Новый дифенилазетидинон с улучшенными физиологическими свойствами, способы его получения, содержащие эти соединения лекарственные средства и их применение - RU2334737C2
Код документа: RU2334737C2
Описание
Изобретение относится к замещенному дифенилазетидинону, его физиологически приемлемым солям, а также физиологически функциональным производным.
Уже описаны дифенилазетидиноны, а также их применение для лечения гиперлипидемии, как и артериосклероза и гиперхолестеринемии (Международная заявка WO-02/50027).
В основу изобретения положена задача получения соединения, которое, в противоположность описанным в Международной заявке WO-02/50027 соединениям, обладает отчетливо повышенной растворимостью в верхнем отделе тонкого кишечника в состоянии до, соответственно, после приема пищи. За счет повышенной растворимости соединения обеспечивается более высокая доступность растворенного вещества в месте действия и вместе с тем улучшенное действие.
Для тестирования этой повышенной растворимости используют среды FaSSIF (Fasted State Simulating Intestinal Fluid) и FeSSIF (Fed State Simulating Intestinal Fluid), которые отражают условия значение рН/солюбилизация в верхней тонкой кишке в состоянии до, соответственно, после приема пищи.
В основу изобретения, далее, положена задача получения соединения, которое, в противоположность описанным в Международной заявке WO-02/50027 соединениям, обладает повышенной стабильностью как в кислой области (желудок), так и также в слабощелочной области (тонкий кишечник). Это свойство приводит к меньшему количеству побочных соединений/продуктов расщепления, которые, со своей стороны, могут оказывать нежелательные побочные действия. Повышенная стабильность в кислой области значений рН, однако, также имеет большое преимущество при получении лекарственной формы, так как не требуется кислотостойкой капсулы/таблетки.
Изобретение, следовательно, относится к соединениям формулы (I):

а также к их фармацевтически приемлемым солям.
Фармацевтически приемлемые соли благодаря своей высокой растворимости в воде, по сравнению с исходными, соответственно, основными соединениями, особенно пригодны для использований в медицине. Эти соли должны содержать фармацевтически приемлемый анион или катион. Пригодными фармацевтически приемлемыми аддитивными солями с кислотами предлагаемого согласно изобретению соединения являются соли с неорганическими кислотами, такими как соляная кислота, бромоводородная кислота, фосфорная кислота, метафосфорная кислота, азотная кислота и серная кислота, а также с органическими кислотами, такими как, например, уксусная кислота, бензолсульфокислота, бензойная кислота, лимонная кислота, этансульфокислота, фумаровая кислота, глюконовая кислота, гликолевая кислота, изетионовая кислота, молочная кислота, лактобионовая кислота, малеиновая кислота, яблочная кислота, метансульфокислота, янтарная кислота, п-толуолсульфокислота и винная кислота. Пригодными фармацевтически приемлемыми основными солями являются соли аммония, соли щелочных металлов (как соли натрия и калия), соли щелочноземельных металлов (как соли магния и кальция), трометамола (2-амино-2-гидроксиметил-1,3-пропандиол), диэтаноламина, лизина или этилендиамина.
Соли с фармацевтически неприемлемым анионом, такие как, например, трифторацетат, также входят в рамки изобретения в качестве полезных промежуточных продуктов для получения или очистки фармацевтически приемлемых солей и/или для использования в случае нетерапевтических, например, in vitro, применений.
Используемое в данном контексте понятие "физиологически функциональное производное" означает любое физиологически приемлемое производное предлагаемого согласно изобретению соединения формулы (I), например, сложный эфир, которое при введении млекопитающему, как, например, человеку, способно (прямо или непрямо) образовывать соединение формулы (I) или его активный метаболит.
К физиологически функциональным производным относятся также пролекарственные формы предлагаемого согласно изобретению соединения, как, например, описывается H. Okada и др., Chem. Pharm. Bull., 42, 57-61 (1994). Такие пролекарственные формы могут метаболизироваться in vivo до предлагаемого согласно изобретению соединения. Эти пролекарственные формы сами могут быть активными или нет.
Предлагаемое согласно изобретению соединение может находиться также в разных полиморфных формах, например в аморфной или кристаллической полиморфной форме. Все полиморфные формы предлагаемого согласно изобретению соединения входят в рамки изобретения и составляют дальнейший аспект изобретения.
В дальнейшем все указания на "соединение (соединения) формулы (I)" относятся к соединению формулы (I), как оно описано выше, а также к его солям, сольватам и физиологически функциональным производным, как описываемые в данном контексте.
Под арильным остатком понимают фенильный, нафтильный, бифенильный, тетрагидронафтильный, альфа- или бета-тетралоновый, инданильный или индан-1-онильный остаток.
Соединение (соединения) формулы (I) можно вводить также в комбинации с другими биологически активными веществами.
Количество соединения формулы (I), которое является необходимым для достижения желаемого биологического эффекта, зависит от ряда факторов, например, выбранного конкретного соединения, предусматриваемого применения, способа введения и клинического состояния пациента. В общем, суточная доза составляет величину в пределах от 0,01 мг до 100 мг (более типично, от 0,05 мг до 50 мг) в сутки на килограмм массы тела, например 0,1-10 мг/кг/сутки.
Перорально вводимые лекарственные формы в виде разовой дозы, как, например, таблетки или капсулы, могут содержать, например, от 1,0 мг до 1000 мг, более типично, от 10 мг до 600 мг, биологически активного вещества. Для лечения вышеуказанных состояний соединения формулы (I) можно использовать индивидуально в виде соединения, предпочтительно, однако они находятся вместе с приемлемым носителем в форме фармацевтической композиции. Носитель, естественно, должен быть приемлем в том смысле, что он совместим с другими компонентами композиции и не угрожает здоровью пациента. Носитель может представлять собой твердое вещество или жидкость или и то и другое и предпочтительно используется вместе с соединением для получения лекарственной формы в виде разовой дозы, например, в виде таблетки, которая может содержать от 0,05 мас.% до 95 мас.% биологически активного вещества. Также могут присутствовать другие фармацевтически активные вещества, включая другие соединения формулы (I). Предлагаемые согласно изобретению фармацевтические композиции можно получать любым из известных фармацевтических способов, которые по существу состоят в том, что компоненты смешивают с фармакологически приемлемыми носителями и/или вспомогательными веществами.
Предлагаемыми согласно изобретению фармацевтическими композициями являются такие, которые пригодны для орального или перорального (например, подъязычного) введения, хотя самый пригодный путь введения в каждом отдельном случае зависит от рода и тяжести подвергамого лечению состояния и от рода используемого в каждом случае соединения формулы (I). В рамки изобретения также входят дражированные готовые лекарственные формы и дражированные готовые лекарственные формы пролонгированного действия. Предпочтительны резистентные к кислоте и желудочному соку готовые лекарственные формы. Пригодные, резистентные к желудочному соку покрытия включают ацетатфталат целлюлозы, поливинилацетатфталат, гидроксипропилметилцеллюлозофталат и анионные полимеры метакриловой кислоты и метилового эфира метакриловой кислоты.
Пригодные фармацевтические соединения для перорального введения могут находиться в виде отдельных разовых форм, как, например, капсулы, оболочки облаток, таблетки для сосания или таблетки, которые, соответственно, содержат определенное количество соединения формулы (I); в виде порошков или гранулятов; в виде раствора или суспензии в водной или неводной жидкости; или в виде эмульсии масло-в-воде или вода-в-масле. Эти композиции, как уже упоминалось, можно получать любым пригодным фармацевтическим способом, включающим стадию, на которой биологически активное вещество и носитель (который может состоять из одного или нескольких дополнительных компонентов) вводят в контакт. В общем, композиции приготовляют путем равномерного и гомогенного смешения биологически активного вещества с жидким и/или высокодисперсным твердым носителем, после чего продукт, если необходимо, формуют. Так, например, таблетки можно получать тем, что порошок или гранулят соединения прессуют или формуют, в случае необходимости, вместе с одним или несколькими дополнительными компонентами. Прессованные таблетки можно получать путем таблетирования в пригодной машине соединения в свободно текучей форме, как, например, в форме порошка или гранулята, в случае необходимости, смешанного со связующим, придающим скользкость (таблеткам) веществом, инертным разбавителем и/или одним (несколькими) поверхностно-активным веществом и/или диспергатором. Формованные таблетки можно получать путем формования в пригодной машине порошкообразного, смоченного инертным жидким разбавителем соединения.
Фармацевтические композиции, которые пригодны для перорального (подъязычного) введения, включают таблетки для сосания, которые содержат соединение формулы (I) вместе с вкусовым веществом, обычно сахарозой и гуммиарабиком или трагантом, и пастилки, которые включают соединение в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик.
В качестве других биологически активных веществ для комбинированных препаратов пригодны:
Все антидиабетические средства, которые указываются в Красном списке 2003, глава 12. Их можно комбинировать с предлагаемыми согласно изобретению соединениями формулы (I), в особенности для синергического повышения действия. Введение комбинации биологически активных веществ можно осуществлять либо путем раздельного введения пациенту биологически активных веществ, либо в виде комбинированных препаратов, где несколько биологически активных веществ находятся в одной фармацевтической композиции. Большинство нижеуказанных биологически активных веществ указываются в фармакопейном справочнике США по USAN и международным названиям лекарственных средств, Фармакопея США, Роквилл, 2001.
Антидиабетические средства включают инсулин и производные инсулина, как, например, Lantus® (см. www.lantus.com) или HMR 1964, быстро действующие инсулины (см. патент США 6221633), производные GLP-1, как, например, таковые, которые описываются в Международной заявке WO-98/08871 на имя фирмы Novo Nordisk A/S, в Международной заявке WO-01/04156 на имя фирмы Zealand или в Международной заявке WO-00/34331 на имя фирмы Beautofour-Ipsen, а также эффективные при пероральном приеме гипогликемические биологически активные вещества.
Эффективные при пероральном приеме гипогликемические биологически активные вещества предпочтительно включают сульфонилмочевины, бигуанидины, меглитиниды, оксадиазолидиндионы, тиазолидиндионы, ингибиторы глюкозидазы, ингибиторы гликогенфосфорилазы, антагонисты глюкагона, агонисты GLP-1, открыватели калиевых каналов, как, например, такие, которые описываются в Международных заявках WO-97/26265 и WO-99/03861 на имя Novo Nordisk A/S, сенсибилизаторы инсулина; ингибиторы ферментов печени, которые принимают участие в стимуляции глюконеогенеза и/или гликогенолиза; модуляторы поглощения глюкозы, транспорта глюкозы и обратной резорбции глюкозы; изменяющие жировой обмен соединения, как антигиперлипидемические биологически активные вещества и антилипидемические биологически активные вещества; соединения, которые способствуют уменьшению потребности в продуктах питания; агонисты PPAR и PXR и биологически активные вещества, которые воздействуют на АТФ-зависимый калиевый канал бета-клеток.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором HMGCoA-редуктазы, такими как симвастатин, флувастатин, правастатин, ловастатин, аторвастатин, церивастатин, розувастатин.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором резорбции холестерина, такими как, например, эзетимиб, тиквесид, памаквесид.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с агонистом PPAR-гамма, таким как, например, розиглитазон, пиоглитазон, JTT-501, GI 262570.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с агонистом PPAR-альфа, таким как, например, GW 9578, GW 7647.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с агонистами смешанного PPAR-альфа/гамма, такими как, например, GW 1536, AVE 8042, AVE 8134, AVE 0847, или как описывается в РСТ/US 11833, РСТ/US 11490, патенте ФРГ 10142734.4.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с фибратом, таким как, например, фенофибрат, клофибрат, безафибрат.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором МТР, таким как, например, имплитапид, BMS-201038, R-103757.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором резорбции желчной кислоты (см., например, патенты США 6245744 или 6221897), такими как, например, HMR 1741.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором СЕТР, таким как, например, JTT-705.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с полимерным адсорбером желчной кислоты, таким как, например, холестирамин, колесевелам.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с активатором рецептора LDL (см. патент США 6342512), таким как, например, HMR 1171, HMR 1586.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором АСАТ, таким как, например, авасимиб.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с антиоксидантом, таким как, например, ОРС-14117.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором липопротеин-липазы, таким как, например, NO-1886.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором АТФ-цитрат-лиазы, таким как, например, SB-204990.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором скваленсинтетазы, таким как, например, BMS-188494.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с антагонистом липопротеина(а), таким как, например, CI-1027 или никотиновая кислота.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором липазы, таким как, например, орлистат.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с инсулином.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с сульфонилмочевиной, такой как, например, толбутамид, глибенкламид, глипизид или глимепирид.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с бигуанидом, таким как, например, метформин.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с меглитинидом, таким как, например, репаглинид.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с тиазолидиндионом, таким как, например, троглитазон, циглитазон, пиоглитазон, розиглитазон, или описанными в Международной заявке WO-97/41097, Dr. Reddy's, Research Foundation, соединениями, в особенности, 5-[[4-[(3,4-дигидро-3-метил-4-оксо-2-хиназолинилметокси]фенил]метил]-2,4-тиазолидиндионом.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с ингибитором α-глюкозидазы, таким как, например, миглитол или акарбоза.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с биологически активным веществом, которое воздействует на АТФ-зависимый калиевый канал бета-клеток, таким как, например, толбутамид, глибенкламид, глипизид, глимепирид или репаглинид.
Согласно варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с более чем одним из вышеуказанных соединений, например в комбинации с сульфонилмочевиной и метформином, сульфонилмочевиной и акарбозой, репаглинидом и метформином, инсулином и сульфонилмочевиной, инсулином и метформином, инсулином и троглитазоном, инсулином и ловастатином, и т.д.
Согласно другому варианту осуществления изобретения, соединения формулы (I) вводят в комбинации с модуляторами CART (см. "Cocaine-amphetamine-regulated transcript influences energy metabolism, anxiety and gastric emptying in mice", Asakawa A. и др., М.: Hormone and Metabolic Research, 33 (9), 554-558 (2001)); антагонистами NPY, такими как, например, {4-[(4-амино-хиназолин-2-иламино)метил]циклогексилметил}амидгидрохлорид нафталин-1-сульфокислоты (CGP 71683A)); антагонистами каннабиноидного рецептора 1 (см., например, Европейский патент 0656354, Международные заявки WO-00/15609 или WO-02/076949); агонистами МС4 (такими как, например, [2-(3а-бензил-2-метил-3-оксо-2,3,3а,4,6,7-гексагидропиразоло[4,3-c]пиридин-5-ил)-1-(4-хлорфенил)-2-оксоэтил]амид 1-амино-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты; (WO-01/91752)); антагонистами орексина (такими как, например, гидрохлорид 1-(2-метилбензоксазол-6-ил)-3-[1,5]нафтиридин-4-ил-мочевины (SB-334867-A)); агонистами Н3 (такими как соль щавелевой кислоты 3-циклогексил-1-(4,4-диметил-1,4,6,7-тетрагидроимидазо-[4,5-c]пиридин-5-ил)пропан-1-она (WO-00/63208)); агонистами фактора некроза опухоли; антагонистами CRF (такими как, например, [2-метил-9-(2,4,6-триметилфенил)-9Н-1,3,9-триазафлуорен-4-ил]-дипропиламин (WO-00/66585)); антагонистами CRF BP (такими как, например, урокортин); агонистами урокортина; агонистами β3 (такими как, например, 1-(4-хлор-3-метансульфонилметилфенил)-2-[2-(2,3-диметил-1Н-индол-6-илокси)этиламино]этанолгидрохлорид (WO-01/83451)); агонистами MSH (меланоцитстимулирующий гормон); антагонистами рецептора МСН (меланинконцентрирующий гормон) (см., например, Международную заявку WO-03/15769); агонистами ССК-А (такими как, например, трифторацетат {2-[4-(4-хлор-2,5-диметоксифенил)-5-(2-циклогексилэтил)тиазол-2-ил-карбамоил]-5,7-диметилиндол-1-ил}уксусной кислоты (WO-99/15525) или SR-146131 (WO-0244150) или SSR-125180); ингибиторами повторного поглощения серотонина (такими как, например, дексфенфлурамин); смешанными серотонин- и норадренергическими соединениями (см., например, WO-00/71549); агонистами 5НТ, такими как, например, соль щавелевой кислоты 1-(3-этилбензофуран-7-ил)пиперазина (WO-01/09111); агонистами бомбезина; антагонистами галанина; гормонами роста (такими как, например, гормон роста человека); высвобождающим гормон роста соединениями (такими как трет-бутиловый эфир 6-бензилокси-1-(2-диизопропиламиноэтилкарбамоил)-3,4-дигидро-1Н-изохинолин-2-карбоновой кислоты (WO-01/85695)); агонистами TRH (см., например, Европейский патент 0462884); 2- и 3-модуляторами разрывающих связь протеинов; агонистами лептина (см., например, Lee Daniel W.; Leinung Matthev C; Rozhavskaya-Arena Marina; Grasso Patricia "Leptin agonists as a potential approach to the treatment of obesity", Drugs of the Future, 26 (9), 873-881 (2001)); агонистами DA (бромкриптин, допрексин); ингибиторами липазы/амилазы (см., например, WO-00/40569); модуляторами PPAR (см., например, WO-00/78312); ингибиторами 11β-HSD1 (11-бета-гидроксистероиддегидрогеназа типа 1) (см., например, WO-01/90094 или T. Barf и др., J. Med. Chem., 45, 3813-3815 (2002)); ингибиторами ацетил-СоА-карбоксилазы (АСС; см., например, WO-99/46262); ингибиторами дипептидпептидазы IV (DPP-IV; см., например, Европейский патент 1259246); модуляторами RXR или агонистами TR-β.
Согласно варианту осуществления изобретения, другим биологически активным веществом является лептин: см., например, "Perspectives in the therapeutic use of leptin", Salvador Javier; Gomez-Ambrosi Javier; Fruhbeck Gema; Exspert Opinion on Pharmacotherapy, 2 (10), 1615-1622 (2001).
Согласно варианту осуществления изобретения, другим биологически активным веществом является дексамфетамин или амфетамин.
Согласно варианту осуществления изобретения, другим биологически активным веществом является фенфлурамин или дексфенфлурамин.
Согласно еще одному варианту осуществления изобретения, другим биологически активным веществом является сибутрамин.
Согласно варианту осуществления изобретения, другим биологически активным веществом является орлистат.
Согласно варианту осуществления изобретения, другим биологически активным веществом является мазиндол или фентермин.
Согласно варианту осуществления, соединения формулы (I) вводят в комбинации с балластными веществами, предпочтительно, с нерастворимыми балластными веществами (см., например, Carob/Caromax® (Zunft H.J. и др., "Carob pulp preperation for treatment of hypercholesterolemia", ADVANCES IN THERAPY, 18 (5), 230-236 (2001, сентябрь-октябрь)). Каромакс представляет собой содержащий кароб продукт фирмы Nutrinova, Nutrition and Food Ingredients GmbH, Industriepark Höchst, 65926, Франкфурт-на-Майне). Комбинацию с Caromax® можно осуществлять в виде композиции или путем раздельного введения соединений формулы (I) и Caromax®. При этом Caromax® можно вводить также в форме пищевых продуктов, например в форме хлебобулочных изделий или батончиков мюсли.
Само собой разумеется, что любая пригодная комбинация предлагаемых согласно изобретению соединений с одним или несколькими вышеуказанными соединениями и, на выбор, с одним или несколькими другими фармакологически активными веществами рассматривается как подпадающая под объем охраны настоящего изобретения.

Изобретение относится, далее, к способам получения соединения формулы (I).
Способ А:
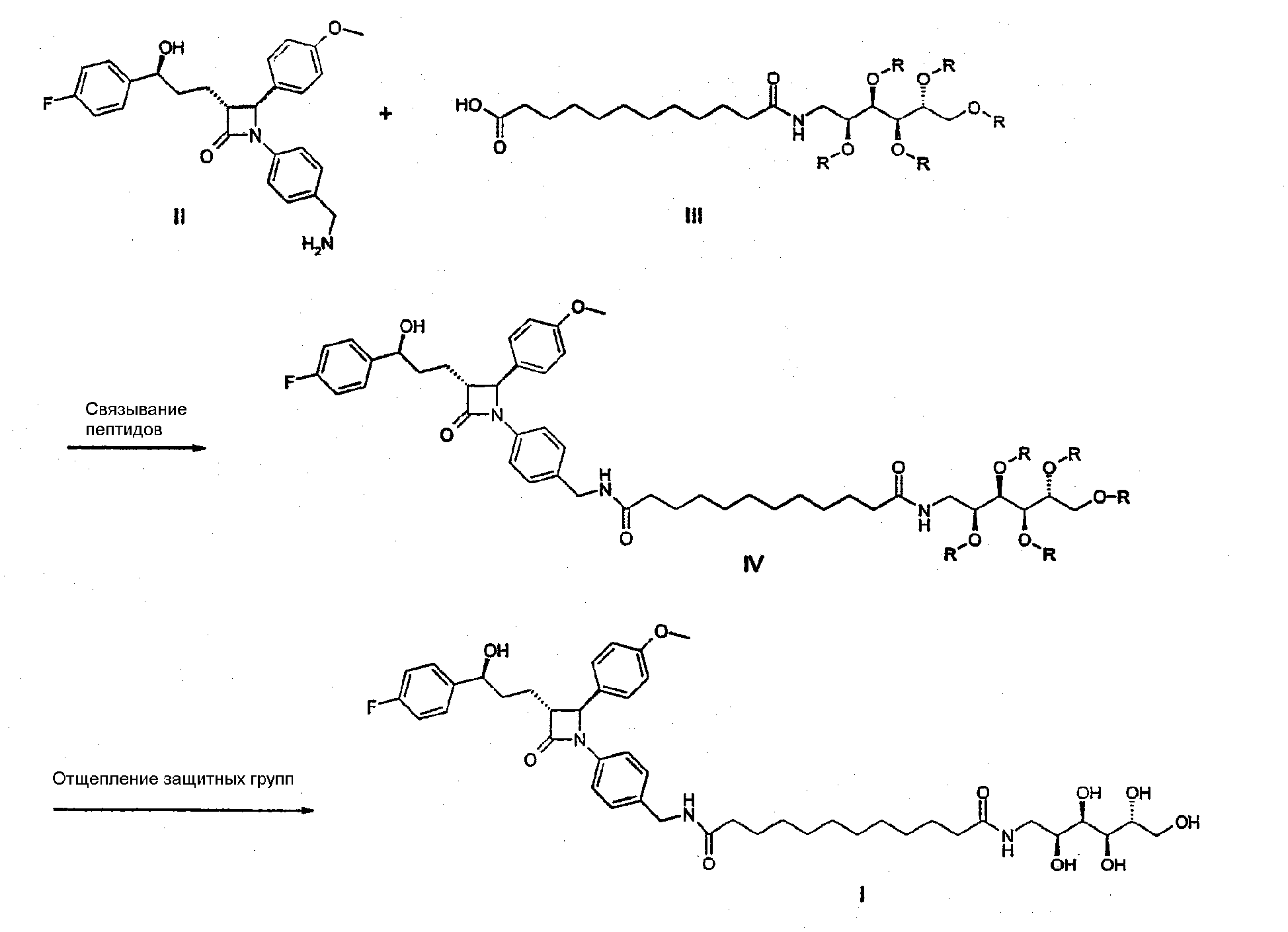
Способ А получения соединения формулы (I) отличается тем, что амин формулы (II) (см. WO-02/50027) вводят во взаимодействие с моноглюкамидом 1,12-додекандикарбоновой кислоты (формула (III)), причем гидроксильные группы глюкаминовой части могут быть защищены, например, ацильными группами, такими как, например, ацетильные группы, или простыми эфирными группами, такими как, например, группы простого бензилового эфира, с получением в результате реакции связывания пептидов соединения формулы (IV). В случае этого взаимодействия можно работать, например, при использовании N-гидроксибензтриазола (HOBt) и N-этил-N'-(3-диметиламинопропил)карбодиимида (EDC) при комнатной температуре, например, в диметилформамиде (ДМФА) в качестве растворителя. Также можно использовать другие реагенты реакции связывания пептидов и растворители или смеси растворителей (см., например, A. Speicher и др., Journal für Praktische Chemie/Chemiker-Zeitung, 340, 581-583 (1998); Y.S. Klausner M. Bodansky, Synthesis, 453 и последующие (1972); K. Ishihara и др., J. Org. Chem., 61, 4196 (1996); M. Kunishima и др., Tetrahedron, 55, 13159-13170 (1999); или также R.C. Larock "Comprehensive Organic Transformations", VCH, Нью-Йорк, 1969, с. 981 и последующие).
Способ В:
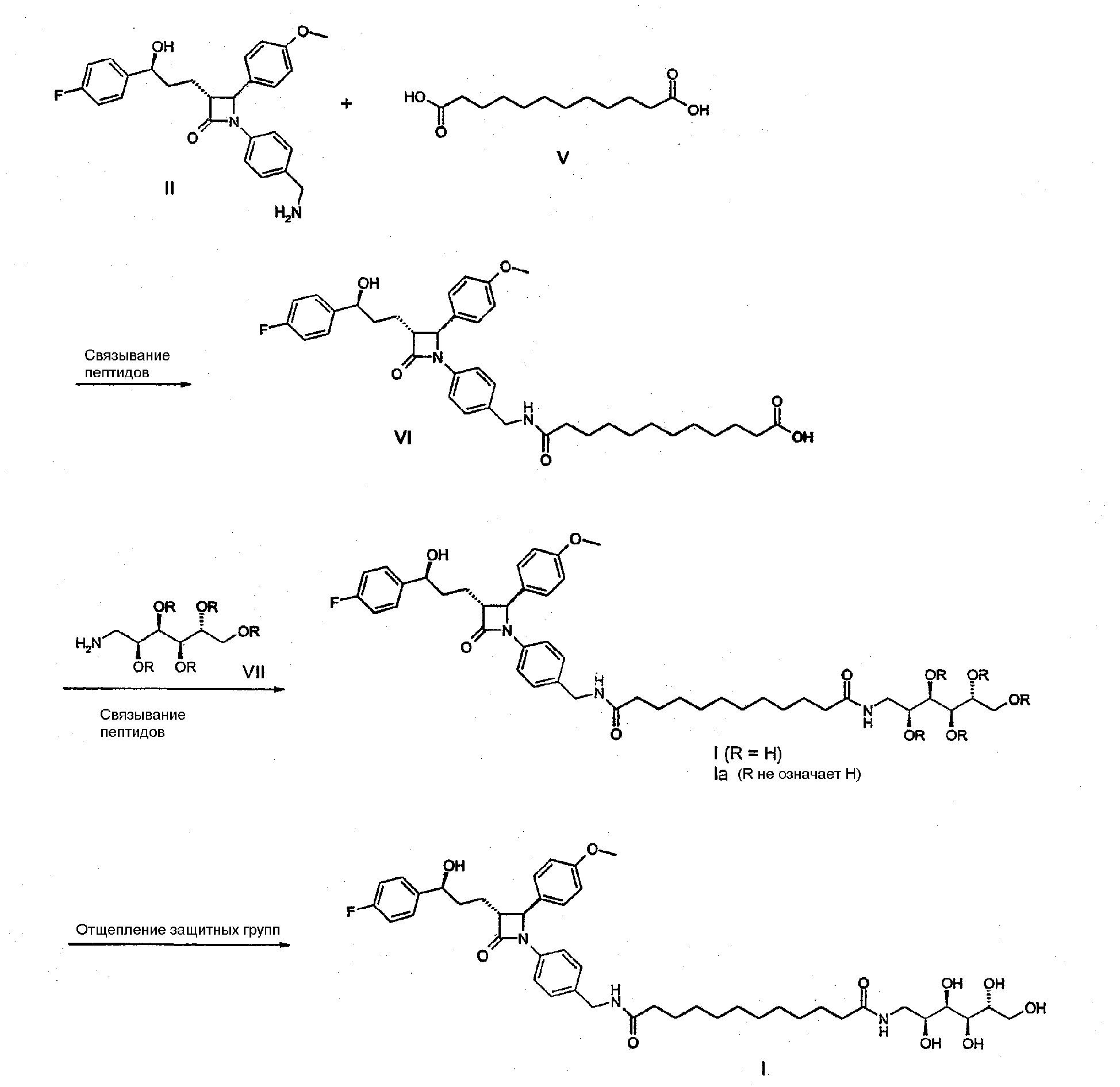
Другой предлагаемый согласно изобретению способ (В) включает введение во взаимодействие амина формулы (II) с 1,12-додекандикарбоновой кислотой формулы (V) в условиях реакции связывания пептидов и дальнейшее взаимодействие продукта формулы (VI) с глюкамином формулы (VII), гидроксильные группы которого могут быть снабжены защитными группами (такими как, например, ацетильные защитные группы или бензильные защитные группы), опять в условиях реакции связывания пептидов, с получением соединения формулы (I) или соответственно снабженного защитными группами соединения формулы (Ia). На следующей стадии защитные группы можно отщеплять либо в слабощелочных условиях, например в разбавленном водном растворе аммиака, либо гидролитически (в случае использования защитных групп простого бензилового эфира) для получения соединения формулы (I).
Способ С:

Согласно другому предлагаемому в изобретении способу С, амин формулы (II) вводят во взаимодействие с галогенангидридом, например хлорангидридом, 11-((4R,6R)-4,5,6-тригидрокси-3-(R)-гидрокси-2-(S)-гидроксигексилкарбамоил)ундекановой кислоты формулы (VIII), например, в пиридине или в дихлорметане, с добавкой первичного амина или без нее, при комнатной температуре. Гидроксильные группы глюкаминной части при этом предпочтительно снабжены вышеуказанными защитными группами и после реакции связывания их можно отщеплять с получением амида формулы (Ia).
Далее, изобретение относится к промежуточным продуктам формул (III), (IV) и (VIII), где R означает ацил, например ацетил или бензоил, или где R означает аралкил, алкил или арил, например бензил.
Пример 1
Способ А1:
1. Монометиловый эфир додекандикарбоновой кислоты

4,6 г (20 ммоль) Додекандикарбоновой кислоты при нагревании растворяют в 40 мл безводного тетрагидрофурана, медленно смешивают с 0,73 мл (10 ммоль) тионилхлорида и перемешивают в течение 30 минут при комнатной температуре. Затем медленно добавляют 0,8 мл (20 ммоль) безводного метанола и перемешивают в течение 4 часов при комнатной температуре; смесь затем выдерживают в течение 4 дней при комнатной температуре. После этого тонкослойная хроматография не показывает никакого дальнейшего превращения; реакционную смесь концентрируют в вакууме, остаток перемешивают с водой (ультразвуковая баня). Осадок отфильтровывают под вакуумом, промывают водой и снова отфильтровывают под вакуумом. Влажный остаток перемешивают с дихлорметаном (ультразвуковая баня), отфильтровывают через складчатый фильтр, промывают дихлорметаном и фильтрат концентрируют в вакууме. Получают монометиловый эфир додекандикарбоновой кислоты (3,09 г) с выходом 63%. Молекулярная масса: 244,34; МС (масс-спектрометрия): 245,4 (М+Н+).
2. Синтез метилового эфира 11-((4R,6R)-4,5,6-тригидрокси-3-(R)-гидрокси-2-(S)-гидроксигексилкарбамоил)ундекановой кислоты
3,07 г (12,6 ммоль) Монометилового эфира додекандикарбоновой кислоты при комнатной температуре растворяют в 30 мл безводного диметилформамида, смешивают с 2,2 г (12,1 ммоль) глюкамина, 1,9 г (12,4 ммоль) N-гидроксибензтриазола (HOBt) и 2,4 г (12,5 ммоль) N-этил-N'-(3-диметиламинопропил)карбодиимида (EDC) и перемешивают в течение 6 часов при комнатной температуре. Выдерживают в течение ночи при комнатной температуре. На следующий день тонкослойная хроматография показывает полное превращение. Реакционную смесь концентрируют в вакууме и высушивают в высоком вакууме. Остаток перемешивают в воде (ультразвуковая баня), отфильтровывают под вакуумом, промывают водой и отфильтровывают под вакуумом. Влажный сырой продукт перемешивают в дихлорметане, отфильтровывают под вакуумом, промывают дихлорметаном и высушивают. Получают метиловый эфир 11-((4R,6R)-4,5,6-тригидрокси-3-(R)-гидрокси-2-(S)-гидроксигексилкарбамоил)-ундекановой кислоты; 4,45 г (выход: 90%). Молекулярная масса: 407,51; МС: 408,20 (М+Н+).
3. Синтез 11-((4R,6R)-4,5,6-тригидрокси-3-(R)-гидрокси-2-(S)-гидроксигексилкарбамоил)ундекановой кислоты (формула (III); R = H):

4,45 г (10,9 ммоль) Метилового эфира 11-((4R,6R)-4,5,6-тригидрокси-3-(R)-гидрокси-2-(S)-гидроксигексилкарбамоил)-ундекановой кислоты при комнатной температуре суспендируют в 75 мл безводного этанола, смешивают с 25 мл воды и 2,2 г КОН (85%-ный раствор) (33 ммоль). После перемешивания в течение 2 часов при температуре 80°С тонкослойная хроматография показывает полное превращение. Реакционную смесь концентрируют в вакууме; остаток растворяют в воде и подкисляют концентрированной соляной кислотой. Выпавший в осадок сырой продукт отфильтровывают под вакуумом, промывают водой и отфильтровывают под вакуумом. Влажный сырой продукт перекристаллизуют из примерно 100 мл этанола, отфильтровывают в горячем состояниии и осаждают на бане со льдом. Осадок отфильтровывают под вакуумом, промывают его этанолом и высушивают его. Получают 2,2 г (51%) 11-((4R,6R)-4,5,6-тригидрокси-3-(R)-гидрокси-2-(S)-гидроксигексилкарбамоил)-ундекановой кислоты. Молекулярная масса: 393,48; МС: 394,28 (М+Н+).
4. Синтез 4-[(2S,3R)-3-[(S)-3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензиламид-((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амид додекандикарбоновой кислоты (формула (I)):

0,63 г (1,45 ммоль) Бензиламина формулы (II) (получение см. патент ФРГ 10064398) и 0,65 г (1,65 ммоль) моноамида дикарбоновой кислоты (см. выше) слегка при нагревании растворяют в 15 мл безводного диметилформамида, смешивают с 0,25 г (1,63 ммоль) HOBt и 0,31 г (1,67 ммоль) EDC и перемешивают в течение 4 часов при комнатной температуре. Реакционную смесь выдерживают в течение ночи при комнатной температуре. На следующее утро тонкослойная хроматография показывает полное превращение. Реакционную смесь концентрируют в вакууме и высушивают в высоком вакууме. Остаток перемешивают в воде (ультразвуковая баня), отфильтровывают под вакуумом, промывают водой и отфильтровывают под вакуумом. Сырой продукт перекристаллизуют из изопропанола. Кристаллизат еще раз перемешивают с водой, отфильтровывают под вакуумом и высушивают. Получают 0,38 г (32%) 4-[(2S,3R)-3-[(S)-3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензиламид-((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амида додекандикарбоновой кислоты (I). Молекулярная масса: 809,97; МС: 810,49 (М+Н+).
Способ А2:
1. 11-((2S,3R,4R,5R)-2,3,4,5,6-Пентаацетоксигексилкарбамоил) ундекановая кислота (формула (III); R = ацетил):

0,4 г 11-((4R,6R)-4,5,6-Тригидрокси-3-(R)-гидрокси-2-(S)-гидроксигексилкарбамоил)ундекановой кислоты (формула (III); R = H) при комнатной температуре смешивают с 3 мл безводного пиридина и 3 мл ангидрида уксусной кислоты и перемешивают в течение 4 часов при комнатной температуре. По окончании реакции реакционную смесь смешивают с водой и концентрируют в вакууме. Остаток перемешивают с небольшим количеством воды и отфильтровывают. Остаток на фильтре промывают водой и затем высушивают в вакууме. Получают 0,56 г 11-((2S,3R,4R,5R)-2,3,4,5,6-пентаацетоксигексилкарбамоил)ундекановой кислоты. Молекулярная масса: 603,66; МС: 604,22 (М+Н+).
2. (2R,3R,4R,5S)-2,3,4,5-Тетраацетокси-6-(11-{4-[(2S,3R)-3-[(S)-3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензилкарбамоил}ундеканоиламино)гексиловый эфир уксусной кислоты (формула (IV); R = ацетил):

87 мг Амина формулы (II) при комнатной температуре растворяют в 3 мл безводного диметилформамида и смешивают с 120 мг вышеуказанной карбоновой кислоты, 31 мг N-гидроксибензтриазола и 39 мг N-этил-N'-(3-диметиламинопропил)карбодиимида. Реакционную смесь перемешивают в течение ночи при комнатной температуре и затем концентрируют в вакууме. Остаток обрабатывают этилацетатом, органическую фазу промывают водой и сушат над сульфатом магния. После этого фильтруют и фильтрат концентрируют в вакууме. Получают 90 мг (2R,3R,4R,5S)-2,3,4,5-тетраацетокси-6-(11-{4-[(2S,3R)-3-[(S)-3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензилкарбамоил}ундеканоиламино)гексилового эфира уксусной кислоты. Молекулярная масса: 1020,16.
3. 4-[(2S,3R)-3-[(S)-3-(4-Фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензиламид((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амид додекандикарбоновой кислоты (формула (I)):

90 мг Вышеуказанного соединения обрабатывают с помощью гуанидина в смеси из метанола и дихлорметана. Получают производное глюкамина формулы (I) с молекулярной массой 809,97.
Способ В:
1. 11-{4-[(2S,3R)-3-[(S)-3-(4-Фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензилкарбамоил}-ундекановая кислота (формула (VI)):

К раствору из 371 мг додекандикарбоновой кислоты, 63 мкл диизопропилкарбодиимида, 55 мг гидроксибензтриазола в 2 мл диметилформамида добавляют раствор из 70 мг амина формулы (I), 23 мкл триэтиламина в 1 мл диметилформамида и перемешивают в течение 12 часов при комнатной температуре. Реакционный раствор концентрируют и разделяют путем высокоэффективной жидкостной хроматографии (ВЭЖХ) (Knauer Eurosper-100-10-C18, вода (0,1% трифторуксусной кислоты)/ацетонитрил (0,1% трифторуксусной кислоты) = 80/20 → 10/90). Получают продукт с молекулярной массой 646,81 (С38Н47F1N2O6); МС (ионизация электронным распылением (ESI)): 647,35 (М+Н+).
2. 4-[(2S,3R)-3-[(S)-3-(4-Фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензиламид-((2S,3R,4R,5R)-2,3,4,5,6-пентагидроксигексил)амид додекандикарбоновой кислоты (формула (I)):

Как описывается в случае других реакций связывания, взаимодействие кислоты формулы (VI) с глюкамином (формула (VII); R = H) и HOBt/EDC в диметилформамиде приводит к получению соединения формулы (I) (R = H). Если вместо глюкамина используют защищенное производное глюкамина, например, формулы (VII) (R = ацетил), то получают соединение формулы (Ia) с R = ацетил.
Способ С:
1. (2R,3R,4R,5S)-2,3,4,5-Тетраацетокси-6-(11-хлоркарбонилундеканоиламино)гексиловый эфир уксусной кислоты (формула (VIII); R = ацетил):

Соединение формулы (III) (R = ацетил) растворяют в тетрагидрофуране и медленно смешивают с тионилхлоридом; перемешивают в течение 1 часа при комнатной температуре. После этого реакционный раствор концентрируют в вакууме и сырой продукт используют на ближайшей стадии.
2. (2R,3R,4R,5S)-2,3,4,5-Тетраацетокси-6-(11-{4-[(2S,3R)-3-[(S)-3-(4-фторфенил)-3-гидроксипропил]-2-(4-метоксифенил)-4-оксоазетидин-1-ил]бензилкарбамоил}ундеканоиламино)гексиловый эфир уксусной кислоты (формула (IV); R = ацетил):

Вышеуказанный хлорангидрид кислоты в смеси из пиридина и дихлорметана при комнатной температуре смешивают с амином и перемешивают в течение ночи при комнатной температуре. Обработка приводит к амиду формулы (IV) с R = ацетил.
Предлагаемое согласно изобретению соединение формулы (I) тестировали на его действие с помощью нижеописанного способа:
Влияние абсорбции холестерина + выделение 3Н-таурохолевой кислоты на основании выделения фекалий мыши, крысы или хомяка
Мышей линии NMRI, крыс линии Wistar или золотистых сирийских хомячков (в группах с n = 4-6) содержат на стандартной диете (альтромин; положение (губа)) в респирационных камерах. Во второй половине дня перед введением радиоактивного индикатора (14С-холестерин) животных выдерживают голодными и адаптируют к предохранительной решетке.
Дополнительно, за 24 часа до перорального введения пробного завтрака (14С-холестерин в Intralipid®20, Pharmacia-Upjohn) животных подкожно метят с помощью3Н-таурохолевой кислоты (3Н-ТСА) (например, от 1 мкКи/мышь до 5 мкКи/крыса).
Тест на абсорбцию холестерина: 0,25 мл/мышь Intralipid®20 (Pharmacia-Upjohn) ((максимально с 0,25 мкКи14С-холестерина в 0,1 мг холестерина) вводят перорально с помощью желудочного зонда.
Тестируемые вещества разводят раздельно в 0,5% метил-целлюлоза (Sigma)/5% солутол (BASF, Людвигсхафен) или в пригодном эксципиенте.
Вводимый объем тестируемого вещества составляет 0,5 мл/мышь. Тестируемое вещество вводят непосредственно перед пробным завтраком (интралипид с меткой в виде14С-холестерина) (тест на абсорбцию холестерина).
Фекалии собирают в течение 24 часов: фекальное элиминирование14С-холестерина и3Н-таурохолевой кислоты (ТСА) определяют спустя 24 часа.
Извлекают печень, гомогенизируют и аликвоты сжигают в оксимате (модель 307, Packard) для определения поглощенного/резорбированного количества14С-холестерина.
Оценка
Образцы фекалиев:
Определяют общую массу, доливают водой до определенного объема, затем гомогенизируют, аликвоту высушивают и сжигают в оксимате (модель 307, Packard, для сжигания радиоактивно меченых образцов): количество радиоактивных3Н-Н2О и14С-СО2 экстраполируют на выделившееся количество3Н-таурохолевой кислоты, соответственно,14С-холестерина (технология бинарных изотопов). Значения ED200 интерполируют в виде дозы из кривой доза-действие как те дозы, которые удваивают выделение ТСА, соответственно, холестерина, в пересчете на одновременно подвергнутую обработке контрольную группу.
Образцы печени:
Поглощенное количество14С-холестерина в печени соотносят к введенной дозе. Значения ED50 интерполируют из кривой доза-действие в качестве той дозы, которая уменьшает вдвое поглощение14С-холестерина в печени (50%), в пересчете на контрольную группу.
Следующее значение ED50 подтверждает активность предлагаемого согласно изобретению соединения формулы (I):
Из таблицы можно видеть, что соединение формулы (I) обладает очень хорошим, снижающим содержание холестерина действием.
Растворимость соединения, а также сравнительного соединения формулы (V1) тестировали следующим образом:
В качестве сравнительного соединения было выбрано структурно подобное соединение, описанное в Международной заявке WO-02/50027:

0,5 мг Тестируемого соединения точно взвешивают в пробирке Эппендорфа и смешивают с 0,5 мл соответствующего растворителя (водный буфер). Пробирку Эппендорфа затем помещают в термомиксер и встряхивают при температуре 25°С в течение 4 часов со скоростью 1400 оборотов в минуту.
Затем пробирку Эппендорфа помещают в центрифугу. После центрифугирования аликвоту супернатанта используют для определения растворенного количества путем ВЭЖХ/УФ-анализа. В следующей таблице представлены достигнутые при этом результаты:
В физиологических растворителях FaSSIF и FeSSIF (состав и способы получения см. Physiologically based dissolution tests - Experiences with poorly soluble drugs) Тесты по растворению на физиологической основе - опыты с плохо растворимыми лекарственными средствами", январь 2000, изд. Shaker, ISBN: 3-8265-6962-8) определяли растворимость соединения примера 1 при использовании в количестве 28 мкг/мл и 454 мкг/мл, в то время как соответствующие значения для соединения формулы (V1) составляли 5 мкг/мл, соответственно, 18 мкг/мл. Эту значительно различающуюся растворимость смогли подтвердить также при повторении теста (43/290 мкг/мл по сравнению с 6/20 мкг/мл).
Предлагаемое согласно изобретению соединение формулы (I), таким образом, в 6-16 раз лучше растворимо, чем сравнительное соединение формулы (VI). Предлагаемое согласно изобретению соединение формулы (I), таким образом, обладает более высокой доступностью в растворенной форме в месте действия. Также, в случае необходимости, более высокие дозы, по сравнению с веществами, обладающими худшей растворимостью, могут быть доступны для взаимодействия с соответствующей транспортной системой. Исходя из имеющегося в распоряжении объема 250 мл (биофармацевтическая система классификации), растворимы дозы вплоть до примерно 100 мг, тогда как соединение формулы (VI) в лучшем случае было бы растворимо только в дозах в области 5 мг (в самом худшем случае даже только 1,25 мг).
Стабильность соединения формулы (I), а также стабильность сравнительного соединения формулы (VI) в растворе тестировали следующим образом:
Стабильность растворенного соединения формулы (I), а также растворенного соединения формулы (VI) определяли в водном буфере в области значений рН 1,2-8,0. 1 мг Соответствующего соединения взвешивали в мерной колбе емкостью 5 мл. Для растворения вещества использовали небольшое количество ацетонитрила. Затем доливали водным буфером вплоть до метки. После центрифугирования выпавшего в осадок соединения прозрачный супернатант в течение 24 часов при температуре 37°С испытывали на стабильность в растворе. Оценку образцов осуществляли путем ВЭЖХ/УФ-анализа. Результаты, достигнутые с соединением примера 1 и соединением формулы (VI), представлены в следующей таблице:
Предлагаемое согласно изобретению соединение формулы (I), таким образом, в зависимости от значения рН по меньшей мере в 2,7 раза стабильнее, чем соединение формулы (VI), и поэтому образует меньше побочных продуктов, чем соединение формулы (VI). Более незначительные количества системно действующих побочных продуктов означают менее значительный потенциал в отношении нежелательных побочных действий.
Реферат
Изобретение относится к новому дифенилазетидинону формулы (I):

а также к его фармацевтически приемлемым солям. Соединение может быть использовано в качестве лекарственного средства для лечения гиперлипидемии. Описан способ его получения и промежуточные соединения для его получения. 8 н. и 1 з.п. ф-лы.
Формула




Документы, цитированные в отчёте о поиске
Производные азетидинона, фармацевтическая композиция с гипохолестеринемической активностью и способ снижения уровня холестерина в сыворотке
Производные азетидинона и их фармацевтически приемлемые соли и фармацевтическая композиция с антиатеросклеротической или гипохолестеринемической активностью
Комментарии