Производные 1-(4,5-дигидроимидазол)-изохромана или 1-(4,5-дигидро)-изотиохромана, полезные в качестве агонистов альфа2 адренорецепторов - RU2642065C2
Код документа: RU2642065C2
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к фармакологически активным изохромановым и изотиохромановым производным или их фармацевтически приемлемым солям и эфирам, а также содержащим их фармацевтическим композициям, и их применению в качестве агонистов альфа2 адренорецепторов, особенно агонистов альфа2A.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Альфа2-адренорецепторы (α2) представляют собой рецепторы клеточных мембран, связанные с G белком, широко распространенные у людей, и их можно разделить на три подтипа у людей: альфа2A, альфа2B и альфа2C (Bylund et al., Mol. Pharmacol., 1992, 42, 1-5). Альфа2-адренорецепторы обладают множеством биологических функций, и соединения, воздействующие на данные рецепторы, представляют собой важные мишени для различных заболеваний (Goodman & Gilman's The Pharmacological Basis of Therapeutics, 12-е издание, 2011, глава 12; Brede et al. Biol. Cell 2004, 96, 343-348). Действительно, получены многие альфа2 активные соединения (Gentili et al., Curr. Top Med. Chem., 2007, 7, 163-186), и они проходят испытания в клинических условиях (Crassous et al., Curr. Top Med. Chem., 2007, 7, 187-194). Например, частичный альфа2A агонист, клонидин, применяют в качестве агента, снижающего кровяное давление, и неселективный полный агонист подтипа не альфа2, дексмедетомидин, применяют в качестве успокаивающего средства в блоках интенсивной терапии.
Патент США №3438995 описывает некоторые изохромановые и изотиохромановые производные и предполагается, что они являются полезными в качестве ускорителей вулканизации каучука, антиоксидантов, ингибиторов коррозии, успокоительных средств центральной нервной системы (ЦНС) и противовоспалительных средств. WO 2007/085558 описывает ряд имидазольных производных, пригодных в качестве лигандов TAAR для лечения ряда расстройств, включая различные расстройства ЦНС.
СУЩНОСТЬ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Цель настоящего изобретения заключается в обеспечении новых соединений, обладающих агонистическими активностями относительно адренергических альфа-рецепторов, особенно относительно альфа2A рецептора. Данные соединения можно применять для лечения расстройств, состояний или заболеваний, подобных бреду (например, гиперактивному бреду), бессоннице, синдрому дефицита внимания с гиперактивностью, абстиненции при отмене приема бензодиазепинов (или алкоголя или опиоидов или табака), преждевременной эякуляции, гипертензии, тахикардии, синдрому беспокойных ног, мышечной спастичности, приступообразному ощущению жара, тревоге, посттравматическому стрессовому расстройству, боли, хроническому тазовому болевому синдрому и приступу неконтролируемой боли при раке и другим подобным заболеваниям, которые можно лечить адренергическими альфа2 агонистами, особенно альфа2A агонистами. Соответственно, настоящее изобретение, кроме того, относится к соединениям, которые будут применять в качестве совместного успокаивающего или болеутоляющего агента в лечении млекопитающих. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим соединения по настоящему изобретению.
Соединения настоящего изобретения представляют собой перорально активные, проникающие в мозг, селективные альфа2A агонисты. Они обладают повышенной альфа2A активностью и/или альфа2A агонистической селективностью относительно других альфа рецепторов и/или повышенной эффективностью, а также улучшенным метаболизмом в гепатоцитах печени in vitro, что все вместе дает среднюю in vivo продолжительность действия. Помимо приведенных выше фармакологических эффектов, соединения настоящего изобретения имеют меньше побочных эффектов и из-за слабого взаимодействия с цитохромом P450.
Приведенные выше, а также другие признаки и преимущества идей настоящего изобретения будут более понятны из следующего описания и формулы изобретения.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым изохромановым и изотиохромановым производным, имеющим общую формулу I,
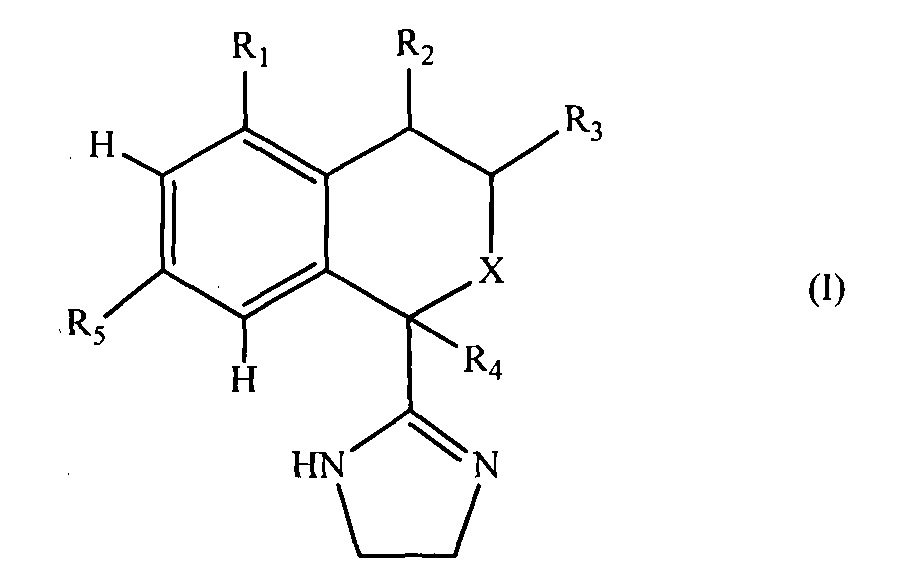
где
X представляет собой O или S;
R1 представляет собой гидрокси, галоген, (C1-C6)алкил, галоген(C1-C6)алкил, (C2-C6)алкенил, (C2-C6)алкинил, цикло(C3-C6)алкил, (C1-C6)алкокси, галоген(C1-C6)алкокси, гидрокси(C1-C6)алкил, циано, (C1-C6)алкокси(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкокси(C1-C6)алкил, гидрокси(C1-C6)алкокси, гидрокси(C1-C6)алкокси(C1-C6)алкил, гидрокси(C2-C6)алкенил, (C2-C6)алкенилокси(C2-C6)алкенил, галоген(C1-C6)алкокси(C1-C6)алкил, галоген(C1-C6)алкокси-галоген(C1-C6)алкил, (C1-C6)алкоксигалоген(C1-C6)алкокси, карбокси, (C1-C6)алкил-(C=O)-, (C1-C6)алкокси-(C=O)-, (С1-C6)алкил-(C=O)-O-, галоген(C1-C6)алкил-(C=O)-, галоген(C1-C6)алкокси-(C=O)-, (R6)2N-, (R6)2N-(С1-С6)алкил, (R6)2N-(C=O)-, R6-(C=O)-N(R6)-(C=O)-, R6-(O=S=O)-N(R6)-(C=O)-, R6-(C=O)-N(R6)-(O=S=O)-, R6-(O=S=O)-N(R6)-(O=S=O)-, (R6)2N-N-, (R6)N=N-, (R6)2N-O-(R6)0-N(R6)-, (C1-C6)алкил-S-, (C2-C6)алкенил-S-(C2-C6)алкенил, гидрокси(C1-C6)алкил-S-, гидрокси(C1-C6)алкил-S-(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил(C1-C6)алкил, галоген(C1-C6)алкил-S-, галоген(C1-C6)алкил-S-(C1-C6)алкил, галоген(C1-C6)алкил-S- галоген(C1-C6)алкил, R6-(O=S)-, (R6)2N-(O=S)-, R6-(O=S=O)-, (R6)2N-(O=S=O)-, фенил, фенил-O-, гетероарил, гетероарил-O-, фенил-N(R6)-, гетероарил-N(R6)- или гетероарил(C1-C6)алкил;
R2 представляет собой H, гидрокси, оксо, фтор, (C1-C6)алкил, галоген(C1-C6)алкил, (C2-C6)алкенил, (C2-C6)алкинил, цикло(C3-C6)алкил, (C1-C6)алкокси, галоген(C1-C6)алкокси, гидрокси(C1-C6)алкил, циано, (C1-C6)алкокси(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкокси(C1-C6)алкил, гидрокси(C1-C6)алкокси, гидрокси(C1-C6)алкокси(Cl-C6)алкил, гидрокси(C2-C6)алкенил, (C2-C6)алкенилокси(C2-C6)алкенил, галоген(C1-C6)алкокси(C1-C6)алкил, галоген(C1-C6)алкоксигалоген(C1-C6)алкил, (C1-C6)алкокси-галоген(C1-C6)алкокси, карбокси, (C1-C6)алкил-(C=O)-, (C1-C6)алкокси-(C=O)-, (C1-C6)алкил-(C=O)-O-, галоген(C1-C6)алкил-(C=O)-, галоген(C1-C6)алкокси-(C=O)-, (R6)2N-, (R6)2N-(C1-C6)алкил, (R6)2N-(C=O)-, R6-(C=O)-N(R6)-(C=O)-, R6-(O=S=O)-N(R6)-(C=O)-, R6-(C=O)-N(R6)-(O=S=O)-, R6-(O=S=O)-N(R6)-(O=S=O)-, (R6)2N-N-, (R6)N=N-, (R6)2N-O-, (R6)0=N-, (С1-С6)алкил-S-, (C2-C6)алкенил-S-(C2-C6)алкенил, гидрокси(C1-C6)алкил-S-, гидрокси(C1-C6)алкил-S-(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил-S-(C1-C6)алкил, галоген(C1-C6)алкил-S-, галоген(C1-C6)алкил-S-(C1-C6)алкил, галоген(C1-C6)алкил-S-галоген(C1-C6)алкил, R6-(O=S)-, (R6)2N-(O=S)-, R6-(O=S=O)-, (R6)2N-(O=S=O)-, фенил, фенил-O-, гетероарил, гетероарил-O, фенил-N(R6)-, гетероарил-N(R6)- или гетероарил(C1-C6)алкил;
R3 представляет собой H, (C1-C6)алкил, галоген(C1-C6)алкил, (С1-C6)алкокси, галоген(C1-C6)алкокси, гидрокси(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил или цикло(C3-C6)алкил;
R4 представляет собой H, гидрокси, галоген, (C1-C2)алкил или галоген(C1-C2)алкил;
R5 представляет собой H, гидрокси, галоген, (C1-C6)алкил, галоген(C1-C2)алкил, цикло(C3-C6)алкил, (C1-C6)алкокси, галоген(C1-C6)алкокси, гидрокси(C1-C6)алкил, фенил или гетероарил; и
R6 представляет собой, независимо в каждом случае, H, (C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкилгидрокси(C1-C6)алкил, галоген(C1-C6)алкил, или R6 и R6 образуют вместе с атомами, к которым они присоединены, конденсированное 4-, 5-, 6- или 7-членное насыщенное или ненасыщенное карбоциклическое кольцо или конденсированное 4-, 5-, 6- или 7-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1 или 2 гетероатома, выбранные из N, O и S, где указанное карбоциклическое или гетероциклическое кольцо является незамещенным или замещенным 1 или 2 заместителями, причем каждый независимо представляет собой гидрокси, оксо, галоген, (C1-C6)алкил, гидрокси(C1-C6)алкил или галоген(C1-C6)алкил-;
или R1 и R2 образуют вместе с атомами углерода кольца, к которым они присоединены, конденсированное 4-, 5-, 6- или 7-членное насыщенное или ненасыщенное карбоциклическое кольцо или конденсированное 4-, 5-, 6- или 7-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N, O и S, где указанное карбоциклическое или гетероциклическое кольцо является незамещенным или замещенным 1 или 2 заместителями, причем каждый независимо представляет собой гидрокси, оксо, галоген, (C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил, гидрокси(C1-C6)алкил или галоген(C1-C6)алкил;
или R2 и R3 образуют вместе с атомами углерода кольца, к которым они присоединены, конденсированное 4-, 5-, 6- или 7-членное насыщенное или ненасыщенное карбоциклическое кольцо или конденсированное 4-, 5-, 6- или 7-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N, O и S, где указанное карбоциклическое или гетероциклическое кольцо является незамещенным или замещенным 1 или 2 заместителями, причем каждый независимо представляет собой гидрокси, оксо, галоген, (C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил, гидрокси(C1-C6)алкил или галоген(C1-С6)алкил;
или их фармацевтически приемлемой соли или сложному эфиру.
В возможной подгруппе соединений формулы I,
R1 представляет собой гидрокси, галоген, (C1-C6)алкил, галоген(C1-C6)алкил, (C2-C6)алкенил, (C2-C6)алкинил, цикло(C3-C6)алкил, (C1-C6)алкокси, галоген(C1-C6)алкокси, гидрокси(C1-C6)алкил, циано, (C1-C2)алкокси(C1-C2)алкил, гидрокси(C1-C2)алкокси, галоген(C1-C2)алкокси(C1-C2)алкил, галоген(C1-C2)алкоксигалоген(C1-C2)алкил, (C1-C2)алкоксигалоген(C1-C2)алкокси, карбокси, (C1-C3)алкил-(C=O)-, (C1-C3)алкокси-(C=O)-, галоген(C1-C3)алкил-(C=O)-, галоген(C1-C3)алкокси-(C=O)-, (R6)2N-(C1-C2)алкил, (R6)2N-(C=O)-, (C1-C6)алкил-S-, R6-(O=S)-, R6-(O=S=O)-, (R6)2N-(O=S=O)-, фенил, фенил-O-, гетероарил, гетероарил-O- или гетероарил(C1-C2)алкил;
R2 представляет собой H, гидрокси, оксо, фтор, (C1-C6)алкил, галоген(C1-C6)алкил, цикло(C3-C6)алкил, (C1-C6)алкокси, галоген(C1-C6)алкокси или циано;
R3 представляет собой H, (C1-C6)алкил, галоген(C1-C6)алкил, (C1-C6)алкокси, гидрокси(C1-C6)алкил, (C1-C6)алкокси(C1-C6)алкил или цикло(C3-C6)алкил;
R4 представляет собой H, фтор, (C1-C2)алкил или галоген(C1-C2)алкил;
R5 представляет собой H, гидрокси, галоген, (C1-C6)алкил, галоген(C1-C6)алкил, цикло(C3-C6)алкил, (C1-C6)алкокси, галоген(C1-C6)алкокси, фенил или гетероарил; и
R6 представляет собой, независимо в каждом случае, H, (C1-C3)алкил, или R6 и R6 образуют, вместе с атомами, к которым они присоединены, конденсированное 5-, 6- или 7-ленное насыщенное или ненасыщенное карбоциклическое кольцо или конденсированное 5-, 6- или 7-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N, O и S, где указанное карбоциклическое или гетероциклическое кольцо является незамещенным или замещенным 1 или 2 заместителями, причем каждый независимо представляет собой гидрокси, оксо, галоген, (C1-C2)алкил или галоген(C1-C2)алкил-;
или R1 и R2 образуют вместе с атомами углерода кольца, к которым они присоединены, конденсированное 5-, 6- или 7-членное насыщенное или ненасыщенное карбоциклическое кольцо или конденсированное 5-, 6- или 7-членное насыщенное или ненасыщенное гетероциклическое кольцо, содержащее 1 или 2 гетероатома, выбранных из N, O и S, где указанное карбоциклическое или гетероциклическое кольцо является незамещенным или замещенным 1 или 2 заместителями, причем каждый независимо представляет собой гидрокси, оксо, галоген, (C1-C2)алкил, (C1-C2)алкокси, гидрокси(C1-C2)алкил или галоген(C1-C2)алкил.
В следующей возможной подгруппе соединений формулы I,
R1 представляет собой гидрокси, галоген, (C1-C6)алкил, галоген(C1-C6)алкил, (C2-C6)алкенил, (C2-C6)алкинил, цикло(C3-C6)алкил, (C1-C6)алкокси, галоген(C1-C6)алкокси, гидрокси(C1-C6)алкил, циано, (R6)2N-(C=O)-, (C1-C6)алкил-S- или гетероарил; и/или
R2 представляет собой H или (C1-C6)алкил; и/или
R3 представляет собой H, (C1-C6)алкил, галоген(C1-C6)алкил или (C1-C6)алкокси(C1-C6)алкил; и/или
R4 представляет собой H или (C1-C2)алкил; и/или
R5 представляет собой H, гидрокси, галоген, (C1-C6)алкил или (C1-C6)алкокси; и/или
R6 представляет собой H; и/или
R1 и R2 образуют вместе с атомами углерода кольца, к которым они присоединены, конденсированное 6- или 7-членное насыщенное или ненасыщенное карбоциклическое кольцо; например,
R1 представляет собой гидрокси, галоген, (C1-C3)алкил, галоген(C1-C3)алкил, (C1-C3)алкокси, галоген(C1-C3)алкокси или гидрокси(C1-C3)алкил; и/или
R2 представляет собой H или (C1-C2)алкил; и/или
R3 представляет собой H, (C1-C3)алкил или галоген(C1-C3)алкил; и/или
R4 представляет собой H или метил; и/или
R5 представляет собой H, галоген или (C1-C2)алкил; и/или
R1 и R2 образуют вместе с атомами углерода кольца, к которым они присоединены, конденсированное 6- или 7-членное насыщенное или ненасыщенное карбоциклическое кольцо; например,
R1 представляет собой гидрокси, галоген, (C1-C3)алкил, галоген(C1-C3)алкил, (C1-C3)алкокси, галоген(C1-C3)алкокси или гидрокси(C1-C3)алкил; и/или
R2 представляет собой H или (C1-C2)алкил; и/или
R3 представляет собой H, (C1-C3)алкил или галоген(C1-C3)алкил; и/или
R4 представляет собой H или метил; и/или
R5 представляет собой H, галоген или (C1-C2)алкил; такую как
R1 представляет собой галоген, (C1-C2)алкил, галоген(C1-C2)алкил, (C1-C2)алкокси или галоген(C1-C2)алкокси; и/или R2 представляет собой H; и/или
R3 представляет собой H или (C1-C2)алкил; и/или
R4 представляет собой H; и/или
R5 представляет собой H.
В следующей возможной подгруппе соединений формулы I, X представляет собой O.
В еще другой возможной подгруппе соединений формулы I, соединение представляет собой 2-(5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазол, 1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-карбонитрил, 2-(5-аллилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-винилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол, гидрохлорид 2-(5-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола, сульфат 2-(5-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола, фумарат 2-(5-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола, 1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-ола, (1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-ил)метанол, 2-(5-бром-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-хлор-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол, более медленно элюирующийся изомер 1-(1-(4,5-дигидро-1H-имидазол-2-ил)-1-метилизохроман-5-ил)-2,2-диметилпропан-1-ола, более быстро элюирующийся изомер 1-(1-(4,5-дигидро-1H-имидазол-2-ил)-1-метилизохроман-5-ил)-2,2-диметилпропан-1-ола, 2-(5-этинилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-3-этил-5-(трифторметил)изохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол, гидрохлорид 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола, сульфат 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола, гемифумарат 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(5-йодизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-3-метил-5-(трифторметил)изохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бром-4-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, более быстро элюирующийся изомер 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазола, более медленно элюирующийся изомер 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-((3R)-1,3,5-триметилизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(5-циклопропилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-хлор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-хлор-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(1,3,5-триметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бром-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-хлор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3S)-5-хлор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3S)-5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3S)-3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-метокси-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бром-3-пропилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-изопропилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-фторизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-метокси-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, гидрохлорид 2-((3R)-5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, гемифумарат 2-((3R)-5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(3-этил-5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-3,5-диэтилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол гидрохлорид, сульфат 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, гемифумарат 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-((3R)-3-метил-5-(трифторметокси)изохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-фтор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-этоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-метил-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3S)-5-метокси-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-(фуран-3-ил)изохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-(проп-1-ин-1-ил)изохроман-1-ил)-4,5-дигидро-1H-имидазол, 1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-карбоксамид, 2-(3,7,8,9,10,10a-гексагидро-1H-циклогепта[de]изохромен-3-ил)-4,5-дигидро-1H-имидазол, более медленно элюирующийся изомер 1-(1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-ил)этанола, 2-(5,7-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(7-бром-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(7-метокси-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(3,5-диметилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бром-3-метилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-метилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бромизотиохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-бром-1-метилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол, гидрохлорид 2-(5,7-дибром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер гидрохлорида 2-5-бром-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(5-метокси-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-метоксиизотиохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-метокси-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-((3R)-5-этил-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, 2-(5-метил-3-(метоксиметил)изохроман-1-ил)-4,5-дигидро-1H-имидазол, гидробромид 1-(4,5-дигидро-1H-имидазол-2-ил)-5-метилизохроман-7-ола, гидрохлорид 1-(4,5-дигидро-1H-имидазол-2-ил)-3-этилизохроман-5-ола, энантиомер 2 2-(5-метокси-3-(2,2,2-трифторэтил)метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(1,5-диметилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол, гидрохлорид 2-(5-(трифторметокси)изохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 1 гидрохлорида 2-(3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола, гидрохлорид 2-(3-(2-фторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2 гидрохлорида 2-5-бром-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола, гидрохлорид 2-(3-(2,2-дифторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(7-метокси-3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол, энантиомер 2 гидрохлорида 2-((3)-5-метил-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(5-(метильтио)изохроман-1-ил)-4,5-дигидро-1H-имидазол, энантиомер 2 гидрохлорида 2-((3)-5-бром-3-пропилизохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2 гидрохлорида 2-((3R)-3-(2,2-дифторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, гидрохлорид 2-(5-(дифторметокси)изохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-((3R)-3-этил-5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол, энантиомер 1 гидрохлорида 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола, гидрохлорид 2-((3R)-5-(дифторметокси)-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2-(5-бром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол, энантиомер 1 гидрохлорида 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2 2-(3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2 гидрохлорида 2-(5-метоксиoизохроман-1-ил)-4,5-дигидро-1H-имидазола, 2-(1-метил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазол, гидрохлорид 2-(5-(дифторметил)изохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2 гидрохлорида 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2 гидрохлорида 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола, гидрохлорид 2-(1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола, энантиомер 2-(1-метил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола, гидрохлорид 2-(3-метил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола, гидрохлорид 2-(3-этил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола или энантиомер 2-(5-бром-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола.
Термины, применяемые в настоящем изобретении, имеют значения, указанные ниже. Термин "по меньшей мере, один", применяемый в значениях ниже, относится к одному или нескольким, таким как один. Например, термин "по меньшей мере, один галоген" относится к одному или нескольким галогенам, например трем, двум или одному галогену, таким как три галогена.
Термин "гидрокси", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к -OH группе.
Термин "галоген", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к фтору, хлору, брому или йоду.
Термин "(C1-C6)алкил", "(C1-C4)алкил", "(C1-C3)алкил" и "(C1-C2)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к насыщенной нормальной или разветвленной углеводородной цепи, содержащей 1-6, 1-4, 1-3 и 1-2 атомов углерода соответственно. Репрезентативные примеры (C1-C6)алкила, (C1-C4)алкила, (C1-C3)алкила и (C1-C2)алкила включают, но не ограничиваются, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил и н-гексил.
Термин "(C2-C6)алкенил" и "(C2-C3)алкенил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к нормальной или разветвленной углеводородной цепи, содержащей 2-6 и 2-3 атомов углерода, соответственно, и содержащей, по меньшей мере, одну углерод-углерод двойную связь. Репрезентативные примеры (C2-C6)алкенила и (C2-C3)алкенила включают, но не ограничиваются, этенил и проп-2-ен-1-ил.
Термин "(C2-C6)алкинил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к нормальной или разветвленной углеводородной цепи, содержащей 2, 3, 4, 5 или 6 атомов углерода и содержащей, по меньшей мере, одну углерод-углерод тройную связь. Репрезентативные примеры (C2-C6)алкинила включают, но не ограничиваются, этинил, проп-1-ин-1-ил и проп-2-инил.
Термин "цикло(C3-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к насыщенной углеводородной группе, содержащей циклическую группу и содержащей 3, 4, 5 или 6 атомов углерода. Репрезентативные примеры цикло(C3-C6)алкила включают, но не ограничиваются, циклопропил, циклобутил, циклопентил и циклогексил.
Термин "(C1-C6)алкокси" и "(C1-C4)алкокси", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к (C1-C6)алкильной или (C1-C4)алкильной группе, соответственно, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через атом кислорода. Репрезентативные примеры (C1-C6)алкокси и (C1-C4)алкокси включают, но не ограничиваются, метокси, этокси, н-пропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, 2,2-диметилпропокси, 3-метилбутокси и н-гексокси.
Термин "галоген(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к, по меньшей мере, одному галогену, как определено в настоящем изобретении, присоединенному к основному молекулярному фрагменту через (C1-C6)алкильную группу, как определено в настоящем изобретении. Когда присутствует несколько галогенов, галогены можно присоединять к одному или различным атомам углерода, и галогены могут быть одинаковыми или отличными. Репрезентативные примеры галоген(C1-C6)алкила включают, но не ограничиваются, фторметил, дифторметил, трифторметил, 2-фторэтил, 2-хлорэтил, 2,2,2-трифторэтил, 1,2,2-трифторэтил, 2-хлорпропил, 3-фторпропил, 3-бромпропил, 1,3-дифторпропил и 3,3,3-трифторпропил.
Термин "галоген(C1-C6)алкокси", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к, по меньшей мере, одному галогену, присоединенному к основному молекулярному фрагменту через (C1-C6)алкокси группу, как определено в настоящем изобретении. Когда присутствует несколько галогенов, галогены можно присоединять к одному или различным атомам углерода, и галогены могут быть одинаковыми или отличными. Репрезентативные примеры галоген(C1-C6)алкокси включают, но не ограничиваются, фторметокси, хлорметокси, дифторметокси, трифторметокси, 2-бромэтокси, 2,2,2-трифторэтокси, 3-фторпропокси, 2-хлорпропокси, 3,3,3-трифторпропокси и 4-фторбутокси.
Термин "галоген(C1-C6)алкокси(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к галоген(C1-C6)алкокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C1-C6)алкильную группу, как определено в настоящем изобретении.
Термин "галоген(C1-C6)алкоксигалоген(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к галоген(C1-C6)алкокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через галоген(C1-C6)алкильную группу, как определено в настоящем изобретении.
Термин "(C1-C6)алкоксигалоген(C1-C6)алкокси", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к (C1-C6)алкокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через галоген(C1-C6)алкокси группу, как определено в настоящем изобретении.
Термин "карбокси", как применяют в настоящем изобретении, как часть другой группы, относится к -COOH группе.
Термин "циано", как применяют в настоящем изобретении, как часть другой группы, относится к -CN группе.
Термин "оксо", как применяют в настоящем изобретении, как часть другой группы, относится к=O группе.
Термин "гидрокси(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к, по меньшей мере, одной гидрокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C1-C6)алкильную группу, как определено в настоящем изобретении. Репрезентативные примеры гидрокси(C1-C6)алкила включают, но не ограничиваются, гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 2,2-дигидроксиэтил, 1-гидроксипропил, 3-гидроксипропил, 1-гидрокси-1-метилэтил, 1-гидрокси-1-метилпропил и 1-гидрокси-2,2-диметилпроп-1-ил.
Термин "гидрокси(C2-C6)алкенил", как применяют в настоящем изобретении, относится к, по меньшей мере, одной гидрокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C2-C6)алкенильную группу, как определено в настоящем изобретении. Репрезентативные примеры гидрокси(C2-C6)алкенила включают, но не ограничиваются, 1-гидроксиэтенил, 2-гидроксиэтенил и 1-гидроксипроп-2-енил.
Термин "(C1-C6)алкокси(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к, по меньшей мере, одной (C1-C6)алкокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C1-C6)алкильную группу, как определено в настоящем изобретении. Когда присутствует несколько (C1-C6)алкокси групп, (C1-C6)алкокси группы могут быть одинаковыми или различными. Репрезентативные примеры (C1-C6)алкокси(C1-C6)алкила включают, но не ограничиваются, метоксиметил, этоксиметил, пропоксиметил, 2-метоксиэтил, 2-этоксиэтил, 2,2-диметоксиэтил, 1-метил-2-пропоксиэтил, 1-метокси-1-метилэтил и 4-метоксибутил.
Термин "гидрокси(C1-C6)алкокси", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к, по меньшей мере, одной гидрокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C1-C6)алкокси группу, как определено в настоящем изобретении. Репрезентативные примеры гидрокси(C1-C6)алкокси включают, но не ограничиваются, гидроксиметокси, дигидроксиметокси, 2-гидроксиэтокси, 2-гидроксипропокси, 3-гидроксипропокси, 2-гидроксибутокси и 2-гидрокси-1-метилэтокси.
Термин "гидрокси(C1-C6)алкокси(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к гидрокси(C1-C6)алкокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C1-C6)алкильную группу, как определено в настоящем изобретении.
Термин "(C1-C6)алкокси(C1-C6)алкокси", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к, по меньшей мере, одной (C1-C6)алкокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C1-C6)алкокси группу, как определено в настоящем изобретении. (C1-C6)алкокси группы могут быть одинаковыми или различными. Репрезентативные примеры (C1-C6)алкокси(C1-C6)алкокси включают, но не ограничиваются, метоксиметокси, пропоксиметокси, 2-метоксиэтокси, 2-этоксиэтокси, 2-бутоксиэтокси, 2,2-диметоксиэтокси, 1-метил-2-пропоксиэтокси, 2-метоксипропокси и 4-метоксибутокси.
Термин "(C1-C6)алкокси(C1-C6)алкокси(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к (C1-C6)алкокси(C1-C6)алкокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C1-C6)алкильную группу, как определено в настоящем изобретении.
Термин "фенил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к 6-членному ароматическому карбоциклическому кольцу, которое может быть незамещенным или замещенным 1 или 2 заместителями, причем каждый независимо представляет собой гидрокси, галоген, (C1-C4)алкил, (С1-С4)алкокси или галоген(C1-C4)алкил.
Термин "гетероарил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к 3-7-членной ароматической моноциклической кольцевой системе, содержащей от одного до трех гетероатомов, выбранных из кислорода, азота и серы. Указанный гетероарил может быть незамещенным или замещенным 1 или 2 заместителями, причем каждый независимо представляет собой гидрокси, галоген, оксо, (C1-C4)алкил, (C1-C4)алкокси или галоген(C1-C4)алкил. Репрезентативные примеры гетероарила включают, но не ограничиваются, фуранил, тиофенил и пиразолил.
Термин "гетероарил(C1-C6)алкил", как применяют в настоящем изобретении, как есть или как часть другой группы, относится к гетероарилу, как определено в настоящем изобретении, присоединенному к основному молекулярному фрагменту через (C1-C6)алкильную группу, как определено в настоящем изобретении.
Термин "(C2-C6)алкенилокси", как применяют в настоящем изобретении как часть другой группы, относится к (C2-C6)алкенильной группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через атом кислорода. Репрезентативные примеры (C2-C6)алкенилокси включают, но не ограничиваются, этенилокси, проп-2-енилокси, бут-2-енилокси и гекс-3-енилокси.
Термин "(C2-C6)алкенилокси(C2-C6)алкенил", как применяют в настоящем изобретении, относится к, по меньшей мере, одной (C2-C6)алкенилокси группе, как определено в настоящем изобретении, присоединенной к основному молекулярному фрагменту через (C2-C6)алкенильную группу, как определено в настоящем изобретении. Когда присутствует несколько (C2-C6)алкенилокси групп, (C2-C6)алкенилокси группы могут быть одинаковыми или различными. Репрезентативные примеры (C2-C6)алкенилокси(C2-C6)алкенила включают, но не ограничиваются, этенилоксиэтенил и проп-2-енилоксиэтенил.
Выражение "соединения настоящего изобретения", как применяют в настоящем изобретении, относится к соединениям формулы I.
"Фармацевтически приемлемые соли" согласно настоящему изобретению включают терапевтически активные, нетоксичные основные и кислые солевые формы, которые способны образовывать соединения формулы I с органическими и неорганическими основаниями и кислотами. Репрезентативные примеры фармацевтически приемлемых основно-аддтивных солевых форм, например солей металла или амина, включают, но не ограничиваются, аммониевые соли, соли лития, натрия, калия, кальция, магния, алюминия и цинка, соли с органическими основаниями, такими как N-метил-D-глюкамин, гидрабаминовые соли и соли с аминокислотами, такими как аргинин, лизин, и подобные. Репрезентативные примеры фармацевтически приемлемых кислотно-аддитивных солей включают, но не ограничиваются, хлориды, бромиды, сульфаты, нитраты, фосфаты, сульфонаты, метансульфонаты, формиаты, тартраты, малеаты, цитраты, бензоаты, салицилаты, аскорбаты, ацетаты, оксалаты, фумараты, гемифумараты и сукцинаты.
Фармацевтически приемлемые сложные эфиры, при необходимости, можно получить известными способами, применяя фармацевтически приемлемые кислоты, которые являются общепринятыми в области фармацевтических средств и которые сохраняют фармакологические свойства свободной формы. Неограничивающие примеры данных эфиров включают эфиры алифатических или ароматических спиртов. Репрезентативные примеры фармацевтически приемлемых эфиров включают, но не ограничиваются, метиловый, этиловый, н-пропиловый, изопропиловый, н-бутиловый, изобутиловый, втор-бутиловый, трет-бутиловый и бензиловый эфиры.
Настоящее изобретение включает в свой объем все возможные геометрические изомеры, например Z и E изомеры (цис- и транс-изомеры), соединений настоящего изобретения, а также все возможные оптические изомеры, такие как диастереомеры и энантиомеры, соединений настоящего изобретения. Кроме того, настоящее изобретение включает в свой объем и отдельные изомеры и любые их смеси, такие как рацемическая смесь. Отдельные изомеры можно получить, применяя соответствующие изомерные формы исходных веществ, или их можно разделить после получения конечного соединения согласно общепринятым способам разделения. Для выделения оптических изомеров, таких как энантиомеры, из их смеси, можно применять общепринятые способы разделения, например фракционную кристаллизацию или препаративную хиральную хроматографию.
Соединения формулы I можно получить рядом путей синтеза, аналогично или согласно способам, известным в литературе, применяя подходящие исходные соединения, например, реакцией 2-фенилэтанола или 2-фенилэтантиола с альдегидом или ацеталем, или согласно другим известным способам (Larghi et al., Synthesis, 2006, 2, 187-220; Ishibashi et al., J Heterocyclic Chem 1985, 22, 1527-1529). Имидазолены можно получить, например, реакцией этан-1,2-диамина с эфиром или альдегидами (Gentili et al., J. Med. Chem., 2003, 46, 2169-2176; Ishihara et al, Synthesis, 2007, 1939-1942).
Исходные соединения, показанные ниже, являются имеющимися в продаже, или их можно получить синтетическими способами, известными в литературе, например восстановлением карбоновой кислоты, эфира карбоновой кислоты или кетонов, раскрытием соответствующего эпоксида металлированными ароматическими соединениями, ферментативным гидролизом или хиральным разделением рацемического спирта (Bunnet et al., J. Org. Chem., 1962, 27, 3836-3843.; Mangas-Sanchez et al., Organic Lett, 2010, 12, 3498-3501; Knolker et al., Tetrahedron Lett, 2000, 41, 1171-1174).
В общем, соединения формулы I можно получить аналогично или согласно следующей схеме 1:
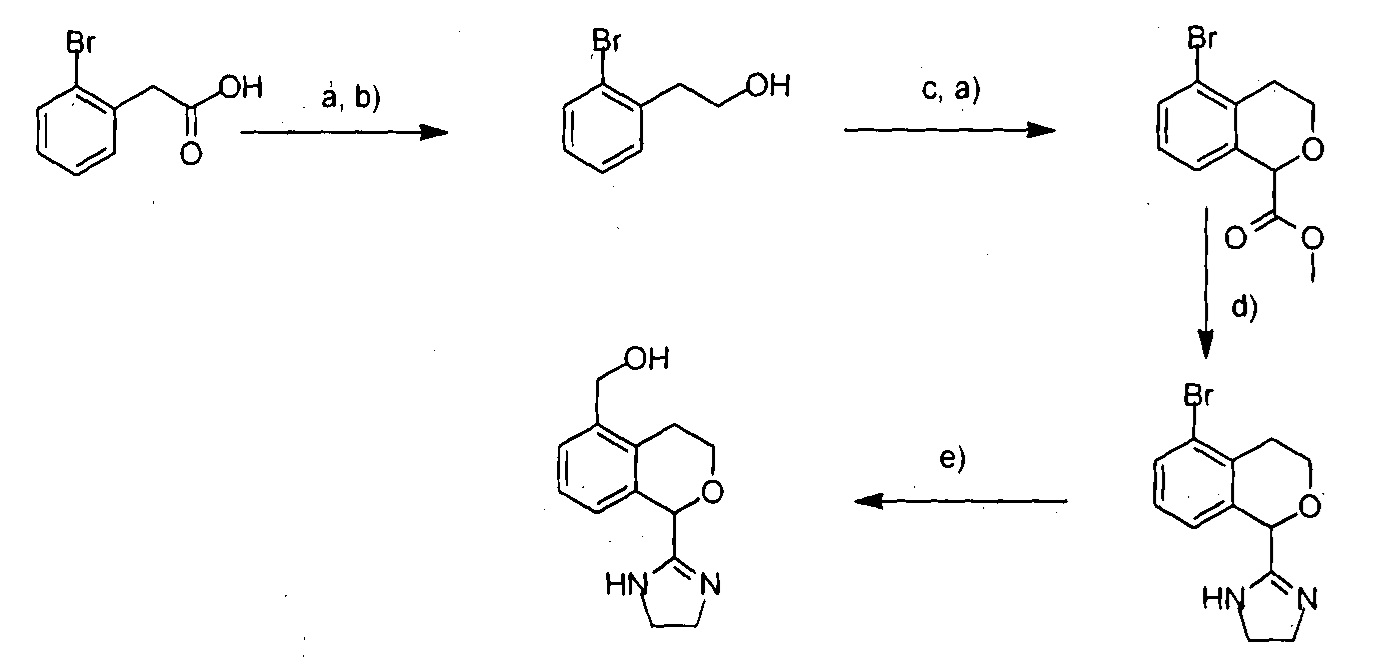
Схема 1
В которой a) SOCl2, MeOH, b) NaBH4, c) TFA, 2,2-дигидроксиуксусная кислота, d) этилендиамин, Me3Al, e) Pd(PPh3)4, HOCH2SnBu3.
Специалист в данной области техники отдает себе отчет в том, что любое исходное соединение или промежуточное соединение в реакциях, описанных выше, можно защищать, при необходимости, способом, известным в данной области техники. Затем любую защищенную функциональную группу можно деблокировать способом, известным в данной области техники.
Синтетические способы, описанные выше, предназначены для иллюстрации получения соединений формулы I, и получение никоим образом не ограничивается ими, то есть имеются также другие возможные способы получения, которые входят в область известных знаний для специалиста в данной области техники.
Соединения формулы I можно превратить, при желании, в их фармацевтически приемлемые солевые или сложноэфирные формы, применяя способы, известные в данной области техники.
Настоящее изобретение будет объяснено более подробно следующими примерами. Примеры предполагаются только для целей иллюстрации и не ограничивают объем настоящего изобретения, определенный формулой изобретения.
Применяют следующие общие сокращения: EtOAc=этиловый эфир уксусной кислоты, DCM=дихлорметан, HCl=хлористоводородная кислота, MeOH=метанол, TFA=трифторуксусная кислота, THF=тетрагидрофуран, Et2O=диэтиловый эфир, SiO2=покупной диоксид кремния для хроматографических целей (CAS 11926-00-8 или аналогичный), hrs=часов, RT=комнатная температура. Микроволновое нагревание проводили, применяя микроволновые реакторы Biotage.
Структуры продуктов подтверждали1H ЯМР.1H ЯМР резонансы регистрировали на Bruker Avance II 400 МГц спектрометре, и химические сдвиги приводили для отобранных соединений в частях на миллион (ppm) в направлении слабого поля относительно тетраметилсилана в качестве внутреннего стандарта.
Способ выделения A
Реакционную смесь разбавляли органическим растворителем (обычно DCM или EtOAc) и промывали водой или водным основанием (обычно NH4OH, NaHCO3 или NaOH) и сушили над осушающим агентом (обычно Na2SO4 или K2CO3), фильтровали и упаривали.
Способ выделения B
Реакционную смесь разбавляли органическим растворителем (обычно DCM или EtOAc) и промывали водой или водной кислотой (обычно HCl или водным KHSO4) и сушили над осушающим агентом (обычно Na2SO4 или K2CO3), фильтровали и упаривали.
Способ выделения C
Неочищенный продукт растворяли в органическом растворителе (обычно DCM или EtOAc) и добавляли HCl раствор в растворителе (обычно EtOAc или Et2O), и растворители упаривали или фильтровали выпавший осадок.
Способ выделения D
Выпавший осадок фильтровали, промывали или перекристаллизовывали из определенного растворителя или смеси растворителей, получая указанное в заголовке соединение.
Способ выделения E
Неочищенный продукт элюировали через колонку (имеющиеся в продаже SiO2 или CombiFlash приборы вместе с одноразовыми Redisep колонками Teledyne ISCO) смесью растворителей, обычно EtOAc в гептане или MeOH в DCM, содержащем триэтиламин, аммиак или другой основный модификатор, с соотношением 0/100/0 - 45/45/10, обычно 5/94/1.
Способ выделения F
Реакционную смесь наносили на кислую ионообменную колонку и колонку промывали MeOH. Соединение элюировали MeOH, содержащим 10% водный NH3, триэтиламин или аналогичное аминовое основание, фильтровали и упаривали.
Способ выделения G
Неочищенный продукт элюировали через обращено-фазовую колонку (обычно combiFlash прибор вместе с одноразовой Redisep Rf 18 колонками Teledyne ISCO) смесью растворителей. Обычно применяли в качестве элюента градиент вода/ацетонитрил или метанол с 0,1% аммиаком или муравьиной кислотой.
Способ выделения H
Разделение проводили препаративной ВЭЖХ Agilent HPLC/UV системой очистки, снабженной Chiracel 1A колонкой или OD-H колонкой. Обычно применяли изократический прогон смеси изопропанол/гептан или гексан при соотношении 70/30-99/1 с 0,1% диэтиламином или 0,1% TFA в качестве элюента.
Способ выделения I
Остаток растворяли в основном водном растворе (обычно NH4OH, NaHCO3 или NaOH) и раствор промывали органическим растворителем (обычно EtOAc, DCM или Et2O). Затем водную фазу делали кислой добавлением кислоты (обычно HCl) и экстрагировали органическим растворителем (обычно Et2O, EtOAc или DCM). Экстракт сушили (обычно Na2SО4 или K2CО3), фильтровали и упаривали.
Способ выделения J
Остаток растворяли в кислом водном растворе (обычно HCl) и раствор промывали органическим растворителем (обычно EtOAc, DCM или Et2O). Затем водную фазу делали основной добавлением основного водного раствора (обычно NH4OH, NaHCO3 или NaOH) и экстрагировали органическим растворителем (обычно Et2О, EtOAc или DCM). Экстракт сушили (обычно Na2SO4 или K2CO3), фильтровали и упаривали.
Способ выделения K
Реакционную смесь упаривали досуха и растворяли в MeOH. Раствор наносили на предварительно промытую (MeOH) колонку с тиомочевиной. Соединение элюировали MeOH и упаривали.
Способ выделения L
Энантиомеры разделяли препаративной ВЭЖХ UV системой очистки, снабженной Phenomenex LUX amylase-2 колонкой. Обычно применяли изократический прогон смеси н-гексан/этанол/муравьиная кислота 70/30/0,1 в качестве элюента и фракции подкисляли сразу же после сбора водной HCl.
Способ выделения M
Разделение проводили Thar SFC 80 препаративной ВЭЖХ системой со сверхкритической жидкостью, обычно снабженной Chiralpak AD-H колонкой. Обычно в качестве элюента применяли изократический прогон диоксид углерода/метанол 93/7 или 90/10.
Получение соединений настоящего изобретения
ПРИМЕР 1: 2-(5-Метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Способ получения A
Способ получения A1: 5-Метилизохроман-1-карбоновая кислота
Смесь 2-(2-метилфенил)этанола (2 г), TFA (10 мл) и 2,2-дигидроксиуксусной кислоты (1,5 г) кипятили с обратным холодильником в течение 23 часов, и летучие компоненты упаривали. Способ выделения I давал указанное в заголовке соединение (2,6 г) в виде грязно-белого твердого остатка. Альтернативно, серную кислоту можно применять в циклизации.
Способ получения A2: Метил 5-метилизохроман-1-карбоксилат
Смесь 5-метилизохроман-1-карбоновой кислоты (1 г), метанола (20 мл) и триметилсилилхлорида (2 мл) перемешивали в течение 1,5 часов и летучие компоненты упаривали. Способ выделения E давал указанное в заголовке соединение (0,5 г) в виде желтоватого масла. Альтернативно, серную кислоту можно применять вместо триметилсилилхлорида.
Способ получения A3: 2-(5-Метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
К охлажденному в течение 10 минут на бане со льдом и перемешиваемому раствору этилендиамина (0,29 мл), триметилалюминия (2M гептановый раствор, 2,2 мл) и толуола (10 мл) добавляли смесь метил 7-бромизохроман-1-карбоксилата (0,5 г) и толуола (10 мл) и реакционную смесь кипятили с обратным холодильником в течение 6 часов. Добавляли воду (2 мл), метанол (5 мл) и DCM (5 мл), смесь кипятили с обратным холодильником в течение 15 минут и осадок отфильтровывали. Органические растворители упаривали и указанное в заголовке соединение (0,38 г) выделяли способом выделения D (2-метокси-2-метилпропан/MeOH).
1H ЯМР (CDCl3) δ м.д. 7,16 (с, 1H), 7,02 (с, 2H), 5,42 (с, 1H), 4,20 (ддд, 1H), 3,94 (шир.с, 1H), 3,84 (тд, 2H), 3,39 (шир.с, 2H), 3,03 (ддд, 1H), 2,68 (д, 1H), 2,30 (с, 3H).
ПРИМЕР 2: 2-(5-Бромизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(2-бромфенил)этанола (200 мг), применяя методику способа получения A и способов выделения A и E (выход 80 мг).
1H ЯМР (CDCl3) δ м.д. 7,45 (д, 1H), 7,30 (д, 1H), 6,99-7,10 (м, 1H), 5,72 (с, 1H), 4,19 (ддд, 1H), 3,67-3,84 (м, 5H), 2,69-2,93 (м, 2H).
ПРИМЕР 3: 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Смесь NaH (0,77 г), метил-5-метилизохроман-1-карбоксилата (3 г, способ получения A) и THF (40 мл) перемешивали 75 минут при температуре бани со льдом, добавляли йодметан (2,3 мл) и смесь перемешивали при температуре окружающей среды в течение 3 часов. Промежуточный метил 1,5-диметилизохроман-1-карбоксилат (2,5 г) очищали способом выделения A и указанное в заголовке соединение получали, применяя методику стадию 3 способа получения A и способа выделения D (2-пропанoл/гептан) (выход 1,0 г).
1H ЯМР (CD3OD) δ м.д. 7,00-7,18 (м, 3H), 3,99 (т, 2H), 3,54 (шир.с, 4H), 2,62-2,87 (м, 2H), 2,24 (с, 3H), 1,70 (с, 3H).
ПРИМЕР 4: 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазол
Способ получения B
Смесь 2-(2-хлорфенил)этанола (1,0 г), этил 2,2-диэтоксиацетата (1,7 г), хлорида титана (IV) (2,0 мл) и 1,2-дихлорэтана (15 мл) кипятили с обратным холодильником в течение 1 часа. Промежуточный этил 5-хлоризохроман-1-карбоксилат очищали способами выделения B и E (0,41 г) и указанное в заголовке соединение получали, применяя методику способа получения A3. Альтернативно, диэтилэфират трифторида бора можно применять вместо хлорида титана (IV) (выход 0,36 г).
1H ЯМР (CDCl3) δ м.д. 7,20-7,40 (м, 2H, CHCl3), 7,09-7,20 (м, 1H), 5,41 (с, 1H), 4,23 (ддд, 1H), 3,77-4,00 (м, 3H), 3,28-3,51 (м, 2H), 2,77-3,01 (м, 2H).
ПРИМЕР 5: 1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-карбонитрил
Смесь 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола (50 мг), дицианоцинка (25 мг), бис(три-трет-бутилфосфин)палладия (0) (3 мг) и DMF (2 мл) перемешивали в микроволновом реакторе при 160°C в течение 30 минут. Указанное в заголовке соединение очищали способами выделения F и G (выход 22 мг).
1H ЯМР (CD3OD) δ м.д. 7,63 (дд, 1H), 7,53 (д, 1H), 7,35 (т, 1H), 4,26 (ддд, 1H), 3,92 (ддд, 1H), 3,51-3,72 (м, 4H), 3,07-3,22 (м, 1H), 2,89-3,03 (м, 1H).
ПРИМЕР 6: 2-(5-аллилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Способ получения C
К смеси 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола (50 мг), аллилтрибутилолова (69,9 мг), CsF (70,6 мг) и бис(три-трет-бутилфосфин)палладия (3,24 мг) добавляли диоксан (2 мл) и DMF (0,5 мл). Реакционную смесь дегазировали N2 и нагревали в микроволновой печи в течение 30 минут при 160°C. Указанное в заголовке соединение очищали, применяя способы F и G (выход 5,8 мг).
1H ЯМР (CD3OD) δ м.д. 6,96-7,21 (м, 3H), 5,94 (ддт, 1H), 4,92-5,08 (м, 2H), 4,22 (ддд, 1H), 3,81 (ддд, 1H), 3,52 (с, 4H), 3,34-3,35 (м, 2H), 2,93 (ддд, 1H), 2,72 (дт, 1H).
ПРИМЕР 7: 2-(5-Винилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (100 мг), применяя методику способа получения C и очищали способами выделения F и G (выход 5 мг).
1H ЯМР (CDCl3) δ м.д. 7,38-7,48 (м, 1H), 7,15-7,30 (м, 1H, CHCl3), 6,80-7,05 (м, 2H), 5,74 (дд, 1H), 5,35 (дд, 1H), 4,25 (ддд, 1H), 3,50-3,85 (м, 5H), 2,80-3,10 (м, 1H), 2,72-2,85 (м, 1H).
ПРИМЕР 8: 2-(5-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Способ получения D
К смеси 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (100 мг), этилбороновой кислоты (56,2 мг), комплекса (1,1'-бис(дифенилфосфино)ферроцен)дихлорпалладия (II) с CH2Cl2 (1:1) (13 мг) и CsF (108 мг) добавляли диоксан (4 мл) и DMF (1 мл). Смесь дегазировали N2 и нагревали в микроволновой печи в течение 30 минут при 100°C и 30 минут при 150°C. Указанное в заголовке соединение очищали способами выделения J и G (выход 6,1 мг).
1H ЯМР (CD3OD) δ м.д. 6,96-7,17 (м, 3H), 5,39 (с, 1H), 4,24 (ддд, 1H), 3,77-3,89 (м, 1H), 3,49-3,69 (м;4H), 2,87-3,01 (м, 1H), 2,73 (дт, 1H), 2,63 (кв., 2H), 1,20 (т, 3H).
ПРИМЕР 8 HCl соль: гидрохлорид 2-(5-Этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Способ получения соли A
К соединению примера 8 (1 г) в этаноле (15 мл) добавляли 4M хлороводород в диоксане (160 мг), и смесь кипятили с обратным холодильником, охлаждали и упаривали, получая указанное в заголовке соединение (1,1 г). Можно применять альтернативные другие растворители, температуры и источники хлороводорода, или продукт фильтровали от раствора или промывали другими органическими растворителями.
1H ЯМР (ДМСO-d6) δ м.д. 10,72 (с, 2H), 7,14-7,24 (м, 3H), 5,91 (с, 1H), 4,16-4,21 (м, 1H), 3,78-3,90 (м, 5H), 2,89-2,96 (м, 1H), 2,69-2,78 (м, 1H), 2,60 (кв., 2H), 1,51 (tr, 3H).
ПРИМЕР 8 сульфатная соль: сульфат 2-(5-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Способ получения соли B
К соединению примера 8 (1,5 г) в этаноле (30 мл) добавляли серную кислоту (440 мг) в этаноле (7,5 мл), и смесь кипятили с обратным холодильником, охлаждали, упаривали и промывали ацетоном, получая указанное в заголовке соединение (1,4 г). Можно применять альтернативные другие растворители и температуры, или продукт фильтровали от раствора или промывали другими органическими растворителями.
1H ЯМР (ДМСO-д6) δ м.д. 7,13-7,25 (м, 2H), 7,05 (дд, 1H), 5,61 (с, 1H), 4,08-4,19 (м, 1H), 3,71 (шир.с, 4H), 3,78-3,93 (м, 1H), 2,78-2,90 (м, 1H), 2,68-2,78 (м, 1H), 2,59 (кв., 2H), 1,07-1,21 (м, 3H).
ПРИМЕР 8 фумаратная соль: фумарат 2-(5-Этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Способ получения соли C
К соединению примера 8 (2,3 г) в этаноле (15 мл) добавляли муравьиную кислоту (1,16 г) в этаноле (5 мл), и смесь кипятили с обратным холодильником, охлаждали, упаривали и промывали TBME, получая указанное в заголовке соединение (3,0 г). Можно применять альтернативные другие растворители и температуры, или продукт фильтровали от раствора или промывали другими органическими растворителями.
1H ЯМР (ДМСO-д6) δ м.д. 7,15-7,20 (м, 2H), 7,05-7,09 (м, 1H), 6,48 (с, 2H), 5,72 (с, 1H), 4,14-4,19 (м, 1H), 3,80-3,86 (м, 1H), 3,71 (с, 4 H), 2,83-2,91 (м, 1H), 2,67-2,75 (м, 1H), 2,58 (кв., 2H), 1,15 (tr, 3H).
ПРИМЕР 9: 1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-ол
Смесь 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (720 мг), бис(пинаколато)дибора (715 мг) бис(три-трет-бутилфосфин)палладия (0) (39 мг), ацетата калия (503 мг), диоксана (9 мл) и DMF (0,75 мл) перемешивали в инертной атмосфере в микроволновом реакторе при 160°C в течение 30 минут. Добавляли дополнительный бис(три-трет-бутилфосфин)палладий (0) (21 мг) и нагревание продолжали в течение 15 минут. Промежуточный 2-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)изохроман-1-ил)-4,5-дигидро-1H-имидазол (680 мг) очищали способом выделения A и перемешивали с EtOAc (2 мл), водой (2 мл) и пероксидом водорода (35%, 0,07 мл) при температуре бани со льдом в течение 2 часов. Указанное в заголовке соединение очищали способами выделения A и E (выход 45 мг).
1H ЯМР (ДМСO-д6) δ м.д. 9,44 (с, 1H), 6,78-7,08 (м, 1H), 6,62 (д, 1H), 6,67 (д, 1H), 5,22 (с, 1H), 4,05-4,15 (м, 1H), 3,70-3,78 (м, 1H), 3,18-3,42 (м, 4H, H2O), 2,53-2,69 (м, 2H).
ПРИМЕР 10: (1-(4,5-Дигидро-1H-имидазол-2-ил)изохроман-5-ил)метанол
Указанное в заголовке соединение получали из 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (50 мг) и (трибутилстаннил)метанола (86 мг), применяя тетракис(трифенилфосфин)палладия (10,28 мг) в качестве катализатора, и методику способа получения C и способов выделения F и G (выход 6,2 мг).
1H ЯМР (CD3OD) δ м.д. 7,31 (д, 1H), 7,11-7,22 (м, 2H), 5,42 (с, 1H), 4,62 (с, 2H), 4,22 (тд, 1H), 3,86 (дд, 1H), 3,54-3,69 (м, 4H), 2,93-3,05 (м, 1H), 2,75-2,86 (м, 1H).
ПРИМЕР 11: 2-(5-бром-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Смесь NaH (0,024 г), этил 5-бромизохроман-1-карбоксилата (0,2 г, способ получения B) и THF (5 мл) перемешивали 90 минут при температуре бани со льдом, добавляли йодметан (0,06 мл), и смесь перемешивали при температуре окружающей среды в течение 16 часов. Промежуточный этил 5-бром-1-метилизохроман-1-карбоксилат (0,11 г) очищали способом выделения A и указанное в заголовке соединение получали, применяя методику способа получения A3 и способа выделения E (DCM/EtOAc/Et3N) (выход 0,07 г).
1H ЯМР (CDCl3) δ м.д. 7,45-7,47 (м, 1H), 7,39-7,41 (м, 1H), 7,07-7,11 (м, 1H), 4,70-5,20 (шир.с, 1H), 3,94-4,05 (м, 2H), 3,22-3,87 (м, 2H), 2,83-2,87 (м, 2H), 1,77 (с, 3H).
ПРИМЕР 12: 2-((3R)-5-хлор-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-хлорфенил)бутан-2-ола (400 мг), применяя методику способов получения B и A3. Промежуточный (3R)-этил 5-хлор-3-этилизохроман-1-карбоксилат очищали способами выделения B (EtOAc/гептан) и E (выход 235 мг).
1H ЯМР (ДМСO-д6) δ м.д. 7,28-7,43 (м, 1H), 7,11-7,26 (м, 2H), 6,12 (шир.с, 1H), 5,35 (с, 1H), 3,53-3,80 (м, 3H), 3,12-3,29 (м, 2H), 2,73-2,92 (м, 1H), 1,67 (м, 2H), 0,99 (т, 3H).
ПРИМЕР 13: более медленно элюирующийся изомер 1-(1-(4,5-дигидро-1H-имидазол-2-ил)-1-метилизохроман-5-ил)-2,2-диметилпропан-1-ола
К раствору 2-(5-бром-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола (пример 2, 0,075 г) в 2 мл THF добавляли 0,33 мл раствора трет-бутиллития в пентане (1,7M) при -78°C, с последующим добавлением 1,2 мл 20,8M раствора триметилацетальдегида в THF. После перемешивания при -78°C в течение 15 минут, реакцию прекращали льдом. Указанное в заголовке соединение выделяли способами выделения A и G (выход 0,003 г).
1H ЯМР (CDCl3) δ м.д. 7,43-7,45 (м, 1H), 7,34-7,36 (м, 1H), 7,22-7,24 (м, 1H), 4,73 (с, 1H), 3,88-4,05 (м, 2H), 3,61 (шир.с, 4H), 2,84-2,91 (м, 2H), 1,78 (с, 3H), 0,97 (с, 9H).
ПРИМЕР 14: более быстро элюирующийся изомер 1-(1-(4,5-дигидро-1H-имидазол-2-ил)-1-метилизохроман-5-ил)-2,2-диметилпропан-1-ола
Указанное в заголовке соединение выделяли в способе получения примера 13 способами выделения A и G (выход 0,007 г).
1H ЯМР (CDCl3) δ м.д. 7,46 (д, 1H), 7,50 (д, 1H), 7,18-7,35 (м, 3H), 4,72 (с, 1H), 4,04 (с, 1H), 3,59-3,81 (м, 3H), 2,81-3,02 (м, 2H), 2,05 (д, 1H), 1,89 (с, 3H), 0,86-1,05 (м, 9H).
ПРИМЕР 15: 2-(5-этинилизохроман-1-ил)-4,5-дигидро-1H-имидазол
К дегазированному раствору 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (300 мг) в Et3N (10 мл) и этинилтриметилсилане (210 мг) добавляли тетракис(трифенилфосфин)палладий (37 мг). Смесь дегазировали N2 и нагревали в микроволновой печи в течение 60 минут при 120°C. Реакционную смесь концентрировали в вакууме и очищали способом выделения K. Добавляли K2CO3 к реакционному остатку (в MeOH), и смесь перемешивали 4 часа при комнатной температуре. Указанное в заголовке соединение выделяли способом выделения G (выход 6,2 мг).
1H ЯМР (CDCl3) δ м.д. 7,42-7,37 (м, 2H), 7,17 (т, 1H), 5,42 (с, 1H), 4,78 (с, 1H), 4,26-4,21 (м, 1H), 4,00-3,73 (м, 4H), 3,08-2,93 (м, 2H).
ПРИМЕР 16: 2-((3R)-3-этил-5-(трифторметил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: (R)-1-(2-(трифторметил)фенил)бутан-2-ол
2-Йодбензотрифторид (3 г) растворяли в THF (18 мл) и охлаждали до -78°C. К реакционной смеси добавляли по каплям н-BuLi (2,5M в гексане, 13,23 мл). Через 1 час добавляли (R)-(+)-1,2-эпоксибутан (1,4 мл) в THF (18 мл). Температуру реакции медленно повышали до комнатной температуры. Смесь выливали на смесь воды со льдом (100 мл) и продукт экстрагировали гептаном. Органическую фазу промывали соляным раствором и водой, сушили (Na2SO4) и упаривали в вакууме. Указанное в заголовке соединение получали способом выделения E (выход 1,05 г).
Стадия 2: 2-((3R)-3-этил-5-(трифторметил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-(трифторметил)фенил)бутан-2-ола (570 мг), применяя методику способов получения B и A3. Промежуточное соединение (3R)-этил 5-трифторметил-3-этилизохроман-1-карбоксилат очищали способами выделения B (EtOAc/гептан) и E (выход 360 мг).
1H ЯМР (CD3OD) δ м.д. 7,59 (д, 1H), 7,50 (д, 1H), 7,25-7,42 (м, 1H), 5,49 (с, 1H), 3,46-3,72 (м, 5H), 2,98 (д, 1H), 2,82 (дд, 1H), 1,59-1,84 (м, 2H), 0,97-1,18 (м, 3H).
ПРИМЕР 17: 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол
Способ получения E
К смеси 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (200 мг), 2-(ди-трет-бутилфосфино)бифенила (5,31 мг), ацетата палладия (II) (3,19 мг) и Cs2CO3 (348 мг) добавляли метанол (1 мл) и толуол (2 мл). Смесь дегазировали N2 и затем нагревали в течение 40 минут при 120°C и 30 минут при 130°C. Альтернативно, другие комплексы Pd-лиганд и условия реакции можно применять в образовании C-O связи. Указанное в заголовке соединение очищали, применяя способы выделения E и G (выход 5 мг).
1H ЯМР (CD3OD) δ м.д. 7,14 (т, 1H), 6,80 (д, 1H), 6,83 (д, 1H), 5,34 (с, 0,3H), 4,16-4,31 (м, 1H), 3,74-3,89 (м, 4H), 3,50-3,68 (м, 4H), 2,61-2,90 (м, 2H).
ПРИМЕР 17 HCl соль: гидрохлорид 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 17 (263 мг), как описано в примере 8 (Et2O, выход 148 мг).
1H ЯМР (CD3OD) δ м.д. 7,28 (т, 1H), 6,97 (д, 1H), 6,80 (д, 1H), 5,72 (с, 1H), 4,12-4,19 (м, 1H), 3,99 (шир.с, 4H), 3,87-3,95 (м, 1H), 3,86 (с, 3H), 2,78-2,84 (м, 2H).
ПРИМЕР 17 сульфатная соль: сульфат 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 17 (50 мг), как описано в примере 8 (EtOH, выход 41,5 мг).
1H ЯМР (ДМСO-д6) δ м.д. 9,37-10,57 (шир., 2H), 7,27 (т, 1H), 6,99 (д, 1H), 6,79 (д, 1H), 5,75 (с, 1H), 4,03-4,20 (м, 1H), 3,85-3,94 (м, 5H), 3,82 (с, 3H), 2,65-2,79 (м, 2H).
ПРИМЕР 17 гемифумаратная соль: гемифумарат 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 17 (50 мг), как описано в примере 8 (EtOH, выход 38 мг).
1H ЯМР (ДМСO-д6) δ м.д. 7,16 (т, 1H), 6,88 (д, 1H), 6,79 (д, 1H), 6,43 (с, 1H), 5,47 (с, 1H), 4,07-4,16 (м, 1H), 3,73-3,82 (м, 4H), 3,40-4,75 (шир.с, 4H), 2,58-2,75 (м, 2H).
ПРИМЕР 18: 2-(5-йодизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(2-йодфенил)этанола (1,5 г), применяя методику способа получения B и способа выделения D (выход 510 мг).
1H ЯМР (CD3OD) δ м.д. 7,72-7,84 (м, 1H), 7,24 (д, 1H), 6,94 (т, 1H), 4,21 (ддд, 1H), 3,83 (ддд, 1H), 3,46-3,68 (м, 4H), 2,80-2,95 (м, 1H), 2,56-2,74 (м, 1H).
ПРИМЕР 19: 2-((3R)-3-метил-5-(трифторметил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-(трифторметил)фенил)пропан-2-ола (570 мг), применяя методику способов получения B и A3. Исходное соединение, (R)-1-(2-(трифторметил)фенил)пропан-2-ол, (1,0 г) получали из 2-йодбензотрифторида и (R)-(+)-пропиленоксида способом, аналогичным способу, описанному в примере 16. Промежуточный (3R)-этил 5-трифторметил-3-этилизохроман-1-карбоксилат очищали способами выделения B (EtOAc/гептан) и E (выход 340 мг).
1H ЯМР (CD3OD) δ м.д. 7,59 (д, 1H), 7,50 (д, 1H), 7,29-7,43 (м, 1H), 5,51 (с, 1H), 3,83-3,97 (м, 1H), 3,49-3,72 (м, 4H), 2,99 (д, 1H), 2,82 (дд, 1H), 1,25-1,48 (м, 3H).
ПРИМЕР 20: 2-(5-Бром-4-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(2-бромфенил)пропан-1-ола (1,55 г), применяя методику способа получения B и способа выделения E (выход 70 мг).
1H ЯМР (CD3OD) δ м.д. 7,43-7,55 (м, 1H), 7,27 (д, 0,6H), 7,04-7,15 (м, 1,4H), 5,34 (с, 0,5H), 3,98-4,10 (м, 1H), 3,75-3,92 (м, 1H), 3,53-3,67 (м, 4H), 2,94-3,06 (м, 1H), 1,45 (д, 1,9H), 1,33 (д, 1,1H).
ПРИМЕР 21: более быстро элюирующийся изомер 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазола (пример 3, 70 мг), применяя методику способа выделения H (выход 22 мг).
1H ЯМР (CDCl3) δ 7,19-7,40 (м, 1H, CHCl3), 7,02-7,19 (м, 2H), 3,90-4,15 (м, 2H), 2,66-2,81 (м, 2H), 2,24 (м, 3H), 1,79 (с, 3H).
ПРИМЕР 22: более медленно элюирующийся изомер 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из 2-(1,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазола (пример 3, 70 мг), применяя методику способа выделения H (выход 9 мг).
1H ЯМР (CDCl3) δ 7,21-7,36 (м, 1H, CHCl3), 7,01-7,18 (м, 2H), 3,91-4,15 (м, 2H), 2,60-2,83 (м, 2H), 2,24 (с, 3H), 1,79 (с, 3H).
ПРИМЕР 23: 2-((3R)-1,3,5-триметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (3R)-метил 3,5-диметилизохроман-1-карбоксилата (454 мг, промежуточное соединение примера 33), применяя методику примера 11 и способа выделения E (DCM/EtOAc/Et3N) (выход 232 мг).
1H ЯМР (ДМСO-d6) δ м.д. 7,12-7,23 (м, 1H), 6,93-7,12 (м, 2H), 6,15 (с, 0,5H), 5,83 (с, 0,5H), 3,87-4,02 (м, 0,5H), 3,61-3,86 (м, 1H), 3,40-3,61 (м, 1,5H), 3,04-3,29 (м, 2H), 2,53-2,71 (м, 1H), 2,27-2,48 (м, 1H), 2,18 (с, 1,5H), 2,16 (с, 1,5H), 1,64 (с, 1,5H), 1,56 (с, 1,5H), 1,23-1,38 (м, 3H).
ПРИМЕР 24: 2-(5-циклопропилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Смесь 2-(5-бром-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола (пример 11, 0,15 г), карбоната натрия (0,27 г), хлорида бис(трифенилфосфин)палладия (II) (0,02 г), циклопропилбороновой кислоты (0,09 г), воды (1 мл) и ацетонитрила (2 мл) нагревали в микроволновой печи при 120°C в течение 15 минут. Указанное в заголовке соединение очищали способами выделения A и G (выход 8 мг).
1H ЯМР (CDCl3) δ 7,26-7,28 (д, 1H), 7,11-7,15 (tr, 1H), 6,92-6,94 (д, 1H), 4,03-4,06 (м, 2H), 3,62 (шир.с, 4H), 2,95-2,98 (м, 2H), 1,80 (с, 3H), 0,87-0,96 (шир.м, 2H), 0,58-0,69 (шир.м, 2H).
ПРИМЕР 25: 2-(3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-o-толилпропан-2-ола (800 мг), применяя методику способа получения B и способов выделения E и D (выход 19,2 мг).
1H ЯМР (CD3OD) δ м.д. 6,98-7,11 (м, 3H), 5,43 (с, 1H), 3,88 (дд, 1H), 3,49-3,67 (м, 4H), 2,64-2,76 (м, 1H), 2,57 (д, 1H), 2,23 (с, 3H), 1,39 (д, 3H).
ПРИМЕР 26: 2-(5-хлор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-(2-хлорфенил)пропан-2-ола (1,0 г), применяя методику способа получения A и способов выделения A и D (Et2O, гептан) (выход 0,077 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,32-7,37 (м, 1H), 7,14-7,23 (м, 2H), 6,23 (шир.с, 1H), 5,36 (с, 1H), 3,83-3,91 (шир.м, 1H), 3,43 (шир.с, 4H), 2,82-2,89 (м, 1H), 2,46-2,53 (м, 1H), 1,29-1,34 (дд, 3H).
ПРИМЕР 27: 2-(3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-o-толилбутан-2-ола (1 г), применяя методику способа получения B и способов выделения E и D (выход 16,4 мг).
1H ЯМР (CD3OD) δ м.д. 6,93-7,11 (м, 3H), 5,41 (с, 1H), 3,45-3,72 (м, 6H), 2,71 (дд, 1 H), 2,55 (дд, 1H), 2,17-2,26 (м, 3H), 1,62-1,78 (м, 2H), 0,98-1,14 (м, 3H).
ПРИМЕР 28: 2-(5-хлор-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Смесь NaH (0,36 г), метил 5-хлор-3-метилизохроман-1-карбоксилата (1,1 г, способ получения A) и THF (20 мл) перемешивали 60 минут при температуре бани со льдом, добавляли йодметан (0,9 мл) и смесь перемешивали при температуре окружающей среды в течение 16 часов. Промежуточный метил 5-хлор-1,3-диметилизохроман-1-карбоксилат (0,95 г) очищали способом выделения A и указанное в заголовке соединение получали, применяя методику способа получения A и способа выделения A (выход 0,1 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,16-7,33 (м, 3H), 6,14 (шир.с, 1H), 3,72-4,02 (шир.м, 1H), 3,40-3,47 (шир.с, 2H), 2,76-2,86 (шир.с, 1H), 2,35-2,54 (шир.м, 1H), 1,60-1,66 (д, 3H), 1,30-1,32 (дд, 3H).
ПРИМЕР 29: 2-(5-Бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-(2-бромфенил)пропан-2-ола (2,5 г), применяя методику способа получения A (43% полученного метил 5-бром-3-метил-изохроман-1-карбоксилата применяли в последней стадии) и способа выделения D (выход 447 мг).
1H ЯМР (CD3OD) δ м.д. 7,42-7,55 (м, 1H), 7,24 (д, 0,8H), 7,02-7,17 (м, 1,2H), 5,41 (с, 0,7H), 4,08 (с, 0,2H), 3,81-3,95 (м, 0,8H), 3,50-3,70 (м, 4H), 2,82-2,93 (м, 1H), 2,40-2,64 (м, 1H), 1,38-1,44 (м, 2,3H), 1,34 (д, 0,7H).
ПРИМЕР 30: 2-(1,3,5-триметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-o-толилпропан-2-ола (0,9 г), применяя способ, как описано в примере 12, за исключением применения способов выделения A и D (Et2O и гептан) на конечной стадии получения (выход 0,012 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,27-7,29 (д, 1H), 7,13-7,17 (tr, 1H), 7,05-7,07 (д, 1H), 4,85 (шир.с, 1H), 3,82-3,90 (м, 1H), 3,64 (шир.с, 4H), 2,59-2,64 (м, 1H), 2,44-2,51 (м, 1H), 2,21-2,23 (м, 3H), 1,72 (с, 3H), 1,37-1,38 (д, 3H).
ПРИМЕР 31: 2-(5-бром-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-(2-бромфенил)пропан-2-ола (2,5 г), применяя методику способа получения A. После первых двух стадий 43% (500 мг) полученного метил 5-бром-3-метил-изохроман-1-карбоксилата растворяли в THF (5 мл). Добавляли к раствору NaH (140 мг) и затем смесь перемешивали при температуре бани со льдом.
Через 60 минут добавляли йодметан (0,325 мл) и смесь перемешивали при температуре окружающей среды в течение 48 часов. Промежуточный метил 5-бром-1,3-диметилизохроман-1-карбоксилат очищали способами выделения A и E (168 мг) и указанное в заголовке соединение получали, применяя методику способа получения A (стадия 3) и способа выделения D (выход 24 мг).
1H ЯМР (CD3OD) δ м.д. 7,41-7,54 (м, 1H), 7,25-7,37 (м, 1H), 7,03-7,17 (м, 1H), 4,04 (ддд, 0,2H), 3,76-3,90 (м, 0,8H), 3,44-3,64 (м, 4H), 2,78-2,95 (м, 1H), 2,37-2,62 (м, 1H), 1,74 (с, 0,6H), 1,68 (с, 2,4H), 1,32-1,41 (м, 3H).
ПРИМЕР 32: 2-((3R)-5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-бромфенил)пропан-2-ола (400 мг), применяя методику способа получения A и способа выделения D (гептан) (выход 75 мг).
1H ЯМР (CD3OD) δ м.д. 7,49 (д, 1H),7,24 (д, 1H), 7,02-7,18 (м, 1H), 5,42 (с, 1H), 3,76-3,97 (м, 1H), 3,51-3,72 (м, 5H), 2,75-2,96 (м, 1H), 2,58 (дд, 1H), 1,22-1,51 (м, 3H).
ПРИМЕР 33: 2-((3R)-5-хлор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-хлорфенил)пропан-2-ола (0,3 г), применяя методику способа получения A и способов выделения A и D (Et2O и гептан) (выход 0,03 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,35-7,37 (д, 1H), 7,17-7,23 (м, 2H), 6,14 (шир.с, 1H), 5,36 (с, 1H), 3,83-3,91 (м, 1H), 3,6 (шир.м, 2H), 3,10-3,30 (шир.с, 2H), 2,82-2,86 (д, 1H), 2,46-2,53 (д, 1H), 1,33-1,34 (д, 3H).
ПРИМЕР 34: 2-((3S)-5-хлор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (S)-1-(2-хлорфенил)пропан-2-ола (0,3 г), применяя методику способа получения A и способов выделения A и E (Et2O и гептан) (выход 0,03 г).
1H ЯМР (CDCl3) δ 7,34-7,36 (д, 1H), 7,28-7,30 (д, 1H), 7,11-7,15 (tr, 1H), 5,54 (шир.с, 1H), 3,86-3,94 (м, 1H), 3,56-3,76 (шир.м, 4H), 2,89-2,94 (дд, 1H), 2,54-2,61 (м, 1H), 1,37-1,43 (м, 3H).
ПРИМЕР 35: 2-((3S)-5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (S)-1-(2-бромфенил)пропан-2-ола (200 мг), применяя методику способа получения A и способов выделения D (Et2O) и G (выход 23 мг).
1H ЯМР (CD3OD) δ м.д. 7,38-7,60 (м, 1H), 7,23 (д, 1H), 7,03-7,18 (м, 1H), 5,42 (с, 1H), 4,03-4,13 (м, 0,3H), 3,79-3,98 (м, 0,7H), 3,43-3,71 (м, 4H), 2,81-2,97 (м, 1H), 2,58 (дд, 0,7H), 2,46 (дд, 0,3H), 1,34-1,40 (м, 3H).
ПРИМЕР 36: 2-((3R)-3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-o-толилпропан-2-ола (400 мг), применяя методику способа получения A и способа выделения E (выход 40 мг).
1H ЯМР (CD3OD) δ м.д. 6,99-7,11 (м, 3H), 5,43 (с, 1H), 3,88 (ддд, 1H), 3,53-3,66 (м, 4H), 2,72 (дд, 1H), 2,47-2,61 (м, 1H), 1,39 (д, 3H).
ПРИМЕР 37: 2-((3S)-3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (S)-o-толилпропан-2-ола (400 мг), применяя методику способа получения A и способов выделения E и D (выход 40 мг).
1H ЯМР (CD3OD) δ м.д. 6,99-7,13 (м, 3H), 5,43 (с, 1H), 3,82-3,93 (м, 1H), 3,53-3,66 (м, 4H), 2,66-2,79 (м, 1H), 2,48-2,62 (м, 1H), 1,39 (д, 3H).
ПРИМЕР 38: 2-(5-Метокси-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола (200 мг), применяя методику способа получения E и способов выделения K и G (выход 5,8 мг).
1H ЯМР (CD3OD) δ м.д. 7,07-7,18 (м, 1H), 6,76-6,87 (м, 2H), 5,41 (с, 1H), 3,76-3,87 (м, 4H), 3,55-3,67 (м, 4H), 2,83 (дд, 1H), 2,42 (дд, 1H), 1,31-1,41 (м, 3H).
ПРИМЕР 39: 2-(5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола (200 мг), применяя методику способа получения D и способов выделения J и G (выход 18,4 мг).
1H ЯМР (CD3OD) δ м.д. 7,04-7,14 (м, 2,6H), 6,96-7,01 (м, 0,4H), 5,45 (с, 0,7H), 4,11 (с, 0,2H), 3,87 (ддд, 0,8H), 3,51-3,66 (м, 4H), 2,75-2,89 (м, 1H), 2,54-2,70 (м, 3H), 1,37-1,43 (м, 2,3H), 1,33 (д, 0,7H), 1,20 (т, 3H).
ПРИМЕР 40: 2-(5-бром-3-пропилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-(2-бромфенил)пентан-2-ола (применяли в последней стадии только 50% полученного эфира), применяя методику способа получения B (кислота Льюиса BF3⋅Et2O в качестве катализатора) (выход 400 мг).
1H ЯМР (CD3OD) δ м.д. 7,41-7,54 (м, 1H), 7,23 (д, 1H), 7,03-7,14 (м, 1H), 5,39 (с, 1H), 3,74 (тд, 1H), 3,50-3,68 (м, 4H), 2,85 (д, 1H), 2,58 (дд, 1H), 1,45-1,75 (м, 4H), 0,92-1,04 (м, 3H).
ПРИМЕР 41: 2-(5-изопропилизохроман-1-ил)-4,5-дигидро-1H-имидазол
К раствору 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (0,25 г, способ получения A), ацетата палладия (II) (0,01 г), три-трет-бутилфосфина (0,044 мл) и толуола (3 мл) при температуре бани со льдом добавляли 6,22 мл 0,5M изопропилбромида цинка в THF, и перемешивали при температуре окружающей среды в течение 3 часов. Реакцию прекращали разбавленной хлористоводородной кислотой и органическую фазу отделяли. Водную фазу подщелачивали 1 M NaOH и очищали способами выделения A и G, получая указанное в заголовке соединение (выход 2 мг).
1H ЯМР (CDCl3) δ м.д. 7,20 (шир.с, 3H), 5,48 (с, 1H), 4,22-4,27 (м, 1H), 3,85-3,91 (м, 1H), 3,63 (шир.с, 4H), 3,05-3,12 (м, 1H), 2,91-2,99 (м, 1H), 2,75-2,80 (м, 1H), 1,21–1,23 (м, 6H).
ПРИМЕР 42: 2-(5-фторизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(2-фторфенил)этанола (0,4 г), применяя методику способа получения B. Конечный продукт очищали промыванием остатка после упаривания холодной водой (выход 0,09 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,17-7,23 (м, 1H), 7,03-7,08 (м, 2H), 6,31 (шир.с, 1H), 5,32 (с, 1H), 4,10-4,15 (м, 1H), 3,79-3,85 (м, 1H), 3,43-3,75 (шир.с, 2H), 3,09-3,30 (шир.с, 2H), 2,67-2,83 (м, 2H).
ПРИМЕР 43: 2-(5-бром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-(2-бромфенил)бутан-2-ола (600 мг), применяя методику способа получения B (диэтилэфират трифторида бора применяли вместо хлорида титана (IV)) и способа выделения D (выход 480 мг).
1H ЯМР (CD3OD) δ м.д. 7,40-7,53 (м, 1H), 7,24 (д, 1H), 7,09 (кв., 1H), 5,39 (с, 1H), 3,52-3,70 (м, 5H), 2,80-2,92 (м, 1H), 2,58 (дд, 1H), 1,56-1,80 (м, 2H), 0,99-1,10 (м, 3H).
ПРИМЕР 44: 2-((3R)-5-метокси-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-((3R)-5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола (200 мг), применяя методику способа получения E (с 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2,4',6'-триизопропил-1,1'-бифенилом) и способов выделения J и G (выход 31,4 мг).
1H ЯМР (CD3OD) δ м.д. 7,08-7,20 (м, 1H), 6,79-6,85 (м, 1,7H), 6,73-6,75 (д, 0,3H), 5,40 (с, 0,7H), 4,07-4,10 (м, 0,2H), 3,80-3,92 (м, 3,8H), 3,46-3,68 (м, 4H), 2,74-2,90 (м, 1H), 2,28-2,47 (м, 1H), 1,35-1,41 (м, 2,3H), 1,31 (д, 0,7H).
ПРИМЕР 45: 2-((3R)-5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-этилфенил)пропан-2-ола (2,2 г), применяя методику способа получения A. Промежуточный (3R)-метил 5-этил-3-метилизохроман-1-карбоксилат очищали способом выделения E (выход 146 мг).
1H ЯМР (CD3OD) δ м.д. 7,01-7,21 (м, 3H), 5,44 (с, 1H), 4,06-4,17 (м, 0,15H), 3,77-3,96 (м, 0,85H), 3,49-3,69 (м, 4H), 2,75-2,91 (м, 1H), 2,54-2,69 (м, 3H), 1,26-1,46 (м, 3H), 1,19 (т, 3H).
ПРИМЕР 45 HCl соль: гидрохлорид 2-((3R)-5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 45 (100 мг), как описано в примере 8 (IPA, выход 71 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,63 (шир.с, 2H), 7,13-7,38 (м, 2H), 6,94-7,13 (м, 1H), 5,87 (шир.с, 1H), 3,88 (шир.с, 4H), 3,61-3,74 (м, 1H), 2,66-2,82 (м, 1H), 2,42-2,65 (м, 1H), 2,14-2,32 (м, 3H), 1,60-1,83 (м, 2H), 0,79-1,07 (м, 3H).
ПРИМЕР 45 гемифумаратная соль: гемифумарат 2-((3R)-5-этил-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 45 (940 мг), как описано в примере 8 (этанол, выход 1,16 г).
1H ЯМР (ДМСO-d6) δ м.д. 6,97-7,20 (м, 3H), 6,43 (с, 1H), 5,59 (с, 0,7H), 5,49 (с, 0,3H), 4,04-4,08 (м, 0,3H), 3,80-3,92 (м, 0,7H), 3,59 (шир.с, 4H), 3,55 (с, 1H), 2,77 (м, 2H), 2,56-2,63 (м, 1H), 1,26-1,37 (м, 3H), 1,15 (т, 3H).
ПРИМЕР 46: 2-(3-этил-5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 1-(2-бромфенил)бутан-2-ола (1,1 г), применяя методику способов получения B (диэтилэфират трифторида бора применяли вместо хлорида титана (IV)) и E (с 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1'-бифенилом) (реакцию конденсации C-O проводили до промежуточного этил 5-бром-3-этилизохроман-1-карбоксилата, только 60% полученного количества применяли в данной стадии). Реакция конденсации C-O давала свободную кислоту, которую затем метилировали (способ получения A) перед конечной стадией реакции. Указанное в заголовке соединение получали концентрированием реакционной смеси (выход 186 мг).
1H ЯМР (CD3OD) δ м.д. 7,12 (т, 1H), 6,82 (д, 2H), 5,38 (с, 1H), 3,82 (с, 3H), 3,48-3,69 (м, 5H), 2,73-2,90 (м, 1H), 2,41 (дд, 1H), 1,70 (дт, 2H), 0,96-1,10 (м, 3H).
ПРИМЕР 47: 2-((3R)-3,5-диэтилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: (R)-1-(2-этилфенил)бутан-1-ол
1-бром-2-этилбензол (2,0 г) растворяли в сухом THF и смесь охлаждали до -78°C. Медленно добавляли 1,6 M н-BuLi (20,26 мл) к реакционной смеси и затем смесь перемешивали при -78°C. Через 1 час добавляли (R)-(+)-1,2-эпоксибутан (1,1 г) в 10 мл THF. Реакционную смесь нагревали до температуры окружающей среды и затем перемешивали в течение ночи. Реакцию прекращали смесью воды со льдом, и продукт экстрагировали в гептан, и, наконец, очищали, применяя способ выделения E, получая указанное в заголовке соединение (1,1 г).
Стадия 2: 2-((3R)-3,5-диэтилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-этилфенил)бутан-1-ола (1,1 г), применяя методику способа получения A и способа выделения D. (выход 660 мг).
1H ЯМР (CD3OD) δ м.д. 6,99-7,19 (м, 3H), 5,43 (с, 1H), 3,49-3,68 (м, 5H), 2,74-2,86 (м, 1H), 2,55-2,66 (м, 3H), 1,54-1,79 (м, 2H), 1,19 (т, 3H), 0,95-1,11 (м, 3H).
ПРИМЕР 48: 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(o-толил)бутан-2-ола (463 мг), применяя методику способа получения B. Диэтилэфират трифторида бора применяли вместо хлорида титана (IV), и промежуточный (3R)-этил 3-этил-5-метилизохроман-1-карбоксилат очищали, применяя способ выделения E. Указанное в заголовке соединение выделяли, применяя способ выделения D с MTBE-гептан в качестве растворителя (выход 287 мг).
1H ЯМР (ДМСO-d6) δ м.д. 6,95-7,12 (м, 3H), 5,98 (шир.с, 1H), 5,32 (с, 1H), 3,49-3,81 (м, 3H), 3,20 (шир.с, 2H), 2,66 (д, 1H), 2,40-2,47 (м, 1H), 2,19 (с, 3H), 1,54-1,76 (м, 2H), 0,99 (т, 3H).
ПРИМЕР 48 HCl соль: гидрохлорид 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 48 (100 мг), как описано в примере 8 (IPA, выход 72 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,60 (шир.с, 2H), 7,15-7,28 (м, 2H), 7,03-7,14 (м, 1H), 5,89 (с, 1H), 3,90 (шир.с, 4H), 3,08 (с, 1H), 2,82 (д, 1H), 2,55-2,70 (м, 3H), 1,36 (д, 3H), 1,15 (т, 3H).
ПРИМЕР 48 сульфатная соль: сульфат 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 48 (1,5 г), как описано в примере 8 (этанол, выход 2,0 г).
1H ЯМР (ДМСO-d6) δ м.д. 10,32 (с, 2H), 7,16-7,22 (м, 2H), 7,00-7,02 (м, 1H), 5,78 (с, 1H), 3,87-3,93 (м, 5H), 2,74 (дд, 1H), 2,50-2,59 (м, 1H), 1,66-1,74 (м, 2H), 1,10 (т, 3H), 1,01 (т, 3H).
ПРИМЕР 48 гемифумаратная соль: гемифумарат 2-((3R)-3-этил-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из соединения примера 48 (100 мг), как описано в примере 8 (этанол, выход 102,3 мг).
1H ЯМР (ДМСO-d6) δ м.д. 7,09-7,19 (м, 2H), 6,99-7,07 (м, 1H), 6,43 (с, 1H), 5,60 (с, 0,7H), 5,50 (с, 0,3H), 4,05-4,08 (м, 0,3H), 3,81-3,92 (м, 0,7H), 3,60 (с, 4H), 2,77 (дд, 1H), 2,56-2,64 (м, 1H), 2,44-2,53 (м, 2H), 1,25-1,37 (м,3H), 1,15 (т, 3H).
ПРИМЕР 49: 2-((3R)-3-метил-5-(трифторметокси)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: (R)-1-(2-(трифторметокси)фенил)пропан-2-ол
К раствору трифторметоксибензола (5,29 мл) и Ν,Ν,Ν',Ν'-тетраметилэтилендиамина (5,96 мл) в тетрагидрофуране (80 мл) при -78°C добавляли втор-бутиллитий (32 мл, 1,4M раствор) в течение 50 минут. Через 2 часа добавляли охлажденный (-78°C) раствор (R)-(+)-пропиленоксида (4,20 мл) в тетрагидрофуране (20 мл) в течение 15 минут, с последующим добавлением диэтилэфирата трифторида бора (1,89 мл) в течение 20 минут. Реакционную смесь перемешивали при -78°C в течение 2 часов, после чего добавляли водный раствор H2SO4 (0,3M, 50 мл) и воду (10 мл). Смесь нагревали до комнатной температуры, после чего ее экстрагировали диэтиловым эфиром (2×100 мл). Объединенные органические фазы промывали соляным раствором, сушили Na2SО4 и концентрировали. Остаток после упаривания очищали способом выделения E (этилацетат - гептан) (выход 1,40 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,24-7,44 (м, 4H), 4,67 (д, 1H), 3,80-3,92 (м, 1H), 2,74 (дд, 1H), 2,65 (дд, 1H), 1,04 (д, 3H).
Стадия 2: 2-((3R)-3-метил-5-(трифторметокси)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-(трифторметокси)фенил)пропан-2-ола (110 мг), применяя методику способа получения B. Диэтилэфират трифторида бора применяли вместо хлорида титана (IV) (выход 36 мг).
1H ЯМР (CD3OD) δ м.д. 7,31-7,16 (м, 3H), 5,45 (с, 1H), 4,12-4,02 (м, 0,25H), 4,94-4,83 (м, 0,75H), 3,70-3,54 (м, 4H), 2,95-2,85 (м, 1H), 2,59 (дд, 0,8H), 2,48 (дд, 0,3H), 1,40 (д, 2,4H), 1,34 (д, 0,9H).
ПРИМЕР 50: 2-((3R)-5-фтор-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-1-(2-фторфенил)пропан-2-ола (170 мг), применяя методику способа получения B. Диэтилэфират трифторида бора применяли вместо хлорида титана (IV). Указанное в заголовке соединение очищали растиранием неочищенного продукта с гептаном (выход 67 мг).
1H ЯМР (CD3OD) δ м.д. 7,22-7,14 (м, 1H), 7,05 (д, 1H), 7,01-6,93 (м, 1H), 5,43 (с, 1H), 4,14-4,04 (м, 0,1H), 3,93-3,83 (м, 0,9H), 3,70-3,52 (м, 4H), 2,93-2,83 (м, 1H), 2,56 (дд, 0,9H), 2,46 (дд, 0,1H), 1,40 (д, 2,6H), 1,33 (д, 0,5H).
ПРИМЕР 51: 2-(5-этоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: 5-этоксиизохроман-1-карбоновая кислота
Смесь этил 5-бромизохроман-1-карбоксилата (1,0 г, способы получения A1 и A2), карбонат цезия (4,57 г), 3,4,7,8-тетраметил-1,10-фенантролина (332 мг), йодида меди (I) (134 мг) и этанола (10 мл) нагревали в микроволновом реакторе при 160°C. Метанол упаривали и указанное в заголовке соединение выделяли и очищали способом выделения B (выход 400 мг).
Стадия 2: 2-(5-этоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали, применяя методику способов получения A2 и A3 и очищали способом выделения D (гептан) (выход 150 мг).
1H ЯМР (CD3OD) δ м.д. 7,11 (т, 1H), 6,80 (т, 2H), 4,11-4,26 (м, 1H), 4,05 (кв.д, 2H), 3,79 (тд, 1H), 3,51-3,66 (м, 4H), 2,62-2,91 (м, 2H), 1,40 (т, 3H).
ПРИМЕР 52: 2-(5-метил-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: 4,4,4-трифтор-1-(o-толил)бутан-2-ол
К смеси гидрохлорида 2,2,2-трифторэтиламина (0,271 г) в смеси дихлорметан/вода (15/0,5 мл) при 0°C добавляли нитрит натрия (0,166 г), и полученный в результате желтый раствор перемешивали в течение 1 часа при 0°C. Затем реакционную смесь охлаждали до -78°C и добавляли 2-o-толилацетальдегид (0,134 г) с последующим добавлением тетрахлорида циркония (IV). После перемешивания в течение 2 часов при -78°C охлаждение прекращали, и добавляли метанол (3 мл) и насыщенный раствор NaHCO3 (10 мл). Смесь экстрагировали дихлорметаном и экстракты сушили и концентрировали. Остаток после упаривания (194 мг) растворяли в метаноле (3 мл) и раствор охлаждали до 0°C. Добавляли боргидрид натрия (44 мг) и реакционную смесь перемешивали в течение 25 минут. Добавляли раствор K2CO3 (2 M, 5 мл) к реакционной смеси с последующим добавлением воды (5 мл) через 5 минут и полученный в результате раствор экстрагировали EtOAc. Экстракты сушили и концентрировали и остаток очищали способом выделения E (EtOAc/гептан) (выход 121 мг).
1H ЯМР (CDCl3) δ м.д. 7,23-7,12 (м, 4H), 4,26-4,17 (м, 1H), 2,90 (дд, 1H), 2,80 (дд, 1H), 2,45-2,27 (м, 2H), 2,34 (с, 3H), 1,86 (д, 1H).
Стадия 2: 2-(5-метил-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 4,4,4-трифтор-1-(o-толил)бутан-2-ола (121 мг), применяя методику способа получения B. Диэтилэфират трифторида бора применяли вместо хлорида титана (IV). Указанное в заголовке соединение очищали способом выделения E (дихлорметан-MeOH-NH4OH) (выход 11 мг).
1H ЯМР (CD3OD) δ м.д. 7,13-7,04 (м, 2,6H), 6,99-6,93 (м, 0,3H), 5,47 (с, 1H), 4,41-4,33 (м, 0,3H), 4,15-4,07 (м, 0,7H), 3,63-3,51 (м, 4H), 2,87-2,46 (м, 4H), 2,25 (с, 3H).
ПРИМЕР 53: 2-((3S)-5-метокси-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-((3S)-5-бром-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола (200 мг), применяя методику способа получения E (с 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1'-фенилом) и способа выделения G (выход 16,3 мг).
1H ЯМР (CD3OD) δ м.д. 7,10-7,24 (м, 1H), 6,82-6,90 (м, 1,6H), 6,78 (д, 0,4H), 5,44 (с, 0,7H), 4,05-4,15 (м, 0,3H), 3,80-3,93 (м, 3,7H), 3,53-3,70 (м, 4H), 2,80-2,92 (м, 1H), 2,31-2,50 (м, 1H), 1,41 (д, 2,1H), 1,34 (д, 0,9H).
ПРИМЕР 54: 2-(5-(фуран-3-ил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
К смеси 2-(2-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (100 мг), бис(три-трет-бутилфосфин)палладия (9 мг), фуран-3-бороновой кислоты (80 мг) и карбоната цезия (202 мг) добавляли диметиловый эфир этиленгликоля (4 мл), этанол (2 мл) и воду (1 мл). Реакционную смесь дегазировали N2 и нагревали в микроволновой печи в течение 30 минут при 100°C. Указанное в заголовке соединение очищали, применяя способы J и G (выход 19 мг).
1H ЯМР (CD3OD) δ м.д. 7,60-7,67 (м, 1H), 7,52-7,60 (м, 1H), 7,10-7,33 (м, 3H), 6,49-6,70 (м, 1H), 4,18 (ддд, 1H), 3,71-3,84 (м, 1H), 3,55-3,67 (м, 4H), 2,97-3,19 (м, 1H), 2,64-2,81 (м, 1H).
ПРИМЕР 55: 2-(5-(проп-1-ин-1-ил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(2-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (100 мг), применяя методику способа получения C и способов выделения F и G (выход 3,7 мг).
1H ЯМР (CD3OD) δ м.д. 7,27 (дд, 1H), 7,12 (м, 2H), 5,34 (с, 0,2H), 4,22 (ддд, 1H), 3,83 (ддд, 1H), 3,59 (м, 4H), 2,94 (м, 2H), 2,07 (с, 3H).
ПРИМЕР 56: 1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-карбоксамид
Смесь 1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-карбонитрила (пример 5,75 мг), ацетамида (39 мг), хлорида цинка (45 мг), THF (0,5 мл) и воды (0,5 мл) нагревали в микроволновом реакторе в течение 50 секунд при 320 Вт. Указанное в заголовке соединение очищали, применяя способ A (выход 15 мг).
1H ЯМР (CDCl3) δ м.д. 8,06 (д, 1H),7,57(д, 1H),7,32(т, 1H), 5,17 (с, 1H), 4,16-4,41 (м, 1H), 3,75-4,03 (м, 1H), 2,93-3,44 (м, 4H), 2,77-2,93 (м, 2H).
ПРИМЕР 57: 2-(3,7,8,9,10,10a-гексагидро-1H-циклогепта[de]изохромен-3-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (6,7,8,9-тетрагидро-5H-бензо[7]аннулен-5-ил)метанола (300 мг), применяя методику способа получения A и способов выделения F и G (выход 3 мг).
1H ЯМР (CDCl3) δ м.д. 6,96-7,24 (м, 3H), 5,32-5,48 (м, 1H), 3,74-4,03 (м, 3H), 3,39-3,60 (м, 4H), 2,65-2,98 (м, 2H), 1,84-2,11 (м, 2H), 1,52-1,82 (м, 2H), 1,19-1,42 (м, 2H).
ПРИМЕР 58: медленно элюирующийся изомер 1-(1-(4,5-дигидро-1H-имидазол-2-ил)изохроман-5-ил)этанола
Указанное в заголовке соединение получали из 2-(2-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола (300 мг), применяя методику примера 13, и очищали, применяя способ выделения G (выход 2 мг).
1H ЯМР (CD3OD) δ м.д. 7,48 (д, 1H), 7,26 (м, 2H), 5,06 (кв., 1H), 3,99 (м, 2H), 3,74 (с, 4H), 2,96 (дт, 1H), 2,84 (дт, 1H), 1,78 (д, 3H).
ПРИМЕР 59: 2-(5,7-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(2,4-диметилфенил)этанола (0,5 г), применяя методику способа получения A. Промежуточную 5,7-диметилизохроман-1-карбоновую кислоту выделяли, применяя способ выделения I, и промежуточный метил 5,7-диметилизохроман-1-карбоксилат очищали способом выделения E. Указанное в заголовке соединение выделяли, применяя способ выделения G (выход 27 мг).
1H ЯМР (CD3OD) δ м.д. 8,45 (шир.с, 1H), 7,04 (с, 1H), 6,85 (с, 1H), 5,68 (с, 1H), 4,11-4,19 (м, 1H), 3,87-4,02 (м, 5H), 2,76-2,88 (м, 1H), 2,64-2,76 (м, 1H), 2,20-2,35 (м, 6H).
ПРИМЕР 60: 2-(7-бром-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(4-бром-2-метилфенил)этанола (4,5 г), применяя методику способа получения A (4-метилбензолсульфокислоту в метаноле применяли на стадии A2), и выделяли, применяя способ выделения D с MTBE-гептан в качестве растворителя (выход 242 мг).
1H ЯМР (ДМСO-d6) δ м.д. 7,29 (с, 1H), 7,20 (с, 1H), 6,32 (шир.с, 1H), 5,28 (с, 1H), 4,09 (дт, 1H), 3,82 (ддд, 1H), 3,63 (дт, 2H), 3,18-3,27 (м, 2H), 2,57-2,72 (м, 2H), 2,20 (с, 3H).
ПРИМЕР 61: 2-(7-метокси-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из метил 7-метокси-5-метилизохроман-1-карбоксилата (0,25 г), применяя методику способа получения A3, и выделяли, применяя способ выделения G (выход 1 13 мг).
1H ЯМР (ДМСO-d6) δ м.д. 6,65-6,75(м, 1H), 6,54-6,65 (м, 1H), 6,18 (шир.с, 1H), 5,23 (с, 1H), 3,98-4,25 (м, 1H), 3,71-3,86 (м, 1H), 3,66 (с, 3H), 3,20-3,30 (м, 4H, Н2О), 2,52-2,72 (м, 2H), 2,17 (с, 3H).
ПРИМЕР 62: 2-(3,5-диметилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из метил 3,5-диметилизотиохроман-1-карбоксилата (0,44 г), применяя методику способа получения A3, и выделяли, применяя способ выделения G. (выход 10 мг).
1H ЯМР (CD3OD) δ м.д. 6,93-7,23 (м, 3H), 3,38-3,68 (м, 5H), 2,98-3,19 (м, 1H), 2,48 (дд, 1H), 2,19-2,37 (м, 3H).
ПРИМЕР 63: 2-(5-Бром-3-метилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из метил 5-бром-3-метилизотиохроман-1-карбоксилата (0,5 г, способы получения A1 и A2), применяя методику способа получения A3, и выделяли, применяя способ выделения E (выход 72 мг).
1H ЯМР (CD3OD) δ м.д. 7,53 (д, 1H), 7,03-7,25 (м, 2H), 3,47-3,69 (м, 5H), 3,37-3,46 (м, 1H), 2,57 (дд, 1H), 1,17-1,46 (м, 3H).
ПРИМЕР 64: 2-(5-метилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из метил 5-метил-изотиохроман-1-карбоксилата (1,3 г, способ получения F), применяя методику способа получения A3, и выделяли, применяя способ выделения D с EtOAc/EtOH в качестве растворителей (выход 110 мг).
1H ЯМР (CDCl3) δ м.д. 6,90-7,17 (м, 3H), 4,63 (с, 1H), 3,52-3,75 (м, 4H), 3,08-3,25 (м, 1H), 2,80-3,02 (м, 3H).
ПРИМЕР 65: 2-(5-бромизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Способ получения F
Способ получения F1: Метил-2-((2-бромфенэтил)тио)ацетат
Смесь метил 2-меркаптоацетата (2,2 г), 2-бромфенэтилметансульфоната (5,7 г), 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-a]азепина (3,7 г) и THF (30 мл) перемешивали при температуре окружающей среды в течение 1,5 часов, и способ выделения A и E давал указанное в заголовке соединение (4,4 г).
Способ получения F2: метил-2-((2-бромфенэтил)тио)-2-хлорацетат
Смесь метил 2-((2-бромфенэтил)тио)ацетата (4,4 г), 1-хлорпирролидин-2,5-диона (2 г) и тетрахлорида углерода (2,4 г) перемешивали в бане со льдом в течение 2,5 часов, фильтровали и упаривали, получая указанное в заголовке соединение (4,7 г).
Способ получения F3: метил-5-бромизотиохроман-1-карбоксилат
Смесь метил 2-((2-бромфенэтил)тио)-2-хлорацетата (4,7 г), трихлорида алюминия (2 г) и DCM (15 мл) перемешивали в бане со льдом и нагревали до температуры окружающей среды и перемешивали в течение 3,5 часов. Способ выделения B и E давал указанное в заголовке соединение (2,9 г).
Способ получения F4: 2-(5-бромизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из метил-5-бром-изотиохроман-1-карбоксилата (0,5 г), применяя методику способа получения A3, и выделяли, применяя способы выделения E и D (2-пропанол, выход 21 мг).
1H ЯМР (ДМСO-d6) δ м.д. 7,52 (дд, 1H), 7,02-7,19 (м, 2H), 4,65 (с, 1H), 3,77 (тд, 1H), 3,17-3,60 (шир.с, 4H), 2,96-3,09 (м, 1H), 2,75-2,95 (м, 2H).
ПРИМЕР 66: 2-(5-бром-1-метилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из метил-5-бромизотиохроман-1-карбоксилата (1,6 г), применяя методики способов получения примера 11 и A3, и выделяли способом выделения E (выход 30 мг).
1H ЯМР (CDCl3) δ м.д. 7,46-7,56 (м, 1H), 7,21-7,39 (м, 1H, CHCl3), 7,02-7,16 (м, 1H), 3,17-4,0 (м, 4H), 2,70-3,10 (м, 3H), 1,48-2,01 (м, 4H).
ПРИМЕР 67: гидрохлорид 2-(5,7-дибром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: Этил 5,7-дибром-3-этилизохроман-1-карбоксилат
В сухую пробирку для микроволновых реакций помещали ди-μ-метоксибис(1,5-циклооктадиен)дииридий (I) (5,0 мг), 4,4'-диметокси-2,2'-дипиридил (3,2 мг), бис(пинаколато)дибор (93 мг) и 2-(5-бром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазол (157 мг, пример 43) и пробирку продували, применяя несколько раз вакуум/N2 цикл. Добавляли тетрагидрофуран (1 мл) и раствор нагревали до 80°C в герметично закрытой емкости. Через 4 часа нагревание прекращали и реакционную смесь концентрировали. Остаток после упаривания растворяли в метаноле (6 мл) и добавляли раствор CuBr2 в воде (6 мл). Полученную в результате гетерогенную смесь нагревали при 80°C в герметично закрытой емкости. Через 8 часов реакционную смесь охлаждали до комнатной температуры и экстрагировали диэтиловым эфиром. Объединенные органические фазы промывали соляным раствором, сушили Na2SO4 и концентрировали. Остаток после упаривания очищали способом выделения E (этилацетат - гептан) (выход 94 мг).
1H ЯМР (CDCl3) δ м.д. 7,63-7,67 (м, 1H), 7,55-7,58 (м, 0,33H), 7,38-7,44 (м, 0,63H), 5,31-5,36 (м, 0,11H) 5,28-5-31 (д, 0,89H), 4,19-4,38 (м, 2H), 4,00-4,09 (м, 0,37H), 3,50-3,60 (м, 0,63H), 2,72-2,83 (м, 1H), 2,56 (ддд, 0,64H), 2,39 (дд, 0,36H), 1,66-1,90 (м, 2H), 1,30-1,36 (м, 3H), 1,02-1,13 (м, 3H).
Стадия 2: гидрохлорид 2-(5,7-дибром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали, применяя способ получения A3, и превращали в HCl-соль, применяя способ получения соли A (выход 60 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,59 (с, 1,57H), 10,08 (с, 0,36H), 7,93-7,96 (м, 1H), 7,59 (д, 0,19Н), 7,48-7,51 (м, 0,77H), 5,92 (шир.с, 0,19H), 5,90 (шир.с, 0,78H), 3,84-4,00 (м, 4H), 3,63-3,78 (м, 1H), 2,74-2,84 (м, 1H), 2,50-2,57 (м, 1H), 1,61-1,77 (м, 2H), 0,94-1,02 (м, 3H).
ПРИМЕР 68: HCl соль энантиомера 2-5-бром-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: 1-(2-Бромфенил)-4,4,4-трифторбутан-2-он
К раствору гидрохлорида 2,2,2-трифторэтанамина (27,1 г, 200 ммоль) в DCM (400 мл) и воде (50 мл) добавляли NaNO2 (15,43 г, 241 ммоль) при 0°C и перемешивали в течение 1 часа. Затем, охлаждали до 78°C и добавляли 2-(2-бромфенил)ацетальдегид (20 г, 100 ммоль) и ZrCl4 (30,4 г, 130 ммоль) и перемешивали в течение 2 часов. Реакцию прекращали MeOH (30 мл). Указанное в заголовке соединение очищали способами выделения A и E (выход 13,0 г).
Стадия 2: энантиомер (1-(2-бромфенил)-4,4,4-трифторбутан-2-ола
К раствору 1-(2-бромфенил)-4,4,4-трифторбутан-2-она (7,0 г, 24,9 ммоль) в MeOH (150 мл) добавляли NaBH4 (1,23 г, 32,3 ммоль) при 0°C и перемешивали в течение 1 часа при комнатной температуре. Затем реакцию прекращали MeOH и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение очищали способами выделения A, E и H (выход 1,6 г энантиомера 1 и 1,4 г энантиомера 2). Энантиомер 1 элюировался при tr=5,67 минут, и энантиомер 2 элюировался при tr=9,57 минут Chiralcel OD-H (4,6X250 мм) 5 мкм, гексан:2-PrOH:TFA (90:10:0,1), 1 мл/минута.
Стадия 3: HCl соль энантиомера 2-5-бром-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из энантиомера 1 этил 1-(2-бромфенил)-4,4,4-трифторбутан-2-ола (500 мг) способом получения B и выделяли способом выделения E. Полученное основание превращали в HCl соль, применяя способ получения соли A (HCl в Et2O и DCM в качестве растворителя) (выход 192 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,73 (шир.с, 1,95H), 10,26 (шир.с, 2,05H), 7,70 (заменяющ. дд, 2H), 7,20-7,42 (заменяющ. м, 4H), 5,99 (шир.с, 1,02H), 5,98 (шир.с, 0,98H), 4,16-4,33 (заменяющ. м, 2H), 3,81-3,96 (м, 8H), 2,59-3,09 (м, 8H).
ПРИМЕР 69: 2-(5-метокси-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Раствор этилглиоксата (50% раствор в толуоле, 11,66 мл) и 2-метоксибензолэтанола (12,73 мл) перемешивали в течение 1 часа, получая этил 2-гидрокси-2-(2-метоксифенэтокси)ацетат. Образование этил 2-гидрокси-2-(2-метоксифенэтокси)ацетата контролировали1H ЯМР и реакционную смесь охлаждали баней со льдом и обрабатывали пиридином (48,4 мл), 4-диметиламинопиридином (0,44 г) и ацетилхлоридом (12,85 мл). После перемешивания при +7°C в течение 1 часа добавляли гептан (50 мл) и охлаждение в бане со льдом прекращали, обеспечивая достижение реакционной смесью температуры окружающей среды с последующим фильтрованием. Фильтрат промывали MTBE (3×50 мл) и объединенные растворители промывали NaHCO3 (насыщенный водный раствор), водой и NaCl (насыщенный водный раствор), сушили над Na2SO4 и фильтровали. Органические растворители упаривали, добавляли толуол и упаривали. Побочные продукты и остаточные растворители отгоняли (0 мбар-1 мбар/72°-105°C) и остаток растворяли в этилацетате. Добавляли силикагель и активированный уголь, раствор перемешивали в течение 10 минут и фильтровали. Органические растворители упаривали, получая этил 2-ацетокси-2-(2-метоксифенэтокси)ацетат (12,3 г), который применяли в следующей стадии.
Раствор этил 2-ацетокси-2-(2-метоксифенэтокси)ацетата (12,3 г) в дихлорметане (415 мл) при -20°C обрабатывали AlCl3 (5,5 г), и перемешивали в течение 1 часа. Добавляли AlCl3 (5,5 г) к раствору и перемешивали в течение 3,5 часов, выливали в смесь воды и льда (400 мл). Применяли способ выделения A, с последующей перегонкой (1-2 мбар/120-130°C) неочищенного продукта, получая этил 5-метоксиизохроман-1-карбоксилат (5,1 г).
Смесь NaH (0,68 г), этил 5-метоксиизохроман-1-карбоксилата (1 г) и DMF (10 мл) перемешивали в течение 2 часов при температуре бани со льдом. Добавляли йодметан (2,6 мл), и смесь перемешивали при температуре окружающей среды в течение 16 часов. Промежуточный этил 5-метокси-1-метилизохроман-1-карбоксилат (0,7 г) очищали способом выделения A, и указанное в заголовке соединение получали, применяя методику способа получения A стадия 3 и способа выделения D (диэтиловый эфир) (выход 0,1 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,10 (tr, 1H), 6,83 (дд, 2H), 6,08 (с, 1H), 3,85 (м, 2H), 3,77 (с, 3H), 3,44-3,70 (шир.м, 2H), 3,07-3,28 (шир.с, 2H), 2,62 (tr, 2H), 1,60 (с, 3H).
ПРИМЕР 70: 2-(5-Метоксиизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-метоксифенэтилметансульфоната (3,58 г), применяя методику способа получения F. Указанное в заголовке соединение очищали, применяя способ выделения E (выход 60 мг).
1H ЯМР (CD3OD) δ м.д. 7,11 (т, 1H), 6,84 (д, 1H), 6,73 (д, 1H), 3,82 (с, 3H), 3,46-3,66 (м, 4H), 3,03-3,17 (м, 2H), 2,75-2,87 (м, 2H).
ПРИМЕР 71: 2-((3R)-5-метокси-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Смесь NaH (0,17 г), (3R)-этил 5-метокси-3-метилизохроман-1-карбоксилата (0,3 г) и DMF (7 мл) перемешивали в течение 2 часов при температуре бани со льдом. Добавляли йодметан (0,7 мл) и смесь перемешивали при температуре окружающей среды в течение 16 часов. Промежуточный (3R)-этил 5-метокси-1,3-диметилизохроман-1-карбоксилат (0,4 г) очищали способом выделения A и указанное в заголовке соединение получали, применяя методику способа получения A стадия 3 и способа выделения E (EtOAc/DCM/триэтиламин 10/20/1) (выход 0,3 г).
1H ЯМР (CDCl3) δ м.д. 7,01-7,24 (м, 2H), 6,71-6,76 (м, 1H), 3,97-4,24 (м, 1H), 3,80-3,84 (м, 3H), 3,37-3,77 (шир., 3H), 2,77-2,90 (м, 1H), 2,32-2,42 (м, 1H), 1,77 (д, 3H), 1,35-1,41 (м, 3H), 1,24-1,29(м, 1H).
ПРИМЕР 72: 2-(5-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: 2-(2-(2,2,2-трифторэтил)фенил)оксиран
Бром-2-(2,2,2-трифторэтил)бензол (0,5 г) растворяли в дегазированном толуоле и добавляли трибутил(винил)олово (0,67 мл), Pd2(dba)3 (0,096 г) и три-трет-бутилфосфин (0,25 мл, 1M раствор). Реакционную смесь нагревали до 40°C. Через 4 часа нагревание прекращали и добавляли 1:1 раствор диэтилового эфира и пентана (10 мл) с последующим добавлением фторида калия (1,0 г). Смесь перемешивали в течение 30 минут, после чего ее фильтровали через слой силикагеля, и фильтрат аккуратно концентрировали при вакууме 500 мбар. Остаток после упаривания очищали колоночной хроматографией (диэтиловый эфир - пентан) и фракции, содержащие продукт, концентрировали вакуумной дистилляцией. Остаток после упаривания (0,389 г), содержащий толуол и требуемый стирольный продукт, растворяли в дихлорметане (12 мл) и добавляли 3-хлорпероксибензойную кислоту (0,70 г).
После перемешивания реакционной смеси в течение ночи при комнатной температуре, ее разбавляли дихлорметаном (20 мл) и добавляли раствор Na2SO3 (10 мл, 1 M). Двухфазную смесь перемешивали в течение 30 минут и фазы разделяли. Водную фазу экстрагировали дихлорметаном (3×10 мл). Все органические фазы объединяли, промывали насыщенным NaHCO3 раствором, сушили Na2SO4 и концентрировали (давление 300 мбар). Остаток после упаривания очищали способом выделения E (диэтиловый эфир - пентан) (выход 220 мг).
1H ЯМР (CDCl3) δ м.д. 7,26-7,38 (м, 4H), 4,01-4,07 (м, 1H), 3,42-3,69 (м, 2H), 3,17 (дд, 1H), 2,69 (дд, 1H).
Стадия 2: 2-(2-(2,2,2-трифторэтил)фенил)этанол
В колбу загружали Pd(0) EnCat 30 NP (0,136 г, промытый этанолом и этилацетатом перед применением) и добавляли раствор 2-(2-(2,2,2-трифторэтил)фенил)оксирана (0,1 10 г) в этилацетате (1,5 мл) с последующим добавлением триэтиламина (0,33 мл) и муравьиной кислоты (90 мкл). Смесь перемешивали при комнатной температуре в течение 26 часов, после чего ее фильтровали. Осадок промывали этилацетатом и фильтрат концентрировали. Остаток после упаривания очищали способом выделения E (этилацетат - гептан) (выход 70 мг).
1H ЯМР (CDCl3) δ м.д. 7,19-7,34 (м, 4H), 3,87 (тд, 2H), 3,50 (кв., 2H), 2,96 (т, 2H).
Стадия 3: 2-(5-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из 2-(2-(2,2,2-трифторэтил)фенил)этанола (70 мг), применяя методику способа получения B. Диэтилэфират трифторида бора применяли вместо хлорида титана (IV) (выход 53 мг).
1H ЯМР (CD3OD) δ м.д. 7,16-7,28 (м, 3H), 5,40 (с, 0,1H, заменяемый протон), 4,23 (ддд, 1H), 3,84 (ддд, 1H), 3,46-3,66 (м, 5H), 3,46-3,53 (м, 1H), 2,99 (ддд, 1H), 2,80 (дт, 1H).
ПРИМЕР 73: 2-((3R)-5-этил-1,3-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Способ получения G
Смесь NaH (0,39 г), (3R)-этил 5-этил-3-метилизохроман-1-карбоксилата (0,6 г) и DMF (7 мл) перемешивали в течение 2 часов при температуре бани со льдом. Добавляли йодметан (1,5 мл), и смесь перемешивали при температуре окружающей среды в течение 16 часов. Промежуточный (3R)-этил 5-этил-1,3-диметилизохроман-1-карбоксилат (0,5 г) очищали способом выделения A, и указанное в заголовке соединение получали, применяя методику способа получения A стадия 3 и способа выделения E (EtOAc/DCM/триэтиламин 10/20/1) (выход 0,06 г).
1H ЯМР (CDCl3) δ м.д. 7,30-7,07 (м, 3H), 4,89 (шир., 1H), 3,81-4,10 (М, 1H), 2,47-2,77 (м, 4H), 1,73-1,83 (д, 3H), 1,38 (тр, 3H), 1,21 (тд 3H).
ПРИМЕР 74: 2-(5-Метил-3-(метоксиметил)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из этил 3-(метоксиметил])-5-метилизохроман-1-карбоксилата (200 мг) способом получения A и выделяли способом выделения D (выход 134 мг).
1H ЯМР (CD3OD) δ м.д. 7,00-7,14 (м, 3H), 5,45 (с, 1H), 3,96 (дд, 1H), 3,50-3,67 (м, 6H), 3,40-3,46 (м, 3H), 2,63-2,74 (м, 2H), 2,24 (с, 3H).
ПРИМЕР 75: гидробромид 1-(4,5-Дигидро-1H-имидазол-2-ил)-5-метилизохроман-7-ола
2-(7-Метокси-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазол (пример 61, 100 мг) растворяли в бромистоводородной кислоте (1,5 мл, 47% по весу в воде) и смесь кипятили с обратным холодильником. Через 5,5 часов смесь охлаждали до комнатной температуры. Растворители упаривали и остаток после упаривания растирали с этанолом, получая указанное в заголовке соединение (выход 27 мг).
1H ЯМР (D2O) δ м.д. 6,84 (д, 1H), 6,57 (д, 1H), 5,76 (с, 1H), 4,06 (т, 2H), 4,00 (с, 4H), 2,74 (т, 2H), 2,23 (с, 3H).
ПРИМЕР 76: гидрохлорид 1-(4,5-дигидро-1H-имидазол-2-ил)-3-этилизохроман-5-ола
Стадия 1: метил 3-этил-5-гидроксиизохроман-1-карбоксилат
К смеси этил 5-бром-3-этилизохроман-1-карбоксилата (500 мг) и KOH (358 мг) добавляли диоксан (4,5 мл) и воду (1,5 мл). Добавляли 2-ди-трет-бутил-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1',1'-бифенил (38 мг), трис(дибензилиденацетон)дипалладий (0) и Pd2(dba)3 (73 мг) и затем смесь нагревали в течение 2,5 часов при 100°C. Затем реакцию реакционную смесь упаривали досуха и применяли непосредственно в следующей стадии.
Остаток из реакции растворяли в MeOH (15 мл). Добавляли хлортриметилсилан (5,9 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь упаривали досуха. Указанное в заголовке соединение очищали способом выделения E (выход 90 мг).
Стадия 2: гидрохлорид 1-(4,5-дигидро-1H-имидазол-2-ил)-3-этилизохроман-5-ола
Метил 3-этил-5-гидроксиизохроман-1-карбоксилат (90 мг) растворяли в дихлорметане (1 мл) и добавляли 3,4-дигидро-2H-пиран (52 мкл) и п-толуолсульфонат пиридиния (9,6 мг). Через 28 часа добавляли дополнительные количества 3,4-дигидро-2H-пирана (102 мкл) и п-толуолсульфоната пиридиния (10 мг) и смесь перемешивали в течение следующих 24 часов. Затем добавляли 3,4-дигидро-2H-пиран (200 мкл) и смесь перемешивали в течение ночи, после чего ее разбавляли диэтиловым эфиром (5 мл) и промывали насыщенным раствором NaHCO3. Органическую фазу сушили Na2SO4 и концентрировали. Очистка остатка после упаривания колоночной хроматографией (этилацетат - гептан) давала 37 мг масла, которое превращали в 2-(3-этил-5-((тетрагидро-2H-пиран-2-ил)окси)изохроман-1-ил)-4,5-дигидро-1H-имидазол (13 мг), применяя способ получения A3. Неочищенный продукт данного способа растворяли в метаноле (1 мл) и добавляли раствор HCl в диэтиловом эфире (2 M, 39 мкл). Через 2,5 часа реакционную смесь концентрировали и остаток растворяли в воде (10 мкл). Раствор промывали этилацетатом и водный слой концентрировали в лиофилизаторе, получая указанное в заголовке соединение (выход 6 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,43 (с, 1,3H), 9,92 (с, 0,45H) 9,80 (с, 0,9H), 7,03-7,14 (м, 1H), 6,78-6,88 (м, 1H), 6,58-6,70 (м, 1H), 5,78 (с, 0,11H), 5,75 (с, 0,61H), 3,84-3,93 (м, 4H), 3,61-3,74 (м, 1H), 2,77 (д, 1H), 2,28-2,44 (м, 1H), 1,53-1,76 (м, 2H), 0,92-1,03 (м, 3H).
ПРИМЕР 77: энантиомер 2 2-(5-метокси-3-(2,2,2-трифторэтил)метил-изохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: (R)-4,4,4-трифтор-1-(2-метоксифенил)бутан-2-ол
К раствору 4,4,4-трифтор-1-(2-метоксифенил)бутан-2-она (6 г, 25,8 ммоль, полученному как в примере 52, стадия 1) в MeOH (100 мл) добавляли NaBH4 (1,47 г, 38,7 ммоль) при 0°C и перемешивали в течение 1 часа при комнатной температуре. Затем реакцию прекращали MeOH и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение очищали способами выделения A, E и H выход (1,7 г). Энантиомер элюировался при tr=12,8 минут Chiralpak 1A (4,6X250 мм) 5 мкм, гексан:2-PrOH:TFA (90:10:0,1), 1 мл/минута.
Стадия 2: энантиомер 2 2-(5-метокси-3-(2,2,2-трифторэтил)метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из энантиомера 2 4,4,4-трифтор-1-(2-метоксифенил)бутан-2-ола (900 мг) способом получения G и выделяли способом выделения E (выход 30 мг).
1H ЯМР (CD3OD) δ м.д. 7,14-7,26 (м, 2H), 6,85 (т, 1H), 5,46 (с, 1H), 4,06 (дд, 1H), 3,81-3,88 (м, 3H), 3,48-3,69 (м, 4H), 2,93 (дд, 1H), 2,45-2,70 (м, 2H).
ПРИМЕР 78: 2-(1,5-диметилизотиохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали, применяя этил 1,5-диметилизотиохроман-1-карбоксилат и методику способа получения A стадия 3 и способов выделения D (диэтиловый эфир), E (EtOAc/DCM/триэтиламин 10/18/1) и D (диэтиловый эфир/гептан) соответственно (выход 0,06 г).
1H ЯМР (ДМСO-d6) δ м.д. 7,04 (м, 3H), 5,91 (с, 1H), 3,45-3,70 (шир., 2H), 3,18-3,30 (шир., 2H), 3,03-3,12 (м, 1H), 2,92-2,81 (м, 3H), 2,20 (с, 3H), 1,77 (с, 3H).
ПРИМЕР 79: HCl соль 2-(5-(трифторметокси)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из 1-(2-(2-(трифторметокси)фенил)этанола (2,2 г), применяя методику способа получения B (диэтилэфират трифторида бора применяли вместо хлорида титана (IV) и только 1/3 промежуточного этил 5-(трифторметокси)изохроман-1-карбоксилата применяли в последней стадии). Полученное основание превращали в HCl соль, применяя способ получения соли A (HCl в Et2O и DCM в качестве растворителя) (выход 192 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,64 (с, 2H), 7,27-7,56 (м, 3H), 5,94 (с, 1H), 4,17 (дт, 1H), 3,83-3,96 (м, 1H), 3,90 (шир., с, 4H), 2,75-2,99 (м, 2H).
ПРИМЕР 80: HCl соль энантиомера 1 2-(3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более быстро элюирующийся энантиомер (138 мг), получали из соединения примера 8 (500 мг) способом выделения L.
1H ЯМР (CD3OD) δ м.д. 7,26 (д, 2H), 7,06 (т, 1H), 5,76 (с, 1H), 4,14-4,25 (м, 1H), 3,89-4,02 (м, 5H), 2,79-3,02 (м, 2H), 2,68 (кв., 2H), 1,22 (т, 3H).
ПРИМЕР 81: HCl соль 2-(3-(2-Фторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: 1-(2-(бензилокси)пент-4-енил)-2-метилбензол
К суспензии NaH (4,9 г, 127,8 ммоль) в THF (150 мл) добавляли 1-o-толилпент-4-ен-2-ол (15 г в THF, 85,22 ммоль) при 0°C и перемешивали в течение 30 минут. Затем добавляли бензилбромид (17,85 г, 102,2 ммоль) при 0°C, затем реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем гасили ледяной H2O и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение выделяли способом выделения A и E (выход 13,0 г).
Стадия 2: 3-(бензилокси)-4-o-толилбутаналь
Через раствор 1-(2-(бензилокси)пент-4-енил)-2-метилбензола (5,0 г, 18,72 ммоль) в DCM (100 мл) пропускали газообразный O3 при -78°C в течение 3 часов. Реакцию прекращали TEA (5 мл) при -78°C и перемешивали при комнатной температуре в течение 5 часов. Указанное в заголовке соединение очищали способами выделения A и E (выход 2,0 г).
Стадия 3: 3-(бензилокси)-4-o-толилбутан-1-ол
К раствору 3-(бензилокси)-4-o-толилбутаналя (4,0 г, 14,81 ммоль) в MeOH (50 мл) добавляли NaBH4 (1,13 г, 29,62 ммоль) при 0°C и перемешивали при комнатной температуре в течение 2 часов. Реакцию прекращали водой и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение очищали способами выделения A и E (выход 3,0 г).
Стадия 4: 1-(2-(Бензилокси)-4-фторбутил)-2-метилбензол
К раствору 3-(бензилокси)-4-o-толилбутан-1-ола (3,0 г, 11,11 ммоль) в DCM (50 мл) добавляли DAST (2,9 мл, 22,22 ммоль) при 0°C и перемешивали при комнатной температуре в течение 5 часов. Затем, охлаждали до 0°C и гасили NH4Cl (10 мл). Указанное в заголовке соединение очищали способами выделения A и E (выход 1,5 г).
Стадия 5: 4-фтор-1-o-толилбутан-2-ол
К раствору 1-(2-(бензилокси)-4-фторбутил)-2-метилбензола (3,0 г, 11,03 ммоль) в MeOH (50 мл) добавляли 10% Pd/C (0,3 г) в атмосфере азота и перемешивали при 40 пси в течение 5 часов. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение очищали способом выделения E (выход 1,5 г).
Стадия 6: Этил 3-(2-фторэтил)-5-метилизохроман-1-карбоксилат
Указанное в заголовке соединение получали способом получения A из 4-фтор-1-o-толилбутан-2-ола (3,0 г, 16,48 ммоль) и очищали способами выделения A и E (выход 1,0 г).
Стадия 7: HCl соль 2-(3-(2-фторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из 3-(2-фторэтил)-5-метилизохроман-1-карбоксилата (200 мг) способом получения A и способом A получения соли и выделяли способом выделения D (выход 50 мг).
1H ЯМР (CD3OD) δ м.д. 7,15-7,34 (м, 2H), 7,01-7,12 (м, 1H), 5,83 (с, 1H), 4,53-4,81 (м, 2H), 3,94-4,10 (м, 5H), 2,30 (с, 3H), 2,79-2,94 (м, 1H), 2,59-2,79 (м, 1H), 1,98-2,27 (м, 2H).
ПРИМЕР 82: энантиомер 2-(5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более быстро элюирующийся энантиомер (77 мг), получали из соединения примера 17 (160 мг) способом выделения L.
1H ЯМР (ДМСO-d6) δ м.д. 10,54 (шир.с, 2H), 7,27 (т, 1H), 6,99 (д, 1H), 6,85 (д, 1H), 5,83 (шир.с, 1H), 4,10-4,17 (м, 1H), 3,87 (шир.с, 4H) 3,82-3,86 (м, 1H), 3,81 (с, 3H), 2,65-2,81 (м, 2H).
ПРИМЕР 83: HCl соль энантиомера 2 2-5-бром-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из энантиомера 2 этил 1-(2-бромфенил)-4,4,4-трифторбутан-2-ола (500 мг, пример 68, стадия 2) способом получения B и выделяли способом выделения E. Полученное основание превращали в HCl соль, применяя способ получения соли A (HCl в Et2O и DCM в качестве растворителя) (выход 290 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,61 (шир.с, 1,24H), 10,50 (шир.с, 0,76H), 7,61-7,79 (заменяющ.м, 1H), 7,21-7,39 (заменяющ.м, 2H), 5,98 (шир.с, 0,62H), 5,97 (шир.с, 0,38H), 4,24 (дд, 1H), 3,83-3,95 (м, 4H), 2,60-3,08 (м, 4H).
ПРИМЕР 84: HCl соль 2-(3-(2,2-дифторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: 1-(2-(бензилокси)-4,4-дифторбутил)-2-метилбензол
К раствору 3-(бензилокси)-4-o-толилбутан-1-ола (10 г, 37,04 ммоль, пример 81, стадия 3) в DCM (100 мл) добавляли DAST (10 мл, в/о) при 0°C и перемешивали при комнатной температуре в течение 5 часов. Затем охлаждали до 0°C и гасили NH4Cl (50 мл). Указанное в заголовке соединение очищали способами выделения A и E (выход 7,0 г).
Стадия 2: 4,4-дифтор-1-o-толилбутан-2-ол
К раствору 1-(2-(бензилокси)-4,4-дифторбутил)-2-метилбензола (10 г, 34,48 ммоль) в MeOH (150 мл) добавляли 10% Pd/C (1,0 г) в атмосфере азота и перемешивали при 40 пси в течение 5 часов. Реакционную смесь фильтровали через целит и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение очищали способом выделения E (выход 6,0 г).
Стадия 3: этил 3-(2,2-дифторэтил)-5-метилизохроман-1-карбоксилат
Указанное в заголовке соединение получали способом получения A из 4,4-дифтор-1-o-толилбутан-2-ола (5,0 г, 25,0 ммоль) и очищали способами выделения A и E (выход 1,5 г).
Стадия 4: HCl соль 2-(3-(2,2-дифторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из этил 3-(2,2-дифторэтил)-5-метилизохроман-1-карбоксилата (200 мг) способом получения A и способом получения соли A, и выделяли способом выделения D (выход 130 мг).
1H ЯМР (CD3OD) δ м.д. 7,15-7,31 (м, 2H), 7,05 (д, 1H), 6,03-6,30 (м, 1H), 5,83 (с, 1H), 4,09 (ддд, 1H), 3,96-4,04 (м, 4H), 2,86 (дд, 1H), 2,73 (дд, 1H), 2,19-2,40 (м, 5H).
ПРИМЕР 85: 2-(7-метокси-3,5-диметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Смесь 2-бром-5-метокситолуола (2 г) и THF (10 мл) при -78°C обрабатывали н-бутиллитием (2,5M раствор в гептане, 8,8 мл) и перемешивали в течение 15 минут с последующим добавлением пропиленоксида (0,9 мл) в THF (5 мл). После перемешивания в течение 30 минут при -78°C реакционную смесь нагревали до температуры окружающей среды и промежуточный 1-(4-метокси-2-метилфенил)пропан-2-ол очищали способами выделения A и E (EtOAc/гептан) соответственно. Смесь этил 2,2-диэтоксиацетата (0,6 мл), 1-(4-метокси-2-метилфенил)пропан-2-ола (0,5 г) и DCE (4 мл) перемешивали при температуре бани со льдом и добавляли диэтилэфират трифторида бора (0,7 мл) с последующим кипячением с обратным холодильником в течение 4 часов. Промежуточный этил 7-метокси-3,5-диметилизохроман-1-карбоксилат (0,05 г) очищали способами выделения B и E (этилацетат/гептан) соответственно. Указанное в заголовке соединение получали, применяя методику стадии 3 способа получения A и способа выделения D (диэтиловый эфир/гептан) (выход 0,01 г).
1H ЯМР (CDCl3) δ м.д. 6,74 (дд, 2H), 5,45 (д, 1h), 3,83-3,92 (м, 1H), 3,75 (д, 3H), 3,40-3,71 (шир.с, 3H), 2,368-2,68 (м, 2H), 2,2 (с, 3H), 1,37 (дд, 3H).
ПРИМЕР 86: гидрохлорид энантиомера 2 2-((3)-5-метил-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: энантиомер (4,4,4-трифтор-1-o-толилбутан-2-oла
К раствору 4,4,4-трифтор-1-o-толилбутан-2-она (7,0 г, 32,4 ммоль, пример 52, стадия 1) в MeOH (150 мл) добавляли NaBH4 (1,84 г, 48,6 ммоль) при 0°C и перемешивали при комнатной температуре в течение 1 часа. Затем реакцию прекращали MeOH и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение очищали способами выделения A, E и H выход (1,4 г). Энантиомер элюировался при tr=7,0 минут Chiralcel OD-H (4,6X250 мм) 5 мкм, гексан: 2-ΡrOΗ: TFA (90:10:0,1), 1 мл/минута.
Стадия 2: гидрохлорид энантиомера 2 2-((3)-5-метил-3-(2,2,2-трифторэтил)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали, как в примере 52, из энантиомера 2 4,4,4-трифтор-1-o-толилбутан-2-ола (1,19 г) и превращали в HCl соль согласно способу получения соли A (выход 74 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,52 (шир.с, 1,5H), 10,13 (шир.с, 2H), 7,16-7,29 (м, 2H), 7,05-7,13 (м, 1H), 5,93 (с, 0,75H), 5,89 (с, 0,22H), 4,14-4,28 (м, 1H), 3,83-3,93 (м, 4H), 2,61-2,91 (м, 4H), 2,23 (с, 3H).
ПРИМЕР 87: 2-(5-(метилтио)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: этил 3,4-дигидро-5-(метилтио)-1H-изохромен-1-карбоксилат
Раствор этил 5-бромизохроман-1-карбоксилата (способ получения A1 и A2, 0,5 г, 1,75 ммоль), NaSMe (0,18, 3,3 ммоль) и CuBr (0,435 г, 1,8 ммоль) в DMF (10 мл) нагревали при 90°C при микроволновом облучении в течение 5 минут. Указанное в заголовке соединение очищали способами выделения D и E (выход 100 мг).
Стадия 2: 2-(5-(метилтио)изохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из этил 5-(метилтио)изохроман-1-карбоксилата (490 мг) способом получения A и выделяли способом выделения G (выход 380 мг).
1H ЯМР (CDCl3) δ м.д. 7,17-7,22 (м, 2H), 7,08-7,13 (м, 1H), 5,44 (с, 1H), 4,24 (ддд, 1H), 3,89 (ддд, 1H), 3,45-3,82 (м, 4H), 2,70-2,93 (м, 2H), 2,46 (с, 3H).
ПРИМЕР 88: гидрохлорид энантиомера 2 2-((3)-5-бром-3-пропилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: энантиомер 2 1-(2-бромфенил)пентан-2-ола
К раствору 2-(2-бромфенил)ацетальдегида (16 г, 80,8 ммоль) в THF (100 мл) добавляли бромид пропилмагния (48 мл (2,0 M) в THF), 97 ммоль) при 0°C, затем перемешивали при комнатной температуре в течение 5 часов. Реакцию прекращали хлоридом аммония, затем указанное в заголовке соединение очищали способами выделения A, E и H (выход 740 мг). Энантиомер 2 элюировался при tr=5,70 минут Chiralcel OD-H (4,6X250 мм) 5 мкм, гексан:2-PrOH: TFA (90:5:0,1), 1 мл/минута.
Стадия 2: гидрохлорид энантиомера 2-((3)-5-бром-3-пропилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из энантиомера 2 1-(2-бромфенил)пентан-2-ола (1 0 г), применяя методику способа получения B и способа получения соли A (выход 510 мг). Диэтилэфират трифторида бора применяли вместо хлорида титана (IV).
1H ЯМР (ДМСO-d6) δ м.д. 10,73 (шир.с, 1,8H), 10,16 (шир.с, 0,1H), 7,70-7-63 (м, 1H), 7,21-7,34 (м, 2H), 5,92 (шир.с, 1H), 3,78-3,94 (м, 5H), 2,78-2,88 (м, 1H), 2,61 (дд, 1H), 1,59-1,72 (м, 2H), 1,35-1,57 (м, 2H), 0,93 (м, 3H).
ПРИМЕР 89: гидрохлорид энантиомера 2 2-(3-(2,2-дифторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: энантиомер 1-(2-(бензилокси)-4,4-дифторбутил)-2-метилбензола
Рацемический 1-(2-(бензилокси)-4,4-дифторбутил)-2-метилбензол (пример 84, стадия 2) разделяли на энантиомеры способом выделения H (время удерживания 8,6 минут, 1 мл/минута, Chiralcel OJ-H, 4,6x250 мм, 90:10:0,1 гексан:iPrOH:TFA) (выход 4,0 г).
Стадия 2: гидрохлорид энантиомера 2 2-(-3-(2,2-дифторэтил)-5-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из энантиомера 2 (3)-этил 3-(2,2-дифторэтил)-5-метилизохроман-1-карбоксилата (250 мг), применяя методику способа получения A3 и способа получения соли A (выход 100 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,67 (с, 2H), 7,09-7,23 (м, 3H), 6,14-6-45 (тт, 1H), 5,93 (с, 1H), 3,97-4,08 (м, 1H), 3,85-3,92 (м, 4H), 2,81 (дд, 1H), 2,63-2,74 (м, 1H), 2,24-2,37 (м, 2H), 2,23 (с, 3H).
ПРИМЕР 90: гидрохлорид 2-(5-(дифторметокси)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из этил 5-(дифторметокси)изохроман-1-карбоксилата (220 мг), применяя методику способа получения A3 и способа получения соли A (выход 180 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,41 (с, 2H), 7,38 (т, 1H), 7,27 (т, 1H), 7,22 (д, 1H), 7,15 (д, 1H), 5,85 (с, 1H), 4,14 (тд, 1H), 3,83-3,95 (м, 5H), 2,72-2,90 (м, 2H).
ПРИМЕР 91: 2-((3R)-3-этил-5-метоксиизохроман-1-ил)-4,5-дигидро-1H-имидазол
Указанное в заголовке соединение получали из (R)-этил 5-бром-3-этилизохроман-1-карбоксилата (700 мг) способами получения E (с 2-ди-трет-бутилфосфино-3,4,5,6-тетраметил-2',4',6'-триизопропил-1,1'-бифенилом, кипячение с обратным холодильником) и A. Реакция конденсации C-O (E) давала свободную кислоту, которую затем метилировали перед конечной стадией реакции (способ получения A). Указанное в заголовке соединение получали концентрированием реакционной смеси (выход 169 мг).
1H ЯМР (CD3OD) δ м.д. 7,00-7,22 (м, 1H), 6,76-6,85 (м, 2H), 5,38 (с, 1H), 3,82 (с, 3H), 3,45-3,71 (м, 4H), 2,82 (дд, 1H),2,41 (дд, 1H), 1,53-1,81 (м, 2H), 0,95-1,12 (м, 3H).
ПРИМЕР 92: HCl соль энантиомера 1 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более быстро элюирующийся энантиомер (43 мг), получали из соединения примера 8 (500 мг) способом выделения L.
1H ЯМР (ДМСO-d6) δ м.д. 10,50 (шир.с, 2H), 7,51 (д, 1H), 7,35 (т, 1H), 7,25 (д, 1H), 5,86 (шир.с, 1H), 4,13-4,22 (м, 1H), 3,83-3,99 (м, 5H), 2,78-2,96 (м, 2H).
ПРИМЕР 93: гидрохлорид 2-((3R)-5-(дифторметокси)-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: (R)-1-(2-бромфенил)пропан-2-ол
К раствору 1-(2-бромфенил)пропан-2-она (30 г, 140 ммоль) в MeOH (100 мл) добавляли NaBH4 (6,09 г, 210 ммоль) при 0°C и перемешивали при комнатной температуре в течение 2 часов. Реакцию прекращали водой и концентрировали при пониженном давлении, получая неочищенное соединение. Указанное в заголовке соединение очищали способами выделения A, E и M (выход 28,0 г). Энантиомер 1 элюировался при tR=7,86 минут Chiralcel OD-H (4,6X250 мм) 5 мкм, гексан:2-PrOH:TFA (90:5:0,1), 1 мл/минута.
Стадия 2: (3R)-этил 5-бром-3-метилизохроман-1-карбоксилат
Указанное в заголовке соединение получали способом получения A1 и A2 из R-2-(2-бромфенила) и очищали способами выделения A и E (выход 2,0 г)
Стадия 3: (3R)-этил 5-гидрокси-3-метилизохроман-1-карбоксилат
К раствору (3R)-этил 5-бром-3-метилизохроман-1-карбоксилата (2,0 г, 5,95 ммоль) в диоксане (50 мл) и воде (10 мл) добавляли KOH (1,37 г, 35,7 ммоль), тетраметил-ди-трет-бутил XPhos (0,28 г, 0,06 ммоль) и Pd2(dba)3 (0,06 г, 0,06 ммоль) при комнатной температуре. Затем реакционную смесь дегазировали три раза N2 и перемешивали при 100°C в течение 16 часов. Реакцию прекращали MeOH и концентрировали при пониженном давлении, получая неочищенное соединение. Неочищенный остаток растворяли в EtOH (80 мл), добавляли H2SO4 (1,0 мл), затем перемешивали при 80°C в течение 5 часов. Растворитель удаляли упариванием, получая неочищенное соединение. Указанное в заголовке соединение очищали способами выделения A и E (выход 100 мг).
Стадия 4: (3R)-этил 5-(дифторметокси)-3-метилизохроман-1-карбоксилат
К раствору (3R)-этил 5-бром-3-метилизохроман-1-карбоксилата (0,25 г, 1,05 ммоль) в DMF (5,0 мл) добавляли K2CO3 (0,184 г, 1,35 ммоль). Затем газообразный дифтормонохлорметан продували при комнатной температуре в течение 2 часов и перемешивали при 50°C в течение 4 часов. Реакцию прекращали водой. Указанное в заголовке соединение очищали способами выделения A и E (выход 40 мг). Энантиомер 1 (т.е. (R)), элюировался при tr=4,71 минут Chiralcel OD-H (4,6X250 мм) 5 мкм, гексан:2-PrOH:TFA (90:5:0,1), 1 мл/минута.
Стадия 5: гидрохлорид 2-((3R)-5-(дифторметокси)-3-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из (3R)-этил 5-(дифторметокси)-3-метилизохроман-1-карбоксилата (250 мг), применяя методику способа получения A3 и способа получения соли A (выход 200 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,81 (с, 1,75H), 10,22 (с, 0,14H), 7,36 (т, 1H), 7,29 (т, 1H), 7,17-7,23 (м, 2H), 5,98 (с, 0,07H), 5,96 (с, 0,88H), 3,84-4,00 (м, 5H, overlaps with H2O signal), 2,81-2,92 (м, 1H), 2,57 (дд, 0,89H), 2,45 (дд, 0,09H), 1,36 (д, 2,65H), 1,33 (д, 0,29H).
ПРИМЕР 94: энантиомер 2-(5-бром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Стадия 1: энантиомер 1-(2-бромфенил)бутан-2-ола
К раствору 2-(2-бромфенил)ацетальдегида (15 г, 75,37 ммоль) в THF (100 мл) добавляли бромид пропилмагния (1,0M в THF) (15,13 г, 113,6 ммоль) при 0°C, затем перемешивали при комнатной температуре в течение 3 часов. Реакцию прекращали хлоридом аммония, затем указанное в заголовке соединение очищали способами выделения A, E и M (выход 7,0 г). Энантиомер элюировался при tr=13,6 минут Chiralcel OJH (4,6X250 мм) 5 мкм, гексан:этанол (98:02), 1 мл/минута.
Стадия 2: энантиомер 2-(-5-бром-3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из энантиомера этил 3-(2,2-дифторэтил)-5-метилизохроман-1-карбоксилата (500 мг) способом получения A и выделяли способом выделения E (выход 470 мг).
1H ЯМР (CD3OD) δ м.д. 7,44-7,58 (м, 1H), 7,24 (д, 1H), 7,00-7,16 (м, 1H), 5,39 (с, 1H), 3,49-3,73 (м, 5H), 2,82-2,90 (м, 1H), 2,42-2,64 (м, 1H), 1,54-1,82 (м, 2H), 0,92-1,12 (м, 3H).
ПРИМЕР 95: HCl соль энантиомера 1 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более быстро элюирующийся энантиомер (28 мг), получали из примера 2 (130 мг) способом выделения L.
1H ЯМР (ДМСO-d6) δ м.д. 10,44 (шир.с, 2H), 7,61-7,77 (м, 1H), 7,17-7,34 (м, 2H), 5,85 (шир.с, 1H), 4,08-4,22 (м, 1H), 3,81-4,03 (м, 5H), 2,70-2,93 (м, 2H).
ПРИМЕР 96: энантиомер 2 2-(3-этилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более медленно элюирующийся энантиомер (110 мг), получали из соединения примера 8 (500 мг) способом выделения L.
1H ЯМР (CD3OD) δ м.д. 7,26 (д, 2H), 7,06 (т, 1H), 5,76 (с, 1H), 4,14-4,25 (м, 1H), 3,89-4,02 (м, 5H), 2,79-3,02 (м, 2H), 2,68 (кв., 2H), 1,22 (т, 3H).
ПРИМЕР 97: HCl соль энантиомера 2 2-(5-метоксиoизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более медленно элюирующийся энантиомер (77 мг), получали из соединения примера 17 (160 мг) способом выделения L.
1H ЯМР (ДМСO-d6) δ м.д. 10,45 (шир.с, 2H), 7,27 (т, 1H), 6,99 (д, 1H), 6,83 (д, 1H), 5,81 (шир.с, 1H), 4,10-4,17 (м, 1H), 3,87 (шир.с, 4H), 3,82-3,86 (м, 1H), 3,81 (с, 3H), 2,65-2,80 (м, 2H).
ПРИМЕР 98: 2-(1-метил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазол
Смесь NaH (0,18 г), метил 1,3-дигидробензо[de]изохромен-1-карбоксилата (0,5 г) и DMF (2 мл), толуола (2 мл) и THF (6 мл) перемешивали при температуре бани со льдом. Добавляли йодметан (0,7 мл) и смесь перемешивали при температуре окружающей среды в течение 3 часов. Промежуточный метил 1-метил-1,3-дигидробензо[de]изохромен-1-карбоксилат (0,3 г) очищали способом выделения A и E (EtOAc/гептан) и указанное в заголовке соединение получали, применяя методику способа получения A стадия 3 и способа выделения E (EtOAc/DCM/триэтиламин 10/20/1) (выход 0,1 г).
1H ЯМР (CDCl3) δ м.д. 7,77 (тд, 2H), 7,41-7,53 (м, 3H), 7,21 (дд, 1H), 5,14 (кв., 2H), 4,92 (шир., 1H), 3,14-4,19 (шир., 4H), 1,90 (с, 3H).
ПРИМЕР 99: гидрохлорид 2-(5-(дифторметилизохроман-1-ил)-4,5-дигидро-1H-имидазол
Стадия 1: этил 5-(дифторметил)изохроман-1-карбоксилат
К раствору этил 5-формилизохроман-1-карбоксилата (200 мг) в дихлорметане (1 мл) добавляли трифторид диэтиламиносеры (275 мг) и раствор перемешивали при комнатной температуре в течение 22 часов. Растворители упаривали и неочищенный продукт очищали способом выделения E (EtOAc-гептан) (выход 153 мг).
1H ЯМР (CDCl3) δ м.д. 7,53 (д, 1H), 7,46 (д, 1H), 7,24-7,33 (м, 1H), 6,72 (т, 1H), 5,36 (с, 1H), 4,21-4,38 (м, 3H), 4,01-4,11 (м, 1H), 2,90-3,06 (м, 2H), 1,32 (т, 3H).
Стадия 2: гидрохлорид 2-(5-(дифторметил)изохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение получали из этил 5-(дифторметил)изохроман-1-карбоксилата (153 мг), применяя методику способа получения A3 и способа получения соли A (выход 87 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,51 (шир.с, 2H), 7,58 (т, 1H), 7,42-7,48 (м, 2H), 7,21 (т, 1H), 5,93 (с, 1H), 4,11-4,20 (м, 1H), 3,82-3,94 (м, 5H), 2,87-3,11 (м, 2H).
ПРИМЕР 100: HCl соль энантиомера 2 2-(5-хлоризохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более медленно элюирующийся энантиомер (40 мг), получали из соединения примера 8 (500 мг) способом выделения L.
1H ЯМР (ДМСO-d6) δ м.д. 10,50 (шир. с, 2H), 7,51 (д, 1H), 7,35 (т, 1H), 7,25 (д, 1H), 5,86 (шир.с, 1H), 4,13-4,22 (м, 1H), 3,83-3,99 (м, 5H), 2,78-2,96 (м, 2H).
ПРИМЕР 101: HCl соль энантиомера 2 2-(5-бромизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более медленно элюирующийся энантиомер, (22 мг) получали из соединения примера 2 (130 мг) способом выделения L.
1H ЯМР (ДМСO-d6)) δ м.д. 10,44 (шир.с, 2H), 7,61-7,77 (м, 1H), 7,17-7,34 (м, 2H), 5,85 (шир.с, 1H), 4,08-4,22 (м, 1H), 3,81-4,03 (м, 5H), 2,70-2,93 (м, 2H).
ПРИМЕР 102: гидрохлорид 2-(1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола
К раствору 1,3-дигидробензо[de]изохромен-1-ола (100 мг) в ацетонитриле (3 мл) при 0°C добавляли йодид Zn (II) (86 мг) и суспензию перемешивали при 0°C в течение 10 минут. Добавляли триметилсилицианид (672 мкл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли раствор насыщенного NaHCO3 (10 мл) и смесь экстрагировали этилацетатом (2×15 мл). Объединенные органические фазы промывали соляным раствором и сушили Na2SO4. Упаривание растворителей давало 110 мг 1,3-дигидробензо[de]изохромен-1-карбонитрила, который смешивали с моносолью п-толуолсульфокислоты и этилендиамина (157 мг) и смесь нагревали до 200°C. Через 3 часа реакционную смесь охлаждали до комнатной температуры и добавляли к смеси дихлорметан (10 мл) и водный NaHCO3 (1:1 насыщенный раствор/вода). Фазы разделяли и водную фазу экстрагировали дихлорметаном (5 мл). Объединенные органические фазы сушили Na2SO4 и концентрировали. Остаток после упаривания очищали способом выделения E (водн. NH3-MeOH-дихлорметан). Полученный 2-(1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазол превращали в указанное в заголовке соединение, применяя способ получения соли A.
1H ЯМР (ДМСO-d6) δ м.д. 10,56 (с, 2H), 7,99 (д, 1H), 7,91 (д, 1H), 7,53-7,62 (м, 2H), 7,41 (д, 2H), 6,25 (с, 1H), 5,22 (AB кв., 2H), 3,90-4,05 (м, 4H).
ПРИМЕР 103: энантиомер 2-(1-метил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более медленно элюирующийся энантиомер (10 мг), получали из соединения примера 98 (1,2 г) способом выделения H.
1H ЯМР (CD3OD) δ м.д. 7,81 (дд, 1H), 7,77 (д, 1H), 7,41-7,53 (м, 3H), 7,24 (дд, 1H), 5,14 (с, 2H), 4,76-4,84 (м, 4H, methanol), 1,85 (с, 3H).
ПРИМЕР 104: гидрохлорид 2-(3-метил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола
К раствору 3-метилбензо[de]изохромен-1(3H)-она (1,38 г) в дихлорметане (34 мл) при -78°C добавляли гидрид диизобутилалюминия (10,5 мл, 20% раствор в толуоле) в течение 25 минут. Через 2 часа добавляли 10% водный раствор лимонной кислоты (20 мл) и смесь перемешивали в течение 25 минут при комнатной температуре. К полученной в результате суспензии добавляли воду (20 мл) и дихлорметан (20 мл) и слои разделяли. Водную фазу экстрагировали этилацетатом (2×20 мл) и объединенные органические фазы промывали соляным раствором, сушили Na2SO4 и концентрировали. Остаток после упаривания, который затвердевал в холодильнике, очищали растиранием с гептаном. Полученный 3-метил-1,3-дигидробензо[de]изохромен-1-ол (0,92 г, смесь диастереомеров) растворяли в дихлорметане (16 мл) и охлаждали до 0°C. Добавляли триметилсилилцианид (1,71 мл) с последующим добавлением триэтилэфирата трифторида бора (1,14 мл). Через 1,5 часа добавляли насыщенный раствор NaHCO3 (10 мл) и смесь экстрагировали дихлорметаном. Объединенные органические фазы промывали соляным раствором, сушили (Na2SO4) и концентрировали. Очистка остатка после упаривания растиранием с гептаном давала 0,68 г 3-метил-1,3-дигидробензо[de]изохромен-1-карбонитрила (смесь диастереомеров).
3-Метил-1,3-дигидробензо[de]изохромен-1-карбонитрил (100 мг) смешивали с моносолью этилендиамина и п-толуолсульфокислоты (111 мг) и смесь нагревали до 200°C. Через 3 часа реакционную смесь охлаждали до комнатной температуры. Очистка смеси колоночной хроматографией с нейтральным оксидом алюминия (этилацетат - гептан) давала 11 мг 2-(3-метил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола, который превращали в указанное в заголовке соединение, применяя способ получения соли A (выход 10 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,76 (с, 1H), 10,25 (с, 0,5H), 7,88-8,05 (м, 2H), 7,52-7,66 (м, 2H), 7,30-7,52 (м, 2H), 6,30 (с, 0,3H), 6,27 (с, 0,6H), 5,35 (кв., 0,6H), 3,92 (кв., 0,3H), 3,86-4,08 (м, 4H), 1,74 (д, 2H), 1,66 (д, 1H).
ПРИМЕР 105: гидрохлорид 2-(3-этил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазола
К раствору 3-этилбензо[de]изохромен-1(3H)-она (0,55 г) в дихлорметане (7 мл) при -78°C добавляли диизобутилалюмогидрид (2,19 мл, 20% раствор в толуоле). Через 3 часа добавляли 10% водный раствор лимонной кислоты (10 мл), и смесь перемешивали в течение 15 минут при комнатной температуре. Фазы разделяли, и водную фазу экстрагировали этилацетатом (2×10 мл). Объединенные органические фазы промывали соляным раствором, сушили Na2SO4 и концентрировали. Очистка остатка после упаривания колоночной хроматографией (этилацетат - гептан) давала 3-этил-1,3-дигидробензо[de]изохромен-1-ол (0,22 г, смесь диастереомеров), который растворяли в дихлорметане (5 мл). Раствор охлаждали до 0°C и добавляли триметилсилилцианид (0,39 мл) с последующим добавлением триэтилэфирата трифторида бора (0,26 мл). Через 2,5 часа добавляли насыщенный раствор NaHCO3 (10 мл) и смесь экстрагировали дихлорметаном. Объединенные органические фазы промывали соляным раствором, сушили (Na2SО4) и концентрировали. Остаток после упаривания, содержащий смесь диастереомеров 3-этил-1,3-дигидробензо[de]изохромен-1-карбонитрила (170 мг), смешивали с диоксаном (2 мл) и добавляли 10% водный раствор NaOH (1,6 мл) и смесь кипятили с обратным холодильником. Через 13 часов реакционную смесь охлаждали до комнатной температуры и добавляли воду (10 мл) и дихлорметан (10 мл). Водную фазу подкисляли 4 M HCl и экстрагировали EtOAc (3×10 мл). Объединенные органические фазы промывали соляным раствором, сушили Na2SO4 и концентрировали. Остаток после упаривания, содержащий 3-этил-1,3-дигидробензо[de]изохромен-1-карбоновую кислоту (80 мг), превращали в указанное в заголовке соединение, применяя способы получения A2 и A3, вместе со способом выделения E в A3. Полученный 2-(3-этил-1,3-дигидробензо[de]изохромен-1-ил)-4,5-дигидро-1H-имидазол превращали в указанное в заголовке соединение, применяя способ получения соли A (выход 5,7 мг).
1H ЯМР (ДМСO-d6) δ м.д. 10,73 (шир.с, 0,9H), 10,42 (шир.с, 0,7H), 7,99 (дд, 1H), 7,92 (дд, 1H), 7,53-7,63 (м, 2H), 7,31-7,50 (м, 2H), 6,28 (с, 0,4H), 6,24 (с, 0,5H), 5,18-5,23 (м, 0,5H), 5,12 (т, 0,4H), 3,90-4,08 (м, 4H), 2,31-2,43 (м, 0,5H), 1,92-2,24 (м, 1,4H), 1,01-1,12 (м, 3H).
ПРИМЕР 106: энантиомер 2-(5-бром-1-метилизохроман-1-ил)-4,5-дигидро-1H-имидазола
Указанное в заголовке соединение, более быстро элюирующийся энантиомер (160 мг), получали из соединения примера 11 (400 мг) способом выделения H.
1H ЯМР (ДМСO-d6) δ м.д. 7,49 (дд, 1H), 7,31 (дд, 1H), 7,11 (дд, 1H), 6,24 (с, H), 3,79-3,98 (м, 2H), 3,41-3,71 (шир.с, 2H), 3,10-3,73 (шир.с, 2H), 1,62 (с, 3H).
Как уже упоминалось в настоящем описании выше, соединения формулы I обладают интересными фармакологическими свойствами, а именно: они обладают агонистическими активностями относительно адренергических альфа2 рецепторов, особенно относительно рецептора альфа2A. Указанная активность продемонстрирована фармакологическим испытанием, приведенным ниже. Иллюстративные примеры настоящего изобретения скринировали на активности относительно адренергических альфа2 рецепторов на основе способов, описанных в литературе (Lehtimaki et al., Eur. J. Pharmacol. 2008, 599, 65-71). Результаты показаны в таблице 1. Метаболическую стабильность измеряли с замороженными гепатоцитами согласно литературному способу (Di et al., Int. J. Pharmaceutics, 2006, 317, 54-60).
Соединения формулы I обладают агонистическими активностями относительно адренергических альфа2 рецепторов, особенно относительно альфа2A рецептора. Таким образом, настоящее изобретение относится к соединениям для применения в качестве лекарственного средства. Настоящее изобретение также относится к соединениям для применения в лечении расстройства, состояния или заболевания, для которого показано, что альфа2 агонист, например альфа2A агонист, является полезным. Кроме того, настоящее изобретение относится к способу лечения расстройства, состояния или заболевания, для которого показано, что альфа2 агонист, например альфа2A агонист, является полезным. В указанном способе эффективное количество, по меньшей мере, одного соединения формулы I вводят млекопитающему, такому как человек, нуждающемуся в данном лечении. Настоящее изобретение также относится к применению соединений формулы I в получении лекарственного средства для лечения расстройства, состояния или заболевания, для которого показано, что альфа2 агонист, например альфа2A агонист, является полезным.
В одном варианте осуществления настоящего изобретения приведенное выше расстройство, состояние или заболевание, для которого показано, что альфа2 агонист, например альфа2A агонист, является полезным, представляет собой бред, гиперактивный бред, бессонницу, синдром дефицита внимания с гиперактивностью, абстиненция при отмене приема бензодиазепинов, или алкоголя, или опиоидов, или табака, преждевременную эякуляцию, гипертензию, тахикардию, синдром беспокойных ног, мышечную спастичность, приступообразное ощущение жара, тревогу, посттравматическое стрессовое расстройство, боль, хронический тазовый болевой синдром и приступ неконтролируемой боли при раке, или заболевание, при котором требуется успокоение или обезболивание; например гиперактивный бред или бессонница.
Настоящее изобретение также относится к соединениям формулы I для применения в качестве успокаивающего или обезболивающего агента.
Соединения настоящего изобретения можно вводить, например, энтерально, местно или парентерально посредством любой фармацевтической композиции для указанного введения, и она содержит, по меньшей мере, одно активное соединение формулы I в фармацевтически приемлемых и эффективных количествах вместе с фармацевтически приемлемыми разбавителями, носителями и/или эксципиентами, известными в данной области техники. Получение данных фармацевтических композиций является известным в данной области техники.
Терапевтическая доза, которую будут вводить нуждающемуся в лечении субъекту, будет изменяться в зависимости от соединения, которое вводят, вида, возраста и пола субъекта, которого лечат, конкретного заболевания, которое лечат, а также пути и способа введения, и ее легко определит специалист в данной области техники. Соответственно, типичная доза для перорального введения составляет от 10 нг/кг до 10 мг/кг в день и для парентерального введения от 1 нг/кг до 10 мг/кг для взрослого млекопитающего.
Соединения настоящего изобретения вводят субъекту в чистом виде или в комбинации с одним или более другими активными ингредиентами, каждый в своей композиции или некоторые или все из активных ингредиентов смешивают в одной композиции и/или с подходящими фармацевтическими эксципиентами. Подходящие фармацевтические эксципиенты включают общепринятые эксципиенты и вспомогательные вещества для получения композиции, такие как наполнители, связующие, дезинтегранты, лубриканты, растворители, агенты, образующие гель, эмульгаторы, стабилизаторы, красители и/или консерванты.
Соединения настоящего изобретения формулируют в виде дозированных форм, применяя общеизвестные фармацевтические способы получения. Дозированные формы могут представлять собой, например, таблетки, капсулы, гранулы, суппозитории, эмульсии, суспензии или растворы. В зависимости от пути введения и галеновой формы количество активного ингредиента в композиции может обычно изменяться от 0,01% до 100% по весу.
Специалисту в данной области техники ясно, что варианты осуществления, описанные в настоящем изобретении, можно модифицировать, не выходя за пределы идеи изобретения. Специалисту в данной области техники также ясно, что настоящее изобретение не ограничивается конкретными описанными вариантами осуществления, но предполагается, что оно также включает модификации вариантов осуществления, которые включены в объем настоящего изобретения.
Реферат
Изобретение относится к области органической химии, а именно к соединению формулы (I) или к его фармацевтически приемлемой соли, где X представляет собой O или S; Rпредставляет собой гидрокси, галоген, (C-C)алкил, галоген(C-C)алкил, (C-C)алкенил, (C-C)алкинил, цикло(C-C)алкил, (C-C)алкокси, галоген(C-C)алкокси, гидрокси(C-C)алкил, циано, (R)N-(C=O)-, (C-C)алкил-S-, или фуранил; Rпредставляет собой H или (C-C)алкил; Rпредставляет собой H, (C-C)алкил, галоген(C-C)алкил или (C-C)алкокси(C-C)алкил; Rпредставляет собой H или (C-C)алкил; Rпредставляет собой H, гидрокси, галоген, (C-C)алкил или (C-C)алкокси; Rпредставляет собой H; или Rи Rобразуют вместе с атомами углерода кольца, к которым они присоединены, конденсированное 6- или 7-членное насыщенное или ненасыщенное карбоциклическое кольцо. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I). Технический результат: получены новые производные изохромана или изотиохромана, полезные в качестве агонистов альфа2 адренорецепторов. 2 н. и 8 з.п. ф-лы, 1 табл., 106 пр.
Формула
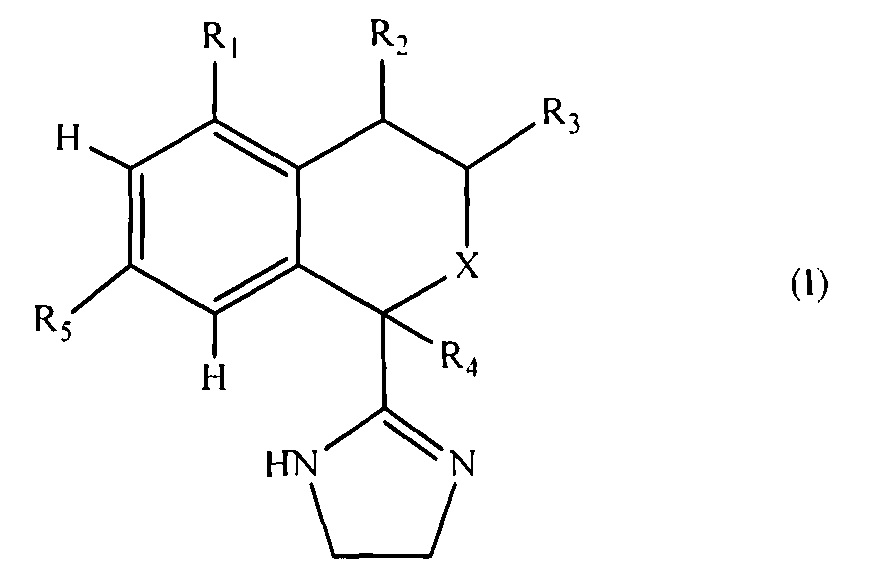
Документы, цитированные в отчёте о поиске
Замещенные производные имидазола, способ введения активного соединения и способ лечения на основе этих соединений
Комментарии