Производные пиперидина, фармацевтическая композиция на их основе и их применение - RU2408591C2
Код документа: RU2408591C2
Описание
Описание
Настоящее изобретение относится к новым антагонистам нейрокининового рецептора 1 (NK1 или NK-1), более конкретно, к производным пипередина, фармацевтическим композициям на их основе и их применению.
Нейрокининовые рецепторы, такие как NK1, NK2 и NK3, участвуют в различных биологических процессах. Их можно обнаружить в нервной системе и системе кровообращения млекопитающих, а также в периферических тканях. По этой причине модуляция этих типов рецепторов исследована с точки зрения возможности лечения или предупреждения различных патологических состояний млекопитающих. В частности, сообщают, что рецепторы NK1 участвуют в капиллярных кровотечениях и выработке слизи. Характерные типы антагонистов нейрокининовых рецепторов и нарушений, которые можно лечит с их помощью, включают, например, сонливость, боль, мигрень, рвоту, ноцицепцию и воспаление; см., например, US 6329401, US 5760018, US 5620989, WO 95/19344, WO 94/13639, WO 94/10165, Wu et al, Tetrahedron, 56, 6279-6290 (2000), Rombouts et al., Tetrahedron, 59, 4721-4731 (2003) и Rogiers et al., Tetrahedron, 57, 8971-8981 (2001).
Наиболее близким изобретению по технической сущности и достигаемому эффекту решением являются описанные в заявке WO 03/051840 производные пиперидина, представляют собой антагонисты нейрокининового рецептора NK1.
Задачей изобретения является расширение арсенала производных пиперидина, являющихся высокоактивными антагонистами нейрокининового рецептора NK1.
Поставленная задача решается предлагаемыми производными пипередина формулы

в которой:
R1 и R2 выбраны из группы, включающей алкил, галогеналкил, алкил, замещенный одной или большим количеством гидроксигрупп, -CN, алкинил, -N(R6)2, -N(R6)-S(O2)-алкил, -N(R6)-C(O)-N(R9)2, -алкилен-CN, -циклоалкилен-CN, -алкилен-O-алкил, -С(O)-алкил, -C(=N-OR5)-aлкил, -С(O)-O-алкил, -алкилен-С(O)-алкил, -алкилен-С(O)-O-алкил, -aлкилeн-C(O)-N(R9)2 и группы




при условии, что по меньшей мере один из R1 и R2 означает -CN или группу




W означает =C(R8)- или =N-;
Х означает -С(O)- или -S(O2)-;
Y выбран из группы, включающей -СН2-, -О- и -N(R6)-C(O)-, при условии, что:
(a) атом азота группы -N(R6)-C(O)- связан с X, и
(b) если R1 и/или R2 означает

Z означает -C(R7)2-, -N(R6)-, или -O-;
R3 выбран из группы, включающей Н и незамещенный алкил;
R4 означает Н;
R5 означает Н или алкил;
R6 выбран из группы, включающей Н, алкил, циклоалкил и арил;
каждый R7 независимо означает Н или алкил; или
каждый R7 совместно с кольцевым атомом углерода, к которым, как показано, они присоединены, образует циклоалкиленовое кольцо;
R8 выбран из группы, включающей Н, алкил, алкил, замещенный одной или большим количеством гидроксигрупп, -N(R6)2, -N(R6)-S(O2)-алкил, -N(R6)-S(O2)-арил, -N(R6)-C(O)-алкил, -N(R6)-С(O)-арил, алкилен-O-алкил и -CN;
R9 выбран из группы, включающей Н, алкил и арил, или каждый R9 совместно с атомом азота, к которому, как показано, они присоединены, образует гетероциклоалкильное кольцо;
Аr1 означает фенил;
Аr2 означает фенил, замещенный 0-3 заместителями, выбранными из группы, включающей галогеналкил;
n равно 0, 1 или 2; и
m равно 1, 2 или 3,
и их фармацевтически приемлемыми солями и гидратами.
Предпочтительной формой выполнения являются производные пиперидина, имеющие структуру IA:

в которой R1, R2, R3, R4, Аr1, Аr2 и n имеют вышеуказанные значения, и их фармацевтически приемлемые соли и гидраты.
Предпочтительные значения радикалов R1-R3, R8, Аr2, X, Y, Z, m и n, приведенные в формуле (I) и формуле (IA), сведены в зависимых пунктах 2-23 формулы изобретения.
Другими предпочтительными производными пиперидина являются соединения формулы (IB)
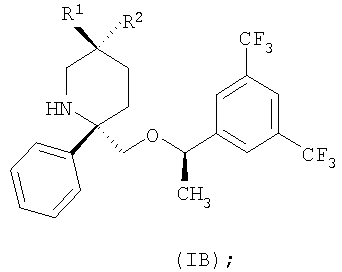
в которой R1 и R2 выбраны из группы, состоящей из:


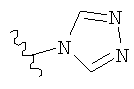




и их фармацевтически приемлемые соли и гидраты.
В частности предпочитаются производные пиперидина формул:
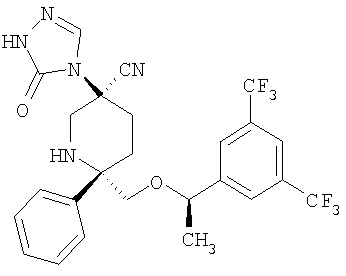

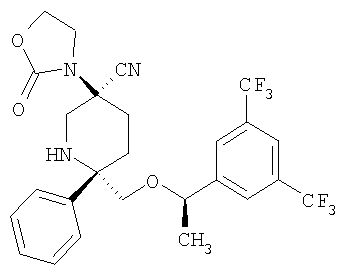

и их фармацевтически приемлемые соли и гидраты.
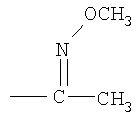
Фрагменты химических формул, заключенные в круглые квадратные скобки, обозначают боковые группы, например, -С(O)- означает карбонильную группу (т.е.


"Алкил" означает алифатическую углеводородную группу, которая может быть линейной или разветвленной и содержать от около 1 до около 20 атомов углерода в цепи. Предпочтительные алкильные группы содержат от около 1 до около 12 атомов углерода в цепи. Более предпочтительные алкильные группы содержат от около 1 до около 6 атомов углерода в цепи. "Разветвленная" означает, что к линейной алкильной цепи присоединена одна или большее количество алкильных групп, таких как метил, этил или пропил. "Низший алкил" означает группу, содержащую от около 1 до около 6 атомов углерода в цепи, которая может быть линейной или разветвленной.
"Алкилен" означает двухвалентную алифатическую углеводородную группу, которая может быть линейной или разветвленной и содержит от около 1 до около 20 атомов углерода в цепи. Предпочтительные алкиленовые группы содержат от около 1 до около 12 атомов углерода в цепи. Более предпочтительные алкиленовые группы содержат от около 1 до около 6 атомов углерода в цепи. Неограничивающие примеры подходящих алкиленовых групп включают метилен (т.е. -CH2-) и этилиден (-CH2CH2- или -СН(СН3)-).
"Алкинил" означает алифатическую углеводородную группу, содержащую не менее одной углерод-углеродной тройной связи, и которая может быть линейной или разветвленной и содержать от около 2 до около 15 атомов углерода в цепи. Предпочтительные алкинильные группы содержат от около 2 до около 12 атомов углерода в цепи; а более предпочтительно - от около 2 до около 4 атомов углерода в цепи. "Разветвленная" означает, что к линейной алкинильной цепи присоединена одна или большее количество алкильных групп, таких как метил, этил или пропил. "Низший алкинил" означает группу, содержащую от 2 до примерно 6 атомов углерода в цепи, которая может быть линейной или разветвленной. Неограничивающие примеры алкенильных групп включают: этинильную, пропинильную, 2-бутинильную и 3-метилбутинильную.
"Арил" означает ароматическую моноциклическую или полициклическую кольцевую систему, включающую от около 6 до около 14 атомов углерода, предпочтительно - от около 6 до около 10 атомов углерода.
"Циклоалкил" означает неароматическую моно- или полициклическую кольцевую систему, содержащую от около 3 до около 10 атомов углерода, предпочтительно - от около 5 до около 10 атомов углерода. Предпочтительные циклоалкильные кольца содержат от около 5 до около 7 кольцевых атомов. Неограничивающие примеры подходящих моноциклических циклоалкилов включают циклопропил, циклопентил, циклогексил, циклогептил и т.п. Неограничивающие примеры подходящих полициклических циклоалкилов включают 1-декалинил, норборнил, адамантил и т.п., а также частично насыщенные системы, такие как, например, инданил, тетрагидронафтил и т.п.
"Циклоалкилен" означает двухвалентную циклоалкильную кольцевую систему, содержащую от около 3 до около 10 атомов углерода, предпочтительно - от примерно до примерно 10 атомов углерода. Предпочтительные циклоалкиленовые кольца содержат от около 5 до около 7 кольцевых атомов. Неограничивающие примеры подходящих моноциклических алкиленов включают циклопропилен (т.е.

"Галоген" означает фтор, хлор, бром или йод. Предпочтительными галогенами являются фтор, хлор и бром. "Галогензамещенные" группы (например, галогеналкильные группы) означают группы, замещенные одним или большим количеством атомов фтора, хлора, брома и/или йода.
"Гетероциклоалкил" означает неароматическую насыщенную моноциклическую или полициклическую кольцевую систему, содержащую от около 3 до около 10 циклических атомов, предпочтительно - от около 5 до около 10 циклических атомов, в которой один или большее количество атомов кольцевой системы представляет собой элемент, не являющийся углеродом, например, азот, кислород или серу, по отдельности или в комбинации. В кольцевой системе не имеется соседних атомов кислорода и/или серы. Предпочтительные гетероциклоалкилы содержат от около 5 до около 6 циклических атомов. Приставка аза-, окса- или тиа- перед названием гетероциклоалкильной системы означает, что в качестве кольцевого атома содержится не менее одного атома азота, кислорода или серы соответственно. Любая группа -NH в гетероциклоалкильном кольце может находиться в защищенном виде, таком как, например, в виде группы -N(Boc), -N(CBz), -N(Tos) и т.п.; такие защищенные функциональные группы также считаются частью настоящего изобретения. Атом азота или серы гетероциклоалкила необязательно может быть окислен с образованием соответствующего N-оксида, S-оксида или S,S-диоксида. Неограничивающие примеры моноциклических гетероциклоалкильных колец включают пиперидил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксанил, тетрагидрофуранил, тетрагидротиофенил, лактам, лактон и т.п.
Термин "в очищенной форме" применительно к соединению согласно изобретению относится к физическому состоянию указанного соединения после получения с помощью способа или способов очистки, описанных в настоящем изобретении или хорошо известных специалисту в данной области техники, при достаточной чистоте, охарактеризованной стандартными способами анализа, описанными в настоящем изобретении или хорошо известными специалисту в данной области техники.
Также следует отметить, что в тексте, на схемах, в примерах и таблицах, приведенных в настоящем изобретении, подразумевается, что любой гетероатом с ненасыщенными валентностями обладает атомом (атомами) водорода, достаточными для насыщения валентностей.
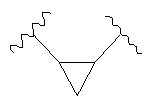
Волнистая линия

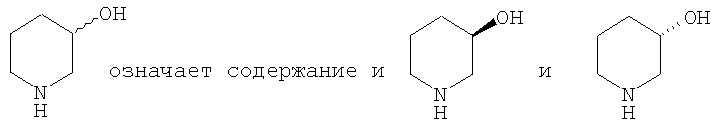
Если стереохимическая конфигурация структуры специально не указана, то структура может соответствовать смеси стереоизомеров или любому возможному отдельному стереоизомеру. Таким образом, если стереохимическая конфигурация структуры специально не указана, то структура включает все стереохимические конфигурации, обладающие указанной системой связей (например, все возможные энантиомеры или диастереоизомеры), а также смеси таких стереоизомеров (например, рацемические смеси), например,


Соединения формулы I образуют соли, которые также входят в объем настоящего изобретения. Если не указано иного, то следует понимать, что ссылка на соединение формулы I означает включение ссылки на его соли. При использовании в настоящем изобретении термин "соль (соли)" означает соли с кислотами, образованные с неорганическими и/или органическими кислотами, а также соли с основаниями, образованные с неорганическими и/или органическими основаниями. Кроме того, если соединение формулы I содержит и фрагмент основания, такой как (без наложения ограничений) пиридин или имидазол, и фрагмент кислоты, такой как (без наложения ограничений) карбоновая кислота, то могут образоваться цвиттерионы ("внутренние соли") и при использовании в настоящем изобретении они включены в содержание термина "соль (соли)". Предпочтительны фармацевтически приемлемые (т.е. нетоксичные, физиологически приемлемые) соли, хотя применимы и другие соли. Соли соединений формулы I могут образовываться, например, с помощью реакции соединения формулы I с количеством кислоты или основания, таким как эквивалентное количество, в среде, такой как среда, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
Примеры солей с кислотами включают ацетаты, адипаты, альгинаты, аскорбаты, аспартаты, бензоаты, бензолсульфонаты, бисульфаты, бораты, бутираты, цитраты, камфораты, камфорсульфонаты, циклопентанпропионаты, диглюконаты, додецилсуль-фаты, этансульфонаты, фумараты, глюкогептаноаты, глицерофосфаты, гемисульфаты, гептаноаты, гексаноаты, гидрохлориды, гидробромиды, гидройодиды, 2-гидрокси-этансульфонаты, лактаты, малеаты, метансульфонаты, метилсульфаты, 2-нафталин-сульфонаты, никотинаты, нитраты, оксалаты, памоаты, пектинаты, персульфаты, 3-фенилпропионаты, фосфаты, пикраты, пивалаты, пропионаты, салицилаты, сукцинаты, сульфаты, сульфонаты (такие как указанные в настоящем изобретении), тартраты, тиоцианаты, толуолсульфонаты (также известные под названием тозилатов), ундеканоаты и т.п.
Примеры солей с основаниями включают соли аммония, соли щелочных металлов, такие как соли натрия, лития и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли алюминия, соли цинка, соли с органическим основаниями (например, органическими аминами), такими как бензатины, диэтиламин, дицикло-гексиламины, гидрабамины (образующиеся с N,N-бис(дегидроабиетил)этилендиамином), N-метил-D-глюкамины, N-метил-D-глюкамиды, трет-бутиламины, пиперазин, фенилциклогексиламин, холин, трометамин, и соли с аминокислотами, такими как аргинин, лизин и т.п. Основные азотсодержащие группы могут быть превращены в четвертичные группы с помощью таких соединений, как галогениды низших алкилов (например, метил-, этил- и бутилхлориды, -бромиды и -йодиды), диалкилсульфаты (например, диметил-, диэтил-, дибутил- и диамилсульфаты), галогениды с длинными цепями (например, децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -йодиды), арилалкилгалогениды (например, бензил- и фенетилбромиды) и др. Кислоты (и основания), которые обычно считаются пригодными для образования фармацевтически применимых солей из основных (кислотных) фармацевтических соединений, обсуждены, например, в публикациях S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1-19; P.Gould, International J. of Pharmaceutics (1986) 33 201-217; Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; in The Orange Book (Food & Drug Administration, Washington, D.C. on their website); и Р. Heinrich Stahl, Camille G. Wermuth (Eds.), Handbook of Pharmaceutical Salts: Properties, Selection, and Use, (2002) Int'l. Union of Pure and Applied Chemistry, p.330-331, и все они включены в настоящее изобретение путем ссылки.
В объеме настоящего изобретения подразумевается, что все такие соли кислот и оснований являются фармацевтически приемлемыми солями и для целей настоящего изобретения все соли кислот и оснований считаются эквивалентными свободным формам соответствующих соединений.
Соединения формулы I и их соли и гидраты могут существовать в своих таутомерных формах (например, в виде простого амидо- или иминоэфира). Подразумевается, что все такие таутомерные формы являются частью настоящего изобретения.
Полиморфные формы соединений формулы I и солей и гидратов соединений формулы I считаются включенными в настоящее изобретение.
В объем настоящего изобретения входят все стереоизомеры (например, геометрические изомеры, оптические изомеры и т.п.) соединений, соответствующих настоящему изобретению (включая изомеры солей, сольватов и пролекарств этих соединений, а также соли и сольваты пролекарств), такие как те, которые могут существовать вследствие наличия асимметрических атомов углерода в различных заместителях, включая энантиомерные формы (которые могут существовать даже при отсутствии асимметрических атомов углерода), ротамерные изомерные формы, атропоизомерные и диастереоизомерные формы. Отдельные стереоизомеры соединений, соответствующих настоящему изобретению, могут, например, в основном не содержать других изомеров или могут быть смешаны, например, как рацематы, или со всеми другими, или с другими выбранными стереоизомерами. Хиральные центры, соответствующие настоящему изобретению, могут обладать S- или R-конфигурацией в соответствии с определением, приведенным в IUPAC 1974 Recommendations. Подразумевается, что при использовании терминов "соль", "гидрат" и т.п. они в равной степени применимы к соли и гидрату энантиомеров, стереоизомеров, ротамеров, таутомеров, позиционных изомеров и рацематов соединений, соответствующих настоящему изобретению. "По меньшей мере один" означает, например, 1-3, 1-2 или 1.
Соединения формулы I являются эффективными антагонистами рецептора NK1 и воздействуют на его эндогенный агонист, вещество Р, на участке рецептора NK1 и поэтому могут применяться при лечении заболеваний, нарушений или патологических состояний, вызывающихся или обостряющихся вследствие активности рецептора.
Поэтому другим объектом изобретения является фармацевтическая композиция, обладающая свойствами антагониста нейрокининового рецептора NK1, включающая по меньшей мере одно соединение вышеприведенной формулы I или его фармацевтически приемлемую соль или гидрат и по меньшей мере один фармацевтически приемлемый носитель.
Дальнейшим объектом изобретения является применение по меньшей мере одного соединения вышеприведенной формулы I или его фармацевтически приемлемой соли или гидрата для получения лекарства для лечения физиологического нарушения, устранения симптома или лечения заболевания, причем физиологическое нарушение, симптом или заболевание выбраны из группы, включающей респираторные заболевания, воспалительные заболевания, кожные нарушения, офтальмологические нарушения, патологические состояния центральной нервной системы, депрессию, тревогу, фобию, биполярное расстройство, аддикции, алкогольную зависимость, злоупотребление психоактивными веществами, эпилепсию, ноцицепцию, психоз, шизофрению, болезнь Альцгеймера, слабоумие при СПИД, болезнь Тауна, связанные со стрессом нарушения, обсессивно-компульсивные нарушения, нарушения питания, булимию, нервную анорексию, переедание, нарушения сна, манию, предменструальный синдром, желудочно-кишечные нарушения, атеросклероз, фиброзные нарушения, ожирение, диабет типа II, связанные с болью нарушения, головную боль, невропатическую боль, послеоперационную боль, хронический болевой синдром, нарушения мочевого пузыря, мочеполовые нарушения, кашель, рвоту и тошноту.
В ниже представленных примерах употребляются следующие условные сокращения.
Ас означает ацетил.
АсОН (или НОАс) означает уксусную кислоту.
Boc означает трет-бутоксикарбонил.
Bu означает бутил.
t-Bu или But означает третичный бутил.
Bn означает бензил.
Cbz означает карбобензоксигруппу (т.е. Ph-CH2-O-C(O)-).
ДХМ означает дихлорметан.
ДИЭА означает диизопропилэтиламин.
ДМФ означает диметилформамид.
ДМАП означает диметиламинопиридин.
ДМПМ означает N,N- диметилпропиленмочевину.
ДМСО означает диметилсульфоксид.
ДФФА означает дифенилфосфоразид.
Et означает этил.
ЭДХ означает 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорид.
ББА означает бомбардировку быстрыми атомами.
HOTs означает п-толуолсульфоновую кислоту.
HATU означает O-(7-азабензотриазол-1-yl)-N,N,N',N'-тетраметилуронийгексафторфосфат.
ВЭЖХ означает высокоэффективную жидкостную хроматографию.
МСВР означает масс-спектроскопию высокого разрешения.
ЖХМС означает жидкостную хроматографию/масс-спектроскопию.
LiГМДС означает гексаметилдисилазид лития.
Me означает метил.
МеОН означает метанол.
МС означает масс-спектроскопию.
Ms или "мезил" означает метансульфонил.
Ni (Ra) означает Ni Ренея.
ОП означает оптическую плотность.
Ph означает фенил.
i-РА (или IPA или iPA) означает изопропил.
ППТС означает пиридиний-п-толуолсульфонат.
ПТСК означает п-толуолсульфоновую кислоту.
PYBOP означает (бензотриазол-1-илокси)трипирролидинофосфонийгексафторфосфат.
КГ означает комнатную температуру.
ТБАФ означает тетрабутиламмонийфторид.
ТБАЙ означает тетрабутиламмониййодид.
ТФК означает трифторуксусную кислоту.
ТГФ означает тетрагидрофуран.
ТСХ означает тонкослойную хроматографию.
ТМС означает триметилсилил.
TMCCI означает триметилсилилхлорид.
"Тозил" означает толуолсульфонил.
Активность соединений формулы I по отношению к NK1, NK2 и NК3 in vitro и in vivo можно определить по различным методикам, известным в данной области техники, таким как исследование их способности ингибировать активность агониста NK1, вещества Р. Выраженная в процентах активность ингибирования агониста нейрокинина является разностью между выраженным в процентах максимальным специфическим связыванием ("МСС") и 100%. Выраженное в процентах МСС определяется приведенным ниже уравнением, в котором "КРМ" означает "количество распадов в минуту":

Концентрацию, при которой соединение приводит к ингибированию, равному 50%, затем используют для определения константы ингибирования ("Ki") по уравнению Чанга-Прусова.
Активность in vivo можно определить путем подавления вызванного агонистом подергивания лапок у карликовой песчанки, как это описано в публикации Science, 281. 1640-1695 (1998), которая во всей своей полноте включена в настоящее изобретение путем ссылки. Следует понимать, что соединения формулы I могут обладать разной степенью антагонистической активности по отношению к NK1. Например, некоторые соединения могут обладать большей антагонистической активностью по отношению к NK1, чем другие.
Соединения, соответствующие настоящему изобретению, обладают высоким средством по отношению к рецептору NK1, которое выражается значениями Ki (в нМ). Активность соединений, соответствующих настоящему изобретению, определяют путем измерения их значений Ki. Чем меньше значение Ki, тем более активное антагонистическое воздействие соединение оказывает на рецептор NK1. Соединения, соответствующие настоящему изобретению, обладают широким диапазоном активности. Средние значения Ki для соединений формулы I по отношению к рецептору NK1 обычно находятся в диапазоне от 0,01 до около 1000 нМ, предпочтительно - от около 0,1 нМ до около 100 нМ, а более предпочтительными являются значения, равные от около 0,1 нМ до около 10 нМ. Еще более предпочтительными являются соединения, обладающие средними значениями Ki по отношению к рецептору NK1, равными от 0,1 до около 5 нМ. Особенно предпочтительные соединения обладают средними значениями Ki по отношению к рецептору МКч, равными от 0,1 до около 1 нМ. Еще более предпочтительные соединения обладают средними значениями Ki по отношению к рецептору NK1, равными от 0,1 до около 0,3 нМ. Соединения 2, 9, 10, 12, 14, 16, 19, 20, 23, 29, 30, 42 и 54 (см. выше таблицу I) обладают значениями Ki, равными соответственно 0,12, 0,18, 0,1, 0,05, 0,1, 0,13, 0,1, 0,11, 0,12, 0,11, 0,54, 0,28 и 0,12 нМ.
Соединения формулы I применяются в целом ряде случаев. Например, соединения, соответствующие настоящему изобретению, можно применять в качестве антагонистов нейрокининовых рецепторов, предпочтительно - рецепторов NKi у млекопитающего, такого как человек. Как таковые, их можно применять при лечении и предупреждении одного или большего количества из множества патологических состояний (физиологических нарушений, симптомов и заболеваний) млекопитающего (человека и животного) у пациента, нуждающегося в таком лечении, при котором патологические состояния выбраны из группы, включающей: (1) респираторные заболевания (например, хроническое заболевание легких, бронхит, пневмония, астма, аллергия, кашель и бронхоспазм), (2) воспалительные заболевания (например, артрит и псориаз), (3) кожные нарушения (например, атопический дерматит и контактный дерматит), (4) офтальмологические нарушения (например, ретинит, гипертензия глаза и катаракты), (5) патологические состояния центральной нервной системы, такие как депрессии (например, невротическая депрессия), тревоги (например, генерализованное тревожное, социализированное тревожное и паническое тревожное расстройство), фобии (например, социофобия) и биполярное расстройство, (6) аддикции (например, алкогольную зависимость и злоупотребление психоактивными веществами), (7) эпилепсию, (8) ноцицепцию, (9) психоз, (10) шизофрению, (11) болезнь Альцгеймера, (12) слабоумие при СПИД, (13) болезнь Тауна, (14) связанные со стрессом нарушения (например, посттравматический стресс), (15) обсессивно/компульсивные нарушения, (16) нарушения питания (например, булимия, нервная анорексия и переедание), (17) нарушения сна, (18) манию, (19) предменструальный синдром, (20) желудочно-кишечные нарушения (например, синдром раздраженной толстой кишки, болезнь Крона, колит и рвота), (21) атеросклероз, (22) фиброзные нарушения (например, фиброз легких), (23) ожирение, (24) диабет типа II, (25) связанные с болью нарушения (например, головная боль, такая как мигрени, невропатическая боль, послеоперационная боль и хронические болевые синдромы), (26) нарушения мочевого пузыря и мочеполовые нарушения (например, интерстициальный цистит и недержание мочи), (27) рвоту (например, вызванная химиотерапией (например, вызванная цисплатином, доксорубицином и таксаном), вызванная радиацией, укачиванием в транспорте, вызванная этанолом и постоперативная тошнота и рвота) и (28) рвоту. Соединения, соответствующие настоящему изобретению, можно предпочтительно применять при лечении и предупреждении одного из указанных ниже патологических состояний млекопитающего (например, человека) у пациента, нуждающегося в таком лечении: респираторные заболевания (например, кашель), депрессия, тревога, фобия и биполярное расстройство, алкогольная зависимость, злоупотребление психоактивными веществами, ноцицепция, психоз, шизофрения, связанные со стрессом нарушения, обсессивно/компульсивное нарушение, булимия, нервная анорексия и переедание, нарушения сна, мания, предменструальный синдром, желудочно-кишечные нарушения, ожирение, связанные с болью нарушения, нарушения мочевого пузыря, мочеполовые нарушения, рвоту и тошноту. В частности, соединения формулы 1 применимы для лечения патологических состояний, связанных с капиллярным кровотечением и выделением слизи. Поэтому соединения, соответствующие настоящему изобретению, особенно полезны при лечении и предупреждении астмы, рвоты, тошноты, депрессий, тревоги, кашля и связанных с болью нарушений, более предпочтительно - рвоты, депрессии, тревоги и кашля.
В другом варианте осуществления настоящее изобретение относится к фармацевтическим композициям, включающим по меньшей мере одно соединение (например, от одного до трех соединений, предпочтительно - одно соединение), описываемое формулой I, и по меньшей мере один фармацевтически приемлемый инертный наполнитель или носитель. Настоящее изобретение также относится к применению таких фармацевтических композиций при лечении патологических состояний млекопитающего (например, человека), таких как перечисленные выше.
В еще одном варианте осуществления настоящее изобретение относится к способу антагонистического влияния на воздействие вещества Р на участке нейрокининового рецептора 1 или блокирования одного или большего количества нейрокининовых рецепторов 1 у млекопитающего (т.е. пациента, например, человека), нуждающегося в таком лечении, включающему введение млекопитающему эффективного количества по меньшей мере одного (например, одного) соединения формулы I.
В другом варианте осуществления настоящего изобретения эффективное количество одного или большего количества антагонистов рецептора NK1, соответствующих настоящему изобретению, можно скомбинировать с эффективным количеством одного или большего количества антидепрессивных средств и/или одного или большего количества успокаивающих средств (например, гепирона, гепиронгидрохлорида, нефазодона или нефазодонгидрохлорида (например, серзона®)) для лечения депрессии и/или тревоги. В патенте США US 6117855 (2000), раскрытие которого включено в настоящее изобретение путем ссылки, раскрыт способ лечения или предупреждения депрессии или тревоги с помощью комбинированного средства, включающего специфический антагонист рецептора NK1 совместно с антидепрессивным средством и/или успокаивающим средством. Таким образом, антидепрессивные и/или успокаивающие средства, такие как раскрытые в патенте США US 6117855 (2000), можно скомбинировать с одним или большим количеством (например, одним) соединений формулы I для лечения депрессивных и/или тревожных патологических состояний у млекопитающего, предпочтительно - человека.
В еще одном варианте осуществления настоящего изобретения эффективное количество одного или большего количества (например, одного) антагонистов рецептора NK1, соответствующих настоящему изобретению, можно скомбинировать с эффективным количеством одного или большего количества (например, одного) селективных ингибиторов повторного поглощения серотонина ("СППС") для лечения у млекопитающего патологических состояний, таких как описанные выше. СППС изменяет пресинаптическую доступность серотонина посредством подавления пресинаптического повторного накопления выделяемого нейронами серотонина. В патенте США US 6162805 (2000), раскрытие которого включено в настоящее изобретение путем ссылки, раскрыт способ лечения ожирения с помощью комбинированного средства, включающего антагонист рецептора NK1 и СППС. Одно или большее количество соединений формулы I, соответствующих настоящему изобретению, можно скомбинировать с одним или несколькими СППС в одной фармацевтической композиции или его можно вводить одновременно, по отдельности или последовательно с СППС. Эта комбинация может быть применима при лечении и предупреждении ожирения или другого из указанных выше патологических состояний человека и млекопитающего. В частности, эффективное количество по меньшей мере одного (например, одного) соединения формулы I, по отдельности или совместно с эффективным количеством по меньшей мере одного (например, одного) селективного ингибитора повторного поглощения серотонина может быть применимо при лечении и предупреждении депрессии и/или тревоги.
Известно, что многие химические вещества изменяют синаптическую доступность серотонина посредством подавления пресинаптического повторного накопления выделяемого нейронами серотонина. Типичные СППС включают, без наложения ограничений, следующие: флуоксетин, флуоксетингидрохлорид (например, прозак®), флувоксамин, флувоксаминмалеат (например, лувокс®), пароксетин, пароксетингидрохлорид (например, паксил®), сертралин, сертралингидрохлорид (например, золофт®), циталопрам, циталопрамгидробромид (например, целекса®), дулоксетин, дулоксетингидрохлорид, венлафаксин и венлафаксингидрохлорид (например, эффексор®). К другим СППС относятся раскрытые в патенте США US 6162805 (2000). Другие соединения можно легко оценить и определить их способность селективно ингибировать повторное поглощение серотонина. Таким образом, в одном варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей по меньшей мере один (например, один) антагонист рецептора NK1, описываемый формулой I, по меньшей мере один (например, один) СППС и по меньшей мере один фармацевтически приемлемый инертный наполнитель или носитель. В другом варианте осуществления настоящее изобретение относится к способу лечения указанных выше патологических состояний млекопитающего (например, человека), способ включает введение пациенту, нуждающемуся в таком лечении, эффективного количества фармацевтической композиции, включающей по меньшей мере один (например, один) антагонист рецептора NK1, описываемый формулой I, в комбинации по меньшей мере с одним (например, с одним) СППС, таким как один из указанных выше, и по меньшей мере с одним фармацевтически приемлемым инертным наполнителем или носителем.
В предпочтительном варианте осуществления настоящее изобретение относится к способу лечения депрессии и тревоги, способ включает введение пациенту, нуждающемуся в таком лечении, эффективного количества по меньшей мере одного (например, одного) антагониста рецептора NK1, описываемого формулой I, в комбинации по меньшей мере с одним (например, с одним) СППС, таким как один из описанных выше. Если антагонист рецептора NK1, соответствующий настоящему изобретению, комбинируют с СППС для введения пациенту, нуждающемуся в таком лечении, то эти два активных ингредиента можно вводить одновременно, по отдельности (один после другого через относительно короткий период времени) или последовательно (сначала один, а затем другой в течение некоторого периода времени). Обычно, если эти два активных ингредиента вводят по отдельности или последовательно, то антагонист рецептора NK1, соответствующий настоящему изобретению, предпочтительно вводить до введения СППС.
Другой вариант осуществления настоящего изобретения относится к лечению пациента, страдающего от нескольких заболеваний, с помощью комбинированной терапии, терапия включает введение пациенту (например, млекопитающему, предпочтительно - человеку), нуждающемуся в таком лечении, по меньшей мере одного соединения формулы I и по меньшей мере одного другого активного ингредиента (т.е. лекарственного средства), применяющегося для лечения одного или большего количества заболеваний, от которых страдает пациент. Соединения формулы I и другие активные ингредиенты можно вводить одновременно, по отдельности и/или последовательно. Соединения формулы I и другие активные ингредиенты можно вводить по отдельности в виде любой дозированной формы. Введение предпочтительно проводить с использованием пероральных дозированных форм или с использованием чрескожных пластырей. Соединения формулы I и другие активные ингредиенты можно объединить друг с другом и вводить в виде комбинированной дозированной формы.
Таким образом, соединения, соответствующие настоящему изобретению, можно использовать по отдельности или в комбинации с другими активными средствами. Комбинированная терапия включает введение двух или большего количества активных ингредиентов пациенту, нуждающемуся в таком лечении. В дополнение к описанной выше комбинированной терапии вида антагонист рецептора NK1 /СППС соединения, описываемые формулой I, можно скомбинировать с одним или большим количеством других активных агентов, таких как следующие: другие типы антагонистов рецептора NK1 (например, раскрытые в указанных выше патентах, относящихся к антагонистам нейрокининового рецептора), простаноиды, антагонисты рецептора H1, агонисты α-адренергического рецептора, агонисты допаминового рецептора, агонисты меланокортинового рецептора, антагонисты эндотелинового рецептора, ингибиторы эндотелинконвертирующего фермента, антагонисты ангиотензинового рецептора II, ингибиторы ангиотензинконвертирующего фермента, ингибиторы нейтральной металлоэндопептидазы, антагонисты ЕТA, ингибиторы ренина, антагонисты серотонинового рецептора 5-НТ3 (например, ондансетрон, ондансетронгидрохлорид (например, золфран®), палоносетрон, гранисетрон и гранисетронгидрохлорид (например, китрил®), агонисты серотонинового рецептора 5-НТ2с, агонисты ноцицептинового рецептора, глюкокорти-коиды (например, дексаметазон), ингибиторы рокиназы, модуляторы кальциевых каналов и/или ингибиторы белка 5 множественной лекарственной устойчивости.
Терапевтическими агентами, особенно подходящими для комбинированной терапии соединениями, соответствующими настоящему изобретению, являются следующие: простаноиды, такие как простагландин E1; α-адренергические агонисты, такие как фентоламинмезилат; агонисты допаминового рецептора, такие как апоморфин; антагонисты ангиотензина II, такие как лосартан, ирбесартан, валсартан и кандесартан; антагонисты ЕТA, такие как босентан и АВТ-627; антагонисты серотонинового рецептора 5-НТ3, такие как ондансетрон; и глюкокортикоиды, такие как дексаметазон. В предпочтительных вариантах осуществления настоящего изобретения соединения, соответствующие настоящему изобретению, можно скомбинировать с другими типами антагонистов рецептора NK1, СППС, агонистами допаминового рецептора, антагонистами серотонинового рецептора 5-НТ3, агонистами серотонинового рецептора 5-НТ2с, агонистами ноцицептинового рецептора, глюкокортикоидами и/или ингибиторами белка 5 множественной лекарственной устойчивости.
Другой вариант осуществления настоящего изобретения относится к способу лечения физиологического нарушения, устранения симптома или лечения заболевания у пациента, нуждающегося в таком лечении, включающему введение пациенту эффективного количества по меньшей мере одного соединения формулы I и эффективного количества по меньшей мере одного активного ингредиента, выбранного из группы, включающей: другие антагонисты рецептора NK1, селективные ингибиторы повторного поглощения серотонина, агонисты допаминового рецептора, антагонисты серотонинового рецептора 5-НТ3, агонисты серотонинового рецептора 5-НТ2с, агонисты ноцицептинового рецептора, глюкокортикоиды и ингибиторы белка 5 множественной лекарственной устойчивости, в котором физиологическое нарушение, симптом или заболевание выбрано из группы, включающей: респираторное заболевание, депрессию, тревогу, фобию, биполярное расстройство, алкогольная зависимость, злоупотребление психоактивными веществами, ноцицепция, психоз, шизофрения, связанное со стрессом нарушение, обсессивно/компульсивное нарушение, булимия, нервная анорексия, переедание, нарушение сна, мания, предменструальный синдром, желудочно-кишечное нарушение, ожирение, головная боль, невропатическая боль, послеоперационная боль, хронический болевой синдром, нарушение мочевого пузыря, мочеполовое нарушение, кашель, рвоту и тошноту.
Фармацевтические композиции могут содержать от около 0,1 до около 99,9 мас.%, или от около 5 до около 95 мас.%, или от около 20 до около 80 мас.% активного ингредиента (соединения формулы I). При изготовлении фармацевтических композиций из соединений, описанных в настоящем изобретении, инертные, фармацевтически приемлемые носители могут быть твердыми или жидкими. К твердым формам препаратов относятся порошки, таблетки, диспергирующиеся гранулы, капсулы, облатки и суппозитории. Порошки и таблетки могут содержать от около 5 до около 95% активного ингредиента. Подходящие твердые носители известны в данной области техники, например, карбонат магния, стеарат магния, тальк, сахар и лактоза. Таблетки, порошки, облатки и капсулы можно использовать в качестве твердых дозированных форм, пригодных для перорального введения. Примеры фармацевтически приемлемых носителей и способов изготовления различных композиций приведены в публикации A.Gennaro (ed.), Remington: The Science and Practice of Pharmacy, 20th Edition, (2000), Lippincott Williams &Wilkins, Baltimore, MD, которая включена в настоящее изобретение путем ссылки.
Жидкие формы препаратов включают растворы, суспензии и эмульсии, например, водные или водно-пропиленгликолевые растворы для парентеральных инъекций или прибавление подсластителей и замутнителей в растворы, суспензии и эмульсии для перорального введения. К жидким формам композиций также могут относиться растворы для внутриназального введения.
Аэрозольные препараты, пригодные для ингаляции, могут включать растворы и твердые вещества в порошкообразной форме, которые могут сочетаться с фармацевтически приемлемым носителем, таким как сжатый инертный газ, например, азот.
В объем настоящего изобретения также включены твердые формы препаратов, которые предназначены для превращения в жидкие формы препаратов, предназначенных для перорального или парентерального введения, которое выполняется незадолго до использования. Такие жидкие формы включают растворы, суспензии и эмульсии.
Соединения, соответствующие настоящему изобретению, также можно вводить чрескожно. Чрескожные композиции могут представлять собой кремы, лосьоны, аэрозоли и/или эмульсии и они могут быть включены в матрицу пластыря чрескожного воздействия или пластыря резервуарного типа, которые обычно используется в данной области техники для такой цели.
Предпочтительно вводить соединение перорально.
Предпочтительно, чтобы фармацевтический препарат находился в виде разовой дозированной формы. В такой форме препараты разделяются на разовые дозы подходящей величины, содержащие соответствующие количества активного компонента, например, количество, достаточное для достижения необходимой цели.
Термин "фармацевтическая композиция" также включает и нерасфасованную композицию, и отдельные дозированные формы в виде любых форм, описанных в настоящем изобретении, содержащие более одного (например, два) фармацевтически активных средства, таких как, например, соединение, соответствующее настоящему изобретению, и дополнительное средство, выбранное из перечня активных средств, описанных в настоящем изобретении, совместно с любыми фармацевтически неактивными инертными наполнителями. Нерасфасованная композиция и каждая отдельная дозированная форма может содержать постоянные количества указанных "более одного фармацевтически активных средств". Термин "нерасфасованная композиция" означает материал, который еще не переработан в отдельные дозированные формы. Примером дозированной формы является пероральная дозированная форма, такая как таблетки, пилюли и т.п. Аналогичным образом, описанный в настоящем изобретении способ лечения пациента путем введения фармацевтической композиции, соответствующей настоящему изобретению, также включает введение указанных нерасфасованной композиции и отдельных дозированных форм.
Количество активного соединения, содержащегося в разовой дозе препарата, в соответствии с конкретным случаем применения обычно может меняться или регулироваться в диапазоне от около 0,01 до около 4000 мг, предпочтительно - от около 0,02 до около 1000 мг, более предпочтительно - от около 0,3 до около 500 мг, а наиболее предпочтительно - от около 0,04 до около 250 мг в соответствии с конкретным применением.
Реальная использующаяся доза может меняться в зависимости от требований пациента и тяжести подвергающегося лечению патологического состояния. Определение надлежащего дозировочного режима для конкретного случая осуществляет специалист в данной области техники. Для удобства полную суточную дозу можно разделять и вводить порциями в течение дня в соответствии с необходимостью.
Количество и частота введения соединений, соответствующих настоящему изобретению, и/или их фармацевтически приемлемых солей будет регулироваться в соответствии с решением лечащего врача, учитывающего такие факторы, как возраст, состояние и массу пациента, а также тяжесть симптомов при лечении. Типичный рекомендованный суточный дозировочный режим для перорального введения может представлять собой введение от около 0,02 до около 2000 мг/сутки, вводимых в виде двух - четырех разделенных доз.
Фармацевтические композиции, соответствующие настоящему изобретению, можно вводить от около 1 до около 5 раз в сутки или, альтернативно, посредством непрерывного вливания. Такое введение можно использовать в качестве длительного или неотложного лечения.
Количество антагониста рецептора NK1 в комбинации с селективным ингибитором повторного поглощения серотонина ("СППС") в разовой дозе препарата может составлять от около 10 до около 300 мг антагониста рецептора NK1 в комбинации с составляющим от около 10 до около 100 мг количеством СППС. В другой комбинации количество антагониста рецептора NK1 в комбинации с СППС в разовой дозе препарата может составлять от около 50 до около 300 мг антагониста рецептора NK1 в комбинации с составляющим от около 10 до около 100 мг количеством СППС. В другой комбинации количество антагониста рецептора NK1 в комбинации с СППС в разовой дозе препарата может составлять от около 50 до около 300 мг антагониста рецептора NK1 в комбинации с составляющим от около 20 до около 50 мг количеством СППС.
Реальная использующаяся доза может меняться в зависимости от требований пациента и тяжести подвергающегося лечению патологического состояния. Определение надлежащего дозировочного режима для конкретного случая осуществляет специалист в данной области техники. Для удобства полную суточную дозу можно разделять и вводить порциями в течение дня в соответствии с необходимостью. После улучшения состояния пациента при необходимости можно вводить поддерживающую дозу соединения, композиции или комбинации, соответствующей настоящему изобретению. Затем дозу или частоту введения, или обе можно снизить в зависимости от симптомов и того, в какой степени сохраняется улучшенное состояние. Если симптомы ослаблены в необходимой степени, то лечение следует прекратить. Однако после любого рецидива симптомов заболевания может потребоваться периодическое лечение пациентов на долговременной основе.
Конкретные дозы и режимы лечения каждого конкретного пациента могут меняться и будут зависеть от различных факторов, включая активность конкретного используемого соединения, возраста, массы тела, общего состояния здоровья, пола и диеты пациента, длительности введения, скорости выведения, конкретной комбинации лекарственных средств, тяжести и проявления симптомов, на устранение которых направлено лечение, предрасположенности пациента к проявлению подвергающегося лечению патологического состояния и решения лечащего врача. Определение надлежащего дозировочного режима для конкретного случая входит в компетенцию специалиста в данной области техники.
Примеры
Описанное изобретение в качестве примеров включает приведенные ниже примеры получения и примеры, которые не следует рассматривать, как ограничивающие объем настоящего описания. Для специалистов в данной области техники могут быть очевидны альтернативные механизмы синтеза и аналогичные структуры.
Пример получения 1

Стадия 1:

В круглодонной колбе объемом 25 мл соединение 42b (0,253 г, 0,42 ммоль, 1,0 экв.) растворяют в 5 мл CH2Cl2 и полученную реакционную смесь охлаждают до 0°С в бане со льдом. После этого к реакционной смеси прибавляют Et3N (0,088 мл, 0,63 ммоль, 1,5 экв.), затем 4-хлорбутирилхлорид (0,065 мл, 0,5 ммоль, 1,2 экв.) и затем ее медленно нагревают до комнатной температуры и перемешивают в течение 14 ч. За протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан) и МС. После завершения реакции реакционную смесь разбавляют с помощью CH2Cl2, реакцию останавливают насыщенным водным раствором NаНСО3, а затем рассолом. Органический слой сушат над Na2SO4 и концентрируют и получают неочищенное соединение 1а (0,3 г), которое используют на следующей стадии без дополнительной очистки.
МС с электрораспылением [М+1] 724,4.
Стадия 2:

В высушенной на огне круглодонной колбе объемом 25 мл соединение 1а (0,3 г, 0,4 ммоль, 1,0 экв.) растворяют в сухом ТГФ. К этой реакционной смеси прибавляют 60% NaH (0,025 г, 0,62 ммоль, 1,5 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. За протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан) и МС. После завершения реакции реакционную смесь разбавляют с помощью ЕtOАс и реакцию останавливают насыщенным водным раствором NаНСО3. Органический слой сушат над Na2SO4 и концентрируют и получают соединение 1b (0,25 г), которое используют на следующей стадии без дополнительной очистки.
Стадия 3:

Соединение 1b (0,25 г, 0,37 ммоль, 1,0 экв.) растворяют в сухом МеОН (2,0 мл) и обрабатывают с помощью 20% Pd(OH)2 (60 мас.%) в инертной атмосфере. Реакционную смесь гидрируют при атмосферном давлении и за протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан). Реакция завершается за 45 мин и затем реакционную смесь фильтруют через целит (диатомовая земля), промывают с помощью ЕtOАс, концентрируют и получают неочищенный продукт. Очистку проводят с помощью препаративной тонкослойной хроматографии (60/40 ЕtOАс/гексан) и получают соединение 1 (0,10 г, 49%).
МС с электрораспылением [М+1] 554,3.
МСВР (ББА) рассчитано для С28Н29F6N3O2(М+1) 554,2242, найдено 554,2249.
Пример получения 2
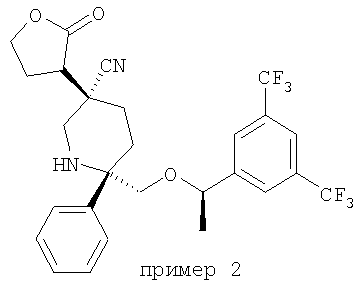
Стадия 1:

В круглодонной колбе объемом 25 мл соединение 42b (0,3264 г, 0,44 ммоль, 1,0 экв.) растворяют в 5 мл ТГФ и реакционную смесь охлаждают до 0°С в бане со льдом. После этого к реакционной смеси прибавляют Et3N (0,073 мл, 0,44 ммоль, 1,2 экв.), затем 2-хлорэтилхлорформиат (0,054 мл, 0,44 ммоль, 1,2 экв.) и ее медленно нагревают до комнатной температуры и перемешивают в течение 14 ч. За протеканием реакции следят с помощью ТСХ (40:60 ЕtOАс/гексан) и МС. Реакция не доходит до конца и поэтому реакционную смесь разбавляют с помощью ЕtOАс и реакцию останавливают насыщенным раствором NaHCO3, а затем рассолом. Органический слой сушат над Na2SO4 и концентрируют и получают (0,3 г) неочищенного продукта, который обрабатывают с помощью хроматографии на колонке BIOTAGE (40:60 ЕtOАс/гексан) и получают соединение 2а (0,125 г).
МС с электрораспылением [М+1] 712,4.
Стадия 2:

В высушенной на огне круглодонной колбе объемом 25 мл соединение 2а (0,125 г, 0,175 ммоль, 1,0 экв.) растворяют в сухом ТГФ. К этой реакционной смеси прибавляют 60% NaH (0,10 г, 0,26 ммоль, 1,5 экв.) и реакционную смесь перемешивают при комнатной температуре в течение ночи. За протеканием реакции следят с помощью ТСХ (40:60 ЕtOАс/гексан) и МС. После завершения реакции реакционную смесь разбавляют с помощью ЕtOАс и реакцию останавливают насыщенным водным раствором NаНСО3. Органический слой сушат над Na2SO4 и концентрируют и получают соединение 2b (0,11 г), которое используют на следующей стадии без дополнительной очистки. МС с электрораспылением [М+1] 676,2.
Стадия 3:

Соединение 2b (0,11 г, 0,16 ммоль, 1,0 экв.) растворяют в сухом МеОН (2,0 мл) и обрабатывают с помощью 20% Pd(OH)2 (60 мас.%) в инертной атмосфере. Реакционную смесь гидрируют при атмосферном давлении и за протеканием реакции следят с помощью ТСХ (40:60 ЕtOАс/гексан). Реакция завершается за 45 мин, реакционную смесь фильтруют через целит, промывают с помощью ЕtOАс и концентрируют и получают неочищенный продукт. Неочищенный продукт очищают с помощью препаративной тонкослойной хроматографии (45/55 ЕtOАс/гексан) и получают соединение 2 (0,04 г, 45%). МС с электрораспылением [М+1] 542,3.
МСВР (ББА) рассчитано для С26Н26FеN3О3(М+1) 542,1897, найдено 542,1878.
Примеры получения 3 и 4
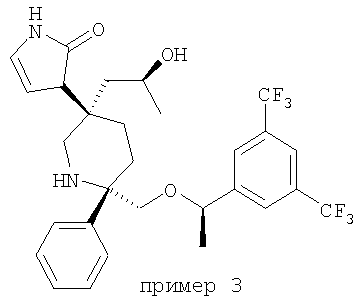

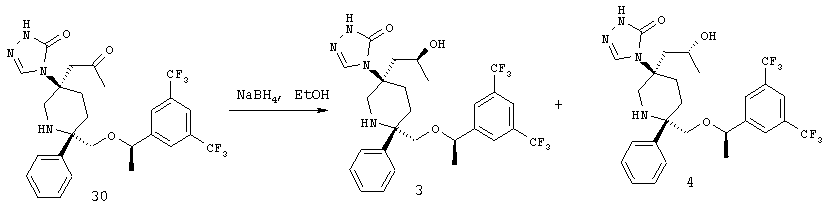
NaBH4 (60 мг, 1,53 ммоль, 8 экв.) при 0°С порциями прибавляют к раствору соединения 30 (109 мг, ~0,19 ммоль, 1 экв.) в абсолютном этаноле (2 мл). После перемешивания при 0°С в течение 30 мин анализ реакционной смеси с помощью ТСХ (MeOH/CH2Cl2=10%) обнаруживает только продукт. Продукт очищают с помощью хроматографии на колонке BIOTAGE (2-10% МеОН в СH2Сl2) и получают чистую смесь двух диастереоизомеров. Эти два диастереоизомера разделяют с помощью хиральной ВЭЖХ (ChialCel OD, IРА/гексан=10%) и получают соединение примера 3, МС [M+1]+ 573,1; и соединение примера 4, МС [М+1]+ 573,1.
Пример получения 5

Стадия А:

MsCl (0,102 мл, 1,32 ммоль) прибавляют к раствору соединения 26а (0,375 г, 0,528 ммоль) и Et3N (0,368 мл, 2,64 ммоль) в CH2Cl2 (5,0 мл) при 0°С. Реакцию останавливают водой (15,0 мл) через 30 мин и затем разбавляют с помощью CH2Cl2 (50 мл). Полученную водную фазу экстрагируют с помощью СН2Сl2 (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный мезилат растворяют в ДМФ (3,0 мл) и обрабатывают с помощью KCN (0,344 г, 5,28 ммоль). Полученную смесь нагревают при 100°С в течение 12 ч и затем ее охлаждают до комнатной температуры. Реакционную смесь разбавляют с помощью ЕtOАс (100 мл) и промывают водой (3×15 мл). Затем органический слой промывают рассолом (25 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=7/1) и получают соединение 5b (0,14 г, 37% за 2 стадии).
Стадия В:

Раствор соединения 5b (0,14 г, 0,195 ммоль) в ТФК (2,5 мл) перемешивают при комнатной температуре в течение 20 мин, а затем растворитель удаляют при пониженном давлении. Остаток растворяют в ЕtOАс (50 мл) и промывают раствором NaOH (4,0 н., 15 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (15 мл), рассолом (15 мл) и затем сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт пропускают через тонкий слой силикагеля с использованием ЕtOАс/МеОН (об./об.=10/1) в качестве элюента и после удаления растворителя получают амин (90 мг). Амин растворяют в пиридине (1,0 мл) и при комнатной температуре в герметичном сосуде обрабатывают с помощью HC(O)NHNHC(O)H (38,3 мг, 0,435 ммоль), TMCCl (0,276 мл, 2,175 ммоль) и Et3N (0,152 мл, 1,088 ммоль). Затем реакционную смесь нагревают при 100°С в течение 2,5 ч, а после этого охлаждают до комнатной температуры. Затем смесь разбавляют с помощью ЕtOАс (40 мл) и промывают с помощью HCl (10 мл, 2,0 н.). Полученную водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (15 мл), рассолом (25 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс/МеОН, об./об.=10/1) и получают соединение 5с (40 мг, 31% за 2 стадии).
Стадия С:
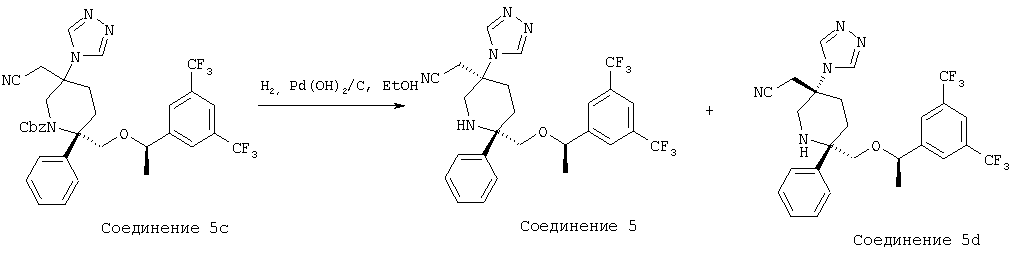
Соединение 5с (40 мг, 0,0595 ммоль) в ЕtOН (2,0 мл) при комнатной температуре обрабатывают с помощью Pd(OH)2/C (8 мг, 10 мас.%) и гидрируют в течение 30 мин с использованием баллона с Н2. Реакционную смесь фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью препаративной ТСХ (ЕtOАс/МеОН, об./об.=40/1) и получают соединение 5 (18 мг, 56%, МС с электрораспылением [М+1]+ 538,1) и соединение 5d (6 мг, 19%, МС с электрораспылением [М+1]+ 538,1).
Пример получения 6

Стадия А:

MsCl (75 мл, 0,969 ммоль) прибавляют к раствору соединения 23d (0,248 г, 0,388 ммоль) и Et3N (0,27 мл, 1,94 ммоль) в CH2Cl2 (3,0 мл) при комнатной температуре. Реакцию останавливают водой (10,0 мл) через 30 мин и разбавляют с помощью CH2Cl2 (30 мл). Водную фазу экстрагируют с помощью CH2Cl2 (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный мезилат растворяют в безводном ДМСО (3,0 мл) и обрабатывают с помощью NaBH4 (59,0 мг, 1,552 ммоль). Реакционную смесь нагревают при 85°С в течение 48 ч, а затем охлаждают до комнатной температуры. Затем смесь разбавляют с помощью ЕtOАс (50 мл) и промывают водным раствором HCl (10 мл, 1,0 М). Полученную водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (3×15 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=5/1) и получают соединение 6а (0,11 г, 45% за 2 стадии).
Стадия В:
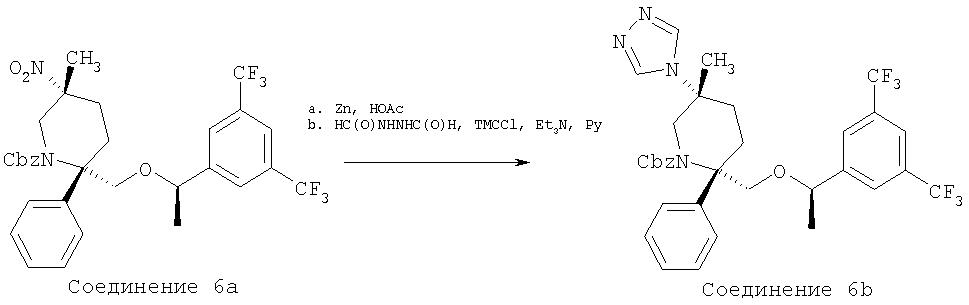
Смесь соединения 6а (0,11 г, 0,176 ммоль) и цинковой пыли (0,114 г, 1,76 ммоль) в НОАс (1,5 мл) нагревают при 60°С в течение 2 ч. Реакционную смесь охлаждают и фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и остаток растворяют в ЕtOАс (25 мл) и промывают раствором NaOH (4,0 н., 10 мл). Полученную водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (15 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный амин (67,1 мг, 0,113 ммоль) растворяют в пиридине (1,0 мл) и при комнатной температуре в герметичном сосуде обрабатывают с помощью HC(O)NHNHC(O)H (29,8 мг, 0,339 ммоль), TMCCl (0,214 мл, 1,69 ммоль) и Et3N (0,118 мл, 0,847 ммоль). Затем смесь нагревают при 100°С в течение 2,5 ч, а после этого охлаждают до комнатной температуры. Затем смесь разбавляют с помощью ЕtOАс (40 мл) и промывают с помощью HCl (10 мл, 2,0 н.). Полученную водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (15 мл), рассолом (15 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс/МеОН, об./об.=20/1) и получают соединение 6b (37 мг, 33% за 2 стадии).
Стадия С:

Соединение 6b (36,5 мг, 0,0565 ммоль) в ЕtOН (2,0 мл) обрабатывают при комнатной температуре посредством Pd(OH)2/C (7,3 мг, 10 мас.%) и гидрируют в течение 30 мин с использованием баллона с Н2. Реакционную смесь фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью препаративной ТСХ (EtOAc/MeOH/Et3N, об./об./об.=40/1/0,1) и получают соединение 6 (20 мг, 69%). МС с электрораспылением [М+1]+ 513,1.
Пример получения 7

Стадия А:

Перйодинан Десса-Мартина (0,114 г, 0,268 ммоль) прибавляют к смеси соединения 12а (70,5 мг, 0,107 ммоль) и NaHCO3 (0,112 г, 1,34 ммоль) в СH2Сl2 (3,0 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют, прибавляя ЕtOАс (30 мл) и воду (10 мл). Органическую фазу промывают насыщенным раствором Nа2S2O3 (3×10 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают раствором NaOH (10 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный альдегид (70,5 мг, 0,107 ммоль) растворяют в ЕtOН (3,0 мл) и обрабатывают с помощью HONH2·HCl (74,4 мг, 1,07 ммоль) и NaOAc (43,9 мг, 0,535 ммоль) при комнатной температуре. Реакционную смесь перемешивают в течение 12 ч и затем ее разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NaHCO3 (10 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования получают неочищенный оксим (63 мг, 0,093 ммоль), который растворяют в бензоле (2,0 мл) и обрабатывают 1,1'-оксалилдиимидазолом (35,4 мг, 0,186 ммоль). Реакционную смесь нагревают при 80°С в течение 3 ч, а затем охлаждают до комнатной температуры и разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором НСl (0,5 н., 5 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс) и получают соединение 7а (39 мг, 55% за 3 стадии).
Стадия В:

Соединение 7а (39 мг, 0,059 ммоль) в ЕtOН (2,5 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (7,8 мг, 10 мас.%) и гидрируют в течение 30 мин с использованием баллона с H2. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью препаративной ТСХ (ЕtOАс/Еt3М, об./об.=100/0,1) и получают соединение 7 (12,2 мг, 40%). МС с электрораспылением [M+]+ 524,3.
Пример получения 8

Стадия А:

Перйодинан Десса-Мартина (0,325 г, 0,767 ммоль) прибавляют к смеси соединения 12а (0,202 г, 0,306 ммоль) и NaHCO3 (0,322 г, 3,83 ммоль) в CH2Cl2 (5,0 мл) при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют с помощью ЕtOАс (50 мл) и воды (10 мл). Органическую фазу промывают насыщенным раствором Nа2S2O3 (3×15 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают раствором NaOH (15 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над МgSO4. После фильтрования и концентрирования неочищенный альдегид (0,202 г) растворяют в трет-бутаноле (4,0 мл) и воде (1,0 мл) и последовательно обрабатывают с помощью NaH2PO4·H2O (84,4 мг, 0,612 ммоль), NaClO2 (96,8 мг, 1,07 ммоль) и 2-метил-2-бутена (0,227 мл, 2,14 ммоль). Реакционную смесь перемешивают в течение 2 ч и затем ее разбавляют с помощью ЕtOАс (30 мл) и промывают водным раствором NH4Cl. Полученную водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенную кислоту растворяют в бензоле (4,0 мл) и МеОН (1,0 мл). Полученный раствор обрабатывают с помощью TMCCHN2 (0,306 мл, 0,612 ммоль) при комнатной температуре и перемешивают в течение 20 мин. Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 5/1 до 1/3) и получают соединение 8а (62 мг, 29% за 3 стадии).
Стадия В:

Соединение 8а (62 мг, 0,090 ммоль) в ЕtOН (3,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (12,4 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с H2. Реакционную смесь фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс/МеОН, об./об.=6/1) и получают соединение 8 (42 мг, 84%). МС с электрораспылением [M+]+ 557,3.
Пример получения 9

Стадия 1:

В круглодонной колбе объемом 25 мл соединение 42b (0,21 г, 0,35 ммоль, 1,0 экв.) растворяют в 2 мл толуола. После этого к реакционной смеси прибавляют 3-хлорпропионилхлорид (0,037 мл, 0,38 ммоль, 1,1 экв.) и ее перемешивают при комнатной температуре в течение 5 ч. За протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан) и МС, которые показывают, что все еще содержится некоторое количество исходного вещества. Затем реакционную смесь нагревают при 80°С. После завершения реакции в результате нагревания в течение еще нескольких часов смесь концентрируют и получают неочищенное соединение 9а (0,2 г), которое используют на следующей стадии без дополнительной очистки.
Стадия 2:

В высушенной на огне круглодонной колбе объемом 25 мл соединение 9а (0,2 г, 0,287 ммоль, 1,0 экв.) растворяют в 0,5 растворе сухой смеси СН2Сl2/ДМФ, соотношение (4/1) (4,59 мл/1,15 мл). К этой смеси с помощью шприцевого насоса в течение 3,5 ч очень медленно прибавляют 0,5 М раствор 60% NaH (0,012 г, 0,316 ммоль, 1,1 экв.) в сухой смеси СН2Сl2/ДМФ (соотношение 4/1; 5,06 мл/1,26 мл) и реакционную смесь перемешивают при комнатной температуре в течение ночи. За протеканием реакции следят с помощью ТСХ (40:60 ЕtOАс/гексан) и МС. Реакция протекает на 60% и затем реакционную смесь разбавляют с помощью СН2Сl2 и реакцию останавливают насыщенным водным раствором NH4Cl. Органический слой сушат над Na2SO4 и концентрируют и получают неочищенный продукт (0,18 г), который очищают с помощью хроматографии на колонке BIOTAGE (30/70 ЕtOАс/гексан) и получают соединение 9b (0,125 г).
МС с электрораспылением [М+1] 660,2.
Стадия 3:

Соединение 9b (0,125 г, 0,189 ммоль, 1,0 экв.) растворяют в сухом МеОН (1,0 мл) и обрабатывают с помощью 20% Pd(OH)2 (60 мас.%) в инертной атмосфере. Реакционную смесь гидрируют при атмосферном давлении и за протеканием реакции следят с помощью ТСХ (60:40 EtOAc/гексан). Реакция завершается за 20 мин и реакционную смесь фильтруют через целит, промывают с помощью ЕtOАс и концентрируют и получают неочищенный продукт. Очистку проводят с помощью препаративной тонкослойной хроматографии (45/55 ЕtOАс/гексан) и получают соединение 9 (0,071 г, 71%).
МС с электрораспылением [М+1] 526,3.
МСВР (ББА) рассчитано для C26H26F6N3O2(M+1) 526,1932, найдено 526,1929.
Пример получения 10

Стадия А:

Раствор соединения 8 (35 мг, 0,063 ммоль) в метанольном растворе аммиака (3,0 мл, 7,0 М) в сосуде высокого давления Парра нагревают при 80°С в течение 5 дней. Систему охлаждают до комнатной температуры и растворитель удаляют при пониженном давлении. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс/МеОН, об./об.=10/1) и получают соединение 10 (26,8 мг, 79%).
МС с электрораспылением [М+1]+ 542,1.
Пример получения 11

Стадия 1:

Раствор метилмагнийбромида в трет-бутиловом эфире (0,42 мл, 1,0 М, 0,42 ммоль, 6,2 экв.) шприцем прибавляют к раствору соединения 30b (48 мг, 0,068 ммоль, 1,0 экв.) в безводном ТГФ (1 мл) при 0°С. Затем реакционную смесь нагревают до комнатной температуры. После того, как ТСХ (элюент -ЕtOАс) показывает, что реакция завершилась, реакционную смесь разбавляют эфиром и промывают насыщенным водным раствором NH4Cl. Объединенные органические слои сушат над МgSО4, фильтруют и концентрируют и получают неочищенный продукт, соединение 11а, которое используют на следующей стадии без очистки.
Стадия 2:

По такой же методике, что и описанная в примере 31, стадия 6, неочищенное соединение 11а гидрируют и получают чистое соединение примера 30b (выход 52,6% из соединения 11). МС [M+]+ 587,1.
Пример получения 12

Стадия А:

HC(O)NHNHC(O)H (0,28 г, 3,18 ммоль), TMCCl (2,0 мл, 15,9 ммоль) и Et3N (1,1 мл, 7,95 ммоль) при комнатной температуре в герметичном сосуде последовательно прибавляют к раствору соединения 23d (0,647 г, 1,06 ммоль) в пиридине (5,0 мл). Затем смесь нагревают при 100°С в течение 2,5 ч, а после этого охлаждают до комнатной температуры. Затем смесь разбавляют с помощью ЕtOАс (100 мл) и промывают с помощью HCl (35 мл, 2,0 н.). Водную фазу экстрагируют с помощью ЕtOАс (3×25 мл) и объединенные органические слои промывают водой (15 мл), рассолом (25 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс/МеОН, об./об.=5/1) и получают соединение 12а (0,48 г, 68%).
Стадия b:

Соединение 12а (32,6 мг, 0,049 ммоль) в ЕtOН (2,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (6,5 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с H2. Затем реакционную смесь фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (элюент -ЕtOАс/МеОН, об./об.=6/1) и получают соединение 12 (17,2 мг, 66%).
МС с электрораспылением [М+1]+ 529,1.
Примеры получения 13 и 14

Стадия А:

HC(O)NHNHC(O)H (67,1 мг, 0,762 ммоль), TMCCl (0,484 мл, 3,81 ммоль) и Et3N (0,266 мл, 1,905 ммоль) при комнатной температуре в герметичном сосуде последовательно прибавляют к раствору соединения 26а (0,155 г, 0,254 ммоль) в пиридине (2,0 мл). Затем смесь нагревают при 100°С в течение 2,5 ч, а после этого охлаждают до комнатной температуры. Смесь разбавляют с помощью ЕtOАс (40 мл) и промывают с помощью HCl (15 мл, 2,0 н.). Водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (15 мл), рассолом (25 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (элюент - ЕtOАс/МеОН, об./об.=10/1) и получают соединение 14а (0,129 г, 75%).
Стадия В:

Соединение 14а (129 мг, 0,19 ммоль) в ЕtOН (4,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (25,8 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с H2. Реакционную смесь фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью препаративной ТСХ (EtOAc/Et3N, об./об.=100/0,1) и получают соединение 13 (36 мг, 35%, МС с электрораспылением [М+1]+ 543,1) и соединение 14 (30 мг, 29%, МС с электрораспылением [M+1]+ 543,1).
Пример получения 15

Стадия А:
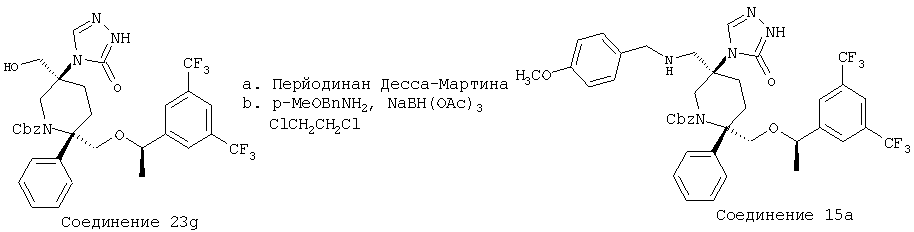
Перйодинан Десса-Мартина (57,7 мг, 0,136 ммоль) при комнатной температуре прибавляют к смеси соединения 23g (46 мг, 0,0678 ммоль) и NaHCO3 (57 мг, 0,678 ммоль) в CH2Cl2 (2,5 мл). Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют с помощью ЕtOАс (20 мл) и воды (10 мл). Органическую фазу промывают насыщенным раствором Na2S2C3 (3×10 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают раствором NaOH (10 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над МgSO4. После фильтрования и концентрирования неочищенный альдегид (46 мг, 0,0679 ммоль) растворяют в ClCH2CH2Cl (1,0 мл) и обрабатывают молекулярными ситами 4Å (15 мг) и пара-метоксибензиламином (26,7 мкл, 0,204 ммоль), а затем прибавляют NаВН(ОАс)3 (86,4 мг, 0,408 ммоль). Полученную реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Затем систему разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NaHCO3 (10 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=2/3) и получают соединение 15а (38 мг, 70% за 2 стадии).
Стадия В:

Смесь соединения 15а (46,6 мг, 0,0584 ммоль), Pd/C (46,6 мг, 10 мас.%) и NH4CO2H (36,8 мг, 0,584 ммоль) в МеОН (2,0 мл) нагревают с обратным холодильником в течение 5 ч. Смесь охлаждают до комнатной температуры и фильтруют через тонкий слой целита, и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и получают неочищенный продукт, который растворяют в ЕtOАс (20 мл) и промывают водным раствором NаНСО3 (10 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (МеОН/ЕtOАс, об./об.=1/10) и получают соединение 15b (18 мг, 57%).
Стадия С:

MsCl (2,5 мкл, 0,0324 ммоль) при 0°С прибавляют к раствору соединения 15b (8,8 мг, 0,0162 ммоль) и Et3N (5,4 мкл, 0,0388 ммоль) в СН2Сl2 (1,0 мл). Реакцию останавливают водой (5,0 мл) через 30 мин и разбавляют с помощью ЕtOАс (15 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=1/5) и получают соединение 15 (7,2 мг, 72%).
МС с электрораспылением [М+1]+ 622,3.
Пример получения 16

Стадия 1:

По такой же методике, что и описанная в примере 30, стадия 1, соединение 16а получают с использованием этиламина вместо N,N-диметиламингидрохлорида и без использования диизопропилэтиламина. Неочищенный продукт используют на следующей стадии без очистки.
Стадия 2:

По такой же методике, что и описанная в примере 31, стадия 6, неочищенное соединение 16а гидрируют и получают чистое соединение примера 16 (выход 70,5% из соединения 16). МС [M+1]+ 600,1.
Пример получения 17

Стадия 1:

Раствор соединения 31h (46,3 мг, 0,066 ммоль, 1,0 экв.) в безводном дихлорметане (1 мл) охлаждают до 0°С. К этому раствору последовательно прибавляют ДМАП (8 мг, 0,066 ммоль, 1,0 экв.) и этанол (36 мкл). Реакционной смеси дают нагреться до комнатной температуры и затем ее концентрируют досуха. Остаток растворяют в ЕtOАс и промывают насыщенным водным раствором NaHCO3. Органический слой сушат над Na2SO4, фильтруют и концентрируют и получают неочищенный продукт, соединение 17а, которое используют на следующей стадии без очистки.
Стадия 2:

По такой же методике, что и описанная в примере 31, стадия 6, неочищенное соединение 17а гидрируют и получают чистое соединение 17 (выход 46% из соединения 31h). MC [M+1]+ 601,1.
Пример получения 18

Стадия А:

Соединение 19 (10 мг, 0,0175 ммоль) в ЕtOН (1,5 мл) при комнатной температуре обрабатывают с помощью HONH2·HCl (12,2 мг, 0,175 ммоль) и NaOAc (7,2 мг, 0,0876 ммоль). Затем реакционную смесь перемешивают при 60°С в течение 12 ч. Смесь разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NаНСО3. Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=2/3) и получают соединение 18 (10 мг, 98%). МС с электрораспылением [M+1]+ 586,1.
Пример получения 19
Стадия А:

Перйодинан Десса-Мартина (0,252 г, 0,595 ммоль) при комнатной температуре прибавляют к смеси соединения 23h (0,202 г, 0,297 ммоль) и NаНСО3 (0,25 г, 2,97 ммоль) в CH2Cl2 (4,0 мл). Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют с помощью ЕtOАс (50 мл) и воды (10 мл). Органическую фазу промывают насыщенным раствором Na2S2O3 (3×15 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают раствором NaOH (15 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный альдегид (0,202 г) растворяют в безводном ТГФ (4,0 мл) и обрабатывают с помощью СН3MgВr (1,19 мл, 1,19 ммоль, 1,0 М в ТГФ) при -78°С. Температуру реакции медленно повышают до комнатной и реакцию останавливают через 2 ч путем медленного прибавления насыщенного водного раствора NH4Cl (10 мл). Затем реакционную смесь разбавляют с помощью ЕtOАс (50 мл) и нейтрализуют с помощью 0,5 н. HCl, пока водная фаза не станет слабокислой. Водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный вторичный спирт (0,21 г) растворяют в CH2Cl2 (5,0 мл) и обрабатывают перйодинаном Десса-Мартина (0,379 г, 0,894 ммоль) и NаНСО3 (0,375 г, 4,47 ммоль) при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют с помощью ЕtOАс (50 мл) и воды (10 мл). Органическую фазу промывают насыщенным раствором Na2S2O3 (3×15 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водным раствором NaOH (15 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/1) и получают соединение 19а (90 мг, 43% за 3 стадии).
Стадия В:

Соединение 19а (57,4 мг, 0,0816 ммоль) в ЕtOН (3,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (11,5 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с Н2. Реакционную смесь фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=2/3) и получают соединение 19 (41 мг, 88%).
МС с электрораспылением [М+1]+ 571,1.
Пример получения 20

Стадия А:
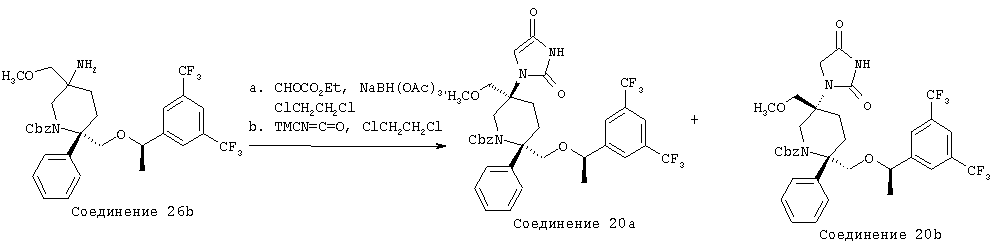
NаВН(ОАс)3 (81,4 мг, 0,384 ммоль) прибавляют при комнатной температуре к раствору соединения 26b (79,9 мг, 0,128 ммоль), CHOCO2Et (37,8 мкл, 0,192 ммоль, 45-50% в толуоле) и 4 Å молекулярным ситам (30 мг) в ClCH2CH2Cl (1,0 мл). Реакционную смесь перемешивают в течение 12 ч и затем ее разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NaHCO3 (10 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт (91 мг, 0,128 ммоль) растворяют в ClCH2CH2Cl (0,5 мл) и обрабатывают с помощью TMCN=C=O (2,5 мл). Реакционную смесь нагревают при 70°С в течение 72 ч, а затем растворитель удаляют при пониженном давлении. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/1) и получают смесь соединения 20а и 20b, которую дополнительно очищают с помощью хиральной ВЭЖХ на колонке OD и получают чистое соединение 20а (30 мг, 33%) и соединение 20b (25 мг, 28%).
Стадия В:

Соединение 20а (23 мг, 0,0325 ммоль) в ЕtOН (2,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (4,6 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с Н2. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOH/Н (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 1/3 до 1/9) и получают соединение 20 (14,3 мг, 77%).
МС с электрораспылением [М+1]+ 574,3.
Примеры получения 21 и 22


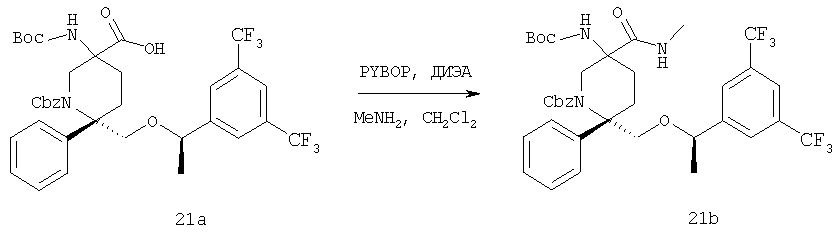
Соединение 21а (1,0 г, 1,4 ммоль, 1,0 экв.) растворяют в CH2Cl2 (16 мл) и раствор охлаждают до 0°С. К реакционной смеси прибавляют диизопропиламин (0,54 г, 4,2 ммоль, 3,0 экв.), затем PYBOP (0,88 г, 1,7 ммоль, 1,2 экв.) и реакционную смесь перемешивают при 0°С в течение 5 мин, затем нагревают до комнатной температуры. Через 20 мин избыток метиламина (7,0 мл, 14 ммоль, 10,0 экв.) прибавляют в виде 2,0 М раствора в ТГФ. Колба немного нагревается и содержимое перемешивают при комнатной температуре в течение ночи. За протеканием реакции следят с помощью ТСХ (элюент - 95/5 ЕtOАс/МеОН). После завершения реакции реакционную смесь разбавляют с помощью Н2O и ЕtOАс, органический и водный слои разделяют и органический слой промывают рассолом, сушат над Na2SO4 и концентрируют и получают неочищенный продукт (1,9 г) в виде белого твердого вещества. Очистку проводят с помощью хроматографии на колонке BIOTAGE (от 1:1 до 2:1 ЕtOАс/гексан) и получают соединение 21b в виде белого твердого вещества (0,72 г, 72%).
МС с электрораспылением [М+1]+ 738,2.

Соединение 21 с (0,7 г, 0,95 ммоль, 1,0 экв.) растворяют в СН2Сl2 (10 мл) в атмосфере N2. К реакционной смеси прибавляют избыток ТФК (2,0 г, 19,4 ммоль, 20,0 экв.) и реакционную смесь перемешивают при комнатной температуре в течение ночи. За протеканием реакции следят с помощью ТСХ (элюент -1/1 ЕtOАс/МеОН), которая показывает, что все еще содержится некоторое количество исходного вещества. Поэтому прибавляют 10,0 экв. ТФК и реакционную смесь перемешивают в течение 3 ч. После завершения реакции реакционную смесь охлаждают до 0°С, реакцию останавливают насыщенным раствором NаНСО3 и разбавляют с помощью ЕtOАс. Органический и водный слои разделяют и органический слой промывают рассолом, сушат над Na2SO4 и концентрируют и получают соединение 21d (0,6 г, 99%) в виде белого вспененного вещества.

Соединение 21с (0,24 г, 0,38 ммоль, 1,0 экв.) растворяют в 5 мл безводного ТГФ в атмосфере азота. Раствор охлаждают до 0°С. В отдельной круглодонной колбе объединяют карбонилдиимидазол (КДИ) (0,15 г, 0,90 ммоль, 2,4 экв.) и трет-бутилкарбазат (0,1 г, 0,76 ммоль, 2,0 экв.) в безводном ТГФ (2 мл). Раствор перемешивают в течение 30 мин и в течение 1 мин прибавляют через канюлю к раствору соединения 21 с. Канюлю промывают безводным ТГФ (1×0,8 мл). Реакционную смесь нагревают с обратным холодильником до израсходования исходного вещества. Затем реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме и получают бесцветное вспененное вещество. Неочищенную смесь очищают с помощью хроматографии на колонке BIOTAGE (2%-5% MeOH/CH2Cl2) и получают соединение 21d (0,22 г, 74%) в виде белого твердого вещества.

Соединение 21d (0,22 г, 0,28 ммоль, 1 экв.) растворяют в 15 мл безводного CH2Cl2 в атмосфере азота. Раствор охлаждают до 0°С. Прибавляют HCl (1,4 мл, 5,6 ммоль, 20 экв., 4 М раствор в диоксане) и раствору дают нагреться до комнатной температуры и его перемешивают в течение ночи. Раствор охлаждают до 0°С и реакцию останавливают насыщенным раствором NаНСО3 (5 мл) и разбавляют с помощью ЕtOАс. Органический и водный слои разделяют и органический слой промывают рассолом (10 мл) и сушат над Na2SO4. Органический слой фильтруют и концентрируют в вакууме и получают белое твердое вещество. Неочищенную смесь очищают с помощью хроматографии на колонке BIOTAGE (5%-8% МеОН/СН3Сl2) и получают соединение 21е (0,15 г, 79%) в виде белого твердого вещества.

Соединение 21e (0,15 г, 0,22 ммоль, 1,0 экв.) растворяют в безводном ДМФ (1 мл). Прибавляют формамидинацетат (0,126 г, 1,2 ммоль, 5,5 экв.), затем уксусную кислоту (0,69 мл, 1,2 ммоль, 5,5 экв.) и реакционную смесь нагревают при 80°С в течение 30 мин. Анализ с помощью ТСХ показывает наличие остатка исходного вещества и реакционную смесь нагревают с обратным холодильником в течение еще 6 ч. За протеканием реакции следят с помощью ТСХ (элюент - 9/1 СН3Сl2/МеОН). После завершения реакции реакционную смесь охлаждают до комнатной температуры, реакцию останавливают с помощью Н2O и разбавляют с помощью ЕtOАс. Органический и водный слои разделяют и органический слой промывают рассолом, сушат над Na2SO4 и концентрируют и получают неочищенный продукт (0,131 г) в виде белого вспененного вещества. Очистку проводят с помощью хроматографии на колонке BIOTAGE (градиентный режим от 100% CH2Cl2 до (95:5) МеОН) и получают соединение 21f в виде белого твердого вещества (0,11 г, 72%).
МС с электрораспылением [М+1] 706,4.
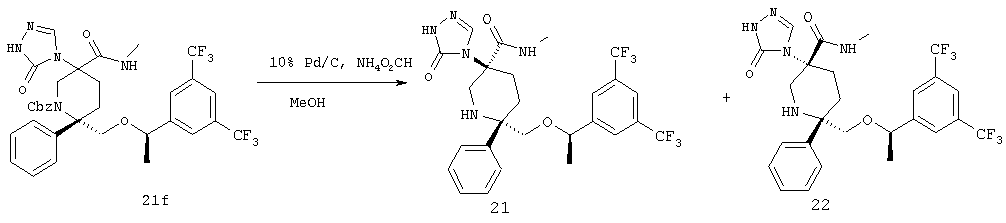
Соединение 21f (0,02 г, 0,028 ммоль, 1,0 экв.) растворяют в сухом МеОН (1,0 мл) и обрабатывают с помощью 10% Pd/C (40 мас.%), затем формиатом аммония (0,09 г, 0,14 ммоль, 5,0 экв.) в инертной атмосфере. Реакционную смесь нагревают с обратным холодильником и за протеканием реакции следят с помощью ТСХ (элюент - 9/1 CH2Cl2/MeOH). Реакция завершается за 1 ч. Реакционную смесь фильтруют через целит, промывают с помощью ЕtOАс и концентрируют в вакууме. Полученный остаток растворяют в ЕtOАс и промывают насыщенным раствором NaHCO3, а затем рассолом и H2O и получают неочищенный продукт (0,019 г) в виде твердой пленки. Очистку проводят с помощью хроматографии на колонке BIOTAGE (градиентный режим от 2% до 6% МеОН/СН3Сl2. Деаэрированный продукт превращают в соль с HCl и получают смесь соединений 21 и 22 (0,014 г) в виде белого твердого вещества. МСВР (ББА) рассчитано для С26Н28F6N3O2 (М+1) 572,2096, найдено 572,2103.
Пример получения 23
Стадия А:

NaBH4 (2,42 г, 64,1 ммоль) 4 порциями прибавляют к раствору соединения 23а в МеОН (160 мл) при 0°С. Реакционную смесь перемешивают в течение 4 ч и температура реакции медленно повышается до КГ. Реакцию останавливают путем медленного прибавления насыщенного водного раствора NH4Cl (50 мл). Затем реакционную смесь разбавляют с помощью ЕtOАс (400 мл) и нейтрализуют с помощью 0,5 н. HCl, пока водная фаза не станет слабокислой. Водную фазу экстрагируют с помощью ЕtOАс (3×100 мл). Объединенные органические слои промывают водой (100 мл), рассолом (100 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт пропускают через тонкий слой силикагеля (гексан/ЕtOАс, об./об.=7/1). Растворитель удаляют при пониженном давлении и получают соединение 23b, 17,4 г (89%) в виде светло-желтого сиропа.
Стадия В:

ТБАФ (2,23 мл, 2,23 ммоль, 1,0 М в ТГФ) по каплям прибавляют к смеси соединения 23b (9,1 г, 14,89 ммоль) с параформальдегидом (3,85 г) в ТГФ (100 мл) при 0°С. Реакционную смесь перемешивают при 0°С в течение 8 ч и затем реакцию останавливают путем прибавления насыщенного водного раствора NH4Cl (50 мл). Затем реакционную смесь разбавляют с помощью ЕtOАс (250 мл) и водную фазу экстрагируют с помощью ЕtOАс (3×50 мл). Объединенные органические слои промывают водой (50 мл), рассолом (100 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают на колонке BIOTAGE (CH2Cl2/EtOAc, об./об.=100/0,5) и получают соединение 23с (6,0 г, 63%) и 23d (2,34 г, 24%).
Стадия С:

Смесь соединения 23 с (7,54 г, 11,76 ммоль) и цинковой пыли (7,68 г, 117,6 ммоль) в НОАс (120 мл) нагревают при 60°С в течение 2 ч. Реакционную смесь охлаждают и фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (50 мл). Растворитель удаляют при пониженном давлении и остаток растворяют в ЕtOАс (250 мл) и промывают раствором NaOH (50 мл, 4,0 н.). Водную фазу экстрагируют с помощью ЕtOАс (3×50 мл). Объединенные органические слои промывают водой (50 мл), рассолом (100 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/3 и ЕtOАс/МеОН, об./об.=10/1) и получают соединение 23е (6,4 г, 89%).
Стадия D:

BnBr (0,668 мл, 5,58 ммоль) при КГ при энергичном перемешивании прибавляют к смеси соединения 23е (3,1 г, 5,07 ммоль) и BU4NHSO4 (0,334 г, 1,014 ммоль) в ТГФ (20 мл) и водного раствора NaOH (20 мл, 50 мас.%). Реакционную смесь перемешивают при комнатной температуре в течение 12 ч и затем ее разбавляют с помощью ЕtOАс (250 мл) и промывают водой (100 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×50 мл). Объединенные органические слои промывают водой (50 мл), рассолом (100 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 1/3 до 1/7) и получают соединение 23f (2,8 г, 79%).
Стадия Е:
Раствор соединения 23f (2,72 г, 3,88 ммоль) и реагента 31с (т.е. метилового эфира N-этоксиметиленгидразинкарбоновой кислоты) (2,83 г, 19,4 ммоль) в ЕtOН (15 мл) нагревают при 60°С в течение 18 ч. Реакционную смесь разбавляют с помощью МеОН (15 мл) и затем обрабатывают с помощью NаОСН3 (7,0 мл, 38,8 ммоль, 30% в МеОН). Полученную реакционную смесь нагревают при 80°С в течение 4 ч и затем ее охлаждают до комнатной температуры. Реакционную смесь разбавляют с помощью ЕtOАс (200 мл) и водного раствора NH4Cl (75 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×50 мл). Объединенные органические слои промывают водой (50 мл), рассолом (100 мл) и сушат над МgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 2,5/1 до 1/1) и получают соединение 23g (2,54 г, 85%).
Стадия F:
ВСl3 (3,26 мл, 3,26 ммоль, 1,0 М в гексане) при перемешивании по каплям прибавляют к раствору соединения 23g (0,502 г, 0,653 ммоль) в СН3Сl2 (45 мл) при
-78°С. Реакцию останавливают через 1 ч путем прибавления водного раствора NаНСО3 (50 мл) при -78°С. Смесь разбавляют с помощью ЕtOАс (100 мл) и энергично перемешивают при комнатной температуре в течение 2 ч. Водную фазу экстрагируют с помощью ЕtOАс (3×30 мл). Объединенные органические слои промывают водой (50 мл), рассолом (50 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 1/3 до 1/9) и получают соединение 23h (0,39 г, 91%).
Стадия G:
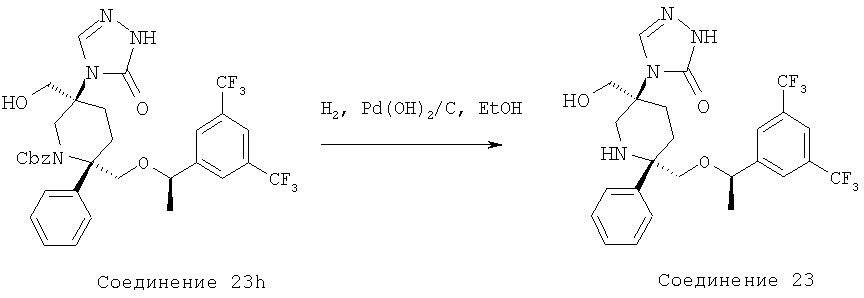
Соединение 23h (100 мг, 0,152 ммоль) в ЕtOН (5,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (20 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с Н2. Реакционную смесь фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/7) и получают соединение 23 (68 мг, 82%).
МС с электрораспылением [М+1]+ 545,1.
Пример получения 24


Раствор соединения 31g (83,3 мг, 0,12 ммоль, 1,0 экв.) в безводном дихлорметане (4 мл) охлаждают до -78°С. Затем О3 пропускают через раствор, пока он не станет синим. Затем раствор продувают с помощью N2 для удаления избытка О3 и реакционную смесь концентрируют досуха. Затем полученный остаток растворяют в этаноле (2 мл) и обрабатывают борогидридом натрия (46 мг, 1,2 ммоль, 10 экв.). Реакционную смесь перемешивают при комнатной температуре, пока ТСХ (50% ЕtOАс/гексаны) не покажет, что исходное вещество полностью израсходовано. Затем реакционную смесь концентрируют досуха. Остаток растворяют в абсолютном этаноле (4 мл) и обрабатывают с помощью Pd(OH)2/C (80 мг, 20 мас.%, 0,11 ммоль, 0,88 экв.), а затем гидрируют водородом из баллона. Реакционную смесь перемешивают при комнатной температуре, пока ТСХ (5% MeOH/CH2Cl2) не покажет, что исходное вещество полностью израсходовано. Реакционную смесь повторно концентрируют досуха. Остаток растворяют в этилацетате, промывают насыщенным водным раствором бикарбоната натрия и водный и органический слои разделяют. Водный слой дополнительно экстрагируют этилацетатом. Объединенные органические слои сушат над безводным Na2SO4, фильтруют, концентрируют и получают неочищенный продукт, который очищают с помощью препаративной ТСХ (MeOH/CH2Cl2=5%) и получают чистое соединение 24 (42 мг, выход 63%). МС [M+1]+ 559,1.
Пример получения 25

К раствору соединения 31g (83,3 мг, 0,12 ммоль, 1,0 экв.) в абсолютном этаноле (3 мл) прибавляют Pd(OH)2/C (20 мг, 20 мас.%, 0,028 ммоль, 0,88 экв.), а затем гидрируют водородом из баллона. Реакционную смесь перемешивают при комнатной температуре, пока ТСХ (50% ЕtOАс/гексаны) не покажет, что исходное вещество полностью израсходовано. Затем реакционную смесь концентрируют досуха. Полученный остаток растворяют в этилацетате, промывают насыщенным водным раствором бикарбоната натрия и водный и органический слои разделяют. Водный слой дополнительно экстрагируют этилацетатом. Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют и получают неочищенный продукт, который очищают с помощью препаративной ТСХ (50% ЕtOАс/гексаны) и получают чистое соединение 25 (15 мг, выход 84%). МС [М+1]+ 557,1.
Пример получения 26

Стадия А:

Et3N (0,129 мл, 0,93 ммоль) прибавляют к раствору соединения 26а (0,472 г, 0,77 ммоль) и Вос2O (0,168 г, 0,77 ммоль) в диоксане (3,0 мл) при комнатной температуре. Полученный раствор перемешивают в течение 8 ч и затем его разбавляют с помощью ЕtOАс (50 мл). Органическую фазу промывают с помощью 0,5 н. HCl (10 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (15 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/1) и получают соединение 26b (0,465 г, 85%).
Стадия В:

СН3I (0,372 мл, 5,98 ммоль) при КТ при энергичном перемешивании прибавляют к смеси соединения 26b (0,425 г, 0,598 ммоль) и BU4NHSO4 (40,6 мг, 0,12 ммоль) в ТГФ (5,0 мл) и водного раствора NaOH (5,0 мл, 50 мас.%). Реакционную смесь перемешивают при комнатной температуре в течение 12 ч и затем ее разбавляют с помощью ЕtOАс (50 мл) и промывают водой (15 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (15 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=5/1) и получают соединение 26с (0,345 г, 80%).
Стадия С:

Раствор соединения 26с (0,345 г, 0,476 ммоль) в ТФК (3,0 мл) перемешивают при комнатной температуре в течение 20 мин, а затем растворитель удаляют при пониженном давлении. Остаток растворяют в ЕtOАс (50 мл) и промывают раствором NaOH (4,0 н., 15 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (15 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный амин (0,29 г, 0,464 ммоль) растворяют в ЕtOН (3,0 мл) и обрабатывают реагентом 31с (0,4,6 г, 2,78 ммоль). Полученный раствор нагревают при 60°С в течение 18 ч. Реакционную смесь разбавляют с помощью МеОН (3,0 мл) и затем обрабатывают с помощью NaOCH3 (0,672 мл, 3,712 ммоль, 30% в МеОН). Полученную реакционную смесь нагревают при 80°С в течение 4 ч и затем ее охлаждают до комнатной температуры. Систему разбавляют путем прибавления ЕtOАс (50 мл) и водного раствора NН4Сl (15 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают водой (15 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/3) и получают смесь соединений 26d и 26е (0,275 г, 83% за 3 стадии), которую разделяют с помощью хиральной ВЭЖХ на колонке OD (гексан/изопропанол об./об.=95/5) и получают чистые соединения 26d и 26е.
Стадия D:

Соединение 26d (38 мг, 0,0548 ммоль) в ЕtOН (3,0 мл) при комнатной температуре обрабатывают с помощью Pd(OH)2/C (7,6 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с Н2. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/4) и получают соединение 26 (25 мг, 82%).
МС с электрораспылением [М+1]+ 559,1.
Пример получения 27

Стадия А:
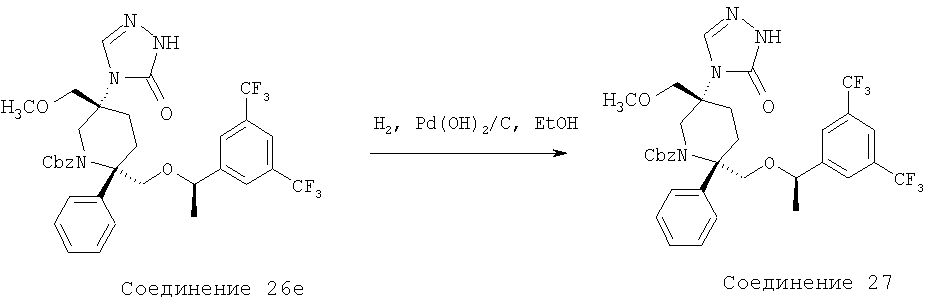
Соединение 26е (41 мг, 0,0592 ммоль) в ЕtOН (3,0 мл) при комнатной температуре обрабатывают с помощью Pd(OH)2/C (8,2 мг, 10 мас.%) и в течение 30 мин гидрируют с использованием баллона с Н2. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/4) и получают соединение 27 (26 мг, 79%).
МС с электрораспылением [М+1]+ 559,1.
Пример получения 28

Стадия А:

В круглодонной колбе объемом 25 мл соединение 44b (0,2 г, 0,45 ммоль, 1,0 экв.) растворяют в ДМФ (5,0 мл). Прибавляют HATU (0,342 г, 0,90 ммоль, 2,0 экв.), ЭДХ (0,172 г, 0,90 ммоль, 2,0 экв.) и ДИЭА (0,118 мл, 0,68 ммоль, 1,5 экв.). Реакционную смесь охлаждают до 0°С и прибавляют Вос-α-метилаланин (0,109 г, 0,54 ммоль, 1,2 экв.). Реакционную смесь перемешивают в течение ночи. Затем реакцию останавливают насыщенным раствором NaHCО3 (5 мл), реакционную смесь разбавляют с помощью ЕtOАс (10 мл) и экстрагируют с помощью ЕtOАс (2×5 мл). Органический слой промывают рассолом (10 мл), сушат над MgSO4 и концентрируют. Полученный остаток очищают с помощью препаративной ТСХ (9/1 гексаны/ЕtOАс) и получают 0,12 г (43%) соединения 28а.
Стадия В:

Соединение 28b по методике, сходной с методикой получения соединения 45с, описанной ниже, в которой для удаления защитной группы Воc раствор соединения 28а в ДХМ вводят в реакцию с ТФК.
Стадия С:
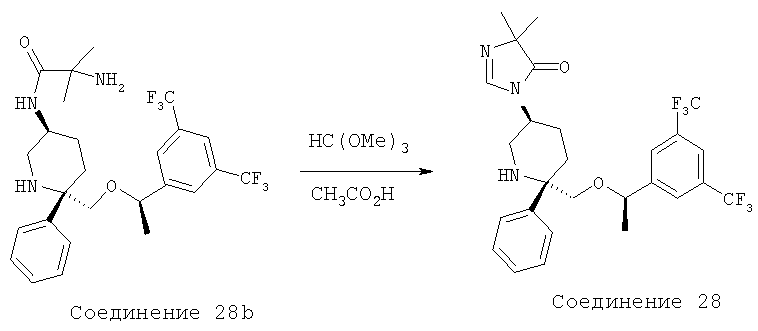
В круглодонной колбе объемом 10 мл соединение 28b (0,050 г, 0,094 ммоль, 1,0 экв.) растворяют в толуоле (1 мл) и затем прибавляют триметилортоформиат (0,012 мл, 0,113 ммоль, 1,2 экв.) и 1 каплю уксусной кислоты. Раствор нагревают при 60°С. Реакционную смесь перемешивают в течение в течение 48 ч. Затем реакционную смесь растворяют в ЕtOАс (5 мл) и промывают насыщенным раствором NаНСО3 (5 мл). Органический слой промывают рассолом (5 мл), сушат над MgSO4 и концентрируют. Неочищенный продукт очищают с помощью препаративной ТСХ (ЕtOАс) и получают 0,010 г соединения 28. МСВР рассчитано для С27Н29F6N3O2 (М+Н) 542,2242, найдено 542,2222.
Пример получения 29


Раствор соединения 31h (39 мг, 0,055 ммоль, 1,0 экв.) в безводном дихлорметане (1 мл) охлаждают до -20°С. Затем прибавляют триэтиламин (10 мл, 0,069 ммоль, 1,25 экв.) и этилхлорформиат (6,5 мл, 0,066 ммоль, 1,2 экв.). Полученный бледно-зеленый раствор перемешивают при -15°С в течение 30 мин. Через раствор в течение 20 мин пропускают газообразный аммиак. ТСХ (ЕtOАс) показывает, что реакция завершилась. Реакционную смесь разбавляют этилацетатом, последовательно промывают 1 н. HCl (1 мл), насыщенным водным раствором карбоната натрия и рассолом. Органический слой сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в абсолютном этаноле (6 мл) и к нему прибавляют Pd(OH)2/C (17 мг, 20 мас.%, 0,024 ммоль, 0,43 экв.), а затем к реакционной колбе присоединяют баллон с водородом. Реакционную смесь перемешивают при комнатной температуре, пока ТСХ (5% МеОН/ЕtOАс) не покажет, что исходное вещество полностью израсходовано. Реакционную смесь концентрируют досуха и остаток растворяют в этилацетате, промывают насыщенным водным раствором бикарбоната натрия и органический и водный слои разделяют. Водный слой дополнительно экстрагируют этилацетатом. Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют и получают неочищенный продукт, который очищают с помощью препаративной ТСХ (5% МеОН/ЕtOАс), и получают чистое соединение примера 29 (18 мг, выход 57%). МС [М+1]+ 572,1.
Пример получения 30

Стадия 1:

К раствору соединения 31h (450 мг, 0,64 ммоль, 1,0 экв.) в безводном ДМФ (3,5 мл) последовательно прибавляют HATU (290,5 мг, 0,764 ммоль, 1,2 экв.), N,N-диметиламингидрохлорид (99 мг, 1,01 ммоль, 1,6 экв.) и диизопропилэтиламин (0,50 мкл, 2,87 ммоль, 4,5 экв.). Полученный оранжевый раствор перемешивают при комнатной температуре, пока ТСХ (5% МеОН/ЕtOАс) не покажет, что исходное вещество полностью израсходовано. Реакционную смесь выливают в дихлорметан (200 мл), последовательно промывают разбавленным вдвое насыщенным водным раствором лимонной кислоты, насыщенным раствором NаНСО3 и рассолом. Органический слой сушат над безводным Na2SO4, фильтруют и концентрируют и получают неочищенный продукт, который очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс/гексан=3:1), и получают соединение 30a в виде коричневого твердого вещества (208 мг, выход 43,6%).
Стадия 2:

Метилмагнийбромид (1,65 мл, 1,0 М в трет-бутиловом эфире, 1,65 ммоль, 6,0 экв.) по каплям прибавляют к раствору соединения 30а (206 мг, 0,275 ммоль, 1,0 экв.) в безводном ТГФ (3 мл). ТСХ (ЕtOАс) показывает, что исходное вещество полностью израсходовано, и затем реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Затем реакционную смесь разбавляют этилацетатом, реакцию останавливают насыщенным водным раствором NH4Cl и экстрагируют с помощью ЕtOАс. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют и получают неочищенный продукт, соединение 30b. Соединение 30b используют на следующей стадии без очистки.
Стадия 3:

По такой же методике, что и описанная в примере 31, стадия 6, неочищенное соединение 30b гидрируют и получают чистое соединение примера 30 (150 мг, выход 95,6% из соединения 30a). МС [М+1]+ 571,1.
Пример получения 31
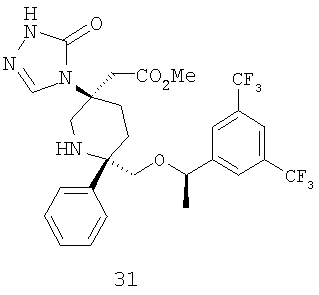
Стадия 1:

Соединение 31b (32 мл) прибавляют к раствору соединения 31а (1,0 г, 11,1 ммоль, 1,0 экв.) в триэтилортоформиате. Раствор нагревают при 88°С в течение 36 ч и затем концентрируют досуха в вакууме. Полученный остаток перекристаллизовывают из ЕtOАс и получают соединение 31с (0,94 г, выход 58%).
Стадия 2:

К раствору соединения 23b (2,5 г, 4,1 ммоль) в ТГФ (20 мл) прибавляют аллилметилкарбонат (0,465 мл, 8,2 ммоль) и Рd(РРh3)4 (236 мг, 0,205 ммоль). Реакционный сосуд трижды продувают азотом и затем раствор перемешивают в течение 16 ч. Затем растворитель удаляют и остаток фильтруют через короткую колонку с диоксидом кремния с использованием смеси 20% ЕtOАС/гексаны в качестве элюента. Фильтрат концентрируют и соединения 31d и 31е разделяют с помощью препаративной ВЭЖХ. МС [М+1]+ 651,1 для обоих соединений.
Стадия 3:

В круглодонную колбу помещают соединение 31d (3,84 г, 5,90 ммоль, 1,0 экв.) и ледяную уксусную кислоту (20 мл). К полученному желтоватому раствору при 0°С несколькими небольшими порциями прибавляют цинковую пыль (3,86 г, 59,0 ммоль, 10 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 6 ч, пока ТСХ (30% ЕtOАс/гексан) не покажет, что исходное вещество, соединение 31d, полностью израсходовано. Затем реакционную смесь разбавляют этилацетатом и пропускают через слой целита в воронке. Слой целита тщательно промывают этилацетатом и промывочный раствор объединяют с фильтратом. Фильтрат концентрируют и получают неочищенный продукт, который очищают с помощью хроматографии на колонке BIOTAGE (30% ЕtOАс/гексаны) и в виде бесцветного масла получают чистый продукт, соединение 31f (3 г, выход 81,9%).

Раствор соединения 31f (33,4 мг, 0,054 ммоль, 1,0 экв.) в этаноле (0,4 мл) обрабатывают реагентом 31с (67,5 мг, 0,46 ммоль, 5 экв.) и перемешивают при комнатной температуре в течение ночи. Затем его разбавляют безводным метанолом (1 мл) и обрабатывают метоксидом натрия, затем нагревают при 88°С, пока ТСХ (ЕtOАс) не показывает, что содержится только продукт. Его концентрируют досуха и затем растворяют в этилацетате, промывают насыщенным раствором бикарбоната натрия и слои разделяют. Водный слой дополнительно экстрагируют этилацетатом. Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют и получают неочищенный продукт, который очищают с помощью хроматографии на колонке BIOTAGE (25-40% ЕtOАс/гексаны) и получают чистое соединение 31g (22,4 мг, выход >60%).
Стадия 4:
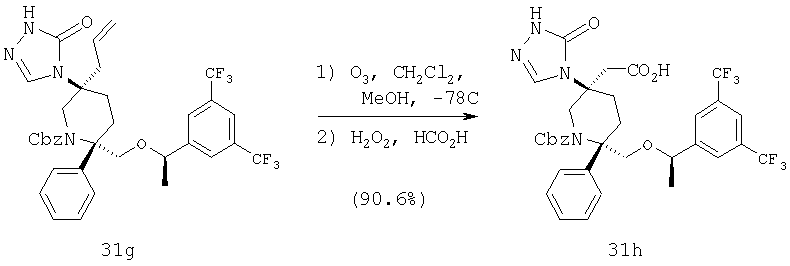
Соединение 31g (306 мг, 0,44 ммоль, 1,0 экв.) растворяют в безводном дихлорметане (5 мл). Полученный бесцветный раствор охлаждают до -78°С, затем продувают с помощью О3, пока раствор не станет пурпурным. Затем раствор продувают с помощью N2 для удаления избытка О3. После этого раствор концентрируют досуха. Полученное белое вспененное вещество растворяют в муравьиной кислоте (1,5 мл) и обрабатывают пероксидом водорода (1,5 мл, 30% водный раствор) и получают белую суспензию, которую нагревают при 80°С в течение ночи. Анализ с помощью ЖХМС обнаруживает пик только продукта. Растворитель удаляют в вакууме и остаток растворяют в этилацетате и промывают разбавленным вдвое насыщенным водным раствором Na2S2O3. Полученные два слоя разделяют и водный слой дополнительно экстрагируют этилацетатом. Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют и получают соединение 31h (284,2 мг, выход 90,6%). Соединение 31h используют на следующей стадии без очистки.
Стадия 5:

К раствору соединения 31h (104 мг, 0,147 ммоль, 1,0 экв.) в бензоле (4 мл) и метаноле (1 мл) по каплям прибавляют 2,0 раствор триметилсилилдиазометана в гексанах (88 мкл, 0,177 ммоль, 1,2 экв.). После перемешивания реакционной смеси при комнатной температуре в течение 30 мин ТСХ (10% MeOH/CH2Cl2) показывает, что исходное вещество полностью израсходовано. Растворитель удаляют и получают неочищенный продукт, который используют на следующей стадии без очистки.
Стадия 6:
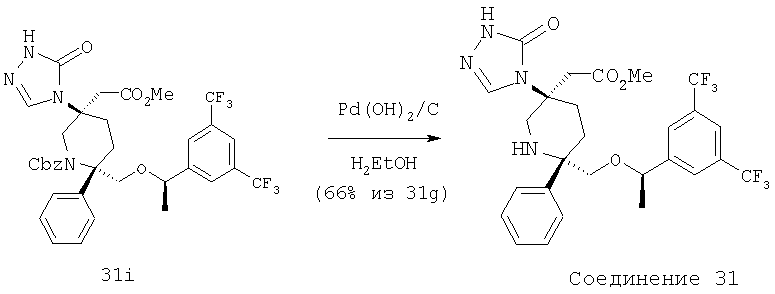
Неочищенный продукт, полученный на стадии 5, соединение 31i, растворяют в абсолютном этаноле (4,5 мл). К этому раствору прибавляют Pd(OH)2/C (46,7 мг, 20 мас.%, 0,067 ммоль, 0,45 экв.) и затем реакционную смесь гидрируют водородом из баллона. Реакцию гидрирования останавливают, когда ТСХ (10% МеОН/СН2Сl2) показывает, что исходное вещество израсходовано. Разбавленную реакционную смесь осторожно пропускают через воронку, заполненную целитом, и слой целита тщательно промывают метанолом. Фильтрат концентрируют досуха. Полученный остаток очищают с помощью препаративной ТСХ (10% MeOH/CH2Cl2) и получают чистое соединение 31 (56,2 мг, выход 65% из соединения 31g), МС [М+1]+ 587,1.
Пример получения 32
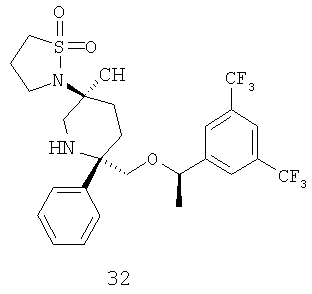
Стадия 1:
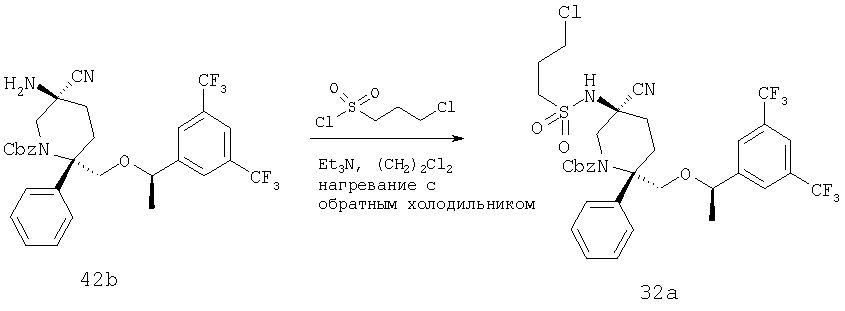
В круглодонной колбе объемом 25 мл соединение 42b (0,142 г, 0,23 ммоль, 1,0 экв.) растворяют в 3 мл дихлорэтана в атмосфере N2 и реакционную смесь обрабатывают с помощью Et3N (0,0,48 мл, 0,34 ммоль, 1,5 экв.), затем 3-хлорсульфонилпропилхлоридом (0,037 мл, 0,3 ммоль, 1,2 экв.). Реакционную смесь перемешивают при комнатной температуре в течение ночи. За протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан) и МС, которые показывают, что ненужный продукт не образовался. Затем реакционную смесь нагревают с обратным холодильником. Через 1 ч нагревания реакция завершается. Затем реакционную смесь охлаждают и разбавляют с помощью CH2Cl2, и реакцию останавливают с помощью 1 н. HCl. Органический слой сушат над Nа2SO4 и концентрируют и получают неочищенное соединение 32а (0,11 г), которое используют на следующей стадии без дополнительной очистки.
Стадия 2:

В высушенной на огне круглодонной колбе объемом 15 мл соединение 32а (0,11 г, 0,23 ммоль, 1,0 экв.) растворяют в сухом ДМФ. К этой реакционной смеси прибавляют 1,8-диазабицикло[5,4,0]ундец-7-ен (ДБУ) (0,044 г, 0,29 ммоль, 1,2 экв.) и реакционную смесь перемешивают при комнатной температуре в течение ночи. За протеканием реакции следят с помощью ТСХ (30:70 ЕtOАс/гексан) и МС. После завершения реакции реакционную смесь разбавляют с помощью ЕtOАс и реакцию останавливают с помощью Н2O. Органический слой сушат над Na2SO4 и концентрируют и получают неочищенное соединение 32b (0,1 г). Очистку проводят с помощью хроматографии на колонке BIOTAGE (30/70 ЕtOАс/гексан) и получают очищенное соединение 32b (0,072 г). МС с электрораспылением [М+1] 710,2.
Стадия 3:

Соединение 32b (0,072 г, 0,1 ммоль, 1,0 экв.) растворяют в сухом МеОН (1,5 мл) и обрабатывают с помощью 20% Рd(ОН)3 (60 мас.%) в инертной атмосфере. Реакционную смесь гидрируют при атмосферном давлении и за протеканием реакции следят с помощью ТСХ (40:60 ЕtOАс/гексан). Реакция завершается за 45 мин и реакционную смесь фильтруют через целит, промывают с помощью ЕtOАс и концентрируют и получают неочищенный продукт. Неочищенный продукт очищают с помощью препаративной хроматографии (60/40 ЕtOАс/гексан) и получают соединение 32 (0,04 г, 70%). МС с электрораспылением [М+1]576,2. МСВР (ББА) рассчитано для С26Н28F6N3O2(М+1) 576,1756, найдено 576,1764.
Пример получения 33

Стадия 1:

В круглодонной колбе объемом 25 мл соединение 42b (0,322 г, 0,53 ммоль, 1,0 экв.) растворяют в 5 мл CH2Cl2 и реакционную смесь охлаждают до 0°С в бане со льдом. После этого к реакционной смеси прибавляю Еt3N (0,111 мл, 0,79 ммоль, 1,5 экв.), затем 4-хлорбутирилхлорид (0,072 мл, 0,64 ммоль, 1,2 экв.) и ее медленно нагревают до комнатной температуры и перемешивают в течение 14 ч. За протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан элюент) и МС. После завершения реакции реакционную смесь разбавляют с помощью CH2Cl2 и реакцию останавливают насыщенным раствором NаНСО3, а затем рассолом. Органический слой сушат над Na2SO4 и концентрируют и получают неочищенное соединение 33а (0,32 г), которое используют на следующей стадии без дополнительной очистки.
Стадия 2:

В высушенной на огне круглодонной колбе объемом 25 мл соединение 33а (0,32 г, 0,45 ммоль, 1,0 экв.) растворяют в сухом ТГФ. К этому раствору прибавляют 60% NaH (0,025 г, 0,68 ммоль, 1,5 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. За протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан) и МС. После завершения реакции реакционную смесь разбавляют с помощью ЕtOАс и реакцию останавливают насыщенным раствором NaHCO3. Органический слой сушат над Na2SO4 и концентрируют и получают соединение 33b (0,4 г) в виде желтого масла, которое используют на следующей стадии без дополнительной очистки.
Стадия 3:

Соединение 33b (0,4 г, 0,59 ммоль, 1,0 экв.) растворяют в сухом МеОН (4,0 мл) и обрабатывают с помощью 20% Pd(OH)2 (60 мас.%) в инертной атмосфере. Реакционную смесь гидрируют при атмосферном давлении и за протеканием реакции следят с помощью ТСХ (элюент - 40:60 ЕtOАс/гексан). Реакция завершается за 45 мин, реакционную смесь фильтруют через целит и промывают с помощью ЕtOАс и концентрируют и получают неочищенный продукт. Очистку неочищенного продукта проводят с помощью хроматографии на колонке BIOTAGE (60/40 EtOAc/гексан) и получают соединение 33 (0,18 г, 59%).
МСВР (ББА) рассчитано для С26Н28F8N3O2 (М+1) 540,2086, найдено 540,2078.
Пример получения 34

Стадия 1:

В круглодонной колбе объемом 25 мл соединение 42 с (0,23 г, 0,38 ммоль, 1,0 экв.) растворяют в 3 мл CH2Cl2 и реакционную смесь охлаждают до 0°С в бане со льдом. После этого к реакционной смеси прибавляют Et3N (0,079 мл, 0,57 ммоль, 1,5 экв.), затем 4-хлорбутирилхлорид (0,051 мл, 0,45 ммоль, 1,2 экв.) и ее медленно нагревают до комнатной температуры и перемешивают в течение 14 ч. За протеканием реакции следят с помощью ТСХ (элюент - 60:40 ЕtOАс/гексан) и МС. После завершения реакции реакционную смесь разбавляют с помощью CH2Cl2 и реакцию останавливают насыщенным раствором NаНСО3, а затем рассолом. Органический слой сушат над Na2SO4 и концентрируют и получают неочищенное соединение 34а (0,23 г), которое используют на следующей стадии без дополнительной очистки.
Стадия 2:

В высушенной на огне круглодонной колбе объемом 25 мл соединение 34а (0,23 г, 0,38 ммоль, 1,0 экв.) растворяют в сухом ТГФ (1 мл). К этой реакционной смеси прибавляют 60% NaH (0,022 г, 0,57 ммоль, 1,5 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. За протеканием реакции следят с помощью ТСХ (60:40 ЕtOАс/гексан элюент) и МС. После завершения реакции реакционную смесь разбавляют с помощью ЕtOАс и реакцию останавливают насыщенным раствором NаНСО3. Органический слой сушат над Na2SO4 и концентрируют и получают соединение 34b (0,21 г) в виде желтого масла, которое используют на следующей стадии без дополнительной очистки.
МС с электрораспылением [М+1] 674,2.
Стадия 3:

Соединение 34b (0,21 г, 0,31 ммоль, 1,0 экв.) растворяют в сухом МеОН (2,0 мл) и обрабатывают с помощью 20% Pd(OH)2 (40 мас.%) в инертной атмосфере. Реакционную смесь гидрируют при атмосферном давлении и за протеканием гидрирования следят с помощью ТСХ (элюент - 40:60 ЕtOАс/гексан). Через 45 мин реакционную смесь фильтруют через целит, промывают с помощью ЕtOАс и концентрируют и получают неочищенный продукт. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (60/40 ЕtOАс/гексан) и получают соединение 34 (0,10 г, 59%).
МСВР (ББА) рассчитано для C26H28F6N3O2 (M+1)540,2086, найдено 540,2078.
Пример получения 35


Соединение 35 получают по методике, сходной с методикой получения соединения 23е в примере 23.

Аналогичным образом соединение 35b получают по методике, сходной с методикой, использованной для получения соединения 23f в примере 23.

Соединение 35с получают по методике, сходной с методикой, использованной для получения соединения 23g в примере 23.

Соединение 35d получают по методике, аналогичной методике получения соединения 23 в примере 23.

Соединение 35е получают по методике, аналогичной методике получения соединения 23h в примере 23.

Соединение 35f получают по методике, аналогичной методике получения соединения 42е в примере 42.

Соединение 35g получают по методике, аналогичной методике получения соединения 42g в примере 42.

Соединение 35 получают по методике, аналогичной методике получения соединения 42 в примере 42. МСВР рассчитано для С25Н23F6N5O2 (М+Н) 540,1834, найдено 540,1822.
Пример 36

Стадия А:
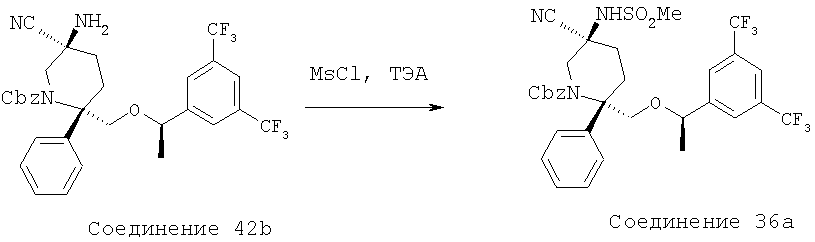
ТЭА - триэтаноламин
Соединение 36а получают по методике, аналогичной методике получения соединения 47 в примере 47.
Стадия В:
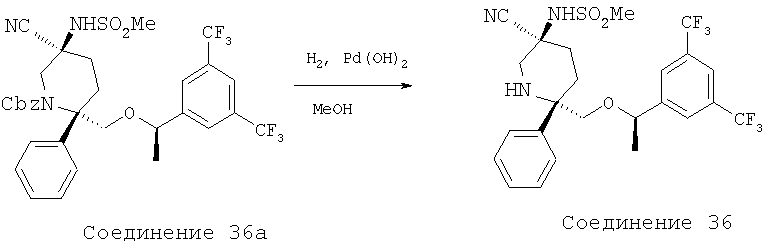
Соединение 36 получают по методике, аналогичной методике получения соединения 23 в примере 23. МСВР рассчитано для С24Н25F6N3О3S (М+Н) 550, 1599, найдено 550,1603.
Пример получения 37

Стадия А:

Соединение 37а получают по методике, аналогичной методике получения соединения 47 в примере 47.
Стадия В:
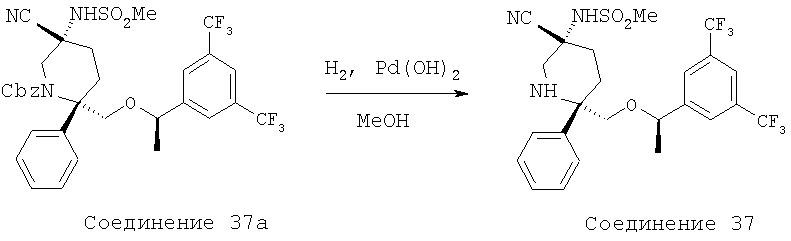
Соединение 37 получают по методике, аналогичной методике получения соединения 23 в примере 23. МСВР рассчитано для С24Н25F6N3О3S (М+Н) 550, 1599, найдено 550,1603.
Пример получения 38

Стадия 1:
Через раствор соединения 31g (640 мг, 0,93 ммоль) в 10 мл CH2Cl2, температуру которого поддерживают равной -78°С, пропускают газообразный О3, пока реакционная смесь не станет синей. Затем реакционную смесь продувают азотом, пока она не станет бесцветной. Затем прибавляют ТБАЙ (412 мг, 1,11 ммоль) и реакционную смесь перемешивают при 20°С в течение 2 ч. Реакционную смесь разбавляют диэтиловым эфиром, промывают насыщенным водным раствором Nа2S2O3, водой и рассолом и сушат и концентрируют. Полученный остаток растворяют в ЕtOН (22 мл) и NaOAc (262,7 мг, 3,2 ммоль) и прибавляют гидроксиламингидрохлорид (222 мг, 3,2 ммоль), и смесь перемешивают в течение ночи. Затем реакционную смесь концентрируют и остаток подвергают распределению между 20 мл ЕtOАс и водой. Органический слой сушат и концентрируют. Неочищенный промежуточный продукт растворяют в толуоле (6,8 мл) и затем прибавляют 1,1'-оксаллилдиимидазол (165 мг, 1,8 ммоль) и смесь нагревают при 80°С в течение 2 ч. После охлаждения реакционной смеси до 23°С толуольный раствор вводят в колонку с силикагелем и элюируют смесью 20-100% ЕtOАс/гексаны и получают соединение 33а. [МС]+ 688,1.

По методике, сходной с методикой примера 31, стадия 6, соединение 38а гидрируют и получают соединение 38. МС [M+1]+ 554,1.
Примеры получения 39 и 40
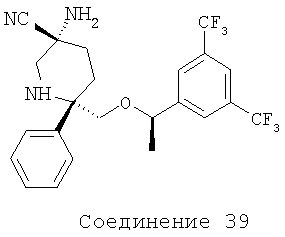

Стадия А:

Соединения 39 и 40 получают по методике, аналогичной методике получения соединения 38. МСВР рассчитано для C23H23F6N3O (М+Н) 472,1824, найдено 472,1820.
Пример получения 41
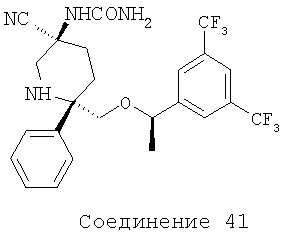
Стадия А:

В круглодонной колбе объемом 25 мл соединение 42b (0,15 г, 0,248 ммоль, 1,0 экв.) растворяют в 6 мл ДХЭ (дихлорэтан). Прибавляют триметилсилилизоцианат (0,51 мл, 3,72 ммоль, 15,0 экв.) и реакционную смесь нагревают с обратным холодильником при 80°С в течение ночи. Реакционную смесь охлаждают и реакцию останавливают насыщенным раствором NаНСО3 (10 мл). Водную фазу экстрагируют с помощью ЕtOАс (2×10 мл). Органические слои промывают рассолом (5 мл), сушат над МgSO4 и концентрируют. Неочищенный продукт очищают с помощью препаративной ТСХ (1:1 ЕtOАс:гексаны) и получают 0,060 г (37%) соединения 41а.
Стадия В:

Соединение 41 получают по методике, аналогичной методике получения соединения 23 в примере 23. МСВР рассчитано для С24Н24F6N4O2 (М+Н) 515,1882, найдено 515,1874.
Пример получения 42
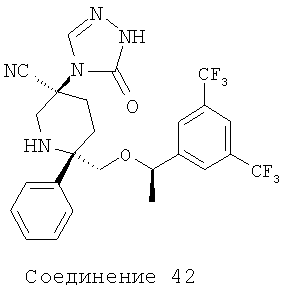
Стадия А:

Соединение 41а (6,87 г, 11,86 ммоль) в ЕtOН (7 мл) при комнатной температуре в герметичном сосуде прибавляют к раствору NaCN (0,767 г), NH4Cl (0,889 г) и NН3·Н2О (3,84 мл) в ЕtOН (7,0 мл) и воде (7,0 мл). Затем герметичный сосуд нагревают при 60°С в течение 12 ч, а после этого охлаждают до комнатной температуры. Реакционную смесь разбавляют с помощью ЕtOАс (200 мл) и промывают водой (50 мл). Водную фазу экстрагируют с помощью ЕtOАс (3×30 мл). Объединенные органические слои промывают рассолом (30 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 7/2 до 5/2) и получают соединение 42b (2,6 г, 36%) и соединение 42с (1,8 г, 25%).
Стадия В:
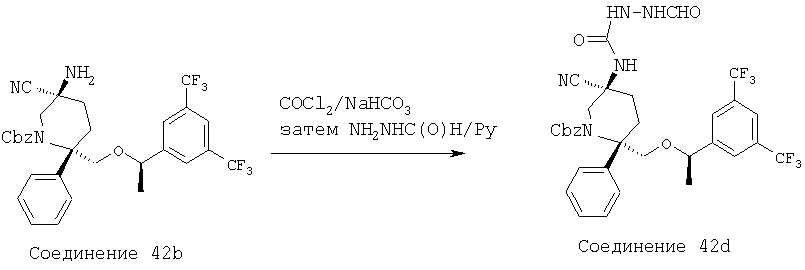
Фосген (6,67 мл, 12,4 ммоль, 20% в толуоле) при 0°С при энергичном перемешивании по каплям прибавляют к смеси соединения 42b (1,5 г, 2,48 ммоль) в CH2Cl2 (30 мл) и насыщенного раствора NаНСО3 (30 мл). Смесь перемешивают при 0°С в течение 3 ч и затем ее разбавляют с помощью CH2Cl2 (50 мл) и водную фазу отделяют от органической фазы. Органическую фазу промывают холодным водным раствором NH4Cl, рассолом и сушат над МgSО4. Для удаления избытка фосгена объем растворителя уменьшают примерно до 5 мл при пониженном давлении при комнатной температуре. Остаток растворяют в CH2Cl2 (15 мл) и обрабатывают с помощью NH2NHC(O)H (0,446 г, 7,44 ммоль) и пиридином (1,2 мл, 14,88 ммоль) при комнатной температуре. Полученный раствор перемешивают при комнатной температуре в течение 12 ч. Затем реакционную смесь разбавляют с помощью ЕtOАс (200 мл) и промывают с помощью HCl (50 мл, 0,5 н.). Водную фазу экстрагируют с помощью ЕtOАс (3×30 мл). Объединенные органические слои промывают рассолом (30 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE, элюируя смесью гексан/ЕtOАс (об./об.=от 1/2 до 1/7), и получают соединение 42d (1,1 г, 64%).
Стадия С:

TMCCl (50 мкл) при перемешивании прибавляют к смеси соединения 42d (15 мг, 0,0217 ммоль) и LiI (2,9 мг, 0,0217 ммоль) в ГМДС (гексаметилдисилазан) (0,5 мл) при комнатной температуре. Полученную реакционную смесь нагревают при 140°С (температура бани) в течение 30 ч, а затем охлаждают до комнатной температуры. Реакционную смесь разбавляют с помощью ЕtOАс (25 мл) и промывают с помощью HCl (5 мл, 1,0 н.). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают рассолом (10 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=6/4) и получают соединение 42 (4 мг, 34%).
Альтернативная методика для примера 42
Альтернативно, соединение 42 также можно получить из соединения 23g следующим образом
Стадия А:

В круглодонной колбе объемом 10 мл соединение 23g (0,02 г, 0,037 ммоль, 1,0 экв.) растворяют в ДХМ (3 мл) и реакционную смесь охлаждают до 0°С. Прибавляют перйодинан Десса-Мартина (0,02 г, 0,048 ммоль, 1,3 экв.) и реакционную смесь перемешивают в атмосфере азота при комнатной температуре в течение 45 мин. За протеканием реакции следят с помощью ТСХ (элюент - 9/1 ЕtOАС/МеОН) и реакцию останавливают через 1,5 ч, выливая реакционную смесь в делительную воронку, содержащую насыщенный раствор Nа2S2О3/NаНСО3 (1:1) (5 мл). Смесь в делительной воронке энергично встряхивают и водный слой экстрагируют с помощью Et2O (2×5), сушат над MgSO4 и концентрируют и получают неочищенное соединение 42е (0,02 г), которое используют на следующей стадии без дополнительной очистки.
Стадия В:

В круглодонной колбе объемом 25 мл соединение 42f (0,09 г, 0,13 ммоль, 1,0 экв.) и ацетат натрия (0,032 г, 0,39 ммоль, 3,0 экв.) растворяют в ЕtOН (6 мл) и прибавляют гидроксиламингидрохлорид (0,056 г, 0,080 ммоль, 6,0 экв.). Реакционную смесь перемешивают в атмосфере азота при комнатной температуре в течение ночи. Затем реакционную смесь разбавляют с помощью ЕtOАс (15 мл), реакцию останавливают насыщенным раствором NаНСО3 (5 мл) и органический слой промывают рассолом (5 мл) и сушат над MgSO4, и получают неочищенное соединение 42д (0,95 г), которое используют на следующей стадии без дополнительной очистки.
Стадия С

В круглодонной колбе объемом 50 мл соединение 42g (1,1 г, 0,59 ммоль, 1,0 экв.) растворяют в бензоле (25 мл). К раствору прибавляют 1,1'-оксаллилдиимидазол (0,302 г, 1,89 ммоль, 1,5 экв.) и реакционную смесь нагревают при 75°С в атмосфере азота в течение 4 ч. Затем реакцию останавливают водой (20 мл), разбавляют с помощью ЕtOАс (30 мл), сушат над MgSO4 и концентрируют и получают неочищенный продукт. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (1/1 ЕtOАс/гексаны) и получают соединение 42h (0,7 г, 66% за 3 стадии).
Стадия D:

В круглодонной колбе объемом 50 мл соединение 42i (0,5 г, 0,742 ммоль, 1,0 экв.) растворяют в ацетонитриле (9 мл). Реакционную смесь охлаждают до 0°С и с помощью шприца по каплям прибавляют TMCl (0,742 мл, 5,19 ммоль, 7,0 экв.). Реакционную смесь перемешивают в течение ночи при комнатной температуре. За протеканием реакции следят с помощью МС, которая показывает, что все еще содержится некоторое количество исходного вещества. Реакцию останавливают насыщенным раствором Nа2S2O3/NаНСО3 (1:1) (10 мл) и разбавляют с помощью ЕtOАс (20 мл). Органический слой промывают рассолом (10 мл), сушат над МgSO4 и концентрируют и получают неочищенный продукт. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (60/40 ЕtOАс/гексаны) и получают соединение 42 (0,4 г). МСВР рассчитано для C25H23F6N5O2 (M+H) 540,1834, найдено 540,1813.
Пример получения 43

Стадия А

Соединение 43а получают по методике, аналогичной методике получения соединения 28а.
Стадия В:
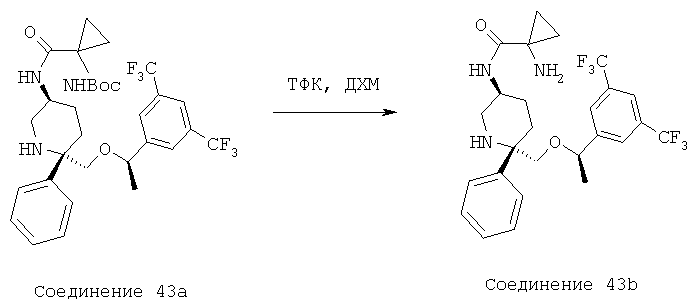
Соединение 43b получают по методике, аналогичной методике получения соединения 45с.
Стадия С:

Соединение 43 получают по методике, аналогичной методике получения соединения 28 (стадия с).
Пример получения 44

Стадия А:
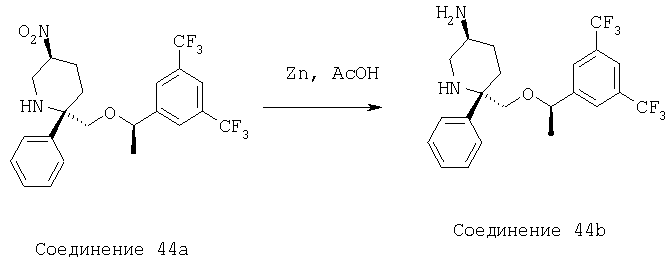
В круглодонной колбе объемом 50 мл соединение 44а (1,1 г, 2,31 ммоль, 1,0 экв.) растворяют в уксусной кислоте (20 мл) и полученную реакционную смесь охлаждают до 0°С. Прибавляют порошкообразный Zn (1,51 г, 23,1 ммоль, 10,0 экв.) и смесь нагревают с обратным холодильником в течение 2,5 ч. Затем реакционную смесь фильтруют через целит, концентрируют, разбавляют с помощью ЕtOАс (30 мл) и нейтрализуют насыщенным раствором NаНСО3 (30 мл). Водную фазу экстрагируют с помощью ЕtOАс (2×10 мл), промывают рассолом (20 мл), сушат над MgSO4 и концентрируют. Неочищенный продукт очищают с помощью фильтровальной колонки и получают 1,0 г (99%) соединения 44b.
Стадия В:

Соединение 44с получают по методике, аналогичной методике получения соединения 45b.
Стадия С:

Соединение 44d получают по методике, аналогичной методике получения соединения 45с.
Стадия D:

Соединение 44е получают по методике, аналогичной методике получения соединения 45d.
Стадия Е:

В круглодонной колбе объемом 10 мл соединение 44е (0,34 г, 0,54 ммоль, 1,0 экв.) растворяют в 5,5 мл МеОН/Н2O (10:1). Круглодонную колбу дегазируют и прибавляют Pd/C (10 мас.%, 0,18 г), затем НСO2NH4 (0,174 г, 2,68 ммоль, 5,0 экв.). Полученную гетерогенную смесь нагревают с обратным холодильником в течение ночи, охлаждают, фильтруют через целит, концентрируют, разбавляют с помощью ЕtOАс (10 мл), промывают насыщенным раствором NaHCO3 (10 мл) и сушат над Na2SO4. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (9:1 ЕtOАс:МеОН) и получают 0,11 г (38%) соединения 44. МСВР рассчитано для С25Н26F6N4O3 (М+Н) 545,1987, найдено 545,1988.
Примеры получения 45 и 46


Стадия А:

Соединение 41а (2,8 г, 4,59 ммоль, 1,0 экв.) растворяют в этаноле (15 мл), к раствору прибавляют никель Ренея и реакционную смесь гидрируют во встряхивающем устройстве Парра при давлении, равном 60 фунт-сила/дюйм2. За протеканием гидрирования следят с помощью ТСХ (4/1 ЕtOАс/гексаны). Через 3 ч реакционную смесь фильтруют через целит, промывают этанолом (30 мл) и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (4/1 ЕtOАс/гексаны) и получают соединение 45а (1,75 г, 65%).
Стадия В:

В круглодонной колбе объемом 50 мл соединение 45а (1,0 г, 1,72 ммоль, 1,0 экв.) растворяют в сухом ТГФ (20 мл) и охлаждают до 0°С. К этому охлажденному раствору через канюлю прибавляют раствор трет-бутилкарбазата (0,228 г, 1,72 ммоль, 1,0 экв.) и карбонилдиимидазола (0,335 г, 2,06 ммоль, 1,2 экв.), которые предварительно перемешивали в сухом ТГФ (10 мл). Реакционной смеси дают нагреться до комнатной температуры и ее перемешивают в течение ночи. Затем реакционную смесь концентрируют и очищают с помощью хроматографии на колонке BIOTAGE (1/1 ЕtOАс/гексаны) и получают соединение 45b (0,85 г, 67%).
Стадия С:

В круглодонной колбе объемом 50 мл соединение 45b (0,39 г, 0,53 ммоль, 1,0 экв.) растворяют в СН2Сl2 (10,0 мл) и охлаждают до 0°С. К раствору прибавляют трифторуксусную кислоту (1,02 мл, 13,2 ммоль, 25,0 экв.) и реакционную смесь перемешивают при комнатной температуре. За протеканием реакции следят с помощью МС (т.е. до исчезновения исходного вещества). Реакционную смесь концентрируют через 7 ч и используют на следующей стадии без какой-либо дополнительной очистки. Неочищенный промежуточный продукт растворяют в ТГФ (5 мл) и охлаждают до 0°С. Прибавляют 20% водный раствор NaOH (5,0 мл), затем метоксиацетилхлорид (0,096 мл, 1,06 ммоль, 2,0 экв.). Реакционную смесь перемешивают при комнатной температуре в течение ночи и затем разбавляют с помощью Н2O (10 мл), экстрагируют с помощью Et2O (2×10 мл), промывают рассолом (10 мл), сушат над MgSO4 и концентрируют и получают неочищенное соединение 45с (0,35 г, 95%), которое используют на следующей стадии без какой-либо дополнительной очистки.
Стадия D:

В круглодонной колбе объемом 25 мл соединение 45с (0,35 г, 0,49 ммоль, 1,0 экв.) растворяют в ЕtOН (3,0 мл). Прибавляют 3,0 мл 6 М раствора NaOH и реакционную смесь нагревают с обратным холодильником в течение ночи. Затем реакционную смесь концентрируют и очищают с помощью препаративной ТСХ (ЕtOАс) и получают 0,145 г (42%) соединения 45d.
Стадия Е:

В круглодонной колбе объемом 10 мл соединение 45d (0,125 г, 0,18 ммоль, 1,0 экв.) растворяют в 3 мл МеОН. Прибавляют Pd(OH)2 (0,010 г, 0,072 ммоль, 40 мас.%) и гетерогенную смесь гидрируют при комнатной температуре. За протеканием гидрирования следят с помощью МС. Реакционную смесь фильтруют через целит, концентрируют и очищают с помощью препаративной ТСХ (ЕtOАс) и получают смесь соединений 45 и 46 (0,008 г, 8%). МСВР рассчитано для C26H28F6N4O3 (M+H) 559,2144, найдено 559,2146.
Примеры получения 47 и 48


Стадия А:

В круглодонной колбе объемом 10 мл смесь соединений 49 и 50 (0,025 г, 0,047 ммоль, 1,0 экв.) растворяют в 2 мл ДХМ и охлаждают до 0°С. Прибавляют триэтиламин (0,0073 мл, 0,052 ммоль, 1,1 экв.), затем MeSO2Cl (0,004 мл, 0,052 ммоль, 1,1 экв.). Реакционную смесь перемешивают в течение ночи. Затем реакционную смесь разбавляют с помощью ЕtOАс (10 мл) и реакцию останавливают насыщенным раствором NаНСО3 (5 мл). Водную фазу экстрагируют с помощью ЕtOАс (2×5 мл), сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают с помощью препаративной ТСХ (4:1 ЕtOАс/гексаны) и получают 0,028 г (100%) смеси соединений 47 и 48. МСВР рассчитано для С25Н27F6N5O4S (M+H) 608,1766, найдено 608,1785.
Примеры получения 49 и 50


Стадия А:

Соединение 49а (0,50 г, 0,77 ммоль, 1,0 экв.) помещают в круглодонную колбу. Затем в колбу прибавляют дымящую НNО3 (3 мл) и полученную реакционную смесь выдерживают в течение 1 ч. После завершения реакции прибавляют лед (10 г). Реакционную смесь разбавляют с помощью ЕtOАс (25 мл) и нейтрализуют насыщенным раствором NaOH (3 мл). Водную фазу экстрагируют с помощью ЕtOАс (2×10 мл). Органические слои промывают рассолом (10 мл), сушат над МgSО4 и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (7:3 ЕtOАс:гексаны) и получают 0,45 г (79%) соединения 49b.
Стадия В:

Соединения 49 и 50 получают по методике, аналогичной методике получения соединения 44b. МСВР рассчитано для С24Н25F6N5O2 (М+Н) 530,1991, найдено 530,1977.
Пример получения 51
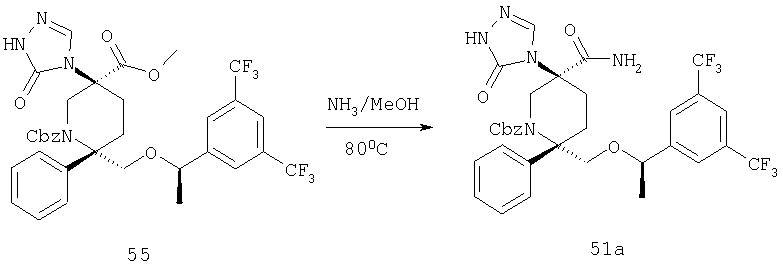
Соединение 55 (0,078 г, 0,11 ммоль, 1,0 экв.) растворяют в 7 М растворе аммиака в МеОН (3,0 мл) и помещают в небольшой сосуд высокого давления Парра, который нагревают при 80°С в течение 2 дней. За протеканием реакции следят с помощью ТСХ (9/1 СН2Сl2/МеОН). После завершения реакции реакционную смесь концентрируют и получают неочищенный продукт в виде белого твердого вещества. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (от 2:1 до 4:1 ЕtOАс/гексан) и получают соединение 51а в виде белого твердого вещества (0,48 г). МС с электрораспылением [М+1] 692,2.
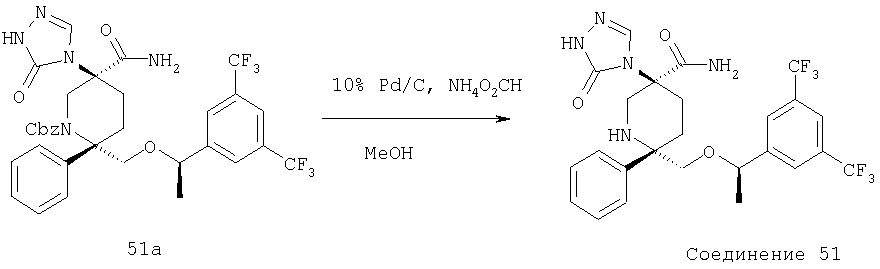
Соединение 51а (0,045 г, 0,065 ммоль, 1,0 экв.) растворяют в сухом МеОН (2,0 мл) и обрабатывают с помощью 10% Pd/C (40 мас.%), затем формиатом аммония (0,02 г, 0,03 ммоль, 5,0 экв.) в инертной атмосфере. Реакционную смесь нагревают с обратным холодильником и за протеканием реакции следят с помощью ТСХ (100% ЕtOАс). Реакция завершается за 1 ч. Реакционную смесь фильтруют через целит, промывают с помощью ЕtOАс и концентрируют в вакууме. Полученный остаток растворяют в ЕtOАс, промывают насыщенным раствором NаНСО3, а затем рассолом и Н2O и получают искомый продукт, соединение 51, в виде белого твердого вещества, которое превращают в его соль с HCl (0,034 г, 94%).
МСВР (ББА) рассчитано для С26Н28F6N3O2 (М+1)558,19242, найдено 558,19398.
Прием получения 52

Стадия А:
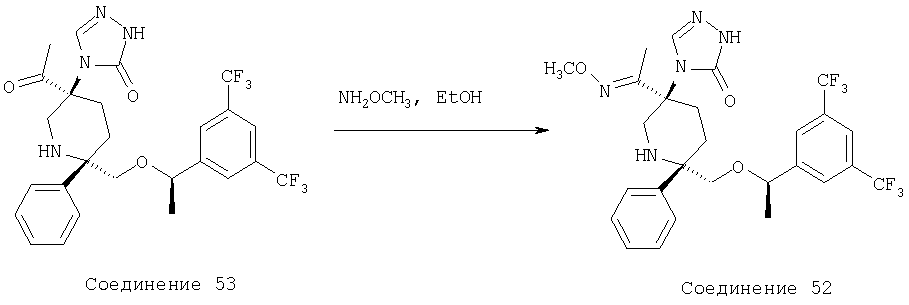
Соединение 53 (18,1 мг, 0,0325 ммоль) в ЕtOН (2,5 мл) обрабатывают с помощью MeONH2·HCl (24,4 мг, 0,292 ммоль) и NaOAc (12,0 мг, 0,146 ммоль) при комнатной температуре. Реакционную смесь перемешивают при 60°С в течение 12 ч, затем разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NаНСО3. Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=от 1/1 до 1/9) и получают соединение 52 (16 мг, 84%). МС с электрораспылением [М+1]+ 586,1.
Пример получения 53

Стадия А:

Перйодинан Десса-Мартина (0,234 г, 0,553 ммоль) при комнатной температуре прибавляют к смеси соединения 23h (0,25 г, 0,369 ммоль) и NaHCO3 (0,232 г, 2,76 ммоль) в СН2Сl2 (5,0 мл). Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют с помощью ЕtOАс (50 мл) и воды (10 мл). Органическую фазу промывают насыщенным раствором Nа2S2O3 раствор (3×15 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают раствором NaOH (15 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный альдегид (0,25 г) растворяют в безводном ТГФ (4,0 мл) и обрабатывают с помощью MeMgBr (0,49 мл, 1,48 ммоль, 3,0 М в Et2O) при -78°С. Температура реакции медленно повышается до комнатной и реакцию останавливают через 2 ч путем медленного прибавления насыщенного водного раствора NH4Cl (10 мл). Затем реакционную смесь разбавляют с помощью ЕtOАс (50 мл) и нейтрализуют с помощью 0,5 н. HCl, пока водная фаза не станет слабокислой. Водную фазу экстрагируют с помощью ЕtOАс (3х15 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный вторичный спирт (0,26 г) растворяют в CH2Cl2 (5,0 мл) и обрабатывают перйодинаном Десса-Мартина (0,468 г, 1,11 ммоль) и с помощью NаНСО3 (0,466 г, 5,55 ммоль) при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют с помощью ЕtOАс (50 мл) и воды (10 мл). Органическую фазу промывают насыщенным раствором Na2S2O3 (3×15 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3×15 мл). Объединенные органические слои промывают раствором NaOH (15 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/1) и получают соединение 53а (0,11 г, 43% за 3 стадии).
Стадия В:

Соединение 53а (107 мг, 0,155 ммоль) в ЕtOН (5,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (21,5 мг, 10 мас.%) и в течение 30 мин гидрируют с помощью Н2 из баллона. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 1/3 до 1/9) и получают соединение 53 (66 мг, 76%). МС с электрораспылением [M+1]+ 557,3.
Пример получения 54


Соединение 53 (14,3 мг, 0,0257 ммоль) в ЕtOН (2,5 мл) обрабатывают с помощью НОNН2·НСl (10,7 мг, 0,154 ммоль) и NaOAc (6,3 мг, 0,077 ммоль) при комнатной температуре. Реакционную смесь перемешивают при 60°С в течение 12 ч, затем разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NаНСО3. Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=1/2) и получают соединение 54 (11 мг, 75%). МС с электрораспылением [М+1]+ 572,1.
Пример получения 55

Стадия А:

Перйодинан Десса-Мартина (0,12 г, 0,284 ммоль) при комнатной температуре прибавляют к смеси соединения 23h (96,3 мг, 0,142 ммоль) и NaHCO3 (0,12 г, 1,42 ммоль) в CH2Cl2 (3,0 мл). Реакционную смесь перемешивают в течение 1 ч и затем ее разбавляют путем прибавления ЕtOАс (50 мл) и воды (10 мл). Органическую фазу промывают насыщенным раствором Nа2S2O3 (3×15 мл). Объединенные водные фазы экстрагируют с помощью ЕtOАс (3х15 мл). Объединенные органические слои промывают раствором NaOH (15 мл, 1,0 н.), водой (10 мл), рассолом (15 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный альдегид (96,3 мг) растворяют в трет-бутаноле (2,0 мл) и воде (0,5 мл) и последовательно обрабатывают с помощью NaH2PO4·H2O (39,2 мг, 0,284 ммоль), NaClO2 (44,9 мг, 0,497 ммоль) и 2-метил-2-бутеном (0,105 мл, 0,994 ммоль). Реакционную смесь перемешивают в течение 2 ч и затем разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NH4Cl. Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенную кислоту (95 мг) растворяют в бензоле (2,8 мл) и МеОН (0,7 мл). Полученный раствор обрабатывают с помощью TMCCHN2 (82,2 мкл, 0,164 ммоль) при комнатной температуре и перемешивают в течение 20 мин. Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=2/3) и получают соединение 55а (70 мг, 35% за 3 стадии).
Стадия В:

Соединение 55а (38 мг, 0,0537 ммоль) в ЕtOН (3,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (7,6 мг, 10 мас.%) и в течение 30 мин гидрируют с помощью Н2 из баллона. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=2/3) и получают соединение 55 (24 мг, 78%). МС с электрораспылением [М+1]+ 573,1.
Пример получения 56

Стадия А:

Соединение 56а (15 мг, 0,0227 ммоль) в ЕtOН (2,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (3,6 мг, 10 мас.%) и в течение 30 мин гидрируют с помощью Н2 из баллона. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт растворяют в Et2O (0,5 мл) и обрабатывают с помощью HCl в эфире (0,23 мл, 0,23 ммоль, 1,0 М в эфире). Смесь перемешивают при комнатной температуре в течение 12 ч. Затем смесь разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NaOH (5 мл, 0,5 н.). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над МgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=1/1) и получают соединение 56 (8,5 мг, 67%). МС с электрораспылением [M+1]+ 563,1.
Пример получения 57

Стадия А:

MsCl (11,7 мкл, 0,151 ммоль) при комнатной температуре прибавляют к раствору соединения 23h (42,8 мг, 0,063 ммоль) и Et3N (26,4 мкл, 0,189 ммоль) в CH2Cl2 (1,0 мл). Реакцию останавливают водой (5,0 мл) через 30 мин и разбавляют с помощью CH2Cl2 (15 мл). Водную фазу экстрагируют с помощью CH2Cl2 (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над МgSО4. После фильтрования и концентрирования неочищенный мезилат (44 мг, 0,0582 ммоль) растворяют в безводном ДМФ (2,0 мл) и обрабатывают с помощью NaBH4 (11,0 мг, 0,291 ммоль). Реакционную смесь нагревают при 90°С в течение 1 ч, а затем охлаждают до комнатной температуры. После этого реакционную смесь разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором HCl (5 мл, 1,0 М). Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (3×10 мл), рассолом (10 мл) и сушат над MgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=3/2) и получают соединение 57а (18 мг, 43%) и соединение 56а (15 мг, 36%).
Стадия B:

Соединение 57а (18 мг, 0,027 ммоль) в ЕtOН (3,0 мл) при комнатной температуре обрабатывают с помощью Pd(OH)2/C (3,6 мг, 10 мас.%) и в течение 30 мин гидрируют с помощью Н2 из баллона. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью ЕtOН (15 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=1/1) и получают соединение 57 (10 мг, 70%). МС с электрораспылением [М+1]+ 529,1.
Пример получения 58


Соединение 19 (10,0 мг, 0,0175 ммоль) в ЕtOН (1,5 мл) при комнатной температуре обрабатывают с помощью МеОNН2·НСl (14,6 мг, 0,175 ммоль) и NaOAc (7,2 мг, 0,0876 ммоль). Реакционную смесь перемешивают при 60°С в течение 12 ч и затем разбавляют с помощью ЕtOАс (20 мл) и промывают водным раствором NaHCO3. Водную фазу экстрагируют с помощью ЕtOАс (3×10 мл). Объединенные органические слои промывают водой (10 мл), рассолом (10 мл) и сушат над МgSO4. После фильтрования и концентрирования неочищенный продукт очищают с помощью препаративной ТСХ (гексан/ЕtOАс, об./об.=2/3) и получают соединение 58 (10,5 мг, 100%). МС с электрораспылением [М+1]+ 600,1.
Пример получения 59

Стадия А:

К раствору соединения 59а (0,53 г, 0,76 ммоль) в СН2Сl2 (4 мл) прибавляют Et3N (0,14 мл, 0,98 ммоль). Реакционную смесь охлаждают до -78°С и прибавляют ацетилхлорид (0,065 мл, 0,91 ммоль). Реакционную смесь медленно нагревают до комнатной температуры и перемешивают в течение 72 ч. К реакционной смеси прибавляют дополнительное количество Et3N (0,068 мл) и ацетилхлорида (0,033 мл) и затем ее перемешивают при комнатной температуре в течение 4 ч. Реакционную смесь концентрируют и очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=3/2) и получают соединение 59b (0,5 г).
Стадия В:

ВСl3 (3,7 мл, 3,7 ммоль, 1,0 М в гексане) при перемешивании по каплям прибавляют к раствору соединения 59b (0,55 г, 0,74 ммоль) в CH2Cl2 (9 мл) при -78°С. Реакцию останавливают через 1 ч путем прибавления водного раствора NаНСО3 (50 мл) при
-78°С. Реакционную смесь разбавляют с помощью ЕtOАс (200 мл) и промывают насыщенным водным раствором NаНСО3 (100 мл) и сушат над Na2SO4. Смесь фильтруют и концентрируют и получают неочищенное соединение 59с (0,4 г), которое используют в следующей реакции без дополнительной очистки.
Стадия С:

Перйодинан Десса-Мартина (0,12 г, 0,28 ммоль) при комнатной температуре прибавляют к смеси соединения 59с (0,12 г, 0,18 ммоль) и NаНСО3 (0,17 г, 2,0 ммоль) в CH2Cl2 (5,0 мл) и перемешивают в течение 45 мин. К реакционной смеси прибавляют дополнительное количество перйодинана Десса-Мартина (50 мг) и перемешивают при комнатной температуре в течение 2 ч. Затем реакционную смесь концентрируют и очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=1/1) и получают соединение 59d (0,1 г).
Стадия D:

Смесь соединения 59d (0,11 г, 0,17 ммоль), бикарбоната калия (26 мг, 0,19 ммоль), тозилметилизоцианида (36 мг, 0,19 ммоль) и метанола (3 мл) нагревают при 80°С в течение 48 ч. Затем реакционную смесь концентрируют и разбавляют с помощью ЕtOАс (200 мл) и промывают насыщенным водным раствором NаНСО3 (2×100 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=от 2/3 до 0/100) и получают соединение 59е (50 мг).
Стадия Е:

Соединение 59d (0,31 мг, 0,45 ммоль) в МеОН (10,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (0,2 г, 20 мас.%) и в течение 2 ч гидрируют с помощью H2 из баллона. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью МеОН (30 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (EtOAc/MeOH, об./об.=9/1) и получают смесь двух изомеров (190 мг), которые дополнительно очищают с помощью ВЭЖХ (хиральная колонка OD) с использованием смеси гексан/IPA (об./об.=9/1) и получают соединение 59 (90 мг).
Пример получения 60

Стадия А:

К раствору соединения 60а (0,26 г, 0,43 ммоль) в CH2Cl2 (4 мл) при 0°С прибавляют Et3N (0,071 мл, 0,51 ммоль), затем этилхлорформиат (0,052 мл, 0,56 ммоль) и реакционную смесь перемешивают в течение 1 ч. Затем к реакционной смеси прибавляют азид натрия (64 мг, 0,98 ммоль) и тетраметиламмонийгидросульфат (43 мг, 0,13 ммоль) и перемешивание продолжают в течение 1 ч. Затем реакционную смесь разбавляют с помощью CH2Cl2 (100 мл) и промывают водой (1×100 мл) и рассолом (1×100 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют. Остаток растворяют в сухом толуоле (4 мл) и нагревают при 80°С в течение 2 ч, а затем охлаждают до комнатной температуры. Прибавляют ТМСN3 (0,13 мл, 0,94 ммоль) и реакционную смесь нагревают при -110°С в течение 18 ч. Затем реакционную смесь охлаждают до комнатной температуры, концентрируют и очищают с помощью хроматографии на колонке BIOTAGE (гексан/ЕtOАс, об./об.=2/1, затем МеОН/ЕtOАс, об./об.=1/99) и получают соединение 60b (0,17 г).
Стадия В:

Соединение 60b (0,17 мг, 0,26 ммоль) в МеОН (10,0 мл) обрабатывают при комнатной температуре с помощью Pd(OH)2/C (15 мг, 20 мас.%) и в течение 2 ч гидрируют с помощью H2 из баллона. Раствор реакционной смеси фильтруют через тонкий слой целита и остаток промывают с помощью МеОН (30 мл). Растворитель удаляют при пониженном давлении и неочищенный продукт очищают с помощью хроматографии на колонке BIOTAGE (ЕtOАс/МеОН, об./об.=98/2) и получают соединение 60 (20 мг).
Хотя настоящее изобретение описано с помощью приведенных выше конкретных вариантов осуществления, для специалистов с общей подготовкой в данной области техники должны быть очевидны их многочисленные альтернативы, модификации и изменения. Подразумевается, что все такие альтернативы, модификации и изменения входят в сущность и объем настоящего изобретения.
Реферат
Изобретение относится к новым производным пиперидина формулы I: ! ! в которой: R1 и R2 выбраны из группы, включающей алкил, галогеналкил, алкил, замещенный одной или большим количеством гидроксигрупп, -CN, алкинил, -N(R6)2, -N(R6)-S(O2)-алкил, -N(R6)-C(O)-N(R9)2, -алкилен-CN, -циклоалкилен-CN, -алкилен-O-алкил, -С(O)-алкил, -C(=N-OR5)-aлкил, -С(O)-O-алкил, -алкилен-С(O)-алкил, -алкилен-С(O)-O-алкил, -алкилен-С(O)-N(R9)2 и группы ! , , , , ! при условии, что по меньшей мере один из R1 и R2 означает -CN или группу ! , , , , ! W означает: =C(R8)- или =N-; X означает -С(O)- или -S(O2)-; Y выбран из группы, включающей -СН2-, -О- и -N(R6)-C(O)-, при условии, что: (а) атом азота группы -N(R6)-С(O)- связан с X, и ! (b) если R1 и/или R2 означает и Y означает -O-, то Х не означает -S(O2)-; Z означает -C(R7)2-, -N(R6)-, или -O-; R3 выбран из группы, включающей Н и незамещенный алкил; R4 означает Н; R5 означает Н или алкил; R6 выбран из группы, включающей Н, алкил, циклоалкил и арил; каждый R7 независимо означает Н или алкил; или каждый R7 совместно с кольцевым атомом углерода, к которым, как показано, они присоединены, образует циклоалкиленовое кольцо; R8 выбран из группы, включающей Н, алкил, алкил, замещенный одной или большим количеством гидроксигрупп, -N(R6)2, -N(R6)-S(O2)-алкил, -N(R6)-S(O2)-apил, -N(R6)-C(O)-aлкил, -N(R6)-C(O)-apил, алкилен-O-алкил и -CN; R9 выбран из группы, включающей Н, алкил и арил, или каждый R9 совместно с атомом азота, к которому, как показано, они присоединены, образует гетероциклоалкильное кольцо; Аr1 означает незамещенный фенил; Аr2 означает фенил, замещенный 0-3 заместителями, выбранными из группы, включающей галогеналкил; n равно 0, 1 или 2; и m равно 1, 2 или 3, и к их фармацевтически приемлемым солям и гидратам. Изобретение также относится к фармацевтической композиции,
Формула

в которой R1 и R2 выбраны из группы, включающей алкил, галогеналкил, алкил, замещенный одной или большим количеством гидроксигрупп, -CN, алкинил, -N(R6)2, -N(R6)-S(O2)-алкил, -N(R6)-C(O)-N(R9)2, -алкилен-CN, -циклоалкилен-CN, -алкилен-O-алкил, -С(O)-алкил, -С(=N-OR5)-алкил, -С(O)-O-алкил, -алкилен-С(O)-алкил, -алкилен-С(O)-O-алкил, -алкилен-C(O)-N(R9)2 и группы
при условии, что по меньшей мере один из R1 и R2 означает -CN или группу
W означает =C(R8)- или =N-;
X означает -С(O)- или -S(O2)-;
Y выбран из группы, включающей -СН2-, -О- и -N(R6)-C(O)-, при условии, что:
(a) атом азота группы -N(R6)-C(O)- связан с X, и
(b) если R1 и/или R2 означает
Z означает -C(R7)2-, -N(R6)-, или -O-;
R3 выбран из группы, включающей H и незамещенный алкил;
R4 означает Н;
R5 означает Н или алкил;
R6 выбран из группы, включающей Н, алкил, циклоалкил и арил;
каждый R7 независимо означает Н или алкил; или
каждый R7 совместно с кольцевым атомом углерода, к которому, как показано, они присоединены, образует циклоалкиленовое кольцо;
R8 выбран из группы, включающей Н, алкил, алкил, замещенный одной или большим количеством гидроксигрупп, -N(R6)2, -N(R6)-S(O2)-алкил, -N(R6)-S(O2)-арил, -N(R6)-C(O)-алкил, -N(R6)-С(O)-арил, алкилен-O-алкил и -CN;
R9 выбран из группы, включающей Н, алкил и арил, или каждый R9совместно с атомом азота, к которому, как показано, они присоединены, образует гетероциклоалкильное кольцо;
Аr1 означает незамещенный фенил;
Аr2 означает фенил, замещенный 0-3 заместителями, выбранными из группы, включающей галогеналкил;
n равно 0, 1 или 2; и
m равно 1, 2 или 3,
и их фармацевтически приемлемые соли и гидраты.

в которой R1, R2, R3, R4, Ar1, Ar2 и n имеют указанные в п.1 значения, и их фармацевтически приемлемые соли и гидраты.
R3 означает незамещенный алкил;
Ar2 означает замещенный фенил;
n равно 1;
и их фармацевтически приемлемые соли и гидраты.
R3 означает незамещенный алкил;
Ar2 означает замещенный фенил;
n равно 1;
и их фармацевтически приемлемые соли и гидраты.
Ar2 означает 3,5-бис(трифторметил)фенил, и их фармацевтически приемлемые соли и гидраты.
R3 означает -СН3;
и его фармацевтически приемлемые соли и гидраты.
один из радикалов R1 или R2 означает

один из радикалов R1 или R2 означает

где W означает =C(R8)-;
и их фармацевтически приемлемые соли и гидраты.
один из радикалов R1 или R2 означает
и их фармацевтически приемлемые соли и гидраты.
один из радикалов R1 или R2 означает -CN,
и их фармацевтически приемлемые соли и гидраты.
Х означает -S(O2)-;
Y означает -СН2-; и
m равно 2,
и их фармацевтически приемлемые соли и гидраты.
Х означает -С(O)-;
Y означает -CH2-; и
m равно 2,
и их фармацевтически приемлемые соли и гидраты.
Х означает -С(O)-;
Y означает -CH2-; и
m равно 3,
и их фармацевтически приемлемые соли и гидраты.
Х означает -С(O)-;
Y означает -O-; и
m равно 2,
и их фармацевтически приемлемые соли и гидраты.
Х означает -С(O)-;
Y означает -СН2-; и
m равно 1,
и их фармацевтически приемлемые соли и гидраты.
Х означает -С(O)-;
Y означает -NH-C(O)-; и
m равно 1,
и их фармацевтически приемлемые соли и гидраты.
Z означает -NH-; и
R8 означает Н,
и их фармацевтически приемлемые соли и гидраты.
Z означает -NH-; и
R8 означает -NH-S(O2)-CH3,
и их фармацевтически приемлемые соли и гидраты.
Z означает -NH-; и
R8 означает -CH2-ОН,
и их фармацевтически приемлемые соли и гидраты.
Z означает -NH-; и
R8 означает -СН2-O-СН3,
и их фармацевтически приемлемые соли и гидраты.
Z означает -NH-; и
R8 означает -NH2,
и их фармацевтически приемлемые соли и гидраты.
Z означает
R8 означает Н;
и их фармацевтически приемлемые соли и гидраты.
Z означает -С(СН3)2-; и
R8 означает Н;
и их фармацевтически приемлемые соли и гидраты.
в которой R1 и R2 выбраны из группы, состоящей из:
и их фармацевтически приемлемые соли и гидраты.
и его фармацевтически приемлемые соли и гидраты.

и его фармацевтически приемлемые соли и гидраты.

и его фармацевтически приемлемые соли и гидраты.
Комментарии