединения, в котором оба атома брома замещены или заменены соединением пиперазина). Например, в одном или нескольких вариантах осуществления реакцию проводят при температуре ниже чем около 60°С, 50°С, 40°С или даже 30°С, причем реакцию в одном конкретном варианте осуществления проводят при около комнатной температуре (например, около 20°С или около 25°С), используя, например, N,N-диизопропилэтиламин (DIEA) в качестве основания (например, приблизительно 1,5, приблизительно 1,75, приблизительно 2 или более мольных эквивалентов) и с ацетонитрилом (CH3CN) в качестве растворителя (отдельно или в сочетании с водой).
В различных вариантах осуществления выход соединения формулы III составляет примерно 85%, примерно 90%, примерно 95% или более, и/или его чистота составляет примерно 90%, примерно 95%, примерно 98% или более.
3. Стадия бромирования
В одном из вариантов осуществления соединение формулы IV или его соль получают путем бромирования соединения формулы V:

или его соли; где каждый Х независимо выбран из хлора и гидроксила. В конкретном варианте осуществления оба заместителя Х представляют собой гидроксил, в то время как в другом варианте оба заместителя Х представляют собой хлор.
В этой связи следует отметить, что в некоторых случаях полученная реакционная смесь может содержать как дибромзамещенное соединение формулы IV, так и бромхлорзамещенное аналогичное соединение, которое, если оно присутствует, будет незначительным продуктом реакции. Как правило, молярное отношение дибром-соединения и бромхлор-соединения будет составлять, например, приблизительно 95:1, приблизительно 96:1, приблизительно 97:1, приблизительно 98:1 или более. Более конкретно, когда дихлор-соединение формулы V подвергают бромированию, полученная реакционная смесь может содержать как соединение формулы IVb (где Y в формуле IV представляет собой бром), так и соединение формулы IVс (где Y в формуле IV представляет собой хлор):


молярное соотношение двух соединений указано выше.
Бромирующий агент для реакции выбран из известных бромирующих агентов, которые при взаимодействии с соединением формулы V в подходящих условиях реакции приводят к образованию легко легколетучего побочного продукта и, более конкретно, побочного продукта, который можно удалить из реакционной смеси путем дистилляции. В этой связи, как правило, считается, что удаление этих побочных продуктов из смеси при проведении реакции позволяет лучше контролировать равновесие реакции, так что реакция сдвигается в сторону замены на галоген (то есть, бром) по обоим положениям, и более конкретно, обе группы X в соединении формулы V (то есть, оба атома хлора в соединении формулы Vс или обе гидроксильных группы в соединении формулы Vb) заменяются на атомы брома. Такой подход обеспечивает превосходное превращение в желаемый продукт реакции и уменьшает количество примесей, которые образуются в противном случае.
Примеры бромирующих агентов включают, но ими не ограничиваются, бром, бромтриметилсилан (или триметилсилилбромид (TMSBr)), оксибромид фосфора (POBr3), N-бромсукцинимид (NBS) и трибромид фосфора (PBr3).
В этой связи следует отметить, что бромирующий агент может быть добавлен в реакционную смесь или, альтернативно, может быть образован in situ, используя способы, хорошо известные в данной области. Например, можно получить TMSBr in situ путем добавления в реакционную смесь триметилсилилхлорида (TMSCl) и бромида натрия (NaBr) или другого бромида щелочного металла (например, KBr, LiBr, MgBr2, ZnBr2 или бромида тетраалкиламмония).
Хотя для оптимизации чистоты и/или выхода продукта реакции могут быть выбраны определенные условия осуществления способа, включая время реакции, температуру, растворитель, реагент, количество реагента(ов), порядок добавления реагентов, рН и тому подобное, в частности, соединение формулы V может быть подвергнуто взаимодействию с примерно от 2 до 7 молярными эквивалентами бромирующего агента и, более конкретно, подвергнуто взаимодействию с примерно от 2,5 до 6, или от примерно 3 до примерно 5 молярных эквивалентов бромирующего агента, причем в одном конкретном варианте осуществления используют примерно 3,5 эквивалента бромирующего агента, причем бромирующий агент добавляют одной аликвотой или несколькими аликвотами в течение определенного периода времени. Кроме того, или альтернативно, реакцию можно проводить при температуре от около 65°С до около 80°С или от около 70°С до около 75°С, используя, например, ацетонитрил (СН3CN) в качестве растворителя в течение от примерно 15 часов до примерно 20 часов или от примерно 16 часов до примерно 18 часов.
В различных вариантах осуществления преобразование соединения формулы V в соединение формулы IV протекает примерно на 85%, примерно на 90%, примерно на 95% или более.
4. Стадия хлорирования
Соединение формулы IV или его соль могут быть получены путем прямого бромирования соединения или соли формулы V с использованием, например, оксибромида фосфора или трибромида фосфора или, альтернативно, соединения формулы Vb или его соли ниже. В одном из конкретных вариантов осуществления, однако, соединение формулы IV или его соль получают путем сначала хлорирования соединения формулы Vb:

или его соли с получением соединения формула Vc:

или его соли, а затем бромирования соединения или соли формулы Vc с получением соединения или соли формулы IV, как описано выше.
Конкретные условия процесса, включая время реакции, температуру, выбор растворителя, реагент, количество реагентов (реагентов), порядок добавления реагентов, рН и тому подобное, могут быть подобраны для оптимизации чистоты и/или выхода. Например, в различных вариантах осуществления соединение формулы Vb будет подвергнуто взаимодействию с примерно от 1,5 до 5 молярных эквивалентов хлорирующего агента, и более типично, подвергнуто взаимодействию с примерно от 2 до 4 или от примерно 2,5 до 3,5 молярными эквивалентами хлорирующего агент, с примерно 3 эквивалентами хлорирующего агента, используемого в одном из конкретных вариантов осуществления. В этих или других вариантах осуществления подходящие хлорирующие агенты включают, среди прочих, например, оксихлорид фосфора (POCl3) и трихлорид фосфора (PCl3). В этих или других вариантах осуществления реакция может быть проведена неразбавленной, хлорирующий агент (например, POCl3) добавлен вместе с соответствующим количеством основания, такого как примерно 1, примерно 1,1, примерно 1,2 или более молярных эквивалентов, например, 2,6-лутидина или N,N-диметиланилина, в отсутствие растворителя. Альтернативно, выбор основания и/или растворителя (например, 2,6-лутидина с толуолом в качестве растворителя) может обеспечить непрерывную обработку, как описано в настоящем изобретении далее.
5. Стадия синтеза пиримидина
В соответствии с настоящим изобретением пиримидиновое соединение формулы Vb или его соль получают путем циклизации соединения формулы VIb:

или его соли. Способ включает осуществление взаимодействия соединения формулы VIb или его соли с формамидином и, более конкретно, его солью, включая, например, ацетатную соль (то есть., формамидин ацетат).
Конкретные условия процесса, включая время реакции, температуру, выбор растворителя, реагент, количество реагентов (реагентов), порядок добавления реагентов, рН и тому подобное, могут быть подобраны для оптимизации чистоты и/или выхода. Например, в различных вариантах осуществления реакцию можно проводить в спиртовом растворителе (например, метаноле). В этих или других вариантах осуществления соединение формулы VIb или его соль подвергают взаимодействию с примерно от 1 до 1,25 молярных эквивалентов формамидина и, более обычно, подвергают взаимодействию с от примерно 1 до примерно 1,15 или от примерно 1 до примерно 1,05 молярных эквивалентов формамидина с примерно 1,05 эквивалента формамидина, используемого в одном из конкретных вариантов осуществления. Кроме того, в реакции можно также использовать примерно 2, примерно 2,5, примерно 3 или более мольных эквивалентов основания, такого как NaOMe.
В различных вариантах осуществления выход соединения формулы VB или его соли составляет примерно 75%, примерно 80%, примерно 85% или более.
6. Стадии добавления конъюгата и ферментативного разделения
Кроме того, соединение формулы VIb или его соль получают путем осуществления взаимодействия кротононитрила с малонатом с образованием изомерной смеси соединения формулы VIа и соединения формулы VIb:


или их солей; и отделения соединения или соли формулы VIb от соединения или соли формулы VIa.
Конкретные условия процесса, включая время реакции, температуру, выбор растворителя, реагент, количество реагентов (реагентов), порядок добавления реагентов, рН и тому подобное, могут быть подобраны для оптимизации чистоты и/или выхода. Например, в различных вариантах осуществления реакцию кротононитрила с малонатом можно проводить в спиртовом растворителе (например, метаноле) или в таком растворителе, как тетрагидрофуран (ТГФ). В этих или других вариантах осуществления кротононитрил подвергают взаимодействию с примерно от 1 до 1,5 молярных эквивалентов малоната и, более обычно, подвергают взаимодействию с примерно от 1,05 до примерно 1,4 или от примерно 1,1 до примерно 1,3 молярных эквивалентов малоната с примерно 1,1 эквивалента малоната, используемого в одном из конкретных вариантов осуществления. Кроме того, может быть добавлено от примерно 0,2 до примерно 0,8 или от примерно 0,4 до примерно 0,6 молярных эквивалентов основания, такого как метоксид натрия (NaOMe), трет-пентоксид натрия (или трет-амилат натрия, трет-AmONa) и трет-пентоксид калия, вместе с приблизительно 0,5 молярными эквивалентами основания, используемого в одном из конкретных вариантов осуществления.
В различных вариантах осуществления выход соединения формулы VIa или его соли составляет примерно 70%, примерно 75%, примерно 80% или более.
Соединение формулы VIb или его соль могут быть отделены от соединения формулы VIa или его соли, используя способы, хорошо известные в области разделения изомеров. В одном из конкретных вариантов осуществления, однако, соединение формулы VIb или его соль отделяют от изомерной смеси, содержащей его и соединение формулы VIa или его соль, с помощью ферментативного разделения. Ферментативное разделение изомерной смеси может быть достигнуто с помощью способов, хорошо известных в данной области, включая, например, осуществление взаимодействия изомерной смеси с подходящим ферментом липазы для селективного гидролиза сложноэфирной группы соединения формулы VIA или его соли, так что соединение формулы VIB или его соль могут быть отделены от гидролизованного соединения. Подходящие ферменты липазы включают, например, ферменты, происходящие из микроорганизма Candida, например, Candida cylindracea, а также Candida rugosa, микроорганизма Chromobacterium chocolatum, свиной печени и термофильного микроорганизма. Другие подходящие ферменты липазы указаны, например, в публикации WO 2013/173736 (содержание которой включено в настоящее описание в качестве ссылки для всех соответственных и соответствующих целей в полном объеме), а также в примерах 2b и 2с настоящего документа. Альтернативно или более конкретно, ферментативное разделение изомерной смеси может быть достигнуто путем осуществления взаимодействия изомерной смеси с подходящим ферментом нитрилазой для селективного гидролиза нитрильной группы соединения формулы VIa или его соли, так что соединение формулы VIb или его соль могут быть отделены от гидролизованного соединения. Подходящие ферменты нитрилазы включают, например, фермент, упомянутый в примерах 2а, 2d и 2е настоящего описания.
Конкретные способы и условия разделения и, в частности, ферментативного разделения соединения или соли формулы VIb и соединения или соли формулы VIa, включая тип фермента, время реакции, температуру, выбор растворителя, реагент, количество реагентов (реагентов), порядок добавления реагента, рН и тому подобное, могут быть подобраны для оптимизации чистоты и/или выхода желаемого продукта и/или времени реакции. Например, в различных вариантах осуществления смесь, содержащая соединения формул IVa и IVb или их соли, фермент нитрилазу, растворитель (такой как вода), основание (такое как NaOH) и/или буфер (такой как KH2SO4, или K2SO4, или Na2B4O7·10H2O), можно использовать для проведения ферментативного разложения при комнатной температуре (например, около 20-25°С) в течение примерно 24 часов, примерно 36 часов, примерно 48 часов или более, причем период от примерно 24 до примерно 48 часов обычно используется в одном или нескольких вариантах осуществления.
В различных вариантах осуществления выход соединения формулы VIb или его соли составляет примерно 30%, примерно 40%, примерно 45% или более.
7. Усовершенствование непрерывных способов и общая эффективность процесса
В целях иллюстрации нижеприведенная схема 4 в общем иллюстрирует типичный вариант осуществления способа по настоящему изобретению, а также различные соединения и промежуточные соединения, охватываемые настоящим изобретением. Более подробные варианты осуществления, включая конкретные условия осуществления способа и регенты, представлены далее в следующих ниже примерах. Специалистам в данной области будет понятно, что другие условия реакции, включая реагенты, концентрации реагентов или молярные эквиваленты, растворители, температуру реакции, продолжительность реакции и тому подобное, а также необходимую обработку (например, кислотная или щелочная обработка), можно использовать в соответствии с настоящим способом для получения желаемых соединений и промежуточных продуктов, не выходя за пределы предполагаемого объема настоящего изобретения. Соответственно, представленное в настоящем документе подробное описание не должно рассматриваться как ограничивающее.
Схема 4

Следует отметить, что способ по настоящему изобретению имеет особенные преимущества, так как две или более стадий способа, как проиллюстрировано, например, на схеме 4 выше, можно проводить по порядку или последовательно, не выделяя промежуточный продукт реакции.
В одном из конкретных вариантов осуществления после осуществления реакции хлорирования соединения формулы Vb или его соли полученное соединение формулы Vс или его соль не выделяют перед осуществлением реакции бромирования с образованием соединений формулы IV (т.е., IVb и IVс) или их солей. В этом или другом конкретном варианте осуществления соединения формулы IV (т.е., IVb и IVc) или их соли не выделяют после осуществления реакции бромирования соединения формулы Vс или его соли перед осуществление взаимодействия с пиперазиновым соединением, как подробно описано выше. В этих вариантах осуществления, в которых эту трехстадийную реакцию проводят без выделения указанных промежуточных продуктов (т.е., реакции хлорирования, бромирования и добавление пиперазина проводят без выделения соединений формул Vс, IVb/IVc и III или их солей, соответственно), средний выход обычно составляет примерно 80%, примерно 85%, примерно 90%, примерно 95% или более. В этом или в еще одном конкретном варианте осуществления изомерную смесь, содержащую соединения формулы VIa и формулы VIb или их соли, не выделяют из смеси продуктов реакции перед последующим отделением соединения формулы VIb или его соли от соединения формулы VIa или его соли; то есть, соединения формулы VIa и формулы VIb или их соли не выделяют перед взаимодействием соединения формулы VIa или его соли с подходящим ферментом (например, ферментом нитрилазой) для выделения соединения формулы VIb или его соли.
Соответственно, настоящее изобретение представляет или обеспечивает возможность удобного проведения вышеуказанных стадий реакции в непрерывном процессе, исключая таким образом необходимость выделения нескольких промежуточных продуктов реакции (например, хлорирование/бромирование осуществляется непрерывным способом, бромирование/добавление пиперазина осуществляется непрерывным способом, хлорирование/бромирование/добавление пиперазина осуществляется непрерывным способом, ферментативное разделение осуществляется непрерывным процессом, или все эти указанные стадии реакций осуществляются непрерывным способом).
В одном из примеров вариантов осуществления настоящего способа используются все вышеописанные стадии способа; то есть стадия ферментативного разделения, а также стадии хлорирования/бромирования/добавления пиперазина осуществляются непрерывным способом, при котором различные продукты реакции, образующиеся на каждой стадии, не выделяют перед проведением следующей стадии реакции. Таким образом, такой способ обладает преимуществом в обеспечении значительного улучшения эффективности процесса. Например, средняя продолжительность всего производственного цикла по настоящему способу (как проиллюстрировано, например, на схеме 4) уменьшается по сравнению, например, со средней продолжительностью всего производственного цикла процесса, при котором не используются такие преимущества непрерывного способа, для получения сложноэфирного замещенного аналога (как показано, например, на схеме 1), примерно на 30%, 40%, 50%, 60%, 70% или более (например, меньшее количество стадий выделения) и/или более короткое время реакции (например, меньшее время, необходимое для ферментативного разделения с использованием фермента нитрилазы, по сравнению, например, с ферментативным разделением с использованием фермента липазы). В еще одном варианте осуществления ферментативное разделение с использованием фермента нитрилазы при повышенном рН (например, рН около 9,2) проявляет примерно двойную скорость реакции и более высокую селективность (Е) по сравнению с разделением с помощью нитрилазы с начальным рН около 7,2 или разделением с использованием фермента липазы.
8. Примерные варианты осуществления
В первом примерном варианте осуществления настоящего изобретения соединение формулы I:
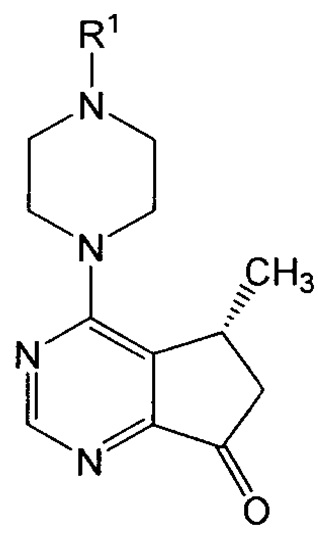
или его соль, где R1 представляет собой водород или аминозащитную группу, получают способом, который включает: (a) взаимодействие соединения формулы IV:
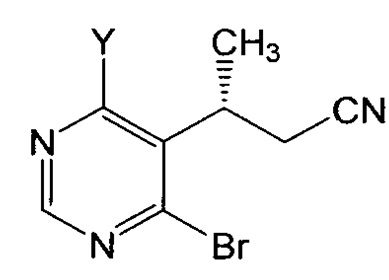
или его соли, где Y выбран из хлора и брома, с пиперазиновым соединением, обладающим структурой:

или его солью, где Lv представляет собой удаляемую группу, и R1 представляет собой аминозащитную группу, с образованием соединения формулы III:

или его соли; и (b) взаимодействие соединения формулы III или его соли с металлирующим агентом с образованием соединения формулы I или его соли.
В одном аспекте первого примерного варианта осуществления R1 представляет собой H или представляет собой аминозащитную группу, выбранную из трет-бутоксикарбонила, бензилоксикарбонила и флуоренилметилоксикарбонила. В другом аспекте Y представляет собой бром. В другом аспекте Lv представляет собой водород или галоген. В другом аспекте соединение формулы IV или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5, или от примерно 1,1 до примерно 1,2, молярных эквивалентов пиперазинового соединения, и в другом аспекте указанную реакцию осуществляют при около комнатной температуре. В другом аспекте соединение формулы III или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,05 до примерно 1,2, молярных эквивалентов металлирующего агента, и в другом аспекте металлирующий агент представляет собой реагент Гриньяра, выбранный из магнийорганического галогенида и литийорганического реагента.
Во втором примерном варианте осуществления настоящего изобретения, соединение формулы I:

или его соль, где R1 представляет собой водород или аминозащитную группу, получают способом, который включает: (a) бромирование соединения формулы V:

или его соли, где каждый X независимо выбран из хлора и гидроксила, с образованием соединения формулы IV:

или его соли, где Y выбран из хлора и брома; (b) взаимодействие соединения формулы IV или его соли, с пиперазиновым соединением, обладающим структурой:

или его соли, где Lv представляет собой удаляемую группу, и R1 представляет собой аминозащитную группу, с образованием соединения формулы III:
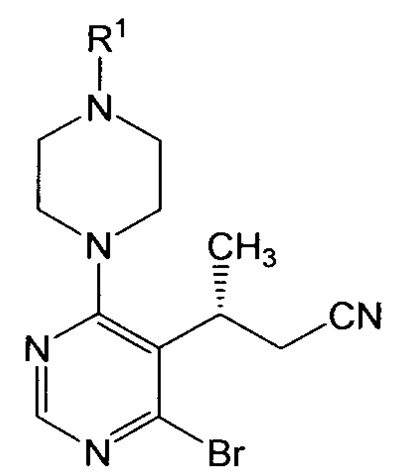
или его соли; и (c) взаимодействие соединения формулы III или его соли с металлирующим агентом с образованием соединения формулы I или его соли.
В одном аспекте второго примерного варианта осуществления R1 представляет собой H или представляет собой аминозащитную группу, выбранную из трет-бутоксикарбонила, бензилоксикарбонила и флуоренилметилоксикарбонила. В другом аспекте X представляет собой хлор. В другом аспекте Y представляет собой бром. В другом аспекте Lv представляет собой водород или галоген. В другом аспекте соединение формулы V или его соль подвергают взаимодействию с от примерно 2 до примерно 7 или от примерно 3 до примерно 5 молярных эквивалентов бромирующего агента, и в другом аспекте бромирующий агент представляет собой триметилсилилбромид. В другом аспекте реакцию бромирования осуществляют при температуре от около 70°C до около 75°C, и в другом аспекте используют дистилляцию для удаления легколетучих продуктов. В другом аспекте соединение формулы IV или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,1 до примерно 1,2 молярных эквивалентов пиперазинового соединения, и в другом аспекте указанную реакцию осуществляют при около комнатной температуре. В другом аспекте соединение формулы III или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,05 до примерно 1,2 молярных эквивалентов металлирующего агента, и в другом аспекте металлирующий агент представляет собой реагент Гриньяра, выбранный из магнийорганического галогенида и литийорганического реагента. В другом аспекте соединение формулы IV или его соль не выделяют после осуществления реакции бромирования и перед осуществлением взаимодействия с пиперазиновым соединением.
В третьем примерном варианте осуществления настоящего изобретения соединение формулы I:

или его соль, где R1 представляет собой водород или аминозащитную группу, получают способом, который включает: (a) хлорирование соединения формулы Vb:

или его соли, с образованием соединения формулы Vc:
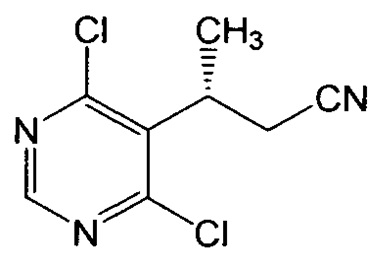
или его соли; (b) бромирование соединение формулы Vc или его соли с образованием соединения формулы IV:

или его соли, где Y выбран из хлора и брома; (c) взаимодействие соединения формулы IV или его соли с пиперазиновым соединением, обладающим структурой:
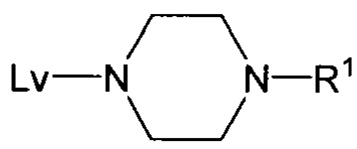
или его солью, где Lv представляет собой удаляемую группу, и R1 представляет собой аминозащитную группу, с образованием соединения формулы III:

или его соли; и (d) взаимодействие соединения формулы III или его соли с металлирующим агентом с образованием соединения формулы I или его соли.
В одном аспекте третьего примерного варианта осуществления R1 представляет собой H или представляет собой аминозащитную группу, выбранную из трет-бутоксикарбонила, бензилоксикарбонила и флуоренилметилоксикарбонила. В другом аспекте Y представляет собой бром. В другом аспекте Lv представляет собой водород или галоген. В другом аспекте соединение формулы Vb или его соль подвергают взаимодействию с от примерно 1,5 до примерно 5 или от примерно 2 до примерно 4 молярных эквивалентов хлорирующего агента, и в другом аспекте хлорирующий агент представляет собой оксихлорид фосфора. В другом аспекте соединение формулы V или его соль подвергают взаимодействию с от примерно 2 до примерно 7 или от примерно 3 до примерно 5 молярных эквивалентов бромирующего агента, и в другом аспекте бромирующий агент представляет собой триметилсилил бромид. В другом аспекте реакцию бромирования осуществляют при температуре от около 70°C до около 75°C, и в другом аспекте используют дистилляцию для удаления легколетучих продуктов. В другом аспекте соединение формулы IV или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,1 до примерно 1,2 молярных эквивалентов пиперазинового соединения, и в другом аспекте указанную реакцию осуществляют при около комнатной температуре. В другом аспекте соединение формулы III или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,05 до примерно 1,2 молярных эквивалентов металлирующего агента, и в другом аспекте металлирующий агент представляет собой реагент Гриньяра, выбранный из магнийорганического галогенида и литийорганического реагента. В другом аспекте соединение формулы IV или его соль, не выделяют после осуществления реакции бромирования и перед осуществлением взаимодействия с пиперазиновым соединением. В другом аспекте соединение формулы Vc или его соль не выделяют после реакции хлорирования и перед реакцией бромирования.
В четвертом примерном варианте осуществления настоящего изобретения соединение формулы I:

или его соль, где R1 представляет собой водород или аминозащитную группу, получают способом, который включает: (a) взаимодействие кротононитрила с малонатом с образованием изомерной смеси, содержащей соединение формулы VIa и соединение формулы VIb:

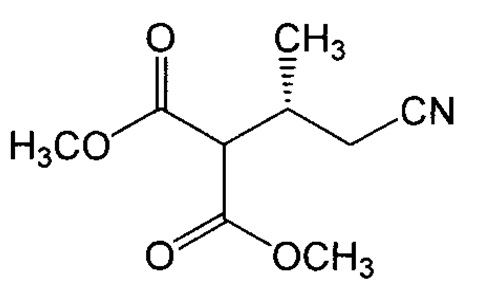
или их соли; (b) отделение соединения формулы VIb или его соли от соединения формула VIa или его соли в изомерной смеси; (c) взаимодействие отделенного соединения формулы VIb, или его соли с помощью формамидиновой соли с образованием соединения формулы Vb:

или его соли; (d) хлорирование соединения формулы Vb или его соли с образованием соединения формулы Vc:

или его соли; (e) бромирование соединение формулы Vc или его соли с образованием соединения формулы IV:
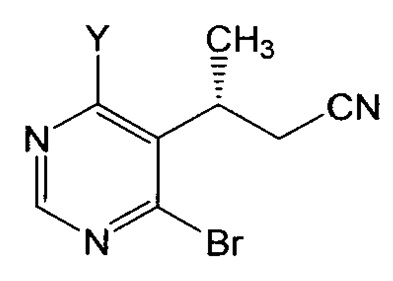
или его соли, где Y выбран из хлора и брома; (f) взаимодействие соединения формулы IV или его соли с пиперазиновым соединением, обладающим структурой:

или его солью, где Lv представляет собой удаляемую группу, и R1 представляет собой аминозащитную группу, с образованием соединения формулы III:

или его соли; и (g) взаимодействие соединения формулы III или его соли с металлирующим агентом с образованием соединения формулы I или его соли.
В одном аспекте четвертого примерного варианта осуществления R1 представляет собой H или представляет собой аминозащитную группу, выбранную из трет-бутоксикарбонила, бензилоксикарбонила и флуоренилметилоксикарбонила. В другом аспекте Y представляет собой бром. В другом аспекте Lv представляет собой водород или галоген. В другом аспекте кротононитрил подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,1 до примерно 1,3 молярных эквивалентов малоната. В другом аспекте соединение формулы VIb или его соль отделяют от соединения формулы VIa или его соли в изомерной смеси путем ферментативного разделения, и в другом аспекте соединение формулы VIb или его соль, отделяют путем взаимодействия изомерной смеси с ферментом нитрилазой. В другом аспекте отделенное соединение формулы VIb или его соль подвергают взаимодействию с от примерно 1 до примерно 1,25 или от примерно 1 до примерно 1,15 молярных эквивалентов формамидиновой соли с образованием соединения формулы Vb, и в другом аспекте формамидиновая соль представляет собой формамидин ацетат. В другом аспекте соединение формулы Vb или его соль подвергают взаимодействию с от примерно 1,5 до примерно 5 или от примерно 2 до примерно 4 молярных эквивалентов хлорирующего агента, и в другом аспекте хлорирующий агент представляет собой оксихлорид фосфора. В другом аспекте соединение формулы V или его соль подвергают взаимодействию с от примерно 2 до примерно 7 или от примерно 3 до примерно 5 молярных эквивалентов бромирующего агента, и в другом аспекте бромирующий агент представляет собой триметилсилил бромид. В другом аспекте реакцию бромирования осуществляют при температуре от около 70°C до около 75°C, и в другом аспекте используют дистилляцию для удаления легколетучих продуктов. В другом аспекте соединение формулы IV или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,1 до примерно 1,2 молярных эквивалентов пиперазинового соединения, и в другом аспекте указанную реакцию осуществляют при около комнатной температуре. В другом аспекте соединение формулы III или его соль подвергают взаимодействию с от примерно 1 до примерно 1,5 или от примерно 1,05 до примерно 1,2 молярных эквивалентов металлирующего агента, и в другом аспекте металлирующий агент представляет собой реагент Гриньяра, выбранный из магнийорганического галогенида и литийорганического реагента. В другом аспекте соединение формулы IV или его соль не выделяют после осуществления реакции бромирования и перед осуществлением взаимодействия с пиперазиновым соединением. В другом аспекте соединение формулы Vc или его соль не выделяют после осуществления реакции хлорирования и перед осуществлением реакции бромирования. В другом аспекте соединения формул VIb и VIa или их соли в изомерной смеси не выделяют перед ферментативным разделением.
B. Синтез ингибитора AKT
Настоящее изобретение, кроме того, относится к применению соединений формулы I или их солей в качестве промежуточного соединения в синтезе ипатасертиба (то есть, (S)-2-(4-хлорфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)-пропан-1-она), как описано, например, в патенте США № 8063050 (смотри, например, пример 14 патента). На схеме 5 далее в общем виде проиллюстрирован вариант осуществления способа по настоящему изобретению, а также различные соединения и промежуточные соединения, охватываемые настоящим изобретением, где соединение формулы I или его соль используется при получении ипатасертиба или его соли в защищенной или незащищенной форме.
Схема 5

В частности, соединения формулы I или его соль могут быть использованы для получения соединения формулы IX или его соли (то есть, ипатасертиба или его соли в защищенной или незащищенной форме):

где R2 представляет собой водород или аминозащитную группу, как определено далее, и, более конкретно, как определено в контексте R1 выше.
В этой связи следует отметить, что в соответствии с настоящим изобретением для преобразования соединения формулы I или его соли в соединение формулы IX или его соль могут быть использованы различные подходящие реакционные схемы и способы. В одном конкретном варианте осуществления, однако, способ включает сначала получение соединения формулы I или его соли, как изложено выше. Соединение формулы I или его соль, затем восстанавливают с получением соединения формулы VIIa:
или его соли. Более конкретно, соединение формулы I или его соль подвергают стереоселективному восстановлению путем его взаимодействия с восстанавливающим агентом, содержащим подходящий фермент, такой как фермент кеторедуктаза, и, необязательно, источник гидразида, как описано, например, в WO 2013/173784 (содержание которого включено в настоящий документ путем ссылки для всех соответственных и соответствующих целей), с получением изомера формулы VIIa.
Если R1 представляет собой защитную группу, соединение формулы VIIa или его соль могут быть подвергнуты удалению защитных групп с использованием средств, обычно известных в данной области техники (например, взаимодействием с подходящей кислотой, такой как хлористоводородная кислота) с образованием соединения формулы VIIb,

или его соли. Соединение формулы VIIb или его соль затем подвергают взаимодействию с соединением формулы VIII:
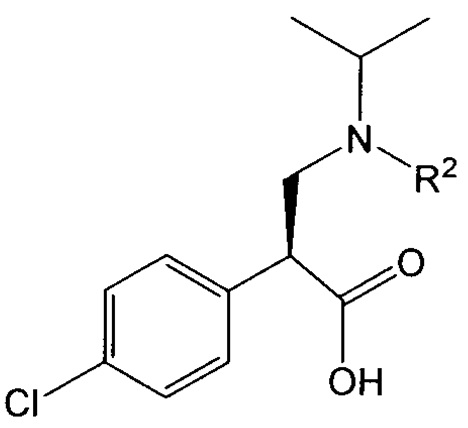
или его солью с получением соединения формулы IX. Конденсация соединения формулы VIIb или его соли с соединением или солью формулы VIII может быть осуществлена с использованием средств, обычно известных в данной области техники, и/или как описано, например, в WO 2013/173784 или WO 2013/173779, и в одном варианте осуществления может быть включено использование подходящего конденсирующего агента, как описано в настоящем документе. Кроме того, способ получения соединения формулы VIII или его соли описан, например, в патенте США № 8063050 и WO 2013/173779. (Полное содержание US 8063050, WO 2013/173784 и WO 2013/17379 включено в настоящий документ путем ссылки для всех соответственных и соответствующих целей.)
C. Продукты реакции и промежуточные соединения
Следует отметить, что настоящее изобретение, кроме того, относится к одному или нескольким нитрил-замещенным продуктам реакции или реакционным промежуточным соединениям или их солям, полученных способами, проиллюстрированными в настоящем документе, включая, например, соединения формул Vb, Vc, IVb и/или III или их соли, описанными в настоящем документе выше.
D. ОПРЕДЕЛЕНИЯ
Что касается настоящего описания, то следующие термины имеют значения, указанные далее.
«Ацил» обозначает карбонил, содержащий заместитель, представленный формулой -C(O)-R, в котором R представляет собой водород, алкил, циклоалкил, гетероциклил, циклоалкил-замещенный алкил или гетероциклил-замещенный алкил, где алкил, алкокси, циклоалкил и гетероциклил являются, независимо, необязательно замещенными и имеют значения, определенные в настоящем документе. Ацильные группы включают алканоил (например, ацетил), ароил (например, бензоил) и гетероароил (например, пиридиноил).
Термин «алкил», как он используется в настоящем документе, относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу, состоящему из от одного до двенадцати атомов углерода, и, в другом варианте осуществления, от одного до шести атомов углерода, где алкильный радикал может быть необязательно замещенным, независимо, одним или несколькими заместителями, описанными в настоящем документе. Примеры алкильных групп включают, но этим не ограничиваются, метил (Me, -CH3), этил (Et, -CH2CH3), 1-пропил (н-Pr, н-пропил, -CH2CH2CH3), 2-пропил (изо-Pr, изопропил, -CH(CH3)2), 1-бутил (н-Bu, н-бутил, -CH2CH2CH2CH3), 2-метил-1-пропил (изо-Bu, изобутил, -CH2CH(CH3)2), 2-бутил (втор-Bu, втор-бутил, -CH(CH3)CH2CH3), 2-метил-2-пропил (трет-Bu, трет-бутил, -C(CH3)3), 1-пентил (н-пентил, -CH2CH2CH2CH2CH3), 2-пентил (-CH(CH3)CH2CH2CH3), 3-пентил (-CH(CH2CH3)2), 2-метил-2-бутил (-C(CH3)2CH2CH3), 3-метил-2-бутил (CH(CH3)CH(CH3)2), 3-метил-1-бутил (-CH2CH2CH(CH3)2), 2-метил-1-бутил (CH2CH(CH3)CH2CH3), 1-гексил (-CH2CH2CH2CH2CH2CH3), 2-гексил (CH(CH3)CH2CH2CH2CH3), 3-гексил (-CH(CH2CH3)(CH2CH2CH3)), 2-метил-2-пентил (C(CH3)2CH2CH2CH3), 3-метил-2-пентил (-CH(CH3)CH(CH3)CH2CH3), 4-метил-2-пентил (CH(CH3)CH2CH(CH3)2), 3-метил-3-пентил (-C(CH3)(CH2CH3)2), 2-метил-3-пентил (CH(CH2CH3)CH(CH3)2), 2,3-диметил-2-бутил (-C(CH3)2CH(CH3)2), 3,3-диметил-2-бутил (CH(CH3)C(CH3)2, 1-гептил, 1-октил и тому подобное.
Термин «алкилен», как он используется в настоящем документе, относится к линейному или разветвленному одновалентному насыщенному углеводородному радикалу, состоящему из от одного до двенадцати атомов углерода, и, в другом варианте осуществления, от одного до шести атомов углерода, где алкиленовый радикал может быть необязательно замещенным, независимо, одним или несколькими заместителями, описанными в настоящем документе. Примеры включают, но этим не ограничиваются, метилен, этилен, пропилен, 2-метилпропилен, пентилен и тому подобное.
Термин «алкенил», как он используется в настоящем документе, относится к линейному или разветвленному одновалентному углеводородному радикалу, состоящему из от двух до двенадцати атомов углерода, и, в другом варианте осуществления, от двух до шести атомов углерода, с, по меньшей мере, одним участком ненасыщенности, то есть, углерод-углерод, sp2 двойной связью, где алкенильный радикал может быть необязательно замещенным, независимо, одним или несколькими заместителями, описанными в настоящем документе, и включает радикалы, имеющие «цис» и «транс» ориентации, или, альтернативно, «E» и «Z» ориентации. Примеры включают, но этим не ограничиваются, этиленил или винил (-CH=CH2), аллил (-CH2CH=CH2), 1-пропенил, 1-бутен-1-ил, 1-бутен-2-ил и тому подобное.
Термин «алкинил», как он используется в настоящем документе, относится к линейному или разветвленному одновалентному углеводородному радикалу, состоящему из от двух до двенадцати атомов углерода, и, в другом варианте осуществления, от двух до шести атомов углерода, с, по меньшей мере, одним участком ненасыщенности, то есть, углерод-углерод, sp тройной связью, где алкинильный радикал может быть необязательно замещенным, независимо, одним или несколькими заместителями, описанными в настоящем документе. Примеры включают, но этим не ограничиваются, этинил (-C=CH) и пропинил (пропаргил, -CH2C=CH).
Термин «алкокси» относится к линейному или разветвленному одновалентному углеводородному радикалу, представленному формулой -OR, в которой R представляет собой алкил, алкенил, алкинил или циклоалкил, которые могут, кроме того, быть необязательно замещенными, как определено в настоящем документе. Алкокси группы включают метокси, этокси, пропокси, изопропокси, моно-, ди- и три-фторметокси и циклопропокси.
«Амино» означает первичные (то есть, -NH2), вторичные (то есть, -NRH), третичные (то есть, -NRR) и четвертичные (то есть, -N+RRRX-) амины, которые являются необязательно замещенными, где R, независимо, представляет собой алкил, алкокси, циклоалкил, гетероциклил, циклоалкил-замещенный алкил или гетероциклил-замещенный алкил, где алкил, алкокси, циклоалкил и гетероциклил имеют значения, определенные в настоящем документе. Конкретными вторичными и третичными аминами являются алкиламин, диалкиламин, ариламин, диариламин, аралкиламин и диаралкиламин, где алкилы и арилы имеют значения, определенные в настоящем документе, и являются, независимо, необязательно замещенными. Конкретные вторичные и третичные амины представляют собой метиламин, этиламин, пропиламин, изопропиламин, фениламин, бензиламин, диметиламин, диэтиламин, дипропиламин и диизопропиламин.
Термины «циклоалкил», «карбоцикл», «карбоциклил» и «карбоциклическое кольцо», как они используются в настоящем документе, используются взаимозаменяемо и относятся к насыщенному или частично ненасыщенному циклическому углеводородному радикалу, имеющему от трех до двенадцати атомов углерода, и, в другом варианте, от трех до восьми атомов углерода. Термин «циклоалкил» включает моноциклические и полициклические (например, бициклические и трициклические) циклоалкильные структуры, где полициклические структуры необязательно содержат насыщенное или частично ненасыщенное циклоалкильное кольцо, конденсированное с насыщенным, частично ненасыщенным или ароматическим циклоалкильным или гетероциклическим кольцом. Примеры циклоалкильных групп включают, но этим не ограничиваются, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклогексенил, циклогексадиенил, циклогептенил и тому подобное. Бициклические карбоциклы включают такие, которые имеют от 7 до 12 кольцевых атомов, расположенных, например, в виде бицикло[4,5], [5,5], [5,6] или [6,6] систем, или в виде мостиковых систем, таких как бицикло[2,2,1]гептан, бицикло[2,2,2]октан и бицикло[3,2,2]нонан. Циклоалкил может быть необязательно замещенным, независимо, одним или несколькими заместителями, описанными в настоящем документе.
Термин «арил», как он используется в настоящем документе, обозначает одновалентный ароматический углеводородный радикал, имеющий 6-20 атомов углерода, образованный путем удаления одного атома водорода от одного атома углерода исходной ароматической кольцевой системы. Арил включает бициклические радикалы, содержащие ароматическое кольцо, конденсированное с насыщенным, частично ненасыщенным кольцом или ароматическим карбоциклическим или гетероциклическим кольцом.
Примеры арильных групп включают, но этим не ограничиваются, радикалы, производные бензола, нафталина, антрацена, бифенила, индена, индана, 1,2-дигидронафталина, 1,2,3,4-тетрагидронафталина и тому подобное. Арильные группы могут быть необязательно замещены, независимо, одним или несколькими заместителями, описанными в настоящем документе.
Термины «гетероцикл», «гетероциклил» и «гетероциклическое кольцо», как они используются в настоящем документе, используются взаимозаменяемо и относятся к насыщенному или частично ненасыщенному карбоциклическому радикалу, состоящему из 3-12 членных кольцевых атомов, в котором, по крайней мере, один кольцевой атом представляет собой гетероатом, независимо выбранный из азота, кислорода и серы, остальные кольцевых атомы представляют собой C, где один или несколько кольцевых атомов могут быть необязательно замещены, независимо, одним или несколькими заместителями, описанными далее. Один вариант осуществления включает гетероциклы, состоящие из от 3 до 7 кольцевых атомов, в котором, по меньшей мере, один кольцевой атом является гетероатомом, независимо выбранным из азота, кислорода и серы, остальные кольцевые атомы представляют собой C, где один или несколько кольцевых атомов могут быть необязательно замещены, независимо, одним или несколькими заместителями, описанными далее. Радикал может представлять собой углеродный радикал или гетероатомный радикал. Термин «гетероцикл» включает гетероциклоалкокси. «Гетероциклил» также включает радикалы, где гетероциклические радикалы конденсированы с насыщенным, частично ненасыщенным или ароматическим карбоциклическим или гетероциклическим кольцом. Примеры гетероциклических колец включают, но этим не ограничиваются, пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, гомопиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 2-пирролинил, 3-пирролинил, индолинил, 2Н-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3-азабицикло[4.1.0]гептанил, азабицикло[2.2.2]гексанил, 3H-индолилхинолизинил и N-пиридилмочевины. Спиро группы также входят в объем данного определения. Гетероцикл может быть С-присоединенным или N-присоединенным, где такое возможно. Например, группа, полученная из пиррола, может представлять собой пиррол-1-ил (N-присоединенный) или пиррол-3-ил (С-присоединенный). Кроме того, группа, получаемая из имидазола, может представлять собой имидазол-1-ил (N-присоединенный) или имидазол-3-ил (С-присоединенный). Примеры гетероциклических групп, где 2 кольцевых атомов углерода замещены оксо (=O), представляют собой изоиндолин-1,3-дионил и 1,1-диоксо-тиоморфолинил. Гетероциклические группы в настоящем документе являются необязательно замещенными, независимо, одним или несколькими заместителями, описанными в настоящем документе.
Термин «гетероарил», как он используется в настоящем документе, относится к одновалентному ароматическому радикалу, состоящему из 5-, 6- или 7-членов кольца и включает конденсированные кольцевые системы (по меньшей мере, одна из которых является ароматической), состоящие из 5-10 атомов, содержащих, по меньшей мере, один гетероатом, независимо выбранный из азота, кислорода и серы. Примеры гетероарильных групп включают, но этим не ограничиваются, пиридинил, имидазолил, имидазопиридинил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Спиро группы также входят в объем данного определения. Гетероарильные группы могут быть необязательно замещены, независимо, одним или несколькими заместителями, описанными в настоящем документе.
«Удаляемая группа» относится к части первого реагента в химической реакции, которая вытесняется из первого реагента в химической реакции. Примеры удаляемых групп включают, но этим не ограничиваются, водород, галоген, гидроксильные группы, сульфгидрильные группы, аминогруппы (например -NRR, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R является независимо необязательно замещенным), силильные группы (например, -SiRRR, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным), -N(R)OR (где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным), алкокси группы (например -OR, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным), тиоловые группы (например -SR, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным), сульфонилокси группы (например -OS(O)1-2R, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным), сульфаматные группы (например -OS(O)1-2NRR, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным), карбаматные группы (например -OC(O)2NRR, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным), и карбонатные группы (например -OC(O)2RR, где R, независимо, представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R, независимо, является необязательно замещенным). Примеры сульфонилокси группы включают, но этим не ограничиваются, алкилсульфонилокси группы (например, метилсульфонилокси (мезилатная группа) и трифторметилсульфонилокси (трифлатная группа)) и арилсульфонилокси группы (например, п-толуолсульфонилокси (тозилатная группа) и п-нитросульфонилокси (нозилатная группа)). Другие примеры удаляемых групп включают замещенные и незамещенные аминогруппы, такие как амино, алкиламино, диалкиламино, гидроксиламино, алкоксиламино, N-алкил-N-алкоксиамино, ациламино, сульфониламино и тому подобное.
Термин «аминозащитная группа», как он используется в настоящем документе, относится к группам, обычно используемым для предохранения амино групп от взаимодействий в процессе проведения реакций с другими функциональными группами. Примеры таких защитных групп включают карбаматные, амидные, алкильные и арильные группы, имины, а также многие N-гетероатомные производные, которые могут быть удалены для восстановления желаемой амино группы. Конкретными аминозащитными группами являются Ac (ацетил), трифторацетил, фталимид, Bn (бензил), Tr (трифенилметил или тритил), бензилиденил, п-толуолсульфонил, Pmb (п-метоксибензил), Boc (трет-бутилоксикарбонил), Fmoc (9-флуоренилметилоксикарбонил) и Cbz (карбобензилокси). Другие примеры указанных групп можно найти в обзоре: Wuts, P. G. M. and Greene, T. W. (2006) Frontmatter, in Greene's Protective Groups in Organic Synthesis, Fourth Edition, John Wiley & Sons, Inc., Hoboken, NJ, USA. Термин «защищенный амино» относится к амино группе, замещенной одной из указанных выше аминозащитных групп.
Термин «замещенный», как он используется в настоящем документе, означает любую из упомянутых выше групп (например, алкил, алкилен, алкенил, алкинил, циклоалкил, арил, гетероциклил и гетероарил), где, по меньшей мере, один атом водорода заменен на заместитель. В случае оксо заместителя (=O) заменены два атома водорода. «Заместители» в контексте данного изобретения включают, но этим не ограничиваются, галоген, гидрокси, оксо, циано, нитро, амино, алкиламино, диалкиламино, алкил, алкенил, алкинил, циклоалкил, алкокси, замещенный алкил, тиоалкил, галогеналкил (включая пергалогеналкил), гидроксиалкил, аминоалкил, замещенный алкенил, замещенный алкинил, замещенный циклоалкил, арил, замещенный арил, гетероарил, замещенный гетероарил, гетероцикл, замещенный гетероцикл, -NReRr, -NReC(=O)Rf, -NReC(=O)NReRf, -NReC(=O)ORf-NReSO2Rr, -ORe, -C(=O)Re-C(=O)ORe, -C(=O)NReRf, -OC(=O)NReRf, -SRe, -SORe, -S(=O)2Re, -OS(=O)2Re, -S(=O)2ORe, где Re и Rf являются одинаковыми или различными и, независимо, представляют собой водород, алкил, замещенный алкил, алкенил, замещенный алкенил, алкинил, замещенный алкинил, циклоалкил, замещенный циклоалкил, арил, замещенный арил, гетероарил, замещенный гетероарил, гетероцикл, замещенный гетероцикл.
Термин «галоген» или «галоген», как он используется в настоящем документе, означает фтор, хлор, бром или йод.
Термин, упомянутый в единственном числе, как он используется в настоящем документе, означает один или несколько.
Указание «примерно» при описании значения или параметра, приведенных в настоящем документе, включает (и описывает) варианты осуществления, которые относятся к этому значению или параметру как таковому и в одном варианте плюс или минус 20% от данного значения. Например, описание, относящееся к «примерно X», включает описание «X».
Термин «соль» для различных соединений и промежуточных соединений, описанных и полученных согласно настоящему документу, обычно относится по существу к любой форме соли, признанной специалистом в данной области техники, подходящей для получения соединений и промежуточных соединений по настоящему изобретению. Термин «соль», как он используется в настоящем документе, подразумевает, но этим не ограничивается, «фармацевтически приемлемые соли» и включает аддитивные соли как с кислотой, так и с основаниями. Типичные соли включают, но не ограничиваются ими, сульфат, цитрат, ацетат, оксалат, хлорид, бромид, иодид, нитрат, бисульфат, фосфат, кислый фосфат, изоникотинат, лактат, салицилат, кислый цитрат, тартрат, олеат, таннат, пантозацетат, битартрат, аскорбат, сукцинат, малеат, гентизат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат (то есть 1,1'-метилен-бис-2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может включать присоединение более тяжелой молекулы, такой как ацетат-ион, сукцинат-ион или другие противоионы. Противоион может представлять собой любой органический или неорганический фрагмент, который стабилизирует заряд на исходном соединении.
Выражение «фармацевтически приемлемая соль присоединения кислоты» относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований и которые не являются биологически или иначе нежелательными, которые образованы с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота и т.п., и с органическими кислотами, которые могут быть выбраны из классов алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоксильных и сульфоновых органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота и тому подобное.
Выражение «фармацевтически приемлемые соли присоединения оснований» включают соли, полученные из неорганических оснований, такие как соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и тому подобное. В частности, солями присоединения основания являются соли аммония, калия, натрия, кальция и магния. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины, и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, трометамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминные смолы и т.п. В частности, органическими нетоксичными основаниями являются изопропиламин, диэтиламин, этаноламин, трометамин, дициклогексиламин, холин и кофеин.
Соединения по настоящему изобретению, если не указано иное, включают соединения, которые отличаются только наличием одного или нескольких изотопно обогащенных атомов. Например, соединения по настоящему изобретению, где один или несколько атомов водорода заменены на дейтерий или тритий, или один или несколько атомов углерода заменены на атом углерода13C или14C, или один или несколько атомов азота заменены на азот атом15N, или один или несколько атомов серы атомы заменены на атом серы33S,34S или36S, или один или несколько атомов кислорода заменены на атом кислорода17O или18O, находятся в рамках данного изобретения.
Следует отметить, что описанные здесь соединения могут содержать один или несколько хиральных центров. Соответственно, при желании, такие соединения могут быть получены или выделены в виде чистых стереоизомеров (таких как отдельные энантиомеры или диастереомеры, или в виде обогащенных стереоизомерами смесей). Все такие стереоизомеры (и обогащенные смеси) включены в объем данного изобретения, если не указано иное. Чистые стереоизомеры (или обогащенные смеси) могут быть получены с использованием, например, оптически активных исходных веществ или стереоселективных реагентов, хорошо известных из уровня техники. Альтернативно, рацемические или обогащенные стереоизомерами смеси таких соединений могут быть разделены с использованием, например, хиральной колоночной хроматографии, хиральных расщепляющих агентов и тому подобное.
Следует также отметить, что все патенты, патентные заявки, документы и статьи, цитируемые в настоящем документе, включены в настоящий документ посредством ссылки во всей их полноте для всех соответственных и соответствующих целей.
Несмотря на то, что способы и соединения во настоящему изобретению описаны в настоящем документе в связи с конкретными представленными вариантами осуществления, следует понимать, что они не предназначены для ограничения объема этих вариантов осуществления. Напротив, описание предназначено для охвата всех альтернатив, модификаций и эквивалентов. Специалисту в данной области известны многие способы и вещества, аналогичные или эквивалентные описанным в настоящем документе, которые могут быть использованы при осуществлении настоящего способа. Соответственно, настоящее изобретение никоим образом не ограничивается описанными здесь способами и материалами.
Кроме того, в случае, если один или несколько включенных литературных и аналогичных материалов отличаются или противоречат этому описанию, включая, но не ограничиваясь ими, определенные термины, понятия, описанные технологии и т.п., данное описание имеет преимущество.
ПРИМЕРЫ
Настоящее изобретение может быть более понятным с отсылкой к следующим примерам, в которых представлены подробные детали получения различных соединений, проиллюстрированных на схеме 4, выше.
Пример 1: Добавление малонатного конъюгата

Диметил-2-(1-цианопропан-2-ил)малонат: трет-Пентоксид натрия (65,7 г) добавляли в ТГФ (900 г), и смесь перемешивали в течение 30 минут (мин). Добавляли диметилмалонат (433 г), и затем смесь нагревали до температуры 60-70°C. Добавляли кротононитрил (200 г), поддерживая внутреннюю температуру при 60-70°C. Смесь перемешивали до завершения реакции и затем охлаждали до комнатной температуры. Добавляли хлористоводородную кислоту (HCl) в метаноле (MeOH) до тех пор, пока смесь не достигала pH между 7-8, и затем фильтровали. Слой на фильтре промывали с помощью ТГФ (360 г). Собранный раствор или фильтрат перегоняли для удаления легколетучих продуктов и получали сырой диметил-2-(1-цианопропан-2-ил)малонат в вид масла от бесцветного до светло-желтого цвета, которое напрямую использовали на следующей стадии.1H ЯМР (500 МГц, ДМСО-d6) δ 3,67 (с, 6H), 3,51 (д, 1H, J=7,5 Гц), 2,68 (дд, 1H, J=5, 17 Гц), 2,54 (дд, 1H, J=8, 17 Гц), 2,44 (м, 1H), 1,04 (д, 3H, J=7 Гц).
Пример 2: Ферментативное разделение
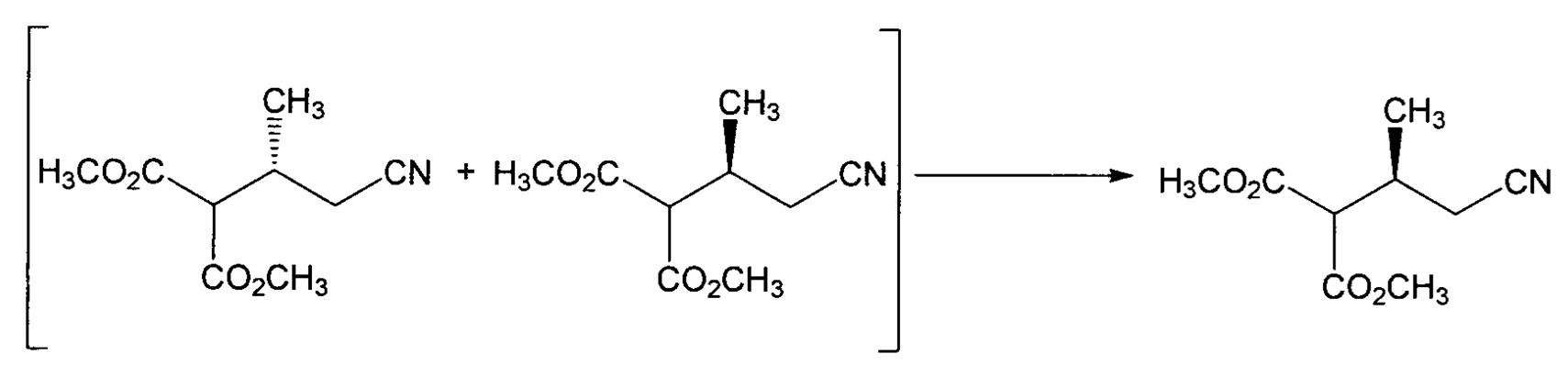
Примеры ферментного разделения были проведены, как подробно описано ниже.
Пример 2а. Диметил-(R)-2-(1-цианопропан-2-ил)малонат посредством нитрилазы. В четырехгорлую круглодонную колбу емкостью 1500 мл, снабженную мешалкой KPG, термометром и капельной воронкой, загружали 69,7 г (400 ммоль) сульфата калия и 5,44 г (40 ммоль) дигидрофосфата калия в 800 мл деионизированной воды (рН 4,85). По каплям при перемешивании добавляли гидроксид натрия (3,87 г, 32% водный раствор) до конечного рН 7,2, а затем раствор перемешивали в течение 15 минут. Добавляли диметил-2-(1-цианопропан-2-ил) малонат (199,5 г, 1,00 моль, 98% масс./масс.), и затем двухфазную эмульсию перемешивали в течение 5 мин при температуре 20-25°С (рН оставался неизменным).
Энантиоселективный гидролиз начинали с добавления 39 мл нитрилазного раствора Nit-BX4-56-H6 (c-Lecta, Лейпциг, Германия, номер в каталоге 10906-3L, 500 ед./мл) в течение 5 мин. Капельную воронку промывали 8 мл деионизированной воды, и реакционную смесь (рН 7,18) перемешивали при температуре 20-25°С. Когда энантиомерный избыток удерживаемого и желаемого нитрила достигал 99,7% эи (после приблизительно 55%-ной конверсии, E приблизительно 50; через 43 часа; pH 6,93), pH реакционной смеси доводили до 2,0 по каплям добавляли приблизительно 96 г 25%-ной соляной кислоты (температура менее 28°С; сильное осаждение белка). Эмульсию/суспензию перемешивали в течение 10 мин и затем доводили рН до 7,5 путем добавления приблизительно 85 г 32%-ного раствора гидроксида натрия (температура менее 35°С). Смесь перемешивали в течение 10 мин и затем добавляли 500 мл этилацетата, и суспензию/эмульсию перемешивали в течение еще 5 мин. Две фазы оставляли разделяться (приблизительно 3 мин; белок осаждался в основном в органической фазе), а затем последовательно фильтровали через фильтровальную ткань (9 см, 20 мкм). Фильтр промывали 500 мл этилацетата, органические фазы в фильтрате объединяли и затем оставляли для отделения водной фазы. Водную фазу снова экстрагировали 1 л этилацетата. Объединенные органические фазы последовательно промывали 200 мл 1М раствора бикарбоната натрия и 100 мл деионизированной воды, соответственно, и упаривали досуха при 50°С/40 мбар/1,5 часа с получением 90,7 г (45%) диметил-(R)-2-(1-цианопропан-2-ил)малоната в виде масла светло-желтого цвета:1H ЯМР (500 МГц, ДМСО-d6) δ 3,67 (с, 6H), 3,51 (д, 1H, J=7,5 Гц), 2,68 (дд, 1H, J=5, 17 Гц), 2,54 (дд, 1H, J=8, 17 Гц), 2,44 (м, 1H), 1,04 (д, 3H, J=7 Гц);13C ЯМР (125 МГц, ДМСО-d6) δ 168,43, 119,30, 55,36, 53,00, 52,95, 30,41, 21,89, 17,17; [α]43620 -2,2 (c=1, MeOH); МСВР, вычислено для C19Н12NO4 [M-H]-: 198,0772; найдено: 198,0770. Анализ: 95,3% ГХ (триметилсилилированный BSТФУ); 99,7% эи (ГХ на BGB-175, 30 м×0,25 мм, 0,25 мкм, H2, 135 кПа, 90°C-150°C при 2°C/мин, до 180°C при 20°C/мин. 200°C; опр. 250°C; инъец. объем 1 мл; разделение 15:1; 30,77 мин (R)-1, 31,09 мин (S)-1; содержащий 2,5% этилацетата и <0,1% воды).
Пример 2b. Диметил-(R)-2-(1-цианопропан-2-ил)малонат посредством гидролиза сложного эфира с использованием липазы CRL3. Диметил-2-(1-цианопропан-2-ил)малонат (3,0 г, 15,1 ммоль) помещали в реактор с последующим добавлением 21 мл 0,03 М ацетатного буфера с pH 5,2, 1,96 г сульфата калия и 6,0 мл гептана. После 5-минутного перемешивания добавление 60,0 мг холестеринэстеразы из Candida cyclindracea [Roche, партия 10347322] (s/e 50) запускало гидролиз сложного эфира при комнатной температуре (температура около 23°С). Поддерживали постоянное значение рН, добавляя 1,0н раствор гидроксида натрия, который расходовался в количестве 7,6 мл (7,6 ммоль) в течение 50 ч при перемешивании. Затем добавляли этилацетат (30 мл), и реакционную смесь интенсивно перемешивали в течение 15 мин с образованием эмульсии. Затем добавляли 1,5 г фильтрационного средства (Dicalite®), и реакционную смесь интенсивно перемешивали в течение еще 15 мин, затем фильтровали через фильтровальный слой (Dicalite®). Две фазы разделяли. Водную фазу дополнительно экстрагировали дважды этилацетатом (50 мл). Органические фазы объединяли и промывали три раза 50 мл 100мМ калий фосфатного буфера с рН 7,2. Органическую фазу сушили над сульфатом магния, фильтровали и упаривали, получая 1,26 г (R)-диметил-2-(1-цианопропан-2-ил)малоната (чистота 99,6%, выход 42%) в виде бесцветного масла. Энантиомерный избыток=100% эи (метод ВЭЖХ: колонка: Chiralpak IA-3 150 мм×4,6 мм, 3 мкм, изократический A: 95% гептан+0,10% ТФУ; B: 5% этанол; скорость потока 2 мл/мин; температура 30°C; 214 нм; 130 бар; время удержания: (S)-нитрил 3,4 мин, (R)-нитрил 3,89 мин).
Пример 2c. Диметил-(R)-2-(1-цианопропан-2-ил)малонат посредством гидролиза сложного эфира с использованием липазы EL030 (III). Диметил-2-(1-цианопропан-2-ил)малонат (300 мг, 1,51 ммоль) помещали в реактор с последующим добавлением 29,7 мл 0,03М буфера MES (pH 6,2) и 300 мг β-циклодекстрина. Через 5 мин перемешивания добавление 60,0 мг EL030 (III) [EUCODIS, лот 04804012SS0911] (s/e 5) запускало гидролиз сложного эфира при комнатной температуре (температура около 23°С). Поддерживали постоянное значение рН, добавляя 1,0н раствор гидроксида натрия, который расходовался в количестве 1,05 мл (1,05 ммоль) в течение 48 ч при перемешивании. Хиральный анализ ВЭЖХ показывал энантиомерный избыток=100% эи диметил-(R)-2-(1-цианопропан-2-ил)малоната.
Пример 2d. Диметил-(R)-2-(1-цианопропан-2-ил)малонат посредством нитрилазы при рН 9. В четырехгорлую круглодонную колбу емкостью 350 мл, снабженную мешалкой KPG, термометром и капельной воронкой, загружали 17,5 г (100 ммоль) сульфата калия, 1,36 г (10 ммоль) дигидрофосфата калия и 1,325 г карбоната натрия (12,5 ммоль) в 200 мл деионизированной воды (рН 9,2), и раствор перемешивали в течение 15 мин. В течение 5 минут добавляли диметил-2-(1-цианопропан-2-ил)малонат (50,0 г, 250 мл, 98% масс./масс.), и затем двухфазную эмульсию перемешивали в течение 5 мин при температуре 20-25°С.
Энантиоселективный гидролиз начинали с добавления 9,75 мл (4875 Ед) нитрилазного раствора Nit-BX4-56-H6 (c-Lecta, Лейпциг, Германия, номер в каталоге 10906-3L, 500 Ед/мл) в течение 2 мин. Капельную воронку промывали 2 мл деионизированной воды, и реакционную смесь перемешивали при температуре 20-25°С. Когда энантиомерный избыток удерживаемого и желаемого нитрила достигал 99% эи (после приблизительно 53%-ной конверсии; E приблизительно 80; через 18 часов; pH 8,2), pH реакционной смеси доводили до 2,0, по каплям добавляя примерно 24,7 г 25%-ной соляной кислоты (температура менее 27°С; сильное осаждение белка). Эмульсию/суспензию перемешивали в течение 10 мин, и затем доводили рН до 7,5 путем добавления примерно 23,1 г 32% раствора гидроксида натрия (температура менее 35°С). Смесь перемешивали в течение 10 мин, и затем добавляли 125 мл этилацетата, и суспензию/эмульсию перемешивали в течение еще 5 мин. Две фазы оставляли разделяться (приблизительно 3 мин; белок осаждался в основном в органической фазе), а затем последовательно фильтровали через фильтровальную ткань (3 см, 20 мкм). Фильтр промывали 125 мл этилацетата, органические фазы в фильтрате объединяли и затем оставляли для отделения водной фазы. Водную фазу снова экстрагировали 250 мл этилацетата. Объединенные органические фазы последовательно промывали 50 мл 1М бикарбоната натрия и 25 мл деионизированной воды, соответственно, и упаривали досуха при 50°С/40 мбар/1,5 часа с получением 23,1 г (46,2%) диметил-(R)-2-(1-цианопропан-2-ил)малоната в виде масла светло-желтого цвета с 99% эи и 94,9% по данным ГХ (сравни с примером 2а).
Пример 2е - Диметил-(R)-2-(1-цианопропан-2-ил)малонат посредством нитрилазы при рН 9. В четырехгорлую круглодонную колбу объемом 350 мл, снабженную мешалкой KPG, термометром и капельной воронкой, содержащей 50,0 г диметил-2-(1-цианопропан-2-ил)малоната (250 ммоль, 98% масс./масс.), добавляли раствор 7,62 г (20 ммоль) декагидрата динатрий тетрабората и 17,5 г (100 ммоль) сульфата калия в 200 мл деионизированной воды (рН 9,3), и двухфазную эмульсию перемешивали в течение 10 мин при температуре 20-25°C.
Энантиоселективный гидролиз начинали с добавления 9,75 мл (4875 Ед) нитрилазного раствора Nit-BX4-56-H6 (c-Lecta, Лейпциг, Германия, номер в каталоге 10906-3L, 500 Ед/мл) в течение 2 мин. Капельную воронку промывали 2 мл деионизированной воды, и реакционную смесь перемешивали при 20-25°С. Когда энантиомерный избыток удерживаемого и желаемого нитрила достигал 99,8% эи (через 18 ч; pH 8,4), pH реакционной смеси доводили до 2,0, по каплям добавляя примерно 27,7 г 25%-ной соляной кислоты (температура менее 27°С; сильное осаждение белка). Эмульсию/суспензию перемешивали в течение 10 мин, и затем рН доводили до 8,0 путем добавления примерно 25,3 г 32%-ного раствора гидроксида натрия (температура менее 32°С). Смесь перемешивали в течение 10 мин, затем добавляли 125 мл метил-трет-бутилового эфира, и суспензию/эмульсию перемешивали в течение еще 5 мин, и затем фильтровали через фильтровальную ткань (5 см, 20 мкм). Реакционный сосуд и фильтр промывали еще 125 мл метил-трет-бутилового эфира, органические фазы в фильтрате объединяли и затем оставляли для отделения водной фазы. Водную фазу снова экстрагировали 250 мл метил-трет-бутилового эфира. Объединенные органические фазы последовательно промывали 50 мл 1М бикарбоната натрия и 25 мл деионизированной воды, соответственно, и упаривали досуха при температуре 50°С/7 мбар/2 часа с получением 21,32 г (42,2%) диметил-(R)-2-(1-цианопропан-2-ил)малоната в виде масла светло-желтого цвета с 99% эи и 95,7% по данным ГХ (сравни с примером 2а).
Пример 3: Синтез пиримидина

(R)-3-(4,6-Дигидроксипиримидин-5-ил)бутаннитрил. В реактор загружали формамидин ацетат (2,9 г) и МеОН (13,0 мл, 2,6 мл/г). Смесь охлаждали до температуры 5°С в атмосфере N2, а затем в реактор загружали NaOMe (17,0 мл), что приводило к небольшой экзотермической реакции. После охлаждения до температуры 5°С к вышеуказанной суспензии медленно добавляли раствор диметил (R)-2-(1-цианопропан-2-ил)малоната (5,0 г) в МеОН (3,0 мл), затем реакционную смесь нагревали до температуры 25°С. Перемешивание продолжали в течение приблизительно 2 ч. и затем добавляли воду (15,0 мл) и доводили рН до 5-7 порционной добавкой концентрированной HCl (4,1 мл), поддерживая температуру ниже 30°С. Реакционную смесь концентрировали в вакууме до приблизительно 1/3 от исходного объема, и затем отбирали образцы для определения содержания MeOH. Когда содержание MeOH составляло 10-20%, взвесь охлаждали до температуры 5°С, и рН доводили до 4-6 добавлением концентрированной HCl (1,25 мл, 0,25 мл/г). После перемешивания в течение 1 ч при температуре 5°С полученные твердые вещества собирали фильтрованием. Осадок на фильтре промывали холодной водой (15,0 мл, 3,0 об., температура 5°С) и сушили в вакуумной печи при температуре 70°С в течение ночи, получая (R)-3-(4,6-дигидроксипиримидин-5-ил)бутаннитрил в виде твердого вещества не совсем белого цвета (3,86 г, выход 86%):1H ЯМР (500 МГц, ДМСО-d6) δ 11,85 (шир. с, 2H), 7,96 (с, 1H), 3,27 (м, 1H), 2,90 (дд, 1H, J=9, 17 Гц), 2,74 (дд, 1H, J=7, 17 Гц), 1,17 (д, 1H, J=7 Гц);13C ЯМР (125 МГц, ДМСО-d6) δ 164,15, 148,16, 120,62, 103,17, 26,86, 21,07, 18,00; [α]43620-69,9 (c=1, N,N-диметилацетамид); МСВР, вычислено для C8H8N3O2 [M-H]-:178,0622, найдено: 178,0622.
Пример 4: Непрерывный способ хлорирования-бромирования-SNAr

трет-Бутил (R)-4-(6-бром-5-(1-цианопропан-2-ил)пиримидин-4-ил)-пиперазин-1-карбоксилат. В реактор, снабженный обратным холодильником, при комнатной температуре в атмосфере N2 помещали (R)-3-(4,6-дигидроксипиримидин-5-ил)бутаннитрил (60,0 г, 334,9 ммоль, 1 экв.), толуол (720 мл, 12 об.) и 2,6-лутидин (39,0 мл, 1,0 экв.). Смесь нагревали до температуры 110°C и медленно добавляли POCl3 (93,4 мл, 3 экв.), поддерживая температуру при 105-115°C. Через 2 часа исходное вещество расходовалось полностью, как определялось методом ВЭЖХ (>99%). Бифазную реакционную смесь охлаждали до внутренней температуры 10-20°C. В отдельный реактор помещали 0,4M буфер K3PO4 с pH 7 (300 мл, 5 об.) при комнатной температуре в атмосфере N2. Хлорированную реакционную смесь медленно добавляли в буферный раствор, поддерживая температуру ниже 30°C. Далее pH гашеной смеси поддерживали при 5-7 с использованием 50%-ного водн. NaOH (266 г, 4,4 масс.). После завершения гашения слои разделяли, и водный слой экстрагировали толуолом (300 мл, 5 об.). Объединенные органические фазы промывали 0,2н водной HCl (250 мл, 4,2 объема). Органический раствор промывали водой (2×200 мл). Полученный раствор перегоняли в вакууме до тех пор, пока уровень толуола в сыром продукте не становился менее 20 масс%, как было определено с помощью ГХ анализа. Аналитический образец получали хроматографией концентрированного образца на силикагеле с использованием в качестве элюента смеси (1:1) EtOAc/гексан. (R)-3-(4,6-дихлорпиримидин-5-ил)бутаннитрил:1H ЯМР (500 МГц, ДМСО-d6) δ 8,83 (с, 1H), 3,93 (м, 1H), 3,11 (м, 2H), 1,43 (д, 3H, J=7 Гц);13C ЯМР (125 МГц, ДМСО-d6) δ 161,40, 157,05, 133,07, 119,25, 32,57, 20,84, 16,80; [α]43620 +16,8 (c=1, MeOH); МСВР, вычисл. для C8H8Cl2N3 [M+H]+: 216,0092, найдено: 216,0092.
К сырому продукту добавляли ацетонитрил (216,0 мл, 3,6 об.). Трубка для подачи азота была установлена таким образом, чтобы газ протекал в свободном пространстве реактора и выходил из дистилляционной головки. Реакционную смесь затем нагревали до температуры 70-80°C (цель=75°C) с получением раствора желтого цвета. Затем при температуре 75°C в течение 10 мин добавляли бромтриметилсилан (90 мл, 1,5 об., 200 моль%). Это приводило к снижению температуры реакции на около 2-5°C и началась медленная дистилляция. После перемешивания в течение 30 мин при температуре 75°C, в течение 90 мин добавляли дополнительное количество бромтриметилсилана (66 мл, 1,1 об., 150 моль%). Этот раствор оставляли перемешиваться при температуре в течение всего 16-18 часов, и затем отбирали образец и анализировали с помощью ВЭЖХ на конверсию. (Если конверсия была неполной, добавляли дополнительное количество бромтриметилсилана в избытке 0,25 экв. до достижения желаемого уровня.) Аналитический образец получали хроматографией концентрированного образца на силикагеле с использованием в качестве элюента смеси (1:1) EtOAc/гексан. (R)-3-(4,6-дибромпиримидин-5-ил)бутаннитрил:1H ЯМР (500 МГц, ДМСО-d6) δ 8,68 (с, 1H), 3,97 (м, 1H), 3,23 (дд, 1H, J=9, 17 Гц), 3,13 (дд, 1H, J=8, 17 Гц), 1,44 (д, 3H, J=7 Гц);13C ЯМР (125 МГц, ДМСО-d6) δ 157,04, 137,23, 119,13, 36,00, 20,77, 16,73; [α]43620+39,5 (c=1, MeOH); МСВР, вычисл. для C8H8Br2N3 [M+H]+: 303,9079, найдено: 303,9087.
Реакционную смесь охлаждали до внутренней температуры в 20-25°C и разбавляли ацетонитрилом (достаточное количество для получения 0,5M раствора). Затем в реакционную смесь медленно добавляли триэтиламин (93,6 мл, 1,56 об., 200 моль%) и воду (36,0 мл, 0,60 об.), и для измерения pH отбирали образец (разбавление 1:100 в воде). Если pH выше чем 10, добавляли N-BOC-пиперазин (71,5 г, 1,19 масс, 115 моль%), и перемешивание продолжали при температуре 20-25°C. Если pH был меньше чем 10, добавляли триэтиламин до достижения желаемого pH. Реакционную смесь перемешивали при температуре 20-25°C в течение 16-18 час. Образец отбирали и анализировали с помощью ВЭЖХ на конверсию (G02855614 ≤2%). Если твердых веществ не было, для затравки использовали трет-бутил (R)-4-(6-бром-5-(1-цианопропан-2-ил)пиримидин-4-ил)пиперазин-1-карбоксилат (6,9 г, 0,12 масс). Для высаживания продукта медленно добавляли воду (720,0 мл, 18,0 об.). Смесь охлаждали до температуры 5°C и перемешивали, по меньшей мере, в течение 2 час и затем фильтровали. Слой на фильтре промывали водой комнатной температуры (360,0 мл, 6,0 об.) и затем помещали в вакуумную печь при температуре 70-80°C до постоянного веса с получением порошка от не совсем белого до белого цвета (123 г, 90%-ный выход) трет-бутил (R)-4-(6-бром-5-(1-цианопропан-2-ил)пиримидин-4-ил)пиперазин-1-карбоксилата:1H ЯМР (500 МГц, ДМСО-d6) δ 8,41 (с, 1H), 3,41 (шир. м, 4H), 3,24 (шир. м, 4H), 3,08 (м, 2H), 1,48 (д, 3H, J=7 Гц), 1,41 (с, 9H);13C ЯМР (125 МГц, ДМСО-d6) δ 168,23, 155,67, 154,44, 152,15, 125,08, 119,51, 79,62, 50,27, 31,37, 28,52, 21,64, 17,66; [α]43620 +78,9 (c=0,3, MeOH); МСВР, вычисл. для C17H25BrN5O2 [M+H]+: 410,1186, найдено: 410,1180.
Пример 5: Циклизация Гриньяра

трет-Бутил (R)-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d] пиримидин-4-ил)пиперазин-1-карбоксилат. Раствор трет-бутил (R)-4-(6-бром-5-(1-цианопропан-2-ил)пиримидин-4-ил)пиперазин-1-карбоксилата (2,00 г) в безводном 2-MeТГФ (5,00 мл) и безводном толуоле (5,00 мл) охлаждали до температуры 5°C в 50 мл-овом сосуде. В течение 4 часов добавляли через шприцевый насос раствор изо-PrMgCl (3,52 мл, 105 моль%, 1,45M в ТГФ), поддерживая температуру бани при температуре 5±3°C. Реакционную смесь оранжевого-коричневого цвета гасили водным раствором NaHSO4 в течение 5 мин, поддерживая температуру бани при температуре 20±5°C. Значение pH водного слоя определяли равным примерно 5 с помощью pH бумаги. Смесь перемешивали в течение 30 мин при температуре 20±5°C, и слои разделяли. Органический слой концентрировали в вакууме (Tj=35°C, 100 мбар) до концентрации раствора 190-210 мг/г. В раствор затем вносили затравку трет-бутил (R)-4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилата (0,10 г, 0,05 г/г), и перемешивали в течение 1 час при температуре 20-25°C. В течение 2 час добавляли дегазированный гептан (14,0 мл, 7,0 мл/г), и затем раствор фильтровали и промывали смесью гептан/толуол комнатной температуры (80:20, 6,0 мл, 3,0 мл/г) и сушили в вакуумной печи при температуре 60°C до постоянного веса с получением порошка коричневато-оранжевого цвета. Высушенный сырой кетон (1,25 г, 1,0 масс), 2-пропанол (5,0 мл, 4,0 мл/г) и воду (1,25 мл, 1,0 мл/г) помещали в сосуд объемом 50 мл. Взвесь дегазировали с помощью N2 в течение, по меньшей мере, 10 мин, затем нагревали при температуре 40°C до растворения всех твердых веществ. Этот раствор затем охлаждали до температуры 25°C и вносили затравку (0,06 г, 0,05 г/г) и перемешивали в течение, по меньшей мере, 1 часа. Взвесь охлаждали до температуры -5°C в течение 1 часа, затем в течение 1 часа добавляли дегазированную воду (6,04 мл, 4,83 мл/г). Эту взвесь оставляли перемешиваться в течение ночи, затем фильтровали и промывали дегазированной, предварительно охлажденной (-5°C) смесью 2-пропанола (1,5 мл, 1,2 мл/г) и воды (3,5 мл, 2,8 мл/г). Твердые продукты сушили в вакуумной печи при температуре 70°C до постоянного веса с получением порошка коричневато-оранжевого цвета. Общий выход по способу составлял приблизительно 68-73% (1,1 г):1H ЯМР (500 МГц, ДМСО-d6) δ 8,60 (с, 1H), 3,82 (м, 2H), 3,72 (м, 1H), 3,65 (м, 2H), 3,50 (м, 2H), 3,42 (м, 2H), 2,90 (дд, 1H, J=7, 19 Гц), 2,25 (дд, 1H, J=2, 19 Гц), 1,41 (с, 9H), 1,19 (д, 3H, J=7 Гц);13C ЯМР (125 МГц, ДМСО-d6) δ 205,75, 161,96, 158,89, 157,97, 154,34, 137,39, 79,67, 45,77, 43,39, 43,25, 31,22, 28,52, 20,40; [α]43620 +453,7 (c=1, MeOH); МСВР, вычисл. для C17H24N4O3 [M+H]+: 333,1921, найдено: 333,1916.
Пример 6: Сравнительный пример - попытка проведения реакции Гриньяра
Циклизация аналогичного хлор-нитрил замещенного соединения
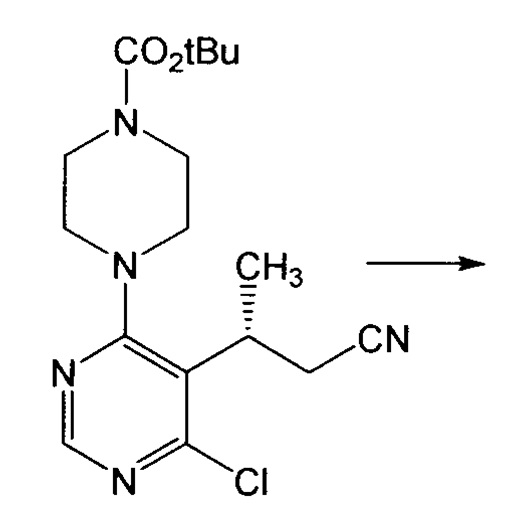
Сначала была предпринята попытка циклизации хлорпириминового соединения, используя катализатор на основе переходного металла (например, Pd, Rh, Ir) с различными фосфиновыми лигандами. Всякий раз главным продуктом в реакционной смеси обнаруживали разложивший хлор-пиримидин. Попытки циклизации соответствующего хлорпиримидиннитрила, такого как описан далее, с кетоном, были проведены с использованием ряда каталитических предшественников на основе переходных металлов, включая Ir, Ni, Pd, Rh и Ru. Эти каталитические предшественники перед началом реакций объединяли в комплекс с моно- и бидендатными фосфиновыми лигандами. Также были испытаны такие добавки, как кислоты Льюиса и Бренстеда, вода, катализаторы на основе переходного металла и катализаторы фазового переноса. Восстановители варьировали от неорганических металлов, таких как цинк, до органических восстановителей, таких как формиатные соли. Наконец, был исследован разнообразный набор растворителей, включая смешиваемые с водой простые эфиры, спирты, углеводороды от полярных апротонных до неполярных и карбонаты. Во всех этих случаях наивысший выход, получаемый в результате отсеивающих экспериментов, составлял приблизительно 20% желаемого кетонового продукта, а остальная часть была продуктами разложения исходного материала. Основным побочным продуктом, образовывавшимся во многих из этих реакций, был продукт обесхлоривания.
Пример 6a: трет-бутил 4-(5-метил-7-оксо-5,6-дигидроциклопента [d]пиримидин-4-ил)пиперазин-1-карбоксилат. Взвесь Pd(OAc)2 (6,1 мг, 0,10 эквив.), диметилфенилфосфина (15,1 мг, 0,40 эквив.) и 1,2-дихлорэтана (2,0 мл, 20,0 мл/г) перемешивали при температуре 20-25°C в течение 15 мин, затем растворитель удаляли в вакууме. Добавляли порошок цинка (35,7 мг, 2,0 эквив.), трет-бутил 4-[6-хлор-5-(2-циано-1-метил-этил)пиримидин-4-ил]пиперазин-1-карбоксилат (100,0 мг, 1,0 эквив.) и ДМФ (6,0 мл, 60,0 мл/г), и взвесь нагревали при температуре 110°C в течение 18 час. Анализ показывал, что выход сырой реакционной смеси составлял 15,8% (14,4 мг).
Пример 6b: трет-бутил 4-(5-метил-7-оксо-5,6-дигидроциклопента [d]пиримидин-4-ил)пиперазин-1-карбоксилат. Взвесь NiCl2(DME) (24,0 мг, 0,40 эквив.), диметилфенилфосфина (15,1 мг, 0,40 эквив.) и 1,2-дихлорэтана (2,0 мл, 20,0 мл/г) перемешивали при температуре 20-25°C в течение 15 мин, затем растворитель удаляли в вакууме. Добавляли порошок цинка (35,7 мг, 2,0 эквив.), трет-бутил 4-[6-хлор-5-(2-циано-1-метил-этил)пиримидин-4-ил]пиперазин-1-карбоксилат (100,0 мг, 1,0 эквив.) и ДМФ (6,0 мл, 60,0 мл/г), и взвесь нагревали при температуре 110°C в течение 18 час. ЖХ анализ показывал, что выход сырой реакционной смеси составлял 14,5% (13,2 мг).
Пример 6c: трет-бутил 4-(5-метил-7-оксо-5,6-дигидроциклопента[d]пиримидин-4-ил)пиперазин-1-карбоксилат. Взвесь NiCl2(DME) (24,0 мг, 0,40 эквив.), диметилфенилфосфина (15,1 мг, 0,40 эквив.) и 1,2-дихлорэтана (2,0 мл, 20,0 мл/г) перемешивали при температуре 20-25°C в течение 15 мин, затем растворитель удаляли в вакууме. Добавляли порошок цинка (35,7 мг, 2,0 эквив.), трет-бутил 4-[6-хлор-5-(2-циано-1-метил-этил)пиримидин-4-ил]пиперазин-1-карбоксилат (100,0 мг, 1,0 эквив.), Mg(OEt)2 (13,2 мг, 0,40 эквив.) и ДМФ (6,0 мл, 60,0 мл/г), и взвесь нагревали при температуре 110°C в течение 18 час. ЖХ анализ показывал, что выход сырой реакционной смеси составлял 19,0% (17,3 мг).
Пример 7: Сравнительный Пример -
Попытка проведения реакции бромирования с использованием HBr
Попытки проведения реакции бромирования дихлорпиримидиннитрила с избытком HBr и уксусной кислотой или бромидом натрия и активированием метансульфоновой кислотой или катализатором на основе меди в различных растворителях не позволяли получить приемлемые количества соответствующего дибромпиримидиннитрила.
Например, бромирование дихлорпиримидиннитрила с использованием избытка HBr в уксусной кислоте или NaBr со стехиометрическим количеством метансульфоновой кислотой в ацетонитриле приводило лишь к неполному преобразованию в желаемый дибромпиримидин с доказательствами обратимости и разложения продукта на основе анализа ЖХВР реакционной смеси. Альтернативно, взаимодействие дихлорпиримидиннитрила с избытком бромида натрия и 5 моль% CuI в качестве катализатора и 10 моль% N,N'-диметилэтилендиамина или 4,4'-ди-трет-бутил-2,2'-бипиридина в качестве лигандов в ацетонитриле, диоксане или 1-бутаноле при температуре 80-120°С, не показывали никакого присутствия желаемого продукта или конкурирующего добавления растворителя или диаминового лиганда к пиримидиновому кольцу.
Кроме того, следует учитывать, что конкретные численные значения, приведенные в настоящем документе, включая, например, концентрации или соотношения реагентов, время реакции, температуру реакции и тому подобное, предназначены для раскрытия и охвата всех значений между ними, а также всех диапазонов, которые могут быть созданы путем выбора любых двух их значений. Например, следует понимать, что ссылка на температуру реакции «около 20°С, 10°С, около 0°С или менее» охватывает температурные диапазоны реакции между около 0°С и около 20°С, между около 0°С и около 10°С, между около 10°С и около 20°С, или любыми двумя значениями между ними.
Что касается использования в данной патентной заявке (включая формулу изобретения) слов «включают», или «включает», или «включающий», то заявители отмечают, что, если контекст не требует иного, эти слова используются на основе четкого понимания того, что они должны толковаться включительно, а не исключительно, и что заявителями каждое из этих слов в конструкции данной патентной интерпретируется таким образом, чтобы толковать эту заявку на патент, включая требования ниже.
Как используется в данном документе, указание термина в единственном числе означает «один или несколько». По всему документу множественное и единственное число должны рассматриваться как взаимозаменяемые, иные, чем указание числа. Например, ссылка на «соединение» включает одно соединение, а также на одно или несколько дополнительных соединений, ссылка на «фармацевтически приемлемый носитель» включает один фармацевтически приемлемый носитель, а также один или несколько дополнительных фармацевтически приемлемых носителей и тому подобное.
Реферат
Изобретение относится к улучшенному способу получения (S)-2-(4-хлорфенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро--циклопента[]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)-пропан-1-она соответствующему формуле IX (ипатасертибу), которое является ингибитором АКТ и может быть использовано при лечении солидных опухолей, рака желудка и рака предстательной железы. Изобретение также относится к способу получения промежуточного соединения формулы I, а также новым промежуточным продуктам для их получения. Способ получения соединения IXIXIIIIгде Rпредставляет собой водород или аминозащитную группу, включает (i) взаимодействие соединения формулы III или его соли с металлирующим агентом, с последующим добавлением водной кислоты с образованием соединения формулы I или его соли; (ii) восстановление соединения формулы I или его соли с образованием соединения формулы VIIформула VII,или его соли;формула VII(iii) необязательное удаление защитных групп у соединения формулы VIIили его соли с образованием соединения формулы VIIили его соли и (iv) взаимодействие соединения формулы VIIили его соли с соединением формулы VIIIформула VIII,или его соли с образованием соединения формулы IX или его соли.7 н. и 28 з.п. ф-лы, 7 пр.
Формула




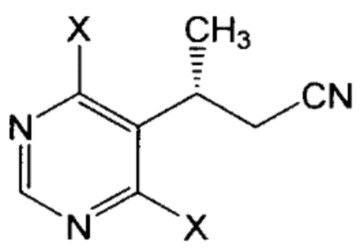



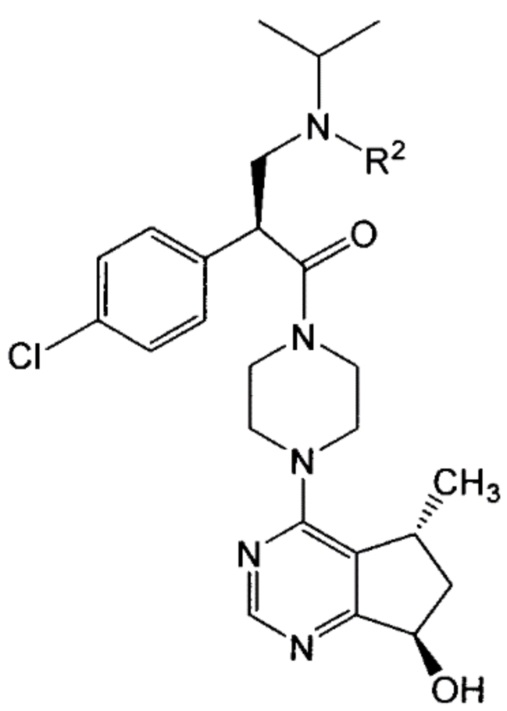




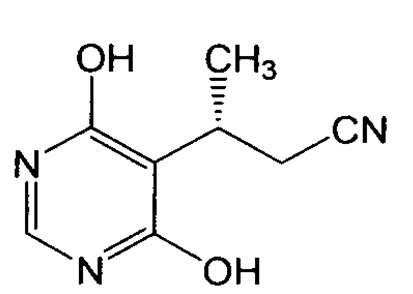
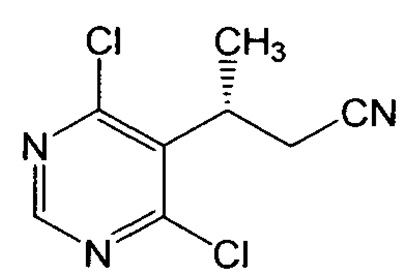
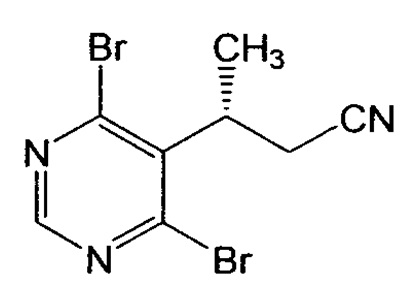

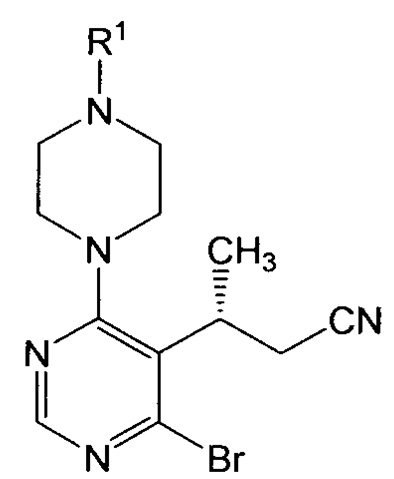
Документы, цитированные в отчёте о поиске
Способ получения гидроксилированных циклопентилпиримидиновых соединений
Способ получения аминокислотных соединений
Комментарии