Производные азетидинона, фармацевтическая композиция с гипохолестеринемической активностью и способ снижения уровня холестерина в сыворотке - RU2138480C1
Код документа: RU2138480C1
Чертежи







Описание
Изобретение относится к новым лактамам с биологической активностью, в частности к новым производным азетидинона, обладающим гипохолестеринемической активностью.
Известны производные азетидинона, которые обладают гипохолестеринемической активностью (см. заявку WO 93/020 48, опуб. 4 февраля 1993 г.).
Задачей изобретения является расширение ассортимента высокоактивных производных азетидинона с гипохолестеринемической активностью.
Данная
задача
решается предлагаемыми производными азетидинона общей формулы (I)

Ar1 и Ar2 независимо друг от друга выбраны из группы, включающей арил и замещенный остатком R4 арил,
Ar3 - арил или замещенный остатком R5 арил,
R и R2 независимо друг от друга выбраны из группы, включающей -OR6 и -O(CO)R6,
R1 и R3 являются водородом или низшим алкилом,
g - 0 или 1,
r - 0 или 1,
m, n и p независимо друг от друга означают число 0, 1 или 2, при этом по меньшей мере один из индексов q и r означает число 1, и сумма индексов m, n, p, q и r равна 2 или 3, и если p - 0, и r - 1, то сумма индексов m, q и n равна 2 или 3,
R4 - независимо выбраны из группы, включающей галоид, -OR6, O(CH2 )1-5OR7
R5 - независимо выбран из группы, включающей - OR6, -O(CO)R6 и, - (низший алкилен)COOR8-CH=CH,
R6 , R7 и R8 независимо выбраны из группы, включающей водород, низший алкил, незамещенный или замещенный арилом.
В первую группу предпочтительных производных
азетидинона входят соединения, у которых
Ar1 - фенил или замещенный остатком R4 фенил,
Ar2 - фенил или замещенный остатком R4 фенил,
Ar3 - замещенный остатком R5 фенил.
Во вторую группу предпочтительных производных азетидинона входят соединения, у которых
Ar1
-замещенный остатком R4 фенил, где R4 означает галоид,
Ar2 -замещенный остатком R4 фенил, где R4 означает галоид или остаток -OR6, где R6 означает низший алкил или водород,
Ar3 -замещенный остатком R5 фенил, где R5 означает остаток - OR6, где R6
означает низший алкил или водород.
В третью группу предпочтительных производных азетидинона входят соединения, у которых
R1 и R3 - водород,
R и
R2 означают группу формулы -OR6, где R6 означает водород, сумма индексов m, n, p, q и r - 2 или 3.
В четвертую группу предпочтительных производных
азетидинона входят соединения, у которых m, n и r - 0,
q - 1,
p - 2.
В пятую группу предпочтительных производных азетидинона входят соединения, у которых
p,
q
и n - 0,
r - 1,
m - 2.
В частности предпочитаются производные азатидинона вышеприведенной общей формулы (I), которые выбраны из группы, включающей
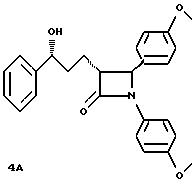
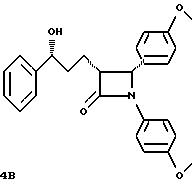
относ.
3(R)-(2(R)-окси-2-фенилэтил)-4(R)-(4- метоксифенил)-1-фенил-2-азетидинон,
относ. 3(R)-(2(R)-окси-2- фенилэтил)-4(S)-(4-метоксифенил)-1-фенил-2-азетидинон,
3(S)-(1(S)- окси-3-фенилпропил)-4(S)-(4-метоксифенил)-1-фенил-2-азетидинон,
3(S)-(1(R)-окси-3-фенилпропил)-4(S)-(4-метоксифенил)-1-фенил-2- азетидинон,
относ.
3(R)-(1(R)-окси-3-фенилпропил)-4(S)-(4- метоксифенил)-1-фенил-2-азетидинон.
относ. 3(R)-[(S)-окси-(2- нафталенил)метил]-4(3)-(4-метоксифенил)-1 -фенил-2-азетидинон,
3(R)-[(R)-окси-(2-нафталенил)метил] -4(S)-(4-метоксифенил)-1-фенил- 2- азетидинон,
3(R)-(3(R)-окси-3-фенилпропил)-1,4(S)-бис-(4- метоксифенил)-2-азетидинон,
3(R)-(3(S)-окси-3-фенилпропил)- 1,4(S)-бис-(4-метоксифенил)-2-азетидинон,
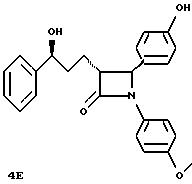
4(3)-(4-оксифенил)-3(R)-(3(R)-окси-3-фенилпропил)-1-(4- метоксифенил)-2-азетидинон,
4(S)-(4-оксифенил)-3(R)-(3(S)-окси-3- фенилпропил)-1-(4-метоксифенил)-2-азетидинон,
3(R)-[3(RS)-окси-[4- (метоксиметокси)-фенил] пропил] -1,4(S)-бис-(4-метоксифенил)-2- азетидинон,
1-(4-фторфенил)-3(R)-[3(S)-(4-фторфенил)-3- оксипропил)]-4(S)-(4-окси-фенил)-2-азетидинон,
1-(4-фторфенил)- 3(R)-[3(R)-(4-фторфенил)-3-оксипропил)]-4(S)-(4-окси-фенил)-2- азетидинон,
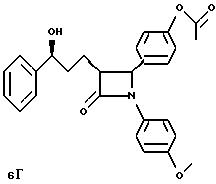
4(S)-[4-(ацетилокси)фенил] -3(R)-(3(R)-окси-3- фенилпропил)-1-(4-метоксифенил)-2-азетидинон,
4(S)-[4- (ацетилокси)фенил]-3(R)-(3(S)-окси-3-фенилпропил)-1-(4- метоксифенил)-2-азетидинон,
1-(4-фторфенил)-3(R)-[3(S)-(4- фторфенил)-3-оксипропил)] -4(S)-[4-(фенилметокси)фенил]-2- азетидинон,
3(R)-[3(R)-(ацетилокси)-3-фенилпропил] -1,4(S)-бис-(4- метоксифенил)-2-азетидинон,
3(R)-[3(S)-(ацетилокси)-3-фенилпропил] -1,4(S)-бис-(4- метоксифенил)-2-азетидинон,
3(R)-[3(R)-(ацетилокси)-3-(4- фторфенил)пропил]
-4(S)-[4-(ацетилокси)-фенил]-1-(4-фторфенил)-2- азетидинон,
3(R)-[3(S)-(ацетилокси)-3-(4-фторфенил)пропил] -4(S)- [4-(ацетилокси)-фенил]-1-(4-фторфенил)-2-азетидинон,
3(R)-[3(R)- (ацетилокси)-3-(4-хлорфенил)пропил] -4(S)-[4-(ацетилокси)-фенил]-1- (4-хлорфенил)-2-азетидинон,
3(R)-[3(S)-(ацетилокси)-3-(4- хлорфенил)пропил]
-4(S)-[4-(ацетилокси)-фенил]-1-(4-хлорфенил)-2- азетидинон,
относ. 1-(4-фторфенил)-4(S)-(4-оксифенил)-3(R)-(1 - (R)-окси-3-фенил-пропил)-2-азетидинон.
Дальнейшим объектом изобретения является фармацевтическая композиция с гипохолестеринемической активностью, которая помимо по меньшей мере одного фармацевтически приемлемого носителя содержит эффективное количество по меньшей мере одного производного азетидинона вышеприведенной обшей формулы (I) или его смеси с ингибитором редуктазы три окситриметилглутарилкоэнцима(HMG CoA), в качестве которого предпочтительно используют ловастатин, правастатин, флувастатин и симвастатин.
Широко известно, что содержание холестерина в плазме крови можно снизить с помощью редуктазы HMG CоА 3-окси-3-метилглутарил- коэнзима A; ЕC1.1.1.34) [см., Witzurn, Circulation, 80, 5 (1989), стр. 1101 -1114].
Еще одним объектом изобретения является способ снижения уровня холестерина в плазме крови путем введения по меньшей мере одного производного азетидинона вышеприведенной общей формулы (I) или его смеси с ингибитором редуктазы HMG CoA в эффективном количестве.
Соединения вышеприведенной общей формулы (I) имеют по меньшей мере один асимметричный атом углерода и поэтому все изомеры, включая энантиомеры и диастереомеры, входят в объем изобретения. Изобретение включает d- и l-изомеры как в чистом виде, так и в виде смеси, включая рацемические смеси. Изомеры можно получать известными приемами, либо путем реакции хиральных исходных соединений, либо путем разделения изомеров соединений вышеприведенной общей формулы (I). Изомеры могут также представлять собой геометрические изомеры, например, в том случае, если имеется двойная связь. Все такие геометрические изомеры входят в объем изобретения.
Приведенная выше формулировка "R6, R7 и R8 независимо выбраны из группы. .." означает не только то, что радикалы R6, R7 и R8 независимо выбраны, но и то, что, если в одной молекуле существуют более одного остатка R6, R7 или R8, соответственно, то эти остатки выбраны независимо друг от друга (т.е. если R - группа - OR6, где R6 означает водород, то R4 может представлять собой группу -OR6, где R6 означает низший алкил).
Применяемый в рамках данной заявки термин "низший алкил" означает неразветвленные или разветвленные алкильные цепи с 1 - 6 атомами углерода.
Под "арилом" понимаются фенил, нафтил, инденил, тетрагидронафтил или инданил.
Под "галоидом" понимаются атомы фтора, хлора, брома или йода.
Определенные соединения настоящего изобретения имеют кислый характер, например, те соединения, которые имеют карбоксильную группу. Эти соединения образуют фармацевтически приемлемые соли с неорганическим или органическим основанием. В качестве примеров таких солей можно назвать натриевую, калиевую, кальциевую, алюминиевую, золотую и серебряную соли. Изобретением также охватываются соли, образованные фармацевтически приемлемыми аминами, такими, как, например, аммиак, алкиламины, оксиалкиламины, N-метилглюкамин и т.п.
Некоторые производные азетидинона вышеприведенной
общей формулы (I) можно получать, например, способом, включающим следующие стадии:
а) обработку сильным основанием лактона формулы


где X, Y и Z означают группу -CH2 -,
R' и R2' - остатки R и R2, соответственно, или защищенные гидроксильные группы,
Ar10 - остаток Ar1 защищенный арил, замещенный гидроксилом, а остальные остатки и индексы имеют вышеуказанные значения, при этом в лактоне формулы Б, если каждый из индексов n и r - 0, то p - число 1 или 2:
б) взаимодействие продукта стадии (а) с имином формулы

где Ar20 - остаток Ar2, защищенный арил, замещенный гидроксилом,
Ar30 - остаток Ar3, защищенный арил, замещенный гидроксилом,
в) прекращение реакции путем добавки кислоты,
г) удаление, если необходимо, защитных групп остатков R', R2', Ar10, Ar20 и Ar30, если такие имеются,
д) введение, если необходимо, функциональных групп в служащие в качестве заместителей гидроксильные группы у остатков R, R2, Ar1, Ar2 и Ar3.
В общем все соединения общей формулы (I) можно получать известными способами, описанными, например, в нижеследующем. В приведенных ниже формулах радикалы X, Y и Z означают группу -CH2-.
Способ A:

Соединения формул (Ia) и (Iб), где Ar1, Ar2, Ar3, X, Y, Z, R, R1, R2, R3, m, n, p, q и r имеют вышеуказанные значения, можно получать путем обработки сложного эфира формулы (III), где R10 означает низший алкил, например этил, или хиральную группу, такую, как, например, ментил или 10- (диизопропилсульфонамидо)изоборнил, а другие остатки и индексы имеют вышеуказанные значения, сильным основанием, таким, как, например, диизопропиламид лития, в среде подходящего растворителя, такого, как, например, тетрагидрофуран, при температуре -78oC. В качестве совместного растворителя при необходимости можно добавлять способствующее растворению вещество, такое, как, например, гексаметилфосфортриамид. Добавляют имин формулы (II), где Ar20 и Ar30 имеют вышеуказанные значения, а реакционную смесь либо нагревают до комнатной температуры, либо оставляют на подходящем низком уровне температуры, например -78oC, в течение соответствующего срока, после чего реакцию прекращают путем добавки пригодной кислоты, такой, как, например, 1н. соляная кислота. Продукт выделяют известными приемами. В случае наличия указанной в таблице 1 (см. в конце описания) защитной группы у одного или нескольких остатков, которые могут быть защищены, необходимо проведение дополнительной стадии, включающей удаление защитной группы известным приемом. Однако в случае соединения формулы (Iа), (Iб) или любого соединения формулы (I), где защищенной гидроксильной группой Ar10, Ar20, Ar30, R' или R2' является алкоксил или бензилокси-группа, защитную группу не надо удалять, чтобы получить соединение формулы (I). При использовании хирального сложного эфира формулы (III) получаемое соединение формулы (Iа) или (Iб) не является рацемическим.
Имины формулы (II) (Ar30-CH=N-Ar20) можно получать из альдегидов формулы Ar30-CHO и аминов формулы Ar20-NH2 известными приемами. Альдегиды формулы Ar30-CHO и амины формулы Ar20-NH2 можно покупать в торговле или получать известными приемами.
Способ A:

Соединения формул (Iв) и (Iг), где остатки и индексы имеют вышеуказанные значения, можно получать способом, включающим следующие стадии:
а) Лактон формулы (IV), где остатки и индексы имеют вышеуказанные значения, обрабатывают сильным основанием, таким, как, например алкиллитий (например, н-бутиллитий), гидрид металла (например, гидрид натрия), алкоголят металла (например, метилат натрия), галоидметаллом (например, четыреххлористым титаном), обмен ионов металлов энолата лития с галоидметаллом (например, хлоридом цинка), обмен ионов металлов энолата лития с алкилом металла (например, 9-борабициклононил-трифлатом), или предпочтительно амидом металла (например, диизопропиламидом лития), в среде пригодного неводного органического растворителя, такого, как, например, сухой тетрагидрофуран, диэтиловый эфир или бензол, в сухой, инертной атмосфере, например в атмосфере азота. Реакцию осуществляют при температуре примерно от 0oC до -85oC, предпочтительно -78oC, в течение примерно 5 - 60 минут, предпочтительно примерно 30 минут. Можно добавить 1 - 50% совместных растворителей, предпочтительно примерно 10% гексаметилфосфортриамида.
(б) В течение 5 - 60 минут, предпочтительно 30 минут, в продукт стадии (а) подают имин формулы (II), где Ar20 и Ar30 имеют вышеуказанные значения, при этом температуру реакционной смеси в течение 1 - 12 часов, предпочтительно примерно 3 часов, сохраняют при примерно от 0oC до -85oC, предпочтительно -78oC, или реакционную смесь в течение указанного времени нагревают на примерно 10oC/час - 70oC/час, предпочтительно примерно 30oC/час, до температуры примерно 20oC.
(в) Реакцию прекращают добавлением пригодной кислоты, такой, как, например, 1 н. соляная кислота.
(г) Защитные группы остатков R', R2', Ar10, Ar20 и Ar30, если имеются, при необходимости удаляют известными приемами, например, силильные защитные группы удаляют путем обработки фторидом.
(д) Соединения формулы (I), где R и R2 означают группы OR6, где R6 - водород, известными приемами можно перевести в другие соединения формулы (I), где R и R2 содержат функциональные группы, т.е. независимо выбраны из группы, включающей OR6a и -O(CO)R6, где R6 имеет вышеуказанные значения, a R6a означает низший алкил, незамещенный или замещенный арилом. Например, в результате взаимодействия спирта с галоидным алкилом в присутствии пригодного основания, такого, как, например, гидрид натрия, получают замещенные алкоксилом соединения (т. е. R или R2 означают OR6, где R6 - низший алкил); в результате взаимодействия спирта с агентом ацилирования, таким, как, например, ацетилхлорид, получают соединения, у которых R или R2 означают -OC(O)R6. В соединения формулы (I), где любой из остатков Ar1, Ar2 или Ar3 содержит гидроксил, или аналогичным приемом можно ввести функциональные группы с получением других соединений формулы (I), т.е. соединений, у которых остатки R4 и R5 независимо выбраны из группы, включающей -OR6a, -O(CO)R6 и -O(CH2)1-5OR7.
Продукты стадий в, г или д выделяют известными приемами, такими, как, например, экстракция, кристаллизация или, предпочтительно, хроматография на силикагеле марки 60. При применении хирального лактона получаемое соединение формулы (Iв) или (Iг) не является рацемическим соединением.
Лактоны формулы IVa можно использовать в качестве исходного вещества в процессе,
описанном стадиями (а) - (д), с получением соединений формул (Iж) и
(Iз), при этом если каждый из индексов n и r - 0, то p должен означать число 1 - 2:

Лактоны формул IV и IVa известны или их можно получать известными приемами.
Способ Б

Азетидиноны формулы (V), где Ar20 и Ar30 имеют вышеуказанные значения, можно подвергать реакции с получением соединений формул (Iд) и (Iе) (т.е. соединений формулы (I), где r - 1, R2 - гидроксил, и p - 0) путем обработки азетидинона (V) сильным основанием, таким, как, например, изопропилциклогексиламид лития, в среде пригодного растворителя, например, тетрагидрофурана, в присутствии или отсутствии гексаметилфосфортриамида при температуре -78oC, с последующей добавкой альдегида или кетона формулы (VI), где Ar10, X, Y, R', R1, R3, m, n и q имеют вышеуказанные значения. Так же, как и в способе A, защитные группы остатков Ar10, Ar20, Ar30, R' и R2' удаляют в случае необходимости.
Данным приемом получают несколько возможных диастереомеров, которые
можно отделять при помощи комбинации известных приемов, включающих кристаллизацию,
хроматографию на силикагеле и высокопроизводительную жидкостную хроматографию. Остальные диастереомеры можно
получать путем реакций транслокации, таких, как, например, нижеприведенная
последовательность реакций по Митсунобу, где изображены частичные структуры соединения формулы (Iе):

DEAD - диэтилазодикарбоксилат
MeOH - метанол
PPh3 - трифенилфосфин
Реагенты перемешивают при комнатной температуре в течение ночи, а получаемый сложный эфир муравьиной кислоты переводят в соответствующее гидроксильное соединение желаемой стереохимии.
Способ
В:

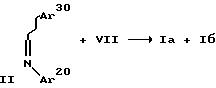
Соединения вышеуказанной формулы (la) можно получать путем взаимодействия хирального вспомогательного вещества, такого, как, например, соединение формулы (VIII), с активированным производным карбоновой кислоты формулы (VII), таким, как, например, хлорангидрид кислоты (L = Cl), смешанный ангидрид, полученный с использованием сложного фенилового эфира дихлорфосфорной кислоты (L = OP(O)(Cl)Oфенил), сложный эфир М-метил-пиридиния, полученный в результате взаимодействия кислоты с йодидом М-метил-2-хлор- пиридиния (L = иодид 2-окси-N-метилпиридиния), и сложный эфир 2- тиопиридила, полученный в результате взаимодействия хлорангидрида кислоты с 2-тиопиридином, где остальные остатки и индексы имеют вышеуказанные значения. Получаемый продукт подвергают энолизации, например, четыреххлористым титаном или тетраметилендиамином, с последующей конденсацией с альдегидом Ar30CHO. Продукт подвергают гидролизу до соответствующей кислоты, с последующим взаимодействием соединения формулы (IX) с амином Ar20NH2, и циклизацией получаемого соединения формулы (X), например, триалкилфосфином и диалкилазодикабоксилатом. Так же, как и в способе A, защитные группы остатков Ar10, Ar20, Ar30, R' и R2' удаляют при необходимости.
Способ Г:
Соединения вышеприведенной формулы (Iа) также можно получать путем обработки имина формулы (II), где Ar20 и Ar30 имеют вышеуказанные значения, активированным производным карбоновой кислоты вышеуказанной формулы (VII), в присутствии основания-трет.амина, такого, как, например, триэтиламин, три-бутиламин или диэтилизопропиламин, в среде инертного растворителя, например дихлорметана. Так же, как и в способе A, защитные группы остатков Ar10, Ar20, Ar30, R' и R2' удаляют при необходимости. Применение других оснований, например пиридина, способствует образованию соединений формулы (Iб).
Способ Д:


TMEDA - тетраметилэтилендиамин
На первой стадии соединение формулы (XII) растворяют в пригодном растворителе, например неводном дихлорметане, и обрабатывают кислотой Льюиса, например четыреххлористым титаном, при температуре примерно -60oC - 0oC, предпочтительно примерно -25o C, в сухой, инертной атмосфере, например в атмосфере аргона. Добавляют основание-трет.амин, такой, как, например, тетраметилэтилендиамин, и смесь перемешивают при температуре примерно от -60oC до 0oC, предпочтительно примерно от -25oC до -15oC, в течение примерно 1 часа. В течение примерно 5 минут подают имин формулы ArCH=NAR либо как таковой, либо в качестве раствора в пригодном растворителе, таком, как, например, неводный дихлорметан, и реакционную смесь интенсивно перемешивают при температуре примерно от -60oC до до 0oC, предпочтительно примерно от -25oC до -15oC, в течение 3 - 6 часов предпочтительно 4 часов, или до того, как результат тонкослойной хроматографии показывает, что реакция окончена. В реакционную смесь при температуре реакции подают кислоту, например уксусную кислоту, и при перемешивании в течение 1 - 3 часов, предпочтительно 2 часов, смеси дают медленно нагреваться до комнатной температуры. Соединение формулы (XIII) выделяют путем экстракции пригодным растворителем, например дихлорметаном, после чего очищают путем кристаллизации или хроматографии на силикагеле.
На второй стадии продукт обрабатывают сильным ненуклеофильным основанием, таким, как, например, бистриметилсилиламид натрия или лития, при температуре примерно от -78oC до 10oC. После реакции смесь подают в водный раствор винной кислоты, а продукт выделяют из органического слоя. Так же, как и в способе А, защитные группы остатков Ar10, Ar20, Ar30, R' и R2' удаляют при необходимости.
Способ Е:

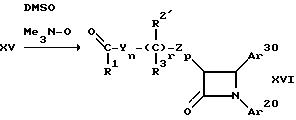
DMSO -диметилсульфоксид

Соединения формулы (Iж') и (Iз) (т.е. соединения формулы (I), где R - гидроксил), где R2 как указано выше означает защищенную гидроксильную группу, а остальные остатки и индексы имеют вышеуказанные значения, можно получать путем взаимодействия имина формулы (II) и производного карбоновой кислоты формулы (XIV), где остатки и индексы имеют вышеуказанные значения, по способу Г, с последующим окислением получаемого галоидного соединения формулы (XV) путем обработки окислителем, таким, как, например, окись триметиламина, трехокись хрома или озон, в среде растворителя, например диметилсульфоксида. Получаемый альдегид или кетон формулы (XVI) подвергают взаимодействию с арил-металлоорганическим реагентом (таким, как, например, Ar10XmMgBr, Ar10XmMgLi, Ar10XmMgCl, Ar10XmCeCl2), с получением соединения формулы (Iж') или (Iз). Как указано выше, заместители Ar10, Ar20, Ar30 и R2 можно переводить в желаемые заместители Ar1, Ar2, Ar3 и R2 известными приемами.
Способ Ж:

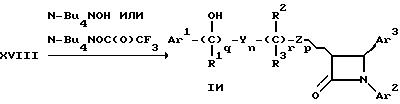
Bu - бутил
Соединения формулы (Iи) с гидроксильным заместителем на боковой цепи, смежной с остатком Ar1, (т.е. соединения формулы I, где m - 0), можно получать путем нагревания соединения формулы. (XVII), полученного по способу Г, где остатки и индексы имеют вышеуказанные значения, в течение примерно 1 - 6 часов до температуры примерно 60oC - 100oC вместе с агентом галогенирования, например N-бромсукцинимидом, в среде пригодного растворителя, такого, как, например, четыреххлористый углерод, в присутствии инициатора, например перекиси бензоила. Получаемое соединение формулы (XVIII), где Hal означает атом хлора, брома или йода, а другие остатки и индексы имеют вышеуказанные значения, нагревают в среде пригодного растворителя, например дихлорметана, вместе с солью тетраалкиламмония, такой, как, например, гидроокись тетра-н- бутил-аммония (n-Bu4NOH), с получением соединения формулы (Iи). В качестве альтернативы, соединение формулы (XVIII) нагревают в среде пригодного растворителя, например дихлорметана, с трифторацетатом тетра-н-бутиламмония (n-Bu4 NO-C(O)CF3), с последующей обработкой слабым основанием, таким, как, например, насыщенный аммиаком этанол, с получением соединения формулы (Iи).
Способ З:


Met - металл
Соединения формулы (Iй) (т.е. соединения формулы (I), где R - гидроксил, R1 - водород, и q - 1) получают из соединений формулы (XIX) через 2 стадии обработки. Первая стадия заключается в том, что соединение формулы (XIX), где остатки и индексы имеют вышеуказанные значения, растворяют в пригодном неводном растворителе, например тетрагидрофуране, при температуре примерно от -20oC до 22oC, предпочтительно примерно 0oC, в сухой, инертной атмосфере, например в атмосфере аргона, и подают донор переходного металла, например тетракис(трифенилфосфин)палладий или смесь ацетата палладия с трифенилфосфином. Металлоорганическое соединение формулы Ar10-Xm-Met, где Ar, X и m имеют вышеуказанные значения, a Met означает, например, монохлорид цинка или -В(OH)2, подают в реакционную смесь при температуре примерно от -20oC до 22oC, предпочтительно примерно 0oC, реакционную смесь перемешивают в течение примерно 15 минут - 4 часов, предпочтительно примерно 1 часа, после чего ей дают нагреваться до температуры примерно 22oC. В результате добавки разбавленной кислоты, например 1 н. соляной кислоты, с последующей экстракцией пригодным органическим растворителем, таким, как, например, этилацетат, получают соединение формулы XX.
Кетон формулы (XX) растворяют в пригодном растворителе, например метаноле. Добавляют катализатор гидрирования, например палладий на угле, и в течение 1 - 24 часов, предпочтительно примерно 16 часов, смесь подвергают воздействию газообразного водорода под давлением примерно 0,984 - 7,31 кг/см3 предпочтительно примерно 4,219 кг/см3. Катализатор гидрирования удаляют путем фильтрации, а растворитель удаляют в вакууме, при этом получают соединение формулы (Iй) в виде смеси спиртовых диастереомеров, которые разделяют известными приемами.
В качестве
альтернативы кетон формулы (XX) растворяют в
пригодном растворителе, например тетрагидрофуране, при температуре примерно от -40oC до 22oC, предпочтительно примерно 0o
C, добавляют подходяющий восстановитель, такой,
как, например, боран натрия, замещенный бороводород (например, [cbz-пролин]3BHNa) или боран, при необходимости в присутствии пригодного
хирального промотора, взятого либо в каталитическом,
либо в стехиометрическом количестве, такого, как, например, хиральный боран следующих структур:

В результате добавки разбавленной кислоты, например 1 н. соляной кислоты, с последующей экстракцией пригодным растворителем получают соединения формулы (Iй). Как и в вышеуказанных приемах, защитные группы остатков Ar10, Ar20, Ar30 и R2' удаляют при необходимости. При применении хирального реагента или хирального промотора получают нерацемический продукт.
Соединения формулы (XIX) можно получать нижеприведенным многостадийным приемом:


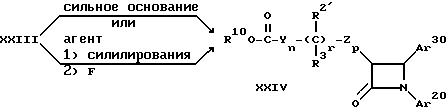

Соединения формулы (XXI), где R10 - низший алкил, а остальные остатки и индексы имеют вышеуказанные значения, являются торговыми продуктами, или их можно получать путем обработки соответствующей карбоновой кислоты (т.е. соединений, где атом хлора замещен гидроксилом) хлорирующим средством, таким, как, например, тионилхлорид или оксалилхлорид, как таковым или в подходящем инертном органическом растворителе, например толуоле, в сухой атмосфере при температуре примерно 40oC - 110oC, предпочтительно примерно 70oC. В качестве альтернативы добавляют катализатор, например диметилформамид, реакцию осуществляют при температуре примерно 22oC, а растворитель и избыточные реагенты удаляют в вакууме. Соединение формулы (XXI) подвергают взаимодействию с хиральным вспомогательным веществом, таким, как, например, (S)-4- фенил-2-оксазолидинон, следующим приемом: хиральное вспомогательное вещество обрабатывают сильным основанием, таким, как, например, алкиллитий, гидридом металла или основанием- трет.амином, например триэтиламином, в среде пригодного неводного органического растворителя, например сухого тетрагидрофурана, в сухой, инертной атмосфере, например в атмосфере аргона, при температуре примерно от -85oC до 22oC, предпочтительно примерно 0oC, в течение примерно 10 - 60 минут, предпочтительно примерно 30 минут. Получаемый анион без выделения подвергают взаимодействию с соединением формулы (XXI) в среде подходящего неводного органического растворителя, например сухого тетрагидрофурана, в сухой, инертной атмосфере, например в атмосфере аргона, при температуре примерно от -85oC до 22oC, предпочтительно примерно 0oC, в течение примерно 30 - 60 минут, предпочтительно примерно 30 минут. Реакционную смесь нагревают до температуры примерно 22oC и реакцию продолжают в течение 1 - 12 часов, предпочтительно 6 часов. Добавляют воду, соединение формулы (XXII) выделяют экстракцией и очищают путем кристаллизации.
Соединение формулы (XXII) обрабатывают приемом, описанным в качестве стадии 1 способа Д, при этом получают соединение формулы (XXIII).
Циклизацию азетидинона можно осуществлять альтернативными приемами. Первый прием заключается в том, что соединение формулы (XXIII) обрабатывают сильным ненуклеофильным основанием, таким, как, например, бистриметилсилиламид натрия или лития, в среде пригодного инертного органического растворителя, таким, как, например, дихлорметан, при температуре примерно от -78oC до 10oC, предпочтительно примерно 0oC. Реакционную смесь перемешивают в течение примерно 1 - 2 часов, при этом постепенно нагревают до температуры 22oC. Соединение формулы (XXIV) выделяют путем стандартной экстрации дихлорметаном. Другой, двухстадийный прием заключается в том, что соединение формулы (XXIII) сперва обрабатывают слабым агентом силилирования, таким, как, например, N, O-бис(триметилсилил)ацетамид, при температуре примерно 0oC - 100oC, предпочтительно примерно 40oC, в течение примерно 10 - 60 минут, предпочтительно 30 минут, после чего обрабатывают донором аниона фторида, таким, как, например, фторид тетрабутиламмония, при температуре примерно 0oC - 100oC, предпочтительно примерно 40oC, и перемешивают в течение 0,5 - 4 часов, предпочтительно 2 часов. Соединение формулы (XXIV) выделяют стандартными методами экстракции.
Соединение формулы (XXIV) подвергают гидролизу пригодным основанием, например гидроокисью лития, в среде подходящего растворителя, такого, как, например, 66%-ный водный раствор метанола, при температуре примерно 0oC - 50oC, предпочтительно примерно 22oC, в течение примерно 1 - 4 часов, предпочтительно 2 часов, после чего экстрагируют пригодным растворителем, например этилацетатом. Получаемую кислоту переводят в соответствующий хлорангидрид кислоты, как описано выше, путем обработки хлорирующим веществом, таким, как, например, оксалилхлорид, с получением соединения формулы (XIX).
Исходные вещества формул (III), (V), (VII), (VIII), (XIV), (XVII), (XXI) и (XXV) являются либо торговыми продуктами, либо широко известными соединениями, которые можно получать известными приемами.
Реакционноспособные группы, которые не участвуют в осуществлении вышеприведенных реакций, можно защищать стандартными защитными группами, которые после завершения реакции опять удаляют известными приемами. В таблице 1 (см. в конце описания) приведены некоторые типичные защитные группы.
Как уже указывалось выше, производные азетидинона согласно изобретению проявляют гипохолестеринемическую активность.
Активность ин виво соединений формулы (I)
можно определять следующим методом:
Исследование ин виво (на гиперлипидемических хомяках)
Хомяки распределяются на группы по 6 и в течение 7 дней им дают пищу Пурина Чау N 5501,
содержащую 0,5% холестерина. Расход пищи регистрируют с тем, чтобы определять общее количество подаваемого
вместе с пищей холестерина. Начиная с началом дачи пищи, раз в день животным орально дают
исследуемое соединение в виде раствора или суспензии в кукурузном масле (0,2 мл). Контрольным животным дают
только 0,2 мл кукурузного масла. Всех животных, умирающих или находящихся в плохом
состоянии, умерщвляют. Через 7 дней животных анестезируют внутримышечным впрыскиванием кетамина и умерщвляют путем
обезглавливания. Кровь собирают в трубках, содержащих этилендиаминотетрауксусную
кислоту, для осуществления анализа липидов в плазме. Кроме того, иссекают печень для осуществления анализа липидов в
ткани. Анализ липидов проводят по опубликованным приемам (Schnitzer-Polokoff. R.,
и др., Comp.Biochem. Physiol. , 99А, 4, 1991 г., стр. 665 - 670), а активность исследуемых соединений определяют как
%-ное уменьшение липидов по сравнению с контрольным опытом.
Результаты опытов сведены в таблице 2 (см. в конце описания).
Если производные азетидинона вышеприведенной общей формулы (I) применять в сочетании с редуктазой HMG CoA, то достигаемое при этом процентное уменьшение уровня холестерина в сыворотке превышает сумму эффектов, получаемых при отдельном применении средств.
Опыты проводили на самцах собак породы Бигл одинакового веса, которые делили на группы по 5 животных. Все собаки проявляли эквивалентный уровень холестерина в крови. Вместе с содержащим мальтодекстрин кормом собакам давали а) 0,007 мг/кг/день соединения примера 6А, б) 5 мг/кг/день ловастатина, в) 2,5 мг/кг/день правастатина, г) 5 мг/кг/день флувастатина, д) 0,25 мг/кг/день симвастатина, е) 0,007 мг/кг/день соединения примера 6А и 5 мг/кг/день ловастатина, ж) 0,007 мг/кг/день соединения примера 6А и 2,5 мг/кг/день правастатина, 3) 0,007 мг/кг/день соединения примера 6А и 5 мг/кг/день флувастатина, и) 0,007 мг/кг/день соединения примера 6А и 0,25 мг/кг/день симвастатина. Опыты проводили в течение 14 дней. В дни 0, 3, 7 и 14 брали на анализ плазму крови.
В каждом опыте определялось процентное снижение уровня холестерина в сыворотке по сравнению с контрольной группой, собакам которой давали корм без добавки соединения примера 6А или указанного статина.
Результаты опытов сведены в таблице 3 (см. в конце описания).
Суточная гипохолестеринемическая доза соединения формулы (I) составляет примерно 0,1 - 30 мг/кг веса тела в день, предпочтительно примерно 0,1 - 15 мг/кг веса тела в день. Таким образом, для среднего веса тела 70 кг, дозировка составляет примерно 5 - 1000 мг медикамента в день, причем это количество дается либо одной дозой, либо 2 - 4 частичными дозами. Однако точная доза определяется лечащим врачом в зависимости от эффективности данного соединения, возраста, веса, состояния и реакции пациента.
В случае комбинированного применения производного азетидинона и ингибитора редуктазы HMG CoA типичная доза ингибитора составляет 0,1 - 80 мг/кг веса тела в день, при этом указанное количество дают либо одной дозой, либо частичными дозами, обычно 1 - 2 раза в день. Точная доза каждого компонента комбинированного средства определяется лечащим врачом в зависимости от эффективности данного соединения, возраста, веса, состояния и реакции пациента.
В случае дачи отдельных компонентов комбинированного средства, количества доз отдельных компонентов в день могут различаться, так как, например, срок активности одного компонента может быть дольше, чем срок активности другого компонента, и поэтому первый компонент можно апплицировать в более широких интервалах по сравнению со вторым компонентом.
В следующих примерах описаны методы получения соединений формулы (I). Приведенная стереохимия является относительной стереохимией, если не указано по-другому. Термины "цис" и "транс", если не указано по-другому, относятся к относительной ориентации в положениях 3 и 4 азетидинона. Термин "J" относится к константе взаимодействия протонной ЯМР, указанной в Гц, между замещенными в положении 3 и замещенными в положении 4 протонами азетидинона. Все данные по ЯМР получены с использованием раствора CDCl3, если не указано по-другому.
Пример 1

Раствор диизопропиламида лития получают путем растворения 1,19 г (11,8 ммоль) диизопропиламина в 20 мл неводного тетрагидрофурана при температуре -78oC в атмосфере аргона. Добавляют 4,9 мл 2,4 м. раствора н-бутиллития (11,8 ммоль) в гексане и перемешивают в течение 0,5 часов при температуре -78oC. В холодный раствор в течение 0,25 часов подают 1,75 г (10,8 ммоль) 4-фенил-бутиролактона в 4 мл тетрагидрофурана, при этом температуру реакции сохраняют ниже -65oC. Перемешивают при температуре -78oC в течение 0,25 часов, после чего в течение 1 часа при температуре -78oC добавляют 2,33г (11,0 ммоль) 4- метоксибензилидин-анизидина в 8 мл тетрагидрофурана. Реакционную смесь медленно в течение 1 часа нагревают до температуры -50oC. Реакцию при низкой температуре прекращают добавкой 12 мл 1 н. соляной кислоты. Реакционную смесь распределяют между диэтиловым эфиром и 1 н. соляной кислотой, эфирный слой промывают водой, эфирные экстракты объединяют, сушат над сульфатом магния и сгущают в вакууме. 3,0 г сырого остатка кристаллизуют из смеси этилацетата с диэтиловым эфиром, при этом получают 1,54 г соединения А. Фильтрат сгущают, очищают путем хроматографии на силикагеле марки 60 с применением в качестве элюента смеси этилацетата с гексаном в соотношении 4: 1 и выделяют дополнительное количество соединения А, равное 0,385 г, а также 0,420 г соединения Б.
Соединение А: точка плавления: 218
- 220oC, инфракрасная спектроскопия (далее:ИК) 1730 см-1, ударная ионизация (далее: УИ) (M+H) 374, J = 5,9 Гц
Соединение Б: точка плавления: 74 - 76oC, ИК 1730 см-1, УИ (M+H) 374, J-2,3 Гц
Аналогичным методом с применением соответствующих исходных веществ получают
соединение 1В:

Пример 2

В раствор 0,5г (1,3 ммоль) соединения А примера 1 в 2,7 мл неводного пиридина подают 0,63 мл (6,7 ммоль) ангидрида уксусной кислоты. Перемешивают в течение 16 часов, разбавляют дихлорметаном и три раза промывают 1 н. соляной кислотой, раз промывают насыщенным раствором хлорида натрия и раз водой. Органический слой сгущают досуха, а остаток выкристаллизуют из этилацетата. При этом получают 0,46 г целевого соединения.
Точка плавл.: 167 - 169oC, ИК 1745 см-1, электр.ионизация (M+) 415, J = 5,9 Гц.
Пример 3

Раствор изопропилциклогексиламида лития получают путем добавки 2,84 мл 1,6-м. раствора н-бутиллития в раствор 0,75 мл изопропилциклогексиламина в 100 мл тетрагидрофурана при температуре -78oC. 1.0 г N-фенил-4-(4-метокси-фенил)-2-азетидинона растворяют в 8 мл тетрагидрофурана и при температуре -78oC медленно подают в раствор изопропилциклогексиламида лития. После перемешивания в течение 20 минут добавляют 0,54 г гидрокоричного альдегида и реакционную смесь перемешивают в течение 4 часов при температуре -78oC. Реакцию прекращают добавкой 10%-ного раствора гидросульфата калия. Продукт экстрагируют этилацетатом. Органический слой отделяют, промывают водой и насыщенным раствором натрия. Экстракт сгущают, а получаемый остаток очищают путем колоночной хроматографии на силикагеле марки 60 с применением в качестве элюента смеси этилацетата с гексаном в соотношении 15:85. При этом получают 1,15 г целевого продукта в виде смеси диастереомеров. Диастереомеры разделяют путем высокопроизводительной жидкостной хроматографии на силикагеле с получением трех диастереомеров 3А, 3Б и 3В:

1H в CDCl3: 7,32-7,18 (м, 11H): 7,08-6,99 (м, 1H); 6,89 (д, J = 9 Гц, 2H); 4,80 (д, J = 2,4 Гц, 1H); 4,10-4,00 (м, 1H); 3,79 (с, 3H); 3,20-3,16 (м, 1H); 2,90-2,67 (м, 2H); 2,15-1,85 (м, 3H)

1H в CDCl3: 7,35-7, 10 (м, 11H); 7,08-6, 99 (м, 1H); 6,89 (д, J = 9 Гц, 2H); 5,09 (д, J = 2,4 Гц, 1H); 4,26-4,14 (м, 1H); 3,79 (с, H); 3,21-3,14 (м, 1H); 2,89-2,57 (м, 2H); 2,10-1,85 (м, 3H)

1 H в CDCl3: 7,30-7,00 (м, 10H); 6,99 (д, J = 8 Гц, 2H); 6,83 (д, J =9 Гц, 2H); 5,12 (д, J = 5, 5 Гц, 1H); 3,82 (с, 3H); 3,75-3,63 (м, 1H); 3,52 (дд, J = 9,5 Гц, 1H); 2,71-2,57 (м, 1H); 2,49-2,33 (м, 1H); 1,68-1,50 (м, 1H); 1,47-1,31 м, 1H)
Диастереомеры 3А, 3Б и 3В дальше разделяют по следующей схеме реакций, в которой изображены частичные структуры:


chiralcel OD HPLC - высокопроизводительная жидкостная хроматография с использованием колонки типа chiralcel OD

chiralcel OD HPLC - высокопроизводительная жидкостная хроматография с использованием колонки типа chiralcel OD
(Следующие данные по спектру CD [θ] получены с использованием метанола)
nM - наномоль дМ - децимоль
3Г) [θ]227nM= +2,0•104 см2/дМ; [θ]241nM= -4,6•104 см2/дМ;
элементарный анализ:
рассчитано для C25H25NO3 •0,25 H2O: C 76,6; H 6,56; N 3,57.
найдено: C 76,66; H 6,49; N 3,64.
3Д) [θ]227nM= -1,95•104 см2/дМ;
[θ]241nM= +4,45•104 см2/дМ:
элементарный анализ:
рассчитано для C25H25NO3•0,5H2O: C
75,73; H 6,61; N 3,53.
найдено: C 75,66; H 6,41; N 3,60.
3Е) [θ]226nM= +1.97•104 см2/дМ; [θ]240nM=
-5.22•104 см2/дМ;
элементарный анализ:
рассчитано для C25H25NO3: C 77,48; H 6,51; N 3,62.
найдено: C 77, 44; H 6,53; N 3,70.
3Ж) [θ ]226nM= -1,78•104 см2/дМ; [θ]241nM= -4,78•104 см2/дМ: (УИМС 388 M+H).
3З) [θ
]226nM= +2,24•104 см2/дМ; [θ]241nM= -5,4•10 см2/дМ:
[α] -54,4o (2,5 мг/мл метанола).
Элементарный анализ:
рассчитано для
C25H25NO3: C 77,
48; H 6,51; N 3,62.
найдено: C 77,11: H 6,50; N 3,72.
3И) [θ]226nM= -2,05•104 см2/дМ; [θ]241nM= +5, 2•104 см2/дМ: (УИМС 388 M+H).
3Й)

0,11 мл диэтилазодикарбоксилата подают в раствор 132 мг соединения 3З, 0,18 г трифенилфосфина и 39 мл муравьиной кислоты в 5 мл тетрагидрофурана. Перемешивают при комнатной температуре в течение ночи, после чего реакционную смесь распределяют между диэтиловым эфиром и водой. Органическую фазу промывают солевым раствором, сушат над сульфатом магния и сгущают досуха. Остаток очищают путем флеш-хроматографии с применением в качестве элюента смеси этилацетата с гексаном в соотношении 1:4, при этом получают сложный эфир муравьиной кислоты, который растворяют в метаноле, а в раствор подают 4 капель концентрированной соляной кислоты. По истечении 4 часов раствор сгущают в вакууме и остаток очищают путем флеш-хроматографии с применением в качестве элюента смеси этилацетата с гексаном в соотношении 1:3. Получают соединение 3Й. [θ]224nM= +2,54•103 см2/дМ; [θ]239nM= +5, 70•10 см2/дМ; [α]= -157,6o (2,5 мг/мл метанола).
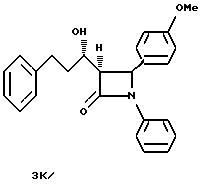
3K)

Соединение 3И обрабатывают способом, описанным для получения соединения 3Й, при этом получают соединение 3К. [θ]222nM= -3, 4•103 см2/дМ: [θ]240nM= -5,6•104 см2/дМ; [α]= +167,2o (2,5 мг/мл метанола).
Способом, описанным выше для получения соединений 3А
и 3Б, N- фенил-4-(4-метоксифенил)-2-азетидинон
последовательно обрабатывают изопропилциклогексиламидом лития и 2-нафтальдегидом, при этом получают диастереомеры 3Л и 3М:

Точка плавления 137 -138oC

Точка плавления 150 - 151oС
Пример 4

Способ 1:
Стадия 1) В раствор 10,0 г (41,5 ммоль) 4-метоксибензилиден-анизидина и 20,8 мл (87 ммоль) трибутиламина в 100 мл толуола при кипячении каплями в течение 2 часов подают 8,5 г (43 ммоль) 5-бромвалероилхлорида в 20 м толуола. Реакционную смесь перемешивают при температуре 80oC в течение 12 часов, охлаждают до комнатной температуры, три раза промывают 1 н. соляной кислотой и раз промывают водой, после чего органическую фазу сушат над сульфатом магния. Очищают путем хроматографии на силикагеле с применением в качестве элюента смеси этилацетата с гексаном в соотношении 4:1, при этом получают 5,1 г (3R, 4S)-1,4-бис-(4-метоксифенил)-3-(3-бромпропил)-2-азетидинон (относительная стереохимия). Точка плавления: 70 - 73oC. Электронная ионизация (M+) 404; J = 2,3 Гц
Стадия 2) В раствор 5,1 г (12,6 ммоль) продукта стадии 1 в 20 мл диметил-сульфоксида подают 2,39 г (31,9 ммоль) триметил-N-оксида. Смесь в течение 3 часов нагревают до температуры 60oC, охлаждают до комнатной температуры, разбавляют этилацетатом и три раза промывают водой. Водные фракции объединяют и экстрагируют этилацетатом. Органические фракции объединяют и сгущают. Сырой продукт очищают путем хроматографии на силикагеле с применением в качестве элюента смеси этилацетата с гексаном в соотношении 1:1. При этом получают 1,4 г (3R, 4S)-1,4-бис-(4-метоксифенил)-2-оксо-3-азетидин-пропаналя (относительн.стереохимия) в виде масла. Электронная ионизация (М+) 339; J =2,3 Гц.
Стадия 3) В раствор 0,734 г (2,2 ммоль) продукта стадии 2 в 4 мл тетрагидрофурана при температуре 0oC в течение 0,25 часов подают 2,4 мл (2,4 ммоль 1,0-м. раствора в тетрагидрофуране) фенилмагния в виде бромида. По истечении 1 часа при температуре 0oC добавляют 5 мл воды, фазы разделяют, органическую фазу раз промывают 1 н. соляной кислотой, сушат над сульфатом магния и сгущают с получением масла, которое очищают путем хроматографии на силикагеле с применением в качестве элюента смеси этилацетата с гексаном в соотношении 2: 1. Получают 0,372 г целевого соединения (в виде смеси диастереомеров) в качестве масла. УИ (M+H) 418.
Разделение диастереомеров: Смесь диастереомеров продукта стадии 3 обрабатывают колоночной хроматографией с использованием колонки типа
"Chiralcel OD" (изготовитель: Chiral Technologies Corp. PA). В
качестве элюента применяют смесь гексана с этанолом в соотношении 9:1. При этом получают следующие энантиомерно чистые (> 98%)
диастереомеры:
масло; [α]= +8,3o, концентрация=3 мг/мл в метаноле; УИ (М+H)418, J=2,1 Гц

масло; [α]= +33,1o, концентрация=3 мг/мл в метаноле; УИ (М+H)418, J= 2,1 Гц
масло; [α]= -8,0o, концентрация=3 мг/мл в метаноле; УИ (М+H)418, J=2,1 Гц

масло; [α]= -29,5o, концентрация=3 мг/мл в метаноле; УИ (М+ H)418, J= 2,1 Гц
Способ 2:
Стадия 1) В раствор 5,04г (0,013 моль) 1, 4-(S)-бис(4- метоксифенил)-3-(3(R)фенилпропил)-2-азетидинона в 20 мл четыреххлористого углерода при температуре 80oC в течение 1 часа 3 равными порциями подают всего 2,76 г (0,0155 моль) N-бромсукцинимида и 0,24 г (1,0 ммоль) перекиси бензоила. Ход реакции контролируют при помощи тонкослойной хроматографии с применением в качестве элюента смеси гексана с этилацетатом в соотношении 4:1. Реакционную смесь охлаждают до температуры 22oC, добавляют бикарбонат натрия, слои разделяют, органический слой три раза промывают водой и сгущают с получением сырого продукта.
УИ (M+H) 480;1H в CDCl3 δ PhCH(OH) - 5,05 ч/милл. (далее: Ph
- фенил)
Стадия 2) Сырой продукт стадии 1 растворяют в 30 мл дихлорметана и
добавляют 30 мл 40%-ного водного раствора н-бутилNOC(O)CF3. Двухфазную реакционную смесь кипятят в
течение 24 часов, охлаждают, слои разделяют, органический слой 6 раз промывают водой.
Органический слой сгущают досуха, а получаемый остаток сразу растворяют в 10 мл насыщенного аммиаком этанола. По
истечении 1 часа реакционную смесь сгущают и частично очищают путем хроматографии на
силикагеле. Дальше очищают путем высокопроизводительной жидкостной хроматографии с получением смеси соединений 4А и
4Б в соотношении 1: 1. Возможна дальнейшая очистка смеси при помощи колонки типа
Chiracel OD, при этом получают вышеуказанные соединения 4А и 4Б в отдельном виде.
Описанным в способе 2
примера 4 приемом с применением в качестве исходного соединения
4(S)-(4-ацетоксифенил)-3(R)-(3-фенилпропил)-1 -(4-метокси-фенил)-2-азетидинона получают следующие соединения:

точка плавления: 87 - 90oС;
ВРМС: рассчитано для C25H25NO4 = 403,1797, найдено 403,1785;1H в CDCl3 δ PhCH(OH) = 4,82 ч/милл.
ВРМС: рассчитано для C25H25NO 403,1797, найдено 403,1787;1H в CDCl3δPhCH(OH) = 4,78 ч/милл
ВРМС - высокоразрешающий масс-спектр
Пример 5

В раствор 0,230 г (0,68 ммоль) продукта стадии 2 примера 4 в 2 мл тетрагидрофурана подают реагент, полученный путем последовательной обработки 0,159 г (0,736 ммоль) 4-метоксиметоксифенилбромида в 4 мл тетрагидрофурана при температуре -78oC 0,6 мл 1,3-м. раствора втор.-бутиллития (0,78 моль) в гексане и 0,186 г (0,75 ммоль) трихлористым церием. По истечении 4 часов продукт экстрагируют и очищают путем хроматографии аналогичным описанному в стадии 3 примера 4 приемом, при этом получают 0,05 г смеси диастереомеров целевого соединения в качестве масла. УИ (M+H) 478.
Пример 6

Стадия 1) В раствор 41 г (0,25 моль) (S)-4-фенил-2-оксазолидинона в 200 мл дихлорметана подают 2,5 г (0, 02 моль) 4- диметиламинопиридина и 84,7 мл (0,61 моль) триэтиламина. Реакционную смесь охлаждают до температуры 0oC. В течение 1 часа по каплям добавляют 50 г (0,3 моль) метил-4-(хлорформил)-бутирата в виде раствора в 375 мл дихлорметана, а реакционной смеси дают нагреваться до температуры 22oC. По истечении 17 часов добавляют 100 мл 2-н. водного раствора серной кислоты, слои разделяют, а органический слой последовательно промывают 10%-ной гидроокисью натрия, насыщенным раствором хлорида натрия и водой. Органическую фазу сушат над сульфатом магния и сгущают с получением полукристаллического продукта.
Стадия 2) В раствор 18,2 мл (0,165 моль) четыреххлористого титана в 600 мл дихлорметана при температуре 0oC подают 16,5 мл (0,055 моль) изопропоксида титана. По истечении 15 минут добавляют 49,0 г (0,17 моль) продукта стадии 1 в виде раствора в 100 мл дихлорметана. После 5 минут подают 65,2 мл (0,37 моль) диизопропилэтиламина и перемешивают при температуре 0oC в течение 1 часа. Реакционную смесь охлаждают до температуры -20oC и подают 114,3 г (0,37 моль) 4-бензилоксибензилидин(4-фтор)анилина в виде твердого вещества. Реакционную смесь при температуре -20oC интенсивно перемешивают в течение 4 часов, после чего в течение 15 минут каплями добавляют раствор уксусной кислоты в дихлорметане. Реакционной смеси дают нагреваться до температуры 0oC и добавляют 2-н. серную кислоту. Реакционную смесь перемешивают в течение еще 1 часа, слои разделяют и промывают водой. Органическую фазу отделяют и сушат. Сырой продукт кристаллизуют из смеси этанола с водой, при этом получают чистый промежуточный продукт.
Стадия 3) В раствор 8,9 г (14,9 ммоль) продукта стадии 2 в 100 мл толуола при температуре 50oC подают 7,50 мл (30,3 ммоль) N, O- бис(триметилсилил)ацетамида. По истечении 0,5 часов подают 0,39 г (1,5 ммоль) фторида тетрабутиламмония и реакционную смесь перемешивают при температуре 50oC в течение дополнительных 3 часов. Реакционную смесь охлаждают до температуры 22oC, добавляют 10 мл метанола, смесь промывают 1 н. соляной кислотой, 1 н. бикарбонатом натрия и насыщенным раствором хлорида натрия. Органическую фазу сушат над сульфатом магния.
Стадия 4) В раствор 0,94 г (2,2 ммоль) продукта стадии 3 в 3 мл метанола подают 1 мл воды и 102 мг (2,4 ммоль) гидроокиси лития в виде моногидрата.
Реакционную смесь перемешивают при температуре 22oC в течение 1 часа, после чего добавляют дополнительное количество 54 мг (1,3 ммоль) гидроокиси лития в виде моногидрата. По истечении всего 2 часов добавляют 1 н. соляную кислоту и этилацетат, слои разделяют, органический слой сушат и сгущают в вакууме. В раствор 0,91 г (2,2 ммоль) получаемого при этом продукта в дихлорметане при температуре 22oC подают 0,29 мл (3,3 ммоль) оксалилхлорида и перемешивают в течение 16 часов. Растворитель удаляют в вакууме.
Стадия 5) В хорошо
перемешанную суспензию 4,4 ммоль хлорида 4- фтор-фенилцинка, получаемого из
4,4 мл 1-м. раствора (4,4 ммоль) 4-фторфенил-магния в виде бромида в тетрагидрофуране и 0,6 г (4,4 ммоль) хлорида цинка,
при температуре 4oC подают 0,25 г (0,21 ммоль)
тетракис(трифенилфосфин)-палладия и раствор 0,94 г (2,2 ммоль) продукта стадии 4 в 2 мл тетрагидрофурана. Реакционную смесь перемешивают при
температуре 0oC в течение 1 часа, а затем при
температуре 22oC в течение 0,5 часов. Добавляют 5 мл 1 н. соляной кислоты и экстрагируют этилацетатом. Органический слой сгущают до
масла и очищают путем хроматографии на силикагеле с
получением 1-(4-фтор-фенил)-4(S)-[4-(фенилметокси)фенил]-3(R)-[3- оксо-3-(4-фторфенил)пропил]-2-азетидинона:
ВРМС: рассчитано для C24H19F2NO3= 408,
1429, найдено 408,1411.
Стадия 6) В 0,95г (1,91 ммоль) продукта стадии 5 в 3 мл тетрагидрофурана подают 120 мг (0,43 ммоль) (R)-тетрагидро-1- метил-3,3-дифенил-1H,3H-пиррол-[1,2-с] [1, 3,2] оксазаборола, смесь охлаждают до температуры -20oC. По истечении 5 минут по каплям в течение 0.5 часов добавляют 0,85 мл 2-м. раствора (1,7 ммоль) комплекса боргидрида и диметилсульфида. По истечении всего 1,5 часов последовательно добавляют метанол и 1 н. соляную кислоту, а реакционную смесь экстрагируют этилацетатом с получением 1-(4- фторфенил)-3(R)-[3(S)-(4-фторфенил)-(3-оксипропил)] -4(S)-[4- (фенилметокси)фенил]-2-азетидинона (соединение 6А-1) в виде масла.1H в CDCl3 δ H3 = 4, 68. J = 2,3 Гц. УИ (M+H) 500.
С использованием в качестве исходного соединения (S)-тетрагидро-1-метил-3,3-дифенил-1 H,3H-пиррол-[1,2-с][1,3,2]оксазаборола получают соответствующее соединение 3(R)-оксипропил-азетидинон (соединение 6Б-1).1H в CDCl3 δ H3 = 4,69. J = 2,3 Гц. УИ (M+H) 500.
В раствор 0,4 г (0,8 ммоль) соединения 6А-1 в 2 мл этанола подают 0,03 г 10%-ного палладия на угле, реакционную смесь перемешивают под давлением водорода, равным 4,219 кг/см2 в течение 16 часов. Реакционную смесь фильтруют, растворитель сгущают с получением соединения 6А.
Точка плавления: 164 - 166oC; УИ (M+H) 410. [α]= -28,1o (с3З, метанол).
Элементарный анализ: рассчитано для C24H21F2NO3 : C 70,41; H 5,17; N 3,42; найдено C 70,25; H 5,19; N 3,54.
Аналогичной обработкой соединения 6Б-1 получают соединение 6Б. Точка плавления: 129,5 - 132,5oC; УИ (M+ H) 410. Элементарный анализ: рассчитано для C24H21F2NO3: C 70,41; H 5,17; N 3,42; найдено C 70,30; H 5,14; N 3,52.
Стадия 6') (альтернатива) В раствор 0,14 г (0,3 ммоль) продукта стадии 5 в 2 мл этанола подают 0,03 г 10%-ного палладия на угле, реакционную смесь перемешивают под давлением водорода, равным 4,219 кг/см2 в течение 16 часов.
Реакционную смесь фильтруют, растворитель сгущают с получением смеси соединений 6А и 6Б в соотношении 1:1.
Приемом,
включающим стадии 1 - 6, с использованием
соответствующих исходных веществ получают следующие соединения:

УИ (M+H) 446; ВРМС: рассчитано для C27H27NO5 = 445,1904, найдено 445,1890
УИ (M+H) 446; ВРМС: рассчитано для C25H25NO4 = 445,1904, найдено 445,1911
Пример 7

Стадия 1):

В раствор 1,0 г (2,1 ммоль) соединения 7а в 10 мл диоксана подают 1,33 г (11,98 ммоль) двуокиси селена и 0,25 мл (14 ммоль) воды. Реакционную смесь нагревают до температуры 100oC. По истечении 1 часа реакционную смесь охлаждают до комнатной температуры, путем экстракции выделяют сырой продукт в виде диастереомерной смеси (в соотношении 1:2) спиртов 76-А и 76-Б. Очищают путем высокопроизводительной жидкостной хроматографии на силикагельной колонке типа "Dynamax" с получением диастереомеров 76-А и 76-Б в отдельном виде.
Диастереомер 76-А (R): масло; J34 = 2,3 Гц,
δ CH(OH) = 4,86 (т); ВРМС C32H29NO4 рассчитано: 491,2097; найдено: 491,2074
Диастереомер 76-Б
(S): масло; J34 = 2,3 Гц, δ CH(OH) = 5,
06 (т); ВРМС C32H29NO4 рассчитано: 491,2097; найдено: 491,2117.
Стадия 2): В раствор 58 мг (0, 12 ммоль) диастереомера А стадии 1 в 2 мл этилацетата подают 20 мг 10%-ного палладия на угле и перемешивают при температуре 22oC в атмосфере водорода под давлением 0,984 кг/см2 в течение 12 часов. Фильтруют и сгущают с получением целевого соединения в качестве полутвердого вещества. Точка плавления: 90 - 92oC. J34 = 2,3 Гц,

Пример 8
В раствор 90 мг (0,2 ммоль) продукта примера 4А в
дихлорметане подают 80 мг (1,0 ммоль) ацетилхлорида и 8 мг (0,1 ммоль) пиридина. Перемешивают в течение 1 часа при комнатной температуре.
Добавляют воду, разделяют слои и выделяют соответствующее
ацетоксильное соединение 8А. Аналогичным приемом обрабатывают продукты примеров 4Б, 6Б и 6А с получением следующих соединений 8Б, 8В и 8Г,
соответственно:
8А:
1.4(S)- бис(4-метоксифенил)-3(R)-(3(R)-ацетокси-3-фенил-пропил)-2- азетидинон. УИ (M+H) 460; ВРМС C28H29NO5, рассчитано: 459,
2044; найдено: 459,2045.
8Б: 1,4(S)-бис(4-метоксифенил)-3(R)-(3(S)- ацетокси-3-фенил-пропил)-2-азетидинон. УИ (MH) 460; ВРМС C28H29NO5, рассчитано: 459,2044; найдено: 459,2048.
8В: 4(S)-(4- ацетилоксифенил)-3(R)-(3(В)-ацетилокси-3-(4-фторфенил)пропил)- 1- (4-фторфенил)-2-азетидинон. MС (бомбардировка быстрыми атомами) 493,4; ВРМС C28H25 NO5, рассчитано: 493,1695; найдено: 493,1701.
8Г: 4(S)-(4-ацетилоксифенил)-3(R)-(3(S)-ацетилокси-3-(4- фторфенил)пропил)-1-(4-фторфенил)-2-азетидинон. MС (бомбардировка быстрыми атомами) 493,4; ВРМС C28H25F2NO5, рассчитано: 493,1695; найдено: 493,1694.
С использованием соответствующих исходных соединений по
способу, описанному в примере 6, получают
1-(4-хлорфенил)-3(R)-(окси-3-(4- хлорфенил)пропил)-4(S)-(4-оксифенил)-2-азетидинон. Описанным в примере 8 приемом получают следующие диацетаты 8Д и 8Е:

УИ (M+H) 527;1H CDCl3 δ H3' = 4,65

УИ (M+H) 527;1H CDCl3 δ H3'= 4,67
Пример 9

и
Me - метил
Стадия 1) В раствор 2,35 г (6,1 ммоль) 1-фенил-3-(3-фенил-1- оксипропил)-4-(4-метоксифенил)-2-азетидинона в дихлорметане подают 2,4 г (11 ммоль) хлорхромата пиридиния и примерно 20 мг ацетата натрия. Перемешивают при комнатной температуре в течение 18 часов, после чего добавляют 40 г силикагеля и сгущают досуха. Остаток подвергают флеш-хроматографии с применением в качестве элюента смеси этилацетата с гексаном в соотношении (1:4), при этом получают 1,98 г масла (выход: 85%).1H ЯМР 2,85 - 2,95 (м, 3H), 3,15 (м, 1H), 3,80 (с, 3H), 4,10 (д, 1H, J 2,6), 5,42 (1H, д, J 2,6), 6,85 (дд, 2H, J 2,8), 7,05 (м, 1H), 7,2 - 7,35 (м, 11H).
Стадия 2) В раствор 1,78 г (4,62 ммоль) продукта стадии 1 в тетрагидрофуране при температуре -10oC подают 115 мг (4,8 ммоль) гидрида натрия. По истечении 15 минут добавляют 865 мг (4,85 ммоль) N-бромсукцинимида и перемешивают в течение 20 минут, после чего добавляют 1 н. соляную кислоту и распределяют между этилацетатом и солевым раствором. Органический слой отделяют, сушат над сульфатом магния и сгущают с получением масла. Масло подвергают флеш-хроматографии с применением в качестве элюента смеси этилацетата с гексаном в соотношении 1:10, при этом получают сперва 830 мг соединения 9а в качестве пенистого твердого вещества (выход: 39%; MС (бомбардировка быстрыми атомами) 466/464, M+H), а затем 1,1 г соединения 9б в качестве бесцветного твердого вещества (выход: 51%; MС (бомбардировка быстрыми атомами) 466/464, M+H).
Стадия 3а) В раствор 0, 68 г (1,46 ммоль) соединения 9а в 5 мл тетрагидрофурана при температуре -50oC подают 7,3 мл 1-м. раствора комплекса трифторуксусной кислоты в виде магниевой соли и трифторуксусной кислоты в диэтиловом эфире. Реакционную смесь перемешивают в течение 5 минут после чего добавляют 254 мг (2,92 ммоль) т-бутил-аминборана. По истечении 15 минут реакционной смеси дают нагреваться в течение 20 минут до температуры 0oC, добавляют 1 н. соляную кислоту и сгущают в вакууме. Остаток распределяют между этилацетатом и солевым раствором. Органические слои сгущают, а получаемое масло растворяют в смеси дихлорметана с метанолом в соотношении 1:1. Добавляют примерно 2 ммоль этаноламина. По истечении 15 минут реакционную смесь сгущают, а остаток распределяют между этилацетатом и 1 н. соляной кислотой. Органическую фазу промывают солевым раствором и сушат над сульфатом магния с получением масла. Масло очищают путем флеш-хроматографии с применением в качестве элюента смеси этилацетата с гексаном в соотношении 1:4, при этом получают 0,52 г соединения 9a-1 в виде бесцветного твердого вещества. Продукт получают в качестве смеси диастереомеров в соотношении 4:1. Выход: 76%. Спектроскопия вторичными ионами 468/466 (M+H).
Стадия 3б) C использованием в качестве исходного вещества соединения 9б способом, аналогичным стадии 3а, с применением дихлорметана в качестве растворителя получают соединение 9б-1 в качестве смеси диастереомеров в соотношении 13:1. Выход: 80%. Спектроскопия втор. ионами 468/466 M+H. Стадия 4а) В раствор 1, 0 мл трис(триметилсилил)силана в толуоле при температуре 80oC по каплям в течение 40 минут подают раствор 0,27 г (0,58 ммоль) соединения 9а-1 и 18 мг (0,12 ммоль) 2, 2'-азобисизобутиронитрила в 40 мл толуола. По истечении 1 часа подают еще 5 мг 2,2'- азобисизобутиронитрила, а температуру в течение дополнительных 1,5 часов сохраняют при 80oC. Реакционную смесь охлаждают и сгущают, остаток растворяют в ацетонитриле и три раза промывают гексаном. Слой ацетонитрила сгущают с получением 0,25 г целевого соединения в качестве рацемической смеси. Получаемое масло очищают путем высокопроизводительной жидкостной хроматографии с использованием колонки типа "Chiralcel OD", при этом получают 3З (основной) и 3Й (неосновной).
Стадия 4б) С использованием в качестве исходного вещества соединения 9б-1 по способу стадии 4а получают масло, которое очищают путем флеш-хроматографии с применением в качестве элюента смеси этилацетата с гексаном в соотношении 1: 3. Получают рацемическое целевое соединение (выход: 70%). Получаемое масло очищают высокопроизводительной жидкостной хроматографией с использованием колонки типа "Chiralcel OD", при этом получают соединения 3Й (основной) и 3З (неосновной).
Пример 10

Стадия 1) По способу примера 3 с применением 1-(4-фторфенил-4-(4- т-бутил-диметилсилилоксифенил)-2-азетидинона получают 1-(4- фторфенил-3-(3-фенил-1-оксипропил)-4-(4-т-бутилдиметилсилил- оксифенил)-2-азетидинон.
Стадия 2) Раствор 0,25 г цис-азетидинона стадии 1 в 21 мл ацетонитрила обрабатывают 2,5 мл 48%-ного водного раствора фтористого водорода. По истечении 18 часов реакционную смесь разбавляют холодной водой и экстрагируют диэтиловым эфиром. Дважды промывают водой, разбавляют бикарбонатом натрия и солевым раствором, сушат над сульфатом магния, сгущают слой диэтилового эфира. Остаток кристаллизуют из смеси этилацетата с гексаном в соотношении 1:2, при этом получают 123 мг целевого соединения в качестве бесцветного игольчатого вещества. Выход: 64%. Точка плавления: 168 - 171oC. Элементарный анализ C24H22O3FN, рассчитано: C 73,64; H 5, 66; N 3,58; найдено C 73,32; H 5,65; N 3,68.
Пример 11
Сложный этиловый эфир 4-[1-(4-фторфенил)-4-оксо-3(R))-(3-ацетокси-3- фенил-пропил)-2(S)-ацетидинил]-бензолуксусной
кислоты
Стадия 1) Аналогично стадиям 1) - 3) примера 6 с применением 5- бромбенимидин-(4-фторанилина), 5-фенилпентаноилхлорида и (S)-4- фенил-2-оксазолидинона получают 1
-(4-фторфенил)-3-(R)-(3- фенилпропил)-4(S)-4-бромфенил)-2-ацетидинон.
C24H21NOBrF
рассчит.: C 65,76; H 4,83; N 3,20
найдено: C 65,66; H 4,84;
N
3,23
Стадия 2) Продукт стадии 1) подвергают взаимодействию с N-бромсукцинимидом и перекисью бензоила аналогично примеру 4. Получаемую при этом смесь диастереомерных спиртов обрабатывают
ацетилхлоридом описанным в примере 8 образом с получением смеси диастереомерных ацетатов, которые используют на стадии 3).
Стадия 3). 0,193 г (0,858 ммоль) бромида цинка нагревают до температуры 140oC в вакууме в течение ночи, после чего охлаждают до комнатной температуры в атмосфере азота. Добавляют смесь 0,327 г (0,66 ммоль) продукта стадии 2) и 0,323 г (0,858 ммоль) 2- (трибутилстаннил)этилацетата в 4 мл диметилформамида, после чего добавляют 0,026 г (0,033 ммоль) дихлорида бис-(три-о- толуил)фосфин-палладия и смесь нагревают до температуры 80oC. Через 3 часа реакционную смесь фильтруют с помощью кизельгура марки (celite и остаток на фильтре промывают этилацетатом. Фильтрат подают в разделительную воронку, промывают водой, сушат над безводным сульфатом натрия и сгущают. Получаемый остаток подвергают хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и гексанов в соотношении 30: 70. При этом получают 0,55 г (56 %) продукта в качестве смеси диастереомеров.
В результате разделения диастереомеров высокопроизводительной жидкостной хроматографией с использованием колонки типа Chiracel OD с применением в качестве элюента смеси изопропанола и гексанов в соотношении 3: 97 получают чистые диастереомеры А и Б.
Диастереомер А
1H ЯМР (CDCl3, 400
МГц) 7,30 (9 H,м), 7,21 (2H, дд, J = 4,8; 9 Гц), 6,92 (2H, т, J= 8,9 Гц), 5,73 (1H, т, J=6,5 Гц), 4,58 (1H, д, J = 2,4 Гц), 4,16 (2H, кв, J = 7,2 Гц), 3,61 (2H, с), 3,06 (1H, м), 2,06 (3H, с), 2,03
(2H, м), 1,88 (2H, м), 1,26 (3H, т, J = 7,0 Гц).
Высокопроизводительная жидкостная хроматография в колонке типа Chiracel OD с применением в качестве элюента смеси изопропанола и гексанов в соотношении 3: 97 со скоростью подачи 1,0 мл/мин: время удерживания: 12,2 мин.
MС (электронная ионизация): 503 (M+, 3), 306 (36), 141 (100).
Диастереомер Б
1H ЯМР (CDCl3, 400 МГц) 7,30 (9 H,м), 7,21 (2H, дд, J = 4,7; 9,1 Гц), 6,92 (2H, т, J = 8,5 Гц), 5,76 (1H, т, J=6,5 Гц), 4,58 (1H, д, J = 2,2 Гц), 4,15 (2H,
кв, J = 7,1 Гц), 3,61 (2H, с), 3,07 (1H, м), 2,06 (3H, с), 1,97 (2H, м), 1,77 (2H, м), 1,26 (3H, т, J = 7,1 Гц).
Высокопроизводительная жидкостная хроматография в колонке типа Chiracel OD с применением в качестве элюента смеси изопропанола и гексанов в соотношении 3:97 со скоростью подачи 1,0 мл/мин: время удерживания: 17,4 мин.
MС (электронная ионизация): 503 (M+, 3), 306 (35), 141 (100).
Пример 12
Сложный этиловый эфир
4-[1-(4-фторфенил)-4-оксо-3(R)-[3(S)- ацетокси-3-(4-фторфенил)-пропил]-2(S)-ацетидинил]-бензолуксусной кислоты
Данное соединение получают в качестве желтого масла аналогично описанному в
примере 11 способу.
1H ЯМР (CDCl3, 400 МГц) 7,25 (8H,м), 7,02 (2H, т, J = 8,7 Гц), 6,92 (2H, т, J = 8,9 Гц), 5,69 (1H, т), 4,58 (1H, д, J = 2,3 Гц), 4,16 (2H, кв, J = 7,1 Гц), 3,61 (2H, с), 3,06 (1H, м), 2,05 (3H, с), 2,02 (2H, м), 1,87 (2H, м), 1,26 (3H, т, J =7,1 Гц).
Пример 13
4-[1-(4-фторфенил)-4-оксо-3(R)-[3(S)-гидрокси-3-(4-фторфенил)- пропил] -2)-ацетидинил]-бензолуксусная кислота
0,225 г (0,43 ммоль) продукта примера 12 растворяют в смеси 11,2 мл метанола и
22,
4 мл триэтиламина. Медленно добавляют 78 мл воды и получаемую смесь перемешивают при комнатной температуре в течение ночи, после чего триэтиламин и метанол удаляют в вакууме. Остаток доводят до pH
добавлением 1 мл 1-м. хлористоводородной кислоты, после чего в смеси разбавляют небольшое количество твердого хлористого натрия, получаемый белый осадок собирают путем вакуумной фильтрации, промывают
водой и сушат в вакууме. Получают 0,175 г (90 %) целевого продукта в качестве белого твердого вещества.
1H ЯМР (CDCl3, 400 МГц) 7,27 (6H,м), 7,22 (2H,м), 7,01 (2H, т, J=8,7 Гц), 6,93 (2H, т, J = 8,4), 4,70 (1H, т), 4,61 (1H, д, J = 2,4 Гц), 3,65 (2H, с), 3,08 (1H,м), 1,94 (4H, м).
ВРМС (бомбардировка быстрыми атомами): рассчитано для
(M+H) C26H24NO4F2: 452,1673;
найдено: 452,1681.
Пример 14
(3R, 4S)-1-(4-фторфенил)-3-[3-(4-фторфенил)-3(R,
S)-гидроксибутил] - 4-(фенил-метокси)фенил]-2-ацетидинон
100 мг (0,2 ммоль) продукта примера 6 в тетрагидрофуране обрабатывают 0,075 мл (0,225 ммоль) 3-м. бромида метилмагния при температуре
-78oC, после чего перемешивают в течение 3 часов. Реакцию прекращают с помощью хлористого аммония, получаемый продукт экстрагируют хлористым метиленом и получаемый экстракт сгущают с
получением масла, которое подвергают хроматографии на силикагеле с применением в качестве элюента смеси гексана и этилацетата. Получают 40 мг целевого продукта в качестве масла.
C32H29NO3F3
рассчит.: C 74,84; H 5,69; N 2,73
найдено: C 74,26; H 5,88; N 2,83.
Далее представлены примеры (см. в конце описания) препаратов предлагаемой фармацевтической композиции, причем под термином "активное вещество" понимается соединение вышеприведенной общей формулы (I).
Реферат
Объектом изобретения являются пригодные в качестве гипохолестеринемического средства замещенные гидроксилом азетидиноны формулы I, где Ar1 и Ar2 - арил или замещенный остатком R4 арил; Ar3 - замещенный остатком R5 арил; R и R2 выбраны из группы, включающей OR6, -O(СО)R6; R1 и R3 выбраны из группы, включающей водород и низший алкил; q = 0 или 1; r = 0 или 1; m, n и p - число 0 - 2, при этом по меньшей мере один из индексов q и r = 1, и сумма индексов m, n, р, q и r равна 2 или 3; если р = 0, и r = 1, то сумма m, q и r равна 2 или 3; R4 выбран из группы, включающей галоид, -OR6, -O(CH2)1-5 OR7; R5 выбран из группы, включающей -OR6, -O(CO)R6 -(низший алкилен)COOR8; R6, R7 и R8 выбраны из группы, включающей водород, низший алкил, не замещенный или замещенный арилом. Дальнейшим объектом изобретения являются способ снижения содержания холестерина в сыворотке путем применения предлагаемых соединений как таковых или в комбинации с ингибитором биосинтеза холестерина, а также содержащие их фармацевтические композиции, обладающие гипохолестеринемической активностью. 3 с. и 7 з.п., 3 табл.
Формула

где Аr1 и Аr2 независимо друг от друга выбраны из группы, включающей арил и замещенный остатком R4 арил;
Аr3 арил или замещенный остатком R5 арил;
R и R2 независимо друг от друга выбраны из групп, включающей -OR6 и -O(CO)R6;
R1 и R3 являются водородом или низшим алкилом;
q - 0 или 1;
r - 0 или 1,
m, n и р независимо друг от друга означают число 0,1 или 2,
при этом по меньшей мере один из индексов q и r означает число 1, и сумма индексов m, n, p, q, r равна 2 или 3, и если р - 0, и r - 1, то сумма индексов m, q и n равна 2 или 3;
R4 независимо выбраны из группы, включающей, галоид, -OR6, O(CH2)1-5OR7;
R5 независимо выбраны из группы, включающей -OR6, -O(CO)R6 и (низший алкилен)COOR8;
R6, R7 и R8 независимо выбраны из группы, включающей водород, низший алкил, незамещенный или замещенный арилом.
относ. 3(R)(2(R)-окси-2-фенилэтил)-4(R)-(4-метоксифенил)-1-фенил-2-азетидинон,
относ. 3(R)-(2(R)-окси-2-фенилэтил)-4 (S)-(4-метонсифенил)-1-фенил-2-азетидинон,
3(S)-(I(S)-окси-3-фенилпропил)-4(S)-(4-метоксифенил)-1-фенил-2-азетидинон,
3(S)-(I(R)-окси-3-фенилпропил)-4(S)-(4-метоксифенил)-1-фенил-2-азетидинон,
относ. 3(R)(I(R)-окси-3-фенилпропил)-4(S)-(4-метоксифенил)-1-фенил-2-азетидинон,
относ. 3(R)-[(S)-окси-(2-нафталенил)метил] -4(S)-(4-метоксифенил) -1-фенил-2-азетидинон,
3(R)-[(R)-окси-(2-нафталенил)метил] -4(S)-(4-метоксифенил)-1-фенил-2-азетидинон,
3(R)(R)-окси-3-фенилпропил)-1,4(S)-бис-(4-метоксифенил)-2-азетидинон,
3(R)(3(S)-окси-3-фенилпропил)-1,4(S)-бис-(4-метоксифенил)-2-азетидинон,
4(S)-(4-оксифенил)-3(R)-(З(R)-окси-3-фенилпропил)-1-(4-метоксифенил)-2-азетидинон,
4(S)-(4-оксифенил)-3(R)-(3(S)-окси-3-фенилпропил)-1-(4-метоксифенил)-2-азетидинон,
3(R)-[3(RS)-окси-[4-(метоксиметокси)-фенил] пропил] -1,4(S)-бис-(4-метоксифенил)-2-азетидинон,
1-(4-фторфенил)-3(R)-[3(S)-(4-фторфенил)-3-оксипропил)] -4(S)-(4-оксифенил)-2-азетидинон,
1-(4-фторфенил)-3(R)-3[(R)-(4-фторфенил)-3-оксипропил)] -4(S)-(4-оксифенил)-2-азетидинон,
4(S)-[4-(ацетилокси)фенил] -3(R)-(3(R)-окси-3-фенилпропил)-1-(4-метоксифенил)-2-азетидинон,
4(S)[4-(ацетилокси)фенил] -3(R)-(3(S)-окси-3-фенилпропил)-1-(4-метоксифенил)-2-азетидинон,
1-(4-фторфенил)-3(R)-[3(S)-(4-фторфенил)-3-оксипропил)] -4(S)-[4-(фенилметокси)фенил]-2-азетидинон,
3(R)-[3(R)-(ацетилокси)-3-фенилпропил] -1,4(S)-бис-(4-метоксифенил)-2-азетидинон,
3(R)-[3(S)-(ацетилокси)-3-фенилпропил] -1, 4(S)-бис-(4-метоксифенил)-2-азетидинон,
3(R)-[3(R)-(ацетилокси)-3-(4-фторфенил)пропил] -4(S)-[4-(ацетилокси)-фенил]-1-(4-фторфенил)-2-азетидинон,
3(R)-[3(S)-(ацетилокси)-3-(4-фторфенил)пропил] -4(S)-[4-(ацетилокси)-фенил]--1-(4-фторфенил)-2-азетидинон,
3(R)-[3(R)-(ацетилокси)-3-(4-хлорфенил)пропил] -4(S)-[(4-(ацетилокси)-фенил]-1-(4-хлорфенил)-2-азетидинон,
3(R)-[3(S)-(ацетилокси)-3-(4-хлорфенил)пропил] -4(S)-[(4-ацетилокси)-фенил]-1-(4-хлорфенил)-2-азетидинон,
относ. 1-(4-фторфенил)-4(S)-(4-оксифенил)-3(R)-(1-(R)-окси-3-фенил-пропил)-2-азетидинон.
21.09.93 - по
а) п. 1, где Аr1 и Аr2 независимо друг от друга выбраны из группы, включающей арил и замещенный остатком R4арил; Ar3 арил или замещенный остатком R5 арил; R и R2 независимо друг от друга выбраны из группы, включающей -OR6 и -O(CO)R6; R1 и R3 являются водородом или низшим алкилом; q - 0 или 1; r - 0 или 1; m, n и p независимо друг от друга означают число 0, 1 или 2, при этом по меньшей мере один из индексов q и r означает число 1 и сумма индексов m, n, p, q и r равна 2 или 3 и, если p - 0, и r - 1, то сумма индексов m, q и n равна 2 или 3; R4 независимо выбран из группы, включающей галоид, -OR6, где R6 имеет указанное в п.1 значение, и - -O(CH2 )1-5OR7, где R7 означает низший алкил, незамещенный или замещенный арилом; R5 независимо выбран из группы, включающей OR6 и -O(CO)R6; R6 имеет указанное в п.1 значение;
б) пп.2-7.
а) п. 1, где R4 - группа -O(CH2)1-5OR7, где R7 означает водород; R5 - группа - (низший алкилен) COOR8, где R8 имеет указанное в п.1 значение.
Комментарии