Композиции для лечения гипертензии и/или фиброза - RU2752088C1
Код документа: RU2752088C1
Чертежи






Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[0001] Формула изобретения настоящей заявки имеет приоритет Австралийской заявки на предварительный патент №2016903804 (поданной 21 сентября 2016 года), полное содержание которой включено в данное описание посредством ссылки.
[0002] Настоящее изобретение относится к новым соединениям и их применению в профилактическом и/или терапевтическом лечении сердечнососудистого заболевания, и в частности для лечения прегипертензии, гипертензии и/или фиброзных состояний.
[0003] Изобретение было разработано в первую очередь для профилактического и/или терапевтического лечения сердечно-сосудистого заболевания, и оно будет описано далее со ссылкой на эту заявку. Однако должно быть понятно, что данное изобретение не ограничивается этой конкретной областью применения.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
[0004] Любое обсуждение предшествующего уровня техники в описании изобретения никоим образом не должно рассматриваться как допущение, что такой предшествующий уровень широко известен или составляет часть общих знаний в данной области.
[0005] Гипертензия (высокое кровяное давление) поражает 26% взрослого населения по всему миру с заболеваемостью 30-33% в западных странах. Ожидается, что заболеваемость гипертензией в мире достигнет 29% к 2025 году вследствие вестернизации Индии и Китая. Текущие исследования показывают, что менее 20% пациентов с гипертензией добиваются установления рекомендованного кровяного давления (ВР), и что для достижения этих целей >75% пациентам требуется терапия множеством антигипертензивных агентов. Прегипертензия (слегка повышенное кровяное давление) поражает 31% взрослых в США и может переходить в гипертензию, если ее не лечить.
[0006] Гипертензия и Прегипертензия являются главными факторами в развитии повреждения кровеносных сосудов в различных органах, приводящего к замещению нормальной функциональной ткани рубцовой тканью или фиброзом. Некоторые современные антигипертензивные агенты способны замедлять прогрессирование замещения функциональной ткани фиброзом, но, как было показано, ни один из них не реверсирует существующий фиброз и не восстанавливает нормальную структуру ткани. Таким образом, существует потребность в агентах, которые обладают значительной эффективностью в снижении ВР и поэтому дают возможность большему проценту пациентов добиться установления целевого ВР с использованием терапии единственным агентом и/или в реверсировании существующего фиброза и/или восстановлении нормальной структуры ткани.
[0007] Задача настоящего изобретения заключается в преодолении или устранении по меньшей мере одного из недостатков предшествующего уровня техники или в предоставлении полезной альтернативы.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0008] Авторы настоящего изобретения неожиданно обнаружили, что некоторые соединения обладают снижающим кровяное давление эффектом и/или антифибротическим эффектом. Эти эффекты можно наблюдать в исследованиях с внутривенным и/или пероральным введением доз.
[0009] Согласно одному из аспектов настоящего изобретения предложено соединение формулы:

где:
Х выбран из группы, состоящей из:


R1-R9 независимо представляют собой С, N, О или S;
R10 независимо выбраны из C1-6алкила, галогена, C0-6алкилкарбоновой кислоты, амино, гидрокси и C1-6алкокси;
Y представляет собой А, СН2-А или СН=А;
А выбран из возможно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; возможно замещенного C1-6алкоксиламина; возможно замещенного C1-6алкиламина; возможно замещенной C0-6алкилкарбоновой кислоты; возможно замещенного C1-6алкилгидроксила; возможно замещенного насыщенного или ненасыщенного C0-6алкил-бициклического гетероциклила; и возможно замещенного насыщенного или ненасыщенного C1-6алкоксил-бициклического гетероциклила;
Z выбран из группы, состоящей из:





R11 независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
R12, R14 и R15 независимо представляют собой С, СН, СН2, О, N, NH или S;
R13 представляет собой С, СН, СН2, N, NH, С-CF3, СН-CF3 или С=O;
m равно 0, 1, 2, 3, 4 или 5; и
n равно 0, 1, 2, 3 или 4, или стереоизомер или фармацевтически приемлемая соль его.
[0010] В одном из воплощений R10 независимо выбран из -СН3, -С(O)ОН, -F, -NH2, -ОН и -ОСН3.
[0011] В одном из воплощений R5-R9 независимо представляют собой С или N.
[0012] В одном из воплощений C0-6алкилкарбоновая кислота представляет собой карбоновую кислоту.
[0013] В одном из воплощений насыщенный, частично насыщенный или ненасыщенный 5- или 6-членный гетероциклил содержит один или более N, S или О, и возможно замещен одним или более заместителями оксо, C1-6алкил, амино, гидроксил или галоген.
[0014] В одном из воплощений насыщенный, частично насыщенный или ненасыщенный 5- или 6-членный гетероциклил выбран из пирролила, пиразолила, имидазолила, триазолила, имидазолидинила, пирролидинила, пирролидинилидена, дигидропирролила, изоксазолила, дигидрооксазолила, изоксазолидинила, оксазолидинила и оксазолила, возможно замещенных одним или более заместителями оксо, C1-6алкил, амино, гидроксил или галоген.
[0015] В одном из воплощений C1-6алкоксиламин представляет собой аминооксиметил.
[0016] В одном из воплощений C1-6алкиламин возможно замещен одним или более чем одним C1-6алкилом, C1-6галогеналкилом, гидроксилом или галогеном, предпочтительно моно-, ди- или три-замещенным галогеналкилом, наиболее предпочтительно трифторметаном.
[0017] В одном из воплощений C1-6алкилгидроксил представляет собой метилгидроксил или пропан-2-ол.
[0018] В одном из воплощений C0-6алкил-бициклический гетероциклил выбран из индолила, изоиндолила, индолинила и изоиндолинила, возможно замещенных одним или более оксо, предпочтительно диоксо.
[0019] В одном из воплощений C1-6алкоксил-бициклический гетероциклил выбран из индолила, изоиндолила, индолинила и изоиндолинила, возможно замещенных одним или более чем одним оксо, и где C1-6алкоксил представляет собой метокси или этокси.
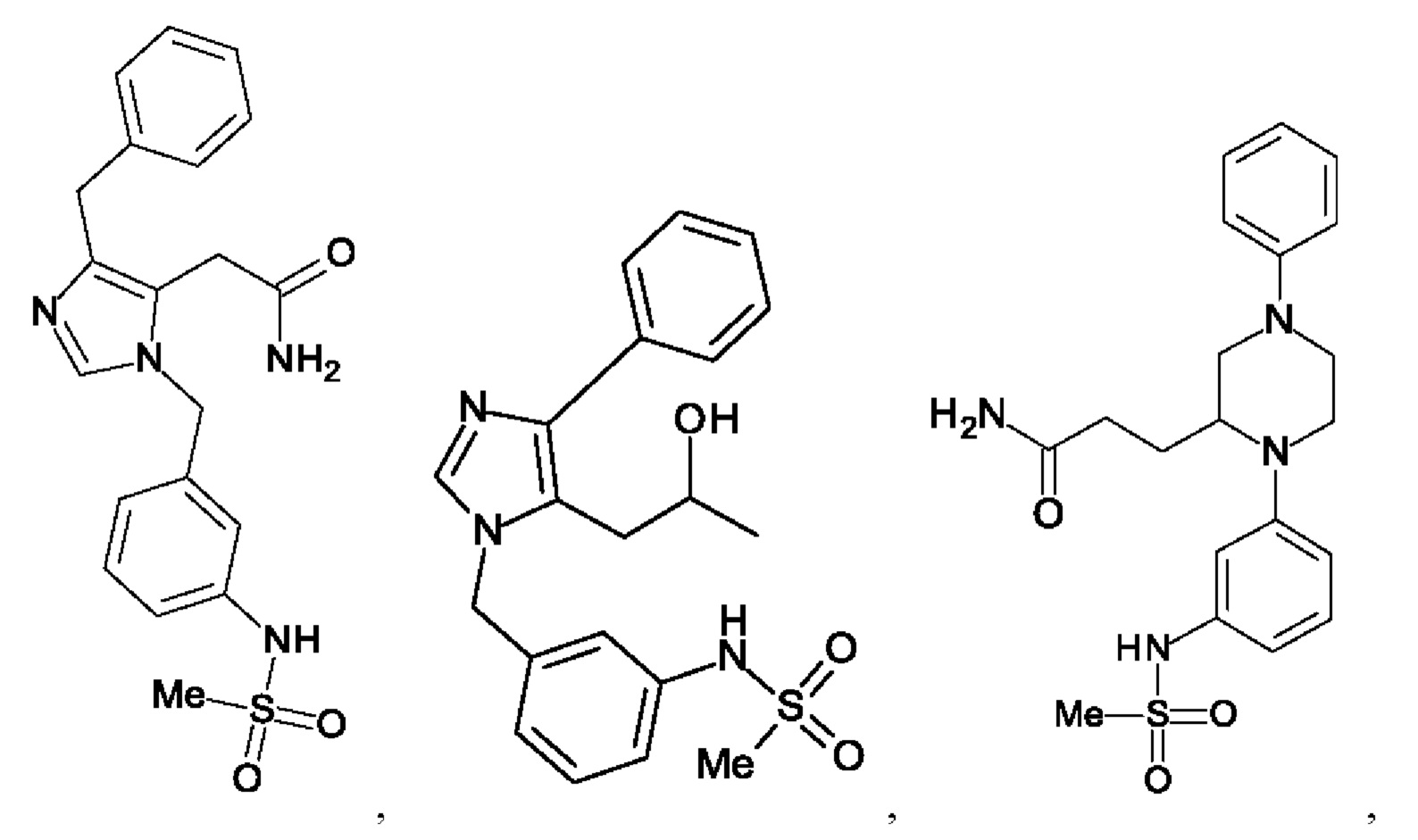
[0020] В одном из воплощений А выбран из:



[0021] В одном из воплощений R11 представляет собой галоген, выбранный из группы; состоящей из F, Cl, Br и I.
[0022] В одном из воплощений R11 представляет собой замещенный амино формулы -NHR16, и где:
R16 Выбран из -CN, -SO2(R17)aR18 И -CO(R17)aR18,
а равно 0 или 1, R17 выбран из -NH- и -O-, и
R18 выбран из -Н, -СН3, -СН2СН3, -CH2OH и -CH2CH2OH.
[0023] В одном из воплощений Рц представляет собой замещенный амино, выбранный из группы, состоящей из -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3, -NHCOCH3, -NHCOOCH3, -NHCOOCH2CH2OH, -NHCONH2 и -NHCN.
[0024] В одном из воплощений R11 представляет собой алкил, выбранный из группы, состоящей из метила, этила, пропила, бутила и пентила.
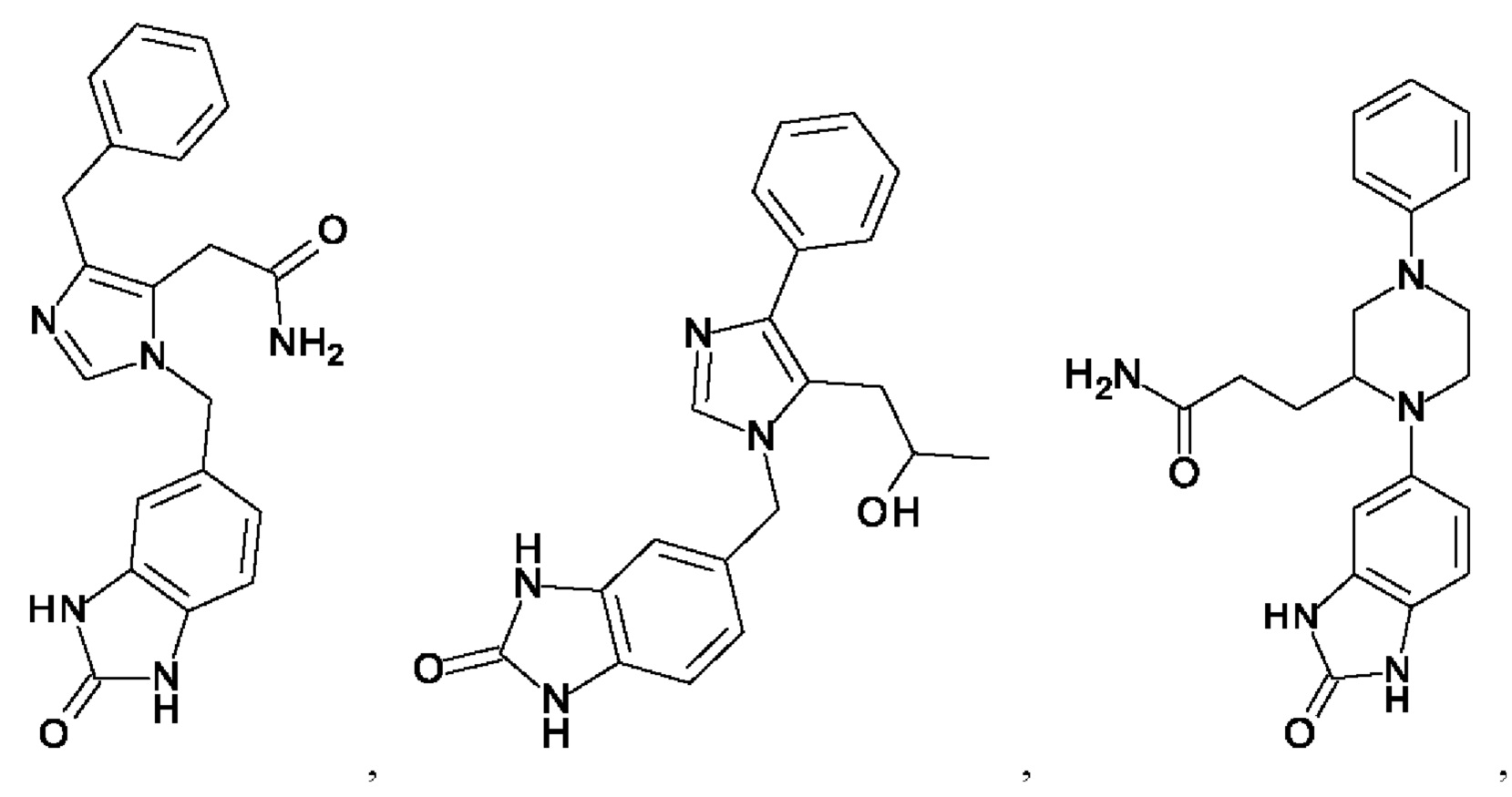
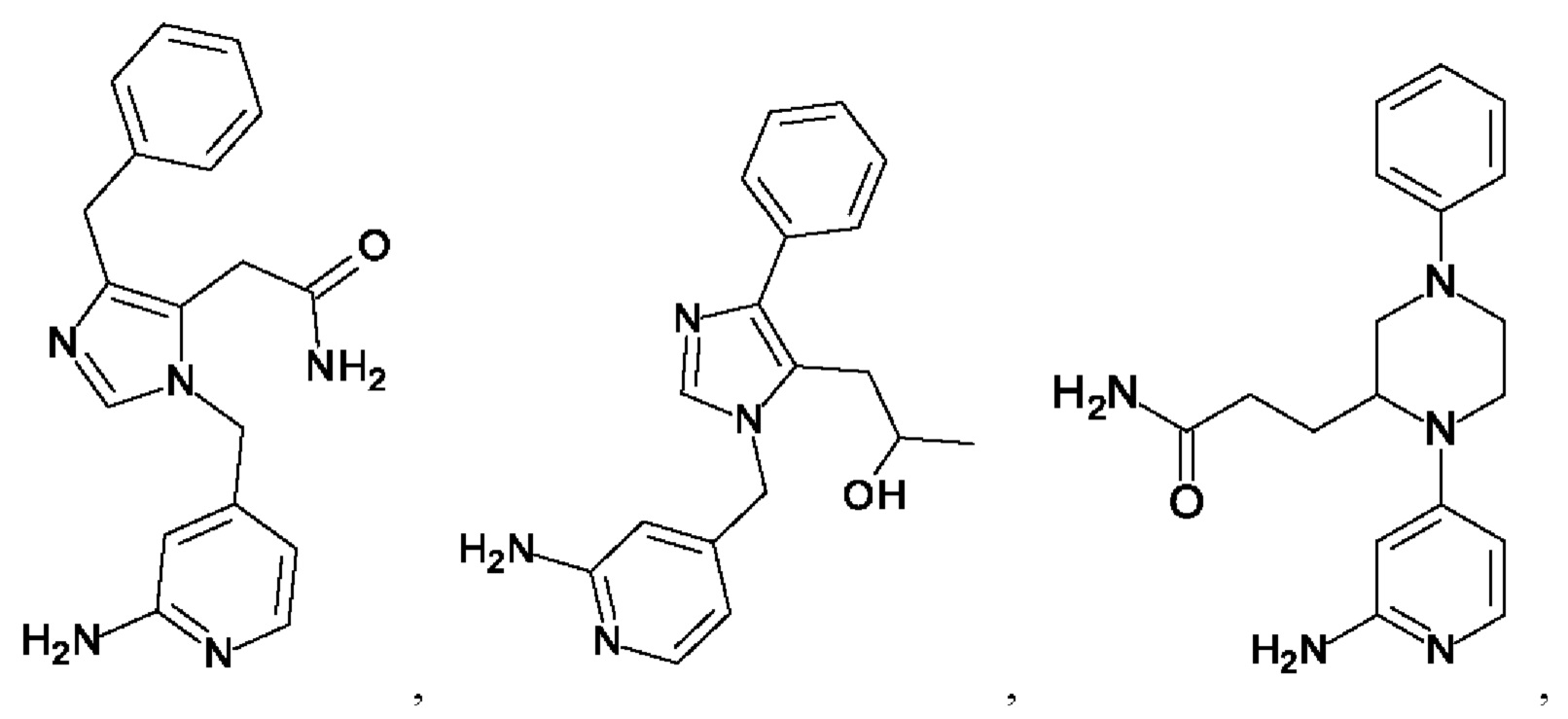
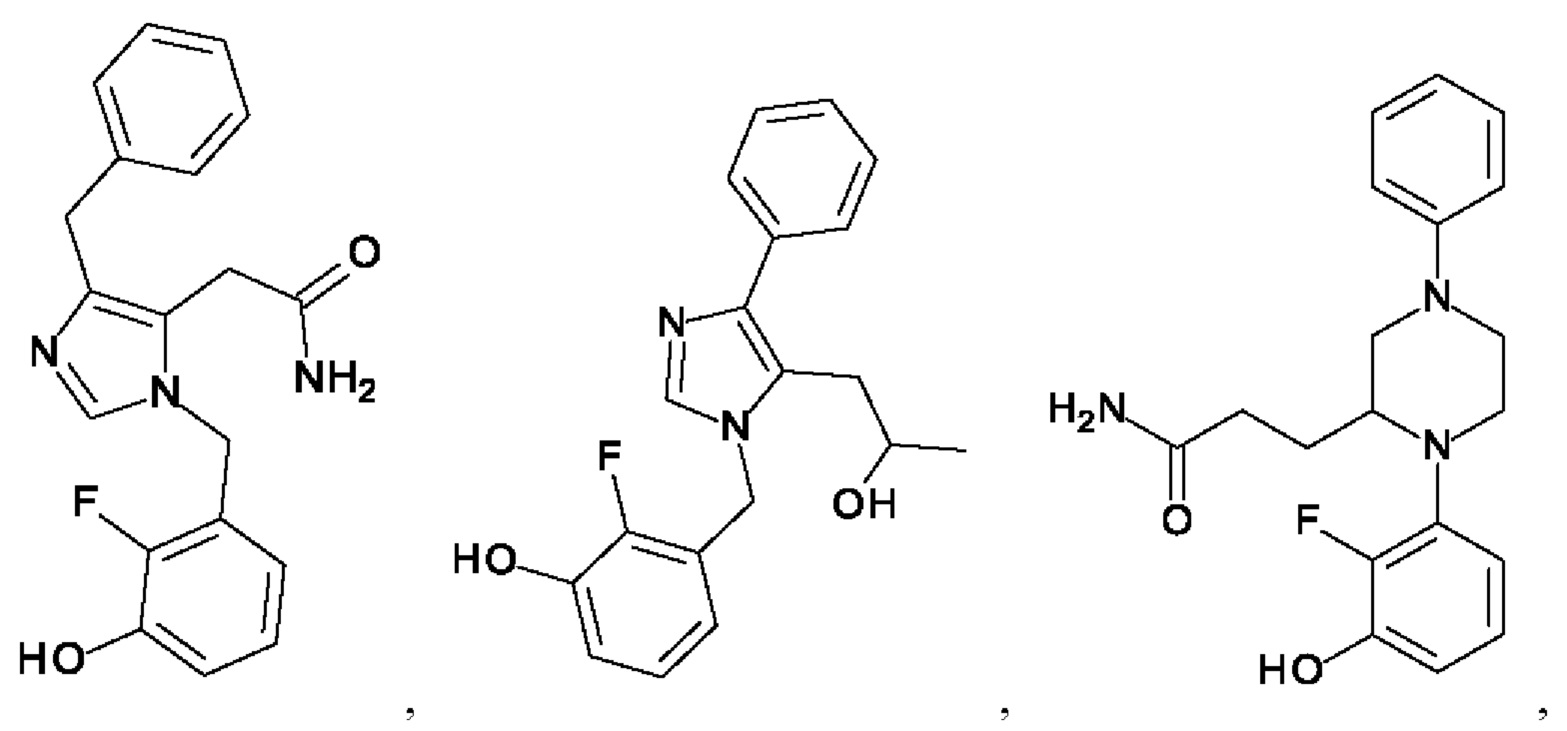
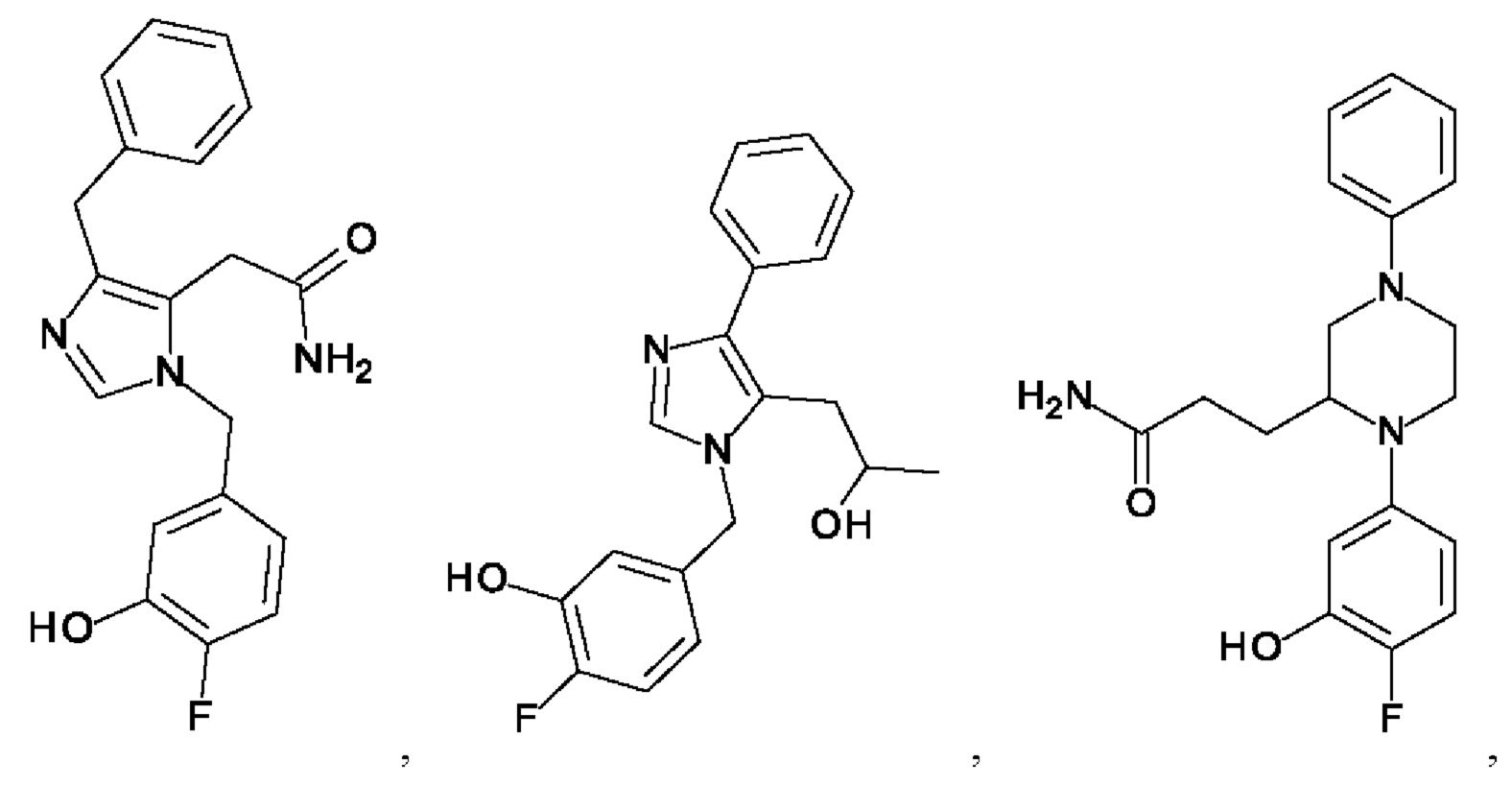
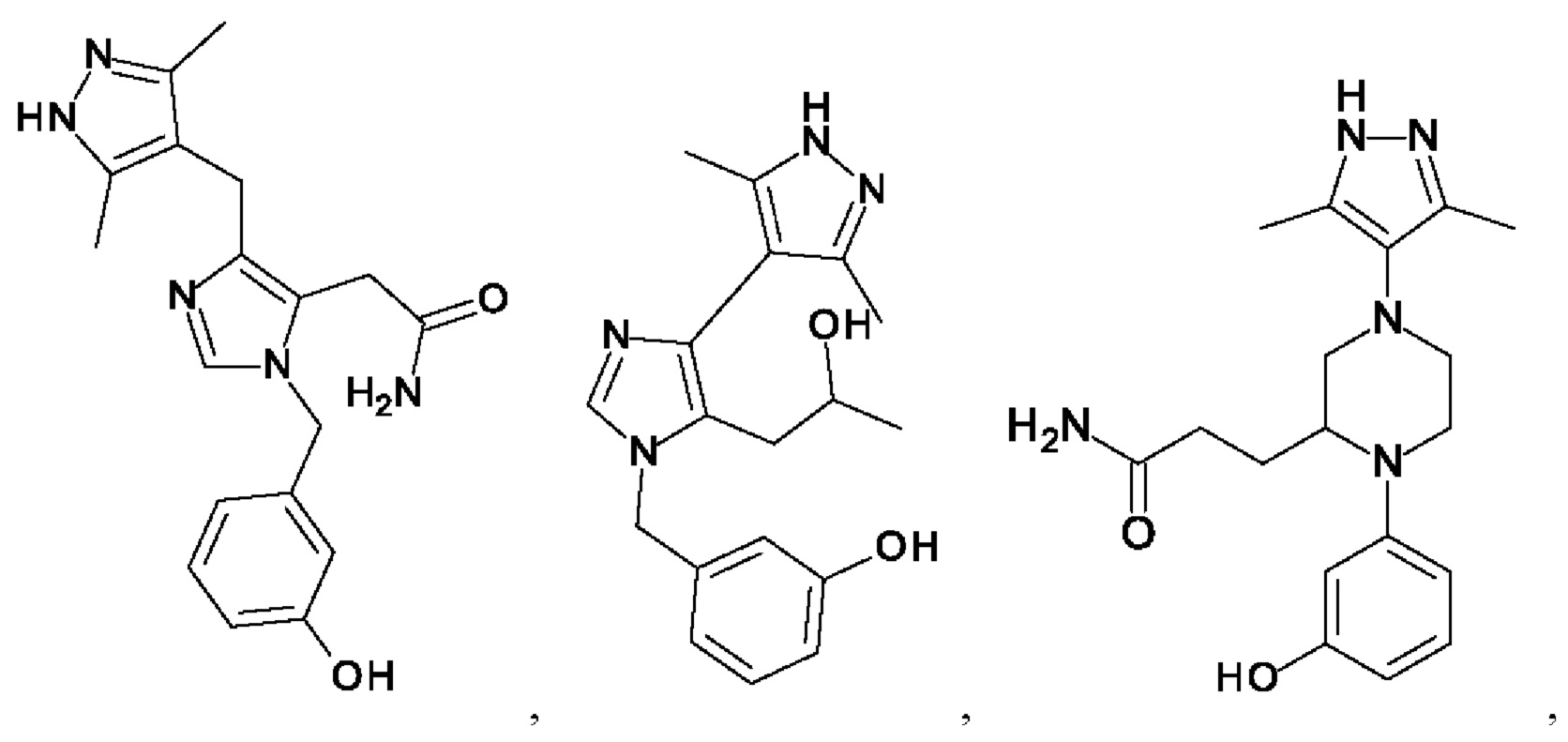
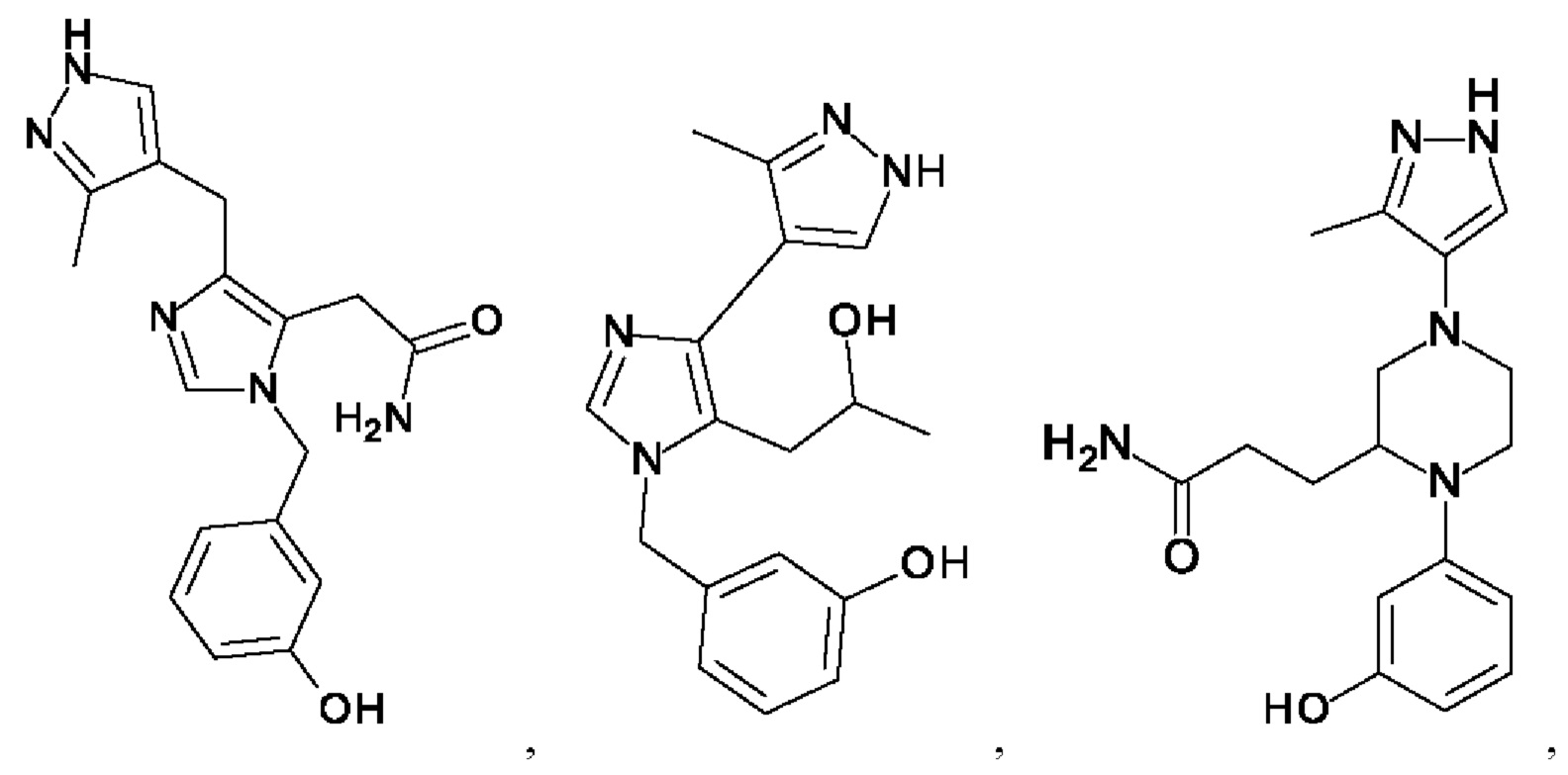
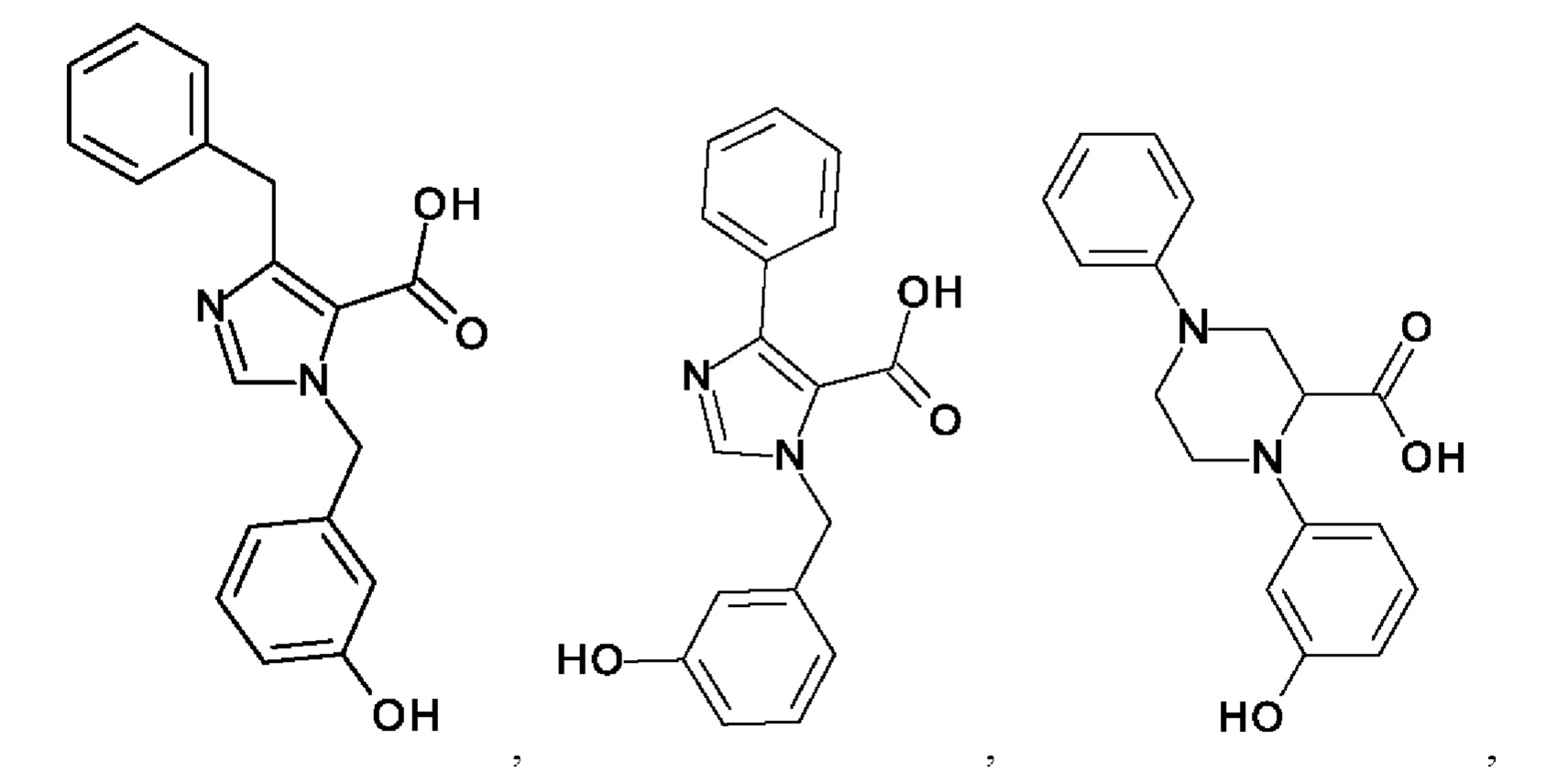
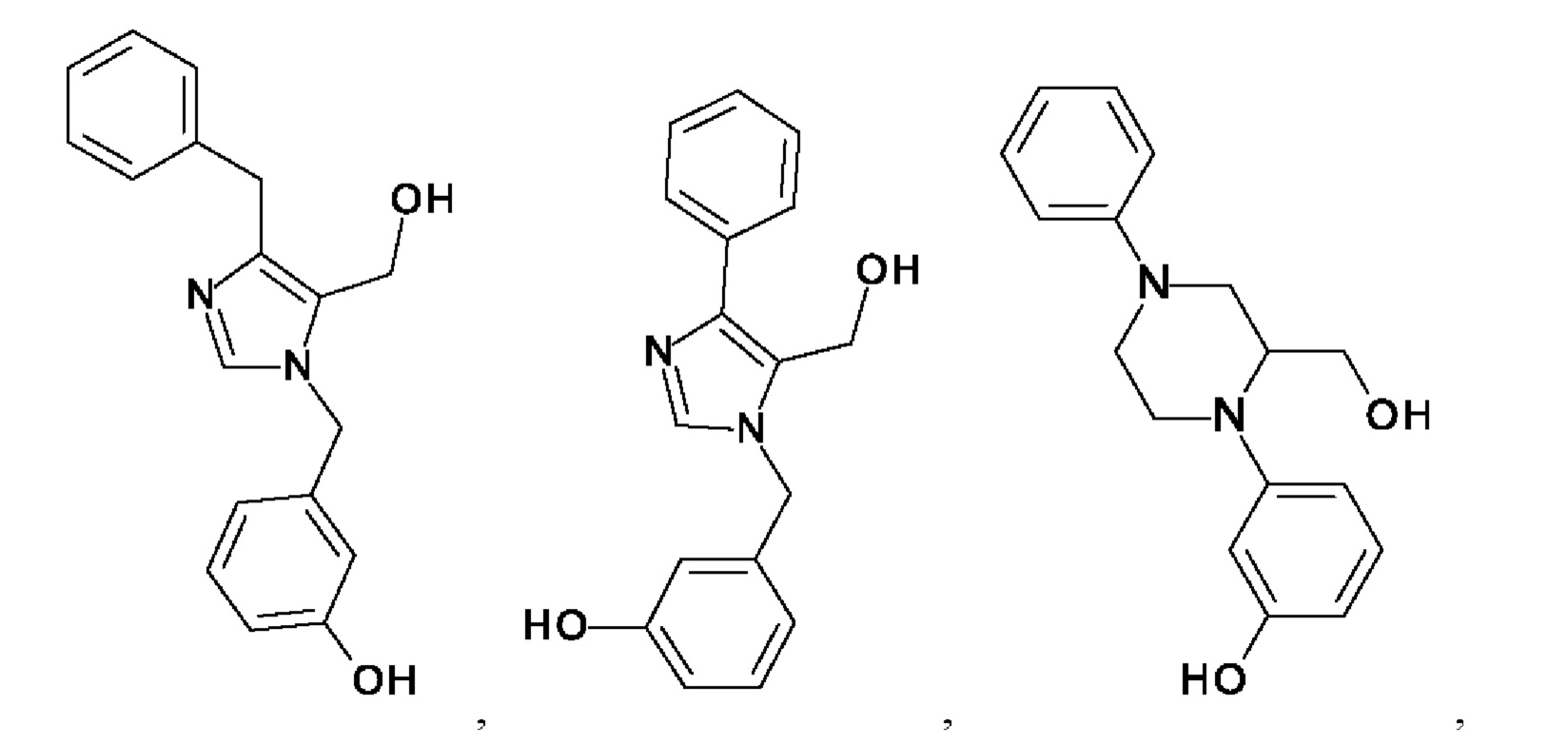
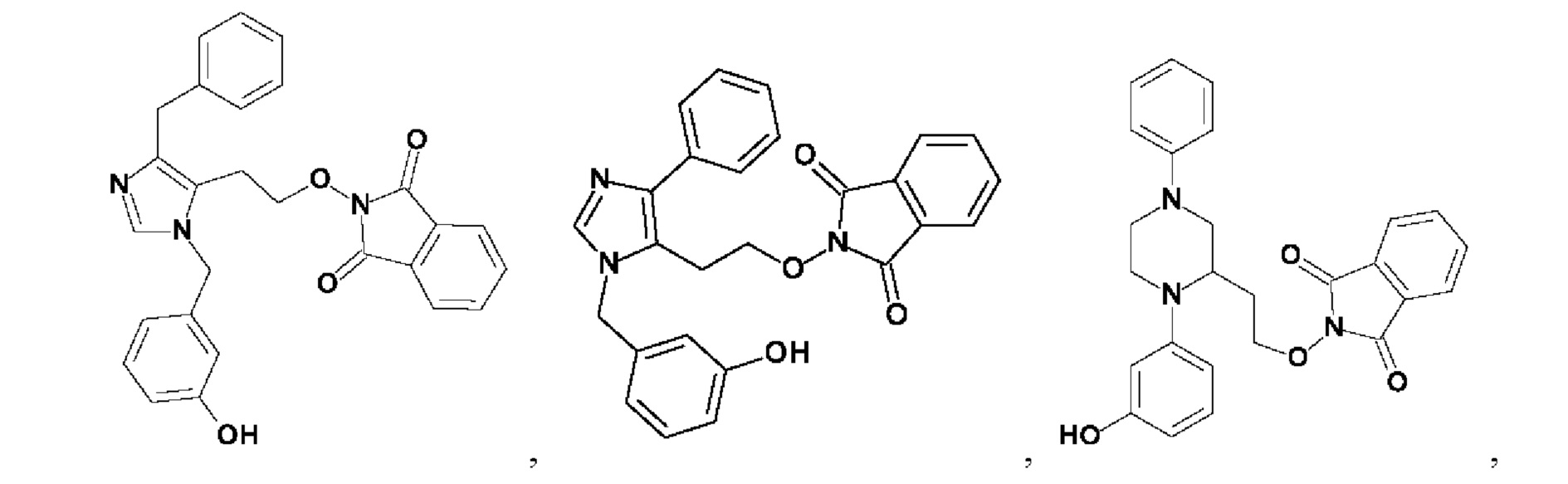
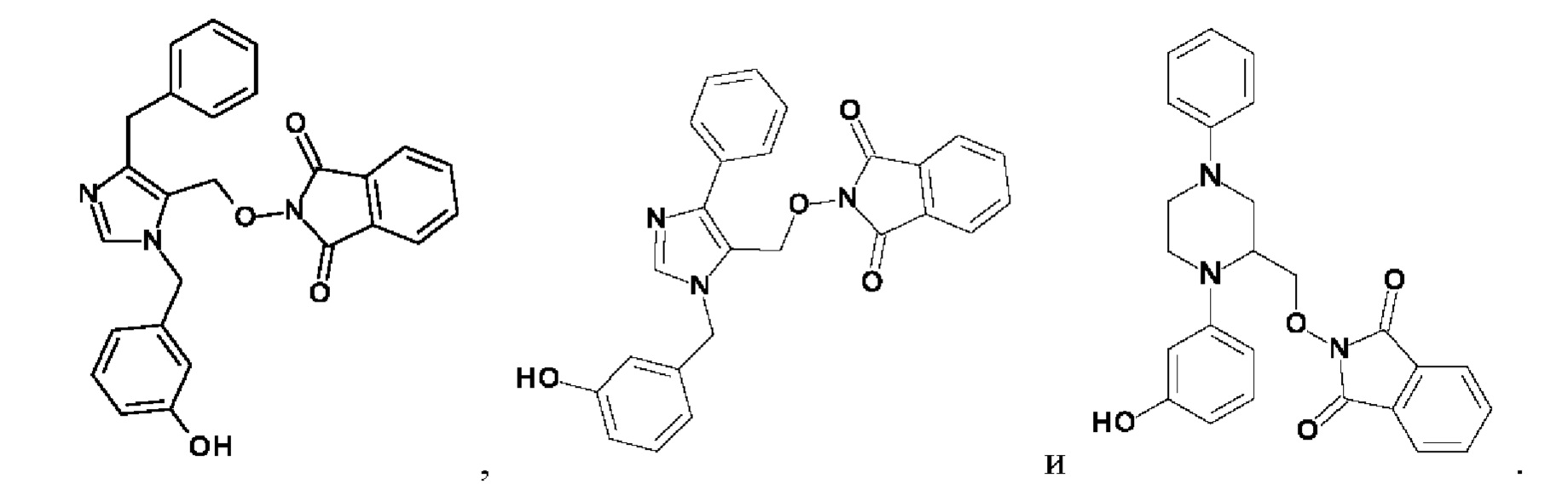
[0025] В одном из воплощений соединение выбрано из группы, состоящей из:


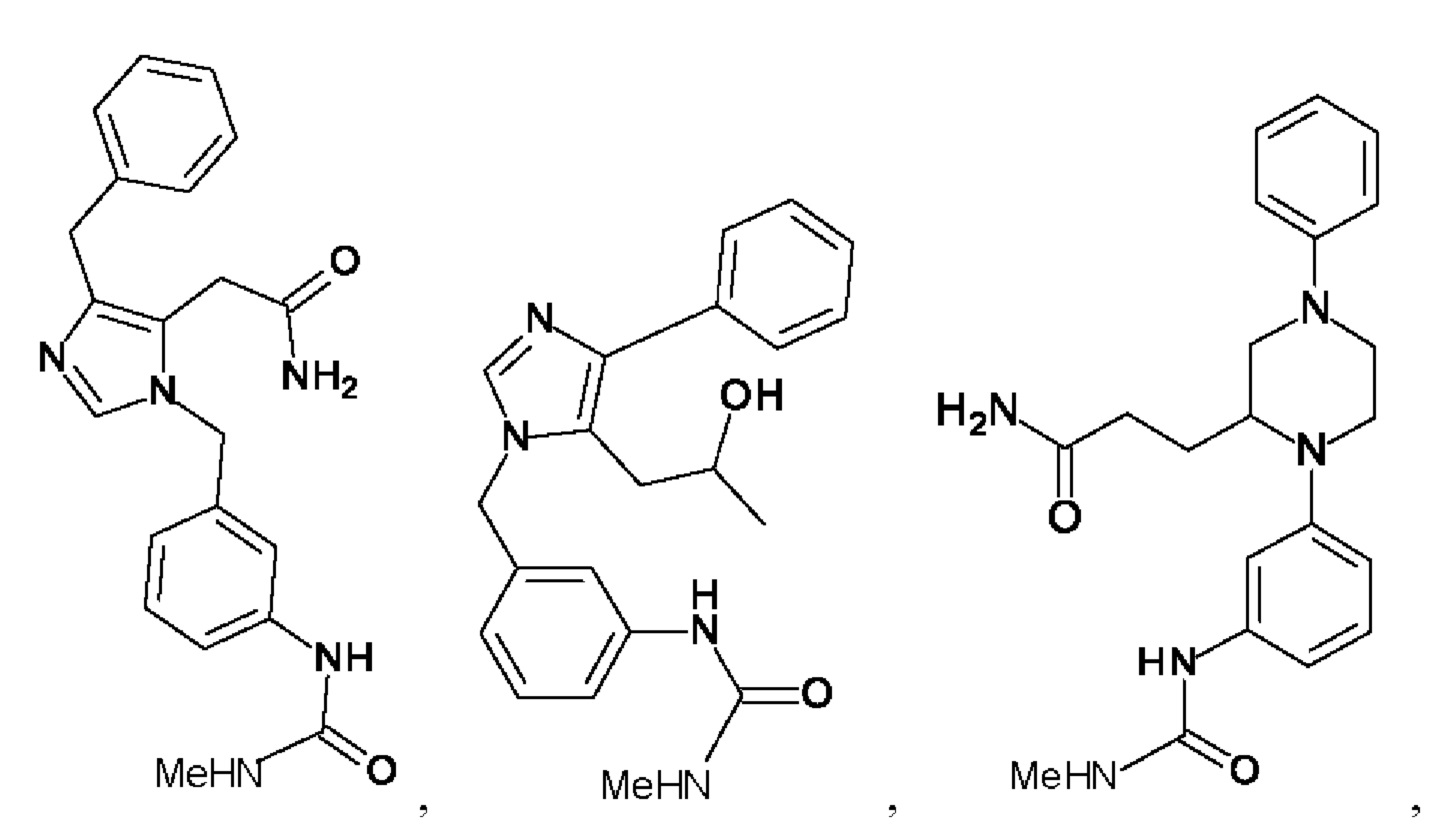


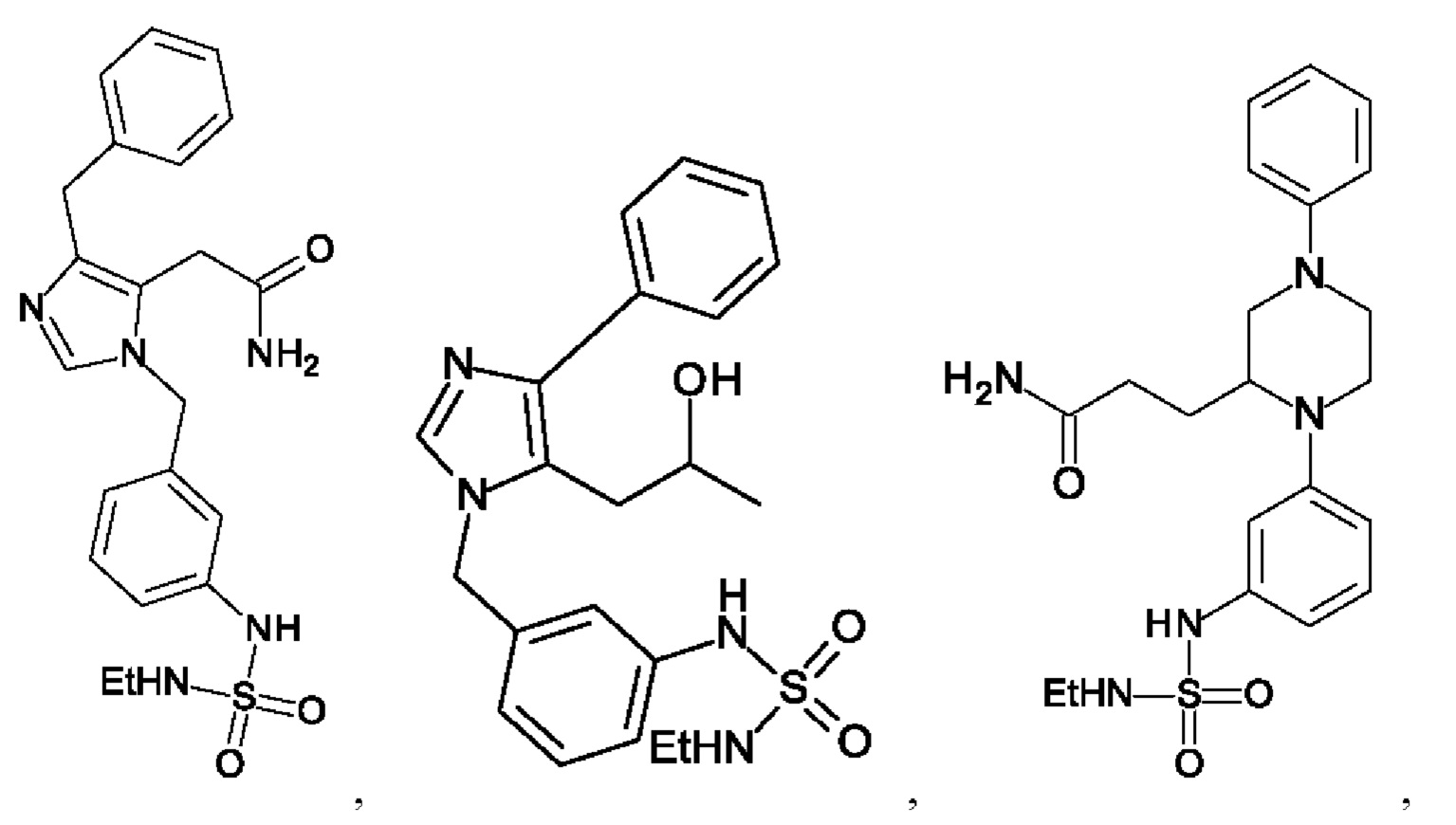







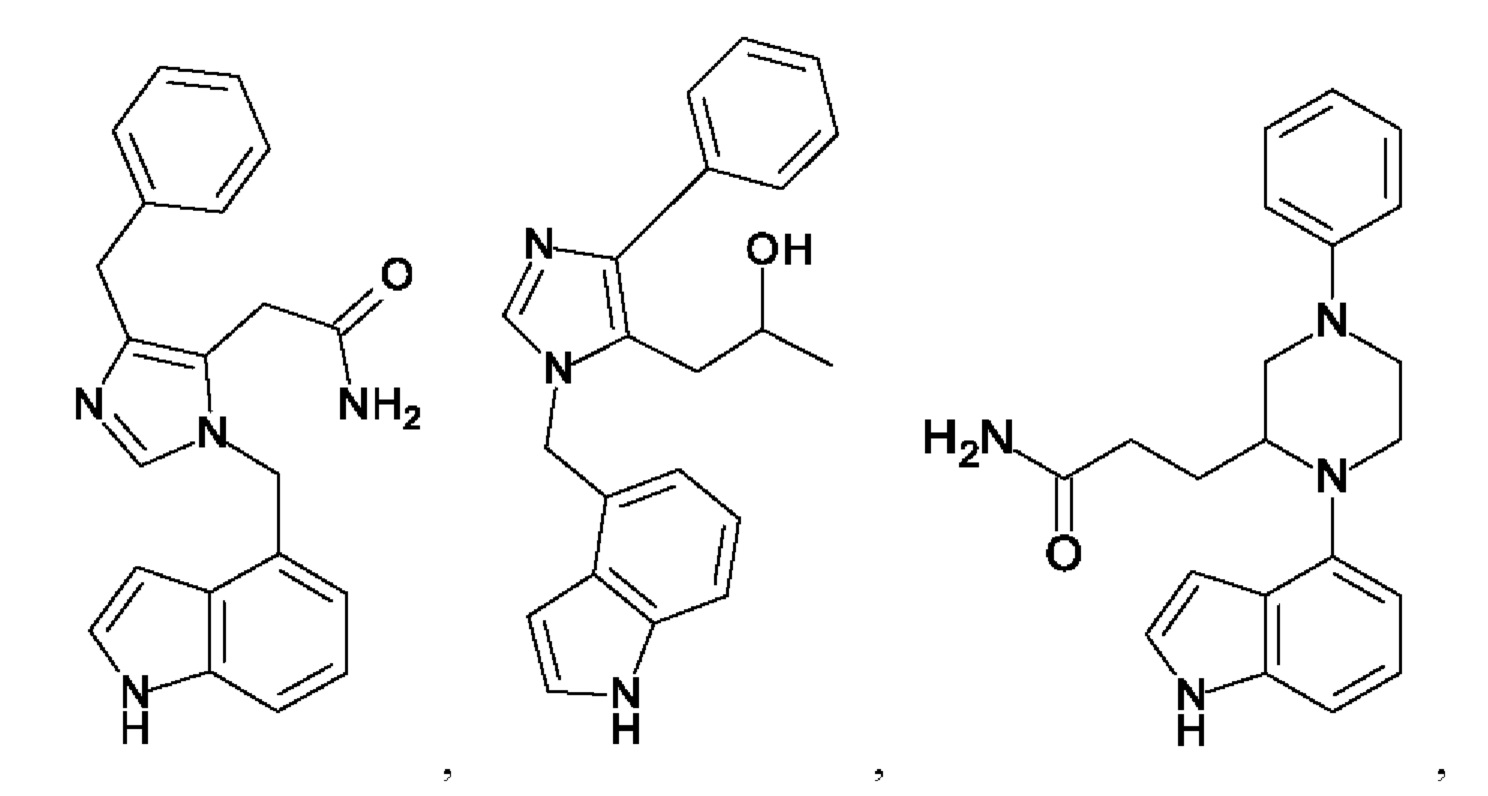
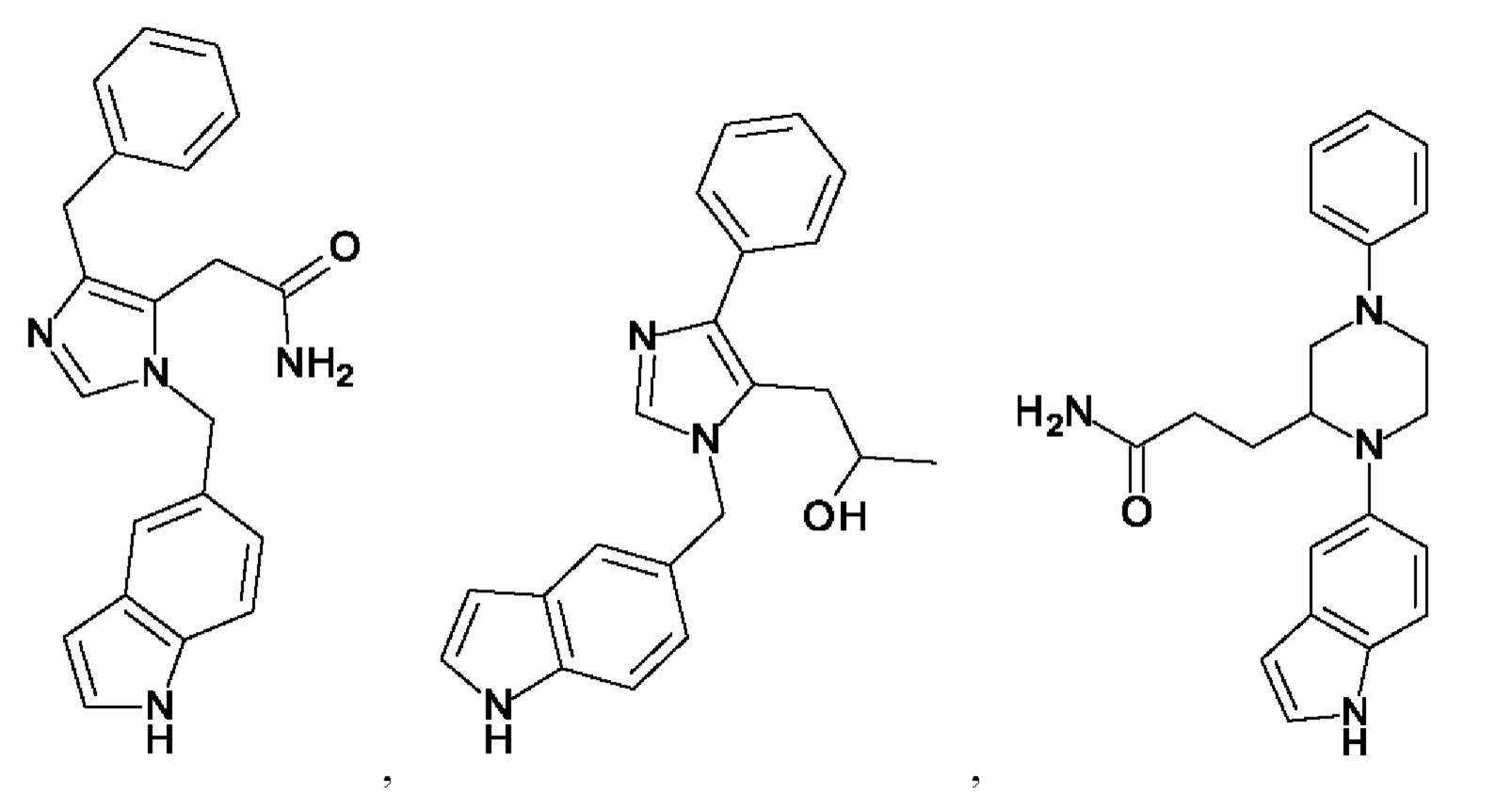
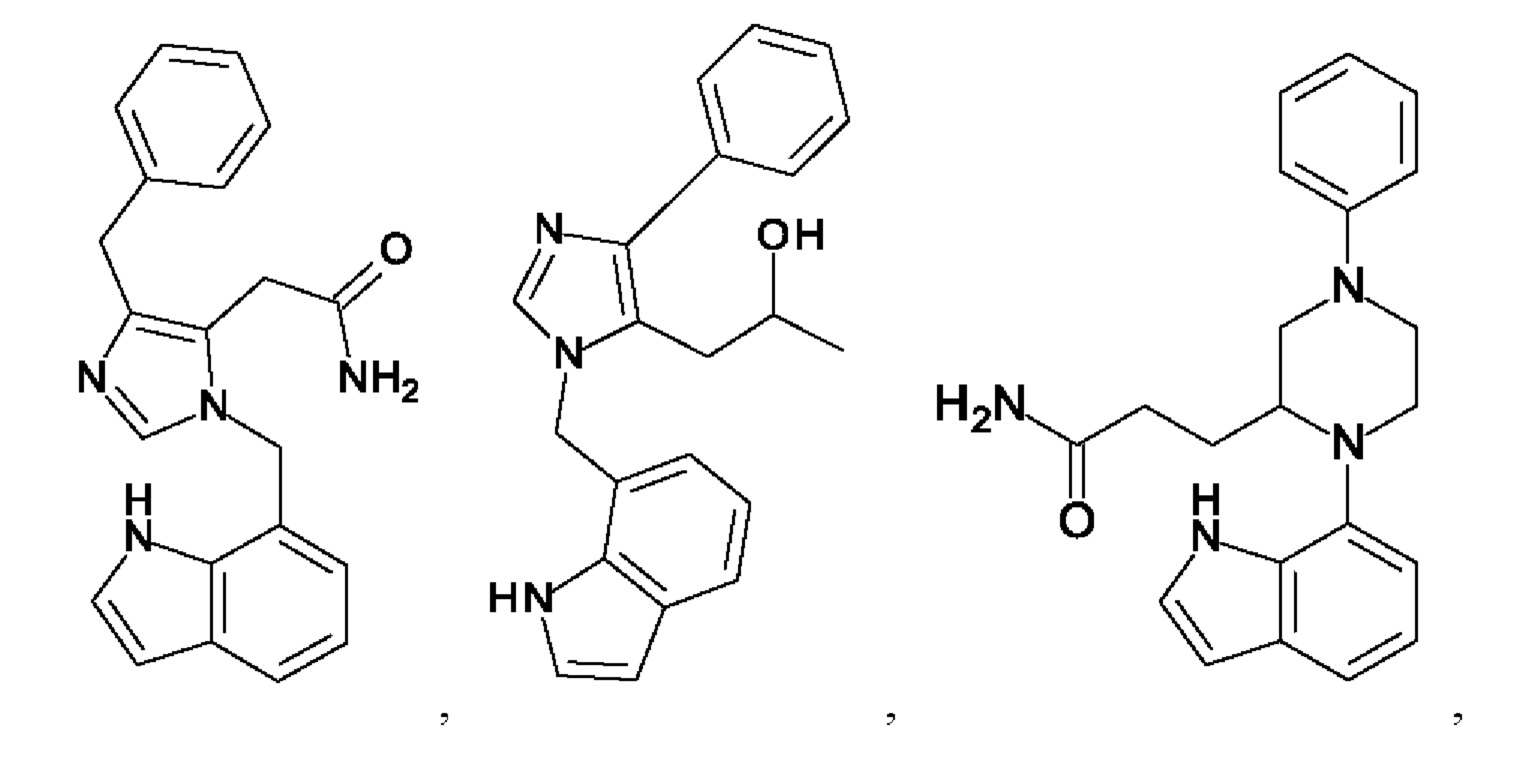
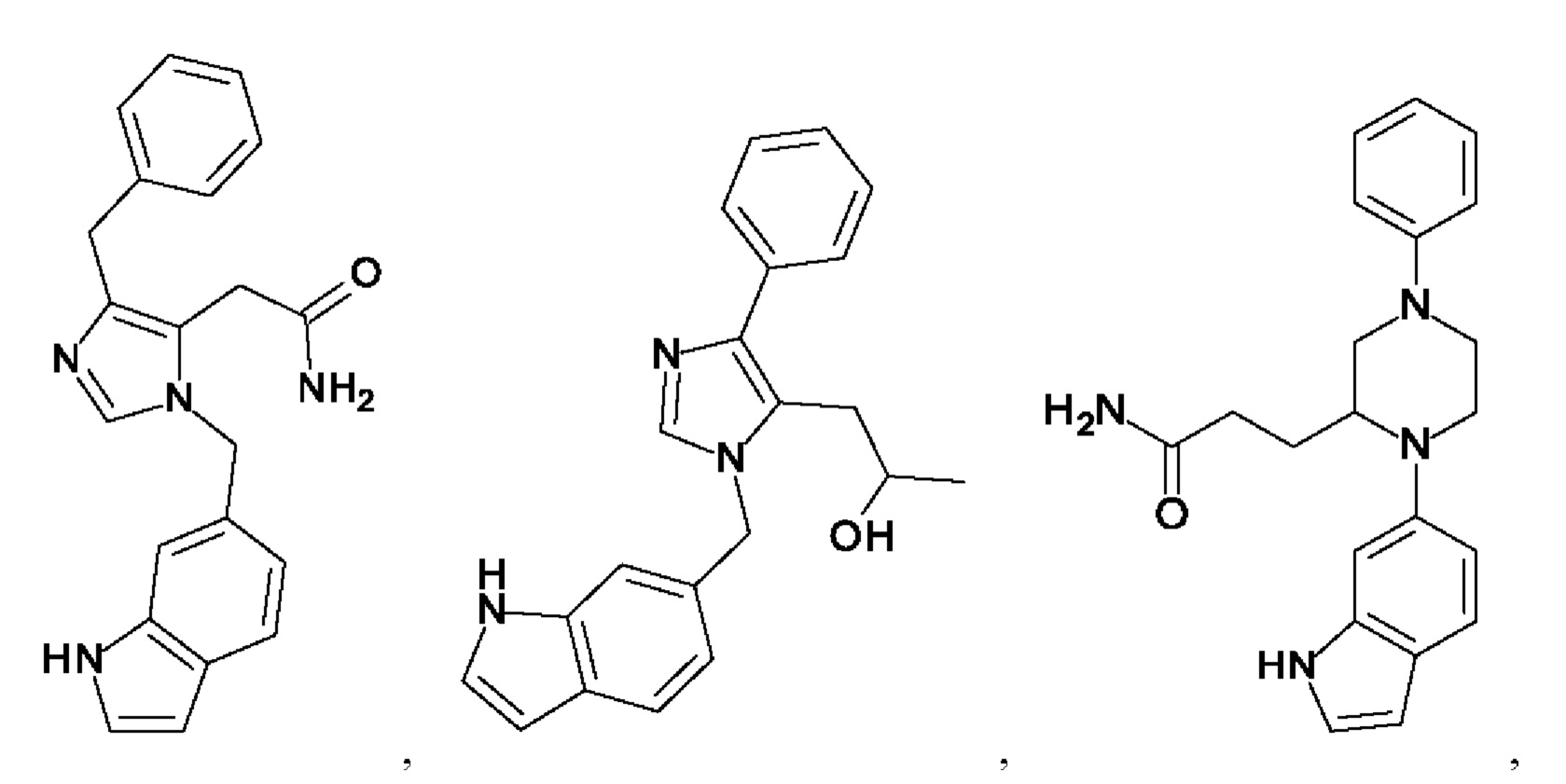

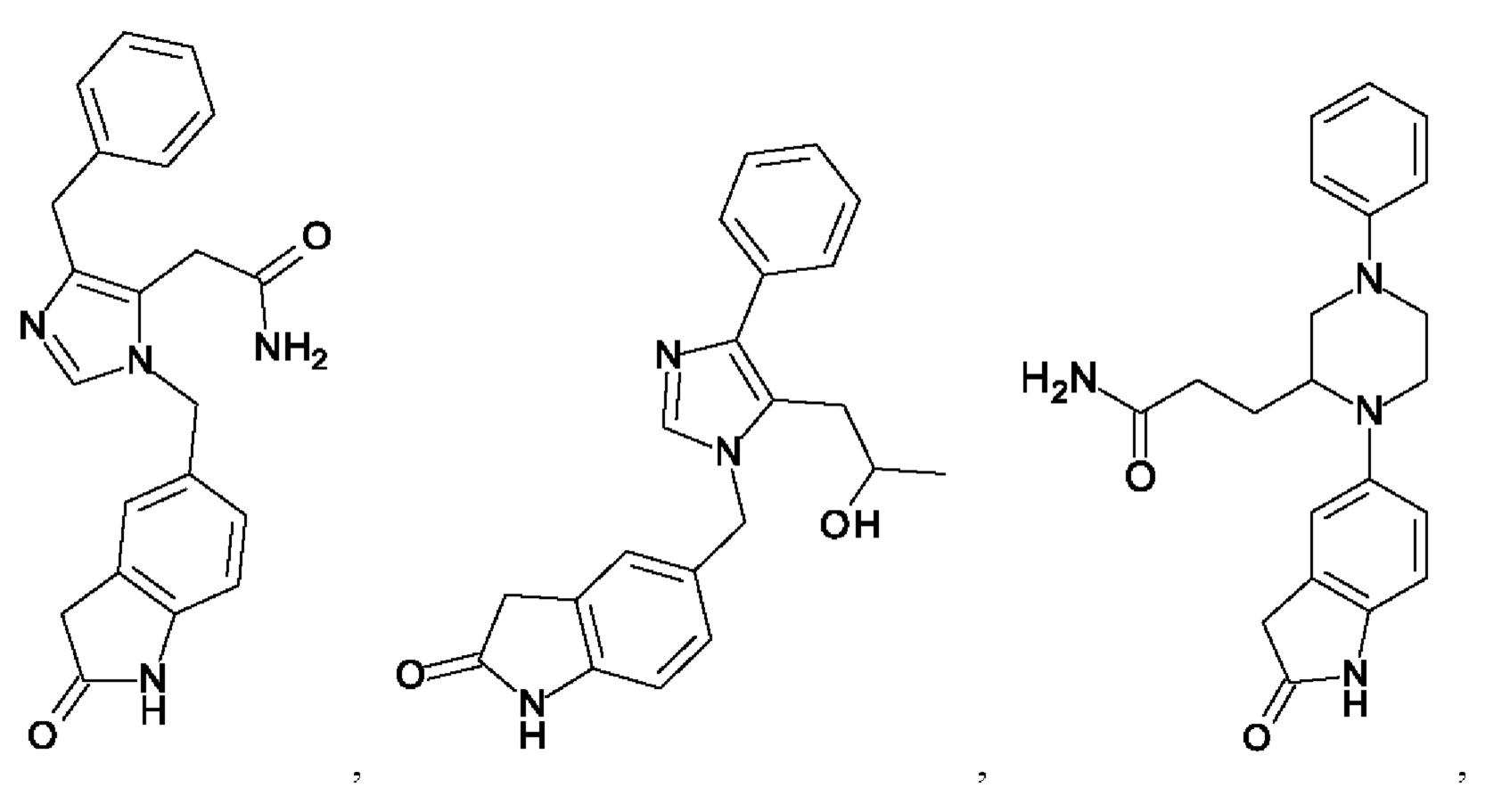











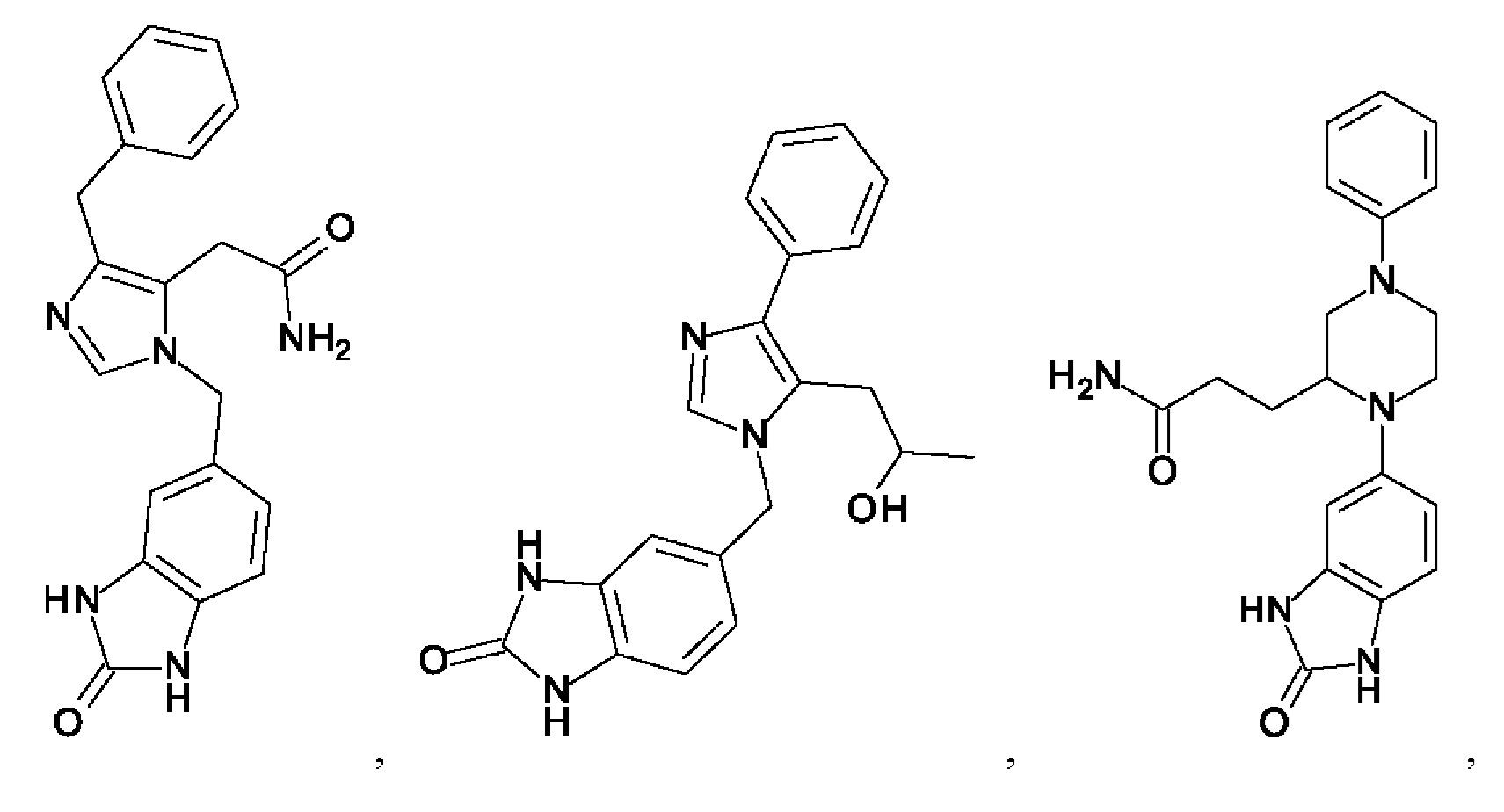




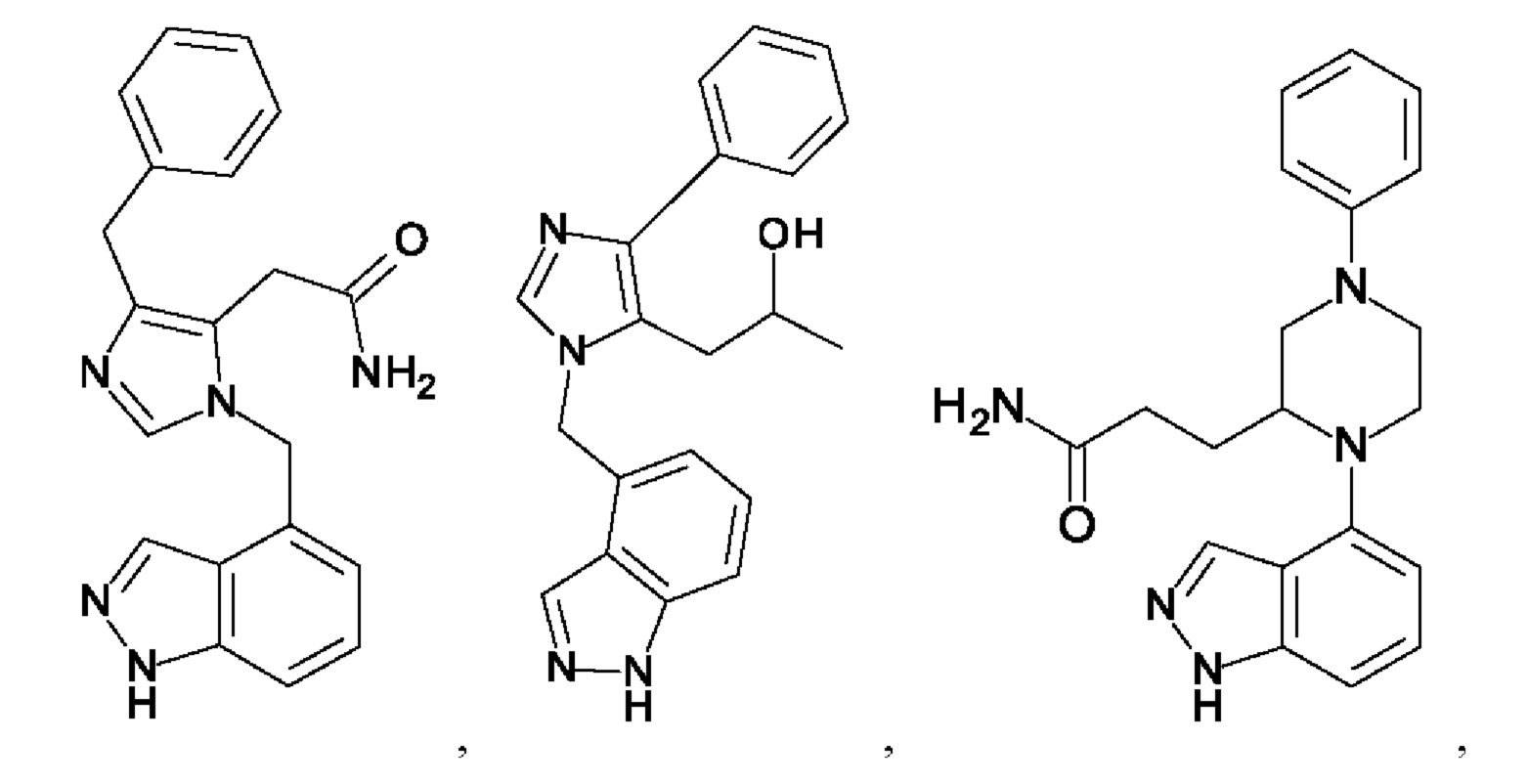

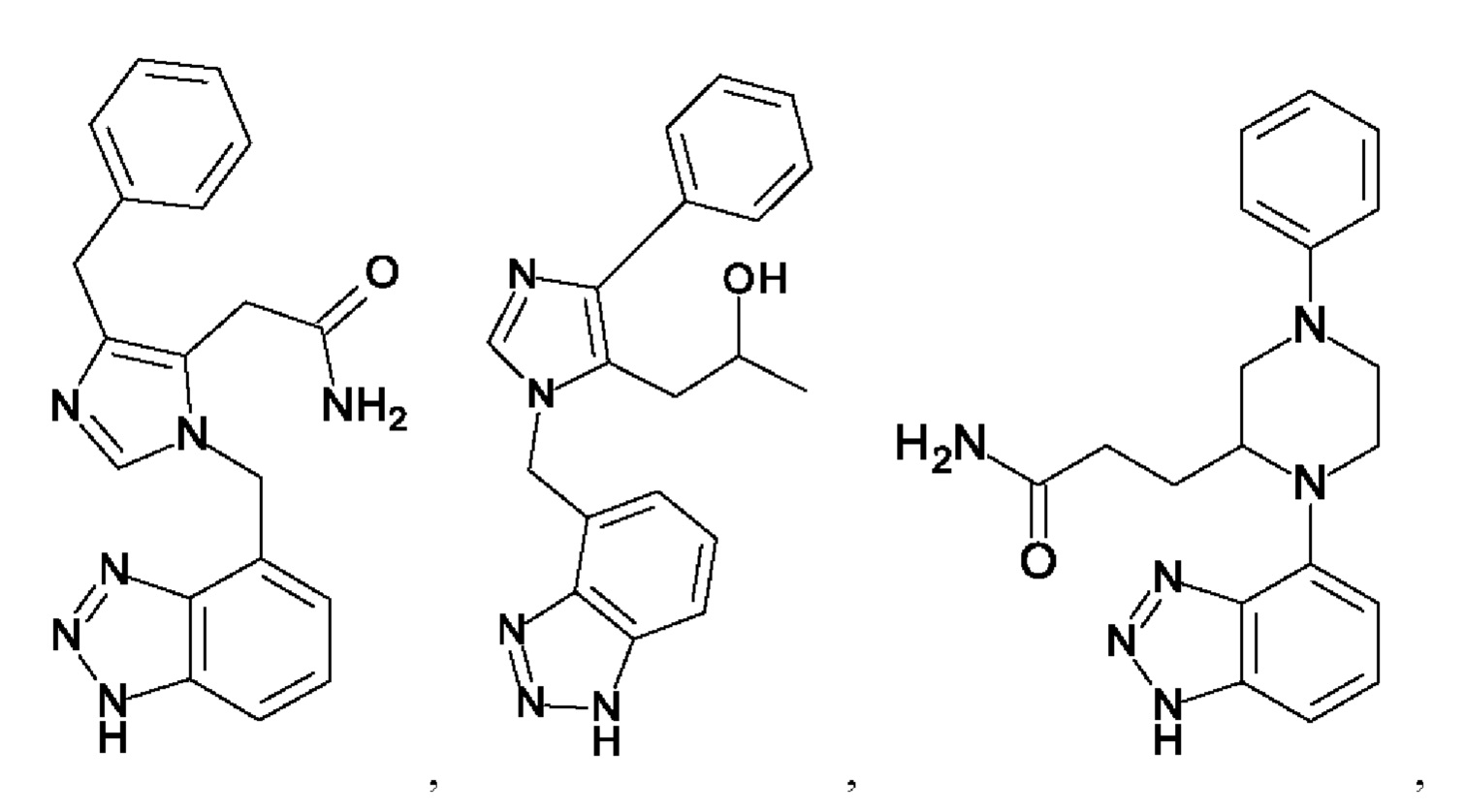


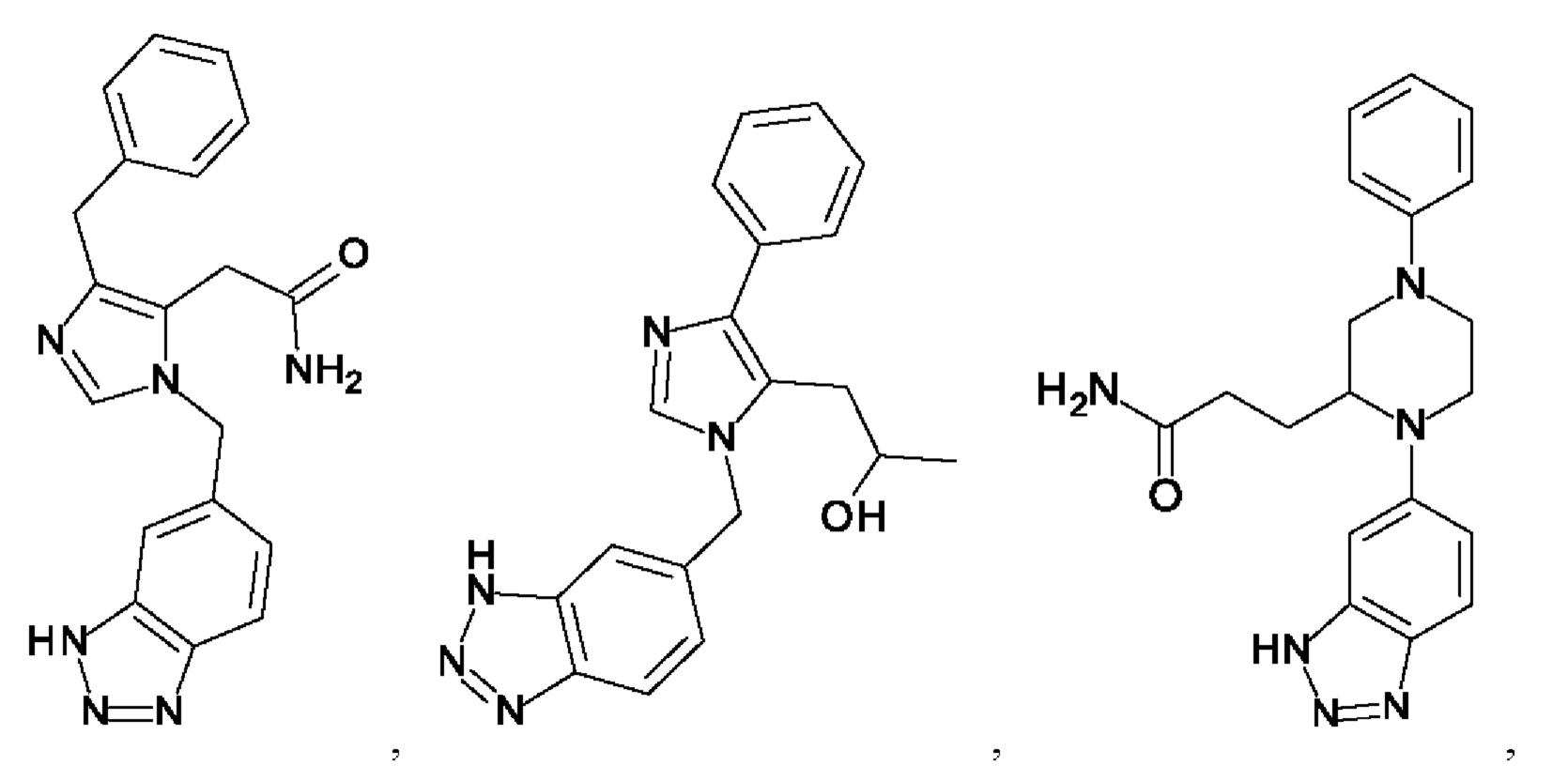



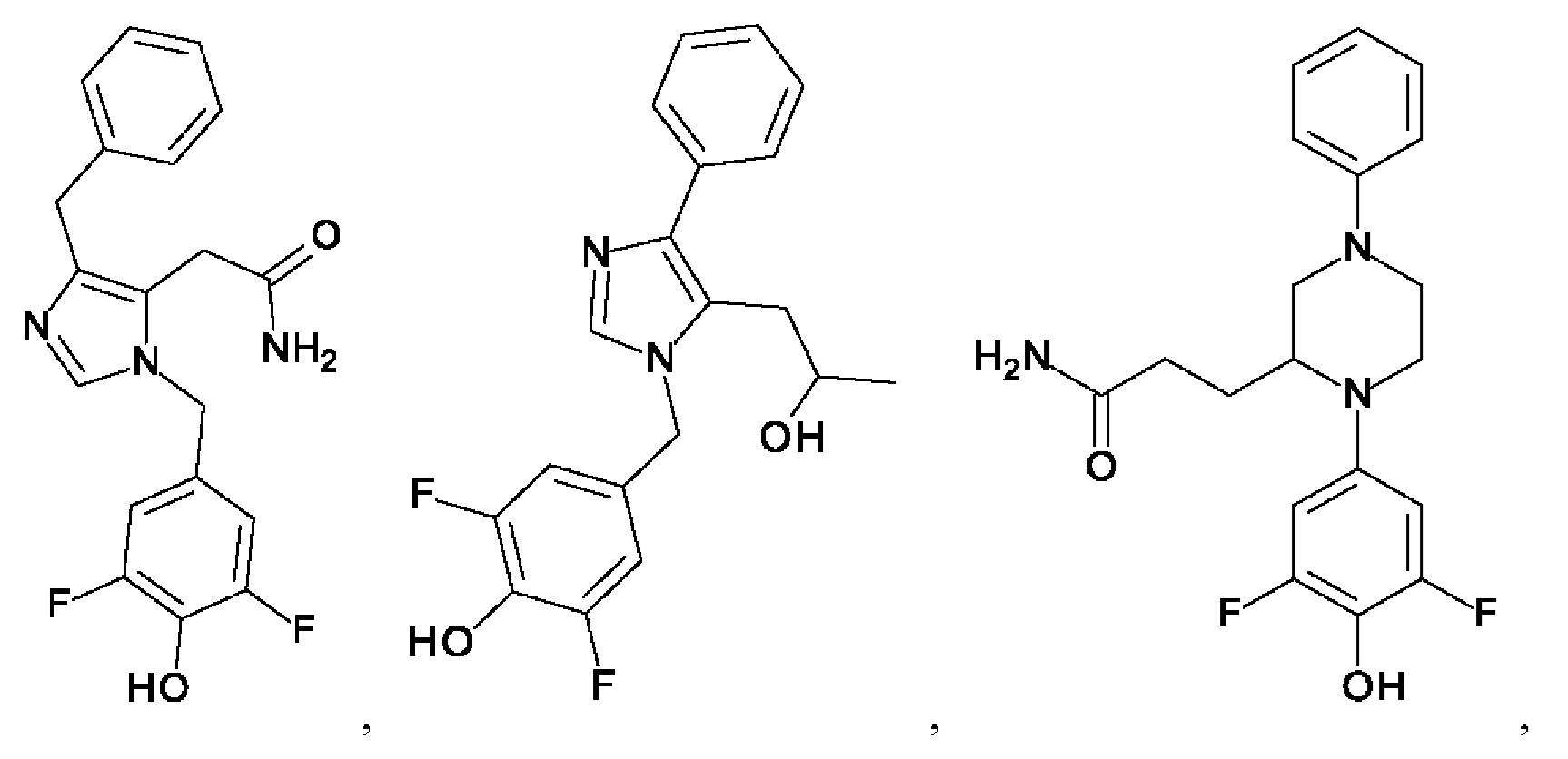
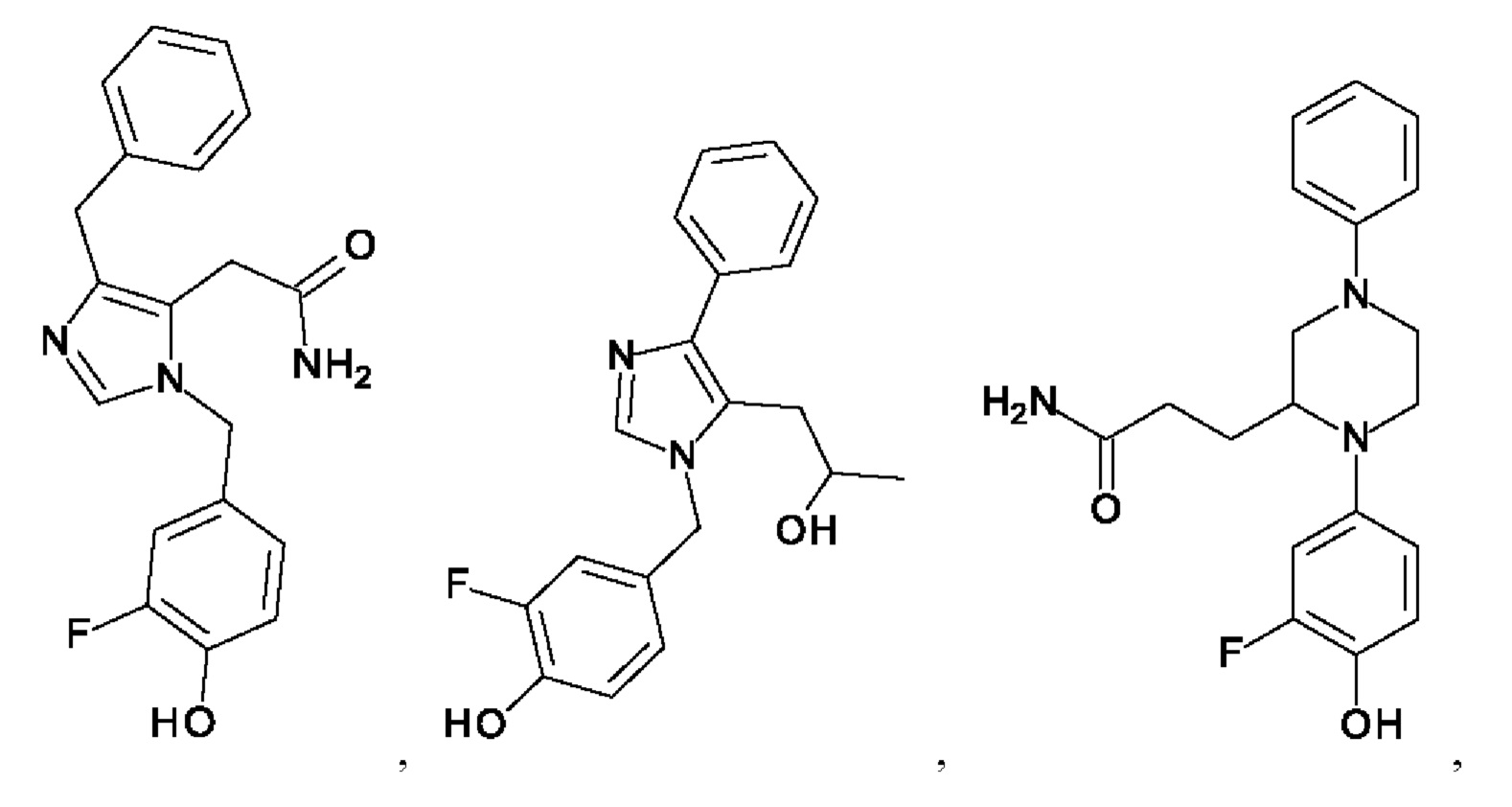

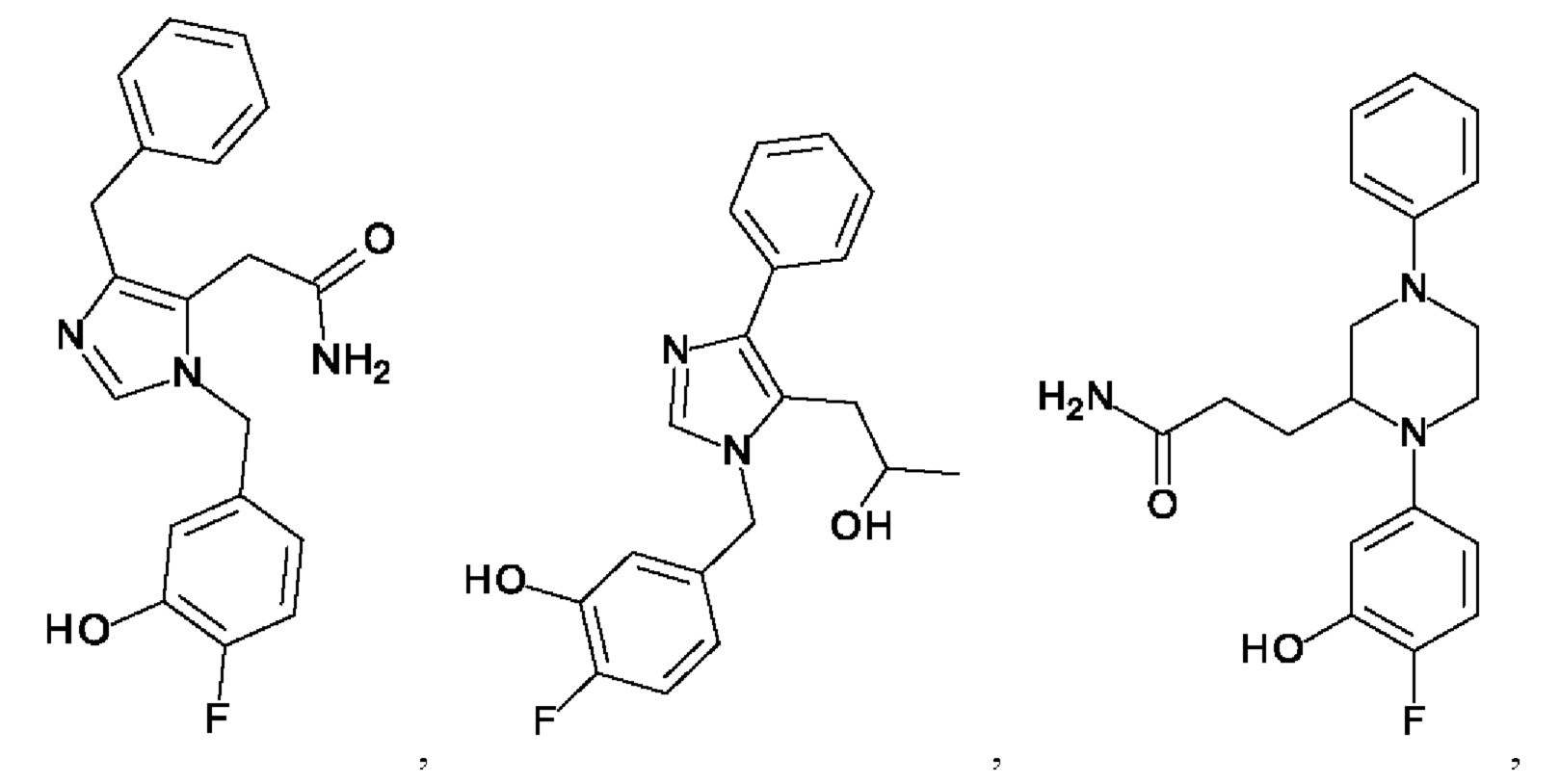














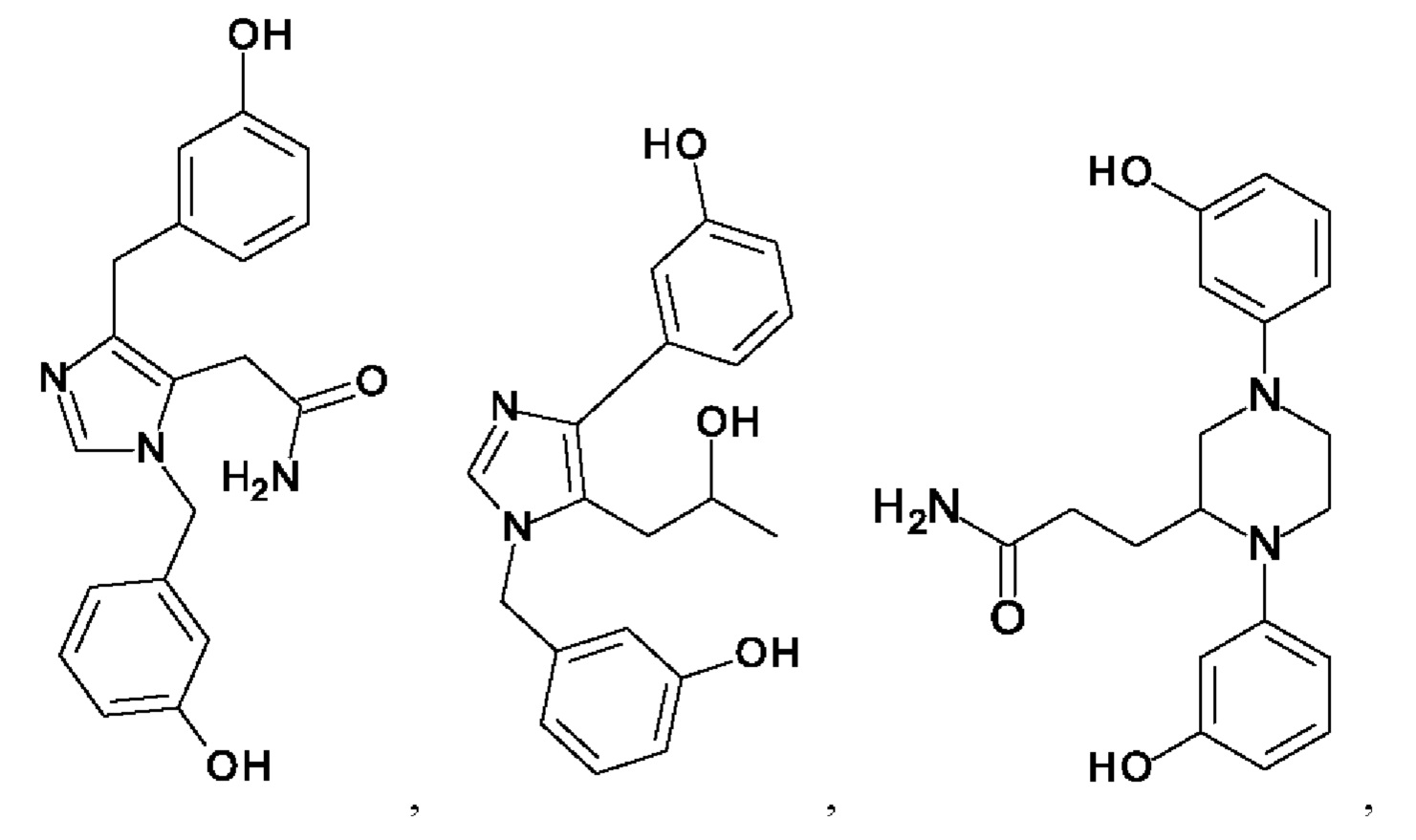








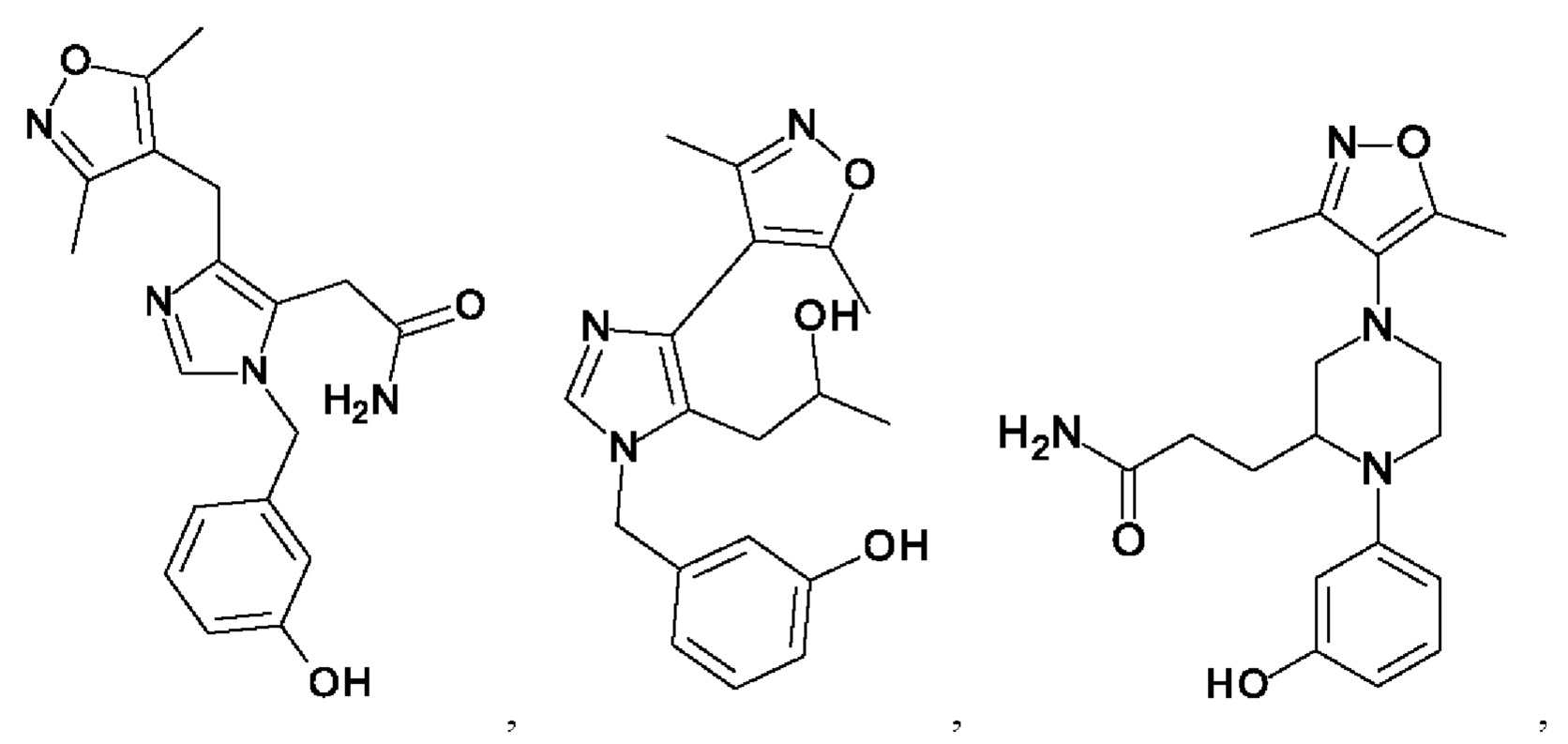











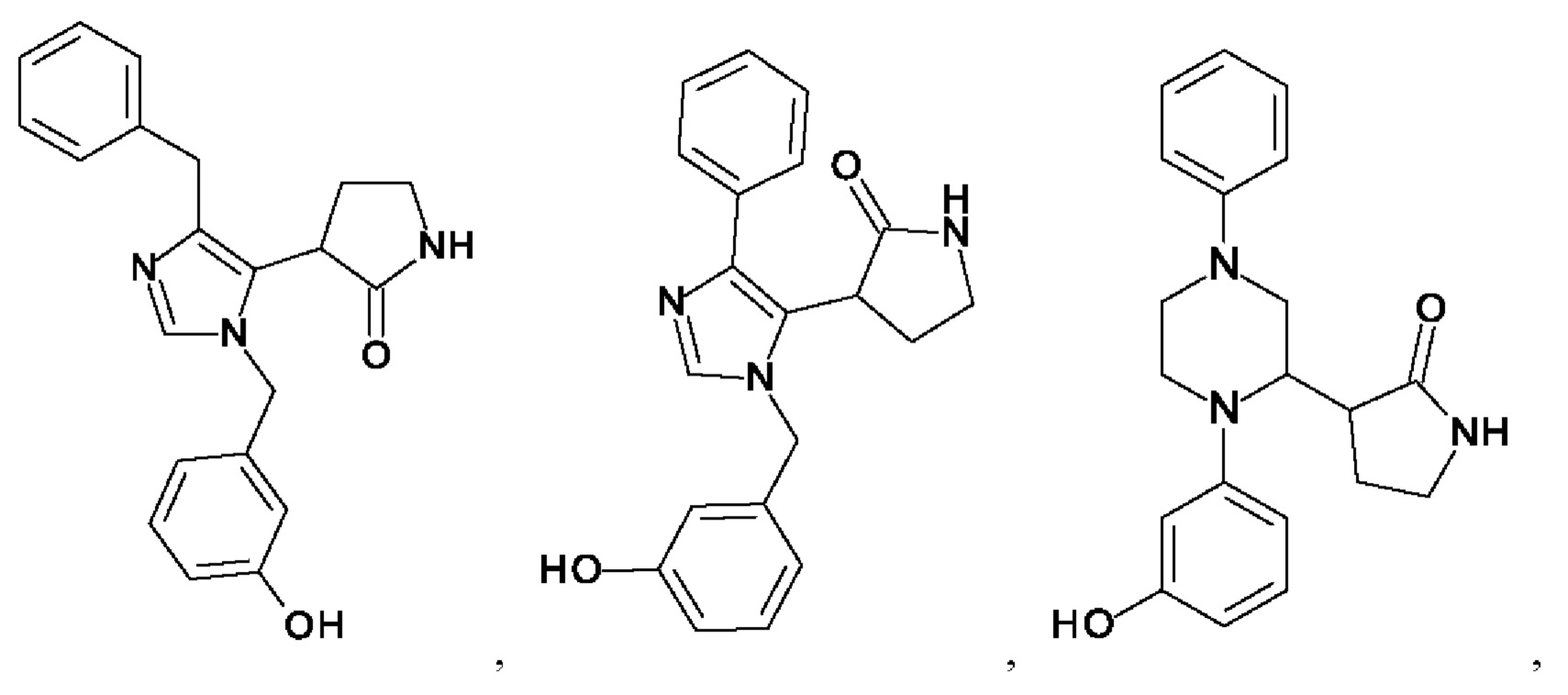
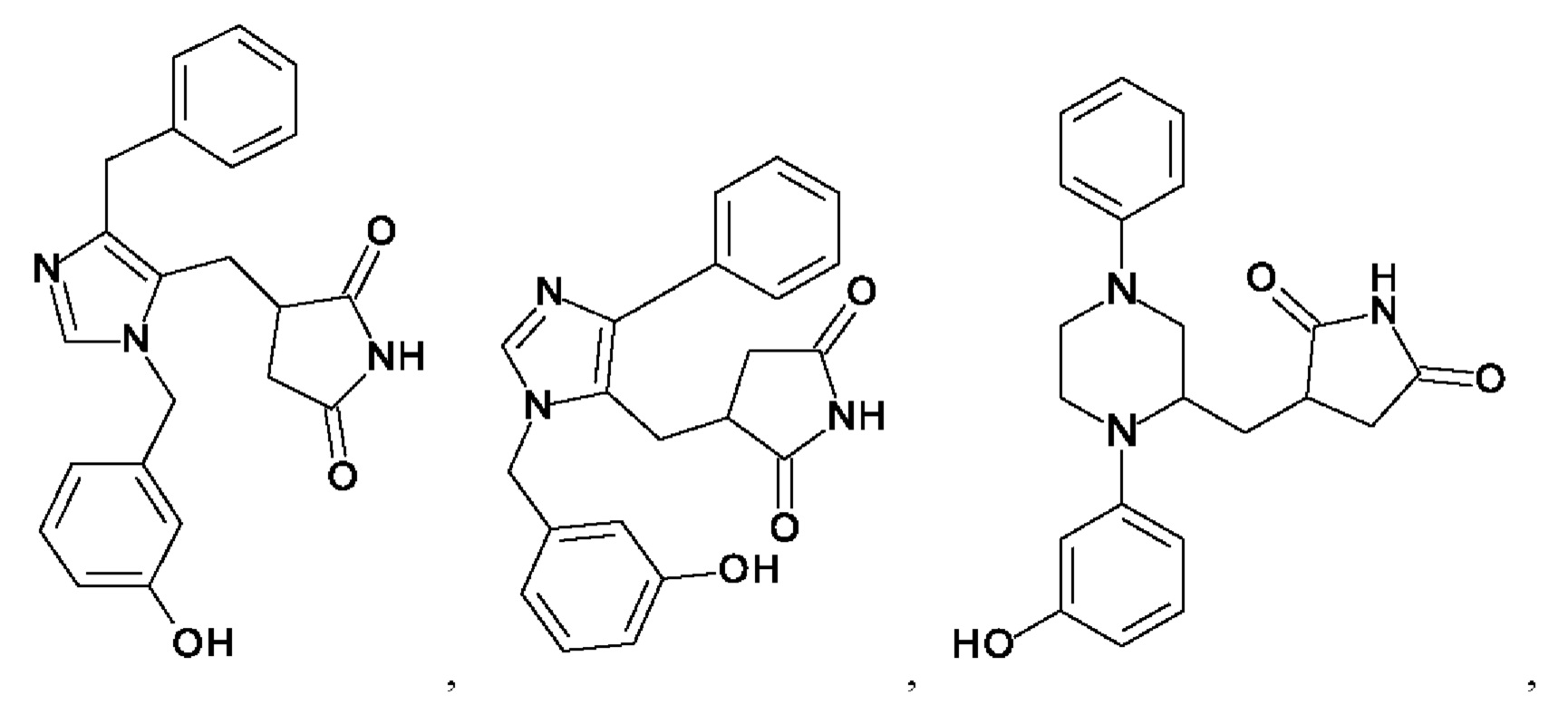















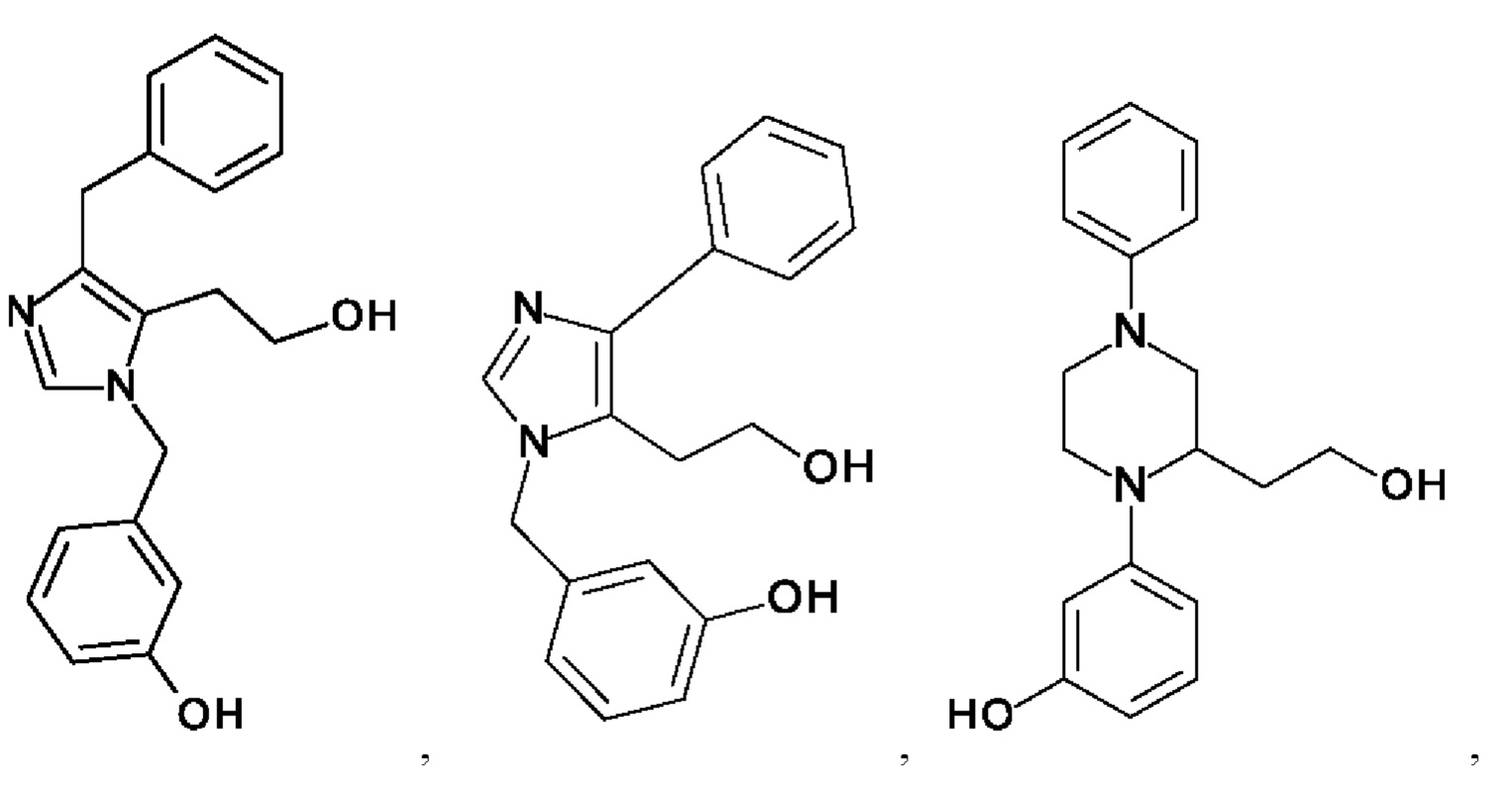


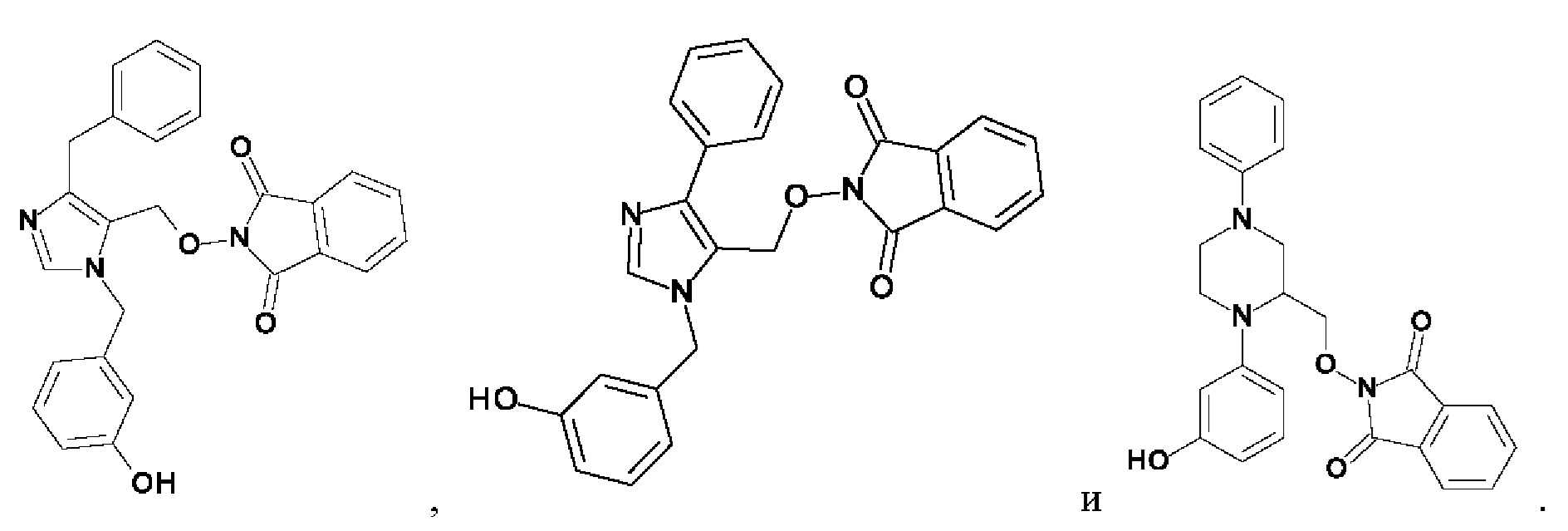
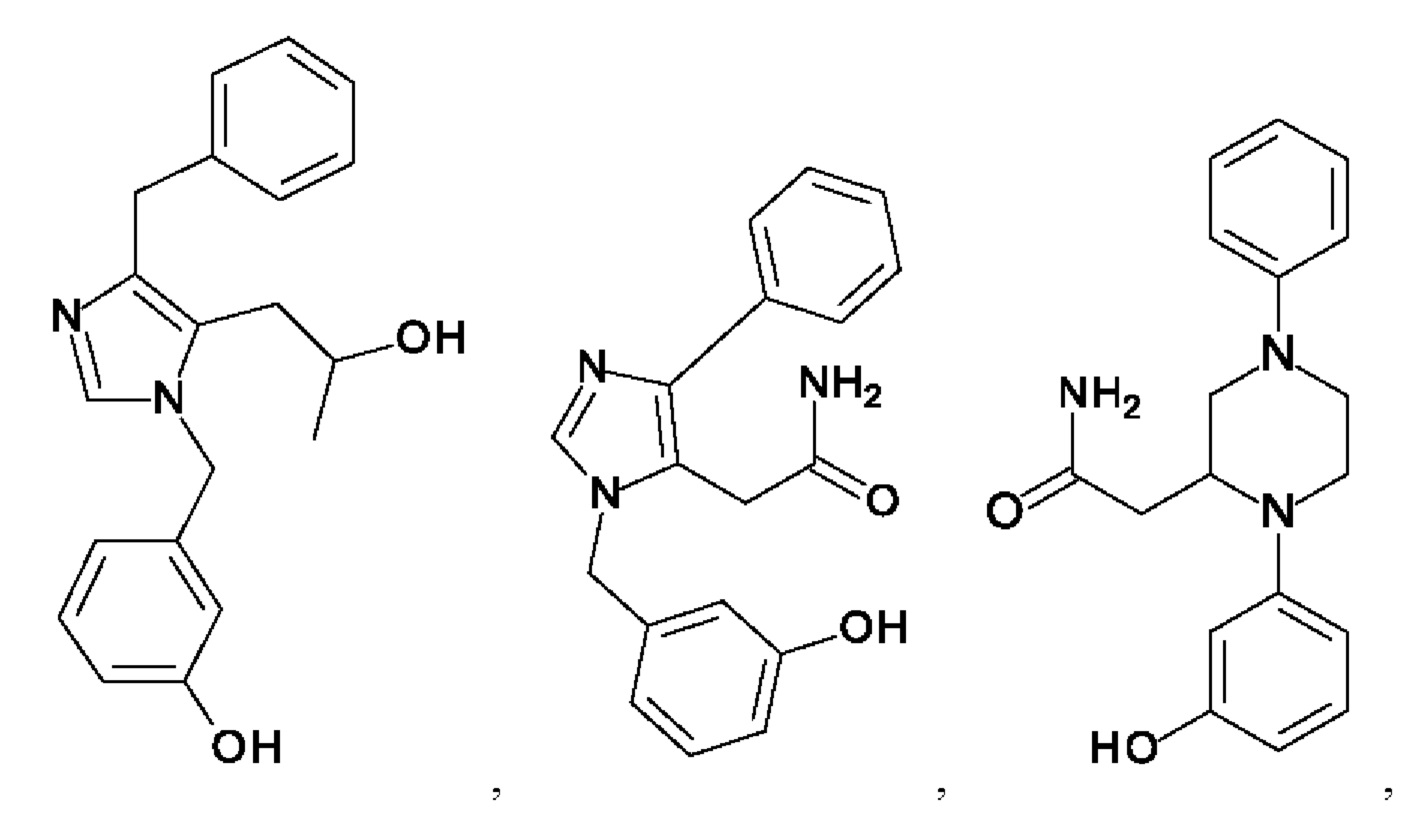
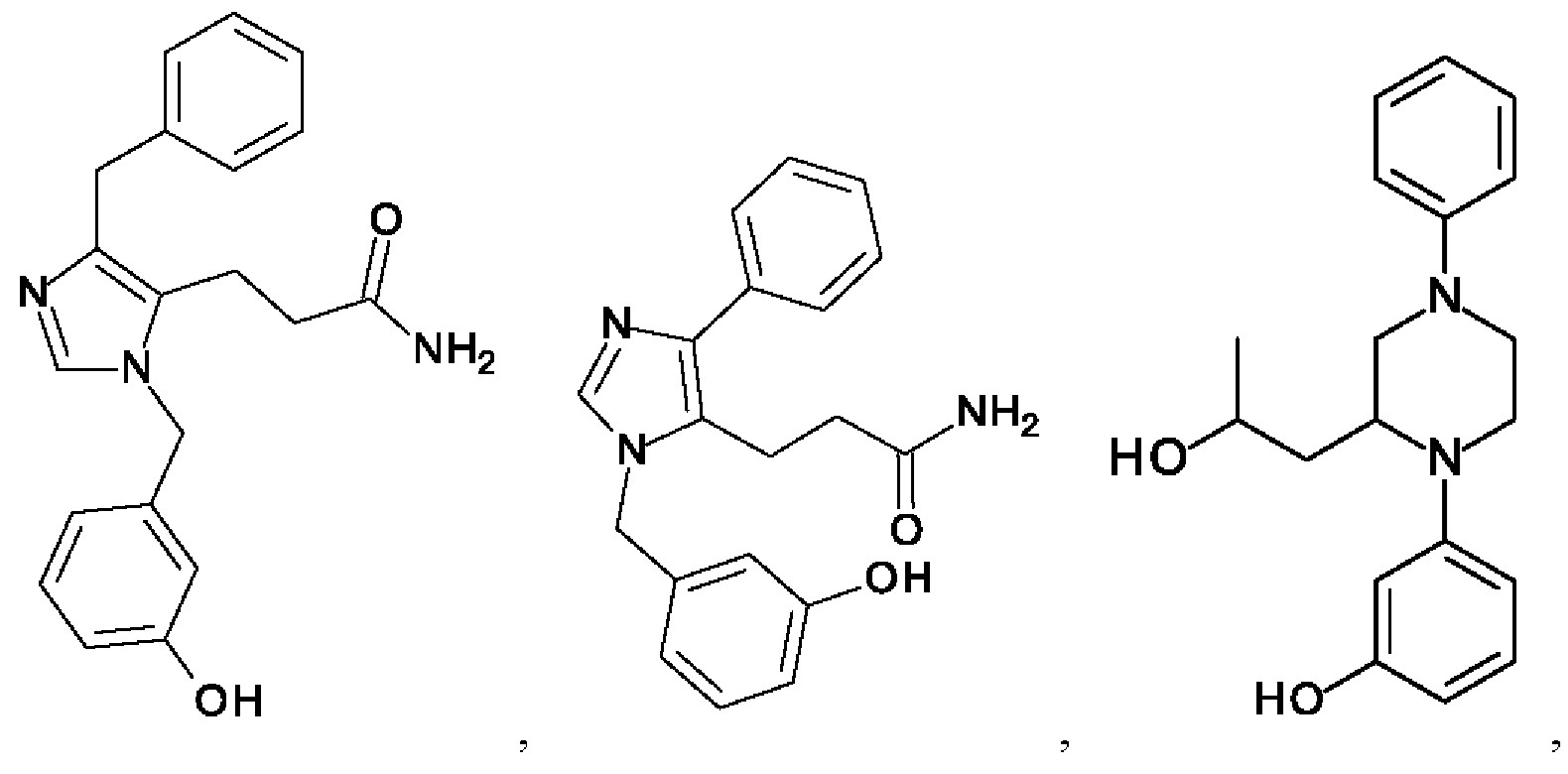
[0026] В одном из воплощений соединение выбрано из:

[0027] Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение по настоящему изобретению и фармацевтически приемлемый эксципиент.
[0028] Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения гипертензии или прегипертензии у субъекта, включающему введение субъекту соединения по настоящему изобретению.
[0029] Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения фиброза у субъекта, включающему введение субъекту соединения по настоящему изобретению.
[0030] Согласно другому аспекту настоящее изобретение относится к способу профилактического лечения фиброза у субъекта, включающему введение субъекту соединения по настоящему изобретению.
[0031] Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения гипертензии и фиброза у субъекта, включающему введение субъекту соединения по настоящему изобретению.
[0032] Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения прегипертензии и фиброза у субъекта, включающему введение субъекту соединения по настоящему изобретению.
[0033] Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении гипертензии или прегипертензии.
[0034] Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении фиброза.
[0035] Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в профилактическом лечении фиброза.
[0036] Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении гипертензии и фиброза.
[0037] Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении прегипертензии и фиброза.
[0038] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного средства для терапевтического лечения гипертензии или прегипертензии.
[0039] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного средства для терапевтического лечения фиброза.
[0040] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного средства для профилактического лечения фиброза.
[0041] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного средства для терапевтического лечения гипертензии и фиброза.
[0042] Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного средства для терапевтического лечения прегипертензии и фиброза.
[0043] В одном из воплощений фиброз представляет собой фиброз миокарда.
[0044] В одном из воплощений фиброз представляет собой фиброз почки.
[0045] В одном из воплощений фиброз представляет собой фиброз печени.
[0046] В одном из воплощений фиброз представляет собой фиброз легкого.
[0047] Если контекст четок не требует иного, везде в описании и формуле изобретения слова "содержат", "содержащий" и т.п.следует понимать в инклюзивном смысле в отличие от эксклюзивного или исчерпывающего смысла, то есть в смысле "включающий, но без ограничения".
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
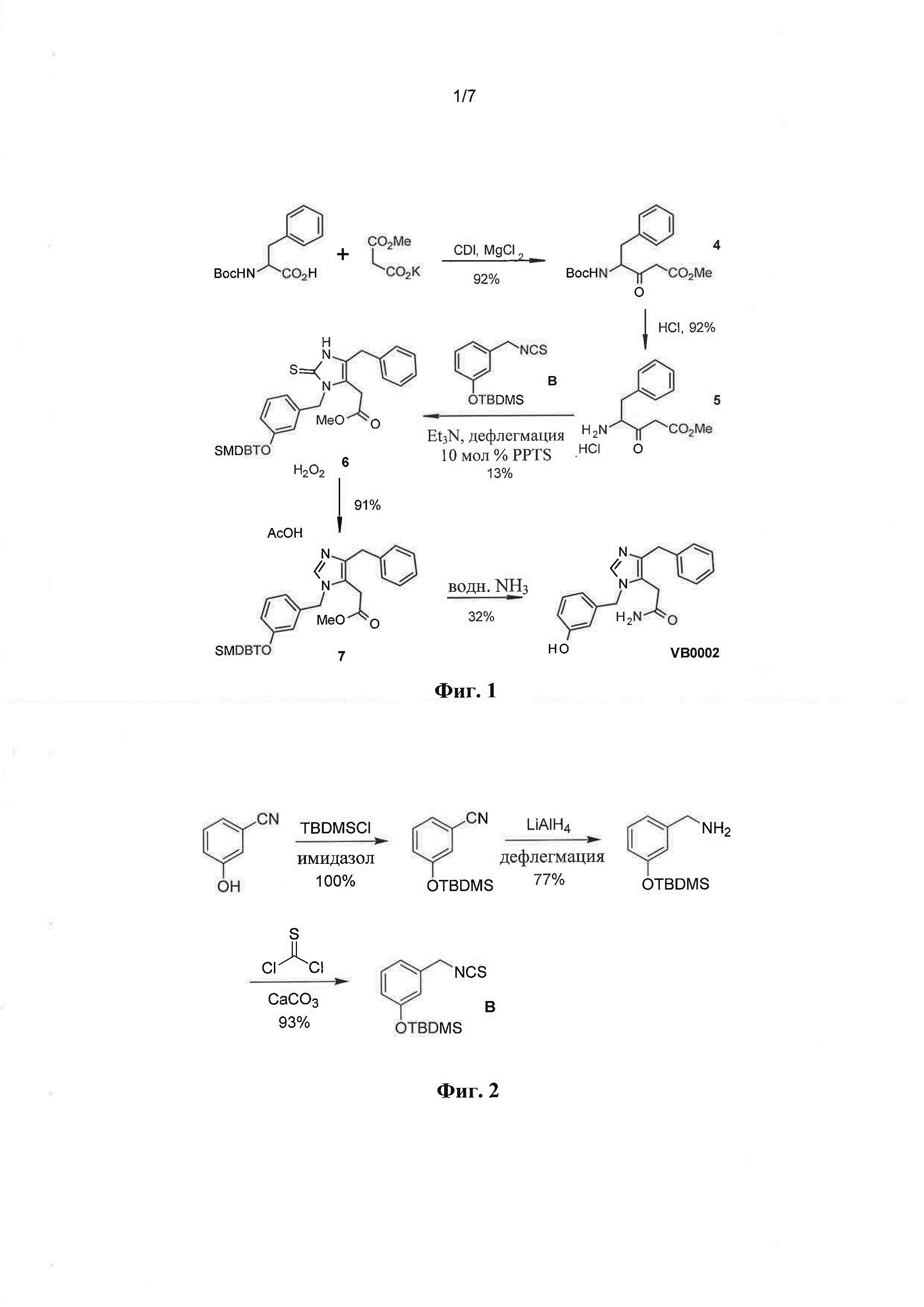
[0048] Фиг. 1: Синтез 2-[4-бензил-1-(3-гидроксибензил)-1Н-имидазол-5-ил]ацетамида (VB0002).
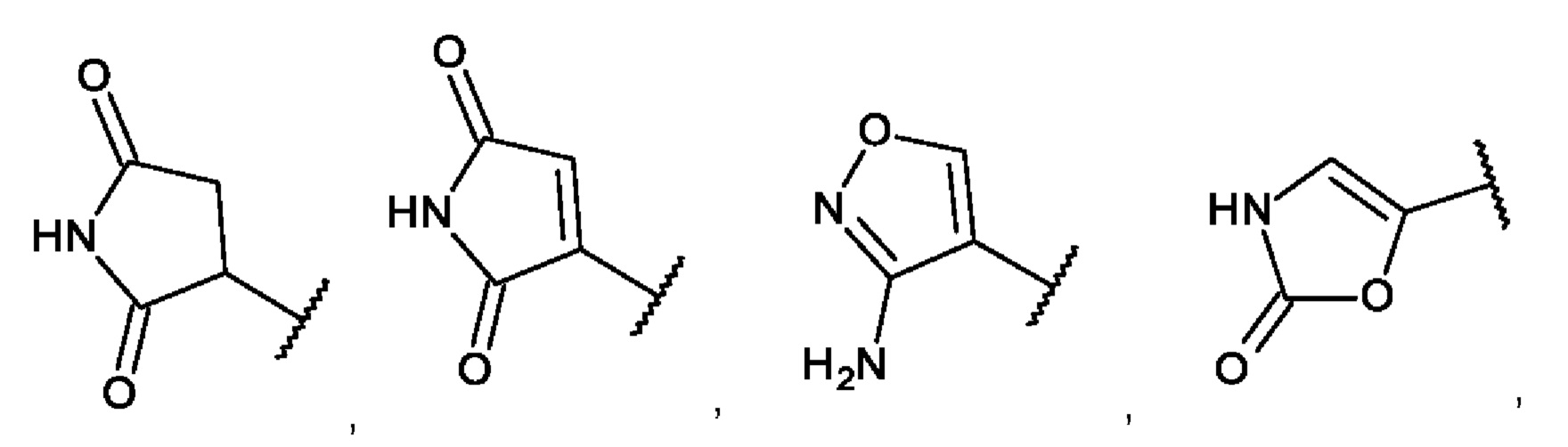
[0049] Фиг. 2: Синтез 3-(трет-бутилдиметилсилилокси)бензилизотиоцианата (соединение В).
[0050] Фиг. 3: Синтез 3-[5-(2-гидроксипропил)-4-фенил-имидазол-1-илметил]-фенола (VB0003).
[0051] Фиг. 4: Синтез α-тозилбензилизоцианида (соединение С).
[0052] Фиг. 5 Синтез 3-(бензилокси)бензиламина (соединение D).
[0053] Фиг. 6: Синтез 3,3-этилендиокси-1-бутаналя (соединение Е).
[0054] Фиг. 7: Синтез (S)-3-[1-(3-гидрокси)фенил-4-фенилпиперазин-2-ил]пропанамида (VB0005).
[0055] Фиг. 8: Систолическое давление крови у SHR на диете с 2,2% соли с использованием VB0002 (в 20% DMSO) в дозе 20 пмоль/кг/мин или являющегося контролем носителя (20% DMSO), которые вводили внутривенно посредством осмотического мининасоса в течение 4 недель. * р<0,005 относительно 18-недельного контроля.
[0056] Фиг. 9: Систолическое давление крови у SHR на диете с 2,2% соли с использованием VB0003 (в 5% этаноле) в дозе 20 пмоль/кг/мин, VB0005 (в 5% этаноле) в дозе 20 пмоль/кг/мин или являющегося контролем носителя (5% этанол), которые вводили в питьевом растворе в течение 4 недель. * р<0,0005 относительно 18-недельного контроля.
[0057] Фиг. 10: Фиброз миокарда у SHR на диете с 2,2% соли с использованием VB0002 (в 20% DMSO) в дозе 20 пмоль/кг/мин или являющегося контролем носителя (20% DMSO), которые вводили внутривенно посредством осмотического мининасоса в течение 4 недель. * р<0,005 относительно 18-недельного контроля, ** р<0,0005 относительно 18-недельного контроля.
[0058] Фиг. 11: Фиброз миокарда у SHR на диете с 2,2% соли с использованием VB0003 (в 5% этаноле) в дозировках 10, 100 и 500 пмоль/кг/мин или являющегося контролем носителя (5% этанол), которые вводили в питьевом растворе в течение 4 недель. * р<0,005 относительно 18-недельного контроля, ** р<0,0005 относительно 18-недельного контроля, # р<0,01 относительно 14-недельного контроля.
[0059] Фиг. 12: Фиброз миокарда у SHR на диете с 2,2% соли с использованием VB0005 (в 5% этаноле) в дозировке 100 пмоль/кг/мин или являющегося контролем носителя (5% этанол), которые вводили в питьевом растворе в течение 4 недель. * р<0,001 относительно 18-недельного контроля, ** р<0,0005 относительно контроля в течение 18 недель, #р<0,01 относительно 14-недельного контроля.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0060] Настоящее изобретение относится к некоторым соединениям, которые демонстрируют снижение давления крови и антифибротические эффекты в исследованиях с пероральным введением в экспериментальной животной модели. В отношении антифибротической активности соединения по настоящему изобретению являются эффективными в предупреждении фиброза, замедлении прогрессирования установленного фиброза и/или снижении степени (реверсировании) установленного фиброза. Эти полученные данные важны в том, что касается области распространения и тяжести состояний, которые можно лечить соединениями по настоящему изобретению.
[0061] Соединения по настоящему изобретению представлены формулами:

где:
Х выбран из группы, состоящей из:
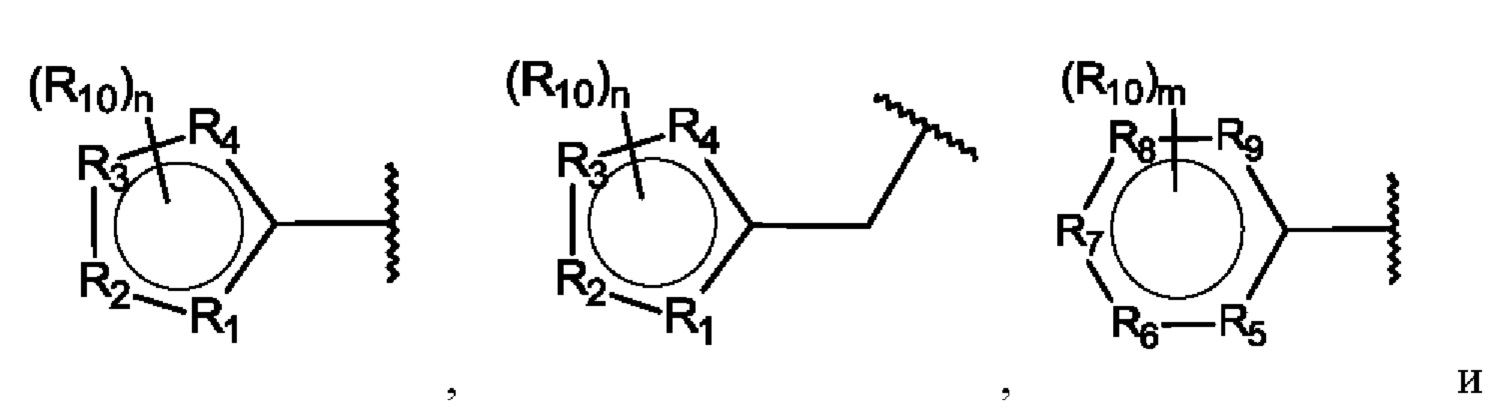

R1-R9 независимо представляют собой С, N, О или S;
R10 независимо выбран из C1-6алкила, галогена, C0-6алкилкарбоновой кислоты, амино, гидрокси и C1-6алкокси;
Y представляет собой А, СН2-А или СН-А;
А выбран из возможно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; возможно замещенного C1-6алкоксиламина; возможно замещенного C1-6алкиламина; возможно замещенной C0-6алкилкарбоновой кислоты; возможно замещенного C1-6алкилгидроксила; возможно замещенного насыщенного или ненасыщенного C0-6алкил-бициклического гетероциклила; и возможно замещенного насыщенного или ненасыщенного C1-6алкоксил-бициклического гетероциклила;
Z выбран из группы, состоящей из:


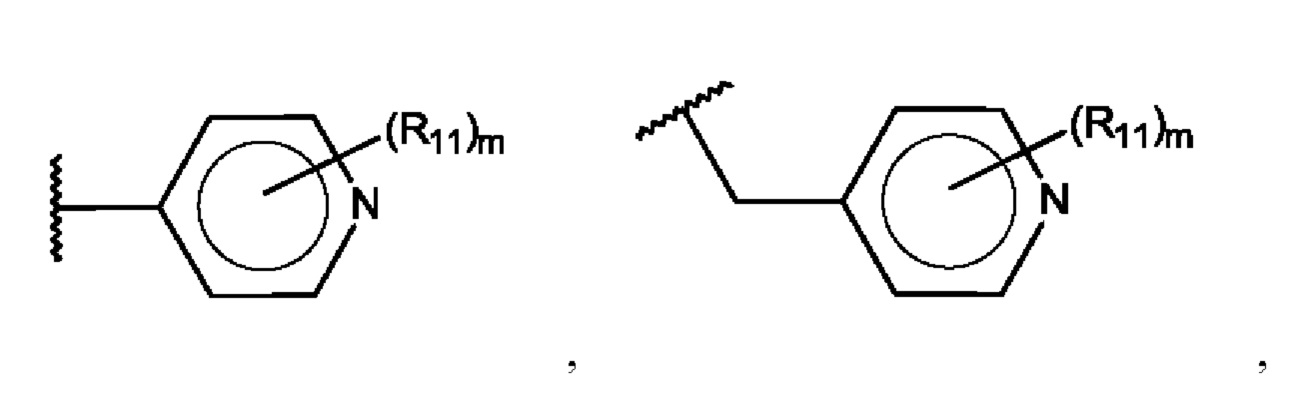


R11 независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
R12, R14 и R15 независимо представляют собой С, СН, СН2, О, N, NH или S;
R13 представляет собой С, СН, СН2, N, NH, С-CF3, СН-CF3 или С=O;
m равно 0, 1, 2, 3, 4 или 5; и
n равно 0, 1, 2, 3 или 4, или его стереоизомер или фармацевтически приемлемая соль.
[0062] Нижеследующие соединения являются конкретными, но не ограничивающими, примерами соединений по настоящему изобретению:

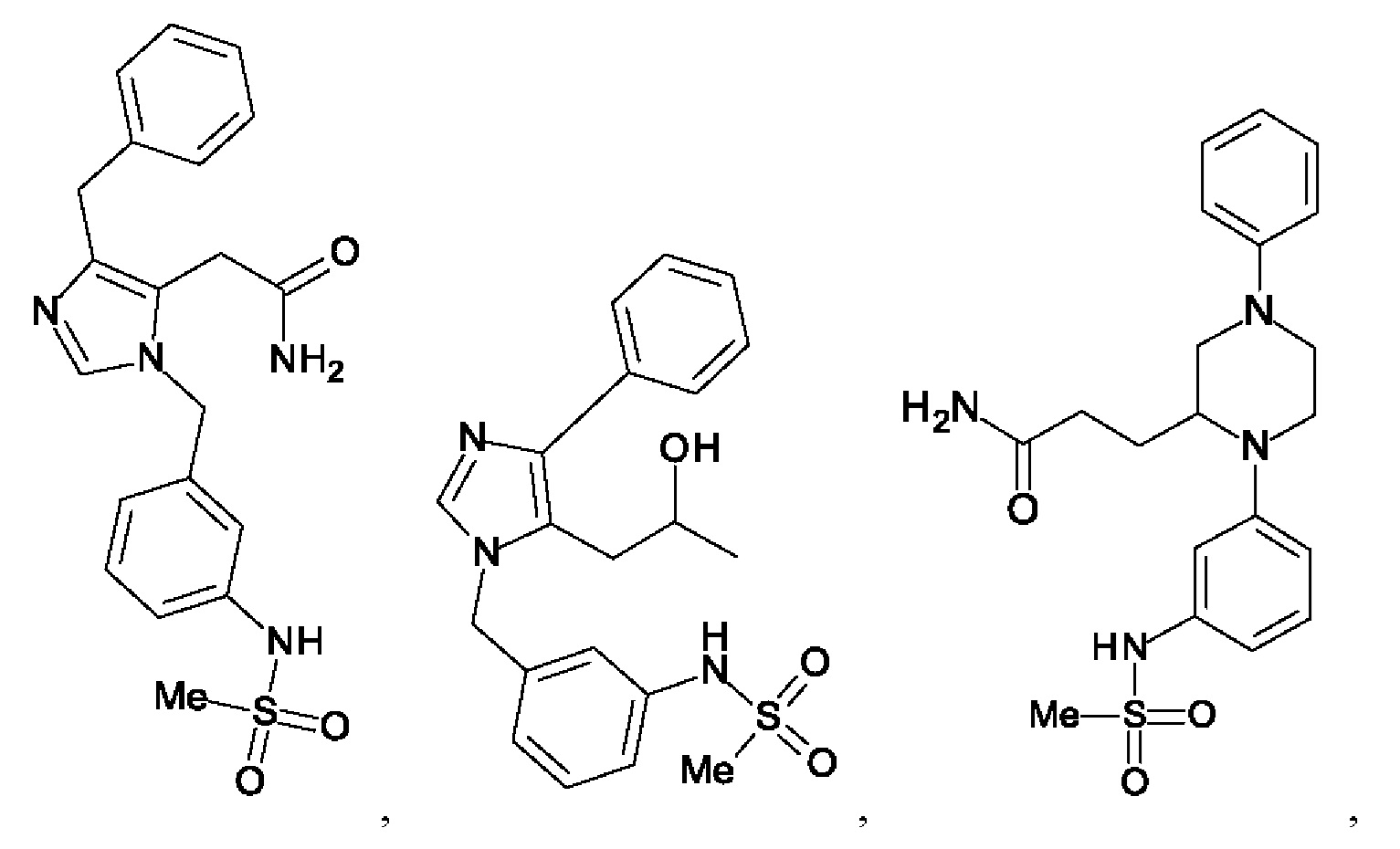


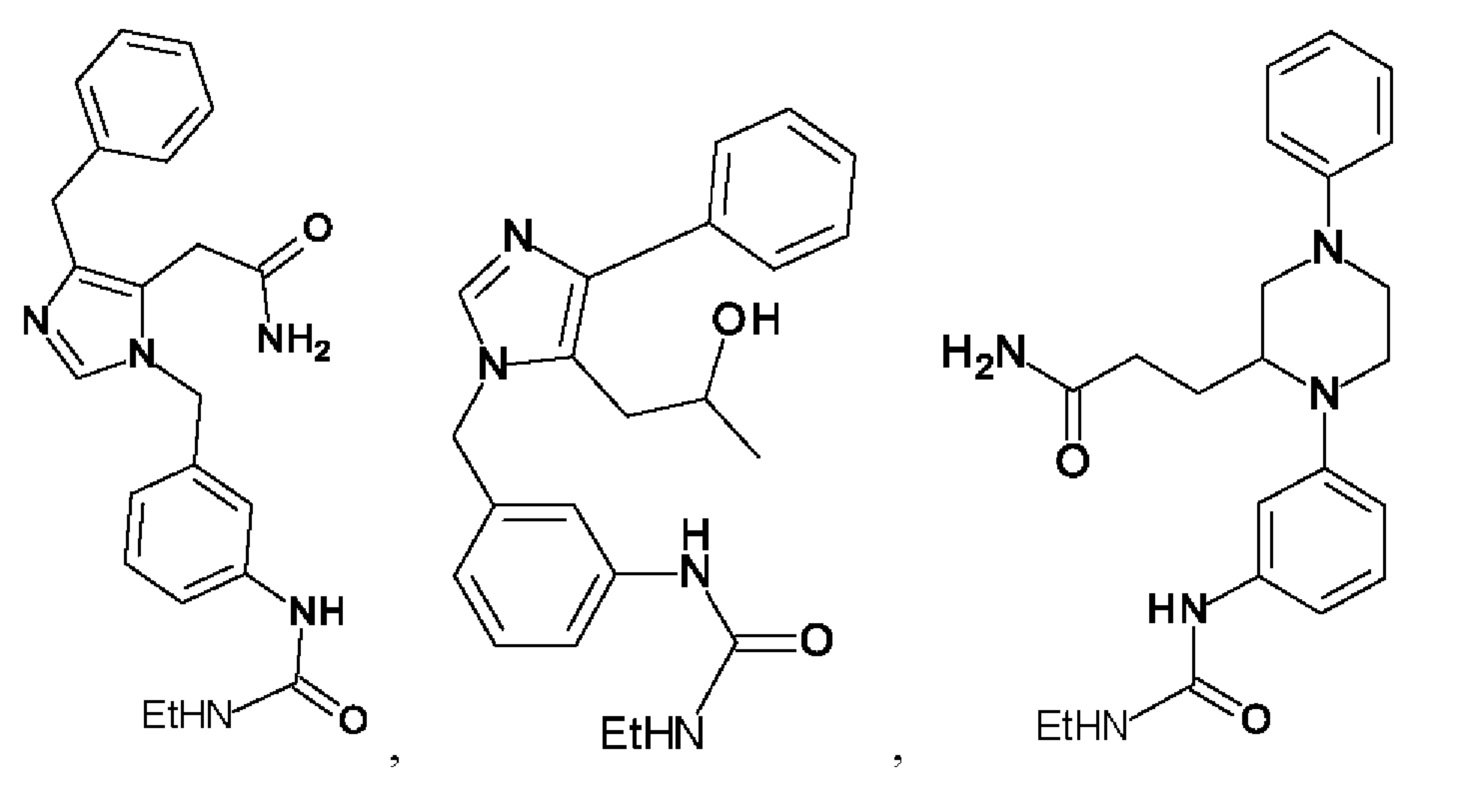
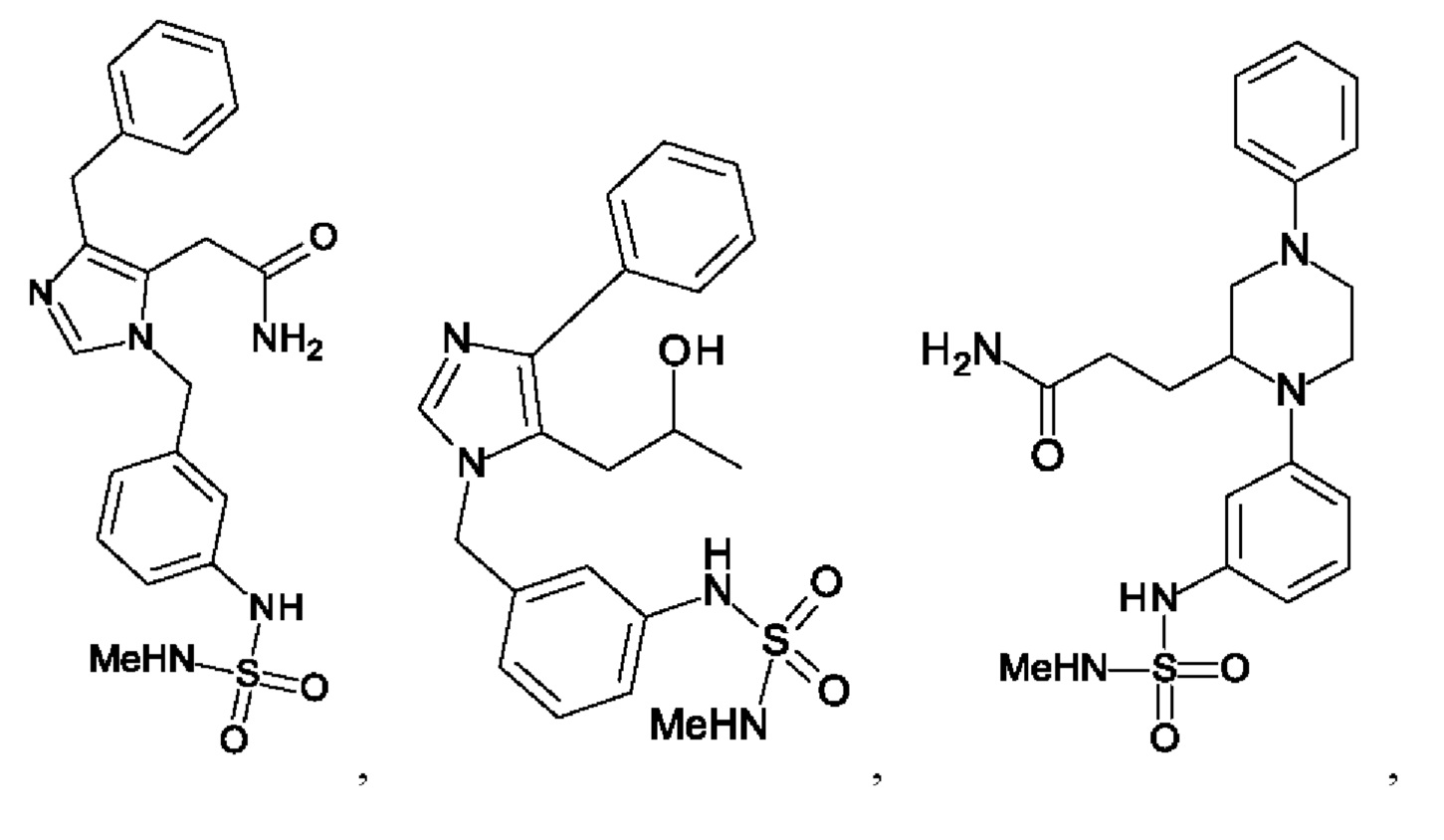

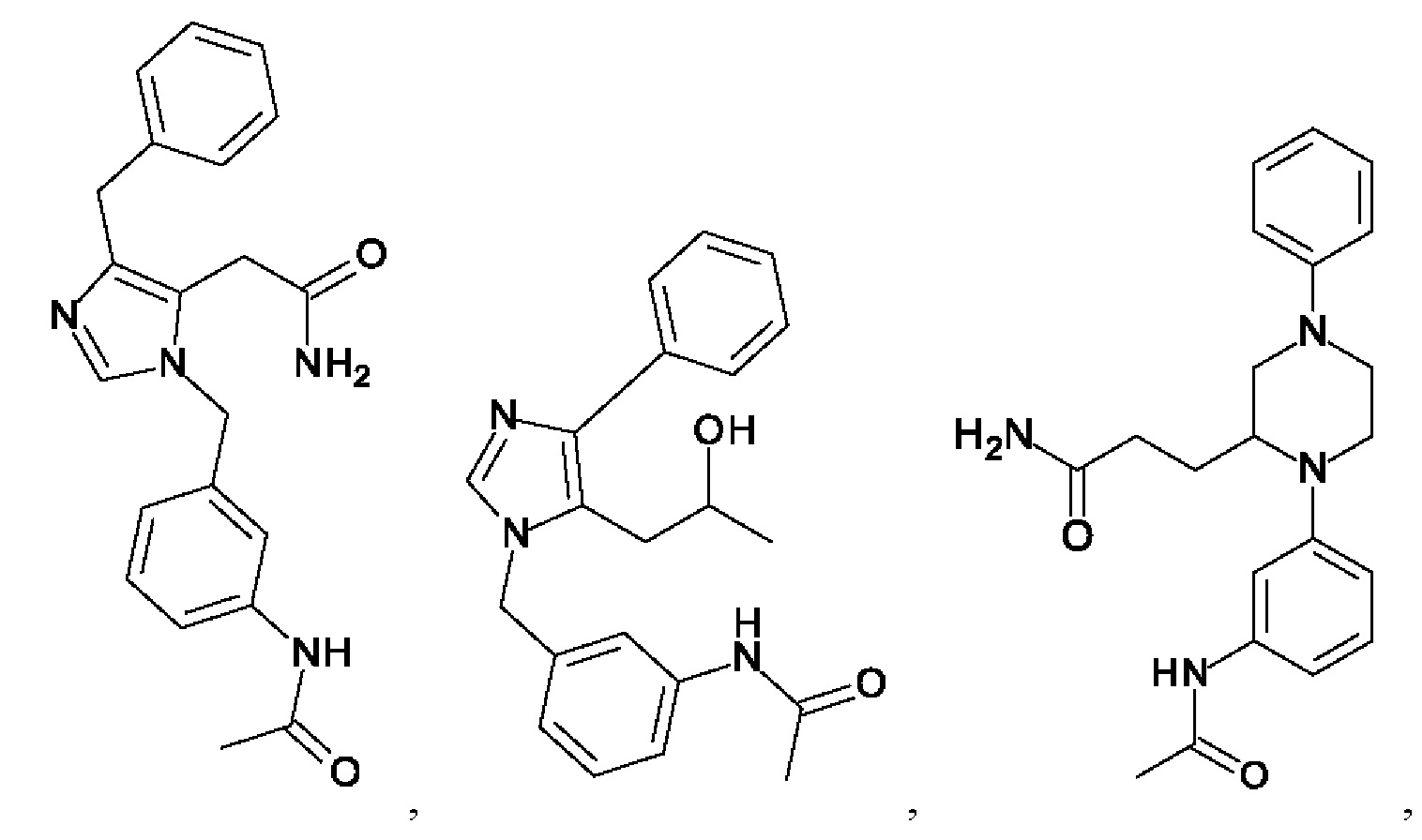




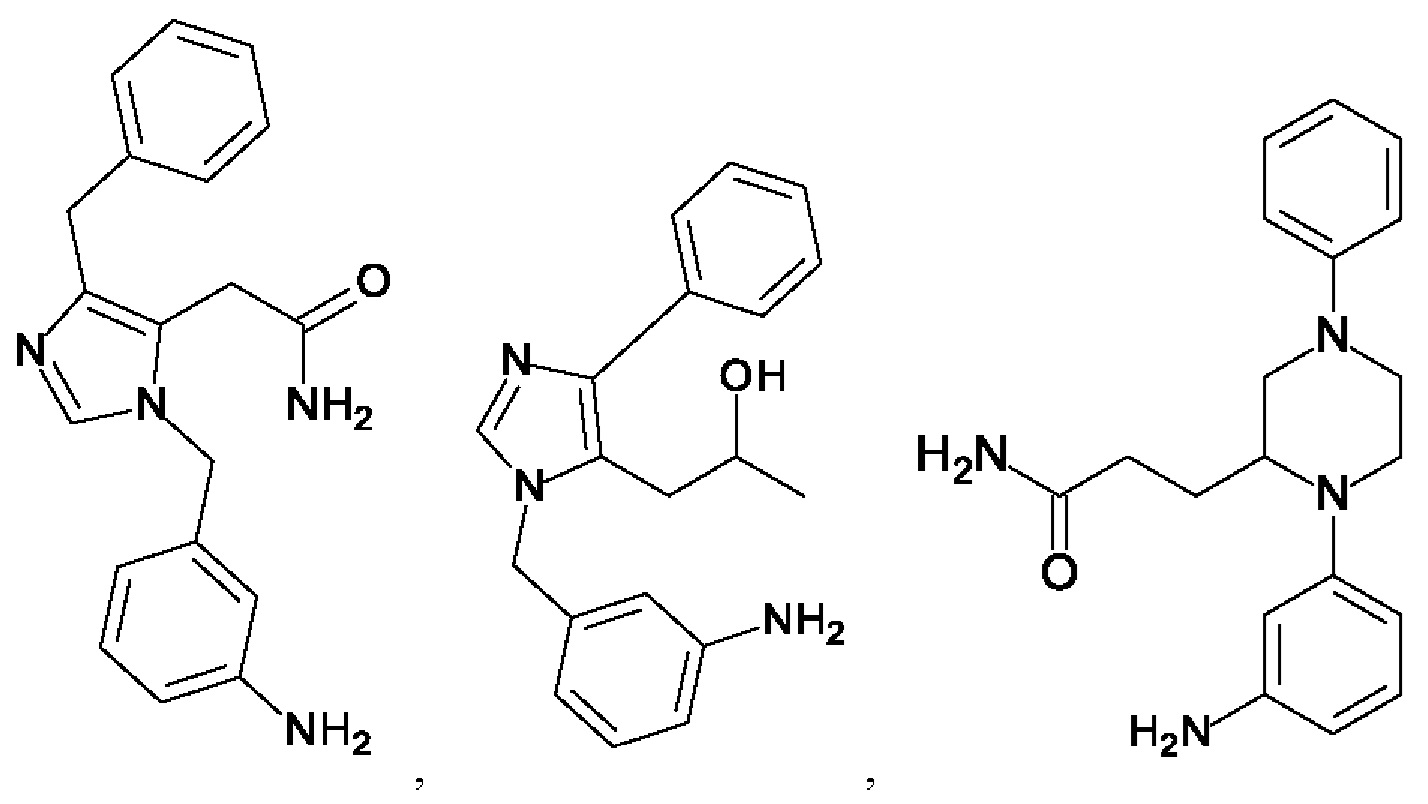
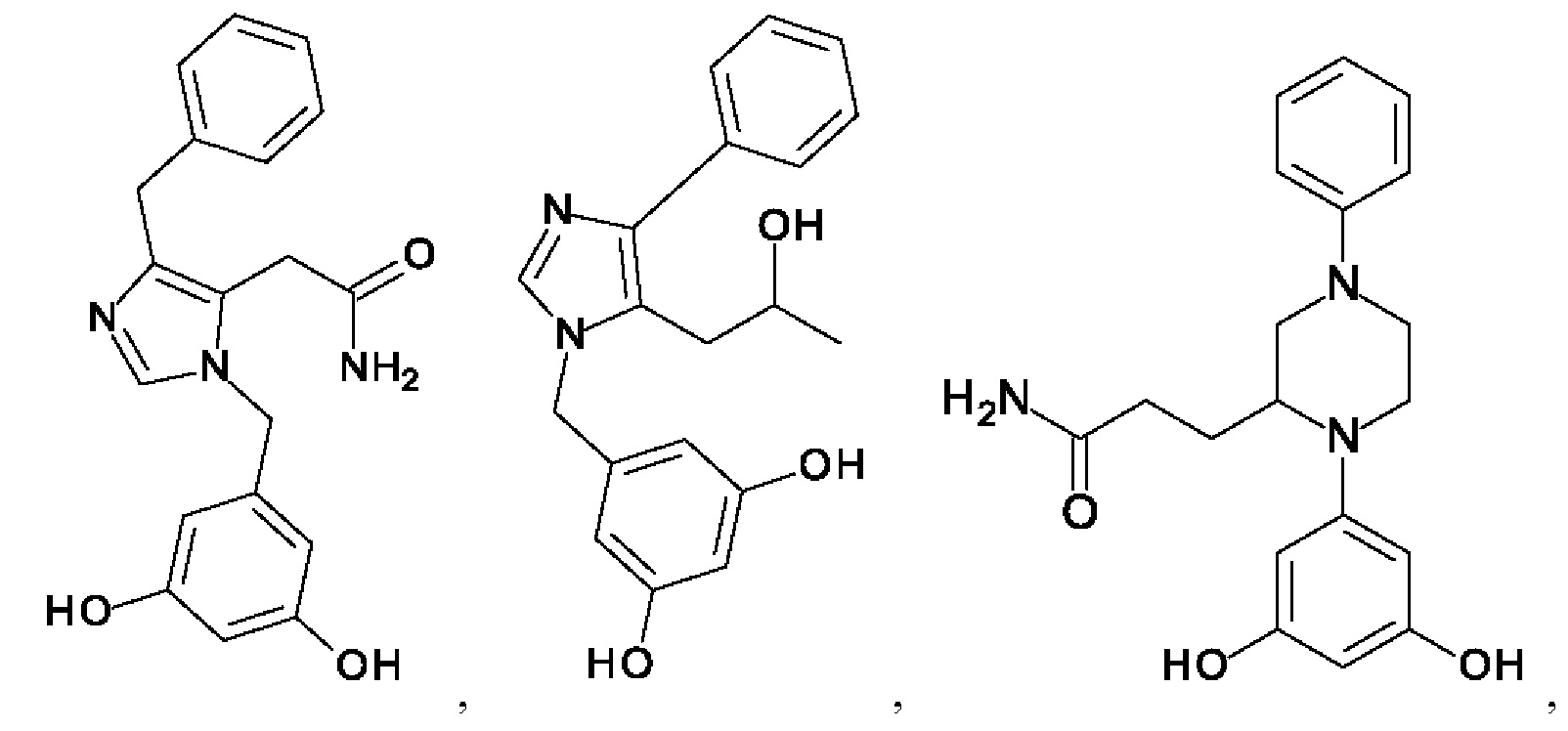
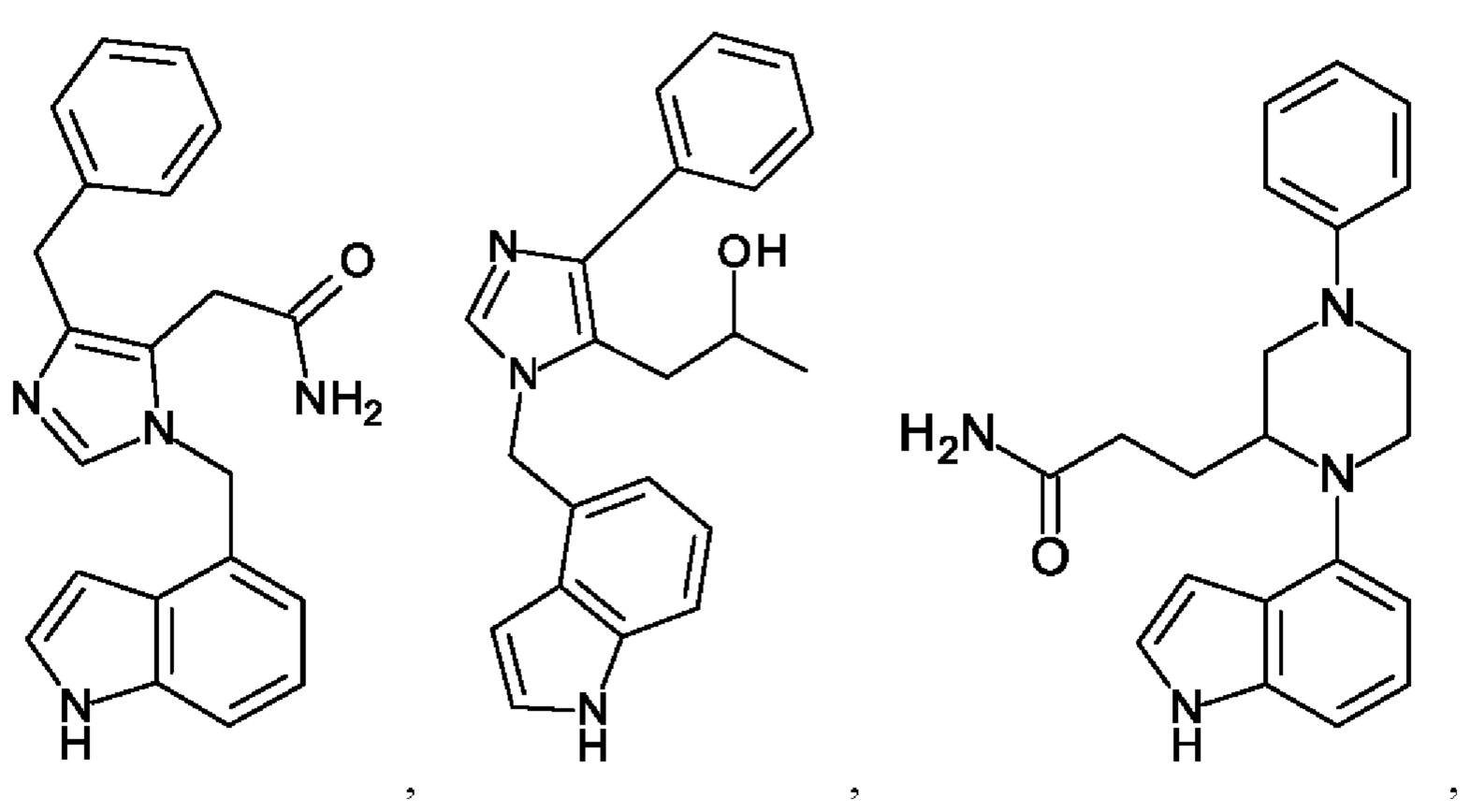
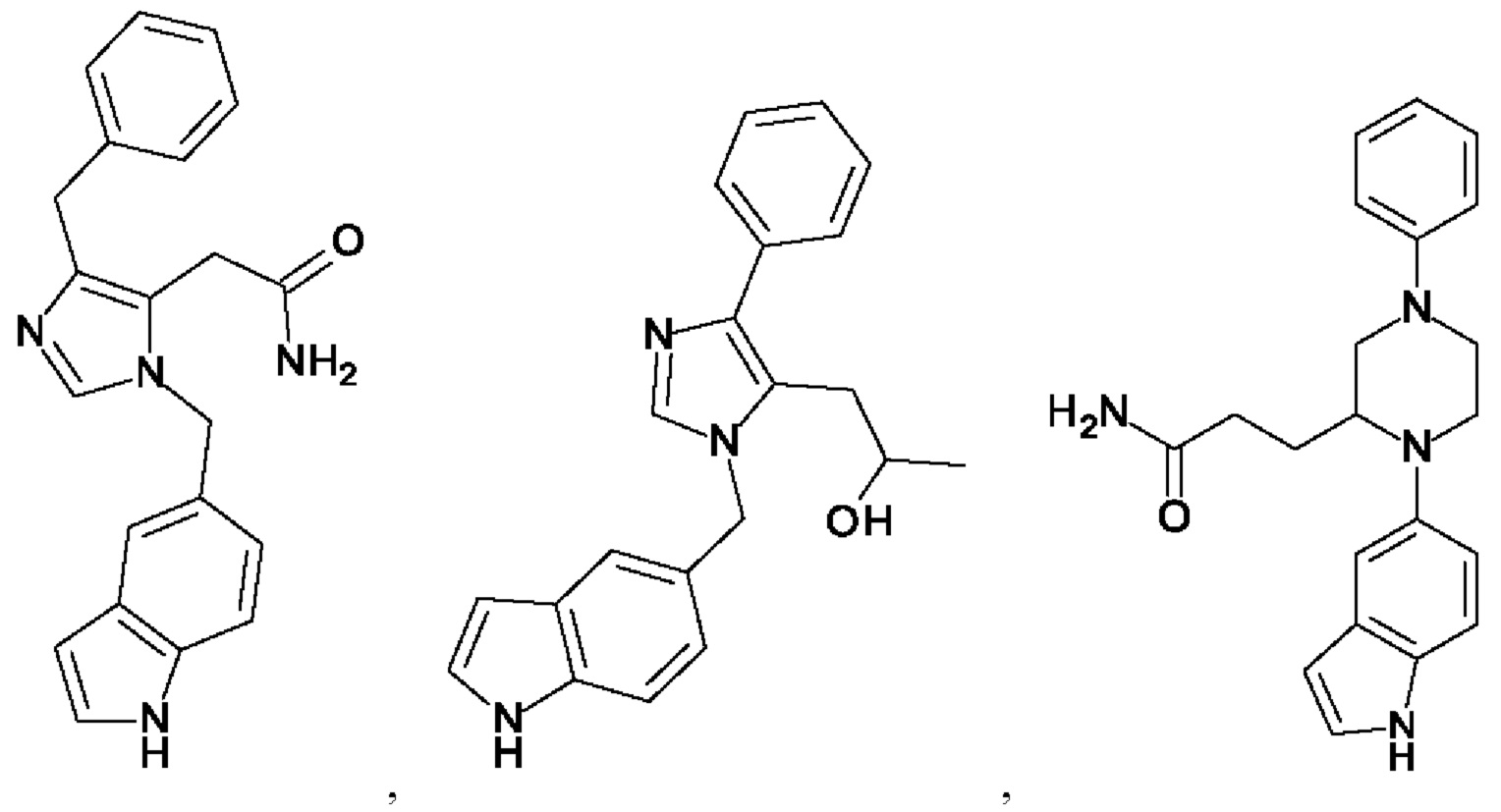







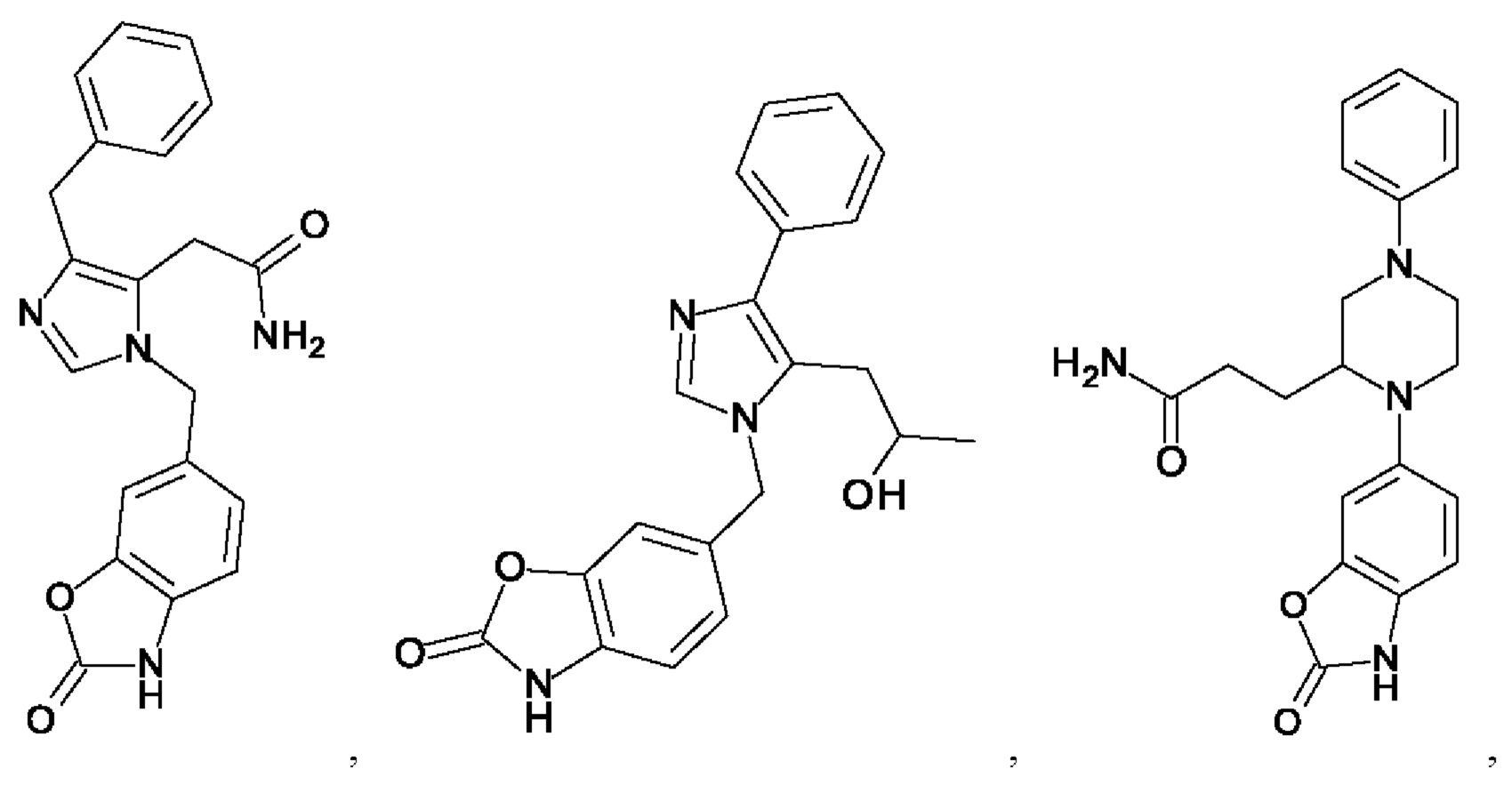

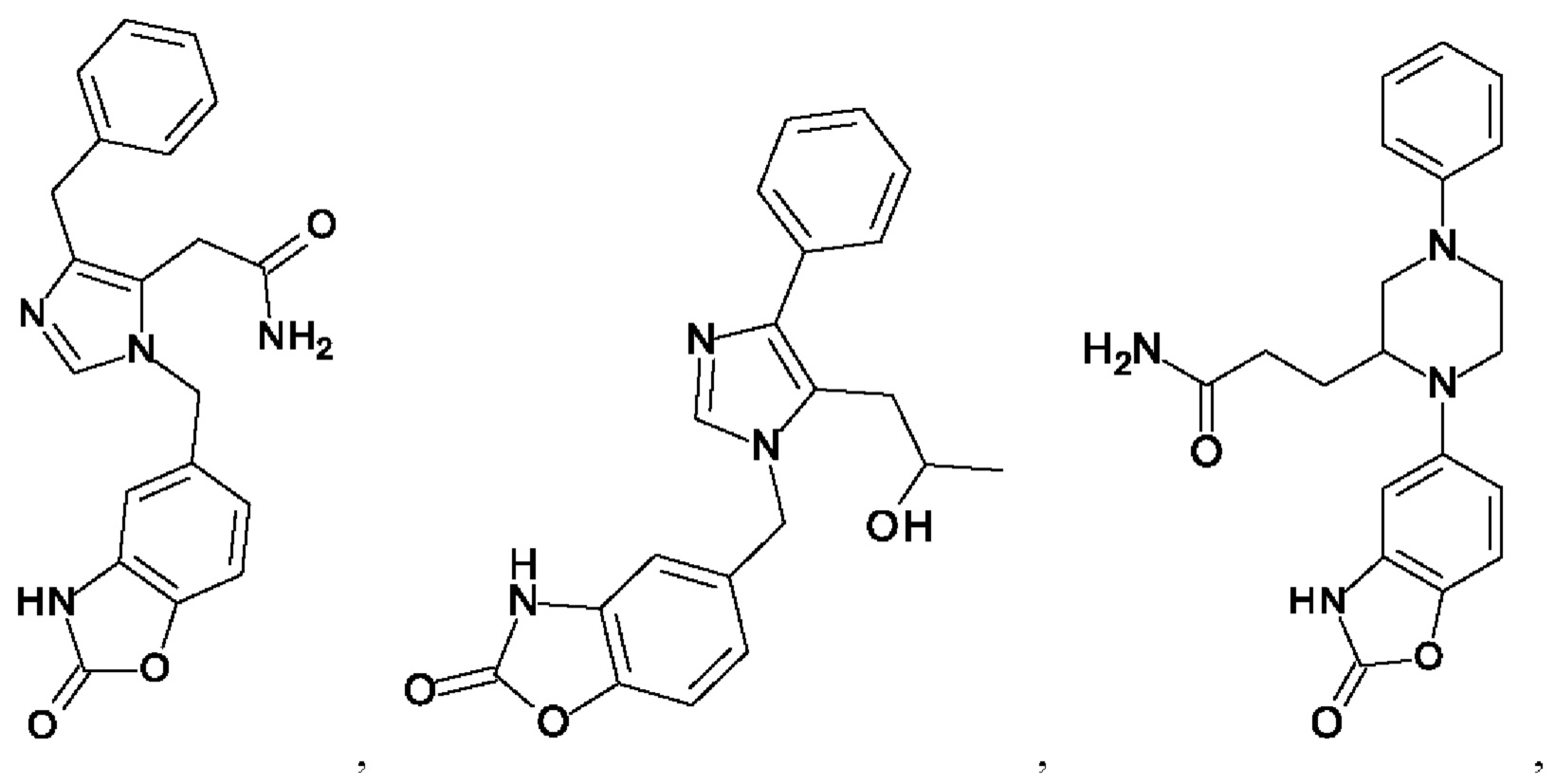




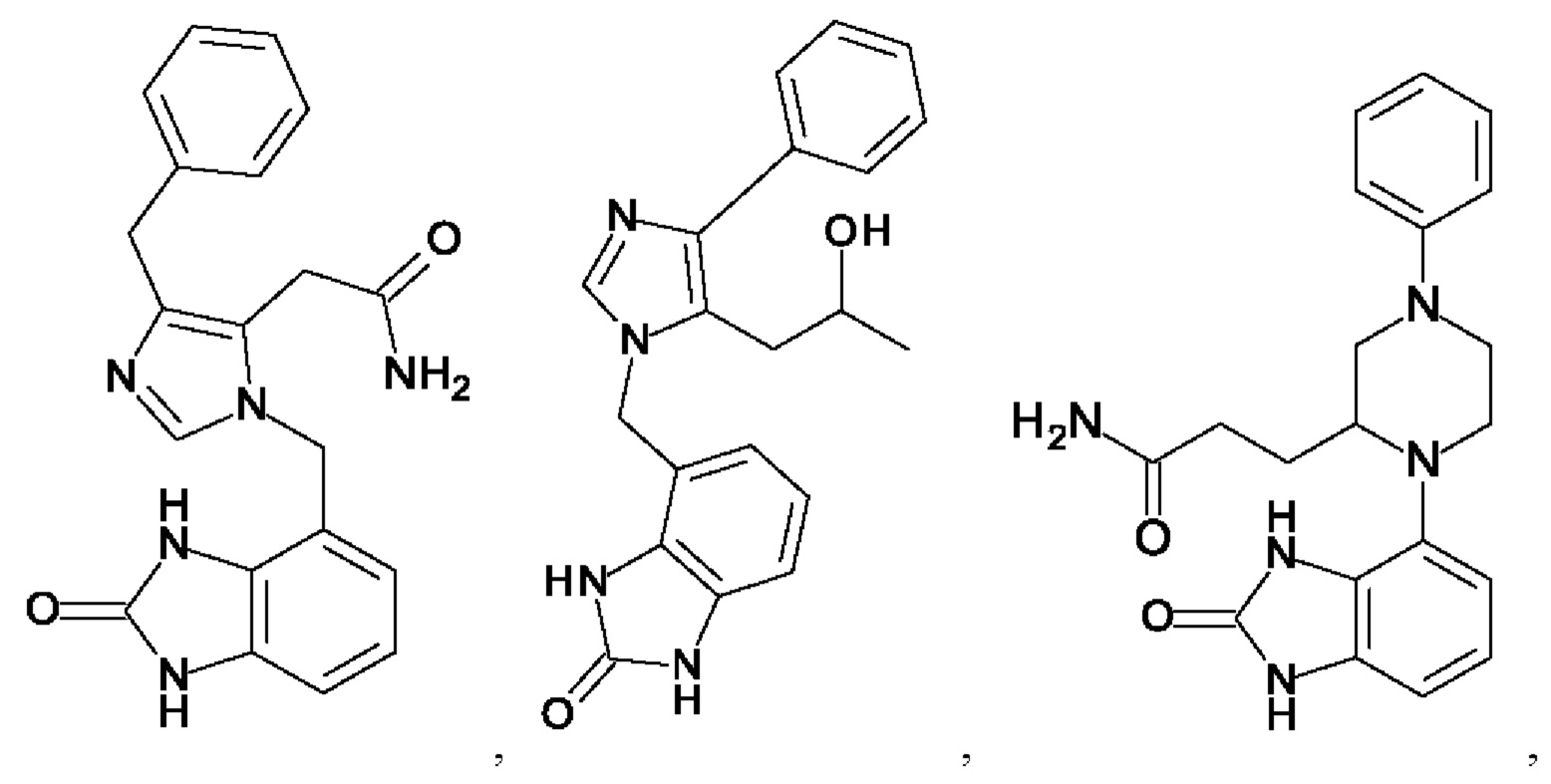
















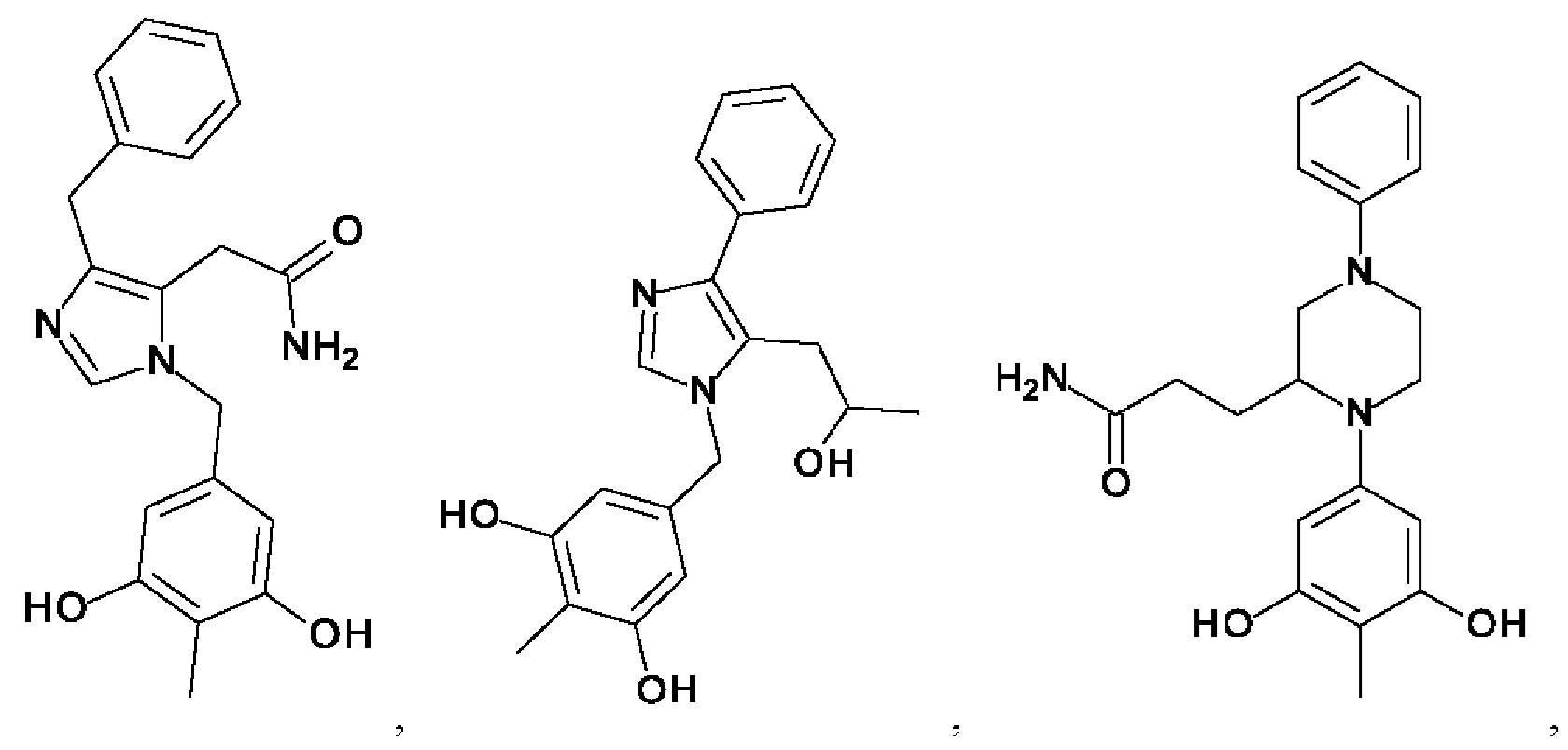

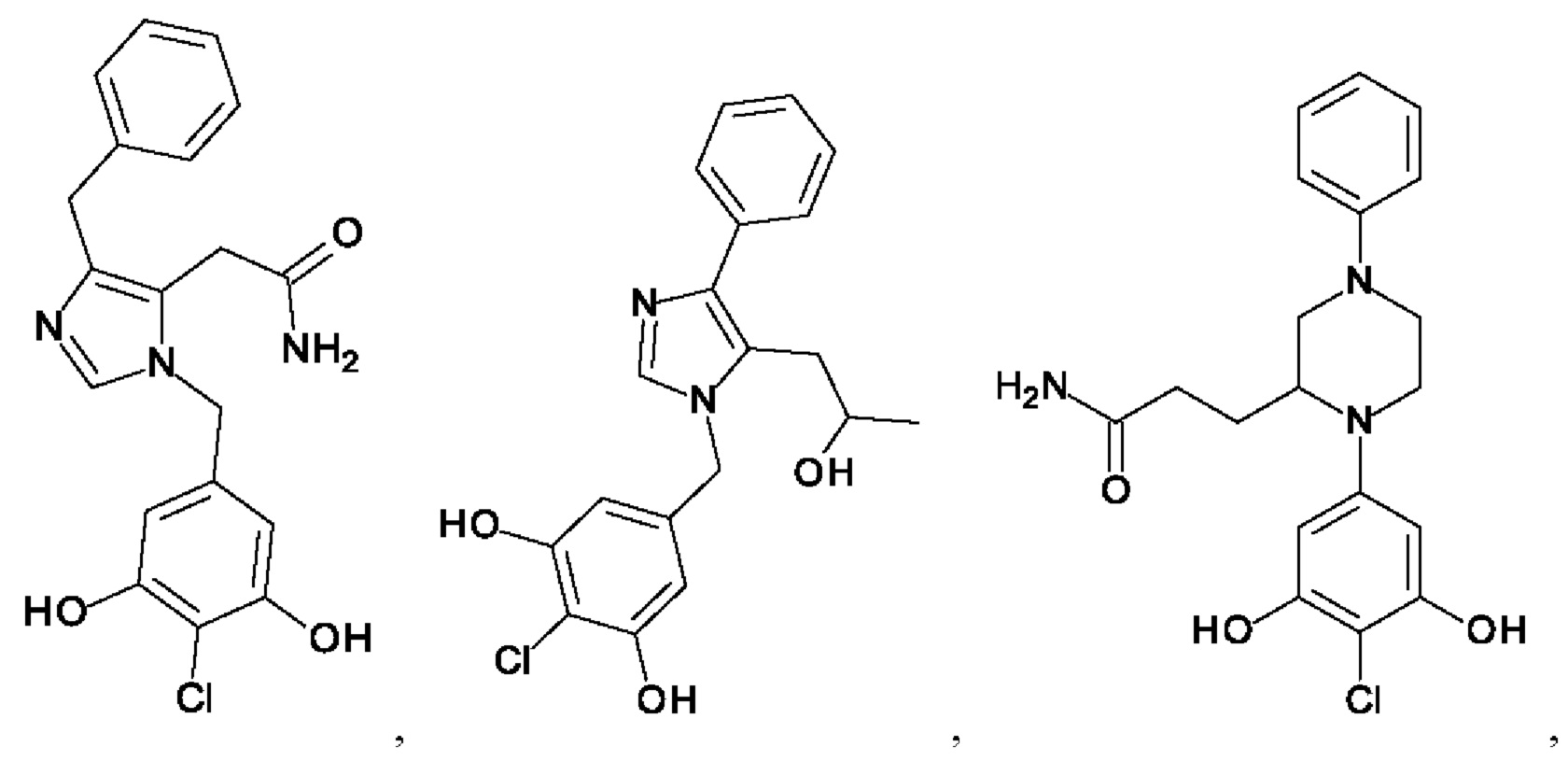

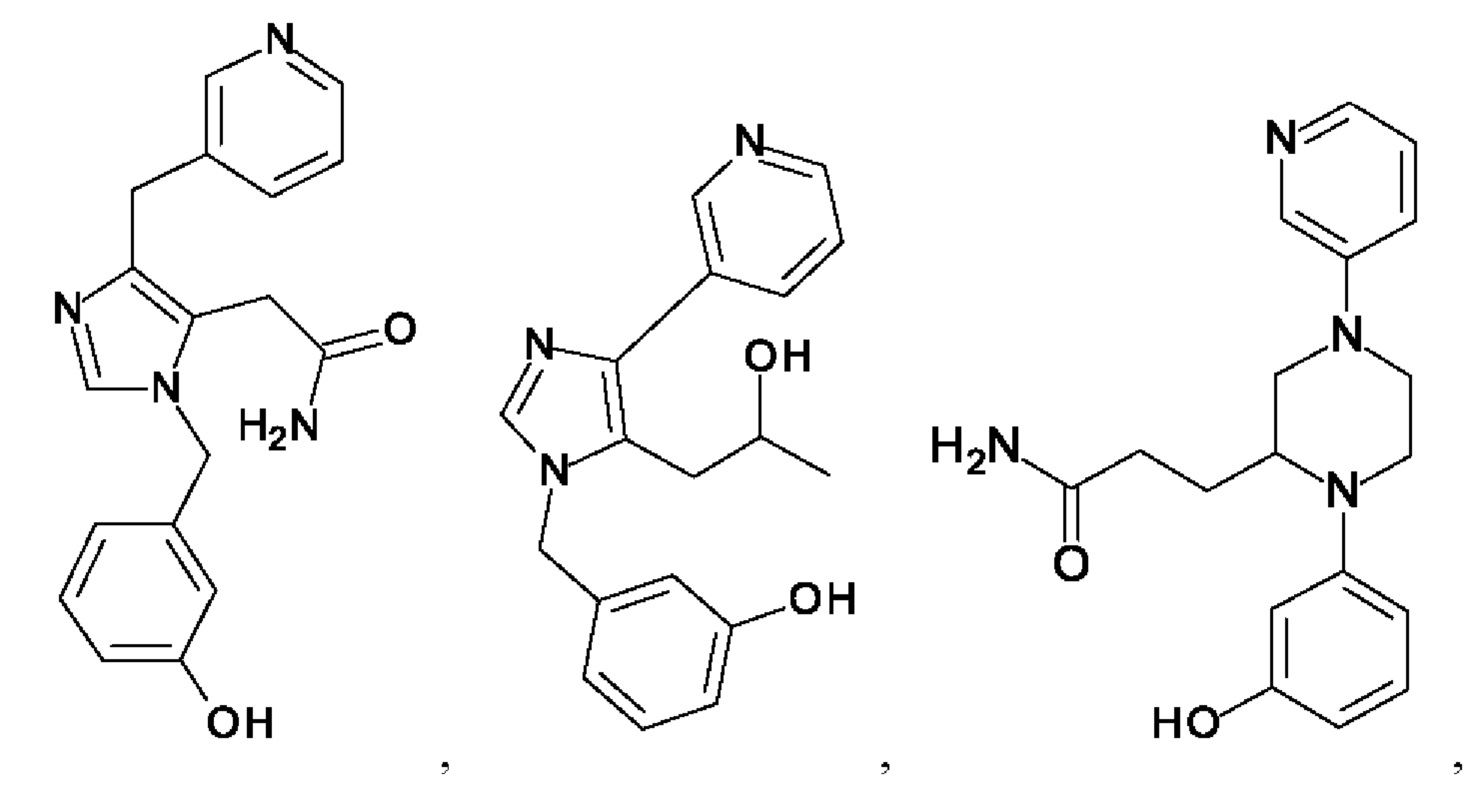

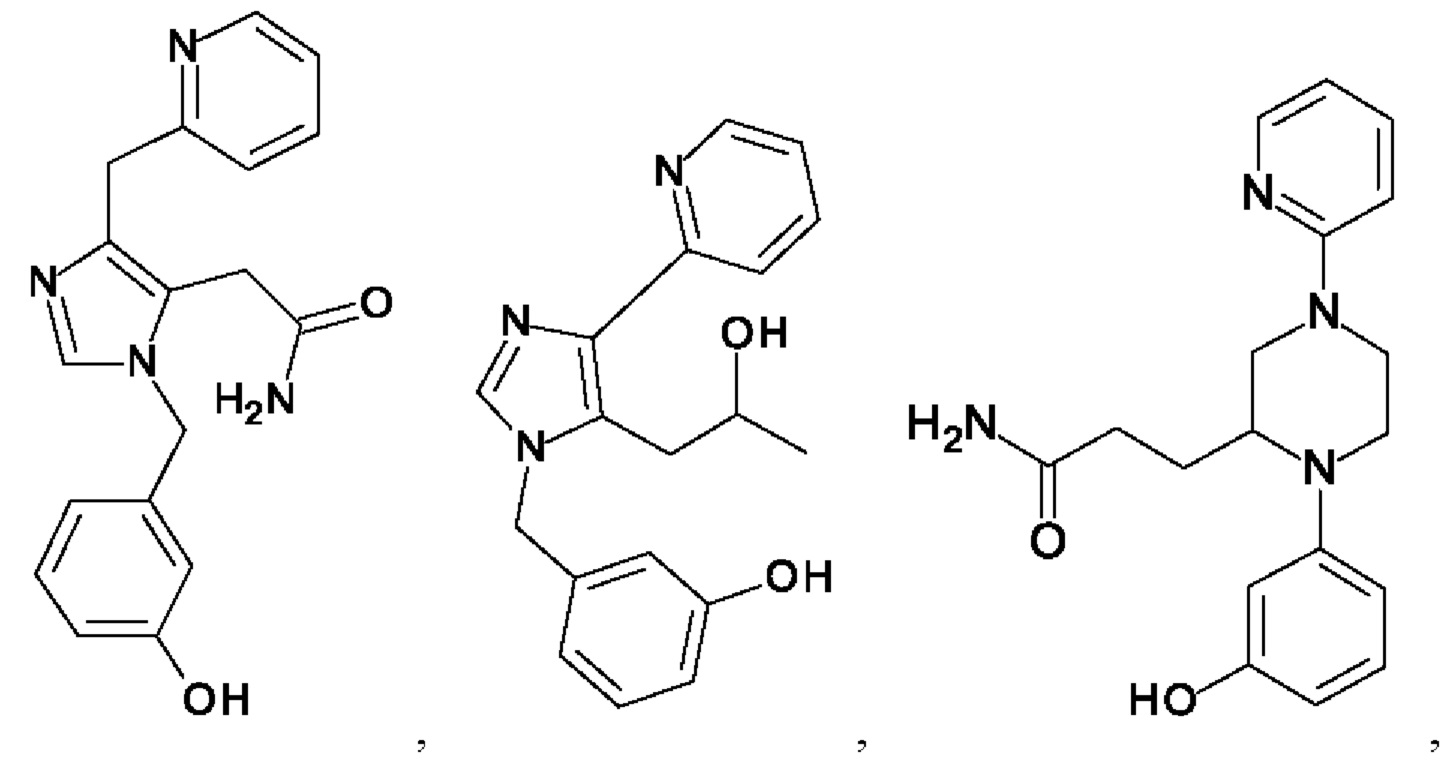
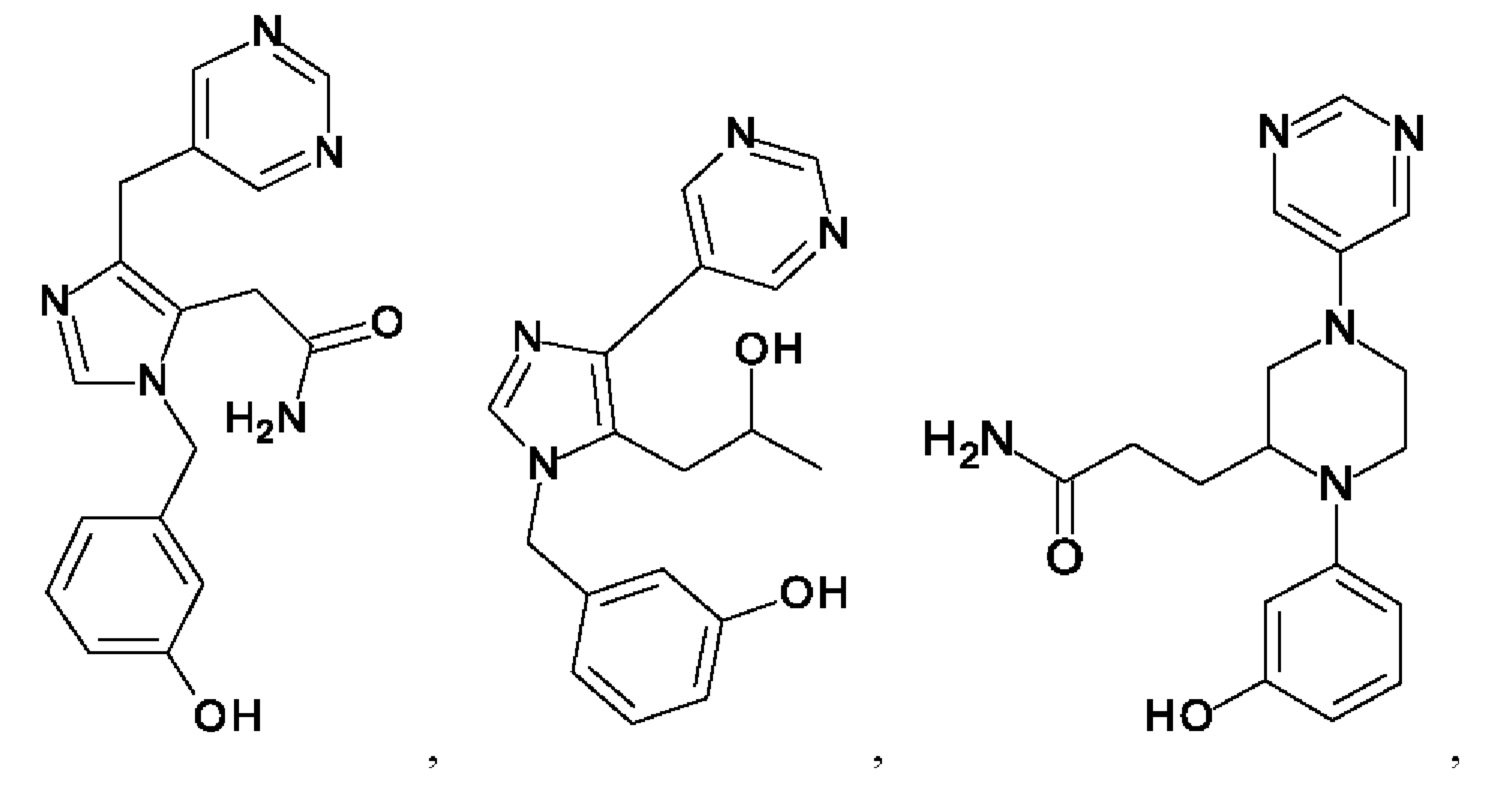

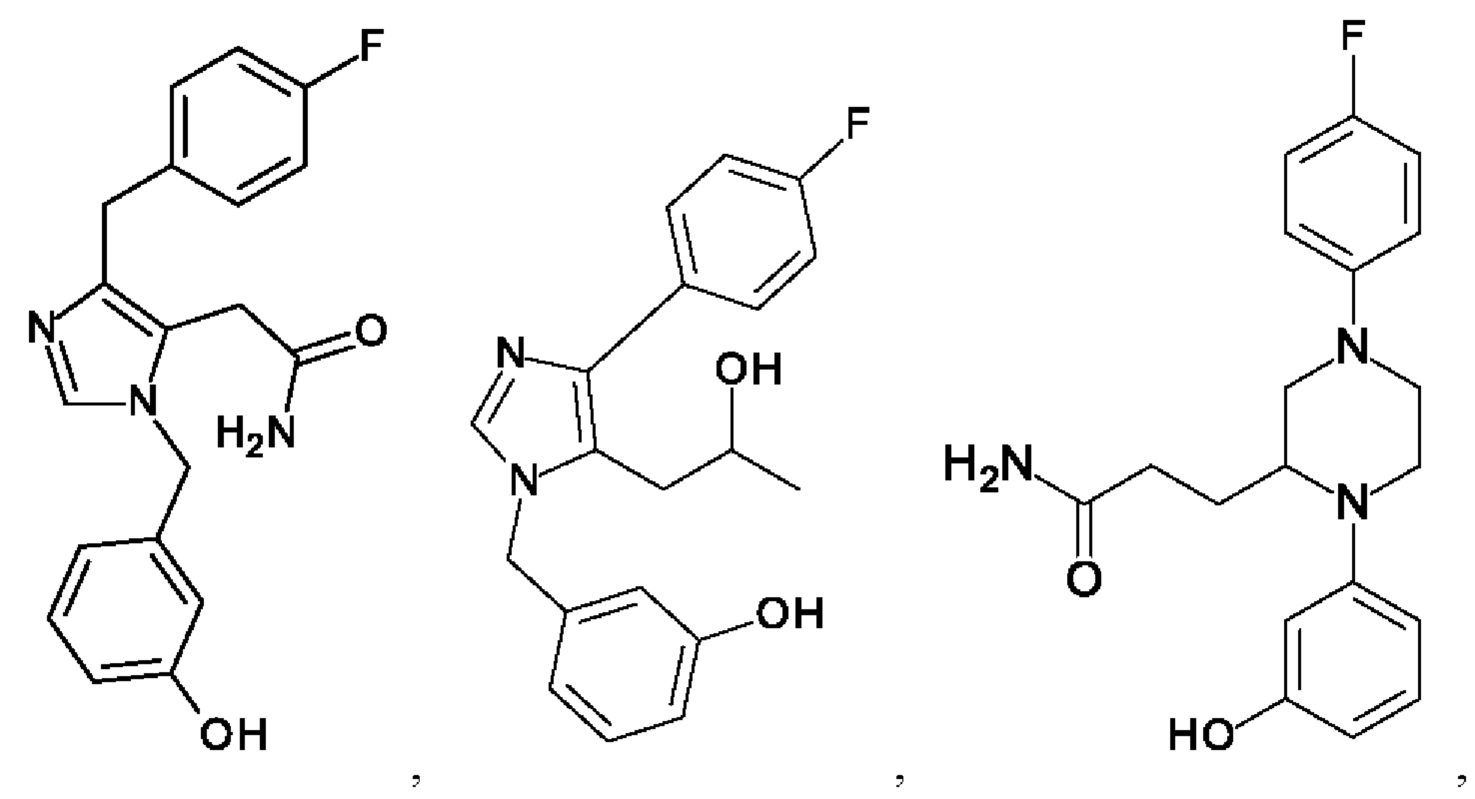





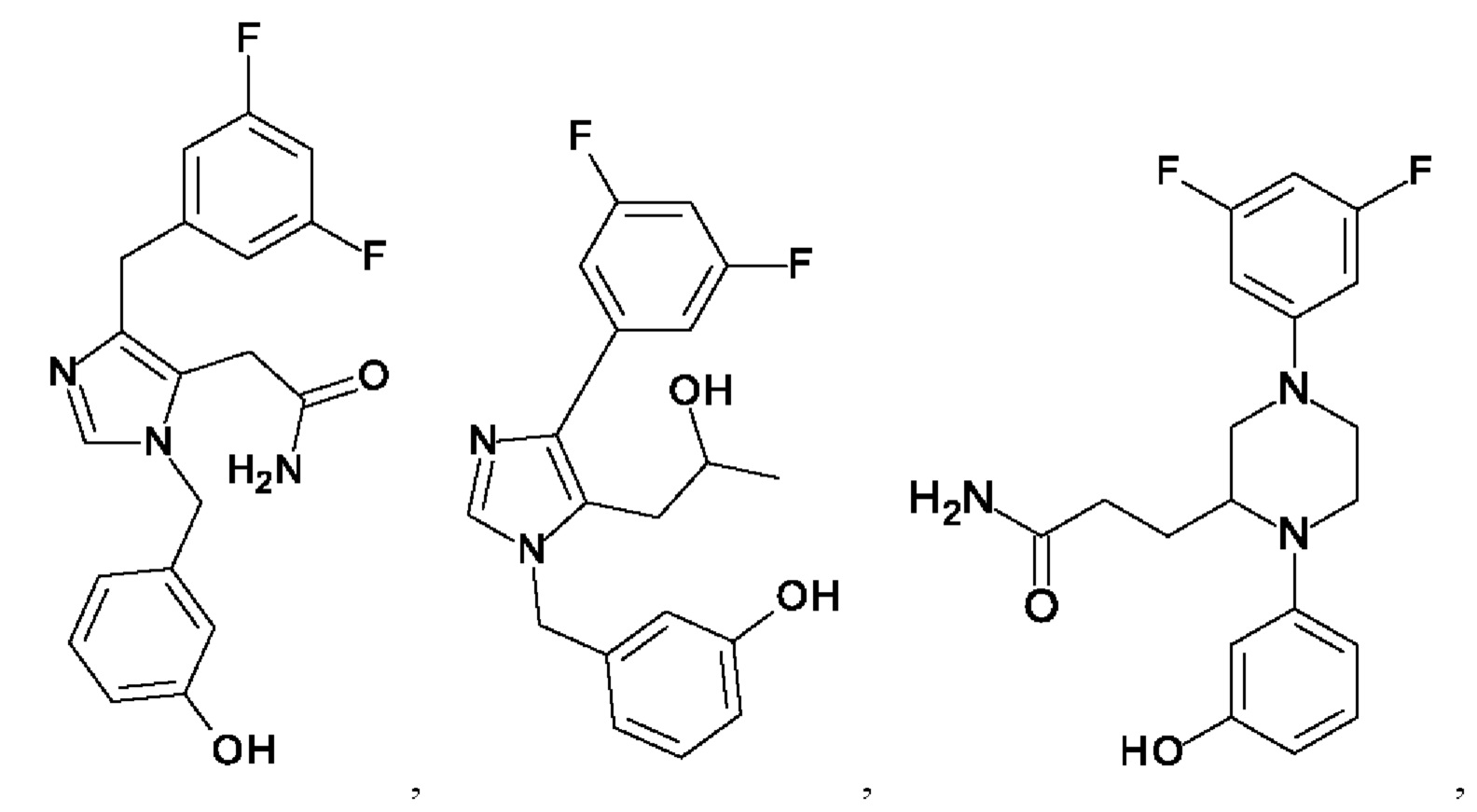




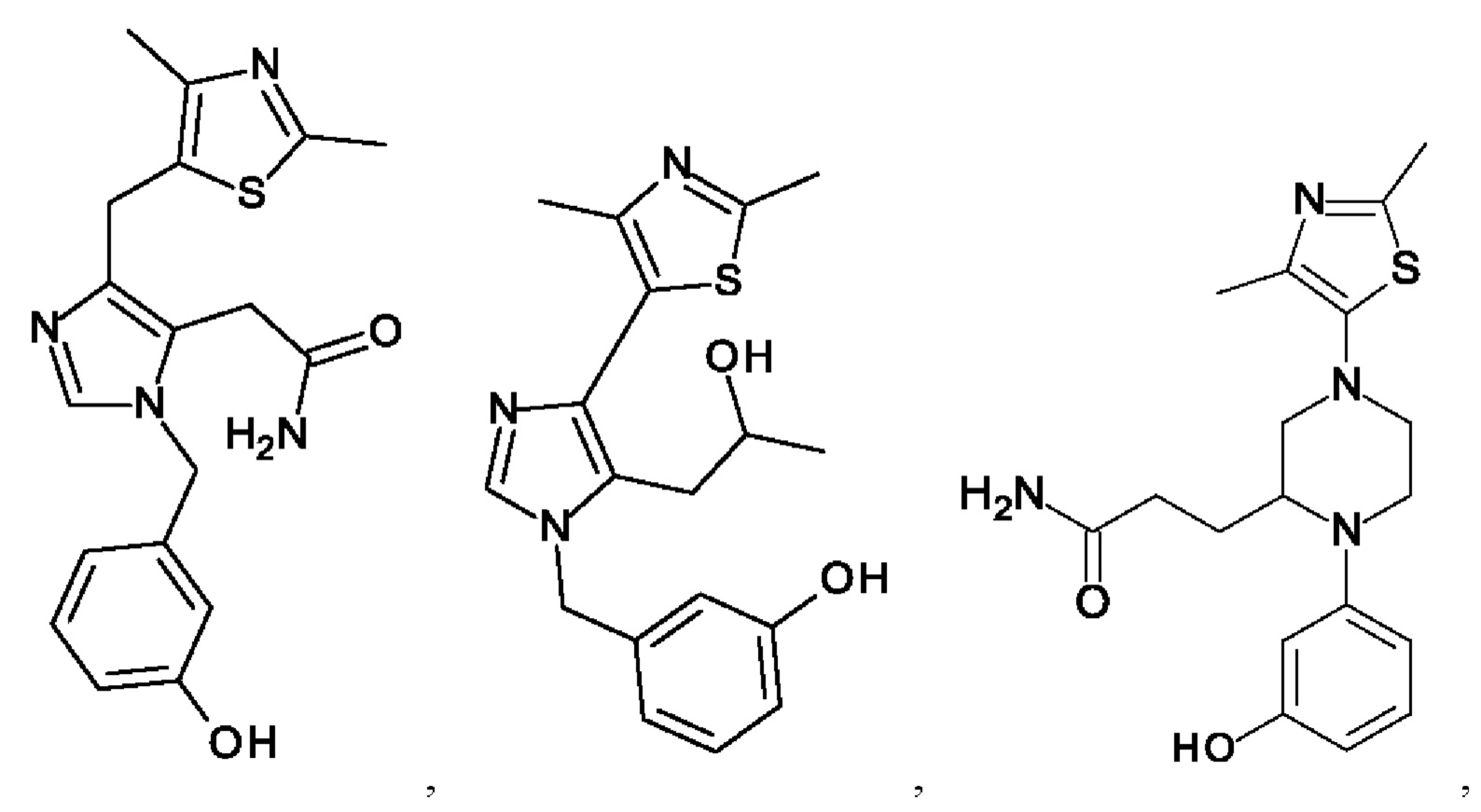














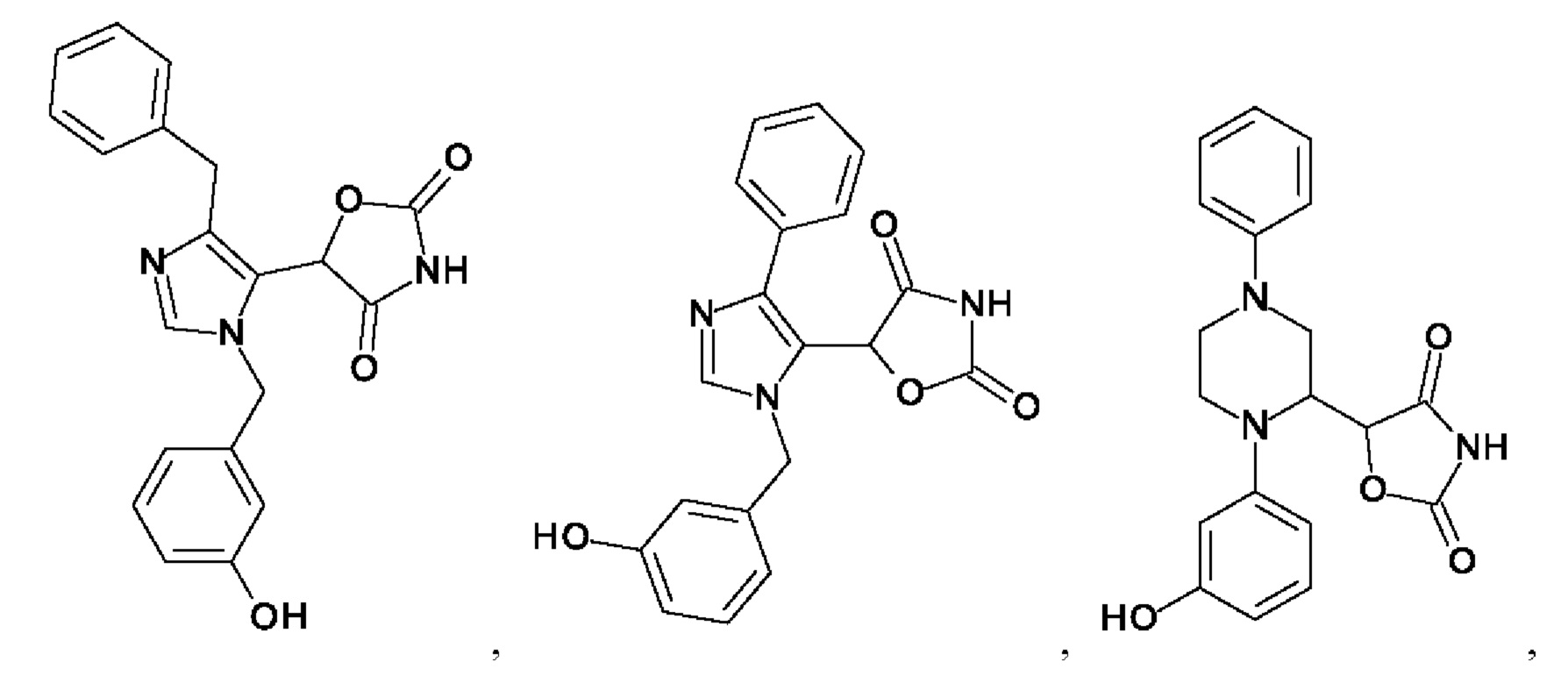

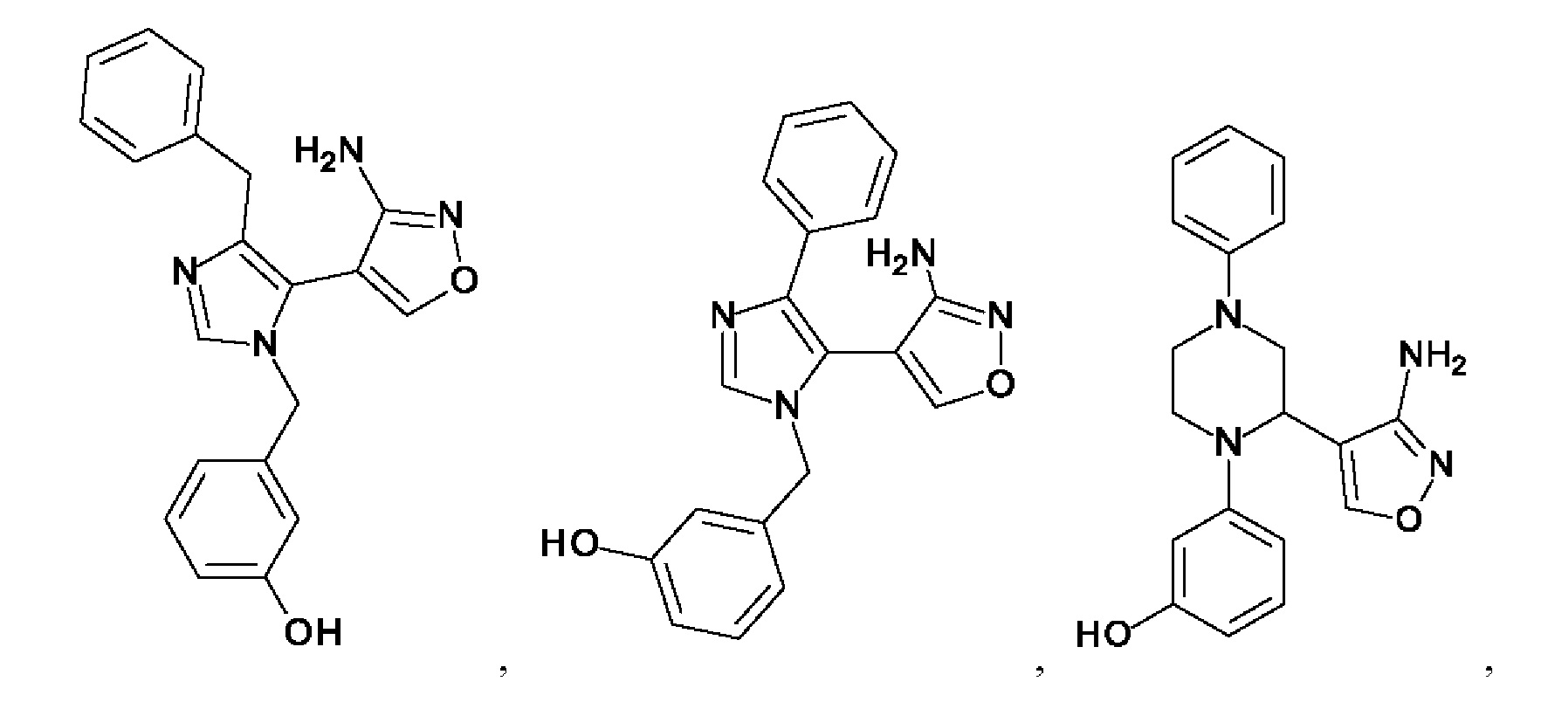

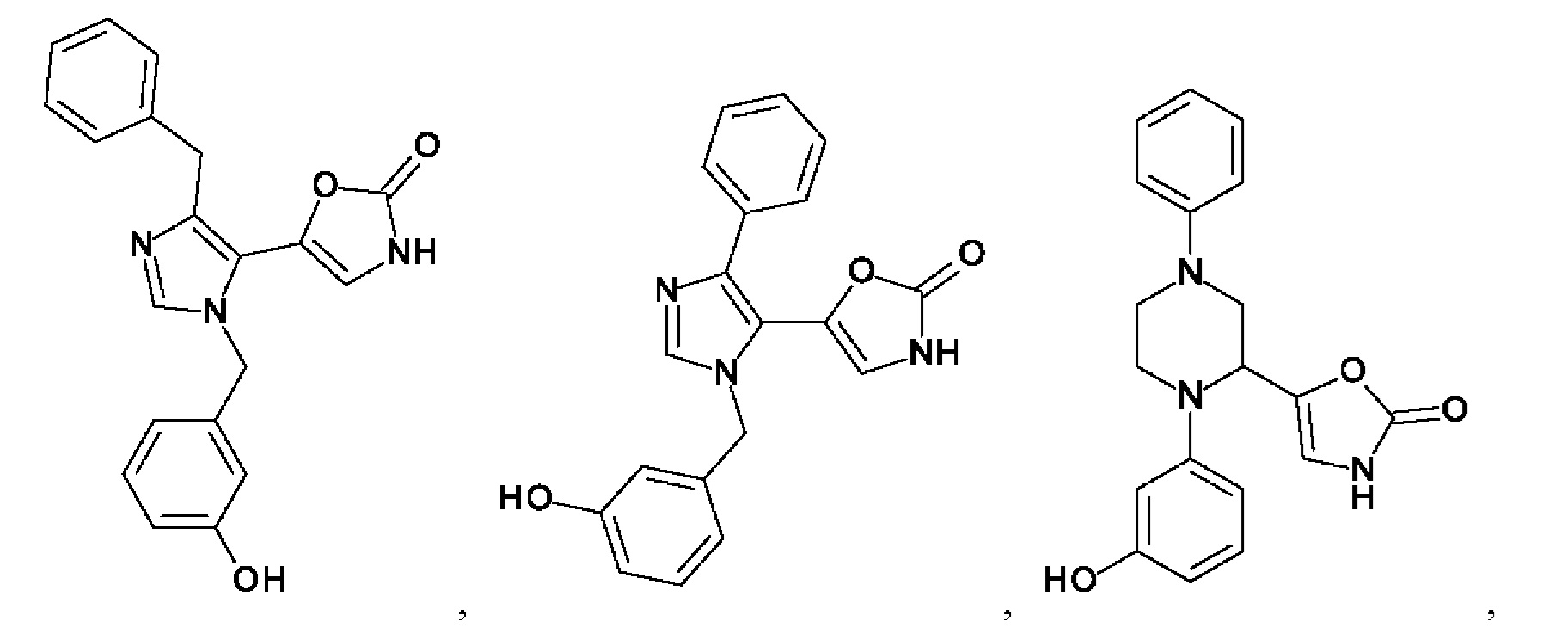


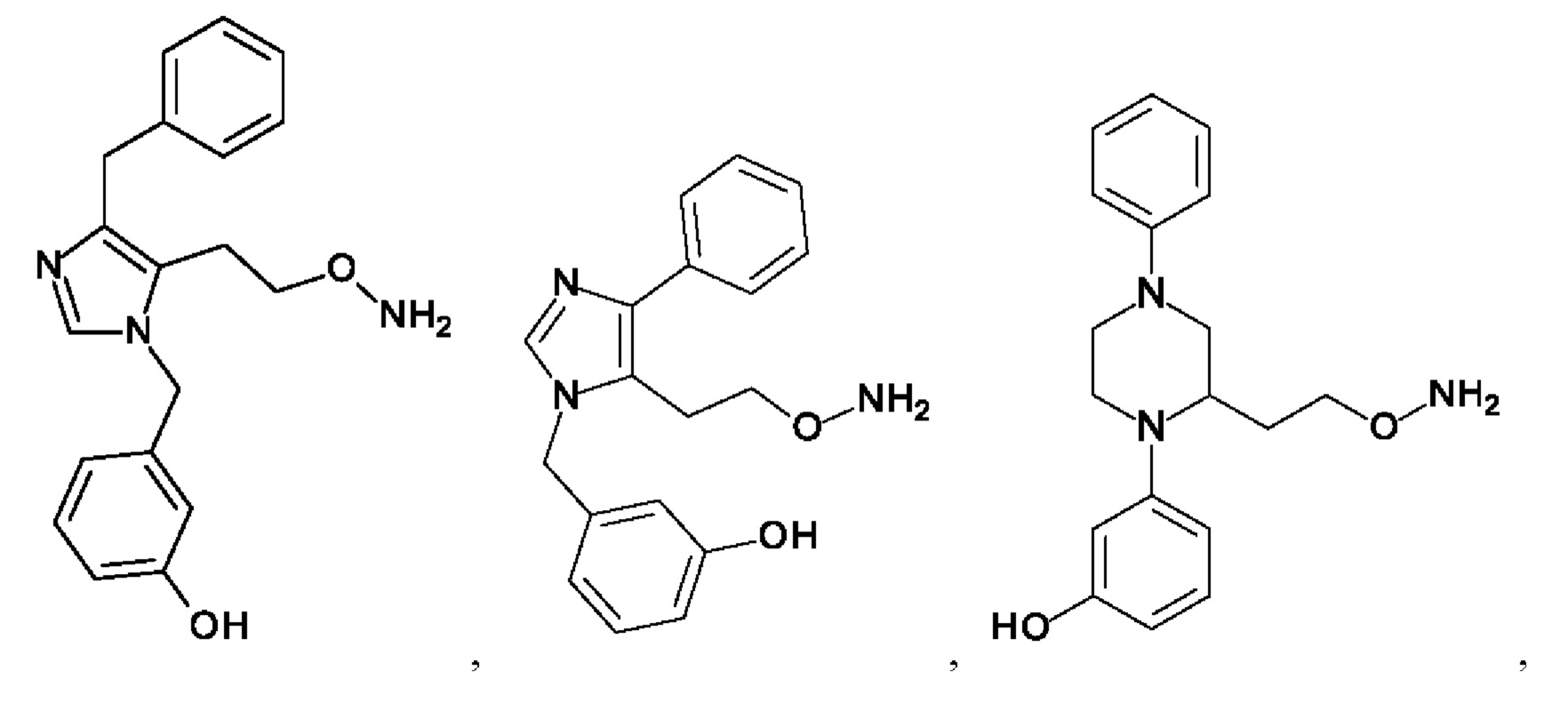



[0063] Используемый в данном документе термин "галоген" означает -F, -Cl, -Br или -I; термин "гидрокси" означает -ОН; термин "амино" означает -NH2; и термин "замещенный амино" включает в себя -NHW, где W выбран из -CN, -SO2(X)aY и -CO(X)aY, а равно 0 или 1, Х выбран из -NH- и -O-, и Y выбран из -Н, -СН3, -СН2СН3, -CH2OH и -CH2CH2OH.
[0064] Используемые в данном документе сокращения Me, Et, Ph, Ms означают метил, этил, фенил и метансульфонил соответственно. Исчерпывающий список сокращений, используемых химиками-органиками обычной квалификации в данной области, приводится в первом выпуске каждого тома Journal of Organic Chemistry; этот список обычно представлен в виде таблицы, озаглавленной Standard List of Abbreviations. Сокращения, содержащиеся в указанном списке, и все сокращения, используемые химиками-органиками обычной квалификации в данной области, включены посредством ссылки.
[0065] Соединения по настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. Настоящее изобретение предусматривает все такие соединения, включая цис- и транс-изомеры, (R)- и (S)-энантиомеры, диастереомеры, (d)-изомеры, (l)-изомеры, их рацемические смеси и другие их смеси, как входящие в объем изобретения. Все такие изомеры, а также их смеси охвачены данным изобретением.
[0066] Если желательным является, к примеру, конкретный энантиомер соединения по настоящему изобретению, то он может быть получен в результате асимметрического синтеза или в результате дериватизации с использованием хирального вспомогательного вещества, когда полученную диастереомерную смесь разделяют, и вспомогательную группу отщепляют с получением желаемых чистых энантиомеров. Альтернативно, могут быть образованы диастереомерные соли с подходящей оптически активной кислотой или оптически активным основанием с последующим разделением образованных таким путем диастереомеров фракционной кристаллизацией или хроматографическими методами, известными в данной области, и последующим выделением чистых энантиомеров.
[0067] Как правило, соединения по настоящему изобретению могут быть получены способами, проиллюстрированными на общих реакционных схемах, например, как описано ниже, или их модификациями с использованием легкодоступных исходных веществ, реагентов и стандартных методик синтеза. В этих реакциях возможно использование вариантов, которые сами являются известными, но здесь не упомянуты.
[0068] Настоящее изобретение также предусматривает фармацевтически приемлемые соли соединений. Термин "фармацевтически приемлемая соль" включает в себя как соли присоединения кислоты, так и соли присоединения основания и относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или кислот, и которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые соли образуются с неорганическими или органическими кислотами или основаниями, и они могут быть получены in situ для конечного выделения и очистки соединений, или в результате отдельного взаимодействия очищенного соединения в форме свободного основания или свободной кислоты с подходящей(им) органической(им) или неорганической(им) кислотой или основанием и выделения образованной таким образом соли.
[0069] Термин "фиброз" в контексте настоящего изобретения включает в себя, но без ограничения, фиброз миокарда, фиброз почек, фиброз печени и/или фиброз легких.
[0070] В дополнение к лечению установленного фиброза соединения по настоящему изобретению можно применять профилактически у субъектов с риском развития фиброза. В качестве примера, субъектами с риском развития фиброза являются субъекты, имеющие гипертензию, диабет, миокардит, ишемическую болезнь сердца, синдром Конна, феохромоцитому, генетическое предрасположение, диету с высоким содержанием соли и/или субъекты, получающие лекарственные средства, применяемые в химиотерапии рака (такие как даунорубицин). Термин "профилактический" в контексте настоящего изобретения охватывает, среди прочего, виды лечения, применяемые для предупреждения или замедления развития фиброза в группе риска. Субъекты, которые могут получать профилактическое лечение, уже могут иметь признаки ранней сердечной недостаточности при эхокардиографии.
[0071] Термин "гипертензия" в контексте настоящего изобретения указывает на то, что давление крови у взрослых выше примерно 139 мм рт.ст. систолическое и/или выше примерно 89 мм рт.ст. диастолическое.
[0072] Термин "прегипертензия" в контексте настоящего изобретения указывает на то, что давление крови у взрослых находится в диапазоне примерно 120-139 мм рт.ст. систолическое и/или примерно 80-89 мм рт.ст. диастолическое.
[0073] Настоящее изобретение также охватывает фармацевтические композиции, содержащие соединения по настоящему изобретению в сочетании с приемлемыми фармацевтическими эксципиентами. Термин "фармацевтически приемлемый эксципиент" в контексте настоящего изобретения означает любой фармацевтически приемлемый неактивный компонент композиции. Как общеизвестно в данной области, эксципиенты включают разбавители, буферы, связывающие вещества, смазывающие вещества, разрыхлители, красители, антиоксиданты/консерванты, рН-регуляторы и т.д. Эксципиенты выбирают исходя из желаемых аспектов конечной формы, например получения таблетки с желаемыми твердостью и рыхлостью, быстрораспадающейся и легко проглатываемой; и т.д. Желаемая скорость высвобождения активного вещества из композиции после ее проглатывания также играет роль в выборе эксципиентов. Фармацевтические композиции могут включать любой тип лекарственной формы, такой как таблетки, капсулы, порошки, жидкие композиции, формы с замедленным или длительным высвобождением, пластыри, порошки для вдыхания через нос, назальные спреи и т.п. Физическая форма и наполнение предусмотренных фармацевтических композиций представляют собой традиционные препараты, которые могут быть приготовлены специалистами в фармацевтической области, и основаны на давно установившихся принципах и композициях, описанных, например, в Remington: The Science and Practice of Pharmacy, 19th Edition, 1995; British Pharmacopoeia 2000, и подобных текстах и руководствах по приготовлению фармацевтических композиций.
[0074] Например, если соединения или композиции предназначены для введения перорально, то они могут быть приготовлены в виде таблеток, капсул, порошков или сиропов; или для парентерального введения они могут быть приготовлены в виде инъекций (внутривенных, внутримышечных или подкожных), препаратов для капельной инфузии или суппозиториев. Для применения офтальмическим путем для нанесения на слизистую оболочку они могут быть приготовлены в виде глазных капель или глазных мазей. Эти формы композиций могут быть получены традиционными способами, и, при необходимости, активный ингредиент может быть смешан с традиционной добавкой, такой как эксципиент, связывающее вещество, разрыхляющий агент, смазывающее вещество, корригент, солюбилизирующий агент, суспендирующий агент, эмульгатор или покрывающий агент.
[0075] Когда соединение(я) по настоящему изобретению вводят в виде фармацевтических средств людям и животным, тогда их вводят сами по себе или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99,5% (более предпочтительно, от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
[0076] Дозировка соединения и частота введения, которые следует использовать, также легко могут быть определены практикующим врачом с целью обеспечения желаемой ответной реакции.
[0077] Хотя дозировка будет зависеть от симптомов, возраста и массы тела пациента, характера и тяжести расстройства, подлежащего лечению или предупреждению, пути введения и формы лекарственного средства, обычно суточная дозировка от 0,0001 мг до 200 мг соединения по настоящему изобретению может представлять собой подходящее эффективное количество для взрослого пациента-человека, и эту дозировку можно вводить в виде однократной дозы или в разделенных дозах.
[0078] "Пациент" или "субъект", подлежащий лечению заявленным способом, может означать либо субъекта-человека, либо субъекта, не являющегося человеком.
[0079] "Эффективное количество" заявленного соединения, по отношению к способу лечения, относится к количеству терапевтического средства в препарате, которое при применении в качестве части желаемого режима дозировки приносит пользу в соответствии с клинически приемлемыми стандартами для лечения или профилактики конкретного расстройства.
[0080] Настоящее изобретение далее описано более подробно со ссылкой на конкретные не ограничивающие примеры, в которых описаны конкретные композиции и способы использования. Следует иметь в виду, однако, что подробное описание конкретных методик, композиций и способов включено исключительно в целях иллюстрации настоящего изобретения. Его никоим образом не следует понимать как ограничение общего описания изобретательского замысла, изложенного выше.
ПРИМЕРЫ
Пример 1 - Синтез соединений
[0081] Путь синтеза, использованный для получения 2-[4-бензил-1-(3-гидроксибензил)-1Н-имидазол-5-ил]ацетамида (VB0002), показан на Фиг. 1. Реакция конденсации между Вос-фенилаланином и калиевой солью метилмалоната, промотиро ванная 1,1'-карбонилдиимидазолом (CDI) приводила к получению промежуточного соединения 4. Удаление Вос-группы затем обеспечивало получение соединения 5 в виде гидрохлоридной соли. Соль 5 подвергали взаимодействию с изотиоцианатом В, и получали циклическую тиомочевину 6 с низким выходом. Превращение циклической тиомочевины 6 в имидазол 7 осуществляли в окислительных условиях. Обработка имидазола 7 водным аммиаком приводила к получению VB0002.
[0082] Соединение В не было коммерчески доступным, и его получали, как показано на Фиг. 2. 3-Цианофенол сначала защищали, преобразуя в трет-бутилдиметилсилиловый эфир, и циано группу затем восстанавливали до соответствующего амина, который подвергали взаимодействию с тиофосгеном с образованием соединения В.
[0083] Путь синтеза, использованный для получения 3-[5-(2-гидроксипропил)-4-фенил-имидазол-1-илметил]-фенола (VB0003), показан на Фиг. 3. Сначала для удобства имидазол 8 собирали путем проведения реакции сочетания трех компонентов: α-тозилбензилизоцианида С, бензиламина D и защищенного альдегида Е. Обе защитные группы затем удаляли с использованием системы тиоанизол-трифторуксусная кислота (TFA) с получением соединения 9, которое восстанавливали с получением VB0003.
[0084] Соединение С получали, как показано на Фиг. 4. 4-Толуолсульфиновую кислоту, полученную из соответствующей натриевой соли, использовали в трехкомпонентной реакционной смеси с бензальдегидом и формамидом с образованием промежуточного соединения, после дегидратирования которого получали соединение С.
[0085] Соединение D не было коммерчески доступным, и его получали, как показано на Фиг. 5. Получение осуществляли путем превращения 3-(бензилокси)бензилового спирта в соответствующий азид с использованием дифенилфосфорилазида и последующего восстановления с получением бензиламина D.
[0086] Соединение Е не было коммерчески доступным, и его получали, как показано на Фиг. 6. Этилацетоацетат сначала защищали в виде ацеталя; сложноэфирную группу затем восстанавливали до первичного спирта, который после этого подвергали реакции окисления по Сверну с образованием защищенного альдегида Е.
[0087] Путь синтеза, использованный для получения (S)-3-[1-(3-гидрокси)фенил-4-фенилпиперазин-2-ил]пропанамида (VB0005), показан на Фиг. 7. В стандартном процессе образования амидной связи 5-трет-бутиловый эфир Fmoc-L-глутаминовой кислоты подвергали взаимодействию с анилином с образованием соответствующего амида, защитную группу которого удаляли трис(2-аминоэтил)амином (ТАЕА) с получением соединения 14. Свободный амин 14 подвергали опосредованной медью реакции перекрестного связывания с 3-(бензилокси)фенилбороновой кислотой с получением N-арилированного продукта 15. Это соединение затем подвергали реакции с бромацетилбромидом с образованием соединения 16, которое превращали в кето-пиперазин 17; после селективного восстановления этого соединения получали пиперазин 18. Последующая стадия снятия двойной защиты с использованием системы тиоанизол-TFA приводила к получению соединения 19, которое подвергали реакции аминолиза с использованием водного аммиака и 1,1'-карбонилдиимидазола (CDI) с получением VB0005.
[0088] Метил-4-(трет-бутоксикарбониламино)-3-оксо-5-фенилпентаноат (4). Суспензию хлорида магния (6,96 г, 73,0 ммоль) и калиевой соли метилмалоната (17,66 г, 113,2 ммоль) перемешивали в THF (280 мл) при 50°С в атмосфере азота в течение 6 ч. К раствору N-(трет-бутоксикарбонил)-L-фенилаланина (20,0 г, 75,4 ммоль) в THF (200 мл) в отдельной колбе под азотом и при 0°С добавляли порциями 1,1'-карбонилдиимидазол (18,34 г, 113,1 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч, затем добавляли к охлажденной суспензии малоната. Реакционную смесь затем перемешивали при комнатной температуре в течение 17 ч. Большую часть THF удаляли в вакууме. К остатку добавляли насыщенный гидросульфат калия (300 мл) и этилацетат (300 мл). Слои разделяли, затем водный слой экстрагировали еще два раза этилацетатом. Объединенные органические слои промывали насыщенным бикарбонатом натрия (2 х 300 мл) и рассолом (300 мл), сушили и концентрировали в вакууме с получением метил-4-(трет-бутоксикарбониламино)-3-оксо-5-фенилпентаноата (22,22 г, 92%) в виде бледного вязкого масла, которое затвердевало.1H ЯМР (400 МГц, CDCl3) δ 7.33-7.08 (m, 5H), 5.20-4.80 (m, 1H), 4.62-4.48 (m, 1H), 3.69 (s, 3H), 3.52 (d, J=16,0 Гц, 1Н), 3.45 (d, J=16,0 Гц, 1H), 3.25-2.88 (m, 2H), 1.39 (s, 9H).13С ЯМР (100 МГц, CDCl3) δ 201.8, 167.3, 155.2, 136.1, 129.2, 128.6, 126.9, 80.1, 60.4, 52.3, 46.5, 36.7, 28.2. EIMS (масс-спектрометрия с ионизацией электронным ударом): m/z 321 [M]+. HRMS (масс-спектрометрия высокого разрешения): вычислено для C17H23NO5 321,1576, найдено 321.1555.
[0089] Метил-4-амино-3-оксо-5-фенилпентаноата гидрохлорид (5). Метил-4-(трет-бутоксикарбониламино)-3-оксо-5-фенилпентаноат (22,20 г, 69,2 ммоль) перемешивали в течение 2 суток с этилацетатом, насыщенным хлористым водородом.
Полученное твердое вещество собирали фильтрованием, промывали диэтиловым эфиром и сушили на воздухе с получением метил-4-амино-3-оксо-5-фенилпентаноата гидрохлорида (16,25 г, 92%) в виде нестабильного кремового твердого вещества.1H ЯМР (400 МГц, DMSO-d6) δ 8.58 (br s, 3H), 7.37-7.20 (m, 5H), 4.49 (t, J=6,6 Гц, 1Н), 3.83 (d, J=17,1 Гц, 1Н), 3.74 (d, J=17,1 Гц, 1Н), 3.62 (s, 3H), 3.22 (dd, J=14.4, 6,4 Гц, 1Н), 3.11 (dd, J=14.4, 7,0 Гц, 1Н).
[0090] Метил-2-{5-бензил-3-[3-(трет-бутилдиметилсилилокси)бензил]-2-тиоксо-2,3-дигидро-1H-имидазол-4-ил}ацетат (6). Смесь метил-4-амино-3-оксо-5-фенилпентаноата гидрохлорида (7,15 г, 27,9 ммоль), 3-(трет-бутилдиметилсилилокси)бензилизотиоцианата (9,34 г, 33,5 ммоль), триэтиламина (5,8 мл, 41,9 ммоль) и 4-толуолсульфоната пиридиния (0,70 г, 2,8 ммоль) в толуоле (100 мл) нагревали при температуре спокойной дефлегмации под азотом в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры, затем распределяли между водой (150 мл) и этилацетатом (150 мл). Слои разделяли, и водный слой экстрагировали еще два раза этилацетатом. Объединенные органические слои промывали водой (2 х 150 мл), сушили и концентрировали в вакууме с получением желто-коричневого масла. Это неочищенное вещество растворяли в диэтиловым эфире, вводили затравку и оставляли кристаллизоваться в течение 20 ч. Метил-2-{5-бензил-3-[3-(трет-бутилдиметилсилилокси)бензил)]-2-тиоксо-2,3-дигидро-1H-имидазол-4-ил}ацетат (2,14 г, 13%) собирали фильтрованием в виде кремовых игл; т.пл. 154,0-156,0°С.1H ЯМР (400 МГц, CDCl3) δ 9.68 (br s, 1Н), 7.34-7.22 (m, 2H), 7.19-7.14 (m, 3H), 6.83-6.71 (m, 3H), 5.32 (s, 2H), 3.79 (s, 2H), 3.61 (s, 3H), 3.33 (s, 2H), 0.96 (s, 9H), 0.18 (s, 6H).13С ЯМР (125 МГц, CDCl3) δ 169.1, 161.5, 156.1, 137.4, 136.3, 129.9, 128.9, 128.6, 127.2, 125.8, 119.8, 119.5, 119.4, 118.6, 52.4, 47.9, 30.1, 29.6, 25.7, 18.2, -4.4. EIMS: m/z 482 [M]+. HRMS: вычислено для C26H34N2O3SSi: 482,2054; найдено: 482,2039.
[0091] Метил-2-{4-бензил-1-[3-(трет-бутилдиметилсилилокси)бензил]-1H-имидазол-5-ил}ацетат (7). К суспензии метил-2-{5-бензил-3-[3-(трет-бутилдиметилсилилокси)бензил]-2-тиоксо-2,3-дигидро-1H-имидазол-4-ил}ацетата (1,00 г, 2,1 ммоль) в уксусной кислоте (3,4 мл) под азотом медленно добавляли 30%-ную перекись водорода (941 мкл). Через 10 мин реакционный раствор охлаждали в ледяной бане и затем гасили 10%-ным карбонатом натрия (20 мл). рН доводили до 9-10 1М гидроксидом натрия, затем экстрагировали этилацетатом (3 х 20 мл), сушили и концентрировали в вакууме с получением метил-2-{4-бензил-1-[3-(трет-бутилдиметилсилилокси)бензил]-1H-имидазол-5-ил}ацетата (0,86 г, 91%) в виде желто-коричневого масла.1H ЯМР (400 МГц, CDCl3) δ 7.50 (s, 1H), 7.27-7.10 (m, 6H), 6.80-6.74 (m, 1H), 6.69-6.60 (m, 1H), 6.50-6.47 (m, 1H), 5.06 (s, 2H), 3.93 (s, 2H), 3.55 (s, 3H), 3.38 (s, 2H), 0.95 (s, 9H), 0.14 (s, 6H).13C ЯМР (50 МГц, CDCl3) δ 170.0, 156.3, 139.9, 139.6, 137.3, 136.2, 130.1, 128.6, 128.4, 126.0, 120.4, 119.8, 118.4, 117.4, 52.2, 48.9, 33.5, 29.1, 25.6, 18.0, -4.5. EIMS: m/z 450 [M]+. HRMS: вычислено для C26H34N2O3Si: 450,2333; найдено: 450,2323.
[0092] 2-[4-Бензил-1-(3-гидроксибензил)-1H-имидазол-5-ил]ацетамид (VB0002). К раствору метил-2-{4-бензил-1-[3-(трет-бутилдиметилсилилокси)бензил]-1H-имидазол-5-ил}ацетата (0,86 г, 1,9 ммоль) в метаноле (2 мл) добавляли 25%-ный водный раствор аммиака. Колбу закупоривали, и реакционную смесь перемешивали при комнатной температуре в течение 3 суток. Снова добавляли метанол и диэтиловый эфир, и реакционную смесь перемешивали в течение 2 ч. Тонкодисперсный осадок собирали фильтрованием, промывая диэтиловым эфиром, затем сушили на воздухе с получением 2-[4-бензил-1-(3-гидроксибензил)-1H-имидазол-5-ил]ацетамида (0,193 г, 32%) в виде бесцветного твердого вещества; т.пл. 249°С (разложение).1Н ЯМР (400 МГц, DMSO-d6) δ 9.44 (s, 1H), 7.52 (s, 1H), 7.37 (br s, 1H), 7.24-7.07 (m, 5H), 6.95 (br s, 1H), 6.66 (m, 1H), 6.53-6.49 (m, 1H), 6.45-6.43 (m, 1H), 5.10 (s, 2H), 3.76 (s, 2H), 3.30 (s, 2H).13C ЯМР (50 МГц, DMSO-d6) δ 170.8, 157.7, 141.1, 138.9, 137.9, 136.9, 129.7, 128.5, 127.9, 125.5, 112.0, 117.3, 114.5, 113.5, 47.7, 32.8, 29.6. EIMS: m/z 321 [M]+. HRMS: вычислено для C19H19N3O2: 321,1477; найдено: 321,1470.
[0093] 3-(трет-Бутилдиметилсилилокси)бензонитрил. К перемешиваемому раствору 3-цианофенола (10,34 г, 86,8 ммоль) и имидазола (14,77 г, 217 ммоль) в DMF (100 мл) под азотом и при 0°С добавляли порциями трет-бутилдиметилсилилхлорид (13,74 г, 91 ммоль) в течение 5 мин. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение еще 2 ч. Реакционную смесь переносили в делительную воронку и распределяли между диэтиловым эфиром (150 мл) и водой (150 мл). Слои разделяли, и водный слой экстрагировали еще два раза диэтиловым эфиром. Объединенные органические экстракты промывали водой (2 х 100 мл), сушили и концентрировали с получением 3-(трет-бутилдиметилсилилокси)-бензонитрила (20,38 г, количественный выход) в виде бледно-желтого масла.1Н ЯМР (400 МГц, CDCl3) δ 7.35-7.29 (m, 1H), 7.27-7.22 (m, 1H), 7.11-7.04 (m, 2H), 0.98 (s, 9H), 0.21 (s, 6H).13C ЯМР (100 МГц, CDCl3) δ 156.0, 130.4, 125.0, 124.9, 123.3, 118.5, 113.2, 25.5, 18.1, -4.6. EIMS: m/z 233 [M]+. HRMS: вычислено для C13H19NOSi: 233, 1236; найдено: 233,1227.
[0094] 3-(трет-Бутилдиметилсилилокси)бензиламин. Смесь алюмогидрида лития (4,97 г, 131 ммоль) и диэтилового эфира (400 мл) кипятили с обратным холодильником в течение 1 ч, затем охлаждали до температуры окружающей среды. Раствор 3-(трет-бутилдиметилсилилокси)бензонитрила (15,25 г, 65,5 ммоль) в диэтиловом эфире (50 мл) добавляли по каплям, так чтобы поддерживалась спокойная дефлегмация. Реакционную смесь затем кипятили с обратным холодильником в течение 3 ч и затем охлаждали в ледяной бане. Осторожно по каплям добавляли воду (5 мл), затем добавляли 15%-ный раствор гидроксида натрия (5 мл) и затем воду (15 мл). Твердое вещество отфильтровывали и тщательно промывали диэтиловым эфиром. Фильтрат промывали водой, сушили и концентрировали в вакууме с получением бледно-желтого масла (14,68 г, 95%). Это неочищенное вещество преадсорбировали на целит, затем подвергали хроматографии методом DCVC (сухая колоночная вакуумная хроматография), элюируя с использованием градиента этилацетата в РЕ (петролейный эфир) (5%-100% этилацетата) с получением 3-(трет-бутилдиметилсилилокси)бензиламина (11,85 г, 77%) в виде бледно-желтого масла.1H ЯМР (400 МГц, CDCl3) δ 7.13 (t, J=7,7 Гц, 1H), 6.84 (d, J=7,7 Гц, 1H), 6.79-6-75 (m, 1H), 6.68 (dd, J=8.0, 2,4 Гц, 1H), 3.75 (s, 2H), 0.97 (s, 9H), 0.18 (s, 6H).13C ЯМР (100 МГц, CDCl3) δ 155.5, 144.7, 129.1, 119.7, 118.5, 118.0, 46.0, 25.4, 17.9, -4.7. EIMS: m/z 237 [M]+. HRMS: вычислено для C13H23NOSi: 237,1543; найдено: 237,1545.
[0095] 3-(трет-Бутилдиметилсилилокси)бензилизотиоцианат (В). Смесь 3-(трет-бутилдиметилсилилокси)бензиламина (13,0 г, 54,8 ммоль), карбоната кальция (5,65 г, 56,4 ммоль), тиофосгена (8,5 мл, 111 ммоль), воды (41 мл) и хлороформа (356 мл) интенсивно перемешивали при комнатной температуре в течение 20 ч в закупоренной колбе. Реакционную смесь промывали водой (2 х 250 мл), сушили и концентрировали в вакууме с получением 3-(трет-бутилдиметилсилилокси)-бензилизотиоцианата (14,29 г, 93%) в виде бледно-рыжего масла.1H ЯМР (400 МГц, CDCl3) δ 7.27-7.21 (m, 1H), 6.92-6.88 (m, 1H), 6.83-6.79 (m, 2H), 4.65 (s, 2H), 1.00 (s, 9H), 0.22 (s, 6H).13С ЯМР (100 МГц, CDCl3) δ 156.1, 135.7, 132.4, 129.9, 120.0, 119.6, 118.5, 48.4, 25.6, 18.2, -4.4. EIMS: m/z 279 [M]+. HRMS: вычислено доя C14H21NOSi: 279,1108; найдено: 279,1105.
[0096] 1-(3-Бензилоксибензил)-5-(2-метил-1,3-Диоксолан-2-ил-метил)-4-фенил-1H-имидазол (8). К раствору 3,3-этилендиокси-1-бутаналя (1,37 г, 10,5 ммоль) в сухом метаноле (60 мл) добавляли 3-бензилоксибензиламин (2,10 г, 10,5 ммоль), и этот прозрачный раствор перемешивали при 25°С в течение 90 мин. Добавляли α-тозилбензилизоцианид (2,86 г, 10,5 ммоль), затем добавляли триэтиламин (2,93 мл, 21,1 ммоль), и реакционную смесь перемешивали и нагревали при 65°С в течение 4 ч. Растворитель затем удаляли, и остаток растворяли в этилацетате (50 мл), промывали водой (50 мл), сушили, и растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией (градиентное элюирование с использованием смеси 20-80% этилацетата/РЕ) с получением 1-(3-бензилоксибензил)-5-(2-метил-[1,3]-диоксалан-2-ил-метил)-4-фенил-1Н-имидазола (2,05 г, 44%) в виде липкого бледно-оранжевого масла.1H ЯМР (200 МГц, CDCl3) δ 7.89 (m, 2H) 7.58 (s, 1H), 7.21-7.7.44 (m, 9H), 6.90 (m, 1H), 6.65 (m, 2H), 5.31 (s, 2H), 5.01 (s, 2H), 3.83 (m, 2H), 3.59 (m, 2H), 3.02 (s, 2H), 1.33 (s, 3Н).13С ЯМР (50 МГц, CDCl3) δ 159.2, 138.5, 137.6, 136.6, 135.3, 130.0, 128.6, 128.3, 128.0, 127.4, 127.2, 126.5, 119.0, 114.0, 113.1, 109.9, 69.9, 65.0, 48.8, 33.9, 29.7, 25.5. ESIMS: m/z 441 [M+H]+. HRMS: вычислено для C28H28N2O3: 440,2094: найдено: 440,2075.
[0097] 1-[3-(3-Гидроксибензил)-5-фенил-3H-имидазол-4-ил]-пропан-2-он (9). К перемешиваемому раствору 1-(3-бензилоксибензил)-5-(2-метил-[1,3]-диоксолан-2-ил-метил)-4-фенил-1H-имидазола (1,12 г, 2,54 ммоль) в TFA (5 мл) добавляли тиоанизол (598 мкл, 5,09 ммоль), и эту реакционную смесь перемешивали при 25°С в течение 15 ч. Реакционную смесь затем охлаждали до 0°С и медленно добавляли воду (30 мл), а затем этилацетат (50 мл). Смесь переносили в делительную воронку, и водный слой удаляли. Органическую фазу промывали водой (2 х 50 мл), сушили, и растворитель удаляли в вакууме. Остаток затем пропускали через небольшой слой силикагеля (90% этилацетата/РЕ), растворитель удаляли при пониженном давлении, и осуществляли перекристаллизацию из смеси хлороформ/РЕ с получением 1-[3-(3-гидроксибензил)-5-фенил-3Н-имидазол-4-ил]-пропан-2-она (419 мг, 54%) в виде белого твердого вещества; т.пл. 173,7-175,5°С.1H ЯМР (200 МГц, CD3CN) δ 8.52 (s, 1H), 7.48 (s, 5H), 7.25 (t, 1H, J=7,8 Гц), 6.79 (m, 3Н), 5.16 (s, 2H), 3.94 (s, 2H), 2.14 (s, 3Н).13С ЯМР (50 МГц, CD3OD) 5204.6, 159.6, 136.7, 136.0, 134.5, 131.7, 131.1, 130.5, 129.2, 128.3, 126.0, 120.0, 117.1, 116.0, 52.1, 38.7, 29.5. ESIMS: m/z 307 [M+H]+. HRMS: вычислено для С19Н18N2O2: 306,1363; найдено: 306,1365.
[0098] 3-[5-(2-Гидроксипропил)-4-фенил-имидазол-1-илметил]-фенол (VB0003). В охлажденный в ледяной бане раствор 1-[3-(3-гидроксибензил)-5-фенил-3H-имидазол-4-ил]пропан-2-она (419 мг, 1,37 ммоль) в сухом метаноле (50 мл) добавляли боргидрид натрия (155 мг, 4,10 ммоль), и эту смесь перемешивали при 25°С в течение 15 ч. Затем добавляли ацетон (20 мл), и реакционную смесь перемешивали в течение еще 2 ч. Растворитель затем удаляли в вакууме, остаток растворяли в этилацетате (50 мл), промывали водой (50 мл), и органическую фазу сушили, и растворитель удаляли при пониженном давлении. Полученное твердое вещество подвергали перекристаллизации из метанола, собирали и промывали холодным ацетонитрилом (4х) с получением 3-[5-(2-гидроксипропил)-4-фенил-имидазол-1-илметил]-фенола (210 мг, 50%) в виде белого твердого вещества; т.пл. 193-194°С.1Н ЯМР (200 МГц, CD3OD) δ 7.72 (s, 1H), 7.62 (m, 2H), 7.39 (m, 2H), 7.28 (d, 1H, J=7,4 Гц), 7.18 (m, 1H), 6.72 (m, 1H), 6.65 (d, 2H, J=7,6 Гц), 6.54 (s, 1H), 5.34 (d, 1H, J=16,0 Гц), 5.24 (d, 1H, J=16,0 Гц), 3.90 (sextet, 1H), 2.82 (d, 2H, J=6,4 Гц), 1.05 (d, 3H, J=6,2 Гц).13С ЯМР (50 МГц, CD3OD) δ 159.3, 140.2, 139.8, 138.7, 136.4, 131.1, 129.5, 128.7, 127.9, 127.4, 118.9, 115.9, 114.6, 68.4, 34.1, 23.3. ESIMS (масс-спектрометрия с электрораспылительной ионизацией): m/z 309 [M+H]+. HRMS: вычислено для C19H20N2O2: 308,1519; найдено: 308,1517. HPLC (высокоэффективная жидкостная хроматография) чистота - 98%.
[0099] 4-Толуолсульфиновая кислота. В колбу Эрленмейера на 500 мл загружали тетрагидрат натриевой соли 4-толуолсульфиновой кислоты (26,82 г, 0,11 моль) и воду (134 мл), и эту смесь перемешивали в течение 30 мин до полного растворения твердого вещества. В перемешиваемый раствор добавляли трет-бутилметиловый эфир (134 мл), затем медленно добавляли 32%-ную HCl (12,2 мл, 0,11 моль) в течение 5 мин. Реакционную смесь перемешивали в течение 30 мин и переносили в делительную воронку, и органическую фазу отделяли и разбавляли толуолом (134 мл). Растворитель выпаривали до примерно 50 мл на роторном испарителе, после чего добавляли гептан (40 мл) с получением твердого вещества, которое собирали с использованием воронки Бюхнера. После последующей промывки гептаном (50 мл) и сушки под вакуумом получили 4-толуолсульфиновую кислоту (15,48 г, 93%) в виде белого твердого вещества.
[00100] N-(α-Тозилбензил)формамид. В трехгорлую колбу на 500 мл, оснащенную дефлегматором, загружали ацетонитрил (35 мл), толуол (35 мл), бензальдегид (6,74 мл, 66 ммоль), формамид (6,57 мл, 166 ммоль) и TMSCl (9,18 мл, 72 ммоль). После нагревания реакционной смеси в течение 4 ч при 50°С (внутренняя температура) добавляли 4-толуолсульфиновую кислоту (15,48 г, 99 ммоль) при перемешивании. В пределах 1 ч образовалось твердое вещество, и реакционную смесь перемешивали с использованием тефлоновой мешалки каждые 30 мин в течение периода времени 4 ч. Реакционную смесь затем оставляли охлаждаться до комнатной температуры, добавляли трет-бутилметиловый эфир (35 мл), и перемешивание продолжали в течение 5 мин, после чего добавляли воду (170 мл). Твердое вещество собирали на воронке Бюхнера, промывали трет-бутилметиловым эфиром (2 х 50 мл) и сушили в вакууме с получением N-(α-тозилбензил)формамида (16,73 г, 82%).1Н ЯМР (200 МГц, DMSO-d6) δ 9.77 (d, 1H, J=10,6 Гц) 7.96 (s, 1H), 7.71 (d, 2H, J=8,2 Гц), 7.54 (m, 2H), 7.42 (m, 5H), 6.38 (d, 1H, J=10,6 Гц), 2.41 (s, 3Н).
[00101] α-Тозилбензилизоцианид (С). В трехгорлую колбу на 500 мл, оснащенную термометром, загружали N-(α-тозилбензил)формамид (16,73 г, 57,8 ммоль) и сухой THF (120 мл). Шприцем добавляли оксихлорид фосфора (10,78 мл, 116,0 ммоль), и полученный раствор перемешивали в течение 5 мин при 25°С. После охлаждения раствора до приблизительно 0°С в бане лед/соль добавляли триэтиламин (48,4 мл, 347,0 ммоль) с помощью капельной воронки в течение 45 мин, поддерживая внутреннюю температуру ниже 10°С. После окончания добавления реакционную смесь перемешивали в течение 45 мин при 5-10°С (ледяная баня). В реакционную смесь добавляли этилацетат (85 мл), затем воду (85 мл), смесь перемешивали в течение 5 мин, и после переноса смеси в делительную воронку водный слой удаляли. Органическую фазу промывали водой (2 х 85 мл), насыщенным раствором бикарбоната натрия (85 мл), рассолом (50 мл) и концентрировали при пониженном давлении до тех пор, пока не осталась суспензия. Этот остаток разбавляли н-пропанолом (85 мл) и концентрировали до половины его первоначального объема. Осадок охлаждали до 0°С в течение 15 мин, собирали на воронке Бюхнера, промывали н-пропанолом (2 х 50 мл) и сушили в вакууме. Получили α-тозилбензилизоцианид (7,47 г, 48%) в виде бежевого твердого вещества.1H ЯМР (200 МГц, CDCl3) δ 7.60 (d, 2H, J=8,2 Гц) 7.30-7.52 (m, 7H), 5.61 (s, 1H), 2.47 (s, 3H).
[00102] 3-(Бензилокси)бензилазид. К перемешиваемому раствору 3-бензилоксибензилового спирта (5,0 г, 23,3 ммоль) и дифенилфосфорилазида (6,03 мл, 28,0 ммоль) в сухом толуоле (40 мл) при 0°С в атмосфере аргона добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (3,83 мл, 25,6 ммоль). Полученную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 18 ч. Реакционную смесь промывали водой (2х) и 5%-ным раствором HCl (1х), затем сушили и концентрировали при пониженном давлении. Неочищенное вещество фильтровали через короткую колонку с диоксидом кремния, элюируя смесью 4/96 этилацетат/пентан, с получением 3-(бензилокси)бензилазида (5,28 г, 95%) в виде прозрачного бесцветного масла.1H ЯМР (200 МГц, CDCl3) δ 7.48-7.22 (m, 6H); 6.99-6.86 (m, 3H); 5.07 (s, 2H); 4.30 (s, 2H).13С ЯМР (50 МГц, CDCl3) δ 159.4, 137.2, 137.0, 130.1, 128.8, 128.3, 127.7, 120.9, 114.9, 114.9, 70.3, 54.9. υN=N=N/см-1 2101.
[00103] 3-(Бензилокси)бензиламин (D). 3-(Бензилокси)бензилазид (1,0 г, 4,2 ммоль) растворяли в безводном THF (20 мл), и раствор охлаждали до -70°С. Твердый алюмогидрид лития (238 мг, 6,3 ммоль) в THF (6 мл) добавляли по каплям в атмосфере аргона. После добавления реакционную смесь оставляли нагреваться до 0°С и перемешивали в течение 1 ч. Реакционную смесь осторожно гасили водой, затем добавляли 1,0 М сегнетову соль и экстрагировали диэтиловым эфиром (3х). Объединенные органические экстракты промывали рассолом (1х), затем сушили и концентрировали с получением 3-(бензилокси)бензиламина (787 мг, 88%) в виде прозрачного бледно-желтого масла. Дополнительная очистка не требовалась.1H ЯМР (200 МГц, CDCl3) δ 7.48-7.17 (m, 6H); 6.98-6.79 (m, 3H); 5.06 (s, 2H); 3.83 (s, 2H), 1.43 (br s, 2H).
[00104] Этил-3,3-этилендиоксибутаноат. Смесь этилацетоацетата (9,72 мл, 76,84 ммоль), этиленгликоля (7,50 мл, 134,50 ммоль) и пара-толуолсульфоновой кислоты (15 мг, 0,08 ммоль) в бензоле (125 мл) кипятили с обратным холодильником в условиях Дина-Старка в течение 24 ч. Реакционную смесь оставляли охлаждаться, затем промывали 5%-ным раствором NaHCO3 (1х) и водой (2х), сушили и концентрировали с получением этил-3,3-этилендиокси-бутаноата (11,67 г, 87%) в виде прозрачного бесцветного масла. Дополнительная очистка не требовалась.1H ЯМР (200 МГц, CDCl3) δ 4.16 (q, 2H, J=7,2 Гц); 3.97 (s, 4H); 2.66 (s, 2H); 1.50 (s, 3H); 1.26 (t, 3Н, J=7,2 Гц).13С ЯМР (100 МГц, CDCl3) δ 169.7, 107.9, 65.0, 60.8, 44.5, 24.7, 14.4. υC=O/см-1 1741.
[00105] 3,3-Этилендиокси-1-бутанол. Раствор этил-3,3-этилендиоксибутаноата (5,0 г, 28,70 ммоль) в безводном THF (10 мл) добавляли по каплям в атмосфере аргона к суспензии алюмогидрида лития (1,10 г, 28,70 ммоль) в THF (50 мл) при -10°С. Эту реакционную смесь перемешивали при 0°С в течение 1,5 ч и затем гасили 1,0 М раствором сегнетовой соли (75 мл). Смесь экстрагировали диэтиловым эфиром (3х), затем объединенные органические экстракты промывали рассолом (1х), затем сушили и концентрировали с получением 3,3-этилендиокси-1-бутанола (3,43 г, 90%) в виде прозрачной бледно-желтой жидкости. Дополнительная очистка не требовалась.1H ЯМР (200 МГц, CDCl3) δ 3.99 (s, 4H), 3.76 (q, 2H, J=5,6 Гц), 2.78 (t, 1H, J=5,6 Гц), 1.95 (m, 2H), 1.36 (s, 3H).
[00106] 3,3-Этилендиокси-1-бутаналь (Е). В капельную воронку добавляли сухой DMSO (4,42 мл, 62,3 ммоль) и сухой дихлорметан (25 мл). Этот раствор добавляли по каплям в течение периода времени 10 мин к перемешиваемому раствору оксалилхлорида (2,64 мл, 31,14 ммоль) в сухом дихлорметане (75 мл) при -78°С. Через 10 минут по каплям добавляли раствор 2-(2-метил-1,3-диоксолан-2-ил)этанола (3,43 г, 26,0 ммоль) в сухом дихлорметане (25 мл) (с помощью капельной воронки) в течение периода времени 10 мин. Полученный раствор перемешивали при -78°С в течение 15 мин и затем шприцем по каплям добавляли триэтиламин (14,47 мл, 103,8 ммоль). Охлаждающую баню удаляли, и реакционную смесь оставляли перемешиваться в течение 30 мин. Затем добавляли воду (70 мл), и перемешивание продолжали в течение еще 10 мин. Органическую фазу отделяли, и водную фазу промывали дихлорметаном (70 мл). Объединенные органические фазы промывали водой (4 х 70 мл), сушили, и растворитель удаляли в вакууме. Остаток очищали фильтрованием через небольшой слой силикагеля (дихлорметан) с получением 2-(2-метил-[1,3]-диоксолан-2-ил)ацетальдегида (1,40 г, 42%) в виде бледно-желтого масла.1H ЯМР (200 МГц, CDCl3) δ 9.75 (t, 1H, J=2,8 Гц), 4.04-3.98 (m, 4H), 2.71 (d, 2H, J=2,8 Гц), 1.43 (s, 3H).
[00107] (S)-трет-Бутил-(4-амино-5-оксо-5-фениламино)пентаноат (14). К перемешиваемому раствору (S)-2-(9H-флуорен-9-илметоксикарбониламино)-5-трет-бутокси-5-оксо-пентановой кислоты (25,3 г, 57 ммоль) в DMF (150 мл) добавляли N-(3-диметиламинопропил)-N'-этилкарбодиимид (12,08 г, 63 ммоль), 1-гидроксибензотриазола гидрат (8,51 г, 63 ммоль) и анилин (5,2 мл; 57 ммоль), и эту реакционную смесь перемешивали при температуре окружающей среды в течение 2 суток. Добавляли воду (300 мл), затем этилацетат (300 мл). Смесь переносили в делительную воронку, и слои разделяли. Водный слой экстрагировали еще два раза этилацетатом. Объединенные органические слои промывали водой (2 х 300 мл), сушили, и растворитель удаляли в вакууме с получением неочищенного (трет-бутил-4-(9H-флуорен-9-илметоксикарбониламино)-5-оксо-5-(фениламино)пентаноата в виде желтого масла. Его использовали на следующей стадии без очистки. К перемешиваемому раствору неочищенного (S)-трет-бутил-4-(9Н-флуорен-9-илметоксикарбониламино)-5-оксо-5-(фениламино)пентаноата в дихлорметане (300 мл) добавляли трис(2-аминоэтил)амин (40 мл, 267 ммоль), и реакционную смесь перемешивали при температуре окружающей среды в течение 1-2 ч. Реакционную смесь затем промывали фосфатным буфером рН 5,5 (2 х 200 мл), затем водой (200 мл), сушили, и растворитель удаляли в вакууме. Остаток растворяли в дихлорметане, и нерастворимый побочный продукт удаляли фильтрованием. Раствор пре адсорбировали на целит, затем подвергали хроматографии (DCVC), элюируя с использованием градиента хлороформа в РЕ, и затем с использованием градиента этилацетата в хлороформе. Аналогичные фракции объединяли с получением (S)-трет-бутил-(4-амино-5-оксо-5-фениламино)пентаноата (10,85 г, 68%) в виде бледно-желтого масла.1H ЯМР (400 МГц, CDCl3) δ 9.41 (br s, 1H), 7.63-7.55 (m, 2H), 7.37-7.28 (m, 2H), 7.12-7.07 (m, 1H), 3.51 (dd, J=7.6, 5,0 Гц, 1H), 2.49-2.34 (m, 2H), 2.26-2.15 (m, 1H), 1.96-1.84 (m, 1H), 1.66 (br s, 2H), 1.45 (s, 9H).13С ЯМР (50 МГц, CDCl3) δ 172.8, 172.6, 137.7, 129.0, 124.1, 119.4, 80.8, 55.2, 32.2, 30.2, 28.1. EIMS: m/z 278 [M]+. HRMS: вычислено для C15H22N2O3: 278,1625; найдено: 278,1612.
[00108] (S)-трет-бутил-4-[(3-бенилокси)фениламино]-5-оксо-5-(фениламино)пентаноат (15). Раствор 3-(бензилокси)фенилбороновой кислоты (7,05 г, 30,0 ммоль) в толуоле (150 мл) подвергали азеотропной перегонке с дефлегмацией течение 2,5 ч под азотом до образования 2,4,6-трис(3-бетилокси)фенил)-1,3,5,2,4,6-триоксатриборинана. Толуол удаляли в вакууме, и к остатку при температуре окружающей среды добавляли раствор (S)-трет-бутил-(4-амино-5-оксо-5-фениламино)пентаноата (4,30 г, 15,5 ммоль) в дихлорметане (100 мл), пиридин (2,5 мл, 30,9 ммоль) и безводный ацетат меди(П) (4,64 г, 23,2 ммоль). Реакционную смесь открывали для доступа воздуха и перемешивали при температуре окружающей среды в течение 2 суток. Реакционную смесь затем выливали на короткую колонку, упакованную силикагелем и покрытую сверху слоем целита. Неочищенный продукт элюировали хлороформом, и фракции, содержащие неочищенный продукт, объединяли. Неочищенное вещество преадсорбировали на целит и затем очищали методом DCVC на силикагеле, элюируя с использованием градиента хлороформа в РЕ (60-100% хлороформа) с получением (S)-трет-бутил-4-[(3-бензилокси)фениламино]-5-оксо-5-(фениламино)пентаноата (3,35 г, 50%) в виде бледно-желто-коричневого масла.1H ЯМР (400 МГц, CDCl3) δ 8.69 (br s, 1H), 7.57-7.48 (m, 2H), 7.46-7.23 (m, 7H), 7.15-7.06 (m, 2H), 6.49-6.42 (m, 1H), 6.33-6.24 (m, 2H), 5.06-5.02 (m, 1H), 5.00 (s, 2H), 3.80-3.72 (m, 1H), 2.67-2.54 (m, 1H), 2.49-2.37 (m, 1H), 2.36-2.24 (m, 1H), 2.21-2.09 (m, 1H), 1.46 (s, 9H).13С ЯМР (50 МГц, CDCl3) δ 173.6, 171.3, 160.0, 148.2, 137.3, 136.9, 130.2, 128.9, 128.5, 127.8, 127.4, 124.4, 119.9, 106.8, 105.5, 100.8, 81.3, 69.8, 61.1, 32.7, 28.0, 27.8. EIMS: m/z 460 [M]+. HRMS: вычислено для C28H32N2O4: 460,2357; найдено 460,2341.
[00109] (S)-трет-бутил-4-[N-(3-бензилокси)фенил-2-бромацетамидо]-5-оксо-5-(фениламино)пентаноат (16). К перемешиваемому раствору (S)-трет-бутил-4-(3-бензилоксифениламино)-5-оксо-5-(фениламино)пентаноата (4,05 г, 8,80 ммоль) в дихлорметане (80 мл) под азотом и при -30°С добавляли последовательно триэтиламин (1,22 мл, 8,80 ммоль), N,N-диметиламинопиридин (20 мг) и бромацетилбромид (941 мкл, 10,80 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и затем перемешивали в течение 30 мин. Реакционную смесь переносили в делительную воронку, промывали 10%-ной лимонной кислотой (50 мл) и рассолом (2 х 50 мл), сушили, и растворитель удаляли в вакууме. Остаток преадсорбировали на целит, затем очищали методом DCVC на силикагеле с получением (S)-трет-бутил-4-[N-(3-бензилокси)фенил-2-бромацетамидо]-5-оксо-5-(фениламино)пентаноата (4,28 г, 83%) в виде бледно-желтого масла.1H ЯМР (400 МГц, CDCl3) δ 8.61 (br s, 1H), 7.63-7.53 (m, 2H), 7.49-7.21 (m, 8H), 7.17-7.09 (m, 1H), 7.08-7.02 (1H), 6.89-6.84 (m, 2H), 5.21 (dd, J=8.8, 6,2 Гц, 1H), 5.04 (br s, 2H), 3.64 (dd, J=19.7, 11,2 Гц, 2H), 2.44-2.19 (m, 2H), 2.05-1.91 (m, 1H), 1.80-1.67 (m, 1H), 1.43 (s, 9H).13С ЯМР (50 МГц, CDCl3) δ 171.8, 168.2, 167.8, 159.4, 138.3, 137.8, 136.1, 130.4, 128.9, 128.5, 128.1, 127.5, 124.3, 121.4, 119.9, 116.6, 115.5, 80.7, 70.2, 59.4, 31.8, 28.0, 27.4, 23.9. EIMS: m/z 581 [M]+. HRMS: вычислено для C30H33BrN2O5: 580,1567; найдено: 580,1547.
[00110] (S)-трет-Бутил-3-[1-(3-бензилокси)фенил-3,6-диоксо-4-фенилпиперазин-2-ил]пропаноат (17). К перемешиваемому раствору (S)-трет-бутил-4-[N-(3-бензилокси)фенил-2-бромацетамидо]-5-оксо-5-(фениламино)пентаноата (4,15 г, 7,15 ммоль) в DMF (70 мл) под азотом и при охлаждении в ледяной бане добавляли карбонат цезия (2,33 г, 7,15 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение 4 ч. Смесь охлаждали в ледяной бане и добавляли воду (100 мл), затем этилацетат (100 мл). Смесь переносили в делительную воронку, и слои разделяли. Водную фазу экстрагировали еще два раза этилацетатом. Объединенные органические слои промывали 10%-ной лимонной кислотой (100 мл), насыщенным раствором бикарбоната натрия (100 мл) и рассолом (100 мл), сушили, и растворитель удаляли в вакууме. Остаток преадсорбировали на целит, затем очищали методом DCVC на силикагеле. Аналогичные фракции объединяли, затем растирали с диэтиловым эфиром с получением (S)-трет-бутил-3-[1-(3-бензилокси)фенил-3,6-диоксо-4-фенилпиперазин-2-ил]пропаноата (4,18 г, 59%) в виде бесцветного твердого вещества; т.пл. 141,6-143,6°С.1H ЯМР (400 МГц, CDCl3) δ 7.48-7.29 (m, 11H), 7.03-6.95 (m, 3H), 5.08 (s, 2H), 4.55 (dd, J=17.6, 17,0 Гц, 2Н), 4.45 (dd, J=9.1, 5,2 Гц, 1Н), 2.52-2.14 (m, 4H), 1.37 (s, 9H).13C ЯМР (50 МГц, CDCl3) δ 171.4, 165.7, 163.8, 159.6, 139.9, 139.8, 136.5, 130.4, 129.3, 128.6, 128.1, 127.6, 127.4, 125.1, 119.3, 114.7, 113.7, 81.0, 70.25, 63.9, 52.9, 30.6, 28.0, 26.9. EIMS: m/z 500 [M]+. HRMS: вычислено для C31H32N2O5: 500,2306; найдено 500,2294.
[00111] (S)-трет-Бутил-3-[1-(3-бензилокси)фенил-4-фенилпиперазин-2-ил]пропаноат (18). К перемешиваемому раствору (S)-трет-бутил-3-[1-(3-бензилокси)фенил-3,6-диоксо-4-фенилпиперазин-2-ил]пропаноата (2,97 г, 5,95 ммоль) в безводном THF (40 мл) под азотом добавляли боран-метилсульфидный комплекс (7,14 мл, 14,30 ммоль, 2М в THF), и эту реакционную смесь перемешивали при температуре окружающей среды в течение 20 ч. Реакционную смесь затем охлаждали в ледяной бане и по каплям добавляли раствор метанола (2 мл) в THF (8 мл). Растворитель удаляли в вакууме, после чего снова добавляли метанол (10 мл). Растворитель удаляли в вакууме, и эту процедуру повторяли еще раз. Остаток преадсорбировали на целит, затем очищали на силикагеле (DCVC), элюируя с использованием градиента этилацетата в РЕ (2%-7% этилацетата), с получением (S)-трет-бутил-3-[1-(3-бензилокси)фенил-4-фенилпиперазин-2-ил]пропаноата (2,08 г, 74%) в виде бледно-желтого масла.1H ЯМР (400 МГц, CDCl3) δ 7.47-7.42 (m, 2H), 7.42-7.35 (m, 2H), 7.35-7.25 (m, 3Н), 7.19-7.13 (m, 1H), 6.97-6.92 (m, 2H), 6.91-6.85 (m, 1H), 6.57-6.52 (m, 2H), 6.48-6.42 (m, 1H), 5.06 (s, 2H), 3.97-3.88 (m, 1H), 3.61-3.50 (m, 2H), 3.50-3.41 (m, 1H), 3.38-3.27 (m, 1H), 3.13-3.04 (m, 1H), 3.03-2.29 (m, 1H), 2.33-2.14 (m, 2H), 2.12-1.90 (m, 2H), 1.41 (s, 9H).13С ЯМР (100 МГц, CDCl3) δ 172.6, 160.1, 151.7, 151.2, 137.3, 13.0, 129.2, 128.5, 127.9, 127.6, 119.9, 116.4, 108.8, 104.5, 103.2, 80.4, 70.0, 55.2, 51.8, 48.8, 43.1, 32.7, 28.1, 22.8. EIMS: m/z 472 [M]+. HRMS: вычислено для C30H36N2O3: 472,2720; найдено: 472,2710.
[00112] (S)-3-[1-(3-Гидрокси)фенил-4-фенилпиперазин-2-ил]пропановая кислота (19). К перемешиваемому раствору (S)-трет-бутил-3-[1-(3-бензилокси)фенил-4-фенилпиперазин-2-ил]пропаноата (0,82 г, 1,75 ммоль) в трифторуксусной кислоте (3,5 мл) добавляли тиоанизол (620 мкл, 5,22 ммоль), и эту реакционную смесь перемешивали при температуре окружающей среды в течение 3 суток. Реакционную смесь затем охлаждали до 0°С и медленно добавляли воду (10 мл), затем добавляли EtOAc (15 мл). Смесь переносили в делительную воронку, слои разделяли, и водную фазу экстрагировали еще два раза EtOAc, сушили, и растворитель удаляли в вакууме. Темно-рыжее масло преадсорбировали на целит, затем подвергали хроматографии (DCVC), элюируя с использованием градиента метанола в хлороформе (0-5% МеОН), с получением (S)-3-[1-(3-гидрокси)фенил-4-фенилпиперазин-2-ил]пропановой кислоты (418 мг, 70%) в виде рыжей пены.1H ЯМР (400 МГц, CDCl3) δ 7.37-7.31 (m, 2Н), 7.30-7.24 (m, 2H), 7.17 (br s, 1H), 7.08-7.00 (m, 3H), 6.92-6.79 (m, 2H), 3.99-3.89 (m, 1H), 3.70-3.37 (m, 6H), 2.42-2.18 (m, 2H), 2.02-1.88 (m, 2H).13С ЯМР (50 МГц, MeOH-d4) δ 175.8, 160.6, 151.0, 144.0, 132.6, 130.6, 123.0, 118.5, 116.7, 112.7, 109.3, 63.7, 55.6, 53.7, 49.2, 30.8, 25.0. EIMS: m/z 326 [M]+. HRMS: вычислено для C19H22N2O3: 326,1625; найдено: 326,1620.
[00113] (S)-3-[1-(3-Бензилокси)фенил-4-фенилпиперазин-2-ил]пропановая кислота (230 мг, 30%) также была получена в результате этой реакции и была выделена в виде бледно-рыжей пены.1H ЯМР (400 МГц, CDCl3) δ 10.59 (br s, 1H), 7.45-7.25 (m, 8H), 7.17-6.90 (m, 6H), 5.07 (s, 2H), 3.99-3.88 (m, 1H), 3.76-3.41 (m, 6H), 2.33-2.15 (m, 2Н).13С ЯМР (100 МГц, CDCl3) δ 176.2 160.1, 148.9, 144.3, 136.2, 131.1, 129.6, 128.7, 128.2, 127.6, 122.8, 117.8, 113.5, 112.6, 107.5, 70.3, 60.5, 52.7, 52.4, 48.9, 29.9, 23.3. EIMS: m/z 416 [M]+. HRMS: вычислено для C26H28N2O3: 416,2094; найдено: 416,2091.
[00114] (S)-3-[1-(3-Гидрокси)фенил-4-фенилпиперазин-2-ил]пропанамид (VB0005). К перемешиваемому раствору (5)-3-[1-(3-гидрокси)фенил-4-фенилпиперазин-2-ил]пропановой кислоты (0,76 г, 2,32 ммоль) в THF (20 мл) добавляли 1,1'-карбонилдиимидазол (0,94 г, 5,80 ммоль), и эту реакционную смесь перемешивали в течение 2 ч. Добавляли водный раствор аммиака (25%-ный) (20 мл), и реакционную смесь перемешивали в течение еще 3 ч. Реакционную смесь переносили в делительную воронку, и слои разделяли. Водную фазу затем экстрагировали этилацетатом (2 х 20 мл). Объединенные органические слои промывали рассолом (20 мл), сушили и концентрировали в вакууме. Неочищенный остаток пропускали через короткую колонку, упакованную Florisil, элюируя с использованием градиента EtOAc в РЕ (50-100% этилацетата). Аналогичные фракции объединяли, растворяли в дихлорметане (20 мл) и промывали водой (3 х 20 мл). Твердое вещество выкристаллизовывалось из влажного дихлорметана. Добавляли петролейный эфир 40-60°С, и смесь оставляли стоять в течение 1 ч. Полученное твердое вещество собирали фильтрованием с получением рацемического продукта (100 мг, 13%). Фильтрат концентрировали досуха с получением (S)-3-[1-(3-гидрокси)фенил-4-фенилпиперазин-2-ил]пропанамида (185 мг, 21%) в виде бледно-рыжей пены.1H ЯМР (400 МГц, CDCl3) δ 7.31-7.24 (m, 2Н), 7.13-7.06 (m, 1H), 6.95-6.84 (m, 4H), 6.47-6.41 (m, 2Н), 6.35-6.30 (m, 1H), 5.74 (brs, 1H), 5.53 (br s, 1H), 4.00-3.91 (m, 1H), 3.60-3.41 (m, 3H), 3.35-3.23 (m, 1H), 3.10-3.00 (m, 1H), 3.00-2.90 (m, 2Н), 2.32-1.96 (m, 4H).13C ЯМР (50 МГц, CDCl3) δ 176.0, 157.3, 151.6, 151.2, 130.4, 129.2, 119.9, 116.4, 107.5, 106.3, 102.9, 54.7, 51.7, 48.8, 42.9, 32.7, 23.4. EIMS: m/z 325 [М]+. HRMS: вычислено для C19H23N3O2: 325,1790; найдено: 325,1776. [α]D = -13,5° (CHCl3, с = 0,005).
[00115] 3-[5-(2-гидроксипропил)-4-фенил-имидазол-1-илметил]фенола гидрохлорид (20). Раствор 3-[5-(2-гидроксипропил)-4-фенил-имидазол-1-илметил] фенола (14 мг, 0,05 ммоль), концентрированной соляной кислоты (3,6 мкл, 0,05 ммоль) и метанола (1 мл) перемешивали при температуре окружающей среды в течение 20 мин. Растворитель затем удаляли в вакууме, остаток затем переносили в диэтиловом эфире (2 мл), растворитель удаляли при пониженном давлении, и эту процедуру повторяли. Полученную белую пену сушили в вакууме при 35°С, растворяли в воде (10 мл), фильтровали через фильтр из бумаги из стекловолокна и подвергали сублимационной сушке с получением 3-[5-(2-гидроксипропил)-4-фенил-имидазол-1-илметил]фенола гидрохлорида в виде рыхлого белого твердого вещества; т.пл. 84-88°С.1H ЯМР (400 МГц, CD3OD) δ 8.84 (s, 1H), 7.63 (m, 2H), 7.53 (m, 3H), 7.25 (m, 1H), 6.81 (m, 1H), 6.77 (d, 1H, J=8,0 Гц), 6.70 (s, 1H), 5.53 (d, 1H, J=16,0 Гц), 5.48 (d, 1H, J=16,0 Гц), 3.92 (sextet, 1H), 2.85 (d, 2H, J=8,0 Гц), 1.12 (d, 3H, J=8,0 Гц).13С ЯМР (125 МГц, CD3OD) δ 159.8, 136.9, 136.3, 133.3, 131.8, 131.0, 130.5, 130.4, 129.7, 129.0, 119.8, 117.0, 115.7, 67.9, 52.3, 33.3, 23.8.
[00116] Схемы синтеза, способы и реагенты для получения других соединений по настоящему изобретению будут очевидны среднему специалисту исходя из приведенной выше информации, коммерчески доступных реагентов и рутинных знаний в области органической химии и синтеза соединений.
Пример 2 - Эксперименты in vivo
[00117] Четырнадцатинедельных спонтанно-гипертензивных крыс (SHR; Australian Animal Resources Centre, WA) на диете с содержанием соли 2,2% (Glenn Forrest Stockfeeders, WA) случайным образом распределяли по следующим группам лечения: 14-недельный контроль, или инфузия VB0002 (20 пмоль/кг/мин в 20% DMSO), или инфузия являющегося контролем носителя (20% DMSO) в течение 4 недель. Инфузии VB0002 и являющегося контролем носителя выполняли с помощью осмотического мининасоса Alzet, который вставляли под общей анестезией (изофлуран 3% в кислороде) через 14 недель.
[00118] Четырнадцатинедельных спонтанно-гипертензивных крыс на диете с содержанием соли 2,2% случайным образом распределяли также по следующим группам лечения: 14-недельный контроль, или введение VB0003 (10, 100 или 500 пмоль/кг/мин в 5% этаноле), или введение VB0005 (100 пмоль/кг/мин в 5% этаноле), или введение питьевого раствора (5% этанол) в течение 4 недель.
[00119] 14-недельную контрольную группу анестезировали с использованием изофлурана (3%), доставляемого в кислороде, затем брали образцы крови, и сердца и почки собирали для количественной оценки фиброза. Остальные группы взвешивали, и давление крови измеряли плетизмографией с помощью хвостовой манжеты (ADI Instruments) дважды в неделю в течение дальнейших 4 недель. После 4-недельного лечения крыс анестезировали, и образцы собирали как в 14-недельной контрольной группе. Результаты представляют собой среднее значение ± sem (стандартная ошибка среднего) для n=5 крыс на группу.
[00120] Для количественной оценки фиброза срезы тканей толщиной ≤3 мм фиксировали в 10% забуференном формалине в течение 24 часов, обрабатывали и заделывали в парафин. Поперечные срезы толщиной три мкм окрашивали с использованием Trichrome по Массону. Минимум 20 случайных областей при увеличении 40х из поперечных срезов (по 5 на каждом из 2 уровней) оцифровывали. Степень фиброза определяли как процент от площади области каждого оцифрованного изображения с использованием Image-Pro Plus V.5 (Media Cybernetics, Bethesda, MD, USA), и затем усредняли для определения уровня фиброза для каждой крысы.
[00121] Систолическое давление крови у крыс, которых лечили VB0002, VB0003 и VB0005, снижалось по сравнению с 18-недельными контролями (Фиг. 8 и Фиг. 9), что свидетельствует о том, что эти соединения эффективны в снижении давления крови.
[00122] Фиброз миокарда у крыс, которых лечили VB0002 при 20 пмоль/кг/мин, сокращался по сравнению с 14-недельными контролями и 18-недельными контролями (Фиг. 10), что свидетельствует о том, что это соединение ослабляет развитие фиброза миокарда и реверсирует установленный фиброз миокарда.
[00123] Фиброз миокарда у крыс, которых лечили VB0003 при 10, 100 и 500 пмоль/кг/мин, сокращался по сравнению с 18-недельными контролями (Фиг. 11), что свидетельствует о том, что эти соединения ослабляют развитие фиброза миокарда. Фиброз миокарда у крыс, которых лечили VB0003 при 100 и 500 пмоль/кг/мин, сокращался по сравнению с 14-недельными контролями (Фиг. 11), что свидетельствует о том, что эти дозировки соединения реверсируют установленный фиброз миокарда.
[00124] Фиброз миокарда у крыс, которых лечили VB0005 при 100 пмоль/кг/мин, сокращался по сравнению с 14-недельными контролями и 18-недельными контролями (Фиг. 10), что свидетельствует о том, что это соединение ослабляет развитие фиброза миокарда и реверсирует установленный фиброз миокарда (Фиг. 12).
Реферат
Изобретение относится к соединениям общих формул, указанных ниже, их стереоизомерам и фармацевтически приемлемым солям, фармацевтической композиции на их основе, их применению и способу терапевтического лечения гипертензии, или прегипертензии, и/или фиброза миокарда и их применению в профилактическом и/или терапевтическом лечении гипертензии и/или фиброза, включающему введение эффективного количества соединения по п. 1.или.В общих формулах X выбран изиY представляет собой А или СН2-А, А выбран изии Z выбран изи.10 н. и 1 з.п. ф-лы, 12 ил., 2 пр.
Формула









Документы, цитированные в отчёте о поиске
Новые пиперазинамидные производные
Комментарии