Феноксиметильные производные - RU2746481C1
Код документа: RU2746481C1
Описание
Настоящее изобретение относится к органическим соединениям, применяемым для терапии или профилактики у млекопитающих, и, в частности, к ингибиторам аутотаксина (ATX), которые являются ингибиторами продукции лизофосфатидиловой кислоты (LPA) и, таким образом, модуляторами уровней LPA и ассоциированного сигналинга, для лечения или профилактики заболеваний почек, заболеваний печени, воспалительных состояний, заболеваний нервной системы, заболеваний дыхательной системы, сосудистых и сердечно-сосудистых заболеваний, фиброзных заболеваний, рака, глазных заболеваний, метаболических нарушений, холестатической и других форм хронического зуда и острого и хронического отторжения трансплантата органов.
В настоящем изобретении предложены новые соединения формулы (I)
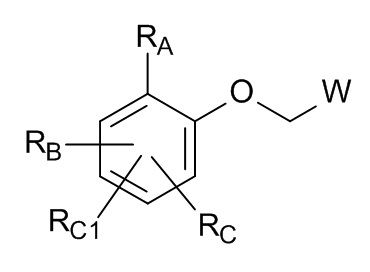
где
RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) арил, замещенный RG, RG1 and RG2,
vii) гетероциклоалкил, замещенный RG, RG1 и RG2, и
viii) гетероарил, замещенный RG, RG1 и RG2;
RB выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C3-C8-циклоалкил,
iii) C1-C6-алкилсульфонил,
iv) C3-C8-циклоалкилсульфонил,
v) C1-C6-алкилсульфониламино,
vi) C3-C8-циклоалкилсульфониламино,
vii) аминокарбонил,
viii) циано,
ix) галоген,
x) гало-C1-C6-алкокси,
xi) гало-C1-C6-алкил,
xii) гетероциклоалкил, и
xiii) гетероарил, замещенный одним H, C1-C6-алкилом или триалкилсилил-C1-C6-алкокси-C1-C6-алкилом;
RC и RC1 независимо выбраны из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил, и
vi) галоген;
или RB и RC, вместе с атомами углерода, которым они присоединены, образуют кольцевую систему, выбранную из группы, состоящей из
i) C3-C8-циклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила,
ii) гетероциклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила,
iii) арил, замещенный одним или двумя заместителями, независимо выбранными из H, галогена, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила, и
iv) гетероарил, замещенный одним или двумя заместителями, независимо выбранными из H, галогена, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила;
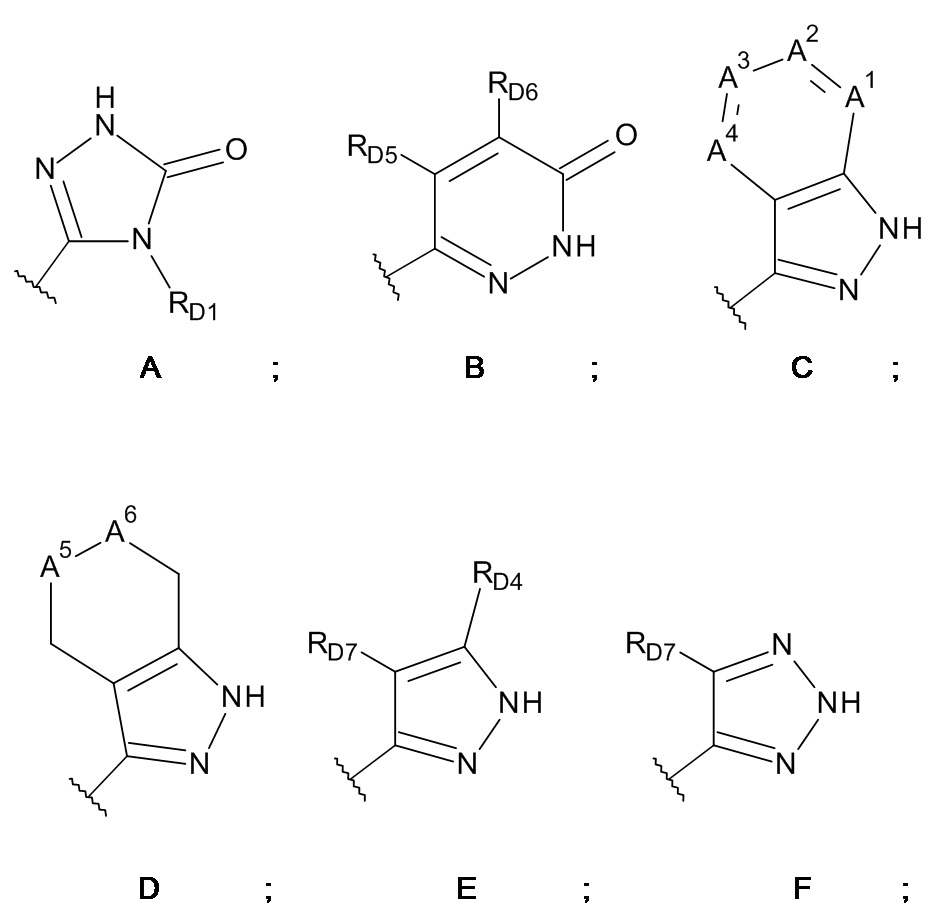
W выбран из кольцевых систем A, B, C, D, E, F и G;

A1, A3 и A4 представляют собой -CH- и A2 представляет собой -CRD2-,
A1 представляет собой -N-, A2 представляет собой -CRD2-, A3 представляет собой -CH- или -N-, и A4 представляет собой -CH-,
A1, A3 и A4 представляют собой -CH- и A2 представляет собой -N-,
A1, A2 и A4 представляют собой -CH- и A3 представляет собой -N-, или
A1 и A3 представляют собой -CH-, A2 представляет собой -CRD2- и A4 представляет собой -N-;
один из A5 и A6 представляет собой -NRD3-, а другой представляет собой -CRLRM-;
RD1 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) гало-C1-C6-алкокси,
iv) гало-C1-C6-алкил, и
v) C3-C8-циклоалкила;
RD2 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iv) C1-C6-алкоксикарбонил,
v) C1-C6-алкилкарбонил,
vi) C3-C8-циклоалкилкарбонил,
vii) C1-C6-алкил,
viii) C3-C8-циклоалкил,
ix) гидрокси-C1-C6-алкокси,
x) гидрокси-C1-C6-алкил(C1-C6-алкил)амино,
xi) гидрокси-C1-C6-алкиламино,
xii) гидрокси-C1-C6-алкил,
xiii) дигидрокси-C1-C6-алкокси,
xiv) дигидрокси-C1-C6-алкил(C1-C6-алкил)амино,
xv) дигидрокси-C1-C6-алкиламино,
xvi) дигидрокси-C1-C6-алкил,
xvii) гало-C1-C6-алкокси,
xviii) гало-C1-C6-алкил,
xix) гетероциклоалкил,
xx) гетероциклоалкилкарбонил, и
xxi) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RD3 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил,
iv) C1-C6-алкилкарбонил,
v) C3-C8-циклоалкилкарбонил,
vi) C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) гало-C1-C6-алкокси,
ix) гало-C1-C6-алкил,
x) гидрокси-C1-C6-алкил, и
xi) дигидрокси-C1-C6-алкил,
RD4 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил,
iv) C1-C6-алкилкарбонил,
v) C3-C8-циклоалкилкарбонил,
vi) C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) гало-C1-C6-алкокси,
ix) гало-C1-C6-алкил,
x) гетероциклоалкилкарбонил,
xi) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами, и
xii) арил, замещенный одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси;
RD5, RD6 и RD7 независимо выбраны из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C1-C6-алкокси
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) C3-C8-циклоалкил, и
vii) C3-C8-циклоалкокси;
RD8 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C1-C6-алкокси,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) C3-C8-циклоалкил,
vii) C1-C6-алкокси-C1-C6-алкилкарбонил,
viii) C1-C6-алкоксикарбонил,
ix) C1-C6-алкилкарбонил,
x) C3-C8-циклоалкилкарбонил,
xi) гетероциклоалкилкарбонил, и
xii) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) C3-C8-циклоалкилсульфонил,
ix) карбокси,
x) циано,
xi) C3-C8-циклоалкил,
xii) C3-C8-циклоалкокси,
xiii) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xiv) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xv) C1-C6-алкилкарбониламино-C1-C6-алкил,
xvi) C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xvii) гало-C1-C6-алкил,
xviii) гало-C1-C6-алкокси,
xix) галоген,
xx) гидрокси,
xxi) аминокарбонил, замещенный по атому азота RN и RO,
xxii) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xxiii) гетероарил, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси,
i) гетероциклоалкил-C1-C6-алкокси, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси, и
ii) гетероциклоалкил-C1-C6-алкил, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси;
RG1 и RG2 независимо выбраны из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил,
iv) C3-C8-циклоалкил,
v) гало-C1-C6-алкокси, и
vi) гало-C1-C6-алкил;
RL и RM независимо выбраны из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
RN выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкил,
iv) C1-C6-алкоксикарбонил-C1-C6-алкил,
v) C1-C6-алкил,
vi) карбокси-C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) C3-C8-циклоалкил-C1-C6-алкил,
ix) гидрокси-C1-C6-алкил,
x) фенил, и
xi) гетероарил-C1-C6-алкил;
RO выбран из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил;
или их фармацевтически приемлемые соли.
Аутотаксин (ATX) представляет собой секретируемый фермент, также называемый эктонуклеотидная пирофосфатаза / фосфодиэстераза 2 или лизофосфолипаза D, которая важна для конвертации лизофосфатидилхолина (LPC) в биоактивную сигнальную молекулу лизофосфатидиловой кислоты (LPA). Было показано, что уровни LPA в плазме хорошо коррелируют с активностью ATX и, следовательно, предполагается, что ATX является важным источником внеклеточной LPA. Ранние эксперименты с прототипами ингибиторов ATX показали, что такое соединение способно ингибировать синтезирующую активность LPA в плазме мышей. Работа, выполненная в 1970-х и начале 1980-х показала, что LPA может вызывать широкий диапазон клеточных ответов; включая сокращение гладкомышечных клеток, активацию тромбоцитов, клеточную пролиферацию, хемотаксис и другие. LPA опосредует свои эффекты через сигналинг к некоторым рецепторам, сопряженным с G-белком (GPCRs); первые члены были изначально обозначены как рецепторы Edg (ген дифференцировки эндотелиальных клеток) или ген-1 вентрикулярной зоны (vzg-1), однако в настоящее время называются рецепторами LPA. Классическая группа в настоящее время состоит из LPA1/Edg-2/VZG-1, LPA2/Edg-4 и LPA3/Edg-7. Недавно были описаны три дополнительных LPA рецептора LPA4/p2y9/GPR23, LPA5/GPR92 и LPA6/p2Y5, которые более тесно связаны с нуклеотид-селективными пуринергическими рецепторами, чем с классическими рецепторами LPA1-3. Сигнальная ось ATX-LPA вовлечена в большой диапазон физиологических и патофизиологических функций, включая, например, функции нервной системы, развитие сосудов, сердечно-сосудистую физиологию, репродукцию, функционирование иммунной системы, хроническое воспаление, метастазирование и прогрессирование опухолей, фиброз органов, а также ожирение и/или другие болезни обмена веществ, такие как сахарный диабет. Таким образом, увеличенная активность ATX и/или увеличенный уровень LPA, изменяет экспрессию рецепторов LPA и измененные ответы на LPA могут способствовать инициации, прогрессированию и/или развитию некоторых различных патофизиологических состояний, связанных с осью ATX/LPA.
В соответствии с настоящим изобретением, соединения формулы (I) или их фармацевтически приемлемые соли и эфиры могут быть использованы для лечения или профилактики заболеваний, нарушений или состояний, которые ассоциированы с активностью аутотаксина и/или биологической активностью лизофосфатидиловой кислоты (LPA).
Соединения формулы (I) или их фармацевтически приемлемые соли и эфиры, раскрытые здесь, ингибируют активность аутотаксина и, таким образом, ингибируют продукцию LPA и модулируют уровни LPA и ассоциированный сигналинг. Ингибиторы аутотаксина, описанные здесь, применяются в качестве агентов для лечения или предотвращения заболеваний или состояний, при которых активность ATX и/или сигналинг LPA участвует, вовлечен в этиологию или патологию заболевания, или другим образом ассоциирован с по меньшей мере одним симптомом заболевания. Ось ATX-LPA вовлечена, например, в ангиогенез, хроническое воспаление, аутоиммунные заболевания, фиброзные заболевания, опухоли и метастазирование и прогрессирование рака, глазные заболевания, заболевания обмена веществ, такие как ожирение и/или сахарный диабет, состояния, такие как холестатический или другие формы хронического зуда, а также острое или хроническое отторжение трансплантата органов.
Объектами настоящего изобретения являются соединения формулы (I) и их вышеуказанные соли и эфиры и их применение в качестве терапевтически активных веществ, способ производства указанных соединений, промежуточных соединений, фармацевтических композиций, лекарственных средств, содержащих указанные соединения, их фармацевтически приемлемые соли или эфиры, применение указанных соединений, солей или эфиров для лечения или профилактики нарушений или состояний, которые ассоциированы с активностью ATX и/или биологической активностью лизофосфатидиловой кислоты (LPA), в частности для лечения или профилактики заболеваний почек, заболеваний печени, воспалительных состояний, заболеваний нервной системы, заболеваний дыхательной системы, сосудистых и сердечно-сосудистых заболеваний, фиброзных заболеваний, рака, глазных заболеваний, метаболических нарушений, холестатической или других форм хронического зуда, и острого или хронического отторжения трансплантата органов, и применение указанных соединений, солей или эфиров для получения лекарственных средств для лечения или профилактики заболеваний почек, заболеваний печени, воспалительных состояний, заболеваний нервной системы, заболеваний дыхательной системы, сосудистых и сердечно-сосудистых заболеваний, фиброзных заболеваний, рака, глазных заболеваний, метаболических нарушений, холестатической и других форм хронического зуда и острого и хронического отторжения трансплантата органов. Более конкретно, соединения формулы (I) и их вышеупомянутые соли и эфиры, и их применение в качестве терапевтически активных веществ, способ получения указанных соединений, промежуточные соединения, фармацевтические композиции, лекарственные средства содержащие указанные соединения, их фармацевтически приемлемые соли или эфиры для лечения или профилактики глазных заболеваний, в частности глаукомы.
Термин " C1-6-алкокси" обозначает группу формулы -O-R ', где R' представляет собой C1-6-алкильную группу. Примеры C1-6-алкоксигруппы включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси и трет-бутокси. Конкретными примерами являются метокси и изопропокси.
Термин “C1-6-алкокси-C1-6-алкил” обозначает C1-6-алкильную группу, где по меньшей мере один из атомов водорода C1-6-алкильной группы заменен на C1-6-алкокси группу. Конкретными примерами являются метоксиметил, метоксиэтил, этоксиметил, этоксиэтил, изо-пропоксиметил и изо-пропоксиэтил. Конкретный пример представляет собой метоксиэтил.
Термин “C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил” обозначает амино-C1-C6-алкил, где атом азота замещен C1-C6-алкильной группой и C1-C6-алкокси-C1-C6-алкилкарбонильной группой. Конкретным примером является аминометил, где атом азота замещен метилом и метоксиметилкарбонилом.
Термин “C1-C6-алкокси-C1-C6-алкилкарбонил” обозначает группу формулы -C(O)-R', где R' представляет собой C1-C6-алкокси-C1-C6-алкильную группу. Примеры C1-C6-алкокси-C1-C6-алкилкарбонильной группы включают группы, где R' представляет собой метоксиметил, метоксиэтил, этоксиметил, этоксиэтил, изо-пропоксиметил и изо-пропоксиэтил. Конкретным примером является группа, где R' представляет собой метоксиметил.
Термин “C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил” обозначает амино-C1-C6-алкильную группу, где атом азота замещен H и C1-C6-алкокси-C1-C6-алкилкарбонильной группой. Конкретным примером является аминометил, где атом азота замещен H и метоксиметилкарбонилом.
Термин “C1-C6-алкоксикарбонил-C1-C6-алкил” обозначает C1-6-алкильную группу, где по меньшей мере один из атомов водорода C1-6-алкильной группы заменен на C1-C6-алкоксикарбонильную группу. Конкретными примерами являются группы, где C1-C6-алкоксикарбонильная группа представляет собой метоксикарбонил или этоксикарбонил и C1-6-алкильная группа представляет собой метил или этил. Более конкретными примерами являются метоксиоксопропил и этоксиоксоэтил.
Термин “C1-C6-алкоксикарбонил” обозначает группу формулы -C(O)-R', где R' представляет собой C1-C6-алкокси группу. Примеры C1-C6-алкоксикарбонильной группы включают группы, где R' представляет собой метокси, этокси, n-пропокси, изопропокси, n-бутокси, изобутокси и трет-бутокси. Конкретным примером являются группы, где R' представляет собой метокси или трет-бутокси.
Термин “C1-6-алкил” обозначает моновалентную линейную или разветвленную насыщенную углеводородную группу из 1-6 атомов углерода. Примеры C1-6-алкила включают метил, этил, пропил, изопропил, n-бутил, изо-бутил, втор-бутил, трет-бутил и пентил. Конкретные алкильные группы включают метил, изопропил и трет-бутил.
Термин “C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил” обозначает амино-C1-C6-алкил, где атом азота замещен C1-C6-алкильной группой и C1-C6-алкилкарбонильной группой. Конкретным примером является аминометил, где атом азота замещен метилом и метилкарбонилом или этилкарбонилом.
Термин “C1-C6-алкилкарбонил” обозначает группу формулы -C(O)-R', где R' представляет собой C1-C6-алкильную группу. Примеры C1-C6-алкилкарбонильной группы включают группы, где R' представляет собой метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси и трет-бутокси. Конкретным примером является группа, где R' представляет собой метил.
Термин “C1-C6-алкилкарбониламино-C1-C6-алкил” обозначает амино-C1-C6-алкил, где атом азота замещен H и C1-C6-алкилкарбонильной группой. Конкретным примером является аминометил, где атом азота замещен H и метилкарбонилом или этилкарбонилом.
Термин “C1-C6-алкилсульфонил” обозначает группу формулы -S(O)2-R', где R' представляет собой C1-C6-алкильную группу. Конкретным примером является группа, где R' представляет собой метил.
Термин “C1-C6-алкилсульфонилamino” обозначает группу формулы -NH-S(O)2-R', где R' представляет собой C1-C6-алкильную группу. Конкретным примером является группа, где R' представляет собой метил.
Термин “амино” обозначает группу -NH2.
Термин “аминоалкил” обозначает C1-6-алкильную группу, где один из атомов водорода C1-6-алкильной группы заменен на аминогруппу. Конкретными примерами являются аминометил, аминоэтил, аминопропил и аминобутил.
Термин “аминокарбонил” обозначает группу формулы -C(O)-NH2.
Термин “аминокарбонил-C1-C6-алкокси” обозначает C1-6-алкокси группу, где один из атомов водорода C1-6-алкокси группы заменен на аминокарбонильную группу. Конкретным примером является группа, где C1-6-алкокси группа представляет собой метокси.
Термин “арил” обозначает фенильную или нафтильную группу. Конкретный пример представляет собой фенил.
Термин “карбокси” обозначает группу -COOH.
Термин “карбокси-C1-C6-алкил” обозначает C1-6-алкильную группу, где один из атомов водорода C1-6-алкильной группы заменен на карбокси группу. Конкретными примерами являются карбоксиметил и карбоксиэтил.
Термин “циано” обозначает -C≡N группу.
Термин “циано-C1-C6-алкил” обозначает C1-6-алкильную группу, где один из атомов водорода C1-6-алкильной группы заменен на цианогруппу. Конкретными примерами являются цианометил, цианоэтил, цианопропил и цианобутил. Конкретный пример представляет собой цианопропил.
Термин “C3-8-циклоалкокси” обозначает группу формулы -O-R', где R' представляет собой C3-8-цикоалкил. Конкретным примером является группа, где R' представляет собой циклопропил.
Термин “C3-8-циклоалкил” обозначает моновалентную насыщенную моноциклическую или бициклическую углеводородную группу из 3 - 8 кольцевых атомов углерода. Бициклическая означает кольцевую систему, состоящую из двух насыщенных карбоциклов, обладающих двумя общими атомами углерода. Примерами моноциклических циклоалкилов являются циклопропил, циклобутанил, циклопентил, циклогексил или циклогептил. Примерами бициклических C3-8-циклоалкилов являются бицикло[2.2.1]гептанил или бицикло[2.2.2]октанил. Конкретными C3-8-циклоалкильными группами являются циклобутил, циклопропил, циклопентил и циклогексил.
Конкретными примерами “C3-8-циклоалкила”, образованного заместителями RB и RC вместе с атомами углерода, которым они присоединены, являются циклопентил или циклогексил, более конкретно циклопентил.
Термин “C3-8-циклоалкил-C1-6-алкил” обозначает C1-6-алкильную группу, где по меньшей мере один из атомов водорода C1-6-алкильной группы заменен на C3-8-циклоалкильную группу. Конкретным примером является циклопропилметил.
Термин “C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил” обозначает амино-C1-C6-алкил, где атом азота замещен C1-C6-алкильной группой и C3-C8-циклоалкилкарбонильной группой. Конкретным примером является аминометил, где атом азота замещен метилом и циклопропилкарбонилом.
Термин “C3-C8-циклоалкилкарбонил” обозначает группу формулы -C(O)-R', где R' представляет собой C3-C8-циклоалкильную группу. Примерами C3-C8-циклоалкилкарбонила являются группы, где R' представляет собой циклопропил.
Термин “C3-C8-циклоалкилкарбониламино-C1-C6-алкил” обозначает амино-C1-C6-алкил, где атом азота замещен H и C3-C8-циклоалкилкарбонильной группой. Конкретным примером является аминометил, где атом азота замещен H и циклопропилкарбонилом.
Термин “C3-C8-циклоалкилсульфонил” обозначает группу формулы -S(O)2-R', где R' представляет собой C3-C8-циклоалкильную группу. Примерами C3-C8-циклоалкилсульфонила являются группы, где R' представляет собой циклопропил.
Термин “C3-C8-циклоалкилсульфониламино” обозначает группу формулы -NH-S(O)2-R', где R' представляет собой C3-C8-циклоалкильную группу. Примеры C3-C8-циклоалкилсульфонила являются группы, где R' представляет собой циклопропил.
Термин “дигидрокси-C1-C6-алкокси” обозначает C1-6-алкокси группу, где два из атомов водорода дигидрокси-C1-C6-алкокси группы, расположенных на разных атомах углерода, каждый заменен на гидроксигруппу. Конкретным примером является дигидроксипропокси. Дополнительным конкретным примером является 2,3-дигидроксипропокси.
Термин “дигидрокси-C1-C6-алкил” обозначает C1-6-алкильную группу, где два из атомов водорода C1-6-алкильной группы, расположенных на разных атомах углерода, каждый заменен на гидроксигруппу. Конкретным примером является дигидроксипропил. Дополнительным конкретным примером является 2,3-дигидроксипропил.
Термин “дигидрокси-C1-C6-алкиламино” обозначает группу формулы -NH-R', где R' представляет собой дигидрокси-C1-C6-алкильную группу. Примеры дигидрокси-C1-C6-алкиламино являются группы, где R' представляет собой дигидроксипропил, более конкретно 2,3-дигидроксипропил.
Термин “дигидрокси-C1-C6-алкил(C1-C6-алкил)амино” обозначает группу формулы -NRR', где R представляет собой C1-C6-алкил и R' представляет собой дигидрокси-C1-C6-алкильную группу. Примером дигидрокси-C1-C6-алкил(C1-C6-алкил)амино является дигидроксипропил(метил)амино, дополнительным конкретным примером является 2,3-дигидроксипропил(метил)амино.
Термин “гало-C1-6-алкокси” обозначает C1-6-алкокси группу, где по меньшей мере один из атомов водорода C1-C6-алкокси группы заменен одинаковыми или различными атомами галогенов. Конкретными примерами являются дифторметокси, трифторметокси, дифторэтокси и трифторэтокси. Более конкретным примером является трифторметокси.
Термин “галоген” и “гало” используются взаимозаменяемо здесь и обозначают фтор, хлор, бром или йод. Конкретными галогенами являются хлор и фтор.
Термин “гало-C1-6-алкил” обозначает C1-6-алкильную группу, где по меньшей мере один из атомов водорода C1-6-алкильной группы одинаковыми или различными атомами галогенов. Конкретными примерами являются дифторметил, трифторметил, дифторэтил и трифторэтил. Более конкретный пример представляет собой трифторметил.
Термин “гетероарил”, самостоятельно или в комбинации, обозначает моновалентную ароматическую гетероциклическую моно- или бициклическую кольцевую системы из 5 - 12 кольцевых атомов, содержащую 1, 2, 3 или 4 гетероатома, выбранных из N, O и S, оставшиеся кольцевые атомы являются углеродом. Примеры гетероарильной группы включают пирролил, фуранил, тиенил, имидазолил, оксазолил, тиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиразинил, пиразолил, пиридинил, пиридазинил, пиримидинил, триазинил, азепинил, диазепинил, изоксазолил, бензофуранил, изотиазолил, бензотиенил , индолил, изоиндолил, изобензофуранил, бензимидазолил, бензоксазолил, бензоизоксазолил, бензотиазол, бензоизотиазолил, бензооксиазолил, бензотиадиазолил, бензотриазолил, пуринил, хинолинил, изохинолинил, хиназолинил, хиноксалинил и бензотиофенил. Конкретными гетероарилгруппами являются пирролил, фуранил, тиенил, имидазолил, оксазолил, тиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиразинил, пиразолил, пиридазинил, пиримидинил, изоксазолил, изотиазолил, бензофуранил и бензотиофенил. Более конкретными гетероарильными группами являются бензоксазолдонил, имидазолил, изоксазолил, оксазолил, тиазолил, оксадиазолил, пиразинил, пиразолил, пиридинил и пиримидинил.
В случае заместителя RA, конкретными гетероарильными группами являются бензоксазолoнил, имидазолил, изоксазолил, оксазолил, пиразинил, пиразолил, пиридинил и пиримидинил. Более конкретными примерами являются изоксазолил и пиридинил.
В случае заместителя RB, конкретными гетероарильными группами являются оксадиазолил, имидазолил, 1,3,4-оксазолил и 1,2,4-оксазолил.
Конкретным примером гетероциклоалкила, образованного заместителями RB и RC вместе с атомами углерода, которым они присоединены, является тиазолил.
В случае заместителя RG, конкретными гетероарильными группами являются изоксазолил, оксазолил и пиразолил. Более конкретными примерами являются изоксазолил и пиразолил.
Термин “гетероарил-C1-C6-алкил” обозначает алкильную группу, где один из атомов водорода C1-C6-алкильной группы заменен на гетероарильную группу.
В случае заместителя RN, конкретными гетероарилалкильными группами являются пиридинилаклил и тиофенилалкил, более конкретно пиридинилметил и тиофенилметил.
Термин "гетероциклоалкил" самостоятельно или в комбинации означает одновалентную насыщенную или частично ненасыщенную моно- или бициклическую кольцевую систему из 4 - 9 кольцевых атомов, содержащую 1, 2, или 3 кольцевых гетероатома, выбранных из N, O и S, остальные кольцевые атомы являются углеродами. Бициклический означает состоящий из двух насыщенных циклов с двумя общими кольцевыми атомами, то есть мост, разделяющий два кольца, представляет собой либо простую связь, либо цепь из одного или двух кольцевых атомов. Примерами моноциклических насыщенных гетероциклоалкилов являются 4,5-дигидро-оксазолил, оксетанил, азетидинил, пирролидинил, 2-оксо-пирролидин-3-ил, тетрагидрофуранил, тетрагидро-тиенил, пиразолидинил, имидазолидинил, оксазолидинил, изоксазолидинил, тиазолидинил, пиперидинил, тетрагидропиранил, тетрагидротиопиранил, пиперазинил, морфолинил, тиоморфолинил, 1,1-диоксо-тиоморфолин-4-ил, азепанил, диазепанил, гомопиперазинил или оксазепанил. Примерами бициклических насыщенных гетероциклоалкилов являются 8-аза-бицикло[3.2.1]октил, хинуклидинил, 8-оксо-3-аза-бицикло[3.2.1]октил, 9-аза-бицикло[3.3.1]нонил, 3-окса-9-аза-бицикло[3.3.1]нонил или 3-тиа-9-аза-бицикло[3.3.1]нонил. Примерами частично ненасыщенных гетероциклоалкилов являются дигидрофурил, имидазолинил, дигидро-оксазолил, тетрагидро-пиридинил или дигидропиранил.
В случае заместителя RD2, конкретным примером гетероциклоалкила является гидроксиазетидинил, более конкретно 3-гидроксиазетидин-1-ил.
В случае заместителя RA, конкретным примером гетероциклоалкила является тетрагидропиранил.
В случае заместителя RB, конкретным примером гетероциклоалкила является морфолинил.
Конкретным примером гетероциклоалкила, образованного заместителем RN и RO вместе с атомом азота, к которому они присоединены, являются пиперидинил, морфолинил, пирролидинил и метилпиперазинонил.
Термин “гетероциклоалкил-C1-6-алкокси” обозначает C1-6-алкокси группу, где по меньшей мере один из атомов водорода алкильной группы заменен на гетероциклоалкильную группу. Конкретными примерами являются тетрагидропиранилметокси и тетрагидрофуранилметокси.
Термин “гетероциклоалкил-C1-6-алкил” обозначает C1-6-алкильную группу, где по меньшей мере один из атомов водорода C1-6-алкильной группы заменен на гетероциклоалкильную группу. Конкертными примерами гетероциклоалкил-C1-6-алкила являются группы, где гетероциклоалкильная группа представляет собой метилпиперазиндионил, пирролидинонил и оксазолидинонил и где C1-6-алкильная группа представляет собой метил.
Термин “гетероциклоалкилкарбонил” обозначает группу формулы -C(O)-R', где R' представляет гетероциклоалкильную группу. Примерами гетероциклоалкилкарбонильной групы являются группы, где R' представляет собой 4,5-дигидро-оксазолил, оксетанил, азетидинил, пирролидинил, 2-оксо-пирролидин-3-ил, тетрагидрофуранил, тетрагидротиенил, пиразолидинил, имидазолидинил, пирролидинил, оксазолидинил, изоксазолидинил, тиазолидинил, пиперидинил, тетрагидропиранил, тетрагидротиопиранил, пиперазинил, морфолинил, тиоморфолинил, 1,1-диоксо-тиоморфолин-4-ил, азепанил, диазепанил, гомопиперазинил, или оксазепанил.
В случае заместителя RD3, конкретным примером гетероциклоалкилкарбонильной группы является группа, где R' представляет собой пирролидинил.
Термин “гидрокси” обозначает группу -OH.
Термин “гидрокси-C1-6-алкокси” обозначает C1-6-алкокси группу, где один из атомов водорода C1-6-алкокси заменен на гидроксигруппу. Конкретными примерами являются гидроксиэтокси и гидроксипропокси. Более конкретным примером является гидроксиэтокси.
Термин “гидрокси-C1-6-алкил” обозначает C1-6-алкильную группу, где один из атомов водорода C1-6-алкильной группы заменен на гидроксигруппу. Конкретными примерами являются гидроксиметил и гидроксиэтил. Более конкретным примером является гидроксиэтил.
Термин “гидрокси-C1-C6-алкиламино” обозначает группу формулы -NH-R', где R' представляет собой гидрокси-C1-C6-алкильную группу. Примеры гидрокси-C1-C6-алкиламино включает группы, где R' представляет гидроксиэтил или гидроксипропил. Конкретным примером является группа, где R' гидроксиэтил.
Термин “гидрокси-C1-C6-алкил(C1-C6-алкил)амино” обозначает группу формулы -NRR', где R представляет собой C1-C6-алкил и R' представляет собой гидрокси-C1-C6-алкильную группу. Примеры гидрокси-C1-C6-алкил(C1-C6-алкил)аминогрупп включают группы, где R представляет собой метил, этил, пропил или изопропил и где R' представляет собой гидроксиэтил или гидроксипропил. Конкретным примером является группа, где R представляет собой метил и R' представляет собой гидроксиэтил.
Термин “триалкилсилил-C1-C6-алкокси-C1-C6-алкил” обозначает C1-C6-алкил, где один из атомов водорода C1-6-алкильной группы заменен на триалкилсилил-C1-C6-алкокси. Конкретным примером является триметилсилилэтоксиметил.
Термин “триалкилсилил-C1-C6-алкокси” обозначает C1-C6-алкокси, где один из атомов водорода C1-6-алкокси группы заменен на триалкилсилил. Конкретным примером является триметилсилилэтоксил.
Термин “триалкилсилил” обозначает группу формулы -Si(R')3, где каждый R' представляет собой независимо выбранную C1-C6-алкильную группу. Конкретным примером являются группы, где R' представляет собой метил, этил, пропил, изопропил, n-бутил, изо-бутил, втор-бутил, трет-бутил и пентил. Более конкретными примерами являются группы, где все R' являются одинаковыми, более конкретно, где R' представляет собой метил.
Термин "фармацевтически приемлемые соли" относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются биологически или иным образом нежелательными. Соли образованы с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т. п., предпочтительно, соляная кислота, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и т. п. Кроме того, эти соли могут быть получены путем добавления неорганического основания или органического основания к свободной кислоте. Соли, полученные из неорганических оснований включают, без ограничения, соли натрия, калия, лития, аммония, кальция и магния и т. п. Соли, полученные из органических оснований, включают, без ограничения, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, лизин, аргинин, N-этилпиперидин, пиперидин, полиаминовые смолы и т. п. Конкретными фармацевтически приемлемыми солями соединений формулы (I) являются соли соляной кислоты, метансульфоновой кислоты и лимонной кислоты.
"Фармацевтически приемлемые эфиры" означает, что соединения общей формулы (I) могут быть дериватизированы по функциональным группам с получением производных, которые способны превратиться обратно в исходные соединения в условиях in vivo. Примеры таких соединений включают физиологически приемлемые и метаболически лабильные эфирные производные, такие как метоксиметиловые эфиры, метилтиометиловые эфиры и пивалоилоксиметиловые эфиры. Дополнительно, любые физиологически приемлемые эквиваленты соединений общей формулы (I), аналогичные метаболически лабильным эфирам, которые способны превращаться в исходные соединения общей формулы (I) in vivo, включены в объем настоящего изобретения.
Термин "защитная группа" обозначает группу, которая селективно блокирует реакционноспособный участок в многофункциональном соединении таким образом, что химическая реакция может быть проведена селективно в другом незащищенном реакционноспособном участке, в значении обычно связанным с ним в синтетической химии. Защитные группы могут быть удалены на соответствующей стадии. Типичные защитные группы представляют собой амино-защитные группы, карбокси-защитные группы или гидрокси-защитные группы. Конкретные защитные группы представляют собой трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBZ или Z), флуоренилметилоксикарбонил (Fmoc) и бензил (Bn). Дополнительно конкретными защитными группами являются трет-бутоксикарбонил (ВОС) и флуоренилметилоксикарбонил (Fmoc). Более конкретнымой защитной группой является трет-бутоксикарбонильная (ВОС) группа.
Сокращение мкМ означает микромоль и является эквивалентным обозначению μM.
Сокращение мкл означает микролитр и является эквивалентным обозначению uL.
Сокращение мкг означает микрограмм и является эквивалентным обозначению μg.
Соединение формулы (I) может содержать несколько асимметричных центров и может присутствовать в форме оптически чистых энантиомеров, смесей энантиомеров, таких как, например, рацематы, смесей диастереоизомеров, диастереоизомерных рацематов или смесей диастереоизомерных рацематов.
Согласно правилам Кана - Ингольда - Прелога асимметричный атом углерода может быть "R" или "S" конфигурации.
Также воплощением настоящего изобретения являются соединения в соответствии с формулой (I), как здесь описано, и их фармацевтически приемлемые соли или эфиры, в частности соединения в соответствии с формулой (I), как здесь описано, и их фармацевтически приемлемые соли, более конкретно соединения в соответствии с формулой (I), как здесь описано.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где
В настоящем изобретении предложены новые соединения формулы (I)
где
RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) арил, замещенный RG, RG1 и RG2,
vii) гетероциклоалкил, замещенный RG, RG1 и RG2, и
viii) гетероарил, замещенный RG, RG1 и RG2;
RB выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C3-C8-циклоалкил,
iii) C1-C6-алкилсульфонил,
iv) C3-C8-циклоалкилсульфонил,
v) C1-C6-алкилсульфониламино,
vi) C3-C8-циклоалкилсульфониламино,
vii) аминокарбонил,
viii) циано,
ix) галоген,
x) гало-C1-C6-алкокси,
xi) гало-C1-C6-алкил,
xii) гетероциклоалкил, и
xiii) гетероарил, замещенный одним H, C1-C6-алкилом или триалкилсилил-C1-C6-алкокси-C1-C6-алкилом;
RC и RC1 независимо выбраны из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил, и
vi) галоген;
или RB и RC вместе с атомами углерода, которым они присоединены, образуют кольцевую систему, выбранную из группы, состоящей из
i) C3-C8-циклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила,
ii) гетероциклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила,
iii) арил, замещенный одним или двумя заместителями, независимо выбранными из H, галогена, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила, и
iv) гетероарил, замещенный одним или двумя заместителями, независимо выбранными из H, галогена, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила;
W выбран из кольцевых систем A, B, C, D, E, F и G;
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -CRD2-,
A1 представляет собой -N-, A2 представляет собой -CRD2-, A3 представляет собой -CH- или -N-, и A4 представляет собой -CH-,
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -N-,
A1, A2 и A4 представляют собой -CH-, и A3 представляет собой -N-, или
A1 и A3 представляют собой -CH-, A2 представляет собой -CRD2- и A4 представляет собой -N-;
один из A5 и A6 представляет собой -NRD3- а другой представляет собой -CRLRM-;
RD1 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) гало-C1-C6-алкокси,
iv) гало-C1-C6-алкил, и
v) C3-C8-циклоалкила;
RD2 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iv) C1-C6-алкоксикарбонил,
v) C1-C6-алкилкарбонил,
vi) C3-C8-циклоалкилкарбонил,
vii) C1-C6-алкил,
viii) C3-C8-циклоалкил,
ix) гидрокси-C1-C6-алкокси,
x) гидрокси-C1-C6-алкил(C1-C6-алкил)амино,
xi) гидрокси-C1-C6-алкиламино,
xii) гидрокси-C1-C6-алкил,
xiii) дигидрокси-C1-C6-алкокси,
xiv) дигидрокси-C1-C6-алкил(C1-C6-алкил)амино,
xv) дигидрокси-C1-C6-алкиламино,
xvi) дигидрокси-C1-C6-алкил,
xvii) гало-C1-C6-алкокси,
xviii) гало-C1-C6-алкил,
xix) гетероциклоалкил,
xx) гетероциклоалкилкарбонил, и
xxi) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RD3 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил,
iv) C1-C6-алкилкарбонил,
v) C3-C8-циклоалкилкарбонил,
vi) C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) гало-C1-C6-алкокси,
ix) гало-C1-C6-алкил,
x) гидрокси-C1-C6-алкил, и
xi) дигидрокси-C1-C6-алкил,
RD4 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил,
iv) C1-C6-алкилкарбонил,
v) C3-C8-циклоалкилкарбонил,
vi) C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) гало-C1-C6-алкокси,
ix) гало-C1-C6-алкил,
x) гетероциклоалкилкарбонил, и
xi) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RD5, RD6 и RD7 независимо выбраны из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C1-C6-алкокси,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) C3-C8-циклоалкил, и
vii) C3-C8-циклоалкокси;
RD8 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C1-C6-алкокси
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) C3-C8-циклоалкил,
vii) C1-C6-алкокси-C1-C6-алкилкарбонил,
viii) C1-C6-алкоксикарбонил,
ix) C1-C6-алкилкарбонил,
x) C3-C8-циклоалкилкарбонил,
xi) гетероциклоалкилкарбонил, и
xii) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) C3-C8-циклоалкилсульфонил,
ix) карбокси,
x) циано,
xi) C3-C8-циклоалкил,
xii) C3-C8-циклоалкокси,
xiii) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xiv) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xv) C1-C6-алкилкарбониламино-C1-C6-алкил,
xvi) C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xvii) гало-C1-C6-алкил,
xviii) гало-C1-C6-алкокси,
xix) галоген,
xx) гидрокси,
xxi) аминокарбонил, замещенный по атому азота RN и RO,
xxii) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xxiii) гетероарил, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси,
i) гетероциклоалкил-C1-C6-алкокси, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси, и
ii) гетероциклоалкил-C1-C6-алкил, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси;
RG1 и RG2 независимо выбраны из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил,
iv) C3-C8-циклоалкил,
v) гало-C1-C6-алкокси, и
vi) гало-C1-C6-алкил;
RL и RM независимо выбраны из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
RN выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкил,
iv) C1-C6-алкоксикарбонил-C1-C6-алкил,
v) C1-C6-алкил,
vi) карбокси-C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) C3-C8-циклоалкил-C1-C6-алкил,
ix) гидрокси-C1-C6-алкил,
x) фенил, и
xi) гетероарил-C1-C6-алкил;
RO выбран из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил;
или их фармацевтически приемлемые соли.
В другом воплощении в настоящем изобретении предложены новые соединения формулы (I)
где
RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) арил, замещенный RG, RG1 и RG2,
vii) гетероциклоалкил, замещенный RG, RG1 и RG2, и
viii) гетероарил, замещенный RG, RG1 и RG2;
RB выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C3-C8-циклоалкил,
iii) C1-C6-алкилсульфонил,
iv) C3-C8-циклоалкилсульфонил,
v) C1-C6-алкилсульфониламино,
vi) C3-C8-циклоалкилсульфониламино,
vii) аминокарбонил,
viii) циано,
ix) галоген,
x) гало-C1-C6-алкокси,
xi) гало-C1-C6-алкил,
xii) гетероциклоалкил, и
xiii) гетероарил, замещенный одним H, C1-C6-алкилом или триалкилсилил-C1-C6-алкокси-C1-C6-алкил;
RC и RC1 независимо выбраны из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил, и
vi) галоген;
или RB и RC вместе с атомами углерода, которым они присоединены, образуют кольцевую систему, выбранную из группы, состоящей из
i) C3-C8-циклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила,
ii) гетероциклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила,
iii) арил, замещенный одним или двумя заместителями, независимо выбранными из H, галогена, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила, и
iv) гетероарил, замещенный одним или двумя заместителями, независимо выбранными из H, галогена, C1-C6-алкила, гало-C1-C6-алкокси, гало-C1-C6-алкила и C3-C8-циклоалкила;
W выбран из кольцевых систем A, B, C, D и E;

A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -CRD2-,
A1 представляет собой -N-, A2 представляет собой -CRD2-, A3 представляет собой -CH- или -N-, и A4 представляет собой -CH-,
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -N-,
A1, A2 и A4 представляют собой -CH-, и A3 представляет собой -N-, или
A1 и A3 представляют собой -CH-, A2 представляет собой -CRD2-, и A4 представляет собой -N-;
один из A5 и A6 представляет собой -NRD3-, а другой представляет собой -CRLRM-;
RD1 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) гало-C1-C6-алкокси,
iv) гало-C1-C6-алкил, и
v) C3-C8-циклоалкила;
RD2 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iv) C1-C6-алкоксикарбонил,
v) C1-C6-алкилкарбонил,
vi) C3-C8-циклоалкилкарбонил,
vii) C1-C6-алкил,
viii) C3-C8-циклоалкил,
ix) гидрокси-C1-C6-алкокси,
x) гидрокси-C1-C6-алкил(C1-C6-алкил)амино,
xi) гидрокси-C1-C6-алкиламино,
xii) гидрокси-C1-C6-алкил,
xiii) дигидрокси-C1-C6-алкокси,
xiv) дигидрокси-C1-C6-алкил(C1-C6-алкил)амино,
xv) дигидрокси-C1-C6-алкиламино,
xvi) дигидрокси-C1-C6-алкил,
xvii) гало-C1-C6-алкокси,
xviii) гало-C1-C6-алкил,
xix) гетероциклоалкил,
xx) гетероциклоалкилкарбонил, и
xxi) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RD3 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил,
iv) C1-C6-алкилкарбонил,
v) C3-C8-циклоалкилкарбонил,
vi) C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) гало-C1-C6-алкокси,
ix) гало-C1-C6-алкил,
x) гидрокси-C1-C6-алкил, и
xi) дигидрокси-C1-C6-алкил,
RD4 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил,
iv) C1-C6-алкилкарбонил,
v) C3-C8-циклоалкилкарбонил,
vi) C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) гало-C1-C6-алкокси,
ix) гало-C1-C6-алкил,
x) гетероциклоалкилкарбонил, и
xi) аминокарбонил , замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RD5, RD6 и RD7 независимо выбраны из группы, состоящей из
i) H,
ii) C1-C6-алкил,
iii) C1-C6-алкокси
iv) гало-C1-C6-алкокси,
v) гало-C1-C6-алкил,
vi) C3-C8-циклоалкил, и
vii) C3-C8-циклоалкокси,
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) C3-C8-циклоалкилсульфонил,
ix) карбокси,
x) циано,
xi) C3-C8-циклоалкил,
xii) C3-C8-циклоалкокси,
xiii) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xiv) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xv) C1-C6-алкилкарбониламино-C1-C6-алкил,
xvi) C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xvii) гало-C1-C6-алкил,
xviii) гало-C1-C6-алкокси,
xix) галоген,
xx) аминокарбонил, замещенный по атому азота RN и RO,
xxi) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xxii) гетероарил, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси,
xxiii) гетероциклоалкил-C1-C6-алкокси, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси, и
xxiv) гетероциклоалкил-C1-C6-алкил, замещенный одним H, C1-C6-алкилом, C3-C8-циклоалкилом, гало-C1-C6-алкилом, гало-C1-C6-алкокси, бензилом или арилом, где бензил и арил замещены одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси;
RG1 и RG2 независимо выбраны из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил,
iv) C3-C8-циклоалкил,
v) гало-C1-C6-алкокси, и
vi) гало-C1-C6-алкил;
RL и RM независимо выбраны из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
RN выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкил,
iv) C1-C6-алкоксикарбонил-C1-C6-алкил,
v) C1-C6-алкил,
vi) карбокси-C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) C3-C8-циклоалкил-C1-C6-алкил,
ix) гидрокси-C1-C6-алкил,
x) фенил, и
xi) гетероарил-C1-C6-алкил;
RO выбран из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил;
или их фармацевтически приемлемые соли.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где
RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) арил, замещенный RG и RG1,
v) гетероциклоалкил, замещенный RG и RG1, и
vi) гетероарил, замещенный RG и RG1;
RB выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C1-C6-алкилсульфонил,
iii) C1-C6-алкилсульфониламино,
iv) аминокарбонил,
v) циано,
vi) галоген,
vii) гетероциклоалкил, и
viii) гетероарил, замещенный одним H, C1-C6-алкилом или триалкилсилил-C1-C6-алкокси-C1-C6-алкилом;
RC1 представляет собой H и RC выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил, и
iii) галоген;
или RB и RC вместе с атомами углерода, которым они присоединены, образуют кольцевую систему, выбранную из группы, состоящей из
i) гетероциклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H и C1-C6-алкила, и
ii) гетероарил, замещенный одним или двумя заместителями, независимо выбранными из H и C1-C6-алкила;
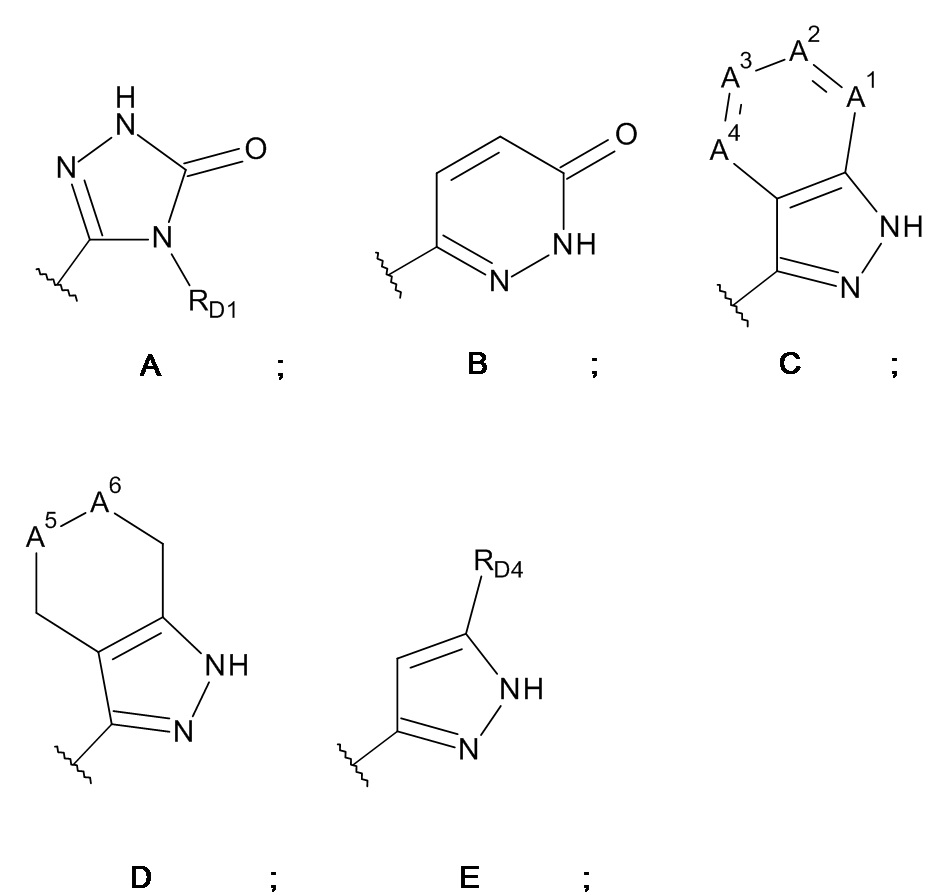
W выбран из кольцевых систем A, B, C, D и E;
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -CRD2-,
A1 представляет собой -N-, A2 представляет собой -CRD2-, A3 представляет собой -CH- или -N-, и A4 представляет собой -CH-,
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -N-, или
A1 и A3 представляют собой -CH-, A2 представляет собой -CRD2-, и A4 представляет собой -N-;
один из A5 и A6 представляет собой -NRD3-, а другой представляет собой -CRLRM-;
RD1 представляет собой C1-C6-алкил;
RD2 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) гидрокси-C1-C6-алкокси,
iv) гидрокси-C1-C6-алкил(C1-C6-алкил)амино,
v) гидрокси-C1-C6-алкиламино,
vi) дигидрокси-C1-C6-алкокси,
vii) дигидрокси-C1-C6-алкил(C1-C6-алкил)амино, и
viii) гетероциклоалкил;
RD3 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил, и
iv) C1-C6-алкилкарбонил;
RD4 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкоксикарбонил,
iii) гетероциклоалкилкарбонил,
iv) аминокарбонил, замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами, и
v) арил, замещенный одним - тремя заместителями, независимо выбранными из H, C1-C6-алкила, C3-C8-циклоалкила, гало-C1-C6-алкила и гало-C1-C6-алкокси;
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) карбокси,
ix) циано,
x) C3-C8-циклоалкокси,
xi) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xii) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xiii) гало-C1-C6-алкил,
xiv) галоген,
xv) гидрокси,
xvi) аминокарбонил, замещенный по атому азота RN и RO,
xvii) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xviii) гетероарил, замещенный одним H или C1-C6-алкилом,
xix) гетероциклоалкил-C1-C6-алкокси, замещенный одним H или C1-C6-алкилом, и
xx) гетероциклоалкил-C1-C6-алкил, замещенный одним H или C1-C6-алкилом;
RG1 м RG2 независимо выбраны из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил, и
iv) гало-C1-C6-алкокси;
RL и RM представляют собой H;
RN выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкил,
iv) C1-C6-алкоксикарбонил-C1-C6-алкил,
v) C1-C6-алкил,
vi) карбокси-C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) C3-C8-циклоалкил-C1-C6-алкил,
ix) гидрокси-C1-C6-алкил,
x) фенил, и
xi) гетероарил-C1-C6-алкил;
RO выбран из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил;
или их фармацевтически приемлемые соли.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где
RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) арил, замещенный RG и RG1,
v) гетероциклоалкил, замещенный RG и RG1, и
vi) гетероарил, замещенный RG и RG1;
RB выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C1-C6-алкилсульфонил,
iii) C1-C6-алкилсульфониламино,
iv) аминокарбонил,
v) циано,
vi) галоген,
vii) гетероциклоалкил, и
viii) гетероарил, замещенный одним H, C1-C6-алкилом или триалкилсилил-C1-C6-алкокси-C1-C6-алкилом;
RC1 представляет собой H, и RC выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил, и
iii) галоген;
или RB и RC вместе с атомами углерода, которым они присоединены, образуют кольцевую систему, выбранную из группы, состоящей из
i) гетероциклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H и C1-C6-алкила, и
ii) гетероарил, замещенный одним или двумя заместителями, независимо выбранными из H и C1-C6-алкила;
W выбран из кольцевых систем A, B, C, D и E;
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -CRD2-,
A1 представляет собой -N-, A2 представляет собой -CRD2-, A3 представляет собой -CH- или -N-, и A4 представляет собой -CH-,
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -N-, или
A1 и A3 представляют собой -CH-, A2 представляет собой -CRD2-, и A4 представляет собой -N-;
один из A5 и A6 представляет собой -NRD3-, а другой представляет собой -CRLRM-;
RD1 представляет собой C1-C6-алкил;
RD2 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) гидрокси-C1-C6-алкокси,
iv) гидрокси-C1-C6-алкил(C1-C6-алкил)амино,
v) гидрокси-C1-C6-алкиламино,
vi) дигидрокси-C1-C6-алкокси,
vii) дигидрокси-C1-C6-алкил(C1-C6-алкил)амино, и
viii) гетероциклоалкил;
RD3 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил, и
iv) C1-C6-алкилкарбонил;
RD4 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкоксикарбонил,
iii) гетероциклоалкилкарбонил, и
iv) аминокарбонил , замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) карбокси,
ix) циано,
x) C3-C8-циклоалкокси,
xi) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xii) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xiii) гало-C1-C6-алкил,
xiv) галоген,
xv) гидрокси,
xvi) аминокарбонил, замещенный по атому азота RN и RO,
xvii) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xviii) гетероарил, замещенный одним H или C1-C6-алкилом,
xix) гетероциклоалкил-C1-C6-алкокси, замещенный одним H или C1-C6-алкилом, и
xx) гетероциклоалкил-C1-C6-алкил, замещенный одним H или C1-C6-алкилом;
RG1 и RG2 независимо выбраны из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил, и
iv) гало-C1-C6-алкокси;
RL и RM представляют собой H;
RN выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкил,
iv) C1-C6-алкоксикарбонил-C1-C6-алкил,
v) C1-C6-алкил,
vi) карбокси-C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) C3-C8-циклоалкил-C1-C6-алкил,
ix) гидрокси-C1-C6-алкил,
x) фенил, и
xi) гетероарил-C1-C6-алкил;
RO выбран из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил;
или их фармацевтически приемлемые соли.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где
RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) арил, замещенный RG и RG1,
v) гетероциклоалкил, замещенный RG и RG1, и
vi) гетероарил, замещенный RG и RG1;
RB выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C1-C6-алкилсульфонил,
iii) C1-C6-алкилсульфониламино,
iv) аминокарбонил,
v) циано,
vi) галоген,
vii) гетероциклоалкил, и
viii) гетероарил, замещенный одним H, C1-C6-алкилом или триалкилсилил-C1-C6-алкокси-C1-C6-алкилом;
RC1 представляет собой H, и RC выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил, и
iii) галоген;
или RB и RC вместе с атомами углерода, которым они присоединены, образуют кольцевую систему, выбранную из группы, состоящей из
i) гетероциклоалкил, замещенный одним или двумя заместителями, независимо выбранными из H и C1-C6-алкила, и
ii) гетероарил, замещенный одним или двумя заместителями, независимо выбранными из H и C1-C6-алкила;
W выбран из кольцевых систем A, B, C, D и E;
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -CRD2-,
A1 представляет собой -N-, A2 представляет собой -CRD2-, A3 представляет собой -CH- или -N-, и A4 представляет собой -CH-,
A1, A3 и A4 представляют собой -CH-, и A2 представляет собой -N-, или
A1 и A3 представляют собой -CH-, A2 представляет собой -CRD2-, и A4 представляет собой -N-;
один из A5 и A6 представляет собой -NRD3- а другой представляет собой -CRLRM-;
RD1 представляет собой C1-C6-алкил;
RD2 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) гидрокси-C1-C6-алкокси,
iv) гидрокси-C1-C6-алкил(C1-C6-алкил)амино,
v) гидрокси-C1-C6-алкиламино,
vi) дигидрокси-C1-C6-алкокси,
vii) дигидрокси-C1-C6-алкил(C1-C6-алкил)амино, и
viii) гетероциклоалкил;
RD3 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил, и
iv) C1-C6-алкилкарбонил;
RD4 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкоксикарбонил,
iii) гетероциклоалкилкарбонил, и
iv) аминокарбонил , замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами;
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) карбокси,
ix) циано,
x) C3-C8-циклоалкокси,
xi) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xii) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xiii) гало-C1-C6-алкил,
xiv) галоген,
xv) аминокарбонил, замещенный по атому азота RN и RO,
xvi) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xvii) гетероарил, замещенный одним H или C1-C6-алкилом,
xviii) гетероциклоалкил-C1-C6-алкокси, замещенный одним H или C1-C6-алкилом, и
xix) гетероциклоалкил-C1-C6-алкил, замещенный одним H или C1-C6-алкилом;
RG1 м RG2 независимо выбраны из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил, и
iv) гало-C1-C6-алкокси;
RL и RM представляют собой H;
RN выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкил,
iv) C1-C6-алкоксикарбонил-C1-C6-алкил,
v) C1-C6-алкил,
vi) карбокси-C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) C3-C8-циклоалкил-C1-C6-алкил,
ix) гидрокси-C1-C6-алкил,
x) фенил, и
xi) гетероарил-C1-C6-алкил;
RO выбран из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил;
или их фармацевтически приемлемые соли.
В другом воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) арил, замещенный RG и RG1,
v) гетероциклоалкил, замещенный RG и RG1, и
vi) гетероарил, замещенный RG и RG1.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) циано-C1-C6-алкил,
iii) C3-C8-циклоалкил,
iv) фенил, замещенный RG и RG1,
v) тетрагидропиранил, замещенный RG и RG1, и
vi) гетероарил, замещенный RG и RG1, где гетероарил выбран из бензоксазолонила, имидазолила, изоксазолила, оксазолила, пиразинила, пиразолила, пиридинила и пиримидинила.
В более конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RA выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C3-C8-циклоалкил,
iii) фенил, замещенный RG и RG1, и
iv) гетероарил, замещенный RG и RG1, где гетероарил выбран из изоксазолила и пиридинила.
В дополнительном конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RA выбран из группы, состоящей из
i) C1-C6-алкил, и
ii) фенил, замещенный RG и RG1.
В другом дополнительном конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RA представляет собой C1-C6-алкил.
В другом дополнительном конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RA представляет собой фенил, замещенный RG и RG1.
В другом воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) карбокси,
ix) циано,
x) C3-C8-циклоалкокси,
xi) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xii) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xiii) гало-C1-C6-алкил,
xiv) галоген,
xv) гидрокси,
xvi) аминокарбонил, замещенный по атому азота RN и RO,
xvii) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xviii) гетероарил, замещенный одним H или C1-C6-алкил,
xix) гетероциклоалкил-C1-C6-алкокси, замещенный одним H или C1-C6-алкил, и
xx) гетероциклоалкил-C1-C6-алкил, замещенный одним H или C1-C6-алкилом.
В другом воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) карбокси,
ix) циано,
x) C3-C8-циклоалкокси,
xi) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xii) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xiii) гало-C1-C6-алкил,
xiv) галоген,
xv) аминокарбонил, замещенный по атому азота RN и RO,
xvi) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xvii) гетероарил, замещенный одним H или C1-C6-алкилом,
xviii) гетероциклоалкил-C1-C6-алкокси, замещенный одним H или C1-C6-алкилом, и
xix) гетероциклоалкил-C1-C6-алкил, замещенный одним H или C1-C6-алкилом.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
iv) C3-C8-циклоалкокси,
v) галоген,
vi) гидрокси,
vii) аминокарбонил, замещенный по атому азота RN и RO,
viii) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
ix) гетероарил, замещенный одним H или C1-C6-алкилом, и
x) гетероциклоалкил-C1-C6-алкил, замещенный одним H или C1-C6-алкилом.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
iv) C3-C8-циклоалкокси,
v) галоген,
vi) аминокарбонил, замещенный по атому азота RN и RO,
vii) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
viii) гетероарил, замещенный одним H или C1-C6-алкилом, и
ix) гетероциклоалкил-C1-C6-алкил, замещенный одним H или C1-C6-алкилом.
В другом воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RG1 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил, и
iv) гало-C1-C6-алкокси.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RG1 выбран из группы, состоящей из
i) H, и
ii) Галогена.
В другом воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RG2 представляет собой H.
В другом воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RL и RM представляют собой H.
В другом воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RN выбран из группы, состоящей из
i) C1-C6-алкокси-C1-C6-алкил, и
ii) C1-C6-алкил.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RO представляет собой C1-C6-алкил.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RN и RO вместе с атомом азота, к которому они присоединены, образуют морфолинил, пирролидинил или метилпиперазинонил.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RB выбран из группы, состоящей из
i) C1-C6-алкил,
ii) C1-C6-алкилсульфонил,
iii) C1-C6-алкилсульфониламино,
iv) аминокарбонил,
v) циано,
vi) галоген,
vii) гетероциклоалкил, где гетероарил выбран из оксадиазолила, имидазолила, 1,3,4-оксазолила и 1,2,4-оксазолила, и
viii) морфолинил, замещенный одним H, C1-C6-алкилом или триалкилсилил-C1-C6-алкокси-C1-C6-алкилом.
В более конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RB выбран из группы, состоящей из
i) циано, и
ii) галоген.
В дополнительном конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RB представляет собой галоген.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RC выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил, и
iii) галоген.
В более конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RC представляет собой C1-C6-алкил.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RC1 представляет собой H.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где W выбран из кольцевых систем A, B и C.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где W выбран из кольцевых систем A и C.
В более конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где W представляет собой кольцевую систему A.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RD1 представляет собой C1-C6-алкил.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RD2 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) гидрокси-C1-C6-алкокси,
iv) гидрокси-C1-C6-алкил(C1-C6-алкил)амино,
v) гидрокси-C1-C6-алкиламино,
vi) дигидрокси-C1-C6-алкокси,
vii) дигидрокси-C1-C6-алкил(C1-C6-алкил)амино, и
viii) гетероциклоалкил.
В более конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RD2 выбран из группы, состоящей из
i) H,
ii) гидрокси-C1-C6-алкокси, и
iii) гидрокси-C1-C6-алкил(C1-C6-алкил)амино.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RD3 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси-C1-C6-алкилкарбонил,
iii) C1-C6-алкоксикарбонил, и
iv) C1-C6-алкилкарбонил.
В другом конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой (I) как здесь описано, где RD4 выбран из группы, состоящей из
i) H,
ii) C1-C6-алкоксикарбонил,
iii) гетероциклоалкилкарбонил, и
iv) аминокарбонил , замещенный по атому азота одним или двумя независимо выбранными C1-C6-алкилами.
В конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой I(a) как здесь описано,

или их фармацевтически приемлемые соли.
В дополнительном конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой I(b) как здесь описано,

где
RA выбран из группы, состоящей из
i) C1-C6-алкил, и
ii) фенил, замещенный RG и RG1;
RB выбран из группы, состоящей из
i) циано, и
ii) галоген;
RC выбран из группы, состоящей из
i) H,
ii) C1-C6-алкил, и
iii) галоген;
RD1 представляет собой C1-C6-алкил;
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
iv) галоген,
v) аминокарбонил, замещенный по атому азота RN и RO,
vi) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
vii) гетероарил, замещенный одним H или C1-C6-алкилом, где гетероарил представляет собой изоксазолил, оксазолил или пиразолил, и
viii) гетероциклоалкил-C1-C6-алкокси, замещенный одним H или C1-C6-алкилом, где гетероциклоалкил-C1-C6-алкокси представляет собой тетрагидропиранилметокси или тетрагидрофуранилметокси;
RG2 представляет собой H, и RG1 выбран из группы, состоящей из
i) H, и
ii) галоген;
RN выбран из группы, состоящей из
i) C1-C6-алкокси-C1-C6-алкил, и
ii) C1-C6-алкил;
RO представляет собой C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют морфолинил, пирролидинил или метилпиперазинонил;
или их фармацевтически приемлемые соли.
В дополнительном конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой I(b) как здесь описано,
где
RA выбран из группы, состоящей из
i) C1-C6-алкил, и
ii) фенил, замещенный RG и RG1;
RB выбран из группы, состоящей из
i) циано, и
ii) галоген;
RD1 представляет собой C1-C6-алкил;
RG выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкилкарбониламино-C1-C6-алкил,
iv) C1-C6-алкокси-C1-C6-алкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
v) C1-C6-алкоксикарбонил,
vi) C1-C6-алкил,
vii) C1-C6-алкилсульфонил,
viii) карбокси,
ix) циано,
x) C3-C8-циклоалкокси,
xi) C3-C8-циклоалкилкарбониламино-C1-C6-алкил,
xii) C3-C8-циклоалкилкарбонил(C1-C6-алкил)амино-C1-C6-алкил,
xiii) гало-C1-C6-алкил,
xiv) галоген,
xv) аминокарбонил, замещенный по атому азота RN и RO,
xvi) аминокарбонил-C1-C6-алкокси, замещенный по атому азота RN и RO,
xvii) гетероарил, замещенный одним H, C1-C6-алкилом, где гетероарил представляет собой изоксазолил, оксазолил или пиразолил,
xviii) гетероциклоалкил-C1-C6-алкокси, замещенный одним H, C1-C6-алкилом, где гетероциклоалкил-C1-C6-алкокси представляет собой тетрагидропиранилметокси или тетрагидрофуранилметокси, и
xix) гетероциклоалкил-C1-C6-алкил, замещенный одним H, C1-C6-алкилом, где гетероциклоалкил-C1-C6-алкил представляет собой метилдиоксопиперазинилметил, оксопирролидинилметил или оксооксазолидинилметил;
RG2 представляет собой H, и RG1 выбран из группы, состоящей из
i) H,
ii) галоген,
iii) C1-C6-алкил,
iv) гало-C1-C6-алкокси, и
RN выбран из группы, состоящей из
i) H,
ii) C1-C6-алкокси,
iii) C1-C6-алкокси-C1-C6-алкил,
iv) C1-C6-алкоксикарбонил-C1-C6-алкил,
v) C1-C6-алкил,
vi) карбокси-C1-C6-алкил,
vii) C3-C8-циклоалкил,
viii) C3-C8-циклоалкил-C1-C6-алкил,
ix) гидрокси-C1-C6-алкил,
x) фенил, и
xi) гетероарил-C1-C6-алкил, где гетероарил-C1-C6-представляет собой пиридинилалкил или тиофенилалкил;
RO выбран из группы, состоящей из
i) H, и
ii) C1-C6-алкил;
или RN и RO вместе с атомом азота, к которому они присоединены, образуют пиперидинил, морфолинил, пирролидинил или метилпиперазинонил;
или их фармацевтически приемлемые соли.
В дополнительном конкретном воплощении настоящего изобретения предложены соединения в соответствии с формулой I(b) как здесь описано,

где
RA представляет собой C1-C6-алкил;
RC выбран из группы, состоящей из
i) C1-C6-алкил, и
ii) галоген;
RD1 представляет собой C1-C6-алкил;
или их фармацевтически приемлемые соли.
Конкретные примеры соединений формулы (I), как здесь описано, выбраны из
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(2-трет-бутил-4-хлор-5-фторфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(3,3-диметил-6-пропан-2-ил-1,2-дигидроинден-5-ил)оксиметил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-(3-метил-1,2,4-оксадиазол-5-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-[1-(2-триметилсилилэтоксиметил)имидазол-2-ил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-(1-метилимидазол-2-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-(1,3-оксазол-2-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(2-трет-бутил-4-морфолин-4-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-(3-метилимидазол-4-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-(5-метил-1,3,4-оксадиазол-2-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-3-пропан-2-илбензонитрил;
2-метил-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-пропан-2-илбензонитрил;
3-трет-бутил-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[(4-хлор-2-циклопропил-5-метилсульфонилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(2-трет-бутил-4-метилсульфонилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
5-трет-бутил-2-метил-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
4-трет-бутил-2-метил-5-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[[2-трет-бутил-4-[3-(2-триметилсилил-этоксиметил)имидазол-4-ил]фенокси]-метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-2-циклопропил-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-2-циклогексил-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(оксан-4-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-4-циклопропил-5-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
N-[2-хлор-4-циклопропил-5-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]метансульфонамид;
4-трет-бутил-3-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
4-трет-бутил-3-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензамид;
5-трет-бутил-2-метил-4-[(5-оксо-4-пропан-2-ил-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
4-метил-3-[(5-метил-2-пропан-2-илфенокси)метил]-1H-1,2,4-триазол-5-он;
3-[(4-хлор-5-метил-2-пропан-2-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-2-пропан-2-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(6-циклопропил-2-метил-1,3-бензотиазол-5-ил)оксиметил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-2-циклобутил-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-(1H-имидазол-2-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[2-трет-бутил-4-(1H-имидазол-5-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-5-метил-2-фенилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(2-хлорфенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(3-хлорфенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(4-хлорфенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензонитрил;
3-[[4-хлор-5-метил-2-(3-метилсульфонилфенил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(2-метилсульфонилфенил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-[3-(пиперидин-1-карбонил)фенил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-циклогексилбензамид;
3-[[4-хлор-5-метил-2-[3-(морфолин-4-карбонил)фенил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензамид;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N,N-диметилбензамид;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-фенилбензамид;
3-хлор-5-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензамид;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-циклопропил-4-фторбензамид;
4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-3-фенилбензонитрил;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-(2-метоксиэтил)бензамид;
3-[[4-хлор-2-(2-хлорпиридин-3-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(6-хлорпиридин-2-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
5-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]пиридин-3-карбоксамид;
3-[[4-хлор-2-(6-метоксипиридин-2-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-5-метил-2-пиразин-2-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-5-метил-2-пиримидин-2-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-[2-метил-5-(трифторметил)пиразол-3-ил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(1,2-оксазол-5-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(1,3-оксазол-5-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(3-метилимидазол-4-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(1H-имидазол-5-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(1,3-оксазол-2-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(2-метилпиразол-3-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-фенилбензонитрил;
2-хлор-5-(4-фторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[4-хлор-5-циано-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-метилбензамид,
2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-[3-(1H-пиразол-3-ил)фенил]бензонитрил;
2-хлор-5-[3-(5-метил-1,2,4-оксадиазол-3-ил)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-(2-гидроксиэтил)бензамид;
2-хлор-5-[3-(5-метил-1,3,4-оксадиазол-2-ил)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фтор-N,N-диметилбензамид;
3-[4-хлор-5-циано-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N,N-диметилбензамид;
3-[[4-хлор-2-[2-фтор-5-(морфолин-4-карбонил)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-[3-(морфолин-4-карбонил)фенил]бензонитрил;
метил 3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензоат;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензойная кислота;
метил 3-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензоил]амино]пропаноат;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-(2-гидроксиэтил)-N-метилбензамид;
этил 2-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензоил]амино]ацетат;
3-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензоил]амино]пропановая кислота;
2-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензоил]амино]уксусная кислота;
метил 3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-метилбензоат;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-метилбензойная кислота
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N,N,4-триметилбензамид;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-(трифторметокси)бензамид;
3-[[4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-5-(2-метоксипиридин-3-ил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[[4-хлор-2-(5-этокси-2-фторфенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(2-метоксифенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(2-фтор-5-пропан-2-илоксифенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-[2-фтор-5-(2-метилпропокси)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-[2-метокси-5-(трифторметил)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(2-метокси-5-пропан-2-илфенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-5-[2-фтор-5-(морфолин-4-карбонил)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[[4-хлор-2-[2-фтор-5-(пирролидин-1-карбонил)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-циклопропил-4-фтор-N-метилбензамид;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фтор-N-(2-гидроксиэтил)-N-метилбензамид;
4-[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторбензоил]-1-метилпиперазин-2-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-циклопентил-4-фтор-N-метилбензамид;
3-[[4-хлор-2-[2-фтор-5-(оксолан-3-илметокси)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фтор-N-метил-N-(тиофен-2-илметил)бензамид;
3-[[4-хлор-2-[2-фтор-5-(пиперидин-1-карбонил)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-(циклопропилметил)-4-фтор-N-метилбензамид;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фтор-N-метил-N-(пиридин-2-илметил)бензамид;
3-[[4-хлор-2-[2-фтор-5-(оксан-4-илметокси)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фтор-N-(2-метоксиэтил)-N-метилбензамид;
3-[[4-хлор-2-[2-фтор-5-(оксолан-2-илметокси)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенокси]-N,N-диметилацетамид;
1-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенил]метил]-4-метилпиперазин-2,5-дион;
7-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-6-фтор-3-метил-1,3-бензоксазол-2-он;
N-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенил]метил]-2-метокси-N-метилацетамид;
N-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенил]метил]циклопропанкарбоксамид;
N-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенил]метил]-N-метилциклопропанкарбоксамид;
3-[[4-хлор-2-[2-фтор-5-[(2-оксопирролидин-1-ил)метил]фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенил]метил]-1,3-оксазолидин-2-он;
N-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенил]метил]-2-метоксиацетамид;
2-хлор-5-(2-фторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-(3-фторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-(трифторметокси)бензонитрил;
2-хлор-5-[2-фтор-5-(оксолан-2-илметокси)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-(2-фтор-3-метоксифенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-(2-фтор-5-пропан-2-илоксифенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[[4-хлор-2-(2-фтор-3-метоксифенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-5-(2,3-дифторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[[4-хлор-2-(5-циклопропилокси-2-фторфенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-5-(5-хлор-2-фторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-(2,5-дифторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-(5-циклопропилокси-2-фторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-[2-фтор-5-(трифторметил)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[(4-хлор-5-метил-2-пропан-2-илфенокси)метил]-1H-пиридазин-6-он;
3-[(4-хлор-2-циклопропил-5-метилфенокси)метил]-1H-пиридазин-6-он;
3-[(4-хлор-5-фтор-2-пропан-2-илфенокси)метил]-1H-пиридазин-6-он;
3-[(5-хлор-4-метил-2-пропан-2-илфенокси)метил]-1H-пиридазин-6-он;
3-[(4-хлор-2-циклобутил-5-метилфенокси)метил]-1H-пиридазин-6-он;
3-[(4-хлор-2-циклогексил-5-метилфенокси)метил]-1H-пиридазин-6-он;
3-[[4-хлор-5-метил-2-(оксан-4-ил)фенокси]метил]-1H-пиридазин-6-он;
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиридазин-6-он;
2-[5-хлор-4-метил-2-[(6-оксо-1H-пиридазин-3-ил)метокси]фенил]-2-метилпропаннитрил;
3-[(2-трет-бутил-4-метилсульфонилфенокси)метил]-1H-индазол;
3-[(4-хлор-5-метил-2-пропан-2-илфенокси)метил]-1H-индазол;
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин;
5-трет-бутил-2-метил-4-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
5-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-1,2-оксазол
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-6-фтор-1H-пиразоло[3,4-b]пиридин;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]амино]этанол;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]-метиламино]этанол;
3-[(2-трет-бутил-5-метил-4-метилсульфонилфенокси)метил]-1H-пиразоло[3,4-b]пиридин;
3-[(2-трет-бутил-5-метил-4-метилсульфонилфенокси)метил]-1H-индазол;
2-[[3-[(2-трет-бутил-4-хлор-5-фторфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]амино]этанол;
3-[(2-трет-бутил-4-хлор-5-фторфенокси)метил]-6-фтор-1H-пиразоло[3,4-b]пиридин;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]окси]этанол;
2-[[3-[(2-трет-бутил-4-хлор-5-фторфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]-метиламино]этанол;
3-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]окси]пропан-1,2-диол;
3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-N-(2-метоксиэтил)бензамид;
[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]phenyl]-морфолин-4-илметанон;
3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-N,N-диметилбензамид;
3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-N-(2-гидроксиэтил)бензамид;
3-[[4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси]метил]-1H-пиразоло[3,4-b]пиридин;
3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-4-фтор-N,N-диметилбензамид;
[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-4-фторфенил]-морфолин-4-илметанон;
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[4,3-b]пиридин;
3-[5-хлор-4-метил-2-(1H-пиразоло[4,3-b]пиридин-3-илметокси)фенил]-N,N-диметилбензамид;
3-[(2-трет-бутил-4-хлор-5-фторфенокси)метил]-1H-пиразоло[4,3-b]пиридин;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]-метиламино]этанол;
3-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]-метиламино]пропан-1,2-диол;
3-[5-хлор-4-метил-2-(1H-пиразоло[4,3-b]пиридин-3-илметокси)фенил]-4-фтор-N,N-диметилбензамид;
1-[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]азетидин-3-ол;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]амино]этанол;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]окси]этанол;
5-трет-бутил-4-[[6-(3-гидроксиазетидин-1-ил)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил;
5-трет-бутил-4-[[6-[2-гидроксиэтил(метил)амино]-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил;
3-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]окси]пропан-1,2-диол;
5-трет-бутил-4-[[6-(2-гидроксиэтиламино)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил;
5-трет-бутил-4-[[6-[2,3-дигидроксипропил(метил)амино]-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил;
5-трет-бутил-4-[[6-(2,3-дигидроксипропокси)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил;
5-трет-бутил-4-[[6-(2-гидроксиэтокси)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил;
4-[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-4-фторбензоил]-1-метилпиперазин-2-он;
3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-4-фтор-N-(2-метоксиэтил)-N-метилбензамид;
4-[3-[5-хлор-2-(1H-индазол-3-илметокси)-4-метилфенил]-4-фторбензоил]-1-метилпиперазин-2-он;
[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]-4-фторфенил]-пирролидин-1-илметанон;
2-хлор-5-[2-фтор-5-(пирролидин-1-карбонил)фенил]-4-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
4-[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-c]пиридин-3-илметокси)фенил]-4-фторбензоил]-1-метилпиперазин-2-он;
3-[(2-трет-бутил-4-метилсульфонилфенокси)метил]-1H-пиразоло[3,4-b]пиридин;
3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-c]пиридин-3-илметокси)фенил]-4-фтор-N-(2-метоксиэтил)-N-метилбензамид;
3-[[4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси]метил]-1H-пиразоло[3,4-c]пиридин;
4-трет-бутил-2-метил-5-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
4-трет-бутил-2-метил-5-(1H-пиразоло[3,4-c]пиридин-3-илметокси)бензонитрил;
трет-бутил 3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилат;
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-ил)этанон;
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-ил)-2-метоксиэтанон;
3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридин;
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5-дигидро-1H-пиразоло[3,4-c]пиридин-6(7H)-ил)этанон;
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5-дигидро-1H-пиразоло[3,4-c]пиридин-6(7H)-ил)-2-метоксиэтанон;
3-((4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридин;
1-(3-((4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси)метил)-4,5-дигидро-1H-пиразоло[3,4-c]пиридин-6(7H)-ил)этанон;
3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-1H-пиразол;
4-((1H-пиразол-3-ил)метокси)-5-трет-бутил-2-метилбензонитрил;
метил 3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-1H-пиразол-5-карбоксилат;
(3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-1H-пиразол-5-ил)(пирролидин-1-ил)метанон;
3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-N,N-диметил-1H-пиразол-5-карбоксамид;
и их фармацевтически приемлемые соли.
Также, конкретные примеры соединений формулы (I), как здесь описано, выбраны из
4-трет-бутил-2-хлор-5-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-[2-фтор-5-(трифторметокси)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-[2-фтор-5-(2,2,2-трифторэтокси)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[[4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси]метил]-1H-пиридазин-6-он;
3-[[4-хлор-2-(2-гидроксипиридин-3-ил)-5-метилфенокси]метил]-1H-пиридазин-6-он;
2-хлор-4-[(6-оксо-1H-пиридазин-3-ил)метокси]-5-фенилбензонитрил;
4-трет-бутил-2-метил-5-[(6-оксо-1H-пиридазин-3-ил)метокси]бензонитрил;
2-хлор-5-(5-циклопропилокси-2-фторфенил)-4-[(6-оксо-1H-пиридазин-3-ил)метокси]бензонитрил;
4-трет-бутил-2-хлор-5-(1H-пиразоло[3,4-c]пиридин-3-илметокси)бензонитрил;
2-хлор-5-(5-циклопропилокси-2-фторфенил)-4-(1H-пиразоло[3,4-c]пиридин-3-илметокси)бензонитрил;
4-трет-бутил-2-хлор-5-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил
2-хлор-5-(5-циклопропилокси-2-фторфенил)-4-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
2-хлор-5-[2-фтор-5-(трифторметокси)фенил]-4-(1H-пиразоло[4,3-c]пиридин-3-илметокси)бензонитрил;
2-хлор-5-[2-фтор-5-(трифторметокси)фенил]-4-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
и их фармацевтически приемлемые соли.
Дополнительные конкретные примеры соединений формулы (I), как здесь описано, выбраны из
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(2-трет-бутил-4-хлор-5-фторфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-трет-бутил-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
5-трет-бутил-2-метил-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
4-трет-бутил-2-метил-5-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[(4-хлор-2-циклогексил-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
5-трет-бутил-2-метил-4-[(5-оксо-4-пропан-2-ил-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[(4-хлор-5-метил-2-пропан-2-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[(4-хлор-2-циклобутил-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-[3-(морфолин-4-карбонил)фенил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-5-метил-2-(1,2-оксазол-5-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-фенилбензонитрил;
2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-[3-(1H-пиразол-3-ил)фенил]бензонитрил;
2-хлор-5-[3-(5-метил-1,2,4-оксадиазол-3-ил)фенил]-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фтор-N,N-диметилбензамид;
3-[[4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-(2-фтор-5-пропан-2-илоксифенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
3-[[4-хлор-2-[2-фтор-5-(пирролидин-1-карбонил)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
4-[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторбензоил]-1-метилпиперазин-2-он;
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фтор-N-(2-метоксиэтил)-N-метилбензамид;
3-[[4-хлор-2-[2-фтор-5-(оксолан-2-илметокси)фенил]-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенокси]-N,N-диметилацетамид;
N-[[3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-фторфенил]метил]-2-метокси-N-метилацетамид;
2-хлор-5-(2-фторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-(2-фтор-3-метоксифенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
2-хлор-5-(2-фтор-5-пропан-2-илоксифенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
3-[[4-хлор-2-(5-циклопропилокси-2-фторфенил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он;
2-хлор-5-(5-циклопропилокси-2-фторфенил)-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
5-трет-бутил-2-метил-4-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]-метиламино]этанол;
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]окси]этанол;
2-хлор-5-[2-фтор-5-(пирролидин-1-карбонил)фенил]-4-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-c]пиридин-3-илметокси)фенил]-4-фтор-N-(2-метоксиэтил)-N-метилбензамид;
и их фармацевтически приемлемые соли.
Также дополнительные конкретные примеры соединений формулы (I), как здесь описано, выбраны из
2-хлор-5-(5-циклопропилокси-2-фторфенил)-4-[(6-оксо-1H-пиридазин-3-ил)метокси]бензонитрил;
4-трет-бутил-2-хлор-5-(1H-пиразоло[3,4-b]пиридин-3-илметокси)бензонитрил;
и их фармацевтически приемлемые соли.
Также конкретные примеры соединений формулы (I), как здесь описано, выбраны из
4-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-2H-триазол;
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-5-фенил-1H-пиразол; и
5-трет-бутил-2-метил-4-[(5-фенил-1H-пиразол-3-ил)метокси]бензонитрил.
Дополнительные конкретные примеры соединений формулы (I), как здесь описано, выбраны из
5-трет-бутил-2-метил-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
5-трет-бутил-2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил; и
5-трет-бутил-2-фтор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил;
и их фармацевтически приемлемые соли.
Способы получения соединений формулы (I) как здесь описано являются объектом настоящего изобретения.
Получение соединений формулы (I) по настоящему изобретению может быть проведено последовательными или конвергентными путями синтеза. Синтезы по изобретению показаны на следующих общих схемах. Навыки, необходимые для проведения реакции и очистки полученных продуктов известны специалистам в данной области техники. В случае, если во время реакции получают смесь энантиомеров или диастереоизомеров, эти энантиомеры или диастереоизомеры могут быть разделены с помощью способов, описанных здесь, или способами, известными специалисту в данной области, такими как, например, (хиральная) хроматография или кристаллизация. Заместители и индексы, используемые в следующем описании способов имеют значения, указанные здесь.
Общее описание изобретения дано в следующих разделах. Для получения соединений формулы (I), либо в незащищенном, либо в защищенном виде (PG = защитная группа), ключевой стадией обычно является реакция связывания между подходящим фенольным структурным элементом A1, где RA представляет собой обязательный ортозаместитель арил-гидрокси группы, RB является заместителем в другом положении фенильного кольца, который обычно необходимо ввести или модифицировать (включая подходящую защиту в случае присутствия любой потенциально мешающей функциональной группы в составе RA и RB), и RC является любой другой функциональной группой, которая обычно присуствует в исходном соединении и остается немодифицированной, с требуемым структруным элементом A2 с получением замещенного фенилэфира формулы A3 (Схема 1).В контексте настоящего изобретения, A2 обычно состоит из 5 или 6-членного гетероцикла, содержащего два смежных атома азота, один из которых несет водород (или подходящую защитную группу PG если азот сохраняется защищенным), и метильную группу, которая замещена подходящей уходящей группой X. X может быть галогеном, таким как например хлор, бром или йод или любой другой уходящей группой, такой как тозилат или мезилат. A2 также может быть бициклическим, где 6 членное кольцо, которое является либо ароматическим, либо насыщенным, и которое может содержать дополнительные атомы азота и дополнительные заместители, конденсировало с первичным 5-членным гетероциклом. Реакция связывания между A1 и A2, которые оба могут содержать независимые друг от друга защитные группы PG, PG', или PG'' в случае необходимости, обычно проводится в присутствии основания, такого как карбонат калия, карбонат цезия или гидрид натрия, подходящего растворителя, такого как ТГФ, ДМФ, CH3CN или аналогичного, при температурах в диапазоне от минус 20°C до температуры кипения растворителя, или при более высоких температурах в закрытых сосудах с получением продукта связывания A3a. В большистве случаев если защита не требуется, A3a является эквивалентным A3 и уже является примером структуры формулы (I), однако, если A3a содержит какую-либо защитную группу PG или PG' или PG'', такую как, например, BOC или тритил, трет-бутил, pMB, MOM, SEM, бензил или аналогичную, они могут быть удалены в известных условиях, которые зависят от природы защитной группы PG. BOC, трет-бутил, pMB и тритил, MOM, SEM, например, могут быть удалены в присутствии кислоты, такой как ТФУ, HCl, HBr, H2SO4 или аналогичной в подходящем растворителе, таком как CH2Cl2, ТГФ, вода, диоксан или аналогичном, где бензил или аналогичная группа могут быть удалены каталитической гидрогенизацией (например, H2, Pd на угле или аналогичные, в этаноле, метаноле, воде, EtOAc или HOAc или аналогичном, при различных температурах). SEM также можно удалить обработкой фторидом (например, тетрабутиламмония фторидом в HMPT или DMPU или аналогичном). Стратегии защитных групп для множества функциональных групп (включая условия для защиты и депротекции) хорошо описаны, например в “Protective Groups in Organic Chemistry” под авторством T. W. Greene и P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y.). Депротекция A3a, если необходимо, которая может содержать несколько стадий, если PG, PG' или PG'' являются отличными друг от друга, даст незащищенное соединение формулы A3, которое соответствует соединениям формулы (I) как заявлено в формуле изобретения.
Схема 1:

Последующие модификации продуктов связывания
В некоторых случаях, в зависимости от природы используемого структурного элемента A2 для реакции связывания, продукт связывания еще не является требуемым соединением и необходимы дополнительные модификации, которые отличаются от простых реакций с защитными группами, для получения соединения формулы A3 (Схема 2).
Для триазолонов: Если, например, коммерческий триазоловый структурный элемент, такой как A4, где RD1 = метил, используется в качестве A2 в реакции связывания с фенолом A1, которая проводится в условиях, описанных выше, тогда подходящие дальнейшие трансформации промеуточного продукта связывания A5 включают: 1) обработку A5 метоксидом натрия в растворителе, таком как метанол, при температурах в диапазоне от 0°C до температуры кипения растворителя с получением гетероарил-метилового эфира A6, и 2) гидролиз соединения A6 в присутствии сильной кислоты, такой как HCl, HBr, H2SO4 или п-толуолсульфоновая кислота или аналогичная, в растворителях, таких как уксусная кислота, содержащая некоторое количество воды при температурах в диапазоне от 0°C до 130°C с получением триазолонов формулы A7, которые являются подмножеством соединений формулы A3, показанныхна Схеме 1.
Схема 2:

Для пиридазинонов: Если, например, коммерчески доступный пиридазиноновый структурный элемент, такой как A8 используется в реакции связывания с фенолом A1, тогда последующие трансформации, которые необходимы для получения пиридазинонов формулы A10, которые являются подмножеством примеров формулы A3, показаны на Схеме 3. В этом случае феноловый структурный элемент формулы A1 обрабатывают подходящим 3-хлор-6-хлорметил-пиридазином A8 с получением хлорпиридазинового промежуточного соединения формулы A9. Хлорметил-пиридазины формулы A9 являются коммерчески доступными или могут быть получены в соответствии с известными способами из литературы. Реакция связывания осуществляется в присутствии основания и в реакционных условиях, описанных выше. Промежуточное соединение A10 может быть превращено в требуемые пиридазиноны формулы A3 использованием различных способов. Способ A, например, основан на обработке соединения A9 подходящим основанием, таким как NaOH, KOH или аналогичным в присутствии воды или растворителя, содержащего воду, при температурах в диапазоне от -20°C до температуры кипения используемых растворителей с получением требуемых пиридазинонов формулы A10. Альтернативно, в соответствии со способом B, конверсия A9 в A10 также возможно, например, обработкой соединения A9 карбоновой кислотой, такой как уксусная кислота, муравьиная кислота или аналогичной, при температурах в диапазоне от 0°C до 150°C, с последующей водной обработкой.
Схема 3:

Дальнейшая разработка подходящих соединений формулы A3
В некоторых случаях, реакция связывания между фенолом A1 и структурным элементом A2 дает бициклическое промежуточное соединение, такое как, например, тиоэфир A11 (Схема 4). A11 может быть использовано для введения, например, подходящей солюбилизирующей группы, которая дополнительно изменяет все свойства соединений. Для их получения защитную группу PG соединения A11 удаляют подходящей обработкой в зависимости от природы PG: например, Boc, тритил или THP группы могут быть удалены посредством обработки сильными кислотами, такими как HBr, HCl, ТФУ или аналогичными, в подходящем растворителе, таком как диоксан, вода, этанол, метанол или ДХМ или аналогичном при температурах в диапазоне от -20°C до температуры кипения растворителя. После удаления PG промежуточное соединение подвергают окислению с получением метилсульфона A12. Подходящие окисляющие реагенты представляют собой, например, m-CPBA в ДХМ, оксон в MeOH/H2O, H2O2, KMnO4 в воде/AcOH или множество других. Обработка соединения A12 подходящим амином NRFRF' как показано на Схеме 4, который может быть в избытке, в растворителе, таком как ТГФ или N-метилпирролидон или ДМСО, или аналогичный, при температурах в диапазоне от 0°C до температуры кипения растворителя или до 200°C в закрытой пробирке или микроволновой печи даст аминопиримидины формулы A13.
Альтернативно, A12 можно обработать подходящим спиртом HO-RH (подходящим образом защищенным, как THP) в присуствии основания, такого как NaH, KH, Cs2CO3, Na2CO3 или аналогичного, в подходящем растворителе, таком как ДМФ, ТГФ, DMA или диоксан или т.п. при температурах в диапазоне от 0°C до температуры кипения растворителя, с получением соединения формулы A14. Любые защитные группы боковой цепи могут быть легко удалены посредством обработки кислотой, такой как ТФУ в ДХМ для группы THP или, например водной HCl для ацетонида, с получением требуемых незащищенных соединений формулы A15.
Схема 4:

Аналогично тому, как это описано выше для пиримидинов, защищенный фторпиридин формулы A16 (PG = тритил или Boc) (Схема 5) может быть обработан подходящим спиртом HO-RH (подходяще защищенным в виде THP эфиров или ацетонидов, если он содержит множество групп OH, как показано на Схеме 5) в присутствии основания, такого как NaH, KH, Cs2CO3, Na2CO3 или т.п., в подходящем растворителе, таком как ДМФ, DMA, ТГФ или т.п., при температурах в диапазоне от 0°C до температуры кипения растворителя, с получением замещенного гидроксипиридина формулы A17. Любые защитные группыпиразола (PG = например, тритил,Boc, PMB) и любые защитные группы боковой цепи (THP, ацетонид или аналогичная) могут быть легко удалены посредством обработки кислотой, такой как ТФУ в ДХМ или, например, водной HCl, HBr или т.п., с получением требуемых незащищенных соединений формулы A18.
Схема 5:

В некоторых случаях, реакция связывания между фенолом A1 и структурным элементом A2 дает подходяще защищённые бициклическое промежуточное соединение, такое как, например, пиразолопиперидины A19 (Схема 6). Эти соединения несут, например, Boc группу по азоту Y1 или Y2 пипердина и возможно защитную группу PG на пиразоле, которая, как описано выше, может представлять собой, например, Boc, тритил, THP или аналогичную. Для дальнейшей обработки все защитные группы удаляются в подходящих условиях, таких как ТФУ в ДХМ или HCl в диоксане или аналогичных, с получением незащищенного производного пиперидина A20. С целью получения соединений, подходящих для настоящего изобретения, A19 обрабатывают хлорангидридом в присутствии основания, такого как NEt3, основание Хунига, пиридин или т.п., в растворителе, таком как ДХМ, ДМФ, диоксан или аналогичный с получением требуемых замещенных пиперидинов формулы A21, которые показаны на Схеме 6. Последующая трансформация также возможна с карбоновыми кислотами, которые являются активированными множеством других различных способов; Подходящие условия хорошо известны квалифицированным специалистам.
Схема 6:

В некоторых случаях, реакция связывания между фенолом A1 и структурным элементом A2 дает подходящим образом защищенный эфир пиразол-карбоновой кислоты A22 (Схема 7). Несмотря на то, что данное соединение само по себе является примером соединения формулы (I) после обычного удаления пиразол защитной группы PG, эфирная группа также является полезной для дополнительной модификации. A22 можно, например. Обработать подходящим амином NRPRP' в присутствии AlMe3 в подходящем растворителе, таком как ДХМ, CH3Cl, ТГФ или диоксан или аналогичном, при температурах в диапазоне от -20°C до температуры кипения растворителя с получением амида карбоновой кислоты формулы A23. Удаление защитной группы пиразола в подходящих условиях, упомянутых выше, даст затем требуемый амид пиразол-карбоновой кислоты A34, который является примером соединения формулы (I) и в котором заместители RP и RP' определены как RD4 в формуле изобретения.
Схема 7:
Общий синтез конкретных подгрупп промежуточных соединений формулы A2, используемых в настоящем изобретении

A) Замещенные триазолоны
Триазолоновые промежуточные соединения формулы B5, в которых RD1 могут быть более сложными, чем метил, являются подгруппой соединений формулы A2 и могут быть получены в соответствии с последовательностью, которая показана на Схеме 8. Подходящим образом N-замещенный гидразинкарбоксамид формулы B1, который является коммерчески доступным или может быть получен в соответствии со способами, известными в литературе, обрабатывают бензилоксиацетилхлоридом B2 и водным основанием, таким как NaOH, KOH, K2CO3 или аналогичным, в подходящем растворителе, таком как ТГФ или т.п. с получением защищенного триазолонового промежуточного соединения формулы B3. Это промежуточное соединение может быть легко дебензилировано с использованием условий, известных в уровне техники, которые включают, например, гидрогенизирование с подходящими катализаторами, такими как Pd/C, PtO2, Pd(OH)2/C или аналогичными, в растворителях, таких как метанол, уксусная кислота, этилацетат или даже вода, с получением гидроксиметильного промежуточного соединения B4. Введение уходящей группы X с получением требуемого структурного элемента B5 происходит с использованием подходящих условий, которые зависят от вводимой уходящей группы. Если, например, X = Cl, промежуточное соединение B4 может быть обработано COCl2 с получением B5, гдеX = Cl. Если, например, X = Br, обработака CBr4 и PPh3 может быть использована. Если X = мезилат или тозилат, тогда B4 может быть обработано подходящим сульфонилхлоридом (метансульфонилхлорид или п-толуолсульфонилхлорид) в присутствии основания, такого как триэтиламин, пиридин, DMAP, основание Хунига или аналогичного, в подходящем растворителе, таком как CH2Cl2 или ТГФ или аналогичного, при температурах в диапазоне от -78°C до температуры кипения растворителя с получением B5, в котором RD1 определено в соответствии с формулой изобретения.
Схема 8:

B) 1H-пиразолопиридины, пиразолопиримидины, индазолы и аналогичные
Подходящим образом функционализированные пиразолопиридины, пиразолопиримидины и индазолы B13 снова являются подгруппой структурных элементов формулы A2, которые применяются в настоящем изобретении для получения соединений формулы (I) (Схема 9A). Подходящие предшественники для синтеза этих бициклических соединений формулы B13 являются коммерчески доступными или могут быть получены в соответствии с методиками, которые описаны в литературе. Например, является возможным применять индазолоны или аза-индазолоны B6 в качестве исходных веществ. Такие соединения являются коммерчески доступными или могут быть созданы заново. Пример создания нового соединения формулы B6 будет дан дополнительно ниже.
Для обработки B6, соединение может быть обработано POBr3 для получения бромированного промежуточного соединения B7. Хлорпроизводное также может быть использовано, хотя оно будет менее реактивным в последующих трансформациях; в этом случае необходимо применять подходящий реагент (например, POCl3 для хлорпроизводного). Промежуточное соединение B7 затем подвергают защите по атому азота с помощью подходящей защитной группы PG с получением B8. PG может быть, например, группой PMB или тритильной группой или аналогичным. Подходящие условия, которые зависят от природы вводимой защитной группы должны применяться; например, для PG = PMB хлорметил-4-метоксибензол в присутствии основания, такого как Cs2CO3, Na2CO3 или аналогичный в растворителе, таком как ДМФ, ТГФ или CH3CN или аналогичный при температурах в диапазоне от 0°C до температуры кипения растворителя, или для PG = тритил хлордифенилметил-бензол в присутствии основания такого как триэтиламин, Основание Хунига, Na2CO3 или CS2CO3 или аналогичный в растворителе, таком как ТГФ, ДМФ или аналогичном, при температурах в диапазоне от 0°C до температуры кипения растворителя. Для получения эфира B10 из бромида B8, соединение обрабатывают монооксидом углерода в присутствии метанола, подходящего Pd катализатора, такого как, например, Pd(OAc)2 и подходящего лиганда, такого как например 1,3-бис(дифенилфосфино)-пропан (dppp) в подходящем растворителе, таком как ДМФ и при подходящей температуре. В зависимости от положения атомов азота и заместителя RE, подходящие эфиры B9 могут быть коммерчески доступны и могут быть превращены в требуемое промежуточное соединение B10 с использованием условий, как описано выше для конверсии B7 в B8. Для возможности введения уходящей группы по экзоциклическому углероду, эфирную группу B10 можно восстановить до спирта B11 с использованием условий, хорошо известных в уровне техники, таких как LiBH4 или NaBH4 в MeOH, EtOH или ТГФ или аналогичном или DIBAL в толуоле или ДХМ или аналогичным при температурах в диапазоне от -20° до температуры кипения растворителя. Необходимо отметить, что в некоторых случаях и в зависимости от реакционных условий и природы гетероцикла B10 может наблюдаться некоторое избыточное восстановление, давая соединение B12. Если это происходит, окисление с подходящим окисляющим агентом [Ox], таким как, например, хлоранилин и безводном растворителе, таком как толуол, бензол или ДХМ может снова дать B11. Для получения требуемого промежуточного соединения B13 , которое приведено здесь в качестве примера с бромидом в качестве уходящей группы, B11 можно обработать, например, с помощью PBr3 или, альтернативно, с помощью CBr4, в присутствии PPh3 в подходящем растворителе, таком как CH3CN или ДХМ или аналогичном. Другие уходящие группы, такие как хлорид или мезилат или тозилат могут также быть возможными; условия для их создания известны квалифицированным специалистам.
Исходные соединения формулы B6 являются либо коммерчески доступными или могут быть получены в соответствии со Схемой 9B, если необходим конкретный участок замещения. В конкретном примере, показанном ниже, подходящим образом замещенный предшественник B14 обрабатывают гидразином в этаноле с получением гидразида B15. Обработка B15 основанием таким как, водный раствор NaOH или KOH с нагреванием даст соответствующее индазолоновое производное B16, которое может быть подвергнуто стадиям синтеза, как показано выше.
Схема 9:
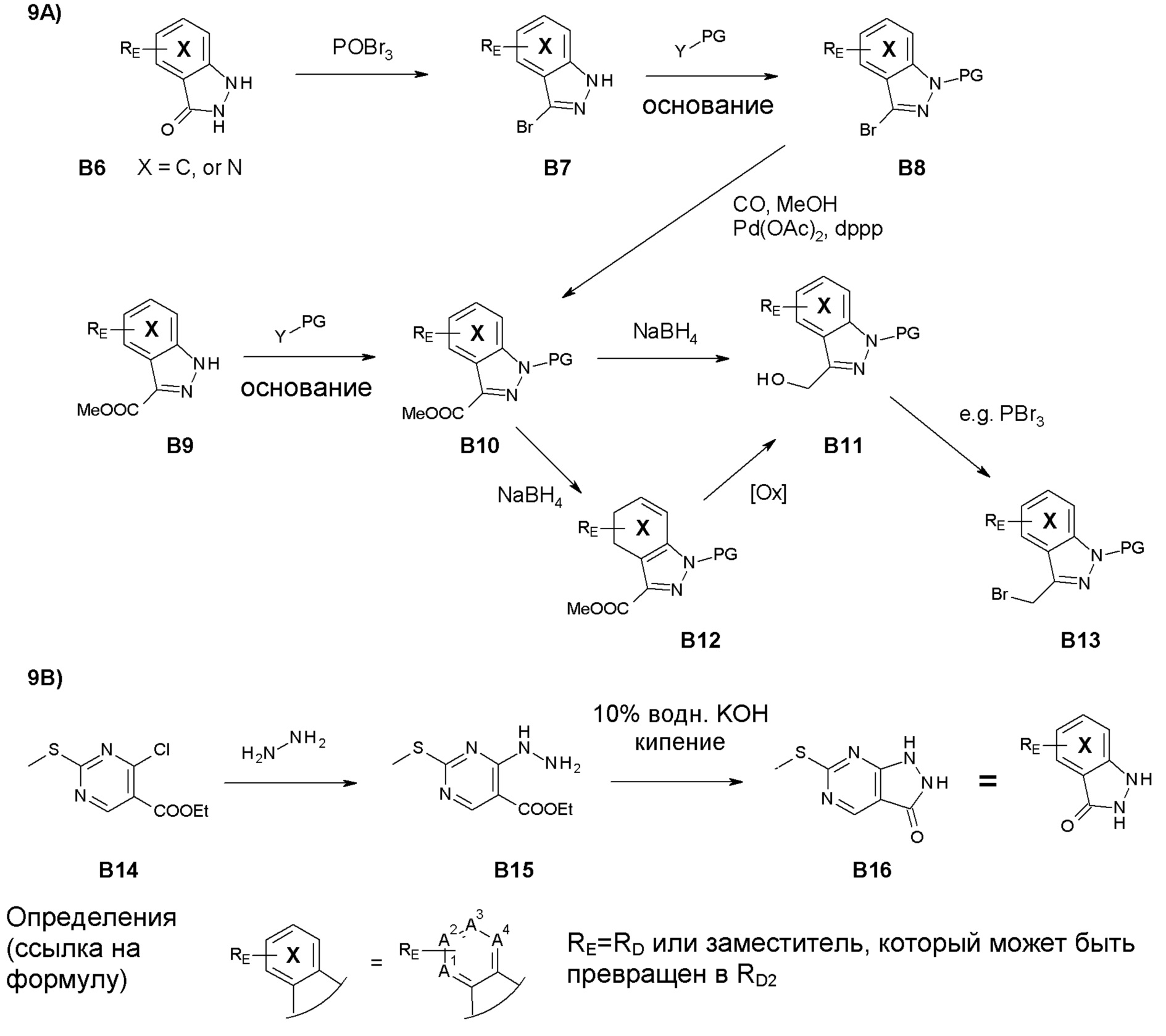
C) Пиразолопиперидины
Подходящим образом функционализированные пиразолопиперидины (один Y=N-Boc) или терагидроиндазолы (оба Y=CH2) B19 являются другой группой структурных элементов формулы A2, которые используются в настоящем изобретении для получения соединений формулы (I) (Схема 10A). Подходящие предшественники для синтеза этих бициклических соединений формулы B19 являются коммерчески доступными или могут быть получены в соответствии с методиками, которые описаны в литературе. Подходящим предшественником или промежуточным соединением является, например, эфир формулы B17, который может быть защищен подходящей защитной группой PG, такой как например Boc или тритил. Такие эфиры могут быть восстановлены до гидроксиметилпроизводных B18 с использованием подходящих восстанавливающих агентов, таких как NaBH4, LiBH4 или DIBAL или аналогичным в растворителях, таких как MeOH, EtOH, ТГФ или аналогичных. Как описано выше, введение уходящей группы X с получением требуемого структурного элемента B19 происходит с использованием подходящих условий которые зависят от вводимой уходящей группы. Если, например, X = Cl, промежуточное соединение B18 может быть обработано COCl2 с получением B19 , где X = Cl. Если, например, X = Br, обработка CBr4 и PPh3 может быть использована. Если X = мезилат или тозилат, тогда B18 может быть обработано подходящим сульфонилхлоридом (метансульфонилхлорид или п-толуолсульфонилхлорид) в присутствии основания, такого как триэтиламин, пиридин, DMAP, Основание Хунига или аналогичным в подходящем растворителе, таком как CH2Cl2 или ТГФ или аналогичным при температурах в диапазоне от -78°C до температуры кипения растворителя с получением структурного элемента B19.
Если подходящий исходный материал B17 не является коммерчески доступным, что может быть в случае некоторых пиразолопиперидинов, где одни Y=N-Boc, тогда возможным является синтетический подход, как показано на Схеме 10B. Коммерчески доступный Boc-пиперидин-4-он B20 подвергают реакции альдольного типа с этилдиазоацетатом и подходящим основанием, таким как LDA, LiHMDS или т.п. в растворителе, таком как ТГФ или другой или аналогичным при температурах в диапазоне от -78°C до температуры кипения растворителя с получением промежуточного соединения B21. B21 может быть дегидратировано с получением B22, стадия, которая проводится посредством обработки распространенным дегидратирующим агентом, таким как POCl3 или аналогичным. Циклизация B22 с получением необходимого пиразолопиперидина B23 достигается, например, посредством нагревания соединения B22 в растворителе, таком как толуол или ксилен или аналогичном вплоть до температуры кипения растворителя. Альтернативно, если растворитель обладает низкой точкой кипения, такой как ТГФ или бензол или аналогичный используемый, трансформация может быть проведена в закрытой пробирке или, например, в микроволновой печи.
Схема 10:

D) Производные пиразолов и пиразолкарбоновой кислоты
Подходящим образом функционализированные пиразолы B26 или B29 являются другой подгруппой подходящих структурных элементов формулы A2, которые применяются в настоящем изобретении для получения соединений формулы (I) (Схема 11). Подходящие предшественники для синтеза соединений формулы B26 и B29, соответственно, являются коммерчески доступными или могут быть получены в соответствии с методиками, которые описаны в литературе. Как показано на Схеме 11A, подходящий предшественник представляет собой метилпиразол B24, который может быть защищен, например, ди-трет-бутил-дикарбонатом в условиях, которые хорошо известны квалифицированным специалистам с получением Boc-защищенного промежуточного соединения B25. Другие защитные группы вместо Boc могут быть использованы в этом превращении, как показано ниже для синтеза соединения B29. Для получения требуемого структурного элемента B26 с уходящей группой, промежуточное соединение B25 обрабатывают NBS и дибензоилпероксидом в CCl4 с получением бромометилового структурного элемента B26.
Схема 11:

Если необходимы пиразолы с некоторыми дополнительными заместителями, они могут быть получены, например из коммерчески доступного диэфира B27 (Схема 11B). B27 может быть защищен, например, в виде геми-аминаля с THP: в этом случае B27 обрабатывают дигидропиранилом и ТФУ или другой подходящей кислотой, такой как п-толуолсульфоновая кислота или аналогичной в растворителе, таком как ДХМ или ТГФ или т.п. с получением защищенного диэфирного промежуточного соединения B28. Одиночная эфирная группа может быть затем восстановлена с помощью DIBAL в растворителе, таком как толуол, бензол или ДХМ при температурах в диапазоне от -78°C до температуры кипения растворителя с получением гидроксиметил производного B29. По аналогии с тем, как описано для других структурных элементов, введение уходящей группы X с получением требуемого структурного элемента B30 происходит с использованием подходящих условий, которые зависят от вводимой уходящей группы. Если, например, X = Cl, промежуточное соединение B29 может быть обработано COCl2 с получением B30, где X = Cl. Если, например, X = Br, обработка CBr4 и PPh3 может быть использована. Если X = мезилат или тозилат, тогда B29 может быть обработано подходящим сульфонилхлоридом (метансульфонилхлорид или п-толуолсульфонилхлорид) в присутствии основания, такого как триэтиламин, пиридин, DMAP, основание Хунига или аналогичным, в подходящем растворителе, таком как CH2Cl2 или ТГФ или аналогичным при температурах в диапазоне от -78°C до температуры кипения растворителя с получением структурного элемента B30.
Общий синтез феноловых структурных элементов формулы A1, применяемых в настоящем изобретении

Введение и обработка ортозамесителя RA, где RA≠арил
В данном разделе описан синтез различных фенольных структурных элементов общей формулы A1. Несмотря на то, что RA обозначает заместитель в ортоположении OH группы, RB представляет собой заместитель, который необходимо ввести, который может нуждаться в дополнительной обработке в течение синтеза, а RC представляет собой один или несколько заместителей, которые присутствуют с самого начала и существуют на протяжении синтеза. Для некоторых предпочтительных фенольных промежуточных веществ с ортозаместителем RA = трет-бутил, доступ возможен на основе соответствующего фенола с отсутствующим заместителем RA. В этих случаях, как показано на Схеме 12, подходящий фенольный предшественник C1 без ортозаместителя RA затем обрабатывают, например трет-бутанолом (C2, X = OH) в качестве предшественника трет-бутилового заместителя в присутствии сильной кислоты, такой как серная кислота или п-толуолсульфоновая кислота, фосфорная кислота или т.п. в подходящем растворителе, таком как уксусная кислота или аналогичная, при температурах в диапазоне от комнатной температуры до температуры кипения растворителя или при более высоких температурах вплоть до 250 °C, например в закрытой пробирке или в микроволновой печи с получением o-трет-бутилфенола формулы C3. Другие источники для трет-бутил заместителя, такого как изобутилен, 2-хлор-2-метилбутан (C2; X = Cl), 2-бромо-2-метилбутан (C2; X = Br), трет-бутилметиловый эфир (C2; X = OMe), или аналогичный также могут быть использованы в такой трансформации и подходящие аддитивы, такие как AlCl3 или ZnCl2 могут быть использованы для ускорения реакции. В зависимости от места замещения исходного соединения, региоизомеры могут быть получены, которые необходимо идентифицировать и разделить.
Схема 12:

Возможный синтез феноловых промежуточных соединений с RA заместителями, которые отличаются от трет-бутила или изо-бутила может быть выполнен, как показано на Схеме 13. Ключевым промежуточным соединением является, например йодид структуры C6 , который является подходяще защищенным с помощью PG (например, PG может быть метильной группой, бензильной группой или группой MOM или аналогичной, в зависимости от совместимости защитной группы с условиями, которые используются для синтеза) (Схема 13A). В контексте настоящего изобретения, C6 может быть получен из коммерчески доступного, подходящим образом защищенного нитрофенола C4, который может быть восставлен до соответствующего анилина C5 с помощью способов и реагентов (показанными [красным]), хорошо известных в литературе, таких как восстановление посредством каталитической гидрогенизации с использованием газообразного водорода и подходящего катализатора, такого как Pd на угле или аналогичным или других восстановительных условий, таких как цинковая пыль или железо в присутствии слабой кислоты, такой как раствор хлорида аммония или уксусная кислота в подходящих растворителях , таких как вода, этанол, метанол или их смеси. Введение йода проводится с использованием хорошо известных условий, таких как, например, обработка C5 нитритом натрия и йодидом калия в воде при нагревании с получением промежуточного соединения C6.
Как показано на Схеме 13B, алифатические или алициклические остатки RA могут быть затем введены посредством обработки предшественника C6 подходящим алифатическим или алициклическим боронатом или бороланом C7 (M = B(OH)2 или B(OR)2) и подходящим катализатором, таким как Pd(OAc)2 или PdCl2 или аналогичным в присутствии подходящего лиганда, такого как трифенилфосфин, трициклогексилфосфин или т.п. в растворителе, таком как ДМФ, вода или толуол или их смесь, при температурах в диапазоне от комнатной температуры до 150 °C с получением продуктов связывания C8. В некоторых случаях, таких как например, если RA = циклопропил, затем защитная группа может быть удалена с непосредственным получением требуемого фенольного структурного элемента C9. Условия для удаления PG будут зависеть от природы PG: Если PG представляет собой метил, тогда удаление может быть выполнено, например, обработкой C8, например, BBr3 в растворителе, таком как ДХМ или т.п., с получением C9. Если, например, PG представляет собой бензильную группу, тогда удаление может быть проведено, например, каталитической гидрогенизацией (например, Pd/C, H2 газ). Если PG = MOM, тогда удаление будет проведено, например, обработкой кислотой (например, водн. HCl в ТГФ или аналогичным).
В других случаях, если C8 содержит двойную связь (например, циклогексенил, дигидропропанил или пропенил), восстановление двойной связи может быть легко проведено каталитической гидрогенизацией в условиях, описанных ранее (например, H2, Pd/C) с получением промежуточных соединений C8A. Затем снова удаление PG из C8A для получения структурных элементов формулы C9 будет выполнено, как описано выше, для промежуточных соединений C8.
Схема 13:

Альтернативный подход для введения некоторых RA-заместителей показан на Схеме 13C). В этом подходе йодид C6 обрабатывают сначала изопропилмагния хлоридом, что приводит к обмену йод-магний, с последующим добавлением подходящего кетона C10 в инертном растворителе, таком как ТГФ или эфир или аналогичном при температурах в диапазоне от -78°C до температуры кипения растворителя, с получением третичного спирта C11. Восстановление третичного спирта C11 с получением алифатического или алициклического промежуточного соединения C12 может быть проведено с применением условий, известных в литературе; например, обработкой BF3OEt2 и триэтилсиланом в растворителях, таких как ДХМ или CHCl3 или т.п., при температурах в диапазоне от -78°C до температуры кипения растворителя. Снова удаление PG из C12 с получением структурных элементов формулы C9 может быть выполнено как описано выше для C8 и C8A, соответственно.
Введение и обработка RB - Синтез различных промежуточных соединений
Во многих случаях в контексте настоящего изобретения, подходящие исходные соединения являются коммерчески доступными или легко доступными, при этом они уже несут требуемый ортозаместитель RA. В этих случаях необходимы введение и обработка требуемого заместителя RB в требуемом положении, хотя другие заместители RC могут присутствовать и обычно они не затрагиваются в процессе синтеза. В зависимости от природы вводимого RB требуются различные предшественники, а фенольные гидроксигруппы нуждаются в соответствующей защите с целью совместимости с используемыми реакционными условиями (Схема 14). Например, подходящими исходными точками являются коммерчески доступные фенилбромиды C13 с RA , который находится в рамках формулы изобретения как определено формулой (I). C13 может быть O-защищен с получением C14: подходящими защитными группами PG являются, например, метил, бензил или MOM. Условия для введения изменяются в зависимости от природы PG, но обычно основание, такое как Cs2CO3, NaH, Na2CO3 или т.п., в подходящем растворителе, таком как ДМФ, ДМСО, ТГФ или CH3CN или т.п., а также реагенты для защитных групп, такие как MeI, бензилхлорид или бромид или MOM-Cl при температуре в диапазоне от -20° до температуры кипения растворителя применяют для этого превращения. Цианогруппа может быть введена с получением арилнитрила C15 с использованием условий, хорошо известных в уровне техники. Zn(CN)2, в присутствии цинка, 1,1'-бис(дифенилфосфино)ферроцен (dppf) и Pd2(dba)3 в растворителях, таких как ДМФ при температурах в диапазоне от комнатной температуры до температуры кипения растворителя или альтернативные условия, такие как CuCN в безводном ДМФ при повышенных температурах вплоть до температуры кипения ДМФ являются примерами подходящих условий для этого превращения.
Циано группа может быть гидролизована с получением свободной карбоновой кислоты C16; подходящие условия включают присутствие водных растворов NaOH или KOH в растворителях, таких как EtOH или MeOH или т.п., при температурах в диапазоне от 0°C до температуры кипения растворителя. Несмотря на то, что карбоновая кислота C16 сама по себе может быть полезной для дальнейшей обработки в отношении RB, эфирная группа может быть предпочтительной. Для получения, например, метилового эфира C17, C16 может быть обработано основанием, таким как Cs2CO3, NaH или K2CO3 или т.п., с последующим MeI в растворителе, таком как ДМФ, ДМСО, ТГФ или CH2CN или т.п., при температурах в диапазоне от 0°C до температуры кипения растворителя. Альтернативно, обработка сильной кислотой (каталитическая вплоть до стехиометрических количеств), такой как H2SO4 или п-толуолсульфоновая кислота в кипящем метаноле с или без водоотделителя может дать такую же трансформацию в эфир C17.
Схема 14:

Введение и обработка RB на основе бромидов формулы C14
Один из путей обработки RB на основе арилбромидов C14 представляет собой применение Pd катализируемого связывания, такого как реакции Сузуки, Стилле или Бухвальда или аналогичные. Подходящие условия для проведения таких превращений были подробно рассмотрены в литературе. Три способа получения промежуточных соединений C18 показаны на Схеме 15, где бромид C14 либо обрабатывают M-HET, который может быть, например, гетероциклическим станнаном, таким как 2-трибутилстаннила оксазол (реакция Стилле) или гетероциклическим боронатом или бороланом, таким как 1-метил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил-1H-имидазол (реакция Сузуки), или, альтернативно, для реакции Бухвальда с азотсодержащим гетероциклом, таким как морфолин. С точки зрения условий, реакции Стилле могут быть проведены с использованием, например, катализаторов, таких как PdCl2(dppf)2 ДХМ комплекс или т.п., в растворителях, таких как диоксан или т.п., где реакции по типу Сузуку могут быть с PdCl2(dppf)2 ДХМ комплексом в присутствии основания, такого как Na2CO3 или K2CO3 или т.п. в растворителях, таких как ДМФ. Реакции Бухвальда могут проводиться с катализаторами, такими как Pd2(dba)3 и подходящим лигандом, таким как ксантфос, в присутствии основания, такого как трет-бутилат калия, в растворителях, таких как безводный толуол. Во всех случаях реакции выполняются при температурах в диапазоне от комнатной температуры до 150°C (или выше в случае использования закрытых пробирок). Удаление защитных групп PG соединения C18 с получением фенолов C19 может быть проведено, как описано выше, для соединений формулы C8 или C8A.
Схема 15:
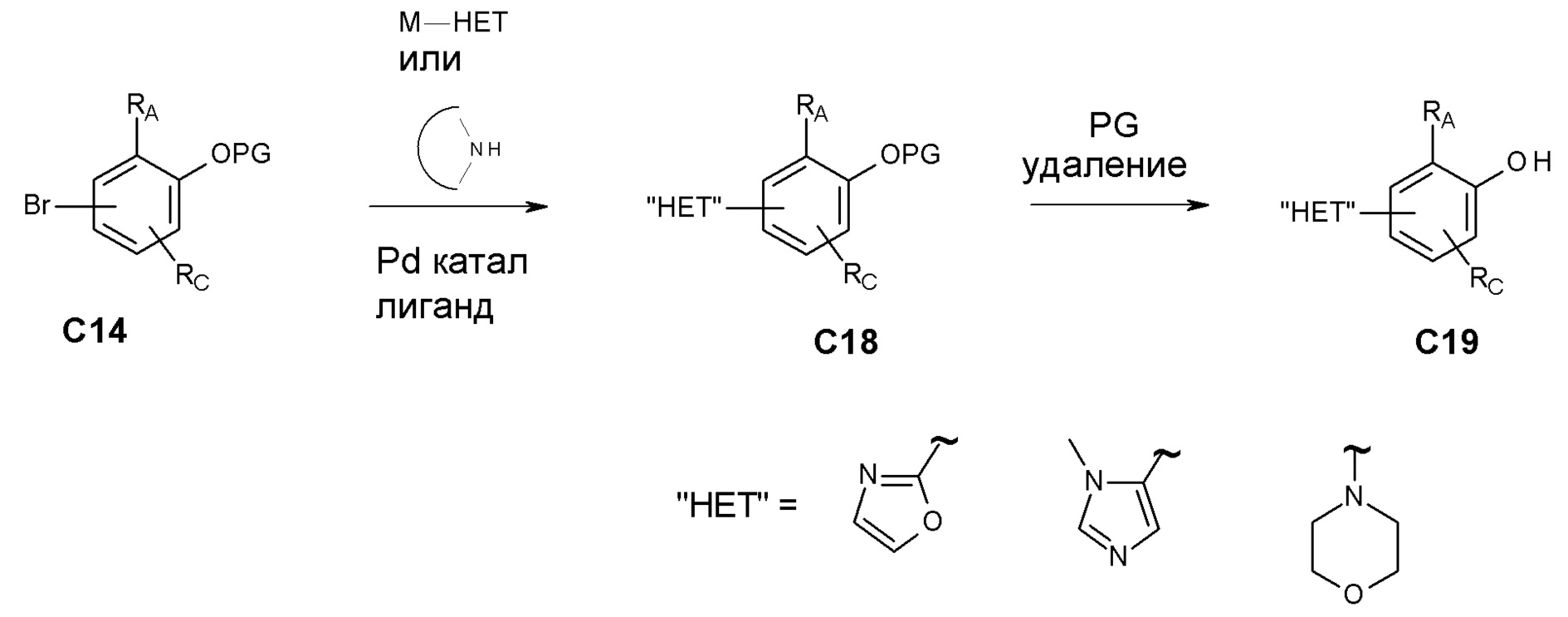
В некоторых особых случаях, когда необходимый гетероциклический станнановый или боронатный структурный элемент является недоступным, может быть необходима постадийная обработка для введения требуемых гетероциклов с использованием в качестве исходного вещества бромида C14. Пример такого постадийного подхода дан на Схеме 16. Обработка бромида C14 в реакции по типу Хека с н-бутилвиниловым эфиром в присутствии Pd катализатора, такого как Pd(OAc)2 или PdCl2 или аналогичным, и бидентантным лигандом, таким как бис(дифенилфосфино)пропан и основанием, таким как K2CO3 или т.п. в растворителе, таком как вода или ДМФ или их смеси, при температурах в диапазоне от комнатной температуры до 200°C с последующей кислотной обработкой с использованием водных минеральных кислот, таких как HCl или HBr или аналогичных, даст ацетилпроизводное C20. Впоследствии, обработка C20 бромом самим по себе или, альтернативно, источником брома, таким как Bu4NBr3 в растворителе, таком как ТГФ, эфир, диоксан или т.п., даст бромацетильное промежуточное соединение C21. Это промежуточное соединение может быть превращено в необходимое имидазольное производное C22 посредством обработки избытком формамида при температурах в диапазоне от 100 - 200°C. В этом случае с целью обеспечения возможности проведения стадии связывания, показанной на Схеме 1, требуется подходящая защита имидазольного гетероцикла. Несмотря на то, что было описано некоторое количество празличных подходящих защитных групп (см.: Green and Wuts., Protecting Groups in Organic Synthesis, J. Wiley & Sons), Схема 16 показывает применение SEM группы, которая может быть введена в C22 посредством обработки SEMCl и подходящим основанием, таким как NaH, KOtBu, LDA или Cs2CO3или аналогичным в ТГФ, эфире или ДМФ или аналогичным при температурах в диапазоне от -20°C до температуры кипения растворителя с получением защищенного имидазола C23. Удаление защитных групп PG соединения C23 с получением фенолов C24, используемых в реакции связывания со структурными элементами формулы A2, может быть выполнено, как описано выше, для соединений формулы C8 или C8A.
Схема 16:
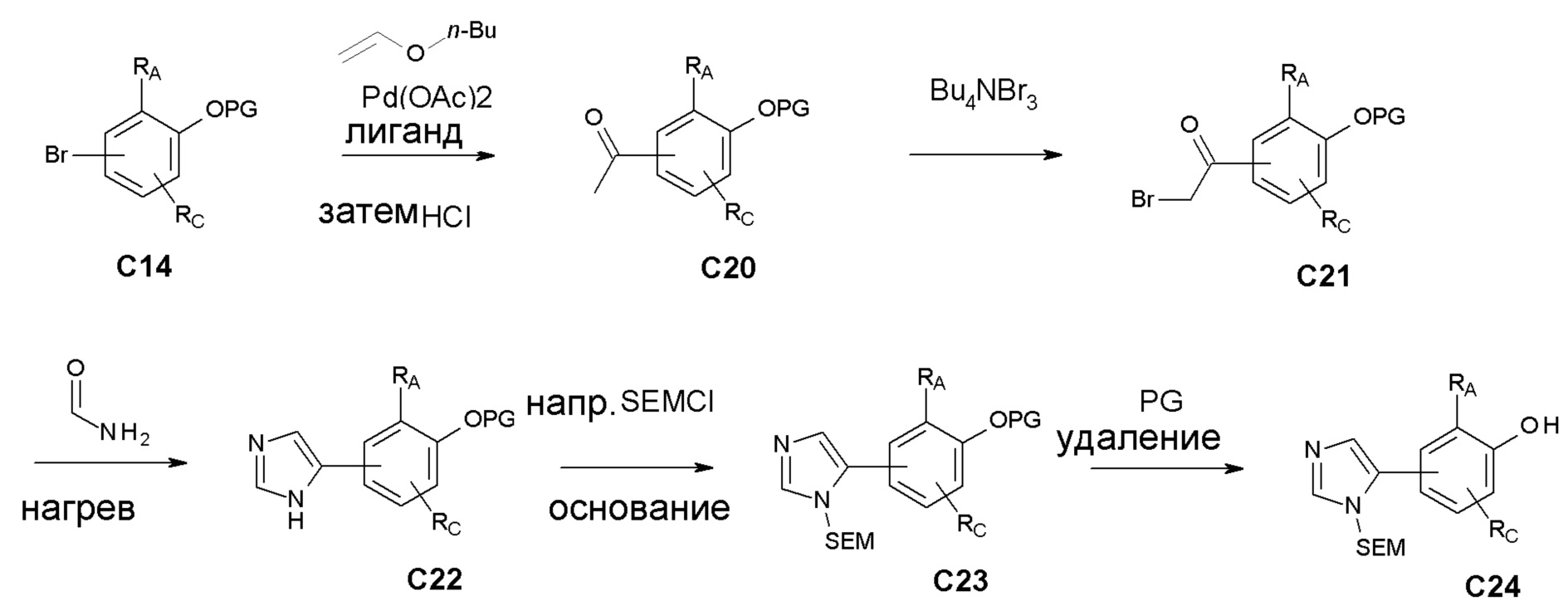
Вариант введения сульфонового остатка RB в бромиды C14 показана на Схеме 16. Для достижения этой трансформации C14 обрабатывают, например, сульфонатом натрия и в присутствии комплекса меди, который получают из йодида меди и L-пролина в присутствии основания гидроксида натрия или т.п.. Реация связывания с получением сульфонов формулы C25 проводится в растворителях, таких как ДМСО или аналогичные, при температурах в диапазоне от комнатной температуры до 130°C. Удаление защитной группы PG соединения C25 с получением фенолов C26 может быть проведено, как описано выше, для соединений формулы C8 или C8A.
Схема 17:

Введение и обработка RB на основе эфиров карбоновой кислоты формулы C17
Эфиры карбоновой кислоты формулы C17 являются другими возможными предшественниками для введения и/или обработки RB, если RB представляет собой, например, один из видов гетероцикла. Пример такого подхода показан на Схеме 18A. В соответствии с этой стратегией, C17 обрабатывают основанием, таким как NaH, Cs2CO3, Na2CO3 или аналогичным, с последующим N-гидроксиацетамидином в растворителе, таком как ДМФ, ТГФ или аналогичном, при температурах в диапазоне от -20°C до температуры кипения растворителя. Эта обработка даст соответствующий 1,2,4-оксадиазол C27, который может подвергнуться депротекции с получением фенола C28 в подходящих условиях, которые зависят от природы PG, как описано выше для C8 и C8A.
Схема 18:

Альтернативный путь для введения гетероциклических заместителей в арилэфирные исходные соединения C17 показан на Схеме 18B. В этом конкретном случае защитная группа PG на феноле уже была удалена ранее, например, на стадии C17, с получением незащищенного эфира C29. Это соединение может быть обработано гидразином в растворителе, таком как метанол, этанол, ДМСО или вода или аналогичном, при температурах вплоть до 100°C с получением ацилгидразида C30. Для получения требуемых 1,3,4-оксадиазолов C31, C30 может быть обработано избытком ортоэфира карбоновой кислоты (например, триметилортоацетатом для RQ = Me) при температурах вплоть до 200°C. Возможным является использование других ортоэфиров карбоновых кислот, что может давать соединения формулы C31 с гетероциклическими заместителями, которые несут RQ отличный от метила.
Введение и обработка RB на основе нитрилов формулы C15
Арилнитрилы формулы C32 являются другими возможными предшественниками для введения и/или обработки RB, если RB является, например, другим типом гетероцикла, т.е. некоторые имидазольные производные. Пример такого подхода показан на Схеме 19. В соответствии с этой стратегией арилнитрил C15 обрабатывают сначала избытком этилендиамина и P2S5 в закрытой пробирке при температурах от комнатной температуры до 150°C с получением дигидроимидазола C32. C32 может быть окислен подходящим оксилительным агентом [Ox] с получением имидазольного производного C33. Подходящими окислительными агентами и условиями являются, например, диазетокси-йодобензол/K2CO3/ДМСО, KMnO4/Al2O3/CH3CN, фульминуровый хлорангидрид/DBU/CH3CN. Азот имидазола соединения C32 затем может быть алкилирован алкилирующим агентом RR-X, где RR представляет собой например алкильную группу и X представляет собой уходящую группу, такую как йод или бром или тозилат, в присутствии основания, такого как NaH, Cs2CO3 или Na2CO3 или аналогичным в растворителе, таком как ДМФ, ТГФ, CH3CN или аналогичным при температурах от -20°C до температуры кипения растворителя с получением N-алкилированного промежуточного соединения C34. Альтернативно, RR также может быть защитной группой, такой как SEM или MOM или т.п. (ортогональной к фенольной защитной группе PG) которую можно снова удалить позже для получения свободного имидазола на необходимой стадии. Удаление защитной группы PG соединения C34 с получением фенолов C35, используемых в реакции связыванияс основной группой формулы A2 может быть выполнено, как описано выше, для соединений формулы C8 или C8A.
Схема 19:

Несмотря на то, что подходы, изображенные выше, показывают предпочтительные пути для множества промежуточных соединений и примеров, показанных в настоящем изобретении, некоторые конкретные соединения с особыми участками замещения могут быть получены альтернативными путями, снова в зависимости от предшественников, которые являются доступными или возможными. Например, введение нитрильной группы может быть проведено не только как показано на Схеме 14 ранее, на также нитрилы могут быть образованы дегидрированием амидов карбоновой кислоты (Схема 20). Если защитную группу удаляют на стадии первичного амида, тогда получившиеся фенолы также могут быть использованы в качестве промежуточных соединений формулы A1.
Схема 20:

Введение и обработка обоих заместителей RA и RB
В некоторых конкретных случаях, и в частности если необходимо ввести и обработать обе группы RA и RB, необходимо применять более сложный путь, который включает стратегии защитных групп в дополнение к таковым, описанным выше, для получения подходящих фенольных промежуточных соединений формулы A1. Такой путь к промежуточному соединению показан на Схеме 21A ниже.
Подходящим исходным соединением является, например, C40, который содержит арил-амин и заместитель X (X= например, Br, I) помимо RC и группы OH. C40 защищают, например, ди-трет-бутил-дикарбонатом в присутствии основания, такого как NEt3, Основание Хунига, DMAP или пиридин или аналогичным, в растворителе, таком как ДХМ, ТГФ или эфир или т.п., при температурах от -20°C до температуры кипения растворителя, давая трис-Boc промежуточное соединение C41. RA теперь может быть введен, например, с помощью подходящей бороновой кислоты или боролана C42 в присутствии подходящего катализатора, такого как Pd(OAc)2 или PdCl2 или аналогичного, в присутствии подходящего лиганда, такого как трифенилфосфин, трициклогексилфосфин или т.п. в растворителе, таком как ДМФ, вода или толуол или их смесь, при температурах в диапазоне от комнатной температуры до 150 °C с получением продуктов связывания C43, несущих подходящий заместитель RA. Удаление защитной группы Boc затем является легко возможным с использованием обычной обработки кислотой, такой как, например, ТФУ в ДХМ, HCl в диоксане или аналогичной, или водн. HCl, при подходящих температурах, с получением амино-гидрокси промежуточного соединения C44. Одним примером дополнительной обработки аминогруппы является обработка нитритом натрия в воде в присутствии кислоты, такой как H2SO4 или HCl или аналогичной, с последующим йодидом калия, с получением йодида C45. Затем возможно ввести, например, цианогруппу, посредством обработки соединения C45 CuCN в растворителе, таком как ДМФ или ДМСО или т.п. при температурах от комнатной температуры до 150 °C с получением нитрила C46, в котором RB=CN.
Альтернативно, аминогруппа промежуточного соединения C44 может быть модифицирована по разному, например посредством ацилирования, как показано на Схеме 21 посредством сульфонилирования. В случае сульфонилирования C44 обрабатывают подходящим сульфонилхлоридом C47 (RF представляет собой, например, небольшой алкил) в присутствии основания, такого как пиридин, DMAP, NEt3 или основание Хунига в растворителе, таком как ДМФ, ДХМ, или ТГФ при температурах в диапазоне от -20°C до температуры кипения растворителя, с последующей обработкой NAOH, KOH или LiOH в воде, с получением сульфонамида C48, где RB=NHSO2RF.
Схема 21:
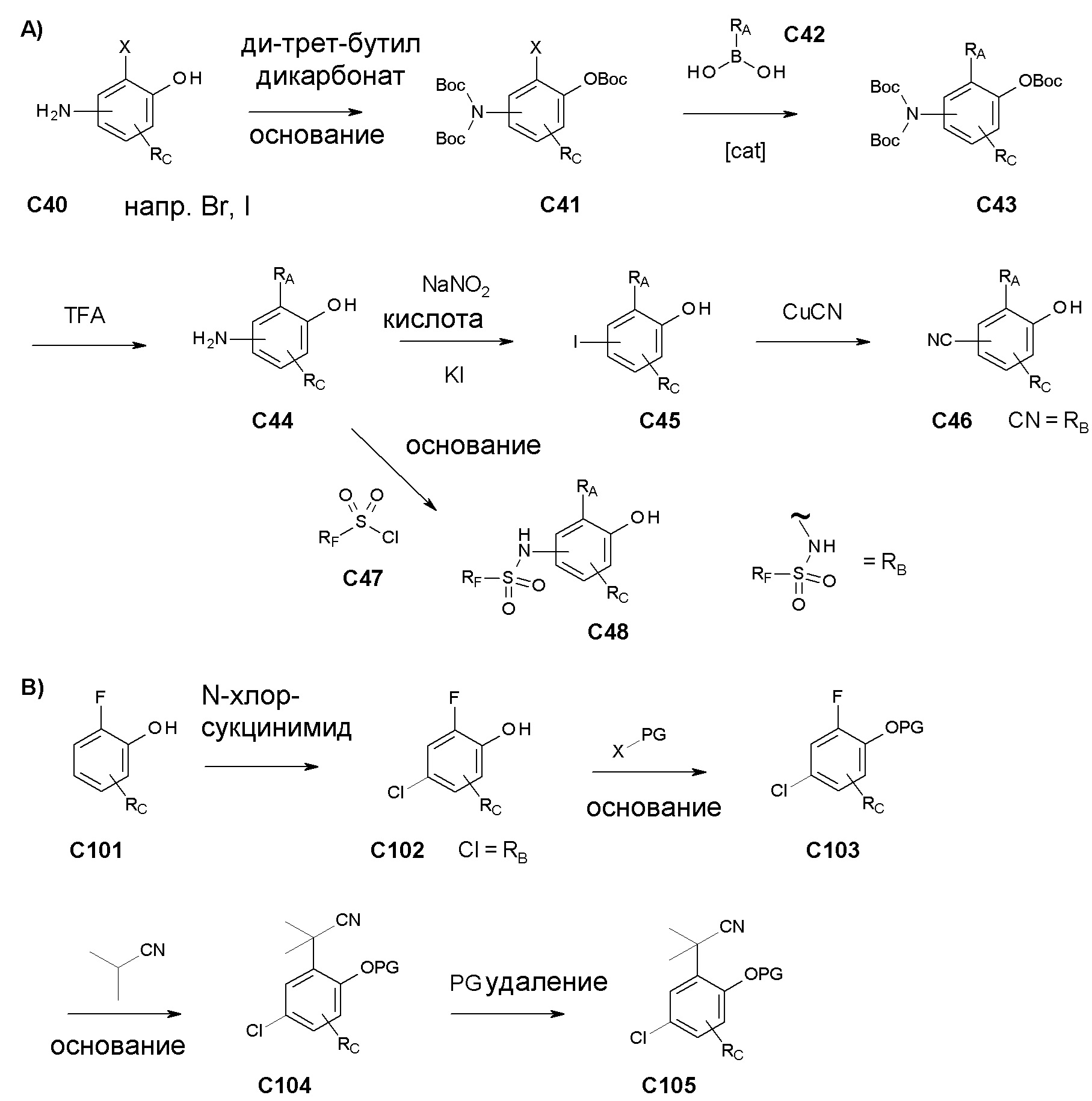
Другой способ введения обоих заместителей RA и RB показан на Схеме 21B. В этом конкретном случае фенол C101 может быть хлорирован, например, в параположении по гидроксильной группе с помощью хлорирующего агента, такого как N-хлор-сукцинимид, в присутствии сильной кислоты, такой как трифлатная кислота или т.п., в кислотном растворителе, таком как уксусная кислота или муравьиная кислота или аналогичном, при температурах в диапазоне от комнатной температуры до температуры кипения растворителя. Квалифицированный специалист определит, что сайт хлорирования может зависеть от других заместителей, присутствующих на арильном кольце соединения C101. Обычно, существует потребность в защите свободной гидроксильной группы продукта C102 для дальнейшей обработки, с получением промежуточного соединения C103. Подходящие защитные группы PG, такие как бензил или MOM или SEM или аналогичные, а также условия для введения обсуждались выше. Введение RA, который соответствует 2-метил-пропионитрил-2-ильной группе в данном случае может быть достигнуто, например, обработкой соединения C103 изобутиронитрилом в присутствии ненуклеофильного сильного основания, такого как KHMDS или NaHMDS в растворителе, таком как толуол или бензол или т.п., в закрытой пробирке при температурах в диапазоне от комнатной температуры до 160°C с получением промежуточного соединения C104, с необходимым участком замещения. Условия для удаления защитной группы PG соединения C104 для получения фенола C105, который используетсяв реакции связыванияс основными группами формулы A2 будут зависеть от природы PG и реакция может быть выполнена, как описано выше, для соединений формулы C8 или C8A.
Введение и обработка ортозаместителя RA для фенолов C49, где RA= замещенный арил

Общая последовательность, изображенная на Схемах 22 - 29 может быть значительной длины и квалифицированный специалист будет понимать, что последовательность реакций, как показано на Схемах 22 - 29 может изменяться в зависимости от реакционной способности и природы промежуточных соединений. Аналогично тому, как описано ранее, если одно из исходных соединений или промежуточных соединений содержит одну или более функциональных групп, которые не являются стабильными или являются реакционноспособными в условиях проведения реакции на одной или более реакционных стадий, подходящие защитные группы (как описано, например, в “Protective Groups in Organic Chemistry” by T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y.) могут быть введены перед критической стадией с применением способов, хорошо известных в уровне техники. Такие защитные группы могут быть удалены при последующих стадиях синтеза с использованием стандартных способов, описанных в литературе.
Синтез фенольных промежуточных соединений C49 в которых RA означает возможно замещенный арильный или гетероарильный заместитель может быть выполнен, например, как показано на Схеме 22 из промежуточных соединений C6, которые являются коммерчески доступными или могут быть получены как описано на Схеме 13. Взаимодействие C6 с (замещенными) бороновыми кислотами RA-B(OH)2 или бороновыми эфирами RA-B(OR')2 (например, пинаколовый или триметиленгликолевый эфир, либо коммерчески доступный, или полученный с использованием способов из литературы, как описано, например, в “Boronic Acids - Preparation and Applications in Organic Synthesis and Medicine” под авторством Dennis G. Hall (ed.) 1st Ed., 2005, John Wiley & Sons, New York) с использованием подходящего катализатора (например, дихлор[1,1'-бис(дифенилфосфино)-ферроцен]палладий(II) дихлорметановый аддукт, тетракис(трифенилфосфин)палладий(0) или ацетат палладия (II) с трифенилфосфином) в подходящем растворителе (например, диоксан, диметоксиэтан, вода, толуол, N,N-диметилформамид или их смеси) и подходящего основания (например, карбонат натрия, гидрокарбонат натрия, фторид калия, карбонат калия или триэтилаамин) при температурах между комнатной температурой и температурой кипения растворителя или смеси растворителей дает промежуточные соединения C50 (стадияa). Реакции Сузуки данного типа хорошо освещены в литературе (например, A. Suzuki, Pure Appl. Chem.1991, 63, 419-422; A. Suzuki, N. Miyaura, Chem. Rev.1995, 95, 2457-2483; A. Suzuki, J. Organomet. Chem.1999, 576, 147-168; V. Polshettiwar et al., Chem. Sus. Chem.2010, 3, 502-522) и хорошо известны квалифицированным специалистам. Альтернативно, арил- или гетероарил-трифторбораты RABF3K могут быть использованы в реакциях кросс-сочетания с применением палладиевого катализатора, такого как, например, тетракис(трифенилфосфин)палладий (0), ацетат палладия (II) или дихлор[1,1'-бис(дифенилфосфино)ферроцен]-палладий(II) дихлорметановый аддукт в присутствии подходящего основания, такого как карбонат цезия или фосфат калия, в растворителях, таких как толуол, ТГФ, диоксан, вода или их смесь, при температурах между комнатной температурой и температурой кипения растворителя или смеси растворителей.
Промежуточные соединения C50 также могут быть синтезированы из C6 с помощью (замещенных) арил- или гетероарил реагентов олова RA-SnR3 (R = например, Me или n-Bu; либо коммерчески доступный или полученный в соответствии со способами, описанными в литературе) в присутствии подходящего катализатора (например, тетракис -(трифенилфосфин)палладий(0), бензилбис(трифенилфосфин)-палладий(II) хлорид, бис(трифенилфосфин)палладий(II) дихлорид или дихлор[1,1'-бис(дифенилфосфино)-ферроцен]палладий(II) дихлорметановый аддукт) в подходящем растворителе, таком как ТГФ, диоксан, ДМФ (N,N-диметилформамид) или HMPA (гексаметилфосфорамид) или их смесь) при температурах между комнатной температурой и температурой кипения растворителя или смеси растворителей, возможно в присутствии хлорида лития. Связывание по Стилле такого типа широко освещено в литературе (например, J. K. Stille, Angew. Chem. Int. Ed. Engl.1986, 25, 508-524; V. Farina et al., J. Org. React.1998, 50, 1-652; T. N. Mitchell, Synthesis1992, 9, 803-815) и хорошо известно квалифицированным специалистам (стадия a).
Альтернативно, промежуточные соединения C50 могут бытьсинтезированы в реакции промежуточного соединения C6 с (замещенным) арил- или гетероарил цинковыми галидами RA-ZnX (X = Cl, Br или I) (либо коммерчески доступный или синтезированный способами, описанными в литературе) с использованием никелевого (например, тетракис(трифенилфосфин)никель (0)) или палладиевого катализатора (например, тетракис(трифенилфосфин)палладий (0)) в подходящем растворителе, таком как, например, ТГФ или DMA в температурном диапазоне между комнатной температурой и точкой кипения растворителя. Связывание по Негиши такого типа широко освещено в литературе (например, “Name Reactions for Homologations-Part I: Negishi cross-coupling reaction”, Li, J. J., Corey, E. J., Eds.; Wiley & Sons, Hoboken, NJ, 2009, 70-99; “Metal-Catalyzed Cross-Coupling Reactions”, Diederich, F.; Stang, P. J., Eds.; Wiley-VCH: Weinheim, Germany, 1998, 1-47; E. Erdik, Tetrahedron 1992, 48, 9577-9648; G. Organ, Eur. J. Org. Chem.2010, 4343-4354) и хорошо известно квалифицированным специалистам (стадия a). Удаление защитной группы PG в промежуточных соединениях C50 способами, известными квалифицированному специалисту, и как описано например в “Protective Groups in Organic Chemistry” by T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y. (например, бензильная группа посредством гидрогенизирования с использованием подходящего катализатора, такого как палладий на угле, в подходящем растворителе, таком как MeOH, EtOH, EtOAc ил их смеси; метильной группы посредством реакции с трехбромистым бором в подходящем растворителе, таком как дихлорметан) дает промежуточные соединения C49 (стадия b), которые являются подгруппой фенольных структурных элементов A1.
Промежуточные соединения C51 также могут быть получены из промежуточных соединений C6 сначала удалением защитной группы PG из промежуточных соединений C6 с использованием условий, описанных ранее (стадия c), и конвертацией получившихся промежуточных соединений C51 в промежуточные соединения C49 посредством применения, например, реакций кросс-сочетания Сузуки, Стилле или Негиши, как описано выше (стадия d).
Схема 22:

Если структурный элемент (т.е. замещенные арил или гетероарил-бороновые кислоты RA-B(OH)2 или бороновые эфиры, арил- или гетероарил олова реагенты RA-SnR3, или (замещенные) арил- или гетероарил цинковые галиды RA-ZnX) для получения промежуточных соединений C49 из промежуточных соединений C6 не являются коммерчески доступными или нестабильны в используемых реакционных условиях, промежуточные соединения C49 могут быть альтернативно получены в соответствии с методиками, описанными ниже.
Например, промежуточные соединения C56 и C58, в которых RA означает фенильное кольцо, подходящим образом замещенное RG1 (см. формулу изобретения) и замещенное вторичной или третичной амидной группой могут быть синтезированы в соответствии со Схемой 23. Реакции кросс-сочетания между фенилбороновыми кислотами или арилбороновыми эфирами C52 замещенными карбоксильной или эфирной группами и RG1 который соответствует заместителю в соответствии с формулой изобретения, либо коммерчески доступными, либо могущими быть синтезированными способами известными квалифицированным специалистам, с промежуточным соединением C6 в реакционных условиях , описанных на Схеме 22, Стадия a, дают биарильные промежуточные соединения C53 (стадия a). Отщепление эфирной группы карбоновой кислоты в промежуточных соединениях C53 в щелочной (например, метиловый или этиловый эфиры с литием или гидроксидом натрия в полярных растворителях, таких как метанол, H2O или ТГФ или смеси указанных растворителей) или нейтральной среде (например, бензильная группа Bn посредством гидрогенизации с использованием подходящего катализатора, такого как палладий на угле, в подходящем растворителе, таком как MeOH, EtOH, EtOAc или их смеси) приводит к промежуточным соединениям C54 (стадия b). Дополнительные эфиры включают, без ограничения, например, аллиловые эфиры, которые могут быть отщеплены способами, известными квалифицированному специалисту, и как описано например в “Protective Groups in Organic Chemistry” под авторством T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y., с применением стратегии ортогональной защитной группы, которая позволит удалить эфирную защитную группу без затрагивания других защитных групп, таких как защитная группа фенола PG. Взаимодействие промежуточных соединений C54 с аминами типа Rc-NH2 дает промежуточные соединения C55 (стадия c). Амидные связывания такого типа широко освещены в литературе и могут быть выполнены использованием связывающих реагентов, таких как, например, CDI, DCC, HATU, HBTU, HOBT, TBTU или реагент Мукаямы в подходящем растворителе, например, ДМФ, DMA, ДХМ или диоксан, возможно в присутствии основания (например, NEt3, DIPEA (Основание Хунига) или DMAP). Альтернативно, в двухстадийном способе, группа карбоновой кислоты в промежуточных соединениях C54 может быть конвертирована в соовтетствующий хлорангидрид посредством обработки, например, тионилхлоридом, чистым или возможно в растворителе, таком как ДХМ. Взаимодействие хлорангидрида с Rc-NH2 в подходящем растворителе, таком как ДХМ или ДМФ и основания, например, NEt3, основание Хунига, пиридин, DMAP или лития бис(триметилсилил)амид при температурах в диапазоне от 0°C до температуры кипения растворителя или смеси растворителей дает промежуточные соединения C55 (стадия c). Удаление защитной группы PG соединения C55 с применением способов, описанных ранее дает промежуточные соединения C56 (стадия d).
Промежуточные соединения C58 могут быть получены также из промежуточных соединений C54 посредством связывания со вторичными аминами вида RcRdNH применением реакционных условий, описанных выше, с получением третичного амида C57 (стадия e). Последующее удаление защитной группы PG применением реакционных условий, описанных ранее, дает промежуточные соединения C58 (стадия f).
Промежуточные соединения C57 альтернативно могут быть получены алкилированием промежуточных соединений C55 соединениями Rd-LG в которых LG означает подходящую уходящую группу, такую как бром (или другую уходящую группу, такую как хлор, йод или OSO2алкил, OSO2фторалкил, OSO2арил) с использованием подходящего основания и растворителя, такого как гидрид натрия в тетрагидрофуране с получением промежуточных соединений C57 (стадия g). Амидные заместители Rc, Rd, используемые в Схеме 23 определены подходящей подгруппой замевстителей RG как определено в формуле изобретения.
Схема 23:

Промежуточные соединения C63, в которых RA означает фенильное кольцо подходящим образом замещенное RG1 и замещенное алкокси, галоалкокси, ариалкокси или гетероарилалкокси заместителем, могут быть синтезированы, например, в соответствии со Схемой 24. Алкилирование бромо-фенолов C59, возможно замещенных RG1, как определено объемом формулы изобретения, которые являются либо коммерчески доступными или могут быть получены способами, известными квалифицированному специалисту, с помощью соединений типа RSLG, в которых RS определен подгруппой объема заместителя RG в формуле изобретения и LG означает подходящую уходящую группу, такую как бром (или другую уходящую группу, такую как хлор, йод или OSO2алкил, OSO2фторалкил, OSO2арил) с использованием подходящего основания и растворителя, такого как карбонат калия в ацетоне или гидрид натрия в тетрагидрофуране, дает промежуточные соединения C60 (стадия a). Промежуточные соединения C60 могут быть превращены в соответствующие бороновые кислоты или эфиры бороновых кислот C61 с использованием литературных способов, как описано например в “Boronic Acids - Preparation and Applications in Organic Synthesis and Medicine” by Dennis G. Hall (ed.) 1st Ed., 2005, John Wiley & Sons, New York, например с использованием 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана (CAS RN 73183-34-3) в присутствии системы подходящего основания, катализатора и растворителя, такой как ацетат калия, [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладиевый (II) комплекс с дихлорметаном в 1,4-диоксане при температурах в диапазоне от комнатной температуры до температуры кипения растворителя или смеси растворителей (стадия b). Подходящие условия для реакции кросс-сочетания промежуточных соединений C61 с промежуточным соединением C6 (стадия c) были описаны выше. Удаление защитной группы PG в промежуточных соединениях C62 применение литературных методик или реакционных условий, показанных ранее дает промежуточные соединения C63 (стадия d).
Промежуточные соединения C62 альтернативно могут быть получены из промежуточных соединений C67 применением стратегии ортогональной защитной группы (удаление одной защитной группы, в любом порядке, с использованием реагентов и условий, которые не затрагивают другие защитные группы в целевом соединении). Гидроксигруппы в возможно замещенных бромо-фенолах C59 могут быть защищены способами, известными квалифицированному специалисту и как описано, например, в “Protective Groups in Organic Chemistry” by T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y. (например, метоксиметильная группа (MOM) посредством взаимодействия возможно замещенных бромо-фенолов с метоксиметилхлоридом в присутствии основания в подходящем растворителе, таком как NaH в ТГФ, при температурах в диапазоне от 0°C до температуры кипения растворителя) с получением промежуточных соединений C64 (стадия e). Промежуточные соединения C64 в свою очередь могут быть конвертированы в промежуточные соединения C116 (стадия f) как показано выше на стадии b. Последующее кросс-сочетание промежуточных соединений C65 с промежуточными соединениями C6 (стадия г) применением реакционных условий, описанных выше для стадии c, дает промежуточные соединения C66. Избирательное удаление защитной группы PG' в промежуточных соединениях C66 на возможно замещенном фенильном заместителе RA дает промежуточные соединения C67 (стадия h). Промежуточные соединения C67 в свою очередь может быть трансформировано в промежуточные соединения C62 посредством алкилирования с соединениями типа RS-LG, в которых RS определен подгруппой объема заместителя RG в формуле изобретения и LG означает подходящую уходящую группу, такую как бром (или другую уходящую группу, такую как хлор, йод или OSO2алкил, OSO2фторалкил, OSO2арил) и с использованием реакционных условий, описанных ранее, дает промежуточные соединения C113 (стадия i).
Схема 24:
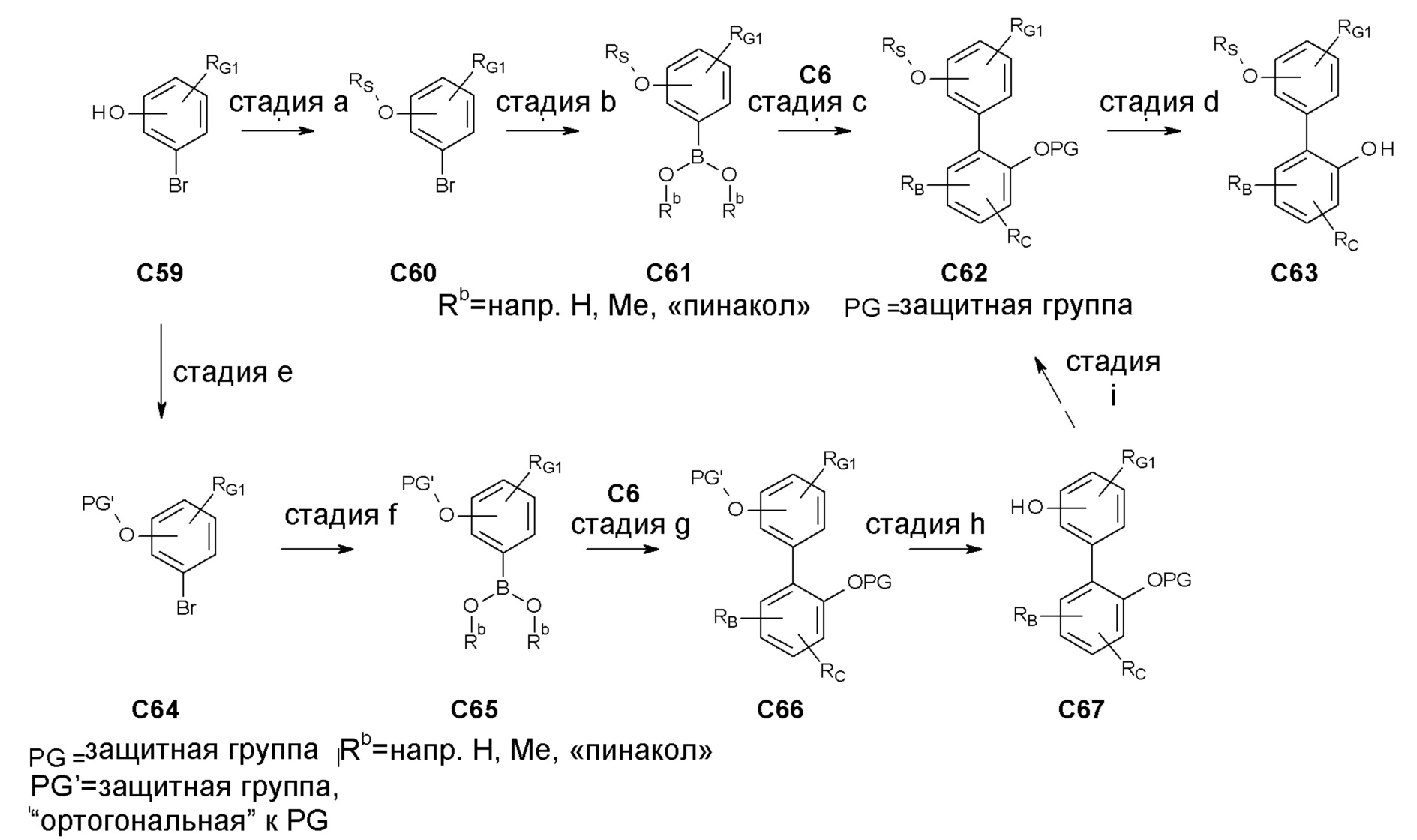
Промежуточные соединения C74 в которых RA означает фенильное кольцо подходящим образом замещенное RG1 и связанное через метиленовый линкер (CH2) с N-связанным лактамом или циклическим уретаном могут быть синтезированы, например, в соответствии со Схемой 25. Группа бензилового спирта в исходном соединении C68, которое является либо коммерчески доступным или может быть легко получено в соответствии с литературными методиками, может быть превращена в подходящую уходящую группу LG, такую как бром (например, посредством взаимодействия промежуточных соединений C119 с тетрабромметаном в присутствии трифенилфосфина в ТГФ в качестве растворителя) или другую уходящую группу, такую как хлор, йод или OSO2алкил, OSO2фторалкил, OSO2арил, с получением промежуточных соединений C69 (стадия a). Взаимодействие промежуточных соединений C69 с лактамами или циклическими уретанами C68 в присутствии системы подходящего основания и растворителя, такой как гидрид натрия в ДМФ дает промежуточные соединения C71 (стадия b). Промежуточные соединения C71 могут быть превращены в их соответствующие бороновые кислоты или бороновые эфиры C72 с использованием обычных литературных методик, как описано, например, в “Boronic Acids - Preparation and Applications in Organic Synthesis and Medicine” под авторством Dennis G. Hall (ed.) 1st Ed., 2005, John Wiley & Sons, New York, например с использованием 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (CAS RN 73183-34-3) в присутствии системы подходящего основания, катализатора и растворителя, такой как ацетат калия, [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладиевый (II) комплекс с дихлорметаном в 1,4-диоксане, при температурах в диапазоне от комнатной температуры до температуры кипения растворителя или смеси растворителей (стадия c). Реакция кросс-сочетания промежуточных соединений C72 с промежуточным соединением C6 в реакционных условиях, описанных ранее, дает промежуточные соединения C73 (стадия d). Удаление защитной группы PG в промежуточных соединениях C73 применением литературных методик или реакционных условий, показанных ранее, дает промежуточные соединения C74 (стадия e). Промежуточные соединения C74 могут быть также получены в результате реакции кросс-сочетания промежуточных соединений C72 со структурным элементом C51 применением способов, описанных ранее (стадия f).
Схема 25:

Промежуточные соединения C79 и C83, в которых RA означает фенильное кольцо подходящим образом замещенное RG1 и которое связано метиленовым линкером (CH2) с N-связанной вторичной или третичной амидной группой карбоновой кислоты могут быть синтезированы, например, в соответствии со Схемой 26. Ацилирование аминогруппы в бензиламинах C75, которые являются либо коммерчески доступными или могут быть легко получены в соответствии с литературными методиками, с помощью карбоновых кислот вида RT-COOH дает промежуточные соединения C76 (стадия a). Амидное связывание этого типа широко освещено в литературе и может быть выполнено применением множества различных связывающих реагентов таких как, например, CDI, DCC, HATU, HBTU, HOBT, TBTU или реагент Мукаямы в подходящем растворителе, например, ДМФ, DMA, ДХМ или диоксан, возможно в присутствии основания (например, NEt3, DIPEA (Основание Хунига) или DMAP). Альтернативно, карбоновые кислоты RT-COOH могут быть конвертированы в их хлорангидриды перед реакцией связывания посредством обработки, например, тионилхлоридом, чистым или возможно в растворителе, таком как ДХМ. Взаимодействие хлорангидрида с промежуточными соединениями C75 в подходящем растворителе, таком как ДХМ или ДМФ и основания, например, NEt3, Основание Хунига, пиридин, DMAP или лития бис(триметилсилил)амида, при температурах в диапазоне от 0°C до температуры кипения растворителя или смеси растворителей дает соединения C76 (стадия a). Промежуточные соединения C76 могут быть конвертированы в их соответствующие промежуточные соединения бороновых кислот или бороновых эфиров C77 с использованием литературных способов, как описано например в “Boronic Acids - Preparation and Applications in Organic Synthesis and Medicine” by Dennis G. Hall (ed.) 1st Ed., 2005, John Wiley & Sons, New York и с применением способов, описанных ранее (стадия b). Кросс-сочетание промежуточных соединений C77 с промежуточными соединениями C6 с использованием реакционных условий, описанных ранее, дает промежуточные соединения C78 (стадия c). Удаление защитной группы PG в промежуточных соединениях C78 производят применением литературных методик или реакционных условий, описанных ранее, с получением промежуточных соединений C79 (стадия d).
Промежуточные соединения C83, в которых RA означат фенильное кольцо, которое соединено через метиленовый линкер (CH2) с N-связанной третичной амидной группой могут быть получены из промежуточных соединений C76, например, N-алкилированием промежуточных соединений C76 с помощью соединений вида RU-LG, в которых LG означает подходящую уходящую группу, такую как бром (или другую уходящую группу, такую как хлор, йод или OSO2алкил, OSO2фторалкил, OSO2арил) с использованием подходящего основания и растворителя, такого как гидрид натрия в тетрагидрофуране с получением промежуточных соединений C80 (стадия e). По аналогии с описанным выше для C76, C77, C78 и C79, промежуточные соединения C80 могут быть конвертированы в промежуточные соединения C81 (стадия f), C82 (стадия г) и в заключение C83 (стадия h) с применением способов, описанных ранее. Промежуточные соединения C81 альтернативно также могут быть получены алкилированием промежуточных соединений C77 с помощью соединений вида RU-LG и применяя условия, описанные на стадии e выше (стадия i). Промежуточные соединения C82 также могут быть получены алкилированием промежуточных соединений C78 с соединениями вида RU-LG и применяя условия, описанные для стадии e выше (стадия j). Оба RT и RU используемые на Схеме 26 определены подходящей подгруппой заместителей в объеме RG, как показано в формуле изобретения.
Схема 26:

Промежуточные соединения C49, в которых RA означает арильное кольцо, которое дополнительно замещено гетероарильным кольцом и для которых подходящие бороновые кислоты RA-B(OH)2 или бороновые эфиры RA-B(OR')2, реагенты олова RA-SnR3 или галиды цинка RA-ZnX для введения заместителей RA в промежуточные соединения CC6 или C51 являются недоступными или нестабильными в используемых реакционных условиях, промежуточные соединения C49 могут быть синтезированы, например, в соответствии со Схемами 27 - 29 и способами, описанными в литературе. Квалифицированные специалисты знают, что эта методология также применима к спектру других гетероарильных систем.
Например, промежуточные соединения C87, в которых RA означает фенильное кольцо подходящим образом замещенное RG1 и замещенное [1.3.4]оксадиазол-2-ильным кольцом, в котором RV представляет собой, например, алкил, циклоалкил, трифторметил, бензил или фенил или аналогичные, могут быть получены из промежуточных соединений C53 (см Схему 23 для получения) в соответствии со Схемой 27. Избирательное отщепление эфирной группы карбоновой кислоты в промежуточных соединениях C53 (например, метиловый или этиловый эфиры с литием или гидроксид натрия в полярных растворителях, таких как метанол, H2O или ТГФ или смеси указанных растворителей; дополнительные эфиры включают, без ограничения, например, бензиловые или аллиловые эфиры, которые могут быть отщеплены способами, известными квалифицированному специалисту, и как описано например в “Protective Groups in Organic Chemistry” под авторством T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y.) и в которых Ra выбирают таким образом, чтобы гидроксильная защитная группа PG не затрагивалась в реакционных условиях, применяемых для отщепления Ra (“стратегия ортогональной защитной группы”) дает промежуточные соединения C84 (стадия a). Взаимодействие промежуточных соединений C84 с ацилгидразинами вида RV-C(O)NHNH2 дает промежуточные соединения C85 (стадия b). Амидные связывания такого типа широко освещены в литературе и могут быть выполнены использованием связывающих реагентов, таких как, например, CDI, DCC, HATU, HBTU, HOBT, TBTU или реагент Мукаямы в подходящем растворителе, например, ДМФ, DMA, ДХМ или диоксан, возможно в присутствии основания (например, NEt3, DIPEA (Основание Хунига) или DMAP). Альтернативно, группа карбоновой кислоты в промежуточных соединениях C84 может быть конвертирована в её хлорангидрид посредством обработки, например, тионилхлоридом, чистым или возможно в растворителе, таком как ДХМ. Взаимодействие хлорангидрида с ацилгидразином вида RV-C(O)NHNH2 в подходящем растворителе, таком как ДХМ или ДМФ и основанием, например, NEt3, Основание Хунига, пиридин, DMAP или лития бис(триметилсилил)амид при температурах в диапазоне от 0°C до температуры кипения растворителя или смеси растворителей дает 1,2-диацилгидразида промежуточные соединения C85 (стадия b). Циклизация диацилгидразидной группы в промежуточных соединениях C135 с использованием подходящего циклодегидратирующего агента, такого как POCl3, SOCl2, реагент Бургесса или диэтиламинодифторсульфиния тетрафторбората (XtalFluor-E®) либо чистого, либо с сипользованием подходящего растворителя, такого как дихлорметан, возможно в присутствии кислоты, такой как уксусная кислота при температурах в диапазоне от комнатной температуры до температуры кипения растворителя или смеси растворителей, дает 1,3,4-оксадиазоловый промежуточные соединения C86 (стадия c). Синтез этого типа 1,3,4-оксадиазолов хорошо известен в уровне техники и также описан в литературе, например, M.-F. Pouliot et al., Org. Biom. Chem. 2012, 10(5), 988. Удаление защитной группы PG соедиения C86 с получением C87 проводитсяспособами, известными квалифицированному специалисту и как описано, например, в “Protective Groups in Organic Chemistry” под авторством T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y. (например, бензильной группы гидрогенизацией с использованием подходящего катализатора, такого как палладий на угле, в подходящем растворителе, таком как MeOH, EtOH, EtOAc или их смеси; метильной группы взаимодействием с трехбромистым бором в подходящем растворителе, таком как дихлорметан) (стадия d).
Схема 27:

Промежуточные соединения C93, в которых RA означает фенильное кольцо, подходящим образом замещенное RG1 и замещенное [1.2.4]оксадиазол-5-иловым кольцом, в котором RW представляет собой например алкил, циклоалкил, трифторметил, бензил или фенил или аналогичный, может быть получено в соответствии со Схемой 28, например, из промежуточных соединений C88. Синтез [1.2.4]оксадиазолов широко описан в литературе, например в R. O. Bora et al., Mini-Reviews Med. Chem. 2013, 13 or K. Hemming, Sci. Synthesis2004, 13, 127. Нитрильные промежуточные соединения C88 , которые являются либо коммерчески доступными или могут быть получены способами, хорошо известными в уровне техники, могут быть конвертированы в их бороновые эфиры, такие как пинаколовые эфиры C89 посредством литературных способов, например, посредством взаимодействия промежуточных соединений C88 с пинаколом в подходящем растворителе, таком как ТГФ (стадия a). Цианогруппы в промежуточных соединениях C89 могут быть превращены в амидоксимовые группы способами, известными в уровне техники, например, взаимодействием с гидроксиламином (возможно в виде его гидрохлоридной соли) в подходящем растворителе, таком как EtOH в присутствии основания, такого как, например, основание Хунига с получением промежуточных соединений C90 (стадия b). Циклизация амидоксимовой группы в промежуточных соединениях C90 с активированным производным карбоновой кислоты, несущим заместитель RW,таким как соответствующий ангидрид, хлорангидрид, эфир кислоты или ортоэфир при температурах в диапазоне от комнатной температуры до температуры кипения растворителя дает промежуточные соединения C91 , которые часто получают без выделения периодически образующегося O-ацилированного промежуточного соединения (стадия c). Промежуточные соединения C91 может взаимодействовать с промежуточными соединениями C6 в реакциях кросс-сочетания с использованием условий, описанных на Схеме 22, Стадия a, с получением промежуточных соединений C92 (стадия d). Удаление защитной группы в промежуточных соединениях C92 способами, известными квалифицированному специалисту, как описано, например, в “Protective Groups in Organic Chemistry” by T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y. и как описано ранее, дает требуемые фенольные промежуточные соединения C93 (стадия e).
Промежуточные соединения C92 альтернативно могут быть получены из промежуточных соединений C88 проведением реакций кросс-сочетания с промежуточным соединением C6 в качестве первой стадии последовательности, как описано на Схеме 22, Стадия a, с получением промежуточных соединений C94 (стадия f), затем конверсией цианогруппы в амидоксимовую группу с использованием способов, описанных выше с получением C95 (стадия г) и затем циклизацией C95 с активированной карбоновой кислотой, несущей заместитель RW в условиях, показанных выше, с получением промежуточных соединений C92 (стадия h). Промежуточные соединения C94 также могут быть получены из промежуточных соединений C89 реакцией кросс-сочетания с промежуточными соединениями C6 применением реакционных условий, описанных ранее (стадия i).
Схема 28:

Промежуточные соединения C100, в которых RA означает фенильное кольцо подходящим образом замещенное RG1 и замещенное трет-бутил защищенной 1H-пиразол-3-ильной группой, могут быть получены, например, как описано на Схеме 29. Кросс-сочетание промежуточных соединений C96, либо коммерчески доступных или полученных в соответствии со способами, описанными в литературе, с промежуточными соединениями C6, применяя способы, описанные на Схеме 22 (стадия a), дает промежуточные соединения C97 (стадия a). Ацетильная группа в промежуточных соединениях C97 может быть трансформирована в 3-диметиламино-акрилоильную группу с применением опубликованных способов, например, взаимодействием с N,N-диметилформамида диметилацеталем, предпочтительно при повышенной температуре, с получением промежуточных соединений C98 (стадия b). Взаимодействие C98 с трет-бутил гидразином в подходящем растворителе, таком как этанол или метанол или аналогичным, предпочтительно при повышенной температуре дает пиразольные промежуточные соединения C99 (стадия c). Хемоселективное удаление фенольной защитной группы PG в промежуточных соединениях C99 применением стратегии ортогональных защитных групп, как показано ранее, дает промежуточные соединения C100 (стадия d).
Алкилирование фенольной группы C100 необходимой основной группой A2, как описано на Схеме 1 и последующее удаление трет-бутильной защитной группы пиразола способами, известными в уровне техники, как описано, например, в “Protective Groups in Organic Chemistry” by T. W. Greene and P. G. M. Wutts, 5th Ed., 2014, John Wiley & Sons, N.Y. (например, a трет-бутильной группы в кислотной среде, такой как HCl в диоксане, в подходящем растворителе, таком как дихлорметан) дает конечные продукты формулы A3.
Схема 29:

Также воплощением настоящего изобретения является способ получения соединения формулы (I), как определено выше, содержащий взаимодействие соединения формулы (II) в присутствии соединения формулы (III);

где RA, RB, RC и W являются такими, как определено здесь, а X представляет собой галоген, мезилат или тозилат.
В частности, в присутствии основания, в частности в присутствии карбоната калия, возможно в присутствии йодида калия, в растворителе, таком как ацетон и при температуре между -78°C и температурой кипения, в частности между комнатной температурой и температурой кипения.
Также объектом настоящего изобретения является соединение формулы (I), как здесь описано, для применения в качестве терапевтически активного вещества.
Аналогично, объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение в соответствии с формулой (I), как здесь описано и терапевтически инертный носитель.
В конкретном воплощении настоящее изобретение представляет собой соединение в соответствии с формулой (I) как здесь описано для лечения или профилактики глазных заболеваний, в частности глаукомы.
Настоящее изобретение также относится к применению соединения согласно формуле (I), как здесь описано для получения лекарственного средства для лечения или профилактики глазных заболеваний, в частности глаукомы.
Также объектом настоящего изобретения является способ лечения или профилактики глазных заболеваний, в частности глаукомы, который включает введение эффективного количества соединения согласно формуле (I) как здесь описано.
Заболевания почек включают, без ограничения, острые повреждения почек и хроническую почечную болезнь с и без протеинурии, в том числе в терминальной стадии почечной недостаточности (ТПН). Более подробно, это включает сниженный клиренс креатинина и сниженную скорость клубочковой фильтрации, микроальбуминурию, альбуминурию и протеинурию, гломерулосклероз с расширением сетчатой мезангиальной матрицы с или без существенной гиперклеточности (в частности, диабетическая нефропатия и амилоидоз), фокальный тромбоз капилляров клубочков (в частности, тромботические микроангиопатии), общий фибриноидный некроз, ишемические поражения, злокачественный нефросклероз (такой как ишемическая ретракция, снижение почечного кровотока и почечная артериопатия), отечность и пролиферация интракапиллярных (эндотелиальных и мезангиальных) и/или экстракапиллярных клеток (серповидных), как при гломерулонефрите, фокальный сегментарный гломерулосклероз, IgA-нефропатии, васкулиты/системные заболевания, а также острое и хроническое отторжение трансплантата почки
Заболевания печени включают, без ограничения, цирроз печени, застой печени, холестатическую болезнь печени, включая зуд, неалкогольный стеатогепатит и острое и хроническое отторжение трансплантата печени.
Воспалительные заболевания включают, без ограничения, артрит, остеоартрит, рассеянный склероз, системную красную волчанка, воспалительное заболевание кишечника, ненормальное нарушение эвакуации и т.д., а также воспалительные заболевания дыхательных путей, такие как идиопатический легочный фиброз (IPF), хроническая обструктивная болезнь легких (ХОБЛ) или хроническим бронхиальная астма.
Другие заболевания дыхательной системы включают, без ограничения, другие диффузные заболевания паренхимы легких различной этиологии, включая ятрогенный медикаментозный фиброз, фиброз, индуцированный профессиональной и/или окружающей средой, системные заболевания и васкулиты, гранулематозные заболевания (саркоидоз, аллергический альвеолит), коллагеноз сосудов, альвеолярный протеиноз, гранулематоз клеток Лангерганса, лимфангиолейомиоматоз, наследственные заболевания (синдром Германски-Пудлака, туберозный склероз, нейрофиброматоз, метаболические расстройства накопления, семейное интерстициальное заболевание легких), индуцированный радиацией фиброз, силикоз, индуцированный асбестом легочный фиброз или острый респираторный дистресс-синдром (РДСВ).
Заболевания нервной системы включают, без ограничения, невропатическую боль, шизофрению, нейровоспаление (например, астроглиоз), периферическую и/или вегетативную (диабетическую) невропатии и т.д.
Сосудистые заболевания включают, без ограничения, атеросклероз, тромботические сосудистые заболевания, а также тромботические микроангиопатии, пролиферативную артериопатию (такую как отек миоинтимальных клеток, окруженных муцинозным внеклеточным матриксом и шаровидное утолщение), атеросклероз, снижение податливости сосудов (например, жесткость, сниженная податливость желудочков и сниженная податливость сосудов), дисфункцию эндотелия и т.д.
Сердечно-сосудистые заболевания включают, без ограничения, острый коронарный синдром, ишемическую болезнь сердца, инфаркт миокарда, артериальную и легочную гипертензию, нарушение сердечного ритма, такие как фибрилляция предсердий, инсульт и другие сосудистые повреждения.
Фиброзные заболевания включают, без ограничения, фиброз миокарда и сосудов, фиброз почек, фиброз печени, фиброз легких, фиброз кожи, склеродермию и инкапсулирующий перитонит.
Рак и метастазы рака включают, без ограничения, рак молочной железы, рак яичников, рак легких, рак простаты, мезотелиомы, глиомы, рак печени, рак желудочно-кишечного тракта и их прогрессирование и метастатическая агрессивность.
Глазные заболевания включают, без ограничения, пролиферативную и непролиферативную (диабетическую) ретинопатия, сухую и влажную возрастная макулярную дегенерацию (ВМД), отек макулы, центральную артериальную/венозную окклюзию, травматическое повреждение, глаукому и т.д. В частности, глазное заболевание представляет собой глаукому.
Метаболические заболевания включают, без ограничения, ожирение и диабет.
Также воплощением настоящего изобретения являются соединения формулы (I), как здесь описано, полученные любым описанным здесь способом.
Методики анализа
Получение полноцепочечного аутотаксина (ATX) человека с и без гистидиновой метки (HIS TAG)
Клонирование аутотаксина (ATX - ENPP2): кДНК получили из коммерческой тотальной РНК гематопоэтических клеток человека и использовали в качестве шаблона в ПЦР с перекрывающимися праймерами для получения полноцепочечной ОРФ ENPP2 человека с или без метки 3'-6xHis. Эти полноцепочечные вставки клонировали в вектор pcDNA3.1V5-His TOPO (Invitrogen). ДНК последовательность нескольких одиночных клонов была подтверждена. ДНК из корректного полноцепочечного клона использовали для трансфекции клеток Hek293 для верификации экспрессии белка. Последовательность кодируемого ENPP2 соответствует последовательности Swissprot Q13822, с или без дополнительной С-концевой метки 6xHis.
Ферментация ATX: Рекомбинантный белок получили крупномасштабной транзиентной трансфекцией в 20 л биореакторах с контролируемым перемешиванием (Sartorius). В течение клеточного роста и трансфекции температура, скорость перемешивания, pH и концентрация растворенного кислорода поддерживались при 37°C, 120 об/мин, 7.1 и 30% DO, соответственно. Клетки FreeStyle 293-F (Invitrogen) культивировали в суспензии в среде FreeStyle 293 (Invitrogen) и трасфицировали при приблизительно 1-1.5 x 10E6 клеток/мл с помощью вышеуказанной плазмидной ДНК с использованием X-tremeGENE Ro-1539 (коммерческий продукт, Roche Diagnostics) в качестве комплексообразующего реагента. Клетки подпитывали концентрированным питательным раствором (J Immunol Methods 194 (1996), 19, 1-199 (с. 193)) и индуцировали бутиратом натрия (2 мМ) через 72 ч после трансфекции и собрали через 96 ч после трансфекции. Экспрессию анализировали с помощью вестерн-блоттинга, ферментативного анализа и/или аналитической аффинной хроматографии на иммобилизованных ионах металла (IMAC). После охлаждения клеточной суспензии до 4°C посредством пропускания через теплообменник, разделение клеток и стерилизующее фильтрование супернатанта проводили посредством фильтрации через фильтры Zeta Plus 60M02 E16 (Cuno) и Sartopore 2 XLG (Sartorius). Супернатант хранили при 4°C перед очисткой.
Очистка ATX: 20 литров культурального супернатанта кондиционировали для ультрафильтрации посредством добавления Brij 35 до конечной концентрации 0.02% и посредством доведения pH до 7.0 с использованием 1 M HCl. Затем супернатант сначала подвергали микрофильтрации через 0.2 мкм фильтр Ultran-Pilot Open Channel PES (Whatman) и затем сконцентрировали до 1 литра через фильтр Ultran-Pilot Screen Channel PES с 30 kDa MWCO (Whatman). Перед IMAC хроматографией добавили NiSO4 до конечной концентрации 1 мМ. Затем очищенный супернатант нанесли на колонку HisTrap (GE Healthcare), предварительно уравновешенную в 50 мМ Na2HPO4 pH 7.0, 0.5 M NaCl, 10% глицерин, 0.3% CHAPS, 0.02% NaN3. Колонку поэтапно промыли таким же буфером, содержащим 20 мМ , 40 мМ и 50 мМ имидазола соответственно. Белок впоследствии элюировали с использованием линейного градиента до 0.5 M имидазола за 15 объемов колонки. Фракции, содержащие ATX, объединили и сконцентрировали с помощью Amicon cell, оснащённым фильтровальной мембраной 30 kDa PES. Белок дополнительно очистили с помощью эксклюзионной хроматографии на Superdex S-200 (XK 26/100) (GE Healthcare) в 20 мМ BICINE pH 8.5, 0.15 M NaCl, 10% глицерин, 0.3% CHAPS, 0.02% NaN3. Конечный выход белка после очистки составил 5-10 мг ATX на литр культурального супернатанта. Белок хранили при -80°C.
Анализ ингибирования фермента ATX человека
Ингибирование ATX измеряли анализом гашения флуоресценции с использованием специфично меченого субстратного аналога (MR121 субстрат). Для получения этого субстрата MR121, BOC и TBS защищенный 6-амино-гексаноевой кислоты (R)-3-({2-[3-(2-{2-[2-(2-амино-этокси)-этокси]-этокси}-этокси)-пропиониламино]-этокси}-гидрокси-фосфорилокси)-2-гидрокси-пропиловый эфир (Ferguson et al., Org Lett 2006, 8 (10), 2023) метили с помощью MR121 флуорофора (CAS 185308-24-1, 1-(3-карбоксипропил)-11-этил-1,2,3,4,8,9,10,11-октагидро-дипиридо[3,2-b:2',3'-i]феноксазин-13-иум) по свободному амину этаноламинового конца и затем, после депротекции, триптофаном со стороны аминогексаноевой кислоты.
Рабочие растворы для анализа готовили следующим образом:
Буфер для анализа (50 мМ Tris-HCl, 140 мМ NaCl, 5 мМ KCl, 1 мМ CaCl2, 1 мМ MgCl2, 0.01% Triton-X-100, pH 8.0;
Раствор ATX: ATX (человека, меченый гистидином) стоковый раствор (1.08 мг/мл в 20 мМ бицине, pH 8.5, 0.15 M NaCl, 10% глицерин, 0.3% CHAPS, 0.02% NaN3), разбавили до 1.4 - 2.5x конечной концентрации в буфере для анализа;
Раствор субстрата MR121: стоковый раствор субстрата MR121 (800 мкМ MR121 субстрат в ДМСО), разбавили до 2 - 5x конечной концентрации в буфере для анализа.
Тестовые соединения (10 мМ сток в ДМСО, 8 мкл) приготовили в 384 луночных планшетах для образцов (Corning Costar #3655) и разбавили 8 мкл ДМСО. Построчные серийные разведения приготовили переносом 8 мкл раствора соединения в следующий ряд до ряда O. Растворы соединений и контроля перемешивали пять раз и 2 мкл перенесли в 384 луночные планшеты для анализа (Corning Costar # 3702). Затем, добавили 15 мкл 41.7 нМ раствора ATX (30 нМ конечная концентрация), перемешали пять раз и затем инкубировали в течение 15 минут при 30°C. Добавили 10 мкл субстрата MR121 (1 мкМ конечная концентрация), смешали 30 раз и затем инкубировали в течение 15 минут при 30 °C. Затем измеряли флуоресценцию каждый 2 минуты в течение 1 часа (планшетный визуальный многорежимный ридер Perkin Elmer); интенсивность света: 2.5%; время экспозиции: 1.4 сек, Фильтр: Fluo_630/690 нм) и из этих считываний рассчитывали значения IC50.
Соединения формулы (I) и их фармацевтически приемлемые соли или эфиры, как здесь описано обладают значениями IC50 между 0.00001 мкМ и 1000 мкМ, конкретные соединения обладают значениями IC50 между 0.0005 мкМ и 500 мкМ, дополнительные конкретные соединения обладают значениями IC50 между 0.0005 мкМ и 50 мкМ, более конкретные соединения обладают значениями IC50 между 0.0005 мкМ и 5 мкМ. Эти результаты были получены с использованием ферментативного анализа, описанного выше.
Соединения формулы I и их фармацевтически приемлемые соли могут применяться в качестве лекарственных средств (например, в форме фармацевтических препаратов). Фармацевтические препараты могут вводиться внутрь, например, перорально (например, в форме таблеток, покрытых оболочкой таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий), назально (например в виде назальных спреев), ректально (например, в форме суппозиториев) или местно через глаза (например, в виде растворов, мазей, гелей или водорастворимых полимерных вкладок). Однако, введение может быть выполнено парентерально, например внутримышечно, внутривенно или внутриглазно (например, в форме стерильных растворов для инъекций).
Соединения формулы I и их фармацевтически приемлемые соли могут производиться вместе с фармацевтически инертными, неорганическими или органическими адъювантами для изготовления таблеток, покрытых оболочкой таблеток, драже, твердых желатиновых капсул, растворов для инъекций или композиций для местного применения. Лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или её соли и т.д. могут применяться, например, в качестве адъювантов для таблеток, драже и твердых желатиновых капсул.
Подходящими адъювантами для мягких желатиновых капсул являются, например, растительные масла, разновидности воска, жиры, полужидкие вещества и жидкие полиолы и т.д.
Подходящими адъювантами для производства растворов и сиропов являются, например, вода, полиолы, сахароза, инвертированный сахар, глюкоза и т.д.
Подходящими адъювантами для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.д.
Подходящими адъювантами для суппозиториев являются, например, натуральные или гидрогенизированные масла, разновидности воска, жиры, полужидкие и жидкие полиолы и т.д.
Подходящими адъювантами для топикальных глазных композиций являются, например, циклодекстрины, маннит или множество других носителей и эксципиентов, известных в данной области техники.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, вещества, повышающие вязкость, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Они также могут содержать другие терапевтически ценные вещества.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, повышающие вязкость вещества, стабилизаторы, увлажняющие средства, эмульгаторы, подсластители, красители, ароматизаторы, соли для регулирования осмотического давления, буферные вещества, маскирующие агенты и антиоксиданты. Они также могут содержать другие терапевтически ценные вещества.
Доза может варьировать в широком диапазоне и будет, естественно, подбираться в соответствии с индивидуальными требованиями в каждом конкретном случае. В общем, в случае перорального введения, подходящей дневной дозой для одного человека будет приблизительно от 0,1 до 20 мг на кг массы тела, предпочтительно приблизительно от 0,5 до 4 мг на кг массы тела (например приблизительно 300 мг на индивидуума), предпочтительно разделенная на 1-3 отдельных дозы, которые могут состоять, например, из одинаковых количеств, если это подходит. В случае местного введения, композиция может содержать 0.001% - 15% по массе медикамента и необходимую дозу, которая может быть между 0.1 и 25 мг и может быть введена либо единичной дозой на день или на неделю или множественными дозами (2 - 4) в день, или множественными дозами в неделю. Однако, ясно, что вышеуказанные верхние и нижние значения могут быть расширены в случае необходимости.
Настоящее изобретение проиллюстрировано здесь с помощью Примеров, которые имеют неограничивающий характер.
В случае, если препаративные примеры получают в виде смеси энантиомеров, чистые энантиомеры могут быть разделены с помощью описанных здесь способов или с помощью способов, известных специалисту в данной области, таких как, например, хиральная хроматография или кристаллизация.
Примеры
Все примеры и промежуточные соединения получили в атмосфере азота, если не указано иного.
Аббревиатуры: водн. = водный; CAS: = регистрационный номер Chemical Abstracts Service; ВЭЖХ = высокоэффективная жидкостная хроматография; MS = месс-спектр; насыщ. = насыщенный; КТ = комнатная температура; ТСХ = тонкослойная хроматография; NMR: спектр ядерного магнитного резонанса;
Промежуточные соединения A
Промежуточное соединение A2:
2-трет-бутил-4-хлор-5-фторфенол

К раствору 4-хлор-3-фторфенола (CAS: 2713-33-9; 1.14 г) в уксусной кислоте (6.0 мл) добавили трет-бутанол (1.73 г), с последующей серной кислотой (1.53 г) при комнатной температуре в атмосфере аргона. Смесь затем нагревали при 80°C в течение 17 часов. ТСХ показала, что взаимодействие было неполным и поэтому реакционную смесь кипятили с обратным холодильником (88°C) в течение дополнительных 30 часов. Несмотря на то, что по данным ТСХ еще оставалось некоторое количество видимого исходного соединения, реакционную смесь охладили до комнатной температуры и влили в воду со льдом. Водную фазу затем экстрагировали два раза этилацетатом и объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенное вещество очистили с помощью флеш-хроматографии на силикагеле при градиенте от 0% до 25% EtOAc в гептане в качестве элюента, с получением соединения, указанного в заголовке, в виде коричневой жидкости (320 мг). MS (m/z): 201.1 [M-H]-.
Промежуточное соединение A4:
2-трет-бутил-4-(3-метил-1,2,4-оксадиазол-5-ил)фенол

Стадия 1: 1-Бензилокси-4-бромо-2-трет-бутил-бензол

Это соединение, которое известно из литературы (CAS: 33839-12-2), получили следующим способом:
К раствору 4-бромо-2-трет-бутил-фенола (CAS: 10323-39-4; 8 г) в безводном ацетонитриле (150 мл) добавили Cs2CO3 (22.7 г) с последующим бензилбромидом (6.26 мл) при 25°C и смесь перемешивали при 85°C в течение 3 ч. Реакционную смесь охладили до 25°C, отфильтровали и фильтрат эвапорировали при пониженном давлении. Получившийся остаток очистили посредством колоночной хроматографии на силикагеле (градиент 3-5% этилацетат в гексанах) с получением 1-бензилокси-4-бромо-2-трет-бутил-бензола (11.0 г) в виде серо-белого осадка.1H-NMR (400MHz, δ, ДМСО-D6): 1.32 (s, 9H), 5.14 (s, 2H), 7.04 (d, 1H), 7.28-7.50 (m, ~7H).
Стадия 2: 4-Бензилокси-3-трет-бутил-бензонитрил

Это соединение, которое известно из литературы (CAS: 847943-59-3) получили следующим способом:
К раствору 1-бензилокси-4-бромо-2-трет-бутил-бензола (4.5 г) в безводном ДМФ (100 мл) добавили Zn(CN)2 (3.30 г), dppf (782 мг) и Zn (229 мг) в атмосфере аргона, и реакционную смесь продули аргоном в течение 10 минут. Затем, Pd2(dba)3 (645 мг) добавили при 25°C, и реакционную смесь снова продули аргоном в течение 10 мин. Смесь затем перемешивали при 110°C в течение 16 ч. Смесь охладили, отфильтровали и фильтрат эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (5-7% этилацетат в гексанах) с получением соединения, указанного в заголовке (3.2 г), в виде серо-белого осадка. MS (m/z): 265.0 [M]+.
Стадия 3: 4-Бензилокси-3-трет-бутил-бензойная кислота

Это соединение, которое известно из литературы (CAS: 146852-63-3), получили следующим способом:
К раствору 4-бензилокси-3-трет-бутил-бензонитрила (800 мг) в MeOH (30 мл) добавили 6 н водн. раствор NaOH (40 мл) при 25°C и реакционную смесь кипятили с обратным холодильником в течение 16 ч. Смесь охладили до 25°C и растворитель эвапорировали при пониженном давлении. Получившийся остаток разбавили водой, подкислили с помощью конц. HCl и экстрагировали EtOAc (2x50мл). Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали при пониженном давлении с получением соединения, указанного в заголовке (830 мг) в виде коричневого осадка. MS (m/z): 285.2 [M+H]+.
Стадия 4: 4-Бензилокси-3-трет-бутил-бензойной кислоты метиловый эфир

Это соединение, которое известно из литературы (CAS: 146852-62-2), получили следующим способом:
К раствору 4-бензилокси-3-трет-бутил-бензойной кислоты (830 мг) в безводном ДМФ (30 мл) при 25°C добавили Cs2CO3 (1.9 г) с последующим йодометаном (0.273 мл) и реакционную смесь перемешивали при 25°C в течение 3ч. Реакционную смесь затем отфильтровали и фильтрат эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 3-5% этилацетат в гексанах) с получением соединения, указанного в заголовке (740 мг), в виде серо-белого осадка. MS (m/z): 299.3 [M+H]+.
Стадия 5: 5-(4-Бензилокси-3-трет-бутил-фенил)-3-метил-[1,2,4]оксадиазол
К раствору 4-бензилокси-3-трет-бутил-бензойной кислоты метилового эфира (740 мг) и N-гидрокси-ацетамидина (229.7 мг) в безводном ДМФ (30 мл) добавили NaH (218 мг) по порциям при 25°C и реакционную смесь перемешивали при 25°C в течение 3 ч. Смесь затем погасили водой и экстрагировали EtOAc (2x50 мл). Объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 7-10% EtOAc в гексане) с получением соединения, указанного в заголовке (650 мг), в виде серо-белого осадка. MS (m/z): 323.0 [M+H]+.
Стадия 6: 2-трет-бутил-4-(3-метил-[1,2,4]оксадиазол-5-ил)-фенол
К раствору 5-(4-бензилокси-3-трет-бутил-фенил)-3-метил-[1,2,4]оксадиазола (300 мг) в безводном ДХМ (30 мл), хранящемуся при -78°C, добавили BBr3 (1M раствор в ДХМ, 2.79 мл) и реакционную смесь перемешивали при -78°C в течение 2 ч. Смесь погасили насыщенным водным раствором NaHCO3 (20 мл) и экстрагировали с помощью ДХМ (2x30 мл). Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 5-20% EtOAc в гексанах) с получением соединения, указанного в заголовке (150 мг), в виде серо-белого осадка. MS (m/z): 233.2 [M+H]+.
Промежуточное соединение A5:
2-трет-бутил-4-[1-(2-триметилсилилэтоксиметил)имидазол-2-ил]фенол

Стадия 1: 2-(4-Бензилокси-3-трет-бутил-фенил)-4,5-дигидро-1H-имидазол

К раствору 4-бензилокси-3-трет-бутил-бензонитрила (1.4 г), полученному в промежуточном соединении A1, Стадия 2, в этилендиамине (20 мл) в закрытой пробирке добавили P2S5 (0.985 мг) при 25°C и реакционную смесь держали под давлением при перемешивании при 120°C в течение 2 ч. Реакционную смесь охладили до 25°C, и затем влили в воду (100 мл). Смесь перемешивали в течение 30 минут и получившийся преципитат собрали посредством фильтрации и высушили под вакуумом с получением соединения, указанного в заголовке (1.45 г), в виде серо-белого осадка. MS (m/z): 308.9 [M+H]+.
Стадия 2: 2-(4-Бензилокси-3-трет-бутил-фенил)-1H-имидазол

К раствору 2-(4-бензилокси-3-трет-бутил-фенил)-4,5-дигидро-1H-имидазола (1.4 г) в ДМСО (50 мл) добавили карбонат калия (690 мг) и диацетокси-йодобензол (1.61 г) и реакционную смесь перемешивали при 25°C в течение 16 ч в темноте. Смесь затем разбавили водой и экстрагировали дихлорметаном (2x40 мл). Объединенные органические слои высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом. Остаток очистили посредством колоночной хроматографии через аминосиликагель (градиент 33-40% EtOAc в гексанах) с получением соединения, указанного в заголовке (805 мг) в виде серо-белого осадка. MS (m/z): 306.8 [M+H]+.
Стадия 3: 2-(4-Бензилокси-3-трет-бутил-фенил)-1-(2-триметилсиланил-этоксиметил)-1H-имидазол
К суспензии NaH (16 мг) в безводном ДМФ (10 мл) при 0°C добавили раствор 2-(4-бензилокси-3-трет-бутил-фенил)-1H-имидазола (500 мг) в безводном ДМФ (5 мл) и реакционную смесь перемешивали при 25°C в течение 30 минут. Затем SEM-хлорид (0.087 мл) добавили по каплям при 25°C и реакционную смесь перемешивали при 25°C в течение 2 ч. Смесь затем погасили водой и экстрагировали EtOAc (2x50 мл). Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали под вакуумом. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 30-40% этилацетат в гексанах) с получением соединения, указанного в заголовке (310 мг) в виде вязкой бесцветной жидкости. MS (m/z): 436.9 [M]+.
Стадия 4: 2-трет-бутил-4-[1-(2-триметилсиланил-этоксиметил)-1H-имидазол-2-ил]-фенол

Раствор 2-(4-бензилокси-3-трет-бутил-фенил)-1-(2-триметилсиланил-этоксиметил)-1H-имидазола (300 мг) в MeOH (20 мл) продули аргоном в течение 10 мин и затем добавили Pd(OH)2 (100 мг). Ввели атмосферу водорода и реакционную смесь перемешивали под H2 при 25°C в течение 6 ч. Смесь отфильтровали и фильтрат эвапорировали под вакуумом с получением соединения, указанного в заголовке (200 мг), в виде серо-белого осадка. MS (m/z): 347.0 [M+H]+.
Промежуточное соединение A6
2-трет-бутил-4-(1-метилимидазол-2-ил)фенол

Стадия 1: 2-(4-Бензилокси-3-трет-бутил-фенил)-1-метил-1H-имидазол
Суспензию NaH (60% в минеральном масле, 31 мг) в безводном ТГФ (15 мл) охладили до 0°C и раствор 2-(4-бензилокси-3-трет-бутил-фенил)-1H-имидазола (200 мг), полученного в промежуточном соединении A5, Стадия 2, в ТГФ (20 мл) затем добавили при 0°C. Реакционную смесь перемешивали при 0°C в течение 30 мин и затем йодометан (0.049 мл) добавили при 0°C. Перемешивание продолжили при 0°C в течение 3ч. Смесь погасили водой и экстрагировали EtOAc (2x50 мл). Объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 60-70% EA в гексанах) с получением соединения, указанного в заголовке (175 мг) в виде вязкого осадка. MS (m/z): 320.8 [M+H]+.
Стадия 2: 2-трет-бутил-4-(1-метилимидазол-2-ил)фенол
Соединение, указанное в заголовке, получили из 2-(4-бензилокси-3-трет-бутил-фенил)-1-метил-1H-имидазола по аналогии с Промежуточным соединением 5, Стадия 4 в виде серо-белого осадка. MS (m/z): 230.7 [M+H]+.
Промежуточное соединение A7
2-трет-бутил-4-(1,3-оксазол-2-ил)фенол

Стадия 1: 2-(3-трет-бутил-4-метоксиметокси-фенил)-оксазол
Раствор 4-бромо-2-трет-бутил-1-метоксиметокси-бензола (500 мг, полученного из 4-бромо-2-трет-бутил-фенола в соответствии с WO2013079223) и 2-трибутилстаннанил-оксазол (0.92 мл) в безводном диоксане (10 мл) в закрытой пробирке продули аргоном в течение 10 мин. Затем PdCl2(dppf)2 CH2Cl2 комплекс (149.5 мг) добавили и реакционную смесь снова продули аргоном в течение 10 мин. Смесь затем перемешивали при 100°C в течение 4 ч и затем охладили до 25°C, отфильтровали и фильтрат эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 5-10% этилацетат в гексанах) с получением соединения, указанного в заголовке (350 мг) в виде светло-коричневой жидкости. MS (m/z): 262.8 [M+H]+.
Стадия 2: 2-трет-бутил-4-оксазол-2-ил-фенол
К раствору 2-(3-трет-бутил-4-метоксиметокси-фенил)-оксазола (200 мг) в безводном ДХМ (10 мл) при 0-5°C добавили 4 н HCl в диоксане (2.5 мл) и реакционную смесь перемешивали при 25°C в течение 28 ч. Растворитель эвапорировали при пониженном давлении и остаток очистили посредством колоночной хроматографии на силикагеле (градиент 8-15% этилацетат в гексанах) с получением соединения, указанного в заголовке (120 мг), в виде серо-белого осадка. MS (m/z): 217.9 [M+H]+.
Промежуточное соединение A8
2-трет-бутил-4-морфолин-4-илфенол
Стадия 1: 4-(3-трет-бутил-4-метоксиметокси-фенил)-морфолин
К раствору 4-бромо-2-трет-бутил-1-метоксиметокси-бензола (900 мг, полученный из 4-бромо-2-трет-бутил-фенола в соответствии с WO2013079223) и морфолина (0.44 мл) в безводном толуоле (10 мл) в закрытой пробирке добавили NaOtBu (475 мг) и ксантфос (76.2 мг) при 25°C. Пробирку продули аргоном в течение 10 мин и затем Pd2(dba)3 (60.3 мг) добавили и пробирку снова продули аргоном в течение 10 мин. Затем, смесь перемешивали под давлением в течение 16ч при 110°C и затем охладили до 25°C и отфильтровали. Фильтрат эвапорировали при пониженном давлении и получившийся неочищенный материал очистили посредством колоночной хроматографии (градиент 7-10% этилацетата в гексанах) с получением соединения, указанного в заголовке (560 мг) в виле вязкой жидкости.1H-NMR (400MHz, δ, CDCl3): 1.38 (s, 9H), 3.04-3.10 (m, 4H), 3.48 (s, 3H), 3.80-3.90 (m. 4H), 5.17 (s, 2H), 6.69 (dd, 1H), 6.93 (d, 1H), 7.04 (d, 1H).
Стадия 2: 2-трет-бутил-4-морфолин-4-ил-фенол

Соединение, указанное в заголовке, получили из 4-(3-трет-бутил-4-метоксиметокси-фенил)-морфолина с использованием условий, описанных для Промежуточного соединения A7, Стадия 2, в виде серо-белого осадка. MS (m/z): 235.9 [M+H]+.
Промежуточное соединение A9
2-трет-бутил-4-(1-метил-1H-имидазол-2-ил)-фенол

К раствору 4-бромо-2-трет-бутил-фенола (CAS: 10323-39-4, 200 мг) и 1-метил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-имидазола (272 мг) в безводном ДМФ (10 мл) добавили K2CO3 (362 мг) при 25°C в атмосфере азота. Реакционную смесь продули аргоном в течение 10 мин, и затем PdCl2(dppf)2 ДХМ комплекс (14.2 мг) добавили. Пробирку снова продули аргоном в течение 10 мин и реакционную смесь перемешивали при 120°C в течение 28 ч. Смесь охладили до 25°C, отфильтровали и фильтрат эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 2-5% MeOH в ДХМ) с получением соединения, указанного в заголовке (124 мг) в виде коричневого осадка. MS (m/z): 230.9 [M+H]+.
Промежуточное соединение A10
2-трет-бутил-4-(5-метил-1,3,4-оксадиазол-2-ил)фенол

Стадия 1: 3-трет-бутил-4-гидрокси-бензойной кислоты метиловый эфир

К раствору 3-трет-бутил-4-гидрокси-бензойной кислоты (CAS: 66737-88-0, 3 г) в безводном MeOH (40 мл) добавили по каплям конц. H2SO4 (0.4 мл) при 25°C и реакционную смесь перемешивали при кипении с обратным холодильником в течение 16 ч. Затем растворитель эвапорировали при пониженном давлении. Остаток разбавили с помощью ДХМ (50 мл) и раствор промыли водой и солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат эвапорировали при пониженном давлении и остаток очистили посредством колоночной хроматографии на силикагеле (градиент 0.5-1% MeOH в ДХМ) с получением соединения, указанного в заголовке (2.8 г), в виде серо-белого осадка. MS (m/z): 209.4 [M+H]+.
Стадия 2: 3-трет-бутил-4-гидрокси-бензойной кислоты гидразид

К раствору 3-трет-бутил-4-гидрокси-бензойной кислоты метилового эфира (3.7 г) в безводном MeOH (40 мл) в закрытой пробирке добавили гидратгидразин (4.36 мл) при 25°C и реакционную смесь нагревали под давлением при 65°C в течение 16 ч. Смесь затем охладили до 25°C и растворитель эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 2-4% MeOH в ДХМ) с получением соединения, указанного в заголовке (2.8 г, 75%), в виде серо-белого осадка. MS (m/z): 209.1 [M+H]+.
Стадия 3: 2-трет-бутил-4-(5-метил-1,3,4-оксадиазол-2-ил)фенол
Суспензию 3-трет-бутил-4-гидрокси-бензойной кислоты гидразида (1.5 г) в триэтилортоацетате (25 мл) нагревали до кипения при 150°C в течение 7 ч. Реакционную смесь затем охладили до 25°C и растворитель эвапорировали при пониженном давлении. Остаток разбавили с помощью ДХМ (50 мл) и промыли водой и солевым раствором. Органический слой высушили над Na2SO4, отфильтровали, и эвапорировали при пониженном давлении. Неочищенное вещество очистили посредством колоночной хроматографии на силикагеле (градиент 2-3% MeOH в ДХМ) с получением соединения, указанного в заголовке (1.15 г), в виде серо-белого осадка. MS: 233.1 [M+H]+.
Промежуточное соединение A14:
4-хлор-2-циклопропил-5-метилсульфонилфенол

Стадия 1: 1-бромо-2-хлор-5-фтор-4-нитробензол
К перемешанному раствору 2-бромо-1-хлор-4-фторбензола (CAS: 201849-15-2; 10 г) в серной кислоте (95-97%; 100 мл) постепенно добавили нитрат калия (5.82 г) при -5°C. Реакционную смесь перемешивали при -5°C в течение 1.5 часов и затем влили в 400 мл воды со льдом. Преципитат отфильтровали и высушили под вакуумом с получением соединения, указанного в заголовке, в виде бесцветного осадка (11.54 г). MS (m/z): 253 [M]+.
Стадия 2: 1-бромо-2-хлор-5-метокси-4-нитробензол

К раствору 1-бромо-2-хлор-5-фтор-4-нитробензола (11.0 г) в метаноле (110 мл) медленно добавили метоксид натрия (2.45 г) при 0°C и реакционную смесь перемешивали при 0°C в течение 1.5 часов. Суспензию влили в воду и экстрагировали с помощью EtOAc. Объединенные органические экстракты высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом с получением соединения, указанного в заголовке, в виде желтого осадка (11.2 г). MS (m/z): 265 [M]+.
Стадия 3: 1-хлор-4-метокси-2-метилсульфонил-5-нитробензол

К раствору L-пролина (61.3 мг) в ДМСО (2 мл) добавили гидроксид натрия (24.0 мг) и смесь перемешивали при комнатной температуре в течение 30 минут. Йодид меди (I) (114 мг), 1-бромо-2-хлор-5-метокси-4-нитробензол (0.20 г) и метансульфинат натрия (173 мг) добавили. Реакционную смесь нагрели до 60°C с получением мутного голубого раствора. Через 5 часов реакционной смеси дали остыть до комнатной температуры. Смесь влили в 50 мл 10% водного раствора NaHCO3 и 50 мл EtOAc и слои отфильтровали для удаления примесей. Фильтрованные слои затем разделили. Водный слой экстрагировали дважды с помощью 50 мл EtOAc и органические слои промыли 50 мл солевого раствора, высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом с получением соединения, указанного в заголовке, в виде желтого осадка (122 мг). MS (m/z): 265 [M]+.
Стадия 4: 5-хлор-2-метокси-4-метилсульфониланилин
К смеси этанола (2.00 мл) и воды (2.00 мл) добавили уксусную кислоту (140 мкл) и эту смесь перемешивали при кипении с обратным холодильником в течение 10 минут. К раствору, полученному таким образом, добавили железо (163 мг) и 1-хлор-4-метокси-2-метилсульфонил-5-нитробензол (200 мг) и смесь перемешивали в течение дополнительных 20 минут. После охлаждения, 20 мл ацетона добавили и железо отфильтровали через слои дикалита и промыли 20 мл ацетона. Фильтрат сконцентрировали под вакуумом и остаток очистили с помощью хроматографии на силикагеле, элюируя при градиенте н-гептана в этилацетате (100/ 0 до 50/50). Это дало соединение, указанное в заголовке, в виде белых кристаллов (177 мг). MS (m/z): 235 [M]+.
Стадия 5: 1-хлор-5-йод-4-метокси-2-метилсульфонилбензол

К суспензии 5-хлор-2-метокси-4-метилсульфониланилина (1.50 г) в полуконцентрированной HCl (18.5%, 15.1 г) добавили по каплям при 0-5°C раствор нитрита натрия (461 мг) в воде (3.66 мл). Первоначальную суспензию постепенно очистили и вернули в раствор. Эту смесь перемешивали в течение 1 часа при 0°C и затем добавили по каплям к перемешанной суспензии йодида калия (3.17 г) в водн. HBr (48%, 21.0 мл) при комнатной температуре. Перемешивание продолжили в течение 30 минут при комнатной температуре. Реакционную смесь влили в 100 мл 2M водного раствора Na2CO3 и 50 мл EtOAc и слои разделили. Водный слой экстрагировали дважды с помощью 100 мл EtOAc. Органические слои промыли Na2S2O3 и солевым раствором (50 мл), высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. Остаток очистили с помощью хроматографии на силикагеле, с элюцией при градиенте н-гептана в этилацетате (100/ 0 до 50/ 50) с получением соединения, указанного в заголовке в виде светло-желтого осадка (950 мг). MS (m/z): 346 [M]+.
Стадия 6: 4-хлор-2-йод-5-метилсульфонилфенол

К раствору 1-хлор-5-йод-4-метокси-2-метилсульфонилбензола (750 мг) в AcOH (15.6 г) добавили водный HBr (48%, 4.7 мл) и прозрачный бесцветный раствор перемешивали при кипении с обратным холодильником в течение 4 дней в закрытой пробирке. Реакционную смесь охладили, влили в 100 мл воды и 100 мл EtOAc и слои разделили. Водный слой экстрагировали дважды с помощью 100 мл EtOAc. Органические слои промыли 100 мл солевого раствора, высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. Соединение очистили с помощью хроматографии на силикагеле с элюцией при градиенте н-гептана в этилацетате (100/0 до 60/40) с получением соединения, указанного в заголовке, в виде светло-желтого осадка (625 мг. MS (m/z): 330.87 [M-H]-.
Стадия 7: 4-хлор-2-циклопропил-5-метилсульфонилфенол
К суспензии 4-хлор-2-йод-5-метилсульфонилфенола (200 мг) в толуоле (3 мл) добавили в атмосфере аргона циклопропилтрифторборат калия (178 мг), воды (0.21 мл), карбонат цезия (490 мг), ацетат палладия (II) (6.75 мг и бутилди-1-адамантилфосфин (21.6 мг) и смесь перемешивали в закрытой пробирке при 125°C в течение 66 часов. Реакционную смесь охладили и влили в насыщенный водный раствор NH4Cl и этилацетат и слои разделили. Водный слой экстрагировали дважды с помощью дополнительного количества этилацетата. Органические слои высушили над MgSO4, отфильтровали, обработали силикагелем и эвапорировали. Соединение очистили с помощью хроматографии на силикагеле с элюцией при градиенте н-гептана в этилацетате (100/ 0 до 50/50) с получением соединения, указанного в заголовке, в виде бесцветного осадка (72 мг). MS (m/z): 245 [M]+.
Промежуточное соединение A15:
2-трет-бутил-4-метилсульфонилфенол

Стадия 1: 4-бромо-2-трет-бутил-1-фенилметоксибензол

К раствору 4-бромо-2-трет-бутилфенола (CAS: 10323-39-4; 500 мг) в ДМФ (5 мл) добавили NaH (114 мг) и реакционную смесь перемешивали в течение 10 минут при комнатной температуре. Затем бензилхлорид (290 мг) добавили и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Смесь влили в 30 мл 10% водного раствора NH4Cl и 30 мл EtOAc и слои разделили. Водный слой экстрагировали дважды с помощью 30 мл EtOAc. Органические слои промыли 30 мл солевого раствора, высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. Остаток очистили с помощью хроматографии на силикагеле с элюцией при градиенте н-гептана в этилацетате (100/0 до 80/20) с получением соединения, указанного в заголовке, в виде бесцветного осадка (583 мг. MS (m/z): 318 [M]+.
Стадия 2: 2-трет-бутил-4-метилсульфонил-1-фенилметоксибензол
К раствору L-пролина (88.3 мг) в ДМСО (10 мл) добавили гидроксид натрия (34.6 мг) и смесь перемешивали при комнатной температуре в течение 30 минут. Йодид меди (I) (165 мг), 4-бромо-2-трет-бутил-1-фенилметоксибензол (345 мг) и метансульфинат натрия (249 мг) добавили. Реакционную смесь нагрели до 60°C с получением мутного голубого раствора и затем реакционную смесь перемешивали в течение 2 часов при 60°C. По данным ТСХ и ЖХ-МС конверсии не произошло. Снова добавили метансульфинат натрия (249 мг) и реакционную смесь перемешивали в течение 2 часов при 60°C. По данным ТСХ и ЖХ-МС конверсии снова не произошло. Реакционную смесь переместили в закрытую пробирку и перемешивали при 135°C в течение ночи. ТСХ и ЖХ-МС показали полную конверсию и ЖХ-МС показала присутствие вещества с требуемой молекулярной массой. Реакционную смесь влили в 50 мл 10% водного раствора NH4Cl и 50 мл EtOAc и слои разделили. Водный слой экстрагировали дважды с помощью 50 мл EtOAc. Органические слои промыли солевым раствором (50 мл), высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. Соединение очистили с помощью хроматографии на силикагеле с элюцией при градиенте н-гептана в этилацетате (100/0 до 60/40) с получением соединения, указанного в заголовке, в виде бесцветного осадка (278 мг). MS (m/z): 317.12 [M-H]-.
Стадия 3: 2-трет-бутил-4-метилсульфонилфенол
К раствору 2-трет-бутил-4-метилсульфонил-1-фенилметоксибензола (250 мг) в MeOH (3 мл) и этилацетате (3 мл) добавили Pd на угле (10% Pd, 25 мг в атмосфере аргона. Реакционную смесь откачали и заполнили водородом. Реакционную смесь перемешивали в течение 18 часов при 1.7 бар в атмосфере H2. Реакционную смесь затем отфильтровали через фильтр (дикалит) и фильтрат сконцентрировали под вакуумом. Остаток очистили с помощью хроматографии на силикагеле с элюцией при градиенте н-гептана в этилацетате (100/0 до 50/50) с получением соединения, указанного в заголовке в виде бесцветного осадка (140 мг). MS (m/z): 227.08 [M-H]-.
Промежуточное соединение A16:
5-трет-бутил-4-гидрокси-2-метилбензонитрил

К раствору 4-бромо-2-трет-бутил-5-метилфенола (CAS: 51345-97-2, 0.45 г), полученного из 4-бромо-3-метилфенола в соответствии с WO2005058798 или WO2008059026, в ДМФ (4 мл) в атмосфере аргона добавили воду (40 мкл), 1,1'-бис(дифенилфосфино)ферроцен (30.8 мг), цианид цинка (120 мг), цинк (4.84 мг), ацетат цинка (13.6 мг) и трис(дибензилиденацетон)-дипалладий (0) (16.9 мг). Реакционную смесь закрыли и нагревали в микроволновой печи в течение 30 минут при 180°C. Реакционную смесь затем влили в насыщенный водный раствор NH4Cl, содержащий небольшой объем воды и этилацетат, и слои разделили. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои высушили над MgSO4, отфильтровали, обработали силикагелем и эвапорировали. Соединение очистили с помощью хроматографии на силикагеле с элюцией при градиенте н-гептана в этилацетате (100/0 до 75/25) с получением соединения, указанного в заголовке (160 мг), в виде светло-коричневого осадка. MS (m/z): 188.11 [M-H]-.
Промежуточное соединение A17:
4-трет-бутил-5-гидрокси-2-метилбензонитрил
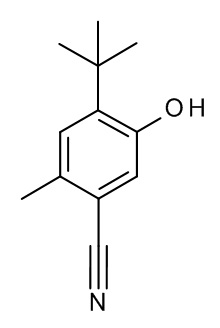
Этот материал получили по аналогии с Промежуточным соединением A16 из 5-бромо-2-трет-бутил-4-метилфенола (CAS 1237614-78-6), полученного из 3-бромо-4-метилфенола в соответствии с WO2005058798 или WO2008059026), в виде светло-желтого осадка. MS (m/z): 188.11 [M-H]-.
Промежуточное соединение A18:
2-трет-бутил-4-[3-(2-триметилсиланил-этоксиметил)-3H-имидазол-4-ил]-фенол
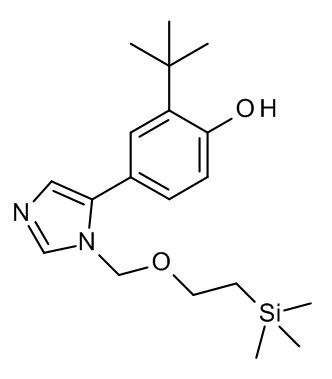
Стадия 1: 1-(4-Бензилокси-3-трет-бутил-фенил)-этанон

К раствору 1-бензилокси-4-бромо-2-трет-бутил-бензола (промежуточное соединение A15, Стадия 1, 5.0 г) в ДМФ (40 мл) и воды (4 мл) добавили n-бутилвиниловый эфир (8.1 мл), 1.3-бис(дифенилфосфино)пропан (1.61 г), K2CO3(2.60 г) и Pd(OAc)2 (351 мг) при 25°C. Затем смесь продули аргоном и перемешивали при 150°C в течение 5 ч. Смесь охладили до 25°C, разбавили с помощью 2 н HCl (25 мл) и перемешивали при 25°C в течение 2 ч. Смесь экстрагировали EtOAc (2x100 мл) и объединенные органические слои промыли насыщенным водным раствором NaHCO3. Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (5% этилацетат в гексанах) с получением соединения, указанного в заголовке (1.6 г) в виде коричневой жидкости. MS (m/z): 283.2 [M+H]+.
Стадия 2: 1-(4-Бензилокси-3-трет-бутил-фенил)-2-бромо-этанон

К раствору 1-(4-бензилокси-3-трет-бутил-фенил)-этанона (1.0 г) в безводном ТГФ (20 мл) и MeOH (10 мл) добавили раствор Bu4NBr3 (1.71 г) в ТГФ (10 мл) при 25°C и реакционную смесь перемешивали при 50°C в течение 5 ч. Смесь охладили до 25°C и растворитель эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (5% этилацетат в гексане) с получением соединения, указанного в заголовке (800 мг) в виде бесцветной жидкости, которую использовали без дополнительной характеризации.
Стадия 3: 5-(4-Бензилокси-3-трет-бутил-фенил)-1H-имидазол

Раствор 1-(4-бензилокси-3-трет-бутил-фенил)-2-бромо-этанона (800 мг) в формамиде (30 мл) перемешивали при 160-170°C в течение 4 ч. Реакционную смесь затем охладили до 25°C и разбавили с помощьюм EtOAc (30 мл) и воды (30 мл). Органический слой разделили и водный слой экстрагировали с дополнительным количеством EtOAc (2x30 мл). Объединенные органические слои высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через аминосиликагель (градиент 5-10% MeOH в ДХМ) с получением соединения, указанного в заголовке (330 мг) в виде светло-желтого осадка. MS (m/z): 306.9 [M+H]+.
Стадия 4: 5-(4-Бензилокси-3-трет-бутил-фенил)-1-(2-триметилсиланил-этоксиметил)-1H-имидазол

К суспензии NaH (51 мг) в безводном ТГФ (20 мл) добавили по каплям раствор 5-(4-бензилокси-3-трет-бутил-фенил)-1H-имидазола (330 мг) в безводном ТГФ (10 мл) при 25°C. Реакционную смесь перемешивали при 25°C в течение 30 мин и затем (2-хлорметьокси-этил)-триметил-силан (0.287 мл) добавили по каплям при 25°C. Реакционную смесь перемешивали при 25°C в течение 3 ч, затем погасили водой и экстрагировали с помощью EtOAc (2x50 мл). Объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (40% этилацетат в гексанах) с получением соединения, указанного в заголовке (310 мг), в виде серо-белого осадка. MS (m/z): 436.9 [M+H]+.
Стадия 5: 2-трет-бутил-4-[3-(2-триметилсиланил-этоксиметил)-3H-имидазол-4-ил]-фенол
К раствору 5-(4-бензилокси-3-трет-бутил-фенил)-1-(2-триметилсиланил-этоксиметил)-1H-имидазола (170 мг) в MeOH (20 мл), который продули аргоном в течение 10 мин, затем Pd(OH)2 (100 мг) добавили при 25°C. Реакционную смесь перемешивали при 25°C в течение 4 ч в атмосфере водорода. Смесь затем отфильтровали и фильтрат эвапорировали при пониженном давлении с получением соединения, указанного в заголовке (170 мг), в виде серо-белого осадка. MS (m/z): 347.0 [M+H]+.
Промежуточное соединение A19
4-хлор-2-циклопропил-5-метилфенол
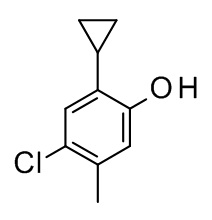
Стадия 1: 5-хлор-2-метокси-4-метил-фениламин

К раствору 1-хлор-4-метокси-2-метил-5-нитро-бензола (CAS: 101080-03-9, 10.64 г) в MeOH (250 мл) и воды (125 мл) добавили Zn пыль (24.15 г) и NH4Cl (31.05 г) при 25 °C и реакционную смесь перемешивали в течение 2 ч при 25 °C. Смесь отфильтровали через слой целита и фильтрат эвапорировали, разбавили с помощью ДХМ (300 мл) и промыли водой (2x200 мл). Органический слой высушили над Na2SO4, отфильтровали и эвапорировали с получением соединения, указанного в заголовке (8.99 г), в виде светло-коричневого осадка.1H NMR (400 MHz, CDCl3): 2.25 (3H, s), 3.80 (3H, s), 6.60 (1H, s), 6.68 (1H, s).
Стадия 2: 1-хлор-5-йод-4-метокси-2-метил-бензол
К суспензии 5-хлор-2-метокси-4-метил-фениламина (8.98 г) в конц. HCl (37%, 84 мл) добавили раствор NaNO2 (7.22 г) в воде (70 мл) at 0 °C. После перемешивания реакционную смесь в течение 30 мин при 0 °C, раствор KI (34.74 г) в воде (176 мл) добавили и реакционную смесь затем перемешивали при 25 °C в течение 16 ч. Реакционную смесь разбавили этилацетатом (250 мл) и органический слой разделили, промыли водой (100 мл), насыщенным раствором тиосульфата натрия (100 мл) и солевым раствором (50 мл). Органический слой высушили над Na2SO4, отфильтровали и эвапорировали с получением неочищенного материала, который очистили с помощью флеш-хроматографии через силикагель (гексаны) с получением соединения, указанного в заголовке (11.9 г), в виде желтого осадка.1H NMR (400 MHz, CDCl3) δ: 2.32 (3H, s), 3.84 (3H, s), 6.65 (1H, s), 7.68 (1H, s).
Стадия 3: 4-хлор-2-йод-5-метил-фенол

К перемешанному раствору 1-хлор-5-йод-4-метокси-2-метил-бензола (3.13 г) в безводном ДХМ (20 мл) при 0 °C добавили раствор 1M трехбромистого бора в ДХМ (44.4 мл) и получившийся раствор перемешивали в течение 2 ч при 25 °C. Реакционную смесь погасили насыщенным водным раствором бикарбоната натрия и разбавили с помощью ДХМ (20 мл). Органический слой разделили и промыли водой (25 мл) и солевым раствором (25 мл). Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили с помощью флеш-хроматографии на силикагеле (градиент 5-10% этилацетата в гексанах) с получением соединения, указанного в заголовке, в виде коричневого осадка (2.89 г).1H NMR (400 MHz, CDCl3) δ: 2.30 (3H, s), 5.12 (1H, s), 6.86 (1H, s), 7.57 (1H, s).
Стадия 4: 4-хлор-2-циклопропил-5-метил-фенол

К раствору 4-хлор-2-йод-5-метил-фенола (500 мг) в толуоле (8.5 мл) и воде (0.5 мл) добавили циклопропилбороновую кислоту (416 мг), K3PO4 (1.31 г), Pd(OAc)2 (33 мг) и трициклогексилфосфин (94 мг) и реакционную смесь нагревали при 100 °C в течение 16 ч. Смесь отфильтровали через целит и фильтрат промыли солевым раствором. Органический слой высушили над Na2SO4 и выпарили под вакуумом. Остаток очистили с помощью флеш-хроматографии на силикагеле (градиент 2-5% этилацетат в гексанах) с получением соединения, указанного в заголовке (210 мг) в виде коричневой жидкости.1H NMR (400 MHz, CDCl3): 0.59-0.62 (2H, m), 0.92-0.96 (2H, m), 1.69-1.76 (1H, m), 5.29 (1H, s), 6.86 (1H, s), 7.57 (1H, s).
Промежуточные соединения A19A и A19B
Следующие промежуточные соединения были синтезированы из 4-хлор-2-йод-5-метил-фенола и подходящих структурных элементов по аналогии с Промежуточным соединением A19, Стадия 4:

Промежуточное соединение A20:
4-хлор-2-циклогексил-5-метил-фенол

К раствору 4-хлор-2-циклогекс-1-енил-5-метил-фенола (промежуточное соединение A19A, 202 мг) в метаноле (10 мл) добавили Pd на угле (10% Pd, 25 мг) и реакционную смесь перемешивали при 25 °C в атмосфере водорода в течение 45 мин. Раствор профильтровали целит и фильтрат эвапорировали при пониженном давлении с получением соединения, указанного в заголовке (190 мг) в виде бесцветной жидкости. Rf= 0.3 (10% этилацетат в гексане)
Следующее промежуточное соединение синтезировали из соответствующего предшественника по аналогии с Промежуточным соединением A20:
Промежуточное соединение A22
2-хлор-4-циклопропил-5-гидрокси-бензонитрил

Стадия 1: 5-амино-2-бромо-4-хлор-фенол

К раствору 4-бромо-2-хлор-5-метокси-фениламина (CAS: 98446-54-9, 2.05 г) в безводном ДХМ (40 мл) при 0 °C добавили раствор BBr3 в ДХМ (1M, 86.7 мл) и реакционную смесь перемешивали при 25 °C в течение 12 ч. Смесь погасили насыщенным водным раствором NaHCO3 (40 мл) и экстрагировали с помощью ДХМ (2x60 мл). Объединенные органические слои высушили над Na2SO4 и эвапорировали при пониженном давлении с получением соединения, указанного в заголовке (1.9 г), в виде коричневого осадка.1H NMR (400 MHz, ДМСО-D6): 5.36 (2H, s), 6.41 (1H, s), 7.21 (1H, s), 9.94 (1H, s).
Стадия 2: Карбоновой кислоты 2-бромо-5-(ди-трет-бутоксикарбонил)амино-4-хлор-фенилового эфира трет-бутиловый эфир

К перемешанному раствору 5-амино-2-бромо-4-хлор-фенола (1.9 г) в безводном ТГФ (40 мл) при 25 °C добавили ди-трет-бутил-дикарбонат (“Boc ангидрид”, 10.96 мл), Et3N (7.12 мл) и DMAP (10 мг) и реакционную смесь кипятили с обратным холодильником в течение 12 ч. Растворитель эвапорировали при пониженном давлении и остаток очистили с помощью флеш-хроматографии на силикагеле (градиент 2-5% этилацетат / гексаны) с получением требуемого соединения (3.82 г) в виде белого осадка. MS (m/z): 522.1 [M]+.
Стадия 3: Карбоновой кислоты 5-(ди-трет-бутоксикарбонил)амино-4-хлор-2-циклопропил-фенилового эфира трет-бутиловый эфир

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением A19, Стадия 4, из карбоновой кислоты 2-бромо-5-(ди-трет-бутоксикарбонил)амино-4-хлор-фенилового эфира трет-бутилового эфира и циклопропилбороновой кислоты в виде бесцветной вязкой жидкости. MS (m/z): 484.0 [M]+.
Стадия 4: 5-амино-4-хлор-2-циклопропил-фенол
К раствору карбоновой кислоты 5-(ди-трет-бутоксикарбонил)амино-4-хлор-2-циклопропил-фенилового эфира трет-бутилового эфира (1.05 г) в ДХМ (20 мл) добавили TFA (6.45 мл) и реакционную смесь перемешивали при 25 °C в течение 2 ч. Растворитель эвапорировали при пониженном давлении и остаток разбавили с помощью ДХМ (50 мл). Раствор промыли насыщенным водным раствором NaHCO3 (30 мл) и водой (20 мл). Органический слой высушили над безводным Na2SO4, отфильтровали и эвапорировали с получением соединения, указанного в заголовке (397 мг) в виде коричневого осадка.1H NMR (400 MHz, ДМСО-D6) δ: 0.42-0.46 (2H, m), 0.70-0.74 (2H, m), 1.80-1.87 (1H, m), 4.94 (2H, s), 6.26 (1H, s), 6.51 (1H, s), 9.11 (1H, s).
Стадия 5: 4-хлор-2-циклопропил-5-йод-фенол

К суспензии 5-амино-4-хлор-2-циклопропил-фенола (395 мг в воде (1 мл) при 0 °C медленно добавили конц. H2SO4(1.29 мл), с последующим раствором NaNO2 (148 мг) в воде (5.5 мл) при 0 °C. После перемешивания реакционной смеси в течение дополнительных 10 минут, раствор KI (714 мг) в воде (1 мл) добавили и реакционную смесь нагревали при 60 °C в течение 2 ч. Смесь охладили до 25 °C и экстрагировали с помощью ДХМ (2x30 мл). Объединенные органические экстракты высушили над Na2SO4, отфильтровали и эвапорировали. Остаток очистили с помощью флеш-хроматографии на силикагеле (градиент 2-5% этилацетат в гексанах) с получением соединения, указанного в заголовке (241 мг), в виде желтой жидкости. MS (m/z): 292.8 [M-H]-.
Стадия 6: 2-хлор-4-циклопропил-5-гидрокси-бензонитрил

К раствору 4-хлор-2-циклопропил-5-йод-фенола (238 мг) в безводном ДМФ (5 мл) добавили CuCN (145 мг) и реакционную смесь нагревали при 100 °C в течение 24 ч. Растворитель эвапорировали под вакуумом и остаток очистили с помощью флеш-хроматографии на силикагеле ( градиент 5-10% этилацетат в гексанах) с получением соединения, указанного в заголовке (120 мг), в виде желтого осадка.1H NMR (400 MHz, CDCl3): 0.68-0.72 (2H, m), 1.06-1.10 (2H, m), 1.86-1.89 (1H, m), 5.64 (1H, s), 7.09 (1H, s), 7.10 (1H, s).
Промежуточное соединение A23
N-(2-хлор-4-циклопропил-5-гидрокси-фенил)-метансульфонамид

К раствору 5-амино-4-хлор-2-циклопропил-фенола (78 мг), полученному в промежуточном соединении A22, Стадия 4, в ДХМ (5 мл) добавили пиридин (0.04 мл) и метансульфонилхлорид (0.03 мл) при 0 °C, и реакционную смесь затем перемешивали при 25 °C в течение 12 ч. Смесь погасили 6 н водным раствором NaOH (5 мл) и водой (10 мл). Органический слой разделили и водную фазу экстрагировали дополнительным количеством ДХМ (20 мл). Водный слой охладили до 0°C, подкислили с помощью конц. HCl и экстрагировали этилацетатом (2x20 мл). Объединенные органические экстракты этилацетата промыли солевым раствором, высушили над Na2SO4, и сконцентрировали при пониженном давлении. Остаток очистили с помощью флеш-хроматографии на силикагеле (градиент 5-20% этилацетат в гексанах) с получением соединения, указанного в заголовке (58 мг), в виде коричневого осадка.1H NMR (400 MHz, ДМСО-D6): 0.62-0.65 (2H, m), 0.84-0.89 (2H, m), 1.97-2.05 (1H, m), 2.95 (3H, s), 6.80 (1H, s), 6.91 (1H, s), 9.19 (1H, s), 9.80 (1H, s).
Промежуточное соединение A24:
4-трет-бутил-3-гидрокси-бензонитрил
Стадия 1: 4-трет-бутил-3-метокси-бензамид

Раствор 4-трет-бутил-3-метокси-бензойной кислоты (CAS: 79822-46-1: 2 г) в SOCl2 (5 мл) кипятили с обратным холодильником в течение 2 ч. Избыток SOCl2 эвапорировали под вакуумом и остаток растворили в ТГФ (5 мл) и конц. водный раствор NH3 (2 мл) добавили по каплям при 0 °C. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем экстрагировали этилацетатом (2x20 мл). Объединенные органические экстракты высушили над Na2SO4 и эвапорировали при пониженном давлении с получением соединения, указанного в заголовке (1.95 г), в виде белого осадка.1H NMR (400 MHz, CDCl3): 1.36 (9H, s), 3.88 (3H, s), 6.0 (2H, bs), 7.20 (1H, dd, J= 4 Hz, 12 Hz), 7.30 (1H, d, J= 12 Hz), 7.42 (1H, d, J = 4Hz).
Стадия 2: 4-трет-бутил-3-метокси-бензонитрил
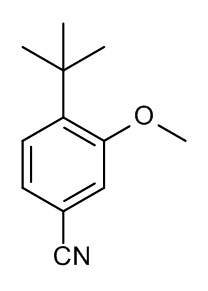
Раствор 4-трет-бутил-3-метокси-бензамида (500 мг, 2.41 ммоль) в POCl3 (5 мл) кипятили с обратным холодильником в течение 6 ч. Избыток POCl3 эвапорировали под вакуумом и остаток растворили в этилацетате (30 мл) и промыли насыщенным водным раствором NaHCO3 (25 мл). Органический слой высушили над Na2SO4, отфильтровали, и затем эвапорировали при пониженном давлении. Неочищенное вещество очистили с помощью флеш-хроматографии через силикагель (5% этилацетат в гексане) с получением соединения, указанного в заголовке (360 мг), в виде бесцветной жидкости. MS (M/z): 189.0 [M]+.
Стадия 3: 4-трет-бутил-3-гидрокси-бензонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением A22, Стадия 1, из 4-трет-бутил-3-метокси-бензонитрила посредством обработки BBr3, в виде желтого масла.1H NMR (400 MHz, CDCl3): 1.39 (9H, s), 5.41 (1H, s), 6.93 (1H, d, J = 4Hz), 7.17 (1H, dd, J= 4Hz, 12 Hz), 7.33 (1H, d, J = 8 Hz).
Следующее Промежуточное соединение синтезировали из соответствующего предшественника по аналогии с Промежуточным соединением A24, Стадия 3:

Промежуточное соединение A30:
6-циклопропил-2-метилбензо[d]тиазол-5-ол

Стадия 1: 6-йодо-5-метокси-2-метилбензо[d]тиазол

Раствор 5-метокси-2-метилбензо[d]тиазол-6-амина (CAS: 89976-71-6; 1.0 г) в воде (2.6 мл) и конц. соляной кислоте (2.54 мл) охладили до 0°C. Затем раствор нитрита натрия (373 мг) в воде (2.6 мл) добавили по каплям в течение 3 минут. Коричневый раствор перемешивали при 0°C в течение 5 минут и затем добавили по каплям к энергично перемешанной суспензии йодида калия (2.56 г) в растворе HBr (48% в воде, 12.2 мл) в течение 5 минут при комнатной температуре. Перемешивание при комнатной температуре продолжили в течение 20 минут. Реакционную смесь влили в 2M водный раствор Na2CO3 (60 мл) и этилацетат (50 мл) и слои разделили. Водный слой экстрагировали дважды с помощью этилацетат (30 мл, каждый). Органические слои промыли один раз водным раствором Na2S2O3 (50 мл) и один раз солевым раствором (20 мл), высушили над MgSO4, отфильтровали, обработали силикагелем и эвапорировали. Соединение очистили дважды с помощью хроматографии на силикагеле на 50 г колонке с использованием системы ЖХСД с элюцией при градиенте н-гептан/этилацетата (100/0 до 50/50) с получением соединения, указанного в заголовке, в виде слегка загрязненного светло-коричневого осадка (0.66 г). MS (m/z): 306.4 [M+H]+.
Стадия 2: 6-йодо-2-метилбензо[d]тиазол-5-ол
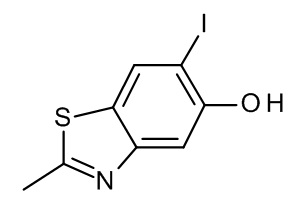
К раствору 6-йод-5-метокси-2-метилбензо[d]тиазола (0.625 г) в дихлорметане (4 мл) в закрытой пробирке в атмосфере аргона при комнатной температуре добавили по каплям трибромборан (1M в ДХМ; 2.25 мл) и светло-коричневую суспензию перемешивали при кипении с обратным холодильником (масляная баня при 60°C) в течение 1.5 часов. После не было обнаружено какого-либо количества продукта. Дополнительное количество ДХМ (4.00 мл) и трибромборана (1M в дихлорметане; 2.25 мл) добавили через 3 часа и перемешивание продолжили при 60°C в течение выходных. Реакционную смесь влили в воду и ДХМ (содержащим небольшое количество метанола) и слои разделили. Водный слой экстрагировали дважды с помощью ДХМ. Органические слои промыли один раз солевым раствором, высушили над MgSO4, отфильтровали и эвапорировали. Остаток растворили в этилацетате и дихлорметане и частично эвапорировали до образования суспензии. Эту суспензию отфильтровали, промыли небольшим количеством этилацетата и высушили под вакуумом с получением соединения, указанного в заголовке, в виде светло-коричневого осадка (455 мг. MS (m/z): 292.3 [M+H]+.
Стадия 3: 6-циклопропил-2-метил-1,3-бензотиазол-5-ол
К суспензии 6-йод-2-метилбензо[d]тиазол-5-ола (0.15 г) в толуоле (3 мл) добавили в атмосфере аргона циклопропилтрифторборат калия (152 мг), воду (0.21 мл), карбонат цезия (420 мг), ацетат палладия (II) (5.78 мг) и бутилди-1-адамантилфосфин (18.5 мг) и смесь перемешивали в закрытой пробирке при 125°C в течение 66 часов. Реакционную смесь влили в насыщенный водный раствор NH4Cl и этилацетат, и слои разделили. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои объединили, высушили над MgSO4, отфильтровали, обработали силикагелем и эвапорировали. Соединение очистили с помощью хроматографии на силикагеле на 10 ш колонке с использованием системы ЖХСД с элюцией при градиенте н-гептана в этилацетате (100/0 до 0/100) с получением соединения, указанного в заголовке, в виде светло-коричневого осадка. MS (m/z): 206.06 [M+H]+.
ПО данным ЖХ-МС, полученное вещество содержало некоторое количество 2-метилбензо[d]тиазол-5-ола в виде примеси.
Промежуточное соединение A31
4-хлор-2-циклобутил-5-метил-фенол

Стадия 1: 1-(5-хлор-2-метокси-4-метил-фенил)-циклобутанол
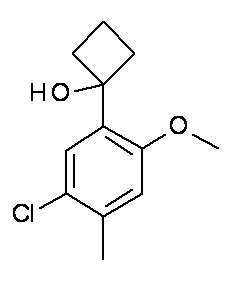
К раствору 1-хлор-5-йод-4-метокси-2-метил-бензола (1 г), Промежуточное соединение A19, Стадия 2, в безводном ТГФ (20 мл) добавили по каплям изопропилмагния хлорид (2M в ТГФ, 2.12 мл) при -40 °C в атмосфере азота и реакционную смесь перемешивали при -40 °C в течение 1 ч. Затем раствор циклобутанона (298 мг) в безводном ТГФ (3 мл) добавили по каплям в реакционную смесь при -40 °C и перемешивание продолжили при 25 °C в течение 11 ч. Реакционную смесь погасили насыщенным водным раствором NH4Cl и экстрагировали этилацетатом (2x30 мл). Объединенные органические слои высушили над Na2SO4, отфильтровали, и эвапорировали при пониженном давлении. Остаток очистили с помощью флеш-хроматографии на силикагеле (градиент 5-15% этилацетат в гексанах) с получением соединения, указанного в заголовке (431 мг), в виде бесцветной жидкости.1H NMR (400 MHz, CDCl3): 1.55-1.66 (1H, m), 1.96-2.06 (1H, m), 2.31-2.41 (5H, m) , 2.43-2.48 (2H, m) 3.48 (1H, s), 3.85 (3H, s), 6.75 (1H, s), 7.23 (1H, s).
Стадия 2: 1-хлор-5-циклобутил-4-метокси-2-метил-бензол

К смеси 1-(5-хлор-2-метокси-4-метил-фенил)-циклобутанола (428 мг) и триэтилсилана (0.362 мл) в безводном ДХМ (20 мл) при -78 °C в атмосфере аргона добавили по каплям эфират трехфтористого бора (0.237 мл) и реакционной смеси дали нагреться -40 °C в течение 3 часов. Перемешивание продолжили при -40 °C в течение дополнительных 2 ч. Реакционную смесь затем влили в 10% водный раствор KHCO3 (30 мл) и экстрагировали с помощью ДХМ (3x30 мл). Объединенные органические слои высушили (Na2SO4), отфильтровали, и эвапорировали. Остаток очистили посредством колоночной хроматографии на силикагеле (градиент 0-5% этилацетат в гексанах) с получением соединения, указанного в заголовке (370 мг), в виде бесцветной жидкости.1H NMR (400 MHz, CDCl3): 1.78-1.81 (1H, m), 1.91-2.06 (3H, m), 2.24-2.32 (5H, m) , 3.62-3.75 (1H, m), 3.38 (3H, s), 6.63 (1H, s), 7.12 (1H, s).
Стадия 3: 4-хлор-2-циклобутил-5-метил-фенол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением A19, Стадия 3, из 4-хлор-2-циклобутил-5-метил-фенола посредством обработки BBr3 в ДХМ в виде коричневого осадка (327 мг).1H NMR (400 MHz, CDCl3): 1.82-1.89 (1H, m), 1.98-2.16 (3H, m), 2.27 (3H, s), 2.31-2.42 (2H, m) 3.53-3.59 (1H, m), 4.50 (1H, s), 6.61 (1H, s), 7.09 (1H, s).
Промежуточное соединение A34
4-(трет-бутил)-2-хлор-5-гидроксибензонитрил

Стадия 1: 5-бромо-2-(трет-бутил)-4-хлорфенол

К раствору 3-бромо-4-хлорфенола (2.0 г, CAS: 13659-24-0) в уксусной кислоте (6.0 мл) добавили трет-бутанол (1.07 г, 1.36 мл) с последующей серной кислотой (946 мг, 517 мкл) при комнатной температуре в атмосфере аргона. Смесь нагревали до 70°C в течение 80 часов. Дополнительное количество трет-бутанола (715 мг, 905 мкл) и серной кислоты (756 мг, 413 мкл) добавили и смесь нагревали до 90°C в течение дополнительных 24 часов. Реакционную смесь охладили до комнатной температуры и затем влили в воду со льдом. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Оставшийся трет-бутанол удалили эвапорацией из толуола (200 мл) досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 20% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-коричневой жидкости (506 мг, 19%). MS (ESI): m/z = 263.1 [M-H]-.
Стадия 2: 4-(трет-бутил)-2-хлор-5-гидроксибензонитрил
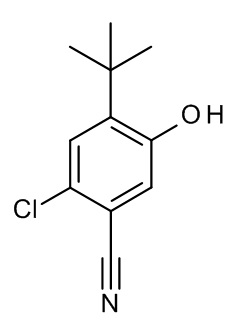
5-бромо-2-(трет-бутил)-4-хлорфенол (501 мг), 1,1'-бис(дифенилфосфино)-ферроцен (31.6 мг), цинк в гранулах (4.97 мг), цианид цинка (123 мг), ацетат цинка (14 мг), трис (дибензилиденацетон)дипалладий (0) (17.4 мг) растворили в диметилформамиде (5.0 мл) и воде (50 мкл) при комнатной температуре. Смесь затем поместили под микроволновое излучение в течение 30 минут при 180°C. Реакционную смесь охладили до комнатной температуры и затем влили в воду со льдом. Водный слой подкислили насыщенным раствором NH4Cl и экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Оставшийся ДМФ удалили эвапорацией из толуола (150 мл) досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 20% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтого осадка (71 мг, 18%). MS (ESI): m/z = 208.1 [M-H]-.
Промежуточное соединение A35
5-трет-бутил-2-хлор-4-гидрокси-бензонитрил

Стадия 1: 4-бромо-2-трет-бутил-5-хлор-фенол

В закрываемую пробирку, содержащую 4-бромо-3-хлорфенол (23 г, 111 ммоль) добавили уксусную кислоту (115 мл), tBuOH (48 мл, 499 ммоль), и серную кислоту (30 мл, 333 ммоль). Пробирку закрыли и нагревали до 90 °C при интенсивном перемешивании. Через 18 ч добавили дополнительную порцию tBuOH (48 мл, 499 ммоль), и серной кислоты (30 мл, 333 ммоль), и нагревание продолжили при 90 °C. Через 5 ч, реакционную смесь охладили до комнатной температуры, затем разделили между этилацетатом и водой. Слои разделили, и водную фазу снова промыли этилацетатом. Объединенные органические фазы промыли дважды с помощью воды, один раз солевым раствором, высушили над MgSO4, отфильтровали и сконцентрировали. Получившуюся неочищенную смесь очистили на SiO2, с элюцией сначала 100% гексанами, затем поднимая до 25% этилацетата в гексанах с получением 9.76 г соединения, указанного в заголовке,котороеиспользовали на следующей стадии без дополнительной очистки (60% чистота, по данным ЯМР).
Стадия 2: 5-трет-бутил-2-хлор-4-гидрокси-бензонитрил
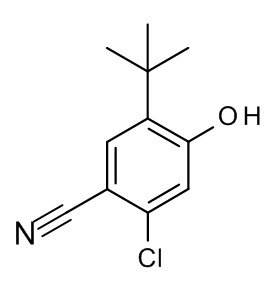
К 4-бромо-2-трет-бутил-5-хлор-фенолу (9.76 г, 60% чистота, 22.2 ммоль) добавили ДМФ (80 мл), затем CuCN (4 г, 44.7 ммоль). Получившуюся смесь нагрели до 170 °C в течение 8 ч. После завершения реакции реакционную смесь охладили, затем разделили между этилацетатом и водой. Получившуюся суспензию отфильтровали через целит и промыли этилацетатом. Слои разделили, и водную фазу снова промыли этилацетатом. Объединенные органические фазы промыли водой, солевым раствором, высушили над MgSO4, отфильтровали и сконцентрировали. Получившуюся неочищенную смесь очистили на SiO2, с элюцией 0-60% этилацетатом в гексанах с получением 2.59 г соединения, указанного в заголовке, (56% выход).
Промежуточное соединение A36
5-трет-бутил-2-фтор-4-гидрокси-бензонитрил

Стадия 1: 2-трет-бутил-4-хлор-5-фтор-фенол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением A35, Стадия 1, из 4-хлор-3-фторфенола (5 г) и получили (4.4 г, 64%).
Стадия 2: 5-трет-бутил-2-фтор-4-гидрокси-бензонитрил
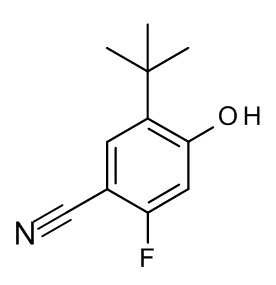
К 2-трет-бутил-4-хлор-5-фтор-фенолу (108 мг, 0.53 ммоль) добавили диоксан/воду (1:1, 2 мл), ацетат калия (10 мг, 0.11 ммоль), и K4[Fe(CN)6]•3H2O (113 мг, 0.27 ммоль). Газообразный азот барботировали через смесь в течение 20 минут. 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (11 мг, 0.027 ммоль) и [(2-ди-трет-бутилфосфино-2,4,6-triisopropyl-1,1-бифенил)-2-(2-амино-1,1-бифенил)]палладия (II) метансульфонат (21 мг, 0.027 ммоль) добавили и смесь нагрели до 100 °C в течение 3 ч. После завершения реакции, смесь охладили, затем разделили между этилацетатом и солевым раствором. Получившуюся суспензию отфильтровали через целит и промыли этилацетатом. Слои разделили, органические фазы промыли дважды солевым раствором, высушили над сульфатом магния, отфильтровали и сконцентрировали. Получившуюся неочищенную смесь очистили на SiO2 с элюцией 0-100% этилацетатом в гексанах) с получением 61.3 мг соединения, указанного в заголовке, (60% выход).
Промежуточные соединения B
Промежуточное соединение B1:
4-хлор-5-метил-2-фенилфенол

Стадия 1: 5-хлор-2-метокси-4-метиланилин

К раствору 1-хлор-4-метокси-2-метил-5-нитробензола (0.15 г, полученного как описано в WO2011/141716) в ТГФ (0.22 мл) добавили Fe(acac)3 (52.6 мг, CAS: 14024-18-1) и 1,1,3,3-тетраметилдисилоксан (300 мг, CAS: 3277-26-7) и реакционную смесь перемешивали при кипении с обратным холодильником в закрытой пробирке в течение 15 часов. Реакционной смеси дали остыть до КТ, обработали EtOAc (5 мл) и экстрагировали водным раствором 25% HCl (2 мл). Водный слой экстрагировали еще раз с помощью EtOAc (5 мл). Водный слой затем довели до приблизительно pH 8 - 9 с использованием твердого NaHCO3 и затем экстрагировали три раза EtOAc. Объединенные органические слои высушили над MgSO4, отфильтровали и эвапорировали с получением соединения, указанного в заголовке, в виде коричневого осадка (0.1 г; 78%). MS (ESI): m/z = 172.4 [M+H]+.
Стадия 2: 1-хлор-5-йод-4-метокси-2-метилбензол

К конц. водной HCl (127 мкл) при 0°C добавили NaNO2 (57.5 мг, CAS: 7632-00-0). К этой смеси добавили по каплям в течение 7 минут раствор 5-хлор-2-метокси-4-метиланилина (0.13 г) в AcOH (1.5 мл) и получившуюся смесь перемешивали при комнатной температуре в течение 30 минут. Эту смесь добавили по каплям с помощью шприца к перемешанному раствору KI (377 мг, CAS: 7681-11-0) в 48% водном HBr (2 мл) при комнатной температуре. Коричневую реакционную смесь перемешивали при комнатной температуре в течение дополнительных 2 часов. Реакционную смесь влили в 2M водный раствор Na2CO3 и EtOAc и слои разделили. Водный слой экстрагировали дважды с помощью EtOAc. Органические слои промыли один раз солевым раствором, высушили над MgSO4, отфильтровали и эвапорировали. Остаток очистили с помощью хроматографии на силикагеле на 10 г колонке с использованием системы ЖХСД (ISCO) при градиенте н-гептана в EtOAc (100/0 до 50/50) с получением соединения, указанного в заголовке, в виде светло-коричневого осадка (0.134 г; 63%). MS (EI): m/z = 282 [M].
Стадия 3: 4-хлор-2-йод-5-метилфенол

К раствору 1-хлор-5-йод-4-метокси-2-метилбензола (1.13 г) в ДХМ (20 мл) добавили по каплям при 0°C трибромборан 1M в ДХМ (4.2 мл, CAS: 10294-33-4) в течение 15 минут. Реакционную смесь перемешивали в течение 4 часов при комнатной температуре. Реакционную смесь влили в H2O (60 мл) и ДХМ (60 мл) и слои разделили. Водный слой экстрагировали второй раз с помощью ДХМ (60 мл). Органические слои промыли солевым раствором (60 мл), высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом с получением соединения, указанного в заголовке, в виде светло-желтого осадка (1.023 г, 95%). MS (ESI): m/z = 266.907 [M-H]-.
Стадия 4: 4-хлор-5-метил-2-фенилфенол

К раствору 4-хлор-2-йод-5-метилфенола (100 мг) в DME (2 мл) добавили фенилбороновую кислоту (45.4 мг, CAS: 98-80-6) и 2M водный Na2CO3 (1 мл). Реакционную смесь перемешивали в течение 15 минут в атмосфере аргона. Ацетат Pd(II) (4.18 мг, CAS: 3375-31-3) и трифенилфосфин (9.77 мг, CAS: 603-35-0) добавили. Реакционную смесь перемешивали в течение 2 часов при 90°C. Реакционную смесь влили в 10% водный раствор NaHCO3 (30 мл) и EtOAc (30 мл) и слои разделили. Водный слой экстрагировали дважды с помощью EtOAc (30 мл). Органические слои промыли солевым раствором (30 мл), высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. Соединение очистили с помощью хроматографии на силикагеле на 20 г колонке с использованием системы ЖХСД (Flashmaster) с элюцией при градиенте н-гептана в EtOAc (100/0 до 70/40) с получением соединения, указанного в заголовке, в виде светло-желтого масла (0.077 г, 95%). MS (EI): m/z = 218 [M]+.
Следующие промежуточные соединения получили по аналогии с Промежуточным соединением B1, Стадия 4, из 4-хлор-2-йод-5-метилфенола (промежуточное соединение B1, Стадия 3) и соответствующей арилбороновой кислоты или арилбороланового структурного элемента, как показано в следующей Таблице:











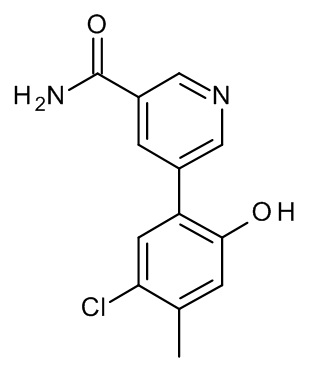



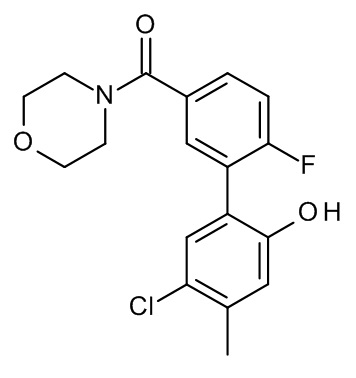
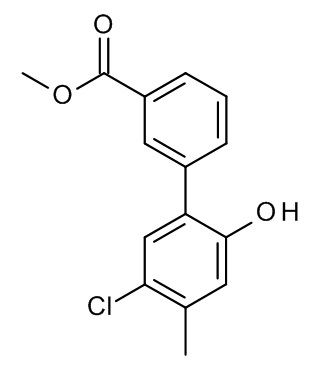
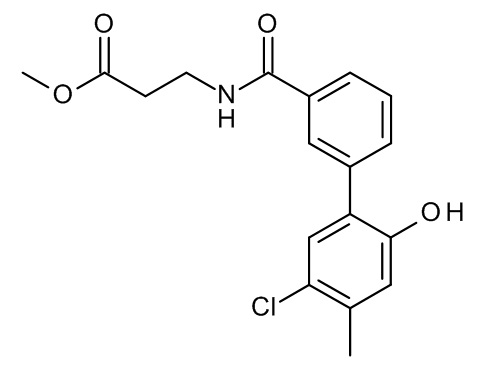
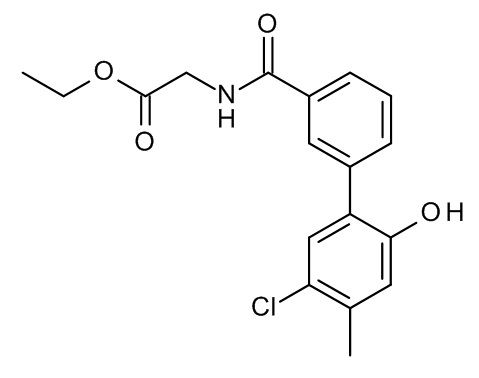
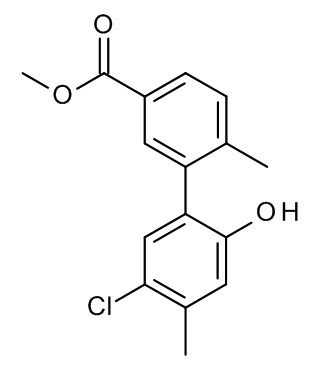

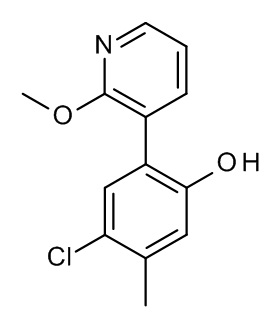
Промежуточное соединение B16:
6-гидрокси-бифенил-3-карбонитрил
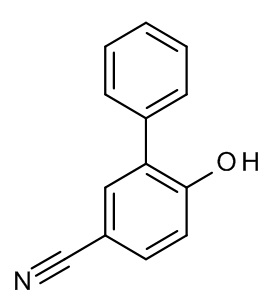
Стадия 1: 4-гидрокси-3-йод-бензонитрил
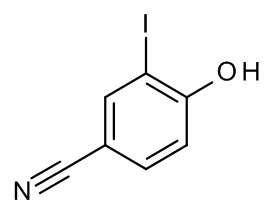
К раствору 4-гидрокси-бензонитрила (5 г, CAS: 767-00-0) в NH4OH (225 мл) добавили раствор KI (34.14 г, CAS: 7681-11-0) и I2 (10.65 г, CAS: 7553-56-2) в H2O (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь отфильтровали и фильтрат эвапорировали. Остаток растворили в ДХМ (250 мл) и промыли с помощью H2O (2x150 мл), насыщенным водным раствором NaS2O3 (100 мл) и солевым раствором (100 мл). Органический слой высушили над безводным Na2SO4, отфильтровали и сконцентрировали при пониженном давлении с получением соединения, указанного в заголовке (8.44 г, 82%), который использовали на следующей стадии без дополнительной очистки.
LC-MS: (ESI): m/z = 244.0 [M-H]-.
Стадия 2: 6-гидрокси-бифенил-3-карбонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B1, Стадия 4, из 4-гидрокси-3-йод-бензонитрила и фенилбороновой кислоты (CAS: 98-80-6) в виде бесцветного осадка (0.580 г, 73%).1H NMR (400 MHz, CDCl3): 5.72 (1H, s), 7.03-7.07 (1H, d, J = 4), 7.40-7.42 (2H, d, J = 8), 7.44-7.56 (5H, m).
Промежуточное соединение B26:
4-хлор-5-метил-2-оксазол-5-ил-фенол

Стадия 1: 5-хлор-2-метокси-4-метил-бензальдегид

К раствору 1-хлор-5-йод-4-метокси-2-метилбензола (1.0 г, промежуточное соединение B1, Стадия 2) в безводном ТГФ (25 мл) добавили n-BuLi (1.7 мл, 2.5M раствор в гексане) по каплям при -78 °C в атмосфере азота и реакционную смесь перемешивали при -78 °C в течение 2 часов. Затем, ДМФ (0.329 мл), растворенный в безводном ТГФ (2 мл), добавили по каплям в реакционную смесь при -78 °C, и раствор перемешивали при -78°C в течение 2 часов. Реакционную смесь погасили насыщенным водным раствором NH4Cl и экстрагировали с помощью EtOAc (2x50 мл). Объединенные органические слои промыли солевым раствором (50 мл), высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении с получением соединения, указанного в заголовке, в виде желтого осадка (0.685 г, 76%). Rf = 0.6 (10% EtOAc/гексан).
Стадия 2: 5-(5-хлор-2-метокси-4-метил-фенил)-оксазол

К перемешанному раствору 5-хлор-2-метокси-4-метил-бензальдегида (500 мг) в MeOH (20 мл) добавили тозилметилизоцианид (582 мг, CAS: 36635-61-7), с последующим K2CO3 (561 мг), и получившуюся смесь кипятили с обратным холодильником в течение 3 часов. Затем MeOH эвапорировали и остаток очистили с помощью флеш-хроматографии через силикагель (5-10% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде серо-белого осадка (294 мг, 49%). MS (ESI): m/z = 224.0 [M+H]+.
Стадия 3: 4-хлор-5-метил-2-оксазол-5-ил-фенол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B1, Стадия 3, из 5-(5-хлор-2-метокси-4-метил-фенил)-оксазола (292 мг) в виде коричневого осадка (0.266 г, 97%). MS (ESI): m/z = 210.2 [M+H]+.
Промежуточное соединение B30:
4-хлор-5-метил-2-(2-метил-2H-пиразол-3-ил)-фенол

Стадия 1: 5-(5-хлор-2-метокси-4-метил-фенил)-1-метил-1H-пиразол
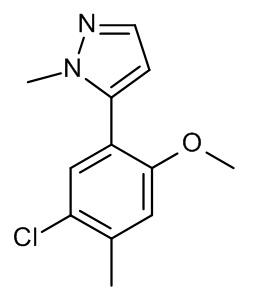
К смеси 1-хлор-5-йод-4-метокси-2-метил-бензола (500 мг, промежуточное соединение B1, Стадия 2), 1-метил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-пиразола (442 мг, CAS: 847818-74-0) и K2CO3 (733 мг) в диоксане (12 мл) и воде (4 мл) добавили PdCl2(PPh3)2-CH2Cl2 (25 мг) и реакционную смесь нагревали до 110°C в течение 16 ч. Реакционную смесь разбавили этилацетатом (50 мл), отфильтровали через целит и фильтрат эвапорировали. Получившийся остаток очистили посредством колоночной хроматографии через силикагель (0-20% EtOAc/гексан) с получением 5-(5-хлор-2-метокси-4-метил-фенил)-1-метил-1H-пиразола (365 мг, 87%) в виде коричневого вязкого осадка. MS (ESI): m/z = 236.7 [M+H]+.
Стадия 2: 4-хлор-5-метил-2-(2-метил-2H-пиразол-3-ил)-фенол

К раствору 5-(5-хлор-2-метокси-4-метил-фенил)-1-метил-1H-пиразола (355 мг в ДХМ (20 мл) при 0°C добавили BBr3 (1M раствор в ДХМ, 3 мл) и реакционную смесь перемешивали при 25°C в течение 3 ч. Затем все летучие компоненты удалили под вакуумом и оставшийся остаток растворили в ДХМ (50 мл) и промыли 10% водным раствором NaHCO3 и солевым раствором. Органический слой высушили над Na2SO4 и сконцентрировали. Получившийся неочищенный материал очистили посредством колоночной хроматографии через силикагель (0-30% EtOAc/гексан) с получением 4-хлор-5-метил-2-(2-метил-2H-пиразол-3-ил)-фенола (138 мг, 41%) в виде желтого осадка. MS (ESI): m/z = 223 [M+H]+.
Промежуточное соединение B31:
4-хлор-6-гидрокси-бифенил-3-карбонитрил

Способ A:
Стадия 1: 5-бромо-2-хлор-4-гидрокси-бензонитрил

К раствору 2-хлор-4-гидрокси-бензонитрила (4.0 г, CAS: 3336-16-1) в безводном ацетонитриле (80 мл) добавили TfOH (2.53 мл) по каплям при -30°C и реакционную смесь перемешивали при -30°C в течение 10 мин. Затем NBS (6.49 г) добавили и смесь перемешивали при -30°C в течение 5 мин. Реакционной смеси дали нагреться до 25°C и перемешивали при 25°C в течение 16 ч. Реакционную смесь погасили насыщенным водным раствором бисульфита натрия и экстрагировали EtOAc. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и отфильтровали. Фильтрат эвапорировали при пониженном давлении и получившийся остаток очистили посредством колоночной хроматографии через силикагель (2-6% EtOAc/гексан) с получением 5-бромо-2-хлор-4-гидрокси-бензонитрила (1.2 г, 20%) в виде серо-белого осадка. MS (ESI): m/z = 231.6 [M+H]+.
Стадия 2: 5-бромо-2-хлор-4-метоксиметокси-бензонитрил

К суспензии NaH (62 мг, 60% в минеральном масле) в безводном ТГФ (10 мл) при 0°C добавили раствор 5-бромо-2-хлор-4-гидрокси-бензонитрила (300 мг) в безводном ТГФ (5 мл) и реакционную смесь перемешивали при 25°C в течение 30 мин. Затем MOM-Cl (0.147 мл) добавили по каплям и реакционную смесь перемешивали при 25°C в течение 3 ч. Реакционную смесь погасили водой и экстрагировали с помощью EtOAc. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали при пониженном давлении. Получившийся неочищенный материал очистили посредством колоночной хроматографии через силикагель (0-3% EtOAc/гексан) с получением 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (315 мг, 88%) в виде серо-белого осадка.1H-NMR (δ, CDCl3): 3.51 (s, 3H), 5.30 (s, 2H), 7.28 (s, 1H), 7.81 (s, 1H).
Стадия 3: 4-хлор-6-гидрокси-бифенил-3-карбонитрил
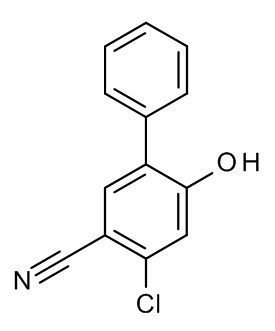
К смеси 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (100 мг) и фенилбороновой кислоты (132.3 мг, CAS: 98-80-6) в безводном ДМФ (50 мл) в закрытой пробирке добавили K2CO3 (149.7 мг) при 25°C и реакционную смесь продули аргоном в течение 10 мин. Затем Pd(dppf)Cl2-CH2Cl2 комплекс (8.85 мг) добавили и смесь снова продули аргоном в течение 10 мин и затем нагревали до 120°C в течение 16 ч. Реакционную смесь охладили до 25°C и отфильтровали. Фильтрат эвапорировали при пониженном давлении и получившийся остаток очистили посредством колоночной хроматографии через силикагель (5-7% EtOAc/гексан) с получением 4-хлор-6-гидрокси-бифенил-3-карбонитрила (43 мг, 44%) в виде серо-белого осадка.1H-NMR (δ, CDCl3): 5.91 (s, 1H), 7.13 (s, 1H), 7.39 (dd, 2H), 7.44-7.56 (m, 4H).
Способ B:
Стадия 1: 5-йод-2-хлор-4-гидрокси-бензонитрил
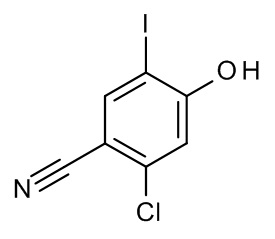
К раствору 2-хлор-4-гидроксибензонитрила (1.18 г, CAS: 3336-16-1) в уксусной кислоте (10 мл) и ДХМ (10 мл) добавили концентрированную серную кислоты (100 мкл) и затем N-йодсукцинимид (1.66 г) за одну порцию при комнатной температуре в атмосфере аргона. Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь затем влили в воду со льдом и экстрагировали два раза этилацетатом. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенное вещество абсорбировали на силикагеле затем очистили с помощью флеш-хроматографии (ISCO, 100 г силикагелевый картридж, градиент от 0% до 20% EtOAc в гептане). Фракции, содержащие требуемый продукт вместе с некоторым количеством ди-йодированных побочных продуктов объединили, эвапорировали и высушили в глубоком вакууме. Остаток (1.42 г) дополнительно очистили с помощью препаративной ВЭЖХ (Колонка: Gemini NX 3u 50x4.6 мм; Элюент: 2% муравьиная кислота, 98% CH3CN) с получением соединения, указанного в заголовке (770 мг), в виде серо-белого осадка. MS (ESI-): m/z = 278.0 [M-H]-.
Стадия 2: 4-хлор-6-гидрокси-бифенил-3-карбонитрил
2-хлор-4-гидрокси-5-йодобензонитрил (350 мг), фенилбороновую кислоту (160 мг) и карбонат натрия (398 мг) объединили с ДМФ (13.0 мл) и водой (2.0 мл) при комнатной температуре в атмосфере аргона. Затем [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладиевый комплекс (II) с ДХМ (133 мг) добавили к оранжевой суспензии. Реакционную смесь три раза эвапорировали и продули аргоном и затем нагревали до 80°C в течение 3.5 часов. Смесь охладили до комнатной температуры, влили в воду со льдом и затем подкислили с помощью насыщенного раствора NH4Cl. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенное вещество очистили с помощью флеш-хроматографии (ISCO, силикагель, 40 г картридж, 100% CH2Cl2 и затем CH2Cl2/CH3CN=96/4). Необходимые фракции объединили и эвапорировали с получением соединения, указанного в заголовке (150 мг), в виде серо-белого осадка.
MS (ESI-): m/z = 228.1 [M-H]-. NMR соответствует материалу, описанному выше из способа A.
Следующие промежуточные соединения получили по аналогии с Промежуточным соединением B31, либо способом A из 5-бромо-2-хлор-4-гидрокси-бензонитрила (промежуточное соединение B31, Стадия 1), или способом B из 5-йод-2-хлор-4-гидрокси-бензонитрил, и бороновокислотных предшественников, как показано в таблице ниже:

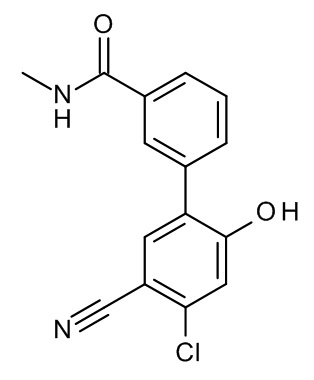


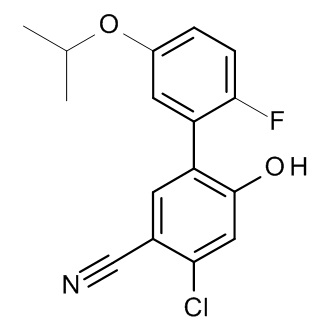
Промежуточное соединение B35:
4-хлор-6-гидрокси-3'-(5-метил-[1,2,4]оксадиазол-3-ил)-бифенил-3-карбонитрил

Стадия 1: 3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензонитрил
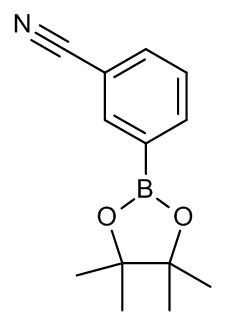
К раствору 3-цианофенилбороновой кислоты (2.5 г, CAS: 150255-96-2) в безводном ТГФ (150 мл) в атмосфере азота добавили пинакол (2.95 г) и Na2SO4 (10 г) при 25°C.Реакционную смесь перемешивали в течение 12 ч при 25°C в атмосфере азота. Затем Na2SO4 отфильтровали и все летучие компоненты эвапорировали. Остаток разделили между этилацетатом и водой. Органический слой затем отделили, высушили над Na2SO4 и сконцентрировали с получением соединения, указанного в заголовке (4.72 г), в виде серо-белого осадка, который сразу использовали на следующей реакционной стадии без дополнительной характеризации.
Стадия 2: N-гидрокси-3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензамидин

К раствору -(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензонитрила (4.65 г) в этаноле (150 мл) добавили DIPEA (7.15 мл) и гидроксиламина гидрохлорид (3.53 г) при 25°C. Реакционную смесь затем кипятили в течение 3 ч. Все летучие компоненты эвапорировали под вакуумом и остаток разделили между этилацетатом и водой. Органический слой разделили, высушили над Na2SO4 и сконцентрировали с получением N-гидрокси-3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензамидина вместе с некоторым количеством примесей (5.7 г) в виде серо-белого вязкого осадка. MS (ESI): m/z = 262.8 [M+H]+.
Стадия 3: 5-метил-3-[3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-[1,2,4]оксадиазол

Раствор N-гидрокси-3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-бензамидина (5.7 г) в уксусном ангидриде (100 мл) кипятили с обратным холодильником в течение 3 ч. Уксусный ангидрид эвапорировали и остаток разделили между водой и этилацетатом. Слой этилацетата отделили, промыли водным раствором NaHCO3, водой и солевым раствором и высушили над Na2SO4 и сконцентрировали. Неочищенный продукт очистили посредством колоночной хроматографии через силикагель (0-5% EtOAc/гексан) с получением соединения, указанного в заголовке (3.5 г) в виде серо-белого осадка. MS (ESI): m/z = 287.0 [M+H]+.
Стадия 4: 4-хлор-6-гидрокси-3'-(5-метил-[1,2,4]оксадиазол-3-ил)-бифенил-3-карбонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 3, из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B32, Стадия 2) (300 мг) и 5-метил-3-[3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-фенил]-[1,2,4]оксадиазола (932 мг) и получили в виде серо-белого осадка (190 мг, 56%). MS (ESI): m/z = 310.2 [M-H]-.
Промежуточное соединение B37:
4-хлор-6-гидрокси-3'-(5-метил-[1,3,4]оксадиазол-2-ил)-бифенил-3-карбонитрил
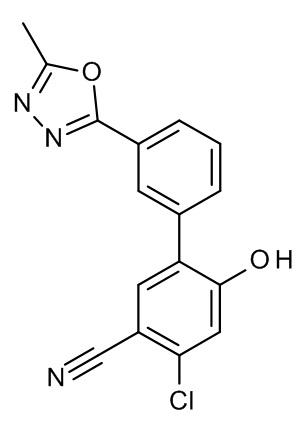
Стадия 1: 4'-хлор-5'-циано-2'-гидрокси-бифенил-3-карбоновой кислоты метиловый эфир

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 3, из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B31, Стадия 2) (1.0 г) и 3-метоксикарбонилфенилбороновой кислоты (973 мг, CAS: 99769-19-4) и получили в виде серо-белого осадка (430 мг, 41%). MS (ESI): m/z = 286 [M-H]-.
Стадия 2: 4'-хлор-5'-циано-2'-метокси-бифенил-3-карбоновой кислоты метиловый эфир
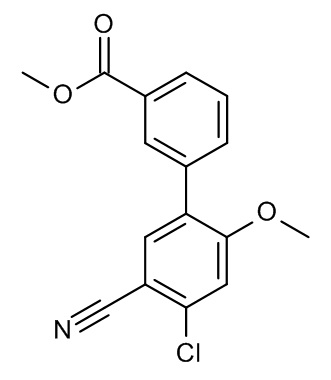
К раствору 4'-хлор-5'-циано-2'-гидрокси-бифенил-3-карбоновой кислоты метилового эфира (650 мг) в безводном ДМФ (30 мл) добавили Cs2CO3 (1.10 г) и метилйодид (0.169 мл) при 25°C. Реакционную смесь перемешивали при 25°C в течение 16 ч и затем отфильтровали. Фильтрат эвапорировали при пониженном давлении и оставшийся остаток очистили посредством колоночной хроматографии через силикагель (10-15% EtOAc/гексан) с получением соединения, указанного в заголовке (490 мг, 71%) в виде серо-белого осадка, который использовали без дополнительной характеризации.
Стадия 3: 4'-хлор-5'-циано-2'-метокси-бифенил-3-карбоновая кислота
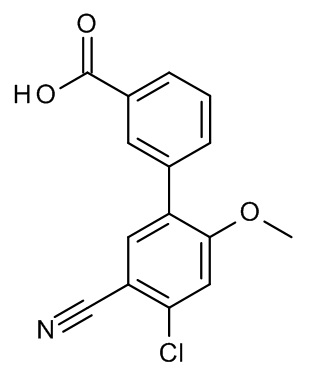
К раствору 4'-хлор-5'-циано-2'-метокси-бифенил-3-карбоновой кислоты метилового эфира (490 мг) в смеси ТГФ и воды (45 мл) добавили LiOH-H2O (136.6 мг) при 25°C и реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь затем разбавили EtOAc (20 мл) и слои разделили. Водный слой подкислили посредством добавления 6 н HCl и экстрагировали с помощью EtOAc. Объединенные экстракты промыли солевым раствором, высушили над Na2SO4 и сконцентрировали с получением соединения, указанного в заголовке (410 мг, 88%), в виде серо-белого осадка. MS (ESI): m/z = 286.1 [M-H]-.
Стадия 4: 4'-хлор-5'-циано-2'-метокси-бифенил-3-карбоновой кислоты N'-ацетил-гидразид

К раствору 4'-хлор-5'-циано-2'-метокси-бифенил-3-карбоновой кислоты (410 мг) в безводном ДМФ (30 мл) добавили HBTU (812.1 мг) и DIPEA (0.76 мл) при 25°C. Реакционную смесь перемешивали в течение 10 мин и затем гидразид уксусной кислоты (211.42 мг) добавили и перемешивание продолжили в течение 16 ч. Растворитель эвапорировали и к оставшемуся остатку добавили EtOAc и воду. Органический слой разделили, высушили над Na2SO4 и сконцентрировали. Остаток очистили посредством колоночной хроматографии через силикагель (2-3% MeOH/ДХМ) с получением соединения, указанного в заголовке (470мг, 96%) в виде серо-белого осадка. MS (ESI): m/z = 344.2 [M+H]+.
Стадия 5: 4-хлор-6-метокси-3'-(5-метил-[1,3,4]оксадиазол-2-ил)-бифенил-3-карбонитрил

Раствор 4'-хлор-5'-циано-2'-метокси-бифенил-3-карбоновой кислоты N'-ацетил-гидразида (400 мг) в POCl3(10 мл) нагрели до 110°C в течение 6 ч. Реакционную смесь затем охладили до 25°C и все летучие компоненты удалили при пониженном давлении. К оставшемуся остатку добавили насыщенный раствор NaHCO3 (15 мл) и смесь экстрагировали EtOAc. Объединенные экстракты высушили над Na2SO4 и сконцентрировали. Остаток очистили посредством колоночной хроматографии через силикагель (10-15% EtOAc/гексан) с получением соединения, указанного в заголовке (130 мг, 34%), в виде серо-белого осадка. MS (ESI): m/z = 326.2 [M+H]+.
Стадия 6: 4-хлор-6-гидрокси-3'-(5-метил-[1,3,4]оксадиазол-2-ил)-бифенил-3-карбонитрил
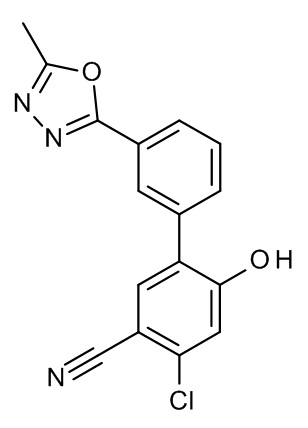
К раствору 4-хлор-6-метокси-3'-(5-метил-[1,3,4]оксадиазол-2-ил)-бифенил-3-карбонитрила (110 мг) в безводном ДХМ (10 мл) добавили BBr3 (0.4 мл) при 0°C. Реакционную смесь перемешивали при 25°C в течение 28 ч. Снова добавили BBr3 (0.5 мл) и реакционную смесь перемешивали в течение дополнительных 16 ч. Все летучие компоненты удалили при пониженном давлении и остаток очистили посредством колоночной хроматографии через силикагель (30-45% EtOAc/гексан) с получением соединения, указанного в заголовке (80 мг, 76%), в виде серо-белого осадка. MS (ESI): m/z = 310.2 [M-H]-.
Промежуточное соединение B39:
3-(4-хлор-5-циано-2-гидрокси-фенил)-N,N-диметил-бензамид

Стадия 1: 3-[4-хлор-5-циано-2-(метоксиметокси)фенил]-N,N-диметил-бензамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 3, из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B31, Стадия 2) (48.9 мг) и 3-(диметилкарбамоил)фенилбороновой кислоты (40.9 мг, CAS: 373384-14-6) и получили в виде светло-желтой смолы (43 мг, 71%). MS (ESI): m/z = 698.3 [2M+H]+.
Стадия 2: 3-(4-хлор-5-циано-2-гидрокси-фенил)-N,N-диметил-бензамид
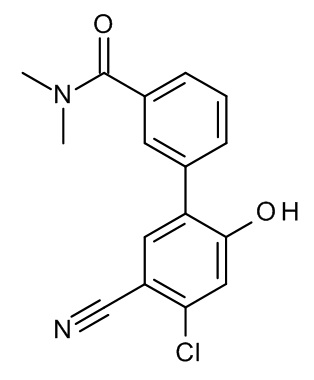
К раствору 3-[4-хлор-5-циано-2-(метоксиметокси)фенил]-N,N-диметил-бензамида (40.0 мг) в диоксане (1 мл) добавили по каплям 4 н HCl в диоксане (150 мкл) при 0°C. Реакционную смесь затем перемешивали при комнатной температуре в течение 4 ч. Дополнительно 4 н HCl в диоксане (100 мкл) добавили, и получившуюся суспензию перемешивали при комнатной температуре в течение ночи. Реакционную смесь сконцентрировали досуха и затем совместно эвапорировали из ДХМ три раза. Получившийся серо-белый осадок (34 мг, 97%) использовали на следующей реакционной стадии без дополнительной очистки. MS (ESI): m/z = 301.1 [M+H]+.
Промежуточное соединение B41:
2-хлор-4-гидрокси-5-[3-(морфолин-4-карбонил)фенил]бензонитрил
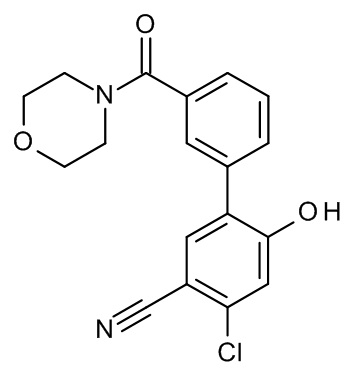
Стадия 1: 2-хлор-4-(метоксиметокси)-5-[3-(морфолин-4-карбонил)фенил]бензонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 3, из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B31, Стадия 2) (50.0 мг) и 3-(морфолин-4-карбонил)фенилбороновой кислоты (51.0 мг, CAS: 723281-55-8) и получили в виде белого осадка (53 мг, 76%). MS (ESI): m/z = 387.2 [M+H]+.
Стадия 2: 2-хлор-4-гидрокси-5-[3-(морфолин-4-карбонил)фенил]бензонитрил
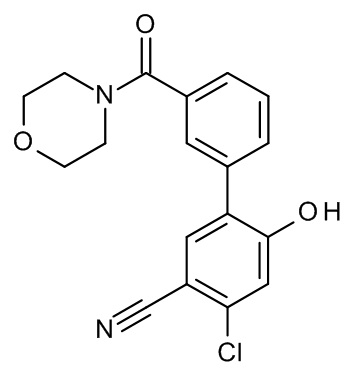
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B39, Стадия 2, из 2-хлор-4-(метоксиметокси)-5-[3-(морфолин-4-карбонил)фенил]бензонитрила (49.9 мг) и получили в виде серо-белого осадка (44 мг, 99%). MS (ESI): m/z = 343.1 [M+H]+.
Промежуточное соединение B54:
2-хлор-4-гидрокси-5-(2-метокси-3-пиридил)бензонитрил

Стадия 1: 2-хлор-4-(метоксиметокси)-5-(2-метокси-3-пиридил)бензонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 3, из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B31, Стадия 2) (50.0 мг) и 2-метоксипиридин-3-илбороновой кислоты (33.2 мг, eMolecules #BB-4033) и получили в виде белого осадка (43 мг, 78%). MS (ESI): m/z = 305.1 [M+H]+.
Стадия 2: 2-хлор-4-гидрокси-5-(2-метокси-3-пиридил)бензонитрил
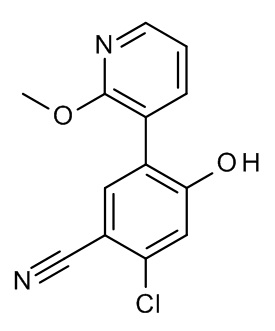
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B39, Стадия 2, из 2-хлор-4-(метоксиметокси)-5-(2-метокси-3-пиридил)бензонитрила (41.1 мг) и получили в виде серо-белого осадка (35.2 мг, 100%). MS (ESI): m/z = 261.1 [M+H]+.
Промежуточное соединение B58
4-хлор-2-[2-фтор-5-(2-метилпропокси)фенил]-5-метилфенол
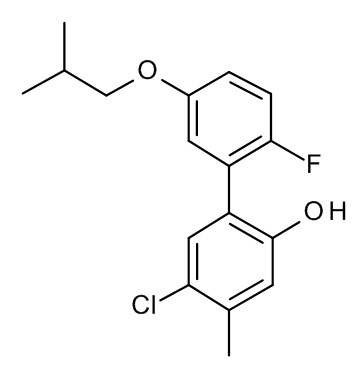
4-хлор-2-йод-5-метилфенол (промежуточное соединение B1, Стадия 3, (80 мг), (2-фтор-5-изобутоксифенил)бороновую кислоту (75.8 мг, CAS: 1217500-65-6) и карбонат натрия (94.7 мг) объединили с ДМФ (5.0 мл) и водой (0.85 мл) при комнатной температуре в атмосфере аргона. Затем [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладиевый (II) комплекс с дихлорметаном (31.6 мг, CAS: 95464-05-4) добавили в оранжевую суспензию. Реакционную смесь нагревали до 80°C в течение 3 часов и затем охладили и хранили при комнатной температуре в течение 64 часов. Реакционную смесь влили в воду со льдом и подкислили насыщенным раствором NH4Cl. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 50% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтой жидкости (62 мг, 65%). MS (ESI): m/z = 307.3 [M-H]-.
Следующие промежуточные соединения получили по аналогии с Промежуточным соединением B58 из 4-хлор-2-йод-5-метилфенола (промежуточное соединение B1, Стадия 3) и соответствующих структурных элементов, указанных в таблице далее:

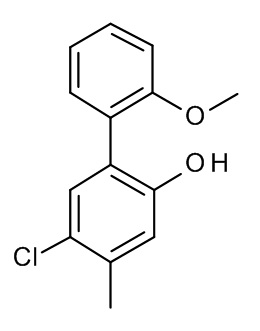



Промежуточное соединение B61
2-хлор-5-[2-фтор-5-(морфолин-4-карбонил)фенил]-4-гидроксибензонитрил

Стадия 1: 2-хлор-5-[2-фтор-5-(морфолин-4-карбонил)фенил]-4-(метоксиметокси)бензонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58 из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B32, Стадия 2) (100 мг) и (2-фтор-5-(морфолин-4-карбонил)фенил)бороновой кислоты (110 мг, CAS: 1072951-41-7) и получили в виде светло-желтой пены (24 мг, 15%). MS (ESI): m/z = 405.2 [M+H]+.
Стадия 2: 2-хлор-5-[2-фтор-5-(морфолин-4-карбонил)фенил]-4-гидроксибензонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B39, Стадия 2 из 2-хлор-5-[2-фтор-5-(морфолин-4-карбонил)фенил]-4-(метоксиметокси)бензонитрила (29 мг) и получили в виде серо-белого осадка (14 мг, 54%). MS (ESI): m/z = 361.1 [M+H]+.
Промежуточное соединение B63
3-(5-хлор-2-гидрокси-4-метилфенил)-N-циклопропил-4-фтор-N-метилбензамид

Стадия 1: 3-(5-хлор-2-метокси-4-метилфенил)-N-циклопропил-4-фторбензамид
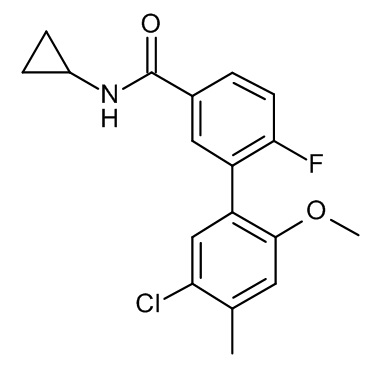
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58, из 1-хлор-5-йод-4-метокси-2-метилбензола (промежуточное соединение B1, Стадия 2) (265 мг) и (5-(циклопропилкарбамоил)-2-фторфенил)бороновой кислоты (251 мг, CAS: 874289-54-0) и получили в виде желтого осадка (163 мг, 52%). MS (ESI): m/z = 334.1 [M+H]+.
Стадия 2: 3-(5-хлор-2-метокси-4-метилфенил)-N-циклопропил-4-фтор-N-метилбензамид

Гидрид натрия, 60% дисперсию в минеральном масле (37.9 мг) добавили в раствор 3-(5-хлор-2-метокси-4-метилфенил)-N-циклопропил-4-фторбензамида (158 мг) в ДМФ (4.0 мл) при комнатной температуре в атмосфере аргона. Смесь перемешивали при комнатной температуре в течение 45 минут. Затем йодометан (87.3 мг) добавили по каплям в течение периода в 2 минуты и смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь затем влили в воду со льдом. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 50% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде белой пены (154 мг, 93%). MS (ESI): m/z = 348.2 [M+H]+.
Стадия 3: 3-(5-хлор-2-гидрокси-4-метилфенил)-N-циклопропил-4-фтор-N-метилбензамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B1, Стадия 3, из 3-(5-хлор-2-метокси-4-метилфенил)-N-циклопропил-4-фтор-N-метилбензамида (151 мг) и получили в виде серо-белой пены (157 мг, 98%). MS (ESI): m/z = 334.1 [M+H]+.
Промежуточное соединение B62
[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]-пирролидин-1-илметанон

Стадия 1: Метил 3-(5-хлор-2-метокси-4-метилфенил)-4-фторбензоат
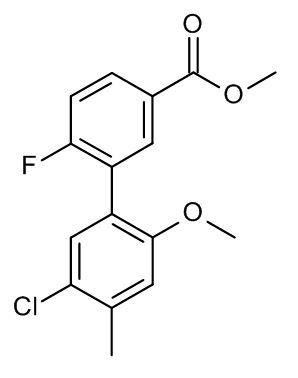
Метил 3-бромо-4-фторбензоат (3.75 г) растворили в циклопентилметиловом эфире (70 мл) при комнатной температуре в атмосфере аргона. Затем ацетат калия (6.32 г), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (5.72 г) и [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладиевый (II) комплекс с дихлорметаном (526 мг CAS: 95464-05-4) добавили в смесь. Реакционную смесь нагревали до 95°C в течение 4 часов и затем охладили до комнатной температуры в течение дополнительного часа. К смеси добавили раствор карбоната натрия (15%, 26.2 мл), 1-хлор-5-йод-4-метокси-2-метилбензол (промежуточное соединение B1, Стадия 2) (6.06 г) и [1,1'-бис(дифенилфосфино)ферроцен] дихлорпалладиевый (II) комплекс с ДХМ (526 мг, CAS: 95464-05-4). Смесь нагревали снова до 85°C в течение 18 часов. Затем реакционную смесь охладили до КТ и влили в воду со льдом. Водный слой экстрагировали дважды с помощью циклопентилметилового эфира. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 50% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтого осадка (4.46 г, 67%).1H NMR (400 MHz, CDCl3): 2.43 (3H, s), 3.76 (3H, s), 3.91 (3H, s), 6.85 (1H, s), 7.14-7.18 (1H, t), 7.24 (1H, s), 8.0-8.07 (2H, m).
Стадия 2: 3-(5-хлор-2-метокси-4-метилфенил)-4-фторбензойная кислота
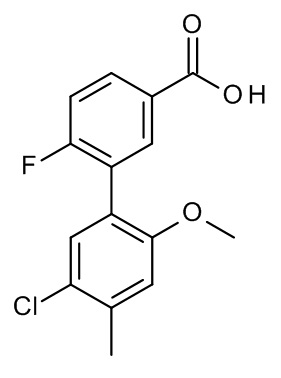
1M раствор LiOH (2.51 мл) добавили в раствор метил 3-(5-хлор-2-метокси-4-метилфенил)-4-фторбензоата (310 мг) в ТГФ (6.0 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь влили в воду со льдом и подкислили раствором HCl 1M до pH=1. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха с получением соединения, указанного в заголовке, в виде белого осадка (309 мг, 99%). MS (ESI): m/z = 293.2 [M-H]-.
Стадия 3: [3-(5-хлор-2-метокси-4-метилфенил)-4-фторфенил]-пирролидин-1-илметанон
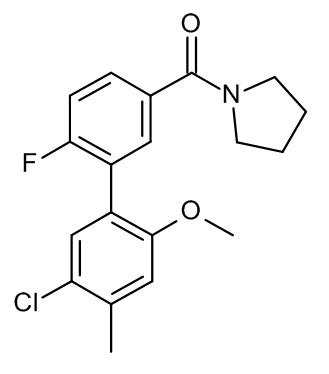
3-(5-хлор-2-метокси-4-метилфенил)-4-фторбензойную кислоту (100 мг), пирролидин (36.2 мг, CAS: 123-75-1) и 4-метилморфолин (51.5 мг растворили в ДМФ (4.0 мл) при комнатной температуре в атмосфере аргона. 1-(3-Диметиламинопропил)-3-этилкарбодиимида гидрохлорид (97.6 мг) и 1-гидроксибензотриазола гидрат (68.8 мг) добавили в светло-желтый раствор. Смесь перемешивали при комнатной температуре в течение 2.5 часов. Реакционную смесь влили затем в воду со льдом и подщелачили раствором 2M Na2CO3. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз 1M раствором HCl и один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 70% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде белого осадка (78 мг, 65%). MS (ESI): m/z = 348.1 [M+H]+.
Стадия 4: [3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]-пирролидин-1-илметанон
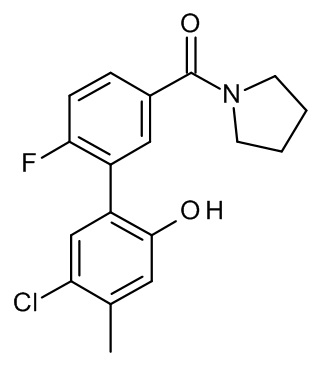
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B1, Стадия 3 из [3-(5-хлор-2-метокси-4-метилфенил)-4-фторфенил]-пирролидин-1-илметанона (74 мг) и получили в виде серо-белого осадка (83 мг, 99%). MS (ESI): m/z = 334.2 [M+H]+.
Следующие промежуточные соединения были получены по аналогии с Промежуточным соединением B62, заменой пирролидина на подходящий амино структурный элемент на Стадии 3, как показано в Таблице ниже:
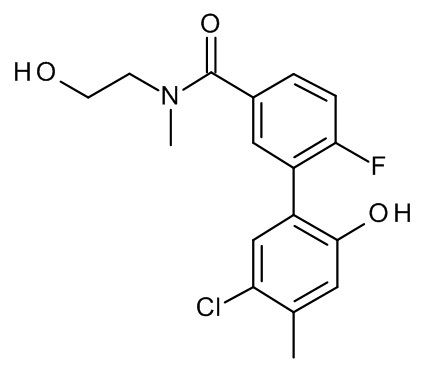
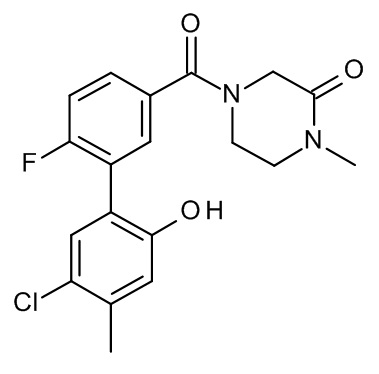
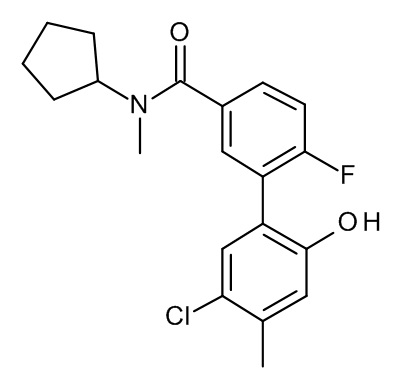

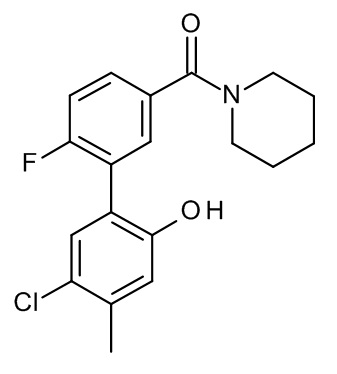


Промежуточное соединение B67
4-хлор-2-[2-фтор-5-(оксолан-3-илметокси)фенил]-5-метилфенол
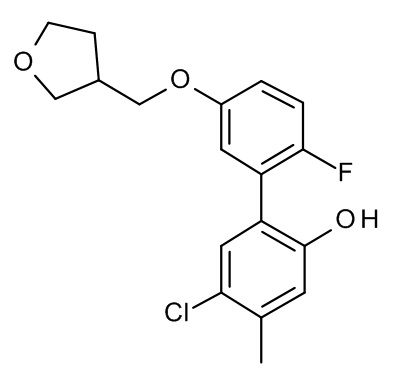
Стадия 1: 3-[(3-бромо-4-фторфенокси)метил]оксолан

Диизопропилазодикарбоксилат (1.22 г) добавили по каплям в течение периода 5 минут в раствор 3-бромо-4-фторфенола (1.05 г, CAS: 27407-11-0), (тетрагидрофуран-3-ил)метанола (674 мг, CAS: 15833-61-1) и трифенилфосфина (1.87 г) в ТГФ (12 мл) при 0°C в атмосфере аргона. Желтый раствор нагрели до КТ и поддерживали при этой температуре в течение 4.5 часов. Реакционную смесь влили в воду со льдом и подщелачили 2M раствором NaOH до достижения pH=10. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 60% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтой жидкости (1.26 г, 79%). MS (EI): m/z = 274.0 [M]+.
Стадия 2: 2-[2-фтор-5-(оксолан-3-илметокси)фенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолан

Смесь 4,4,4',4',5,5,5',5'-октаметил-2,2'-би (1,3,2-диоксаборолана) (129 мг), 3-[(3-бромо-4-фторфенокси)метил]оксолана (100 мг), ацетата калия (107 мг) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладиевый (II) комплекс дихлорметаном (14.8 мг, CAS: 95464-05-4) в 1,4-диоксане (4.0 мл) продули три раза аргоном и затем нагревали при 85°C в течение 20 часов в атмосфере аргона. Реакционную смесь охладили до комнатной температуры и затем влили в воду со льдом. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 20% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде бесцветной жидкости (90 мг, 62%, чистота приблизит. 80%). MS (EI): m/z = 322.0 [M]+.
Стадия 3: 4-хлор-2-[2-фтор-5-(оксолан-3-илметокси)фенил]-5-метилфенол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58 из 4-хлор-2-йод-5-метилфенола (промежуточное соединение B1, Стадия 3) (58 мг) и 2-[2-фтор-5-(оксолан-3-илметокси)фенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолана (83.5 мг) и получили в виде светло-желтого масла (29 мг, 39%). MS (ESI): m/z = 337.1 [M+H]+.
Следующие промежуточные соединения были получены по аналогии с Промежуточным соединением B67, посредством замены (тетрагидрофуран-3-ил)метанола на стадии 1 на подходящий спиртовой структурный элемент, как показано в Таблице ниже:

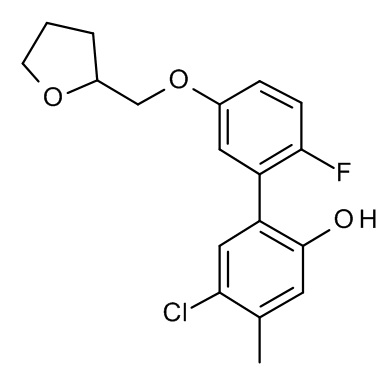
Промежуточное соединение B75
2-[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенокси]-N,N-диметилацетамид

Стадия 1: 2-(3-бромо-4-фторфенокси)-N,N-диметилацетамид
3-бромо-4-фторфенол (500мг, CAS 27407-11-0), 2-хлор-N,N-диметилацетамид (573 мг) и карбонат калия (832 мг) объединили с ацетоном (15 мл) при комнатной температуре в атмосфере аргона. Смесь нагрели до кипения в течение 18 часов и затем поддерживали при комнатной температуре в течение 1 часа. Реакционную смесь затем влили в воду со льдом и водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 80% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтой жидкости (934 мг, 91%, чистота приблизит. 70%). MS (ESI): m/z = 278.1 [M+H]+.
Стадия 2: 2-[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]-N,N-диметилацетамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B67, Стадия 2 из 2-(3-бромо-4-фторфенокси)-N,N-диметилацетамида (930 мг) и получили в виде бесцветного масла (188 мг, 18%, чистота 70%). MS (ESI): m/z = 324.2 [M+H]+.
Стадия 3: 2-[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенокси]-N,N-диметилацетамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58 из 4-хлор-2-йод-5-метилфенола (промежуточное соединение B1, Стадия 3) (108 мг) и 2-[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенокси]-N,N-диметилацетамида (186 мг, чистота 70%) и получили в виде светло-коричневой пены (63 мг, 46%). MS (ESI): m/z = 338.1 [M+H]+.
Промежуточное соединение B73
3-(5-хлор-2-гидрокси-4-метилфенил)-4-фтор-N-(2-метоксиэтил)-N-метилбензамид

Стадия 1: 3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторбензойная кислота

3-(5-хлор-2-метокси-4-метилфенил)-4-фторбензойную кислоту (промежуточное соединение B62, Стадия 2) (145 мг объединили дихлорметаном (5.0 мл) при 0°C в атмосфере аргона. Затем 1M раствор трехфтористого бора в дихлорметане (1.23 мл) добавили по каплям в течение периода 2 минуты. Получившийся желтый раствор затем поддерживали при комнатной температуре в течение 6 часов. Реакционную смесь влили затем в воду со льдом и водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха с получением соединения, указанного в заголовке, в виде светло-коричневого осадка (146 мг, 100%). MS (ESI): m/z = 559.2 [2M-H]-.
Стадия 2: 3-(5-хлор-2-гидрокси-4-метилфенил)-4-фтор-N-(2-метоксиэтил)-N-метилбензамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B62, Стадия 3 из 3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторбензойной кислоты (50 мг) с использованием 2-метокси-N-метилэтанамина (24 мг, CAS: 38256-93-8) вместо пирролидина и получили в виде белой пены (40 мг, 63%). MS (ESI): m/z = 352.1 [M+H]+.
Промежуточное соединение B76
1-[[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]метил]-4-метилпиперазин-2,5-дион

Стадия 1: 2-бромо-4-(бромометил)-1-фторбензол

Тетрабромметан (7.28 г) добавили в раствор (3-бромо-4-фторфенил)метанола (3.0 г, CAS: 77771-03-0) в ТГФ (50 мл) при 0°C в атмосфере аргона. Затем раствор трифенилфосфина (5.76 г) в ТГФ (30 мл) добавили по каплям в течение периода 40 минут. Смесь нагрели до КТ и поддерживали при этой температуре в течение 65 часов. Реакционную смесь влили затем в воду со льдом и экстрагировали два раза этилацетатом. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 10% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтой жидкости (5.10 г, 95%, чистота 70%). MS (EI): m/z = 267.9 [M]+.
Стадия 2: 1-[(3-бромо-4-фторфенил)метил]-4-метилпиперазин-2,5-дион

1-Метилпиперазин-2,5-дион (150 мг, CAS: 5625-52-5) растворили в ДМФ (5.0 мл) при комнатной температуре в атмосфере аргона. Смесь охладили до 0°C и 60% дисперсию гидрида натрия в минеральном масле (103 мг) добавили за одну порцию. Суспензию перемешивали при 0°C в течение 10 минут и затем нагрели до КТ в течение 45 минут. Затем 2-бромо-4-(бромометил)-1-фторбензол (627 мг) добавили по каплям в течение периода 5 минут. Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь влили затем в воду со льдом и водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 10% метанол в дихлорметане) с получением соединения, указанного в заголовке в виде светло-коричневой смолы (121 мг, 32%). MS (ESI): m/z = 315.0 [M+H]+.
Стадия 3: 1-[[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метил]-4-метилпиперазин-2,5-дион

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B67, Стадия 2, из 1-[(3-бромо-4-фторфенил)метил]-4-метилпиперазин-2,5-диона (120 мг) и получили в виде светло-коричневой смолы (102 мг, 52%, чистота 70%). MS (ESI): m/z = 363.2 [M+H]+.
Стадия 4: 1-[[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]метил]-4-метилпиперазин-2,5-дион
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58 из 4-хлор-2-йод-5-метилфенола (промежуточное соединение B1, Стадия 3) (52 мг) и 1-[[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метил]-4-метилпиперазин-2,5-диона (100 мг, чистота ~70%) и получили в виде светло-коричневой пены (28 мг, 37%). MS (ESI): m/z = 377.1 [M+H]+.
Следующие промежуточные соединения были получены по аналогии с Промежуточным соединением B76, посредством замены 1-метилпиперазин-2,5-диона на Стадии 2 на подходящий амидный структурный элемент, как показано в Таблице ниже:


Промежуточное соединение B78
N-[[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]метил]-2-метокси-N-метилацетамид

Стадия 1: N-[(3-бромо-4-фторфенил)метил]-2-метоксиацетамид
2-метоксиуксусную кислоту (165 мг, CAS: 625-45-6), (3-бромо-4-фторфенил)-метанамина гидрохлорид (529 мг, CAS: 202865-68-7) и 4-метилморфолин (741 мг) растворили в ДМФ (6.0 мл) при комнатной температуре в атмосфере аргона. 1-(3-Диметиламинопропил)-3-этилкарбодиимида гидрохлорид (527 мг) и 1-гидроксибензотриазола гидрат (371 мг) добавили к светло-желтому раствору. Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь влили затем в воду со льдом и подщелачили раствором 2M Na2CO3. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 80% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде белого осадка (503 мг, 98%). MS (ESI): m/z = 278.0 [M+H]+.
Стадия 2: N-[(3-бромо-4-фторфенил)метил]-2-метокси-N-метилацетамид
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B63, Стадия 2, из N-[(3-бромо-4-фторфенил)метил]-2-метоксиацетамида (290 мг) и получили в виде бесцветной жидкости (198 мг, 62%). MS (ESI): m/z = 290.0 [M+H]+.
Стадия 3: N-[[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метил]-2-метокси-N-метилацетамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B67, Стадия 2, из N-[(3-бромо-4-фторфенил)метил]-2-метокси-N-метилацетамида (199 мг) и получили в виде светло-желтой жидкости (262 мг, 57%, чистота ~50%). MS (ESI): m/z = 338.2 [M+H]+.
Стадия 4: N-[[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]метил]-2-метокси-N-метилацетамид
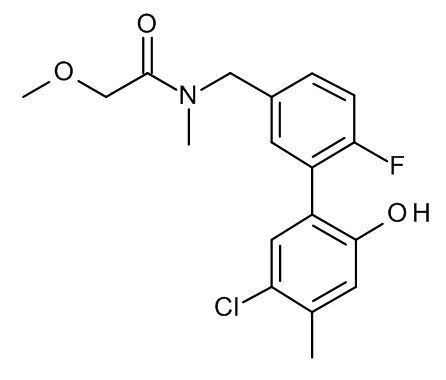
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58 из 4-хлор-2-йод-5-метилфенола (промежуточное соединение B1, Стадия 3) (102 мг) и N-[[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метил]-2-метокси-N-метилацетамида (256 мг, чистота ~50%) и получили в виде желтой смолы (106 мг, 64%, чистота ~80%). MS (ESI): m/z = 352.1 [M+H]+.
Следующие промежуточные соединения были получены по аналогии с Промежуточным соединением B78, посредством замены 2-метоксиуксусной кислоты на Стадии 1 на подходящий карбоновокислотный структурный элемент, как показано в Таблице ниже:

Промежуточное соединение B79
N-[[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]метил]циклопропан-карбоксамид
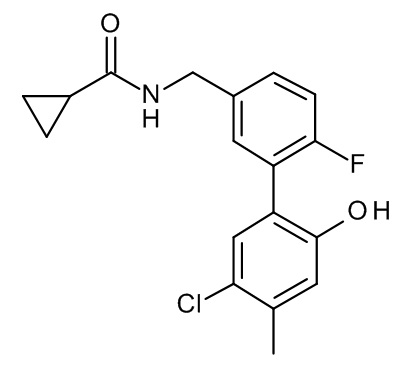
Стадия 1: N-[(3-бромо-4-фторфенил)метил]циклопропанкарбоксамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B78, Стадия 1, из циклопропанкарбоновой кислоты (127 мг, CAS: 1759-53-1) и получили в виде белого порошка (444 мг, 100%). MS (ESI): m/z = 272.0 [M+H]+.
Стадия 2: N-[[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метил]-циклопропанкарбоксамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B67, Стадия 2, из N-[(3-бромо-4-фторфенил)метил]циклопропанкарбоксамида (217 мг) и получили в виде светло-желтого масла (257 мг, 61%, чистота ~60%). MS (ESI): m/z = 320.2 [M+H]+.
Стадия 3: N-[[3-(5-хлор-2-гидрокси-4-метилфенил)-4-фторфенил]метил]циклопропан-карбоксамид

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58 из 4-хлор-2-йод-5-метилфенола (промежуточное соединение B1, Стадия 3) (119 мг) и N-[[4-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]метил]-циклопропанкарбоксамида (236 мг, чистота ~60%) и получили в виде светло-желтой пены (63 мг, 42%). MS (ESI): m/z = 334.1 [M+H]+.
Следующие промежуточные соединения были получены по аналогии с Промежуточным соединением B79, посредством замены циклопропанкарбоновой кислоты на стадии 1 на подходящий карбоновокислотный структурный элемент, как показано в Таблице ниже:

Промежуточное соединение B77
7-(5-хлор-2-гидрокси-4-метилфенил)-6-фтор-3-метил-1,3-бензоксазол-2-он
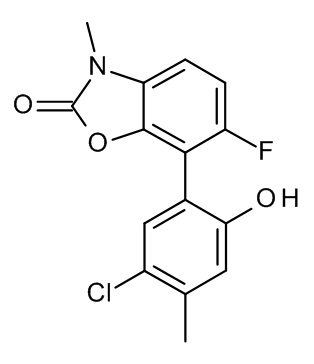
Стадия 1: 7-бромо-6-фтор-3H-1,3-бензоксазол-2-он

Раствор 1,1´-карбонилдиимидазола (378 мг) в тетрагидрофуране (6.0 мл) добавили к суспензии 6-амино-2-бромо-3-фторфенола (400 мг, CAS: 1257535-00-4) в ТГФ (5.0 мл) при комнатной температуре в атмосфере аргона. Смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь влили затем в воду со льдом и водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали досуха. Неочищенное вещество очистили с помощью флеш-хроматографии (0% до 100% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде оранжевого осадка (298 мг, 65%). MS (ESI): m/z = 230.0 [M-H]-.
Стадия 2: 7-бромо-6-фтор-3-метил-1,3-бензоксазол-2-он

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B63, Стадия 2, из 7-бромо-6-фтор-3H-1,3-бензоксазол-2-она (290 мг) и получили в виде светло-коричневой пены (263 мг, 84%). MS (EI): m/z = 245.0 [M]+.
Стадия 3: 2-(5-хлор-2-метокси-4-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
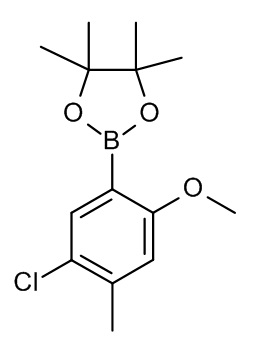
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B67, Стадия 2, из 1-хлор-5-йод-4-метокси-2-метилбензола (промежуточное соединение B1, Стадия 2) (300 мг) и получили в виде серо-белого осадка (75 мг, 21%, чистота ~80%). MS (ESI): m/z = 311.1 [M+H]+.
Стадия 4: 7-(5-хлор-2-метокси-4-метилфенил)-6-фтор-3-метил-1,3-бензоксазол-2-он

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B58 из 7-бромо-6-фтор-3-метил-1,3-бензоксазол-2-она (промежуточное соединение B77, Стадия 2) (38 мг) и 2-(5-хлор-2-метокси-4-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (71 мг, чистота ~80%) и получили в виде серо-белого осадка (18 мг, 36%). MS (ESI): m/z = 322.0 [M+H]+.
Стадия 5: 7-(5-хлор-2-гидрокси-4-метилфенил)-6-фтор-3-метил-1,3-бензоксазол-2-он
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B1, Стадия 3, из 7-(5-хлор-2-метокси-4-метилфенил)-6-фтор-3-метил-1,3-бензоксазол-2-она (18 мг) и получили в виде светло-коричневого осадка (15 мг, 79%, чистота 90%). MS (ESI): m/z = 306.1 [M-H]-.
Промежуточное соединение B86
3-(5-хлор-2-гидрокси-4-метилфенил)-4-(трифторметокси)бензонитрил

Циклопентилметиловый эфир (5 мл) продули три раза аргоном и затем объединили с 3-бромо-4-(трифторметокси)бензонитрилом (130 мг, CAS: 191602-89-8), ацетатом калия (144 мг), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксабороланом) (130 мг) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) ДХМ комплексом (20 мг). Смесь нагревали до 95 °C в течение 4 часов в атмосфере аргона. Реакционной смеси дали остыть до КТ и затем объединили с продутым аргоном раствором карбоната натрия (15% в воде, 600 мкл), 4-хлор-2-йод-5-метилфенолом (131 мг, полученный в способе для Промежуточного соединения B1, Стадия 3) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II) ДХМ комплексом (20 мг). Реакционную смесь снова нагревали до 86 °C в течение 18 часов. Реакционную смесь охладили до КТ и влили затем в воду со льдом. Водный слой экстрагировали дважды с помощью этилацетата и органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Остаток очистили с помощью флеш-хроматографии (силикагель, 20 г картридж, градиент от 0% до 20% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтого осадка (48 мг). MS (ESI-): m/z = 326.3 [M-H]-.
Следующие промежуточные соединения получили по аналогии с Промежуточным соединением B86 посредством замены 3-бромо-4-(трифторметокси)бензонитрила на соответствующий арил-бромид и, если необходимо, 4-хлор-2-йод-5-метилфенол на соответствующий арил-йодид, как описано в следующей таблице:








Промежуточное соединение B87
2-хлор-5-[2-фтор-5-(оксолан-2-илметокси)фенил]-4-гидроксибензонитрил

Это соединение получили по аналогии с Промежуточным соединением B67, Стадии 1-3, посредством замены на стадии 1 (тетрагидрофуран-3-ил)метанола на (тетрагидрофуран-2-ил)метанол (CAS: 97-99-4), и на стадии 3 4-хлор-2-йод-5-метилфенола на 2-хлор-4-гидрокси-5-йодобензонитрил (полученный в способе для Промежуточного соединения B31, Способ B, Стадия 1). Светло-коричневое масло: MS (ESI): 348.1 [M+H]+.
Промежуточное соединение B97
2-хлор-5-[2-фтор-5-(трифторметокси)фенил]-4-гидроксибензонитрил

Этот материал получили по аналогии с Промежуточным соединением B86 из 2-бромо-1-фтор-4-(трифторметокси)бензола (210 мг, CAS: 286932-57-8) и 2-хлор-4-гидрокси-5-йодобензонитрила (204 мг, промежуточное соединение B31, Способ B, Стадия 1) с получением соединения, указанного в заголовке, в виде бесцветного осадка (130 мг, 47%). MS (m/z): 330.1 [M-H]-.
Промежуточное соединение B98
2-хлор-5-[2-фтор-5-(2,2,2-трифторэтокси)фенил]-4-гидроксибензонитрил

Стадия 1: 2-бромо-1-фтор-4-(2,2,2-трифторэтокси)бензол

3-бромо-4-фторфенол (500 мг, CAS: 27407-11-0), 1,1,1-трифтор-2-йодэтан (824 мг, CAS: 353-83-3) и карбонат калия (1.09 г) объединили с ДМФ (10.0 мл) при комнатной температуре в атмосфере аргона. Смесь нагревали до 80°C в течение 2 часов и затем при 60°C в течение дополнительных 16 часов. ТСХ показала большое количество исходного соединения. Дополнительное количество 1,1,1-трифтор-2-йодэтана (824 мг) и карбоната калия (724 мг) добавили и смесь нагревали снова при 80°C в течение 64 часов. Реакционную смесь затем охладили и влили в воду со льдом. Водный слой подщелачили насыщенным раствором Na2CO3 и экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Оставшийся ДМФ удалили совместной эвапорацией с толуолом. Неочищенный материал очистили с помощью флеш-хроматографии (ISCO, силикагель, 25 г картридж, 0% до 20% этилацетат в гептане) с получением соединения, указанного в заголовке, в виде светло-желтой жидкости (601 мг, 80%). MS (EI, m/z): 272.0 [M]+.
Стадия 2: 2-хлор-5-[2-фтор-5-(2,2,2-трифторэтокси)фенил]-4-гидроксибензонитрил
Этот материал получили по аналогии с Промежуточным соединением B86 из 2-бромо-1-фтор-4-(2,2,2-трифторэтокси)бензола (220 мг) и 2-хлор-4-гидрокси-5-йодобензонитрила (203 мг, промежуточное соединение B31, Способ B, Стадия1) с получением соединения, указанного в заголовке в виде серо-белого осадка (8 мг, 3%). (MS (m/z): 346.1 [M+H]+.
Промежуточные соединения C
Промежуточное соединение C3
4-хлор-5-фтор-2-изопропил-фенол
Стадия 1: 4-хлор-5-фтор-2-изопропенил-фенол

К раствору 4-хлор-5-фтор-2-йод-фенола (500 мг, CAS: 1235407-15-4) в ДМФ (8 мл) добавили изопропенилбороновой кислоты пинаколовый эфир (771 мг, CAS: 126726-62-3) и K2CO3 (761 мг) и реакционную смесь продули аргоном в течение 30 мин. Затем добавили Pd(dppf)Cl2 ДХМ комплекс (45 мг, CAS: 14221-01-3) и реакционную смесь перемешивали при 90 °C в течение 16 часов. Растворитель эвапорировали, остаток разбавили H2O (40 мл) и экстрагировали с помощью EtOAc (3x25 мл). Объединенные органические слои промыли солевым раствором (30 мл), высушили над безводным Na2SO4, отфильтровали и сконцентрировали при пониженном давлении. Неочищенное вещество очистили с помощью флеш-хроматографии через силикагель (2-4% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде бесцветной жидкости (263 мг, 76%).1H-NMR (400 MHz, CDCl3) 2.07 (3H, s), 5.15 (1H, s), 5.42 (1H, s), 5.73 (1H, bs), 6.73 (1H, d, J = 8), 7.12 (1H, d, J = 8).
Стадия 2: 4-хлор-5-фтор-2-изопропил-фенол
Раствор 4-хлор-5-фтор-2-изопропенил-фенола (524 мг) в MeOH (30 мл) продули аргоном в течение 15 мин. Затем, 10% палладий на угле (130 мг, CAS: 7440-05-3) добавили и реакционную смесь продули аргоном в течение дополнительных 5 мин. Реакционную смесь перемешивали под водородом (баллон) в течение 16 часов при комнатной температуре. Реакционную смесь отфильтровали через слой целита, слой целита промыли с помощью EtOAc (20 мл) и фильтрат эвапорировали с получением соединения, указанного в заголовке, в виде зеленой жидкости (513 мг, 97%).1H-NMR (400 MHz, CDCl3) 1.25 (6H, m), 3.07-3.14 (1H, m), 4.99 (1H, s), 6.58 (1H, d, J = 12), 7.13 (1H, d, J = 8).
Промежуточное соединение C4:

5-хлор-2-изопропил-4-метил-фенол
Стадия 1: 1-хлор-4-йод-5-метокси-2-метил-бензол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением A14, Стадия 5, из 4-хлор-2-метокси-5-метил-фениламина (CAS: 62492-42-6) в виде коричневой жидкости.1H-NMR (400 MHz, CDCl3): 2.25 (3H, s), 3.83 (3H, s), 6.79 (1H, s), 7.60 (1H, s).
Стадия 2: 5-хлор-2-йод-4-метил-фенол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B1, Стадия 3, из 1-хлор-4-йод-5-метокси-2-метил-бензола в виде желтой жидкости.1H-NMR (400 MHz, CDCl3): 2.25 (3H, s), 5.17 (1H, s), 6.99 (1H, s), 7.49 (1H, s).
Стадия 3: 5-хлор-2-изопропенил-4-метил-фенол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением C3, Стадия 1, из 5-хлор-2-йод-4-метил-фенола и изопропенилбороновой кислоты пинаколового эфира (CAS: 126726-62-3) в виде желтой жидкости.1H-NMR (400 MHz, CDCl3): 2.20 (3H, s), 2.26 (3H, s) 5.10 (1H, s), 5.38 (1H, s), 6.93 (1H, s), 6.94 (1H, s).
Стадия 4: 5-хлор-2-изопропил-4-метил-фенол
Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением C3, Стадия 2, из 5-хлор-2-изопропенил-4-метил-фенола посредством гидрогенизации в виде бесцветной жидкости.1H-NMR (400 MHz, CDCl3) 1.22 (6H, m), 2.27 (3H, s), 3.08-3.11 (1H, m), 4.63 (1H, s), 6.75 (1H, s), 6.93 (1H, s).
Промежуточное соединение C9:
2-(5-хлор-2-гидрокси-4-метил-фенил)-2-метил-пропионитрил
Стадия 1: 4-хлор-2-фтор-5-метил-фенол

К раствору 2-фтор-5-метил-фенола (5.0 г, CAS: 63762-79-8) в AcOH (50 мл) добавили NCS (5.82 г, CAS: 128-09-6) и трифторметансульфокислоту (1.76 мл, CAS: 1493-13-6) и реакционную смесь кипятили с обратным холодильником в течение 24 часов. Растворитель эвапорировали при пониженном давлении и соединение очистили с помощью флеш-хроматографии через силикагель (40-50% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде бесцветной жидкости (3.5 г, 55%).1H-NMR (400 MHz, CDCl3): 2.26 (3H, s), 6.85 (1H, d, J = 8), 7.07 (1H, d, J = 8).
Стадия 2: 1-Бензилокси-4-хлор-2-фтор-5-метил-бензол
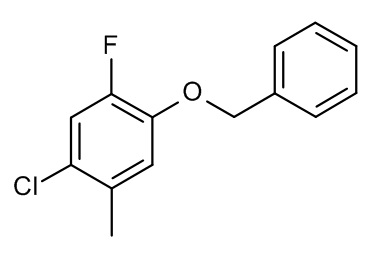
К перемешанному раствору 4-хлор-2-фтор-5-метил-фенола (3.25 г) в CH3CN (50 мл) добавили Cs2CO3 (13.19 г, CAS: 534-17-8) и бензилбромид (2.66 мл, CAS: 100-39-0) и реакционную смесь кипятили с обратным холодильником в течение 2 часов. Реакционную смесь отфильтровали через слой целита и фильтрат эвапорировали. Соединение очистили посредством колоночной хроматографии (10% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде белого осадка (3.94 г, 78%).1H-NMR (400 MHz, CDCl3): 2.27 (3H, s), 5.09 (2H, s), 6.85(1H, d, J = 8), 7.09 (1H, d, J = 12), 7.30-7.42 (5H, m).
Стадия 3: 2-(2-Бензилокси-5-хлор-4-метил-фенил)-2-метил-пропионитрил

К перемешанному раствору 1-бензилокси-4-хлор-2-фтор-5-метил-бензола (1.0 г) в безводном толуоле (10 мл) в закрытой пробирке добавили изобутиронитрил (1.43 мл, CAS: 78-82-0) и KHMDS (6 мл, CAS: 40949-94-8, 1M раствор в толуоле) и реакционную смесь перемешивали при 60 °C в течение 12 часов. Реакционную смесь влили в H2O (20 мл) и экстрагировали EtOAc (2x30 мл). Объединенный органический экстракт высушили над Na2SO4 и эвапорировали при пониженном давлении. Соединение очистили с помощью флеш-хроматографии через силикагель (3-20% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде коричневого осадка (167 мг, 14%).1H-NMR (400 MHz, CDCl3): 1.74 (6H, s) ,2.32 (3H, s), 5.15 (2H, s), 6.85 (1H, s), 7.28 (1H, s), 7.31-7.51 (5H, m).
Стадия 4: 2-(5-хлор-2-гидрокси-4-метил-фенил)-2-метил-пропионитрил
К перемешанному раствору 2-(2-бензилокси-5-хлор-4-метил-фенил)-2-метил-пропионитрила (420 мг) в безводном ДХМ (20 мл) при -78 °C добавили BCl3 (2.8 мл, 1M раствор в ДХМ, CAS: 10294-34-5) и реакционную смесь перемешивали при 25 °C в течение 4 часов. Реакционную смесь погасили насыщенным водным раствором Na2CO3 и экстрагировали с помощью ДХМ (2x20 мл). Объединенные органические слои высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили с помощью флеш-хроматографии через силикагель (0-10% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде коричневого осадка (50 мг, некоторые примеси).1H-NMR (400 MHz, CDCl3): 1.76 (6H, s), 2.28 (3H, s), 6.57 (1H, s), 7.29 (1H, s).
Промежуточное соединение C1-A:
3-хлор-6-[(4-хлор-2-изопропил-5-метил-фенокси)метил]пиридазин

К раствору 3-хлор-6-хлорметил-пиридазина (81 мг, CAS: 120276-59-7) в ацетоне (5 мл) добавили 4-хлор-2-изопропил-5-метил-фенол (92 мг, CAS: 89-68-9) и K2CO3 (104 мг) и реакционную смесь нагревали при кипении с обратным холодильником в течение 16 часов. Реакционную смесь влили в H2O (50 мл) и EtOAc (75 мл) и слои разделили. Органический слой промыли H2O (50 мл), высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом с получением соединения, указанного в заголовке, в виде светло-желтого осадка (0.072 г, 46%). MS (ESI): m/z = 311.07 [M]+.
Промежуточное соединение C2-A:
3-хлор-6-(4-хлор-2-циклопропил-5-метил-феноксиметил)-пиридазин
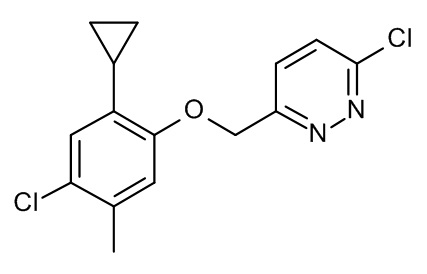
К раствору 3-хлор-6-хлорметил-пиридазина (200 мг, CAS: 120276-59-7) в CH3CN (10 мл) добавили 4-хлор-2-циклопропил-5-метил-фенол (82 мг, Промежуточное соединение A19), K2CO3 (109 мг, CAS: 584-08-7) и тетрабутиламмония йодид (16 мг, CAS: 311-28-4) и реакционную смесь нагревали до кипения в течение 16 часов. Растворитель эвапорировали при пониженном давлении и продукт очистили с помощью флеш-хроматографии через силикагель (30-40% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде желтого осадка (48 мг, 35%).1H-NMR (400 MHz, CDCl3): 0.61-0.65 (2H, m), 0.90-0.95 (2H, m), 2.07-2.09 (1H, m), 2.28 (3H, s), 5.41 (2H, s), 6.73 (1H, s), 6.83 (1H, s), 7.56 (1H, d, J = 8), 7.74 (1H, d, J = 8).
Следующие промежуточные соединения получили по аналогии с Промежуточным соединением C2-A из коммерчески доступного 3-хлор-6-хлорметил-пиридазина (CAS: 120276-59-7) и подходящего фенольного структурного элемента, как показано в следующей Таблице:
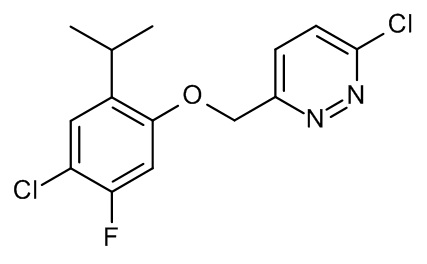
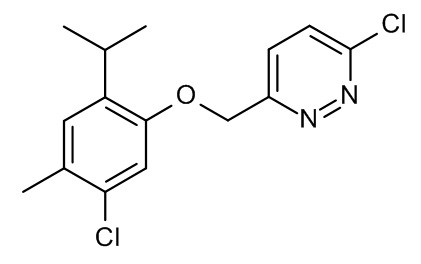


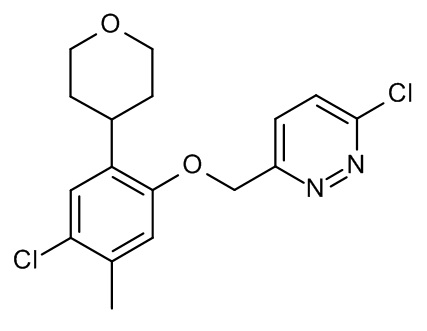


Следующие промежуточные соединения типа C были получены по аналогии с Промежуточным соединением C2-A из коммерчески доступного 3-хлор-6-хлорметил-пиридазина (CAS: 120276-59-7) и подходящего фенольного структурного элемента, как показано в следующей Таблице:



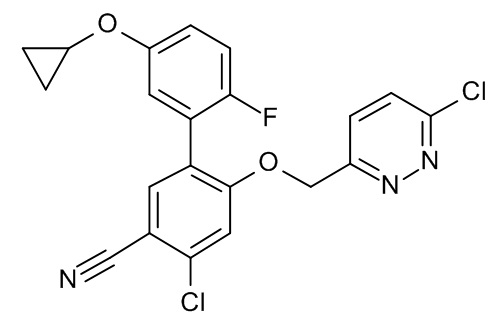
Промежуточные соединения D
Промежуточное соединение D9:
2-трет-бутил-5-метил-4-метилсульфонил-фенол

Стадия 1: 4-бромо-2-трет-бутил-5-метил-фенол
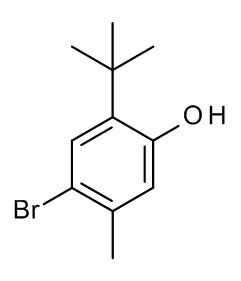
К раствору 2-трет-бутил-5-метилфенола (1.0 г, CAS: 88-60-8) добавили N-бромсукцинимид (1.19 г) и реакционную смесь перемешивали в течение 15 ч . Реакционную смесь затем влили в насыщенный раствор NH4Cl и экстрагировали с помощью EtOAc. Объединенные органические слои высушили над Na2SO4 и сконцентрировали под вакуумом. Остаток очистили посредством хроматографии (силикагель, 20 г, градиент EtOAc в гептане) с получением соединения, указанного в заголовке (1.4 г, 93%) в виде желтого масла. MS (ESI): m/z = 244.2 [M+H]+.
Стадия 2: 1-Бензилокси-4-бромо-2-трет-бутил-5-метил-бензол
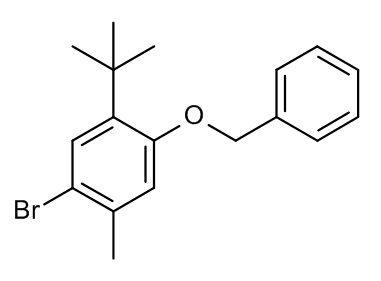
К раствору 4-бромо-2-трет-бутил-5-метил-фенола (1.4 г, в ДМФ (15 мл) медленно добавили NaH (302 мг, 55% в минеральном масле) и реакционную смесь перемешивали в течение 30 минут при комнатной температуре. Затем добавили бензилхлорид (765 мг, 696 мкл) и реакционную смесь перемешивали в течение 2 ч. Реакционную смесь влили в насыщенный раствор NH4Cl и экстрагировали с помощью EtOAc. Объединенные органические слои высушили над Na2SO4 и сконцентрировали под вакуумом с получением соединения, указанного в заголовке (1.9 г, 97%) в виде белого порошка, который использовали на следующей реакционной стадии без дополнительной характеризации.
Стадия 3: 1-Бензилокси-2-трет-бутил-5-метил-4-метилсульфонил-бензол
К смеси 1-бензилокси-4-бромо-2-трет-бутил-5-метил-бензола (1.9 г), L-пролина (525 мг и йодида меди (I) (869 мг) в ДМФ (10 мл) добавили гидроксид натрия (182 мг) и метансульфинат натрия (1.16 г) и реакционную смесь нагревали до 135°C в закрытой пробирке в течение 15 ч. Реакционную смесь влили в насыщенный раствор NH4Cl и экстрагировали с помощью EtOAc. Объединенные органические слои высушили над Na2SO4и сконцентрировали под вакуумом. Остаток очистили посредством хроматографии (силикагель, 20 г, 0% до 50% EtOAc в гептане) с получением соединения, указанного в заголовке (1.33 г, 69%) в виде белого осадка. MS (ESI): m/z = 331.13 [M-H]-.
Стадия 4: 2-трет-бутил-5-метил-4-метилсульфонил-фенол

К раствору 1-бензилокси-2-трет-бутил-5-метил-4-метилсульфонил-бензола (1.2 г) в смеси MeOH (100 мл) и EtOAc (100 мл) добавили Pd(C) (10% на угле, 200 мг) в атмосфере аргона. Реакционную пробирку затем откачали и продули водородом и реакционную смесь перемешивали в течение 18 ч. Реакционную смесь отфильтровали через дикалит и фильтрат сконцентрировали под вакуумом с получением соединения, указанного в заголовке (865 мг, 99%) в виде белого осадка. MS (ESI): m/z = 241.09 [M-H]-.
Промежуточное соединение D26-B:
3-Бромометил-1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин
Стадия 1: 4-Гидразино-2-метилсульфанил-пиримидин-5-карбоновой кислоты этиловый эфир
К раствору гидратгидразина (6.28 мл) в этаноле (100 мл) добавили по каплям раствор 4-хлор-2-метилсульфанил-пиримидин-5-карбоновой кислоты этилового эфира (10 г, CAS: 5909-24-0) в этаноле (250 мл) при 0°C и реакционную смесь перемешивали при 25°C в течение 1 ч. Все летучие компоненты удалили при пониженном давлении с получением соединения, указанного в заголовке (12.0 г) в виде серо-белого осадка, который использовали на следующей реакционной стадии без дополнительной очистки. MS (ESI): m/z = 229.5 [M+H]+.
Стадия 2: 6-метилсульфанил-1,2-дигидро-пиразоло[3,4-d]пиримидин-3-он

Раствор 4-гидразино-2-метилсульфанил-пиримидин-5-карбоновой кислоты этилового эфира (12.0 г) в 10% водном растворе KOH (300 мл) кипятили с обратным холодильником в течение 15 мин. Реакционную смесь затем охладили до 0°C и подкислили 25% водным AcOH. Получившийся преципитат отфильтровали, высушили при пониженном давлении и эвапорировали совместно с толуолом с получением соединения, указанного в заголовке (6.3 г, 66%), в виде серо-белого осадка. MS (ESI): m/z = 183.2 [M+H]+.
Стадия 3: 3-бромо-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин

Смесь 6-метилсульфанил-1,2-дигидро-пиразоло[3,4-d]пиримидин-3-она (4.5 г) и POBr3 (21.21 г) нагревали в закрытой пробирке до 170°C в течение 8 ч. Реакционную смесь охладили до 25°C, разбавили водой и подщелачили 25% водным раствором аммония и затем экстрагировали EtOAc. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении с получением соединения, указанного в заголовке (2.41 г, 40%) в виде светло-коричневого осадка. MS (ESI): m/z = 245.1 [M+H]+.
Стадия 4: 3-бромо-1-(4-метокси-бензил)-6-метилсульфанил-1I-пиразоло[3,4-d]пиримидин
К раствору 3-бромо-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидина (3.6 г) в безводном ДМФ (100 мл) добавили Cs2CO3(7.16 г) при 25°C и реакционную смесь перемешивали при 25°C в течение 15 мин. Затем 1-(хлорметил)-4-метокси-бензол (2.39 мл) добавили по каплям при 25°C и реакционную смесь перемешивали при 25°C в течение 16 ч. Реакционную смесь отфильтровали и фильтрат разбавили водой и экстрагировали EtOAc. Объединенные органические экстракты высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (3-6% EtOAc в гексане) с получением соединения, указанного в заголовке (4 г, 75%), в виде серо-белого осадка. MS (ESI): m/z = 367.1 [M+H]+.
Стадия 5: 1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты метиловый эфир
К раствору 3-бромо-1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидина (2.0 г) в смеси безводного ДМФ (30 мл) и MeOH (25 мл) добавили Pd(OAc)2 (98 мг), DPPP (135 мг) с последующим Et3N (2.34 мл) при 25°C. Реакционную смесь перемешивали в атмосфере монооксида углерода при 70 фунт/кв. дюйм и при 70°C в течение 16 ч. Реакционную смесь отфильтровали через целит и растворитель эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (15-20% EtOAc в гексане) с получением соединения, указанного в заголовке (1.38 г, 73%), в виде серо-белого осадка. MS (ESI): m/z = 345.3 [M+H]+.
Стадия 6: [1-(4-метокси-бензил)-6-метилсульфанил-4,7-дигидро-1H-пиразоло[3,4-d]пиримидин-3-ил]-метанол

Суспензию NaBH4 (2.64 г) и CaCl2 (3.87 г) в смеси ТГФ (90 мл) и EtOH (90 мл) охладили до 0°C и раствор -(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-карбоновой кислоты метилового эфира (3.0 г) в безводном ТГФ (60 мл) добавили по каплям. Реакционную смесь перемешивали при 0°C в течение 2 ч и затем погасили медленным добавлением воды при 0°C, с последующим разбавлением с помощью EtOAc. Органический слой разделили и водный слой экстрагировали EtOAc. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали с получением соединения, указанного в заголовке (2.67 г, 97%), в виде серо-белого осадка. MS (ESI): m/z = 319.2 [M+H]+.
Стадия 7: [1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-ил]-метанол
Суспензию [1-(4-метокси-бензил)-6-метилсульфанил-4,7-дигидро-1H-пиразоло[3,4-d]пиримидин-3-ил]-метанола (2.67 г) и хлоранила (2.064 г) в безводном толуоле (100 мл) кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охладили до 25°C и добавили EtOAc и воду. Органический слой разделили, промыли солевым раствором, высушили над Na2SO4 и эвапорировали при пониженном давлении. Получившийся остаток очистили посредством колоночной хроматографии через силикагель (50-70% EtOAc в гексане) с получением соединения, указанного в заголовке (2.2 г, 83%), в виде серо-белого осадка. MS (ESI): m/z = 317.0 [M+H]+.
Стадия 8: 3-Бромометил-1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин

К раствору [1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-ил]-метанола (1.5 г) в безводном ацетонитриле (100 мл) добавили PBr3 (0.586 мл) по каплям при 25°C и реакционную смесь перемешивали при 25°C в течение 2 ч. Смесь разбавили EtOAc и промыли насыщенным водным раствором NaHCO3 и солевым раствором. Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Получившийся остаток очистили посредством колоночной хроматографии через силикагель (10-14% EtOAc в гексане) с получением соединения, указанного в заголовке (1.26 г, 70%), в виде серо-белого осадка. MS (ESI): m/z = 381.0 [M+H]+.
Промежуточное соединение D3-A:
Трет-бутил 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]пиразоло[3,4-b]пиридин-1-карбоксилат

Суспензию 2-трет-бутил-4-хлор-5-метил-фенола (61.0 мг, CAS: 30894-16-7), трет-бутил 3-(бромометил)пиразоло[3,4-b]пиридин-1-карбоксилата (95.8 мг, CAS: 174180-76-8, полученного в соответствии с WO200876223A1) и карбоната калия (106 мг) в ацетоне (2.5 мл) нагревали до 50°C в течение 3 ч. Ацетон удалили под вакуумом и остаток разбавили насыщенным раствором NH4Cl. Смесь экстрагировали EtOAc и объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством хроматографии (20 г силикагель; гептан/EtOAc 90:10 - 75:25) с получением соединения, указанного в заголовке, в виде белого осадка (89 мг, 67%). MS (ESI): m/z = 428.4 [M-H]-.
Следующие промежуточные соединения были синтезированы из подходящих структурных элементов по аналогии с Промежуточным соединением D3-A:












Промежуточное соединение D43:
2-хлор-5-[2-фтор-5-(пирролидин-1-карбонил)фенил]-4-гидрокси-бензонитрил

Стадия 1: 2-хлор-5-[2-фтор-5-(пирролидин-1-карбонил)фенил]-4-(метоксиметокси)бензонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 3, из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B31, Стадия 2) (80 мг) и (2-фтор-5-(пирролидин-1-карбонил)фенил)бороновой кислоты (82.3 мг, CAS: 874289-42-6) и получили в виде белой пены (60 мг, 52%). MS (ESI): m/z = 389.2 [M+H]+.
Стадия 2: 2-хлор-5-[2-фтор-5-(пирролидин-1-карбонил)фенил]-4-гидрокси-бензонитрил
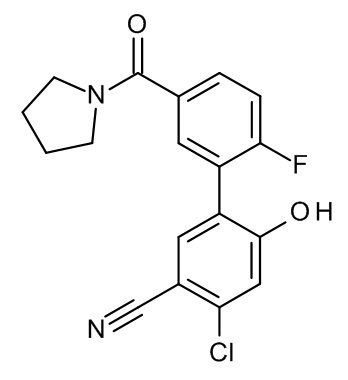
К суспензии 4-хлор-2'-фтор-6-(метоксиметокси)-5'-(пирролидин-1-карбонил)-[1,1'-бифенил]-3-карбонитрила (50 мг) в диоксане (3 мл) добавили 4M HCl в диоксане (257 мкл) по каплям в течение периода 2 минут при комнатной температуре. Получившийся раствор перемешивали при комнатной температуре в течение 2 ч. Затем снова 4M HCl в диоксане (129 мкл) добавили и перемешивание продолжили в течение 5 ч. Реакционную смесь эвапорировали досуха и остаток очистили перекристаллизации из ДХМ и гептана с получением соединения, указанного в заголовке (40 мг, 88%) в виде белого осадка. MS (ESI): m/z = 386.2 [M+CH3CN+H]+.
Промежуточное соединение D44-B:
3-(Хлорметил)-1-тритилпиразоло[3,4-c]пиридин

Стадия 1: Этил 1-тритилпиразоло[3,4-c]пиридин-3-карбоксилат

В 20 мл круглодонной колбе, этил 1H-пиразоло[3,4-c]пиридин-3-карбоксилат (CAS: 1053656-33-9, 500 мг) объединили с ДМФ (3 мл). Триэтиламин (397 мг) добавили с получением светло-коричневого раствора. Смесь охладили при 0°C и затем медленно добавили [хлор(дифенил)метил]-бензол (802 мг). Реакционную смесь перемешивали в течение 4 h. Реакционную смесь влили в 20 мл этилацетат и органический раствор промыли с помощью H2O (2 x 10 мл). Водные слои снова экстрагировали этилацетатом. Органические слои объединили, высушили над Na2SO4 и сконцентрировали под вакуумом. Остаточный ДМФ удалили посредством добавления толуола с последующей эвапорацией под вакуумом. Неочищенное вещество очистили с помощью флеш-хроматографии (силикагель, 20 г, градиент от 0% до 60% этилацетата в гептане) с получением соединения, указанного в заголовке, в виде бесцветной пены (700 мг). MS (m/z): 434.18 [M+H]+.
Стадия 2: (1-Тритилпиразоло[3,4-c]пиридин-3-ил)метанол

В 20 мл круглодонной колбе, этил 1-тритил-1H-пиразоло[3,4-c]пиридин-3-карбоксилат (700 мг) объединили с CH2Cl2 (3 мл) и затем охладили до -78°C. Раствор гидрида диизобутилалюминия (1 M в CH2CL2, 3.2 мл) медленно добавили и смесь оставили перемешиваться при -78°C в течение 2 ч. Реакционную смесь влили в 20 мл этилацетат и раствор сегнетовой соли (10 мл) и экстрагировали этилацетатом (2 x 10 мл).Органические слои объединили, высушили над Na2SO4 и сконцентрировали под вакуумом с получением белой пены (500 мг), которую использовали без дополнительной очистки. MS (m/z): 392.17 [M+H]+.
Стадия 3: 3-(Хлорметил)-1-тритилпиразоло[3,4-c]пиридин

В 20 мл круглодонной колбе, (1-тритил-1H-пиразоло[3,4-c]пиридин-3-ил)метанол (55 мг объединили с CH2Cl2 (3 мл) и смесь охладили до 0°C. Тионилхлорид (20.4 мкл) затем добавили по каплям. Ледяную ванну убрали и реакционную смесь оставили перемешиваться при комнатной температуре в течение 1 ч. Реакционную смесь погасили водным раствором бикарбоната натрия и органический слой разделили, промыли солевым раствором и высушили над Na2SO4. Раствор сконцентрировали под вакуумом с получением соединения, указанного в заголовке, в виде бесцветной пены (53 мг). MS (m/z): 410.14 [M+H]+.
Промежуточное соединение D54-B
3-(Хлорметил)-1-тритилпиразоло[4,3-c]пиридин
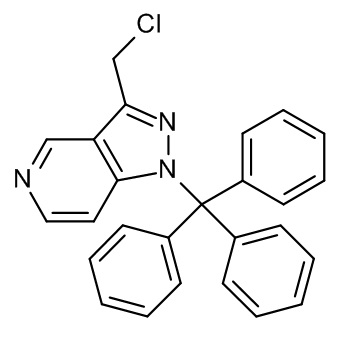
Стадия 1: Этил 1H-пиразоло[4,3-c]пиридин-3-карбоксилат
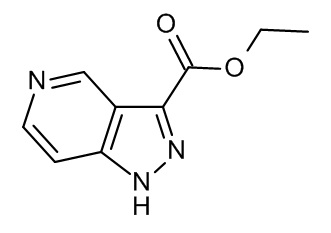
В атмосфере аргона, тионилхлорид (241 мг, 147 мкл) добавили в раствор 1H-пиразоло[4,3-c]пиридин-3-карбоновой кислоты (300 мг, CAS: 932702-11-9) в этаноле (20 мл). Смесь затем нагревали при кипении и оставили перемешиваться в течение 2 часов. Смесь охладили до комнатной температуры и погасили насыщенным раствором Na2CO3. Водный слой экстрагировали дважды с помощью этилацетата (80 мл). Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали, эвапорировали и высушили в глубоком вакууме с получением соединения, указанного в заголовке, в виде светло-желтого порошка (228 мг, 62%). MS (m/z): 192.1 [M+H]+.
Стадии 2 - 4: 3-(Хлорметил)-1-тритилпиразоло[4,3-c]пиридин
Этот материал получили в виде осадка в точном соответствии с Промежуточным соединением D44-B, Стадии 1 - 3, из этил 1H-пиразоло[4,3-c]пиридин-3-карбоксилата (полученного выше на стадии 1) сначала тритилированием, затем восстановлением до гидроксиметильного производного, и, в заключение хлорированием. MS (m/z): 410.2 [M+H]+.
Промежуточные соединения F
Промежуточное соединение F1:
трет-бутил 3-(бромометил)пиразол-1-карбоксилат
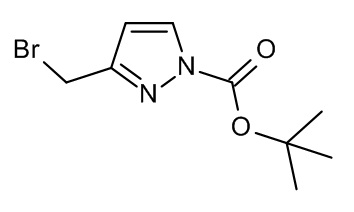
Стадия 1: трет-бутил 3-метилпиразол-1-карбоксилат

К раствору 3-метил-1H-пиразола (500 мг) в ацетонитриле (10 мл) добавили ди-трет-бутил дикарбонат (1.59 г, 1.7 мл) и DMAP (74.4 мг) при 0°C. Смеси дали нагреться до КТ и перемешивали в течение 2 ч. Затем EtOAc добавили и смесь промыли 0.1 н HCl, насыщенным раствором NaHCO3 и солевым раствором, высушили над Na2SO4 и эвапорировали. Неочищенный продукт (содержащий ~15% трет-бутил 5-метилпиразол-1-карбоксилата) использовали на следующей реакционной стадии без дополнительной очистки.
Стадия 2: трет-бутил 3-(бромометил)пиразол-1-карбоксилат

Смесь трет-бутил 3-метилпиразол-1-карбоксилата (1.24 г), NBS (1.7 г) и бензоилпероксида (308 мг) в CCl4 (50 мл) нагревали до кипения в течение 4 ч. Смесь затем охладили до 0°C, отфильтровали и эвапорировали. Остаток очистили посредством колоночной хроматографии (100 г SiO2, н-гептан/EtOAc 100/0 до 90/10) с получением соединения, указанного в заголовке (481 мг, 27%) в виде бесцветного масла. MS (ESI): m/z = 161.0 [M-CO2tBu+H]+.
Промежуточное соединение F3:
Метил 5-(гидроксиметил)-2-тетрагидропиран-2-ил-пиразол-3-карбоксилат

Стадия 1: Диметил 1-тетрагидропиран-2-илпиразол-3,5-дикарбоксилат

ТФУ (74.0 мг, 50 мкл) добавили к суспензии диметил 1H-пиразол-3,5-дикарбоксилата (1.546 г, CAS: 4077-76-3) в толуоле (15 мл) и смесь нагрели до 80 °C. Затем, 3,4-дигидро-2H-пиран (828 мг, 900 мкл) добавили и реакционную смесь кипятили с обратным холодильником в течение ночи. Смесь охладили до КТ и растворитель эвапорировали. Остаток растворили в воде и экстрагировали с помощью EtOAc. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Остаток очистили посредством хроматографии (50 г силикагель; гептан/EtOAc 100/0 до 50/50) с получением соединения, указанного в заголовке (1.95 г, 87%) в виде белого осадка. MS (ESI): m/z = 185.1 [M-THP+H]+.
Стадия 2: Метил 5-(гидроксиметил)-2-тетрагидропиран-2-ил-пиразол-3-карбоксилат
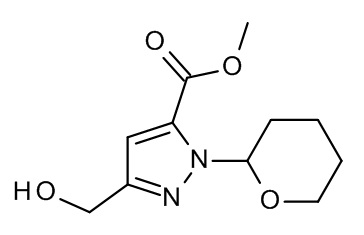
К раствору диметил 1-тетрагидропиран-2-илпиразол-3,5-дикарбоксилата (1.95 г) в смеси ТГФ (40 мл) и диэтилового эфира (80 мл) добавили 1M раствор DIBAL-H в толуоле (16.0 мл) по каплям при -78°C и смесь перемешивали в течение 3 ч при -78°C. Затем дополнительное количество DIBAL-H 1 M в толуоле (8 мл) добавили и смесь перемешивали в течение дополнительного часа. Смесь нагревали до 0 °C и воду (15 мл) добавили по каплям. Белую суспензию эвапорировали и оставшийся белый остаток суспендировали в MeOH (120 мл), продули CO2 (газ) в течение 10 минут и затем кипятили в течение 5.5 ч. Смесь отфильтровали и фильтрат сконцентрировали. Оставшийся материал очистили посредством хроматографии (50 г силикагель, гептан/EtOAc 100/0 до 1/2) с получением соединения, указанного в заголовке (810 мг, 47%) в виде белого осадка. MS (ESI): m/z = 157.0 [M-THP+H]+.
Примеры A: Триазолоны, где RA ≠ арил
Пример A1:
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он

2-(трет-бутил)-4-хлор-5-метилфенол (CAS: 30894-16-7, 250 мг), 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-он (CAS: 1338226-21-3; 279 мг), карбонат калия (348 мг) и йодид калия (20.9 мг) объединили в ацетоне (10.0 мл) при комнатной температуре в атмосфере аргона. Смесь затем нагревали при кипении в течение 3 часов и затем поддерживали при комнатной температуре в течение дополнительных 16 часов. ТСХ показала отсутствие исходного соединения в это время. Реакционную смесь затем влили в воду со льдом и водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенное вещество очистили с помощью флеш-хроматографии на силикагеле с 0% до 70% этилацетата в гептане в качестве элюента с получением соединения, указанного в заголовке, в виде бесцветного осадка (60 мг). MS (m/z): 310.2 [MH]+.
Следующие примеры были синтезированы из подходящих структурных элементов/промежуточных соединений и известного 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-она по аналогии с примером A1:


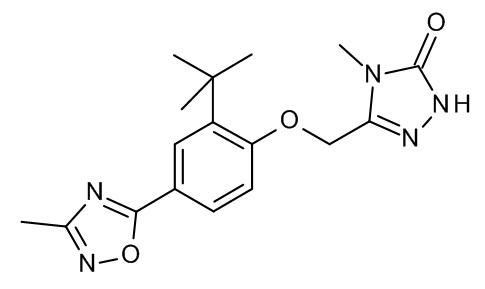


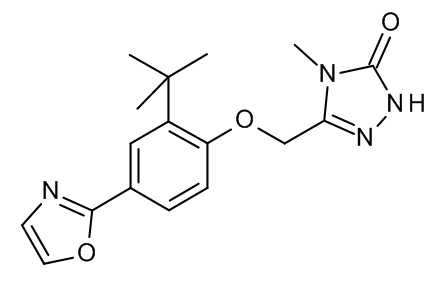

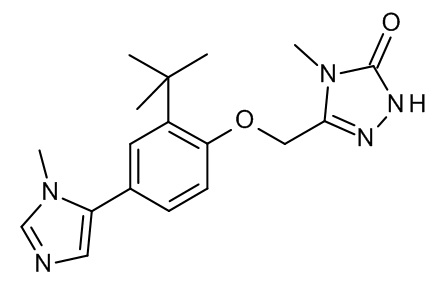
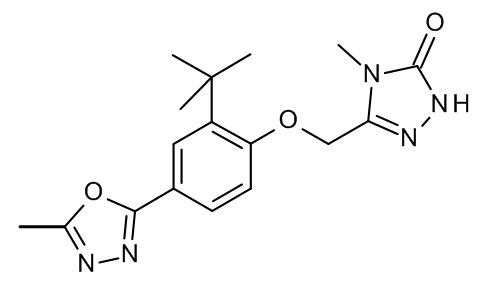







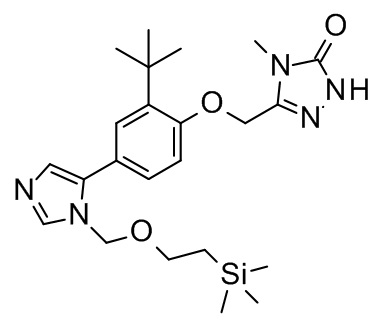



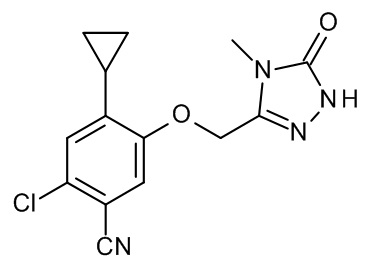


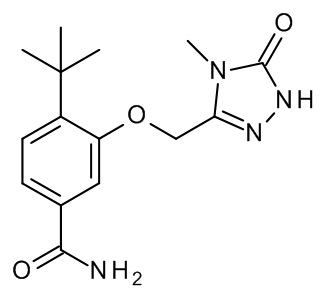
Пример A26
5-трет-бутил-2-метил-4-[(5-оксо-4-пропан-2-ил-1H-1,2,4-триазол-3-ил)метокси]бензонитрил

Стадия 1: 3-(Бензилоксиметил)-4-изопропил-1H-1,2,4-триазол-5(4H)-он
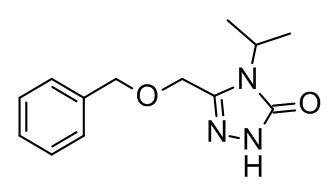
К суспензии N-изопропилгидразинкарбоксамида гидрохлорида (CAS: 35578-82-6, 1.0 г) в ТГФ (12.5 мл) добавили 2-(бензилокси)ацетилхлорид (1.2 г) и смесь охладили до 0°C. К указанной смеси затем добавили по каплям 5M водн. гидроксид натрия (2.67 мл) в течение 3 минут и реакционную смесь тщательно перемешали при комнатной температуре в течение 2.5 часов. Затем NUA удалили эвапорацией под вакуумом. Оставшуюся коричневую вязкую суспензию обработали 2M водным гидроксидом натрия (6.51 мл) и нагрели до 95°C (температура масляной бани) в течение 16 часов. После охлаждения, мутный раствор довели до pH 4 с использованием 25% водн. HCl. Этилацетат добавили и слои разделили. Водный слой экстрагировали дважды с помощью этилацетата. Органические слои промыли солевым раствором, высушили над MgSO4, отфильтровали, обработали силикагелем и эвапорировали. Остаток очистили с помощью хроматографии на силикагеле при градиенте н-гептана в этилацетате (100/0 до 0/100) с получением соединения, указанного в заголовке (0.705 г). MS (m/z): 248.14 [M+H]+
Стадия 2: 3-(гидроксиметил)-4-пропан-2-ил-1H-1,2,4-триазол-5-он

К раствору 3-(бензилоксиметил)-4-изопропил-1H-1,2,4-триазол-5(4H)-она (0.70 г) в этаноле (12 мл) в атмосфере аргона добавили гидроксид палладия на угле (20%, 298 мг) и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре и 0.5 давления в течение 18 часов. Реакционную смесь затем отфильтровали и осадок промыли этанолом (10 мл). Фильтрат эвапорировали с получением соединения, указанного в заголовке, в виде бесцветного осадка (0.391 г). MS (m/z): 157.0 [M]+.
Стадия 3: 3-(Хлорметил)-4-пропан-2-ил-1H-1,2,4-триазол-5-он

К суспензии 3-(гидроксиметил)-4-изопропил-1H-1,2,4-триазол-5(4H)-она (0.30 г) в ацетонитриле (6 мл) добавили тионилхлорид (261 мг) и суспензия быстро превратилась в раствор. Смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь эвапорировали и остаток дважды обработали толуолом и эвапорировали снова для удаления оставшегося тионилхлорида. Остаток серо-белого осадка тритурировали с н-гептаном в ультразвуковой ванне. Суспензию затем отфильтровали, промыли н-гептаном и осадок высушили под вакуумом с получением соединения, указанного в заголовке (0.37 г), в виде бесцветного осадка. MS (m/z): 175.0 [M]+.
Стадия 4: 5-трет-бутил-2-метил-4-[(5-оксо-4-пропан-2-ил-1H-1,2,4-триазол-3-ил)метокси]бензонитрил
Это соединение получили по аналогии с примером 1 из 3-(хлорметил)-4-пропан-2-ил-1H-1,2,4-триазол-5-она и 5-трет-бутил-4-гидрокси-2-метилбензонитрила (промежуточное соединение A16) с получением соединения, указанного в заголовке в виде светло-коричневого осадка. MS (m/z): 329.20 [M+H]+.
Пример A27:
4-метил-3-[(5-метил-2-пропан-2-илфенокси)метил]-1H-1,2,4-триазол-5-он

Стадия 1: 4-метил-3-[(5-метил-2-пропан-2-илфенокси)метил]-5-метилсульфонил-1,2,4-триазол

К раствору 2-изопропил-5-метилфенола (300мг, CAS: 89-83-8) в ацетоне (20 мл) добавили K2CO3 (387 мг) и 3-(йодометил)-4-метил-5-(метилсульфонил)-4H-1,2,4-триазол (783 мг, CAS: 1068603-49-5, полученный в соответствии с WO2008119662). Реакционную смесь перемешивали в течение ночи при 60°C и затем экстрагировали водой (150 мл) и EtOAc (150 мл). Органический слой промыли водой (150 мл), высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенное вещество очистили посредством колоночной хроматографии (градиент вплоть до 50% EtOAc в ДХМ) с получением соединения, указанного в заголовке, в виде желтой смолы (0.241 г). MS (m/z): 324.14 [M+H]+.
Стадия 2: 3-метокси-4-метил-5-[(5-метил-2-пропан-2-илфенокси)метил]-1,2,4-триазол
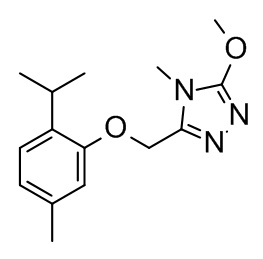
К суспензии 4-метил-3-[(5-метил-2-пропан-2-илфенокси)метил]-5-метилсульфонил-1,2,4-триазола (241 мг) в метаноле (8 мл) добавили метоксид натрия в метаноле (0.6 мл, 5.4 M). Реакционную смесь нагревали при 85°C при кипении с обратным холодильником. Через 2ч реакционную смесь остановили и охладили. Смесь разбавили EtOAc (50 мл), промыли насыщенным водным раствором NaHCO3(50мл) и насыщенным раствором NaCl (20 мл) и затем высушили над Na2SO4 и сконцентрировали под вакуумом. Требуемый продукт получили как таковой в виде светло-желтого осадка и использовали без дополнительной очистки (0.2 г, содержащих некоторое количество исходного соединения). MS (m/z): 276.17 [M+H]+.
Стадия 3: 4-метил-3-[(5-метил-2-пропан-2-илфенокси)метил]-1H-1,2,4-триазол-5-он

К раствору 3-метокси-4-метил-5-[(5-метил-2-пропан-2-илфенокси)метил]-1,2,4-триазола (113 мг) в AcOH (29 мл) добавили HBr (19.5 мл 48% раствора) при комнатной температуре и реакционную смесь перемешивали в течение ночи. Раствор сконцентрировали (при температуре водяной бани 50-70°C) и затем разбавили с помощью ДХМ (50мл) и промыли с помощью 2M раствора KHCO3 (30 мл). Органический слой высушили над Na2SO4, отфильтровали и сконцентрировали с получением соединения, указанного в заголовке, в виде светло-желтого осадка (0.094 г). MS (m/z): 262.2 [M+H]+.
Следующие примеры были синтезированы из подходящих структурных элементов/промежуточных соединений по аналогии с примером A1, Стадии 1-3:

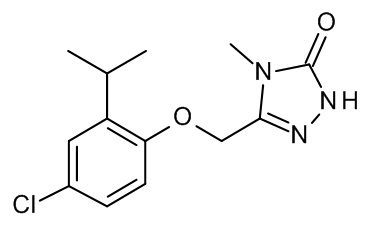


Пример A32:
3-[[2-трет-бутил-4-(1H-имидазол-2-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он
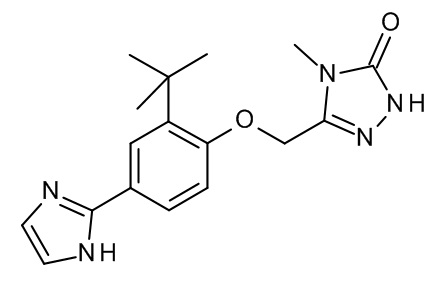
К раствору 3-[[2-трет-бутил-4-[1-(2-триметилсилилэтоксиметил)имидазол-2-ил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-она (45 мг, полученного в примере A5, в безводном ТГФ (10 мл) при 0°C добавили 2 н водную HCl (3 мл) при 0°C и реакционную смесь перемешивали при 25°C в течение 8 ч. Растворитель эвапорировали при пониженном давлении и остаток разбавили водой, подщелачили с помощью насыщенного водного раствора NaHCO3 и экстрагировали с помощью ДХМ (2x30 мл). Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали при пониженном давлении. Остаток очистили колоночной хроматографией на силикагеле ( градиент 3-5% MeOH в ДХМ) с получением соединения, указанного в заголовке (12 мг), в виде серо-белого осадка. MS (m/z): 328.1 [M+H]+.
Следующий пример синтезировали из подходящих структурных элементов/промежуточных соединений по аналогии с примером A32, за исключением того, что растворитель ТГФ заменили на ДХМ, а 2н HCl заменили на трифторуксусную кислоту.

Пример A34
4-трет-бутил-2-хлор-5-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил

Этот материал получили по аналогии с примером A1 из Промежуточного соединения A34 и коммерчески доступного 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-она (CAS: 1338226-21-3) в виде бесцветного осадка. MS (m/z): 321.2 [M+H]+.
Пример A35
5-трет-бутил-2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил
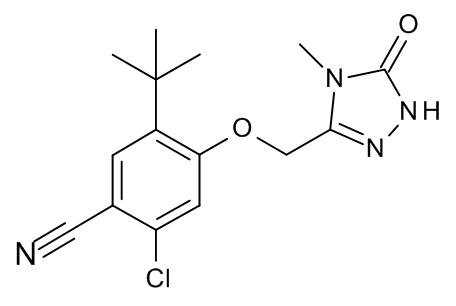
К 5-трет-бутил-2-хлор-4-гидрокси-бензонитрилу (2.29 г, 10.9 ммоль) в N-метил-пирролидиноне (55 мл) добавили NaH (568 мг, 14.2 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. 3-(Хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-он (1.93 г, 13.11 ммоль) растворили в N-метил-пирролидиноне (20 мл) и добавили по каплям через капельную воронку в течение 30 мин. После завершения добавления, воду (160 мл) медленно добавили в реакционную смесь. После перемешивания до охлаждения до комнатной температуры, реакционную смесь отфильтровали и осадки высушили с получением 3.15 г, указанного в заголовке, в виде белого осадка (90% выход). MS (m/z): 321.5 [M+H]+.
Пример A36
5-трет-бутил-2-фтор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]бензонитрил
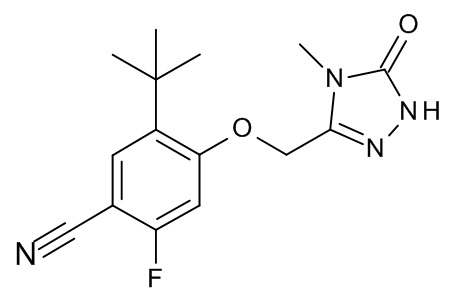
Соединение, указанное в заголовке, получили по аналогии с примером A35, из 5-трет-бутил-2-фтор-4-гидрокси-бензонитрила (0.118 г) и 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-она (0.108 г) и получили (17 мг, 9% ) в виде белого осадка. MS (m/z): 305.5 [M+H]+.
Примеры B: Триазолоны, где RA = арил и гетероарил
Пример B1:
3-[(4-хлор-5-метил-2-фенилфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он

К раствору 4-хлор-5-метил-2-фенилфенола (62.2 мг, промежуточное соединение B1) в ацетоне (2 мл) добавили K2CO3 (42.6 мг) и 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-он (35 мг, CAS: 1338226-21-3). Реакционную смесь перемешивали в течение 2 часов при кипении с обратным холодильником. Реакционную смесь влили в 10% водный раствор NH4Cl (30 мл) и EtOAc (30 мл) и слои разделили. Водный слой экстрагировали дважды с помощью EtOAc (30 мл). Органические слои промыли солевым раствором (30 мл), высушили над MgSO4, отфильтровали и сконцентрировали под вакуумом. Остаток очистили с помощью хроматографии на силикагеле на 20 г колонке, с использованием ЖХСД системы Flashmaster с элюцией при градиенте ДХМ: MeOH (100/0 до 50/50) с получением соединения, указанного в заголовке, в виде серо-белого осадка (7 мг, 9%). MS (ESI): m/z = 330.100 [M+H]+.
Следующие примеры были синтезированы из подходящих структурных элементов/промежуточных соединений и известного 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-она по аналогии с примером B1:








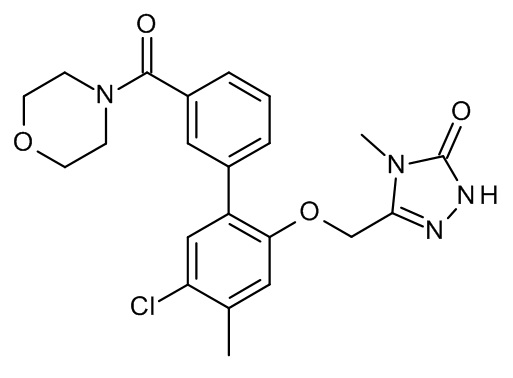

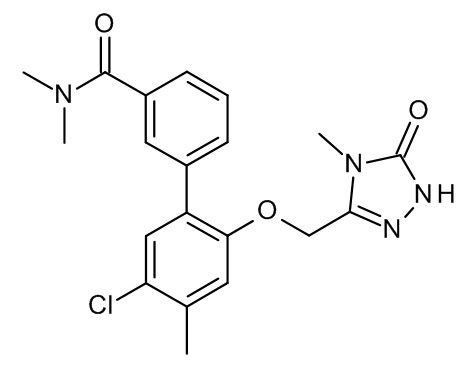








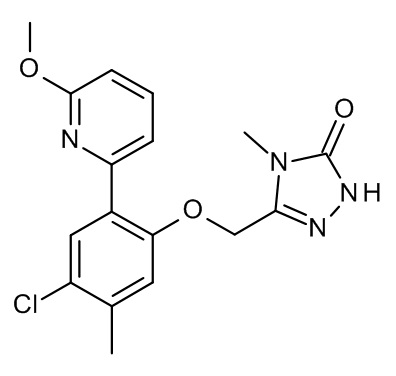
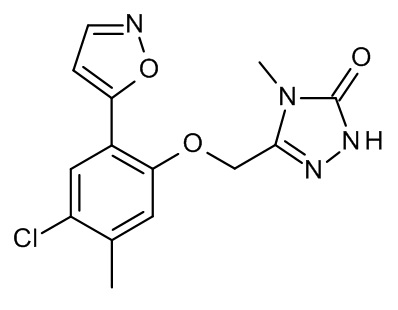


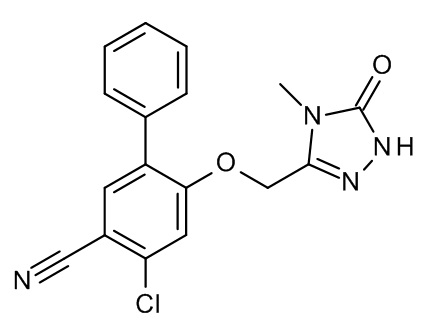
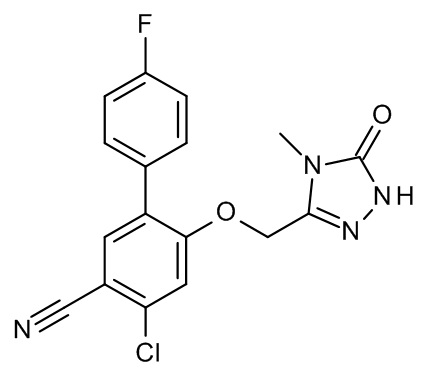
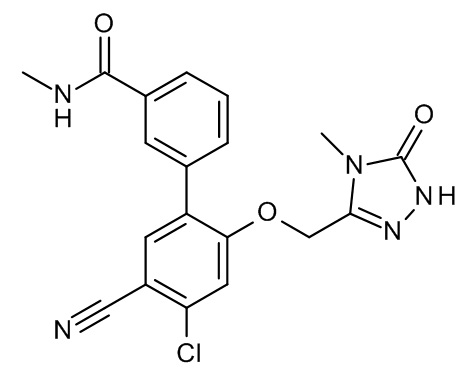

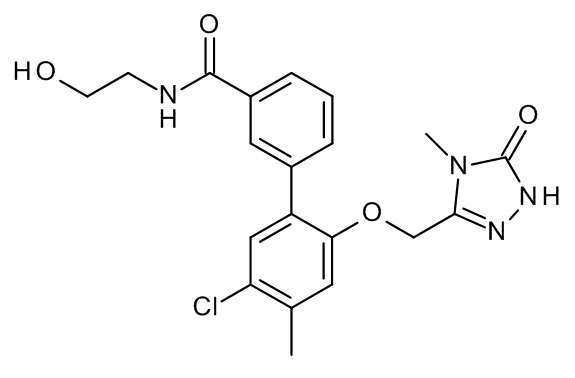


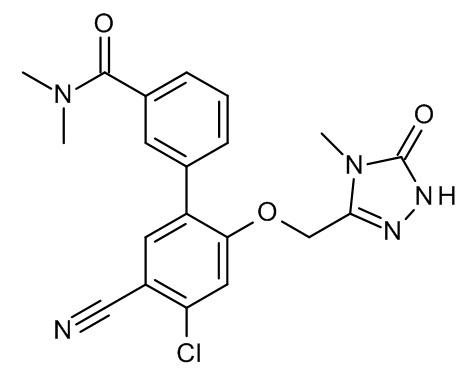




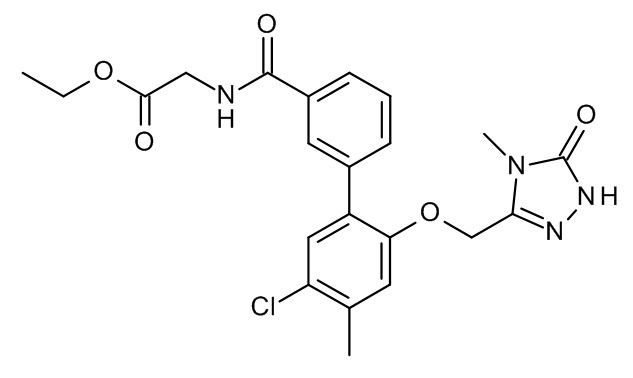









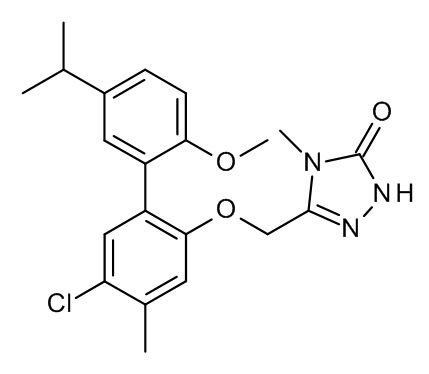

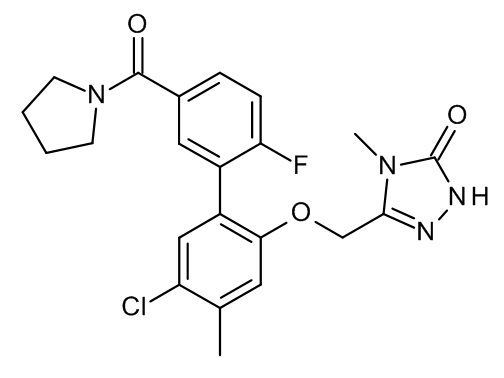
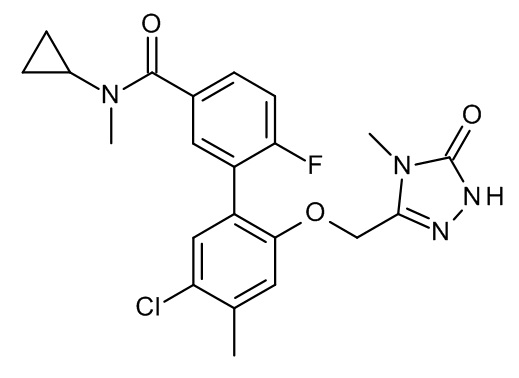



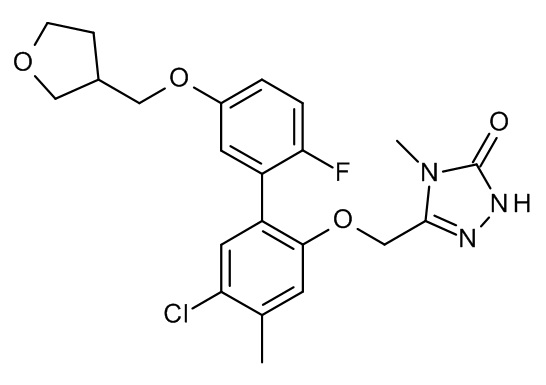



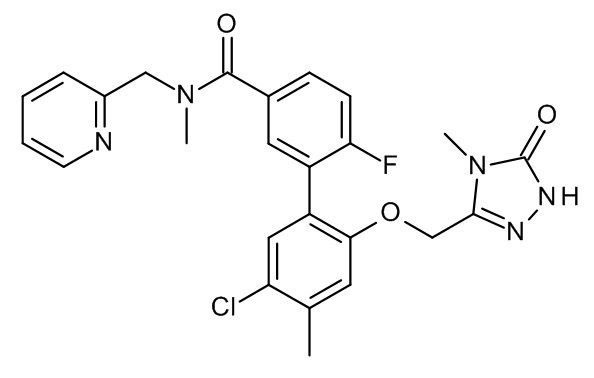

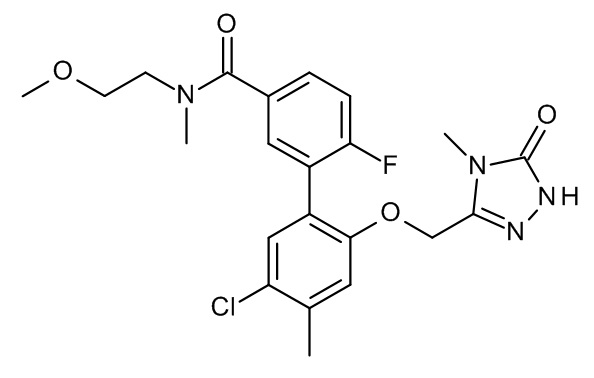







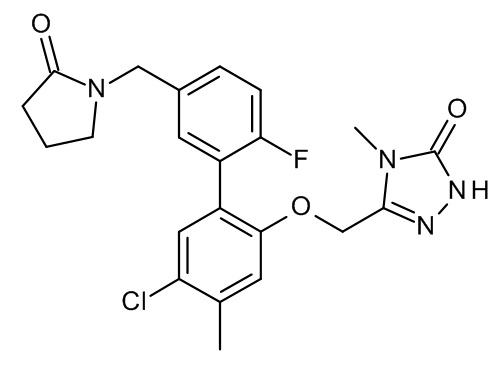






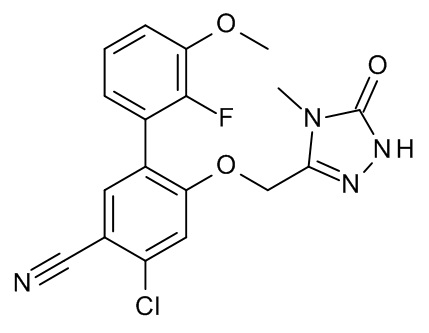

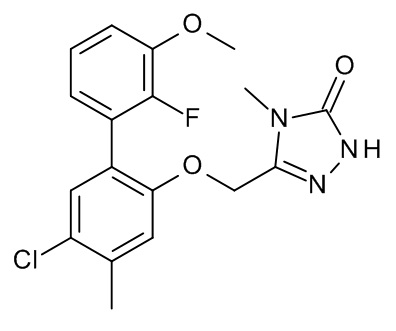
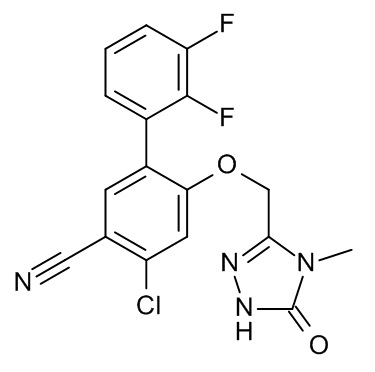



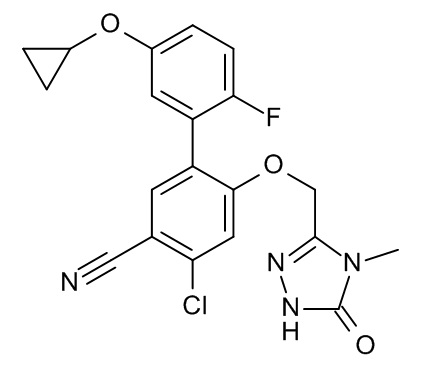

Следующие примеры типа B синтезировали из подходящих структурных элементов/промежуточных соединений и известного 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-она по аналогии с примером B1:
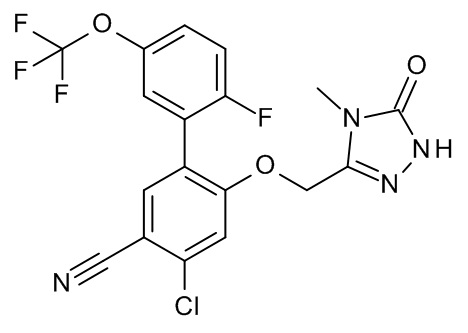

Пример B22:
3-[(4-хлор-5-метил-2-пиразин-2-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он

Стадия 1: 5-(4-хлор-2-йод-5-метил-феноксиметил)-4-метил-2,4-дигидро-[1,2,4]триазол-3-он
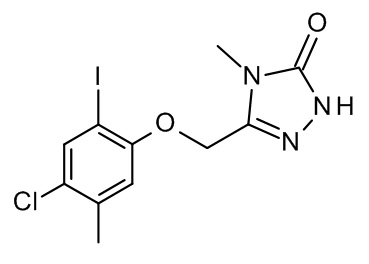
Соединение, указанное в заголовке, получили по аналогии с примером B1 из 4-хлор-2-йод-5-метил-фенола (900 мг, промежуточное соединение B1, Стадия 3) и 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-она (35 мг, CAS: 1338226-21-3) в виде серо-белого осадка (1.07 г, 84%). MS (ESI): m/z = 380.1 [M+H]+.
Стадия 2: 5-[(4-хлор-2-йод-5-метил-фенокси)метил]-4-метил-2-(2-триметилсилилэтоксиметил)-1,2,4-триазол-3-он

К суспензии NaH (192 мг, 60% в минеральном масле, CAS: 7646-69-7) в безводном ДМФ (10 мл) добавили раствор 5-(4-хлор-2-йод-5-метил-феноксиметил)-4-метил-2,4-дигидро-[1,2,4]триазол-3-она (1.21 г) в безводном ДМФ (20 мл) при комнатной температуре и получившуюся реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Затем 2-(триметилсилил)этоксиметилхлорид (0.85 мл, CAS: 76513-69-4) добавили по каплям в реакционную смесь и смесь перемешивали при комнатной температуре в течение 16 часов. Смесь погасили H2O при 0 °C и растворитель эвапорировали досуха с получением остатка, который растворили в EtOAc (30 мл). Органический слой промыли H2O (30 мл) и солевым раствором (30 мл), высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Продукт очистили с помощью флеш-хроматографии (5-25% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде коричневой жидкости (0.735 г, 45%). MS (ESI): m/z = 509.8 [M+H]+.
Стадия 3: 5-[(4-хлор-5-метил-2-пиразин-2-ил-фенокси)метил]-4-метил-2-(2-триметилсилилэтоксиметил)-1,2,4-триазол-3-он
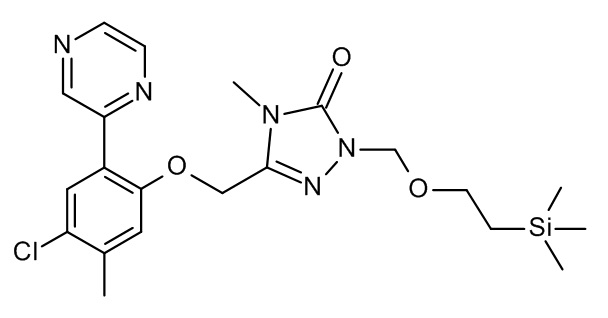
Смесь 5-[(4-хлор-2-йод-5-метил-фенокси)метил]-4-метил-2-(2-триметилсилилэтоксиметил)-1,2,4-триазол-3-она (312 мг), 2-(трибутилстаннил)пиразина (339 мг, CAS: 205371-27-3) в безводном ДМФ (6 мл) продули аргоном в течение 30 мин. Затем Pd(PPh3)4(14 мг, CAS: 14221-01-3) добавили и реакционную смесь нагревали при 120 °C в течение 16 часов в атмосфере аргона. Реакционную смесь отфильтровали через слой целита. Фильтрат эвапорировали с получением остатка, который растворили в EtOAc (40 мл). Органический слой промыли H2O (40 мл) и солевым раствором (40 мл), высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Неочищенный продукт очистили с помощью флеш-хроматографии через силикагель (15-35% EtOAc/гексан) с получением соединения, указанного в заголовке, в виде светло-желтого масла (0.187 г, 66%). MS (ESI): m/z = 461.9 [M+H]+.
Стадия 4: 3-[(4-хлор-5-метил-2-пиразин-2-илфенокси)метил]-4-метил-1H-1,2,4-триазол-5-он

К раствору 5-[(4-хлор-5-метил-2-пиразин-2-ил-фенокси)метил]-4-метил-2-(2-триметилсилилэтоксиметил)-1,2,4-триазол-3-она (210 мг) в ДХМ (5 мл) добавили ТФУ (2 мл, CAS: 76-05-1) и смесь перемешивали при 25 °C в течение 3 часов. Растворитель эвапорировали с получением остатка, который растворили в EtOAc (30 мл) и промыли насыщенным водным раствором NaHCO3 (25 мл), H2O (25 мл) и солевым раствором (25 мл). Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Получившийся неочищенный продукт очистили с помощью препаративной ВЭЖХ (NH4OAc/CH3CN) с получением соединения, указанного в заголовке, в виде белого осадка (0.052 г, 35%). LC-MS: (ESI): m/z = 332.1 [M+H]+.
Пример B23:
3-[(4-хлор-5-метил-2-пиримидин-2-ил-фенокси)метил]-4-метил-1H-1,2,4-триазол-5-он
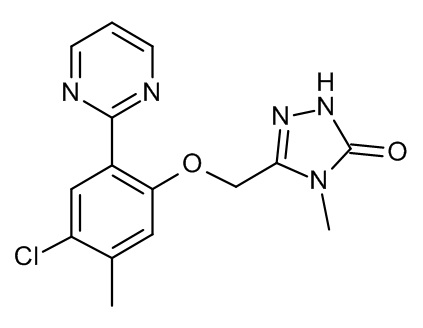
К раствору 5-(4-хлор-2-йод-5-метил-феноксиметил)-4-метил-2,4-дигидро-[1,2,4]триазол-3-она (150 мг, пример B22, Стадия 1) в диоксане (5 мл) добавили 2-трибутилстаннанил-пиримидин (364 мг, CAS: 153435-63-3) и Pd(PPh3)4(13 мг, CAS: 14221-01-3) и реакционную смесь нагревали при 100 °C в течение 16 часов. Растворитель эвапорировали и получившийся неочищенный продукт очистили с помощью препаративной ВЭЖХ (NH4OAc/CH3CN) с получением соединения, указанного в заголовке, в виде белого осадка (0.020 г, 15%). MS (ESI): m/z = 332.3 [M+H]+.
Пример B24:
3-[[4-хлор-5-метил-2-[2-метил-5-(трифторметил)пиразол-3-ил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он

Стадия 1: 3-(4-хлор-2-йод-5-метил-феноксиметил)-5-метансульфонил-4-метил-4H-[1,2,4]триазол
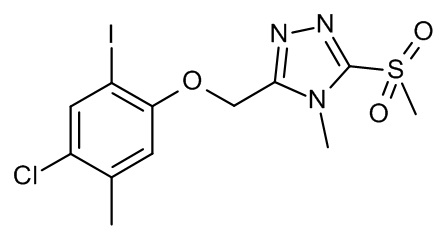
Соединение, указанное в заголовке, получили по аналогии с примером B1 из 4-хлор-2-йод-5-метил-фенола (0.1 г, промежуточное соединение B1, Стадия 3) и 3-(йодометил)-4-метил-5-(метилсульфонил)-4H-1,2,4-триазола (118 мг, полученного как описано в US2008249151) в виде бесцветного осадка (0.145 г; 88%). MS (ESI): m/z = 441.95 [M+H]+.
Стадия 2: 3-[[4-хлор-5-метил-2-[2-метил-5-(трифторметил)пиразол-3-ил]фенокси]метил]-4-метил-5-метилсульфонил-1,2,4-триазол

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B1, Стадия 4, из 3-(4-хлор-2-йод-5-метил-феноксиметил)-5-метансульфонил-4-метил-4H-[1,2,4]триазола (0.066 г) и 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3-(трифторметил)-1H-пиразола (0.049 г, CAS: 1025719-23-6) в виде светло-коричневой смолы (0.038 г; 55%). MS (ESI): m/z = 464.08 [M+H]+.
Стадия 3: 3-[[4-хлор-5-метил-2-[2-метил-5-(трифторметил)пиразол-3-ил]фенокси]метил]-5-метокси-4-метил-1,2,4-триазол
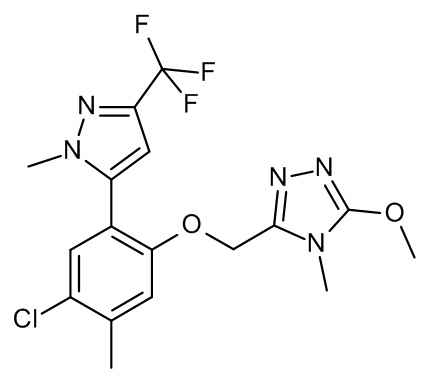
К раствору 3-[[4-хлор-5-метил-2-[2-метил-5-(трифторметил)пиразол-3-ил]фенокси]метил]-4-метил-5-метилсульфонил-1,2,4-триазола (0.084 г) в MeOH (1 мл) добавили 5.4M раствор метоксида натрия в MeOH (134 мкл, CAS: 124-41-4) и раствор нагревали при кипении с обратным холодильником в течение 15 минут. Реакционную смесь влили в насыщенный водный раствор NH4Cl и EtOAc и слои разделили. Водный слой экстрагировали дважды с помощью EtOAc. Органические слои высушили над MgSO4, отфильтровали, обработали силикагелем и эвапорировали. Соединение очистили с помощью хроматографии на силикагеле на 5 г колонке с использованием системы ЖХСД с элюцией при градиенте ДХМ : MeOH (100/0 до 90/10) с получением соединения, указанного в заголовке, в виде бесцветного осадка (0.054 г; 72%). MS (ESI): m/z = 416.11 [M+H]+.
Стадия 4: 3-[[4-хлор-5-метил-2-[2-метил-5-(трифторметил)пиразол-3-ил]фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он
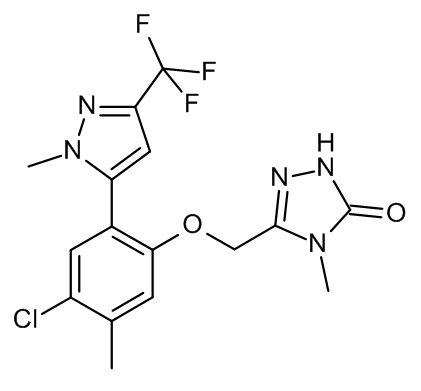
К раствору 3-[[4-хлор-5-метил-2-[2-метил-5-(трифторметил)пиразол-3-ил]фенокси]метил]-5-метокси-4-метил-1,2,4-триазола (0.05 г) в AcOH (826 мкл) добавили HBr 48% в H2O (261 мкл) и прозрачный бесцветный раствор перемешивали при кипении с обратным холодильником в течение 15 минут. Затем реакционную смесь эвапорировали. Остаток растворили в водном насыщенном растворе NaHCO3 и EtOAc и слои разделили. Водный слой экстрагировали дважды с помощью EtOAc. Органические слои промыли солевым раствором, высушили над MgSO4, отфильтровали и эвапорировали. Остаток очистили с помощью хроматографии на силикагеле на 5 г колонке с использованием системы ЖХСД (ISCO) с элюцией при градиенте н-гептан : EtOAc (100/0 до 0/100) с получением соединения, указанного в заголовке, в виде бесцветной смолы (0.036 г; 75%). MS (ESI): m/z = 402.10 [M+H]+.
Пример B27:
3-[[4-хлор-5-метил-2-(3-метилимидазол-4-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он

Стадия 1: 5-[4-хлор-5-метил-2-(3-метил-3H-имидазол-4-ил)-феноксиметил]-4-метил-2-(2-триметилсиланил-этоксиметил)-2,4-дигидро-[1,2,4]триазол-3-он

Смесь 5-(4-хлор-2-йод-5-метил-феноксиметил)-4-метил-2-(2-триметилсиланил-этоксиметил)-2,4-дигидро-[1,2,4]триазол-3-она (220 мг, пример 22, Стадия 2), 1-метил-1H-имидазол-5-бороновой кислоты пинаколового эфира (180 мг, CAS: 942070-72-6), трехосновного фосфата калия (183 мг, CAS: 7778-53-2), трициклогексилфосфина (2 мг, CAS: 2622-14-2) в диоксане (4 мл) и H2O (20 мл) продули аргоном в течение 20 мин. Затем трис(дибензилиденацетон)дипалладий(0) (4мг, CAS: 52409-22-0) добавили и смесь нагревали при 120 °C в микроволновой печи в течение 1 часа. Смесь отфильтровали через слой целита. Фильтрат эвапорировали с получением остатка, который растворили в EtOAc (30 мл) и промыли H2O (40 мл) и солевым раствором (40 мл). Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Получившийся неочищенный материал очистили с помощью флеш-хроматографии через силикагель (1-5% MeOH/ДХМ) с получением соединения, указанного в заголовке, в виде a коричневой жидкости (180 мг, 90%). MS (ESI): m/z = 464.4 [M+H]+.
Стадия 2: 3-[[4-хлор-5-метил-2-(3-метилимидазол-4-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B22, Стадия 4, из 5-[4-хлор-5-метил-2-(3-метил-3H-имидазол-4-ил)-феноксиметил]-4-метил-2-(2-триметилсиланил-этоксиметил)-2,4-дигидро-[1,2,4]триазол-3-она (0.160 г) в виде серо-белого осадка (28 мг, 24%). MS (ESI): m/z = 334.0 [M+H]+.
Пример B28:
3-[[4-хлор-2-(1H-имидазол-5-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он
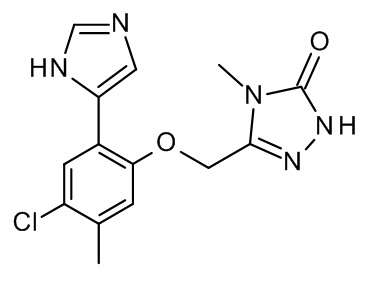
Стадия 1: 5-йодо-1-тритил-1H-имидазол
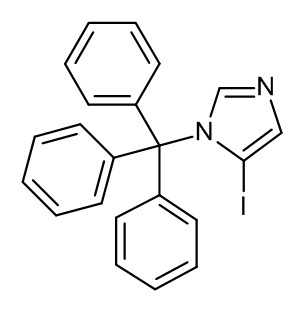
К раствору 4-йод-1H-имидазола (2.05 г, CAS: 71759-89-2) и тритилхлорида (4.4 г, CAS: 76-83-5) в ДМФ (35 мл) добавили триэтиламин (3.04 мл, CAS: 121-44-8) при 0 °C. Реакционную смесь медленно нагрели до КТ и перемешивали в течение 48 часов. Реакционную смесь затем влили в H2O (150 мл). Осадок отфильтровали, промыли H2O (60 мл) и высушили при пониженном давлении. Получившийся неочищенный материал очистили с помощью флеш-хроматографии с использованием силикагеля (EtOAc) с получением соединения, указанного в заголовке, в виде белого осадка (4.3 г, 93%). Rf= 0.50 (10% EtOAc/гексан).
Стадия 2: 5-Трибутилстаннанил-1-тритил-1H-имидазол

К раствору 5-йод-1-тритил-1H-имидазола (2.02 г) в ДХМ (50 мл) добавили этил магния бромид (1.8 мл, 3M в диэтиловом эфире, CAS: 925-90-6). Реакционную смесь перемешивали в атмосфере аргона при комнатной температуре в течение 1 часа. Хлорид трибутилолова (1.5 мл, CAS: 1461-22-9) добавили в реакционную смесь и получившуюся смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавили с помощью ДХМ (100 мл) и тщательно промыли насыщенным водным раствором NH4Cl (100 мл), H2O (100 мл) и солевым раствором (100 мл). Органическую фазу высушили над Na2SO4, отфильтровали и эвапорировали с получением соединения, указанного в заголовке, в виде белого осадка (2.7 г, 97%), который использовали на следующей стадии без дополнительной очистки. Rf= 0.60 (10% EtOAc/гексан).
Стадия 3: 5-[4-хлор-5-метил-2-(3-тритил-3H-имидазол-4-ил)-феноксиметил]-4-метил-2,4-дигидро-[1,2,4]триазол-3-он

Соединение, указанное в заголовке, получили по аналогии с примером 23, из 5-(4-хлор-2-йод-5-метил-феноксиметил)-4-метил-2,4-дигидро-[1,2,4]триазол-3-она (Пример 22, Стадия 1) и 5-трибутилстаннанил-1-тритил-1H-имидазола в виде желтого осадка. MS (EI): m/z = 562.3 [M+H]+.
Стадия 4: 3-[[4-хлор-2-(1H-имидазол-5-ил)-5-метилфенокси]метил]-4-метил-1H-1,2,4-триазол-5-он

К раствору 5-[4-хлор-5-метил-2-(3-тритил-3H-имидазол-4-ил)-феноксиметил]-4-метил-2,4-дигидро-[1,2,4]триазол-3-она (80 мг) в ДХМ (5 мл) добавили HCl (0.5 мл, 4 н в диоксане, CAS: 7647-01-0) при 25 °C и получившуюся смесь перемешивали в течение 4 часов. Реакционную смесь эвапорировали и получившееся соединение очистили с помощью флеш-хроматографии с использованием силикагеля (10% MeOH/ДХМ) с получением соединения, указанного в заголовке, в виде белого осадка (12 мг, 26%). MS (ESI): m/z = 320.2 [M+H]+.
Пример B29:
3-[[4-хлор-5-метил-2-(1,3-оксазол-2-ил)фенокси]метил]-4-метил-1H-1,2,4-триазол-5-он

Соединение, указанное в заголовке, получили по аналогии с примером B23 из 5-(4-хлор-2-йод-5-метил-феноксиметил)-4-метил-2,4-дигидро-[1,2,4]триазол-3-она (150 мг, пример B22, Стадия 1) и 2-трибутилстаннанилоксазола (207 мг, CAS: 145214-05-7). Соединение получили в виде белого осадка (9 мг, 7%). MS (ESI): m/z = 320.9 [M+H]+.
Пример B34:
2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-[3-(1H-пиразол-3-ил)фенил]бензонитрил

Стадия 1: 3'-ацетил-4-хлор-6-гидрокси-бифенил-3-карбонитрил and 3'-ацетил-4-хлор-6-метоксиметокси-бифенил-3-карбонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 3, из 5-бромо-2-хлор-4-метоксиметокси-бензонитрила (промежуточное соединение B31, Стадия 2) (1.0 г) и 3-ацетил-фенилбороновой кислоты (886 мг, CAS: 204841-19-0). Необходимо отметить, что в данном случае в качестве исходного соединения использовали MOM-защищенный фенол.
Эта реакция дала MOM-защищенный фенол 3'-ацетил-4-хлор-6-метоксиметокси-бифенил-3-карбонитрила (270 мг, 24%,1H-NMR (400 MHz, ДМСО-D6): 2.63 (s, 3H), 3.36 (s, 3H), 5.37 (s, 2H), 7.55 (s, 1H), 7.62 (t, 1H, J=7.6), 7.80 (d, 1H, J=7.6), 7.98 (d, 1H, J=7.6), 8.03 (s, 1H), 8.10 (s, 1H)), а также свободный фенол 3'-ацетил-4-хлор-6-гидрокси-бифенил-3-карбонитрила (190 мг, 19%):1H-NMR (400 MHz, ДМСО-D6): 2.63 (s, 3H), 7.19 (s, 1H), 7.53-7.63 (m, ~1H), 7.83 (d, 1H, J=8); 7.95 (d, 1H, J=7.8), 7.97 (s, 1H), 8.11 (s, 1H), 11.48 (s, 1H).
В зависимости от условий реакции MOM группа может быть частично утеряна в данной реакции с получением свободного фенол 3'-ацетил-4-хлор-6-гидрокси-бифенил-3-карбонитрила. Это соединение можно подвергнуть стадии 2, как показано ниже, для повторного введения MOM группы. MOM защищенный материал со стадии 1 может быть использован сразу на стадии 3.
Стадия 2: 3'-ацетил-4-хлор-6-метоксиметокси-бифенил-3-карбонитрил

Соединение, указанное в заголовке, получили по аналогии с Промежуточным соединением B31, Стадия 2, из 3'-ацетил-4-хлор-6-гидрокси-бифенил-3-карбонитрила (250 мг) посредством взаимодействия с NaH (44 мг, 60% в минеральном масле) и MOM-Cl (0.14 мл) и получили в виде серо-белого осадка (183 мг, 63%).1H-NMR (400 MHz, ДМСО-D6): 2.63 (s, 3H), 3.36 (s, 3H), 5.37 (s, 2H), 7.55 (s, 1H), 7.62 (t, 1H, J=7.6), 7.80 (d, 1H, J=7.6), 7.98 (d, 1H, J=7.6), 8.04 (s, 1H), 8.10 (s, 1H).
Стадия 3: 4-хлор-3'-((E)-3-диметиламино-акрилоил)-6-метоксиметокси-бифенил-3-карбонитрил
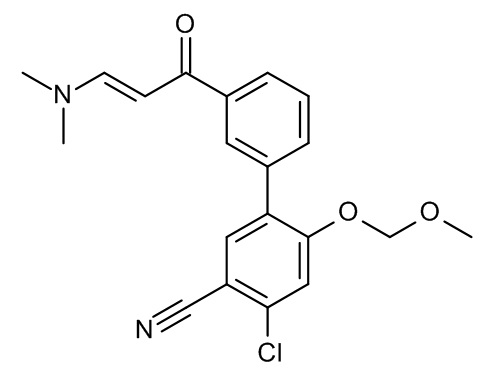
Раствор 3'-ацетил-4-хлор-6-метоксиметокси-бифенил-3-карбонитрила (180 мг) в ДМФ-DMA (2.5 мл) нагревали до 80°C в течение 16 ч. Реакционную смесь охладили до 25°C и все летучие компоненты эвапорировали при пониженном давлении. Оставшийся остаток очистили посредством колоночной хроматографии через силикагель (80-100% EtOAc/гексан) с получением соединения, указанного в заголовке (140 мг, 66%), в виде серо-белого осадка. MS (ESI): m/z = 370.9 [M+H]+.
Стадия 4: 3'-(1-трет-бутил-1H-пиразол-3-ил)-4-хлор-6-метоксиметокси-бифенил-3-карбонитрил

К раствору 4-хлор-3'-((E)-3-диметиламино-акрилоил)-6-метоксиметокси-бифенил-3-карбонитрила (140 мг) в EtOH (20 мл) добавили трет-бутил-гидразин (70.37 мг, CAS: 7400-27-3) при 0°C. Реакционную смесь нагревали до кипения в течение 12 ч. Смесь затем охладили до 25°C и все летучие компоненты удалили при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (10-15% EtOAc/гексан) с получением соединения, указанного в заголовке (110 мг, 73%), в виде серо-белого осадка. MS (ESI): m/z = 396.0 [M+H]+.
Стадия 5: 3'-(1-трет-бутил-1H-пиразол-3-ил)-4-хлор-6-гидрокси-бифенил-3-карбонитрил

К раствору 3'-(1-трет-бутил-1H-пиразол-3-ил)-4-хлор-6-метоксиметокси-бифенил-3-карбонитрила (110 мг) в безводном ДХМ (10 мл) добавили HCl в диоксане (1 мл) при 0°C и реакционную смесь перемешивали при 25°C в течение 32 ч. Смесь разбавили с помощью ДХМ (30 мл) и промыли насыщенным раствором NaHCO3 и солевым раствором, высушили над Na2SO4 и сконцентрировали. Остаток очистили посредством колоночной хроматографии через силикагель (1-2% MeOH/ДХМ) с получением соединения, указанного в заголовке (45 мг, 46%), в виде серо-белого осадка. MS (ESI): m/z = 352.2 [M+H]+.
Стадия 6: 3'-(1-трет-бутил-1H-пиразол-3-ил)-4-хлор-6-(4-метил-5-оксо-4,5-дигидро-1H-[1,2,4]триазол-3-илметокси)-бифенил-3-карбонитрил

Соединение, указанное в заголовке, получили по аналогии с примером B1 из 3'-(1-трет-бутил-1H-пиразол-3-ил)-4-хлор-6-гидрокси-бифенил-3-карбонитрила (80 мг) и 3-(хлорметил)-4-метил-1H-1,2,4-триазол-5(4H)-она (34 мг) в виде серо-белого осадка (45 мг, 43%). MS (ESI): m/z = 463.0 [M+H]+.
Стадия 7: 2-хлор-4-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]-5-[3-(1H-пиразол-3-ил)фенил]бензонитрил

Раствор 3'-(1-трет-бутил-1H-пиразол-3-ил)-4-хлор-6-(4-метил-5-оксо-4,5-дигидро-1H-[1,2,4]триазол-3-илметокси)-бифенил-3-карбонитрила (45 мг в муравьиной кислоте (4 мл) нагревали до 85°C в течение 6 ч. Затем, все летучие компоненты удалили и остаток разбавили с помощью ДХМ (20 мл) и промыли водой и солевым раствором, высушили над Na2SO4 и сконцентрировали. Получившийся материал очистили посредством колоночной хроматографии через силикагель (2-3% MeOH/ДХМ) с получением соединения, указанного в заголовке (15 мг, 37%), в виде серо-белого осадка. MS (ESI): m/z = 407.2 [M+H]+.
Пример B43:
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензойная кислота
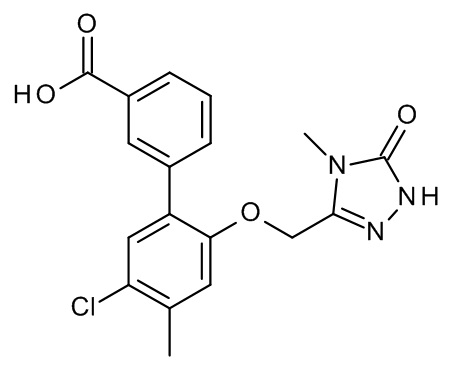
Метил 3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензоат (36.0 мг, пример B42) суспендировали в ТГФ (1.5 мл) при комнатной температуре. MeOH добавили по каплям до растворения всех веществ. Затем раствор моногидрата гидроксида лития (11.7 мг) в воде (280 мкл) добавили по каплям. Небольшое количество белого осадка преципитировалось. Снова добавили MeOH по каплям до полного растворения. Бесцветный раствор затем перемешивали при комнатной температуре в течение ночи. Летучие компоненты удалили и оставшийся остаток растворили в воде. pH довели до 2 посредством добавления 1 н HCl. Преципитат отфильтровали, промыли небольшим количеством воды и высушили с получением соединения, указанного в заголовке в виде белого осадка (31 мг, 89%). MS (ESI): m/z = 374.1 [M+H]+.
Следующие примеры были синтезированы по аналогии с примером B43:



Пример B45:
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N-(2-гидроксиэтил)-N-метилбензамид

3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]бензойную кислоту (23.0 мг, пример B43) растворили в N,N-диметилформамиде (0.5 мл). Затем HATU (35.1 мг) и основание Хунига (19.9 мг, 26.9 мкл) добавили при комнатной температуре, с последующим раствором 2-(метиламино)этанола (6.47 мг) в N,N-диметилформамиде (0.5 мл). Светло-желтый раствор перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавили насыщенным раствором NH4Cl и экстрагировали с помощью EtOAc. Объединенные органические экстракты промыли водой и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством ВЭЖХ (Gemini NX колонка, ацетонитрил/вода (содержащая 0.05% муравьиной кислоты) 85:15) с получением соединения, указанного в заголовке, в виде белого осадка (10 мг, 38%). MS (ESI): m/z = 431.2 [M+H]+.
Пример B51:
3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-N,N,4-триметилбензамид

Соединение, указанное в заголовке, получили по аналогии с примером B47 из 3-[5-хлор-4-метил-2-[(4-метил-5-оксо-1H-1,2,4-триазол-3-ил)метокси]фенил]-4-метилбензойной кислоты (23 мг, пример B50) и гидрохлорида диметиламина (6.77 мг) в виде белого осадка (14 мг, 57%). MS (ESI): m/z = 415.2 [M+H]+.
Примеры C: Соединения с пиридазиноновой основной группой
Пример C1:
3-[(4-хлор-5-метил-2-пропан-2-илфенокси)метил]-1H-пиридазин-6-он

К раствору 3-хлор-6-[(4-хлор-2-изопропил-5-метил-фенокси)метил]пиридазина (60 мг, промежуточное соединение C1-A) в EtOH (3 мл) добавили водный 3M NaOH (0.642 мл) и реакционную смесь нагревали при кипении с обратным холодильником в течение 16 часов. Реакционную смесь влили в H2O и EtOAc и слои разделили. Органический слой высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом. Материал очистили с помощью флеш-хроматографии через силикагель (0-3% MeOH/ДХМ) с получением с получением соединения, указанного в заголовке, в виде серо-белого осадка (0.005 г, 9%). MS (ESI): m/z = 293.11 [M]+.
Пример C2:
3-[(4-хлор-2-циклопропил-5-метилфенокси)метил]-1H-пиридазин-6-он

Раствор 3-хлор-6-(4-хлор-2-циклопропил-5-метил-феноксиметил)-пиридазина (45 мг, промежуточное соединение C2-A) кипятили с обратным холодильником в ледяной уксусной кислоте (5 мл) при 120 °C в течение 16 часов. Затем растворитель удалили при пониженном давлении. Остаток растворили в ДХМ и органическую часть промыли насыщенным раствором NaHCO3 и солевым раствором, высушили над безводным Na2SO4, отфильтровали и сконцентрировали при пониженном давлении. Продукт очистили посредством препаративной ВЭЖХ (NH4OAc/CH3CN) с получением соединения, указанного в заголовке, в виде белого осадка (30 мг, 71%). MS: (ESI): m/z = 291.4 [M+H]+.
Следующие примеры были синтезированы из подходящих структурных элементов/промежуточных соединений по аналогии с примером C2:

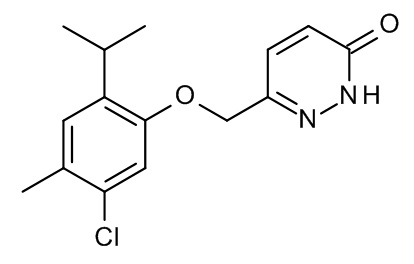


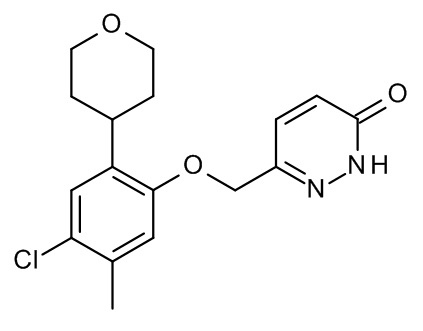
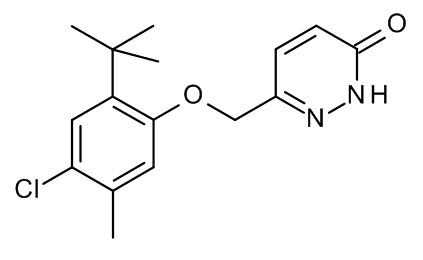

Следующие примеры типа C были получены из подходящих структурных элементов/промежуточных соединений по аналогии с примером C2:

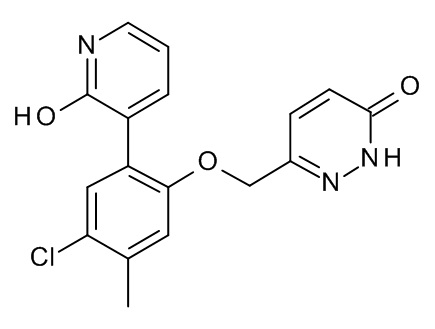
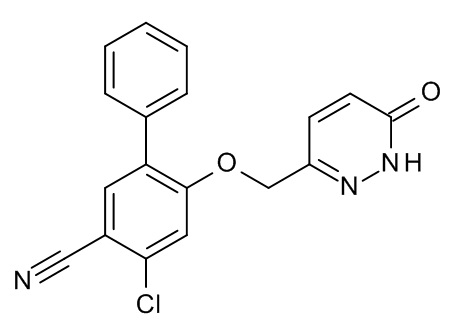
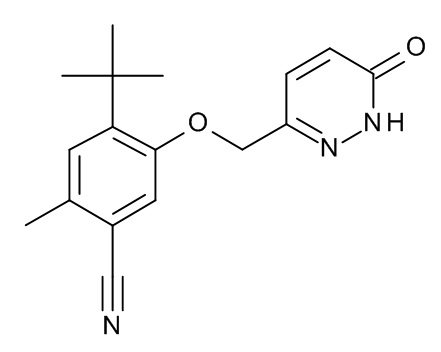

Примеры D: Соединения с основными группами, относящимися к индазолу, аза-индазолу и аналогичным
Пример D2:
3-[(4-хлор-5-метил-2-пропан-2-илфенокси)метил]-1H-индазол

К раствору 4-хлор-2-изопропил-5-метилфенола (0.15 г, CAS: 89-68-9) в ДМФ (1.5 мл) добавили гидрид натрия (42.5 мг, 55-60% в минеральном масле) и смесь перемешивали при комнатной температуре в течение 15 минут. Суспензию трет-бутил 3-(бромометил)-1H-индазол-1-карбоксилата (253 мг CAS: 174180-42-8) в ДМФ (2.5 мл) затем добавили по каплям. После перемешивания при комнатной температуре в течение 2.5 часов, реакционную смесь влили в смесь насыщенного водного раствора NH4Cl и этилацетат и слои разделили. Водный слой экстрагировали дважды с помощью этилацетата. Объединенные органические слои промыли дважды с помощью воды и один раз солевым раствором, высушили над MgSO4, отфильтровали, и эвапорировали. Остаток очистили с помощью хроматографии на силикагеле с использованием системы ЖХСД (с элюцией градиентом н-гептан : этилацетат от 100/0 до 60/40). Получившееся светло-коричневое масло (0.157 г) растворили в дихлорметане (1.5 мл). Раствор охладили до 0°C и добавили трифторуксусную кислоту (1.85 г, 1.25 мл). После перемешивания при комнатной температуре в течение 1.25 часов, реакционную смесь влили в смесь насыщенного водного раствора NaHCO3 и дихлорметан и слои разделили. Водный слой экстрагировали дважды дихлорметаном. Объединенные органические слои промыли один раз солевым раствором, высушили над MgSO4, отфильтровали и эвапорировали. Остаток очистили с помощью хроматографии на силикагеле с использованием системы ЖХСД (с элюцией градиентом н-гептан : этилацетат от 100/0 до 60/40) с получением соединения, указанного в заголовке, в виде бесцветного масла (48 мг; 19%). MS (ESI): m/z = 315.13 [M+H]+.
Пример D3:
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин

К раствору Промежуточного соединения D3-A (85.1 мг) в диоксане (3 мл) добавили 4 н HCl в диоксане (495 мкл) и раствор перемешивали при комнатной температуре. Белый осадок преципитировался и суспензию перемешивали при комнатной температуре в течение ночи. Затем дополнительное количество 4 н HCl в диоксане (495 мкл) добавили и через 4 ч при комнатной температуре снова добавили дополнительное количество 4 н HCl в диоксане (495 мкл) и смесь перемешивали при комнатной температуре в течение дополнительных 2 дней. Белую суспензию влили в насыщенный раствор NaHCO3 и получившуюся смесь экстрагировали с помощью ДХМ. Объединенные органические слои высушили над Na2SO4 и эвапорировали. Остаток очистили посредством хроматографии (10 г силикагель; гептан/EtOAc 90/10 - 70/30) с получением соединения, указанного в заголовке (54 мг, 83%) в виде белого осадка. MS (ESI): m/z = 330.2 [M+H]+.
Следующие примеры были синтезированы по аналогии с примером D3.


Пример D6:
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-6-фтор-1H-пиразоло[3,4-b]пиридин

Раствор Промежуточного соединения D6-A (55.1 мг) в смеси ДХМ (1 мл) и трифторуксусной кислоты (0.4 мл) перемешивали при комнатной температуре в течение 1 ч. Затем раствор разбавили с помощью ДХМ и его медленно добавили в насыщенный раствор Na2CO3. Смесь экстрагировали с помощью ДХМ и объединенные органические слои промыли солевым раствором, высушили над Na2SO4, отфильтровали, и эвапорировали. Оставшийся остаток очистили посредством хроматографии (5г силикагель; ДХМ/MeOH 98:2) с получением соединения, указанного в заголовке (40 мг, 94%), в виде белого осадка. MS (ESI): m/z = 348.1 [M+H]+.
Следующие примеры были синтезированы по аналогии с примером D6.

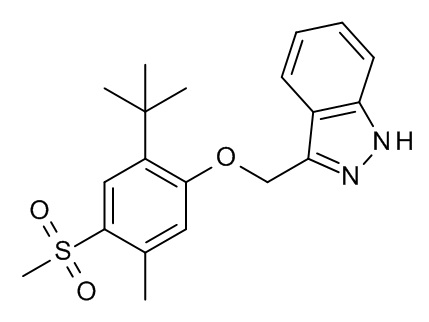








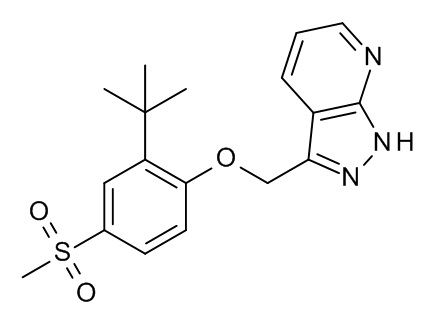
Пример D7:
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]амино]этанол
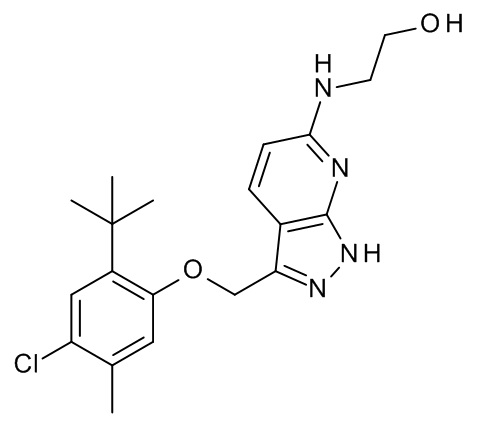
Раствор Промежуточного соединения D6-A (99.9 мг) и этаноламина (136 мг, 135 мкл) в N-метил-2-пирролидиноне (1.8 мл) нагревали до 95°C в течение ночи. Раствор охладили до КТ, разбавили полунасыщенным солевым раствором и смесь экстрагировали EtOAc. Объединенные органические слои промыли водой и солевым раствором, высушили над Na2SO4 и эвапорировали. Неочищенное вещество очистили посредством хроматографии (20г силикагель; ДХМ/MeOH 100/0 - 95/5) с получением белого осадка, который дополнительно очистили препаративной ВЭЖХ с получением соединения, указанного в заголовке (38 мг, 44%), в виде белого осадка. MS (ESI): m/z = 389.2 [M+H]+.
Пример D8:
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]-метиламино]этанол

Раствор Промежуточного соединения D6-A (99.9 мг) и 2-(метиламино)этанола (167 мг, 178 мкл) в N-метил-2-пирролидиноне (1.8 мл) нагревали до 50°C в течение 5 ч. Раствор охладили до КТ, разбавили полунасыщенным солевым раствором и экстрагировали EtOAc. Объединенные органические слои промыли водой и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток растворили в ДХМ (2 мл) и добавили трифторуксусную кислоту (0.4 мл). Раствор перемешивали при комнатной температуре в течение 30 мин и затем влили в насыщенный раствор Na2CO3 и смесь экстрагировали с помощью ДХМ. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (5г силикагель; ДХМ/MeOH 98/2 - 95/5) с получением соединения, указанного в заголовке (41 мг, 46%), в виде белого осадка. MS (ESI): m/z = 403.2 [M+H]+.
Следующие примеры были синтезированы по аналогии с примером D8.


Пример D13:
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]окси]этанол

Стадия 1: 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-6-фтор-1-тритил-пиразоло[3,4-b]пиридин

Гидрид натрия (23.2 мг, 55% в минеральном масле) суспендировали в ДМФ (1 мл) и раствор 3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-6-фтор-1H-пиразоло[3,4-b]пиридина (148 мг, пример D6) в ДМФ (1 мл) добавили по каплям при 0°C. Получившуюся коричневую суспензию перемешивали при 0 °C в течение 10 мин и затем при комнатной температуре в течение 20 мин. Раствор тритилхлорида (= [хлор(дифенил)метил]бензол) (125 мг в ДМФ (1 мл) затем добавили при 0°C и реакционную смесь перемешивали при комнатной температуре в течение ночи. Осторожно добавили воду и смесь экстрагировали EtOAc. Объединенные органические слои промыли водой и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством хроматографии (20г силикагель; гептан/EtOAc 98/2 - 90/10) с получением соединения, указанного в заголовке (186 мг, 74%), в виде белого осадка.1H-NMR (300 MHz, CDCl3): 1.29 (s, 9H), 2.25 (s, 3H), 5.34 (s, 2H), 6.64 (dd, 1H), 6.92 (s, 1H), 7.19 (s, 1H), 7.20-7.35 (m, ~15H), 8.10 (dd, 1H).
Стадия 2: 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-6-(2-тетрагидропиран-2-илоксиэтокси)-1-тритил-пиразоло[3,4-b]пиридин

Гидрид натрия (7.68 мг, 55% в минеральном масле) суспендировали в DMA (1 мл). Затем раствор 2-(тетрагидро-2H-пиран-2-илокси)этанола (25.7 мг, 23.9 мкл, CAS: 2162-31-4) в DMA (1.5 мл) добавили по каплям при 0 °C и получившуюся суспензию перемешивали при комнатной температуре в течение 30 мин. Затем белый мутный раствор 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-6-фтор-1-тритил-пиразоло[3,4-b]пиридина (83.2 мг) в DMA (1.5 мл) добавили по каплям и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь осторожно разбавили водой и экстрагировали с помощью EtOAc. Объединенные органические слои промыли водой и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили с помощью флеш-хроматографии (10г силикагель; гептан/EtOAc 98/2 - 95/5) с получением соединения, указанного в заголовке (91 мг, 90%), в виде бесцветного масла.1H-NMR (300 MHz, CDCl3): 1.29 (s, ~9H); 1.45-1.88 (m,~6H), 2.24 (s, 3H), 3.35-3.51 (m, 2H), 3.60-3.70 (m, 1H), 3.75-3.88 (m, 2H), 4.50 (m, 1H), 5.30 (s, 2H), 6.50 (d, 1H, J=8.4), 6.94 (s, 1H), 7.18 (s, 1H), 7.20-7.30 (m, ~15H), 7.86 (d, 1H, J=8.7).
Стадия 3: 2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]окси]этанол
К раствору 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-6-(2-тетрагидропиран-2-илоксиэтокси)-1-тритил-пиразоло[3,4-b]пиридина (83.8 мг) в ДХМ (1.5 мл) добавили трифторуксусную кислоту (0.4 мл) по каплям и смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь разбавили с помощью ДХМ и осторожно добавили в насыщенный раствор Na2CO3. Получившуюся смесь экстрагировали с помощью ДХМ и объединенные экстракты промыли насыщенным раствором Na2CO3 и солевым раствором, высушили над Na2SO4, и эвапорировали. Остаток очистили посредством хроматографии (10г силикагель; гептан/EtOAc 90/10 - 50/50) с получением соединения, указанного в заголовке (36 мг, 79%), в виде белого осадка. MS (ESI): m/z = 390.2 [M+H]+.
Пример D15:
3-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-b]пиридин-6-ил]окси]пропан-1,2-диол

Соединение, указанное в заголовке, получили из 3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-6-фтор-1H-пиразоло[3,4-b]пиридина (Пример D6) по аналогии с примером D13 с использованием (2,2-диметил-1,3-диоксолан-4-ил)метанола вместо 2-(тетрагидро-2H-пиран-2-илокси)этанола на стадии 2 и получили в виде белого осадка. MS (ESI): m/z = 420.2 [M+H]+.
Пример D17:
[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-b]пиридин-3-илметокси)фенил]фенил]-морфолин-4-илметанон

Суспензию Промежуточного соединения B10 (40.1 мг), трет-бутил 3-(бромометил)пиразоло[3,4-b]пиридин-1-карбоксилат (37.8 мг, CAS: 174180-76-8) и карбонат калия (41.7 мг) в ацетоне (1.5 мл) нагревали до 50°C в течение 5.5 ч. Реакционную смесь охладили до КТ, разбавили водой и экстрагировали EtOAc. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток растворили в ДХМ (2 мл) и трифторуксусную кислоту (0.2 мл) добавили и раствор перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь погасили осторожно посредством добавления насыщенного раствора Na2CO3 и экстрагировали с помощью ДХМ. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством хроматографии (10г силикагель; гептан/EtOAc 70/30 - 0/100) с получением соединения, указанного в заголовке (40 мг, 71%), в виде белого осадка. MS (ESI): m/z = 463.2 [M+H]+.
Следующие примеры были синтезированы по аналогии с примером D17.
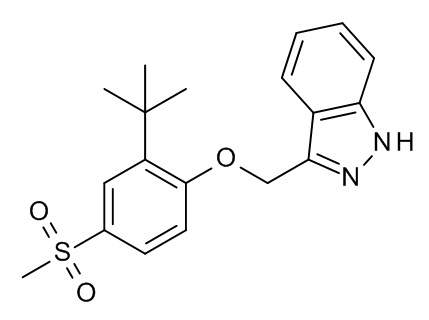

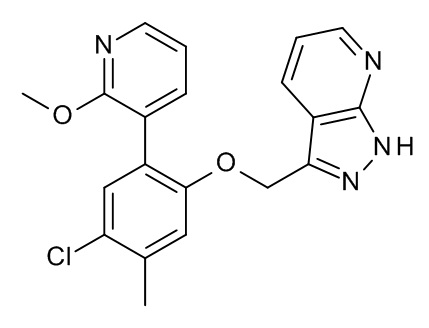


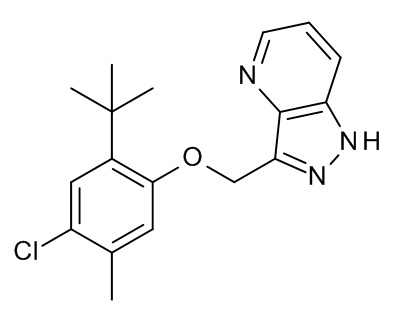




Пример D26:
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]-метиламино]этанол

Стадия 1: 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-]пиримидин

К раствору 2-трет-бутил-4-хлор-5-метил-фенола (851 мг, CAS: 30894-16-7) в безводном ДМФ (50 мл) добавили Cs2CO3(1.61 г) и TBAI (122 мг) при 25°C и реакционную смесь перемешивали при 25°C в течение 15 мин. Затем раствор 3-бромометил-1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидина (1.26 г, промежуточное соединение D26-B) в безводном ДМФ (10 мл) добавили при 25°C и реакционную смесь перемешивали в течение 16 ч при 25°C. Смесь отфильтровали и фильтрат разбавили EtOAc и промыли водой и солевым раствором. Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (5-10% EtOAc в гексане) с получением соединения, указанного в заголовке (1.41 г, 86%), в виде серо-белого осадка. MS (ESI): m/z = 497.1 [M+H]+.
Стадия 2: 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин

Раствор 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-]пиримидина (1.4 г) в 30% HBr в AcOH (30 мл) нагревали до 80°C в течение 2 ч. Реакционную смесь охладили до 25°C, разбавили EtOAc и промыли насыщенным раствором NaHCO3 и солевым раствором. Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (15-50% EtOAc в гексане) с получением соединения, указанного в заголовке (750 мг, 71%), в виде серо-белого осадка. MS (ESI): m/z = 377.2 [M+H]+.
Стадия 3: 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-метансульфонил-1H-пиразоло[3,4-d]пиримидин

К раствору 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидина (750 мг) в безводном ТГФ (100 мл) добавили m-CPBA (1.03 г) при 25°C и реакционную смесь перемешивали при 25°C в течение 16 ч. Смесь разбавили EtOAc и промыли насыщенным водным раствором тиосульфата натрия и солевым раствором. Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Получившийся остаток очистили посредством колоночной хроматографии через силикагель ( 30-50% EtOAc в гексане) с получением соединения, указанного в заголовке (580 мг, 71%), в виде серо-белого осадка. MS (ESI): m/z = 409.3 [M+H]+.
Стадия 4: 2-{[3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-1H-пиразоло[3,4-d]пиримидин-6-ил]-метил-амино}-этанол

К раствору 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-метансульфонил-1H-пиразоло[3,4-d]пиримидина (45 мг в диоксане (10 мл) при 25°C добавили 2-метиламино-этанол (24.82 мг) с последующим Et3N (0.03 мл) и реакционную смесь нагревали до кипения в течение 3 ч. Смесь охладили до 25°C и все летучие компоненты удалили при пониженном давлении. Остаток очистили посредством колоночной хроматографии через силикагель (60-70% EtOAc в гексане) с получением соединения, указанного в заголовке (12 мг, 27%) в виде серо-белого осадка. MS (ESI): m/z = 404.4 [M+H]+.
Следующие примеры были синтезированы по аналогии с примером D26 из 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-метансульфонил-1H-пиразоло[3,4-d]пиримидина (Пример D26, Стадия 3) в аналогичных реакционных условиях, как описано в примере D26, Стадия 4, с подходящим аминореагентом:



Пример D31:
2-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]окси]этанол
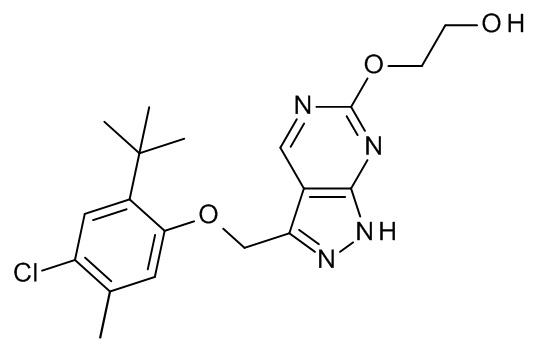
Смесь 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-метансульфонил-1H-пиразоло[3,4-d]пиримидина (150 мг, пример D26, Стадия 3), этан-1,2-диола (1 мл) и Et3N (0.103 мл) нагревали до 100°C в течение 6 ч. Реакционную смесь охладили до 25°C, разбавили EtOAc и промыли водой и солевым раствором. Органический слой высушили над Na2SO4, отфильтровали и эвапорировали при пониженном давлении. Остаток очистили препаративной ВЭЖХ с получением соединения, указанного в заголовке (29 мг, 20%) в виде серо-белого осадка. MS (ESI): m/z = 391.2 [M+H]+.
Пример D32:
5-трет-бутил-4-[[6-(3-гидроксиазетидин-1-ил)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил
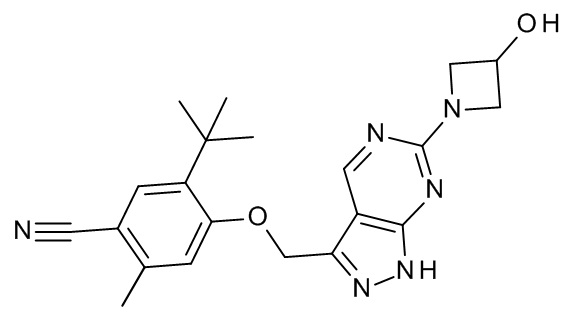
Стадия 1: 5-трет-бутил-4-[1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-илметокси]-2-метил-бензонитрил

Соединение, указанное в заголовке, получили по аналогии с примером D26, Стадия 1 из 3-бромометил-1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидина (400 мг, промежуточное соединение D26-B) и 5-трет-бутил-4-гидрокси-2-метил-бензонитрила (259.31 мг, Промежуточное соединение A16) и получили в виде серо-белого осадка (450 мг, 87%). MS (ESI): m/z = 488.5 [M+H]+.
Стадия 2: 5-трет-бутил-2-метил-4-(6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-илметокси)-бензонитрил

Соединение, указанное в заголовке, получили по аналогии с примером D26, Стадия 2 из 5-трет-бутил-4-[1-(4-метокси-бензил)-6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-илметокси]-2-метил-бензонитрила (450 мг) и получили в виде серо-белого осадка (225 мг, 66%). MS (ESI): m/z = 368.2 [M+H]+.
Стадия 3: 5-трет-бутил-4-(6-метансульфонил-1H-пиразоло[3,4-d]пиримидин-3-илметокси)-2-метил-бензонитрил

Соединение, указанное в заголовке, получили по аналогии с примером D26, Стадия 3, из 5-трет-бутил-2-метил-4-(6-метилсульфанил-1H-пиразоло[3,4-d]пиримидин-3-илметокси)-бензонитрила (700 мг) и получили в виде серо-белого осадка (580 мг, 76%). MS (ESI): m/z = 400.2 [M+H]+.
Стадия 4: 5-трет-бутил-4-[[6-(3-гидроксиазетидин-1-ил)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил
Соединение, указанное в заголовке, получили по аналогии с примером D26, Стадия 4, из 5-трет-бутил-4-(6-метансульфонил-1H-пиразоло[3,4-d]пиримидин-3-илметокси)-2-метил-бензонитрила (100 мг) и 3-(гидрокси)азетидина гидрохлорида (82.0 мг) и получили в виде серо-белого осадка (35 мг, 36%). MS (ESI): m/z = 393.3 [M+H]+.
Следующие примеры были синтезированы по аналогии с примером D32 из 5-трет-бутил-4-(6-метансульфонил-1H-пиразоло[3,4-d]пиримидин-3-илметокси)-2-метил-бензонитрила (Пример D32, Стадия 3) в аналогичных реакционных условиях, как описано в примере D26, Стадия 4, с подходящим аминореагентом:
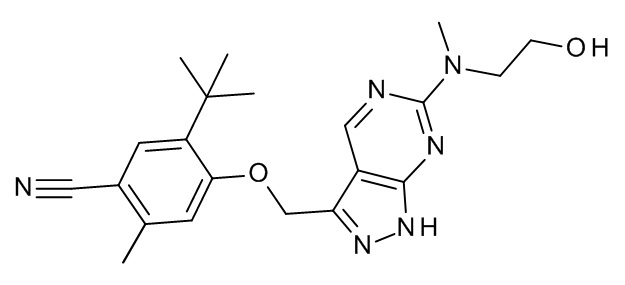


Пример D34:
3-[[3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-1H-пиразоло[3,4-d]пиримидин-6-ил]окси]пропан-1,2-диол
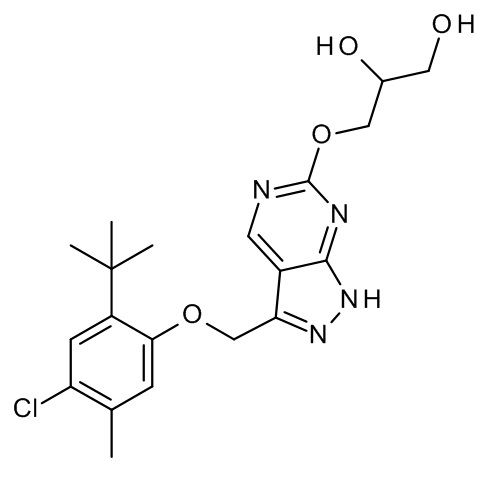
Стадия 1: 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-(2,2-диметил-[1,3]диоксолан-4-илметокси)-1H-пиразоло[3,4-d]пиримидин

Смесь 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-метансульфонил-1H-пиразоло[3,4-d]пиримидина (150 мг, пример D26, Стадия 3), (2,2-диметил-[1,3]диоксолан-4-ил)-метанола (1 мл) и Et3N (0.103 мл) нагрели до 100°C в течение 3 ч. Реакционную смесь охладили до 25°C и все летучие компоненты эвапорировали при пониженном давлении. Остаток разбавили EtOAc и органическую фазу промыли водой и солевым раствором. Органический слой высушили над Na2SO4 и эвапорировали при пониженном давлении с получением соединения, указанного в заголовке, которое использовали на следующей реакционной стадии без дополнительной очистки. MS (ESI): m/z = 461.1 [M+H]+.
Стадия 2: 3-[3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-1H-пиразоло[3,4-d]пиримидин-6-илокси]-пропан-1,2-диол

Раствор 3-(2-трет-бутил-4-хлор-5-метил-феноксиметил)-6-(2,2-диметил-[1,3]диоксолан-4-илметокси)-1H-пиразоло[3,4-d]пиримидина (135 мг в 2 н водной HCl (15 мл) перемешивали при 25°C в течение 16 ч. Реакционную смесь разбавили с помощью ДХМ и органический слой разделили, промыли насыщенным водным раствором NaHCO3 и солевым раствором, высушили над Na2SO4 и эвапорировали при пониженном давлении. Остаток очистили посредством ВЭЖХ с получением соединения, указанного в заголовке (36 мг, 29%), в виде серо-белого осадка. MS (ESI): m/z = 421.2 [M+H]+.
Пример D37:
5-трет-бутил-4-[[6-(2,3-дигидроксипропокси)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил
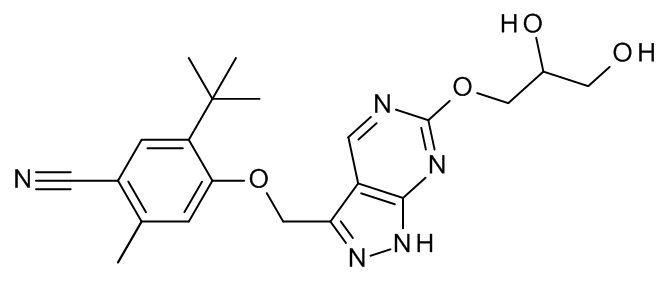
Соединение, указанное в заголовке, получили по аналогии с примером D34, Стадии 1 и 2, из 5-трет-бутил-4-(6-метансульфонил-1H-пиразоло[3,4-d]пиримидин-3-илметокси)-2-метил-бензонитрила (150 мг, пример D32, Стадия 3) и получили в виде серо-белого осадка (26 мг, 18%). MS (ESI): m/z = 412.2 [M+H]+.
Пример D38:
5-трет-бутил-4-[[6-(2-гидроксиэтокси)-1H-пиразоло[3,4-d]пиримидин-3-ил]метокси]-2-метилбензонитрил

Соединение, указанное в заголовке, получили по аналогии с примером D31 из 5-трет-бутил-4-(6-метансульфонил-1H-пиразоло[3,4-d]пиримидин-3-илметокси)-2-метил-бензонитрила (105 мг, пример D32, Стадия 3) и получили в виде серо-белого осадка (16 мг, 16%). MS (ESI): m/z = 382.2 [M+H]+.
Пример D44:
4-[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-c]пиридин-3-илметокси)фенил]-4-фторбензоил]-1-метилпиперазин-2-он
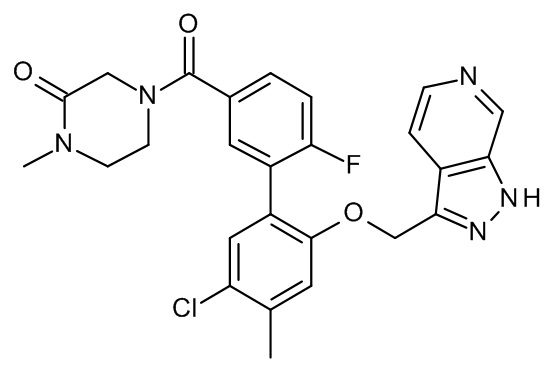
Стадия 1: 4-[3-[5-хлор-4-метил-2-[(1-тритилпиразоло[3,4-c]пиридин-3-ил)метокси]фенил]-4-фторбензоил]-1-метилпиперазин-2-он
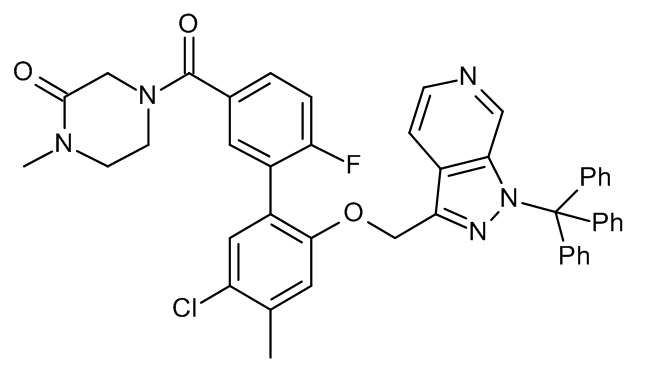
В 20 мл круглодонной колбе, 4-(5'-хлор-6-фтор-2'-гидрокси-4'-метил-[1,1'-бифенил]-3-карбонил)-1-метилпиперазин-2-он (промежуточное соединение B65, 50 мг), 3-(хлорметил)-1-тритил-1H-пиразоло[3,4-c]пиридин (промежуточное соединение D44-B, 54.4 мг) и карбонат калия (40.3 мг) объединили с ацетоном (6 мл) с получением белой суспензии. Смесь затем нагревали при 50°C и перемешивали в течение 4 ч, однако взаимодействия не наблюдалось. Таким образом, ацетон удалили под вакуумом и ацетонитрил (6 мл) и карбонат цезия (95 мг) добавили. Смесь снова нагревали при 80°C в течение 4 часов. Растворитель удалили под вакуумом и в светло-коричневй остаток добавили насыщенный водный раствор NH4Cl. Смесь экстрагировали 3 раза этилацетатом и объединенные органические слои высушили над Na2SO4 и сконцентрировали под вакуумом с получением светло-коричневой пены. Неочищенное вещество очистили с помощью флеш-хроматографии (силикагель, 20 г, градиент от 0% до 100% этилацетат в гептане, с последующим от 0% до 10% MeOH в ДХМ) с получением соединения, указанного в заголовке, в виде бесцветной пены (53 мг). MS (m/z): 750.26 [M+H]+.
Стадия 2: 4-[3-[5-хлор-4-метил-2-(1H-пиразоло[3,4-c]пиридин-3-илметокси)фенил]-4-фторбензоил]-1-метилпиперазин-2-он
В 10 мл круглодонной колбе, 4-(5'-хлор-6-фтор-4'-метил-2'-((1-тритил-1H-пиразоло[3,4-c]пиридин-3-ил)метокси)-[1,1'-бифенил]-3-карбонил)-1-метилпиперазин-2-он (50 мг) растворили в CH2Cl2 (2.9 мл) с получением бесцветного раствора. ТФУ (444 мкл) добавили при комнатной температуре и реакционную смесь перемешивали в течение 30 мин. ТСХ анализ подтвердил израсходование исходного материала и образование единого продукта. Реакционную смесь затем погасили осторожным добавлением насыщенного раствора Na2CO3. Смесь экстрагировали с помощью ДХМ (2 x 25 мл) и объединенные органические экстракты промыли солевым раствором, высушили над Na2SO4, и эвапорировали с получением соединения, указанного в заголовке, в виде бесцветной пены (27.0 мг). MS (m/z): 508.15 [M+H]+.
Следующие примеры были синтезированы из подходящих структурных элементов/промежуточных соединений по аналогии с примером D44, Стадии 1-2:



Следующие дополнительные примеры типа D были синтезированы из подходящих структурных элементов/промежуточных соединений по аналогии с примером D44, Стадии 1-2:

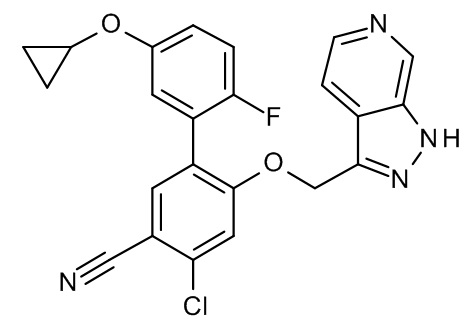

Следующие дополнительные примеры типа D были синтезированы из подходящих структурных элементов/промежуточных соединений по аналогии с примером D17:


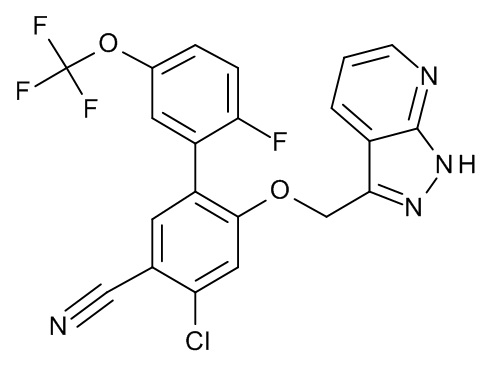
Примеры E: Примеры, относящиеся к тетрагидро-пиразолопиридиновым основным группам
Пример E1:
трет-бутил 3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилат

Стадия 1: 5-трет-бутил 3-этил 6,7-дигидро-1H-пиразоло[4,3-c]пиридин-3,5(4H)-дикарбоксилат
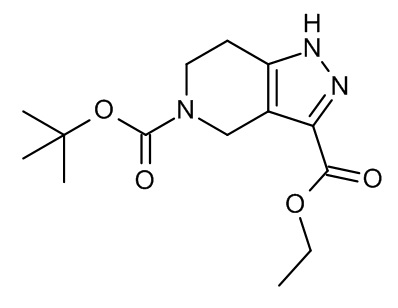
Ди-трет-бутил-дикарбонат (1.04 г) добавили к суспензии этил 4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридин-3-карбоксилата (930 мг, CAS: 926926-62-7) в диэтиловом эфире (45 мл) при 0°C. После добавления реагентов, реакционную смесь перемешивали при комнатной температуре в течение ночи. Воду добавили и смесь экстрагировали EtOAc. Объединенные органические слои промыли водой и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (50 г силикагель; гептан/EtOAc 70/30 - 45/55) с получением соединения, указанного в заголовке, в виде белой пены (1.079 г, 77%). MS (ESI): m/z = 294.2 [M-H]-.
Стадия 2: трет-бутил 3-(гидроксиметил)-1,4,6,7-тетрагидропиразоло[4,3-c]пиридин-5-карбоксилат

Раствор 5-трет-бутил 3-этил 6,7-дигидро-1H-пиразоло[4,3-c]пиридин-3,5(4H)-дикарбоксилата (60.0 мг) в ТГФ (0.5 мл) добавили по каплям при 0°C к белой суспензии хлорида кальция (90.1 мг) и борогидрида натрия (61.4 мг) в смеси EtOH (1 мл) и ТГФ (0.5 мл). Получившуюся белую суспензию перемешивали при 0°C в течение ночи. Затем 0.1 н HCl добавили осторожно и смесь экстрагировали EtOAc. Объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (5 г силикагель; ДХМ/MeOH 98/2 - 95/5) с получением соединения, указанного в заголовке, в виде белого осадка (47 мг, 92%). MS (ESI): m/z = 254.2 [M+H]+.
Стадия 3: трет-бутил 3-(хлорметил)-1,4,6,7-тетрагидропиразоло[4,3-c]пиридин-5-карбоксилат

трет-бутил 3-(гидроксиметил)-1,4,6,7-тетрагидропиразоло[4,3-c]пиридин-5-карбоксилат (94.0 мг) суспендировали в смеси ДХМ (1 мл), ацетонитрила (1 мл) и ТГФ (1 мл). Трифенилфосфин (102 мг) и тетрахлорид углерода (285 мг, 179 мкл) добавили к белой суспензии и смесь перемешивали при комнатной температуре. После этого реакционная смесь превратилась в бесцветный раствор, который перемешивали при комнатной температуре в течение 2 дней. Дополнительное количество тетрахлорида углерода (285 мг, 179 мкл) добавили и раствор перемешивали в течение еще 1 дня при комнатной температуре. Затем смесь сконцентрировали досуха и остаток очистили посредством колоночной хроматографии (20 г силикагель; ДХМ/MeOH 98/2 - 90/10) с получением соединения, указанного в заголовке, в виде белого осадка (37 мг, 35%). MS (ESI): m/z = 272.2 [M+H]+.
Стадия 4: трет-бутил 3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-карбоксилат

Смесь трет-бутил 3-(хлорметил)-1,4,6,7-тетрагидропиразоло[4,3-c]пиридин-5-карбоксилата (45.1 мг), 2-трет-бутил-4-хлор-5-метил-фенола (82.5 мг, CAS: 30894-16-7) и карбоната калия (57.4 мг) в ацетонитриле (2 мл) нагревали до кипения в течение 5 ч. Реакционную смесь охладили до КТ и затем сконцентрировали. Остаток разделили между EtOAc и водой. Водный слой экстрагировали EtOAc и объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Оставшееся масло очистили посредством колоночной хроматографии (10г силикагель; гептан/EtOAc 70/30 - 10/90) с получением соединения, указанного в заголовке, в виде белого осадка (19 мг, 26%). MS (ESI): m/z = 434.3 [M+H]+.
Пример E2:
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-ил)этанон
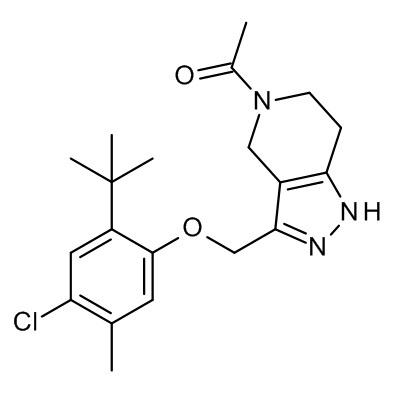
Стадия 1: 5-трет-Бутил 3-этил 1-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-3,5-дикарбоксилат и 5-трет-Бутил 3-этил 2-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-3,5-дикарбоксилат


Раствор 5-трет-бутил 3-этил 6,7-дигидро-1H-пиразоло[4,3-c]пиридин-3,5(4H)-дикарбоксилата (1.17 г, пример E1, Стадия 1) в ДМФ (7 мл) добавили по каплям К суспензии гидрида натрия (216 мг, 55% в минеральном масле) в ДМФ (7 мл) при 0°C. Реакционную смесь перемешивали при 0°C в течение 15 мин и затем при комнатной температуре в течение 30 мин. Раствор снова охладили до 0°C и раствор [хлор(дифенил)метил]бензола (1.16 г) в ДМФ (7 мл) добавили. Получившуюся суспензию перемешивали при комнатной температуре в течение 2 дней. Воду добавили осторожно и смесь экстрагировали EtOAc. Объединенные экстракты промыли водой (3 раза) и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (120 г силикагель; гептан/EtOAc 95/5 - 25/75) с получением 5-трет-бутил 3-этил 1-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-3,5-дикарбоксилата (серо-белый осадок, 790 мг, 37%, MS (ESI): m/z = 560.4 [M+Na]+) а также 5-трет-бутил 3-этил 2-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-3,5-дикарбоксилата (белый осадок, 450 мг, 21%). MS (ESI): m/z = 560.4 [M+Na]+).
Стадия 2: трет-бутил 3-(гидроксиметил)-1-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилат

К раствору 5-трет-бутил 3-этил 1-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-3,5-дикарбоксилата (200 мг) в ТГФ (3 мл) добавили по каплям диизобутилалюминия гидрид (818 мкл, 1M в толуоле) при 0°C. После добавления, бесцветный раствор перемешивали при комнатной температуре в течение ночи. 0.5 мл MeOH добавили с интенсивным перемешиванием и получившийся раствор влили в смесь 20 мл 10 мас. % раствора сегнетовой соли и 20 мл EtOAc. Эту смесь перемешивали при комнатной температуре в течение 1 ч и слои затем разделили. Водный слой экстрагировали EtOAc и объединенные органические слои промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (20 г силикагель; гептан/EtOAc 80/20 - 50/50) с получением соединения, указанного в заголовке, в виде белого осадка (156 мг, 85%). MS (ESI): m/z = 518.4 [M+Na]+).
Стадия 3: трет-бутил 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-1-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилат
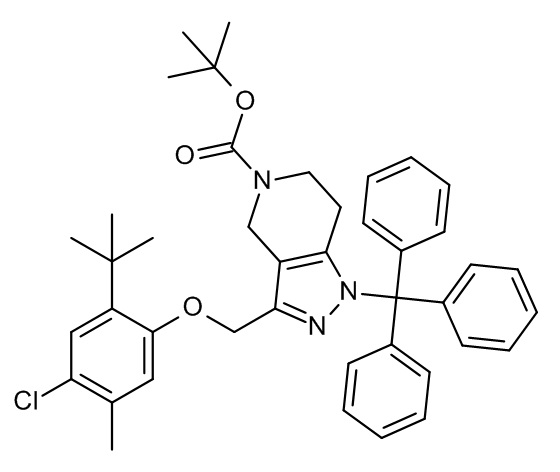
К раствору трет-бутил 3-(гидроксиметил)-1-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилата (100 мг) в 2-метилтетрагидрофуране (1 мл) добавили триэтиламин (22.5 мг, 30.9 мкл) при 0°C. К этой смеси добавили по каплям метансульфонилхлорид (24.3 мг, 16.5 мкл) и получившуюся смесь перемешивали при 0°C в течение 1 ч. Преципитировался белый осадок, который отфильтровали и промыли 2-метилтетрагидрофураном. Фильтрат сконцентрировали и оставшееся бесцветное масло растворили в DMA (0.5 мл) и получившийся раствор добавили по каплям к суспензии 2-трет-бутил-4-хлор-5-метил-фенола (40.1 мг, CAS: 30894-16-7), калия йодида (56.9 мг) и фторида цезия (153 мг) в DMA (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Воду добавили и смесь экстрагировали EtOAc. Объединенные органические слои промыли водой (3 раза) и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (20 г силикагель; гептан/EtOAc 98/2 - 50/50) с получением соединения, указанного в заголовке, в виде серо-белого осадка (49 мг, 36%). MS (ESI): m/z = 674.5 [M-H]-.
Стадия 4: 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридин
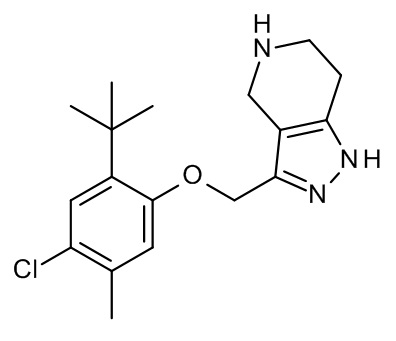
Трифторуксусную кислоту (0.2 мл) добавили в раствор трет-бутил 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-1-тритил-6,7-дигидро-4H-пиразоло[4,3-c]пиридин-5-карбоксилата (47.0 мг) в ДХМ (1 мл) при комнатной температуре. Через 30 мин осторожно добавили насыщенный раствор Na2CO3 и смесь экстрагировали с помощью ДХМ. Объединенные экстракты промыли небольшим количеством солевого раствора, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (5 г силикагель; ДХМ/MeOH 95/5 - 85/15) с получением соединения, указанного в заголовке, в виде белого осадка (18 мг, 78%). MS (ESI): m/z = 334.2 [M+H]+.
Стадия 5: 1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-ил)этанон
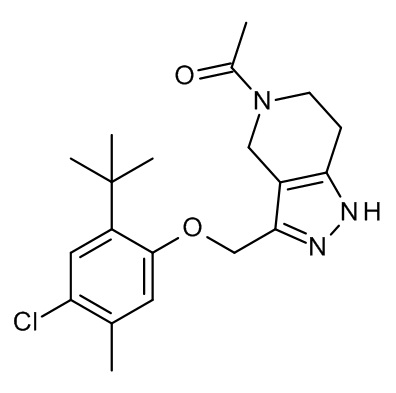
Смесь 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридина (15.0 мг), триэтиламина (9.09 мг, 12.5 мкл) и ацетилхлорида (3.52 мг, 3.19 мкл) в ТГФ (0.5 мл) перемешивали при комнатной температуре в течение ночи. Затем добавили воду и смесь экстрагировали EtOAc и затем с помощью ДХМ. Объединенные экстракты промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (5 г силикагель; ДХМ/MeOH 19/1) с получением соединения, указанного в заголовке, в виде белого осадка (13 мг, 77%). MS (ESI): m/z = 376.2 [M+H]+.
Пример E3:
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-6,7-дигидро-1H-пиразоло[4,3-c]пиридин-5(4H)-ил)-2-метоксиэтанон

Соединение, указанное в заголовке, получили по аналогии с примером E2, Стадия 5, из 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-4,5,6,7-тетрагидро-1H-пиразоло[4,3-c]пиридина (15.0 мг) и 2-метоксиацетилхлорида (5.11 мг, 4.27 мкл) и получили в виде белого осадка (13 мг, 71%). MS (ESI): m/z = 406.3 [M+H]+.
Пример E4:
3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридин

Стадия 1: трет-Бутил 4-(1-диазо-2-этокси-2-оксо-этил)-4-гидрокси-пиперидин-1-карбоксилат

Раствор диизопропиламида лития получили посредством покапельного добавления nBuLi (19.1 мл, 1.6 M в гексане) в раствор диизопропиламина (3.12 г, 4.4 мл) в безводном ТГФ (77 мл) при -78 °C. Этот раствор диизопропиламида лития затем добавили по каплям в течение 1 часа в раствор трет-бутил 4-оксопиперидин-1-карбоксилата (3.854 г) и этилдиазоацетата (2.31 г, 2.1 мл) в безводном ТГФ (115 мл) при -78 °C. Смесь перемешивали в течение 2 часов при -78 °C. Затем AcOH (5.81 г, 5.54 мл) добавили при -78 °C и смесь затем держали при комнатной температуре в течение ночи. Растворитель удалили при пониженном давлении до примерно 1/10 объема и диэтиловый эфир (400 мл) добавили. Смесь промыли насыщенным раствором NaHCO3 и затем высушили над Na2SO4, отфильтровали и эвапорировали. Оставшееся оранжево-коричневое вязкое масло (6.46 г) использовали без дополнительного анализа на следующей реакционной стадии.
Стадия 2: трет-бутил 4-(1-диазо-2-этокси-2-оксо-этил)-3,6-дигидро-2H-пиридин-1-карбоксилат

Пиридин (30.5 г, 31.2 мл) добавили в раствор трет-бутил 4-(1-диазо-2-этокси-2-оксо-этил)-4-гидрокси-циклогексанкарбоксилата (6.05 г) в MTBE (120 мл). Смесь охладили до -10 °C и оксихлорид фосфора (5.92 г, 3.6 мл) добавили по каплям в течение 8 минут при интенсивном перемешивании. Смесь медленно нагрели до КТ и перемешивали в течение ночи. Смесь охладили снова до -10°C и дополнительное количество оксихлорида фосфора (592 мг, 360 мкл) добавили по каплям. Реакционную смесь медленно нагрели до КТ и перемешивали в течение 3 ч перед тем, как 0.1M NaOH (193 мл) медленно добавили. Смесь экстрагировали EtOAc и объединенные экстракты промыли солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Остаток (6.2 г) использовали на следующей реакционной стадии без дополнительной очистки. MS (ESI): m/z = 294.2 [M-H]-.
Стадия 3: 6-трет-Бутил 3-этил 1,4,5,7-тетрагидропиразоло[3,4-c]пиридин-3,6-дикарбоксилат

В двухгорловой колбе, оснащенной капельной воронкой и дистиляционной колонной, толуол (80 мл) нагрели до кипения. Раствор трет-бутил 4-(1-диазо-2-этокси-2-оксо-этил)-3,6-дигидро-2H-пиридин-1-карбоксилата (5.7 г) в смеси EtOAc (20 мл) и пиридина (8 мл) добавили через капельную воронку при скорости аналогичной скорости дистилляции. Дополнительное количество толуола (7 мл) добавили. Темно-коричневую смесь перемешивали в течение часа и was затем дали остыть до КТ. EtOAc (250 мл) добавили и смесь промыли водой (3 раза) и солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенное вещество очистили посредством колоночной хроматографии (50 г силикагель; гептан/EtOAc 9/1 до 1/1) с получением соединения, указанного в заголовке, в виде светло-коричневого осадка (3.12 г, 55%). MS (ESI): m/z = 294.2 [M-H]-.
Стадия 4: 6-трет-Бутил 3-этил 1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-3,6-дикарбоксилат, и 6-трет-Бутил 3-этил 2-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-3,6-дикарбоксилат


По аналогии с примером E2, Стадия 1, 6-трет-бутил 3-этил 1,4,5,7-тетрагидропиразоло[3,4-c]пиридин-3,6-дикарбоксилат (980 мг) конвертировали в 6-трет-бутил 3-этил 1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-3,6-дикарбоксилат (белый осадок, 762 мг, 43%, MS (ESI): m/z = 1097.8 [2M+Na]+) и 6-трет-бутил 3-этил 2-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-3,6-дикарбоксилат (белый осадок, 510 мг, 29%). MS (ESI): m/z = 560.4 [M+Na]+).
Стадия 5: трет-бутил 3-(гидроксиметил)-1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-6-карбоксилат

Из 6-трет-бутил 3-этил 1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-3,6-дикарбоксилата (715 мг) соединение, указанное в заголовке, получили по аналогии с примером E2, Стадия 2, в виде белого осадка (581 мг, 88%). MS (ESI): m/z = 496.3 [M+H]+.
Стадия 6: трет-бутил 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-6-карбоксилат

Из трет-бутил 3-(гидроксиметил)-1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-6-карбоксилата (100 мг) соединение, указанное в заголовке, получили по аналогии с примером E2, Стадия 3, в виде светло-желтой пены (58 мг, 43%). MS (ESI): m/z = 698.5 [M+Na]+.
Стадия 7: 3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридин

Из трет-бутил 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-6-карбоксилата (75.7 мг) Соединение, указанное в заголовке, получили по аналогии с примером E2, Стадия 4, в виде светло-желтого осадка (29 мг, 78%). MS (ESI): m/z = 334.2 [M+H]+.
Пример E5:
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5-дигидро-1H-пиразоло[3,4-c]пиридин-6(7H)-ил)этанон

Соединение, указанное в заголовке, получили по аналогии с примером E2, Стадия 5, из 3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридина (25.0 мг, пример 4) и ацетилхлорида (5.88 мг, 5.33 мкл) и получили в виде белого осадка (19 мг, 67%). MS (ESI): m/z = 376.2 [M+H]+.
Пример E6:
1-(3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5-дигидро-1H-пиразоло[3,4-c]пиридин-6(7H)-ил)-2-метоксиэтанон
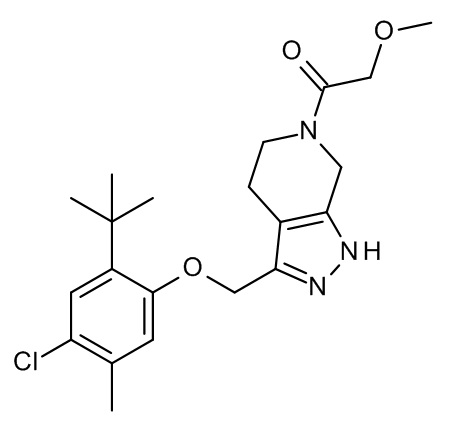
Соединение, указанное в заголовке, получили по аналогии с примером E2, Стадия 5, из 3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридина (22.0 мг, пример 4) и 2-метоксиацетилхлорида (7.51 мг, 6.27 мкл) и получили в виде белого осадка (17 мг, 64%). MS (ESI): m/z = 406.2 [M+H]+.
Пример E7:
3-((4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридин

Триэтиламин (26.9 мг, 37.1 мкл) добавили в раствор трет-бутил 3-(гидроксиметил)-1-тритил-5,7-дигидро-4H-пиразоло[3,4-c]пиридин-6-карбоксилата (120 мг, полученный в примере E4, Стадия 5) в 2-метилтетрагидрофуране (1.5 мл) при 0°C. К указанной смеси добавили по каплям метансульфонилхлорид (29.1 мг, 19.7 мкл) и получившуюся смесь перемешивали при 0°C в течение 1 ч. Суспензию затем отфильтровали, остаток на фильтре промыли 2-метилтетрагидрофураном и фильтрат сконцентрировали. Остаток растворили в DMA (1.5 мл) и этот раствор добавили по каплям к суспензии 4-хлор-2-(2-метокси-3-пиридил)-5-метил-фенола (60.4 мг, промежуточное соединение B53), калия йодида (68.2 мг) и цезия фторида (184 мг) в DMA (1.5 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем воду добавили и смесь экстрагировали EtOAc. Объединенные органические слои промыли водой (3 раза) и солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (20 г силикагель; гептан/EtOAc 90/10 - 50/50) с получением желтой пены (81 мг), которую растворили в смеси ДХМ (2 мл) и трифторуксусной кислоты (0.2 мл). Эту смесь перемешивали при комнатной температуре в течение 1 ч. Затем насыщенный раствор Na2CO3 добавили осторожно и смесь экстрагировали с помощью ДХМ. Объединенные органические слои промыли очень небольшим количеством солевого раствора, высушили над Na2SO4 и эвапорировали. Оставшийся остаток очистили посредством колоночной хроматографии (5 г силикагель; ДХМ/MeOH 95/5 + ДХМ/MeOH/NH4OH 8/1.9/0.1) с получением соединения, указанного в заголовке в виде белой пены (21 мг, 23%). MS (ESI): m/z = 385.2 [M+H]+.
Пример E8:
1-(3-((4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси)метил)-4,5-дигидро-1H-пиразоло[3,4-c]пиридин-6(7H)-ил)этанон
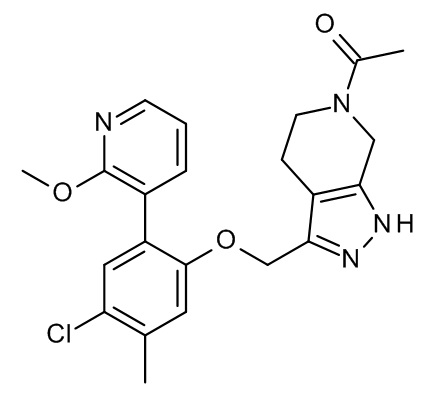
Соединение, указанное в заголовке, получили по аналогии с примером E2, Стадия 5, из 3-((4-хлор-2-(2-метоксипиридин-3-ил)-5-метилфенокси)метил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-c]пиридина (25.0 мг, пример E7) и ацетилхлорида (5.88 мг, 5.33 мкл) и получили в виде белого осадка (8 мг, 42%). MS (ESI): m/z = 427.2 [M+H]+.
Примеры F: Примеры, относящиеся к пиразоловым основным группам
Пример F1:
3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-1H-пиразол

Стадия 1: трет-бутил 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]пиразол-1-карбоксилат
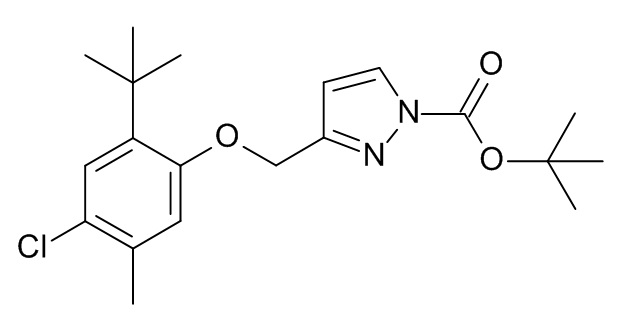
Смесь трет-бутил 3-(бромометил)пиразол-1-карбоксилата (65.8 мг, промежуточное соединение F1), 2-трет-бутил-4-хлор-5-метил-фенола (50 мг, CAS: 30894-16-7) и карбоната калия (87.1 мг) в ацетоне (5 мл) нагрели до 50°C. Через 6 ч и после 7.5 ч, дополнительное количество 2-трет-бутил-4-хлор-5-метилфенола (10 мг и 22 мг, соответственно) добавили и нагревание продолжили в течение 4 ч. Насыщенный раствор NH4Cl добавили и смесь экстрагировали EtOAc. Объединенные экстракты промыли солевым раствором, высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии (20 г SiO2, н-гептан/EtOAc 100/0 до 65/35) с получением соединения, указанного в заголовке (71 мг, 74%), в виде бесцветного масла. MS (ESI): m/z = 279.2 [M-CO2tBu+H]+.
Стадия 2: 3-((2-трет-бутил-4-хлор-5-метилфенокси)метил)-1H-пиразол
Раствор трет-бутил 3-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]пиразол-1-карбоксилата (65 мг в смеси ДХМ (1 мл) и ТФУ (1 мл) перемешивали при комнатной температуре в течение 4 ч. Затем насыщенный раствор NaHCO3 добавили и смесь экстрагировали EtOAc. Объединенные экстракты промыли солевым раствором, высушили Na2SO4 и эвапорировали с получением соединения, указанного в заголовке (47 мг, 98%), в виде вязкого бесцветного масла. MS (ESI): m/z = 279.2 [M+H]+.
Пример F2:
4-((1H-пиразол-3-ил)метокси)-5-трет-бутил-2-метилбензонитрил
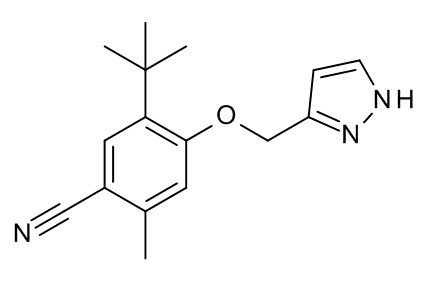
Соединение, указанное в заголовке, получили по аналогии с примером F1, Стадии 1 и 2, из трет-бутил 3-(бромометил)пиразол-1-карбоксилата (промежуточное соединение F1) и 5-трет-бутил-4-гидрокси-2-метил-бензонитрила (промежуточное соединение A16) и получили в виде белой пены. MS (ESI): m/z = 270.2 [M+H]+.
Пример F3:
Метил 3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-1H-пиразол-5-карбоксилат

Стадия 1: Метил 5-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-2-тетрагидропиран-2-ил-пиразол-3-карбоксилат

Триэтиламин (29 мг, 40 мкл) добавили в раствор метил 5-(гидроксиметил)-2-тетрагидропиран-2-ил-пиразол-3-карбоксилата (63 мг, промежуточное соединение F3) в 2-метилтетрагидрофуране (1.5 мл) при 0 °C. Затем метансульфонилхлорид (30.9 мг, 21 мкл) добавили по каплям и реакционную смесь перемешивали при 0 °C в течение 2 ч. Белый преципитат отфильтровали и промыли 2-метилтетрагидрофураном. Фильтрат сконцентрировали и остаток суспендировали в DMA (2.4 мл) и добавили к суспензии 2-трет-бутил-4-хлор-5-метил-фенола (52 мг, CAS: 30894-16-7), йодида калия (76 мг) и фторида цезия (197 мг) в DMA (1.5 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем добавили воду и смесь экстрагировали EtOAc. Объединенные экстракты промыли водой и солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Остаток очистили посредством хроматографии (20 г силикагель; гептан/EtOAc 100/0 до 1/2) с получением соединения, указанного в заголовке (22 мг, 20%) в виде воскообразного осадка. MS (ESI): m/z = 337.2 [M-THP+H]+.
Стадия 2: Метил 3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-1H-пиразол-5-карбоксилат
Метил 5-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-2-тетрагидропиран-2-ил-пиразол-3-карбоксилат (22 мг) суспендировали в MeOH (1 мл) и HCl (4 M в 1,4-диоксане, 70 мкл) добавили. Бесцветный раствор перемешивали при комнатной температуре в течение 2 ч перед тем, как добавили насыщенный раствор NaHCO3 (5 мл). Смесь эвапорировали до половины от объема и затем экстрагировали EtOAc. Объединенные органические слои промыли солевым раствором (10 мл), высушили над Na2SO4, отфильтровали и эвапорировали. Остаток очистили посредством хроматографии (10 г силикагель; гептан/EtOAc 100/0 до 70/30) с получением соединения, указанного в заголовке (16 мг, 91%), в виде белого порошка. MS (ESI): m/z = 337.2 [M+H]+.
Пример F4:
(3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-1H-пиразол-5-ил)(пирролидин-1-ил)метанон
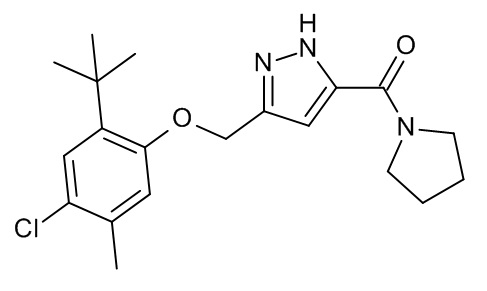
Стадия 1: [5-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-2-тетрагидропиран-2-ил-пиразол-3-ил]-пирролидин-1-ил-метанон
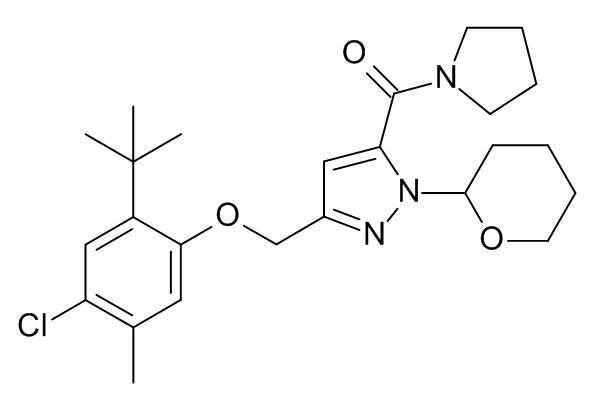
К раствору пирролидина (12.9 мг, 15 мкл) в ДХМ (0.3 мл) добавили 2M раствор триметилалюминия в гексане (90 мкл) по каплям при комнатной температуре и смесь перемешивали в течение 15 минут. Затем раствор метил 5-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-2-тетрагидропиран-2-ил-пиразол-3-карбоксилата (50 мг, пример F3, Стадия 1) в ДХМ (0.6 мл) медленно добавили. Реакционную смесь нагревали до кипения в течение ночи. Смесь осторожно разбавили 1 M HCl, экстрагировали с помощью ДХМ, и объединенные органические слои высушили над Na2SO4, отфильтровали и эвапорировали. Остаток очистили посредством хроматографии (20 г силикагель; гептан/EtOAc 100/0 до 1/1) с получением соединения, указанного в заголовке (36 мг, 66%), в виде бесцветного масла. MS (ESI): m/z = 376.3 [M-THP+H]+.
Стадия 2: (3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-1H-пиразол-5-ил)(пирролидин-1-ил)метанон
Соединение, указанное в заголовке, получили по аналогии с примером F3, Стадия 2, из [5-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-2-тетрагидропиран-2-ил-пиразол-3-ил]-пирролидин-1-ил-метанона (36 мг) и получили в виде белого осадка (21 мг, 71%). MS (ESI): m/z = 376.3 [M+H]+.
Пример F5:
3-((2-(трет-бутил)-4-хлор-5-метилфенокси)метил)-N,N-диметил-1H-пиразол-5-карбоксамид
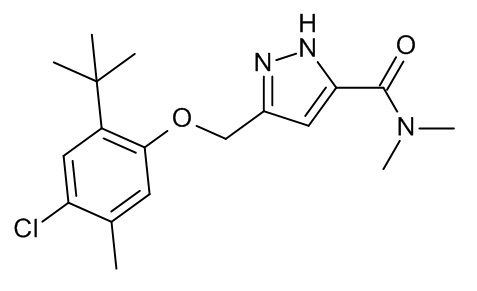
Соединение, указанное в заголовке, получили по аналогии с примером F4, Стадии 1 и 2, из метил 5-[(2-трет-бутил-4-хлор-5-метил-фенокси)метил]-2-тетрагидропиран-2-ил-пиразол-3-карбоксилата (Пример F3, Стадия 1) и гидрохлорида диметиламина и получили в виде белого осадка. MS (ESI): m/z = 350.3 [M+H]+.
Пример F6
4-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-2H-триазол
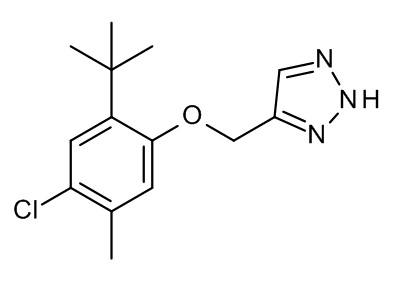
Стадия 1: 1-трет-бутил-5-хлор-4-метил-2-проп-2-иноксибензол

В 25 мл круглодонной колбе, 2-(трет-бутил)-4-хлор-5-метилфенол (CAS: 30894-16-7, 200 мг), 3-бромпроп-1-ин (80% в толуоле) (225 мг, 163 мкл) и карбонат калия (209 мг) объединили с CH3CN (4 мл) с получением белой суспензии. Реакционную смесь перемешивали в течение 15 ч и затем сконцентрировали под вакуумом. Реакционную смесь влили в этилацетат (25 мл) и органический слой промыли H2O (1 x 10 мл). Органический слой высушили над Na2SO4 и сконцентрировали под вакуумомс получением соединения, указанного в заголовке (180 мг, 74%). MS (m/z): 236.1 [M]+.
Стадия 2: 4-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-2H-триазол
В 25 мл круглодонной колбе, 1-(трет-бутил)-5-хлор-4-метил-2-(проп-2-ин-1-илокси)бензол (180 мг) объединили с ДМФ (4 мл) и водой (4 мл) с получением белой суспензии. Пентагидрат сульфата меди (II) (38 мг) и натриевую соль L-аскорбиновой кислоты (304 мг) добавили. Реакционную смесь откачали дважды и продули азотом. Триметилсилилазид (746 мг) добавили и реакционную смесь нагрели до 90°C в течение 2 ч при перемешивании. Смесь разбавили H2O (50 мл) и этилацетатом (100 мл) и слои разделили. Водную фазу экстрагировали дополнительным количеством этилацетата (2 x 50 мл). Органические слои высушили над Na2SO4 и сконцентрировали под вакуумом с получением неочищенного масла. Неочищенное вещество очистили с помощью флеш-хроматографии (20 г силикагелевый картридж, 0% до 10% MeOH в CH2Cl2) с получением соединения, указанного в заголовке, в виде бесцветной пены (80 мг, 37%). MS (m/z): 280.2 [M+H]+.
Пример F7
3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-5-фенил-1H-пиразол

Стадия 1: 3-(Хлорметил)-5-фенил-1H-пиразол

Это известное промежуточное соединение (CAS: 755700-32-4) получили из (5-фенил-1H-пиразол-3-ил)метанола, который является коммерчески доступным (CAS: 179057-19-3), следующим образом: В 25 мл круглодонной колбе, (5-фенил-1H-пиразол-3-ил)метанол (50 мг) объединили с CH2Cl2 (3 мл) и смесь охладили до 0°C в атмосфере аргона. Затем, тионилхлорид (68.3 мг, 41.6 мкл) добавили по каплям в течение периода 2 минут и через 30 минут ледяную ванну убрали. Реакционную смесь оставили перемешиваться при комнатной температуре в течение дополнительных 2 часов. Смесь влили в лед и раствор NaHCO3. Водный слой затем экстрагировали дважды с помощью этилацетата и органические слои промыли солевым раствором, высушили над Na2SO4 и сконцентрировали под вакуумом с получением неочищенного светло-желтого осадка (55 мг, 95%), который использовали без дополнительной очистки. MS (m/z): 193.0 [M+H]+.
Стадия 2: 3-[(2-трет-бутил-4-хлор-5-метилфенокси)метил]-5-фенил-1H-пиразол
2-(трет-бутил)-4-хлор-5-метилфенол (CAS: 30894-16-7, 62.4 мг), карбонат калия (233 мг) и 3-(хлорметил)-5-фенил-1H-пиразол (55 мг) объединили с ацетонитрилом (4 мл) при комнатной температуре в атмосфере аргона. Реакционную смесь затем нагревали при 50°C в течение 16 часов, после чего ТСХ подтвердила образование некоторого количества продукта. Реакционную смесь влили в воду со льдом и водный слой подщелачили с помощью 2M раствора NaOH и экстрагировали дважды с помощью этилацетата. Органические слои промыли один раз солевым раствором, высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенное вещество очистили с помощью флеш-хроматографии (25 г силикагелевый картридж, 0% до 40% этилацетат в гептане) с получением - помимо некоторых других неидентифицированных соединений- соединения, указанного в заголовке, в виде бесцветного осадка (16 мг, 16%). MS (m/z): 355.2 [M+H]+.
Следующие примеры типа F были синтезированы из подходящих фенольных структурных элементов/промежуточных соединений по аналогии с примером F7, Стадия 2:

Пример A
Пример B
Реферат
Настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли, а также к конкретному феноксиметильному производному или его фармацевтически приемлемой соли, обладающим способностью ингибировать АТХ, к содержащей их фармацевтической композиции, к их применению и к способу ингибирования АТХВ формуле I W выбран из кольцевых систем А, В, С, D и Е:где RA, RB, RC, RC1, RD1,RD4, A1-A6являются такими, как определено в формуле изобретения. 6 н. и 32 з.п. ф-лы, 30 табл., 221 пр.
Формула

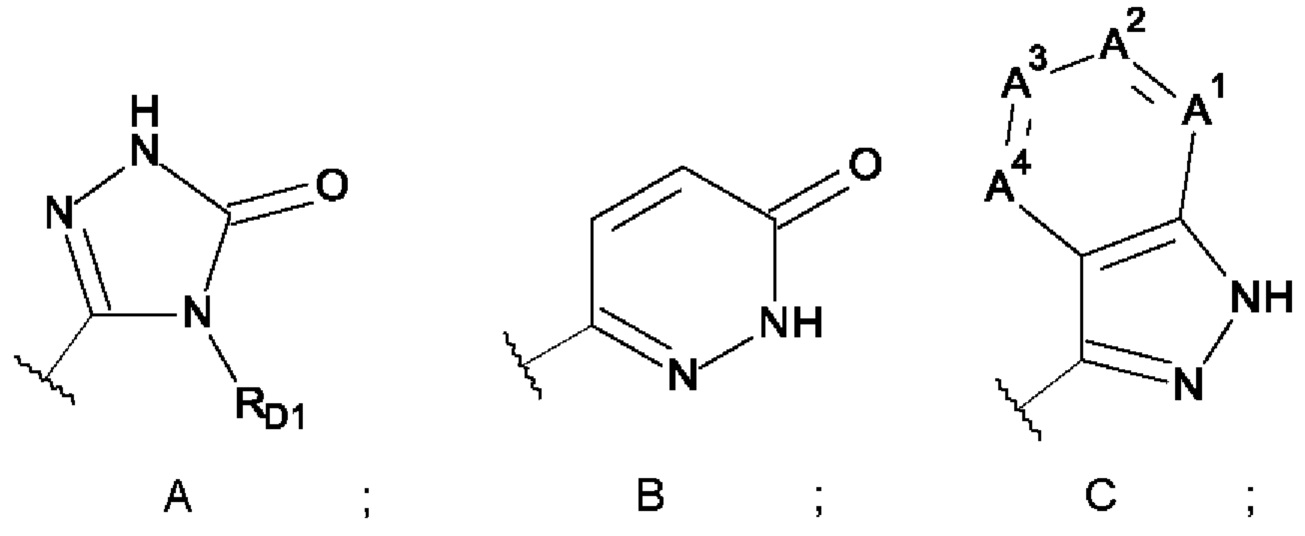


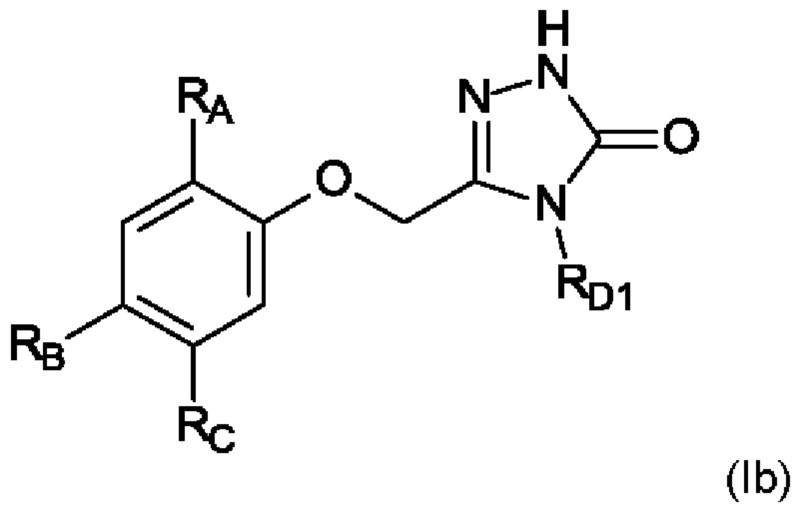

Документы, цитированные в отчёте о поиске
Пиридилокси производные, полезные в качестве активатора/модулятора гамма-рецептора, активируемого пролифератором пероксисом (ppar) гамма
Комментарии