Производные тетрагидропиридо[3,4-b]индола, полезные при лечении рака - RU2664109C2
Код документа: RU2664109C2
Чертежи










Описание
Изобретение относится к некоторым новым производным индола или их фармацевтически приемлемым солям, которые обладают противораковой активностью и, соответственно, являются полезными в способах лечения человека или животного. Изобретение также относится к способам получения указанных производных индола, фармацевтических композиций, содержащих такие производные, и их применению в терапевтических способах, например, при изготовлении лекарственных средств для применения в предупреждении или лечении злокачественных новообразований у теплокровного животного, например, человека, включая применение в предупреждении или лечении рака.
Настоящее изобретение также относится к производным индола, которые являются селективными негативными регуляторами рецептора эстрогенов.
Рецептор эстрогенов альфа (ERα, ESR1, NR3A (ядерный рецептор А подсемейства 3)) и рецептор эстрогенов бета (ERβ, ESR2, NR3b) представляют собой рецепторы стероидных гормонов, которые являются членами большого семейства ядерных рецепторов. Схожий по структуре со всеми ядерными рецепторами, ERα состоит из шести функциональных доменов (обозначенных как A-F) (Dahlman-Wright, et al., Pharmacol. Rev.. 2006, 58: 773-781) и классифицируется как лиганд-зависимый транскрипционный фактор, поскольку после его ассоциации со специфическим лигандом, женским половым стероидным гормоном 17b-эстрадиолом (Е2), полученный комплекс связывается с геномными последовательностями, называемыми элементами рецептора эстрогенов (ERE; estrogen receptor elements), и взаимодействует с корегуляторами, модулируя транскрипцию генов-мишеней. Ген ERα локализован на 6q25.1 и кодирует состоящий из 595 аминокислот (АА) белок, и в результате альтернативного сплайсинга и наличия альтернативных сайтов начала трансляции могут продуцироваться многочисленные изоформы. Помимо ДНК-связывающего домена (домена С) и лиганд-связывающего домена (домена Е), данный рецептор содержит N-концевой (А/В) домен, шарнирный (D) домен, который соединяет C и Е домены, и удлинение на С-конце (F домен). В то время как домены C и E у ERα и ERβ являются довольно консервативными (идентичность аминокислот составляет 96% и 55%, соответственно), консервативность доменов A/B, D и F является низкой (идентичность аминокислот составляет ниже 30%). Оба рецептора вовлечены в регуляцию и развитие женских половых путей и помимо этого принимают участие в работе центральной нервной системы, сердечно-сосудистой системы и в костном метаболизме. Геномное действие ER осуществляется в ядре клетки, когда рецептор связывается с ERE непосредственно (прямая активация или классический путь) или опосредованно (непрямая активация или неклассический путь). В отсутствие лиганда молекулы ER образуют ассоциаты с белками теплового шока, Hsp90 и Hsp70, и объединенная шаперонная машина стабилизирует лиганд-связывающий домен (LBD), делая его доступным для лиганда. Связавшийся с лигандом ER диссоциирует из комплекса с белками теплового шока, вызывая конформационное изменение в рецепторе, что способствует димеризации, связыванию с ДНК, взаимодействию с коактиваторами или корепрессорами и модулированию экспрессии генов-мишеней. В неклассическом пути, последовательности АР-1 (активаторный белок 1) и Sp-1 (specificity protein 1) представляют собой альтернативные регуляторные последовательности ДНК, используемые обеими изоформами рецептора для модулирования генной экспрессии. В этом примере ER не взаимодействует с ДНК напрямую, а взаимодействует посредством ассоциации с другими связанными с ДНК транскрипционными факторами, например c-Jun или c-Fos (Kushner et al., Pure Applied Chemistry, 2003, 75: 1757-1769). Точный механизм, посредством которого ER воздействует на транскрипцию генов, изучен плохо, но по-видимому он опосредуется многочисленными ядерными факторами, которые рекрутируются связанным с ДНК рецептором. Этот рекрутмент корегуляторов главным образом опосредуется двумя участками белковой поверхности, AF2 (функциональный активатор транскрипции; activation function 2) и AF1, которые расположены в Е-домене и А/В-домене, соответственно. AF1 регулируется факторами роста, и его активность зависит от клеточного окружения и окружения промотора, в то время как активность AF2 полностью зависит от связывания с лигандом. Несмотря на то, что эти два домена могут действовать независимо, максимальная транскрипционная активность ER достигается в результате синергического взаимодействия этих двух доменов (Tzukerman, et al., Mol. Endocrinology, 1994, 8: 21-30). Несмотря на то, что ER считаются транскрипционными факторами, они также могут действовать через негеномные механизмы, о чем свидетельствуют быстрые эффекты ER в тканях после введения Е2 в сроки, которые считаются слишком быстрыми для проявления геномного действия. До сих пор неясно, являются ли рецепторы, ответственные за быстрое действие эстрогенов, теми же самыми ядерными ER или особыми G-белок-связанными стероидными рецепторами (Warner, et al., Steroids, 2006, 71: 91-95), тем не менее идентифицировано возрастающее число Е2-индуцируемых сигнальных путей, например, путь MAPK/ERK (митоген-активируемая протеинкиназа/внеклеточная сигнал-регулируемая киназа), и путь активации эндотелиальной синтазы оксида азота, и путь PI3K/Akt (фосфоинозитид-3-киназа/протеинкиназа В). Помимо участия в лиганд-зависимых путях, было показано, что ERα обладают не зависимой от лиганда активностью через AF-1, что ассоциируется со стимуляцией МАРK в сигнальном пути через ростовой фактор, например инсулиноподобный фактор роста 1 (IGF-1) и эпидермальный фактор роста (EGF). Активность AF-1 зависит от фосфорилирования Ser118, и примером взаимодействия между сигнальными путями через ER и ростовой фактор, является фосфорилирование Ser118 под действием МАРK в ответ на такие факторы роста, как IGF-1 и EGF (Kato, et al., Science, 1995, 270: 1491-1494).
Было показано, что с ER связывается большое количество различных по структуре соединений. Некоторые соединения, такие как эндогенный лиганд Е2, действуют как агонисты рецепторов, в то время как другие конкурентно ингибируют связывание с Е2 и действуют как антагонисты рецепторов. Эти соединения можно разделить на 2 класса в зависимости от их функциональных проявлений. Селективные модуляторы рецепторов эстрогенов (SERM), такие как тамоксифен, обладают способностью действовать в качестве как агонистов, так и антагонистов рецепторов в зависимости от клеточного окружения и окружения промотора, а также от целевой изоформы ER. Например, тамоксифен действует в качестве антагониста в молочной железе, но действует в качестве частичного агониста в кости, сердечно-сосудистой системе и матке. Все SERM, по-видимому, действуют как антагонисты AF2, а посредством AF1 проявляют свои свойства как частичные агонисты. Представители второй группы, примером которой является фулвестрант, классифицируются как полные антагонисты, способные блокировать активность эстрогенов посредством полного ингибирования доменов AF1 и AF2 в результате индуцирования уникального конформационного изменения в лиганд-связывающем домене (LBD) при связывании с соединением, что приводит к полному упразднению взаимодействия между спиралью 12 и остальной частью LBD, блокируя рекрутмент кофакторов (Wakeling, et al., Cancer Res., 1991, 51: 3867-3873; Pike, et al., Structure, 2001, 9: 145-153).
Внутриклеточные уровни ERα снижаются в присутствии Е2 по пути убиквитин/протеасома (Ub/26S). Полиубиквитинилирование связанного с лигандом ERα катализируется по меньшей мере тремя ферментами; убиквитин, активированный убиквитин-активирующим ферментом Е1, под действием Е2 соединяется изопептидной связью с остатками лизина с участием убиквитинлигазы Е3, и полиубиквитинилированный ERα затем направляется в протеасому для деградации. Несмотря на то что ER-зависимая регуляция транскрипции и протеасома-опосредуемая деградация ER связаны друг с другом (Lonard, et al., Mol. Cell, 2000, 5: 939-948), самой по себе транскрипции не требуется для деградации ERα, и достаточно сборки комплекса инициации транскрипции, чтобы направить ERα в ядро для протеасомной деградации. Считается, что этот Е2-индуцированный процесс деградации необходим для его способности быстро активировать транскрипцию в ответ на требования, касающиеся пролиферации, дифференцировки и метаболизма клеток (Stenoien, et al., Mol. Cell Biol., 2001, 21: 4404-4412). Фулвестрант также классифицируют как селективный негативный регулятор рецепторов эстрогенов (SERD), разновидность антагонистов, которые также могут индуцировать быструю отрицательную регуляцию ERα посредством связанного с 26S-протеасомой пути. В противоположность этому, SERM, такой как тамоксифен, может приводить к повышению уровней ERα, хотя влияние на транскрипцию аналогично тому, которое наблюдается для SERD.
Приблизительно в 70% случаев рака молочной железы экспрессируются ER и/или рецепторы прогестерона, что указывает на зависимость роста этих опухолевых клеток от гормонов. Также полагают, что рост других раковых опухолей, например яичника и эндометрия, зависит от передачи сигнала через ERα. Терапевтические средства для таких пациентов могут ингибировать передачу сигнала через ER, либо антагонизируя связывание лиганда с ER, например, как тамоксифен, который применяют для лечения раннего и распространенного ER-положительного рака молочной железы как в пред-, так и в постменопаузальном состоянии; либо антагонизируя и отрицательно регулируя ERα, как например фулвестрант, который применяют для лечения рака молочной железы у женщин, прогрессирующего, несмотря на терапию тамоксифеном или ингибиторами ароматазы; либо блокируя синтез эстрогенов, как например ингибиторы ароматазы, которые применяют для лечения раннего и распространенного ER-положительного рака молочной железы. Хотя эти терапии оказали чрезвычайно положительное влияние на лечение рака молочной железы, значительное количество пациентов, опухоли у которых экспрессируют ER, демонстрируют de novo устойчивость к существующим ER-опосредованным терапиям, или устойчивость к этим терапиям у них развивается со временем. Для объяснения устойчивости к первичной терапии тамоксифеном был описан несколько отличный механизм, который в основном связан с переключением действия тамоксифена как антагониста к его действию как агониста либо посредством снижения аффинности связывания определенных кофакторов с комплексом тамоксифен-ERα вследствие сверхэкспрессии этих кофакторов, либо посредством образования вторичных сайтов, которые облегчают взаимодействие комплекса тамоксифен-ERα с кофакторами, которые в норме не связываются с этим комплексом. Поэтому устойчивость могла возникать в результате разрастания клеток, экспрессирующих специфические кофакторы, которые управляют активностью комплекса тамоксифен-ERα. Также существует вероятность того, что сигнальные пути, опосредованные другими факторами роста, непосредственно активируют рецептор ER или коактиваторы для управления клеточной пролиферацией независимо от лиганд-опосредстванной передачи сигнала.
Совсем недавно были идентифицированы мутации в ESR1 с частотами в диапазоне 17-25%, которые возможно обуславливают механизм устойчивости в образцах от пациентов с метастатическими ER-положительными опухолями и в моделях ксенотрансплантатов, выделенных из пациентов (PDX). Эти мутации в большинстве случаев, но не исключительно, располагаются в лиганд-связывающем домене, что приводит к образованию мутированных функциональных белков; примеры аминокислотных замен включают Ser463Pro, VaI543Glu, Leu536Arg, Tyr537Ser, Tyr537Asn и Asp538Gly, при этом замены по аминокислотам 537 и 538 составляют большинство описанных в настоящее время замен. Эти мутации прежде не обнаруживали в геномах из образцов первичного рака молочной железы, охарактеризованных в базе данных Атласа ракового генома. Из 390 образцов первичного рака молочной железы, положительных в отношении экспрессии ER, не обнаружено ни одной мутации в ESR1 (Cancer Genome Atlas Network, 2012, Nature, 490: 61-70). Полагают, что мутации в лиганд-связывающем домене развиваются как устойчивый ответ на эндокринные терапии ингибиторами ароматазы, поскольку эти мутантные рецепторы демонстрируют базальную транскрипционную активность в отсутствие эстрадиола. Анализ кристаллической структуры ER, имеющего мутации в аминокислотах 537 и 538, показал, что наличие обеих мутаций способствовало образованию конформации ER, благоприятной для агониста, в результате смещения положения спирали 12, предоставляя возможность для рекрутмента коактиваторов и тем самым имитируя активированный агонистом ER дикого типа. Опубликованные данные показали, что эндокринные терапевтические средства, такие как тамоксифен и фулвестрант, все еще могут связываться с мутантным ER и ингибировать до некоторой степени активацию транскрипции, и что фулвестрант способен привести к расщеплению рецептора с мутацией Try537Ser, однако для полного ингибирования рецептора могут потребоваться более высокие дозы (Toy et al., Nat. Genetics, 2013, 45: 1439-1445; Robinson et al., Nat. Genetics, 2013, 45: 144601451; Li, S. et al. Cell Rep., 4, 1116-1130 (2013). Поэтому возможно, что некоторые соединения формулы (I) или их фармацевтически приемлемые соли будут способны отрицательно регулировать и антагонизировать мутантный ER, хотя неизвестно, связаны ли мутации в ESR1 на этой стадии с измененным клиническим исходом.
Независимо от того, какой механизм устойчивости или какая комбинация механизмов имеет место, в основе многих из них тем не менее лежат ER-зависимые активности, и такое удаление рецептора посредством механизма с использованием SERD является наилучшим путем удаления рецептора ERα из клетки. В настоящее время единственным SERD, одобренным для клинического применения, является фулвестрант, но несмотря на его механизм действия, фармакологические свойства этого лекарственного средства лимитируют его эффективность ввиду существующего ограничения в 500 мг для месячной дозы, что приводит к обновлению рецептора менее чем на 50% в образцах от пациентов в сравнении с полной отрицательной регуляцией рецептора, наблюдаемой в экспериментах in vitro на линиях клеток молочной железы (Wardell, et al., Biochem. Pharm., 2011, 82: 122-130). Поэтому существует потребность в новых ER-направляющих агентах, имеющих необходимые фармацевтические свойства и демонстрирующих механизм SERD, для обеспечения улучшенной эффективности на ранних стадиях заболевания, стадиях с метастазированием и приобретением устойчивости.
Было установлено, что соединения по изобретению обладают сильной противоопухолевой активностью, являясь полезными для ингибирования неконтролируемой клеточной пролиферации, которая является результатом злокачественного заболевания. Соединения по изобретению обеспечивают противоопухолевый эффект посредством, как минимум, действия в качестве SERD.
Согласно одному из аспектов изобретения, предложены соединение формулы (I) или его фармацевтически приемлемая соль

где:
R1 и R2 каждый независимо представляет собой Н или F;
R3 представляет собой Н или метил; и
либо:
а) R4 представляет собой Н, a R5 представляет собой F; либо
б) R4 представляет собой F, a R5 представляет собой Н.
Согласно другому аспекту изобретения, предложено соединение формулы (I), которое определено выше.
Соединения формулы (I) имеют один, два или три хиральных центра, и изобретение охватывает чистые хиральные формы или их смеси в любой пропорции. Синтез оптически активных форм может быть проведен с использованием стандартных методов органической химии, хорошо известных в данной области техники, например, путем синтеза из оптически активных исходных веществ или путем выделения из рацемической формы. Аналогично, вышеупомянутая активность может быть оценена с использованием стандартных лабораторных методов.
Конкретный энантиомер или диастереоизомер соединения, описанного в данной заявке, может быть более активным, чем другие энантиомеры или диастереоизомеры того же соединения.
Согласно другому аспекту изобретения, предложено соединение формулы (I) или его фармацевтически приемлемая соль, которое является отдельным энантиомером, находящимся в энантиомерном избытке (% ее), составляющем не менее 95, не менее 98% или не менее 99%. Подходящим образом отдельный энантиомер присутствует в энантиомерном избытке (% ее) не менее 99%.
Согласно другому аспекту изобретения, предложена фармацевтическая композиция, содержащая соединение формулы (I), которое является отдельным энантиомером, находящимся в энантиомерном избытке (% ее), составляющем не менее 95, не менее 98% или не менее 99%, или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем. Подходящим образом отдельный энантиомер присутствует в энантиомерном избытке (% ее), составляющем не менее 99%.
Согласно другому аспекту изобретения, предложено соединение формулы (I) или его фармацевтически приемлемая соль, которое является отдельным диастереоизомером, находящимся в диастереомерном избытке (% de), составляющем не менее 95, не менее 98% или не менее 99%. Подходящим образом отдельный диастереоизомер присутствует в диастереомерном избытке (% de), составляющем не менее 99%.
Согласно другому аспекту изобретения, предложена фармацевтическая композиция, содержащая соединение формулы (I), которое является отдельным диастереоизомером, находящимся в диастереомерном избытке (% de), составляющем не менее 95, не менее 98% или не менее 99%, или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем. Подходящим образом отдельный диастереоизомер присутствует в диастереомерном избытке (% de), составляющем не менее 99%.
Согласно одному конкретному аспекту, соединение формулы (I) представляет собой соединение формулы (IA):

Согласно другому аспекту, соединение формулы (I) представляет собой соединение формулы (IB):

Ссылку в данном описании на соединения формулы (I) следует понимать, как упоминание соединений формулы (IA) и/или (IB), если не указано иное.
Например, соединение из примера 1, (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота, представляет собой пример соединения формулы (IA).

Его изомер, (Е)-3-(3,5-дифтор-4-((1S,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота, представляет собой пример соединения формулы (IB).

Эти два изомера являются примерами (Е)-3-(3,5-дифтор-4-((3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая представляет собой пример соединения формулы (I).

Некоторые соединения формулы (I) могут быть кристаллическими и могут существовать более чем в одной кристаллической форме. Следует понимать, что настоящее изобретение охватывает любую кристаллическую либо аморфную форму или их смеси, при этом форма обладает свойствами, полезными для проявления активности в качестве SERD, причем в данной области техники хорошо известно, как определить эффективность кристаллической или аморфной формы в отношении проявления активности в качестве SERD, используя стандартные тесты, описанные далее.
Общеизвестно, что анализ кристаллических веществ можно провести, используя традиционные методы, такие как анализ с применением дифракции рентгеновских лучей на порошке (далее XRPD), дифференциальная сканирующая калориметрия (далее DSC), термогравиметрический анализ (далее TGA), инфракрасная спектроскопия диффузного отражения с преобразованием Фурье (DRIFT), спектроскопия в ближней инфракрасной области (NIR), спектроскопия ядерного магнитного резонанса в растворе и/или твердом состоянии. Содержание воды в таких кристаллических веществах можно определить посредством анализа по методу Карла Фишера.
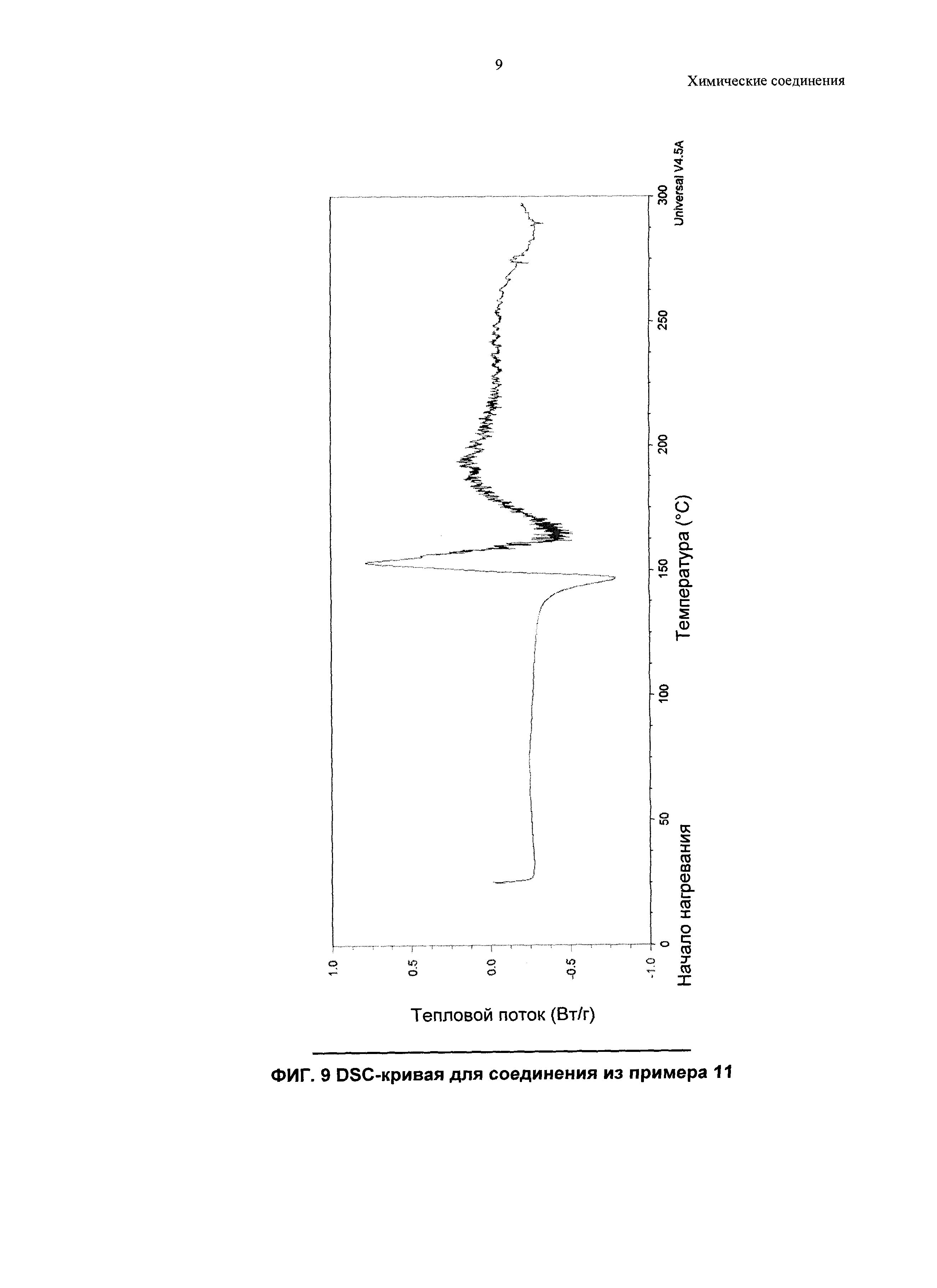
В качестве примера, соединение из примера 7 демонстрирует кристалличность, и была идентифицирована одна его кристаллическая форма.
Соответственно, другой аспект изобретения относится к Форме А (Е)-3-(4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты (пример 7).
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 4,5°.
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 10,8°.
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 4,5 и 10,8°.
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с характерными пиками при 2-тета, приблизительно составляющих 4,5; 4,8; 6,1; 7,9; 9,9; 10,8; 13,4; 14,0; 14,3 и 18,5°.
Согласно настоящему изобретению предложена, кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке, по существу аналогичную картине дифракции рентгеновских лучей на порошке, показанной на Фиг. 1.
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 4,5°±0,2° 2-тета.
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 10,8°±0,2° 2-тета.
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 4,5 и 10,8°±0,2° 2-тета.
Согласно другому аспекту настоящего изобретения, предложена кристаллическая форма, Форма A соединения из примера 7, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 4,5; 4,8; 6,1; 7,9; 9,9; 10,8; 13,4; 14,0; 14,3 и 18,5°±0,2° 2-тета.
Кроме того, соединение из примера 1 также является кристаллическим.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 8,4°.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, приблизительно составляющем 10,9°.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, приблизительно составляющих 8,4° и 10,9°.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 8,4; 10,9; 18,3; 24,0 и 14,0°.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 8,4; 10,9; 18,3; 24,0; 14,0; 19,0; 14,4; 13,0; 15,3; 20,6°.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке по существу аналогичную картине дифракции рентгеновских лучей на порошке, показанной на Фиг.2.
Согласно настоящему изобретению предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 8,4°±0,2° 2-тета.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с одним характерным пиком при 2-тета, составляющем 10,9°±0,2° 2-тета.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке по меньшей мере с двумя характерными пиками при 2-тета, составляющих 8,4° и 10,9°, где указанные значения могут отклоняться на величину ±0,2° 2-тета.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, приблизительно составляющих 8,4; 10,9; 18,3; 24,0 и 14,0°, где указанные значения могут отклоняться на величину ±0,2° 2-тета.
Согласно настоящему изобретению, предложена кристаллическая форма, Форма В (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, которая имеет картину дифракции рентгеновских лучей на порошке с характерными пиками при 2-тета, составляющих 8,4; 10,9; 18,3; 24,0; 14,0; 19,0; 14,4; 13,0; 15,3; 20,6°, где указанные значения могут отклоняться на величину ±0,2° 2-тета.
Когда указано, что настоящее изобретение относится к кристаллической форме соединения из примера 1, Форме B, степень кристалличности подходящим образом составляет более чем примерно 60%, еще более подходящим образом более чем примерно 80%, предпочтительно более чем примерно 90% и более предпочтительно более чем примерно 95%. Наиболее предпочтительно, если степень кристалличности составляет более чем примерно 98%.
Кроме того, когда указано, что настоящее изобретение относится к кристаллической форме соединения из примера 1, Форме B, то это вещество предпочтительно по существу не содержит других кристаллических форм или аморфного вещества. Под термином «по существу не содержит» авторы подходящим образом понимают более чем примерно 60%, еще более подходящим образом более чем примерно 80%, предпочтительно более чем примерно 90%, более предпочтительно более чем примерно 95% и наиболее предпочтительно более чем примерно 99% отдельного полиморфа.
Будет понятно, что значения 2-тета на картинах дифракции рентгеновских лучей на порошке могут незначительно изменяться от одного устройства к другому или от одного образца к другому, и поэтому приведенные значения не следует истолковывать как абсолютные.
Известно, что можно получить картину дифракции рентгеновских лучей на порошке, которая имеет одну или более ошибок измерения в зависимости от условий измерения (таких как используемое оборудование или устройство). В частности, общеизвестно, что интенсивности на картине дифракции рентгеновских лучей на порошке могут колебаться в зависимости от условий измерения. Таким образом, следует понимать, что кристаллические формы по настоящему изобретению, описанные выше, если не указано иное, не ограничиваются кристаллами, которые обеспечивают получение картин дифракции рентгеновских лучей на порошке, идентичных картине дифракции рентгеновских лучей на порошке, показанной на релевантных Фигурах, и любые кристаллы, обеспечивающие получение картин дифракции рентгеновских лучей на порошке, по существу аналогичных показанным на этих Фигурах, попадают в объем настоящего изобретения. Специалист в области дифракции рентгеновских лучей на порошке в состоянии иметь суждение, насколько существенно идентичны картины дифракции рентгеновских лучей на порошке.
Специалистам в области дифракции рентгеновских лучей на порошке также понятно, что на относительную интенсивность пиков могут оказывать влияние, например, гранулы в размере свыше 30 микрон и с неунитарными соотношениями сторон, что может отразиться на анализе образцов. Специалисту также понятно, что на положение отражений может влиять точность размещения по высоте, на которой образец находится в дифрактометре, и калибровка нуля дифрактометра. Плоскостность поверхности образца также может производить небольшой эффект. Следовательно, представленные данные дифракционных картин не должны приниматься за абсолютные значения (см. Jenkins, R. and Snyder, R.L. "Introduction to X-Ray Powder Diffractometry", John Wiley and Sons, 1996; Bunn, C.W. (1948), Chemical Crystallography, Clarendon Press, London; Klug, H.P. and Alexander, L.E. (1974), X-Ray Diffraction Procedures).
Обычно ошибка измерения угла дифракции на картине дифракции рентгеновских лучей на порошке составляет приблизительно ±0,2° 2-тета, и такую величину ошибки измерения необходимо учитывать при рассмотрении данных дифракции рентгеновских лучей на порошке. Кроме того, следует понимать, что интенсивности могут колебаться в зависимости от условий эксперимента и подготовки образца (предпочтительной ориентации).
Конкретные соединения по изобретению представляют собой соединения из примеров, каждое из которых составляет дополнительный независимый аспект изобретения. Другими конкретными соединениями по изобретению являются фармацевтически приемлемая(ые) соль(и) тех соединений из примеров, каждое из которых обеспечивает дополнительный независимый аспект изобретения.
Согласно другому аспекту изобретения, предложено соединение формулы (I), которое получают, следуя любому из примеров, описанных в данном изобретении.
Следующим признаком является любой из объемов изобретения, определенных в данном описании, при условии, что конкретные соединения из примеров, таких как пример 1, 2, 3 и т.д., каждый в отдельности, исключены из притязаний.
Специалистам в данной области техники будет очевидно, что определенные соединения формулы (I) содержат асимметрические замещенные атомы углерода и соответственно могут существовать и могут быть выделены в оптически активных и рацемических формах. Некоторые соединения формулы (I) могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любую рацемическую, оптически активную, полиморфную или стереоизомерную форму или их смеси, при этом форма обладает свойствами, полезными для использования ее в качестве SERD, причем в данной области техники хорошо известно, как получить оптически активные формы (например, путем выделения из рацемической формы методами перекристаллизации, путем синтеза из оптически активных исходных веществ, посредством хирального синтеза, путем разделения с использованием ферментов, путем биотрансформации или путем хроматографического разделения с использованием хиральной неподвижной фазы) и как определить эффективность в качестве SERD, используя стандартные тесты, описанные далее.
Следует понимать, что некоторые соединения формулы (I), определенные выше, могут демонстрировать явление таутомерии. Следует понимать, что настоящее изобретение включает в своем определении любую такую таутомерную форму или их смесь, которая обладает активностью в качестве SERD, и не должно быть ограничено только какой-либо одной таутомерной формой, использованной в изображениях формул или приведенной в названиях примеров. В общем случае, только одну из всех таких таутомерных форм приводят в названиях примеров, следующих далее, или представляют в любых релевантных изображениях формул, следующих далее.
Подразумевается, что настоящее изобретение включает все изотопы атомов, имеющихся в соединениях по настоящему изобретению. Понятно, что изотопы включают такие атомы, которые имеют один и тот же атомный номер, но разные массовые числа. Например, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают13С и14С. Дейтерированный вариант соединения из примера 1 описан в примере 10.
Подходящей фармацевтически приемлемой солью соединения формулы (I) является, например, соль щелочного или щелочноземельного металла, как например, соль натрия, кальция или магния, или соль аммония, или соль с органическим основанием, таким как метиламин, диметиламин, триметиламин, пиперидин, морфолин или трис-(2-гидроксиэтил)амин. Другими подходящими фармацевтически приемлемыми солями соединения формулы (I) могут быть соли других металлов, например, соли, имеющие катионы калия, цинка или других таких металлов, известные в данной области техники. Согласно одному из аспектов изобретения фармацевтически приемлемая соль соединения формулы (I) представляет собой соль с катионом металла, соль аммония или соль с органическим основанием.
Другой подходящей фармацевтически приемлемой солью соединения формулы (I) является, например, соль, образуемая в организме человека или животного после введения соединения формулы (I).
Подходящей фармацевтически приемлемой солью соединения формулы (I) также может быть, например, полученная присоединением кислоты соль соединения формулы (I), например соль присоединения сильной неорганической или органической кислоты, такой как соляная, бромистоводородная, серная или трифторуксусная кислота. Другой возможной подходящей фармацевтически приемлемой солью соединения формулы (I) также может быть соль, описанная ниже для примера 1. Согласно другому аспекту изобретения фармацевтически приемлемая соль соединения формулы (I) представляет собой соль присоединения кислоты.
В экспериментах, относящихся к образованию солей соединений формулы (I), исследовали способность соединения из примера 1 ((Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты) образовывать кристаллические соли. Исследовали следующие кислоты и основания:
уксусную кислоту, адипиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, коричную кислоту, лимонную кислоту, D,L-молочную кислоту, этандисульфоновую кислоту, этансульфоновую кислоту, фумаровую кислоту, соляную кислоту, L-винную кислоту, малеиновую кислоту, яблочную кислоту, малоновую кислоту, метансульфоновую кислоту, 1,5-нафталиндисульфоновую кислоту (napadisylic acid), фосфорную кислоту, сахарин, янтарную кислоту, серную кислоту, толуолсульфоновую кислоту, ацетат кальция, диэтиламин, этаноламин, этилендиамин, гидроксиэтилпирролидин, ацетат магния, меглумин, пиперазин, гидроксид калия, гидроксид натрия, трет-бутиламин, триэтаноламин, трис(гидроксиметил)аминометан (трис) и N,N-диэтилэтаноламин.
С использованием вышеупомянутых кислот и оснований не всегда получали или вообще не получали соли, которые в применяемых экспериментальных условиях можно было выделить в твердой кристаллической форме. Предпочтительные соли соединения из примера 1 включают соли, которые могут быть выделены в кристаллической форме, например, соль бензолсульфоновой кислоты (безилатную соль), соль янтарной кислоты (сукцинатную соль) и соль малеиновой кислоты (малеатную соль).
Согласно одному из аспектов подходящие соли соединения из примера 1 могут включать безилат, сукцинат и малеат. Согласно другому аспекту подходящей солью соединения из примера 1 может быть малеатная соль, которая описана в примере 11.
Кроме этого следует понимать, что подходящий фармацевтически приемлемый сокристалл соединения формулы (I) также образует аспект настоящего изобретения. Во избежание неясности, термин «со-кристалл (или сокристалл)» относится к многокомпонентной системе, в которой имеются молекула или молекулы хозяина - активного фармацевтического ингредиента (API) и молекула или молекулы гостя (или коформера). В сокристалле, и молекула API, и молекула гостя (или коформера) по отдельности существуют в виде твердого вещества при комнатной температуре, когда они находятся в своей чистой форме (чтобы отличать сокристалл от сольватов или гидратов). Соли, в которых происходит значительный или полный обмен протонами между молекулой API и молекулой гостя, исключены из этого конкретного определения. В сокристалле, молекулы API и коформера взаимодействуют посредством образования водородных связей и возможно других нековалентных взаимодействий. Фармацевтически приемлемые коформеры включают электронейтральные молекулы, такие как никотинамид, резорцин и ксиленолы, равно как и ионизируемые молекулы, такие как щавелевая кислота, 3,5-дигидроксибензойная кислота и изохинолин (при этом степень обмена протонами является определяющей в отношение того, образуется ли соль или сокристалл). Можно отметить, что сокристалл сам может образовывать сольваты, включая гидраты.
Кроме этого следует понимать, что подходящий фармацевтически приемлемый сольват соединения формулы (I) также образует аспект настоящего изобретения. Подходящим фармацевтически приемлемым сольватом является, например, гидрат, как например полугидрат, моногидрат, дигидрат или тригидрат, или его альтернативное количество.
Кроме этого следует понимать, что подходящее фармацевтически приемлемое пролекарство соединения формулы (I) также образует аспект настоящего изобретения. Соответственно, соединения по изобретению можно вводить в форме пролекарства, представляющего собой соединение, которое распадается в организме человека или животного с высвобождением соединения по изобретению. Пролекарство можно применять для изменения физических свойств и/или фармакокинетических свойств соединения по изобретению. Пролекарство может образовываться, если соединение по изобретению содержит подходящую группу или подходящий заместитель, к которой (которому) может присоединяться группа, модифицирующая какое-либо свойство. Примеры пролекарств включают расщепляемые in vivo производные сложных эфиров, которые могут образовываться по карбоксильной группе в соединении формулы (I).
Соответственно, настоящее изобретение включает те соединения формулы (I), как определено выше, которые становятся доступными посредством органического синтеза и которые становятся доступными в организме человека или животного в результате расщепления их пролекарственной формы. Соответственно, настоящее изобретение включает те соединения формулы (I), которые получают методами органического синтеза, а также те соединения, которые образуются в организме человека или животного в ходе метаболизма соединения-предшественника, то есть соединение формулы (I) может представлять собой полученное синтетическим путем соединение или полученное метаболическим путем соединение.
Подходящим фармацевтически приемлемым пролекарством соединения формулы (I) является пролекарство, которое, согласно обоснованному медицинскому мнению считается подходящим для введения в организм человека или животного без нежелательных фармакологических активностей и без чрезмерной токсичности.
Различные формы пролекарства описаны, например, в следующих документах:
а) Methods in Enzymoloqy, Vol. 42, p. 309-396, под ред. K. Widder и др. (Academic Press, 1985);
б) Design of Pro-drugs, под ред. H. Bundgaard, (Elsevier, 1985);
в) A Textbook of Drug Design and Development, под ред. Krogsgaard-Larsen и H. Bundgaard, Chapter 5 "Design and Application of Pro-drugs", by H. Bundgaard p.113-191 (1991);
г) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992);
д) H. Bundgaard ef al., Journal of Pharmaceutical Sciences, 77, 285 (1988);
е) N. Kakeya et al., Chem. Pharm. Bull. 32, 692 (1984);
ж) Т. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems", A.C.S. Symposium Series, Volume 14; и
з) E. Roche (редактор), "Bioreversible Carriers in Drug Design", Pergamon Press, 1987.
Подходящим фармацевтически приемлемым пролекарством соединения формулы (I), которое имеет карбоксильную группу, является, например, его сложный эфир, расщепляемый in vivo. Расщепляемым in vivo сложным эфиром соединения формулы (I), содержащего карбоксильную группу, является, например, фармацевтически приемлемый сложный эфир, который расщепляется в организме человека или животного с получением исходной кислоты. В случае карбоксильной группы подходящие фармацевтически приемлемые сложные эфиры включают (1-6С)алкиловые эфиры, такие как метиловый, этиловый и трет-бутиловый, (1-6С)алкоксиметиловые эфиры, такие как метоксиметиловые эфиры, (1-6С)алканоилоксиметиловые эфиры, такие как пивалоилоксиметиловые эфиры, 3-фталидиловые эфиры, (3-8С)циклоалкилкарбонилокси-(1-6С)алкиловые эфиры, такие как циклопентилкарбонилоксиметиловый и 1-циклогексил-карбонилоксиэтиловый эфиры, 2-оксо-1,3-диоксоленилметиловые эфиры, такие как 5-метил-2-оксо-1,3-диоксолен-4-илметиловые эфиры, и (1-6С)алкоксикарбонилокси-(1-6С)алкиловые эфиры, такие как метоксикарбонилоксиметиловый и 1-метоксикарбонилоксиэтиловый эфиры.
Подходящим фармацевтически приемлемым пролекарством соединения формулы (I), которое имеет карбоксильную группу, является, например, расщепляемый in vivo амид, такой как N-C1-6алкил- и N,N-ди-(C1-6алкил)амид, например N-метил-, N-этил-, N-пропил-, N,N-диметил-, N-этил-N-метил- или N,N-диэтиламид.
Действия in vivo соединения формулы (I) могут проявляться частично посредством одного или более метаболитов, которые образуются в организме человека или животного после введения соединения формулы (I). Как указано ранее, действия in vivo соединения формулы (I) также могут проявляться в процессе метаболизма соединения-предшественника (пролекарства).
Из in vitro систем клеток человека были идентифицированы две изомерные формы активного метаболита соединения из примера 1, которые показаны ниже (при этом данные изомеры являются диастереомерными соединениями вследствие существования обеих конфигураций при атоме углерода, отмеченном *), и синтез обоих изомеров приведен в данном описании в примерах 14А и B:
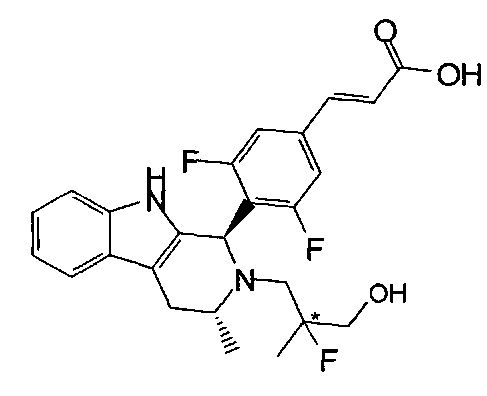
Помимо этого, считается, что приведенное ниже соединение представляет собой активный метаболит у некоторых видов, например у мышей:

Такие активные метаболиты образуют дополнительные независимые аспекты изобретения.
Во избежание неясности следует понимать, что там, где в данном описании группа квалифицируется как «ранее определенная» или «определенная ранее», указанная группа охватывает первое упоминаемое и самое широкое определение, а также каждое и все конкретные определения для этой группы.
Конкретные новые соединения по изобретению включают, например, соединения формулы (I) или их фармацевтически приемлемые соли, где, если не указано иное, каждый из R1 и R2 имеет любое из значений, определенных ранее или в приведенных ниже утверждениях.
Согласно одному из аспектов R1 представляет собой водород. Согласно другому аспекту R1 представляет собой фтор.
Согласно одному из аспектов R2 представляет собой водород. Согласно другому аспекту R2 представляет собой фтор.
Согласно одному из аспектов оба R1 и R2 представляют собой водород. Согласно другому аспекту оба R1 и R2 представляют собой фтор. Согласно другому аспекту R1 представляет собой водород, a R2 представляет собой фтор.
Согласно одному из аспектов R3 представляет собой водород. Согласно другому аспекту R3 представляет собой метил.
Согласно одному из аспектов R4 представляет собой водород, a R5 представляет собой фтор. Согласно другому аспекту R5 представляет собой водород, a R4 представляет собой фтор.
Конкретными соединениями по изобретению являются, например, соединения формулы (I), раскрытые в Примерах, которые приведены далее.
Например, конкретным соединением по изобретению является соединение формулы (I), выбранное из любого соединения, приведенного ниже:
(Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3,5-дифтор-4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3,5-дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3-фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
или его фармацевтически приемлемая соль.
Другим конкретным соединением по изобретению является соединение формулы (I), выбранное из любого соединения, приведенного ниже:
(Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3,5-дифтор-4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3,5-дифтор-4(1R)-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3,5-дифтор-4(1R)-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4(1R)-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(4(1R)-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты;
(Е)-3-(3-фтор-4(1R)-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты; и
(Е)-3-[4-[(1R,3R)-1-дейтеро-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-1-ил]-3,5-дифтор-фенил]проп-2-еновой кислоты; или его фармацевтически приемлемая соль.
Конкретной фармацевтически приемлемой солью по изобретению является (1R,3R)-1-{4-[(Е)-2-карбоксиэтенил]-2,6-дифторфенил}-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-бета-карболин-2-ия малеат.
Согласно другому аспекту настоящего изобретения предложен способ получения соединения формулы (I) или его фармацевтически приемлемой соли. Подходящий способ проиллюстрирован следующими репрезентативными вариантами способа, в которых, если не указано иное, R1-R5 принимают любое из значений, определенных ранее. Необходимые исходные вещества могут быть получены по стандартным методикам органической химии. Получение таких исходных веществ описано вместе со следующими далее репрезентативными вариантами способа и в сопроводительных примерах. Альтернативно, необходимые исходные вещества получают по методикам, аналогичным приведенным с целью иллюстрации, которые известны специалисту в области органической химии.
Соединения формулы (I) подходящим образом получают путем гидролиза сложноэфирного производного формулы (II), где R6 представляет собой (1-6С)алкил, такой как метил. Гидролиз подходящим образом осуществляют в присутствии основания, например, с использованием гидроксида натрия в подходящем растворителе (таком как водный THF (тетрагидрофуран) и МеОН (или другой аналогичный спирт) или таком как водный спирт, например водный изопропанол) и при подходящей температуре, подходящим образом, комнатной температуре.

Соединения формулы (II) могут быть получены, например:
а) в результате взаимодействия соединения формулы (III) с соединением формулы (IV) в условиях, известных в данной области техники как подходящие для реакций Пикте-Шпенглера (как например, в присутствии кислоты (такой как уксусная кислота) и в подходящем растворителе (например, толуоле) и при подходящей температуре (такой как 80°С)); или


б) в результате взаимодействия соединения формулы (V) с соединением формулы (VI), где LG представляет собой уходящую группу, известную в данной области техники, такую как галогенид или трифторметансульфонат (трифлат), подходящим образом трифлат, в присутствии основания (например, аминного основания, такого как N-этил-N-изопропилпропан-2-амин) и подходящего полярного растворителя (такого как диоксан) при подходящей температуре (например, от комнатной температуры до 90°С).


Соединения формулы (III) могут быть получены в результате взаимодействия соединения формулы (VII) с соединением формулы (VI) в условиях, которые описаны выше для взаимодействия соединений формул (V) и (VI).

Соединения формулы (IV) могут быть получены в результате взаимодействия соединения формулы (VIII) со сложным алкилакрилатным эфиром (таким как метилакрилат, если R6 представляет собой метил) в условиях, известных в данной области техники для реакции Хека; то есть в присутствии арилфосфина (например, три-о-толилфосфина), палладиевого катализатора (такого как ацетат палладия(II)) и основания (такого как триэтиламин) в подходящем растворителе (таком как DMA (диметилацетамид)) и при подходящей температуре (например, 80°С).

Соединения формулы (V) могут быть получены в результате взаимодействия соединения формулы (VII) с соединением формулы (IV) с использованием условий, аналогичных описанным для взаимодействия приведенных выше соединений формул (III) и (IV).
Соединения формулы (VI), где LG представляет собой трифлат, могут быть получены так, как показано ниже на схемах 1 и 2. Другое соединение формулы (VI), где LG представляет собой группу, отличную от трифлата, могут быть получены аналогичными способами, известными в данной области техники.
Схема 1

Стадия 1: фторирующий агент, например N,N-диэтил-1,1,2,3,3,3-гексафторпропан-1-амин/DCM(дихлорметан)/КТ(комнатная температура), затем восстанавливающий агент, например алюмогидрид лития/THF/KT.
Стадия 2: трифторметансульфоновый ангидрид/основание, например 2,6-лутидин/ОСМ/0°С.
Схема 2
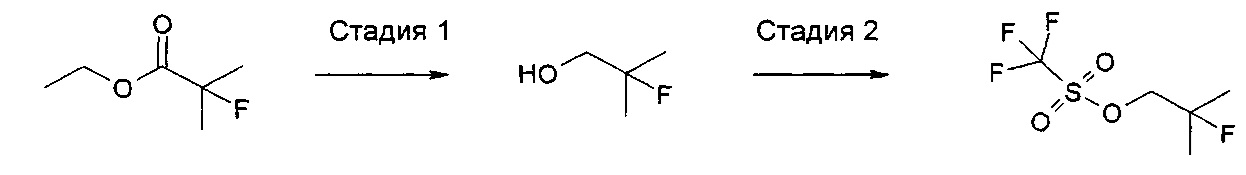
Стадия 1: восстанавливающий агент, например алюмогидрид лития/эфир/0°С.
Стадия 2: трифторметансульфоновый ангидрид/основание, например 2,6-лутидин/DCM/-10°С.
Соединения формулы (I) являются хиральными. Специалисту будет очевидно, что для получения желаемых изомеров можно использовать стереоизбирательные взаимодействия. Альтернативно, соотношение стереоизомеров (стереохимию) можно откорректировать подходящим образом, например, путем эпимеризации цис-изомеров в транс-изомеры посредством подкисления промежуточного соединения с защищенной аминогруппой, как проиллюстрировано в данном изобретении в примере 4 (и описано, например, в J. Org. Chem., 2009, 74, 2771-2779).
Согласно другому аспекту изобретения предложен способ получения соединения формулы (I), включающий гидролиз соединения формулы (II), подходящим образом в присутствии основания.
Следует понимать, что также возможны другие изменения стадий способа в описанных выше вариантах способа.
Следует понимать, что при необходимости любое соединение формулы (I), полученное любым из способов, изложенных ранее, может быть преобразовано в другое соединение формулы (I).
Когда требуется фармацевтически приемлемая соль соединения формулы (I), ее можно получить, например, путем взаимодействия указанного соединения с подходящим основанием.
Когда требуется фармацевтически приемлемое пролекарство соединения формулы (I), его можно получить с использованием традиционной методики. Например, расщепляемый in vivo сложный эфир соединения формулы (I) может быть получен, например, путем взаимодействия соединения формулы (I), содержащего карбоксильную группу, с фармацевтически приемлемым спиртом. Дополнительная информация по пролекарствам приведена ранее.
Также будет очевидно, что в некоторых из упомянутых ранее реакций необходимой или желательной может оказаться защита всех чувствительных групп в соединениях. Случаи, где необходима или желательна защита, и подходящие для этого методы известны специалистам в данной области техники. В соответствии с общепринятой практикой можно использовать традиционные защитные группы (для иллюстрации см. T.W. Green, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991). Так, если реагенты включают такие группы, как амино, карбокси или гидрокси, может оказаться желательным защитить данную группу в некоторых из упомянутых в данном описании реакций.
Подходящей защитной группой для амино- или алкиламиногруппы является, например, ацильная группа, например алканоильная группа, такая как ацетил, алкоксикарбонильная группа, например метоксикарбонильная, этоксикарбонильная или трет-бутоксикарбонильная группа, арилметоксикарбонильная группа, например бензилоксикарбонильная, или ароильная группа, например бензоильная. Условия удаления защиты для вышеупомянутых защитных групп, несомненно, меняются в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или алкоксикарбонильная группа, или ароильная группа могут быть удалены, например путем гидролиза в присутствии подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно, ацильная группа, такая как трет-бутоксикарбонильная группа, может быть удалена, например, путем обработки подходящей кислотой, такой как соляная, серная или фосфорная кислота либо трифторуксусная кислота, арилметоксикарбонильная группа, такая как бензилоксикарбонильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле, или путем обработки кислотой Льюиса, например трис(трифторацетатом) бора. Подходящей альтернативной защитной группой для первичной аминогруппы является, например, фталоильная группа, которая может быть удалена путем обработки алкиламином, например диметиламинопропиламином, или гидразином.
Подходящей защитной группой для гидроксильной группы является, например, ацильная группа, например алканоильная группа, такая как ацетил, ароильная группа, например бензоил, или арилметильная группа, например бензил. Условия удаления защиты для вышеупомянутых защитных групп, несомненно, будут меняться в зависимости от выбора защитной группы. Так, например, ацильная группа, такая как алканоильная или ароильная группа, может быть удалена, например, путем гидролиза в присутствии подходящего основания, такого как гидроксид щелочного металла, например гидроксид лития или натрия. Альтернативно, арилметильная группа, такая как бензильная группа, может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле.
Подходящей защитной группой для карбоксильной группы является, например, этерифицирующая группа, например метильная или этильная группа, которая может быть удалена, например, путем гидролиза в присутствии основания, такого как гидроксид натрия, или, например, трет-бутильная группа, которая может быть удалена, например, путем обработки кислотой, например органической кислотой, такой как трифторуксусная кислота, или, например, бензильная группа, которая может быть удалена, например, путем гидрирования над катализатором, таким как палладий-на-угле.
Защитные группы можно удалить на любой соответствующей стадии синтеза, применяя традиционные методы, хорошо известные в области химии.
Некоторые из промежуточных соединений (например, соединения формул II, III, IV, V, VI, VII и VIII, в частности, соединения формулы II, III и/или V) определенных в данном описании, являются новыми, и они предложены в качестве следующего признака данного изобретения.
Биологические анализы
Приведенные далее анализы использовали для измерения эффектов соединений по настоящему изобретению.
Анализ связывания с ERα
Способность соединений связываться с выделенным лиганд-связывающим доменом рецептора эстрогенов альфа (ERальфа-LBD (GST (глутатион-3-трансфераза))) оценивали в конкурентных анализах с использованием метода резонансного переноса энергии флуоресценции с временным разрешением (TR-FRET) с детекцией по конечной точке (LanthaScreen™). Для анализа LanthaScreen с применением TR-FRET с детекцией по конечной точке приобретали у Invitrogen подходящий флуорофор (Fluormone ES2, код продукта Р2645) и рекомбинантный лиганд-связывающий домен рецептора эстрогенов альфа человека (код продукта PV4543) и использовали для измерения связывания с соединениями. Принцип анализа состоит в том, что ERальфa-LBD (GST) добавляют к флуоресцентному лиганду с образованием комплекса рецептор/флуорофор. Меченное тербием антитело (Ab) к GST (код продукта PV3551) используют для осуществления непрямого мечения рецептора в результате связывания с его GST-меткой, и детекцию конкурентного связывания осуществляют по способности тестируемых соединений вытеснять флуоресцентный лиганд, что приводит к потере сигнала TR-FRET между Tb-антителом к GST и индикатором. Данный анализ проводили, как описано ниже, при этом добавление всех реагентов выполняли с использованием рабочей станции для микрожидкостных систем BioRAPTR FRD от Beckman Coulter:
1) с помощью акустического диспенсера добавить по 120 нл раствора тестируемого соединения в используемые в анализе черные 384-луночные планшеты с лунками малого объема;
2) приготовить раствор 1х ERальфa-LBD/Tb-Ab к GST в буфере для скрининга по ES2 и выполнить инкубирование в течение 20 минут;
3) перед применением добавить 1х флуорофор к раствору 1х ERaльфa-LBD/Tb-Ab к GST;
4) внести по 12 мкл реагента 1х AR-LBD/Tb-Ab к GST/флуорофор в каждую лунку используемого в анализе планшета;
5) закрыть используемый в анализе планшет для защиты реагентов от света и испарения и выполнить инкубирование при комнатной температуре в течение 1 часа;
6) осуществить возбуждение флуоресценции излучением при 337 нм и провести измерение сигнала эмиссии флуоресценции для каждой лунки при 490 нм и 520 нм, используя PheraSTAR от BMG.
Соединения вносили непосредственно из микропланшета с исходными растворами соединений, содержащего соединения в серийных разведениях (4 лунки, содержащие соединения в конечной концентрации 10 мМ, 0,1 мМ, 1 мкМ и 10 нМ, соответственно), в микропланшет для анализа, используя Echo 550 от Labcyte. Echo 550 представляет собой устройство для дозирования жидкостей, в котором применяется основанная на акустических волнах технология для осуществления непосредственных переносов из-микропланшета-в-микропланшет растворов соединений в DMSO (диметилсульфоксид), и эта система может быть запрограммирована для переноса большого числа проб соединений небольшого объема (нл) из разных лунок планшета с исходными растворами соединений с целью получения в этом анализе желаемого серийного разведения соединений, которые затем наполняют для нормирования концентрации DMSO во всем диапазоне разведений. В общей сложности в каждую лунку добавляли по 120 нл соединения плюс DMSO, и соединения тестировали в формате концентрация-ответ по 12 точкам в диапазоне конечных концентраций соединений 100; 29,17; 10,42; 2,083; 1; 0,292; 0,104; 0,02083; 0,01; 0,002917; 0,001042; 0,0001 мкМ, соответственно. Данные TR-FRET в формате доза-ответ, полученные для каждого соединения, экспортировали в подходящий пакет программ (такой как Origin или Genedata) для проведения анализа с приненением аппроксимации кривых. Конкурентное связывание с ERальфa выражали в виде величины IC50. Ее определяли, рассчитывая концентрацию соединения, необходимую для получения уменьшения на 50% связывания меченого соединения с ER альфа-LBD.
Анализ отрицательной регуляции ER в MCF-7
Способность соединений отрицательно регулировать численность рецепторов эстрогенов (ER) оценивали в основанном на использовании клеток иммунофлуоресцентном анализе с применением линии клеток протоковой карциномы молочной железы человека MCF-7. Клетки MCF-7 реактивировали непосредственно из криофлакона (приблизительно 5×106 клеток) в среде для анализа (модифицированной Дульбекко среде Игла (DMEM) без фенолового красного (Sigma D5921), содержащей 2 мМ L-глутамин и 5% (об./об.) фетальной телячьей сыворотки, обработанной активированным углем/декстраном). Клетки вводили с помощью шприца один раз, используя стерильную иглу калибра 18G×1,5 дюйма (1,2×40 мм) широкого спектра применения, и плотность клеток измеряли, используя счетчик Коултера (Beckman). Клетки разбавляли далее в среде для анализа до плотности 3,75×104 клеток в одном мл и добавляли по 40 мкл на одну лунку в черные, обработанные для культур тканей 384-луночные планшеты с прозрачным дном (Costar, №3712), используя Thermo Scientific Matrix WellMate или Thermo Multidrop. После посева клеток планшеты инкубировали в течение ночи при 37°C, 5% CO2 (вращающийся инкубатор от Liconic). Данные тестирования получали, используя устройство для переформирования смесей (compound reformatter) Echo®, модель 555, от LabCyte, которое является частью автоматизированного производственного модуля (объединенного производственного модуля Echo 2). Чтобы сформировать 384-луночный планшет для дозирования соединений (Labcyte, Р-05525-CV1), использовали концентрированные 10 мМ растворы тестируемых соединений. По 40 мкл каждого из концентрированных 10 мМ растворов соединений вносили в лунки первого квадранта и затем выполняли постадийные серийные разведения 1:100 в DMSO, используя устройство для дозирования жидкостей Hydra II (MATRIX, UK) с получением по 40 мкл разбавленных растворов соединения в лунках 2 (0,1 мМ), 3 (1 мкМ) и 4 (0,01 мкМ) квадрантов, соответственно. В лунки ряда Р планшета с исходными растворами соединений вносили по 40 мкл DMSO для нормирования концентрации DMSO во всем диапазоне доз. Чтобы заполнить контрольные лунки, в планшет с исходными растворами соединений в лунки ряда O1 добавляли по 40 мкл DMSO, а в лунки ряда O3 добавляли по 40 мкл 100 мкМ Faslodex® в DMSO. В Echo 550 применяется основанная на акустических волнах технология для осуществления непосредственных переносов из-микропланшета-в-микропланшет растворов соединений в DMSO в используемые в анализе планшеты. Эта систему можно запрограммировать для переноса между микропланшетами проб объемом всего лишь 2,5 нл с многократными приращениями дозы, и тем самым можно получать серийное разведение соединения в лунках используемого в анализе планшета, которые затем наполняют для нормирования концентрации DMSO во всем диапазоне разведений. В планшеты с клетками вносили соединения из планшета с исходными растворами соединений, подготовленного, как описано выше, с получением 12 дублированных проб в диапазоне доз от 3 мкМ до 3 пМ с применением 3-кратных разведений и одной пробы в конечном 10-кратном разведении, используя объединенный производственный модуль Echo 2. В контрольные лунки для получения максимального сигнала вносили DMSO, получая конечную концентрацию 0,3%, а в контрольные лунки для получения минимального сигнала вносили Faslodex®, получая конечную концентрацию 100 нМ, соответственно. Далее планшеты инкубировали в течение 18-22 часов при 37°С, 5% CO2 и затем фиксировали путем добавления 20 мкл 11,1%-ного (об./об.) раствора формальдегида (в забуференном фосфатом физиологическом растворе (PBS)), получая конечную концентрацию формальдегида 3,7% (об./об.). Клетки фиксировали при комнатной температуре в течение 20 мин, после чего промывали дважды, используя 250 мкл смеси PBS/ProClin (PBS с биоцидным консервантом), используя устройство для промывки планшетов от BioTek, затем во все лунки добавляли по 40 мкл смеси PBS/ProClin и планшеты хранили при 4°С. Описанный выше метод фиксирования осуществляли на объединенном производственном модуле Echo 2. Иммуноокрашивание проводили с использованием автоматизированного производственного модуля AutoElisa. Смесь PBS/ProClin отсасывали из всех лунок и клетки подвергали пермеабилизации, используя 40 мкл PBS, содержащего 0,5% Tween™ 20 (об./об.), в течение 1 часа при комнатной температуре. Планшеты трижды промывали, используя по 250 мкл смеси PBS/0,05% (об./об.) твина 20 с ProClin (PBST с биоцидным консервантом) и затем добавляли по 20 мкл раствора кроличьего моноклонального антитела к ERα (SP1) (Thermofisher) в разведении 1:1000 в смеси PBS/Tween™/3% (масс./об.) бычьего сывороточного альбумина. Планшеты инкубировали в течение ночи при 4°С (вращающийся инкубатор от Liconic) и затем трижды промывали, используя по 250 мкл смеси PBS/0,05% (об./об.) Tween™ 20 с ProClin (PBST). Затем планшеты инкубировали после добавления по 20 мкл/лунка антител козы к IgG кролика, конъюгированных с AlexaFluor 594, или антител козы к IgG кролика, конъюгированных с AlexaFluor 488 (Molecular Probes), с красителем Хехст (Hoechst) в разведении 1:5000 в смеси PBS/Tween™/3% (масс./об.) бычьего сывороточного альбумина в течение 1 ч при комнатной температуре. Затем планшеты трижды промывали, используя по 250 мкл смеси PBS/0,05% (об./об.) Tween™ 20 с ProClin (PBST с биоцидным консервантом). В каждую лунку добавляли по 20 мкл PBS, планшеты закрывали крышкой для черных планшетов и хранили при 4°C, после чего прочитывали. Планшеты прочитывали с использованием Cellomics ArrayScan, определяя флуоресценцию на 594 нм (момент времени 24 ч) или на 488 нм (момент времени 5 ч) для измерения уровня рецептора ERα в каждой лунке. Среднее значение общей интенсивности нормировали на количество клеток, получая значение общей интенсивности от одной клетки. Данные экспортировали в подходящий пакет программ (такой как Origin) для проведения анализа с приненением аппроксимации кривых. Отрицательную регуляцию рецептора ERα выражали в виде величины IC50, и ее определяли, рассчитывая концентрацию соединения, которая была необходима для получения уменьшения на 50% среднего значения максимальной общей интенсивности сигнала.
Хотя, как и ожидалось, фармакологические свойства соединений формулы (I) меняются в зависимости от структурных изменений, в большинстве случаев активность, которой обладают соединения формулы (I), может быть продемонстрирована для приведенных ниже концентраций или доз в одном или более чем одном из приведенных выше тестов.
Приведенные ниже данные получали для соединений из примеров (приведенные ниже данные могут быть результатом одного эксперимента или представлять собой среднее значение для многократно повторенных экспериментов).


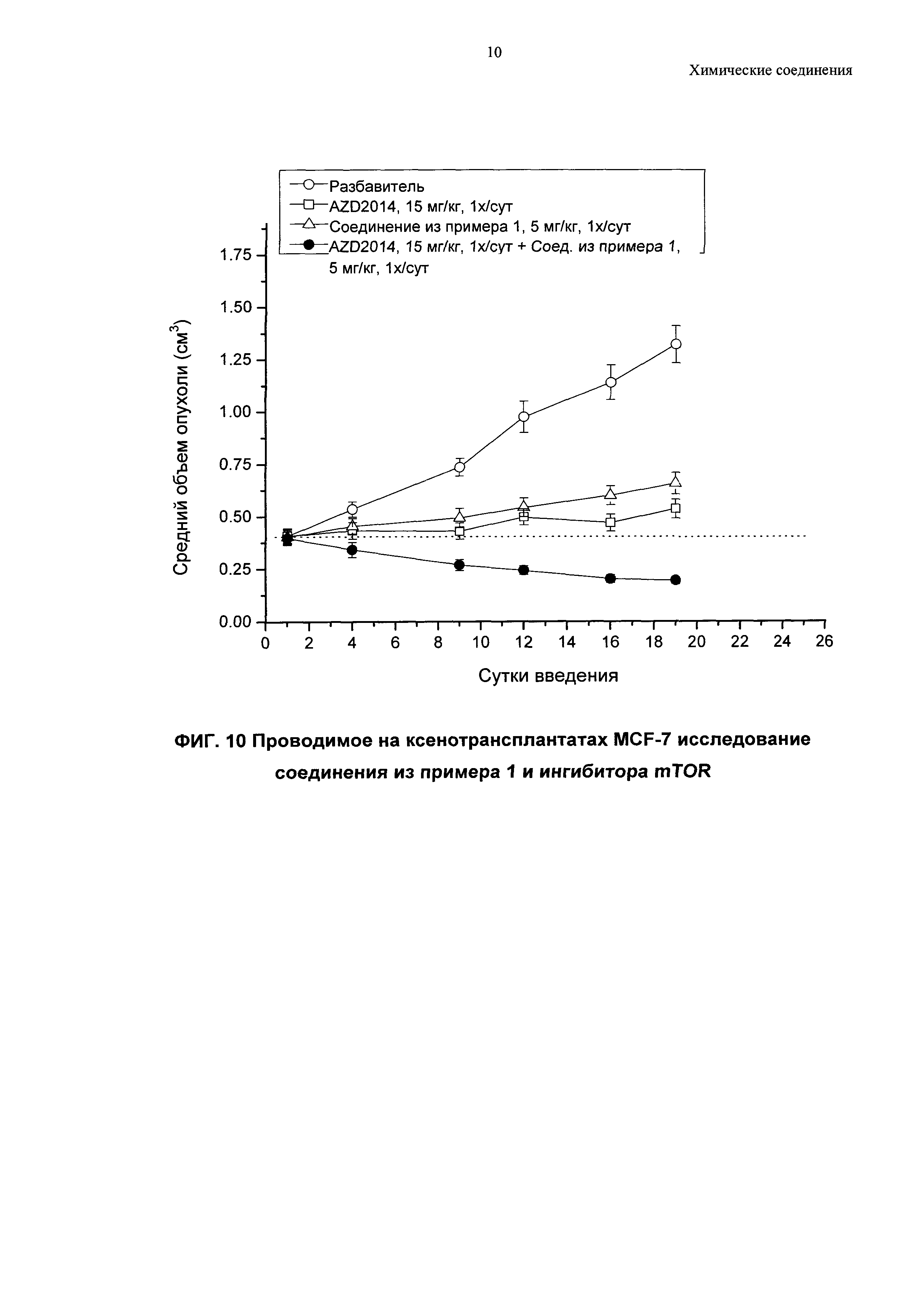
Проводимое in vivo на ксенотрансплантатах MCF-7 исследование соединения из примера 1 в виде единственного агента и в комбинации с ингибитором mTOR (мишень рапамицина у млекопитающих)
Клетки MCF7 (5×106 клеток, суспендированных в 100 мкл питательной среды RPMI (среда 1640 от Мемориального института Розуэлла Парка (Roswell Park Memorial Institute))) имплантировали подкожно в задний пах мышам с тяжелым комбинированным иммунодефицитом (SCID); спустя одни сутки каждой мыши хирургическим путем имплантировали гранулу эстрогена 0,5 мг с высвобождением в течение 21 суток (Innovative Research, USA). Опухоли измеряли два раза в неделю, и изменения в объеме опухолей и ингибировании роста определяли с помощью измерения двусторонним штангенциркулем (длина × ширина), при этом за длину принимали наибольший диаметр по всей опухоли, а за ширину - соответствующий перпендикуляр. Объем опухолей рассчитывали, используя формулу (длина × ширина) × √ (длина × ширина) × (π/6).
Опухоли измеряли через 13 суток после имплантации клеток, чтобы иметь возможность провести рандомизацию мышей в тестируемых группах. Обработку соединениями начинали на следующие сутки (т.е. через 14 суток после имплантации клеток).
Ингибитор mTOR AZD2014 вводили перорально (п.о.) разным группам мышей в дозе 15 мг/кг один раз в сутки ежедневно в объеме 0,1 мл на 10 г. Соединение из примера 1 вводили перорально в дозе 5 мг/кг один раз в сутки в объеме 0,1 мл/10 г. Одной группе животных в качестве контроля вводили п.о. разбавитель. По девять мышей на одну группу использовали для групп с введением активных агентов и для контрольной группы.
Данные, полученные в этом исследовании, показаны на Фиг. 10.
Эффект комбинации соединения формулы (I) с ингибитором РI3Кα/δ можно исследовать аналогично тому, как описано выше для комбинации с ингибитором mTOR.
Исследование эффективности на ксенотрансплантатах НСС1428 после длительной эстрогенной депривации (НСС1428 LTED)
По окончании подходящего периода клеточного культивирования LTED-клетки НСС1428 (1×106) имплантировали подкожно в задний пах самок мышей NSG с ослабленным иммунитетом (Jackson Labs, USA), которых подвергали овариэктомии. Опухоли измеряли два раза в неделю, и изменения в объеме опухолей и ингибировании роста определяли с помощью измерения двусторонним штангенциркулем (длина × ширина), при этом за длину принимали наибольший диаметр по всей опухоли, а за ширину - соответствующий перпендикуляр. Объем опухолей рассчитывали, используя формулу (длина × ширина) × √ (длина × ширина) × (π/6). Опухоли измеряли один раз в неделю после имплантации клеток, пока средний размер не достигал 150 мм3, тогда мышей разделяли на рандомизированные тестируемые группы, при этом каждая группа содержала по 10 мышей. Обработку соединениями начинали на следующие сутки (62-е сутки в этом исследовании) и опухоли продолжали измерять один раз в неделю. Соединение из примера 1 вводили перорально (п.о.) в дозе 25 мг/кг один раз в сутки ежедневно в объеме 0,1 мл на 10 г. Другой группе животных в качестве контроля вводили п.о. разбавитель.
По окончании 28 суток введения обработанные разбавителем контрольные опухоли выросли в среднем на 220 мм3 (согласно среднегеометрическим значениям), в то время как опухоли у мышей, обработанных соединением из примера 1, уменьшились в размере на 46 мм3, что составляет ингибирование опухолевого роста на 121% (P<0,001 согласно t-критерию Стьюдента для непарных выборок).
Чтобы провести измерение уровней рецептора эстрогенов по белку в ксенотрансплантатных опухолях, отбирали образцы опухолей через 24 ч после заключительного введения разбавителя или соединения из примера 1 и мгновенно замораживали в жидком азоте. Чтобы экстрагировать белки, фрагменты опухоли добавляли к 700 мкл буфера для экстракции из клеток (Invitrogen, FNN0011) с добавлением ингибиторов фосфатаз от Sigma (№2 (Р5726) и 3 (Р0044) в разведении 1 к 100) и полного коктейля ингибиторов протеаз от Roche (11836145001) (1 таблетка на 50 мл), 1 мМ дитиотреита (DTT) в пробирки для образцов емкостью 2 мл, помещенные в воду со льдом. Гомогенизацию образца проводили, используя Mixer/Mill (уровень 27/с) и 3 цикла гомогенизации по 2 мин. Образцы центрифугировали в течение короткого промежутка времени, чтобы удостовериться в завершении гомогенизации опухолей. Гомогенат обрабатывали ультразвуком в течение 10 секунд и затем центрифугировали на высокоскоростной центрифуге (13000 об/мин) в течение 15 мин. Измеряли уровни белка в супернатанте и приблизительно по 45 мкг белка вносили в 15 лунок для разделения на гелях в бис-трис-буферной системе (гелях с градиентом 4-12%), используя стандартные методы. После разделения белков и переноса на нитроцеллюлозный фильтр добавляли антитело к альфа-пептиду (68 кДа) рецептора эстрогенов (ThermoFisher, SP1, №9101S), разбавленное 1:400 в смеси молоко/PBS/T, и инкубировали в течение ночи при 4°С. Фильтр промывали 3×5 мин приблизительно в 20 мл TBS/0,05% Т, добавляли вторичное детектирующее анти-кроличье антитело, разбавленное 1:2000 в 5%-ном молоке Marvel в TBS/T (забуференный трисом физиологический раствор/твин), и инкубировали в течение 1 ч при КТ. Детекцию сигнала осуществляли, используя хемилюминесцентный субстрат с увеличенной продолжительностью хемилюсценции SuperSignal West Dura, и выполняли количественное определение, используя программное обеспечение Syngene. Уровни белка винкулина измеряли в качестве контроля нагрузки, используя антитело V931 от Sigma, разбавленное 1:10000 в Marvel, и детектирующее анти-мышиное антитело. Результаты, приведенные на Фиг. 11 и 12, показывают, что уменьшение уровней ER на 60% наблюдали после обработки соединением из примера 1 относительно разбавителя как контроля.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль, которые определены ранее, вместе с фармацевтически приемлемым разбавителем или носителем.
Подходящие фармацевтически приемлемые эксципиенты для композиции в форме таблетки включают, например, инертные разбавители, гранулирующие и разрыхляющие агенты, связывающие вещества, смазывающие вещества, консерванты и антиоксиданты. Другим подходящим фармацевтически приемлемым эксципиентом может быть хелатирующий агент. Композиции в форме таблетки могут быть без оболочки или покрыты оболочкой либо для модификации их распадаемости и последующего всасывания активного ингредиента в желудочно-кишечном тракте, либо для улучшения их стабильности и/или внешнего вида, в любом случае с использованием традиционных покрывающих агентов и методик, хорошо известных в данной области техники.
Композиции для перорального применения, альтернативно, могут быть представлены в форме твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, или в форме мягких желатиновых капсул, в которых активный ингредиент смешан с водой или маслом.
Водные суспензии обычно содержат активный ингредиент в тонкоизмельченной форме вместе с одним или более чем одним суспендирующим агентом, диспергирующим или увлажняющим агентом. Водные суспензии также могут содержать один или более консервантов, антиоксидантов, красителей, корригентов и/или подсластителей.
Масляные суспензии могут быть изготовлены путем суспендирования активного ингредиента в растительном масле или минеральном масле. Кроме того, масляные суспензии также могут содержать загуститель. Для обеспечения приятного на вкус препарата для перорального приема можно добавить такие подсластители, как указанные выше, и корригенты. В эти композиции в качестве консерванта может быть добавлен антиоксидант.
Диспергируемые порошки и гранулы, подходящие для приготовления водной суспензии путем добавления воды, обычно содержат активный ингредиент вместе с диспергирующим или увлажняющим агентом, суспендирующим агентом и одним или более консервантами. Также могут присутствовать такие дополнительные эксципиенты, как подсластители, корригенты и красители.
Фармацевтические композиции по изобретению также могут быть представлены в форме эмульсий типа масло-в-воде. Масляной фазой может быть растительное масло или минеральное масло либо смесь любого из них. Эмульсии также могут содержать подсластители, корригенты и консерванты.
Сиропы и эликсиры могут быть изготовлены с использованием подсластителей и также могут содержать уменьшающее раздражение средство, консервант, корригент и/или краситель.
Фармацевтические композиции также могут быть представлены в форме стерильной водной или масляной суспензии для инъекций, которая может быть изготовлена согласно известным методикам с использованием одного или более чем одного соответствующего диспергирующего или увлажняющего агента и суспендирующего агента, которые упомянуты выше. Стерильный препарат для инъекций также может представлять собой стерильный раствор или стерильную суспензию для инъекций в нетоксичной приемлемой для парентерального введения системе разбавителей или растворителей.
Композиции для введения путем ингаляции могут быть представлены в форме традиционного находящегося под давлением аэрозоля, приспособленного для распределения активного ингредиента, либо в виде аэрозоля, содержащего тонкоизмельченное твердое вещество, либо в виде жидких капель. Можно использовать традиционные пропелленты для аэрозолей, как например, летучие фторированные углеводороды или углеводороды, и аэрозольное устройство удобным образом приспособлено для распределения отмеренного количества активного ингредиента. Сухие порошковые ингаляторы также могут быть приемлемы.
Для дополнительной информации по технологии изготовления лекарственных средств читатель отсылается к главе 25.2 тома 5 «Комплексной медицинской химии» (Comprehensive Medicinal Chemistry; Corwin Hansch; Chairman of Editorial Board), Pergamon Press, 1990.
Согласно одному из аспектов изобретения описанная выше фармацевтическая композиция содержит соединение из примера 1 ((Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту) или его фармацевтически приемлемую соль. Подходящим образом соединение из примера 1 представлено в виде своего полиморфа, описанного в данном изобретении как кристаллическая Форма В.
Для способа синтеза (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты, представленного в примере 1, рекомендуется его проведение при отсутствии освещения и в атмосфере азота, чтобы избежать образования продукта разложения
Упомянутый продукт разложения, который представляет собой (R,E)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилат, имеет следующую структуру:

и считается, что он может образовываться из соединения примера 1 ((Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты) в результате автоокисления на воздухе по свободнорадикальному механизму роста цепи. Во избежание неясности, считается, что этот продукт разложения не обладает существенной активностью в качестве SERD.
Это соединение также можно обозначить как (Е)-3-[3,5-дифтор-4-[(3R)-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил]фенил]проп-2-еноат, и способ синтеза для его получения приведен в примере 13.
Специалисту будет очевидно, что контроль процесса образования продуктов разложения является существенной мерой обеспечения безопасного производства и хранения фармацевтических средств. Кроме того, специалисту очевидно, что некоторые соединения могут разлагаться при хранении даже после изготовления их в составе фармацевтической композиции, и что такое разложение в некоторых случаях можно регулировать путем применения соответствующих эксципиентов в фармацевтической композиции и/или путем соответствующего упаковывания конечного продукта. Специалисту также будет очевидно, что окончательная композиция, разработанная для коммерческого применения, должна быть оптимизирована по ряду характеристик, включая химическую стабильность, и также включая, например, характеристики, относящиеся к физической стабильности и растворимости. Следовательно, такие композиции будут разработаны с соблюдением баланса ряда разных факторов.
Соответственно, композиции на основе (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты или ее фармацевтически приемлемой соли включают в себя соединение, действующее в качестве антиоксиданта.
Соединения-антиоксиданты, которые подходят для применения в фармацевтических композициях, известны в данной области техники и включают, например, ацетона-бисульфит натрия; альфа-липоевую кислоту; альфа-токоферол; аскорбиновую кислоту; аскорбилпальмитат; бутил-гидроксианизол; бутил-гидрокситолуол; каротины; моногидрат лимонной кислоты; додецилгаллат; эриторбовую кислоту; фумаровую кислоту; глутатион; гистидин; гипофосфористую кислоту; лактобионовую кислоту; липоевую кислоту; яблочную кислоту; мелатонин; метионин; d-маннозу; монотиоглицерин; октилгаллат; метабисульфит калия; пропионовую кислоту; пропилгаллат; аскорбат натрия; бисульфит натрия; формальдегидсульфоксилат натрия; метабисульфит натрия; сульфит натрия; тиосульфат натрия; хлорид олова; двуокись серы; тимол; токоферол; токотриенолы; убихинол; мочевую кислоту; витамин Е и полиэтиленгликольсукцинат витамина Е. Такие соединения могут проявлять свое антиоксидантное действие посредством разнообразных механизмов, и один или более из этих механизмов могут быть более эффективными по сравнению с другими в случае любого конкретного соединения. Некоторые соединения-антиоксиданты, такие как ВНА (бутилгидроксианизол), действуют как поглотители свободных радикалов. Другие антиоксиданты, такие как метабисульфит натрия и аскорбиновая кислота, легко окисляются и поэтому скорее могут быть подвергнуты окислению, чем активный ингредиент.
Например, если в механизм окисления вовлечено индуцируемое металлами образование перекисей, то применение хелатирующего агента, такого как, например, EDTA (этилендиаминтетрауксусная кислота), может быть полезным для удаления любых примесей металлов и тем самым для достижения непосредственно эффекта стабилизации. В данной области техники известны и другие металл-хелатирующие агенты, и они включают, например, натриевую соль сульфобутилового эфира бетадекса; ацетат кальция; моногидрат лимонной кислоты; циклодекстрины; эдетат динатрия; эдетовую кислоту; фумаровую кислоту; галактозу; глутаминовую кислоту; гистидин; гидроксипропилбетадекс; яблочную кислоту; триамин пентауксусной кислоты; фитохелатин; поли(метилвиниловый эфир/малеиновый ангидрид); цитрат калия; дигидрат цитрата натрия; двухосновный фосфат натрия; одноосновный фосфат натрия; винную кислоту и трегалозу.
Например, в типичные композиции на основе соединения из примера 1 включали пропилгаллат, метабисульфит натрия, аскорбиновую кислоту и бутил-гидроксианизол. Также включали EDTA. Пример такого изготовления композиций приведен в примере 12. Среди них композиции, содержащие метабисульфит натрия, оказались менее стабильными, чем композиции без какого-либо антиоксиданта, после четырех недель хранения в условиях различных температур и влажности. Композиции, содержащие аскорбиновую кислоту, оказались самыми стабильными спустя 4 недели хранения в условиях различных температур и влажности (что определено в анализе с использованием жидкостной хроматографии, например, UHPLC (ультра высокоэффективная жидкостная хроматография)).
Другие подходящие вспомогательные вещества для композиций, содержащих соединение из примера 1, включают эксципиенты с низким содержанием металлов, эксципиенты с низким содержанием перекисей, такие эксципиенты, как маннит, представляющий собой поглотитель свободных радикалов, а также наполнитель. Способ изготовления такой композиции также может влиять на стабильность. Например, что касается некоторых активных ингредиентов, то обеспечение тщательного перемешивания активного ингредиента с обуславливающими стабильность эксципиентами может быть важным фактором обеспечения максимальной стабильности. На результат тщательного перемешивания могут влиять, например, скорость перемешивания, размеры частиц и процессы влажного или сухого смешивания/гранулирования. Активный ингредиент можно подвергнуть гранулированию вместе с антиоксидантом и затем смешать с другими эксципиентами. Антиоксиданты также можно добавлять в любую оболочку, покрывающую фармацевтическую композицию снаружи.
Согласно одному из аспектов изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым разбавителем или носителем.
Согласно одному из аспектов изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль и также содержащая антиоксидант. Соответственно, в качестве антиоксиданта можно использовать аскорбиновую кислоту.
Согласно одному из аспектов изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль и также содержащая металл-хелатирующий агент. Соответственно, в качестве металл-хелатирующего агента можно использовать EDTA.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтop-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль, антиоксидант и возможно дополнительно содержащая металл-хелатирующий агент.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым разбавителем или носителем, причем композиция содержит менее 5% (масс./масс.) (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилата.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтop-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым разбавителем или носителем, причем композиция содержит менее 2% (масс./масс.) (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилата.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтop-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым разбавителем или носителем, причем композиция содержит менее 1% (масс./масс.) (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилата.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтop-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым разбавителем или носителем, причем композиция содержит менее 0,5% (масс./масс.) (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилата.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтop-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым разбавителем или носителем, причем композиция содержит менее 0,1% (масс./масс.) (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилата.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтop-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту или ее фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым разбавителем или носителем, причем композиция содержит менее 0,05% (масс./масс.) (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилата.
В упомянутых выше аспектах, в тех случаях, когда композиция описана как содержащая менее 5% (масс./масс.), 2% (масс./масс.), 1% (масс./масс.), 0,5% (масс./масс.), 0,1% (масс./масс.) или 0,05% (масс./масс.) (R,E)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилата, специалисту будет понятно, что это подразумевает значение, приведенное в процентах по массе, в сравнении с (Е)-3-(3,5-дифтop-4-((1R,3R)-2-(2-фтop-2-мeтилпpoпил)-3-мeтил-2,3,4,9-тeтpaгидpo-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислотой, присутствующей в этой композиции.
Таким образом, продукт разложения (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилат может быть использован в качестве маркера сравнения или стандарта сравнения в таких аналитических методах, как HPLC, с целью мониторинга стабильности соединения из примера 1 или содержащих его фармацевтических композиций.
Количество активного ингредиента, комбинированного с одним или более чем одним эксципиентом для изготовления лекарственной формы для разового введения, обязательно будет меняться в зависимости от подвергаемого лечению пациента и конкретного пути введения. Например, для перорального введения людям обычно необходимо, например, от 1 мг до 2 г подлежащего введению активного агента (более приемлемо от 100 мг до 2 г, например, от 250 мг до 1,8 г, как например, от 500 мг до 1,8 г, в частности, от 500 мг до 1,5 г, подходящим образом от 500 мг до 1 г) в смеси с соответствующим и удобным количеством эксципиентов, которое может изменяться от примерно 3 до примерно 98 процентов от массы всей композиции. Будет очевидно, что в случае необходимости в большой дозировке может потребоваться много лекарственных форм, например две или более таблеток или капсул, содержащих активный ингредиент в дозе, удобно поделенной между ними. В типичном случае, стандартные лекарственные формы будут содержать примерно от 10 мг до 0,5 г соединения по данному изобретению, хотя стандартная лекарственная форма может содержать до 1 г. Подходящим образом твердая лекарственная форма для разового введения может содержать от 1 до 300 мг активного ингредиента.
Размер дозы соединений по настоящему изобретению с точки зрения терапевтических или профилактических целей будет непременно меняться в зависимости от природы и тяжести болезненного состояния, возраста и пола животного или пациента и пути введения, согласуясь с хорошо известными принципами медицины.
В случае применения соединений по настоящему изобретению для терапевтических или профилактических целей обычно его будут вводить так, чтобы получить суточную дозу в диапазоне, например, от 1 мг/кг до 100 мг/кг массы тела, введенную при необходимости разделенными дозами. В общем случае, при использовании парентерального пути будут введены более низкие дозы. Так, например, в случае внутривенного введения обычно будет применена доза в диапазоне, например, от 1 мг/кг до 25 мг/кг массы тела. Подобно этому, в случае введения посредством ингаляции будет применена доза в диапазоне, например, от 1 мг/кг до 25 мг/кг массы тела. Однако пероральное введение, в частности в форме таблетки, является предпочтительным.
Согласно одному из аспектов изобретения соединения по настоящему изобретению или их фармацевтически приемлемые соли вводят в виде таблеток, содержащих от 10 мг до 100 мг соединения формулы (I) (или его фармацевтически приемлемой соли), при этом одну или более таблеток вводят так, как необходимо для достижения желаемой дозы.
Как указано выше, известно, что передача сигнала посредством ERα является причиной онкогенеза путем оказания одного или более действий по опосредованию пролиферации раковых и других клеток, опосредованию ангиогенных событий и опосредованию подвижности, миграции и инвазивности раковых клеток. Авторы изобретения обнаружили, что соединения по настоящему изобретению обладают сильной противоопухолевой активностью, которая, как полагают, возникает в результате антагонизма и отрицательной регуляции ERα, вовлеченного в стадии передачи сигнала, приводящие к пролиферации и выживаемости опухолевых клеток и инвазивной и миграционной способности метастазирующих опухолевых клеток.
Соответственно, соединения по настоящему изобретению могут быть полезны в качестве противоопухолевых агентов, в частности в качестве селективных ингибиторов пролиферации, выживаемости, подвижности, распространения и инвазивности раковых клеток млекопитающих, обуславливая ингибирование опухолевого роста и выживаемости и ингибирование роста метастатической опухоли. В частности, соединения по настоящему изобретению могут быть полезны в качестве антипролиферативных и антиинвазивных агентов для сдерживания распространения и/или лечения солидного опухолевого заболевания. В частности, соединения по настоящему изобретению могут быть полезны в предупреждении или лечении тех опухолей, которые чувствительны к ингибированию ERα и которые вовлечены в стадии передачи сигнала, приводящие к пролиферации и выживаемости опухолевых клеток и миграционной способности и инвазивности метастазирующих опухолевых клеток. Кроме того, соединения по настоящему изобретению могут быть полезны для предупреждения или лечения тех опухолей, воздействие на которые опосредовано исключительно или частично антагонизмом и отрицательной регуляцией ERα, т.е. соединения можно использовать для получения эффекта ингибирования ERα у теплокровного животного, нуждающегося в таком лечении.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в качестве лекарственного средства у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в получении антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения у теплокровного животного, такого как человек, в качестве антиинвазивного агента для сдерживания распространения и/или лечения солидного опухолевого заболевания.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, для получения антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в получении антипролиферативного эффекта у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения у теплокровного животного, такого как человек, в качестве антиинвазивного агента для сдерживания распространения и/или лечения солидного опухолевого заболевания.
Согласно другому аспекту изобретения предложен способ получения антипролиферативного эффекта у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложен способ получения антиинвазивного эффекта посредством сдерживания распространения и/или лечения солидного опухолевого заболевания у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в предупреждении или лечении рака у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в предупреждении или лечении рака у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложен способ предупреждения или лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в предупреждении или лечении солидного опухолевого заболевания у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в предупреждении или лечении солидного опухолевого заболевания у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложен способ предупреждения или лечения солидного опухолевого заболевания у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в предупреждении или лечении тех опухолей, которые чувствительны к ингибированию ERα, вовлеченных в стадии передачи сигнала, приводящие к пролиферации, выживаемости, инвазивности и миграционной способности опухолевых клеток.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в предупреждении или лечении тех опухолей, которые чувствительны к ингибированию ERα, вовлеченных в стадии передачи сигнала, приводящие к пролиферации, выживаемости, инвазивности и миграционной способности опухолевых клеток.
Согласно другому аспекту изобретения предложен способ предупреждения или лечения тех опухолей, которые чувствительны к ингибированию ERα, вовлеченных в стадии передачи сигнала, приводящие к пролиферации, выживаемости, инвазивности и миграционной способности опухолевых клеток, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в получении эффекта ингибирования ERα.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в получении эффекта ингибирования ERα.
Согласно другому аспекту изобретения также предложен способ получения эффекта ингибирования ERα, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены ранее, для применения в получении эффекта, селективного в отношении ингибирования ERα.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены ранее, в изготовлении лекарственного средства для применения в получении эффекта, селективного в отношении ингибирования ERα.
Согласно другому аспекту изобретения также предложен способ получения эффекта, селективного в отношении ингибирования ERα, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
В данном изобретении описаны соединения, которые могут связываться с лиганд-связывающим доменом ERα и которые являются селективными веществами, способствующими деградации рецепторов эстрогенов. Согласно биохимическим и основанным на использовании клеток анализах показано, что соединения по настоящему изобретению являются эффективными связывающимися с рецепторами эстрогенов веществами, и что они снижают уровни ERα в клетках и поэтому могут быть использованы в лечении эстроген-чувствительных заболеваний или состояний (включая заболевания, при лечении которых развивается устойчивость к эндокринным терапиям), т.е. использованы для применения в лечении рака молочной железы и гинекологических раковых заболеваний (включая рак эндометрия, яичников и шейки матки) и раковых опухолей, экспрессирующих белки с мутацией по ERα, которые могут содержать de novo мутации или в которых мутации могут возникать в результате предшествующего лечения с использованием эндокринной терапии, как например, ингибитора ароматазы.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены в данном описании ранее, для применения в лечении рака молочной железы или гинекологических раковых заболеваний.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены в данном описании ранее, для применения в лечении рака молочной железы, эндометрия, яичника или шейки матки.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены в данном описании ранее, для применения в лечении рака молочной железы.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены в данном описании ранее, для применения в лечении рака молочной железы, при котором у данного рака развивается устойчивость к одной или более чем одной другой эндокринной терапии.
Согласно другому аспекту изобретения предложен способ лечения рака молочной железы или гинекологических раковых заболеваний, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложен способ лечения рака молочной железы, эндометрия, яичника или шейки матки, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложен способ лечения рака молочной железы, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложен способ лечения рака молочной железы, при котором у данного рака развивается устойчивость к одной или более чем одной другой эндокринной терапии, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены в данном описании ранее, в изготовлении лекарственного средства для применения в лечении рака молочной железы или гинекологических раковых заболеваний.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены в данном описании ранее, в изготовлении лекарственного средства для применения в лечении рака молочной железы, эндометрия, яичника или шейки матки.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены в данном описании ранее, в изготовлении лекарственного средства для применения в лечении рака молочной железы.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены в данном описании ранее, в изготовлении лекарственного средства для применения в лечении рака молочной железы, при котором у данного рака развивается устойчивость к одной или более чем одной другой эндокринной терапии.
Согласно одному признаку данного изобретения подлежащий лечению рак представляет собой рак молочной железы. Согласно другому аспекту этого признака рак молочной железы является раком, положительным (+ve) по рецептору эстрогенов (ER+ve). В одном из воплощений этого аспекта соединение формулы (I) вводят в комбинации с другим противораковым средством, таким как антигормональный агент, как определено здесь.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль, которые определены в данном описании ранее, для применения в лечении ER+ve рака молочной железы.
Согласно другому аспекту изобретения предложен способ лечения ER+ve рака молочной железы, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, как определено ранее.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли, которые определены в данном описании ранее, в изготовлении лекарственного средства для применения в лечении ER+ve рака молочной железы.
Как указано ранее, эффекты, оказываемые соединением формулы (I) in vivo, частично могут быть обусловлены одним или более метаболитами, которые образуются в организме человека или животного после введения соединения формулы (I).
Таким образом, согласно настоящему изобретению также предположен способ ингибирования ER-α у пациента, включающий введение пациенту количества соединения формулы (I) или его фармацевтически приемлемой соли, эффективного для ингибирования ER-α у пациента.
Таким образом, согласно настоящему изобретению также предположен способ ингибирования ER-α у пациента, включающий введение пациенту количества соединения формулы (I) или его фармацевтически приемлемой соли, эффективного для ингибирования ER-α у пациента.
Во всех приведенных выше применениях и способах подходящим соединением формулы (I) является соединение из примера 1 или его фармацевтически приемлемая соль. Согласно одному из аспектов соединение из примера 1 находится в кристаллической форме В, как изложено в данном описании.
В лечении рака, определенном в данном описании, можно применять монотерапию или можно, помимо введения соединения по изобретению, включать проведение стандартных хирургических операций, лучевую терапию или химиотерапию. Такая химиотерапия может включать введение одного или более противоопухолевых агентов следующих категорий:
(1) другие антипролиферативные/антинеопластические лекарственные средства и их комбинации, которые применяют в терапевтической онкологии, такие как алкилирующие агенты (например, цисплатин, оксалиплатин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан, темозоламид и нитрозомочевины); антиметаболиты (например, гемцитабин и антифолаты, такие как фторпиримидины типа 5-фторурацила и тегафура, ралтитрексед, метотрексат, цитозинарабинозид и гидроксимочевина); противоопухолевые антибиотики (например, антрациклины, такие как адриамицин, блеомицин, доксорубицин, дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин и митрамицин); антимитотические агенты (например, алкалоиды барвинка, такие как винкристин, винбластин, виндезин и винорелбин, и таксоиды, такие как таксол и таксотер, и ингибиторы полокиназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан и камптотецин);
(2) антигормональные агенты, такие как антиэстрогены (например, тамоксифен, фулвестрант, торемифен, ралоксифен, дролоксифен и иодоксифен), прогестогены (например, мегестрола ацетат), ингибиторы ароматазы (например, такие как анастрозол, летрозол, воразол и эксеместан;
(3) ингибиторы функции факторов роста и их расположенных ниже сигнальных путей: включены Ab модуляторы к любым целевым факторам роста или рецепторам факторов роста, обзор по которым представлен в Stern et al., Critical Reviews in Oncology/Haematology, 2005, 54, pp. 11-29; также включены низкомолекулярные ингибиторы таких мишеней, например, ингибиторы киназ: примеры включают антитело к erbВ2 трастузумаб (Herceptin™), антитело к рецептору эпидермального фактора роста (EGFR) панитумумаб, антитело к EGFR цетуксимаб (эрбитукс, С225) и ингибиторы тирозинкиназ, в том числе ингибиторы erbB-семейства рецепторов, как например, ингибиторы тирозинкиназ семейства рецепторов эпидермального фактора роста (EGFR/erbB1), такие как гефитиниб или эрлотиниб, ингибиторы тирозинкиназы erb2, такие как лапатиниб, и смешанные ингибиторы erb1/2, такие как афатиниб; подобные стратегии имеются для других классов факторов роста и их рецепторов, например, ингибиторов семейства факторов роста гепатоцитов или их рецепторов, включая c-met и ron; ингибиторы инсулина и семейства инсулиноподобных факторов роста или их рецепторов (IGFR (рецептор инсулиноподобного фактора роста), IR (инсулиновый рецептор)), ингибиторов семейства факторов роста тромбоцитов или их рецепторов (PDGFR (рецептор фактора роста тромбоцитов)) и ингибиторов передачи сигнала, опосредуемой другими рецепторными тирозинкиназами, такими как c-kit, киназа анапластической лимфомы (AnLK) и рецептор колониестимулирующего фактора-1 (CSF-1R);
также включены модуляторы, направленно воздействующие на белки передачи сигнала в РI3-киназа-опосредованном сигнальном пути, например, ингибиторы изоформ РI3-киназы, таких как РI3K-α/β/γ, и ser/thr-киназ, таких как АКТ, mTOR (такие как AZD2014), РI3K-зависимая киназа (PDK), сыворотка- и глюкокортикоид-индуцибельная киназа (SGK), фосфатидилинозитол-4-киназа (PI4K) или фосфатидилинозитол-4-фосфат-5-киназа (PIP5K);
также включены ингибиторы серин/треониновых киназ, не приведенных выше, например, ингибиторы raf-киназ, такие как вемурафениб, ингибиторы киназы митоген-активируемой протеинкиназы (МЕК), такие как селуметиниб (AZD6244), ингибиторы тирозинкиназы Абельсона (Abl), такие как иматиниб или нилотиниб, ингибиторы тирозинкиназы Бруттона (Btk), такие как ибрутиниб, ингибиторы тирозинкиназы из селезенки (Syk), такие как фостаматиниб, ингибиторы аврора-киназ (например, AZD1152), ингибиторы других ser/thr-киназ, таких как янус-киназа (JAK), передатчики сигнала и активаторы транскрипции (STAT) и киназа, ассоциированная с рецептором интерлейкина-4 (IRAK4), и ингибиторы циклин-зависимых киназ, такие как палбоциклиб;
(4) модуляторы сигнальных путей повреждения ДНК, например ингибиторы поли(АДФ-рибоза)-полимеразы (PARP) (например, олапариб), ингибиторы АТР(атаксия-телеангиэктазия + Rab3-родственной)-киназы или ингибиторы атаксия-телеангиэктазия-мутированной (ATM) киназы;
(5) модуляторы апоптотического и других путей гибели клетки, такие как модуляторы Bcl-семейства (например, АВТ-263/навитоклакс, АВТ-199);
(6) антиангиогенные агенты, например агенты, ингибирующие действие сосудистого эндотелиального фактора роста (например, антитело к фактору роста эндотелиальных клеток сосудов бевацизумаб (Avastin™) и, например, ингибитор рецепторной тирозинкиназы VEGF (сосудистый эндотелиальный фактор роста), такой как сорафениб, акситиниб, пазопаниб, сунитиниб и вандетаниб, и соединения, которые действуют по другим механизмам (например, линомид, ингибиторы функции интегрина αvβ3 и ангиостатин));
(7) агенты, повреждающие сосуды, такие как комбретастатин А4;
(8) антиинвазивные агенты, например ингибиторы c-Src-киназного семейства, такие как дазатиниб (J. Med. Chem., 2004, 47, 6658-6661) и босутиниб (SKI-606), и ингибиторы металлопротеиназ, такие как маримастат, ингибиторы функции рецептора урокиназного активатора плазминогена или антитела к гепараназе;
(9) иммунотерапевтические подходы, включая, например, подходы для повышения иммуногенности опухолевых клеток пациента в условиях ex vivo и in vivo, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или гранулоцитарно-макрофагальный колониестимулирующий фактор, подходы для снижения Т-клеточной анергии, подходы с использованием трансфицированных иммунных клеток, таких как трансфицированные цитокинами дендритные клетки, подходы с использованием трансфицированных цитокинами линий опухолевых клеток и подходы с использованием антиидиотипических антител. Конкретные примеры включают моноклональные антитела, направленно воздействующие на белок 1 программируемой (клеточной) смерти (PD-1) (например, BMS-936558) или цитотоксический Т-лимфоцитарный антиген 4 (CTLA4) (например, ипилимумаб и тремелимумаб);
(10) средства, используемые в антисмысловой терапии или терапии, основанной на РНК-интерференции (RNAi), например, такие, которые направлены на перечисленные выше мишени;
(11) генотерапевтические подходы, включая, например, подходы с заменой аберрантных генов, такие как подходы, касающиеся аберратного гена р53 или аберрантного гена 1 рака молочной железы (BRCA1) или BRCA2, с применением пролекарственной терапии ген-направленным ферментом (GDEPT), например, с использованием цитозиндезаминазы, тимидинкиназы или бактериального фермента нитроредуктазы, и подходы, применяемые для повышения толерантности пациента к химиотерапии или лучевой терапии, такие как терапия с использованием генов множественной лекарственной устойчивости.
Согласно этому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединения по настоящему изобретению, определенные в данном описании, или их фармацевтически приемлемую соль и другой противоопухолевый агент, в частности любой из противоопухолевых агентов, перечисленных выше в пунктах (1)-(11). В частности, применение противоопухолевого агента, приведенного выше в пунктах (1)-(11), представляет собой стандартное лечение для конкретного подлежащего лечению рака; специалисту в данной области техники будет понятен смысл выражения «стандартное лечение». Согласно одному из аспектов соединением по настоящему изобретению является соединение из примера 1 или его фармацевтически приемлемая соль.
Таким образом, согласно другому аспекту изобретения предложены соединения по настоящему изобретению или их фармацевтически приемлемая соль в комбинации с другим противоопухолевым агентом, в частности противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту изобретения предложено соединение из примера 1 ((Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота) или его фармацевтически приемлемая соль в комбинации с другим противоопухолевым агентом, в частности противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту изобретения предложены соединения по настоящему изобретению или их фармацевтически приемлемая соль в комбинации с другим противоопухолевым агентом, в частности противоопухолевым агентом, выбранным из агентов, перечисленных выше в пункте (1).
Согласно другому аспекту изобретения предложены соединение по настоящему изобретению, определенное в данном описании ранее, или его фармацевтически приемлемая соль и любой из противоопухолевых агентов, перечисленных выше в пункте (1).
Согласно другому аспекту изобретения предложено соединение из примера 1 или его фармацевтически приемлемая соль в комбинации с любым из противоопухолевых агентов, перечисленных выше в пункте (1).
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединения по настоящему изобретению, определенные ранее, или их фармацевтически приемлемую соль и таксоид, такой как, например, таксол или таксотер, подходящим образом таксотер. Например, подходящим соединением по изобретению в комбинации с таксоидом, таким как, например, таксол или таксотер, подходящим образом таксотер, является соединение из примера 1 или его фармацевтически приемлемая соль.
Согласно другому аспекту изобретения предложены соединения по настоящему изобретению или их фармацевтически приемлемая соль в комбинации с другим противоопухолевым агентом, в частности противоопухолевым агентом, выбранным из агентов, перечисленных в данном описании выше в пункте (2).
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединения по настоящему изобретению, определенные в данном описании ранее, или их фармацевтически приемлемую соль и любой из антигормональных агентов, перечисленных выше в пункте (2), например любой из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитор ароматазы, перечисленный выше в пункте (2).
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и любой из антигормональных агентов, перечисленных выше в пункте (2), например любой из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитор ароматазы, перечисленный выше в пункте (2).
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и ингибитор mTOR, такой как AZD2014 (см., например, WO 2008/023161).

Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и ингибитор PI3Kα, такой как ингибиторы РI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163. Одним из примеров подходящего ингибитора PI3Kα/δ является соединение примера 3 из PCT/GB2014/050163, представляющее собой соединение 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он или его фармацевтически приемлемую соль. Способ получения соединения примера 3 из PCT/GB2014/050163 приведен в ссылочном примере 1 в данном описании.
Согласно другому аспекту изобретения предложена комбинация, подходящая для применения в лечении рака, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и палбоциклиб.
Согласно одному из аспектов вышеупомянутая комбинация соединения формулы (I) или его фармацевтически приемлемой соли, в частности, соединения из примера 1 или его фармацевтически приемлемой соли, с противоопухолевым агентом, перечисленным выше в пункте (2), или ингибитором mTOR (таким как AZD2014), или ингибитором PI3K-α (таким как ингибиторы PI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163, в частности, приведенное там соединение из примера 3), или палбоциклибом подходит для применения в лечении рака молочной железы или гинекологических раковых заболеваний, таких как рак молочной железы, эндометрия, яичника или шейки матки, в частности, рак молочной железы, такой как ER+ve рак молочной железы.
Там, где в данном описании используется термин «комбинация», следует понимать, что он относится к одновременному, раздельному или последовательному введению. Согласно одному из аспектов изобретения «комбинация» относится к одновременному введению. Согласно другому аспекту изобретения «комбинация» относится к раздельному введению. Согласно другому аспекту изобретения «комбинация» относится к последовательному введению. Если введение является последовательным или раздельным, то задержка во введении второго компонента не должна быть такой, чтобы привести к потере благоприятного эффекта от действия комбинации. Если комбинацию двух или более компонентов вводят раздельно или последовательно, то будет очевидно, что режим дозирования для каждого компонента может быть разным и независимым от других компонентов. Подходящим образом соединения по настоящему изобретению вводят один раз в сутки.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 ((Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с любым из антигормональных агентов, перечисленных выше в пункте (2), например, любым из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитором ароматазы, перечисленным выше в пункте (2), вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и любой из антигормональных агентов, перечисленных выше в пункте (2), например любой из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитор ароматазы, перечисленный выше в пункте (2), вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и ингибитор mTOR, такой как AZD2014 (см., например, WO 2008/023161), вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и ингибитор PI3Kα, такой как ингибиторы РI3α/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163, вместе с фармацевтически приемлемым разбавителем или носителем. Одним из примеров подходящего ингибитора PI3Kα/δ является соединение примера 3 из PCT/GB2014/050163, описанное ранее.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и палбоциклиб вместе с фармацевтически приемлемым разбавителем или носителем.
Согласно одному из аспектов вышеупомянутые фармацевтические композиции соединения формулы (I) или его фармацевтически приемлемой соли, в частности, соединения из примера 1 или его фармацевтически приемлемой соли, с противоопухолевым агентом, перечисленным выше в пункте (2), или ингибитором mTOR (таким как AZD2014), или ингибитором PI3K-α (таким как ингибиторы РI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163, в частности, приведенное там соединение из примера 3), или палбоциклибом, подходят для применения в лечении рака молочной железы или гинекологических раковых заболеваний, таких как рак молочной железы, эндометрия, яичника или шейки матки, в частности, рак молочной железы, такой как ER+ve рак молочной железы.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с любым из антигормональных агентов, перечисленных выше в пункте (2), например, любым из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитором ароматазы, перечисленным выше в пункте (2), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и любой из антигормональных агентов, перечисленных выше в пункте (2), например любой из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитор ароматазы, перечисленный выше в пункте (2), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и ингибитор mTOR, такой как AZD2014 (см., например, WO 2008/023161), вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и ингибитор PI3Kα, такой как ингибиторы PI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163, вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака. Одним из примеров подходящего ингибитора РI3Kα/δ является соединение примера 3 из PCT/GB2014/050163, описанное ранее.
Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение из примера 1 или его фармацевтически приемлемую соль и палбоциклиб вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении рака.
Согласно одному из аспектов вышеупомянутые фармацевтические композиции соединения формулы (I) или его фармацевтически приемлемой соли, в частности, соединения из примера 1 или его фармацевтически приемлемой соли, с противоопухолевым агентом, перечисленным в пункте (2) выше, или ингибитором mTOR (таким как AZD2014), или ингибитором PI3K-α (таким как ингибиторы РI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163, в частности, приведенное там соединение из примера 3), или палбоциклибом, подходят для применения в лечении рака молочной железы или гинекологических раковых заболеваний, таких как рак молочной железы, эндометрия, яичника или шейки матки, в частности, рак молочной железы, такой как ER+ve рак молочной железы.
Согласно другому признаку данного изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), в изготовлении лекарственного средства для применения при лечении рака у теплокровного животного, такого как человек.
Согласно другому признаку данного изобретения предложено применение соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11), в изготовлении лекарственного средства для применения при лечении рака у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с любым из антигормональных агентов, перечисленных выше в пункте (2), например, любым из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитором ароматазы, перечисленным выше в пункте (2), в изготовлении лекарственного средства для применения в лечении рака у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с любым из антигормональных агентов, перечисленных выше в пункте (2), например, любым из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитором ароматазы, перечисленным выше в пункте (2), в изготовлении лекарственного средства для применения в лечении рака у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с ингибитором mTOR, таким как AZD2014 (см., например, WO 2008/023161), в изготовлении лекарственного средства для применения в лечении рака у теплокровного животного, такого как человек.
Согласно другому аспекту изобретения предложено применение соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с ингибитором PI3Kα, таким как ингибиторы Р13Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163, в изготовлении лекарственного средства для применения в лечении рака у теплокровного животного, такого как человек. Одним из примеров подходящего ингибитора PI3Kα/δ является соединение примера 3 из PCT/GB2014/050163, описанное ранее.
Согласно другому аспекту изобретения предложено применение соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с палбоциклибом в изготовлении лекарственного средства для применения в лечении рака у теплокровного животного, такого как человек.
Согласно одному из аспектов вышеупомянутые применения соединения формулы (I) или его фармацевтически приемлемой соли, в частности, соединения из примера 1 или его фармацевтически приемлемой соли, в комбинации с противоопухолевым агентом, перечисленным выше в пункте (2), или ингибитором mTOR (таким как AZD2014), или ингибитором PI3K-α (таким как ингибиторы РI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163, в частности, приведенное там соединение из примера 3), или палбоциклибом подходят для применения в изготовлении лекарственного средства для лечения рака молочной железы или гинекологических раковых заболеваний, таких как рак молочной железы, эндометрия, яичника или шейки матки, в частности, рак молочной железы, такой как ER+ve рак молочной железы.
Таким образом, согласно другому признаку данного изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с любым из антигормональных агентов, перечисленных выше в пункте (2), например, любым из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитором ароматазы, перечисленным выше в пункте (2).
Согласно другому аспекту изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с любым из антигормональных агентов, перечисленных выше в пункте (2), например, любым из антиэстрогенов, перечисленных выше в пункте (2), или, например, ингибитором ароматазы, перечисленным выше в пункте (2).
Согласно другому аспекту изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с ингибитором mTOR, таким как AZD2014 (см., например, WO 2008/023161).
Согласно другому аспекту изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с ингибитором PI3Kα, таким как ингибиторы РI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163. Одним из примеров подходящего ингибитора РI3Kα/δ является соединение примера 3 из PCT/GB2014/050163, описанное ранее.
Согласно другому аспекту изобретения предложен способ лечения рака у теплокровного животного, такого как человек, нуждающегося в таком лечении, включающий введение указанному животному эффективного количества соединения из примера 1 или его фармацевтически приемлемой соли в комбинации с палбоциклибом.
Согласно одному из аспектов вышеупомянутые способы лечения рака представляют собой способы лечения рака молочной железы или гинекологических раковых заболеваний, таких как рак молочной железы, эндометрия, яичника или шейки матки, в частности, рак молочной железы, такой как ER+ve рак молочной железы.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту настоящего изобретения предложен набор, содержащий соединение формулы (I) или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1) или (2).
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) противоопухолевый агент, выбранный из агентов, перечисленных выше в данном описании в пунктах (1)-(11), во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение формулы (I) или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) противоопухолевый агент, выбранный из агентов, перечисленных выше в данном описании в пунктах (1)-(2), во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий соединение из примера 1 или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1)-(11).
Согласно другому аспекту настоящего изобретения предложен набор, содержащий соединение из примера 1 или его фармацевтически приемлемую соль в комбинации с противоопухолевым агентом, выбранным из агентов, перечисленных выше в данном описании в пунктах (1) или (2).
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение из примера 1 или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) противоопухолевый агент, выбранный из агентов, перечисленных выше в данном описании в пунктах (1)-(11), во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение из примера 1 или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) противоопухолевый агент, выбранный из агентов, перечисленных выше в данном описании в пунктах (1)-(2), во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
Согласно другому аспекту настоящего изобретения предложен набор, содержащий:
а) соединение из примера 1 или его фармацевтически приемлемую соль в первой стандартной лекарственной форме;
б) противоопухолевый агент, выбранный из AZD2014, ингибитора PI3Kα (такого как ингибиторы PI3Kα/δ, которые приведены в одной из заявок авторов настоящего изобретения, находящихся одновременно на рассмотрении патентного ведомства (РСТ), под номером PCT/GB2014/050163) и палбоциклиба, во второй стандартной лекарственной форме; и
в) контейнерные устройства для размещения в них указанных первой и второй лекарственных форм.
В приведенных выше способах, применениях и других аспектах, где используется соединение из примера 1, его соответственно используют в виде кристаллической формы В.
Описанная выше комбинированная терапия может быть добавлена на фоне стандартного лечения, в типичном случае осуществляемой по своей обычно предписанной схеме.
Несмотря на то, что соединения формулы (I) прежде всего имеют значение как терапевтические агенты для применения в случае теплокровных животных (включая человека), они также являются полезными всякий раз, когда необходимо ингибировать ER-α. Таким образом, они полезны в качестве фармакологических стандартов для применения в разработке новых биологических тестов и в поиске новых фармакологических агентов.
Персонализированная медицинская помощь
Другой аспект настоящего изобретения основан на идентификации связи между статусом гена, кодирующего ERα, и чувствительностью к лечению соединением формулы (I). В частности, статус гена ERα может указывать на то, что пациент с меньшей долей вероятности поддается лечению существующей гормонотерапией (например, ингибиторами ароматазы), отчасти по меньшей мере ввиду того, что некоторые затрагивающие ERα мутации, как полагают, являются результатом действия механизмов устойчивости к существующим схемам лечения. SERD, в частности, SERD, который можно вводить перорально в возможно более высоких дозах без чрезмерного неудобства, тогда можно успешно использовать для лечения пациентов с мутациями по ERα, которые могут быть устойчивы к другим терапиям. Таким образом, эта идентификация предоставляет возможности, способы и средства для отбора пациентов с целью лечения соединением формулы (I), в частности, раковых пациентов. Настоящее изобретение относится к средствам и способам отбора пациентов (включая персонализированную медицину). Отбор основан на определении того, ген ERα какого типа, дикого или мутантного, имеют подлежащие лечению опухолевые клетки. Таким образом, статус гена ERα может быть использован в качестве биомаркера, указывающего, что выбранное лечение с применением SERD может иметь преимущество. Во избежание неясности, полагают, что соединения формулы (I), описанные в данном изобретении, являются одинаково активными в отношении гена ERα дикого и мутантного типов, по меньшей мере в отношении тех мутаций в гене ERα, которые идентифицированы на момент подачи данной заявки.
Существует очевидная потребность в биомаркерах, с помощью которых будет расширен круг или будут отобраны пациенты, у которых опухоли будут поддаваться лечению SERD, таким как соединение формулы (I). Биомаркеры для отбора пациентов, с помощью которых идентифицируют пациентов, с наибольшей вероятностью поддающихся лечению тем или иным агентом, а не другим, являются идеальными биомаркерами для лечения рака, поскольку, применяя их, можно отказаться от нецелесообразного лечения пациентов, имеющих не поддающиеся лечению опухоли, и снизить возможные побочные эффекты таких агентов.
Биомаркер можно описать как «отличительный признак, который объективно определяют и оценивают в качестве индикатора нормальных биологических процессов, патогенных процессов или фармакологических ответов на терапевтическое вмешательство». Биомаркером является любой поддающийся определению и количественной оценке индикатор, ассоциированный с конкретным состоянием или заболеванием, где имеет место корреляция между наличием или уровнем этого биомаркера и некоторым аспектом данного состояния или заболевания (включая наличие, уровень или изменение уровня, тип, стадию состояния или заболевания, чувствительность к состоянию или заболеванию или восприимчивость к лекарственному средству, используемому для лечения данного состояния или заболевания). Корреляция может быть качественной, количественной или качественной и количественной. Обычно биомаркером является соединение, фрагмент соединения или группа соединений. Такими соединениями могут быть любые соединения, обнаруженные в организме или продуцируемые организмом, включая белки (и пептиды), нуклеиновые кислоты и другие соединения.
Биомаркеры могут обладать прогностической способностью, и в силу этого их можно использовать для предсказания или обнаружения присутствия, определения уровня, типа или стадии конкретных состояний или заболеваний (включая присутствие или уровень конкретных микроорганизмов или токсинов), предрасположенности (включая генетическую предрасположенность) к конкретным состояниям или заболеваниям или ответной реакции на конкретные виды лечения (включая лечения лекарственными средствами). Считается, что биомаркеры будут играть все более важную роль в будущем в открытии и разработке лекарственных средств, повышая эффективность программ научно-исследовательских работ и программ развития. Биомаркеры можно использовать в качестве диагностических агентов, средств мониторинга прогрессирования заболеваний, средств мониторинга лечения и средств прогноза клинического исхода. Например, различные научные проекты, касающиеся биомаркеров, представляют собой попытки идентификации маркеров конкретных видов рака и конкретных сердечно-сосудистых и иммунных заболеваний. Полагают, что разработка новых одобренных биомаркеров будет приводить к значительным сокращениям затрат в области здравоохранения и разработки лекарственных средств, а также к значительным улучшениям в лечении широкого спектра заболеваний и состояний.
Для оптимального планирования клинических испытаний и получения наиболее полной информации по результатам этих испытаний может потребоваться биомаркер. Маркер может поддаваться количественной оценке в суррогатных и опухолевых тканях. В идеале эти маркеры также будут коррелировать с эффективностью лечения и, таким образом, могут в конечном итоге быть использованы для отбора пациентов.
Таким образом, технической задачей, лежащей в основе этого аспекта настоящего изобретения, является идентификация средств стратификации пациентов с целью лечения соединением формулы (I). Данная техническая проблема решается путем применения воплощений, охарактеризованных в формуле изобретения и/или в описании данной заявки.
Считается, что опухоли, которые содержат ERα дикого типа, чувствительны к лечению соединением формулы (I), например, в качестве терапии «первой линии». Опухоли также могут поддаваться лечению соединением формулы (I) в качестве терапии «второй линии», «третьей линии» или последующей терапии, и такое лечение может быть полезным, в частности, если опухоли содержат ERα мутантного типа и, таким образом, могут быть устойчивыми к существующим терапиям, таким как аутоиммунные (Al) терапии. В случае устойчивых опухолей может потребоваться более высокая дозировка соединения формулы (I), чем в случае опухолей дикого типа.
Согласно изобретению предложен способ определения чувствительности клеток к соединению формулы (I). Способ включает определение статуса гена ERα в указанных клетках. Клетку определяют как чувствительную к соединению формулы (I), если оно ингибирует увеличение количества клеток в анализе клеточного роста (или посредством ингибирования клеточной пролиферации, и/или посредством усиления гибели клеток). Способы по изобретению полезны для предсказания того, какие клетки с большей долей вероятности реагируют ингибированием роста на соединение формулы (I).
«Относящийся к опухоли» образец может представлять собой образец, непосредственно выделенный из опухоли, или может представлять собой образец, который был подвергнут дополнительной обработке, например, образец ПЦР-амплифицированной нуклеиновой кислоты из опухолевого образца.
Определения
В этом разделе под заголовком «Персонализированная медицинская помощь» использованы следующие термины.
«Аллель» относится к особой форме генетического локуса, отличающейся от других форм своей особой нуклеотидной или аминокислотной последовательностью.
«Реакции амплификации» представляют собой реакции с участием нуклеиновых кислот, которые приводят к специфическому многократному увеличению числа копий целевых нуклеиновых кислот по сравнению с нецелевыми нуклеиновыми кислотами. Полимеразная цепная реакция (ПЦР) представляет собой хорошо известную реакцию амплификации.
Термин «рак» используется в данном описании для обозначения неопластического роста, вызываемого трансформацией клеток с приобретением ими неопластического фенотипа. Такая трансформация клеток часто подразумевает генетическую мутацию.
«Ген» представляет собой сегмент ДНК, который содержит всю информацию относительно регулируемого биосинтеза продукта РНК, включая промотор, экзоны, интроны и другие элементы последовательности, которые могут быть локализованы между 5'- или 3'-фланкирующими участками (не между транскрибируемыми частями гена), которые регулируют экспрессию.
«Статус гена» относится к тому, является ли данный ген геном дикого типа или нет (т.е. мутантным геном).
«Метка» относится к композиции, способной давать детектируемый сигнал, указывающий на присутствие целевого полинуклеотида в анализируемом образце. Подходящие метки включают радиоактивные изотопы, нуклеотидные хромофоры, ферменты, субстраты, флуоресцентные молекулы, хемилюминесцентные группировки, магнитные частицы, биолюминесцентные группировки и тому подобное. Таким образом, метка представляет собой любую композицию, которую можно обнаружить спектроскопическим, фотохимическим, биохимическим, иммунохимическим, электрическим, оптическим или химическим способами.
«Несинонимичное изменение» относится к изменению (вариации) в кодирующей последовательности или к перекрыванию кодирующей последовательности гена, которое приводит к образованию отличающейся (измененной) полипептидной последовательности. Эти изменения могут влиять или могут не влиять на функционирование белка и включают миссенс-варианты (в результате замены одной аминокислоты на другую), нонсенс-варианты (в результате укорачивания полипептида ввиду образования преждевременного стоп-кодона) и варианты со вставкой/делецией.
«Синонимичное изменение» относится к изменению (вариации) в кодирующей последовательности гена, которое не влияет на последовательность кодируемого полипептида. Эти изменения могут влиять на функционирование белка опосредованно (например, путем изменения экспрессии гена), но, в отсутствие доказательств обратного, как правило, считаются безвредными.
Термин «нуклеиновая кислота» относится к одноцепочечным или двухцепочечным молекулам ДНК и РНК, в том числе обычным нуклеиновым кислотам, обнаруженным в природе, и/или модифицированным, искусственным нуклеиновым кислотам, имеющим модифицированные остовы или основания, которые известны в данной области техники.
«Праймер» относится к олигонуклеотидной последовательности одноцепочечной ДНК, способной действовать в качестве точки инициации синтеза продукта удлинения праймера, который комплементарен копируемой цепи нуклеиновой кислоты. Длина и последовательность праймера должны быть такими, чтобы способствовать праймированию синтеза продуктов удлинения. Типичный праймер содержит последовательности длиной по меньшей мере примерно 7 нуклеотидов, по существу комплементарные последовательности-мишени, однако предпочтительны несколько более длинные праймеры. Обычно праймеры содержат примерно 15-26 нуклеотидов, тем не менее также можно использовать более длинные или более короткие праймеры.
«Полиморфный сайт» представляет собой положение в пределах локуса, в котором в популяции обнаружены по меньшей мере две альтернативные последовательности.
«Полиморфизм» относится к изменению последовательности в полиморфном сайте, наблюдаемому у индивидуума. Полиморфизмы включают нуклеотидные замены, вставки, делеции и микросателлитные локусы и могут, но не обязательно, приводить к детектируемым различиям в экспрессии генов или функционировании белка. В отсутствие данных о влиянии на экспрессию или функционирование белка обычные полиморфизмы, включая несинонимичные варианты, обычно считаются включенными в определение последовательности гена дикого типа. Каталог полиморфизмов у человека и связанная с ним аннотация, включая валидацию, наблюдаемые частоты встречаемости и связь с заболеваниями, поддерживается Национальным центром биотехнологической информации (NCBI) (база данных по однонуклеотидным полиморфизмам (dbSNP): http://www.ncbi.nlm.nih.gov/projects/SNP/). Необходимо отметить, что термин «полиморфизм» при его использовании в контексте последовательностей генов, не следует путать с термином «полиморфизм», когда его используют в контексте твердой формы соединения, то есть кристаллической или аморфной природы соединения. Специалисту будет понятно предполагаемое значение по его контексту.
«Зонд» относится к одноцепочечным олигонуклеотидам со специфической последовательностью, точно комплементарной последовательности-мишени аллеля, подлежащего обнаружению.
«Ответ» определяют по результатам измерений, проведенных в соответствии с «Критериями оценки ответа при солидных опухолях» (RECIST; Response evaluation criteria in solid tumours), включая разделение пациентов на две основные группы: на тех, которые демонстрируют частичный ответ или стабильное заболевание, и тех, которые демонстрируют признаки прогрессирующего заболевания.
Термин «строгие условия гибридизации» относится к инкубированию в течение ночи при 42°C в растворе, содержащем 50% формамида, 5х SSC (раствор хлорида и цитрата натрия) (750 мМ NaCl, 75 мМ трехзамещенный цитрат натрия), 50 мМ фосфат натрия (рН 7,6), 5х раствор Денхардта, 10% декстрансульфата и денатурированную, деградированную в результате гидродинамического сдвига ДНК из молок лососевых рыб в концентрации 20 пг/мл, с последующей промывкой фильтров в 0,1х SSC при примерно 65°С.
Термин «выживаемость» охватывает общую выживаемость и выживаемость пациентов без прогрессирования заболевания.
«Общая выживаемость» (OS) определяется как промежуток времени от начала введения лекарственных средств до момента смерти по любой причине. «Выживаемость без прогрессирования заболевания» (PFS) определяется как промежуток времени от начала введения лекарственных средств до первого проявления прогрессирующего заболевания или смерти по любой причине.
Согласно одному аспекту изобретения предложен способ отбора пациента для лечения соединением формулы (I), включающий получение содержащего опухолевые клетки образца от пациента; определение того, является ли ген ERα в содержащем опухолевые клетки образце от пациента геном дикого типа или мутантным геном; и на основании этого отбор пациента для лечения соединением формулы (I).
Способ может включать или не включать фактическую стадию выделения образца от пациента. Таким образом, согласно одному аспекту изобретения предложен способ отбора пациента для лечения соединением формулы (I), включающий определение того, является ли ген ERα в содержащем опухолевые клетки образце, предварительно полученном от пациента, геном дикого типа или мутантным геном; и на основании этого отбор пациента для лечения соединением формулы (I).
В одном из воплощений пациента отбирают для лечения соединением формулы (I), если ДНК из опухолевых клеток имеет мутантный ген ERα. В других воплощениях, пациента, у которого ДНК из опухолевых клеток содержит ген ERα дикого типа, отбирают для лечения соединением формулы (I).
В контексте данного изобретения под статусом гена дикого типа подразумевается указание на обычную или соответствующую экспрессию гена и обычное функционирование кодируемого белка. И наоборот, под мутантным статусом подразумевается указание на экспрессию белка с измененной функцией, соответствующую известной роли мутантных генов ERα при раке (как описано в данной заявке). Любое число генетических или эпигенетических изменений, включая, но не ограничиваясь этим, мутацию, амплификацию, делецию, геномную перегруппировку или изменения в профиле метилирования, может приводить к формированию мутантного статуса. Однако, если такие изменения, тем не менее, приводят к соответствующей экспрессии нормального белка или функционально эквивалентного варианта, тогда статус данного гена считается статусом гена дикого типа. Примеры вариантов, которые, как правило, не приводят к формированию у гена функционального мутантного статуса, включают варианты, кодирующие синонимичные изменения, и общие полиморфизмы (синонимичные или несинонимичные). Как осуждается ниже, статус гена можно оценить в функциональном анализе, или он может быть выведен из природы детектируемых отклонений от ссылочной последовательности.
В некоторых воплощениях статус дикого типа или мутантный статус гена ERα определяют по присутствию или отсутствию несинонимичных изменений нуклеиновой кислоты в генах. Наблюдаемые несинонимичные изменения, соответствующие известным общим полиморфизмам без каких-либо аннотированных функциональных эффектов, не придают гену статуса мутантного гена.
Другие изменения в гене ERα, являющиеся признаком мутантного статуса, включают изменения в сайте сплайсинга, которые ухудшают распознавание мест соединения интрон/экзон во время процессинга пре-мРНК в мРНК. Это может приводить к пропуску/прерыванию экзона или включению в норме интронной последовательности в подвергнутую сплайсингу мРНК (удерживанию интрона или использованию скрытых границ сплайсинга). Это может, в свою очередь, приводить к получению аберрантного белка со вставками и/или делециями по сравнению с нормальным белком. Таким образом, в других воплощениях ген имеет мутантный статус, если имеется вариант, в котором изменена последовательность распознавания сайта сплайсинга в месте соединения интрон/экзон.
В случае ESR1 ссылочные последовательности для этого гена (номер доступа в Genbank: NG_008493), мРНК (номер доступа в Genbank: NM_000125) и белка (номер доступа в Genbank: NP_000116 или номер доступа в Swiss-Prot: Р03372) находятся в свободном доступе. Специалист в данной области техники будет в состоянии определить статус гена ESR1, т.е. то, является ли конкретный ген ESR1 геном дикого типа или мутантным геном, основываясь на сравнении последовательности ДНК или белка с последовательностями, отнесенными к дикому типу.
Очевидно, что последовательности гена и мРНК, описанные для гена ERα представляют собой репрезентативные последовательности. У здоровых индивидуумов имеется две копии каждого гена, материнская и отцовская копия, которые вероятно будут несколько отличаться по последовательности, кроме того, в пределах популяции будут существовать многочисленные аллельные варианты генной последовательности. Другие последовательности, отнесенные к дикому типу, включают последовательности, имеющие одно или более синонимичных изменений в нуклеиновокислотной последовательности (изменений, не влияющих на кодируемую белковую последовательность), несинонимичных общих полиморфизмов (например, полиморфизмов, относящихся к зародышевой линии), которые изменяют белковую последовательность, но не влияют на функцию белка, и изменений в интронах, не затрагивающих последовательность сайтов сплайсинга.
Существуют многочисленные методы, доступные специалисту в данной области техники, для определения статуса гена PIK3CA. Статус гена может быть установлен посредством определения нуклеиновокислотной последовательности. Это можно осуществить путем прямого секвенирования полноразмерного гена или анализа специфических сайтов в данном гене, например, сайтов, чаще всего подвергающихся мутациям.
Образцы
Образцом от пациента, предназначенным для тестирования на статус гена, может быть любая опухолевая ткань или образец, содержащий клетки опухоли, полученный или который может быть получен от индивидуума. Подходящим образом тестируемый образец представляет собой образец крови, мазка из ротовой полости, биоптата либо другой жидкости или ткани, полученный от индивидуума. Конкретные примеры включают: циркулирующие опухолевые клетки, циркулирующую в плазме или сыворотке крови ДНК, клетки, выделенные из асцитной жидкости пациентов с раком яичника, мокроту из легких от пациентов с опухолями в легких, тонкоигольный пунктат т пациента с раком молочной железы, мочу, периферическую кровь, клеточный соскоб, волосяной фолликул, взятый пункционной биопсией образец кожи или буккальный мазок.
Очевидно, что помимо этого тестируемый образец может содержать нуклеиновую кислоту с последовательностью, соответствующей последовательности в тестируемом образце, то есть весь участок или часть участка нуклеиновой кислоты в образце перед анализом можно сначала амплифицировать, используя любой удобный метод, например полимеразную цепную реакцию (ПЦР). Нуклеиновая кислота может представлять собой геномную ДНК либо фракционированную или суммарную клеточную РНК. В конкретных воплощениях РНК представляет собой суммарную клеточную РНК, и ее используют непосредственно в качестве матрицы для мечения первой цепи «ДНК с применением случайных праймеров или поли(А)-содержащих праймеров. Находящиеся в тестируемом образце нуклеиновую кислоту или белок можно экстрагировать из этого образца в соответствии со стандартными методологиями (см. Green & Sambrook, Eds., Molecular Cloning: A Laboratory Manual (2012, 4th edition, Vol. 1-3, ISBN 9781936113422), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.).
Могут быть применены диагностические способы по изобретению с использованием образца, предварительно взятого у индивидуума или пациента. Такие образцы можно сохранять путем замораживания или можно фиксировать в формалине и заливать парафиновой или другими средами. Альтернативно, можно получить и использовать свежеприготовленный образец, содержащий опухолевые клетки.
Способы по изобретению могут быть применены для клеток из любой опухоли. Опухоли, подходящие для лечения соединением формулы (I), описаны ранее.
Способы обнаружения нуклеиновых кислот
Обнаружение мутантных по ERα нуклеиновых кислот может быть применено, в контексте настоящего изобретения, для выбора лекарственного средства для лечения. Поскольку мутации в этих генах присутствуют на уровне ДНК, способы по изобретению могут быть основаны на обнаружении мутаций или вариаций в геномной ДНК, а также в транскриптах и самих белках. Возможно будет необходимо подтвердить наличие мутаций в геномной ДНК путем анализа транскриптов и/или полипептидов, чтобы убедиться, что обнаруженная мутация на самом деле проявляется у субъекта.
Специалисту в данной области техники будет очевидно, что имеется большое число аналитических методик, которые можно использовать для обнаружения присутствия или отсутствия измененных нуклеотидов в одном или более положениях в гене. В общем случае, обнаружение аллельной вариации требует применения метода дискриминации мутаций, возможно реакции амплификации (например, основанной на полимеразной цепной реакции) и возможно системы генерирования сигнала. Существует большое число методов обнаружения мутаций, доступных в данной области техники, и они могут быть использованы в сочетании с системой генерирования сигнала, одной из многочисленных в данной области техники. Обзор многих методов обнаружения аллельной вариации приведен в Nollau et al., Clin. Chem., 1997, 43, 1114-1120; Anderson SM. Expert Rev. Mol. Diaqn., 2011, 11, 635-642; Meyerson M. et al., Nat. Rev. Genet., 2010, 11, 685-696 и в стандартных учебниках, например, "Laboratory Protocols for Mutation Detection" ("Лабораторные протоколы обнаружения мутаций"), под ред. U. Landegren, Oxford University Press, 1996 и "PCR", 2oe издание под ред. Newton & Graham, BIOS Scientific Publishers Limited, 1997.
Как отмечалось выше, определение присутствия или отсутствия конкретной вариации или множества вариаций в гене ERα у пациента с раковым заболеванием может быть выполнено различными путями. Подобные тесты обычно проводят, используя ДНК или РНК, собранные из биологических образцов, например, биоптатов тканей, образцов мочи, кала, мокроты, крови, клеток, соскобов ткани, пунктатов молочной железы или другого клеточного материала, и они могут быть осуществлены различными способами, включая, но не ограничиваясь этим, ПЦР, гибридизацию с аллель-специфичными зондами, обнаружение мутаций с использованием ферментов, химическое расщепление ошибочных спариваний, масс-спектрометрию или секвенирование ДНК, в том числе минисеквенирование.
Подходящие методы обнаружения мутаций включают метод с использованием амплификационной системы рефракторных мутаций (ARMS™), амплификационной системы рефракторных мутаций с линейным расширением (ALEX™), системы праймирования конкурентными олигонуклеотидами (COPS), Taqman, метод молекулярных маячков, метод оценки полиморфизма длин фрагментов рестрикции (RFLP) и ПЦР с использованием сайтов рестрикции и метод резонансного переноса энергии флуоресценции (FRET).
В конкретных воплощениях метод, используемый для определения нуклеотида(ов) в гене-биомаркере, выбран из: аллель-специфической амплификации (аллель-специфической ПЦР), например, с использованием амплификационной системы рефракторных мутаций (ARMS), секвенирования, аллельного дискриминантного анализа, гибридизации, полиморфизма длины фрагментов рестрикции (RFLP) или лигирования олигонуклеотидных зондов (OLA).
В конкретных воплощениях гибридизация с аллель-специфическими зондами может быть проведена с применением: (1) аллель-специфических олигонуклеотидов, связанных с твердой фазой (например, стеклом, кремнием, нейлоновыми мембранами), с меченым образцом в растворе, например, как во многих системах использования ДНК-чипов; или (2) связанного образца (часто клонированной ДНК или ПЦР-амплифицированной ДНК) и меченых олигонуклеотидов в растворе (либо аллель-специфических, либо настолько коротких, что они позволяют осуществлять секвенирование посредством гибридизации). Диагностические тесты могут включать применение панели вариаций, часто на твердой подложке, что дает возможность для одновременного определения более чем одной вариации. Такие гибридизационные зонды хорошо известны в данной области техники (см. например, Green & Sambrook, Eds., Molecular Cloning: A Laboratory Manual (2012, 4th edition, Vol. 1-3, ISBN 9781936113422), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.) и могут перекрывать два или более сайтов с вариациями.
Таким образом, в одном из воплощений обнаружение присутствия или отсутствия по меньшей мере одной мутации обеспечивается путем приведения в контакт нуклеиновой кислоты ERα, содержащей сайт предполагаемой мутации, по меньшей мере с одним нуклеиновокислотным зондом. Зонд предпочтительно гибридизуется с нуклеиновокислотной последовательностью, содержащей сайт с вариацией и содержащей комплементарные нуклеотидные основания в сайте с вариацией, в выбранных условиях гибридизации. Обнаружение наличия гибридизации можно осуществить с помощью детектируемой метки, используя метки, известные специалисту в данной области техники. Такие метки включают, но не ограничиваются этим, радиоактивные, флуоресцентные метки, красители и ферментативные метки.
В другом воплощении обнаружение присутствия или отсутствия по меньшей мере одной мутации обеспечивается путем приведения в контакт нуклеиновой кислоты ERα, содержащей сайт предполагаемой мутации, по меньшей мере с одним нуклеиновокислотным праймером. Праймер предпочтительно гибридизуется с нуклеиновокислотной последовательностью, содержащей сайт с вариацией и содержащей комплементарные нуклеотидные основания в сайте с вариацией, в выбранных условиях гибридизации.
Олигонуклеотиды, используемые в качестве праймеров для специфической амплификации, могут содержать нуклеотидное основание, комплементарное представляющей интерес мутации, в центре молекулы (так что амплификация зависит от дифференциальной гибридизации; см., например, Gibbs, et al., 1989, Nucl. Acids Res., 17, 2437-248) или на самом 3'-конце одного праймера, где в соответствующих условиях ошибочное спаривание может предотвращать или ослаблять удлинение под действием полимеразы (см., например, Prossner, 1993, Tibtech., 11, 238).
В еще одном воплощении обнаружение присутствия или отсутствия по меньшей мере одной мутации включает секвенирование по меньшей мере одной нуклеиновокислотной последовательности и сравнение полученной последовательности с известной нуклеиновокислотной последовательностью дикого типа.
Альтернативно, обнаружение присутствия или отсутствия по меньшей мере одной мутации включает масс-спектрометрическое определение по меньшей мере одной нуклеиновокислотной последовательности.
В одном из воплощений обнаружение присутствия или отсутствия по меньшей мере одной нуклеиновокислотной вариации включает проведение полимеразной цепной реакции (ПЦР). Нуклеиновокислотную последовательность-мишень, содержащую гипотетическую вариацию, амплифицируют и определяют нуклеотидную последовательность амплифицированной нуклеиновой кислоты. Определение нуклеотидной последовательности амплифицированной нуклеиновой кислоты включает секвенирование по меньшей мере одного сегмента нуклеиновой кислоты. Альтернативно, продукты амплификации можно анализировать, используя любой метод, с помощью которого можно провести разделение продуктов амплификации в соответствии с их размером, включая автоматизированный и ручной варианты гель-электрофореза и тому подобное.
Обнаружение мутаций в геномной нуклеиновой кислоте предпочтительно осуществляют методами, основанными на изменении подвижности амплифицированных фрагментов нуклеиновой кислоты. Например, в Chen et al., Anal. Biochem., 1996, 239, 61-9 описывается обнаружение точечных мутаций с использованием конкурентного анализа изменения подвижности (competitive mobility shift assay). Кроме того, коммерчески доступными являются анализы, основанные на методе Marcelino и др. (BioTechniques, 1999, 26, 1134-1148).
В конкретном примере, для обнаружения присутствия мутаций можно использовать гетеродуплексный анализ с применением капиллярного электрофореза, основанного на изменении подвижности в капиллярных системах дуплексов нуклеиновых кислот, образованных в результате наличия ошибочных спариваний.
Получение нуклеиновых кислот из образцов для проведения анализа, как правило, требует амплификации нуклеиновой кислоты. Многие методы амплификации основаны на применении ферментативной цепной реакции (такой как полимеразная цепная реакция, лигазная цепная реакция или самоподдерживающаяся репликация последовательностей) или репликации всего или части вектора, использованного для клонирования. Предпочтительно, амплификация, соответствующая данному изобретению, представляет собой экспоненциальную амплификацию, как демонстрируется, например, в полимеразной цепной реакции.
Многие методы амплификации мишеней и усиления сигналов описаны в литературе, например, общие обзоры этих методов представлены в Landegren, U., et al., Science, 1988, 242, 229-237 и Lewis, R., Genetic Engineering News, 1990, 10, 54-55. Эти методы амплификации можно использовать в способах по настоящему изобретению, и они включают полимеразную цепную реакцию (ПЦР), ПЦР in situ, амплификацию в лигазной цепной реакции (LAR), лигазную гибридизацию, метод с применением репликазы бактериофага QB, систему транскрипционно-опосредованной амплификации (TAS), геномную амплификацию с секвенированием транскриптов (GAWTS), амплификацию, основанную на последовательности нуклеиновых кислот (NASBA) и гибридизацию in situ. Праймеры, подходящие для применения в различных методах амплификации, могут быть получены в соответствии со способами, известными в данной области техники.
Полимеразная цепная реакция (ПЦР) представляет собой метод амплификации нуклеиновых кислот, описанный среди прочего в патентах США №№4683195 и 4683202. ПЦР состоит из повторяющихся циклов генерируемых ДНК-полимеразой реакций удлинения праймеров. ДНК-мишень подвергают термоденатурации и осуществляют гибридизацию двух олигонуклеотидов, которые окружают последовательность-мишень на противоположных нитях предназначенной для амплификации ДНК. Эти олигонуклеотиды становятся праймерами для применения вместе с ДНК-полимеразой. ДНК копируют посредством удлинения праймеров, создавая вторую копию обеих нитей. В результате повторения цикла термоденатурации, гибридизации праймеров и удлинения ДНК-мишень может быть амплифицирована миллион или более раз в промежуток времени примерно от двух до четырех часов. ПЦР представляет собой инструмент молекулярной биологии, который должен быть использован совместно с методом обнаружения для определения результатов амплификации. Преимущество ПЦР заключается в том, что она повышает чувствительность посредством увеличения количества ДНК-мишени в число раз от 1 миллиона до 1 миллиарда приблизительно за 4 часа. ПЦР можно использовать в диагностике для амплификации любой известной нуклеиновой кислоты (Мок et al., Gynaecoloqic Oncology. 1994, 52: 247-252).
Метод аллель-специфической амплификации, такой как амплификационная система рефракторных мутаций (ARMS™) (Newton et al., Nucleic Acids Res., 1989, 17, 2503-2516), также может быть использован для обнаружения точечных мутаций. В соответствующих условиях ПЦР-амплификации ошибочного спаривания одного основания на 3'-конце праймера достаточно для предпочтительной амплификации полностью комплементарного аллеля (Newton и др., 1989, выше), что дает возможность для различения близкородственных видов. Основой амплификационной системы, использующей описанные выше праймеры, является то, что олигонуклеотиды с ошибочно спаренным 3'-остатком не будут действовать как праймеры в ПЦР в соответствующих условиях. Такая амплификационная система позволяет осуществлять генотипирование только путем исследования реакционных смесей после проведения электрофореза в агарозном геле.
Анализ продуктов амплификации может быть осуществлен с использованием любого метода, с помощью которого можно провести разделение продуктов амплификации в соответствии с их размером, включая автоматизированный и ручной варианты гель-электрофореза, масс-спектрометрию и тому подобное.
Методы выделения, амплификации и анализа нуклеиновых кислот являются рутинными для специалиста в данной области техники, и примеры протоколов можно найти, например, в Green & Sambrook, Eds., Molecular Cloning: A Laboratory Manual (2012, 4th edition, Vol. 1-3, ISBN 9781936113422), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.) Особенно полезным источником протоколов для методов, используемых в ПЦР-амплификации, является PCR (Basics: From Background to Bench) под ред. M.J. McPherson, S.G. Mailer, R. Beynon, C. Howe, Springer Verlag, 1-е издание (15 октября 2000 г.), ISBN: 0387916008.
Согласно настоящему изобретению также предложены наборы для прогнозирования и диагностики, содержащие вырожденные праймеры для амплификации нуклеиновой кислоты-мишени в гене ERα, и инструкции, содержащие протокол для амплификации и для анализа результатов. Набор также может альтернативно содержать буферы, ферменты и контейнеры для проведения амплификации и анализа продуктов амплификации. Данный набор также может являться компонентом набора для скрининга или диагностики, содержащего другие средства, такие как ДНК-микрочипы или другие подложки. Предпочтительно, в наборе также имеется одна или более чем одна контрольная матрица, например нуклеиновые кислоты, выделенные из нормального образца ткани и/или из серии образцов, представляющих разные вариации в генах сравнения (reference genes).
В одном из воплощений в наборе содержится две или более пар праймеров, каждая из которых позволяет амплифицировать свой участок в гене (ERα) сравнения (каждый участок представляет собой сайт возможной вариации), тем самым предоставляется набор для анализа экспрессии нескольких генных вариаций в биологическом образце в одной реакции или нескольких параллельных реакциях.
Праймеры в наборах могут быть мечеными, например, меченными флуоресцентными метками, для облегчения обнаружения продуктов амплификации и проведения последующего анализа вариаций в нуклеиновой кислоте. С помощью этого набора также можно осуществлять обнаружение более чем одной вариации в одном анализе. Таким образом, комбинированный набор будет содержать праймеры, позволяющие осуществлять амплификацию разных сегментов гена сравнения. Праймеры могут быть помечены разными метками, например, с использованием разных флуоресцентных меток, чтобы различить такие вариации.
Согласно другому аспекту изобретения предложен способ лечения пациента, страдающего раковым заболеванием, включающий: определение для гена ERα мутантного статуса или статуса дикого типа в опухолевых клетках пациента, и, если ген ERα является мутантным, введение пациенту эффективного количества соединения формулы (I).
Использованные в данном описании термины «эффективный» и «эффективность» включают как фармакологическую эффективность, так и физиологическую безопасность. Фармакологическая эффективность относится к способности получения в результате такого лечения желаемого биологического эффекта у пациента. Физиологическая безопасность относится к уровню токсичности или другим неблагоприятным физиологическим эффектам на уровне клетки, органа и/или организма (часто называемым как побочные эффекты) в результате применения такого лечения. «Менее эффективный» означает, что в результате лечения наблюдается терапевтически значительно более низкий уровень фармакологической эффективности и/или терапевтически более высокий уровень неблагоприятных физиологических эффектов.
Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли для лечения пациента с раковым заболеванием, у которого опухолевые клетки идентифицированы как содержащие мутантный ген ERα. В одном из воплощений соединением формулы (I) является соединение из примера 1.
Согласно другому аспекту изобретения предложены соединение формулы (I) или его фармацевтически приемлемая соль для лечения случаев рака с опухолевыми клетками, идентифицированными как несущие мутантный ген ERα. В одном из воплощений соединением формулы (I) является соединение из примера 1.
Согласно другому аспекту изобретения предложен способ лечения случаев рака с опухолевыми клетками, идентифицированными как несущие мутантный ген ERα, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном из воплощений соединением формулы (I) является соединение из примера 1.
В следующих дополнительных воплощениях изобретение относится к фармацевтической композиции, содержащей соединение формулы (I), для применения в предупреждении и лечении рака с опухолевыми клетками, идентифицированными как несущие мутантный ген ERα. В одном из воплощений соединением формулы (I) является соединение из примера 1.
Для всех изложенных выше аспектов определенные/идентифицированные мутантные формы ERα содержат мутации, расположенные в любых положениях по всему гену.
Для всех изложенных выше аспектов с использованием опухолей, таких как, в качестве примера, рак молочной железы, конкретные определенные/идентифицированные мутантные формы ERα содержат мутации, расположенные в положениях Ser463Pro, Val543Glu, Leu536Arg, Tyr537Ser, Tyr537Asn и Asp538Gly.
Краткое описание фигур
На Фиг. 1 показана картина дифракции рентгеновских лучей на порошке для Формы А соединения из примера 7.
На Фиг. 2 показана картина дифракции рентгеновских лучей на порошке для Формы В соединения из примера 1.
На Фиг. 3 показана термограмма DSC для Формы В соединения из примера 1.
На Фиг. 4 показана картина дифракции рентгеновских лучей на порошке для Формы А соединения из примера 1.
На Фиг. 5 показана термограмма TGA для Формы А соединения из примера 1.
На Фиг. 6 показана картина дифракции рентгеновских лучей на порошке для Формы С соединения из примера 1.
На Фиг. 7 показана термограмма TGA для Формы С соединения из примера 1.
На Фиг. 8 показана картина дифракции рентгеновских лучей на порошке для соединения из примера 11.
На Фиг. 9 показана кривая DSC для соединения из примера 11.
На Фиг. 10 показаны результаты исследования соединения из примера 1 и AZD2014 на ксенотрансплантатах MCF-7.
На Фиг. 11 и 12 показаны результаты исследования эффективности соединения из примера 1 на ксенотрансплантатах НСС1428 после длительной эстрогенной депривации (LTED).
Примеры
Теперь изобретение будет проиллюстрировано следующими далее примерами, в которых, как правило:
(1) операции осуществляли при температуре окружающей среды, т.е. в диапазоне 17-25°C, и в атмосфере инертного газа, такого как азот, если не указано иное;
(2) упаривание осуществляли на роторном испарителе или с использованием оборудования Genevac в вакууме, и процедуры обработки выполняли после удаления фильтрованием оставшихся твердых веществ;
(3) флэш-хроматографические очистки осуществляли на автоматизированной системе CombiFlash® Rf (Teledyne Isco) или CombiFlash® Companion® (Teledyne Isco), используя предварительно упакованные колонки с диоксидом кремния RediSep Rf Gold™ (20-40 мкм; сферические частицы), картриджи GraceResolv™ (диоксид кремния Davisil®) или картриджи Silicycle (40-63 мкм);
(4) препаративную хроматографию осуществляли на приборе для препаративной HPLC от Gilson со сбором фракций по детекции в УФ (ультрафиолет);
(5) хиральную препаративную хроматографию осуществляли на приборе от Gilson со сбором фракций по детекции в УФ (инжектор/коллектор фракций 233, насосы 333 и 334, УФ-детектор 155) или приборе PrepStar от Varian (2 насоса SD1, УФ-детектор 325, коллектор фракций 701), используя для хроматографирования инжекционный насос Gilson 305;
(6) выходы, где они приведены, необязательно являются максимально достижимыми;
(7) в общем случае структуры конечных продуктов формулы I подтверждали с применением спектроскопии ядерного магнитного резонанса (ЯМР); величины химического сдвига при проведении ЯМР измеряли по шкале дельта (спектры протонного магнитного резонанса определяли, используя прибор Avance 500 (500 МГц) от Bruker или Avance 400 (400 МГц) от Bruker); измерения проводили при температуре окружающей среды, если не указано иное; использовали следующие сокращения: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; dd, дублет дублетов; ddd, дублет дублетов дублетов; dt, дублет триплетов; bs, уширенный сигнал;
(8) в общем случае конечные продукты формулы I также охарактеризовывали посредством жидкостной хроматографии с последующей масс-спектроскопией (LCMS или UPLC (сверхэффективная жидкостная хроматография)); UPLC осуществляли, используя прибор для UPLC от Waters, оснащенный масс-спектрометром SQ от Waters (температура колонки 40°C, УФ: 220-300 нм, масс-спектрометрия: электрораспылительная ионизация (ESI) с переключением режима положительных/отрицательных ионов), при скорости потока 1 мл/мин, применяя систему растворителей от 97% А+3% В до 3% А+97% В в течение 1,50 мин (общее время хроматографирования с обратным уравновешиванием к исходным условиям и т.п. составляет 1,70 мин), где A представляет собой 0,1%-ную муравьиную кислоту в воде (для работы в кислотных условиях) или 0,1%-ный аммиак в воде (для работы в щелочных условиях), В представляет собой ацетонитрил. Для анализа в кислотных условиях использовали колонку Acquity HSS Т3 (1,8 мкм; 2,1×50 мм) от Waters, для анализа в щелочных условиях использовали колонку Acquity ВЕН (1,7 мкм; 2,1×50 мм) от Waters; LCMS осуществляли, используя прибор Alliance НТ (2795) от Waters, оснащенный масс-спектрометром ZQ ESCi от Waters и колонкой Phenomenex Gemini-NX (50×2,1 мм; 5 мкм), при скорости потока 1,1 мл/мин, применяя систему растворителей от 95% А до 95% В в течение 4 минут, затем выдерживая в течение 0,5 мин. Концентрацию модификатора поддерживают постоянной и равной 5% С (смесь 50:50 ацетонитрил : вода с 0,1% муравьиной кислоты) или D (смесь 50:50 ацетонитрил:вода с 0,1% гидроксида аммония (удельная плотность (SG) 0,88)) в зависимости от того, в каких условиях применяют метод, кислотных или щелочных;
(9) очистку ионообменной хроматографией обычно осуществляли с использованием SCX-2 картриджа (Biotage, функционализированный пропилсульфоновой кислотой диоксид кремния. Изготовлен с использованием трехфункционального силана. Концевые группы не блокированы);
(10) чистоту промежуточных соединений оценивали посредством анализа с применением тонкослойной хроматографии, масс-спектрального анализа, высокоэффективной жидкостной хроматографии (HPLC) и/или ЯМР-анализа;
(11) для проведения XRPD-анализа соединения из примера 7 образцы помещали на кремниевые пластины с нулевым уровнем фона и анализировали, используя дифрактометр CubiX PRO от PANalytical (l (длина волны) 1.5418

(12) для проведения XRPD-анализа соединения из примера 1 картины дифракции рентгеновских лучей на порошке определяли путем помещения образца кристаллического вещества на держатель пластины из монокристалла кремния (SSC) от PANalytical, и распределения образца в виде тонкого слоя с помощью предметного стекла микроскопа. Образец вращали при 30 оборотах в минуту (для улучшения статистики счета) и облучали рентгеновскими лучами, генерируемыми длиннофокусной трубкой с медным анодом, работающей при 45 кВ и 40 мА, с длиной волны 1,5418 ангстрем. Пучок рентгеновских лучей пропускали через щель Соллера 0,04 радиана, затем через автоматически изменяющуюся щель расходимости, установленную на 20 мм и окончательно через отверстие 20 мм для диафрагмирования пучка (beam mask). Отраженное излучение направляли через антирассеивающую щель 20 мм и щель Соллера 0,04 радиана. Образец подвергали воздействию в течение 1,905 секунды с шагом 0,0025067° 2-тета (непрерывный режим сканирования) в диапазоне от 2 градусов до 40 градусов 2-тета в режиме тета-тета. Продолжительность испытания составляла 3 минуты и 36 секунд. Прибор был оснащен детектором X-Celerator. Получение контрольных и экспериментальных данных осуществляли с помощью дисплейного терминала (Workstation) Dell Pentium 4НТ, работающего с программным обеспечением X'Pert Industry.
(13) дифференциальную сканирующую калориметрию проводили, используя прибор для DSC Q1000 от ТА Instruments. Обычно менее 5 мг вещества, содержащегося в стандартном алюминиевом тигле, снабженном крышкой, нагревали в температурном диапазоне от 25°C до 300°C с постоянной скоростью нагревания 10°C в минуту. В качестве продувочного газа использовали азот при скорости потока 50 мл в минуту; и
(14) термогравиметрический анализ проводили, используя аналитический прибор для TGA Q5000 от ТА Instruments. Обычно менее 10 мг размещают на 100 мкл платиновом тигле и нагревают в температурном диапазоне от 30°C до 150°C с постоянной скоростью нагревания 10°C в минуту;
(15) использованы следующие сокращения:
Пример 1. (Е)-3-(3,5-Дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пирид[3,4-b]индол-1-ил)фенил)акриловая кислота

Указанные далее способы следует осуществлять в атмосфере азота в отсутствие освещения, поскольку под действием света может образовываться продукт разложения ((R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилат).
(Е)-Метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (350 г; 766,69 ммоль) загружали в закрепленный сосуд емкостью 5 л. В сосуд добавляли изопропиловый спирт (2,80 л). Добавляли гидроксид натрия (5 М; 460 мл; 2,30 моль) в виде одной порции и смесь перемешивали при 21°C в течение 16 ч. Темный раствор пропускали через фильтр для удаления твердых частиц. Фильтрат возвращали в сосуд реактора. Фильтр и сосуд для сбора фильтрата промывали изопропанолом (700 мл) и промывки добавляли в сосуд реактора. Реакционную смесь перемешивали и добавляли воду (1,75 л). В сосуд загружали концентрированную соляную кислоту (37%-ную (масс./масс.); 165 мл; 1,92 моль). В сосуд добавляли дополнительное количество соляной кислоты (21,5 мл) для подведения рН до значения от 4,0 до 4,5. Раствор нагревали до 50°С. В сосуд в течение 1 часа добавляли воду (1,92 л), поддерживая внутреннюю температуру 50-53°С. Температуру рубашки поднимали до 70°С для поддержания температуры в реакторе в этом диапазоне в процессе добавления. В течение 10 минут после завершения добавления воды в смеси самопроизвольно образовывалась затравка, и начиналась кристаллизация. Смесь выдерживали при 50-52°C в течение 1,5 часа (температура рубашки 58°С). Полученную желтую суспензию охлаждали до 5°C (температура рубашки) в течение 6 часов. Суспензию выдерживали при 5°C (температура рубашки) в течение 11 ч. Полученное желтое твердое вещество выделяли фильтрованием. Осадок на фильтре растирали до пастообразного состояния с помощью шпателя, чтобы предотврать его растрескивание. Сосуд промывали водой (1,05 л). Промывки использовали для промывания осадка на фильтре. Осадок на фильтре подсушивали на воздухе, затем сушили в вакууме до постоянной массы в течение 4 суток (температура в сушильном шкафу 30°С). Таким образом выделяли (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту в виде желтого кристаллического вещества, «Формы В» (319,45 г; 94%).1Н ЯМР (500 МГц, DMSO, 27°С) 1.05 (3Н, d), 1.08-1.28 (6Н, m), 2.35 (1Н, dd), 2.58 (1Н, dd), 2.8-2.97 (2Н, m), 3.47-3.57 (1Н, m), 5.22 (1Н, s), 6.67 (1Н, d), 6.91-7.06 (2Н, m), 7.19 (1Н, d), 7.41 (1Н, d), 7.46 (2Н, d), 7.54 (1Н, d), 10.58 (1Н, s), 12.62 (1Н, s).
Ниже приведен альтернативный способ синтеза соединения из примера 1, в результате которого образуется кристаллическое вещество в виде Формы В.
Указанные далее способы следует осуществлять в атмосфере азота в отсутствие освещения, поскольку под действием света может образовываться продукт разложения (как описано выше).
(Е)-Метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (50,0 г; 109,53 ммоль) перемешивали в изопропиловом спирте (450 мл). Добавляли гидроксид натрия (68,34 г; 65,72 мл; 328,58 ммоль) в виде одной порции и смесь перемешивали при 20°C в течение 16 ч. Реакционную смесь разбавляли водой (250 мл), рН подводили до 4 конц. соляной кислотой (27,28 мл; 317,63 ммоль) и смесь нагревали до 50°С. В течение 30 минут добавляли дополнительное количество воды (225 мл), поддерживая температуру выше 45°С. В процессе добавления вещество начинало кристаллизоваться. Смесь охлаждали от 50°C до 5°C в течение 5 часов, затем суспензию выдерживали при 0°C в течение еще 11 часов. Желтое твердое вещество выделяли фильтрованием. Осадок на фильтре промывали водой (100 мл), сушили на фильтре в течение еще 20 минут, затем сушили в вакуумном сушильном шкафу в течение 16 часов до постоянной массы (30°C, отвод воздуха), получая указанное в заголовке соединение (46,52 г) в виде кристаллического вещества (Формы В).
1Н ЯМР (500 МГц, DMSO, 27°С) 1.02-1.09 (3Н, m), 1.17 (6Н, dd), 2.37 (1Н, dd), 2.59 (1Н, dd), 2.8-2.98 (2Н, m), 3.47-3.58 (1Н, m), 5.24 (1Н, s), 6.68 (1Н, d), 6.9-7.06 (2Н, m), 7.20 (1Н, d), 7.38-7.51 (3H, m), 7.55 (1H, d), 10.59 (1H, s), 12.60 (1H, br).
Кристаллическую Форму В также можно выделять из смесей этанол/вода и смесей этанол/МТВЕ.
Согласно другому аспекту изобретения предложена кристаллическая Форма B соединения из примера 1, выделенная из смеси изопропанол/вода.
Согласно другому аспекту изобретения предложен способ выделения кристаллической Формы В соединения из примера 1, включающий гидролиз (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата в присутствии изопропилового спирта и основания, затем подкисление и выделение кристаллического продукта из водного изопропанола.
Форма B соединения из примера 1 характеризуется наличием по меньшей мере одного из следующих значений 2θ, измеренных с использованием CuKa-излучения: 8,4 и 10,9. Форма B соединения из примера 1 характеризуется наличием картины дифракции рентгеновских лучей на порошке, по существу такой, как показано на Фиг. 2. Десять пиков дифракции рентгеновских лучей на порошке показаны в Таблице А.

DSC-анализ Формы B соединения из примера 1 демонстрирует, что эта форма представляет собой высокоплавкое твердое вещество с эндотермой, показывающей начало плавления при 188,6°C (Фиг. 3). Соединение из примера 1 демонстрирует разложение при плавлении, что может привести к изменению начала плавления, поэтому значение 188,6°C не следует принимать за абсолютное.
Также были обнаружены две сольватированные формы соединения из примера 1.
Форма A представляет собой моно-сольват простого метил-трет-бутилового эфира. Картина дифракции рентгеновских лучей на порошке показана на Фиг. 4. TGA демонстрирует ассоциированное уменьшение массы на 14,7% (масс./масс.) в диапазоне 55-150°C (Фиг. 5). Теоретический расчет потери для моно-сольвата простого метил-трет-бутилового эфира составляет 16,6%.
Форма С представляет собой ацетон моно-сольват, картина дифракции рентгеновских лучей на порошке показана на Фиг. 6. TGA демонстрирует ассоциированное уменьшение массы на 10,0% (масс./масс.) в диапазоне 50-150°C (Фиг. 7). Теоретический расчет потери для ацетон моно-сольвата составляет 11,0%.
(Е)-Метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат, используемый в качестве исходного вещества, получали так, как указано ниже.
Получение 2-фтор-2-метилпропилтрифторметансульфоната

В атмосфере азота в сосуд емкость 20 л загружали 2-фтор-2-метил-пропан-1-ол (1000 г; 10,86 моль). В сосуд добавляли DCM (8,5 л). Смесь перемешивали и охлаждали до 1°С. К смеси добавляли 2,6-лутидин (1395 г; 13,02 моль). В течение 1 часа добавляли раствор трифторметансульфонового ангидрида (3220 г; 11,41 моль) в DCM (1 л), поддерживая температуру реакционной смеси ниже 5°C (в процессе добавления температуру рубашки понижали до -20°С). Этот дополнительный сосуд и подводящие линии промывали DCM (0,5 л) и промывки добавляли в основной сосуд в виде одной порции. Смесь перемешивали при 0°C в течение 1 часа, получая красный раствор.
Добавляли раствор концентрированной соляной кислоты (1,23 л 37%-ной (масс./масс.); 16,3 моль) в воду (7 л). Этот разбавленный раствор соляной кислоты добавляли к красному раствору и перемешиваемую смесь нагревали до 25°С. Смесь отставляли для разделения слоев, и верхний водный слой отбрасывали. Органический слой промывали водой (2×5 л). Органический раствор концентрировали при пониженном давлении, получая красное масло. Это красное масло очищали дистилляцией с использованием пленочного испарителя (4,5 мбар (450 Па), температура рубашки 50°C, температура в холодильнике 4°С), получая 2-фтор-2-метилпропилтрифторметансульфонат (1,69 кг; 69%) в виде бледно-красного масла.1Н ЯМР (500 МГц, DMSO-d6, 27°С) δ 1.40 (6Н, d), 4.79 (2Н, d).
Получение (R)-N-(1-(1Н-индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина

(2R)-1-(1Н-Индол-3-ил)пропан-2-амин (3,81 кг; 21,21 моль) добавляли в атмосфере азота в облицованный стеклом сосуд емкостью 100 л с рубашкой. Добавляли 1,4-диоксан (23 л) и включали устройство для перемешивания. К перемешиваемой суспензии добавляли диизопропилэтиламин (5,55 л; 31,82 моль), затем (2-фтор-2-метил-пропил)трифторметансульфонат (5,55 кг; 23,77 моль). В сосуд добавляли 1,4-диоксан (4 л) и смесь нагревали до 75°С. Нагревание продолжали в течение 24 часов, после чего выполняли охлаждение смеси до 25°С. В сосуд добавляли воду (30,5 л), затем толуол (30,5 л). Через 40 минут устройство для перемешивания отключали, и слои оставляли разделяться. Водный слой удаляли и к органическому раствору добавляли воду (30,5 л). Смесь перемешивали в течение 15 минут, после чего слои оставляли разделяться. Водный слой удаляли из сосуда. Органический раствор концентрировали вакуумной дистилляцией (в рубашке: температура 65°C, давление 110 мбар (11 кПа)) до удаления приблизительно 27 л дистиллята. Оставшийся в сосуде раствор охлаждали, получая (R)-N-(1-(1Н-индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин в виде раствора в толуоле (33% (масс./масс.)) (15,4 кг; 97%).1Н ЯМР (500 МГц, DMSO, 27°С) 0.98 (3Н, d), 1.26 (3Н, d), 1.30 (3Н, d), 2.57-2.75 (3Н, m), 2.81 (1Н, dd), 2.84-2.92 (1Н, m), 6.97 (1Н, t), 7.06 (1Н, t), 7.11-7.22 (1Н, мультиплет скрыт сигналами от толуола), 7.34 (1Н, d), 7.52 (1H, d), 10.80 (1H, s).
Получение {2,6-дифтор-4-[(1Е)-3-метокси-3-оксопроп-1-ен-1-ил]фенил}(гидрокси)метансульфоната натрия

2,6-Дифтор-4-бромбензальдегид (1000 г; 4,39 моль) и дихлорид 1,1-бис(ди-трет-бутилфосфино)ферроценпалладия (57,2 г; 87,76 ммоль) загружали в сосуд емкостью 20 л. Добавляли тетра-н-бутилхлорид аммония (122 г; 438,97 ммоль), затем диметилацетамид (5 л). Сосуд продували струей газообразного азота. В сосуд добавляли диизопропилэтиламин (1,5 л; 8,78 моль), затем метилакрилат (0,435 л; 4,82 моль). Смесь перемешивали и нагревали до 60°C. Смесь выдерживали при этой температуре в течение 20 часов. К смеси добавляли этилацетат (10 л) и нагревание прекращали. В сосуд добавляли воду (5 л). Перемешиваемую смесь охлаждали до 25°C и перемешивание продолжали в течение 10 минут. Перемешивание останавливали и оставляли разделяться слои. Водный слой удаляли и отбрасывали. Органический слой промывали последовательно соляной кислотой (2,2 М; 6 л) и водой (5 л). В сосуд добавляли поглотитель металлов Phosphonics SPM32 (1050 г; 1050 моль), и смесь перемешивали в течение 3 суток при 25°С. Твердое вещество удаляли фильтрованием. Осадок на фильтре промывали этанолом (5 л), и объединенные фильтраты концентрировали при пониженном давлении, получая твердое вещество. Это твердое вещество растворяли в этаноле (9 л) и раствор перемешивали в сосуде емкостью 20 л. Раствор нагревали до 50°С. Добавляли раствор бисульфита натрия (460 г; 4,42 моль) в воде (2,5 л) в течение 30 минут. Получали густую суспензию, которую перемешивали в течение 4 часов при 50°C. Суспензию охлаждали до 20°C в течение 2 часов. Твердое вещество выделяли фильтрованием и сосуд и осадок на фильтре промывали МТВЕ (2×3 л). Полученное твердое вещество сушили в вакууме, получая {2,6-дифтор-4-[(1Е)-3-метокси-3-оксопроп-1-ен-1-ил]фенил}(гидрокси)метансульфонат натрия (1035 г; 71%) в виде светло-коричневого твердого вещества.1Н ЯМР (500 МГц, DMSO, 27°С) δ 3.73 (3Н, s), 5.32 (1Н, d), 5.94 (1Н, d), 6.76 (1Н, d), 7.40 (2Н, d), 7.60 (1Н, d).
Получение (Е)-метил-3-(3.5-дифтор-4-формилфенил)акрилата

В сосуд емкостью 100 л в атмосфере азота загружали {2,6-дифтор-4-[(1Е)-3-метокси-3-оксопроп-1-ен-1-ил]фенил}(гидрокси)метансульфонат натрия (1,211 кг; 3,52 моль), затем карбонат калия (0,974 кг; 7,05 моль). Добавляли воду (9,1 л) и включали устройство для перемешивания. Добавляли этилацетат (9,1 л). Смесь перемешивали при 25°C в течение 5 часов. Устройство для перемешивания отключали, и смесь оставляли стоять в течение 14 часов при 25°С. Нижнюю водную фазу удаляли и отбрасывали. Верхнюю органическую фазу концентрировали при пониженном давлении, получая бледно-коричневое твердое вещество. Твердое вещество сушили в вакууме, получая (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат в виде коричневого твердого вещества (608 г; 76%).1Н ЯМР (500 МГц, DMSO, 27°С) δ 3.77 (3Н, s), 6.94 (1Н, d), 7.66 (1Н, d), 7.71 (2Н, d), 10.20 (1Н, s).
Получение (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата
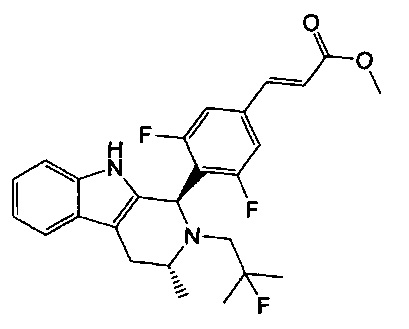
В сосуд емкостью 20 л в атмосфере азота загружали (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат (0,606 кг; 2,65 моль) и (R)-N-(1-(1Н-индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин (33%-ный (масс./масс.) раствор в толуоле; 2,0 кг; 2,65 моль), затем толуол (4,22 л). В сосуд добавляли уксусную кислоту (304 мл; 5,31 моль). Смесь перемешивали и нагревали до 80°С. Смесь перемешивали при 80°C в течение ночи, после чего охлаждали до 20°С. К смеси добавляли раствор карбоната калия (0,916 кг; 6,63 моль) в воде (3,3 л). Смесь перемешивали в течение 10 минут, после чего устройство для перемешивания отключали, и слои оставляли разделяться. Водный слой удаляли и отбрасывали. В реактор загружали воду (3,3 л). Смесь перемешивали в течение 10 минут, затем оставляли стоять в течение 10 минут. Нижнюю водную фазу удаляли, а органический слой оставляли стоять при комнатной температуре в течение ночи. Эту порцию нагревали до 80°С. К горячему раствору в течение 35 минут добавляли гептан (4,61 л). Перемешиваемую смесь выдерживали при приблизительно 80°C в течение 1 часа. Смесь охлаждали до 30°C в течение 2 часов, за это время происходила кристаллизация продукта. Суспензию перемешивали при 30°C в течение 2,5 часа. Твердое вещество выделяли фильтрованием. Стенки реакционного сосуда промывали гептаном и промывки использовали для промывания осадка на фильтре. Твердое вещество сушили в вакууме, получая (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат в виде розового твердого вещества (0,763 кг; 61%).1Н ЯМР (400 МГц, DMSO, 27°С) δ 1.06 (3Н, d), 1.13 (3Н, d), 1.21 (3Н, d), 2.35 (1Н, dd), 2.58 (1Н, dd), 2.8-2.98 (2Н, m), 3.44-3.61 (1Н, m), 3.74 (3Н, s), 5.24 (1Н, s), 6.80 (1Н, d), 6.9-7.05 (2Н, m), 7.19 (1Н, d), 7.41 (1Н, d), 7.50 (2Н, d), 7.63 (1Н, d), 10.58 (1Н, s). m/z: ES+ [М+Н]+ 457.
Альтернативное получение соединения из примера 1: (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты

7,5 М Гидроксид натрия (32,9 мл; 247,10 ммоль) добавляли к раствору (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (11,28 г; 24,71 ммоль) в THF (143 мл) и метаноле (71,4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Значение pH водного слоя подводили приблизительно до 6,5 путем добавления 2 н. раствора HCl, затем раствор экстрагировали диэтиловым эфиром (3×150 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния с элюированием градиентом 0-20% метанола в DCM, получая желтое твердое вещество. Попытка растирания со смесью ацетон/гептан не удалась ввиду более высокой, чем ожидалось, растворимости. Растворители удаляли, получая желтое твердое вещество, которое растирали в изогексане (50 мл), содержащем несколько капель диэтилового эфира; полученное твердое вещество отфильтровывали и сушили, получая неочищенный продукт (11,14 г) в виде желтого порошка. Это твердое вещество растворяли в этаноле (100 мл) в атмосфере азота и в темноте. Затем раствор упаривали, создавая вакуум (5 мбар (500 Па)) с использованием вакуумного насоса, при 62°C в темноте. Эту процедуру повторяли дважды и полученное в результате желтое стеклообразное вещество растирали шпателем в тонкоизмельченный порошок и создавали вакуум (5 мбар (500 Па)) с использованием вакуумного насоса при 62°C в течение 60 мин, получая желтый порошок. Затем порошок оставляли в вакуумном сушильном шкафу над Р2О5 при 62°C и 300 мбар (30 кПа) в течение ночи, получая указанный в заголовке продукт (9,77 г; 89%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, DMSO, 27°С) δ 1.07-1.16 (3Н, m), 1.18-1.29 (6Н, m), 2.39 (1Н, dd), 2.62 (1Н, dd), 2.92 (2Н, dd), 3.56 (1Н, d), 5.26 (1Н, s), 6.70 (1H, d), 7.02 (2H, dd), 7.22 (1H, d), 7.47 (3Н, dd), 7.58 (1H, d), 10.60 (1H, s), 12.60 (1H, s). m/z: ES+ (электрораспыление+) [M+H]+ 443.
(Е)-Метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтop-2-мeтилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат, используемый в качестве исходного вещества, получали так, как указано ниже.
Получение 2-фтор-2-метилпропан-1-ола

Алюмогидрид лития (3,37 г; 88,56 ммоль) добавляли порциями в течение 15 мин к охлажденному раствору этил-2-фтор-2-метилпропаноата (9,9 г; 73,80 ммоль) в диэтиловом эфире (184 мл) при 0°С. Реакционную смесь перемешивали в течение 1 ч, затем последовательно добавляли воду (3,3 мл), далее 15%-ный раствор NaOH (3,3 мл) и воду (6,7 мл). Суспензию перемешивали в течение 15 мин, затем фильтровали и твердые вещества промывали диэтиловым эфиром. Фильтрат упаривали, получая 2-фтор-2-метилпропан-1-ол (5,90 г; 87%) в виде бесцветного масла.
1Н ЯМР (400 МГц, CDCl3, 27°С) δ 1.37 (6Н, d), 3.56 (2Н, d), сигнала от ОН не наблюдали.
Альтернативное получение 2-фтор-2-метилпропилтрифторметан-сульфоната

Трифторметансульфоновый ангидрид (12,06 мл; 71,24 ммоль), затем 2,6-лутидин (11,42 мл; 81,42 ммоль) добавляли к раствору 2-фтор-2-метилпропан-1-ола (6,25 г; 67,85 ммоль) в DCM (146 мл) при -10°С. Реакционную смесь перемешивали в течение 1 ч, затем промывали 2 н. HCl (2×100 мл) и насыщенным раствором NaНСО3 (2×100 мл). Органическую фазу затем сушили над Na2SO4 и концентрировали, получая 2-фтор-2-метилпропилтрифторметан-сульфонат (12,89 г; 85%) в виде красного масла.
1Н ЯМР (400 МГц, CDCl3, 27°С) δ 1.46 (6Н, d), 4.41 (2Н, d).
Это промежуточное соединение может быть очищено вакуумной дистилляцией. DSC-анализ показал, что данное вещество обладало способностью к самопроизвольному выделению тепла. Из соображений безопасности способа, для проведения циклической дистилляции предпочтительным может быть использование пленочного или ему подобного испарителя.
Альтернативное получение (R)-N-(1-(1Н-индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина

2-Фтор-2-метилпропилтрифторметансульфонат (8,04 г; 35,87 ммоль) добавляли к раствору (R)-1-(1Н-индол-3-ил)пропан-2-амина (5,00 г; 28,70 ммоль) и N-этил-N-изопропилпропан-2-амина (7,44 мл; 43,04 ммоль) в диоксане (50 мл). Реакционную смесь нагревали до 90°C в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь разбавляли EtOAc (200 мл) и промывали насыщенным раствором NaHCO3 (2×00 мл). Водную фазу экстрагировали EtOAc (150 мл), затем объединенные органические экстракты сушили над MgSO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя 100%-ным EtOAc. Фракции чистого продукта упаривали досуха, получая (R)-N-(1-(1H-индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин (6,49 г; 91%) в виде коричневого масла.1Н ЯМР (400 МГц, CDCl3, 27°С) δ 1.14 (3Н, d), 1.31 (3Н, d), 1.37 (3Н, d), 1.94 (1Н, s), 2.63-2.87 (3Н, m), 2.92 (1Н, dd), 3.07 (1Н, h), 7.07 (1Н, d), 7.08-7.15 (1Н, m), 7.16-7.24 (1Н, m), 7.37 (1Н, d), 7.62 (1Н, d), 8.04 (1Н, s). m/z: ES+ [М+Н]+ 249.
Альтернативное получение (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилата

4-Бром-2,6-дифторбензальдегид (9,99 г; 45,20 ммоль) и метилакрилат (6,14 мл; 67,81 ммоль) переносили в тщательно дегазированный DMA (100 мл) и добавляли три-о-толилфосфин (1,376 г; 4,52 ммоль), ацетат палладия(II) (0,507 г; 2,26 ммоль) и триэтиламин (12,60 мл; 90,41 ммоль). Реакционную смесь перемешивали и нагревали до 80°C в течение 6 часов. Реакционную смесь охлаждали и фильтровали через слой целита и промывали метанолом (50 мл). Неочищенный продукт предварительно абсорбировали на диоксид кремния и очищали хроматографией с использованием вакуумной системы, элюируя смесью 0-10% диэтиловый эфир/дихлорметан. Фракции, содержащие желаемый продукт, упаривали и растирали с диэтиловым эфиром (50 мл), получая желтое твердое вещество, которое растирали с водой (50 мл) и сушили под высоким вакуумом при 50°C, получая (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат (8,85 г; 87%) в виде желтого твердого вещества.1Н ЯМР (400 МГц, DMSO, 27°С) δ 3.75 (3Н, s), 6.93 (1Н, d), 7.52-7.81 (3Н, m), 10.18 (1Н, s). При проведении LCMS никакого молекулярного иона не наблюдали.
Альтернативное получение (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата

(Е)-Метил-3-(3,5-дифтор-4-формилфенил)акрилат (6,58 г; 29,09 ммоль) добавляли к суспензии (R)-N-(1-(1Н-индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амина (6,02 г; 24,24 ммоль) в толуоле (51,1 мл) и уксусной кислоте (2,78 мл; 48,48 ммоль). Реакционную смесь нагревали до 80°C в течение 5 часов. Реакционную смесь очищали ионообменной хроматографией, используя SCX-2 колонку. Желаемый продукт элюировали с колонки, используя 7 М NH3/метанол, и фракции чистого продукта упаривали досуха, получая коричневое твердое вещество. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-30% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая указанный в заголовке продукт (7,52 г; 68,0%) в виде желтого твердого вещества.1Н ЯМР (400 МГц, DMSO, 100°С) δ 1.10 (3Н, d), 1.12-1.31 (6Н, m), 2.28-2.72 (2Н, m), 2.84-3.09 (2Н, m), 3.52-3.69 (1Н, m), 3.76 (3Н, s), 5.30 (1Н, s), 6.64 (1Н, d), 6.9-7.11 (2Н, m), 7.21 (1Н, d), 7.32 (2Н, d), 7.42 (1Н, d), 7.58 (1Н, d), 10.14 (1Н, s). m/z: ES+ [М+Н]+ 457.
Пример 2. (Е)-3-(4-((1R,3R)-2-(2-Фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота

7,5 М раствор гидроксида натрия (0,983 мл; 7,37 ммоль) добавляли к раствору (Е)-метил-3-(4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (310 мг; 0,74 ммоль) в метаноле (5 мл). Смесь перемешивали при 20°C в течение 2 часов. Реакционную смесь очищали ионообменной хроматографией, используя SCX-2 колонку. Фракции, содержащие желаемый продукт, элюировали с колонки с использованием смеси 7 М NH3/метанол и фракции чистого продукта упаривали досуха, получая желтое твердое вещество. Неочищенный продукт очищали препаративной HPLC (колонка SunFire от Waters, диоксид кремния (5 мкм), диаметр 50 мм, длина 100 мм), используя в качестве элюентов смеси воды (содержащей 0,1% муравьиной кислоты) и MeCN с уменьшающейся полярностью. Фракции, содержащие желаемое соединение, упаривали досуха, затем загружали на SCX-2 колонку и элюировали 7 н. раствором аммиака в метаноле, получая указанный в заголовке продукт (63,0 мг; 21,02%) в виде желтого твердого вещества.1Н ЯМР (400 МГц, DMSO, 30°С) δ 1.06 (3Н, d), 1.30 (3Н, d), 1.47 (3Н, d), 2.53-2.64 (2Н, m), 2.79 (2Н, s), 3.10 (1Н, d), 5.08 (1Н, s), 6.47 (1Н, d), 6.98 (1Н, t), 7.06 (1Н, t), 7.19-7.37 (3Н, m), 7.44 (1Н, d), 7.56 (1Н, d), 7.63 (2Н, d), 10.81 (1Н, s), 12.30 (1Н, s). m/z: ES+ [М+Н]+ 407.
(Е)-Метил-3-(4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат, используемый в качестве исходного вещества, получали так, как указано ниже.
Получение (Е)-метил-3-(4-формилфенил)акрилата

4-Бромбензальдегид (30 г; 162,15 ммоль) и метилакрилат (20,94 г; 243,22 ммоль) переносили в тщательно дегазированный DMA (300 мл), обрабатывали три-о-толилфосфином (4,94 г; 16,21 ммоль), ацетатом палладия(II) (1,820 г; 8,11 ммоль) и триэтиламином (45,2 мл; 324,29 ммоль) и нагревали до 110°C в течение 16 часов. По прошествии этого времени реакция, по-видимому, завершалась. Реакционную смесь выливали в воду (4 л) и полученный осадок отфильтровывали и сушили. Твердое вещество хроматографировали на диоксиде кремния, элюируя градиентом от 100%-ного гептана до 30%-ного EtOAc в гептане. Релевантные фракции объединяли и упаривали досуха, получая желтый твердый продукт, который растирали с гептаном, фильтровали и промывали холодным гептаном. Твердое вещество сушили, получая (Е)-метил-3-(4-формилфенил)акрилат (25,6 г; 83%) в виде желтого кристаллического продукта.1Н ЯМР (400 МГц, DMSO, 30°С) δ 3.75 (3Н, s), 6.79 (1Н, d), 7.72 (1Н, d), 7.93 (4Н, s), 10.03 (1Н, s). При проведении LCMS никакого молекулярного иона не наблюдали.
Получение (Е)-метил-3-(4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3.4-b]индол-1-ил)фенил)акрилата
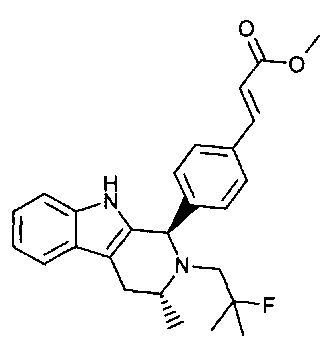
(R)-N-(1-(1Н-Индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин (полученный, как описано в примере 1, получение исходных веществ) (450 мг; 1,81 ммоль) и (Е)-метил-3-(4-формилфенил)акрилат (345 мг; 1,81 ммоль) растворяли в толуоле (15 мл), добавляли уксусную кислоту (5 мл) и молекулярные сита. Реакционную смесь перемешивали при 110°C в течение 16 часов в атмосфере азота, затем охлаждали до комнатной температуры. Неочищенный продукт очищали ионообменной хроматографией, используя SCX-2 колонку. Желаемый продукт элюировали с колонки с использованием смеси 7 М NH3/метанол и фракции чистого продукта упаривали досуха, получая неочищенный продукт. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-30% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (317 мг; 41,6%) в виде желтого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) δ 1.09 (3Н, d), 1.30 (3Н, d), 1.47 (3Н, d), 2.48-2.78 (4Н, m), 3.30 (1Н, m), 3.79 (3Н, s), 5.09 (1Н, s), 6.40 (1Н, d), 7.12 (1Н, td), 7.17 (1Н, td), 7.29 (1Н, d), 7.34 (2Н, d), 7.43 (2Н, d), 7.54 (1Н, d), 7.66 (2Н, m). m/z: ES+ [М+Н]+ 421.
Пример 3. (Е)-3-(3,5-Дифтор-4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота

2 М Гидроксид натрия (3,0 мл; 6,00 ммоль) добавляли к раствору (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (275 мг; 0,60 ммоль) в смеси THF (1,5 мл)/метанол (1,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли EtOAc (15 мл) и воду (15 мл), затем значение рН водного слоя подводили примерно до 7 путем добавления 2 н. раствора HCl. Проводили разделение слоев и водный слой экстрагировали EtOAc (15 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-10% метанола в DCM. Фракции чистого продукта упаривали досуха, получая указанный в заголовке продукт (250 мг; 94%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, DMSO, 30°С) δ 0.77 (3Н, d), 1.06 (3Н, d), 1.93 (1Н, m), 2.18 (1Н, dd), 2.58 (1Н, dd), 2.65 (1Н, dd), 2.84 (1Н, dd), 3.35 (1Н, dd), 4.32 (1Н, d), 4.44 (1Н, d), 5.16 (1Н, s), 6.67 (1Н, d), 6.93-7.04 (2Н, m), 7.21 (1Н, d), 7.42 (1Н, d), 7.46 (2Н, m), 7.54 (1Н, d), 10.57 (1Н, s), 12.51 (1Н, s). m/z: ES+ [М+Н]+ 443.
(Е)-Метил-3-(3,5-дифтор-4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат, используемый в качестве исходного вещества, получали так, как указано ниже.
Получение (S)-3-фтор-2-метилпропан-1-ола

N,N-Диэтил-1,1,2,3,3,3-гексафторпропан-1-амин (18,25 мл; 100,57 ммоль) по каплям добавляли к раствору (R)-метил-3-гидрокси-2-метилпропаноата (9,25 мл; 83,81 ммоль) в DCM (77 мл) (реакционная смесь нагревалась до приблизительно 40°С). Реакционную смесь перемешивали в течение 1 ч при температуре окружающей среды, затем нагревали до температуры дефлегмации в течение 4 ч, после чего охлаждали до комнатной температуры в течение ночи. Реакционную смесь выливали на лед и проводили разделение слоев. Водный слой экстрагировали DCM (2×150 мл), затем объединенные органические экстракты сушили и концентрировали с осторожностью. Остаток растворяли в THF (200 мл) и охлаждали в ледяной бане. Порциями в течение 15 мин добавляли алюмогидрид лития (6,45 г; 167,61 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 ч и нагревали до комнатной температуры в течение еще 1 ч. После охлаждения в ледяной бане реакцию гасили, добавляя воду (7 мл), затем 15%-ный раствор NaOH (7 мл) и в конце воду (21 мл). Добавляли MgSO4 до образования гранул твердого вещества. Твердое вещество отфильтровывали через целит и промывали диэтиловым эфиром (50 мл). Фильтрат промывали 2 н. раствором HCl (2×100 мл), затем органическую фазу сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-10% EtOAc в DCM. Фракции чистого продукта упаривали досуха, получая (S)-3-фтор-2-метилпропан-1-ол (6,42 г; 83%) в виде масла желтоватой окраски.1Н ЯМР (400 МГц, CDCl3, 30°С) δ 0.97 (3Н, dd), 1.96-2.14 (1Н, m), 3.64 (2Н, d), 4.3-4.42 (1Н, m), 4.42-4.54 (1Н, m), сигнала от ОН не наблюдали.
Получение (S)-3-фтор-2-метилпропилтрифторметансульфоната

К перемешиваемому раствору (S)-3-фтор-2-метилпропан-1-ола (7,9 г; 85,77 ммоль) в DCM (140 мл) при 0°C по каплям добавляли трифторметансульфоновый ангидрид (17,31 мл; 102,92 ммоль), затем по каплям добавляли 2,6-диметилпиридин (11,95 мл; 102,92 ммоль). Реакционную смесь перемешивали при 0°C в течение 45 мин и при комнатной температуре в течение 30 мин. Реакционную смесь разбавляли DCM (60 мл) и промывали последовательно 1 М раствором HCl (3×100 мл), насыщенным раствором бикарбоната натрия (100 мл) и насыщенным рассолом (50 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали почти досуха. Раствор фильтровали через набивку диоксида кремния, ее промывали DCM (50 мл) и раствор упаривали, получая (S)-3-фтор-2-метилпропилтрифторметансульфонат (14,38 г; 74,8%) в виде коричневого масла.1Н ЯМР (400 МГц, CDCl3, 27°С) δ 1.09 (3Н, dd), 2.24-2.44 (1Н, m), 4.30 (0,5Н, dd), 4.37-4.46 (1Н, m), 4.52 (2,5Н, tt).
Получение (S)-N-((R)-1-(1Н-индол-3-ил)пропан-2-ил)-3-фтор-2-метилпропан-1-амина

(S)-3-Фтор-2-метилпропилтрифторметансульфонат (666 мг; 2,97 ммоль) добавляли к раствору (R)-1-(1Н-индол-3-ил)пропан-2-амина (470 мг; 2,7 ммоль) и N-этил-N-изопропилпропан-2-амина (0,700 мл; 4,05 ммоль) в 1,4-диоксане (6,05 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли EtOAc (20 мл) и промывали водой (20 мл). Водный слой экстрагировали EtOAc (2×20 мл), затем объединенные органические экстракты сушили (MgSO4) и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-10% метанола в DCM. Фракции чистого продукта упаривали досуха, получая (S)-N-((R)-1-(1Н-индол-3-ил)пропан-2-ил)-3-фтор-2-метилпропан-1-амин (590 мг; 88%) в виде коричневого масла.1Н ЯМР (400 МГц, CDCl3, 30°С) δ 0.86 (3Н, dd), 1.20 (3Н, d), 1.94-2.11 (1Н, m), 2.64-2.74 (2Н, m), 2.85-2.98 (2Н, m), 3.05-3.15 (1Н, m), 4.13-4.39 (2Н, m), 7.09 (1Н, d), 7.12 (2Н, ddd), 7.20 (1Н, ddd), 7.33-7.41 (1Н, m), 7.56-7.65 (1Н, m), 8.10 (1Н, s). m/z: ES+ [М+Н]+ 249.
Получение (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата

Уксусную кислоту (2,0 мл) добавляли к раствору (S)-N-((R)-1-(1Н-индол-3-ил)пропан-2-ил)-3-фтор-2-метилпропан-1-амина (273 мг; 1,10 ммоль) и (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилата (полученного, как описано в примере 1, получение исходных веществ) (226 мг; 1 ммоль) в толуоле (8,0 мл). Реакционную смесь нагревали до 95°C в течение 2,5 ч. Летучие вещества удаляли под вакуумом, после чего остаток пропускали через SCX-2 колонку. Затем колонку промывали смесью 7 М NH3/метанол для элюирования продукта. Фильтрат концентрировали и неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-10% метанола в DCM. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (282 мг; 61,8%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) δ 0.82 (3Н, d), 1.12 (3Н, d), 1.81-1.99 (1Н, m), 2.24 (1Н, ddd), 2.64 (1Н, ddd), 2.71 (1Н, dd), 2.93-3.01 (1Н, ddd), 3.42 (1Н, dq), 3.81 (3Н, s), 4.25-4.39 (1Н, m), 4.38-4.53 (1Н, m), 5.20 (1Н, s), 6.39 (1Н, d), 6.99 (2Н, m), 7.06-7.16 (2Н, m), 7.21-7.25 (1Н, m), 7.52 (3Н, m). m/z: ES+ [M+H]+ 457.
Пример 4. (Е)-3-(4-((1R,3R)-2-((S)-3-Фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота

7,5 М раствор гидроксида натрия (0,904 мл; 6,78 ммоль) добавляли к раствору (Е)-метил-3-(4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (285 мг; 0,68 ммоль) в метаноле (3 мл). Смесь перемешивали при 20°C в течение 2 часов. Реакционную смесь очищали ионообменной хроматографией, используя SCX-2 колонку. Фракции, содержащие желаемый продукт, элюировали с колонки, используя 7 М NH3/метанол, и фракции чистого продукта упаривали досуха, получая желтое твердое вещество. Неочищенный продукт очищали препаративной LCMS (препаративная колонка Gemini-NX axia С18 OBD от Phenomenex, диоксид кремния (5 мкм), диаметр 50 мм, длина 100 мм), используя в качестве элюентов смеси воды (содержащей 1% NH3) и MeCN с уменьшающейся полярностью. Фракции, содержащие желаемое соединение, упаривали досуха, получая указанный в заголовке продукт (80 мг; 29,0%) в виде желтого твердого вещества.1Н ЯМР (500 МГц, DMSO, 33°С) δ 0.87 (3Н, d), 1.04 (3Н, d), 1.91-2.27 (2Н, m), 2.50 (1Н, р), 2.57-2.75 (2Н, m), 3.13 (1Н, s), 4.51 (2Н, dd), 4.86 (1Н, s), 6.47 (1Н, d), 6.92-6.99 (1Н, m), 7-7.09 (1Н, m), 7.15-7.35 (3Н, m), 7.35-7.5 (2Н, m), 7.57 (2Н, d), 10.64 (1Н, d), сигнала от CO2H не наблюдали, m/z: ES+ [М+Н]+ 407.
(Е)-Метил-3-(4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат, используемый в качестве исходного вещества, получали так, как указано ниже.
Получение (Е)-метил-3-(4-(1S,3R)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата

(Е)-Метил-3-(4-формилфенил)акрилат (полученный, как описано в примере 2, получение исходных веществ) (18,56 г; 97,57 ммоль) добавляли к перемешиваемому раствору (R)-1-(1Н-индол-3-ил)пропан-2-амина (17 г; 97,57 ммоль) в уксусной кислоте (250 мл) при 23°C в атмосфере азота. Полученный раствор перемешивали при 80°C в течение 2 часов. Реакционную смесь упаривали досуха, перерастворяли в DCM (500 мл) и промывали последовательно насыщенным раствором NaHCO3 (300 мл×2), 2 М раствором NaOH (водн.) (300 мл), водой (300 мл) и насыщенным рассолом (300 мл). Органический слой сушили над MgSO4, фильтровали и упаривали, получая неочищенный продукт. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 1-7% метанола в DCM. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(4-((1S,3R)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (25,1 г; 74,3%) в виде бежевой пены. Продукт представлял собой в основном цис-изомер, содержащий примерно 12% транс-изомера, который было невозможно отделить.1Н ЯМР (400 МГц, DMSO, 30°С) δ 1.25 (3Н, d), 2.37-2.48 (1Н, m), 2.74 (1Н, d), 3.12 (1Н, s), 3.73 (3Н, s), 5.18 (1Н, s), 6.64 (1Н, d), 6.97 (2Н, dd), 7.19 (1Н, d), 7.36-7.46 (3Н, m), 7.64-7.75 (3Н, m), 10.19 (1Н, s), никакого сигнала от NH не наблюдали, m/z: ES+ [М+Н]+ 347.
Получение (Е)-метил-3-(4-((1S,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата

(Е)-Метил-3-(4-((1S,3R)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (35 г; 101,03 ммоль), 3-бромпроп-1-ен (9,62 мл; 111,14 ммоль) и N-этил-N-изопропилпропан-2-амин (19,36 мл; 111,14 ммоль) суспендировали в ацетонитриле (160 мл), через эту смесь в течение 5 мин барботировали азот и затем смесь герметично закрывали в пробирке для микроволнового реактора. Реакционную смесь нагревали до 140°C в течение 3,5 часа в микроволновом реакторе и охлаждали до комнатной температуры.
Реакционную смесь упаривали досуха, перерастворяли в DCM (100 мл) и промывали последовательно 1 М раствором лимонной кислоты (100 мл), водой (100 мл) и насыщенным рассолом (100 мл). Органический слой сушили над MgSO4, фильтровали и упаривали, получая неочищенный продукт. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-20% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая смесь 50:50 (Е)-метил-3-(4-((1R,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата и (Е)-метил-3-(4-((1S,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (10,00 г; 25,6%) в виде бледно-желтого твердого вещества, m/z: ES+ [М+Н]+ 387.
Получение (Е)-метил-3-(4-((1R,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата

Трифторуксусную кислоту (5,59 мл; 75,29 ммоль) добавляли к (Е)-метил-3-(4-((1S,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилату (9,7 г; 25,10 ммоль) в DCM (100 мл) при 20°C в атмосфере азота. Полученный раствор перемешивали при 20°C в течение 3 суток. Реакционную смесь осторожно разбавляли насыщенным раствором NaHCO3 (250 мл) и слой в DCM промывали последовательно водой (250 мл) и насыщенным рассолом (250 мл). Органический слой сушили над MgSO4, фильтровали и упаривали, получая неочищенный продукт. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 10-20% EtOAc в гептане. Фракции упаривали досуха, получая смесь 65:35 (Е)-метил-3-(4-((1R,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата и (Е)-метил-3-(4-((1S,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (7,99 г; 82%) в виде бледно-желтого твердого вещества, m/z: ES+ [М+Н]+ 387.
Получение (Е)-метил-3-(4-((1R,3R)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата

7 отдельных порций смеси 65:35 (Е)-метил-3-(4-((1R,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата и (Е)-метил-3-(4-((1S,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (2,00 г; 5,17 ммоль) приводили во взаимодействие следующим образом.
(Е)-Метил-3-(4-((1R,3R)-2-аллил-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (2,00 г; 5,17 ммоль) и хлортрис(трифенилфосфин)родий(I) (катализатор Уилкинсона) (2,346 г; 2,54 ммоль) суспендировали в ацетонитриле (12 мл) и воде (2,4 мл), и через эту суспензию в течение 5 мин барботировали азот, после чего ее герметично закрывали в пробирке для микроволнового реактора. Реакционную смесь нагревали до 100°C в течение 60 мин в микроволновом реакторе и охлаждали до комнатной температуры. Реакционные смеси объединяли, упаривали досуха, перерастворяли в DCM (200 мл) и добавляли насыщенный раствор NaНСО3 (200 мл). Органический слой промывали последовательно водой (200 мл) и насыщенным рассолом (200 мл), после чего сушили над MgSO4, фильтровали и упаривали, получая неочищенный продукт. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-5% метанола в DCM. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(4-((1R,3R)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (7,02 г; 55,9%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, DMSO, 30°С) δ 1.14 (3Н, d), 2.27-2.4 (1Н, m), 2.81 (1Н, dd), 2.94-3.05 (1Н, m), 3.72 (3Н, s), 5.19 (1Н, s), 6.60 (1Н, d), 6.94-7 (1Н, m), 7.01-7.09 (1Н, m), 7.26 (3Н, d), 7.43 (1Н, d), 7.59-7.68 (3Н, m), 10.70 (1Н, s), сигнала от NH не наблюдали, m/z: ES+ [М+Н]+ 347.
Получение (Е)-метил-3-(4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата

(S)-3-Фтор-2-метилпропилтрифторметансульфонат (полученный, как описано в примере 3, получение исходных веществ) (291 мг; 1,30 ммоль) добавляли к раствору (Е)-метил-3-(4-((1R,3R)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (300 мг; 0,87 ммоль) и N,N-диизопропилэтиламина (0,226 мл; 1,30 ммоль) в 1,4-диоксане (5 мл). Смесь перемешивали при 90°C в течение 1 часа, затем смесь упаривали досуха и остаток распределяли между DCM (30 мл) и водой (30 мл). Водный слой экстрагировали DCM (30 мл) и экстракты объединяли с органическим слоем. Объединенные экстракты фильтровали через бумагу для разделения фаз и упаривали. Остаток очищали флэш-хроматографией на диоксиде кремния с элюированием растворителем 15%-ным EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(4-((1R,3R)-2-((S)-3-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (314 мг; 86%) в виде беловатого твердого вещества.1Н ЯМР (400 МГц, DMSO, 30°С) δ 0.87 (3Н, d), 1.06 (3Н, d), 1.9-2.28 (2Н, m), 2.55-2.8 (3Н, m), 2.97-3.21 (1Н, m), 3.72 (3Н, s), 4.31-4.69 (2Н, m), 4.88 (1Н, s), 6.60 (1Н, dd), 6.98 (1Н, t), 7.04 (1Н, t), 7.17-7.35 (3Н, m), 7.44 (1Н, d), 7.55-7.76 (3Н, m), 10.65 (1Н, s). m/z: ES+ [M+H]+ 421.
Пример 5. (Е)-3-(3.5-Дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота (изомер 1)*

Гидроксид натрия (184 мг; 4,61 ммоль) добавляли к (Е)-метил-3-(3,5-дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилату (изомеру 1) (217 мг; 0,46 ммоль) в смеси THF (1 мл)/метанол (1 мл). Полученный раствор перемешивали при 20°C в течение 16 часов. Реакционную смесь разбавляли водой (10 мл) и рН подводили до 7, добавляя 2 н. раствор HCl. Раствор экстрагировали EtOAc (2х 20 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-50% EtOAc в гептане. Фракции чистого продукта упаривали, получая неочищенное вещество. Неочищенный продукт растирали с использованием смеси диэтиловый эфир/изогексан, получая указанный в заголовке продукт (53,0 мг; 25,2%) в виде желтого твердого вещества.1Н ЯМР (400 МГц, DMSO, 27°С) δ 0.54 (3Н, d), 1.06 (3Н, s), 1.34 (3Н, s), 2.14 (1Н, dd), 2.66 (1Н, d), 2.83 (1Н, d), 3.03 (1Н, dd), 4.21 (1Н, t), 4.33 (1Н, t), 5.12 (1Н, s), 6.73 (1Н, d), 7.01 (2Н, dtd), 7.14-7.28 (1Н, m), 7.43 (1Н, d), 7.54 (2Н, s), 7.59 (1Н, d), 10.50 (1Н, s), 12.61 (1Н, s), сигнала от СO2Н не наблюдали, m/z: ES+ [М+Н]+ 457.
* Предполагается, что стереохимия при неидентифицированном центре соответствует (R) по аналогии с соединениями из других примеров, т.е. предполагается, что это соединение представляет собой: (Е)-3-(3,5-дифтор-4-(1R)-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту.
3-(3,5-Дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1), используемый в качестве исходного вещества, получали так, как указано ниже.
Получение (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)-3,5-дифторфенил)акрилата

1-(1Н-Индол-3-ил)-2-метилпропан-2-амин (807 мг; 4,29 ммоль) и (Е)-метил-3-(3,5-дифтор-4-формилфенил)акрилат (полученный, как описано в примере 1, получение исходных веществ) (970 мг; 4,29 ммоль) объединяли в уксусной кислоте (15 мл) и смесь нагревали до 80°C в течение 2 часов. Реакционную смесь очищали ионообменной хроматографией, используя SCX-2 колонку. Желаемый продукт элюировали с колонки, используя 2 М NH3 в метаноле, и содержащие продукт фракции упаривали досуха, получая (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)-3,5-дифторфенил)акрилат (1610 мг; 95%) в виде желтой пены.1Н ЯМР (500 МГц, DMSO, 20°С) δ 1.15 (3Н, s), 1.27 (3Н, s), 2.23 (1Н, s), 2.61 (2Н, s), 3.75 (3Н, s), 5.46 (1Н, s), 6.84 (1Н, d), 6.97 (2Н, dtd), 7.18 (1Н, d), 7.39 (1Н, d), 7.58 (2Н, s), 7.67 (1Н, d), 10.60 (1Н, s). m/z: ES- [М-Н]- 395.
Получение (Е)-метил-3-(3,5-дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-]индол-1-ил)фенил)-акрилата (изомера 1)

(S)-3-Фтор-2-метилпропилтрифторметансульфонат (полученный, как описано в примере 3, получение исходных веществ) (0,339 г; 1,51 ммоль) добавляли к раствору (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)-3,5-дифторфенил)акрилата (0,3 г; 0,76 ммоль) и N-этил-N-изопропилпропан-2-амина (0,458 мл; 2,65 ммоль) в 1,4-диоксане (2 мл). Перемешивание продолжали в течение 24 часов, затем летучие вещества удаляли под вакуумом и неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-25% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(3,5-дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (0,202 г; 36%) в виде белого твердого вещества. Это вещество объединяли с другой порцией (0,36 г) и очищали препаративной HPLC (колонка Chiralpak IA, диоксид кремния (20 мкм), диаметр 20 мм, длина 250 мм), элюируя смесью 70:30 гептан:IPA со скоростью 80 мл/мин (4 инжекции). Фракции, содержащие желаемые соединения, упаривали, получая (Е)-метил-3-(3,5-дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1, элюируемый первым, 217 мг) и (Е)-метил-3-(3,5-дифтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 2, элюируемый вторым, 165 мг). Анализ выполняли на колонке Chiralpak IA (диоксид кремния, 5 мкм; диаметр 4,6 мм, длина 50 мм), элюируя смесью 70:30 гептан:IPA со скоростью 2 мл/мин.
1Н ЯМР (400 МГц, DMSO, 30°С) δ 0.50 (3Н, d), 1.02 (3Н, s), 1.30 (3Н, s), 2.11 (1Н, dd), 2.62 (2Н, d), 2.80 (1Н, d), 2.99 (1Н, dd), 3.74 (3Н, s), 4.09-4.23 (1Н, m), 4.29 (1Н, d), 5.09 (1Н, s), 6.81 (1Н, d), 6.97 (2Н, dt), 7.16 (1Н, d), 7.39 (1Н, d), 7.53 (2Н, d), 7.64 (1Н, d), 10.44 (1Н, s). m/z: ES+ [М+Н]+ 471.
Пример 6. (Е)-3-(3,5-Дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота (изомер 1)*

2 М гидроксид натрия (1,6 мл; 3,20 ммоль) добавляли к раствору (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомер 1) (148 мг; 0,31 ммоль) в смеси THF (0,8 мл)/метанол (0,8 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли EtOAc (15 мл) и воду (15 мл) и pH водного слоя подводили примерно до 7, добавляя 2 н. раствор HCl. Проводили разделение слоев и водный слой экстрагировали EtOAc (15 мл). Объединенные органические экстракты сушили над Na2SO4 и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 25-100% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая указанный в заголовке продукт (изомер 1) (124 мг; 86%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) δ 1.03 (3Н, d), 1.08 (3Н, s), 1.16 (3Н, d), 1.35 (3Н, s), 2.56 (1Н, dd), 2.66 (1Н, d), 3.02 (1Н, d), 3.17 (1Н, dd), 5.24 (1Н, s), 6.39 (1Н, d), 7.00 (2Н, d), 7.05-7.16 (2Н, m), 7.20 (1Н, dd), 7.28 (1Н, s), 7.49 (1Н, dd), 7.61 (1Н, d), сигнала от CO2H не наблюдали. m/z: ES+ [М+Н]+ 457.
* Предполагается, что стереохимия при неидентифицированном центре соответствует (R) по аналогии с соединениями из других примеров.
(Е)-Метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1), используемый в качестве исходного вещества, получали так, как указано ниже.
Получение (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1)
2-Фтор-2-метилпропилтрифторметансульфонат (полученный, как описано в примере 1, получение исходных веществ) (679 мг; 3,03 ммоль) добавляли к раствору (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)-3,5-дифторфенил)акрилата (полученного, как описано в примере 5, получение исходных веществ) (600 мг; 1,51 ммоль) и N-этил-N-изопропилпропан-2-амина (0,915 мл; 5,30 ммоль) в 1,4-диоксане (2,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Затем реакционную смесь нагревали до 105°C в течение 88 часов. Летучие вещества удаляли под вакуумом и неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-50% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (430 мг; 60,4%) в виде белого твердого вещества. Рацемический продукт очищали препаративной HPLC (колонка Chiralpak AD, диоксид кремния, 20 мкм; диаметр 50 мм, длина 250 мм), элюируя смесью 90:10 гептан : этанол со скоростью 90 мл/мин. Фракции, содержащие желаемые соединения, упаривали досуха, получая (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1, элюируемый первым, 149 мг; 34,6%) и (Е)-метил-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 2, элюируемый вторым; 143 мг; 33,3%) в виде твердых веществ кремовой окраски.1Н ЯМР (400 МГц, CDCl3, 30°С) δ 1.01 (3Н, d), 1.08 (3Н, s), 1.15 (3Н, d), 1.34 (3Н, s), 2.55 (1Н, dd), 2.66 (1Н, d), 2.98-3.06 (1Н, m), 3.17 (1Н, dd), 3.80 (3Н, s), 5.23 (1Н, s), 6.38 (1Н, d), 6.97 (2Н, d), 7.07-7.12 (2Н, m), 7.17-7.22 (1Н, m), 7.32 (1Н, s), 7.46-7.5 (1Н, m), 7.52 (1Н, d). m/z ES- [М-Н]- 469.
Пример 7. (Е)-3-(4-(2-((S)-3-Фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота (изомер 1)*

2 М раствор гидроксида натрия (20,42 мл; 40,85 ммоль) добавляли к раствору (Е)-метил-3-(4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1) (3,55 г; 8,17 ммоль) в метаноле (100 мл), THF (100 мл) и воде (75 мл). Смесь нагревали при 40°C в течение 16 часов. Смесь разбавляли водой (100 мл) и концентрировали до такого объема, при котором органические растворители были удалены. Полученный водный раствор подкисляли до pH 6, используя 2 М HCl. Полученную водную суспензию экстрагировали DCM (500 мл) (добавляя рассол для облегчения разделения образовавшейся эмульсии), фильтровали через бумагу для разделения фаз, сушили над MgSO4, затем фильтровали через целит и упаривали, получая приблизительно 3,5 г бледно-желтого твердого вещества. Неочищенный продукт обрабатывали смесью диэтиловый эфир/DCM (1:1, 150 мл) и ультразвуком. Образовавшуюся тонкодисперсную суспензию пропускали через набивку диоксида кремния (примерно 100 г) и элюировали с диоксида кремния диэтиловым эфиром (примерно 2 л). Фракции, содержащие продукт, объединяли, упаривали и сушили под вакуумом при 50°C, получая указанный в заголовке продукт (2,305 г; 64,5%) в виде бежевого твердого вещества.1Н ЯМР (400 МГц, DMSO, 30°С) δ 0.52 (3Н, d), 1.03 (3Н, s), 1.05-1.16 (1Н, m), 1.23 (3Н, s), 2.21 (1Н, dd), 2.65 (1Н, d), 2.84 (1Н, d), 2.89 (1Н, dd), 4.19 (1Н, ddd), 4.31 (1Н, ddd), 4.66 (1Н, s), 6.50 (1Н, d), 6.93 (1Н, ddd), 6.98 (1Н, ddd), 7.18 (1Н, d), 7.38 (2Н, d), 7.40 (1Н, d), 7.58 (1Н, d), 7.63 (2Н, d), 10.18 (1Н, s), 12.30 (1Н, s). m/z: ES+ [М+Н]+ 421.
* Предполагается, что стереохимия при неидентифицированном центре соответствует (R) по аналогии с соединениями из других примеров.
Продукт (9,0 г; 21,40 ммоль) суспендировали в ацетонитриле (150 мл) в атмосфере азота в темноте в течение 1 часа в закрытой круглодонной колбе емкостью 250 мл. Смесь перемешивали в течение выходных при комнатной температуре, затем фильтровали и промывали холодным ацетонитрилом (60 мл), получая белое твердое вещество, которое сушили под высоким вакуумом при 40°C в течение 5 часов, получая кристаллическую Форму A указанного в заголовке продукта (7,81 г; 87%).
XRPD-кривая кристаллической Формы A включает следующие пики и показана на Фиг. 1.

(Е)-Метил-3-(4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1), используемый в качестве исходного вещества, получали так, как указано ниже.
Получение (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (рацемата)

(Е)-Метил-3-(4-формилфенил)акрилат (41,8 г; 219,62 ммоль) (полученный, как описано в примере 2, получение исходных веществ) добавляли в виде одной порции к 1-(1Н-индол-3-ил)-2-метилпропан-2-амину (43,8 г; 219,62 ммоль) в уксусной кислоте (314 мл) в атмосфере азота. Полученный раствор перемешивали при 80°C в течение 5 часов. Реакционную смесь концентрировали в вакууме. Добавляли толуол (200 мл) и остаток упаривали досуха.
Азеотропную обработку повторяли еще дважды, получая коричневое твердое вещество. Его перемешивали в смеси 1:1 EtOAc/гептан (500 мл) в течение 30 мин, после чего фильтровали и промывали смесью 1:1 EtOAc/гептан. Соединение сушили на воздухе, получая белое твердое вещество. Неочищенное вещество суспендировали в 2-метилтетрагидрофуране (750 мл) и к перемешиваемой смеси добавляли в течение 10 мин насыщенный раствор бикарбоната натрия (выделение газа), смесь перемешивали до полного растворения вещества, и водная фаза оставалась щелочной. Фазы разделяли, и органическую фазу промывали рассолом, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая бледно-желтую пену (приблизительно 78 г). Это вещество растворяли в диэтиловом эфире (200 мл) и концентрировали досуха (повторяли дважды). После второго добавления получали соответствующее твердое вещество. Его перемешивали в диэтиловом эфире и упаривали досуха, получая (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (рацемат) (73,6 г; 89%).1Н ЯМР (400 МГц, CDCl3, 30°С) δ 1.26 (3Н, s), 1.35 (3Н, s), 1.42 (1Н, br s), 2.69-2.82 (2Н, m), 3.80 (3Н, s), 5.12 (1Н, s), 6.41 (1Н, d), 7.06-7.16 (2Н, m), 7.21 (1Н, dd), 7.37 (2Н, d), 7.46-7.54 (4Н, m), 7.67 (1Н, d). m/z: ES+ [М+Н]+ 361.
Получение (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1)

(Е)-Метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (рацемат) (65 г) очищали, используя семь инжекций, как приведено ниже.
Рацемическое вещество очищали препаративной HPLC (колонка Chiralpak OD, диоксид кремния, 20 мкм; диаметр 100 мм, длина 250 мм), элюируя смесью 50:50 гептан:IPA. Фракции, содержащие желаемые соединения, упаривали досуха, получая (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1, элюируемый первым; 30,3 г; 93%) и (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 2, элюируемый вторым; 28,2 г; 86%).1Н ЯМР (400 МГц, CDCl3, 30°С) δ 1.27 (3Н, s), 1.36 (3Н, s), 2.69-2.82 (2Н, m), 3.80 (3Н, s), 5.14 (1Н, s), 6.43 (1Н, d), 7.12 (2Н, pd), 7.2-7.24 (1Н, m), 7.39 (3Н, d), 7.51 (3Н, d), 7.68 (1Н, d), сигнала от NH не наблюдали. m/z: ES+ [М+Н]+ 361.
Получение (Е)-метил-3-(4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1)

(S)-3-Фтор-2-метилпропилтрифторметансульфонат (полученный, как описано в примере 3, получение исходных веществ) (5,32 г; 21,36 ммоль) добавляли к раствору (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1) (3,5 г; 9,71 ммоль) и N-этил-N-изопропилпропан-2-амина (6,34 мл; 36,41 ммоль) в 1,4-диоксане (17,5 мл). Смесь перемешивали при 22°C в течение 3 суток. Смесь упаривали и остаток распределяли между DCM (150 мл) и водой (150 мл). Водный слой экстрагировали DCM (50 мл), и экстракты объединяли с органическим слоем. Объединенные экстракты фильтровали через бумагу для разделения фаз и упаривали. Остаток очищали флэш-хроматографией на диоксиде кремния, элюируя растворителем 15%-ным EtOAc в гептане. Во фракциях, содержащих значительные количества продукта, начиналось образование кристаллов; пробирки встряхивали для содействия дальнейшей кристаллизации. Кристаллы собирали фильтрованием и промывали небольшим количеством 15%-ного EtOAc в гептане, получая (Е)-метил-3-(4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1) (2,91 г; 69,0%) в виде белого кристаллического вещества. Маточные жидкости после кристаллизации и другие содержащие продукт фракции объединяли и упаривали. Остаток перекристалл изовывал и из смеси EtOAc/гептан, получая дополнительное количество 3-(4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1) в виде белого кристаллического вещества (635 мг; 15,1%).1Н ЯМР (400 МГц, CDCl3, 30°С) δ 0.53 (3Н, d), 0.95-1.07 (1Н, m), 1.09 (3Н, s), 1.32 (3Н, s), 2.16 (1Н, dd), 2.66 (1Н, d), 2.94 (1Н, d), 2.97 (1Н, d), 3.80 (3Н, s), 4.14 (1Н, ddd), 4.31 (1Н, ddd), 4.59 (1Н, s), 6.42 (1Н, d), 7.05-7.11 (2Н, m), 7.13 (1Н, s), 7.17 (1Н, dd), 7.35 (2Н, d), 7.45 (2Н, d), 7.50 (1Н, dd), 7.67 (1Н, d). m/z: ES+ [М+Н]+ 435.
Пример 8. (Е)-3-(4-(2-(2-Фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота (изомер 1)*

2 М раствор гидроксида натрия (0,782 мл; 1,56 ммоль) добавляли к раствору (Е)-метил-3-(4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1) (68 мг; 0,16 ммоль) в метаноле (5 мл), THF (5,00 мл) и воде (5,00 мл), и смесь перемешивали в течение 40 часов при 22°С. Смесь концентрировали до такого объема, при котором весь органический растворитель был удален, и подкисляли до рН 6, используя 2 М HCl. Добавляли концентрированный водный аммиак (2 капли), затем метанол (приблизительно 2 мл), получая бледно-желтый раствор. Неочищенный продукт очищали препаративной HPLC (препаративная колонка XBridge С18 OBD от Waters, диоксид кремния, 5 мкм; диаметр 50 мм, длина 100 мм), используя в качестве элюентов смеси воды (содержащей 1% NH3) и MeCN с уменьшающейся полярностью. Фракции, содержащие желаемое соединение, упаривали досуха, получая указанный в заголовке продукт (изомер 1) (50,0 мг; 76%) в виде желтого твердого вещества.1Н ЯМР (500 МГц, DMSO, 30°С) δ 0.93 (3Н, s), 1.19 (3Н, d), 1.23 (3Н, s), 1.51 (3Н, d), 2.56-2.64 (2Н, m), 2.75 (1Н, d), 3.04 (1Н, dd), 5.06 (1Н, s), 6.40 (1Н, d), 7.03 (1Н, ddd), 7.09-7.15 (2Н, m), 7.35 (1Н, dd), 7.44-7.51 (5Н, m), 10.87 (1Н, s), сигнала от СO2Н не наблюдали. m/z: ES+ [М+Н]+ 421.
* Предполагается, что стереохимия при неидентифицированном центре соответствует (R) по аналогии с соединениями из других примеров.
(Е)-Метил-3-(4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1), используемый в качестве исходного вещества, получали так, как указано ниже.
Получение (Е)-метил-3-(4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (изомера 1)

2-Фтор-2-метилпропилтрифторметансульфонат (полученный, как описано в примере 1, получение исходных веществ) (389 мг; 1,73 ммоль) добавляли к раствору (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (рацемата) (полученного, как описано в примере 7, получение исходных веществ) (250 мг; 0,69 ммоль) и N,N-диизопропилэтиламина (0,453 мл; 2,60 ммоль) в 1,4-диоксане (1,25 мл). Смесь перемешивали при 95°C в течение 64 часов и затем распределяли между DCM (30 мл) и водой (30 мл). Водный слой экстрагировали DCM (20 мл) и экстракты объединяли с органическим слоем. Объединенные экстракты фильтровали через бумагу для разделения фаз и упаривали. Остаток очищали флэш-хроматографией на диоксиде кремния, элюируя растворителем 15%-ным EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (рацемат) (178 мг; 59,1%) в виде бежевого твердого вещества. Рацемический продукт очищали препаративной HPLC (колонка Chiralpak AD, диоксид кремния, 20 мкм; диаметр 50 мм, длина 250 мм), элюируя смесью 80:20 гептан : этанол. Фракции, содержащие желаемые соединения, упаривали досуха, получая (Е)-метил-3-(4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1, элюируемый первым; 70 мг) и (Е)-метил-3-(4-(2-(2-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 2, элюируемый вторым; 66 мг).1Н ЯМР (400 МГц, CDCl3, 30°С) δ 1.00 (3Н, s), 1.13 (3Н, d), 1.20 (3Н, s), 1.44 (3Н, d), 2.57-2.78 (3Н, m), 2.93 (1Н, dd), 3.80 (3Н, s), 5.05 (1Н, s), 6.41 (1Н, d), 7.13 (1Н, ddd), 7.18 (1Н, ddd), 7.32 (1Н, d), 7.43 (2Н, d), 7.51 (2Н, d), 7.54 (1Н, d), 7.64 (1Н, s), 7.67 (1Н, d). m/z: ES+ [М+Н]+ 435.
Пример 9. (Е)-3-(3-Фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловая кислота (изомер 1)*

2 М гидроксид натрия (3,31 мл; 6,63 ммоль) добавляли к раствору (Е)-метил-3-(3-фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (600 мг; 1,33 ммоль) (смеси диастереоизомеров) в метаноле (5 мл) и THF (20 мл) и смесь перемешивали при температуре окружающей среды в течение 4 часов. Смесь концентрировали до такого объема, при котором весь органический растворитель был удален, разбавляли водой (50 мл), подкисляли разбавленной HCl до pH 6 и экстрагировали этилацетатом (2×50 мл). Экстракты объединяли и упаривали при пониженном давлении. Остаток очищали препаративной HPLC (колонка Chiralpak IA, диоксид кремния, 20 мкм; диаметр 20 мм, длина 250 мм), элюируя смесью 90:10 гептан:IPA, содержащей 0,2% уксусной кислоты, со скоростью 80 мл/мин. Фракции, содержащие желаемые соединения, объединяли и анализировали, получая (Е)-3-(3-фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту (изомер 1, элюируемый первым; 130 мг; 22,36%) и (Е)-3-(3-фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту (изомер 2, элюируемый вторым; 30,0 мг; 5,16%).1Н ЯМР (500 МГц, DMSO, 30°С) δ 0.49 (3Н, d), 1.03 (3Н, s), 1.14-1.21 (1H, m), 1.28 (3Н, s), 2.17 (1H, dd), 2.59-2.71 (1H, m), 2.8-2.99 (2H, m), 4.09-4.34 (2H, m), 4.99 (1H, s), 6.59 (1H, d), 6.86-7.07 (2H, m), 7.1-7.19 (1H, m), 7.18-7.29 (1H, m), 7.36-7.49 (2H, m), 7.49-7.66 (2H, m), 10.31 (1H, s), сигнала от CO2H не наблюдали. m/z: ES+ [М+Н]+ 439.
* Предполагается, что стереохимия при неидентифицированном центре соответствует (R) по аналогии с соединениями из других примеров.
(Е)-Метил-3-(3-фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (смесь диастереомеров), использованный в качестве исходного вещества, получали так, как указано ниже.
Получение (Е)-метил-3-(3-фтор-4-формилфенил)акрилата

4-Бром-2-фторбензальдегид (20,88 г; 102,87 ммоль) и метилакрилат (13,98 мл; 154,30 ммоль) переносили в тщательно дегазированный DMA (150 мл) и добавляли три-о-толилфосфин (3,13 г; 10,29 ммоль), ацетат палладия(II) (1,155 г; 5,14 ммоль) и триэтиламин (28,7 мл; 205,74 ммоль). Реакционную смесь перемешивали и нагревали до 100°C в течение 16 часов. Добавляли дополнительное количество три-о-толилфосфина (3,13 г; 10,29 ммоль) и ацетата палладия(II) (1,155 г; 5,14 ммоль) и реакционную смесь нагревали до 110°C в течение еще 2 часов. Добавляли воду (1 л) и реакционную смесь экстрагировали DCM (2×500 мл). Объединенные органические экстракты сушили (MgSO4), фильтровали и упаривали, получая коричневое твердое вещество. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-25% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(3-фтор-4-формилфенил)акрилат (15,20 г; 71,0%) в виде желтого твердого вещества.
1Н ЯМР (400 МГц, CDCl3, 30°С) δ 3.83 (3Н, s), 6.53 (1Н, d), 7.31 (1Н, dd), 7.41 (1Н, d), 7.65 (1Н, d), 7.79-8 (1Н, m), 10.25-10.41 (1Н, m). При проведении LCMS никакого молекулярного иона не наблюдали.
Получение (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)-3-фторфенил)акрилата (рацемата)

1-(1Н-Индол-3-ил)-2-метилпропан-2-амин (1 г; 5,31 ммоль) и (Е)-метил-3-(3-фтор-4-формилфенил)акрилат (1,106 г; 5,31 ммоль) в уксусной кислоте (15 мл) перемешивали при 80°C в течение 2 часов в атмосфере азота. Неочищенный продукт очищали ионообменной хроматографией, используя SCX-2 колонку. Желаемый продукт элюировали с колонки, используя 7 М NH3/метанол, и фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)-3-фторфенил)акрилат (рацемат) (2,000 г; 99%).1Н ЯМР (400 МГц, DMSO, 30°С) δ 1.14 (3Н, s), 1.27 (3Н, s), 2.52-2.74 (2Н, m), 3.74 (3Н, s), 5.12 (1Н, s), 6.69 (1Н, d), 6.86-7.05 (2Н, m), 7.20 (1Н, d), 7.25-7.33 (2Н, m), 7.39 (1Н, d), 7.73 (1Н, d), 7.85 (1Н, t), 10.29 (1Н, s), сигнала от NH не наблюдали. m/z: ES+ [М+Н]+ 379.
Получение (Е)-метил-3-(3-фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата (смеси диастереомеров)

(S)-3-Фтор-2-метилпропилтрифторметансульфонат (полученный, как описано в примере 3, получение исходных веществ) (1,259 г; 5,62 ммоль) добавляли к раствору (Е)-метил-3-(4-(3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)-3-фторфенил)акрилата (рацемата) (850 мг; 2,25 ммоль) и N-этил-N-изопропилпропан-2-амина (1,467 мл; 8,42 ммоль) в 1,4-диоксане (5 мл). Смесь нагревали при 60°C в течение 4 часов, затем перемешивали при температуре окружающей среды в течение 12 часов. Смесь распределяли между этилацетатом (25 мл) и водой (25 мл). Водный слой экстрагировали этилацетатом (2×25 мл) и экстракты объединяли с органическим слоем. Объединенные экстракты упаривали под вакуумом. Остаток очищали флэш-хроматографией на диоксиде кремния, элюируя растворителем 10%-ным EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(3-фтор-4-(2-((S)-3-фтор-2-метилпропил)-3,3-диметил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (смесь диастереоизомеров) (600 мг; 59,0%). m/z: ES+ [М+Н]+ 453.
Пример 10. (Е)-3-[4-[(1R,3R)-1-Дейтеро-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-1-ил]-3,5-дифтор-фенил]проп-2-еновая кислота

Метил-(Е)-3-[4-[(1R,3R)-1-дейтеро-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-1-ил]-3,5-дифтор-фенил]проп-2-еноат (4,70 г; 10,27 ммоль) растворяли в iPrOH (42,8 мл) и добавляли в виде одной порции 5 М раствор гидроксида натрия (6,16 мл; 30,82 ммоль), реакционную смесь затем перемешивали при комнатной температуре в течение 4 часов. Добавляли воду (100 мл) и pH подводили до приблизительно 5, добавляя 2 н. раствор HCl. Раствор экстрагировали EtOAc (×2), объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Остаток пропускали через набивку диоксида кремния, элюируя сначала DCM, затем до 5%-ного МеОН в DCM. Фракции, содержащие продукт, упаривали до желтого твердого вещества (приблизительно 4,2 г). Остаток (4,2 г) растворяли в EtOH (20 мл) и нагревали до 35°C. Медленно приблизительно в течение 40 мин добавляли воду (30 мл). Далее смесь перемешивали в течение еще 30 минут, затем медленно охлаждали до комнатной температуры. Добавляли дополнительное количество воды (30 мл) и реакционную смесь затем охлаждали до 0°С. Смесь фильтровали и твердые вещества промывали водой, после чего сушили под вакуумом при 35°С в течение ночи, получая (Е)-3-[4-[(1R,3R)-1-дейтеро-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-1-ил]-3,5-дифтор-фенил]проп-2-еновую кислоту (3,34 г; 73,3%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) 1.12 (3Н, d), 1.19 (3Н, d), 1.26 (3Н, d), 2.43 (1Н, dd), 2.63 (1Н, dd), 2.87 (1Н, dd), 3.07 (1Н, dd), 3.65 (1Н, q), 6.41 (1Н, d), 7.02 (2Н, d), 7.06-7.16 (2Н, m), 7.19-7.25 (1Н, m), 7.41 (1Н, s), 7.48-7.57 (1Н, m), 7.63 (1Н, d), сигнала от CO2H не наблюдали. m/z: ES+ [М+Н]+ 444.
Это соединение альтернативно может быть названо как (E)-3-[4-[(1R,3R)-1-дейтеро-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-1-ил]-3,5-дифтор-фенил]акриловая кислота.
Получение (4-бром-2,6-дифтор-фенил)-дидейтеро-метанола

Бордейтерид лития (0,497 г; 17,25 ммоль) добавляли порциями к раствору метил-4-бром-2,6-дифторбензоата (2,89 г; 11,5 ммоль) в THF (46,0 мл). Реакционную смесь нагревали до 50°C в течение 2 часов. После охлаждения осторожно добавляли 2 н. HCl (30 мл). Проводили разделение слоев и водный слой экстрагировали EtOAc (2×50 мл). Объединенные органические экстракты промывали рассолом, сушили (MgSO4) и концентрировали, получая (4-бром-2,6-дифтор-фенил)-дидейтеро-метанол (2,250 г; 87%) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) 1.96 (1Н, s), 7.07-7.13 (2Н, m).
Получение 4-бром-2,6-дифтор-1-дейтеробензальдегида

Реагент Десса-Мартина (4,98 г; 11,73 ммоль) добавляли к (4-бром-2,6-дифтор-фенил)-дидейтеро-метанолу (2,20 г; 9,78 ммоль) в DCM (39,1 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 1 часа, затем гасили, добавляя насыщ. раствор NaHCO3 (50 мл), содержащий 10% тиосульфата натрия. Проводили разделение слоев и водную фазу экстрагировали DCM (2×50 мл). Органические экстракты сушили (MgSO4) и концентрировали, затем неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-25% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая 4-бром-2,6-дифтор-1-дейтеробензальдегид (2,040 г; 94%) в виде белого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) 7.18-7.25 (2Н, m). Никакого молекулярного иона не наблюдали.
Получение метил-(Е)-3-(4-дейтерокарбонил-3,5-дифтор-фенил)проп-2-еноата

4-Бром-2,6-дифтор-1-дейтеробензальдегид (3,33 г; 15,0 ммоль), триэтиламин (4,18 мл; 30,00 ммоль), ацетат палладия(II) (0,168 г; 0,75 ммоль) и тритолилфосфин (0,457 г; 1,50 ммоль) растворяли в DMF (36,6 мл), который был дегазирован. Затем добавляли метилакрилат (2,026 мл; 22,50 ммоль) и реакционную смесь нагревали до 80°C в течение 4 часов. После охлаждения смесь добавляли к воде (150 мл) и экстрагировали EtOAc (2х 150 мл). Объединенные органические экстракты промывали 2 н. HCl (100 мл), затем рассолом (100 мл), после чего сушили (MgSO4) и концентрировали. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-40% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая метил-(Е)-3-(4-дейтерокарбонил-3,5-дифтор-фенил)проп-2-еноат (2,93 г; 86%) в виде желтого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) 3.83 (3Н, d), 6.51 (1Н, d), 7.12 (2Н, m), 7.57 (1Н, d). m/z (ES+), [М+Н]+=228.
(R)-N-(1-(1Н-Индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин
(2R)-1-(1Н-Индол-3-ил)пропан-2-амин (3,81 кг; 21,21 моль) добавляли в атмосфере азота в облицованный стеклом сосуд емкостью 100 л с рубашкой. Добавляли 1,4-диоксан (23 л) и включали устройство для перемешивания. К перемешиваемой суспензии добавляли диизопропилэтиламин (5,55 л; 31,82 моль), затем (2-фтор-2-метил-пропил)трифторметансульфонат (5,55 кг; 23,77 моль). В сосуд добавляли 1,4-диоксан (4 л), и смесь нагревали до 75°С. Нагревание продолжали в течение 24 часов, после чего выполняли охлаждение смеси до 25°С. В сосуд добавляли воду (30,5 л), затем толуол (30,5 л). Через 40 минут устройство для перемешивания отключали, и слои оставляли разделяться. Водный слой удаляли и к органическому раствору добавляли воду (30,5 л). Смесь перемешивали в течение 15 минут, после чего слои оставляли разделяться. Водный слой удаляли из сосуда. Органический раствор концентрировали вакуумной дистилляцией (в рубашке: температура 65°C, давление 110 мбар (11 кПа)) до удаления приблизительно 27 л дистиллята. Оставшийся в сосуде раствор охлаждали, получая (R)-N-(1-(1H-индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин в виде раствора в толуоле (33% (масс./масс.)) (15,4 кг; 97%).1Н ЯМР (500 МГц, DMSO, 27°С) 0.98 (3Н, d), 1.26 (3Н, d), 1.30 (3Н, d), 2.57-2.75 (3Н, m), 2.81 (1Н, dd), 2.84-2.92 (1Н, m), 6.97 (1Н, t), 7.06 (1Н, t), 7.11-7.22 (1Н, мультиплет скрыт сигналами от толуола), 7.34 (1Н, d), 7.52 (1H,d), 10.80 (1Н, s).
Получение метил-(Е)-3-[4-[(1R,3R]-дейтеро-2-(2-фтор-2-метил-
пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-1-ил]-3,5-дифтор-фенил]проп-2-еноата

(R)-N-(1-(1Н-Индол-3-ил)пропан-2-ил)-2-фтор-2-метилпропан-1-амин (33%-ный (масс./масс.) в толуоле) (11,26 г; 14,97 ммоль) и (Е)-3-(4-дейтерокарбонил-3,5-дифтор-фенил)проп-2-еноат (3,40 г; 14,97 ммоль) нагревали в смеси толуола (55,6 мл) и уксусной кислоты (4,28 мл; 74,83 ммоль) при 80°C в течение 5 ч. После охлаждения летучие вещества удаляли под вакуумом. Остаток переносили в DCM (200 мл) и промывали насыщ. раствором NaНСО3 (200 мл). Водную фазу экстрагировали DCM (100 мл), затем объединенные органические экстракты промывали рассолом, сушили и концентрировали в вакууме. Неочищенное вещество загружали на SCX-2 колонку, элюируя метанолом для удаления непрореагировавшего альдегида. Затем колонку промывали смесью 7 М NH3-МеОН для элюирования продукта. Основный фильтрат упаривали и неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-40% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая метил-(Е)-3-[4-[(1R,3R)-1-дейтеро-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-1-ил]-3,5-дифтор-фенил]проп-2-еноат (4,70 г; 68,6%) в виде бледно-желтого твердого вещества.1Н ЯМР (400 МГц, CDCl3, 30°С) 1.11 (3Н, d), 1.19 (3Н, d), 1.25 (3Н, d), 2.42 (1Н, dd), 2.62 (1Н, dd), 2.87 (1Н, dd), 3.07 (1Н, dd), 3.65 (1Н, q), 3.81 (3Н, s), 6.39 (1Н, d), 6.99 (2Н, d), 7.06-7.17 (2Н, m), 7.23 (1Н, dd), 7.45 (1Н, s), 7.49-7.6 (2Н, m). m/z: ES+ [М+Н]+ 458.
Пример 11. Получение (1R,3R)-1-(4-[(Е)-2-карбоксиэтенил1-2,6-дифторфенил}-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-бета-карболин-2-ий-малеата
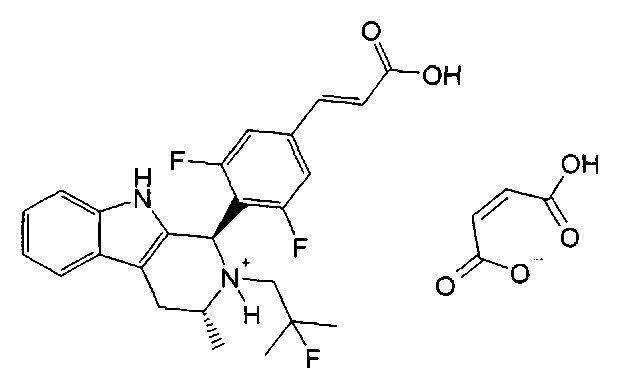
Раствор малеиновой кислоты (1,31 г; 11,29 ммоль) в ацетоне (15 мл) перемешивали в атмосфере азота. К этому раствору малеиновой кислоты добавляли раствор (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловой кислоты (пример 1) (5,00 г; 11,3 ммоль) в ацетоне (25 мл), получая желтый раствор. Реакционный сосуд заворачивали в фольгу для защиты от света и содержимое продували струей газообразного азота в течение ночи, пока растворитель не испарялся. Получали твердое вещество, которое сушили в вакууме в течение 2 часов, получая указанное в заголовке соединение в виде кремового твердого вещества (6,23 г; 98%).1Н ЯМР (500 МГц, DMSO, 27°С) 0.95-1.34 (9Н, m), 2.24-2.45 (1Н, m), 2.54-2.66 (1Н, m), 2.8-2.99 (2Н, m), 3.52 (1Н, s), 5.22 (1Н, s), 6.26 (2Н, s), 6.67 (1Н, d), 6.89-7.07 (2Н, m), 7.19 (1Н, а), 7.39-7.51 (3Н, m), 7.55 (1Н, d), 10.59 (1Н, s).
XRPD-кривая для этой малеатной соли показана на Фиг. 8, а DSC-кривая показана на Фиг. 9.
Пример 12
Типичные композиции на основе соединения из примера 1 готовили в масштабе 75 г, используя процесс влажного гранулирования. Активный ингредиент, маннит, микрокристаллическую целлюлозу и натрия крахмала гликолят взвешивали в количествах, указанных в приведенной ниже таблице, переносили в смеситель-гранулятор Р1-6 от Diosna и смешивали (с измельчением) со скоростью 600 об/мин в течение 6 минут. В случае композиции А смешивание продолжали при одновременном добавлении 30 мл воды в виде двух аликвот со скоростью приблизительно 1 мл в секунду, прекращая перемешивание в промежутке между добавлением аликвот, в то время как в случае композиции В добавляли, используя аналогичный способ, раствор, приготовленный путем перемешивания необходимых количеств EDTA и аскорбиновой кислоты с 20 мл воды при 50°С в течение 20 минут (с защитой от света), причем вторая аликвота в этом случае содержала приблизительно 10 мл промывочной жидкости. Смешивание с увлажнением продолжали в общей сложности в течение 1,5 минуты. Влажные гранулы пропускали через сито (1,5 мм), затем сушили под вакуумом при 50-60°С до содержания влаги меньше 2% (масс./масс.). Полученные гранулы измельчали, используя сито (1 мм), затем перемешивали со смазывающим веществом в течение 5 минут со скоростью 32 об/мин, используя смеситель Turbula. Выполняли формование таблеток, содержащих 10 мг активного ингредиента, путем прессования гранул до номинального компрессионного веса в 100 мг, используя мини-пресс от Riva, оснащенный 6 мм стандартным углублением для получения двояковыпуклой таблетки.

Стабильность композиций А и В с точки зрения образования продукта разложения (где «продукт разложения» означает (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилат) оценивали для таблеток, упакованных в герметично закрытые посредством индукционного запаивания флаконы из полиэтилена высокой плотности (HDPE) емкость 75 сс (кубических сантиметров), или подвергнутых воздействию атмосферного воздуха в чашках Петри, которые хранили в темноте с контролем температуры и влажности, как показано ниже в таблице. Очевидно, что композиция B, которая содержит и хелатирующий агент, и антиоксидант, более стабильна в отношении химической деградации, чем композиция A, представляющая собой стандартную композицию в форме таблетки.
Стабильность типичных композиций с точки зрения образования продукта разложения (%)

Пример 13. (Е)-3-[3,5-Дифтор-4-[(3R)-2-(2-фтор-2-метил-пропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил]фенил]проп-2-еноат

(Е)-3-(3,5-Дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту (200 мг; 0,45 ммоль) добавляли к раствору аммонийного нитрата церия (248 мг; 0,45 ммоль) в смеси ацетонитрила (6 мл) и воды (1,500 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 2 ч и добавляли дополнительное количество аммонийного нитрата церия (248 мг; 0,45 ммоль). Раствор перемешивали при 25°C в течение еще 15 минут. Реакционную смесь подкисляли, используя 2 М HCl (3 мл), и экстрагировали DCM (2×10 мл). Органические экстракты далее концентрировали в вакууме, и неочищенный продукт очищали препаративной HPLC (колонка SunFire от Waters, диоксид кремния, 5 мкм; диаметр 50 мм, длина 100 мм), используя в качестве элюентов смеси воды (содержащей 0,1% муравьиной кислоты) и MeCN с уменьшающейся полярностью. Фракции, содержащие желаемое соединение, упаривали досуха, получая (R,Е)-3-(3,5-дифтор-4-(2-(2-фтор-2-метилпропил)-3-метил-4,9-дигидро-3Н-пиридо[3,4-b]индол-2-ий-1-ил)фенил)акрилат (35,0 мг; 17,58%) в виде оранжевого стеклообразного вещества.1Н ЯМР (500 МГц, DMSO, 30°С) 1.27 (3Н, d), 1.43-1.55 (6Н, m), 3.51 (1Н, d), 3.72 (1Н, dd), 3.96 (1Н, dd), 4.18-4.32 (1Н, m), 4.67 (1Н, s), 6.59 (1Н, s), 7.09-7.3 (2Н, m), 7.42 (1Н, t), 7.52-7.73 (3Н, m), 7.78 (1Н, d), сигнала от NH не наблюдали. HRMS (ESI): [М+Н]+, обнаружено: 441,17831, C23H24F2N2O2 соответствует: 441,17844.
Получение (Е)-3-[3,5-дифтор-4-[2-(2-фтор-2-метил-пропил)-3-метил-9Н-пиридо[3,4-b]индол-2-ий-1-ил]фенил]проп-2-еноата

(Е)-3-(3,5-Дифтор-4-((1R,3R)-2-(2-фтор-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту (0,500 г; 1,13 ммоль) растворяли в DMSO (10 мл) и нагревали до 120°C на воздухе и на свету в течение 16 ч. Затем реакционную смесь нагревали до 180°C в течение 2,5 ч. Реакционную смесь охлаждали и очищали препаративной HPLC (препаративная колонка XBridge С18 OBD от Waters, диоксид кремния, 5 мкм; диаметр 50 мм, длина 100 мм), используя в качестве элюентов смеси воды (содержащей 1% NH3) и MeCN с уменьшающейся полярностью. Фракции, содержащие желаемое соединение, упаривали досуха, получая (Е)-3-[3,5-дифтор-4-[2-(2-фтор-2-метил-пропил)-3-метил-9Н-пиридо[3,4-b]индол-2-ий-1-ил]фенил]проп-2-еноат (0,047 г; 9,40%) в виде оранжевого твердого вещества.1Н ЯМР (400 МГц, DMSO, 27°С) 1.27 (3Н, s), 1.33 (3Н, d), 3.09 (3Н, s), 4.59-4.8 (1Н, m), 5.14-5.37 (1Н, m), 6.54 (1Н, d), 7.19 (1Н, d), 7.39 (1Н, t), 7.63 (2Н, d), 7.74 (1Н, t), 7.91 (1Н, d), 8.45 (1Н, d), 8.88 (1Н, s), 10.79 (1Н, s). m/z: ES+ [М+Н]+ 439.
Пример 14А. Получение (Е)-3-[3,5-дифтор-4-[(1R,3R)-2-(2-фтор-3-гидрокси-2-метил-пропил)-3-метил-1,3,4,9-тетрагидропиридо[3,4-b]индол-1-ил]фенил]проп-2-еновой кислоты (изомера 1)

2 М Гидроксид натрия (1,27 мл; 2,54 ммоль) добавляли к раствору (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата - изомера 1 (120 мг; 0,25 ммоль) в смеси THF (0,635 мл) и метанола (0,635 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем разбавляли EtOAc и водой. Значение pH водного слоя подводили до 6, добавляя 2 М HCl, и проводили разделение слоев. Водный слой экстрагировали EtOAc, затем объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-10% МеОН в DCM. Фракции чистого продукта упаривали досуха, получая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту - изомер 1 (85 мг; 72,9%) в виде бежевого твердого вещества.1Н ЯМР (400 МГц, DMSO, 27°С) 0.97 (3Н, d), 1.05 (3Н, d), 2.32-2.4 (1Н, m), 2.44-2.54 (1Н, m), 2.73-2.93 (3Н, m), 3-3.14 (2Н, m), 3.32-3.49 (2Н, m), 4.73 (1Н, s), 5.14 (1Н, s), 6.58 (1Н, d), 6.82-6.96 (2Н, m), 7.10 (1Н, d), 7.33 (1Н, d), 7.36 (1Н, d), 7.45 (1Н, d), 10.49 (1Н, s). m/z (ES+), [М+Н]+=459.
Пример 14В. Получение (Е)-3-[3,5-дифтор-4-[(1R,3R)-2-(2-фтор-3-гидрокси-2-метил-пропил)-3-метил-1,3,4,9-тетрагидропиридо[3,4-b]индол-1-ил]фенил]проп-2-еновой кислоты (изомера 2)

2 М Гидроксид натрия (1,27 мл; 2,54 ммоль) добавляли к раствору (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилата - изомера 2 (110 мг; 0,23 ммоль) в смеси THF (0,529 мл) и метанола (0,529 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем разбавляли EtOAc и водой. Значение pH водного слоя подводили до 6, добавляя 2 М HCl, и проводили разделение слоев. Водный слой экстрагировали EtOAc, затем объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-10% МеОН в DCM. Фракции чистого продукта упаривали досуха, получая (Е)-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акриловую кислоту - изомер 2 (81 мг; 76%) в виде бежевого твердого вещества.1Н ЯМР (400 МГц, DMSO, 27°С) 1.02 (2Н, s), 1.05 (3Н, d), 1.23 (1Н, s), 1.90 (3Н, s), 2.28-2.46 (1Н, m), 2.53-2.7 (1Н, m), 2.86-3.03 (2Н, m), 3.56 (1Н, d), 4.83 (1Н, s), 5.17 (1Н, s), 6.66 (1Н, d), 6.86-7.08 (2Н, m), 7.17 (1Н, d), 7.40 (1Н, d), 7.44 (2Н, d), 7.53 (1Н, d), 10.54 (1Н, s). m/z (ES+), [М+Н]+ = 459.
(Е)-Метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат, изомеры 1 и 2, использованные в качестве исходных веществ, получали так, как указано ниже.
2-Фтор-2-метилпропан-1,3-диол

LiAlH4 (0,741 г; 19,25 ммоль) добавляли порциями к охлажденному раствору диэтил-2-фтор-2-метилмалоната (1,345 г; 7,00 ммоль) в THF (35,0 мл). Реакционную смесь оставляли нагреваться до комнатной температуры в течение 1 ч. После охлаждения до 0°C реакцию гасили, добавляя воду (0,75 мл), 15%-ный NaOH (0,75 мл), затем воду (1,5 мл). Суспензию перемешивали в течение 30 мин, затем фильтровали и твердые вещества промывали THF. Фильтрат упаривали, получая 2-фтор-2-метилпропан-1,3-диол (0,745 г; 98%) в виде бесцветного масла.1Н ЯМР (400 МГц, CDCl3, 27°С) 1.34 (3Н,d), 2.12-2.27 (2Н, m), 3.75 (4Н, d).
3-(((R)-1-(1Н-Индол-3-ил)пропан-2-ил)амино)-2-фтор-2-метилпропан-1-ол

Трифторметансульфоновый ангидрид (1,151 мл; 6,80 ммоль) добавляли к раствору 2-фтор-2-метилпропан-1,3-диола (0,70 г; 6,47 ммоль) в DCM (17,85 мл) при 0°C, затем добавляли 2,6-лутидин (0,908 мл; 7,77 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры в течение 30 мин, затем промывали 2 М раствором HCl. Органическую фазу пропускали через картридж для разделения фаз и концентрировали в вакууме. Остаток растворяли в диоксане (12 мл), затем добавляли (R)-1-(1Н-индол-3-ил)пропан-2-амин (1,128 г; 6,47 ммоль) и диизопропилэтиламин (DIPEA) (1,678 мл; 9,71 ммоль) и реакционную смесь нагревали до 90°C в течение 2 ч. После охлаждения реакционную смесь разбавляли DCM и промывали водой. Водный слой экстрагировали DCM, затем органические экстракты концентрировали в вакууме. Неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-10% МеОН в DCM. Фракции чистого продукта упаривали досуха, получая 3-(((R)-1-(1Н-индол-3-ил)пропан-2-ил)амино)-2-фтор-2-метилпропан-1-ол (0,815 г; 47,6%) в виде коричневого смолообразного вещества, m/z (ES+), [М+Н]+ = 265.
(Е)-Метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомеры 1 и 2)
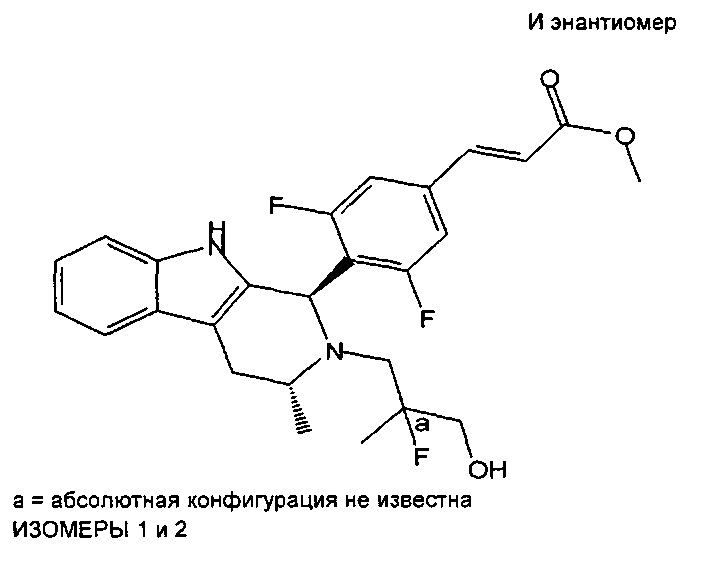
(Е)-Метил-3-(3,5-дифтор-4-формилфенил)акрилат (565 мг; 2,50 ммоль) добавляли к суспензии 3-(((R)-1-(1Н-индол-3-ил)пропан-2-ил)амино)-2-фтор-2-метилпропан-1-ола (661 мг; 2,50 ммоль) в смеси толуола (11,3 мл) и уксусной кислоты (1,25 мл). Реакционную смесь нагревали до 90°C в течение 5 ч. После охлаждения летучие вещества удаляли под вакуумом, затем остаток пропускали через SCX-2 колонку, элюируя метанолом для удаления непрореагировавшего альдегида. Затем колонку промывали смесью Nb3/MeOH для элюирования продуктов. Основный фильтрат упаривали, затем неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом 0-50% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 1-122 мг; 10,3%) в виде желтого твердого вещества и (Е)-метил-3-(3,5-дифтор-4-((1R,3R)-2-(2-фтор-3-гидрокси-2-метилпропил)-3-метил-2,3,4,9-тетрагидро-1Н-пиридо[3,4-b]индол-1-ил)фенил)акрилат (изомер 2-129 мг; 11%) в виде желто-оранжевого твердого вещества. Изомер 1 -1Н ЯМР (400 МГц, CDCl3, 27°С) 1.10 (3Н, d), 1.14 (3Н, d), 2.54 (1Н, dd), 2.62-2.73 (1Н, m), 3.06-3.3 (2Н, m), 3.40 (1Н, dd), 3.56 (1Н, t), 3.80 (3Н, s), 3.87-4.08 (1Н, m), 4.27 (1Н, s), 5.16 (1Н, s), 6.37 (1Н, d), 7.01 (2Н, d), 7.07-7.15 (2Н, m), 7.14-7.24 (1Н, m), 7.42-7.56 (2Н, m), 7.71 (1Н, s). m/z (ES+), [М+Н]+=473. Изомер 2 -1Н ЯМР (400 МГц, CDCl3, 27°С) 1.15 (3Н, d), 1.20 (3Н, d), 2.65 (1Н, dd), 2.79 (1Н, t), 2.93-3.09 (2Н, m), 3.57 (1Н, dt), 3.70 (1Н, dd), 3.78 (3Н, s), 4.2-4.67 (1Н, m), 5.42 (1Н, s), 6.32 (1Н, d), 6.94 (2Н, d), 7.06-7.15 (2Н, m), 7.19-7.27 (2Н, m), 7.42 (1Н, s), 7.51 (1Н, dd), 8.02 (1Н, s). m/z (ES+), [М+Н]+=473.
Ссылочный пример 1. 1-(4-(5-(5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он

4-Метилбензолсульфонат пиридина (11,62 г; 46,24 ммоль) добавляли к суспензии 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-она (128 г; 231,19 ммоль) в метаноле (1 л) в атмосфере азота. Смесь перемешивали при 60°C в течение 1,5 часа. Через 5 минут смесь превращалась в раствор. Смесь выдерживали при 50°C в течение ночи, за это время образовывался осадок. Твердое вещество выделяли фильтрованием и промывали водой и ацетонитрилом. Это вещество все еще содержало небольшое количество примесей с предыдущей стадии и требовало дополнительной очистки. Вещество растворяли в дихлорметане и очищали флэш-хроматографией на силикагеле (от 0% метанола/DCM до 10% метанола/DCM). Таким образом выделяли желаемый продукт, 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (ссылочный пример 1) (92 г; 85%), в виде кремового твердого вещества (Форма A):1Н ЯМР-спектр (DMSO-d6) 1.4-1.51 (12Н, m), 1.51-1.78 (2Н, m), 1.89-2.05 (2Н, m), 2.72-2.86 (1Н, m), 2.91-3.05 (1Н, m), 3.12-3.24 (1Н, m), 3.64 (2Н, q), 3.83-4.01 (1Н, m), 4.29-4.41 (1Н, m), 4.47 (1Н, t), 4.58 (2Н, q), 8.26 (2Н, s), 8.85 (1Н, s); масс-спектр: [М+Н]+=470.
1-(4-(5-(5-Амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-он получали так, как указано ниже.
1,8-Диазабицикло[5.4.0]ундец-7-ен (76 мл; 511,14 ммоль) добавляли к суспензии трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (150 г; 319,46 ммоль) в 2-метил-ТНF (1,2 л). Добавляли иодэтан (46 мл; 575,03 ммоль) и смесь перемешивали в течение 16 часов при 35°С. Еще раз добавляли иодэтан (46 мл; 575,03 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ен (76 мл; 511,14 ммоль) и перемешивание продолжали в течение 24 часов при 35°С. Смесь выливали в воду, и нерастворенное вещество выделяли фильтрованием, промывали водой и МТВЕ и сушили в вакууме, получая трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (116 г; 73,0%) в виде желтого твердого вещества. Фильтрат экстрагировали DCM и органический раствор сушили, используя сульфат магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией на диоксиде кремния, используя градиентное элюирование (от 30% МТВЕ/гептан до 100% МТВЕ). Таким образом выделяли вторую порцию желаемого продукта (12 г; 24,12 ммоль; 7,55%) в виде желтого твердого вещества, которое позже объединяли с первой порцией:1Н ЯМР-спектр (DMSO-d6) 1.41 (9Н, s), 1.44 (9Н, s), 1.48 (3Н, t), 1.52-1.69 (2Н, m), 1.87-2.04 (2Н, m), 2.79-3.03 (3Н, m), 3.86-4.03 (2Н, m), 4.59 (2Н, q), 7.89 (2Н, s), 8.85 (1Н, s); масс-спектр: [М+Н]+=498.
Суспензию трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилата (3009,5 г; 6,05 моль) в DCM (9 л) охлаждали до 5-10°C в атмосфере N2. К суспензии порциями добавляли TFA (9 л), поддерживая температуру меньше 30°C. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали, полученный остаток растворяли в воде (30 л) и медленно добавляли к 35%-ному водному раствору аммиака (12 л) при 0-5°C. Суспензию перемешивали в течение 30 мин, затем продукт отфильтровывали и промывали водой (2×6 л). Продукт сушили при 50°С в вакууме в течение 2 суток, получая 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин (2496 г):1Н ЯМР-спектр: (DMSO-d6) 1.4-1.52 (12Н, m), 1.57-1.73 (2Н, m), 1.83-1.93 (2Н, m), 2.57-2.7 (2Н, m), 2.71-2.84 (1Н, m), 2.96-3.09 (2Н, m), 4.58 (2Н, q), 8.06 (2Н, s), 8.84 (1H, s); масс-спектр [M+H]+=398.
К раствору 3-(тетрагидро-2Н-пиран-2-илокси)пропановой кислоты (48,80 г; 0,2774 моль) и N-этил-N-изопропилпропан-2-амина (86,96 мл; 0,4993 моль) в THF (552 мл) порциями при кт в атмосфере азота добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU; 115,73 г; 0,3051 моль). Полученную смесь перемешивали в течение 20 мин, затем порциями в течение 1 ч добавляли 3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)-5-(1-этил-3-(пиперидин-4-ил)-1Н-1,2,4-триазол-5-ил)пиразин-2-амин (122,5 г (110,25 г активного вещества); 0,2774 моль). Через 3,5 ч смесь концентрировали и остаток суспендировали в MeCN (275 мл) в течение 15 мин при комнатной температуре. Продукт отфильтровывали, промывали MeCN (3×110 мл) и сушили в течение ночи при 50°C в вакууме. В результате получали 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-он (131,9 г; 96%).1Н ЯМР-спектр: (DMSO-d6) 1.29-1.48 (16Н, m), 1.48-1.75 (4Н, m), 1.83-1.99 (2Н, m), 2.48-2.68 (2Н, m), 2.68-2.79 (1Н, m), 2.87-2.99 (1Н, m), 3.07-3.19 (1Н, m), 3.32-3.42 (1Н, m), 3.47-3.6 (1Н, m), 3.64-3.75 (1Н, m), 3.75-3.84 (1Н, m), 3.84-3.95 (1Н, m), 4.24-4.39 (1Н, m), 4.47-4.6 (3Н, m), 7.84 (2Н, s), 8.79 (1Н, s); масс-спектр [M+Na]+=577.
Альтернативное получение соединения из ссылочного примера 1
К суспензии 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-(тетрагидро-2Н-пиран-2-илокси)пропан-1-она (131,9 г; 0,2382 моль) в метаноле (1045 мл) добавляли п-толуолсульфонат пиридиния (11,97 г; 47,7 ммоль) в атмосфере N2. Реакционную смесь перемешивали при 60°C в течение 5,5 ч, затем при 50°C в течение ночи. Реакционную смесь охлаждали до 0°C и твердое вещество отфильтровывали. Продукт суспендировали в воде (250 мл) в течение 20 мин при комнатной температуре, отфильтровывали, промывали водой (3×40 мл) и сушили при 50°C в вакууме. В результате получали 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (21,4 г) в виде Формы A (см. ниже).
Концентрировали метанольные растворы и полученное твердое вещество суспендировали в воде (0,6 л) в течение 20 мин при комнатной температуре. Твердое вещество выделяли фильтрованием и промывали водой (3×100 мл). Осадок на фильтре суспендировали второй раз в воде (0,5 л) в течение еще 20 минут. Продукт выделяли фильтрованием, промывали водой (100 мл) и сушили при 50°C в вакууме. В результате получали 81,9 г 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она (81,9 г) в виде Формы A.
Обе порции объединяли (103,3 г), вносили затравку Формы B (16,68 г) и суспендировали в MeCN (826 мл) при комнатной температуре в течение ночи. В результате этого получали 117,4 г 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она в виде бледно-желтого твердого вещества (117,4 г), Форму B (см. ниже). Это вещество далее очищали, суспендируя в в гептане (7,5 относительных объемов) в течение 1 часа. Смесь фильтровали, подсушивали на фильтре и сушили при 50°С в вакуумном сушильном шкафу в течение ночи, получая 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он (102,5 г) в виде Формы В.
Форма B также может быть получена путем суспендирования Формы A в MeCN без внесения затравки.
Форма A или B также может быть превращена в Форму C, как указано ниже.
Суспензию 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-она (например, Формы B, полученной способами, приведенными выше) в IPA (12 об.) нагревали при температуре дефлегмации до растворения твердого вещества. Раствор фильтровали в теплом состоянии, затем охлаждали до комнатной температуры. В результате получали 1-(4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1-этил-1Н-1,2,4-триазол-3-ил)пиперидин-1-ил)-3-гидроксипропан-1-он в виде бледно-желтого твердого вещества (99,3 г; 97%) в виде Формы С.
Форма C также может быть превращена в Форму B, как указано ниже.
В колбе с патрубком емкостью 10 л нагревали до 110-115°C Форму C (1 порцию 377,8 г) в MIBK (метилизобутилкетон) (7900 мл), получая раствор. Раствор оставляли охлаждаться до 97-103°C и незамедлительно выполняли осветляющую фильтрацию в сосуд емкостью 50 л, содержащий затравку Формы B (0,8 г) в ацетонитриле (8220 мл), с перемешиванием при -15°C. Во время добавления температуру в сосуде емкостью 50 л поддерживали в диапазоне от -15 до 25°C, используя для охлаждения рубашку. Три следующих порции соединения, растворенного в MIBK, добавляли тем же самым способом. К полученной суспензии добавляли затравку Формы B (0,8 г) и затем смесь перемешивали при 10-20°C в течение ночи. Анализом в ходе процесса подтверждали наличие желаемой формы (Формы В) без заметных количеств Формы C или аморфной формы. Смесь фильтровали и промывали ацетонитрилом (3340 мл). Твердое вещество сушили в сушильном шкафу в течение 2 суток (твердое вещество измельчали во время сушки до порошка и смеси небольших комков размеров приблизительно от 1 мм до 3-4 мм) до получения постоянной массы. Выход 1532,8 г (93,5%).
3-(Тетрагидро-2Н-пиран-2-илокси)пропановую кислоту получали так, как указано ниже.
К перемешиваемому раствору метанола (2,4 л) и концентрированной серной кислоты (44,4 мл; 832,61 ммоль) при 0°C в атмосфере азота по каплям добавляли бета-пропиолактон (175 мл; 2,78 моль). Этот раствор оставляли перемешиваться при комнатной температуре в течение 2 суток. Реакционную смесь охлаждали до 10°C, затем порциями добавляли бикарбонат натрия (145 г; 1,72 моль), полученную суспензию оставляли перемешиваться при комнатной температуре в течение 75 минут. Этот раствор фильтровали, осадок на фильтре промывали метанолом (800 мл). Фильтрат упаривали до состояния масла, которое перерастворяли в дихлорметане (1,2 л) и перемешивали в течение 60 минут, затем повторно фильтровали. Этот раствор фильтровали, после чего упаривали, получая метил-3-гидроксипропаноат (219 г; 76%) в виде масла:1Н ЯМР-спектр (CDCl3) 2.50 (2Н, t), 3.63 (3Н, s), 3.78 (2Н, t).
пара-Толуолсульфонат пиридиния (7,65 г; 30,45 ммоль) добавляли к прозрачному раствору метил-3-гидроксипропаноата (63,4 г; 609,00 ммоль) и 3,4-дигидро-2Н-пирана (78 мл; 852,60 ммоль) в дихлорметане (650 мл) при комнатной температуре в атмосфере азота, получая мутный раствор. Его оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь промывали водой (250 мл) и рассолом (250 мл), после чего сушили (MgSO4) и упаривали до состояния масла. Этот неочищенный продукт очищали флэш-хроматографией на диоксиде кремния, элюируя градиентом от 15 до 30% EtOAc в гептане. Фракции чистого продукта упаривали досуха, получая метил-3-(тетрагидро-2Н-пиран-2-илокси)пропаноат (67,7 г; 59,0%) в виде бесцветного масла:1Н ЯМР-спектр (CDCl3) 1.47 (4Н, dddd), 1.55-1.84 (2Н, m), 2.55 (2Н, t), 3.33-3.53 (1Н, m), 3.53-3.7 (4Н, m), 3.78 (1Н, ddd), 3.93 (1Н, dt), 4.42-4.72 (1Н, m); масс-спектр: [МН]+=189.
Гидроксид натрия (2 М; 349 мл; 697,58 ммоль) добавляли к раствору метил-3-(тетрагидро-2Н-пиран-2-илокси)пропаноата (67,68 г; 359,58 ммоль) в THF (680 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. THF удаляли в вакууме, затем водный слой промывали этилацетатом (260 мл), после чего охлаждали до 0°C и осторожно подкисляли до pH 5, добавляя соляную кислоту (2 М). Продукт экстрагировали этилацетатом (3×250 мл), после чего сушили (MgSO4) и упаривали, получая 3-(тетрагидро-2Н-пиран-2-илокси)пропановую кислоту (57,0 г; 91%) в виде прозрачного масла. Это вещество растворяли в этилацетате (750 мл), затем промывали водой (3×250 мл) и рассолом (250 мл) для удаления оставшейся уксусной кислоты. Органический раствор сушили (MgSO4) и упаривали, получая 3-(тетрагидро-2Н-пиран-2-илокси)пропановую кислоту (45,67 г; 72,9%) в виде бесцветного масла:1Н ЯМР-спектр (CDCl3) 1.43-1.67 (4Н, m), 1.65-1.95 (2Н, m), 2.68 (2Н, t), 3.48-3.58 (1Н, m), 3.73 (1Н, dt), 3.88 (1Н, ddd), 4.02 (1Н, dt), 4.59-4.7 (1Н, m); масс-спектр: [М-Н]-=173.
трет-Бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат получали так, как указано ниже.
Гидрат гидразина (23,59 мл; 480,75 ммоль) по каплям добавляли к перемешиваемой смеси метил-3-амино-6-бромпиразин-2-карбоксилата (100 г; 418,04 ммоль) в EtOH (2 л). Смесь нагревали при 50°C в атмосфере азота. Полученную густую суспензию перемешивали при 50°C в течение 16 часов. Добавляли дополнительно гидразин (2,5 мл) в виде одной порции, и суспензию перемешивали при 50°C в течение еще 24 часов. В густую реакционную смесь загружали этанол (500 мл), и смесь оставляли охлаждаться до комнатной температуры. Полученную суспензию фильтровали, твердое вещество промывали этанолом (1 л) и сушили в вакууме, получая 3-амино-6-бромпиразин-2-карбогидразид (98 г; количественный выход) в виде кремового твердого вещества:1Н ЯМР-спектр: (DMSO-d6) 4.52 (2Н, s), 7.59 (2Н, s), 8.30 (1Н, s), 9.74 (1Н, s); масс-спектр [М+Н]+=232.
Ангидрид пивалевой кислоты (165 мл; 815,38 ммоль) добавляли к перемешиваемой смеси 3-амино-6-бромпиразин-2-карбогидразида (172 г; 741,26 ммоль) в ацетонитриле (1,8 л) и смесь нагревали при 80°C в течение 1 часа. Реакционную смесь оставляли перемешиваться в течение 16 часов. Необходимое желтое твердое вещество выделяли фильтрованием. Фильтрат распределяли между EtOAc (2 л) и водным раствором бикарбоната натрия (2 л). Органический слой промывали насыщенным рассолом и сушили над MgSO4. Раствор фильтровали и концентрировали, получая оранжевое вязкое твердое вещество, которое растирали с МТВЕ (250 мл). Нерастворенное желтое твердое вещество выделяли фильтрованием, и было показано, что это вещество идентично первому твердому веществу. Объединенные твердые вещества сушили в вакуумном сушильном шкафу при 50°C в течение 3 суток, получая 3-амино-6-бром-N'-пивалоилпиразин-2-карбогидразид (224 г; 96%) в виде желтого твердого вещества:1Н ЯМР-спектр: (DMSO-d6) 1.17 (9Н, s), 7.62 (2Н, s), 8.37 (1Н, s), 9.42-9.56 (1Н, m), 10.09-10.23 (1Н, m); масс-спектр [М+Н]+=318.
К 3-амино-6-бром-N'-пивалоилпиразин-2-карбогидразиду (2301 г; 7,28 моль) в MeCN (10,8 л) добавляли DIPEA (3,044 л; 17,48 моль) и п-толуолсульфонилхлорид (1665 г; 8,73 моль) порциями (приблизительно по 280 г×6) при 50°C в течение 30 мин. Температуру реакционной смеси поддерживали в диапазоне 65-70°C, контролируя скорость добавления. По завершении добавления реакционную смесь перемешивали при 70°C в течение 1 ч. Смесь охлаждали до комнатной температуры и гасили 5%-ным NaHCO3 (водным; 24,2 л). Полученную суспензию перемешивали в течение 30 мин, затем фильтровали. Продукт промывали водой (14,8 л), подсушивали и сушили при 50°C в течение 16 ч. Продукт растворяли в DCM (12 л) и проводили разделение фаз. Органическую фазу загружали на набивку диоксида кремния (6 кг) и продукт элюировали 20% EtOAc/DCM (8×10 л). После концентрирования фракций, содержащих продукт, получали 1987 г 5-бром-3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амина (36 г; 17%; выход 92%) с чистотой 99,8% по данным HPLC:1Н ЯМР-спектр: (DMSO-d6) 1.43 (9Н, s), 7.70 (2Н, s), 8.39 (1Н, s); масс-спектр [М+Н]+=298.
Через раствор 5-бром-3-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-амина (89,35 г; 239,75 ммоль) в DMA (1,2 л) в течение 20 минут пропускали струю газообразного азота. К перемешиваемой смеси последовательно добавляли дициклогексил(2',4',6'-триизопропилбифенил-2-ил)фосфин (11,43 г; 23,98 ммоль), трис(дибензилиденацетон)дипалладий(0) (5,49 г; 5,99 ммоль), цинк (1,568 г; 23,98 ммоль) и дицианоцинк (16,89 г; 143,85 ммоль). Смесь нагревали до 100°C и перемешивали в течение 1 часа. Смесь охлаждали и распределяли между DCM (3 л) и водой (1 л). Черную смесь фильтровали через целит, и органический слой отделяли. Раствор промывали водой, затем рассолом. Раствор сушили, используя сульфат магния, и концентрировали при пониженном давлении. Остаток растирали с МТВЕ и выделяли фильтрованием, промывая МТВЕ. Осадок на фильтре сушили в вакууме, получая 5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбонитрил (55,7 г; 95%) в виде бледно-оранжевого твердого вещества:1Н ЯМР-спектр (DMSO-d6) 1.46 (9Н, s), 6.02 (1Н, s), 8.38 (2Н, s); масс-спектр: [М-Н]-=242.
Гидрат гидразина (82 мл; 1,69 моль) добавляли к 5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбонитрилу (55 г; 225,18 ммоль) в IPA (200 мл) и смесь нагревали при 50°C в атмосфере азота в течение 16 часов. Смесь охлаждали в ледяной бане. Полученный осадок собирали фильтрованием, промывали IPA и диэтиловым эфиром и сушили до постоянной массы, получая (Z)-5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбогидразонамид (49,2 г; 79%) в виде желтого твердого вещества:1Н ЯМР-спектр (DMSO-d6) 1.45 (9Н, s), 5.26 (2Н, s), 5.58 (2Н, s), 7.56 (2Н, s), 8.75 (1Н, s); масс-спектр: [М+Н]+=277.
Гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (74,3 г; 195,44 ммоль) добавляли к раствору N-Boc-изонипекотиновой кислоты (41,1 г; 179,15 ммоль) и 4-метилморфолина (35,9 мл; 325,74 ммоль) в DMA (800 мл). Смесь перемешивали в течение 10 минут, затем к раствору добавляли (Z)-5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбогидразонамид (45 г; 162,87 ммоль) в виде одной порции (наблюдали экзотермический эффект в диапазоне от 22°C до 27°С). Через несколько минут продукт кристаллизовался из реакционной смеси. Реакционную смесь извлекали из сосуда и фильтровали через фильтр из пористого стекла. Чтобы смыть продукт со стенок сосуда, добавляли дополнительное количество DMA (150 мл) и все это выливали на осадок на фильтре. В сосуд добавляли изопропанол (600 мл), и оставшуюся в сосуде часть продукта суспендировали в этом растворителе, используя энергичное перемешивание. После удаления DMA отсасыванием эту суспензию в изопропаноле использовали для промывки осадка на фильтре. Осадок на фильтре сушили отсасыванием, затем промывали МТВЕ и еще раз сушили отсасыванием. Осадок на фильтре сушили в вакууме, получая (Z)-трет-бутил 4-(2-(амино(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)метилен)гидразинкарбонил)пиперидин-1-карбоксилат (76 г; 95%) в виде желтого твердого вещества:1Н ЯМР-спектр (DMSO-d6) 1.40 (9Н, s), 1.46 (9Н, s), 1.63-1.9 (2Н, m), 2.33-2.6 (2Н, m, скрыт сигналом от DMSO), 2.63-3.03 (2Н, m), 3.18-3.48 (4Н, m, скрыт сигналом от воды), 3.88-4.11 (2Н, m), 6.43 (2Н, s), 7.76 (2Н, br), 8.84 (0,5Н, s), 8.87 (0,5Н, s), 9.85 (1Н, s): масс-спектр: [М+Н]+=488.
При альтернативном получении N-Boc-изонипекотиновая кислота может быть получена in situ так, как указано ниже. Изонипекотиновую кислоту (858 г; 3,74 моль) растворяли в DMA (25,3 л) и добавляли 4-метилморфолин (393 мл; 3,74 моль). Перемешивали в течение 5 мин и добавляли изобутилхлорформиат (489 мл; 3,74 моль). Реакционную смесь перемешивали при 25°С в течение 2 ч и охлаждали до 15°С, после чего порциями в течение 10 мин добавляли (Z)-5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-карбогидразонамид (940 г; 3,4 моль). Реакционную смесь перемешивали в течение 1-2 ч при 15°С. Добавляли порциями в течение 1 ч воду (20,5 л) и перемешивали в течение еще 1 ч, после чего фильтровали. Затем осадок на фильтре промывали водой (4×4 л) и подсушивали на фильтре, после чего сушили досуха в вакуумном сушильном шкафу при 50°C, получая желаемый продукт.
В закрепленный сосуд с двойной рубашкой емкостью 3 л, содержащий диоксан (500 мл), добавляли уксусную кислоту (200 мл) и раствор нагревали до 70°C в атмосфере азота. К этой нагретой смеси порциями добавляли (Z)-трет-бутил-4-(2-(амино(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)метилен)-гидразинкарбонил)-пиперидин-1-карбоксилат (74,5 г; 152,80 ммоль). Через 10 минут температуру повышали до 100°C (слабая дефлегмация). Реакционную смесь перемешивали при 100°C в течение 1,5 часа (суспензию), затем выдерживали при 80°C в течение ночи (после выдерживания в течение ночи образовывался раствор). Полученный раствор концентрировали при пониженном давлении, затем разбавляли толуолом, упаривали досуха, переносили в толуол и концентрировали еще раз. Оставшееся масло смешивали с некоторым количеством этилацетата и концентрировали досуха. Из раствора кристаллизовалось твердое вещество, которое растирали с МТВЕ (200 мл) и выделяли фильтрованием. Осадок на фильтре промывали водой и МТВЕ, получая трет-бутил-4-(5-(5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил)-1Н-1,2,4-триазол-3-ил)пиперидин-1-карбоксилат (50 г; 70%) в виде серого твердого вещества. Фильтрат концентрировали при пониженном давлении, получая желтое твердое вещество. Это вещество растирали с МТВЕ и проводили фильтрование. Осадок на фильтре промывали этилацетатом и затем МТВЕ, получая вторую порцию в виде бледно-желтого твердого вещества (4,93 г; 7%). Это вещество было идентично веществу из первой порции:1Н ЯМР-спектр (DMSO-d6) 1.17 (9Н, s), 1.22 (9Н, s), 1.29-1.47 (2Н, m), 1.67-1.78 (2Н, m), 2.57-2.87 (3Н, m), 3.57-3.92 (2Н, m), 7.56 (2Н, br), 8.56 (1Н, s), 13.47 (2Н, br s); масс-спектр: [М+Н]+=470.
Реферат
Изобретение относится к области органической химии, а именно к соединению формулы (I) или к его изомеру или к его фармацевтически приемлемой соли, где Rи Rкаждый независимо представляет собой Н или F; Rпредставляет собой Н или метил; и либо а) Rпредставляет собой Н, и Rпредставляет собой F; либо б) Rпредставляет собой F, и Rпредставляет собой Н. Также изобретение относится к способу предупреждения или лечения рака, фармацевтической композиции и комбинации на основе соединения формулы (I). Технический результат: получено новое гетероциклическое соединение, полезное при лечении рака. 5 н. и 9 з.п. формулы, 12 ил., 2 табл., 15 пр.
Формула


Документы, цитированные в отчёте о поиске
Замещенные бета-карболины
Комментарии