Производные с азотсодержащим шестичленным ароматическим кольцом и содержащие их фармацевтические продукты - RU2470927C2
Код документа: RU2470927C2
Чертежи













Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится кновому соединению и его фармацевтически приемлемой соли, которые обладают способностью стимулировать аксональный рост в сочетании со способностью стимулировать ангиогенез и, таким образом, являются эффективными в уменьшении или лечении поражений центральной нервной системы, таких как поражение головного мозга и поражение спинного мозга, ишемический инсульт, ишемические заболевания сердца, такие как инфаркт миокарда и стенокардия, обусловленная органическим поражением, периферические окклюзионные поражения артерий, такие как критическая ишемия конечностей, или последствий данных заболеваний или других заболеваний, против которых соединения настоящего изобретения считаются эффективными.
Уровень техники
В опубликованной японской заявке на патент под номером 2005-239711 (патентный документ 1) изложено следующее:
"Достижения в исследовании регуляторных факторов ангиогенеза привели к терапевтическому применению данных факторов. Среди факторов, известных как стимулирующие ангиогенез, находятся фактор роста эндотелия сосудов (VEGF), основной фактор роста фибробластов (bFGF) и фактор роста гепатоцитов (HGF). Данные факторы роста и их гены применяются в настоящее время для лечения заболеваний, при которых необходимо улучшение в кровообращении (таких как облитерация при артериосклерозе и ишемические заболевания сердца).
Однако данные факторы роста являются белками и поэтому являются трудными для введения перорально. Они также приводят к другим проблемам, связанным с анафилактическими реакциями, вызванными повторным введением, защите вирусных векторов, применяемых в генной терапии, и побочным эффектам, таким как отек. Следовательно, существует необходимость в развитии новых способов лечения".
Известно, что некоторые заболевания вызываются органическим нарушением, связанным с ретракцией (втягиванием) аксона и потерей синапсов, хотя их этиология может варьироваться от заболевания к заболеванию. Такие заболевания включают болезнь Альцгеймера, мультиинфарктную деменцию, цереброваскулярную деменцию, старческое слабоумие, болезнь телец Леви, болезнь Паркинсона и болезнь Хантингтона.
Поражения центральной нервной системы, такие как кровоизлияние в мозг, ишемический инсульт, опухоль головного мозга, поражение головного мозга и поражение спинного мозга, могут также вызываться органическим нарушением, связанным с ретракцией аксона и потерей синапсов.
Были разработаны различные лекарственные средства для данных заболеваний, действие которых основано на защите нейронов при участии различных механизмов.
Ни одно из данных лекарственных средств не обеспечивает принципиального лечения данных заболеваний и является менее чем удовлетворительным, хотя они могут до некоторой степени задерживать развитие заболевания. В частности, не существует эффективного лечения ишемического инсульта, которое в настоящее время применяют по всему миру, за исключением тканевого активатора плазминогена (tPA, ТАП).
Хотя находящиеся в настоящее время в разработке несколько лекарственных средств сконструированы, чтобы оказывать защитное действие в отношении нейронов, ни одно из них не направлено на активное содействие восстановлению функционирования нервной системы после ишемического инсульта.
Большое внимание привлекает регенерация нейрональных стволовых клеток, и проводится много исследований в попытке имплантировать клетки. Что касается лечения ишемического инсульта, имплантированные нейрональные стволовые клетки, однако, не в состоянии работать в качестве нейронов для формирования нейрональных сетей, так как нейрональные стволовые клетки обладают небольшим шансом на выживание после имплантации или могут не дифференцироваться в нейроны.
Недавние исследования предполагают, что сосудистое ремоделирование, такое как ангиогенез, имеет существенное значение для образования и регенерации, в том числе для последующей дифференцировки и созревания нейрональных стволовых клеток и других клеток после ишемического инсульта (непатентный документ 1: J. Clin. Invest., 114, 2004). Таким образом, эффективное лечение ишемического инсульта требуется не только для обеспечения непосредственной защиты нейронов, чтобы предотвратить развитие нейронального повреждения, но также для стимулирования аксонального роста, необходимого для регенерации/ремоделирования сосудистых сетей, и воссоздания новых нейрональных сетей в пораженной «ишемической полутени» (пенумбре) (непатентный документ 2: Science, 3:272 (5262), pp.664-666 (1996)).
При таком положении дел низкомолекулярные соединения, которые могут стимулировать аксональный рост и стимулировать ангиогенез и которые могут быть введены перорально, считают потенциальными лекарственными средствами, эффективными в уменьшении или лечении поражения центральной нервной системы, такого как поражение головного мозга и поражение спинного мозга, ишемический инсульт, ишемические заболевания сердца, такие как инфаркт миокарда и стенокардия, обусловленная органическим поражением, периферические окклюзионные поражения артерий, такие как критическая ишемия конечностей, и последствий данных заболеваний, а также других заболеваний, против которых соединения настоящего изобретения считаются эффективными. Такое соединение также рассматривается как потенциальное лекарственное средство, эффективное в уменьшении или лечении симптомов, возникающих в результате функционального или органического нарушения в головном мозге, включая ишемические поражения головного мозга, такие как последствия ишемического инсульта, кровоизлияние в мозг и артериосклероз головного мозга, а также заболевания, связанные с органическим нарушением, возникающим вследствие старческого слабоумия, болезни Альцгеймера, болезни Паркинсона и последствий травмы головного мозга, повреждения спинного мозга и операции на головном мозге.
Полагают, что описанное выше соединение, которое стимулирует ангиогенез, может быть эффективным при патологических изменениях проходимости сосудов, обнаруживаемых при периферических окклюзионных поражениях артерий, таких как облитерация при артериосклерозе, болезнь Бюргера и болезнь Рейно. Полагают, что соединение является особенно эффективным в отношении критической ишемии конечностей и других тяжелых симптомов, при которых традиционные лекарственные средства являются неэффективными.
Патентный документ 1: опубликованная японская заявка на патент под номером 2005-239711.
Непатентный документ 1: A. Taguchi, et al., J. Clin. Invest., 114:3, pp.330-338 (2004).
Непатентный документ 2: M. Barinaga, Science, 3:272 (5262), pp.664-666 (1996).
Описание изобретения
Проблемы, решаемые с помощью изобретения
В связи с этим целью настоящего изобретения является обеспечение терапевтического агента для лечения или уменьшения заболеваний, включая поражения центральной нервной системы, такие как поражение головного мозга и поражение спинного мозга, ишемический инсульт, ишемические заболевания сердца, такие как инфаркт миокарда и стенокардия, обусловленная органическим поражением, периферические окклюзионные поражения артерий, такие как критическая ишемия конечностей, и последствий данных заболеваний, где терапевтический агент является в высокой степени безопасным, обладает способностью стимулировать аксональный рост в сочетании со способностью стимулировать ангиогенез и является пригодным для составления в лекарственный препарат для перорального введения, такой как таблетки и порошки, лекарственный препарат для парентерального введения, такой как инъекционный препарат, и лекарственный препарат для наружного применения, такой как мази и катаплазмы (припарки).
Средства для решения задач
Для достижения описанной выше цели настоящее изобретение обеспечивает соединение, представленное следующей формулой (I):
(Химическая формула 1)

где
группа Nx представляет собой 6-членное ароматическое кольцо, содержащее 1 или 2 атома азота;
R0, R1 и R2 каждый независимо представляет собой атом водорода, атом галогена, гидроксильную группу, неразветвленную или разветвленную алкокси-группу с 1-5 атомами углерода, ацетильную группу, карбамоильную группу, карбоксильную группу, неразветвленную или разветвленную сложноэфирную группу с 1-5 атомами углерода, незамещенную или замещенную галогеном неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода или группу -NR3R4, где R3 и R4 каждый независимо представляет собой атом водорода, атом кислорода, незамещенную или замещенную галогеном неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода или неразветвленную или разветвленную алкилоксикарбонильную группу с 2-10 атомами углерода;
R5 и R6 каждый независимо представляет собой атом водорода или незамещенную или замещенную галогеном неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода;
R7 представляет собой неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода;
E представляет собой атом кислорода или группу -NR8 (где R8 представляет собой атом водорода или неразветвленную или разветвленную алкильную группу с 1-5 атомами углерода);
n представляет собой целое число из 0-5;
X и Y каждый независимо представляет собой соединяющую связь; неразветвленную или разветвленную алкиленовую группу с 1-5 атомами углерода, либо незамещенную, либо замещенную 1-4 гидроксильными или алкокси-группами; циклоалкиленовую группу с 3-6 атомами углерода, либо незамещенную, либо замещенную 1-4 гидроксильными группами, атомами кислорода или алкильными группами; гетероциклоалкиленовую группу, либо незамещенную, либо замещенную 1-4 гидроксильными группами, атомами кислорода или алкильными группами; алкениленовую группу с 2-4 атомами углерода, либо незамещенную, либо замещенную 1-4 алкильными группами с 1-5 атомами углерода; -NHCO-; -CONH-; -CO- или -SO2- и
Q представляет собой атом водорода; фенильную группу, либо незамещенную, либо замещенную атомом галогена, гидроксильной группой, неразветвленной или разветвленной алкокси-группой с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой; тиофенильную группу, либо незамещенную, либо замещенную атомом галогена, гидроксильной группой, неразветвленной или разветвленной алкокси-группой с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой; феноксигруппу, либо незамещенную, либо замещенную атомом галогена, гидроксильной группой, неразветвленной или разветвленной алкокси-группой с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой; бензоильную группу, либо незамещенную, либо замещенную атомом галогена, гидроксильной группой, неразветвленной или разветвленной алкокси-группой с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой; пиридильную группу, либо незамещенную, либо замещенную атомом галогена, гидроксильной группой, неразветвленной или разветвленной алкокси-группой с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой; хинолильную группу, либо незамещенную, либо замещенную атомом галогена, гидроксильную группу, неразветвленную или разветвленную алкокси-группу с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой; изохинолильную группу, либо незамещенную, либо замещенную атомом галогена, гидроксильной группой, неразветвленной или разветвленной алкокси-группой с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой; или бензимидазолильную группу, либо незамещенную, либо замещенную атомом галогена, гидроксильной группой, неразветвленной или разветвленной алкокси-группой с 1-5 атомами углерода, незамещенной или замещенной галогеном неразветвленной или разветвленной алкильной группой с 1-5 атомами углерода, нитрильной группой, аминогруппой, карбоксильной группой, карбамоильной группой, ацетильной группой, метилсульфонильной группой или фенильной группой, при условии, что, когда R7 представляет собой циклопропильную группу и E представляет собой атом кислорода, группа Nx не является 3-пиридинильной группой;
и его фармацевтически приемлемую соль.
Эффекты изобретения
Обеспечиваемые настоящим изобретением соединения, являющиеся производными с азотсодержащим 6-членным ароматическим кольцом, представленные формулой (I), являются новыми соединениями, которые обладают способностью стимулировать аксональный рост в сочетании со способностью стимулировать ангиогенез. Доказано, что соединения являются высокоэффективными и безопасными в различных фармакологических тестах и поэтому могут применяться в качестве фармацевтического продукта. Они также применимы для составления в фармацевтические препараты.
Лучший вариант осуществления изобретения
Настоящее изобретение далее описываться здесь детально.
В соединениях по настоящему изобретению, представленных формулой (I), азотсодержащее 6-членное ароматическое кольцо, содержащее один атом азота, служащее в качестве группы Nx, может представлять собой пиридинильную группу. Азотсодержащее 6-членное ароматическое кольцо, содержащее два атома азота, служащее в качестве группы Nx, может представлять собой пиримидинильную группу, пиразинильную группу или пиридазинильную группу.
Конкретные примеры таких азотсодержащих 6-членных ароматических колец включают 6-членные ароматические кольца, которые могут быть либо замещенными, либо незамещенными группами R0, R1, R2 и R10, включая следующие заместители:
(Химическая формула 2)
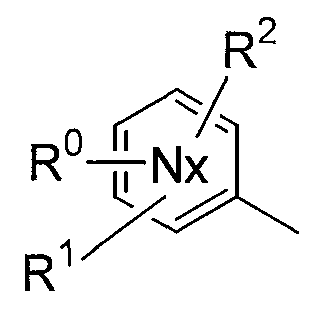
представляет следующее:
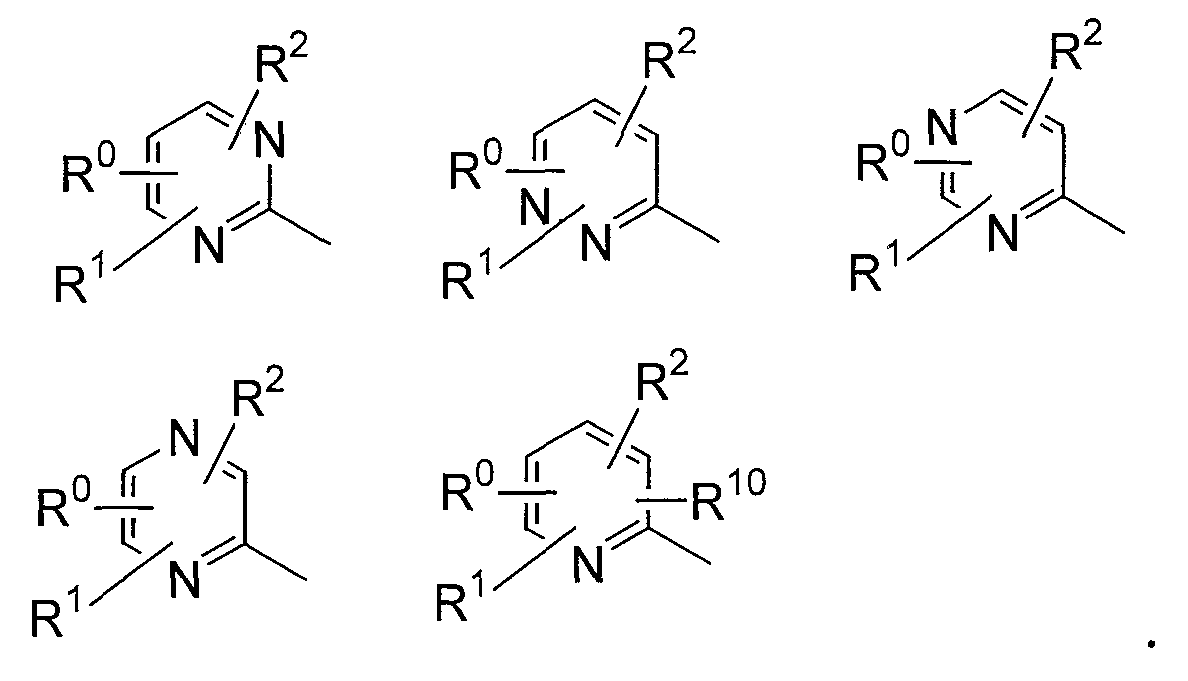
В данной формуле R0, R1, R2 и R10 каждый независимо представляет собой атом водорода, атом галогена, гидроксильную группу, алкокси-группу, ацетильную группу, карбамоильную группу, карбоксильную группу, сложноэфирную группу, незамещенную или замещенную галогеном неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода или -NR3R4 группу (где R3 и R4 каждый независимо представляет собой атом водорода, атом кислорода, или незамещенную, или замещенную галогеном неразветвленную, разветвленную или циклическую алкильную или алкилоксикарбонильную группу с 1-5 атомами углерода).
Что касается определения заместителей R0-R4 и R10 выше, "атом галогена" включает атом фтора, атом хлора и атом брома. "Алкокси-группа" включает неразветвленную или разветвленную алкокси-группу с 1-5 атомами углерода, такую как метокси-группа и этокси-группа. "Алкильная группа" включает неразветвленную или разветвленную алкильную группу с 1-5 атомами углерода, такую как метильная группа, этильная группа, пропильная группа и трифторметильная группа, которая может быть либо незамещенной, либо замещенной 1-3 атомами галогена, такими как атом фтора, атом хлора и атом брома.
"Сложноэфирная группа" включает неразветвленную или разветвленную сложноэфирную группу с 1-5 атомами углерода, такую как сложный метиловый эфир, сложный этиловый эфир и сложный пропиловый эфир.
"Алкилоксикарбонильная группа" включает неразветвленную или разветвленную алкилоксикарбонильную группу с 2-10 атомами углерода, такую как метилоксикарбонильную группу, этилоксикарбонильную группу, трет-бутилоксикарбонильную группу и бензилоксикарбонильную группу.
R5 и R6 каждый независимо представляет собой атом водорода или незамещенную или замещенную галогеном неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода. R7 представляет собой алкильную группу.
Что касается определения заместителей R5 и R6 выше, "алкильная группа" включает неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода, такую как метильная группа, этильная группа, пропильная группа, циклопропильная группа и трифторметильная группа, которая может быть либо незамещенной, либо замещенной 1-3 атомами галогена, такими как атом фтора, атом хлора и атом брома.
Что касается определения заместителя R7 выше, "алкильная группа" включает неразветвленную, разветвленную или циклическую алкильную группу с 1-5 атомами углерода, такую как метильная группа, этильная группа, пропильная группа и циклопропильная группа.
E представляет атом кислорода или группу -NR8 (где R8 представляет собой атом водорода или алкильную группу).
Что касается группы -NR8, "алкильная группа", представленная R8, представляет собой неразветвленную или разветвленную алкильную группу с 1-5 атомами углерода, такую как метильная группа, этильная группа, пропильная группа и трифторметильная группа. Алкильная группа может быть замещена 1-3 атомами галогена, такими как атом фтора, атом хлора и атом брома.
X и Y каждый независимо представляет собой соединяющую связь; алкиленовую группу, либо незамещенную, либо замещенную 1-4 гидроксильными или алкокси-группами; циклоалкиленовую группу, либо незамещенную, либо замещенную 1-4 гидроксильными группами, атомами кислорода или алкильными группами; гетероциклоалкиленовую группу, либо незамещенную, либо замещенную 1-4 гидроксильными группами, атомами кислорода или алкильными группами; алкениленовую группу, либо незамещенную, либо замещенную 1 или 2 алкильными группами с 1-5 атомами углерода; -NHCO-; -CONH-; -CO- или -SO2-.
В отношении определения X и Y выше алкиленовая группа включает неразветвленную или разветвленную алкиленовую группу с 1-5 атомами углерода, такую как метиленовая группа, метилметиленовая группа, этиленовая группа, триметиленовая группа и тетраметиленовая группа.
Циклоалкиленовая группа включает циклоалкиленовую группу с 3-6 атомами углерода, такую как 1,1-циклопропиленовая группа 1,2-циклопропиленовая группа, 1,1-циклобутиленовая группа, 1,2-циклобутиленовая группа, 1,1-циклопентиленовая группа, 1,2-циклопентиленовая группа, 1,1-циклогексиленовая группа, 1,2-циклогексиленовая группа, 2-гидрокси-1,1-циклопентиленовая группа и 3-гидрокси-1,2-циклопентиленовая группа.
Гетероциклоалкиленовая группа включает гетероциклоалкиленовую группу, которая имеет 3-6 атомов углерода и может содержать один или несколько атомов кислорода или азота, такую как тетрагидро-2H-пиранильная группа, пиперидинильная группа, пиперазинильная группа, морфолинильная группа, 2-оксопирролидинильная группа и азетидинильная группа.
Алкениленовая группа включает алкениленовую группу с 2-4 атомами углерода, такую как виниленовая группа и бутадиеновая группа. Алкильная группа с 1-5 атомами углерода, чтобы служить в качестве заместителя алкениленовой группы, включает неразветвленную или разветвленную алкильную группу, такую как метильная группа, этильная группа, пропильная группа и изопропильная группа.
Используемое здесь "соединяющая связь" означает непосредственную связь. В частности, если "X" и "Y" каждый представляет собой соединяющую связь, два соседних заместителя при "X" и "Y" являются непосредственно связанными друг с другом: ни "X", ни "Y" не существует как группа.
Заместитель "Q" является таким, как определено выше. Предпочтительно заместители фенильной группы, тиофенильной группы, фенокси-группы, бензоильной группы, пиридильной группы, хинолильной группы, изохинолильной группы или бензимидазолильной группы включают атом галогена, такой как атом фтора, атом хлора и атом брома; гидроксильную группу; неразветвленную или разветвленную алкокси-группу с 1-5 атомами углерода, такую как метокси-группа и этокси-группа; неразветвленную или разветвленную алкильную группу с 1-5 атомами углерода, замещенную 1-3 атомами галогена; нитрильную группу; аминогруппу; карбоксильную группу; карбамоильную группу; ацетильную группу и метилсульфонильную группу.
Атом галогена в замещенной галогеном неразветвленной или разветвленной алкильной группе с 1-5 атомами углерода включает атом фтора, атом хлора и атом брома.
Хотя соединения по настоящему изобретению являются новыми соединениями, соединения, имеющие до некоторой степени сходный скелет, описаны, например, в опубликованном японском переводе международной заявки под номером 2003-507456, WO 01/79170 и WO 00/23076. В отличие от соединений, описанных в опубликованном японском переводе международной заявки под номером 2003-507456, которые действуют как модуляторы активности хемокинового рецептора, соединения, описанные в примерах 65, 85, 91, 102, 109, 130 и 135 настоящего изобретения обнаруживают аффинитет к хемокиновому рецептору CCR3 0,0, 2,4, 18, 0,0, 16, 3,5 и 22% при концентрации 10 мкМ соответственно, не демонстрируя аффинитет к хемокиновому рецептору CCR3. Это предполагает, что соединения по настоящему изобретению не вызывают возможных побочных эффектов, которые могут быть связаны с хемокиновым рецептором.
Соединения по настоящему изобретению, представленные общей формулой (I), могут существовать в виде изомеров (таких как таутомеры, энантиомеры, геометрические изомеры или диастереомеры). Поэтому настоящее изобретение охватывает любые такие изомеры и смеси, содержащие данные изомеры в любом соотношении.
Соединения по настоящему изобретению, представленные общей формулой (I), могут быть получены с применением известного метода или любой подходящей комбинации известных методов.
В частности, соединения могут быть получены путем следующих реакционных процессов (a), (b), (c) или (d).
(a) Соединения могут быть получены путем взаимодействия соединения следующей общей формулы (IV-b):
(Химическая формула 3)

с соединением следующей общей формулы (V)
(Химическая формула 4)

или его солью.
(b) Или соединения могут быть получены путем взаимодействия соединения следующей общей формулы (II-b)
(Химическая формула 5)

с соединением следующей общей формулы (VI)
(Химическая формула 6)

или его солью.
(с) Или соединения могут быть получены путем взаимодействия соединения следующей общей формулы (VII)
(Химическая формула 7)

с соединением следующей общей формулы (VIII)
(Химическая формула 8)
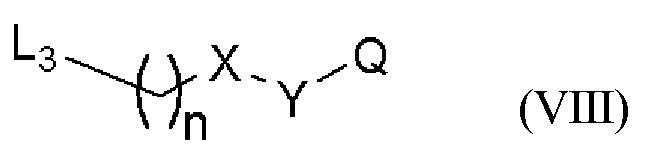
или его солью.
(d) Или соединения могут быть получены путем взаимодействия соединения следующей общей формулы (II-a)
(Химическая формула 9)

с соединением следующей общей формулы (IX)
(Химическая формула 10)

Каждое из соединений формул (IIa)-(IX), представленное выше, является коммерчески доступным или может быть легко получено известным методом.
Специалистам в данной области техники следует понимать, что функциональные группы, такие как гидроксильные группы и аминогруппы, исходных реагентов или промежуточных соединений процесса по настоящему изобретению могут быть защищены защитными группами, и получение соединений формулы (I) охватывает присоединение одной или нескольких таких защитных групп в соответствующий момент и удаление защитной группы в подходящий момент любого последующего процесса.
Методы введения защиты и удаления защиты функциональных групп описаны, например, в «Protective Groups in Organic Chemistry», J. W. F. McOmie ed., Plenum Press (1973), «Protective Groups in Organic Synthesis», 2nd Edition, T. W. Greene и P. G. M. Wuts, Wiley-Interscience (1991), and «Greene's Protective Groups in Organic Synthesis», 4th. Edition, T. W. Greene и P. G. M. Wuts, Wiley-Interscience (2006).
Защитной группой для функциональной гидроксильной группы может быть любая защитная группа, обычно используемая для защиты гидроксильной группы. Примеры защитных групп включают алкоксикарбонильную группу, такую как бензилоксикарбонил, 4-нитробензилоксикарбонил, 4-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил, 1,1-диметилпропоксикарбонил, изопропоксикарбонил, изобутилоксикарбонил, дифенилметоксикарбонил, 2,2,2-трихлорэтоксикарбонил, 2-(триметилсилил)этоксикарбонил, винилоксикарбонил и аллилоксикарбонил; ацильную группу, такую как ацетил, формил, хлорацетил, дихлорацетил, трихлорацетил, трифторацетил, метоксиацетил, феноксиацетил, пивалоил и бензоил; низшую алкильную группу, такую как метил, трет-бутил, 2,2,2-трихлорэтил и 2-триметилсилилэтил; группу арил(низший алкил), такую как бензил, 4-метоксибензил, 3,4-диметоксибензил и тритил; низшую алкенильную группу, такую как аллил; низшую алкинильную группу, такую как пропаргил; содержащую азот или серу гетероциклическую кольцевую группу, такую как тетрагидрофурил, тетрагидропиранил и тетратиопиранил; низшую алкокси или алкилтиоалкильную группу, такую как метоксиметил, метилтиометил, бензилоксиметил, 2-метоксиэтоксиметил, 1-этоксиэтил и 1-(метил)метоксиэтил; низший алкил или арилсульфонильную группу, такую как метансульфонил и п-толуолсульфонил; и замещенную силильную группу, такую как триметилсилил, триэтилсилил, триизопропилсилил, трет-бутилдиметилсилил и трет-бутилдифенилсилил.
Защитная группа для аминогруппы может быть защитной группой, обычно используемой для защиты аминогруппы. Примеры таких групп включают алкоксикарбонильную группу, такую как бензилоксикарбонил, 4-нитробензилоксикарбонил, 4-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, метоксикарбонил, трет-бутоксикарбонил, 1,1-диметилпропоксикарбонил, 2,2,2-трихлорэтоксикарбонил, 2-(триметилсилил)этоксикарбонил, 9-флуоренилметилоксикарбонил, винилоксикарбонил и аллилоксикарбонил; ацильную группу, такую как ацетил, формил, хлорацетил, дихлорацетил, трихлорацетил, трифторацетил, фенилацетил, фталоил, сукцинил, аланил, лейцил и бензоил; группу арил(низший алкил), такую как бензил, 4-метоксибензил, 3,4-диметоксибензил, дифенилметил и тритил; арилтиогруппу, такую как 2-нитрофенилтио и 2,4-динитрофенилтио; низший алкил или арилсульфонильную группу, такую как метансульфонил и п-толуолсульфонил; группу ди(низший алкиламино)низший алкилиден, такую как N,N-диметиламинометилен; группу арил(низший алкилиден), такую как бензилиден, 2-гидроксибензилиден и 2-гидрокси-5-хлорбензилиден; содержащую азот гетероциклическую алкилиденовую группу, такую как 3-гидрокси-4-пиридилметилен; циклоалкилиденовую группу, такую как циклогексилиден, 2-этоксикарбонилциклогексилиден и 2-этоксикарбонилциклопентилиден; фосфорильную группу, такую как дифенилфосфорил; и замещенную силильную группу, такую как триметилсилил.
Соединения и промежуточные соединения по настоящему изобретению могут быть выделены из реакционной смеси с применением стандартных методов и, если необходимо, могут быть дополнительно очищены.
Каждый реакционный процесс далее будет описан детально.
[Реакционный процесс (a)]
Данный реакционный процесс подробно представлен на следующей реакционной схеме:
(Химическая формула 11)

В реакционной схеме выше, R0-R7, E, Nx, n, X, Y и Q являются такими, как описано выше.
Заместитель R9 представляет собой алкильную группу, такую как метильная группа, этильная группа, трет-бутокси-группу и бензильную группу. Заместители L1 и L2 каждый представляет собой уходящую группу, которая может быть легко заменена на аминогруппу или гидроксильную группу. Их конкретные примеры включают атом галогена, такой как атом хлора, атом брома и атом йода; группу алкилсульфонилокси, такую как группа метансульфонилокси и группа трифторметансульфонилокси; и группу арилсульфонилокси, такую как группа п-толуолсульфонилокси и группа 3-нитробензолсульфонилокси.
Настоящий процесс, в общем, предполагает взаимодействие соединения (II-a) со сложноэфирным производным (III-a) с получением соединения (IV-a), которое в свою очередь гидролизуют с получением карбоксильного соединения (IV-b). Затем полученное соединение (IV-b) подвергают реакции конденсации с аминопроизводным (V) с получением желаемого соединения (I), которое составляет один аспект настоящего изобретения.
Альтернативно, соединение (IV-a) может быть получено путем превращения соединения (II-a) в соединение (II-b), с последующим его взаимодействием со сложноэфирным производным (III-b).
В тех случаях, когда необходимо защитить функциональные группы, такие как гидроксильная группа и аминогруппа, в данном реакционном процессе, процесс может включать процедуру введения одной или нескольких таких защитных групп на соответствующей стадии и процедуру удаления защитной группы на последующей стадии.
Например, когда R0 в соединении (I) представляет собой -NR3R4, с R3 или R4 являющимся метилоксикарбонильной группой, этилоксикарбонильной группой, трет-бутилоксикарбонильной группой, бензилоксикарбонильной группой, бензильной группой или каждый атомом кислорода, чтобы служить в качестве защитной группы атома азота, защитная группа может быть удалена или превращена в другие функциональные группы с получением желаемого соединения (I), у которого R3 и/или R4 превращен в атом водорода. Данный реакционный процесс может быть предусмотрен в настоящих реакционных процессах.
Соответствующие процессы описываются более подробно ниже.
Процесс 1:
Соединения формулы (II-a) и сложноэфирные производные формулы (III-a), которые служат в качестве исходных веществ настоящего процесса, являются коммерчески доступными, или они могут быть получены с помощью известных методов.
Соединение (II-a) может быть получено в соответствии с методом или комбинацией методов или применяя методы, описанные, например, в «Heterocyclic соединение New Edition, Introduction», Yamanaka Hiroshi, Sakamoto Norio et al., Kodansha Scientific (2004) и «Heterocyclic compound New Edition, Application», Yamanaka Hiroshi, Sakamoto Norio et al., Kodansha Scientific (2004).
Примеры сложноэфирного производного (III-а) включают этилбромацетат, этил-2-бромпропионат и этил-2-бром-2-метилпропионат.
В настоящем процессе, в качестве первой его стадии, соединение (II-a) вводят во взаимодействие со сложноэфирным производным (III-а) с получением соединения (IV-a).
Реакция может проводиться путем смешивания соединения (II-a) с 1,0-1,5 эквивалентами сложноэфирного производного (III-а) при -20°C - 150°C, и предпочтительно при 0°C - 100°C, в инертном растворителе, таком как бензол, толуол, тетрагидрофуран, диоксан, диметилформальдегид, диметилсульфоксид, ацетонитрил, ацетон, метанол, этанол, изопропиловый спирт, трет-бутиловый спирт, диэтиловый эфир, этиленгликоль, метиленхлорид или хлороформ, и если необходимо, в присутствии органического основания, такого как триэтиламин, диизопропилэтиламин или пиридин, или неорганического основания, такого как натрий, гидрид натрия, калий, гидрид калия, этилат натрия, трет-бутилат калия, карбонат натрия, карбонат калия, карбонат цезия, фторид цезия, бикарбонат натрия или бикарбонат калия.
Если необходимо, в реакции может применяться комбинация органических оснований или неорганических оснований. Альтернативно, может быть добавлен йодид натрия, йодид калия, йодид тетрабутиламмония или краунэфир.
Процессы 2 и 3
Данные процессы используются в качестве альтернативного процесса для синтеза соединения (IV-a). В частности, соединение (II-b) сначала превращают из соединения (II-a), и полученный продукт вводят во взаимодействие со сложноэфирным производным (III-b) с получением соединения (IV-a).
Превращение соединения (II-a) (процесс 2) может проводиться различными методами в зависимости от типа заместителя L2 соединения (II-b). Например, когда L2 представляет собой атом галогена, такой как атом хлора и атом брома, соединение (II-a) вводят во взаимодействие в присутствии как хлорокиси фосфора (POCl3), так и пятихлористого фосфора (PCl5), или в присутствии одной хлорокиси фосфора, пятихлористого фосфора, бромокиси фосфора (POBr3), пятибромистого фосфора (PBr5) и тому подобного. Если необходимо, реакция может проводиться в инертном растворителе, таком как бензол, толуол, этилацетат, диоксан, хлороформ или метиленхлорид.
Когда уходящая группа L2 представляет собой группу алкилсульфонилокси, такую как группа метансульфонилокси или группа трифторметансульфонилокси; или группу арилсульфонилокси, такую как группа п-толуолсульфонилокси или группа 3-нитробензолсульфонилокси, реакцию проводят путем смешивания соединения (II-а) с 1,0-1,5 экв. метансульфонилхлорида (MsCl), п-толуолсульфонилхлорида (TsCl) или ангидрида трифторметансульфоновой кислоты (Tf20) при -20°C-150°C, и предпочтительно при 0°C-100°C, в инертном растворителе, таком как толуол, этилацетат, тетрагидрофуран, диоксан, хлороформ или ацетонитрил, и если необходимо, в присутствии органического основания, такого как триэтиламин, диизопропилэтиламин или пиридин, или неорганического основания, такого как натрий, гидрид натрия, калий, гидрид калия, этилат натрия, трет-бутилат калия, карбонат натрия, карбонат калия, карбонат цезия, фторид цезия, бикарбонат натрия или бикарбонат калия.
Поскольку полученное соединение может непосредственно использоваться в последующем процессе, оно может быть очищено, по желанию, с помощью известного метода очистки, такого как перекристаллизация и колоночная хроматография, до последующего процесса.
Соединение (II-b), полученное, как описано выше, затем вводят во взаимодействие со сложноэфирным производным (III-b), чтобы получить соединение (IV-a) (процесс 3).
Реакция может проводиться путем смешивания соединения (II-b) с 1,0-1,5 экв. сложноэфирного производного (III-b) при -20°C-150°C, и предпочтительно при 0°C-100°C, в инертном растворителе, таком как бензол, толуол, тетрагидрофуран, диоксан, диметилформальдегид, диметилсульфоксид, ацетонитрил, ацетон, метанол, этанол, изопропиловый спирт, трет-бутиловый спирт, диэтиловый эфир, этиленгликоль, метиленхлорид или хлороформ, и если необходимо, в присутствии органического основания, такого как триэтиламин, диизопропилэтиламин или пиридин, или неорганического основания, такого как натрий, гидрид натрия, калий, гидрид калия, этилат натрия, трет-бутилат калия, карбонат натрия, карбонат калия, карбонат цезия, фторид цезия, бикарбонат натрия или бикарбонат калия.
Если необходимо, в реакцию может быть добавлен йодид натрия, йодид калия, йодид тетрабутиламмония или краунэфир.
Соединение (III-b), применяемое в реакции, может быть коммерческим продуктом или известным соединением или, альтернативно, может быть легко синтезировано известными методами.
Конкретные примеры таких соединений (III-b) включают гликолевую кислоту, метилгликолят, этилгликолят, трет-бутилгликолят, бензилгликолят, молочную кислоту, метиллактат, этиллактат, трет-бутиллактат, бензиллактат, 2-гидроксиизомасляную кислоту, метил-2-гидроксиизобутират, этил-2-гидроксиизобутират, трет-бутил-2-гидроксиизобутират, глицин, метиловый эфир глицина, этиловый эфир глицина, трет-бутиловый эфир глицина, бензиловый эфир глицина, саркозин, метиловый эфир саркозина, этиловый эфир саркозина, метиловый эфир саркозина, аланин, метиловый эфир аланина, этиловый эфир аланина, трет-бутиловый эфир аланина, бензиловый эфир аланина, N-метилаланин, 2-аминоизомасляная кислота, метил-2-аминоизобутират, этил-2-аминоизобутират, трет-бутил-2-аминоизобутират, бензил-2-аминоизобутират и 2-(метиламино)изомасляная кислота.
Процессы 4 и 5
Соединение (IV-a), полученное, как описано выше, гидролизуют известными методами (процесс 4) для превращения в карбоновую кислоту (IV-b), которую, в свою очередь, подвергают конденсации с аминопроизводным (V) для получения амида (I).
Соединение (V), которое может быть использовано в реакции конденсации с соединением (IV-b), может быть известным соединением или, альтернативно, может быть легко синтезировано известными методами.
Условия реакции амидирования могут быть основаны на методах, описанных в «Compendium for Organic Synthesis» (Wiley-5 Interscience: Division of John Wiley & Sons).
Например, производное карбоновой кислоты (IV-b) обрабатывают диэтилфосфорцианидом (DEPC), дифенилфосфоразидом (DPPA), дициклогексилкарбодиимидом (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлоридом, 2-йод-1-метилпиридинием, ангидридом пропанфосфоновой кислоты, 2-хлор-l,3-диметилимидазолинийхлоридом, 2-хлор-1,3-диметилимидазолинийгексафторфосфатом или (бензотриазол-1-илокси)трис(диметиламино)фосфонийгексафторфосфатом (реагент BOP). Когда необходимо, реакция может проводиться в присутствии органического или неорганического основания. Аминопроизводное (V) добавляется либо после, либо до реакции для получения амида (I). Альтернативно, производное карбоновой кислоты (IV-b) может быть превращено в активированное сложноэфирное соединение, такое как галогенид кислоты, симметричный ангидрид кислоты или смешанный ангидрид кислоты, которое, в свою очередь, вводится во взаимодействие с аминопроизводным (V) для получения амидного производного (I).
Когда R0 в конечном амидном производном (I) представляет собой -NR3R4 и R3 или R4 является метилоксикарбонильной группой, этилоксикарбонильной группой, трет-бутилоксикарбонильной группой, бензилоксикарбонильной группой, бензильной группой или каждый атомом кислорода, чтобы служить в качестве защитной группы атома азота, защитная группа может быть удалена или превращена в другие функциональные группы, чтобы получить желаемое соединение (I), у которого R3 и/или R4 превращены в атом водорода. Это может быть альтернативным реакционным процессом.
Реакция может проводиться различными методами в зависимости от типа защитных групп у атома азота соединения (I). Например, соединение (I), в котором R0 представляет собой группу -NR3R4 с R3 или R4, представляющем собой бензильную группу, 4-метоксибензильную группу или бензилоксикарбонильную группу, или R3 и R4, каждым представляющим собой атом кислорода, вместе образующие нитрогруппу, может гидрироваться в присутствии катализатора, такого как палладий на угле, гидроксид палладия на угле, платина и оксид платины, в инертном растворителе, таком как метанол, этанол, изопропиловый спирт, толуол, этилацетат, тетрагидрофуран, диоксан, хлороформ или уксусная кислота. Альтернативно, соединение (I) может быть восстановлено в кислых условиях с применением цинка или хлорида олова.
Соединение (I), в котором защитная группа R3 или R4 представляет собой трет-бутоксикарбонильную группу, этоксикарбонильную группу, 4-метоксибензильную группу, 3,4-диметоксибензильную группу, ацетильную группу или формильную группу, может подвергаться удалению защиты путем обработки кислотой, такой как трифторуксусная кислота, хлороводородная кислота, бромоводородная кислота или серная кислота, в инертном растворителе, таком как метанол, этанол, изопропиловый спирт, толуол, этилацетат, тетрагидрофуран, диоксан, хлороформ или ацетонитрил.
Если необходимо, полученное в результате соединение (I) может очищаться известным методом очистки, таким как перекристаллизация или колоночная хроматография.
[Реакционный процесс (b)]
Данный процесс подробно представлен на следующей химической реакционной схеме:
(Химическая формула 12)

В реакционной схеме выше, R0-R2, R5-R7, E, Nx, n, X, Y, L2 и Q являются такими, как описано выше. Заместитель R9 представляет собой атом водорода и P является защитной группой.
Примеры защитной группы включают бензильную группу, п-метоксибензильную группу, трет-бутоксикарбонильную группу, этоксикарбонильную группу, бензилоксикарбонильную группу и п-метоксибензилоксикарбонильную группу.
В частности, соединение (V) вводят во взаимодействие со сложноэфирным производным (III-b), чтобы получить соединение (VI).
В альтернативном процессе, для получения соединения (VI) соединение (V) вводят во взаимодействие со сложноэфирным производным (III-c) для получения соединения (X), которое, в свою очередь, превращают в соединение (VI). Затем полученное в результате соединение (VI) вводят во взаимодействие с соединением (II-b) для получения желаемого соединения (I), которое составляет один аспект настоящего изобретения.
Когда R0 в полученном в результате амидном производном (I) представляет собой -NR3R4 с R3 или R4, представляющим собой метилоксикарбонильную группу, этилоксикарбонильную группу, трет-бутилоксикарбонильную группу, бензилоксикарбонильную группу или бензильную группу или каждый является атомом кислорода, чтобы служить в качестве защитной группы атома азота, защитная группа может быть удалена или превращена в другие функциональные группы с получением желаемого соединения (I), у которого R3 и/или R4 превращен в атом водорода. Это может являться альтернативным реакционным процессом.
Соответствующие процессы описываются более подробно ниже.
Процессы 6 и 7:
Соединение (III-b) или (III-c), которые служат исходным веществом настоящего процесса, являются коммерчески доступными веществами, или они могут быть получены с применением известных методов.
В данном процессе, в качестве его первой стадии, карбоновую кислоту (III-b) или (III-c) подвергают амидной конденсации с аминопроизводным (V) для получения амида (VI) или (X) соответственно.
Условия для реакции амидирования могут базироваться на методах, описанных в «Compendium for Organic Synthesis» (Wiley-Interscience: Division of John Wiley & Sons).
Например, производное карбоновой кислоты (III-b) или (III-c) обрабатывают диэтилфосфорилцианидом (DEPC), дифенилфосфорилазидом (DPPA), дициклогексилкарбодиимидом (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлоридом, 2-йод-1-метилпиридинием, ангидридом пропанфосфоновой кислоты, 2-хлор-1,3-диметилимидазолинийхлоридом, 2-хлор-1,3-диметилимидазолинийгексафторфосфатом или (бензотриазол-1-илокси)трис(диметиламино)фосфонийгексафторфосфатом (реагент BOP). Когда необходимо, реакция может проводиться в присутствии органического или неорганического основания. Аминопроизводное (III) добавляется либо после, либо до реакции для получения амида (VI) или (X).
Альтернативно, производное карбоновой кислоты (III-b) или (III-c) может быть превращено в активированное сложноэфирное соединение, такое как галогенид кислоты, симметричный ангидрид кислоты или смешанный ангидрид кислоты, которое в свою очередь вводится во взаимодействие с аминопроизводным (V) для получения амида (VI) или (X).
Процесс 8:
Когда полученный продукт является соединением (X), он подвергается удалению защитной группы и превращается в соединение (VI).
Удаление защиты может осуществляться различными методами в зависимости от типа защитной группы P соединения (X).
Например, когда P представляет собой бензильную группу, 4-метоксибензильную группу или бензилоксикарбонильную группу, соединение (X) гидрируют в присутствии катализатора, такого как палладий на угле, гидроксид палладия на угле, платина или оксид платины, в инертном растворителе, таком как метанол, этанол, изопропиловый спирт, толуол, этилацетат, тетрагидрофуран, диоксан, хлороформ или уксусная кислота. Когда защитная группа P представляет собой трет-бутоксикарбонильную группу, этоксикарбонильную группу, 4-метоксибензильную группу или 3,4-диметоксибензильную группу, у соединения (X) удаляют защитную группу обработкой кислотой, такой как трифторуксусная кислота, хлороводородная кислота, бромоводородная кислота или серная кислота, в инертном растворителе, таком как метанол, этанол, изопропиловый спирт, толуол, этилацетат, тетрагидрофуран, диоксан, хлороформ или ацетонитрил.
Поскольку полученное в результате соединение может непосредственно использоваться в последующем процессе, оно может по желанию очищаться с помощью известного метода очистки до последующего процесса.
Процесс 9:
Соединение (VI), полученное, как описано выше, вводят во взаимодействие с соединением (II-b) для получения соединения, представленного формулой (I), которое является соединением по настоящему изобретению.
Соединение (II-b), используемое в настоящем процессе, является таким, как описано в процессе 2. В данной реакции соединение (I) может быть синтезировано так же, как и в процессе 3.
Если необходимо, защитная группа на атоме азота, полученного в результате амидного производного (I), может быть удалена или превращена в функциональные группы, так что амидное производное (I) может быть превращено в соединение, представленное формулой (I), которое является соединением по настоящему изобретению.
В данной реакции соединение (I) может быть синтезировано так же, как и в реакционном процессе (a).
Если необходимо, соединения, полученные в описанных выше реакциях, могут очищаться с помощью известного метода очистки, такого как перекристаллизация и колоночная хроматография.
[Реакционный процесс (c)]
Данный процесс подробно представлен на следующей химической реакционной схеме:
(Химическая формула 13)
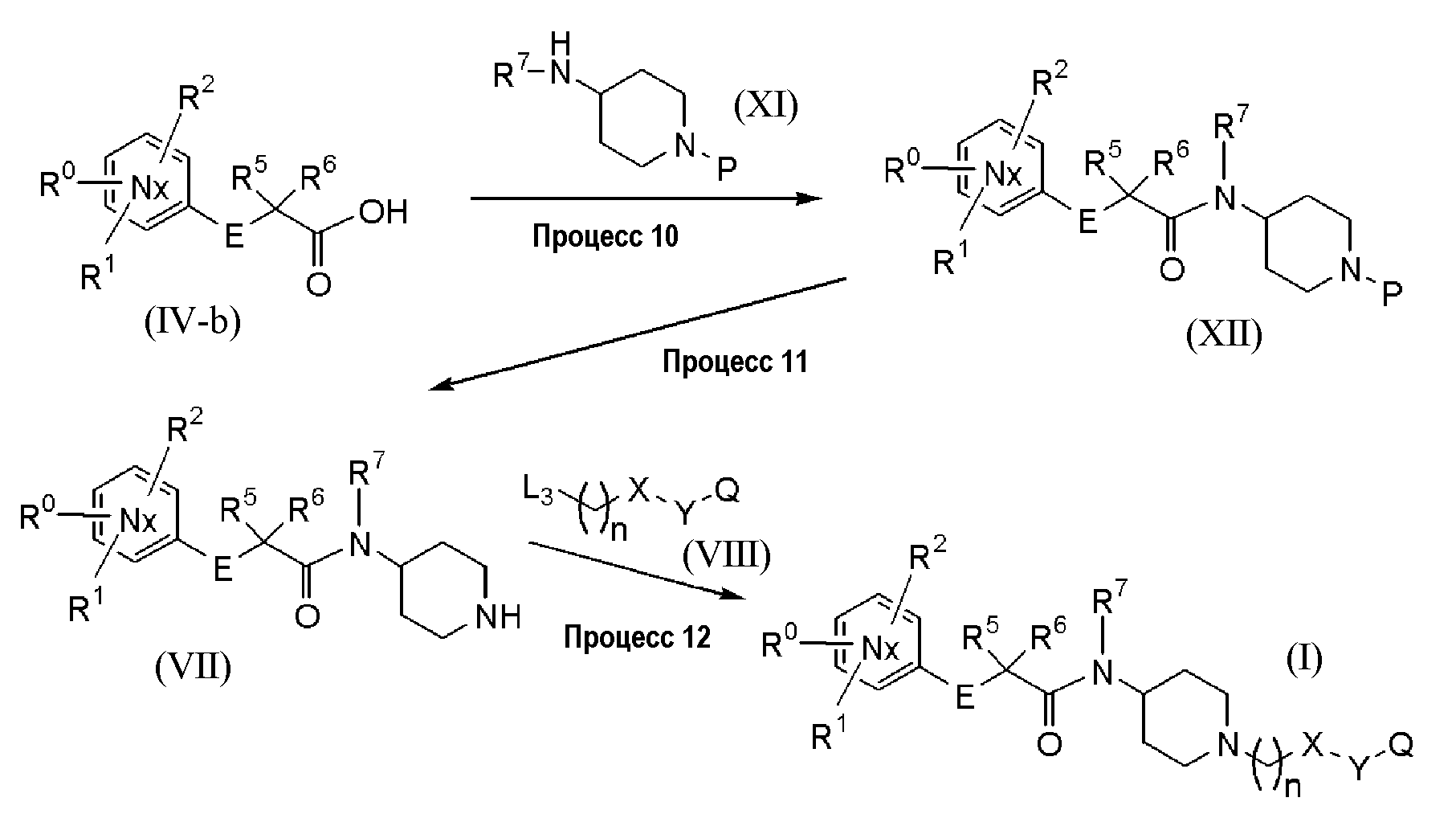
В реакционной схеме выше R0-R2, R5-R7, E, Nx, n, X, Y и Q являются такими, как описано выше, и P представляет собой защитную группу.
Примеры защитной группы включают бензильную группу, п-метоксибензильную группу, трет-бутоксикарбонильную группу, этоксикарбонильную группу, бензилоксикарбонильную группу и п-метоксибензилоксикарбонильную группу.
Заместитель L3 является уходящей группой, которая может быть легко заменена на аминогруппу. Конкретные примеры групп включают атом галогена, такой как атом хлора, атом брома и атом йода; группу алкилсульфонилокси, такую как группа метансульфонилокси и группа трифторметансульфонилокси; и группу арилсульфонилокси, такую как группа п-толуолсульфонилокси и группа 3-нитробензолсульфонилокси.
Настоящий процесс, в частности, охватывает конденсацию карбоксильного производного (IV-b), описанного в процессе 4 выше, с соединением (XI) для получения амидного соединения (XII). Затем у амидного соединения (XII) удаляют защиту для получения соединения (VII). Полученное в результате соединение (VII) затем вводят во взаимодействие с соединением (VIII) для получения желаемого соединения (I).
Данные процессы описаны более подробно далее.
Процесс 10:
Соединение (XI), которое служит исходным веществом настоящего процесса, может быть коммерчески доступным продуктом, может быть известно из литературы (J. Med. Chem., 36:3707(1993) [R.H. Mach et al.], EP 0184257-Al [R. A. Stokbroekx et al.]) или может быть получено известным методом.
Амидирование для получения соединения (XII) может осуществляться в тех же самых условиях, как в процессе 5.
Процесс 11:
Соединение (XII), полученное выше, подвергают удалению защитной группы для получения соединения (VII).
Данная реакция может проводиться таким же образом, как в процессе 8, для получения соединения (VII).
Процесс 12:
Соединение (VII), полученное в процессе 11 выше, вводится во взаимодействие с соединением (VIII) для получения соединения, представленного формулой (I), которое является соединением по настоящему изобретению.
В частности, в настоящем процессе соединение (VII) вводится во взаимодействие с 1,0-1,5 экв. соединения (VIII) при приблизительно -50°C - приблизительно 120°C, и предпочтительно при приблизительно -20°C - приблизительно 80°C, в инертном растворителе, таком как бензол, толуол, тетрагидрофуран, диоксан, диметилформамид, диметилсульфоксид, ацетонитрил, ацетон, диэтиловый эфир, метиленхлорид, хлороформ или тетрахлорид углерода, в присутствии органического основания, такого как триэтиламин, диизопропилэтиламин или пиридин, или неорганического основания, такого как натрий, гидрид натрия, калий, гидрид калия, этилат натрия, трет-бутилат натрия, карбонат натрия, карбонат калия, карбонат цезия, фторид цезия, бикарбонат натрия или бикарбонат калия.
Если необходимо, к реакции может быть добавлен йодид натрия, йодид калия, йодид тетрабутиламмония или краунэфир.
Когда необходимо, защитная группа у атома азота, полученного в результате амидного производного (I), может быть удалена или превращена в другие функциональные группы, так что амидное производное (I) может давать соединение, представленное формулой (I), которое является соединением по настоящему изобретению.
Реакция может проводиться таким же образом, как описано в реакционном процессе (a), для получения соединения (I).
Когда необходимо, соединения, полученные в описанных выше реакциях, могут очищаться с помощью известного метода очистки, такого как перекристаллизация и колоночная хроматография.
[Реакционный процесс (d)]
Данный процесс подробно представлен на следующей химической реакционной схеме:
(Химическая формула 14)

В реакционной схеме выше R0-R2, R5-R7, E, Nx, n, X, Y, L3 и Q являются такими, как описано выше, и заместитель R9 является атомом водорода.
Настоящий процесс, в частности, охватывает конденсацию соединения (V) с соединением (III-а) для получения амидного соединения (IX). Полученное соединение (IX) затем вводят во взаимодействие с соединением (II-а) для получения желаемого соединения (I).
Данные процессы описываются далее более детально.
Процесс 13:
В настоящем процессе, в качестве первой стадии, карбоновую кислоту (III-а) подвергают амидной конденсации с аминопроизводным (V) для получения амида (IX).
Реакция амидирования может проводиться таким же образом, как в процессе 5 для получения соединения (IX).
Процесс 14:
В данном процессе 14, соединение (IX), полученное выше в процессе 13, вводится во взаимодействие с соединением (II-а) для получения соединения, представленного формулой (I) - желаемое соединение по настоящему изобретению.
Реакция может проводиться таким же образом, как в процессе 1, для получения соединения (I).
Когда необходимо, защитная группа у атома азота, полученного в результате амидного производного (I), может быть удалена или превращена в другие функциональные группы, так что амидное производное (I) может давать соединение, представленное формулой (I), которое является соединением по настоящему изобретению.
Реакция может проводиться таким же образом, как описано в реакционном процессе (a), для получения соединения (I).
Когда необходимо, соединения, полученные в упомянутых выше реакциях, могут очищаться известным методом чистки, таким как перекристаллизация и колоночная хроматография.
Изомеры, присутствующие в соединении по настоящему изобретению, представленные общей формулой (I), могут быть разделены с применением известного метода, такого как перекристаллизация, колоночная хроматография, тонкослойная хроматография и высокоэффективная жидкостная хроматография, или подобного метода с применением оптически активных реагентов.
Соединения по настоящему изобретению, представленные общей формулой (I), могут быть превращены в соответствующие соли путем их растворения в подходящем органическом растворителе, таком как вода, метанол, этанол, изопропанол, диэтиловый эфир, диизопропил диэтиловый эфир, тетрагидрофуран, метиленхлорид, хлороформ, бензол или толуол, и обработки органической или неорганической кислотой.
Неорганические кислоты, используемые для этой цели, включают хлороводородную кислоту, бромоводородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и периодную кислоту. Органические кислоты включают муравьиную кислоту, уксусную кислоту, масляную кислоту, щавелевую кислоту, малоновую кислоту, пропионовую кислоту, валериановую кислоту, янтарную кислоту, фумаровую кислоту, малеиновую кислоту, винную кислоту, лимонную кислоту, яблочную кислоту, бензойную кислоту, бензолсульфоновую кислоту, п-толуолсульфоновую кислоту, метансульфоновую кислоту и этансульфоновую кислоту.
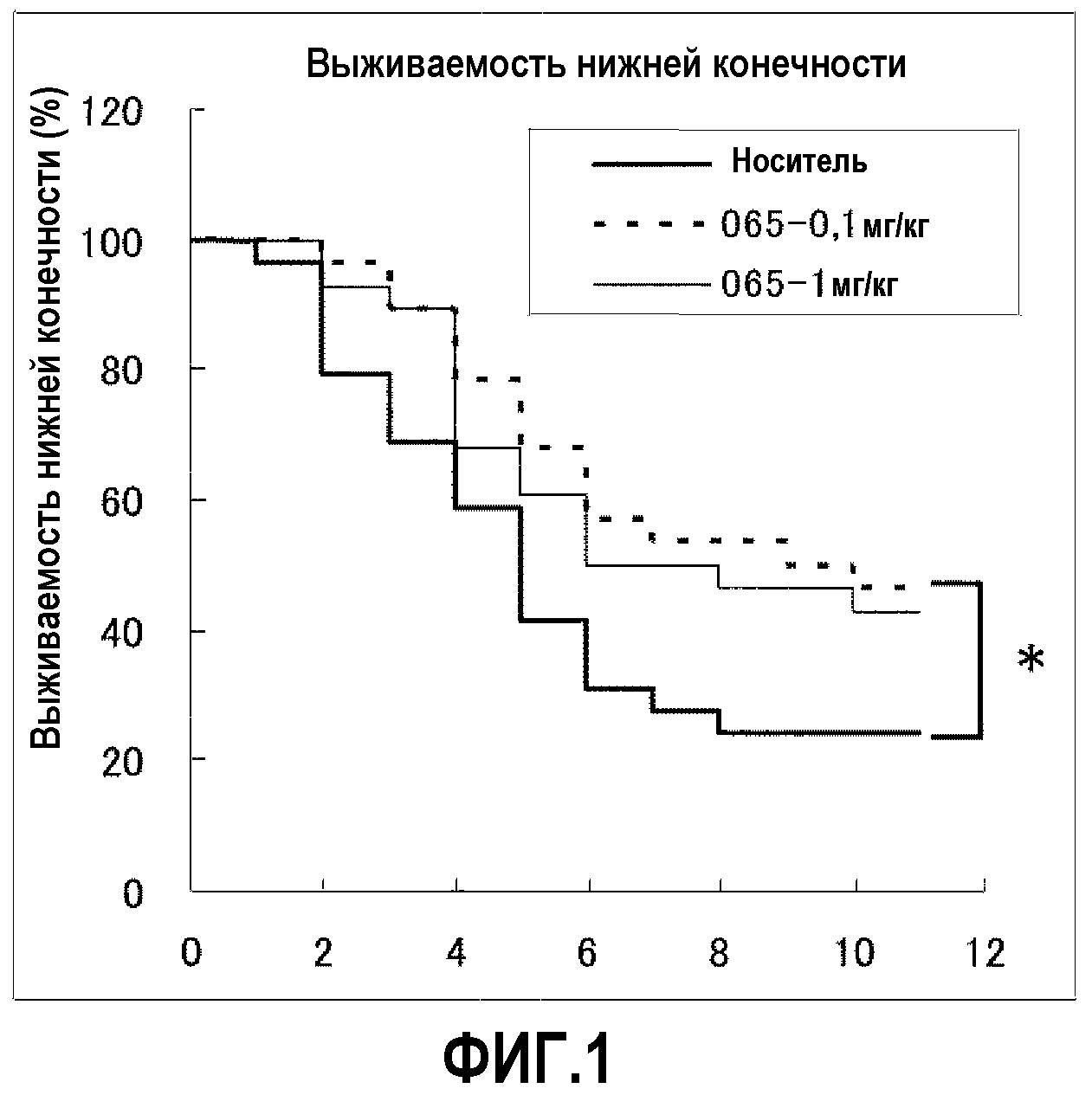
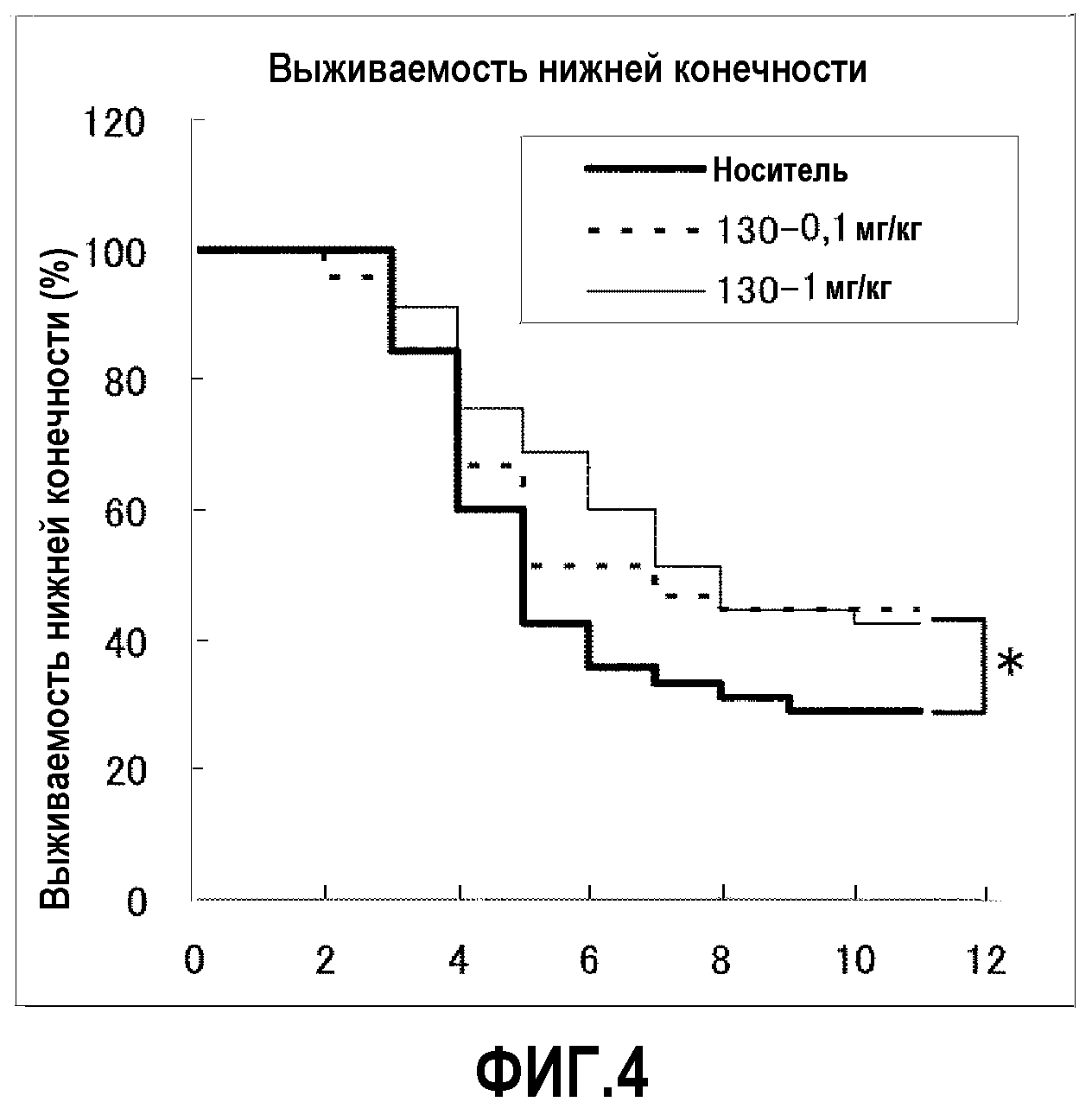
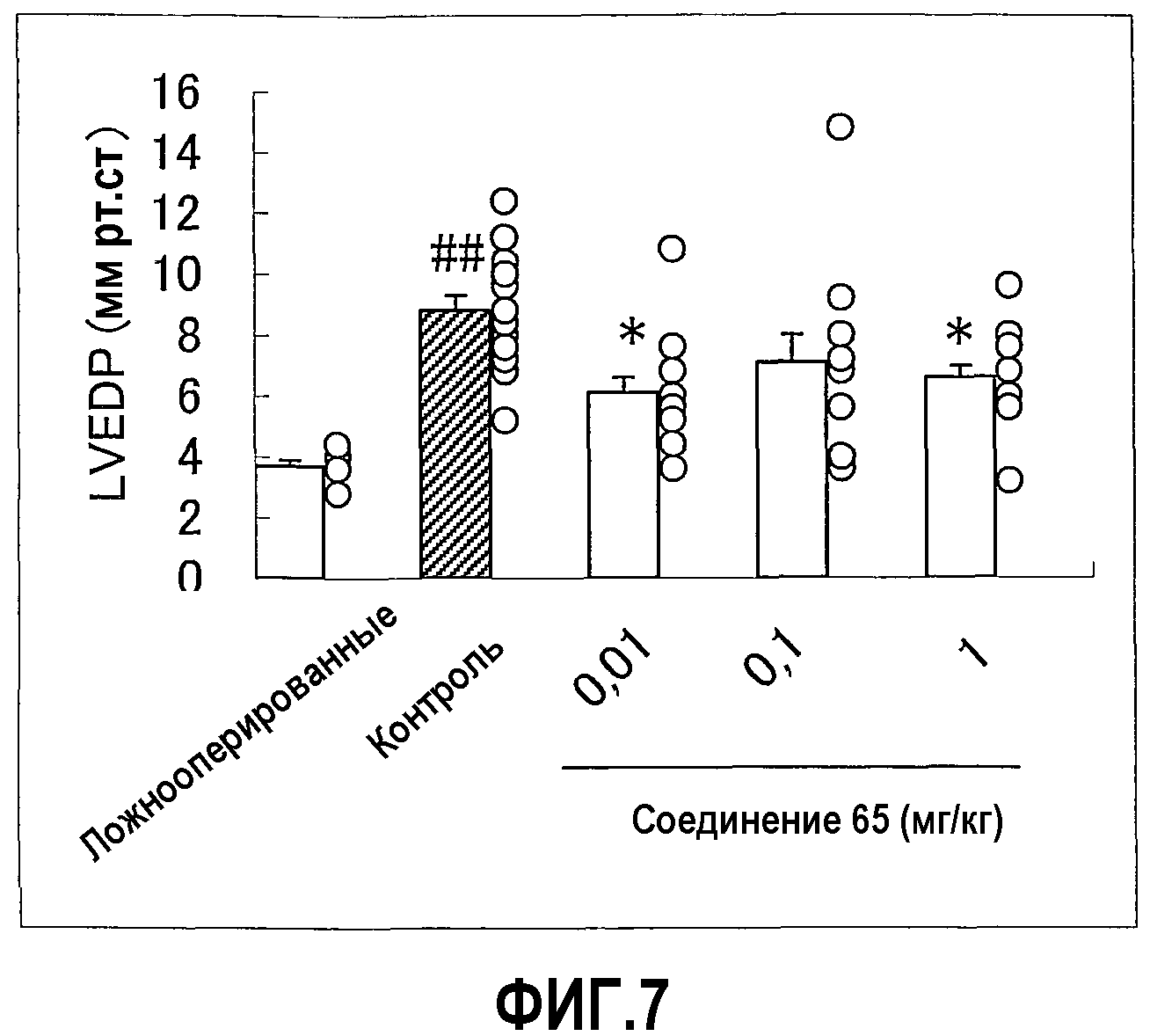
Соединения по настоящему изобретению, представленные общей формулой (I), и их соли демонстрируют пониженную токсичность. В эксперименте, в котором многократно перорально вводили крысам соединение Примера 65 по настоящему изобретению один раз в день в течение недели, соединение не показало токсичности в дозе 150 мг/кг/день.
В то время как соединения по настоящему изобретению, представленные общей формулой (I), и их соли могут применяться сами по себе, они могут быть составлены, по желанию, с другими фармацевтически приемлемыми, широко используемыми носителями в фармацевтические препараты, которые предназначены для уменьшения или лечения заболеваний, включая поражения центральной нервной системы, такие как поражение головного мозга и поражение спинного мозга, ишемический инсульт, ишемические заболевания сердца, такие как инфаркт миокарда и стенокардия, обусловленная органическим поражением, периферические окклюзионные поражения артерий, такие как критическая ишемия конечностей, и последствий данных заболеваний путем стимулирования аксонального роста стимулирования ангиогенеза. Препарат может быть получен с применением наполнителя, разрыхлителя, связывающего вещества, увлажнителя, агента, способствующего распадаемости, поверхностно-активного вещества, смазывающего вещества и других традиционно применяемых разбавителей и вспомогательных веществ. Фармацевтические препараты могут быть обеспечены в различных формах в зависимости от цели лечения, включая таблетки, пилюли, порошки, жидкости, суспензии, эмульсии, гранулы, капсулы, суппозитории, инъекционные (такие как жидкости и суспензии), мази, припарки, ингаляторы и другие приемлемые формы.
Таблетки могут быть сформированы путем использования вспомогательного вещества, такого как лактоза, сахароза, хлорид натрия, глюкоза, мочевина, крахмал, карбонат кальция, каолин, кристаллическая целлюлоза и кремниевая кислота; связывающего вещества, такого как вода, этанол, пропанол, простой сироп, раствор глюкозы, раствор крахмала, раствор желатина, карбоксиметилцеллюлоза, щеллак, метилцеллюлоза, фосфат калия и поливинилпирролидон; способствующего распадаемости агента, такого как сухой крахмал, альгинат натрия, порошкообразный агар-агар, порошкообразный ламинаран, бикарбонат натрия, карбонат кальция, полиоксиэтиленовые эфиры сорбита и жирной кислоты, лаурилсульфат натрия, моноглицерид стеариновой кислоты, крахмал и лактоза; препятствующего распадаемости агента, такого как сахароза, стеарин, какао-масло и гидрированное масло; ускоряющего абсорбцию агента, такого как четвертичное аммониевое основание и лаурилсульфат натрия; смачивающего вещества, такого как глицерин и крахмал; увлажнителя, такого как глицерин и крахмал; адсорбента, такого как крахмал, лактоза, каолин, бентонит и коллоидная кремниевая кислота; смазывающего вещества, такого как очищенный тальк, стеарат, порошкообразная борная кислота и полиэтиленгликоль; и других носителей.
Когда необходимо, таблетки могут быть сформированы путем применения общеизвестного покрытия, такого как покрытие сахаром, покрытие желатином, энтеросолюбильное покрытие, пленочное покрытие, или, альтернативно, таблетки могут быть сформированы в двухслойные пилюли или многослойные пилюли.
Пилюли могут быть сформированы с применением вспомогательного вещества, такого как глюкоза, лактоза, крахмал, какао-масло, гидрированное растительное масло, каолин и тальк; связывающего вещества, такого как порошкообразный гуммиарабик, порошкообразный трагакант, желатин и этанол; способствующего распадаемости агента, такого как ламинаран и агар-агар, и других носителей.
Суппозитории могут быть сформированы с применением полиэтиленгликоля, какао-масла, высшего спирта, сложного эфира высшего спирта, желатина, полусинтетического глицерида и других носителей.
Капсулы могут быть традиционно получены путем смешивания соединения по настоящему изобретению с различными носителями, описанными выше, и заключения смеси в твердую желатиновую капсулу, мягкую желатиновую капсулу или другие капсулы, используя известные технологии.
Когда соединение по настоящему изобретению получают в виде инъекционной формы, такой как раствор, эмульсия или суспензия, инъекционная форма является предпочтительно стерильной и является изотоничной крови. Инъекционные формы могут быть сформированы с применением разбавителя, такого как вода, этиловый спирт, макрогол, пропиленгликоль, этоксилированный изостеариловый спирт, полиоксиизостеариловый спирт и полиоксиэтиленовый эфир сорбита и жирной кислоты.
Инъекционная форма может содержать соль, глюкозу или глицерин в достаточных количествах для формирования изотоничных растворов, а также известный солюбилизирующий агент, буфер или успокаивающий агент.
Когда необходимо, фармацевтический препарат может содержать краситель, консервант, ароматизирующее вещество, вкусовое вещество, подсластитель или другие приемлемые фармацевтические продукты.
Пасты, кремы и гели могут быть сформированы с применением разбавителя, такого как белый вазелин, парафин, глицерин, производное целлюлозы, полиэтиленгликоль, силикон и бентонит.
Описанный выше фармацевтический препарат может вводиться любым путем, определяемым формой препарата, возрастом, полом и другими состояниями пациентов, а также тяжестью заболевания. Например, таблетки, пилюли, растворы, суспензии, эмульсии, гранулы и капсулы вводятся перорально. Инъекционные формы вводятся внутривенно либо одни, либо в виде смеси с известным восстановителем потерянной жидкости, таким как глюкоза и аминокислоты. Когда необходимо, инъекционные формы вводятся одни либо внутримышечно, внутрикожно, подкожно, либо интраперитонеально. Суппозитории вводятся ректально. Мази и припарки вводятся трансдермально. Формы для ингаляции вводятся через слизистую носовой полости или легких.
Обладая способностью стимулировать аксональный рост и стимулировать ангиогенез и направленные на уменьшение или лечение заболеваний, включая поражения центральной нервной системы, такие как поражение головного мозга и поражение спинного мозга, ишемический инсульт, ишемические заболевания сердца, такие как инфаркт миокарда и стенокардия, обусловленная органическим поражением, периферические окклюзионные поражения артерий, такие как критическая ишемия конечностей, и последствий данных заболеваний, соединения по настоящему изобретению вводятся в варьированной дозе, определяемой симптомами, тяжестью заболевания или возрастом пациентов, которых лечат, и имеет ли пациент осложнения. Доза может также варьироваться от пути введения, лекарственной формы и частоты введения дозы. В случае перорального введения доза обычно представляет собой от 0,1 до 1000 мг/день/пациента, и предпочтительно от 1 до 500 мг/день/пациента, что определяется количеством активного компонента. В случае парентерального введения доза может составлять от одной сотой до половины дозы для перорального введения. Однако предпочтительная доза по желанию может варьироваться в зависимости от возраста, симптомов и других состояний пациентов.
ПРИМЕРЫ
Настоящее изобретение описывается далее со ссылкой на Примеры, которые не предназначены для ограничения объема изобретения.
Порядковые номера, присвоенные соединениям в следующих Примерах, соответствуют порядковым номерам соединений в представленных потом таблицах.
Пример 1
Получение 4,6-диметил-5-нитропиримидин-2-ола (соединение 1)
4,6-Диметилпиримидин-2-ола гидрохлорид (20,0 г) добавляли к концентрированной серной кислоте (94,2 г), в то же время охлаждая льдом. К данной смеси добавляли при перемешивании и охлаждении льдом при 5°C или ниже дымящую азотную кислоту (15,7 г: d=1,52). Полученной смеси позволяли медленно нагреваться до комнатной температуры (20-30°C) и затем продолжали перемешивать при комнатной температуре (20-30°C) в течение ночи. Реакционную смесь выливали на лед (340 г) и нейтрализовали 10 н. водным раствором гидроксида натрия до pH приблизительно 2,5 (при 20°C или ниже). Затем смесь экстрагировали дважды изопропанолом (225 мл) и органический слой концентрировали при пониженном давлении для получения 33,1 г остатка. К полученному остатку добавляли 660 мл хлороформа и 66 мл метанола и смесь кипятили в течение 30 мин, с последующим перемешиванием при 50°C в течение 30 мин. Затем отделяли нерастворимое вещество фильтрованием. Фильтрат концентрировали при пониженном давлении до 210 г, с последующим добавлением 100 мл хлороформа и концентрации до 133 г. Полученный в результате остаток перемешивали при комнатной температуре (20-30°C) в течение 30 мин и затем охлаждали льдом в течение 2 часов. Отделяемые кристаллы собирали фильтрованием, промывали холодным хлороформом и сушили с получением 13,1 г желаемого продукта.
Когда необходимо, некоторое количество продукта очищали колоночной хроматографией на силикагеле (метиленхлорид:метанол=50:1) и перекристаллизовывали из метиленхлорида с получением очищенного продукта.
Пример 2
Получение 2-хлор-4,6-диметил-5-нитропиримидина (соединение 2)
Смесь соединения 1 (500 мг) и хлорокиси фосфора (3,89 г) перемешивали в течение 3 часов при кипении. После завершения реакции смесь концентрировали при пониженном давлении. К полученному остатку добавляли хлороформ и воду и смесь охлаждали и нейтрализовали 2 н. водным раствором гидроксида натрия до pH 5-7. Затем смесь экстрагировали хлороформом и органический слой концентрировали при пониженном давлении с получением 411 мг желаемого продукта. Когда необходимо, некоторое количество продукта очищали колоночной хроматографией на силикагеле (этилацетат:гексан=1:1) с получением очищенного продукта.
Пример 3
Получение 5-амино-4,6-диметилпиримидин-2-ола (соединение 3)
Соединение 1 (1,0 г), 5% Pd-C (131 мг) суспендировали в метаноле (60 мл). Повторяли откачку и замещение водородом три раза. Затем суспензию интенсивно перемешивали при комнатной температуре (20-30°C) в течение 8 часов в атмосфере водорода. После завершения реакции смесь фильтровали через целит и отфильтрованный продукт промывали метанолом. Фильтрат упаривали при пониженном давлении с получением 853 мг неочищенного желаемого продукта соединения 3 в виде желтого твердого вещества.
Пример 4
Получение 4,6-диизопропилпиримидин-2-ола (соединение 4)
Трет-бутил-4,6-диизопропилпиримидин-2-илкарбонат (42 мг) растворяли в метиленхлориде (4 мл). По мере охлаждения раствора льдом к раствору добавляли трифторуксусную кислоту (1 мл). Затем смесь перемешивали при комнатной температуре (20-30°C) в течение 1 часа. После завершения реакции смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (метиленхлорид:метанол=10:1) с получением 8,5 мг (выход 30%) желаемого продукта в виде аморфного вещества коричневого цвета.
Пример 5
Получение 4,6-диизопропил-5-нитропиримидин-2-ола (соединение 5)
Соединение 4 (20 мг) суспендировали в смеси концентрированной серной кислоты (1 мл) и хлороформа (1 мл). К полученной в результате смеси добавляли дымящую азотную кислоту (166 мкл: d=1,50) при перемешивании и охлаждении льдом до 5°C или ниже. Полученной в результате смеси позволяли медленно нагреваться до комнатной температуры (20-30°C) и затем продолжали перемешивать при комнатной температуре (20-30°C) в течение ночи. Реакционную смесь выливали на лед и нейтрализовали 10 н. водным раствором гидроксида натрия до pH приблизительно 5 (при 20°C или ниже). Смесь экстрагировали дважды хлороформом и органический слой концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле (метиленхлорид:метанол=50:1 - 10:1) с получением 17,8 мг (выход 49%) желаемого продукта в виде аморфного вещества коричневого цвета.
Пример 6
Получение этил-2-(4,6-диметил-5-нитропиримидин-2-илокси)ацетат (соединение 6)
Соединение 1 (111,86 г) и карбонат калия (274,21 г: 3 экв.) суспендировали в ацетоне (2 л) и добавляли этилбромацетат (165,67 г: 1,5 экв.) при комнатной температуре (20-30°C). Устройство, использованное для добавления этилбромацетата, тщательно промывали ацетоном (237 мл) и перемешивали при 50°C в течение 8 часов. Затем смесь охлаждали до 35°С и концентрировали при пониженном давлении. К полученному остатку добавляли толуол (1120 мл) и смесь перемешивали при комнатной температуре в течение ночи. Затем смесь фильтровали на вакууме и отфильтрованный продукт промывали толуолом (560 мл). Отфильтрованный продукт отжимали и снова промывали толуолом (450 мл). Фильтрат упаривали при пониженном давлении и полученный в результате неочищенный продукт очищали колоночной хроматографией на силикагеле (гексан:этилацетат=1:0 - 4:1) с получением 75,98 г (выход 45%) желаемого продукта в виде желтого твердого вещества.
Пример 7
Получение этил-2-(5-амино-4,6-диметилпиримидин-2-илокси)ацетата (соединение 7)
Соединение 6 (75,98 г) и 5% Pd-C (7,598 г, N. E. CHEMCAT, STD Type) суспендировали в этаноле (760 мл). Повторяли откачку и замещение водородом три раза. Затем суспензию интенсивно перемешивали при комнатной температуре (20-30°C) в течение 4,5 часов в атмосфере водорода. После завершения реакции смесь подвергали тонкому фильтрованию под давлением (0,2 мкм, PTFE) и отфильтрованный продукт промывали этанолом (474 мл). Фильтрат упаривали при пониженном давлении с получением 66,99 г (выход 99,9%) желаемого продукта в виде бледно-желтого твердого вещества.
Пример 8
Получение этил-2-(5-трет-бутоксикарбониламино)-4,6-диметилпиримидин-2-илокси)ацетата (соединение 8)
Соединение 7 (рассчитанное, чтобы составлять 75,98 г, как соединение 6, полученное в предыдущем процессе) и ди-трет-бутилдикарбонат (77,97 г) суспендировали в этилацетате (250 мл). Смесь перемешивали при 70°C в течение ночи. К данной реакционной смеси порциями добавляли гексан (576 мл), с последующим добавлением небольшого количества соединения 8 для затравки, и порциями гексан (288 мл). Затем смесь оставляли охлаждаться и перемешивали в течение ночи. Далее смесь перемешивали и охлаждали льдом в течение 2 часов и затем фильтровали с помощью вакуума. Полученное в результате твердое вещество промывали гексаном (288 мл) и сушили с получением 92,41 г (выход 95% на 2 стадии) желаемого продукта в виде твердого белого вещества.
Пример 9
Получение 2-(5-(трет-бутоксикарбониламино)-4,6-диметилпиримидин-2-илокси)уксусной кислоты (соединение 9)
Соединение 8 (92,4 г) суспендировали в этаноле (127 мл) и добавляли к суспензии 2 н. водным раствором гидроксида натрия (284 мл) при комнатной температуре (20-30°C). Смесь перемешивали в течение 2 часов при комнатной температуре (20-30°C) и добавляли порциями к реакционной смеси 2 н. водный раствор HCl (148 мл) при охлаждении смеси. Для затравки кристаллизации добавляли небольшое количество соединения 9, далее порциями 2 н. водный раствор HCl (119 мл) (внутренняя температура = 10°C или ниже). Смесь перемешивали при комнатной температуре (20-30°C) в течение ночи. Затем смесь перемешивали и охлаждали на льду в течение 3 часов и далее фильтровали с помощью вакуума. Полученное в результате твердое вещество промывали холодной водой (193 мл) и сушили с получением 74,8 г (выход 89%) желаемого продукта в виде белого твердого вещества.
Пример 10
Получение этил-2-(4,6-диметил-5-нитропиримидин-2-иламино)ацетата (соединение 10)
Раствор соединения 2 (69 мг), сложного этилового эфира глицина гидрохлорида (102 мг) и триэтиламина (103 мкл) в этаноле перемешивали в течение 3 часов при кипении с обратным холодильником. После завершения реакции смесь концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле (этилацетат:гексан=1:3) с получением 82 мг (выход 86%) желаемого продукта в виде бледно-желтого аморфного вещества.
Пример 11
Получение 2-(4,6-диметил-5-нитропиримидин-2-иламино)уксусной кислоты (соединение 11)
Соединение 10 (80 мг) суспендировали в 1,4-диоксане (1,5 мл) и добавляли 2 н. водный раствор гидроксида натрия (1,5 мл) при комнатной температуре (20-30°C). Смесь перемешивали при комнатной температуре (20-30°C) в течение 8 часов. Затем смесь промывали диэтиловым эфиром. При охлаждении реакционной смеси порциями добавляли 2 н. водный раствор HCl для нейтрализации смеси до pH 3. Смесь экстрагировали дважды хлороформом и органический слой концентрировали при пониженном давлении с получением 52 мг (выход 75%) желаемого продукта в виде бледно-желтого аморфного вещества.
Пример 12
Получение 2-(5-(трет-бутоксикарбониламино)-4,6-диметилпиримидин-2-илокси)пропановой кислоты (соединение 12)
Смесь Соединения 3 (653 мг) и ди-трет-бутилдикарбоната (1,02 г) в N,N-диметилформамида (25 мл) перемешивали при 50°C в течение ночи. Затем реакционную смесь охлаждали до комнатной температуры (20-30°C). Последовательно добавляли карбонат калия (972 мг) и этил-2-бромпропионат (609 мкл) и смесь перемешивали при комнатной температуре (20-30°C) в течение ночи. Затем к реакционной смеси добавляли воду и смесь экстрагировали дважды этиловым эфиром. Органический слой концентрировали при пониженном давлении. К полученному в результате остатку добавляли 1,4-диоксан (15 мл) и добавляли 2 н. водный содержащий натрий раствор (15 мл) при комнатной температуре (20-30°C). Смесь перемешивали при комнатной температуре (20-30°C) в течение 2 часов. Затем реакционную смесь промывали диэтиловым эфиром. При охлаждении реакционной смеси порциями добавляли 2 н. водный раствор HCl для нейтрализации смеси до pH 3. Далее смесь дважды экстрагировали хлороформом и органический слой концентрировали при пониженном давлении с получением 708 мг (выход 48%, за 3 стадии от примера 3) желаемого продукта в виде бледно-желтого аморфного вещества.
Пример 13
Получение этил-2-(5-амино-4,6-диизопропилпиримидин-2-илокси)ацетата (соединение 13)
Соединение 5 (6,7 мг) и 5% Pd-C (1 мг, N. E. CHEMCAT, STD Type) суспендировали в метаноле (1 мл). Повторяли откачку и замещение водородом три раза. Далее суспензию интенсивно перемешивали при комнатной температуре (20-30°C) в течение 1 часа в атмосфере водорода. После завершения реакции смесь фильтровали через целит и отфильтрованный продукт промывали метанолом. Фильтрат концентрировали при пониженном давлении. К полученному в результате остатку добавляли карбонат калия (6,2 мг) и диметилформамид (1 мл). К смеси добавляли этилбромацетат (3,3 мкл) при комнатной температуре (20-30°C). Далее смесь перемешивали при комнатной температуре (20-30°C) в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали методом колоночной хроматографии на силикагеле (этилацетат:гексан=1:1) с получением 0,7 мг (выход 8%) желаемого продукта в виде бледно-желтого аморфного вещества.
Пример 14
Получение этил-2-(2-хлор-5-метилпиримидин-4-иламино)ацетата (соединение 14)
Этиловый эфир глицина гидрохлорид (157 мг) добавляли к раствору 2,4-дихлор-5-метилпиримидина (184 мг) и диизопропилэтиламина (486 мкл) в ацетонитриле (3 мл) при охлаждении раствора льдом. Смесь перемешивали при 40°C в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле (этилацетат:гексан=1:3 - 1:1) с получением 205 мг (выход 81%) желаемого продукта в виде белого аморфного вещества.
Пример 15
Получение этил-2-(2-хлор-5-метилпиримидин-4-илокси)-2-метилпропионата (соединение 15)
Гидрид натрия (52 мг: 60%) суспендировали в тетрагидрофуране (3 мл). Охлаждая суспензию льдом, добавляли этил-альфа-гидроксиизобутилат (145 мкл). Смесь перемешивали при комнатной температуре (20-30°C) в течение 30 мин. При охлаждении данной смеси льдом добавляли порциями 2,4-дихлор-5-метилпиримидин (176 мг) и смесь перемешивали при комнатной температуре (20-30°C) в течение 4 дней. Затем реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле (этилацетат:гексан=1:49 - 1:4.5) с получением 78 мг (выход 30%) желаемого продукта в виде маслянистого вещества.
Пример 16
Получение 2-(2-хлор-5-метилпиримидин-4-илокси)-2-метилпропановой кислоты (соединение 16)
Названное в заголовке соединение синтезировали из соединения 15 таким же образом, как в примере 11.
Пример 17
Получение этил-2-(2-амино-4,6-диметилпиримидин-5-илокси)ацетата (соединение 17)
Названное в заголовке соединение синтезировали из 2-амино-4,6-диметилпиримидин-5-ола таким же образом, как в примере 6.
Пример 18
Получение этил-2-(4-амино-5-фтор-2-оксопиримидин-1(2H)-ил)ацетата (соединение 18)
5-Фторцитозин (500 мг) и карбонат калия (803 мг) суспендировали в N,N-диметилформамиде (5 мл) и добавляли к суспензии этилбромацетат (430 мкл). Смесь перемешивали при 100°C в течение ночи. Полученный в результате осадок удаляли фильтрованием и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле (метиленхлорид:метанол=99:1 - 92:9) с получением 329 мг (выход 39%) желаемого продукта в виде белого аморфного вещества.
Пример 19
Получение этил-2-(3-метил-5-нитропиридин-2-илокси)ацетата (соединение 19)
Гидрид натрия (116 мг: 60%) суспендировали в тетрагидрофуране (4 мл). Добавляли этилгликолят (273 мкл) при охлаждении суспензии льдом. Смесь перемешивали при комнатной температуре (20-30°C) в течение 30 мин. Затем добавляли к смеси порциями 2-хлор-3-метил-5-нитропиридин (200 мг) при комнатной температуре (20-30°C) и смесь перемешивали при комнатной температуре (20-30°C) в течение 3 часов. Затем к реакционной смеси добавляли воду и смесь дважды экстрагировали хлороформом. Органический слой концентрировали при пониженном давлении с обеспечением 310 мг неочищенного желаемого продукта соединения 19 в виде аморфного вещества коричневого цвета.
Пример 20
Получение этил-2-(5-(трет-бутоксикарбониламино)-3-метилпиридин-2-илокси)ацетата (соединение 20)
Соединение 19 (319 мг) и 5% Pd-C (5mg, N. E. CHENCAT, STD Type) суспендировали в метаноле. Повторяли откачку и замещение водородом три раза. Затем суспензию интенсивно перемешивали при комнатной температуре (20-30°C) в течение 1 часа в атмосфере водорода. После завершения реакции смесь фильтровали через целит и отфильтрованный продукт промывали метанолом. Фильтрат концентрировали при пониженном давлении. К полученному в результате остатку добавляли диметилформамид и к смеси добавляли ди-трет-бутилдикарбонат (253 мг) при комнатной температуре. Далее смесь перемешивали при 50°C в течение ночи. Затем к реакционной смеси добавляли воду и смесь дважды экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением 368 мг неочищенного желаемого продукта соединения 20 в виде желтого маслянистого вещества.
Пример 21
Получение 2-(5-(трет-бутоксикарбониламино)-3-метилпиридин-2-илокси)уксусной кислоты (соединение 21)
Названное в заголовке соединение синтезировали из соединения 20 таким же образом, как в примере 11 (выход 70%, за 3 стадии от примера 19).
Пример 22
Получение этил-2-(3-хлорпиразин-2-иламино)ацетата (соединение 22)
К этанольному раствору (10 мл) 2,3-дихлорпиразина (1,0 г) добавляли этиловый эфир глицина гидрохлорид (940 мг) и триэтиламин (1,9 мл) и смесь подвергали микроволновому облучению (150°C, мин). Реакционную смесь концентрировали при пониженном давлении. К полученному в результате остатку добавляли насыщенный водный раствор бикарбоната натрия и продукт экстрагировали хлороформом. Органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате остаток очищали методом колоночной хроматографии на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; NH-DM1020) (гексан:этилацетат=10:1) с получением 212 мг (выход 15%) желаемого продукта.
Пример 23
Получение 2-(3-хлорпиразин-2-иламино)уксусной кислоты (соединение 23)
2 н. Водный раствор гидроксида натрия (0,6 мл) добавляли к этанольному раствору (0,3 мл) соединения 22 (194 мг) и смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли к реакционной смеси 2 н. хлороводородную кислоту при охлаждении льдом и смесь нейтрализовали. Продукт экстрагировали хлороформом (5 раз). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 38 мг (выход 23%) желаемого продукта.
Пример 24
Получение этил-2-(3-хлорпиразин-2-илокси)ацетата (соединение 24)
Названное в заголовке соединение синтезировали из 2,3-дихлорпиразина и этил-2-гидроксиацетата таким же образом, как в примере 19.
Пример 25
Получение этил-2-(6-хлорпиразин-2-илокси)ацетата (соединение 25)
Названное в заголовке соединение синтезировали из 2,6-дихлорпиразина и этилового эфира глицина гидрохлорида таким же образом, как в примере 19.
Пример 26
Получение 2-(3-хлорпиразин-2-илокси)уксусной кислоты (соединение 26)
Названное в заголовке соединение синтезировали из соединения 24 таким же образом, как в примере 23.
Пример 27
Получение 2-(6-хлорпиразин-2-илокси)уксусной кислоты (соединение 27)
Названное в заголовке соединение синтезировали из соединения 25 таким же образом, как в примере 23.
Пример 28
Получение этил-2-(5,6-дихлорпиридазин-4-иламино)ацетата (соединение 28)
Раствор 3,4,5-трихлорпиридазина (300 мг), этилового эфира глицина гидрохлорида (228 мг) и диизопропилэтиламина (844 мкл) в этаноле перемешивали в течение 3 часов при кипении. После завершения реакции смесь концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле (этилацетат:гексан=1:9-1:1) с получением 126 мг (выход 31%) указанного в заголовке соединения в виде бледно-розового аморфного вещества.
Пример 29
Получение этил-2-(3,5-дихлорпиридазин-4-иламино)ацетата (соединение 29)
В синтезе примера 28 также получали 78 мг (выход 19%) указанного в заголовке соединения в виде бледно-розового аморфного вещества.
Пример 30
Получение трет-бутил-2-(2-((1-бензилпиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 30)
Соединение 9 (74,74 г), 1-бензил-N-метилпиперидин-4-амин (66,78 г: 1,3 экв.) и триэтиламин (127,20 г: 5 экв.) суспендировали в ацетонитриле (800 мл). Добавляли порциями 50% этилацетатный раствор ангидрида пропанфосфоновой кислоты (191,99 г) при охлаждении суспензии льдом. Устройство, используемое для добавления 50% этилацетатного раствора ангидрида пропанфосфоновой кислоты, тщательно промывали ацетонитрилом (192 мл) и осуществляли перемешивание при комнатной температуре (20-30°C) в течение ночи. Затем смесь концентрировали при пониженном давлении. К полученному в результате остатку последовательно добавляли хлороформ (250 мл) и насыщенный водный раствор бикарбоната натрия (140 мл) и смесь переносили на делительную воронку. Реакционный сосуд тщательно промывали хлороформом (175 мл) и осуществляли разделение. После отделения органического слоя к водному слою добавляли хлороформ (175 мл) и снова осуществляли разделение. Объединенный органический слой сушили над сульфатом магния и фильтровали с помощью вакуума. Фильтрат упаривали при пониженном давлении с получением 183,27 г неочищенного желаемого продукта Соединения 30 в виде желтого аморфного вещества.
Пример 31
Получение трет-бутил-4,6-диметил-2-(2-(метил(пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 31)
К раствору соединения 30 (3,7 г) в метаноле (111 мл) добавляли Pd-C (371 мг) и затем осуществляли гидрирование при перемешивании смеси при атмосферном давлении и комнатной температуре в течение ночи. Затем катализатор отделяли фильтрованием и фильтрат концентрировали при пониженном давлении с получением 2,9 г (выход 96%) указанного в заголовке соединения.
Пример 32
Получение трет-бутил-2-(2-((1-(циклогексилметил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 32)
Бромметилциклогексан (25 мкл) и диизопропилэтиламин (63 мкл) добавляли к раствору соединения 31 (71 мг) в диметилформамиде (1 мл) и смесь перемешивали при 120°C в течение 8 часов. Затем к реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия и продукт экстрагировали этилацетатом. Органический слой промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали методом колоночной хроматографии (гексан:этилацетат=1:1) на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; NH-DM1020) с получением 40 мг (выход 46%) желаемого продукта.
Пример 33
Получение трет-бутил-2-(2-((1-(4-хлорбензил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 33)
Названное в заголовке соединение синтезировали из соединения 31 и 4-хлорбензилбромида таким же образом, как в примере 32.
Пример 34
Получение трет-бутил-2-(2-((1-изобутилпиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 34)
Названное в заголовке соединение синтезировали из соединения 31 и изобутилбромида таким же образом, как в примере 32.
Пример 35
Получение трет-бутил-2-(2-((1-бензоилпиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамат (соединение 35)
Названное в заголовке соединение синтезировали из соединения 31 и бензоилхлорида таким же образом, как в примере 32.
Пример 36
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-фенэтилпиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 36)
Названное в заголовке соединение синтезировали из соединения 31 и фенэтилбромида таким же образом, как в примере 32.
Пример 37
Получение трет-бутил-2-(2-((1-(циклогексанкарбонил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 37)
Названное в заголовке соединение синтезировали из соединения 31 и циклогексанкарбонилхлорида таким же образом, как в примере 32.
Пример 38
Получение трет-бутил-2-(2-((1-ацетилпиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 38)
Названное в заголовке соединение синтезировали из соединения 31 и ацетилхлорида таким же образом, как в примере 32.
Пример 39
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-(фенилсульфонил)пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 39)
Названное в заголовке соединение синтезировали из соединения 31 и бензолсульфонилхлорида таким же образом, как в примере 32.
Пример 40
Получение трет-бутил-2-(2-((1-циклогексилпиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 40)
Названное в заголовке соединение синтезировали из соединения 31, циклогексанона и триацетоксиборгидрида натрия таким же образом, как в примере 32.
Пример 41
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-(пиперидин-1-карбонил)пиперидин-4-ил)амино)-2-оксоэтокси)-пиримидин-5-илкарбамата (соединение 41)
Названное в заголовке соединение синтезировали из соединения 31 и 1-пиперидинкарбонилхлорида таким же образом, как в примере 32.
Пример 42
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-(2-метилбензил)пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 42)
Названное в заголовке соединение синтезировали из соединения 31 и 2-метилбензилбромида таким же образом, как в примере 32.
Пример 43
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-фенилпиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 43)
Названное в заголовке соединение синтезировали из соединения 31, фенилборной кислоты, ацетата меди(II) и пиридина таким же образом, как в примере 32.
Пример 44
Получение трет-бутил-2-(2-((1-(2-метоксиэтил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 44)
Названное в заголовке соединение синтезировали из соединения 31 и бромэтилметилового эфира таким же образом, как в примере 32.
Пример 45
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-(пиридин-3-илметил)пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 45)
Названное в заголовке соединение синтезировали из соединения 31 и 3-(бромметил)пиридина гидробромида таким же образом, как в примере 32.
Пример 46
Получение трет-бутил-2-(2-((1-(4-фторбензил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 46)
Названное в заголовке соединение синтезировали из соединения 31 и 4-фторбензилбромида таким же образом, как в примере 32.
Пример 47
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-((тетрагидрофуран-2-ил)метил)пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 47)
Названное в заголовке соединение синтезировали из соединения 31 и тетрагидрофурфурилбромида таким же образом, как в примере 32.
Пример 48
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-(пиридин-3-ил)пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 48)
Названное в заголовке соединение синтезировали из соединения 31, 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина, ацетата меди(II) и пиридина таким же образом, как в примере 32.
Пример 49
Получение трет-бутил-2-(2-((1-(циклопропилметил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 49)
Соединение 9 (100 мг), 1-(циклопропилметил)-N-метилпиперидин-4-амин (56,6 мг) и триэтиламин (234 мкл) суспендировали в ацетонитриле (2 мл). Добавляли порциями 50% этилацетатный раствор ангидрида пропанфосфоновой кислоты (273 мг) при охлаждении суспензии льдом. Устройство, используемое для добавления 50% этилацетатного раствора ангидрида пропанфосфоновой кислоты, тщательно промывали ацетонитрилом (0,5 мл) и осуществляли перемешивание при комнатной температуре (20-30°C) в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении, полученный в результате остаток очищали методом колоночной хроматографии (метиленхлорид:метанол=30:1) на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; NH-DM1020) с получением 79 мг (выход 52%) желаемого продукта в виде бледно-желтого аморфного вещества.
Пример 50
Получение трет-бутил-2-(2-((1-(3-метоксибензил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 50)
Названное в заголовке соединение синтезировали из соединения 31 и 1-(бромметил)-3-метоксибензола таким же образом, как в примере 32.
Пример 51
Получение трет-бутил-2-(2-((1-(4-цианобензил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 51)
Названное в заголовке соединение синтезировали из соединения 31 и 4-(бромметил)бензонитрила таким же образом, как в примере 32.
Пример 52
Получение трет-бутил-4,6-диметил-2-(2-метил(1-(3-(трифторметил)бензил)пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 52)
Названное в заголовке соединение синтезировали из соединения 31 и 1-(бромметил)-3-(трифторметил)бензола таким же образом, как в пример 32.
Пример 53
Получение трет-бутил-4,6-диметил-2-(2-(метил(1-(3,4,5-трифторбензил)пиперидин-4-ил)амино)-2-оксоэтокси)пиримидин-5-илкарбамата (соединение 53)
Названное в заголовке соединение синтезировали из соединения 31 и 5-(бромметил)-1,2,3-трифторбензола таким же образом, как в примере 32.
Пример 54
Получение трет-бутил-2-(2-((1-(циклопропанкарбонил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 54)
Названное в заголовке соединение синтезировали из соединения 31 и циклопропанкарбонилхлорида таким же образом, как в примере 32.
Пример 55
Получение трет-бутил-2-(2-((1-(бифенил-4-ил)(метил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 55)
Названное в заголовке соединение синтезировали из соединения 31 и 4-(бромметил)бифенила таким же образом, как в примере 32.
Пример 56
Получение трет-бутил-2-(1-((1-бензилпиперидин-4-ил)(метил)амино)-1-оксопропан-2-илокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 56)
Названное в заголовке соединение синтезировали из соединения 12 и 1-бензил-N-метилпиперидин-4-амина таким же образом, как в примере 49.
Пример 57
Получение трет-бутил-4,6-диметил-2-(2-оксо-2-(пиперидин-4-иламино)этокси)пиримидин-5-илкарбамата (соединение 57)
Трет-бутил-2-(2-(1-бензилпиперидин-4-иламино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамат синтезировали из соединения 9 и 4-амино-1-бензилпиперидина таким же образом, как в примере 49. Затем указанное в заголовке соединение синтезировали из трет-бутил-2-(2-(1-бензилпиперидин-4-иламино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата таким же образом, как в примере 31.
Пример 58
Получение трет-бутил-2-(2-(1-(циклопропилметил)пиперидин-4-иламино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 58)
Названное в заголовке соединение синтезировали из соединения 57 и бромметилциклопропана таким же образом, как в примере 32.
Пример 59
Получение трет-бутил-2-(2-(1-(4-фторбензоил)пиперидин-4-иламино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 59)
Названное в заголовке соединение синтезировали из соединения 57 и 4-фторбензоилхлорида таким же образом, как в примере 32.
Пример 60
Получение трет-бутил-2-(2-((1-бензилпиперидин-4-ил)(циклопропил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамата (соединение 60)
Названное в заголовке соединение синтезировали из соединения 9 и 1-бензил-N-циклопропилпиперидин-4-амина таким же образом, как в примере 49.
Пример 61
Получение трет-бутил-6-(2-((1-бензилпиперидин-4-ил)(метил)амино)-2-оксоэтокси)-5-метилпиридин-3-илкарбамата (соединение 61)
Названное в заголовке соединение синтезировали из соединения 21 и 1-бензил-N-метилпиперидин-4-амина таким же образом, как в примере 49.
Пример 62
Получение N-(1-бензилпиперидин-4-ил)-2-(4,6-диметил-5-нитропиримидин-2-иламино)-N-метилацетамида (соединение 62)
Названное в заголовке соединение синтезировали из соединения 11 и 1-бензил-N-метилпиперидин-4-амина таким же образом, как в примере 49.
Пример 63
Получение 2-(5-амино-4,6-диметилпиримидин-2-иламино)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида (соединение 63)
Соединение 62 (93 мг) и цинк (147 мг) суспендировали в уксусной кислоте (2 мл) и суспензию перемешивали при комнатной температуре (20-30°C) в течение 3 часов. Затем реакционную смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали методом колоночной хроматографии на силикагеле (этилацетат:гексан=5:1-3:1) с получением 25,4 мг (выход 29%) желаемого продукта в виде бледно-желтого аморфного вещества.
Пример 64
Получение 4-((4-(2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метилацетамид)пиперидин-1-ил)метил)бензамида (соединение 64)
Трет-бутил-2-(2-((1-(4-карбамоилбензил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамат синтезировали из соединения 9 и 4-((4-(метиламино)пиперидин-1-ил)метил)бензамида таким же образом, как в примере 49. Затем трет-бутил-2-(2-((1-(4-карбамоилбензил)пиперидин-4-ил)(метил)амино)-2-оксоэтокси)-4,6-диметилпиримидин-5-илкарбамат растворяли в метиленхлориде (4 мл). При охлаждении раствора льдом добавляли трифторуксусную кислоту (1 мл). Смесь перемешивали при комнатной температуре (20-30°C) в течение 4-5 часов. После завершения реакции реакционную смесь концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; NH-DM1020) методом колоночной хроматографии (метиленхлорид:метанол=10:1) с получением 28,1 мг (выход 36%: на 2 стадии) желаемого продукта в виде белого твердого вещества.
Пример 65
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида (соединение 65)
Соединение 30 (рассчитанное, предполагая, что соединение 9 в предыдущем процессе составляло 74,74 г и выход амидирования составлял 80%) растворяли в хлороформе (140 мл). Раствор добавляли к 6 н. водному раствору HCl (530 мл) при комнатной температуре (20-30°C). Устройство, используемое для добавления соединения 30, тщательно промывали хлороформом (240 мл) и затем осуществляли перемешивание при комнатной температуре (20-30°C) в течение 2,5 часов. После отделения слоя хлороформа к водному слою добавляли хлороформ (300 мл). Добавляли порциями 4 н. водный раствор гидроксида натрия (805 мл), добавляя по мере необходимости лед (всего 740,7 г). Добавляли дополнительное количество 4 н. водного раствора гидроксида натрия (25 мл для регулирования pH водного слоя до 8,5), с последующим добавлением хлороформа (80 мл), и осуществляли разделение. После отделения слоя хлороформа к водному слою добавляли хлороформ (200 мл) и снова осуществляли разделение. Слой хлороформа, собранный в двух процессах экстракции, сушили над сульфатом магния и фильтровали отсасыванием. Затем фильтрат упаривали при пониженном давлении с получением 100,15 г (выход 103,9%, за 2 стадии) неочищенного желаемого продукта в виде бледно-желтого твердого вещества.
К полученному в результате твердому веществу добавляли изопропанол (1020 мл), и смесь нагревали при 85°C для растворения твердого вещества. Смесь перемешивали, позволяя ей охладиться. Когда температура нагревательной бани достигала 67°C, осуществляли введение затравки и смесь продолжали перемешивать в течение ночи. Затем смесь перемешивали в течение 2 часов при охлаждении льдом и далее фильтровали отсасыванием. Полученное в результате твердое вещество промывали холодным изопропанолом (150 мл) и сушили с получением 89,60 г (выход 92,9%, за 2 стадии) желаемого соединения (перекристаллизованное из изопропанола) в виде бесцветных кристаллов.
К перекристаллизованному из изопропанола продукту (50,00 г) добавляли этанол (220 мл) и смесь нагревали до 85°C для растворения продукта. Смесь перемешивали, позволяя ей охладиться. Когда температура нагревательной бани достигала 65°C, осуществляли введение затравки и смесь продолжали перемешивать в течение ночи. Затем смесь перемешивали в течение 2 часов при охлаждении с помощью льда и далее фильтровали отсасыванием. Полученное в результате твердое вещество промывали холодным изопропанолом (100 мл) и сушили с получением 48,18 (выход 96%) желаемого продукта в виде белых кристаллов.
Пример 66
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилацетамида дигидрохлорида (соединение 66)
К хлороформному раствору (1 мл) соединения 49 (78 мг) добавляли 6 н. HCl (1 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли 4 н. раствор гидроксида натрия, при охлаждении реакционной смеси с помощью льда, для нейтрализации смеси. Полученный в результате продукт экстрагировали хлороформом и органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; NH-DM1020) методом колоночной хроматографии (этилацетат) с получением 37,5 мг (выход 62%) 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилацетамида. Затем к метанольному раствору 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилацетамида добавляли 2 экв. 4 н. раствора HCl/1,4-диоксан и смесь концентрировали при пониженном давлении. Желаемый продукт обеспечивали путем перекристаллизации из метанола/этилацетата в виде белых кристаллов.
Пример 67
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклопентил)пиперидин-4-ил)-N-метилацетамида (соединение 67)
Названное в заголовке соединение синтезировали из соединения 9 и 1-циклопентил-N-метилпиперидин-4-амина таким же образом, как в примере 64.
Пример 68
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(2-циклогексилэтил)пиперидин-4-ил)-N-метилацетамида дигидрохлорида (соединение 68)
2-(5-Амино-4,6-диметилпиримидин-2-илокси)-N-(1-(2-циклогексилэтил)пиперидин-4-ил)-N-метилацетамид синтезировали из соединения 9 и N-метил-1-(2-циклогексилэтил)пиперидин-4-амина таким же образом, как в примере 64. Затем к метанольному раствору 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(2-циклогексилэтил)-пиперидин-4-ил)-N-метилацетамида добавляли 2 эквивалента 4 н. раствора HCl/1,4-диоксан и смесь концентрировали при пониженном давлении. Желаемый продукт обеспечивали путем перекристаллизации из метанола/диэтилового эфира в виде белых кристаллов.
Пример 69
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(4-(трифторметил)бензил)пиперидин-4-ил)ацетамида (соединение 69)
Названное в заголовке соединение синтезировали из соединения 9 и N-метил-1-(4-(трифторметил)бензил)-пиперидин-4-амина таким же образом, как в примере 64, и перекристаллизовывали из метиленхлорида.
Пример 70
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклобутанкарбонил)пиперидин-4-ил)-N-метилацетамида (соединение 70)
Названное в заголовке соединение синтезировали из соединения 9 и циклобутил(4-(метиламино)пиперидин-1-ил)метанона таким же образом, как в примере 64.
Пример 71
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклогексилметил)пиперидин-4-ил)-N-метилацетамида (соединение 71)
К хлороформному раствору (0,5 мл) соединения 32 (39 мг) добавляли 6 н. HCl (0,7 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли 4 н. водный раствор гидроксида натрия, при охлаждении реакционной смеси с помощью льда, для нейтрализации смеси. Полученный в результате продукт экстрагировали хлороформом и органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd., NH-DM1020) методом колоночной хроматографии (этилацетат) с получением 23 мг (выход 76%) указанного в заголовке соединения.
Пример 72
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(4-хлорбензил)пиперидин-4-ил)-N-метилацетамида (соединение 72)
Названное в заголовке соединение синтезировали из соединения 33 таким же образом, как в примере 71.
Пример 73
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-изобутилпиперидин-4-ил)-N-метилацетамида (соединение 73)
Названное в заголовке соединение синтезировали из соединения 34 таким же образом, как в примере 71.
Пример 74
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензоилпиперидин-4-ил)-N-метилацетамида (соединение 74)
Названное в заголовке соединение синтезировали из соединения 35 таким же образом, как в примере 71.
Пример 75
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-фенэтилпиперидин-4-ил)ацетамида (соединение 75)
Названное в заголовке соединение синтезировали из соединения 36 таким же образом, как в примере 71.
Пример 76
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклогексанкарбонил)пиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 76)
2-(5-Амино-4,6-диметилпиримидин-2-илокси)-N-(1-циклогексанкарбонил)пиперидин-4-ил)-N-метилацетамид синтезировали из соединения 37 таким же образом, как в примере 71. Затем к метанольному раствору 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-циклогексанкарбонил)пиперидин-4-ил)-N-метилацетамида добавляли 1 экв. 4 н. раствор HCl/1,4-диоксан и смесь концентрировали при пониженном давлении. Желаемый продукт обеспечивали путем перекристаллизации из метанола/диэтилового эфира в виде бледно-желтого твердого вещества.
Пример 77
Получение N-(1-ацетилпиперидин-4-ил)-2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метилацетамида гидрохлорид (соединение 77)
Названное в заголовке соединение синтезировали из соединения 38 таким же образом, как в примере 76.
Пример 78
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(фенилсульфонил)пиперидин-4-ил)ацетамида (соединение 78)
Названное в заголовке соединение синтезировали из соединения 39 таким же образом, как в примере 71.
Пример 79
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-циклогексилпиперидин-4-ил)-N-метилацетамида (соединение 79)
Названное в заголовке соединение синтезировали из соединения 40 таким же образом, как в примере 71.
Пример 80
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(пиперидин-1-карбонил)пиперидин-4-ил)ацетамида (соединение 80)
Названное в заголовке соединение синтезировали из соединения 41 таким же образом, как в примере 71.
Пример 81
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(2-метилбензил)пиперидин-4-ил)ацетамида (соединение 81)
Названное в заголовке соединение синтезировали из соединения 42 таким же образом, как в примере 71.
Пример 82
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-фенилпиперидин-4-ил)ацетамида (соединение 82)
Названное в заголовке соединение синтезировали из соединения 43 таким же образом, как в примере 71.
Пример 83
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(2-метоксиэтил)пиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 83)
Названное в заголовке соединение синтезировали из соединения 44 таким же образом, как в примере 76.
Пример 84
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(пиридин-3-илметил)пиперидин-4-ил)ацетамида гидрохлорид (соединение 84)
Названное в заголовке соединение синтезировали из соединения 45 таким же образом, как в примере 76.
Пример 85
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(4-фторбензил)пиперидин-4-ил)-N-метилацетамида (соединение 85)
Названное в заголовке соединение синтезировали из соединения 46 таким же образом, как в примере 71.
Пример 86
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-((тетрагидрофуран-2-ил)метил)пиперидин-4-ил)ацетамида (соединение 86)
Названное в заголовке соединение синтезировали из соединения 47 таким же образом, как в примере 71.
Пример 87
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(пиридин-3-ил)пиперидин-4-ил)ацетамида гидрохлорид (соединение 87)
Названное в заголовке соединение синтезировали из соединения 48 таким же образом, как в примере 76.
Пример 88
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(3-метоксибензил)пиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 88)
Названное в заголовке соединение синтезировали из соединения 50 таким же образом, как в примере 76.
Пример 89
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(4-цианобензил)пиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 89)
Названное в заголовке соединение синтезировали из соединения 51 таким же образом, как в примере 76.
Пример 90
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(3-(трифторметил)бензил)пиперидин-4-ил)ацетамида (соединение 90)
Названное в заголовке соединение синтезировали из соединения 52 таким же образом, как в примере 71.
Пример 91
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(3,4,5-трифторбензил)пиперидин-4-ил)ацетамида (соединение 91)
Названное в заголовке соединение синтезировали из соединения 53 таким же образом, как в примере 71.
Пример 92
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклопропанкарбонил)пиперидин-4-ил)-N-метилацетамида (соединение 92)
Названное в заголовке соединение синтезировали из соединения 54 таким же образом, как в примере 71.
Пример 93
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(бифенил-4-илметил)пиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 93)
Названное в заголовке соединение синтезировали из соединения 55 таким же образом, как в примере 76.
Пример 94
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(пиперидин-4-ил)ацетамида (соединение 94)
Названное в заголовке соединение синтезировали из соединения 31 таким же образом, как в примере 71.
Пример 95
Получение N-(1-бензилпиперидин-4-ил)-2-(4,6-диметилпиримидин-2-илокси)-N-метилацетамида гидрохлорид (соединение 95)
N-(1-бензилпиперидин-4-ил)-2-(4,6-диметилпиримидин-2-илокси)-N-метилацетамид (147 мг: выход 62%) синтезировали из 2-(4,6-диметилпиримидин-2-ил)оксиуксусной кислоты (117 мг) и 1-бензил-N-метилпиперидин-4-амина (131 мг) таким же образом, как в примере 49. Затем к метанольному раствору N-(1-бензилпиперидин-4-ил)-2-(4,6-диметилпиримидин-2-илокси)-N-метилацетамида добавляли 1 экв. 4 н. раствор HCl/этилацетата. Затем смесь концентрировали при пониженном давлении с получением желаемого продукта в виде белого аморфного вещества.
Пример 96
Получение 2-(4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(4-(трифторметил)бензил)пиперидин-4-ил)ацетамида малеат (соединение 96)
2-(4,6-Диметилпиримидин-2-илокси)-N-метил-N-(1-(4-(трифторметил)бензил)пиперидин-4-ил)ацетамид синтезировали из 2-(4,6-диметилпиримидин-2-ил)оксиуксусной кислоты и N-метил-1-(4-(трифторметил)бензил)пиперидин-4-амина таким же образом, как в примере 49. Затем к метанольному раствору 2-(4,6-диметил-пиримидин-2-илокси)-N-метил-N-(1-(4-(трифторметил)бензил)пиперидин-4-ил)ацетамида добавляли 1 эквивалент малеиновой кислоты и смесь концентрировали при пониженном давлении. Желаемый продукт обеспечивали путем перекристаллизации из 2-пропанола/диизопропилового эфира в виде белого твердого вещества.
Пример 97
Получение 2-(5-амино-4,6-диметил-2-оксопиримидин-1(2H)-ил)-N-(1-бензилпиперидин-4-ил)-N-метилацетамид (соединение 97)
Этил-2-(4,6-диметил-5-нитро-2-оксопиримидин-1(2H)-ил)ацетат (500 мг), полученный в примере 6 побочный продукт, и 5% Pd-C (40 мг) суспендировали в метаноле (20 мл). Повторяли откачку и замещение водородом три раза. Затем суспензию интенсивно перемешивали при комнатной температуре (20-30°C) в течение 1 часа в атмосфере водорода. После завершения реакции смесь фильтровали через целит и фильтрат упаривали при пониженном давлении. Полученный в результате остаток и ди-трет-бутилдикарбонат (427 мг) растворяли в диметилформамиде (15 мл) и раствор перемешивали при 50°C в течение ночи. Реакционную смесь распределяли между этилацетатом и водой. Водный слой концентрировали при пониженном давлении. К полученному в результате остатку добавляли 2 н. водный раствор гидроксида натрия (4 мл) и смесь перемешивали при комнатной температуре (20-30°C) в течение 6 часов. Затем порциями добавляли 2 н. водный раствор HCl (4 мл) при охлаждении реакционной смеси и смесь концентрировали при пониженном давлении. Далее добавляли метанол и нерастворимые продукты удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении. 9,5 мг (выход 1,3%, на 5 стадий) названного в заголовке соединения синтезировали из полученного в результате остатка и 1-бензил-N-метилпиперидин-4-амина таким же образом, как в примере 64.
Пример 98
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилпропанамида (соединение 98)
Названное в заголовке соединение синтезировали из соединения 12 и 1-(циклопропилметил)-N-метилпиперидин-4-амина таким же образом, как в примере 64.
Пример 99
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилпропанамида (соединение 99)
Названное в заголовке соединение синтезировали из соединения 56 таким же образом, как в примере 71.
Пример 100
Получение N-(1-бензилпиперидин-4-ил)-2-(4,6-диметил-5-(метиламино)пиримидин-2-илокси)-N-метилпропанамида (соединение 100)
К раствору в тетрагидрофуране (1 мл) соединения 56 (72 мг) добавляли бис(триметилсилил)амид калия (0,5M раствор в толуоле: 273 мкл) при -78°C. Смесь перемешивали при -78°C в течение 30 мин и добавляли порциями метилйодид (9 мкл). Затем смесь перемешивали в течение ночи, позволяя медленно нагреваться до комнатной температуры (20-30°C). Затем к реакционной смеси добавляли насыщенный водный раствор хлорида аммония и смесь дважды экстрагировали хлороформом. Органический слой концентрировали при пониженном давлении и полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; NH-DM1020) методом колоночной хроматографии (этилацетат:гексан=1:19-1:1) с получением 7,7 мг (выход 10%) трет-бутил-2-(1-((1-бензилпиперидин-4-ил)(метил)амино)-1-оксопропан-2-илокси)-4,6-диметилпиримидин-5-ил(метил)карбамата. 2,6 мг (выход 42%) указанного в заголовке соединения синтезировали из трет-бутил-2-(1-((1-бензилпиперидин-4-ил)(метил)амино)-1-оксопропан-2-илокси)-4,6-диметилпиримидин-5-ил(метил)карбамата таким же образом, как в примере 71.
Пример 101
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклопропилметил)пиперидин-4-ил)ацетамида (соединение 101)
Названное в заголовке соединение синтезировали из соединения 58 таким же образом, как в примере 71.
Пример 102
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(4-фторбензоил)пиперидин-4-ил)ацетамида (соединение 102)
Названное в заголовке соединение синтезировали из соединения 59 таким же образом, как в примере 71.
Пример 103
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-циклопропилацетамида (соединение 103)
Названное в заголовке соединение синтезировали из соединения 60 таким же образом, как в примере 71.
Пример 104
Получение 2-(5-амино-4,6-диизопропилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида (соединение 104)
Так же, как в примере 11, карбоксильное производное синтезировали из соединения 13. Затем синтезировали названное в заголовке соединение из полученного карбоксильного производного и 1-бензил-N-метилпиперидин-4-амина таким же образом, как в примере 49.
Пример 105
Получение 2-(5-амино-4,6-диизопропилпиримидин-2-илокси)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилацетамида (соединение 105)
Так же, как в примере 11, карбоксильное производное синтезировали из соединения 13. Затем синтезировали названное в заголовке соединение из полученного карбоксильного производного и 1-(циклопропилметил)-N-метилпиперидин-4-амина таким же образом, как в примере 49.
Пример 106
Получение 2-(2-хлор-5-метилпиримидин-4-иламино)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилацетамида (соединение 106)
Названное в заголовке соединение синтезировали из соединения 14 таким же образом, как в примере 105.
Пример 107
Получение N-(1-(циклопропилметил)пиперидин-4-ил)-2-(2-(4-метоксибензиламино)-5-метилпиримидин-4-иламино)-N-метилацетамида (соединение 107)
Раствор соединения 106 (93 мг), 4-метоксибензиламина (345 мкл) и диизопропилэтиламина (45,5 мкл) в н-бутаноле (1,5 мл) перемешивали в течение ночи при нагревании до кипения. Затем реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле (метиленхлорид:метанол=50:1 - 4:1) с получением 43 мг (выход 36%) желаемого продукта в виде белого аморфного вещества.
Пример 108
Получение 2-(2-амино-5-метилпиримидин-4-иламино)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилацетамида (соединение 108)
Соединение 107 (24,9 мг) растворяли в метиленхлориде (1 мл). Добавляли трифторуксусную кислоту (3 мл) при охлаждении раствора льдом. Реакционную смесь перемешивали при комнатной температуре (20-30°C) в течение ночи. После завершения реакции реакционную смесь концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле (метиленхлорид:метанол=10:1-4:1) с получением 12,6 мг (выход 68%) желаемого продукта в виде белого аморфного вещества.
Пример 109
Получение N-(1-бензилпиперидин-4-ил)-2-(2-хлор-5-метилпиримидин-4-иламино)-N-метилацетамида (соединение 109)
Названное в заголовке соединение синтезировали из соединения 14 таким же образом, как в примере 104.
Пример 110
Получение N-(1-бензилпиперидин-4-ил)-2-(2-(4-метоксибензиламино)-5-метилпиримидин-4-иламино)-N-метилацетамида (соединение 110)
Названное в заголовке соединение синтезировали из соединения 109 таким же образом, как в примере 107.
Пример 111
Получение 2-(2-амино-5-метилпиримидин-4-иламино)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида малеат (соединение 111)
2-(2-Амино-5-метилпиримидин-4-иламино)-N-(1-бензилпиперидин-4-ил)-N-метилацетамид синтезировали из соединения 110 таким же образом, как в примере 108. Затем к метанольному раствору 2-(2-амино-5-метилпиримидин-4-иламино)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида добавляли 1 эквивалент малеиновой кислоты и смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения.
Пример 112
Получение N-(1-бензилпиперидин-4-ил)-2-(2-хлор-5-метилпиримидин-4-илокси)-N,2-диметилпропанамида малеат (соединение 112)
Названное в заголовке соединение синтезировали из соединения 16 и 1-бензил-N-метилпиперидин-4-амина таким же образом, как в примере 96.
Пример 113
Получение 2-(2-амино-4,6-диметилпиримидин-5-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида (соединение 113)
Названное в заголовке соединение синтезировали из соединения 17 таким же образом, как в примере 104.
Пример 114
Получение 2-(4-амино-5-фтор-2-оксопиримидин-1(2H)-ил)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида (соединение 114)
Названное в заголовке соединение синтезировали из соединения 18 таким же образом, как в примере 104.
Пример 115
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида гидробромид (соединение 115)
Соединение 65 (198 мг) суспендировали в метаноле (1 мл) и добавляли к суспензии бромоводородную кислоту (89 мг: 47% водный раствор) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. К полученному в результате остатку добавляли этанол (3 мл) и смесь перемешивали при кипении. Затем порциями добавляли воду (0,6 мл). Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали этанолом и сушили с получением 189 мг (выход 78%) желаемого продукта в виде белых кристаллов.
Пример 116
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 116)
Соединение 65 (200 мг) суспендировали в метаноле (1 мл) и к суспензии добавляли 4 н. раствор HCl/1,4-диоксана (130 мкл) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. Затем к полученному в результате остатку добавляли изопропанол (2,0 мл) и смесь перемешивали при кипении. Затем порциями добавляли воду (0,2 мл). Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали изопропанолом и сушили с получением 162 мг (выход 74%) желаемого продукта в виде бледно-желтых кристаллов.
Пример 117
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида малеат (соединение 117)
Соединение 65 (1,0 г) суспендировали в метаноле (5,0 мл) и к суспензии добавляли малеиновую кислоту (307 мг) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. Затем к полученному в результате остатку добавляли изопропанол (5,0 мл) и смесь перемешивали при кипении. Затем порциями добавляли воду (0,4 мл). Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали изопропанолом и сушили с получением 1,1 г (выход 86%) желаемого продукта в виде белых кристаллов.
Пример 118
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида метансульфонат (соединение 118)
Соединение 65 (202 мг) суспендировали в метаноле (1 мл) и к суспензии добавляли метансульфоновую кислоту (50,6 мг) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. Затем к полученному остатку добавляли изопропанол (1,0 мл) и смесь перемешивали при кипении. Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали изопропанолом и сушили с получением 232 мг (выход 92%) желаемого продукта в виде белых кристаллов.
Пример 119
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида нитрата (соединение 119)
Соединение 65 (186 мг) суспендировали в метаноле (1 мл) и добавляли к суспензии азотную кислоту (44,3 мг: d=1,42) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. Затем к полученному в результате остатку добавляли изопропанол (2,0 мл) и смесь перемешивали при кипении. Затем порциями добавляли воду (240 мкл). Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали изопропанолом и сушили с получением 116 мг (выход 54%) желаемого продукта в виде белых кристаллов.
Пример 120
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида тозилат (соединение 120)
Соединение 65 (201 мг) суспендировали в метаноле (1 мл) и к суспензии добавляли п-толуолсульфоновой кислоты моногидрат (99,3 мг) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. Затем к полученному в результате остатку добавляли изопропанол (2,0 мл) и смесь перемешивали при кипении. Затем порциями добавляли воду (150 мкл). Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали изопропанолом и сушили с получением 247 мг (выход 84%) желаемого продукта в виде белых кристаллов.
Пример 121
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида этансульфонат (соединение 121)
Соединение 65 (202 мг) суспендировали в метаноле (1 мл) и добавляли к суспензии этансульфоновую кислоту (58,8 мг) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. Затем к полученному в результате остатку добавляли этанол (2,0 мл) и смесь перемешивали при кипении. Затем порциями добавляли воду (100 мкл). Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали этанолом и сушили с получением 181 мг (выход 70%) желаемого продукта в виде белых кристаллов.
Пример 122
Получение 2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида бензолсульфонат (соединение 122)
Соединение 65 (200 мг) суспендировали в метаноле (1 мл) и к суспензии добавляли бензолсульфоновой кислоты моногидрат (83,4 мг) при комнатной температуре (20-30°C). После растворения соединения смесь концентрировали при пониженном давлении. Затем к полученному в результате остатку добавляли изопропанол (2,0 мл) и смесь перемешивали при кипении. Затем порциями добавляли воду (230 мкл). Когда осадок растворялся, смесь перемешивали в течение ночи, позволяя ей охладиться. Затем смесь перемешивали и охлаждали льдом в течение 1 часа и фильтровали отсасыванием. Полученное в результате твердое вещество промывали изопропанолом и сушили с получением 194 мг (выход 69%) желаемого продукта в виде белых кристаллов.
Пример 123
Получение N-(1-бензилпиперидин-4-ил)-N-метил-2-(3-метил-5-нитропиридин-2-иламино)ацетамида (соединение 123)
Названное в заголовке соединение синтезировали из 2-хлор-3-метил-5-нитропиридина и 2-амино-N-(1-бензилпиперидин-4-ил)-N-метилацетамида таким же образом, как в примере 10.
Пример 124
Получение 2-(5-амино-3-метилпиридин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 124)
Названное в заголовке соединение синтезировали из соединения 61 таким же образом, как в примере 76.
Пример 125
Получение N-(1-бензилпиперидин-4-ил)-2-(5-хлорпиридин-3-илокси)-N-метилацетамида гидрохлорид (соединение 125)
Названное в заголовке соединение синтезировали из 2-(5-хлорпиридин-3-илокси)уксусной кислоты таким же образом, как в примере 95.
Пример 126
Получение 2-(5-хлорпиридин-3-илокси)-N-(1-(циклопропилметил)пиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 126)
Названное в заголовке соединение синтезировали из 2-(5-хлорпиридин-3-илокси)уксусной кислоты и 1-(циклопропилметил)-N-метилпиперидин-4-амина таким же образом, как в примере 95.
Пример 127
Получение N-(1-бензилпиперидин-4-ил)-2-(3-хлорпиразин-2-иламино)-N-метилацетамида гидрохлорид (соединение 127)
Названное в заголовке соединение синтезировали из соединения 23 таким же образом, как в примере 95.
Пример 128
Получение N-(1-бензилпиперидин-4-ил)-2-(3-хлорпиразин-2-илокси)-N-метилацетамида гидрохлорида (соединение 128)
Названное в заголовке соединение синтезировали из соединения 26 таким же образом, как в примере 95.
Пример 129
Получение N-(1-бензилпиперидин-4-ил)-2-(6-хлорпиразин-2-илокси)-N-метилацетамида гидрохлорид (соединение 129)
Названное в заголовке соединение синтезировали из соединения 27 таким же образом, как в примере 95.
Пример 130
Получение N-(1-бензилпиперидин-4-ил)-N-метил-2-(3-(метиламино)пиразин-2-илокси)ацетамида гидрохлорид (соединение 130)
30% Раствор метиламина в этаноле (120 мг) добавляли к раствору в этаноле (1 мл) соединения 128 (100 мг) и смесь подвергали микроволновому облучению (160°C, 30 мин). Реакционную смесь концентрировали при пониженном давлении. К полученному в результате остатку добавляли воду и продукт экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; MH-DM1020) методом колоночной хроматографии (гексан:этилацетат=1:2) и превращали в гидрохлорид с получением 68,1 мг (выход 63%) указанного в заголовке соединения.
Пример 131
Получение N-(1-бензилпиперидин-4-ил)-N-метил-2-(6-(метиламино)пиразин-2-илокси)ацетамида гидрохлорид (соединение 131)
Названное в заголовке соединение синтезировали из соединения 129 таким же образом, как в примере 130.
Пример 132
Получение N-(1-бензилпиперидин-4-ил)-2-(3-(диметиламино)пиразин-2-илокси)-N-метилацетамида малеат (соединение 132)
Названное в заголовке соединение синтезировали из соединения 128 и диметиламин гидрохлориада таким же образом, как в примере 130.
Пример 133
Получение N-(1-бензилпиперидин-4-ил)-2-(6-(диметиламино)пиразин-2-илокси)-N-метилацетамида гидрохлорид (соединение 133)
Названное в заголовке соединение синтезировали из соединения 129 и диметиламин гидрохлорида таким же образом, как в примере 130.
Пример 134
Получение 2-(3-аминопиразин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамида гидрохлорид (соединение 134)
(4-Метоксифенил)метиламин (40 мг) добавляли к раствору в этаноле (1 мл) соединения 128 (100 мг) и смесь подвергали микроволновому облучению (160°C, 100 мин). Реакционную смесь концентрировали при пониженном давлении. К полученному в результате остатку добавляли воду и продукт экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; MH-DM1020) методом колоночной хроматографии (гексан:этилацетат=1:1) с получением бесцветного маслянистого вещества (46,7 мг). Затем данный продукт растворяли в хлороформе (1 мл) и добавляли к раствору трифторуксусную кислоту (1 мл). Смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении и к остатку добавляли воду. Полученный в результате продукт экстрагировали хлороформом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; MH-DM1020) методом колоночной хроматографии (этилацетат) и превращали в гидрохлорид с получением 21,5 мг (выход 21%) указанного в заголовке соединения.
Пример 135
Получение 2-(6-хлорпиридазин-3-илокси)-N-(1-(циклопропанкарбонил)пиперидин-4-ил)-N-метилацетамида (соединение 135)
Названное в заголовке соединение синтезировали из этил-2-(6-хлорпиридазин-3-илокси)ацетата и циклопропил-(4-(метиламино)пиперидин-1-ил)метанона таким же образом, как в примере 105 (выход 63%, на 2 стадии).
Пример 136
Получение N-(1-(циклопропанкарбонил)пиперидин-4-ил)-2-(6-(диметиламино)пиридазин-3-илокси)-N-метилацетамида (соединение 136)
Смесь соединения 135 (60 мг), 2M раствора диметиламина/тетрагидрофурана (766 мкл), йодида калия (2,82 мг), триэтиламина (23,7 мкл) и н-бутанола (1,0 мл) перемешивали при 110°C в течение 2 дней в запаянной пробирке. Затем реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле (этилацетат:гексан=1:3-99:1) с получением 12,1 мг (выход 19%) желаемого продукта в виде бледно-желтого аморфного вещества.
Пример 137
Получение N-(1-бензилпиперидин-4-ил)-2-(6-хлорпиридазин-3-илокси)-N-метилацетамида (соединение 137)
Названное в заголовке соединение синтезировали из этил-2-(6-хлорпиридазин-3-илокси)ацетата таким же образом, как в примере 104 (выход 48%, на 2 стадии).
Пример 138
Получение N-(1-бензилпиперидин-4-ил)-2-(6-хлорпиридазин-3-илокси)-N-циклопропилацетамида (соединение 138)
Названное в заголовке соединение синтезировали из этил-2-(6-хлорпиридазин-3-илокси)ацетата и 1-бензил-N-циклопропилпиперидин-4-амина таким же образом, как в примере 104.
Пример 139
Получение N-(1-бензилпиперидин-4-ил)-2-(3,5-дихлорпиридазин-4-иламино)-N-метилацетамида (соединение 139)
Названное в заголовке соединение синтезировали из соединения 29 таким же образом, как в примере 104.
Пример 140
Получение N-(1-бензилпиперидин-4-ил)-2-(3-хлор-5-метоксипиридазин-4-иламино)-N-метилацетамида (соединение 140)
Раствор соединения 139 (10 мг) и метилата натрия (1,3 мг) в метаноле (1 мл) перемешивали в течение ночи при кипении. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали на силикагеле с привитым амином (Fuji Sylysia Chemical Ltd.; NH-DM1020) методом колоночной хроматографии (этилацетат:гексан=1:9 - 1:1) с получением 3,0 мг (выход 30%) желаемого продукта в виде белого аморфного вещества.
Пример 141
Получение N-(1-бензилпиперидин-4-ил)-2-(5,6-дихлорпиридазин-4-иламино)-N-метилацетамида (соединение 141)
Названное в заголовке соединение синтезировали из соединения 28 таким же образом, как в примере 104.
Физические свойства соединений, полученных в примере выше, представлены в таблицах 1, 25-27.





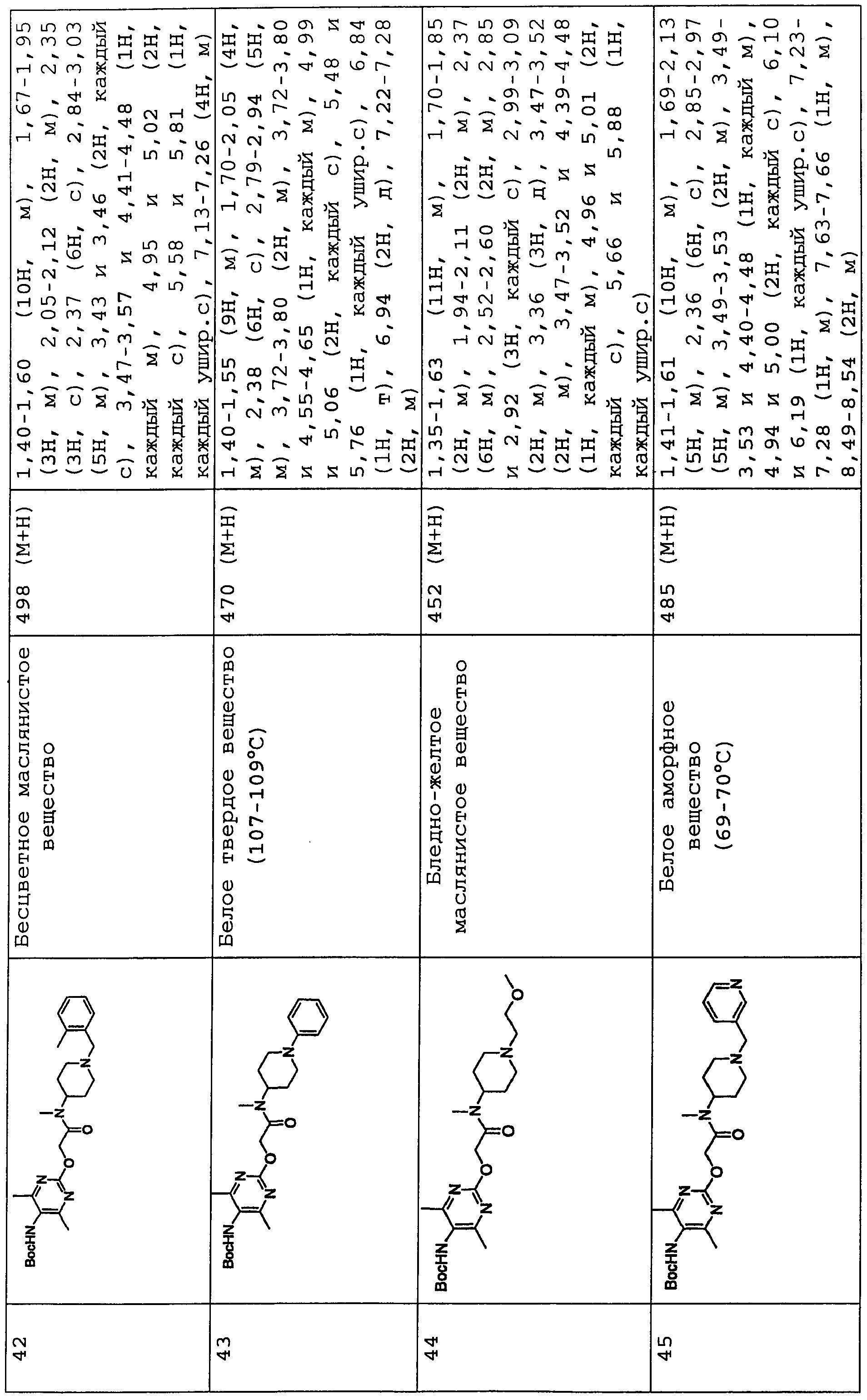

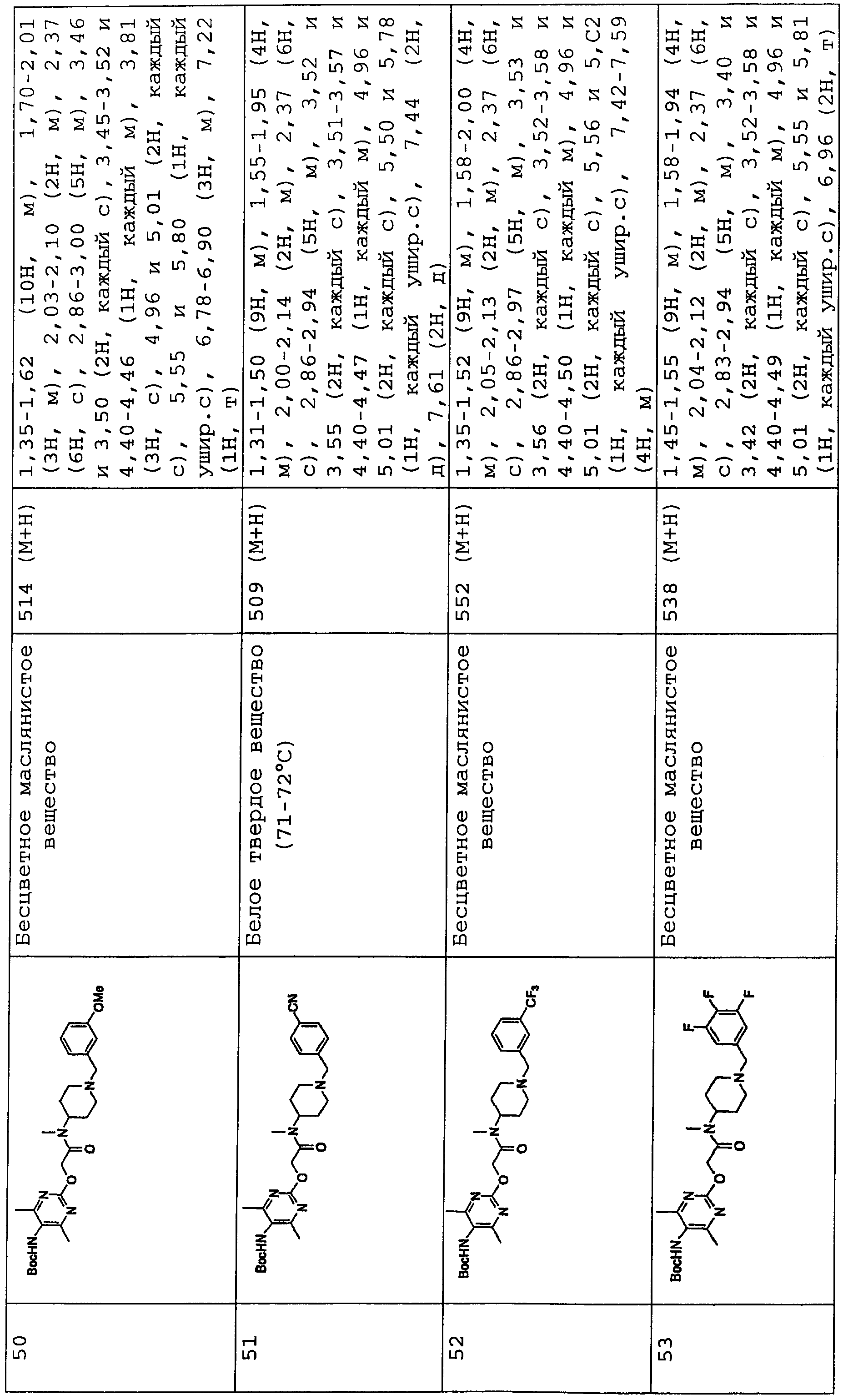
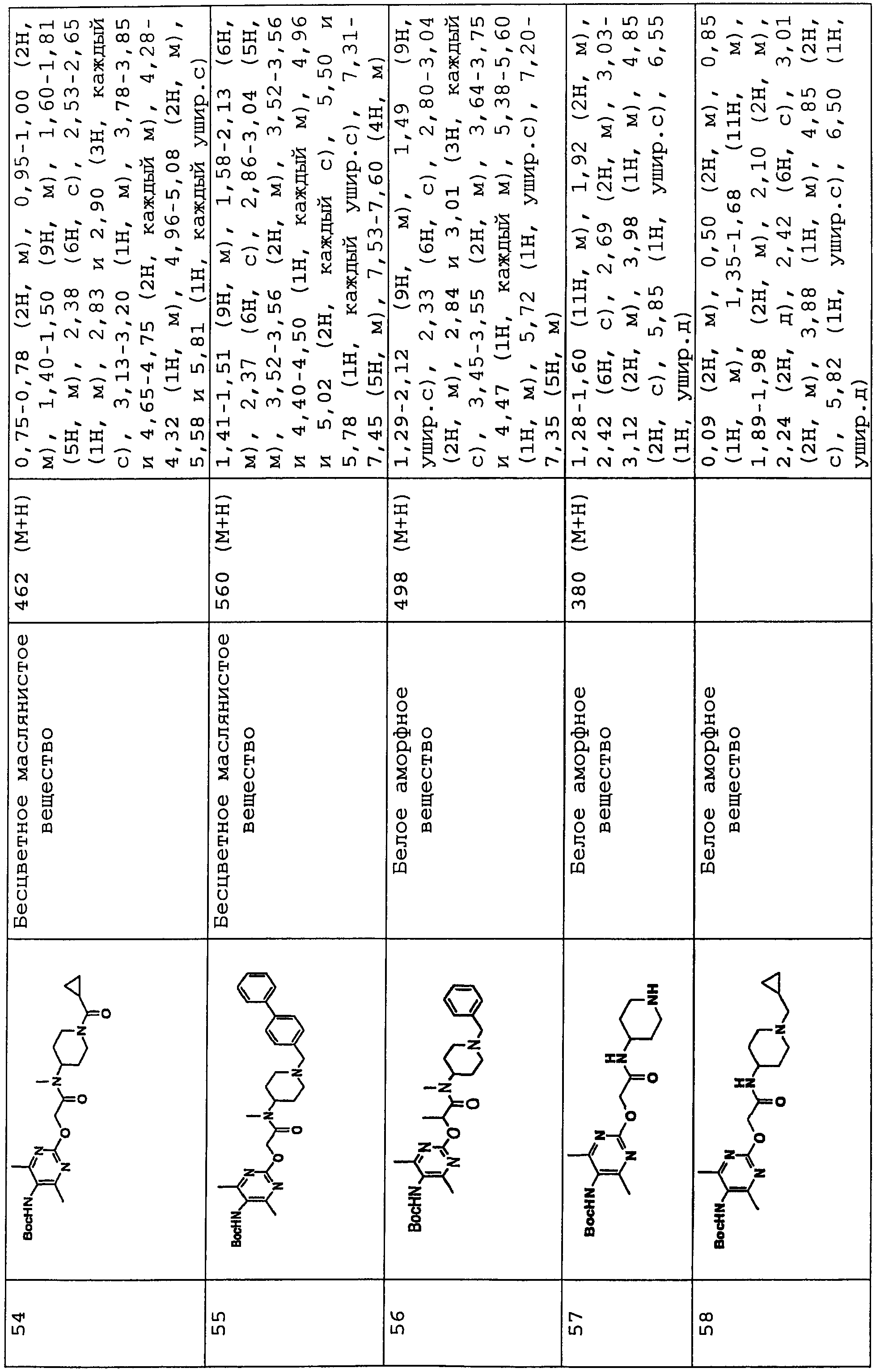

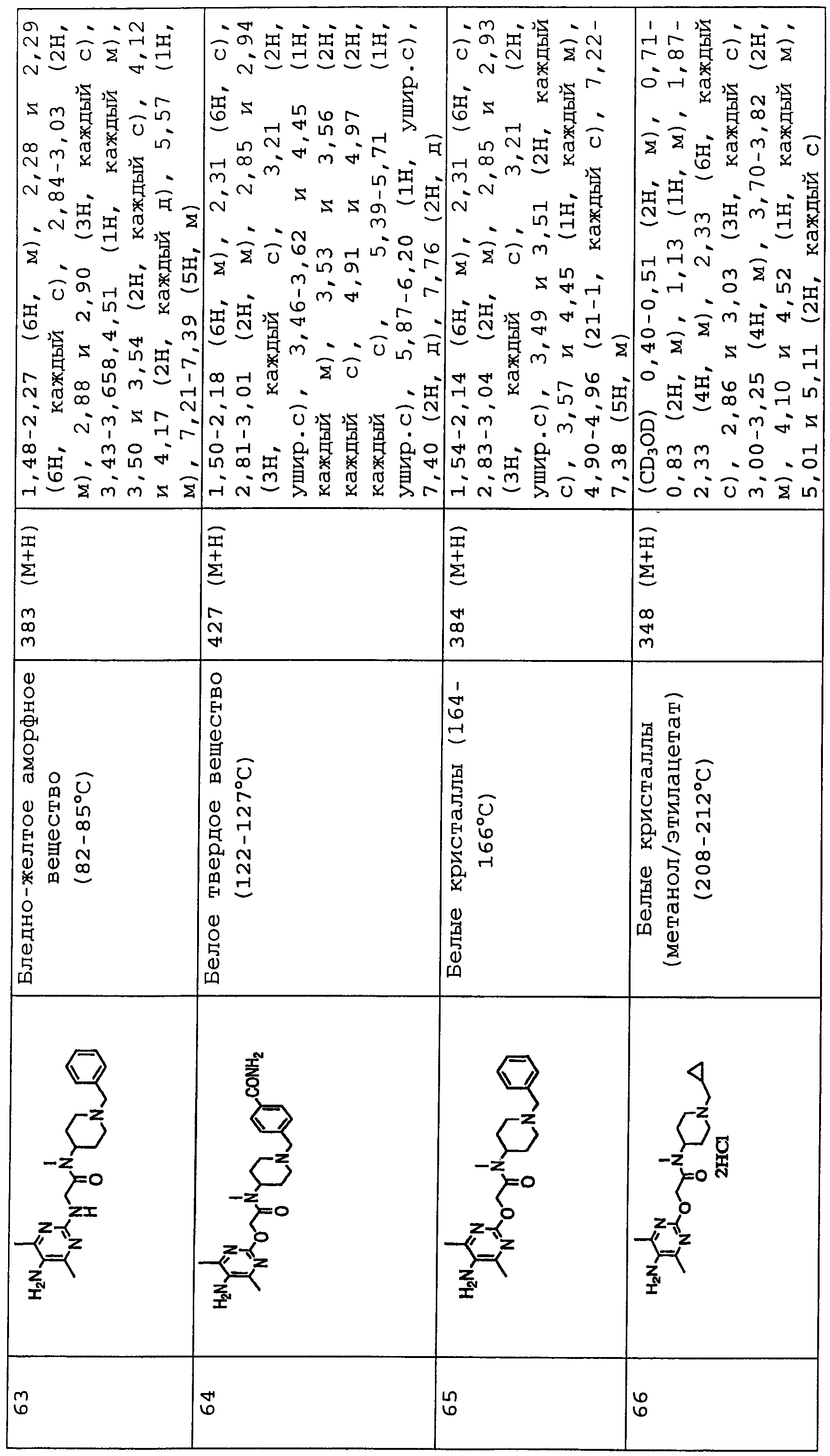
















Производные по настоящему изобретению с азотсодержащим шестичленным ароматическим кольцом, представленные формулой (I), оценивали на различную активность в следующих тестах 1-7.
Тест 1: Способность стимулировать аксональный рост.
Тест 2: Способность стимулировать ангиогенез.
Тест 3: Способность стимулировать рост нейритов нейронов спинного мозга.
Тест 4: Оценка ангиогенеза на модели ишемизированной нижней конечности у мыши (критическая ишемия конечностей).
Тест 5: Способность улучшать сердечные функции на модели инфаркта миокарда у крыс.
Тест 6: Способность улучшать функции на модели, индуцированной микросферами (микроэмболии), у крыс.
Тест 7: Способность улучшать функции на модели поражения спинного мозга у крыс.
Конкретные методы для оценки описаны ниже.
Пример теста 1: Способность стимулировать аксональный рост
В соответствии с методом М.П. Маттсона [M. P. Mattson, Brain Res. Rev., 13, 179, (1988)] у 18-дневных эмбрионов самок крыс Вистар (Wistar) выделяли область гиппокампала и нейроны отделяли друг от друга с помощью системы диссоциации с папаином. Гиппокампальные нейроны суспендировали в 5% Nu-serum + B27 supplement + среда Neurobasal при 5×104 клеток/лунку/2 мл. 2 мл суспензии клеток засевали на покрытую поли-D-лизином чашку диаметром 36 мм и инкубировали нейроны при 37°С в инкубаторе с 5% CO2.
На третий день инкубационного периода 1 мл культуральной среды заменяли на 1 мл 5% Nu-serum + B27 supplement + среда Neurobasal, содержащей 2 мМ AraC. Каждое тестируемое соединение добавляли к культуре на 3 день и 7 день инкубационного периода. На 9 день клетки фиксировали 4% раствором параформальдегида. Аксоны нейронов фиксированных клеток помечали поликлональным антителом кролика против белка 43, ассоциированного с ростом нейронов, и помеченные клетки количественно определяли с помощью программного обеспечения Kurabo для подсчета отростков нейронов.
Способность (%) каждого тестируемого соединения стимулировать аксональный рост рассчитывали по следующему уравнению:
Способность стимулировать аксональный рост (%)=[(результат измерения для представляющего интерес соединения)/(результаты измерения для контроля)]×100
Соединения примеров, представленные в таблице 25, использовали в качестве тестируемых соединений и каждое добавляли в концентрациях 3 мкМ и 0,3 мкМ.
Результаты представлены в таблице 25. Хотя результаты измерений, представленные в таблице 25, главным образом приведены для концентрации 3 мкМ, результаты измерений, полученные в случае концентрации 0,3 мкМ, представлены для соединений, которые давали результат измерений 120 (величина, отражающая значительный эффект) или выше лишь при концентрации 0,3 мкМ, или соединений, которые давали значительно более высокий результат измерений при 0,3 мкМ, чем при 3 мкМ.
Так как bFGF (основной фактор роста фибробластов) приводит в данном тесте к результату измерений приблизительно 120, 120 использовали в качестве пороговой величины для определения, обладает ли соединение способностью стимулировать аксональный рост.
Как может быть видно из таблицы 25, каждое из соединений примеров по настоящему изобретению показывало результат приблизительно 120 (величина, отражающая значительный эффект) или выше, демонстрируя значительную способность стимулировать аксональный рост.
Пример теста 2: способность стимулировать ангиогенез
Используя набор для тестирования ангиогенеза, содержащий смесь эндотелиальных клеток сосудов человека и фибробластов [KZ-1000; Kurabo], оценивали способность тестируемых соединений стимулировать ангиогенез.
Каждое тестируемое соединение добавляли на 1, 4 и 7 дни инкубационного периода. На 9 день клетки подвергали иммунному окрашиванию с помощью CD31 и фотографировали. Контрольные клетки культивировали в тех же самых условиях, только без добавления тестируемого соединения, и фотографировали.
Размер просвета кровеносного сосуда количественно определяли из полученных изображений с помощью программного обеспечения для количественного анализа ангиогенеза. Способность (%) каждого тестируемого соединения стимулировать ангиогенез рассчитывали в следующем уравнении:
Способность стимулировать ангиогенез (%)=[(результат измерения для представляющего интерес соединения)/(результаты измерения для контроля)]×100
Соединения Примеров, представленные в таблице 26, использовали в качестве тестируемых соединений.
Результаты приведены в таблице 26. Хотя результаты измерений, представленные в таблице 26, главным образом приведены для концентрации 3 мкМ, результаты измерений, полученные в случае концентрации 0,3 мкМ, представлены для соединений, которые давали результат измерений 120 (величина, отражающая значительный эффект) или выше лишь при концентрации 0,3 мкМ, или соединений, которые давали значительно более высокий результат измерений при 0,3 мкМ, чем при 3 мкМ.
Так как bFGF приводит в данном Тесте к результату измерений приблизительно 120, 120 использовали в качестве пороговой величины для определения, обладает ли данное соединение способностью стимулировать ангиогенез.
Как может быть видно из таблицы 26, каждое из соединений примеров по настоящему изобретению показывало результат приблизительно 120 (величина, отражающая значительный эффект) или выше, демонстрируя значительную способность стимулировать ангиогенез.
Пример теста 3: Способность стимулировать рост нейритов нейронов спинного мозга
В соответствии с методом Мартина Г. Хансона младшего [Martin G. Hanson Jr., J. Neurosci., 1998 Sep. 15; 18(18): 7361-71] у 15-дневных эмбрионов самок крыс Вистар (Wistar) выделяли область спинного мозга и клетки отделяли друг от друга с помощью системы диссоциации с папаином. Нейроны собирали в градиенте плотности 6,8% метризамида и суспендировали в 2% ФБС + среда L-15 (Leibovitz), так что плотность клеток была 2×103 клеток/лунку/200 мкл. 200 мкл клеточной суспензии засевали на покрытые поли-D-лизин/ламинином 96-луночный планшет и планшет инкубировали в инкубаторе при 37°C. Тестируемые соединения добавляли через 1 час после осаждения клеток.
На 3 день инкубационного периода нейроны окрашивали Кальцеином АМ и определяли длину нейритов с помощью анализатора клеток IN (GE Healthcare Bioscience).
Способность (%) каждого тестируемого соединения стимулировать рост нейритов рассчитывали по следующему уравнению:
Способность стимулировать рост нейритов (%)=[(результат измерения для представляющего интерес соединения)/(результаты измерения для контроля)]×100
Соединения примеров 65, 71, 74, 92, 130 и 135 использовали в качестве тестируемых соединений и каждое добавляли в концентрации 0,1 мкМ.
Результаты представлены в таблице 27. bFGF приводит в данном тесте к результату измерений приблизительно 130. Значение 120 использовали в качестве пороговой величины для определения, что данное соединение обнаруживает значительный эффект.
Как может быть видно из таблицы 27, каждое из соединений примеров по настоящему изобретению показывало результат приблизительно 120 (величина, отражающая значительный эффект) или выше, демонстрируя значительную способность стимулировать рост нейритов нейронов спинного мозга.
Пример теста 4: Оценка ангиогенеза на модели ишемизированной нижней конечности у мыши (критическая ишемия конечностей)(тест in vivo)
В соответствии с методом Ичиро Масаки [Ichiro Masaki, Circ Res.90; 966-973 (2002)] у мыши Balb/c иссекали левую бедренную артерию для создания модели критической ишемии конечностей (n=29-30).
Используемыми тестируемыми соединениями были соединения примера 65, 92, 114, 130, 135 и 136, каждое из которых перорально вводили один раз в день в дозе 0,1 мг/кг или 1,0 мг/кг в течение 10 дней, начиная со дня иссечения бедренной артерии. При наблюдении некроза в нижней конечности животного наблюдение за особью останавливали и следили за выживаемостью нижней конечности с течением времени.
Результаты представлены на фиг.1-6, где на фиг.1, 2, 3, 4, 5 и 6 представлены результаты для соединений примеров 65, 92, 114, 130, 135 и 136 соответственно.
На фигурах толстой сплошной линией обозначена группа с носителем, тонкой пунктирной линией обозначена группа, которой вводили 0,1 мг/кг/день каждого соединения, и тонкой сплошной линией обозначена группа, которой вводили 1,0 мг/кг/день каждого соединения.
*: P<0,05, **: P<0,01, ***: P<0,001 относительно носителя в тесте Вилкоксона.
Результаты указывают, что каждое из соединений примера 65, 92, 114, 130, 135 и 136 по настоящему изобретению приводит в результате к увеличению выживаемости бедренной артерии/вены нижней конечности и поэтому является высокоэффективным в улучшении состояния при критической ишемии конечностей.
Пример теста 5: Способность улучшать сердечные функции на модели инфаркта миокарда у крыс
В соответствии с методом Масакатсу Вакено и др. [Masakatsu Wakeno et al., Circulation 114: 1923-1932 (2006)], самцов крыс Slc:SD 10-недельного возраста анестезировали пентобарбиталом и вскрывали в грудной области. Полностью окклюдировали переднюю межжелудочковую ветвь левой коронарной артерии (LAD) для создания модели инфаркта миокарда. Ложнооперированную группу подвергали только операции на грудной клетке.
Используемым тестируемым соединением было соединение Примера 65, которое вводили перорально один раз в день в дозе 0,01 мг/кг, 0,1 мг/кг или 1,0 мг/кг в течение 28 дней, начиная со дня создания модели инфаркта миокарда (после выведения из анестезии).
Через 4 недели после создания инфаркта миокарда животных анестезировали пентобарбиталом и контролировали их сердечную гемодинамику с помощью оптического катетера. Затем собирали образцы крови из брюшной аорты, определяли величину отека легких и измеряли уровни МНП (BNP, мозговой натрийуретический пептид) в плазме крови (RIA) после забора образцов артериальной крови.
Результаты представлены на фиг.7-10.
На фиг.7, 8, 9 и 10 показаны результаты в случае LVEDP (КДД ЛЖ, конечное диастолическое давление в левом желудочке), LVdP/dt max, массы легкого и уровней МНП в плазме крови соответственно.
#: P<0,05, ##: P<0,01 между ложнооперированными и контролем по t-критерию Стьюдента.
*: P<0,05, **: P<0,01: относительно контроля по критерию Даннетта.
Соединение примера 65 сдерживало снижение максимального сокращения левого желудочка и диастолической скорости после инфаркта миокарда, увеличение конечного диастолического давления и увеличение величины отека легких, признак застоя крови в легких. Соединение также сдерживало увеличение уровней МНП в плазме крови. Данные результаты демонстрируют, что соединение Примера 65 по настоящему изобретению является высокоэффективным в улучшении состояния снижения сердечных функций и сердечной недостаточности, связанных с инфарктом миокарда.
Пример теста 6: Способность улучшать функции на модели, индуцированной микросферами (микроэмболии), у крыс
В соответствии с методом Ичиро Датэ [Ichiro Date et al., Journal of Neuroscience Research 78; 442-453 (2004)] использовали самцов крыс Crl:CD(SD) в возрасте 8-недель. Животных подвергали анестезии с помощью галотана и делали срединный разрез. Правую сонную артерию отделяли и обматывали нитью общую сонную артерию и наружную сонную артерию. Крылонебную артерию временно зажимали с помощью клипсы для аневризмы. Ток крови через общую сонную артерию и наружную сонную артерию временно останавливали и инъецировали в общую сонную артерию 0,1 мл суспензии микросфер, состоящей из микросфер, диспергированных в 30% растворе сахарозы (1800 микросфер/0,1 мл). После инъекции суспензии укол от инъекционной иглы заполняли мгновенно схватывающимся клеем, пока ток крови был остановлен. Затем ток крови восстанавливали и операционную рану зашивали. Наблюдали неврологические симптомы на следующий день после операции. Животных определяли в каждую группы лечения случайным образом с учетом оценки неврологических симптомов, снижения массы тела с предыдущего дня и массы тела на данный день.
Используемыми тестируемыми соединениями были соединения примеров 65, 108, 130, 135 и 137, каждое из которых вводили в дозе 1,0 мг/кг. Соединение примера 65 также вводили в дозе 0,1 мг/кг. Каждое соединение вводили перорально один раз в день в течение 10 дней, начиная со следующего после операции дня. Координированные двигательные функции оценивали 5-кратно на ускоряющемся вращающемся стержне (4-40 (об/мин)/5 мин) 2 недели после операции.
В качестве стандартного параметра на фиг.11-15 суммировано общее время 5 измерений на вращающемся стержне.
В частности, на фиг.11-15 демонстрируется эффект различных соединений 2 недели после инъекции микросфер (стандартный параметр теста «вращающийся стержень»). На фигурах представлены результаты тестов «вращающегося стержня» 2 недели после события ишемии, в которых каждое соединение вводили перорально при 1,0 мг/кг один раз в день в течение 10 дней (соединение примера 65 вводили при 0,1 мг/кг или 1,0 мг/кг). То, что представлено на фигурах, является общим временем 5 измерений на вращающемся стержне в качестве стандартного параметра.
На фиг.11, 12, 13, 14 и 15 показаны результаты для соединений примеров 65, 108, 130, 135 и 137 соответственно.
***: p<0,0001, по методу множественных сравнений Даннетта относительно носителя.
Данные результаты свидетельствуют о том, что каждое соединение примеров 65, 108, 130, 135 и 137 по настоящему изобретению является высокоэффективным в улучшении состояния при ишемическом инфаркте мозга и его последствий.
В соответствии со статьей Крафта с сотрудниками (Kraft et al., Eur. J. Neurosci. 2005 Jun; 21(11):3117-32), эксперимент примера теста 6, использующий микросферы для достижения инфаркта мозга (ишемического инсульта), вызванного эмболией, служит подходящей моделью для микрососудистых нарушений - признака когнитивного расстройства. Таким образом, результаты примера теста 6 могут указывать на то, что соединения по настоящему изобретению являются эффективными в улучшении состояния при деменции.
Пример теста 7: Способность улучшать функции в модели поражения спинного мозга (ПСМ, SCI) у крыс
В соответствии с методом Дасари [Venkata Ramesh Dasari et al., J Neurotrauma. 2007 Feb; 24 (2):391-410] использовали самок крыс Slc: SD 9-недельного возраста. Животных анестезировали с помощью пентобарбитала и делали разрез вдоль средней линии спины. После того как обнажился позвоночник при разрезе средней линии спины, удаляли дугу 9-го грудного позвоночника с помощью костных ножниц, сосредоточив внимание на том, чтобы не повредить твердую оболочку. Верхнюю и нижнюю кости закрепляли в таком положении, чтобы поместить оголенный участок позвоночника прямо под устройством, осуществляющим ударную нагрузку (где прикладывается ударная нагрузка), и затем к этому месту прикладывали силу 200 кдин. Контролировали тоническое вытягивание нижних конечностей при поражении спинного мозга.
Используемым тестируемым соединением было соединение примера 65, которое вводили в дозе 1 мг/кг. В частности, соединение вводили через хвостовую вену один раз в день в течение 10 дней, начиная с 90 мин после операции. Моторную функцию нижней конечности оценивали в шкале BBB (Basso DM, Beattie MS, Bresnahan JC:J Neurotrauma, 1995; 12:1-21) раз в неделю до 5 недели после операции. Результаты представлены на фиг.16.
Результаты свидетельствуют о том, что соединение примера 65 по настоящему изобретению является высокоэффективным в улучшении состояния при поражении спинного мозга.
Промышленная применимость
Соединения по настоящему изобретению обладают способностью стимулировать аксональный рост в сочетании со способностью стимулировать ангиогенез и поэтому являются эффективными в уменьшении или лечении поражения центральной нервной системы, такого как травма головы и поражение спинного мозга, ишемический инсульт, ишемические заболевания сердца, такие как инфаркт миокарда и стенокардия, обусловленная органическим поражением, периферические окклюзионные поражения артерий, такие как критическая ишемия конечностей, или последствий данных заболеваний или других заболеваний, против которых соединения настоящего изобретения считаются эффективными. Настоящее изобретение поэтому имеет большое медицинское значение.
Краткое описание чертежей
Фиг.1 представляет собой диаграмму, демонстрирующую результаты для соединения примера 65 в примере теста 4.
Фиг.2 представляет собой диаграмму, демонстрирующую результаты для соединения примера 92 в примере теста 4.
Фиг.3 представляет собой диаграмму, демонстрирующую результаты для соединения примера 114 в примере теста 4.
Фиг.4 представляет собой диаграмму, демонстрирующую результаты для соединения примера 130 в примере теста 4.
Фиг.5 представляет собой диаграмму, демонстрирующую результаты для соединения примера 135 в примере теста 4.
Фиг.6 представляет собой диаграмму, демонстрирующую результаты для соединения примера 136 в примере теста 4.
Фиг.7 представляет собой диаграмму, демонстрирующую результаты в случае LVEDP в примере теста 5.
Фиг.8 представляет собой диаграмму, демонстрирующую результаты в случае LVdP/dt max в примере теста 5.
Фиг.9 представляет собой диаграмму, демонстрирующую результаты для измерения массы легких в примере теста 5.
Фиг.10 представляет собой диаграмму, демонстрирующую результаты в случае уровней МНП в плазме крови в примере теста 5.
Фиг.11 представляет собой диаграмму, демонстрирующую результаты для соединения примера 65 в примере теста 6.
Фиг.12 представляет собой диаграмму, демонстрирующую результаты для соединения примера 108 в примере теста 6.
Фиг.13 представляет собой диаграмму, демонстрирующую результаты для соединения примера 130 в примере теста 6.
Фиг.14 представляет собой диаграмму, демонстрирующую результаты для соединения примера 135 в примере теста 6.
Фиг.15 представляет собой диаграмму, демонстрирующую результаты для соединения примера 137 в примере теста 6.
Фиг.16 представляет собой диаграмму, демонстрирующую результаты примера теста 7.
Реферат
Изобретение относится к новым производным пиримидина общей формулы (I-а) и его фармацевтически приемлемой соли, которые обладают способностью стимулировать аксональный рост в сочетании со способностью стимулировать ангиогенез и могут быть использованы при лечении поражения спинного мозга, поражения центральной нервной системы в результате травмы головы, ишемического инсульта, ишемического заболевания сердца, периферического окклюзионного поражения артерий, мультиинфарктной деменции, цереброваскулярной деменции или старческого слабоумия. В соединении формулы (I-а):Rпредставляет собой группу -NRR, где Rи Rпредставляют собой атом водорода; Rпредставляет собой метильную группу; Rпредставляет собой метильную группу; Rпредставляет собой атом водорода; Rпредставляет собой атом водорода; Rпредставляет собой метильную группу; Е представляет собой атом кислорода;представляет собой бензильную группу, циклогексилметильную группу, изобутильную группу, циклогексанкарбонил, ацетильную группу, фенилсульфонильную группу, циклогексильную группу, пиперидин-1-карбонильную группу, метилбензильную группу, фенильную группу, фторбензильную группу, метоксибензильную группу. 3 н. и 1 з.п. ф-лы, 16 ил., 27 табл., 148 пр.
Формула

где
R0 представляет собой группу -NR3R4, где R3 и R4 представляют собой атом водорода;
R1 представляет собой метильную группу;
R2 представляет собой метильную группу;
R5 представляет собой атом водорода;
R6 представляет собой атом водорода;
R7 представляет собой метильную группу;
Е представляет собой атом кислорода;

или его фармацевтически приемлемая соль.
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-бензилпиперидин-4-ил)-N-метилацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклогексилметил)пиперидин-4-ил)-N-метилацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-изобутилпиперидин-4-ил)-N-метилацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(циклогексанкарбонил)пиперидин-4-ил)-N-метилацетамида гидрохлорид,
N-(1-ацетилпиперидин-4-ил)-2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метилацетамида гидрохлорид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(фенилсульфонил)пиперидин-4-ил)ацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-циклогексилпиперидин-4-ил)-N-метилацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(пиперидин-1-карбонил)пиперидин-4-ил)ацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(2-метилбензил)пиперидин-4-ил)ацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-фенилпиперидин-4-ил)ацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(4-фторбензил)пиперидин-4-ил)-N-метилацетамид,
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-(1-(3-метоксибензил)пиперидин-4-ил)-N-метилацетамида гидрохлорид, и
2-(5-амино-4,6-диметилпиримидин-2-илокси)-N-метил-N-(1-(3,4,5-трифторбензил)пиперидин-4-ил)ацетамид.
Документы, цитированные в отчёте о поиске
N-замещенные производные пиперидина в качестве агентов серотонинового рецептора
Комментарии