Новые пиррольные ингибиторы s-нитрозоглутатионредуктазы в качестве терапевтических агентов - RU2500668C2
Код документа: RU2500668C2
Описание
Заявки-аналоги
Данная заявка является продолжением заявки US 61/089,313, поданной 15.08.2008, и заявки US 61/116,982, поданной 21.11.2008. Каждая из этих заявок включена полностью в качестве ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым пиррольным ингибиторам S-нитрозоглутатионредуктазы, фармацевтическим композициям, содержащим такие ингибиторы, и способам их получения и применения.
Уровень техники
Химическое соединение оксид азота представляет собой газ химической формулы NO. NO является одной из нескольких газообразных сигнальных молекул, известных в биологических системах, и играет важную роль при контроле различных биологических процессов. Например, эндотелий использует NO для сигнала окружающих гладких мышц в стенках артериол расслабляться, что приводит к вазодилатации и повышению потока крови в гипоксические ткани. NO также участвует в регулировании пролиферации гладких мышц, функции тромбоцитов, нейротрансмиссии, и играет роль при защите хозяина. Хотя оксид азота является высокореакционным и имеет время жизни несколько секунд, он может проходить свободно через мембраны и связываться со многими молекулярными мишенями. Эти свойства делают NO идеальной сигнальной молекулой, способной контролировать биологические процессы между соседними клетками и в клетках.
NO представляет собой газ свободного радикала, который делает его реакционным и нестабильным, таким образом NO недолго существует in vivo, имея время полураспада 3-5 секунд в физиологических условиях. В присутствии кислорода NO может объединяться с тиолами с получением биологически важного класса стабильных NO аддуктов, называемых S-нитрозотиолами (SNO). Этот стабильный резерв NO, как было установлено, выступает в качестве источника биоактивного NO, и как таковой оказывается очень важным для здоровья и заболевания, обеспечивая центральное место NO в клеточном гомеостазе (статья Stamler и др., Proc. Natl. Acad. Sci. USA, 1992, 89, cc.7674-7677). Белок SNO играет важные роли в функциях сердечнососудистой, дыхательной, метаболической, желудочно-кишечной, иммунной и центральной нервной систем (книга Foster и др., 2003, Trends in Molecular Medicine, Т.9, Выпуск 4, апрель 2003, cc.160-168). Одним из наиболее исследуемых SNO в биологических системах является 5-нитрозоглутатион (GSNO) (статья Gaston и др., Proc. Natl. Acad. Sci. USA, 1993, 90, cc.10957-10961), выявленный ключевой регулятор пути передачи сигнала NO, поскольку он является эффективным транс-нитрозирующим агентом, и как оказалось, поддерживает равновесие с другими S-нитрозирующими белками (статья Liu и др., 2001) в клетках. Учитывая это граничное положение в NO-SNO континууме, GSNO обеспечивает терапевтически ценную мишень для рассмотрения, когда модулирование NO является фармакологически оправданным.
В свете понимания того, что GSNO является ключевым регулятором NO гомеостаза и клеточных уровней SNO, исследования были сфокусированы на изучении эндогенного продуцирования белков GSNO и SNO, которые встречаются вниз по ходу относительно продуцирования NO радикала ферментами синтетазы оксида азота (NOS). Недавно стал более понятен ферментативный катаболизм GSNO, который играет важную роль в управлении доступными концентрациями GSNO, и следовательно, доступными NO и SNO.
В центре этого исследования катаболизма GSNO исследователи недавно идентифицировали высоко консервативную 5-нитрозоглутатионредуктазу (GSNOR) (статьи Jensen и др., Biochem J., 1998, 331, cc.659-668; Liu и др., Nature, 2001, 410, cc.490-494). GSNOR также известна как глутатион-зависимая формальдегидная дегидрогеназа (GS-FDH), алкогольдегидрогеназа 3 (ADH-3) (книга Uotila и Koivusalo, Coenzymes and Cofactors., под ред. D. Dolphin., cc.517-551 (New York, John Wiley & Sons, 1989)), и алкогольдегидрогеназа 5 (ADH-5). Важно, что GSNOR показывает большую активность в отношении GSNO по сравнению с другими субстратами (статьи Jensen и др., 1998; Liu и др., 2001), и как оказалось, опосредует важную белковую и пептидную денитрозирующую активность в бактериях, растениях и животных. GSNOR, как оказалось, является основным GSNO-метаболизирующим ферментом у эукариот (статья Liu и др., 2001). Так, GSNO может накапливаться в биологических составляющих, в которых активность GSNOR является низкой или отсутствует (например, жидкость дыхательных путей) (статья Gaston и др., 1993).
Дрожжи с недостатком GSNOR аккумулируют S-нитрозилированные белки, которые не являются субстратами фермента, что явно предполагает, что GSNO существует в равновесии с SNO-белками (статья Liu и др., 2001). Точный ферментативный контроль окружающих уровней GSNO, и таки образом SNO-белков, повышает возможность того, что GSNO/GSNOR могут играть роли в совокупности с физиологическими и патологическими функциями, включая защиту от нитрозативного стресса, где NO продуцируется в избытке от физиологических уровней. Действительно, GSNO в особенности участвует в физиологических процессах от пищеварения до дыхания (статья Lipton и др., Nature, 2001, 413, cc.171-174), при регулировании трансмембранного регулятора кистозного фиброза (статья Zaman и др., Biochem Biophys Res Commun, 2001, 284, cc.65-70, при регулировании сосудистого тонуса, тромбоза и функции тромбоцитов (статьи de Belder и др., Cardiovasc Res., 1994, May; 28(5), cc.691-4. (1994); Z. Kaposzta, А и др.. Circulation; 2002, 106(24), cc.3057-3062), а также для защитных сил организма (статья de Jesus-Berrios и др., Curr. Biol., 2003, 13, cc.1963-1968). Другие исследования показали, что GSNOR защищает дрожжевые клетки от нитрозативного стресса как in vitro (статья Liu и др., 2001), так и in vivo (статья de Jesus-Berrios и др., 2003).
Общие данные предполагают, что GSNOR является первичным физиологическим лигандом для фермента S-нитрозоглутатионредуктазы (GSNOR), который катаболизирует GSNO, и следовательно снижает доступные SNO и NO в биологических системах (статья Liu и др., 2001), (статья Liu и др., Cell, 2004, 116(4), cc.617-628) и (статья Que и др., Science, 2005, 308, (5728), cc.1618-1621). Как таковой этот фермент играет центральную роль в регулировании локального и системного биоактивного NO. Поскольку нарушение биодоступности NO связано с патогенезом многочисленных заболеваний, включая гипертензию, атеросклероз, тромбоз, астму, желудочно-кишечные нарушения, воспаление и рак, агенты, которые регулируют активность GSNOR, являются кандидатами терапевтических агентов для лечения заболеваний, связанных с нарушением баланса оксида азота.
В настоящее время существует большая потребность в диагностике, профилактике, облегчении и лечении медицинских состояний, связанных с повышенным синтезом NO и/или повышенной биодоступностью NO. Кроме того, существует существенная потребность в новых соединениях, композициях и способах профилактики, облегчения или излечения других связанных с NO нарушений. Настоящее изобретение удовлетворяет эти потребности.
Сущность изобретения
Настоящее изобретение относится к новым пиррольным соединениям, полезным в качестве ингибиторов S-нитрозоглутатионредуктазы ("GSNOR"). Настоящее изобретение включает фармацевтически приемлемые соли, пролекарства и метаболиты описанных ингибиторов GSNOR. В изобретение также включены фармацевтические композиции, содержащие по крайней мере один ингибитор GSNOR и по крайней мере один фармацевтически приемлемый носитель.
Композиции настоящего изобретения могут быть получены в любой подходящей фармацевтически приемлемой форме дозировки.
Настоящее изобретение относится к способу ингибирования S-нитрозоглутатионредуктазы у субъекта, нуждающегося в этом. Такой способ включает введение фармацевтической композиции, содержащей по крайней мере один ингибитор GSNOR или его фармацевтически приемлемую соль, его пролекарство или метаболит в терапевтически эффективном количестве, в комбинации по крайней мере с одним фармацевтически приемлемым носителем. Ингибитором GSNOR может быть новое соединение по изобретению, или им может быть известное соединение, которое ранее не было известно как ингибитор GSNOR.
Настоящее изобретение также относится к способу лечения нарушения, излечиваемого NO-донорной терапией у субъекта, нуждающегося в этом. Такой способ включает введение фармацевтической композиции, содержащей по крайней мере один ингибитор GSNOR или его фармацевтически приемлемую соль, его пролекарство или метаболит в терапевтически эффективном количестве, в комбинации по крайней мере с одним фармацевтически приемлемым носителем. Ингибитором GSNOR может быть новое соединение по изобретению, или им может быть известное соединение, которое ранее не было известно как ингибитор GSNOR.
Настоящее изобретение также относится к способу лечения клеточного пролиферативного нарушения у субъекта, нуждающегося в этом. Такой способ включает введение фармацевтической композиции, содержащей по крайней мере один ингибитор GSNOR или его фармацевтически приемлемую соль, его пролекарство или метаболит в терапевтически эффективном количестве, в комбинации по крайней мере с одним фармацевтически приемлемым носителем. Ингибитором GSNOR может быть новое соединение по изобретению, или им может быть известное соединение, которое ранее не было известно как ингибитор GSNO.
Способы по изобретению включают введение с одним или несколькими вторичными активными агентами. Такое введение может быть последовательным или в комбинированной композиции.
Хотя способы и материалы, аналогичные или эквивалентные описанным здесь способам и материалам, могут использоваться для осуществления или тестирования настоящего изобретения, подходящие способы и материалы описаны ниже. Все указанные здесь публично доступные публикации, заявки на патенты, патенты и другие ссылки включены полностью в качестве ссылки. В случае конфликта будет преобладать настоящее описание, включая определения.
Вышеизложенная сущность изобретения и следующее подробное описание являются примерными и поясняющими, и предназначены для обеспечения других подробностей заявленных композиций и способов. Другие объекты, преимущества и новые особенности станут понятны специалисту в данной области техники из вышеизложенного подробного описания.
Подробное описание предпочтительных вариантов осуществления
А. Обзор изобретения
До последнего времени было известно, что 5-нитрозоглутатионредуктаза (GSNOR) окисляет формальдегидный глутатионовый аддукт, S-гидроксиметилглутатион. Поэтому GSNOR обнаружен в различных бактериях, дрожжах, растениях и животных, и является высоко консервативным. Белки из Е. coli, S. cerevisiae и макрофагов мыши показывают идентичность аминокислотной последовательности более 60%. Активность GSNOR (то есть разрушение S-нитрозоглутатиона, когда NADH присутствует как необходимый кофактор) определена в Е. coli, в макрофагах мыши, в эндотелиальных клетках мыши, в клетках гладкой мускулатуры мыши, в дрожжах и в HeLa человека, эпителиальных и моноцитных клетках. Информация о нуклеотиде GSNOR человека и аминокислотной последовательности может быть получена из базы данных National Center for Biotechnology Information (NCBI) под номером M29872, NM_000671. Информация о нуклеотиде GSNOR мыши и аминокислотной последовательности может быть получена из базы данных NCBI под номером NM_007410. В нуклеотидной последовательности стартовый участок и концевой участок подчеркнуты. CDS разработал кодирующую последовательность. SNP разработал единичный нуклеотидный полиморфизм. Другие родственные GSNOR нуклеотидные и аминокислотные последовательности, включая нуклеотидные и аминокислотные последовательности других видов, могут быть обнаружены в заявке на патент US 2005/0014697.
В соответствии с настоящим изобретением было показано, что GSNOR функционирует in vivo и in vitro для метаболизма S-нитрозоглутатиона (GSNO) и белков S-нитрозотиолов (SNO) для модулирования биоактивности NO, путем контроля внутриклеточных уровней низкомолекулярных NO-донорных соединений и предотвращения достижения белка нитрозилирования токсичных уровней.
На этом основании следует, что ингибирование этого фермента потенциирует биодоступность при всех болезнях, при которых показана NO-донорная терапия, ингибирует пролиферацию патологически пролиферирующих клеток и повышает биодоступность NO при заболеваниях, при которых это выгодно.
Настоящее изобретение относится к фармацевтическим агентам, которые являются мощными ингибиторами GSNOR. В частности, описаны замещенные пиррольные аналоги, которые являются ингибиторами GSNOR структуры, приведенной ниже (формулы I и II), или их фармацевтически приемлемые соли, стереоизомеры или пролекарства.


Тризамещенные пиррольные аналоги являются мощными ингибиторами GSNOR. Как используется в данном контексте, термин "аналог" относится к соединению, имеющему сходную химическую структуру или функцию с соединениями формулы I-II, которые содержат пиррольное кольцо.
Некоторые пиррольные аналоги по изобретению также могут существовать в различных изомерных формах, включая конфигурационные, геометрические и конформационные изомеры, а также существуя в различных таутомерных формах, особенно отличающихся по точке присоединения атома водорода. Как здесь используется, термин "изомер" предназначен для включения всех изомерных форм соединения, включая таутомерные формы соединения.
Иллюстрирующие соединения, имеющие асимметричные центры, могут существовать в различных энантиомерных и диастереомерных формах. Соединение может существовать в форме оптического изомера или диастереомера. Соответственно, настоящее изобретение включает соединения в формах их оптических изомеров, диастереомеров и смесей, включая рацемические смеси.
Следует отметить, что если существует различие между приведенной структурой и названием, данной этой структуре, преимущество имеет приведенная структура. Кроме того, если стереохимия структуры или часть структуры не показана, например, с помощью жирных, клиноподобных или штриховых линий, структура или часть структуры должна рассматриваться как включающая все стереоизомеры описанного соединения.
В соответствии с изобретением, уровни S-нитрозоглутатионредуктазы в биологическом образце могут определяться способами, описанными в заявке на патент US 2005/0014697. Термин "биологический образец" включает, но не ограничивается ими, образцы крови (например, сыворотка, плазма или цельная кровь), мочу, слюну, пот, грудное молока, секрет вагинальной полости, сперму, волосяные фолликулы, кожу, зубы, кости, ногти или другие секреты, жидкости организма, ткани или клетки.
В. Определения
Как здесь используется, термин "около" является понятным специалистам в данной области техники и варьируется в некоторой степени в зависимости от содержания, в котором используется. Если встречаются применения термина, которые не понятны специалисту в данной области техники в используемом контексте, "около" обозначает плюс или минус 10% от конкретного термина.
Термин "ацил" включает соединения и группы, которые содержат ацетильный радикал (СН3СО-) или карбонильную группу, к которой присоединен линейный или разветвленный низший алкильный остаток.
Термин "алкил" как здесь используется обозначает линейный или разветвленный насыщенный углеводород с указанным количеством атомов углерода. Например, (С1-С6)алкил включает, но не ограничивается ими, метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, гексил, изогексил и неогексил. Алкильная группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как здесь описано.
Термин "алкенил" как здесь используется обозначает линейный или разветвленный ненасыщенный углеводород с указанным количеством атомов водорода и по крайней мере одну двойную связь. Примеры (C2-C8) алкенильной группы включают, но не ограничиваются ими, этилен, пропилен, 1-бутилен, 2-бутилен, изобутилен, втор-бутилен, 1-пентен, 2-пентен, изопентен, 1-гексен, 2-гексен, 3-гексен, изогексен, 1-гептен, 2-гептен, 3-гептен, изогептен, 1-октен, 2-октен, 3-октен, 4-октен и изооктен. Алкенильная группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как здесь описано.
Термин "алкинил" как здесь используется обозначает линейный или разветвленный ненасыщенный углеводород с указанным количеством атомов водорода и по крайней мере одну тройную связь. Примеры (C2-C8) алкинильныой группы включают, но не ограничиваются ими, ацетилен, Пронин, 1-бутин, 2-бутин, 1-пентин, 2-пентин, 1-гексин, 2-гексин, 3-гексин, 1-гептин, 2-гептин, 3-гептин, 1-октин, 2-октин, 3-октин и 4-октин. Алкинильная группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как здесь описано.
Термин "алкоксигруппа" как здесь используется обозначает -O-алкильную группу, содержащую указанное количество атомов углерода. Например, (С1-С6)алкоксигруппа включает -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-втор-бутил, -O-трет-бутил, -O-пентил, -O-изопентил, -O-неопентил, -O-гексил, -O-изогексил и -O-неогексил.
Термин "аминоалкил" как здесь используется обозначает алкильную группу (обычно содержащую от одного до шести атомов углерода), где один или несколько атомов водорода С1-С6 алкильной группы замещены амином формулы -N(Rc)2, где в каждом случае Rc независимо представляет собой -Н или (C1-С6)алкил. Примеры аминоалкильных групп включают, но не ограничиваются ими, -CH2NH2, -CH2CH2NH2-, -CH2CH2CH2NH2, -CH2CH2CH2CH2NH2, -CH2CH2CH2CH2CH2NH2, -CH2CH2CH2CH2CH2CH2NH2, -CH2CH2CH2N(CH3)2, трет-бутиламинометил, изопропиламинометил и им подобные.
Термин "арил" как здесь используется обозначает 5-14-членную моноциклическую, бициклическую или трициклическую ароматическую кольцевую систему. Примеры арильной группы включают фенил и нафтил. Арильная группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как здесь описано. Примеры арильных групп включают фенильные или арильные гетероциклы, такие как, пиррол, фуран, тиофен, тиазол, изотиазол, имидазол, триазол, тетразол, пиразол, оксазол, изоксазол, пиридин, пиразин, пиридазин и пиримидин, и им подобные.
Как здесь используется, термин "биоактивность" показывает действие на один или несколько клеточных или внеклеточных процессов (например, путем связывания, передачи сигнала и т.д.), которое может влиять на физиологические или патофизиологические процессы.
Термин "карбонил" или "карбоксигруппа" или "карбоксил" включает соединения и группы, которые содержат атом углерода, связанный двойной связью с атомом кислорода. Примеры групп, содержащих карбонил, включают, но не ограничиваются ими, альдегиды, кетоны, карбоновые кислоты, амиды, сложные эфиры, ангидриды и т.д.
Термин "Cm-Cn" обозначает количество атомов углерода "m" до количества атомов углерода "n". Например, термин "C1-С6" обозначает от одного до шести атомов углерода (C1, C2, С3, С4, C5 или C6). Термин "С2-С6" включает от двух до шести атомов углерода (С2, С3, C4, C5 или С6). Термин "С3-С6" включает от трех до шести атомов углерода (С3, С4, C5 или С5).
Термин "циклоалкил" как здесь используется обозначает 3-14-членную насыщенную или ненасыщенную неароматическую моноциклическую, бициклическую или трициклическую углеводородную кольцевую систему. В данные класс включены циклоалкильные группы, которые конденсированы с бензольным кольцом. Примеры циклоалкильных групп включают, но не ограничиваются ими, циклопропил, циклобутил, циклобутенил, циклопентил, циклопентенил, циклопентадиенил, циклогексил, циклогексенил, 1,3-циклогексадиенил, циклогептил, циклогептенил, 1,3-циклогептадиенил, 1,4-циклогептадиенил, -1,3,5-циклогептатриенил, циклооктил, циклооктенил, 1,3-циклооктадиенил, 1,4-циклооктадиенил, -1,3,5-циклооктатриенил, декагидронафтален, октагидронафтален, гексагидронафтален, октагидроинден, гексагидроинден, тетрагидроинден, декагидробензоциклогептен, октагидробензоциклогептен, гексагидробензоциклогептен, тетрагидробензоциклогептен, додекагидрогептален, декагидрогептален, октагидрогептален, гексагидрогептален и тетрагидрогептален, (1s,3s)-бицикло[1.1.0]бутан, бицикло[1.1.1]пентан, бицикло[2.1.1]гексан, бицикло[2.2.1]гептан, бицикло[2.2.2]октан, бицикло[3.1.1]гептан, бицикло[3.2.1]октан, бицикло[3.3.1]нонан, бицикло[3.3.2]декан, бицикло[3.3]ундекан, бицикло[4.2.2]декан, бицикло[4.3.1]декан. Циклоалкильная группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как здесь описано.
Термин "галоген" включает фтор, бром, хлор, йод и т.д.
Термин "галогеналкил" как здесь используется обозначает C1-С6 алкильную группу, в которой один или несколько атомов водорода C1-С6 алкильной группы замещены атомами галогена, которые могут быть одинаковыми или различными. Примеры галогеналкильных группы включают, но не ограничиваются ими, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил, пентахлорэтил и 1,1,1-трифтор-2-бром-2-хлорэтил.
Термин "гетероалкил" отдельно или в комбинации с другим термином, обозначает, если не укзано иное, стабильный линейный или разветвленный алкил или их комбинации, состоящий из атомов углерода и из 1-3 гетероатомов, выбранных из группы, состоящей из О, N и S, и где атомы азота и серы необязательно могут быть окислены, и гетероатом азота необязательно может быть кватернизирован. Гетероатом (гетероатомы) О, N и S может быть помещен в любое положение гетероалкильной группы. Примеры включают -CH2-CH2-O-СН3, -СН2-СН2-NH-СН3, -СН2-СН2-N(СН3)-СН3, -СН2-S-СН2-СН3, -СН2-СН2-S(O)-СН3, -СН2-СН2-S(O)2-СН3 и -СН2-СН=N-ОСН3. Вплоть до двух гетероатомов могут располагаться последовательно, например, -СН2-NH-ОСН3. Когда приставка, такая как (C2-C8), используется для обозначения гетероалкильной группы, количество атомов углерода (от 2 до 8 в данном примере) включает также гетероатомы. Например, С2-гетероалкильная группа включает, например, -СН2ОН (один атом углерода и один гетероатом, замещающий атом углерода) и -CH2SH.
Для дальнейшей иллюстрации определения гетероалкильной группы, где гетероатом представляет собой кислород, гетероалкильная группа может быть оксиалкильной группой. Например, (С2-С5) оксиалкил включает, например, -СН2-O-СН3 (С3-оксиалкильную группу с двумя атомами углерода и с одним атомом кислорода вместо атома углерода), -СН2СН2СН2СН2ОН, -ОСН2СН2ОСН2СН2ОН, -ОСН2СН(ОН)CH2OH и им подобные.
Термин "гетероарил" как здесь используется обозначает ароматическое гетероциклическое кольцо, содержащее от 5 до 14 членов и по крайней мере один гетероатом, выбранный из азота, кисолорода и серы, и содержащий по крайней мере 1 атом углерода, включающий моноциклические, бициклические и трициклические кольцевые системы. Некоторыми гетероарилами являются триазолил, тетразолил, оксадиазолил, пиридил, фурил, бензофуранил, тиенил (тиофенил), бензотиенил, хинолинил, пирролил, индолил, оксазолил, бензоксазолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изоксазолил, пиразолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил, хиназолинил, пиримидил, азепинил, оксепинил, хиноксалинил и оксазолил. Гетероарильная группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как здесь описано.
Как здесь используется, термин "гетероатом" включает кислород (О), азот (N) и серу (S).
Как здесь используется, термин "гетероцикл" обозначает 3-14-членные кольцевые системы, которые являются насыщенными, ненасыщенными или ароматическими, и которые содержат от 1 до 4 гетероатомов, независимо выбранных из азота, кислорода и серы, и где гетероатомы азота и серы необязательно могут быть окислены, и гетероатом азота может быть необязательно кватернизирован, включая моноциклические, бициклические и трициклические кольцевые системы. Бициклические и трициклические кольцевые системы могут включать гетероцикл или гетероарил, конденсированный с бензольным кольцом. Гетероцикл может быть присоединен через любой гетероатом или атом углерода, химически приемлемый. Гетероциклы включают гетероарилы как определено выше. Некоторые примеры гетероциклов включают, но не ограничиваются ими, азиридинил, оксиранил, трииранил, триазолил, тетразолил, азиринил, диазиридинил, диазиринил, оксазиридинил, азетидинил, азетидинонил, оксетанил, тиетанил, пиперидинил, пиперазинил, морфолинол, пирролил, оксазинил, тиазинил, диазинил, диоксанил, триазинил, тетразинил, имидазолил, тетразолил, пирролидинил, изоксазолил, фуранил, фуразанил, пиридинил, оксазолил, бензоксазолил, бензизоксазолил, тиазолил, бензтиазолил, тиенил, пиразолил, триазолил, пиримидинил, бензимидазолил, изоиндолил, индазолил, бензодиазолил, бензотриазолил, бензоксазолил, бензизоксазолил, пуринил, индолил, изохинолинил, хинолинил и хиназолинил. Гетероциклическая группа может быть незамещенной или необязательно замещенной одним или несколькими заместителями, как здесь описано.
Термин "гетероциклоалкил" отдельно или в комбинации с другими терминами представляет собой, если не указано иное, циклические версии "гетероалкила". Дополнительно, гетероатом может занимать положение, в котором гетероцикл присоединен к остатку молекулы. Примеры гетероциклоалкила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиен-2-ил, тетрагидротиен-3-ил, 1-пиперазинил, 2-пиперазинил и им подобные.
Термин "гидроксиалкил" как здесь используется обозначает алкильную группу, содержащую указанное количество атомов углерода, где один или несколько атомов водорода в алкильной группе замещены -ОН группой. Примеры гидроксиалкильных групп включают, но не ограничиваются ими, -CH2OH, -CH2CH2OH, -CH2CH2CH2OH, -CH2CH2CH2CH2OH, -CH2CH2CH2CH2CH2OH, -CH2CH2CH2CH2CH2CH2OH и их разветвленные версии.
Термин "гидроксигруппа" или "гидроксил" включает группы с -ОН или -О-.
Как здесь используется и если не указано иное, термин "стереоизомер" обозначает один стереоизомер соединения, который по существу свободен от других стереоизомеров этого соединения. Например, стереомерно чистое соединение с одним хиральным центром по существу свободно от противоположного энантиомера соединения. Стереомерно чистое соединение с двумя хиральными центрами по существу свободно от других диастереомеров соединения. В некоторых вариантах осуществления, стереомерно чистое соединение включает более чем около 80% по весу одного стереоизомера соединения и менее чем около 20% по весу других стереоизомеров соединения, например, более чем около 90% по весу одного стереоизомера соединения и менее чем около 10% по весу других стереоизомеров соединения, или более чем около 95% по весу одного стереоизомера соединения и менее чем около 5% по весу других стереоизомеров соединения, или более чем около 97% по весу одного стереоизомера соединения и менее чем около 3% по весу других стереоизомеров соединения.
Как здесь используется, "белок" используется в качестве синонима "пептиду", "полипептиду" или "пептидному фрагменту". "Очищенный" полипептид, белок, пептид или пептидный фрагмент по существу свободен от клеточного материала или других загрязняющих белков клетки, ткани или бесклеточного источника, из которого получена аминокислотная последовательность, или по существу свободен от химических предшественников или других химических веществ при химическом синтезе.
Как здесь используется, "модулировать" обозначает повышать или понижать уровни пептида или полипептида, или повышать или понижать стабильность или активность пептида или полипептида. Термин "ингибировать" обозначает понижать уровни пептида или полипептида или понижать стабильность или активность пептида или полипептида. В предпочтительных вариантах осуществления, модулируемый или ингибируемый пептид представляет собой S-нитрозоглутатион (GSNO) или протеин S-нитрозотиолы (SNO).
Как здесь используется, термины "оксид азота" и "NO" обозначает незаряженный оксид азота и заряженные виды оксида азота, особенно включающие ион нитрозония (NO+) и ион нитроксила (NO-). Реакционная форма оксида азота может быть представлена газообразным оксидом азота. Соединения структуры X-NOy, где Х представляет собой группу, высвобождающую, доставляющую или переносящую оксид азота, включая любое и все такие соединения, которые доставляют оксид азота в их участок предназначенного действия в форме, активной для этой предназначенной цели, и Y имеет значение 1 или 2.
Как здесь используется, термин "фармацевтически приемлемый" обозначает одобренный регулирующим органом федерального или государственного управления или представленный в U.S. Pharmacopoeia или других общепризнанных фармакопеях для применения для животных и, более конкретно, для людей. Термин "носитель" обозначает разбавитель, адъювант, эксципиент или связующее, которое пригодно для терапевтического введения, и включает, но не ограничивается ими, такие стерильные жидкости, как вода и масла.
"Фармацевтически приемлемая соль" или "соль" ингибитора GSNOR представляет собой продукт описанного соединения, который содержит ионную связь, и обычно получается реакцией описанного соединения либо с кислотой, либо с основанием, подходящим для введения субъекту. Фармацевтически приемлемая соль может включать, но не ограничивается ими, кислотные аддитивные соли, включающие гидрохлориды, гидробромиды, фосфаты, сульфаты, гидросульфаты, алкилсульфонаты, арилсульфонаты, арилалкилсульфонаты, ацетаты, бензоаты, цитраны, малеаты, фумараты, сукцинаты, лактаты и тартраты; катионы щелочного металла, такие как соли с Li, Na, K, щелочноземельным металлом, таким как Mg или Са, или соли органического амина.
"Фармацевтическая композиция" представляет собой состав, включающий описанные соединения в форме, подходящей для введения субъекту. Фармацевтическая композиция по изобретению предпочтительно составляется в совместимости с предназначенным способом введения. Примеры способов введения включают, но не ограничиваются ими, пероральное и парентеральное, например, внутривенное, интрадермальное, подкожное, ингаляционное, местное, трансдермальное, трансмукозальное и ректальное введение.
Термин "замещенный" как здесь используется обозначает, что любой один или несколько атомов водорода при обозначенном атоме замещены выбранной указанной группой, при условии, что обычная валентность обозначенного атома не превышается, и что замещение приводит к образованию стабильного соединения. Когда заместителем является кетогруппа (то есть =O), тогда 2 атома водорода при атоме замещены. Кольцевые двойные связи, как здесь используется, являются двойными связями, которые образуются между двумя соседними кольцевыми атомами (например, С=С, C=N или N=N).
Заместители для групп, обозначенных как алкил, гетероалкил, алкилен, алкенил, алкинил, циклоалкил, гетероциклоалкил, циклоалкенил и гетероциклоалкенил, могут быть выбраны из различных групп, включающих -ORd′, =O, =NRd′, =N-ORd′, -NRd′Rd′′, -SRd′, -галоген, -SiRd′Rd′′Rd′′′, -ОС(O)Rd′, -С(O)Rd′, -CO2Rd′, -CONRd′Rd′′, -OC(O)NRd′Rd′′, -NRd′′C(O)Rd′, -NRd′′′C(O)NRd′Rd′′, -NRd′′′SO2NRd′Rd′′′, -NRd′′CO2Rd′, -NHC(NH2)=NH, -NRd′C(NH2)=NH, -NHC(NH2)=NRd′, -S(O)Rd′, -SO2Rd′, -SO2NRd′Rd′′′, -NRd′′SO2Rd', -CN и -NO2, в числовом значении от нуля до трех, примеры этих групп содержат ноль, один или два заместителя.
Rd′, Rd′′ и Rd′′′ каждый независимо обозначает водород, незамещенный (C1-C8)алкил, незамещенный гетеро(С1-С8)алкил, незамещенный арил и арил, замещенный 1-3 заместителями, выбранными из -галогена, незамещенного алкила, незамещенной алкоксигруппы, незамещенной тиоалкоксигруппы и незамещенного арил (С1-С4)алкила. Когда Rd′ и Rd′′ присоединены к одному атому азота, они могут быть объединены с атомом азота с получением 5-, 6- или 7-членного кольца. Например, -NRd′Rd′′ может представлять собой 1-пирролидинил или 4-морфолинил.
Обычно, алкильная или гетероалкильная группа содержат от 0 до 3 заместителей, примеры этих групп содержат два или более заместителей настоящего изобретения. Аалкильный или гетероалкильный радикал может быть незамещенным или монозамещенным. В некоторых вариантах осуществления, алкильный или гетероалкильный радикал является незамещенным.
Примеры заместителей для алкильных и гетероалкиьных радикалов включают, но не ограничиваются ими, -ORd′, =O, =NRd′, =N-ORd′, -NRd′Rd′′, -SRd′, -галоген, -SiRd′Rd′′Rd′′′, -ОС(O)Rd′, -С(O)Rd′, -CO2Rd′, -CONRd′Rd′′, -OC(O)NRd′Rd′′, -NRd′′C(O)Rd′, -NRd′′′C(O)NRd′Rd′′, -NRd′′′SO2NRd′Rd′′′, -NRd′′CO2Rd′, -NHC(NH2)=NH, -NRd′C(NH2)=NH, -NHC(NH2)=NRd′, -S(O)Rd′, -SO2Rd′, -SO2NRd′Rd′′′, -NRd′′SO2Rd′, -CN и -NO2, где Rd′, Rd′′, Rd′′′ являются такими, как описано выше. Обычные заместители могут быть выбраны из: -ORd′, =O, =NRd′Rd′′, -галогена, -ОС(O)Rd′, -CO2Rd′, -C(O)NRd′Rd′′, -OC(O)NRd′Rd′′, -NRd′′C(O)Rd′, -NRd′′CO2Rd′, -NRd′′′SO2NRd′Rd′′, -SO2Rd′, -SO2NRd′Rd′′, -NRd′′SO2Rd′, -CN и -NO2.
Аналогично, заместители для арильных и гетероарильных групп варьируются и выбраны из: -галогена, -ORe′, -OC(O)Re′, -NRe′Re′′, -SRe′, -Re′, -CN, -NO2, -CO2Re′, -C(O)NRe′Re′′, -C(O)Re′, -OC(O)NRe′Re′′, -NRe′′C(O)Re′, -NRe′′CO2Re′, -NRe′′′C(O)NRe′Re′′, -NRe′′′SO2NRe′Re′′, -NHC(NH2)=NH, -NRe′C(NH2)=NH, -NH-C(NH2)=NRe′, -S(O)Re′, -SO2Re′, -SO2NRe′Re′′, -NRe′′SO2Re′, -N3, -CH(Ph)2, перфторалкоксигруппы и перфтор(С1-С4)алкила, в числовом диапазоне от 0 до общего количества открытых валентностей в ароматической циклической системе.
Re′, Re′′ и Re′′′ независимо выбраны из водорода, незамещенного (C1-C8)алкила, незамещенного гетеро(C1-C8)алкила, незамещенного арила, незамещенного гетероарила, незамещенного арил(С1-С4)алкила и незамещенного арилокси(С1-С4)алкила. Обычно, арильная или гетероарильная группа содержит от 0 до 3 заместителей, примерами этих групп являются группы с двумя или более заместителями настоящего изобретения. В одном варианте осуществления по изобретению, арильная или гетероарильная группа является незамещенной или монозамещенной. В другом варианте осуществления, арильная или гетероарильная группа является незамещенной.
Два заместителя при соседних атомах арильного или гетероарильного кольца в арильной или гетероарильной группе, как здесь описано, необязательно могут быть замещены заместителем формулы -T-C(O)-(CH2)q-U-, где Т и U независимо представляют собой -NH-, -O-, -СН2- или простую связь, и q имеет значении от 0 до 2. Альтернативно, два заместителя при соседних атомах арильного или гетероарильного кольца необязательно могут быть замещены заместителем формулы -J-(CH2)r-K-, где J и К независимо представляют собой -СН2-, -O-, -NH-, -S-, -S(O)-, -S(O)2-, -S(O)2NRf′- или простую связь, и r имеет значение от 1 до 3. Одна из простых связей нового полученного таким образом кольца необязательно может быть замещена двойной связью. Альтернативно, два заместителя при соседних атомах арильного или гетероарильного кольца необязательно могут быть замещены заместителем формулы -(СН2)s-Х-(СН2)t, где s и t независимо имеют значение от 0 до 3, и Х представляет собой -O-, -NRf'-, -S-, -S(O)-, -S(O)2- или -S(O)2NRa'-. Заместитель Rf' в -NRf'- и -S(O)2NRf'- выбран из водорода или незамещенного (С1-С6) алкила.
"Стабильное соединение" и "стабильная структура" обозначают соединение, которое является достаточно прочным для выделения в полезной степени чистоты из реакционной смеси, и введения в эффективный терапевтический агент.
Как здесь используется, термин "терапевтически эффективное количество" обычно обозначает количество, необходимое для облегчения по крайней мере одного симптома предотвращаемого нарушения, снижения или излечивания, как здесь описано. Фраза "терапевтически эффективное количество", относящееся к ингибиторам GSNOR настоящего изобретения, обозначает дозировку ингибитора GSNOR, которая обеспечивает конкретный фармакологический отклик, для которого ингибитор GSNOR вводят в значительное количество субъектов, нуждающихся в таком лечении. Подразумевается, что терапевтически эффективное количество ингибитора GSNOR, которое вводят конкретному субъекту в конкретном варианте, не всегда эффективно для лечения описанных здесь состояний/заболеваний, даже если такая дозировка считается терапевтически эффективным количеством специалистом в данной области техники.
С. Ингибиторы S-нитрозоглутатионредуктазы
1. Соединения по изобретению
В одном из вариантов осуществления настоящее изобретение относится к соединению структурной формулы I или его фармацевтически приемлемой соли, стереоизомеру или пролекарству:
где:
Ar выбран из группы, состоящей из фенила и тиофенила;
R1 выбран из группы, состоящей из незамещенного имидазолила, замещенного имидазолила, хлора, брома, фтора, гидроксигруппы и метоксигруппы;
R2 выбран из группы, состоящей из водорода, метила, хлора, фтора, гидроксигруппы, метоксигруппы, этоксигруппы, пропоксигруппы, карбамоила, диметиламиногруппы, аминогруппы, формамидогруппы и трифторметила; и
Х выбран из группы, состоящей из СО и SO2.
В другом варианте осуществления изобретения, подходящие группы для R1 включают, но не ограничиваются ими, незамещенный имидазолил и замещенный имидазолил. Подходящие заместители для замещенной имидазолильной группы включают, но не ограничиваются ими, C1-C6алкил.
В другом варианте осуществления изобретения группы ArR1R2 включают, но не ограничиваются ими:



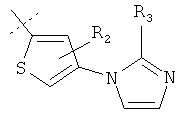

где R3 выбран из Н, метила и этила.
В другом варианте осуществления изобретения группы ArR1 включают, но не ограничиваются ими, 4-хлорфенил, 3-хлорфенил, 4-бромфенил, 3-бромфенил, 4-фторфенил, 3-фторфенил, 4-гидроксифенил, 4-метоксифенил, 3-метоксифенил, 2-метоксифенил, 4-хлортиофен-2-ил, 5-хлортиофен-2-ил, 3-бромтиофен-2-ил, 4-бромтиофен-2-ил, 5-бромтиофенил-2-ил и 5-бромтиофен-3-ил.
В одном из вариантов осуществления настоящего изобретения описано соединение структурной формулы II или его фармацевтически приемлемая соль, стереоизомер или пролекарство:
где:
Ar выбран из группы, состоящей из фенила и тиофенила;
R4 выбран из группы, состоящей из незамещенного имидазолила и замещенного имидазолила;
R5 выбран из группы, состоящей из водорода, фтора, гидроксигруппы и метоксигруппы;
R6 выбран из группы, состоящей из водорода, хлора, брома и фтора;
R7 выбран из группы, состоящей из водорода и метила; и
R8 выбран из группы, состоящей из CONH2, SO2NH2 и NHSO2CH3.
В другом варианте осуществления изобретения, подходящие группы для ArR4R5 включают, но не ограничиваются ими:


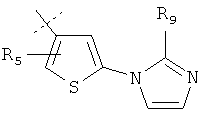


где R9 выбран из Н, метила и этила.
Когда связь с заместителем показана с пересечением связи, соединяющей два атома в кольце, тогда такой заместитель может быть связан с любым атомом в кольце. Когда заместитель показан без указания атома, с которым такой заместитель связан с остатком соединения указанной формулы, тогда такой заместитель может быть связан через любой атом в таком заместителе. Комбинации заместителей и/или переменных возможны, но только если такие комбинации приводят к образованию стабильных соединений.
Описанные здесь соединения могут иметь асимметричные центры. Соединения настоящего изобретения, содержащие асимметрично замещенный атом, могут быть выделены в оптически активных или рацемических формах. Из предшествующего уровня техники хорошо известно, как получить оптически активные формы, такие как расщепление рацемических форм или синтез из оптически активных исходных материалов. Многие геометрические изомеры олефинов, C=N двойных связей и им подобных также могут присутствовать в описанных здесь соединениях, и все такие стабильные изомеры входят в настоящее изобретение. Описаны цис- и транс- геометрические изомеры соединений настоящего изобретения, и они могут быть выделены в виде смеси изомеров или в виде отдельных изомерных форм. Все хиральные, диастереомерные, рацемические и геометрические изомерные формы структуры включены, если не указана конкретная стереохимия или изомерная форма. Все таутомеры показанных или описанных соединений также составляют часть настоящего изобретения.
Следует понимать, что изомеры вследствие асимметрии (например, все энантиомеры и диастереомеры) включены в объем изобретения, если не указано иное. Такие изомеры могут быть получены по существу в чистой форме классическими методиками разделения и стереохимически контролируемым синтезом. Кроме того, структуры и другие соединения и группы, описанные в настоящем описании, также включают все их таутомеры. Алкены могут включать Е- или Z-геометрию, если возможно.
2. Некоторые ингибиторы GSNOR
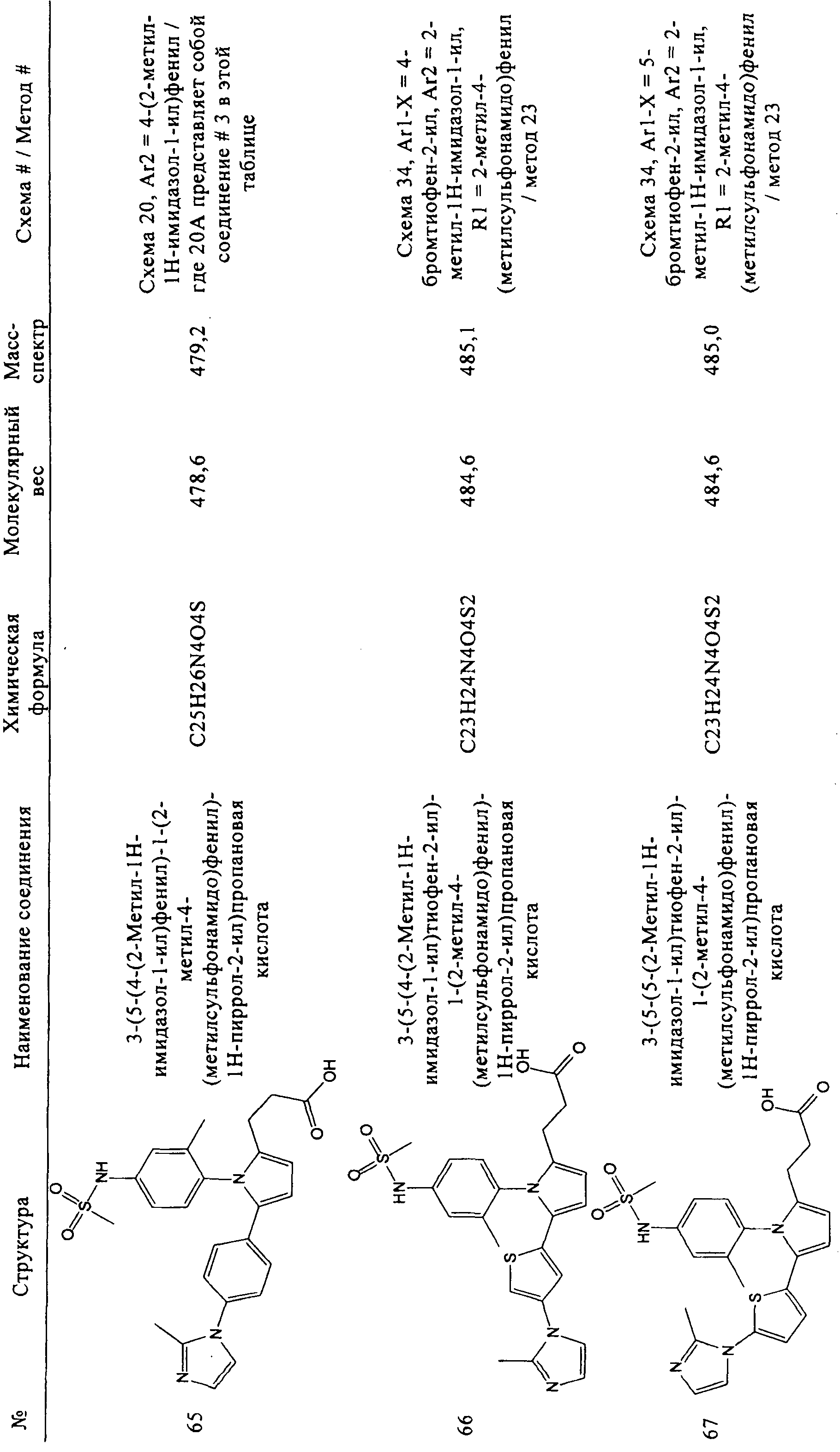
В таблице 1 ниже показаны некоторые новые пиррольные аналоги формулы I и формулы II, полезные в качестве ингибиторов GSNOR по изобретению. Методики синтеза, которые могут использоваться для получения каждого соединения, показаны более подробно ниже в таблице 1 (то есть схема 1, схема 2 и т.д.). В некоторых случаях, если исходный материал или промежуточное соединение схемы не является коммерчески доступным, тогда соответствующий метод описывает синтез этого исходного материала или промежуточного соединения (то есть метод 1, метод 2 и т.д.). В таблице 1 приведен номер схемы, определенные исходные материалы, показанные на схемах, и при необходимости, приведен номер метода, который соответствует подробному синтезу промежуточного соединения или исходного материала. Подтверждающие данные масс-спектрометрии для каждого соединения также включены в таблицу 1. Ингибирующая активность GSNOR определена с помощью анализа, описанного в примере 2, и получены значения IC50. Соединения-ингибиторы GSNOR 1-70 в таблице 1 имеют значения IC50 около <15 мкМ. Соединения-ингибиторы GSNOR 1-12, 14-15, 17-19, 22-36, 38-42, 44-56, 58-69 в таблице 1 имеют значения IC50 менее 1,0 мкМ.








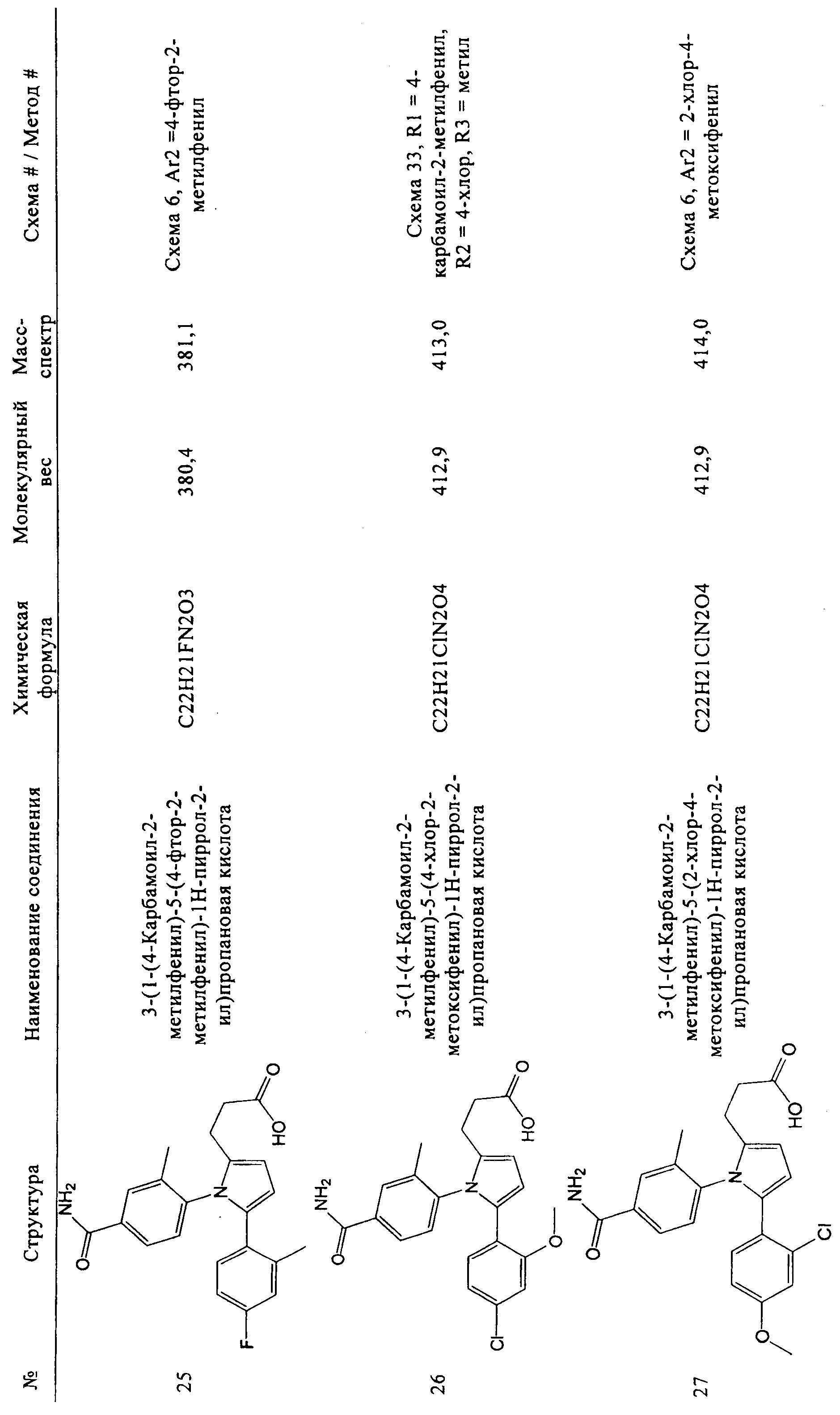






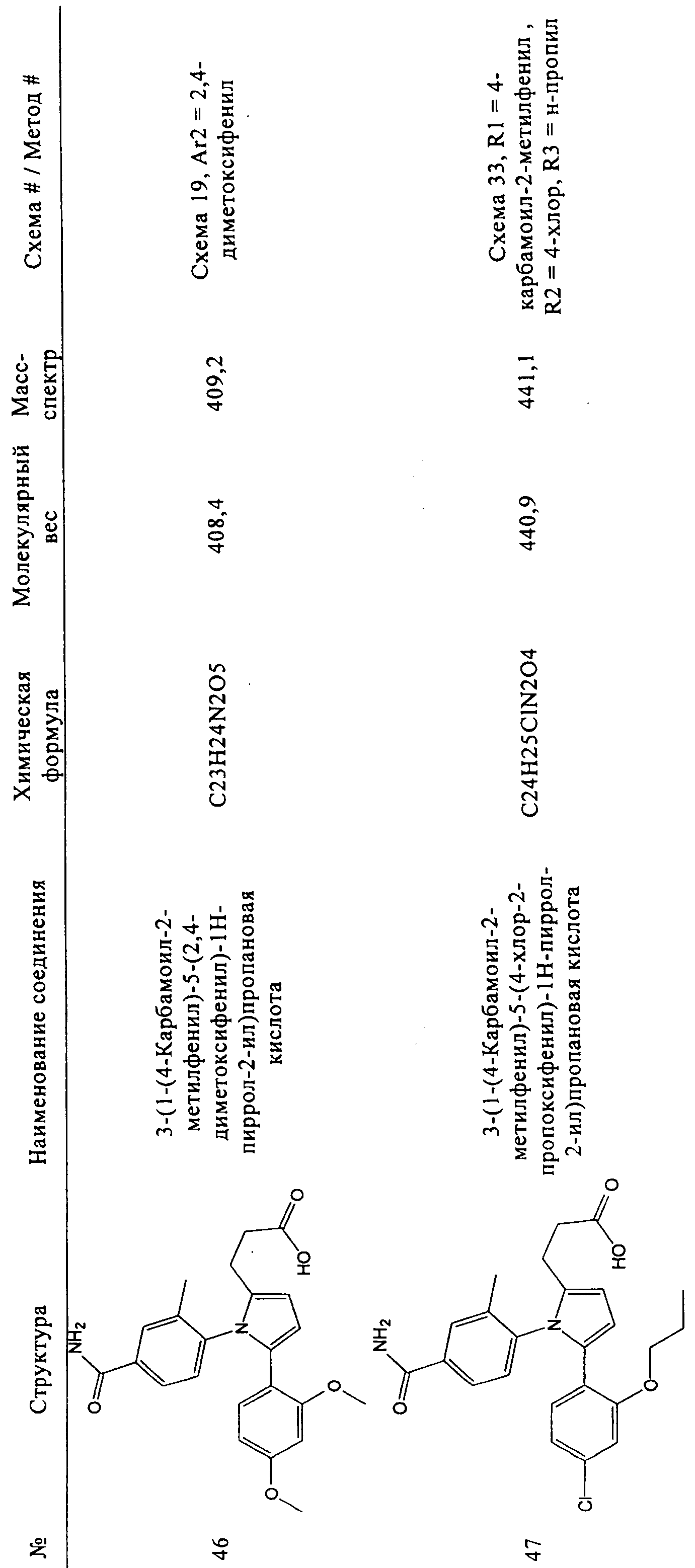








D. фармацевтические композиции, содержащие ингибитор GSNOR
Настоящее изобретение включает фармацевтические композиции, содержащие по крайней мере один описанный здесь ингибитор GSNOR и по крайней мере один фармацевтически приемлемый носитель. Подходящие носители описаны в книге "Remington: The Science and Practice, Twentieth Edition," опубликованной Lippincott Williams & Wilkins, которая включена в качестве ссылки. Фармацевтические композиции в соответствии с изобретением также могут включать один или несколько не-GSNOR ингибирующих активных агентов.
Фармацевтические композиции по изобретению могут включать новые описанные здесь ингибиторы GSNOR, фармацевтические композиции могут включать известные соединения, которые ранее не были известны как ингибиторы GSNOR, или их комбинации.
Ингибиторы GSNOR могут использоваться в любой фармацевтически приемлемой дозированной форме, включая, но не ограничиваясь ими, инъекционные дозированные формы, жидкие дисперсии, гели, аэрозоли, мази, кремы, лиофилизованные составы, сухие порошки, таблетки, капсулы, составы контролируемого высвобождения, быстро плавящиеся составы, составы с отсроченным высвобождением, составы длительного высвобождения, составы с пульсирующим высвобождением, составы смешанного немедленного высвобождения и контролируемого высвобождения и т.д. Конкретно, описанные здесь ингибиторы GSNOR могут быть составлены: (а) для введения, выбранного из группы, состоящей из перорального, легочного, внутривенного, внутриартериального, интратекального, внутриглазного, ректального, офтальмического, ободочного, парентерального, интрацистернального, внутривагинального, внутрибрюшинного, локального, буккального, назального и местного введения; (b) в дозированной форме, выбранной из группы, состоящей из жидких дисперсий, гелей, аэрозолей, мазей, кремов, таблеток, саше и капсул; (с) в дозированной форме, выбранной из группы, состоящей из лиофилизованных составов, сухих порошков, быстро плавящихся составов, составов с контролируемым высвобождением, составов с отсроченным высвобождением, составов длительного высвобождения, составов с пульсирующим высвобождением и составов смешанного немедленного высвобождения и контролируемого высвобождения.
Для дыхательных инфекций ингаляционный состав может использоваться для достижения высоких местных концентраций. Составы, подходящие для ингаляции, включают сухой порошок или аэрозольные или распыляемые растворы, дисперсии или суспензии, способные распределяться ингалятором или небулайзером в эндобронхиальную или назальную полость зараженных пациентов для лечения бактериальных инфекций верхних и нижних дыхательных путей.
Растворы или суспензии, используемые для парентерального, интрадермального или подкожного применения, могут включать один или несколько из следующих компонентов: (1) стерильный разбавитель, такой как вода для инъекций, соляный раствор, фиксированные масла, полиэтиленгликоли, глицерин, пропилен гликоль или другие синтетические растворители; (2) антибактриальные агенты, такие как бензиловый спирт или метилпарабены; (3) антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; (4) хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; (5) буферы, такие как ацетаты, цитраты или фосфаты; и (5) агенты для регулирования тоничности, такие как хлорид натрия или декстроза. Значение рН может регулироваться кислотами или основаниями, такими как хлористоводородная кислота или гидроксид натрия. Парентеральный состав может быть включен в ампулы, наполняемые шприцы или сосуды с несколькими дозами из стекла или пластика.
Фармацевтические композиции, подходящие для инъекционного применения, могут включать стерильные водные растворы (для водорастворимых) или дисперсии и стерильные порошки для получения перед применением стерильных инъекционных растворов или дисперсий. Для внутривенного введения подходящие носители включают физиологический соляной раствор, бактериостатическую воду, Cremophor EL (BASF, Parsippany, N.J.) или забуференный фосфатом соляной раствор (PBS). Во всех случаях композиция должна быть стерильной и должна быть жидкостью в той степени, чтобы ею можно было легко наполнить шприц. Фармацевтическая композиция должна быть стабильной в условиях изготовления и хранения, и должна защищаться от нежелательного действия микроорганизмов, таких как бактерии и грибки.
Носителем может быть растворитель или дисперсионная среда, включающая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль, и им подобные), и их подходящие смеси. Нужная текучесть может обеспечиваться, например, с помощью покрытий, таких как лецитин, обеспечением необходимого размера частиц для дисперсии и с помощью поверхностно-активных веществ. Предотвращение действия микроорганизмов может достигаться различными антибактериальными и противогрибковыми агентами, например, парабенами, хлорбутанолом, фенолом, аскорбиновой кислотой, тимеросалом и им подобными. Во многих случаях они предпочтительно включают изотонические агенты, например, сахара, полиспирты, такие как маннит или сорбит, и неорганические соли, такие как хлорид натрия, в композиции. Пролонгированная абсорбция инъекционных композиций может достигаться включением в композицию агенты, который замедляет абсорбцию, например, моностеарат алюминия и желатин.
Стерильные инъекционные растворы могут быть получены введением активного реагента (например, ингибитор GSNOR) в необходимом количестве в подходящем растворителе с одним или несколькими ингредиентами, перечисленными выше, при необходимости, с последующей фильтрационной стерилизацией. Обычно, дисперсии получают введением по крайней мере одного ингибитора GSNOR в стерильный носитель, который содержит основную дисперсионную среду и любые другие необходимые ингредиенты. Для стерильных порошков для получения стерильных инъекционных растворов, примерные методы получения включают вакуумную сушку и сушку с замораживанием, которые приводят к получению порошка ингибитора GSNOR плюс любой дополнительный нужный ингредиент из заранее стерильно-отфильтрованного раствора.
Пероральные композиции обычно включают инертный разбавитель или съедобный носитель. Они могут быть включены, например, в желатиновые капсулы или спрессовываться в таблетки. Для перорального терапевтического введения ингибитор GSNOR может быть составлен с эксципиентами и использоваться в форме таблеток, лепешек или капсул. Пероральные композиции также могут быть получены с помощью жидкого носителя для применения в качестве раствора для полоскания полости рта, где соединение в жидком носителе используется перорально и смывается и сплевывается или проглатывается. Фармацевтически совместимые связующие агенты и/или адъюванты могут быть включены в композицию.
Для введения ингаляцией соединения доставляют в форме аэрозольного спрея из контейнера под давлением или диспенсера, который содержит подходящий газ-вытеснитель, например, газ, такой как диоксид углерода, аэрозольной жидкости или сухого порошка из подходящего устройства. Для трансмукозального или трансдермального введения в составе используются вещества, способствующие проникновению, подходящие для проникновения через барьер. Такие вещества, способствующие проникновению, обычно известны из предшествующего уровня техники, и включают, например, для трансмукозального введения, детергенты, соли желчной кислоты и производные фусидовой кислоты. Трансмукозальное введение может осуществляться с помощью применения назальных спреев или суппозиториев. Для трансдермального введения активные реагенты составляют в мази, бальзамы, гели или кремы, известные из предшествующего уровня техники. Реагенты также могут быть получены в форме суппозиториев (например, с обычными основами для суппозиториев, такими как масло какао и другие глицериды) или удерживающих клизм для ректальной доставки.
В одном варианте осуществления, ингибиторы GSNOR получают с носителями, которые защищают от быстрого расщепления в организме. Например, может использоваться состав с контролируемым высвобождением, включая импланты и микроинкапсулированные системы доставки. Биодеградируемые, биосовместимые полимеры могут использоваться, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких составов ясны специалисту в данной области техники.
Липосомные суспензии (включающие липосомы, направленные на инфицированные клетки с моноклональными антителами к вирусным антигенам) также могут использоваться в качестве фармацевтически приемлемых носителей. Они могут быть получены в соответствии со способами, известными специалисту в данной области техники, например, как описано в патенте US 4,522,811.
Дополнительно, суспензии ингибиторов GSNOR могут быть получены в виде подходящих масляных инъекционных суспензий. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или синтетические эфиры жирных кислот, такие как этилолеат, триглицериды или липосомы. Нелипидные поликатионные аминополимеры также могут использоваться для доставки. Необязательно, суспензия также может включать подходящие стабилизаторы или агенты для повышения растворимости соединений и получения высоко концентрированных растворов.
Особенно предпочтительны для составления пероральные или парентеральные композиции в единичной дозированной форме для легкости введения и единства дозировки. Единичная дозированная форма как здесь используется обозначает физически дискретные единицы, подходящие в качестве унитарных дозировок для излечиваемого субъекта; причем каждая единица содержит предварительно определенное количество ингибитора GSNOR, рассчитанное для получения нужного терапевтического действия в смеси с нужным фармацевтическим носителем. Перечень единичных дозированных форм по изобретению предписан и прямо зависит от конкретных характеристик ингибитора GSNOR и конкретного достигаемого терапевтического действия, и ограничений, известных из уровня техники для составления, таких как активный агент для лечения пациентов.
Фармацевтические композиции в соответствии с изобретением, содержащие по крайней мере один ингибитор GSNOR, могут включать один или несколько фармацевтических эксципиентов. Примеры таких эксципиентов включают, но не ограничиваются ими, связующие агенты, наполнители, лубриканты, суспендирующие агенты, подсластители, отдушки, консерванты, буферы, увлажняющие агенты, разрыхлители, агенты для получения шипучей смеси и другие эксципиенты. Такие эксципиенты известны из предшествующего уровня техники. Примерные эксципиенты включают: (1) связующие агенты, которые включают различные целлюлозы и поперечно сшитый поливинилпирролидон, микрокристаллическую целлюлозу, такую как Avicel® PH101 и Avicel® PH102, кремниевую микрокристаллическую целлюлозу (ProSolv SMCC™), смолу трагаканта и желатин; (2) наполнители, такие как различные крахмалы, лактоза, моногидрат лактозы и безводная лактоза; (3) разрыхлители, такие как альгиновая кислота, Primogel, кукурузный крахмал, легкий поперечно-сшитый поливинилпирролидон, картофельный крахмал, кукурузный крахмал и модифицированные крахмалы, кросскармелоза натрия, кросс-повидон, натрийгликолированный крахмал и их смеси; (4) лубриканты, включая агенты, которые действуют на текучесть спрессовываемого порошка, включают стеарат магния, коллоидный диоксид кремния, такой как Aerosil® 200, тальк, стеариновую кислоту, стеарат кальция и силикагель; (5) глиданты, такие как коллоидный диоксид кремния; (6) консерванты, такие как сорбат калия, метилпарабен, пропилпарабен, бензойная кислота и ее соли, другие сложные эфиры парагидроксибензойной кислоты, такие как бутилпарабен, спирты, такие как этиловый или бензиловый спирт, фенольные соединения, такие как фенол, или четвертичные соединения, такие как хлорид бензалкония; (7) разбавители, такие как фармацевтически приемлемые инертные наполнители, такие как микрокристаллическая целлюлоза, лактоза, диосновный фосфат кальция, сахариды и/или смеси любого из вышеизложенных; примеры разбавителей включают микрокристаллическую целлюлозу, такую как Avicel® PH101 и Avicel® PH102; лактозу, такую как моногидрат лактозы, безводную лактозу и Pharmatose® DCL21; диосновный фосфат кальция, такой как Emcompress®; маннит; крахмал; сорбит; сахароза и глюкоза; (8) подсластители, включая любой природный или искусственный подсластитель, такой как сахароза, сахарин, ксилит, сахарин натрия, цикламат, аспартам и ацесульфам; (9) отдушки, такие как мята, метилсалицилат, апельсиновая отдушка, Magnasweet® (товарный знак MAFCO), отдушка жевательной резинки, фруктовые отдушки и им подобные; и (10) агенты для получения шипучей смеси, включая соединения для получения шипучих смесей, такие как органические кислота и карбонат или бикарбонат. Подходящие органические кислоты включают, например, лимонную, винную, яблочную, фумаровую, адипиновую, янтарную и альгиновые кислоты и ангидриды и соли кислот. Подходящие карбонаты и бикарбонаты включают, например, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия, карбонат магния, карбонат натрия глицина, карбонат L-лизина и карбонат аргинина. Альтернативно, может присутствовать только компонент бикарбоната натрия в соединении для получения шипучих смесей.
Е. Наборы, включающие композиции по изобретению
Настоящее изобретение также включает наборы, содержащие композиции по изобретению. Такие наборы могут включать, например, (1) по крайней мере один ингибитор GSNOR; и (2) по крайней мере один фармацевтически приемлемый носитель, такой как растворитель или разбавитель. Дополнительные компоненты набора необязательно могут включать, например: (1) любой из описанных здесь фармацевтически приемлемых эксципиентов, такой как стабилизаторы, буферы и т.д., (2) по крайней мере одну емкость, сосуд или аналогичное устройство для хранения и/или смешения компонентов набора; и (3) устройство доставки, такое как ингалятор, небулайзер, шприц и т.д.
F. Способы получения ингибиторов GSNOR
Ингибиторы GSNOR по изобретению могут быть легко синтезированы известными методиками синтеза или модификацией известных методик синтеза. Специалисту в данной области техники понятно, что описанные ниже методики позволяют осуществить синтез пирролов с различными заместителями. Примерные методы синтеза описаны ниже в примерах.
В соответствии с одной методикой синтеза, реакция 2-фуральдегида с подходящим образом замещенным ацетофеноном с последующей обработкой сильной кислотой приводят к получению подходящим образом замещенного 1,4,7-триона. Циклизация триона в соответствующий 1,2,5-тризамещенный пиррол легко достигается реакцией триона с первичным амином в присутствии п-толуолсульфоновой кислоты. В одном варианте осуществления настоящего изобретения, легко осуществляют дальнейшее получение производных фенильного кольца в С5 пиррола, например, различными реакциями перекрестной конденсации. Например, синтез тризамещенных пирролов реакцией 1-(4-хлорфенил)этанона и 2-фуральдегида приводит к получению целевого пиррола с 4-хлорфенильной группой в С5. Из арилхлорида могут быть получены производные реакцией с борной кислотой в условиях реакции конденсации Сузуки. Такие обычные методики получения производных позволяют быстро получить библиотеки соединений для исследования ингибирования GSNOR in vitro. Различные дополнительные способы описаны в примере 1 данного документа.
При необходимости, дальнейшая очистка и разделение энантиомеров и диастереомеров может осуществляться стандартными методиками, известными из предшествующего уровня техники. Так, например, разделение энантиомеров соединения может осуществляться с помощью хиральной ВЭЖХ и аналогичных хроматографических методик. Диастереомеры могут разделяться аналогично. В некоторых вариантах, однако, диастереомеры могут просто разделяться физически, например, контролируемым осаждением или кристаллизацией.
Способ по изобретению, при осуществлении как здесь описано, может удобно проводиться при температурах, которые используются в уровне техники. В одном варианте осуществления, способ осуществляют при температуре в диапазоне от около 25°С до около 110°С. В другом варианте осуществления, температуру поддерживают в диапазоне от около 40°С до около 100°С. В другом варианте осуществления, температуру поддерживают в диапазоне от около 50°С до около 95°С.
Стадии синтеза с использованием основания осуществляют с помощью любого обычного органического или неорганического основания. Обычно, основание не является нуклеофильным. Так, в одном варианте осуществления основание выбрано из карбонатов, фосфатов, гидроксидов, алкоксидов, солей дисилазанов и третичных аминов.
Способ по изобретению, при осуществлении как здесь описано, может по существу заканчиваться через несколько минут или через несколько часов в зависимости от природы и количества реагентов и температуры реакции. Определение того, что реакция по существу закончилась, может обычно оцениваться стандартными методиками, известными из предшествующего уровня техники, такими как, например, ВЭЖХ, LCMS, ТСХ и1Н ЯМР.
G. Способ лечения
Настоящее изобретение относится к способам профилактики или лечения (например, облегчения одного или нескольких симптомов) медицинских состояний путем применения одного или нескольких из описанных соединений. Способы включают введение ингибитора GSNOR в терапевтически эффективном количестве пациенту, нуждающемуся в этом. Композиции по изобретению также могут включать применение для профилактической терапии.
Ингибитором GSNOR, используемым в способах лечения в соответствии с изобретением, может быть: (1) описанный здесь новый ингибитор GSNOR или его фармацевтически приемлемая соль, его пролекарство или его метаболит; (2) соединение, которое было известно до настоящего изобретения, но оно не было известно как соединение-ингибитор GSNOR, или его фармацевтически приемлемая соль, его пролекарство или его метаболит; или (3) соединение, которое было известно до настоящего изобретения, и было известно как соединение-ингибитор GSNOR, но где не было известно, что соединение полезно для описанных здесь способов лечения, или его фармацевтически приемлемая соль, его пролекарство или его метаболит.
Пациентом может быть любое животное, домашнее, крупный рогатый скот или дикое животное, включая, но не ограничиваясь ими, кошек, собак, лошадей, свиней и крупный рогатый скот, и предпочтительно людей. Как здесь используется, термины пациент и субъект могут использоваться взаимозаменяемо.
У субъектов с крайне высокими уровнями GSNOR или активности GSNOR, модулирование может достигаться, например, введением одного или нескольких описанных соединений, которые нарушают или регулируют далее функцию GSNOR, или снижают уровень GSNOR. Эти соединения могут вводиться с другими агентами-ингибиторами GSNOR, такими как анти-GSNOR антитела или фрагменты антител, антисмословые GSNOR, и-РНК или низкомолекулярные молекулы, или другими ингибиторами, отдельно или в комбинации с другими агентами, описанными здесь более подробно.
Настоящее изобретение относится к способу лечения субъекта, страдающего нарушением, облегчаемым донорной терапией N0. Такой способ включает введение субъекту терапевтически эффективного количества ингибитора GSNOR.
Как здесь используется, "лечение" обозначает управление и защиту пациента в целях сохранения от заболевания, состояния или нарушения, и включает введение соединения настоящего изобретения для предотвращения возникновения симптомов или осложнений, облегчения симптомов или осложнений или устранения заболевания, состояния или нарушения. Более конкретно, "лечение" включает изменение, смягчение, облегчение, снижение, подавление или остановку по крайней мере одного неблагоприятного симптома или действия заболевания (нарушения), развития заболевания, возбудителя заболевания (например, бактерии или вирусы) или другого нарушенного состояния. Лечение продолжается настолько долго, пока улучшаются симптомы и/или патология.
Нарушения могут включать легочные нарушения, связанные с гипоксемией и/или сокращением гладкой мускулатуры в легких и/или легочной инфекцией и/или легочной травмой (например, легочная гипертензия, ARDS, астма, пневмония, легочный фиброз/интерстициальные легочные заболевания, кистозный фиброз, COPD), сердечно-сосудистое заболевание и болезнь сердца, включая состояния, такие как гипертензия, ишемические коронарные синдромы, атеросклероз, глаукому, заболевания, характеризующиеся ангиогенезом (например, заболевание коронарной артерии), нарушения, при которых существует риск развития тромбоза, нарушения, при которых существует риск развития рестеноза, хронические воспалительные заболевания (например, AID слабоумие и псориаз), нарушения, при которых существует риск развития апоптоза (например, сердечная недостаточность, атеросклероз, сердечная недостаточность, дегенеративные неврологические нарушения, артрит и повреждение печени (ишемическое или алкогольное)), импотенцию, ожирение, вызванное питанием с потребностью в пище, инсульт, реперфузионную травму (например, травматическое мышечное повреждение при сердечной или легочной травме или повреждении раздавливанием) и нарушения, в которых желательна предварительная защита NO сердца или головного мозга против последующих ишемических приступов.
В одном варианте осуществления, соединения настоящего изобретения или их фармацевтически приемлемые соли или пролекарства или метаболиты могут вводиться в комбинации с донором NO. Донор NO дает оксид азота или связанные типы восстановления-окисления, и обычно обеспечивают биоактивность оксида азота, которая идентична оксиду азота, например, вазорелаксация или стимулирование или ингибирование белка рецептора, например, ras-белок, адренергический рецептор, NFκB. Полезные доноры NO, включающие S-нитрозо-, O-нитрозо-, С-нитрозо- и N-нитрозосоединения и их нитропроизводные и комплексы NO с металлом, но не исключающие другие соединения, генерирующие биоактивность NO, описаны в книге "Methods in Nitric Oxide Research," под ред. Feelisch и др., сс.71-115 (J.S., John Wiley & Sons, New York, 1996), которая включена в качестве ссылки. Доноры NO, которые представляют собой С-нитрозосоединения, где нитрозогруппа присоединена к третичному атому углерода, которые являются полезными, включают доноры, описанные в патенте US 6,359,182 и международной заявке на патент WO 02/34705. Примеры S-нитрозосоединений, включая полезные S-нитрозотиолы, включают, например, S-нитрозоглутатион, S-нитрозо-N-ацетилпеницилламин, S-нитрозоцистеин и его этиловый эфир, S-нитрозоцистеинглицин, S-нитрозо-гамма-метил-L-гомоцистеин, S-нитрозо-L-гомоцистеин, S-нитрозо-гамма-тио-L-лейцин, S-нитрозо-дельта-тио-L-лейцин и S-нитрозоальбумин. Примерами других полезных доноров NO являются нитропруссид натрия (ниприд), этилнитрит, изосорбид, нитроглицерин, SIN 1, который представляет собой мольсидомин, фуроксамины, N-гидрокси (N-нитроsамин) и перфторуглеводы, которые насыщены NO, или гидрофобный донор NO.
Комбинация ингибитора GSNOR с R(+) энантиомером амлодипина, известного вещества, высвобождающего NO (статья Zhang X.P и др., 2002, J. Cardiovascular Pharmacology, 39, сс.208-214) является также вариантом осуществления настоящего изобретения.
Настоящее изобретение также относится к способу лечения субъекта, страдающего патологически пролиферирующими клетками, который включает введение указанному субъекту ингибитора GSNOR в терапевтически эффективном количестве. Ингибиторы GSNOR представляют собой описанные выше соединения или их фармацевтически приемлемые соли, или пролекарства или метаболит, в комбинации с фармацевтически приемлемым носителем. Лечение продолжают настолько долго, пока облегчаются симптомы и/или патология.
В другом варианте осуществления, патологически пролиферирующими клетками могут быть патологически пролиферирующие микробы. Микробами могут быть микробы, в которых GSNOR экспрессируется для защиты микроба от нитрозативного стресса, или в которых клетки пациента, инфицированные микробом, экспрессируют фермент, тем самым защищая микроб от нитрозативного стресса. Термин "патологически пролиферирующие микробы" как здесь используется обозначает патологические микроорганизмы, включающие, но не ограничиваясь ими, патологические бактерии, патологические вирусы, патологические хламидии, патологические протозойи, патологические Rickettsia, патологические грибки и патологические микоплазматы. Более подробное описание микробов приведено в колонках 11 и 12 патента US 6,057,367. Термин "клетки хозяина, инфицированные патологическими микробами", включает не только клетки млекопитающего, инфицированного патологическими вирусами, но также клетки млекопитающего, содержащие внутриклеточные бактерии или протозойи, например, макрофаги, содержащие Mycobacteriwn tuberculosis, Mycobacterium leper (проказа) или Salmonella typhi (брюшной тиф).
В другом варианте осуществления, патологически пролиферирующими клетками могут быть патологические гельминты. Термин "патологические гельминты" используется здесь для обозначения патологическим нематод, патологических трематод и патологических цистод. Более подробно по применению гельминтов см. в колонке 12 патента US 6,057,367.
В другом варианте осуществления, патологически пролиферирующими клетками могут быть патологически пролиферирующие клетки млекопитающего. Термин "патологически пролиферирующие клетки млекопитающего" как здесь используется обозначает клетки млекопитающего, которые увеличиваются в размере или количестве в указанном млекопитающем, оказывая нежелательное на млекопитающего или его органы. Термин включает, например, патологически пролиферирующие или разрастающиеся клетки, вызывающие рестеноз, патологически пролиферирующие или разрастающиеся клетки, вызывающие доброкачественную гипертрофию простаты, патологически пролиферирующие клетки, вызывающие гипертрофию миокарда, и пролиферирующие клетки в участках воспаления, такие как синовиальные клетки при артрите или клетки, связанные с клеточным пролиферативным нарушением.
Как здесь используется, термин "клеточное пролиферативное нарушение" обозначает состояния, в которых нерегулируемый и/или нарушенный рост клеток может приводить к развитию нежелательного состояния или заболевания которое может быть раковым или нераковым, например, псориатическое состояние. Как здесь используется, термин "псориатические состояние" обозначает нарушения, включающие гиперпролиферацию кератиноцитов, инфильтрацию воспалительных клеток и деформацию цитокинов. Клеточным пролиферативным нарушением может быть предраковое состояние или рак. Раком может быть первичный рак или метастатический рак, или они оба.
Как здесь используется, термин "рак" включает солидные опухоли, такие как опухоль легких, груди, толстой кишки, яичников, поджелудочной железы, простаты, аденокарцинома, плоскоклеточная карцинома, саркома, злокачественная глиома, лейомиосаркома, гепатома, рак головы и шеи, злокачественная меланома, немеланомные раки кожи, а также гематологические опухоли и/или злокачественные образования, такие как лейкемия, детская лейкемия и лимфома, множественная миелома, болезнь Ходжкина, лимфомы лимфоцитного и кожного происхождения, острая и хроническая лейкемия, такая как острая лимфобластомная, острая миелоцитная или хроническая миелоцитная лейкемия, неоплазм плазматических клеток, лимфоидный неоплазм и виды рака, связанные с AIDS.
В дополнение к псориатическим состояниям, типами пролиферативных заболеваний, которые могут излечиваться композициями настоящего изобретения, являются эпидермальные и дермоидные кисты, липомы, аденомы, капиллярные и кожные гемангиомы, лимфангиомы, травмы невуса, тератомы, нефромы, миофиброматоз, остеогенные опухоли и другие диспластические массы и им подобные. В одном варианте осуществления, пролиферативные заболевания включают дисплазии и подобные им нарушения.
В одном варианте осуществления лечение рака включает снижение размера опухоли, снижение количества опухолей, остановку развития опухоли, снижение метастатических участков в других тканях или органах, отдаленных от первичного участка опухоли, улучшение жизнеспособности пациента или улучшение качества жизни пациента, или по крайней мере два пункта из вышеизложенных.
В другом варианте осуществления лечение клеточного пролиферативного нарушения включает снижение скорости клеточной пролиферации, снижение доли пролиферирующих клеток, снижение размера площади или зоны клеточной пролиферации или снижение количества или доли клеток, имеющих нарушенное появление или морфологию, или по крайней мере два пункта из вышеизложенных.
В другом варианте осуществления, соединения настоящего изобретения или их фармацевтически приемлемые соли, их пролекарства или их метаболиты могут вводиться в комбинации со вторым химиотерапевтическим агентом. В другом варианте осуществления, второй химиотерапевтический агент выбран из группы, состоящей из тамоксифена, ралоксифена, анастрозола, эксеместана, летрозола, цисплатина, карбоплатина, паклитакселя, циклофосфамида, ловастатина, минозина, гемцитабина, араС, 5-фторурацила, метотрексата, доцетакселя, госерелина, винкристина, винбластина, нокодазола, тенипозида, этопозида, эпотилона, навельбина, камптотецина, даунонибицина, дактиномицина, митоксантрона, амсакрина, доксорубицина, эпирубицина, идарубицина, иматиниба, гефитибина, эрлотиниба, сорафениба, малата сунитиниба, трастузумаба, ритуксимаба, цетуксимаба и бевацизумаба.
В другом варианте осуществления, соединения настоящего изобретения или их фармацевтически приемлемые соли, их пролекарства или их метаболиты могут вводиться в комбинации с агентом, которые вызывают нитрозатирование или окислительный стресс. Агенты для селективного вызванного нитрозативного стресса для ингибирования пролиферации патологически пролиферирующих клеток в комбинированной терапии с описанными ингибиторами GSNOR и дозировки и способы их введения включают те, которые описаны во включенном патенте US 6,057,367. Вспомогательные агенты для вызывания окислительного стресса (то есть агенты, которые повышают соотношение GSSG (окисленный глутатион) относительно GSH (глутатион) или соотношение NAD(P) относительно NAD(P)H или повышают производные тиобарбитуровой кислоты) в комбинированной терапии с описанными ингибиторами GS-FDH включают, например, L-бутионин-3-сульфоксимин (BSO), ингибиторы глутатионредуктазы (например, BCNU), ингибиторы или вещества, разъединяющие митохондриальное дыхание, и лекарственные препараты, которые повышают виды реакционного кислорода (ROS), например, адриамицин, в стандартных дозировках со стандартными способами введения.
Ингибиторы GSNOR также могут совместно вводиться с ингибиторами фосфодиэстеразы (например, ролипрам, циломиласт, рофлумиласт, Viagra (сильденифил цитрат), Cialis® (тадалафил), Levitra® (варденифил) и т.д.), β-агонистом, стероидом или антагонистом лейкотриена (LTD4). Специалист в данной области техники легко определит подходящее терапевтически эффективное количество в зависимости от излечиваемого заболевания.
Ингибиторы GSNOR могут использоваться как средства для улучшения β-адренергического пути передачи сигнала. В частности, ингибиторы GSNOR отдельно или в комбинации с β-агонистами могли бы использоваться для лечения или защиты от сердечной недостаточности или других васкулярных нарушений, таких как гипертензия и астма. Ингибиторы GSNOR также могут использоваться для модулирования рецепторов, связанных с G-белком (GPCR) путем потенциирования Gs G-белка, что приводит к релаксации гладких мышц (например, сосудов дыхательных путей и кровеносных сосудов), и снижением Gq G-белка, и тем самым предотвращением сокращения гладких мышц (например, сосудов дыхательных путей и кровеносных сосудов).
Терапевтически эффективное количество для лечения субъекта, страдающего нарушением, облегчающимся NO-донорной терапией, представляет собой ингибирующее GSNOR количество in vivo, которое вызывает облегчение излечиваемого нарушения или защищает от риска, связанного с нарушением. Например, для астмы терапевтически эффективным количеством является бронхорасширяющее эффективное количество; для кистозного фиброза терапевтически эффективным количеством является эффективное количество, облегчающее обструкцию дыхательных путей; для ARDS терапевтически эффективным количеством является эффективное количество, облегчающее гипоксемию; для сердечного заболевания терапевтически эффективным количеством является эффективное количество, ослабляющие стенокардию или индуцирующие ангиогенез; для гипертензии терапевтически эффективным количеством является эффективное количество, снижающее кровяное давление; для ишемических коронарных нарушений терапевтически эффективным количеством является эффективное количество, повышающее поток крови; для атеросклероза терапевтически эффективным количеством является эффективное количество, останавливающее эндотелиальную дисфункцию; для глаукомы терапевтически эффективным количеством является эффективное количество, снижающее внутриглазное давление; для заболеваний, характеризующихся ангиогенезом, терапевтически эффективным количеством является эффективное количество, ингибирующее ангиогенез; для нарушений, при которых существует риск тромбоза, терапевтически эффективным количеством является эффективное количество, предотвращающее тромбоз; для нарушений, при которых существует риск рестеноза, терапевтически эффективным количеством является эффективное количество, ингибирующее рестеноз; для хронических воспалительных заболеваний терапевтически эффективным количеством является эффективное количество, снижающее воспаление; для нарушений, при которых существует риск развития апоптоза, терапевтически эффективным количеством является эффективное количество, предотвращающее апоптоз; для импотенции терапевтически эффективным количеством является эффективное количество, вызывающее или продлевающее эрекцию; для ожирения терапевтически эффективным количеством является эффективное количество, вызывающее чувство насыщения; для инсульта терапевтически эффективным количеством является эффективное количество, повышающее поток крови или защищающее TIA; для реперфузионной травмы терапевтически эффективным количеством является эффективное количество, повышающее функцию; и для предварительной обработки сердца и головного мозга терапевтически эффективным количеством является эффективное количество, защищающее клетку, например, при измерении трипонином или СРК.
Терапевтически эффективное количество для лечения субъекта, страдающего патологически пролиферирующими клетками, обозначает ингибирующее GSNOR количество in vivo, которое является антипролиферативным эффективным количеством. Такое антипролиферативное эффективное количество как здесь используется обозначает количество, вызывающее снижение скорости пролиферации по крайней мере на 20%, по крайней мере на 10%, по крайней мере на 5% или по крайней мере на 1%.
Обычно дозировка, то есть терапевтически эффективное количество, находится в диапазоне от 1 мкг до 10 г/кг, и часто в диапазоне от 10 мкг до 1 г/кг, или от 10 мкг до 100 мг/кг веса тела излечиваемого пациента в день.
Н. Другие применения
Соединения настоящего изобретения или их фармацевтически приемлемые соли или их пролекарства или метаболиты могут использоваться в различных аппаратах при обстоятельствах, при которых присутствие таких соединений является желательным. Таким аппаратом может быть любое устройство или емкость, например, имплантируемые устройства, в которых ингибитор GSNOR может использоваться для покрытия хирургического отверстия или сердечнососудистого стента до имплантации пациенту. Ингибиторы GSNOR настоящего изобретения также могут использоваться для различных аппаратов в целях анализов in vitro или для культурирования клеток.
Соединения настоящего изобретения или их фармацевтически приемлемые соли или их пролекарства или метаболиты также могут использоваться в качестве агента для разработки, выделения или очистки связывающих партнеров соединений-ингибиторов GSNOR, таких как антитела, природные лиганды и им подобные. Специалист в данной области техники легко определит аналогичные применения для соединений настоящего изобретения.
Примеры
Следующие примеры приведены для иллюстрации настоящего изобретения. Следует отметить, однако, что изобретение не ограничено конкретными условиями или деталями, описанными в этих примерах. В описании любые и все ссылки на публично доступные документы, включая патенты США, включены конкретно в качестве ссылки.
Пример 1: Общие и конкретные способы получения новых пиррольных ингибиторов GSNOR
В этом примере описаны схемы получения ингибиторов GSNOR, описанных в таблице 1. Некоторые схемы являются специфичными для конкретного соединения, тогда как другие являются общими схемами, которые включают примерный способ получения представительного соединения. Далее на схемах представлены способы, которые раскрывают получение промежуточных соединений, которые используются в выбранных схемах.
Схема 1: Общая схема получения ингибиторов GSNOR структуры 1D
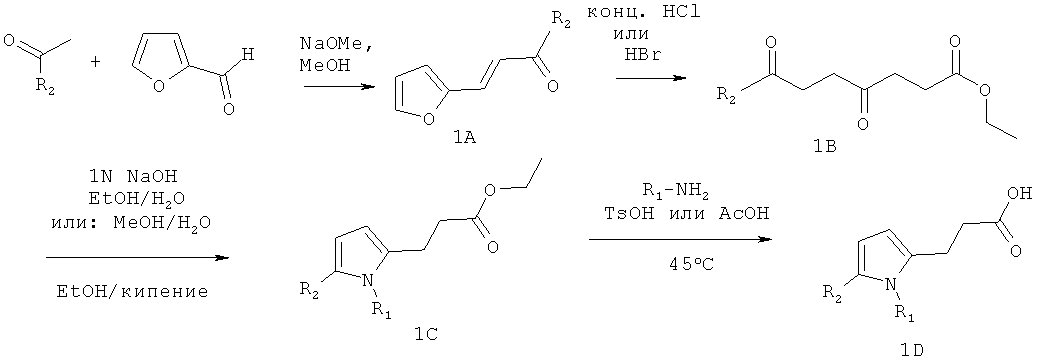
Примерная методика для схемы 1: Синтез 3-[1-(4-карбамоил-2-метилфенил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропановой кислоты
Стадия 1: Синтез (Е)-3-фуран-2-ил-1-(4-метоксифенил)пропенона. Раствор 2-фуральдегида (5,85 г, 60,92 ммоля) добавляли к метанольному раствору (120 мл) 4-метоксиацетофенона (8,5 г, 56,6 ммоля), затем добавляли метоксид натрия (3,1 г, 56,6 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, затем растворитель удаляли в вакууме. Полученную смесь разбавляли водой (130 мл) и экстрагировали этилацетатом (350 мл). Водный слой повторно экстрагировали этилацетатом (100 мл). Объединенные органические слои сушили безводным Na2SO4, и растворитель удаляли в вакууме с получением продукта (Е)-3-фуран-2-ил-1-(4-метоксифенил)пропенона в виде оранжевого твердого вещества (12,6 г, 97%).
Стадия 2: Синтез 1-(4-метоксифенил)декан-1,4,7-триона. Конц. HCl (59 мл) добавляли к раствору (Е)-3-фуран-2-ил-1-(4-метоксифенил)пропенона (12,6 г, 55,2 ммоля) в этаноле (237 мл). Реакционную смесь нагревали при кипении с обратным холодильником в течение 16 ч, концентрировали и разбавляли дихлорметаном (250 мл), и полученный органический слой промывали водой (25 мл). После разделения фаз органический слой сушили безводным Na2SO4, и растворитель удаляли в вакууме с получением сырой смеси, которую очищали ускоренной хроматографией на силикагеле с получением 1-(4-метоксифенил)декан-1,4,7-триона (6,89 г, 43%).
Стадия 3: Синтез этилового эфира 3-[1-(4-карбамоил-2-метилфенил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропановой кислота. 4-Амино-3-метилбензамид (180 мг, 1,2 ммоля) добавляли к раствору 1-(4-метоксифенил)декан-1,4,7-триона (350 мг, 1,2 ммоля) в этаноле (6 мл), затем добавляли моногидрат п-толуолсульфоновой кислоты (сокращение TsOH или пTsOH) (23 мг, 0,12 ммоля). Реакционную смесь нагревали при кипении с обратным холодильником в течение 16 ч, и растворитель удаляли в вакууме с получением сырого продукта, который после очистки ускоренной хроматографией на силикагеле приводил к получению этилового эфира 3-[1-(4-карбамоил-2-метилфенил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропановой кислоты (147 мг, 30%).
Стадия 4: Синтез 3-[1-(4-карбамоил-2-метилфенил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропановой кислоты. Этиловый эфир 3-[1-(4-карбамоил-2-метилфенил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропановой кислоты (86 мг, 0,216 ммоля) растворяли в этаноле (4 мл). Добавляли воду (0,5 мл) к этанольному раствору, затем добавляли 1N NaOH (0,51 мл, 0,51 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем при 45°С еще в течение часа. После удаления растворителя в вакууме, остаток разбавляли водой (6 мл) и экстрагировали этилацетатом (2×6 мл). Значение рН водного слоя доводили до 2 с помощью 1N HCl, и затем экстрагировали этилацетатом (6 мл). Объединенные органические слои сушили безводным Na2SO4, и растворитель удаляли в вакууме с получением 3-[1-(4-карбамоил-2-метилфенил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропановой кислоты в качестве продукта (68 мг, 85%).
Схема 1А: Альтернативные условия

Примерная методика схемы 1А, альтернативные условия: Синтез 3-[1-(4-карбамоилтиазол-2-ил)-5-(4-метоксифенил)-1Н-пиррол-2-ил1пропионовой кислоты
Стадия 3: Синтез этилового эфира 3-[1-(4-карбамоилтиазол-2-ил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 1C, R1=4-карбамоилтиазол-2-ил, R2=4-метоксифенил): К раствору этилового эфира 7-(4-метоксифенил)-4,7-диоксогептановой кислоты (0,5 ммоля), см. схему 1, в этаноле (2 мл) добавляли амин (1,5 эквив.) и моногидрат п-толуолсульфоновой кислоты (0,5 эквив.). Реакцию проводили с помощью Biotage Microwave Initiator в течение 1-3 часов при 150°С. Растворитель удаляли в вакууме с получением сырой смеси, которую очищали преп.хроматографией на пластине с силикагелем с получением конечного продукта (70 мг, 38%).
Стадия 4: Синтез 3-[1-(4-карбамоилтиазол-2-ил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 1D, R1=4-карбамоилтиазол-2-ил, R2=4-метоксифенил): К этиловому эфиру 3-[1-(4-карбамоилтиазол-2-ил)-5-(4-метоксифенил)-1Н-пиррол-2-ил]пропионовой кислоты (0,15 ммоля) в смеси 2:1 метанол/ТГФ добавляли 2М LiOH (0,30 ммоля). Реакционную смесь перемешивали в течение 24 часов. Растворитель удаляли в вакууме. Остаток разбавляли водой (2 мл) и экстрагировали этиловым эфиром. Значение рН водного слоя доводили до 2 с помощью 1N HCl. Полученную суспензию отфильтровывали; твердое вещество промывали водой и сушили с получением конечного соединения. Выход: 36 мг, 69%.
Схему 2 - схему 4 умышленно пропускали.
Схема 5: Общая схема получения ингибиторов GSNOR структуры 5Е

Примерная методика схемы 5: Синтез 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 5Е, Ar1=4-карбамоил-2-метилфенил, R=Н)
Стадия 1: Синтез 1-(4-бромфенил)-3-(фуран-2-ил)проп-2-ен-1-она (соединение 5А). К раствору 4-бромфенилэтанона (112,6 г, 570 ммолей) и фуран-2-карбальдегида (58,5 г, 610 ммолей) в метаноле (1,5 л) добавляли CH3ONa (31 г, 570 ммолей) в течение 10 мин, и реакционный раствор перемешивали при комнатной температура в течение ночи. Реакционную смесь нейтрализовали конц. HCl до значения рН=7, и растворитель удаляли при пониженном давлении. К полученному остатку добавляли ЕА и воду. Водный слой экстрагировали ЕА 3 раза. Объединенные слои промывали насыщенным раствором хлорида натрия, сушили MgSO4, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ (петролейный эфир): ЕА (этилацетат)=10:1) с получением 1-(4-бромфенил)-3-(фуран-2-ил)проп-2-ен-1-она (соединение 5А) в виде желтого твердого вещества (90,2 г, 65%).
Стадия 2: Синтез этил 7-(4-бромфенил)-4,7-диоксогептаноата (соединение 5В). К раствору соединения 1-(4-бромфенил)-3-(фуран-2-ил)проп-2-ен-1-она (соединение 5А) (20,0 г, 72,2 ммоля) в этаноле (160 мл) добавляли HBr (48% в воде, 40 мл). Полученную смесь перемешивали при кипении с обратным холодильником в течение 8 ч, и затем реакционный раствор концентрировали в вакууме. К остатку добавляли насыщ. NaHCO3 до значения рН=7 и экстрагировали ЕА. Объединенные органические слои промывали насыщенным раствором хлорида натрия, сушили MgSO4, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=5:1) с получением этил 7-(4-бромфенил)-4,7-диоксогептаноата (соединение 5В) в виде желтого твердого вещества (7,0 г, 28%).
Стадия 3: Синтез этил 3-(5-(4-бромфенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (соединение 5С, Ar1=4-карбамоил-2-метилфенил). К раствору этил 7-(4-бромфенил)-4,7-диоксогептаноата (соединение 5В) (3,41 г, 10 ммолей) и 4-амино-3-метилбензамида (1,65 г, 11 ммолей) в 50 мл этанола добавляли TsOH·Н2О (570 мг, 3 ммоля). Реакционный раствор перемешивали при кипении с обратным холодильником в течение ночи, и затем концентрировали в вакууме. Полученный остаток нейтрализовали насыщ. NaHCO3 и экстрагировали этилацетатом. Органические слои промывали насыщенным раствором хлорида натрия, концентрировали и очищали колоночной хроматографией на силикагеле (ДХМ:РЕ=1:1) с получением этил 7-(4-бромфенил)-4,7-диоксогептаноата в виде бледного твердого вещества (2,80 г, 61%).
Стадия 4: Синтез этил 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (соединение 5D, Ar1=4-карбамоил-2-метилфенил, R=Н). К смеси этил 7-(4-бромфенил)-4,7-диоксогептаноата (4,54 г, 10 ммолей) и имидазола (2,04 г, 30 ммолей) в ДМСО (50 мл) добавляли L-пролин (0,345 г, 3 ммоля), CuI (1,14 г, 6 ммолей) и К2СО3 (2,76 г, 20 ммолей). Полученную смесь перемешивали в N2 при 100°С в течение ночи, охлаждали до комнатной температуры, отфильтровывали и концентрировали в вакууме. Остаток растворяли в этилацетате, и добавляли насыщенный водный раствор NaHCO3 до значения рН=8,5. Смесь отфильтровывали, и полученный водный слой экстрагировали ЕА (5 раз). Объединенные органические слои промывали насыщенным раствором хлорида натрия, сушили MgSO4, концентрировали и очищали колоночной хроматографией на силикагеле (ДХМ:МеОН=30:1-20:1) с получением 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропаноата в виде бледного твердого вещества (1,6 г, 36%).
Стадия 5: Синтез 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 5Е, Ar1=4-карбамоил-2-метилфенил, R=Н). К раствору соединения 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (22,0 г, 48,3 ммоля) в смеси ТГФ/H2O (об/об=1/1, 220 мл) добавляли LiOH·H2O (4,15 г, 96,6 ммоля). Реакционный раствор перемешивали при комнатной температуре в течение 5 ч. ТГФ удаляли при пониженном давлении, и водный раствор подкисляли 10% HCl до значения рН=5. Твердое вещество отфильтровывали и перекристаллизовывали из ТГФ и воды [1:1 (об/об)] с получением 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты в виде желтого твердого вещества (11,35 г, 55%).
Схема 6: Общая схема получения ингибиторов GSNOR структуры 6Н

Примерная методика схемы 6: Синтез 3-(5-(бензо[(1][1,31диоксол-5-ил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты
Стадия 1: Синтез 3-метил-4-(1Н-пиррол-1-ил)бензамида (соединение 6А). 2,5-Диметокситетрагидрофуран (106 г, 80 ммолей) добавляли к раствору 4-амино-3-метилбензамида (100 г, 66,7 ммоля) в АсОН (300 мл). Смесь перемешивали при 80°С в течение 1,5 ч, и затем охлаждали до комнатной температуры. Добавляли по каплям раствор Na2CO3 при 0°С, и три раза экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором хлорида натрия, сушили Na2SO4, концентрировали и промывали петролейным эфиром. Полученное твердое вещество отфильтровывали и сушили с получением 3-метил-4-(1Н-пиррол-1-ил)бензамида в виде бледного твердого вещества (89,7 г, выход 67%).
Стадия 2: Синтез 4-(2-формил-1Н-пиррол-1-ил)-3-метилбензонитрила (соединение 6В). POCl3 (65 г, 427 ммолей) добавляли к DMF (34 мл) при 0°С в течение 30 мин. После добавления смесь перемешивали при комнатной температуре в течение 1,5 ч, и затем охлаждали до 0°С. Добавляли раствор 3-метил-4-(1Н-пиррол-1-ил)бензамида (соединение 6А) (42,7 г, 213,5 ммоля) в DMF (150 мл) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 20 мин, и затем нагревали при 80°С в течение 1 ч. Раствор охлаждали до комнатной температуры, и затем добавляли насыщ. раствор Na2CO3 при 0°С до значения рН=8. Смесь экстрагировали этилацетатом три раза. Объединенные органические слои промывали насыщ. раствором NaHCO3 и насыщенным раствором хлорида натрия, сушили Na2S04, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=10:1) с получением 4-(2-формил-1Н-пиррол-1-ил)-3-метилбензонитрила в виде желтого твердого вещества (30,5 г, выход 68%).
Стадия 3: Синтез этил 3-(1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)акрилата (соединение 6С).
Метод А: Смесь 4-(2-формил-1Н-пиррол-1-ил)-3-метилбензонитрила (15 г, 71,4 ммоля) и (карбэтоксиметилен)трифенилфосфорана (27,5 г, 78,6 ммоля) в толуоле нагревали при 100°С в течение ночи. Затем смесь охлаждали до комнатной температуры, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=5:1) с получением этил 3-(1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)акрилата в виде желтого масла (19,8 г, 98%).
Метод В: К смеси 4-(2-формил-1Н-пиррол-1-ил)-3-метилбензонитрила (24,5 г, 116,7 ммоля), ДМАП (2,9 г, 23,3 ммоля) и моноэтилмалоната калия (99,2 г, 583,3 ммоля) в DMF (600 мл) добавляли АсОН (35,0 г, 583,3 ммоля) и пиперидин (29,8 г, 350 ммолей). Полученную смесь нагревали при 80°С, и перемешивали в течение 48 ч. Реакционную смесь выливали в охлажденную воду и экстрагировали этилацетатом (800 мл × 3). Объединенные органические слои промывали насыщ. раствором NaHCO3 и насыщенным раствором хлорида натрия, сушили Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=5:1) с получением этил 3-(1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)акрилата в виде желтого масла (21,8 г, 67%).
Стадия 4: Синтез этил 3-(1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (соединение 6D). К раствору этил 3-(1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)акрилата (соединение 6С) (8,0 г, 28,6 ммоля) в этаноле добавляли 10% Pd/C (0,8 г). Смесь перемешивали при 1 атм Н2 в течение 30 мин при комнатной температуре и отфильтровывали. Полученный фильтрат концентрировали досуха с получением сырого продукта этил 3-(1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (7,5 г), который использовали на следующей стадии без дополнительной очистки: LC-MS m/z 283,0 [М+Н]+, чистота 68%.
Стадия 5: Синтез этил 3-(5-бром-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (соединение 6Е). NBS (4,76 г, 1 эквив.) порциями добавляли к раствору этил 3-(1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата в DMF при 0°С в течение 45 мин. После добавления смесь перемешивали при комнатной температуре в течение 30 мин, затем выливали в воду, и три раза экстрагировали этилацетатом. Объединенные органические слои промывали насыщенным раствором хлорида натрия, сушили Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=15:1) с получением этил 3-(5-бром-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата в виде белого твердого вещества.
Стадия 6: Синтез этил 3-(5-(бензо[d][1,3]диоксол-5-ил)-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата. К суспензии этил 3-(5-бром-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (400 мг, 0,665 ммоля), 3,4-метилендиоксилфенилборной кислоты (143 мг, 0,864 ммоля), бикарбоната натрия (560 мг, 5,32 ммоля) в растворителе (4 мл) добавляли Pd(PPh3)4 (60 мг, 0,199 ммоля). Реакционную смесь дегазировали и нагревали при кипении с обратным холодильником в течение 5 ч. ТСХ показала, что реакция завершилась. Добавляли воду (4 мл), и смесь экстрагировали этилацетатом (5 мл × 3). Объединенные органические слои сушили сульфатом магния, отфильтровывали и упаривали с получением коричневого масла, которое очищали колоночной хроматографией на силикагеле с получением этил 3-(5-(бензо[d][1,3]диоксол-5-ил)-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата в виде бесцветного масла (308 мг, 69%).
Стадия 7 и стадия 8: Синтез 3-(5-(бензо[d][1,3]диоксол-5-ил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты. К смеси этил 3-(5-(бензо[d][1,3]диоксол-5-ил)-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (100 мг, 0,249 ммоля) и карбоната калия (52 мг, 0,373 ммоля) в ДМСО (1 мл) добавляли 30% водный раствор H2O2 (28,2 мг, 0,249 ммоля). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. ТСХ показала, что реакция завершилась. Добавляли воду (7 мл), и осаждалось белое твердое вещество. Суспензию центрифугировали, и водную фазу выгружали. Полученное твердое вещество сушили в вакууме с получением амидного промежуточного соединения в виде белого твердого вещества (85 мг, выход 81%). К смеси этого промежуточного соединения в Н2О (0,6 мл) и ТГФ (0,6 мл) добавляли LiOH·Н2О (10 мг, 0,238 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение ночи. ТГФ упаривали в вакууме. Остаток подкисляли до значения рН=4 с помощью 5% хлористоводородной кислоты, центрифугировали и сушили с получением 3-(5-(бензо[d][1,3]диоксол-5-ил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты в виде белого твердого вещества (46 мг, общий выход 47%).
Схему 7 - схему 8 умышленно пропускали.
Схема 9а: Общая схема получения ингибиторов GSNOR структуры 9а-С
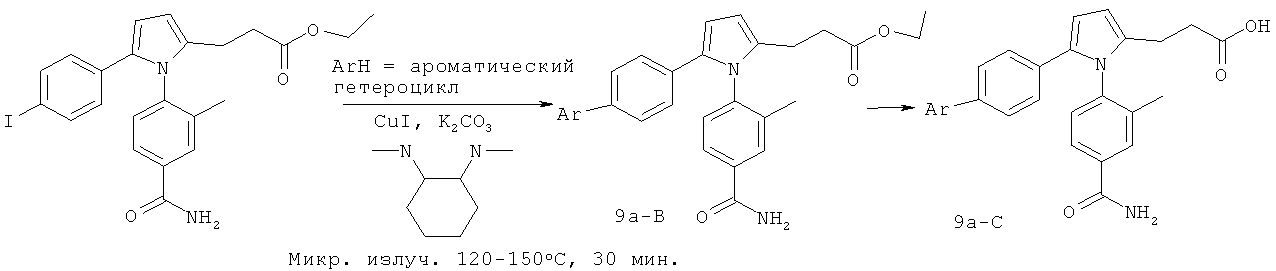
Примерная методика схемы 9а: Синтез 3-[1[(4-карбамоил-2-метилфенил)-5-(4-пиразол-1-ил)-1Н-пиррол-2-ил]пропионовой кислоты
Синтез этилового эфира 3-[1-(4-карбамоил-2-метилфенил)-5-(4-пиразол-1-илфенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 9а-В, Ar=1Н-пиразол-1-ил). N,N-Диметилциклогексан-1,2-диамин (11 мг, 0,08 ммоля) растворяли в ДМСО и дегазировали пропусканием аргона через раствор в течение 2 минут. Полученный раствор затем добавляли к смеси этилового эфира 3-[1-(4-карбамоил-2-метилфенил)-5-(4-йодфенил)-1Н-пиррол-2-ил]пропионовой кислоты (который получали в соответствии с первыми 3 стадиями схемы 1, R2=4-йодфенил, и R1=4-карбамоил-2-метилфенил) (150 мг, 0,29 ммоля), пиразола (500 мг, 7,5 ммоля), йодида меди (11 мг, 0,06 ммоля) и карбоната калия (86 мг, 0,61 ммоля), и полученную реакционную смесь снова дегазировали в течение 2 минут пропусканием аргона через раствор. Реакционную смесь затем подвергали микроволновому излучению в течение 30 минут при 120°С. Реакционную смесь затем добавляли в воду (10 мл), экстрагировали в этилацетат (3×10 мл). Этилацетатные экстракты объединяли, промывали водой (5 мл) и затем насыщенным раствором хлорида натрия (5 мл). Органический слой затем сушили MgSO4. Хроматография (картридж с 5 г силикагеля sep-pak) дихлорметаном, затем 1% метанолом в дихлорметане приводила к получению чистого промежуточного соединения этилового эфира 3-[1[(4-карбамоил-2-метилфенил)-5-(4-пиразол-1-ил)-1Н-пиррол-2-ил]пропионовой кислоты (26 мг, 20%).
Синтез 3-(5-(4-(1Н-пиразол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 9а-С, Ar=1Н-пиразол-1-ил). Этиловый эфир 3-[1-(4-карбамоил-2-метилфенил)-5-(4-пиразол-1-илфенил)-1Н-пиррол-2-ил]пропионовой кислоты (24 мг, 0,06 ммоля) подвергали гидролизу методикой, описанной выше на конечной стадии схемы 1 с получением указанного в заголовке соединения, 3-[1[(4-карбамоил-2-метилфенил)-5-(4-пиразол-1-ил)-1Н-пиррол-2-ил]пропионовой кислоты (18 мг, 75%).
Схема 9b: Общая схема получения ингибиторов GSNOR структуры 9b-С

Примерная методика схемы 9b: Синтез 3-[1-(4-карбамоил-2-метилфенил)-5-(5-имидазол-1-илтиофен-2-ил)-1Н-пиррол-2-ил]пропионовой кислоты
Синтез этилового эфира 3-[1-(4-карбамоил-2-метилфенил)-5-(5-имидазол-1-илтиофен-2-ил)-1Н-пиррол-2-ил]пропионовой кислоты. Получали, используя ту же методику, что и на стадии 1 схемы 9а, за исключением того, что начинали с этил 3-(5-(5-бромтиофен-2-ил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (который получали в соответствии с первыми 3 стадиями схемы 1, R2=5-бромтиофен-2-ил, и R1=4-карбамоил-2-метилфенил).
Синтез 3-[1-(4-карбамоил-2-метилфенил)-5-(5-имидазол-1-илтиофен-2-ил)-1Н-пиррол-2-ил]пропионовой кислоты. Этиловый эфир 3-[1-(4-карбамоил-2-метилфенил)-5-(5-имидазол-1-илтиофен-2-ил)-1Н-пиррол-2-ил]пропионовой кислоты подвергали гидролизу в соответствии с методикой, описанной на первой стадии схемы 1, с получением указанного в заголовке соединения 3-[1-(4-карбамоил-2-метилфенил)-5-(5-имидазол-1-илтиофен-2-ил)-1Н-пиррол-2-ил]пропионовой кислоты.
Схему 10 - схему 18 умышленно пропускали.
Схема 19: Общая схема получения ингибиторов GSNOR структуры 19F

Примерная методика схемы 19: Синтез 3-[5-бензотиазол-6-ил-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 19F, Ar2=бензотиазол-6-ил)
Синтез бензотиазол-6-карбонилхлорида (соединение 19А, Ar2=бензотиазол-6-ил). В атмосфере азота бензотиазол-6-карбоновую кислоту (1,014 г, 5,6 ммоля) растворяли в метиленхлориде (25 мл). Добавляли пять капель N,N-диметилформамида. Медленно добавляли оксалилхлорид (0,5 мл, 5,6 ммоля). Через 2 ч реакционную смесь нагревали при 30°С в течение 16 ч. Реакционную смесь концентрировали в вакууме с получением бензотиазол-6-карбонилхлорида (1,665 г, колич., светло-желтый порошок).
Синтез 7-(бензотиазол-6-карбонил)-1,4-диоксаспиро[4.5]декан-8-она (соединение 19В, Ar2=бензотиазол-6-ил). В атмосфере азота гексаметилдисилазид лития (2,4 мл, 2,4 ммоля) смешивали с ТГФ (5 мл). Реакционную смесь охлаждали до -78°С. Через капельную воронку медленно добавляли моноэтиленацеталь 1,4-циклогександиона (374 мг, 2,4 ммоля), растворенного в ТГФ (2 мл). Реакционную смесь перемешивали в течение 20 мин при -78°С. Смесь затем переносили пипеткой в колбу, охлаждали до -78°С, содержащийся бензотиазол-6-карбонилхлорид (498 мг, 2,52 ммоля) растворяли в ТГФ (5 мл). После добавления реакционную смесь перемешивали при -78°С в течение 1 ч, и затем оставляли нагреваться до комнатной температуры. Через 12 ч добавляли воду (30 мл) и экстрагировали этилацетатом (3×20 мл). Объединенные органические слои промывали 10% раствором лимонной кислоты (20 мл), водой (20 мл), бикарбонатом натрия (20 мл) и насыщенным раствором хлорида натрия (20 мл). Смесь затем сушили Na2SO4, отфильтровывали и концентрировали в вакууме. Сырой материал очищали на колонке с силикагелем (1:1 EtOAc/гексан) с получением 7-(бензотиазол-6-карбонил)-1,4-диоксаспиро[4.5]декан-8-она (271 мг, 35%, светло-желтое твердое вещество).
Синтез этилового эфира 3-[2-(3-бензотиазол-6-ил-3-оксопропил)-[1,3]диоксолан-2-ил]пропионовой кислоты (соединение 19С, Ar2=бензотиазол-6-ил). В атмосфере азота 7-(бензотиазол-6-карбонил)-1,4-диоксаспиро[4.5]декан-8-он (271 мг, 0,85 ммоля) растворяли в этаноле (1 мл). Добавляли 2,43 М раствор этоксида натрия (0,01 мл, 0,03 ммоля). Через 12 ч реакционную смесь концентрировали в вакууме. Остаток разбавляли 10 мл EtOAc/5 мл 10% лимонной кислоты. Слои разделяли. Водный слой затем экстрагировали EtOAc (3×3 мл). Объединенные органические слои промывали водой (5 мл) и насыщенным раствором хлорида натрия (5 мл), сушили Na2SO4, отфильтровывали и концентрировали в вакууме. Сырой материал очищали на колонке с силикагелем (40% EtOAc/гексан) с получением этилового эфира 3-[2-(3-бензотиазол-6-ил-3-оксопропил)-[1,3]диоксолан-2-ил]пропионовой кислоты (100 мг, 38%, светло-желтое масло).
Синтез этилового эфира 7-бензотиазол-6-ил-4,7-диоксогептановой кислоты (соединение 19D, Ar2=бензотиазол-6-ил). В атмосфере азота этиловый эфир 3-[2-(3-бензотиазол-6-ил-3-оксопропил)-[1,3]диоксолан-2-ил]пропионовой кислоты (соединение 19С) (100 мг, 0,28 ммоля) растворяли в ТГФ (1 мл). Добавляли 3N HCl и перемешивали при комнатной температуре. Через 12 ч реакционную смесь разбавляли водой и экстрагировали EtOAc (3 раза). Объединенные органические слои промывали насыщенным раствором хлорида натрия, сушили Na2SO4, отфильтровывали и концентрировали в вакууме с получением этилового эфира 7-бензотиазол-6-ил-4,7-диоксогептановой кислоты (52 мг, 58%, темно-красное твердое вещество; 2/3 в виде этилового эфира, 1/3 в виде карбоновой кислоты).
Синтез этилового эфира 3-[5-бензотиазол-6-ил-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 19Е, Ar2=бензотиазол-6-ил). В колбе объемом 4 мл, насыщенной азотом, этиловый эфир 7-бензотиазол-6-ил-4,7-диоксогептановой кислоты (52 мг, 0,16 ммоля) растворяли в 2 мл этанола. Добавляли п-толуолсульфоновую кислоту (pTSA) (9,9 мг, 0,05 ммоля) и 4-амино-3-метилбензамид (37 мг, 0,24 ммоля). Сосуд плотно закрывали и нагревали при 80°С в масляной бане. Через 12 ч реакционную смесь охлаждали и концентрировали в вакууме. Сырой материал растворяли в N,N-диметилформамиде (1 мл). Добавляли карбонат калия (44 мг, 0,32 ммоля). Затем добавляли йодэтан (0,01 мл, 0,17 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенные органические слои промывали водой, насыщенным раствором хлорида натрия и сушили Na2SO4, отфильтровывали и концентрировали в вакууме. Сырой продукт очищали на колонке с силикагелем (5% IPA/CH2Cl2) с получением этилового эфира 3-[5-бензотиазол-6-ил-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 19Е, Ar2=бензотиазол-6-ил) (42 мг, 73% с 2 стадий, красное твердое вещество).
Синтез 3-[5-бензотиазол-6-ил-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 19F, Ar2=бензотиазол-6-ил). Этиловый эфир 3-[5-бензотиазол-6-ил-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил]пропионовой кислоты (соединение 19Е) (42 мг, 0,10 ммоля) подвергали гидролизу в соответствии с методикой, описанной выше на последней стадии схемы 4, с получением указанного в заголовке соединения 3-[5-бензотиазол-6-ил-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил]пропионовой кислоты (23 мг, 59%, светлый желто-коричневый порошок).
Схема 20: Общая схема получения ингибиторов GSNOR структуры 20С
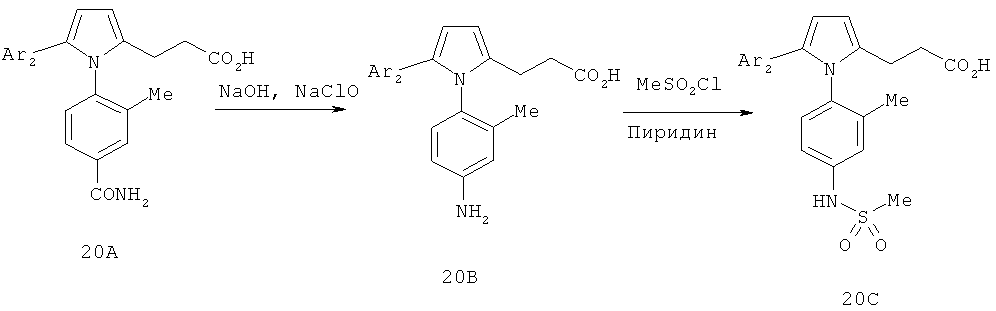
Примерная методика схемы 20: Синтез 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(2-метил-4-(метилсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 20С, Ar2=4-(1Н-имидазол-1-ил)фенил)
Синтез 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-амино-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 20В, Ar2=4-(1Н-имидазол-1-ил)фенил). 3-(5-(4-(1Н-Имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановую кислоту (соединение 20А, полученное в соответствии со схемой 5, Ar2=4-карбамоил-2-метилфенил) (3,88 г, 9,37 ммоля) добавляли к водн. раствору NaOH (4,12 г, 103,09 ммоля, растворенный в 50 мл). Затем добавляли по каплям 11% водн. NaClO (28,83 г, 42,17 ммоля). Полученную смесь выдерживали при 0~10°С в течение 1 ч, при 35°С в течение 1 ч и при 75°С в течение 30 мин. После охлаждения до комнатной температуры реакционную смесь подкисляли 10% хлористоводородной кислотой до значения рН=7,0, и отфильтровывали для удаления твердых примесей. Фильтрат далее подкисляли 10% хлористоводородной кислотой до значения рН=5,0, и появлялся новый осадок. Осадок отфильтровывали и сушили с получением соединения 20В, Ar2=4-(1Н-имидазол-1-ил)фенил, в виде серого порошка (3,20 г, 88%).
Синтез 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(2-метил-4-(метилсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 20С, Ar2=4-(1Н-имидазол-1-ил)фенил). К раствору пиридина (2 мл) и CH3SO2Cl/ДХМ (об/об=1/100, 5 мл) добавляли раствор 3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-амино-2-метилфенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 20В) (250 мг, 0,74 ммоля) в пиридине (2 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 1 ч. Растворители удаляли при пониженном давлении, и полученное твердое вещество подкисляли 10% хлористоводородной кислотой до значения рН=5,0. Полученный осадок выделяли с помощью центрифуги, промывали водой, сушили при пониженном давлении с получением соединения 20С, Ar2=4-(1Н-имидазол-1-ил)фенил, в виде коричневого порошка (40 мг, 13%).
Схему 21 - схему 32 умышленно пропускали.
Схема 33: Общая схема получения ингибиторов GSNOR структуры 33С

Примерная методика схемы 33: Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-метоксифенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение ЗЗС, R1=4-карбамоил-2-метилфенил, R2=4-хлор, R3=метил)
Синтез этил 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-гидроксифенил)-1Н-пиррол-2-ил)пропаноата (соединение 33А, R1=4-карбамоил-2-метилфенил, R2=4-хлор). Получали в соответствии со схемой 1 для 1C, R1=4-карбамоил-2-метилфенил, R2=4-хлор-2-гидроксифенил.
Синтез этил 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-метоксифенил)-1Н-пиррол-2-ил)пропаноата (соединение 33В, R1=4-карбамоил-2-метилфенил, R2=4-хлор, R3=метил). Этил 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-гидроксифенил)-1Н-пиррол-2-ил)пропаноат (300 мг, 0,704 ммоля) растворяли в ацетоне. Добавляли карбонат калия (146 мг, 1,056 ммоля) и метил йодид (299 мг, 2,112 ммоля), и перемешивали при комнатной температуре в течение ночи. Когда ТСХ показала, что реакция завершилась, смесь отфильтровывали, упаривали в вакууме. Остаток разделяли между этилацетатом (20 мл) и водой (5 мл). Органическую фазу сушили сульфатом магния, отфильтровывали и концентрировали с получением соединения 33В, R1=4-карбамоил-2-метилфенил, R2=4-хлор, R3=метил в виде желтого масла (295 мг, выход 95%).
Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-метоксифенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение ЗЗС, R1=4-карбамоил-2-метилфенил, R2=4-хлор, R3=метил). Гидролиз проводили с помощью конечной стадии схемы 5 с получением указанного в заголовке соединения.
Схема 34: Общая схема получения ингибиторов GSNOR структуры 34С

Примерная методика схемы 34: Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-циклопропил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 34С. Ar1-X=4-бромфенил, Ar2 представляет собой 2-циклопропил-1Н-имидазол-1-ил, R1=4-карбамоил-2-метилфенил)
Синтез этил 3-(5-(4-бромфенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (соединение 34A, R1=4-карбамоил-2-метилфенил, Ar1-X=4-бромфенил). Получали в соответствии со схемой 1, стадии 1-3.
Синтез этил 3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-циклопропил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропаноата (соединение 34В, Ar1-X=4-бромфенил, Ar2 представляет собой 2-циклопропил-1Н-имидазол-1-ил, R1=4-карбамоил-2-метилфенил. К смеси соединения 34A (Ar2=4-бромфенил) (455 мг, 1,0 ммоль) и 2-циклопропил-1H-имидазола (см. метод 14 для синтеза) (324 мг, 3,0 ммоля, 3,0 эквив.) в NMP (4 мл) добавляли 8-гидроксихинолин (22 мг, 0,15 ммоля, 0,15 эквив.), Cu2O (282 мг, 0,1 ммоля) и К2СО3 (166 мг, 1,2 ммоля) и PEG-2000 (50 мг). Полученную смесь в N2 подвергали микроволновому излучению при 128°С в течение 6,0 ч, охлаждали до комнатной температуры и разбавляли ТГФ (10 мл) и водой (10 мл). Смесь отфильтровывали, и полученный водный слой экстрагировали ЕА (30 мл × 5). Объединенные органические слои промывали насыщенным раствором хлорида натрия (20 мл), сушили MgSO4, отфильтровывали, концентрировали и очищали колоночной хроматографией на силикагеле (МеОН:CH2Cl2=1:15) с получением целевого соединения в виде желтого твердого вещества (190 мг, выход 39%).
Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-циклопропил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 34С Ar1-X=4-бромфенил, Ar2 представляет собой 2-циклопропил-1Н-имидазол-1-ил, R1=4-карбамоил-2-метилфенил). Гидролиз осуществляли в соответствии с конечной стадией схемы 5 с получением указанного в заголовке соединения.
Схему 35 умышленно пропускали.
Схема 36: Общая схема получения ингибиторов GSNOR структуры 36D

Примерная методика схемы 36: 3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-оксооксазолидин-3-ил)фенил)-1Н-пиррол-2-ил)пропановая кислота
Синтез этил 4,7-диоксо-7-(4-(2-оксооксазолидин-3-ил)фенил)гептаноата. К смеси этил 7-(4-бромфенил)-4,7-диоксогептаноата ((соединение 36А, где Ar1-Br=4-бромфенил, также см. соединение 5В, схема 5) (1,50 г, 4,4 ммоля) и оксазолидин-2-она (575 мг, 6,6 ммоля) в диоксане (5 мл) добавляли L-пролин (50 мг, 0,44 ммоля), CuI (42 мг, 0,22 ммоля) и К2СО3 (1,22 г, 8,8 ммоля). Полученную смесь перемешивали в атмосфере N2 при 110°С в течение 48 ч, и затем упаривали. Остаток разбавляли смесью ЕА/вода (40 мл/40 мл). Смесь отфильтровывали, и полученный водный слой экстрагировали ЕА (30 мл × 5). Объединенные органические слои промывали насыщенным раствором хлорида натрия, сушили NaSO4, концентрировали и очищали колоночной хроматографией на силикагеле (от чистого ДХМ до смеси ДХМ:МеОН=30:1) с получением указанного в заголовке соединения в виде белого твердого вещества (158 мг, выход 10%).
Синтез этил 3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-оксооксазолидин-3-ил)фенил)-1Н-пиррол-2-ил)пропаноата. К раствору этил 4,7-диоксо-7-(4-(2-оксооксазолидин-3-ил)фенил)гептаноата (158 мг, 0,43 ммоля) и 4-амино-3-метилбензамида (130 мг, 0,68 ммоля) в EtOH (1 мл) добавляли Zn(OTf)2 (313 мг, 0,86 ммоля). Смесь нагревали при 120°С при микроволновом излучении в течение 2 ч. После упаривания при пониженном давлении сырой продукт очищали колоночной хроматографией на силикагеле (ДХМ:МеОН=20:1) с получением указанного в заголовке соединения в виде желтого твердого вещества (77 мг, выход 39%).
Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-оксооксазолидин-3-ил)фенил)-1Н-пиррол-2-ил)пропановой кислоты. К раствору этил 3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-оксооксазолидин-3-ил)фенил)-1Н-пиррол-2-ил)пропаноата (67 мг, 0,15 ммоля) в смеси ТГФ/Н2О (1 мл, об/об=1/1) добавляли моногидрат гидроксида лития (7 мг, 0,15 ммоля). Смесь перемешивали при комнатной температуре в течение 6 ч. ТГФ упаривали в вакууме. Остаток подкисляли до значения рН=5 с помощью 5% хлористоводородной кислоты, концентрировали и очищали преп-ТСХ с получением указанного в заголовке соединения в виде коричневого твердого вещества (24 мг, выход 39%).
Схема 36А: Альтернативные условия для получения промежуточных соединений типа соединения 36В (схема 36 выше).
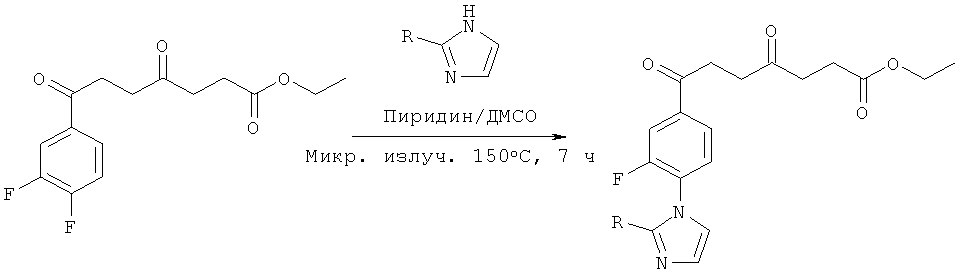
Примерная методика схемы 36А: Синтез этил 7-(3-фтор-4-(1Н-имидазол-1-ил)фенил)-4,7-диоксогептаноата (R=Н). Этил 7-(3,4-дифторфенил)-4,7-диоксогептаноат (351 мг) обрабатывали имидазолом (241 мг) и пиридином (395 мг) в ДМСО (3 мл) при 150°С в течение 7 ч при микроволновом излучении. Полученную смесь разбавляли водой (12 мл) и экстрагировали EtOAc (20 мл × 3). После удаления растворителей смесь очищали ускоренной хроматографией на силикагеле, элюируя EtOAc, с получением целевого продукта - этил 7-(3-фтор-4-(1Н-имидазол-1-ил)фенил)-4,7-диоксогептаноата (279 мг, 68%) в виде светло-коричневого твердого вещества.
Схему 37 - схему 38 умышленно пропускали.
Схема 39: Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-(диметиламино)фенил)-1Н-пиррол-2-ил)пропановой кислоты
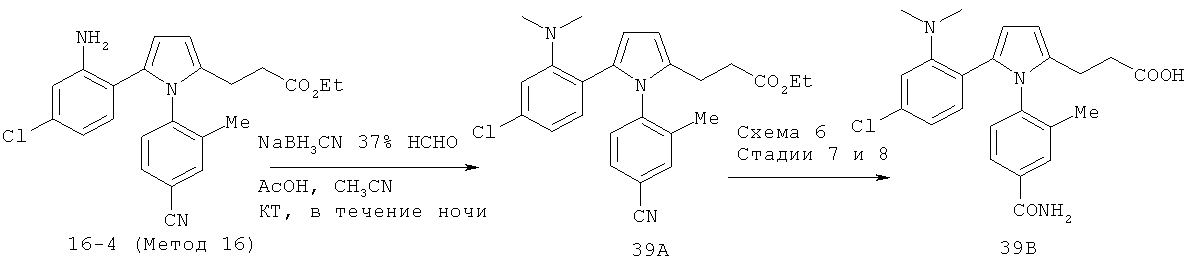
Синтез соединения 39А. К смеси соединения 16-4 (метод 16) (200 мг, 0,419 ммоля). NaBH3CN (53 мг, 0,838 ммоля), 37%HCHO (1,5 мл, 2,095 ммоля) в CH3CN (5 мл) добавляли АсОН (0,5 мл). После перемешивания при комнатной температуре в течение ночи раствор концентрировали и разбавляли водой (15 мл), экстрагировали этилацетатом (10 мл × 4). Органическую фазу отделяли и сушили, очищали преп-ТСХ (РЕ:ЕА=1: 1) с получением соединения 39А в виде желтого масла (97 мг, 49%).
Синтез соединения 39В. Следуя методике, описанной на последних двух стадиях схемы 6 (стадии 7 и 8), с очисткой конечного продукта преп-ВЭЖХ.
Схема 40: Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-формамидофенил)-1Н-пиррол-2-ил)пропановой кислоты

Синтез этил 3-(5-(4-хлор-2-формамидофенил)-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата (соединение 40А): Смесь Ас2О (301 мг, 2,948 ммоля) и НСО2Н (226 мг, 4,914 ммоля) перемешивали при 55°С в течение 5 мин. Смесь добавляли к раствору соединения 16-4 (см. метод 16 для синтеза) (300 мг, 0,737 ммоля) в ТГФ (6 мл), и перемешивали при 55°С в течение 10 мин. ТСХ показала окончание реакции. Летучие примеси удаляли при пониженном давлении, и остаток растворяли в ЕА (50 мл), промывали насыщ. раствором NaHCO3 (10 мл × 3) и насыщенным раствором хлорида натрия (10 мл). Органический слой сушили безводным Na2SO4, отфильтровывали и концентрировали с получением сырого продукта в виде желтого твердого вещества (320 мг, выход: 99%), которое непосредственно использовали на следующей стадии.
Синтез 3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-формамидофенил)-1Н-пиррол-2-ил)пропановой кислоты (соединение 40В): См. методику, описанную на последних стадиях схемы 6 (6F→6H).
Следующие методы использовали для получения промежуточных соединений, которые использовались на приведенных выше схемах, как показано в таблице.
Метод 1 - метод 11 умышленно пропускали.
Метод 12: Синтез 2-хлор-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина
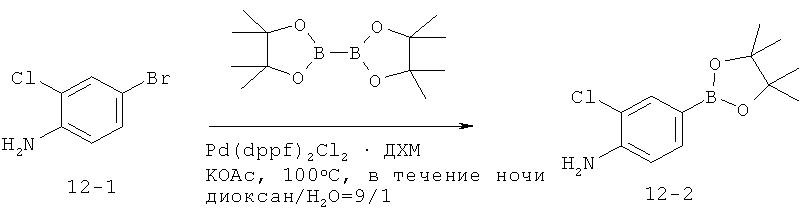
Соединение 12-2. Раствор соединения 12-1 (12,3 г, 0,06 ммоля), бис(пинаколято)диборона (18,3 г, 0,072 моля), КОАс (11,75 г, 0,12 ммоля) и Pd(dppf)2Cl2 ДХМ (2,0 г, 2,45 ммоля) в смеси диоксан/Н2О (об/об=9/1, 100 мл) перемешивали при 80°С в течение ночи. ТСХ показала, что реакция завершилась. Смесь упаривали с получением коричневого масла. Добавляли воду (60 мл), и смесь экстрагировали этилацетатом (60 мл × 3). Объединенные органические слои сушили MgSO4, отфильтровывали, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=10:1) с получением соединения 12-2 в виде желтого твердого вещества (9,1 г, 60%).
Метод 13 - метод 14 умышленно пропускали.
Метод 15: Синтез 1-(4-бром-2-метоксифенил)этанона

Соединение 15-1. К перемешиваемой суспензии 3-бромфенола (50 г, 0,29 моля) в пиридине (200 мл) и дихлорметане (100 мл) добавляли по каплям ацетилхлорид (25 мл, 0,35 моля) при 0°С, и смесь перемешивали в течение 18 ч при комнатной температуре. LC-MS показала, что реакция завершилась. Пиридин и дихлорметан упаривали в вакууме. Добавляли воду (600 мл) и подкисляли хлористоводородной кислотой до значения рН 2. Реакционную смесь экстрагировали этилацетатом (500 мл × 3), и органическую фазу сушили безводным сульфатом натрия, отфильтровывали, концентрировали и очищали колоночной хроматографией (РЕ:ЕА=60:1) с получением соединения 15-1 в виде бесцветной жидкости (46 г, 74%).
Соединение 15-2. Перемешиваемую суспензию соединения 15-1 (46 г, 0,0,21 моля) и безводного порошка хлорида алюминия (57 г, 0,42 моля) нагревали при 160°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и выливали в лед (200 г) и воду (800 мл), и очищали хлористоводородной кислотой при рН 7. Реакционную смесь экстрагировали этилацетатом (500 мл × 3), и органическую фазу промывали насыщенным раствором бикарбоната натрия, сушили безводным сульфатом натрия, отфильтровывали, концентрировали и очищали колоночной хроматографией (РЕ:ЕА=60:1) с получением соединения 15-2 в виде светло-зеленого твердого вещества (35,1 г, 76%).
Соединение 15-3. К суспензии соединения 15-2 (25 г, 0,12 моля) и карбоната калия (24 г, 0,18 моля) в безводном DMF (20 мл) добавляли MeI (22,6 мл, 0,23 моля), и реакционную смесь перемешивали при комнатной температуре в течение ночи. LCMS показала, что реакция завершилась. Затем выливали в воду (300 мл), и смесь экстрагировали этилацетатом, и органическую фазу (200 мл × 3) промывали насыщенным раствором хлорида натрия, сушили безводным сульфатом натрия, отфильтровывали, концентрировали с получением соединения 15-3 в виде бесцветного твердого вещества (26,1 г, 98%).
Метод 16: Синтез этил 3-(5-(2-амино-4-хлорфенил)-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноата
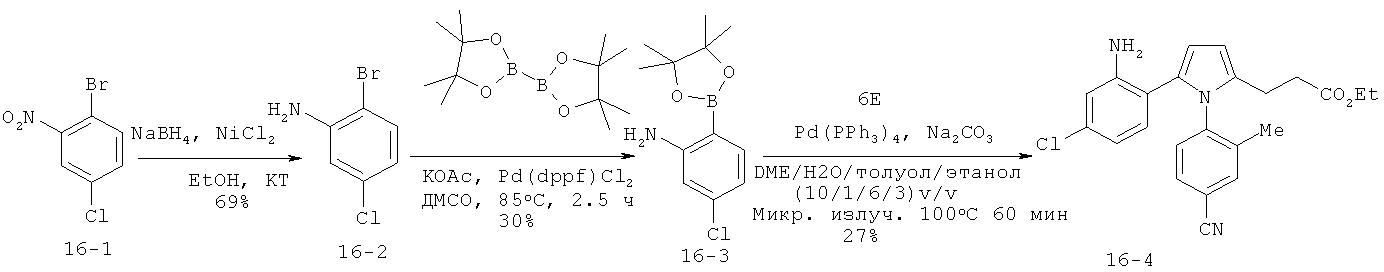
Соединение 16-2. К раствору соединения 16-1 (6,50 г, 27,66 ммоля) и NiCl2 (7,80 г, 55,3 ммоля) в EtOH (50 мл) медленно добавляли NaBH4 (5,60 г, 138,3 ммоля). Полученную смесь перемешивали при 0°С в течение 2 ч, отфильтровывали и концентрировали при пониженном давлении. Остаток растворяли в этилацетате (200 мл), промывали водой (50 мл × 3), сушили Na2SO4, концентрировали и очищали на колонке с силикагелем (РЕ:ЕА=5:1) с получением соединения 16-2 в виде темного твердого вещества (3,778 г, выход 67%).
Соединение 16-3. Раствор соединения 16-2 (3,778 г, 18,43 ммоля), бис(пинаколято)диборона (8,5 г, 33,17 моля), КОАс (3,2 г, 36,86 ммоля) и Pd(dppf)2Cl2·ДХМ (500 мг, 0,92 ммоля) в ДМСО (50 мл) перемешивали при 85°С в течение 2,5 ч. ТСХ показала, что реакция завершилась. Добавляли воду (60 мл), и смесь экстрагировали этилацетатом (60 мл × 3). Объединенные органические слои сушили Na2SO4, отфильтровывали, концентрировали и очищали на колонке с силикагелем (РЕ:ЕА=10:1) с получением соединения 16-3 в виде желтого твердого вещества (5,0 г, выход 100%).
Соединение 16-4. К раствору соединения 16-3 (7,0 г, 27,7 ммоля), Na2CO3 (11,75 г, 110,8 ммоля) и соединения 6Е (этил 3-(5-бром-1-(4-циано-2-метилфенил)-1Н-пиррол-2-ил)пропаноат, см. схему 6) (10 г, 21,4 ммоля) в ДМСО (30 мл) добавляли Pd(PPh3)4 (3,0 г, 8,31 ммоля). После дегазирования и насыщения азотом реакционную смесь перемешивали при 80°С в течение ночи. ТСХ показала, что реакция завершилась. После охлаждения до комнатной температуры добавляли воду (50 мл), и экстрагировали этилацетатом (50 мл × 4). Объединенные органические слои сушили Na2SO4, отфильтровывали, концентрировали и очищали на колонке с силикагелем (РЕ:ЕА=3:1) с получением соединения 16-4 в виде желтого твердого вещества (3,10 г, выход 27%).
Метод 17 умышленно пропускали.
Метод 18: Синтез 3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенола

Соединение 18-2. Получали той же методикой, как описано в методе 12, с диоксаном в качестве растворителя, и очищали колоночной хроматографией (РЕ:ЕА=5:1) с получением целевого соединения с выходом 35%.
Метод 19: Синтез 1-(5-бромтиофен-3-ил)этанона

Соединение 19-2. К раствору 3-ацетилтиофена (2,52 г, 20 ммолей, 1,0 эквив.) в НОАс (50 мл) добавляли NaOAc (2,46 г, 30 ммолей, 1,5 эквив.), затем по каплям бром (3,2 г, 20 ммолей, 1,0 эквив.) в течение 30 мин. Смесь оставляли перемешиваться при комнатной температуре в течение ночи. Добавляли воду (150 мл), и реакционную смесь перемешивали в течение 2 ч. Полученное твердое вещество собирали фильтрацией, промывали водой (10 мл) и РЕ (20 мл), и сушили с получением соединения 19-2 в виде коричневого твердого вещества (1,52 г, выход 37%).
Метод 20 - метод 22 умышленно пропускали.
Метод 23: Синтез N-(4-аминофенил)метансульфонамида (соединение 23-3, R=Н) и N-(4-амино-3-метилфенил)метансульфонамида (соединение 23-3, R=СН3)

Примерный пример метода 23: Синтез N-(4-аминофенил)метансульфонамида (соединение 23-3, R=Н)
Соединение 23-2, R=Н. К раствору пиридина (50 мл) и MsCl (15,86 г, 139,13 ммоля) в ДХМ (150 мл) добавляли раствор 4-нитробензоламина (16,0 г, 115,94 ммоля) в пиридине (100 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 4 ч. Летучие примеси удаляли при пониженном давлении. Остаток промывали водой (200 мл × 3) и сушили при пониженном давлении с получением указанного в заголовке соединения в виде желтого порошка (23,20 г, выход 95%).
Соединение 23-3, R=Н. К раствору соединения 23-2, R=Н (23,0 г, 106,48 ммоля) в МеОН (100 мл) добавляли 10% Pd/C (3,0 г), насыщали N2. Затем постепенно добавляли раствор HCO2NH4 (67,0 г, 1,06 моля) в МеОН (500 мл) в бане лед-вода в течение 5 мин. После добавления смесь нагревали до 45°С и перемешивали в течение ночи и отфильтровывали. Фильтрат упаривали при пониженном давлении с получением желтого твердого вещества, которое промывали ЕА (500 мл × 3). Объединенные органические слои упаривали при пониженном давлении, очищали колоночной хроматографией на силикагеле (РЕ:ЕА=1:2) с получением N-(4-аминофенил)метансульфонамида в виде желтого твердого вещества (9,80 г, выход 49%).
Метод 24: Синтез 1-(3-хлортиофен-2-ил)этанона

Соединение 24-1. К раствору 3-хлортиофена (4,80 г, 40,48 ммоля) в ТГФ (50 мл) добавляли BuLi (2,5N в гексане, 17,9 мл) при -30°С. После добавления смесь перемешивали в течение 30 мин при -10°С, и затем охлаждали до -45°С. Добавляли N-метокси-N-метилацетамид (55,0 г, 48,8 ммоля), и оставляли нагреваться до комнатной температуры в течение 40 мин, и оставляли еще на 20 мин. Добавляли насыщенный раствор хлорида натрия (80 мл) для того, чтобы погасить реакцию, экстрагировали ЕА (60 мл × 3). Объединенные органические слои сушили безводным Na2SO4, отфильтровывали, концентрировали с получением 24-1 (~80% чистота) в виде желтого масла (6,80 г), которое непосредственно использовали на следующей стадии.
Метод 25: Синтез 1-(3-бром-5-метокситиофен-2-ил)этанона
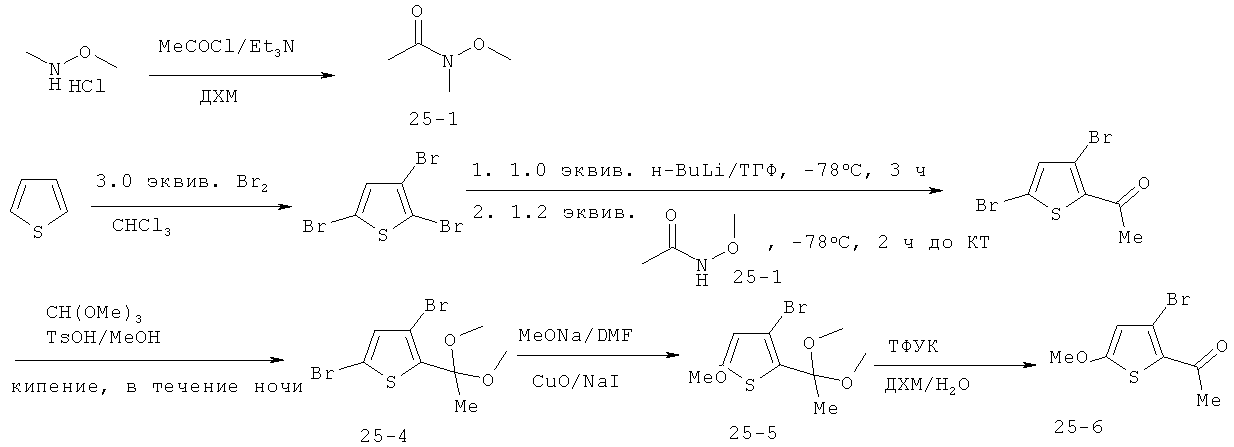
Соединение 25-1. К суспензии гидрохлорида М,0-диметилгидроамина (100 г, 1026 ммолей) в ДХМ (1000 мл) добавляли триэтиламин (300 мл, 2052 ммоля) при 0°С. К суспензии по каплям добавляли ацетилхлорид в течение 2 ч при 0°С. После окончания добавления смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 2 ч. Смесь промывали насыщенным раствором хлорида натрия (1 л), 1 N HCl (500 мл), насыщенным раствором хлорида натрия (200 мл) соответственно и сушили сульфатом магния, отфильтровывали и концентрировали с получением коричневого масла, которое очищали перегонкой с получением соединения 25-1 в виде прозрачной жидкости (65 г, 61%).
Соединение 25-2. К раствору тиофена (84 г, 1,0 моль) в хлороформе (34 мл) добавляли по каплям бром при комнатной температуре в течение 3 ч. После окончания добавления смесь перемешивали при комнатной температуре в течение ночи. Смесь нагревали при 50°С в течение 3 ч. Реакционную смесь промывали 1М NaOH (водн. 100 мл), насыщенным раствором хлорида натрия (100 мл × 2) соответственно. Органическую фазу сушили безводным сульфатом натрия, отфильтровывали и концентрировали с получением светло-желтого масла, которое затвердевало в метаноле (100 мл). Твердое вещество отфильтровывали и сушили в вакууме с получением соединения 25-2 (89 г, 56%).
Соединение 25-3. 25-2 (9,5 г, 30 ммолей) растворяли в безводном ТГФ (100 мл) и охлаждали до -78°С. К указанному раствору по каплям добавляли н-BuLi (8 мл, 21 ммоль) в течение 30 мин, и перемешивали в течение 30 мин. По каплям добавляли соединение 25-1 при -78°С, перемешивали в течение 30 мин и оставляли нагреваться до комнатной температуры, затем гасили насыщенным раствором хлорида аммония. Органическую фазу отделяли и промывали насыщенным раствором хлорида натрия, сушили безводным Na2SO4, отфильтровывали и концентрировали с получением желтого масла, которое очищали колоночной хроматографией (элюирование: РЕ/ЕА=10/1) с получением соединения 25-3 в виде желтого твердого вещества (2,3 г, 28%).
Соединение 25-4. К раствору соединения 25-3 (2,4 г, 8,5 ммоля) в метаноле (35 мл) добавляли триметилортоформиат (15 мл) и TsOH (300 мг, 1,7 ммоля). Раствор нагревали при кипении с обратным холодильником в течение 10 ч. Метанол упаривали в вакууме, и остаток разделяли между ЕА (300 мл) и 5% бикарбонатом натрия (100 мл). Органическую фазу отделяли, сушили безводным сульфатом натрия, отфильтровывали и концентрировали с получением соединения 25-4 в виде желтого масла, которое использовали непосредственно на следующей стадии (2,3 г, 82%).
Соединение 25-5. К раствору соединения 25-4 (6,0 г, 18,3 ммоля) в DMF (75 мл) добавляли метоксид натрия (9,9 г, 183 ммоля), оксид меди (1,5 г, 18,3 ммоля) и йодид натрия (2,8 г, 18,3 ммоля). Смесь нагревали при 100°С в течение 4 ч. ТСХ показала, что реакция завершилась, и реакцию гасили насыщенным раствором хлорида натрия (250 мл). Твердое вещество отфильтровывали, и фильтрат экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои сушили безводным сульфатом натрия, отфильтровывали и концентрировали с получением коричневого масла, которое очищали колоночной хроматографией (элюирование: РЕ/ЕА=3/1) с получением соединения 25-5 в виде светло-желтого масла (1,2 г, 23%).
Соединение 25-6. К раствору соединения 25-5 (1,2 г, 4,29 ммоля) в ДХМ (8 мл) и воде (10 мл) добавляли трифторуксусную кислоту (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Добавляли насыщенный раствор бикарбоната натрия (10 мл), и органическую фазу отделяли, сушили безводным сульфатом натрия, отфильтровывали и концентрировали с получением коричневого масла, которое очищали колоночной хроматографией (элюируя: РЕ/ЕА=10/1) с получением соединения 25-6 в виде светло-желтого твердого вещества (750 г, 74%).
Метод 26 умышленно пропускали.
Метод 27: Синтез 4-амино-3-метилбензолсульфонамида

Соединение 27-2: Ас2О (16 мл, 0,16 моля) добавляли к раствору соединения 27-1 (20 г, 0,107 моля) в 80 мл пиридина. Смесь перемешивали при комнатной температуре в течение 2 часов. Затем добавляли EtOH (40 мл), и твердое вещество выделяли фильтрацией и промывали EtOH с получением соединения 27-2 в виде коричневого твердого вещества (10,3 г, выход 56%).
Соединение 27-3: Соединение 27-2 (10 г, 43,6 ммоля) добавляли в колбу, содержащую 1N NaOH (36 мл), и смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель удаляли, и остаток промывали EtOH. Соединение 27-3 выделяли фильтрацией в виде бледного твердого вещества (8,8 г, выход 88%).
Соединение 27-4: Соединение 27-3 (16 г, 63,7 ммоля) и DMF (20 мл) добавляли в колбу, и затем добавляли по каплям SOCl2 (18,4 г, 155 моля) при -30-40°С. Когда добавление завершали смесь перемешивали при комнатной температуре в течение 2 часов. Затем смесь добавляли медленно в лед, и появлялось твердое вещество. Твердое вещество выделяли фильтрацией и сушили с получением соединения 27-4 в виде бледного твердого вещества (6,0 г, выход 38%).
Соединение 27-5: Раствор соединения 27-4 (6,0 г, 24,2 ммоля) в 50 мл ТГФ по каплям добавляли к 50 мл NH4OH при 0°С. Смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли при пониженном давлении, и остаток экстрагировали ЕА (30 мл × 4). Органический слой сушили Na2SO4 и отфильтровывали, концентрировали с получением соединения 27-5 в виде бледного твердого вещества (5,1 г, выход 93%).
Соединение 27-6: Смесь соединения 27-5 (5,1 г, 22,3 ммоля), HCl (2 N, 76,5 мл) и EtOH (100 мл) кипятили с обратным холодильником в течение ночи. Затем смесь нейтрализовали Na2CO3 (водн.) до значения рН=8. Смесь экстрагировали ЕА (80 мл × 4), сушили Na2SO4, и концентрировали с получением соединения 27-6 в виде бледного твердого вещества (4,9 г, выход 100%).
Метод 28: Синтез 1-(5-бром-4-хлортиофен-2-ил)этанона (соединение 28-2) и 1-(4-хлортиофен-2-ил)этанона (соединение 28-3)

Соединение 28-1. К раствору 3-хлортиофена (6,52 г, 55 ммолей) в CHCl3 (30 мл) и АсОН (30 мл) добавляли NBS (9,80 г, 55 ммолей). Смесь нагревали при кипении с обратным холодильником в течение 1,5 ч, затем охлаждали до комнатной температуры. Добавляли воду (70 мл), и смесь экстрагировали CHCl3 (30 мл × 2). Объединенные органические слои промывали насыщ. раствором NaHCO3 (40 мл) и насыщенным раствором хлорида натрия (30 мл), сушили безводным Na2SO4, отфильтровывали, концентрировали с получением соединения 28-1 в виде коричневого масла (10,02 г, количественный выход), которое использовали непосредственно на следующей стадии.
Синтез 1-(5-бром-4-хлортиофен-2-ил)этанона (соединение 28-2). К смеси соединения 28-1 (10,0 г, 50,6 ммоля) и AlCl3 (8,09 г, 60,7 ммоля) в ДХМ (120 мл) добавляли по каплям ацетилхлорид (4,76 г, 60,7 ммоля) в течение 5 мин при 0°С. После окончания добавления смесь перемешивали в течение ночи при комнатной температуре, промывали разбавленной хлористоводородной кислотой (1,2N, 150 мл) и насыщенным раствором хлорида натрия (150 мл), сушили безводным Na2SO4, отфильтровывали, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ/ЕА = от 20/1 до 3/1) с получением соединения 28-2 в виде коричневого твердого вещества (8,0 мг, выход: 66%).
Синтез 1-(4-хлортиофен-2-ил)этанона (соединение 28-3). К раствору соединения 28-2 (3,20 мг, 13,36 ммоля) в EtOH (70 мл) добавляли 10% Pd/C (2,50 г) и AcONa (1,10 г, 13,36 ммоля). Реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 3 ч, отфильтровывали, и фильтрат концентрировали. Полученный остаток растворяли в ЕА (100 мл) промывали насыщ. раствором NaHCO3 (40 мл) и насыщенным раствором хлорида натрия (30 мл), сушили безводным Na2SO4, отфильтровывали, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ/ЕА = от 30/1 до 5/1) с получением соединения 28-3 в виде желтого масла (1,32 г, выход: 62%).
Метод 29 - метод 32 умышленно пропускали.
Метод 33: Синтез N-(3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)формамида
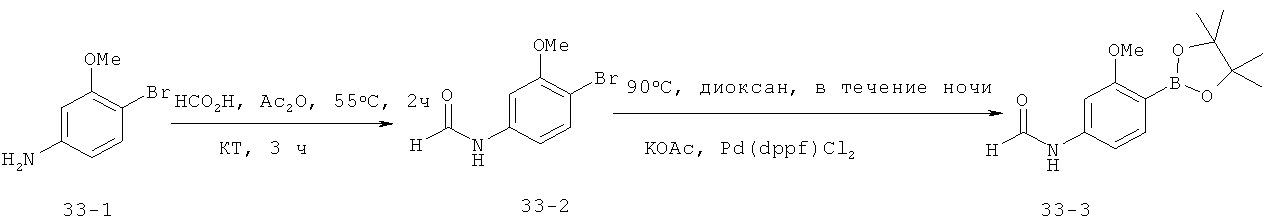
Синтез соединения 33-2: смесь НСО2Н (644 мг, 14 ммолей) и Ас2О (1,16 г, 11,4 ммоля) нагревали при 55°С в течение 2 ч, и затем охлаждали до комнатной температуры. По очереди добавляли ТГФ (1 мл) и соединение 33-1 (880 мг, 4,38 ммоля) в ТГФ (1 мл), и полученную смесь непрерывно перемешивали при комнатной температуре в течение 3 ч. После упаривания остаток экстрагировали ЕА (5 мл × 3). Органическую фазу последовательно промывали насыщ. водным раствором бикарбоната натрия (10 мл) и хлорида натрия (10 мл), сушили безводным сульфатом натрия, отфильтровывали и концентрировали с получением соединения 33-2 в виде жидкости (845 мг, выход: 85%), которую использовали непосредственно на следующей стадии.
Синтез соединения 33-3: к смеси соединения 33-2 (845 мг, 4,38 ммоля), KOAc (726 мг, 7,4 ммоля), B(pin)2 (1,41 г, 5,6 ммоля) и диоксана добавляли Pd(dppf)2Cl2 (20 мг, 0,02 ммоля). После дегазирования и насыщения азотом смесь кипятили с обратным холодильником при 90°С в течение ночи. ТСХ показала, что реакция завершилась. Добавляли воду (10 мл), и смесь экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои сушили Na2SO4, отфильтровывали, концентрировали и очищали колоночной хроматографией на силикагеле (РЕ:ЕА=4:1) с получением соединения 33-3 в виде бесцветного твердого вещества (220 мг, выход: 29%).
Метод 34 умышленно пропускали.
Метод 35: Синтез 4-аминобензолсульфонамида
Следуя методике/схеме, описанной в методе 27, для синтеза 4-амино-3-метилбензолсульфонамида.
Метод 36-40 умышленно пропускали.
Метод 41: Синтез 1-(5-бром-2-метоксифенил)этанона

Соединение 41-2. К раствору соединения 41-1 (2,0 г, 13,32 ммоля) в ацетоне (25 мл) добавляли NBS (2,37 г, 13,32 ммоля) и 1М водн. HCl (0,13 мл, 0,13 ммоля). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, и затем концентрировали досуха при пониженном давлении. Остаток растворяли в РЕ (40 мл), полученный осадок отфильтровывали и сушили в вакууме с получением соединения 41-2 в виде белого твердого вещества (2,90 г, выход: 95%).
Пример 2: Анализы с GSNOR
Различные соединения тестировали in vitro на их способность ингибировать активность GSNOR. Примерные соединения и их соответствующая активность в отношении GSNOR описаны выше в параграфе перед таблицей 1. Экспрессия и очистка GSNOR описана в статье Biochemistry, 2000, 39, сс.10720-10729.
Ферментация GSNOR: Предварительные культуры высаживали из срезов стержня GSNOR в глицерине в 2XYT среде, содержащей 100 ug/мл ампициллина после инкубирования в течение ночи при 37°С. Клетки затем добавляли в свежий 2XYT (4 л), содержащий ампициллин, и выращивали до OD (А600) 0,6-0,9 при 37°С перед введением. Экспрессию GSNOR индуцировали 0,1% арабинозой при инкубировании в течение ночи при 20°С.
Очистка GSNOR: Пасту клеток Е. coli подвергали лизису при кавитации азотом, и свободный лизат очищали Ni аффинной хроматографией на АКТА FPLC (Amersham Pharmacia). Колонку элюировали 20 мМ Tris pH 8,0/250 мМ NaCl с градиентом 0-500 мМ имидазола. Элюированные фракции GSNOR, содержащие слитой Smt-GSNOR, настаивали в течение ночи с Ulp-1 при 4°С для удаления аффиности к мишени, затем повторно запускали на Ni колонку в тех же условиях. GSNOR извлекали в поточной фракции, и для кристаллографии затем очищали с помощью Q-сефарозы и гепариновой поточной хроматографии в 20 мМ Tris рН 8,0, 1 мМ DTT, 10 мкМ ZnSO4.
Анализ с GSNOR: Методика: Растворы GSNO и фермент/NADH получали свежими каждый день. Растворы отфильтровывали и оставляли нагреваться до комнатной температуры. Раствор GSNO: 100 мМ NaPO4 (рН 7,4), 0,480 мМ GSNO. 396 мкл раствора GSNO добавляли в кювету, затем 8 мкл тестируемого соединения в ДМСО (или только ДМСО для полного контроля реакции), и смешивали наконечником пипетки. Тестируемые соединения получали в конечной концентрации 10 мМ в 100% ДМСО. 2-кратные серийные разбавления осуществляли в 100% ДМСО. 8 мкл каждого разбавления добавляли в анализ так, чтобы конечная концентрация ДМСО в анализе составляла 1%. Концентрации тестируемых соединений составляли в диапазоне от 100 до 0,003 мкМ. Раствор фермент/NADH: 100 мМ NaPO4 (рН 7,4), 0,600 мМ NADH, 1,0 мкг/мл GSNO редуктазы. 396 мкл раствора фермент/NADH добавляли в кювету для инициирования реакции. Кювету помещали в УФ/оптический спектрофотометр Cary 3Е, и изменение поглощения при 340 нм/мин при 25°С записывали в течение 3 минут. Анализы осуществляли трижды для каждой концентрации соединения. Значения IC50 для каждого соединения рассчитывали, используя стандартный анализ построения кривых в Enzyme Kinetics Module of SigmaPlot.
Условия конечного анализа: 100 мМ NaP04, рН 7,4, 0,240 мМ GSNO, 0,300 мМ NADH, 0,5 мкг/мл GSNO редуктазы и 1% ДМСО. Конечный объем: 800 мкл/кювета.
Пример 3: Анализ ингибирования GSNOR на модели животного in vivo
Для демонстрации влияния на ингибирование GSNOR использовали модель мыши с астмой, которая аналогична модели, предварительно описанной для влияния GSNO редуктазы и биодоступности SNO (статья Que и др.. Science, 2005). Que и др. Показали, что после введения ova-альбумина (OVA) мыши дикого типа проявляют бронхиальную реактивность с повышенными уровнями GSNOR, и имеют легкие, которые расщепляют SNO. В противоположность мышам дикого типа, Que и др. показали, что мыши с генетическим расщеплением GSNOR повышают легочные SNO и защищают от вызванной OVA гиперреактивности дыхательных путей.
Для определения того, будут ли аналогичные наблюдения проявляться, если GSNOR ингибируется фармакологически ингибитором GSNOR, использовали модель мыши OVA (то есть модель мыши дикого типа Que и др.). В этом анализе OVA-чувствительным мышам вводили 1 мг/кг, 10 мг/кг или 30 мг/кг соединения 1 внутривенно за 24 часа до помещения в плетизмограф всего тела (Buxco Research Systems, Wilmington, NC), и обеспечивали свежий воздух.
Исследуемым животным затем вводили аэрозоль повышенных дозировок бронхорасширяющего агента метахолина, фармакологического агента, общеиспользуемого для определения степени бронхиальной гиперреактивности у экспериментальных субъектов. В этом анализе мышам вводили повышенные концентрации метахолина, каждую дозу сохраняли в течение 3 минут, после чего проводили измерения. Дозы метахолина составляли 0 мг/мл, 5 мг/мл, 20 мг/мл и 50 мг/мл. Степень бронхиальной гиперреактивности измеряли как ′повышенная пауза′ (Penh), единичный индекс гиперреактивности дыхательных путей (статья Dohi и др., Lab Invest., 1999, 79(12), сс.1559-1571).
Введение соединения 1 производило низкие бронхорасширяющие отклики у этих тестируемых животных по сравнению с животными, обработанными только носителем. Эти результаты сочетаются с большими уровнями биоактивности SNO, доступными для бронхорасширяющего метахолинового изменения.
Пример 4: Эффективность GSNORi в эксперименте с астмой
Экспериментальная модель астмы:
Модель мыши с вызванной овальбумином (OVA) астмой использовали для скрининга ингибиторов GSNOR на эффективность в отношении вызванной метахолином (MCh) гиперреактивности бронхоконстрикции/дыхательных путей. Эта широко используемая и хорошо характеризуемая модель, которая представлена в фенотипе острой, аллергической астмы с аналогиями астмы человека. Эффективность ингибиторов GSNOR оценивали с помощью профилактической методики, в которой ингибиторы GSNOR вводили до введения MCh. Бронхоконстрикцию в ответ на введение повышенных доз MCh оценивали с помощью плетизмографии всего тела (Penh; Buxco). Количество эозинофильного инфильтрата в бронхиальной промывочной жидкости (BALF) также определяли как значение легочного воспаления. Действие ингибиторов GSNOR по сравнению с носителями и с Combivent (ингаляцией; IH) в качестве положительного контроля.
Материалы и методы
Чувствительность к аллергену и методика введения
OVA (500 мкг/мл) в PBS смешивали с равными объемами 10% (вес/об) сульфата алюминия калия в дистиллированной воде, и инкубировали в течение 60 мин при комнатной температуре, затем значение рН доводили до 6,5 с помощью 10 N NaOH. После центрифугирования при 750×g в течение 5 мин остаток OVA/alum повторно суспендировали до начального объема в дистиллированной воде. Мышам вводили внутрибрюшинной (IP) инъекцией 100 мкг OVA (0,2 мл 500 мкг/мл в обычном соляном растворе) в комплексе с alum на день 0. Мышей анестезировали IP инъекцией 0,2 мл смеси кетамина и ксилазина (0,44 и 6,3 мг/мл, соответственно) в обычном соляном растворе, и помещали на доску в положении лежа на спине. 250 мкг (100 мкл 2,5 мг/мл) OVA (на день 8) и 125 мкг (50 мкл 2,5 мг/мл) OVA (в дни 15, 18 и 21) помещали на заднюю сторону языка каждого животного.
Тестирование легочной функции (Penh)
Реактивность дыхательных путей in vivo на метахолин измеряли через 24 ч после последнего введения OVA у мышей в сознании, свободно двигающихся, спонтанно дышащих, с помощью плетизмографии всего тела в камере Buxco (Wilmington, NC). Мышам вводили аэрозолем соляной раствор или повышенные дозы метахолина (5, 20 и 50 мг/мл), полученным в ультразвуковом небулайзере в течение 2 мин. Степень бронхоконстрикции выражали как повышение паузы (Penh), рассчитанной безразмерной величины, которая соответствует измерению резистентности дыхательных путей, полного сопротивления и внутриплеврального давления на тех же мышах. Значения Penh брали и усредняли в течение 4 мин после каждого введения аэрозоля. Penh рассчитывали следующим образом: Penh=[(Те/Tr-1)×(PEF/PIF)], где Те представляет собой время выдоха, Tr представляет собой время расслабления, PEF представляет собой пик потока выдоха, и PIF представляет собой пик потока вдоха × коэффициент 0,67. Время для полного давления для изменения с максимального на пользовательское процентное соотношение максимума представляет собой время релаксации. Измерение Tr начинается с максимального полного давления и заканчивается на 40%.
Инфильтрат эозинофилов в BALF
После измерения гиперреактивности дыхательных путей мышей обескровливали пункцией сердца, и затем BALF собирали из обоих легких или правого легкого после отсоединения левого легкого от бронхов. Общее количество клеток BALF определяли из аликвоты 0,05 мл, и остальную жидкость центрифугировали при 200×g в течение 10 мин при 4°С. Остаток клеток повторно суспендировали в соляном растворе, содержащем 10% BSA, мазками, полученными на стеклянных пластинах. Эозинофилы окрашивали в течение 5 мин 0,05% водным эозином и 5% ацетоном в дистиллированной воде, промывали дистиллированной водой и повторно окрашивали 0,07% метиленовым голубым.
Ингибиторы GSNOR и контроли
Ингибиторы GSNOR восстанавливали в забуференном фосфатом соляном растворе (PBS), рН 7,4, в концентрациях в диапазоне от 0,00005 до 3 мг/мл. Ингибиторы GSNOR вводили мышам (10 мл/кг) в виде одной дозы внутривенно (IV) или перорально. Введение дозировки осуществляли от 30 мин до 24 ч до введения MCh. Действие ингибиторов GSNOR сравнивали с носителем PBS, введенным таким же способом.
Combivent использовали в качестве положительного контроля во всех испытаниях. Combivent (Boehringer Ingelheim) вводили в легкое с помощью ингаляционного устройства, содержащего продукт, но подгоняли для введения мыши, используя наконечник для пипетки. Combivent вводили за 48 ч, 24 ч и 1 ч до введения MCh. Каждое вдыхание (или доза) Combivent обеспечивало дозу 18 мкг ипратропия бромида (IpBr) и 103 мкг сульфата альбутерола, или около 0,9 мг/кг IpBr и 5 мг/кг альбутерола.
Статистические анализы
Значения площади под кривой для Penh введения базового вещества, соляного раствора и повышенных доз MCh рассчитывали с помощью GraphPad Prism 5,0 (San Diego, CA), и выражали как процентное соотношение соответствующего (IV или перорально вводимого) контрольного носителя. Статистические различия между обрабатываемыми группами и соответствующей группой контрольного носителя в каждом исследовании рассчитывали с помощью одностадийного ANOVA, Dunnetts (JMP 8,0, SAS Institute, Cary, NC). Значение р<0,05 для обрабатываемых групп и соответствующей группой контрольного носителя считалось существенным различием.
Результаты
Результаты для соединения 1
Соединение 1, вводимое внутривенно (IV), было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции и легочного воспаления. Значительная эффективность соединения 1 наблюдалась для единичной IV дозы 0,01 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 42,1±2,8% (р<0,0001). Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 98% (р<0,0001). Значительная эффективность соединения 1 также наблюдалась через 1 ч (AUC=76,4±6,6; р=0,0082) и до 48 ч (AUC=64,4±55; р=<0,0001) до MCh в единичной IV дозе 0,1 мг/кг. Значение ED50, доза соединения 1, показывающая 50% снижения отклика Penh, составляло 0,011±0,003 мг/кг.
Результаты для соединения 2
Соединение 2, вводимое внутривенно (IV), было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции. Значительная эффективность соединения 2 наблюдалась для единичной IV дозы 0,01, 0,1 и 1 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 65,3±6,5% (р=0,0002); 50,5±6,3% (р<0,0001) и 41,7±5,2% (р<0,0001) для 0,01, 0,1 и 1 мг/кг, соответственно, для соединения 2. Результаты для соединения 3
Соединение 3, вводимое внутривенно (IV), было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции и легочного воспаления. Значительная эффективность соединения 3 наблюдалась для единичной IV дозы 1 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 71,0±8,6% (р=0,0051). Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 46% (р=0,0002).
Результаты для соединения 6
Соединение 6, вводимое внутривенно (IV) или перорально, было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции и легочного воспаления. Значительная эффективность соединения 6 наблюдалась для единичной IV дозы 1 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 65,3±5,9% (р=0,0001). Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 92% (р<0,0001). Значительная эффективность соединения 6 наблюдалась также для единичной пероральной дозы 30 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 24,6±3,0% (р<0,0001). Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 100% (р=0,0004).
Результаты для соединения 7
Соединение 7, вводимое внутривенно (IV), было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции. Значительная эффективность соединения 7 наблюдалась для единичной IV дозы 0,1 и 1 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 56,1±2,2% (р<0,0001) и 50,4±3,7% (р<0,0001) для 0,1 и 1 мг/кг соединения 7, соответственно.
Результаты для соединения 26
Соединение 26, вводимое внутривенно (IV) или перорально, было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции и легочного воспаления. Значительная эффективность соединения 26 наблюдалась для единичной IV дозы 0,1, 1 и 10 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 64,2±7,6% (р=0,0007); 60,2±7,9% (р=0,0002) и 40,7±2,4% (р<0,0001) для 0,1 мг/кг, 1 мг/кг и 10 мг/кг, соответственно, для соединения 26. Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 79% (р=0,0064); 100% (р=0,0007); и 100% (р=0,0007) для 0,1 мг/кг, 1 мг/кг и 10 мг/кг, соответственно, для соединения 26. Значительная эффективность соединения 26 также наблюдалась через 30 мин (AUC=35,2±9,3; р<0,0001) до MCh в единичной IV дозе 10 мг/кг. Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 94% (р<0,0001). Значительная эффективность соединения 26 также наблюдалась после единичной пероральной дозы 30 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 26,7±1,4% (р<0,0001). Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 100% (р=0,0019).
Результаты для соединения 33
Соединение 33, вводимое внутривенно, было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции и легочного воспаления. Значительная эффективность соединения 33 наблюдалась для единичной IV дозы 1 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 72,9±8,7% (р=0,0089). Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 61% (р<0,0001).
Результаты для соединения 67
Соединение 67, вводимое внутривенно (IV), было эффективным в отношении экспериментальной астмы, что определено по смягчению вызванной метахолином (MCh) бронхоконстрикции и легочного воспаления. Значительная эффективность соединения 67 наблюдалась для единичной IV дозы 1 мг/кг за 24 ч до MCh. Площадь под кривой (AUC) для отклика Penh, описанная как процентное соотношение контрольного носителя (AUC=100%), составляла 78,7±8,1% (р=0,0323). Эозинофильная инфильтрация в бронхиальную промывочную жидкость (BALF) снижалась на 63% (р<0,0001).
Специалисту в данной области техники ясно, что различные модификации и изменения могут быть сделаны в методах и композициях настоящего изобретения без выхода за рамки или объем настоящего изобретения.
Реферат
Изобретение относится к новым пиррольным соединениям формулы I или его фармацевтически приемлемым солям:где Ar означает фенил, тиофенил; Rозначает имидазолил, имидазолил замещенный С-Салкилом, хлор, бром, фтор, гидроксигруппу, метоксигруппу; Rозначает Н, СН, Cl, F, ОН, ОСН, ОСН, пропоксигруппу, карбамоил, диметиламиногруппу, NH, формамидогруппу, CF; X означает СО и SO. Соединения ингибируют S-нитрозоглутатионредуктазу (GSNOR), что позволяет использовать их для получения фармацевтической композиции и для лечения астмы. 8 н. и 9 з.п. ф-лы, 1 табл., 14 схем, 4 пр.
Формула

где Ar выбран из группы, состоящей из фенила и тиофен-ила;
R1 выбран из группы, состоящей из незамещенного имидазолила, имидазолила замещенного С1-С6алкилом, хлора, брома, фтора, гидроксигруппы и метоксигруппы;
R2 выбран из группы, состоящей из водорода, метила, хлора, фтора, гидроксигруппы, метоксигруппы, этоксигруппы, пропоксигруппы, карбамоила, диметиламиногруппы, аминогруппы, формамидогруппы и трифторметила; и
X выбран из группы, состоящей из СО и SO2.

где R3 выбран из Н, метила и этила.
3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(5-(1Н-имидазол-1-ил)тиофен-2-ил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-метил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-(4-метил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-этил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-(1Н-имидазол-1-ил)тиофен-2-ил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(5-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3-фтор-4-(1Н-имидазол-1-ил)фенил)-1 Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3-фтор-4-(2-метил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(2-метокси-4-(2-метил-1Н-имидазол-1-ил)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-(1Н-имидазол-1-ил)-2-метоксифенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(5-(2-метил-1Н-имидазол-1-ил)тиофен-3-ил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(5-(2-этил-1Н-имидазол-1-ил)тиофен-2-ил)-1Н-пиррол-2-ил)пропановая кислота и
3-(5-(5-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1-(2-метил-4-сульфамоилфенил)-1Н-пиррол-2-ил)пропановая кислота.
3-(1-(4-карбамоил-2-метилфенил)-5-(4-гидроксифенил)-1H-пиррол-2-ил)пропановая кислота;
3-(5-(5-бромтиофен-2-ил)-1-(4-карбамоил-2-метилфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-бромфенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3-хлор-4-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3-фтор-4-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3-хлор-4-гидроксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-метокси-3-метилфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-амино-3-хлорфенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3,4-дифторфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(2,4-дифторфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлорфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-бромтиофен-2-ил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-фтор-3-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-карбамоил-3-фторфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-метокси-2-метилфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-фторфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-фторфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-фтор-2-метилфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(2-хлор-4-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(2-этокси-4-фторфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-метокси-2-(трифторметил)фенил)-1 Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-фтор-2-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-3-фторфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-этоксифенил)-1H-пиррол-2-ил)пропановая кислота;
3-(5-(5-бром-2-метоксифенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-бром-2-метоксифенил)-1-(4-карбамоил-2-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-гидроксифенил)-1H-пиррол-2-ил)пропановая кислота;
3-(5-(5-бромтиофен-3-ил)-1-(4-карбамоил-2-метилфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-гидрокси-3-метилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(2-карбамоил-4-хлорфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(2-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(2,4-диметоксифенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-пропоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-гидрокси-2-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-(диметиламино)фенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(5-хлортиофен-2-ил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлор-2-формамидофенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(3-хлортиофен-2-ил)-1Н-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-формамидо-2-метоксифенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(3-бром-5-метокситиофен-2-ил)-1-(4-карбамоил-2-метилфенил)-1H-пиррол-2-ил)пропановая кислота;
3-(1-(4-карбамоил-2-метилфенил)-5-(4-хлортиофен-2-ил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(5-бром-4-хлортиофен-2-ил)-1-(4-карбамоил-2-метилфенил)-1H-пиррол-2-ил)пропановая кислота и
3-(5-(4-бромтиофен-2-ил)-1-(2-метил-4-сульфамоилфенил)-1Н-пиррол-2-ил)пропановая кислота.

где Ar выбран из группы, состоящей из фенила и тиофен-ила;
R4 выбран из группы, состоящей из незамещенного имидазолила и имидазолила, замещенного С1-С6алкилом;
R5 выбран из группы, состоящей из водорода, фтора, гидроксигруппы и метоксигруппы;
R6 выбран из группы, состоящей из водорода, хлора, брома и фтора;
R7 выбран из группы, состоящей из водорода и метила; и
R8 выбран из группы, состоящей из CONH2, SO2NH2 и NHSO2CH3.

где R9 выбран из Н, метила и этила.
3-(5-(5-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1-(4-сульфамоилфенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(5-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1-(2-метил-4-(метилсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-(1Н-имидазол-1-ил)фенил)-1-(2-метил-4-(метилсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-(2-метил-1Н-имидазол-1-ил)фенил)-1-(2-метил-4-(метилсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1-(2-метил-4-(метилсульфонамидо)фенил)-1H-пиррол-2-ил)пропановая кислота;
3-(5-(5-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1-(2-метил-4-(метидсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(4-(2-метил-1Н-имидазол-1-ил)тиофен-2-ил)-1-(4-(метилсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановая кислота;
3-(5-(2-метокси-4-(2-метил-1Н-имидазол-1-ил)фенил)-1-(4-(метилсульфонамидо)фенил)-1Н-пиррол-2-ил)пропановая кислота и
3-(5-(4-(2-метил-1Н-имидазол-1-ил)фенил)-1-(4-(метилсульфонамидо)фенил)-1H-пиррол-2-ил)пропановая кислота.
Документы, цитированные в отчёте о поиске
Производные сульфонилпирролидина
Комментарии