Способ получения no-донорных соединений, таких как no-донорный диклофенак - RU2322434C2
Код документа: RU2322434C2
Чертежи

Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому способу получения NO-донорных соединений, то есть соединений, высвобождающих оксид азота, с помощью сульфонированного промежуточного соединения. Изобретение относится к полученным таким образом новым промежуточным соединениям, пригодным для получения большого числа NO-донорных соединений. Изобретение далее относится к применению новых промежуточных соединений для получения фармацевтически активных NO-донорных соединений.
Изобретение далее относится по существу к кристаллической форме NO-донорных NSAID, особенно 2-[2-(нитроокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил} ацетата, способам их получения и к фармацевтическим составам, содержащим указанную кристаллическую форму, и к применению указанной кристаллической формы для получения лекарственного средства.
УРОВЕНЬ ТЕХНИКИ
NO-донорные соединения являются соединениями, имеющими NO или NO2 группу, связанную с фармацевтически активным соединением. Между фармацевтически активным соединением и NO или NO2 группой может использоваться линкер.
Преимуществом NO-донорных соединений по сравнению с близким соединением является, среди прочего, хорошая толерантность и снижение желудочно-кишечных побочных эффектов. Это является особенно очевидным для NO-донорных аналогов NSAID, таких как диклофенак и кетопрофен. NO-донорные аналоги NSAID известны благодаря их фармацевтической активности в качестве противовоспалительных и/или анальгетических агентов.
В уровне техники описаны различные способы получения NO-донорных соединений.
Cainelli и др. (Tetrahedron Lett., 1985, 28, 3369-3372) и Cainelli и др. (Tetrahedron 1985, 41, 1385-1392) описывают замещение сульфонатных эфиров нитратом тетрабутиламмония или ионообменную реакцию с нитратными ионами в растворителе, таком как пентан, толуол или бензол. В этом способе применяются высокие температуры, что делает способ небезопасным для широкомасштабного применения.
Cainelli и др. (J. Chem. Soc. Perkin Trans. I, 1987, 2637-2642) описывает нитратное замещение сульфонатных эфиров реакцией алкилметансульфонатов с нитратом тетрабутиламмония в толуоле.
Kawamura и др. (Chem. Parm. Bull, 1990, 38, 2092-2096) описывает реакцию, в которой алкилфенилсульфонат реагирует с нитратом тетрабутиламмония в толуоле.
Стоимость исходного нитрата тетраалкиламмония, применяемого в стехиометрических количествах, как описано в документах предшествующего уровня техники, является высокой для широкомасштабного получения NO-донорных соединений. По экономически причинам предпочтительными являются способы, в которых могут использоваться более дешевые нитраты и нитраты щелочного металла с низким молекулярным весом. Однако нитраты тетраалкиламмония могут использоваться в качестве катализаторов фазового переноса в количествах, ниже стехиометрических.
У Hwu и др. (Synthesis, 1994, 471-474) описано получение нитратных эфиров из эфиров сульфоновой кислоты. Слишком высокие температуры и долгая продолжительность реакции в сочетании с низкой стабильностью полученных конечных продуктов делает этот способ менее пригодным для широкомасштабного производства. Кроме того, молярный избыток нитрата натрия почти вдвое больше по сравнению с настоящим изобретением, что повышает стоимость и может обеспечить проблемы с отходами. Кроме того, сырой продукт, полученный способом согласно Hwu и др., требует очистки либо хроматографией, либо перегонкой для обеспечения фармацевтически приемлемой чистоты. Никакой из этих двух способов очистки не подходит для широкомасштабного получения соединений.
ES 2073995 описывает синтезы алкилнитратных эфиров из алкилсульфонатов или 4-толуолсульфонатов и нитратов металла с помощью растворителей, таких как диметилформамид, диметилацетамид, ацетонитрил или диметилсульфоксид. Используя диметилацетамид или диметилсульфоксид в качестве растворителя в синтезе NO-донорных соединений исходя из сульфонированных промежуточный соединений, получают сырой продукт, который нуждается в очистке либо хроматографией, либо перегонкой для достижения фармацевтически приемлемой чистоты.
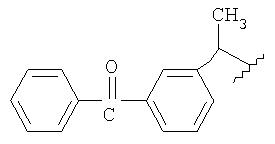
Примерами NSAID является диклофенак (соединение формулы Ia) и кетопрофен (соединение формулы Id):
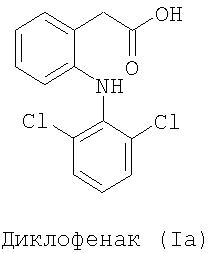
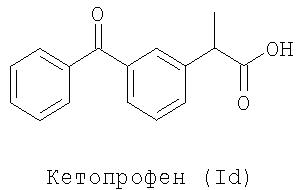
WO 94/04484 и WO 94/12463 описывают способы получения NO-донорных аналогов диклофенака и кетопрофена соответственно. В указанных способах дигалогенидные производные подвергают реакции с солью карбоновой кислоты ДМФА. Продукты реакции превращают в конечные продукты реакцией с AgNO3 в ацетонитриле способом, известным из уровня техники.
Способ настоящего изобретения включает использование сульфонированного промежуточного соединения. Это промежуточное соединение может быть легко получено и является высокореакционным в реакциях с нитратными ионами для получения соответствующего нитрооксиалкильного эфира.
Таким образом, существует потребность в более удобном и экономически выгодном широкомасштабном способе получения фармацевтически чистых NO-донорных соединений, и их сульфонированных промежуточных соединений, где такие факторы, как стоимость, время получения, применение более безопасных для окружающей среды растворителей, являются важными для коммерческого использования. Настоящее изобретение обеспечивает такой способ.
В составе лекарственных композиций важно, чтобы соединение находилось в форме, в которой его можно легко обрабатывать и использовать. Это важно для обеспечения коммерчески приемлемого способа получения и для изготовления фармацевтических составов, включающих активное соединение.
Далее, при изготовлении лекарственных композиций важно, чтобы при введении пациенту обеспечивался надежный, воспроизводимый и постоянный профиль концентрации соединения в плазме.
Химическая стабильность и физическая стабильность соединений являются важными факторами. Соединение и содержащие его составы должны эффективно храниться в течение длительных периодов времени, без существенных изменений физико-химических характеристик активного соединения, таких как его химический состав, плотность, гигроскопичность и растворимость.
Кроме того, важно получить соединение в форме, которая является как можно более химически чистой.
Аморфные материалы могут создавать существенные проблемы в этом отношении. Такие материалы трудно обрабатывать и вводить в составы, они показывают плохую растворимость и часто оказывается, что они являются нестабильными и химически загрязненными.
Таким образом, при получении коммерчески выгодных и фармацевтически приемлемых составов важно, по возможности, получить лекарственное средство по существу в кристаллической и стабильной форме.
Следует отметить, однако, что эта цель не всегда достижима. Действительно, обычно только из молекулярной структуры невозможно предсказать, каким будет поведение соединения при кристаллизации. Обычно это может быть определено только экспериментально.
Неожиданно было обнаружено, что 2-[2-(нитроокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил}-ацетат (соединение IVa) может быть получено в форме, которая является по существу кристаллической и стабильной.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому способу получения NO-донорных соединений. Кроме того, оно относится к новым промежуточным соединениям и к способу получения указанных промежуточных соединений, особенно к широкомасштабному способу получения.
Новый способ получения NO-донорных соединений описан ниже.
Одно воплощение изобретения относится к способу получения NO-донорных соединений, включающему:
стадию 1

с помощью кислотного или дегидрирующего агента и растворителя, необязательно с последующей очисткой экстракцией или кристаллизацией, и
стадию 2

с помощью растворителя, основания и, необязательно, катализатора, с последующей очисткой экстракцией и кристаллизацией, и
стадию 3

с помощью растворителя и, необязательно, катализатора,
необязательно с последующей кристаллизацией для получения соединения формулы IV по существу в кристаллической форме, и
где:
М представляет собой радикал физиологически активного соединения;
L представляет собой О, S, (CO)O, (CO)NH, (CO)NR1, NH, NR1, где R1 представляет собой линейную или разветвленную алкильную группу, или
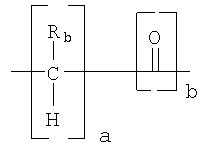

где Rb представляет собой Н, С1-12алкил или С2-12алкенил;
R2 представляет собой (CO)NH, (CO)NR1, (CO)O или CR1 и а и b независимо имеют значение 0 или 1;
А представляет собой замещенную или незамещенную линейную или разветвленную алкильную цепь;
Х представляет собой углеродный линкер;
R выбран из группы, состоящей из C1-C8алкила, фенила, фенилметила, C1-C4алкилфенила, галогенфенила, нитрофенила, ацетиламинофенила, галогена, CF3 и н-C4F9;
Y-NO3 представляет собой нитрат лития, нитрат натрия, нитрат калия, нитрат магния, нитрат кальция, нитрат железа, нитрат цинка или нитрат тетраалкиламмония (где алкил представляет собой C1-C18-алкил, который может быть линейным или разветвленным);
m имеет значение 1 или 2; и
Т1 и Т2 каждый независимо имеет значение 0, 1, 2 или 3;
при условии, что
когда MLT1АT2-СООН представляет собой напроксен, X не является (СН2)4.
Другое воплощение изобретения относится к способу получения промежуточных соединений формулы III, которые могут использоваться для получения NO-донорных соединений, включающему:
стадию 1,

с помощью кислотного или дегидрирующего агента и растворителя, необязательно с последующей очисткой экстракцией или кристаллизацией, и стадию 2,
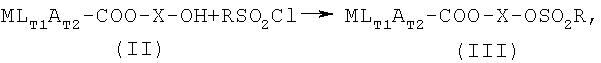
с помощью растворителя, основания и, необязательно, катализатора, с последующей очисткой экстракцией и кристаллизацией, и где М, L, A, T1, T2, X и R имеют указанные выше значения.
Термин "C1-C8алкил" обозначает алкил с 1-8 атомами углерода и включает алкильные группы как с линейной, так и с разветвленной цепью, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и т.д.
Термин "C1-C4алкилфенил" обозначает метилфенил, этилфенил, н-пропилфенил, изопропилфенил, н-бутилфенил, изобутилфенил и трет-бутилфенил.
Термин "фенилметил" обозначает бензил.
Термин "гало" и "галоген" обозначает фтор, хлор или бром.
Термины "галогенфенил", "нитрофенил" и "ацетиламинофенил" относятся к фенильным группам, замещенным одним или несколькими группами галогена, нитро или ацетиламино.
Термин "широкомасштабный" обозначает получение количества в области "от килограмма до мультитонны".
М может представлять собой любой радикал любого физиологически активного соединения.
MLT1АT2-СООН может быть любая физиологически активная карбоновая кислота.
В одном воплощении изобретения группа М является частью молекулы NSAID, ингибитора СОХ1 или СОХ2.
В другом воплощении изобретения группа М выбрана из группы, состоящей из
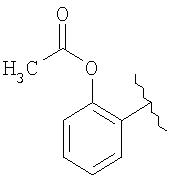

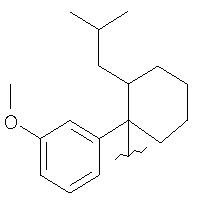


как описано в WO 00/51988, и


как описано в US 3641127, и
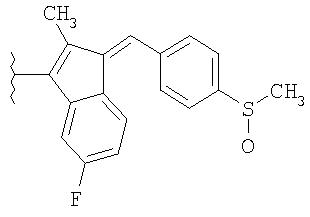
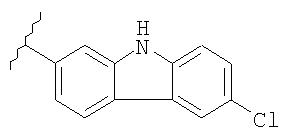





как описано в WO 96/32946, и
циклоалкилов, описанных в WO 98/25918, таких как 2,2-диметил-циклопропан-1-метанол, и

как описано в CN 1144092, и




как описано в WO 95/09831, и









как описано в WO 95/30641, и





как описано в WO 02/30866, и
















как описано в US 6297260.
В одном воплощении изобретения L выбран из группы, состоящей из О, S, NH, NR1, где R1 представляет собой линейную или разветвленную алкильную группу, как описано в WO 95/09831, и (СО) или (CO)O, как описано в WO 95/30641, и
где Rb представляет собой Н, С1-12 алкил или C2-12 алкенил и а и b независимо имеют значения 0 или 1, как описано в WO 02/053188,

и
где Rb, а и b имеют указанные выше значения; и
R2 представляет собой (CO)NH, (CO)NR1, (CO)O или CR1.
В другом воплощения изобретения А выбран из группы, состоящей из -(CH2)n-, где n имеет значение 0, 1, 2, 3 или 4,



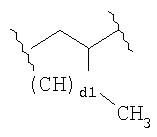





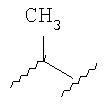


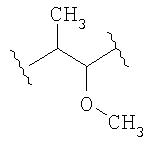
где d1 имеет значение 1, 2 или 3.
В другом воплощении изобретения А выбран из группы, состоящей из




где d1 имеет значение 1, 2 или 3.
Углеродный линкер X может быть выбран из группы, состоящей из

где А′ и В выбраны из водорода, линейной или разветвленной или циклической замещенной или незамещенной алкильной группы, и v1 имеет значение от 1 до 10 как описано в WO 95/09831, и
-(СН2-СН2-O)2-, или циклоалкил, имеющий 5-7 атомов углерода, необязательно замещенных, и

где m1 имеет значение от 0 до 3, и
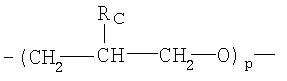
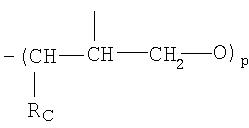
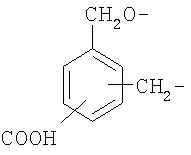


где Rc представляет собой Н или метил, и р имеет значение от 0 до 6, как описано в WO 95/30641 и WO 02/92072, и
-(CH2)q-OCO-(CH2)r, где q и r каждый независимо имеет значение от 0 до 6, и

где Z представляет собой О, SO, S или насыщенное, ненасыщенное или ароматическое 5 или 6-членное кольцо или 5 или 6-членное гетероциклическое кольцо, содержащее один или несколько гетероатомов, независимо выбранных из N, О и S, где указанное кольцо необязательно может быть замещено, и v2 и v3 независимо имеют значения от 0 до 4.
В одном воплощении изобретения X выбран из группы, состоящей из линейной, разветвленной или циклической -(CH2)w1-, где w1 имеет значение от 2 до 10; -(CH2)w2 -O-(CH2)w3-, где w2 и w3 имеют значения от 2 до 10; и -СН2-С6Н4-СН2-.
В другом воплощении изобретения X выбран из группы, состоящей из линейной -(CH2)w1-, где w1 имеет значение от 2 до 6; -(СН2)2-O-(СН2)2- и -СН2-C6H4-CH2-.
В другом воплощении изобретения R выбран из группы, состоящей из C1-C8алкила, фенила, фенилметила, C1-C4алкилфенила, галогенфенила, нитрофенила, ацетиламинофенила и галогена.
В одном воплощении изобретения группа MLT1АT2 выбрана из группы, состоящей из













В другом воплощении изобретения группа MLT1АT2 выбрана из группы, состоящей из
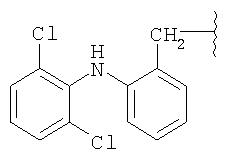
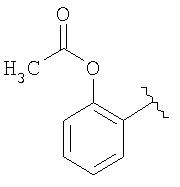

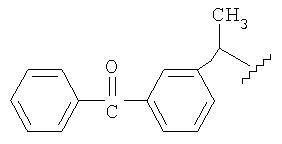
В конкретном воплощении группа MLT1АT2 представляет собой

Описание способа
Стадия 1

где М, L, A, T1, T2 и X имеют значения, указанные выше.
MLT1АT2-СООН может быть этерифицирован на стадии 1 реакции с помощью кислотной катализируемой этерификации в присутствии диэтиленгликоля, как описано в DE 88-3811118, при использовании п-толуолсульфоновой кислоты.
Стадия 1 этерификации может осуществляться способом, известным специалисту в данной области техники, например, обработкой соединения формулы I, например диклофенака и диэтиленгликоля, кислотным или дегидрирующим агентом.
Одно воплощение относится к способу по изобретению, в котором кислотный или дегидрирующий агент на стадии 1 выбирают из группы, состоящей из серной кислоты или ее солей, перхлорной кислоты (например, 70%) или других подходящих кислот, таких как полистиролсульфоновые кислоты, цеолиты, кислые глины, песок в комбинации с сильными гидрофильными кислотами, такими как перхлорная кислота или газообразный хлорид водорода, и монтмориллониты.
Соединения формулы II могут быть также получены тем же способом с помощью 1,4-бутандиола, 1,3-пропандиола и триэтиленгликоля, соответственно. В ES 85-548226 для катализирования этерификации используют тионилхлорид.
Кислоты могут использоваться в виде газа, жидкости или в твердой форме.
Гетерогенные кислоты могут быть относительно легко отфильтрованы из реакционного раствора и повторно использоваться в широкомасштабных способах получения.
Примерами других конденсирующих реагентов, используемых на стадии 1 этерификации, являются карбодиимиды, такие как N,N'-дициклогексилкарбодиимид (DCC), хлориды кислот, такие как оксалилхлорид, хлорформаты, такие как изобутилхлорформат, или другие реагенты, такие как цианурилхлорид, N, N'-карбонилдиимидазол, диэтилхлорфосфит, 2-хлор-1-метил-пиридинйодид и 2,2′-дипиридилдисульфид.
Одно воплощение относится к способу по изобретению, в котором растворитель на стадии 1 является неполярным и/или некислотным растворителем.
Стадия 1 реакции может проводиться в растворителе, выбранном из группы, включающей ароматические углеводороды, такие как бензол или толуол, алифатические углеводороды, такие как н-гептан, кетоны, такие как метилизобутилкетон, эфиры, такие как тетрагидрофуран или диметиловый эфир диэтиленгликоля и хлорированные углеводороды, такие как дихлорметан или хлорбензол, или их смеси.
Альтернативно, избыток соответствующего диола может использоваться в качестве растворителя, необязательно в смеси с любыми другими органическими растворителями, упомянутыми выше.
Соединения формулы II, полученные на стадии 1, могут быть очищены экстракцией, партиями или непрерывно, получая раствор, содержащий соединение формулы II, имеющий хроматографическую чистоту по крайней мере 92% и, предпочтительно, более 97% (после стадии i экстракции) и содержание алкилендиола или алкиленгликоля ниже 0.5% (вес.) (после на стадии ii экстракции).
Стадия i) экстракции
На этой стадии экстракции повышается хроматографическая чистота. Раствор, используемый на этой стадии экстракции, может включать смесь i) алкилендиола или алкиленгликоля, ii) воды и/или алифатического спирта с низким молекулярным весом и iii) углеводородный растворитель или их смеси или смеси органических растворителей с углеводородными растворителями.
Алифатические спирты с низким молекулярным весом могут быть выбраны из группы, состоящей из метанола, этанола и пропанола или их смеси.
Углеводородные растворители, используемые на стадии i) экстракции, могут быть выбраны из группы, включающей толуол, кумол, ксилол, лигроин, петролейный эфир, галогенбензолы, гептаны, гексаны, октаны, циклогексаны, циклогептаны и им подобные, или их смеси.
Подходящие органические растворители, используемые на стадии i) экстракции, могут быть выбраны из группы, включающей кетоны, такие как метилизобутилкетон, эфиры, такие как ди-н-бутиловый эфир или трет-бутилметиловый эфир, и алифатические эфиры, такие как этилацетат или н-бутилацетат, и галогеналканы, такие как дихлорметан, или их смеси.
Очищенное соединение формулы II получают в виде раствора в смеси с алкилендиолом или алкиленгликолем с водой и/или алифатическим спиртом с низким молекулярным весом.
Стадия ii) экстракции
Эта экстракция осуществляется для снижения содержания алкилендиола или алкиленгликоля и проводится после стадии i) экстракции, на которой повышается хроматографическая чистота, как описано выше. Раствор может включать i) смесь воды и/или алифатического спирта с низким молекулярным весом, ii) органический растворитель или смеси органических растворителей. Алифатические спирты с низким молекулярным весом могут быть выбраны из группы, состоящей из метанола, этанола и пропанола или их смесей.
Подходящие органические растворители, используемые на стадии ii) экстракции, могут быть выбраны из группы, включающей ароматические углеводороды, такие как толуол, кумол или ксилолы, кетоны, такие как метилизобутилкетон, эфиры, такие как ди-н-бутиловый эфир или трет-бутилметиловый эфир и алифатические эфиры, такие как этилацетат или н-бутилацетат и галогеналканы, такие как дихлорметан, или их смеси.
Общее количество растворителей, используемых на стадии 1 процесса этерификации, может изменяться от 0 до 100 частей по объему от веса исходного сырья.
Температура стадии 1 этерификации может изменяться от -100°С до +130°С, предпочтительно от 0°С до +120°С.
Стадия 2

где М, L, A, T1, T2, X и R имеют указанные выше значения.
Условия реакции стадии 2 включают избыток RSO2Cl в органическом растворителе или смеси органических растворителей.
Подходящие растворители на стадии 2 могут быть выбраны из группы, включающей ароматические углеводороды, такие как толуол, кумол или ксилолы, кетоны, такие как метилизобутилкетон, эфиры, такие как ди-н-бутиловый эфир или трет-бутилметиловый эфир или тетрагидрофуран, алифатические нитрилы, такие как ацетонитрил, и алифатические эфиры, такие как этилацетат или н-бутилацетат, и галогеналканы, такие как дихлорметан, или их смеси.
Одно воплощение относится к способу по изобретению, в котором растворители на стадии 2 выбраны из группы, состоящей из толуола, кумола, ксилолов, этилацетата, ацетонитрила, бутилацетата и изопропилацетата.
На стадии 2 может быть добавлено основание. В одном воплощении изобретения основание на стадии 2 может быть выбрано из группы, состоящей из триэтиламина, пиридина, N-метилморфолина, диизопропилэтиламина, трибутиламина и N-метилпиперидина.
Другое воплощение относится к способу изобретения, в котором основание на стадии 2 представляет собой триэтиламин или N-метилморфолин.
Другое воплощение относится к способу по изобретению, в котором на стадии 2 необязательно может использоваться катализатор, такой как 4-(диметиламино)пиридин.
Соединения формулы III, полученные на стадии 2, могут быть очищены кристаллизацией из органического растворителя для получения кристаллического твердого вещества, имеющего химическую чистоту около 95% и особенно около 98%.
Другое воплощение относится к способу по изобретению, в котором на стадии 2 для перекристаллизации соединения формулы III используется осаждающий растворитель.
В другом воплощении изобретения растворитель, используемый для кристаллизации, может быть выбран из группы, включающей ароматические углеводороды, такие как толуол, кумол или ксилолы, кетоны, такие как метилизобутилкетон, эфиры, такие как ди-н-бутиловый эфир, трет-бутилметиловый эфир или тетрагидрофуран, алифатические нитрилы, такие как ацетонитрил, и алифатические эфиры, такие как этилацетат или бутилацетат, или их смеси.
Другое воплощение относится к способу по изобретению, в котором растворитель, используемый для кристаллизации на стадии 2, выбран из группы, состоящей из толуола, кумола, ксилолов, этилацетата, ацетонитрила, бутилацетата и изопропилацетата, или их смесей.
Другое воплощение относится к способу по изобретению, в котором осаждающий растворитель, используемый для кристаллизации на стадии 2, выбран из группы, включающей лигроин, петролейный эфир, галогенбензолы, гептаны, гексаны, октаны, такие как изооктан, циклогексаны, циклогептаны и спирты, или их смеси.
Стадия 3

где M, L, A, T1, T2, X, R, m и Y являются такими, как определено выше.
На стадии 3 способа получения соединение формулы IV получают по реакции соединения формулы III с источником нитрата (Y-NO3), необязательно в присутствии растворителя.
Эта реакция может осуществляться с источником нитрата Y-NO3, выбранным из группы, состоящей из нитрата лития, нитрата натрия, нитрата калия, нитрата магния, нитрата кальция, нитрата железа, нитрата цинка и нитрата тетраалкиламмония (где алкил представляет собой C1-C18-алкил, который может быть линейным или разветвленным).
Одно воплощение относится к способу по изобретению, в котором источники нитрата Y-NO3 на стадии 3 выбраны из группы, состоящей из нитрата лития, нитрата натрия, нитрата калия, нитрата магния и нитрата кальция, или их смесей.
Другое воплощение относится к способу по изобретению, в котором органический растворитель на стадии 3 является полярным апротонным растворителем.
В другом воплощении по изобретению полярные апротонные растворители, используемые на стадии 3, могут быть выбраны из группы, включающей N-метилпирролидинон, N,N-диметилацетамид, сульфолан, тетраметилмочевину, 1,3-диметил-2-имидазолидинон и нитрилы, такие как ацетонитрил, или их смеси.
Другими растворителями могут быть ароматические углеводороды, такие как толуол, алифатические углеводороды, такие как н-гептан, кетоны, такие как метилэтилкетон, метилизобутилкетон, эфиры, такие как тетрагидрофуран или диметиловый эфир диэтиленгликоля, хлорированные углеводороды, такие как хлорбензол, алифатические эфиры, такие как этилацетат, бутилацетат или изопропилацетат, нитроуглеводороды, такие как нитрометан, этиленгликоли, такие как полиэтиленгликоль и их смеси, необязательно с дополнительными алифатическими спиртами, такими как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол или трет-бутанол.
Одно воплощение изобретения относится к способу по изобретению, в котором органический растворитель на стадии 3 выбран из группы, состоящей из N-метилпирролидинона, сульфолана, тетраметилмочевины, 1,3-диметил-2-имидазолидинона, ацетонитрила, метилизобутилкетона, этилацетата, бутилацетата и изопропилацетата, или их смесей.
Стадия 3 нитрования может также осуществляться в воде, необязательно в комбинации с любыми указанными выше органическими растворителями.
Стадия 3 нитрования может необязательно проводиться в присутствии катализатора фазового переноса.
Одно воплощение относится к способу по изобретению, в котором катализатор фазового переноса на стадии 3 выбран из группы, состоящей из соли тетраалкиламмония, соли арилалкиламмония, соли тетраалкилфосфония, соли арилалкилфосфония, краунэфира, пентаэтиленгликоля, гексаэтиленгликоля и полиэтиленгликолей, или их смесей.
Кристаллизация соединений формулы IV
Соединения формулы IV, полученные на стадии 3, могут быть очищены кристаллизацией из органического растворителя, необязательно с помощью углеводородов, спиртов или воды в качестве осаждающего растворителя, для получения кристаллического твердого продукта химической чистоты 90% и особенно около 95%.
Одно воплощение относится к способу по изобретению, в котором соединение формулы IV на стадии 3 экстрагируют порциями или непрерывно и кристаллизуют из органического растворителя, необязательно с помощью осаждающего растворителя, для получения кристаллического твердого вещества, имеющего химическую чистоту по крайней мере 95%.
Предпочтительно кристаллизацию осуществляют в подходящей системе растворителя. Кристаллизация может также осуществляться без системы растворителя. Другие примеры перекристаллизации включают перекристаллизацию из расплава, в надкритических условиях или при сублимации.
Кристаллизация соединений формулы IV из подходящей системы растворителя может проводиться при достижении сверхнасыщения в системе растворителя, которая включает соединение формулы IV. Это может быть достигнуто охлаждением системы растворителя, упариванием растворителя, добавлением подходящего осаждающего растворителя или любой комбинацией этих методов. Кристаллизация может также достигаться при снижении растворимости соединения путем добавления соли, такой как, например, NaCl.
Способ кристаллизации может применяться к реакционному раствору, включающему соединение формулы IV, полученному после приготовления указанного соединения.
Способ кристаллизации также может применяться к сухому соединению формулы IV. Альтернативно способ кристаллизации может применяться после экстракции соединения формулы IV из реакционного раствора.
Одно воплощение настоящего изобретения относится к описанному выше способу, в котором способ кристаллизации соединения формулы IV включает следующие стадии:
a) i) растворение соединения в растворителе;
или
ii) экстракция соединения из реакционного раствора в растворитель;
или
iii) из реакционного раствора, включающего указанное соединение;
b) упаривание растворителя;
c) добавление осаждающего растворителя и/или охлаждение;
d) выделение полученных кристаллов, и необязательно:
e) перекристаллизация кристаллов, полученных на стадии с); или выделенных на стадии d).
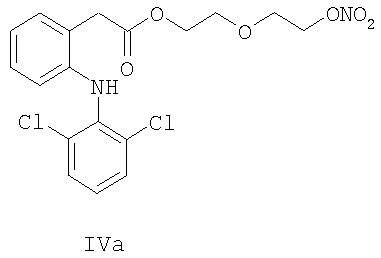
Другое воплощение изобретения относится к описанному выше способу, в котором способ кристаллизации соединения 2-[2-(нитроокси)-этокси]этил{2-[(2,6-дихлорфенил)амино]фенил}ацетата (IVa) включает следующие стадии:
a) экстракция соединения из реакционного раствора в растворитель;
b) упаривание растворителя;
c) добавление осаждающего растворителя и/или охлаждение;
d) выделение полученных кристаллов и, необязательно:
e) перекристаллизация кристаллов, полученных на стадии с); или выделенных на стадии d).
По существу кристаллическая форма 2-[2-(нитроокси)-этокси]этил{2-[(2,6-дихлорфенил)амино] фенил} ацетата обозначена далее как "Форма А соединения IVa".
Другое воплощение изобретения включает способ получения Формы А соединения IVa, который включает кристаллизацию 2-[2-(нитроокси)-этокси]этил{2-[(2,6-дихлорфенил)амино]фенил} ацетата.
Подходящие растворители, используемые для способа перекристаллизации, могут быть выбраны из группы, включающей низшие алкилацетаты, например линейные или разветвленные C1-6алкилацетаты, такие как этилацетат, изопропилацетат или бутилацетат, низшие линейные или разветвленные С2-6алкиловые спирты, предпочтительно C2-4алкильные спирты, такие как этанол или изопропанол, алифатические и ароматические углеводороды, например C5-12алифатические углеводороды или С6-10ароматические углеводороды, такие как изооктан, кумол, ксилолы, н-гептан, 1-метил-2-пирролидинон или толуол; диалкилкетоны, например ди-С1-6 алкилкетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон или 4-метил-2-пентанон, диалкиловые эфиры, например ди-С1-6алкиловые эфиры, такие как диизопропиловый эфир, ди-н-бутиловый эфир, трет-бутилметиловый эфир или тетрагидрофуран, алифатические нитрилы, такие как ацетонитрил, и воду, или их смеси.
Одно воплощение изобретения относится к описанному выше способу кристаллизации, в котором растворитель на стадии а) выбран из группы, включающей низшие алкилацетаты, низшие алкиловые спирты, алифатические углеводороды, ароматические углеводороды, гетероароматические углеводороды, диалкилкетоны, диалкилэфиры, нитрилы и воду, или их смеси.
Другое воплощение изобретения относится к описанному выше способу кристаллизации, в котором растворитель на стадии а) выбран из группы, состоящей из этилацетата, изопропилацетата, бутилацетата, этанола, изопропанола, изооктана, н-гептана, толуола, 1-метил-2-пирролидинона, метилэтилкетона, метилизобутилкетона, диизопропилового эфира, трет-бутилметилового эфира, ацетонитрила и воды, или их смеси.
Другое воплощение относится к описанному выше способу кристаллизации, в котором растворитель выбран из группы, состоящей из бутилацетата, изопропанола, изооктана, ацетона, ацетонитрила и воды, или их смеси.
Растворители могут также использоваться как "осаждающие растворители" (т.е. растворитель, в котором соединение плохо растворимо), и могут с этой целью использоваться в процессе кристаллизации.
В одном воплощении изобретения осаждающий растворитель на стадии b) способа кристаллизации выбран из группы, состоящей из этанола или 2-пропанола, толуола, кумола, ксилолов, лигроина, петролейного эфира, галогенбензолов, гептанов, гексанов, октанов, циклогексанов и циклогептанов, или их смесей.
Другая очистка соединения может осуществляться перекристаллизацией и/или высаживанием. Перекристаллизация может осуществляться из подходящей системы растворителя, например линейных или разветвленных алкилацетатов, таких как этилацетат, изопропилацетат и бутилацетат, кетонов, таких как ацетон и 4-метил-2-пентанон, ароматических углеводородов, таких как толуол и 1-метил-2-пирролидинон, которые могут включать осаждающий растворитель, например воду или низшие алкиловые спирты, такие как этанол и изопропанол, или алифатические углеводороды, такие как изооктан и н-гептан, или комбинацию этих растворителей.
Другое воплощение изобретения относится к описанному выше способу кристаллизации, в котором растворитель на стадии d) выбран из группы, состоящей из ароматических углеводородов, таких как толуол, кумол или ксилолы, кетонов, таких как метилизобутилкетон, эфиров, таких как ди-н-бутиловый эфир, трет-бутилметиловый эфир или тетрагидрофуран, алифатических нитрилов, таких как ацетонитрил, и алифатических эфиров, таких как этилацетат или изобутилацетат, и галогеналканов, таких как дихлорметан, или их смесей, необязательно вместе с осаждающим растворителем, выбранным из группы, состоящей из воды, этанола, изопропанола, изооктана и н-гептана, или их смесей.
Другое воплощение изобретения относится к описанному выше способу кристаллизации, в котором растворитель на стадии d) выбран из группы, состоящей из толуола, кумола, ксилолов, метилизобутилкетона, ди-н-бутилового эфира, трет-бутилметилового эфира, тетрагидрофурана, ацетонитрила, н-бутилацетата и дихлорметана, или их смесей, необязательно вместе с осаждающим растворителем, выбранным из группы, состоящей из воды, этанола, изопропанола, изооктана и н-гептана, или их смесей.
Соединения формулы IV для перекристаллизации могут быть, например, сначала растворены в органическом растворителе, таком как ацетон, и затем промыты осаждающим растворителем, таким как вода, с последующим охлаждением и отфильтровыванием полученных кристаллов. После фильтрации кристаллы могут быть далее промыты жидкостью, затем жидкость может быть упарена и кристаллы высушены.
Кристаллические формы соединений формулы IV могут быть выделены обычными методами, такими как декантирование, фильтрация или центрифугирование.
Изобретение относится к соединению IV, полученному описанными выше способами.
Одно воплощение изобретения относится к Форме А соединения IVa, кристаллизуемой в соответствии с описанными выше способами, в которой химическая чистота Формы А соединения IVa составляет около 95%, предпочтительно около 98%, более предпочтительно около 99%.
При кристаллизации и/или перекристаллизации Формы А соединения IVa, как описано выше, предполагается, что полученный кристалл имеет повышенную химическую, физическую и твердофазную стабильность.
В соответствии с одним воплощением изобретения предлагается 2-[2-(нитроокси)этокси]-этил{2-[(2,6-дихлорфенил)амино]фенил} ацетат (IVa) по существу в кристаллической форме.
Другое воплощение изобретения относится к безводной форме соединения IVa. Получение и характеристики безводной формы описаны далее.
Хотя было обнаружено, что возможно получить 2-[2-(нитроокси)этокси]-этил{2-[(2,6-дихлорфенил)амино]фенил} ацетат в форме, которая является кристаллической более чем на 90%, под "по существу кристаллической" формой следует понимать форму, которая является кристаллической более чем на 50%, предпочтительно более чем на 60% и более предпочтительно более чем на 70%.
"Степень (%) кристаллизации" может быть определена с помощью рентгеновской дифракции на порошке (XRPD). В качестве вспомогательных методов могут также использоваться другие методы, такие как ЯМР твердой фазы, FT-IR, Raman спектроскопия, дифференциальная сканирующая калориметрия (ДСК) и микрокалориметрия.
Одно воплощение изобретения относится к Форме А соединения IVa, характеризуемой основными пиками на рентгеновской дифрактограмме на порошке, как показано в Таблице 1 Примера 5а.
Форма А соединения IVa может характеризоваться ее элементарной ячейкой кристаллов.
Другое воплощение изобретения относится к Форме А соединения IVa, характеризуемой наличием моноклинной элементарной ячейки кристалла с параметрами а=13.79 Å, b=11.90 Å, с=13.01 Å, α =90°, β=94.0°, γ=90°.
Предполагается, что форма А соединения IVa является химически и физически стабильной в течение длительного периода времени при условиях хранения, описанных ниже.
Приведенный здесь термин "стабильность" и "стабильный" обозначает химическую стабильность и физическую стабильность.
Термин "химическая стабильность" обозначает, что Форма А соединения IVa может храниться в выделенной твердой форме или в форме твердого состава, необязательно в смеси с фармацевтически приемлемыми носителями, разбавителями или адъювантами, в условиях хранения, с незначительной степенью химической деградации или разрушения.
Термин "физическая стабильность" обозначает, что Форма А соединения IVa может храниться в выделенной твердой форме или в форме твердого состава, необязательно в смеси с фармацевтически приемлемыми носителями, разбавителями или адъювантами, в условиях хранения, с незначительной степенью физической деградации (например, кристаллизации, перекристаллизации, твердофазного перехода, гидратации, дегидратации, сольватизации или десольватизации).
Предполагается, что Форма А соединения IVa имеет улучшенные химические и физические характеристики, такие как улучшенная растворимость, термическая стабильность, стабильность на свету, гигроскопическая стабильность и т.д.
Изобретение также относится к способу получения соединений формул IVa, IVb, IVc и IVd. Соединения диклофенака a, b и с отличаются друг от друга различными линкерами X.
В соединениях формул IIa, IIIa и IVa линкер X представляет собой С2Н4OC2Н4.
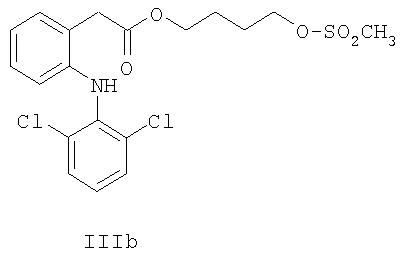
В соединениях формул IIb, IIIb и IVb линкер X представляет собой C4H8.
В соединениях формул IIc, IIIc и IVc линкер X представляет собой С2Н4OC2 Н4OC2Н4.
Соединения IId, IIId и IVd являются соединениями кетопрофена, в которых линкер X представляет собой С3H6.
Одно воплощение изобретения относится к способу получения NO-донорного диклофенака формул IVa, IVb или IVc, включающему:
стадию 1, реакцию соединения формулы Ia с НО-Х-ОН, где×представляет собой C2H4OC2H4, C4H8 или C2H4OC2H4OC2H4, с получением соединений формул IIa, IIb или IIc,

с последующей
стадией 2, реакцией соединений формул IIa, IIb или IIc с RSO2Cl, где R является таким, как определено выше, с получением соединений формул IIIa, IIIb или IIIc,

с последующей
стадией 3, реакцией соединений формул IIIa, IIIb или IIIc с нитратным источником Y-NO3, где Y является таким, как описано выше, с получением соединений формул IVa, IVb или IVc.

с последующей
кристаллизацией соединений формул IVa, IVb или IVc с помощью следующих стадий:
a) экстракция соединения из реакционного раствора в растворитель;
b) упаривание растворителя;
c) добавление осаждающего растворителя и/или охлаждение;
d) выделение полученных кристаллов и, необязательно:
e) перекристаллизация полученных на стадии с) кристаллов; или выделение на стадии d).
Другое воплощение изобретения относится к способу получения NO-донорного диклофенака формулы IVa, включающему:
стадию 1, реакцию соединения формулы Ia с диэтиленгликолем с получением соединения формулы IIa

с последующей стадией 2, реакцией соединения формулы IIa с RSO2Cl, где R является таким, как определено выше, с получением соединения формулы IIIa,
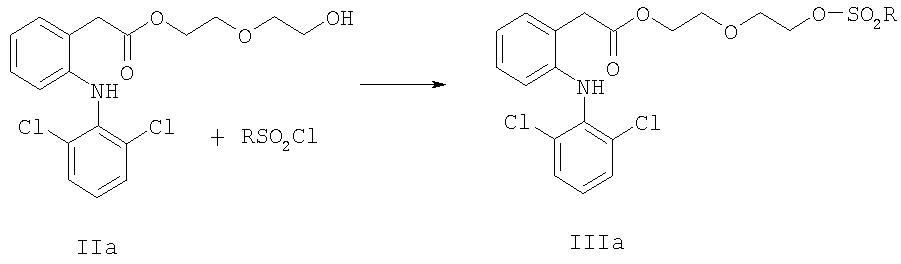
стадию 3, реакцию соединения формулы IIIa с источником нитрата Y-NO3, где Y является таким, как определено выше, с получением соединения формулы IVa,

с последующей кристаллизацией соединения формулы IVa с помощью следующих стадий:
a) экстракция соединения из реакционного раствора в растворитель;
b) упаривание растворителя;
c) добавление осаждающего растворителя и/или охлаждение;
d) выделение полученных кристаллов и, необязательно:
e) перекристаллизация полученных на стадии с) кристаллов; или выделение на стадии d).
Другое воплощение изобретения относится к способу получения NO-донорного кетопрофена формулы IVd, включающему:
стадию 1, реакцию соединения формулы Id с 1,3-пропандиолом с получением соединения формулы IId,
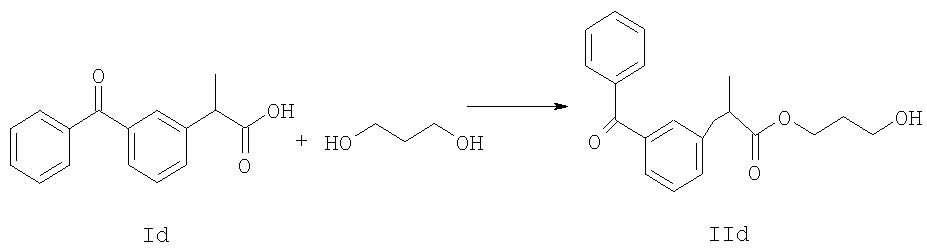
с последующей
стадией 2, реакцией соединения формулы IId с RSO2Cl, где R является таким, как определено выше, с получением соединения формулы IIId,

стадию 3, реакцию соединения формулы IIId с источником нитрата Y-NO3, где Y является таким, как определено выше, с получением соединения формулы IVd,

Одно воплощение изобретения относится к описанному выше способу для получения S-энантиомера NO-донорного кетопрофена формулы IVd.
Температура, используемая на стадиях 1 и 2 способа, может поддерживаться между -100°С и +130°С. Температура предпочтительно поддерживается ниже 130°С, поскольку стабильность конечного продукта может быть нарушена при высоких температурах. Стадия 3 реакции предпочтительно проводится при температуре ниже 90°С. Температура, используемая с способе кристаллизации, может поддерживаться ниже 0°С, например около -40°С.
Одно воплощение относится к способам изобретения, в которых температура поддерживается между -40°С и 120°С.
Комнатная температура обозначает температуру между 18°С и 25°С.
Общее количество растворителей может изменяться от 0 до 100 объемных частей от веса исходного реагента.
Разным стадиям реакции может требоваться различное время для реакции.
В способах изобретения исключено применение взрывчатых промежуточных соединений, таких как нитрооксиалканолы. Кроме того, новые способы являются коммерчески приемлемыми и безопасными для окружающей среды по сравнению с известными способами.
Другое преимущество способов изобретения заключается в том, что энантиомерная чистота исходного реагента по крайней мере сохраняется в конечных продуктах (IV), в которых присутствуют асимметричные атомы углерода.
Промежуточные соединения
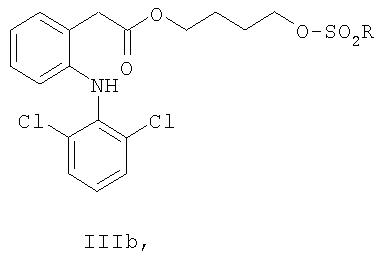
Одно воплощение изобретения относится к промежуточным соединениям формулы III, MLT1AT2-X-O-SO2R, где М, L, A, T1, T2, X и R являются такими, как определено выше.
Другое воплощение изобретения относится к соединениям формул IIIa, IIIb, IIIc и IIId:
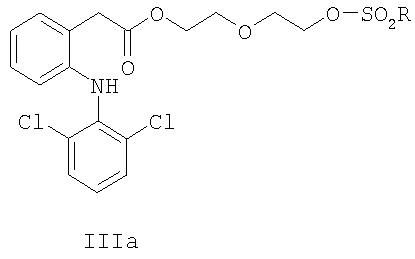
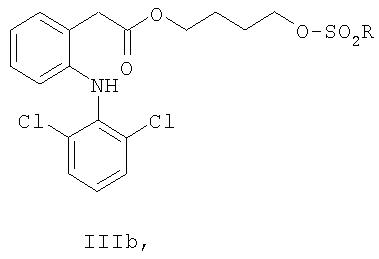


где R выбран из группы, состоящей из C1-C8алкила, фенила, фенилметила, C1-C4алкилфенила, галогенфенила, нитрофенила, ацетиламинофенила, галогена, CF3 и н-С4H9.
Другое воплощение изобретения относится к S-энантиомеру соединения формулы IIId:

где R выбран из группы, состоящей из C1-C8алкила, фенила, фенилметила, C1-C4алкилфенила, галогенфенила, нитрофенила, ацетиламинофенила, галогена, CF3 и н-С4F9.
Другое воплощение изобретения относится к соединениям формулы IIIa,
где R выбран из группы, состоящей из C1-C8алкила, фенила, фенилметила, C1-C4алкилфенила, галогенфенила, нитрофенила, ацетиламинофенила, галогена, CF3 и n-C4 F9.
Применение
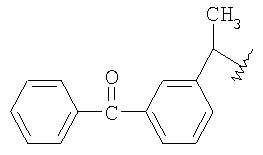
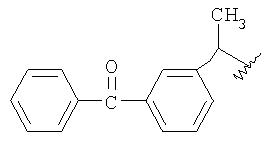
Одно воплощение изобретения относится к применению описанных выше соединений формул IIIa, IIIb, IIIc и IIId в качестве промежуточного соединения для получения 2-[2-(нитроокси)этокси]этил {2-[(2,6-дихлорфенил)амино] фенил} ацетата, 4-(нитроокси)бутил {2-[(2,6-дихлорфенил)амино]фенил} ацетата, 2-{2-[2-(нитроокси)этокси]этокси}этил{2-[(2,6-дихлорфенил)амино]фенил}ацетата, 3-(нитроокси)пропил-2-(2-бензоилфенил)-пропаноата и 3-(нитроокси)пропил(2S)-2-(2-бензоилфенил)пропаноата.
Другое воплощение изобретения относится к применению описанного выше способа для широкомасштабного получения NO-донорных соединений формулы IV.
Другое воплощение изобретения относится к применению описанного выше способа для широкомасштабного получения соединений формул IVa, IVb, IVc и IVd.
Применение в медицине
Одно воплощение изобретения относится к применению соединений формулы III, MLT1АT2-Х-O-SO2R, где М, L, A, T1, T2, X и R являются такими, как определено выше, в качестве промежуточного соединения для получения фармацевтически активного соединения.
Другое воплощение изобретения относится к применению описанных выше промежуточных соединений формул IIIa, IIIb, IIIc и IIId, полученных описанным выше способом на стадиях 1 и 2, для получения лекарственного средства для лечения боли и/или воспаления.
Другое воплощение изобретения относится к применению Формы А соединения IVa для получения лекарственного средства.
Форма А соединения IVa может использоваться для лечения боли и/или воспаления.
Другое воплощение изобретения относится к применению Формы А соединения IVa для получения лекарственного средства для лечения боли и/или воспаления.
Другое воплощение изобретения относится к способу лечения боли и/или воспаления, включающему введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества Формы А соединения IVa.
Фармацевтические составы
Соединения формулы IV могут обычно вводиться орально, ректально или парентерально в фармацевтически приемлемой дозированной форме. Дозированная форма может быть твердым, полутвердым или жидким составом. Обычно активное соединение содержится в количестве от 0.1 до 99% от веса дозированной формы, предпочтительно от 0.5 до 20% от веса дозированной формы для инъекций и от 0.2 до 80% от веса дозированной формы для орального введения.
Фармацевтический состав, включающий соединения формулы IV, может быть получен обычными методами.
Подходящие ежедневные дозы соединений формулы IV для терапевтического лечения человека составляют около 0.001-100 мг/кг веса тела для парентерального введения и около 0.01-100 мг/кг веса тела для других способов введения.
Одно воплощение изобретения обеспечивает фармацевтический состав, включающий в качестве активного соединения терапевтически эффективные количества Формы А соединения IVa, необязательно в смеси в разбавителями, эксципиентами или носителями.
Другое воплощение изобретения относится к составу, включающему водный раствор, содержащий Форму А соединения IVa.
Другое воплощение изобретения относится к фармацевтическому составу, включающему Форму А соединения IVa, необязательно в смеси с разбавителями, эксципиентами или носителями.
Другое воплощение изобретение относится к фармацевтическому составу для применения для лечения боли и/или воспаления.
Термин "боль" включает, но не ограничивается ими, ноцицептивную и нейропатическую боль или их комбинации; острую, прерывистую и хроническую боль; боль при раке; мигрень и головную боль подобного происхождения.
Термин "воспаление" включает, но не ограничивается ими, ревматоидный артрит; остеоартрит и juvenile артрит.
В контексте настоящего описания термин "терапевтический" и "лечение" включают предотвращение и профилактику, если конкретно не указано иное.
Краткое описание чертежей
На чертеже представлена рентгеновская дифрактограмма на порошке кристаллической формы 2-[2-(нитроокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил}ацетата, полученного способом, описанным в Примере 5 (Форма А соединения IVa).
Следующие примеры далее иллюстрируют получение соединений формулы IV, особенно Формы А соединения IVa, описанными выше способами. Эти примеры не предназначены для ограничения объема описанного здесь изобретения или описанного далее в формуле изобретения.
Примеры
Пример 1
Синтез 2-[2-(нитроокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил}ацетата (соединение формулы IVa).
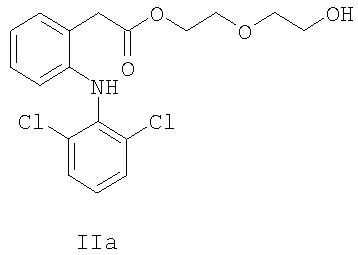
2-(2-Гидроксиэтокси)этил{2-[(2,6-дихлорфенил)амино]фенил} ацетат (соединение формулы IIa).
Диклофенак натрия (20 г, 63 ммоль) растворяли в диэтиленгликоле (67 г, 0.63 моль) при 60°С. После растворения твердых веществ добавляли толуол (170 мл) и концентрированную серную кислоту (4.5 мл, 81.7 ммоль). Реакционную смесь нагревали при 60°С в течение 14 ч перед добавлением К2СО3 (1 М, 120 мл). После разделения фаз водную фазу отделяли и органическую фазу промывали водой (100 мл). Органическую фазу концентрировали в вакууме с получением 23 г IIa в виде коричневого масла (выход 85%, 90% чистота согласно ВЭЖХ), используемого на следующей стадии. MS [M+]=384;1Н-ЯМР (CDCl3) δ 7.34 (арр d, J=8 Гц, 2Н), 7.24 (арр d, J=8 Гц, 1Н), 7.12 (app t, J=7 Гц, 1Н), 6.92-7.05 (m, 2H), 6.88 (br s, 1H), 6.54 (app d, J=8 Гц, 1H), 4.32 (app t, J=4 Гц, 2Н), 3.85 (s, 2Н), 3.64-3.76 (m, 4Н), 3.50-3.58 (m, 2H), 2.08 (brs, 1H);13C-ЯМР (CDCl3) δ 172.8, 143.1, 138.2, 131.1, 129.9, 129.4, 128.5, 124.6, 124.5, 123.5, 122.4, 118.7, 72.8, 69.3, 64.7, 62.10, 53.9, 38.9.
2-(2-Гидроксиэтокси)этил{2-[(2,5-дихлорфенил)амино]фенил}ацетат (соединение формулы IIa).
Смесь Диклофенака Ia (450 г, 1.52 моль) и диэтиленгликоля (2.42 кг, 22.8 моль) перемешивали при 30°С. Добавляли тионилхлорид (90.1 г, 0.757 моль) в течение 30 мин. После перемешивания в течение 6.5 ч при 30°С добавляли толуол (2.20 л) и водный карбонат калия (168.1 г растворяли в 1800 мл воды, 1.22 моль), продолжая перемешивание. Через 0.5 ч перемешивания при температуре 29-30°С водный слой отделяли. Органическую фазу промывали три раза водой (1.8 л на одну промывку) при температуре 54-56°С для улучшения разделения. Органическую фазу концентрировали в вакууме до объема 1900 мл. Перед применением на следующей стадии сульфирования (см. ниже) добавляли толуол (0.70 л), и содержание воды полученного раствора, измеренное титрованием Карла-Фишера, составляло 0.07 вес.%. Чистота по ВЭЖХ: 92%.
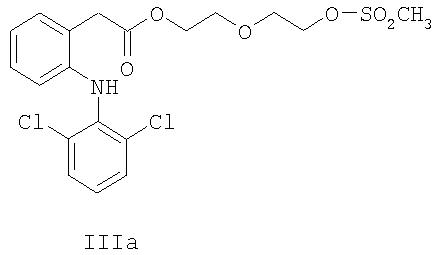
2-{2-[(Метилсульфонил)окси]этокси}этил{2-[(2, 6-дихлорфенил)амино]фенил}ацетат (соединение формулы IIIa).
Гидроксиэфир IIa (23 г, 0.16 моль), выделенный на предыдущей стадии, растворяли в толуоле (300 мл) и N-метилморфолине (16.9 г, 157 ммоль) при 30°С. К реакции по каплям добавляли метансульфонилхлорид (18.0 г, 157 ммоль), растворенный в толуоле (50 мл). Реакцию нагревали при 60°С в течение 2 ч, после чего реакционную смесь промывали 0.1 М серной кислотой (200 мл) и водой (2×200 мл). Органическую фазу концентрировали при пониженном давлении, и полученное масло растворяли в толуоле (200 мл) и опять концентрировали. Сырой продукт растворяли в толуоле (150 мл) при 30°С и добавляли изооктан (150 мл) в течение 1 ч перед охлаждением до 5°С. После перемешивания полученной мутной жидкости в течение ночи кристаллы отфильтровывали, промывали изооктаном (100 мл) и затем сушили при 40°С в вакууме. Получали 52.4 г (71%) указанного в заголовке соединения в виде белых кристаллов (98.0% чистота по ВЭЖХ). Тпл=87°С; MS [М+]=462;1Н-ЯМР (CDCl3) δ 7.34 (app d, J=8 Гц, 2H), 7.23 (app d, J=7 Гц, 1H), 7.13 (app t, J=7 Гц, 1H), 6.97 (app q, J=8 Гц, 2H), 6.85 (br s, 1H), 6.54 (app d, J=8 Гц, 1Н), 4.26-4.36 (m, 4Н), 3.84 (s, 2H), 3.68-3.78 (m, 4H), 2.99 (s, 3H);13С-ЯМР (CDCl3) δ 172.2, 142.7, 137.7, 130.9, 129.5, 128.9, 128.1, 124.2, 124.1, 122.1, 118.3, 100.0, 69.1, 69.0, 64.1, 38.5, 37.6.
2-{2-[(Метилсульфонил)окси]этокси)этил{2-[(2, 6-дихлорфенил)амино]фенил)-ацетат (соединение формулы IIIa).
Раствор гидроксиэфира IIa (2.6 л), полученный на предыдущей стадии, смешивали с N-метилморфолином (154 г, 1.52 моль), затем по каплям добавляли метансульфонилхлорид (174 г, 1.52 моль) при 30°С в течение 25 мин при интенсивном перемешивании. Температуру поднимали до 41°С в процессе добавления. Реакцию перемешивали при 30°С еще в течение 40 мин перед повышением температуры до 60°С. После перемешивания в течение 3 ч 40 мин добавляли еще N-метилморфолин (7.7 г, 76 ммоль) и метансульфонилхлорид (8.7 г, 76 ммоль), и перемешивание при 60°С затем продолжали в течение 54 мин. Добавляли водную серную кислоту (0.10 М, 1.8 л) при 60°С, и полученную двухфазную систему перемешивали около 20 мин перед разделением фаз. Органический слой промывали дважды при 60°С водой (2×1.8 л) и затем концентрировали при пониженном давлении до конечного объема 1.4 л. Добавляли изооктан (1.35 л) в течение 30 мин при 60°С, затем охлаждали до 30°С. После перемешивания полученной мутной жидкости в течение ночи при 30°С кристаллы отфильтровывали и промывали изооктаном (0.20 л). Полученные кристаллы перекристаллизовывали один раз как описано выше из толуола (1.35 л) и изооктана (1.35 л). После фильтрации и промывки изооктаном (0.90 л) кристаллы сушили при 40°С в вакууме. Получали 610.2 г (86.3% за две стадии) указанного в заголовке соединения в виде белых кристаллов (>99% чистота по ВЭЖХ).
2-[2-(Нитроокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил}ацетат (соединение формулы IVa).
Мезилат IIIa (461 г, 0.997 моль) и нитрат лития (293 г, 4.25 моль) растворяли в N-метилпирролидиноне (1800 мл) и температуру поддерживали при 75°С. Через 3.5 ч добавляли другую партию нитрата лития (146 г, 2.11 моль). Реакцию проводили в течение ночи (всего 27 ч), затем реакцию останавливали понижением температуры до 35°С и добавлением толуола (1800 мл) и воды (1000 мл). Водную фазу отделяли и органическую фазу промывали водой (1000 мл). Органическую фазу упаривали досуха с получением 513 г IVa, который затвердевал при хранении. Аналитический образец (10 г) перекристаллизовывали из н-бутилацетата (30 мл) и изооктана (60 мл). Тпл=73°С; MS [М+]=429;1H-ЯМР (CDCl3) δ 7.34 (арр d, J=8 Гц, 2Н), 7.24 (арр d, J=8 Гц, 1Н), 7.12 (app t, J=8 Гц, 1H), 6.97 (app q, J=8 Гц, 2Н), 6.86 (br s, 1H), 6.55 (d, J=8 Гц, 1H), 4.54 (t, J=4 Гц, 2Н), 4.30 (t, J=5 Гц, 2Н), 3.84 (s, 2H), 3.66-3.74 (m, 4H);13С-ЯМР (CDCl3) δ 171.7, 142.2, 137.2, 130.4, 129.0, 128.4, 127.5, 123.7, 123.6, 121.5, 117.7, 71.4, 68.7, 66.6, 63.6, 38.0.
2-[2-(Нитроокси)этокси)этил{2-[(2,6-дихлорфенил)амино]фенил}ацетат (соединение формулы IVa).
Мезилат IIIa (471 г, 1.02 моль) смешивали с н-бутилацетатом (1.9 л) при 60°С. Добавляли нитрат тетрабутиламмония (62.3 г, 0.204 моль) и нитрат натрия (355 г, 5.15 моль), измельченные с помощью мешалки, при 60°С и полученную мутную жидкость перемешивали при температуре 60°С в течение 10 мин. Добавляли воду (45.9 мл) и температуру поднимали до 90°С, и через 51 ч смесь охлаждали до 50°С. Добавляли воду (1.9 л), и полученную двухфазную систему перемешивали при 50°С в течение 5 мин. Водную фазу отделяли и органическую фазу промывали дважды водой (2×1.9 л) при 50°С. Органическую фазу затем упаривали до объема 1.0 л. Добавляли изопропанол (2.36 л) при 50°С и полученный раствор охлаждали до температуры -11°С в течение 15 ч. Полученные кристаллы отфильтровывали и промывали изопропанолом (1.0 л) и затем сушили в вакууме при 40°С, получая 361.6 г (82.7%) чистого IVa. Чистота согласно ВЭЖХ составляла 98%.
2-[2-(Нитроокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил}ацетат (соединение формулы IVa).
Мезилат IIIa (608.8 г, 1.317 моль) и нитрат тетрабутиламмония (120.8 г, 0.397 моль) смешивали с н-бутилацетатом (1.7 л) при 60°С. Добавляли ацетонитрил (0.70 л) и нитрат натрия (459.7 г, 6.668 моль) при 60°С, и полученную мутную жидкость перемешивали при температуре 87°С в течение 50 ч. Добавляли воду (2.4 л), и температуру понижали до 50°С. Через 10 мин перемешивания водную фазу отделяли и органическую фазу промывали дважды водой (2×2.4 л) при 50°С. Органическую фазу затем упаривали до объема 1.5 л. Добавляли изопропанол (3.1 л) при 50°С и полученный раствор охлаждали до температуры -12°С в течение 15 ч. Через 7 ч перемешивания при -12°С полученные кристаллы отфильтровывали и промывали изопропанолом (0.84 л) и затем сушили в вакууме при 40°С с получением 527.7 г (93.4%) чистого IVa. Чистота согласно ВЭЖХ составляла >99%.
Пример 2
Синтез 4-(нитроокси)бутил{2-[(2,6-дихлорфенил)амино]фенил}ацетата (соединение формулы IVb)
4-Гидроксибутил {2-[(2,6-дихлорфенил)амино]фенил}ацетат (соединение формулы IIb).
К смеси диклофенака натрия (20.0 г, 62.9 ммоль) и 1,4-бутандиола (56.6 г, 629 ммоль) в толуоле (120 мл) при 65°С добавляли серную кислоту (4.5 мл, 84.5 ммоль). Полученный чистый раствор перемешивали при 65°С в течение 6 ч, затем охлаждали до 50°С. Реакционную смесь промывали водным бикарбонатом калия (0.2 М, 120 мл) и водой (2×120 мл). После разделения фаз толуол упаривали с получением 22.9 г IIb в виде коричневого масла (88%, чистота ВЭЖХ по меньшей мере 89%), которое использовали на следующей стадии.1Н-ЯМР (CDCl3) δ 7.34 (арр d J=8 Гц; 2Н), 7.23 (арр d, J=8 Гц, 1Н), 7.13 (арр t, J=7 Гц, 1H), 6.97 (арр q, J=8 Гц, 2Н), 6.56 (арр d, J=8 Гц, 1Н), 4.19 (t, J=7 Гц, 2Н), 3.82 (s, 2Н), 3.63 (t, J=7 Гц, 2H), 1.71-1.80 (m, 2 H), 1.55-1.64 (m, 2H);13С-ЯМР (CDCl3) δ 172.4, 142.6, 137.7, 130.8, 129.4, 128.8, 127.9, 124.4, 124.0, 121.9, 118.2, 65.1, 62.1, 38.6. 28.9, 25.0.
4-[(Метилсульфонил)окси]бутил{2-[(2,6-дихлорфенил)амино]фенил}ацетат (соединение формулы IIIb).
Эфир IIb (20 г, 54 ммоль) с предыдущей стадии и метансульфонилхлорид (7.5 г, 65.1 ммоль) растворяли в толуоле (100 мл) при 20°С. По каплям добавляли N-метилморфолин (6.0 г, 59.7 ммоль). После окончания добавления раствор (слегка мутный) нагревали при 40°С в течение 5 ч. Добавляли толуол (40 мл), и реакцию нагревали при 60°С в течение 0.5 ч, затем добавляли серную кислоту (водн.) (0.1 M, 80 мл). Водный слой отделяли, и толуольную фазу промывали водным карбонатом калия (0.6 M, 40 мл), затем упаривали толуол с получением 35 г масла. Полученное масло растворяли в толуоле (60 мл) при комнатной температуре, и добавляли изооктан. Полученную мутную жидкость охлаждали до 5°С, кристаллы отфильтровывали и промывали изооктаном. Кристаллы сушили при пониженном давлении в течение 1 ч. Получали 19.0 г IIIb в виде белых кристаллов (79% выход с чистотой ВЭЖХ 98.9%). Тпл=57-58°С.1H-ЯМР (CDCl3) δ 7.35 (арр d, J=8 Гц, 2H), 7.22 (арр d, J=8 Гц, 1H), 7.13 (арр t, J=7 Гц, 1H), 6.93-7.01 (m, 2H), 6.88 (br s, 1H), 6.55 (арр d, J=8 Гц, 1 H), 4.15-4.28 (m, 4Н), 3.81 (s, 2H), 2.99 (s, 3H), 1.74-1.84 (m, 4Н);13С-ЯМР (CDCl3) δ 172.3, 142.7, 137.7, 130.8, 129.5, 128.9, 128.0, 124.2, 124.1, 122.0, 118.3, 69.1, 64.3, 38.6, 64.3, 38.6, 37.4, 25.8, 24.8.
4-(Нитроокси)бутил{2-[(2, 6-дихлорфенил)амино]фенил}ацетат (соединение формулы IVb).
Соединение IIIb (5.0 г, 11 ммоль) и нитрат лития (2.2 г, 32 ммоль) растворяли в N-метилпирролидоне (15 мл) при 70°С. Через 23 ч реакцию охлаждали до 35°С, добавляли толуол (20 мл) и реакцию промывали водой (2×30 мл). Органический слой сушили над Na2SO4 и упаривали досуха. Полученное масло очищали хроматографией на силикагеле (EtOAc:гексан; 80:20) и собирали 4.02 г IVb в виде бесцветного масла.1H-ЯМР (CDCl3) δ 7.34 (арр d, J=8 Гц, 2H), 7.22 (арр d, J=7 Гц, 1H), 7.08-7.19 (m, 1H), 6.91-7.02 (m, 2H), 6.88 (br s, 1H), 6.55 (арр d, J=7 Гц, 1H), 4.38-4,46 (m, 2H), 4.14-4.21 (m, 2H), 3.81 (s, 2H), 1.71-1.82 (m, 4H);13С-ЯМР (CDCl3) δ 172.3, 142.7, 137.8, 130.8, 129.5, 128.9, 128.1, 124.2, 124.1, 122.1, 118.3, 72.5, 64.3, 38.6, 25.0, 23.5.
Пример 3
Синтез 2-{2-[2-(нитроокси)этокси]этокси}этил{2-[(2,6-дихлорфенил)амино]-фенил}ацетата (соединение формулы IVc).
2-[2-(2-Гидроксиэтокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил}ацетат (соединение формулы IIc).
Тионилхлорид (1.2 мл, 16.9 ммоль) добавляли к суспензии диклофенака (10 г, 33.8 ммоль) и триэтиленгликоля (90 мл, 676 ммоль) при 30°С. Реакцию перемешивали в течение 7 ч, затем добавляли водный карбонат калия (0.27 M, 100 мл) и толуол (100 мл). Температуру повышали до 60°С, и водную фазу отделяли. Органическую фазу промывали водой (3×100 мл) и концентрировали с получением 14.4 г IIc в виде масла. Это масло непосредственно использовали на следующей стадии.1Н-ЯМР (CDCl3) δ 7.33 (арр d, J=8 Гц, 2Н), 7.23 (арр d, J=7 Гц, 1Н), 7.08-7.20 (m, 1Н), 6.85-7.07 (m, 3H), 6.54 (арр d, J=8 Гц, 1Н), 4.31 (арр t, J=5 Гц, 2Н), 3.85 (s, 2H), 3.71 (m, 4 Гц, 4H), 3.54-3.64 (m, 4H), 2.50 (арр br s, 1H);13С-ЯМР (CDCl3) δ 172.4, 142.8, 137.8, 130.9, 129.6, 128.9, 128.01, 124.2, 124.1, 122.0, 118.2, 72.5, 70.6, 70.3, 69.0, 64.3, 61.7, 38.5.
10,10-Диоксидо-3,6, 9-триокса-10-тиаундец-1-ил{1-[(2,6-дихлорфенил)амино] фенил) ацетат (соединение формулы IIIc).
Гидроксиэфир IIc (13.4 г, 31.3 ммоль) с предыдущей стадии растворяли в толуоле (80 мл) вместе с N-метилморфолином (3.5 г, 34.4 ммоль) при 30°С. Добавляли метансульфонилхлорид (3.9 г, 34.4 ммоль) в толуоле (10 мл) в течение 15 мин. После окончания добавления температуру повышали до 60°С в течение 2 ч и затем снова понижали до 30°С в течение ночи. Добавляли водную серную кислоту (0.1 M, 40 мл), и температуру повышали до 60°С для экстракции. Водную фазу отделяли, и органическую фазу промывали водой (2×100 мл). Органическую фазу концентрировали с получением масла (15.3 г). Это масло очищали хроматографией на силикагеле (EtOAc/гексан; от 30/70 до 50/50) с получением 13.8 г IIIc в виде коричневого масла.1H-ЯМР (CDCl3) δ 7.34 (арр d, J=8 Гц, 2H), 7.23 (арр d, J=7 Гц, 1H), 7.12 (арр t, J=7 Гц, 1H), 6.88-7.02 (m, 2H), 6.54 (d, J=8 Гц, 1H), 4.75-4.36 (m, 4H), 3.84 (s, 2H), 3.67-3.74 (m, 4H) 3.6 (арр br s, 4 H), 3.04 (s, 3H);13С-ЯМР (CDCl3) δ 172.2, 142.6, 137.6, 130.8, 129.4, 128.8, 127.9, 124.1, 124.0, 121.9, 118.1, 70.4, 69.1, 68.91, 68.87, 64.2, 60.2, 38.4, 37.5.
2-{2-[2-(Нитроокси)этокси]этокси]этил{2-[(2,6-дихлорфенил)амино1фенил}ацетат (соединение формулы IVc).
Добавляли нитрат натрия к раствору мезилата IIIc с предыдущей стадии (12.7 г, 25.1 ммоль) и нитрат тетрабутиламмония (2.3 г, 7.6 ммоль) в н-бутилацетате (50 мл) и воде (1.7 мл) при 60°С. Полученную суспензию нагревали при 85°С в течение 41 ч, затем охлаждали до 60°С и добавляли воду (100 мл). После экстракции водную фазу отделяли, и органическую фазу промывали дважды водой (2×100 мл). Органическую фазу упаривали досуха и остаток кристаллизовали из н-бутилацетата (26 мл) и 2-пропанола (110 мл). Кристаллы отфильтровывали, промывали 2-пропанолом (25 мл) и сушили при пониженном давлении при 40°С с получением 9.3 г IVc в виде кристаллов. Тпл=68°С.1H-ЯМР (CDCl3) δ 7.34 (арр d, J=8 Гц, 2Н), 7.23 (арр d, J=7 Гц, 1Н), 7.12 (app t, J=7 Гц, 1Н), 6.91-7.02 (m, 3H), 6.55 (app d, J=8 Гц, 1H), 4.58 (app t, J=5 Гц, 2Н), 4.31 (арр t, J=4 Гц, 2Н), 3.85 (s, 2H), 3.67-3.78 (m, 4H), 3.60 (app s, 4H);13С-ЯМР (CDCl3) δ 172.4, 142.8, 137.8, 130.9,129.5, 128.9, 128.0, 124.3, 124.0, 122.0, 118.3, 72.2, 70.8, 70.6, 69.1, 67.2, 64.3, 38.5.
Пример 4
Синтез 3-(нитроокси)пропил2-(2-бензоилфенил)пропаноата (соединение формулы IVd).
3-Гидроксипропил(2S)-2-(2-бензоилфенил)пропаноат (соединение формулы IId)
Смесь (S)-кетопрофена (10.0 г, 39.3 ммоль), 1,3-пропандиола (29.9 г, 393 ммоль), толуола (40 мл) и концентрированной серной кислоты (0.3 г, 3.06 ммоль) нагревали при 80-95°С в течение 28 ч, затем охлаждали до 45°С и добавляли 5% водный раствор карбоната калия (50 мл). Нижний водный слой отделяли, и верхний органический слой промывали водой (2×50 мл). Органический слой концентрировали досуха при пониженном давлении с получением 11.9 г IId в виде бесцветного масла (чистота ВЭЖХ 96%). Энантиомерная чистота >99.5%. MS [М+]=312,1H-ЯМР (CDCl3) δ 7.78 (арр t, J=7 Гц, 3H), 7.41-7.68 (m, 6Н), 4.30-4.79 (m, 2H), 3.81 (q, J=7 Гц, 1H), 3.51 (t, J=6 Гц, 2H), 2.35 (br s, 1H), 1.82 (quin, J=7 Гц, 2H), 1.53 (d, J=7 Гц, 3H);13С-ЯМР (CDCl3) δ 196.7, 174.4, 140.9, 137.9, 137.4, 132.6, 131.5, 130.1, 129.1, 128.6, 128.3, 61.9, 58.9, 45.4, 31.5, 18.4, 14.2.
S-[(Метилсульфонил)окси]пропил(2S)-2-(2-бензоилфенил)пропаноат (соединение формулы IIId)
Гидроксиэфир IId (5.0 г, 16 ммоль) с предыдущей стадии растворяли в толуоле (25 мл). К смеси добавляли метансульфонилхлорид (2.2 г, 19.2 ммоль), затем по каплям добавляли N-метилморфолин (1.78 г, 17.6 ммоль). Реакционную смесь нагревали при 40°С в течение 1 ч и затем нагревали до 60°С, добавляли водную серную кислоту (0.1 М, 20 мл) и толуол (10 мл). После экстракции смесь отделяли, и органический слой промывали водным карбонатом калия (0.93 г в 20 мл воды). Органический слой концентрировали в вакууме с получением 5.6 г IIId в виде масла. MS [M+]=391;1H-ЯМР (300 МГц, CDCl3) δ 7.78 (арр t, J=7 Гц, 3H), 7.41-7.69 (m, 6Н), 4.21 (арр t, J=6 Гц, 2Н), 4.18 (арр t, J=6 Гц, 2Н), 3.82 (q, J=7 Гц, 1Н), 2.94 (s, 3H), 2.04 (quin, J=7 Гц, 2Н), 1.55 (d, J=7 Гц, 3H);13С-ЯМР (100 МГц, CDCl3) δ 196.4, 173.8, 140.7, 138.0, 132.5, 131.4, 130.0, 129.1, 129.0, 128.6, 128.3, 66.0, 60.4, 45.3, 37.2, 28.4, 18.2.
3-(Нитроокси)пропил(2S)-2-(2-бензоилфенил)пропаноат (соединение формулы IVd)
Смесь мезилата IIId (5.0 г, 12.8 ммоль) с предыдущей стадии и нитрат лития (2.65 г, 38.5 ммоль) в N-метилпирролидиноне (15 мл) нагревали при 70°С в течение 9 ч. Нагревание прекращали и реакционную смесь охлаждали до комнатной температуры, затем добавляли толуол (30 мл) и воду (20 мл). Слои разделяли и органический слой промывали водой (20 мл). Концентрация досуха приводила к получению IVd в виде масла (5.0 г). Энантиомерная чистота 99.5%. MS [М+] =357;1Н-ЯМР (300 МГц, CDCl3) δ 7.73-7.84 (m, 3H), 7.67 (арр d, J=1 Гц, 1Н), 7.38-7.64 (m, 5Н), 4.40 (t, J=6 Гц, 2Н), 4.18 (t, J=6 Гц, 2Н), 3.81 (q, J=7 Гц, 1H), 2.94 (s, 3H), 2.01 (quin, J=6 Гц, 2Н), 1.55 (d, J=7 Гц, 3H),13С-ЯМР (100 МГц, CDCl3) δ 196.4, 173.8, 140.7, 138.0, 137.5, 132.6, 131.4, 130.0, 129.2, 129.1, 128.6, 128.3, 69.6, 60.8, 45.3, 26.3, 18.3.
Пример 5
Анализ дифракции рентгеновских лучей на порошке (XRPD) проводили стандартными методами, например, описанными Giacovazzo, С. и др. (1995), рр 287-301, Fundamentals of Crystallography, Oxford University Press; Jenkins, R. и Snyder, R.L. (1996), Introduction to X-Ray Powder Diffractometry, John Wiley &. Sons, New York; Bunn, C.W. (1948), pp 103-127, Chemical Crystallography, Clarendon Press, London; или Klug, H.P. & Alexander, L.E. (1974), X-ray Diffraction Procedures, second edition, John Wiley and Sons, New York.
Анализ рентгеновских лучей проводили с помощью дифрактометра Philips X′Pert MPD. Дифференциальную сканирующую калориметрию (ДСК) осуществляли с помощью аппарата Perkin Elmer DSC7, стандартными методами, например, описанными Höhne, G. W. H. и др. (1996), Differential Scanning Calorimetry, Springer, Berlin. Термогравиметрический анализ (TGA) осуществляли с помощью аппарата Perkin Elmer TGA7.
Кристаллическая форма, полученная в соответствии с приведенным ниже Примером 1, по существу имеет тот же образец дифракции XRPD и термограмму ДСК и TGA, что и кристаллические формы, полученные в соответствии с другими Примерами, описанными ниже, с погрешностью эксперимента. Границы погрешности эксперимента для температуры ДСК могут находиться в области ±5°С (например, ±2°С) и для значений XRPD могут находиться в области ±2 от последнего десятичного значения.
Синтез безводного 2-[2-(нитроокси)этокси]этил {2-[(2,6-дихлорфенил)амино]фенил}-ацетата
Пример 5а
0.3 г 2-[2-(нитроокси)этокси]этил{2-[(2,6-дихлорфенил)амино]фенил} ацетата IVa загружали вместе с 0.9 мл толуола в пробирку для анализа на 4 мл. Пробирку для анализа помещали на магнитную мешалку при температуре окружающей среды. После растворения всего соединения добавляли 1.8 мл изооктана порциями по 0.3 мл. Кристаллизация начиналась после добавления всего изооктана. Через 4.5 ч от начала кристаллизации кристаллы отфильтровывали в вакууме. Пробирку промывали 0.3 мл изооктана. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (от количества, оставшегося в маточном растворе) 80.6%.
Кристаллы анализировали с помощью XRPD, ДСК и TGA. Результаты XRPD представлены в Таблице 1 и показаны на чертеже. Термограмма ДСК показала температуру плавления 72°С и термограмма TGA показала, что кристаллы не содержат существенного количества примесей растворителей.
Основные пики с положением (D/Å) и относительные интенсивности взяты из дифрактограммы на чертеже. Относительные интенсивности приведены как VS= очень сильная, S = сильная, М = средняя, W = слабая. Включены только пики менее 2θ=40°. Некоторые дополнительные очень слабые пики, обнаруженные на дифрактограмме, исключены из таблицы, но представлены на чертеже.
Все пики помечены моноклинной элементарной ячейкой кристалла: а=13.79 Å, b=11.90 Å, с=13.01 Å, α=90°, β=94.0°, γ=90°.
Пример 5b
0.3 г соединения IVa загружали вместе с 0.9 мл метилизобутилкетона в пробирку для анализа на 4 мл. Пробирку для анализа помещали на магнитную мешалку при температуре окружающей среды. Для растворения всего соединения необходимы дополнительные 0.3 мл 4-метил-2-пентанона. Затем добавляли 1.8 мл изооктана порциями по 0.3 мл. Кристаллизация начиналась после добавления всего изооктана. Через 4 ч после начала кристаллизации кристаллы отфильтровывали в вакууме. Пробирку промывали 0.3 мл изооктана. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (основываясь на количестве, оставшемся в маточном растворе) 44.1%.
Кристаллы анализировали с помощью XRPD, ДСК и TGA. Результаты являются по существу такими же, которые описаны для формы, полученной в Примере 5а.
Пример 5с
2.5 г соединения IVa загружали вместе с 7.5 мл бутилацетата в реактор с рубашкой на 100 мл. Реактор нагревали до 35°С до растворения всего соединения. Затем применяли следующий температурный профиль: температуру понижали до 20°С в течение 1.5 ч и затем оставляли на 0.5 ч при 20°С. По каплям при 20°С добавляли 15 мл изооктана. Кристаллизация начиналась после добавления 12 мл изооктана. Температуру снижали в течение 3 ч до 0°С. Через 0.5 ч при 0°С кристаллы отфильтровывали в вакууме. Реактор промывали 7.5 мл охлажденного изооктана. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (основываясь на количестве, оставшемся в маточном растворе) 91.6%.
Кристаллы анализировали с помощью XRPD, ДСК, TGA, LC и GC. Результаты XRPD, ДСК и TGA являются по существу такими же, которые описаны для формы, полученной в Примере 5а. LC показала чистоту 99.12%, GC показала 0.01 вес.% изооктана и 0.10 вес.% бутилацетата. Исходный реагент имел чистоту 98.42% и содержал 0.13 вес.% этилацетата.
Пример 5d
0.5 г соединения IVa загружали вместе с 1.5 мл трет-бутилметилового эфира в пробирку для анализа на 4 мл. Пробирку помещали в масляную баню. Магнитной мешалкой обеспечивали перемешивание. Масляную баню нагревали до получения чистого раствора в пробирке для анализа. Это происходило при 40°С. Затем температуру масляной бани снова понижали до 20°С. Смесь перемешивали в течение ночи с образованием кристаллов. Кристаллы отфильтровывали в вакууме. Пробирку промывали 0.3 мл трет-бутилметиловым эфиром. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (основываясь на количестве, оставшемся в маточном растворе) 77%.
Кристаллы анализировали с помощью XRPD, ДСК и TGA. Результаты являются по существу такими же, которые описаны для формы, полученной в Примере 1. Результаты показали по существу тот же образец XRPD, что и образец формы, полученной в Примере 5а.
Пример 5е
0.5 г соединения IVa загружали вместе с 1.5 мл бутанола в пробирку для анализа на 4 мл. Пробирку помещали в масляную баню. Магнитной мешалкой обеспечивали перемешивание. Масляную баню нагревали до получения в пробирке для анализа чистого раствора. Это происходило при 60°С. Затем пробирку для анализа помещали на магнитную мешалку при температуре окружающей среды. Кристаллизация начиналась немедленно. Через 2.5 ч кристаллы отфильтровывали в вакууме. Пробирку промывали 0.3 мл бутанола. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (основываясь на количестве, оставшемся в маточном растворе) 94%.
Кристаллы анализировали XRPD, ДСК и TGA. Результаты являются по существу такими же, которые описаны для формы, полученной в Примере 5а.
Пример 5f
0.5 г соединения IVa загружали вместе с 1.5 мл изопропанола в пробирку для анализа на 4 мл. Пробирку помещали в масляную баню. Магнитной мешалкой обеспечивали перемешивание. Масляную баню нагревали до получения в пробирке для анализа чистого раствора. Это происходило при 60°С. Затем пробирку для анализа помещали на магнитную мешалку при температуре окружающей среды. Кристаллизация начиналась немедленно. Через 2.5 ч кристаллы отфильтровывали в вакууме. Пробирку промывали 0.3 мл изопропанола. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (основываясь на количестве, оставшемся в маточном растворе) 96%.
Кристаллы анализировали XRPD, ДСК и TGA. Результаты являются по существу такими же, которые описаны для формы, полученной в Примере 5а.
Пример 5g
0.5 г соединения IVa загружали вместе с 2.5 мл этанола в пробирку для анализа на 4 мл. Пробирку для анализа помещали на магнитную мешалку при температуре окружающей среды. Мутную жидкость в пробирке для анализа перемешивали в течение ночи. Кристаллы отфильтровывали в вакууме. Пробирку промывали 0.6 мл этанола. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (основываясь на количестве, оставшемся в маточном растворе) 93.4%.
Кристаллы анализировали XRPD, ДСК и TGA. Результаты являются по существу такими же, которые описаны для формы, полученной в Примере 5а.
Пример 5h
0.5 г соединения IVa загружали вместе с 2.5 мл изооктана в пробирку для анализа на 4 мл. Пробирку для анализа помещали на магнитную мешалку при температуре окружающей среды. Мутную жидкость в пробирке для анализа перемешивали в течение ночи. Кристаллы отфильтровывали в вакууме. Кристаллы затем сушили в вакуумной печи при 35°С. Выход (основываясь на количестве, оставшемся в маточном растворе) 99.1%.
Кристаллы анализировали XRPD, ДСК и TGA. Результаты являются по существу такими же, которые описаны для формы, полученной в Примере 5а.
Пример 5i
Соединение IVa (4.0 г) смешивали с ацетоном (8.0 мл) и полученную смесь перемешивали при 40°С. После получения чистого раствора добавляли изопропанол (40 мл) и раствор оставляли перемешиваться в течение ночи при температуре окружающей среды. В раствор затем добавляли затравочные кристаллы при температуре окружающей среды, и приблизительно через 30 мин затравочные кристаллы все еще не растворялись. Температуру затем понижали с 20°С до -5°С в течение 12 часов. Кристаллы отфильтровывали и сушили в вакууме при 40°С с получением 3.55 г (88.8%) чистого IVa. Кристаллы анализировали с помощью XRPD и ВЭЖХ, и результаты имели по существу тот же XRPD образец, что и для формы, полученной в Примере 5а. ВЭЖХ показала чистоту 98.2%.
Пример 5j
Соединение IVa (10.0 г) смешивали с ацетонитрилом (62 мл), и полученную смесь перемешивали при комнатной температуре. После получения чистого раствора добавляли воду (14 мл), и в полученный раствор затем добавляли затравочные кристаллы при температуре окружающей среды. Добавляли воду (2 мл), и приблизительно через 1 ч 30 мин перемешивания затравочные кристаллы все еще не растворялись. Раствор перемешивали в течение двух дней при температуре окружающей среды, и после этого температуру понижали до -10°С в течение 24 часов. Кристаллы отфильтровывали, промывали водой (20 мл) и сушили в вакууме при 40°С с получением 7.98 г (79.8%) чистого IVa. Кристаллы анализировали с помощью XRPD и ВЭЖХ, и результаты имели по существу тот же XRPD образец, что и для формы, полученной в Примере 5а. ВЭЖХ показала чистоту 99.0%.
Пример 5k
Соединение IVa (10.3 г) смешивали с этилацетатом (20 мл), и полученную смесь перемешивали при 40°С. После получения чистого раствора добавляли изопропанол (80 мл) и температуру понижали с 40°С до -10°С в течение 15 часов. Кристаллы отфильтровывали, промывали изопропанолом (20 мл) и сушили в вакууме при 40°С с получением 9.37 г (91%) чистого IVa. Кристаллы анализировали с помощью XRPD и ВЭЖХ, и результаты имели по существу тот же XRPD образец, что и для формы, полученной в Примере 5а. ВЭЖХ показала чистоту 99%.
Пример 5l
Соединение IVa (438.9 г) смешивали с ацетоном (4.0 л), и полученную смесь перемешивали при 30°С до получения чистого раствора. После получения чистого раствора добавляли воду (1.3 л) и температуру понижали с 30°С до -3°С в течение 8 часов. После перемешивания при -3°С в течение 10 ч температуру далее понижали до -12°С в течение 5 ч. Кристаллы затем отфильтровывали, промывали водой (0.90 л) и сушили в вакууме при 40°С с получением 392 г (89.2%) чистого IVa. Кристаллы анализировали с помощью XRPD и ВЭЖХ, и результаты имели по существу тот же XRPD образец, что и для формы, полученной в Примере 5а. ВЭЖХ показала чистоту >99%.
Сокращения:
D - расстояние, измеренное в Å [Ангстрем],
ДСК - дифференциальная сканирующая калориметрия,
FT-IR - Фурье-трансформированная инфракрасная спектроскопия,
ЯМР - ядерный магнитный резонанс,
TGA - термогравиметрический анализ,
XRDP - рентгеновская дифракция на порошке
Чертеж: Дифракция рентгеновских лучей на порошке по существу кристаллического 2-[2-(нитроокси)этокси]-этил{2-[(2,6-дихлорфенил)амино]фенил}ацетата.
Реферат
Настоящее изобретение относится к новому способу получения NO-донорных соединений формулы (IV), включающему: стадию 1) реакцию соединения формулы (I)

с соединением формулы НО-Х-ОН с помощью кислотного или дегидрирующего агента и первого растворителя, необязательно с последующей очисткой экстракцией или кристаллизацией, чтобы получить соединение формулы (II)

стадию 2) реакцию соединения формулы (II) с соединением формулы RSO2Cl, с помощью второго растворителя, основания и, необязательно, катализатора, с последующей очисткой экстракцией и кристаллизацией, чтобы получить соединение формулы III,

стадию 3) реакцию соединения формулы (III) с соединением формулы Y-NO3 с помощью третьего растворителя и, необязательно, катализатора, необязательно с последующей кристаллизацией для получения соединения формулы IV

в котором MLT1AT2 выбран из группы, состоящей из










в котором X выбран из группы, состоящей из линейного -(CH2)w1-, где w1 имеет значение от 2 до 10; -(СН2-СН2-О)2-СН2-СН2- и -СН2-С6H4-CH2-. R выбран из группы, состоящей из С1-С8алкила, фенила, фенилметила, С1-С4алкилфенила, галогенфенила, нитрофенила, ацетиламинофенила, галогена, CF3 и n-C4F9; Y-NO3 представляет собой нитрат лития, нитрат натрия, нитрат калия, нитрат магния, нитрат кальция, нитрат железа, нитрат цинка и нитрат тетраалкиламмония, где алкил представляет собой C1-C18-алкил, который может быть линейным или разветвленным и их смесью; m имеет значение 2; и при условии, что когда MLT1AT2-COOH представляет собой напроксен, X не является (СН2)4. Изобретение также относится к применению способа для получения соединений формул IVa, IVb, IVc и IVd, к новым промежуточным соединениям формул IIIa, IIIb, IIIc и IIId, к кристаллической форме 2-[2-(нитроокси)этокси]этил{2-[(2, 6-дихлорфенил)амино]фенил}ацетата и способу ее получения, и к ее применению для получения лекарственного средства для лечения боли и/или воспаления, к применению соединений формулы III, в качестве промежуточных соединений. А также к применению промежуточных соединений формул IIIa, IIIb, IIIc и IIId для получения соединений формулы IVa, IVb, IVc и IVd. Технический результат - новый способ получения соединений формулы (IV). 8 н. и 24 з.п. ф-лы, 1 ил., 1 табл.
Формула

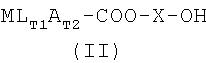
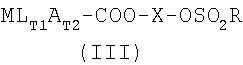





















Документы, цитированные в отчёте о поиске
Нитросоединения и фармацевтическая композиция, обладающие противовоспалительной и антитромботической активностями
Комментарии