Ксантиновое производное - RU2635109C2
Код документа: RU2635109C2
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области фармацевтической химии, в частности, к классу замещенных ксантиновых производных, способу его получения и его применению в качестве терапевтических средств, в частности, в качестве ингибиторов дипептидилпептидазы IV (DPP-IV).
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Диабет представляет собой заболевание обмена веществ, вызываемое многими причинами, которое характеризуется хроническим высоким уровнем глюкозы в крови, сопровождающееся нарушением обмена Сахаров, жиров и белков, вызванным недостатком секреции и/или действия инсулина. Диабет также является давно существующим заболеванием, которое вследствие относительного или абсолютного недостатка инсулина в организме человека вызывает повышенную концентрацию глюкозы в крови, что приводит к выделению сахара в больших количествах с мочой, и сопровождается патологически усиленной жаждой, полиурией, полифагией, потерей веса, головокружением, утомляемостью и другими симптомами.
При лечении диабета лечебная физкультура и диетотерапия являются двумя основными методами лечения диабета. Если эти два метода лечения не являются эффективными для контроля болезненного состояния, могут применяться инсулин или пероральные гипогликемические средства. Однако, поскольку данные существующие гипогликемические средства обладают слишком большим количеством побочных эффектов, особо важным является разработка нового лекарственного средства с меньшим количеством побочных эффектов и большим количеством терапевтических эффектов при лечении диабета.
Дипептидилпептидаза IV (DPP-IV) представляет собой сериновую протеазу, которая может избирательно расщеплять N-концевой дипептид пептидной цепи, содержащий один остаток пролина в предпоследнем положении от N-конца. Несмотря на то что физиологический эффект DPP-IV у млекопитающих не был полностью подтвержден, он играет важную роль в нейропептидном обмене, активации Т-клетки, адгезии раковых клеток и эндотелия, процессе вхождения ВИЧ в лимфоциты и других процессах (см. WO 98/19998).
Исследования показали, что DPP-IV может привести к снижению глюкагоноподобного пептида (GLP-1), т.е., путем расщепления дипептида на основе гистидин-аланин на N-конце GLP-1, при этом GLP-1 в его активной форме может разлагаться до неактивного амида GLP-1-(7-36), который дополнительно разлагается до неактивного амида GLP-1-(9-36) (см. Hansen L, Deacon CF, Orskov С, et al, Endocrinology, 1999, 140: 5356-5363). В физиологических условиях период полувыведения исходного GLP-1 в кровоток является очень коротким, а неактивные метаболиты, полученные после распада GLP-1 посредством DPP-IV, могут связываться с рецептором GLP-1 с порождением антагонизма GLP-1 с тем, чтобы сократить физиологические реакции рецептора GLP 1 на GLP-1, при этом ингибиторы DPP-IV могут полностью защитить эндогенный и даже экзогенный GLP-1 от инактивации посредством DPP-IV, и таким образом могут значительно повышать физиологическую активность GLP-1 (в 5-10 раз). GLP-1 является важным стимулирующим веществом для секреции панкреатического инсулина и может непосредственно влиять на распределение глюкозы, следовательно, ингибиторы DPP-IV играют очень положительную роль при лечении пациентов с инсулиннезависимым диабетом (US 6110949).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
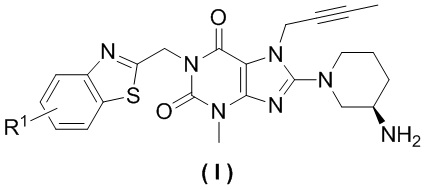
Настоящее изобретение относится к замещенным ксантиновым производным, а также к способу их получения и их медицинскому применению, в частности, к замещенным ксантиновым производным, представленным общей формулой (I), или их фармацевтически приемлемой соли, а также к их применению при получении лекарственного препарата для лечения заболеваний, связанных с DPP-IV. Более конкретно, указанное применение заключается в получении лекарственного препарата для лечения диабета II типа или заболеваний, связанных с нарушением толерантности к глюкозе. Одной из целей настоящего изобретения является обеспечение замещенного ксантинового производного, характеризующегося структурой, представленной следующей общей формулой (I), или его фармацевтически приемлемой соли,

где R1 выбран из атома водорода, атома фтора, атома хлора, атома брома, атома йода или цианогруппы.
При этом R1 предпочтительно представляет собой заместитель в положении 5 (1,3-бензотиазол-2-ил)метила и R1 дополнительно предпочтительно выбран из атома водорода, атома фтора или атома хлора.
Замещенное ксантиновое производное по настоящему изобретению предпочтительно характеризуется структурой, представленной следующей общей формулой (II),
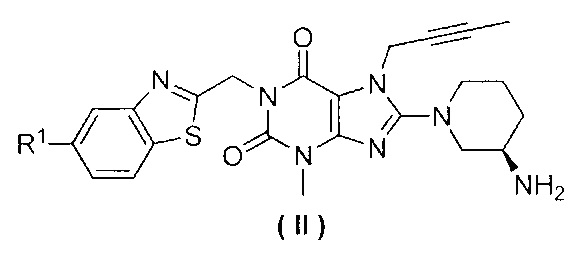
При этом R1 выбран из атома водорода, атома фтора или атома хлора.
Наиболее предпочтительно замещенное ксантиновое производное по настоящему изобретению представляет собой следующее соединение,


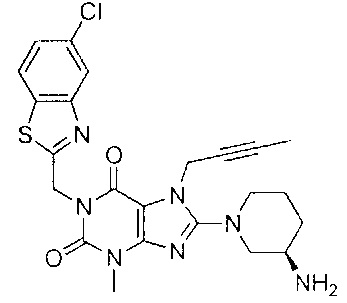
Фармацевтически приемлемые соли по настоящему изобретению представляют собой соли, образованные соединениями по настоящему изобретению и кислотами, выбранными из следующих: соляной кислоты, п-толуолсульфоновой кислоты, винной кислоты, малеиновой кислоты, молочной кислоты, метансульфоновой кислоты, серной кислоты, фосфорной кислоты, лимонной кислоты, уксусной кислоты или трифторуксусной кислоты; при этом предпочтительно кислота представляет собой соляную кислоту, п-толуолсульфоновую кислоту, трифторуксусную кислоту или винную кислоту.
Более конкретно, замещенные ксантиновые производные по настоящему изобретению или их фармацевтически приемлемые соли представляют собой:
1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидии-1-ил]-ксантин;
1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантин;
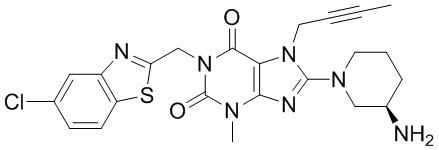
1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантин;
1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина гидрохлорид;
1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидип-1-ил]-ксантина гидрохлорид.
Другой целью настоящего изобретения является обеспечение способа получения указанных выше замещенных ксантиновых производных или их фармацевтически приемлемой соли, включающего следующие стадии.
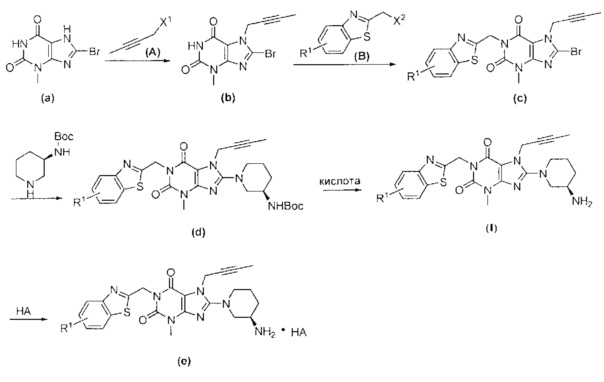
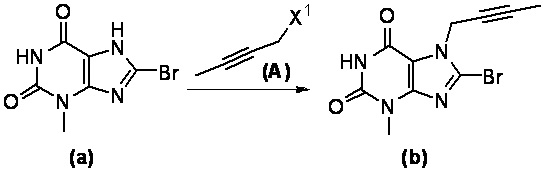
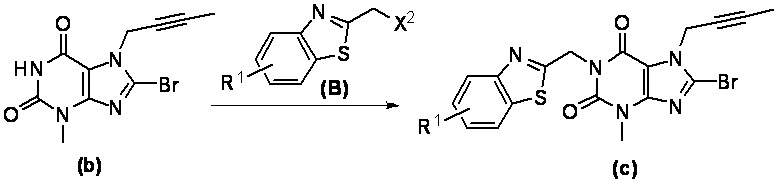
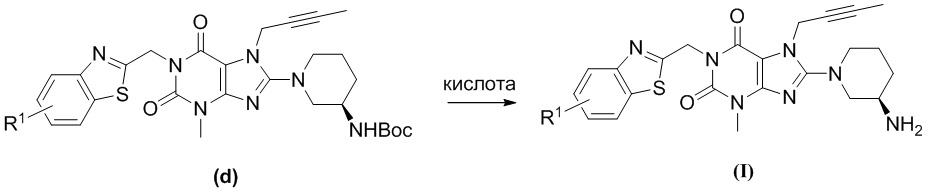
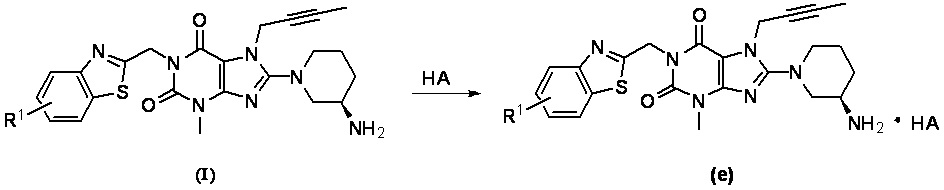
При комнатной температуре (10~25°C) осуществляют реакцию исходного сырьевого материала а с сырьевым материалом А; образованное промежуточное соединение b дополнительно подвергают реакции замещения с сырьевым материалом В с образованием промежуточного соединения с; промежуточное соединение с и (R)-3-трет-бутоксикарбонил-аминопиперидин подвергают реакции при условии нагревания (50~100°C) с образованием промежуточного соединения d; промежуточное соединение d подвергают снятию защитной группы в кислых условиях с получением целевого соединения I в форме свободного основания и необязательно целевое соединение 1 дополнительно подвергают реакции с кислотой с получением соответствующей соли е.
При этом в сырьевом материале А X1 представляет собой уходящую группу, причем указанный X1 предпочтительно представляет собой Cl, Br или I; при этом в сырьевом материале В X2 представляет собой уходящую группу, причем указанный X2 предпочтительно представляет собой Cl, Br или I; при этом кислота, применяемая для удаления защитной группы Вое предпочтительно представляет собой соляную кислоту или трифторуксусную кислоту.
Другой целью настоящего изобретения является обеспечение применения описанных выше замещенных ксантиновых производных или их фармацевтически приемлемой соли в качестве терапевтического средства, в частности, в качестве активного ингибитора DPP-IV в области медицины.
Конкретно настоящее изобретение относится к применению указанных выше замещенных ксантиновых производных или их фармацевтически приемлемой соли при получении лекарственного препарата для лечения заболеваний, связанных с DPP-IV. Более конкретно, настоящее изобретение относится к применению указанных выше замещенных ксантиновых производных или их фармацевтически приемлемой соли при получении лекарственного препарата для лечения диабета II типа или заболеваний, связанных с нарушением толерантности к глюкозе.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет описано более подробно с помощью примеров, но описание не предназначено для ограничения объема правовой охраны настоящего изобретения, поскольку какой-либо эквивалент в данной области в соответствии с раскрытием настоящего изобретения входит в объем настоящего изобретения.
Структуры соединений подтверждены с помощью масс-спектрометрии (MS) или с помощью ядерно-магнитного резонанса (1Н ЯМР). Смещение (5) ядерно-магнитного резонанса (1Н ЯМР) приведено в единице частей на миллион (ppm); измерение с помощью ядерно-магнитного резонанса (1Н ЯМР) проводят на ЯМР-анализаторе Bruker AVANCE-300, при этом растворитель для измерения представляет собой гексадейтерированный диметилсульфоксид (DMSO-d6), а внутренний стандарт представляет собой тетраметилсилан (TMS).
Измерение с помощью масс-спектрометрии (MS) проводят на масс-спектрометре FINNIGAN LCQAd (ESI) (производитель: Therm, тип: Finnigan LCQ advantage MAX).
Значения IC50 определяют посредством En Vision (PerkinElmer Corporation).
Пластины силикагеля Yantai Huanghai HSGF254 или Qingdao GF254 применяются в качестве тонкого слоя силикагеля.
Если не указано иное, реакции, указанные в настоящем изобретении, проводят в атмосфере азота.
В настоящем изобретении выражение "атмосфера азота" относится, например, к соединению реакционной колбы с баллоном с азотом с объемом в 1 л.
Если не указано иное, растворы, указанные в реакции в соответствии с настоящим изобретением, относятся к водным растворам.
В настоящем изобретении выражение "комнатная температура" относится к температуре от 10°C до 25°C.
В одном варианте осуществления настоящее изобретение относится к замещенным ксантиновым производным, характеризующимся структурой, представленной общей формулой (I), или их фармацевтически приемлемой соли,
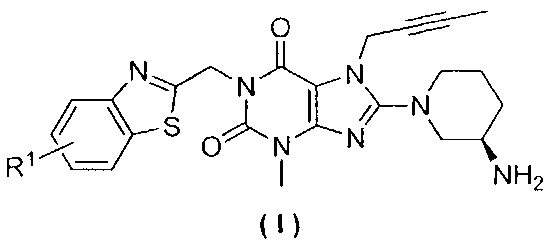
где R1 выбран из атома водорода, атома фтора, атома хлора, атома брома, атома йода или цианогруппы, при этом R1 предпочтительно находится в положении 5 (1,3-бензотиазол-2- ил)метила, причем R1 дополнительно предпочтительно выбран из атома водорода, атома фтора или атома хлора.
В предпочтительном варианте осуществления упомянутые выше фармацевтически приемлемые соли образованы замещенными ксантиновым производным по настоящему изобретению и одной или несколькими кислотами, выбранными из следующих: соляной кислоты, п-толуолсульфоновой кислоты, винной кислоты, малеиновой кислоты, молочной кислоты, метансульфоновой кислоты, серной кислоты, фосфорной кислоты, лимонной кислоты, уксусной кислоты или трифторуксусной кислоты. Более предпочтительно кислота выбрана из соляной кислоты, п-толуолсульфоновой кислоты, трифторуксусной кислоты, винной кислоты или их смесей.
В дополнительном предпочтительном варианте осуществления замещенное ксантиновое производное по настоящему изобретению или его фармацевтически приемлемая соль выбраны из:
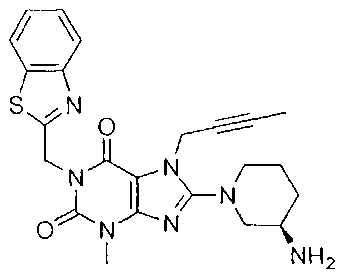

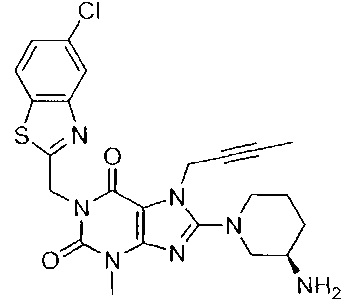
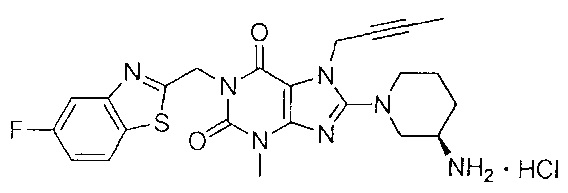
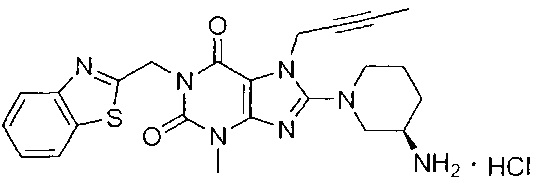
В другом варианте осуществления, настоящее изобретение относится к способу получения замещенных ксантиновых производных, характеризующихся структурой, представленной следующей общей формулой (I), или их фармацевтически приемлемой соли, при этом способ включает следующие стадии:
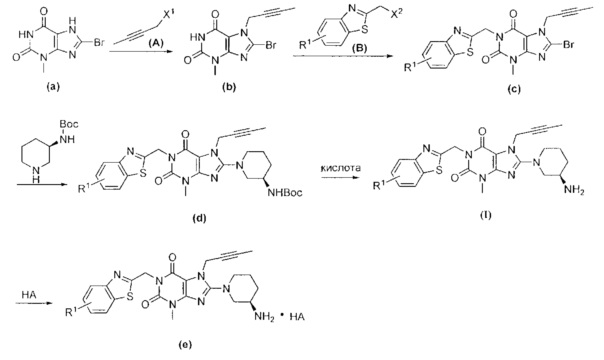
(1) при комнатной температуре в течение ночи осуществляют реакцию сырьевого материала а с сырьевым материалом А в N,N-диметилформамиде; после завершения реакции, полученный реакционный раствор выливают в воду, фильтруют с отсасыванием, промывают водой и высушивают с получением промежуточного соединения b; где X1 в сырьевом материале А представляет собой уходящую группу, при этом указанный X1 предпочтительно представляет собой Cl, Br или I;
(2) полученное промежуточное соединение b подвергают реакции в течение ночи с сырьевым материалом В и основанием в N,N-диметилформамиде при комнатной температуре; после завершения реакции полученную реакционную смесь выливают в воду, фильтруют с отсасыванием, промывают водой и сушат с получением промежуточного соединения с; где X2 в сырьевом материале В представляет собой уходящую группу, причем указанный X2 предпочтительно представляет собой Cl, Br или I; при этом основание предпочтительно представляет собой карбонат калия, карбонат натрия, гидроксид натрия или гидрид натрия;
(3) полученное промежуточное соединение с подвергают реакции с (R)-3-трет-бутоксикарбонил-аминопиперидином и основанием в N,N-диметилформамиде при условиях нагревания (50~100°C) в течение 2~8 ч; после реакции раствор охлаждают до комнатной температуры, полученный реакционный раствор выливают в воду, фильтруют с отсасыванием, промывают водой и сушат с получением промежуточного соединения d;
при этом основание предпочтительно представляет собой карбонат калия, карбонат натрия, гидроксид натрия или гидрид натрия;
(4) полученное промежуточное соединение d подвергают реакции с кислотой в органическом растворителе при комнатной температуре в течение 2~10 ч; после завершения реакции рН остаточного раствора регулируют до 7-8 водным раствором карбоната калия и затем экстрагируют органическим растворителем; полученную органическую фазу высушивают, фильтруют и концентрируют с получением неочищенного продукта; неочищенный продукт дополнительно очищают посредством хроматографии с получением целевого соединения I; при этом кислота, применяемая для удаления защитной группы Воc предпочтительно представляет собой соляную кислоту или трифторуксусную кислоту; использованный органический растворитель предпочтительно представляет собой дихлорметан, хлороформ, этилацетат или тетрагидрофуран; и
необязательно (5) полученное целевое соединение I подвергают реакции с раствором кислоты в органическим растворителе и перемешивают в течение соответствующего времени; затем растворитель выпаривают и остаток промывают и высушивают с получением соответствующей соли е; при этом использованный органический растворитель предпочтительно представляет собой дихлорметан, хлороформ, этилацетат или тетрагидрофуран.
В другом варианте осуществления настоящее изобретение относится к применению указанных выше замещенных ксантиновых производных или их фармацевтически приемлемой соли при получении лекарственного препарата для лечения заболеваний, связанных с DPP-IV.
В предпочтительном варианте осуществления настоящее изобретение относится к применению указанных выше замещенных ксантиновых производных или их фармацевтически приемлемой соли при получении лекарственного препарата для лечения диабета 11 типа или заболеваний, связанных с нарушением толерантности к глюкозе.
ПРИМЕРЫ
Пример 1. Получение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина

Схема получения показана ниже.

Стадия 1. Получение 3-метил-7-(2-бутин-1-ил)-8-бромксантина
При использовании хорошо известного способа 8-бром-3-метил-ксантин (5 г, 20,4 ммоль) растворяли в N,N-диметилформамиде (30 мл). Добавляли N,N-диизопропилэтиламин (2,633 г, 20,4 ммоль) и 1-бром-2-бутин (2,714 г, 20,4 ммоль) с получением реакционной смеси. Реакционную смесь подвергали реакции в течение ночи при комнатной температуре и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь выливали в воду, фильтровали с отсасыванием и полученное твердое вещество промывали водой три раза, высушивали с получением 3-метил-7-(2-бутин-1-ил)-8-бром-ксантина 1а (5,15 г, светло-желтое твердое вещество), выход: 85%.
MS масса/заряд (ES): 297, 299 [М+1].
Стадия 2. Получение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантина
При использовании хорошо известного способа 3-метил-7-(2-бутин-1-ил)-8-бромксантин 1а (156 мг, 0,53 ммоль) растворяли в N,N-диметилформамиде (3 мл). Добавляли 2-бромметил-5-фтор-1,3-бензотиазол (140 мг, 0,57 ммоль), карбонат калия (118 мг, 0,79 ммоль) с получением реакционной смеси. Полученную реакционную смесь подвергали реакции в течение ночи при комнатной температуре и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь выливали в воду, фильтровали с отсасыванием и полученное твердое вещество промывали водой, высушивали с получением 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантина 1b (240 мг, грязно-белое твердое вещество), выход: 99%.
MS масса/заряд (ES): 462, 464 [М+1].
Стадия 3. Получение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантина
При использовании хорошо известного способа 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантин 1b (240 мг, 0,51 ммоль) растворяли в N,N-диметилформамиде (5 мл). Добавляли (R)-3-трет-бутоксикарбонил-аминопиперидин (130 мг, 0,66 ммоль) и карбонат калия (107 мг, 0,78 ммоль) с получением реакционной смеси. Реакционную смесь подвергали реакции при 75°C в течение 2 часов и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь охлаждали до комнатной температуры. Охлажденный реакционный раствор выливали в холодную воду, фильтровали с отсасыванием и полученное твердое вещество промывали водой, высушивали с получением 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидии-1-ил]-ксантина 1с (230 мг, желтое твердое вещество), выход: 77,6%.
MS масса/заряд (ES): 582 [М+1].
Стадия 4. Получение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина
Соединение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантин 1с (230 мг, 0.396 ммоль) растворяли в дихлорметане (5 мл). Трифторуксусную кислоту (0,7 мл) добавляли по каплям при комнатной температуре с получением реакционной смеси. Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученный реакционный раствор концентрировали с использованием роторного испарителя при 30°C для удаления трифторуксусной кислоты. Остаток растворяли в дихлорметане (5 мл) и водный раствор карбоната калия с рН=10 применяли для регуляции рН до 7-8 для получения смешанного раствора. Смешанный раствор экстрагировали дихлорметаном и полученную органическую фазу высушивали над безводным сульфатом магния, а затем фильтровали и концентрировали. Остаток отделяли и очищали посредством тонкослойной хроматографии (дихлорметан : метанол = 10:1 в качестве элюирующей системы) с получением соединения 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина 1 (153 мг, желтое твердое вещество), выход: 80%.
MS масса/заряд (ES): 482 [М+1].
1Н ЯМР (300 МГц, DMSO) δ 8,16-8,03 (m, 1Н), 7,87-7,74 (m, 1H), 7,42-7,26 (m, 1H), 5,45 (s, 2Н), 4,93 (s, 2Н), 3,74-3,53 (m, 2Н), 3,41 (s, 3Н), 3,14-2,95 (m, 2Н), 2,95-2,80 (m, 1H), 1,98-1,73 (m, 5Н), 1,72-1,53 (m, 1H), 1,44-1,24 (m, 1Н).
Пример 2. Получение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина
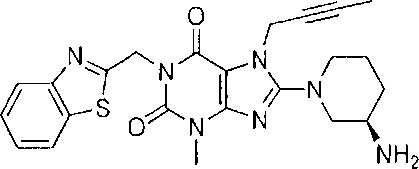
Схема получения показана ниже.
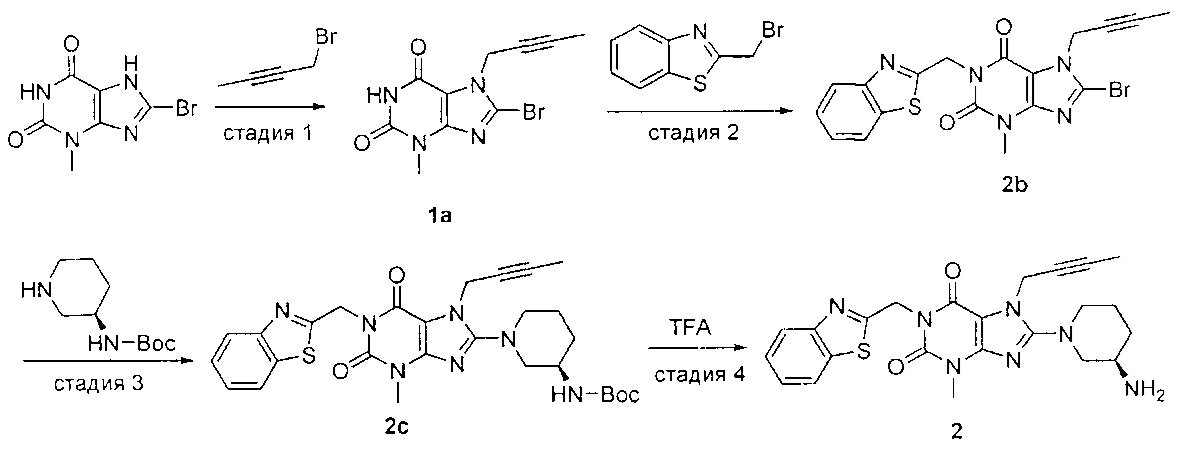
Стадию 1 проводили таким же образом, как и стадию 1 в примере 1.
Стадия 2. Получение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бромксантина
При использовании хорошо известного способа 3-метил-7-(2-бутин-1-ил)-8-бром-ксантин 1а (327 мг, 1 ммоль) растворяли в N,N-диметилформамиде (5 мл). Добавляли карбонат калия (221 мг, 1.6 ммоль) и 2-бромметил-1,3-бензотиазол (228 мг, 1 ммоль) с получением реакционной смеси. Реакционную смесь подвергали реакции в течение ночи при комнатной температуре и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь выливали в воду, фильтровали с отсасыванием и полученное твердое вещество промывали водой, высушивали с получением 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бром-ксантина 2b (400 мг, желтое твердое вещество), выход: 90%.
MS масса/заряд (ES): 444, 446 [М+1].
Стадия 3. Получение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ид)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантина
При использовании хорошо известного способа 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бром-ксантин 2b (400 мг, 0,9 ммоль) растворяли в N,N-диметилформамиде (6 мл). Добавляли (R)-3-трет-бутоксикарбонил-аминопиперидин (180 мг, 0,9 ммоль) и карбонат калия (186,5 мг, 1,35 ммоль) с получением реакционной смеси. Реакционную смесь подвергали реакции при 75°C в течение 2 часов и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь охлаждали до комнатной температуры. Охлажденный реакционный раствор выливали в воду, фильтровали с отсасыванием и полученное твердое вещество промывали водой, высушивали с получением 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантина 2 с (460 мг, желтое твердое вещество), выход: 90,8%.
MS масса/заряд (ES): 564 [М+1].
Стадия 4. Получение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина
При использовании хорошо известного способа соединение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантин 2 с (460 мг, 0,82 ммоль) растворяли в дихлорметане (8 мл). Трифторуксусную кислоту (0,8 мл) добавляли по каплям при комнатной температуре с получением реакционной смеси. Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь концентрировали с использованием роторного испарителя при 30°C для удаления трифторуксусной кислоты. Остаток растворяли в дихлорметане (5 мл) и водный раствор карбоната калия с рН=10 применяли для регуляции рН до 7-8 для получения смешанного раствора. Смешанный раствор экстрагировали дихлорметаном и полученную органическую фазу высушивали над безводным сульфатом магния, а затем фильтровали и концентрировали. Остаток отделяли и очищали посредством тонкослойной хроматографии (дихлорметан: метанол = 10:1 в качестве элюирующей системы) с получением соединения 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина 2 (210 мг, желтое твердое вещество), выход: 55,4%.
MS масса/заряд (ES): 464 [М+1].
1Н ЯМР (300 МГц, DMSO) δ 8,04 (d, J=7,7 Гц, 1H), 7,94 (d, J=7,9 Гц, 1Н), 7,57-7,35 (m, 2Н), 5,45 (s, 2Н), 4,91 (s, 2Н), 3,75-3,55 (m, 2Н), 3,41 (s, 3Н), 3,09-2,93 (m, 1Н), 2,89-2,70 (m, 2Н), 1,92-1,73 (m, 5Н), 1,70-1,53 (m, 1H), 1,32-1,15 (m, 1H).
Схема получения показана ниже.
Пример 3. Получение 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина
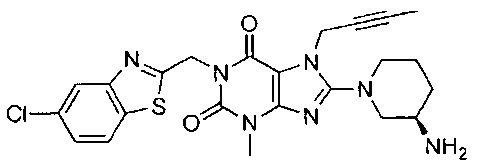
Схема получения показана ниже.

Стадию 1 проводили таким же образом, как и стадию 1 в примере 1.
Стадия 2. Получение 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бром-ксантина
При использовании хорошо известного способа 3-метил-7-(2-бутин-1-ил)-8-бром-ксантин 1а (297 мг, 1 ммоль) растворяли в N,N-диметилформамиде (8 мл). Добавляли 2-бромметил-5-хлор-1,3-бензотиазол (263 мг, 1 ммоль) и карбонат калия (213 мг, 1,5 ммоль) с получением реакционной смеси. Реакционную смесь подвергали реакции в течение ночи при комнатной температуре и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь выливали в воду, фильтровали с отсасыванием и полученное твердое вещество промывали водой, высушивали с получением 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бром-ксантина 3b (460 мг, светло-желтое твердое вещество), выход: 96%.
MS масса/заряд (ES): 478, 480 [М+1].
Стадия 3. Получение 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантина
При использовании хорошо известного способа 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-бром-ксантин 3b (460 мг, 0,96 ммоль) растворяли в N,N-диметилформамиде (12 мл). Добавляли (R)-3-трет-бутоксикарбонил-аминопиперидин (193 мг, 0,96 ммоль) и карбонат калия (200 мг, 1,44 ммоль) с получением реакционной смеси. Реакционную смесь подвергали реакции при 75°C в течение 2 часов и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь охлаждали до комнатной температуры. Охлажденный реакционный раствор выливали в холодную воду, фильтровали с отсасыванием и полученное твердое вещество промывали водой, высушивали с получением 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантина 3c (417 мг, серое твердое вещество), выход: 72,6%.
MS масса/заряд (ES): 598 [М+1].
Стадия 4. Получение 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина
При использовании хорошо известного способа 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-трет-бутоксикарбонил-аминопиперидин-1-ил]-ксантина 3с (417 мг, 0,7 ммоль) растворяли в дихлорметане (10 мл). Трифторуксусную кислоту (1,5 мл) добавляли по каплям при комнатной температуре с получением реакционной смеси. Реакционную смесь подвергали реакции при комнатной температуре в течение 2 часов и применяли TLC для наблюдения за ходом реакции. После завершения реакции полученную реакционную смесь концентрировали с использованием роторного испарителя при 30°C для удаления трифторуксусной кислоты. Остаток растворяли в дихлорметане (5 мл) и водный раствор карбоната калия с рН=10 применяли для регуляции pH до 7-8 для получения смешанного раствора. Смешанный раствор экстрагировали дихлорметаном и полученную органическую фазу высушивали над безводным сульфатом магния, а затем фильтровали и концентрировали. Остаток отделяли и очищали посредством тонкослойной хроматографии (дихлорметан: метанол = 10:1 в качестве элюирующей системы) с получением соединения 1-[(5-хлор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина 3 (310 мг, светло-желтое твердое вещество), выход: 88,9%.
MS масса/заряд (ES): 498 [М+1].
1Н ЯМР (300 МГц, DMSO) δ 8,17-7,98 (m, 2Н), 7,54-7,42 (m, 1Н), 5,46 (s, 2Н), 5,07-4,80 (m, 2Н), 3,80-3,48 (m, 2Н), 3,41 (s, 3Н), 3,19-2,99 (m, 3Н), 2,02-1,75 (m, 5Н), 1,72-1,59 (m, 1Н), 1,57-1,43 (m, 1Н).
Пример 4. Получение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина гидрохлорида

Схема получения показана ниже.
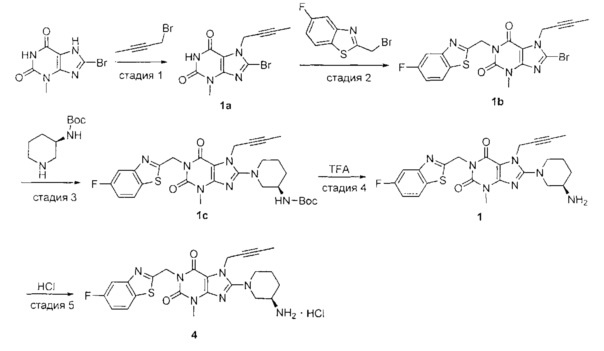
Стадию 1 проводили таким же образом, как и стадию 1 в примере 1.
Стадию 2 проводили таким же образом, как и стадию 2 в примере 1.
Стадию 3 проводили таким же образом, как и стадию 3 в примере 1.
Стадию 4 проводили таким же образом, как и стадию 4 в примере 1.
Стадия 5. Получение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина гидрохлорида
Соединение 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутии-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантин 1 (60 мг, 0,124 ммоль) растворяли в дихлорметане (2 мл). Добавляли 0,14 мл раствора хлористого водорода в дихлорметане (1 моль/л) с получением реакционной смеси. Реакционную смесь перемешивали в течение 10 минут и растворитель отгоняли. Остаток промывали этилацетатом и высушивали с получением целевого соединения 1-[(5-фтор-1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина гидрохлорида 4 (47 мг, желтое твердое вещество), выход: 76%.
1Н ЯМР (300 МГц, DMSO) δ 8,55 (s, 3Н), 8,09 (dd, J=8,6, 5,4 Гц, 1Н), 7,87-7,75 (m, 1H), 7,34 (t, J=8,0 Гц, 1Н), 5,46 (s, 2Н), 5,14-4,84 (m, 2Н), 3,75 (d, J=11,0 Гц, 1H), 3,50 (d, J=12,3 Гц, 1Н), 3,42 (s, 4H), 3,23 (dd, J=19,4, 10,9 Гц, 2H), 2,14-1,88 (m, 2H), 1,81 (s, 3Н), 1,77-1,62 (m, 2H).
Пример 5. Получение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина гидрохлорида
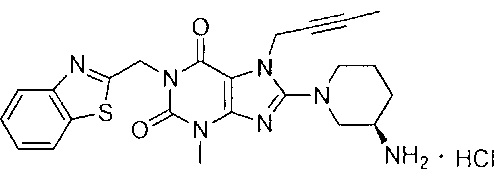
Схема получения показана ниже.
Стадию 1 проводили таким же образом, как и стадию 1 в примере 1.
Стадию 2 проводили таким же образом, как и стадию 2 в примере 2.
Стадию 3 проводили таким же образом, как и стадию 3 в примере 2.
Стадию 4 проводили таким же образом, как и стадию 4 в примере 2.
Стадия 5. Получение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-амино-пиперидин-1-ил]-ксантина гидрохлорида
Соединение 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантин 2 (100 мг, 0,216 ммоль) растворяли в дихлорметане (3 мл).
Добавляли 0,24 мл раствора хлористого водорода в дихлорметане (1 моль/л) с получением реакционной смеси. Реакционную смесь перемешивали в течение 10 минут и растворитель отгоняли. Остаток промывали этилацетатом и высушивали с получением целевого соединения 1-[(1,3-бензотиазол-2-ил)метил]-3-метил-7-(2-бутин-1-ил)-8-[(R)-3-аминопиперидин-1-ил]-ксантина гидрохлорида 5 (85 мг, желтое твердое вещество), выход: 79%.
1Н ЯМР (300 МГц, DMSO) δ 8,47 (s, 3Н), 8,05 (d, J=7,7 Гц, 1H), 7,94 (d, J=7,9 Гц, 1Н), 7,56-7,34 (m, 2Н), 5,46 (s, 2Н), 5,11-4,84 (m, 2Н), 3,74 (d, J=11,0 Гц, 1H), 3,50 (d, J=11,3 Гц, 1H), 3,42 (s, 4Н), 3,31-3,13 (m, 2Н), 2,12-1,86 (m, 2Н), 1,80 (s, 3Н), 1,76-1,60 (m, 2Н).
Экспериментальный пример 1. Анализ in vitro ингибирующей активности в отношении DPP-IV
1. Цель эксперимента
Ингибирующие способности в отношении дипептидилпептидазы IV (DPP-1V) соединений, полученных в описанных выше примерах, подлежали рассмотрению с целью оценки ингибирующего эффекта соединений, полученных в описанных выше примерах.
2. Экспериментальные материалы
2.1 Дипептидилпептидаза IV (DPP-IV): SIGMA Products, № изделия D4943-1VL.
2.2 Субстрат: раствор Gly-Pro-7-амидо-4-метилкумарина, SIGMA Products, № изделия G2761-25 мг, FW=41,03.
2.3 Буфер для DPP-IV, содержащий 25 ммоль HEPES, 140 ммоль NaCl, 1% BSA, 80 ммоль MgCl2, рН которого регулировали до 8,0.
2.4 Лекарственное средство положительного контроля (линаглиптин): предоставленное Shanghai Yingrui Chemical Technology Co., Ltd., характеристика: 2 г, CAT: YRY0687, LCT №: YR111130, с молекулярной массой 472,54, растворенное в DMSO в виде 10 мМ исходного раствора, разбавленное дистиллированной водой до 10 мкМ в качестве рабочего раствора, с конечной концентрацией 1 мкМ.
2.5 Оборудование для испытаний: En Vision (PerkinElmer Company).
3. Принцип эксперимента
Gly-Pro-7-амидо-4-метилкумарин может быть гидролизован посредством дипептидилпептидазы IV (DPP-IV) при комнатной температуре с образованием 7-амидо-4-метилкумарина, который может излучать флуоресценцию с длиной волны 460 нм при длине волны возбуждения 355 нм. Изменение количества продукта может быть определено посредством изменения интенсивности флуоресценции для отражения уровня активности фермента.
4. Экспериментальный метод
Дипептидилпептидазу IV (DPP-IV), буфер для DPP-IV и образцы для испытаний использовали для получения 200 мкл реакционной смеси, при этом устанавливали холостой контроль (без фермента и образцов) и отрицательный контроль (без образцов) с одинаковым объемом. Реакционную систему и контроли подвергали реакции при комнатной температуре в течение 10 мин, а затем в них добавляли субстрат дипептидилпептидазы IV, соответственно, затем подвергали реакции при комнатной температуре в течение 30 мин. Определяли интенсивность флуоресценции F (длина волны возбуждения 355 нм, длина волны излучения 460 нм). Степень ингибирования рассчитывали в соответствии со значением интенсивности флуоресценции F, при этом степень ингибирования = [1-(Fобразца-Fхолостого контроля)/(Fотрицательного контроля-Fхолостого контроля)]⋅100. Если каждый из образцов при разных концентрациях был предварительно представлен в двух повторностях, с образцами со степенью ингибирования более 70% проводили эксперименты на основе метода исключения ошибочно положительных образцов. Относительно образцов, подтвержденных как положительные, определяли значения IC50, при этом каждый образец последовательно разбавляли (в 3 раза) до шести концентраций и для каждой концентрации получали второй образец. В соответствии со степенью ингибирования для расчета IC50 применяли модель вероятности с логистическим распределением с 4 параметрами с помощью программного обеспечения XLfit.
5. Результаты эксперимента
Данные измеренной IC50 каждого соединения по настоящему изобретению в описанных выше примерах были следующими:

Из данных анализа in vitro ингибирующей активности в отношении DPP-IV в приведенной выше таблице может быть известно, что по сравнению с лекарственным средством положительного контроля линаглиптином соединения по настоящему изобретению в примерах обладают значительной ингибирующей активностью в отношении DPP-IV.
Экспериментальный пример 2. Влияние на толерантность к глюкозе у нормальных мышей
1. Экспериментальный материал
1.1 Лекарственные средства
Рабочее лекарственное средство: глюкоза, , предоставленное компанией SIGMA, № партии 101021941, характеристики: 100 г/бутылка; исследуемое лекарственное средство: соединение примера 1, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., желтый порошок, № партии: 20120315;
исследуемое лекарственное средство: соединение примера 2, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., желтый порошок, № партии: 20120320;
исследуемое лекарственное средство: соединение примера 3, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., светло-желтый порошок, № партии: 20120323;
исследуемое лекарственное средство: соединение примера 4, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., желтый порошок, № партии: 20120401;
положительный контроль: линаглиптин, предоставленный Shanghai Yingrui Chemical Technology Co., Ltd., характеристики: 2 г, CAT: YRY0687, LCT №: YR111130.
1.2 Экспериментальное оборудование
Электронные весы FA2204B, предоставленные Shanghai Precision Instruments Scientific Instrument Co., Ltd.;
аналитические весы от METTLER-TOLEDO, тип XS-105, предоставленные компанией Mettler-Toledo, Швейцария;
тест-полоски на глюкозу в крови: активные тест-полоски на глюкозу ACCU-CHEK, характеристики: 50 полосок, № партии: 23435532, предоставленные Roche Diagnostics (Шанхай) Co., Ltd;
хирургические ножницы, шприцы и т.д.
1.3 Экспериментальные животные
Мыши КМ, 6-недельного возраста, с весом 18~22 г, половина самцы и половина самки, 60 мышей, предоставленные Chengdu Dashuo Biological Technology Co., Ltd., лицензия на производство: SCXK (Chuan) 2008-24. Животных содержали в помещении для животных после приобретения, адаптивно наблюдали в течение по меньшей мере трех дней и использовали для анализов за исключением случаев, когда они не соответствовали требованиям в отношении стандартов карантина.
2. Экспериментальный метод
2.1 Животных не кормили в течение по меньшей мере 12 часов перед началом анализа.
2.2 Группировка. Измеряли значения уровня глюкозы в крови натощак у мышей, которые не получали пищу, и мышей произвольно группировали согласно таблице 1 с незначительным отличием между группами.

2.3 Измерение значения глюкозы в крови
Животным в каждой группе вводили соответствующие исследуемые соединения согласно таблице 1 посредством внутрижелудочного введения (i.g.), затем вводили глюкозу (8 г/кг) соответственно посредством внутрижелудочного введения через 30 мин после введения лекарственных средств и для них измеряли значения глюкозы в крови соответственно через 30 мин, 60 мин и 120 мин после введения глюкозы (с нагрузкой глюкозы).
3. Статистический метод
Для статистических расчетов применяли Excel, экспериментальные данные выражали в виде и применяли двусторонний t-критерий Стьюдента для статистического сравнения экспериментальных данных среди нескольких групп.
4. Экспериментальные результаты

5. Выводы
(1) Как можно увидеть из таблицы 2, при сравнении с контрольной группой через 30 мин, 60 мин и 120 мин после нагрузки глюкозы значения глюкозы в крови в группе прим. 1, группе прим. 2, группе прим. 3, группе прим. 4 и группе положительного контроля есть существенное различие (**Р<0,01), что указывает на то, что все из соединения примера 1, соединения примера 2, соединение примера 3, соединения примера 4 и лекарственного средства положительного контроля (линаглиптин) могут весьма значительно снижать уровни глюкозы в крови;
(2) при сравнении с лекарственным средством положительного контроля (линаглиптином) через 30 мин, 60 мин и 120 мин после нагрузки глюкозы значение глюкозы в крови в группе прим. 1 весьма значительно снижалось (▲▲P<0,01), при этом значение глюкозы в крови в группе прим. 2, что в группе прим. 3 и что в группе прим. 4 значительно снижалось (▲Р<0,05), что указывает на то, что гипогликемические эффекты соединений по настоящему изобретению в примерах являются существенными.
Экспериментальный пример 3. Влияние на глюкозу в крови у мышей со спонтанным сахарным диабетом
1. Экспериментальные материалы
1.1 Лекарственные средства
Рабочее лекарственное средство: глюкоза, , предоставленное компанией SIGMA, № партии 101021941, характеристики: 100 г/бутылка;
исследуемое лекарственное средство: соединение примера 1, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., желтый порошок, № партии: 20120315;
исследуемое лекарственное средство: соединение примера 2, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., желтый порошок, № партии: 20120320;
исследуемое лекарственное средство: соединение примера 3, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., светло-желтый порошок, № партии: 20120323;
исследуемое лекарственное средство: соединение примера 4, предоставленное лабораторией синтеза Chengdu Easton Pharmaceutical Co., Ltd., желтый порошок, № партии: 20120401;
положительный контроль: линаглиптин, предоставленный Shanghai Yingrui Chemical Technology Co., Ltd., характеристики: 2 г, CAT: YRY0687, LCT №: YR111130.
1.2 Экспериментальное оборудование
Электронные весы FA2204B: предоставленные Shanghai Precision Instruments Scientific Instrument Co., Ltd.;
аналитические весы METTLER-toledo, тип XS-105, предоставленные компанией Mettler-Toledo, Швейцария;
тест-полоски на глюкозу в крови: активные тест-полоски на глюкозу ACCU-CHEK, характеристики: 50 полосок, № партии: 23435532, предоставленные Roche Diagnostics (Шанхай) Co., Ltd;
хирургические ножницы, шприцы и т.д.
1.3 Экспериментальные животные
Страдающие ожирением мыши ККАу со спонтанным диабетом 11 типа, 60 мышей, 14-недельного возраста, половина самцы и половина самки, приобретенные в Институте изучения лабораторных животных, Академия медицинских наук Китая (квалификационный номер: SCXK (Jing) 2009-0004). Животных содержали в помещении для животных после приобретения, адаптивно наблюдали в течение по меньшей мере трех дней и использовали для анализов за исключением случаев, когда они не соответствовали требованиям в отношении стандартов карантина.
2. Экспериментальный метод
2.1 Животных не кормили в течение по меньшей мере 12 часов перед началом анализа.
2.2 Значения уровня глюкозы в крови натощак у мышей, которые не получали пищу, измеряли с помощью активных тест-полосок на глюкозу ACCU-CHEK и мышей произвольно группировали согласно таблице 3. Кроме того, для группы холостого контроля использовали мышей C57BL/6J, при этом мышей ККАу со спонтанным диабетом II типа использовали для модельной группы.


2.3 Измерение значения глюкозы в крови
Животным в каждой группе вводили соответствующие исследуемые соединения согласно таблице 3 посредством внутрижелудочного введения (i.g.), затем вводили глюкозу (8 г/кг) соответственно посредством внутрижелудочного введения через 30 мин после введения лекарственных средств и для них измеряли значения глюкозы в крови соответственно через 30 мин, 60 мин и 120 мин после введения глюкозы (с нагрузкой глюкозы).
3. Статистический метод
Для статистических расчетов применяли Excel, экспериментальные данные выражали в видеи применяли двусторонний t-критерий Стьюдента для статистического сравнения экспериментальных данных среди нескольких групп.
4. Экспериментальные результаты

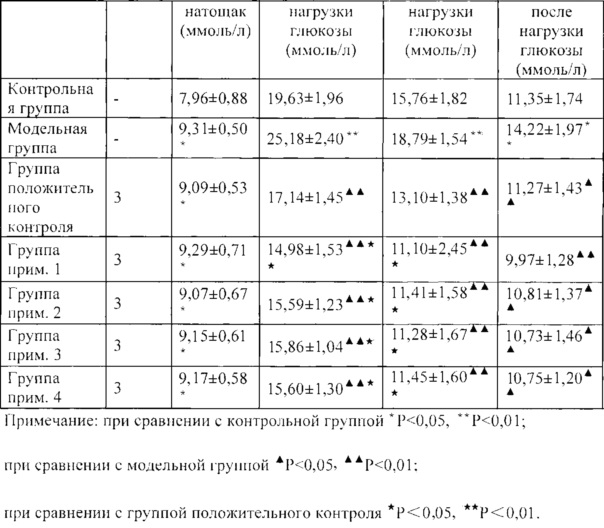
5. Вывод
Как можно увидеть из таблицы 4, при сравнении с контрольной группой как значение уровня глюкозы в крови натощак, так и значение глюкозы в крови после нагрузки глюкозы в модельной группе значительно повышались (*Р<0,05, **Р<0,01), что указывает на то, что мышиная модель со спонтанным диабетом была устойчива;
при сравнении с модельной группой через 30 мин, 60 мин и 120 мин после нагрузки глюкозы значения глюкозы в крови в каждой группе введения лекарственного средства значительно снижались (▲▲<0,01), что указывает на то, что все из соединений из примеров 1-4 и лекарственного средства положительного контроля линаглиптина могут весьма значительно снижать уровни глюкозы в крови;
(3) при сравнении с лекарственным средством положительного контроля линаглиптином через 30 мин после нагрузки глюкозы значение глюкозы в крови в группе прим. 1 весьма значительно снижалось (**Р<0,01), при этом значение глюкозы в крови в группе прим. 2, что в группе прим. 3 и что в группе прим. 4 значительно снижалось (*Р<0,05); через 60 мин после нагрузки глюкозы значения глюкозы в крови в каждой группе примера значительно снижались (*Р<0,05), что указывает на то, что гипогликемические эффекты соединений по настоящему изобретению в примерах являются существенными.
Вышеприведенные результаты указывают на то, что соединения по настоящему изобретению в примерах проявляют значительную ингибирующую активность в отношении DPP-IV и гипогликемический эффект.
Специалисту в данной области будет очевидно, что без отступления от сущности и объема настоящего изобретения возможны различные модификации и вариации по отношению к соединениям, композициям и способам получения по настоящему изобретению, следовательно, объем правовой охраны настоящего изобретения охватывает различные модификации и вариации, примененные по отношению к нему, при условии, что модификации и вариации подпадают под действие объема правовой охраны, охватываемого формулой изобретения и ее эквивалентными вариантами осуществления.
Реферат
Изобретение относится к соединениям, представленным общей формулой I, или их фармацевтически приемлемым солям, которые могут найти применение при лечении заболеваний, связанных с дипептидилпептидазой IV. В формуле I Rвыбран из атома водорода, атома фтора, атома хлора, атома брома, атома йода или цианогруппы. Изобретение относится также к способу получения указанных соединений и их применению при получении лекарственного препарата для лечения заболеваний, связанных с дипептидилпептидазой IV. 3 н. и 9 з.п. ф-лы, 4 табл., 8 пр.
Формула



Документы, цитированные в отчёте о поиске
Гетероциклические соединения, которые являются ингибиторами фермента dpp-iv
Комментарии