Замещенные производные имидазола, способ введения активного соединения и способ лечения на основе этих соединений - RU2235092C2
Код документа: RU2235092C2
Описание
1. Область изобретения
Настоящее изобретение относится к способу лечения глаукомы или повышенного внутриглазного давления и других заболеваний, при котором сердечно-сосудистые или седативные побочные эффекты существенно снижаются, путем введения млекопитающим, в том числе людям, соединений, которые являются селективными агонистами адренергических рецепторов только подтипа α2В или подтипов α2В и α2С и которые не имеют значительной активности по отношению к рецепторам подтипа α2А. Настоящее изобретение также относится к новым соединениям и фармацевтическим композициям, адаптированным для введения указанных соединений млекопитающим, в том числе людям.
2. Краткое описание уровня техники
Соединения, которые обладают адренергической активностью, широко известны в данной области и описаны в многочисленных патентах США и других государств и научных изданиях. В данной области техники общеизвестно и общепризнанно, что адренергическая активность полезна для лечения млекопитающих, в том числе людей, в целях излечивания или облегчения симптомов и состояний ряда заболеваний и состояний. Другими словами, в данной области общепризнанно, что фармацевтические композиции, имеющие в своем составе адренергическое соединение или соединения в качестве активных ингредиентов, полезны для лечения глаукомы, хронической боли, назальной гиперемии, высокого кровяного давления, застойной сердечной недостаточности и для анестезирования.
В данной области два основных семейства адренергических рецепторов называются альфа-адренергическими рецепторами и бета-адренергическими рецепторами, и известно, что каждое из этих двух семейств имеет подтипы, которые обозначают буквами алфавита, например α2А, α2В (Bylund et al., Pharmacol. Rev. 46, pp. 121-136 (1994)).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению было обнаружено, что адренергические соединения, которые действуют селективно и предпочтительно еще специфично как агонисты рецепторов подтипов α2В или α2В/α2С (именуемых здесь ниже как α2В или α2В/α2С) в предпочтение рецепторам подтипа α2А, обладают требуемыми терапевтическими свойствами, связанными с адренергией, но не имеют нежелательных побочных эффектов, таких как изменения кровяного давления или седативное действие. В целях настоящего изобретения соединение определено как специфичный или по меньшей мере селективный агонист рецепторов подтипа(ов) α2В или α2В/α2С, если это соединение по меньшей мере примерно в десять раз сильнее как агонист по отношению либо к α2В и α2С, либо к обоим подтипам рецепторов, чем по отношению к рецепторам подтипа α2А, или если различие в эффективности этого соединения в отношении α2В- и α2В/2С-рецептора относительно α2А-рецептора превышает 0,3, а его эффективность в отношении α2А-рецептора составляет ≤0,4.
Соответственно, настоящее изобретение относится к способам лечения животных, относящихся к млекопитающим, в том числе людей, фармацевтической композицией, содержащей в качестве активного ингредиента одно или более чем одно соединение, представляющее собой специфичный или селективный α2В- или α2В/2С-адренергический агонист, для лечения многих заболеваний и состояний, против которых используют альфа-адренергические соединения, включая, без ограничений, глаукому, снижение повышенного внутриглазного давления, хроническую боль, диарею и назальную гиперемию. Кроме того, соединения по этому изобретению пригодны для лечения мышечной спастичности, в том числе гиперактивного мочеиспускания, диареи, диуреза, синдромов отмены, боли, в том числе невропатической боли, нейродегенеративных заболеваний, в том числе глазной невропатии, ишемии спинного мозга и инсульта, дефицита памяти и познавательной способности, дефицита внимания, психозов, в том числе маниакальных расстройств, тревожности, депрессии, гипертензии, застойной сердечной недостаточности, ишемии сердца и назальной гиперемии.
Настоящее изобретение также относится к фармацевтическим композициям, используемым в вышеуказанных способах лечения.
В частности, настоящее изобретение охватывает способы лечения заболеваний и состояний, при которых для лечения эффективны адренергические соединения, но их применение ограничено из-за их общеизвестных побочных эффектов.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения, которые используют в фармацевтических композициях и способах лечения по настоящему изобретению, являются селективными или специфичными агонистами адренергических рецепторов подтипов α2В или α2В/2С в предпочтение рецепторам подтипа α2А. Согласно настоящему изобретению соединение считается селективным α2В- или α2В/2С-агонистом, если различие в эффективности этого соединения в качестве агониста α2В- или α2В/2С-рецепторов и в качестве агониста α2А-рецепторов превышает 0,3, а его эффективность по отношению к рецепторам подтипа α 2А составляет ≤0,4 и/или он по меньшей мере в 10 раз сильнее действует. Соединения, используемые согласно настоящему изобретению, предпочтительно являются специфичными агонистами рецепторов подтипов α2В или α2В/2С. Конкретно, в этом отношении специфичный агонист определяют в том смысле, что специфичный α-адренергический агонист не действует как агонист рецепторов подтипа α2А до какой-либо измеримой или биологически значимой степени.
Было обнаружено множество агентов, которые функционально являются селективными по отношению к α 2В- или α2В/2С-подтипам указанных адренергических рецепторов. Эта преимущественная активность может быть определена посредством ряда функциональных анализов, таких как продукция циклического АМФ [Shimizu et al., J. Neurochem. 16, pp. 1609-1619 (1969)], ТСАР (технология селекции и амплификации рецепторов) [Messier et al., Pharmacol. Toxicol. 76, pp. 308-311 (1995)] и цитосенсорный микрофизиометр [Neve et al., J. Biol. Chem. 267, pp. 25748-25753 (1992)], с использованием клеток, которые естественным путем экспрессируют индивидуальные подтипы или в которые введен один из этих подтипов. Используемые клетки или рекомбинантные рецепторы должны быть человеческими или из видов, для которых было показано, что у них аналогичная фармакология. В описанном ниже исследовании использовали ТСАР-анализ на клетках, которые на короткий срок трансфецировали человеческим α2А-рецептором (ген с10), крысиным α2В-рецептором (ген RNG) и человеческим α 2С-рецептором (ген с4). Было показано, что фармакология крысиного α2В-рецептора соответствует человеческому α2В-рецептору [Bylund et al., Pharmacol. Rev. 46, pp. 127-129 (1994)].
При лечении в частности глаукомы можно использовать местное введение. На глаз при глаукоме и на кожу при лечении по другим показаниям можно наносить любой обычный препарат для местного применения, такой как раствор, суспензия, гель, мазь или бальзам и тому подобное. Приготовление таких препаратов для местного применения хорошо описано в уровне техники фармацевтических препаратов, например в Remington's Pharmaceutical Science, Edition 17, Mack Publishing Company, Easton, Pennsylvania.
Если лекарственное средство требуется вводить системно, оно может быть изготовлено в виде порошка, пилюли, таблетки и тому подобного или в виде сиропа или эликсира для перорального введения. Для внутривенного, внутрибрюшинного, внутриоболочкового или эпидурального введения соединение готовят в виде раствора или суспензии, которые можно вводить инъекцией. В некоторых случаях может быть полезным включать эти соединения в состав суппозитория или препарата пролонгированного высвобождения, в том числе в форме дермального пластыря, для размещения на коже или под кожей или для внутримышечной инъекции.
Лечение глаукомы или по любым другим показаниям, относительно которых известно или обнаружено, что они поддаются лечению адренергическими соединениями, осуществляют введением терапевтически эффективной дозы одного или более чем одного соединения по настоящему изобретению. Терапевтической концентрацией является такая концентрация, которая осуществляет редукцию конкретного состояния или замедляет его распространение. В некоторых случаях лекарственное средство потенциально можно использовать в целях профилактики для предупреждения возникновения какого-либо конкретного состояния. Назначаемая терапевтическая концентрация будет изменяться от состояния к состоянию и в определенных случаях может варьировать в зависимости от тяжести состояния, подлежащего лечению, и податливости пациента лечению. Соответственно, назначаемую терапевтическую концентрацию лучше всего определять на месте и на тот момент времени рутинным экспериментированием. Однако предполагается, что при лечении, например, глаукомы препарат, содержащий между 0,001 и 5% мас., предпочтительно примерно от 0,01 до 3% мас., обычно будет представлять собой терапевтически эффективную концентрацию. При системном ведении в большинстве случаев терапевтический результат будет давать количество между 0,001 и 50 мг на 1 кг веса тела в день, предпочтительно между 0,001 и 10 мг на 1 кг массы тела в день и наиболее предпочтительно примерно от 0,01 до 1,0 мг на 1 кг массы тела в день.
Поскольку соединения, представляющие собой α2В- и α2В/С-специфичные селективные агонисты, не имеют существенных побочных эффектов, обусловленных действием на α2А-рецепторы, лечение заболеваний или состояний такими соединениями по настоящему изобретению создает преимущества, особенно когда лечат человека с сердечно-сосудистыми проблемами.
Общие структуры примеров соединений, представляющих собой специфичные α2В- и α2С-агонисты или селективные α2В- и α2В/С-агонисты, которые используют в фармацевтических композициях и способах лечения по настоящему изобретению, представлены ниже общими формулами.
В одном из аспектов данного изобретения соединение, имеющее активность селективного агониста по отношению к адренергическим рецепторам подтипа(ов) α2В или α2В/С по сравнению с адренергическими рецепторами подтипа α2А, представлено общей формулой
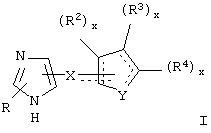
где пунктирные линии представляют собой возможные двойные связи; R представляет собой Н или низший алкил; Х представляет собой S или C(H)R1, где R1 представляет собой Н или низший алкил, либо R1 отсутствует, когда Х представляет собой S или когда связь между Х и кольцом, которое представляет собой
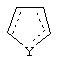
является двойной связью; Y представляет собой О, N, S, (CR)y, где у является целым числом от 1 до 3, -СН=СН- или -Y1CH2-, где Y1 представляет собой О, N или S; х является целым числом от 1 до 2, причем х равно 1, когда R2, R3 или R4 связан с ненасыщенным атомом углерода, и х равно 2, когда R2, R3 или R4 связан с насыщенным атомом углерода; R2 представляет собой Н, низший алкил, галоген, гидрокси или низший алкокси либо, будучи присоединенным к насыщенному атому углерода, R2 может представлять собой оксо; R3 и R4 каждый представляет собой Н, низший алкил, гидрокси, низший алкокси или фенил или вместе представляют собой -(C(R2)x)z-, -Y1(C(R2)x)z’-, -Y1(C(R2)x)yY1-, -(C(R2)x)-Y1-(C(R2)x)-, -(C(R2)x)-Y1-(C(R2)x)-(C(R2)x)- и -Y1-(C(R2)x)-Y1-(C(R2)x)-, где z является целым числом от 3 до 5, z' является целым числом от 2 до 4, а х и у такие, как определено выше, и, далее, каждый конец каждой из этих двухвалентных группировок может быть присоединен либо по R3, либо по R4 с образованием конденсированной кольцевой структуры
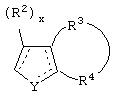
и образованные таким образом кольца могут быть полностью ненасыщенными, частично ненасыщенными или полностью насыщенными при условии, что кольцевой углерод имеет не более 4 валентностей, азот имеет не более трех, а О и S имеют не более двух.
В другом аспекте изобретения вышеуказанное соединение представлено формулой
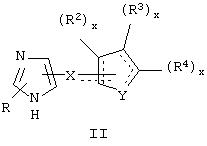
где X может представлять собой C(H)R1, a R1 представляет собой Н.
В указанном соединении формулы II R2 может представлять собой Н, а
может представлять собой фуранильный радикал.
В таких фуранильных производных формулы II R3 и R4 вместе могут представлять собой (СН)4, или R3 может представлять собой Н, а R4 может быть трет-бутилом, или R3 и R4 могут представлять собой Н, или R3 может представлять собой Н, а R4 может быть метилом или этилом.
Альтернативно в соединении формулы I R1 может быть метилом, а
может представлять собой фуранильный радикал.
Альтернативно в указанных соединениях формулы II R2 может представлять собой Н, а
может представлять собой тиенильный радикал.
В таких тиенильных производных формулы II R3 и R4 вместе могут представлять собой (CH2)4, или R3 может быть фенилом, а R4 может представлять собой Н, или R3 и R4 могут представлять собой (СН2)3S, или R3 и R4 могут представлять собой Н, или R3 и R4 вместе могут представлять собой (СН)4, или R3 может представлять собой Н, а R4 может быть метилом, или R3 может представлять собой бромо, а R4 может представлять собой Н, или R3 может быть водородом, а R4 может представлять собой хлоро, или R3 может быть метилом, а R4 может быть водородом.
Альтернативно в соединениях формулы II
может представлять собой циклогексильный радикал.
В таких циклогексильных производных формулы II R2 может быть водородом, а R3 и R4 могут вместе представлять собой (СН)4, или R2 может представлять собой оксо, а R3 и R4 могут вместе представлять собой (СН)4, или R2 может быть водородом или оксо, а R3 и R4 могут вместе представлять собой (СН)2S, или R2 может быть водородом, а R3 и R4 могут вместе представлять собой (СН2)4, образуя октагидронафталин, или R2 может представлять собой оксо, а R3 и R4 могут вместе представлять собой (СН2)4, R2 может представлять собой оксо, а R3 и R4 могут вместе представлять собой (СН2)С(СН3)(СН), или R2 может быть водородом, а R3 и R4 могут вместе представлять собой S(CH2)2, или R2, R3 и R4 могут представлять собой Н, или R2 может представлять собой оксо, а R3 и R4 могут вместе представлять собой (СН2 )С(ОСН3)СН, или R3 и R4 могут вместе представлять собой -Y1-C(R2)x-C(R2)x-Y1-, где Y1 представляет собой N, образуя тетрагидрохиноксалин, где R2 может быть водородом или оксо.
Альтернативно в соединениях формулы II
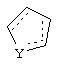
может представлять собой тетрагидрохинолиновый радикал, где R3 и R4 вместе представляют собой -Y1-C(R2)x-C(R2)x-C(R2)x-, где Y1 представляет собой N. В таких тетрагидрохинолиновых производных (R2 )x может быть водородом или оксо или может представлять собой тетрагидроизохинолиновый радикал, где R3 и R4 вместе представляют собой -C(R2)x -Y1-C(R2)x-C(R2)x-, где Y1 представляет собой N, a (R2)x может быть водородом или оксо.
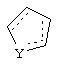
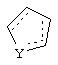
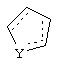
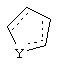
Альтернативно в соединениях формулы II
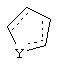
может представлять собой циклопентильный радикал.
В таких циклопентильных производных формулы II R2 может представлять собой Н, а R3 и R4 могут вместе представлять собой (СН)4, или R2может представлять собой оксо, а R3 и R4 могут вместе представлять собой (СН)4, или R2 может быть водородом, а R3 и R4 могут вместе представлять собой (CH2)3.
В еще одном аспекте изобретения Y представляет собой (СН2)3, Х может представлять собой СН, а R2 может представлять собой оксо, или Х может представлять собой СН2, а R2 может представлять собой Н. Или же R3 и R4 вместе могут представлять собой (СН)4, Y может представлять собой CH2C(CR)2, где R1 является водородом, или Y может представлять собой -СН2С(Ме)-, а R2 может быть водородом или оксо.
Наконец, в соединениях формулы II
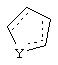
может представлять собой фенильный радикал.
В таких фенильных производных формулы I, Х может представлять собой СН2, R может представлять собой Н или СН3, R2, R3 и R4 могут представлять собой Н, или R3 и R4 вместе представляют собой O(CR2)2O с образованием 1,4-бензодиоксанового производного, или же Х может представлять собой S, а R2, R3 и R4 могут представлять собой Н.
В еще одном аспекте изобретения указанное соединение имеет формулу
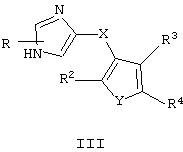
где Y представляет собой S или О.
В таком соединении формулы III X может представлять собой C(H)R1, R, R1, R2, R3 и R4 могут представлять собой Н, а Y может представлять собой О или S.
В еще одном аспекте изобретения указанное соединение имеет формулу

и R3 и R4 вместе представляют собой (СН)4.
В таких соединениях формулы IV Y1 может представлять собой О, R2может представлять собой оксо, а Х представляет собой СН или СН2, или один из R2 представляет собой гидрокси, а другой может представлять собой Н, или R2 может представлять собой Н.
В таких соединениях формулы IV Y1 может представлять собой S, X может представлять собой СН2, а R2 может представлять собой оксо, или R2может представлять собой Н, Х может представлять собой СН, а R2 может представлять собой оксо.
В еще одном аспекте изобретения соединение, обладающее селективной активностью по отношению к адренергическим рецепторам подтипа(ов) α2В или α2В/С по сравнению с адренергическими рецепторами подтипа α2А, представлено формулой
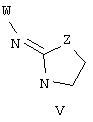
альтернативно W является бициклическим радикалом, выбранным из группы, состоящей из
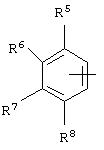
где R5, R6, R7 и R8 выбраны из группы, состоящей из Н и низшего алкила, при условии, что по меньшей мере один из R5 и R6 или R6 и R7 представляет собой OC(R9)C(R9)N(R) с образованием конденсированного кольца с
где R9 представляет собой Н, низший алкил или оксо,
и
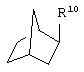
где R10 представляет собой Н, низший алкил, фенил или фенил, замещенный низшим алкилом, a Z представляет собой О или NH. Соединения, где W является норборнилом, раскрыты и заявлены в находящейся на совместном рассмотрении заявке 09/003902, поданной 7 января 1998, которая во всей полноте включена сюда ссылкой.
В одном из аспектов изобретения Z может представлять собой О, W может представлять собой
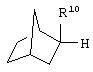
a R10 может быть выбран из группы, состоящей из Н, фенила и о-метилфенила, например R10 может быть о-метилфенилом.
В еще одном аспекте изобретения W может представлять собой
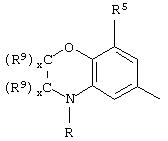
где Z может представлять собой NR, R может быть метилом или водородом, один из (R9)x может представлять собой Н, а R5 может представлять собой Н.
Альтернативно W может представлять собой
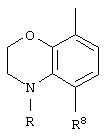
где R может представлять собой Н, а R8 может быть метилом.
Данное изобретение дополнительно иллюстрируется следующими примерами (включая общие схемы синтеза), которые являются иллюстрациями разных аспектов изобретения и не предназначены для того, чтобы ограничивать объем изобретения, как он определен прилагаемой формулой изобретения.
Пример А
Синтез 1-диметилсульфамоил-2-трет-бутилдиметилсилил-5-имидазолкарбоксальдегида:
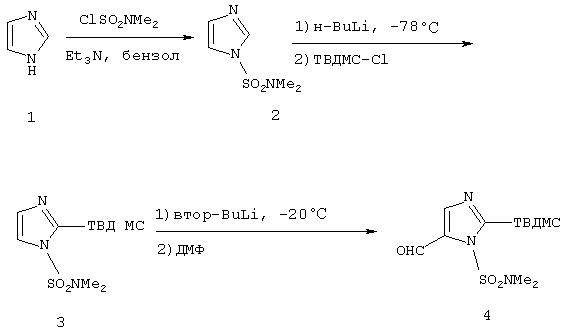
Процедура
К 320 мл бензола добавляли имидазол (1) (20,0 г, 0,29 моль), триэтиламин (41,0 мл, 0,29 моль) и N,N-диметилсульфамоилхлорид (31,6 мл, 0,29 моль). Реакционную смесь перемешивали в течение 48 часов при комнатной температуре (к.т.), а затем фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Вакуумная дистилляция неочищенного продукта (≈ 0,5 мм рт.ст., 115-118°С) давала 38,7 г (76%) прозрачного и бесцветного масла. При охлаждении этот продукт затвердевает с образованием белых кристаллов (2). 1-(Диметилсульфамоил)имидазол (2) (18,8 г, 0,11 моль) добавляли к 430 мл тетрагидрофурана (ТГФ). Этот раствор охлаждали до -78°С. В реакционную колбу по каплям добавляли раствор н-бутиллития (н-BuLi) в гексане (1,6 М, 70,9 мл, 0,11 моль). После окончания добавления реакционную смесь перемешивали в течение 1 часа при -78°С. В реакционную смесь через канюлю добавляли трет-бутилдиметилсилилхлорид (17,8 г, 0,12 моль) в 50 мл ТГФ. После того как добавление заканчивали, реакционную смесь медленно нагревали до к.т., а затем перемешивали в течение 24 часов. Реакционную смесь разбавляли водой и отделяли органический слой. Органическую фазу промывали рассолом, а затем сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Колоночная хроматография (20% этилацетата/гексан в качестве элюента) давала светло-желтое твердое вещество. Перекристаллизация из пентана давала 30 г (94%) белых кристаллов (3).
1-Диметилсульфамоил-2-трет-бутилдиметилсилилимидазол (3) (5,0 г, 17,3 ммоль) добавляли к 100 мл ТГФ. Этот раствор охлаждали до -20°С. В реакционную колбу по каплям добавляли раствор вторичного бутиллития (втop-BuLi) в гексане (1,3 М; 14,6 мл; 19 ммоль). После этого реакционную смесь перемешивали в течение 1 часа при -20°С. К реакционной смеси добавляли 8 мл диметилформамида (ДМФ), а затем перемешивали ее при к.т. в течение 3,5 часов. Реакционную смесь разбавляли водой и органический слой отделяли. Органическую фазу промывали рассолом, а затем сушили над сульфатом натрия. Эту смесь фильтровали и фильтрат концентрировали при пониженном давлении. Колоночная хроматография (20% этилацетата/гексан) давала светло-желтое масло. При охлаждении этот продукт затвердевает с образованием желтых кристаллов 1-диметилсульфамоил-2-трет-бутилдиметилсилил-5-имидазолкарбоксальдегида (4).
Пример А-0
Методика получения 3-(1Н-имидазол-4-илметил)-1-метил-1Н-индола фумарата А
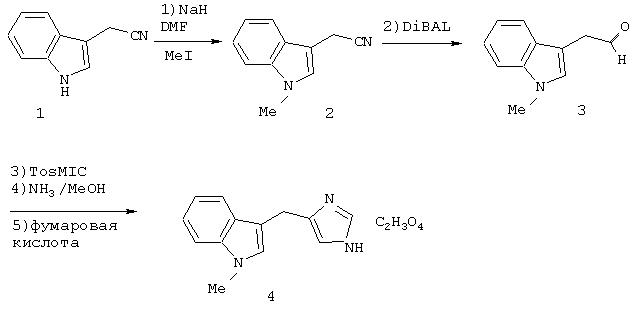
Раствор имидазола (1) (1,08 г, 6,9 ммоль) в DMF (диметилформамид) (5 мл) при 20°С добавили к суспензии NaH (370 мг, 9.25 ммоль) в DMF (5 мл). Через 30 мин смесь охлаждали до 0°С. Добавили раствор MeI (5,2 мл, 2 М в трет-бутилметиловом эфире) и раствор оставили реагировать при комнатной температуре в течение 18 часов. Добавили воду (10 мл) и ЕtOАс (15 мл). Органический слой отделили, высушили над МgSO4, отфильтровали и выпарили досуха. Остаток очищали хроматографией на SiO2 с помощью ЕtOАсНх с получением 0,65 г (55%) метилированного индола (2).
(1-Метил-1Н-индол-3-ил)-ацетонитрил (2) в СН2Сl2 (10 мл) восстанавливали DiBAL (диизобутилалюминийгидрид) (7 мл, 1 М в циклогексане) при -78°С в течение 1 часа. Смесь добавили к раствору сегнетовой соли и перемешивали в течение 0,5 часа. Органический слой отделили, высушили над MgSO4, отфильтровали и выпарили досуха. Сырой альдегид (3) использовали на следующей стадии без дополнительной очистки.
Альдегид (3) подвергали "протоколу Бучи" ("Buchi protocol", см. Horne, D.A.; Yakushijin К.; Buchi G. Heterocycles, 1994, 39, 139). Раствор альдегида 3 (1 ммоль) в EtOH (5 мл) обработали тозилметилизоцианидом (TosMIC) (0,9 ммоль) и NaCN (~2 мг, кат.). Эту смесь оставляли перемешиваться при комнатной температуре в течение 20 мин. Растворитель удалили in vacuo и остаток растворили в ~ 7 М NН3 в МеОН и перенесли в запечатываемую пробирку. Эту смесь нагревали при 100°С в течение 15 часов. Смесь сконцентрировали и очищали хроматографией на SiO2 с помощью 5% МеОН (нас. масс/NН3) CH2Cl2. Соединение имидазола дополнительно очищали в виде соли фумарата 4 путем рекристаллизации из МеОН с помощью 20% Et2O/Hx 3-(1Н-имидазол-4-илметил)-1-метил-1Н-индола фумарата А.
1H ЯМР (300 МГц, DMSO-d6w/TMS) δ: 7.81 (s, 1H), 7.79 (s, 1H), 7.56 (s, 1H), 7.40-7.39 (m, 2H), 7.20-7.00 (m, 2H), 6.47 (s, 2H), 3.76 (s, 3H), 3.14 (s, 2H).
Пример А-1
Применение (1Н-индол-3-ил)ацетонитрила (имеется в продаже от Aldrich) в способе А (стадии 2-5) для получения 3-(1Н-имидазол-4-илметил)-1Н-индола фумарата.
1H ЯМР (500 МГц, DMSO-d6w/TMS) δ: 7.85 (s, 1H), 7.46 (d, J=7.5 Гц, 1Н), 7.33 (d, J=7.5 Гц, 1H), 7.13 (s, 1H), 7.05 (t, J=7.3 Гц, 1H). 6.86 (s, 1H), 6.61 (s, 2H), 3.99 (s, 2H).
Пример А-2
Применение (1-метил-1Н-пиррол-2 ил)уксусной кислоты метилового эфира (от Aldrich) в способе А (стадии 2-5) для получения 4-(1-метил-1Н-пиррол-2-илметил)-1Н-имидазола фумарата.
1Н ЯМР (300 МГц, MeOD-d4) δ: 8.44 (s, 1H), 7.07 (s, 1H), 6.72 (s, 2H), 6.61 (s, 1H), 5.97-5.86 (m, 2H), 4.02 (s, 2H), 3.53 (s, 3H).
Процедура
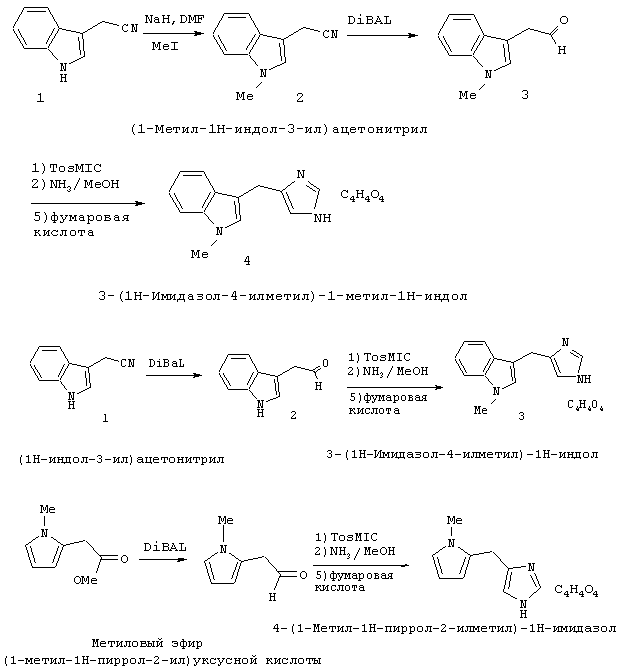
К 8,5 мл 40% раствора серной кислоты добавляли 7-метокси-1-тетралон (1) (1,5 г; 8,5 ммоль) и 1-диметилсульфамоил-2-трет-бутилдиметилсилил-5-имидазолкарбоксальдегид (2) (2,7 г; 8,5 ммоль). Эту реакционную смесь нагревали в течение 24 часов при 90°С. После охлаждения до к.т. реакционную смесь делали щелочной с использованием избытка концентрированного гидроксида аммония. Эту смесь дважды экстрагировали тетрагидрофураном. Органические слои объединяли и промывали рассолом. Органический слой отделяли и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением 2,7 г желтого твердого вещества (3), состоящего из 3-(3Н-имидазол-4(5)-илметилен)-7-метоксихроман-4-она. Неочищенный продукт суспендировали в 100 мл этанола и добавляли катализатор - палладий на углероде (10%, 0,27 г). Эту смесь встряхивали в аппарате для гидрирования Парра под давлением водорода 40 фунтов на квадратный дюйм (880 кПа). Через 19 часов реакционную смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. Колоночная хроматография с использованием 7% метанола в хлороформе давала 1,05 г (46%) твердого вещества цвета бронзы, состоящего из 2-[3Н-имидазол-4(5)-илметил]-7-метокси-3, 4-дигидро-2Н-нафталин-1-она (4) (Б-1а). Соединение (4) (0,5 г; 1,95 ммоль) добавляли к 20 мл метанола. К этому раствору добавляли борогидрид натрия (74 мг; 1,95 ммоль). После перемешивания в течение 2, 5 часов при к.т. реакционную смесь гасили водой. Затем реакционную смесь дважды экстрагировали этилацетатом. Органические слои объединяли и промывали рассолом. Органический слой отделяли и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением 0,5 г белого твердого вещества (5), состоящего из 2-[3Н-имидазол-4(5)-илметил]-7-метокси-3, 4-дигидро-2Н-нафталин-1-ола. Этот неочищенный продукт растворяли в 26 мл дихлорметана. Добавляли триэтилсилан (2,5 мл; 15,6 ммоль) и трифторуксусную кислоту (4,8 мл; 62,3 ммоль) и перемешивали реакционную смесь при к.т. в течение 22 часов. Реакционную смесь подщелачивали 2 н. NaOH и органический слой отделяли и промывали рассолом. Раствор сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Колоночная хроматография с использованием 7% метанола в хлороформе давала 0,39 г (83%) желто-коричневого масла (6). Этот продукт растворяли в метаноле и добавляли избыток хлороводорода (HCl) в эфире. Этот раствор концентрировали при пониженном давлении с получением 0,3 г твердого вещества цвета бронзы. Колоночная хроматография с использованием 7% метанола в хлороформе давала после перекристаллизации из смеси ацетона и метанола 0,25 г (46%) гидрохлоридной соли 4(5)-(7-метокси-1,2,3,4-тетрагидронафталин-2-илметил)-1Н-имидазола в виде белых кристаллов (7).
1H-ЯМР (300 Мгц, СD3OD): 8.83 (s, 1H), 7.38 (s, 1H), 6.95 (d, 1H, J=8.5 Гц), 6.66 (d, 1H, J=8.4 Гц), 6.57 (s, 1H), 3,73 (s, 3H), 2.71-2.81 (m, 5H), 2.43-2.52 (m, 1H), 1.90-2.14 (m, 2H), 1.40-1.51 (m, 1H).
Для получения перечисленных ниже производных имидазола, следуя процедуре примера Б-1, взаимодействию подвергали разные соединения с конденсированными кольцами.
Пример Б-2 (а-г)
4-хроманон
(2а) 3-(3Н-имидазол-4(5)-илметилен)хроман-4-он
(2б) 3-(3Н-имидазол-4(5)-илметил)хроман-4-он
(2в) 3-(3Н-имидазол-4(5)-илметил)хроман-4-ол
(2г) 4(5)-хроман-3-илметил-1Н-имидазол
Пример Б-3 (а-б)
1-тетралон
(3а) 2-(3Н-имидазол-4(5)-илметил)-3,4-дигидро-2Н-нафталин-1-он
(3б) 4(5)-(1,2,3,4-тетрагидронафталин-2-илметил)-1Н-имидазол
Пример Б-4 (а-б)
4-метил-1-тетралон
(4а) 4(5)-(4-метил-1,2,3,4-тетрагидронафталин-2-илметил)-1Н-имидазол
(4б) 2-(3Н-имидазол-4(5)-илметил)-4-метил-3, 4-дигидро-2Н-нафталин-1-он
Пример Б-5 (а-б)
тиохроман
(5а) 3-(3Н-имидазол-4(5)-илметилен)тиохроман-4-он
(5б) 3-(3Н-имидазол-4(5)-илметил)тиохроман-4-он
Пример Б-6
Гидрохлоридную соль предыдущего соединения получают по стадии 5 способа примера Б-1 выше.
тиохроман
4(5)-тиохроман-3-илметил-1Н-имидазол
Пример Б-7 (а-в)
1-инданон
(7а) 2-(3Н-имидазол-4(5)-илметилен)индан-1-он
(7б) 2-(3Н-имидазол-4(5)-илметил)индан-1-он
(7в) 4(5)-индан-2-илметил-1 Н-имидазол
Пример Б-8 (а-б)
7-метил-1-тетралон
(8а) 2-(3Н-имидазол-4(5)-илметил)-7-метил-3,4-дигидро-2Н-нафталин-1-он
(8б) 4(5)-(7-метил-1,2,3,4-тетрагидронафталин-2-илметил)-1Н-имидазол
Гидрохлоридную соль этого соединения получали способом
Примера Б-6
Пример Б-9 (а-в)
4-кето-4,5,6,7-тетрагидротианафтен
(9а) 4(5)-(4,5,6, 7-тетрагидробензо[b]тиофен-5-илметил)-1Н-имидазол
Гидрохлоридную соль этого соединения получали способом примера Б-6
(9б) 5-(3Н-имидазол-4(5)-илметил)-6, 7-дигидро-5Н-бензо[b]тиофен-4-он
Гидрохлоридную соль этого соединения получали способом примера Б-6
(9в) 5-(октагидробензо[b]-5-илметил)-1Н-имидазол
Пример Б-10
4,4-Диметил-1-тетралон
4(5)-(4,4-диметил-1,2,3,4-тетрагидронафталин-2-илметил)-1Н-имидазол
1-Бензосуберон
Пример Б-11 (а-б)
(11а) 4(5)-(6,7,8,9-тетрагидро-5Н-бензоциклогептен-6-илметил)-1Н-имидазол
(11б) 6-(1Н-имидазол-4(5)-илметилен)-6,7,8,9-тетрагидробензоциклогептен-5-он
Пример В-1
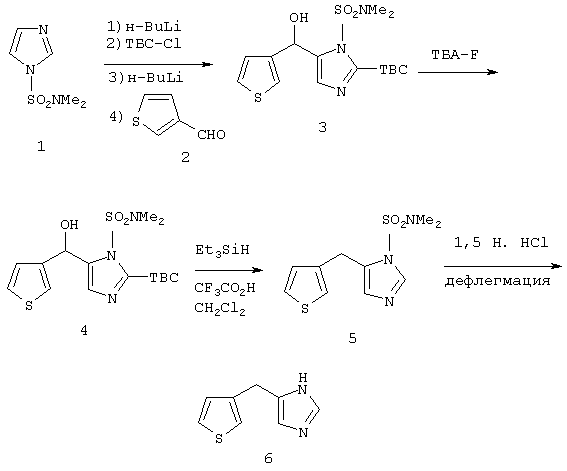
Методика получения (4(5)-тиофен-3-илметил-1Н-имидазола:
Процедура
1-(Диметилсульфамоил)имидазол (1) (2,0 г; 11,4 ммоль) переносят в 42 мл безводного ТГФ и охлаждают до -78°С. К раствору соединения (1) по каплям добавляют н-BuLi (6,6 мл; 10,6 ммоль). Полученный раствор перемешивают при -78°С в течение 30 минут. К реакционной смеси добавляют трет-бутилдиметилсилилхлорид (ТБС-Сl) (1,6 г; 10,6 ммоль) в 8 мл ТГФ. Эту реакционную смесь нагревают до к.т. и перемешивают в течение ночи. На следующий день реакционную смесь охлаждают до -20°С и добавляют н-BuLi (7,3 мл; 11,6 ммоль). После перемешивания при -20°С в течение 45 минут к реакционной смеси добавляют 3-тиофенкарбоксальдегид (2) (1,0 мл; 11,6 ммоль). Затем реакционную смесь нагревают до к.т. и перемешивают в течение ночи. На следующий день реакционную смесь гасят водой и разбавляют этилацетатом. Органический слой промывают водой, а затем рассолом. Органическую фазу сушат над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография (2:5, этилацетат/гексан) дает 3,0 г (7,5 ммоль) диметиламида 2-(трет-бутилдиметилсилил)-5-(гидрокситиофен-2-илметил)имидазол-1-сульфоновой кислоты (3). Соединение (3) (1,5 г; 3,74 ммоль) переносят в 37 мл ТГФ. К раствору соединения (3) добавляют по каплям 1 М раствор тетра-н-бутиламмонийфторида (ТБА-F) в ТГФ (4,1 мл; 4,1 ммоль). Эту реакционную смесь перемешивают в течение ночи при к.т. На следующий день реакционную смесь гасят водой, а затем экстрагируют этилацетатом. Органический слой промывают водой, а затем рассолом. Органическую фазу сушат над сульфатом натрия и удаляют растворитель при пониженном давлении. Выделяют 0,94 г (3,3 ммоль) диметиламида 5-(гидрокситиофен-2-илметил)имидазол-1-сульфоновой кислоты (4). Соединение (4) (0,5 г; 1,74 ммоль) переносят в 23 мл дихлорметана, к этому раствору добавляют 2,2 мл (13,9 ммоль) триэтилсилана и 4,3 мл (55,7 ммоль) трифторуксусной кислоты. Эту реакционную смесь перемешивают при к.т. в течение ночи, а затем гасят водой и нейтрализуют твердым бикарбонатом натрия. Органический слой промывают водой, а затем рассолом. Органическую фазу сушат над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография с использованием смеси этилацетата и гексана (1:1) дает 0,42 г (1,55 ммоль) диметиламида 5-(тиофен-2-илметил)имидазол-1-сульфоновой кислоты (5). Соединение (5) переносят в 10 мл 1,5 н. HCl и нагревают с обратным холодильником в течение 3 часов, а затем перемешивают при к.т. в течение ночи. Реакционную смесь разбавляют этилацетатом, нейтрализуют твердым бикарбонатом натрия, а затем подщелачивают 2 н. NaOH.
Органический слой промывают водой, а затем рассолом. Органическую фазу сушат над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография с использованием смеси хлороформа и метанола (10:1) дает 0,17 г (1,0 ммоль) 4(5)-тиофен-3-илметил-1Н-имидазола (6) (В-1).
1H-ЯМР (300 Мгц, СD3ОD): 7.52 (s, 1H), 7.25-7.27 (m, 1H), 6.97-7.01 (m, 2H), 6.77 (s, 1H), 3.98 (s, 2H).
Пример В-2
В способе примера В-1 вместо 3-тиофенкарбоксальдегида используют 2-карбоксальдегидный изомер с получением 4(5)-тиофен-2-илметил-1Н-имидазола.
Пример В-3
В способе примера В-1 вместо 3-тиофенкарбоксальдегида используют 5-метил-2-тиофенкарбоксальдегид с получением 4(5)-(5-метилтиофен-2-илметил)-1Н-имидазола.
Пример В-4
В способе примера В-1 вместо 3-тиофенкарбоксальдегида используют 5-хлор-2-тиофенкарбоксальдегид с получением 4(5)-(5-хлортиофен-2-илметил)-1Н-имидазола.
Пример В-5
В способе примера В-1 используют 2-фуранкарбоксальдегид с получением 4(5)-фуран-2-илметил-1Н-имидазола.
Пример В-6
В способе примера В-1 используют 3-фуранкарбоксальдегид с получением 4(5)-фуран-3-илметил-1Н-имидазола.
Пример В-7
В способе примера В-1 используют 5-метил-2-фуранкарбоксальдегид с получением 4(5)-(5-метилфуран-2-илметил)-1Н-имидазола.
Пример В-8
В способе примера В-1 используют бензальдегид с получением 4(5)-бензил-1Н-имидазола.
Пример В-9
В способе примера В-1 используют 2-тианафтенкарбоксальдегид с получением 4(5)-бензо[b]тиофен-2-илметил-1Н-имидазола.
Пример В-10
В способе примера В-1 используют 2-бензофуранкарбоксальдегид с получением 4(5)-бензофуран-2-илметил-1Н-имидазола.
Пример В-11
В способе примера В-1 используют 5-этил-2-фуранкарбоксальдегид с получением 4(5)-(5-этилфуран-2-илметил)-1Н-имидазола.
Пример В-12
В способе примера В-1 используют 4-бром-2-тиофенкарбоксальдегид с получением 4(5)-(4-бромтиофен-2-илметил)-1Н-имидазола.
Пример В-13
В способе примера В-1 используют 4-фенил-2-тиофенкарбоксальдегид с получением 4(5)-(4-фенилтиофен-2-илметил)-1Н-имидазола.
Пример В-14
В способе примера В-1 используют 4-метил-2-тиофенкарбоксальдегид с получением гидрохлоридной соли 4(5)-(4-метилтиофен-2-илметил)-1Н-имидазола.
Пример Г-1
Методика получения оксазолидин-2-илиден-(3-фенилбицикло[2.2.1]гепт-2-ил)амина:
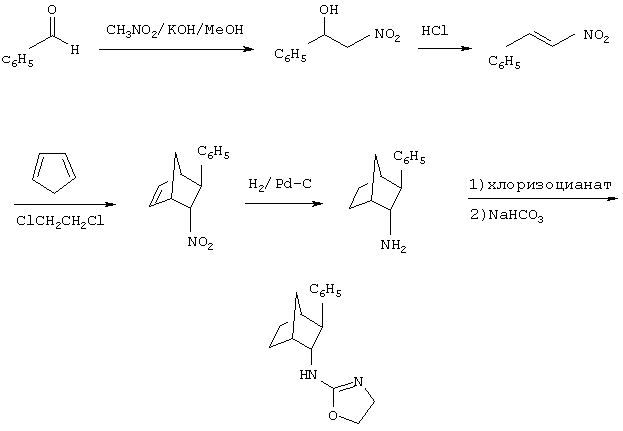
Процедура
Соединение, имеющее эндо, экзо относительную стереохимическую конфигурацию, получали, синтезируя β-нитростирол, как показано выше. Обработка метанольного раствора бензальдегида (10 г; 94,3 ммоль) нитрометаном (51 мл; 943 ммоль) в присутствии гидроксида натрия (3 н. в метаноле, до рН 8) давала нитроспирт с выходом 60%. Дегидратацию спирта осуществляли путем обработки метансульфонилхлоридом (3,5 г; 31,1 ммоль), а затем триэтиламином (6,3 г; 62,2 ммоль) в дихлорметане (35 мл) с получением продукта с выходом 97%. Соединение очищали перегонкой в шариковом дистилляторе. Построение бицикло[2.2.1]гептанового скелета выполняли в одну стадию. Реакцию Дильса-Альдера проводили нагреванием нитростирола (4,5 г; 30,2 моль) с циклопентадиеном (3,98 г; 60,4 ммоль) в 1,2-дихлорэтане (10 мл). Реакция Дильса-Альдера протекает при приблизительном соотношении эндо : экзо по нитро 3:1. Как это соотношение, так и относительная стереохимическая конфигурация были подтверждены данными рентгеноструктурного анализа. Восстановление как нитрогруппы, так и олефина проводили в атмосфере водорода в присутствии 10 % мас. палладия на угле. Разделение изомеров на этой стадии осуществляли флэш-хроматографией с использованием 5% метанола, насыщенного аммиаком, в дихлорметане. Амин (0,7 г; 3,74 ммоль) обрабатывали сначала хлорэтилизоцианатом (0,38 мл; 4,49 ммоль) с получением хлорэтилмочевины, которую затем нагревали в присутствии водного раствора NaHCO3 с получением оксазолидин-2-илиден-(3-фенилбицикло[2.2.1]гепт-2-ил)амина (Г-1) с выходом 51%.
1H-ЯМР (300 МГц, CDCl3) d: 1.36-1.80 (m, 6H), 2.14 (d, 1H, J=4.40 Гц), 2.37 (s, 1Н), 2.65 (s, 1H), 3.71-3.78 (m, 2Н), 3.95-3.98 (m, 1H), 4.19-4.25 (t, 2H, J=17.15 Гц), 7.17-7.29 (m, 5H).
Пример Г-2
Оксазолидин-2-илиден-(3-о-толилбицикло[2.2.1]гепт-2-ил)амин получают, используя о-метил-β-нитростирол в способе Г-1.
Пример Г-3
Бицикло[2.2.1]гепт-2-илоксазолидин-2-илиденамин получают, используя нитроэтен в способе Г-1.
Пример Д-1
Методика получения имидазолидин-2-илиден-(4-метил-3, 4-дигидро-2Н-бензо[1,4]оксазин-6-ил)амина:
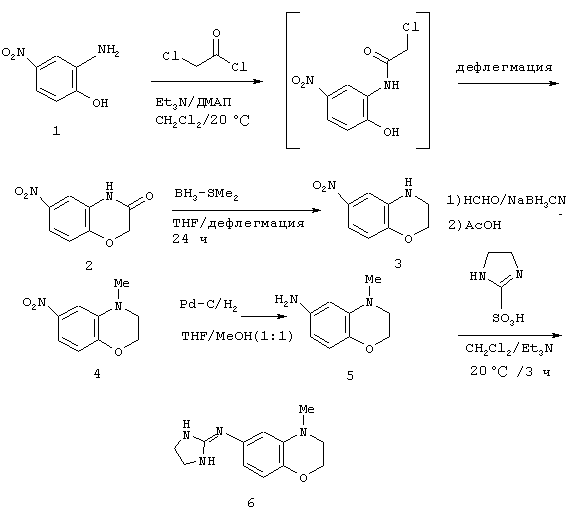
Процедура
К 2-амино-4-нитофенолу (1) (4,00 г; 25,95 ммоль), триэтиламину (15,20 мл; 109,0 ммоль) и 4-диметиламинопиридину (0,063 г; 0,52 ммоль) в виде взвеси в безводном СН2Сl2 (250 мл) при 0°С под аргоном через шприц добавляли хлорацетилхлорид (2,27 мл; 28,55 ммоль). После нагревания с обратным холодильником в течение 72 часов чистый продукт отфильтровывали и промывали водой. Маточную жидкость последовательно промывали фосфорной кислотой (0,5 М), насыщенным бикарбонатом натрия, водой и рассолом, а затем сушили над МgSО4. Этот раствор наносили на диоксид кремния и очищали флэш-хроматографией на диоксиде кремния с использованием смеси гексан/этилацетат (4:6) с получением добавочного продукта. Объединенные твердые вещества сушили в вакууме с получением чистого 6-нитро-4Н-бензо[1,4]оксазин-3-она (2) (4,12 г) с выходом 82%. К взвеси соединения (2) (1,49 г, 7,65 моль) в безводном ТГФ (40 мл) под аргоном в двугорлой круглодонной колбе, снабженной дефлегматором, добавляли боран-диметилсульфидный комплекс (15,3 мл; 30,62 ммоль). Эту смесь грели при температуре образования флегмы до тех пор, пока посредством тонкослойной хроматографии не будет определено отсутствие исходного материала (2 часа). Реакционную смесь охлаждали до к.т. и осторожно гасили добавлением метанола по каплям. Полученную смесь грели при температуре образования флегмы еще 10 минут. Неочищенную реакционную смесь концентрировали в вакууме и очищали флэш-хроматографией на диоксиде кремния, используя смесь гексан/этилацетат (8:2), с получением чистого 6-нитро-3,4-дигидро-2Н-бензо[1, 4]оксазина (3) (1,36 г) в виде оранжевого твердого вещества с выходом 99%. К соединению (3) (0,032 г; 0,178 ммоль) и формалину (37% в Н2О; 0,20 мл; 2,67 ммоль) в безводном ацетонитриле (1,5 мл) при температуре окружающей среды добавляли цианоборогидрид натрия (0,034 г; 0,534 ммоль). Этот раствор перемешивали в течение 30 минут, затем добавляли ледяную уксусную кислоту (0,032 мл; 0,534 моль). Полученную смесь перемешивали еще 16 часов. Органические вещества переносили в диэтиловый эфир и последовательно промывали NaOH (2 н.) и рассолом, сушили над MgSO4 и концентрировали в вакууме. Полученное твердое веществ очищали флэш-хроматографией на диоксиде кремния, используя смесь гексан/этилацетат (7:3), с получением чистого 4-метил-6-нитро-3,4-дигидро-2Н-бензо[1,4]оксазина (4) (0,031 г) с выходом 93%. К соединению (4) (2,16 г; 11,12 ммоль) и 10% палладию на углероде (0,216 г; 10 масс.) под аргоном добавляли метанол (МеОН) (30 мл), а затем ТГФ (30 мл). Через полученную смесь пропускали водород до тех пор, пока посредством тонкослойной хроматографии не будет определено отсутствие соединения (4) (2 часа). Добавляли целит и фильтровали эту смесь через слой целита с последующей промывкой метанолом. Полученный раствор концентрировали в вакууме с получением чистого 4-метил-3,4-дигидро-2Н-бензо[1,4]оксазин-6-иламина (5) (1,86 г) в виде бледно-пурпурного масла с выходом 100%, причем без дополнительной очистки. К соединению (5) (1,86 г; 11,34 ммоль) и имидазолин-2-сульфоновой кислоте (1,84 г; 12,24 ммоль) в безводном ацетонитриле (50 мл) под аргоном при 0°С добавляли триэтиламин (3,26 мл; 23,36 ммоль). Этот раствор постепенно нагревали до температуры окружающей среды и перемешивали в течение 16 часов. В этот момент добавляли дополнительное количество имидазолин-2-сульфоновой кислоты (0,86 г; 5,55 моль) и перемешивали полученную смесь еще 5 часов. Этот раствор концентрировали в вакууме и остаток переносили в Н2О. Органические вещества экстрагировали в CH2Cl2 и дважды промывали NaOH и рассолом, сушили над MgSO4 и концентрировали в вакууме. Полученную пену очищали флэш-хроматографией на диоксиде кремния, используя 20% метанола (насыщенного аммиаком) в хлороформе, с получением чистого имидазолидин-2-илиден-(4-метил-3,4-дигидро-2Н-бензо[1,4]оксазин-6-ил)амина (6) (Д-1) (0,905 г) с выходом 34%.
1H-ЯМР (СDСl3): 2.81 (s, 3Н); 3.26 (t, J=8.9 Гц, 2Н); 3.60 (s, 4H); 4.26 (m, 2Н); 4.60 (vbrs, 2H); 6.34 (dd, J=8.2 Гц, J=2.4 Гц, 1Н); 6.39 (d, J=2.4 Гц, 1Н); 6.68 (d, J=8.2H4, 1H).
Примеры Е и Ж
Методика получения 6-(имидазолидин-2-илиденамино)-5-метил-4Н-бензо[1,4]оксазин-3-она (Е) и имидазолидин-2-илиден-(5-метил-3,4-дигидро-2Н-бензо[1,4]оксазин-8-ил)амина (Ж):
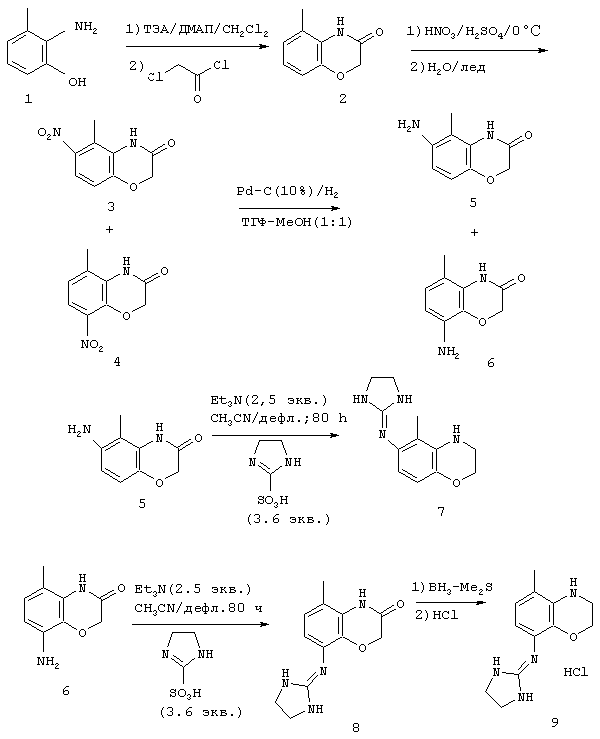
Процедура
К 2-амино-3-метилфенолу (1) (14,72 г; 0,120 моль), триэтиламину (35,0 мл; 0,251 моль) и 4-диметиламинпиридину (0,29 г; 2,39 ммоль) в безводном CH2Cl2 (100 мл) при 0°С под аргоном через шприц по каплям добавляли хлорацетилхлорид (10,0 мл; 0,126 моль). После завершения добавления полученный раствор нагревали с обратным холодильником в течение 24 часов. Органические вещества последовательно промывали фосфорной кислотой (0,5 М), насыщенным бикарбонатом натрия, водой и рассолом, а затем сушили над МgSO4. Полученный раствор концентрировали и переносили в ТГФ, к которому был добавлен эфир. Полученные кристаллы отфильтровывали с получением чистого 5-метил-4Н-бензо[1,4]оксазин-3-она (2) (12,30 г) с выходом 63%. К соединению (2) (14,64 г; 89,72 ммоль), растворенному в концентрированной Н2SO4 (65 мл), при -10°С добавляли 70% концентрированную НNО3 (8,08 г; 89,72 ммоль) и концентрированную H2SO4 (25 мл) при быстром механическом перемешивании со скоростью, при которой внутренняя температура поддерживалась ниже -5°С. Как только добавление завершали, смесь выливали на колотый лед (500 мл) и полученные твердые вещества отфильтровывали и суспендировали в холодной воде (300 мл), добавляя в то же время достаточно NaOH, чтобы довести рН до 7, 0. Полученный желтый порошок растворяли в ТГФ, наносили на диоксид кремния и очищали флэш-хроматографией, используя смесь 60% гексана и этилацетата, с получением нитрованного продукта в виде смеси двух региоизомеров, а именно желаемого 6-замещенного ароматического вещества, состоящего из 6-нитро-5-метил-4Н-бензо[1,4]оксазин-3-она (3) (55%) и 8-замещенного побочного продукта, состоящего из 8-нитро-5-метил-4Н-бензо[1,4]оксазин-3-она (4) (22%). На этой стадии эти изомеры разделяются с трудом, и их в виде смеси направляли на следующую стадию. К смеси соединений (3) (1,93 г; 9,27 ммоль) и (4) (0,48 г; 2,32 ммоль), растворенной в растворе МеОН (300 мл) и ТГФ (300 мл), под аргоном добавляли 10% палладий на углероде (1,20 г). Полученный раствор подвергали действию Н2 при атмосферном давлении. Через 16 часов катализатор отфильтровывали, полученный раствор концентрировали в вакууме и очищали флэш-хроматографией на диоксиде кремния, используя смесь 50% гексана и этилацетата, с получением 6-амино-5-метил-4Н-бензо[1,4]оксазин-3-она (5) (0,96 г) с выходом 46% и 8-амино-5-метил-4Н-бензо[1,4]оксазин-3-она (6) (0,17 г) с выходом 8%. Соединение (5) (1,20 г; 6,74 ммоль), имидазолин-2-сульфоновую кислоту (2,02 г; 13,48 ммоль) и триэтиламин (2,45 г; 16,85 ммоль) нагревали при температуре образования флегмы в безводном ацетонитриле (50 мл) под аргоном в течение 48 часов. В это время добавляли дополнительное количество имидазолин-2-сульфоновой кислоты (1,01 г; 6,74 ммоль) и триэтиламина (1,41 мл; 10,12 ммоль) и перемешивали полученную смесь еще 24 часа. Этот раствор концентрировали в вакууме, остаток переносили в раствор СНСl3/изопропиловый спирт (3:1) и последовательно промывали гидроксидом натрия (1 н.) и рассолом, сушили над MgSO4 и концентрировали в вакууме. Полученную пену очищали флэш-хроматографией на диоксиде кремния, используя смесь 20% метанола, насыщенного аммиаком, в хлороформе, с получением 6-(имидазолидин-2-илиденамино)-5-метил-4Н-бензо[1,4]оксазин-3-она (7) (0,42 мг) в виде пены с выходом 27% одновременно с 55% вновь полученного исходного материала. HCl-соль перекристаллизовывали из смеси этанола и диэтилового эфира (EtOH/Et2O) с получением тонких белых игл.
1H-ЯМР (ДМСО): 2.10 (s, 3H), 3.59 (s, 4H), 4.53 (s, 2H), 6.83 (d, J=8.6 Гц, 1Н), 6.90 (d, J=8.6 Гц, 1Н), 8.07 (brs, 2H), 10.15 (vbrs, 1H), 10.42 (s, 1H).
Соединение (6), имидазолин-2-сульфоновую кислоту (0,223 г; 1,49 ммоль) и триэтиламин (0,415 мл; 2,98 ммоль) нагревали при 95°С в безводном ацетонитриле (10 мл) в закрытой пробирке в течение 2 часов. По истечении этого времени добавляли дополнительное количество имидазолин-2-сульфоновой кислоты (0,12 мг; 0,75 ммоль) и продолжали реакцию еще 16 часов. Этот раствор концентрировали в вакууме и остаток переносили в раствор СНСl3/изопропиловый спирт (3:1) и последовательно промывали гидроксидом натрия (2 н.) и рассолом, сушили (MgSO4) и концентрировали в вакууме. Полученное масло перекристаллизовывали из СНСl3 с получением чистого 6-(имидазолидин-2-илиденамино)-5-метил-4Н-бензо[1,4]оксазин-3-она (8) (Е) (0,048 г) в виде белого порошка с выходом 15% одновременно с 35% вновь полученного исходного материала. К взвеси соединения (8) (0,08 г; 0,321 ммоль) в безводном ТГФ (50 мл) в трехгорлой круглодонной колбе, снабженной дефлегматором, под аргоном добавляли боран-диметилсульфидный комплекс (0,48 мл; 0,936 мл). Эту смесь грели при температуре образования флегмы, пока исходный материал не переставал обнаруживаться посредством тонкослойной хроматографии (3 часа). Реакционную смесь охлаждали до комнатной температуры и осторожно гасили добавлением метанола по каплям. Неочищенную смесь концентрировали в вакууме и очищали флэш-хроматографией на диоксиде кремния, используя смесь 20% метанола, насыщенного аммиаком/хлороформом, с получением имидазолин-2-илиден-(5-метил-3,4-дигидро-2Н-бензо[1,4]оксазин-8-ил)амина (9) (Ж) (0,03 г) в виде HCl-соли с выходом 37%.
1H-ЯМР (СDСl3): 2.07 (s, 3Н); 3.46 (t, J=4.3 Гц, 2Н); 3.55 (s, 4H), 4.24 (t, J=4.3 Гц, 2Н); 5.60-5.95 (vbrs, 2H); 6.44 (d, J=8.0 Гц, 1Н); (d, J=8.0 Гц, 1Н).
Пример 3
Методика получения 4(5)-фенилсульфанил-1Н-имидазола:
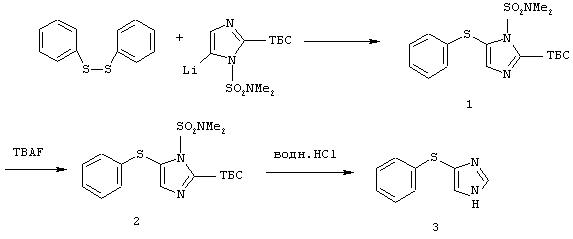
Процедура
1-(N,N-Диметилсульфамоил)имидазол (1,5 г; 8,6 ммоль) переносили в 28 мл ТГФ. Этот раствор охлаждали до -78°С и по каплям шприцом добавляли н-BuLi (5,4 мл; 8,6 ммоль). После перемешивания при -78°С в течение 1 часа добавляли ТБС-Сl (1,3 г; 8,56 моль) в 10 мл ТГФ. Баню убирали и реакционной смеси давали нагреться до к.т. Реакционную смесь перемешивали в течение ночи. Реакционную смесь охлаждали до -20°С и добавляли н-BuLi (5,4 мл; 8,6 ммоль). Через 45 мин добавляли фенилдисульфид (1,9 г; 8,6 ммоль) в 8 мл ТГФ. Реакционную смесь перемешивали при к.т. в течение 48 часов. Реакционную смесь гасили насыщенным хлоридом аммония и экстрагировали этилацетатом. Органический слой собирали и промывали водой, а затем рассолом. Этот раствор сушили над сульфатом натрия и удаляли растворитель при пониженном давлении. Флэш-хроматография (2,5% ЕtOАс/гексан) дала 2,8 г (7,0 ммоль) диметиламида 2-(трет-бутилдиметилсилил)-5-фенилсульфанилимидазол-1-сульфоновой кислоты (1) в виде масла желтого цвета. Соединение (1) (2,8 г; 7,0 ммоль) растворяли в ТГФ и этот раствор охлаждали до 0°С. К раствору по каплям добавляли ТБА-F (7,0 мл; 7,0 ммоль). Эту реакционную смесь перемешивали в течение ночи при к.т. На следующий день реакционную смесь гасили водой и экстрагировали этилацетатом. Органический слой промывали водой, а затем рассолом. Этот раствор сушили над сульфатом натрия и удаляли растворитель при пониженном давлении. Флэш-хроматография (50% ЕtOАс/гексан) дала 474 мг диметиламида 5-фенилсульфанилимидазол-1-сульфоновой кислоты (2) и 290 мг 5-фенилсульфанил-1Н-имидазола (3) (3). 478 мг соединения (2) добавляли к 2 н. HCl и этот раствор грели при температуре образования флегмы в течение 2 часов. Реакционную смесь делали щелочной 2 н. гидроксидом натрия и экстрагировали этилацетатом. Органический слой промывали водой, а затем рассолом. Этот раствор сушили над сульфатом натрия и удаляли растворитель при пониженном давлении. Флэш-хроматография (ЕtOАс) дала (3) в виде белого кристаллического вещества. Суммарный выход (3) составил 360 мг (2,0 ммоль).
1H-ЯМР (300 МГц, СD3ОD): 7.91 (s, 1H), 7.37 (s, 1H), 7.19-7.23 (m, 2H), 7.07-7.11 (m, 3H).
Пример И
Методика получения соли 4(5)-(1,2,3,4-тетрагидронафталин-2-илметил)-4,5-дигидро-1Н-имидазола, соль метансульфоновой кислоты:
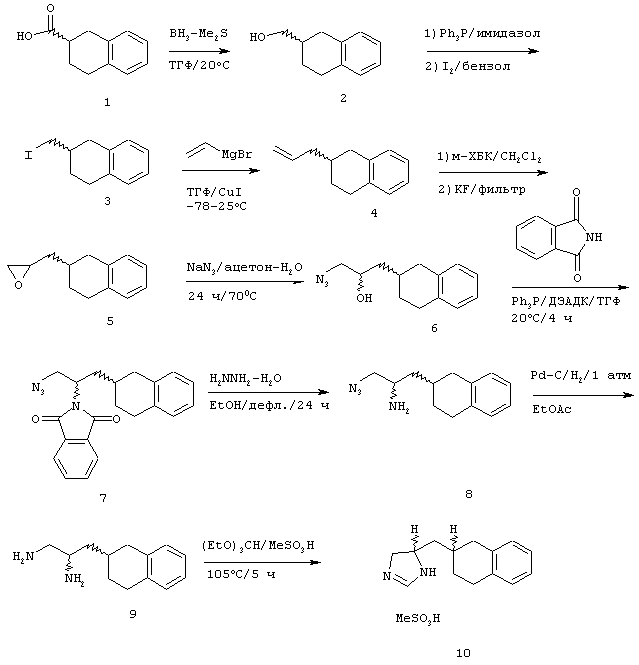
Процедура
К 1,2,3, 4-тетрагидронафталин-2-карбоновой кислоте (1) (4,93 г; 27,42 ммоль) в безводном ТГФ (250 мл) при 20°С под аргоном шприцом добавляли 3,26 мл (32,90 ммоль) боран-диметилсульфида (ВН3 -Ме2S). После перемешивания в течение 16 часов добавляли МеОН (4 мл) и нагревали смесь до 55°С, пока не переставал выделяться газ. Смесь концентрировали до масла, переносили в Et2O и последовательно промывали 2 М фосфорной кислотой, насыщенным бикарбонатом натрия, водой и рассолом, а затем сушили над МgSO4 и повторно концентрировали. Полученное масло очищали перегонкой в шариковом дистилляторе под высоким вакуумом при 150°С с получением чистого спирта (1,2,3,4-тетрагидронафталин-2-ил)метанола (2) (4,09 г) с выходом 93%. К трифенилфосфину (10,179 г; 38,809 ммоль) и имидазолу (2,64 г; 38,809 ммоль) в безводном бензоле (175 мл) при быстром перемешивании добавляли йод (8,60 г; 33,865 ммоль) и бензол (75 мл), а затем соединение (2) в бензоле (50 мл). Через 3 часа отфильтровывали твердые вещества и упаривали фильтрат в вакууме до объема 50 мл, к которому добавляли гексан (200 мл). Полученные твердые вещества отфильтровывали, фильтрат промывали последовательно водой и рассолом, сушили над MgSO4 и концентрировали в вакууме. Полученное масло очищали флэш-хроматографией на диоксиде кремния, используя гексан, с получением чистого 2-йодметил-1,2,3,4-тетрагидронафталина (3) (6,239 г) с выходом 90%. К соединению (3) (10,02 г; 36,85 ммоль) и CuI (1,41 г; 7,37 ммоль) в безводном ТГФ (50 мл) при -78°С под аргоном медленно, со скоростью, при которой не появлялась окраска, добавляли винилмагнийбромид (1М в ТГФ; 73,70 мл; 73,70 ммоль). Этому раствору давали нагреться до 0°С и перемешивали его в течение 6 часов. Полученную смесь снова охлаждали до -40°С и гасили осторожным добавлением 2 М фосфорной кислоты (35 мл). Этот раствор разбавляли водой (100 мл) и экстрагировали гексанами. Органические фракции промывали последовательно водой и рассолом, сушили над MgSO4 и концентрировали в вакууме. Полученное масло очищали флэш-хроматографией на диоксиде кремния, используя гексан, с получением 2-аллил-1,2,3,4-тетрагидронафталина (4) (5,618 г) с выходом 88%. Соединение (4) (5,615 г; 32,645 ммоль) и м-хлорбензойную кислоту (м-ХБК) (14,08 г; 81,613 ммоль) перемешивали в безводном хлориде метилена (50 мл) в течение 16 часов. Отфильтровывали твердые вещества, добавляли фторид калия KF (5,11 г; 88,142 ммоль) и перемешивали эту смесь еще 1 час. Отфильтровывали твердые вещества и концентрировали реакционную смесь в вакууме. Полученное масло очищали флэш-хроматографией на диоксиде кремния, используя 5% этилацетата в гексане, с получением 2-(1,2,3, 4-тетрагидронафталин-2-илметил)оксирана (5) (5,41 г) с выходом 88%. К соединению (5) (1,62 г; 8,649 ммоль) в растворе ацетона (20 мл) и воды (5 мл) добавляли азид натрия (1,97 г; 30,271 ммоль). Этот раствор нагревали до 85°С и перемешивали в течение 48 часов. Раствор концентрировали в вакууме, остаток переносили в СН3Сl и промывали последовательно водой и рассолом, сушили над МgSO4 и концентрировали в вакууме. Полученное масло очищали флэш-хроматографией на диоксиде кремния, используя смесь 30% этилацетата в гексане, с получением чистого 1-азидо-3-(1,2,3, 4-тетрагидронафталин-2-ил)пропан-2-ола (6) (1,762) с выходом 88%. Смесь соединения (6) (1,88 г; 8,140 ммоль), трифенилфосфина (2,67 г; 10,173 ммоль), фталимида (1,50 г; 10,173 ммоль), диэтилазодикарбоксилата (ДЭАДК) (1,77 г; 10,173 ммоль) перемешивали в безводном ТГФ (50 мл) в течение 4 часов. Этот раствор концентрировали в вакууме, переносили в раствор гексана (25 мл) и эфира (25 мл) и перемешивали в течение 16 часов. Отфильтровывали твердые вещества и концентрировали фильтрат в вакууме. Полученное масло очищали флэш-хроматографией на диоксиде кремния со смесью 20% этилацетата в гексане с получением 2-[1-азидометил-2-(1,2,3,4-тетрагидронафталин-2-ил)этил]изоиндол-1,3-диона (7) (2,487 г), загрязненного небольшим количеством примесей, который использовали дальше без дополнительной очистки. Смесь соединения (7) (3,93 г; 10,917 ммоль) и гидразина (0,680 мл; 21,833 ммоль) грели в этаноле (60 мл) при температуре образования флегмы в течение 16 часов. Отфильтровывали твердые вещества и концентрировали фильтрат в вакууме. Остаток очищали флэш-хроматографией на диоксиде кремния со смесью 5% МеОН в CH2Cl2 с получением 1-азидометил-2-(1,2,3, 4-тетрагидронафталин-2-ил)этиламина (8) (2,057 г) с выходом 88%. Смесь соединения (8) (2,056 г; 8,940 ммоль) и 10% палладия на углероде (0,260 г) перемешивали в МеОН (30 мл) под водородом при атмосферном давлении в течение 16 часов. Отфильтровывали твердые вещества и концентрировали фильтрат в вакууме. Остаток очищали флэш-хроматографией на диоксиде кремния со смесью 10% метанола, насыщенного аммиаком, в CH2Cl2 с получением 3-(1,2,3,4-тетрагидронафталин-2-ил)пропан-1,2-диона (9) (1,557 мг) с выходом 85%. Смесь соединения (9) (0,590 г; 2,892 ммоль) и метансульфоновой кислоты (0,980 мл; 14,460 ммоль) нагревали в триэтилортоформиате (10 мл) при 105°С в течение 3 часов. Концентрировали реакционную смесь в вакууме и отфильтровывали твердые вещества. Последующая перекристаллизация этих твердых веществ из смеси МеОН и эфира давала чистую соль метансульфоновой кислоты 4(5)-(1,2,3,4-тетрагидронафталин-2-илметил)-4,5-дигидро-1Н-имидазола (И) (0,435 г) с выходом 48%.
1H-ЯМР (CDCl3): 1.37-1.56 (m, 1H); 1.56-1.70 (m, 1H); 1.80-2.02 (m, 2H); 2.32-2.55 (m, 2H); 2.72 (s, 3Н), 2.75-2.95 (m, 3H), 3.38-3.59 (m, 1H), 3.93-4.08 (m, 1H); 4.31-4.47 (m, 1H); 7.00-7.20 (m, 4Н); 8.46 (s, 1H); 10.04 (s, 1H); 10.35 (brs, 1H).
Пример К-1
Методика получения 4(5)-циклогексилметил-1Н-имидазола:
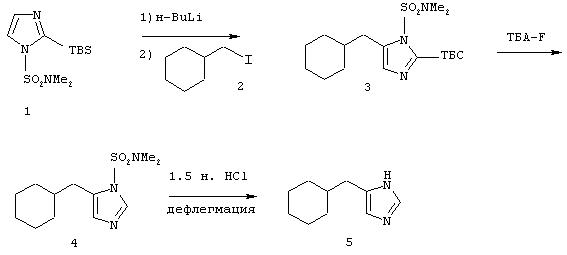
Процедура
2-трет-Бутилдиметилсилил-1-диметилсульфамоилимидазол (1) (4,1 г; 14,2 ммоль) переносят в 47 мл безводного ТГФ и охлаждают до -20°С. К раствору соединения (1) по каплям добавляют н-BuLi (8,9 мл; 14,2 ммоль). Полученный раствор перемешивают при -20°С в течение 45 минут. Затем к реакционной смеси по каплям добавляют циклогексилметилйодид (2) (3,13 мг; 14 ммоль). Затем нагревают реакционную смесь до к.т. и перемешивают в течение ночи. На следующий день гасят реакционную смесь насыщенным хлоридом аммония и разбавляют водой. Эту смесь экстрагируют этилацетатом (3×100 мл). Объединяют органические слои и промывают их водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография (4:1, этилацет/гексан) дает 2,26 г (5,6 ммоль) 5-циклогексилметил-2-трет-бутилдиметилсилил-1-диметилсульфамоилимидазола (3). Соединение (3) (2,26 г; 5,6 ммоль) переносят в 56 мл ТГФ и охлаждают до 0° С. К раствору соединения (3) по каплям добавляют 1 М раствор ТБА-F в ТГФ (5,6 мл; 5,6 ммоль). Эту реакционную смесь нагревают до к.т. и перемешивают в течение ночи. На следующий день реакционную смесь гасят водой, а затем экстрагируют этилацетатом. Органический слой промывают водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография (1:1, этилацетат/гексан) дает 1,2 г (4,42 ммоль) 5-циклогексилметил-1-диметилсульфамоилимидазола (4). Соединение (4) (1,2 г; 4,42 ммоль) переносят в 25 мл 1,5 н. раствора HCl и греют при температуре образования флегмы в течение 2 часов. Доводят рН этой смеси до 13 гидроксидом натрия (2 н.), а затем экстрагируют ее хлороформом (4×100 мл). Органические слои объединяют и промывают водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография (9:1, хлороформ/метанол) дает 700 мг (4,27 ммоль) 4(5)-циклогексилметил-1Н-имидазола (5) (К-1).
1H-ЯМР (CDCl3): 0.92-1.0 (m, 2Н); 1.16-1.26 (m, 3H); 1.57-1.73 (m, 6H); 2.48 (d, J=6.9 Гц, 2H); 6.77 (s, 1Н); 7.56 (s, 1H).
Пример К-2
В способе примера К-1 используют (S)-2-иодметил-1,2,3,4-тетрагидронафталин с получением (S)-4(5)-(1,2,3, 5)-тетрагидронафталин-2-илметил)-1Н-имидазола. (S)-2-Иодметил-1,2,3,4-тетрагидронафталин получали из (S)-1,2,3,4-тетрагидро-2-нафтоевой кислоты. (S)-1,2,3,4-Тетрагидро-2-нафтоевую кислоту получали в результате разделения 1,2,3,4-тетрагидро-2-нафтоевой кислоты (J. Med. Chem. 1983, 26, 328-334).
Пример К-3
В способе примера К-1 использовали (R)-2-иодметил-1,2,3, 4-тетрагидронафталин с получением (R)-4(5)-(1,2,3,5)-тетрагидронафталин-2-илметил)-1Н-имидазола. (R)-2-Иодметил-1,2,3,4-тетрагидронафталин получали из (R)-1,2,3,4-тетрагидро-2-нафтоевой кислоты. (R)-1, 2,3,4-Тетрагидро-2-нафтоевую кислоту получали в результате разделения 1,2,3,4-тетрагидро-2-нафтоевой кислоты (J. Med. Chem. 1983, 26, 328-334).
Пример Л-1
Методика получения 4(5)-(4,5,6,7-тетрагидробензо[b]тиофен-2-илметил)-1Н-имидазола:
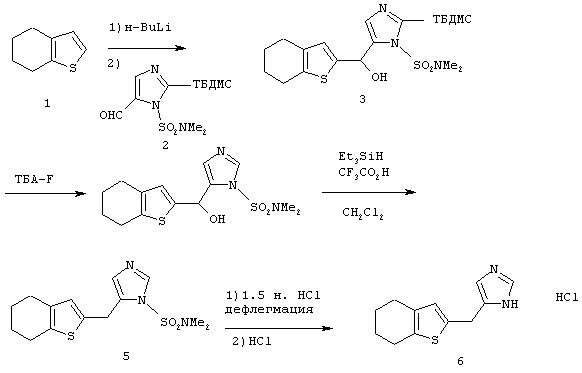
Процедура
4,5,6,7-Тетрагидробензо[b]тиофен (1) (2,1 г; 15 ммоль) переносят в 75 мл безводного ТГФ и охлаждают до -78°С. К раствору соединения (1) по каплям добавляют н-BuLi (6,0 мл; 15 ммоль). Полученный раствор перемешивают при -78°С в течение 60 минут. К этой реакционной смеси добавляют 1-диметилсульфамоил-2-трет-бутилдиметилсилил-5-имидазолкарбоксальдегид (2) (4,8 г; 15 ммоль) в 25 мл ТГФ. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 2 часов, а затем гасят водой и разбавляют этилацетатом. Органический слой промывают водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография (1:3, этилацетат/гексан) дает 5,2 г (11 ммоль) диметиламида 2-(трет-бутилдиметилсилил)-5-[гидрокси-(4,5,6,7-тетрагидробензо[b]тиофен-2-ил)метил]имидазол-1-сульфоновой кислоты (3). Соединение (3) (5,2 г; 11,3 ммоль) переносят в 57 мл ТГФ. К раствору соединения (3) по каплям добавляют 1 М раствор тетра-н-бутиламмонийфторида (ТБА-F) в ТГФ (11,3 мл; 11,3 ммоль). Реакционную смесь перемешивают в течение 1 часа 15 минут, а затем гасят водой, после чего экстрагируют этилацетатом. Промывают органический слой водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Перекристаллизация из смеси гексан/этилацетат дает диметиламид 5-[гидрокси-(4,5,6,7-тетрагидробензо[b]тиофен-2-ил)метил]имидазол-1-сульфоновой кислоты (4) (2,1 г; 6,2 ммоль). К тому же дополнительно выделяют 2 г неочищенного продукта. Соединение (4) (2,0 г; 5,9 ммоль) переносят в 78 мл дихлорметана, добавляют к раствору 7,5 мл (46,9 ммоль) триэтилсилана и 14,4 мл (0,19 моль) трифторуксусной кислоты. Эту реакционную смесь перемешивают в течение ночи, а затем гасят водой и нейтрализуют гидроксидом натрия (2 н.). Органический слой промывают водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография с использованием смеси этилацетата и гексана (1:1) дает 0,75 г (2,3 ммоль) диметиламида 5-(4,5,6, 7-тетрагидробензо[b]тиофен-2-илметил)имидазол-1-сульфоновой кислоты (5). Соединение (5) (0,42 г; 1,55 ммоль) переносят в 15 мл 1,5 н. раствора HCl и нагревают при температуре образования флегмы в течение 2 часов, а затем перемешивают при к.т. в течение ночи. Эту реакционную смесь разбавляют этилацетатом и нейтрализуют гидроксидом натрия (2 н.). Органический слой промывают водой, а затем рассолом. Органическую фазу сушат над сульфатом натрия и растворитель удаляют при пониженном давлении. Неочищенный продукт растворяют в метаноле и добавляют избыток HCl в эфире. Растворитель удаляют при пониженном давлении с получением 0,6 г (2,3 ммоль) 4(5)-(4,5,6,7-тетрагидробензо[b]тиофен-2-илметил)-1Н-имидазола (6)(Л-1).
1H-ЯМР (CD3OD): 8.80 (s, 1H); 7.34 (s, 1H); 6.57 (s, 1H); 4.18 (s, 2H); 2.65-2.69 (m, 2H); 2.51-2.55 (m, 2H); 1.74-1.83 (m, 4H).
Пример Л-2
В способе примера Л-1 использовали 2-(трет-бутил)фуран с получением 4(5)-(5-трет-бутилфуран-2-илметил)-1Н-имидазола.
Пример Л-3
В способе примера Л-1 использовали 5,6-дигидро-4Н-тиено[2,3-b]тиопиран с получением 4(5)-(5,6-дигидро-4Н-тиено[2,3-b]тиопиран-2-илметил)-1Н-имидазола.
Пример М
Методика получения 4(5)-(1-фуран-2-илэтил)-1Н-имидазола:
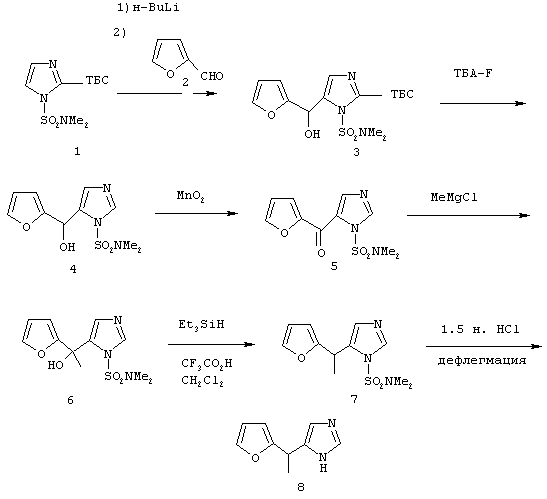
Процедура
2-(трет-Бутилдиметилсилил)-1-(диметилсульфамоил)имидазол (1) (3,3 г; 11, 4 ммоль) переносят в 75 мл безводного ТГФ и охлаждают до -78°С. К раствору соединения (1) по каплям добавляли н-BuLi (7,2 мл; 11,4 ммоль). Полученный раствор перемешивают при -78°С в течение 30 минут. К этой реакционной смеси добавляют 2-фурфурол (2) (0,94 мл; 11,4 ммоль). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. На следующий день реакционную смесь гасят насыщенным хлоридом аммония и разбавляют этилацетатом. Промывают органический слой водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография (4:1, этилацетат/гексан) дает 4,4 г (11,4 ммоль) диметиламида 2-(трет-бутилдиметилсилил)-5-(фуран-2-илгидроксиметил)имидазол-1-сульфоновой кислоты (3). Соединение (3) (4,4 г, 11,4 ммоль) переносят в 110 мл ТГФ и охлаждают до 0°С. К раствору соединения (3) по каплям добавляют 1 М раствор тетра-н-бутиламмонийфторида (ТБА-F) в ТГФ (11,4 мл, 11,4 ммоль). Реакционную смесь перемешивают при к.т. в течение ночи. На следующий день реакционную смесь гасят водой, а затем экстрагируют этилацетатом. Промывают органический слой водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Выделяют 3,9 г неочищенного диметиламида 5-(фуран-2-илгидроксиметил)имидазол-1-сульфоновой кислоты (4). Соединение (4) (1,0 г; 3,7 ммоль) переносят в 37 мл дихлорметана, к раствору добавляют 1,6 г (18,5 ммоль) диоксида магния. Реакционную смесь перемешивают в течение ночи, а затем фильтруют через целит. Собирают элюент и удаляют растворитель при пониженном давлении. Флэш-хроматография с использованием смеси этилацетата и гекасна (1:1) дает 0,69 г (2,6 ммоль) диметиламида 5-(фуран-2-илкарбонил)имидазол-1-сульфоновой кислоты (5). Соединение (5) (0,69 г; 2,6 ммоль) переносят в 26 мл ТГФ. Этот раствор охлаждают до -78°С. Добавляют 1,7 мл (5,1 ммоль) 3 М раствора метилмагнийхлорида. После перемешивания при -78°С в течение 1,5 часов реакционную смесь нагревают до к.т. и перемешивают еще 1 час. Реакционную смесь гасят водой, а затем экстрагируют этилацетатом. Промывают органический слой водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Кристаллизация из смеси эфир/гексан дает 0,39 г (1,4 ммоль) диметиламида 5-(1-фуран-2-ил-1-гидроксиэтил)имидазол-1-сульфоновой кислоты (6). Выделяют дополнительно 0,19 г соединения (6). Соединение (6) (0,58 г; 2,0 ммоль) переносят в 27 мл дихлорметана, к этому раствору добавляют 2,6 мл (16,3 ммоль) триэтилсилана и 5,5 мл (71,4 ммоль) трифторуксусной кислоты. Эту реакционную смесь перемешивают при к.т. в течение ночи и нейтрализуют твердым бикарбонатом натрия. Промывают органический слой водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении.
Флэш-хроматография с использованием смеси этилацетата и гексана (2:1) дает 0,53 г (2,0 ммоль) диметиламида 5-(1-фуран-2-илэтил)имидазол-1-сульфоновой кислоты (7). Соединение (7) переносят в 10 мл 1,5 н. раствора HCl и греют при температуре образования флегмы в течение 30 минут, а затем перемешивают при к.т. в течение ночи. Реакционную смесь разбавляют этилацетатом, а затем делают щелочной гидроксидом натрия (1 н.). Промывают органический слой водой, а затем рассолом. Сушат органическую фазу над сульфатом натрия и удаляют растворитель при пониженном давлении. Флэш-хроматография (10:1, хлороформ/метанол) дает 0,1 г (0,62 ммоль) 4(5)-(1-фуран-2-илэтил)-1Н-имидазола (8) (М).
1H-ЯМР (300 МГц, CDCl3): 7.56 (m, 1H), 7.33-7.34 (m, 1H), 6.81 (m, 1H), 6.29-6.31 (т, 1H), 6.06-6.07 (т, 1H), 4.22 (q, J=7.2 Гц, 1H), 1.63 (d, J=7.2 Гц, 3Н).
Пример Н
Методика получения 4(5)-(2,3-дигидробензо[1, 4]диоксин-6-илметил)-4-метил-1Н-имидазола:
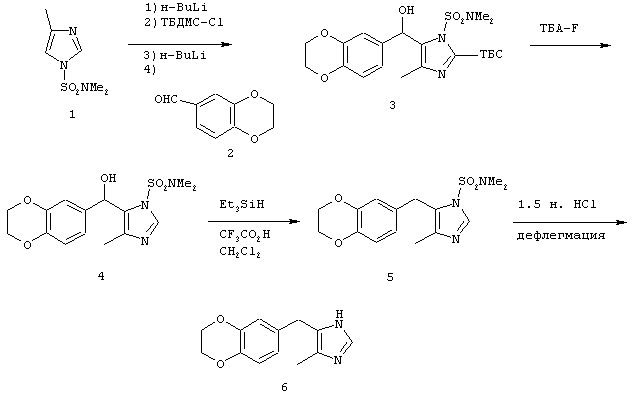
Процедура
4-Метил-1-(диметилсульфамоил)имидазол (1) (2,0 г; 10,6 ммоль) переносили в 42 мл безводного ТГФ и охлаждали до -78°С. К раствору соединения (1) по каплям добавляли н-BuLi (6,6 мл; 10,6 ммоль). Полученный раствор перемешивали при -78°С в течение 30 минут. К реакционной смеси добавляли тpeт-бутилдиметилсилилхлорид (ТБС-Сl) (1,6 г; 10,6 ммоль). Реакционную смесь нагревали до к.т. и перемешивали в течение ночи. На следующий день реакционную смесь охлаждали до -20°С и добавляли н-BuLi (7,3 мл; 11,6 ммоль). После перемешивания при -20°С в течение 30 минут к реакционной смеси добавляли 1,4-бензодиоксан-6-карбоксальдегид (2) (1,92 г; 11,7 ммоль) в 10 мл ТГФ. Затем реакционную смесь нагревали до к.т. и перемешивали в течение 3 часов. Реакционную смесь гасили водой и разбавляли этилацетатом. Промывали органический слой водой, а затем рассолом. Сушили органическую фазу над сульфатом натрия и удаляли растворитель при пониженном давлении. Флэш-хроматография (1:2, этилацетат/гексан) давала 3,9 г (8,4 ммоль) диметиламида 2-(трет-бутилдиметилсилил)-5-[(2,3-дигидробензо[1,4]диоксин-6-ил)гидроксиметил]-4-метилимидазол-1-сульфоновой кислоты (3). Соединение (3) (1,0 г; 2,14 ммоль) переносили в 21 мл ТГФ. К раствору соединения (3) по каплям добавляли 1 М раствор тетра-н-бутиламмонийфторида (ТБА-F) в ТГФ (2,35 мл, 2,35 ммоль). Реакционную смесь перемешивали при к.т. в течение 30 минут. Реакционную смесь гасили водой, а затем экстрагировали этилацетатом. Промывали органический слой водой, а затем рассолом. Сушили органическую фазу над сульфатом натрия и удаляли растворитель при пониженном давлении. Флэш-хроматография с использованием этилацетата в качестве элюента давала 0,75 г (2,12 ммоль) диметиламида 5-[(2,3-дигидробензо[1, 4]диоксин-6-ил)гидроксиметил]-4-метилимидазол-1-сульфоновой кислоты (4). Соединение (4) (0,75 г; 2,12 ммоль) переносили в 28 мл дихлорметана и добавляли к этому раствору 2,7 мл (17 ммоль) триэтилсилана и 5,2 мл (67,8 ммоль) трифторуксусной кислоты. Эту реакционную смесь перемешивали в течение ночи при к.т., а затем гасили водой и нейтрализовали твердым бикарбонатом натрия. Органический слой промывали водой, а затем рассолом. Сушили органическую фазу над сульфатом натрия и удаляли растворитель при пониженном давлении. Флэш-хроматография с использованием смеси этилацетата и гекасна (3:1) давала 0,63 г (1,87 ммоль) диметиламида 5-(2,3-дигидробензо[1,4]диоксин-6-илметил)-4-метилимидазол-1-сульфоновой кислоты (5). Соединение (5) (0,63 г; 1,87 ммоль) переносили в 10 мл 1,5 н. раствора НСl и грели при температуре образования флегмы. Эту реакционную смесь разбавляли этилацетатом и нейтрализовали твердым бикарбонатом натрия. Промывали органический слой водой, а затем рассолом. Сушили органическую фазу над сульфатом натрия и удаляли растворитель при пониженном давлении. Кристаллизация из смеси эфир/гексан давала 0,33 г (1,43 ммоль) 4(5)-(2,3-дигидробензо[1, 4]диоксин-6-илметил)-4-метил-1Н-имидазола (6) (Н).
1H-ЯМР (300 МГц, ацетон-d6): 7.37 (s, 1H); 6.66-6.67 (m, 3Н); 4.18 (s, 4H), 3.73 (s, 1H),2.13 (s, 3H).
Пример О
Методика получения 2-(3Н-имидазол-4(5)-илметил)-3,4,5,6,7,8-гексагидро-2Н-нафталин-1-она (O-1), 4(5)-(2,3,4,4а,5,6,7, 8-октагидронафталин-2-илметил)-1Н-имидазола (O-2) и 4(5)-(1,2,3,4,5,6,7,8-октагидронафталин-2-илметил)-1Н-имидазола (О-3):
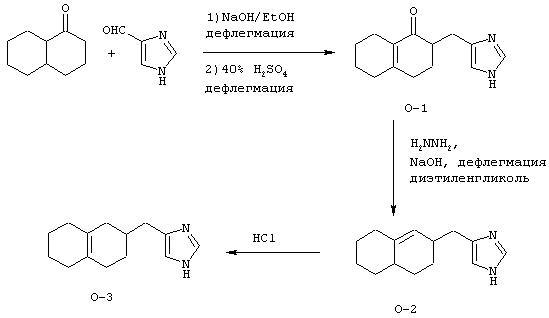
Процедура
К 100 мл этанола добавляли 1-декалон (10,6 г; 66 ммоль) и 4(5)-имидазолкарбоксальдегид (6,3 г; 66 ммоль). К этому раствору добавляли NaOH (5,2 г; 130 ммоль) в 20 мл воды. Реакционную смесь грели при температуре образования флегмы в течение 5 дней. Реакционную смесь охлаждали до к.т. и делали щелочной водным HCl. Этот раствор экстрагировали смесью ТГФ/этилацетат. Объединяли органические слои и промывали рассолом. Органическую фазу сушили над сульфатом магния и растворитель удаляли при пониженном давлении с получением неочищенного продукта. Этот неочищенный продукт в течение 1 дня нагревали в 40% H2SO4 при температуре образования флегмы. Реакционную смесь охлаждали до к.т. и делали щелочной насыщенным К2СО3. Этот раствор экстрагировали смесью ТГФ/этилацетат. Объединяли органические слои и промывали рассолом. Органическую фазу сушили над сульфатом магния и растворитель удаляли при пониженном давлении. Очистка флэш-хроматографией (15:1, СН3Сl/МеОН) давала O-1 (4,9 г; выход 32%).
1H-ЯМР: 7.55 (s, 1H), 6.77 (s, 1H), 3.08-3.14 (m, 2H), 1.52-2.46 (m, 13H).
С использованием NaOH из гидрохлоридной соли O-1 получали свободное основание и добавляли его к диэтиленгликолю (100 мл). К этому раствору добавляли гидразингидрат (3,2 мл; 100 ммоль) и оставляли реакционную смесь на ночь перемешиваться при к.т. Добавляли NaOH (3,1 г; 77 ммоль) и грели раствор при температуре образования флегмы в течение 5 дней. Эту реакционную смесь охлаждали до к.т. и разбавляли водой. Этот раствор экстрагировали смесью ТГФ/этилацетат. Объединяли органические слои и промывали рассолом. Органическую фазу сушили над сульфатом магния и растворитель удаляли при пониженном давлении. Очистка флэш-хроматографией (8:1, СН3Сl/МеОН) давала O-2 (0,64 г; выход 27%).
1H-ЯМР: 7.58 (s, 1H), 6.76 (s, 1H), 5.24 (d, J=4.3 Гц, 1H), 0.91-2.58 (m, 16H).
Соединение O-2 (1,0 г; 4,6 ммоль) добавляли к 10 мл концентрированной HCl. Этот раствор перемешивали при к.т. в течение 30 мин, а затем нейтрализовали карбонатом калия. Раствор экстрагировали смесью ТГФ/этилацетат. Объединяли органические слои и промывали рассолом. Органическую фазу сушили над сульфатом магния и растворитель удаляли при пониженном давлении. Очистка флэш-хроматографией (15:1, СН3Сl/МеОН) давала O-3.
1H-ЯМР: 7.54 (s, 1H), 6.74 (s, 1H), 2.45-2.52 (m, 3Н), 1.46-1.97 (m, 14H).
Пример П
Методика получения 4(5)-октагидропентален-2-илметил)-1Н-имидазола гидрохлорида:
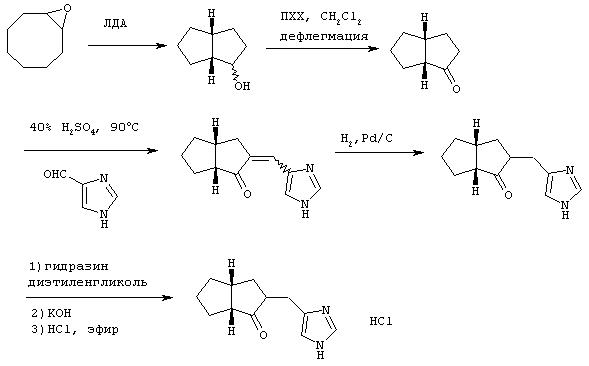
Процедура
А. Следуя синтезу Уайта и Уайтселла (White and Whitesell, Synthesis, pp. 602-3, 1975), в просушенную пламенем колбу, охлажденную до 0°С и находящуюся в атмосфере аргона, добавляли эфир (10 мл). Затем медленно добавляли н-бутиллитий (35 мл 2,5 М раствора в гексане; 2,2 экв.), а затем диизопропиламин (14 мл, 2,5 экв.) и смесь оставляли перемешиваться при 0°С в течение 30 минут. К этому полученному раствору диизопропиламида лития добавляли оксид циклооктена (5,0 г; 1,0 экв.). Смесь перемешивали при к.т. в течение одного дня, а затем нагревали до температуры образования флегмы в атмосфере аргона в течение 2 дней. Реакционную смесь гасили добавлением NH4Cl. Раствор экстрагировали смесью ТГФ/ЕtOАс. Объединяли органические экстракты, промывали их рассолом, сушили над сульфатом магния и концентрировали с получением желто-коричневого масла, которое представляло собой 1-гидроксиоктагидропентален. Это соединение использовали на следующей стадии без дополнительной очистки.
Б. Полученный таким образом спирт (5,0 г; 1 экв.) растворяли в дихлорметане (200 мл), добавляли к этому раствору хлорхромат пиридиния (13 г; 1,5 экв.) и перемешивали смесь при к.т. в течение одного дня. Затем фильтровали этот раствор через короткую колонку с SiO2, используя в качестве элюента диэтиловый эфир. Полученный раствор концентрировали в вакууме с получением бледного желто-зеленого масла, которое использовали на следующей стадии без дополнительной очистки.
В. Октагидропентален-1-он (5,0 г; 1,0 экв.) из предыдущей стадии добавляли к 4(5)-имидазолкарбоксальдегиду (3,8 г; 1,0 экв.) и 40% Н2SO4 (20 мл), эту смесь выдерживали при 90°С в течение 3 дней. Затем гасили реакцию добавлением гидроксида аммония и экстрагировали реакционную смесь смесью тетрагидрофуран/этилацетат. Объединяли органические экстракты, промывали их рассолом и сушили над сульфатом магния. Полученный органический слой нейтрализовали смесью HCl/NH4Cl. Повторно экстрагировали водный слой, как описано выше, и концентрировали объединенные органические фракции в вакууме с получением оранжевого твердого вещества.
Г. Это оранжевое твердое вещество растворяли в этаноле, к которому был добавлен палладий на углероде (0,5 г). Реакционную колбу на один день помещали под водород при давлении 40 фунтов на квадратный дюйм (≈). Реакционный раствор фильтровали через целит с дополнительным этанолом в качестве элюента. Раствор концентрировали в вакууме с получением желто-коричневого масла. Очистка колоночной хроматографией с использованием смеси хлороформ/метанол (17:1) давала продукт, кетон, в несколько загрязненном виде.
Д. Кетонную функциональную группу удаляли добавлением продукта с предыдущей стадии (8,2 г; 1,0 экв.) к диэтиленгликолю (80 мл) и гидразингидрату (13,0 г; 1,0 экв.). Эту смесь перемешивали в течение ночи, а затем добавляли гидроксид калия (1,0 г; 5,0 экв.) и нагревали раствор при температуре образования флегмы в течение одного дня. Реакционный раствор охлаждали до к.т. и промывали водой. Раствор экстрагировали смесью ТГФ/ЕtOАс, объединенные фракции промывали рассолом, сушили над сульфатом магния и концентрировали с получением желтого масла. Соль, моногидрохлорид, получали растворением этого масла в безводном этаноле, насыщенном НСl, и нагреванием.
Пример Р
Методика получения 7-(3Н-имидазол-4(5)-илметил)-6,7-дигидро-5Н-изохинолин-8-она (Р-1) и 7-(3Н-имидазол-4(5)-илметил)-5,6,7,8-тетрагидроизохинолина (Р-2):

Процедура
А. 3,4-Лютидин (21,44 г; 1 экв.) растворяли в 200 мл воды при 20°С и добавляли перманганат калия порциями по 6,32 г дважды в день в течение 5 дней (всего 63,2 г; 2 экв.). Через 5 дней раствор выдерживали в морозильнике, затем размораживали и фильтровали через целит. Полученный бесцветный раствор концентрировали при 90°С в роторном испарителе до получения белого твердого вещества. Это твердое вещество перекристаллизовывали из 5 н. НСl с получением 9,56 г белых кристаллов. ЯМР показал наличие смеси двух региоизомеров, причем основным продуктом был желаемый изомер.
Б. Эти кристаллы в течение 6 часов нагревали в безводном этаноле, насыщенном НСl, под аргоном и при температуре образования флегмы. Затем этанол удаляли из раствора на роторном испарителе, остаток переносили в 100 мл воды и доводили рН до 7-8 твердым бикарбонатом натрия. Трижды экстрагировали водную фазу диэтиловым эфиром, органические фракции промывали рассолом, сушили над сульфатом магния, а затем фильтровали и концентрировали с получением бесцветного масла (5,36 г; выход 10,8%).
В. К н-BuLi (11,21 мл; 1,3 экв.) в 100 мл безводного ТГФ под аргоном при -78°С добавляли через шприц диизопропиламин (2,84 г; 1,3 экв.) с получением in situ диизопропиламида лития. К этому раствору шприцом добавляли продукт вышеописанной стадии Б (3,56 г; 1 экв.) в 20 мл тетрагидрофурана и перемешивали смесь при -78°С в течение 20 минут. В этот момент добавляли по каплям через канюлю метилакрилат (4,85 мл; 2,5 экв.) в 20 мл тетрагидрофурана. Раствор перемешивали еще 2 часа, а затем гасили добавлением 40 мл 10% ацетата калия. Раствору давали нагреться до 20°С, а затем концентрировали его на роторном испарителе. Водный остаток трижды экстрагировали хлороформом. Объединенные фракции промывали рассолом и сушили над сульфатом магния, фильтровали и концентрировали до черного твердого вещества, которое хранили под глубоким вакуумом. Хроматография на силикагеле со смесью гексаны/этилацетат (7/3→6/4) давала 2,41 г (58,2%) желаемого продукта, который использовали на следующей стадии без дополнительной очистки.
Г. Вещество со стадии В (0,48 г; 1 экв.) растворяли в 1 мл 6 М HCl и грели при 105°С в течение 16 часов, после чего концентрировали раствор на роторном испарителе при 80°С до твердого вещества. Остаток переносили в 2 мл воды и нейтрализовали твердым бикарбонатом натрия. Нейтрализованный раствор трижды экстрагировали хлороформом, объединенные фракции промывали рассолом, сушили над сульфатом магния и концентрировали до бесцветного масла (0,456 г; 93,4%).
Д. Изохинолон (1,91 г; 1 экв.), полученный на стадии Г выше, нагревали с 4(5)-имидазолкарбоксальдегидом (1,25 г; 1 экв.) при 110°С в 15 мл 40% серной кислоты в течение 30 часов. Эту реакционную смесь хранили в течение нескольких дней при 0°С под аргоном. Затем раствор разбавляли водой (20 мл) и подщелачивали до рН 8,9 гидроксидом аммония. Твердые вещества собирали фильтрацией и сушили с использованием высокого вакуума. Продуктом было желтое твердое вещество (2,81 г; 96,1%), состоящее из обоих позиционных изомеров по экзо-двойной связи.
Е. Продукт стадии Д выше растворяли в 150 мл метанола и к этому раствору добавляли Pd/C (0,412 г; 0,15 масс. экв.). Этот метанольный раствор затем насыщали водородом посредством многократных откачек и повторных закачек водорода. Раствор перемешивали под водородом при давлении 1 атм в течение 20 часов, пока ТСХ не выявляла, что исходного ненасыщенного вещества не осталось. Этот раствор фильтровали через целит и концентрировали до масла. Хроматография на диоксиде кремния с использованием дихлорметана и метанола (9:1) дала чистый продукт (1,835 г; 65,04%) в виде белой пены. Ее переносили в метанол, к которому была добавлена фумаровая кислота (0,4817 г; 1,5 экв.), при нагревании с целью растворения твердых веществ. Медленно охлаждали этот раствор и получали не совсем белые кристаллы (0,826 г; 74%), которые представляли собой соединение Р-1. Соединение Р-2 получали путем восстановления гидразином таким же образом, как это описано в стадии Д примера П выше.
Пример С
Методика получения (Z)-6-(3Н-имидазол-4(5)-илметилен)-7,8-дигидро-6Н-хинолин-5-она (С-1), (Е)-6-(3Н-имидазол-4(5)-илметилен)-7,8-дигидро-6Н-хинолин-5-она (С-2), 6-(3Н-имидазол-4(5)-илметил)-7,8-дигидро-6Н-хинолин-5-она (С-3), дигидрохлорида 6-(3Н-имидазол-4(5)-илметил)-5,6,7,8-тетрагидрохинолина (С-4) и 6-(3Н-имидазол-4(5)-илметил)октагидрохинолин-5-она (С-5)
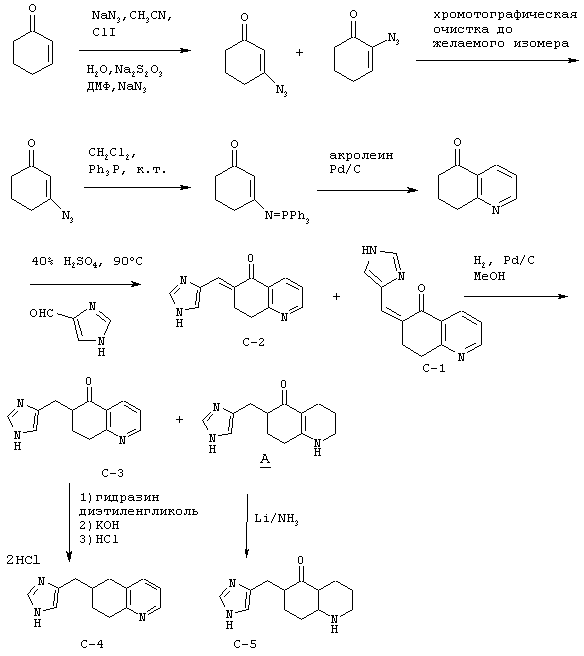
Процедура
А. Реактивный азидореагент первой стадии образовывался in situ при добавлении по каплям через капельную воронку монохлорида йода (67,6 г; 1,15 экв.) в 50 мл ацетонитрила к перемешиваемой взвеси азида натрия (58,84 г; 2,5 экв.) в 350 мл безводного ацетонитрила при -10°С под аргоном. Добавление завершали через 30 минут, смесь перемешивали еще 30 минут и к ней шприцом добавляли циклогексенон (34,81 г; 1,0 экв.), а затем перемешивали при 20°С еще 20 часов. Затем вливали эту смесь в литр воды и экстрагировали тремя порциями по 200 мл диэтилового эфира. Объединенные фракции промывали 5% раствором тиосульфата натрия, а затем рассолом. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали в вакууме при 20°С. Остаток переносили в 1 л диметилсульфоксида при 0°С, добавляли вторую порцию NаN3 и перемешивали смесь, подогревая до температуры окружающей среды. Затем разбавляли эту смесь ледяной водой (2,5 л) и десять раз экстрагировали дихлорметаном (10×250 мл). Концентрировали объединенные органические фракции на роторном испарителе до объема около 1 л и экстрагировали этот концентрат тремя порциями по 250 мл воды, затем рассолом, а затем сушили над сульфатом магния и концентрировали до темного масла (39,5 г) и хранили его при -40°С.
Это масло очищали хроматографией на диоксиде кремния, используя смесь гексана и этилацетата (градиент от 9:1 до 8:2). Выделяли два изомера, причем первый с азидогруппой в положении α относительно кетонной функциональной группы получали с выходом 13,22 г, 26,6%. β-Изомер получали с выходом 15,825, 32,0%.
Б. Трифенилфосфин растворяли в 20 мл дихлорметана и помещали в атмосферу аргона при 20°С. К этому перемешиваемому раствору добавляли через канюлю β-изомер, полученный, как описано выше, и выдерживали в течение 2 часов при 20°С. По мере протекания реакции из раствора выделялся азот, и после 2 часов ТСХ показала, что исходного материала не осталось. Этот раствор концентрировали и пропускали через колонку с силикагелем, используя в качестве элюента градиент от дихлорметана до смеси дихлорметана с метанолом в соотношении 95/5. Амидофосфонатный промежуточный продукт получали с выходом 2,139 г, 65,1%.
В. Амидофосфонат растворяли в 100 мл безводного о-ксилола, а затем при перемешивании добавляли 10% Pd/C. Затем к этой смеси шприцом добавляли свежеперегнанный акролеин и в течение 4 часов нагревали до температуры образования флегмы, после чего добавляли остальной акролеин и продолжали нагревание при температуре образования флегмы в течение 44 часов под пальцевым дефлегматором под аргоном. В этой точке ТСХ показывала, что остается некоторое количество промежуточного продукта, поэтому добавляли еще 0,5 г Pd/C и снова в течение 8 часов нагревали смесь до температуры образования флегмы. Смесь охлаждали до к.т. и концентрировали на роторном испарителе для удаления избытка акролеина, пока не оставалось около 100 мл о-ксилольного раствора. Этот раствор охлаждали добавлением льда и трижды экстрагировали 1 н. соляной кислотой. Объединенные водные фракции трижды экстрагировали Et2O. Затем водную фазу охлаждали до 0°С и доводили рН до примерно 10, используя концентрированный NaOH. Затем водную фазу экстрагировали пять раз по 100 мл хлороформа. Объединенные хлороформные фракции промывали водой, а затем рассолом и сушили над сульфатом магния, фильтровали и, наконец, концентрировали с получением 3,51 г 7,8-дигидро-6Н-хинолин-5-она в виде масла с выходом 84,4%.
Г. 4(5)-Имидазолкарбоксальдегид конденсировали с этим хинолиноном, как описано в стадии Д примера Р, и получали как С-1, так и С-2.
Д. Экзо-двойную связь затем восстанвливали палладием на углероде, как описано в стадии Е примера П выше, с получением двух продуктов, которые разделяли хроматографией с получением С-3 и А.
Е. Кетогруппу удаляли такой же процедурой восстановления гидразином, которая описана выше в стадии Д примера П, с получением С-4.
Ж. Продукт С-5 с полностью восстановленным хинолиновым кольцом получали стандартным восстановлением соединения А литием/аммиаком (Li, 10 экв., в NН3 при -78°С в течение 10 минут, гашеный NH4OH, постепенный нагрев с выпариванием NН3).
Пример Т-1
Методика получения (Е)-6-(3Н-имидазол-4(5)-илметилен)-7,8-дигидро-6Н-хиноксалин-5-она
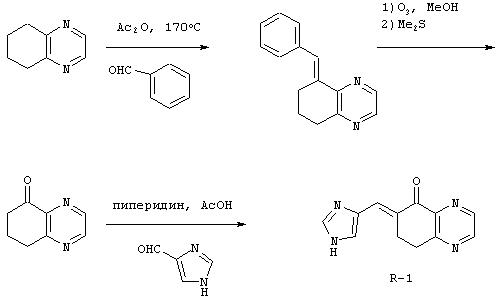
Процедура
А. Смесь 5,6,7,8-тетрагидрохиноксалина (23,75 г, 1 экв.), бензальдегида (19,81 мл; 1,1 экв.) и уксусного ангидрида (33,4 мл; 2,0 экв.) перемешивали при 150°С под аргоном в течение 15 часов, после чего ТСХ показывала наличие главным образом желаемого продукта с остатками некоторого количества исходных веществ. Исходные вещества удаляли вакуумной дистилляцией, используя колонку Vigreux, при 170°С. Затем остаток подвергали дистилляции в шариковом дистилляторе при температуре от 170 до 220°С. Первая фракция была слегка загрязнена исходными веществами (4,71 г). Вторая фракция была чистой (18,9 г). После применения высокого вакуума к первой фракции она кристаллизовалась. Суммарный выход фракций составил 20,11 г (51%).
Б. Продукт из вышеуказанной стадии А растворяли в 100 мл метанола и слегка подогревали, затем охлаждали до температуры от -35 до -40°С и через этот раствор пропускали озон. Через несколько минут исходное вещество начинало выкристаллизовываться из раствора, раствор подогревали и добавляли к нему еще 200 мл метанола, а затем возобновляли реакцию. Примерно через 30 минут раствор становился бледно-голубым. Затем в него вводили азот пропусканием через раствор в течение 30 минут, а затем впрыскивали в этот раствор метилсульфид (3,5 мл), после чего перемешивали раствор еще 30 минут при -30°С, после чего давали ему нагреться при перемешивании до температуры окружающей среды. После примерно 48 часов при 20°С эту смесь перегоняли с паром для удаления растворителей с получением остатка в виде 8,4 г желто-коричневого масла. Этот остаток переносили в диэтиловый эфир и экстрагировали тремя порциями по 25 мл 1 н. HCl. Объединенные водные фракции трижды промывали диэтиловым эфиром. Водный раствор постепенно подщелачивали до рН 8 концентрированным NaOH. Затем свободный амин трижды экстрагировали из водной фазы хлороформом. Объединенные хлороформные экстракты дважды промывали рассолом, сушили над MgSO4 и концентрировали до желтого масла (3,01 г). После выдерживания под высоким вакуумом в течение 1 часа оставалось 2,97 г. Это количество перекристаллизовывали из диэтилового эфира с получением 2,35 г ярко-желтого твердого вещества. Выход 67,5%.
В. 7,8-Дигидрохиноксалин-5-он и 4(5)-имидазолкарбоксальдегид (Aldrich Chemicals) суспендировали в 75 мл безводного тетрагидрофурана при 20°С под аргоном с последующим добавлением пиперидина, а затем уксусной кислоты. Эту смесь перемешивали в течение 16 часов при 20°С. Через 20 часов, как показывала ТСХ, следов хиноксалона не оставалось. Твердые вещества собирали фильтрацией и промывали небольшим количеством тетрагидрофурана, а затем хлороформа. Твердые вещества сушили под высоким вакуумом с получением 6,85 г Т-1. Выход 90,3%.
Пример Т-2 и Т-3
Так же, как для получения Т-1, 5,6,7,8-тетрагидроизохинолин (5,42 г; 1 экв., Aldrich) перемешивали с бензальдегидом (5,182 г; 1,2 экв.) и уксусным ангидридом (6,309 г; 2,0 экв.) с последующей дистилляцией в вакууме и использовали на следующей стадии без дополнительной очистки. Выход (неочищенного) 8,28 г.
Неочищенный продукт (7,96 г) из вышеуказанной стадии подвергали озонолизу, как описано выше в стадии Б. После обработки и хроматографии получали 5, 18 г бледного масла. Выход 97,8%, если исходить из того, что исходное вещество чистое.
Полученный 7,8-дигидро-6Н-изохинолин-5-он (1,692 г, 1 экв.) конденсировали с 4(5)-имидазолкарбоксальдегидом, как описано выше в стадии В, с получением 2,23 г ненасыщенного соединения, аналогичного соединению Т-1 на указанной выше схеме, с выходом 92,8%. Этот продукт обрабатывали палладием на углероде, как описано в стадии Е примера Р, с целью восстановления экзо-двойной связи с получением 6-(3Н-имидазол-4(5)-илметил)-7,8-дигидро-6Н-изохинолин-5-она (Т-2) с выходом 52%.
Указанный выше кетон восстанавливали гидразином и превращали в соль, фумарат, как подробно изложено в примере Р, стадия Е. Выход при восстановлении 62%. Выход фумарата после перекристаллизации 30,4% 6-(3Н-имидазол-4(5)-илметил)-5,6,7,8-тетрагидроизохинолина (Т-3).
Пример У
Способ измерения селективности α-агониста включает в себя анализ на основе технологии селекции и амплификации рецептора (ТСАР), как описано в Messier et al. (1995). "High throughput assays of cloned adrenergic, muscarinic, neurokinin and neurotrophin receptors in living mammalian cells", Pharmacol. Toxicol. 76:308-11, адаптированный для использования с альфа2-рецепторами. Анализ позволяет измерять опосредованную рецепторами потерю контактного ингибирования, которое приводит к селективной пролиферации клеток, содержащих рецепторы, в смешанной популяции конфлюэнтных клеток. Увеличение числа клеток оценивают с использованием соответствующего трансфецированного гена-маркера, такого как β-галактозидаза, активность которой легко можно измерить в планшете на 96-лунок. Эту реакцию вызывают рецепторы, которые активируют белок G, Gq. Альфа2-рецепторы, которые обычно связываются с Gi, активируют ТСАР-реакцию, когда ко-экспрессируются с гибридным белком Gq, у которого есть домен распознавания Gi рецептора, называемый G [Conklin et al. (1993) "Substitution of three amino acids switches receptor specificity of Gqa to that of Сi а." Nature 363:274-6].
Высевают клетки NIH-3Т3 при плотности 2×106 клеток в 15-сантиметровые чашки и выдерживают в модифицированной по Дульбекко среде Игла с добавлением 10% телячьей сыворотки. Спустя один день клетки ко-трансфецируют преципитацией фосфата кальция плазмидами для экспрессии у млекопитающих, кодирующими p-SV-β-галактозидазу (5-10 мг), рецептор (1-2 мг) и белок G (1-2 мг). В смесь для трансфекции может быть включено также 40 мг ДНК спермы лосося. На следующий день и через 1-2 дня добавляют свежую среду, клетки собирают и замораживают аликвотами на 50 анализов. В планшетах на 96 лунок клетки оттаивают и их добавляют по 100 мкл к аликвотам по 100 мкл лекарственных средств разных концентраций, каждая в трех экземплярах. Инкубационные периоды продолжаются 72-96 часов при 37°С. После промывания забуференным фосфатом физиологическим раствором определяют ферментативную активность β-галактозидазы путем добавления 200 мл хромогенного субстрата (состоящего из 3,5 мМ о-нитрофенил-β-D-галактопиранозида и 0, 5% нонидета Р-40 в забуференном фосфатом физиологическом растворе), инкубирования в течение ночи при 30°С и измерения оптической плотности при 420 нм. Оптическое поглощение является мерой ферментативной активности, которая зависит от числа клеток и отражает опосредованную рецептором пролиферацию клеток. Определяют EC50 и максимальный эффект каждого лекарственного средства на каждый альфа2-рецептор. Эффективность или внутреннюю активность вычисляют как отношение максимального эффекта лекарственного средства к максимальному эффекту стандартного полного агониста для рецептора каждого подтипа. В качестве стандартного агониста для рецепторов альфа2A и альфа2C используют бримонидин, называемый также UK14,304-18. Оксиметазолин является стандартным агонистом, используемым для альфа2B-рецептора.
В приведенной ниже таблице 1 представлены значения внутренней активности в отношении подтипов α 2-адренорецепторов, как они определены ТСАР-анализом для соединений приведенных выше примеров от Б до Т и некоторых адренергических соединений, не имеющих активности селективных агонистов по отношению к подтипу(ам) α2В или α2В/С. По отношению к подтипу α2А соединения этих примеров не активны или показывают низкую эффективность (≤0,4). Они имеют более высокую эффективность по отношению к подтипам α2В и α2С, чем к подтипу α2А. Поэтому в отличие от офтальмологических α2-адренергических соединений, таких как клонидин и бримонидин, соединения примеров от Б до Т могут селективно активировать α2-адренорецепторы
Пример Ф
Снижение внутриглазного давления (ВГД) и седативные побочные эффекты
ВГД измеряли у находящихся в полном сознании самок обезьян cynomolgus весом 3-4 кг с устойчивым повышенным ВГД, которое вызывали в правом глазе фотокоагуляцией трабекулярной сети посредством аргонового лазера. Животные были пригодны для экспериментов примерно в течение 2 месяцев после операции. Во время экспериментов обезьяны сидели в креслах специальной конструкции (Primate Products, San Francisco) и когда нужно их кормили апельсиновым соком и фруктами. Для измерения ВГД использовали пневмоманометр Digilab, модель 30 R (Alcon, Texas).
Перед измерениями ВГД каждой обезьяне местно наносили 25 мкл анестетика (пропаракаина), чтобы минимизировать вызываемый тонометрией дискомфорт в глазу. Перед инстилляцией делали два измерения исходного давления, после чего делали периодические измерения в течение срока до 6 часов после инстилляции. Тестируемые соединения вводили в один глаз одной каплей объемом 50 мкл, другой глаз получал равный объем физиологического раствора.
На обезьянах были испытаны многие α2В- или α2В/С-селективные соединения приведенных примеров. Неожиданно оказалось, как показывает таблица 2, что все эти структурно разные соединения снижают ВГД в обработанном глазе.
Одновременно измеряли и оценивали седативное действие согласно следующей системе баллов: 0 = оживленное состояние, типичные звуки, движения и т.п.; 1 = спокойное состояние, меньше движений; 2 = слабо седативное состояние, издаются некоторые звуки, есть реакции на стимуляцию; 3 = седативное состояние, звуки не издаются, есть некоторые реакции на стимуляцию; 4 = сон.
Соединения по настоящему изобретению также не оказывали седативного действия. Это контрастирует с действием клонидина и бримонидина, которые оказывают седативное действие.
Таблица 2. Воздействие агонистов α2-адренорецепторов на ВГД и на седативное состояние у находящихся в сознании обезьян cynomolgus после введения в глаза, в одном из которых повышенное давление вызвано фотокоагуляцией посредством аргонового лазера. Седативное состояние оценивали субъективно во время экспериментов по ВГД, используя следующую систему баллов: 0 = оживленное состояние, типичные звуки, движения и т.п.; 1 = спокойное состояние, меньше движений; 2 = слабо седативное состояние, издаются некоторые звуки, есть реакции на стимуляцию; 3 = седативное состояние, звуки не издаются, есть некоторые реакции на стимуляцию; 4 = сон. Число животных в группе = (6-9).
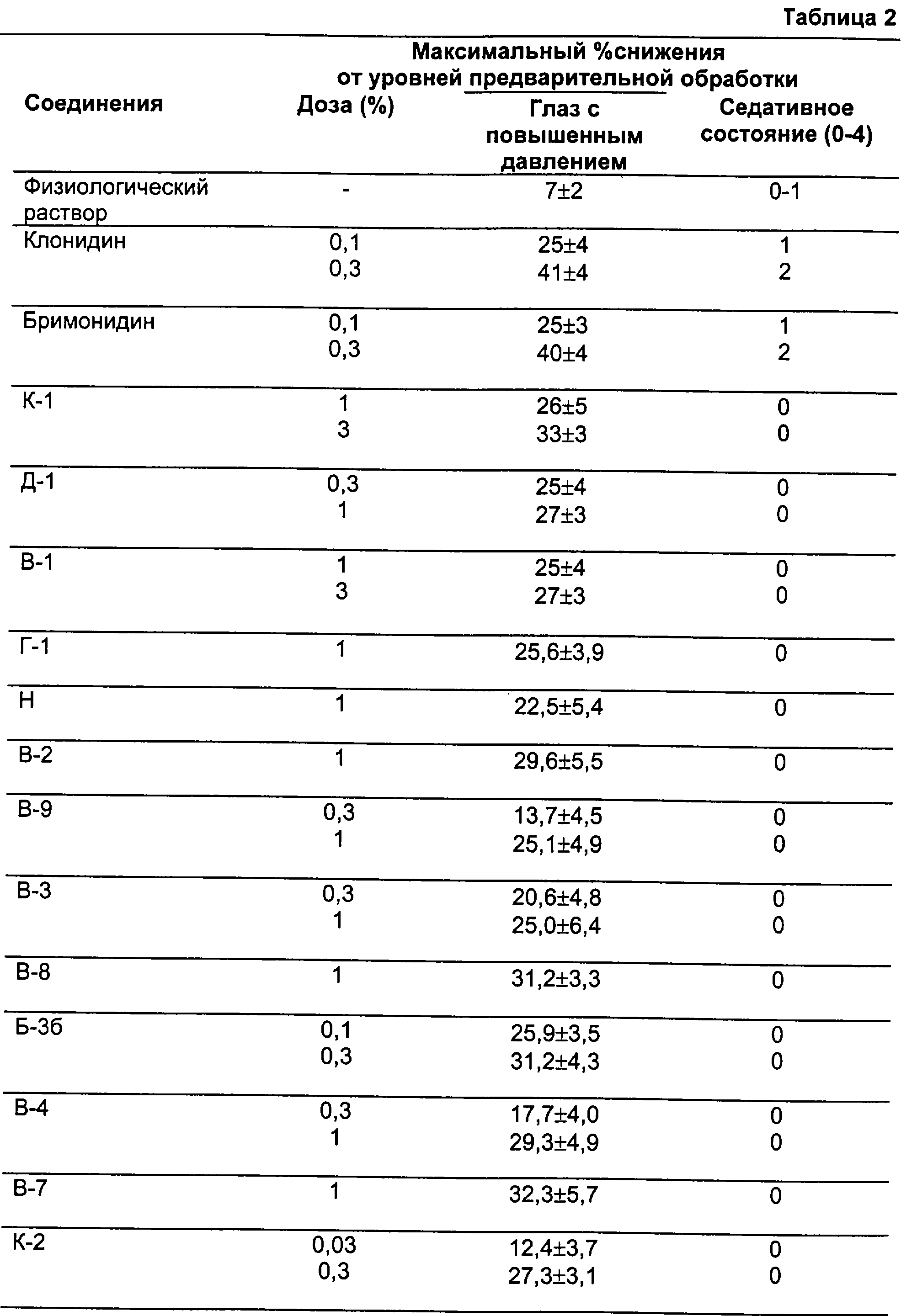

Пример Х
Измерение сердечно-сосудистых побочных эффектов
Сердечно-сосудистые измерения делали в разных группах обезьян, используя автоматический сфигмоманометр ВР 100 S (Nippon Colin., Япония). Внутривенное (в.в.) введение некоторых соединений по настоящему изобретению в дозах, которые в 10-30 раз превышают дозы для клонидина и бримонидина, не снижали частоту сердечных сокращений или не понижали давление крови. Интересно, что соединение 4(5)-3-метилтиофен-2-илметил)-1Н-имидазол, которое имеет внутреннюю активность 0,43 по отношению к α2А-подтипу, оказывало слабое действие на частоту сердечных сокращений. Клонидин и бримонидин оказывали еще более сильные эффекты на частоту сердечных сокращений. Смотри таблицу 3 ниже.
Таблица 3. Воздействие агонистов α2-адренорецепторов на сердечно-сосудистые показатели у находящихся в сознании обезьян cynomolgus после в.в. ведения. Измерения делали периодически в течение срока до 6 часов. Число животных на группу = (6-10).
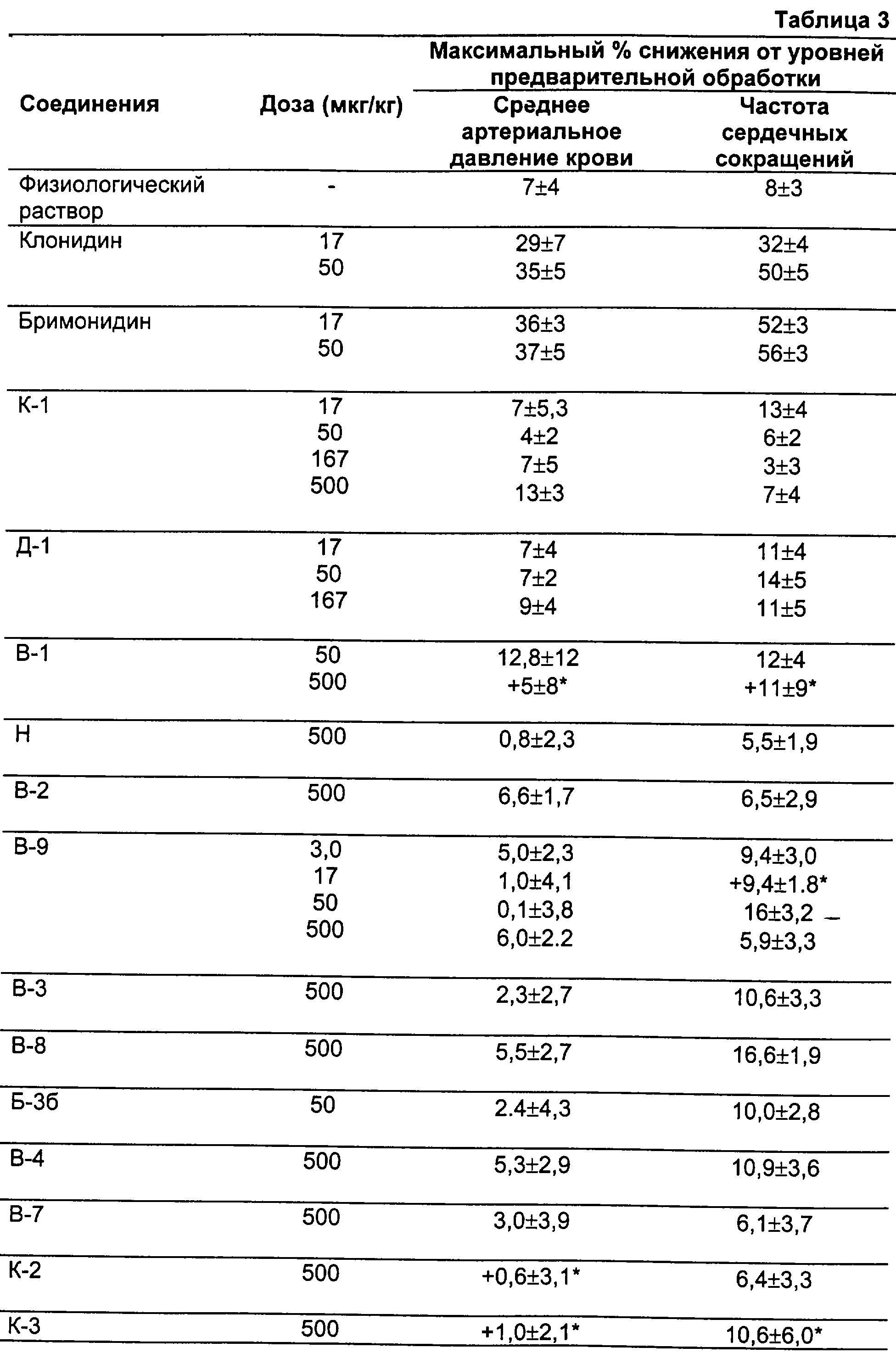

ПРИМЕР Ц
Исследования в вышеуказанных примерах Ф и Х показывают, что терапевтический эффект альфа2-агонистов может иметь место отдельно от седативного и сердечно-сосудистого побочных эффектов. Такое отделение осуществляют с использованием соединений, общим свойством которых является то, что они активны преимущественно по отношению к рецепторам подтипов альфа2В и альфа2В/альфа2С по сравнению с рецепторами подтипа альфа2А.
Альфа2-адренергические агонисты, известные из уровня техники, которые активируют все три подтипа альфа2-рецепторов, оказывают седативное действие, гипотензию и брадикардию, что препятствует или сильно ограничивает их применение для лечения заболеваний и расстройств, которые, как известно, облегчаются этими агонистами. Такие заболевания и расстройства включают в себя мышечную спастичность, в том числе гиперактивное мочеиспускание, диарею, диурез, синдромы отмены, боль, в том числе невропатическую боль, нейродегенеративные заболевания, в том числе глазную невропатию, ишемию спинного мозга и инсульт, дефицит памяти и познавательной способности, дефицит внимания, психозы, в том числе маниакальные расстройства, тревожность, депрессию, гипертензию, застойную сердечную недостаточность, ишемию сердца и назальную гиперемию [Hieble et al. "Therapeutic application of agents interacting with alpha-adrenoreceptors, in Alpha-adrenoreceptors: molecular biology, biochemistry and pharmacology". Prog. Basic Clin. Pharmacol. (Basel, Karger) 8, pp. 180-220 (1991)]. Например, было показано, что клонидин клинически эффективен в обезболивании при послеоперационной, связанной с раком и нейрогенной боли. Но, как было установлено [Maze M.B. and Tranquilli W. "Alpha-2 adrenoreceptor agonists: defining the role in clinical anesthesia". Anesthesiology 74, 581-605 (1991)], "полная клиническая перспективность" этого и других альфа2-агонистов требует разработки соединений, которые не оказывают седативного действия, гипотензии и брадикардии.
Перечисленные выше заболевания и расстройства можно лечить путем активации рецепторов подтипов α2В или α2В/С. Поэтому альфа2-соединения, описанные выше, которые, как было показано, не оказывают седативного действия и сердечно-сосудистых эффектов, пригодны и обладают преимуществом в лечении этих состояний.
Еще одним примером новой полезности соединений по изобретению является улучшение, касающееся нейрональной дегенерации при глаукомной невропатии. Недавние исследования показали, что клонидин и другие альфа2-агонисты являются нейропротективными по отношению к клеткам сетчатки в нескольких моделях нейрональной дегенерации у крыс. Эти модели включают в себя вызываемую светом дегенерацию фоторецепторов у белых крыс, как описано в Wen et al., "Alpha2-adrenergic agonists induce basic fibroblast growth factor expression in photoreceptors in vivo and ameliorate light damage." J.Neurosci. 16, 5986-5992, и калиброванное повреждение зрительного нерва, которое приводит к вторичной потере клеток ганглионов сетчатки, как описано в Yoles et al., "Injury-induced secondary degeneration of rat optic nerve can be attenuated by alpha-2-adrenergic agonists AGN 191103 and brimonidine". Invest. Ophthalmol. Vis. Sci. 37, 540, S114. Однако в отличие от соединений по настоящему изобретению дозы, использованные в этих исследованиях (от 0,1 до >1 мг/кг при внутрибрюшинном и внутримышечном введении) также оказывают седативное действие и сердечно-сосудистые эффекты. Чувствительным показателем активации альфа2-рецепторов в сетчатке считается индукция экспрессии основного фактора роста фибробластов (оФРФ) (см. Wen et al. выше), и измерение индукции оФРФ после местного введения альфа2-агонистов в глаза крысы показывает, что примерно 1% доза необходима для того, чтобы индуцировать увеличение уровней оФРФ в 2-3 раза, что соответствует опосредованному альфа2-агонистами нейропротекторному действию (см. Wen et al. выше и Lai et al., "Neuroprotective effect of ocular hypotensive agent brimonidine", in Proceedings of XIth Congress of the European Society of Ophthalmology (Bologna, Monduzzi Editore), 439-444). Известно, что эти местные дозы существующих альфа2-агонистов, таких как клонидин, приводят к системным побочным эффектам, таким как седативное состояние и гипотензия, которые будут препятствовать их применению в качестве глазных нейропротективных агентов. Кроме того, применение некоторых неселективных α2-адренергических агентов в лечении нейронального повреждения раскрыто и заявлено в находящейся на совместном рассмотрении заявке 08/496292, поданной 28 июня 1995 г, содержание которой во всей полноте включено сюда ссылкой.
Соединения по настоящему изобретению не оказывают седативного действия и сердечно-сосудистых эффектов у обезьян после местного введения в дозах по меньшей мере 3%. Таким образом, можно достичь нейропротективных концентраций этих соединений у людей, не вызывая побочных эффектов. Фактически, как указано ниже, было показано, что соединение примера Б-9(б) является нейропротективным в модели калиброванного повреждения зрительного нерва у крысы, которая описана в указанной выше работе Yoles et al. См. таблицу 4 ниже.

Этот уровень нейрозащиты сравним с эффектом, который наблюдали в предыдущих исследованиях со стандартным агонистом альфа2-адренорецепторов, бримонидином, и нейропротективным агентом МК801.
Пример Ч
Облегчение боли, в том числе невропатической боли, которая является еще одним примером расстройства, при котором полезны и имеют преимущества соединения по данному изобретению, поскольку боль облегчается без нежелательных побочных эффектов. Клонидин, агонист, который активирует все три подтипа альфа2-рецепторов, применяли для лечения хронической боли клинически, но его применимость по этому показанию ограничена, поскольку он оказывает седативные и сердечно-сосудистые побочные эффекты. Соединения по настоящему изобретению сравнивали с клонидином и бримонидином в модели невропатической боли у грызунов, которая, как известно, дает возможность прогнозировать клиническую активность [Kim S. And Chung J. "An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat." Pain 50, pp. 355-363 (1992)]. После перевязки двух спинно-мозговых нервов у животных развивалась чувствительность к обычно не болевым стимулам, таким как прикосновение. Способность соединений, являющихся альфа2-агонистами, обращать эту чувствительность, называемую аллодинией, проверяли через 30 минут после введения дозы в мозговые оболочки или в брюшную полость. Также измеряли седативную активность каждого соединения, используя камеру для определения активности.
Соединения по данному изобретению, представленные в качестве примера соединением O-1, способны облегчать аллодинию, не оказывая седативного действия даже при очень высоких дозах. Это контрастирует с клонидином и бримонидином, которые оказывают седативное действие в дозах, которые лишь немого выше их антиаллодинийных доз. См. таблицы 5 и 6 ниже.


Результаты этих примеров демонстрируют, что обычные побочные эффекты α2-адреноцепторных лекарственных средств опосредованы рецепторами подтипа α2А и что их действие против повышенного внутриглазного давления и другие терапевтические действия могут быть опосредованы подтипом, иным чем α2А. Таким образом, α 2-адреноцепторные соединения неродственных структурных классов, которые имеют общую низкую функциональную активность по отношению к рецепторам подтипа α2А, снижают ВГД и оказывают другие терапевтические эффекты без ограничивающих дозу побочных эффектов.
Пример фармацевтической композиции по изобретению
Раствор, %:
Активное соединение
(5-фенилсульфанил-1Н-имидазол
в виде белого кристаллического вещества) 2
Физиологический раствор
(0,9% раствор NaCl в воде) 98
Активное соединение растворяют в физиологическом растворе и наливают в стерильную тару.
Несмотря на то, что конкретные воплощения данного изобретения были описаны, должно быть понятно, что изобретение ими не ограничено, поскольку возможны многие очевидные модификации, и это изобретение будет включать в себя любую такую модификацию, которая будет подпадать под объем прилагаемой формулы.
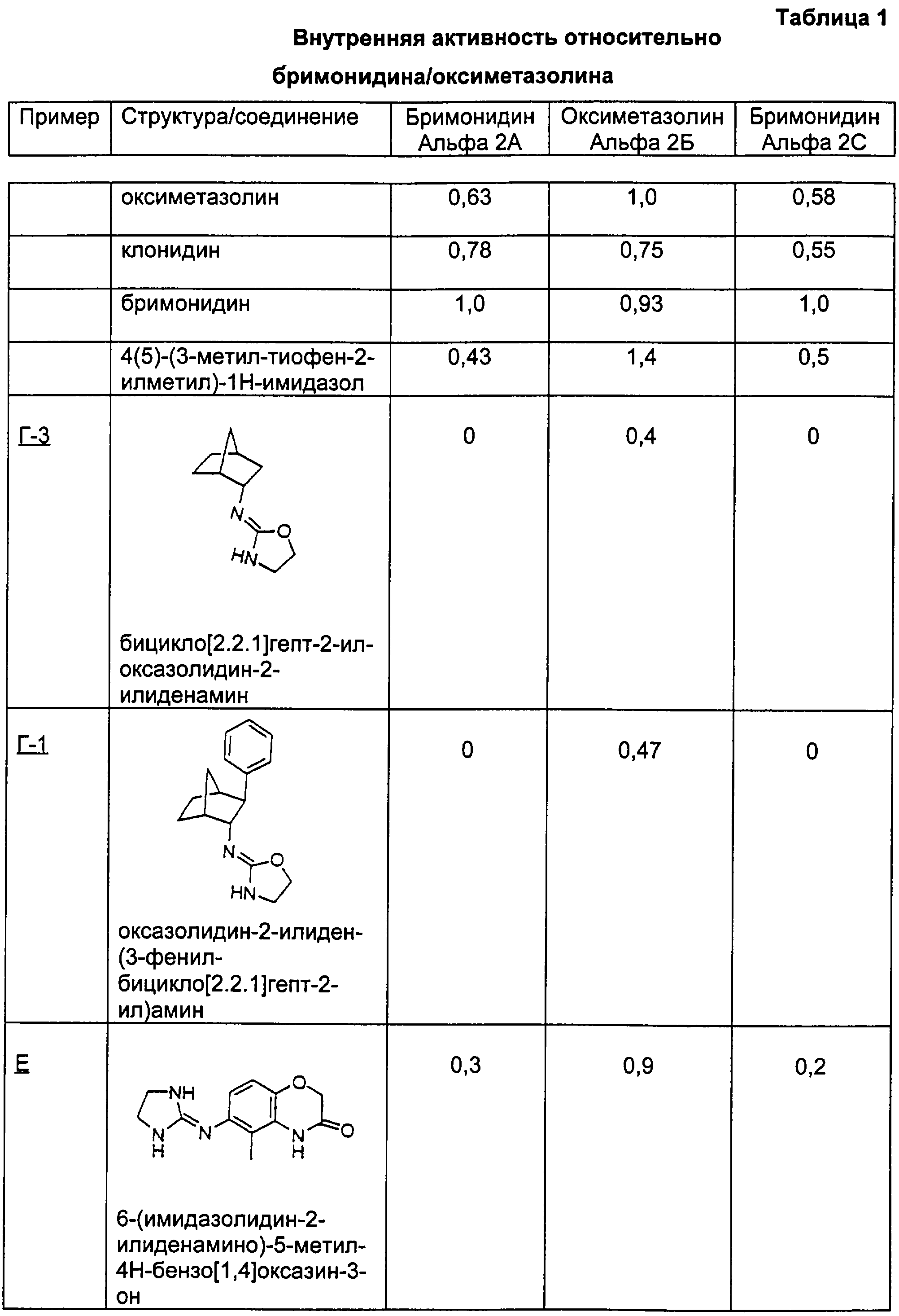
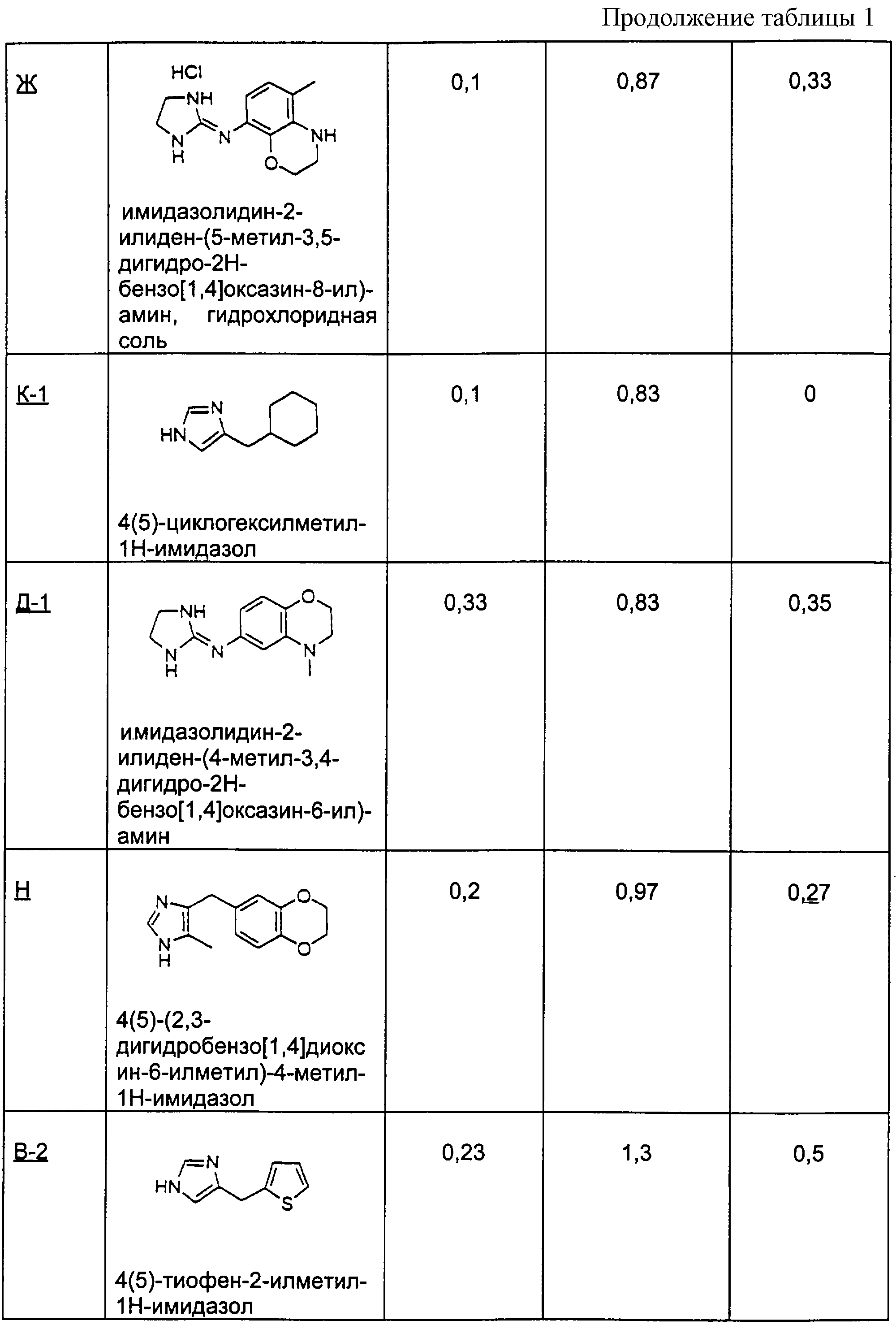
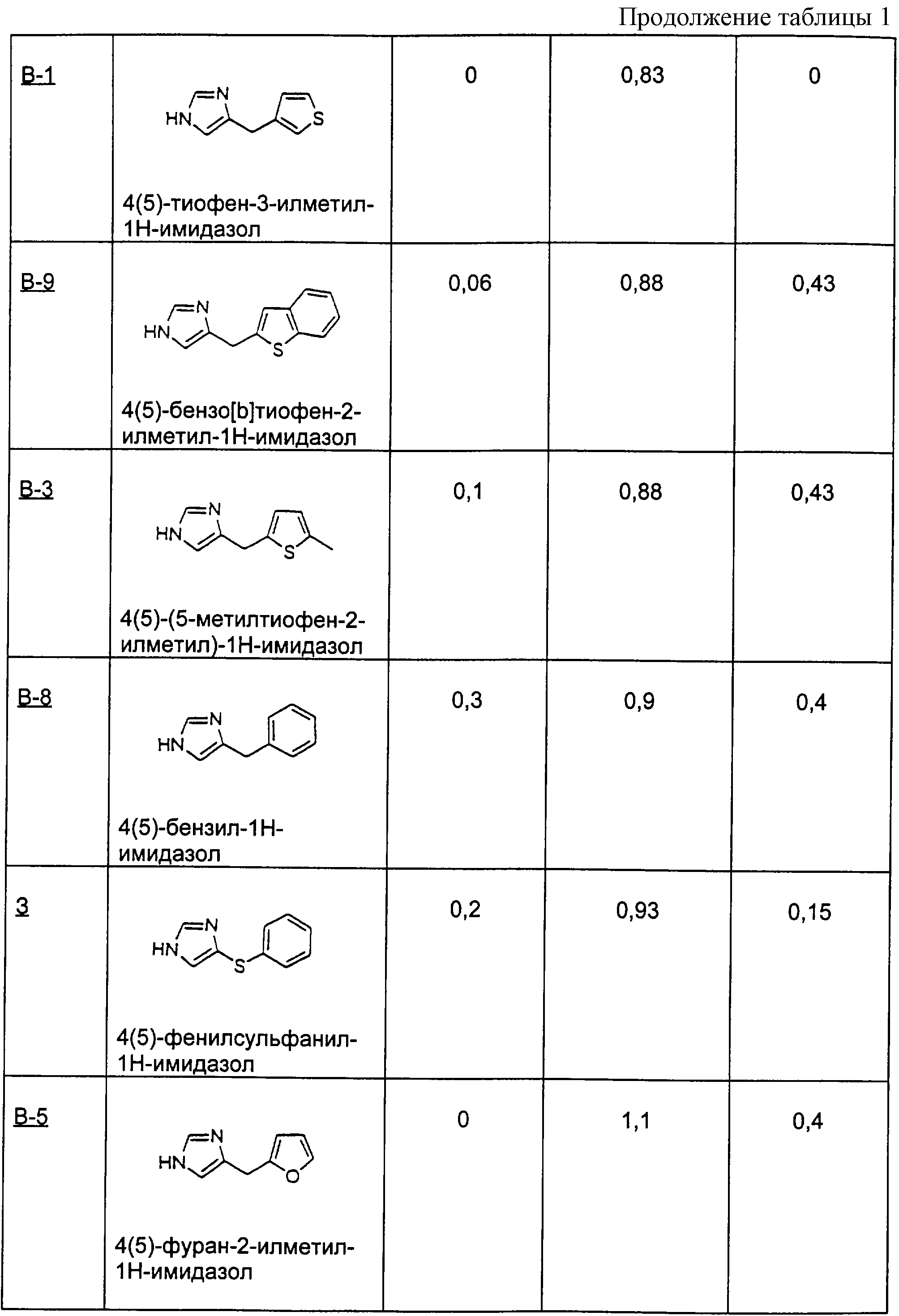
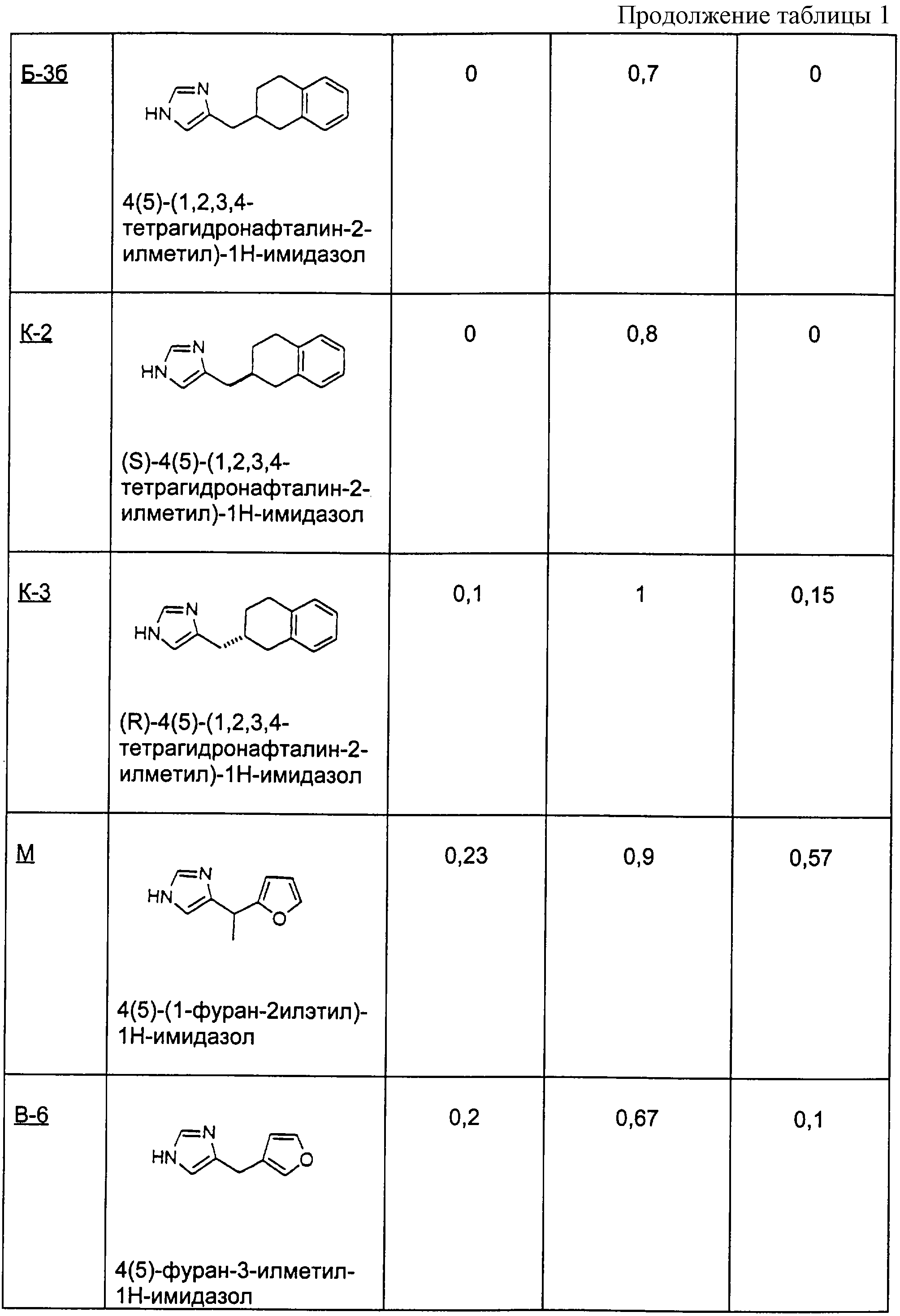
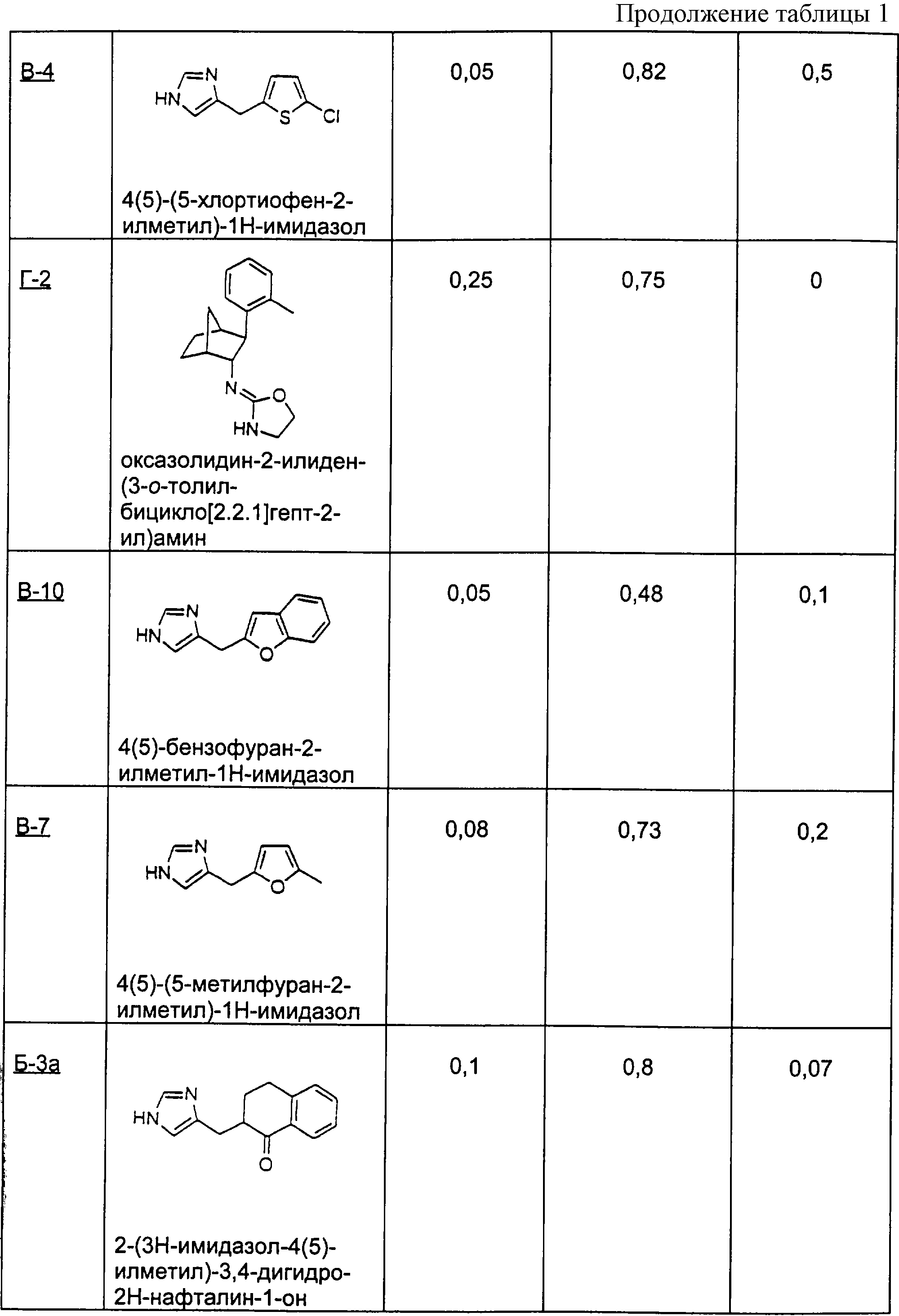

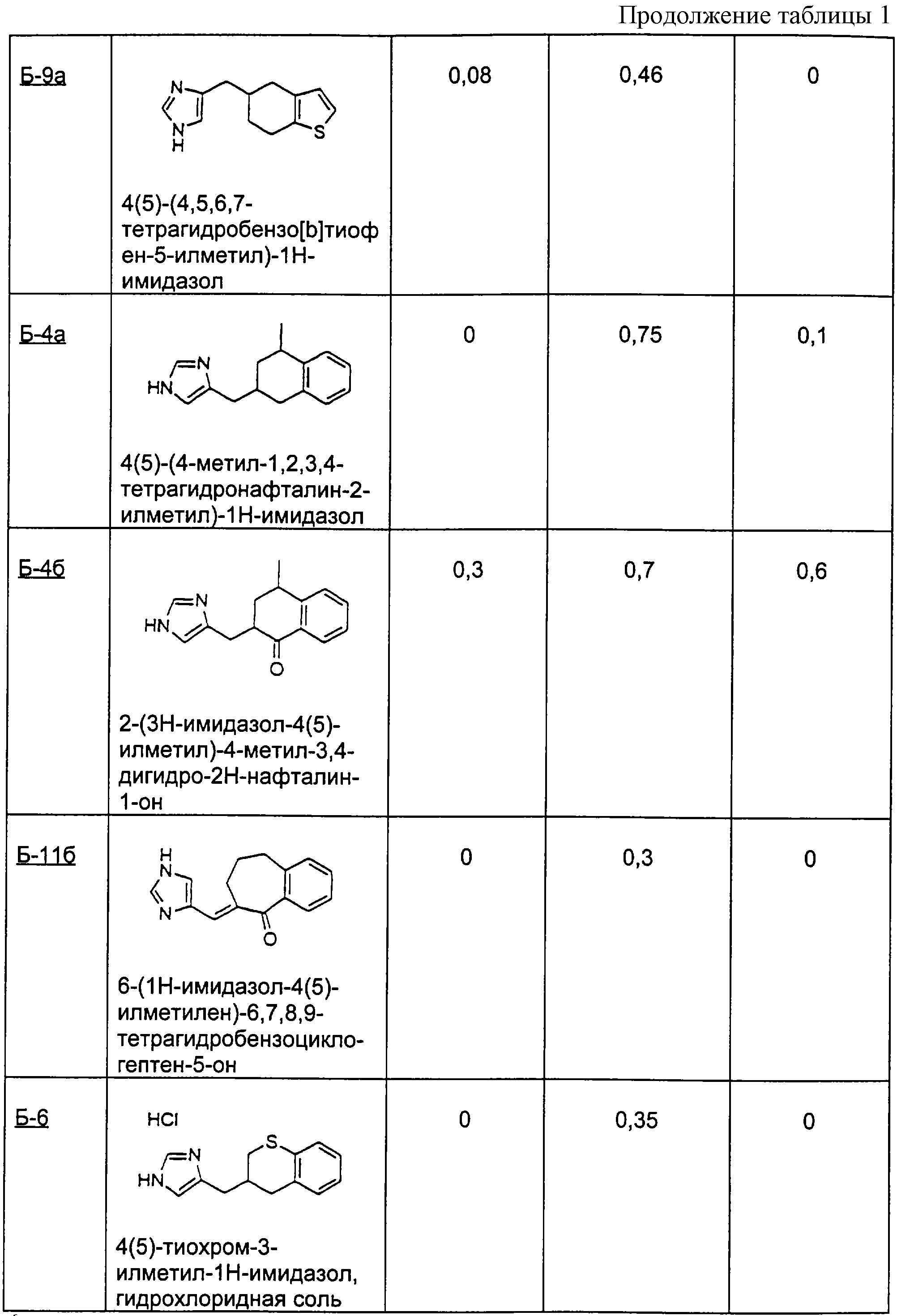
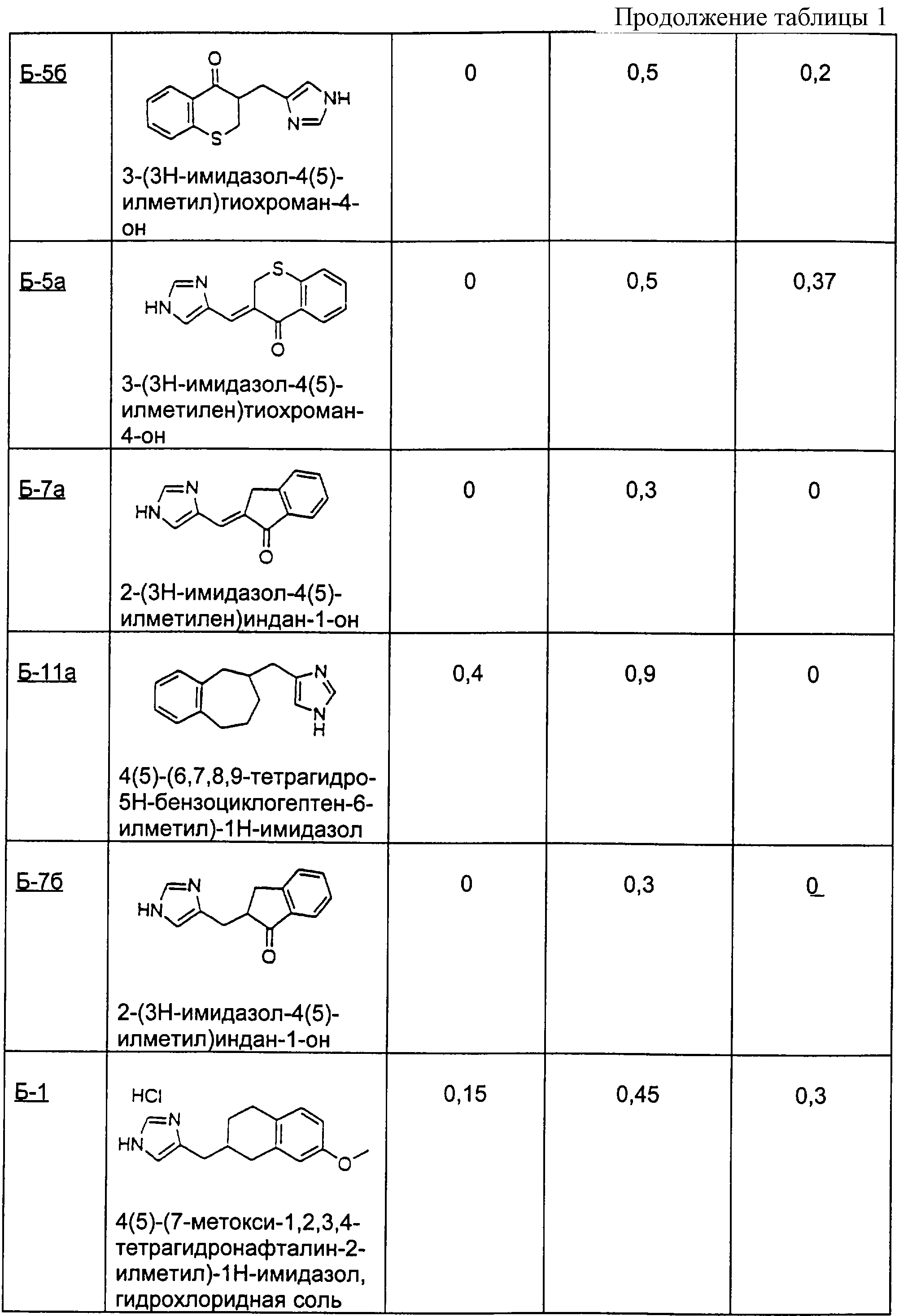
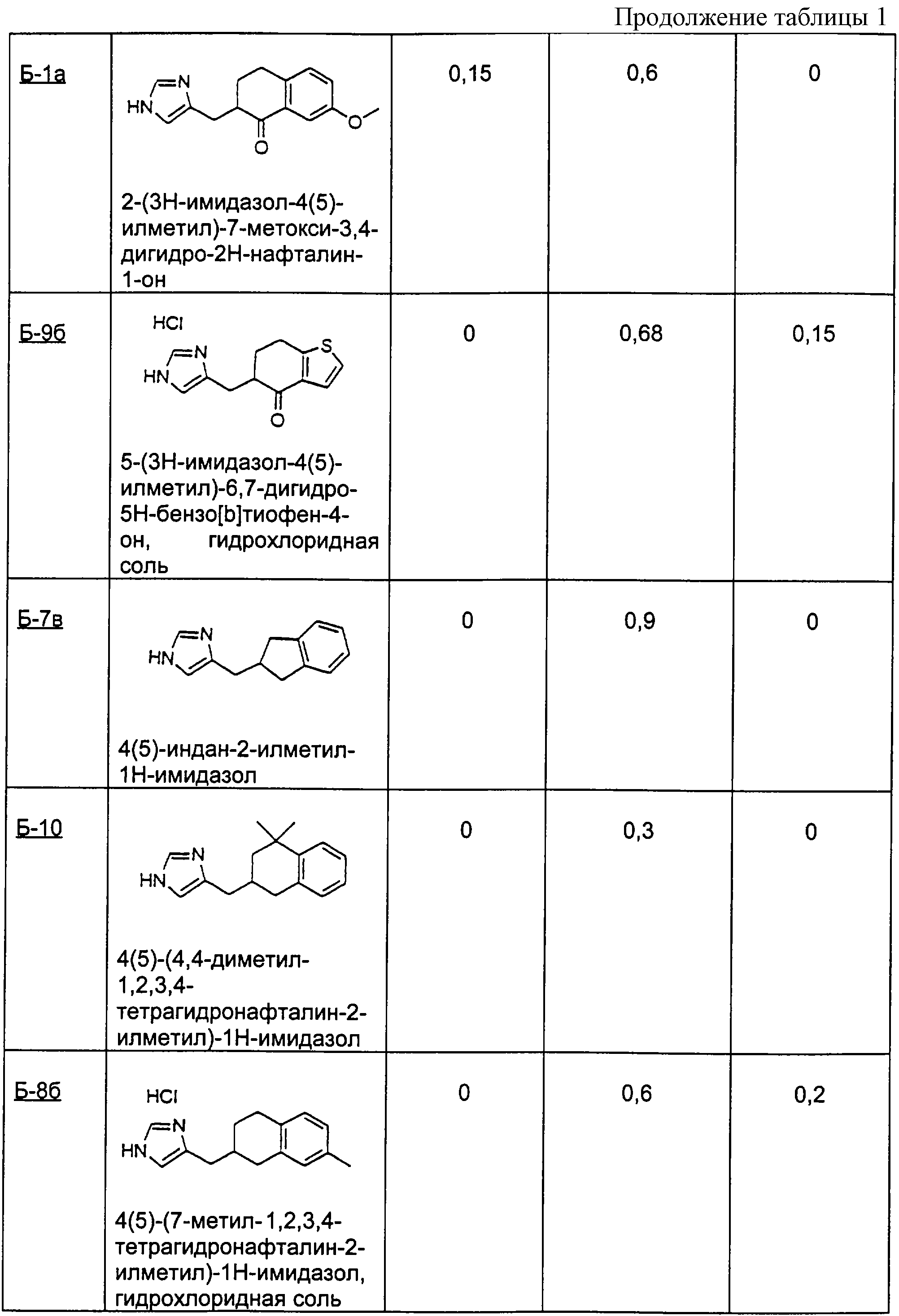
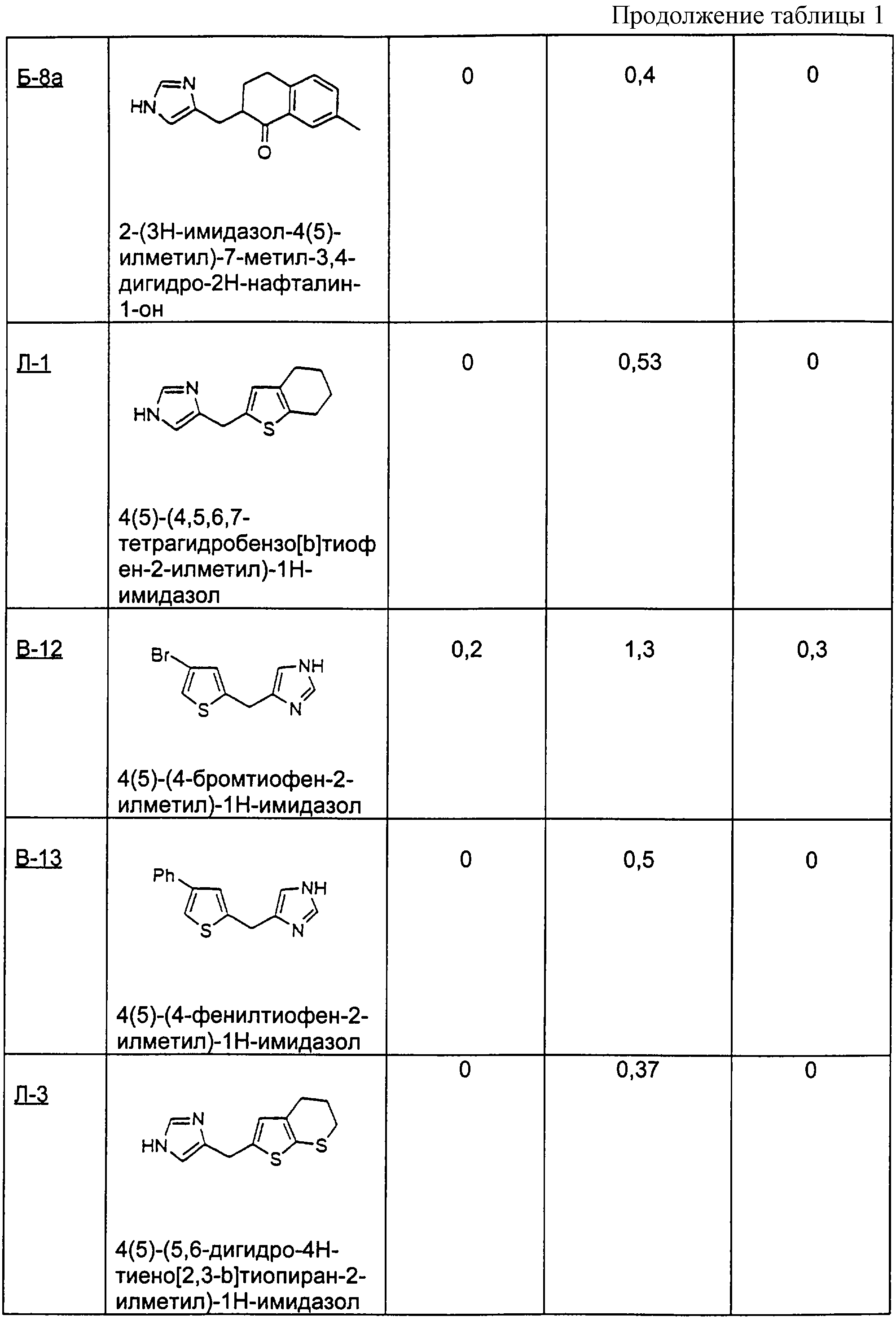
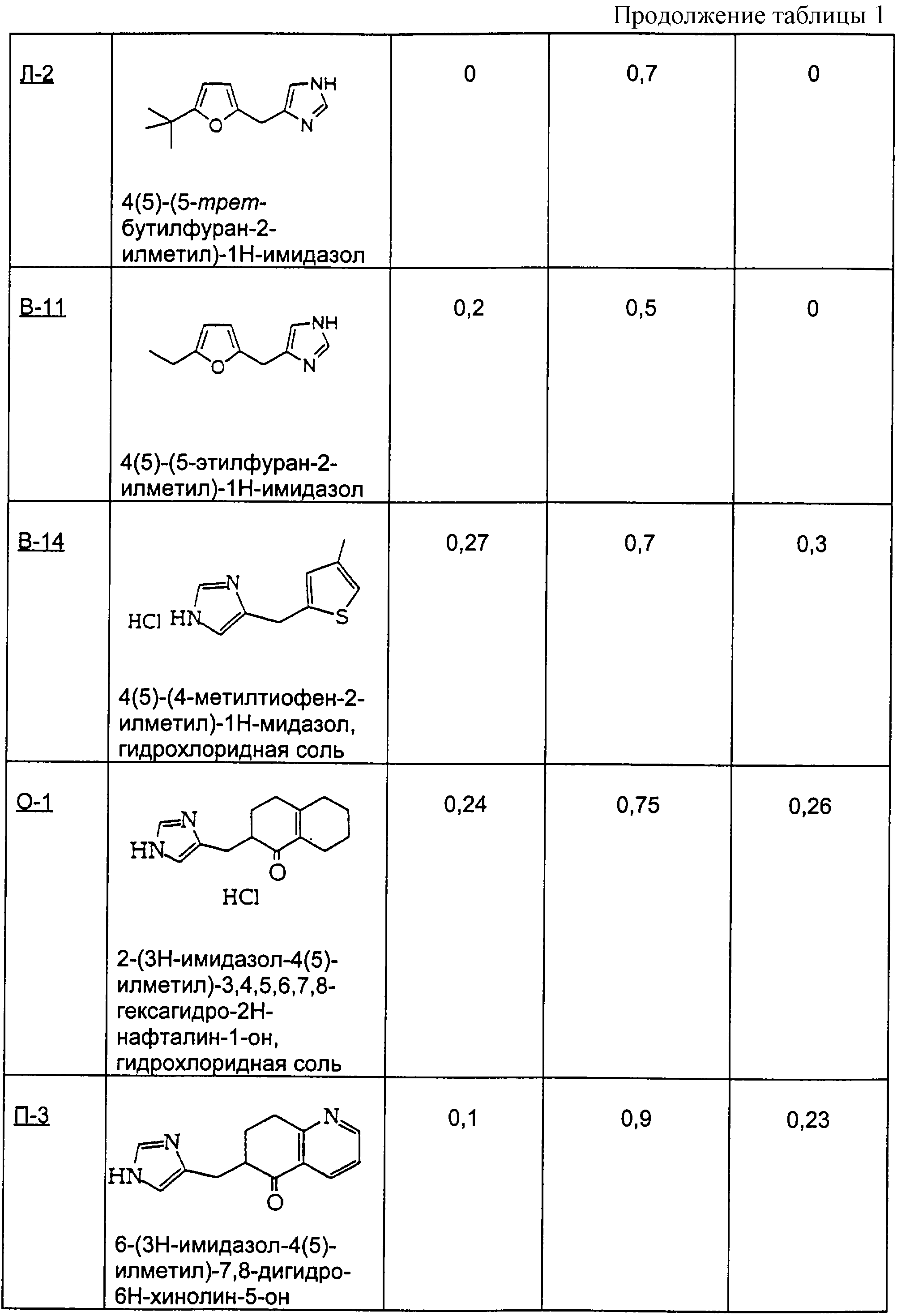
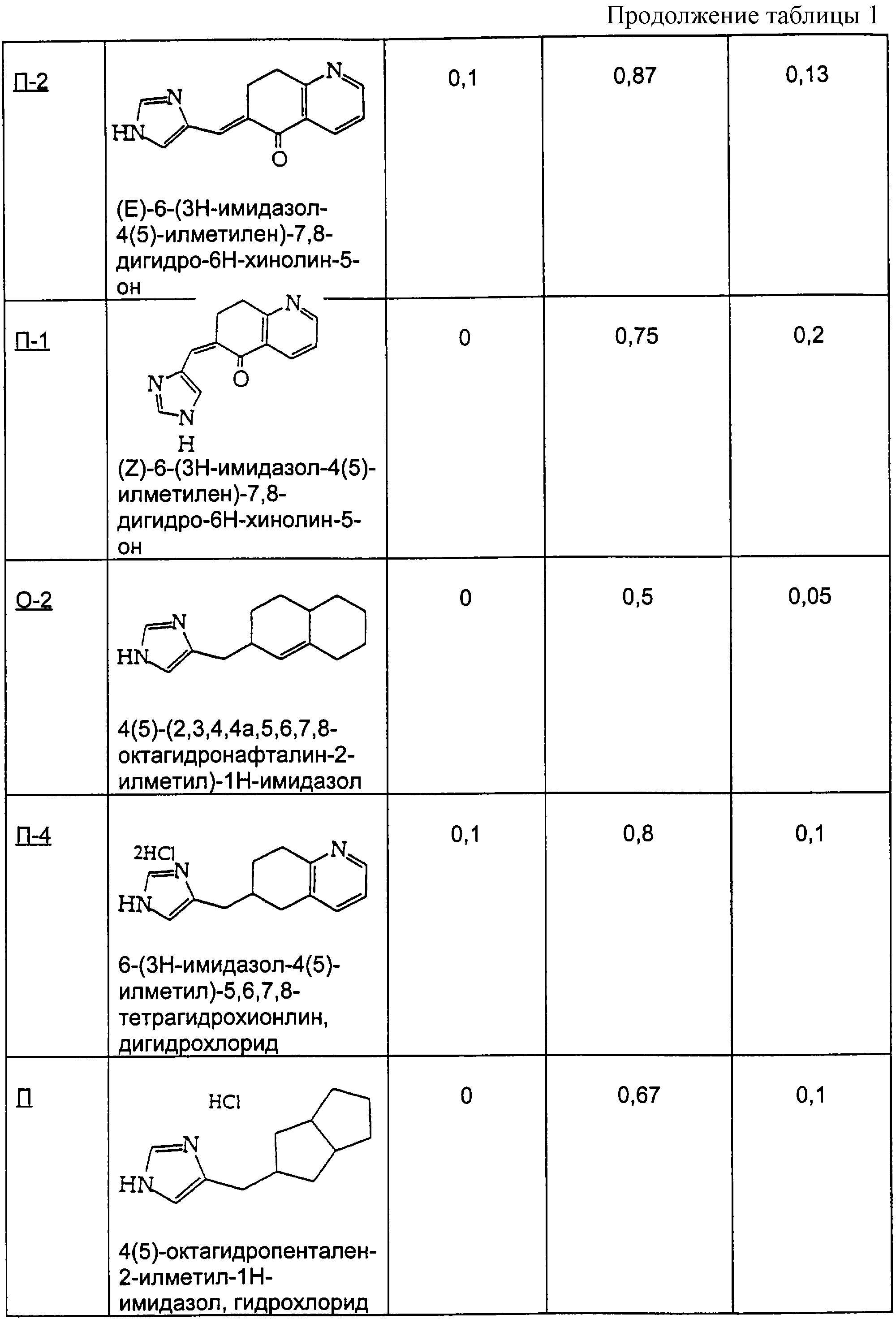
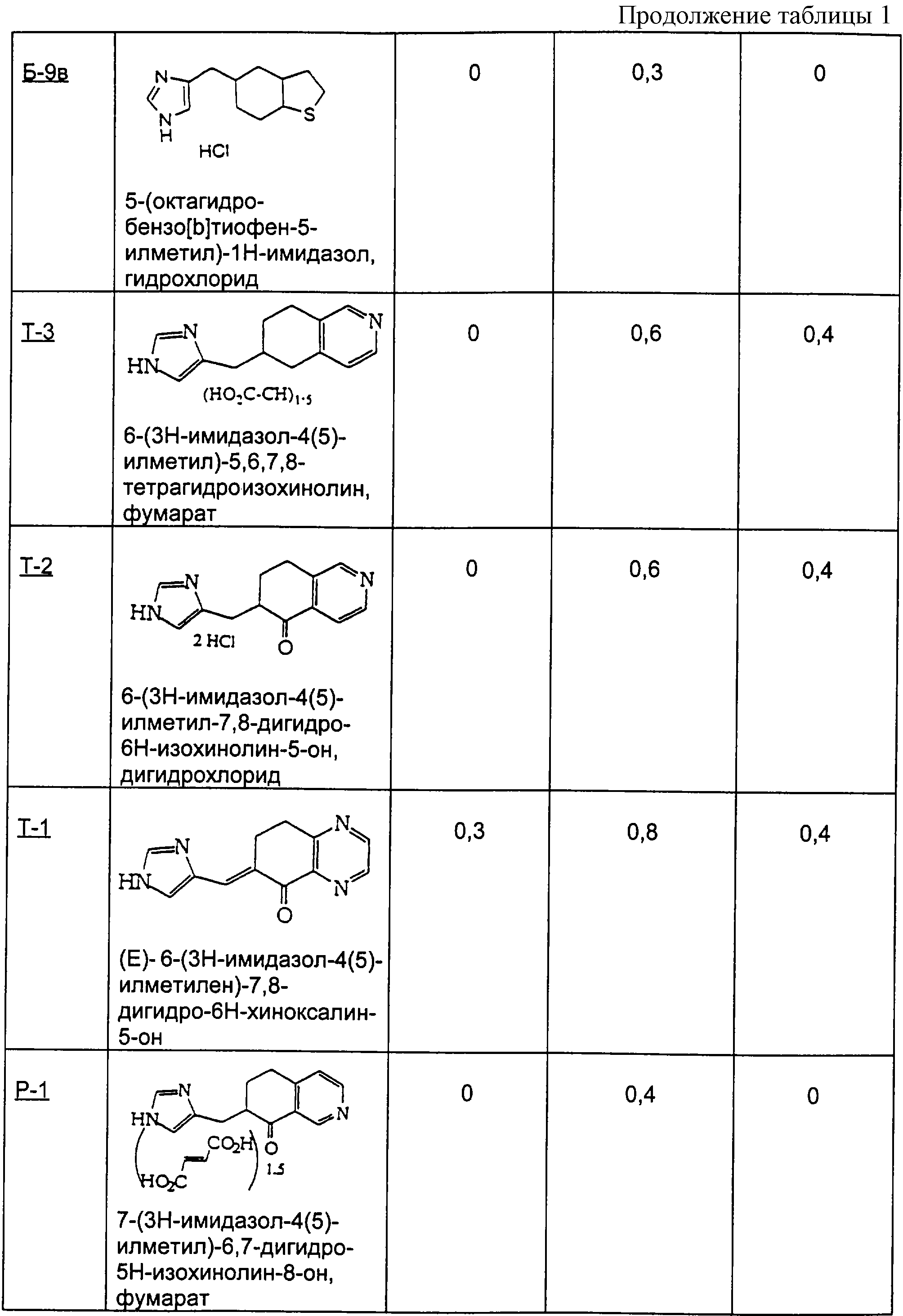
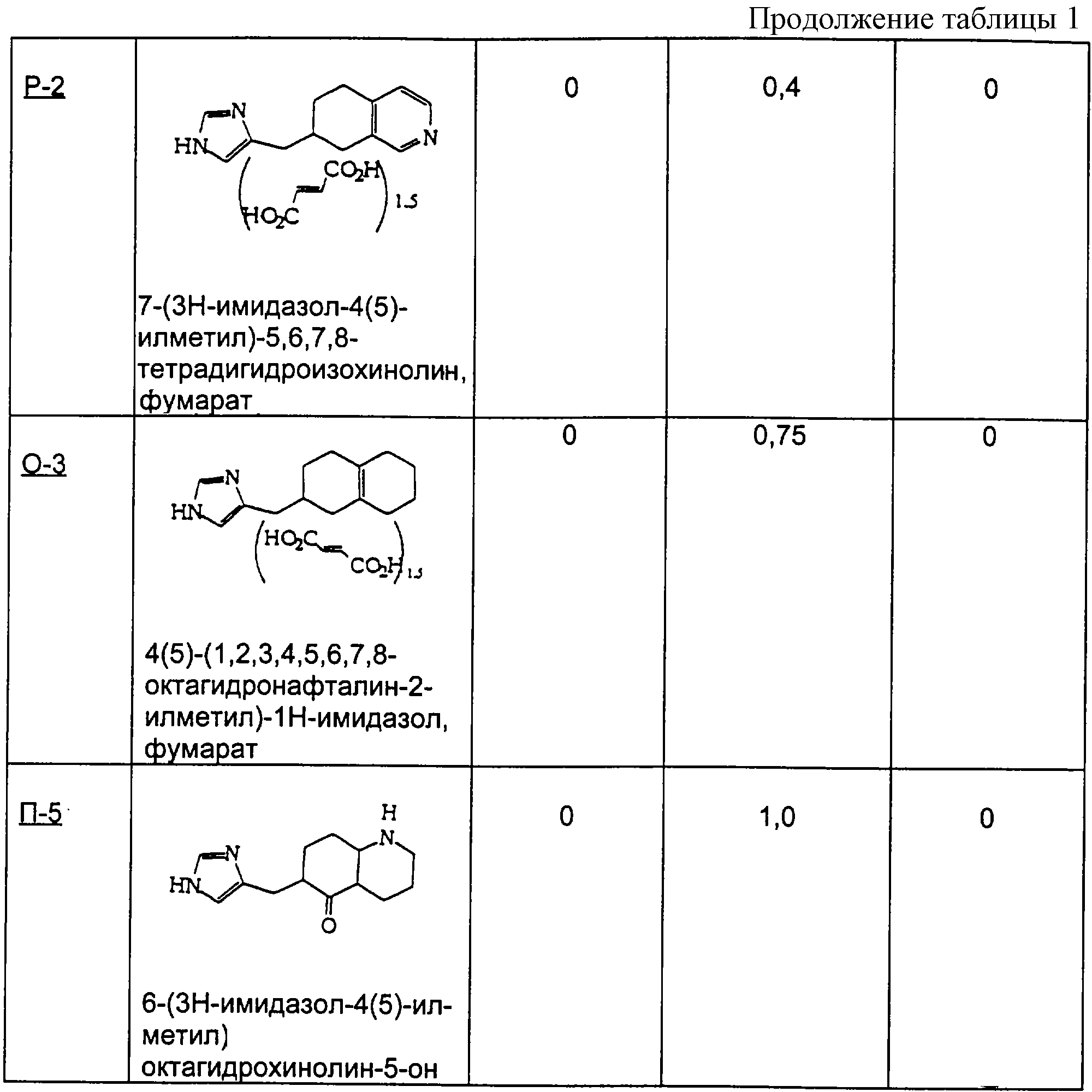
Реферат
Изобретение относится к производным имидазола формулы (I), где X, Y, R, R2, R3 и R4 такие, как определено в формуле изобретения. Эти соединения являются селективными агонистами по отношению к адренергическим рецепторам подтипа(ов) α2В или α2B/α2C. Также описан способ введения фармацевтической композиции на основе этих соединений. 13 н. и 76 з.п. ф-лы, 6 табл.
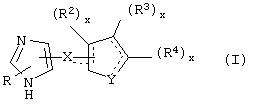
Формула
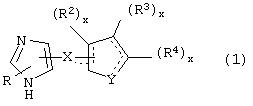
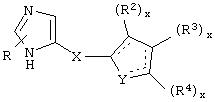
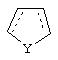
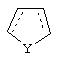
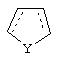

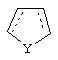
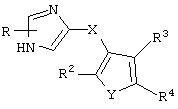
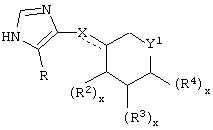
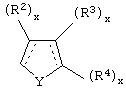
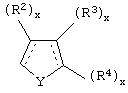
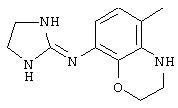
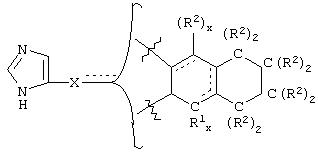
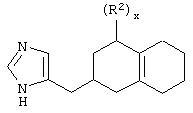
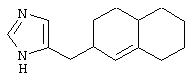
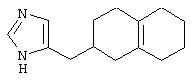
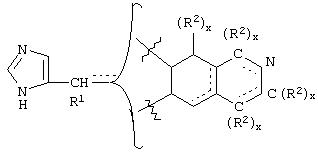
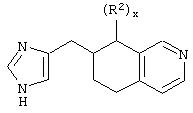
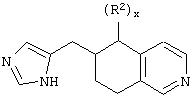
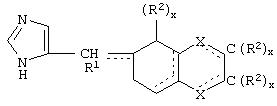
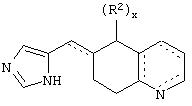
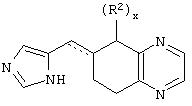
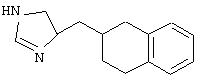
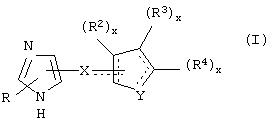
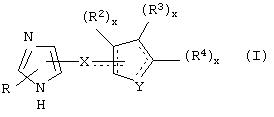
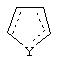
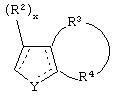
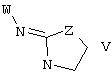
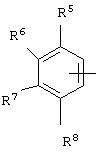
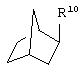
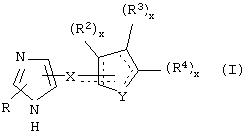
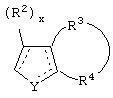
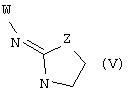
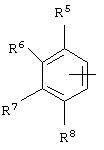
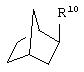
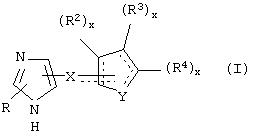
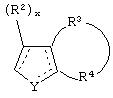
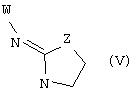
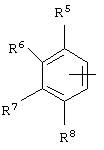
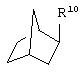
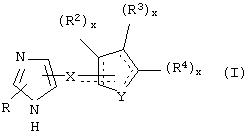
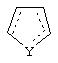
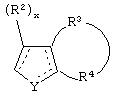
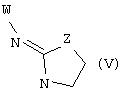
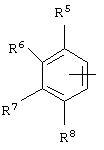
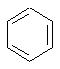
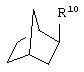
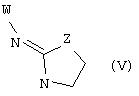
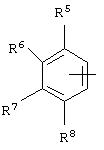
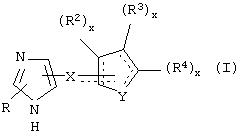
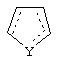
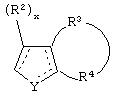
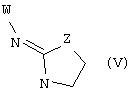
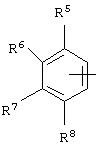
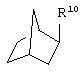
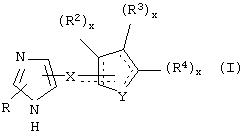
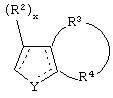
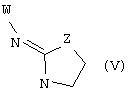
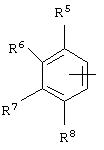
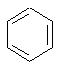
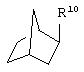
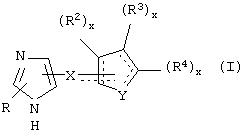
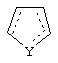
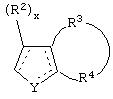
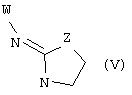
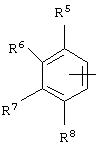
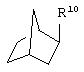
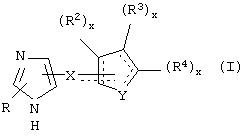
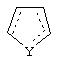
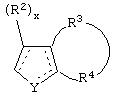
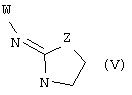
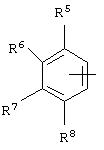
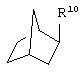
Комментарии