Анальгетические соединения и способы их применения - RU2772045C2
Код документа: RU2772045C2
Чертежи






Описание
Настоящее изобретение относится к области биохимии, ветеринарии, медицины, химико-фармацевтической промышленности и касается биологически активных низкомолекулярных соединений органической природы, обладающих противовоспалительной активностью и обезболивающим действием на млекопитающих. Более конкретно изобретение относится к способу получения биологически активных соединений методом химического синтеза и способу применения этих соединений для получения устойчивого обезболивающего и противовоспалительного эффекта.
В настоящее время одной из основных причин обращения людей за врачебной помощью является боль. Патологическая боль, появляющаяся при различных состояниях, представляет собой нежелательное явление, резко снижающее качество жизни, работоспособность и вызывающее страдания. До 40% взрослого населения развитых стран страдает от хронической боли. Наиболее широко в качестве анальгетических препаратов традиционно используются: нестероидные противовоспалительные средства, опиоиды и разнообразные вещества неспецифического действия (антиконвульсанты, антидепрессанты). В последнее время в качестве анальгетических препаратов предлагалось использовать ингибиторы активности протон-чувствительных ионных каналов (ASIC).
Четыре гена кодируют шесть изоформ ASIC у млекопитающих: ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3 и ASIC4. ASIC каналы экспрессируются многими типами нервных клеток, главным образом, периферическими и сенсорными нейронами центральной нервной системы (ЦНС), в том числе нейронами, чувствительными к болевым стимулам. Многочисленные исследования показали, что эти ионные каналы играют важную роль во многих патологических процессах помимо разнообразных болевых и воспалительных синдромов. В ЦНС широко распространены каналы ASIC1a, которые участвуют в синаптической пластичности и обучении [Du, J., Reznikov, L. R., Price, M. P., Zha, X. -m., Lu, Y., Moninger, T. O., et al. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala.//Proc. Natl. Acad. Sci.2014. 111, 8961–8966; Wemmie, J. A., Chen, J., Askwith, C. C., Hruska-Hageman, A. M., Price, M. P., Nolan, B. C., et al. The Acid-Activated Ion Channel ASIC Contributes to Synaptic Plasticity, Learning, and Memory. //Neuron 2002. 34, 463–477]. При этом эти каналы вовлечены в процессы нейродегенрации. Блокада ASIC1a каналов амилоридом дает хороший нейропротекторный эффект [Friese, M. A., Craner, M. J., Etzensperger, R., Vergo, S., Wemmie, J. A., Welsh, M. J., Vincent, A. and Fugger, L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. // Nat. Med. 2007. 13, 1483–1489]. Аналогично было продемонстрировано, что амилорид защищает как миелин, так и сами нейроны от повреждений в моделях острой боли [Vergo, S., Craner, M. J., Etzensperger, R., Attfield, K., Friese, M. A., Newcombe, J., Esiri, M. and Fugger, L. Acid-sensing ion channel 1 is involved in both axonal injury and demyelination in multiple sclerosis and its animal model // Brain 2011. 134, 571–584]. Фармакологическое ингибирование ASIC1a с помощью PcTx1 или генетическая делеция ASIC1a уменьшает гибель нейронов и объем инфаркта в модели ишемического инсульта [McCarthy, C. A., Rash, L. D., Chassagnon, I. R., King, G. F., and Widdop, R. E. PcTx1 affords neuroprotection in a conscious model of stroke in hypertensive rats via selective inhibition of ASIC1a. // Neuropharmacology 2015. 99, 650–657], тогда как повышенная активность ASIC1a способствует повреждению нейронов во время ишемии у животных [Duan, B., Wang, Y.-Z., Yang, T., Chu, X.-P., Yu, Y., Huang, Y., et al. Extracellular Spermine Exacerbates Ischemic Neuronal Injury through Sensitization of ASIC1a Channels to Extracellular Acidosis.//J. Neurosci. 2011. 31, 2101–2112]. Кроме того, ингибитор PcTx1 проявляет нейропротекторную активность в модели болезни Паркинсона [Dwyer, J. M., Rizzo, S. J. S., Neal, S. J., Lin, Q., Jow, F., Arias, R. L., et al. Acid sensing ion channel (ASIC) inhibitors exhibit anxiolytic-like activity in preclinical pharmacological models. // Psychopharmacology (Berl).2009. 203, 41–52].
Эти же каналы вовлечены активно в процессы формирования страха и проявления тревожного поведения [Wemmie, J. A., Askwith, C. C., Lamani, E., Cassell, M. D., Freeman, J. H., and Welsh, M. J. Acid-Sensing Ion Channel 1 Is Localized in Brain Regions with High Synaptic Density and Contributes to Fear Conditioning. // J. Neurosci. 2003. 23, 5496–5502; Wemmie, J. A., Coryell, M. W., Askwith, C. C., Lamani, E., Leonard, A. S., Sigmund, C. D., et al. Overexpression of acid-sensing ion channel 1a in transgenic mice increases acquired fear-related behavior. // Proc. Natl. Acad. Sci. 2004. 101, 3621–3626]. Использование ингибиторов ASIC1a, таких как PcTX1, A-317567 и амилорид, улучшает как параметры, связанные с вегетативной нервной системой, так и поведенческие параметры в моделях тревоги у грызунов [Dwyer, J. M.etal.Acid sensing ion channel (ASIC) inhibitors exhibit anxiolytic-like activity in preclinical pharmacological models. // Psychopharmacol. 2009. 203, 41–52.].
Оказывается, что опухолевые клетки экспрессируют в том числе и ASIC каналы. ASIC1a физически взаимодействует с Ca2+ кальмодулин-зависимой протеинкиназой II (CaMKII) и интегрином-β1 [Xu, S., Liu, C., Ma, Y., Ji, H.-L., and Li, X. Potential Roles of Amiloride-Sensitive Sodium Channels in Cancer Development. // Biomed Res. Int. 2016, 1–6], которые являются важными регуляторами внутриклеточной передачи сигналов и адгезии. Поэтому, ASIC влияют на пролиферацию клеток глиобластомы с помощью различных механизмов. В дополнение к глиомам, ASIC участвуют в регуляции роста, миграции, а также лекарственной устойчивости других карцином [Gupta, S. C., Singh, R., Asters, M., Liu, J., Zhang, X., Pabbidi, M. R., et al. Regulation of breast tumorigenesis through acid sensors. // Oncogene2016. 35, 4102–4111; Zhang, Y., Zhang, T., Wu, C., Xia, Q., and Xu, D.ASIC1a mediates the drug resistance of human hepatocellular carcinoma via the Ca2+/PI3-kinase/AKT signaling pathway.// Lab. Investig. 2017.97, 53–69]. Ингибиторы бензамил и PcTx1 продемонстрировали антипролиферативную активность против клеток глиобластомы [Rooj, A. K., McNicholas, C. M., Bartoszewski, R., Bebok, Z., Benos, D. J., and Fuller, C. M. Glioma-specific Cation Conductance Regulates Migration and Cell Cycle Progression.// J. Biol. Chem. 2012. 287, 4053–4065], а ингибитор мамбалгин-2 против клеток лейкемии [Bychkov, M. L. etal. ASIC1a inhibitor mambalgin-2 suppresses growth of leukemia cells by cell cycle arrest. // Acta Naturae, 2020, 12, 111–116].
В итоге, широкий круг патологий предлагается лечить с применением ингибиторов ASIC различной структуры. Предложен способ лечения наркомании, тревожности или тревожного расстройства, включающий: введение нуждающемуся в этом пациенту терапевтически эффективного количества антагониста ASIC (US2007197583); способ уменьшения аксональной дегенерации у индивидуума, страдающего демиелинизирующим заболеванием, где демиелинизирующим заболеванием является рассеянный склероз, включающий введение антагониста рецептора ASIC (US2010015127); способ лечения ишемической боли из-за сердечно-сосудистых заболеваний, инсульт-индуцированного повреждения, боли из-за артрита, ишемической боли из-за рака, боли из-за воспаления или инфекции, острого и хронического кашля, боли из-за желудочно-кишечных расстройств, расстройств центральной нервной системы и психических заболеваний введением эффективного количества блокатора ASIC (US2008293740); способ устранения последствий мышечной и сердечной ишемии, вызванной переломами, гематомами, отеками, инфекциями, поражениями тканей, включая хирургические вмешательства, травмами глаза и опухолями, включая опухоли костей и метастазы (US2011152197).
Одним из ингибиторов ASIC является растительный лигнан Севанол с подтвержденной анальгетической активностью (RU2491950), который было предложено использовать как перспективный инструмент исследования функциональной активности ионного канала в научных исследованиях, а также в медицине как средство, направленное против боли. Были разработаны способы получения Севанола из растительного сырья (RU2491950) или за счет химического синтеза (RU2698725). Однако, как конкретно можно использовать Севанол в медицине, его побочные свойства применения и возможность его применения неинвазивными способами описана не была. Изобретение раскрывает возможности использования Севанола различными способами введения, для лечения разных патологических процессов и показывает отсутствие основных побочных действий - возникновения зависимости и седативного влияния, которое так характерно для опиоидных анальгетиков (конкурентов Севанола по эффективности обезболивающего действия).
Севанол - природная молекула, построенная на основе эпифиловой кислоты, с расчетной средней молекулярной массой 706,52 Да, содержит 6 хиральных центров, причем один стереоизомер молекулы, такой как изосеванол, также обладает анальгетической активностью и может быть использован как анальгетический препарат [Д. И. Осмаков, С. Г. Кошелев, О. А. Белозерова, В. С. Кублицкий, Я. А. Андреев, Е. В. Гришин, С. А. Козлов Биологическая активность севанола и его аналогов // Биоорганическая химия, 2015, т 41, с. 606–611]. Получение Севанола путем химического синтеза за 9 стадий (RU2698725) приводит к целевому продукту с выходом 2,2%. Себестоимость анальгетического средства можно уменьшить, если упростить структуру и снизить количество хиральных центров, что приводит соответственно к уменьшению стадий химического синтеза и большему выходу активного вещества. Изобретение раскрывает структуру активного центра молекулы Севанол, который сохраняет способность ингибировать токи через ионные каналы ASICs и вызывать обезболивающий эффект у животных. На основе этого активного центра может быть синтезирована более простая молекула – s590, s414 и ЕА. ЕА – эпифиловая кислота, биологическая активность и методика синтеза которой не была ранее раскрыта, s414 и s590 – новые молекулы с анальгетическим действием.
Изобретение решает задачу расширения арсенала анальгетических и противовоспалительных лекарственных препаратов, имеющих направленное действие на клеточную мишень ионные каналы ASIC, задачу разработки способов применения препаратов для лечения заболеваний и задачу разработки способов химического синтеза биологически активных веществ.
Поставленная задача решается за счет применения биологически активных соединений (1R, 2S) – 1-(3,4-дигидроксифенил)-6,7-дигидрокси-1,2-дигидронафтален-2,3-дикарбоксилатов, представленных формулой (I),
где R может представлять собой водород, этильную группу, остаток сложного эфира (L)-яблочной кислоты, остаток сложного эфира изолимонной кислоты или их ближайшие структурные аналоги.
Если R является водородом, то активная молекула представляет собой эпифиловую кислоту (EA) (1R, 2S) – 1-(3,4-дигидроксифенил)-6,7-дигидрокси-1,2-дигидронафтален-2,3-дикарбоновая кислота (ФИГ. 1А), если R – этильная группа, то активная новая молекула s414 (1R,2S)-диэтил-1-(3,4-дигидроксифенил)-6,7-дигидрокси-1,2-дигидронафтален-2,3-дикарбоксилат (ФИГ. 1Б), если R - сложный эфир (L)-яблочной кислоты, то активная новая молекула s590 (2S,2’S)-2,2’-(((1R,2S)-1-(3,4-дигидроксифенил)-6,7-дигидрокси-1,2-дигидронафтален-2,3-дикарбонил)бис(окси))дисукциновая кислота (ФИГ. 1В), если R - сложный эфир изолимонной кислоты, то это Севанол (s706), известный как (1R,2S)-1-[(1R,2S)-1-(3,4-дигидроксифенил)-6,7-дигидрокси-3-[(1R,2S)-1,2,3-трикарбоксипропокси]карбонил-1,2-дигидронафтален-2-карбонил]оксипропан-1,2,3-трикарбоновая кислота (ФИГ. 1Г).
Заявляемые анальгетические соединения кроме s706 и EA являются синтетическими молекулами, получение которых из природных источников невозможно. Соединение s706 может быть выделено хроматографическими методами из уксуснокислого экстракта молодых побегов тимьяна (RU2491950) (этот способ требователен к качеству сырья), или за счет химического синтеза (RU2698725). Указанный способ синтеза используется для синтеза активного соединения s590 в 6 этапов с суммарным выходом целевого продукта s590 - 11%.
Первая стадия синтеза соединения s590 заключается в синтезе ди-третбутилового эфира (L)-яблочной кислоты в две стадии согласно методике, разработанной ранее [Calo, F., Richardson, J., Barrett, A.G.M. Total synthesis of Citrafungin A. J.Org.Chem. 2008, 73, 9692-9697] и оптимизированной в патенте (RU2698725):

Исходным соединением для синтеза ди-третбутилового эфира (L)-яблочной кислоты 5 является коммерчески доступная (L)-яблочная кислота 4, которая вступает во взаимодействие с О-третбутилдиизопропилизомочевиной 3. После проведения стандартных процедур ключевой промежуточный продукт бис-третбутиловый эфир (L)-яблочной кислоты 5 получается с общим выходом 59% (ФИГ. 2).
Второй этап синтеза активного соединения s590 (ФИГ. 4) связан с ацилированием промежуточного продукта хлорангидрида бисацетилкофейной кислоты, полученного по способу [M. Sefkow, First efficient synthesis of chlorogenic acid // Eur. J. Org. Chem., i.6, p. 1137-1141]:

и синтезированного ранее ди-третбутилового эфира (L)-яблочной кислоты 5:

аналогично методике, описанной ранее в патенте (RU2698725) (ФИГ. 3). Дальнейшее снятие ацетильных защит с использованием N-метилпиперазина с промежуточного продукта 9 и последующая ключевая стадия окислительной димеризации сложного эфира соответствующей кофейной кислоты 10 в присутствии хлорида железа (III) в качестве реагента протекает с соблюдением условий, подробно ранее изложенных в (RU2698725) (ФИГ. 4). Сущностью способа является реакция сочетания двух молекул производного кофейной кислоты (ФИГ. 4), которая протекает в течение двух часов в отсутствии доступа источников света и при охлаждении до 0оС. Выход продукта димеризации на данной стадии составляет 46%. После достаточно простого способа выделения продукта димеризации 11:
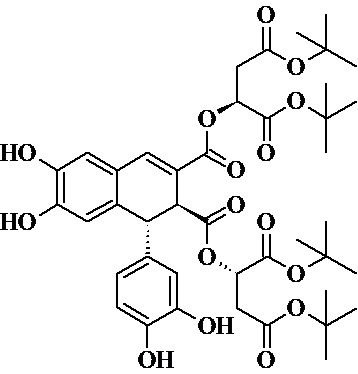
с использованием методики очистки на силикагеле полученный остаток подвергают обработке смесью ТФУ – вода (4:1) при нагревании до 50оС в течение одного часа с целью снятия третбутильных защит с карбоксильных групп (ФИГ. 4).
Далее полученную реакционную смесь упаривают и подвергают очистке при помощи обращеннофазной хроматографии. В результате получают необходимый продукт –s590 (ФИГ. 3) с суммарным выходом 11% и чистотой 98%.
Заявленный способ получения s590 не позволяет получать активные молекулы EA и s414 для которых предлагается использовать другой способ. Способ получения соединения ЕА состоит из трех стадий с суммарным выходом 13%. Соединение s414 является ключевым промежуточным продуктом синтеза эпифиловой кислоты.
Первая стадия синтеза включает в себя получение этилового эфира кофейной кислоты,
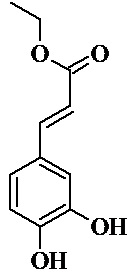
согласно стандартной процедуре этерификации кофейной кислоты в этаноле в присутствии каталитического количества серной кислоты (ФИГ. 5):
Ключевой стадией и одновременно стадией получения активного соединения s414 является окислительная димеризация по методике, описанной ранее в патенте (RU2698725) для получения молекулы севанола (ФИГ. 5). Выход синтеза молекулы s414 на данной стадии окислительного сочетания составляет 37%. Последующее снятие этильных групп с s414 приводит к образованию ЕА. После обработки ключевого промежуточного соединения s414 4М раствором HCl в диоксане в течение 16 часов при температуре 100оС, упаривании на водоструйном насосе и последующей очистки при помощи обращеннофазной препаративной хроматографии получают необходимое соединение ЕА с выходом 37% и чистотой 98% (ФИГ. 6).
Заявляемые соединения, получаемые вышеперечисленными способами и соответствующие представленной формуле (I), проявляют анальгетическую и противовоспалительную активность на млекопитающих, что показывается в тестах на мышах линии ICR: как для модели стимуляции «уксусных корчей» (ФИГ. 7-9), так и на модели тепловой гиперчувствительности после введения адъюванта Фрейнда (ФИГ. 10-14). В этих тестах анальгетическая и противовоспалительная активность заявляемых соединений проявляется при использовании парентеральных способов введения: внутривенном (ФИГ. 7), внутримышечном (ФИГ. 10) или при использовании неинвазивных способов применения: перорально (ФИГ. 8,11), интраназально (ФИГ. 9,12).
Заявляемые соединения оказывают выраженный блокирующий эффект на пиковый ток через протон-активируемые ионные каналы типа ASIC1a и ASIC3 (ФИГ. 15), возникающий после применения быстрого закисления внешнего раствора клеток, в которых эти каналы гетерологически экспрессируются.
Заявляемые соединения могут быть использованы как лекарственные средства или как их компоненты для одиночного применения или в комбинации с другими компонентами для облегчения патологических состояний, обусловленных излишней активностью ионных каналов подтипа ASIC1a и ASIC3, которые широко распространены в центральной и периферической нервной системах, и которые отвечают за возникновение болевых синдромов разной этиологии, развитие воспалительных реакций, а также за инсульт-индуцированные повреждения, нейродегенеративные заболевания, фибромиалгию, синдром раздраженной толстой кишки, острый и хронический кашель, абстинентный синдром, депрессию, тревожное состояние и некоторые другие.
Изобретение иллюстрируют фигуры:
ФИГ. 1. Пространственная структура анальгетических соединений: (А) эпифиловой кислоты, (Б) s414, (В) s590, (Г) Севанола.
ФИГ. 2. Синтез ди-третбутилового эфира (L)-яблочной кислоты.
ФИГ. 3. Синтез хлорангидрида бис-ацетилкоричной кислоты.
ФИГ. 4. Заключительный этап синтеза соединения s590.
ФИГ. 5. Синтез соединения s414.
ФИГ. 6. Синтез эпифиловой кислоты (ЕА).
ФИГ. 7. Исследование влияния анальгетических соединений на внутрибрюшинное введение уксусной кислоты в животной модели «Уксусные корчи» n=7 для всех образцов кроме s414, где n=8. По оси ординат приводят среднее значение и стандартное отклонение для количества корчей в группе, измеренное сразу после введения кислоты за 15 минутный интервал наблюдения. Внутривенное введение соединений в трех дозах: 1, 0,1 и 0,01 мг/кг, проводят за 30 минут до начала измерения, контрольной группе вводят физраствор. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, получавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 8. Исследование влияния анальгетических соединений на внутрибрюшинное введение уксусной кислоты в животной модели «Уксусные корчи» n=8. По оси ординат приводят среднее значение и стандартное отклонение для количества корчей в группе, измеренное сразу после введения кислоты за 15 минутный интервал наблюдения. Пероральное введение соединений в трех дозах: 1, 0,1 и 0,01 мг/кг, проводят за 30 минут до начала измерения, контрольной группе вводят физраствор. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, получавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 9. Исследование влияния анальгетических соединений на внутрибрюшинное введение уксусной кислоты в животной модели «Уксусные корчи» n=8 для всех образцов кроме s590, где n=7. По оси ординат приводят среднее значение и стандартное отклонение для количества корчей в группе, измеренное сразу после введения кислоты за 15 минутный интервал наблюдения. Интраназальное введение соединений в трех дозах: 1, 0,1 и 0,01 мг/кг, проводят за 30 минут до начала измерения, контрольной группе вводят физраствор. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, получавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 10. Исследование влияния анальгетических соединений на тепловую гиперчувствительность, вызванную введением полного адьюванта Фрейнда (ПАФ), n=8. По оси ординат приводят среднее значение и стандартное отклонение для времени отдергивания соответствующей лапы от горячей пластины (t=530C) в секундах. Внутримышечное введение анальгетических соединений в дозах 1, 0,1 и 0,01 мг/кг или физраствора проводят за 30 минут до начала измерения. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, получавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 11. Исследование влияния анальгетических соединений на тепловую гиперчувствительность, вызванную введением полного адьюванта Фрейнда (ПАФ), n=8. По оси ординат приводят среднее значение и стандартное отклонение для времени отдергивания соответствующей лапы от горячей пластины (t=530C) в секундах. Пероральное введение анальгетических соединений в дозах 1, 0,1 и 0,01 мг/кг или физраствора проводят за 30 минут до начала измерения. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, получавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 12. Исследование влияния анальгетических соединений на тепловую гиперчувствительность, вызванную введением полного адьюванта Фрейнда (ПАФ), n=8 для всех образцов кроме s590, где n=6. По оси ординат приводят среднее значение и стандартное отклонение для времени отдергивания соответствующей лапы от горячей пластины (t=530C) в секундах. Интраназальное введение анальгетических соединений в дозах 0,5, 0,05 и 0,005 мг/кг или физраствора проводят за 120 минут до начала измерения. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, получавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 13. Противовоспалительный эффект S706 в тесте воспаления, вызванном введением ПАФ, n=8. По оси ординат приводят среднее значение и стандартное отклонение воспалительного отека лапы, рассчитанного как изменение диаметра конечности до введения ПАФ к наблюдаемой в процентах. Изменение диаметра лапы контролируют через 2, 4 и 24 часа после перорального приема препарата в дозах 1, 0,1 и 0,01 мг/кг. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, принимавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 14. Противовоспалительный эффект EA в тесте воспаления, вызванном введением ПАФ, n=8. По оси ординат приводят среднее значение и стандартное отклонение воспалительного отека лапы, рассчитанного как изменение диаметра конечности до введения ПАФ к наблюдаемой в процентах. Изменение диаметра лапы контролируют через 2, 4 и 24 часа после перорального приема препарата в дозах 1, 0,1 и 0,01 мг/кг. Статистически значимые различия: * p<0,01; ** p<0,005; *** p<0,001 рассчитывают относительно контрольной группы, принимавшей физраствор, (ANOVA-1, Duncan test).
ФИГ. 15. Ингибирующая активность на экспрессированных каналах приведена как зависимость величины амплитуды регистрируемого тока, в присутствии различных концентраций s706, s590, s414 и EA молекул, к контрольному току в процентах через каналы ASIC1a (панель А) и ASIC3 (панель Б). Специфические токи через гетерологически экспрессированные ионные каналы ASIC1a и ASIC3 получают быстрым закислением внешнего раствора с рН 7,4 до 5,5; потенциал удержания составляет -50 мВ. Кривая строится подгонкой с помощью логистического уравнения. Каждая точка представляет собой среднее значение ± SEM для 5–6 измерений.
Изобретение иллюстрируют примеры.
Пример 1.
Получение О-трет-бутилдиизопропилизомочевины (3). М=228, m = 16,5 г (бесцветная жидкость)
В круглодонную колбу объемом 500 мл помещают соединение 2 (0,08 моль, 10,1 г) и 0,01 эквивалента CuCl (0,56 ммоль, 55 мг), затем добавляют 1 эквивалент трет-бутилового спирта 1 (0,08 моль, 5,9 г) и оставляют перемешиваться при комнатной температуре в течение 72 часов. Полученную реакционную смесь фильтруют через пористый стеклянный фильтр и перегоняют при температуре 70-75оС/15 мм. рт. ст. В результате перегонки получают 16,5 г целевого соединения О-трет-бутилдиизопропилизомочевины 3 с выходом 83%.
Пример 2.
Получение бис-трет-бутилового эфира (L)-яблочной кислоты (5): М=248, m = 3 г (бесцветное масло)
В круглодонную колбу объемом 500 мл, снабженную капельной воронкой и внутренним термометром помещают соединение 4 (0,014 моль, 1,9 г), прибавляют 500 мл хлористого метилена. Реакционную смесь охлаждают в ледяной бане до температуры 0-5оС и постепенно прикапывают 5 эквивалентов соединения 3 (0,072 моль, 16,5 г) растворенного в хлористом метилене (120 мл). Реакционную смесь отогревают до комнатной температуры и перемешивают в течение 16 часов. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:4. Далее выпавший осадок фильтруют, маточный раствор упаривают, остаток очищают на силикагеле в системе этилацетат-гексан в соотношении 1:4. Целевое соединение 5 получают с выходом 71% (2,4 г).
Пример 3.
Получение 3,4-бисацилкоричной кислоты (7): М=264, m = 18,7 г (порошок белого цвета)
В круглодонную колбу объемом 500 мл помещают 3,4-дигидроксикоричную кислоту 6 (0,08 моль, 14,4 г) и 120 мл хлористого метилена, охлаждают до -20оС, затем добавляют 5 эквивалентов ацетилхлорида (0,4 моль, 28,5 мл) и при помощи капельной воронки постепенно прикапывают 10 эквивалентов пиридина (0,8 моль, 64,4 мл). Отогревают до комнатной температуры и далее перемешивают в течение 16 часов. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:2. Полученную реакционную смесь упаривают на роторном испарителе, растворяют полученный остаток в хлористом метилене (100 мл), обрабатывают охлажденной льдом разбавленной 1М соляной кислотой до рН = 4. Органический слой сушат Na2SO4 и упаривают на роторном испарителе. Затем добавляют гексан (50 мл), перемешивают 10 минут и полученный осадок фильтруют через пористый стеклянный фильтр. В результате перекристаллизации получают целевой бис-ацетил-защищенный продукт 7 с выходом 91% (18,7 г).
Пример 4.
Получение хлорангидрида 3,4-бисацилкоричной кислоты (8): М=282,5, m = 19,8 г (светло-розовый порошок)
В круглодонную колбу объемом 500 мл помещают соединение 7 (0,07 моль, 18,7 г) и толуол (150 мл). Далее при помощи капельной воронки, постепенно прикапывают 1,2 эквивалента тионилхлорида (0,084 моль, 6,1 мл). Добавляют каплю ДМФ (10 мкл) в качестве катализатора, снабжают колбу обратным холодильником с отводом газа и перемешивают при температуре 80оС в течение 2 часов. Затем реакционную смесь упаривают на роторном испарителе. Целевое соединение 8 получено количественно. (19,8 г).
Пример 5.
Получение эфира бис-трет-бутилового эфира (L)-яблочной кислоты 3,4-бисацилкоричной кислоты (8): М=492 m = 3,8 г (порошок белого цвета)
В круглодонную колбу объемом 250 мл, снабженную капельной воронкой, помещают соединение 5 (9,75 ммоль, 2,4 г), хлористый метилен (20 мл) и добавляют 1,2 эквивалента пиридина (11,7 ммоль, 1 мл). Полученную реакционную смесь охлаждают в ледяной бане до 0-5оС и постепенно прикапывают растворенное в хлористом метилене (200 мл) соединение 8 (10,7 ммоль, 3 г). Перемешивают в течение 4 часов. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:4. Затем реакционную смесь обрабатывают 1М соляной кислотой до рН=3. Полученный органический раствор сушат Na2SO4 и упаривают на роторном испарителе. Далее соединение выделяют при помощи прямофазной силикагельной хроматографии в системе этилацетат-гексан в соотношении 1:4. Целевое соединение 8 получают с выходом 79% (3,8 г).
Rf (33% ЭА/гексан) 0.3;1H ЯМР (600 MHz, CDCl3) δH: 7.69 (1H, д, J = 16.0 Hz), 7.42 (1H, дд, J = 8.4, 2.1 Hz), 7.37 (1H, д, J = 2.1 Hz), 7.24 (1H, д, J = 8.4 Hz), 6.45 (1H, д, J = 16.0 Hz), 5.45 (1H, дд, J = 7.9, 4.7 Hz), 2.88 – 2.80 (2H, м), 2.32 (3H, с), 2.32 (3H, с), 1.49 (9H, с), 1.47 (9H, с);13C ЯМР (600 MHz, CDCl3): 167.9, 167.5, 167.5, 164.9, 143.4, 143.1, 141.9, 132.64, 126.02, 123.5, 122.3, 117.8, 82.2, 81.1, 68.8, 37.1, 27.5, 27.5, 20.1, 20.1; ВРМС: MH+ теор. C25H33O10 493.2068 (MH)+: фактическая 493.2065.
Пример 6.
Получение эфира бис-трет-бутилового эфира (L)-яблочной кислоты 3,4-дигидроксикоричной кислоты (10): М=408 m = 2,7 г (масло светло-желтого цвета)
В круглодонную колбу объемом 250 мл помещают соединение 9 (7,7 ммоль, 3,8 г) и растворяют в ТГФ (50 мл). Реакционную смесь охлаждают в ледяной бане до 0-5оС и затем прикапывают при помощи капельной воронки 2,2 эквивалента N-метилпиперазина (17 ммоль, 2 мл), растворенного в ТГФ (10 мл). Реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:4. Далее упаривают ТГФ на роторном испарителе до объема ~ 10 мл, добавляют этилацетат (30 мл) и обрабатывают 1М соляной кислотой до рН=4. Органический раствор сушат Na2SO4 и упаривают на роторном испарителе. Далее соединение выделяют при помощи прямофазной силикагельной хроматографии в системе этилацетат-гексан в соотношении 1:4. Целевое соединение 10 получают с выходом 85% (2,7 г).
Rf (33% ЭА/гексан) 0.25;1H ЯМР (600 MHz, CDCl3) δH: 7.53 (1H, д, J = 15.9 Hz), 7.00 (1H, д, J = 1.7 Hz), 6.87 (1H, дд, J = 8.2, 1.8 Hz), 6.84 (1H, д, J = 8.2), 6.71 (1H, с, OH), 6.28 (1H, с, OH), 6.17 (1H, д, J = 15.9 Hz), 5.45 (1H, дд, J = 6.7, 5.8 Hz), 2.89 – 2.84 (2H, м), 1.52 (9H, с), 1.49 (9H, с);13C NMR (600 MHz, CDCl3) δ: 168.5, 165.9, 146.2, 145.9, 143.7, 126.7, 121.9, 114.9, 113.9, 113.4, 82.8, 81.6, 68.5, 37.1, 27.5, 27.5; ВРМС: MH+ теор. C21H29O8 409.1857 (MH)+: фактическая 409.1861.
Пример 7.
Получение бис-эфира эпифиловой кислоты и бис-третбутилового эфира (L)-яблочной кислоты (11): М=814 (масло темно-желтого цвета), m = 1.2 г
В круглодонную колбу объемом 250 мл, снабженную капельной воронкой и помещенную в темное помещение при температуре 5оС, помещают соединение 10 (6,6 ммоль, 2,7 г) и ацетонитрил (27 мл). При температуре не выше 5оС и отсутствии света по каплям прибавяют 10% водный раствор 2,5 эквивалентов FeCl3(16,5 ммоль, 2,7 г в 27 мл воды). Перемешивают при данной температуре 2 часа. Далее реакционную смесь обрабатывают 1М соляной кислотой до рН=4 и экстрагируют толуолом (50мл). Полученный раствор в толуоле сушат Na2SO4 и упаривают на роторном испарителе при температуре не выше 40оС в отсутствии света. Полученный остаток очищают на силикагеле в системе этилацетат-толуол = 4:1 с 4% уксусной кислоты. Целевое соединение 11 получают с выходом 46% (1,2 г). Соединение 11 используется в следующей стадии без дополнительной процедуры очистки.
Пример 8.
Получение s590: М=706, m = 1,5 г (стеклообразная масса коричневого цвета)
В круглодонную колбу объемом 50 мл, снабженную обратным холодильником, помещают соединение 11 (1,5 ммоль, 1,2 г) и добавляют смесь ТФУ-вода в соотношении 4:1 (20 мл ТФУ : 5 мл вода). Перемешивают при температуре 50оС в течение 45 минут. Полученную реакционную смесь упаривают на роторном испарителе при температуре не выше 40оС. Полученный остаток массой 1,5 г очищают на препаративной ВЭЖХ.
Хроматографирование s590 осуществляют на колонке LPS-500 (13 мкм, 50 х 250 мм) на приборе Waters Prep LC 2000. Водный раствор s590 (1,5 г в 6 мл воды) наносят на колонку с помощью одного из насосов. Хроматографию проводят в следующих условиях (буфер А – 0,1% трифторуксусная кислота в воде, буфер В – ацетонитрил):
1) 2% буфера В - 5 мин. со скоростью 10 мл/мин,
2) линейный градиент от 10% до 60% буфера В за 60 мин. со скоростью 10 мл/мин.
После очистки и лиофильной сушки фракций получают соединение s590 с выходом 25% в количестве (275 мг).
Пример 9.
Подтверждение структуры s590:
Достоверность структуры полученного соединения подтверждают данными спектроскопии ЯМР1H и13С.
1H ЯМР (600 MHz, D2O) δH: 7.69 (1H, дд, J = 5.3, 2.1 Hz), 6.92 (1H, д, J = 2.3 Hz), 6.65 (1H, д, J = 2.1 Hz), 6.61 (1H, дд, J = 23.3, 2.3 Hz), 6.55 (1H, дд, J = 6.3, 2.2 Hz), 6.37 (1H, дд, J = 8.4, 2.0 Hz), 5.34 (1H, дд, J = 7.0, 4.2 Hz), 5.25 (1H, дд, J = 7.4, 3.7 Hz), 4.46 – 4.37 (1H, м), 4.03 – 3.94 (1H, м), 2.98 – 2.88 (2H, м), 2.84 – 2.73 (2H, м);13C ЯМР (600 MHz, D2O) δ: 173.4, 172.7, 173.0, 166.9, 147.3, 143.9, 143.2, 142.8, 141.1, 134.8, 134.4, 130.7, 123.6, 119.8, 119.5, 117.3, 116.5, 116.3, 116.1, 115.3, 115.1, 69.4, 69.2, 46.5, 46.4, 44.5, 44.2, 35.7, 35.4; ВРМС: MH+ теор. C26H23O16591.0980 (MH)+: фактическая 591.0982.
Пример 10.
Получение этилового эфира 3,4-дигидроксикоричной кислоты (13): М=208, m=11,5 г (осадок белого цвета)
В круглодонную колбу объемом 500 мл, снабженную обратным холодильником, помещают соединение 6 (0,055 моль, 10 г) и абсолютный этанол (200 мл). К полученному раствору добавляют каталитическое количество H2SO4 (100 мкл) и перемешивают реакционную смесь при температуре 80оС в течение 16 часов. Затем упаривают на роторном испарителе этанол до объема ~ 30 мл, добавляют этилацетат (80 мл) и водный раствор NaHCO3 (75 мл). Органический слой сушат Na2SO4 и упаривают на роторном испарителе. Целевое соединение 13 получено количественно (11,5 г).
Пример 11.
Получение этилового эфира эпифиловой кислоты s414: M=414, m=0,75 г (осадок светло-желтого цвета)
В круглодонную колбу объемом 100 мл, снабженную капельной воронкой и помещенную в темное помещение при температуре 5оС, помещают соединение 13 (9.8 ммоль, 2 г) и ацетонитрил (30 мл). При температуре не выше 5оС и отсутствии света по каплям прибавляют 10% водный раствор 2,5 эквивалентов FeCl3 (24,5 ммоль, 4 гр в 40 мл воды). Перемешивают при данной температуре 2 часа. Далее реакционную смесь обрабатывают 1М соляной кислотой до рН=4 и экстрагируют толуолом (25 мл). Полученный раствор в толуоле сушат Na2SO4 и упаривают на роторном испарителе при температуре не выше 40оС в отсутствии света. Полученный остаток очищают на силикагеле в системе этилацетат-гексан = 1:1. Целевое соединение s414 получают методом перекристаллизации с выходом 37% (0,75 г).
Пример 12.
Подтверждение структуры s414:
Достоверность структуры полученного соединения подтверждают данными спектроскопии ЯМР1H и13С.
Rf (50% ЭА/гексан) 0.3;1H ЯМР (600 MHz, acetone-d6) δH: 7.57 (1H, с), 6.96 (1H, с), 6.71 (1H, д, J = 8.1 Hz), 6.63 (1H, с), 6.51 (1H, д, J = 2.2 Hz), 6.46 (1H, дд, J = 8.2, 2.2 Hz), 4.44 (1H, д, J = 3.3 Hz), 4.20 – 4.12 (2H, м), 4.09 – 3.99 (2H, м), 3.89 (1H, д, J = 3.2 Hz), 1.26 (3H, т, J = 7.1 Hz), 1.13 (3H, т, J = 7.1 Hz);13C ЯМР (600 MHz, acetone-d6) δ: 147.2, 144.7, 144.1, 143.7, 137.1, 135.2, 130.0, 123.9, 122.6, 118.8, 116.1, 115.8, 115.0, 114.6, 60.3, 59.9, 47.6, 45.4, 29.4, 29.3, 29.1, 13.6, 13.5; ВРМС: MH+ теор C22H23O8 415.1387 0980 (MH)+: фактическая 415.1390.
Пример 13.
Получение эпифиловой кислоты EA: М=358, m=0,24 г (осадок светло-желтого цвета)
В круглодонную колбу объемом 100 мл помещают соединение s414 (0,75 г, 1,8 ммоль) и 40 мл диоксана. Далее к реакционной смеси добавляют 20 мл 4М HCl в диоксане и перемешивают при температуре 100оС в течение 16 часов. Полученный раствор упаривают на роторном испарителе и получают остаток массой 0,5 г, который далее претерпевает очистку на препаративной ВЭЖХ.
Хроматографирование ЕА осуществляют на тандеме из двух колонок 11AD2 11 микрон и LPS-500 70 микрон (20 х 250 мм) на приборе Waters 515. Хроматографию проводят в следующих условиях (буфер А – 0,2% трифторуксусная кислота в воде, буфер В – этанол):
линейный градиент от 20% до 60% буфера В за 60 мин. со скоростью 10 мл/мин.
После очистки и лиофильной сушки фракций получают соединение ЕА с выходом 37 % (0,24 г).
Пример 14.
Подтверждение структуры ЕА:
Достоверность структуры полученного соединения подтверждают данными спектроскопии ЯМР1H и13С.
1H ЯМР (600 MHz, CDCl3) δH: 7.64 (1H, с), 6.94 (1H, с), 6.68 (1H, д, J = 8.3 Hz), 6.65 (1H, с), 6.59 (1H, д, J = 2.2 Hz), 6.43 (1H, дд, J = 8.3, 2.2 Hz), 4.41 (1H, д, J = 3.2 Hz), 3.83 (1H, д, J = 3.1 Hz);13C NMR (600 MHz, CDCl3) δ: 170.0, 146.4, 143.4, 142.7, 142.3, 139.1, 134.8, 130.0, 123.5, 121.4, 119.1, 116.5, 115.9, 115.6, 114.7, 46.7, 44.3; ВРМС: MH+ теор C18H15O8359.0761 (MH)+: фактическая 359.0762.
Пример 15.
Тестирование анальгетической активности в тесте кислотной стимуляции боли.
Тесты проводят на самцах мышей ICR массой 20-30 г. Мышей делят на группы по 7-8 в каждой. Анальгетические соединения растворяют в стерильном физиологическом растворе и вводят по 100 мкл раствора внутривенно, перорально или интраназально за 30 мин до введения 0,75% раствора уксусной кислоты. Используют три дозы препарата 1, 0,1 и 0,01 мг/кг. Для контрольной группы животных вводят просто 100 мкл физиологического раствора (ФИГ. 7-9). Анальгетический эффект определяют на основании подсчета корчей, вызываемых внутрибрюшинной инъекцией 100 мкл раствора 0,75% уксусной кислоты за 15 минут наблюдения. Корчи – специфическая болевая реакция, сопровождающаяся характерными движениями животных, которые включают сокращения брюшных мышц, чередующиеся с их расслаблением, вытягивание задних конечностей и изгибание спины. Результаты обрабатывают статистически, достоверность отличий результатов контрольной и экспериментальной группы определяют с помощью ANOVA и теста Дункана.
Анальгетический эффект измеряют по снижению болевой чувствительности к висцеральному и воспалительному типу боли, что выражается в снижении количества корчей за измеряемый промежуток времени. При парентеральном способе введения в варианте внутривенной инъекции (ФИГ. 7) все изучаемые молекулы достоверно вызывают анальгезию у животных в дозе 1 и 0,1 мг/кг, а в дозе 0,01 мг/кг анальгезию вызывают все молекулы, кроме s706. В абсолютном значении количество корчей наиболее интенсивно снижают две молекулы – s706 и EA. При неинвазивном способе введения наиболее интенсивно снижает количество корчей также s706 и молекула s590, а наименьшую активность проявляет молекула s414, как при пероральном (ФИГ. 8), так и при интраназальном (ФИГ. 9) способе введения. При неивазивном введении группы с дозами 1 и 0,1 мг/кг для всех веществ, кроме s414 интраназальное введение, достоверно отличаются по наблюдаемому эффекту от контрольной группы.
Пример 16.
Тестирование анальгетической активности в тесте тепловой гиперчувствительности.
Тесты проводят на мышах ICR массой 20-30 г. Мышей делят на группы по 6 или 8 в каждой. Воспаление у мышей вызывают введением в подушечку задней лапы воспалительного агента, представляющего собой 20 мкл смеси полного адьюванта Фрейнда/физраствор 1:1 (v/v). Через 24 часа анальгетические соединения растворяют в стерильном физиологическом растворе и вводят по 100 мкл раствора внутримышечно, перорально или интраназально, а также вводят 100 мкл физиологического раствора животным группы «физ. р-р» аналогичным методом введения. Для анальгетических соединений изучают три дозы. Измерение проводят через 30 мин после введения образцов, кроме способа интраназального введения, для которого время увеличивают до 120 мин. Фиксируют латентное время отдергивания лапы, подвергшейся действию воспалительного агента, от горячей пластины (t=530C). Результаты обрабатывают статистически, достоверность отличий результатов контрольной и экспериментальной группы определяют с помощью ANOVA и теста Дункана.
Анальгетический эффект измеряют по увеличению времени, прошедшего от момента посадки животного на пластину до момента отдергивания воспаленной лапы (ФИГ. 10-12). При парентеральном способе введения в варианте внутримышечной инъекции (ФИГ. 10) s414 достоверно вызывают анальгезию у животных в дозах 1 - 0,01 мг/кг с одинаковым количественным показателем, тогда как s706 демонстрирует дозо-зависимое падение активности, так что в дозе 0,01 мг/кг анальгезию уже не вызывает. При пероральном (ФИГ. 11) способе введения эти вещества ведут себя сходным образом, а ещё одно соединение EA показывает колоколообразную зависимость анальгетической активности с максимумом в дозе 0,1 мг/кг. При этом как s414, так и EA достоверно отличаются от контроля во всех используемых дозах. Дозы для молекул при интраназальном (ФИГ. 12) способе введения уменьшают в два раза. В наибольшей дозе 0,5 мг/кг все изучаемые вещества достоверно проявляют анальгетический эффект (кроме s590 для которого активная доза 0,05 мг/кг). s706 и EA демонстрируют дозо-зависимое падение активности и неактивны в двух более низких дозах. s414 в случае интраназального введение снова достоверно вызывает анальгезию у животных во всех дозах 0,5 - 0,005 мг/кг с одинаковым количественным показателем.
Пример 17.
Тестирование противовоспалительной активности соединений в тесте воспаления, вызванном введением CFA.
Тесты проводят на мышах ICR массой 20-30 г. Мышей делят на 4 группы по 8 в каждой. Воспаление у мышей вызывают введением в подушечку задней лапы воспалительного агента, представляющего собой 20 мкл смеси полного адьюванта Фрейнда/физраствор 1:1 (v/v). Через 24 часа анальгетические соединения растворяют в стерильном физиологическом растворе и вводят по 100 мкл раствора перорально в желудок с помощью зонда в трех дозах: 1, 0,1 и 0,01 мг/кг, а 4 группе животных вводят 100 мкл физиологического раствора. Измерение диаметра конечности проводят до введения CFA, через 24 часа после введения CFA перед введением образцов, после введения образцов через 2, 4 и 24 часа (ФИГ. 13,14). Результаты обрабатывают статистически, достоверность отличий результатов контрольной и экспериментальной группы определяют с помощью ANOVA и теста Дункана.
Две наиболее активные анальгетические молекулы s706 и EA показывают достоверное уменьшение воспалительного отека у мышей через сутки относительно группы, употребляющей физраствор. s706 более эффективен в дозе 1 мг/кг и уже неактивен в дозе 0,01 мг/кг, EA достоверно активен в дозе 0,1 мг/кг, а в других дозах показывает слишком большой разброс, но снижает среднее значение по группе.
Пример 18.
Экспрессия каналов ASIC1a и ASIC3 в ооцитах лягушки.
РНК, кодирующую ионный канал ASIC1a и ASIC3, получают при помощи набора реагентов RiboMAX Large Scale RNA Production System (Promega) для чего к 33 мкл расщепленной и очищенной плазмиды, содержащей Т7 промотер перед кодирующей областью гена канала, добавляют 20 мкл 5-кратного буфера для транскрипции (400 мМ HEPES-KOH, 120 мМ MgCl2, 10 мМ спермедина, 200 мМ дитиотриэтола), 20 мкл смеси рибонуклеозидтрифосфатов (25 мМ ATP, CTP, UTP и 2 мМ GTP), 7 мкл Сар-аналога (в концентрации 40 мМ) и 10 мкл Т7 РНК полимеразы (Promega). Реакционную смесь инкубируют 3 часа при 370С. Очистку РНК проводят методом «фенол/хлороформом». Полученные образцы растворяют в воде, анализируют в агарозном гель-электрофорезе и хранят при температуре –80оС несколько недель.
Каналы ASIC1a и ASIC3 получают в результате их экспрессии в мембранах ооцитов лягушки Xenopus laevis. Для этого выделенные из яичника самки ооциты в стадии созревания IV и V обрабатывают коллагеназой типа I или типа II (Sigma-Aldrich, США) в концентрации 1 мг/мл в течение 2 часов для снятия фолликулярной оболочки. Дефолликулированные ооциты помещают в стерильную среду ND96 (NaCl 96 мM, KCl 2 мM, CaCl2 1.8 мM, MgCl2 1 мM, HEPES 5 мM титрованный NaOH до значения pH 7.4) и выдерживают ночь при температуре 15–16°С. Инъекцию 2.5 – 10 нг мРНК каналов ASIC1a и ASIC3 крысы производят под бинокулярным микроскопом МБС-10 (Россия) с помощью микроинъектора Eppendorf 5242 (Германия). После инъекции ооциты хранят в течение 2-3 дней при температуре 17-19°C, а затем до 7 дней при температуре 15°C в ND-96 среде, которую предварительно титруют NaOH до рН 7.4 и в которую дополнительно добавляют антибиотик гентамицин до концентрации 50 мкг/мл и пируват до концентрации 5 мМ.
Пример 19.
Измерение ингибирования ионных токов через ASIC каналы.
Двухэлектродную систему фиксации мембранного потенциала используют для измерения проводимости каналов. Разность потенциалов -50 мВ поддерживают цифровым усилителем GeneClamp 500 (Axon Instruments, CA), сигналы регистрируют с частотой 10 Гц. Микроэлектроды заполняют раствором 3 M KCl. Используют буферный раствор ND-96 (96 мМ NaCl, 2 мМ KCl, 0.1 мМ ВaCl2, 1 мМ MgCl2, HEPES 5мМ титрованный NaOH до значения pH 7,4).
Измерение тока проводят в рабочей камере со свободным объемом 80 мкл. в ламинарном потоке раствора ND96 (pH 7.4) со скоростью 1 мл/мин. ASIC каналы активируют резким изменением рН среды от 7.4 до 5.5 за счет быстрой замены рабочего раствора в камере на раствор со значением pH 5.5, в котором в качестве буфера вместо 5 мМ HEPES применяют 10 мМ MES. Длина импульса изменения значения pH составляет 1 секунду. Для уменьшения неспецифического связывания с каналом навески тестируемых образцов растворяют в буферных растворах, содержащих 0.1% BSA. Аппликацию тестируемого образца начинают за 15 сек до подачи в измерительную камеру активационного буфера. Процент ингибирования наблюдаемых токов вычисляют как отношение пиковой амплитуды тока при аппликации образца к усредненной амплитуде пика контрольных токов до и после аппликации образца. Ингибирование обратимо, что проявляется в полном восстановлении амплитуды вызванных закислением токов через каналы после полной отмывки испытуемого соединения. Всего используют 5-6 различных концентраций образца не менее чем в пяти повторах и строят кривую зависимости ингибирования тока от использованной концентрации (ФИГ. 15). Применяют наибольшую концентрацию образца 3 мМ, при которой наблюдают снижение амплитуды быстрой компоненты тока менее чем до 10% от контроля во всех случаях кроме s414, который ингибирует ток ASIC1a на 50% и не ингибирует ток ASIC3, и EA, который ингибирует ток ASIC3 до 15% от контроля.
На основании кривой определяют значения концентрации, ингибирующей ток на 50% (IC50), и коэффициент Хилла (nH) для каждого типа ионного канала. Для ASIC1a значения IC50 227,5 ± 37,4 мкM и nH 1,6 ± 0,3 получают для молекулы s706; IC50 297 ± 41 мкM и nH1,4 ± 0,1 получают для молекулы s590; IC50 415 ± 66 мкM и nH1,5 ± 0,2 получают для молекулы ЕА; IC50 3,4 ± 1,3 мM и nH0,35 ± 0,04, получают для молекулы s414. Для ASIC3 значения IC50 175 ± 18 мкM и nH 1,7 ± 0,2 получают для молекулы s706; IC50 241 ± 25 мкM и nH1,75 ± 0,15 получают для молекулы s590; IC50 424 ± 33 мкM и nH1,9 ± 0,2 получают для молекулы ЕА.
Таким образом, предлагаемый способ получения молекул, представленных формулой (I), позволяет получать соединения проявляющие сильный обезболивающий (анальгетический) и противовоспалительный эффект на животных. Эти соединения имеют явную клеточную мишень – ионные каналы ASIC, ингибирование проводимости которых в нейронах животных и человека приводит к положительным терапевтическим эффектам. Для достижения положительного эффекта такие молекулы могут применяться перорально, интраназально или парентерально.
Реферат
Изобретение относится к соединению общей формулы (I), которое обладает анальгетическим или противовоспалительным действием. В формуле (I) оба R одинаковы и представляют собой этильную группу или остаток сложного эфира (L)-яблочной кислоты. Изобретение относится также к применению соединений общей формулы (I), где оба R одинаковы и представляют собой водород, этильную группу или остаток сложного эфира (L)-яблочной кислоты, в качестве анальгетического или противовоспалительного средства, в качестве ингибиторов ASIC1a и ASIC3 ионных каналов или в качестве лекарственного средства для профилактики или лечения боли и воспаления. 4 н. и 3 з.п. ф-лы, 15 ил., 19 пр.

Формула

Документы, цитированные в отчёте о поиске
Способ получения лигнана, обладающего анальгетическим действием
Патенты аналоги
Способ получения лигнана, обладающего анальгетическим действием
Комментарии