Способ получения лигнана, обладающего анальгетическим действием - RU2698725C1
Код документа: RU2698725C1
Чертежи



Описание
Изобретение относится к области органической химии, конкретно, к способу получения биологически активного соединения органической природы, обладающего анальгетическим и противовоспалительным действием, а также к способу получения структурных аналогов этого соединения для применения в медицине, ветеринарии или научных исследованиях.
Севанол - это растительный лигнан из экстракта тимьяна Thymus armeniacus, структура которого описывается как: 1,2,3-пропантрикарбоксиловая кислота, 1,1'-[[(1R,2S)-1-(3,4-дигидроксифенил)-1,2-дигидро-6,7-дигидрокси-2,3-нафталенедиил]бис(карбонилокси)]бис-,(1S,1'S,2R,2'R)-(+)-; или ICPAC наименование: (1R,2S)-1-[(1R,2S)-1-(3,4-дигидроксифенил)-6,7-дигидрокси-3-[(1R,2S)-1,2,3-трикарбоксипропокси]карбонил-1,2-дигидронафтален-2-карбонил]оксипропан-1,2,3-трикарбоксиловая кислота (ФИГ. 1).
Это соединение имеет ярко выраженный анальгетический и противовоспалительный эффект и может быть выделено хроматографическими методами из уксуснокислого экстракта молодых побегов тимьяна (патент РФ 2491950). Недостаток хроматографического метода выделения из природного сырья заключается в ограниченности запасов молодых побегов лекарственного растения Thymus armeniacus (регион сбора высокогорное озеро Севан, Армения), и невозможности использования других видов тимьяна из других регионов, так как эти растения не синтезируют эту молекулу.
Севанол - природная молекула, построенная на основе эпифиловой кислоты, с расчетной средней молекулярной массой 706,52 Да содержит 6 хиральных центров, причем стереоизомеры молекулы такие как изосеванол (ФИГ. 2) также обладают анальгетической активностью и могут быть использованы как анальгетические препараты (Д.И. Осмаков, С.Г. Кошелев, О.А. Белозерова, В.С. Кублицкий, Я.А. Андреев, Е.В. Гришин, С.А. Козлов Биологическая активность севанола и его аналогов // Биоорганическая химия, 2015, т 41, с. 606-611).
Предложенный способ синтеза заключается в параллельном получении три-трет-бутилового эфира изолимонной кислоты и защищенной кофейной кислоты, с последующим синтезом соответствующего эфира дигидроксикоричной кислоты. Полученный эфир кофейной кислоты впоследствии димеризуется в окислительных условиях, образующийся продукт димеризации, содержащий защищенные карбоксильные группы, подвергается кислотно-катализируемому деблокированию с образованием целевого продукта - севанола.
Первая ключевая стадия данного подхода заключается в синтезе трет-бутилового эфира изолимонной кислоты за три стадии.
Известен способ получения трет-бутилового эфира изолимонной кислоты с общим выходом 33%, включающий в себя 7 синтетических стадий из исходной (L)-яблочной кислоты, основанный на методе, описанном в статье (F. Calo, J. Richardson, А. Barrett, Total Synthesis of Citrafungin A //, J. Org. Chem., 2008, v. 73, p. 9692-9697). Основным недостатком данного способа является многостадийность синтеза и использование большого количества дорогостоящих реагентов.
Вторая ключевая стадия синтеза целевой молекулы севанола заключается в окислительной димеризации сложного эфира дигидрокоричной кислоты (кофейной кислоты).
Известно несколько способов проведения данного процесса, которые разнятся выбором окислителя. Предложено использовать такие окислительные реагенты, как: MnO2 (С. Daquino, A. Rescifina, С. Spatafora, С. Tringali, Biomimetic Synthesis of Natural and "Unnatural" Lignans by Oxidative Coupling of Caffeic Esters // Eur. J. Org. Chem, 2009, p. 6289-6300), K3[Fe(CN)6] (S. Maeda, H. Masuda, T. Tokoroyama, Studies on the Preparation of Bioactive Lignans by Oxidative Coupling Reaction II. Oxidative Coupling Reaction of Methyl (E)-3-(4,5-Dihydroxy-2-methoxyphenyl)propionate and Lipid Peroxidation Inhibitory Effect of the Produced Lignans // Chem. Pharm. Bull., 1994, v. 42, p. 2506-2513), ферментные системы (H. Takahashi, K. Matsumoto, M. Ueda, Y. Miyake, Y. Fukuyama, Biomimetic Synthesis of Neurotrophic Americanol A and Isoamericanol A by Horseradish Peroxidase (HRP) Catalysed Oxidative Coupling // Heterocycles, 2002, v. 56, p. 245-256). Основным недостатком использования этих окислительных реагентов является образование большого количества побочных продуктов, и, как следствие, низкий выход целевого продукта димеризации.
Наиболее близким к предлагаемому изобретению является способ с использованием FeCl3 (хлорида железа(III)) в водно-ацетоновом растворе, для окисления производных кофейной кислоты, описанный в работах (D.E. Bogucki, J.L. Charlton, A non-enzymatic synthesis of (S)-(-)-rosmarinic acid and a study of a biomimetic route to (+)-rabdosiin // Can. J. Chem. 1997, v. 75, p. 1783-1794; Zhi-Hong Jiang, TakashiTanaka, IsaoKouno Two diastereomeric triterpene-lignan esters having dimeric structure and their biosynthetically related triterpene caffeate from Rhoiptelea cheliantha // Tetrahedron Lett, 1994, v. 35, p. 2031-2034; Zhi-Hong Jiang, Takashi Tamaka and Isao Kouno, Chilianthins A-F, six triterpene esters having dimeric structures from Rhoiptelea cheliantha diels et Hand.-Mazz // Chem. Pharm. Bull., 1996, v. 44, p. 1669-1675). Основным недостатком данного способа является сравнительно низкие выходы целевых продуктов реакции 5-30%.
Задачей предлагаемого изобретения является разработка технологичного и малостадийного способа синтеза природного лигнана - севанола, содержащего дигидронафталиновую систему, протекающего с высоким выходом по исходному производному кофейной кислоты.
Техническим результатом предлагаемого изобретения является технологичный и малостадийный способ синтеза природного лигнана - севанола и способ его последующего выделения и очистки с суммарным выходом 2,2% целевого продукта.
Поставленный технический результат достигается в два этапа. Первый этап (ФИГ. 3) связан с разработкой нового способа получения три-трет-бутилового эфира изолимонной кислоты следующей формулы:

Сущностью данного способа является прямое введение трет-бутилацетатного фрагмента в молекулу (L)-яблочной кислоты с карбоксильными группами, защищенными трет-бутиловыми эфирами.
Преимуществом данного способа является высокий выход 61%, одностадийность процесса, простота выделения продукта реакции, малое количество реагентов для синтеза.
Способ заключается в металлировании ди-третбутилового эфира (L)-яблочной кислоты по ранее известной методике (М. F. A. Amer, K. Takahashi, J. Ishihara and S. Hatakeyama, Total synthesis of Citrafungin A// Heterocycles 2007, v. 72, p. 181-185) диизопропиламидом лития (ЛДА) с последующим алкилированием данного субстрата трет-бутиловым эфиром бромуксусной кислоты при температуре -78°С. При этом используется 2.5 кратный избыток литирующего реагента - ЛДА и 1.2 кратный избыток алкилирующего вещества. Обнаруживается, что уменьшение или увеличение количества диизопропиламида лития не улучшает выход продукта реакции, а увеличение или уменьшение температуры реакции приводит к значительному уменьшению выхода реакции.
Промежуточные стадии проводят с использованием стандартных процедур для получения двух основных промежуточных продуктов: эфира кофейной кислоты с три-трет-бутиловым эфиром изолимонной кислоты

и хлорангидрид бисацетикофейной кислоты
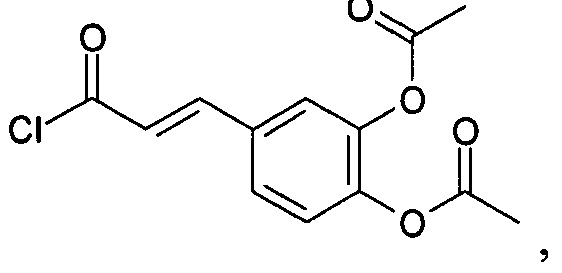
которую получают по способу (М. Sefkow, First efficient synthesis of chlorogenic acid // Eur. J. Org. Chem., 2001, i. 6, p. 1137-1141) (ФИГ. 4).
На стадии реакции ацилирования эфира кофейной кислоты с три-трет-бутиловым эфиром изолимонной кислоты хлорангидридом бисацетикофейной кислоты (ФИГ. 5) в качестве основания используют пиридин, что является ключевым моментом на этом этапе. Необходимым условием проведения синтеза во избежание образования побочных продуктов является проведение реакции при температурах близким к 0°С. При проведении данного процесса синтеза используют охлаждение реакционной смеси и порционное добавление хлорангдрида бисацетилкофейной кислоты, растворенного в хлористом метилене, при помощи капельной воронки. Такая процедура увеличивает эффективность реакции до 82% выхода.
Известен способ снятия ацетильных групп с эфиров кофейной кислоты с использованием метиламина в метаноле как растворителя (Патент WO 2015154721). Для соединения (10 на ФИГ. 5) этот способ дает очень низкий выход продукта, поэтому для удаления ацетильных групп используется N-метилпиперазин в абсолютном ТГФ. Это позволяет получать ключевой интермедиат (11 на ФИГ. 5) с выходом близким к количественному (96%). При масштабировании способа получения эфира три-третбутилового эфира изолимонной кислоты 3,4-дигидроксикоричной кислоты соблюдают температурный режим, а именно, реакцию деблокирования проводят при охлаждении до 0°С, а также исключают доступ кислорода (проведение реакции под аргоном).
Второй принципиальный этап заключается в окислительной димеризации соответствующего эфира кофейной кислоты, с использованием хлорида железа(III) в качестве реагента. Реакция димеризации требует соблюдения условий на предмет количества FeCl3, используемых растворителей и температурного режима проведения реакции. Сущностью способа является реакция сочетания двух молекул производного кофейной кислоты (ФИГ. 5 стадия А8). Преимуществом данного способа является высокий выход 51%, небольшая продолжительность реакции 1 час, простота выделения продукта реакции, а также возможность получения различных производных эпифиловой кислоты, с разнообразной биологической активностью.
Способ заключается во взаимодействии эфира (три-трет-бутилового эфира изолимонной кислоты) кофейной кислоты с хлоридом железа(III), при мольном соотношении реагентов 1:2, в смеси растворителей ацетонитрил-вода в соотношении 4:1, при температуре 5°С, в течение 1 часа, в отсутствии света, с последующей экстракцией продукта реакции толуолом. При уменьшении продолжительности реакции не достигается хороший выход продукта. Увеличение продолжительности реакции является нецелесообразным с экономической точки зрения. Увеличение или уменьшение температуры реакции приводит к значительному уменьшению выхода реакции. Присутствие света приводит к образованию продуктов хлорирования целевого вещества. Оптимальным условием проведения реакции окислительной димеризации является ее осуществление при мольном соотношении производного коричной кислоты к хлориду железа(III), 1:2 соответственно. Меньший избыток FeCl3 приводит к снижению выхода целевого продукта, а увеличение избытка к значительному количеству побочных, трудноотделяемых примесей. Синтезированное соединение после обработки реакционной смеси подвергается очистки на силикагеле. После кислото-катализируемого (80% водная трифторуксусная кислота) деблокирования карбоксильных функций изолимонной кислоты, упаривания и очистки при помощи обращеннофазной хроматографии, получается продукт - севанол с суммарным выходом 2.2% и чистотой 98%.
Севанол проявляет анальгетическую активность на млекопитающих, что показывается в тестах на мышах ICR: как для модели тепловой гиперчувствительности после введения адъювант Фрейнда (ФИГ. 6), так и на модели стимуляции «уксусных корчей» (ФИГ. 7).
Изобретение иллюстрируют фигуры:
ФИГ. 1. Структура севанола.
ФИГ. 2. Структура изосеванола.
ФИГ. 3. Синтез три-трет-бутилового эфира изолимонной кислоты.
ФИГ. 4. Синтез хлорангидрида бис-ацетилкоричной кислоты.
ФИГ. 5. Заключительный этап синтеза севанола.
ФИГ. 6. Исследование влияния севанола на тепловую гиперчувствительность вызванную введением полного адьюванта Фрейнда (CFA) n=8. По оси ординат показано время отдергивания соответствующей лапы от горячей пластины (t=53°C). Внутримышечное введение севанола в различных дозах проводят за 120 минут до начала измерения. Статистически значимые различия от группы контроль отмечены * Р≤0,05 относительно контрольной группы (ANOVA-1, Duncan test).
ФИГ. 7. Исследование влияния севанола на внутрибрюшинное введение уксусной кислоты в животной модели «Уксусные корчи» n=8. По оси ординат показано количество корчей, измеренное сразу после введения кислоты за 15 минутный интервал наблюдения. Внутримышечное введение севанола в различных дозах проводят за 120 минут до начала измерения. Статистически значимые различия: * Р≤0,05 относительно контрольной группы (ANOVA-1, Duncan test); # Р≤0,05 относительно группы «0,1 мг/кг» (ANOVA-1, Duncan test).
Изобретение иллюстрируют примеры.
Пример 1.
Получение О-трет-бутилдиизопропилизомочевины (3). М=228, m=330 гр (бесцветная жидкость)
В круглодонную колбу объемом 1 л помещают соединение 1 (1,65 моль, 208 гр) и 0,01 эквивалента CuCl (20 ммоль, 1 гр), затем добавляют 1 эквивалент трет-бутилового спирта 2 (1,65 моль, 122 гр) и оставляют перемешиваться при комнатной температуре в течение 72 часов. Полученную реакционную смесь фильтруют через пористый стеклянный фильтр и перегоняют при температуре 70-75°С/15 мм. рт.ст. В результате перегонки получают 330 гр целевого соединения О-трет-бутилдиизопропилизомочевины 3 с выходом 83%.
Пример 2.
Получение бис-трет-бутилового эфира (L)-яблочной кислоты (5): М=248, m=60 гр (бесцветное масло)
В круглодонную колбу объемом 2 л, снабженную капельной воронкой и внутренним термометром помещают соединение 4 (0,34 моль, 45 гр), прибавляют 500 мл хлористого метилена. Реакционную смесь охлаждают в ледяной бане до температуры 0-5°С и постепенно прикапывают 5 эквивалентов соединения 3 (1,7 моль, 330 гр) растворенного в хлористом метилене (500 мл). Реакционную смесь отогревают до комнатной температуры и перемешивают в течение 16 часов. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:4. Далее выпавший осадок фильтруют, маточный раствор упаривают, остаток очищают на силикагеле в системе этилацетат-гексан в соотношении 1:4. Целевое соединение 5 получают с выходом 71% (60 гр).
Пример 3.
Получение три-трет-бутилового эфира изолимонной кислоты (6): М=360, m=53 гр (бесцветное масло)
В круглодонную колбу объемом 2 л, снабженную септой, переходом для ввода аргона и низкотемпературным термометром, помещают 2,5 эквивалента диизопропиламина (0,6 моль, 85 мл) растворенного в ТГФ (500 мл). Полученный раствор охлаждают при помощи жидкого азота до -70°С и, используя шприц, через септ постепенно прикапывают 2,5 эквивалента 2,5М бутиллития в гексане (0,6 моль, 240 мл). Реакционную смесь перемешивают при -50°С в течение 30 минут. Далее реакционную смесь охлаждают до -78°С и постепенно прикапывают соединение 5 (0,24 моль, 60 гр), растворенное в ТГФ (200 мл). Реакционную смесь отогревают до -60°С и перемешивают при данной температуре в течение 20 минут. Далее реакционную смесь охлаждают до -78°С и постепенно прикапывают 1,2 эквивалента трет-бутилбромацетата (0,29 моль, 42 мл), растворенного в ТГФ (100 мл). Реакционную смесь отогревают до -20°С и перемешивают при данной температуре в течение 3 часов. Прохождение реакции контролируют по ТСХ в системе этилацетат-гексан 1:9. Полученную реакционную смесь обрабатывают 1М соляной кислотой до рН=3. Полученный органический раствор сушат Na2SO4 и упаривают на роторном испарителе. Целевое соединение выделяют при помощи прямофазной силикагельной хроматографии в системе этилацетат-гексан 1:9. Целевое соединение 6 получают с выходом 61% (53 гр).
Пример 4.
Получение 3,4-бисацилкоричной кислоты (8): М=264, m=37,4 гр (порошок белого цвета)
В круглодонную колбу объемом 500 мл помещают 3,4-дигидроксикоричную кислоту 7 (0,15 моль, 28 гр) и 150 мл хлористого метилена, охлаждают до -20°С, затем добавляют 5 эквивалентов ацетилхлорида (0,77 моль, 55,5 мл) и при помощи капельной воронки постепенно прикапывают 10 эквивалентов пиридина (1,5 моль, 120,7 мл). Отогревают до комнатной температуры и далее перемешивают в течение 16 часов. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:2. Полученную реакционную смесь упаривают на роторном испарителе, растворяют полученный остаток в хлористом метилене (150 мл), обрабатывают охлажденной льдом разбавленной 1М соляной кислотой до рН=4. Органический слой сушат Na2SO4 и упаривают на роторном испарителе. Затем добавляют гексан (100 мл), перемешивают 10 минут и полученный осадок фильтруют через пористый стеклянный фильтр. В результате перекристаллизации получают целевой бис-ацетил-защищенный продукт 8 с выходом 91% (37,4 гр).
Пример 5.
Получение хлорангидрида 3,4-бисацилкоричной кислоты (9): М=282,5, m=40 гр (светло-розовый порошок)
В круглодонную колбу объемом 500 мл помещают соединение 8 (0,14 моль, 37,4 гр) и толуол (300 мл). Далее при помощи капельной воронки, постепенно прикапывают 1,2 эквивалента тионилхлорида (0,17 моль, 12,2 мл). Добавляют каплю ДМФ (20 мкл) в качестве катализатора, снабжают колбу обратным холодильником с отводом газа и перемешивают при температуре 80°С в течение 2 часов. Затем реакционную смесь упаривают на роторном испарителе. Целевое соединение 9 получено количественно. (40 гр).
Пример 6.
Получение эфира три-трет-бутилового эфира изолимонной кислоты 3,4-бисацилкоричной кислоты (10): М=606, m=64,6 гр (порошок белого цвета)
В круглодонную колбу объемом 500 мл, снабженную капельной воронкой, помещают соединение 6 (0,13 моль, 46 гр), хлористый метилен (300 мл) и добавляют 1,2 эквивалента пиридина (0,16 моль, 12,5 мл). Полученную реакционную смесь охлаждают в ледяной бане до 0-5°С и постепенно прикапывают растворенное в хлористом метилене (200 мл) соединение 9 (0,14 моль, 40 гр). Перемешивают в течение 4 часов. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:4. Затем реакционную смесь обрабатывают 1М соляной кислотой до рН=3. Полученный органический раствор сушат Na2SO4 и упаривают на роторном испарителе. Далее соединение выделяют при помощи прямофазной силикагельной хроматографии в системе этилацетат-гексан в соотношении 1:4. Целевое соединение 10 получают с выходом 82% (64,6 гр).
Пример 7.
Получение эфира три-трет-бутилового эфира изолимонной кислоты 3,4-дигидроксикоричной кислоты (11): М=522, m=55 гр (масло светло-желтого цвета)
В круглодонную колбу объемом 1 л помещают соединение 10 (0,11 моль, 64,6 гр) и растворяют в ТГФ (500 мл). Реакционную смесь охлаждают в ледяной бане до 0-5°С и затем прикапывают при помощи капельной воронки 2,2 эквивалента N-метилпиперазина (0,23 моль, 26 мл), растворенного в ТГФ (100 мл). Реакционную смесь перемешивают в течение 4 часов при комнатной температуре. Прохождение реакции контролируют при помощи ТСХ в системе этилацетат-гексан в соотношении 1:4. Далее упаривают ТГФ на роторном испарителе до объема ~100 мл, добавляют этилацетат (300 мл) и обрабатывают 1М соляной кислотой до рН=4. Органический раствор сушат Na2SO4 и упаривают на роторном испарителе. Далее соединение выделяют при помощи прямофазной силикагельной хроматографии в системе этилацетат-гексан в соотношении 1:4. Целевое соединение 11 получают с выходом 96% (55 гр).
Rf (35% ЭА/гексан) 0.3;1Н ЯМР (300 MHz, CDCl3): δН 7.54 (1Н, д, J=15.9 Hz), 6.99 (1Н, с), 6.86 (1Н, с), 6.85 (1H, с), 6.59 (1Н, с, ОН), 6.17 (1Н, д, J=15.9 Hz), 6.03 (1H, с, ОН), 5.31 (1Н, д, J 3.3 Hz), 3.45-3.51 (1Н, м), 2.77 (1H, дд, J=16.8, 9.7 Hz), 2.47 (1H, дд, J 16.8, 5.2 Hz), 1.52 (9H, c), 1.51 (9H, c), 1.48 (9H, с);13C ЯМР (300 MHz, CDCl3): 170.7, 169.5, 167.6, 166.3, 146.6, 146.3, 144.1, 127.1, 122.4, 115.5, 114.4, 113.9, 83.4, 82.3, 81.3, 71.9, 43.9, 34.0, 28.0; ИК (KBr): 1721, 2936, 2980, 3391; BPMC: MH+ теор. C27H39O10 523.2537 (MH)+: фактическая 523.2536.
Пример 8.
Получение бис-эфира эпифиловой кислоты и три-третбутилового эфира изолимонной кислоты (12): М=1042 (масло желтого цвета), m=28 г
В круглодонную колбу объемом 1 л, снабженную капельной воронкой и помещенную в темное помещение при температуре 5°С, помещают соединение 11 (0,1 моль, 55 гр) и ацетонитрил (550 мл). При температуре не выше 5°С и отсутствии света по каплям прибавяют 10% водный раствор 2,5 эквивалентов FeCl3 (0,25 моль, 40,6 гр в 400 мл воды). Перемешивают при данной температуре 2 часа. Далее реакционную смесь обрабатывают 1М соляной кислотой до рН=4 и экстрагируют толуолом (300 мл). Полученный раствор в толуоле сушат Na2SO4 и упаривают на роторном испарителе при температуре не выше 40°С в отсутствии света. Полученный остаток очищают на силикагеле в системе этилацетат-толуол = 4:1 с 3% уксусной кислоты. Целевое соединение 12 получают с выходом 51% (28 гр).
Пример 9.
Получение севанола: М=706, m=15 гр (стеклообразная массы коричневого цвета)
В круглодонную колбу объемом 1 л, снабженную обратным холодильником, помещают соединение 12 (21 ммоль, 22,5 гр) и добавляют смесь ТФУ-вода в соотношении 4:1 (400 мл ТФУ : 100 мл вода). Перемешивают при температуре 50°С в течение 1 часа. Полученную реакционную смесь упаривают на роторном испарителе при температуре не выше 40°С. Полученный остаток массой 15 г очищают на препаративной ВЭЖХ.
Хроматографирование севанола осуществляют на колонке LPS-500 (13 мкм, 50×250 мм) на приборе Waters Prep LC 2000. Водный раствор севанола (5 г в 20 мл воды) наносят на колонку с помощью одного из насосов. Хроматографию проводят в следующих условиях (буфер А - 0,1% трифторуксусная кислота в воде, буфер В - ацетонитрил):
1) 2% буфера В - 5 мин. со скоростью 80 мл/мин,
2) линейный градиент от 2% до 30% буфера В за 60 мин. со скоростью 80 мл/мин.
После очистки и лиофильной сушки фракций получают смесь севанола и изосеванола в соотношении 75% и 25% соответственно с выходом 30% (4,5 гр).
Пример 10.
Очистка севанола от изосеванола.
Разделение этой смеси осуществляют на колонке_Phenomenex (20×250 мм, 10 mkm, 300А, С18) на приборе Waters 515. Водный раствор смеси севанола и изосеванола в соотношении 75% и 25% соответственно (100 мг в 2 мл воды) наносят на колонку с помощью насоса. Хроматографию проводят в следующих условиях:
1) изократика: 7% буфер В за 90 мин. со скоростью 10 мл/мин.
После разделения и лиофильной сушки получают севанол с выходом 75% (75 мг).
Пример 11.
Подтверждение структуры севанола
Достоверность структуры полученного соединения подтверждают данными спектроскопии ЯМР1Н и13С. Химические сдвиги и мультиплетность сигналов в спектрах ЯМР1Н и13С полученного образца севанола полностью совпадают с природным образцом севанола.1Н ЯМР (700 MHz, D2O): δН 7.75 (1H, с), 7.01 (1Н, с), 6.72 (1H, с), 6.68 (1Н, д, J=8.3 Hz), 6.62, (1Н, д, J=1.7 Hz), 6.43 (1H, дд, J=1.7 Hz, 8.3 Hz), 5.38 (1H, д, J=4.0 Hz), 5.33 (1H, д, J=3.4 Hz), 4.5 (1H, д, J=1.9 Hz), 4.08 (1H, д, J=1.9 Hz), 3.56-3.59 (1H, м), 3.45-3.47 (1H, м), 2.78 (1H, дд, J=9.3 Hz, 17.2 Hz), 2.6 (1H, дд, J=5.2 Hz, 17.2 Hz), 2.50 (1H, дд, J=9.6 Hz, 17.3 Hz), 2.24 (1H, дд, J=9.6 Hz, 17.3 Hz);13C ЯМР (300 MHz, D2O): 175.4, 175.2, 174.2, 173.7, 172.8, 172.7, 171.8, 166.9, 147.5, 143.9, 143.3, 142.8, 141.4, 134.5, 130.7, 123.7, 119.8, 119.6, 117.4, 116.6, 116.1, 115.3, 73.2, 73.2, 46.4, 44.0, 43.2, 42.2, 32.3, 31.8; ИК (KBr): 1709, 2648, 3253; MCBP: MH- теор. C30H25O20 705.0939 (MH)-: фактическая 705.0941.
Измеренная молекулярная масса методом хромато-масс-спектрометрии равняется 706.31 Да, что соответствует расчетной массе. Спектр УФ-поглощения имеет два максимума 253 и 344 нм и один минимум 279 нм, что соответствует ранее полученному спектру УФ-поглощения для природной молекулы.
Пример 12.
Тестирование анальгетической активности севанола в тесте тепловой гиперчувствительности.
Тесты проводят на мышах ICR массой 20-30 г. Мышей делят на 4 группы по 8 в каждой. Воспаление у мышей, кроме контрольной группы, вызывают введением в подушечку задней лапы воспалительного агента, представляющего собой 20 мкл смеси полного адьюванта Фрейнда/физраствор 1:1 (v/v). Через 24 часа внутримышечно вводят 100 мкл физиологического раствора животным группы «физ. p-p», остальным группам внутримышечно вводят 100 мкл раствора севанола в дозе 10, 1 и 0,1 мг/кг. Измерение проводят через 120 мин после внутримышечного введения. Фиксируют латентное время отдергивания лапы, подвергшейся действию воспалительного агента, от горячей пластины (t=53°C). Результаты обрабатывают статистически, достоверность отличий результатов контрольной и экспериментальной группы определяют с помощью ANOVA и теста Дункана. Анальгетический эффект измеряют по увеличению времени, прошедшего от момента посадки животного на пластину до момента отдергивания воспаленной лапы (ФИГ. 6).
Пример 13.
Тестирование анальгетической активности севанола в тесте кислотной стимуляции боли.
Тесты проводят на самцах мышей ICR массой 20-30 г. Мышей делят на 4 группы по 8 в каждой. Севанол растворяют в стерильном физиологическом растворе и вводят по 100 мкл раствора внутримышечно за 120 мин до введения 0,75% раствора уксусной кислоты. Используют три дозы препарата 10, 1 и 0,1 мг/кг. Для контрольной группы животных вводят просто 100 мкл физиологического раствора (ФИГ. 7). Анальгетический эффект определяют на основании подсчета корчей, вызываемых внутрибрюшинной инъекцией 100 мкл раствора 0,75% уксусной кислоты за 15 минут наблюдения. Корчи - специфическая болевая реакция, сопровождающаяся характерными движениями животных, которые включают сокращения брюшных мышц, чередующиеся с их расслаблением, вытягивание задних конечностей и изгибание спины. Результаты обрабатывают статистически, достоверность отличий результатов контрольной и экспериментальной группы определяют с помощью ANOVA и теста Дункана. Анальгетический эффект измеряют по снижению болевой чувствительности к висцеральному и воспалительному типу боли, что выражается в снижении количества корчей за измеряемый промежуток времени.
Таким образом, предлагаемый способ получения севанола позволяет получать соединение заявленной структурной формулы с выполнением следующих совокупных условий:
а) целевое соединение получают в девять стадии с суммарным выходом 2,2%.
б) исходные соединения являются продажными реагентами и имеют низкую стоимость.
в) условия проведения реакций отличаются простотой и не требуют специального оборудования.
Реферат
Изобретение относится к области органической химии, преимущественно связанной с синтезом биологически активных природных соединений - лигнанов и их синтетических аналогов. Конкретным конечным продуктом является молекула севанол, которая представляет собой перспективное терапевтическое средство для купирования боли, имеющая общую формулу, приведенную ниже. Заявляемый способ химического синтеза состоит из 9 стадий синтеза, среди которых есть две принципиальные химические реакции: получение три-трет-бутилового эфира изолимонной кислоты, при помощи прямого алкилирования ди-трет-бутилового эфира (L)-яблочной кислоты трет-бутилбромацетатом с использованием диизопропиламида лития в качестве основания при температуре -78°С в тетрагидрофуране и окислительная димеризация соответствующего производного кофейной кислоты при использовании двухкратного избытка хлорида железа(III) в смеси растворителей ацетонитрил-вода в соотношении 4:1, соответственно, при температуре 5°С в отсутствие света в течение 1 часа, что обеспечивает выход конечного продукта после стадии финальной хроматографической очистки на уровне 2,2%. Помимо этого предложенный способ получения приводит к преимущественному образованию полного аналога севанола над изосеванолом 75% к 25% соответственно. Получаемый предлагаемым способом полный аналог севанола в экспериментах in vivo не уступает по биологической активности природной молекуле, выделяемой из природного сырья. Разработанный способ синтеза может быть использован для создания модифицированных аналогов севанола, для получения новых биологически активных лигнанов. 13 пр., 7 ил.
Формула





Комментарии