Аналоги cyp-эйкозаноидов для применения в лечении или предотвращении нарушения, связанного с неоваскуляризацией и/или воспалением - RU2776153C2
Код документа: RU2776153C2
Чертежи





Описание
Настоящее изобретение относится к соединениям в соответствии с общей формулой (I), которые являются метаболически устойчивыми аналогами биологически активных липидных медиаторов, являющихся производными полиненасыщенных жирных кислот омега-3 (ПНЖК n-3), для применения в лечении или снижении риска развития, или предотвращении: (i) неоваскуляризации и/или (ii) воспалительного нарушения, в частности офтальмологических нарушений, связанных с неоваскуляризацией и/или воспалением.
Уровень техники
Полиненасыщенные жирные кислоты омега-6 и омега-3 (ПНЖК n-6 и n-3) являются необходимыми компонентами рациона млекопитающего. Наиболее важными с биологической точки зрения ПНЖК n-3 являются эйкозапентаеновая кислота (ЕРА, 20:5 n-3) и докозагексаеновая кислота (DHA, 22:6 n-3). Поступающие с пищей ПНЖК n-3 влияют на различные физиологические процессы, обуславливающие нормальное состояние здоровья и хронические заболевания, такие как регуляция уровней липидов в плазме, функционирование сердечно-сосудистой и иммунной системы, воспаление, действие инсулина, а также развитие нейронов и зрительная функция.
Прием ПНЖК n-3 будет способствовать их распространению практически в каждую клетку организма, что влияет на состав и функционирование мембран, синтез эйкозаноидов и передачу сигнала, а также на регуляцию экспрессии генов.
Эпидемиологические и экспериментальные исследования показали, что потребление ПНЖК n-3 ассоциировано со сниженным риском дегенерации желтого пятна. Основной общий механизм защиты от дегенерации желтого пятна и рака состоит в способности ПНЖК n-3 подавлять патологический ангиогенез. ЕРА и DHA подавляют аномальную неоваскуляризацию сетчатки, проницаемость сосудов и воспаление. Ангиогенез представляет собой важный этап роста и метастазирования опухоли, стимулируемый ПНЖК n-6 и метаболитами, образующимися из ПНЖК n-6, но подавляется ПНЖК n-3 и метаболитами, образующимися из ПНЖК n-3.
Симопулос (Simopoulos) и его коллеги обобщили данные экспериментов на животных и клинических исследований вмешательств, указывающие на то, что ПНЖК n-3 обладают противовоспалительными свойствами и поэтому могут быть полезны для контроля воспалительных и аутоиммунных заболеваний (Simopoulos АР. Omega-3 fatty acids in inflammation and autoimmune diseases. J Am. Coll. NutL 2L 495-505 (2002)). Среди ПНЖК n-3 важную и значимую роль в противовоспалительных эффектах играют ЕРА и DHA (Calder С.Р., Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance, Biochimica et Biophsica Acta - Molecular and Cell Biology of Lipids, Volume 1851 (4), April 2015, 469-484).
Koto (Koto) и его коллеги показали, что ЕРА обладает противовоспалительной активностью в модели хориоидальной неоваскуляризации (ХНВ) у мышей (Koto et al. Eicosapentaenoic Acid Is Anti-Inflammatory in Preventing Choroidal Neovascularization in Mice. Invest Ophthalmol Vis Sci. 2007;48:4328-4334). Они продемонстрировали, что обогащенный ЕРА рацион приводил к значительному подавлению выработки связанных с ХНВ молекул воспаления, таких как ICAM-1 и МСР-1 в эндотелиальных клетках и VEGF и ИЛ-6 в макрофагах, как in vivo, так и in vitro. Янаи (Yanai) и его коллеги продемонстрировали, что обогащение рациона ПНЖК n-3 подавляет хориоидальную неоваскуляризацию в модели возрастной макулярной дегенерации (ВМД) у мышей (Yanai et al. Cytochrome P450-generated metabolites derived from ω-3 fatty acids attenuate neovascularization. Proc Natl Acad Sci USA. 2014 Jul 1;111(26):9603-8). Кроме того, они показали, что ПНЖК n-3 обладают противовоспалительными свойствами в этой модели. Об этом свидетельствовало значительное снижение системного накопления иммунных клеток и отрицательная регуляция экспрессии Icam-1 и Е-селектина на эндотелиальных клетках и лиганда ICAM-1, CD11b-CD18, на поверхности циркулирующих лейкоцитов. ПНЖК n-3 также вызывали подавление проникновения макрофагов в очаги ХНВ. Также было показано, что этот эффект опосредуют образующиеся с участием CYP биологически активные липидные медиаторы, образующиеся из длинноцепочечных полиненасыщенных жирных кислот (ДЦПНЖК) ω-3, в частности основные метаболиты эпоксигеназы CYP, образующиеся из ЕРА (17,18-EEQ) и DHA (19,20-EDP) (Yanai et al. Cytochrome P450-generated metabolites derived from ω-3 fatty acids attenuate neovascularization Proc Natl Acad Sci USA. 2014 Jul 1;111(26):9603-8 and WO 2014/110261 A1).
Модель индуцированной лазером ХНВ у мышей является признанной моделью для исследования потенциальных лекарственных средств в отношении их эффективности для лечения офтальмологических нарушений, связанных с неоваскуляризацией и/или воспалением, в частности ВМД. Кроме того, предполагается, что неоваскулярные заболевания глаза, такие как ВМД, имеют выраженный воспалительный компонент (Lopez et al. Pathologic features of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1991;112(6):647-656.; Grossniklaus et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002 Apr 21;8:119-2б; Lopez et al. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996, 37(5):855-868; Tezel et al. Pathogenesis of age-related macular degeneration. Trends Mol Med. 2004;10(9):417-420.; Schlingemann RO- Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2004; 242(1):91-101). Гроссниклаус (Grossniklaus) и его коллеги с использованием образцов хориоидальной мембраны пациентов с ВМД показали, что прогрессирование ХНВ представляет собой динамический процесс, включающий не только ангиогенез, но также сильный воспалительный компонент, в частности, связанный с макрофагами. На основании работы Амбати (Ambati) и его коллег (Ambati et al. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat. Med. 2003, 9, 1390-1397), связывающей патогенез ВМД с системой комплемента и макрофагами, был дополнительно интерпретирован воспалительный компонент ВМД, и представления о патогенезе ВМД значительно изменились. Впоследствии многие исследовательские группы дополнительно изучали роль воспалительных процессов в патогенезе ВМД и ХНВ, например, в недавней работе был проведен анализ поляризации макрофагов при экспериментальной и клинической хориоидальной неоваскуляризации (Yang et al. Macrophage polarization in experimental and clinical choroidal neovascularization. Sci Rep., 2016 Aug 4, 6:30933). В недавних обзорах, сделанных группой Дэвида Хинтона (David Hinton), основоположника исследований ХНВ, и Campa et al. (Inflammatory mediators and angiogenic factors in choroidal neovascularization: pathogenetic interactions and therapeutic implications. Mediators of Inflammation, 2010), подробно охарактеризовано влияние воспаления на патогенез ВМД и последующий фиброз (Ishikawa et al. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp Eye Res. 2016 Jan, 142:19-252016) и сделано заключение, что ХНВ вызывает гетерогенное заболевание, поражающее задний сегмент глаза, которое более точно определяется как аберрантная инвазия ткани эндотелиальными и воспалительными клетками, в которую вовлечены как ангиогенез, так и воспаление.
Становится очевидным, что модель индуцированной лазером ХНВ, которая широко используется для прояснения патобиологии хориоидального ангиогенеза и выявления новых терапевтических средств (Grossniklaus et al. 2010), включает сильный воспалительный стимул вследствие повреждения после лазерного ожога мембраны Бруха. Таким образом, наряду с антиангиогенными соединениями, многие противовоспалительные соединения демонстрируют активность широкого спектра, и указанную модель можно также рассматривать как модель воспаления глаза и ответа на лечение, и модель воспаления глаза может рассматриваться специалистом в качестве доказательства пригодности соединения для лечения воспаления как такового.
В процессе воспаления увеличивается выход циркулирующих моноцитов в сосудистое русло и миграция в ткани, где после кондиционирования местными факторами роста, провоспалительными цитокинами и продуктами жизнедеятельности микроорганизмов, они дифференцируют в популяции макрофагов или дендритных клеток. В модели на крысах, приведенной в примере 5, этот процесс виден при окрашивании инфильтрированных ED1-положительных моноцитов/макрофагов в срезах сердца и почки с помощью соответствующего антитела. В целом, привлечение (рекрутинг) моноцитов необходим для эффективной борьбы и устранения вирусных, бактериальных, грибковых и протозойных инфекций, но рекрутируемые моноциты также участвуют в патогенезе воспалительных и дегенеративных заболеваний (Shi С, et al., Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011 Oct 10; 11 (11) 762-74). Известно, что, помимо участия в развитии атеросклероза, рекрутируемые моноциты/макрофаги вносят вклад в патогенез острых и хронических воспалительных заболеваний сердца и почки (Ingersoll et al., Monocyte trafficking in acute and chronic inflammation. Trends Immuno 2011 Oct. 32(10) 470-7; Hansson G., Inflammation, Atherosclerosis, and Coronary Artery Disease. New Engl Jour Med 2005, (352) 1685-95; Kinsey et al, Imflammation in Acute Kidney Injury. Experim Nephro 2008, (109) e102-e107; Bonventre, J. Cellular pathophysiology of ischemic acute kidney injury. J Clinic Invest 2011 Nov., (121) 4210-4221; Guiteras R., et al., Macrophage in chronic kidney disease. Cli Kid j 2016, vol. 9, no 6, 765-771).
Выработка ФНО-альфа играет важную роль в хронических воспалительных состояниях, промежуточном метаболизме и риске развития сердечно-сосудистых заболеваний (Рора С. et al., The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res 2007, (48) 752-761). Изменения выработки ФНО-альфа и сигнализации рецептора ФНО связывают с патогенезом нескольких хронических воспалительных заболеваний (Parameswaran N. et al., Tumor Necrosis Factor-a Signaling in Macrophages. Crit Rev Eukaryot Gene Expr 2010 20(2) 87-103). ФНО-альфа выполняет разнонаправленные и потенциально противоположные роли в функционировании и патологии сердца (Sack М., Tumor necrosis factor-alpha in cardiovascular biology and the potential role for anti-tumor necrosis factor-alpha therapy in heart disease. Pharmacol Ther 2002 Apr-May, 94(1-2) 123-135). Также, как показано в примере 4, ФНО-альфа может продуцироваться самими кардиомиоцитами в ответ на провоспалительные стимулы. После высвобождения в окружающую ткань ФНО-альфа совместно с широким спектром дополнительных медиаторов вызывает активацию и привлечение лейкоцитов (Ghigo A. et al., Myocyte signalling in leucocyte recruitment to the heart. Cardicovasc Res 2014 May. 102(2) 270-280).
Одна из наиболее важных биологических ролей ПНЖК заключается в поставке предшественников для выработки биологически активных метаболитов жирных кислот, которые могут модулировать большое количество функций. Например, арахидоновая кислота (АА; 20:4, n-6) метаболизируется ферментами цитохрома Р450 (CYP) с образованием оксигенированныхметаболитов различных классов с высокой биологической активностью. Основные метаболиты включают 20-гидроксиэйкозатетраеновую кислоту (20-НЕТЕ) и разновидности регио- и стереизомерных эпоксиэйкозатриеновых кислот (ЕЕТ). Изоформы CYP4A и CYP4F позволяют получить 20-НЕТЕ, а изоформы CYP2C и CYP2J позволяют получить изоформы EETs.
Известно, что ЕРА (20:5, n-3) и ОНА (22:6, n-3) могут выполнять функцию альтернативных субстратов для метаболизирующих арахидоновую кислоту изоформ CYP (Arnold С. et al., J Biol Chem. 2010 Oct 22;285(43):32720-33.; Fischer R. et al., J Lipid Res. 2014 Mar 16;55(6):1150-1164.). Представители подсемейств CYP2C и CYP2J, которые эпоксидируют арахидоновую кислоту с образованием ЕЕТ, метаболизируют ЕРА с образованием эпоксиэйкозатетраеновых кислот (EEQ) и DHA с образованием эпоксидокозапентаеновых кислот (EDP). Двойная связь ω-3, которая отличает ЕРА и DHA от арахидоновой кислоты, является предпочтительным местом воздействия большинства эпоксигеназ, в результате которого образуются 17,18-EEQ и 19,20-EDP в качестве основных метаболитов. Изоформы CYP4A и CYP4F, гидроксилирующие арахидоновую кислоту с получением 20-НЕТЕ, метаболизируют ЕРА в 20-гидроксиэйкозапентаеновую кислоту (20-НЕРЕ) и DHA в 22-гидроксидокозагексаеновую кислоту (22-HDHA). CYP1A1, CYP2E1 и другие изоформы, превращающие арахидоновую кислоту предпочтительно в 19-НЕТЕ, демонстрируют выраженную ω-3 эпоксигеназную активность по отношению к ЕРА и DHA. Варианты CYP1A1 человека позволяют получить различные метаболические варианты эйкозапентаеновой кислоты. Зависимые от цитохрома Р450 метаболиты эйкозапентаеновой кислоты представляют собой новые активаторы ВК-каналов. Отличительным признаком CYP-зависимого метаболизма ПНЖК n-3 является предпочтительное эпоксидирование двойной связи n-3, которая отличает ЕРА и DHA от арахидоновой кислоты. Полученные метаболиты -17,18-EEQ из ЕРА и 19,20-EDP из DHA являются уникальными, поскольку они не имеют гомологов среди продуктов арахидоновой кислоты. Наряду с субстратной специфичностью изоформ CYP добавка EPA/DHA в пищу обеспечивает основательный сдвиг от метаболитов арахидоновой кислоты к производным от ЕРА и DHA эпокси- и ω-гидроксиметаболитам в большинстве органов и тканей у крыс и, предположительно, у человека.
ЕЕТ и 20-НЕТЕ играют важную роль в регуляции различных функций сердечнососудистой системы (Roman RJ., Physiol Rev. 2002;82:131-85). Было показано, что индуцируемая ангиотензином II (Ang II) гипертензия связана с отрицательной регуляцией CYP-зависимого метаболизма арахидоновой кислоты (Kaergel et I., Hypertension. 2002;40:273-9) в модели индуцированной Ang II гипертензии и повреждения органов-мишеней на дважды трансгенных крысах (dTGR, англ.: double-transgenic rat) (Luft et al., Hypertension. 1999;33:212-8). У трансгенных крыс есть гены ренина и ангиотензиногена человека, у них местно вырабатывается Ang II и развивается значительная гипертензия, инфаркт миокарда и альбуминурия. Животные погибают от миокардиальной и почечной недостаточности в возрасте менее восьми недель. Данная модель демонстрирует тяжелые признаки индуцируемого Ang II воспаления. Образуются активные формы кислорода, факторы транскрипции NF-κВ и АР-1 активируются, и активируются гены, содержащие сайты связывания этих факторов транскрипции.
Недавно было показано, что добавка эйкозапентаеновой кислоты (ЕРА) значительно снижала смертность у dTGR (Theuer et al., Kidney Int. 2005;67:248-58). Кроме того, было показано, что у dTGR развиваются желудочковые аритмии вследствие индуцируемого Ang II электрического ремоделирования (Fischer et si. Am J Physiol Heart Ore Physiol. 2007; 293:H1242-1253). Лечение крыс dTGR активатором PPAR-альфа вызвало интенсивную СУР2С23-зависимую выработку ЕЕТ и защищало от гипертензии и повреждения органов-мишеней (Muller et al., Am J Pathol. 2004;164:521-32).
Длительное кормление dTGR (в возрасте 4-7 недель) смесью чистых этиловых эфиров ЕРА и ОНА (Omacor from Solvay Arzneimittel, Hannover, Germany) улучшало состояние при электрическом ремоделировании сердца в этой модели индуцируемой ангиотензином II гипертензии. В частности, ЕРА и DHA снижали смертность, подавляли индуцибельность сердечных аритмий и защищали от ремоделирования, связанного со щелевыми контактами, содержащими коннексин 43 (Fischer et al., Hypertension. 2008 Feb; 51(2):540-6). В целом, CYP-зависимые эйкозаноиды должны считаться вторичными мессенджерами: EETs и 20-НЕТЕ вырабатываются ферментами CYP после индуцируемого внеклеточным сигналом высвобождения арахидоновой кислоты из мембранных фосфолипидов (посредством фосфолипазы А2) и выполняют свою функцию в контексте сигнальных путей, обеспечивающих модулирование транспорта ионов, пролиферации клеток и воспаления. В зависимости от рациона ПНЖК n-3 частично замещают арахидоновую кислоту в положении sn2 фосфолипидов. и могут, таким образом, быть вовлечены в качестве альтернативных молекул в последующие сигнальные пути.
В нескольких исследованиях биологической активности CYP-зависимых эйкозаноидов в сердце была показана важная роль ЕЕТ и 20-НЕТЕ в регуляции Са2+ каналов L-типа и чувствительных к АТФ калиевых (KАТР) каналов в сакролемме и митохондриях. В сердечных миоцитах ток Са2+ через каналы L-типа и сокращение клеток (cell shorting) уменьшаются в результате ингибирования выработки ЕЕТ, и на эти эффекты может быть оказано обратное действие путем добавления 11,12-ЕЕТ (Xiao et al., J Physiol. 1998;508 (Pt 3):777-92). Также было показано, что ЕЕТ активируют KАТР каналы в сердце. Данный эффект является высокостереоселективным: только S,R-энантиомер 11,12-ЕЕТ был эффективен, a R,S-энантиомер эффективности не проявил (Lu et al., Mol Pharmacol. 2002;62:1076-83). Сверхэкспрессия ЕЕТ-образующего CYP2J2 человека обеспечивала улучшенное функциональное восстановление сердца трансгенных мышей после ишемии благодаря активации KАТР каналов (Seubert et al., Ore Res. 2004;95:506-14). 20-ETE, как представляется, играет обратную роль, активируя эндогенный блокатор KАТР каналов (Gross et al., J Mol Cell Cardiol. 2004;37:1245-9; Nithipatikom etal., Ore Res. 2004;95:e65-71).
Несмотря на то, что метаболиты, образующиеся из ПНЖК n-3 при помощи CYP, такие как 17,18-EEQ и 19,20-EDP, играют важную роль в опосредовании положительных эффектов ПНЖК n-3 в организме млекопитающего, их не используют в качестве терапевтических средств из-за их ограниченной биодоступности, а также химической и метаболической нестабильности. Эти эпоксиметаболиты ПНЖК n-3 склонны к автоокислению, быстро инактивируются растворимой эпоксидгидролазой и разлагаются по механизму β-окисления.
Таким образом, задача настоящего изобретения состоит в обеспечении улучшенных аналогов метаболитов ПНЖК n-3 для лечения, или снижения риска развития, или предотвращения нарушений, связанных с неоваскуляризацией и/или воспалением, в частности офтальмологических нарушений, связанных с неоваскуляризацией и/или воспалением.
В первом аспекте указанная выше задача решается путем обеспечения соединений общей формулы (I):

или фармацевтически приемлемой соли указанного соединения, где
Р представляет собой группу, представленную общей формулой (II):

где
n равен 0 или представляет собой целое число от 3 до 8, то есть 3, 4, 5, 6, 7 или 8, предпочтительно 3; и
k равен 0, 1 или 2; предпочтительно при условии, что, если n равен 0, k равен 1, наиболее предпочтительно k равен 1;
X представляет собой СН2ОН, СН2ОАс, СН(О) или группу, выбранную из группы, состоящей из:
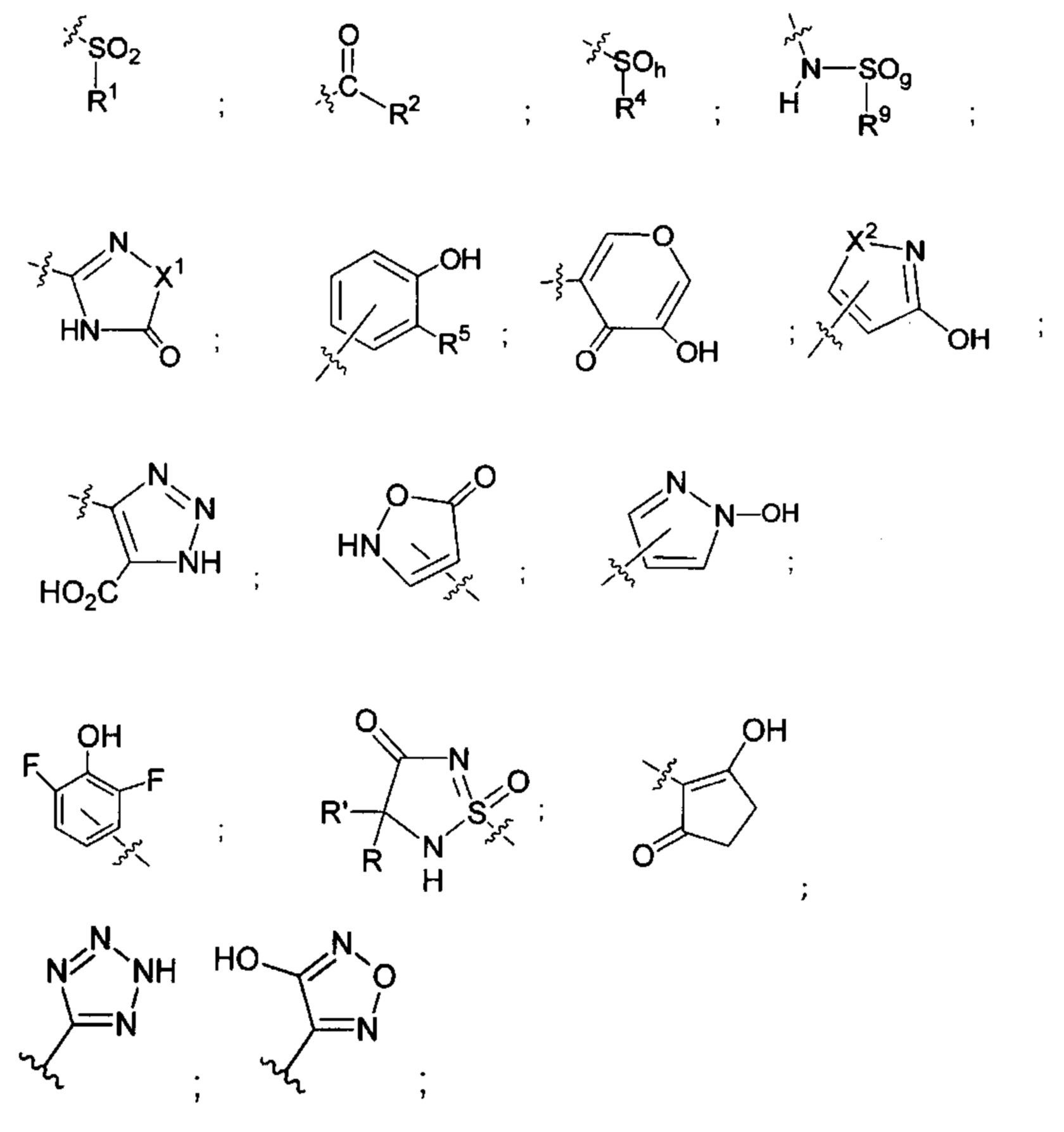

предпочтительно X представляет собой

где
каждый из R и R1 независимо представляет собой атом водорода или C1-С6алкильную группу, которая может быть замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами);
R1 представляет собой гидроксильную группу, C1-С6алкокси, -NHCN, -NH(С1-С6алкил), -NH(С3-С6циклоалкил), -NH(арил) или -O(С1-С6алкилдиил)O(С=O)R11; R11 представляет собой C1-С6алкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора, или С3-С6циклоалкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами);
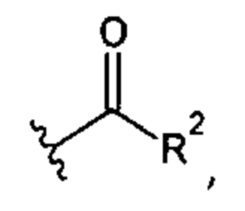
R2 представляет собой -NHR3; -NR20R21; -OR22; -(OCH2-CH2)i-R23; -С3-С10-гетероциклил, необязательно замещенный одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из гидроксильной группы, C1-С6алкокси, C1-С6алкила и оксо; -(Хаа)o; моно- или дисахарид или их производное, которые присоединены к -С(О) сложноэфирной связью через 1-O-, 3-O- или 6-О-положение сахарида;
или выбран из группы, состоящей из:
где
R3 представляет (SO2R30); (OR31); -С1-С6алкандиил(SO2R32); -С1-С6алкандиил(СО2Н), арильную группу, гетероарильную группу, циклоалкильную группу или гетероциклоалкильную группу, причем указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-С6алкила), -N(С1-С6)диалкила и -C(=O)OR51; причем указанная гетероарильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила), -N(С1-С6)диалкила и -C(=O)OR51; причем указанная циклоалкильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-С6алкила), -N(С1-С6)диалкила и -C(=O)OR51; а указанная гетероциклоалкильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила), -N(С1-С6)диалкила и -C(=O)OR51;
R30 представляет собой C1-С6алкильную или арильную группу, причем указанная C1-С6алкильная группа необязательно замещена -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, С1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонилом-, ди(С1-С6)алкиламинокарбонилом-, одним, двумя или тремя атомами фтора или хлора или гидроксильной группой; а указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила) и -N(С1-С6)диалкила;
R31 представляет собой C1-С6алкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами); или С3-С6циклоалкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами);
R32 представляет собой C1-С6алкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами); или С3-С6циклоалкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами);
каждый из R20 и R21 независимо представляет атом водорода; C1-С6алкильную группу, которая может быть замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами); С3-С6циклоалкильную группу, которая может быть замещена одним или более атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами); -С1-С6алкилдиил(СО2Н); или R20 и R21 совместно образуют С3-С10-гетероциклоалкил, который может быть замещен одной или более C1-С6алкильной (-ыми) группой (-ами), C1-С6алкоксигруппой (-ами), атомом (-ами) фтора или хлора или гидроксильной (-ыми) группой (-ами);
R22 представляет собой атом водорода, С1-С6алкильную группу или С3-С6циклоалкильную группу; причем указанная C1-С6алкильная группа или указанная С3-С6циклоалкильная группа необязательно замещены -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(C1-С6)алкилдиил-С1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидроксилом или C1-С6алкокси, аралкильной группой, гетероалкильной группой или гетероалкилциклоалкильной группой;
R23 представляет собой -ОН, -O(С1-С3)алкил или -N(С1-С3)диалкил;
i представляет собой целое число от 1 до 10;
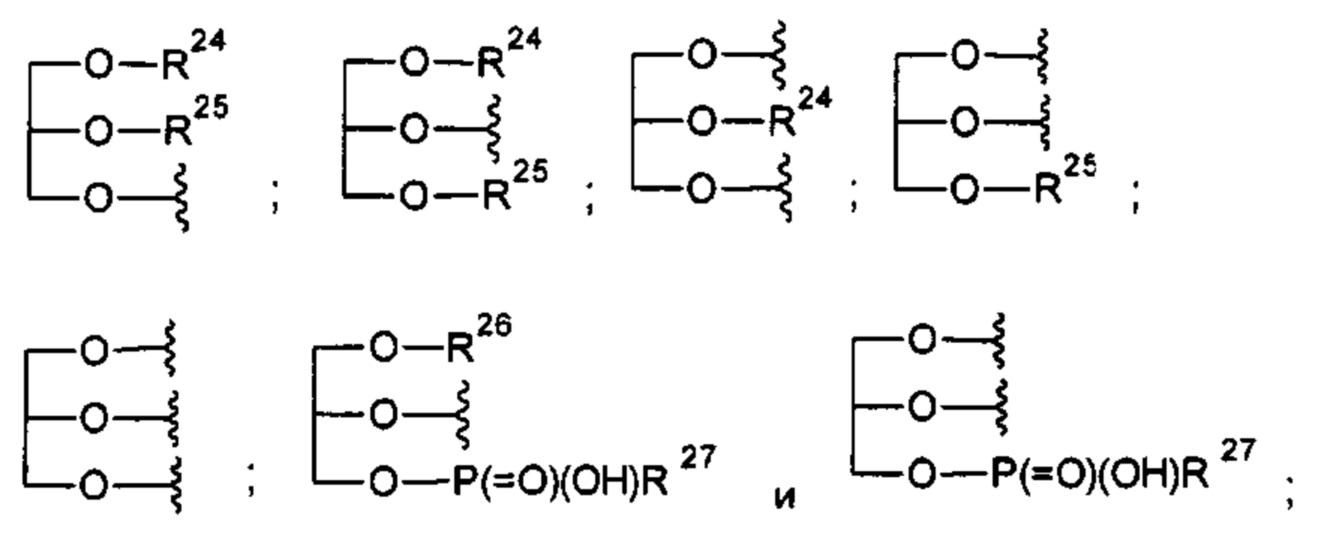
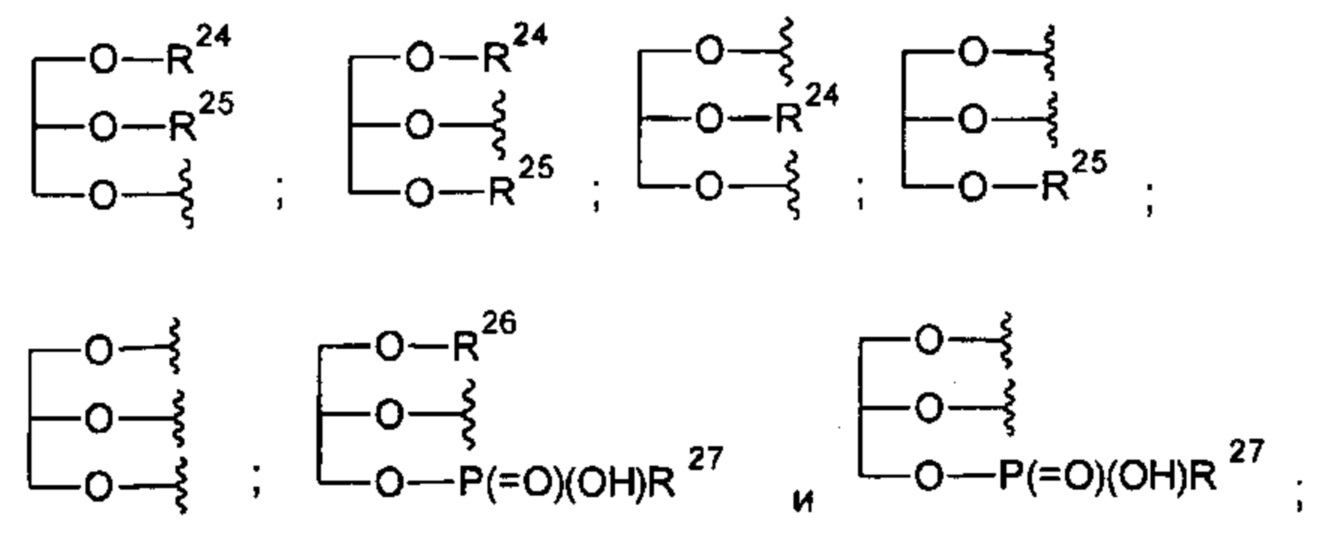
каждый из R24, R25 и R26 независимо представляет атом водорода; -С(=O)С11-С21алкил или -С(=O)С11-С21алкенил;
R27 представляет собой -ОН; -O(CH2)2NH2, -OCH2-[CH(NH2)(CO2H)], -O(CH2)2N(CH3)3 или
Хаа представляет Gly, стандартную D,L-, D- или L-аминокислоту, нестандартную D,L-, D- или L-аминокислоту или 2-10-мерный пептид; и присоединена к -С(=O) амидной связью;
о представляет собой целое число от 1 до 10;
R4 выбран из группы, состоящей из:
h равен 0, 1 или 2;
R5 представляет атом водорода; атом фтора или хлора; -CF3; -C(=O)OR51; -NHC(=O)R52; -C(=O)NR53R54 или -S(O2)OH;
R51 представляет атом водорода; С1-С6алкильную группу или С3-С6циклоалкильную группу; причем указанная C1-С6алкильная группа или указанная С3-С6циклоалкильная группа необязательно замещены -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(C1-С6)алкилдиил-С1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидроксилом или C1-С6алкокси;
каждый из R52, R53 и R54 независимо представляет собой C1-С6алкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора; С3-С6циклоалкильную группу, которая необязательно замещена одним или более атомом (-ами) фтора или хлора; или арильную группу, которая необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6галогеналкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила), -N(С1-С6)диалкила и оксозаместителя;
каждый из R6 и R7 независимо представляет собой гидроксильную группу; -O(С1-С6)алкильную группу, -O(С2-С6)алкенильную группу, -O(С1-С6)алкилдиилО(С=O)(С1-С6)алкильную группу или -O(С1-С6)алкилдиилО(С=O)(С2-С6)алкенильную группу; причем указанная C1-С6алкильная группа и указанная С2-С6алкенильная группа могут быть замещены NH2, -NH(С1-С6)алкилом, -N(C1-С6)диалкилом, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6 алкиламинокарбонилом-, ди(С1-С6)алкиламинокарбонилом- или одним, двумя или тремя атомами фтора или хлора; или
R6 представляет собой гидроксильную группу и R7 представляет собой группу:

R9 представляет собой C1-С6алкил или арил; причем указанный C1-С6алкил необязательно замещен -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(С1-С6)алкилдиил-С1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидрокси, C1-С6алкокси, арилом, арилокси, -С(=O)-арилом, -С(=O)С1-С6алкокси; и причем указанная арильная группа необязательно замещена одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила), -N(С1-С6)диалкила и оксозаместителя;
g равен 1 или 2, предпочтительно 2;
X1 представляет собой атом кислорода; атом серы или NH;
X2 представляет собой атом кислорода; атом серы; NH или N(СН3);
X3 представляет собой атом кислорода; атом серы; атом азота; атом углерода или С-ОН; и пунктирная линия представляет собой связь углерод-углерод или двойную углерод-углеродную связь;
Е представляет собой группу, представленную общей формулой (III) или (IV):

где R12 и R13 предпочтительно находятся в цис-конфигурации, и где
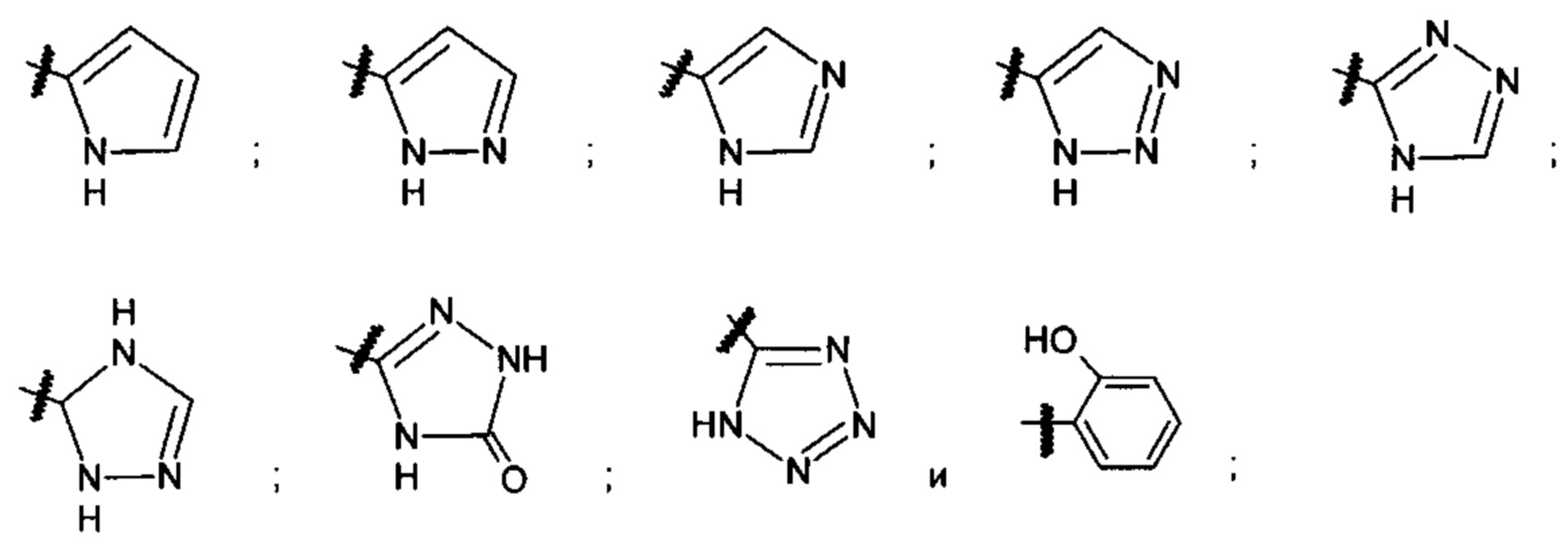
кольцо А в формуле (III) представляет собой 5-членное или 6-членное карбоциклическое или гетероциклическое кольцо, содержащее по меньшей мере одну двойную связь, включая ароматическое карбоциклическое или гетероциклическое кольцо, которое может содержать от одного до трех или от одного до четырех заместителей, независимо выбранных из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила) и -N(С1-С6)диалкила; и каждый из L и Т независимо представляет собой атом кольца, где L и Т являются соседними по отношению друг к другу;
каждый из R12 и R13 независимо представляет собой атом водорода, атом фтора, гидроксил, -NH2, C1-С6алкил, C1-С6алкокси, -С(=O)-арил, -С(=O)С1-С6алкил или -SO2(С1-С6алкил); или -SО2арил; причем любой из указанных C1-С6алкила, C1-С6алкокси или арила необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(С1-С6)диалкила, С1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, С1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонила-, ди(С1-С6)алкиламинокарбонила-, атома фтора или хлора и гидроксила; или R12 и R13 объединены с образованием 5-членного или 6-членного кольца, которое необязательно замещено одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(С1-С6)диалкила, C1-С6алкилкарбонилокси-, С1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонила-, ди(С1-С6)алкиламинокарбонила-, атома фтора или хлора и гидроксила;
I представляет собой -(CH2)m-Y, где
m представляет собой целое число от 3 до 6, то есть 3, 4, 5 или 6, при условии, что m представляет собой целое число от 3 до 5, если Е представляет собой группу в соответствии с общей формулой (III);
Y представляет собой -U-V-W-(CH2)p-(CH3)q, где р представляет собой целое число от 0 до 6; q равен 0 или 1; U отсутствует или выбран из группы, состоящей из СН, СН2 и NR40, при условии, что U представляет собой только СН, если он образует эпоксигруппу совместно с V и W; V выбран из группы, состоящей из -С(О)-, -С(O)-С(O)-, -О-, и -S-; W выбран из группы, состоящей из СН, СН2 и NR40, при условии, что W представляет собой только СН, если он образует эпоксигруппу совместно с U и V;
или Y представляет собой группу, выбранную из группы, состоящей из:


где
каждый из R40, R41, R43, R44, R46, R48 и R49 независимо представляет собой атом водорода, -C1-С6алкил, -С3-С6циклоалкил, -C1-С6алкокси, -С(=O)арил или -С(=O)С1-С6алкил, причем любой из указанных C1-С6алкила, С3-С6циклоалкила, C1-С6алкокси или арила необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(С1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонила-, ди(С1-С6)алкиламинокарбонила-, атома фтора или хлора и гидрокси; или R40 и R41, или R43 и R44 объединены с образованием 5-членного или 6-членного кольца, которое может быть замещено одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(С1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонила-, ди(С1-С6)алкиламинокарбонила-, атома фтора или хлора и гидроксила;
каждый из R42, R45, R47 и R50 независимо представляет собой -C1-С3алкил, причем указанный C1-С3алкил может быть замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С3)алкила, -N(С1-С3)диалкила, C1-С3алкилкарбонилокси-, C1-С3алкоксикарбонилокси-, С1-С3алкилкарбонилтио-, C1-С3алкиламинокарбонила-, ди(С1-С3)алкиламинокарбонила-, атома фтора или хлора и гидроксила; или R40 и R41; R43 и R44; R49 и R50 объединены с образованием 5-членного или 6-членного кольца, которое может быть замещено одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из -NH2, -NH(С1-С6)алкила, -N(C1-С6)диалкила, C1-С6алкилкарбонилокси-, C1-С6алкоксикарбонилокси-, C1-С6алкилкарбонилтио-, C1-С6алкиламинокарбонила-, ди(С1-С6)алкиламинокарбонила-, атома фтора или хлора и гидроксила;
f представляет собой целое число от 0 до 2;
при условии, что
если X не содержит фрагмент -С(=O)O- с карбонильным углеродом в альфа- или бета-положении по отношению к атому кислорода в общей формуле (II), Y представляет собой оксамид, карбамат или карбамид, предпочтительно Y представляет собой оксамид, определенный выше,
для применения в лечении, снижении риска развития или предотвращении нарушения, связанного с неоваскуляризацией и/или воспалением.
В предпочтительном варианте реализации соединения согласно настоящему изобретению представляют собой соединения формулы (I), описанные выше, при условии, что
если X не содержит фрагмент -С(=O)O- с карбонильным углеродом в альфа- или бета-положении по отношению к атому кислорода в общей формуле (II), Y представляет собой оксамид, карбамат или карбамид, предпочтительно Y представляет собой оксамид, определенный выше.
В предпочтительном варианте реализации соединения формулы (I) представляют собой соединения, описанные выше, при дополнительном условии, что
если n равен 3, 5, 6, 7 или 8, предпочтительно 3, k равен 1 и Е представляет собой группу в соответствии с общей формулой (III) или общей формулой (IV), где каждый из R12 и R13 представляет атом водорода;
Р представляет собой группу:

где
X81 представляет собой группу, выбранную из группы, состоящей из:

R1' такой, как определено для R1 выше;
R2' представляет собой -NHR3'; -OR22'; -(OCH2-CH2)i-R23; моно- или дисахарид или их производное, которые присоединены к -С(=O) сложноэфирной связью через 1-O-, 3-O- или 6-О-положение сахарида;
или где R2 выбран из группы, состоящей из:

где
R3' представляет (SO2R30); (OR31); -С1-С6алкандиил(SO2R32) или -С2-С6алкандиил(CO2H);
R22' представляет собой атом водорода или С3-С6циклоалкильную группу, которая необязательно замещена -NH2, -NH(С1-С6)алкилом, -N(С1-С6)диалкилом, -NH(C1-С6)алкилдиил-C1-С6алкокси, одним, двумя или тремя атомами фтора или хлора, гидрокси или C1-С6алкокси;
R23 и i такие, как определено выше;
R24, R25, R26 и R27 такие, как определено выше;
R4' такой, как определено для R4 выше; и h такой, как определено выше;
R6' и R7' такие, как определено для R6 и R7 выше;
R8'' и R8'' такие, как определено для R8 и R8' выше;
R9' такой, как определено для R9 выше; R9'' представляет собой арил, который необязательно замещен одним, двумя или тремя заместителями, независимо выбранными из группы, состоящей из C1-С6алкила, C1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(С1-С6алкила), -N(С1-С6)диалкила и оксозаместителя.
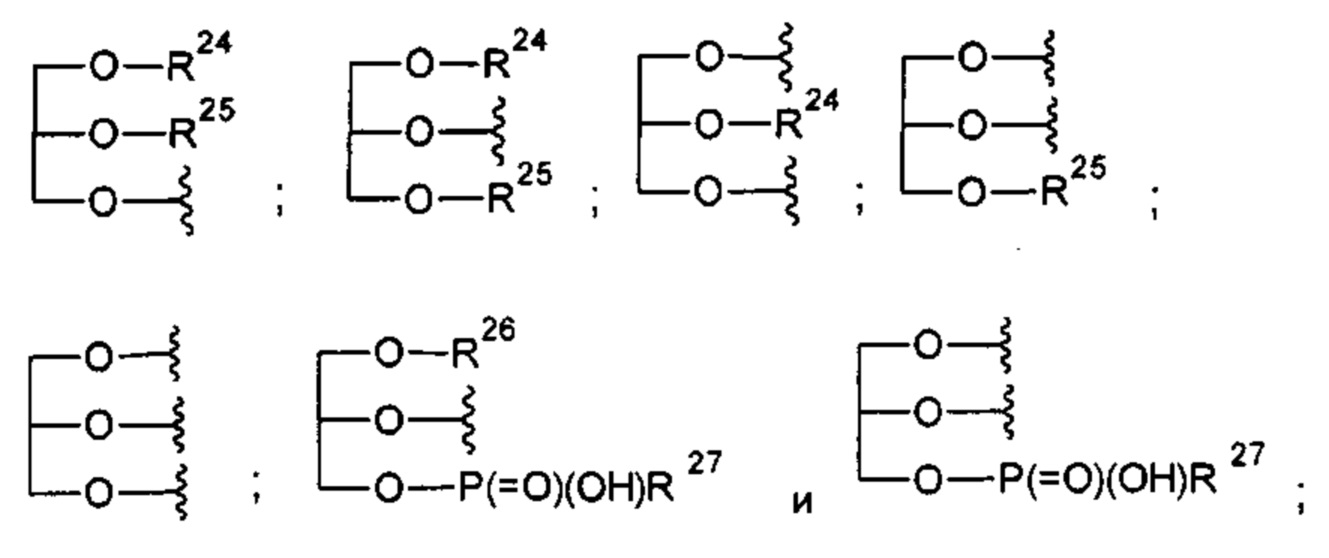
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, где X представляет собой

где R2 представляет собой -OR22; -(OCH2-CH2)i-R23; моно- или дисахарид или их производное, которые присоединены к -С(=O) сложноэфирной связью через 1-O-, 3-O- или 6-O-положение сахарида;
или где R2 выбран из группы, состоящей из:
где R23 и i такие, как определено выше, предпочтительно i равен 3;
и где R22 и R23-R27 такие, как определено в п. 1 формулы изобретения, предпочтительно R22 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода.
В другом более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, где X представляет собой -С(=O)ОН или подходящую соль карбоновой кислоты, предпочтительно свободную карбоновую кислоту.
В другом более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, где Y представляет собой один из оксамидов, определенных выше.
Кроме того, предпочтительно, чтобы соединение согласно настоящему изобретению представляло собой соединение, где X представляет собой
где R2 представляет собой -OR22; -(OCH2-CH2)i-R23; моно- или дисахарид или их производное, которые присоединены к -С(=O) сложноэфирной связью через 1-O-, 3-O- или 6-O-положение сахарида; или где R2 выбран из группы, состоящей из:
где R22, R23-R27 и i такие, как определено выше, предпочтительно R22 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода, предпочтительно i составляет от 2 до 4, более предпочтительно 3, и где Y предпочтительно представляет собой один из оксамидов, определенных выше.
В более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение, где X представляет собой С(=O)ОН, предпочтительно свободную карбоновую кислоту, и Y предпочтительно представляет собой один из оксамидов, определенных выше.
В другом более предпочтительном варианте реализации соединение согласно настоящему изобретению представляет собой соединение со следующей формулой (V):

где
R55 представляет собой -ОН; -OR22; -(OCH2-CH2)i-R23; моно- или дисахарид или их производное, которые присоединены к -С(=O) сложноэфирной связью через 1-O-, 3-O- или 6-О-положение сахарида;
R22, R23 и i такие, как определено выше, предпочтительно R22 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода, и i предпочтительно составляет от 2 до 4, более предпочтительно 3;
Y представляет собой группу, выбранную из группы, состоящей из:

где предпочтительными являются
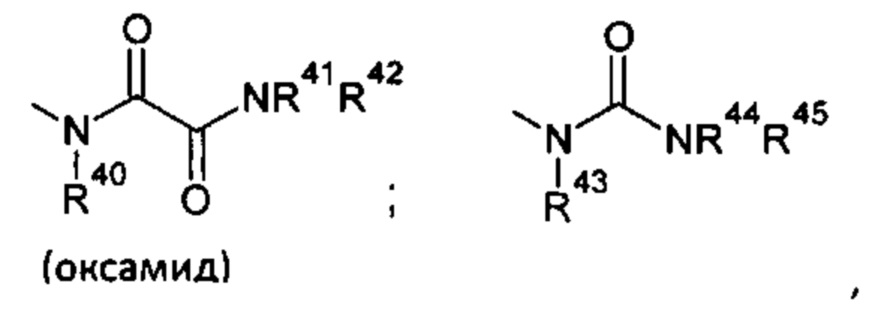

где R40-R50 такие, как определено выше, предпочтительно R40 представляет собой атом водорода или C1-С6алкильную группу, более предпочтительно атом водорода;
R57 и R58 представляют собой атом водорода или совместно образуют пяти- или шестичленное кольцо, предпочтительно ароматическое кольцо, необязательно содержащее от одного до трех или от одного до четырех заместителей, независимо выбранных из группы, состоящей из C1-С6алкила, С1-С6алкокси, C1-С6алкилтио, атома фтора или хлора, гидроксильной группы, аминогруппы, -NH(C1-С6алкила), -N(С1-С6)диалкила и оксозаместителя;
s равен 0, 1 или 2, при условии, что s равен 0, если R57 и R58 совместно образуют пяти- или шестичленное кольцо;
двойная связь в формуле (V) представляет собой двойную углерод-углеродную связь в цис-конфигурации, если R57 и R58 представляют собой водород, или указанная двойная связь является частью пяти- или шестичленного кольца, совместно образуемого R57 и R58.
В другом наиболее предпочтительном варианте реализации соединения формулы (V) представляют собой соединения, где
R55 представляет собой -ОН или -(OCH2-CH2)i-R23; i составляет от 2 до 4, предпочтительно i равен 3; R23 предпочтительно представляет собой ОН;
Y представляет собой оксамид, карбамид или карбамат, предпочтительно C1-С6алкилзамещенный оксамид, карбамид или карбамат;
R57 и R58 оба представляют собой Н или совместно образуют замещенное или незамещенное пяти- или шестичленное ароматическое кольцо, предпочтительно образуют замещенное или незамещенное бензильное кольцо; и
s равен 1 или s равен 0, если R57 и R58 совместно образуют замещенное или незамещенное пяти- или шестичленное ароматическое кольцо.
Конкретными наиболее предпочтительными соединениями согласно настоящему изобретению являются соединения, выбранные из группы, состоящей из:


или фармацевтически приемлемой соли указанных соединений.
Среди указанных выше соединений наиболее предпочтительным является соединение со следующей формулой (VI)

или его фармацевтически приемлемая соль.
Соединения согласно настоящему изобретению имеют преимущество, которое, как показано ниже в экспериментальной части, заключается в том, что они являются эффективными в лечении, снижении риска развития или предотвращении нарушения, связанного с неоваскуляризацией и/или воспалением, в частности офтальмологического нарушения, связанного с неоваскуляризацией и/или воспалением. В то же время они являются метаболически устойчивыми для получения фармацевтического состава и введения нуждающимся в этом субъектам.
Соединения, описанные в настоящем документе, в целом описаны с использованием стандартной номенклатуры. Для соединений, имеющих центры асимметрии, следует понимать, что если не указано иное, настоящее изобретение включает все их оптические изомеры и их смеси. Соединения с двумя или более элементами асимметрии могут присутствовать в виде смесей диастереомеров. Помимо этого, соединения с двойными связями углерод-углерод могут существовать в Z- и Е-формах, и все изомерные формы соединений включены в настоящее изобретение, если не указано иное. Если соединение существует в различных таутомерных формах, такое соединение не ограничено каким-либо конкретным таутомером, а, наоборот, предполагается, что они включают все таутомерные формы. Также предполагается, что указанные соединения включают соединения, в которых один или более атомов заменены изотопом, то есть атомом, имеющим тот же атомный номер, но другое массовое число. В качестве общего примера и без ограничения, изотопы водорода включают тритий и дейтерий, а изотопы углерода включают11C,13С и14С.
Соединения согласно формулам, представленным в настоящем описании, которые имеют один или более стереогенный (-ых) центр (-ов), имеют энантиомерный избыток по меньшей мере 50%. Например, такие соединения могут иметь энантиомерный избыток по меньшей мере 60%, 70%, 80%, 85%, 90%, 95% или 98%. В некоторых вариантах реализации соединения имеют энантиомерный избыток по меньшей мере 99%. Будет очевидно, что отдельные энантиомеры (оптически активные формы) могут быть получены путем асимметрического синтеза из оптически чистых предшественников, биосинтеза, например с применением модифицированного CYP102 (CYP ВМ-3), или путем разделения рацематов, например путем ферментативного разделения или разделения традиционными способами, такими как кристаллизация в присутствии разделяющего агента или хроматография, с использованием, например, колонки для хиральной ВЭЖХ.
Некоторые соединения описаны в настоящем документе с использованием общей формулы, которая включает переменные, такие как, например, Р, Е, I, R1-R50, Х-Х81 и Y. Если не указано иное, каждая переменная в такой формуле определена независимо от любой другой переменной, и любая переменная, которая встречается более одного раза в формуле, в каждом случае определена независимо. Таким образом, например, если показано, что группа замещена посредством 0-2 R*, указанная группа может быть незамещенной или может содержать в качестве заместителей до двух групп R*, и R* в каждом случае выбран независимо из определения R*. Также допустимы комбинации заместителей и/или переменных, только если в результате таких комбинаций получаются стабильные соединения, то есть соединения, которые могут быть выделены, охарактеризованы и протестированы на биологическую активность.
«Фармацевтически приемлемая соль» соединения, раскрытого в настоящем описании, представляет собой соль кислоты или основания, которая в данной области техники в общем считается подходящей для применения в контакте с тканями человека или животных без избыточной токсичности или канцерогенности, и предпочтительно без раздражения, аллергической реакции или других проблем или осложнений. Такие соли включают соли, образованных минеральными и органическими кислотами и основными остатками, такими как амины, а также соли, образованные щелочами или органическими частями и кислотными остатками, таких как карбоновые кислоты.
Подходящие фармацевтические соли включают, но не ограничиваются ими, соли кислот, таких как хлористоводородная, фосфорная, бромистоводородная, яблочная, гликолевая, фумаровая, серная, сульфаминовая, сульфаниловая, муравьиная, толуолсульфоновая, метансульфоновая, бензолсульфоновая, этандисульфоновая, 2-гидроксиэтилсульфоновая, азотная, бензойная, 2-ацетоксибензойная, лимонная, винная, молочная, стеариновая, салициловая, глутаминовая, аскорбиновая, памовая, янтарная, фумаровая, малеиновая, пропионовая, гидроксималеиновая, иодистоводородная, фенилуксусная, алкановая, такая как уксусная, НООС-(СН2)n-СООН, где n представляет собой любое целое число от 0 до 6, то есть 0, 1, 2, 3, 4, 5 или 6 и тому подобное. Аналогичным образом, фармацевтически приемлемые катионы включают, но не ограничиваются ими, натрий, калий, кальций, алюминий, литий и аммоний. Специалисты в данной области техники смогут определить другие фармацевтически приемлемые соли для соединений, представленных в настоящем описании. В общем, фармацевтически приемлемая соль кислоты или основания может быть синтезирована из исходного соединения, которое содержит основной или кислотный фрагмент, любые обычным химическим способом. Вкратце, такие соли могут быть получены путем реакции форм свободной кислоты или основания указанных соединений со стехиометрическим количеством подходящего основания или кислоты в воде или органическом растворителе, или их в смеси. Как правило, применение неводных сред, таких как эфир, этилацетат, этанол, изопропанол или ацетонитрил, является предпочтительным.
Будет очевидно, что каждое соединение формулы (I) может, но необязательно, присутствовать в форме гидрата, сольвата или нековалентного комплекса. Кроме того, в объем настоящего изобретения входят различные кристаллические формы и полиморфы, которые являются пролекарствами соединений формулы (I), предложенных в настоящем описании.
«Пролекарство» представляет собой соединение, которое может не полностью удовлетворять структурным требованиям, предъявляемым к предложенным в настоящем описании соединениям, но после введения субъекту или пациенту подвергается модификации in vivo с образованием соединения формулы (I), предложенного в настоящем описании. Например, пролекарство может представлять собой ацилированное производное соединения, предложенного в настоящем описании. Пролекарства включают соединения, в которых гидрокси-, карбокси-, амино- или сульфгидрильные группы связаны с любой группой, которая при введении субъекту, являющемуся млекопитающим, подвергается расщеплению с образованием свободной гидрокси-, карбокси-, амино- или сульфгидрильной группы, соответственно. Примеры пролекарств включают, но не ограничиваются ими, ацетатные, формиатные, фосфатные и бензоатные производные спирта и аминных функциональных групп в структуре предложенных в настоящем описании соединений. Пролекарства предложенных в настоящем описании соединений могут быть получены путем модификации функциональных групп, присутствующих в указанных соединениях, таким образом, что модифицированные группы подвергаются расщеплению in vivo с образованием исходных соединений.
Термин «заместитель» в настоящем описании относится к молекулярному фрагменту, который ковалентно связан с атомом, находящимся в данной молекуле. Например, «заместитель кольца» может представлять собой фрагмент, такой как галоген, алкильная группа, галогеналкильная группа или другой заместитель, описанный в настоящем документе, который ковалентно связан с атомом, предпочтительно атомом кислорода или азота, который входит в состав кольца. Термин «замещенный» в настоящем описании означает, что любой один или более атомов водорода при указанном атоме заменен заместителем, выбранным из указанных заместителей, при условии, что нормальная валентность указанного атома не превышена, и что указанное замещение приводит к получению стабильного соединения, то есть соединения, которое может быть выделено, охарактеризовано, и его биологическая активность может быть протестирована. Если заместитель представляет собой оксо, то есть =O, 2 атома водорода при указанном атоме могут быть заменены. Оксо-группа, представляющая собой заместитель при ароматическом атоме углерода, приводит в превращению -СН- в -С(=O)- и потере ароматичности. Например, пиридильная группа, замещенная посредством оксо, представляет собой пиридон.
Выражение «необязательно замещенный» относится к группе, в которой один, два, три или более атома водорода заменены независимо друг от друга соответствующими заместителями.
В настоящем описании термин «аминокислота» относится к любой органической кислоте, содержащей один или более аминозаместителей, например к α-, β- или γ-аминопроизводным алифатических карбоновых кислот. При используемом в настоящем описании упоминании полипептидов, например Xaa5, то есть Xaa1Xaa2Xaa3Xaa4Xaa5, где каждая из аминокислот с Xaa1 по Xaa5 независимо выбрана из аминокислот согласно определению, слева находится аминоконец, а справа находится карбоксиконец, в соответствии со стандартными правилами употребления данных терминов и договоренностями.
Термин «стандартная аминокислота» относится к двадцати существующим в природе аминокислотам, и включает все стереомерные изоформы, то есть их D,L-, D- и L-аминокислоты. Эти стандартные аминокислоты могут быть указаны в настоящем описании путем ссылки на их традиционные трехбуквенные или однобуквенные сокращения или сокращения согласно их обычному применению (см., например, Immunology-A Synthesis, 2nd Edition, Е.S. Golub, and D.R. Gren, Eds., Sinauer Associates, Sunderland Mass. (1991)).
Термин «нестандартная аминокислота» относится к не встречающимся в природе аминокислотам или химическим аналогам аминокислот, например α,α-дизамещенным аминокислотам, N-алкиламинокислотам, гомоаминокислотам, дегидроаминокислотам, ароматическим аминокислотам (отличным от фенилаланина, тирозина и триптофана) и орто-, мета- или пара-аминобензойной кислоте. Нестандартные аминокислоты также включают соединения, которые содержат аминную или карбоксильную функциональную группу, разделенные друг от друга с нахождением в положениях 1,3 или дальше, такие как β-аланин, γ-аминомасляная кислота, лактам Фрейдингера (Freidinger lactam), бициклический дипептид (BTD), аминометилбензойная кислота и другие, хорошо известные в данной области техники. Могут применяться статиноподобные изостеры, гидроксиэтиленовые изостеры, изостеры с восстановленной амидной связью, тиоамидные изостеры, изостеры мочевины, карбаматные изостеры, тиоэфирные изостеры, виниловые изостеры и другие изостеры по амидной связи, известные в данной области техники. Применение аналогов или нестандартных аминокислот может улучшать стабильность и биологическое время полувыведения добавленного пептида, поскольку они более устойчивы к разрушениям в физиологических условиях. Специалист в данной области техники будет осведомлен о подобных типах замещений, которые могут быть сделаны. Следующий неограничивающий перечень нестандартных аминокислот, которые могут быть использованы в качестве подходящих блоков для получения пептида, и их стандартные сокращения (в скобках) включает: α-аминомасляная кислота (Abu), L-N-метилаланин (Nmala), α-амино-α-метилбутират (Mgabu), L-N-метиларгинин (Nmarg), аминоциклопропан (Cpro), L-N-метиласпарагин (Nmasn), карбоксилат L-N-метиласпарагиновой кислоты (Nmasp), аминоизомасляная кислота (Aib), L-N-метилцистеин (Nmcys), аминонорборнил (Norb), L-N-метилглутамин (Nmgln), карбоксилат L-N-метилглутаминовой кислоты (Nmglu), циклогексилаланин (Chexa), L-N-метилгистидин (Nmhis), циклопентилаланин (Cpen), L-N-метилизолейцин (Nmile), L-N-метиллейцин (Nmleu), L-N-метиллизин (Nmlys), L-N-метилметионин (Nmmet), L-N-метилнорлейцин (Nmnle), L-N-метилнорвалин (Nmnva), L-N-метилорнитин (Nmorn), L-N-метилфенилаланин (Nmphe), L-N-метилпролин (Nmpro), L-N-метилсерин (Nmser), L-N-метилтреонин (Nmthr), L-N-метилтриптофан (Nmtrp), D-орнитин (Dorn), L-N-метилтирозин (Nmtyr), L-N-метилвалин (Nmval), L-N-метилэтилглицин (Nmetg), L-N-метил-трет-бутилглицин (Nmtbug), L-норлейцин (Nle), L-норвалин (Nva), α-метил-аминоизобутират (Maib), α-метил-γ-аминобутират (Mrabu), D-α-метилаланин (Dmala), α-метилциклогексилаланин (Mchexa), D-α-метиларгинин (Dmarg), α-метилциклопентилаланин (Mcpen), D-α-метиласпарагин (Dmasn), α-метил-α-нафтилаланин (Manap), D-α-метиласпартат (Dmasp), α-метилпеницилламин (Mpen), D-α-метилцистеин (Dmcys), N-(4-аминобутил)глицин (Nglu), D-α-метилглутамин (DMrln), N-(2-аминоэтил)глицин (Naeg), D-α-метилгистидин (Dmhis), N-(3-аминопропил)глицин (Norn), D-α-метилизолейцин (Dmile), N-амино-α-метилбутират (Nmaabu), D-α-метиллейцин (Dmleu), α-нафтилаланин (Anap), D-α-метиллизин (Dmlys), N-бензилглицин (Nphe), D-α-метилметионин (Dmmet), N-(2-карбамилэтил)глицин (Ngln), D-α-метилорнитин (Dmorn), N-(карбамилметил)глицин (Nasn), D-α-метилфенилаланин (Dmphe), N-(2-карбоксиэтил)глицин (Nglu), D-α-метилпролин (Dmpro), N-(карбоксиметил)глицин (Nasp), D-α-метилсерин (Dmser), N-циклобутилглицин (Ncbut), D-α-метилтреонин (Dmthr), N-циклогептилглицин (Nchep), D-α-метилтриптофан (Dmtrp), N-циклогексилглицин (Nchex), D-α-метилтирозин (Dmty), N-циклодецилглицин (Ncdec), D-α-метилвалин (Dmval), N-циклододецилглицин (Ncdod), D-N-метилаланин (Dnmala), N-циклооктилглицин (Ncoct), D-N-метиларгинин (Dnmarg), N-циклопропилглицин (Ncpro), D-N-метиласпарагин (Dnmasn), N-циклоундецилглицин (Ncund), D-N-метиласпартат (Dnmasp), N-(2,2-дифенилэтил)глицин (Nbhm), D-N-метилцистеин (Dnmcys), N-(3,3-дифенилпропил)глицин (Nbhe), D-N-метилглутамин (Dnmgln), N-(3-гуанидинопропил)глицин (Narg), D-N-метилглутамат (Dnmglu), N-(1-гидроксиэтил)глицин (Ntbx), D-N-метилгистидин (Dnmhis), N-(гидроксиэтил))глицин (Nser), D-N-метилизолейцин (Dnmile), N-(имидазолилэтил))глицин (Nhis), D-N-метиллейцин (Dnmleu), N-(3-индолилэтил)глицин (Nhtrp), D-N-метиллизин (Dnnilys), N-метил-γ-аминобутират (NMrabu), N-метилциклогексилаланин (Nmchexa), D-N-метилметионин (Dnmmet), D-N-метилорнитин (Dnmorn), N-метилциклопентилаланин (Nmcpen), N-метилглицин (Nala), D-N-метилфенилаланин (Dnmphe), N-метиламиноизобутират (Nmaib), D-N-метилпролин (Dnmpro), N-(1-метилпропил)глицин (Nile), D-N-метилсерин (Dnmser), N-(2-метилпропил)глицин (Nleu), D-N-метилтреонин (Dnmthr), D-N-метилтриптофан (Dnmtrp), N-(1-метилэтил)глицин (Nval), D-N-метилтирозин (Dnmtyr), N-метилнафтилаланин (Nmanap), D-N-метилвалин (Dnmval), N-метилпеницилламин (Nmpen), γ-аминомасляная кислота (Gabu), N-(p-гидроксифенил)глицин (Nhtyr), L-/-бутилглицин (Tbug), N-(тиометил)глицин (Ncys), L-этилглицин (Etg), пеницилламин (Pen), L-гомофенилаланин (Hphe), L-α-метилаланин (Mala), L-α-метиларгинин (Marg), L-α-метиласпарагин (Masn), L-α-метиласпартат (Masp), L-α-метил-трет-бутил глицин (Mtbug), L-α-метилцистеин (Mcys), L-метилэтилглицин (Metg), L-α-метилглутамин (Mrln), L-α-метилглутамат (Mrlu), L-α-метилгистидин (Mhis), L-α-метилгомофенилаланин (Mhphe), L-α-метилизолейцин (Mile), N-(2-метилтиоэтил)глицин (Nmet), L-α-метиллейцин (Mleu), L-α-метиллизин (Mlys), L-α-метилметионин (Mmet), L-α-метилнорлейцин (Mnle), L-α-метилнорвалин (Mnva), L-α-метилорнитин (Morn), L-α-метилфенилаланин (Mphe), L-α-метилпролин (Mpro), L-α-метилсерин (Mser), L-α-метилтреонин (Mthr), L-α-метилтриптофан (Mtrp), L-α-метилтирозин (Mtyr), L-α-метилвалин (Mval), L-N-метилгомофенилаланин (Nmhphe), N-(N-(2,2-дифенилэтил)карбамилметил)глицин (Nnbhm), N-(N-(3,3-дифенилпропил)карбамилметил)глицин (Nnbhe), 1-карбокси-1-(2,2-дифенил-этиламино)циклопропан (Nmbc), L-O-метилсерин (Omser), L-O-метилгомосерин (Omhser).
Термин «алкил» относится к насыщенной углеводородной группе с неразветвленной или разветвленной цепью, которая содержит от 1 до 20 атомов углерода, предпочтительно от 1 до 10 атомов углерода, например н-октильной группе, в частности от 1 до 6, то есть 1, 2, 3, 4, 5 или 6 атомов углерода, например метилу, этилу, пропилу, изопропилу, н-бутилу, изобутилу, emop-бутилу, mpem-бутилу, н-пентилу, изопентилу, н-гексилу или 2,2-диметилбутилу.
Термин «алкенил» относится к по меньшей мере частично ненасыщенной углеводородной группе с неразветвленной или разветвленной цепью, которая содержит от 2 до 21 атомов углерода, предпочтительно от 2 до 6 атомов углерода, то есть 2, 3, 4, 5 или 6 атомов углерода, например этенильной (винильной), пропенильной (аллильной) изопропенильной, бутенильной, изопренильной или гекс-2-енильной группе, или от 11 до 21 атомов углерода, то есть 11, 12, 13, 14, 15, 16, 17, 18, 19, 20 или 21 атомов углерода, например углеводородной группе, содержащей цепь из метиленовых звеньев, в промежутке между которыми находится одна двойная связь, как, например, встречается в мононенасыщенных жирных кислотах, или углеводородной группе, содержащей полиены, в промежутке между которыми находится метилен, например углеводородной группе, содержащей два или более структурных звеньев -[СН=СН-СН2]-, как, например, встречается в полиненасыщенных жирных кислотах. Алкенильные группы содержат одну или более, предпочтительно 1, 2, 3, 4, 5 или 6, двойную (-ых) связь (-ей).
Термин «алкинил» относится к по меньшей мере частично ненасыщенным углеводородным группам с прямой или разветвленной цепью, которые содержат от 2 до 20 атомов углерода, предпочтительно от 2 до 10 атомов углерода, в частности от 2 до 6, то есть 2, 3, 4, 5 или 6 атомов углерода, например, этинильной, пропинильной, бутинильной, ацетиленильной или пропаргильной группе. Предпочтительно алкинильные группы содержат одну или две (в частности, предпочтительно одну) тройную (-ых) связь (связи).
Кроме того, термины «алкил», «алкенил» и «алкинил» относятся к группам, в которых один или более атом (-ов) водорода заменены, например, атомом галогена, предпочтительно F или Cl, таким как, например, 2,2,2-трихлорэтильная или трифторметильная группа.
Термин «гетероалкил» относится к алкильной, алкенильной или алкинильной группе, в которой один или более, предпочтительно 1, 2 или 3, атом (-ов) углерода независимо друг от дргуга заменены атомом кислорода, азота, фосфора, бора, селена, кремния или серы, предпочтительно атомом кислорода, серы или азота. Термин «гетероалкил» также может относиться к карбоновой кислоте или группе, полученной из карбоновой кислоты, такой как, например, ацил, ацилалкил, алкоксикарбонил, ацилокси, ацилоксиалкил, карбоксиалкиламид или алкоксикарбонилокси.
Предпочтительно гетероалкильная группа содержит от 1 до 10 атомов углерода и от 1 до 4 гетероатомов, выбранных из кислорода, азота и серы (в частности кислорода и азота). Особенно предпочтительно гетероалкильная группа содержит от 1 до 6, то есть 1, 2, 3,4, 5 или 6, атомов углерода и 1, 2 или 3, в частности 1 или 2, гетероатома, выбранных из кислорода, азота и серы, в частности кислорода и азота.
Примерами гетероалкильных групп являются группы формул: Ra-O-Ya-, Ra-S-Ya-, Ra-N(Rb)-Ya-, Ra-O-Ya-, Ra-O-CO-Ya-, Ra-CO-O-Y3-, Ra-CO-N(Rb)-Ya-, Ra-N(Rb)-CO-Ya-, Ra-O-CO-N(Rb)-Ya-, Ra-N(Rb)-CO-O-Ya-, Ra-N(Rb)-CO-N(Rc)-Ya-, Ra-O-CO-O-Ya-, Ra-N(Rb)-C(=NRd)-N(Rc)-Ya-, Ra-CS-Ya-, Ra-O-CS-Ya-, Ra-CS-O-Ya-, Ra-CS-N(Rb)-Ya-, Ra-N(Rb)-CS-Ya-, Ra-O-CS-N(Rb)-Ya-, Ra-N(Rb)-CS-O-Ya-, Ra-N(Rb)-CS-N(Rc)-Ya-, Ra-O-CS-O-Ya-, Ra-S-CO-Ya-, Ra-CO-S-Ya-, Ra-S-CO-N(Rb)-Ya-, Ra-N(Rb)-CO-S-Ya-, Ra-S-CO-O-Ya-; Ra-O-CO-S-Ya-, Ra-S-CO-S-Ya-, Ra-S-CS-Ya-, Ra-CS-S-Ya-, Ra-S-CS-N(Rb)-Ya-, Ra-N(Rb)-CS-S-Ya-, Ra-S-CS-O-Ya-, Ra-O-CS-S-Ya-, где Ra представляет собой атом водорода, C1-С6 алкильную, С2-С6 алкенильную или С2-С6 алкинильную группу; Rb представляет собой атом водорода, C1-С6 алкильную, С2-С6 алкенильную или С2-С6 алкинильную группу; Rc представляет собой атом водорода, C1-С6 алкильную, С2-С6 алкенильную или С2-С6 алкинильную группу; Rd представляет собой атом водорода, C1-С6 алкильную, С2-С6 алкенильную или С2-С6 алкинильную группу, и Ya представляет собой простую связь, C1-С6 алкиленовую, С2-С6 алкениленовую или С2-С6 алкиниленовую группу, где каждая гетероалкильная группа содержит по меньшей мере один атом углерода и один или более атом (-ов) водорода могут быть заменены атомами фтора или хлора.
Конкретными примерами гетероалкильных групп являются метокси, трифторметокси, этокси, н-пропилокси, изопропилокси, бутокси, трет-бутилокси, метоксиметил, этоксиметил, -СН2СН2ОН, -СН2ОН, метоксиэтил, 1-метоксиэтил, 1-этоксиэтил, 2-метоксиэтил или 2-этоксиэтил, метиламино, этиламино, пропиламино, изопропиламино, диметиламино, диэтиламино, изопропилэтиламино, метиламинометил, этиламинометил, диизопропиламиноэтил, метилтио, этилтио, изопропилтио, енольно-эфирная группа, диметиламинометил, диметиламиноэтил, ацетил, пропионил, бутирилокси, ацетилокси, метоксикарбонил, этоксикарбонил, пропионилокси, ацетиламино или пропиониламино, карбоксиметил, карбоксиэтил или карбоксипропил, N-этил-N-метилкарбамоил или N-метилкарбамоил. Дополнительно примерами гетероалкильных групп являются нитрильная, изонитрильная, цианатная, тиоцианатная, изоцианатная, изотиоцианатная и алкилнитрильная группы.
Термин «алкокси» относится к алкильной группе, соединенной одинарной связью с кислородом.
Термин «алкилтио» относится к алкильной группе, соединенной одинарной связью с серой.
Выражения «циклоалкил» и «карбоциклическое кольцо» относятся к насыщенной циклической углеводородной группе, которая содержит одно или более колец, предпочтительно 1 или 2, и содержит от 3 до 14 атомов углерода в кольце, предпочтительно от 3 до 10, в частности 3, 4, 5, 6 или 7 атомов углерода в кольце, примерами являются циклопропильная, циклобутильная, циклопентильная, спиро[4,5]деканильная, норборнильная, циклогексильная, декалинильная, бицикло[4.3.0]нонильная, тетралиновая или циклопентилциклогексильная группы. Кроме того, термин «циклоалкил» относится к группам, в которых один или более атом (-ов) водорода заменены атомами фтора, хлора, брома или иода или группами ОН, =O, SH, NH2, =NH, N3 или NO2, таким как, например, циклические кетоны, такие как, например, циклогексанон, 2-циклогексенон или циклопентанон. Дополнительно конкретными примерами циклоалкильных групп являются циклопропильная, циклобутильная, циклопентильная, спиро[4,5]деканильная, норборнильная, циклогексильная, циклопентенильная, циклогексадиенильная, декалинильная, бицикло[4.3.0]нонильная, тетралиновая, циклопентилциклогексильная, фторциклогексильная или циклогекс-2-енильная группы.
Термин «арил» относится к ароматический группе, которая содержит одно или более колец, содержащих от 6 до 14 атомов углерода в кольце, предпочтительно от 6 до 10, в частности 6, атомов углерода в кольце.
Термин «гетероарил» относится к ароматической группе, которая содержит одно или более колец, содержащих от 5 до 14 атомов в кольце, предпочтительно от 5 до 10, в частности 5 или 6, атомов в кольце, и содержит один или более, предпочтительно 1, 2, 3 или 4, атомов кислорода, азота, фосфора или серы в кольце, предпочтительно О, S или N. Примерами являются пиридильная (например, 4-пиридильная) имидазолильная (например, 2-имидазолильная), фенилпирролильная (например, 3-фенилпирролильная), тиазолильная, изотиазолильная, 1,2,3-триазолильная, 1,2,4-триазолильная, оксадиазолильная, тиадиазолильная, индолильная, индазолильная, тетразолильная, пиразинильная, пиримидинильная, пиридазинильная, оксазолильная, изоксазолильная, триазолильная, тетразолильная, изоксазолильная, индазолильная, индолильная, бензимидазолильная, бензоксазолильная, бензизоксазолильная, бензтиазолильная, пиридазинильная, хинолинильная, изохинолинильная, пирролильная, пуринильная, карбазолильная, акридинильная, пиримидильная, 2,3'-бифурильная, пиразолильная (например, 3-пиразолильная) и изохинолинильная группы. Термин «гетероциклоалкил» относится к циклоалкильной группе, определенной выше, в которой один или более (предпочтительно 1, 2 или 3) атомов углерода кольца, каждый независимо, заменены атомом кислорода, азота, кремния, селена, фосфора или серы (предпочтительно атомом кислорода, серы или азота). Гетероциклоалкильная группа содержит предпочтительно 1 или 2 кольца, содержащих от 3 до 10 (в частности, 3, 4, 5, 6 или 7) атомов кольца (предпочтительно выбранных из С, О, N и S). Кроме того, термин «гетероциклоалкил» относится к группам, в которых один или более атомов водорода заменены атомами фтора, хлора, брома или иода или группами ОН, =O, SH, =S, NH2, =NH, N3 или NO2. Примерами являются пиперидильная, пролинильная, имидазолидинильная, пиперазинильная, морфолинильная, уротропинильная, пирролидинильная, тетрагидротиофенильная, тетрагидропиранильная, тетрагидрофурильная или 2-пиразолинильная группы, а также лактамы, лактоны, циклические имиды и циклические ангидриды.
Термин «алкилциклоалкил» относится к группе, которая содержит как циклоалкильную, так и алкильную, алкенильную или алкинильную группы в соответствии с указанными выше определениями, например, алкилциклоалкильной, циклоалкилалкильной, алкилциклоалкенильной, алкенилциклоалкильной и алкинилциклоалкильной группам. Алкилциклоалкильная группа предпочтительно содержит циклоалкильную группу, которая содержит одну или две кольцевых системы, содержащих от 3 до 10 (в частности, 3, 4, 5, 6 или 7) атомов углерода в кольце, и одну или две алкильные, алкенильные или алкинильные группы, содержащие 1 или от 2 до 6 атомов углерода. Термин «аралкил» относится к группе, содержащей как арильную, так и алкильную, алкенильную, алкинильную и/или циклоалкильную группы в соответствии с указанными выше определениями, такой как, например, арилалкильная, арилалкенильная, арилалкинильная, арилциклоалкильная, арилциклоалкенильная, алкиларилциклоалкильная и алкиларилциклоалкенильная группы. Конкретными примерами аралкилов являются толуол, ксилол, мезителен, стирол, бензилхлорид, о-фтортолуол, 1Н-инден, тетралин, дигидронафталин, инданон, фенилциклопентил, кумол, циклогексилфенил, флуорен и индан. Аралкильная группа предпочтительно содержит одну или две ароматических кольцевых системы (1 или 2 кольца), содержащих от 6 до 10 атомов углерода, и одну или две алкильные, алкенильные и/или алкинильные группы, содержащие от 1 или 2 до 6 атомов углерода, и/или циклоалкильную группу, содержащую 5 или 6 атомов углерода в кольце.
Термин «гетероалкилциклоалкил» относится к алкилциклоалкильным группам, определенным выше, в которых один или более, предпочтительно 1, 2 или 3, атомов углерода независимо друг от друга заменены атомом кислорода, азота, кремния, селена, фосфора или серы (предпочтительно атомом кислорода, серы или азота). Гетероалкилциклоалкильная группа предпочтительно содержит 1 или 2 кольцевых системы, содержащих от 3 до 10 (в частности, 3, 4, 5, 6 или 7) атомов в кольце, и одну или две алкильные, алкенильные, алкинильные или гетероалкильные группы, содержащие от 1 или 2 до 6 атомов углерода. Примерами таких групп являются алкилгетероциклоалкил, алкилгетероциклоалкенил, алкенилгетероциклоалкил, алкинилгетероциклоалкил, гетероалкилциклоалкил, гетероалкилгетероциклоалкил и гетероалкилгетероциклоалкенил, указанные циклические группы могут быть насыщенными или моно-, ди- или триненасыщенными.
Выражение «гетероциклическое кольцо» относится к гетероарильной группе, определенной выше, циклоалкильной группе или карбоциклическому кольцу, определенным выше, в которых один или более (предпочтительно 1, 2 или 3) атомов углерода в кольце, каждый независимо, заменены атомом кислорода, азота, кремния, селена, фосфора или серы, предпочтительно атомом кислорода, серы или азота. Гетероциклическое кольцо предпочтительно содержит 1 или 2 кольца, содержащие от 3 до 10, в частности 3, 4, 5, 6 или 7 атомов в кольце, предпочтительно выбранных из С, О, N и S. Примерами являются азиридинильная, оксиранильная, тииранильная (thiiranyl), оксазиридинильная, диоксиранильная, азетидинильная, оксетанильная, тиетанильная, диазетидинильная, диоксетанильная, дитиетанильная, пирролидинильная, тетрагидрофуранильная, тиоланильная, фосфоланильная, силоланильная, азолильная, тиазолильная, изотиазолильная, имидазолидинильная, пиразолидинильная, оксазолидинильная, изоксазолидинильная, тиазолидинильная, изотиазолидинильная, диоксоланильная, дитиоланильная, пиперазинильная, морфолинильная, тиоморфолинильная, триоксанильная, азепанильная, оксепанильная, тиепанильная, гомопиперазинильная или уротропинильная группы.
Термин «гетероаралкил» относится к аралкильной группе, определенной выше, в которой один или более (предпочтительно 1, 2, 3 или 4) атомов углерода, каждый независимо, заменены атомом кислорода, азота, кремния, селена, фосфора, бора или серы (предпочтительно кислорода, серы или азота), то есть группе, содержащей как арильную или гетероарильную, соответственно, так и алкильную, алкенильную, алкинильную и/или гетероалкильную и/или циклоалкильную и/или гетероциклоалкильную группы в соответствии с указанными выше определениями. Гетероаралкильная группа предпочтительно содержит одну или две ароматических кольцевых системы (1 или 2 кольца), содержащих от 5 или 6 до 10 атомов углерода в кольце, и одну или две алкильные, алкенильные и/или алкинильные группы, содержащие от 1 или 2 до 6 атомов углерода, и/или циклоалкильную группу, содержащую 5 или 6 атомов углерода в кольце, где 1, 2, 3 или 4 из этих атомов углерода заменены атомами кислорода, серы или азота.
Примерами являются арилгетероалкильная, арилгетероциклоалкильная, арилгетероциклоалкенильная, арилалкилгетероциклоалкильная, арилалкенил-гетероциклоалкильная, арилалкинилгетероциклоалкильная, арилалкилгетероциклоалкенильная, гетероарилалкильная, гетероарилалкенильная, гетероарилалкинильная, гетероарилгетероалкильная, гетероарилциклоалкильная, гетероарилциклоалкенильная, гетероарилгетероциклоалкильная, гетероарилгетероциклоалкенильная, гетероарилалкил-циклоалкильная, гетероарилалкилгетероциклоалкенильная, гетероарилгетероалкил-циклоалкильная, гетероарилгетероалкилциклоалкенильная и гетероарилгетероалкил-гетероциклоалкильная группы, указанные циклические группы могут быть насыщенными или моно-, ди- или триненасыщенными. Конкретными примерами являются тетрагидроизохинолинильная, бензоильная, 2- или 3-этилиндолильная, 4-метилпиридино, 2-, 3- или 4-метоксифенильная, 4-этоксифенильная, 2-, 3- или 4-карбоксифенилалкильная группы.
Как уже было указано выше, термины «циклоалкил», «гетероциклоалкил», «алкилциклоалкил», «гетероалкилциклоалкил», «арил», «гетероарил», «аралкил» и «гетероаралкил» также относятся к группам, в которых один или более атомов водорода таких групп независимо друг от друга заменены атомами фтора, хлора, брома или иода или группами ОН, =O, SH, =S, NH2, =NH, N3 или NO2.
Общий термин «кольцо» в настоящем описании, если не указано иное, включает циклоалкильные группы или карбоциклические кольца, гетероциклические кольца, арильные группы и гетероарильные группы.
Термины «гало», «галоген» или «атом галогена» в настоящем описании означают фтор, хлор, бром или йод, предпочтительно фтор и/или хлор.
Выражение «моно- или дисахарид и их производные» в настоящем описании означает углеводород или сахар, принадлежащий к или полученный из группы моносахаридов или дисахаридов.
Примеры моно-, дисахаридов и производных включают глюкозу, 3-O-метилглюкозу, 1-дезоксиглюкозу, 6-дезоксиглюкозу, галактозу, маннозу, фруктозу, ксилозу, рибозу, целлобиозу, мальтозу, лактозу, гентиобиозу, сахарозу, трегалозу и маннитол, сорбитол и рибитол. Предпочтительно сахариды представляют собой сахариды в D-форме, например D-глюкозу, 3-О-метил-D-глюкозу, 1-дезокси-D-глюкозу или 6-дезокси-D-глюкозу, D-галактозу, D-маннозу.
В настоящем описании выражение, определяющее пределы диапазона длины, такое как, например, «от 1 до 5», означает любое целое число от 1 до 5, то есть 1, 2, 3, 4 и 5. Другими словами, подразумевается, что любой диапазон, определенный двумя явно указанными целыми числами, включает и раскрывает любое целое число, определяющее указанные пределы и любое целое число, входящее в указанный диапазон.
Выражение «фрагмент -С(=O)O-» в настоящем описании используется для того, чтобы четко определить группу, содержащую sp2-гибридизованный карбонильный атом углерода, присоединенный (i) к любому атому углерода или гетероатому и (ii) к атому кислорода, который в свою очередь может быть присоединен к атому водорода или атому любого другого химического элемента. Термин «карбоксильная группа» не используется для описания «фрагмента -С(=O)O-», поскольку он может быть неправильно понят как описывающий только карбоновую кислоту.
Термин «в альфа-положении» используется для описания непосредственно соседнего положения, в то время как термин «в бета-положении» указывает на соседнее положение атома или группы А и атома или группы В, характеризующееся тем, что между А и В расположен еще один атом или группа.
В настоящем описании термин «оксамид» относится к произвольно замещенному органическому соединению, содержащему 2 карбонильных атома углерода и два атома азота, которое представляет собой произвольно замещенный диамид, полученный из любого производного щавелевой кислоты.
Специалисты в данной области техники легко поймут, что некоторые из аналогов ПНЖК n-3 общей формулы (I) согласно настоящему изобретению представляют собой «биоизостеры» встречающихся в природе эпоксиметаболитов, получаемых посредством ферментов цитохрома Р450 (CYP) из полиненасыщенных жирных кислот (ПНЖК) омега-3 (n-3). Биоизостер представляет собой соединение, получаемое в результате обмена атома или группы атомов на альтернативный, в широком смысле аналогичный, атом или группу атомов с получением, тем самым, нового соединения с биологическими свойствами, аналогичными свойствам исходного соединения. Биоизостерия используется, например, медицинскими химиками для улучшения желаемых биологических или физических свойств соединения, например, для ослабления токсичности, изменения активности, изменения фармакокинетики и/или метаболизма соединения. Например, замена атома водорода атомом фтора в месте метаболического окисления соединения может предотвратить протекание такого метаболизма. Поскольку атом фтора по размеру аналогичен атому водорода, на общую топологию молекулы это существенно не влияет, и необходимая биологическая активность остается неизменной. Тем не менее, с заблокированным путем для метаболизма указанное соединение может иметь более длительный период полувыведения. Другим примером является биоизостерная замена групп карбоновой кислоты, в результате которой получаются аналоги, демонстрирующие улучшенную биодоступность, улучшенное проникновение через гематоэнцефалический барьер, повышенную активность, лучшую химическую стабильность и/или селективность по отношению к мишени (см., например, учебник "The practice of medicinal chemistry", edited by Camille Georges Wermuth, 3rd edition, Academic Press, 2008, например, с. 303-310; Ballatore С. et al. "Carboxylic Acid (Bio)lsosteres in Drug Design", ChemMedChem 8, 385-395 (2013)). Кроме того, биоизостеризм также может быть использован для получения пролекарства соединения, то есть соединения, которое первоначально вводят субъекту или пациенту в неактивной (или менее активной) форме, а затем оно модифицируется in vivo в его активную форму путем обычных метаболических процессов, протекающих в организме. Например, конъюгация соединения с частями липидов и/или Сахаров позволила получить аналоги (пролекарства), демонстрирующие усиленную доставку лекарственного средства по сравнению с исходным соединением (см., например, Wong A. and Toth I. "Lipid, Sugar and Liposaccharide Based Delivery Systems", Current Medicinal Chemistry 8,1123-1136 (2001)).
Аналоги ПНЖК n-3 общей формулы (I) согласно настоящему изобретению могут быть получены рядом способов, хорошо известных специалистам в области органического синтеза. Например, соединения согласно настоящему изобретению могут быть синтезированы в соответствии с представленными ниже общими схемами реакций с использованием способов синтеза, известных в области синтетической органической химии или их вариантов, очевидных специалистам в данной области техники. Если не указано иное, все переменные, например n, k, R2 (обозначаемый также R2), R6, R7, R8, R41, R42, R44 и R45, имеют вышеуказанное определенное значение. В качестве исходных материалов можно использовать реагенты стандартного коммерческого класса без дополнительной очистки, или реагенты могут быть легко получены из таких материалов рутинными способами. Специалистам в области органического синтеза будет понятно, что исходные материалы и условия реакций могут быть различными, включая дополнительные стадии, используемые для получения соединений для применения, охватываемых настоящим изобретением.
Соединения согласно настоящему изобретению являются эффективными в лечении, снижении риска развития или предотвращении нарушения, связанного с неоваскуляризацией и/или воспалением. В одном из вариантов реализации указанное нарушение представляет собой нарушение, связанное с неоваскуляризацией. В другом варианте реализации указанное нарушение представляет собой нарушение, связанное с воспалением.
Примеры нарушения, связанного с воспалением, включают воспалительные нарушения, воспаление, вызванное другими заболеваниями независимо от типа, этиологии или патогенеза, воспаление, вызванное воспалительными заболеваниями, примеры которых приведены ниже, и иммунологические нарушения.
В одном из вариантов реализации нарушение, связанное с воспалением, представляет собой воспалительное нарушение. Примерами воспалительных нарушений являются реакция острой фазы, местное и системное воспаление.
В одном из вариантов реализации нарушение, связанное с воспалением, представляет собой иммунологическое нарушение. Примерами иммунологических нарушений являются гиперестезия, аутоиммунные нарушения, отторжение трансплантата при трансплантации, токсичность трансплантата, гранулематозное воспаление/ремоделирование тканей, миастения гравис, иммуносупрессия, болезни иммунных комплексов, избыточная и недостаточная выработка антител и васкулит.
В одном из вариантов реализации нарушение, связанное с воспалением, представляет собой воспаление, вызванное другими заболеваниями независимо от типа, этиологии или патогенеза, или воспаление, вызванное воспалительными заболеваниями. Примеры таких состояний и заболеваний включают воспалительное заболевание кишечника, включая болезнь Крона и язвенный колит, синдром раздраженного кишечника, энтероколит, заболевания печени, панкреатит, нефрит, цистит (интерстициальный цистит), отит среднего уха, периодонтит, воспалительные заболевания кожи, такие как псориаз, экзема, атопические заболевания, дерматит, ревматоидный артрит и подагрический артрит с началом в юношеском или взрослом возрасте, анкилозирующий спондилит, болезнь Стилла с началом во взрослом возрасте или у детей (ювенильный идиопатический артрит с системным началом), псориатический артрит, остеоартрит и отек, связанные с ожогами, растяжениями или переломом, отек головного мозга, ангионевротический отек, васкулит, диабетическую васкулопатию, диабет типа I, диабетическую нефропатию, диабетическую нейропатию, диабетическую ретинопатию, посткапиллярное сопротивление или диабетические синдромы, связанные с инсулитом (например, гипергликемию, диурез, протеинурию и повышенную экскрецию с мочой нитритов и калликреина), заболевания желчного пузыря, рассеянный склероз, эпилепсию, боковой амиотрофический склероз, синдром системного воспалительного ответа (ССВО), ишемически-реперфузионное повреждение и атеросклероз, септический шок, воспаление, вызванное антигиповолемическими и/или антигипотензивными агентами, мигрень, гингивит, остеопороз, доброкачественную гиперплазию предстательной железы, гиперактивный мочевой пузырь, фиброзные заболевания, такие как фиброз легких, фиброз почек, прогрессирующий склероз и образование рецидивирующих стриктур при болезни Крона, нарушения дыхательных путей при астме, атопическую или неатопическую астму, профессиональную астму, вызванный физической нагрузкой бронхоспазм, бронхит, пневмокониоз, включая алюминоз, антракоз, асбестоз, халикоз, птилоз, сидероз, силикоз, табакоз и биссиноз, хроническую обструктивную болезнь легких, включая эмфизему, респираторный дистресс-синдром взрослых, пневмонию, аллергический ринит, вазомоторный ринит и плеврит, аутовоспалительные заболевания, такие как семейная средиземноморская лихорадка (ССЛ), периодический синдром, связанный с рецептором фактора некроза опухолей (TRAPS, англ.: tumor-necrosis factor receptor associated periodic syndrome), мультисистемное воспалительное заболевание неонатального возраста (NOMID, англ.: neonatal onset multisystem inflammatory disease), семейный холодовой аутовоспалительный синдром (FCAS, англ.: familial cold autoinflammatory syndrome), включая семейную холодовую крапивницу (FCU, англ.: familial cold urticaria), синдром стерильного гнойного артрита, гангренозной пиодермии и акне (PAPA, англ.: pyogenic arthritis pyoderma gangrenosum acne) и болезнь Макла-Уэлса.
В предпочтительном варианте реализации нарушение, связанное с неоваскуляризацией и/или воспалением, представляет собой офтальмологическое нарушение, связанное с неоваскуляризацией, например, связанное с неоваскуляризацией роговицы, сетчатки, хориоидальной, увеальной неоваскуляризацией или неоваскуляризацией радужной оболочки, и/или офтальмологическое нарушение, связанное с воспалением. Предпочтительно офтальмологическое нарушение, связанное с неоваскуляризацией, представляет собой возрастную макулярную дегенерацию, например, неоваскулярную (влажную) ВМД или атрофическую (сухую) ВМД. В этом варианте реализации лечение приводит к регрессии кровеносных сосудов. В некоторых вариантах реализации офтальмологическое нарушение, связанное с неоваскуляризацией, представляет собой ретинопатию, предпочтительно ретинопатию недоношенных (РН); диабетическую ретинопатию; пролиферативную диабетическую ретинопатию, окклюзию вены сетчатки; например, окклюзию ветви вены сетчатки, окклюзию центральной вены сетчатки; серповидно-клеточную ретинопатию и радиационную ретинопатию; болезнь Беста или болезнь Штаргардта. Предпочтительно офтальмологическое нарушение, связанное с воспалением, представляет собой возрастную макулярную дегенерацию.
В одном из вариантов реализации нарушение, связанное с неоваскуляризацией и/или воспалением, не является сердечно-сосудистым заболеванием.
В предпочтительном варианте реализации соединение или композицию для применения согласно настоящему изобретению вводят перорально, местно, подкожно, интравитреально, внутримышечно, внутрибрюшинно, внутривенно или интраназально, предпочтительно перорально или внутривенно, более предпочтительно перорально или внутрибрюшинно. Предпочтительными путями доставки офтальмологических лекарственных средств к глазу для лечения офтальмологического нарушения являются местный, местный глазной (например, субконъюктивальный, интравитреальный, ретробульбарный, интракамеральный) и системный. Последнее предпочтительно достигается путем перорального, внутримышечного или внутривенного введения.
Также предпочтительно, чтобы соединение или композиция для применения согласно настоящему изобретению представляла собой лекарственную форму, выбранную из группы, состоящей из спрея, аэрозоля, пены, формы для ингаляций, порошка, таблетки, капсулы, мягкой желатиновой капсулы, чая, сиропа, гранулы, жевательной таблетки, мази (salve), крема, геля, суппозитория, пастилки, липосомальной композиции и раствора, подходящего для инъекций.
Соединение или композиция для применения в соответствии с настоящим изобретением могут дополнительно содержать по меньшей мере одно соединение формулы (I) и, необязательно, одно или более веществ-носителей, например, циклодекстрины, такие как гидроксипропил-β-циклодекстрин, мицеллы или липосомы, вспомогательные вещества и/или адъюванты. Дополнительно могут содержаться, например, один или более из воды, буферов, таких как, например, нейтральный солевой буферный раствор или фосфатно-солевой буферный раствор, этанол, минеральное масло, растительное масло, диметилсульфоксид, углеводы, такие как, например, глюкоза, манноза, сахароза или декстраны, маннитол, белки, адъюванты, полипептиды или аминокислоты, такие как глицин, антиоксиданты, хелатирующие агенты, такие как ЭДТА или глутатион, и/или консерванты. Кроме того, в композиции для применения, предложенные в настоящем описании, могут, но необязательно, быть включены один или более других активных ингредиентов. Например, соединения согласно настоящему изобретению могут быть успешно применены в комбинации с антибиотиком, противогрибковым или противовирусным агентом, антигистаминным агентом, нестероидным противовоспалительным лекарственным средством, противоревматическим лекарственым средством, модифицирующим течение заболевания, противовоспалительным лекарственным средством для лечения аутоиммунного заболевания, цитостатическим лекарственным средством, антиангиогенным лекарственным средством или смесями вышеуказанных агентов. Предпочтительно лекарственное средство представляет собой антиангиогенное лекарственное средство, более предпочтительно ингибитор фактора роста эндотелия сосудов (VEGF) или рецептора фактора роста эндотелия сосудов (VEGFR).
Композиции для применения могут быть приготовлены для введения любым подходящим путем, включая, например, местное введение, такое как трансдермальное или глазное, пероральное, трансбуккальное, назальное, вагинальное, ректальное или парентеральное введение. Термин «парентеральный» в настоящем описании включает подкожную, внутрикожную, внутрисосудистую, такую как, например, внутривенную, внутримышечную, спинальную, внутричерепную, интратекальную, интраокулярную, периокулярную, интраорбитальную, внутрисуставную и внутрибрюшинную и местную глазную (например, субконъюктивальную, интравитреальную, ретробульбарную, интракамеральную) инъекцию, а также любую подобную инъекцию или инфузию. В некоторых вариантах реализации предпочтительными являются композиции в форме, подходящей для перорального применения. Такие формы включают, например, таблетки, троше, пастилки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, сиропы или эликсиры. В рамках других вариантах композиции, предложенные в настоящем описании, могут быть приготовлены в виде лиофилизата.
Композиции, предназначенные для перорального применения, могут дополнительно содержать один или более компонентов, таких как подслащивающие агенты, ароматизирующие агенты, окрашивающие агенты и/или консервирующие агенты, для создания привлекательных на вид и приятных на вкус препаратов. Таблетки содержат активный ингредиент в смеси с физиологически приемлемыми вспомогательными веществами, которые являются подходящими для получения таблеток. Такие вспомогательные вещества включают, например, инертные разбавители, такие как, например, карбонат кальция, карбонат натрия, лактозу, фосфат кальция или фосфат натрия, гранулирующие и разрыхляющие агенты, такие как, например, кукурузный крахмал или альгиновая кислота, связующие агенты, такие как, например, крахмал, желатин или аравийская камедь, и смазывающие агенты, такие как, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут не содержать покрытия, или они могут быть покрыты известными способами для обеспечения задержки разложения и всасывания в желудочно-кишечном тракте и тем самым обеспечения замедленного действия в течение более длительного периода времени. Например, может быть использован материал, обеспечивающий задержку во времени, такой как глицерилмоностеарат или глицерилдистеарат. Способы получения таких композиций известны (см., например, Н.С. Ansel and N.G. Popovish, Pharmaceutical Dosage Forms and Drug Delivery Systems, 5th ed., Lea and Febiger (1990)).
Составы для перорального применения могут быть также представлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, таким как, например, карбонат кальция, фосфат кальция или каолин, или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляной средой, такой как, например, арахисовое масло, жидкий парафин или оливковое масло.
Водные суспензии содержат активный(-ые) ингредиент(-ы) в смеси со вспомогательными веществами, подходящими для получения водных суспензий. Такие вспомогательные вещества включают суспендирующие агенты, такие как, например, натрий карбоксиметилцеллюлоза, метилцеллюлоза, гидропропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; и диспергирующие или смачивающие агенты, такие как, например, существующие в природе фосфатиды, такие как лецитин, продукты конденсации алкиленоксида с жирными кислотами, такие как полиоксиэтиленстеарат, продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, такими как гептадексэтиленоксицетанол, продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситола, такие как полиоксиэтиленсорбитолмоноолеат, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и ангидридов гекситола, такие как полиэтиленсорбитанмоноолеат. Водные суспензии также могут один или более консервантов, например этил- или н-пропил-n-гидроксибензоат, один или более окрашивающих агентов, один или более ароматизирующих агентов и один или более подслащивающих агентов, таких как сахароза или сахарин.
Масляные суспензии могут быть приготовлены путем суспендирования активных ингредиентов в растительном масле, таком как, например, арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Для получения приятных на вкус препаратов для перорального введения могут быть добавлены подслащивающие агенты, такие как указано выше, и/или ароматизирующие агенты. Такие суспензии могут быть сохранены путем добавления антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для получения водной суспензии путем добавления воды, обеспечивают активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и один или консервантов. Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты представлены агентами, уже указанными выше. Также могут присутствовать дополнительные вспомогательные вещества, такие как подслащивающие, ароматизирующие и окрашивающие агенты.
Композиции для применения также могут находиться в форме эмульсий масло-в-воде. Масляная фаза может представлять собой растительное масло, такое как, например, оливковое масло или арахисовое масло, минеральное масло, такое как, например, жидкий парафин или их смесь. Подходящие эмульгирующие агенты включают существующие в природе смолы, такие как, например, аравийская камедь или трагакантовая камедь, существующие в природе фосфатиды, такие как, например, соевый лецитин, и сложные эфиры или частичные сложные эфиры, полученные из жирных кислот и гекситола, ангидридов, такие как, например, сорбитанмоноолеат, и продукты конденсации частичных сложных эфиров, полученных из жирных кислот и гекситола с этиленоксидом, такие как, например, полиоксиэтиленсорбитанмоноолеат. Эмульсия может также содержать один или более подслащивающих и/или ароматизирующих агентов.
Сиропы и эликсиры может быть приготовлены с подслащивающими агентами, такими как глицерин, пропиленгликоль, сорбит или сахароза. Такие составы могут также содержать один или более смягчающих агентов, консервантов, ароматизирующих агентов и/или окрашивающих агентов.
Соединения для применения в соответствии с настоящим изобретением могут быть включены в составы для локального или местного введения, такого как местное нанесение на кожу или слизистые оболочки, такие как находятся в глазу. Композиции для местного введения, как правило, содержат местный носитель, объединенный с активным(-ыми) агентом(-ами), с дополнительными необязательными компонентами или без них. Подходящие местные носители и дополнительные компоненты хорошо известны в уровне техники, и будет очевидно, что выбор носителя будет зависеть от конкретной физической формы и способа доставки. Местные носители включают воду; органические растворители, такие как спирты, такие как, например, этанол, или изопропиловый спирт, или глицерин; гликоли, такие как, например, бутилен, изопрен или пропиленгликоль; алифатические спирты, такие как, например, ланолин; смеси воды и органических растворителей и смеси органических растворителей, таких как спирт и глицерин; материалы на основе липидов, такие как жирные кислоты, ацилглицерины, включая масла, такие как, например, минеральное масло, и жиры натурального или синтетического происхождения, фосфоглицериды, сфинголипиды и воски; материалы на основе белка, такие как коллаген и желатин; материалы на основе кремния, как нелетучие, так и летучие; и материалы на основе углеводородов, такие как микрогубки и полимерные матрицы. Композиция может дополнительно содержать один или более компонентов, адаптированных для повышения стабильности или эффективности применяемого состава, такие как стабилизирующие агенты, суспендирующие агенты, эмульгирующие агенты, регуляторы вязкости, желирующие агенты, консерванты, антиоксиданты, агенты, способствующие проникновению через кожу, увлажнители и материалы, обеспечивающие замедленное высвобождение. Примеры таких компонентов описаны в источнике Martindale - The Extra Pharmacopoeia (Pharmaceutical Press, London, 1993) и Martin (ed.), Remington's Pharmaceutical Sciences. Составы могут содержать микрокапсулы, микрокапсулы гидроксиметилцеллюлозы или желатина, липосомы, микросферы альбумина, микроэмульсии, наночастицы или нанокапсулы.
Состав для местного применения может быть приготовлен в различных физических формах, включая, например, твердые вещества, пасты, кремы, пены, лосьоны, гели, порошки, водные жидкости, эмульсии, спреи, глазные капли и накожные пластыри. Физический внешний вид и вязкость таких форм может определяться присутствием и количеством эмульгатора(-ов) и регулятора(-ов) вязкости, присутствующих в составе. Твердые вещества, как правило, являются твердыми и нетекучими, и их часто готовят в виде брусков или стиков, или в форме частиц; твердые вещества могут быть непрозрачными или прозрачными и необязательно могут содержать растворители, эмульгаторы, увлажнители, смягчающие агенты, отдушки, красители/окрашивающие агенты, консерванты и другие активные ингредиенты, которые увеличивают или усиливают эффективность конечного продукта. Кремы и лосьоны часто похожи друг на друга, отличаясь главным образом по вязкости; и лосьоны, и кремы могут быть непрозрачными, полупрозрачными или прозрачными и часто содержат эмульгаторы, растворители и агенты, регулирующие вязкость, а также увлажнители, смягчающие агенты, отдушки, красители/окрашивающие агенты, консерванты и другие активные ингредиенты, которые увеличивают или усиливают эффективность конечного продукта. Гели могут быть получены с диапазоном вязкостей, от густого, или геля высокой вязкости, до жидкого, или геля низкой вязкости. Эти составы, подобно составам лосьонов и кремов, могут также содержать растворители, эмульгаторы, увлажнители, смягчающие агенты, отдушки, красители/окрашивающие агенты, консерванты и другие активные ингредиенты, которые увеличивают или усиливают эффективность конечного продукта. Жидкости имеют меньшую вязкость, чем кремы, лосьоны или гели, и часто не содержат эмульгаторов. Жидкие продукты для местного применения часто содержат растворители, эмульгаторы, увлажнители, смягчающие агенты, отдушки, красители/окрашивающие агенты, консерванты и другие активные ингредиенты, которые увеличивают или усиливают эффективность конечного продукта.
Подходящие эмульгаторы для применения в составах для местного применения включают, но не ограничиваются ими, ионные эмульгаторы, цетеариловый спирт, неионные эмульгаторы, такие как полиоксиэтиленолеиловый эфир, ПЭГ-40-стеарат, цетеарет-12, цетеарет-20, цетеарет-30, цетеаретовый спирт, ПЭГ-100-стеарат и глицерилстеарат. Подходящие агенты, регулирующие вязкость включают, но не ограничиваются ими, защитные коллоиды или неионные камеди, такие как гидроксиэтилцеллюлоза, ксантановая камедь, силикат магния-алюминия, диоксид кремния, микрокристаллический воск, пчелиный воск, парафин и цетилпальмитат. Гелевая композиция может быть получена путем добавления желирующего агента, такого как хитозан, метилцеллюлоза, этилцеллюлоза, поливиниловый спирт, поликватерниумы, гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбомер или глицирризинат аммония. Подходящие поверхностно-активные вещества включают, но не ограничиваются ими, неионные, амфотерные, ионные и анионные поверхностно-активные вещества. Например, в составах для местного применения может быть использован один или более из диметиконсополиола, полисорбата 20, полисорбата 40, полисорбата 60, полисорбата 80, лаурамида ДЭА, кокамида ДЭА, и кокамида МЭА, олеилбетаина, кокамидопропилфосфатидил-ПГ-димония хлорида и лауретсульфата аммония.
Подходящие консерванты включают, но не ограничиваются ими, противомикробные агенты, такие как метилпарабен, пропилпарабен, сорбиновая кислота, бензойная кислота и формальдегид, а также физические стабилизаторы и антиоксиданты, такие как витамин Е, аскорбат натрия/аскорбиновая кислота и пропилгаллат. Подходящие увлажнители включают, но не ограничиваются ими, молочную кислоту и другие гидроксикислоты и их соли, глицерин, пропиленгликоль и бутиленгликоль. Подходящие смягчающие агенты включают ланолиновый спирт, ланолин, производные ланолина, холестерин, петролатум, изостеарилнеопентаноат и минеральные масла. Подходящие отдушки и окрашивающие агенты включают, но не ограничиваются ими, FD&C Red No. 40 и FD&С Yellow No. 5. Другие подходящие дополнительные ингредиенты, которые могут быть включены в состав для местного применения, включают, но не ограничиваются ими, абразивы, абсорбенты, агенты, препятствующие слипанию, антивспениватели, антистатики, вяжущие агенты, такие как, например, гамамелис (witch hazel), спирт и растительные экстракты, такие как экстракт ромашки, связующие агенты/вспомогательные вещества, буферные агенты, хелатирующие агенты, пленкообразующие агенты, кондиционирующие агенты, пропелленты, замутнители, регуляторы рН и защитные агенты.
Примером подходящего носителя для приготовления состава для местного применения в виде геля является: гидроксипропилцеллюлоза (2,1%); 70/30 изопропиловый спирт/вода (90,9%); пропиленгликоль (5,1%) и полисорбат 80 (1,9%). Примером подходящего носителя для приготовления состава для местного применения в виде пены является: цетиловый спирт (1,1%); стеариловый спирт (0,5%); Кватерниум 52 (1,0%); пропиленгликоль (2,0%); этанол 95 PGF3 (61,05%); деионизированная вода (30,05%); Р75 углеводородный пропеллент (4,30%). Все проценты являются массовыми.
Типичные способы доставки композиций для местного применения включают нанесение с использованием пальцев; нанесение с использованием физического аппликатора, такого как салфетка (cloth), ткань (tissue), тампон (swab), стик или кисть; распыление, включая распыление распрыскивателя (mist), аэрозоля или пены; нанесения через капельницу; разбрызгивание; пропитка и ополаскивание (rinsing). Также могут быть использованы носители, обеспечивающие контролируемое высвобождение, и композиции могут быть приготовлены для трансдермального введения в виде трансдермального пластыря.
Композиция для применения может быть приготовлена в виде составов для ингаляций, включая спреи (sprays), распрыскиватели (mists) или аэрозоли (aerosols). Такие составы являются особенно подходящими для лечения астмы или других состояний, связанных с дыхательными путями. Для составов для ингаляции соединения, предложенные в настоящем описании, могут быть доставлены любыми ингаляционными способами, известными специалистам в данной области техники. Такие ингаляционные способы и устройства включают, но не ограничиваются ими, дозированные ингаляторы с пропеллентами, такими как CFC или HFA, или пропеллентами, которые являются физиологически и экологически приемлемыми. Другими подходящими устройствами являются ингаляторы, приводимые в действие дыханием, многодозовые ингаляторы сухих порошков и аэрозольные небулайзеры. Составы в виде аэрозолей для применения в рассматриваемом способе, как правило, включают пропелленты, поверхностно-активные вещества и сорастворители, и могут быть помещены в обычные емкости для аэрозолей, которые закрываются с помощью соответствующего дозирующего клапана.
Композиции для ингаляций могут содержать жидкие или порошкообразные композиции, содержащие активный ингредиент, которые подходят для пульверизации и интрабронхиального применения, или композиции в виде аэрозолей, вводимые с помощью устройства, дозирующего отмеренные дозы аэрозоля. Подходящие жидкие композиции включают активный ингредиент в водном, фармацевтически приемлемом растворителе для ингаляций, например, изотоническом солевом растворе или бактериостатической воде. Растворы вводят с помощью насоса или активируемого надавливанием дозатора распыляемого спрея или любыми другими обычными средствами, предназначенными для того, чтобы позволить или обеспечить попадание необходимого дозированного количества жидкой композиции в легкие пациента путем ингаляции. Подходящие составы, в которых указанный носитель представляет собой жидкость, для введения, в виде, например, спрея для носа или в виде капель для носа, включают водные или маслянистые растворы активного ингредиента.
Составы или композиции, подходящие для назального введения, в которых носитель представляет собой твердое вещество, включают крупный порошок, имеющий размер частиц, например, в диапазоне от 20 до 500 микрон, который вводят таким же образом, как вводят нюхательный табак, то есть путем быстрого вдыхания через носовой проход из емкости с порошком, подносимой близко к носу. Подходящие порошкообразные композиции включают, в качестве иллюстрации, порошкообразные составы активных ингредиентов, тщательно смешанные с лактозой или другими инертными порошками, приемлемыми для внутрибронхиального введения. Порошкообразные композиции могут быть введены с помощью аэрозольного распылителя или заключены в разрушаемую капсула, которую пациент может вставить в устройство, которое прокалывает капсулу, и из нее выдувается порошок в устойчивый поток, подходящий для ингаляции.
Композиции для применения также могут быть приготовлены в форме суппозиториев, таких как, например, для ректального введения. Такие композиции могут быть получены путем смешивания лекарственного средства с подходящим нераздражающим вспомогательным веществом, которое является твердым при обычных температурах и жидким при температуре в прямой кишке и, следовательно, будет плавиться в прямой кишке с высвобождением лекарственного средства. Подходящие вспомогательные вещества включают, например, масло какао и полиэтиленгликоли.
Композиции для применения может быть приготовлены в виде составов с замедленным высвобождением, таких как, например, состав, такой как капусла, которая обеспечивает медленное высвобождение модулятора после введения. Такие составы обычно могут быть получены с использованием хорошо известной технологии и введены путем, например, пероральной, ректальной или подкожной имплантации или путем имплантации в желаемом участке-мишени. Носители для применения в таких составах являются биологически совместимыми и могут быть также биоразлагаемыми; предпочтительно состав обеспечивает относительно постоянный уровень высвобождения модулятора. Количество модулятора, содержащееся в составе с замедленным высвобождением, зависит от, например, места имплантации, скорости и ожидаемой продолжительности высвобождения, а также природы состояния, подлежащего лечению или предотвращению.
Следует понимать, однако, что конкретный уровень дозы для любой конкретного пациента будет зависеть от множества факторов, включая активность конкретного используемого соединения, возраст, массу тела, общее состояния здоровья, пол, диету, время введения, путь введения и скорость выведения, комбинацию лекарственных средств, то есть других лекарственных средств, используемых для лечения пациента, и тяжесть конкретного заболевания, подвергаемого терапии.
Предпочтительные соединения согласно настоящему изобретению будут иметь определенные фармакологические свойства. Такие свойства включают, но не ограничиваются ими, биодоступность при пероральном введении, такую, что предпочтительные пероральные лекарственные формы, описанные выше, могут обеспечить терапевтически эффективные уровни соединения in vivo.
Производные ПНЖК n-3, предложенные в настоящем описании, предпочтительно вводят пациенту, такому как, например, человек, перорально или парентерально, и они присутутсвуют по меньшей мере в одной биологической жидкости или ткани пациента.
В настоящем описании термин «лечение» включает любой тип лечения, модифицирующего заболевание, и включает симптоматическое лечение, то есть лечение после появления симптомов, и каждое такое лечение может быть профилактическим. Однако, лечение, модифицирующее заболевание, может включать введение до появления симптомов, чтобы предотвратить, по меньшей мере отсрочить или уменьшить тяжесть симптомов после их появления. Лечение, модифицирующее заболевание, также может быть терапевтическим, то есть проводиться после появления симптомов, с целью уменьшить их тяжесть и/или продолжительность. Лечение после появления симптомов может просто включать остановку прогрессирования заболевания (обеспечить стабильность заболевания). В некоторых вариантах реализации производные ПНЖК n-3, предложенные в настоящем описании, вводят профилактически, то есть до начала заболевания и/или симптомов, в идеале, но не обязательно, чтобы фактически предотвратить заболевания и/или симптомы. Следует понимать, что термин «профилактика» и «профилактический» в контексте настоящего изобретения, служит для простого описания того, что соединение(-я) согласно настоящему изобретению вводят до появления симптомов. Профилактическое введение может представлять собой введение до появления симптомов, которые четко связаны с заболеванием, обсуждаемым в настоящем описании: производные ПНЖК n-3, предложенные в настоящем описании, могут, например, быть введены субъекту профилактически, когда у него или у нее проявляются различные состояния, которые могут говорить о склонности к развитию одного из состояний или заболеваний, которые могут быть подвержены лечению одним из производных ПНЖК n-3 согласно настоящему описанию. Такими показательными 5 состояниями являются, например, высокое кровяное давление или диабет. Такое профилактическое лечение называется первичной профилактикой. В другом варианте реализации производные ПНЖК n-3, предложенные в настоящем описании, могут быть введены субъекту профилактически, когда он или она ранее страдал(-а) от состояния или заболевания, которое может быть подвержено лечению производными ПНЖК n-3 согласно настоящему изобретению, но сейчас у него (нее) не проявляются какие-либо симптомы. Такое профилактическое лечение называется вторичной профилактикой. Пациенты, получающие ПНЖК n-3, для цели первичной или вторичной профилактики считаются нуждающимися в таком лечении. Пациенты могут включать, но не ограничиваются ими, млекопитающих, в частности людей, одомашненных животных-компаньонов, таких как собаки, кошки, лошади и скот, такой как крупный рогатый скот, свиньи, овцы, в дозах согласно настоящему описанию.
Активность аналогов ПНЖК n-3 в соответствии с настоящим изобретением может, например, быть определена путем соответствующих анализов in vitro и/или in vivo. Например, биологическая активность аналогов ПНЖК n-3 в соответствии с настоящим изобретением может быть определена с применением установленной клеточной модели согласно Kang и 20 Leaf (Proc Natl Acad Sci USA, 1994. 91(21): p. 9886-90.), известной специалистам в данной области техники.
Следующие чертежи и примеры служат для иллюстрации настоящего изобретения и не предназначены для ограничения объема настоящего изобретения, описанного в прилагаемой формуле изобретения.
Описание чертежей
Фиг. 1: Эффективность соединения со структурой в соответствии с формулой (VI), обозначенного как «Соединение 1», в модели индуцированной лазером хориоидальной неоваскуляризации у крысы оценивали путем определения повышенной проницаемости 30 сосудов с помощью флюоресцентной ангиографии:
Приведены данные оценки с помощью флюоресцентной ангиографии (среднее ± СО.) на 14 и 21 дни после индукции хориоидальной неоваскуляризации (лазерного ожога), шкала оценки: от 0 до 3 условных единиц. Утечка флюоресцеина на флюоресцентных ангиограммах была оценена двумя независимыми экспертами с ослеплением в отношении исследуемых групп и следующей шкалой оценки: значение 0: утечка отсутствует; значение 1: слабое окрашивание; значение 2: умеренное окрашивание; значение 3: сильное окрашивание, n=10 животных в группе с 6 повреждениями у каждого животного, статистический анализ: критерий Краскела-Уоллиса (Kruskal-Wallis) и критерий Даннета (Dunnett) для множественных сравнений, *p<0,05, п/о: пероральное введение, в/б: внутрибрюшинное введение, и/в: интравитреальное введение, ФА: флюоресцентная ангиография.
Фиг. 2: На фигуре 2 показано противовоспалительное действие метаболически устойчивого аналога 17,18-EEQ (соед. 02) в отношении кардиомиоцитов HL-1. Использовали клеточную линию кардиомиоцитов (иммортализованные кардиомиоциты мыши, клетки HL-1). Клетки обрабатывали либо носителем (0,01% этанолом), либо различными концентрациями исследуемого соединения (соед. 02: сЕ=10 нМ, 100 нМ или 1 мкМ). Одновременно указанные клетки подвергали воздействию 1 мкг/мл липополисахарида (ЛПС). После 24 ч инкубирования клетки подвергали обработке для определения жизнеспособности.
Фиг. 3: На фигуре 3 показано противовоспалительное действие метаболически устойчивого аналога 17,18-EEQ (соед. 02) в отношении кардиомиоцитов HL-1. Использовали клеточную линию кардиомиоцитов (иммортализованные кардиомиоциты мыши, клетки HL-1). Клетки обрабатывали либо носителем (0,01% этанолом), либо различными концентрациями исследуемого соединения (соед. 02: сЕ=10 нМ, 100 нМ или 1 мкМ). Одновременно указанные клетки подвергали воздействию 1 мкг/мл липополисахарида (ЛПС). После 24 ч инкубирования клетки подвергали обработке для определения высвобождения провоспалительного цитокина ФНО-альфа.
Фиг. 4: На фигуре 4 показано подавляющее действие метаболически устойчивого аналога 17,18-EEQ (соед. 02) в отношении воспаления тканей сердца в модели тяжелой гипертензии и поражения органов-мишеней у крысы. Модуляцию воспаления определяли по инфильтрации макрофагов посредством окрашивания ED1 в качестве маркера воспаления. Приведенные значения представлены в виде среднего и межквартильного размаха. Значения встречаемости объединены в ячейки по 20 полей зрения. Полученные результаты показывают, что у двойных трансгенных (dTGR) животных наблюдали значительно большее количество инфильтрированных макрофагов в ткани сердца по сравнению с не получавшими лечения животными Спрег-Доули (СД). Однако лечение с применением соед. 02 приводило к значительному уменьшению инфильтрации макрофагов (ED1-положительных клеток) у dTGR животных по сравнению с dTGR животными, получавшими носитель.
Фиг. 5: На фигуре 5 показано подавляющее действие метаболически устойчивого аналога 17,18-EEQ (соед. 02) в отношении воспаления тканей почки в модели тяжелой гипертензии и поражения органов-мишеней у крысы. Модуляцию воспаления определяли по инфильтрации макрофагов посредством окрашивания ED1 в качестве маркера воспаления. Приведенные значения представлены в виде среднего и межквартильного размаха. Значения встречаемости объединены в ячейки по 20 полей зрения. Полученные результаты показывают, что у dTGR животных наблюдали значительно большее количество инфильтрированных макрофагов в ткани почки по сравнению с не получавшими лечения животными СД. Однако лечение с применением соед. 02 приводило к значительному уменьшению инфильтрации макрофагов (ED1-положительных клеток) у dTGR животных по сравнению с dTGR животными, получавшими носитель.
Пример 1: Синтез соединений
Далее описан синтез выбранных соединений согласно настоящему изобретению.
Соединение 1 (соед. 01)
Синтез соединения 1 (соед. 01) аналогичен синтезу соединения 3 (соед. 03), при этом введение фрагмента мочевины осуществляли в соответствии со способом синтеза, описанным в патентной заявке WO 2010/081683 (пример 13).
Соединение 2 (соед. 02)
Общая схема синтеза

Общий способ
Спектры ЯМР регистрировали с помощью спектрометра Bruker Avance 400 МГц для1Н ЯМР и 100 МГц для13С ЯМР. ЖХМС проводили с помощью квадрупольного масс-спектрометра Shimadzu LCMS 2010 (колонка: sepax ODS 50×2,0 мм, 5 мкм) или хроматографа Agilent 1200 HPLC, 1956 MSD (колонка: Shim-pack XR-ODS 30×3,0 мм, 2,2 мкм), работая в режиме регистрации положительных ионов при ионизации электрораспылением. Хроматографическую очистку проводили с помощью флэш-хроматографии с использованием силикагеля 100~200 меш. Безводные растворители перед использованием предварительно обрабатывали с помощью колонки 3А MS. Все коммерчески доступные реагенты использовали без дополнительной обработки, если не указано иное.
Общая методика получения соединения 2
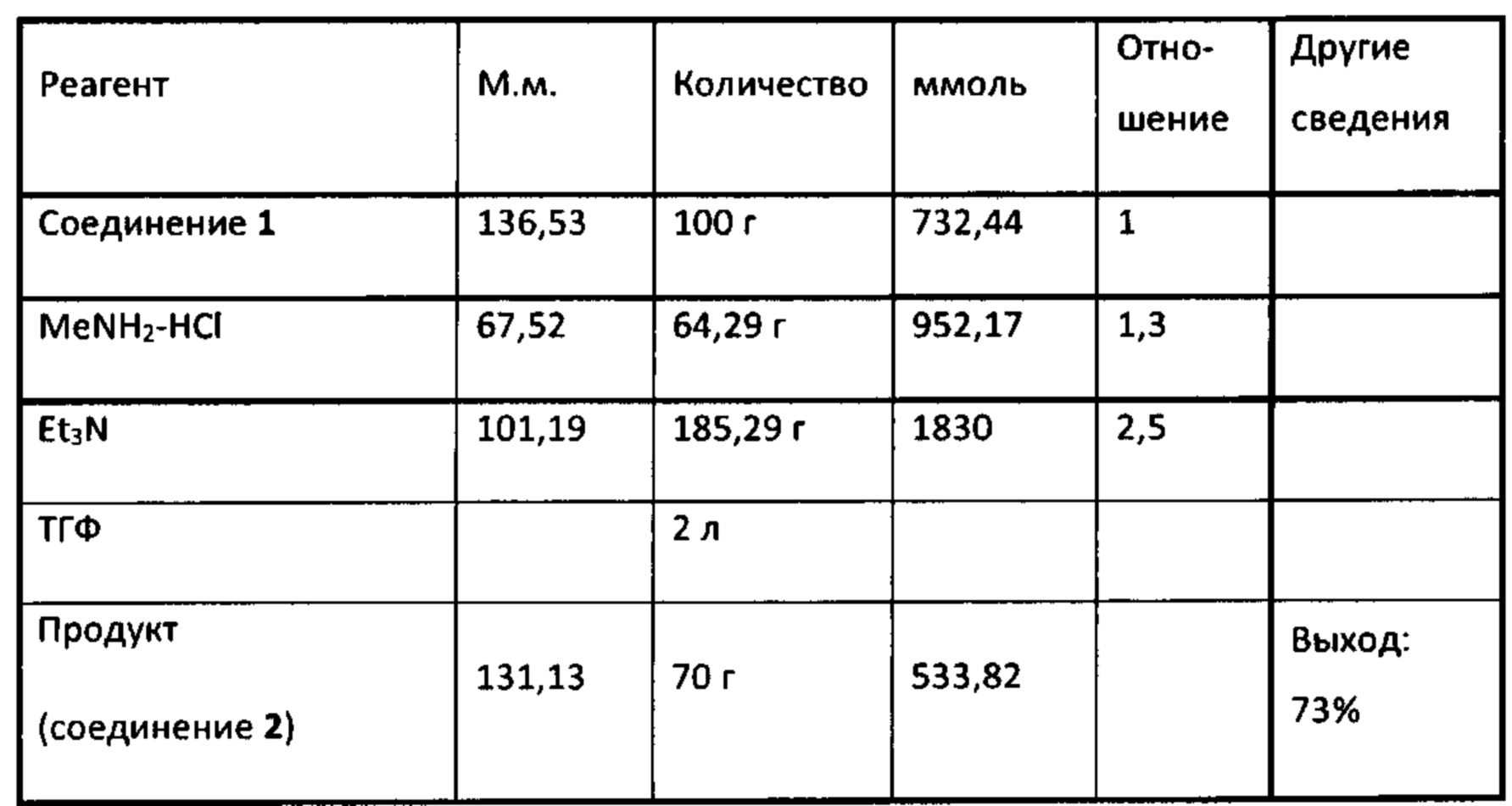
К метанамину (64,29 г, 952,17 ммоль, 1,30 экв.) в 500 мл ТГФ добавляли Et3N (75 г, 732,44 ммоль), полученный раствор добавляли к соединению 1 (100,00 г, 732,44 ммоль, 1,00 экв.), Et3N (111 г, 1,1 моль) в ТГФ (1,5 л) при -10°C. Полученную смесь перемешивали при 25°C в течение 16 ч. Затем указанную смесь фильтровали, фильтрат промывали 2 н. HCl (500 мл), подвергали экстракции с помощью ЭА (300 мл*4), концентрировали и очищали с помощью силикагеля (ПЭ : ЭА = 3:1-1:1) с получением соединения 2 (70,00 г, 533,82 ммоль, выход 72,88%) в виде масла желтого цвета.
Данные ТСХ (ПЭ : EtOAc = 2:1): Rf (соед. 02) = 0,39; ЖХМС: ЕТ2662-1-Р1А (М+Н+): 131,7;1Н ЯМР (CDCl3, 400 МГц) 4,36~4,24 (q, J=8 Гц, 2Н), 2,93~2,85 (d, J=4 Гц, 3Н), 1,38~1,30 (t, J=8 Гц, 3Н).
Общая методика получения соединения 4


К раствору соединения 4 (78,33 г, 532,40 ммоль, 1,10 экв.) и PPh3 (133,30 г, 508,20 ммоль, 1,05 экв.) в безводном ТГФ (100 мл) с температурой 0°C медленно добавляли через трубку раствор соединения 3 (47,50 г, 484,00 ммоль, 1,00 экв.) и DIAD (107,66 г, 532,40 ммоль, 1,10 экв.) в безводном ТГФ (50 мл). Колбу и трубку промывали дополнительной порцией сухого ТГФ (30 мл) для обеспечения полного добавления. Реакционную смесь оставляли для постепенного нагревания до 25°C и перемешивали в течение 18 ч. Затем добавляли Н2О (1000 мл), подвергали экстракции с помощью ЭА (500 мл*2), концентрировали и очищали с помощью силикагеля (ПЭ : ЭА = 0-10:1) с получением соединения 5 (42,5 г, 374,02 ммоль, 77,28% выход) в виде твердого вещества белого цвета.
Данные ТСХ (ПЭ : EtOAc = 5:1): Rf (соед. 03) = 0,2; Rf (соед. 05) = 0,5;1Н ЯМР (CDCl3, 400 МГц) 7,86~7,79 (m, 2Н), 7,72~7,67 (m, 2Н), 3,73~3,66 (t, J=8 Гц, 2Н), 2,27~2,20 (m, 2Н), 1,95~1,91 (t, J=4 Гц, 1Н), 1,85~1,75 (m, 2Н), 1,61~1,52 (m, 2Н).
Общая методика получения соединения 6


К раствору соединения 5 (88,00 г, 387,22 ммоль, 1,00 экв.) и AgNO3 (16,44 г, 96,81 ммоль, 0,25 экв.) в безводном ТГФ (1600 мл) добавляли NIS (130,68 г, 580,83 ммоль, 1,50 экв.) одной порцией при 25°С. В пространство над реакционной смесью нагнетали N2, и реакционную смесь защищали от света с помощью алюминиевой фольги и перемешивали в течение 16 ч. Полученную смесь вливали в воду (1000 мл), подвергали экстракции с помощью ЭА (600 мл*3), концентрировали и очищали с помощью диоксида кремния (ПЭ : ЭА = 10:1-2:1) с получением соединения 6 (118,6 г, 1,01 моль, выход 86,78%) в виде твердого вещества белого цвета.
Данные ТСХ (ПЭ : EtOAc = 20:1): Rf (соед. 05) = 0,22; Rf (соед. 6) = 0,21;1Н ЯМР (CDCl3, 400 МГц) 7,87~7,82 (m, 2Н), 7,74~7,69 (m, 2Н), 3,74~3,67 (t, J=8 Гц, 2Н), 2,45~2,39 (t, J=8 Гц, 2Н), 1,84-1,74 (m, 2Н), 1,61~1,52 (m, 2Н).
Общая методика получения соединения 7

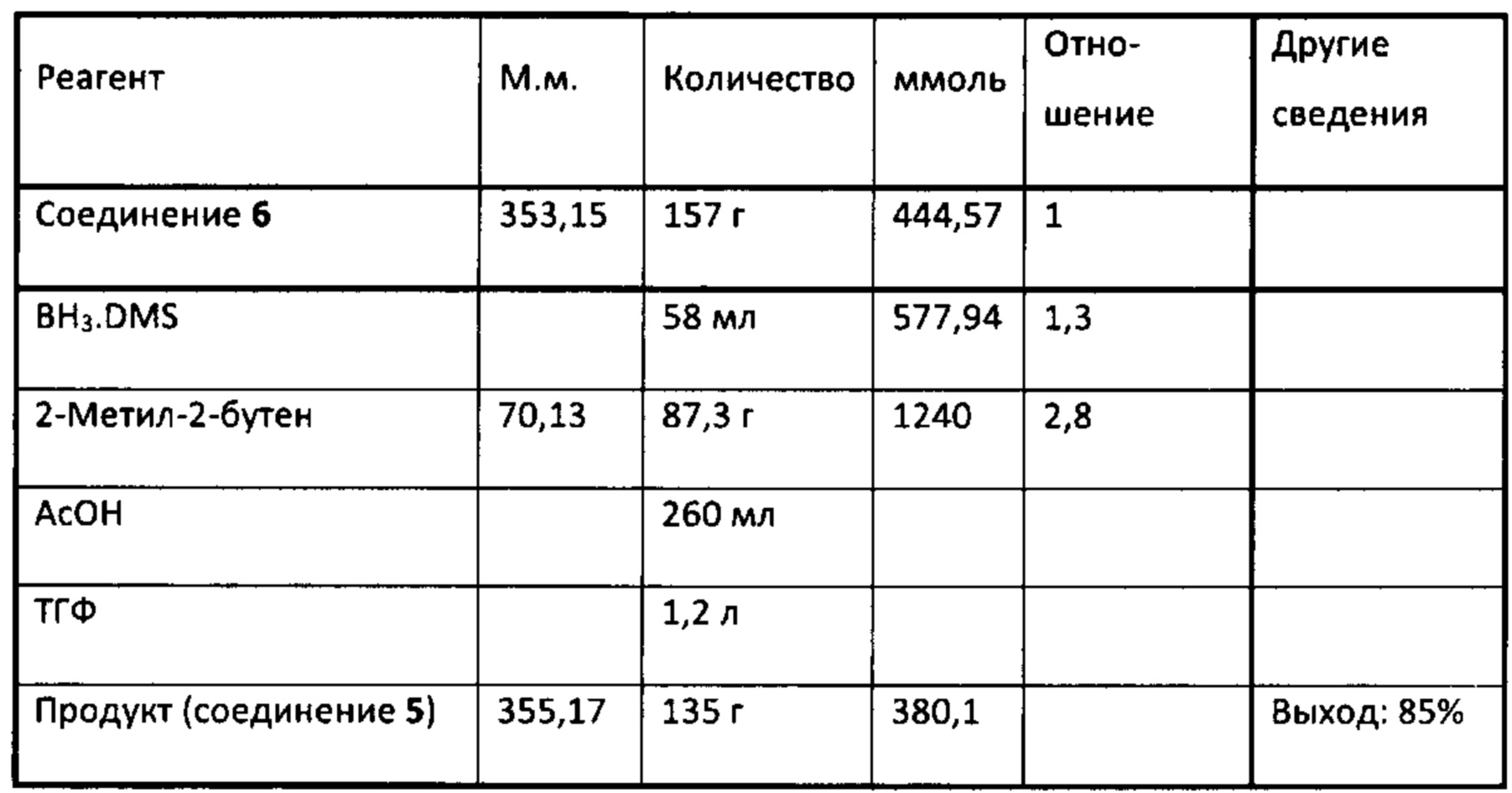
К раствору BH3.Me2S (43,91 г, 577,94 ммоль, 1,30 экв.) в ТГФ (300 мл) с температурой 0°С добавляли 2-метилбут-2-ен (87,30 г, 1,24 моль, 2,80 экв.) в течение 30 мин. Через 1 ч реакционную смесь нагревали до 25°С и перемешивали в течение 90 мин. После повторного охлаждения до 0°С медленно добавляли раствор соединения 6 (157,00 г, 444,57 ммоль, 1,00 экв.) в ТГФ (900 мл) в течение 1 ч. После окончания добавления холодную баню удаляли и реакционную смесь перемешивали при 25°С. Через 2 ч реакционную смесь снова охлаждали до 0°С, после чего медленно добавляли ледяную АсОН (260 мл) в течение 30 мин (осторожно: выделение газа) и перемешивали при 25°С в течение 16 ч. После окончания реакции по данным ТСХ (ПЭ : ЭА = 10:1) полученную смесь вливали в воду (1 л), подвергали экстракции с помощью ЭА (300 мл*2), концентрировали и очищали с помощью силикагеля (ПЭ : ЭА = 0-10:1) с получением соединения 7 (135 г, 380,1 ммоль, выход 85,50%) в виде масла желтого цвета.
Данные ТСХ (ПЭ : EtOAc = 10:1): Rf (соед. 6) = 0,5; Rf (соед. 7) = 0,55;1Н ЯМР: (CDCl3, 400 МГц) 7,88~7,80 (m, 2Н), 7,75~7,67 (m, 2Н), 6,24~6,11 (m, 2Н), 3,74~3,66 (t, J=8 Гц, 2Н), 2,24~2,15 (q, J=8 Гц, 2Н), 1,78~1,67 (m, 2Н), 1,55~1,44 (m, 2Н)
Общая методика получения соединения 8
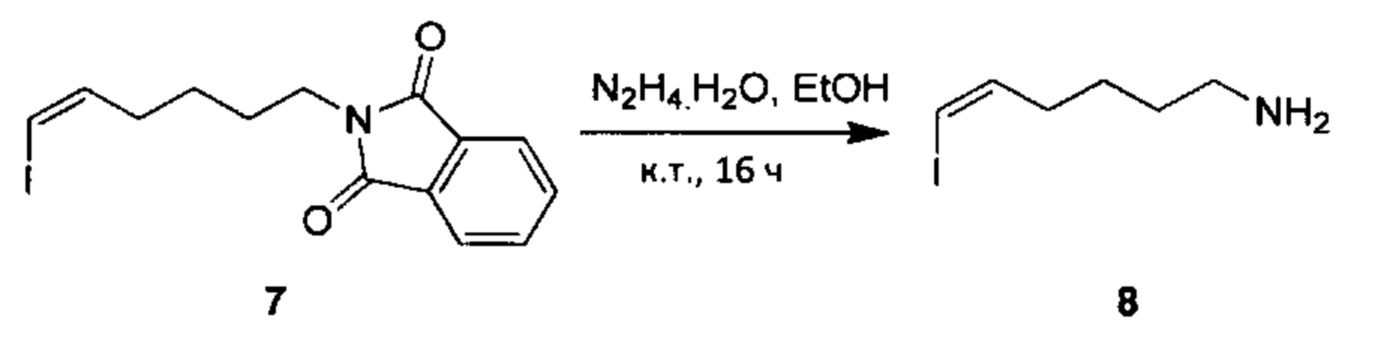
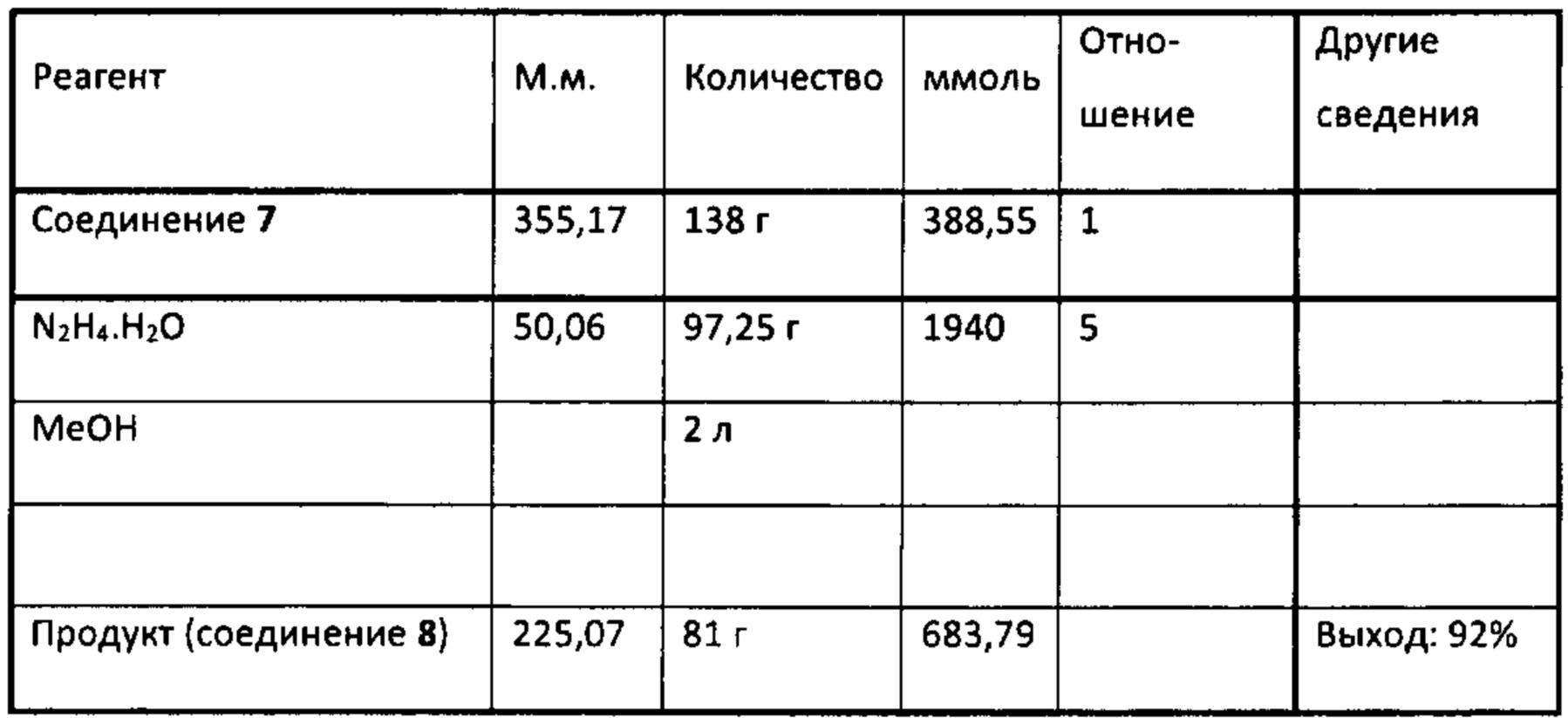
К раствору соединения 7 (138,00 г, 388,55 ммоль, 1,00 экв.) в безводном МеОН (2,00 л) добавляли N2H4⋅H2O (97,25 г, 1,94 моль, 5,00 экв.) при 0°С и перемешивали при 25°С в течение 18 ч, после окончания реакции по данным ТСХ (ПЭ : ЭА = 10:1) реакционную смесь концентрировали, остаток вливали в ДХМ (5000 мл) и перемешивали в течение 30 мин. Фильтровали, и осадок на фильтре промывали ДХМ (1 л*2), фильтрат концентрировали с получением соединения 8 (162,00 г неочищенного) в виде масла желтого цвета.
Данные ТСХ (ПЭ : EtOAc = 10:1): Rf (соед. 7) = 0,5; Rf (соед. 8) = 0; данные ТСХ (ДХМ : МеОН = 10:1): Rf (соед. 7) = 1; Rf (соед. 8) = 0,2;1Н ЯМР: (CDCl3, 400 МГц) 6,19~6,07 (m, 2Н), 2,73~2,59 (m, 2Н), 2,20~2,05 (m, 2Н), 1,75~1,55 (m, 2Н), 1,51~1,36 (m, 4Н).
Общая методика получения соединения 9
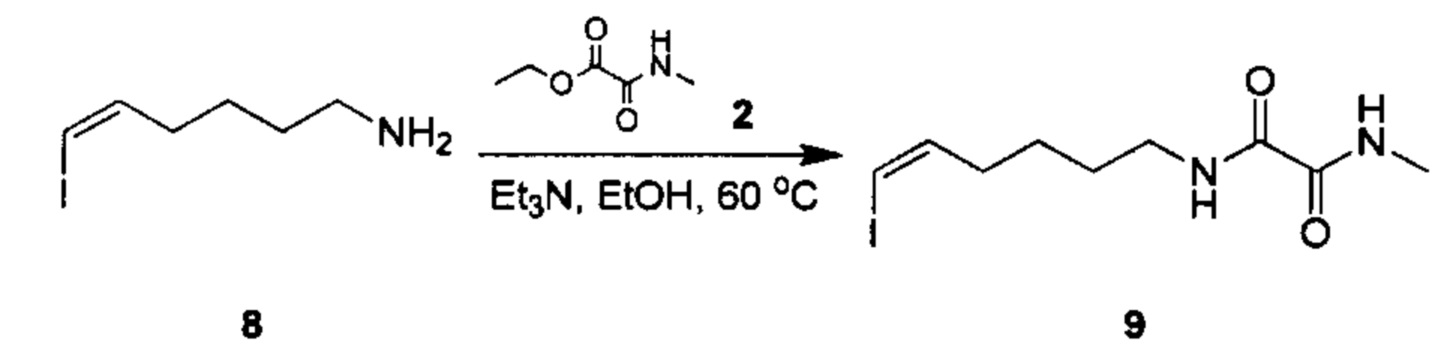


Соединение 8 (92,00 г, 408,76 ммоль, 1,00 экв.), соединение 2 (53,60 г, 408,76 ммоль, 1,00 экв.) и Et3N (49,64 г, 490,51 ммоль, 1,20 экв.) в безводном этаноле (1,5 л) нагревали при 60°С в течение 20 ч. После окончания реакции по данным ТСХ (ДХМ : МеОН = 10:1) смесь концентрировали до примерно 300 мл. Фильтровали и концентрировали с получением соединения 9 (90 г, 232,16 ммоль, выход 57%) в виде твердого вещества белого цвета.
Данные ТСХ (ДХМ : МеОН = 10:1): Rf (соед. 8) = 0,2; Rf (соед. 9) = 0,5;1Н ЯМР: (CDCl3, 400 МГц) 7,57~7,37 (s, 2Н), 6,25~6,20 (d, J=8 Гц, 1Н), 6,18~6,11 (q, J=8 Гц, 1Н), 3,37-3,30 (q, J=8 Гц, 2Н), 2,93~2,88 (d, J=4 Гц, 3Н), 2,21~2,13 (m, 2Н), 1,66~1,56 (m, 2Н), 1,53~1,43 (m, 2Н)
Общая методика получения соединения 12
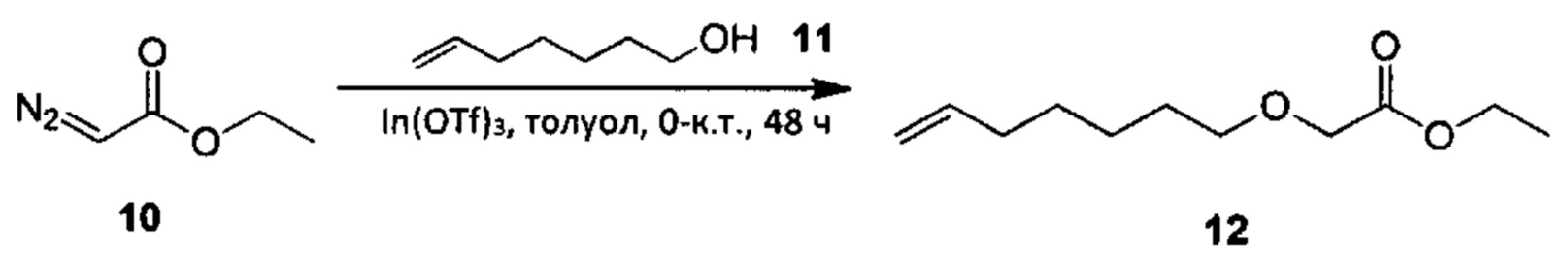

К раствору соединения 11 (27,02 г, 236,63 ммоль, 1,20 экв.), In(OTf)3 (22,09 г, 39,44 ммоль, 0,20 экв.) в толуоле (275 мл) добавляли соединение 10 (25,00 г, 197,20 ммоль, 1,00 экв.) в толуоле (75 мл) в течение 20 мин. Затем смесь перемешивали при 25°С в течение 48 ч. Полученную смесь концентрировали и очищали с помощью силикагеля (ПЭ : ЭА = 20:1) с получением этилированного соединения 12 (35,00 г, 139,81 ммоль, выход 70,90%, чистота 80%) в виде масла желтого цвета.
Данные ТСХ (ПЭ : EtOAc = 10:1): Rf (соед. 11) = 0,21; Rf (соед. 12) = 0,55;1Н ЯМР: (CDCl3,400 МГц) 5,86~5,72 (m, 1Н), 5,03~5,86 (m, 2Н), 4,24~4,17 (q, J=8 Гц, 2Н), 4,07~4,01 (s, 2Н), 3,54~3,47 (t, J=8 Гц, 2Н), 2,09~1,98 (m, 2Н), 1,68~1,55 (m, 2Н), 1,45~1,32 (m, 4Н), 1,30~1,25 (t, J=8 Гц, 3Н).
Общая методика получения соединения 13

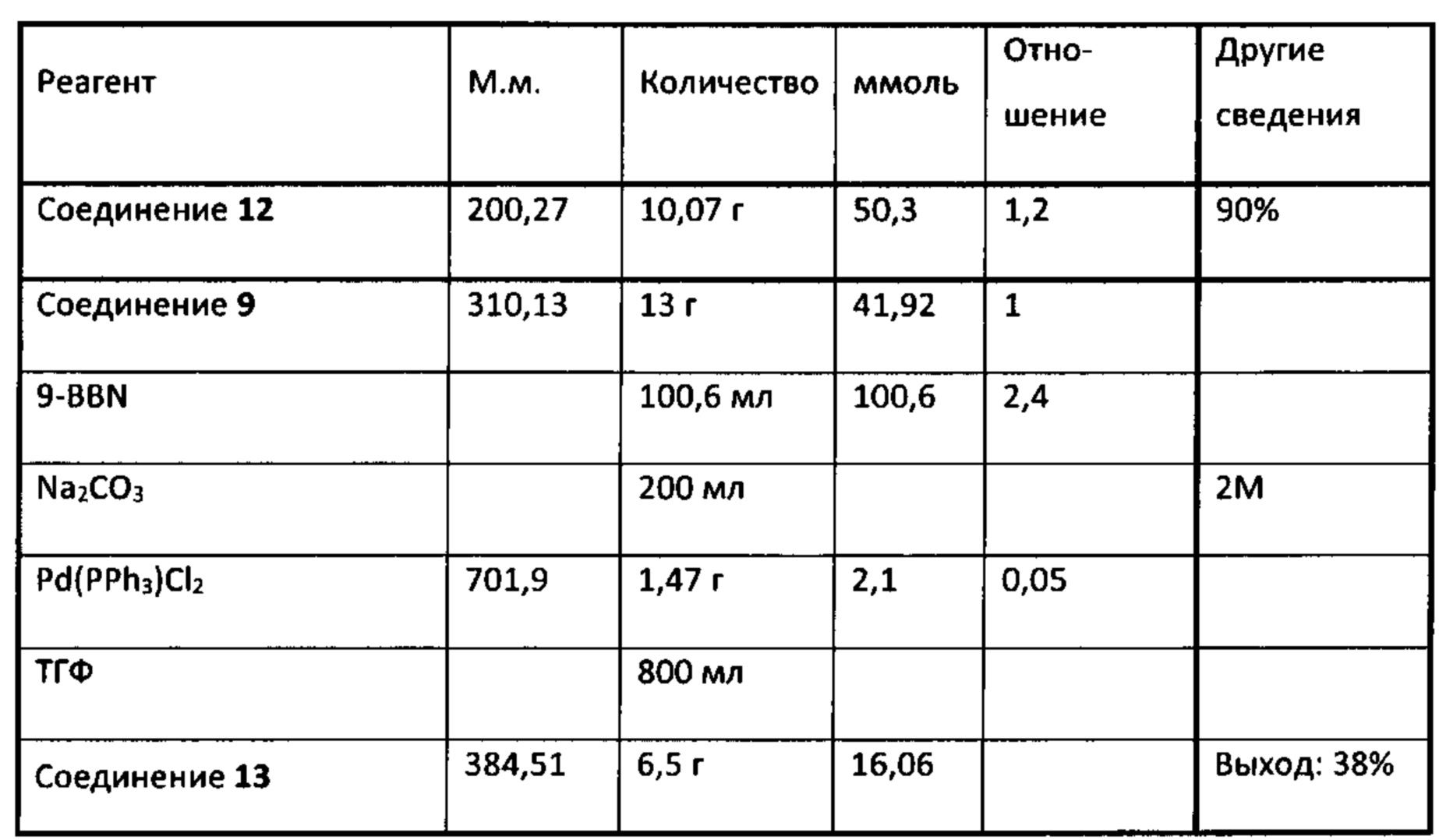
В колбу, высушенную в сушильном шкафу, содержащую 9-BBN (17,53 г, 100,60 ммоль, 2,40 экв.) в ТГФ (540 мл), добавляли раствор соединения 12 (10,07 г, 50,30 ммоль, 1,20 экв.) в ТГФ (60 мл) при 0°С. После перемешивания в течение 16 ч при 25°С добавляли водный раствор Na2CO3 (200 мл 2 М раствора, приготовленного с использованием барботированной аргоном Н2О). Через 2 ч добавляли Pd(PPh3)2Cl2 (1,47 г, 2,10 ммоль, 0,05 экв.), после чего добавляли соединение 9 (13,00 г, 41,92 ммоль, 1,00 экв.), растворенное в ТГФ (200 мл). Полученный раствор красного цвета защищали от света. Реакционную смесь перемешивали при 50°С в течение 5 ч. Окончание реакции определяли по данным ЖХМС. После охлаждения до 25°С реакционную смесь концентрировали под вакуумом и остаток очищали с помощью силикагеля (ПЭ : ЭА = 10:1-3:1) с получением соединения 13 (6,5 г, 16,06 ммоль, выход 38,31%, чистота 95%) в виде твердого вещества желтого цвета.
Данные ТСХ (ПЭ : EtOAc = 2:1): Rf (соед. 12) = 0,3; Rf (соед. 13) = 0,3; ЖХМС: ET2662-38-P1D (М+Н+): 385,1;1H ЯМР: (CDCl3, 400 МГц) 7,57~7,38 (s, 1Н), 5,41~5,25 (m, 2Н), 4,25~4,17 (q, J=8 Гц, 2Н), 4,07~4,02 (s, 2Н), 3,54~3,47 (t, J=8 Гц, 2Н), 3,34~3,26 (q, J=8 Гц, 2Н), 2,92~2,87 (d, J=8 Гц, 3Н), 2,08~1,94 (m, 4Н), 1,65~1,51 (m, 4Н), 1,43~1,23 (m, 13Н).
Общая методика получения соед. 02

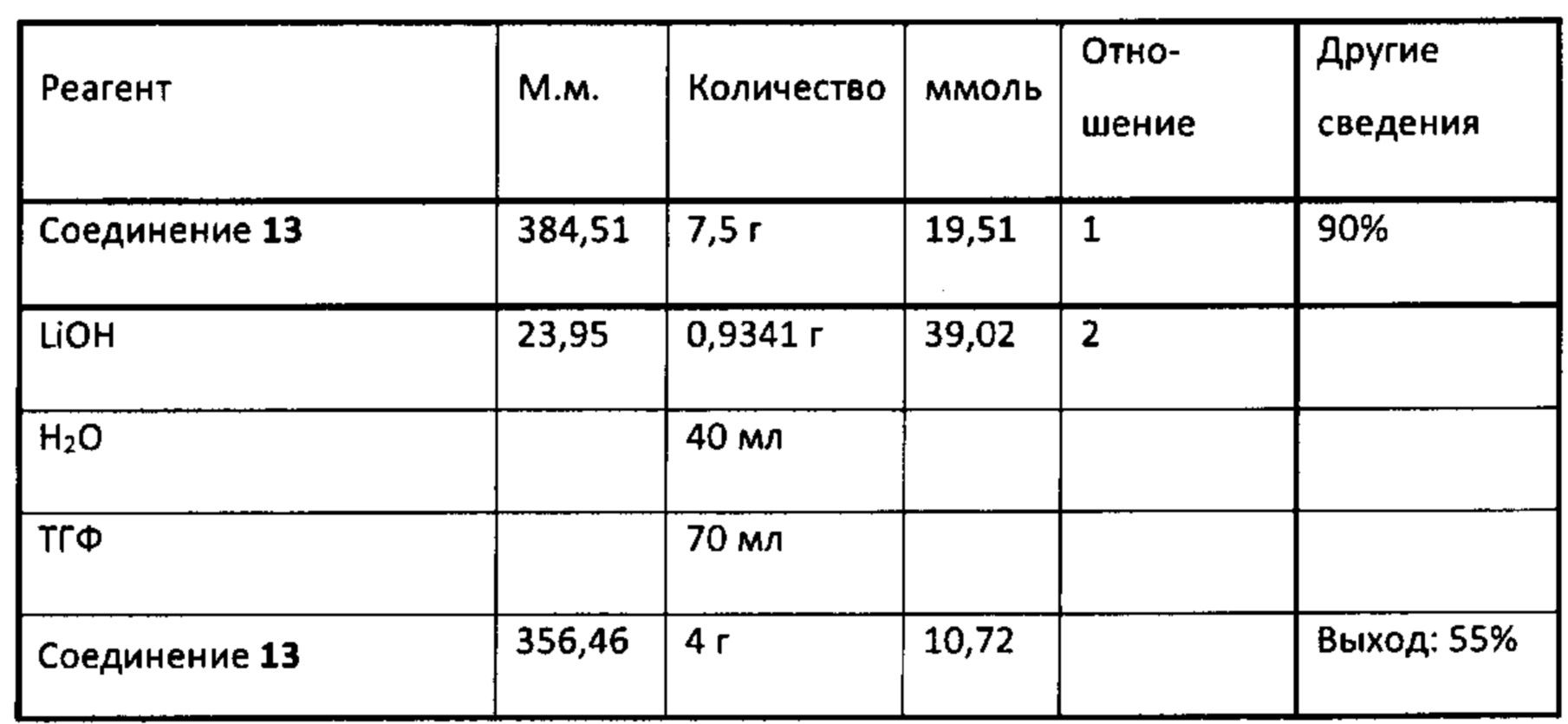
К раствору соединения 13 (7,50 г, 19,51 ммоль, 1,00 экв.) в ТГФ (70,00 мл) добавляли LiOH (934,31 мг, 39,02 ммоль, 2,00 экв.) в H2O (40,00 мл) при 0°С и затем реакционную смесь перемешивали при 0-25°С в течение 1 ч. Окончание реакции определяли по данным ЖХМС. Затем к реакционной смеси добавляли Н2О (60 мл), водную фазу обрабатывали 3 н. HCl (10 мл) до рН=3-4, подвергали экстракции с помощью ЭА (100 мл*3), сушили, органическую фазу концентрировали с получением неочищенного продукта. Остаток очищали с помощью геля на колонке (ПЭ : ЭА = 5:1-ЭА) с получением соед. 02 (4,00 г, 10,72 ммоль, 54,95% выход, чистота 95,51%).
Данные ТСХ (ДХМ : МеОН = 10:1): Rf (соед. 13) = 0,9; Rf (соед. 02) = 0,4; MS: ЕТ2662-43-Р1С (M+Na+): 379,2;1Н ЯМР (CDCl3, 400 МГц) 7,84 (s, 1Н), 7,74 (s, 1H), 5,40~5,32 (m, 2H), 4,11 (s, 2H), 3,59~3,55 (t, J=6,4 Гц, 2H), 3,35~3,32 (t, J=6,8 Гц, 2H), 2,92~2,91 (d, J=5,2 Гц, 3H), 2,07~2,00 (m, 4H), 1,64-1,59 (m, 4H), 1,42~1,32 (m, 10Н);13C ЯМР (CDCl3, 100 МГц) δ 173,7, 160,7, 159,8, 130,5, 129,0, 72,0, 67,8, 39,7, 29,4, 29,3, 29,0, 29,0, 28,6, 27,1, 26,8, 26,7, 25,8.
Соединение 3 (соед. 03)
Общая схема синтеза

Синтез 2-(гекс-5-ин-1-ил)-изоиндолин-1,3-диона (2): Согласно описанному в литературе1, к раствору фталимида (7,5 г, 51 ммоль) и трифенилфосфина (ТРР, 13,4 г, 1 эквив.) в безводном ТГФ (50 мл) с температурой 0°С медленно добавляли через трубку раствор 5-гексин-1-ола (1) (5 г, 1 эквив.) и диизопропилазодикарбоксилата (DIAD, 10,5 г, 1,02 эквив.) в безводном ТГФ (30 мл). Колбу и трубку промывали дополнительной порцией сухого ТГФ (20 мл) для обеспечения полного добавления. Реакционную смесь оставляли для постепенного нагревания до комнатной температуры в течение ночи. Через в общей сложности 18 ч все летучие вещества упаривали и остаток очищали с помощью хроматографической системы Teledyne Isco Combiflash• RF (колонка с 80 г SiO2, элюирование: гексан, 2 мин; 0-20% EtOAc/гексан, 12 мин; 20% EtOAc/гексан, 6 мин) с получением 2 (8,3 г, 72%) в виде твердого вещества белого цвета, спектральные значения которого были идентичны указанным значениям2.
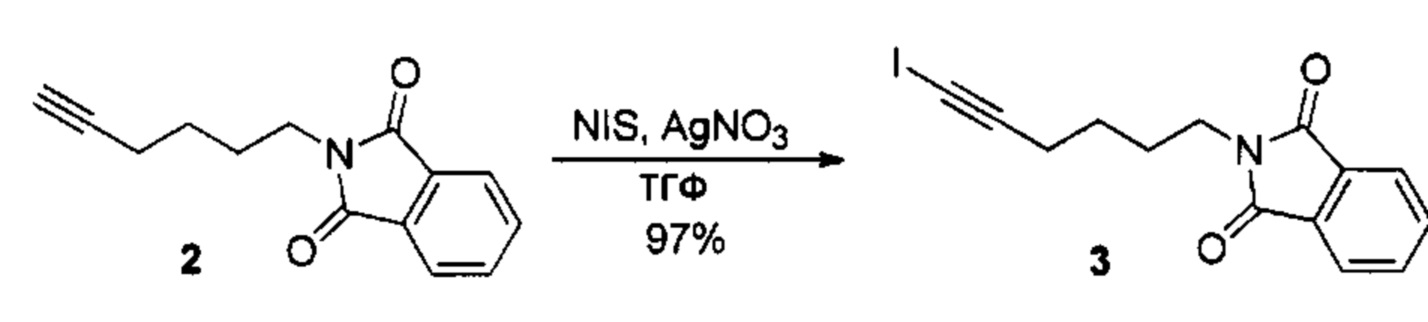
Синтез 2-(6-иодгекс-5-ин-1-ил)-изоиндолин-1,3-диона (3): Согласно описанному в литературе3, к раствору алкина 2 (5,0 г, 22 ммоль) и AgNO3 (0,93 мг, 0,25 эквив.) в безводном ТГФ (120 мл) при к.т. добавляли N-иодсукцинимид (NIS, 7,4 г, 1,5 эквив.) одной порцией. В пространство над реакционной смесью нагнетали аргон и реакционную смесь защищали от света с помощью алюминиевой фольги. Через 4 ч реакционную смесь вливали в Н2О (200 мл) и подвергали экстракции с помощью Et2O (2×50 мл). Эфирные экстракты промывали солевым раствором (3×60 мл) (примечание: двухфазная смесь приобретала коричневый цвет). Объединенные водные фазы повторно подвергали экстракции с помощью Et2O (2×50 мл). Объединенные эфирные экстракты сушили над Na2SO4, фильтровали и концентрировали на ротационном испарителе. Остаток очищали использованием хроматографической системы Teledyne Isco Combiflash• RF (колонка с 80 г SiO2, элюирование: гексан, 2 мин; 0-40% EtOAc/гексан, 8 мин; 40% EtOAc/гексан, 10 мин; 40-100% EtOAc/гексан, 5 мин; 100%, EtOAc, 3 мин) с получением 3 (97%) в виде твердого вещества белого цвета, точка плавления 132,5-132,7°C.1H ЯМР (500 МГц, CDCl3) δ 7,85 (ddd, J=5,4, 3,0, 1,0 Гц, 2Н), 7,72 (ddd, J=5,5, 3,0, 1,0 Гц, 2Н), 3,71 (t, J=7,1 Гц, 2Н), 2,42 (t, J=7,0 Гц, 2Н), 1,83-1,73 (m, 2Н), 1,61-1,51 (m, 2Н);13С ЯМР (100 МГц, CDCl3) δ 168,62, 134,14, 132,30, 123,44, 94,04, 37,60, 27,91, 25,89, 20,60, -6,27.

Синтез 2-(6-иодгекс-5(Z)-ен-1-ил)-изоиндолин-1,3-диона (4): Согласно описанному в литературе4, к раствору BH3⋅Me2S (2,0 М в ТГФ, 9,2 мл, 1,3 эквив.) в ТГФ (3 мл) с температурой 0°С добавляли чистый (neat) 2-метил-2-бутен (4,2 мл, 2,8 эквив.) в течение 5 мин. Через 1 ч реакционную смесь нагревали до комнатной температуры и перемешивали в течение 90 мин. После повторного охлаждения до 0°С медленно добавляли раствор иодалкина 3 (5 г, 1 эквив.) в ТГФ (30 мл) в течение 5 мин. После окончания добавления холодную баню удаляли и реакционную смесь перемешивали при к.т. Через 2 ч реакционную смесь снова охлаждали до 0°С, после чего медленно добавляли ледяную АсОН (8,5 мл) в течение 5 мин (осторожно: выделение газа). После перемешивания в течение ночи (14 ч) реакционную смесь разбавляли Н2О (20 мл), затем осторожно вливали в насыщенный раствор бикарбоната натрия (40 мл) при перемешивании. Полученную двухфазную смесь подвергали экстракции эфиром (2×40 мл) и объединенные эфирные экстракты промывали водой, солевым раствором, сушили над безводным MgSO4, фильтровали и концентрировали под вакуумом. Остаток очищали с использованием хроматографической системы Teledyne Isco Combiflash• RF (колонка с 40 г SiO2, элюирование: 0-20% EtOAc/гексан, 8 мин; 20% EtOAc/гексан, 6 мин) с получением смеси (4,52 г) 4 и боранового побочного продукта. Дополнительную очистку проводили на следующей стадии.

Синтез 6-иодгекс-5(Z)-ен-1-амина (5): Согласно описанному в литературе5, к раствору неочищенного 4 (4,52 г) в безводном EtOH (20 мл) при к.т. добавляли 40% (масс.) MeNH2 в Н2О (15 мл). После перемешивания в течение ночи (18 ч) реакционную смесь вливали в ледяную воду (100 мл) и подвергали экстракции с помощью Et2O (30 мл × 2). Объединенные эфирные экстракты промывали холодным 1 н. раствором HCl (20 мл × 2). рН объединенных водных промывочных растворов доводили до 8 с помощью разбавленного водн. NaOH. Раствор подвергали экстракции с помощью Et2O (30 мл × 2), сушили над безводным Na2SO4, фильтровали и концентрировали под вакуумом с получением неочищенного 5 (1,12 г) в виде масла коричневого цвета, которое использовали на следующей стадии без дополнительной очистки.1Н ЯМР (500 МГц, CDCl3) δ 6,29-6,08 (m, 2Н), 2,71 (tt, J=7,0, 1,8 Гц, 2Н), 2,16 (каж. q, J=6,5 Гц, 2Н), 1,78-1,52 (m, 2Н).

Синтез N1-(6-иодгекс-5(Z)-ен-1-ил)-N2-метилоксаламида (7): Согласно описанному в литературе6, раствор иодалкена 5 (1,12 г, 4,98 ммоль), этил-2-(метиламино)-2-оксоацетата (6) (0,62 г, 1,2 эквив.) и триэтиламина (0,83 мл, 1,2 эквив.) в безводном этаноле (10 мл) нагревали при 60°С. Через 20 ч полученный раствор коричневого цвета охлаждали до к.т. и концентрировали под вакуумом. Очистка остатка с использованием хроматографической системы Teledyne Isco Combiflash• RF (колонка с 25 г SiO2, элюирование: 0-50% EtOAc/гексан, 10 мин; 50% EtOAc/гексан, 10 мин) приводила к получению 7 (0,93 г, 60%) в виде твердого вещества белого цвета, 99,7-99,8°С.1Н ЯМР (500 МГц, CDCl3) δ 7,46 (ушир. s, 2Н), 6,32-6,02 (m, 2Н), 3,34 (каж. q, J=6,9 Гц, 2Н), 2,91 (d, J=5,3 Гц, 3Н), 2,18 (dt, J=7,5, 7,0 Гц, 2Н), 1,68-1,59 (m, 2Н), 1,54-1,42 (m, 2Н);13С ЯМР (100 МГц, CDCl3) δ 160,47, 159,70, 140,43, 83,07, 39,40, 34,11, 28,61, 26,15, 25,11.
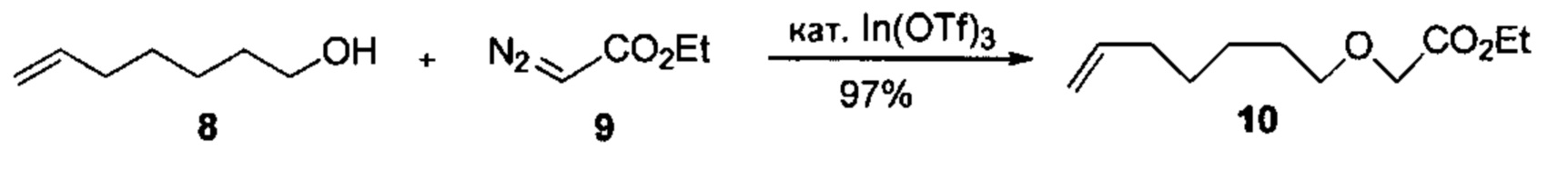
Синтез этил-2-(окт-7-ен-1-илокси)-ацетата (10); Согласно описанному в литературе7, к суспензии In(OTf)3 (1,57 г, 20 мол. %) в безводном толуоле (20 мл) при к. т.добавляли чистый 8 (1,92 г, 1,2 эквив.). В атмосфере аргона медленно добавляли этилдиазоацетат (9) (1,60 г, 14 ммоль) в течение 5 мин (осторожно: экзотермическая реакция) с получением раствора желтого цвета. Через 2 дня реакционную смесь концентрировали под вакуумом и остаток очищали с использованием хроматографической системы Teledyne Isco Combiflash• RF (колонка с 25 г SiO2, элюирование: 0-10% EtOAc/гексан, 5 мин; 10% EtOAc/гексан, 8 мин) с получением 10 (2,72 г, 97%) в виде бесцветного масла.1Н ЯМР (500 МГц, CDCl3) δ 5,80 (ddt, J=16,9, 10,2, 6,6 Гц, 1Н), 5,08-4,84 (m, 2Н), 4,22 (q, J=7,1 Гц, 2Н), 4,06 (s, 2Н), 3,52 (t, J=6,7 Гц, 2Н), 2,13-1,96 (m, 2Н), 1,72-1,52 (m, 2Н), 1,48-1,33 (m, 4Н), 1,28 (t, J=7,1 Гц, 3Н);13С ЯМР (100 МГц, CDCl3) δ 170,70, 138,99, 114,48, 71,97, 68,48, 60,86, 33,84, 29,55, 28,84, 25,63, 14,34.
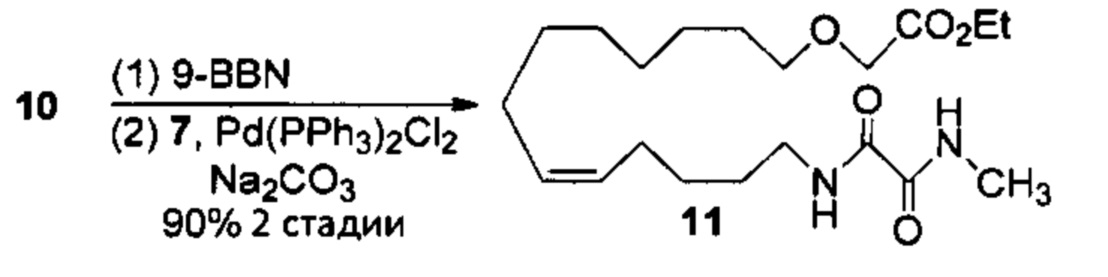
Синтез этил-2-((13-(2-(метиламино)-2-оксоацетамидо)-тридец-8(Z)-ен-1-ил)-окси)-ацетата (11): В колбу, высушенную в сушильном шкафу, содержащую этил-2-(окт-7-ен-1-илокси)-ацетат (10) (220 мг, 1,2 эквив.), добавляли раствор 9-BBN (0,5 М в ТГФ, 2,4 эквив., 4,40 мл). После перемешивания при к. т.в течение 3 ч добавляли водный раствор Na2CO3 (1,5 мл 2 М раствора, приготовленного с использованием барботированной аргоном Н2О). Через 5 мин добавляли Pd(PPh3)2Cl2 (33 мг, 5 мол. %), затем добавляли 7 (284 мг, 0,92 ммоль), растворенный в ТГФ (4 мл). Полученный раствор красного цвета защищали от света, при этом добавляли вторую порцию водн. Na2CO3 (0,5 мл 2 М раствора). Реакцию продолжали в течение ночи (14 ч) при к.т., затем при 50°С в течение 4 ч. После охлаждения до к. т.реакционную смесь концентрировали под вакуумом и остаток очищали с использованием хроматографической системы Teledyne Isco Combiflash• RF (колонка с 24 г SiO2, элюирование: 0-40% EtOAc/гексан, 6 мин; 40% EtOAc/гексан, 8 мин; 40-100% EtOAc/гексан, 4 мин) с получением простого эфира 11 (330 мг, 90%) в виде твердого вещества грязно-белого цвета. Аналитический образец очищали с помощью препаративной ТСХ с получением 11 в виде легкоплавкого твердого вещества белого цвета.
ТСХ: 50% EtOAc/гексан, Rf ~ 0,49.1Н ЯМР (500 МГц, CDCl3) δ 7,45 (ушир. s, 2Н), 5,42-5,26 (m, 2Н), 4,22 (q, J=7,2 Гц, 2Н), 4,06 (s, 2Н), 3,52 (t, J=6,7 Гц, 2Н), 3,31 (dt, J=7,0, 6,5 Гц, 2Н), 2,91 (d, J=5,1 Гц, 3Н), 2,15-1,91 (m, 4Н), 1,70-1,50 (m, 2Н), 1,44-1,31 (m, 12Н), 1,29 (t, J=7,1 Гц, 3Н);13С ЯМР (100 МГц, CDCl3) δ 170,62, 160,55, 159,66, 130,58, 128,86, 71,96, 68,64, 39,55, 29,61, 29,51, 29,30, 29,19, 27,20, 26,83, 26,67, 26,15, 25,93, 14,21.
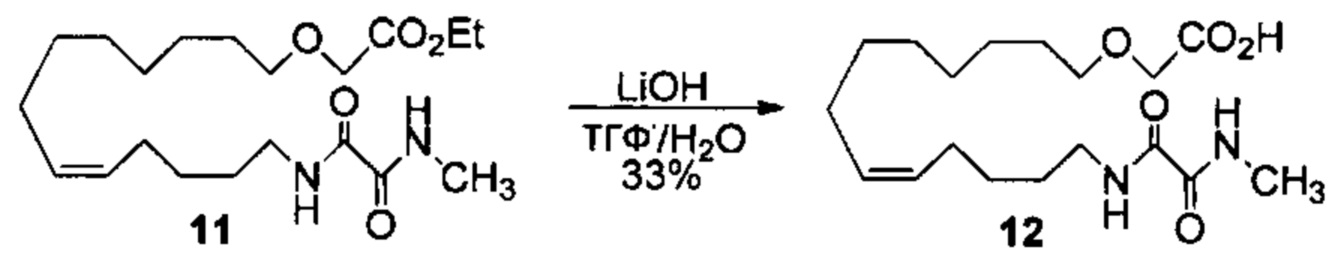
Синтез 2-((13-(2-(метиламино)-2-оксоацетамидо)-тридец-8(Z)-ен-1-ил)-окси)-уксусной кислоты (12): К раствору 11 (720 мг, 1,87 ммоль) в ТГФ (44 мл) при к.т. добавляли LiOH (9 мл 1,0 М водн. раствора). Через 48 ч реакционную смесь охлаждали до 4°С и подкисляли до рН 4 с помощью водн. 2 н. HCl. Смесь разбавляли Н2О (10 мл) и подвергали экстракции с помощью EtOAc (15 мл × 3). Объединенные органические экстракты сушили над Na2SO4, фильтровали через воронку с пористым стеклянным фильтром и концентрировали под вакуумом. Неочищенный продукт очищали с использованием хроматографической системы Teledyne Isco Combiflash• RF (колонка с 12 г SiO2, элюирование: 0-80% EtOAc/гексан, 15 мин; 80% EtOAc/гексан, 5 мин) с получением 12 (232 мг, 33%) в виде твердого вещества белого цвета, точка плавления 94,6-94,7°С.
1Н ЯМР (500 МГц, CDCl3) δ 7,90 (s, 1Н), 7,66 (s, 1Н), 5,48-5,22 (m, 2Н), 4,10 (s, 2Н), 3,58 (t, J=6,5 Гц, 2Н), 3,32 (dt, J=7,0, 6,5 Гц, 2Н), 2,91 (d, J=5,2 Гц, 3Н), 2,16-1,90 (m, 4Н), 1,71-1,48 (m, 4Н), 1,45-1,18 (m, 10Н);13С ЯМР (75 МГц, CD3OD) δ 176,96, 160,32, 160,12, 130,65, 129,99, 72,51, 69,84, 39,45, 29,82, 29,58, 29,15, 27,71, 27,38, 27,24, 27,08, 26,83, 25,84, 25,03.

Синтез соед. 03: Смесь EDCl (275 мг, 1,3 эквив.) и триэтиленгликоля (1,5 мл, 10 эквив.) сушили под высоким вакуумом в течение 90 мин. В колбу для проведения реакции нагнетали аргон и добавляли DMAP (175 мг, 1,3 эквив.), ацетонитрил (50 мл) и кислоту 12 (395 мг, 1,1 ммоль), растворенную в CH2Cl2 (20 мл). Через 3 дня реакционную смесь концентрировали под вакуумом, неочищенный остаток растворяли в EtOAc (20 мл) и промывали 1 н. HCl (20 мл) и солевым раствором (20 мл). Водные промывочные растворы повторно подвергали экстракции с помощью EtOAc (20 мл × 2). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали с использованием хроматографической системы Teledyne Isco Combiflash• RF (колонка с 12 г SiO2, элюирование: 0-80% EtOAc/гексан, 8 мин; 80% EtOAc/гексан, 4 мин; 80-100% EtOAc/гексан, 3 мин; 100% EtOAc, 15 мин; 10% МеОН/CH2Cl2, 5 мин) с получением аналога 13 (174 мг, 32%) в виде твердого вещества белого цвета, точка плавления 65,3-65,8°С.
1Н ЯМР (500 МГц, CDCl3) δ 7,46 (s, 2Н), 5,41-5,27 (m, 2Н), 4,33 (t, J=4,7 Гц, 2Н), 4,11 (s, 2Н), 3,77-3,70 (m, 4Н), 3,70-3,64 (m, 4Н), 3,61 (каж. t, J=4,5 Гц, 2Н), 3,52 (t, J=6,7 Гц, 2Н), 3,42 (t, J=6,1 Гц, ОН), 3,31 (dt, J=7,0, 6,5 Гц, 2Н), 2,91 (d, J=5,2 Гц, 3Н), 2,44 (s, 1Н), 2,05 (dt, J=7,5, 7,0 Гц, 2Н), 2,00 (dt, J=7,0, 6,5 Гц, 2Н), 1,62-1,50 (m, 4Н), 1,45-1,21 (m, 10Н);13С ЯМР (125 МГц, CDCl3) δ 170,86, 160,83, 159,95, 130,76, 129,12, 72,76, 72,21, 70,77, 70,52, 69,19, 68,34, 63,84, 61,92, 39,78, 29,84, 29,73, 29,54, 29,42, 29,02, 27,44, 27,09, 26,92, 26,42, 26,15.
Соединение 4 (соед. 04)
Синтез соединения 4 (соед. 04) аналогичен синтезу соединения 2 (соед. 02), при этом введение фрагмента мочевины осуществляли в соответствии со способом синтеза, описанным в патентной заявке WO 2010/081683 (пример 6).
Соединение 5 (соед. 05)
Общая схема синтеза

Общая методика получения соединения 4-2


К смеси соед. 4-1 (30,0 г, 137 ммоль, 1,0 экв.) в ТЭА (480 мл) добавляли соед. 1 (13,4 г, 137 ммоль, 1,0 экв.), Cul (522 мг, 2,74 ммоль, 0,02 экв.), Pd(PPh3)4 (1,58 г, 1,37 ммоль, 0,01 экв.) в атмосфере N2 при 25°С и перемешивали при 25°С в течение 16 ч. Окончание реакции определяли по данным ТСХ (петролейный эфир/этилацетат = 1/1, Rf = 0,5). Полученный раствор вливали в водн. NH4Cl (1,0 л), подвергали экстракции с помощью ДХМ (200 мл*5), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с элюированием смесью петролейного эфира : EtOAc (10:1, 1:1) с получением соед. 4-2 (21,0 г, выход 73%) в виде масла желтого цвета.
1Н ЯМР: ЕТ5008-6-P1b1 400 МГц CDCl3; 7,30-7,24 (m, 1Н), 7,10 (t, J=7,6 Гц, 1H), 6,73-6,65 (m, 2H), 4,19 (ушир., 2H), 3,74 (m, 2H), 2,54 (t, J=6,0 Гц, 2H), 1,87-1,68 (m, 4H), 1,50-1,45 (m, 1H).
Общая методика получения соед. 4-3
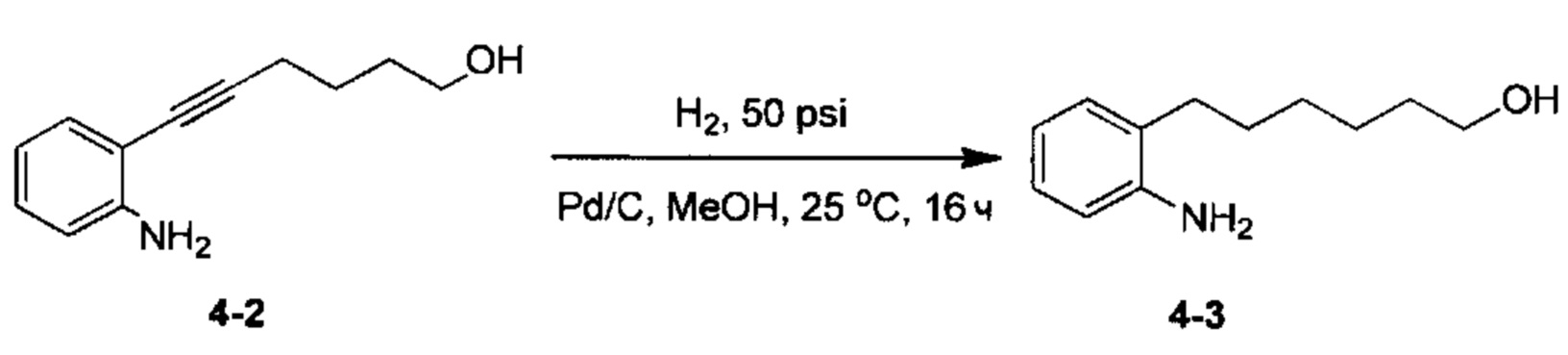

К смеси соед. 4-2 (21,0 г, 111 ммоль, 1,0 экв.) в МеОН (500 мл) добавляли Pd/C (500 мг) и перемешивали при 25°С в атмосфере Н2 под давлением примерно 0,34 МПа (50 psi) в течение 16 ч. Окончание реакции определяли по данным ЖХМС (ЕТ5008-10-Р1А5, продукт: время удерживания = 1,10 мин). Затем полученный раствор фильтровали и концентрировали с получением соед. 4-3 (17,0 г, выход 75%) в виде масла желтого цвета.
1Н ЯМР: ЕТ5008-10-P1B1 400 МГц CDCl3; 7,08-7,03 (m, 2Н), 6,78-6,69 (m, 2Н), 3,69-3,62 (m, 4Н), 2,52 (t, J=8,0 Гц, 2Н), 1,68-1,59 (m, 4Н), 1,47-1,42 (m, 4Н), 1,31-1,27 (m, 1Н).
Общая методика получения соед. 4-4


К соед. 4-3 (17,0 г, 88,0 ммоль, 1,0 экв.) в H2O (500 мл) добавляли конц. H2SO4 (30,2 г, 308 ммоль, 3,5 экв.) при 0°C в атмосфере N2. К полученному раствору добавляли раствор NaNO2 (6,07 г, 88,0 ммоль, 1,0 экв.) в Н2О (30,0 мл) при 0°С и перемешивали при 0°С в течение 15 мин. Добавляли раствор Kl (43,8 г, 264 ммоль, 3,0 экв.) в H2O (30,0 мл) при 0°С и полученную в результате суспензию нагревали до 25°С и перемешивали в течение 45 мин. Окончание реакции определяли по данным ТСХ (петролейный эфир/этилацетат = 1/1, Rf = 0,9). Добавляли Н2O (400 мл), осуществляли экстракцию с помощью EtOAc (350 мл*3), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с элюированием смесью петролейного эфира : EtOAc (100:1, 10:1) с получением соед. 4-4 (17,0 г, выход 57%) в виде масла коричневого цвета.
1Н ЯМР: ЕТ5008-22-P1b1 400 МГц CDCl3; 7,80 (d, J=7,2 Гц, 1Н), 7,28-7,23 (m, 1Н), 7,21-7,18 (m, 1Н), 6,89-6,85 (m, 1Н), 3,65 (t, J=6,8 Гц, 2Н), 2,71 (t, J=8,0 Гц, 2Н), 1,61-1,50 (m, 4Н), 1,45-1,40 (m, 4Н), 1,31-1,28 (m, 1Н).
Общая методика получения соединения 4-5'
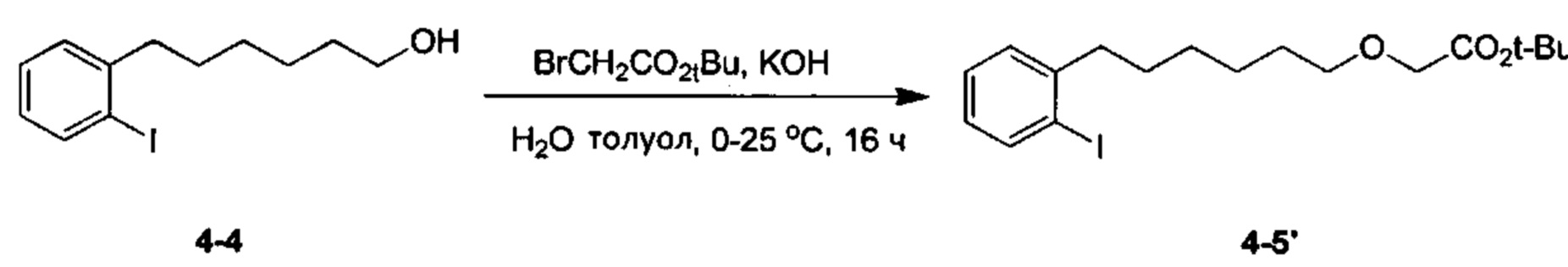
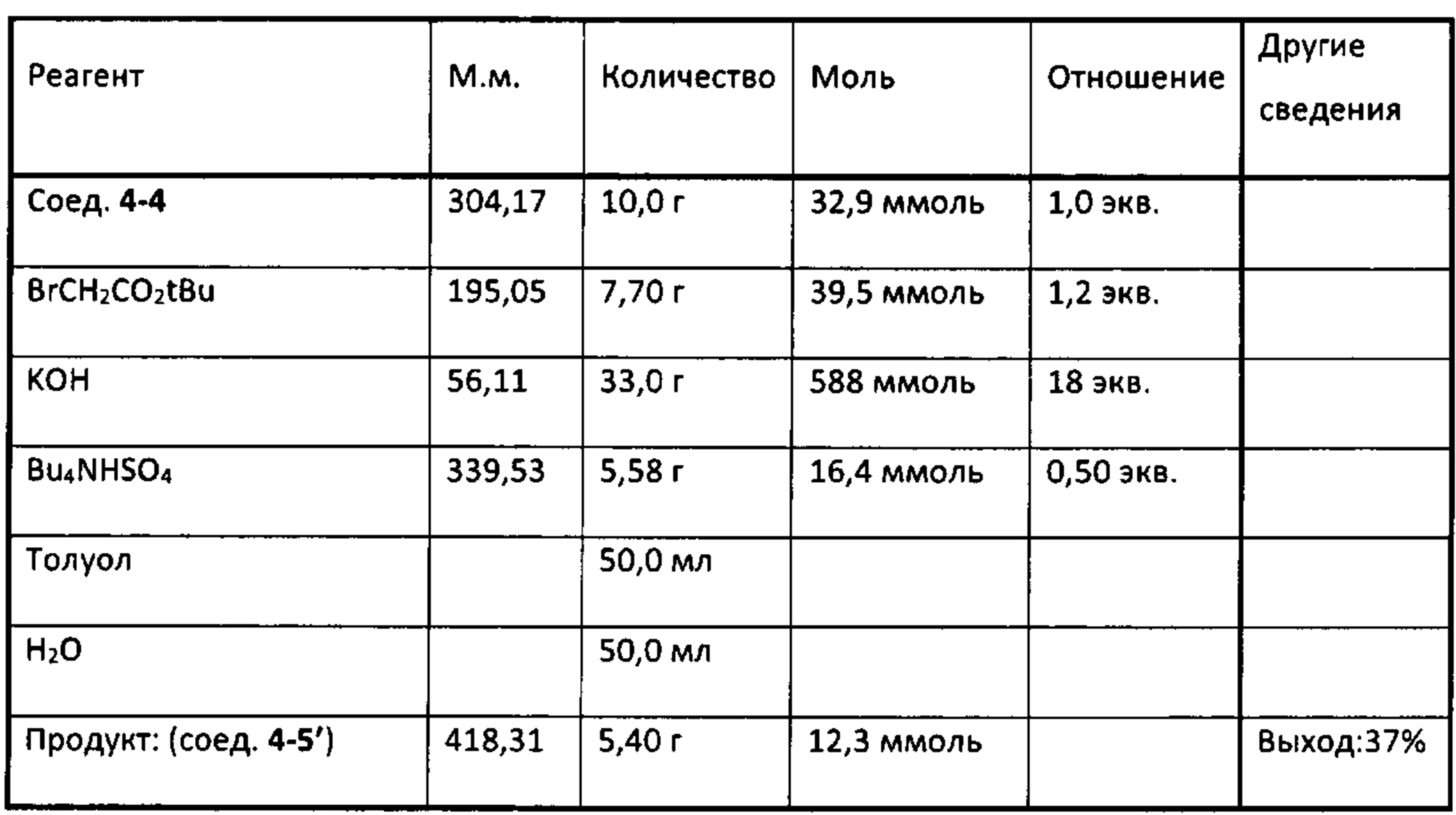
К смеси BrCH2CO2tBu (7,70 г, 39,5 ммоль, 1,2 экв.) и соед. 4-4 (10,0 г, 32,9 ммоль, 1,0 экв.) в толуоле (50,0 мл) добавляли Bu4NHSO4 (5,58 г, 16,4 ммоль, 0,50 экв.), KOH (33,0 г, 588 ммоль, 17,9 экв.) в Н2О (50,0 мл) при 0°С, затем полученную смесь перемешивали при 25°С в течение 16 ч. По данным ТСХ (петролейный эфир/этилацетат = 10/1, Rf = 0,62) оставалось 40% исходного вещества. Добавляли Н2О (200 мл), осуществляли экстракцию с помощью ДХМ (200 мл*2), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с элюированием смесью петролейного эфира : EtOAc (40:1) с получением соед. 4-5' (5,40 г, выход 37%) в виде масла желтого цвета.
1H ЯМР: ЕТ5008-26-P1b1 400 МГц CDCl3; 7,82 (d, J=7,2 Гц, 1Н), 7,30-7,26 (m, 1H), 7,23-7,20 (m, 1H), 6,91-6,88 (m, 1H), 3,97 (s, 2H), 3,53 (t, J=6,8 Гц, 2Н), 2,72 (t, J=8,0 Гц, 2Н), 1,69-1,59 (m, 4H), 1,58-1,43 (m, 13H).
Общая методика получения соед. 4-6

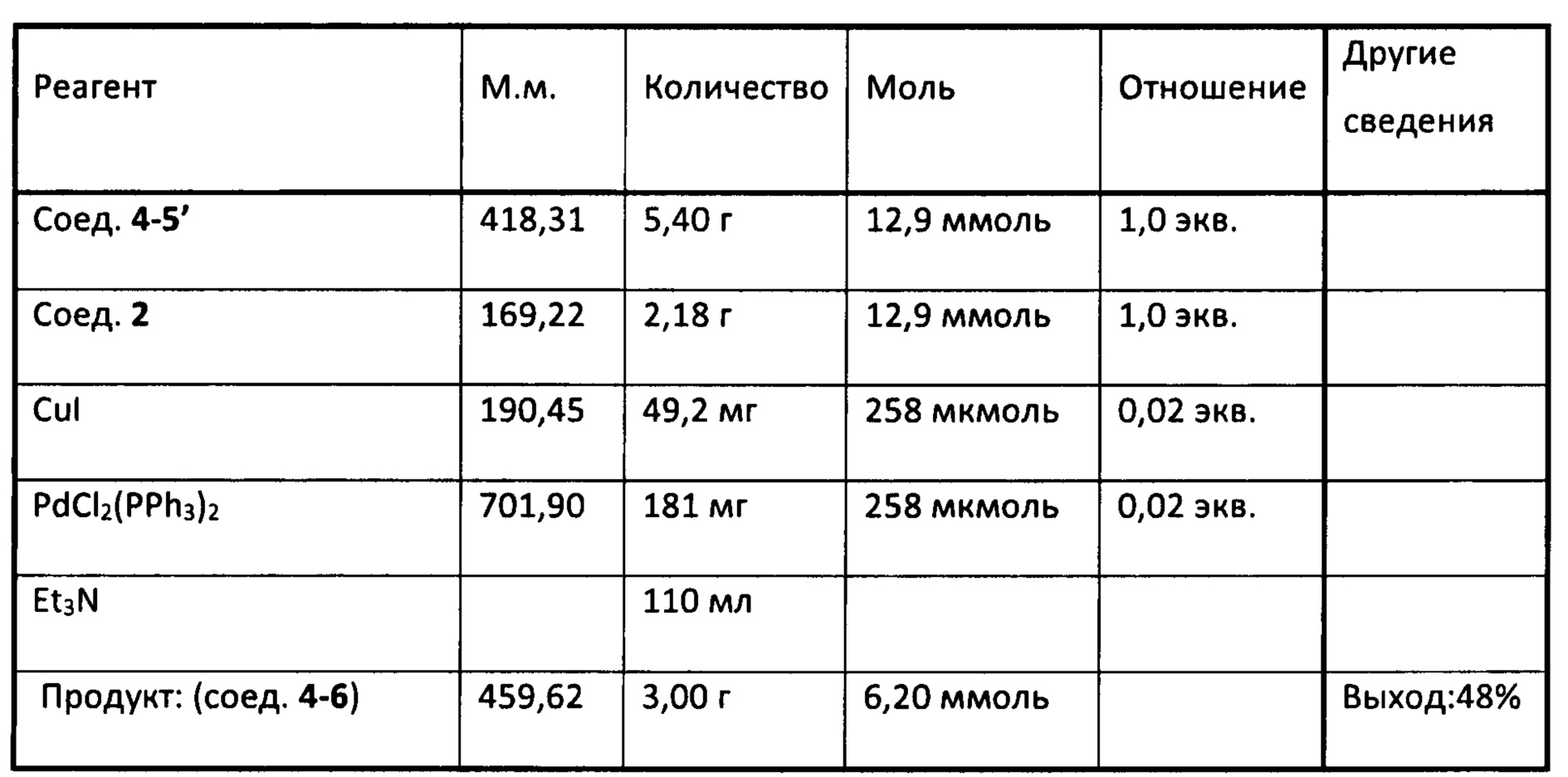
К смеси соед. 4-5' (5,40 г, 12,9 ммоль, 1,0 экв.) и соед. 2 (2,18 г, 12,9 ммоль, 1,0 экв.) в Et3N (110 мл) добавляли Cul (49,2 мг, 258 мкмоль, 0,02 экв.), PdCl2(PPh3)2 (181 мг, 258 мкмоль, 0,02 экв.) при 25°С в атмосфере N2 и перемешивали при 25°С в течение 16 ч. Окончание реакции определяли по данным ТСХ (петролейный эфир/этилацетат = 1/1, Rf=0,3). Затем добавляли водн. NH4Cl (200 мл), осуществляли экстракцию с помощью EtOAc (200 мл*3), объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали при пониженном давлении. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с элюированием смесью петролейного эфира : EtOAc (10:1, 1:1) с получением соед. 4-6 (3,00 г, 48% выход) в виде масла желтого цвета.
1H ЯМР: ЕТ5008-32-P1b1 400 МГц CDCl3; 7,41-7,34 (m, 1H), 7,23-7,06 (m, 3H), 4,97-4,87 (m, 1H), 3,97 (s, 2Н), 3,53 (t, J=6,8 Гц, 2Н), 3,43-3,33 (m, 2Н), 2,72 (t, J=8,0 Гц, 2Н), 2,64 (J=8,0 Гц, 2Н), 1,69-1.59 (m, 4H), 1,55-1,43 (m, 22H).
Общая методика получения соед. 4-7


К смеси соед. 4-6 (3,00 г, 6,53 ммоль, 1,0 экв.) в МеОН (20,0 мл) добавляли Pd/C (200 мг) и перемешивали при 25°С в атмосфере H2 под давлением примерно 0,34 МПа (50 psi) в течение 5 ч. Окончание реакции определяли по данным ЖХМС (ЕТ5008-33-Р1А4, продукт: время удерживания = 1,04 мин). Затем раствор фильтровали и концентрировали с получением соед. 4-7 (2,50 г, выход 75%) в виде масла желтого цвета.
1H ЯМР: ЕТ5008-33-P1b1 400 МГц CDCl3; 7,13 (s, 4Н), 4,54 (s, 1Н), 3,96 (s, 2H), 3,52 (t, J=6,8 Гц, 2H), 3,18-3,14 (m, 2H), 2,65-2,57 (m, 4Н), 1,75-1,54 (m, 10Н), 1,53-1,37 (m, 20H).
Общая методика получения соед. 4-10
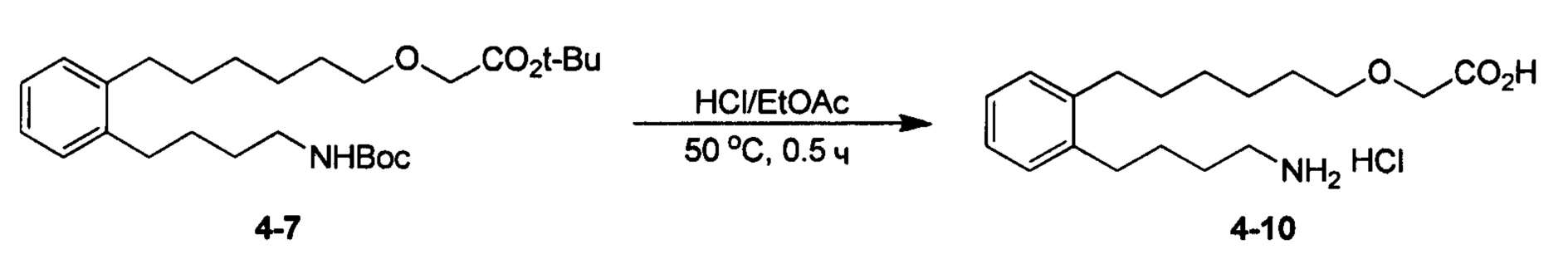

Смесь соед. 4-7 (1,00 г, 2,16 ммоль, 1,0 экв.) в HCl/EtOAc (30,0 мл) нагревали при 50°С и перемешивали при 50°С в течение 0,5 ч. Окончание реакции определяли по данным ЖХМС (ЕТ5008-34-Р1А4, продукт: время удерживания = 0,698 мин). Полученную смесь концентрировали с получением неочищенного соед. 4-10 (800 мг) в виде твердого вещества желтого цвета.
Общая методика получения соед. 05

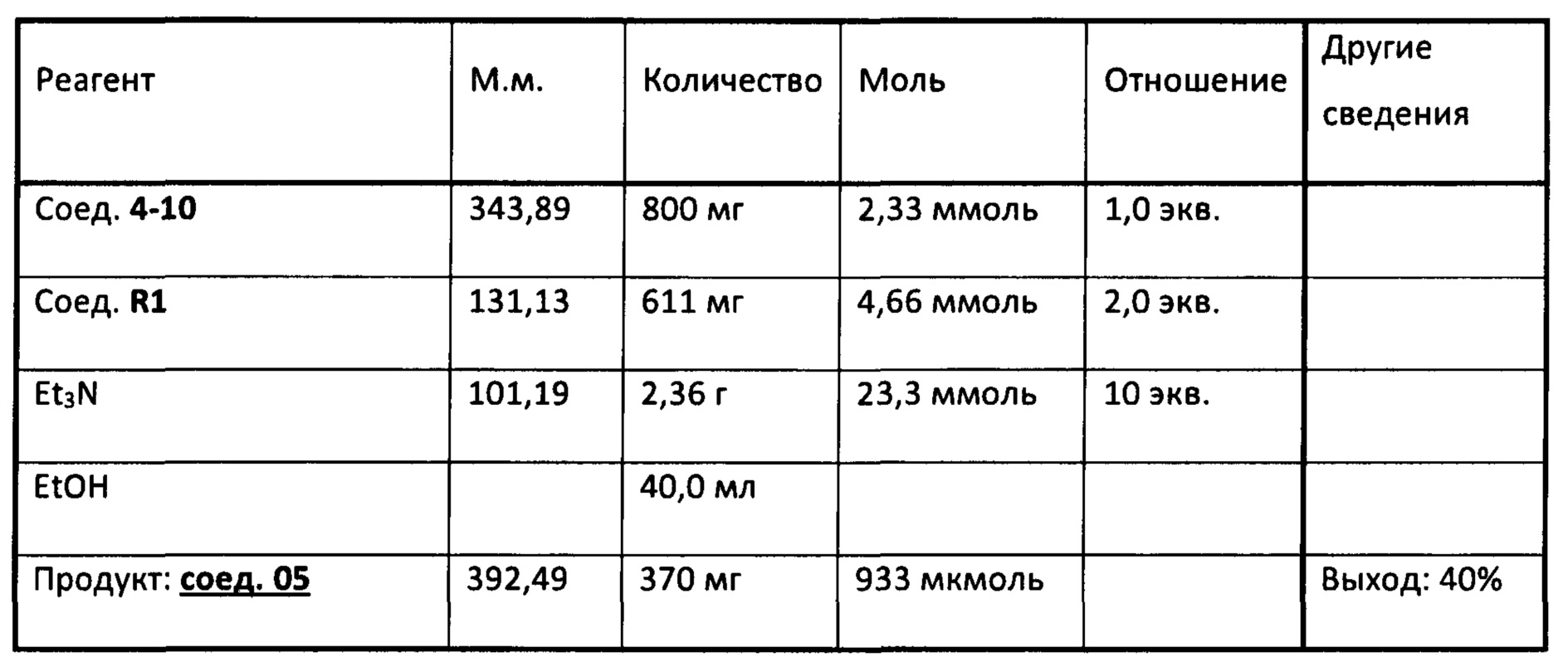
К смеси соед. 4-10 (800 мг, 2,33 ммоль, 1,0 экв.) в EtOH (40,0 мл) добавляли Et3N (2,36 г, 23,3 ммоль, 10,0 экв.) и соед. R1 (611 мг, 4,66 ммоль, 2,0 экв.) при 25°С. Затем полученный раствор перемешивали при 60°С в течение 20 ч. Окончание реакции определяли по данным ЖХМС (ЕТ5008-35-Р1А1, продукт: время удерживания = 0,81 мин). Полученный раствор концентрировали. Остаток очищали с помощью препаративной ВЭЖХ (в присутствии ТФУ) с получением соед. 05 (370 мг, выход 40%) в виде твердого вещества белого цвета.
Способ разделения с помощью ВЭЖХ:

1H ЯМР: ЕТ5008-35-P1b1 400 МГц CDCl3; 10,46 (ушир., 1Н), 8,35 (s. 1H), 7,74 (s, 1H), 7,12 (s. 4H), 4,12 (s, 2H), 3,59 (t, J=6,0 Гц, 2H 2H). 3,35 (q, J=7,2 Гц, 2Н), 2,92 (d, J=5,2 Гц. 3Н), 2,65-2,57 (m, 4H), 1,68-1,44 (m, 14H).
Для синтеза соединений с соед. 14 по соед. 32 предварительно синтезировали общие структурные блоки:
Структурный блок 1 (СБ-1)
N'-[(5Z)-6-иодгекс-5-ен-1-ил]-N-метилэтандиамид
Стадия 1:
PPh3 (140 г) и фталимид (82,5 г) суспендировали в сухом ТГФ (500,0 мл) и охлаждали до 0°С. Затем по каплям добавляли раствор 5-гексин-1-ола (50,0 г) и диизопропилазодикарбоксилата (110 мл) в сухом ТГФ (100 мл) в течение 45 мин. Полученную смесь перемешивали при 0°С в течение 1 ч и затем при к.т. в течение ночи.
ТГФ удаляли под вакуумом в максимально возможной степени. Остаток суспендировали в ПЭ/EtOAc=9:1 (700 мл) и энергично перемешивали. Растворитель отделяли декантацией от осажденного OPPh3. При этом в декантированном растворителе образовывались игольчатые кристаллы белого цвета (продукт), которые отфильтровали и оставляли (F1).
Затем осадок OPPh3 дополнительно несколько раз промывали ПЭ/EtOAc=9:1. Затем все фильтраты объединяли и упаривали под вакуумом (F2). Игольчатые кристаллы из F1 растворяли в EtOAc (200 мл) и промывали 1 н. NaOH (2×75 мл) и солевым раствором (50 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток фильтровали через слой SiO2 (элюент - CH2Cl2). Растворитель удаляли под вакуумом и маслянистый остаток оставляли в холодильнике в течение выходных, после чего образовывались игольчатые кристаллы белого цвета. Смесь разбавляли ПЭ, затем продукт отфильтровывали, промывали ПЭ и сушили под вакуумом с получением F1 в виде игольчатых кристаллов белого цвета. Маточный раствор объединяли с F2.
Масло желтого цвета из F2 растворяли в EtOAc (400 мл) и промывали 1 н. NaOH (3×150 мл) и солевым раствором (50 мл). Органический слой сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент - CH2Cl2). Фракции, содержащие продукт, объединяли и упаривали. К маслянистому остатку желтого цвета добавляли ПЭ, после чего образовывался осадок. Смесь охлаждали до 0°C, затем твердое вещество отфильтровывали и промывали ПЭ с получением F2 в виде твердого вещества белого цвета. Маточный раствор упаривали. К маслянистому остатку добавляли ПЭ, после чего образовывался осадок. Смесь оставляли в холодильнике на 2 ч, затем осадок отфильтровывали, промывали ПЭ и сушили под вакуумом с получением F3 в виде твердого вещества бледно-желтого цвета.
Стадия 2:
2-(Гекс-5-ин-1-ил)-2,3-дигидро-1Н-изоиндол-1,3-дион (46,3 г), AgNO3 (8,65 г) и NIS (68,8 г) помещали в колбу объемом 1 л. Добавляли сухой ТГФ (500 мл), в колбу нагнетали аргон и оборачивали ее алюминиевой фольгой для защиты реакционной смеси от света. Затем смесь перемешивали в атмосфере Ar при к.т. в течение 16 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь отделяли декантацией от полученного осадка, разбавляли водой (400 мл) и подвергали экстракции с помощью EtOAc (3×200 мл). Объединенные органические слои промывали водой (100 мл), насыщенным Na2SO3 (3×100 мл) и солевым раствором (100 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток перекристаллизовывали из EtOH с получением F1 в виде твердого вещества белого цвета. Маточный раствор упаривали и повторно перекристаллизовывали из EtOH с получением F2 в виде твердого вещества желтого цвета.
Стадия 3:
К охлажденному до 0°С раствору ВН 3*SMe2 (2,00 М в ТГФ, 64,4 мл) по каплям добавляли 2-метил-2-бутен (29,4 мл) и перемешивали при 0°С в течение 1 ч и затем при к.т. в течение 1 ч. Затем полученную смесь по каплям добавляли к охлажденной до 0°С суспензии 2-(6-иодгекс-5-ин-1-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона (17,5 г) в ТГФ (200 мл). После добавления полученную смесь перемешивали при к.т. в течение 1 ч. Согласно данным ЖХ/МС исходное вещество было полностью израсходовано. Реакционную смесь охлаждали до 0°С, затем по каплям добавляли НОАс (30,0 мл), перемешивали в течение 30 мин при 0°С и затем при к.т. в течение ночи. Образование продукта регистрировали поданным ЖХ/МС.
ТГФ удаляли под вакуумом в максимально возможной степени. Затем остаток медленно вливали в раствор NaOH (15,0 г) в H2O (200 мл) и подвергали экстракции с помощью CH2Cl2 (3×100 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток использовали для дальнейших превращений без дополнительной обработки.
Стадия 4:
2-[(5Z)-6-иодгекс-5-ен-1-ил]-2,3-дигидро-1Н-изоиндол-1,3-дион (17,6 г, неочищенный IK-0353/4) растворяли в МеОН (150 мл). Добавляли гидразингидрат (6,00 мл) и полученную смесь перемешивали при к.т. в течение 16 ч. Образование продукта регистрировали поданным ЖХ/МС.
МеОН удаляли под вакуумом. Остаток суспендировали в CH2Cl2 (300 мл). Твердое вещество отфильтровывали и промывали CH2Cl2 (2×100 мл). Затем объединенные фильтраты промывали водой (2×100 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением неочищенного продукта в виде масла оранжевого цвета, которое использовали для дальнейших превращений без дополнительной обработки.
Стадия 5:
Этилхлорформилформиат (10,0 г) растворяли в ТГФ (50 мл) и охлаждали до 0°С. По каплям добавляли пиридин (7,70 мл) и полученную смесь перемешивали при 0°С в течение 30 мин. Затем по каплям добавляли метиламин (2,0 М в ТГФ, 47,6 мл). Продолжали перемешивание при 0°С в течение 3 ч. Образование продукта регистрировали поданным ТСХ (ПЭ/EtOAc=1:3).
Осажденную соль отфильтровывали и фильтрат упаривали. Остаток растворяли в EtOAc (200 мл), промывали 1 н. HCl (2×50 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде масла коричневого цвета.
Стадия 6:
(5Z)-6-иодгекс-5-ен-1-амин (11,15 г) растворяли в EtOH (200 мл). Добавляли этил-(метилкарбамоил)-формиат (6,50 г) и NEt3 (8,26 мл) и полученную смесь перемешивали при 50°С в течение 24 ч. Согласно данным ЖХ/МС превращение было неполным. Добавляли дополнительную порцию (метилкарбамоил)-формиата (1,00 г) и NEt3 (4,00 мл) и продолжали перемешивание при 50°С в течение 24 ч. Образование продукта регистрировали по данным ЖХ/МС.
EtOH удаляли под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (CH2Cl2

Выход: 10,3 г (67%) твердого вещества бледно-желтого цвета.
Структурный блок 2 (СБ-2)
N'-[4-(2-иодфенил)-бутил]-N-метилэтандиамид
Стадия 1:
PPh3 (95,5 г), фталимид (56,1 г) и 3-бутен-1-ол (25,0 г) суспендировали в сухом ТГФ (250 мл) и охлаждали до 0°С. Затем по каплям добавляли диизопропилазодикарбоксилат (75,1 мл) в течение 20 мин. Полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение ночи. Образование продукта регистрировали поданным ЖХ/МС.
ТГФ удаляли под вакуумом в максимально возможной степени. Маслянистый остаток разбавляли ПЭ/EtOAc=9:1 (400 мл) и энергично перемешивали до появления осадка. Осажденный OPPh3 отфильтровывали и тщательно промывали ПЭ/EtOAc=9:1. Объединенные фильтраты фильтровали через слой SiO2 и затем упаривали. Остаток разбавляли ПЭ (200 мл), энергично перемешивали и помещали на ледяную баню. Затем осажденный продукт отфильтровывали и промывали ПЭ с получением указанного продукта с достаточной степенью чистоты в виде твердого вещества бледно-желтого цвета.
Стадия 2:
2-(Бут-3-ен-1-ил)-2,3-дигидро-1Н-изоиндол-1,3-дион (22,1 г) помещали в колбу объемом 1 л в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 273 мл) при 0°С и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение ночи. Затем добавляли раствор Na2CO3 (48,4 г) в воде (250 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли 2-иодфениламин (20,0 г) и PdCl2(PPh3)2 (2,80 г) и полученную смесь нагревали до 50°С в течение 4 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь разбавляли EtOAc (200 мл) и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (300 мл) и объединенные органические слои промывали солевым раствором (200 мл) и сушили над Na2SO4. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=6:4).
Стадия 3:
2-[4-(2-Аминофенил)-бутил]-2,3-дигидро-1Н-изоиндол-1,3-дион (22,0) растворяли в ацетоне (100 мл). Затем добавляли воду (200 мл) и конц. H2SO4 (13,9 мл) и полученную в результате суспензию охлаждали до 0°С. По каплям добавляли раствор NaNO2 (5,23 г) в воде (50 мл) и полученную смесь перемешивали при 0°С в течение 30 мин. Затем по каплям добавляли раствор KI (37,2 г) в воде (50 мл), реакционную смесь нагревали до к.т. и перемешивали в течение 20 ч. Образование продукта регистрировали поданным ЖХ/МС.
Реакционную смесь разбавляли насыщенным Na2SO3 (200 мл) и подвергали экстракции с помощью EtOAc (3×200 мл). Объединенные органические слои промывали солевым раствором (150 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=8:2).
Стадия 4:
2-[4-(2-Иодфенил)-бутил]-2,3-дигидро-1Н-изоиндол-1,3-дион (21,2 г) суспендировали в МеОН (300 мл). Добавляли гидразингидрат (5,10 мл) и полученную смесь перемешивали при к.т. в течение 3 дней. Образование продукта регистрировали по данным ЖХ/МС.
МеОН удаляли под вакуумом. Остаток суспендировали в CH2Cl2 (200 мл). Твердое вещество отфильтровывали и промывали CH2Cl2 (100 мл). Затем объединенные фильтраты промывали водой (2×100 мл). Объединенные водные слои повторно подвергали экстракции с помощью CH2Cl2 (50 мл), и затем объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде масла желтого цвета.
Стадия 5:
Этилхлорформилформиат (10,0 г) растворяли в ТГФ (50 мл) и охлаждали до 0°С. По каплям добавляли пиридин (7,70 мл) и полученную смесь перемешивали при 0°С в течение 30 мин. Затем по каплям добавляли метиламин (2,0 М в ТГФ, 47,6 мл). Продолжали перемешивание при 0°С в течение 3 ч. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=1:3).
Осажденную соль отфильтровывали, и фильтрат упаривали. Остаток растворяли в EtOAc (200 мл), промывали 1 н. HCl (2×50 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде масла коричневого цвета.
Стадия 6:
4-(2-Иодфенил)-бутан-1-амин (11,0 г, неочищенный IK-0355710) растворяли в EtOH (100 мл). Добавляли этил-(метилкарбамоил)-формиат (5,76 г) и NEt3 (6,67 мл), и полученную смесь перемешивали при 50°С в течение 18 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь охлаждали до к.т. и удаляли EtOH под вакуумом. Остаток фильтровали через слой SiO2 (CH2Cl2/MeOH=98:2). Дополнительную очистку проводили путем перекристаллизации из EtOAc.
Выход: 7,76 г (54%) твердого вещества бежевого цвета.
Структурный блок 4 (СБ-4)
2-{[(8Z)-13-[(метилкарбамоил)-формамидо]-тридец-8-ен-1-ил]окси}-уксусная кислота
Стадия 1
NaH (60% в минеральном масле, 771 мг) суспендировали в сухом ТГФ (20,0 мл). Смесь охлаждали до 0°С, затем добавляли 6-гептен-1-ол (1,18 мл). Продолжали перемешивание при 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (1,34 г) в ТГФ (10,0 мл). После окончания добавления ледяную баню удаляли и продолжали перемешивание в течение 15 мин, затем смесь нагревали до 70°С в течение 1,5 ч. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=1:1).
Реакционную смесь вливали в 1 н. NaOH (50 мл) и подвергали экстракции с помощью EtOAc (2×30 мл). Объединенные органические слои не содержали продукта, и их отбрасывали. Водный слой осторожно подкисляли с помощью конц. HCl и затем снова подвергали экстракции с помощью EtOAc (3×30 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде бесцветного масла.
Стадия 2
1,1'-Карбонилдиимидазол (15,6 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли раствор 2-(гепт-6-ен-1-илокси)-уксусной кислоты (15,1 г) в ТГФ (20 мл) и полученную смесь перемешивали при к.т. в течение 6 ч. Затем ТГФ удаляли под вакуумом и к остатку добавляли МеОН (200 мл). Полученную смесь перемешивали при к.т. в течение 3 дней. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=9:1).
МеОН удаляли под вакуумом. К остатку добавляли ПЭ (200 мл) и энергично перемешивали в течение 5 мин. Затем растворитель отделяли декантацией от густого маслянистого остатка, который дополнительно промывали ПЭ (2×100 мл) и затем отбрасывали. Объединенные ПЭ фракции промывали 1 н. HCl (100 мл) и 1 н. NaOH (100 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде бесцветной жидкости.
Стадия 3
Метил-2-(гепт-6-ен-1-илокси)-ацетат (2,88 г) помещали в колбу объемом 100 мл и охлаждали до 0°С в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 38,7 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч. Затем добавляли раствор Na2CO3 (6,84 г) в воде (30,0 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[(5Z)-6-иодгекс-5-ен-1-ил]-N-метилэтандиамид (СБ-1, 4,00 г) и PdCl2(PPh3)2 (453 мг) и полученную смесь нагревали до 50°С в течение 1,5 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь охлаждали до к.т. и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc 1:1).
Стадия 4
Метил-2-{[(8Z)-13-[(метилкарбамоил)-формамидо]-тридец-8-ен-1-ил]-окси}-ацетат (400 мг) суспендировали в МеОН (20,0 мл). Добавляли NaOH (3 н., 5,00 мл) и полученную смесь перемешивали при к.т. в течение 15 мин. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь вливали в 1 н. HCl (30 мл). Осажденный продукт отфильтровывали, промывали водой и сушили под вакуумом.
Выход: 869 мг (86%) твердого вещества бежевого цвета.
Структурный блок 6 (СБ-6)
2-[3-(2-{4-[(Метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-уксусная кислота
Стадия 1:
NaH (60% в минеральном масле, 15,2 г) суспендировали в сухом ТГФ (250 мл). Смесь охлаждали до 0°С, затем добавляли аллиловый спирт (11,8 мл). Продолжали перемешивание при 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (26,3 г) в ТГФ (50,0 мл). После окончания добавления ледяную баню удаляли и продолжали перемешивание в течение 15 мин, затем смесь нагревали до 70°С в течение 3 ч и перемешивали при к.т. в течение ночи.
Реакционную смесь вливали в воду (250 мл) и подвергали экстракции с помощью EtOAc (2×100 мл). Объединенные органические слои не содержали продукта, и их отбрасывали. Водный слой осторожно подкисляли с помощью конц. HCl и затем снова подвергали экстракции с помощью EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде жидкости бледно-коричневого цвета.
Стадия 2:
1,1'-Карбонилдиимидазол (30,7 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли 2-(проп-2-ен-1-илокси)-уксусную кислоту (неочищенная IK-0352/9) и полученную смесь перемешивали при к.т. в течение 7 ч. Затем ТГФ удаляли под вакуумом и к остатку добавляли МеОН (200 мл). Смесь перемешивали при к.т. в течение ночи. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=8:2).
МеОН удаляли под вакуумом. К остатку добавляли ПЭ (200 мл) и энергично перемешивали в течение 5 мин. Затем растворитель отделяли декантацией от густого маслянистого остатка, который дополнительно промывали ПЭ (2×100 мл). Согласно данным ТСХ, большая часть продукта оставалась в маслянистом остатке, который поэтому промывали МТБЭ (4×100 мл). ПЭ и МТБЭ слои объединяли и промывали 1 н. HCl (3×100 мл) и солевым раствором (50 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде жидкости бледно-желтого цвета.
Стадия 3:
Метил-2-(проп-2-ен-1-илокси)-ацетат (1,30 г) помещали в колбу объемом 100 мл и охлаждали до 0°С в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 25,0 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч. Затем добавляли раствор Na2CO3 (4,41 г) в воде (25,0 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[4-(2-иодфенил)-бутил]-N-метилэтандиамид (СБ-2, 3,00 г) и PdCl2(PPh3)2 (292 мг) и смесь нагревали до 50°С в течение 4 ч и затем перемешивали при к.т. в течение ночи. Согласно данным ЖХ/МС превращение было неполным. В атмосфере Ar в отдельную колбу помещали дополнительную порцию метил-2-(проп-2-ен-1-илокси)-ацетата (650 мг). Добавляли 9-BBN (0,5 М в ТГФ, 12,5 мл) при к.т. и полученную смесь перемешивали при к.т. в течение 2 ч. Добавляли насыщенный раствор Na2CO3 (10 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем полученную смесь добавляли к указанной выше реакционной смеси. После добавления свежего PdCl2(PPh3)2 (200 мг) смесь перемешивали при 50°С в течение 2 ч. Образование продукта регистрировали поданным ЖХ/МС.
Реакционную смесь охлаждали до к.т. и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc 3:7).
Стадия 4:
Метил-2-[3-(2-{4-[(метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-ацетат (2,04 г) растворяли в ТГФ (30 мл). Добавляли NaOH (3 н., 30 мл) и МеОН (20 мл) и полученную смесь перемешивали при к.т. в течение 5 мин. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь подкисляли с помощью 6 н. HCl и подвергали экстракции с помощью EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью короткой колонки на SiO2 (CH2Cl2/MeOH=9:1).
Выход: 1,56 г (80%) твердого вещества бежевого цвета.
Структурный блок 8 (СБ-8)
N'-[(5Z)-13-гидрокситридец-5-ен-1-ил]-N-метилэтандиамид
Стадия 1:
6-Гептен-1-ол (3,00 г) и имидазол (3,57 г) растворяли в ДМФА (20,0 мл). Добавляли TIPSCI (6,18 мл) и полученную смесь перемешивали при 60°С в течение 6 ч. Согласно данным ТСХ (ПЭ/EtOAc=8:2) превращение было почти полным.
Реакционную смесь разбавляли водой (100 мл) и подвергали экстракции с помощью МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. HCl (2×50 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=95:5).
Стадия 2:
(Гепт-6-ен-1-илокси)-трис-(пропан-2-ил)-силан (1,57 г) помещали в колбу объемом 100 мл и охлаждали до 0°С в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 14,5 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч. Затем добавляли раствор Na2CO3 (2,56 г) в воде (15,00 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[(5Z)-6-иодгекс-5-ен-1-ил]-N-метилэтандиамид (СБ-1, 1,50 г) и PdCl2(PPh3)2 (170 мг) и смесь нагревали до 50°С в течение 2 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь охлаждали до к.т. и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (2×50 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток фильтровали через слой SiO2 (ПЭ/EtOAc=4:6). Полученный таким образом неочищенный продукт использовали для дальнейших превращений без дополнительной обработки.
Стадия 3:
(Гепт-6-ен-1-илокси)-трис-(пропан-2-ил)-силан (2,20 г, неочищенный IK-0357/16) растворяли в ТГФ (50 мл) и охлаждали до 0°С. Добавляли TBAF*3H2O (2,29 г) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 6 ч. Согласно данным ТСХ (ПЭ/EtOAc=1:1) и ЖХ/МС превращение было полным.
Реакционную смесь вливали в воду (100 мл) и подвергали экстракции с помощью EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток пропускали через короткую колонку с SiO2 (ПЭ/EtOAc = 1:1
Выход: 1,11 г (77%) твердого вещества бежевого цвета.
Структурный блок 9 (СБ-9)
N'-{4-[2-(3-гидроксипропил)-фенил]-бутил}-N-метилэтандиамид
Стадия 1:
2-Пропен-1-ол (3,00 г) и имидазол (7,03 г) растворяли в ДМФА (20,0 мл). Добавляли TIPSCI (14,4 мл) и полученную смесь перемешивали при 60°С в течение 6 ч. Согласно данным ТСХ (ПЭ/EtOAc=8:2) превращение было почти полным. Реакционную смесь разбавляли водой (100 мл) и подвергали экстракции с помощью МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. HCl (2×50 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=95:5).
Стадия 2:
(Проп-2-ен-1-илокси)-трис-(пропан-2-ил)-силан (1,33 г) помещали в колбу объемом 100 мл и охлаждали до 0°С в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 14,2 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч.
Затем добавляли раствор Na2CO3 (2,21 г) в воде (15,0 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[4-(2-иодфенил)-бутил]-N-метилэтандиамид (1,50 г) и PdCl2(PPh3)2 (146 мг) и смесь нагревали до 50°С в течение 3 ч. Образование продукта регистрировали по данным ЖХ/МС. Реакционную смесь охлаждали до к.т. и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (100 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток пропускали через короткую колонку с SiO2 (ПЭ/EtOAc 1:1). Оставшийся неочищенным продукт затем использовали для дальнейших превращений без дополнительной обработки.
Стадия 3:
N-метил-N'-{4-(2-(3-{[трис-(пропан-2-ил)-силил]-окси}-пропил)-фенил]-бутил}-этандиамид (1,87 г, неочищенный IK-0357/17) растворяли в ТГФ (50 мл) и охлаждали до 0°С. Добавляли TBAF*3H2O (1,97 г) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 16 ч. Согласно данным ЖХ/МС превращение было полным. Реакционную смесь вливали в воду (100 мл) и подвергали экстракции с помощью EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток пропускали через короткую колонку с SiO2 (ПЭ/EtOAc = 1:1
Выход: 911 мг (75%) твердого вещества бежевого цвета.
Структурный блок 11 (СБ-11)
N'-[(5Z)-13-(2-аминоэтокси)-тридец-5-ен-1-ил]-N-метилэтандиамид
Стадия 1:
NaH (60% в минеральном масле, 7,71 г) суспендировали в сухом ТГФ (200 мл). Смесь охлаждали до 0°С, затем добавляли 6-гептен-1-ол (11,8 мл). Продолжали перемешивание при 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (13,4 г) в ТГФ (100 мл). После окончания добавления ледяную баню удаляли и продолжали перемешивание в течение 15 мин, затем смесь нагревали до 70°С в течение 3 ч. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=1:1).
Реакционную смесь вливали в 1 н. NaOH (300 мл) и подвергали экстракции с помощью EtOAc (2×100 мл). Объединенные органические слои не содержали продукта, и их отбрасывали. Водный слой осторожно подкисляли с помощью конц. HCl и затем снова подвергали экстракции с помощью EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде масла бледно-коричневого цвета.
Стадия 2:
1,1'-Карбонилдиимидазол (15,6 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли раствор 2-(гепт-6-ен-1-илокси)-уксусной кислоты (15,1 г) в ТГФ (20 мл) и полученную смесь перемешивали при к.т. в течение 6 ч. Затем ТГФ удаляли под вакуумом и к остатку добавляли МеОН (200 мл). Полученную смесь перемешивали при к.т. в течение 3 дней. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=9:1).
МеОН удаляли под вакуумом. К остатку добавляли ПЭ (200 мл) и энергично перемешивали в течение 5 мин. Затем растворитель отделяли декантацией от густого маслянистого остатка, который дополнительно промывали ПЭ (2×100 мл) и затем отбрасывали. Объединенные ПЭ фракции промывали 1 н. HCl (100 мл) и 1 н. NaOH (100 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде бесцветной жидкости.
Стадия 3:
Метил-2-(гепт-6-ен-1-илокси)-ацетат (5,00 г) растворяли в CH2Cl2 (100 мл) и охлаждали до 0°С. По каплям добавляли DIBALH (1,00 М в CH2Cl2, 61,7 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение ночи. Согласно данным ТСХ (ПЭ/EtOAc=8:2) превращение было полным.
Реакционную смесь охлаждали до 0°С и осторожно гасили насыщенным водным Na2SO4. Затем смесь разбавляли CH2Cl2 (100 мл), энергично перемешивали в течение 20 мин и затем фильтровали через целит. Осадок на фильтре несколько раз промывали CH2Cl2. Объединенные фильтраты концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде бесцветной жидкости.
Стадия 4:
PPh3 (7,17 г), фталимид (4,21 г) и 2-(гепт-6-ен-1-илокси)-этан-1-ол (4,12 г) суспендировали в сухом ТГФ (100 мл) и охлаждали до 0°С. Затем по каплям добавляли диизопропилазодикарбоксилат (5,79 мл) в течение 20 мин. Полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение ночи.
ТГФ удаляли под вакуумом в максимально возможной степени. Маслянистый остаток разбавляли ПЭ/EtOAc=9:1 (200 мл) и энергично перемешивали до появления осадка. Осажденный OPPh3 отфильтровывали и тщательно промывали ПЭ/EtOAc=9:1. Объединенные фильтраты фильтровали через слой SiO2 (элюент - ПЭ/EtOAc=9:1) и упаривали. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=8:2).
Стадия 5:
2-[2-(Гепт-6-ен-1-илокси)-этил]-2,3-дигидро-1Н-изоиндол-1,3-дион (2,22 г) помещали в колбу объемом 100 мл и охлаждали до 0°С в атмосфере Ar. Затем по каплям добавляли 9-BBN (0.5 М в ТГФ, 19,3 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч. Затем добавляли раствор Na2CO3 (3,42 г) в воде (20,0 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[(5Z)-6-иодгекс-5-ен-1-ил]-N-метилэтандиамид (СБ-1, 2,00 г) и PdCl2(PPh3)2 (226 мг) и смесь нагревали до 50°С в течение 1,5 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь охлаждали до к.т. и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (2×30 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=4:6).
Стадия 6:N'-[(5Z)-13-[2-(1,3-диоксо-2,3-дигидро-1Н-изоиндол-2-ил)-этокси]-тридец-5-ен-1-ил]-N-метилэтандиамид (2,36 г) суспендировали в МеОН (100 мл). Добавляли гидразингидрат (486 мкл) и полученную смесь перемешивали при к.т. в течение 16 ч. Образование продукта регистрировали поданным ЖХ/МС.
МеОН удаляли под вакуумом. Остаток суспендировали в CH2Cl2/7 н. NH3 в МеОН=9:1 (100 мл) и фильтровали через слой SiO2 и дополнительно элюировали CH2Cl2/7 н. NH3 в МеОН=9:1 (300 мл). Фильтрат концентрировали под вакуумом с получением 1,68 г неочищенного продукта. 60 мг подвергали очистке с помощью препаративной ТСХ (CH2Cl2/7 н. NH3 в МеОН=9:1). Остаток неочищенного вещества использовали для дальнейших превращений без дополнительной обработки.
Выход: 41 мг (2%) твердого вещества бледно-желтого цвета (очищенного).
Соединение 14 (соед. 14)
N-метил-N'-[(5Z)-13-[(1H-1,2,3,4-тетразол-5-ил)-метокси]-тридец-5-ен-1-ил]-этандиамид
Стадия 1
NaH (60% в минеральном масле, 7,71 г) суспендировали в сухом ТГФ (200 мл). Смесь охлаждали до 0°С, затем добавляли 6-гептен-1-ол (11,8 мл). Продолжали перемешивание при 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (13,4 г) в ТГФ (100 мл). После окончания добавления ледяную баню удаляли и продолжали перемешивание в течение 15 мин, затем смесь нагревали до 70°С в течение 3 ч. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=1:1). Реакционную смесь вливали в 1 н. NaOH (300 мл) и подвергали экстракции с помощью EtOAc (2×100 мл), Объединенные органические слои не содержали продукта, и их отбрасывали. Водный слой осторожно подкисляли с помощью конц. HCl и затем снова подвергали экстракции с помощью EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде масла бледно-коричневого цвета.
Выход: 14,1 г (93%) масла бледно-коричневого цвета.
Стадия 2
Смесь 2-(гепт-6-ен-1-илокси)-уксусной кислоты (3,00 г) и SOCl2 (15,00 мл) нагревали до 70°С в течение 1 ч. Затем избыток SOCl2 удаляли под вакуумом и остаток растворяли в дихлорэтане (15,0 мл). Затем через раствор медленно барботировали аммиак в течение 5 мин. Реакционную смесь разбавляли водой (50 мл) и подвергали экстракции с помощью CH2Cl2 (3×30 мл). Объединенные органические слои промывали насыщенным NaHCO3 (30 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде твердого вещества белого цвета. m=2,16 г (выход = 62%). Аналог IK-0367/1 по данным ТСХ.
Стадия 3
2-(Гепт-6-ен-1-илокси)-ацетамид (2,61 г) растворяли в CH2Cl2 (50 мл). Добавляли NEt3 (6,35 мл) и полученную смесь охлаждали до 0°С. Медленно добавляли раствор POCl3 (1,54 мл) в CH2Cl2 (4 мл). Затем продолжали перемешивание при 0°С в течение 15 мин. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=8:2).
Добавляли насыщенный NaHCO3 (5,00 мл) при 0°С и перемешивали в течение 30 мин при той же температуре. Смесь доводили до к.т., разбавляли водой (15,0 мл) и подвергали экстракции с помощью CH2Cl2 (3×20 мл). Объединенные органические слои промывали насыщенным NaHCO3 (10,0 мл) и солевым раствором (10,0 мл), сушили над Na2SO4 и затем фильтровали через слой SiO2 (элюент - CH2Cl2). После упаривания получали продукт с достаточной степенью чистоты в виде бесцветного масла, m=2,08 г, выход = 89%.
Стадия 4
2-(Гепт-6-ен-1-илокси)-ацетонитрил помещали в колбу объемом 10 мл и охлаждали до 0°C в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 1,63 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч. Затем добавляли раствор Na2CO3 (288 мг) в дегазированной воде (1 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[(5Z)-6-иодгекс-5-ен-1-ил]-N-метилэтандиамид (168 мг) и PdCl2(PPh3)2 (19 мг) и смесь нагревали до 50°С в течение ночи. Добавляли воду и полученную смесь подвергали экстракции с помощью ДХМ. Органический слой сушили над MgSO4, фильтровали и растворитель упаривали. Смесь очищали с помощью препаративной ТСХ (ДХМ/МеОН 95/5). m=70 мг, выход = 38%.
Стадия 5
N'-[(5Z)-13-(цианометокси)-тридец-5-ен-1-ил]-N-метилэтандиамид, азид натрия и триэтиламина гидрохлорид растворяли в ТГФ и полученную реакционную смесь перемешивали при 70°С в течение ночи.
Добавляли воду и этилацетат. Смесь подкисляли с помощью 3 н. HCl. Затем водный слой (кислый рН) подвергали экстракции с помощью этилацетата (×3) и объединенный органический слой промывали солевым раствором. Органический слой сушили над MgSO4, фильтровали и растворитель удаляли под вакуумом, m=82 мг. Продукт очищали с помощью препаративной ТСХ (ДХМ/МеОН 95/5).
Выход: 8 мг (10%), в виде порошка белого цвета.
Соединение 15 (соед. 15)
N'-(4-{2-[3-(карбамоилметокси)-пропил]-фенил}-бутил)-N-метилэтандиамид
250 мг (0,72 ммоль) 2-[3-(2-{4-[(метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-уксусной кислоты (СБ-6) и 164,1 мг (0,86 ммоль) EDCI растворяли в 20 мл ДХМ. Добавляли 36,5 мг (2,14 ммоль, 5,35 мл) аммиака (0,4 М в ТГФ) и полученную смесь перемешивали при к.т. в течение выходных.
Смесь вливали в 50 мл воды и подвергали экстракции с помощью ДХМ (3×50 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали.
Выход; 50 мг (20%) твердого вещества белого цвета.
Соединение 16 (соед. 16)
N-Метил-N'-[(5Z)-13-[(5-оксо-4,5-дигидро-1Н-1,2,4-триазол-3-ил)-метокси]-тридец-5-ен-1-ил]-этандиамид
Стадия 1:
NaH (60% в минеральном масле, 7,71 г) суспендировали в сухом ТГФ (200 мл). Смесь охлаждали до 0°С, затем добавляли 6-гептен-1-ол (11,8 мл). Продолжали перемешивание при 0°С в течение 30 мин, затем по каплям добавляли раствор бромуксусной кислоты (13,4 г) в ТГФ (100 мл). После окончания добавления ледяную баню удаляли и продолжали перемешивание в течение 15 мин, затем смесь нагревали до 70°С в течение 3 ч. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=1:1). Реакционную смесь вливали в 1 н. NaOH (300 мл) и подвергали экстракции с помощью EtOAc (2×100 мл). Объединенные органические слои не содержали продукта, и их отбрасывали. Водный слой осторожно подкисляли с помощью конц. HCl и затем снова подвергали экстракции с помощью EtOAc (3×100 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде масла бледно-коричневого цвета.
Стадия 2:
1,1'-Карбонилдиимидазол (15,6 г) суспендировали в ТГФ (200 мл). Затем по каплям добавляли раствор 2-(гепт-6-ен-1-илокси)-уксусной кислоты (15,1 г) в ТГФ (20 мл) и полученную смесь перемешивали при к.т. в течение 6 ч. Затем ТГФ удаляли под вакуумом и к остатку добавляли МеОН (200 мл). Смесь перемешивали при к.т. в течение 3 дней. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=9:1). МеОН удаляли под вакуумом. К остатку добавляли ПЭ (200 мл) и энергично перемешивали в течение 5 мин. Затем растворитель отделяли декантацией от густого маслянистого остатка, который дополнительно промывали ПЭ (2×100 мл) и затем отбрасывали. Объединенные ПЭ фракции промывали 1 н. HCl (100 мл) и 1 н. NaOH (100 мл), сушили над Na2SO4 и концентрировали под вакуумом с получением продукта с достаточной степенью чистоты в виде бесцветной жидкости.
Стадия 3:
500 мг (2,68 ммоль) и 1,34 г (26,9 ммоль, 1.30 мл) гидразингидрата растворяли в 5 мл EtOH и перемешивали при 70°С в течение 4,5 ч (

Смесь упаривали до сухого состояния.
Стадия 4:
500 мг (2,68 ммоль) 2-(гепт-6-ен-1-илокси)-ацетогидразида (GH-0513/1) растворяли в 3 мл АсОН. Добавляли 653,3 мг (8,05 ммоль) цианата калия, растворенного в 3 мл воды, и полученную смесь перемешивали при к.т. в течение 1,5 ч (до получения раствора желтого цвета). Смесь упаривали до сухого состояния.
Стадия 5:
Маслянистый остаток растворяли в 10 мл 2М NaOH и нагревали до появления конденсата в течение 2 ч.
Стадия 6:
В атмосфере аргона к раствору 88,5 мг (0,73 ммоль, 1,45 мл) 9BBN (0,5М в ТГФ) добавляли 92,0 мг (0,44 ммоль) 3-[(гепт-6-ен-1-илокси)-метил]-4,5-дигидро-1Н-1,2,4-триазол-5-она (GH-0515/1), растворенного в 2 мл безводного ТГФ, и смесь перемешивали при к.т. в течение ночи. Добавляли раствор 153,8 мг (1,45 ммоль) Na2CO3 в 1 мл воды и продолжали перемешивание при к.т. в течение 15 мин. Затем добавляли 90,0 мг (0,29 ммоль) N'-[(5Z)-6-иодгекс-5-ен-1-ил]-метилэтандиамида (IK-0356/2), растворенного в 2 мл ТГФ, и 10,2 мг (14,5 мкмоль) PdCl2(PPh3)2 и смесь нагревали до 50°С в течение 4 ч (до получения двухфазной смеси желтого цвета).
Органический слой отделяли с помощью пипетки и упаривали до сухого состояния. Неочищенный продукт очищали с помощью колоночной флэш-хроматографии на силикагеле (ДХМ/МеОН от 20:1 до 9:1, Rf возможного продукта: 0,62). Перекристаллизация из CAN.
Выход: 51 мг (0,13 ммоль, 45%).
Соединение 17 (соед. 17)
N-метил-N'-[(5Z)-13-[(фенилкарбамоил)-метокси]-тридец-5-ен-1-ил]-этандиамид
2-{[(8Z)-13-[(метилкарбамоил)-формамидо]-тридец-8-ен-1-ил]-окси}-уксусную кислоту (СБ-4, 50,0 мг, 140,3 мкмоль), анилин (26 мкл, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при к.т. в течение 16 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь разбавляли водой (20 мл) и подвергали экстракции с помощью Et2O (3×20 мл). Объединенные органические слои промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Продукт подвергали лиофилизации.
Выход: 52 мг (87%) твердого вещества белого цвета.
Соединение 18 (соед. 18)
N-метил-N'-[(5Z)-13-{[(оксан-4-ил)-карбамоил]-метокси}-тридец-5-ен-1-ил]-этандиамид
2-{[(8Z)-13-[(метилкарбамоил)-формамидо]-тридец-8-ен-1-ил]-окси}-уксусную кислоту (СБ-4, 50,0 мг, 140,3 мкмоль), 4-аминотетрагидропиран (29 мкл, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при к.т. в течение 16 ч. Образование продукта регистрировали поданным ЖХ/МС. Реакционную смесь разбавляли водой (20 мл) и подвергали экстракции с помощью Et2O (3×20 мл). Объединенные органические слои промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Продукт подвергали лиофилизации.
Выход: m=58 мг (94%) твердого вещества белого цвета.
Соединение 19 (соед. 19)
N-метил-N'-[(5Z)-13-{[(1,3-оксазол-2-ил)-карбамоил]-метокси}-тридец-5-ен-1-ил]-этандиамид
2-{[(8Z)-13-[(метилкарбамоил)-формамидо]-тридец-8-ен-1-ил]-окси}-уксусную кислоту (СБ-4, 50,0 мг, 140,3 мкмоль). 1,3-оксазол-2-амин (24 мг, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при к.т. в течение 16 ч. Образование продукта регистрировали поданным ЖХ/МС.
Реакционную смесь разбавляли водой (20 мл) и подвергали экстракции с помощью Et2O (3×20 мл). Объединенные органические слои промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (ДХМ/МеОН=95:5).
Выход: 11 мг (19%) твердого вещества белого цвета.
Соединение 20 (соед. 20)
N'-[(5Z)-13-{[(4-метоксифенил)-карбамоил]-метокси}-тридец-5-ен-1-ил]-N-метилэтандиамид
2-{[(8Z)-13-[(метилкарбамоил)-формамидо]-тридец-8-ен-1-ил]-окси}-уксусную кислоту (СБ-4, 50,0 мг, 140,3 мкмоль), п-анизидин (35 мг, 280,5 мкмоль), HBTU (53,4 мг, 140,3 мкмоль) и DMAP (1,7 мг, 14,0 мкмоль) помещали во флакон G16. Добавляли ДМФА (2,00 мл) и NEt3 (78,0 мкл, 561,1 мкмоль) и полученную смесь перемешивали при к.т. в течение 16 ч. Образование продукта регистрировали поданным ЖХ/МС.
Реакционную смесь разбавляли водой (20 мл) и подвергали экстракции с помощью Et2O (3×20 мл). Объединенные органические слои промывали насыщенным NaHCO3 (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (ДХМ/МеОН=98:2).
Выход: 18 мг (28%) твердого вещества бежевого цвета.
Соединение 21 (соед. 21)
N-метил-N'-[4-(2-{3-[(фенилкарбамоил)-метокси]-пропил}-фенил)-бутил]-этандиамид
40 мг (0,11 ммоль) 2-[3-(2-{4-[(метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-уксусной кислоты (СБ-6), 26,3 мг (0,14 ммоль) EDCI и 11,7 мг (0,13 ммоль, 11,5 мкл) анилина растворяли в 3 мл ДХМ и перемешивали при к.т. в течение выходных (прозрачный раствор).
Смесь упаривали до сухого состояния и очищали с помощью препаративной ТСХ (1 мм, ДХМ/МеОН 20:1, Rf возможного продукта: 0,54).
Выход: 24 мг (51%) твердого вещества белого цвета.
Соединение 22 (соед. 22)
N-метил-N'-[4-(2-{3-[2-оксо-2-(пирролидин-1-ил)-этокси]-пропил}-фенил)-бутил]-этандиамид
50 мг (0,14 ммоль) 2-[3-(2-{4-[(метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-уксусной кислоты (СБ-6), 32,8 мг (0,17 ммоль) EDCI и 20,3 мг (0,29 ммоль, 23,4 мкл) пирролидина растворяли в 3 мл ДХМ и перемешивали при к.т. в течение 1,5 ч (прозрачный раствор).
Смесь упаривали до сухого состояния и очищали с помощью препаративной ТСХ (1 мм, ДХМ/МеОН 10:1, Rf возможного продукта: 0,46).
Выход: 20 мг (35%) твердого вещества белого цвета.
Соединение 23 (соед. 23)
N-метил-N'-[4-(2-{3-[2-(морфолин-4-ил)-2-оксоэтокси]-пропил}-фенил)-бутил]-этандиамид
50 мг (0,14 ммоль) 2-[3-(2-{4-[(метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-уксусной кислоты (СБ-6, IK-0358/6), 32,8 мг (0,17 ммоль) EDCI и 24,9 мг (0,29 ммоль, 24,9 мкл) морфолина растворяли в 3 мл ДХМ и перемешивали при к.т. в течение выходных (прозрачный раствор).
Смесь упаривали до сухого состояния и очищали с помощью препаративной ТСХ (1 мм, ДХМ/МеОН 10:1, Rf возможного продукта: 0,48).
Выход: 34 мг (58%) твердого вещества белого цвета.
Соединение 24 (соед. 24)
4-{2-[3-(2-{4-[(Метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-ацетамидо}-бензойная кислота
СТАДИЯ 1:
50 мг (0,14 ммоль) 2-[3-(2-{4-[(метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-уксусной кислоты (СБ-6, IK-0358/6), 32,8 мг (0,17 ммоль) EDCI и 43,1 мг (0,29 ммоль) метил-4-аминобензоата растворяли в 3 мл ДХМ и перемешивали при к.т. в течение 1,5 ч (прозрачный раствор).
Смесь упаривали до сухого состояния и очищали с помощью препаративной ТСХ (1 мм, ЭА/ПЭ 4:1, Rf возможного продукта: 0,31).
СТАДИЯ 2:
К раствору 48 мг (0,10 ммоль) N-метил-N'-{4-[2-(3-{[(пиридин-2-ил)-карбамоил]-метокси}-пропил)-фенил]-бутил}-этандиамида (GH-0498/1) в 2 мл ТГФ добавляли 16,7 мг (0,40 ммоль) моногидрата LiOH, растворенного в 0,5 мл воды, и полученную смесь перемешивали при к.т. в течение ночи (до получения двухфазной смеси).
Смесь вливали в 1 н. раствор HCl (10 мл) и подвергали экстракции с помощью ДХМ (3×20 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали до сухого состояния. Неочищенный продукт очищали с помощью препаративной ТСХ (ДХМ/MeOH/FA 100:10:1, Rf возможного продукта: 0,43).
Выход: 10 мг (21%) твердого вещества белого цвета.
Соединение 25 (соед. 25)
N'-[(5Z)-13-[2-(4-гидрокси-2-оксо-2,5-дигидрофуран-3-ил)-2-оксоэтокси]-тридец-5-ен-1-ил]-N-метилэтандиамид
2-{[(8Z)-13-[(метилкарбамоил)-формамидо]-тридец-8-ен-1-ил]-окси}-уксусную кислоту (СБ-4, 50 мг), DCC (34,7 мг), DМАР (22,3 мг) и 2,4-(3Н,5Н)-фурандион (15,4 мг) помещали во флакон G16. Добавляли CH2Cl2 (3,00 мл) и полученную смесь перемешивали при к.т. в течение 18 ч. Образование продукта регистрировали поданным ЖХ/МС.
Реакционную смесь разбавляли водой (30 мл) и подвергали экстракции с помощью CH2Cl2 (3×20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (CH2Cl2/MeOH=9:1).
Выход: 20 мг (33%) твердого вещества бежевого цвета.
Соединение 26 (соед. 26)
N'-[4-(2-(3-[2-(гидроксиметил)-феноксил]-пропил)-фенил)-бутил]-N-метилэтандиамид
Стадия 1:
Салицилальдегид (2,00 г) и имидазол (2,79 г) растворяли в ДМФА (20,0 мл). Добавляли TIPSCI (5,96 мл) и полученную смесь перемешивали при 60°С в течение 2 дней. Согласно данным ТСХ (ПЭ/EtOAc=95:5) и ЖХ/МС превращение было неполным. Добавляли дополнительную порцию TIPSCI (2,00 мл) и продолжали перемешивание при 60°С в течение 3 дней. Согласно данным ТСХ (ПЭ/EtOAc=95:5) и ЖХ/МС превращение было почти полным. Реакционную смесь разбавляли водой (100 мл) и подвергали экстракции с помощью МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. NaOH (30 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=95:5).
Выход: 3,54 г (78%) жидкости бледно-желтого цвета.
Стадия 2:
2-{[Трис-(пропан-2-ил)-силил]-окси}-бензальдегид (3,54 г) растворяли в EtOH (30,0 мл) и охлаждали до 0°С. Добавляли NaBH4 (481 мг) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 18 ч. Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=8:2) и ЖХ/МС. Реакционную смесь разбавляли водой (100 мл) и подвергали экстракции с помощью EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=8:2).
Выход: 2,73 г (77%) масла желтого цвета
Стадия 3:
(2-{[Трис-(пропан-2-ил)-силил]-окси}-фенил)-метанол (400 мг) растворяли в сухом ТГФ (15 мл). Добавляли NaH (60% в минеральном масле, 85,6 мг) и полученную смесь перемешивали при к.т. в течение 15 мин. Затем добавляли аллилбромид (309 мкл) и полученную смесь перемешивали при к.т. в течение ночи. Образование продукта регистрировали по данным ЖХ/МС. Реакционную смесь разбавляли водой (50 мл) и подвергали экстракции с помощью CH2Cl2 (3×30 мл). Объединенные органические слои промывали солевым раствором (20 мл), сушили над Na2SO4 и концентрировали под вакуумом. Полученный таким образом неочищенный продукт использовали для дальнейших превращений без дополнительной обработки.
Выход: 480 мг (неочищенного) масла желтого цвета.
Стадия 4:
{2-[(Проп-2-ен-1-илокси)-метил]-фенокси}-трис-(пропан-2-ил)-силан (178 мг) помещали в колбу объемом 10 мл и охлаждали до 0°С в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 1,38 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч. Затем добавляли раствор Na2CO3 (147 мг) в воде (1,50 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[4-(2-иодфенил)-бутил]-N-метилэтандиамид (СБ-2, 100 мг) и PdCl2(PPh3)2 (9,7 мг) и полученную смесь нагревали до 50°С в течение 3 ч. Образование продукта регистрировали по данным ЖХ/МС. Реакционную смесь охлаждали до к.т., разбавляли водой (20 мл) и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (15 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток пропускали через короткую колонку с SiO2 (ПЭ/EtOAc 1:1). Оставшийся неочищенным продукт затем использовали для дальнейших превращений без дополнительной обработки.
Выход: 235 мг (неочищенного) масла желтого цвета.
Стадия 5:
N-метил-N'-[4-(2-{3-[(2-{[трис-(пропан-2-ил)-силил]-окси}-фенил)-метокси]-пропил}-фенил)-бутил]-этандиамид (154 мг, неочищенный IK-0357/19) растворяли в ТГФ (5,00 мл). Добавляли TBAF*3H2O (87,0 мг) и полученную смесь перемешивали при к.т. в течение 30 мин. Согласно данным ЖХ/МС превращение было полным. Реакционную смесь вливали в воду (20 мл) и подвергали экстракции с помощью EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (CH2Cl2/МеОН=95:5).
Выход: 75 мг (66%) твердого вещества белого цвета.
Соединение 27 (сред. 27)
N'-[(5Z)-13-(2-гидроксифенокси)-тридец-5-ен-1-ил]-N-метилэтандиамид
Стадия 1:
N'-[(5Z)-13-гидрокситридец-5-ен-1-ил]-N-метилэтандиамид (СБ-8, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (598 мг) и CBr4 (756 мг) и полученную смесь перемешивали при к.т. в течение 1,5 ч. Согласно данным ТСХ (ПЭ/EtOAc=1:1) и ЖХ/МС превращение было полным.
Реакционную смесь концентрировали под вакуумом и остаток очищали с помощью препаративной ТСХ (ПЭ/EtOAc=1:1).
Стадия 2:
N'-[(5Z)-13-бромтридец-5-ен-1-ил]-N-метилэтандиамид (50 мг), пирокатехин (76,2 мг) и K2CO3 (57,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при 60°С в течение 2,5 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь разбавляли водой (40 мл) и подвергали экстракции с помощью МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (CH2Cl2/МеОН=95:5).
Выход: 43 мг (80%) твердого вещества белого цвета.
Соединение 28 (сред. 28)
N-метил-N'-[4-(2-{3-[2-(морфолин-4-ил)-2-оксоэтокси]-пропил}-фенил)-бутил]-этандиамид
50 мг (0,14 ммоль) 2-[3-(2-{4-[(метилкарбамоил)-формамидо]-бутил}-фенил)-пропокси]-уксусной кислоты (СБ-8, IK-0358/6), 32,8 мг (0,17 ммоль) EDCI и 24,9 мг (0,29 ммоль, 24,9 мкл) морфолина растворяли в 3 мл ДХМ и перемешивали при к.т. в течение выходных (прозрачный раствор).
Смесь упаривали до сухого состояния и очищали с помощью препаративной ТСХ (1 мм, ДХМ/МеОН 10:1, Rf возможного продукта: 0,48).
Выход: 34 мг (58%) твердого вещества белого цвета.
Соединение 29 (соед. 29)
N'-(4-{2-[3-(3-гидроксифенокси)-пропил]-фенил}-бутил)-N-метилэтандиамид
Стадия 1:
N'-{4-[2-(3-гидроксипропил)-фенил]-бутил}-N-метилэтандиамид (СБ-9, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (610 мг) и CBr4 (771 мг) и полученную смесь перемешивали при к.т. в течение 30 мин. Согласно данным ТСХ (ПЭ/EtOAc=1:1) и ЖХ/МС превращение было полным. Реакционную смесь концентрировали под вакуумом и остаток очищали с помощью препаративной ТСХ (ПЭ/EtOAc=1:1).
Стадия 2:
N'-{4-[2-(3-бромпропил)-фенил]-бутил}-N-метилэтандиамид (50 мг), 1,3-бензолдиол (77,5 мг) и K2CO3 (58,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при 60°С в течение 1 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь разбавляли водой (40 мл) и подвергали экстракции с помощью МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (CH2Cl2/MeOH=95:5).
Выход: 40 мг (74%) твердого вещества белого цвета.
Соединение 30 (соед. 30)
N'-[(5Z)-13-(4-гидроксифенокси)-тридец-5-ен-1-ил]-N-метилэтандиамид
Стадия 1:
N'-[(5Z)-13-гидрокситридец-5-ен-1-ил]-N-метилэтандиамид (СБ-8, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (598 мг) и CBr4 (756 мг) и полученную смесь перемешивали при к.т. в течение 1,5 ч. Согласно данным ТСХ (ПЭ/EtOAc=1:1) и ЖХ/МС превращение было полным.
Реакционную смесь концентрировали под вакуумом и остаток очищали с помощью препаративной ТСХ (ПЭ/EtOAc=1:1).
Стадия 2:
N'-[(5Z)-13-бромтридец-5-ен-1-ил]-N-метилэтандиамид (50 мг), гидрохинон (76,2 мг) и K2CO3 (57,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при 60°С в течение 2,5 ч. Образование продукта регистрировали по данным ЖХ/МС.
Реакционную смесь разбавляли водой (40 мл) и подвергали экстракции с помощью МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (CH2Cl2/MeOH=95:5).
Выход: 43 мг (80%) твердого вещества белого цвета.
Соединение 31 (соед. 31)
N'-(4-{2-[3-(4-гидроксифенокси)-пропил]-фенил}-бутил)-N-метилэтандиамид
Стадия 1:
N'-{4-[2-(3-гидроксипропил)-фенил]-бутил}-N-метилэтандиамид (СБ-9, 400 мг) суспендировали в CH2Cl2 (20 мл). Добавляли PPh3 (610 мг) и CBr4 (771 мг) и полученную смесь перемешивали при к.т. в течение 30 мин. Согласно данным ТСХ (ПЭ/EtOAc=1:1) и ЖХ/МС превращение было полным.
Реакционную смесь концентрировали под вакуумом и остаток очищали с помощью препаративной ТСХ (ПЭ/EtOAc=1:1).
Стадия 2:
N'-{4-[2-(3-бромпропил)-фенил]-бутил}-N-метилэтандиамид (50 мг), гидрохинон (77,5 мг) и K2CO3 (58,4 мг) помещали во флакон G16. Добавляли ДМФА (3,00 мл) и полученную смесь перемешивали при 60°С в течение 1 ч. Образование продукта регистрировали поданным ЖХ/МС.
Реакционную смесь разбавляли водой (40 мл) и подвергали экстракции с помощью МТБЭ (3×20 мл). Объединенные органические слои промывали водой (20 мл) и солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (CH2Cl2/МеОН=95:5).
Выход: 39 мг (72%) твердого вещества белого цвета.
Соединение 3'' (соед. 3'')
N'-[4-(2-{3-[(4-гидроксифенил)-метокси]-пропил}-фенил)-бутил]-N-метилэтандиамид
Стадия 1
4-Гидроксибензальдегид (2,00 г) и имидазол (2,79 г) растворяли в ДМФА (20,0 мл). Добавляли TIPSCl (5,96 мл) и полученную смесь перемешивали при 60°С в течение 2 дней.
Согласно данным ТСХ (ПЭ/EtOAc=95:5) и ЖХ/МС превращение было полным.
Реакционную смесь разбавляли водой (100 мл) и подвергали экстракции с помощью МТБЭ (3×40 мл). Объединенные органические слои промывали 1 н. NaOH (30 мл) и солевым раствором (20 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=95:5).
Выход: 3,94 г (86%) масла бледно-желтого цвета.
Стадия 2
4-{[Трис-(пропан-2-ил)-силил]-окси}-бензальдегид (3,94 г) растворяли в EtOH (30,0 мл) и охлаждали до 0°С. Добавляли NaBH4 (535 мг) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 18 ч.
Образование продукта регистрировали по данным ТСХ (ПЭ/EtOAc=8:2) и ЖХ/МС.
Реакционную смесь разбавляли водой (100 мл) и подвергали экстракции с помощью EtOAc (3×40 мл). Объединенные органические слои промывали солевым раствором (30 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью колоночной хроматографии на SiO2 (ПЭ/EtOAc=8:2).
Стадия 3
(4-{[Трис-(пропан-2-ил)-силил]-окси}-фенил)-метанол (300 мг) растворяли в сухом ТГФ (5,00 мл). Добавляли NaH (60% в минеральном масле, 64,2 мг) и полученную смесь перемешивали при к.т. в течение 15 мин. Затем добавляли аллилбромид (231 мкл) и полученную смесь перемешивали при к.т. в течение 2,5 ч. Согласно данным ЖХ/МС превращение было полным.
Реакционную смесь вливали в воду (30 мл) и подвергали экстракции с помощью EtOAc (3×10 мл). Объединенные органические слои промывали солевым раствором, сушили над Na2SO4 и концентрировали под вакуумом. Остаток использовали для дальнейших превращений без дополнительной обработки.
Выход: 372 мг (неочищенного) масла желтого цвета.
Стадия 4
{4-[(Проп-2-ен-1-илокси)-метил]-фенокси}-трис-(пропан-2-ил)-силан (356 мг) помещали в колбу объемом 10 мл и охлаждали до 0°С в атмосфере Ar. Затем по каплям добавляли 9-BBN (0,5 М в ТГФ, 3,33 мл) и полученную смесь перемешивали при 0°С в течение 30 мин и затем при к.т. в течение 2 ч.
Затем добавляли раствор Na2CO3 (147 мг) в воде (3,00 мл) и продолжали перемешивание при к.т. в течение 30 мин. Затем добавляли N'-[4-(2-иодфенил)-бутил]-N-метилэтандиамид (СБ-2, 100 мг) и PdCl2(PPh3)2 (9,7 мг) и полученную смесь нагревали до 50°С в течение 2 ч. Образование продукта регистрировали по данным ЖХ/МС. Реакционную смесь охлаждали до к.т., разбавляли водой (20 мл) и разделяли слои. Водный слой подвергали экстракции с помощью EtOAc (15 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток пропускали через короткую колонку с SiO2 (ПЭ/EtOAc 1:1). Оставшийся неочищенным продукт затем использовали для дальнейших превращений без дополнительной обработки.
Выход: 248 мг (неочищенного) масла желтого цвета.
Стадия 5
N-метил-N'-[4-(2-{3-[(4-{[трис-(пропан-2-ил)-силил]-окси}-фенил)-метокси]-пропил}-фенил)-бутил]-этандиамид (154 мг, неочищенный IK-0357/20) растворяли в ТГФ (5,00 мл), Добавляли TBAF*3H2O (131 мг) и полученную смесь перемешивали при к.т. в течение 30 мин. Согласно данным ЖХ/МС превращение было полным.
Реакционную смесь вливали в воду (40 мл) и подвергали экстракции с помощью EtOAc (3×20 мл). Объединенные органические слои промывали солевым раствором (10 мл), сушили над Na2SO4 и концентрировали под вакуумом. Остаток очищали с помощью препаративной ТСХ (CH2Cl2/MeOH=95:5).
Выход: 76 мг (68%) твердого вещества белого цвета.
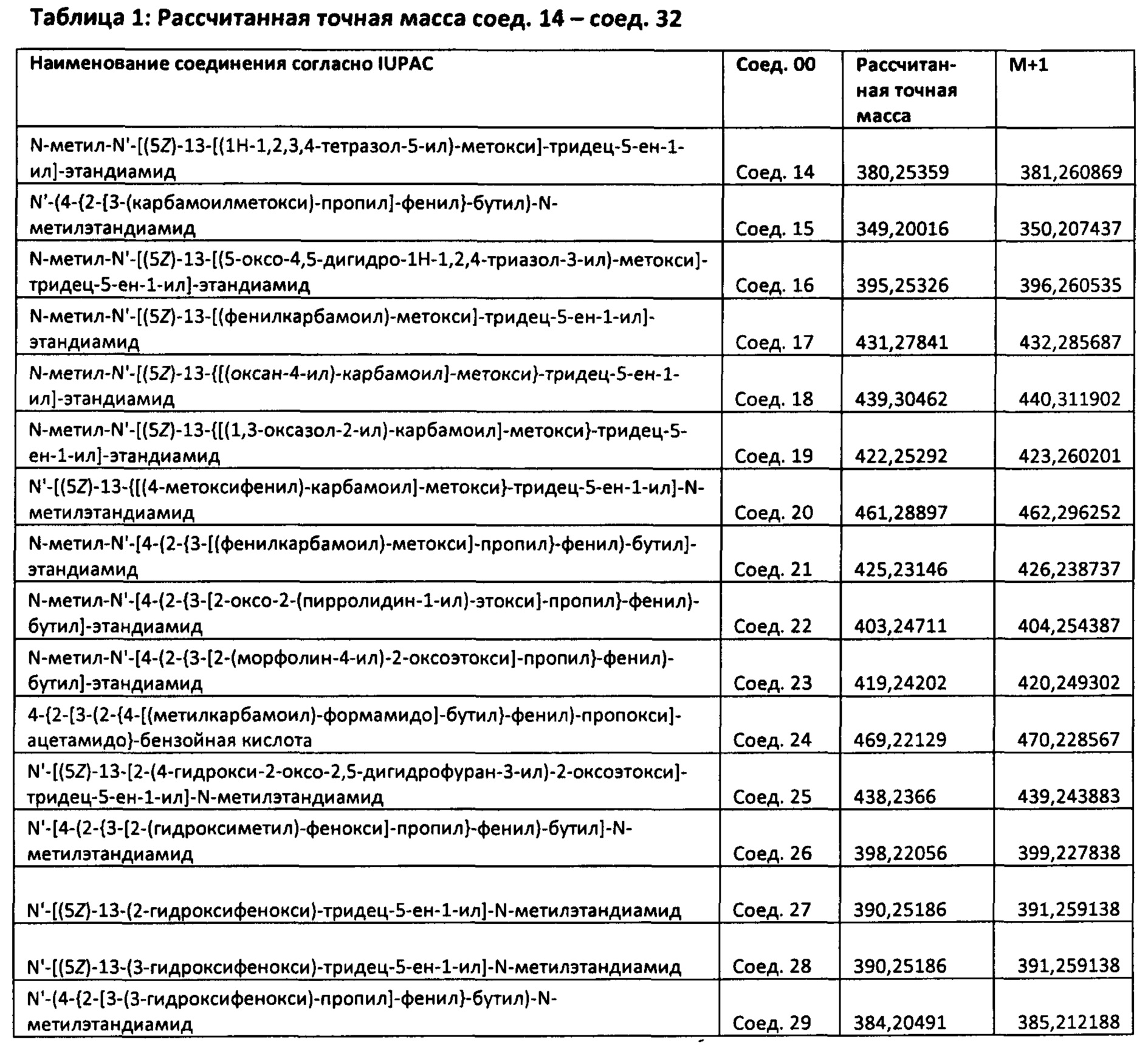

Пример 2: Эффективность соединения 1 в модели индуцированной лазером хориоидальной неоваскуляризации у крысы при оценке путем определения повышенной проницаемости сосудов с помощью флюоресцентной ангиографии
Модель индуцированной лазером ХНВ у крысы является широко используемой моделью для доказательства терапевтической эффективности лекарственных средств для лечения отдельных заболеваний глаза, характеризующихся ангиогенезом и воспалением в глазу, таких как влажная форма возрастной макулярной дегенерации (ВМД). В этой модели ожог сфокусированным лазерным лучом в слое мембраны Бруха вызывает нарушение этой мембраны, локальное повреждение с последующим воспалением и ростом новых кровеносных сосудов. Эти вновь образованные кровеносные сосуды как правило имеют повышенную проницаемость. Эту повышенную проницаемость определяют по экстравазации флюоресцентного красителя (флюоресцеина) в задней части глаза, и она является общепринятым маркером повышенной проницаемости сосудов, пропорциональным количеству вновь образованных кровеносных сосудов. Этот способ также является современной методикой в клинической практике у человека.
Все стандартные рабочие методики и протоколы, описанные в плане этого исследования, были рассмотрены Этическим комитетом. Все эксперименты с животными проводили в соответствии с Директивой 2010/63/UE Европейской конвенции о защите позвоночных животных, используемых в экспериментальных и иных научных целях, и положениями Ассоциации исследований в области зрения и офтальмологии (ARVO) по использованию животных в исследованиях в области зрения и офтальмологии.
Животные: серые крысы, достигшие возраста 8-10 недель на день индукции.
Индукция неоваскуляризации: Животных подвергали анестезии путем внутримышечной инъекции смеси ксилазина и кетамина. Вызывали расширение зрачка правого глаза путем инстилляции одной капли 0,5% тропикамида перед воздействием. У животных под анестезией вызывали шесть ожогов в правом глазу путем воздействия лазерного излучения с длиной волны 532 нм мощностью 170-180 мВт (лазер Viridis, Quantel, Франция) на область площадью 75 мкм вокруг зрительного нерва, между главными ветвями сосудов сетчатки, в течение 0,1 с через щелевую лампу и контактную линзу. Появление пузырька в момент применения лазера подтверждало разрыв мембраны Бруха.
Лечение: Контрольной группе, получающей носитель, в 0 день сразу после индукции неоваскуляризации вводили носитель путем внутривенной инъекции (5 мкл) в правый глаз под операционным микроскопом с использованием иглы размера 30-G, установленной на шприц Гамильтона объемом 100 мкл. Введение животным проводили под анестезией (такая же анестезия, как для индукции неоваскуляризации). В первой группе лечения соединение 1, которое имеет структуру, указанную в формуле (VI), вводили внутрибрюшинно (100 мкл/100 г) один раз в день с 0 дня (за 3 часа ± 15 мин до индукции) до 23 дня. В другой группе лечения соединение 1 вводили перорально через зонд (1 мл/100 г) два раза в день с 0 дня (за 3 часа ± 15 мин до индукции) до 22 дня (каждые 7 ч ± 30 мин).
Ангиография с флюоресцеином: Ангиографию с флюоресцеином проводили для обработанного лазером правого глаза на 14 и 21 дни с использованием гейдельбергского ретинального ангиографа (HRA). После анестезии (см. выше) будет введено 250 мкл/100 г массы тела 10% флюоресцеина натрия путем подкожной инъекции, и флюоресцентные изображения будут получены через 10 минут после инъекции красителя. Для оценки с помощью ангиографии с флюоресцеином утечка флюоресцеина на ангиограммах будет оценена двумя независимыми экспертами с ослеплением в отношении исследуемых групп и следующей шкалой оценки: значение 0: утечка отсутствует; значение 1: слабое окрашивание; значение 2: умеренное окрашивание; значение 3: сильное окрашивание.
Окончание прижизненной фазы: В конце исследования на 23 день все животные будут подвергнуты анестезии (см. выше) с последующей эвтаназией путем внутрисердечной инъекции высокой дозы пентобарбитала. Этот способ является одним из рекомендованных европейскими уполномоченными органами способов.
Статистический анализ: статистический анализ проводили с использованием программного обеспечения Prism. Проводили анализ Краскела-Уоллиса (Kruskal-Wallis) и оценивали действие лекарственного средства с использованием критерия Даннета (Dunnett) для множественных сравнений; каждую получавшую лечение группу сравнивали с группой, получавшей носитель, в каждый момент времени. Значимыми будут являться р-значения менее 0,05.
Результаты представлены на фиг. 1.
Пример 3: Распределение соед. 02 после перорального, местного или интравитреального введения в различных отделах глаза кролика
Этот пример показывает, что наибольшая величина воздействия соед. 02 на глаз в задней части глаза достигается после интравитреальной инъекции.
Материалы и методы
План исследования: Для получения сведений о характеристиках распределения синтетических агонистов 17,18-EEQ проводили офтальмологические фармакокинетические исследования у пигментированных кроликов HY79b. Кратко, 15 самцов кроликов для каждого пути введения подвергали анестезии в моменты времени для получения образцов, обозначенные в таблицах 2-4, путем внутримышечной инъекции смешанного раствора ксилазина и кетамина. Взятие крови производили в пробирки с K3EDTA путем пункции сердца и центрифугировали при 2000 g 10 мин при 4°С. Отбирали примерно 3 мл плазмы, помещали в пластиковые пробирки, быстро замораживали в жидком азоте и хранили при -80°С до анализа. Затем животных подвергали эвтаназии путем внутрисердечной инъекции высокой дозы пентобарбитала. Этот способ является одним из рекомендованных европейскими уполномоченными органами способов эвтаназии [French decree No. 2013-118, dated February 01, 2013 publishing the European directive 2010/63/UE. J. Offic. Rp. Fr. 2013; Text 24 out of 130). Сразу после эвтаназии быстро и осторожно отбирали образцы из обоих глаз, помещали их в пластиковые пробирки, взвешивали, быстро замораживали в жидком азоте и хранили при -80°С до анализа. Содержание соед. 02 в образцах из глаз и плазме определяли в соответствии с известным способом ЖХМС/МС быстрого разрешения (RRLCMS/MS). Результаты представлены в таблицах 5-7 и выражены в виде средних значений для различных фармакокинетических параметров. Интегрирование хроматограмм выполняли с помощью программного обеспечения MassHunter. Кажущиеся Cmax и Tmax, T1/2, AUC1-48 ч рассчитывали с помощью программного обеспечения Excel®. Концентрации выражены в нг/г или нг/мл ткани/отдела глаза.
Результаты
После перорального введения концентрация соед. 02 была меньше нижнего предела обнаружения (при инъекционном введении 0,1 нг/10 мкл) как в образцах водянистой влаги (за исключением одного из 15 животных), так и в образцах сетчатки (за исключением двух из 15 животных). Однако пероральное введение соед. 02 приводило к достижению концентрации, соответствующей эффективной дозе (246 нг/г), через 1 ч после введения 2 мг/кг (0,2 мг/мл) в ткани сосудистой оболочки, а также значимой концентрации в плазме (1213 нг/мл через 1 ч после введения). В соответствии с максимальной концентрацией значимую величину воздействия наблюдали только в сосудистой оболочке и плазме (AUC1-48 часов (нг*ч/мл): 1387 и 6331).
Для местного введения было обнаружено, что состав, содержащий соед. 02, характеризуется очень хорошей переносимостью на макроскопическом уровне при оценке с помощью офтальмоскопии. В таблице 6 показано, что, при очень низкой концентрации в целом, концентрацию, соответствующую максимальной дозе, наблюдали в водянистой влаге (88 нг/мл через 1 ч после введения), затем в сосудистой оболочке (79 нг/г через 1 ч после введения). Как в сетчатке, так и в плазме была достигнута только очень низкая концентрация (24 нг/г через 1 ч после введения и 31 нг/г через 0,5 ч после введения, соответственно). После местного введения значения AUC1-48 часов были низкими во всех исследованных тканях глаза (AUC1-48 часов (нг*ч/мл): 324 для водянистой влаги, 117 для сосудистой оболочки и 21 для сетчатки), а также в плазме (262 нг*ч/мл).
Для интравитреальной инъекции также было обнаружено, что состав, содержащий соед. 02, характеризуется очень хорошей переносимостью на макроскопическом уровне при оценке путем осмотра глаза с помощью щелевой лампы. В таблице 6 показано, что значимая концентрация соед. 02 была обнаружена во всех тканях глаза и в плазме. Наивысшая концентрация была обнаружена в сосудистой оболочке (14957 нг/г через 1 ч после инъекции), затем в сетчатке и стекловидном теле (10052 нг/г и 6647 нг/мл через 1 ч после инъекции). Величина воздействия составляла >55000 (AUC1-48 часов (нг*ч/мл): 55401 для стекловидного тела, 63283 для сосудистой оболочки и 79866 для сетчатки), за исключением плазмы (287 нг*ч/мл)).
В итоге, наибольшая величина воздействия соед. 02 на глаз в задней части глаза достигалась после интравитреальной инъекции по сравнению с пероральным и местным введением.




Cmax = наибольшее определенное среднее значение (нг/г или мл ткани)
Tmax = момент времени определения наибольшего среднего значения (часы)
AUC от момента времени 1 до момента времени 2= площадь под кривой в нг/г или мл ткани
t1/2 = ln(2)/(-a), где а = угол наклона ln (концентрации) = f(t)
t1/2 = рассчитан на основании значения Tmax
н/п = не применимо, недостаточно данных

Cmax = наибольшее определенное среднее значение (нг/г или мл ткани)
Tmax = момент времени определения наибольшего среднего значения (часы)
AUC от момента времени 1 до момента времени 2= площадь под кривой в нг/г или мл ткани
t1/2 = ln(2)/(-a), где а = угол наклона ln (концентрации) = f(t)
t1/2 = рассчитан на основании значения Tmax
н/п = не применимо, недостаточно данных
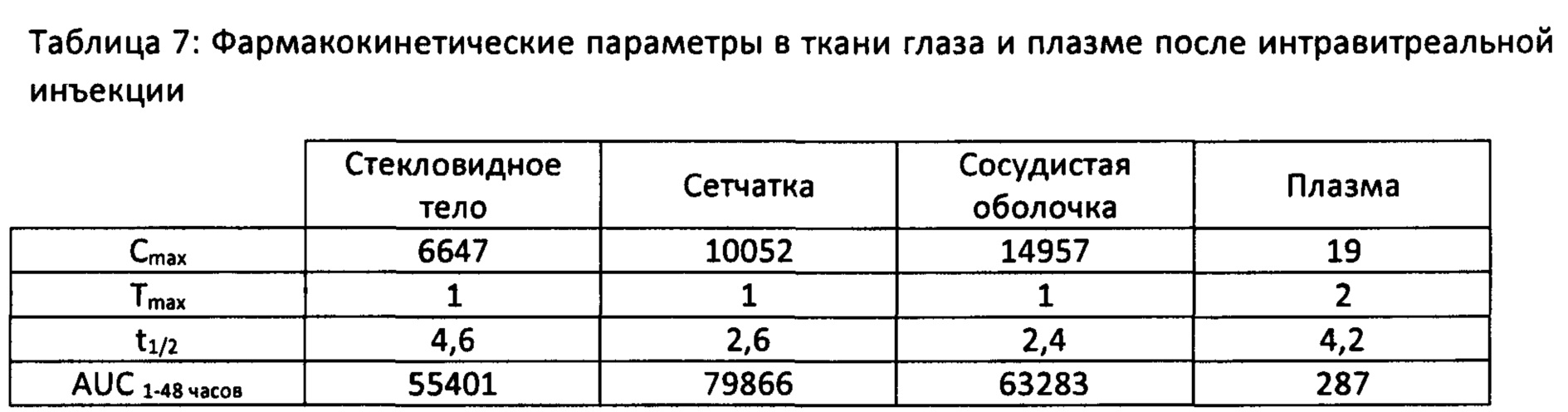
Cmax = наибольшее определенное среднее значение (нг/г или мл ткани)
Tmax = момент времени определения наибольшего среднего значения (часы)
AUC от момента времени 1 до момента времени 2= площадь под кривой в нг/г или мл ткани
t1/2 = ln(2)/(-a), где а = угол наклона ln (концентрации) = f(t)
t1/2 = рассчитан на основании значения Tmax
н/п = не применимо, недостаточно данных
Пример 4: Противовоспалительное действие метаболически устойчивого аналога 17,18-EEQ (соед. 02) в отношении кардиомиоцитов HL-1
Материалы и способы
Для того, чтобы исследовать возможное противовоспалительное действие соединений, являющихся частью настоящего изобретения, in vitro, использовали клеточную линию кардиомиоцитов (иммортализованные кардиомиоциты мыши, клетки HL-1). Клетки обрабатывали либо носителем (0,01% этанолом), либо различными концентрациями исследуемого соединения (соед. 02: cE=10 нМ, 100 нМ или 1 мкМ). Одновременно указанные клетки подвергали воздействию 1 мкг/мл липополисахарида (ЛПС). После 24 ч инкубирования клетки подвергали обработке для определения жизнеспособности (фиг. 2) и высвобождения провоспалительного цитокина ФНО-альфа (фиг. 3).
Результаты
Результаты представлены на фиг. 2 и фиг. 3. Воспалительный стимул - ЛПС - приводит к значительному снижению жизнеспособности клеток. Этот цитотоксический эффект был дозозависимым образом обращен под действием соед. 02, фиг. 2. Кроме того, инкубирование с ЛПС в значительной степени индуцирует выработку провоспалительного цитокина ФНО-альфа (фиг. 3). Соед. 02 отдельно не оказывает влияния на выработку ФНО-альфа. Индуцированное ЛПС высвобождение ФНО-альфа из клеток HL-1 в значительной степени и дозозависимым образом снижалось под действием соед. 02 (фиг. 3).
Пример 5: Подавляющее действие метаболически устойчивого аналога 17,18-EEQ (соед. 02) в отношении воспаления тканей почки и сердца в модели тяжелой гипертензии и поражения органов-мишеней у крысы
Материалы и способы
Задача данного исследования заключалась в том, чтобы оценить и сравнить влияние непрерывного лечения с пероральным введением метаболически устойчивого аналога 17,18-EEQ (соед. 02) или носителя (изотонического солевого раствора) на физиологические параметры, клинические лабораторные показатели и поражение органов-мишеней у самцов двойных трансгенных (dTGR) крыс. Не получавших лечения крыс Спрег-Доули (СД) с таким же генетическим окружением использовали в качестве контроля. У двойных трансгенных крыс, характеризующихся сверхэкспрессией генов ренина и ангиотензиногена человека, развивается тяжелая гипертензия, и они являются моделью для индуцированного ангиотензином-II (Ang-II) поражения органов-мишеней с гипертрофией сердца и почечной недостаточностью. Более того, модель dTGR характеризуется тяжелой артериальной гипертензией, гипертрофией сердца и нефросклерозом с выраженным фиброзом и воспалением.
Перед началом эксперимента животных случайным образом распределяли в группы лечения с применением соединения либо носителя. Корм для всех групп лечения, а также для контрольных животных СД, заменяли на корм, богатый омега-6, затем проводили эксперимент. Получавшим соединение dTGR животным вводили через зонд 1,33 мг/кг два раза в день в течение 21 дня. Для введения соед. 02 готовили в 61,6 ppm Na2CO3 и готовом к применению растворе 0,9% NaCl, в то время как dTGR животным два раза в день вводили готовый к применению раствор 0,9% NaCl в качестве контроля с применением носителя. Применяемый объем составлял 5,0 мл/кг м.т. После 21 дня лечения, в возрасте семи недель, животных умерщвляли путем удаления сердца под пентобарбиталовой анестезией (20 мг/кг м.т., гепарин 500 МЕ/мл) и забирали кровь и органы. Сердце и почку удаляли и взвешивали. Сердце и почку разделяли и быстро замораживали в жидком азоте для молекулярно-биологических исследований, замораживали в изопентане при -40°С и хранили при -80°С для получения замороженных срезов. Из замороженных почек и сердец, заключенных под пленку TissueTek, изготавливали замороженные срезы толщиной 5 мкм. Замороженные срезы фиксировали охлажденным на льду ацетоном, промывали ФСБ и блокировали нормальной сывороткой осла при комнатной температуре. Затем срезы инкубировали в темной влажной камере в течение ночи при 4°C с первичным моноклинальным антителом мыши к ED1 (cD68) (AbD serotec). После промывки ФСБ срезы инкубировали со вторичным антителом осла против иммуноглобулинов мыши (Cy3) в течение 90 минут в темной влажной камере при комнатной температуре с последующим заключением в среду Vectashield. Образцы анализировали с использованием микроскопа Zeiss Axioplan-2 imaging с цифровой программой обработки изображений AxioVision 4.8. Все оценки проводились односторонне-ослепленным исследователем. Анализ инфильтрированных макрофагов проводили путем подсчета ED1-положительных клеток в срезах сердца (желудочка) и ткани почки (внешнего мозгового слоя и коры). Данные выражали в виде суммы 20 случайным образом выбранных неперекрывающихся полей зрения на каждый срез.
Статистический анализ проводили с использованием программного обеспечения R для иммуногистохимии. Поскольку данные являются непараметрическими (встречаемость), применяли критерии Краскела-Уоллиса (Kruskall-Wallis) для различий между группами в целом, тогда как для анализа различий между отдельными группами использовали парные U-критерии с коррекцией FDR. Значимыми являлись р-значения менее 0,05.
Результаты
На фиг. 4 и фиг. 5 продемонстрирована способность устойчивого аналога 17,18-ЕЕQ, (соед. 02) модулировать воспаление у dTGR животных. Указанная способность была определена для тканей сердца (фиг. 4) и почки (фиг. 5) по инфильтрации макрофагов путем окрашивания ED1, используемого в качестве маркера воспаления. Приведенные значения представлены в виде среднего и межквартильного размаха. Значения встречаемости объединены в ячейки по 20 полей зрения. Полученные результаты показывают, что у dTGR животных имеется значительно большее количество инфильтрированных макрофагов в ткани сердца или почки сравнению с не получавшими лечения животными СД (фиг. 4 и фиг. 5). Однако лечение с применением соед. 02 приводило к значительному уменьшению инфильтрации макрофагов (ED1-положительных клеток) у dTGR животных по сравнению с dTGR животными, получавшими носитель.
Реферат
Изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли, которые являются метаболически устойчивыми аналогами биологически активных липидных медиаторов, образующихся из полиненасыщенных жирных кислот омега-3 (ПНЖК n-3), для лечения, снижения риска развития или предотвращения нарушения, связанного с неоваскуляризацией и/или воспалением. Технический результат: лечение, снижение риска развития или предотвращение нарушения, связанного с неоваскуляризацией и/или воспалением. В общей формуле (I) P представляет собой группу, представленную общей формулой (II), E представляет собой группу, представленную общей формулой (III) или (IV), I представляет собой -(CH2)m-Y. Остальные значения переменных указаны в формуле изобретения. 14 з.п. ф-лы, 5 ил., 7 табл., 5 пр.
P-E-I (I)
-(CH2)n-O-(CH2)k-X (II),

Формула
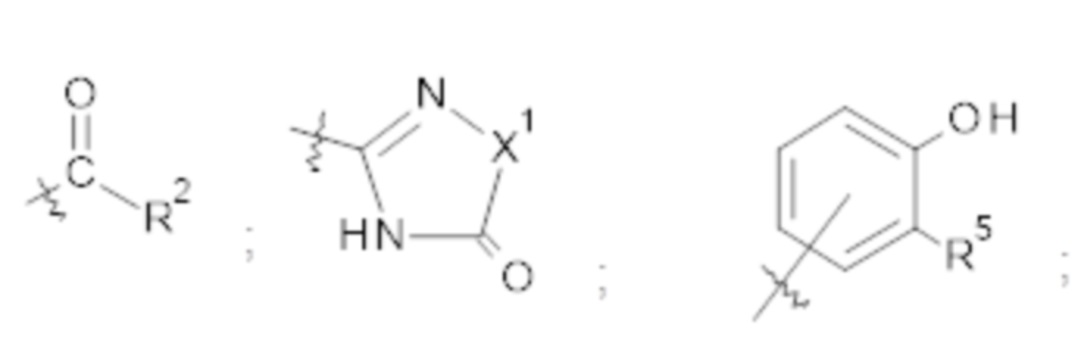




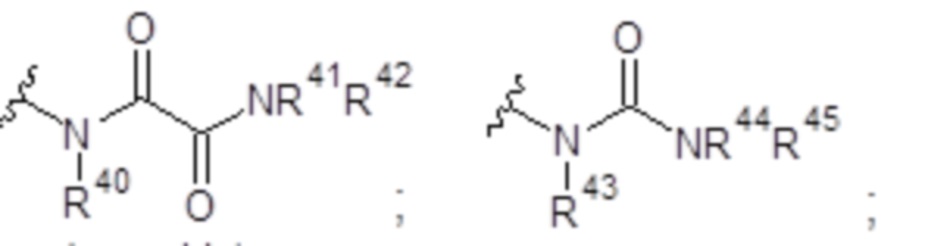

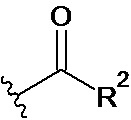

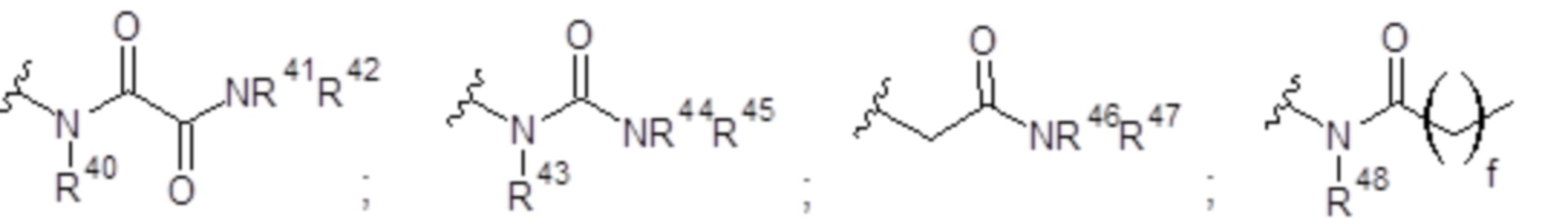
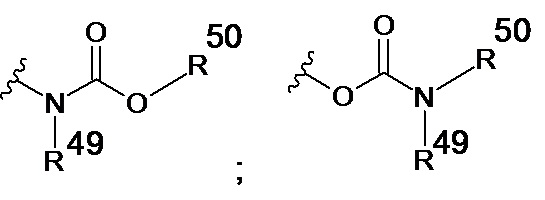


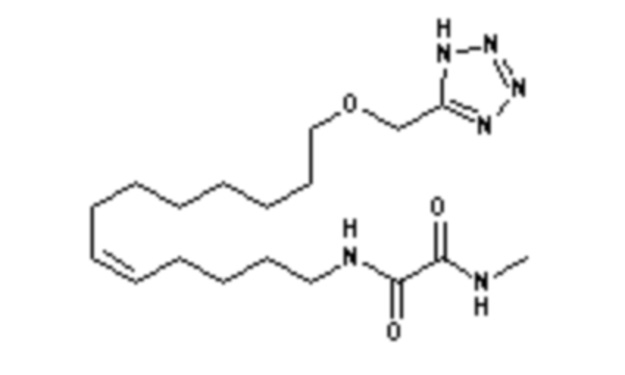

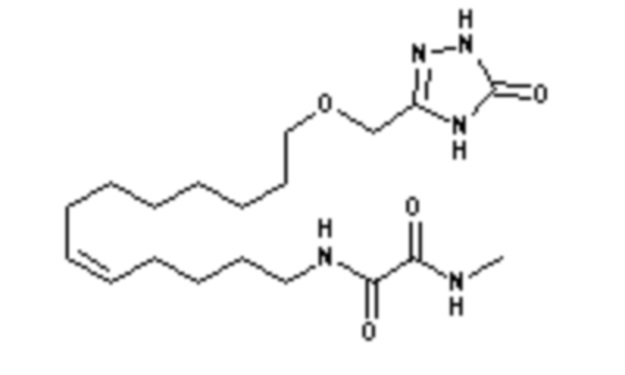
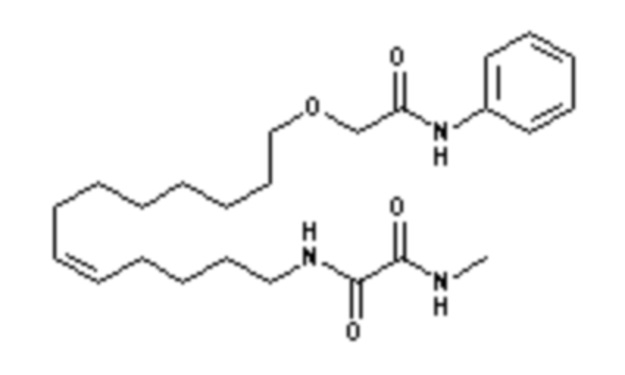
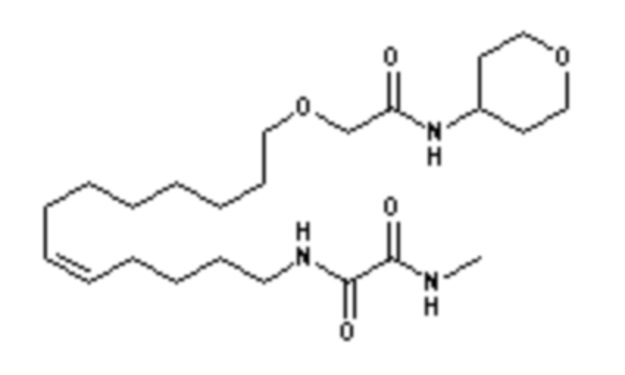

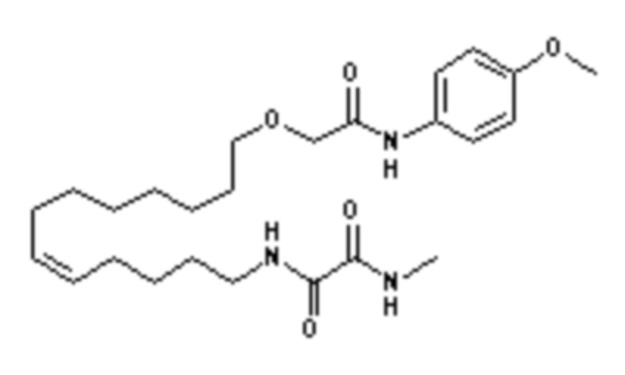







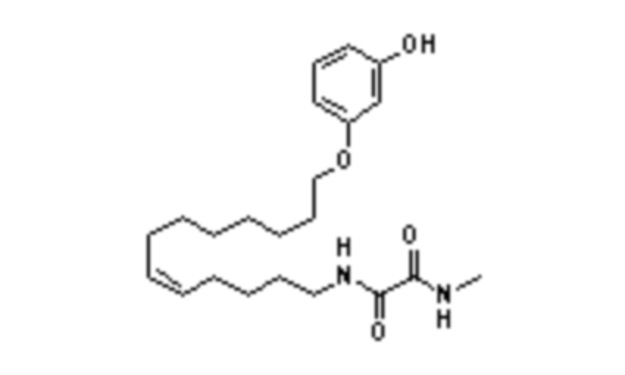



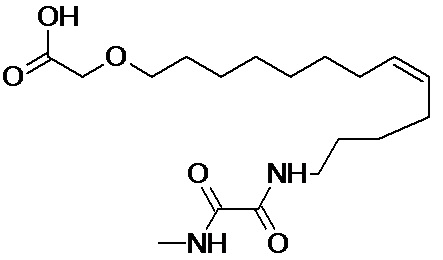
Документы, цитированные в отчёте о поиске
Офтальмическая композиция на основе полиненасыщенных жирных кислот омега-3 и омега-6
Комментарии