Новые конъюгаты связывающее соединение - активное соединение (adc) и их применение - RU2610336C2
Код документа: RU2610336C2
Чертежи






















































































































































Описание
Настоящая заявка относится к новым содержащим N,N-диалкилауристатины конъюгатам связывающее соединение - активное соединение (ADC), направленным против мишени С4.4а, к активным метаболитам таких конъюгатов ADC, к способам получения этих конъюгатов ADC, к применению этих конъюгатов ADC для лечения и/или предотвращения заболеваний, а также к применению этих конъюгатов ADC для получения лекарственного средства для лечения и/или предотвращения заболеваний, в частности, гиперпролиферативных и/или ангиогенных заболеваний, таких как, например, раковые заболевания. Такие средства лечения могут применяться в форме монотерапии или в комбинации с другими лекарствами или терапевтическими мерами.
Раковые заболевания являются следствием неконтролируемого роста клеток в различных тканях. Во многих случаях новые клетки проникают в существующую ткань (инвазивный рост) или метастазируют в отдаленные органы. Раковые заболевания возникают в различных тканях и органах, и заболевание часто протекает ткенеспецифическим образом. Соответственно обозначение "раковое заболевание" как родовое понятие охватывает большую группу определенных заболеваний различных органов, тканей и типов клеток.
На ранних стадиях опухоли поддаются удалению хирургическими и химиотерапевтическими средствами. При метастазирующих опухолях обычно можно лишь назначить паллиативное лечение химиотерапевтическими агентами. В этом случае целью является улучшение качестве жизни и увеличение продолжительности жизни.
Большинство химиотерапевтических агентов, которые в настоящее время вводят парентеральным путем, часто не оказывает направленного действия на мишень в опухолевой ткани или в опухолевых клетках, а из-за системного введения распределяется в организме неспецифичным образом, попадая, соответственно, в участки, где воздействие лекарственного средства нежелательно, такие как здоровые клетки, ткани и органы, например. Это может привести к нежелательным побочным эффектам и даже к тяжелым эффектам общей токсичности, что часто сильно ограничивает терапевтически применимые диапазоны доз лекарственных средств или является причиной необходимости полного прекращения лечения.
Таким образом, улучшенная и селективная доставка этих химиотерапевтических агентов в опухоль или ткани в непосредственной близости от нее и соответственное усиление эффекта, с одной стороны, и минимизация побочных токсических эффектов с другой стороны, на протяжении нескольких лет являются центральной задачей разработки новых химиотерапевтических агентов. К настоящему времени уже было предпринято много попыток разработать эффективный способ введения активного соединения в клетку-мишень. Оптимизация связывания активного соединения и внутриклеточной мишени и минимизации внутриклеточного распределения активного соединения, например, в соседних клетках, все еще составляют трудную задачу.
Моноклональные антитела, например, подходят для мишень-направленного воздействия на опухолевые ткани или опухолевые клетки. За последние годы значение таких антител для клинического лечения раковых заболеваний значительно возросло благодаря активности таких агентов как трастузумаб (Herceptm), ритуксимаб (Rituxan), цетуксимаб (Erbitux) и бевацизумаб (Avastin), которые одобрены для лечения отдельных конкретных опухолевых заболеваний [см., например, G.P. Adams and L.M. Weiner, Nat. Biotechnol. 23, 1147-1157 (2005)]. Соответственно, сильно повысился интерес к так называемым иммуноконъюгатам, таким как, например, указанные выше конъюгаты связывающее соединение - активное соединение (ADC), в которых интернализующееся антитело к опухоль-ассоццированному антигену ковалентно связано связывающим фрагментом ("линкером") с цитотоксическим агентом. После введения ADC в опухолевую клетку и последующего расщепления конъюгата, либо сам цитотоксический агент, либо другой метаболит, образованный из цитотоксического агента и обладающий цитотоксической активностью, высвобождается внутри опухолевой клетки, где действует прямо и селективно. Это позволяет удерживать повреждение нормальной ткани в существенно более узких рамках по сравнению с обычной химиотерапией раковых заболеваний [см., например, J. M. Lam-bert, Curr. Opin. Pharma-col. 5, 543-549 (2005); A.M. Wu and P.D. Senter, Nat. Biotechnol. 23, 1137-1146 (2005); P.D. Senter, Curr. Opin. Chem. Biol. 13, 235-244 (2009); L. Ducry and B. Stump, Bioconjugate Спет.21, 5-13 (2010)].
Вместо антител также возможно использовать связывающие соединения из области низкомолекулярных лекарственных средств, которые специфично связываются с мишенью, расположенной в определенной области ("мишенью"), например, с рецептором [см., например, Е. Ruoslahti et al., Science 279. 377-380 (1998); D. Karkan et al., PLoS ONE 3 (6), e2469 (June 25, 2008)]. Также известны конъюгаты цитотоксических активных соединений и нацеливающих лигандов, которые характеризуются определенно и точкой расщепления между лигандом и лекарственны средством для высвобождения активного соединения. "Заранее определенная точка расщепления" этого типа может существовать, например, в пептиде, который может селективно расщепляться в определенном участке под действием фермента в месте действия активного соединения [см., например, R.A. Firestone and L.A. Telan, Заявка на патент США US 2002/0147138].
Особенно подходящими для мишень-направленного воздействия на опухолевые ткани и опухолевые клетки являются антитела против антигена С4.4а. С4.4а (ген: LYPD3) впервые был описан как ассоциированный с метастазированием белок поверхности клетки в клетках опухолей поджелудочной железы крыс (Rosel M. et al., Oncogene 1998, 17(15):1989-2002). Человеческий С4.4а выл выделен из бублиотеки кДНК плаценты (Wurfel, J. et al., Gene 2001, 262:35-41). С4.4а характеризуется структурной гомологией с рецептором uPA и содержит два домена LY6, которые демонстрируют типичный паттерн связывания в виде трех пальце и связан девятью дисульфидными мостиками (Jacobsen В. & Ploug M., Current Medicinal Chemistry 2008, 15:2559-2573). С4.4а заякорен в клетке гликофосфатидилинозитолом (GPI). Белок в высокой степени гликозилирован и содержит многочисленные сайты N- и O-гликозилирования. С4.4а активно экспрессируется в опухолевых клетках рака легких, рака толстого кишечника, рака груди, рака яичника, рака почки, опухолей головы и шеи и меланом. Анализы РНК продемонстрировали экспрессию С4.4а в ~50% первичных опухолей легких и в ~75% метастазов рака легкого, в то время как в здоровых тканях экспрессия не определялась (Wurfel J. et al., Gene 2001, 262:35-41). С4.4а может быть использован в качестве прогностического маркера при мелкоклеточном раке легкого - высокий уровень экспрессии С4.4а коррелирует с плохим прогнозом (Hansen L. et al., Lung Cancer 2007, 58:260-266). То же касается рака толстого кишечника. С4.4а отщепляется от поверхности опухолевой клетки и может использоваться в качестве сывороточного прогностического маркера (K. Konishi et al., Cancer Science 2010). Детальный анализ меланом показал, что экспрессируется в ~60% первичных злокачественных меланом и в 100% метастазов в лимфатические узлы и кожу (Seiter S. et al., J Invest Dermatol. 2001, 116(2):344-347). В тканях рака груди наблюдается повышенная экспрессия гена С4.4а наблюдается по сравнению с прилегающими нормальными тканями (Fletcher G.C., Ушир. J. Cancer 2003, 88(4):579-585). С4.4а является идеальным белком-мишенью для терапии рака, поскольку экспрессия С4.4а в здоровых тканях ограничена кератиноцитами кожи и эпителиальными клетками пищевода, а также клетками плаценты (Wurfel J. et al., Gene 2001, 262:35-41). В WO 01/23553 описано применение ингибитора С4.4а (например, анти-С4.4а антитело), которое является средством противораковой терапии, способным ингибировать экспрессию или активность С4.4а.
Точная функция С4.4а неизвестна. В ходе заживления ра, его экспрессия повышена в мигрирующих кератиноцитах (Hansen L. et al., Biochem J. 2004, 380:845-857). Считается, что С4.4а участвует в инвазии клеток, предположительно путем взаимодействия с внеклеточным матриксом (Rosel M. et al., Oncogene 1998, 17(15): 1989-2002; Paret С.et al., British Journal of Cancer 2007, 97:1146-1156). Потенциальными лигандами являются ламинин 1 и 5, а также галектин 3 (Paret С., Int. J. Cancer 2005, 115:724-733).
Ауристатин Е (АЕ) и монометилауристатин Е (ММАЕ) являются синтетическими аналогами доластатинов, специфичной группы линейных псевдопептидов, которые первоначально были выделены из морских источников и в некоторых случаях обладают очень высокой цитотоксической активностью в отношении опухолевых клеток [обзор можно найти, например, в G.R. Pettit, Prog. Chem. Org. Nat. Prod. 70, 1-79 (1997); G.R. Pettit et al., Анти-Cancer Drug Design 10, 529-544 (1995); G.R. Pettit et al., Анти-Cancer Drug Design 13, 243-277 (1998)].

MMAE, однако, обладает недостатком, заключающимся в относительно высокой системной токсичности. Для улучшения селективности в отношении опухоли MMAE используют в комбинации с ферментативно расщепляемыми линкерами валин-цитрулин в конъюгате связывающее соединение - активное соединение для более направленной терапии опухоли [WO 2005/081711-А2; S.О. Doro-nina et al., Bio-conjugate Chem. 17, 114-124 (2006)]. После протеолитического расщепления MMAE высвобождается из соответствующего конъюгата связывающее соединение - активное соединение внутри клетки.
Однако, при применении в форме конъюгатов антитело-активное соединение (ADC),, MMAE не совместим со связывающими фрагментами (линкерами) между антителом и лекарственным средством, которые не содержат точки ферментативного расщепления [S.О. Doro-nina et al., Bio-conjugate Chem. 17, 114-124 (2006)].
Монометилауристатин F (MMAF) представляет собой производное ауристатина, содержавшее С-концевой фенилаланиновый фрагмент, которое демонстрирует лишь умеренную антипролиферативную активность по сравнению с MMAE. Этот факт возможно обусловлен наличием свободной карбоксильной группы, полярность и заряд которой негативно влияют на способность этого соединения проникать в клетки. В этом отношении был описан метиловый эфир MMAF (MMAF-OMe) как нейтрально заряженное пролекарственное производное, который, по сравнению с MMAF, обладает способностью, и обладает in vitro цитотоксичностью в отношении различных линий карциномы, повышенной на несколько порядков величины [S.О. Doro-nina et al., Bio-con-jugate Chem. 17. 114-124 (2006)]. Можно предположить, что этот эффект обеспечивает сам MMAF, который, который после поглощения пролекарства клеткой, быстро высвобождается за счет внутриклеточного гидролиза эфира.

Однако, лекарственные соединения на основе простых эфиров обычно подвержены риску химической нестабильности из-за неспецифического гидролиза эфиров, вне зависимости от предполагаемого места действия, например, под действием эстераз, присутствующих в плазме крови; такой неспецифический гидролиз может существенно ограничивать применимость таких соединений в терапии.
Монометилауристатин F (MMAF) а также его различные эфирные производные и амидные производные раскрыты в WO 2005/081711-А2. Другие аналоги ауристатина, содержащие на С-конце, замещенный амидом фенилаланиновой группой раскрыты в в WO 01/18032-A2. В WO 02/088172-А2 и WO 2007/008603-A1 описаны аналоги MMAF, в которых боковая цепь фенилаланина замещена амидом, а в WO 2007/008848-A2 описаны аналоги, в которых карбоксильная группа фенилаланина модифицирована. Конъюгаты ауристатина, связанного по С-концу были недавно раскрыты в WO 2009/117531-А1 [см. также S.О. Doroniria et al., Bioconjugate Chem. 19, 1960-1963 (2008)].
Кроме того, производные ауристатина, такие как ММАЕ и MMAF также являются субстратами белков-переносчиков, экспрессируемых опухолевыми клетками, что может привести к развитию устойчивости к этим лекарственным средствам.
Задачей настоящего изобретения является обеспечение новых конъюгатов связывающее соединение - активное соединение (ADC), которые, благодаря комбинации новых производных N,N-диалкилауристатина с новыми, подходящими линкерами и связывающим соединением, демонстрируют очень привлекательные профили активности, например, в терминах специфичного воздействия на опухоль и/или пониженной эффективности образующихся внутри клетки метаболитов в качестве субстратов для белков-переносчиков, и которые, соответственно, подходят для лечения и/или профилактики гиперпролиферативных и/или ангиогенных заболеваний, таких как раковые заболевания, например.
Согласно настоящему изобретению предложен конъюгат связывающее соединение активное соединение общей формулы (Ia)

где
n представляет собой число от 1 до 50,
АК представляет собой связывающее соединение,
группа §-G-L1-B-L2-§§ представляет собой линкер,
где
§ обозначает точку связывания с группой AK и
§§ обозначает точку связывания с атомом азота,
D представляет собой группу формулы
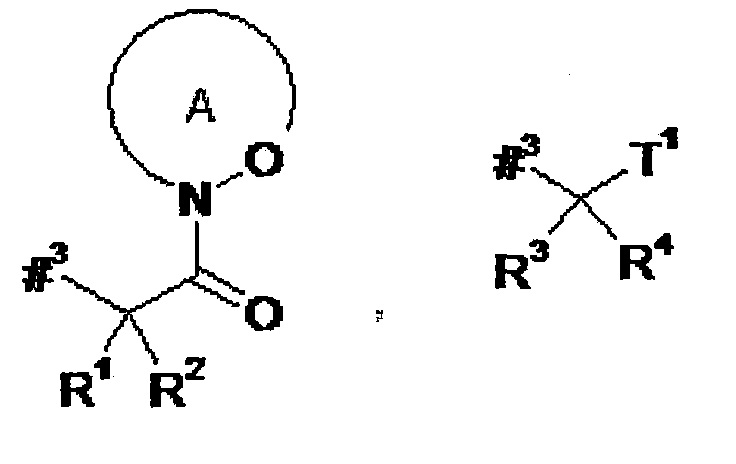

где
#3 обозначает точку связывания с атомом азота,
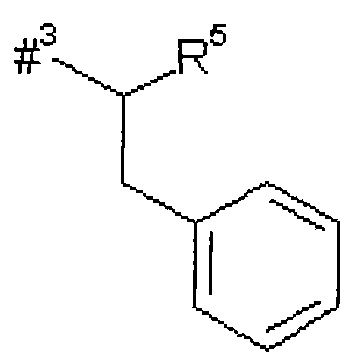
R1 представляет собой водород или метил,
R2 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород или метил,
R4 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=О)-NR8R9, -С(=O)-NH-NH-R10 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил, или
R8 и R9, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
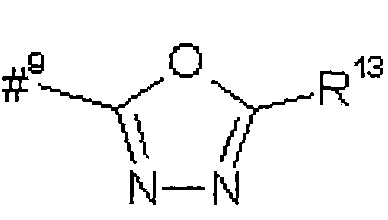
R5 представляет собой водород, метил или группу формулы



где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил,
R35 представляет собой метил или гидрокси,
или соль, сольват или сольват соли указанного соединения.
Соединения согласно настоящему изобретению представляют собой соединения формулы (Ia) и (I), а также соли, сольваты и сольваты солей таких соединений, соединения формул, определенных ниже, и охватываемые формулами (Ia) и (I), а также соли, сольваты и сольваты солей таких соединений, а также соединения приведенные ниже в качестве рабочих примеров и охватываемые формулами (Ia) и (I), и также их соли, сольваты и сольваты солей, если соединения, определенные ниже охватываются и подпадающие по формулы (Ia) и (I), не являются сами по себе солями, сольватами и сольватами солей.
В зависимости от своей структуры соединения согласно настоящему изобретению могут существовать в различных стереоизомерных формах, т.е. в форме конфигурационных изомеров, или, в других случаях, где это возможно, конформационных изомеров (энантиомеров и/или диастереомеров, включая атропоизомеры). Соответственно, настоящее изобретение включает энантиомеры и диастереомеры и их соответствующие смеси. Стереомерно гомогенные составляющие могут быть выделены из таких смесей известным образом, для этой цели предпочтительно использовать хроматогрфические методы, более конкретно ВЭЖХ-хроматогрфию на ахиральной или хиральной фазе.
В тех случаях, когда соединения согласно настоящему изобретению могут существовать в таутомерных формах, настоящее изобретения включает все таутомерные формы.
Настоящее изобретение также включает все возможные варианты соединений согласно настоящему изобретению, содержащие изотопы. В настоящей заявке под вариантами настоящего изобретения, содержащими изотопы, понимают соединение, где по меньшей мете один атом в составе соединения согласно настоящему изобретению заменен на другой атом, характеризующийся тем же атомным числом, но имеющий другую атомную массу, который обычно или преимущественно встречаются в природных условиях. Примеры изотопов, которые могут быть включены в соединения согласно настоящему изобретению включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора, брома и йода, такие как2Н (дейтерий),3H (тритий),13С,14С,15N,17О,18О,32Р,33Р,33S,34S,35S,36S,18F,36Cl,82Br,123I,124I,129I и131I. Конкретные содержащие изотопы варианты соединений согласно настоящему изобретению, такие как, в частности, варианты, в которые включены один или более радиоактивных изотопов, могут быть полезны, например, для исследования механизма действия активного соединения или его распределения в организме; благодаря сравнительной легкости получения и детектирования, для этих целей особенно подходят соединения, меченные изотопами3H или14С. Кроме того, включение изотопов, таких как, например, дейтерий, может обеспечить некоторые терапевтические преимущества вследствие повышенной устойчивости таких соединений к метаболизму; такие терапевтические преимущества включают увеличенный период полужизни в организме или снижение необходимой активной дозы, например; соответственно, такие модификации соединений согласно настоящему изобретению в соответствующих случаях могут представлять собой предпочтительные варианты реализации настоящего изобретения. Содержащие изотопы соединений согласно настоящему изобретению могут быть получены методами, известными специалисту, например, в соответствиями со способами, описанными ниже, и процедурами, приведенными в рабочих примерах, путем осуществления соответствующих модификаций изотопами соответствующих реагентов и/или исходных соединений.
Предпочтительные соли в контексте настоящего изобретения представляют собой физиологически приемлемые соли соединений согласно настоящему изобретению. Также настоящее изобретение включает соли, которые, сами по себе, не будучи приемлемыми для фармацевтического применения, могут, тем не менее, использоваться, например, для выделения или очистки соединения согласно настоящему изобретению.
Физиологически приемлемые соли согласно настоящему изобретению включают соли присоединения органических солей, карбоновых кислот и сульфоновых кислот, например, представлять собой соли хлороводородной кислоты, бромоводородная кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты, толуолсульфоновой кислоты, нафталиндисульфокислотой кислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты, молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты и бензойной кислоты.
Физиологически приемлемые соединения согласно настоящему изобретению также включают соли обычных оснований, таких как, в качестве примера, соли щелочных металлов (например, соли натрия и калия), соли щелочноземельных металлов (например, соли кальция и магния) и аммониевые соли - производные аммиака или органических аминов, содержащих от 1 до 16 атомов углерода, такие как, в качестве примера и предпочтительно, этиламин, диэтиламин, триэтиламин, этилдиизопропиламин, моноэтаноламин, диэтаноламин, триэтаноламин, дициклогексиламин, диметиламиноэтанол, прокаин, дибензиламин, N-метил-пиперидин, N-метил-морфолин, аргинин, лизин и 1,2-этилендиамин.
Сольваты в контексте настоящего изобретения представляют собой такие соединения согласно настоящему изобретению, которые образуют комплекс в твердом или жидком состоянии за счет координирования с молекулами растворителя. Гидраты представляют собой одну из форм сольватов, в которых происходит координирование с молекулами воды. Предпочтительными сольватами в контексте настоящего изобретения являются гидраты.
Далее, настоящее изобретение также включает пролекарства соединений согласно настоящему изобретению. Термин "пролекарства" в данном случае относится к соединениям, которые сами по себе могут обладать или не обладать биологической активностью, но в процессе нахождения в организме превращаются в соединения согласно настоящему изобретению (например, в результате метаболизма или гидролиза).
В контексте настоящего изобретения заместители, если не указано другое, имеют следующие значения:
(С1-С4)-алкил в контексте настоящего изобретения представляет собой линейный или разветвленный алкильный радикал, содержащий от 1 до 4 атомов углерода. В качестве примеров и предпочтительных вариантов реализации могут быть упомянуты: метил, этил, n-пропил, изопропил, n-бутил, изобутил, 1-метилпропил и трет-бугил.
Алкандиил в контексте настоящего изобретения представляет собой линейный, α,ω-двухвалентный радикал, содержащий определенное указанное число атомов углерода. В качестве примеров и предпочтительных вариантов реализации могут быть упомянуты:
метилен, этан-1,2-диил (1,2-этилен), пропан-1,3-диил (1,3-пропилен), бутан-1,4-диил (1,4-бутилен), пентан-1,5-диил (1,5-пентилен), гексан-1,6-диил (1,6-гексилен), цептан-1,7-диил (1,7-гексилен), октан-1,8-диил (1,8-октилен), нонан-1,9-диил (1,9-нонилен), декан-1,10-диил (1,10-децилен).
(С3-С7)-циклоалкил и 3-7-членный карбоцикл. соответственно, в контексте настоящего изобретения представляет собой моноциклическую насыщенную циклоалкильну группу, содержащую от 3 до 7 атомов углерода. В качестве примеров и предпочтительных вариантов реализации могут быть упомянуты: циклопропил, циклобутил, циклопентил, циклогескил и циклогептил.
Боковая группа α-аминокислоты в определении R19 включает не только боковые группы природных α-аминокислот, но также боковые группы гомологов и изомеров этих α-аминокислот. В данном случае α-аминокислота может быть в L- или D-конфигурации, или же может представлять собой смесь L- и D-форм. Примерами боковых групп могут служить следующие: метил (аланин), пропан-2-ил (валин), пропан-1-ил (норвалин), 2-метилпропан-1-ил (лейцин), 1-метилпропан-1-ил (изолейцин), бутан-1-ил (норлейцин), трет-бутил (2-отреот-бутил-глицин), фенил (2-фенилглицин), бензил (фенил-аланин), р-гидрокси-бензил (тирозин), индол-3-илметил (триптофан), имидазол-4-илметил (гистидин), гидроксиметил (серии), 2-гидроксиэтил (гомосерин), 1-гидроксиэтил (треонин), меркаптометил (цистеин), метил-тио-метил (S-метил-цистеин), 2-меркаптоэтил (гомоцистеин), 2-метил-тиоэтил (метионин), карбамоил-метил (аспарагин), 2-карбамоил-этил (глютамин), карбоксиметил (аспарагиновая кислота), 2-карбоксиэтил (глютаминовая кислота), 4-аминобутан-1-ил (лизин), 4-амино-3-гидрокси-бутан-1-ил (гидрокси-лизин), 3-амино-пропан-1-ил (орнитин), 2-аминоэтил (2,4-диаминомасляная кислота), аминометил (2,3-ди-амино-пропионовая кислота), 3-гуанидинопропан-1-ил (аргинин), 3-уреидо-пропан-1-ил (цитрулин). Предпочтительными боковыми группами α-аминокислот в определении R19 являются метил (аланин), пропан-2-ил (валин), 2-метилпропан-1-ил (лейцин), бензил (фенил-аланин), имидазол-4-илметил (гистидин), гидрокси-метил (серии), 1-гидроксиэтил (треонин), 4-аминобутан-1-ил (лизин), 3-амино-пропан-1-ил (орнитин), 2-аминоэтил (2,4-ди-амино-масляная кислота), аминометил (2,3-ди-амино-пропионовая кислота), 3-гуанидинопропан-1-ил (аргинин). В каждом случае предпочтительной является L-конфигурация.
4-7-членный гетероцикл в контексте настоящего изобретения представляет собой моноциклический насыщенный гетероцикл, содержащий в целом от 4 до 7 атомов в кольце, который содержит один или два гетероатома в кольце, выбранных из ряда N, О, S, SO и/или SO2, и связан через атом углерода в кольце или атом азота в кольце. Предпочтительными являются 5--7-членные гетероциклы, содержащие в кольце один или два гетероатома, выбранные из ряда N, О и/или S, более предпочтительно 5- или 6-членные гетероциклы, содержащие в кольце один или два гетероатома, выбранные из ряда Н и/или О. В качестве примеров могут быть упомянуты: азетидинил, оксетанил, пирролидинил, пиразолидинил, тетрагидрофуранил, тиоланил, пиперидинил, пиперазинил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, гексагидроазеинил и гексагидро-1,4-диазепирин. Предпочтительными являются пирролидинил, тетрагидрофуранил, пиперидинил, пиперазинил, тетрагидропиранил и морфолинил.
В формуле группы, которая может быть представлены А, В, D, G, L1, L2, L4, R1, R2, R3, R4 и R5, соответственно, конечная точка линии, на которой расположен символ #6, *, **, #3, #1 #2, ##1, ##2, ##3, ##4, ***, ****, #4, #5, #6, #7, #8 или #9, не является атомом углерода или группой 0%, а является частью связи с атомом, указанным в каждом случае, с которым связана группа А, В, D, G, L1, L2, L4, R1, R2, R3, R4 или R5.
В контексте настоящего изобретения, все радикалы, которые встречаются больше одного раза, имеют независимые друг от друга значения. Если радикалы в соединениях согласно настоящему изобретению содержат заместители («замещены»), эти радикалы, если не указано другое, могут содержать один или более заместителей. Предпочтительным являются замещения одним или двумя идентичными заместителями. Особенно предпочтительно замещение одним заместителем.
В контексте настоящего изобретения используемые формулы, если не указано другое, имеют следующие значения:
Термин "линкер" понимается в самом широком смысле как химический фрагмент, который содержит ковалентную связь или цепочку атомов, которые ковалентно соединяют связывающее соединение с активным соединением. В предпочтительном варианте термин "линкер" обозначает цепочку атомов согласно настоящему изобретению, которая ковалентно соединяет связывающее соединение с активным соединением. Кроме того линкеры могут быть представлены, например, двухвалентными химическими фрагментами, такими как алкилдиилы, арилдиилы, гетероарилдиилы, гетероциклилдиилы, эфиры дикарбоновой кислоты, амиды дикарбоновой кислоты.
Термин "связывающее соединение" понимается в самом широком смысле как молекула, которая связывается с молекулой-мишенью, которая присутствует на конкретной популяции клеток-мишеней, на которую нужно направить конъюгат связывающее соединение - активное соединение. Термин "связывающее соединение" следует понимать в самом широком смысле, он включает, например, пектины - белки, способные связываться с цепями определенных Сахаров, или фосфолипид-связывающие белки. Такие связывающие соединения содержат, например, высокомолекулярные белки (связывающие белки), полипептиды или пептиды (связывающие пептиды), непептидные (например, аптамеры (US 5,270,163) (обзорная статься Keefe AD., et al., Nat. Rev. Drag discov. 2010; 9:537-550) или витамины) и другие связывающиеся с клетками молекулы или вещества. Связывающими белками являются, например, антитела и фрагменты антител или миметики антител, такие как, например, аффитела, аднектины, антикалины, ДАРПины (искусственные белки с анкириновыми повторами), авимеры, нанотела (обзорная статься Gebauer M. et al., Curr. Opinion in Chem. Biol. 2009; 13:245-255; Nuttall S.D. et al., Curr. Opinion in Pharmacology 2008; 8:608-617). Связывающими пептидами являются, например, лиганды пар лиганд-рецептор, как VEGF (фактор роста эндотелия сосудов) пары VEGF/рецептор KDR, трансферин в лиганд-рецепторной паре трансферин/рецептор трансферина или цитокин/цитокиновый рецептор, как ФНО альфа в лиганд-рецепторной паре ФНОальфа/рецептор ФНОальфа.
Предпочтительными связывающими соединениями в соответствии с настоящим изобретением являются (более конкретно, человеческие, моноклональные) антитела или антигенсвязывающие фрагменты антител, которые связываются с С4.4а. В случае анти-С4.4а антител, n, т.е. число молекул токсофора на молекулу антитела, предпочтительно лежит в диапазоне от 1 до 10, более предпочтительно от 2 до 8.
Термин "молекула-мишень" понимается в самом широком смысле как молекула, которая присутствует в популяции-мишени, и может представлять собой белок (например, рецептор фактора роста) или непептидную молекулу (например, сахар или фосфолипид). В предпочтительном случае она является рецептором или антигеном.
Термин "внеклеточная" молекула-мишень обозначает молекулу-мишень, которая присоединена к клетке, и которая находится вне клетки, или часть молекулы-мишени, которая расположена вне клетки, т.е. связывающее соединение может связываться с интактной клеткой через находящуюся на ней внеклеточную молекулу-мишень. Внеклеточная молекула-мишень может быть заякорена в клеточной мембране или являться частью клеточной мембраны. Специалистам известны способы идентификации внеклеточных молекул-мишеней. Для белков это может быть осуществлено путем определения трансмембранного домена (доменов) и ориентации белка в мембране. Эти данные обычно содержатся в базах данных по белкам (например, SwissProt).
Термин "раковая молекула-мишень" обозначает молекулу-мишень, которая присутствует на клетках одного или более типов рака в большем количестве, чем на нераковых клетках того же типа ткани. В предпочтительном случае раковая молекула-мишень присутствует предпочтительно на клетках одного или более типов рака, по сравнению с нераковыми клетками того же типа ткани, где "предпочтительно" (селективно) обозначает по меньшей мере двукратно повышенное содержание в раковых клетках по сравнению с нераковыми клетками того же типа ткани ("селективно раковая молекула-мишень"). Применение раковых молекул-мишеней обеспечивает возможность селективного лечения раковых клеток конъюгатами согласно настоящему изобретению.
Связывающее соединение может быть присоединено посредством связи с линкером. Из литературы известны разнообразные варианты ковалентного связывания (конъюгирования) органических молекул с антителом. Связывающее соединение может быть присоединено через гетероатом в составе связывающего соединения. В соответствии с настоящим изобретением гетероатомы, которые могут быть использованы для связывания, представляют собой серу (в одном варианте реализации используется связывание через сульфгидрильную группу связывающего соединения), кислород (в соответствии с настоящим изобретением используется связывание через карбоксильную или гидроксильную группу связывающего соединения) и азот (в одном варианте реализации используется связывание через первичную или вторичную аминную группу или амидную группу связывающего соединения). В соответствии с настоящим изобретением предпочтительным является конъюгирование токсофоров с антителом через один или более атомов серы в остатках цистеина в составе антитела и/или через одну или более групп NH остатков лизина в составе антитела. Эти гетероатомы могут присутствовать в природном связывающем соединении или могут быть встроены химическими методами или методами молекулярной биологии. В соответствии с настоящим изобретением, связывание связывающего соединения с токсофором не оказывает большого влияния на активность связывания связывающего соединения с молекулой-мишенью. В предпочтительном варианте реализации влияние на активность связывания связывающего соединения с молекулой-мишенью отсутствует.
Термин "антитело" в соответствии с настоящим изобретением понимается в самом широком свясле и включает молекулы иммуноглобулинов, примерами которых являются интактные или модифицированные моноклональные антитела, поликлональные антитела и мультиспецифичные антитела (например, биспецифичные антитела). Молекула иммуноглобулина предпочтительно включает молекулу, содержащую четыре полипептидные цепи, две тяжелые цепи (Н-цепи) и две легкие цепи (L-цепи), которые обычно связаны дисульфидными мостиками. Каждая тяжелая цепь содержит вариабельную область тяжелой цепи (сокращенно, VH) и константную область тяжелой цепи. Константная область тяжелой цепи может включать, например, три домена СН1, СН2 и СН3. Каждая легкая цепь содержит вариабельную область (сокращенно, VL) и константную область. Константная область легкой цепи содержит один домен (сокращенно, CL). Области VH и VL могут далее быть разделены на участки, характеризующиеся гипервариабельностью, называемые также участками, определяющими комплиментарность (сокращенно, CDR), и участки с низкой вариабельностью последовательности ("каркасные участки", сокращенно, FR). Каждая из областей VH VL обычно состоит из трех участков CDR и до четырех участков FR, расположенных, например, в следующем порядке от амино-конца к карбокси-концу: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. Антитело может быть получено из организма любого вида, такого как, например, кролик, лама, верблюд, мышь или крыса. В одном варианте реализации антитело является антителом человека или мыши. Антитело может быть, например, человеческим, гуманизированными или химерным.
Термин "моноклональное" антитело обозначает антитела, которые получены из популяции по существу однородных антител, т.е. отдельные антитела в популяции идентичны за исключением природных мутаций, которые могут присутствовать в небольшом количестве. Моноклональные антитела распознают один сайт связывания антигена с высокой специфичностью. Термин "моноклональное антитело" не относится к конкретному способу получения.
Термин "интактное" антитело относится к антителам, которые содержат не только антигенсвязывающую область, но также константную область легкой и тяжелой цепи. Константная область может представлять собой природную область или ее вариант, в котором изменены одно или более положений аминокислот.
Термин "модифицированное интактное" антитело относится к интактным антителам, связанным с другим пептидом или белком, происходящим не из антитела, по амино-концу или карбоксильному концу, посредством ковалентной связи (например, пептидной связи). Далее, антитела могут быть модифицированы путем встраивания в некоторых положениях реакционноспособных остатков цистеина для облегчения связывания с токсофором (см. Junutula et al., Nat Biotechnol. 2008 Aug; 26(8):925-32).
Термин "человеческое" антитело обозначает антитела, которые могут быть получены из организма человек, или синтетические антитела. "Синтетическое" человеческое антитело представляет собой антитело, которое частично или целиком получено из синтетических последовательностей in silico, основанных не анализе последовательностей человеческих антител. Человеческое антитело может быть закодировано, например, нуклеиновой кислотой, выделенной из библиотеки последовательностей антител человеческого происхождения. Один пример таких антител описан в Soderlind et al., Nature Biotech. 2000, 18:853-856.
Термин "гуманизированное" или "химерное" антитело относится к антителам, которые состоят их компонентов человеческого происхождения и компонентов не из тела человека. В этих антителах часть последовательности человеческого иммуноглобулина (реципиента) заменена компонентами последовательности иммуноглобулина, не являющегося человеческим (донора). Во многих случаях донором является мышиный иммуноглобулин. В случае гуманизированных антител аминокислоты участков CDR в реципиенте заменяют на аминокислоты донора. В некоторых случаях аминокислоты каркасного участка также заменяют на аминокислоты донора. В некоторых случаях гуманизированное антитело содержит аминокислоты, которых нет ни в реципиенте, ни в доноре, и которые были встроены в ходе оптимизации антитела. В случае химерных антител, например, вариабельные области иммуноглобулина-донора, или, в другом случае, весь фрагмент, Fab т.е. VL-CL и VH+СН1, соединены с константными областями человеческого антитела.
Термин гипервариабельный (определяющий комплементарность) участок (CDR) в настоящей заявке обозначает те аминокислоты в вариабельной области антитела, которые необходимы для связывания с антигеном. Каждая вариабельная область обычно содержит три участка CDR, обозначаемые CDR1, CDR2 и CDR3. Каждый участок CDR может содержать аминокислоты, определенные согласно Kabat, и/или аминокислот гипервариабельной петли, определенные согласно Chotia. Определение по Kabat включает, например, участок, приблизительно соответствующий положениям аминокислот 24-34 (CDR1), 50-56 (CDR2) и 89-97 (CDR3) вариабельной области легкой цепи и 31-35 (CDR1), 50-65 (CDR2) и 95-102 (CDR3) вариабельной области тяжелой цепи (Kabat et al., Sequences of Proteins of Immulological Interest, 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD. (1991)). Определение согласно Chotia включает, например, участок, приблизительно соответствующий положениям аминокислот 26-32 (CDR1), 50-52 (CDR2) и 91-96 (CDR3) вариабельной области легкой цепи и 26-32 (CDR1), 53-55 (CDR2) и 96-101 (CDR3) вариабельной области тяжелой цепи Chothia and Lesk; J Mol Biol 196: 901-917 (1987)). В некоторых случаях CDR может содержать аминокислоты одного участка CDR, определенного согласно Kabat и Chotia.
В зависимости от аминокислотной последовательности константной области тяжелой цепи антитела могут быть разделены на несколько классов. Существует пять классов интактных антител: IgA, IgD, IgE, IgG и IgM, часть их которых может быть далее разделена на подклассы (изотипы), например, IgG1, IgG2, IgG3, IgG4, IgA1 и IgA2. Константные области тяжелой цепи, соответствующие различным классам, обозначаются [альфа/α], [дельта/δ], [эпсилон/ε], [гамма/γ] и [мю/μ]. Известны как трехмерная структура, так и субъединичная структура антител.
Термин "функциональный фрагмент" или "антигенсвязывающие фрагменты антитела" антитела/иммуноглобулина определяется как фрагмент антитела/иммуноглобулина (например, вариабельные области IgG) который включает антигенсвязывающие области антитела/иммуноглобулина. "Антигенсвязывающая область" антитела обычно включает один или более гипервариабельных участков антитела, например, участки CDR1, CDR2 и/или CDR3. Однако, "каркасный» или "рамочный" участок антитела также может иметь значения для связывания антитела с антигеном. Каркасный участок образует каркас для участков CDR. Антигенсвязывающая область обычно содержит по меньшей мере аминокислоты 4-103 вариабельной области легкой цепи и аминокислоты с 5 по 109 вариабельной области тяжелой цепи, более предпочтительно аминокислоты с 3 по 107 вариабельной области легкой цепи и с 4 по 111 вариабельной области тяжелой цепи, особенно предпочтительными являются полные легкая и тяжелая цепи, т.е. аминокислоты 1 - 109 в VL и с 1 по 113 в VH (нумерация в соответствии с WO 97/08320).
"Функциональные фрагменты" или "антигенсвязывающие фрагменты антитела" согласно настоящему изобретению включают, но не ограничиваются перечисленными: фрагменты Fab, Fab', F(ab')2 и Fv, диатела, однодоменные антитела (Dab), линейные антитела, отдельные цепи антител (одноцепочечные Fv, сокращенно, ScFv); и мультспецифичные антитела, такие как би- и -триспецифичные антитела, образованные фрагментами антител. А. К Borrebaeck, editor (1995) Antibody Engineering (Breakthroughs in Molecular Biology), Oxford University Press; R. Kontermann & S. Duebel, editors (2001) Antibody Engineering (Springer Laboratory Manual), Springer Verlag). Антител, не являющиеся "мультиспецифичными" или "мультифункциональными", представляют собой антитела, содержащие идентичные сайты связывания. Мультиспецифичные антитела могут обладать специфичностью к эпитопам более чем одного антигена (см., например, WO 93/17715; WO 92/08802; WO 91/00360; WO 92/05793; Tutt, et al., 1991, J. Immunol. 147:60 69; U. S. Pat. Nos. 4,474,893; 4,7 14,68 1; 4,925,648; 5,573,920; 5,601,8 19; или Kostelny et al., 1992, J. Immunol. 148: 1547 1553). Молекула F(ab')2 или Fab может быть сконструирована таким образом, чтобы обеспечить возможность восстановления или полного устранения ряда внутримолекулярных диеульфидных взаимодействий, присутствующих между доменами Ch1 и CL.
"Функциональные фрагменты" или "антигенсвязывающие фрагменты антитела" могут быть соединены с другим пептидом или белком, происходящим не из антитела, по амино-концу или карбоксильному концу, посредством ковалентной связи (например, пептидной связи). Кроме того, антитела и антигенсвязывающие фрагменты могут быть модифицированы путем встраивания в некоторых положениях реакционноспособных остатков цистеина для облегчения связывания с токсофором (см. Junutula et al., Nat Biotechnol. 2008 Aug; 26(8):925-32).
Поликлональные антитела могут быть получены методами, известными среднему специалисту в данной области. Моноклональные антитела могут быть получены методами, известными среднему специалисту в данной области (Kohler and Milstein, Nature, 256, 495-497, 1975). Человеческие и гуманизированные моноклональные антитела могут быть получены методами, известными среднему специалисту в данной области (Olsson et al., Meth Enzymol. 92, 3-16, или Cabilly et al., US 4,816,567, или Boss et al., US 4,816,397).
Специалисту известны разнообразные способы получения человеческих антител и их фрагментов, такие как, например, с использованием трансгенных мышей (N Lonberg and D Huszar, hit Rev Immunol. 1995; 13(1):65-93) или технологий фагового дисплея (Clackson et al., Nature. 1991 Aug 15; 352(6336):624-8). Антитела согласно настоящему изобретению могут быть получены из рекомбинантных библиотек антител, состоящих, например, из аминокислотных последовательностей множества антител, полученных от большого количества здоровых добровольцев. Антитела также могут быть получены с использованием известных технологий рекомбинантной ДНК. Нуклеотидные последовательности антител могут быть получены путем рутинного секвенирования или доступны в общедоступных базах данных.
"Изолированное" антитело или связывающее соединение очищено таким образом, чтобы удалить другие компоненты клетки. Составляющими клетки, которые могут препятствовать диагностическому или терапевтическому применению, являются, например, ферменты, гормоны и другие пептидные или непептидные составляющие клетки. Предпочтительными являются антитело или связывающее соединение, очищенное до степени более 95% по антителу или связывающему соединению (определенной, например, методом Лоури, спектроскопией в УФ-видимом диапазоне или капиллярным электрофорезом в SDS-геле), степень очистки которых позволяет определить по меньшей мере 15 аминокислот амино-конца или внутреннюю аминокислотную последовательность, или очищенные до гомогенного состояния, где гомогенность определяют электрофорезом на SDS-ПААГ в восстанавливающих или невосстанавливающих условиях (детектирование может осуществляться путем окрашивания Кумасси голубым или, предпочтительно, серебром). Однако при получении антитела обычно используют одну или более стадий очистки.
Термин "специфично связывающийся" или " специфично связывает" относится к антителу или связывающему соединению, которое связывается с определенным антигеном/молекулой-мишенью. Специфичное связывание антитела или связывающего соединения обычно подразумевает, что антитело или связывающее соединение характеризуется аффинностью 10-7 М (выраженную как значение Kd, т.е. предпочтительно, те антитела, значение Kd которых ниже 10-7 М), причем аффинность антитела или связывающего соединения в отношении указанного определенного антигена/молекулы-мишени по меньшей мере в два раза выше, чем аффинность к неспецифичному антигену /молекуле-мишени (например, к бычьему сывороточному альбумину или казеину), отличному от указанного определенного антигена/молекулы-мишени, или к родственному антигену /молекуле-мишени.
Антитела, специфичные к антигенам раковых клеток, могут быть изготовлены средним специалистом в данной области известными ему/ей методами (такими как, например, рекомбинантная экспрессия) или могут быть приобретены у коммерческих поставщиков (таких как Merck KGaA, Германия). Примеры известных коммерчески доступных антител для лечения рака включают Е Erbitux® (цетуксимаб, Merck KGaA), Avastm® (бевацизумаб, Roche) и Herceptin® (трастузумаб, Genentech). Трастузумаб представляет собой рекомбинантное гуманизированное моноклональное антитело типа IgG1 каппа, которое в клеточном анализе (Kd=5 нМ) связывается с внеклеточными доменами челвеческого рецептора эпидермального фактора роста с высокой аффинностью. Это антитело получают рекомбинантным путем в клетках СНО.
Соединения формулы (I) представляют подгруппу соединений формулы represent a subgroup (Ia).
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia)
где
n представляет собой число от 1 до 50,
AK представляет собой AK1 или AK2 где
AK1 представляет собой связывающее соединение (предпочтительно анти-С4.4а антитело), которое связано через атом серы связывающего соединения с группой G,
AK2 представляет собой связывающее соединение (предпочтительно анти-С4.4а антитело), которое связано через атом азота связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

или

где
#1 обозначает точку связывания с атомом серы в связывающем соединении,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С1-С10)-алкандиил, группу формулы


где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
L1 представляет собой линейный (С2-С10)-алкандиил,
В1 представляет собой группу формулы

или
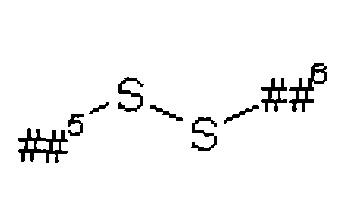
где
##5 обозначает точку связывания с группой L1A,
##6 обозначает точку связывания с группой L1B,
L5 представляет собой связь или (С2-С4)-алкандиил,
L6 представляет собой связь или группу формулы


где
##7 обозначает точку связывания с карбонильной группой,
##8 обозначает точку связывания с L1B,
R33 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутил-оксикарбонил или бензилоксикарбонил,
R34 представляет собой водород или метил, R29 представляет собой водород или (С)-С4)-алкил, R30 представляет собой водород или (С1-С4)-алкил, или
R29 и R30 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R31 представляет собой водород или (С1-С4)-алкил,
R32 представляет собой водород или (С1-С4)алкил, или
R31 и R32 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
L13 представляет собой линейный (С2-С10)-алкандиил, и
где (С1-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
где два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
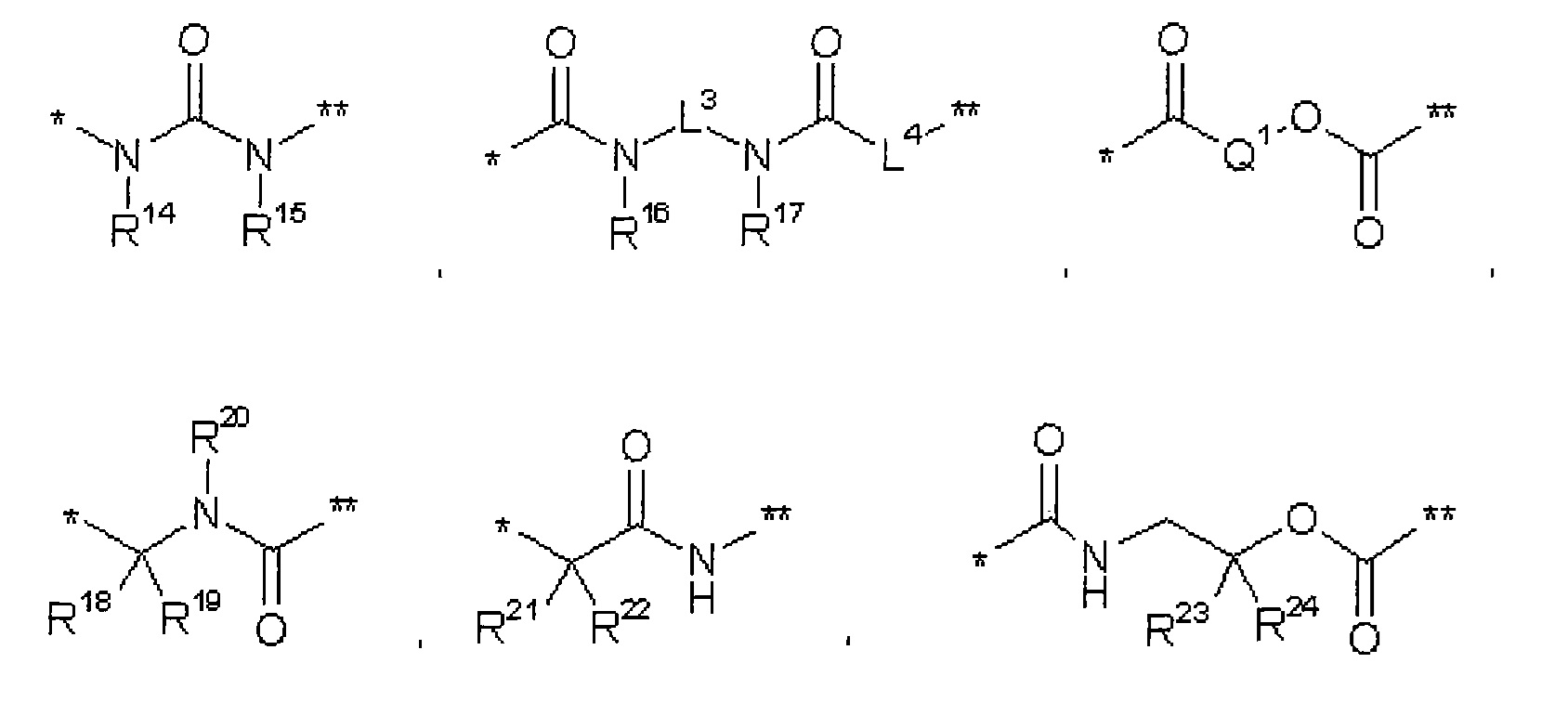
В представляет собой связь или группу формулы


или

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
L3 представляет собой связь или (С2-С4)-алкандиил,
L4 представляет собой связь или группу формулы
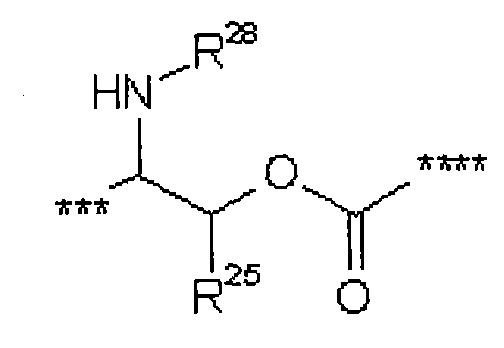

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
R28 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил или бензилоксикарбонил,
Q1' представляет собой 4-7-членный гетероцикл,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный гетероцикл,
R14 представляет собой водород или (С1-С4)-алкил,
R15 представляет собой водород или (С1-С4)-алкил,
или
R14 и R15 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R16 представляет собой водород или (С1-С4)-алкил, R17 представляет собой водород или (С1-С4)-алкил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R18 представляет собой водород или (С1-С4)-алкил,
R19 представляет собой водород или боковую группу природной α-аминокислоты, или ее гомолог или изомер,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или (С1-С4)-алкил,
R22 представляет собой водород или (С1-С4)-алкил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют 3-7-членный карбоцикл,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
R27 представляет собой водород или (С1-С4)-алкил,
R36 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил или бензил-1-оксикарбонил,
R37 представляет собой водород или метил,
или
R36 и R37 вместе с атомом, с которым они связаны, образуют пирролидиновое кольцо,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы
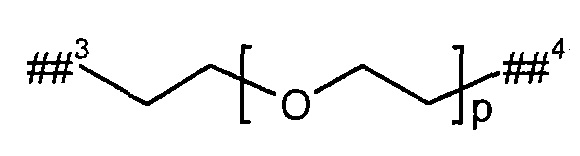
где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R' представляет собой водород или метил,
R2 представляет собой изобутил, ceN-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород или метил,
R4 представляет собой изобутил, ceN-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,1R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т',
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=O)-NR8-R9, -C(=O)-NH-NH-R10 или -CH2-О-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9 вместе с атомом азота, с которым они связаны, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы


где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1-й-индол-3-илметил,
R35 представляет собой метил или гидрокси,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным вариантом реализации настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia), описанный выше, где
n представляет собой число от 1 до 50,
АК представляет собой AK1 или AK2
где
AK1 представляет собой связывающее соединение (предпочтительно анти-С4.4а антитело), которое связано через атом серы связывающего соединения с группой G,
AK2 представляет собой связывающее соединение (предпочтительно анти-С4.4а антитело), которое связано через атом азота связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

или

где
#1 обозначает точку связывания с атомом серы в связывающем соединении,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С1-С10)-алкандиил, группу формулы


где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
L1A представляет собой линейный (С2-С10)-алкандиил,
В1 представляет собой группу формулы

или
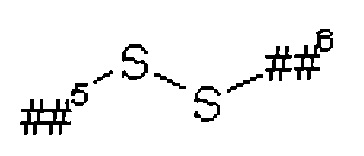
где
##5 обозначает точку связывания с группой L1A,
##6 обозначает точку связывания с группой L1B,
L5 представляет собой связь или (С2-С4)-алкандиил,
L6 представляет собой связь или группу формулы


где
##7 обозначает точку связывания с карбонильной группой,
##8 обозначает точку связывания с L1B,
R33 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил или бензилоксикарбонил,
R34 представляет собой водород или метил,
R29 представляет собой водород или (С1-С4)-алкил,
R30 представляет собой водород или (С1-С4)-алкил,
или
R29 и R30 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R31 представляет собой водород или (С1-С4)-алкил, R32 представляет собой водород или (С1-С4)-алкил, или
R31 и R32 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
L1B представляет собой линейный (С2-С10)-алкандиил, и
где (С1-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
В представляет собой связь или группу формулы



где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
L3 представляет собой связь или (С2-С4)-алкандиил,
L4 представляет собой связь или группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
R28 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил или бензилоксикарбонил,
Q1 представляет собой 4-7-членный гетероцикл,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный гетероцикл,
R14 представляет собой водород или (С1-С4)-алкил,
R15 представляет собой водород или (С1-С4)-алкил,
или
R14 и R15 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R16 представляет собой водород или (С1-С4)-алкил,
R17 представляет собой водород или (С1-С4)алкил,
или
R16 и R7 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R18 представляет собой водород или (С1-С4)-алкил,
К19 представляет собой водород или боковую группу природной ос-аминокислоты, или ее гомолог или изомер,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или (С1-С4)-алкил,
R22 представляет собой водород или (С1-С4)-алкил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют 3-7-членный карбоцикл,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
R27 представляет собой водород или (С1-С4)-алкил,
R36 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил или бензил-1-оксикарбонил,
R37 представляет собой водород или метил,
или
R36 и R37 вместе с атомом, с которым они связаны, образуют пирролидиновое кольцо,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород или метил,
R2 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3 -илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,1R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси, R3 представляет собой водород или метил,
R4 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,1R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=O)-NH-NH-R9, -C(=O)-NH-NH-R10 или -СН2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы



где
#9 обозначает точку связывания с -CHC(R26)T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил,
R35 представляет собой метил или гидрокси,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia), где
n представляет собой число от 1 до 20,
AK представляет собой AK1 или AK2,
где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С2-С6)-алкандиил, группу формулы


где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
L1A представляет собой линейный (С2-С6)-алкандиил,
B1B представляет собой группу формулы

или

где
##5 обозначает точку связывания с группой L1A,
##6 обозначает точку связывания с группой L1B,
L5 представляет собой связь,
L6 представляет собой связь или группу формулы


где
##7 обозначает точку связывания с карбонильной группой,
##8 обозначает точку связывания с L1B,
R33 представляет собой водород, метилкарбонил или трет-бутоксикарбонил,
R34 представляет собой водород или метил,
R29 представляет собой водород,
R30 представляет собой водород,
R31 представляет собой водород или метил,
R32 представляет собой водород или метил,
L1B представляет собой линейный (С2-С6)-алкандиил, и
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
В представляет собой связь или группу формулы



где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы
где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
Q1 представляет собой 4-7-членный гетероцикл,
R14 представляет собой водород,
R15 представляет собой водород,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R18 представляет собой водород,
R19 представляет собой водород, метил, пропан-2-ил, 2-метилпропан-1-ил или 1-метилпропан-1-ил,
R20 представляет собой водород или метил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или метил,
R22 представляет собой водород или метил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют циклопропильное кольцо,
R23 представляет собой метил,
R24 представляет собой водород или метил,
R27 представляет собой водород,
R36 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R37 представляет собой водород или метил,
или
R36 и R37 вместе с атомом, с которым они связаны, образуют пирролидиновое кольцо,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы
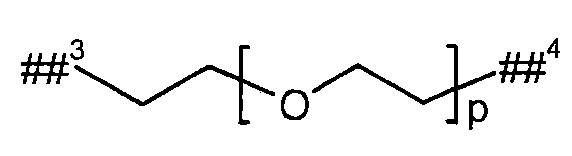
где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, где (С2-С10)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -C(=O)-NR8R9, -C(=O)-NH-NH-R10 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9' вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы



где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил,
R35 представляет собой метил или гидрокси, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia), описанный выше, где
n представляет собой число от 1 до 20,
AK. представляет собой AK1 или AK2
где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С2-С6)-алкандиил, группу формулы
где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
LlA представляет собой линейный (С2-С6)-алкандиил,
В1 представляет собой группу формулы
или
где
##5 обозначает точку связывания с группой L1A,
##6 обозначает точку связывания с группой L1B,
L5 представляет собой связь,
L6 представляет собой связь или группу формулы
где
##7 обозначает точку связывания с карбонильной группой,
##8 обозначает точку связывания с L1B,
R33 представляет собой водород, метилкарбонил или трет-бутоксикарбонил,
R34 представляет собой водород или метил,
R29 представляет собой водород,
R30 представляет собой водород,
R31 представляет собой водород или метил,
R32 представляет собой водород или метил,
L1B представляет собой линейный (С2-С6)-алкандиил,
и
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
В представляет собой связь или группу формулы


где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы
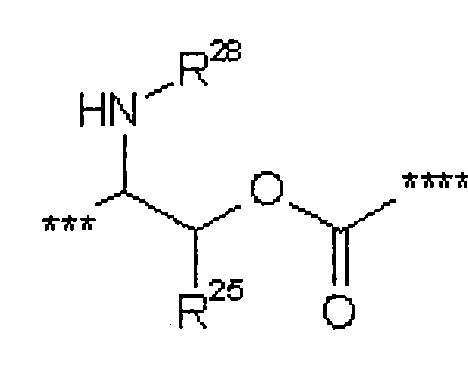

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
Q1 представляет собой 4-7-членный гетероцикл,
R14 представляет собой водород,
R15 представляет собой водород,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R18 представляет собой водород,
R19 представляет собой водород, метил, пропан-2-ил, 2-метилпропан-1-ил или 1-метилпропан-1-ил,
R20 представляет собой водород или метил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или метил,
R22 представляет собой водород или метил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют циклопропильное кольцо,
R23 представляет собой метил,
R24 представляет собой водород или метил,
R27 представляет собой водород,
R36 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил, или бензилоксикарбонил,
R37 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, D представляет собой группу формулы

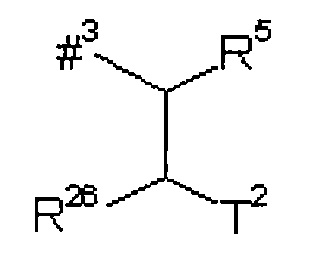
где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3 -илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1 H-индол-3 -илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=О)-NR8R9, -C(=O)-NH-NH-R10 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы



где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил,
R35 представляет собой метил или гидрокси,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia), где
n представляет собой число от 1 до 10,
AK представляет собой AK1 или AK2 где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если АК=AK1, представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой Ь1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С2-С6)-алкандиил, группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
В представляет собой связь или группу формулы

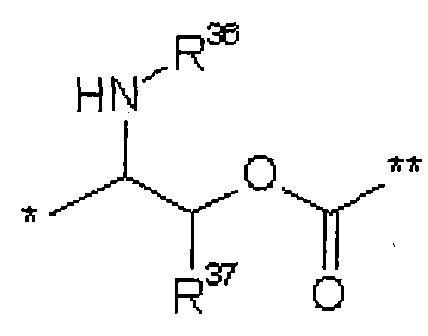

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы
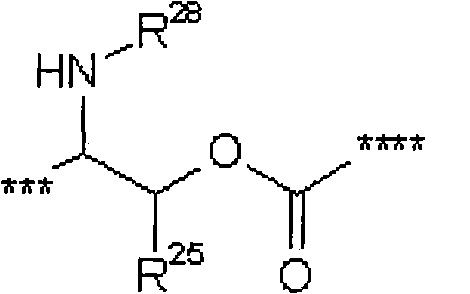

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
Q1 представляет собой пиперидин-1,4-диил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R21 представляет собой водород или метил,
R22 представляет собой водород или метил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют циклопропильное кольцо,
R23 представляет собой метил,
R24 представляет собой водород,
R36 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R37 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D представляет собой группу формулы

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой бензил, 4-гидроксибензил, 1-фенилэтил или 1Я-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -C(=O)-NR8R9 или -СН2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы


где
#9 обозначает точку связывания с -СНСН2фенилом,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R35 представляет собой метил или гидрокси,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia), описанное выше, где
n представляет собой число от 1 до 10,
AK представляет собой AK1 или AK2 где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С2-С6)-алкандиил, группу формулы

где m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы



где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
Q1 представляет собой пиперидин-1,4-диил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R21 представляет собой водород или метил,
R22 представляет собой водород или метил, или
R21 и R22 вместе с атомом, с которым они связаны, образуют циклопропильное кольцо,
R23 представляет собой метил,
R24 представляет собой водород,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D представляет собой группу формулы

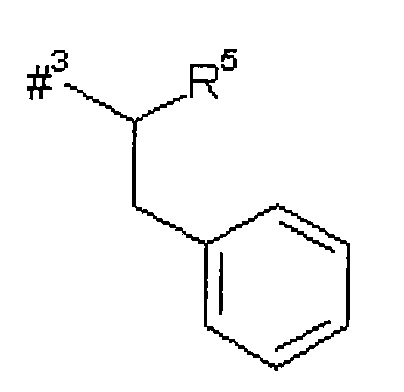
где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 1-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой бензил, 1-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы
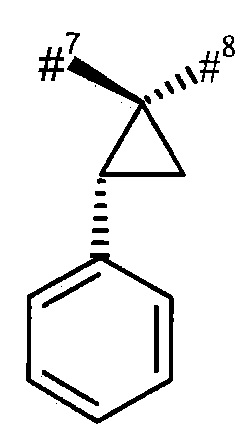
где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т' представляет собой группу формулы -C(=O)-OR7, -C(=O)-NR8R9 или -СН2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы
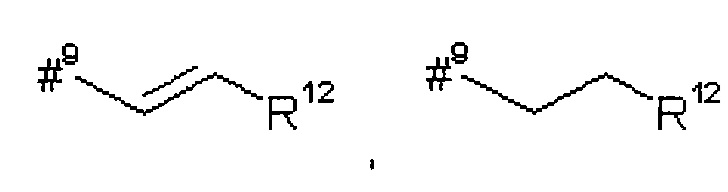

где
#9 обозначает точку связывания с -СНСН2фенилом,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R35 представляет собой метил или гидрокси, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia), описанное выше, где
n представляет собой число от 1 до 10,
AK представляет собой AK2,
где
AK2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
L1 представляет собой связь,
В представляет собой связь,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D представляет собой группу формулы

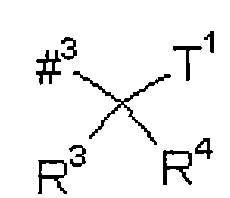
где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой бензил или 1H-индол-3-илметил, или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород,
R9 представляет собой водород или бензил,
R35 представляет собой метил,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia) описанный выше, где
n представляет собой число от 1 до 10,
AK представляет собой AK2,
где
AK2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела ВО 1-3, ВО 1-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела ВО 1-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
L1 представляет собой связь,
В представляет собой связь,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы
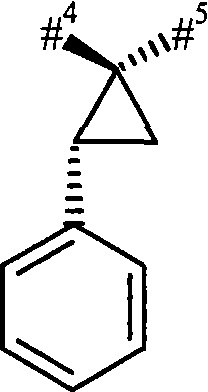
где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил, или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород,
R9 представляет собой водород или бензил, R35 представляет собой метил, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia), описанный выше, где
n представляет собой число от 1 до 10,
AK представляет собой AK1,
где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой связь, линейный (С3-С5)-алкандиил или группу формулы

где m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В, где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
В представляет собой связь или группу формулы
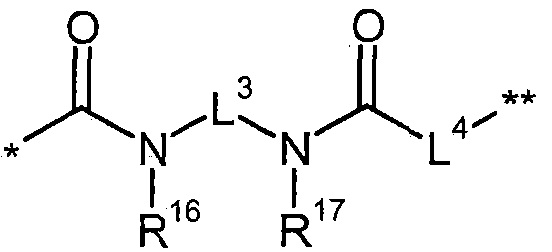
где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
or
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
L2 представляет собой линейный (С3-С5)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы
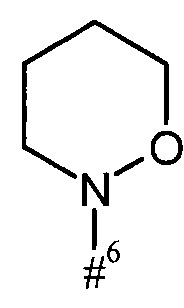
где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой бензил или 1H-индол-3-илметил, или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород,
R9 представляет собой водород или бензил,
R35 представляет собой метил, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (Ia) описанный выше, где
n представляет собой число от 1 до 10,
AK представляет собой AK1, где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, М31-В01 или D02-6, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой связь, линейный (С3-С5)-алкандиил или группу формулы
где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
L2 представляет собой линейный (С3-С5)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D представляет собой группу формулы
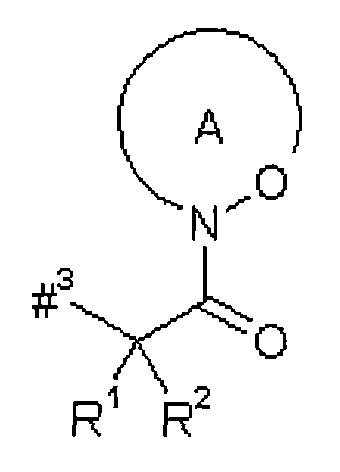

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
or
R' и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил, или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород,
R9 представляет собой водород или бензил,
R35 представляет собой метил,
или соль, сольват или сольват соли указанного соединения.
Дополнительно согласно настоящему изобретению предложены соединения формулы (ХХХа)

где
Cys представляет собой остаток цистеина, который через атом серы боковой цепи связан с атомом углерода сукцинимида,
L1 представляет собой связь, линейный (С1-С10)-алкандиил, группу формулы


где m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##1 обозначает точку связывания с группой В,
L1A представляет собой линейный (С2-С10)-алкандиил,
В1 представляет собой группу формулы


где
##5 обозначает точку связывания с группой L1A,
##6 обозначает точку связывания с группой L1B,
L5 представляет собой связь или (С2-С4)-алкандиил,
L6 представляет собой связь,
R29 представляет собой водород или (С1-С4)-алкил,
R30 представляет собой водород или (С1-С4)-алкил,
или
R29 и R30 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R31 представляет собой водород или (С1-С4)-алкил,
R32 представляет собой водород или (С1-С4)-алкил, или
R31 и R32 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
L18 представляет собой линейный (С2-С10)-алкандиил, и
где (С1-С10)-алкандиил может содержать от 1 до 4 заместителей, независимо выбранных из группы, состоящей из метила, гидрокси и бензила,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
В представляет собой связь или группу формулы



где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
L3 представляет собой связь или (Сг-С4)-алкандиил,
L4 представляет собой связь,
Q* представляет собой 4-7-членный гетероцикл,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный гетероцикл,
R14 представляет собой водород или (С1-С4)-алкил,
R15 представляет собой водород или (С1-С4)-алкил,
или
R14 и R15 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R16 представляет собой водород или (С1-С4)-алкил,
R17 представляет собой водород или (С1-С4)-алкил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R18 представляет собой водород или (C1-C4)-алкил,
R19 представляет собой водород или боковую группу природной α-амино кислоты, или его гомологи или изомеры,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или (С1-С4)-алкил,
R22 представляет собой водород или (С1-С4)-алкил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют 3-7-членный карбоцикл,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
R27 представляет собой водород или (С1-С4)-алкил,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород или метил,
R2 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород или метил,
R4 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т' представляет собой группу формулы -C(=O)-OR7, -С(=O)-NR8R9, -C(=O)-NH-NH-R10 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9 вместе с атомом азота, с которым они связаны, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы



где #9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил,
R35 представляет собой метил или гидрокси, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является соединение формулы (ХХХа), описанное выше, где
Cys представляет собой остаток цистеина, который через атом серы боковой цепи связан с атомом углерода сукцинимида,
L1 представляет собой связь, линейный (С2-С6)-алкандиил, группу формулы


где
m представляет собой число 2 или 3,
## ' обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
L1A представляет собой линейный (С2-С6)-алкандиил,
В1 представляет собой группу формулы


где
##5 обозначает точку связывания с группой L1A,
##6 обозначает точку связывания с группой L1B,
L5 представляет собой связь,
L6 представляет собой связь,
R29 представляет собой водород,
R30 представляет собой водород,
R31 представляет собой водород или метил,
R32 представляет собой водород или метил,
L1B представляет собой линейный (С2-С6)-алкандиил,
и
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

или

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь,
R14 представляет собой водород,
R15 представляет собой водород,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R23 представляет собой метил,
R24 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1 Д-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -C(=O)-NR8R9, -C(=O)-NH-NH-R10 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы

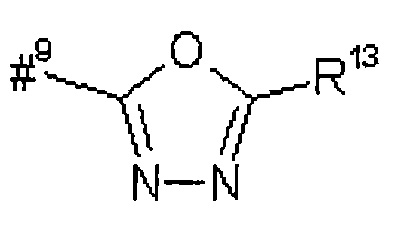
где
#9 обозначает точку связывания с -СНСН2фенилом,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R35 представляет собой метил или гидрокси, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является соединение формулы (ХХХа) описанный выше, где
Cys представляет собой остаток цистеина, который через атом серы боковой цепи связан с атомом углерода сукцинимида,
L1 представляет собой связь или линейный (С2-С6)-алкандиил,
В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь,
L4 представляет собой связь,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы
где р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D представляет собой группу формулы
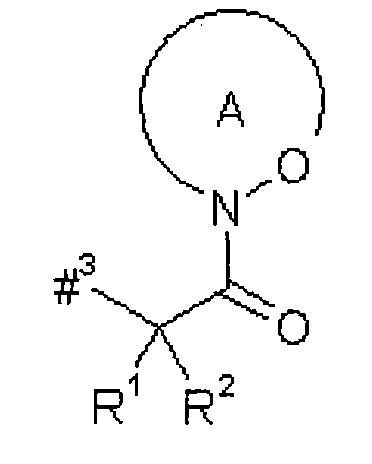

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой бензил или 1H-индол-3-илметил, или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы
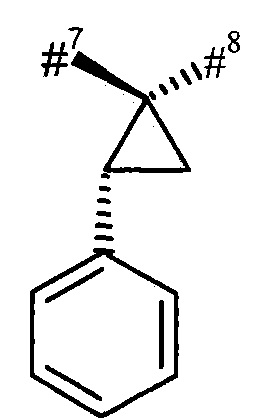
где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9,
где
R7 представляет собой водород,
R8 представляет собой водород,
R9 представляет собой водород,
R35 представляет собой метил,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является соединение формулы (ХХХа), описанное выше, где
Cys представляет собой остаток цистеина, который через атом серы боковой цепи связан с атомом углерода сукцинимида,
L1 представляет собой связь или линейный (С2-С6)-алкандиил,
В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь,
L4 представляет собой связь,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы
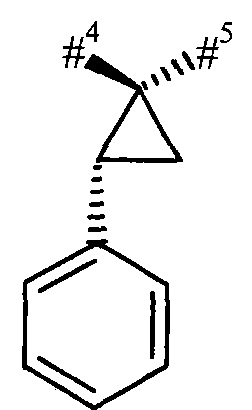
где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил, или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
# обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9, где
R7 представляет собой водород,
R8 представляет собой водород,
R9 представляет собой водород,
R35 представляет собой метил, или соль, сольват или сольват соли указанного соединения.
Дополнительно согласно настоящему изобретению предложены соединения формулы (XXXI)

где
L1 представляет собой связь, линейный (С1-С10)-алкандиил, группу формулы


где m представляет собой число от 2 до 6,
##1] обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
L1A представляет собой линейный (С2-С10)-алкандиил,
В1 представляет собой группу формулы


где
##5 обозначает точку связывания с группой L1A,
##6 обозначает точку связывания с группой L1B,
L5 представляет собой связь или (С2-С4)-алкандиил,
L6 представляет собой связь,
R29 представляет собой водород или (С1-С4)-алкил,
R30 представляет собой водород или (С1-С4)-алкил,
или
R29 и R30 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R31 представляет собой водород или (С1-С4)-алкил,
R32 представляет собой водород или (С1-C4)-алкил,
или
R31 и R32 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
L18 представляет собой линейный (С2-С10)-алкандиил,
и
где (С1-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
В представляет собой связь или группу формулы



где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
Q1 представляет собой 4-7-членный гетероцикл,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный гетероцикл,
R18 представляет собой водород или (С1-С4)-алкил,
R19 представляет собой водород или боковую группу природной α-аминокислоты, или ее гомолог или изомер,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или (С1-С4)-алкил,
R22 представляет собой водород или (С1-С4)-алкил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют 3-7-членный карбоцикл,
R27 представляет собой водород или (С1-С4)-алкил,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород или метил,
R2 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1Н-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород или метил,
R4 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидроксил-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,1R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=О)-NR8R9, -C(=O)-NH-NH-R10 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9 вместе с атомом азота, с которым они связаны, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы



где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил,
R35 представляет собой метил или гидрокси,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является соединение формулы (XXXI), описанное выше, где
L1 представляет собой связь, линейный (С2-С6)-алкандиил или группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы



где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
R18 представляет собой водород,
R19 представляет собой метил, пропан-2-ил, 2-метилпропан-1-ил или 1-метилпропан-1-ил,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или метил,
R22 представляет собой водород или метил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют циклопропильное кольцо,
R27 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
и
где два атома углерода цепи алкандиила в положении 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием фенильного кольца,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой 1-гидроксиэтил, бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -C(=O)-NR8R9, -C(=O)-NH-NH-R10 или -СН2-О-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9 вместе с атомом азота, с которым они связаны, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы


где
#9 обозначает точку связывания с -СНСН2фенилом,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R35 представляет собой метил или гидрокси, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является соединение формулы (XXXI), описанное выше, где
L1 представляет собой связь,
В представляет собой связь,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно-или бициклический, возможно содержащий заместители гетероцикл формулы
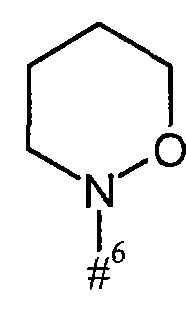
где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой бензил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)NR8R9,
где
R7 представляет собой водород,
R8 представляет собой водород,
R9 представляет собой водород,
R35 представляет собой метил, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является соединение формулы (XXXI), описанное выше, где
L1 представляет собой связь, В представляет собой связь,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 4-гидроксибензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1-№индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1, Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9, где
R7 представляет собой водород,
R8 представляет собой водород,
R9 представляет собой водород,
R35 представляет собой метил,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является соединение формул (ХХХа) и (XXXI), выбранное из следующей группы:
N-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S}-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(1H-идол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид,
N-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-идол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид,
N-(6-{[(5S)-5-амино-5-карбоксипентил]амино}-6-оксогекеил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-идол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат,
N-(6-{[(5S)-5-амино-5-карбоксипентил]амино}-6-оксогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(1H-идол-3-ил)этил]амино}-1-мктокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид,
или соль, сольват или сольват соли указанного соединения.
Согласно настоящему изобретению также предложен конъюгат связывающее соединение -активное соединение общей формулы (I)
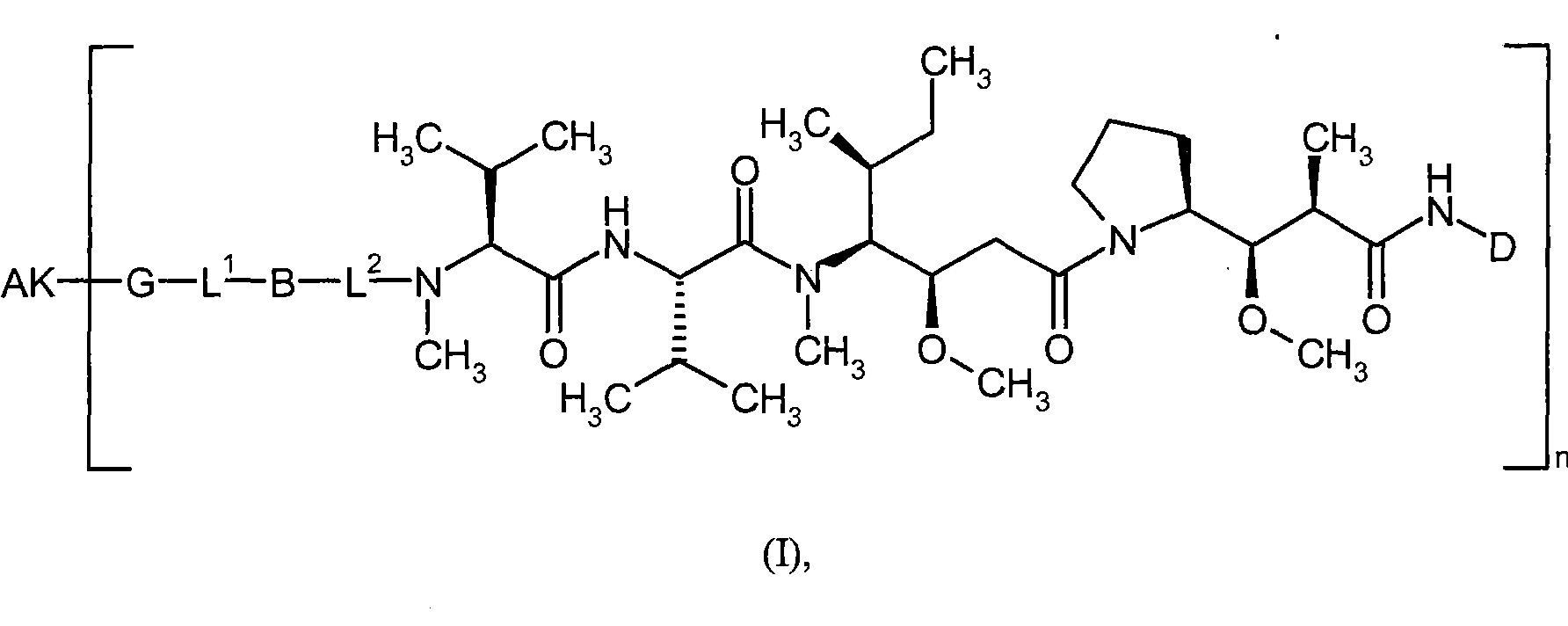
где
n представляет собой число от 1 до 50,
AK представляет собой связывающее соединение,
группа §-G-L1-B-L2-§§ представляет собой линкер, где
§ обозначает точку связывания с группой AK and §§ обозначает точку связывания с атомом азота,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 1-гидроксиэтил, бензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=О)-NR8R9, -C(=O)-NH-NH-R10 или -CH2-О-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы
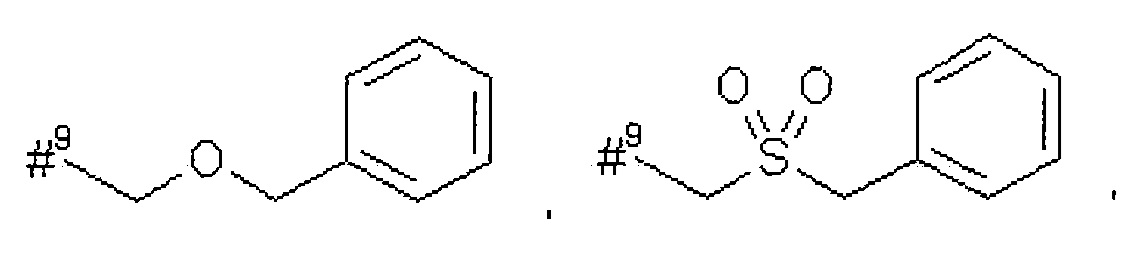


где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (I), где
n представляет собой число от 1 до 50,
AK представляет собой AK1 или AK2 где
AK1 представляет собой связывающее соединение, которое через атом серы в связывающем соединении связано с группой G,
AK2 представляет собой связывающее соединение, которое через атом азота в связывающем соединении связано с группой G,
G если AK=AK1, представляет собой группу формулы


где
#1 обозначает точку связывания с атомом серы в связывающем соединении,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С1-С10)-алкандиил или представляет собой группу формулы

где m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С1-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
где два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
В представляет собой связь или группу формулы

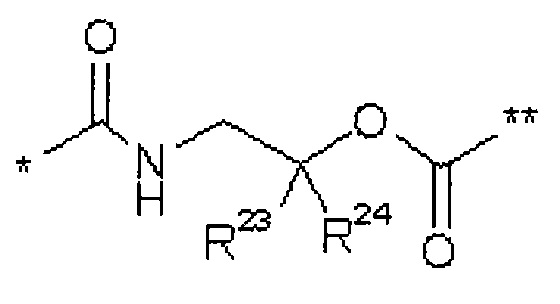

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
L3 представляет собой связь или (С2-С4)-алкандиил,
L4 представляет собой связь или группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
Q1 представляет собой 4-7-членный гетероцикл,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный гетероцикл,
R14 представляет собой водород или (С1-С4)-алкил,
R15 представляет собой водород или (С1-С4)-алкил, или
R14 и R15 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R16 представляет собой водород или (С1-С4)-алкил, R17 представляет собой водород или (С1-С4)-алкил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R18 представляет собой водород или (С1-С4)-алкил,
R19 представляет собой водород или боковую группу природной α-аминокислоты, или ее гомолог или изомер,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или (С1-С4)-алкил,
R22 представляет собой водород или (С1-С4)-алкил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют 3-7-членный карбоцикл,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
R27 представляет собой водород или (С1-С4)-алкил,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
где два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
D имеет значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (I),
где
n представляет собой число от 1 до 50,
AK представляет собой AK1 или AK2
где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела и через атом серы связан с группой G,
AK2 представляет собой антитело или антигенсвязывающий фрагмент антитела и через атом азота связан с группой G,
G, L1, В, L2 и D имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (I), в котором
n представляет собой число от 1 до 20,
AK представляет собой AK1 или AK2
где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы
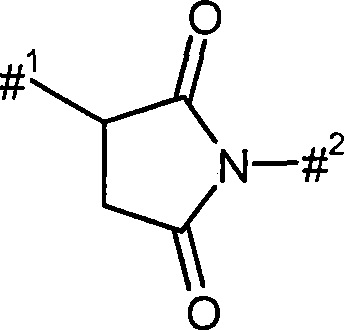
где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы


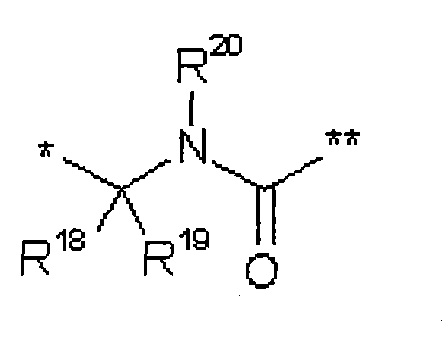
где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
Q1 представляет собой 4-6-членный карбоцикл или пиперидин-1,4-диил,
Q2 представляет собой циклопентил или циклогексил,
R14 представляет собой водород,
R15 представляет собой водород,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R18 представляет собой водород,
R19 представляет собой водород, метил, пропан-2-ил, 2-метилпропан-1-ил или 1-метилпропан-1-ил,
R20 представляет собой водород или метил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1Д-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=O)NR8P9, -C(=O)-NH-NH-R10 или -СН2-О-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9' вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы



где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил, или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (I), где
n представляет собой число от 1 до 10,
AK представляет собой AK1 или AK2 где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антител, приведенных в Таблице 2, вариабельную область легкой цепи и вариабельную область тяжелой цепи антитела, приведенного в Таблице 2, или легкую цепь и тяжелую цепь антитела, приведенного в Таблице 2, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антител, приведенных в Таблице 2, вариабельную область легкой цепи и вариабельную область тяжелой цепи антитела, приведенного в Таблице 2, легкую цепь и тяжелую цепь антитела, приведенного в Таблице 2, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L' представляет собой связь, линейный (С2-С6)-алкандиил илипредставляет собой группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1',
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы
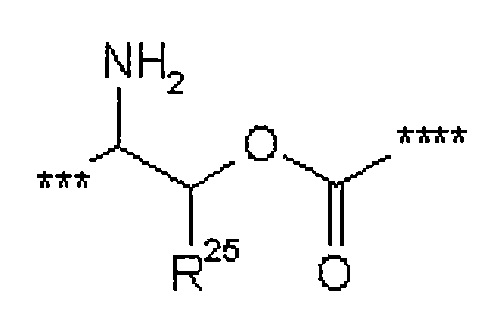

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
L2 представляет собой линейный (С2-С6)-алкандиил,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1Я-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой бензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=O)NR8R9 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, n-пропил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород или группу формулы


где
#9 обозначает точку связывания с -СНС(R26) фенил,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительным объектом настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (I), где
n представляет собой число от 1 до 10, AK представляет собой AK1 или AK2 где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В0-3, В01-10 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10 или D02-6, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AN-2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В0-10, М31-В01 или D02-6 или тяжелую и легкую цепь антитела В0-3, В01-10, М31-В01 или D02-6, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С2-С6)-алкандиил или представляет собой группу формулы
где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила, L2 представляет собой линейный (С2-С6)-алкандиил,
D представляет собой группу формулы
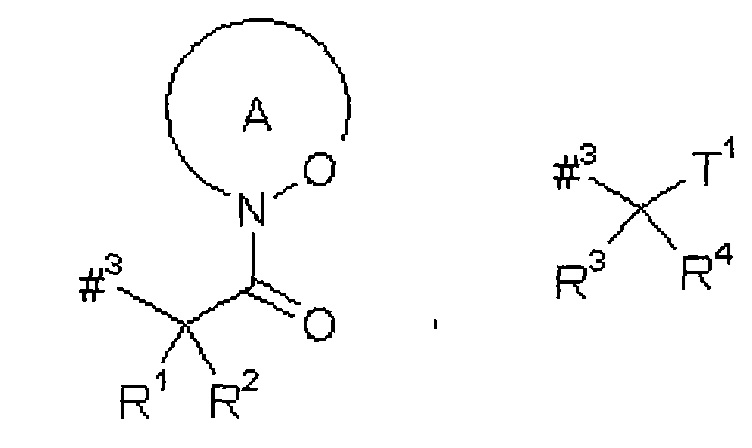

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1H-индол-3-илметил,
or
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой бензил, 1-фенилэтил или 1H-индол-3-илметил, или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т, Т' представляет собой группу формулы -C(=O)-OR7, -С(=O)-NR8R9 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, n-пропил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород или группу формулы


где
#9 обозначает точку связывания с -СНС(R26)фенил,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
или соль, сольват или сольват соли указанного соединения.
Дополнительно согласно настоящему изобретению предложены соединения формулы (XXX)

где
Cys представляет собой остаток цистеина, который через атом серы боковой цепи связан с атомом углерода сукцинимида.
L1 представляет собой связь, линейный (С1-С10)-алкандиил или представляет собой группу формулы

где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С1-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
В представляет собой связь или группу формулы


где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
L3 представляет собой связь или (С2-С4)-алкандиил,
L4 представляет собой связь или группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил
Q1 представляет собой 3-7-членный карбоцикл или 4-7-членный аза-гетероцикл,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный аза-гетероцикл,
R14 представляет собой водород или (С1-С4)-алкил,
R15 представляет собой водород или (С1-С4)-алкил, или
R14 и R15 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R16 представляет собой водород или (С1-С4)-алкил,
R17 представляет собой водород или (С1-С4)-алкил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R18 представляет собой водород или (С1-С4)-алкил,
R19 представляет собой водород или боковую группу природной α-аминокислоты, или ее гомолог или изомер,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или (С1-С4)-алкил,
R22 представляет собой водород или (С1-С4)-алкил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют 3-7-членный карбоцикл,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
R27 представляет собой водород или (С1-С4)-алкил,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит от 1 до 4 заместителей,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
D представляет собой группу формулы
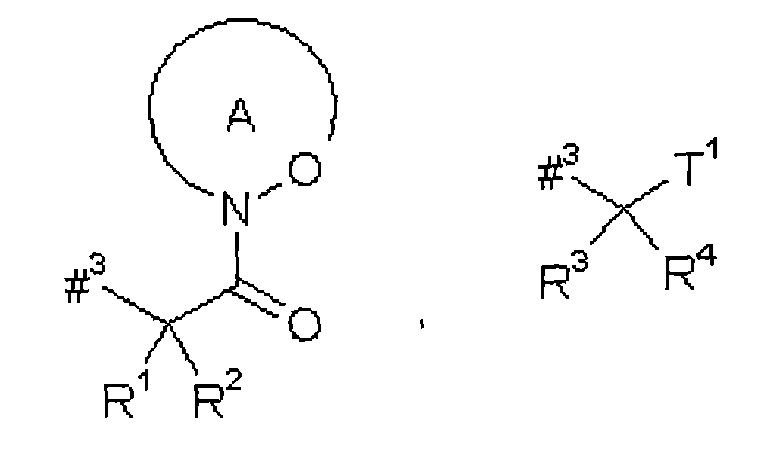

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1H-индол-3-илметил,
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

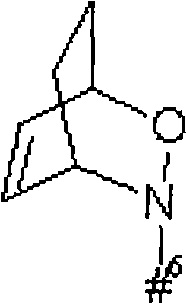
где
#6 обозначает точку связывания с карбонильной группой, R6 представляет собой водород, гидрокси или бензилокси, R3 представляет собой водород,
R4 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы
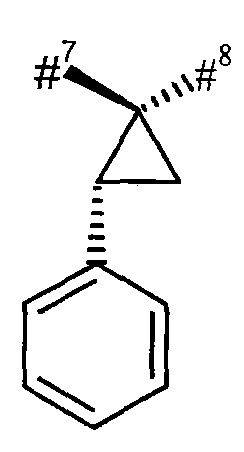
где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=O)-NR8R9, -C(=O)-NH-NH-R10 или -СН2-О-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил, или
R8 и R9, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород, метил или группу формулы



где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород или гидрокси,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил, или соль, сольват или сольват соли указанного соединения.
Дополнительно, в контексте настоящего изобретения особенно предпочтительны также соединения формулы (XXX), где
Cys представляет собой остаток цистеина, который через атом серы боковой цепи связан с атомом углерода сукцинимида,
L1 представляет собой связь, линейный (С2-С6)-алкандиил или представляет собой группу формулы
где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

или

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь,
R14 представляет собой водород,
R15 представляет собой водород,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R23 представляет собой метил,
R24 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 1-гидроксиэтил, бензил, 1-фенилэтил или 1Я-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где #4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород,
R4 представляет собой бензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т,
Т1 представляет собой группу формулы -C(=O)-OR7, -С(=O)-NR8R9, -C(=O)-NH-NH-R10 или-СН2-О-R11
где
R7 представляет собой водород, метил, этил, n-пропил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил,
R5 представляет собой водород или группу формулы


где
#9 обозначает точку связывания с -СНС(R26)фенил,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
n=1-20, более предпочтительно Н=1-10, и в очень предпочтительном варианте Н=2-8.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
AK представляет собой AK1 где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через атом серы в остатке цистеина связывающего соединения с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
и
n, L1, В, L2, D и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
AK представляет собой AK2
где
AK2 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
и
n, L1, В, L2, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
В контексте настоящего изобретения предпочтительными также являются соединения формулы (Ia), где
AK представляет собой AK1 где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, ВО 1-10 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10 или D02-6, и которое присоединено через атом серы остатка цистеина в связывающем соединении с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1, и
n, L1, В, L2, D и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
В контексте настоящего изобретения предпочтительными также являются соединения формулы (Ia), где
AK представляет собой AK2 где
AK2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, М31-В01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, M31-B01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, M31-B01 или D02-6, и которое связано через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
и
n, L1, В, L2, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Предпочтительными в контексте настоящего изобретения также являются соединения общей формулы (Ia), где
AK представляет собой AK2 где
AK2 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10, M31-B01 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10, M31-B01 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10, M31-B01 или D02-6, и которое связано через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
L1 представляет собой связь, В представляет собой связь,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
n, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Предпочтительными в контексте настоящего изобретения также являются соединения общей формулы (Ia), где
AK представляет собой AK1
где
AK1 представляет собой антитело, которое содержит шесть последовательностей гипервариабельных участков (CDR) антитела В01-3, В01-10 или D02-6, вариабельную легкую и вариабельную тяжелую цепь антитела В01-3, В01-10 или D02-6 или тяжелую и легкую цепь антитела В01-3, В01-10 или D02-6, и которое присоединено через атом серы остатка цистеина в связывающем соединении с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой связь, линейный (С3-С5)-алкандиил или группу формулы

где
m представляет собой число 2 или 3,
##* обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
L2 представляет собой линейный (С3-С5)-алкандиил или представляет собой группу формулы
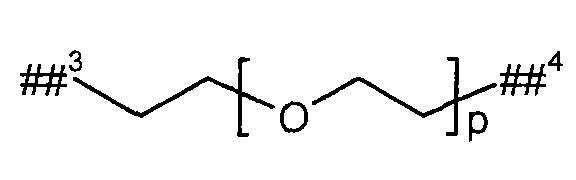
где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
и
n, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
L1 представляет собой связь,
В представляет собой связь,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, и
n, AK, Cys, G, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
L1 представляет собой линейный (С1-С10)-алкандиил или группу формулы

где
m представляет собой число от 2 до 6,
##' обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С1-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
В представляет собой связь или группу формулы

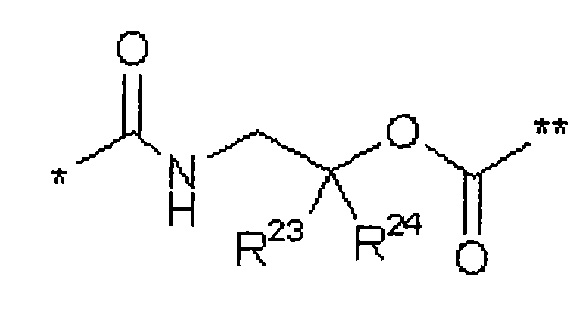

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или (С2-С4)-алкандиил,
L4 представляет собой группу формулы
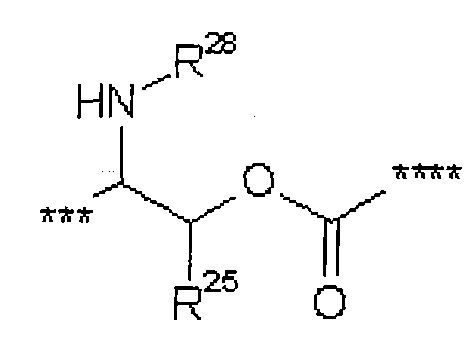

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
R28 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил или бензилоксикарбонил,
Q1 представляет собой 4-7-членный гетероцикл,
R16 представляет собой водород или (С1-С4)-алкил,
R17 представляет собой водород или (С1-С4)-алкил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
R36 представляет собой водород, (С1-С4)-алкилкарбонил, трет-бутилоксикарбонил или бензилоксикарбонил,
R37 представляет собой водород или метил,
или
R36 и R37 вместе с атомом, с которым они связаны, образуют пирролидиновое кольцо,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит от 1 до 4 заместителей, независимо друг от друга выбранных из группы, состоящей из метила, гидроксила и бензила,
и
n, AK, G, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
L1 представляет собой линейный (С2-С6)-алкандиил или группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
В представляет собой связь или группу формулы


где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R36 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R37 представляет собой водород или метил,
или
R36 и R37 вместе с атомом, с которым они связаны, образуют пирролидиновое кольцо,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, и
n, AK, G, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia) и (ХХХа), где
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой линейный (С3-С5)-алкандиил или группу формулы

где m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь,
L2 представляет собой линейный (С3-С5)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, and
n, AK1, Cys, D, R16 и R17 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia) и (ХХХа), где
В представляет собой связь или группу формулы
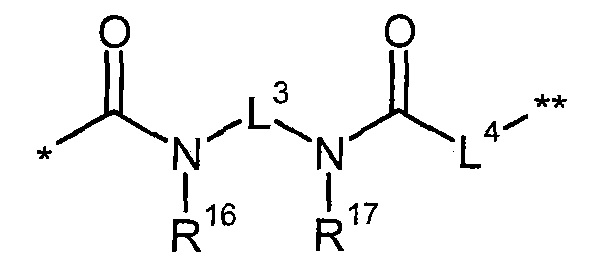
где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь,
n, AK, Cys, G, L1', L2, D, R16, R17 и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
L1' представляет собой связь, линейный (С3-С5)-алкандиил или группу формулы
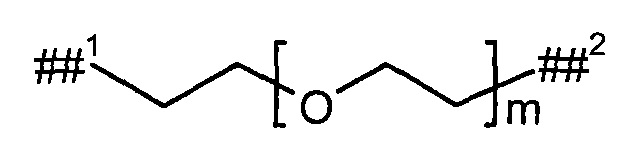
где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь,
L4 представляет собой связь
R16 представляет собой водород,
R17 представляет собой водород,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
n, AK, Cys, G, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
L1 представляет собой связь,
В представляет собой связь,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
n, AK, Cys, G, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
L1 представляет собой линейный (С3-С5)-алкандиил или группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
В представляет собой группу формулы
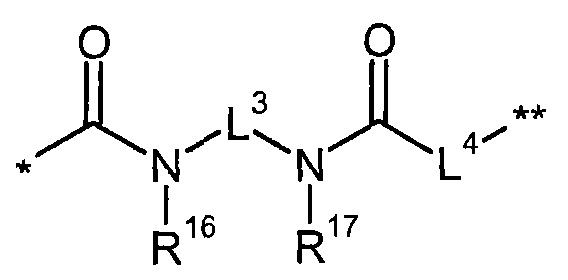
где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь,
L4 представляет собой связь,
R16 представляет собой водород,
R17 представляет собой водород,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
n, AK, Cys, G, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
АК представляет собой AK1 или AK2 где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы
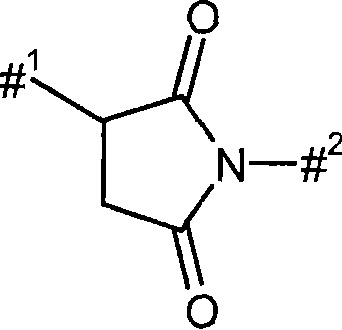
где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С3-С5)-алкандиил или группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь,
L4 представляет собой связь, R16 представляет собой водород,
R17 представляет собой водород,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, и
D и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
AK представляет собой AK1 или AK2, где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L',
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, В представляет собой связь,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
AK представляет собой AK1, где
AK1 представляет собой антитело или антигенсвязывающий фрагмент антитела, который связывается с С4.4а и связан через атом серы в остатке цистеина связывающего соединения с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой линейный (С3-С5)-алкандиил или группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С3-С5)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила, В представляет собой группу формулы
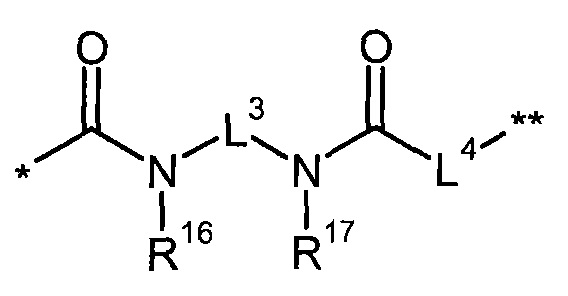
где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь,
L4 представляет собой связь,
R16 представляет собой водород,
R17 представляет собой водород,
L2 представляет собой линейный (С3-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
L1 представляет собой связь, линейный (С3-С5)-алкандиил, группу формулы


где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
L1A представляет собой линейный (С3-С6)-алкандиил,
В1 представляет собой группу формулы

или
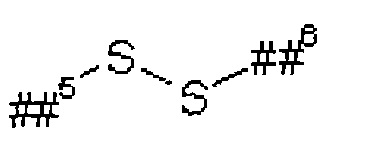
где
##5 обозначает точку связывания с группой L1A,
#6 обозначает точку связывания с группой L1B,
L5 представляет собой связь или этан-1,2-диил,
L6 представляет собой связь или группу формулы


где
##7 обозначает точку связывания с карбонильной группой,
##8 обозначает точку связывания с L1B,
R33 представляет собой водород, (С1-С4)-алкилкарбонил или трет-бутилоксикарбонил,
R34 представляет собой водород или метил,
R29 представляет собой водород или метил,
R30 представляет собой водород или метил,
R31 представляет собой водород или метил,
R32 представляет собой водород или метил,
L13 представляет собой линейный (С3-С6)-алкандиил,
В представляет собой связь или группу формулы

или

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р is О,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
R28 представляет собой водород, (С1-С4)-алкилкарбонил или трет-бутилоксикарбонил,
Q1 представляет собой 4-7-членный гетероцикл,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный гетероцикл,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
R36 представляет собой водород, (С1-С4)-алкилкарбонил или трет-бутилоксикарбонил,
R37 представляет собой водород или метил,
или
R36 и R37 вместе с атомом, с которым они связаны, образуют пирролидиновое кольцо,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), где
L1 представляет собой линейный (С3-С5)-алкандиил или группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
В представляет собой группу формулы

или

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
Q1 представляет собой пиперидин-1,4-диил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
R23 представляет собой метил,
R24 представляет собой водород,
R36 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
R37 представляет собой водород или метил,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы
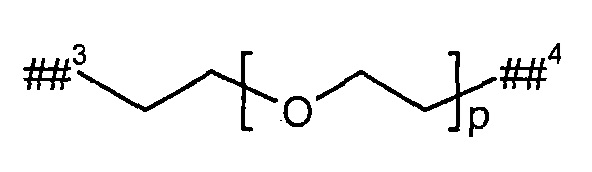
где
р представляет собой число 2 или 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород или метил,
R2 представляет собой изобутил, ceN-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидрокси-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-идол-3 -илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенил-циклопропан-1,1 -диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
R3 представляет собой водород или метил,
R4 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидроксил-3-нитробензил, 4-гидрокси-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3-илметил,
или
R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют (1S,2H)-2-фенилциклопропан-1,1-диил формулы

где
#7 обозначает точку связывания с соседним атомом азота,
#8 обозначает точку связывания с группой Т1,
Т1 представляет собой группу формулы -C(=O)-OR7, -C(=O)-NR8R9, -C(=O)-NH-NH-R10 или -CH2-O-R11,
где
R7 представляет собой водород, метил, этил, н-пропил, трет-бутил, бензил или адамантилметил,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, этил, н-пропил или бензил,
или
R8 и R9, вместе с атомом азота, к которому они присоединены, образуют 4-7-членный гетероцикл,
R10 представляет собой бензоил,
R11 представляет собой бензил, фенильная группа которого возможно содержит в качестве заместителя метоксикарбонил или карбоксил
R5 представляет собой водород, метил или группу формулы


где
#9 обозначает точку связывания с -CHC(R26)-T2,
R12 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил, карбоксил или группу формулы -S(O)2OH,
R13 представляет собой фенил, который возможно содержит в качестве заместителей метоксикарбонил или карбоксил,
R26 представляет собой водород,
Т2 представляет собой фенил, бензил, 1H-индол-3-ил или 1H-индол-3-илметил, и
n, AK, Cys, G, L1, В, L2, D и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил, 4-гидроксибензил, 1-фенилэтил или 1Я-индол-3-илметил,
кольцо А с присутствующей на нем группой N-O представляет собой гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
Т' представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9,
где
R7 представляет собой водород,
R8 представляет собой водород,
R9 представляет собой водород,
n, AK, Cys, G, L1, В, L2, D и R33 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы
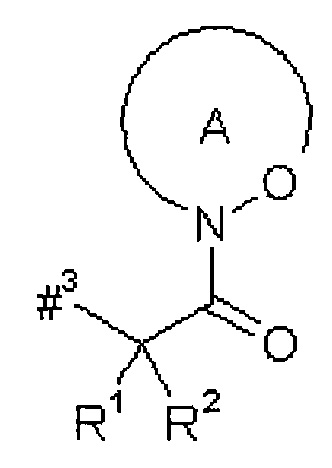

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
кольцо А с присутствующей на нем группой N-O представляет собой гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
Т1 представляет собой группу формулы -C(=O)-NR8R9,
где
R8 представляет собой водород,
R9 представляет собой водород,
n, AK, Cys, G, L1, В, L2, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
Р1 представляет собой водород,
R2 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
кольцо А с присутствующей на нем группой N-O представляет собой гетероцикл формулы
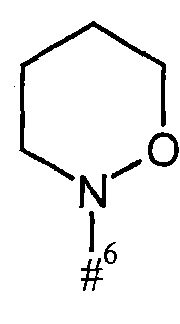
где
#6 обозначает точку связывания с карбонильной группой, R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
Т1 представляет собой группу формулы -C(=O)-NR8R9,
где
R8 представляет собой водород,
R9 представляет собой водород,
n, AK, Cys, G, L1, В, L2, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы

где
#3 обозначает точку связывания с атомом азота,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
Т1 представляет собой группу формулы -C(=O)-OR7 или -C(=O)-NR8R9,
где
R7 представляет собой водород,
R8 представляет собой водород,
R9 представляет собой водород,
n, AK, Cys, G, L1, В, L2, D и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы

где
#3 обозначает точку связывания с атомом азота,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
Т1 представляет собой группу формулы -C(=O)-NR8R9,
где
R8 представляет собой водород,
R9 представляет собой водород,
n, AK, Cys, G, L1, В, L2, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород или метил,
R2 представляет собой изобутил, сек-бутил, трет-бутил, фенил, бензил, 1-гидроксиэтил, 4-гидроксибензил, 4-гидрокси-3-нитробензил, 4-гидрокси-3-аминобензил, 1-фенилэтил, дифенилметил, 1H-имидазол-4-илметил или 1Н-индол-3 -илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенилциклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
and
n, AK, Cys, G, L1, В, L2 и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
D представляет собой группу формулы

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил, 4-гидроксибензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенилциклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой, и
n, AK, Cys, G, L1, В, L2 и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
R35 представляет собой гидроксил, и
n, AK, Cys, G, L1, B, L2, D и R35 имеют значения, определенные выше, или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (Ia), (ХХХа) и (XXXI), где
R35 представляет собой метил,
и
n, AK, Cys, G, L1, В, L2, D и R35 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Соответственно, особенно предпочтительными в контексте настоящего изобретения являются соединения формулы (ХХХа), где
Cys представляет собой остаток L-цистеина, который связан через атом серы боковой цепи с атомом углерода сукцинимида,
или соль, сольват или сольват соли указанного соединения.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (I) и (XXX), где
D представляет собой группу формулы

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил, 1-фенилэтил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенилциклопропан-1,1-диил формулы
где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой моно- или бициклический, возможно содержащий заместители гетероцикл формулы


где
#6 обозначает точку связывания с карбонильной группой,
R6 представляет собой водород, гидрокси или бензилокси,
n, AK, Cys, G, L1, L2 и В имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительными в контексте настоящего изобретения также являются соединения формулы (I) и (XXX), где
D представляет собой группу формулы

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой бензил или 1H-индол-3-илметил,
или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют (1S,2R)-2-фенилциклопропан-1,1-диил формулы

где
#4 обозначает точку связывания с соседним атомом азота,
#5 обозначает точку связывания с карбонильной группой,
кольцо А с присутствующей на нем группой N-O представляет собой гетероцикл формулы

где
#6 обозначает точку связывания с карбонильной группой,
n, AK, Cys, G, L1, L2 и В имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Более конкретно, предпочтительным объектом настоящего изобретения являются соединения формулы (I), где
D представляет собой группу формулы

Т1 представляет собой -С(=O)-ОН или -C(=O)-NH2, и
n, AK, G, L1, В, L2, #3, R3 и R4 имеют значения, определенные выше.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (I), где
n=1-20, более предпочтительно Н=1-10, и в очень предпочтительном варианте Н=2-8.
Также в контексте настоящего изобретения предпочтительными являются соединения формулы (I) и (XXX), где
В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь,
n, AK, Cys, G, L1, L2, D, R16 и R17 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также особенно предпочтительными в контексте настоящего изобретения являются соединения формулы (I) и (XXX), где
В представляет собой связь или группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2, L3 и L4 представляет собой связь,
n, AK, Cys, G, L1, L2, D, R16 и R17 имеют значения, определенные выше,
или соль, сольват или сольват соли указанного соединения.
Также предпочтительным в контексте настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (I), где
AK представляет собой AK1,
где
AK1 представляет собой связывающее соединение, которое связано через атом серы в остатке цистеина связывающего соединения с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой связь, линейный (С1-С10)-алкандиил или представляет собой группу формулы

где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С1-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
В представляет собой связь или группу формулы

или

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или (С2-С4)-алкандиил,
L4 представляет собой связь или группу формулы

где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
Q1 представляет собой 4-7-членный гетероцикл,
R14 представляет собой водород или (С1-С4)-алкил,
R15 представляет собой водород или (С1-С4)-алкил, или
R14 и R15 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
R16 представляет собой водород или (С1-С4)-алкил,
R17 представляет собой водород или (С1-С4)-алкил, или
R16 и R17 вместе с атомом, с которым они связаны, образуют 5- или 6-членный гетероцикл,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
или соль, сольват или сольват соли указанного соединения.
Также предпочтительным в контексте настоящего изобретения является конъюгат связывающее соединение - активное соединение общей формулы (I), где
AK представляет собой AK2,
где
AK2 представляет собой связывающее соединение, которое связано через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
L1 представляет собой связь, линейный (С1-С10)-алкандиил или представляет собой группу формулы

где m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С1-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
В представляет собой связь или группу формулы

или

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
Р представляет собой О или NH,
Q2 представляет собой 3-7-членный карбоцикл или 4-7-членный гетероцикл,
R18 представляет собой водород или (С1-С4)-алкил,
R19 представляет собой водород или боковую группу природной α-аминокислоты, или ее гомолог или изомер,
R20 представляет собой водород или (С1-С4)-алкил,
или
R19 и R20 вместе с атомом, с которым они связаны, образуют кольцо пирролидина,
R21 представляет собой водород или (С1-С4)-алкил,
R22 представляет собой водород или (С]-С4)-алкил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют 3-7-членный карбоцикл,
R23 представляет собой (С1-С4)-алкил,
R24 представляет собой водород или (С1-С4)-алкил,
L2 представляет собой линейный (С2-С10)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
где (С2-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (Сз-Сб)-циклоалкила или фенила,
или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение формулы (Ia), где
n представляет собой число от 2 до 8,
AK представляет собой AK1 или AK2,
где
AK1 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
AK2 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G если AK=AK1, представляет собой группу формулы

где
#1' обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
или
если AK=AK2, представляет собой карбонил,
L1 представляет собой связь, линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
m представляет собой число 2 или 3,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С2-С6)-алкандиил возможно содержит в качестве заместителей 1 или 2 метила,
В представляет собой связь или группу формулы



где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь или этан-1,2-диил,
L4 представляет собой связь или группу формулы


где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой метил,
R28 представляет собой водород, метилкарбонил или трет-бутилоксикарбонил,
Q1 представляет собой пиперидин-1,4-диил,
R16 представляет собой водород или метил,
R17 представляет собой водород или метил,
или
R16 и R17 вместе с атомом, с которым они связаны, образуют кольцо пиперазинила,
R21 представляет собой водород или метил,
R22 представляет собой водород или метил,
или
R21 и R22 вместе с атомом, с которым они связаны, образуют циклопропильное кольцо,
R23 представляет собой метил,
R24 представляет собой водород,
L2 представляет собой линейный (С2-С6)-алкандиил или представляет собой группу формулы

где
р представляет собой число от 2 до 6,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота, D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
Р1 представляет собой водород,
R2 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
кольцо А с присутствующим в нем группой N-O
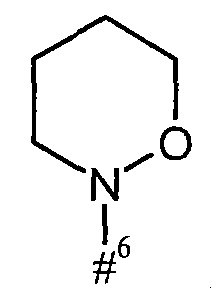
где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
Т1 представляет собой группу формулы -C(=O)-NR8R9,
R8 представляет собой водород или метил,
R9 представляет собой водород, метил, или этил,
R35 представляет собой метил, или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение формулы (Ia), где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
AK представляет собой AK1, где
AK1 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой пентан-1,5-диил,
В представляет собой группу формулы

где
* обозначает точку связывания с L1,
** обозначает точку связывания с L2,
L3 представляет собой связь,
L4 представляет собой связь,
R16 представляет собой водород,
R17 представляет собой водород,
L2 представляет собой пропан-1,3-диил,
D представляет собой группу формулы


где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
кольцо А с присутствующим в нем группой N-O

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
Т1 представляет собой группу формулы -C(=O)-NR8R9,
R8 представляет собой водород,
R9 представляет собой водород,
R35 представляет собой метил,
или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение формулы (Ia), где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
AK представляет собой AK1,
где
AK1 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
G представляет собой группу формулы

где
#1 обозначает точку связывания с остатком цистеина связывающего соединения,
#2 обозначает точку связывания с группой L1,
L1 представляет собой связь, В представляет собой связь,
L2 представляет собой гексан-1,6-диил,
и D имеет значение, определенное выше, или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение формулы (Ia), где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
AK представляет собой AK2,
где
AK2 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
L1 представляет собой связь,
В представляет собой связь,
L2 представляет собой пентан-1,5-диил,
D представляет собой группу формулы
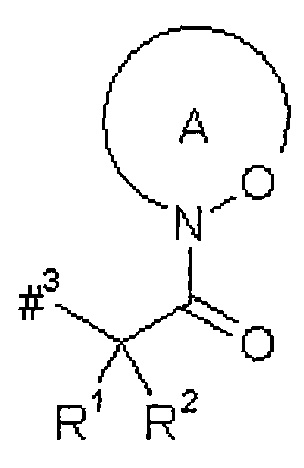

где
#3 обозначает точку связывания с атомом азота,
R1 представляет собой водород,
R2 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
кольцо А с присутствующим в нем группой N-O

где
#6 обозначает точку связывания с карбонильной группой,
R3 представляет собой водород,
R4 представляет собой 4-гидроксибензил или 1H-индол-3-илметил,
Т1 представляет собой группу формулы -C(=O)-NR8R9,
R8 представляет собой водород,
R9 представляет собой водород, R35 представляет собой метил, или соль, сольват или сольват соли указанного соединения.
Особенно предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение формулы (Ia), где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
AK представляет собой AK2, где
AK2 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через боковую группу NH остатка лизина связывающего соединения с группой G,
G представляет собой карбонил,
L1 представляет собой связь,
В представляет собой связь,
L2 представляет собой группу формулы
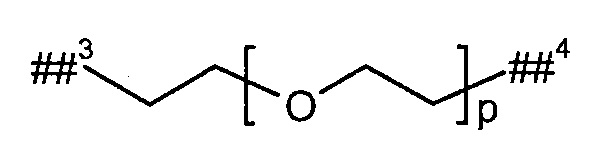
где
р представляет собой число 3,
##3 обозначает точку связывания с группой В,
##4 обозначает точку связывания с атомом азота,
и D имеет значение, определенное выше,
или соль, сольват или сольват соли указанного соединения.
Также особенно предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение, выбранные из следующих соединений:


где в каждом случае
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
AK1 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через атом серы в остатке цистеина связывающего соединения с группой G,
и
AK2 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связан через боковую группу NH остатка лизина связывающего соединения с группой G.
Более конкретно, предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение, выбранные из следующих соединений:

где в каждом случае
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
и
AK представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которые связываются с С4.4а.
В этих формулах AK1F, AK1B, AK2B могут быть заменены другими человеческими или гуманизированными анти-С4.4а антителами.
Более конкретно, предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение, выбранные из следующих соединений:


где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
и
AK1 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, связанные через остаток цистеина с токсофорным линкерным фрагментом,
AK2 представляет собой человеческое или гуманизированное антитело или антигенсвязывающий фрагмент антитела, которое связывается с С4.4а, и связанные через остаток лизина с токсофорным линкерным фрагментом.
Более конкретно, предпочтительными в контексте настоящего изобретения являются конъюгаты связывающее соединение - активное соединение, выбранные из следующих соединений:


где
n представляет собой число от 2 до 8, предпочтительно от 2 до 5,
и
AK1B и AK2B представляют собой В01-3.
Определения радикалов, приведенные отдельно в соответствующих комбинациях и предпочтительных комбинациях радикалов также можно произволно заменять определениями радикалов из других комбинаций, вне зависимости от соответствующих комбинаций радикалов, которые указаны.
Особенно предпочтительны комбинации двух или трех указанных выше диапазонов.
Далее согласно настоящему изобретению предложены способы получения соединений согласно настоящему изобретению формулы (Ia), в которых раствор связывающего соединения в буфере (предпочтительно в таком буфере как, например фосфатно-буферный раствор (ФБР)
[А] смешивают с подходящим восстановителем, таким как, например, дитиотреитол или трис(2-карбоксиэтил)фосфин гидрохлорид, и затем осуществляют реакцию с соединением формулы (IIа)
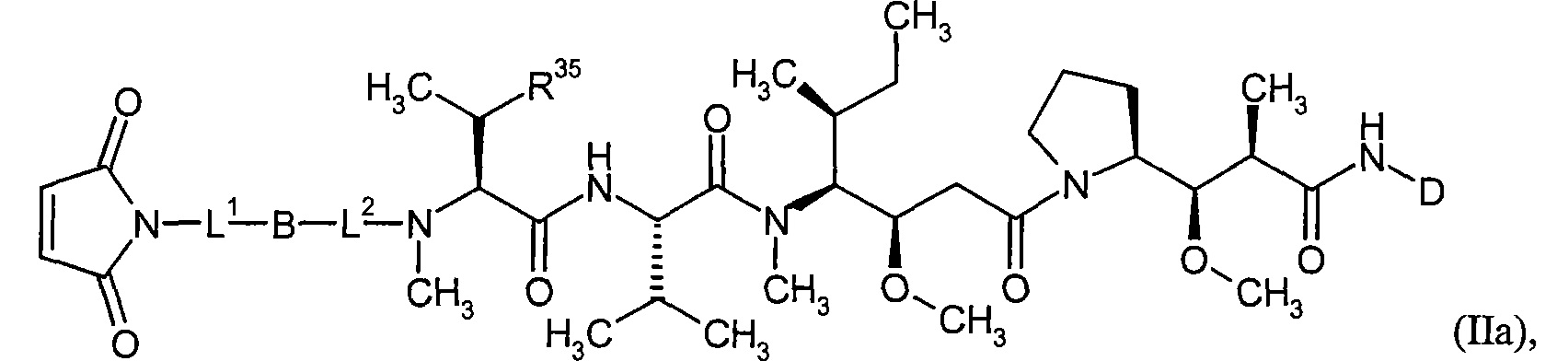
где каждый из D, L1, В, L2 и R35 имеет значения, определенные выше, с получением соединения формулы (I-A)

где каждый из n, AK2, D, L1, В, L2 и R35 имеет значения, определенные выше, или
[В] осуществляют реакцию указанного раствора с соединением формулы (IIIa)
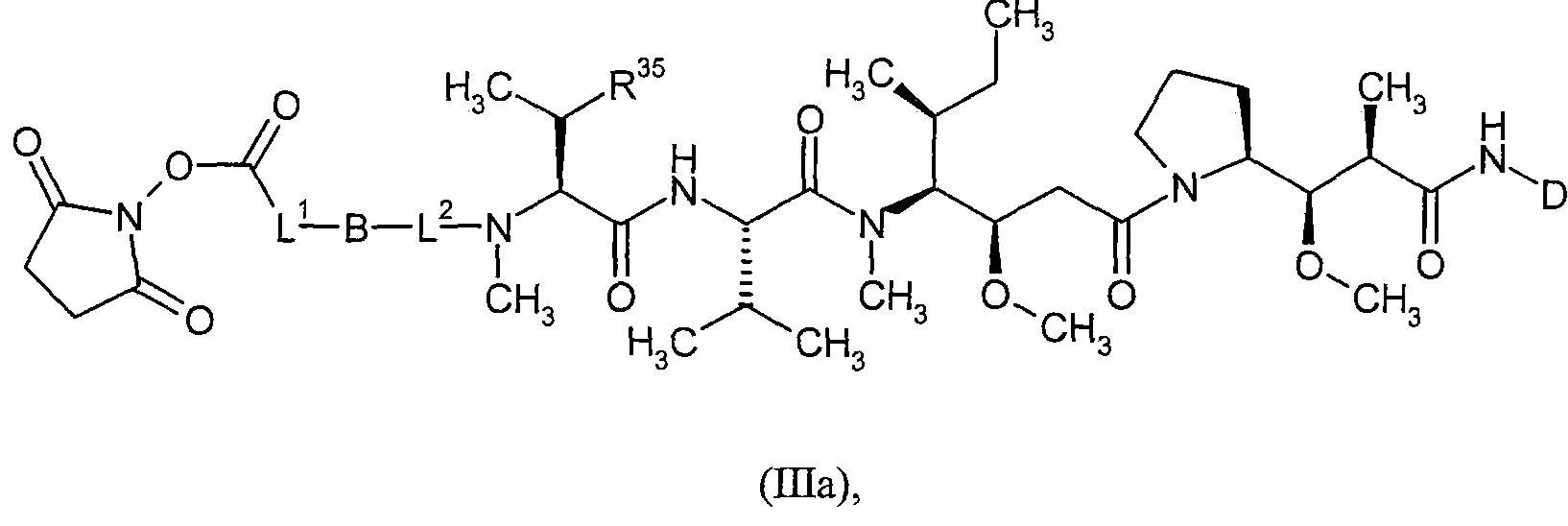
где каждый из D, L1, В, L2 и R35 имеет значения, определенные выше, с получением соединения формулы (Ia-В)

где каждый из n, AK2, D, L1, В, L2 и R35 имеет значения, определенные выше.
Далее согласно настоящему изобретению предложены способы получения соединений согласно настоящему изобретению формулы (I), в которых раствор связывающего соединения в фосфатно-буферном растворе
[А] смешивают с подходящим восстановителем, таким как, например, дитиотреитол или трис(2-карбоксиэтил)фосфин гидрохлорид, и затем осуществляют реакцию с соединением формулы (П)
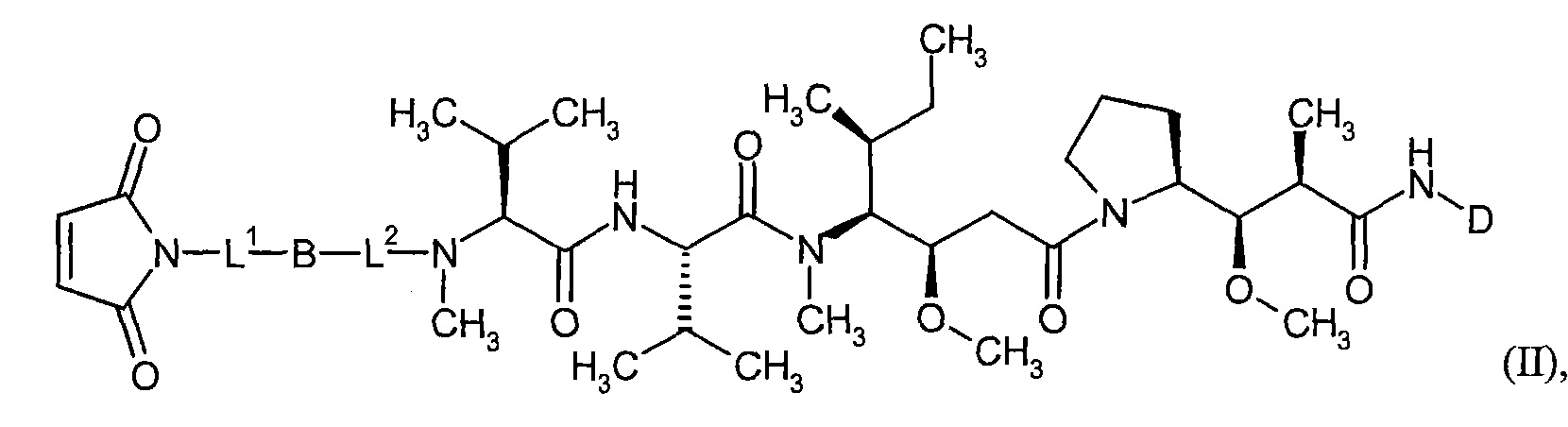
где каждый из D, L, В и L имеют значения, определенные выше, с получением соединения формулы (I-A)

где каждый из n, AK1, D, L1, В и L2 имеет значения, определенные выше, или
[В] осуществляют реакцию указанного буфера с соединением формулы (III)

где каждый из D, L1, В и L2 имеет значения, определенные выше, с получением соединения формулы (I-B)

где каждый из n, AK2, D, L1, В и L2 имеет значения, определенные выше.
Связывание по цистеину;
Частичное восстановление антитела, а также последующую конъюгацию (частично) восстановленного антитела с соединением формулы (II) или (IIa) осуществляют в соответствии с известными специалисту методами, см., например Ducry et al., Bioconj. Chem. 2010, 21, 5 и цитируемые там источники, Klussman et al., Bioconj. Chem. 2004, 15(4), 765-773. Мягкое восстановление антитела осуществляют предпочтительно путем добавления 2-6 эквивалентов трифторэтилфосфата (ТСЕР) к антителу, находящемуся в подходящем буферном растворе, предпочтительно в фосфатном буфере и путем перемешивания в течение 30-180 мин при температуре от 15 до 40°С, предпочтительно при комнатной температуре. После этого осуществляют конъюгацию путем добавления раствора соединения формулы (II) или (IIa) в ДМСО, ацетонитриле или ДМФ к раствору (частично) восстановленного антитела в фосфатно-буферном растворе, с последующим проведением реакции при температуре от 0°С до +40°С, более конкретно, от +10°С до +30°С, в течение периода от 30 мин до 6 часов, более конкретно от 1 до 2 часов.
Связывание по лизину:
Сначала соединения формулы (III) или (IIa) или аналогичные карбоксил-активированные компоненты готовят методами, обычно используемыми в химии пептидов. Затем их разбавляют в растворителях, таких как ДМСО или ДМФ, например, и добавляют к антителу, которое предпочтительно находится в фосфатном буфере с нейтральным рН. Раствор перемешивают в течение 1-16 ч при температуре от 15 до 40°С, предпочтительно при комнатной температуре.
Способы получения, описанные выше, поясняются примерами и схемами, представленными ниже (Схемы 1 и 2):

[а): 1. АК (антитело), ТСЕР, фосфатно-буферный раствор, комнатная температура; 2. Добавление производного малеимида в ДМСО, RT].

[а): АК (антитело), фосфатно-буферный раствор, смешивание при комнатной температуре с активированным карбоксил-производным компонентов линкер-активное соединение].
Соединения формулы (II) где L1 и В представляют собой связь, могут быть получены путем осуществления с соединением формулы (IV)

где D имеет значение, определенное выше, восстановительного аминирования в инертном растворителе с соединением формулы (V)

где
L2A имеет определенное выше значение L2, но с укороченной на один атом углерода алкильной цепью,
PG1 представляет собой амино-защитную группу, такую как,, например, (9H-флуорен-9-илметокси)карбонил, трет-бутоксикарбонил или бензилоксикарбонил,
с получением соединения формулы (VI)

где D, L2 и PG1 имеют определенные выше значения,
удаления защитной группы PG1 из этого соединения известными специалисту методами, и Осуществления реакции соединения без защитной группы в инертном растворителе в присутствии подходящего основания с метил 2,5-диоксо-2,5-дигидро-1Н-пиррол-1-карбоксилатом с получением соединения формулы (II-А)

где каждый из D и L2 имеют значения, определенные выше.
Соединения формулы (II) где В представляет собой группу формулы (В1)

где каждый из *, **, R14 и R15 имеют значения, определенные выше,
могут быть получены путем удаления защитной группы PG1 из соединения формулы (VI) известными специалисту методами и осуществления реакции соединения без защитной группы в инертном растворителе в присутствии подходящего основания с соединением формулы (VII)
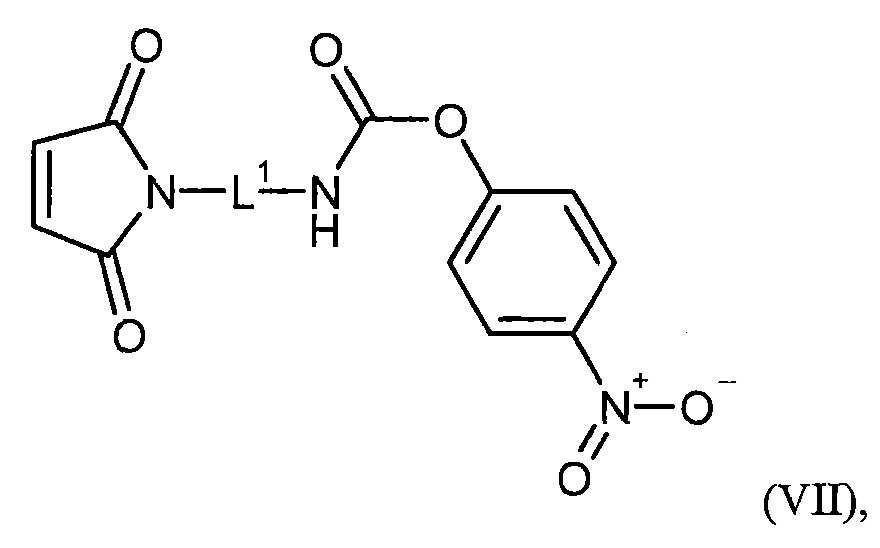
где L1 имеет значение, определенное выше,
с получением соединения формулы (II-В)

где каждый из D, L1 и L2 имеет значения, определенные выше.
Соединения формулы (П) где В представляет собой группу формулы (В2)

где каждый из *, **, L3, R16 и R17 имеет значения, указанные выше, могут быть получены путем осуществления восстановительного аминирования соединения формулы (IV)
в инертном растворителе с соединением формулы (VIII)

где
L2A имеет определенное выше значение L2, с укороченной на один атом углерода алкильной цепью,
с получением соединения формулы (IX)

где D и L2 имеют значения, определенные выше,
и осуществления реакции этого соединения в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с соединением формулы (X)

где каждый из L1 и L3 имеет значения, определенные выше,
с получением соединения формулы (II-С)

где каждый из D, L1, L2 и L3 имеет значения, определенные выше.
Соединение формулы (II), где В представляет собой группу формулы (В3)

где каждый из *, **, L3, R16 и R17 имеет значения, определенные выше, и
L4A представляет собой группу формулы

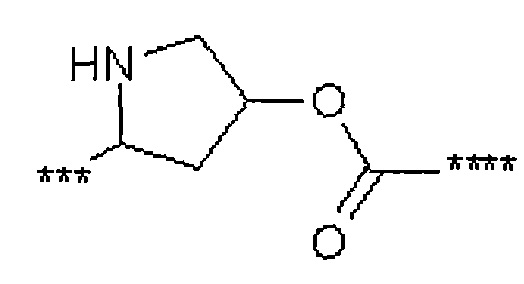
где
*** обозначает точку связывания с карбонильной группой,
**** обозначает точку связывания с L2,
R25 представляет собой водород или метил,
может быть получено путем осуществления реакции соединения формулы (IX) в инертном растворителе в присутствии подходящего основания и подходящего связывающего реагента с соединением формулы (XI-A) или (XI-B)
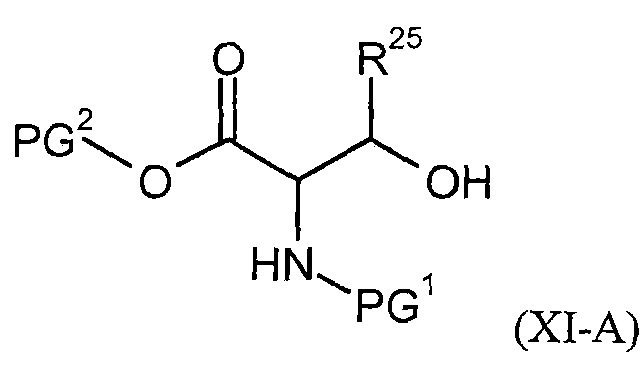

где каждый из R25 и PG'1 имеют значения, определенные выше, и
PG2 представляет собой подходящую карбоксил-защитную группу, более конкретно, бензил,
с получением соединения (XII-А) или (XII-В)

или

где D, PG1, PG2 и L2 имеют значения, определенные выше,
последующего устранения защитной группы PG2 из этого соединения, известными специалисту методами и осуществления реакции соединения без защитной группы в инертном растворителе в присутствии подходящего связывающего агента подходящего основания с соединением формулы (X), и наконец, устранения защитной группы PG1 из этого соединения, известными специалисту методами, с получением соединения формулы (II-D-A) или (II-D-B)

или

где D, L1, L2 и L3 имеют значения, определенные выше. Соединение формулы (П), где В представляет собой группу формулы (В4)

где каждый *, ** имеет указанные выше значения, и
Q1A представляет собой N-связанный 4--7-членный гетероцикл,
может быть получено путем осуществления реакции соединения формулы (IX) в инертном растворителе в присутствии подходящего основания и подходящего связывающего реагента с соединением формулы (XXI)

где каждый из PG1 и Q1A имеет значения, определенные выше, с получением соединения формулы (XXII)

где PG1, Q1A, D и L2 имеют значения, определенные выше,
устранения защитной группы PG' из этого соединения, известными специалисту методами, и последующего осуществления реакции соединения без защитной группы в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с соединением формулы (XXIII)
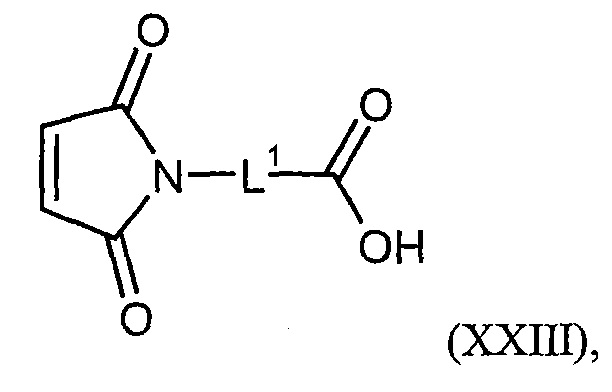
где L1 имеет значение, определенное выше,
с получением соединения формулы (II-D)

где Q1A, D, L1' и L2 имеют значения, определенные выше.
Соединения формулы (III), где L1 и В представляют собой связь, может быть получено путем осуществления реакции соединения формулы (DC) в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с N-гидроксисукцинимидом с получением соединения формулы (III-А)

где каждый из D и L имеют значения, определенные выше.
Соединения формулы (III), где L1 представляет собой связь, и В представляет собой группу формулы (B5A)

где каждый из *, ** и Р имеет значения, определенные выше, т
Q2A представляет собой 3-7-членный карбоцикл,
может быть получено путем осуществления реакции соединения формулы (IX) в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с соединением формулы (ХШ)

где каждый из Р, Q2A и PG2 имеет значения, определенные выше, с получением соединения формулы (XIV)
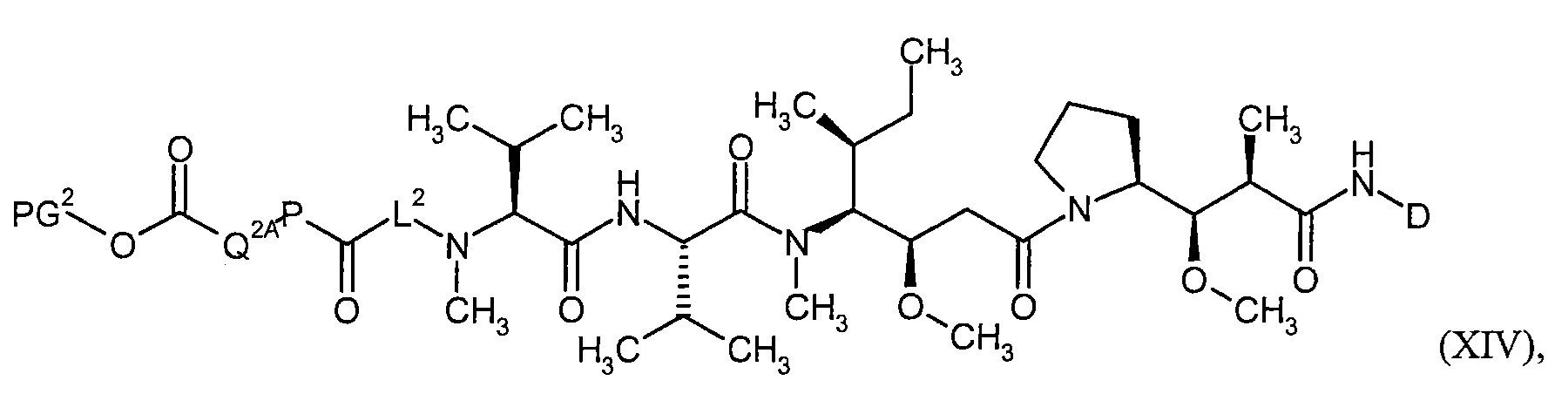
где каждый из D, Р, Q2A, L2 и PG2 имеет значения, определенные выше,
устранения защитной группы PG2 из этого соединения известными специалисту методами и последующего осуществления реакции соединения без защитной группы в инертном растворителе в присутствии подходящего основания с N-гидроксисукцинимидом с получением соединения формулы (III-В)

где каждый из D, Р, Q2A, и L2 имеют значения, определенные выше.
Соединения формулы (III), где L' представляет собой связь, и В представляет собой группу формулы (В6)

где каждый из *, **, R18, R19 и R20 имеет значения, определенные выше,
может быть получено путем осуществления реакции соединения формулы (IX) в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с соединением формулы (XV)

где каждый из R18, R19, R20 и PG2 имеет значения, определенные выше, с получением соединения формулы (XVI)

где каждый из D, R18, R19, R20, L2 и PG2 имеет значения, определенные выше,
устранения защитной группы PG2 из этого соединения известными специалисту методами и последующего осуществления реакции соединения без защитной группы в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с N-гидроксисукцинимида с получением соединения формулы (III-С)

где каждый из D, R18, R19, R20 и L2 имеет значения, определенные выше.
Соединения формулы (III), где L1 представляет собой связь, и В представляет собой группу формулы (В7)

где каждый из *, **, R21 и R22 имеет значения, определенные выше,
могут быть получены путем удаления защитной группы PG1 из соединения формулы (VI) известными специалисту методами и осуществления реакции полученного соединения без защитной группы в инертном растворителе в присутствии подходящего основания с соединением формулы (XVII)

где каждый из R21 и R22 имеет значения, определенные выше,
с получением соединения формулы (Ш-D)

где каждый из D, R21, R22 и L2 и имеет значения, определенные выше.
Соединения формулы (III), где В представляет собой группу формулы (В8)

где каждый из *, **, R23 и R24 имеет значения, определенные выше,
может быть получено путем осуществления реакции соединения формулы (IX) в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с соединением формулы (XVIII)

где каждый из R23, R24 и PG1 имеет значения, определенные выше,
с получением соединения формулы (XIX)

где каждый из D, R23, R24, L2 и PG1 имеет значения, определенные выше,
устранения защитной группы PG1 из этого соединения известными специалисту методами и последующего осуществления реакции соединения без защитной группы в инертном растворителе в присутствии подходящего связывающего агента и подходящего основания с соединением формулы (XX)
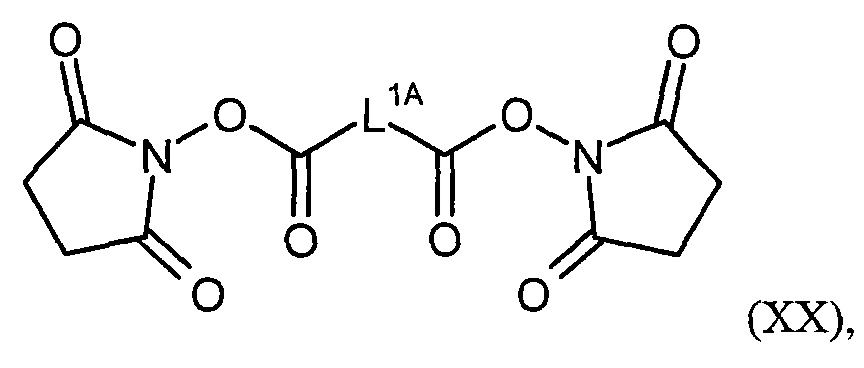
где
L1A представляет собой линейный (С1-С10)-алкандиил или представляет собой группу формулы

где
m представляет собой число от 2 до 6,
##1 обозначает точку связывания с группой G,
##2 обозначает точку связывания с группой В,
где (С1-С10)-алкандиил возможно содержит в качестве заместителей от 1 до 4 метилов,
и
при этом два атома углерода цепи алкандиила в положении 1,2, 1,3 или 1,4 относительно друг друга, а также любые расположенные между ними атомы углерода, возможно связаны с образованием (С3-С6)-циклоалкила или фенила,
с получением соединения формулы (III-Е)

где каждый из D, R23, R24, L1A и L2 имеет значения, определенные выше.
Соединения формулы (III), где В представляет собой группу формулы (В53)

где каждый из * и ** и имеет значения, определенные выше, и
Q2B представляет собой N-связанный 4--7-членный гетероцикл,
может быть получено путем осуществления реакции соединения формулы (DC) в инертном растворителе в присутствии подходящего основания и подходящего связывающего реагента с соединением формулы (XXIV)

где каждый из PG1 и Q2B имеет значения, определенные выше,
с получением соединения формулы (XXV)

где каждый из PG1, Q2B, D и L2 имеет значения, определенные выше,
устранения защитной группы PG1 из этого соединения известными специалисту методами,
и последующего превращения соединения без защитной группы в инертном растворителе в присутствии подходящего основания с соединением формулы (XX) в соединение формулы (III-F)

где Q2B, D, L1A и L2 имеют значения, определенные выше.
Реакции (IV)+(V)→(VI) и (IV)+(VIII)→(DC) протекают в растворителях, которые обычно используются для восстановительного аминирования и инертны в условиях данной реакции, необязательно в присутствии кислоты и/или агента, удаляющего воду, в качестве катализатора. Такие растворители включают, например, спирты, такие как метанол, этанол, n-пропанол, изопропанол, n-бутанол или трет-бутанол, эфиры, такие как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан или бис(2-метоксиэтиловый) эфир, или другие растворители, такие как дихлорметан, 1,2-дихлорэтан, N,N-диметилформамид или, в альтернативном варианте, вода. Также возможно использовать смеси указанных растворителей. В качестве растворителя предпочтительно использовать смесь 1,4-диоксан/вода с добавлением уксусной кислоты или разбавленной соляной кислоты в качестве катализатора.
Восстанавливающими агентами, пригодными для этих реакций являются, в частности, сложные борогидриды, такие как, например, борогидрид натрия, цианоборогидрид натрия, триацетоксиборогидрид натрия, тетра-n-бутиламмония борогидрид или комплекс боран-пиридин. В предпочтительном варианте используют цианоборогидрид натрия или комплекс боран-пиридин.
Реакции (IV)+(V)→(VI) и (IV)+(VIII)→(DC) обычно протекают в температурном диапазоне от 0°С до +120°С, предпочтительно от+50°С до +100°С. Реакции могут быть осуществлены при атмосферном, повышенном или пониженном давлении (например, от 0,5 до 5 бар); обычно работают при атмосферном давлении.
Описанные выше реакции связывания (IX)+(X)→(II-С), (XII-А) или (XII-В)+(X)→(II-D-А) или (II-D-B), (IX)+(XIII)→(XIV), (IX)+(XV)→(XVI) и (XXII)+(XXIII)→(II-D) (образование амида из аминного компонента или компонента карбоновой кислоты, соответственно) осуществляют стандартными методами химии пептидов [см, например,. М. Bodanszky, Principles of Peptide Synthesis, Springer-Verlag, Berlin, 1993; М. Bodanszky and A. Bodanszky, The Practice of Peptide Synthesis, Springer-Verlag, Berlin, 1984; H.-D. Jakubke и Н. Jeschkeit, Аминозаигеп, Peptide, Proteine, Verlag Chemie, Weinheim, 19 82].
Примерами инертных растворителей для этих реакций связывания являются эфиры, такие как диэтиловый эфир, дипропиловый эфир, отрети-бутилметиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан или бис(2-метоксиэтил) эфир, углеводороды, такие как бензол, толуол, ксилен, пентан, гексан, цептан, циклогексан или нефтяные фракции, галогенированные углеводороды, такие как дихлорметан, трихлорметан, тетрахлорметан, 1,2-дихлорэтан, трихлорэтилен или хлорбензол, или диполярные апротонные растворителия, такие как ацетон, метил этил-кетон, ацетонитрил, этилацетат, пиридин, диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФ), N,N-диметилацетамид (DMA), N,N'-диметилпропиленмочевина (DMPU) или N-метилпирролидинон (NMP). Также можно использовать смеси этих растворителей. Предпочтение отдается применению N,N-диметилформамида.
Примеры подходящих активирующих/конденсирующих агентов для этих реакций связывания включают карбодиимиды, такие как N,N'-диэтил-, N,N'-дипропил-, N,N'-диизопропил-, N,N'-дициклогексилкарбодиимид (DCC) или N-(3-диметиламиноизопропил)-N-этилкарбодиимид гидрохлорид (EDC), производные фосгена, такие как N,N'-карбонилдиимидазол (CDI) или изобутил хлорформат, соединения 1,2-оксазола, такие как 2-этил-5-фенил-1,2-оксазола 3-сульфат или 2-отреот-бутил-5-метилизоксазоле перхлорат, ациламино-соединения, такие как 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин, соединения фосфора, такие как пропанфосфорный ангидрид, диэтилцианофосфонат, бис(2-оксо-3-оксазолидинл)фосфорил хлорид, бензотриазол-1-илокситрис(диметиламино)фосфония гексафторфосфонат или бензотриазол-1-ил-окситрис(пирролидино)фосфония гексафторфосфонат (РуВОР), или соединения урония, такие как O-(бензотриазол-1-ил)-N,N,N',N'-метраметилурония тетрафторборат (TBTU), O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфонат (HBTU), 2-(2-оксо-1-(2H)-пиридил)-1,1,3,3-тетраметилурония тетрафторборат (TPTU), (9-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфонат (HATU) или O-(1Н-6-хлоробензотриазол-1-ил)-1,1,3,3-тетраметилурония-тетрафторборат (TCTU), возможно в комбинации с другими вспомогательными соединениями, такими как 1-гидроксибензотриазол (HOBt) или N-гидроксисукцинимид (HOSu), а также, в качестве оснований, карбонаты щелочных металлов, например, карбонат натрия или калия, или основания третичных аминов, такие как триэтиламин, N-метилморфолин, N-метилпиперидин, N,N-диизопропилэтиламина, пиридин или 4-N,N-диметиламинопиридин.
В контексте настоящего изобретения в качестве активирующих агентов/агентов конденсации для таких реакций связывания предпочтительно использовать N-(3-диметиламиноизопропил)-N'-этилкарбодиимид гидрохлорид (EDC) в комбинации с 1-гидроксибензотриазолом (HOBt) и N,N-диизопропилэтиламином или O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфонат (HATU), также в комбинации с N,N-диизопропилэтиламином.
Реакции связывания (IX)+(X)→(II-С), (XII-А) или (XII-В)+(X)→(II-D-A) или (II-D-B), (IX)+(XIII)→(XIV), (IX)+(XV)→(XVI) и (XXII)+(XXIII)→(II-D) обычно осуществляют в температурном диапазоне от -20°С до +60°С, предпочтительно от 0°С до+40°С. Реакции могут протекать при атмосферном, повышенном или пониженном давлении (например, от 0,5 до 5 бар); обычно работают при атмосферном давлении.
Реакции этерификации (IX)+(XVIII)→(XII) и (IX)+(XI-A) или (XI-B)→(XII-А) или (XII-В), (IX)+(XXIV)→(XXV), а также (DC)+(XXI)→(XXII) протекают аналогично описанным выше реакциям амидного связывания. Эти реакции осуществляют предпочтительно в дихлорметане с использованием N-(3-диметиламиноизопропил)-N'-этилкарбодиимид гидрохлорида (EDC) и 4-диметиламино-пиридин при температуре от +50°С до 100°С при атмосферном давлении.
В соединениях могут присутствовать функциональные группы, такие как амино-, гидроксильная и карбоксильная группы, в частности, могут также присутствовать во временно защищенной форме на протяжении описанных выше этапов, если это полезно или необходимо. В этих случаях такие защитные группы вводят и удаляют в соответствии в обычными методами, известными из области химии пептидов [см., например, T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 1999; M. Bodanszky and A. Bodanszky, The Practice of Peptide Synthesis, Springer-Verlag, Berlin, 1984]. Если присутствуют две или более защитных групп, они могут быть снова высвобождены, возможно одновременно в одной реакции, или могут высвобождаться на разных этапах реакции.
В качестве амино-защитной группы PG' предпочтительно использовать трет-бутоксикарбонил (Вое), бензилоксикарбонил (Z) или (9H-флуорен-9-илметокси)карбонил (Fmoc); для гидроксильной или карбоксильной функциональных групп предпочтительно использовать трет-бутил или бензил в качестве защитной группы PG2. Удаление трет-бутиловой или трет-бутоксикарбониловой группы обычно осуществляют путем обработки сильной кислотой такой как хлороводородная, бромоводородная или трифторуксусная кислота, в инертном растворителе, таком ка диэтиловый эфир, 1,4-диоксан, дихлорметан или уксусная кислота; эту реакцию также можно осуществить без добавления инертного растворителя. В случае использования бензила или бензилоксикарбонила в качестве защитной группы, эту группу удаляют предпочтительно путем гидролиза в присутствии подходящего палладиевого катализатора, такого как, например, палладий на активированном угле. (9H-флуорен-9-илметокси)карбонильную группу обычно удаляют с использованием вторичного аминного основания, такого как диэтиламин или пиперидин.
Реакция (VI)→(II-А) протекает в растворителе, который инертен в условиях данной реакции, таком как например, эфиры, такие как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан или бис(2-метоксиэтиловый) эфир, спирты, такие как этанол, метанол, изопропанол, n-бутанол или трет-бутанол, или диполярные апротонные растворители, такие как ацетон, метилэтиловый кетон, ацетонитрил, этилацетат, пиридин, диметилсульфоксид (ДМСО), N-диметилформамид (ДМФ), N-диметилацетамид (DMA), N,N-диметилпропиленмочевина (DMPU) или N-метилпирролидинон (NMP), или вода. Также можно использовать смеси этих растворителей. Предпочтение отдается использованию смеси 1,4-диоксана и воды.
Подходящими основаниями для реакции (VI)→(II-А) являются, например, карбонаты щелочных металлов, такие как карбонат калия, карбонат натрия или карбонат лития, гидрокарбонаты щелочных металлов, такие как гидрокарбонат калия или лития, или алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия или трет-бутоксид калия. Предпочтительно использовать гидрокарбонат натрия.
Реакция (VI)→(II-A) протекает в диапазоне температур от 0°С до 50°С, предпочтительно от 10°С до 30°С. еакция может протекать при атмосферном, повышенном или пониженном давлении (например, от 0,5 до 5 бар); обычно работают при атмосферном давлении.
Реакция (VI)+(VII)→(II-В) протекает в растворителе, который инертен в условиях данной реакции, таком как, например, эфиры, такие как тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан или бис(2-метоксиэтил)овый эфир, спирты, такие как метанол, этанол, изопропанол, n-бутанол или трет-бутаиол, или диполярные апротонные растворители, такие как ацетон, метил-этиловый кетон, ацетонитрил, этилацетат, пиридин, диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФ), N-диметилацетамид (DMA), N,N-диметилпропиленмочевина (DMPU) или N-метилпирролидинон (NMP) или вода. Также можно использовать смеси этих растворителей. Предпочтение отдается использованию ДМФ.
Подходящими основаниями для реакции (VI)+(VII)→(II-В) являются, например, основания третичных аминов, такие как триэтиламин, N-метилморфолин, N-метилпиперидин, N-диизопропилэтиламина, пиридин или 4-N,N-диметиламинопиридин. Предпочтение отдается использованию N,N-диизопропилэтиламина.
Реакция (VI)+(VII)→(II-В) протекает в диапазоне температур от 0°С до +50°С, предпочтительно от +10°С до +30°С.Реакция может протекать при атмосферном, повышенном или пониженном давлении (например, от 0,5 до 5 бар); обычно работают при атмосферном давлении.
Реакции (IX)→(III-А), (XIV)→(III-В) и (XVI)→(III-С), а также (VI)+(XVII)→(III-D), (XIX)+(XX)→(III-Е) и (XXV)+(XX)→(III-F), протекают в растворителе, который инертен в условиях данной реакции. Примерами подходящих растворителей являются эфиры, такие как диэтиловый эфир, дипропиловый эфир, трет-бутилметиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан или бис(2-метоксиэтил)овый эфир, углеводороды, такие как бензол, толуол, ксилен, пентан, гексан, цептан, циклогексан или фракции нефти, галогенированные углеводороды, такие как дихлорметан, трихлорметан, тетрахлорметан, 1,2-дихлорэтан, трихлорэтилен или хлорбензол, или диполярные апротонные растворители, такие как ацетон, метил-этиловый кетон, ацетонитрил, этилацетат, пиридин, диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФ), N,N-диметилацетамид (DMA), N,N'-диметилпропиленмочевина (DMPU) или N-метилпирролидинон (NMP). Также можно использовать смеси этих растворителей. Предпочтение отдается использованию N,N-диметилформамид.
Подходящими основаниями для этих реакций являются, например, третичные амины, такие как триэтиламин, N-метилморфолин, N-метилпиперидин, N,N-диизопропилэтиламина, пиридин или 4-N,N-диметиламинопиридин. Предпочтение отдается использованию N,N-диизопропилэтиламина, возможно с добавлением 4-N,N-диметиламинопиридина.
Реакции (IX)→(III-А), (XIV)→(III-В) и (XVI)→(III-С), а также (VI)+(XVII)→(III-D) и (XIX)+(XX)→(III-Е) протекают в диапазоне температур от 0°С до +50°С, предпочтительно от +10°С до +30°С. Реакция может протекать при атмосферном, повышенном или пониженном давлении (например, от 0,5 до 5 бар); обычно работают при атмосферном давлении.
Соединения формул (II), (III), (I-A) и (I-B) являются подгруппами соединений формул (IIa), (IIIa), (Ia-А) и (Ia-В), соответственно, где R35 представляет собой метил. Получение соединений (IIa) и (IIIa) осуществляют аналогично получению соединения формул (II) и (III), как описано выше.
Описанные выше способы иллюстрирует представленные ниже примеры и схемы синтеза (Схемы 3-13, 18):

TFA = трифторуксусная кислота
[а): 1. Вода/диоксан, 1Н HCl, 100°С; 2. H2, Pd/C, метанол, комнатная температура; b): NaHCO3, H2O, диоксан, комнатная температура].

[а): Диизопропилэтиламин, ДМФ, комнатная температура].

TFA = трифторуксусная кислота
[а): Вода/диоксан, 1Н HCl, 100°С; b): HATU, диизопропиламин, ДМФ, комнатная температура].
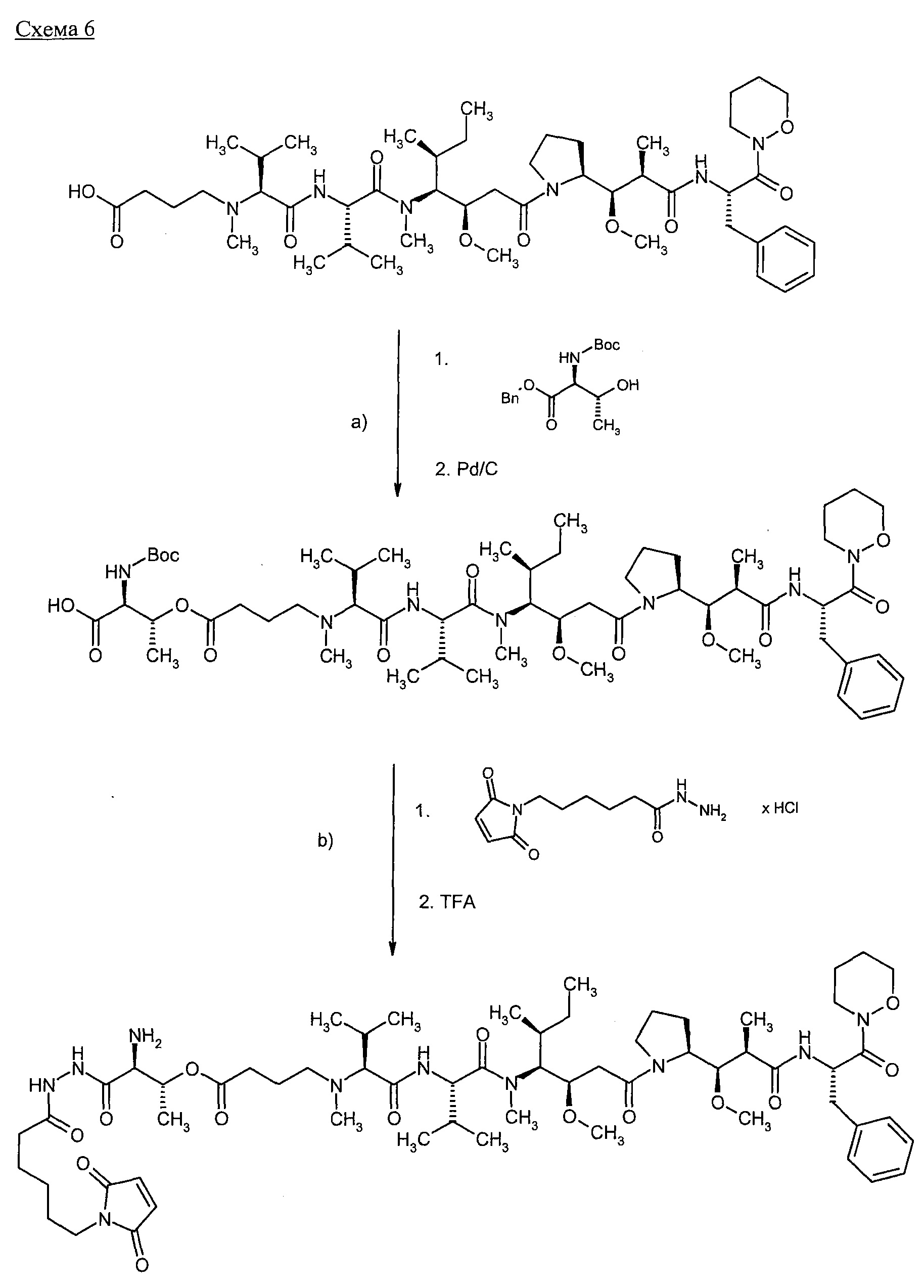
TFA = трифторуксусная кислота
[а): 1. EDCI, DMAP, дихлорметан, комнатная температура; 2. Н2, метанол, комнатная температура; b): I. EDCI, HOBt, диизопропиламин, ДМФ, комнатная температура; 2. дихлорметан, комнатная температура].

[а): 1. EDCI, DMAP, дихлорметан, комнатная температура; 2. Н2, метанол, комнатная температура; b): HATU, диизопропиламин, ДМФ, комнатная температура].

[a): EDCI, дихлорметан, комнатная температура].

[a): HATU, диизопропилэтиламин, ДМФ, комнатная температура; 2. Н2, метанол, комнатная температура; b): EDCI, DMAP, дихлорметан, комнатная температура].

TFA = трифторуксусная кислота
[а): 1. HATU, диизопропилэтиламин, ДМФ, комнатная температура; 2. дихлорметан, комнатная температура; b): EDCI, DMAP, дихлорметан, комнатная температура].

[а): Диизопропилэтиламина, ДМФ, комнатная температура].

TFA = трифторуксусная кислота
[а): 1. EDCI, DMAP, дихлорметан, комнатная температура; 2. дихлорметан, комнатная температура; b): диизопропиламин, DMAP, дихлорметан, комнатная температура].

[a): DMAP, диизопропиламин, дихлорметан, комнатная температура].

TFA = трифторуксусная кислота
[а): 1. Вода/диоксан, 1Н HCl, 100°С; 2. H2, Pd/C, метанол, комнатная температура; b); HATU, диизопропилэтиламин, комнатная температура].
Соединения формулы (IV) могут быть получены из коммерчески доступных аминокислот в качестве структурных блоков или из известных из литературы (см., например, Pettit et al., Synthesis 1996, 719; Shioiri et al., Tetrahedron Lett. 1991, 32, 931; Shioiri et al., Tetrahedron 1993, 49, 1913; Koga et al., Tetrahedron Lett. 1991, 32, 2395; Vidal et al., Tetrahedron 2004, 60, 9715; Poncet et al., Tetrahedron 1994, 50, 5345. Pettit et al., J. Org. Chem. 1994, 59, 1796) аналогично со способом, известным из литературы, в соответствии с обычными методами химии пептидов, как описано ниже в экспериментальном разделе. Приведенные ниже схемы синтеза (Схемы 14 -16) иллюстрируют процесс получения примерами.

[а): Гидроксиламин гидрохлорид, КОН, МеОН, 0°С → комнатная температура; b): BrCH2(СН2)2CH2Br, K2CO3, ацетон, нагревание с обратным холодильником].

[а): 1. Диизопропилэтиламин, ВЕР, дихлорметан, -10°С → комнатная температура; 2. МеОН].

ТРА = трифторуксусная кислота
[а): 1. Диизопропилэтиламин, ВЕР, ДМФ, комнатная температура; 2. дихлорметан; b): 1. HATU, Диизопропилэтиламин, ДМФ, комнатная температура; 2. дихлорметан, комнатная температура; с): 1. HATU, Диизопропилэтиламин, ДМФ, комнатная температура; 2. пиперидин, ДМФ, комнатная температура].
Соединения формул (XI), (XIII), (XV), (XVII) и (XXI), включая, где это возможно, их хиральные и диастереомерные формы, доступны для приобретения или описаны в литературе, либо они могут быть получены способами, очевидными для специалиста, по аналогии с методами, описанными в литературе. Многочисленные подробные инструкции, а также литературная информация по получению исходных материалов также приведена в экспериментальном разделе, в разделе, относящемся к получению исходных соединений и промежуточных соединений (интермедиатов).
Соединения формул (V), (VII), (VIII), (X), (XVIII), (XX) и (ХХШ), включая, где это возможно, их хиральные и диастереомерные формы, доступны для приобретения или описаны в литературе, либо они могут быть получены способами, очевидными для специалиста, по аналогии с методами, описанными в литературе. Многочисленные подробные инструкции, а также литературная информация по получению исходных материалов также приведена в экспериментальном разделе, в разделе, относящемся к получению исходных соединений и промежуточных соединений (интермедиатов).
В качестве альтернативы, отдельные этапы последовательности получения могут быть осуществлены в другом порядке. Это подход иллюстрируют примеры в приведенных ниже схемах синтеза (Схемы 17, 19 и 20).

ТРА = трифторуксусная кислота
[а): Комплекс боран-пиридин, уксусная кислота, МеОН; b): I. HOBt, EDCI, диизопропилэтиламин, ДМФ, комнатная температура; 2. TFA, дихлорметан, комнатная температура; с): HATU, диизопропилэтиламин, ДМФ, комнатная температура; d): 1. Pd/C, МеОН, комнатная температура; 2. NaHCO3, диоксан, вода].

TFA-трифторуксусная кислота
[а): 1. HATU, диизопропилэтиламин, ДМФ, комнатная температура; 2. TFA, дихлорметан, комнатная температура; 3. ((Н3С)3С(С=O))2O, ДМФ, диизопропилэтиламин; b): диизопропилэтиламин, ВЕР, ДМФ, комнатная температура; с): 1. H2, Pd/C (10%), метанол, комнатная температура; 2. HATU, диизопропилэтиламин, ДМФ, комнатная температура; 3. TFA, дихлорметан, комнатная температура].

[а): Комплекс боран-пиридин, уксусная кислота, МеОН; b): 1. HOBt, EDCI, диизопропилэтиламин, ДМФ, комнатная температура; 2. TFA, дихлорметан, комнатная температура; с): 1. H2, Pd/C, МеОН, комнатная температура; 2. NaHCO3, диоксан, вода; d): HATU, диизопропилэтиламин, ДМФ, комнатная температура;].
В одном варианте реализации связывающее соединение связывается с молекулой-мишенью, которая расположена на раковой клетке. В предпочтительном варианте реализации связывающее соединение связывается с раковой молекулой-мишенью.
В другом предпочтительном варианте реализации молекула - мишень представляет собой селективную раковую молекулу-мишень.
В одном особенно предпочтительном варианте реализации молекула-мишень представляет собой белок.
В одном варианте реализации молекула-мишень представляет собой внеклеточную молекулу-мишень. В предпочтительном варианте реализации внеклеточная молекула-мишень представляет собой белок.
Раковые молекулы-мишени известны специалистам. Их примеры приведены ниже. Ниже приведены примеры раковых молекул-мишеней:
(1) Рецептор эпидермального фактора роста EGF (ссылочный номер последовательности в NCBINP_005219.2)
Последовательность (1210 аминокислот):

Внеклеточный домен выделен подчеркиванием.
(2) Мезотелин (номер в базе SwissProt Q 13421-3)
Последовательность (622 аминокислот):

Мезотелин кодируется аминокислотами 296-598. Аминокислоты 37-286 кодируют "фактор потенцирования мегакариоцитов". Мезотелин удерживается в клеточной мембране якорем GPI и располагается во вне клетки.
(3) Карбоангидраза IX (номер в базе SwissProt Q16790) Последовательность (459 аминокислот):

Внеклеточный домен выделен подчеркиванием.
(4) С4.4а (номер в базе NCBINP_055215.2; Синоним LYPD3)
Последовательность (346 аминокислот):

Зрелый внеклеточный домен выделен подчеркиванием (SEQ ID NO:1). (5) CD52 (номер в базе NCBINP_001794,2)

(6) Her2 (номер в базе NCBI NP_004439,2)

(7) CD20 (номер в базе NCBI NP_068769,2)

(8) Лимфоцит-активирующий антиген CD30 (Идентификационный номер в базе Swiss Prot: Р28908)

(9) Молекула адгезии лимфоцитов CD22 (Идентификационный номер в базе Swiss Prot: Р20273)

(10) поверхностный антиген CD33 миелоидных клеток (Идентификационный номер в базе Swiss Prot: P20138)

(11) Трансмембранный гликопротеин NMB (Идентификационный номер в базе Swiss Prot: Q14956)

(12) Молекула адгезии CD56 (Идентификационный номер в базе Swiss Prot: P13591)


(13) Поверхностная молекула CD70 (Идентификационный номер в базе Swiss Prot: P32970)

(14) Поверхностная молекула CD74 (Идентификационный номер в базе Swiss Prot: P04233)

(15) Антиген В-лимфоцитов CD19 (Идентификационный номер в базе Swiss Prot: P15391)

(16) Поверхностный белок муцин-1 (Идентификационный номер в базе Swiss Prot: P15941)

(17) Поверхностный белок CD138 (Идентификационный номер в базе Swiss Prot: P18827)

(18) Интегрин альфа V (Номер доступа в базе Genbank: NP_002201,1)

(19) Белок фактора роста 1, выделенный из тератокарциномы 1, TDGF1 (Номер доступа в базе Genbank: NP_003203,1)

(20) Простат-специфический мембранный антиген PSMA (Идентификационный номер в базе Swiss Prot: Q04609)


(21) Тирозин-протеинкиназа ЕРНА2 (Идентификационный номер в базе Swiss Prot: P29317)

(22) Поверхностный белок SLC44A4 (№доступа в базе Genbank: NP_001171515)

(23) Поверхностный белок BMPR1B (SwissProt: 000238)
(24) Транспортный белок SLC7A5 (SwissProt: Q01650)
(25) Антиген эпителия простаты STEAP1 (SwissProt: Q9UHE8)
(26) Антиген карциномы яичника MUC16 (SwissProt: Q8WXI7)
(27) Транспортный белок SLC34A2 (SwissProt: 095436)
(28) Поверхностный белок SEMA5b (SwissProt: Q9P283)
(29) Поверхностный белок LYPD1 (SwissProt: Q8N2G4)
(30) Рецептор В эндотелиного ranaEDNRB (SwissProt: P24530)
(31) Белок с доменом типа кольцо-палец RNF43 (SwissProt: Q68DV7)
(32) Белок, ассоциированный с карциномой простаты STEAP2 (SwissProt: Q8NFT2)
(33) Катионный канал TRPM4 (SwissProt: Q8TD43)
(34) Рецептор комплемента CD21 (SwissProt: P20023)
(35) Белок, ассоциированный с комплексом антигенных рецепторов В-клеток CD79b (SwissProt: P40259)
(36) Антиген клеточной адгезии СЕАСАМ6 (SwissProt: P40199)
(37) Дипептидаза DPEP1 (SwissProt: P16444)
(38) Рецептор интерлейкина ЕЬ20Кальфа (SwissProt: Q9UHF4)
(39) Протеогликан BCAN (SwissProt: Q96GW7)
(40) Рецептор эфрина ЕРНВ2 (SwissProt: P29323)
(41) Белок, ассоциированный со стволовыми клетками простаты PSCA (№доступа в базе Genbank: NP_005663,2)
(42) Поверхностный белок LHFPL3 (SwissProt: Q86UP9)
(43) Рецепторный белок TNFRSF13C (SwissProt: Q96RJ3)
(44) Белок, ассоциированный с комплексом антигенных рецепторов В-клеток CD79a (SwissProt: PI 1912)
(45) Рецепторный белок CXCR5 (SwissProt: P32302)
(46) Ионный канал Р2Х5 (SwissProt: Q93086)
(47) Антиген лимфоцитов CD180 (SwissProt: Q99467)
(48) Рецепторный белок FCRL1 (SwissProt: Q96LA6)
(49) Рецепторный белок FCRL5 (SwissProt: Q96RD9)
(50) антиген Ia ГКГС класса Ia HLA-DOB (№ доступа в базе Genbank: NP_002111,1)
(51) Т-клеточный белок VTCN1 (SwissProt: Q7Z7D3).
В одном предпочтительном варианте реализации изобретения раковая молекула-мишень выбрана из группы, состоящей из раковых молекул-мишеней consisting of the cancer молекула-мишень (1)-(51).
В другом особенно предпочтительном варианте реализации настоящего изобретения связывающее соединение связывается с внеклеточной раковой молекулой-мишенью, которая выбрана из группы, состоящей из раковых молекул-мишеней (1)-(51).
В другом особенно предпочтительном варианте реализации настоящего изобретения связывающее соединение специфично связывается с внеклеточной раковой молекулой-мишенью, выбранной из группы, состоящей из раковых молекул-мишеней (1)-(51).
В одном особенно предпочтительном варианте реализации изобретения раковая молекула-мишень выбрана из группы, состоящей из рецептора эпидермального фактора рост (EGF) (NP_005219.2), мезотелина (Q13421-3), С4.4а (NP_055215.2) и карбоангидразы IX (СА IX; NP_001207,2), более конкретно, С4.4а (NP_055215.2).
В другом особенно предпочтительном варианте реализации настоящего изобретения связывающее соединение связывается с внеклеточной раковой молекулой-мишенью, выбранной из группы, состоящей из рецептора эпидермального фактора рост (EGF) (NP_005219.2), мезотелина (Q13421-3), С4.4а (NP_055215.2) и карбоангидразы DC (СА IX; Q16790)), более конкретно С4.4а (NP_055215.2).
В другом особенно предпочтительном варианте реализации настоящего изобретения связывающее соединение специфично связывается с внеклеточной раковой молекулой-мишенью, выбранной из группы, состоящей из рецептора эпидермального фактора рост (EGF) (NP_005219.2), мезотелина (Q13421-3), С4.4а (NPJ355215.2) и карбоангидразы IX (СА IX; Q16790)), более конкретно С4.4а (NP_055215.2).
В предпочтительном варианте реализации связывающее соединение после связывания со своей внеклеточной молекулой-мишенью на клетке-мишени интернализуется клеткой-мишенью в результате связывания. В результате этого конъюгат связывающее соединение -активное соединение, которое может представлять собой иммуноконъюгат или ADC, поглощается клеткой-мишенью.
В одном варианте реализации связывающее соединение представляет собой связывающий белок. В предпочтительном варианте реализации связывающее соединение представляет собой антитело, антигенсвязывающий фрагмент антитела, мультиспецифичное антитело или миметик антитела.
Предпочтительными миметиками антител являются аффктины, аднектины, антикалины DARPin, авимеры или нанотела. Предпочтительными мультиспецифичными антителами являются биспецифичные антитела и триспецифичные антитела.
В предпочтительном варианте реализации связывающее соединение представляет собой антитело или антигенсвязывающий фрагмент антитела, более предпочтительно выделенное антитело или выделенный антигенсвязывающий фрагмент антитела.
Предпочтительными антигенсвязывающими фрагментами антител являются фрагменты Fab, Fab', F(ab')2 и Fv, диатела, доменные антитела (Dab), линейные антитела и одноцепочечные антитела (scFv). Особенно предпочтительны Fab, диатела и scFv.
В одном особенно предпочтительном варианте реализации связывающее соединение представляет собой антитело. Особенно предпочтительны моноклональные антитела или антигенсвязывающие фрагменты антител. Более конкретно, предпочтительны человеческие, гуманизированные или химерные антитела или антигенсвязывающие фрагменты антител.
Антитела или антигенсвязывающие фрагменты антител, которые связывают раковые молекулы-мишени, могут быть получены специалистом с использованием известных методов, таких как, например, химический синтез или рекомбинантная экспрессия. Соединения, связывающие раковые молекулы-мишени, могут быть приобретены в коммерческих источниках или приготовлены средним специалистом в данной области с использованием известных методов, таких как, например, химический синтез или рекомбинантная экспрессия. Дополнительные способы получения антител или антигенсвязывающих фрагментов антител описаны в WO 2007/070538 (см. стр.22 "Antibodies"). Специалисту известно как модифицировать такие методы как библиотека на фаговом дисплее (например, Morphosys HuCAL Gold) и применять для обнаружения антител или антигенсвязывающих фрагментов антител (см. WO 2007/070538, стр.24 и Пример 1 на странице 70, Пример 2 на странице 72). Другое способы получения антител с использованием ДНК-библиотек из В-клеток описаны, например, на странице 26 (WO 2007/070538). Способы гуманизации антител описаны на страницах 30-32 в W02007070538 и подробно в источнике Queen, et al., Pros. Natl. Acad. Sci. USA 86:10029-10033, 1989 или в WO 90/0786. Далее, способы рекомбинантной экспрессии булков в целом и антител в частности известны специалисту (см., например, in Berger and Kimmel (Guide to Molecular Cloning Techniques, Methods in Enzymology, Vol.152, Academic Press, Inc.); Sambrook, et al., (Molecular Cloning: A Laboratory Manual, (Second Edition, Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.; 1989) Vol.1-3); Current Protocols in Molecular Biolony, (F.M. Ausabel et al. [Eds.], Current Protocols, Green Publishing Associates, hic./John Wiley & Sons, Inc.); Harlow et al., (Monoclonal Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press (19881, Paul [Ed.]); Fundamental Immunology, (Lippincott Williams & Wilkins (1998)); и Harlow, et al., (Using Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press (1998)). Специалисту известны соответствующие векторы, промоторы и сигнальные пептилы, которые необходимы для экспрессии белка/антитела. Обычные способы описаны также в WO 2007/070538 на страницах 41-45. Способы получения антитела описаны, например, в WO 2007/070538 в Примере 6 на странице 74 ff. Processes, обеспечивающие определение интернализации антитела после связывания с соответствующим антитгеномизвестны специалисту и описаны, например, в WO 2007/070538 на странице 80. Специалист сможет применить способы, описанные в WO 2007/070538, использующиеся для получения антител к карбоангидразе IX (Mn) по аналогии с получением антител, специфичным к различным молекулам-мишеням.
Антитела к EGFR (рецептору эпидермального фактора роста)
Примерами антител, которые свзывают раковую молекулу-мишень EGFR являются цетуксимаб (номер INN 7906), панитумумаб (номер INN 8499) и нимотузумаб (номер INN 8545). Цетуксимаб (номер доступа в базе Drug DB00002) представляет собой химерное анти-EGFR1 антитело, продуцируемое клетками SP2/0 миеломы мыши и продаваемое компаниями ImClone Systems Inc/Merck KgaA/Bristol-Myers Squibb Co. Цетуксимаб назначают для лечения метастазирующей колоректальной карциномы, экспрессирующей EGFR, с диким типом гена K-Ras. Он обладает аффинностью 10-10 M.
Последовательность:
Легкая цепь Цетуксимаба (каппа):

Тяжелая цепь Цетуксимаба:

Панитумумаб (номер INN 8499) (Drug Bank Accession Number DB01269) представляет собой рекомбинантное человеческое антитело IgG2, которое специфично связывается с человеческим рецептором 1 эпидермального фактора роста и которое продает компания Abgenix/Amgen. Панитумумаб получают путем иммунизации трансгенных мышей (XenoMouse). Эти мыши обладают способностью продуцировать человеческий иммуноглобулин (легкую и тяжелую цепи). Был отобран специфичный клон В-клеток, который продуцирует антитела к EGFR, и этот клон иммортализовали с клетками СНО (клетки яичника китайского хомячка). Сейчас эти клетки используются для продукции на 100% человеческого антитела. Панитумумаб назначают при лечении экспрессирующей EGFR метастазирующей колоректальной карциномы, устойчивой к химиотерапевтическому лечению фторпиримидином, оксалиплатином и иринотеканом. Его аффинность составляет 10-11 М.
Последовательность:
Легкая цепь панитумумаба (каппа);

Тяжелая цепь панитумумаба:

Нимотузумаб (номер INN 8545) (ЕР 00586002, ЕР 00712863) представляет собой гуманизирванное IgGI, которое специфично связывается с человеческим рецептором 1 эпидермального фактора роста и которое продает компания YM BioScienecs Inc. (Mississauga Canada). Оно продуцируется несекретирующими клетками NSO (линия клеток человека). Нимотузумаб одобрен для лечения опухолей головы и шеи, высоко злокачественной астроцитомы и полиморфной глиобластомы (не в Евросоюзе и США) и карциномы поджелудочной железы (орфанное лекарственное средство, ЕМА). Его аффинность составляет 10-8 М.
Ниже приведены другие варианты антител к EGFR:
- Залутумумаб / 2F8 / HuMax-EGFr, распространяемый компанией Genmab A/S (WO 02/100348, WO 2004/056847, номер INN 8605)
- Нектимумаб / 11F8, ImClone / IMC-11F8, от компании ImClone Systems Inc. [Eli Lilly & Co] (WO 2005/090407 (ЕР 01735348-А1, US 2007/0264253-A1, US 7,598,350, WO 2005/090407-A1), номер INN 9083)
- Матузумаб / анти-EGFR MAT, Merck KGaA / анти-EGFR MAT, Takeda / EMD 72000 / EMD-6200 / EMD-72000 и EMD-55900 / MAb 425 / моноклоналыюе антитело 425, распространяемое компаниями Merck KGaA / Takeda (WO 92/15683, номер INN 8103 (Matuzumab))
- RG-7160 / GA-201 / GA201 / R-7160 / R7160 / RG7160 / RO-4858696 / RO-5083945 / R04858696 / R05083945, от Glycart Biotechnology AG (Roche Holding AG) (WO 2010/112413-A1, WO 2010/115554)
- GT-MAB 5,2-GEX / CetuGEX, от Glycotope GmbH (WO 2008/028686-A2 (EP 01900750-A1, EP 01911766-A1, EP 02073842-A2, US 2010/0028947-A1)
- ISU-101, от Isu Abxis Inc (ISU Chemical Co Ltd) / Scancell (WO 2008/004834-A1)
- ABT-806 / mAb-806 / ch-806 / анти-EGFR моноклональное антитело 806, от Института исследований в области рака Людвига (Ludwig Institute for Cancer Research) / Abbott / Life Science Pharmaceuticals (WO 02/092771, WO 2005/081854 и WO 2009/023265)
- SYM-004 (состоит из двух химерных антител IgG1 (992 и 1024)), распространяется компанией SymphogenA/S (WO 2010/022736-A2)
- MR1-1 /MR1-1KDEL, распространяется компанией IVAX Corp (Teva Pharmaceutical Industries Ltd) (Duke University), (Patent: W02001/062931 - A2)
- Антитело против мутантного варианта с делецией EGFRvin, распространяется компанией Amgen/Abgenix (WO 2005/010151, US 7,628,986)
- SC-100, распространяется компанией Scancell Ltd (WO 01/088138-A1)
- MDX-447 / EMD 82633 / BAB-447 / H 447 / MAb, EGFR, Medarex/Merck KgaA, распространяется компанией Bristol-Myers Squibb (US) / Merck KGaA (DE) / Takeda (JP), (WO 91/05871, WO 92/15683)
- Анти-EGFR-MAT, распространяется компанией Xencor (WO 2005/056606)
- DXL-1218 / анти-EGFR моноклональное антитело (рак), InNexus, распространяется компанией hiNexus Biotechnology Inc., Pharmaprojects PH048638
В предпочтительном варианте реализации анти-EGFR антитела выбраны из группы, состоящей из цетуксимаба, панитумумаба, нимотузумаба, залутумумаба, нектимумаба, матузумаба, RG-716, GT-MAB 5,2-GEX, ISU-101, ABT-806, SYM-004, MR1-1, SC-100, MDX-447 и DXL-1218.
В одном особенно предпочтительном варианте реализации -EGFR антитела выбраны из группы, состоящей из цетуксимаба, панитумумаба, нимотузумаба, залутумумаба, нектимумаба и матузумаба.
Специалисту известны способа получения других антител из участков CDR указанных выше антител посредством изменения последовательности, чтобы эти дргие антитела обладали близкой или повышенной аффинностью и/или специфичностью в отношении молекулы-мишени.
В другом варианте реализации анти-EGFR антитела или антигенсвязывающие фрагменты антител выбраны из группы, состоящей из:
антитела или антигенсвязывающие фрагмента антитела, содержащего три участка CDR легкой цепи и три участка CDR тяжелой цепи одного из следующих антител: цетуксимаб, панитумумаб, нимотузумаб, залатумумаб, нектимумаб, матузумаб, RG-716, GT-MAB 5,2-GEX, ISU-101, ABT-806, SYM-004, MR1-1, SC-100, MDX-447 и DXL-1218.
В другом варианте реализации анти-EGFR антитела или антигенсвязывающие фрагменты антител выбраны из группы, состоящей из
антител или антигенсвязывающих фрагментов антител, содержащих три участка CDR легкой цепи три участка CDR тяжелой цепи одного из следующих антител: цетуксимаб, панитумумаб, нимотузумаб, залутумумаб, нектимумаб, матузумаб.
Антитела к карбоангидразе IX
Примерами антител, которые связывают раковую молекулу-мишень карбоангидразу DC являются антитела, описанные в WO 2007/070538-A2 (например, пункты формулы изобретения 1-16).
В предпочтительном варианте реализации антитела к карбоангидразе DC или их антигенсвязывающие фрагменты выбраны из группы, состоящей из следующих антител к карбоангидразе DC антитела или антигенсвязывающие фрагменты антител 3ее9 (пункт формулы изобретения 4 (а) в WO 2007/070538-A2), 3ef2 (пункт формулы изобретения 4 (b) в WO 2007/070538-A2), 1е4 (пункт формулы изобретения 4 (с) в WO 2007/070538-A2), 3а4 (пункт формулы изобретения 4 (d) в WO 2007/070538-A2), 3ab4 (пункт формулы изобретения 4 (е) в WO 2007/070538-A2), 3ар10 (пункт формулы изобретения 4 (f) в WO 2007/070538-A2), 3bb2 (пункт формулы изобретения 4 (g) в WO 2007/070538-A2), 1aa1 (пункт формулы изобретения 4 (h) в WO 2007/070538-A2), 5а6 (пункт формулы изобретения 4 (i) в WO 2007/070538-A2) и 5аа3 (пункт формулы изобретения 4 (j) в WO 2007/070538-A2).
В предпочтительном варианте реализации антитела к карбоангидразе IX или их антигенсвязывающие фрагменты выбраны из группы, состоящей из следующих:
антитела к карбоангидразе IX или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 3ее9 (из источника WO 2007/070538-A2),
антитела к карбоангидразе IX или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 3ef2 (из источника WO 2007/070538-A2),
антитела к карбоангидразе ГХ или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 1е4 (из источника WO 2007/070538-A2),
антитела к карбоангидразе DC или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 3а4 (из источника WO 2007/070538-A2),
антитела к карбоангидразе DC или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 3ab4 (из источника WO 2007/070538-A2),
антитела к карбоангидразе DC или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 3ah10 (из источника WO 2007/070538-A2),
антитела к карбоангидразе DC или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 3bb2 (из источника WO 2007/070538-A2),
антитела к карбоангидразе DC или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 1aa1 (из источника WO 2007/070538-A2),
антитела к карбоангидразе DC или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 5а6 (из источника WO 2007/070538-A2), and
антитела к карбоангидразе IX или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела 5аа3 (из источника WO 2007/070538-A2).
Указанные здесь последовательности участков CDR раскрыты на Фигурах 2а-2с, стр.128-130 в WO 2007/070538-A2.
В предпочтительном варианте реализации антитела к карбоангидразе IX или их антигенсвязывающие фрагменты выбраны из группы, состоящей из следующих:
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 3ее9, описанные в WO 2007/070538-A2 на Фигуре 4b на странице 137,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 3ef2, описанные в WO 2007/070538-A2 на Фигуре 4с на странице 138 и на Фигуре 4b на странице 137,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 1е4, описанные в WO 2007/070538-A2 на Фигуре 4а на странице 136,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 3а4, описанные в WO 2007/070538-A2 на Фигуре 4а на странице 136,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 3ab4, описанные в WO 2007/070538-A2 на Фигуре 4а на странице 136,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 3ah10, описанные в WO 2007/070538-A2 на Фигуре 4а на странице 136,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 3bb2, описанные в WO 2007/070538-A2 на Фигуре 4b на странице 137,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 1aa1, описанные в WO 2007/070538-A2 на Фигуре 4а на странице 136,
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабелыюй области тяжелой цепи антитела 5а6, описанные в WO 2007/070538-A2 на Фигуре 4b на странице 137, and
антитело или антигенсвязывающий фрагмент, который содержит аминокислотную последовательность вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела 5аа3, описанные в WO 2007/070538-A2 на Фигуре 4b на странице 137.
В одном особенно предпочтительном варианте реализации антитело к карбоангидразе IX представляет собой антитело 3ее9 из источника WO 2007/070538-A2.
В одном особенно предпочтительном варианте реализации антитело к карбоангидразе IX или антигенсвязывающий фрагмент содержит аминокислотные последовательности участков CDR вариабельной области тяжелой цепи 3ее9
(VH3-CDR1: GFTFSSYGMS; VH3-CDR2: GISSLGSTTYYADSVKG; VH3-CDR3: TGSPGTFMHGDH, см. Фигуру 2а, стр.128 в WO 2007070538-A2) и аминокислотные последовательности участков CDR вариабельной области легкой цепи антитела 3ее9 (VLk1-CDR1: RASQDINNYLS; VLk1-CDR2: YGASNLQS; VLk1-CDR3: QQYYGRPT, см. Фигуру 2b, стр.129 в WO 2007/070538-A2).
В одном особенно предпочтительном варианте реализации антитело к карбоангидразе IX или антигенсвязывающий фрагмент содержит аминокислотную последовательность вариабельной области тяжелой цепи антитела 3ее9
(VH3:ELVESGGGLVQPGGSLRLSCAASGFTFSSYGMSWVRQAPGKGLEWVSGISSLGSTT YYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARTGSPGTFMHGDHWGQGTL VTVSS, см. Фигуру 4b, стр.137 в WO 2007070538-A2) и аминокислотную последовательность вариабельной области легкой цепи антитела 3ее9
(VLk1:DIQMTQSPSSLSASVGDRVTITCRaSQDINNYLSWYQQKPGKAPKLLIYGASNLQSG VPSRFSGSGSGTDFTLTISSLQPEDFAVYYCQQYYGRPTTFGQGTKVEIKRT, см. Фигуру 4b, стр.137 в WO 2007070538-A2).
В предпочтительном варианте реализации антитело к карбоангидразе DC 3ее9 представляет собой антитело класса IgG.
В одном особенно предпочтительном варианте реализации антитело к карбоангидразе IX 3ее9 представляет собой антитело IgG1 (3ee9-IgG1),
где аминокислотная последовательность тяжелой цепи содержит следующую последовательность:

и аминокислотная последовательность легкой цепи содержит следующую последовательность:

Антитело к карбоангидразе DC 3ee9-IgG1:
Дальнейший вариант реализации настоящего изобретения относится к обеспечению антитела к карбоангидразе IX 3ee9-IgG1.
Антитела к С4.4а:
Связывающими соединениями, особенно предпочтительными в соответствии с настоящим изобретением являются анти-С4.4а антитела, более конкретно, человеческие и гуманизированные анти-С4.4а антитела. Антитела предпочтительно обладают аффинностью по меньшей мере 10-7 М (выраженной в форме значения Kd; другими словами, предпочтительными являются антитела со значениями Kd ниже 10-8 М), предпочтительно по меньшей мере 10-8 М, более предпочтительно в диапазоне от 10-9 - М до 10-11 М. Значения Kd могут быть определены, например посредством спектроскопии плазменного резонанса.
Конъюгаты связывающее соединение - активное соединение согласно настоящему изобретению также демонстрируют значение аффинности в этих диапазонах. На аффинность предпочтительно не оказывает существенного влияния конюгирования с активным соединением (обычно аффинность снижается менее чем на один порядок величины, другими словами, максимум с 10-8 М до 10-7 М).
Антитела, используемые в соответствии с настоящим изобретением также предпочтительно обладают высокой селективностью. Высокая селективность имеет место, если антитело согласно настоящему изобретению демонстрирует аффинность к белку-мишени, которая по меньшей мере в 2 раза, предпочтительно в 5 раз, или более предпочтительно в 10 раз выше аффинности в отношении другого независимого антигена, например сывороточного альбумина человека (аффинность может быть определена, например, посредством спектроскопии плазменного резонанса).
Далее, используемые антитела согласно настоящему изобретению предпочтительно являются кросс-реактивными. Для облегчения проведения и улучшения интерпретации доклинических испытаний, например, испытаний токсичности или активности (например, на мышах с ксенографтами), полезно, если антитело, используемое в соответствии с настоящим изобретением не только связывает человеческий белок-мишень, но также связывает соответствующий вариант белка-мишени у вида, используемого в исследованиях. В одном варианте реализации антитело, используемое в соответствии с настоящим изобретением, помимо человеческого белка-мишени, обладает кросс-реактивностью в отношении белка по меньшей мере одного другого вида. Для испытаний токсичности и активности предпочтительно использовать виды из семейств грызунов, собак и приматов, не являющихся человеком. Предпочтительными грызунами являются мыши и крысы. Предпочтительными приматами, не являющимися человеком, являются макаки резус, шимпанзе и длиннохвостые макаки.
В одном варианте реализации антитело, используемое в соответствии с настоящим изобретением, помимо человеческого белка-мишени, обладает кросс-реактивностью в отношении белка по меньшей мере одного другого вида, выбранного из группы, состоящей из мыши, крысы и длиннохвостой макаки (Масаса fascicularis). Особенно предпочтительными антителами для использования в соответствии с настоящим изобретением являются те, которые помимо человеческого белка-мишени, обладает кросс-реактивностью по меньшей мере в отношении белка мыши. Предпочтение отдается кросс-реактивым антителам, аффинность которых в отношении целевого белка другого вида, отличного от человека, отличается не более чем в 50 раз, более конкретно, не более чем в десять раз, от аффинности в отношении человеческого белка-мишени.
Анти-С4.4а антитела описаны, например, в WO 01/23553 или WO 2011070088. Эти антитела могут использоваться в соответствии с настоящим изобретением.
Примеры антител к С4.4а и их антигенсвязывающих фрагментов описаны ниже. Последовательности антител приведены в Таблице 1, причем каждая строка воспроизводит аминокислотные последовательности участков CDR вариабельной области легкой цепи и вариабельной области тяжелой цепи, соответственно, антитела, приведенного в столбце 1. Также приведены аминокислотные последовательностей вариабельной области легкой цепи и вариабельной области тяжелой цепи, и нуклеотидные последовательности антитела, указанного в столбце 1 в каждом случае.
В одном варианте реализации анти-С4.4а антитела или их антигенсвязывающие фрагменты антител связываются с доменом (положения аминокислот 1-85 в SEQ ID NO:1) молекулы С4.4а.
В одном варианте реализации анти-С4.4а антитела или их антигенсвязывающие фрагменты антител обладают кросс-реактивностью с человеческим С4.4а (SEQ ID NO:1) и мышиным С4.4а (SEQ ID NO:2).
В одном варианте реализации анти-С4.4а антитела или их антигенсвязывающие фрагменты антител после связывания с клетками, которые экспрессируют С4.4а, интернализуются в клетку.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител конкурируют с антителом М31-В01 и/или антителом M20-D02-S-A за связывание с С4.4а. Антитела М31-В01 и M20-D02-S-A конкурируют за связывание с С4.4а. Антитела В01-1 - В01-12 получены из М31-В01 методом созревания аффинности и конкурируют с М31-В01 за связывание с С4.4а. Антитела D02-1-D02-13 получены из M20-D02-S-A методом созревания аффинности и конкурируют с M20-D02-S-A за связывание с С4.4а.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат по меньшей мере одну, две или три аминокислотных последовательности CDR, приведенные в Таблице 1 или Таблице 2.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат по меньшей мере одну, две или три аминокислотных последовательности CDR антитела, приведенного в Таблице 1 или Таблице 2.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат по меньшей мере одну, две или три аминокислотных последовательности CDR вариабельной области легкой цепи и по меньшей мере одну, две или три аминокислотных последовательности CDR вариабельной области тяжелой цепи антитела, приведенного в Таблице 1 или Таблице 2.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат последовательности, которые по меньшей мере на 50%, 60%, 70%, 80%, 90% или 95% идентичны аминокислотным последовательностям CDR вариабельной области легкой цепи и аминокислотным последовательностям CDR вариабельной области тяжелой цепи, антитела, приведенного в Таблице 1 или Таблице 2.
В другом варианте реализации последовательности CDR анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат
последовательности CDR тяжелой цепи, соответствующие последовательностям SEQ ID NO:297 (CDR H1), SEQ ID NO:298 (CDR H2) и SEQ ID NO:299 (CDR Н3), и последовательности CDR легкой цепи, соответствующие последовательностям SEQ ID NO:300 (CDR L1), SEQ ID NO:22 (CDR L2) и SEQ ID NO:301 (CDR L3), или
последовательности CDR тяжелой цепи, соответствующие последовательностям SEQ ID NO:302 (CDR H1), SEQ ID NO:303 (CDR H2) и SEQ ID NO:304 (CDR НЗ), и последовательности CDR легкой цепи, соответствующие последовательностям SEQ ID NO:305 (CDR L1), SEQ ID NO:306 (CDR L2) и SEQ ID NO:307 (CDR L3).
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат последовательности, которые по меньшей мере на 50%, 60%, 70%, 80%, 90% или 95% идентичны аминокислотным последовательностям CDR вариабельной области легкой цепи и вариабельной области тяжелой цепи антитела, приведенного в Таблице 1 или Таблице 2.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат аминокислотные последовательности трех CDR вариабельной области легкой цепи и аминокислотные последовательности трех CDR вариабельной области тяжелой цепи антитела, приведенного в Таблице 1 или Таблице 2.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат вариабельную область легкой цепи и/или вариабельную область тяжелой цепи антитела, приведенного в Таблице 1 или Таблице 2.
В другом варианте реализации анти-С4.4а антитела или антигенсвязывающие фрагменты антител содержат вариабельную область легкой цепи и вариабельную область тяжелой цепи антитела, приведенного в Таблице 1 или Таблице 2.
В предпочтительном варианте реализации анти-Са.а антитела и антигенсвязывающие фрагменты антител выбраны из группы, состоящей из
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:75-77, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:78-80 (В01-10),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:5, 9 и 13, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:17, 21 и 25 (М31-В01),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:6, 10 и 14, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:18, 22 и 26 (M20-D02-S-A),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ Id NO:7, 11 и 15, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:19, 23 и 27 (M60-G03),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:8, 12 и 16, и которое содержи последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:20, 24 и 28 (36-Н02),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:45-47, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:48-50 (В01-3),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:55-57, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:58-60 (В01-5),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:65-67, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ IDNO:68-70 (В01-7),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:85-87, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ IDNO:88-90 (В01-12),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:95-97, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:98-100 (D02-4),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:105-107, и которое содержит последовательности CDR легкой цепи, представленные последовательностями SEQSEQ ID NO:108-110 (D02-6),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:115-117, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:118-120 (D02-7),
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:125-127, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:128-130 (D02-11), и
антитела, которое содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ID NO:135-137, и которое содержит последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ID NO:138-140 (D02-13).
В предпочтительном варианте реализации анти-Са.а антитела и антигенсвязывающие фрагменты антител выбраны из группы, состоящей из
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ Ш NO:81, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:82 (В01-7),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:33, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:29 (М31-В01),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:34, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:30 (M20-D02 S-A),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:35, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:31 (M60-G03),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:36, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:32 (М36-Н02),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:51, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:52 (В01-3),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:61, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:62 (В01-5),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:71, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:72 (В01-7)
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:91, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:92 (В01-12),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:101, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:102 (D02-4),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:111, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:112 (D02-6),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:121, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:122 (D02-7),
антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:131, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:132 (D02-11),
and антител, которые содержат аминокислотную последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:141, и которые содержат аминокислотную последовательность легкой цепи, представленную последовательностью SEQ ID NO:142 (D02-13).
В другом варианте реализации анти-С4.4а антитела содержат тяжелую цепь и легкую цепь антитела, приведенного в Таблице 2.
В предпочтительном варианте реализации анти-С4.4а антитела содержат тяжелую цепь и легкую цепь антитела, приведенного в Таблице 2.
В одном особенно предпочтительном варианте реализации антитело к С4.4а выбрано из группы, состоящей из
антитела, которое содержит аминокислотную последовательность легкой цепи, представленную SEQ ID NO:346, и которое содержит аминокислотную последовательность тяжелой цепи, представленную SEQ ID NO:347 (М31-В01),
антитела, которое содержит аминокислотную последовательность легкой цепи, представленную SEQ ID NO:352, и которое содержит аминокислотную последовательность тяжелой цепи, представленную SEQ ID NO:353 (В01-3),
антитела, которое содержит аминокислотную последовательность легкой цепи, представленную SEQ ID NO:364, и которое содержит аминокислотную последовательность тяжелой цепи, представленную SEQ ID NO:365 (В01-10), и
антитела, которое содержит аминокислотную последовательность легкой цепи, представленную SEQ ID NO:382, и которое содержит аминокислотную последовательность тяжелой цепи, представленную SEQ ID NO:383 (D02-6).
Анти-С4.4а антитело IgG:
Другой аспект настоящего изобретения заключается в обеспечении анти-С4.4а антитела класса IgG1, которое содержит аминокислотную последовательность легкой цепи и аминокислотную последовательность тяжелой цепи антитела, представленного в Таблице 2.
Антитело к мезотелину
Другой аспект настоящего изобретения заключается в обеспечении нового антитела к мезотелину (MF-Ta), аминокислотная последовательность которого содержит последовательности CDR вариабельной области тяжелой цепи, представленные последовательностями SEQ ГО NO:398 (HCDR1), SEQ ID NO:399 (HCDR2) и SEQ ID NO:400 (HCDR3), и последовательности CDR вариабельной области легкой цепи, представленные последовательностями SEQ ГО NO:401 (LCDR1), SEQ ID NO:402 (LCDR2) and SEQ ГО NO:403 (LCDR3).
В предпочтительном варианте реализации аминокислотная последовательность антитела к мезотелину MF-Ta или его антигенсвязывающих фрагментов содержит последовательность вариабельной области тяжелой цепи, представленную последовательностью SEQ ID NO:404, и последовательность вариабельной области легкой цепи, представленную последовательностью SEQ ID NO:405. В предпочтительном варианте реализации аминокислотная последовательность антитела к мезотелину MF-Та или его антигенсвязывающих фрагментов содержит последовательность вариабельной области тяжелой цепи, кодируемую нуклеотидной последовательностью SEQ ID NO:406, и последовательность вариабельной области легкой цепи, кодируемую нуклеотидной последовательностью SEQ ID NO:407.
В одном особенно предпочтительном варианте реализации аминокислотная последовательность антитела к мезотелину MF-Ta содержит последовательность тяжелой цепи, представленную последовательностью SEQ ID NO:408, и последовательность легкой цепи, представленную последовательностью SEQ ID NO:409.
В одном особенно предпочтительном варианте реализации аминокислотная последовательность антитела к мезотелину MF-Ta содержит последовательность тяжелой цепи, кодируемую нуклеотидной последовательностью SEQ ID NO:410, и последовательность легкой цепи, кодируемую нуклеотидной последовательностью SEQ ID NO:411.
Другие примеры антител, которые связывают раковую молекулу-мишень мезотелин известны специалисту и описаны, например, в WO 2009/068204, и могут использоваться для конъюгатов связывающее соединение - активное соединение согласно настоящему изобретению.
В одном варианте реализации конъюгатов связывающее соединение - активное соединение, связывающее соединение представляет собой антитело к мезотелину или его антигенсвязывающий фрагмент, где антитело связывается с мезотелином и демонстрирует инвариантное связывание.
В одном варианте реализации конъюгатов связывающее соединение - активное соединение, антитело к мезотелину или его антигенсвязывающий фрагмент содержит аминокислотные последовательности трех участков CDR легкой цепи и аминокислотные последовательности трех участков CDR тяжелой цепи антитела, описанного в W02009/068204-A1 (Таблица 7; стр.61-63).
В предпочтительном варианте реализации антитела к мезотелину или их антигенсвязывающие фрагменты выбраны из группы, состоящей из следующего:
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела MF-Ta,
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела MF-J (WO 2009068204-A1; Таблица 7; стр.61),
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела MOR06640 (WO 2009/068204-A1; Таблица 7; стр.61),
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела MF-226 (WO 2009/068204-A1; Таблица 7; стр.61) и
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательности трех участков CDR легкой цепи и последовательности трех участков CDR тяжелой цепи антитела MOR06626 (WO 2009/068204-A1; Таблица 7; стр.61).
В одном особенно предпочтительном варианте реализации антитела к мезотелину или их антигенсвязывающие фрагменты выбраны из группы, состоящей из следующего:
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательность вариабельной области легкой цепи и последовательность вариабельной области тяжелой цепи MF-Ta,
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательность вариабельной области легкой цепи и последовательность вариабельной области тяжелой цепи MF-J (WO 2009/068204-A1; Таблица 7; стр.61),
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательность вариабельной области легкой цепи и последовательность вариабельной области тяжелой цепи MOR06640 (WO 2009/068204-A1; Таблица 7; стр.61),
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательность вариабельной области легкой цепи и последовательность вариабельной области тяжелой цепи MF-226 (WO 2009/068204-A1; Таблица 7; стр.61),
антитела к мезотелину или их антигенсвязывающие фрагменты, которые содержат последовательность вариабельной области легкой цепи и последовательность вариабельной области тяжелой цепи MOR06626 (WO 2009/068204-A1; Таблица 7; стр.61).
Другие антитела:
Примером антитела, которое связывает раковую молекулу-мишень Нег2, является трастузумаб (Genentech). Трастузумаб представляет собой гуманизированное антитело, которое используется для лечения, среди прочего, рака груди. Одним примером антитела, которое связывает раковую молекулу-мишень CD20, является ритуксимаб (Genentech). Ритуксимаб (номер CAS: 174722-31-7) представляет собой химерное антитело, которое используется для лечения неходжкинской лимфомы. Одним примером антитела, которое связывает раковую молекулу-мишень CD52, является алетузумаб (Genzyme). Алетузумаб (номер CAS: 216503-57-0) представляет собой гуманизированное антитело, которое используется для лечения хронического лимфоцитарного лейкоза.
Другими примерами антител, которые связывают HER2, помимо трастузумаба (INN 7637, №CAS: RN: 180288-69-1) и пертузумаба (№CAS: 380610-27-5), являются антитела, раскрытые в WO 2009/123894-A2, WO 200/8140603-А2, или в WO 2011/044368-A2. Примером анти-HER2 конъюгата является трастузумаб-эмтанзин (INN No.9295).
Примерами антител, которые связывают раковую молекулу-мишень CD30 и могут быть использовано в лечении рака, например, лимфомы Хлджкина, являются брентуксимаб, иратумумаб и антитела, раскрытые в WO 2008/092117, WO 2008/036688 или WO 2006/089232. Примером анти-CD30 конъюгата является брентуксимаб-ведотин (№ INN. 9144).
Примерами антител, которые связывают раковую молекулу-мишень CD22 и могут быть использованы в лечении рака, например, лимфомы, являются инотузумаб или эпратузумаб. Примерами анти-С022 конъюгатов являются анотузумаб озагамицин (№INN 8574), или анти-С022-ММАЕ и анти-CD22-МС-ММАЕ (CAS RN: 139504-50-0 и 474645-27-7).
Примерами антител, которые связывают раковую молекулу-мишень CD33 и могут быть использованы в лечении рака, например, лейкоза, являются гемтузумаб или линтузумаб (INN 7580). Примером анти-CD33 конъюгата является гемтузумаб-озагамицин.
Примером антитела, которое связывает раковую молекулу-мишень NMB и может быть использовано в лечении рака, например, меланомы или рака груди, является глембатумумаб (INN 9199). Примером анти-NMB конъюгата является глембатумумаб ведотин (CAS RN: 474645-27-7).
Примером антитела, которое связывает раковую молекулу-мишень CD56 и может быть использовано в лечении рака, например, множественной миеломы, мелкоклеточной карциному легкого, карциномы клеток Меркеля или карциномы яичника, является лорвотузумаб. Примером анти-СВ56 конъюгата является лорвотузумаб мертанзин (CAS RN: 139504-50-0).
Примеры антител, которые связывают раковую молекулу-мишень CD70 и могут быть использованы в лечении рака, например, неходжкинской лимфомы или почечноклеточного рака, раскрыты в WO 2007/03 8 63 7-А2 или WO 2008/070593-A2. Примером анти-CD70 конъюгата является SGN-75 (CD70 MMAF).
Примером антитела, которое связывает раковую молекулу-мишень CD74 и может быть использовано в лечении рака, например, множественной миеломы, является милатузумаб. Примером анти-CD74 конъюгата является милатузумаб -доксорубицин (CAS RN: 23214-92-8).
Пример антитела, которое связывает раковую молекулу-мишень CD 19 и может быть использовано в лечении рака, например, неходжкинской лимфомы, раскрыт в WO 2008/031056-А2. Другие антитела и примеры анти-CD19 конъюгата (SAR3419) раскрыты в WO 2008/047242-A2.
Примерами антител, которые связывают раковую молекулу-мишень муцин-1 и могут быть использованы в лечении рака, например, неходжкинской лимфомы, являются кливатузумаб или антитела, раскрытые в WO 2003/106495-А2, WO 2008/028686-A2. Примеры конъюгатов, направленных против муцина, раскрыты в WO 2005/009369-A2.
Примерами антител, которые связывают раковую молекулу-мишень CD 13 8 и их конъюгатов, которые могут быть использованы для лечения рака, например, множественной миеломы, раскрыты в WO 2009/080829-A1, WO 2009/080830-A1.
Примерами антител, которые связывают раковую молекулу-мишень интегрин-альфа V и могут быть использованы в лечении рака, например, меланомы, саркомы или карциномы, являются интетумумаб (Cas RN: 725735-28-4), абциксимаб (Cas-RN: 143653-53-6), этарацизумаб (Cas-RN: 892553-42-3) или антитела, раскрытые в US 7,465,449, ЕР 19859-А1, WO 2002/012501-А1 или WO 2006/062779-A2. Примерами конъюгатов, направленных против интегрина-альфаУ, являются интетумумаб-ОМ4 и другие ADC, раскрытые в WO 2007/024536-А2.
Примерами антител, которые связывают раковую молекулу-мишень TDGF1 и могут быть использованы в лечении рака, являются антитела, раскрытые в WO 02/077033-A1, US 7,318,924, WO 2003/083041-А2 и WO 2002/088170-A2. Примеры анти-TDGFl конъюгатов раскрыты в WO 2002/088170-A2.
Примерами антител, которые связывают раковую молекулу-мишень PSMA и могут быть использованы в лечении рака, например, карциномы простаты, являются антитела, раскрытые в WO 97/35616-A1, WO 99/47554-A1, и WO 01/009192-A1. Примеры анти-PSMA конъюгатов раскрыты в WO 2009/026274-A1.
Примеры антител, которые связывают раковую молекулу-мишень ЕРНА2, могут быть использованы в приготовлении конъюгатов и могут быть использованы в лечении рака раскрыты в WO 2004/091375-A2.
Примеры антител, которые связывают раковую молекулу-мишень SLC44A4, могут быть использованы в приготовлении конъюгатов и могут быть использованы в лечении рака, например, например, карциномы поджелудочной или предстательной железы, раскрыты в WO 2009/033094-A2 и US 2009/0175796-А1.
Примером антитела, которое связывает раковую молекулу-мишень HLA-DOB является антитело lym-1 (Cas-RN: 301344-99-0), которое может быть использовано для лечения рака, например, неходжкинской лимфомы. Примеры анти-HLA-DOB конъюгатов раскрыты, например, в WO 2005/081711-А2.
Примерами антител, которые связывают раковую молекулу-мишень VTCN1, могут быть использованы в приготовлении конъюгатов и могут быть использованы в лечении рака, например, карциномы яичников, рака поджелудочной железы, легкого или груди, раскрыты в WO 2006/074418-А2.
Соединения согласно настоящему изобретеию обладают ценными фармакологическими свойствами и могут быть использованы для предотвращения и лечения заболеваний человека и животных.
Конъюгаты связывающее соединение - активное соединение (ADC) согласно настоящему изобретению формулы (Ia) проявляют высокую и специфичную цитотоксическую активность в отношении опухолевых клеток, что может быть продемонстрировано на основании ряда анализов, описанных в экспериментальном разделе настоящей заявки (С-1. по С-6.). Эта высокая и специфичная цитотоксическая активность конъюгатов связывающее соединение - активное соединение (ADC) согласно настоящему изобретению, имеющих формулу (Ia), достигается благодаря соответствующей комбинации нового производного N,N-диалкилауристатина и связывающего соединения с линкерами, которые могут как поддаваться ферментативному, гидролитическому или восстановительному расщеплению в заранее определенной точке для высвобождения токсофоров, так и не содержать такой точки заранее определенного разрыва. Более конкретно, применение стабильныз линкеров, которые не содержат заранее определенных точек ферментативного, гидролитического или восстановительного расщепления для высвобождения токсофором, и которые после поглощения конъюгата связывающее соединение - активное соединение опухолевой клеткой и после полного внутриклеточного ферментативного разрушения антитела, все еще остаются полностью или частично интактными, обеспечивают очень специфичное нацеливание этой активности на опухолевую клетку. Совместимость конъюгатов связывающее соединение - активное соединение и стабильных линкеров предполагает, среди прочего, что метаболиты, образующиеся внутри клетки, могут образовываться с достаточной эффективностью, способны достигать своей мишени и способны проявлять так свою антипролиферативную активность в отношении мишени с достаточной эффективностью, и при этом транспортные белки не уносят их от опухолевых клеток раньше. Метаболиты, образующиеся внутри клеток после поглощения соединения формулы (Ia) согласно настоящему изобретению, проявляют пониженную эффективность в качестве субстрата для транспортных белков, подавляя таким образом их перераспределение в системном кровотоке и, соответственно, запуская потенциальные побочные эффекты самого токсофора.
Совместимость конъюгатов связывающее соединение - активное соединение со стабильным линкером и с рассматриваемой мишенью, в соединении с метаболитами, являющимися относительно неэффективными субстратами белков-переносчиков обеспечивают расширенное терапевтическое окно.
Более конкретно, конъюгаты связывающее соединение - активное соединение согласно настоящему изобретению, имеющие формулу (Ia), проявляют высокую и специфичную активность в отношении опухолевых клеток, которые экспрессируют С4.4а. В то же время, активность в отношении опухолевых клеток, которые не экспрессируют С4.4а, значительно слабее.
Благодаря этому профилю свойств, соединения согласно настоящему изобретению пригодны для лечения определенных гиперпролиферативных заболеваний у людей и млекопитающих в целом. Соединения с одной стороны способны ингибировать, блокировать, уменьшать или снижать пролиферацию и деление клеток, а с другой стороны, повышают апоптоз.
Гиперпролиферативные заболевания, для лечения которых можно применять соединения согласно настоящему изобретению включают, в частности группу раковых и опухолевых заболеваний. В контексте настоящего изобретения это означает, в частности, следующие заболевания, но приведенный вписок не является ограничивающим: карциномы и опухоли молочных желез (протоковая и дольковая форма, также in situ), опухоли дыхательных путей (мелкоклеточная и немелкоклеточная карцинома, карцинома бронхов), опухоли головного мозга (например, опухоли ствола головного мозга и гапоталамуса, астоцитома, медуллобластома, эпендимома и нейро- и эктодермальные опухоли гипофиза), опухоли органов желудочно-кишечного тракта (пищевода, желудка, желчного пузыря, тонкой кишки, толстой кишки, прямой кишки), опухоли печени (включая гепатоцеллюлярную карциному, холангиоцеллюлярную карциному и смешанную гепатоцеллюлярную и холангиоцеллюлярную карциному), опухоли области головы и шеи (гортани, глотки, носоглотки, ротоглотки, губ и ротовой полости), опухоли кожи (плоскоклеточную эпителиальную карциному, саркому Калоши, злокачественную меланому, рак кожи из клеток Меркеля и немеланоматозный рак кожи), опухоли мягких тканей (включая саркомы мягких тканей, остеосаркомы, злокачественный фиброзные гистиоцитомы, лимфосаркомы и и рабдомиосаркомы), опухоли глаз (включая внутриглазную меланому и ретинобластому), опухоли эндокринных и экзокринных желез (например, щитовидной и паращитовидной желез, поджелудочной железы и слюнной железы), опухоли мочевыводящих путей (опухоли мочевого пузыря, пениса, почки, почечной лоханки и уретры) и опухоли репродуктивных органов (карциномы эндометрия, шейки матки, яичника, влагалища, вульвы и матки у женщин и карциномы простаты и яичников у мужчин). К ним также относятся пролиферативные заболевания крови в солидной форме, такие как лимфомы, лейкозы и миелопролиферативные заболевания, например, острый миелоидный, острый лимфобластический, хронический лимфоцитарный, хронический миелогенный лейкоз и лейкоз волосатых клеток, а также коррелированные со СПИД лимфомы, лимфомы Ходжкина, неходжкинские лимфомы, кожные Т-клеточные лимфомы, лимфомы Беркитта и лимфомы центральной нервной системы.
Предпочтительные пролиферативные заболевания для использования анти-СА9 конъюгатов связывающее соединение - активное соединение
Гиперпролиферативнми заболеваниями, для лечении которых может быть предпочтительно применять соединения согласно настоящему изобретению являются опухоли с гиперэкспрессией СА9, карциномы и опухоли молочных желез (протоковая и дольковая форма, также in situ); опухоли дыхательных путей (мелкоклеточная и немелкоклеточная карцинома, карцинома бронхов), включая предпочтительно немелкоклеточную карциному легкого; опухоли голвного мозга (например, опухоли ствола головного мозга и гапоталамуса, астоцитома, медуллобластома, эпендимома и нейро- и эктодермальные опухоли гипофиза); опухоли органов желудочно-кишечного тракта (пищевода, желудка, желчного пузыря, тонкой кишки, толстой кишки, прямой кишки), включая, более предпочтительно, опухоли желудка и опухоли кишечника; опухоли печени (включая гепатоцеллюлярную карциному, холангиоцеллюлярную карциному и смешанную гепатоцеллюлярную и холангиоцеллюлярную карциному); опухоли области головы и шеи (гортани, глотки, носоглотки, ротоглотки, губ и ротовой полости языка и пищевода); опухоли мочевыводящих путей (опухоли мочевого пузыря, пениса, почки, почечной лоханки и уретры), включая более предпочтительно опухоли почек и мочевого пузыря; и/или опухоли репродуктивных органов (карциномы эндометрия, шейки матки, яичника, влагалища, вульвы и матки у женщин и карциномы простаты и яичников у мужчин), включая более предпочтительно карциномы шейки матки и матки.
Предпочтительные гиперпролиферативные заболевания для анти-EGFR конъюгатов связывающее соединение - активное соединение
Гиперпролиферативными заболеваниями, для лечении которых может быть предпочтительно применять соединения согласно настоящему изобретению являются опухоли с гиперэкспрессией, опухоли дыхательных путей (например, мелкоклеточная и немелкоклеточная карцинома, карцинома бронхов), включая предпочтительно немелкоклеточная карциному легкого; опухоли органов пищеварения (например, пищевода, желудка, желчного пузыря, тонкого кишечника, толстого кишечника, прямой кишки), включая, в частности, опухоли кишечника; опухоли желез внутренней и внешней секреции (например, щитовидной и паращитовидной желез, поджелудочной железы и слюнной железы), включая предпочтительно опухоли поджелудочной железы; опухоли области головы и шеи (например, глотки, подглоточника, носоглотки, ротоглотки, губ, ротовой полости, языка и пищевода); и/или глиомы.
Предпочтительные гиперпролиферативные заболевания анти-мезотелиновых конъюгатов связывающее соединение - активное соединение
Гиперпролиферативными заболеваниями, для лечении которых может быть предпочтительно применять соединения согласно настоящему изобретению являются опухоли, гиперэкспрессирующие мезотелин, опухоли репродуктивных органов (карциномы эндометрия, шейки матки, яичника, влагалища, вульвы и матки у женщин и карциномы простаты и яичников у мужчин), включая предпочтительно карциному яичников; опухоли желез внутренней и внешней секреции (например, щитовидной и паращитовидной желез, поджелудочной железы и слюнной железы), включая предпочтительно опухоли поджелудочной железы; опухоли дыхательных путей (например, мелкоклеточная и немелкоклеточная карцинома, карцинома бронхов), включая предпочтительно немелкоклеточную карциному легкого; и/или мезотелиомы.
Предпочтительные гиперпролиферативные заболевания для анти-С4.4а конъюгатов связывающее соединение - активное соединение
Гиперпролиферативные заболевания, для лечении которых может быть предпочтительно применять соединения согласно настоящему изобретению являются опухоли с гиперэкспрессией С4.4а-, плоскоклеточные карциномы эпителия (например, шейки матки, вульвы, влагалища, анальных протоков, эндометрия, фаллопиевых труб, пениса, мошонки, пищевода, молочной железы, мочевого пузыря, желчного протока, слизистой оболочки матки, матки и яичников); карциномы и опухоли молочных желез (протоковая и дольковая форма, также in situ); опухоли дыхательных путей (мелкоклеточная и немелкоклеточная карцинома, карцинома бронхов), включая предпочтительно немелкоклеточную карциному легкого, плоскоклеточную карциному и аденокарциному легкого; опухоли области головы и шеи (например, глотки, подглоточника, носоглотки, ротоглотки, губ, ротовой полости, языка и пищевода, карциномы плоского эпителия области головы и шеи); опухоли мочевыводящих путей (опухоли мочевого пузыря, пениса, почек, почечной лоханки и мочеточника, карциномы плоского эпителия мочевого пузыря), включая более предпочтительно опухоли почек и мочевого пузыря; опухоли кожи (плоскоклеточная эпителиальная карцином, саркома Капоши, злокачественная меланома, рак кожи клеток Меркеля и немелмноматозный рак кожи), включая более предпочтительно меланомы; опухоли желез внутренней и внешней секреции (например, щитовидной и паращитовидной желез, поджелудочной железы и слюнной железы), включая предпочтительно опухоли поджелудочной железы; опухоли органов пищеварения (например, пищевода, желудка, желчного пузыря, тонкого кишечника, толстого кишечника, прямой кишки), включая, в частности, колоректальные карциному; и/или опухоли репродуктивных органов (карциномы эндометрия, шейки матки, яичника, влагалища, вульвы и матки у женщин и карциномы простаты и яичников у мужчин), включая более предпочтительно карциномы матки.
Эти подробно описанные для человека заболевания могут развиваться и у других млекопитающих по сопоставимой этиологии, и в таком случае может проводиться их лечение соединениями согласно настоящему изобретению.
В контексте настоящего изобретения термин "лечение" или "лечить" употребляется в общепринятом смысле и означает посещение и лечение пациента и уход за пациентом с целью противодействия, уменьшения, ослабления или облегчения заболевания или расстройства здоровья и улучшения качества жизни, сниженного указанным заболеванием, например, при раковом заболевании.
В настоящем изобретении предложено также применение соединений согласно настоящему изобретению для лечения и/или предотвращения заболеваний, в частности, вышеупомянутых заболеваний.
В настоящем изобретении предложено также применение соединений согласно настоящему изобретению для получения медикамента для лечения и/или предотвращения заболеваний, в частности, вышеупомянутых заболеваний.
В настоящем изобретении предложено также применение соединений согласно настоящему изобретению в способе лечения и/или предотвращения заболеваний, в частности, вышеупомянутых заболеваний.
В настоящем изобретении предложен также способ лечения и/или предотвращения заболеваний, в частности, вышеупомянутых заболеваний, включающий применение эффективного количества по меньшей мере одного соединения согласно настоящему изобретению.
Конъюгат анти-С4.4а связывающее соединение - активное соединение согласно настоящему изобретению применяют предпочтительно для лечения рака у пациента, причем что в раковых клетках указанного пациента, лечение которого необходимо осуществить, экспрессируется С4.4а. Лечение проводят более предпочтительно у пациентов, в раковых клетках которых экспрессия С4.4а выше, чем в здоровых клетках.
Один из способов идентификации пациентов, у которых наблюдается благоприятный ответ на конъюгат анти-С4.4а связывающего соединения - активного соединения для лечения рака, включает определение экспрессии С4.4а в раковых клетках указанного пациента. В одном варианте реализации экспрессию С4.4а определяют посредством анализа экспрессии гена С4.4а. Специалистам известны способы анализа генной экспрессии, такие как, например, детекция РНК, количественная или качественная полимеразная цепная реакция или флуоресцентная гибридизация in situ (FISH). В другом предпочтительном варианте реализации экспрессия С4.4а определяют иммуногистохимическими методами с применением антитела против С4.4а. Иммуногистохимическое исследование проводят предпочтительно на фиксированной формальдегидом ткани. Антитело для применения при иммуногистохимическом исследовании представляет собой то же антитело, что и используемое в конъюгате. Указанное антитело для применения при иммуногистохимическом исследовании представляет собой второе антитело, которое -предпочтительно специфично - распознает белок-мишень С4.4а.
Соединения согласно настоящему изобретению могут применяться отдельно или, при необходимости, в комбинации с одним или более другим фармакологически активным веществом, при условии, что указанная комбинация не приводит к нежелательным и неприемлемым побочным эффектам. Таким образом, в настоящем изобретении предложены также медикаменты, содержащие по меньшей мере одно соединение согласно настоящему изобретению и одно или более другое лекарственное средство, в частности, для лечения и/или предотвращения вышеупомянутых заболеваний.
Например, соединения согласно настоящему изобретению могут быть скомбинированы с известными анти-гиперпролиферативными, цитостатическими или цитотоксическими веществами для лечения раковых заболеваний. Подходящими лекарственными средствами для указанной комбинации, которые можно упомянуть в качестве примеров, являются следующие:
алдеслейкин, алендроновая кислота, альфаферон, алитретиноин, аллопуринол, алоприм, алокси, алтретамин, амино-глютетимид, амифостин, амрубицин, амсакрин, анастрозол, анземет, аранесп, арглабин, триоксид мышьяка, аромазин, 5-азацитидин, азатиоприн, БЦЖ или Tice-БЦЖ, бестатин, бетаметазона ацетат, бетаметазон фосфат натрия, бексаротен, блеомицина сульфат, броксуридин, бортезомиб, бусульфан, кальцитонин, кэмпас, капецитабин, карбоплатин, касодекс, цефезон, целмолейкин, церубидин, хлорамбуцил, цисплатин, кладрибин, клодроновая кислота, циклофосфамид, цитарабин, дакарбазин, дактиномицин, дауноксом, декадрон, декадрона фосфат, делестроген, денилейкин дифтитокс, депомедрол, деслорелин, дексразоксан, диэтилстильбэстрол, дифлюкан, доцетаксел, доксифлуридин, доксорубицин, дронабинол, DW-166HC, элигард, элитек, элленс, эменд, эпирубицин, эпоэтин-альфа, эпоген, эптаплатин, эргамизол, эстрейс, эстрадиол, эстрамустина натрия фосфат, этинилэстрадиол, этиол, этидроновая кислота, этопофос, этопозид, фадрозол, фарстон, филграстим, финастерид, флиграстим, флоксуридин, флюконазол, флударабин, 5-фтордезоксиуридина монофосфат, 5-фторурацил (5-FU), фтороксиместерон, флутамид, форместан, фостеабин, фотемустин, фулвестрант, гаммагард, гемцитабин, гемтузумаб, гливек, глиадел, госерелин, гранисетрона гидрохлорид, гистрелин, гикамтин, гидрокортизон, эритро-гидроксинониладенин, гидроксимочевина, ибритутомаб тиуксетан, идерубицин, ифосфамид, интерферон-альфа, интерферон-альфа-2, интерферон-альфа-2α, интерферон-альфа-2β, интерферон-альфа-n1, интерферон-альфа-n3, интерферон-бета, интерферон-гамма-1α, интерлейкин-2, интрон А, иресса, иринотекан, китрил, лентинана сульфат, летрозол, лейковорин, лейпролид, лейпролида ацетат, левамизол, кальциевая соль левофолиевой кислоты, левотроид, левоксил, ломустин, лонидамин, маринол, мехлорэтамин, мекобаламин, медроксипрогестерона ацетат, мегестрола ацетат, мелфалан, менест, 6-меркаптопурин, месна, метотрексат, метвикс, милтефозин, миноциклин, митомицин С, митотан, митоксантрон, модренал, миоцет, недаплатин, невласта, ньюмега, нейпоген, нилутамид, нолвадекс, NSC-631570, ОСТ-43, октреотид, ондансетрона гидрохлорид, орапред, оксалиплатин, паклитаксел, педиапред, пегаспаргаза, пегасис, пентостатин, пицибанил, пилокарпина гидрохлорид, пирарубицин, пликамицин, порфимер натрия, преднимустин, преднизолон, преднизон, премарин, прокарбазин, прокрит, ралтитрексед, ребиф, рения-186 этидронат, ритуксимаб, роферон-А, ромуртид, салаген, сандостатин, сарграмостим, семустин, сизофиран, собузоксан, солу-медрол, стрептозоцин, стронция-89 хлорид, синтроид, тамоксифен, тамсулозин, тазонермин, тастолактон, таксотер, тецелейкин, темозоломид, тенипозид, тестостерона пропионат, тестред, тиогуанин, тиотепа, тиротропин, тилудроновая кислота, топотекан, торемифен, тозитумомаб, тастузумаб, теосульфан, третиноин, трексалл, триметилмеламин, триметрексат, трипторелина ацетат, трипторелина памоат, UFT (УФТ), уридин, вальрубицин, веснаринон, винбластин, винкристин, виндезин, винорелбин, вирулизин, зинекард, зиностатин-стималамер, зофран; ABI-007, аколбифен, актиммун, аффинитак, аминоптерин, арзоксифен, асоприснил, атаместан, атрасентан, авастин, BAY 43-9006 (сорафениб), CCI-779, CDC-501, целебрекс, цетуксимаб, криснатол, ципротерона ацетат, децитабин, DN-101, доксорубицин-МТС, dSLIM, дутастерид, эдотекарин, эфлорнитин, экзатекан, фенретинид, гистамина дигидрохлорид, гидрогелевый имплантат с гистрелином, гольмий-166 DOTMP, ибандроновая кислота, интерферон-гамма, интрон-ПЭГ, иксабепилон, гемоцианин фисуреллы, L-651582, ланреотид, лазофоксифен, либра, ионафарниб, мипроксифен, минодронат, MS-209, липосомальный МТР-РЕ, МХ-6, нафарелин, неморубицин, неовастат, нолатрексед, облимерсен, Онко-TCS, осидем, паклитаксела полиглутамат, динатрия памидронат, PN-401, QS-21, квазепам, R-1549, ралоксифен, ранпирназа, 13-cis-ретиноевая кислота, сатраплатин, сеокальцитол, Т-13 8067, тарцева, таксопрексин, тимозин-альфа-1, тиазофурин, типифарниб, тирапазамин, TLK-286, торемифен, TransMID-107R, валсподар, вапреотид, ваталаниб, вертепорфин, винфлунин, Z-100, золедроновая кислота и их комбинации.
В предпочтительном варианте реализации соединения согласно настоящему изобретению можно комбинировать с анти-гиперпролиферативными агентами, которые могут представлять собой, в качестве примера - указанный список не является окончательным -следующие:
аминоглютетимид, L-аспарагиназу, азатиоприн, 5-азацитидин, блеомицин, бусульфан, карбоплатин, кармустин, хлорамбуцил, цисплатин, коласпазу, циклофосфамид, цитарабин, дакарбазин, дактиномицин, даунорубицин, диэтилстильбэстрол, 2',2'-дифтордезоксицитидин, доцетаксел, доксорубицин (адриамицин), эпирубицин, эпотилон и его производные, эритро-гидроксинониладенин, этинилэстрадиол, этопозид, флударабина фосфат, 5-фтордезоксиуридин, 5-фтордезоксиуридина монофосфат, 5-фторурацил, фтороксиместерон, флутамид, гексаметилмеламин, гидроксимочевина, гидроксипрогестерона капроат, идерубицин, ифосфамид, интерферон, иринотекан, лейковорин, ломустин, мехлорэтамин, медроксипрогестерона ацетат, мегестрола ацетат, мелфалан, 6-меркаптопурин, месна, метотрексат, митомицин С, митотан, митоксантрон, паклитаксел, пентостатин, TV-фосфоноацетил L-аспартат (PALA), пликамицин, преднизолон, преднизон, прокарбазин, ралоксифен, семустин, стрептозоцин, тамоксифен, тенипозид, тестостерона пропионат, тиогуанин, тиотепа, топотекан, триметилмеламин, уридин, винбластин, винкристин, виндезин и винорелбин.
Очень перспективными могут быть комбинации соединения согласно настоящему изобретению могут также быть скомбинированы с биологическими терапевтическими средствами, такими как антитела (например, Авастин, Ритуксан, Эрбитукс, Герцептин). Соединения согласно настоящему изобретению могут также давать положительный эффект при комбинировании с терапией, направленной против ангиогенеза, в частности, например, с авастином, акситинибом, рецентином, регорафенибом, сорафенибом или сунитинибом. Также подходят, в частности, комбинации с ингибиторами протеасомы и mTOR, а также с антигормонами и ингибиторами ферментов метаболизма стероидов, благодаря благоприятному профилю побочных эффектов.
В целом, комбинирование соединений согласно настоящему изобретению с другими агентами, обладающими цитостатическим или цитотоксическим действием, может обеспечивать достижение следующих целей:
- повышенная активность в отношении замедления роста опухоли, снижения ее размеров или даже полной элиминации, по сравнению с лечением индивидуальным активным соединением;
- возможность применения химиотерапевтических средств в более низкой дозировке, чем при монотерапии;
- возможность проводить лучше переносимую терапию с незначительным числом побочных эффектов по сравнению с индивидуальным введением;
- возможность лечения более широкого спектра опухолевых заболеваний;
- достижение более быстрого ответа на терапию;
- более длительное время выживания пациента по сравнению с современной стандартной терапией.
Соединения согласно настоящему изобретению могут, кроме того, применяться в комбинации с радиационной терапией и/или хирургическим вмешательством.
В настоящем изобретении предложены также медикаменты, которые содержат по меньшей мере одно соединение согласно настоящему изобретению, обычно вместе с одним или более инертным нетоксичным фармацевтически подходящим вспомогательным веществом, и их применение в вышеупомянутых целях.
Соединения согласно настоящему изобретению могут действовать системно и/или местно. Они могут вводиться подходящим для этой цели способом, например, перорально, парентерально, в легкие, назально, сублингвально, лингвально, буккально, ректально, кожно, чрескожно, в конъюнктиву, в глазное яблоко или в виде импланта или стента.
Соединения согласно настоящему изобретению могут вводиться в формах, подходящих для указанных путей введения.
Формы для введения, функционирующие в соответствии с существующим уровнем техники, высвобождающие соединения согласно настоящему изобретению быстро и/или модифицированным образом и содержащие соединения согласно настоящему изобретению в кристаллической и/или аморфной и/или растворенной форме, подходят для перорального введения, как, например, таблетки (таблетки без покрытия или с покрытием, например, с покрытиями, которые устойчивы к желудочному соку, или с отложенным растворением, или нерастворимы и контролируют высвобождение соединения согласно настоящему изобретению), пленки/облатки или таблетки, которые быстро распадаются в ротовой полости, пленки/лиофилизаты, капсулы (например, твердые или мягкие желатиновые капсулы), таблетки в пленочной оболочке, гранулы, пеллеты, порошки, эмульсии, суспензии, аэрозоли или растворы.
Парентеральное введение можно осуществлять, минуя этап всасывания (например, внутривенно, внутриартериально, внутрисердечно, интраспинально или интралюмбально), либо с включением абсорбции (например, внутримышечно, подкожно, внутрикожно, чрескожно или внутрибрюшинно). Формы для введения, подходящие для парентерального введения, включают инъекционные и инфузионные составы в форме растворов, суспензий, эмульсий, лиофилизатов или стерильных порошков.
Для других путей введения подходят, например, лекарственные формы для ингаляции (включая порошковые ингаляторы, небулайзеры), назальные капли, растворы или спреи, таблетки, пленки/облатки или капсулы для лингвального, сублингвального или буккального введения, суппозитории, ушные или глазные составы, вагинальные капсулы, водные суспензии (лосьоны, суспензии), липофильные суспензии, мази, кремы, трансдермальные терапевтические системы (например, пластыри), молочко, пасты, пены, присыпки, импланты или стенты. Предпочтительны пероральное и парентеральное введение, в частности, пероральное и внутривенное введение.
А. Примеры Аббревиатуры и сокращения:
Методы HPLC и ЖХ-МС:
Способ 1 ОКХ-МС):
Прибор: Система Waters Acquity SQD UPLC System; колонка Waters Acquity UPLC HSS T3 1,8 мк 50 мм × 1 мм; элюент A: 1 л воды + 0,25 мл 99% сильной муравьиной кислоты, элюент В; 1 л ацетонитрила + 0,25 мл 99% сильной муравьиной кислоты; градиент: 0,0 мин 90% А →1,2 мин 5% А→2,0 мин 5% А; скорость потока: 0,40 мл/мин; печь: 50°С; УФ детекция: 210-400 нм.
Метод 2 (-ЖХ-MC):
Прибор: Micromass QuattroPremier с Waters UPLC Acquity; колонка Thermo Hypersil GOLD 1,9 мк 50 мм × 1 мм; элюент A: 1 л воды+0,5 мл 50% сильной муравьиной кислоты, элюент В: 1 л ацетонитрила+0,5 мл 50% сильной муравьиной кислоты; градиент: 0,0 мин 90% А→0,1 мин 90% А→1,5 мин 10% А→2,2 мин 10% А; скорость потока: 0,33 мл/мин; печь: 50°С; УФ детекция: 210 нм.
Метод 3 (ЖХ-МС):
Прибор: Micromass Quattro Micro MS with HPLC Agilent Series 1100; колонка Thermo Hypersil GOLD 3 мк 20 мм x 4 мм; элюент А: 1 л воды+0,5 мл 50% сильной муравьиной кислоты, элюент В: 1 л ацетонитрила+0,5 мл 50% сильной муравьиной кислоты; градиент: 0,0 мин 100% А→3,0 мин 10% А→4,0 мин 10% А→4,01 мин 100% А (скорость потока 2,5 мл/мин)→5,00 мин 100% А; печь: 50°С; скорость потока: 2 мл/мин; УФ детекция: 210 нм.
Метод 4 (ЖХ-МС):
MS Прибор: Micromass ZQ; HPLC Прибор: HP 1100 Series; УФ-детектор на диодной матрице; колонка Phenomenex Gemini 3 мк 30 мм × 3,00 мм; элюент А: 1 л воды +0,5 мл 50% сильной муравьиной кислоты, элюент В: 1 л ацетонитрила +0,5 мл 50% сильной муравьиной кислоты; градиент: 0,0 мин 90% А→2,5 мин 30% А→3,0 мин 5% А→4,5 мин 5% А; скорость потока: 0,0 мин 1 мл/мин→2,5 мин/3,0 мин/4,5 мин 2 мл/мин; печь: 50°С;
УФ детекция: 210 нм.
Метод 5 СВЭЖХ):
Прибор: HP 1090 Series П; колонка Merck Chromolith SpeedROD RP-18e, 50 мм × 4,6 мм; первичная колонка Merck Chromolith Guard Cartridge Kit RP-18e, 5 мм × 4,6 мм; инъекционный объем: 5 мкл; элюент А: 70% НСЮ4 в воде (4 мл/литр), элюент В: ацетонитрил; градиент: 0,00 мин 20% В→0,50 мин 20% В→3,00 мин 90% В→3,50 мин 90% В→3,51 мин 20% В→4,00 мин 20% В; скорость потока: 5 мл/мин; температура колонки:40°С.
Метод 6 ГВЭЖХ):
Прибор: Waters 2695 с DAD 996; колонка Merck Chromolith SpeedROD RP-18e, 50 мм × 4,6 мм; № заказа: 1,51450,0001, первичная колонка Merck Chromolith Guard Cartridge Kit RP-18e, 5 мм × 4,6 мм; №заказа: 1,51470,0001, элюент А: 70% HC104 в воде (4 мл/литр), элюент В:
ацетонитрил; градиент: 0,00 мин 5% В→0,50 мин 5% В→3,00 мин 95% В→4,00 мин 95% В; скорость потока: 5 мл/мин.
Метод 7 (ЖХ-МС):
MS Прибор: Waters ZQ; HPLC Прибор: Agilent 1100; УФ-детектор на диодной матрице; колонка Thermo Hypersil GOLD 3 мк 20 мм × 4 мм; элюент А: 1 л воды +0,5 мл 50% сильной муравьиной кислоты, элюент В: 1 л ацетонитрила+0,5 мл 50% сильной муравьиной кислоты; градиент: 0,0 мин 100% А→3,0 мин 10% А→4,0 мин 10% А→4,1 мин 100% А (скорость потока 2,5 мл/мин); печь: 55°С; скорость потока: 2 мл/мин; УФ детекция: 210 нм.
Метод 8 (ЖХ-МС):
MS Прибор: Waters ZQ; HPLC Прибор: Agilent 1100; УФ-детектор на диодной матрице;
колонка Thermo Hypersil GOLD 3 мк 20 мм × 4 мм; элюент А: 1 л воды +0,5 мл 50% сильной муравьиной кислоты, элюент В: 1 л ацетонитрила+0,5 мл 50% сильной муравьиной кислоты; градиент: 0,0 мин 100% А→2,0 мин 60% А→2,3 мин 40% А→3,0 мин 20% А→4,0 мин 10% А→4,2 мин 100% А (скорость потока 2,5 мл/мин); печь: 55°С; скорость потока: 2 мл/мин; УФ детекция: 210 нм.
Метод 9 (ЖХ-МС):
Прибор: Система Waters Acquity SQD UPLC System; колонка Waters Acquity UPLC HSS T3 1,8 мк 50 мм × 1 мм; элюент A: 1 л воды +0,25 мл 99% сильной муравьиной кислоты, элюент В: 1 л ацетонитрила +0,25 мл 99% сильной муравьиной кислоты; градиент: 0,0 мин 95% А →6,0 мин 5% А→7,5 мин 5% А; печь: 50°С; скорость потока: 0,35 мл/мин; УФ детекция: 210-400 нм.
Метод 10 ГВЭЖХ):
Прибор: Agilent 1200 Series; колонка Agilent Eclipse XDB-C18 5 мк 4,6 мм × 150 мм; первичная колонка Phenomenex KrudKatcher disposable Pre-Column; инъекционный объем: 5 мкл; элюент А: 1 л воды+0,01% трифторуксусной кислота; элюент В: 1 л ацетонитрила+0,01% трифторуксусной кислоты; градиент: 0,00 мин 10% В→1,00 мин 10% В↓1,50 мин 90% В→5,5 мин 10% В; скорость потока: 2 мл/мин; температура колонки: 30°С.
Все исходные компоненты и реагенты, которые не описаны ниже в явном виде, могут быть получены из общедоступных коммерческих источников. Для всех других исходных компонентов или реагентов, которые также не описаны ниже, и которые не могут быть получены в коммерческих источниках, дана ссылка на опубликованную литературу, где описано их получение.
Метод 11 (ЖХ-МС):
Прибор: Система Waters Acquity SQD UPLC System; колонка Waters Acquity UPLC HSS T3 1,8 mk 30 × 2 мм; элюент A: 1 л воды +0,25 мл 99% сильной муравьиной кислоты, элюент В; 1 л ацетонитрила+0,25 мл 99% сильной муравьиной кислоты; градиент: 0,0 мин 90% А→1,2 мин 5% А→2,0 мин 5% А печь: 50°С; скорость потока: 0,60 мл/мин; УФ детекция: 208-400 нм.
Метод 12 (ВЭЖХ:
Прибор: Agilent 1200 с колоночным термостатом и детектором на диодной матрице; колонка Merck Chromolith SpeedROD RP-18e, 50 мм × 4,6 мм; № заказа: 1,51450,0001; первичная колонка Merck Chromolith Guard Cartridge Kit RP-18e, 5 мм x 4,6 мм; № заказа; 1,51470,0001; элюент А: 70% HClO4 в воде (4 мл/литр), элюент В: ацетонитрил; градиент: 0,00 мин 5% В→0,50 мин 5% В→3,00 мин 95% В→4,00 мин 95% В; скорость потока: 5 мл/мин; температура колонки: 30°С.
Метод 13 (ЖХ-МС):
MS Прибор: Waters (Micromass) Quattro Micro; прибор ВЭЖХ: Agilent 1100 Series; колонка YMC-Triart C18 3 мк 50 × 3 мм; элюент А: 1 л воды+0,01 мол карбоната алюминия, элюент В: 1 л ацетонитрила; градиент: 0,0 мин 100% А→2,75 мин 5% А→4,5 мин 5% А; печь: 40°С; скорость потока: 1,25 мл/мин; УФ детекция: 210 нм.
Исходные соединения и промежуточные соединения (интермедиаты):
Исходное соединение 1
(2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановая кислота (Вос-долапролин)
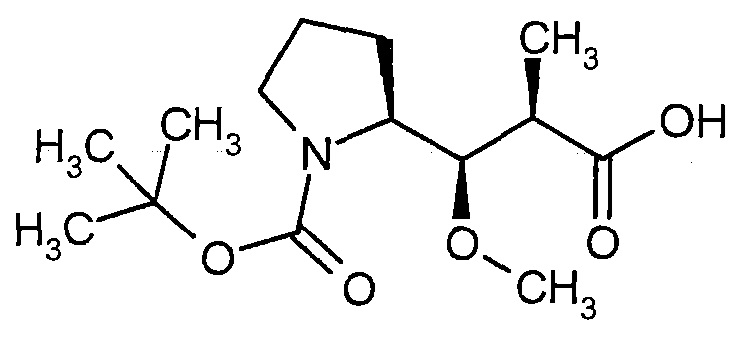
Указанное в названии соединение может быть получено различными путями в соответствии с описанными в литературе методами; см., например, Pettit et al., Synthesis 1996, 719; Shioiri et al., Tetrahedron Lett. 1991, 32, 931; Shioiri et al., Tetrahedron 1993, 49, 1913; Koga et al., Tetrahedron Lett. 1991, 32, 2395; Vidal et al., Tetrahedron 2004, 60, 9715; Poncet et al., Tetrahedron 1994, 50, 5345. Его получали либо в виде свободной кислоты, либо в форме 1:1 соли с дициклогексиламином.
Исходное соединение 2а
Трет-бутип (3R,4S,5S)-3-метокси-5-метил-4-(метиламино)гептаноата гидрохлорид (долаизолейцин-OtBu×HCl)

Указанное в названии соединение может быть получено различными путями в соответствии с описанными в литературе методами; см., например, Pettit et al., J, Org. Chem. 1994, 59, 1796; Koga et al., Tetrahedron Lett. 1991, 32, 2395; Shioiri et al., Tetrahedron Lett. 1991, 32, 931; Shioiri et al., Tetrahedron 1993, 49, 1913.
Исходное соединение 2b
Трет-бугил (3R,4S,5S)-3-метокси-5-метил-4-(метиламино)гептаноат(долаизолейцин-O'Bu)

Это соединение получали аналогично исходному соединению 2а, за тем исключением, что гидрогенирование осуществляли без добавления 1 н. соляной кислоты.
Исходное соединение 3
Nα-(трет-бутоксикарбонил)-N-гидрокси-L-фенилаланинамид

Указанное в названии соединение получали описанным в литературе методом (A. Ritter et al., J. Org. Chem. 1994, 59, 4602).
Выход: 750 мг (75% теоретического)
ЖХ-МС (Метод 3): R,=1,67 мин; MS (ESIпоз): m/z=281 (М+Н)+.
Исходное соединение 4
1,2-оксазолидин гидрохлорид

Указанное в названии соединение может быть получено описанными в литературе методами (см., например, Н. King, J. Chem. Soc. 1942, 432); также оно доступно для приобретения.
Исходное соединение 5
1,2-оксазинан гидрохлорид

Указанное в названии соединение может быть получено описанными в литературе методами (см., например, Н. King, J. Chem. Soc. 1942, 432).
Исходное соединение 6
2-окса-3-азабицикло[2,2,2]окт-5-ен
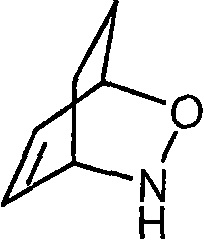
Указанное в названии соединение может быть получено в Вос-защищенной форме описанным в литературе методом (см., например, С. Johnson et al., Tetrahedron Lett. 1998, 39, 2059); удаление защитной группы осуществляли обычным способом путем обработки трифторуксусной кислотой с последующей нейтрализацией.
Выход: 149 мг (89% теоретического) Исходное соединение 7
Трепг-бутил (1S,2R)-1-(гидроксикарбамоил)-2-фенилциклопропил карбамат
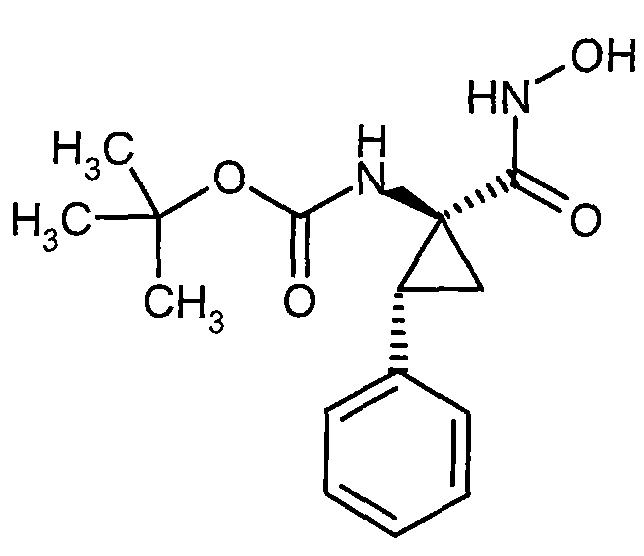
Указанное в названии соединение получили описанным в литературе методом (A. Ritter et al., J. Org. Chem. 1994, 59, 4602) из коммерчески доступной (1S,2R)-1-[(трет-бутоксикарбонил)амино]-2-фенилциклопропанкарбоновой кислоты (С. Cativiela et al., Chirality 1999, 11, 5S3).
Выход: 339 мг (59% теоретического)
ЖХ-МС (Метод 1): Rt=0,82 мин; MS (ESIпоз): m/z=293 (M+H)+.
Промежуточное соединение 1
mpem-бутил (3R,4S,5S)-4-[{N-[(бензилокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноат

10,65 г (41,058 ммоль) трет-бугил (3R,4S,5S)-3-метокси-5-метил-4-(матиламино)гептаноата (исходное соединение 2b) помещали в 250 мл дихлорметана и охлаждали раствор до -10°С. Затем, при помешивании, добавляли 10.317 г (41,058 ммоль) N-[(бензилокси)карбонил]-L-валина, 16.866 г (61.586 ммоль) 2-бромо-1-этилпиридиния тетрафторбората (ВЕР) и 28,6 мл N,N-диизопропилэтиламина и постепенно перемешивали реакционную смесь при комнатной температуре в течение 20 часов. Затем реакционную смесь разбавляли дихлорметаном, и дважды встряхивали с насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали методом флэш-хроматографии на силикагеле используя смесь 4:1 петролейный эфир/этилацетат в качестве элюента. Соответствующие фракции концентрировали, сушили осадок в высоком вакууме в течение ночи. В результате получали 10.22 г (51% от теоретического количества) указанного в названии соединения в виде желтоватого масла.
ЖХВД (Метод 5): Rt=2.3 мин;
ЖХ-МС (Метод 2): Rt=1.59 мин; MS (ESIпоз): m/z=493 (M+H)+.
Промежуточное соединение 2
трет-бутил (3R,4S,5S)-3-метокси-5-метил-4-[метил(L-валил)амино]гептаноат

500 мг (1 ммоль) трет-бутил (3R,4S,5S)-4-[{N-[(бензилокси)карбонил]-L-валил}(метил)амино]-3-метокси-5-метилгептаноата (промежуточного соединения 1) растворяли в 50 мл метанола, затем добавляли 100 мг 10% палладия на активированном угле, после чего гидрогенировали при стандартном давлении водорода при комнатной температуре в течение 1 часа. Затем катализатор отфильтровывали и удаляли растворитель при пониженном давлении. Процедура давала 370 мг (колич.) указанного в названии соединения в виде практически бесцветного масла.
ЖХВД (Метод 5): Rt=1.59 мин;
ЖХ-МС (Метод 1): Rt=0,74 мин; MS (ESIпоз): m/z=359 (М+Н)+.
Промежуточное соединение 3
N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

4,64 г (13,13 ммоль) N-[(9H-флуорен-9-илметокси]карбонил]-N-метил-L-валина растворяли в 20 мл DMF, и последовательно добавляли в смесь 4.28 г (11,94 ммоль) трет-бутил (3R,4S,5S)-3-метокси-5-метил-4-[метил(L-валил)амино]гептаноата (промежуточное соединение 2), 2,75 г (14.33 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 2.2 г (14.33 ммоль) 1-гидрокси-1H-бензотиазол гидрата. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь наливали в смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали последовательно насыщенным раствором бикарбоната натри, и насыщенным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали. Осадок непосредственно использовали на следующем этапе, без дальнейшей очистки.
Выход: 9,1 г (колич., 60% чистота)
ЖХВД (Метод 5): Rt,=2,7 мин;
ЖХ-МС (Метод 2): Rt=1,99 мин; MS (ESIпоз): m/z=694 (M+H)+.
Промежуточное соединение 4
N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамид

9,1 г сырого продукта N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 3) помещали в 56,6 мл дихлорметана, добавляли 56,6 мл трифторуксусной кислоты и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Далее реакционную смесь концентрировали при пониженном давлении, очищали оставшийся осадок методом флэш-хроматографии с использованием дихлорметана, смеси 3:1 дихлорметан/этилацетат и смеси 15:5:0.5 дихлорметан/этилацетат/метанол в качестве элюента. После очистки концентрировали соответствующие фракции с получением 5,8 г (86% от теоретического) указанного в названии соединения в виде бесцветной пены.
ЖХВД (Метод 5): Rt=2,2 мин;
ЖХ-МС (Метод 1): Rt=1,3 мин; MS (ESIпоз): m/z=638 (М+Н)+.
Промежуточное соединение 5
трет-бутил (2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил карбамат

500 мг (1,9 ммоль) N-(трет-бутоксикарбонил)-L-фенилаланин растворяли в 10 мл ДМФ и последовательно добавляли в смесь 466 мг (3,8 ммоль) 1,2-оксазинан гидрохлорида (Исходное соединение 5), 433 мг (2,3 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 382 мг (2,8 ммоль) 1-гидрокси-1H-бензотиазол гидрата и 731 мг (5,7 ммоль) N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь наливали в смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали. В результате получали 620 мг (98% от теоретического количества) указанного в названии соединения.
ЖХВД (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 2): Rt=1,62 мин; MS (ESIпоз): m/z=235 (M-C4H8-CO2+H)+.
Промежуточное соединение 6
(2S)-2-амино-1-(1,2-оксазинан-2-ил)-3-фенилпропан-1-онтрифторацетат

620 мг (1,85 ммоль) трет-бугип (2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил карбамата (промежуточное соединение 5) помещали в 5 мл дихлорметана, добавляли 10 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Далее, реакционную смесь концентрировали при пониженном давлении, и лиофилизировали полученный осадок из воды/ацетонитрила. Таким образом, получали 779 мг (91% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ЖХВД (Метод 5): Rt=0.45 мин;
ЖХ-МС (Метод 3): Rt=1,09 мин; MS (ESIпоз): m/z=235 (М+Н)+.
Промежуточное соединение 7
(2R,3R)-3-метокси-2-метил-N-[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]-3-[(2S)-пирролидин-2-ил]пропанамидтрифторацетат

360 мг (1,25 ммоль) (2R,3R)-3-[(25)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановой кислоты (Исходное соединение 1) помещали в 10 мл ДМФ и последовательно добавляли в смесь 579,2 мг (1,25 ммоль) (25)-2-амино-1-(1,2-оксазинан-2-ил)-3-фенилпропан-1-он трифторацетата (промежуточное соединение 6), 714.5 мг (1,88 ммоль) О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 655 мкЛ N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем реакционную смесь концентрировали, и помещали осадок в этилацетат, после чего экстрагировали встряхиванием, на первом этапе синтеза - с 5% водным раствором лимонной кислоты, а затем - с 5% водным раствором бикарбоната натрия, и затем с насыщенным раствором хлорида натрия. Органическую фазу концентрировали, и очищали осадок методом флэш-хроматографии на силикагеле с 16:4 смесью дихлорметан/метанол в качестве элюента. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении. После сушки осадка в высоком вакууме, получали 503.5 мг (74% от теоретического количества) защищенного группой Boc промежуточного вещества трет-бугил (2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-карбоксилата.
ЖХВД (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=1,12 мин; MS (ESIпоз): m/z=504 (M+H)+.
503 мг (1 ммоль) этого промежуточного вещества помещали в 20 мл дихлорметана, добавляли 10 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Далее, реакционную смесь концентрировали при пониженном давлении, и повторно перегоняли дихлорметаном. Оставшийся осадок выпадал в растворе этилацетата с n-пентаном, после чего растворитель удаляли декантированием. Осадок, полученный таким образом, растворяли в воде и экстрагировали встряхиванием с этилацетатом, после чего водную фазу лиофилизировали. Таким образом, получали 462 мг (89% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ЖХВД (Метод 12): Rt=1.53 мин;
ЖХ-МС (Метод 11): Rt=0.57 мин; MS (ESIпоз): m/z=404 (M+H)+.
Промежуточное соединение 8
N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамид

51 мг (0,08 ммоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) растворяли в 10 мл ДМФ, после чего добавляли 0.5 мл пиперидина. Реакционную смесь перемешивали при комнатной температуре в течение 10 мин, после чего концентрировали при пониженном давлении, и перемешивали осадок с диэтиловым эфиром. Нерастворимые составляющие отфильтровывали, и несколько раз промывали реакционную смесь с диэтиловым эфиром. Затем остаток фильтрации помещали в 5 мл раствора диоксан/вода, после чего доводили раствор до уровня рН 11 с помощью 1 н. раствора гидроксида натрия. Под воздействием ультразвука, несколькими порциями добавляли в общей сложности 349 мг (1,6 ммоль) ди-отреот-бутилдикарбоната, в процессе чего рН раствора поддерживали на уровне 11. После завершения реакции, диоксан выпаривали, и доводили водный раствор до уровня рН 2-3 с помощью лимонной кислоты. Реакционную смесь дважды экстрагировали, каждый раз с использованием 50 мл этилацетата. Органические фазы объединяли, сушили над сульфатом магния, и концентрировали при пониженном давлении. Осадок помещали в диэтиловый эфир, после чего указанное в названии соединение выпадало в осадок с пентаном. Растворитель удаляли декантированием. Осадок еще несколько раз дигидрировали с пентаном и в конце сушили в высоком вакууме. Таким образом получали 40 мг (97% от теоретического количества) указанного в названии соединения.
ЖХВД (Метод 6): Rt=2,2 мин;
ЖХ-МС (Метод 2): Rt=1,32 мин; MS (ESIпоз): m/z=516 (М+H)+.
Промежуточное соединение 9
трет-бутил (2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-карбоксилат

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточных соединений 5, 6 и 7 в три этапа, осуществляя реакцию сочетания коммерчески доступной (1S,2R)-1-[(отреот-бутоксикарбонил)амино]-2-фенилциклопропанкарбоновой кислоты с 1,2-оксазинан гидрохлоридом (Исходное соединение 5), последующим удалением защиты с использованием трифторуксусной кислоты, и осуществляя реакцию сочетания с исходным соединением 1. Конечный продукт очищали методом препаративной ВЭЖХ.
ВЭЖХ (Метод 5): Rt=2,12 мин;
ЖХ-МС (Метод 2): Rt=1,25 мин; MS (ESIпоз): m/z=516 (М+Н)+.
Промежуточное соединение 10
N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1 -метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
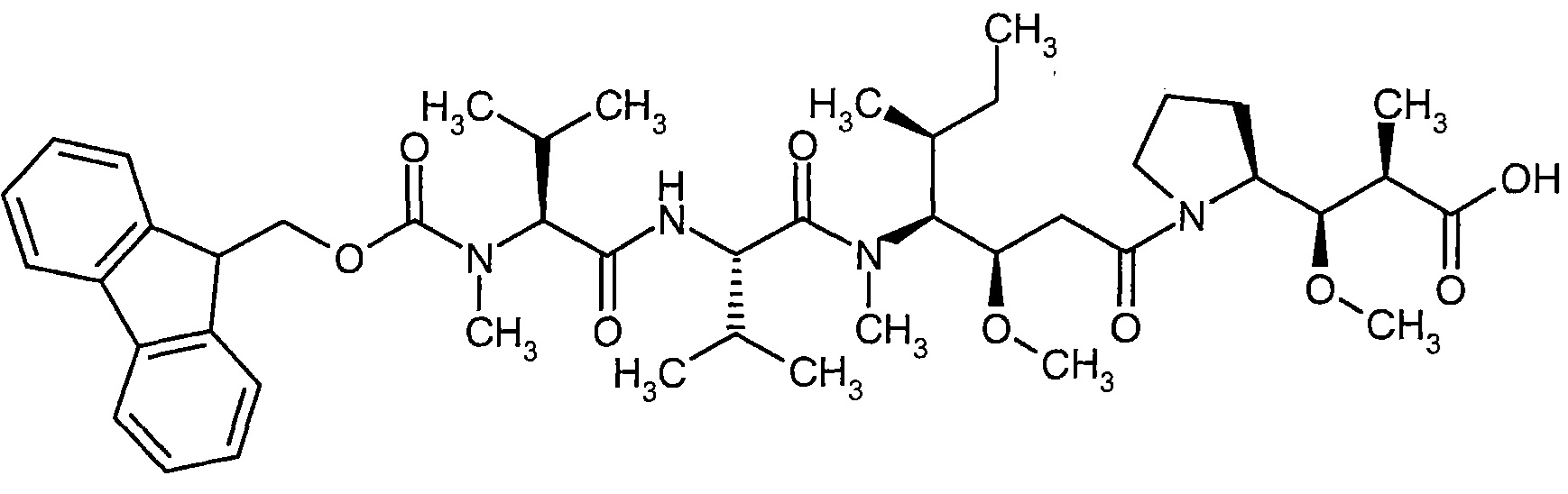
315 мг (0.494 ммоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида
(промежуточное соединение 4) растворяли в 12 мл ДМФ, и добавляли к смеси 104 мг (0.543 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 83 мг (0.543 ммоль) 1-гидрокси-1H-бензотиазол гидрата, после чего перемешивали реакционную смесь при комнатной температуре в течение 90 мин. Затем добавляли 112 мкл N,N-диизопропилэтиламина и 149 мг (0.494 ммоль) (2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропановой кислоты трифторацетата, который получали заранее из исходного соединения 1 удалением защитной группы Boc трифторуксусной кислотой. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, и затем концентрировали в высоком вакууме. Оставшийся осадок очищали дважды методом препаративной ВЭЖХ. В результате было получено 140 мг (35% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2.40 мин;
ЖХ-МС (Метод 1): Rt=1,38 мин; MS (ESIпоз): m/z=807 (M+H)+.
Промежуточное соединение 11
N-[(бензилокси)карбонил]-N-метил-L-треонил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамид

На первом этапе синтеза N-[(бензилокси)карбонил]-N-метил-L-треонин получали из 237 мг (0,887 ммоль) его соли дициклогексиламина, поместив ее в этилацетат и экстрагировав встряхиванием с 5% водной серной кислотой. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Осадок помещали в 16 мл ДМФ, и последовательно добавляли в смесь 365 мг (1 ммоль) трет-бугил (3R,4S,5S)-3-метокси-5-метил-4-[метил(Ь-валил)амино]гептаноата (промежуточное соединение 2), 185 мг (0,967 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 148 мг (0,967 ммоль) 1-гидрокси-1H-бензотиазол гидрата. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь наливали в смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали последовательно насыщенным раствором бикарбоната натри, и насыщенным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали. Осадок очищали методом препаративной ВЭЖХ. Таким образом получали 283 мг (53% от теоретического количества) трет-бутил эфира промежуточного соединения N-[(бензилокси)карбонил]-N-метил-L-треонил-N-[(3R,4S,5S)-1-трет-бутокси-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида.
ВЭЖХ (Метод 5): Rt=2,17 мин.
283 мг (0.466 ммоль) этого промежуточного вещества помещали в 5 мл дихлорметана, добавляли 5 мл безводной трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Затем реакционную смесь концентрировали в высоком вакууме, и очищали оставшийся осадок методом препаративной ВЭЖХ. Процедура давала 156 мг (61% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1.50 мин;
ЖХ-МС (Метод 2): Rt=1,09 мин; MS (ESIпоз): m/z=552 (М+Н)+.
Промежуточное соединение 12
Бензил N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-фенилаланинат трифторацетат

На первом этапе процедуры исходное соединение 1 получали из 600 мг (1,28 ммоль) соответствующей соли дициклогексиламмония растворением этой соли в 100 мл этилацетата и экстрагирование при встряхивании, сначала с 50 мл 0.5% серной кислоты и затем с насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния, фильтровали, концентрировали, и немедленно осуществляли реакцию с бензил L-фенилаланинатом, по аналогии с синтезом промежуточного соединения 7, после чего удаляли защитные группы.
Выход: 650 мг (94% в 2 этапа)
ВЭЖХ (Метод 6): Rt=1,76 мин;
ЖХ-МС (Метод 2): Rt=1,68 мин; MS (ESIпоз): m/z=425 (M+H)+.
Промежуточное соединение 13
Бензил (βS)-N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-β-метил-L-фенилаланинат трифторацетат

На первом этапе синтеза (2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановую кислоту получали из 351 мг (0,75 ммоль) ее соли дициклогексиламина (Исходное соединение 1), поместив ее в этилацетат и экстрагировав встряхиванием с водным 5% раствором кислого сульфата калия. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Осадок помещали в 10 мл ДМФ и последовательно добавляли в смесь 373 мг (0,75 ммоль) бензил (βS)-β-метил-L-фенилаланинат трифторацетата [приготовленного из коммерчески доступного (βS)-N-(трет-бутоксикарбонил)-β-метил-L-фенилаланина путем EDC/DMAP-опосредуемой этерификации бензиловым спиртом и последующим удалением защитной группы Boc трифторуксусной кислотой], 428 мг (1,125 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 392 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь наливали в смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, и затем концентрировали. Осадок очищали методом препаративной ВЭЖХ. Процедура давала 230 мг (57% от теоретического количества) защищенного группой Boc промежуточного соединения бензил (βS)-N-{(2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропаноил}-β-метил-L-фенилаланината.
ВЭЖХ (Метод 6): Rt=2,3 мин;
ЖХ-МС (Метод 1): Rt=1,36 мин; MS (ESIпоз): m/z=539 (М+Н)+.
230 мг (0.42 ммоль) этого промежуточного вещества помещали в 5 мл дихлорметана, добавляли 5 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении. Оставшийся осадок реакционной смеси затем сушили при пониженном давлении, и лиофилизировали из ацетонитрила/воды. Таким образом, получали 230 мг (колич.) указанного в названии соединения.
ВЭЖХ (Метод 6): Rt=1,6 мин.
Промежуточное соединение 14
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

143 мг (0,223 ммоль) N-[(9-H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) помещали в 15 мл ДМФ, и последовательно добавляли в смесь 141 мг (0,22 ммоль) (2R,3R)-3-метокси-2-метил-N-[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]-3-[(2S)-пирролидин-2-ил]пропанамид трифторацетата (промежуточное соединение 7), 102 мг (0,27 ммоль) О-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 128 мкл (0,74 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Затем реакционную смесь наливали в смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали. Процедура давала 275 мг (колич.) защищенного группой Fmoc промежуточного соединения N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида.
ВЭЖХ (Метод 5): Rt=2,73 мин;
ЖХ-МС (Метод 4): Rt=3,19 мин; MS (ESIпоз): m/z=1023 (M+H)+.
46 мг (0,045 ммоль) этого промежуточного вещества растворяли в 4 мл ДМФ. Затем добавляли 1 мл пиперидина, после чего реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении, очищали осадок методом препаративной ВЭЖХ (элюент: ацетонитрил+0,01% TFA/вода+0,01% TFA). В результате получали 22 мг (54% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,68 мин;
ЖХ-МС (Метод 2): Rt=1,03 мин; MS (ESIno3): m/z=801 (M+H)
1Н ЯМР (600 МГц, ДМСО-d6): δ=8,8 (m, 2H), 8,7 (m, 1H), 8.42 и 8,15 (2d, 1H), 7,3-7,1 (m, 5H), 5,12 и 4,95 (2m, 1H), 4,70 и 4,62 (2m, 1H), 4,62 и 4,50 (2t, 1H), 4,1-3,9 (m, 3H), 3,85 (m, 1H), 3,75-3,6 (m, 2H), 3,23, 3,18, 3,17, 3,14, 3,02 и 2,96 (6s, 9H), 3,1-2,9 и 2,75 (2m, 2H), 2.46 (m, 3H), 2.4-2,1 (m, 2H), 2,05 (ушир. m, 2H), 1,85-1,55 (ушир. m, 6H), 1,5-1,2 (ушир. m, 3H), 1,1-0,8 (m, 18H), 0,75 (t, 3H) [другие сигналы перекрыты пиками H2O].
Промежуточное соединение 15
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

126 мг (0,198 ммоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) помещали в 10 мл ДМФ и последовательно добавляли в смесь 105 мг (0,198 ммоль) (2R,3R)-3-метокси-2-метил-N-[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]-3-[(2S)-пирролидин-2-ил]пропанамид трифторацетата (промежуточное соединение 17), 41,6 мг (0,217 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 33 мг (0,217 ммоль) 1-гидрокси-1H-бензотиазол гидрата и 79 мкл (0.454 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь наливали в смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали. Процедура давала 220 мг (колич.) защищенного группой Fmoc промежуточного соединения N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ВЭЖХ (Метод 5): Rt=2,77 мин;
ЖХ-МС (Метод 1): Rt=1,5 мин; MS (ESIпоз): m/z=1037 (М+Н)+.
220 мг (0,212 ммоль) этого промежуточного вещества растворяли в 5 мл ДМФ. Затем добавляли 1 мл пиперидина, после чего реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении, очищали осадок методом препаративной ВЭЖХ (элюент: ацетонитрил+0,01% TFA/вода+0,01% TFA). Процедура давала 91 мг (46% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,71 мин;
ЖХ-МС (Метод 1): Rt=0,9 мин; MS (ESIпоз): m/z=815 (M+H)+.
1Н ЯМР (600 МГц, ДМСО-d6): δ=8,87 и 8,80 (2d, 2H), 8,75 (m, 1H), 8.40 и 7,98 (2d, 1H), 7,3-7,1 (m, 5H), 5.45 и 5,2 (2t, 1H), 4,78 и 4,62 (2m, 1H), 4,73 и 4,58 (2t, 1H), 4,2-4,0 (m, 3Н), 3,7-3,6 (m, 1H), 3,35, 3,20, 3,18, 3,14, 3,12 и 3,00 (6s, 9H), 3,1 и 2,95 (2m, 2H), 2.46 (m, 3H), 2.4-2,0 (m, 4H), 1,9-1,6 (m, 4H), 1,6-1,2 (m, 5H), 1,1-0,75 (m, 21H), 0,80 (t, 3H) [другие сигналы перекрыты пиками H2O peak].
Промежуточное соединение 16
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

617 мг (1,2 ммоль) трет-бутил (2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-карбоксилата (промежуточное соединение 24) помещали в 44 мл дихлорметана, добавляли 4.4 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении, и лиофилизировали полученный осадок из диоксана/воды. В результате получали 702 мг (колич.) незащищенного соединения (2R,3R)-3-метокси-2-метил-N-[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]-3-[(2S)-пирролидин-2-ил]пропанамид трифторацетат в виде сырого продукта, который использовали на следующем этапе синтеза без дальнейшей очистки.
470 мг (0,74 ммоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамид (промежуточное соединение 4) помещали в 57 мл ДМФ и последовательно добавляли в смесь 390 мг (примерно 0,74 ммоль) полученного ранее (2R,3R)-3-метокси-2-метил-N-[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]-3-[(2S)-пирролидин-2-ил]пропанамид трифторацетата, 336 мг (0,88 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 423 мкл (2.4 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь наливали в смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали последовательно насыщенным раствором бикарбоната натрия и насыщенным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали. Осадок очищали методом препаративной ВЭЖХ. Процедура давала 453 мг (59% от теоретического количества) защищенного группой Fmoc промежуточного соединения N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)4-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил] пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ВЭЖХ (Метод 5): Rt=2,58 мин;
ЖХ-МС (Метод 1): Rt=3,10 мин; MS (ESIпоз): m/z=1035 (М+Н)+.
453 мг (0.438 ммоль) этого промежуточного вещества растворяли в 24 мл ДМФ. Затем добавляли 2.4 мл пиперидина, после чего реакционную смесь перемешивали при комнатной температуре в течение 30 мин.Затем реакционную смесь концентрировали при пониженном давлении, очищали осадок методом препаративной ВЭЖХ (элюент:ацетонитрил/0,1% TFA в воде). В результате получали 260 мг (64% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,64 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=813 (M+W)+.
1Н ЯМР (400 МГц, ДМСО-d6): δ=8,8 (m, 2Н), 8,65 (m, 2H), 7,3-7,1 (m, 5H), 4,8-4,05 (m, 2H), 4,0 и 3,82 (2m, 2H), 3,8-3,5 (m, 8H), 3,32, 3,29, 3,20, 3,19, 3,12 и 3,00 (6s, 9H), 2,65 (t, 1H), 2,5-2.45 (m, 3H), 2.4-1,3 (m, 15H), 1,15-0,85 (m, 18H), 0,8 и 0,75 (2d, 3H) [другие сигналы перекрыты пиками НзО peak].
Промежуточное соединение 17
N-бензил-N-метил-L-фенилаланинамидтрифторацетат

1000 мг (3,77 ммоль) N-(трет-бутоксикарбонил)-L-фенилаланина растворяли в 10 мл ДМФ и добавляли к смеси 457 мг (3,77 ммоль) N-метилбензиламина, 2150 мг (5,65 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 657 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, и затем концентрировали при пониженном давлении. Осадок помещали в дихлорметан, и экстрагировали трижды встряхнув с водой. Органическую фазу сушили над сульфатом магния и концентрировали. Осадок очищали методом флэш-хроматографии на силикагеле с использованием смеси 3:1 петролейный эфир/этилацетат в качестве элюента. Фракции полученного продукта концентрировали, и сушили осадок в высоком вакууме. Процедура давала 1110 мг (75% от теоретического количества) защищенного группой Boc промежуточного соединения N-бетил-Nα-(трет-бутоксикарбонил)-N-метил-L-фенилаланинамид.
ВЭЖХ (Метод 6): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,14 мин; MS (ESIпоз): m/z=369 (М+Н)+.
1108 мг (3,007 ммоль) этого промежуточного вещества помещали в 30 мл дихлорметана, добавляли 10 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении, полученный осадок перемешивали с дихлорметаном, после чего удаляли растворитель дистилляцией. Осадок еще два раза перемешивали с пентаном, каждый раз удаляя растворитель декантированием, после чего указанное в названии соединение сушили в высоком вакууме. Таким образом получали 1075 мг (93% от теоретического количества) указанного в названии соединения в виде смолы.
ВЭЖХ (Метод 6): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,6 мин; MS (ESIпоз): m/z=269 (М+Н)+.
Промежуточное соединение 18
N-бензил-Nα-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-N-метил-L-фенилаланинамид трифторацетат

На первом этапе синтеза (2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановой кислоты (Исходное соединение 1) получали из 141 мг (0.491 ммоль) его соли дициклогексиламина, поместив ее в этилацетат и экстрагировав встряхиванием с 5% водной серной кислотой. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Осадок помещали в 10 мл ДМФ и добавляли 187,6 мг (0.49 ммоль) N-бензил-ТУ-метил-L-фенилаланинамид трифторацетата (промежуточное соединение 9), 190,3 мг (1.47 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 256 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали, осадок помещали в этилацетат, после чего раствор экстрагировали встряхиванием последовательно с насыщенным раствором хлорида аммония, насыщенным раствором бикарбоната натрия и водой. Органическую фазу сушили над сульфатом магния и концентрировали. Осадок очищали методом флэш-хроматографии на силикагеле с 30:1 смесью ацетонитрил/вода в качестве элюента. Фракции полученного продукта концентрировали, и сушили осадок в высоком вакууме. Процедура давала 168 мг (64% от теоретического количества) защищенного группой Boc промежуточного соединения трет-бутил (2S)-2-[(1R,2R)-3-({(2S)-1-[бензил(метил)амино]-1-оксо-3-фенилпропан-2-ил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-карбоксилат.
ВЭЖХ (Метод 6): Rt=2,2 мин;
ЖХ-МС (Метод 2); Rt=1,22 мин; MS (ESIпоз): m/z=538 (M+H)+.
168 мг (0,312 ммоль) этого промежуточного вещества помещали в 15 мл дихлорметана, добавляли 3 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин.Затем реакционную смесь концентрировали при пониженном давлении. Оставшийся осадок перемешивали сначала с дихлорметаном, затем с диэтиловым эфиром, после каждого раза удаляя растворитель дистилляцией. После сушки в высоком вакууме, получали 170 мг (99% от теоретического количества) указанного в названии соединения в виде смолы.
ВЭЖХ (Метод 6): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,73 мин; MS (ESIпоз): m/z=438 (М+Н)+.
Промежуточное соединение 19
Метил N-{(2R,3R)-3-метокси-2-метил-3-[(25)-пирролидин-2-ил]пропаноил}-L-фенилаланинат трифторацетат

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 18, в качестве исходного соединения брали (2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановую кислоту (Исходное соединение 1), которую получали из соли дициклогексиламина, и метил L-фенилаланинат гидрохлорида.
ВЭЖХ (Метод 5): Rt=0,6 мин;
ЖХ-МС (Метод 3): Rt=1,17 мин; MS (ESIпоз): ra/z=349 (М+Н)+.
Промежуточное соединение 20
Бензил N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-триптофанат трифторацетат

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 18, в качестве исходного соединения брали(2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановую кислоту (Исходное соединение 1), которую получали из соли дициклогексиламина, и бензил L-триптофанат.
ВЭЖХ (Метод 6): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,8 мин; MS (ESIпоз): m/z=464 (M+H)+.
Промежуточное соединение 21
Бензил (1S,2R)-1-({(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}амино)-2-фенилциклопропанкарбоксилат трифторацетат

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 18, в качестве исходного соединения брали(2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановую кислоту (Исходное соединение 1), которую получали из соли дициклогексиламина, и бензил (1S,2R)-1-амино-2-фенилциклопропанкарбоксилат. Бензил (1S,2R)-1-амино-2-фенилциклопропанкарбоксилат получали заблаговременни стандартными методами, этерификацией коммерчески доступной (1S,2R)-1-[(трет-бутоксикарбонил)амино]-2-фенилциклопропанкарбоновой кислоты с бензиловым спиртом и последующим удалением группы Boc трифторуксусной кислотой.
ВЭЖХ (Метод 5): Rt=1,5 мин;
ЖХ-МС (Метод 2): Rt=0,93 мин; MS (ESIпоз): m/z=437 (М+Н)+.
Промежуточное соединение 22
6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N'-метилгексангидразидтрифторацетат

100 мг (473 мкМоль) 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)капроновой кислоты растворяли в 71 мкл ДМФ, после чего добавляли к смеси 139 мг (947 мкМоль) трет-бутил 1-метилгидразинкарбоксилата, 182 мг (947 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 145 мг (947 мкМоль) 1-гидрокси-1H-бензотиазол гидрата. Реакционную смесь перемешивали при комнатной температуре в течение ночи, и затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана/воды, получали 129 мг (80% от теоретического количества) защищенного промежуточного соединения в виде бесцветной пены.
Затем эти 129 мг (380 мкМоль) деблокировали, используя 2 мл трифторуксусной кислоты в 8 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, реакционную смесь концентрировали при пониженном давлении. Осадок лиофилизировали из ацетонитрила/воды, с получением 125 мг (83% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ЖХ-МС (Метод 1): Rt=0,38 мин; MS (ESIпоз): m/z=240 (М+Н)+.
Промежуточное соединение 23
N-(2-аминоэтил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-метилбутанамид трифторацетат
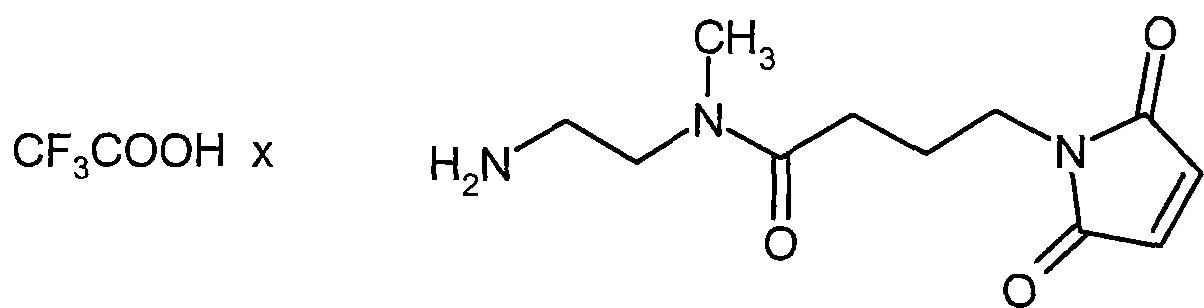
На первом этапе синтеза 35 мг (164 мкМоль) трет-бутил 2-(матиламино)этил карбамат гидрохлорид трифторацетата, 30 мг (164 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутановой кислоты, 75 мг (197 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 57 мкл N,N-диизопропилэтиламина объединяли в 5 мл ДМФ, и перемешивали при комнатной температуре в течение ночи. Затем удаляли растворитель при пониженном давлении, и очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции концентрировали, и лиофилизировали из диоксана/воды, с получением 35 мг (63% от теоретического количества) защищенного промежуточного соединения.
ВЭЖХ (Метод 12): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,71 мин; MS (ESIпоз): m/z=340 (М+Н)+.
Затем все полученное защищенное промежуточное соединение деблокировали с использованием 1 мл трифторуксусной кислоты в 5 мл дихлорметана, с получением 28 мг (77% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 3): Rt=0,75 мин; MS (ESIпоз): m/z=240 (M+H)+.
Промежуточное соединение 24
4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-[2-(матиламино)этил]бутанамид трифторацетат

На первом этапе синтеза 35 мг (164 мкМоль) трет-бутил (2-аминоэтил)метил карбамат гидрохлорид трифторацетата, 30 мг (164 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутановой кислоты, 75 мг (197 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 57 мкл N,N-диизопропилэтиламина объединяли в 5 мл ДМФ и перемешивали при комнатной температуре в течение 30 мин. Затем растворитель удаляли при пониженном давлении, и очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции концентрировали с получением после лиофилизации из диоксана/воды 51 мг (91% от теоретического количества) защищенного промежуточного соединения.
ВЭЖХ (Метод 12): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,77 мин; MS (ESIпоз): m/z=340 (М+Н)+.
Затем у всего полученного количества удаляли защитные группы с использованием 1 мл трифторуксусной кислоты в 5 мл дихлорметана, и получали 45 мг (69% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,19 мин; MS (ESIпоз): m/z=240 (М+Н)+.
Промежуточное соединение 25
Бензил (2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноаттрифторацетат

На первом этапе синтеза (2R,3R)-3-[(2S)-1-(трет-б^отоксмкарбонил)пирролидин-2-ил]-3-метокси-2-метилпропановую кислоту получали из 1,82 г (388 ммоль) ее соли дициклогексиламина, поместив ее в этилацетат и экстрагировав встряхиванием с 100 мл 0,5% серной кислоты. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Осадок помещали в 10 мл диоксана и 10 мл воды, добавляли 1517 мг (4,66 ммоль) карбоната цезия, и оборабатывали реакционную смесь в ультразвуковой ванне в течение 5 мин, после чего концентрировали при пониженном давлении, и повторно перегоняли один раз с ДМФ. Осадок помещали в 15 мл дихлорметана, и добавляли 1990 мг (11,64 ммоль) бензил бромида. Реакционную смесь обрабатывали в ульультразвуковой ванне в течение 15 мин, и затем концентрировали при пониженном давлении. Осадок разделяли на этилацетат и воду, и отделяли органическую фазу, после чего экстрагировали встряхиванием с насыщенным раствором хлорида натрия, а затем концентрировали. Осадок затем очищали методом препаративной ВЭЖХ. Процедура давала 1170 мг (80% от теоретического количества) защищенного группой Boc промежуточного соединения.
Затем у этих 1170 мг немедленно удаляли защитные группы с использованием 5 мл трифторуксусной кислоты в 15 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин, затем реакционную смесь концентрировали при пониженном давлении. Осадок лиофилизировали из диоксана. После сушки в высоком вакууме, получали 1333 мг (84% от теоретического количества) указанного в названии соединения в виде желтого масла.
ВЭЖХ (Метод 6): Rt=1,5 мин;
ЖХ-МС (Метод 1): Rt=0,59 мин; MS (ESIпоз): m/z=278 (М+Н)+.
Промежуточное соединение 26
N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

1200 мг (2,33 ммоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(2R,3S,4S-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 5) соединяли с 910,8 мг (2,33 ммоль) бензил (2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноат трифторацетата (промежуточное соединение 14), 1327 мг (3.49 ммоль) О-(7-азабензотриазол-1-и)-N,N,N',N'-тетраметилуроний гексафторфосфата и 2027 мкл N,N-диизопропилэтиламина в 50 мл ДМФ, и перемешивали реакционную смесь при комнатной температуре в течение 5 мин. После этого растворитель удаляли при пониженном давлении. Оставшийся осадок помещали в этилацетат, и экстрагировали встряхиванием последовательно с 5% водным раствором лимонной кислоты, и насыщенным раствором бикарбоната натрия. Органическую фазу отделяли и концентрировали. Осадок очищали методом препаративной ВЭЖХ. Объединенные фракции полученного продукта концентрировали, и сушили осадок в высоком вакууме. Процедура давала 1000 мг (55% от теоретического количества) бензилового эфира промежуточного соединения N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-(бензилокси)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде смолы.
ЖХ-МС (Метод 1): Rt=1,56 мин; MS (ESIпоз): m/z=775 (М+Н)+.
Все полученное количество этого промежуточного вещества помещали в 25 мл смеси метанола и дихлорметана (20:1), и удаляли группу бензилового эфира гидрогенизацией при стандартном давлении водорода с использованием 10% палладия на активированном угле в качестве катализатора. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, катализатор отфильтровывали, и концентрировали фильтрат при пониженном давлении. Процедура давала 803 мг (91% от теоретического количества) указанного в названии соединения в виде белого твердого вещества.
ВЭЖХ (Метод 6): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,24 мин; MS (ESIпоз): m/z=685 (М+Н)+.
Промежуточное соединение 27
(1S,2R)-1-амино-2-фенил-N-пропилциклопропанкарбоксамидтрифторацетат

Указанное в названии соединение получали, осуществляя реакцию сочетания коммерчески доступной (1S,2R)-1-[(трет-бутоксикарбонил)амино]-2-фенилциклопропанкарбоновой кислоты с n-пропиламином в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и последующим отсоединением группы Boc трифторуксусной кислотой (выход: 85% от теоретического количества за два этапа).
ВЭЖХ (Метод 6): Rt=1,2 мин;
ЖХ-МС (Метод 1): Rt=0,52 мин; MS (ESIпоз): ш/z=219 (M+H)+.
Промежуточное соединение 28
Этил (1S,2R)-1-амино-2-фенилциклопропанкарбоксилат трифторацетат
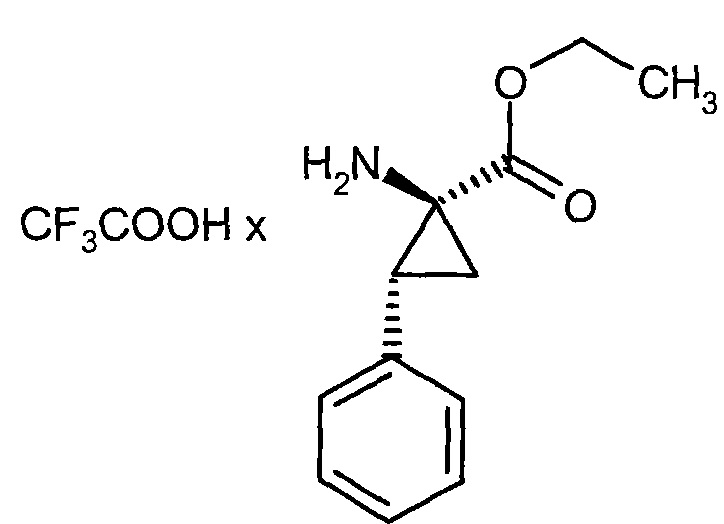
Указанное в названии соединение получали стандартными методами, этерификацией коммерчески доступного (1S,2R)-1-[(трет-йуотоксукарбонил)амино]-2-фенилциклопропанкарбоновой кислоты с этанолом и последующим отсоединением группы Boc трифторуксусной кислотой.
ЖХ-МС (Метод 1): Rt=0,50 мин; MS (ESIпоз): m/z=206 (М+Н)+.
Промежуточное соединение 29
4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутановая кислота

К раствору 1,39 г (8,95 ммоль) N-метоксикарбонилмалеимида в 44 мл насыщенного раствора бикарбоната натрия добавляли, при температуре 0°С, 1,5 г (8,95 ммоль) 4-амино-2,2-диметилмасляной кислоты, и перемешивали реакционную смесь в течение 40 мин. Затем охлаждающую ванну удаляли, и перемешивали реакционную смесь в течение еще одного часа. При охлаждении льдом, реакционную смесь доводили до уровня рН 3 добавлением серной кислоты, и экстрагировали этилацетатом. Объединенные оганические фазы сушили над сульфатом магния и концентрировали с получением 1,17 г (79% чистоты, 49% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1); Rt=0,64 мин; m/z=212 (М+Н)+.
Промежуточное соединение 30
трет-бутил 2-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутаноил]гидразинкарбоксилат

К раствору 50 мг (237 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутановой кислоты в 2 мл ТГФ при температуре 0°С добавляли сначала 26 мкл (237 мкМоль) 4-метилморфолина и затем 31 мкл (237 мкМоль) изобутил хлорформата. Затем удаляли охлаждающую баню, и перемешивали при комнатной температуре в течение еще 15 мин, добавляли 31,3 мг (237 мкМоль) трет-бутилоксикарбонил гидразида. Реакционную смесь перемешивали в течение ночи, и затем концентрировали. Осадок очищали методом препаративной ВЭЖХ, с получением 50,8 мг (66% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,71 мин; m/z=324 (N-Н)+.
Промежуточное соединение 31
4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутангидразидтрифторацетат

50 мг (154 ммоль) трет-бутил 2-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутаноил]гидразинкарбоксилат растворяли в 2 мл дихлорметана, и добавляли 0.4 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем концентрировали с получением 55,2 мг (93% чистоты, 99% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,36 мин; m/z=226 (М+Н)+.
Промежуточное соединение 32
Адамантан-1-илметил N-(трет-бутоксикарбонил)-L-фенилаланинат

К раствору 500 мг (1,89 ммоль) N-Boc-L-фенилаланина в 25 мл дихлорметана при комнатной температуре добавляли 1192 мг (6,2 ммоль) EDC, 578 мкл (4,1 ммоль) триэтиламина, 345 мг (2,8 ммоль) DMAP и 345 мг (2,1 ммоль) 1-адамантилметанол. Реакционную смесь перемешивали в течение ночи, затем разбавляли 50 мл дихлорметана, и последовательно промывали 10% водным раствором лимонной кислоты, водой и насыщенным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния, затем концентрировали, и очищали осадок методом препаративной ВЭЖХ, с получением 769 мг (90% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 2): Rt=1,84 мин; m/z=414 (М+Н)+.
Промежуточное соединение 33
Адамантан-1-илметил L-фенилаланинат гидрохлорид

769 мг (1,86 ммоль) адамантан-1-илметил N-(трет-бутоксикарбонил)-L-фенилаланината (промежуточное соединение 13) растворяли в 25 мл 4 н. раствора соляной кислоты в диоксане и перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали, и сушили осадок при пониженном давлении, с получением 619 мг (95% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,82 мин; m/z=314 (М+Н)+.
Промежуточное соединение 34
N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(Адамантан-1-илметокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 20 мг (29 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в 1 мл ДМФ при комнатной температуре добавляли 15,3 мкл (88 мкМоль) N,N-диизопропилэтиламина, 6,7 мг (44 мкМоль) HOBt и 6,7 мг (35 мкМоль) EDC, и перемешивали реакционную смесь в течение 30 мин. Затем добавляли 10,1 мг (32 мкМоль) Адамантан-1-ил L-фенилаланинат гидрохлорида. Перемешивали в течение ночи, после чего реакционную смесь непосредственно разделяли на составляющие методом препаративной ВЭЖХ, с получением 27,5 мг (93% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=1,70 мин; m/z=980 (М+Н)+.
Промежуточное соединение 35
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(Адамантан-1-илметокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамидтрифторацетат

27,5 мг (28 мкМоль) N-трет-бутоксикарбонил)-N-метил-L-валил-N-(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(Адамантан-1-илметокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 1,8 мл дихлорметана, после чего добавляли 361 мкл TFA. Реакционную смесь перемешивали в течение 30 мин и затем концентрировали. Осадок помещали в воду и лиофилизировали, с получением 22,7 мг (81% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=1,14 мин; m/z=880 (М+Н)+.
Промежуточное соединение 36
трет-бутип (2S)-1-(бензилокси)-3-фенилпропан-2-ил карбамат

В атмосфере аргона 500 мг (1,99 ммоль) N-Boc-L-фенилаланинол растворяли в 5 мл ДМФ и охлаждали до 0°С. Затем добавляли 159 мг (3,98 ммоль) 60% суспензии гидрида натрия в параффиновом масле. Реакционную смесь перемешивали до тех пор, пока не прекратится выделение газа, и затем добавляли 260 мкл (2,19 ммоль) бензил бромида. Охлаждающую баню удаляли, и перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Затем реакционную смесь концентрировали, осадок помещали в ледяную воду, и экстрагировали реакционную смесь дихлорметаном. Органическую фазу промывали насыщенным раствором хлорида натрия, сушили над сульфатом магния и концентрировали. Осадок очищали методом препаративной ВЭЖХ, с получением 226 мг (33% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=1,28 мин; m/z=342 (М+Н)+.
Промежуточное соединение 37
(2S)-1-(бензилокси)-3-фенилпропан-2-амин гидрохлорид

220 мг (644 мкМоль) трет-бутил (2S)-1-(бензилокси)-3-фенилпропан-2-ил карбамата растворяли в 11 мл 4 н. раствора соляной кислоты в диоксане, и перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали, и сушили осадок при пониженном давлении, с получением 138 мг (77% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,65 мин; m/z=242 (М+Н)+.
Промежуточное соединение 38
N-(трет-буотоксукарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 20 мг (29 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в 1 мл ДМФ при комнатной температуре добавляли 15,3 мкл (88 мкМоль) N,N-диизопропилэтиламина, 6,7 мг (44 мкМоль) HOBt и 6,7 мг (35 мкМоль) EDC, и перемешивали реакционную смесь в течение 30 мин. Затем добавляли 7,8 мг (32 мкМоль) (2S)-1-(бензилокси)-3-фенилпропан-2-амин гидрохлорида. Перемешивали в течение ночи, после чего реакционную смесь непосредственно разделяли на составляющие методом препаративной ВЭЖХ, с получением 26 мг (98% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=1,51 мин; m/z=909 (М+Н)+.
Промежуточное соединение 39
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-фенилпропан-2-ил]амино}-1-метокеи-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

26 мг (29 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 1,8 мл дихлорметана, и добавляли 370 мкл TFA. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем концентрировали. Осадок помещали в воду и лиофилизировали, с получением 26.4 мг (колич.) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,97 мин; m/z=809 (М+Н)+.
Промежуточное соединение 40
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

50 мг (70 мкМоль) промежуточного соединения 26 и 11 мг (70 мкМоль) of (1S,2R)-2-амино-1-фенилпропан-1-ола в 10 мл ДМФ добавляли к смеси 42 мг (0,11 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 25 мкл N,N-диизопропилэтиламина, и перемешивали реакционную смесь при комнатной температуре в течение 5 мин. Затем осадок концентрировали и очищали методом препаративной ВЭЖХ. Соответствующие фракции объединяли, концентрировали, и сушили в высоком вакууме, с получением 49 мг (81%) защищенного промежуточного соединения. Затем защитную группу Boc удаляли при хорошо известных условиях трифторуксусной кислотой в дихлорметане. Концентрировали, после чего очищали указанное в названии соединение методом препаративной ВЭЖХ, и получали 26 мг (52%) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,65 мин;
ЖХ-МС (Метод 1): Rt=0,77 мин; MS (ESIпоз): m/z=718 (М+Н)+.
Промежуточное соединение 41
3-{2-[2-(2-аминоэтокси)этокси]этокси}пропановой кислоты трифторацетат

150 мг (541 мкМоль) трет-бутил 3-{2-[2-(2-аминоэтокси)этокси]этокси}пропаноата растворяли в 3 мл дихлорметана, добавляли 1,5 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 1 часа, после чего концентрировали.
Процедура давала 181 мг (100% от теоретического количества) указанного в названии соединения.
MS (EI): m/z 222 (М+Н)+.
Промежуточное соединение 42
3-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этокси]этокси}этокси)пропановая кислота

186 мг (555 мкМоль) 3-{2-[2-(2-аминоэтокси)этокси]этокси}пропановой кислоты трифторацетата растворяли в 2,6 мл насыщенного раствора бикарбоната натрия и добавляли к смеси при температуре 0°С 86 мг (555 мкМоль) N-метоксикарбонилмалеимида. Реакционную смесь перемешивали при температуре 0°С в течение 40 мин, и при комнатной температуре в течение 1 часа, затем опять охлаждали до 0°С, доводили до уровня рН 3 с использованием серной кислоты, и экстрагировали 3 раза 25 мл этилацетата. Объединенные оганические фазы сушили над сульфатом магния и концентрировали, с получением 126 мг (75% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,53 мин; m/z=302 (М+Н)+.
Промежуточное соединение 43
трет-бутил 15-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-4-оксо-7,10,13-триокса-2,3-диазапентадекан-1-оат

125 мг (417 мкМоль) 3-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этокси]этокси}этокси) пропановой кислоты растворяли при температуре 0°С в 2,1 мл ТГФ и добавляли к смеси 46 мкл (417 ммоль) 4-метилморфолина и 54,5 мкл (417 мкМоль) изобутил хлорформата. Ледяную баню удаляли, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем при температуре 0°С добавляли 55 мг (417 мкМоль) трет-бутилоксикарбонил гидразида. Реакционную смесь нагревали до комнатной температуры в течение ночи, концентрировали очищали методом препаративной ВЭЖХ.
В результате получали 60 мг (33% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,66 мин; m/z=416 (М+Н)+.
Промежуточное соединение 44
3-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этокси]этокси}этокси)пропангидразид трифторацетат

60 мг (145 мкМоль) трет-бутил 15-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-4-оксо-7,10,13-триокса-2,3-диазапентадекан-1-оата растворяли в 1 мл дихлорметана, и добавляли 0,2 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем концентрировали.
В результате получали 62 мг (100% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,35 мин; m/z=316 (М+Н)+.
Промежуточное соединение 45
Бензил (1S,2R)-1-амино-2-фенилциклопропанкарбоксилат трифторацетат

Указанное в названии соединение получали стандартными методами, этерификацией коммерчески доступной (1S,2R)-1-[(трет-бутоксикарбонил)амино]-2-фенилциклопропанкарбоновой кислоты с бензиловым спиртом и последующим отсоединением группы Boc трифторуксусной кислотой.
ЖХ-МС (Метод 1): Rt=0,72 мин; MS (ESIпоз): m/z=268 (М+Н)+.
Промежуточное соединение 46
N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

383 мг (0,743 ммоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 8) объединяли с 485 мг (0,743 ммоль) бензил N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил-L-фенилаланинат трифторацетата (промежуточное соединение 12), 424 мг (1,114 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 388 мкл N,N-диизопропилэтиламина в 15 мл ДМФ, и перемешивали реакционную смесь при комнатной температуре в течение 10 мин. Затем удаляли растворитель при пониженном давлении. Оставшийся осадок помещали в этилацетат и экстрагировали встряхиванием последовательно с 5% водным раствором лимонной кислоты, и насыщенным раствором бикарбоната натрия. Органическую фазу отделяли и концентрировали, затем очищали осадок методом препаративной ВЭЖХ. Объединенные фракции полученного продукта концентрировали, после чего сушили осадок в высоком вакууме. В результате поучали 335 мг (48% от теоретического количества) бензил эфира промежуточного соединения в виде пены.
ЖХ-МС (Метод 1): Rt=1.49 мин; MS (ЕSIпо): m/z=922 (М+Н)+.
100 мг (0,11 ммоль) этого промежуточного вещества помещали в 15 мл метанола и удаляли группу бензилового эфира гидрогенизацией при стандартном давлении водорода с 10% палладием на активированном угле в качестве катализатора. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, катализатор отфильтровывали, и концентрировали фильтрат при пониженном давлении. После лиофилизации из диоксана, получали 85 мг (94% от теоретического количества) указанного в названии соединения в виде твердого вещества.
ВЭЖХ (Метод 12): Rt=2.4 мин;
ЖХ-МС (Метод 1): Rt=1,24 мин; MS (ESIпоз): m/z=832 (М+Н)+.
Промежуточное соединение 47
N-бензил-L-триптофанамидтрифторацетат

202 мг (0,5 ммоль) 2,5-диоксопирролидин-1-ил N-(трет-бутоксикарбонил)-L-триптофаната и 45 мг (0.42 ммоль) бензиламина растворяли в 10 мл ДМФ, и добавляли 110 мкл (630 мкМоль) N,N-диизопропилэтиламин. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Затем реакционную смесь концентрировали при пониженном давлении, очищали осадок методом флэш-хроматографии на силикагеле (элюент: 20:0,5:0,05 дихлорметан/метанол/17% вод. амиак). Соответствующие фракции объединяли и концентрировали. Полученный осадок дегидрирование диэтиловым эфиром, и затем сушили в высоком вакууме. Затем этот осадок помещали в 10 мл дихлорметана, и добавляли 3 мл безводной трифторуксусной кислоты. Перемешивали при комнатной температуре в течение 45 мин, после чего реакционную смесь концентрировали очищали осадок методом препаративной ВЭЖХ. После сушки в высоком вакууме, получали 117 мг (57% то теоретического количества после двух этапов синтеза) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,66 мин; MS (ESIпоз): m/z=294 (М+Н)+.
Промежуточное соединение 48
(1S,2R)-1-амино-2-фенилциклопропанкарбоксамид трифторацетат

50 мг (180 мкМоль) коммерчески доступной (1S,2R)-1-[(терете-бутоксикарбонил)амино]-2-фенилциклопропанкарбоновой кислоты растворяли в 5 мл ДМФ, добавляли 94 мкл (541 мкМоль) N,N-диизопропилэтиламина, 31 мг (270 мкМоль) N-гидроксисукцинимида и 41,5 мг (216 мкМоль) EDC, и затем смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали, осадок помещали в диоксан, добавляли 71 мг (901 мкМоль) бикарбоната аммония, и оставляли стоять реакционную смесь при комнатной температуре в течение 3 дней. Затем реакционную смесь разбавляли 1:1 смесью этилацетата и воды. Органическую фазу отделяли, сушили над сульфатом магния и концентрировали. Полученный осадок последовательно помещали в 3 мл дихлорметана, и добавляли 3 мл безводной трифторуксусной кислоты. Перемешивали при комнатной температуре в течение 1 часа, после чего концентрировали. Осадок перемешивали с пентаном, отфильтровывали с отсасыванием, и лиофилизировали из диоксана. Таким образом, получали 32 мг (62% от теоретического количества после двух этапов синтеза) указанного в названии соединения.
ВЭЖХ (Метод 6): Rt=0,38 мин;
ЖХ-МС (Метод 1): Rt=0,20 мин; MS (ESIпоз): m/z=177 (М+Н)+.
Промежуточное соединение 49
Nα-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-триптофанамид трифторацетат
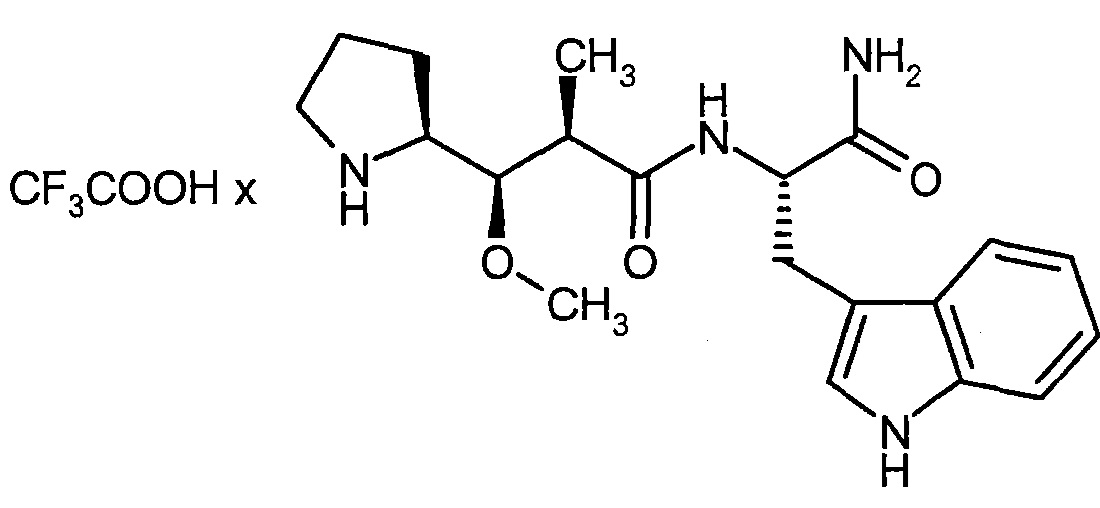
Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 13 из исходного соединения 1 и L-триптофанамид гидрохлорида.
ВЭЖХ (Метод 5): Rt=1.4 мин;
ЖХ-МС (Метод 1): Rt=0,92 мин; MS (ESIпоз): m/z=473 (М+Н)+.
Промежуточное соединение 50
4-нитрофенил 2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил карбамат

813 мг (3,1 ммоль) трифенилфосфина растворяли в 25 мл ТГФ и охлаждали до -70°С в атмосфере аргона. Затем ао каплям добавляли 627 мг (3,1 ммоль) диизопропил азодикарбоксилата, после чего смесь перемешивали в течение 5 мин.Затем по каплям добавляли 500 мг (3,1 ммоль) трет-бутил 2-аминоэтил карбамат растворенного в 5 мл ТГФ, и перемешивали реакционную смесь при температуре -70°С в течение еще 5 мин. Затем добавляли 136,6 мг (1,55 ммоль) 2,2-диметил-1-пропанола растворенного в 1 мл ТГФ и 301 мг (3,1 ммоль) малеимида, реакционную смесь перемешивали при температуре -70°С в течение еще 10 мин и затем смесь нагревали до комнатной температуры. Перемешивали при комнатной температуре в течение еще 16 часов, и удаляли растворитель при пониженном давлении, после чего очищали осадок методом препаративной ВЭЖХ. Процедура давала 463 мг (62%) защищенного промежуточного соединения.
Защитную группу Boc удаляли при стандартных условиях, с получением 652 мг 1-(2-аминоэтил)-1H-пиррол-2,5-диона в виде трифторацетата.
112,9 мг (543 мкМоль) нитрофенил хлорформата растворяли в 30 мл ТГФ и затем добавляли 100 мг (271,6 мкМоль) 1-(2-аминоэтил)-1H-пиррол-2,5-дион трифторацетата, смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь фильтровали, и концентрировали фильтрат до сухого состояния, после чего раежирали с диэтиловым эфиром. После фильтрации с отсасыванием и сушки, получали 60 мг (95% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=0,65 мин;
ЖХ-МС (Метод 1): Rt=0,74 мин; MS (ESIпоз): m/z=306 (М+Н)+.
Промежуточное соединение 51
(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этанаминтрифторацетат

200 мг (0,75 ммоль) N-(трет-бутоксикарбонил)-L-фенилаланин в начале процедуры помещали в реакционный сосуд с 5,5 мл дихлорметана при температуре 0°С, после чего добавляли 128 мг (0,79 ммоль) 1,1'-карбонилдиимидазол. Через 30 мин добавляли 103 мг (0,75 ммоль) бензоил гидразида. По прошествии еще 45 мин, при температуре 0°С добавляли 500 мг (1,5 ммоль) тетрабромметана и, в конце, 395 мг (1,5 ммоль) трифенилфосфина. Реакционную смесь перемешивали сначала при температуре 0°С в течение 2 часов, и затем при комнатной температуре в течение ночи. Реакционную смесь концентрировали в роторном испарителе, после чего сушили осадок в высоком вакууме. Сырой продукт, полученный таким образом, очищали методом препаративной ВЭЖХ. В результате получали 217 мг (78% от теоретического количества) защищенного группой Boc промежуточного соединения трет-бутил (15)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил карбамат.
ЖХ-МС (Метод 12): Rt=1,15 мин; MS (ESIпоз): m/z=366 (M+H)+
217 мг (0,59 ммоль) этого промежуточного вещества помещали в 3 мл дихлорметана, добавляли 0,6 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении. Затем реакционную смесь сушили при пониженном давлении, после чего полученный осадок лиофилизировали из диоксана. Таким образом получали 214 мг (90% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 11): Rt=0,62 мин; MS (ESIпоз): m/z=266 (M+H)+
Промежуточное соединение 52
(1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этанаминтрифторацетат

200 мг (0,75 ммоль) N-(трет-бутоксикарбонил)-D-фенилаланина в начале процедуры помещали в реакционный сосуд с 5,5 мл дихлорметана при температуре 0°С, и добавляли 128,3 мг (0,79 ммоль) 1,1'-карбонилдиимидазола. Через 30 мин добавляли 103 мг (0,75 ммоль) бензоил гидразида. По прошествии еще 45 мин при температуре 0°С добавляли 500 мг (1,5 ммоль) тетрабромметана, и, наконец, 395 мг (1,5 ммоль) трифенилфосфина. Реакционную смесь перемешивали сначала при температуре 0°С в течение 2 часов, и затем при комнатной температуре в течение ночи. После этого реакционную смесь концентрировали в роторном испарителе, и сушили осадок в высоком вакууме. Сырой продукт, полученный таким образом, очищали методом препаративной ВЭЖХ. Таким образом получали 219 мг (80% от теоретического количества) защищенного группой Boc промежуточного соединения трет-бутил (1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил карбамат.
ЖХ-МС (Метод 2): Rt=1,36 мин; MS (ESIпоз): m/z=366 (М+Н)+
219 мг (0,6 ммоль) этого промежуточного вещества помещали в 3 мл дихлорметана, добавляли 0,6 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении. Затем реакционную смесь сушили при пониженном давлении, после чего полученный осадок лиофилизировали из диоксана. Таким образом получали 196 мг (86% от теоретического количества) указанного в названии соединения в виде твердого вещества.
ВЭЖХ (Метод 10): Rt=2.41 мин
Промежуточное соединение 53
(2S)-1-(бензилсульфонил)-3-фенилпропан-2-амин

200 мг (1,13 ммоль) (4S)-4-бензил-1,3-оксазолидин-2-она в начале процедуры помещали в реакционный сосуд с 3 мл трет-бутанола, после чего добавлляи 280 мг (2,26 ммоль) бензил меркаптана. После этого реакционную смесь нагревали с обратным холодильником в течение 2 дней. Затем реакционную смесь концентрировали в роторном испарителе, после чего полученный (2S)-1-(бензилсульфанил)-3-фенилпропан-2-амин промежуточного соединения использовали на следующем этапе без дополнительной обработки.
ВЭЖХ (Метод 10): Rt=2,63 мин
ЖХ-МС (Метод 1): Rt=0,67 мин; MS (ESIпоз): m/z=258 (М+Н)+
Сырое промежуточное соединение полученное, как описано выше, растворяли в 2 мл 30% раствора перекиси водорода и 5 мл муравьиной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 12 часов. Затем реакционную смесь добавляли к насыщенному раствору сульфата натрия, и экстрагировали три раза с этилацетатом. Органическую фазу сушили над сульфатом магния, и концентрировали при пониженном давлении. Полученный сырой продукт очищали методом препаративной ВЭЖХ. Таким образом получалии 343 мг (61% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 10): Rt=2.40 мин;
ЖХ-МС (Метод 12): Rt=0,65 мин; MS (ESIпоз): m/z=290 (M+H)+
Промежуточное соединение 54
(2S,3E)-1,4-дифенилбут-3-ен-2-амин

552,7 мг (9,85 ммоль) гидроксида калия растворяли в метаноле, адсорбировали на 1,1 г нейтрального оксида алюминия, и затем сушили в высоком вакууме. К раствору 240 мг (0,82 ммоль) (2S)-1-(бензилсульфонил)-3-фенилпропан-2-амина и 1,56 г гидроксида калия на оксиде алюминия, приготовленного таким образом, в 6,2 мл n-бутанола капельно, при температуре 5-10°С, добавляли 307 мкл (3,3 ммоль) дибромдифторметана. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем фильтровали через целит, и тщательно промывали осадок дихлорметаном. Фильтрат концентрировали, и сушили полученный осадок при пониженном давлении. Сырой продукт, полученный таким образом, очищали методом препаративной ВЭЖХ. Получали 98 мг (35% от теоретического количества) указанного в названии соединения с соотношением E/Z диастереомеров 4:1.
ВЭЖХ (Метод 10): Rt=2.46 мин;
ЖХ-МС (Метод 12): Rt=0,75 мин; MS (ESIпоз): m/z=224 (M+H)+
Смесь E/Z диастереомеров, полученных, как описано выше, растворяли в 2 мл этанола и 0,2 мл N,N-диизопропилэтиламина, и сепарировали посредством хиральной ВЭЖХ [колонка: Daicel Chiralpak AD-H, 5 мкм 250 мм × 20 мм, элюент: гексан/(этанол+0,2% диэтиламин) 50:50 объем/объем; УФ детектирование: 220 нм; температура: 30°С]. Нужные фракции концентрировали в роторном испарителе, и сушили осадок при пониженном давлении. В результате получали 45 мг указанного в названии соединения.
1Н ЯМР (400 МГц, ДМСО-d6) δ [ppm]=2,62-2,83 (m, 2H) 3,52-3,71 (m, 1H) 6,18-6,30 (m, 1H) 6,34-6.46 (m, 1H) 6,98-7,57 (m, 10H) [другие сигналы перекрыты пиками от растворителей].
Промежуточное соединение 55
N-метил-L-валил-Н-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

20 мг (29 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 1 мл ДМФ, добавляли 13,3 мг (35 мкМоль) HATU и 15,3 мкл (88 мкМоль) N,N-диизопропилэтиламина, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем добавляли 12,2 мг (32 мкМоль) (1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этанамин трифторацетата. Реакционную смесь перемешивали при комнатной температуре в течение ночи, и затем сепарировали методом препаративной ВЭЖХ. Процедура давала 22 мг (81% от теоретического количества) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида.
ЖХ-МС (Метод 12): Rt=1.45 мин; MS (ESIпоз): m/z=933 (М+Н)+
При последующем удалении защитной группы ВОС трифторуксусной кислотой, получали 22.4 мг (98% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 11): Rt=0,85 мин; MS (ESIпоз): m/z=833 (M+H)+
Промежуточное соединение 56
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокеи-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид получали, следуя процедуре, аналогичной синтезу промежуточного соединения 55, реакцией 20 мг (29 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1 -метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида с 12,2 мг (32 мкМоль) (1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этанамин трифторацетата.
Выход: 17 мг (64% от теоретического количества)
ВЭЖХ (Метод 10): Rt=3,74 мин;
ЖХ-МС (Метод 1): Rt=1.45 мин; MS (ESIпоз): m/z=933 (M+H)+
При последующем удалении защитной группы ВОС трифторуксусной кислотой, получали 17,1 мг (99% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 10): Rt=2,55 мин;
ЖХ-МС (Метод II): Rt=0,85 мин; MS (ESIпоз): m/z=833 (М+Н)+
Промежуточное соединение 57
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилсульфонил)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилсульфонил)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид получали, следуя процедуре, аналогичной синтезу промежуточного соединения 55, реакцией 20 мг (29 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида с 9,3 мг (20 мкМоль) (25)-1-(бензилсульфонил)-3-фенилпропан-2-амина.
Выход: 19,2 мг (68% от теоретического количества)
ВЭЖХ (Метод 10): Rt=3,5 мин;
ЖХ-МС (Метод 12): Rt=1.41 мин; MS (ЕЗЪпоз): m/z=957 (M+H)+
При последующем удалении защитной группы ВОС трифторуксусной кислотой, получали 19,3 мг (99% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 10): Rt=2,52 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=857 (M+Н)+
Промежуточное соединение 58
N-метил-L-валил-N-(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3Е)-1,4-дифенилбут-3-ен-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3 -метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3Е)-1,4-дифенилбут-3-ен-2-ил]амино}-1 -метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид получали, следуя процедуре, аналогичной синтезу промежуточного соединения 55, осуществляя реакцию 20 мг (29 мкМоль) N-(трет-бутоксикарбонил-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[{1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида с 7,1 мг (32 мкМоль) (2S,3E)-1,4-дифенилбут-3-ен-2-амина.
Выход: 15,1 мг (58% от теоретического количества)
ВЭЖХ (Метод 10): Rt=4,2 мин;
ЖХ-МС (Метод 12): Rt=1,51 мин; MS (ESIпоз): m/z=891 (M+H)+
При последующем удалении защитной группы ВОС трифторуксусной кислотой, получали 15,7 мг (99% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 10): Rt=2,62 мин;
ЖХ-МС (Метод 12): Rt=0,97 мин; MS (ESIпоз): m/z=791 (M+H)+
Промежуточное соединение 61
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
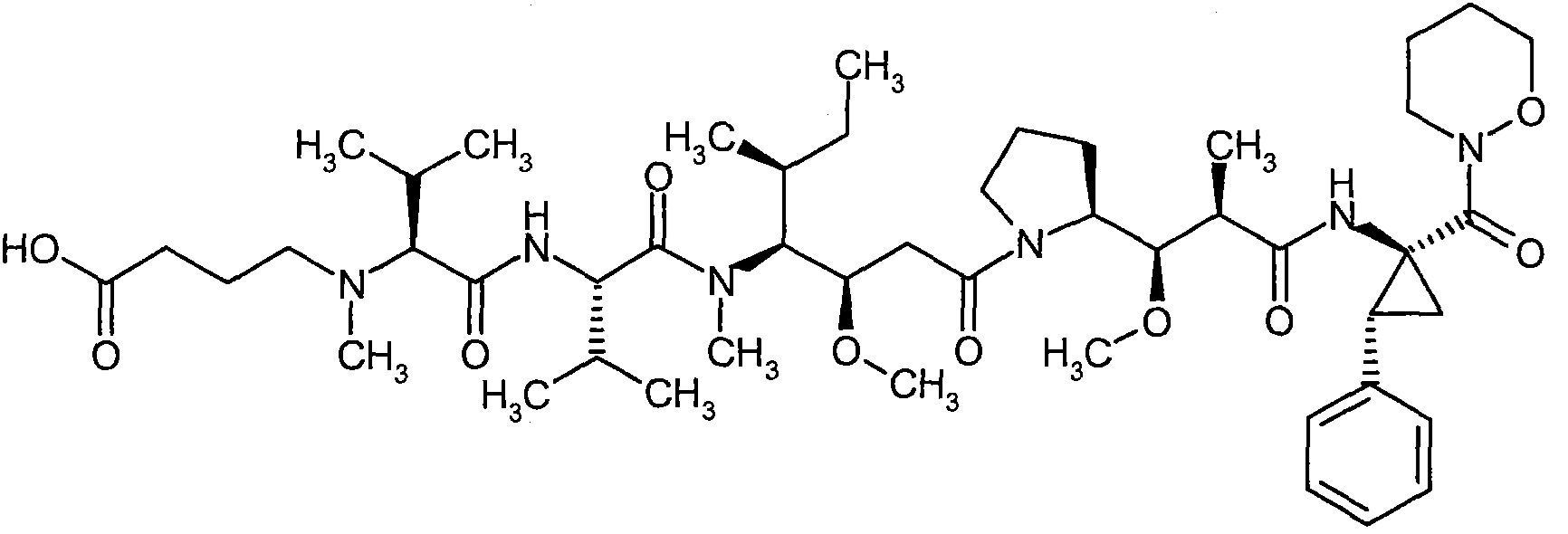
50 мг (0,054 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 16) растворяли в 8 мл раствора диоксан/вода, после чего добавляли добавляли 70 мл (0,108 ммоль) 15% раствора 4-оксобутановой кислоты в воде. После этого реакционную смесь перемешивали при температуре 100°С в течение 1 часа. Затем охлаждали до комнатной температуры, после чего добавляли 3,7 мг (0,059 ммоль) цианоборогидрида натрия, и доводили реакционную смесь до уровня рН 3 добавлением примерно 300 мкл 0,1 н. соляной кислоты. Затем реакционную смесь перемешивали при температуре 100°С в течение еще 2 часов. После охлаждения, добавляли еще 70 мл (0,108 ммоль) 15% раствора 4-оксобутановой кислоты, и снова перемешивали реакционную смесь при температуре 100°С в течение 1 часа. Затем добавляли еще 3,7 мг (0,059 ммоль) цианоборогидрида натрия, после чего использовали примерно 300 мкл 0,1 н. соляной кислоты до достижения уровня рН, снова равного 3. Затем реакционную смесь перемешивали при температуре 100°С в течение еще 2 часов. Пока превращение еще не завершено, указанную процедуру повторяли в третий раз. После этого реакционную смесь концентрировали очищали осадок методом препаративной ВЭЖХ. Таким образом, получали 32 мг (65% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,64 мин;
ЖХ-МС (Метод 9): Rt=4,76 мин; MS (ESIпоз): m/z=899 (М+Н)+
1Н ЯМР (500 МГц, ДМСО-d6): δ=8,95 и 8,8 (2m, 1Н), 8,88 и 8,65 (2s, 1H), 7.4-7,1 (m, 5H), 5,0, 4,78, 4,65 и 4,55 (4m, 2H), 4,1-3,7 (m, 5H), 3,32, 3,29, 3,20, 3,12, 3,1 и 3,0 (6s, 9H), 2,75 (m, 2H), 2,63 (t, 1H), 2.4-2,2 (m, 4H), 2,1-1,2 (m, 12H), 1,2-0,8 (m, 16H), 0,75 (m, 3H) [другие сигналы перекрыты пиками H2O и ДМСО].
Промежуточное соединение 62
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 61, осуществляя реакцию 50 мг N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 14) с 4-оксобутановой кислотой.
Выход: 34 мг (70% от теоретического количества)
ВЭЖХ (Метод 5): Rt=1,64 мин;
ЖХ-МС (Метод 9): Rt=4,77 мин; MS (ESIпоз): m/z=887 (M+H)+.
Промежуточное соединение 63
N-(4-карбоксибензил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 61, осуществляя реакцию 15 мг N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 16) с 4-формилбензойной кислотой.
Выход: 7,5 мг (48% от теоретического количества)
ВЭЖХ (Метод 5): Rt=1,75 мин;
ЖХ-МС (Метод 1): Rr=0,97 мин; MS (ESIпоз): m/z=947 (М+Н)+.
Промежуточное соединение 64
N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил] амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
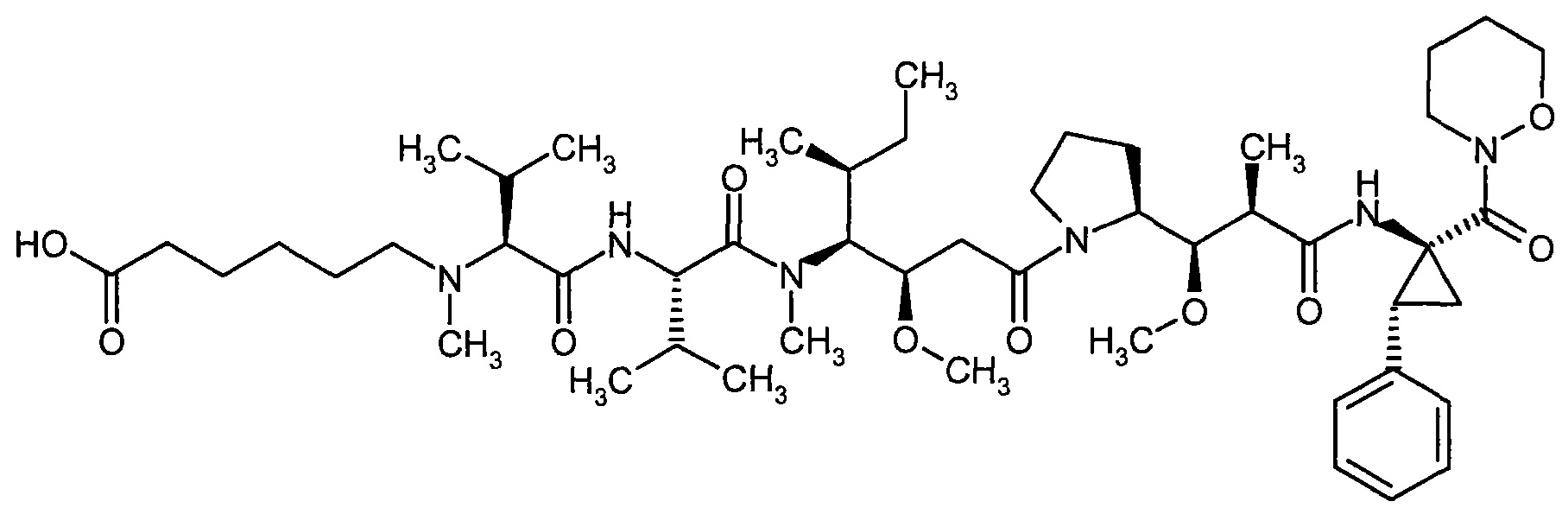
10 мг (0,011 ммоль) N-метил-L-валил-N-(3R,4S,5S)-3-метокси-1-(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 16) растворяли в 2 мл раствора диоксан/вода, и добавляли 2,8 мг (0,022 ммоль) 6-оксокапроновой кислоты. После этого, реакционную смесь перемешивали при температуре 100°С в течение 1 часа. Затем охлаждали до комнатной температуры, добавляли 0,75 мг (0,012 ммоль) цианоборогидрида натрия, и доводили реакционную смесь до уровня рН 3 добавлением 0,1 н. соляной кислоты. Затем реакционную смесь перемешивали при температуре 100°С в течение еще часа. После охлаждения, добавляли еще 2,8 мг (0,022 ммоль) 6-оксокапроновой кислоты, и снова перемешивали реакционную смесь при температуре 100°С в течение 1 часа. Добавляли еще 0,75 мг (0,012 ммоль) цианоборогидрида натрия, после чего использовали 0,1 н. соляной кислоты до достижения уровня рН снова равного 3. Затем реакционную смесь перемешивали при температуре 100°С в течение еще 1 часа. Описанную процедуру затем повторяли в третий раз. После этого, реакционную смесь концентрировали, очищали сырой продукт методом препаративной ВЭЖХ. Процедура давала 6.4 мг (64% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,68 мин;
ЖХ-МС (Метод 9): Rt=4,86 мин; MS (ESIпоз): m/z=927 (М+Н)+.
Промежуточное соединение 65
N-(2-аминоэтил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид бистрифторацетат

Указанное в названии соединение получали осуществляя реакцию 68 мг N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[{1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 14) с трет-бутш 2-оксоэтил карбаматом, и последующим удалением защитной группы Boc трифторуксусной кислотой.
Выход: 49 мг (62% от теоретического количества после двух этапов синтеза)
ВЭЖХ (Метод 5): Rt=1,58 мин;
ЖХ-МС (Метод 2): Rt=1,05 мин; MS (ESIпоз): m/z=844 (M+H)+
1Н ЯМР (600 МГц, ДМСО-d6): δ=8,25 (m, 1Н), 8.45 и 8,15 (2d, 1H), 7,65-7,55 (m, 3H), 7,23-7,1 (m, 5H), 5,12 и 4,95 (2m, 1H), 4,72 и 4,62 (2m, 1H), 4,6 и 4,52 (2t, 1H), 4,2-3,8 (m, 4H), 3,7 (d, 1H), 3,23, 3,20, 3,19, 3,18, 3,03 и 2,98 (6s, 9H), 3,0-2,7 (m, 6H), 2.4-1,2 (m, 15H), 1,05, 1,0, 0,88 и 0,82 (4d, 6H), 0,92 (m, 6H), 0,73 (m, 6H) [другие сигналы перекрыты пиками Н2О peak].
Промежуточное соединение 66
N-(3-аминопропил)-N-метил-L-валил-N-[(3R,4S,5S)-N-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино} -3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
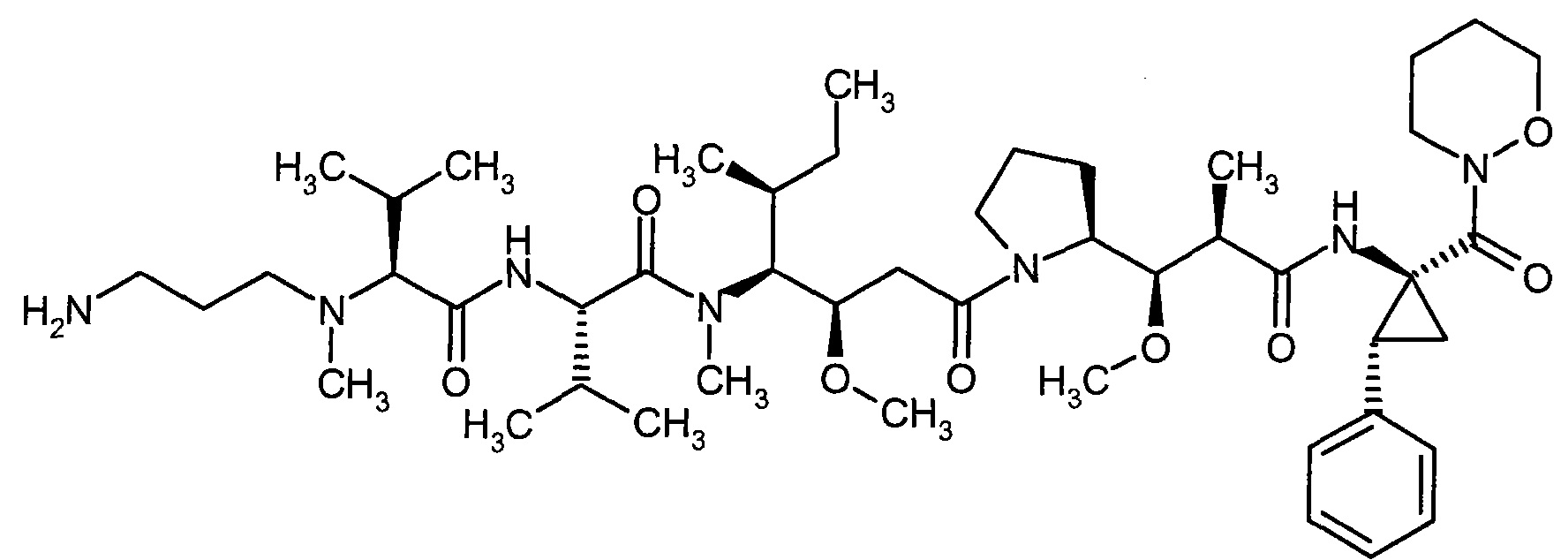
Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 65, осуществляя реакцию 25 мг (0,027 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 16) с бензил 3-оксопропил карбаматом и последующим гидролитическим удалением защитной группы Z (с 10% палладием на угле в качестве катализатора, в этаноле в качестве растворителя).
Выход: 11 мг (41% от теоретического количества после двух этапов синтеза)
ВЭЖХ (Метод 5): Rt=1,53 мин;
ЖХ-МС (Метод 1): Rt=0,72 мин; MS (ESIпоз): m/z=870 (M+H)+.
Промежуточное соединение 67
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(Адамантан-1-илметокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

26 мг (26 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(Адамантан-1-илметокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата и 33,9 мкл 15% водного раствора кислого сукцинальдегида (53 мкМоль) растворяли в 957 мкл 1:1-смеси диоксан/вода и нагревали до 100°С в течение 1 часа. После непродолжительного охлаждения, добавляли 1,81 мг (29 мкМоль) цианоборогидрида натрия. Реакционную смесь доводили до уровня рН 3 добавлением 0,1 н. соляной кислоты, и нагревали реакционную смесь до 100°С в течение еще 2 часов. После повторного добавления того же количества раствора кислого сукцинальдегида, цианоборогидрида натрия и соляной кислоты, реакционную смесь снова нагревали до 100°С в течение 2 часов. Затем реакционную смесь непосредственно разделяли на составляющие методом препаративной ВЭЖХ. Таким образом получали 18,5 мг (73% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=1,17 мин; m/z=967 (M+H)+.
Промежуточное соединение 68
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

24 мг (26 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата и 33,7 мкл 15% водного раствора кислого сукцинальдегида (52 мкМоль) растворяли в 953 мкл 1:1-смеси диоксан/вода и нагревали до 100°С в течение 1 часа. После непродолжительного охлаждения, добавляли 1,80 мг (29 мкМоль) цианоборогидрида натрия. Реакционную смесь доводили до уровня рН 3 добавлением 0,1 н. соляной кислоты, и нагревали до 100°С в течение еще 2 часов. После повторного добавления тех же количеств раствора кислого сукцинальдегида, цианоборогидрида натрия и соляной кислоты, реакционную смесь снова нагревали до 100°С в течение 2 часов. Затем реакционную смесь непосредственно разделяли на составляющие методом препаративной ВЭЖХ. Таким образом получали 15,2 мг (65% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=1,01 мин; m/z=895 (M+H)+
Промежуточное соединение 69
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

53 мг (84 мкМоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) и 45 мг (84 мкМоль) бензил N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-фенилаланинат трифторацетата (промежуточное соединение 12) помещали в 2 мл ДМФ, добавляли 19 мкл N,N-диизопропилэтиламин, 14 мг (92 мкМоль) HOBt и 17,6 мг (92 мкМоль) EDC, и затем смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали, и очищали осадок методом препаративной ВЭЖХ. Процедура давала 59 мг (68% от теоретического количества) защищенного группой Fmoc промежуточного соединения N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ЖХ-МС (Метод 1): Rt=1,55 мин; m/z=1044 (М+Н)+.
57 мг (0,055 ммоль) этого промежуточного вещества обрабатывали 1,2 мл пиперидина в 5 мл ДМФ для удаления защитной группы Fmoc. После концентрирования и очистки методом препаративной ВЭЖХ, получали 39 мг (76% от теоретического количества) свободного амина промежуточного соединения N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в виде трифторацетата.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=1,01 мин; m/z=822 (М+Н)+.
37 мг (0,045 ммоль) этого промежуточного вещества растворяли в 5 мл раствора диоксан/вода и, следуя процедуре аналогичной получению соединения в примере Промежуточного соединения 66, осуществляли реакцию с 15% водным раствором 4-оксобутановой кислоты в присутствии цианоборогидрида натрия. В результате получали 1 6 мг (39% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,01 мин; MS (ESIпоз): m/z=908 (M+H)+.
Промежуточное соединение 70
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3S)-1-(бензилокси)-1-оксо-3-фенилбутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 14, брали промежуточные соединения 4 и 26, и получали амин N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3S)-1-(бензилокси)-1-оксо-3-фенилбутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
30 мг (0,032 ммоль) этого соединения растворяли в 6 мл раствора диоксан/вода, и добавляли 41 мкл (0,063 ммоль) 15% водного раствора 4-оксобутановой кислоты. После этого реакционную смесь перемешивали при температуре 100°С в течение 1 часа. Затем охлаждали до комнатной температуры, добавляли 2,2 мг (0,035 ммоль) цианоборогидрида натрия, и доводили реакционную смесь до уровня рН 3 добавлением примерно 300 мкл 0,1 н. соляной кислоты. Затем реакционную смесь перемешивали при температуре 100°С в течение еще 2 часов. После охлаждения добавляли еще 41 мкл (0,063 ммоль) 15% раствора 4-оксобутановой кислоты, и снова перемешивали реакционную смесь при температуре 100°С в течение 1 часа. Затем добавляли еще 2,2 мг (0,035 ммоль) цианоборогидрида натрия, после чего использовали примерно 300 мкл 0,1 н. соляной кислоты до достижения уровня рН снова равного 3. После этого, реакционную смесь перемешивали при температуре 100°С в течение еще 2 часов. Пока превращение еще не завершено, указанную процедуру повторяли в третий раз. После этого реакционную смесь концентрировали, и очищали сырой продукт методом препаративной ВЭЖХ. Процедура давала 24 мг (82% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 9): Rt=5,15 мин; MS (ESIпоз): m/z=922 (M+H)+.
Промежуточное соединение 71
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
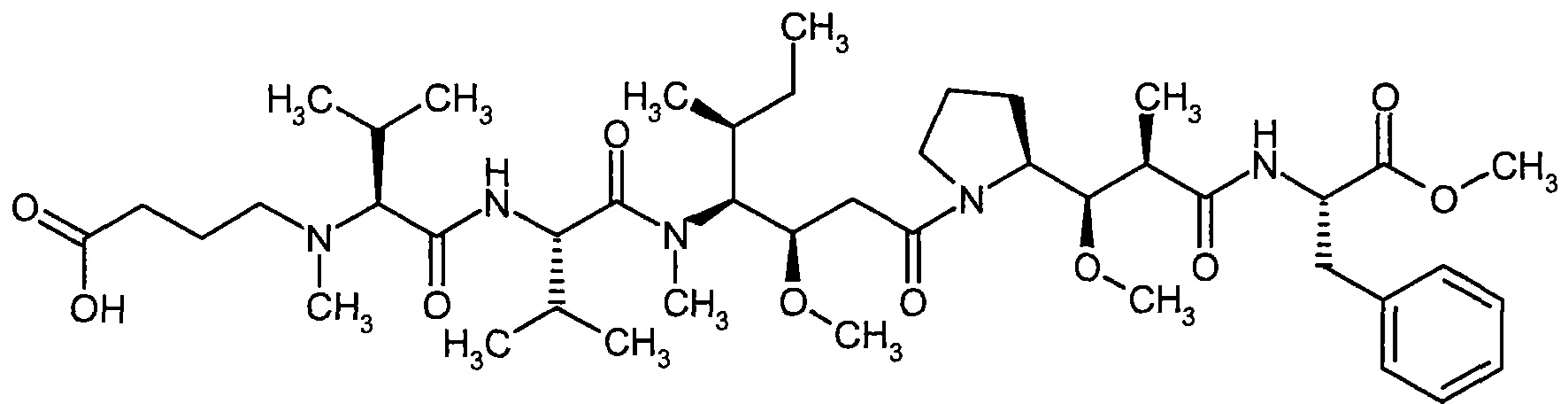
На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 14, вначале брали промежуточные соединения 4 и 39, и получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-3-{[(2S)-1-метокси-1-оксо-3-фенилпропан-2-ил]амино}-2-метил-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид. 7 мг (0,009 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 2 мг (22% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=1,9 мин;
ЖХ-МС (Метод 2): Rt=1,06 мин; MS (ESIпоз): m/z=832 (М+Н)+.
Промежуточное соединение 72
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

212 мг (411 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 8) и 237 мг (411 мкМоль) бензил-N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-триптофанат трифторацетата (промежуточное соединение 20) помещали в 30 мл ДМФ, и добавляли 188 мг (493 мкМоль) 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 215 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, затем концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. Объединенные фракции полученного продукта концентрировали, после чего сушили осадок в высоком вакууме. Процедура давала 315 мг (80% от теоретического количества) защищенного группой Boc промежуточного соединения N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде бесцветной пены.
ЖХ-МС (Метод 1): Rt=1.45 мин; m/z=961 (M+H)+.
50 мг (52 мкМоль) этого промежуточного вещества обрабатывали 1 мл трифторуксусной кислоты в 9 мл дихлорметана для удаления защитной группы Boc. После концентрирования и очистки методом препаративной ВЭЖХ, получали 29 мг (57% от теоретического количества) свободного амина промежуточного соединения N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата.
ЖХ-МС (Метод 1): Rt=0,99 мин; m/z=861 (М+Н)+.
29 мг (0,03 ммоль) этого промежуточного вещества растворяли в 6 мл раствора диоксан/вода, и добавляли 39 мкл (0,059 ммоль) 15% водного раствора 4-оксобутановой кислоты. После этого, реакционную смесь перемешивали при температуре 100°С в течение 1 часа. Затем охлаждали до комнатной температуры, добавляли 2 мг (0,033 ммоль) цианоборогидрида натрия, и доводили реакционную смесь до уровня рН 3 добавлением примерно 300 мкл 0,1 н. соляной кислоты. Затем реакционную смесь перемешивали при температуре 100°С в течение еще 2 часов. После охлаждения добавляли еще 39 мкл (0,059 ммоль) 15% раствора 4-оксобутановой кислоты, и снова перемешивали реакционную смесь при температуре 100°С в течение 1 часа. Затем добавляли еще 2 мг (0,033 ммоль) цианоборогидрида натрия, после чего использовали примерно 300 мкл 0,1 н. соляной кислоты до достижения уровня рН снова равного 3. Затем реакционную смесь перемешивали при температуре 100°С в течение еще 2 часов. После этого реакционную смесь наливали на 1:1 смесь полунасыщенного водного раствора хлорида аммония и этилацетата. Органическую фазу отделяли, промывали с насыщенным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали. Осадок лиофилизировали из воды/ацетонитрила. Процедура давала 27 мг (94% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2,2 мин;
ЖХ-МС (Метод 9): Rt=5,04 мин; MS (Е8 шоз): m/z=947 (М+Н)+.
Промежуточное соединение 73
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(2S)-1-[бензил(метил)амино]-1-оксо-3-фенилпропан-2-ил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 14, вначале брали промежуточные соединения 4 и 38, и получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(2S)-1-[бензил(метил)амино]-1-оксо-3-фенилпропан-2-ил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид. 25 мг (0,026 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 13 мг (54% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 12): Rt=2,2 мин;
ЖХ-МС (Метод 9): Rt=5,01 мин; MS (ESIпоз): m/z=921 (М+Н)+.
Промежуточное соединение 74
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(1S,2R)-1-[(бензилокси)карбонил]-2-фенилциклопропил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

50 мг (73 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и 28 мг (73 мкМоль) бензил (1S,2R)-1-амино-2-фенилциклопропанкарбоксилат трифторацетата (промежуточное соединение 45) помещали в 5 мл ДМФ, и добавляли 42 мг (110 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфоефата и 38 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, затем концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. Объединенные фракции полученного продукта концентрировали. После лиофилизации из диоксана/воды, получали 35 мг (51% от теоретического количества) защищенного группой Boc промежуточного соединения N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(1S,2R)-1-[(бензилокси)карбонил]-2-фенилциклопропил}амино)-1-метокеи-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде бесцветной пены.
ЖХ-МС (Метод 1): Rt=1,52 мин; m/z=934 (М+Н)+.
35 мг этого промежуточного вещества обрабатывали 1 мл трифторуксусной кислоты в 5 мл дихлорметана для удаления защитной группы Вое. После концентрирования и лиофилизации из диоксана/воды, получали 34 мг (97% от теоретического количества) свободного амина промежуточного соединения N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(1S,2R)-1-[(бензилокси)карбонил]-2-фенилциклопропил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата.
ЖХ-МС (Метод 1): Rt=0,91 мин; m/z=834 (М+Н)+.
11 мг (0,011 ммоль) этого промежуточного вещества затем использовали по аналогии с приготовлением промежуточного соединения 66 в реакции с 4-оксобутановой кислоой в присутствии цианоборогидрида натрия, с получением 2,5 мг (24% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 12): Rt=2,2 мин;
ЖХ-МС (Метод 9): Rt=5,1 мин; MS (ESIпоз): m/z=920 (М+Н)+.
Промежуточное соединение 75
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S,2R)-2-фенил-1-(пропилкарбамоил)циклопропил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 74, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и (1S,2R)-1-амино-2-фенил-N-пропилциклопропанкарбоксамид трифторацетата (промежуточное соединение 27) в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S,2R)-2-фенил-1-(пропилкарбамоил)циклопропил]амино}пропил] пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 14 мг (0,016 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 11,3 мг (83% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 6): Rt=1,9 мин;
ЖХ-МС (Метод 2): Rt=1,27 мин; MS (ESIпоз): m/z=871 (M+R)+.
Промежуточное соединение 76
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-(этоксикарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза осуществляя реакцию сочетания промежуточного соединения 46 (N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид) с промежуточным соединением 48 (этил (1S,2R)-1-амино-2-фенилциклопропанкарбоксилат трифторацетат) в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и последующим отсоединением группы Boc, получали исходное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-(этоксикарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат. 70 мг (0,079 ммоль) этого исходного соединения затем использовали в реакции с 4-оксобутановой кислотой, по аналогии с промежуточным соединением 61, с получением 46 мг (68% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 6): Rt=1,9 мин;
ЖХ-МС (Метод 2): Rt=1,28 мин; MS (ESIпоз): m/z=858 (М+Н)+
Промежуточное соединение 77
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 75, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-3-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и L-фенилаланинамид гидрохлорида в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 47 мг (0,049 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 39 мг (96% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 6): Rt=1,7 мин;
ЖХ-МС (Метод 9): Rt=4.44 мин; MS (ESIпоз): m/z=817 (М+H)+
1Н ЯМР (500 МГц, ДМСО-d6): δ=8,95 и 8,8 (2m, 1H), 8,25 и 8,0 (2d, 1H), 7.45, 7,35 и 7,0 (3s, ушир, 2H), 7,3-7,1 (m, 5H), 4,8-4.4 (2m, 3H), 3,95 (m, 1H), 3,82 (m, 1H), 3,72 (d, 1H), 3,22, 3,18, 3,15, 3,05 и 3,00 (5s, 9H), 2,85-2,7 (m, 4H), 2.45-1,6 (m, 12H), 1,5-1,2 (m, 3H), 1,1-0,7 (m, 21H) [другие сигналы перекрыты пиками от растворителей].
Промежуточное соединение 78
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 66 в 2 этапа, в качестве исходного соединения брали20 мг (16 мкМоль) вещества из примера для промежуточного соединения 14 и бензил 6-оксогексил карбамат, и проводя гидрогенезацию в метаноле в качестве растворителя.
Выход: 7,6 мг (55% от теоретического количества в 2 этапа)
ВЭЖХ (Метод 6): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,7 мин; MS (ESIпоз): m/z=901 (M+H)+.
Промежуточное соединение 79
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил] амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
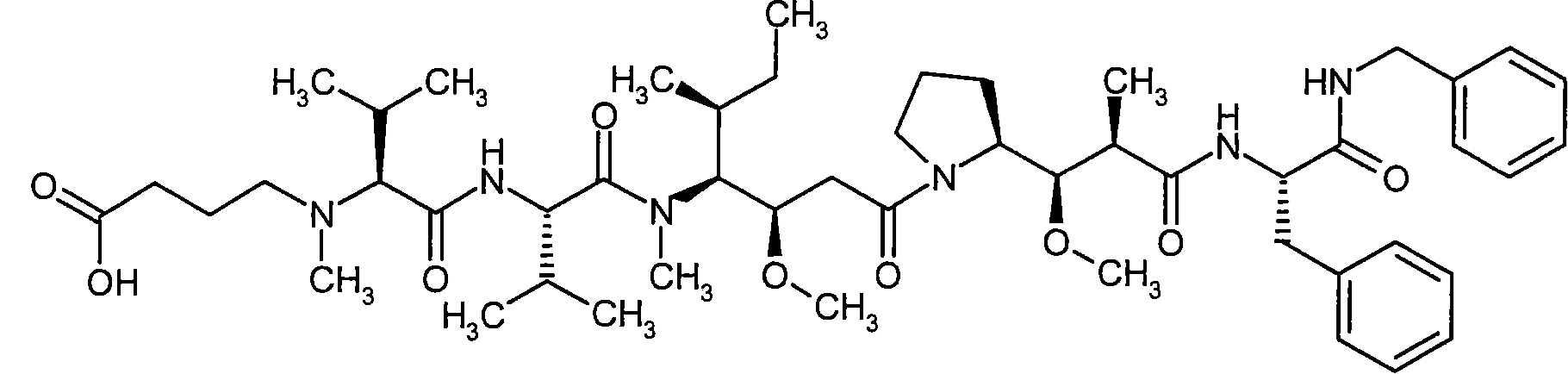
36 мг (43 мкМоль) N-(трет-бутоксмкарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид (промежуточное соединение 46) и 4,6 мг (43 мкМоль) бензиламина помещали в 5 мл ДМФ, добавляли 7,5 мкл (88 мкМоль) N,N-диизопропилэтиламина, 10 мг (65 мкМоль) HOBt и 10 мг (52 мкМоль) EDC, и затем смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали, и очищали осадок методом препаративной ВЭЖХ. В результате получали 29 мг (73% от теоретического количества) защищенного группой Boc промежуточного соединения N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ЖХ-МС (Метод 1): Rt=1.43 мин; m/z=921 (М+Н)+.
29 мг этого промежуточного вещества обрабатывали 1 мл трифторуксусной кислоты в 6 мл дихлорметана для удаления защитной группы Вое. После концентрирования и лиофилизации из диоксана/воды, получали 30 мг (колич.) свободного амина промежуточного соединения N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата.
ЖХ-МС (Метод 1): Rt=0,95 мин; m/z=821 (М+Н)+.
17 мг (0,018 ммоль) этого промежуточного вещества затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 13 мг (80% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5); Rt=1,7 мин;
ЖХ-МС (Метод 9): Rt=4,97 мин; MS (ESIпоз): m/z=907 (М+Н)+.
Промежуточное соединение 80
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 74, осуществляя реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2Ы)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и N-бензил-L-триптофанамид трифторацетата (промежуточное соединение 47) в присутствии О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 10 мг (0,01 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 2,5 мг (26% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 2); Rt=1,13 мин; MS (ESIпоз): m/z=946 (М+Н)+.
Промежуточное соединение 81
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-карбамоил-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1 -оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом, описанным в примере промежуточного соединения 74, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и (1S,2R)-1-амино-2-фенилциклопропанкарбоксамид трифторацетата (промежуточное соединение 48) в присутствии 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и с последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-карбамоил-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 14 мг (0,0163 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 8 мг (57% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 9): Rt=4,64 мин; MS (ESIпоз): m/z=829 (М+Н)+.
Промежуточное соединение 82
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 69, осуществляли реакцию сочетания N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) и Nα-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-триптофанамид трифторацетата (промежуточное соединение 49) в присутствии О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и с последующим удалением защитной группы Fmoc с помощью пиперидина, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 78 мг (0,088 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 68 мг (90% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 9): Rt=4.49 мин; MS (ESIпоз): m/z=856 (М+Н)+.
Промежуточное соединение 83
N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, описанной для промежуточного соединения 82, брали в качестве исходного соединения 20 мг (26 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат и осуществляли реакцию с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия.
Выход: 5 мг (25% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 11): Rt=0,72 мин; MS (ESIпоз): m/z=884 (M+H)+.
Промежуточное соединение 84
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(морфолин-4-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1 -ил} -5 -метил-1 -оксогептан-4-ил] -Л^-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 79, осуществляли реакцию сочетания N-(трет-бутоксикарбоннл)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N- метил-L-валинамида (промежуточное соединение 46) и морфолина в присутствии EDC и НОВТ, и с последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(морфолин-4-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 30 мг (0,033 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 22 мг (76% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 9): Rt=4,58 мин; MS (ESIпоз): m/z=887 (М+Н)+.
Промежуточное соединение 85
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3R)-1-(бензиламино)-3-гидрокси-1-оксобутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 79, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 46) и N-бензил-L-треонинамид трифторацетата в присутствии HATU, и последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3R)-1-(бензиламино)-3-гидрокси-1-оксобутан-2-ил]амино}-1-метокси-2-метил-З -оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 21 мг (0,024 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 20 мг (97% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,54 мин;
ЖХ-МС (Метод 9): Rt=4.49 мин; MS (ESIпоз): m/z=861 (М+Н)+.
Промежуточное соединение 86
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогепган-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 74, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и трет-бутил-L-фетлалгатнат гидрохлорида в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и последующим удалением защитной группы Boc трифторуксусной кислотой с получением трет-бутил эфира (при перемешивании с трифторуксусной кислотой в дихлорметане в течение 40 мин), получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетат. 22 мг (0,02 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 16 мг (94% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=2,0 мин;
ЖХ-МС (Метод 9): Rt=5,05 мин; MS (ESIпоз): m/z=874 (M+Н)+.
Промежуточное соединение 87
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 86, брали в качестве исходного соединения 230 мг (336 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и трет-бутил-L-тритггофанат гидрохлорида в три этапа.
Выход: 95 мг (31% от теоретического количества в три этапа)
ВЭЖХ (Метод 5): Rt=2,0 мин;
ЖХ-МС (Метод 9): Rt=5,05 мин; MS (EShios): m/z=913 (M+H)+.
Промежуточное соединение 88
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
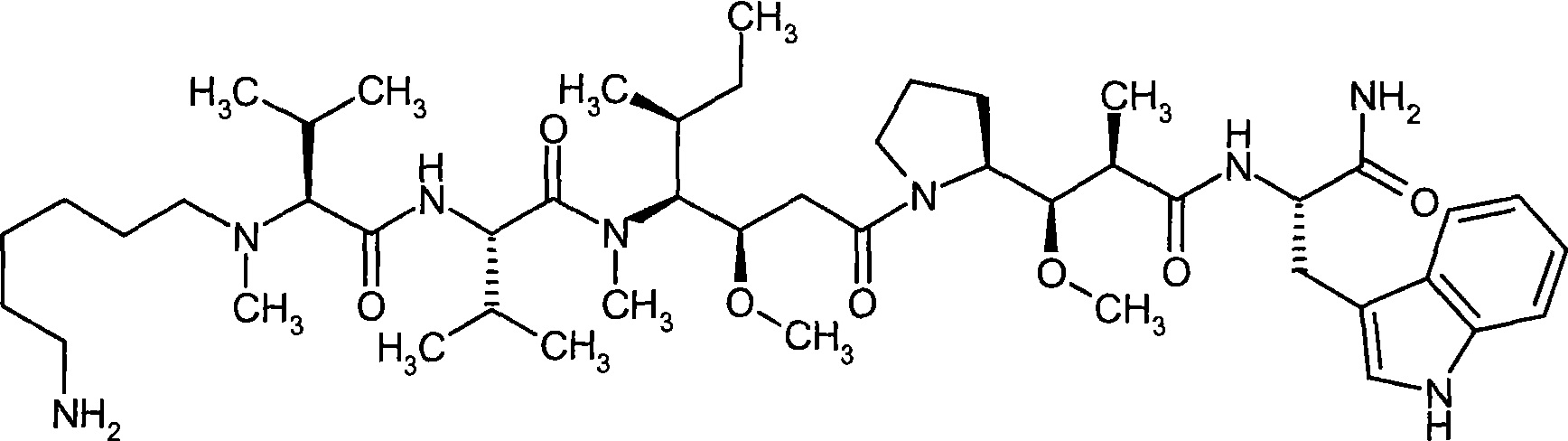
На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 69, осуществляли реакцию сочетания N-[(9Н-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) и Nα-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-триптофанамид трифторацетата (промежуточное соединение 49) в присутствии О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и последующим удалением защитной группы Fmoc с помощью пиперидина, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 30 мг (0,03 ммоль) этого соединения затем использовали по аналогии с приготовлением соединения описанного в примере промежуточного соединения 61 в реакции с бензил 6-оксогексил карбаматом, который получали заблаговременно окислением бензил 6-гидроксигексил карбамата, в присутствии цианоборогидрида натрия, с получением 17 мг (45% от теоретического количества) соединения защищенного группой Z. Затем гидрогенизацией в метаноле над 10% палладием на активированном угле получали указанное в названии соединение.
Выход: 14 мг (95% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,5 мин;
ЖХ-МС (Метод 1): Rt=0,73 мин; MS (ESIпоз): m/z=869 (М+Н)+.
Промежуточное соединение 89
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-отреот-бутокси-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 86, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и трет-бутил-L-тритофанат гидрохлорида в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и последующим удалением защитной группы Boc трифторуксусной кислотой с получением тпрет-бутш эфира (перемешиванием с 1:10 трифторуксусной кислотой/дихлорметаном в течение 30 мин), получали аминное соединение N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 71 мг (0,075 ммоль) этого соединения затем использовали по аналогии с приготовлением соединения описанного в примере промежуточного соединения 61 в реакции с бензил 6-оксогексил карбаматом, который получали заблаговременно окислением бензил 6-гидроксигексил карбамата, в присутствии цианоборогидрида натрия, с получением 35 мг (44% от теоретического количества) соединения защищенного группой Z. Затем гидрогенизацией в метаноле над 10% палладием на активированном угле получали указанное в названии соединение.
Выход: 30 мг (98% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,77 мин; MS (ESIпоз): m/z=926 (М+Н)+.
Промежуточное соединение 90
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(1H-индол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 74, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и 2-(1H-индол-3-ил)этанамина в присутствии 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(1H-индол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата. 100 мг (0,119 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 50 мг (49% от теоретического количества) указанного в названии соединения. Указанное в названии соединение очищали методом флэш-хроматографии на силикагеле с дихлорметан/метанол/17% аммиаком в качестве элюента, в процессе чего соотношение смеси менялось от исходного 15/2/02 до 15/4/0,5.
ВЭЖХ (Метод 6): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=813 (M+R)+.
Промежуточное соединение 91
N-(3-карбоксипропил)-N-метил-L-валил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 74, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и фенилэтиламина в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и последующим удалением защитной группы Boc трифторуксусной кислотой, получали аминное соединение N-метил-L-валил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил)амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамида в виде трифторацетата. 57 мг (0,071 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 44 мг (80% от теоретического количества) указанного в названии соединения. Указанное в названии соединение также может быть очищено в данном примере методом флэш-хроматографии на силикагеле с дихлорметан/метанол/17% аммиаком в качестве элюента (15/2/02→15/4/0,5).
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 9): Rt=4,64 мин; MS (ESIпоз): m/z=774 (M+H)+.
Промежуточное соединение 92
N-(3-карбоксипропил)-N-метил-L-валил-N-[(2R,4S,5S)-1-{(2S)-2-[(1S,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

100 мг (0,139 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 40) использовали по аналогии с приготовлением промежуточного соединения 61 в реакции с 4-оксобутановой кислотой в присутствии цианоборогидрида натрия, с получением 94 мг (84% от теоретического количества) указанного в названии соединения. Указанное в названии соединение очищали методом препаративной ВЭЖХ.
ВЭЖХ (Метод 5): Rt=1,5 мин;
ЖХ-МС (Метод 9): Rt=4.46 мин; MS (ESIпоз): m/z=804 (М+H)+.
Промежуточное соединение 93
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

22.4 мг (24 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино} пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата растворяли в 1.4 мл раствора диоксан/вода и, аналогично процедуре приготовления промежуточного соединения 61, осуществляли реакцию с 15% водным раствором 4-оксобутановой кислоты в присутствии цианоборогидрида натрия. После лиофилизации из диоксана, получали 8,2 мг (38% от теоретического количества) указанного в названии соединения в виде белого твердого вещества.
ВЭЖХ (Метод 10): Rt=2,54 мин
ЖХ-МС (Метод 12): Rt=0,94 мин; MS (ESIпоз): m/z=919 (M+H)+.
Промежуточное соединение 94
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

17,1 мг (18 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата растворяли в 1,1 мл раствора диоксан/вода и, аналогично процедуре приготовления промежуточного соединения 61, осуществляли реакцию с 15% водным раствором 4-оксобутановой кислоты в присутствии цианоборогидрида натрия. После лиофилизации из диоксана, получали 14,8 мг (89% от теоретического количества) указанного в названии соединения в виде белого твердого вещества.
ВЭЖХ (Метод 10):Rt=2,54 мин;
ЖХ-МС (Метод 12): Rt=0,92 мин; MS (ESIпоз): m/z=919 (M+H)+
Промежуточное соединение 95
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилсульфонил)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

19,3 мг (20 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилсульфонил)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата растворяли в 1,2 мл раствора диоксан/вода и, аналогично процедуре приготовления промежуточного соединения 61, осуществляли реакцию с 15% водным раствором 4-оксобутановой кислоты в присутствии цианоборогидрида натрия. После лиофилизации из диоксана, получали 8,6 мг (45% от теоретического количества) указанного в названии соединения в виде твердого вещества.
ЖХ-МС (Метод 11): Rt=0,85 мин; MS (ESIпоз): m/z=943 (M+H)+
Промежуточное соединение 96
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3S)-1,4-дифенилбут-3-ен-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

15,5 мг (10 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3E)-1,4-дифенилбут-3-ен-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата растворяли в 1,0 мл раствора диоксан/вода и, аналогично процедуре приготовления промежуточного соединения 61, осуществляли реакцию с 15% водным раствором 4-оксобутановой кислоты в присутствии цианоборогидрида натрия. После лиофилизации из диоксана, получали 10,3 мг (68% от теоретического количества) указанного в названии соединения в виде белого твердого вещества.
ВЭЖХ (Метод 10): Rt=2,59 мин;
ЖХ-МС (Метод 11): Rt=0,94 мин; MS (ESIпоз): m/z=877 (M+H)+
Промежуточное соединение 97
N-(6-аминогексил)-N-метил-L-валил-N-(3R,4S,5S}-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 66, осуществляли реакцию 200 мг (0,108 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 16) с бензил 6-оксогексил карбаматом, и последующим гидролитическим удалением защитной группы Z (с 5% палладием на угле в качестве катализатора, в метаноле в качестве растворителя).
Выход: 69 мг (65% от теоретического количества после двух этапов синтеза) ВЭЖХ (Метод 5): R,=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,76 мин; MS (ESIпоз): m/z=912 (М+Н)+.
Промежуточное соединение 98
N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 80. Очистку проводили методом препаративной ВЭЖХ.
Выход: 40 мг (29% от теоретического количества в три этапа)
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,92 мин; MS (ESIпоз): m/z=974 (М+H)+.
Промежуточное соединение 99
(2S)-2-амино-3-(1H-индол-3-ил)-1 -(1,2-оксазинан-2-ил)пропан-1-он трифторацетат

324 мг (0,81 ммоль) 2,5-диоксопирролидин-1-ил N-(трет-бутоксикарбонил)-L-триптофаната растворяли в 20 мл ДМФ, добавляли и 200 мг (1,62 ммоль) of 1,2-оксазинан гидрохлорида (Исходное соединение 5) и 850 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при 50°С в течение ночи, затем концентрировали при пониженном давлении. Осадок помещали в дихлорметан и экстрагоировали водой. Органическую фазу сушили над сульфатом магния и концентрировали. Осадок очищали методом флэш-хроматографии на силикагеле с 4:1 дихлорметан/этилацетатом в качестве элюента. Фракции полученного продукта концентрировали, сушили осадок в высоком вакууме. Процедура давала 147,5 мг (48% от теоретического количества) защищенного группой Boc промежуточного соединения.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=1,03 мин; MS (ESIпоз): m/z=374 (М+Н)+.
Брали 166 мг (444,5 мкМоль) этого промежуточного вещества, при стандартных условиях с помощью 3 мл трифторуксусной кислоты в 20 мл дихлорметана удаляли защитную группу Boc, и, после очистки методом ВЭЖХ, получали 155 мг (86% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1.43 мин;
ЖХ-МС (Метод 11); Rt=0,56 мин; MS (ESIпоз): m/z=274 (M+H)+.
Промежуточное соединение 100
N-(6-{[(бензилокси)карбонил]амино}гексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1R-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

177 мг (260 мкМоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и 100 мг (260 мкМоль) (2S)-2-амино-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)пропан-1-он трифторацетата (промежуточное соединение 99) помещали в 15 мл ДМФ, и добавляли 118 мг (310 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 140 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. Фракции полученного продукта объединяли и концентрировали. После лиофилизации из диоксана, получали 170 мг (68% от теоретического количества) защищенного группой Boc промежуточного соединения.
ЖХ-МС (Метод 1): Rt=1,36 мин; m/z=940 (М+Н)+.
170 мг этого промежуточного вещества обрабатывали 3 мл трифторуксусной кислоты в 30 мл дихлорметана в течение 30 мин для удаления защитной группы Вое. Затем реакционную смесь концентрировали при пониженном давлении, очищали осадок методом препаративной ВЭЖХ с получением 155 мг (86% от теоретического количества) незащищенного промежуточного соединения N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ВЭЖХ (Метод 12): Rt=1,85 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=840 (M+H)+.
50 мг (0,052 ммоль) этого промежуточного вещества затем использовали по аналогии с приготовлением промежуточного соединения 97, в реакции с бензил 6-оксогексил карбаматом в присутствии цианоборогидрида натрия, с последующим гидролитическим удалением защитной группы Z (с 5% палладием на угле в качестве катализатора, в метаноле в качестве растворителя), с получением указанного в названии соединения.
Выход: 21 мг (37% от теоретического количества) ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,02 мин; MS (ESIпоз): m/z=1073 (М+Н)+.
Промежуточное соединение 101
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

26,7 мг (24,87 мкМоль) промежуточного соединения 100 растворяли в 10 мл метанола и гидрогенировали над подложкой палладий/активированный уголь (5%) при стандартном давлении водорода в течение 30 мин. Катализатор отфильтровывали, растворитель выпаривали при пониженном давлении. После сушки осадка в высоком вакууме, получали 22,5 мг (96% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,76 мин; MS (ESIпоз): m/z=939 (М+Н)+.
Промежуточное соединение 102
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(морфолин-4-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 157 из N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(морфолин-4-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1 -ил)гексангидразида.
Выход: 8 мг (71% от теоретического количества)
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=1094 (M+H)+.
Промежуточное соединение 103
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3R)-1-(бензиламино)-3-гидрокси-1-оксобутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
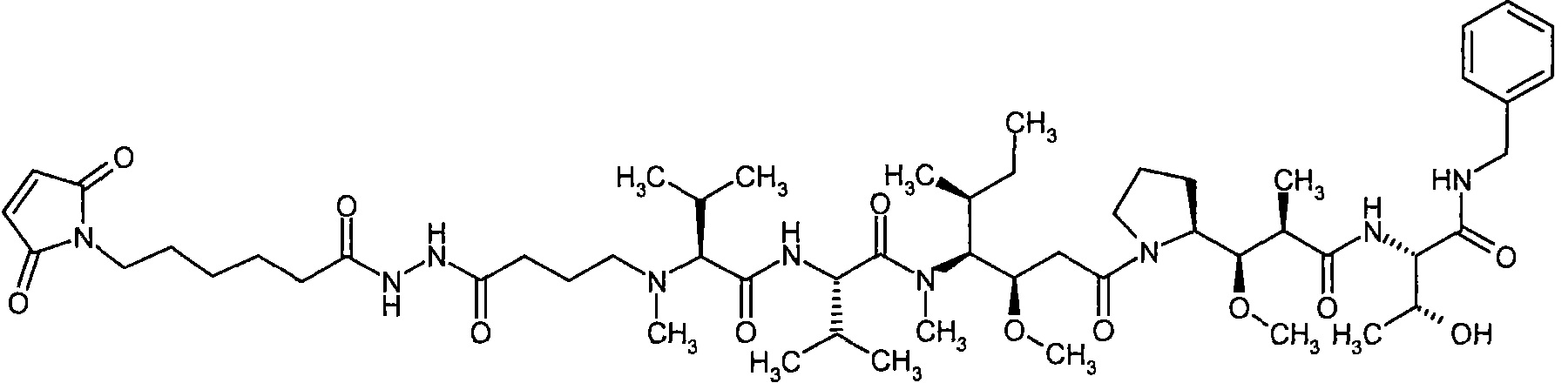
Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 157 из N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3R)-1-(бензиламино)-3-гидрокси-1-оксобутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида.
Выход: 3 мг (22% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,78 мин; MS (ESIпоз): m/z=1069 (M+H)+.
Промежуточное соединение 104
N-{4-[(транс-4-{[(2,5-диоксопирролидин-1-ил)окси]карбонил}циклогексил)амино]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-3-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза бензил транс-4-аминоциклогексанкарбоксилат трифторацетат получали из отрянс-4-аминоциклогексанкарбоновой кислоты введя защитную группу Вое, после чего вводили защитную группу бензил эфира, и затем концентрировали, удалив защитную группу Boc любым стандартным методом химии пептидов.
Затем 15 мг (18 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 5 мл диметилформамида, после чего концентрировали, добавляли к смеси 13 мг (35 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 9 мкл N,N-диизопропилэтиламина и 15 мг (44 мкМоль) бензил транс-4-аминоциклогексанкарбоксилат трифторацетата. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. Соответствующие фракции объединяли, растворитель выпаривали при пониженном давлении. После сушки осадка в высоком вакууме, получали 14,7 мг (78% от теоретического количества) защищенного промежуточного соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,95 мин; MS (ESIпоз): m/z=1072 (M+H)+.
Из этого защищенного промежуточного соединения, сначала гидролитическими средствами удаляли бензил эфир, с образованием свободной карбоксильной компоненты с количественным выходом. 14 мг (14 мкМоль; 1 эквив.) незащищенного соединения помещали в 5 мл ДМФ и добавляли к смеси 3,3 мг (29 мкМоль; 2,1 эквив.) N-гидроксисукцинимида в присутствии 4,1 мг (21 мкМоль; 1,5 эквив.) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 7,5 мкл (44 мкМоль; 3,1 эквив.) N,N-диизопропилэтиламина и 0,9 мг (7 мкМоль; 0,5 эквив.) 4-диметиламинопиридина, и перемешивали реакционную смесь при комнатной температуре в течение ночи. Затем добавляли еще 10 эквив. N-гидроксисукцинимида, 5 эквив. 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 5 эквив. N,N-диизопропилэтиламина и 0,5 эквив. 4-диметиламинопиридина, и обрабатывали реакционную смесь в ультразвуковой ванне 5 часов. Затем растворитель выпаривали, осадок очищали методом препаративной ВЭЖХ, после чего соответствующие фракции объединяли и концентрировали. После лиофилизации осадка из диоксана, получали 9,7 мг (62% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=1,8 мин;
ЖХ-МС (Метод 11): Rt=0,77 мин; MS (ESIпоз): m/z=1078 (М+Н)+.
Промежуточное соединение 105
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 157, брали в качестве исходного соединения 4-{[(2S)-1-{[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)амино}-3-метилбутан-2-ил]амино}-3-метил-1-оксобутан-2-ил](метил)амино}бутановую кислоту и коммерчески доступный 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразид. Эфир промежуточного соединения получали с 42% выходом. На втором этапе, 6 мг (6 мкМоль) этого промежуточного вещества расщепляли трет-бутиловым эфиром трифторуксусной кислоты. После очистки методом ВЭЖХ, получали 3.4 мг (48% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,66 мин;
ЖХ-МС (Метод 2): Rt=1,04 мин; MS (ESIпоз): m/z=1025 (М+Н)+.
Промежуточное соединение 106
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

14 мг (16 мкМоль) N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 88) помещали в 750 мкл диоксана и добавляли к смеси 1,5 мл насыщенного растворабикарбоната натрия, и затем 3,2 мг (21 мкМоль) метил 2,5-диоксо-2,5-дигидро-1H-пиррол-1-карбоксилата. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 5,5 мг (36% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,84 мин; MS (ESIпоз): m/z=949 (М+Н)+.
Промежуточное соединение 107
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(1H-индол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

38 мг (47 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[2-(1H-индол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 37 мл ДМФ, и затем добавляли к смеси 71 мг (187 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 33 мкл N,N-диизопропилэтиламина и 37 мг (140 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали в высоком вакууме и очищали полученный осадок методом препаративной ВЭЖХ. Таким образом получали 12,2 мг (26% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): rt=0,85 мин; MS (ESIпоз): m/z=1020 (М+Н)+.
Промежуточное соединение 108
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-{(3R,4S,5S)-3-метокси-1-[(2S)-2-{(1R,2R)-1-метокси-2-метил-3-оксо-3-[(2-фенилэтил) амино]пропил}пирролидин-1-ил]-5-метил-1-оксогептан-4-ил}-N-метил-L-валинамид

Соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 107.
Выход: 2,5 мг (30% от теоретического количества)
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,9 мин; MS (ESIпоз): m/z=981 (М+Н)+.
Промежуточное соединение 109
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-L-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 107 из соединения, описанного в процедуре синтеза промежуточного соединения 92.
Выход: 35 мг (65% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 11): Rt=0,76 мин; MS (ESIпоз): m/z=1011 (М+Н)+.
Промежуточное соединение 110
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 147 из соединения, описанного в процедуре синтеза промежуточного соединения 83.
Выход: 2.4 мг (24% от теоретического количества) ВЭЖХ (Метод 6): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=981 (М+H)+.
Промежуточное соединение 111
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-1-метилгидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 140 из промежуточного соединения 82 и промежуточного соединения 22.
Выход: 6,5 мг (51% от теоретического количества)
ВЭЖХ (Метод 6): R1=1,8 мин;
ЖХ-МС (Метод 1): Rt=4,71 мин; MS (ESIпоз): m/z=1077 (M+H)+.
Промежуточное соединение 112
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-карбамоил-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 157 из соединения, описанного в процедуре синтеза промежуточного соединения 81.
Выход: 5,7 мг (57% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIno3): m/z=1036 (M+H)+.
Промежуточное соединение 113
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(1H-индол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

95 мг (104 мкМоль) 4-{[(2S)-1-{[(2S)-1-{[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил](метил)амино}-3-метилбутан-2-ил]амино}-3-метил-1-оксобутан-2-ил](метил)амино} бутановой кислоты растворяли в ДМФ и затем добавляли к смеси 79,5 мг (209 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 73 мкл N,N-диизопропилэтиламин и 68 мг (261 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем реакционную смесь концентрировали в высоком вакууме, и очищали полученный осадок методом препаративной ВЭЖХ. Таким образом получали 104 мг (89% от теоретического количества) трет-бутилового эфира указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,93 мин; MS (ESIпоз): m/z=1121 (M+H)+.
Полученное промежуточное соединение помещали в 33.4 мл дихлорметана, добавляли 17 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ.
Таким образом получали 61 мг (62% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1064 (М+Н)+.
Промежуточное соединение 114
N-[6-({[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил]карбамоил}амино)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

5 мг (5 мкМоль) N-(6-аминогексил)-N-метил-L-валил-N-(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида помещали в 885 мкл ДМФ и добавляли к смеси 5,3 мг (8 мкМоль) 4-нитрофенил 2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил карбамата и 2,8 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, и затем концентрировали до сухого состояния. Осадок очищали методом препаративной ВЭЖХ.
Выход: 0,58 мг (11% от теоретического количества) бесцветной пены ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,83 мин; MS (ESIпоз): m/z=1035 (M+H)+.
Промежуточное соединение 115
N-{4-[(2,5-диоксопирролидин-1-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
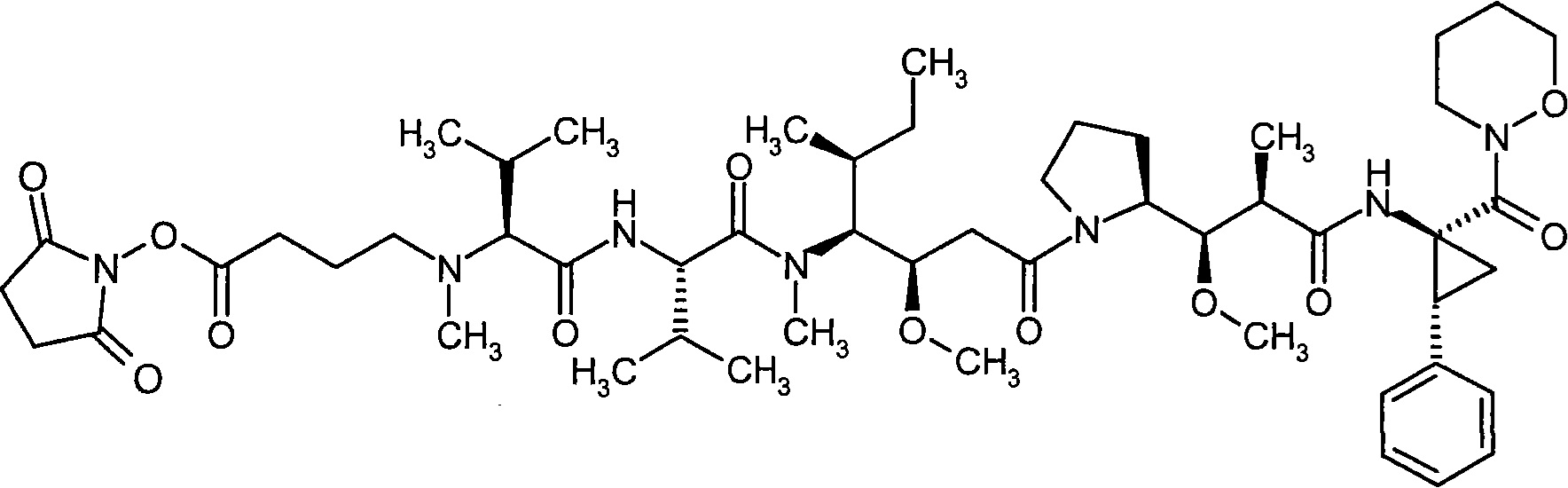
Данное соединение получали, следуя процедуре, описанной для промежуточного соединения 147, брали в качестве исходного соединения 8 мг (9 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида. Затем концентрировали, и очищали активированный эфир методом препаративной ВЭЖХ и, после удаления растворителя при пониженном давлении, немедленно осуществляли реакцию с антителом.
Выход: 3 мг (27% от теоретического количества) (чувствительное к гидролизу)
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=996 (M+H)+.
Промежуточное соединение 116
N-{4-[(2,5-диоксопирролидин-1-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, описанной для промежуточного соединения 147, брали в качестве исходного соединения 5 мг (6 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида. Затем концентрировали и очищали активированный эфир методом препаративной ВЭЖХ и, после удаления растворителя при пониженном давлении, немедленно осуществляли реакцию с антителом.
Выход: 3,2 мг (43% от теоретического количества) (чувствительное к гидролизу)
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,92 мин; MS (ESIпоз): m/z=984 (М+Н)+.
Промежуточное соединение 117
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 157 из соединения, описанного в процедуре синтеза промежуточного соединения 86.
Выход: 7 мг (42% от теоретического количества)
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,94 мин; MS (ESIпоз): m/z=1081 (М+Н)+.
Промежуточное соединение 118
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2R)-1-(бензилокси)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
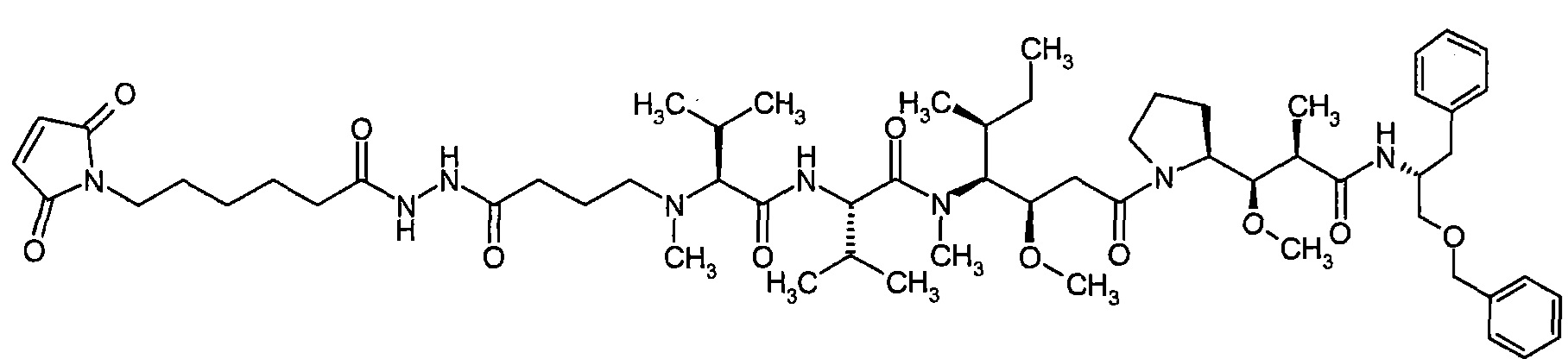
Нужное соединение получали аналогично процедуре, описанной для промежуточного соединения 157 из 7 мг (7,8 мкМоль) соединения, описанного в примере промежуточного соединения 68. Выход: 6,3 мг (53% от теоретического количества)
ЖХ-МС (Метод 1): Rt=1,00 мин; MS (ESIпоз): m/z=1102 (M+H)+.
Промежуточное соединение 119
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли реакцию сочетания 7.4 мг (8,1 ммоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и 6,3 мг (24,2 ммоль) 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразид гидрохлорида, после чего действовали аналогично процедуре, описанной для промежуточного соединения 157. В результате получали 1,6 мг (13% от теоретического количества) указанного в названии соединения в виде твердого вещества.
ЖХ-МС (Метод 11): Rt=0,89 мин; MS (ESIпоз): m/z=1126 (M+H)+
Промежуточное соединение 120
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли реакцию сочетания 12,8 мг (13,9 ммоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1R)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино} пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и 10,9 мг (41,8 ммоль) 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразид гидрохлорида, после чего действовали аналогично процедуре, описанной для промежуточного соединения 157. В результате получали 10,8 мг (59% от теоретического количества) указанного в названии соединения в виде твердого вещества.
ЖХ-МС (Метод 11): Rt=0,90 мин; MS (ESIпоз): m/z=1126 (M+H)+
Промежуточное соединение 121
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилсульфонил)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли реакцию сочетания 7.4 мг (7,9 ммоль) N-(3-карбоксипропил)-L-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилсульфонил)-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и 6,2 мг (23,5 ммоль) 6-(2,5-диоксо-2,5-дигидро-1Л-пиррол-1-ил)гексангидразид гидрохлорида, действовали аналогично процедуре, описанной для промежуточного соединения 157. В результате получали 6,9 мг (74% от теоретического количества) указанного в названии соединения в виде твердого вещества.
ЖХ-МС (Метод II): Rt=0,87 мин; MS (ESIпоз): m/z=1150 (M+H)+
Промежуточное соединение 122
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(0R,2R)-3-{[(2S,3E)-1,4-дифенилбут-3-ен-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли реакцию сочетания 8 мг (9,1 ммоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3E)-1,4-дифенилбут-3-ен-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и 7,2 мг (27.4 ммоль) 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразид гидрохлорид, после чего действовали аналогично процедуре, описанной для промежуточного соединения 157. В результате получали 8,2 мг (82% от теоретического количества) указанного в названии соединения в виде белого твердого вещества.
ЖХ-МС (Метод 11): Rt=0,95 мин; MS (ESIпоз): m/z=1083 (M+H)+
Промежуточное соединение 123
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

30 мг (30 мкМоль) промежуточного соединения 89 помещали в 2 мл 1,4-диоксана, и добавляли к смеси 4 мл насыщенного раствора бикарбоната натрия, и затем 7,5 мг (50 мкМоль) метил 2,5-диоксо-2,5-дигидро-1H-пиррол-1-карбоксилата. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 24 мг (74% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=2,2 мин;
ЖХ-МС (Метод 1): Rt=1,01 мин; MS (ESIпоз): m/z=1006 (M+H)+.
Промежуточное соединение 124
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(1H-индол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли реакцию 22 мг (20 мкМоль) промежуточного соединения 123 с 4 мл трифторуксусной кислоты в 8 мл дихлорметана при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 11 мг (54% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 11): Rt=0,85 мин; MS (ESIпоз): m/z=950 (М+Н)+.
Промежуточное соединение 125
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

22,5 мг (20 мкМоль) промежуточного соединения 101 помещали в 2 мл 1:1 смеси диоксан/вода, и затем добавляли к смеси 5,6 мг (40 мкМоль) метил 2,5-диоксо-2,5-дигидро-1H-пиррол-1-карбоксилата и 0,25 мл насыщенного раствора бикарбоната натрия. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли еще 0,25 мл ненасыщенного раствора бикарбоната натрия, и перемешивали реакционную смесь при комнатной температуре в течение еще 15 мин, после чего концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 12,8 мг (50% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,95 мин; MS (ESIпоз): m/z=1019 (М+Н)+.
Промежуточное соединение 126
N-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

64 мг (70 мкМоль) N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил] амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 97) помещали в 3 мл смеси 1:1 диоксан/вода, доводили до уровня рН 9 с помощью 4 мл насыщенного раствора бикарбоната натрия и затем концентрировали, добавляли к смеси 16,3 мг (110 мкМоль) метил 2,5-диоксо-2,5-дигидро-Ш-пиррол-1-карбоксилат. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем концентрировали при пониженном давлении. Затем добавляли еще 8 мг (55 мкМоль) метил 2,5-диоксо-2,5-дигидро-1Н-пиррол-1-карбоксилат, и снова доводили реакционную смесь до уровня рН 9 и перемешивали при комнатной температуре в течение еще часа. Полученное вещество затем концентрировали, очищали полученный осадок методом препаративной ВЭЖХ. На первом этапе синтеза получали 31 мг дециклизованного таким образом промежуточного соединения. 27 мг этого промежуточного вещества снова помещали в 2 мл 1:1 смеси диоксан/вода и затем добавляли к смеси 250 мкл насыщенного раствора бикарбоната натрия. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего концентрировали, и очищали осадок методом препаративной ВЭЖХ. После лиофилизации, получали 20 мг (29% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,96 мин;
ЖХ-МС (Метод 1): Rt=0,97 мин; MS (ESIпоз): m/z=992 (М+Н)+.
Промежуточное соединение 127
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

17 мг (18 мкМоль) N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 98) растворяли в 2,8 мл дихлорметана и добавляли к смеси 20 мг (174 ммоль) 1-гидроксипирролидин-2,5-диона, и затем 10 мг (52 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 0,21 мг (0,17 мкМоль) DMAP. Перемешивали при комнатной температуре в течение 4 часов, после чего реакционную смесь концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 8,2 мг (43% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,98 мин; MS (ESIпоз): m/z=1071 (M+H)+.
Промежуточное соединение 128
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

5 мг (5,6 мкМоль) N-(3-Kap6oKCHnponHn)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 845 мкл ДМФ и затем добавляли к смеси 3,2 мг (17 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 2,6 мг (17 мкМоль) 1-гидрокси-1H-бензотиазол гидрата, 1,96 мкл N,N-диизопропилэтиламина и 5,9 мг (22,5 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 2,2 мг (36% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,88 мин; MS (ESIпоз): m/z=1094 (M+H)+.
Промежуточное соединение 129
N-(6-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-6-оксогексил)-N-метил-L-валил-N-(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1 -(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

4 мг (4,3 мкМоль) N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 646 мкл ДМФ, и затем добавляли к смеси 2,5 мг (13 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 2,0 мг (13 мкМоль) 1-гидрокси-1H-бензотиазол гидрата, 2,25 мкл N,N-диизопропилэтиламина и 4,5 мг (17 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, и затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 1,9 мг (39% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 9): Rt=4,9 мин; MS (ESIпоз): m/z=1134 (M+H)+.
Промежуточное соединение 130
N-(4-{[(2R)-1-({5-[(2,5-диокеопирролидин-1-ил)окси]-5-оксопентаноил}амино)пропан-2-ил]окси}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10,5 мг (11,7 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил] амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 3,7 мл дихлорметана, и затем добавляли к смеси 6,7 мг (35 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 0,7 мг (5,8 мкМоль) 4-диметиламинопиридина и 8,2 мг (47 мкМоль) коммерчески доступного трет-бутил (2R)-2-гидроксипропил карбамата. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 7,5 мг (61% от теоретического количества) защищенного группой Boc промежуточного соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=1,03 мин; MS (ESIпоз): m/z=1056 (М+Н)+.
Затем удаляли защитную группу Boc трифторуксусной кислотой. 4,9 мг (0,005 ммоль) незащищенного сырого продукта затем, без дальнейшей очистки, помещали в 1,8 мл дихлорметана и добавляли к смеси 3,7 мг (0,011 ммоль) 1,1'-[(1,5-диоксопентан-1,5-диил)бис(окси)]дипирролидин-2,5-диона, 2.4 мкл (0,014 ммоль) N,N-диизопропилэтиламина и 0,6 мг (5 мкМоль) 4-диметиламинопиридина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, и затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 0,77 мг (15% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5); Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,93 мин; MS (ЕВЬюз): m/z=1167 (М+Н)+.
Промежуточное соединение 131
N-{4-[(1-{5-[(2,5-диоксопирролидин-1-ил)окси]-5-оксопентаноил}пиперидин-4-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (11 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил] амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 2 мл дихлорметана и затем добавляли к смеси 4,3 мг (22 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 0,88 мг (6 мкМоль) 4-диметиламинопиридина и 5,2 мг (22 мкМоль) коммерчески доступного бензил 4-гидроксипиперидин-1-карбоксилата. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом, получали 5 мг (40% от теоретического количества) защищенного Z-группой промежуточного соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,04 мин; MS (ESIпоз): m/z=1116 (M+H)+.
Затем защитную Z-группу отделяли гидролитическим путем в этаноле над подложкой палладий/активированный уголь. После этого 4,6 мг (0,005 ммоль) незащищенного сырого продукта, без дальнейшей очистки, помещали в 1,8 мл дихлорметана и добавляли к смеси 3,8 мг (0,012 ммоль) 1,1'-[(1,5-диоксопентан-1,5-диил)бис(окси)]дипирролидин-2,5-диона, 0,8 мкл (0,005 ммоль) N,N-диизопропилэтиламина и 0,6 мг (5 мкМоль) 4-диметиламинопиридина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 0,96 мг (16% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,94 мин; MS (ESIпоз): m/z=1193 (М+Н)+.
Промежуточное соединение 132
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразинил}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

15 мг (16,7 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 2500 мкл ДМФ, и затем добавляли к смеси 9,6 мг (50 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 7,6 мг (50 мкМоль) 1-гидрокси-1H-бензотиазол гидрат, 5,8 мкл N,N-диизопропилэтиламина и 17.4 мг (67 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 11,2 мг (52% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 2): Rt=1,09 мин; MS (ESIпоз): m/z=1106 (M+H)+.
Промежуточное соединение 133
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразинил}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3S)-1-(бензилокси)-1-оксо-3-фенилбутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
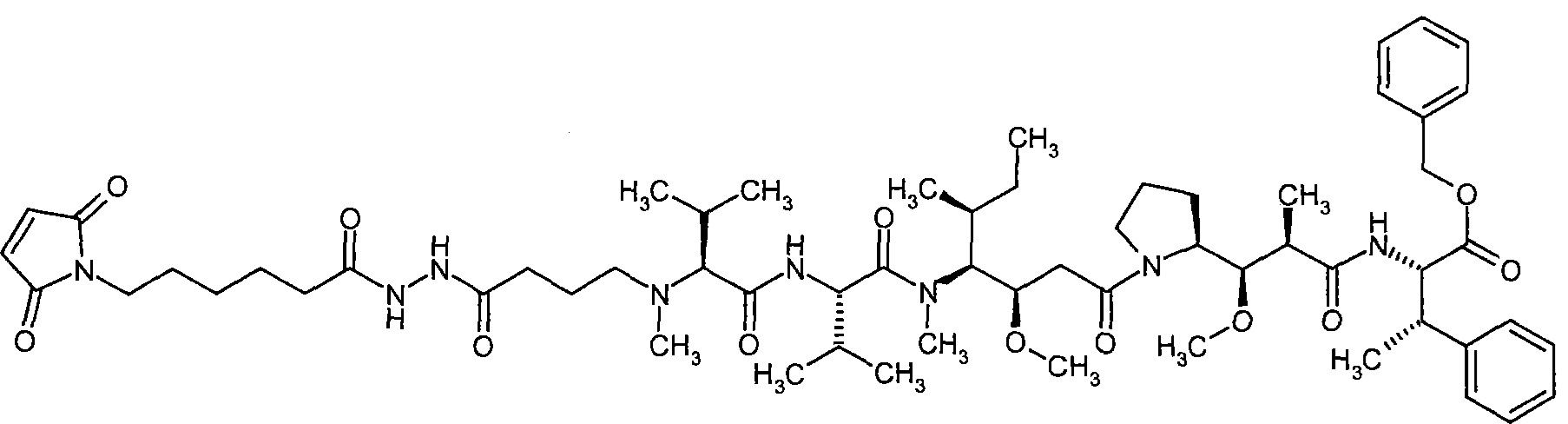
5,8 мг (6,3 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3S)-1-(бензилокси)-1-оксо-3-фенилбутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 943 мкл ДМФ, и затем добавляли к смеси 3,6 мг (19 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 2,9 мг (19 мкМоль) 1-гидрокси-1Д-бензотиазол гидрат, 2,2 мкл N,N-диизопропилэтиламина и 6,6 мг (25 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 4,5 мг (64% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=1,03 мин; MS (ESIпоз): m/z=1129 (M+H)+.
Промежуточное соединение 134
N-[3-({[2-(2,5-диоксо-2,5-дигидро-1Н-тшррол-1-ил)этил]карбамоил} амино)пропил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1S,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза 4-нитрофенил 2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил карбамат получали при стандартных условиях, брали в качестве исходного соединения коммерчески доступного 1-(2-аминоэтил)-1H-пиррол-2,5-дион трифторацетат и 4-нитрофенил хлоркарбоната.
5 мг (6 мкМоль) N-(3-аминопропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S}-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 1000 мкл ДМФ, и затем добавляли к смеси 2 мкл N,N-диизопропилэтиламина и 2,2 мг (9 мкМоль) 4-нитрофенил 2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этил карбамата. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 1,6 мг (23% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 2): Rt=1,09 мин; MS (ESIпоз): m/z=1036 (M+H)+.
Промежуточное соединение 135
N(4-{2-[6-(2,5-диоксо-2,5-1H-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (11 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R(2R)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 4000 мкл ДМФ, и затем добавляли к смеси 6,3 мг (33 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 4,5 мг (33 мкМоль) 1-гидрокси-1H-бензотиазол гидрата, 5,7 мкл N,N-диизопропилэтиламин и 11,5 мг (44 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом, получали 2,6 мг (14% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,01 мин; MS (ESIпоз): m/z=1115 (M+H)+.
Промежуточное соединение 136
N-(4-{4-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил]пиперазин-1-ил}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза 1-[4-оксо-4-(пиперазин-1-ил)бутил]-1H-пиррол-2,5-дион трифторацетат получали при стандартных условиях, брали в качестве исходного соединения трет-бутил пиперазин-1-карбоксилата и 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутановой кислоты, в 2 этапа.
5 мг (5,6 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил] амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 1000 мкл ДМФ, и затем добавляли к смеси 2,1 мг (11 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 1,7 мг (11 мкМоль) 1-гидрокси-1H-бензотиазол гидрата, 2 мкл N,N-диизопропилэтиламина и 3,5 мг (5,6 мкМоль) 1-[4-оксо-4-(пиперазин-1-ил)бутил]-1H-пиррол-2,5-дион трифторацетата. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли 2,1 мг (5,6 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и перемешивали реакционную смесь при комнатной температуре в течение еще 3 часов. Затем при пониженном давлении удаляли растворитель, и очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции концентрировали и, методом лиофилизации из воды, получали 0,6 мг (10% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,9 мин; MS (ESIпоз): m/z=1132 (М+Н)+.
Промежуточное соединение 137
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-1-метилгидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза при стандартных условиях получали 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N'-метилгексангидразид трифторацетат, брали в качестве исходного соединения коммерчески доступную 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)капроновую кислоту и трет-бутил 1-метилгидразинкарбоксилат. Синтез проводили в 2 этапа.
6,9 мг (8 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 2540 мкл ДМФ, и затем добавляли к смеси 3,6 мг (9 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 3 мкл N,N-диизопропилэтиламина и 4,1 мг (12 мкМоль) 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N'-метилгексангидразид трифторацетата. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем при пониженном давлении удаляли растворитель, и очищали оставшийся осадок методом препаративной ВЭЖХ. Таким образом получали 3,9 мг (45% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,93 мин; MS (ESIпоз): m/z=1108 (М+Н)+.
Промежуточное соединение 138
N-{4-[(2-{[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил](метил)амино}этил)(метил)амино]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

В начале синтеза брали трет-бутилметил 2-(матиламино)этил карбамат и 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутановую кислоту, и в 2 этапа получали 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-метил-N-[2-(матиламино)этил] бутанамид трифторацетат.
6,6 мг (7,3 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 2000 мкл ДМФ, и затем добавляли к смеси 5,6 мг (14,7 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата, 2,6 мкл N,N-диизопропилэтиламина и 4,1 мг (9 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-метил-N-[2-(матиламино)этил]бутанамид трифторацетата. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, еще раз добавляли те же количества HATU и N,N-диизопропилэтиламина, и перемешивали реакционную смесь при комнатной температуре в течение ночи. Затем удаляли растворитель при пониженном давлении, и очищали оставшийся осадок методом препаративной ВЭЖХ. Таким образом получали 4 мг (44% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,91 мин; MS (ESIпоз): m/z=1134 (M+H)+.
Промежуточное соединение 139
(2R,3S)-3-амино-4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутан-2-ил(3R,4S,7S,10S)-4-[(2S)-бутан-2-ил]-7,10-диизопропил-3-(2-{(2S)-2-[(1R,2S)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил} -2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазапентадекан-15-оат

13 мг (14,7 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 10 мл дихлорметана, и затем добавляли к смеси 8.4 мг (44 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 5.4 мг (44 мкМоль) 4-диметиламинопиридина и 9 мг (29,3 мкМоль) коммерчески доступного бензил N-(mpem-буотоксмкарбонил)-L-треонината. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Затем реакционную смесь дважды экстрагировали встряхиванием с водой, и сушили органичекую фазу над сульфатом натрия, после чего концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана/воды, получали 14 мг (81% от теоретического количества) защищенного промежуточного соединения в виде бесцветной пены.
ВЭЖХ (Метод 12): Rt=2,3 мин;
ЖХ-МС (Метод 1): Rt=1,13 мин; MS (ESIпоз): m/z=1178 (M+H)+.
Затем защитную Z-группу отделяли гидролитическим путем в метаноле над 10% палладием на активированном угле. Затем 9,5 мг (0,0087 ммоль) незащищенного сырого продукта, без дальнейшей очистки, помещали в 5 мл ДМФ, и добавляли к смеси 5 мг (26,2 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 4 мг (26,2 мкМоль) 1-гидрокси-1H-бензотиазол гидрата, 54,6 мкл N,N-диизопропилэтиламин и 9,1 мг (34,9 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана, получали 9,5 мг (84% от теоретического количества) защищенного группой Boc промежуточного соединения.
ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=0,97 мин; MS (ESIпоз): m/z=1295 (M+H)+.
Затем у 9,5 мг (7,3 мкМоль) защищенного группой Boc промежуточного соединения удаляли защиту с использованием 0,5 мл трифторуксусной кислоты в 2 мл дихлорметана и, после лиофилизации из диоксана, получали 9 мг (82% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=0,84 мин; MS (ESIпоз): m/z=1195 (M+H)+.
Промежуточное соединение 140
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]-1-метилгидразино}-4-оксобутил-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
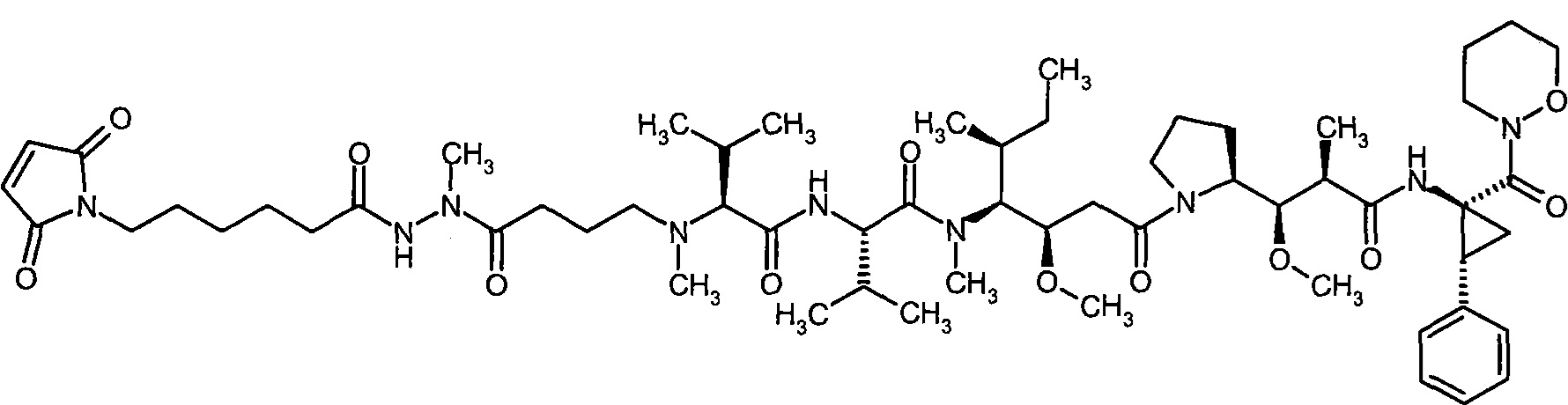
4,1 мг (12 мкМоль) 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N'-метилгексангидразид трифторацетата (промежуточное соединение 22) растворяли вместе с 6,9 мг (8 мкМоль) вещества из примера промежуточного соединения 61 в 2,5 мл ДМФ, и затем добавляли к смеси 3,5 мг (9 мкМоль) О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 3 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана, получали 2,6 мг (30% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,90 и 0,91 мин; MS (ESIпоз): m/z=1120 (M+H)+
Промежуточное соединение 141
N-[4-({1-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил]пиперидин-4-ил}окси)-4-оксобутил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

44 мг (49 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 2 мл дихлорметана, и затем добавляли к смеси 18,8 мг (98 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 3,8 мг (24 мкМоль) 4-диметиламинопиридина и 23 мг (98 мкМоль) коммерчески доступного бензил 4-гидроксипиперидин-1-карбоксилата. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 22 мг (40% от теоретического количества) защищенного Z-группой промежуточного соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,04 мин; MS (ESIпоз): m/z=1116 (M+H)+.
Затем защитную Z-группу отделяли гидролитическим путем в этаноле над подложкой палладий/активированный уголь.
Затем 19 мг (19 мкМоль) незащищенного сырого продукта, без дальнейшей очистки, помещали в 4 мл ДМФ, и добавляли к смеси 7 мг (39 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутановой кислоты, 11 мг (29 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 5 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана получали 7,5 мг (34% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,94 мин; MS (ESIпоз): m/z=1147 (M+H)+.
Промежуточное соединение 142
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

9 мг (9,5 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 72) растворяли в 1000 мкл ДМФ, и затем добавляли к смеси 10 мг (38 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-177-пиррол-1-ил)гексангидразида, 7,2 мг (19 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 8 мкл N,N-диизопропилэтиламина, после чего перемешивали реакционную смесь при комнатной температуре в течение 1 часа. Затем при пониженном давлении удаляли растворитель, и очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции концентрировали и, после лиофилизации, получали 6.4 мг (58% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1); Rt=0,99 мин; MS (ESIпоз): m/z=1154 (M+H)+.
Промежуточное соединение 143
N-(4-{2-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-l-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
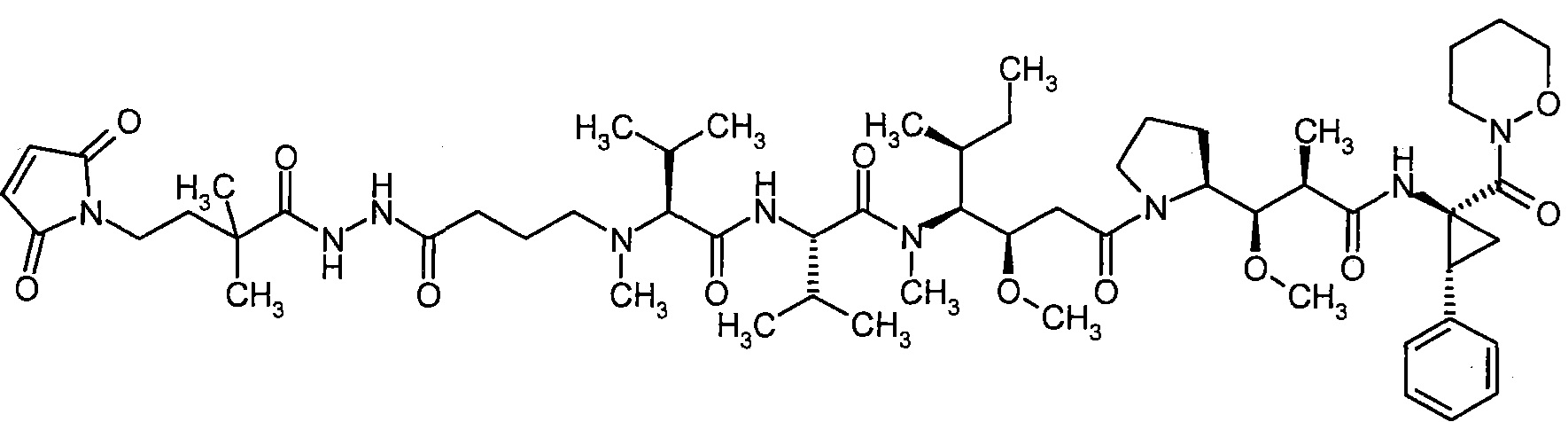
Осуществляли реакцию 6 мг (6,7 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил] амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 61) с 3 мг (8,7 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутангидразид трифторацетата аналогично процедуре, описанной для промежуточного соединения 142, с получением 2 мг (27% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод З): Rt=1,92 мин; MS (ESIпоз): m/z=1106 (M+H)+.
Промежуточное соединение 144
N-(4-{2-[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-2,2-диметилбутаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 5 мг (5,6 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в 1 мл ДМФ добавляли 7,65 мг (22,5 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)-2,2-диметилбутангидразид трифторацетата, 3,2 мг (16,9 мкМоль) EDC, 1,96 мкл (11,3 мкМоль) диизопропилэтиламина и 2,6 мг (16,9 мкМоль) НОВТ. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Затем добавляли еще 0,95 мг (2,8 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)-2,2-диметилбутангидразид трифторацетата. Перемешивали в течение ночи, после чего реакционную смесь концентрировали и очищали методом препаративной ВЭЖХ. Процедура давала 3,5 мг (85% чистота, 48% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод З): Rt=1,86 мин; m/z=1094 (М+Н)+.
Промежуточное соединение 145
N-[3-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)пропил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенил циклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

12 мг (14 мкМоль) N-(3-аминопропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 66) помещали в 750 мкл диоксана и добавляли к смеси 1,5 мл насыщенного раствора бикарбоната натрия, и затем 3,2 мг (21 мкМоль) метил 2,5-диоксо-2,5-дигидро-1H-пиррол-1-карбоксилата. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 4,2 мг (32% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,94 мин; MS (ESIпоз): m/z=950 (М+Н)+.
Промежуточное соединение 146
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(2S)-1-[бензил(метил)амино]-1-оксо-3-фенилпропан-2-ил}амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли реакцию 9 мг (9,8 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-({(2S)-1-[бензил(метил)амино]-1-оксо-3-фенилпропан-2-ил} амино)-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 73) аналогично процедуре, описанной для промежуточного соединения 133, с 10 мг (39 мкМоль) 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида, с получением 1,8 мг (15% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,2 мин;
ЖХ-МС (Метод 9): Rt=5,11 мин; MS (ESIпоз): m/z=1128 (M+H)+.
Промежуточное соединение 147
N-{4-[(2,5-диоксопирролидин-1-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3S)-1-(бензилокси)-1-оксо-3-фенилбутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

16 мг (17 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S,3S)-1-(бензилокси)-1-оксо-3-фенилбутан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 70) растворяли в 2 мл дихлорметана и добавляли к смеси 2,6 мг (23 ммоль) 1-гидроксипирролидин-2,5-диона, и затем 4 мг (21 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида. Реащионную смесь перемешивали при комнатной температуре в течение 2 часов, еще раз добавляли те же количества 1-гидроксипирролидин-2,5-диона и 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид. Затем перемешивали при комнатной температуре в течение ночи, после чего реакционную смесь концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 10 мг (56% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=2,0 мин;
Промежуточное соединение 148
N-{4-[(2-{[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил](метил)амино}этил)амино]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-N-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
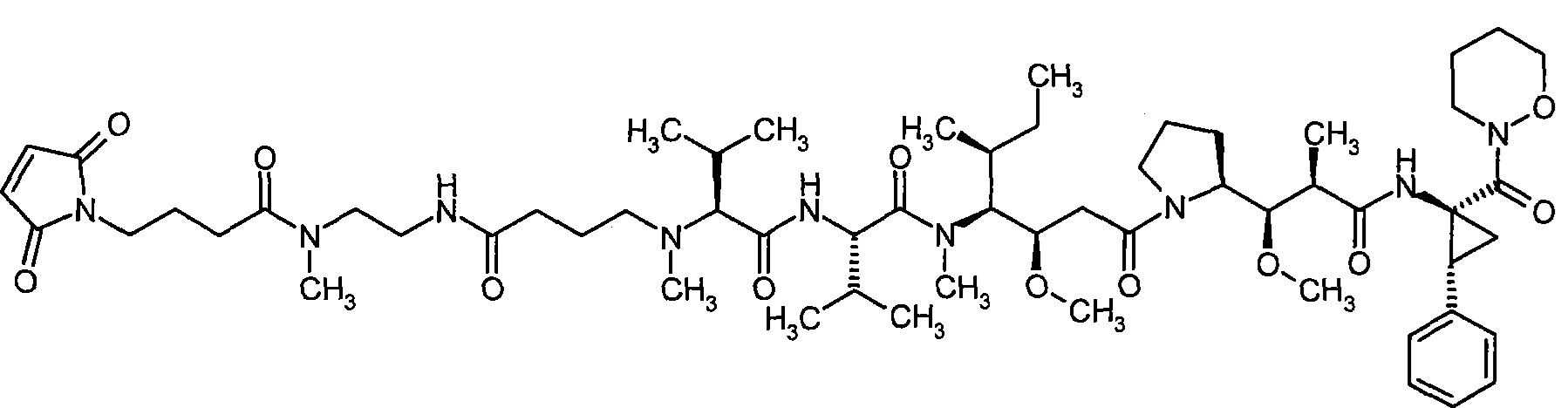
6 мг (7 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил] амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 61) соединяли с 2,8 мг (8 мкМоль) N-(2-аминоэтил)-4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-метилбутанамид трифторацетата, 10,1 мг (27 мкМоль) О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 5 мкл N,N-диизопропилэтиламина в 2 мл ДМФ, и перемешивали при комнатной температуре в течение ночи. Затем добавляли еще 5 мг (23,5 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 3 мкл N,N-диизопропилэтиламина. Перемешивали при комнатной температуре в течение еще 5 часов, и удаляли растворитель при пониженном давлении, после чего очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции концентрировали и, после лиофилизации из диоксана, получали 1,3 мг (15% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод 2): Rt=1,21 мин; MS (ESIпоз): m/z=1120 (M+H)+.
Промежуточное соединение 149
N-{4-[(2-{[4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)бутаноил]амино}этил)(метил)амино]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

6 мг (7 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 61) соединяли с 3,1 мг (9 мкМоль) 4-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-N-[2-(матиламино)этил] бутанамид трифторацетата, 10,1 мг (27 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата и 5 мкл НТУ-диизопропилэтиламина в 2 мл ДМФ, и перемешивали реакционную смесь при комнатной температуре в течение 4 часов. Затем удаляли растворитель при пониженном давлении, и очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции концентрировали и, после лиофилизации из диоксана, получали 1 мг (13.4% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=0,89 мин; MS (ESIпоз): m/z=1121 (M+H)+.
Промежуточное соединение 150
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-((2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S,2R)-2-фенил-1-(пропилкарбамоил)циклопропил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
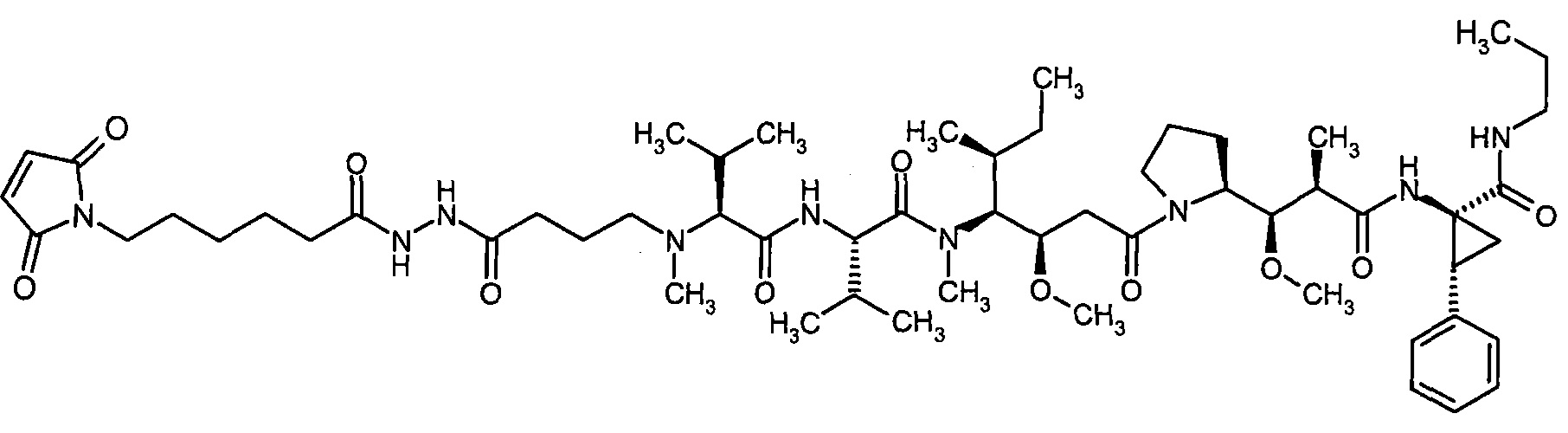
7,9 мг (9 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S,2R)-2-фенил-1-(пропилкарбамоил)циклопропил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 3 мл ДМФ, и затем добавляли к смеси 10.4 мг (54 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 8,3 мг (54 мкМоль) 1-гидрокси-1H-бензотиазол гидрата, 9 мкл N,N-диизопропилэтиламина, и 9,5 мг (36 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом, получали 4,3 мг (22% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=1,9 мин;
ЖХ-МС (Метод 9): Rt=4,93 мин; MS (ESIпоз); m/z=1078 (M+H)+.
Промежуточное соединение 151
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-карбамоил-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
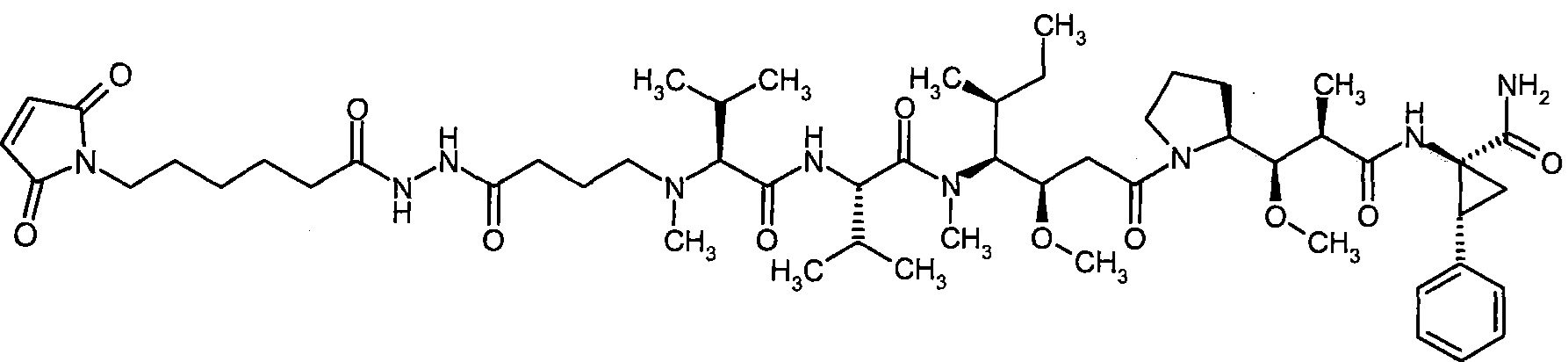
Соединение получали аналогично процедуре, описанной для промежуточного соединения 150, в качестве исходного соединения брали соединение, описанное в процедуре для промежуточного соединения 81.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=1036 (M+H)+.
Промежуточное соединение 152
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-(этоксикарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (12 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-(этоксикарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 3 мл ДМФ и затем добавляли к смеси 8,9 мг (23 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 10 мкл N,N-диизопропилэтиламина и 12 мг (47 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После этого полученное соединение концентрировали в высоком вакууме, и очищали полученный осадок методом препаративной ВЭЖХ. Таким образом получали 5,8 мг (37% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=2,0 мин;
ЖХ-МС (Метод 9): Rt=4,99 мин; MS (ESIпоз): m/z=1066 (М+Н)+.
Промежуточное соединение 153
N-[1-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)-12,15-диоксо-3,6,9-триокса-13,14-диазаоктадекан-18-ил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил] пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 5 мг (5,6 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил] пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в 1 мл ДМФ добавляли 9,7 мг (22,5 мкМоль) 3-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)этокси]этокси}этокси)пропангидразид трифторацетата, 3,2 мг (16,9 мкМоль) EDC, 1,96 мкл (11,3 мкМоль) N,N-диизопропилэтиламина и 2,6 мг (16,9 мкМоль) НОВТ. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Затем добавляли еще 1,2 мг (2,8 мкМоль) 3-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этокси]этокси}этокси)пропангидразид трифторацетата. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем очищали методом препаративной ВЭЖХ.
Процедура давала 3,6 мг (51% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,90 мин; m/z=1185 (М+H)+.
Промежуточное соединение 154
(2R,3S)-3-амино-4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутан-2-ил (3R,4S,7S,10S)-4-[(2S)-бутан-2-ил]-7,10-диизопропил-3-(2-{(2S)-2-[(1S,2S)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазапентадекан-15-оат

15 мг (17 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S)-1-(1,2-оксазинан-2-ил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 10 мл дихлорметана и затем добавляли к смеси 12,8 мг (67 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 10 мг (83 мкМоль) 4-диметиламинопиридина и 10,3 мг (33 мкМоль) коммерчески доступного бензил N-(трет-бутоксикарбонил)-L-треонината. Реакционную смесь нагревали с обратным холодильником в течение 4 часов. Затем еще раз добавляли те же количества связывающего реагента и 4-диметиламинопиридина, и нагревали реакционную смесь с обратным холодильником в течение ночи. Затем реакционную смесь разбавляли дихлорметаном и экстрагировали однократным встряхиванием с водой, после чего отделяли органическую фазу, и концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 7,7 мг (37% от теоретического количества) защищенного промежуточного соединения в виде бесцветной пены.
ВЭЖХ (Метод 12): Rt=2,5 мин;
ЖХ-МС (Метод 1): Rt=1,13 мин; MS (ESIпоз): m/z=1190 (M+H)+.
Затем защитную группу бензилового эфира удаляли гидрогенизацией при стандартном давлении водорода в метаноле над 10% палладием на активированном угле, после чего осуществляли реакцию присоединения кислоты, полученной как описано в примере промежуточного соединения 151, и 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. На последнем этапе, защитную группу Boc удаляли трифторуксусной кислотой. Оставшийся осадок очищали методом препаративной ВЭЖХ. Таким образом получали 0,22 мг (2,5% от теоретического количества в три этапа) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,81 мин; MS (ESIпоз): m/z=1207 (М+Н)+.
Промежуточное соединение 155
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-n-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 152, из N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,82 мин; MS (ESIпоз): m/z=1024 (М+Н)+.
Промежуточное соединение 156
N-(3-{[(1-{[(2,5-диоксопирролидин-1-ил)окси]карбонил}циклопропил)карбонил]амино}пропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу, описанному в качестве последнего этапа синтеза промежуточного соединения 131, из N-(3-аминопропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и 1,1'-[циклопропан-1,1-диилбис(карбонилокси)]дипирролидин-2,5-диона, который заблаговременно получали из соответствующей дикарбоновой кислоты.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,92 мин; MS (ESIпоз): m/z=1080 (M+H)+.
Промежуточное соединение 157
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
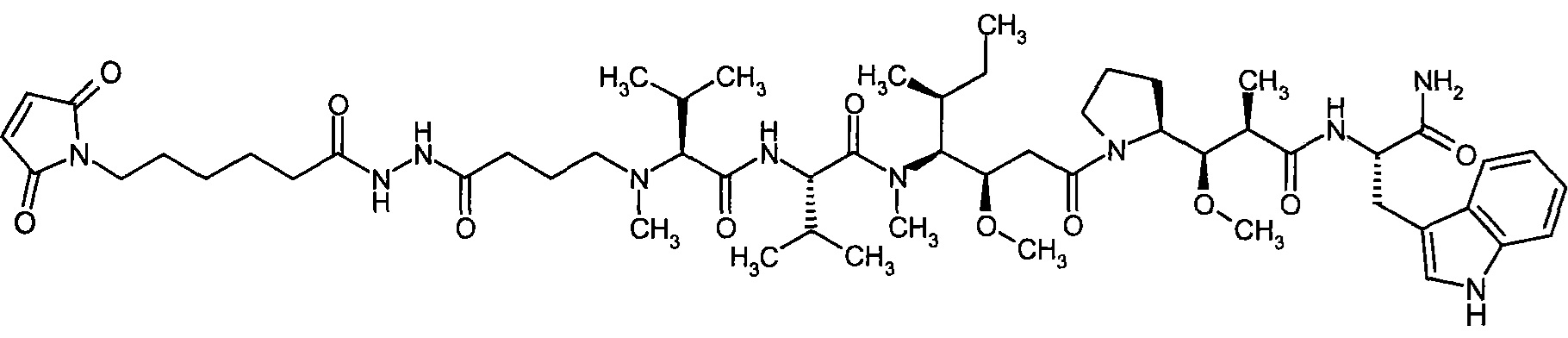
15 мг (18 мкМоль) (N-(3-карбоксипропил)-N-метил-L-валил-N-(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 3,8 мл ДМФ, и затем добавляли к смеси 27 мг (70 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 12 мкл N,N-диизопропилэтиламина и 14 мг (53 мкМоль) коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После этого полученное соединение концентрировали в высоком вакууме, и очищали полученный осадок методом препаративной ВЭЖХ. Таким образом получали 6,2 мг (33% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,85 мин; MS (ESIпоз): m/z=1063 (М+Н)+.
1Н-ЯМР (500 МГц, ДМСО-d6, характеристические сигналы): 5=10,8 (d, 1Н), 9,8-9,7 (m, 2H), 9,6 и 9.4 (2m, 1Н), 8,9, 8,88, 8,78 и 8,75 (4d, 1Н), 8,08 и 7,85 (2d, 1Н), 7,6-6,9 (m, 9H), 4,7-4.4 (m, 3H), 3.4 (t, 2H), 3,23, 3,2, 3,18, 3,0, и 2,99 (5s, 9H), 2,8 (m, 3H), 2,1 (t, 2H), 1,06 и 1,01 (2d, 3Н), 0,95-0,8 (m, 15H), 0,8-0,75 (dd, 3H).
Промежуточное соединение 158
N-[4-({(2R)-1-[(2,5-диоксопирролидин-1-ил)окси]-4-метил-1-оксопентан-2-ил}амино)-4-оксобутил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N--метил-L-валинамид

13 мг (14,7 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 4 мл диметилформамида, и затем добавляли к смеси 9.4 мг (25 мкМоль) О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 6 мкл N,N-диизопропилэтиламина и 7 мг (31 мкМоль) коммерчески доступного трет-бутил D-лейцинат гидрохлорида. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, и затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана/воды, получали 6,5 мг (49% от теоретического количества) защищенного промежуточного соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=2,2 мин;
ЖХ-МС (Метод 1): Rt=1,21 мин; MS (ESIпоз): m/z=1076 (M+H)+.
Сначала для удаления защитной группы Boc у этого защищенного промежуточного соединения, использовали трифторуксусную кислоту в дихлорметане, с получением 6,2 мг (99% от теоретического количества) незащищенного соединения. 5,2 мг (5 мкМоль) этого промежуточного соединения помещали в 1,5 мл дихлорметана, и осуществляли его реакцию с 0,8 мг (7 мкМоль) N-гидроксисукцинимида в присутствии 1,2 мг (6 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 0,16 мг (1 мкМоль) 4-диметиламинопиридина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали и очищали методом препаративной ВЭЖХ. В результате получали 1,3 мг указанного в названии соединения, некоторая часть которого была гидролизована до исходного соединения.
Промежуточное соединение 159
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-]-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 157, из N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида.
Выход: 6 мг (53% от теоретического количества)
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,94 мин; MS (ESIпоз): m/z=1114 (M+H)+.
Промежуточное соединение 160
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 157, из 20 мг (21 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида.
Выход: 13 мг (52% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,92 мин; MS (ESIпоз): m/z=1153 (М+Н)+.
Промежуточное соединение 161
N-(6-{2-[6-2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексаноил]гидразино}-6-оксогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу описанному в примере промежуточного соединения 157, из N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1H-пиррол-1-ил)гексангидразида.
Выход: 0,8 мг (16% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,78 мин; MS (ESIпоз): m/z=1092 (M+H)+.
Промежуточное соединение 162
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-(2R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
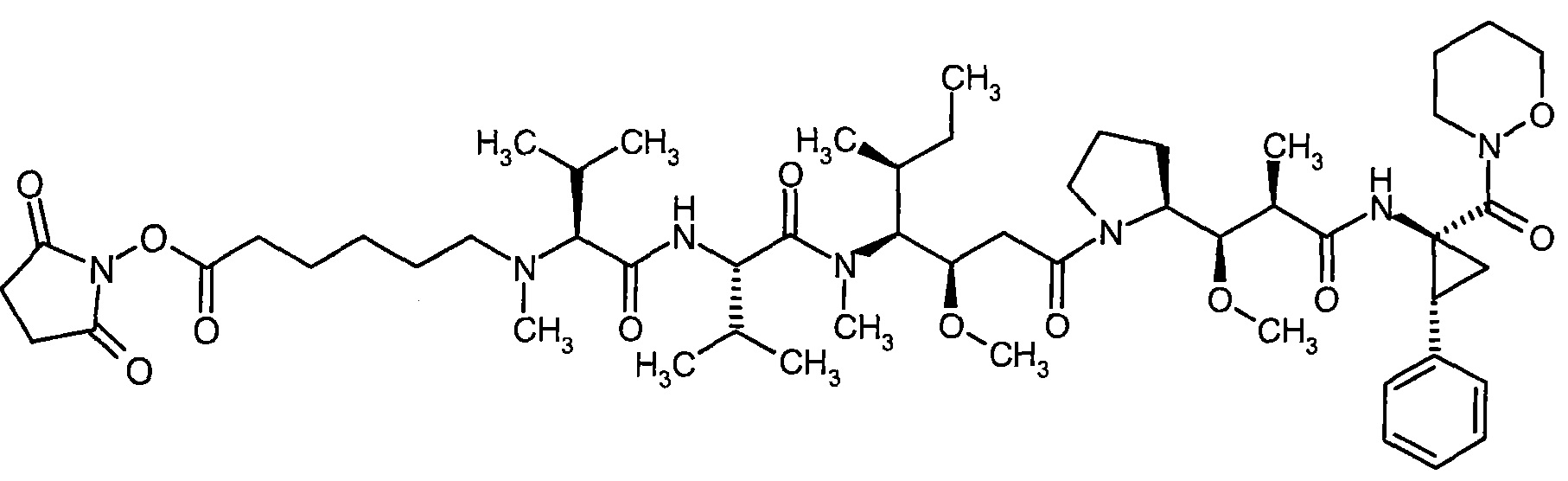
18 мг (20 мкМоль) N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 64) растворяли в 3,2 мл дихлорметана, и добавляли к смеси 22 мг (190 ммоль) 1-гидроксипирролидин-2,5-диона и затем 11 мг (60 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 0,24 мг (0,17 мкМоль) DMAP. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, добавляли еще 22 мг (190 ммоль) 1-гидроксипирролидин-2,5-диона, 11 мг (60 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 0,24 мг (0,17 мкМоль) DMAP, и перемешивали реакционную смесь при комнатной температуре в течение еще часа. После этого, полученное соединение концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 8,2 мг (41% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=2,0 мин;
ЖХ-МС (Метод 11): Rt=0,9 мин; MS (ESIпоз): m/z=1024 (M+H)+.
Промежуточное соединение 163
[(1S,2R)-1-амино-2-фенилциклопропил](1,4-дигидро-3H-2,3-бензоксазин-3-ил)метанон трифторацетат
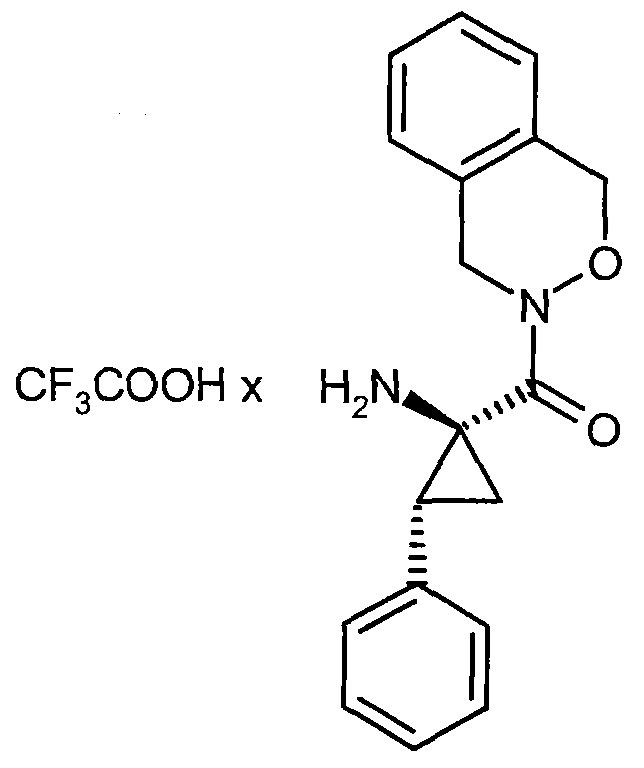
На первом этапе синтеза брали в качестве исходного соединения 265 мг (0,82 ммоль) трет-бутил (1S,2R)-1-(гидроксикарбамоил)-2-фенилциклопропил карбамата (Исходное соединение 7), и осуществляли реакцию с 1,2-бис(бромметил)бензолом, по аналогии с описанным в литературе методом (см. Н. King, J. Chem. Soc. 1942, 432), и получали промежуточное соединение - защищенный группой Boc трет-бутил (1S,2R)-1-(1,4-дигидро-3Н-2,3-бензоксазин-3-илкарбонил)-2-фенилциклопропил карбамат.
Выход: 108 мг (34% от теоретического количества)
ЖХ-МС (Метод 2): Rt=1,3 мин; MS (ESIпоз): m/z=395 (М+Н)+.
108 мг (0,27 ммоль) этого промежуточного вещества помещали в 3,7 мл дихлорметана, добавляли 1,8 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 15 мин. После этого полученное соединение концентрировали при пониженном давлении, и лиофилизировали полученный осадок из диоксана. В результате получали 112 мг указанного в названии соединения с количественным выходом в виде бесцветной пены.
ЖХ-МС (Метод 1): Rt=0,7 мин; MS (ESIпоз): m/z=295 (М+Н)+.
Промежуточное соединение 164
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1S,2R)-3-{[(1S,2R)-1-(1,4-дигидро-3H-2,3-бензоксазин-3-илкарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

166 мг (0,196 ммоль) N[(9Я-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 10) помещали в 40 мл ДМФ и последовательно добавляли в смесь 80 мг (0,196 ммоль) [(1S,2R)-1-амино-2-фенилциклопропил](1,4-дигидро-3H-2,3-бензоксазин-3-ил)метанон трифторацетата (промежуточное соединение 163), 112 мг (0,294 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 682 мкл (3,9 ммоль) N,N-диизопропилэтиламина. После этого, реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении, осадок помещали в этилацетат, и промывали раствор насыщенным водным раствором хлорида натрия. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Осадок окончательно очищали методом препаративной ВЭЖХ. Таким образом, получали 19 мг (9% от теоретического количества) защищенного группой Fmoc промежуточного соединения N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-(1,4-дигидро-3H-2,3-бензоксазин-3-илкарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ВЭЖХ (Метод 5): Rt=1,68 мин;
ЖХ-МС (Метод 1): Rt=1,51 мин; MS (ESIпоз): m/z=1083 (М+Н)+.
19 мг (0,015 ммоль) этого промежуточного вещества растворяли в 4 мл ДМФ. После добавления 817 мкл пиперидина, реакционную смесь перемешивали при комнатной температуре в течение 5 мин. После этого полученное соединение концентрировали при пониженном давлении и дегидрировали осадок диэтиловым эфиром, после чего очищали методом препаративной ВЭЖХ (элюент: ацетонитрил+0,1% TFA / 0,1% вод. TFA). Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении, затем осадок лиофилизировали из диоксана/воды. В результате получали 12 мг (92% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 6): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,94 мин; MS (ESIпоз): m/z=861 (М+Н)+.
Промежуточное соединение 165
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-(1,4-дигидро-3Н-2,3-бензоксазин-3-илкарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
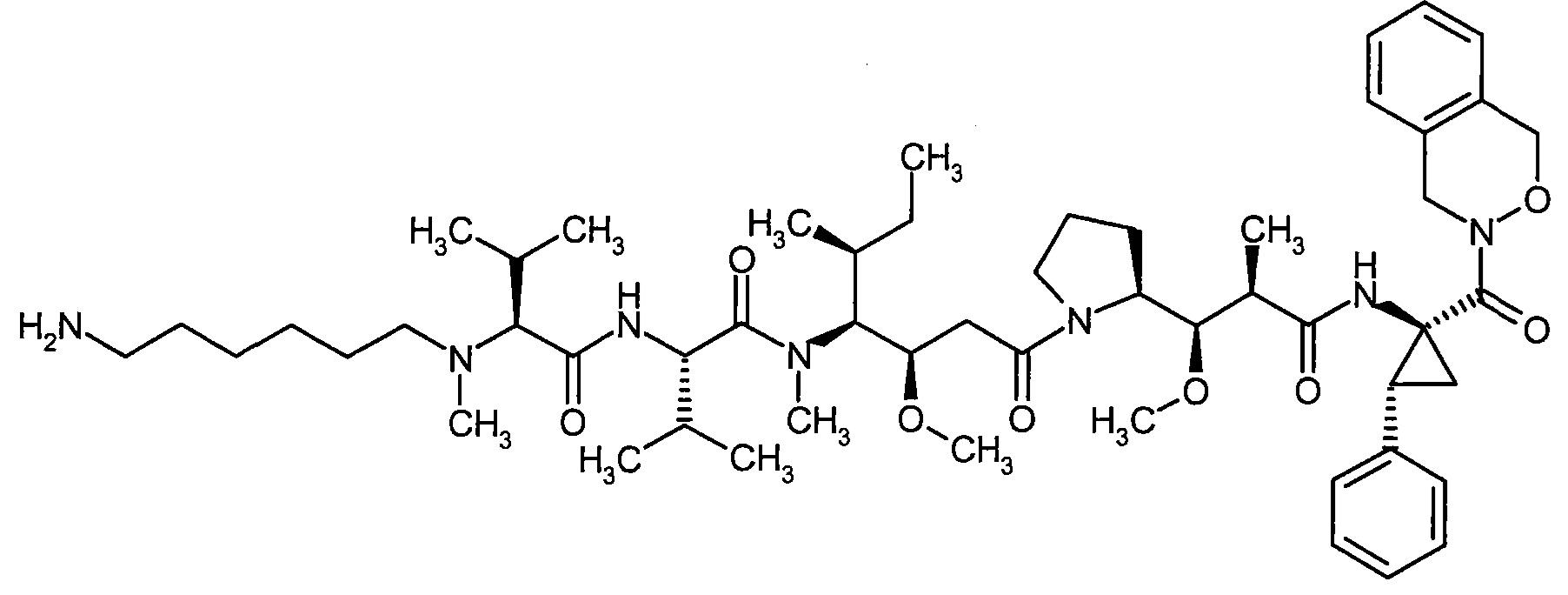
Для приготовления указанного в названии соединения, брали 20 мг (0,021 ммоль) промежуточного соединения 164 по аналогии с приготовлением промежуточного соединения 97, использовали бензил 6-оксогексил карбамат в присутствии цианоборогидрида натрия, после чего гидрогенолитически удаляли защитную группу Z (с 5% палладием на угле в качестве катализатора, в метаноле в качестве растворителя).
Выход: 4,5 мг (23% от теоретического количества в 2 этапа)
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,9 мин; MS (ESIпоз): m/z=960 (М+Н)+.
Промежуточное соединение 166
N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-(1,4-дигидро-3Н-2,3-бензоксазин-3-илкарбонил)-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

4.4 мг (4,5 мкМоль) промежуточного соединения 165 помещали в 1 мл смеси 1:1 диоксан/вода, и затем добавляли к смеси 1 мг (6,8 мкМоль) метил 2,5-диоксо-2,5-дигидро-1H-пиррол-1-карбоксилата и 50 мкл насыщенного водного раствора бикарбоната натрия. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли еще 50 мкл насыщенного водного раствора бикарбоната натрия, и перемешивали реакционную смесь при комнатной температуре в течение еще 15 мин, после ченго концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации, получали 1 мг (21% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,08 мин; MS (ESIпоз): m/z=1040 (M+H)+.
Промежуточное соединение 167
Бензил 3-{2-[2-(2-оксоэтокси)этокси]этокси}пропаноат

Указанное в названии соединение получали из 6 г (21,55 ммоль) коммерчески доступной 3-{2-[2-(2-гидроксиэтокси)этокси]этокси}пропановой кислоты при стандартных условиях, этерификацией хлоридом бензила и карбонатом цезия, и последующим окислением комплексом серный ангидрид-пиридин.
Выход: 611 мг (10% от теоретического количества в 2 этапа)
ЖХ-МС (Метод 2): Rt=1,69 мин; MS (ESIпоз): m/z=311 (M+H)+.
Промежуточное соединение 168
N-(2-{2-[2-(2-карбоксиэтокси)этокси]этокси}этил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 69, осуществляли реакцию сочетания N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) и Nα-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-триптофанамид трифторацетата (промежуточное соединение 49) в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, с последующим удалением защитной группы Fmoc с помощью пиперидина, и получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата.
25 мг (0,028 ммоль) этого соединения и 17,5 мг (0,06 ммоль) промежуточного соединения 167 объединяли в 2 мл метанола и добавляли к смеси 12,6 мг (0,14 ммоль) косплекса боран-пиридин и 2,5 мл уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем еще раз добавляли те же количества комплекса боран-пиридин и уксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение еще 24 часов. После этого полученное соединение концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. После концентрирования соответствующих фракций и лиофилизации из 1:1 диоксана/воды, получали 26,5 мг (88% от теоретического количества) защищенного Z-группой промежуточного соединения.
ВЭЖХ (Метод 12): Rt=2,04 мин;
ЖХ-МС (Метод 1): Rt=0,97 мин; MS (ESIпоз): m/z=1064 (M+H)+.
25 мг (0,024 ммоль) этого промежуточного вещества помещали в 10 мл метанола и гидрогенировали над подложкой 10% палладий на активированном угле при стандартном давлении водорода, при комнатной температуре в течение 45 мин. Затем катализатор отфильтровывали и удаляли растворитель при пониженном давлении. После лиофилизации из диоксана, получали 19,7 мг (85% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,83 мин; MS (ESIno3): m/z=974 (M+H)+.
Промежуточное соединение 169
N-{2-[2-(2-{3-[(2,5-диоксопирролидин-1-ил)окси]-3-оксоргорокси}этокси)этокси]этил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1H-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (10 мкМоль) промежуточного соединения 168 растворяли в 3 мл ДМФ и добавляли к смеси 3,5 мг (30 ммоль) 1 -гидроксипирролидин-2,5-дион, и затем 2.4 мг (10 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 5 мкл N,N-диизопропилэтиламин. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, добавляли 8 мг (0,02 ммоль) HATU и перемешивали реакционную смесь еще раз при комнатной температуре в течение ночи, затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана, получали 8,6 мг (64% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 11): Rt=0,81 мин; MS (ESIпоз): m/z=1071 (M+H)+.
Промежуточное соединение 170
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 101 в 2 этапа, в качестве исходного соединения брали 26 мг (0,028 ммоль) промежуточного соединения 15.
Выход: 16,7 мг (63% от теоретического количества в 2 этапа)
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,81 мин; MS (ESIпоз): m/z=914 (M+H)+.
Промежуточное соединение 171
N-(6-{[4-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)бутаноил]амино}гексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

6,7 мг (7,3 мкМоль) соединения, полученного из промежуточного соединения 170 и 3 мг (14,7 мкМоль) коммерчески доступной 4-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)бутановой кислоты, помещали в 2 мл ДМФ, и добавляли к смеси 5,6 мг (14,7 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 2 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали и очищали осадок методом препаративной ВЭЖХ. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении, затем осадок лиофилизировали из диоксана. Таким образом, получали 4,5 мг (56% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=1,12 мин; MS (ESIпоз): m/z=1079 (М+Н)+.
Промежуточное соединение 172
бензил 2-{2-[2-(2-оксоэтокси)этокси]этокси}этил карбамат

Указанное в названии соединение получали из коммерчески доступного 2-{2-[2-(2-аминоэтокси)этокси]этокси}этанола при стандартных условиях, сначала введением защитной группы Z, и затем окислением комплексом серный ангидрид-пиридин.
ВЭЖХ (Метод 12): Rt=1.4 мин;
ЖХ-МС (Метод 11): Rt=0,65 мин; MS (ESIпоз): m/z=326 (М+Н)+.
Промежуточное соединение 173
бензил {2-[2-(2-оксоэтокси)этокси]этил карбамат

Указанное в названии соединение получали аналогично процедуре, описанной для промежуточного соединения 172 из коммерчески доступного 2-[2-(2-аминоэтокси)этокси]этанола при стандартных условиях, на первом этапе введением защитной группы Z, и затем окислением комплексом серный ангидрид-пиридин.
ВЭЖХ (Метод 12): Rt=1,3 мин;
ЖХ-МС (Метод 11): Rt=0,68 мин; MS (ESIпоз): m/z=282 (M+H)+.
Промежуточное соединение 174
N-(2-{2-[2-(2-аминоэтокси)этокси]этокси}этил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенил циклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли восстановительное аминирование 47 мг (0,05 ммоль) промежуточного соединения 16, по аналогии с приготовлением промежуточного соединения 167, с бензил 2-{2-[2-(2-оксоэтокси)этокси]этокси}этил карбаматом в присутствии комплекса боран-пиридин. Затем защитную группу Z удаляли гидролитическим путем с 5% палладием на угле в качестве катализатора и в метаноле в качестве растворителя, и получали 38 мг (66% от теоретического количества в 2 этапа) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,8 мин; MS (ESIпоз): m/z=988 (M+H)+.
Промежуточное соединение 175
N-[2-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этокси]этокси}этокси)этил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(18,2К)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Соединение готовили аналогично процедуре синтеза промежуточного соединения 166, в качестве исходного соединения брали 34 мг (0,03 ммоль) промежуточного соединения 174.
Выход: 8,3 мг (23% от теоретического количества)
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,97 мин; MS (ESIпоз): m/z=1068 (M+H)+.
Промежуточное соединение 176
N-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этокси]этокси}этил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили аналогично процедуре, описанной для промежуточных соединений 174 и 175, начиная с восстановительного аминирования промежуточного соединения 16 промежуточным соединением 173, с последующим удалением защиты и образованием малеимида.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 11); Rt=0,8 мин; MS (ESIпоз): m/z=981 (М+Н)+.
Промежуточное соединение 177
N-[2-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этокси]этокси}этокси)этил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили аналогично процедуре, описанной для промежуточных соединений 174 и 175, начиная с восстановительного аминирования промежуточного соединения 16 промежуточным соединением 172, с последующим удалением защиты и образованием малеимида.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1025 (М+Н)+.
Промежуточное соединение 178
N-{4-[(2,5-диоксопирролидин-1-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили аналогично процедуре, описанной для промежуточного соединения 162, в качестве исходного соединения брали 6 мг промежуточного соединения 82.
ЖХ-МС (Метод 1): Rt=0,82 мин; MS (ESIпоз): m/z=953 (M+H)+.
Промежуточное соединение 179
4-[(1Е,3S)-3-амино-4-фенилбут-1-ен-1-ил]бензолсульфокислотытрифторацетат
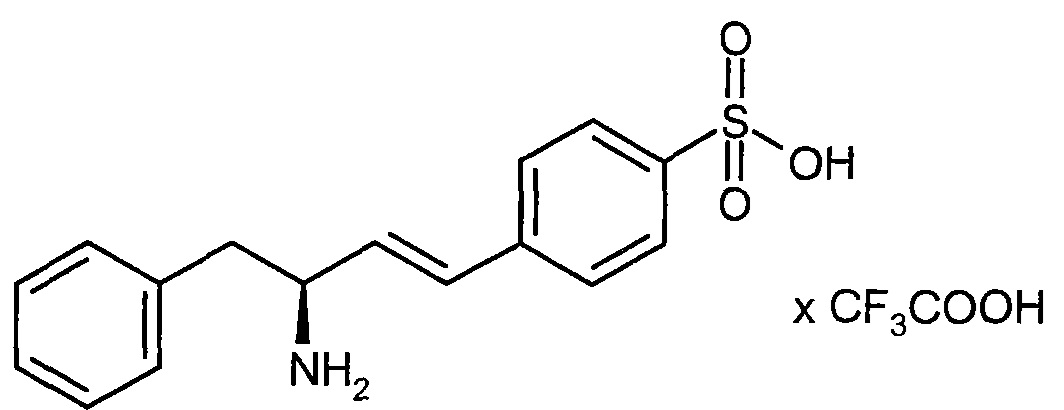
Смесь 13,6 мг (0,06 ммоль) ацетата палладия (II), 469 мг (1.46 ммоль) 4-йодобензолсульфоната калия, 300 мг (1,21 ммоль) (S)-трет-бутил 1-фенилбут-3-ен-2-ил карбамата, 16,5 мг (0,12 ммоль) фенилмочевины и 167,6 мг (1,21 ммоль) карбоната калия в 7,5 мл ДМФ нагревали до 160°С в микроволновой печи в течение 15 мин. Сырой продукт затем непосредственно очищали методом препаративной ВЭЖХ. Процедура давала 312 мг смеси 31% ВОС-защищенного соединения и 69% свободного амина.
Эту смесь затем помещали в 30 мл дихлорметана, добавляли к смеси 1 мл трифторуксусной кислоты, и перемешивали при комнатной температуре в течение 20 часов. После концентрирования при пониженном давлении, осадок перемешивали с диэтиловым эфиром, и отфильтровывали образовавшийся осадок с отсасыванием, после чего промывали с диэтиловым эфиром. Процедура давала 200 мг (62% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 11): Rt=0.44 мин; MS (ESIпоз): m/z=304 (М+Н)+.
Промежуточное соединение 180
4-[(3R)-3-амино-4-фенилбутил]бензолсульфокислота

100 мг (0,25 ммоль) 4-[(1Е,3S)-3-амино-4-фенилбут-1-ен-1-ил]бензолсульфокислоты трифторацетата суспендировали в 10 мл уксусной кислоты, нескольких каплях ДМФ и воды, добавляли к смеси 70 мг (0,07 ммоль) палладия на угле (10%) и гидрогенировали при давлении водорода 2,2 бар в течение 24 часов. Реакционный раствор фильтровали, и очищали фильтрат методом препаративной ВЭЖХ.
29 мг (76% чистота, 21% от теоретического количества) продукта.
ЖХ-МС (Метод 1): Rt=0.46 мин; MS (ESIпоз): m/z=306 (M+H)+.
Промежуточное соединение 181
N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2s)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S,3Е)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 90 мг (0,13 ммоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамидв в 4 мл ДМФ добавляли 60 мг (0,16 ммоль) HATU и 69 мкл (0,39 ммоль) основания Хунига. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем добавляли к смеси 60 мг (0,15 ммоль) 60,3 мг (0,13 ммоль) 4-[(1Е,3S)-3-амино-4-фенилбут-1-ен-1-ил]бензолсульфокислоты трифторацетата. Перемешивали в течение ночи, после чего реакционную смесь очищали методом препаративной ВЭЖХ. Процедура давала 127 мг 44:56 смеси указанного в названии соединения и амина с уже удаленной защитной группой.
ЖХ-МС (Метод 1): Rt=1,21 мин; MS (ESIпоз): m/z=971 (М+Н)+; Rt=0,84 мин; MS (ESIпоз): m/z=871 (М+Н)+ для соединения с удаленной защитой.
Промежуточное соединение 182
N-метил-L-валил-N-(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S,3E)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

90 мг промежуточного соединения 180 растворяли в 4,6 мл дихлорметана, и добавляли 0,92 мл трифторуксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем концентрировали. Полученный сырой продукт очищали методом препаративной ВЭЖХ.
Процедура давала 91 мг (98% от теоретического количества) нужного соединения.
ЖХ-МС (Метод 1): Rt=0,85 мин; MS (ESIпоз): m/z=871 (M+H)+.
Промежуточное соединение 183
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S,3Е)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино} пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-R-метил-L-валинамид

16,7 мкл (0,03 ммоль) 15% водного раствора сукцинальдегида в начале процедуры помещали в реакционный сосуд с 943 мкл метанола, и добавляли к смеси 17 мг (0,02 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S,3Е)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 181) и 1,1 мкл (0,02 ммоль) уксусной кислоты. Реакционную смесь перемешивали в течение 5 мин при комнатной температуре, и затем добавляли 2,9 мкл (0,02 ммоль) комплекса боран-пиридин. Через 1 час, добавляли еще по два эквивалента сукцинальдегида и уксусной кислоты, после чего добавляли комплекс боран-пиридин и перемешивали реакционную смесь при комнатной температуре в течение 20 часов. Затем реакционную смесь очищали методом препаративной ВЭЖХ.
Процедура давала 20 мг (83% чистота, 80% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=957 (М+Н)+.
Промежуточное соединение 184
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S,3E)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

8 мг (7,5 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3Е,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3- {[(2S,3E)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида, 2,8 мг (8,2 мкМоль) 6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексангидразид трифторацетата, 3.4 мг (9 мкМоль) HATU и 3,9 мкл основания Хунига перемешивали в 0,77 мл ДМФ при комнатной температуре в течение 20 часов. Затем реакционную смесь очищали методом препаративной ВЭЖХ.
В результате получали 3 мг (31% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,90 мин; MS (ESIпоз): m/z=1164 (М+Н)+.
Промежуточное соединение 185
N-{4-[(2,5-диоксопирролидин-1-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S,3Е)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 8 мг (7,5 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2S,3Е)-1-фенил-4-(4-сульфофенил)бут-3-ен-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в 2 мл ДМФ добавляли 8,6 мг (74,8 мкМоль) N-гидроксисукцинимида, 8,5 мг (22.4 мкМоль) EDCI и 0,1 мг (0,75 мкМоль) DMAP. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Затем добавляли 1,3 мкл (7,5 мкМоль) основания Хунига, и перемешивали реакционную смесь в течение 1 часа. Затем реакционную смесь очищали методом препаративной ВЭЖХ. В результате получали 2,6 мг (72% чистота, 21% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,89 мин; MS (ESIпоз): m/z=1054 (M+H)+
Промежуточное соединение 186
N-(трет-бутоксикарбонил)-N-метил-L-валил-n-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 43 мг (0,06 ммоль) N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в 1,9 мл ДМФ добавляли 29 мг (0,07 ммоль) HATU и 33 мкл (0,19 ммоль) основания Хунига. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем добавляли к смеси 29 мг (0,07 ммоль) 4-[(3R)-3-амино-4-фенилбутил]бензолсульфокислота трифторацетата. Перемешивали в течение ночи, после чего реакционную смесь очищали методом препаративной ВЭЖХ. Процедура давала 58 мг 45:55 смеси указанного в названии соединения и амина с уже удаленной защитной группой.
ЖХ-МС (Метод 1): Rt=1,09 мин; MS (ESIпоз): m/z=973 (М+Н)+; Rt=0,87 мин; MS (ESIпоз): m/z=873 (M+H)+ для соденинения с удаленной защитой.
Промежуточное соединение 187
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

58 мг промежуточного соединения 186 растворяли в 4,1 мл дихлорметана, добавляли 0.41 мл трифторуксусной кислоты и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. После концентрирования при пониженном давлении, сырой продукт очищали методом препаративной ВЭЖХ.
В результате получали 50 мг (90% чистота, 85% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=873 (М+Н)+
Промежуточное соединение 188
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

171 мкл (0,26 ммоль) 15% водного раствора сукцинальдегида в начале процедуры помещали в реакционный сосуд с 2,5 мл метанола, и добавляли к смеси 50 мг (0,05 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата и 11,6 мкл (0,2 ммоль) уксусной кислоты. Реакционную смесь перемешивали в течение 5 мин при комнатной температуре и добавляли затем 30 мкл (0,24 ммоль) комплекса боран-пиридин. Перемешивали в течение 24 часов, добавляли еще эквивалент комплекса боран-пиридин, и перемешивали реакционную смесь в течение еще 2 часов. Затем реакционную смесь очищали методом препаративной ВЭЖХ.
В результате получали 40 мг (90% чистота, 66% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,91 мин; MS (ESIпоз): m/z=959 (М+Н)+
Промежуточное соединение 189
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (9,3 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида, 3,5 мг (10,3 мкМоль) 6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексангидразид трифторацетата, 4,3 мг (11,2 мкМоль) HATU и 4,9 мкл (28 мкМоль) основания Хунига перемешивали в 1 мл ДМФ при комнатной температуре в течение 20 часов. Затем реакционную смесь очищали методом препаративной ВЭЖХ.
В результате получали 4,2 мг (92% чистота, 33% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,91 мин; MS (ESIпоз): m/z=1166 (М+Н)+
Промежуточное соединение 190
N-{4-[(2,5-диоксопирролидин-1-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К раствору 10 мг (9,3 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(2R)-1-фенил-4-(4-сульфофенил)бутан-2-ил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в 2,5 мл ДМФ добавляли 10,7 мг (93 мкМоль) N-гидроксисукцинимида, 10,6 мг (28 мкМоль) EDCI и 0,12 мг (0,9 мкМоль) DMAP. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, и затем очищали методом препаративной ВЭЖХ.
Процедура давала 3,8 мг (72% чистота, 25% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,90 мин; MS (ESIпоз): m/z=1055 (М+Н)+
Промежуточное соединение 191
(2R,3R)-N-[(2S)-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропанамидтрифторацетат

Указанное в названии соединение получали, следуя процедуре, аналогичной синтезу промежуточного соединения 7 после двух этапов синтеза из исходного соединения 1 и (2S)-2-амино-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)пропан-1-он трифторацетата (промежуточное соединение 99).
Выход в 2 этапа: 62 мг (67% от теоретического количества) ВЭЖХ (Метод 6): Rt=1,65 мин;
ЖХ-МС (Метод 1): Rt=0,7 мин; MS (ESIпоз): m/z=443 (М+Н)+.
Промежуточное соединение 192
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

1015 мг (1,59 ммоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) помещали в 50 мл ДМФ, добавляли к смеси 654 мг (2,39 ммоль) 2-бромо-1-этилпиридиний тетрафторбората (ВЕР) и 2,8 мл N,N-диизопропилэтиламина, и перемешивали при комнатной температуре в течение 10 мин. Затем добавляли 1083 мг (1,75 ммоль) (2R,3R)-N-[(2S)-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропанамид трифторацетата (промежуточное соединение 191), после чего реакционную смесь обрабатывали в ульультразвуковой ванне при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении, и помещали осадок в 300 мл этилацетата. Органическую фазу промывали последовательно 5% водным раствором лимонной кислоты и 5% водным раствором бикарбоната натрия, сушили над сульфатом магния, фильтровали и концентрировали. Сырой продукт, полученный таким образом, (1684 мг), без дальнейшей очистки помещали в 20 мл ацетонитрила, добавляли 2 мл пиперидина, и перемешивали реакционную смесь при комнатной температуре в течение 10 мин. После этого реакционную смесь концентрировали при пониженном давлении, к осадку добавляли диэтиловый эфир. Растворитель повторно концентрировали выпариванием, и очищали осадок методом флэш-хроматографии на силикагеле (элюент:15:1:0,1→15:2:0,2 дихлорметан/метанол/17% водный раствор аммиака). Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении, после чего осадок лиофилизировали из ацетонитрила/воды. Таким образом получали 895 мг (67% в 2 этапа) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,84 мин; MS (ESIпоз): m/z=840 (M+H)+.
1Н ЯМР (500 МГц, ДМСО-d6): δ=10,8 (d, 1Н), 8,3 и 8,05 (2d, 1H), 8,0 (d, 1H), 7,5 (m, 1H), 7,3 (m, 1H), 7,15 и 7,08 (2s, 1H) 7,05-6,9 (m, 2H), 5,12 и 4,95 (2m, 1H), 4,65 (m, 1H), 4,55 (m, 1H), 4,1-3,8 (m, 4H), 3,75 (d, 1H), 3,23, 3,18, 3,17, 3,12, 2,95 и 2,88 (6s, 9H), 3,1-3,0 и 2,85 (2m, 2H), 2,65 (d, 1H), 2.4-2,2 (m, 3H), 2,15 (m, 3H), 1,95 (ушир. m, 2H), 1,85-0,8 (ушир. m, 11H), 1,08 и 1,04 (2d, 3H), 0,9-0,75 (m, 15H), 0,75-0,65 (dd, 3H) [другие сигналы перекрыты пиками Н2О peak].
Промежуточное соединение 193
N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

50 мг (0,052 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 192) и 204 мкл 15% водного раствора 4-оксобутановой кислоты объединяли в 2 мл метанола, и добавляли к смеси 23.4 мг (0,252 ммоль) комплекса боран-пиридин и 6 мкл уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После этого полученное соединение концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. После концентрирования соответствующих фракций, получали 38 мг (78% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 9): Rt=4,7 мин; MS (ESIпoз): m/z=926 (М+H)+.
Промежуточное соединение 194
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной синтезу, описанному в примере промежуточного соединения 157 из 10 мг (11 мкМоль) N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида и коммерчески доступного 6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексангидразида.
Выход: 4.4 мг (35% от теоретического количества) ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,90 мин; MS (ESIпоз): m/z=1133 (M+R)+.
Промежуточное соединение 195
N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гекспл]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 166, в качестве исходного соединения брали 9 мг (0,010 ммоль) промежуточного соединения 170.
Выход: 1,1 мг (10% от теоретического количества)
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,99 мин; MS (ESIпоз): m/z=994 (M+H)+.
Промежуточное соединение 196
(2S)-2-амино-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-3-фенилпропан-1-он трифторацетат

41 мг (0,37 ммоль) 2,5-диоксопирролидин-1-ил N-(трет-бутоксикарбонил)-L-фенилаланината помещали в 10 мл ДМФ, и добавляли к смеси 149 мг (0.41 ммоль) 2-окса-3-азабицикло[2,2,2]окт-5-ена (Исходное соединение 6) и 72 мкл (0.41 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли при пониженном давлении, и помещали осадок в этилацетат, после чего экстрагировали встряхиванием с 5% водным раствором лимонной кислоты, и затем с 5% водным раствором бикарбоната натрия. Органическую фазу концентрировали, и очищали осадок методом флэш-хроматографии на силикагеле с 10:1 смесью толуол/этанол в качестве элюента. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении. После сушки осадка в высоком вакууме, получали 69 мг (47% от теоретического количества) защищенного группой Boc промежуточного соединения трет-бутил (2S)-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил карбамата в виде смеси диастереомеров.
ЖХ-МС (Метод 1): ri=1,1 мин; MS (ESIпоз): ш/z=359 (M+H)+.
64 мг (0,18 ммоль) этого промежуточного вещества помещали в 10 мл дихлорметана, 1 мл трифторуксусной кислоты добавляли, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин.После этого полученное соединение концентрировали при пониженном давлении, и лиофилизировали полученный осадок из воды/диоксана. Таким образом получали 66 мг (колич.) указанного в названии соединения в виде пены.
ВЭЖХ (Метод 6): Rt=1.45 мин;
ЖХ-МС (Метод 3): Rt=1,12 мин; MS (ESIпоз): m/z=259 (М+Н)+.
Промежуточное соединение 197
(2R,3R)-3-метокси-2-метил-N-[(2S)-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил]-3-[(2S)-пирролидин-2-ил]пропанамидтрифторацетат

На первом этапе синтеза из 83 мг (0,18 ммоль) из соотвестсвующей соли дициклогексиламина получали (2R,3R)-3-[(2S)-1-(трет-бутоксикарбонил)пирролидин-1-ил]-3-метокси-2-метилпропановую кислоту (Исходное соединение 1), поместив ее в этилацетат, и экстрагировав встряхиванием с 5% водным раствором кислого сульфата калия. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Осадок помещали в 10 мл ДМФ и последовательно добавляли в смесь 66 мг (0,18 ммоль) (2S}-2-амино-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-3-фенилпропан-1-он трифторацетата (промежуточное соединение 196), 101 мг (0,266 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 93 мкл (0,53 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали, очищали осадок методом препаративной ВЭЖХ. Процедура давала 52 мг (56% от теоретического количества) защищенного группой Boc промежуточного соединения трет-бутил (2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1 -(2-окса-3-азабицикло [2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-карбоксилата.
ВЭЖХ (Метод 6): Rt=2,13 мин;
ЖХ-МС (Метод 1): Rt=1,13 мин; MS (ESIпоз): m/z=528 (М+Н)+.
52 мг (0,1 ммоль) этого промежуточного вещества помещали в 10 мл дихлорметана, добавляли 1 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 20 мин. После этого полученное соединение концентрировали при пониженном давлении, перемешивали полученный осадок с 20 мл диэтилового эфира. Через 10 мин реакционную смесь фильтровали, и сушили остаток фильтраци в высоком вакууме. Таким образом получали 39 мг (72% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 6):Rt=1,62 мин;
ЖХ-МС (Метод 1): Rt=0,68 мин; MS (ESIпоз): m/z=428 (М+Н)+.
Промежуточное соединение 198
N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

44,5 мг (0,071 ммоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) помещали в 10 мл ДМФ, и последовательно добавляли в смесь 38,6 мг (0,071 ммоль) (2R,3R)-3-метокси-2-метил-N-[(2S)-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил]-3-[(2S)-пирролидин-2-ил]пропанамид трифторацетата (промежуточное соединение 197), 32,5 мг (0,086 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 41 мкл (0,235 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали при пониженном давлении, и помещали осадок в этилацетат. Органическую фазу промывали последовательно 5% водным раствором лимонной кислоты и 5% водным раствором бикарбоната натрия, сушили над сульфатом магния, фильтровали и концентрировали. Процедура давала 73 мг (98% от теоретического количества) защищенного группой Fmoc промежуточного соединения N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ВЭЖХ (Метод 6): Rt=2,78 мин;
ЖХ-МС (Метод 3): Rt=2,96 мин; MS (ESIпоз): m/z=1047 (М+Н)+.
73 мг (0,071 ммоль) этого промежуточного вещества растворяли в 5 мл ДМФ. После добавления 0,5 мл пиперидина, реакционную смесь перемешивали при комнатной температуре в течение 10 мин. После этого, полученное соединение концентрировали при пониженном давлении, и осадок дегидрировали несколько раз диэтиловым эфиром. После удаления диэтилового эфира декантированием, осадок очищали методом препаративной ВЭЖХ (элюент:ацетонитрил/0,1% вод. TFA). В результате получали 16 мг (26% от теоретического количества) указанного в названии соединения в виде пены.
ВЭЖХ (Метод 6): Rt=1,94 мин;
ЖХ-МС (Метод 3): Rt=1,71 мин; MS (ESIпоз): m/z=825 (М+Н)+.
1Н ЯМР (400 МГц, ДМСО-d6): δ=8,9-8,6 (m, 3Н), 8.4, 8,3, 8,1 и 8,0 (4d, 1H), 7,3-7,1 (m, 5H), 6,7-6,5 (m, 2H), 5,2-4,8 (m, 3Н), 4,75-4,55 (m, 3Н), 4,05-3,95 (m, 1H), 3,7-3.4 (m, 4H), 3,22, 3,17, 3,15, 3,05, 3,02 и 2,95 (6s, 9H), 3,0 и 2,7 (2 ушир. m, 2H), 2.46 (m, 3Н), 2.4-1,2 (ушир. m, 13Н), 1,1-0,85 (m, 18H), 0,75 (m, 3Н) [другие сигналы перекрыты пиками Н2О].
Промежуточное соединение 199
N-(4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Указанное в названии соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточных соединений 193 и 194, в качестве исходного соединения брали 23 мг (24 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S)-1-(2-окса-3-азабицикло[2,2,2]окт-5-ен-3-ил)-1-оксо-3-фенилпропан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 198).
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 2): Rt=2,1 мин; MS (ESIпоз): m/z=1118 (М+Н)+.
Промежуточное соединение 200
N-[2-(2-{2-[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этокси]этокси}этокси)этил]-N-метил-L-валил-R-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили аналогично процедуре, описанной для промежуточных соединений 174 и 175, начиная с восстановительного алкилирования промежуточного соединения 192 промежуточным соединением 172, с последующим удалением защиты и образованием малеимида.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1025 (M+H)+.
Промежуточное соединение 201
N-{6-[(бромацетил)амино]гексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2s)-3-(1Н-индол-3-ил)-1 -(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

22 мг (0,023 ммоль) N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил] -N-метил-L-валинамида (промежуточное соединение 101) растворяли в 9,5 мл ТГФ, и добавляли к смеси при температуре 0°С 4,2 мкл триэтиламина. ао каплям добавляли раствор бромацетил хлорида в ТГФ и перемешивали реакционную смесь при температуре 0°С в течение 30 мин. Реакционную смесь концентрировали, очищали осадок методом препаративной ВЭЖХ. Таким образом получали 6,9 мг (26% от теоретического количества) указанного в названии соединения в виде пены.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 11): Rt=0,9 мин; MS (ESIпоз): m/z=1059 и 1061 (М+Н)+.
Промежуточное соединение 202
N-(2-[2-(2-{3-[(2,5-диоксопирролидин-1-ил)окси]-3-оксоргорокси}этокси)этокси]этил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Процедуру приготовления начинали аналогично процедуре, описанной для промежуточного соединения 168, сначала проводили восстановительное алкилирование промежуточного соединения 192 промежуточным соединением 167, после чего гидрогенолитически отщепляли бензиловый эфир от N-(2-{2-[2-(2-карбоксиэтокси)этокси]этокси}этил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида.
13 мг (10 мкМоль) этого промежуточного вещества растворяли в 5 мл ДМФ, и добавляли к смеси 2,1 мг (20 ммоль) 1-гидроксипирролидин-2,5-диона, 6,5 мкл N,N-диизопропилэтиламина и 7,1 мг (0,02 ммоль) HATU. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из ацетонитрила/воды, получали 9,2 мг (62% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 2): Rt=2,1 мин; MS (ESIпоз): m/z=1141 (М+Н)+.
Промежуточное соединение 203
трет-бутил 6-гидразино-6-оксогексил карбамат

Данное соединение получали стандартными методами химии пептидов, осуществляя реакцию сочетания 6-[(трет-бутоксикарбонил)амино]капроновой кислоты с бензил гидразинкарбоксилатом в присутствии EDCI и НОВТ, после чего гидрогенолитически расщепляли защитную группу бензилоксикарбонила.
ЖХ-МС (Метод 11): Rt=0,59 мин; MS (ESIпоз): m/z=246 (М+Н)+.
Промежуточное соединение 204
N-{4-[2-(6-аминогексаноил)гидразино]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

146 мг (50 мкМоль) (N-(3-карбоксипропил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида растворяли в 5 мл ДМФ, и затем добавляли к смеси 30,6 мг (80 мкМоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, 19 мкл N,N-диизопропилэтиламина и 22.4 мг (60 мкМоль) трегг-бутш 6-гидразино-6-оксогексил карбамата. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. После этого полученное соединение концентрировали в высоком вакууме, и очищали полученный осадок методом препаративной ВЭЖХ. Таким образом получали 43 мг (68% от теоретического количества) защищенного промежуточного соединения, которое затем помещали в 10 мл дихлорметана, и удаляли защиту с использванием 1 мл трифторуксусной кислоты. Реакционную смесь концентрировали, перемешивали осадок дихлорметаном, и повторно удаляли растворитель при пониженном давлении. Таким образом получали 45 мг (68% от теоретического количества в 2 этапа) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,6 мин;
ЖХ-МС (Метод 11): Rt=0,66 мин; MS (ESIпоз): m/z=983 (M+H)+.
Промежуточное соединение 205
N-(4-{2-[6-({[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этил]карбамоил}амино)гексаноил] гидразино}-4-оксобутил)-Н-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 114, в качестве исходных соединений используя промежуточные соединения 50 и 204.
Выход: 4 мг (78% от теоретического количества) ВЭЖХ (Метод 12): Rt=1,7 мин;
ЖХ-МС (Метод 11): Rt=0,73 мин; MS (ESIпоз): m/z=1149 (M+H)+.
Промежуточное соединение 206
N-(6-{[3-({3-[(2,5-диоксопирролидин-1-ил)окси]-3-оксопропил}дисульфанил)пропаноил]амино}гексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

8 мг (10 мкМоль) промежуточного соединения 101 растворяли в 2 мл ДМФ, и добавляли к смеси 8,6 мг (20 мкМоль) 1,1'-{дисульфандиилбис[(1-оксопропан-3,1-диил)окси]}дипирролидин-2,5-диона и 3,7 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, и затем выпаривали растворитель при пониженном давлении, после чего очищали осадок методом препаративной ВЭЖХ. В результате получали 7,2 мг (68% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 11): Rt=0,94 мин; MS (ESIпоз): m/z=615 [½(М+2Н+]
Промежуточное соединение 207
(1S,2R)-1-амино-2-фенилциклопропанкарбоновой кислоты трифторацетат
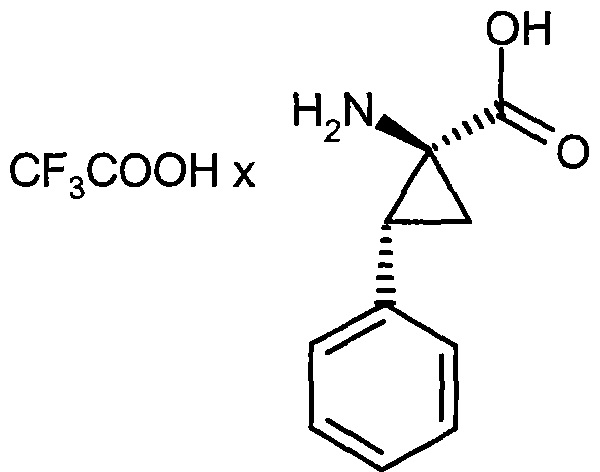
Указанное в названии соединение получали с количественным выходом удалением защиты у 210 мг (0,76 ммоль) коммерчески доступной (1S,2R)-1-[(трет-бутоксикарбонил)амино]-2-фенилциклопропанкарбоновой кислоты трифторуксусной кислотой.
ЖХ-МС (Метод 1): Rt=0,23 мин; MS (ESIпоз): m/z=178 (М+H)+.
Промежуточное соединение 208 9Н-флуорен-9-илметил 6-оксогексил карбамат
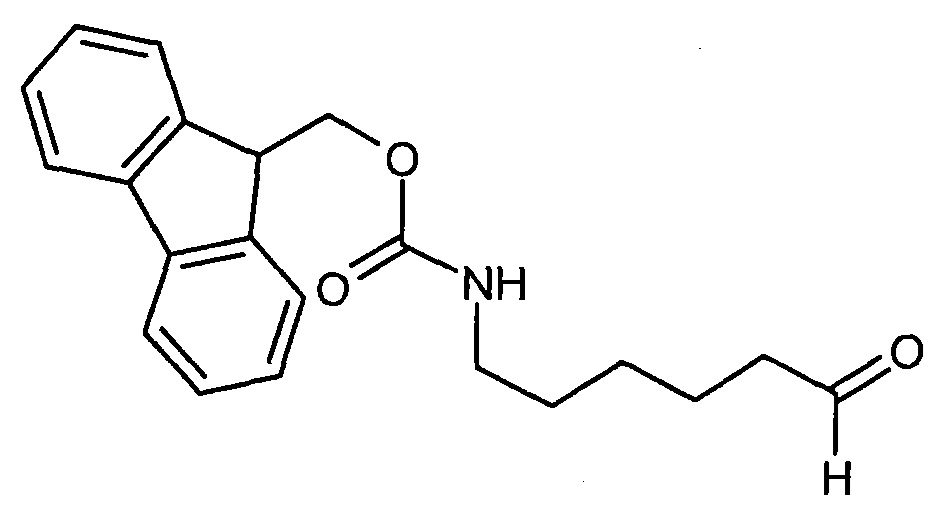
Указанное в названии соединение получали из 1 г (2,95 ммоль) коммерчески доступного 9Н-флуорен-9-илметил 6-гидроксигексил карбамата при стандартных условиях, окислением комплексом серный ангидрид-пиридин. В результате получали 840 мг (85% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=1,1 мин; MS (ESIпоз): m/z=338 (М+Н)+.
Промежуточное соединение 209
N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-карбокси-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 75, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и (1S,2R)-1-амино-2-фенилциклопропанкарбоновой кислоты трифторацетата (промежуточное соединение 207) в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, с последующим удалением защитной группы Boc трифторуксусной кислотой, в результате чего получали аминное соединение N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-карбокси-2-фенилциклопропил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата.
К 22 мг (0,026 ммоль) этого соединения в 10 мл метанола затем добавляли 17 мг (0,05 ммоль) 9Н-флуорен-9-илметил 6-оксогексил карбамата (промежуточное соединение 208) и 2,3 мг уксусной кислоты, а также 11.4 мг (0,12 ммоль) комплекса боран-пиридин. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем еще раз добавляли те же количества комплекса боран-пиридин и уксусной кислоты, а также 8 мг флуорен-9-илметил 6-оксогексил карбамата, после чего перемешивали реакционную смесь при комнатной температуре в течение еще 24 часов. После этого, полученное соединение концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. После концентрирования соответствующих фракций, продукт немедленно использовали на следующем этапе синтеза.
33 мг все еще неочищенного промежуточного соединения помещали в 5 мл ДМФ, и добавляли 1 мл пиперидина. Реакционную смесь перемешивали при комнатной температуре в течение 15 мин, затем концентрировали полученный осадок, и очищали методом препаративной ВЭЖХ. Таким образом получали 11 мг (55% от теоретического количества в 2 этапа) промежуточной аминокарбоновой кислоты.
ВЭЖХ (Метод 12): Rt=1,7 мин;
ЖХ-МС (Метод 11): Rt=0,7 мин; MS (ESIпоз): m/z=843 (M+H)+.
6 мг (7,12 мкМоль) этого промежуточного вещества помещали в 1 мл диоксана, и затем добавляли к смеси 6,6 мг (42,7 мкМоль) метил 2,5-диоксо-2,5-дигидро-1Н-пиррол-1-карбоксилата и 5 мкл насыщенного водного раствора бикарбоната натрия. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли еще 3 порции, каждая по 50 мкл, насыщенного водного раствора бикарбоната натрия, и перемешивали реакционную смесь при комнатной температуре в течение еще 30 мин. Затем реакционную смесь окисляли до уровня рН 2 трифторуксусной кислотой, после чего концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из ацетонитрила/воды, получали 4 мг (60% от теоретического количества) указанного в названии соединения в виде пены.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 11): Rt=0,88 мин; MS (ESIпоз): m/z=923 (М+H)+.
Промежуточное соединение 210
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза описанным в литературе методом (J. Org. Chem. 58, 1993, 2196-2200) получали 6-оксокапроновую кислоту.
80 мг (0,08 ммоль) N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 192) и 65.4 мг (0,5 ммоль) 6-оксокапроновой кислоты объединяли в 9 мл метанола и добавляли к смеси 10 мкл уксусной кислоты и 37.4 мг (0.4 ммоль) комплекса боран-пиридин. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После этого, полученное соединение концентрировали при пониженном давлении, и помещали осадок в 1:1 смесь ацетонитрил/вода, и затем доводили до уровня рН 2 трифторуксусной кислотой. Реакционную смесь повторно концентрировали, и очищали осадок методом препаративной ВЭЖХ. После концентрирования соответствующей фракции, получали 70 мг (86% от теоретического количества) N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3- {[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида в виде трифторацетата.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=955 (M+H)+.
1Н ЯМР (500 МГц, ДМСО-d6, характеристические сигналы): δ=12,0 (ушир. М, 1Н), 10,8 (s, 1Н), 9.4 (m, 1Н), 8,9 и 8,8 (2d, 1Н), 8,3 и 8,02 (2d, 1Н), 7,5 (m, 1Н), 7,3 (m, 1Н), 7,15 и 7,1 (2s, 1Н) 7,05-6,9 (m, 2H), 5,12 и 4,95 (2m, 1Н), 4,7-4,5 (m, 2H), 4,1-3,8 (m, 4H), 3,75 (d, 1Н), 3,25, 3,2, 3,18, 3,13, 2,98 и 2,88 (6s, 9H), 2,8 (m, 3H), 1,08 и 1,04 (2d, 3Н), 0,95-0,8 (m, 15H), 0,8-0,65 (dd, 3H).
22 мг (23 мкМоль) этого промежуточного вещества растворяли в 1,8 мл дихлорметана и добавляли к смеси 13,2 мг (70 мкМоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида, 26,5 мг (230 мкМоль) 1-гидроксипирролидин-2,5-диона и 0,28 мг (2 мкМоль) диметиламинопиридина, после чего перемешивали реакционную смесь при комнатной температуре в течение 2 часов. Затем реакционную смесь концентрировали при пониженном давлении, и очищали оставшийся осадок методом препаративной ВЭЖХ. После лиофилизации из ацетонитрила/воды, получали 21,3 мг (88% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,94 мин; MS (ESIпоз): m/z=1052 (M+H)+.
Промежуточное соединение 211
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(2R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Осуществляли восстановительное алкилирование 15 мг (20 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата (промежуточное соединение 15) 6-оксокапроновой кислотой, по аналогии с промежуточным соединением 210.
Выход: 9,2 мг (61% от теоретического количества) ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=929 (M+H)+.
9 мг (10 мкМоль) этого промежуточного вещества растворяли в 3 мл ДМФ, и добавляли к смеси 5,6 мг (48 мкМоль) 1-гидроксипирролидин-2,5-диона, 5 мкл N,N-диизопропилэтиламина и 5,5 мг (0,015 ммоль) HATU, после чего реакционную смесь обрабатывали в ультразвуковой ванне в течение 6 часов. В ходе процесса, каждый час добавляли по 5,5 мг HATU. Затем реакционную смесь концентрировали при пониженном давлении, помещали осадок в ацетонитрил/воду, и доводили до уровня рН 2 трифторуксусной кислотой. После повторного концентрирования при пониженном давлении, оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из ацетонитрила/воды, получали 5,8 мг (57% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,95 мин; MS (ESIno3): m/z=1027 (M+H)+.
Промежуточное соединение 212
N-{2-[2-(2-{3-[(2,5-диоксопирролидин-1-ил)окси]-3-оксоргорокси}этокси)этокси]этил}-N-метил-L-валил-N-[(3R,4S8,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролвдин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Процедуру приготовления начинали аналогично процедуре, описанной для промежуточного соединения 168, сначала проводили восстановительное алкилирование промежуточного соединения 15 промежуточным соединением 167, и затем гидролизом отщепляли бензиловый эфир от N-(2-{2-[2-(2-карбоксиэтокси)этокси]этокси}этил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(2S,3S)-1-(1,2-оксазинан-2-ил)-1-оксо-3-фенилбутан-2-ил]амино}-3-оксопропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида.
8.4 мг (8 мкМоль) этого промежуточного вещества растворяли в 3 мл ДМФ, и добавляли к смеси 9,5 мг (80 мкМоль) 1-гидроксипирролидин-2,5-диона, 10 мкл 'N,N-диизопропилэтиламина и 9.4 мг (25 мкМоль) HATU, после чего перемешивали реакционную смесь при комнатной температуре в течение ночи, и затем концентрировали при пониженном давлении. Затем реакционную смесь концентрировали при пониженном давлении, помещали осадок в ацетонитрил/воду, и доводили до уровня рН 2 трифторуксусной кислотой. После повторного концентрирования при пониженном давлении, оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из ацетонитрила/воды, получали 4 мг (32% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12); Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,96 мин; MS (ESIпоз): ш/z=1117 (M+H)+.
Промежуточное соединение 213
N-{6-[(транс-4-{[(2,5-диоксопирролидин-1-ил)окси]карбонил}циклогексил)амино]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 104, в качестве исходного соединения брали N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид, синтез которого описан в примере промежуточного соединения 210. В результате получали 9,3 мг указанного в названии соединения (37% от теоретического количества в три этапа).
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,9 мин; MS (ESIпоз): m/z=1177 (M+H)+.
Промежуточное соединение 214
N-{4-[(2,5-диоксопирролидин-1-ил)окси]-4-оксобутил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 210, превращением промежуточного соединения 92 в активный эфир.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 11): Rt=0,82 мин; MS (ESIпоз): m/z=901 (M+H)+.
Промежуточное соединение 215
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза промежуточного соединения 40, по аналогии с промежуточным соединением 183, для приготовления N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3- {[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида использовали комплекс боран-пиридин. Из данного соединения, по аналогии с промежуточным соединением 210, затем получали активный эфир. Процедура давала 34 мг (36% от теоретического количества в 2 этапа) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,85 мин; MS (ESIпоз): m/z=930 (М+Н)+.
Промежуточное соединение 216
N-(4-{[(2,5-диоксопирролидин-1-ил)окси]карбонил}бензил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1 -метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с приготовлением промежуточного соединения 183, осуществляли реакцию промежуточного соединения 192 с 4-формилбензойной кислотой в присутствии комплекса боран-пиридин, с получением N-(4-карбоксибензил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида. Данное соединение затем использовали, по аналогии с промежуточным соединением 210, с получением 11 мг (68% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=1,13 мин; MS (ESIпоз): m/z=1072 (М+Н)+.
Промежуточное соединение 217
N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3 -оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

53 мг (84 мкМоль) N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(2R,3S,4S)-1-карбокси-2-метокси-4-метилгексан-3-ил]-N-метил-L-валинамида (промежуточное соединение 4) и 45 мг (84 мкМоль) бензил N-{(2R,3R)-3-метокси-2-метил-3-[(2S)-пирролидин-2-ил]пропаноил}-L-фенилаланинат трифторацетата (промежуточное соединение 12) помещали в 2 мл ДМФ, добавляли 19 мкл N,N-диизопропилэтиламина, 14 мг (92 мкМоль) HOBt и 17,6 мг (92 мкМоль) EDC, и затем смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали, и очищали осадок методом препаративной ВЭЖХ. Процедура давала 59 мг (68% от теоретического количества) защищенного группой Fmoc промежуточного соединения N-[(9H-флуорен-9-илметокси)карбонил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид.
ЖХ-МС (Метод 1): Rt,=1,55 мин; m/z=1044 (M+H)+.
57 мг (0,055 ммоль) этого промежуточного вещества обрабатывали 1,2 мл пиперидина в 5 мл ДМФ для удаления защитной группы Fmoc. После концентрирования и очистки методом препаративной ВЭЖХ, получали 39 мг (76% от теоретического количества) свободного амина промежуточного соединения N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид в виде трифторацетата.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1); Rt=1,01 мин; m/z=822 (М+Н)+.
Осуществляли реакцию 60 мг (0,06 ммоль) этого промежуточного вещества, по аналогии с промежуточным соединением 210, с 6-оксокапроновой кислотой в присутствии комплекса боран-пиридин. Процедура давала 45 мг (75% от теоретического количества) указанного в названии соединения в виде пены.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,97 мин; MS (ESIпоз): m/z=9936 (M+H)+.
Промежуточное соединение 218
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
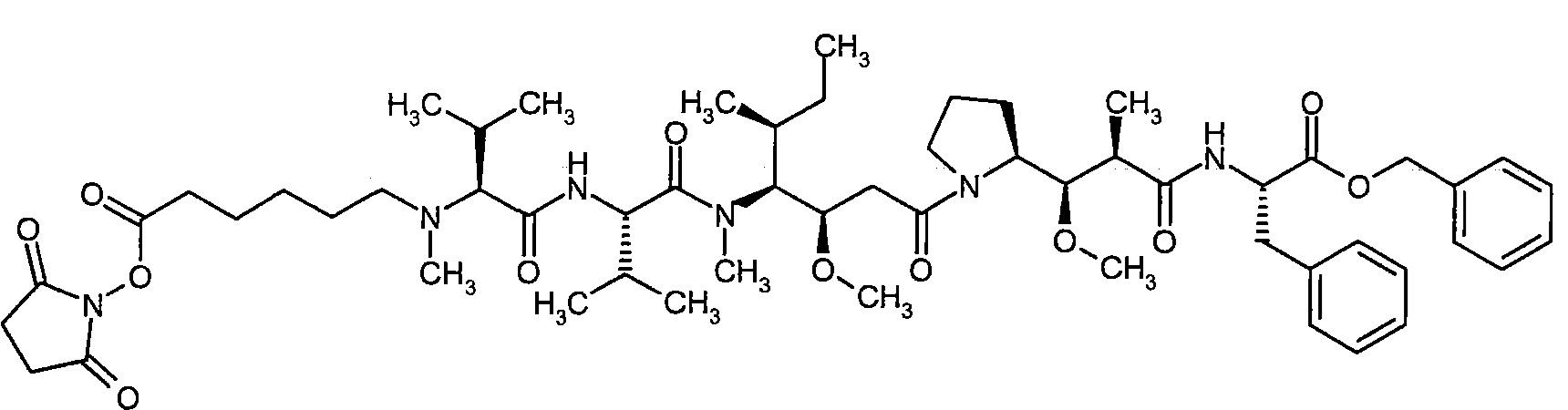
Данное соединение получали превращением 42 мг (0,05 ммоль) промежуточного соединения 217 в активный эфир.
Выход: 26 мг (54%) ВЭЖХ (Метод 5): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=1,01 мин; MS (ESIпоз): m/z=1034 (M+H)+.
Промежуточное соединение 219
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(18)-1-карбокси-2-фенилэтил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
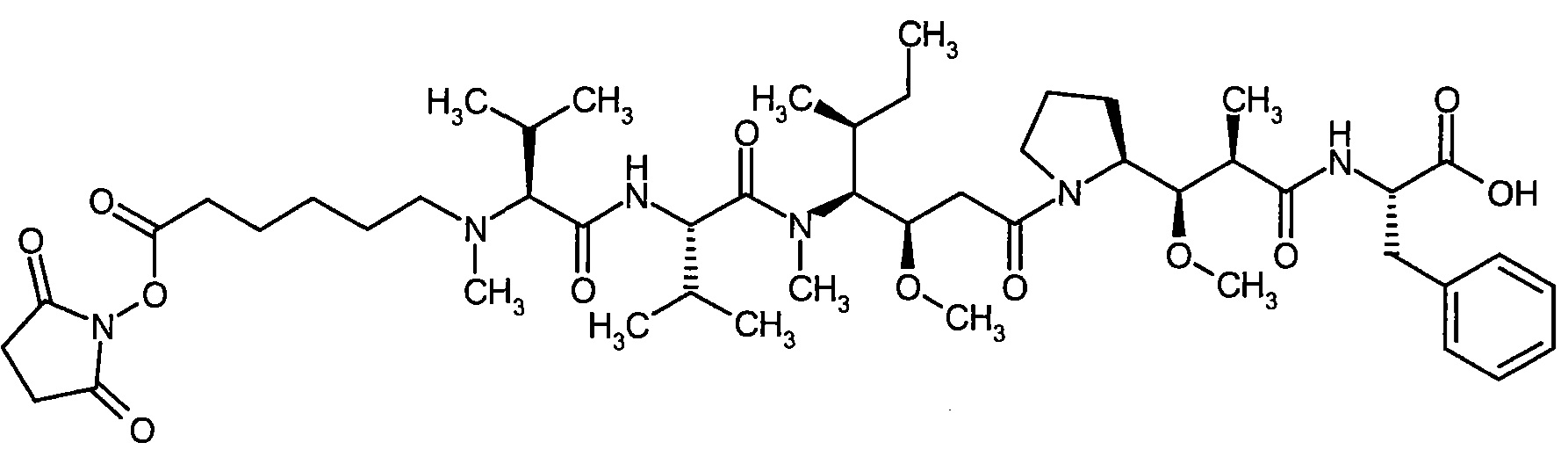
20 мг (0,02 моль) вещества из примера для промежуточного соединения 218 помещали в 2.4 мл метанола и гидрогенировали над подложкой 5% палладий на активированном угле при стандартном давлении водорода при комнатной температуре в течение 30 мин.Затем катализатор отфильтровывали, и удаляли растворитель при пониженном давлении. Осадок лиофилизировали из 1:1 смеси ацетонитрил/вода. Процедура давала 14 мг (92% от теоретического количества) указанного в названии соединения в виде бесцветной пены.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=944 (М+Н)+.
Промежуточное соединение 220
N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

К 0,5 г (1,01 ммоль) промежуточного соединения 1 в 10 мл дихлорметана добавляли 1 мл трифторуксусной кислоты. После обработки в ульультразвуковой ванне в течение 30 мин, реакционную смесь концентрировали, и повторно перегоняли сначала с DCM и затем с диэтиловым эфиром, после чего сушили в высоком вакууме. Маслянистый осадок использовали на следующем этапе без дальнейшей очистки.
500 мг этого промежуточного вещества растворяли в 20 мл ДМФ, и добавляли к смеси 466 мг (3,8 ммоль) промежуточного соединения 191, 382 мг (1,01 ммоль) O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 440 мкл (2,5 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали. Осадок помещали в дихлорметан и экстрагировали встряхиванием сначала дважды с 5% водным раствором лимонной кислоты, и затем с насыщенным водным раствором бикарбоната натрия. Органическую фазу концентрировали, и очищали осадок методом флэш-хроматографии на силикагеле с 95:5 дихлорметан/метанолом в качестве элюента. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении. После сушки осадка в высоком вакууме, получали 562 мг (65% от теоретического количества после двух этапов синтеза) защищенного Z-группой промежуточного соединения.
562 мг (0,57 ммоль) этого промежуточного вещества помещали в 50 мл метанола и гидрогенировали с 155 мг 10% палладием на активированном угле при стандартном давлении водорода при комнатной температуре в течение 20 мин. Затем катализатор отфильтровывали, и удаляли растворитель при пониженном давлении. Осадок очищали методом препаративной ВЭЖХ. Соответствующие фракции объединяли, растворитель выпаривали при пониженном давлении, осадок лиофилизировали из диоксана. Процедура давала 361 мг (87% от теоретического количества) указанного в названии соединения в виде пены.
ВЭЖХ (Метод 5): двойной пик с Rt=1,75 и 1,86 мин;
ЖХ-МС (Метод 1): двойной пик с Rt=0,84 мин и 0,91 мин с той же массой; MS (ESIпоз): m/z=944 (M+R)+.
Промежуточное соединение 221
N-{(2S)-2-[(трет-бутоксикарбонил)амино]-3-фенилпропил}-N-метил-L-валин

100 мг (0,76 ммоль) коммерчески доступного N-метил-L-валина и 285 мг (1,14 ммоль) коммерчески доступного трет-бутил (25)-1-оксо-3-фенилпропан-2-ил карбамата объединяли в 22 мл метанола, и добавляли к смеси 340 мг (3,66 ммоль) комплекса боран-пиридин и 70 мкл уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После этого, полученное соединение концентрировали при пониженном давлении, и очищали осадок методом флэш-хроматографии на силикагеле с дихлорметан/метанол/17% водным раствором аммиака в качестве элюента. После концентрирования соответствующих фракций и лиофилизации из 1:1 диоксан/воды, получали 259 мг (93% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,6 мин;
ЖХ-МС (Метод 11): Rt=0,76 мин; MS (ESIпоз): m/z=365 (М+Н)+.
Промежуточное соединение 222
N-[(2S)-2-амино-3-фенилпропил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

40 мг (0,11 ммоль) N-{(2S)-2-[(трет-бутоксикарбонил)амино]-3-фенилпропил}-N-метил-L-валина (промежуточное соединение 221) растворяли в 5 мл ДМФ, и добавляли к смеси 80 мг (0,11 ммоль) N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 220), 50 мг (0,13 ммоль) О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 57 мкл (2,5 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и затем концентрировали. Осадок помещали в этилацетат, и промывали сначала с 5% водным раствором лимонной кислоты и затем водой. Органическую фазу концентрировали, и очищали осадок методом препаративной ВЭЖХ. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении. После лиофилизации из диоксана, получали 60 мг (50% от теоретического количества) защищенного промежуточного соединения.
ВЭЖХ (Метод 12): Rt=2,2 мин;
ЖХ-МС (Метод 1): Rt=1,17 мин; MS (ESIпоз): m/z=1073 (M+H)+.
60 мг (0,05 ммоль) этого промежуточного вещества помещали в 10 мл дихлорметана, добавляли 2 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 1,5 часов. Затем реакционную смесь концентрировали при пониженном давлении, и очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении, осадок лиофилизировали из диоксана/воды. Таким образом получали 25 мг (42% от теоретического количества) указанного в названии соединения в виде пены.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,95 мин; MS (ESIпоз): m/z=974 (М+Н)+.
Промежуточное соединение 223
N-[(2S)-2-({[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этил]карбамоил}амино)-3-фенилпропил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили аналогично процедуре, описанной для промежуточного соединения 134, в качестве исходного соединения брали 5 мг (4,6 мкМоль) промежуточного соединения 222. Процедура давала 3.4 мг (65% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=0,99 мин; MS (ESIпоз): m/z=1140 (M+H)+.
Промежуточное соединение 224
N-[(2S)-2-({[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этил]карбамоил}амино)пропил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили по аналогии с синтезом промежуточного соединения 223.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,92 мин; MS (ESIпоз): m/z=1064 (М+Н)+.
Промежуточное соединение 225
N-(2-аминоэтил)-N-метил-L-валил-N-[(3R,4S,5s)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

100 мг (0,76 ммоль) коммерчески доступного N-метил-L-валина и 182 мг (1,14 ммоль) коммерчески доступного трет-бутил 2-оксоэтил карбамата объединяли в 20 мл метанола, и добавляли к смеси 340 мг (3,66 ммоль) комплекса боран-пиридин и 65 мкл уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в течение ночи. После этого полученное соединение концентрировали при пониженном давлении, и очищали осадок методом флэш-хроматографии на силикагеле с дихлорметан/метанол/17% водным раствором аммиака (15/4/0,5) в качестве элюента. После концентрирования соответствующих фракций и лиофилизации из 1:1 диоксан/воды, получали 190 мг 39% чистоты (35% от теоретического количества) промежуточного соединения, которые использовали на следующем этапе без дальнейшей очистки.
50 мг (0,07 ммоль) этого промежуточного вещества растворяли в 10 мл ДМФ, и добавляли к смеси 52 мг (0,07 ммоль) N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1H-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 220), 32 мг (0,09 ммоль) О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 37 мкл (0,2 ммоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали. Осадок помещали в этилацетат, и экстрагировали встряхиванием сначала с 5% водным раствором лимонной кислоты и затем с водой. Органическую фазу концентрировали, очищали осадок методом препаративной ВЭЖХ. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении. После лиофилизации из диоксана, получали 53 мг (76% от теоретического количества) защищенного промежуточного соединения.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=1,02 мин; MS (ESIпоз): m/z=984 (М+H)+.
53 мг (0,05 ммоль) этого промежуточного вещества помещали в 10 мл дихлорметана, добавляли 2 мл трифторуксусной кислоты, и перемешивали реакционную смесь при комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении, и очищали оставшийся осадок методом препаративной ВЭЖХ. Соответствующие фракции объединяли, и удаляли растворитель при пониженном давлении, после чего осадок лиофилизировали из диоксана/воды. Таким образом получали 21 мг (40% от теоретического количества) указанного в названии соединения 65% чистоты.
ВЭЖХ (Метод 12): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз); m/z=884 (М+Н)+.
Промежуточное соединение 226
N-[2-({[2-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)этил]карбамоил}амино)этил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили, по аналогии с синтезом промежуточного соединения 134, в качестве исходного соединения брали промежуточное соединение 225. Процедура давала 11,6 мг (59% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,90 мин; MS (ESIпоз): m/z=1050 (М+Н)+.
Промежуточное соединение 227
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензилокси)-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Данное соединение получали аналогично процедуре, описанной для промежуточного соединения 218, превращением в активный эфир.
Выход: 18 мг (51% от теоретического количества)
ВЭЖХ (Метод 5): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=0,98 мин; MS (ESIпоз): m/z=1073 (М+Н)+.
Промежуточное соединение 228
(2R,3S)-3-[(трет-бутоксикарбонил)амино]-4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]гидразино}-4-оксобутан-2-ил(3R,4S,7S,10S)-4-[(2S)-бутан-2-ил]-7,10-диизопропил-3-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазапентадекан-15-оат

Указанное в названии соединение получали, осуществляя реакцию сочетания защищенного группой Boc промежуточного соединения, полученного в процессе синтеза промежуточного соединения 154, с коммерчески доступным 6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексангидразидом.
ВЭЖХ (Метод 12): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=0,97 мин; MS (ЕSIпоз): m/z=1308 (М+Н)+.
Промежуточное соединение 229
(2R,3S)-3-ацетамидо-4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]гидразино}-4-оксобутан-2-ил(3R,4S,7S,10S)-4-[(2S)-6yraH-2-ил]-7,10-диизопропил-3-(2-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазапентадекан-15-оат

Указанное в названии соединение получали из 7,5 мг (2,5 мкМоль) промежуточного соединения 154 ацетилированием с 2,3 мкл уксусного ангидрида в 1 мл ДМФ в присутствии 0.4 мкл N,N-диизопропилэтиламина.
Выход: 1.4 мг (40% от теоретического количества)
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1250 (M+H)+.
Промежуточное соединение 230
(2R,3S)-3-[(трет-бутоксикарбонил)амино]-4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил] гидразино}-4-оксобутан-2-ил (3R,4S,7S,10S)-4-[(2S)-бутан-2-ил]-3-(2-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-7,10-диизопропил-5,11-диметил-6,9-диоксо-2-окса-5,8,11 -триазапентадекан-15-оат

Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 228, в качестве исходного соединения брали промежуточное соединение 193. В результате получали 16 мг (30% от теоретического количества в три этапа) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=2,0 мин;
ЖХ-МС (Метод 1): Rt=1,02 мин; MS (ESIпоз): m/z=1335 (M+H)+.
Промежуточное соединение 231
(2R,3S)-3-ацетамидо-4-{2-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексаноил]гидразино}-4-оксобутан-2-ил (3R,4S,7S,10S)-4-[(2S)-бутан-2-ил]-3-(2-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1 Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-7,10-диизопропил-5,11-диметил-6,9-диоксо-2-окса-5,8,11-триазапентадекан-15-оат

Данное соединение получали из 8 мг (6 мкМоль) промежуточного соединения 230, сначала удалением защитной группы трифторуксусной кислотой, и последующим ацетилированием уксусным ангидридом в ДМФ в присутствии N,N-диизопропилэтиламина. В результате получали 2 мг (37% от теоретического количества в 2 этапа) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,88 мин; MS (ESIпоз): m/z=1277 (M+H)+.
Промежуточное соединение 232
бензил N-[(4-нитрорЬепокси)карбонил]-бета-аланинат
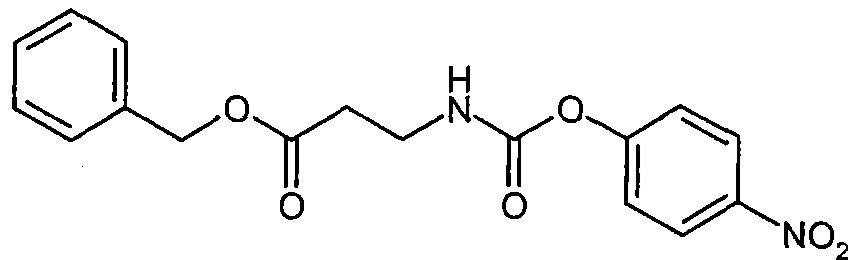
200 мг (0,57 ммоль) коммерчески доступного 4-метилбензолсульфокислота-бензил бета-аланината и 229 мг (1,14 ммоль) 4-нитрофенил хлоркарбоната помещали в 15 мл тетрагидрофурана, и нагревали реакционную смесь с обратным холодильником в течение 30 мин. Затем реакционную смесь концентрировали при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. После концентрирования соответствующих фракций и сушки осадка в высоком вакууме, получали 86 мг (44% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=1,07 мин; MS (ESIпоз): m/z=345 (M+H)+.
Промежуточное соединение 233
N-{2-[({3-[(2,5-диоксопирролидин-1-ил)окси]-3-оксопропил}карбамоил)амино]этил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

13 мг (10 мкМоль) промежуточного соединения 225 и 6,7 мг (20 мкМоль) промежуточного соединения 232 растворяли в 3 мл ДМФ, и затем добавляли 7 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в высоком вакууме. Оставшийся осадок очищали методом препаративной ВЭЖХ. После концентрирования соответствующих фракций и сушки осадка в высоком вакууме, получали 5.4 мг (38% от теоретического количества) защищенного промежуточного соединения.
ВЭЖХ (Метод 5): Rt=2,1 мин;
ЖХ-МС (Метод 1): Rt=0. 6in; MS (ESIпоз): m/z=1089 (M+H)+.
5.4 мг (5 мкМоль) этого промежуточного вещества растворяли в 5 мл метанола и, затем добавляли 2 мг 10% палладия на активированном угле, после чего гидрогенировали при стандартном давлении водорода при комнатной температуре в течение 20 мин. Затем катализатор отфильтровывали, и удаляли растворитель при пониженном давлении. После сушки осадка в высоком вакууме, получали 5 мг (колич.) промежуточной кислоты.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,84 мин; MS (ESIпоз): m/z=999 (M+H)+.
5 мг (10 мкМоль) этого промежуточного вещества растворяли в 1 мл ДМФ, и добавляли к смеси 5,8 мг (50 ммоль) 1-гидроксипирролидин-2,5-диона, и затем 2,6 мкл N,N-диизопропилэтиламина и 3,8 мг (10 мкМоль) HATU. Реакционную смесь перемешивали при комнатной температуре в течение 20 часов, после чего концентрировали при пониженном давлении. Оставшийся осадок очищали методом препаративной ВЭЖХ. После лиофилизации из 1:1 диоксана/воды, получали 1,1 мг (20% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=1096 (М+Н)+.
Промежуточное соединение 234
N-(6-{[(бензилокси)карбонил]амино}гексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
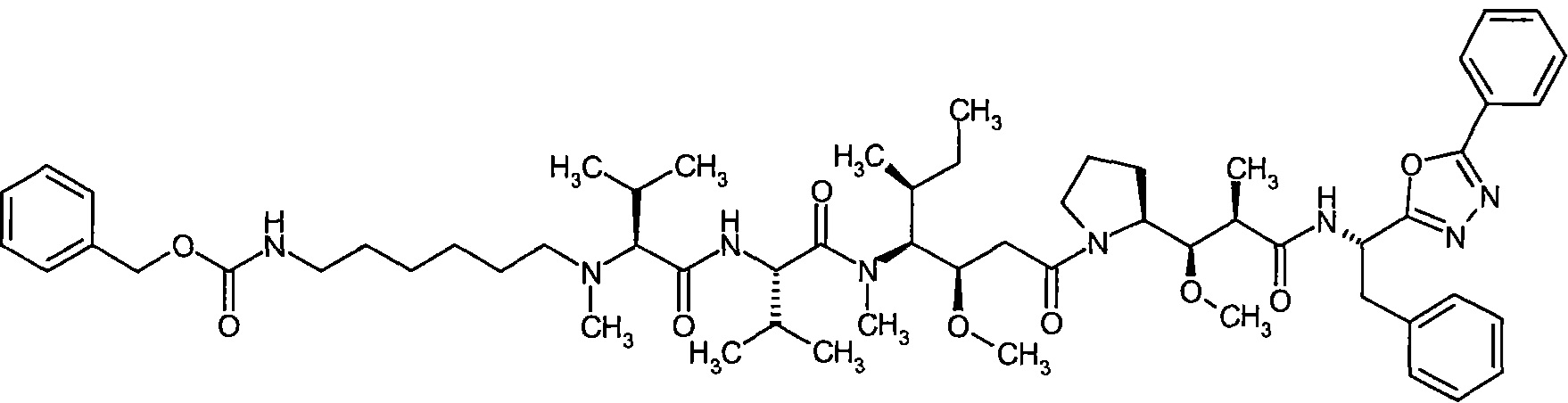
25 мг (30 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил] амино} пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 55) и 45 мг (180 мкМоль) бензил 6-оксогексил карбамата помещали в 3 мл метанола и подкисляли уксусной кислотой. Далее при комнатной температуре добавляли 15 мкл (144 мкМоль; 9.4М) комплекса боран-пиридин. После этого реакционную смесь перемешивали при комнатной температуре в течение 24 часов, и через 8 часов повторно добавляли уксусную кислоту и 15 мкл (144 мкМоль; 9.4М) комплекса боран-пиридин. После этого, реакционную смесь доводили до уровня рН 2 с помощью TFA, и очищали методом препаративной ВЭЖХ. Объединенные фракции полученного продукта концентрировали, и сушили осадок в высоком вакууме. Процедура давала 15 мг (46% от теоретического количества) указанного в названии соединения в виде пены.
ЖХ-МС (Метод 1): Rt=1,03 мин; m/z=1066 (М+Н)+.
Промежуточное соединение 235
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
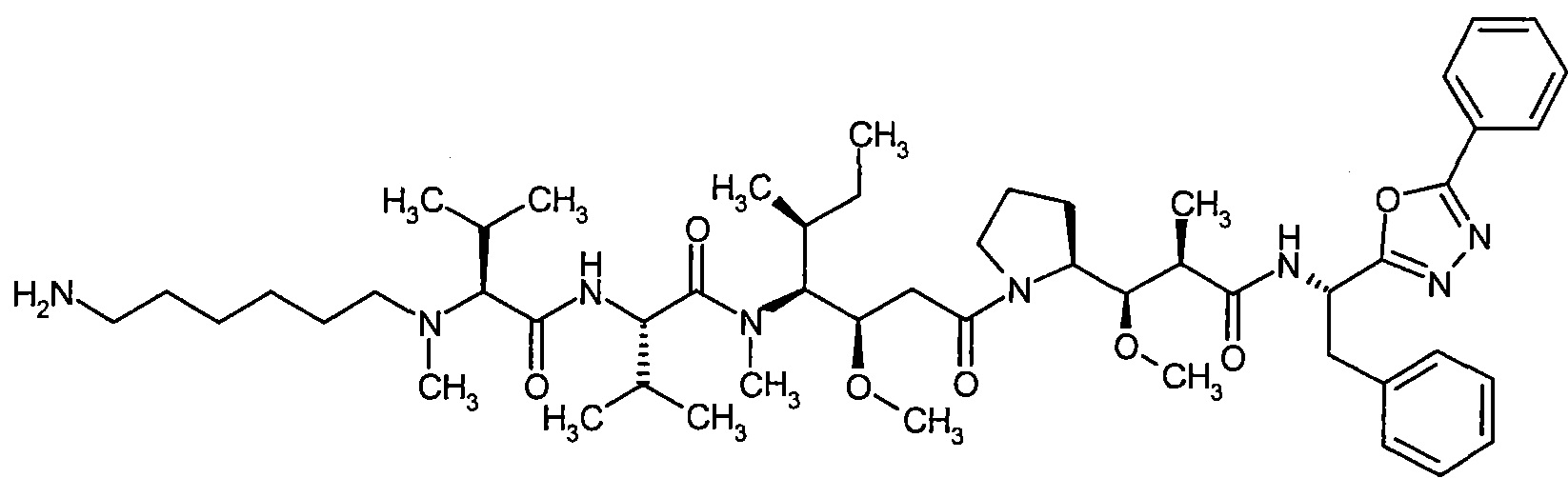
15 мг (14 мкМоль) N-(6-{[(бензилокси)карбонил]амино}гексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 234) помещали в 3 мл метанола, и добавляли 1,8 мг палладия на угле (5%). После этого реакционную смесь гидрогенировали при стандартном давлении водорода при комнатной температуре в течение 2 часов. Затем катализатор отфильтровывали, и удаляли растворитель при пониженном давлении. Осадок лиофилизировали из 1:1 смеси ацетонитрил/вода. В результате получали 11 мг (86% от теоретического количества) указанного в названии соединения в виде пены.
ЖХ-МС (Метод 1): Rt=0,81 мин; m/z=932 (М+Н)+.
Промежуточное соединение 236
N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1s)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил] амино} пропил] пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

11 мг (12 мкМоль) N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3- {[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино} пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 235) помещали в 500 мкл смеси 1:1 диоксан/вода и добавляли к смеси 253 мкл 1М водного раствора бикарбоната натрия, и затем 2,8 мг (18 мкМоль) метил 2,5-диоксо-2,5-дигидро-1Н-пиррол-1-карбоксилата. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем подкисляли трифторуксусной кислотой. Реакционную смесь очищали методом препаративной ВЭЖХ. После лиофилизации, получали 0,8 мг (7% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=1,01 мин; m/z=1012 (М+Н)+.
Промежуточное соединение 237
N-(5-карбоксипентил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

25 мг (30 мкМоль) N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил]амино}пропил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 55) и 23 мг (180 мкМоль) 6-оксокапроновой кислоты помещали в 3 мл метанола и подкисляли уксусной кислотой. После этого при комнатной температуре добавляли 15 мкл (144 мкМоль; 9.4М) комплекса боран-пиридин. После этого реакционную смесь перемешивали при комнатной температуре в течение 20 часов, и через 8 часов повторно добавлляи уксусную кислоту и 15 мкл (144 мкМоль; 9.4М) комплекса боран-пиридин. После этого, реакционную смесь доводили до уровня рН 2 трифторуксусной кислотой, и очищали методом препаративной ВЭЖХ. Объединенные фракции полученного продукта концентрировали, и лиофилизировали осадок. Процедура давала 21 мг (74% от теоретического количества) указанного в названии соединения в виде пены.
ЖХ-МС (Метод 1): Rt=0,91 мин; ш/z=947 (М+Н)+.
Промежуточное соединение 238
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-оксо-3-{[(1S)-2-фенил-1-(5-фенил-1,3,4-оксадиазол-2-ил)этил] амино} пропил] пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

21 мг (22 мкМоль) промежуточного соединения 237 растворяли в 1 мл ДМФ и добавляли к смеси 38 мг (333 мкМоль) 1-гидроксипирролидин-2,5-диона и затем 2.4 мг (10 мкМоль) 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата (HATU) и 19 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем очищали методом препаративной ВЭЖХ. После лиофилизации из диоксана, получали 22 мг (96% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,95 мин; m/z=1044 (М+Н)+.
Промежуточное соединение 239
N-метил-L-треонил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил] пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

На первом этапе синтеза N-[(бензилокси)карбонил]-N-метил-L-треонин получали из 237 мг (0,887 ммоль) соответствующей соли дициклогексиламина, поместив ее в этилацетат и экстрагировав встряхиванием с 5% водной серной кислотой. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. 14,7 мг (0,055 ммоль) N-[(бензилокси)карбонил]-N-метил-L-треонина помещали в 3 мл ДМФ и последовательно добавляли в смесь 40 мг (0,055 ммоль) промежуточного соединения 220, 12,7 мг (0,066 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида и 10 мг (0,066 ммоль) 1-гидрокси-1H-бензотиазол гидрата. После этого, реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель затем удаляли при пониженном давлении, и очищали осадок методом препаративной ВЭЖХ. Таки образом получали 29 мг (54% от теоретического количества) защищенного Z-группой промежуточного соединения.
ЖХ-МС (Метод 1): Rt=1,15 мин; MS (ESIпоз): m/z=976 (M+H)+.
29 мг (0,003 ммоль) этого промежуточного вещества растворяли в 5 мл метанола и гидрогенировали над 5 мг 5% палладий/угля при комнатной температуре и стандартном давлении в течение 1 часа. После этого катализатор отфильтровывали, а растворитель выпаривали. Оставшийся осадок очищали методом препаративной ВЭЖХ. Процедура давала 17 мг (54% от теоретического количества) указанного в названии соединения.
ЖХ-МС (Метод 1): Rt=0,77 мин; MS (ESIпоз): m/z=842 (M+H)+.
Промежуточное соединение 240
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-треонил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид
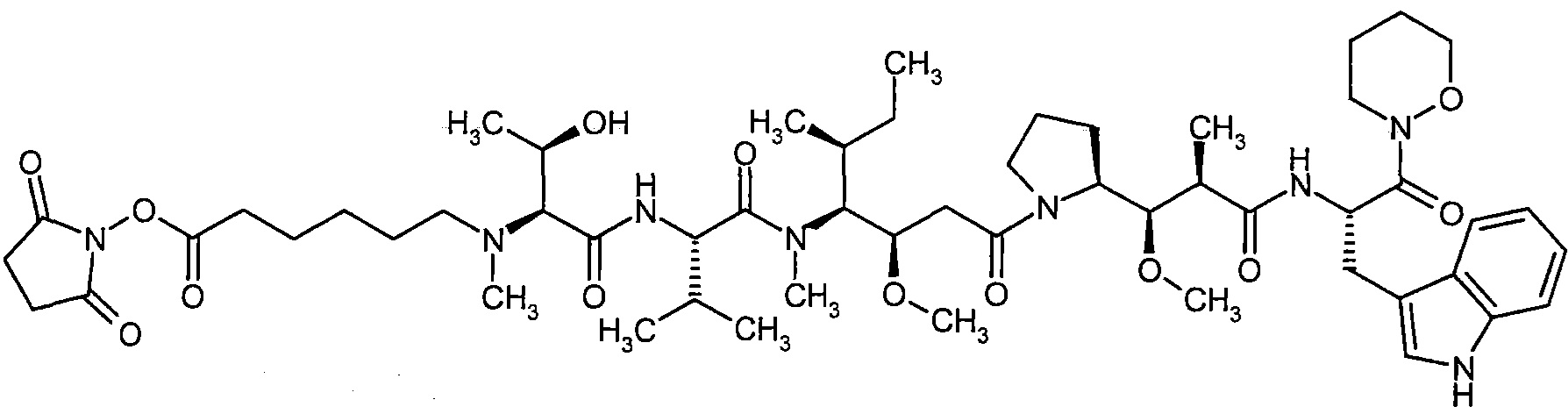
Данное соединение получали, следуя процедуре, аналогичной процедуре, описанной для промежуточного соединения 210 из 15,6 мг (0,016 ммоль) промежуточного соединения 239. В результате получали 10,8 мг (67% от теоретического количества в 2 этапа) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,85 мин; MS (ESIпоз): m/z=1053 (М+Н)+.
Промежуточное соединение 241
N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-гидроксифенил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат
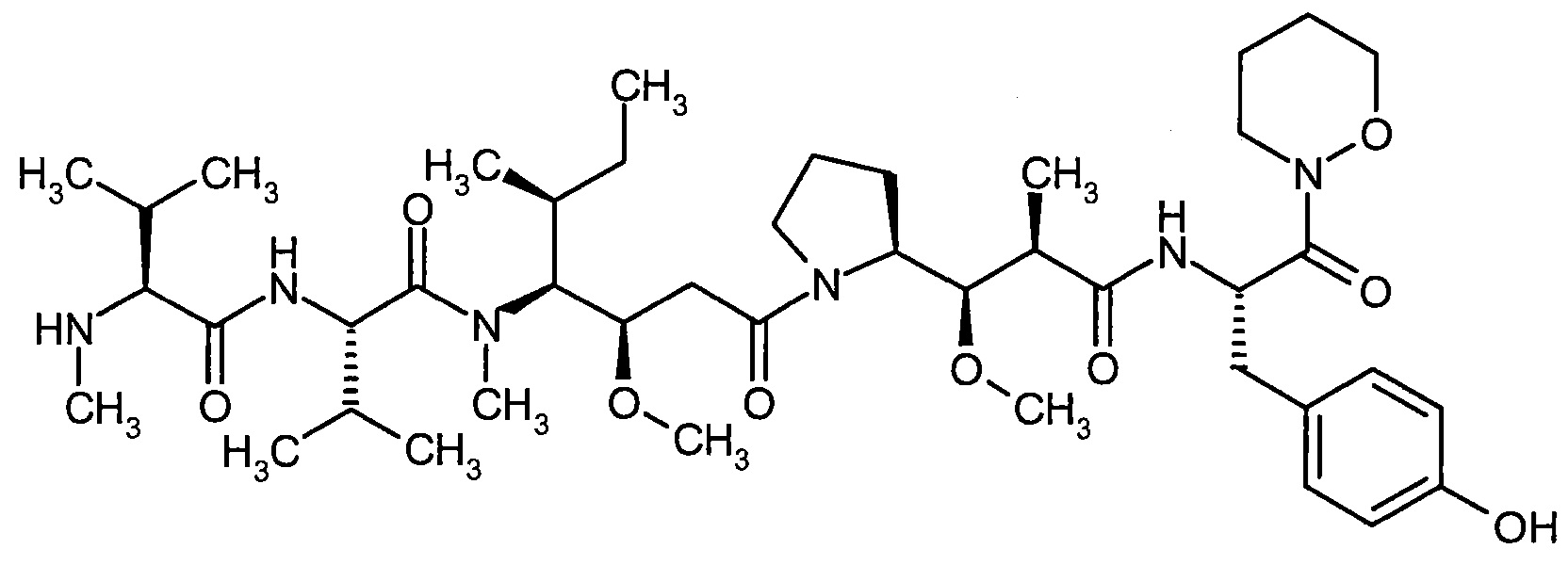
На первом этапе синтеза аналогично процедуре, описанной для промежуточного соединения 5, получали соединение трифторуксусная кислота-(2S)-2-амино-3-(4-гидроксифенил)-1-(1,2-оксазинан-2-ил)пропан-1-он (1:1). Этот реагент затем использовали по аналогии с синтезом описанным в примере промежуточного соединения 75, а именно осуществляли реакцию сочетания с N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1 -метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамидом (промежуточное соединение 26) в присутствии O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, с последующим удалением защитной группы Boc трифторуксусной кислотой, с получением указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,85 мин; MS (ESIпоз): m/z=817 (M+H)+.
Промежуточное соединение 242
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-гидроксифенил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-Н-метил-L-валинамид

Осуществляли реакцию 50 мг (0,05 ммоль) промежуточного соединения 241, по аналогии с промежуточным соединением 210, с 6-оксокапроновой кислотой в присутствии комплекса боран-пиридин. Затем 22,5 мг (0,02 ммоль) полученной кислоты превращали в активированный эфир. Процедура давала 13,5 мг (36% от теоретического количества в 2 этапа) указанного в названии соединения.
ВЭЖХ (Метод 12): R,=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1028 (М+Н)+.
Промежуточное соединение 243
N-(6-аминогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-гидроксифенил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили аналогично процедуре, описанной для промежуточного соединения 78, восстановительным алкилированием промежуточного соединения 241 бензил 6-оксогексил карбаматом и комплексом боран-пиридин, и последующей гидрогенезацией в метаноле в качестве растворителя.
Выход: 17,5 мг (34% от теоретического количества в 2 этапа)
ВЭЖХ (Метод 12): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,63 мин; MS (ESIпоз): m/z=916 (M+H)+.
Промежуточное соединение 244
N-[6-(2,5-диоксо-2,5-дигидро-1Н-пиррол-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-гидроксифенил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

Синтез соединения проводили аналогично процедуре, описанной для промежуточного соединения 166, в качестве исходного соединения брали промежуточное соединение 243.
Выход: 1,3 мг (12% от теоретического количества)
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,89 мин; MS (ESIпоз): m/z=996 (M+H)+.
Промежуточное соединение 245
2,5-диоксопирролидин-1-ил O-[(3R,4S,7S,10S)-4-[(2S)-бутан-2-ил]-3-(2-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-2-оксоэтил)-7,10-диизопропил-5,11-диметил-6,9,15-триоксо-2-окса-5,8,11-триазапентадекан-15-ил]-N-(трет-бутоксикарбонил)-L-треонил-бета-аланинат

На первом этапе синтеза, как описано для промежуточного соединения 154, осуществляли реакцию промежуточного соединения 193 с бензил N-(трет-бутоксикарбонил)-L-треонинатом, после чего гидролизом удаляли бензиловый эфир. Затем осуществляли реакцию сочетания полученных таким образом 30 мг (0,027 ммоль) N-[4-({(1S,2R)-1-[(трет-бутоксикарбонил)амино]-1-карбоксипропан-2-ил}окси)-4-оксобутил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-индол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метип-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида с 4-метилбензолсульфокислота-бензил бета-аланинатом в присутствии HATU, после чего снова удаляли бензил методом гидролиза (выход: 24 мг (71% от теоретического количества в 2 этапа)). Окончательно, 10 мг (0,008 ммоль) полученной кислоты превращали в активированный эфир. После очистки методом ВЭЖХ, получали 2,7 мг (23% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=1,01 мин; MS (ESIпоз): m/z=1295 (M+H)+
Промежуточное соединение 246а
(2S)-2-амино-1-(4-гидрокси-1,2-оксазолидин-2-ил)-3-(1Н-индол-3-ил)пропан-1-он трифторацетат (Диастереомер 1)

1,6 г (3,982 ммоль) 2,5-диоксопирролидин-1-ил]-L-(трет-бутоксикарбонил)-L-триптофаната растворяли в 15 мл ДМФ, и добавляли к смеси 500 мг (3,982 ммоль) 1,2-оксазолидин-4-ола и 100 мкл ЦТУ-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем добавляли еще 100 мкл N,N-диизопропилэтиламина, и обрабатывали реакционную смесь в ультразвуковой ванне 5 часов, после чего перемешивали при комнатной температуре в течение ночи, и затем концентрировали при пониженном давлении. Оставшийся осадок помещали в этилацетат и экстрагировали сначала дважды с 5% водным раствором лимонной кислоты, затем с насыщенным водным раствором бикарбоната натрия и наконец с водой. Органическую фазу концентрировали, осадок разделяли на диастереомеры методом флэш-хроматографии на силикагеле с 95:5 дихлорметан/метанолом в качестве элюента. Соответствующие фракции обоих диастереомеров соединяли, и удаляли растворитель при пониженном давлении. После сушки осадка в высоком вакууме, получали 272 мг (18% от теоретического количества) Диастереомера 1 (Rf=0,18 (95:5 дихлорметан/метанол) и 236 мг (16% от теоретического количества) Диастереомера 2 (Rf=0,13 (95:5 дихлорметан/метанол), а также 333 мг (22% от теоретического количества) смешанной фракции защищенного группой Boc промежуточного соединения.
При стандартных условиях использовали 5 мл трифторуксусной кислоты в 20 мл дихлорметана для удаления защитной группы Boc из 272 мг (725 мкМоль) Диастереомера 1 этого промежуточного вещества и, после лиофилизации из диоксана/воды, получали 290 мг (колич.) указанного в названии соединения 75% чистоты, которые использовали на следующем этапе без дальнейшей очистки.
ВЭЖХ (Метод 12): Rt=1,1 мин;
ЖХ-МС (Метод 13); Rt=1,80 мин; MS (ESIпоз): m/z=276 (M+H)+
Промежуточное соединение 246b
(2S)-2-амино-1-(4-гидрокси-1,2-оксазолидин-2-ил)-3-(1Н-индол-3-ил)пропан-1-он трифторацетат (Диастереомер 2)

При стандартных условиях использовали 5 мл трифторуксусной кислоты в 20 мл дихлорметана для удаления защитной группы Boc из 236 мг (630 мкМоль) Диастереомера 2 промежуточного соединения описанного в 246а и, после концентрирование, перемешивания с диэтиловым эфиром, после чего сушили осадок в высоком вакууме, и получали 214 мг (76%) указанного в названии соединения.
ЖХ-МС (Метод 13): Rt=1,84 мин; MS (ESIпоз): m/z=276 (M+H)+
Промежуточное соединение 247а
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогексил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2 В)-1-(4-гидрокси-1,2-оксазолидин-2-ил)-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид (Диастереомер 1)

Для получения данного соединения, осуществляли реакцию сочетания промежуточных соединения 26 и 246а с последующим удалением защитной группы Вое, как описано для промежуточного соединения 74. Затем аликилированием с 6-оксокапроновой кислотой в присутствии комплекса боран-пиридин и последующим превращением кислоты в активный эфир, как описано для промежуточного соединения 210, и очистки методом препаративной ВЭЖХ, получали указанное в названии соединение.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1053 (М+H)+
Промежуточное соединение 247b
N-{6-[(2,5-диоксопирролидин-1-ил)окси]-6-оксогекеил}-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1 -(4-гидрокси-1,2-оксазолидин-2-ил)-3-(1Н-индол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид (Диастереомер 2)

Для синтеза данного соединения, осуществляли реакцию сочетания промежуточных соединений 26 и 246b с последующим удалением защитной группы Вое, как описано для промежуточного соединения 74. Затем алкилированием с 6-оксокапроновой кислотой в присутствии комплекса боран-пиридин и последующим превращением кислоты в активный эфир, как описано для промежуточного соединения 210, получали указанное в названии соединение, которое очищали методом препаративной ВЭЖХ.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1053 (M+H)+
Промежуточное соединение 248
N-(5-карбоксипентил)-Н-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-трет-бутокси-3-(4-гидроксифенил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

На первом этапе синтеза по аналогии с синтезом описанным в примере промежуточного соединения 86, осуществляли реакцию сочетания N-(трет-бутоксикарбонил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-2-карбокси-1-метоксипропил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамида (промежуточное соединение 26) и трет-бутил L-тирозината в присутствии O-(7-азабенгзотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфата, и последующим удалением защитной группы Boc трифторуксусной кислотой, получали трет-бутиловый эфир (перемешиванием трифторуксусной кислоты в дихлорметане в течение 40 мин), аминное соединение трет-бутил N-[(2R,3R)-3-метокси-3-{(2S)-1-[(3R,4S,5S)-3-метокси-5-метил-4-(метил{(2S)-3-метил-2-[(N-метил-L-валил)амино]бутил}амино)heptanoyl]пирролидин-2-ил}-2-метилпропаноил]-L-тирозинат в виде трифторацетата. 38 мг (0,04 ммоль) этого соединения затем использовали по аналогии с приготовлением промежуточного соединения 210 в реакции с 6-оксокапроновой кислотой в присутствии комплекса боран-пиридин, с получением 31 мг (99% от теоретического количества) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,88 мин; MS (ESIпоз): m/z=918 (М+Н)+.
В: Получение конъюгатов антитело-активное соединение (ADC)
В-1. Общий способ получения анти-С4.4а антител
Анти-С4.4а антитела, характеризующиеся последовательностями, приведенными в Таблице 1 и Таблице 2 получали путем скрининга библиотеки на фаговом дисплее рекомбинантного человеческого С4.4а с последовательностью SEQ ID NO:1 и сышиного С4.4а с последовательностью SEQ ID NO:2 и клеток, экспрессирующих С4.4а. Антитела, полученные таким путем, переводили в формат IgGI человека и сипользовали в описанных ниже рабочих примерах.
В-2. Общий способ получения анти-С4.4а антител в клетках млекопитающих
Антитела, например М31-В01 (легкая цепь SEQ ID NO:346 и тяжелая цепь SEQ ID NO:347), или дополнительно антитела из Таблицы 2 получали в культуре клеток млекопитающих. С этой целью клетки клетки HEK293 6Е временно трансфецировали подходящей экспрессионной плазмидой на основе промотора цитомегаловируса. Тяжелую и легкую цепи антитела клонировали либо вместе в одновекторную систему, либо раздельно в систему с двумя векторами. Масштаб культуры клеток был либо 1,5 л во встряхиваемом флаконе, либо 10 л в одноразовом реакторе WAVE Biotech ". "Экспрессию осуществляли при 37°С в течение 5-6 дней в среде F17 (Invitrogen), дополненной Триптоном TN1 (Organotechnie) с 1% фетальной бычьей сыворотки "PCS ultra low IgG" (Invitrogen) и 0,5 мл вальпроевой кислоты. Выход экспрессии составлял от 100 до 600 мг/л.
В-3. Общий способ очистки антител от клеточного супернатанта
Антитела, например М31-В01 (легкая цепь SEQ ID NO:346 и тяжелая цепь SEQ ID NO:347), или дополнительные антитела из Таблицы 2, получали из супернатантов культур клеток. Эти клеточные супернатанты осветляли путем центрифугирования для удаления клеток. Затем супернатантны клеток очищали аффинной хроматографией на хроматографической колонке MabSelect Sure (GE Healthcare). Для этого колонку уравновешивали в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4 (Sigma/Aldrich), наносили супернатант клеток, и промывали колонку приблизительно 10 объемами колонки буфера DPBS рН 7,4+500 MM хлорида натрия. Антитела элюировали в 50 мм ацетата натрия при рН 3,5+500 MM хлорида натрия, а затем очищали эксклюзионной хроматографией на колонке Superdex 200 (GE Healthcare) в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4.
В-4. Общий способ связывания с боковыми цепями цистеина
Для связывания использовали следующие антитела:
анти-С4.4а антитело М31-В01
анти-С4.4а антитело В01-3
анти-С4.4а антитело В01-10
анти-С4.4а В01-7 анти-С4.4а
антитело D02-4 анти-С4.4а
антитело D02-6 анти-С4.4а
антитело D02-7
К раствору соответствующего антитела в фосфатно-солевом буфере при концентрации антитела в диапазоне от 1 мг/мл до 15 мг/мл добавляли 3 эквивалента трис(2-карбоксиэтил)фосфин гидрохлорида (ТСЕР) в виде раствора в фосфатно-солевом буфере и перемешивали смесь при комнатной температуре в течение 1 часа. Затем в зависимости от желаемой загрузки добавляли от 2 до 10 эквивалентов соединения - предшественника малеимида или соединения-предшественника галида для связывания (Промежуточное соединение 102, 103, 105-109, 111-114, 117-126, 128, 129, 132-146, 148-155, 157, 159-161, 166, 171, 175-177, 184, 189, 194-195, 199-201, 205, 209, 223-224, 226, 228-231, 236 и 244) в виде раствора в ДМСО. В этом случае количество ДМСО не должно превышать 10% общего объема. Смесь перемешивали при комнатной температуре в течение 60-120, а затем наносили на колонки PD 10 (Sephadex® G-25, GE Healthcare), уравновешенные фосфатно-буферным раствором, и элюировали фосфатно-буферным раствором. Опционально осуществляли процедуру концентрировалия путем ультрацентрифугирования. В случае необходимости более эффективного удаления низкомолекулярных компонентов повторяли концентрирования путем ультрацентрифугирования после повторного разбавления фосфатно-буферным раствором.
Обычно, если не указано иное, для восстановления и последующего связывания использовали 5 мг соответствующего антитела в фосфатно-солевом буфере. После очистки с использованием колонки PD10 получали, в каждом случае, растворы соответствующих конъюгатов связывающее соединение - активное соединение в 3,5 мл фосфатно-солевого буфера. Для этих растворов определяли конкретные концентрации белка. Затем определяли нагрузку антителом (отношение лекарственное средство/МАТ) в соответствии с методами, указанными ниже.
Это способ использовали для получения иммуноконъюгатов, представленных в Примерах 1 -3, 5-30, 32-36, 38-59, 61-66, 68-70, 80, 82-85, 87, 88, 92-95, 97, 98, 107, 109-114, 119 и 122.
В приведенных структурных формулах AK1A-AK1G имеют следующие значения
AK1A=анти-С4.4а антитело М31-В01 (частично восстановленное)-S§1
AK1B=анти-С4.4а антитело В01-3 (частично восстановленное)-S§1
AK1C=анти-С4.4а антитело В01-10 (частично восстановленное)-S§1
AK1D=анти-С4.4а антитело В01-7 (частично восстановленное)-S§1
AK1E=анти-С4.4а антитело D02-4 (частично восстановленное)-S§1
AK1F=анти-С4.4а антитело D02-6 (частично восстановленное)-S§1
AK1G=анти-С4.4а антитело D02-7 (частично восстановленное)-S§1
где
§1 обозначает связь с сукцинимидной группой,
и
S обозначает атом серы остатка цистеина частично восстановленного антитела.
В-5. Общий способ связывания с боковыми цепями лизина
В реакциях связывания использовали следующие антитела:
анти-С4.4а антитело М31-В01
анти-С4.4а антитело В01-3
К раствору соответствующего антитела в фосфатно-солевом буфере при концентрации антитела в диапазоне от 1 мг/мл до 15 мг/мл добавляли, в зависимости от желательной нагрузки, от 2 до 5 эксивалентов соединения-предшественника для связывания (Промежуточные соединения 104, 110, 115, 116, 127, 130, 131, 147, 156, 158, 162, 169, 178, 185, 190, 202, 206, 210-216, 218, 219, 227, 233, 238, 240, 242, 245, 247а и 247b)) в виде раствора в ДМСО. После 30 мин перемешивания при комнатной температуре снова добавляли то же количество предшественника в ДМСО. В качестве альтернативы можно добавлять 4-10 эквивалентов соединения-предшественника для связывания за один раз. В этом случае количество ДМСО не должно превышать 10% общего объема. После дополнительных 30 мин перемешивания при КТ, смесь наносили на колонку PD 10, (Sephadex® G-25, GE Healthcare) уравновешенную фосфатно-буферным раствором, и элюировали фосфатно-буферным раствором. Опционально осуществляли процедуру концентрировали путем ультрацентрифугирования. В случае необходимости более эффективного удаления низкомолекулярных компонентов повторяли концентрирования путем ультрацентрифугирования после повторного разбавления фосфатно-буферным раствором.
Обычно, если не указано иное, для связывания использовали 5 мг соответствующего антитела в фосфатно-солевом буфере. После очистки с использованием колонки PD10 получали, в каждом случае, растворы соответствующих конъюгатов связывающее соединение - активное соединение в 3,5 мл фосфатно-солевого буфера. Для этих растворов определяли конкретные концентрации белка. Затем определяли нагрузку антителом (отношение лекарственное средство/МАТ) в соответствии с методами, указанными ниже.
Это способ использовали для получения иммуноконъюгатов, представленных в Примерах 4, 31, 37, 60, 67, 81, 86, 89-91, 96, 99-106, 108, 118, 120, 121 и 123-125.
В приведенных структурных формулах AK2A и A2B имеют следующие значения
AK2A=анти-С4.4а антитело M31-B01-NH§2
AK2B=анти-С4.4а антитело B01-3-NH§2
где
§2 обозначает связь с карбонильной группой,
и
NH обозначает аминогруппу боковой цепи остатка лизина антитела.
В-6. Общий способ получения продуктов присоединения по цистеину:
10 мкмоль описанного выше соединения-предшественника малеимида разбавляли в 3 мл ДМФ и смешивали с 2,1 мг (20 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ.
В приведенной структурной формуле Cys имеет следующее значение

где
§3 обозначает связь с линкерным фрагментом токсофора.
Дальнейшая очистка и исследование конъюгатов согласно настоящему изобретению
После осуществления реакции реакционную смесь в некоторых случаях концентрировали, например, путем ультрафильтрации, а затем обессоливали и очищали посредством хроматографии, например, с использованием Sephadex® G-25. Элюирование осуществляли например, фосфатно-солевым буфером (ФБР). Затем раствор подвергали стерилизующей фильтрации и замораживанию. Альтернативу составляет лиофилизация конъюгата.
В-7. Определение нагрузки токсофором
Токсофорную нагрузку полученных раствором конъюгатов, описанных в рабочих примерах, в фосфатно-солевом буфере, определяли следующим образом:
Для определения токсофорной нагрузки конъюгатов ADC, связанных по лизину определяли методом масс-спектрометрии измеряли молекулярных масс отдельных молекул конъюгатов. В этом случае конъюгаты антител вначале гликозилировали с использованием фермента PNGaseF, после чего образец подкисляли, а затем, после разделения методом ВЭЖХ, анализировали методом масс-спектрометрии на приборе ESI-MicroTofQ (Broker Daltonik). Все спектры посредством сигнала добавляли в TIC (полную ионную хроматограмму) и рассчитывали молекулярные массы различных видов на основе обратной свертки методом максимальной энтропии. После интегрирования сигнала по различным видам рассчитывали (отношение лекарственное средство/антитело).
Для идентификации пептидов в дополнение к определению молекулярной массы осуществляли расщепление трипсином после дегликозилирования и/или денатурации, и это расщепление после денатурации, восстановления и дериватизации, подтверждало идентичность белка на основании детектируемых продуктов расщепления трипсином.
Нагрузку токсофором связанных через цистеин конъюгатов определяли путем обращено-фазовой хроматографии восстановленного и денатурированного конъюгата ADC. Раствор конъюгата связывающее соединение - активное соединение (1 мг/мл, 50 мкл) смешивали с гуанидина гидрохлорид (GuHCl) (28,6 mg) и раствором DL-дитиотреитола (DTT) (500 мМ, 3 мкл). Смесь инкубировали при 55°С в течении одного часа и анализировали методом ВЭЖХ.
Анализ методом ВЭЖХ проводили на системе Agilent 1260 HPLC System с детектированием на 220 нм. Использовали колонку PLRP-S Polymeric Reversed Phase производства Polymer Laboratories (номер по каталогу PL1912-3802) (2,1×150 мм, размер частиц 8 мкм, 1000 Å) со скоростью потока 1 мл/мин, с использованием следующего градиента: 0 мин, 25% В; 3 мин, 25% В; 28 мин, 50% В. Элюент А состоял из 0,05% трифторуксусной кислоты (TFA) в воде, элюент В состоял их 0,05% трифторуксусной кислоты в ацетонитриле.
Детектируемые пики связывали по времени удердивания с легкой (LO) и тяжелой цепью (НО) неконъюгированного антитела. Пики, детектируемые исключительно в конъюгированном образце, связывали с легкой цепью, с таксофором (L1), и с тяжелыми цепями с одним, двумя и тремя таксофорами (H1, H2, Н3).
Среднюю нагрузку антитела токсофором рассчитывали следующим образом: сначала, рассчитывали нагрузку легкой цепи по площадям пиков, определенным путем интегрирования, для пиков L0 и L1, соответствующих легким цепям, как сумму разельтатов интегрирования чисел молекул токсофора для L0 и L1, разделенную на сумму отдельно взвешенных результатов интегрирования отдельно определенных масс L0 и L1. Аналогичным образом нагрузку тяжелой цепи рассчитывали по площадям пиков, определенным путем интегрирования, для пиков Н0, H1, H2 Н3 соответствующим тяжелым цепям, как сумму результатов интегрирования для масс с токсофором для Н0, H1, H2 и Н3, разделенную на сумму отдельно взвешенных результатов интегрирования отдельно определенных масс Н0, H1, H2 и Н3. DAR дано по нагрузке легкой цепи и нагрузке тяжелой цепи. Множитель 2 учитывает тот факт, что антитело состоит из двух легких и двух тяжелых цепей. В некоторых отдельных случаях может быть невозможно точно определить нагрузку токсофором из-за совместного элюирования некоторых пиков.
В-8. Тестирование конъюгатов связывающее соединение - активное соединение на связывание антигенов
Способность антитела связываться с молекулой-мишенью тестировали после осуществления связывания. Специалисту известны разнообразные методы осуществления таких тестов, например, можно определять аффинность конъюгатов по методике твердофазного иммуноферментного анализа ELISA или посредством анализа поверхностного плазменного резонанса (измерительные приборы BIAcore™). Концентрация конъюгата может быть измерена специалистом с использованием обычных методов, например, в случае конъюгатов с антителами, путем определения белка (см. также Doronina et al.; Nature Biotechnol. 2003; 21:778-784 и Poison et al., Blood 2007; 1102:616-623).
Рабочие примеры - иммуноконъюгаты
Пример 1

В этом случае реакцию сочетания осуществляли с использованием 70 мг анти-С4.4а антитела М31-В01 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 12,2 мг/мл
Отношение лекарственное средство/МАТ: 1,5
Пример 2

Концентрация белка: 0,87 мг/мл
Отношение лекарственное средство/МАТ: 5,8
Пример 3

Концентрация белка: 1,16 мг/мл
Отношение лекарственное средство/МАТ: 3,1
Пример 4

Концентрация белка: 1,24 мг/мл
Отношение лекарственное средство/МАТ: 1,6
Пример 5

Концентрация белка: 0,88 мг/мл
Отношение лекарственное средство/МАТ: 6,9
Пример 6

Концентрация белка: 1,2 мг/мл
Отношение лекарственное средство/МАТ: 2,8
Пример 7

Концентрация белка: 0,9 мг/мл
Отношение лекарственное средство/МАТ: 3,9
Пример 8

Концентрация белка: 0,52 мг/мл
Отношение лекарственное средство/МАТ: 1,6
Пример 9

Концентрация белка: 0,47 мг/мл
Отношение лекарственное средство/МАТ: 6,6
Пример 10

Концентрация белка: 0,77 мг/мл
Отношение лекарственное средство/МАТ: 6,9
Пример 11

Концентрация белка: 0,47 мг/мл
Отношение лекарственное средство/МАТ: 4,0
Пример 12

Концентрация белка: 1,46 мг/мл
Отношение лекарственное средство/МАТ: 2,5
Пример 13

Концентрация белка: 0,45 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 14

Концентрация белка: 0,98 мг/мл
Отношение лекарственное средство/МАТ: 3,6
Пример 15

Реакцию сочетания осуществляли с использованием 70 мг анти-С4.4а антитела М31-В01 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 9,42 мг/мл
Отношение лекарственное средство/МАТ: 4,1
Пример 16

Концентрация белка: 0,65 мг/мл
Отношение лекарственное средство/МАТ: 1,8
Пример 17

Концентрация белка: 1,07 мг/мл
Отношение лекарственное средство/МАТ: не определено
Пример 18

Концентрация белка: 0,47 мг/мл
Отношение лекарственное средство/МАТ: 4,4
Пример 19

Концентрация белка: 0,43 мг/мл
Отношение лекарственное средство/МАТ: 4,8
Пример 20

Концентрация белка: 1,01 мг/мл
Отношение лекарственное средство/МАТ: 2,6
Пример 21

Концентрация белка: 0,53 мг/мл
Отношение лекарственное средство/МАТ: 0,6
Пример 22

Концентрация белка: 0,55 мг/мл
Отношение лекарственное средство/МАТ: 1,3
Пример 23

Концентрация белка: 0,65 мг/мл
Отношение лекарственное средство/МАТ: 1,1
Пример 24

Концентрация белка: 1,04
Отношение лекарственное средство/МАТ: 3,5
Пример 25

Концентрация белка: 0,62 мг/мл
Отношение лекарственное средство/МАТ: 2,4
Пример 26

Реакцию сочетания осуществляли с использованием 90 мг анти-С4.4а антитела М31-В01 в 5 буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 11,2 мг/мл
Отношение лекарственное средство/МАТ: 2,3
Пример 27

Концентрация белка: 1,11 мг/мл
Отношение лекарственное средство/МАТ: 2,4
Пример 28

Реакцию сочетания осуществляли с использованием 70 мг анти-С4.4а антитела М31-В01 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 10,7 мг/мл
Отношение лекарственное средство/МАТ: 2,2
Пример 29

Концентрация белка: 0,87 мг/мл
Отношение лекарственное средство/МАТ: 1,8
Пример 30

Концентрация белка: 1,3 мг/мл
Отношение лекарственное средство/МАТ: 2,1
Пример 31

Концентрация белка: 1,3 мг/мл
Отношение лекарственное средство/МАТ: 0,3
Пример 32

Реакцию сочетания осуществляли с использованием 70 мг анти-С4.4а антитела М31-В01 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 12,0 мг/мл
Отношение лекарственное средство/МАТ: 3,2
Пример 33

Реакцию сочетания осуществляли с использованием 90 мг анти-С4.4а антитела М31-В01 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 10,2 мг/мл
Отношение лекарственное средство/МАТ: 4,3
Пример 34

Концентрация белка: 1,37 мг/мл
Отношение лекарственное средство/МАТ: 2,6
Пример 35

Концентрация белка: 1,14 мг/мл
Отношение лекарственное средство/МАТ: 2,0
Пример 36

Концентрация белка: 1,07 мг/мл
Отношение лекарственное средство/МАТ: 3,5
Пример 37

Концентрация белка: 1,14 мг/мл
Отношение лекарственное средство/МАТ: 1,9
Пример 38

Концентрация белка: 1,22 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 39

Концентрация белка: 1,3 мг/мл
Отношение лекарственное средство/МАТ: 3,2
Пример 40

Концентрация белка: 1,23 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 41

Концентрация белка; 1,64 мг/мл
Отношение лекарственное средство/МАТ: 1,8
Пример 42

Концентрация белка: 1,07 мг/мл
Отношение лекарственное средство/МАТ: 3,1
Пример 43

Концентрация белка: 1,14 мг/мл
Отношение лекарственное средство/МАТ: 2,3
Пример 44

Концентрация белка: 1,23 мг/мл
Отношение лекарственное средство/МАТ: 3,4
Пример 45
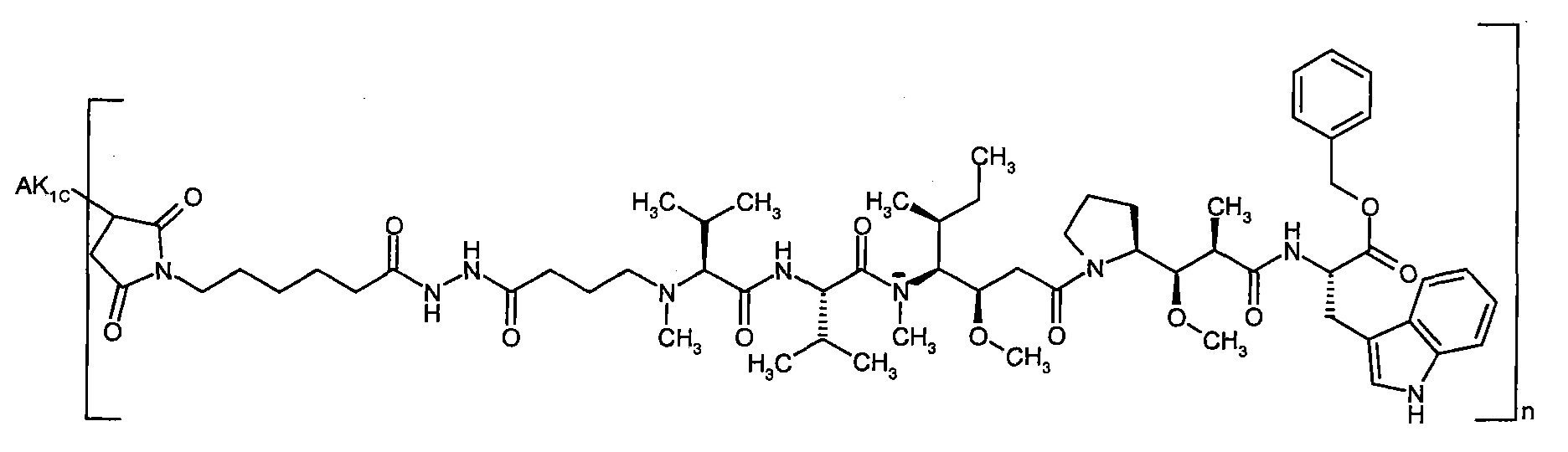
Концентрация белка: 1,22 мг/мл
Отношение лекарственное средство/МАТ: 2,5
Пример 46

Концентрация белка: 1,22 мг/мл
Отношение лекарственное средство/МАТ: 2,4
Пример 47

Концентрация белка: 1,32 мг/мл
Отношение лекарственное средство/МАТ: не определено
Пример 48

Концентрация белка: 1,44 мг/мл
Отношение лекарственное средство/МАТ: 2,3
Пример 49
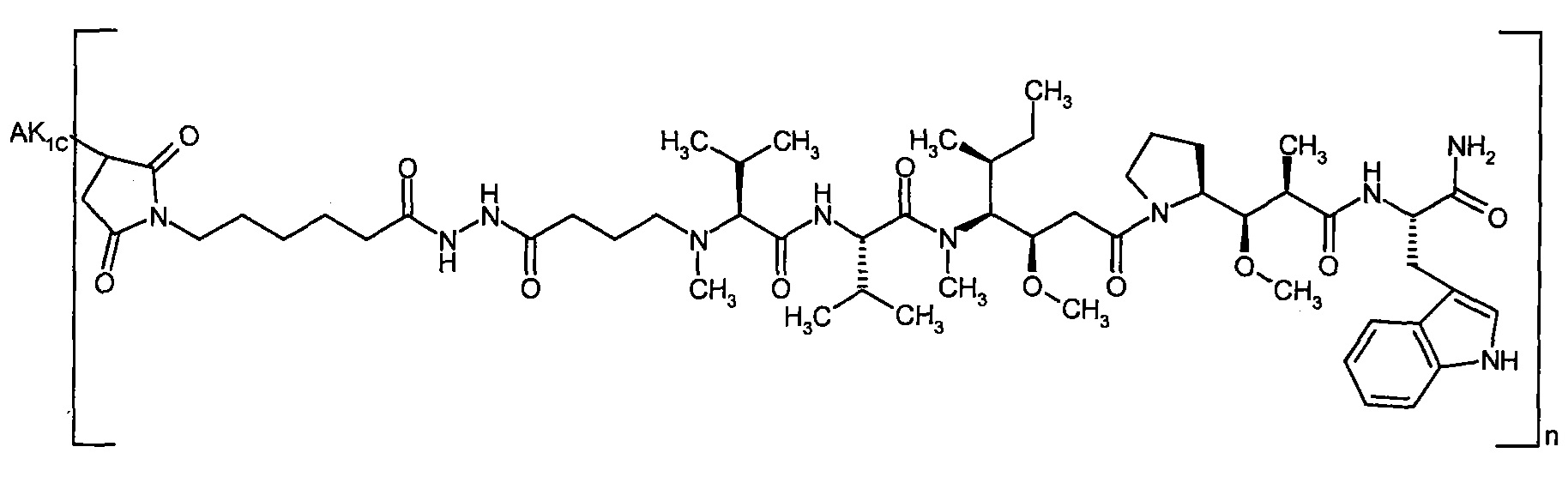
Реакцию сочетания осуществляли с использованием 250 мг анти-С4.4а антитела В01-10 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 12,8 мг/мл
Отношение лекарственное средство/МАТ: 5,2
Пример 50

Концентрация белка: 0,9 мг/мл
Отношение лекарственное средство/МАТ: 2
Пример 51

Реакцию сочетания осуществляли с использованием 250 мг анти-С4.4а антитела В01-3 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 8,0 мг/мл
Отношение лекарственное средство/МАТ: 4,5
Пример 52

Реакцию сочетания осуществляли с использованием 250 мг анти-С4.4а антитела В01-10 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 12,3 мг/мл
Отношение лекарственное средство/МАТ: 5,2
Пример 53

Реакцию сочетания осуществляли с использованием 250 мг анти-С4.4а антитела В01-10 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 10,2 мг/мл
Отношение лекарственное средство/МАТ: 4,4
Пример 54

Реакцию сочетания осуществляли с использованием 50 мг анти-С4.4а антитела В01-3 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 11,5 мг/мл
Отношение лекарственное средство/МАТ: 5,2
Пример 55

Реакцию сочетания осуществляли с использованием 250 мг анти-С4.4а антитела D02-6 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 13 мг/мл
Отношение лекарственное средство/МАТ: 5,2
Пример 56

Реакцию сочетания осуществляли с использованием 250 мг анти-С4.4а антитела ВО 1-3 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 10,3 мг/мл
Отношение лекарственное средство/МАТ: 4,9
Пример 57

Концентрация белка: 0,88 мг/мл
Отношение лекарственное средство/МАТ: 3,2
Пример 58

Концентрация белка: 1,18 мг/мл
Отношение лекарственное средство/МАТ: 3,4
Пример 59

Концентрация белка: 1,23 мг/мл
Отношение лекарственное средство/МАТ: 3,0
Пример 60

Концентрация белка: 1,3 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 61

Концентрация белка: 1,11 мг/мл
Отношение лекарственное средство/МАТ: не определено
Пример 62

Концентрация белка: 1,25 мг/мл
Отношение лекарственное средство/МАТ: 2,4
Пример 63
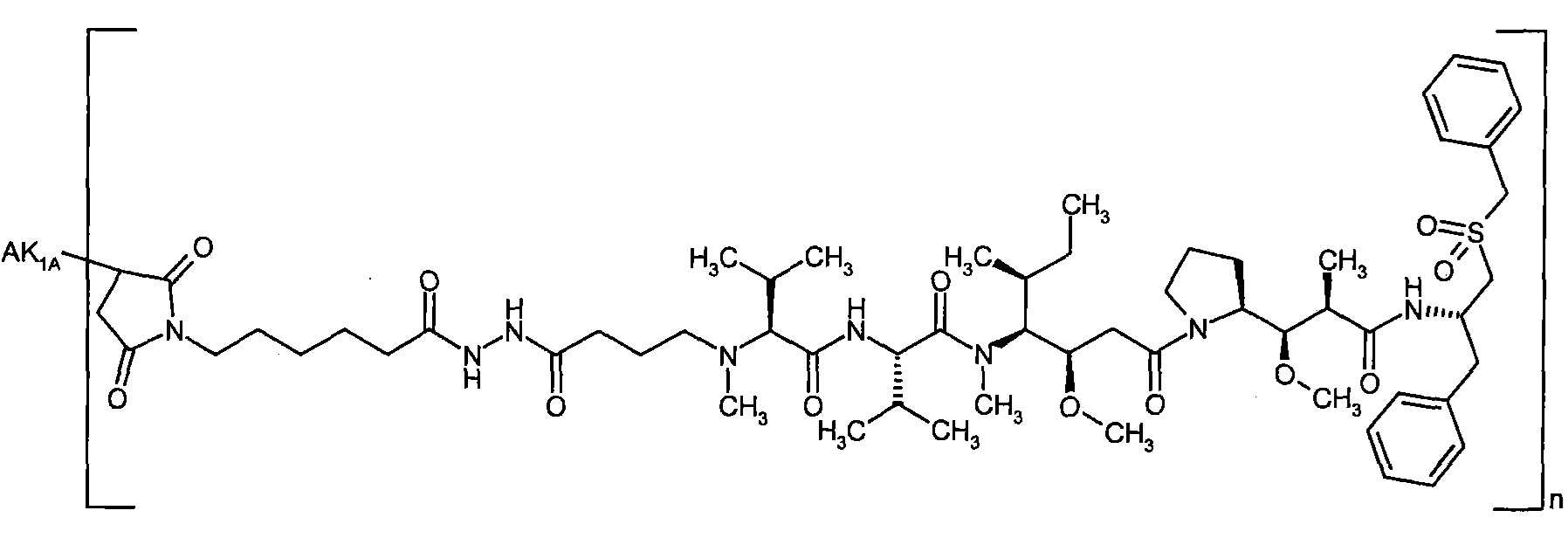
Концентрация белка: 0,88 мг/мл
Отношение лекарственное средство/МАТ: 5,0
Пример 64

Концентрация белка: 1,23 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 65

Концентрация белка: 0,93 мг/мл
Отношение лекарственное средство/МАТ: 1,8
Пример 66

Концентрация белка: 0,85 мг/мл
Отношение лекарственное средство/МАТ: 5,3
Пример 67

Концентрация белка: 1,51 мг/мл
Отношение лекарственное средство/МАТ: 1,4
Пример 68

Реакцию сочетания осуществляли с использованием 150 мг анти-С4.4а антитела В01-3 в буфере DPBS (фосфатно-солевой буфер в модификации Дульбекко) с рН 7,4, и после очистки на сефадексе смесь концентрировали путем ультрацентрифугирования.
Концентрация белка: 11,0 мг/мл
Отношение лекарственное средство/МАТ: 4,5
Пример 69

Концентрация белка: 1,2 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 70

Концентрация белка: 1,25 мг/мл
Отношение лекарственное средство/МАТ: 3,1
Пример 71
N-(4-{2-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1 -амино-3-(1Н-идол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (10 мкмоль) Промежуточного соединения 157 разбавляли в 5,2 мл ДМФ и смешивали с 2,28 мг (20 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 5,8 мг (48% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,45 мин;
ЖХ-МС (Метод 1): Rt=0,74 мин; MS (ESIпоз): m/z=1184 (M+H)+.
Пример 72
N-(4-{2-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(1Н-идол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил} -3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (10 мкмоль) Промежуточного соединения 113 разбавляли в 5,2 мл ДМФ и смешивали с 2,28 мг (20 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 6 мг (54% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,5 мин;
ЖХ-МС (Метод 1): Rt=0,77 мин; MS (ESIпоз): m/z=1185 (M+H)+.
Пример 73
N-(4-{2-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопролил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

9 мг (8,3 мкмоль) Промежуточного соединения 132 разбавляли в 4 мл ДМФ и смешивали с 3 мг (24.4 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 6,8 мг (68% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 12); Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,78 мин; MS (ESIпоз): m/z=1227 (M+H)+.
Пример 74
N-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-амино-3-(1Н-идол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (10 мкмоль) Промежуточного соединения 106 разбавляли в 5,8 мл ДМФ и смешивали с 2,5 мг (20 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 5,2 мг (46% теоретического указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,5 мин;
ЖХ-МС (Метод 11): Rt=0,71 мин; MS (ESIпоз): m/z=1070 (M+H)+.
Пример 75
N-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(1Н-идол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (10 мкмоль) Промежуточного соединения 124 разбавляли в 4 мл ДМФ и смешивали с 2,5 мг (20 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 7,2 мг (64% теоретического указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,8 мин; MS (ESIпоз): m/z=1071 (М+Н)+.
Пример 76
N-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-идол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (10 мкмоль) Промежуточного соединения 125 разбавляли в 4 мл ДМФ и смешивали с 2,4 мг (20 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 7,7 мг (69% теоретического указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 2): Rt=1,91 мин; MS (ESIпоз): m/z=1140 (М+Н)+.
Пример 77
N-(4-{2-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-3-(1Н-идол-3-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

10 мг (10 мкмоль) Промежуточного соединения 160 разбавляли в 3 мл ДМФ и смешивали с 2,1 мг (20 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 8,1 мг (73% теоретического указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,86 мин; MS (ESIпоз): m/z=1274 (М+Н)+.
Пример 78
N-(4-{2-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексаноил]гидразино}-4-оксобутил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-1-(бензиламино)-1-оксо-3-фенилпропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

3,5 мг (3 мкмоль) Промежуточного соединения 159 разбавляли в 1 мл ДМФ и смешивали с 0,76 мг (6 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 2,6 мг (65% теоретического указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,75 мин;
ЖХ-МС (Метод 1): Rt=0,85 мин; MS (ESIпоз): m/z=1235 (М+Н)+.
Пример 79
N-(6-{2-[6-(3-{[(2R)-2-амино-2-карбоксиэтил]сульфанил}-2,5-диоксопирролидин-1-ил)гексаноил]гидразино}-6-оксогексил)-N-метил-L-валил-N-[(3R,4S,5S)-3-метокси-1-{(2S)-2-[(1R,2R)-1-метокси-2-метил-3-{[(1S,2R)-1-(1,2-оксазинан-2-илкарбонил)-2-фенилциклопропил]амино}-3-оксопролил]пирролидин-1-ил}-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

3,6 мг (3 мкмоль) Промежуточного соединения 129 разбавляли в 1 мл ДМФ и смешивали с 0,77 мг (6 мкмоль) L-цистеина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 1,55 мг (39% теоретического указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,6 мин;
ЖХ-МС (Метод 1): Rt=0,87 мин; MS (ESIпоз): m/z=1255 (М+Н)+.
Пример 80

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 0,83 мг/мл
Отношение лекарственное средство/МАТ: 1,6
Пример 81

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,59 мг/мл
Отношение лекарственное средство/МАТ: 3,1
Отношение лекарственное средство/МАТ: 2,9
Пример 82

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,25 мг/мл
Отношение лекарственное средство/МАТ: 4,0
Пример 83

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,27 мг/мл
Отношение лекарственное средство/МАТ: 3,6
Пример 84

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,54 мг/мл
Отношение лекарственное средство/МАТ: 4,7
Пример 85

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,73 мг/мл
Отношение лекарственное средство/МАТ: 4,7
Пример 86

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,66 мг/мл
Отношение лекарственное средство/МАТ: 1,3
Пример 87

Концентрация белка: 2,11 мг/мл
Отношение лекарственное средство/МАТ: 5,5
Пример 88

Концентрация белка: 1,53 мг/мл
Отношение лекарственное средство/МАТ: 3,4
Пример 89

Концентрация белка: 1,5 мг/мл
Отношение лекарственное средство/МАТ: 0,2
Пример 90

Концентрация белка: 1,32 мг/мл
Отношение лекарственное средство/МАТ: 0,1
Пример 91

Реакцию сочетания осуществляли с использованием 80 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования, повторно разбавляли в ФБР и снова концентрировали.
Концентрация белка: 10,3 мг/мл
Отношение лекарственное средство/МАТ: 3,1
Пример 92

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,09 мг/мл
Отношение лекарственное средство/МАТ: 1,8
Пример 93

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,52 мг/мл
Отношение лекарственное средство/МАТ: 4,2
Пример 94

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,1 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 95

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,43 мг/мл
Отношение лекарственное средство/МАТ: 4,8
Пример 96

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе, концентрировали путем ультрацентрифугирования, повторно разбавляли в ФБР и снова концентрировали.
Концентрация белка: 1,36 мг/мл
Отношение лекарственное средство/МАТ: 4,6
Пример 97

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,33 мг/мл
Отношение лекарственное средство/МАТ: 4,0
Пример 98

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,33 мг/мл
Отношение лекарственное средство/МАТ: 4,6
Пример 99
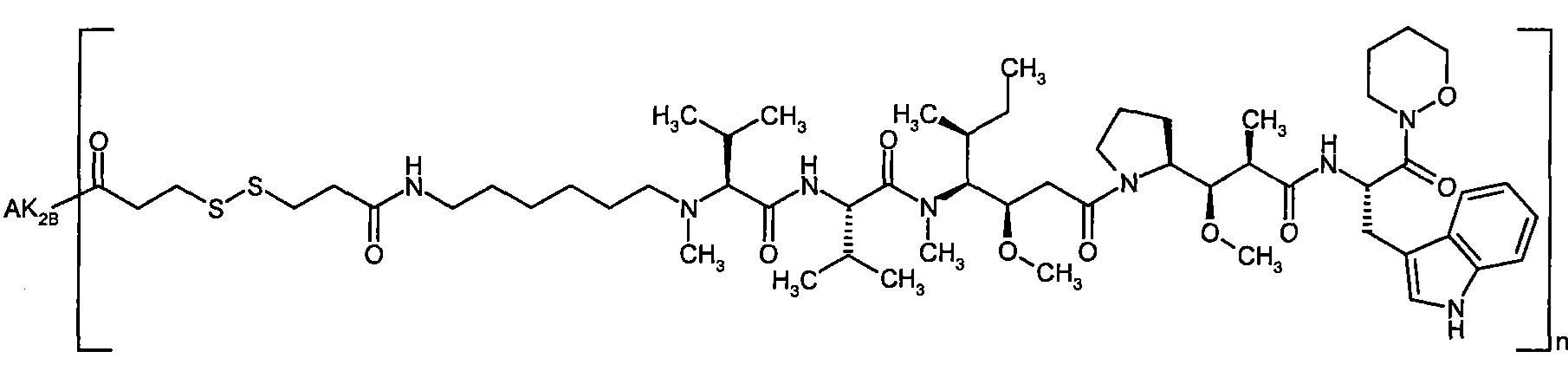
Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,47 мг/мл
Отношение лекарственное средство/МАТ: 1,6
Пример 100

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,49 мг/мл
Отношение лекарственное средство/МАТ: 4,5
Пример 101

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,29 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 102

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,74 мг/мл
Отношение лекарственное средство/МАТ: 3,5
Пример 103
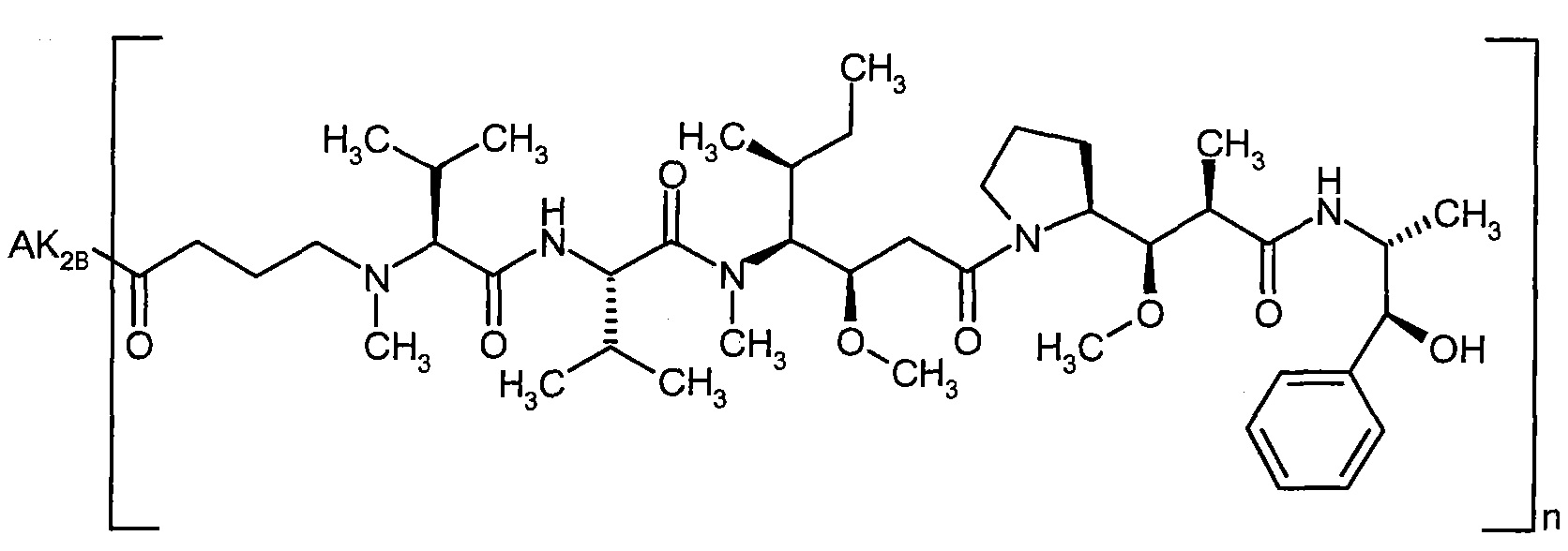
Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,09 мг/мл
Отношение лекарственное средство/МАТ: 3,2
Пример 104

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,63 мг/мл
Отношение лекарственное средство/МАТ: 0,2
Пример 105

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,41 мг/мл
Отношение лекарственное средство/МАТ: 7,6
Пример 106

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 2,0 мг/мл
Отношение лекарственное средство/МАТ: 1,6
Пример 107

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,67 мг/мл
Отношение лекарственное средство/МАТ: 2,8
Пример 108
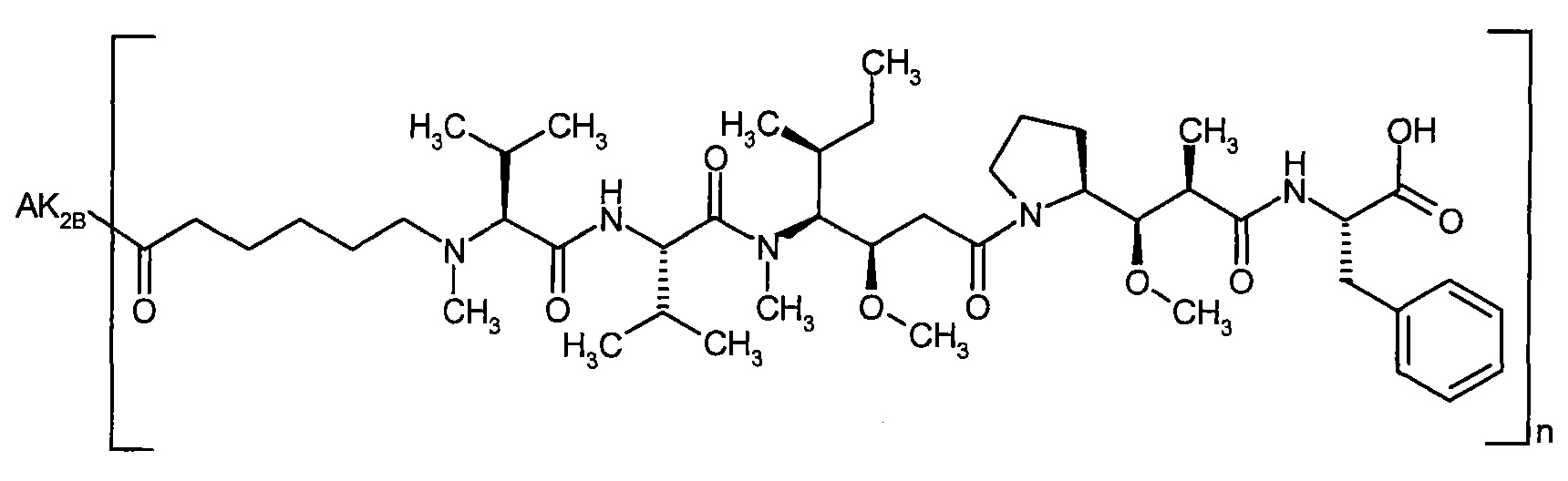
Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,91 мг/мл
Отношение лекарственное средство/МАТ: 5,3
Пример 109

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,82 мг/мл
Отношение лекарственное средство/МАТ: 4,6
Пример 110

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела ВО 1-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,9 мг/мл
Отношение лекарственное средство/МАТ: 4,2
Пример 111

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,89 мг/мл
Отношение лекарственное средство/МАТ: 2,7
Пример 112

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,73 мг/мл
Отношение лекарственное средство/МАТ: 2,3
Пример 113

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,71 мг/мл
Отношение лекарственное средство/МАТ: 3,3
Пример 114

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,47 мг/мл
Отношение лекарственное средство/МАТ: 3,9
Пример 115
N-(6-{[(5S)-5-амино-5-карбоксипентил]амино}-6-оксогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-идол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамидтрифторацетат

15,5 мг (15 мкмоль) Промежуточного соединения 210 разбавляли в 5 мл ДМФ и смешивали с 4.4 мг (18 мкмоль) N2-(трет-бутоксикарбонил)-L-лизина и 7,7 мкл (44 мкмоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем концентрировали при пониженном давлении. Затем остаток очищали путем препаративной ВЭЖХ. В результате получали 14 мг (81% теоретического выхода) защищенного интермедиата указанного в названии соединения, которое затем разбавляли в 1 мл дихлорметана и с которого снимали защиту с использованием 1 мл трифторуксусной кислоты. Смесь концентрировали и после лиофилизации остатка из смеси ацетонитрил/вода (1:1), получали 15 мг (97% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,8 мин;
ЖХ-МС (Метод 1): Rt=0,79 мин; MS (ESIпоз): m/z=1083 (М+Н)+.
Пример 116
N-(6-{[(5S)-5-амино-5-карбоксипентил]амино}-6-оксогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(1H-идол-3-ил)этил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид

40 мг (40 мкмоль) Промежуточного соединения 227 разбавляли в 5 мл ДМФ и смешивали с 11,5 мг (40 мкмоль) N2-[(бензилокси)карбонил]-L-лизина и 13 мкл (80 мкмоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали при пониженном давлении и после этого очищали препаративной ВЭЖХ. В результате получали 32,5 мг (70% теоретического выхода) защищенного интермедиата указанного в названии соединения.
32,5 мг этого интермедиата растворяли в 10 мл метанола и после добавления 2 мг 10% палладия на активированном угле гидрогенировали при стандартном давлении водорода при комнатной температуре в течение 30 мин. Затем катализатор удаляли путем фильтрации, а растворитель удаляли при пониженном давлении. В результате лиофилизации остатка из смеси диоксан/вода 1:1 получали 26 мг (99% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,76 мин; MS (ESIпоз): m/z=1014 (М+Н)'+.
Пример 117
N-[(18S)-18-амино-18-карбокси-12-оксо-3,6,9-триокса-13-азаоктадек-1-ил]-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-идол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетата
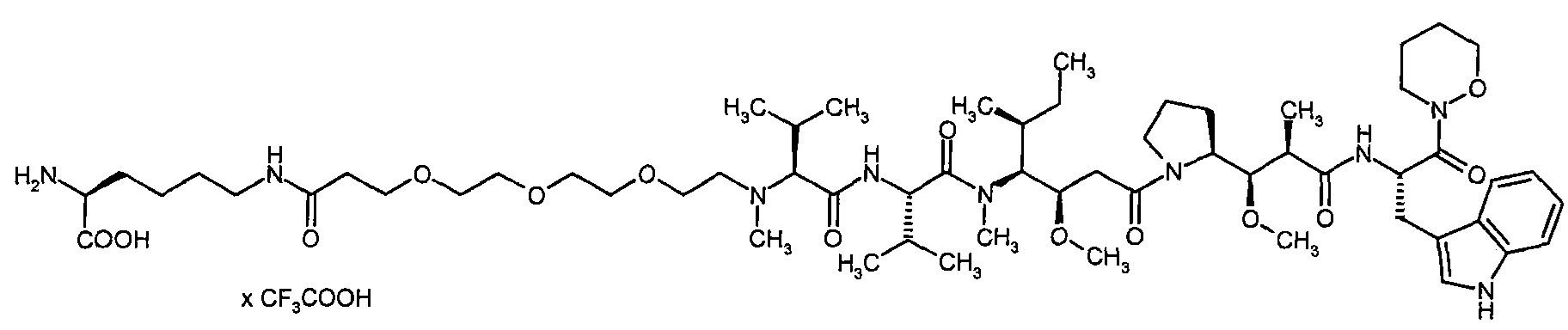
3,5 мг (3 мкмоль) Промежуточного соединения 202 разбавляли в 2 мл ДМФ и смешивали с 0,8 мг (3 мкмоль) N2-(трет-бутоксикарбонил)-L-лизина и 1,6 мкл (10 мкмоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали при пониженном давлении. Остаток разбавляли в смеси ацетонитрил/вода: (1:1), доводили рН до 2 трифторуксусной кислотой, а затем очищали преаративной ВЭЖХ. В результате получали 1 мг (25% теоретического выхода) защищенного интермедиата указанного в названии соединения, которое затем разбавляли в 500 мкл дихлорметана, снимали защиту 500 мкл трифторуксусной кислоты. Смесь концентрировали и после лиофилизации остатка из смеси ацетонитрил/вода (1:1) получали 1 мг (89% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,9 мин;
ЖХ-МС (Метод 1): Rt=0,82 мин; MS (ESIпоз): m/z=1173 (М+Н)+.
Пример 118

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР, и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 0,89 мг/мл
Отношение лекарственное средство/МАТ: 1,8
Пример 119

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 0,57 мг/мл
Отношение лекарственное средство/МАТ: 1,5
Пример 120

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3, реакционную смесь после очистки на Сефадекса концентрировали путем ультрацентрифугирования и снова разбавляли фосфатно-буферным раствором.
Концентрация белка: 1,39 мг/мл
Отношение лекарственное средство/МАТ: 7,1
Пример 121

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 реакционную смесь после очистки на Сефадекса концентрировали путем ультрацентрифугирования и снова разбавляли фосфатно-буферным раствором.
Концентрация белка: 1,54 мг/мл
Отношение лекарственное средство/МАТ: 2,4
Пример 122

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР, и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,48 мг/мл
Отношение лекарственное средство/МАТ: 2,4
Пример 123

Реакцию сочетания осуществляли с использованием 5 мг анти-С4.4а антитела В01-3 в ФБР, и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли.
Концентрация белка: 1,43 мг/мл
Отношение лекарственное средство/МАТ: 3,6
Пример 124 Диастереомер 1

Реакцию сочетания осуществляли с использованием промежуточного соединения 247а и 5 мг анти-С4.4а антитела В01-3 в ФБР, и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли фосфатно-буферным раствором.
Концентрация белка: 1,45 мг/мл
Отношение лекарственное средство/МАТ: 3,8
Пример 125
Диастереомер 2

Реакцию сочетания осуществляли с использованием Промежуточного соединения 247а и 5 мг анти-С4.4а антитела В01-3 в ФБР, и смесь после очистки на Сефадексе концентрировали путем ультрацентрифугирования и снова разбавляли фосфатно-буферным раствором.
Концентрация белка: 1,42 мг/мл Отношение лекарственное средство/МАТ: 4,0
Пример 126 N-(6-{[(5S)-5-амино-5-карбоксипентил]амино}-6-оксогексил)-N-метил-L-threonyl-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(2S)-3-(1Н-идол-3-ил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамидтрифторацетат

8,6 мг (8 мкмоль) Промежуточного соединения 240 разбавляли в 5 мл ДМФ и смешивали с 4,0 мг (16 мкмоль) N2-(трет-бутоксикарбонил)-L-лизина и 2 мкл (16 мкмоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем снова смешивали с теми же количествами N2-(трет-бутоксикарбонил)-L-лизина и N,N-диизопропилэтиламина и перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь концентрировали при пониженном давлении. Затем остаток очищали препаративной ВЭЖХ. В результате получали 7 мг (72% теоретического выхода) защищенного интермедиата указанного в названии соединения, который затем разбавляли в 1 мл дихлорметана, и снимали защиту 0,5 мл трифторуксусной кислоты. Реакционную смесь концентрировали и очищали остаток путем препаративной ВЭЖХ. В результате сушки под вакуумом получали 3,3 мг (47% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 5): Rt=1,5 мин;
ЖХ-МС (Метод 1): Rt=0,8 мин; MS (ESIпоз): m/z=1084 (М+Н)+.
Пример 127
N-(6-{[(5S)-5-амино-5-карбоксипентил]амино}-6-оксогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-{[(2S)-3-(4-гидроксифенил)-1-(1,2-оксазинан-2-ил)-1-оксопропан-2-ил]амино}-1-метокси-2-метил-3-оксопролил1пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамидтрифторацетат

8 мг (8 мкмоль) Промежуточного соединения 242 разбавляли в 3 мл ДМФ и смешивали с 2,9 мг (12 мкмоль) N2-(трет-бутоксикарбонил)-L-лизина и 2,7 мкл (16 мкмоль) N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем снова смешивали с теми же количествами N2-(трет-бутоксикарбонил)-L-лизина и N,N-диизопропилэтиламина, и перемешивали при комнатной температуре в течение еще 4 часов. Затем реакционную смесь концентрировали при пониженном давлении. Затем остаток очищали препаративной ВЭЖХ. Лиофилизация из смеси ацетонитрил/вода давала 6,5 мг (72% теоретического выхода) защищенного интермедиата указанного в названии соединения, который затем разбавляли в 5 мл дихлорметана, защиту снимали с использованием 0,75 мл трифторуксусной кислоты. Смесь концентрировали, и лиофилизация остатка из смеси диоксан/вода давала 5 мг (76% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,7 мин;
ЖХ-МС (Метод 1): Rt=0,69 мин; MS (ESIпоз): m/z=1059 (М+Н)+.
Пример 128
N-(6-{[(5S)-5-амино-5-карбоксипентил]амино}-6-оксогексил)-N-метил-L-валил-N-[(3R,4S,5S)-1-{(2S)-2-[(1R,2R)-3-{[(1S)-1-карбокси-2-(4-гидроксифенил)этил]амино}-1-метокси-2-метил-3-оксопролил]пирролидин-1-ил}-3-метокси-5-метил-1-оксогептан-4-ил]-N-метил-L-валинамид трифторацетат

38 мг (41 мкмоль) Промежуточного соединения 248 сначала превращали в N-гидроксисукцинимидный эфир. 72 мг полученного сырого продукта разбавляли в 5 мл ДМФ и смешивали с 24 мг (100 мкмоль) N2-(трет-бутоксикарбонил)-L-лизина и 23 мкл N,N-диизопропилэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение ночи, и затем снова смешивали с 16 мг N2-(трет-бутоксикарбонил)-L-лизина и 12 мкл N,N-диизопропилэтиламина, после чего обрабатывали в ультразвуковой ванне в течение еще 2 часов. Затем смесь концентрировали при пониженном давлении и очищали остаток путем препаративной ВЭЖХ. Лиофилизации из смеси ацетонитрил/вода давала 20 мг (50% теоретического выхода) защищенного интермедиата указанного в названии соединения.
Затем 15 мг (12 мкмоль) этого интермедиата разбавляли в 3 мл дихлорметана и смешивали с 1 мл трифторуксусной кислоты. После 40 мин перемешивания при комнатной температуре добавляли еще 1,5 мл трифторуксусной кислоты и обрабатывали смесь в ультразвуковой ванне в течение 1 часа. Затем реакционную смесь концентрировали, лиофилизация из смеси диоксан/вода давала 13 мг (90% теоретического выхода) указанного в названии соединения.
ВЭЖХ (Метод 12): Rt=1,5 мин;
ЖХ-МС (Метод 1): Rt=0,68 мин; MS (ESIпоз): m/z=990 (М+Н)+.
С: Оценка биологической активности:
Биологический эффект соединений согласно настоящему изобретению был продемонстрирован в тестах, описанных ниже
С-1. Анализ цитотоксического эффекта ADC, направленных против С4.4а
Цитотоксическое действие анти-С4.4а ADC исследуют в различных клеточных линиях:
- А549 (CCL-185, ATCC), трансфицированных последовательностью для получения целого рецептора С4.4а,
- А549, трансфицированных фиктивными последовательностями,
- А549 дикий тип (DSMZ, лот 11),
- NCI-H292, эндогенно экспрессирующая С4.4а клеточная линия рака легких (CRL-1848, ATCC),
- SCC-4 эндогенно экспрессирующая С4.4а клеточная линия плоскоклеточный эпителиальной карциномы (CRL-1624, ATCC),
- SCC-9 эндогенно экспрессирующая С4.4а клеточная линия плоскоклеточный эпителиальной карциномы (CRL-1629, ATCC),
- НСТ-116 эндогенно экспрессирующая С4.4а клеточная линия карциномы толстой кишки (CCL-247, ATCC),
- НСТ-116/VM46, НСТ-116, трансфицированные VM46,
- A431NS (CRL-2592, ATCC)
Клетки культивируют стандартным методом, как указано в Американской коллекции типа тканей (ATCC) для соответствующих клеточных линий. Для указанной процедуры, клетки отделяют с использованием раствора трипсина (0,05%) и ЭДТА (0,02%) в ФБР (Biochrom AG #L2143), гранулируют, ресуспендируют в культуральной среде, подсчитывают и высевают в 96-луночный планшет для культивирования с белым основанием (Costar #3610) (2500 клеток в 100 мкл/лунку) и инкубируют в термостате при 37°С с 5% диоксидом углерода. Через 24 часа конъюгаты антитела и активного соединения в 100 мкл культуральной среды добавляют к клеткам в концентрации от 10-7 М до 10-11 М (в двух повторах) и инкубируют в инкубаторе при 37°С с 5% диоксидом углерода. После 72 часов, жизнеспособность клеток определяют с помощью анализа жизнеспособности клеток по титру свечения люминесценции (Promega #G7573 и G7571#). Для этого 100 мкл субстрата добавляют в ячейку с пробой, затем планшеты покрывают алюминиевой фольгой, встряхивают при 180 оборотах в минуту в шейкере в течение 2 мин, выдерживают на лабораторном столе в течение 8 мин, а затем измеряют с использованием Victor X2 (Perkin Elmer). Субстрат детектирует содержание АТФ живых клеток, производя люминесцентный сигнал, степень которого прямо пропорциональна жизнеспособности клеток. Данные измерений используют для расчета 1С50 использованием программного обеспечения Graph Pad Prism Laboratory.
В Таблице 3 приведены значения IC501) репрезентативных рабочих примеров из этого анализа:
1) Данные об активности относятся к рабочим примерам, описанным в настоящей экспериментальной части, с указанными соотношениями соединение/моноклональное антитело. Значения могут отклоняться возможности для различных соотношений соединений/моноклональное антитело
С-2. Определение влияния на полимеризацию тубулина
Раковые клетки являются неестественными клетками, которые часто приводят к образованию опухолей также в результате усиленного клеточного деления. Микротрубочки образуют волокна веретена в аппарате веретена и являются существенной частью клеточного цикла. Регулируемое образование и разрыв микротрубочек делает возможным четкое разделение хромосом между дочерними клетками и представляет собой непрерывный динамический процесс. Нарушение этого динамического процесса приводит к неправильному делению клеток и в конечном итоге к гибели клеток. Усиленное клеточное деление опухолевых клеток, однако, также делает их особенно чувствительными к ядам, действующим на волокна веретена, которые представляют собой неотъемлемую часть химиотерапии. Яды, действующие на волокна веретена, такие как паклитаксел или эпотилоном, приводят к резкому увеличению скорости полимеризации микротрубочек, в то время как алкалоиды барвинка или еще монометилауристатин-Е (ММАЕ) приводят к резкому уменьшению скорости полимеризации микротрубочек. В обоих случаях необходимый динамизм клеточного цикла нарушается критическим образом. Соединения, исследованные в контексте настоящего изобретения, способствуют уменьшению скорости полимеризации микротрубочками.
Полимеризацию тубулина исследовали с помощью "Набора для анализа полимеризации микротрубочек на основе флуоресценции" компании Cytoskeleton (Денвер, Колорадо, США, номер для заказа: ВК011). В этом анализе ГТФ добавляют к неполимеризованному тубулину, что делает возможным спонтанную полимеризацию. Анализ основан на связывании флюорофора 4',6-ди-амидино-2-фенилиндола (DAPI) с тубулином. Свободный и связанный DAPI может быть дифференцирован на основе различных спектров излучения. Поскольку DAPI обладает значительно более высоким сродством к полимеризованному тубулину по сравнению с неполимеризованным тубулином, полимеризация тубулина может наблюдаться при увеличении флуоресценции связанных флуорофоров DAPI.
Для осуществления этого анализа соединения согласно настоящему изобретению в виде раствора в ДМСО разбавляли от их исходной концентрации от 10 мМ до 1 мкМ в воде. В дополнение к контрольному буферу дополнительно были использованы паклитаксел, с эффектом увеличения полимеризации, и винбластин, с эффектом ингибирования полимеризации, в качестве контроля анализа. Измерение проводили с использованием 96-луночных планшетов с половинной площадью базы. Кинетику полимеризации тубулина наблюдали с использованием флуориметра при 37°С в течение 1 часа. Длина волны возбуждения была 355 нм, излучение контролировали при 460 нм. Для области линейного увеличения в течение первых 10 мин расчет был выполнен на основе изменения флуоресценции в минуту (ΔF/мин), которая представляет скорость полимеризации микротрубочек. Активность тестируемых веществ была количественно оценена на основе их соответствующего снижени\скорости полимеризации. Значение для ингибирования MMAF при концентрации 1 мкМ принимали за 100%.
В таблице 4 приведены данные по ингибированию полимеризации тубулина соединениями из репрезентативных рабочих примеров.
Токсофор MMAF и соединения из рабочих примеров ингибируют полимеризацию тубулина в зависимости от их концентрации. При 100 мкм MMAF полимеризацию тубулина ингибируется полностью. Соединение из рабочего примера 115 ингибирует скорость полимеризации тубулина в концентрации 1 мкМ до 45% от значения, измеренного при 1 мкМ MMAF.
С-3. Тесты in vitro для определения проницаемости в клетки
Проницаемость вещества в клетки может быть исследована с помощью тестирования in vitro в потоковом анализе с использованием клеток Сасо-2 [M.D. Troutman и D.R. Thakker, Pharm. Res. 20 (8), 1210-1224 (2003)]. Для этого клетки культивировали в течение 15-16 дней в 24-луночных фильтровальных планшетах. Для определения проницаемости, вещество из соответствующего рабочего примера в буфере HEPES добавляли к клетками либо у вершины (А) или у основания (В) и инкубировали в течение 2 часов. После 0 часов и через 2 часа из цис-и транс отсеков отбирали образцы. Образцы разделяли с помощью ВЭЖХ (Agilent 1200, Беблингер, Германия) с использованием колонок с обратной фазой. Система ВЭЖХ была соединена через интерфейс Turbo Ion Spray к масс-спектрометру Triple Qua-dro-pol API 4000 (Applied Biosystems Applera, Дармштадт, Германия). Проницаемость оценивали на основании значения Рарр, которое было рассчитано по формуле, опубликованной Schwab et al. [D. Schwab et al., J. Med. Chem. 46, 1716-1725 (2003)].
Решающее значение для токсофоров, которые высвобождаются внутриклеточно, является проницаемость от В к А [Папп (ВА)]: чем ниже эта проницаемость, тем дольше время пребывания соединения из рабочего примера в клетке после внутриклеточного высвобождения, а следовательно, и больше время для взаимодействия с биохимической целью (в данном случае: тубулином).
В таблице 5 ниже приводятся данные по проницаемости соединения из репрезентативных рабочих примеров в этом анализе:
Рабочие примеры демонстрируют низкую проницаемость от В к А [Рарр (В-А) и поэтому имеют длительное время пребывания в клетках СаСо-2. Для сравнения, монометилауристатин Е (ММАЕ) и монометилауристатинп F (MMAF) в данном тесте демонстрируют значение Рарр (В-А) 73 нм/с, и, следовательно, имеют значительно более короткое время пребывания в клетках Сасо-2.
С-4. Тесты in vitro для определения свойств субстрата для Р-гликопротеина (P-gp)
Многие опухолевые клетки экспрессируют белки, транспортирующие вещества, и это часто сопровождается развитием резистентности к цитостатикам. Вещества, которые не являются субстратами таких белков транспортеров, таких как Р-гликопротеин (P-gp) или BCRP, могут обладать улучшенным профилем активности.
Свойства вещества в качестве субстрата для P-gp (ABCB1) определяли с помощью поточного анализа с использованием клеток LLC-PK1, которые в избытке экспрессируют Р-гликопротеин (клетки L-MDR1) [А.Н. Schinkel et al., J. Clin. Invest. 96, 1698-1705 (1995)]. Для этой цели клетки LLC-PK1 или клетки L-MDR1 культивировали в 96-луночных фильтровальных планшетах в течение 3-4 дней. Для определения проникновения, соответствующее тестовое вещество, по отдельности или в присутствии ингибитора (например, ивермектина или верапамила), в буфере HEPES добавляли к клеткам либо у вершины (А) или у основания (В) и инкубировали течение 2 часов. После 0 часов и через 2 часа отбирали пробы из цис-и транс отсеков. Образцы разделяли с помощью ВЭЖХ с использованием колонок с обратной фазой. Система ВЭЖХ была соединена через интерфейс Turbo Ion Spray к масс-спектрометру Triple Qua-dro-pol API 4000 (Applied Biosystems Applera, Дармштадт, Германия). Проницаемость оценивали на основании значения Рарр, которое было рассчитано по формуле, опубликованной Schwab et al. [D. Schwab et al., J. Med. Chem. 46, 1716-1725 (2003)].
Решающее значение для токсофоров, которые высвобождаются внутриклеточно, является проницаемость от В к А [Рарр (В-А)]: чем ниже эта проницаемость, тем дольше время пребывания вещества рабочего примера в клетке после внутриклеточного высвобождения, а, следовательно, и больше время для взаимодействия с биохимической целью (в данном случае: тубулином).
В Таблице 6 перечислены данные по проницаемости веществ репрезентативных рабочих примеров из этого анализа, который был проведен в клетках L-MDR1:
Рабочие примеры демонстрируют низкую проницаемость от В к А [Рарр (В-А)] и поэтому имеют длительное время пребывания в клетках L-MDR1.
С-5. Тест на активность in vivo
Активность конъюгатов согласно настоящему изобретению испытывали в естественных условиях с помощью, например, в модели ксенотрансплантата. Специалисту в данной области известны методы из предшествующего уровня техники для тестирования активности конъюгата согласно настоящему изобретению (см., например, WO 2005/081711; Poison и др., Cancer Res 15 марта 2009, 69 (6): 2358 - 64). С этой целью, например, в грызунов (например, мышей) имплантировали линии опухолевых клеток, которые экспрессируют целевую для связывающей части молекулу. Этим грызунам с опухолью вводили либо конъюгат согласно настоящему изобретению или контрольный конъюгат с антителом, или изотонический солевой раствор. Введение имело место один раз или чаще. Рост опухоли определяли дважды в неделю с помощью штангенциркуля. После роста опухоли в течение нескольких недель, размер опухоли животных, обрабатываемых конъюгатом, сравнивали с контрольной группой. У животных, обрабатываемых конъюгатом, был значительно меньший размер опухоли.
С-5а. Проверка ADC в экспериментальных опухолях у мышей
Предсказательная сила модели ксенотрансплантата опухоли мышей по отношению к клинической ситуацией в случае терапии с иммунотоксином часто ограничена, с одной стороны ввиду дефицитной перекрестной реактивностью терапевтических антител с видом мышей, а с другой стороны, снижением количества антител против активного соединения (ADAs) в организме человека при введении мышиных или химерных антител. Для того чтобы использовать весь потенциал специфической экспрессии С4.4а для терапии рака, для подхода с иммуноконъюгатами, например, существует потребность в человеческих антителах, которые обладают высоким сродством, являются селективными и проявляют межвидовую перекрестную реактивность, подобную предпочтительно используемой в соответствии с настоящим изобретением. С такими антителами модели ксенотрансплантата опухоли мыши позволяет получить значимые результаты по отношению к клинической ситуации.
Опухолевые клетки человека, которые экспрессируют С4.4а, прививали подкожно в бок мышей с подавленным иммунитетом, таких как голым мышам или мышам SCID. 1-10 миллионов клеток, отделенных от клеточной культуры, центрифугировали и ресуспендировали в 100 мкл среды или 50% среда/50% Матригель. Клеточную суспензию вводили под кожу мыши.
В течение нескольких дней, опухоль растет.Лечение начинается не ранее, чем размер опухоли достигнет 25 мм2.
Лечение с помощью ADC происходит путем внутривенного введения в хвостовую вену мыши. ADC растворяют в ФБР и вводят в объеме 10 мл/кг.
Схема лечения регулируется фармакокинетикой антитела. В качестве стандарта лечение происходит три раза после каждого четвертого дня. Лечение, однако, также может быть продолжено Затем или второй цикл с тремя днями лечения может следовать после более позднего момента времени.
В качестве стандартной основы, используют на группу для лечения из 8 животных. Это число может быть выше, особенно если ожидаются сильные колебания в росте опухоли или после лечения. Так же, как группы, которые получают активные вещества, одну группу, в качестве контрольной группы, обрабатывают только буфером, в соответствии с той же схеме.
В ходе эксперимента площадь опухоли измеряют регулярно с помощью штангенциркуля в двух измерениях (длина/ширина).
В конце эксперимента, опухоли извлекали и взвешивали. Отношение среднего веса опухоли для группы, подвергавшейся лечению (Т), к контрольной группе (С) выражается в виде Т/С.Если эксперимент для контрольных групп и групп, подвергавшихся лечению, заканчиваются в разное время, значение Т/С вычисляется на основе площадей опухоли при последнем совместном измерении всех групп, подвергавшихся лечению, и контрольных групп.
1 млн клеток SCC-4 инокулировали подкожно в бок самок голых мышей NMRI.
Внутривенное введение ADC начинали при среднем размере опухоли 30-35 мм2. Когда размер опухоли у контрольных групп достигал максимального возможного размера, эксперимент заканчивали, опухоли извлекали и взвешивали. Все проверенные ADC, нацеленные на С4.4а, ингибировали рост опухоли в зависимости от дозы. При дозе 30 мг/кг, для каждого из примера 54, примера 49, примера 51 и примера 53 Т/С было<0,1. Значительная противоопухолевая активность в сравнении с контрольной была достигнута для веществ из примеров 49, 52, 53, 54 и 56 при дозе до 15 мг/кг, достигая значения Т/С≤0,29.
1 млн клеток NCI-H292 инокулировали подкожно в бок самок голых мышей NMRI.
Внутривенное введение ADC начинали при среднем размере опухоли 30-35 мм2. Эксперименты с контрольными группами и группами, подвергавшимися лечению, заканчивали, когда достигался максимально возможный размер опухоли. Таким образом, различие в дальнейшем росте опухоли после окончания лечения может способствовать дальнейшей характеристики ADC. Следовательно, площадь опухоли при последнем совместном моменте в ходе измерений был использован для определения противоопухолевой активностью по сравнению с контролем (Т/С). В использованной мышиной модели NCI-H292, показано, что все изученные ADC уменьшали рост опухоли в зависимости от дозы по сравнению с контролем. Значительный противоопухолевый эффект был получен в Примере 54 при дозе до 1,9 мг/кг, а в Примере 49 при дозе до 3,75 мг/кг. Минимальные значения Т/С, полученные в этой модели, являются следующими: Т/С 0,16 при 30 мг/кг в Примере 54, Т/С 0,17 при дозе 30 мг/кг в Примере 49, Т/С 0,16 при дозе 30 мг/кг в Примере 53, Т/С 0,17 при дозе 15 мг/кг в Примере 51, и Т/С 0,19 при дозе 3,75 мг/кг в Примере 70. В сравнительном введении ADC АЦП с постоянной дозой 7,5 мг/кг, можно было достичь Т/С от 0,20 в каждом из Примеров 49 и 54, Т/С 0,27 в Примере 51, Т/С 0,22 в Примере 52, Т/С 0,23 в Примере 53, Т/С 0,24 в Примере 55, Т/С 0,21 в Примере 56 и Т/С 0,17 в Примере 70.
С-6. Фармакокинетика в модель опухоли А549 с трансфицированными С4.4а и нетрансфицированными клетками А549
После внутривенного введения 7-30 мг/кг различных ADC, измеряли концентрации ADC в плазме и опухоли, а также потенциальных метаболитов, и рассчитывали фармакокинетические параметры, такие как клиренс (CL), площадь под кривой (AUC) и половину полураспада (tl/2).
Анализ количественного определения потенциальных метаболитов
Измерение соединений в плазме и опухоли было проведено после осаждения белков метанолом, с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) в сочетании с тандемным масс-спектрометром (MS).
Для обработки 100 мкл плазмы, ее смешивали с 400 мкл метанола и 10 мкл внутреннего стандарта (ISTD, 50 нг/мл в метаноле) и встряхивали в течение 10 секунд. После центрифугирования в течение 5 мин при 16 ООО g переносили 250 мкл супернатанта в ампулы автоматического пробоотборника, который был дополнен 250 мкл аммоний-ацетатного буфера (ААС, 10 мМ, рН 6,8) и встряхивали снова.
Для обработки опухоли ее смешивали с 4-х кратным количеством метанола. В TissueLyser II (Quiagen) образец измельчали при 30 ударах в минуту в течение 6 мин, а затем центрифугировали при 16 ООО g в течение 5 мин. 50 мкл супернатанта переносили в ампулы пробоотборника и дополняли 50 мкл аммоний-ацетатного буфера (10 мМ, рН 6,8) и 5 мкл ISTD. После того, как образец снова перемешивали, образец опухоли был готов к измерению.
Измерение обоих образцов матриц имело место, наконец, с помощью ВЭЖХ, связанной с тандемным масс-спектрометром при ионизации при атмосферном давлении с помощью интерфейса турбоионного распыления (TISP) на инструменте API4000 из SCIEX.
HPLC/LC-MSMS (TISP) анализ проводили с использованием насоса HP 1100 (Agilent) с колонкой Оемиш (5 мкм С18 110 А, 50 3 мм, Phenomenex).
Для калибровки образцы плазмы смешивали с концентрацией 0,5-2000 мкг/л Предел обнаружения (ПКО) составлял около 2 мкг/л. Линейный диапазон простирался от 2 до 1000 мкг/л.
Для калибровки образцов опухоли, супернатант необработанных опухолей смешивали с концентрацией 0,5-200 мкг/л. Предел обнаружения составлял 5 мкг/л. Линейный диапазон простирался от 5 до 200 мкг/л.
Качественный контроль для проверки достоверности содержал 5 и 50 мкг/л с дополнительными 500 мкг/л в плазме. Концентрации, обнаруженные для этих образцов, отклоняются до 20% от предполагаемого значения (данные не прилагается).
D. Рабочие примеры для фармацевтических композиций
Соединения согласно настоящему изобретению могут быть преобразованы следующим образом в фармацевтические препараты:
в/в Раствор:
Соединение согласно настоящему изобретению растворяют при концентрации ниже растворимости насыщения в физиологически приемлемом растворителе (например, изотонический солевой раствор, D-PBS, или композиции с глицином и хлоридом натрия в цитратном буфере с добавлением полисорбата 80). Раствор подвергают стерильной фильтрации и разливают в стерильные и апирогенные контейнеры для инъекции.
в/в Раствор:
Соединения согласно настоящему изобретению могут быть преобразованы в процитированные формы введения. Это может быть осуществлено известным способом посредством "смешивания с" или "растворения в" инертных, нетоксичных, фармацевтически приемлемых носителях (например, буферные вещества, стабилизаторы, солюбилизаторы, консерванты). Могут присутствовать следующие соединения, например, аминокислоты (глицин, гистидин, метионин, аргинин, лизин, лейцин, изолейцин, треонин, глутаминовая кислота, фенилаланин и др.), сахар и связанные соединения (глюкоза, сахароза, маннит, трегалоза, сахароза, манноза, лактоза, сорбит), глицерин, соли натрия калия, аммония и соли кальция (например, хлорид натрия, хлорид калия или двухосновный фосфат натрия и многие другие), буферные системы этилацетат/уксусная кислота, системы фосфатного буфера, лимонную кислоту и системы цитратного буфера, трометамол (Трис и соли Трис), полисорбаты (например, полисорбат 80 и полисорбат 20), полоксамеры (например, полоксамер 188 и полоксамер 171), макрогол (ПЭГ производные, например, 3350), Тритон Х-100, соли ЭДТА, глутатион, альбумины (например, человека), мочевина, бензиловый спирт, фенол, хлоркрезол, крезола, хлорид бензалкония и многие другие.
Лиофилизат для последующего преобразования в IV, SC или внутримышечный раствор:
Альтернативно, соединения согласно настоящему изобретению могут быть превращены в стабильный лиофилизат (возможно, с помощью вышеуказанных наполнителей), и, прежде чем быть введенным, разводиться подходящим растворителем (например, инъекционная вода, изотонический солевой раствор) и вводиться.
Реферат
Настоящее изобретение относится к новым конъюгатам связывающее соединение - активное соединение (ADC) N,N-диалкилауристатинов, которые направлены против мишени С4.4а, к активным метаболитам указанных конъюгатов ADC, к способу получения этих конъюгатов ADC, к применению этих конъюгатов ADC для лечения и/или предотвращения заболеваний, а также к применению этих конъюгатов ADC для получения лекарственных средств для лечения и/или предотвращения заболеваний, более конкретно гиперпролиферативных и/или ангиогенных заболеваний, таких как, например, онкологические заболевания. Такие средства лечения могут применяться в виде монотерапии или, в альтернативном варианте, в комбинации с другими лекарственными средствами или дополнительными другими терапевтическими мерами. 9 н. и 43 з.п. ф-лы, 6 табл., 128 пр.
Формула

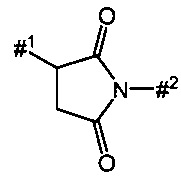














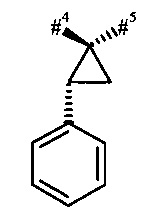




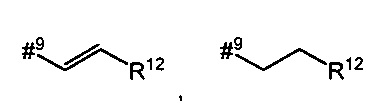











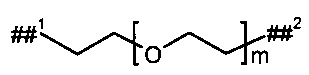

















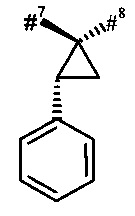



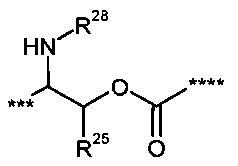
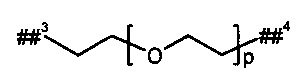


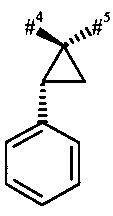





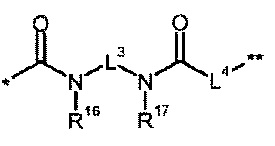



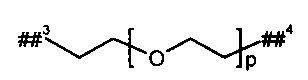

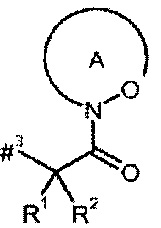





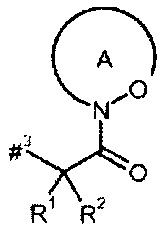



























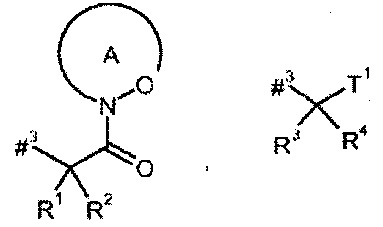










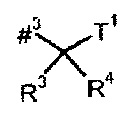

















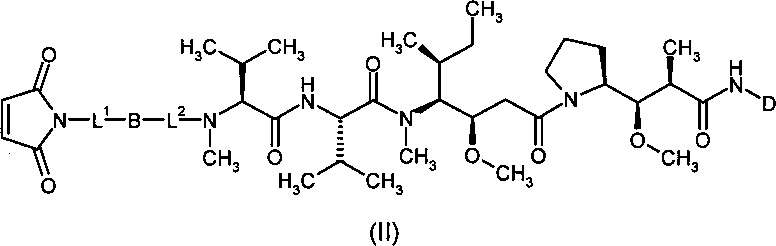





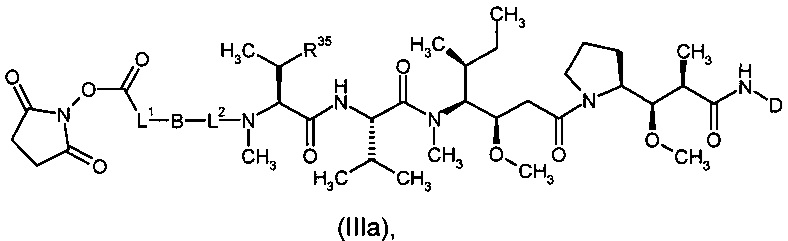

Документы, цитированные в отчёте о поиске
Антитела и иммуноконъюгаты и их применения