Ингибиторы лизинспецифичного гингипаина - RU2728785C2
Код документа: RU2728785C2
Чертежи




Описание
ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] По настоящей заявке испрашивается приоритет временной заявки на патент США № 62/060483, поданной 6 октября 2014 года, которая полностью включена в настоящее описание в качестве ссылки.
УРОВЕНЬ ТЕХНИКИ
[0002] Заражение бактериями Porphyromonas gingivalis было связано с развитием заболевания пародонта, болезни Альцгеймера и нарушений мозговой деятельности, сердечно-сосудистого заболевания, диабета, рака, заболевания печени, заболевания почек, преждевременными родами, развитием артрита, пневмонии и других заболеваний. Р. gingivalis является анаэробной несахаролитической грамотрицательной палочкой, которая, как известен, заражает ротовую полость и транслоцируется системно в коронарные артерии, аорту, плацентарную ткань, мозг, почки и печень. Бактерии также были идентифицированы в раковых тканях, и было сделано предположение о механизме, посредством которого гингипаины могут вызвать иммортализацию и метастазирование. См: Gandhimadhi,et al. Journal of Indian Society of Periodontology. 2010;14(2):114-120; Liao, et al., Med Hypotheses, 2009. 72(6): 732-5; Byrne, et al., ORal Microbiol Immunol, 2009. 24(6): 469-77; Mahendra, et al., J Maxillofac Oral Surg, 2009. 8(2): 108-13; Stelzel, et al., J Periodontol, 2002. 73(8): 868-70; Katz, et al., Journal of Dental Research, 2009. 88(6): 575-578; Poole, et al., J Alzheimers Dis, 2015, 43(1): 67-80; Ishikawa, et al., Biochim Biophys Acta, 2013. 1832(12): 2035-2043; Inaba, et al., Cellular Microbiology, 2014. 16(1): 131-145.
[0003]Р. gingivalis продуцирует внеклеточные протеазы, называемые гингипаинами, в том числе аргинин-специфичный гингипаин А (RgpA), аргинин-специфичный гингипаин B (RgpB) и лизин-специфичныЙ гингипаин (Kgp). Гингипаины участвуют во многих функциях организма, включая его выживание и вирулентность. Гингипаины могут секретироваться, транспортироваться к наружным поверхностям мембран P. gingivalis или высвобождаться в везикулы наружных мембран бактериями. Гингипаины разлагают широкий спектр белков (например, иммуноглобулины, ингибиторы протеиназы, актин и коллаген), что может привести к коллапсу цитоскелета и апоптозу в клетках многих типов. Недавние исследования показали, что небольшие продуцированные пептидами ингибиторы Kgp могут предотвращать гингипаин-индуцированную гибель клеток эпителия. См: Travis, et al., Adv. Exp. Med. Biol., 2000. 477: 455-65; Sheets, et al., Infect. Immun., 2005. 73(3): 1543-52; Sheets, et al., Infect. Immun., 2006. 74(10): 5667-78; Stathopoulou, et al., BMC Microbiol., 2009. 9: 107. Существует потребность в новых соединениях для ингибирования гингипаиновой активности и лечения заболеваний, связанных с активностью гингипаинов и инфекцией P. gingivalis. Настоящее изобретение относится к этой и другим потребностям.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0004] В одном аспекте настоящее изобретение относится к соединению формулы I
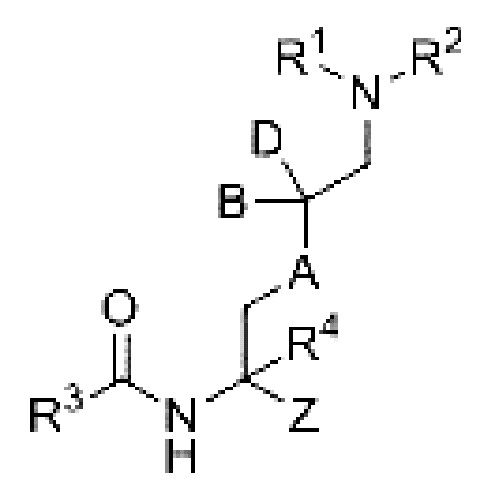
или его фармацевтически приемлемой соли, где
Z представляет собой тиол-реакционноспособную группу или защищенную тиол-реакционноспособную группу;
А выбран из -CH2- и -О-;
В и D независимо выбраны из водорода, галогена, C1-4 галогеналкила и C1-4 галогеналкокси;
R1 выбран из водорода и защитной группы аминогруппы;
R2 представляет собой водород; и
R3 выбран из C6-10 арила, 5-12-членного гетероарила, C1-8 алкила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила, -L-R5 и -OR6, где
L выбран из O, NR, C1-4 алкилена и 2-4-членного гетероалкилена, где R выбран из водорода и C1-8алкила,
R5 выбран из С6-10арила, 5-12-членного гетероарила, С3-8 циклоалкила и 5-12-членного насыщенного гетероциклила, и
-OR6 и карбонил, к которому он присоединен, образуют защитную группу аминогруппы,
и где R3 является необязательно замещенным одним или несколькими заместителями, выбранными из галогена, CN, NO2, N3, OH, Ra, Rb, ORa, ORb, (CH2)kC(O)Rc, NRd(CH2)uC(O)Rc, O(CH2)uC(O)Rc, (CH2)kCONRdRd, (CH2)kNRdC(O)Rc, -NRd(CH2)uCONRdRd, -NRd(CH2)uNRdC(O)Rc, -O(CH2)uCONRdRd, O(CH2)uNRdC(O)Rc, (CH2)kS(O)2NRdRd, (CH2)kNRdS(O)2Rc, (CH2)kS(O)2Rc, (CH2)kS(O)Rc, (CH2)kSRd, NRd(CH2)uS(O)2NRdRd, NRd(CH2)uNRdS(O)2Rc, NRd(CH2)uS(O)2Rc, NRd(CH2)uS(O)Rc, NRd(CH2)uSRd, O(CH2)uS(O)2NRdRd, O(CH2)uNRdS(O)2Rc, O(CH2)uS(O)2Rc, O(CH2)uS(O)Rc, -О(СН2)uS(О)RCи O(CH2)uSRc, где:
каждый Rа независимо выбран из C1-4 алкила и C1-4 галогеналкила,
каждый Rb независимо выбран из C3-6 циклоалкила, C3-6 галогенциклоалкила, С6-10 арила, 5 12-членного гетероарила и 5 12-членного насыщенного гетероциклила,
каждый Rc независимо выбран из -ОН, C1-8 алкила, C1-8 галогеналкила, С3-8 циклоалкила, С3-8 галогенциклоалкила, C6-10 арила, (C6-10 арил)-(C1-8 алкила), 5-12-членного гетероарила и 5 12 членного насыщенного гетероциклила,
каждый Rd независимо выбран из водорода и C1-8 алкила,
каждый подстрочный индекс k независимо выбран из 0, 1, 2, 3, 4, 5 и 6;
каждый подстрочный индекс u независимо выбран из 1, 2, 3, 4, 5 и 6; и
R4 выбран из водорода, галогена, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила и C1-4 галогеналкокси;
при условии, что когда Z представляет собой бензотиазол-2-илкарбонил, А представляет собой -CH2-, а B, D и R1 представляют собой водород, тогда R3 не является бензилоксигруппой, замещенной бензилоксигруппой или 1-(3-фенилпропаноил)пиперидин-3-илом, и
при условии, что когда Z представляет собой феноксиметилкарбонил или замещенный феноксиметилкарбонил, А представляет собой -CH2-, а В и D представляют собой водород, тогда R3 не является (2-фенил)этилом или замещенным (2-фенил)этилом.
[0005] В некоторых вариантах осуществления настоящего изобретения структура соединения соответствует формуле Ic:

где R3 выбран из С6-10 арила, 5 12 членного гетероарила, C3-8 циклоалкила, 5 12 членного насыщенного гетероциклила и L-R5,
где L представляет собой C1-4 алкилен.
[0006] В некоторых вариантах осуществления настоящего изобретения структура соединения соответствует формуле Id:

где R3 выбран из С6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и -L-R5,
где L представляет собой C1-4 алкилен.
[0007] В другом аспекте настоящее изобретение относится к фармацевтической композиции, включающей соединение формулы I и фармацевтически приемлемый эксципиент.
[0008] В еще одном аспекте настоящее изобретение относится к способу лечения заболевания или состояния, связанного с инфекцией P. gingivalis. Способ включает введение пациенту эффективного количества соединения формулы Ie:

или его фармацевтически приемлемой соли, где
Z представляет собой тиол-реакционноспособную группу или защищенную тиол-реакционноспособную группу;
А выбран из -CH2- и -О-;
В и D независимо выбраны из водорода, галогена, C1-4 галогеналкила и C1-4 галогеналкокси;
R1 выбран из водорода и защитной группы аминогруппы;
R2 представляет собой водород; и
R3 выбран из С6-10 арила, 5 12 членного гетероарила, C1-8 алкила, C3-8 циклоалкила, 5 12 членного насыщенного гетероциклила, -L-R5 и -OR6, где
L выбран из -О-, -NR-, C1-4 алкилена и 2-4-членного гетероалкилена, где R выбран из водорода и C1-8 алкила,
R5 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила и 5-12-членного насыщенного гетероциклила, и
-OR6 и карбонил, к которому он присоединен, образуют защитную группу аминогруппы,
и где R3 является необязательно замещенным одним или несколькими заместителями, выбранными из галогена, CN, NO2, N3, OH, Ra, Rb, ORa, ORb, (CH2)kC(O)Rc, NRd(CH2)uC(O)Rc, O(CH2)uC(O)Rc, (CH2)kCONRdRd, (CH2)kNRdC(O)Rc, NRd(CH2)uCONRdRd, NRd(CH2)uNRdC(O)Rc, O(CH2)uCONRdRd, O(CH2)uNRdC(O)Rc, (CH2)kS(O)2NRdRd, (CH2)kNRdS(O)2Rc, (CH2)kS(O)2Rc, (CH2)kS(O)Rc, (CH2)kSRd, NRd(CH2)uS(O)2NRdRd, NRd(CH2)uNRdS(O)2Rc, NRd(CH2)uS(O)2Rc, NRd(CH2)uS(O)Rc, NRd(CH2)uSRd, O(CH2)uS(O)2NRdRd, O(CH2)uNRdS(O)2Rc, O(CH2)uS(O)2Rc, O(CH2)uS(O)Rcи O(CH2)uSRc, где:
каждый Ra независимо выбран из C1-4 алкила и C1-4 галогеналкила,
каждый Rb независимо выбран из C3-6 циклоалкила, C3-6 галогенциклоалкила, C6-10 арила, 5-12-членного гетероарила и 5-12 -членного насыщенного гетероциклила,
каждый Rc независимо выбран из -ОН, C1-8 алкила, C1-8 галогеналкила, C3-8 циклоалкила, C3-8 галогенциклоалкила, C6-10 арила, (C6-10 арил)-(C6-10 алкила), 5-12-членного гетероарила и 5-12-членного насыщенного гетероциклила,
каждый Rd независимо выбран из водорода и C1-8 алкила,
каждый подстрочный индекс k независимо выбран из 0, 1, 2, 3, 4, 5 и 6, и
каждый подстрочный индекс u независимо выбран из 1, 2, 3, 4, 5 и 6; и
R4 выбран из водорода, галогена, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила и C1-4 галогеналкокси.
[0009] В некоторых вариантах осуществления настоящего изобретения заболевание или состояние, связанное с инфекцией P. gingivalis, представляет собой нарушение мозговой деятельности, выбранное из болезни Альцгеймера, синдрома Дауна, эпилепсии, аутизма, болезни Паркинсона, эссенциального тремора, фронто-темпоральной деменции, прогрессирующего надъядерного паралича, бокового амиотрофического склероза, болезни Гантингтона, рассеянного склероза, умеренных когнитивных нарушений, возрастного нарушения памяти, хронической травматической энцефалопатии, инсульта, болезни телец Леви, множественной системной атрофии, шизофрении и депрессии. В некоторых вариантах осуществления настоящего изобретения заболевание или состояние, связанное с инфекцией P. gingivalis, представляет собой болезнь Альцгеймера.
КРАТКОЕ ОПИСАНИЕ РИСУНКОВ
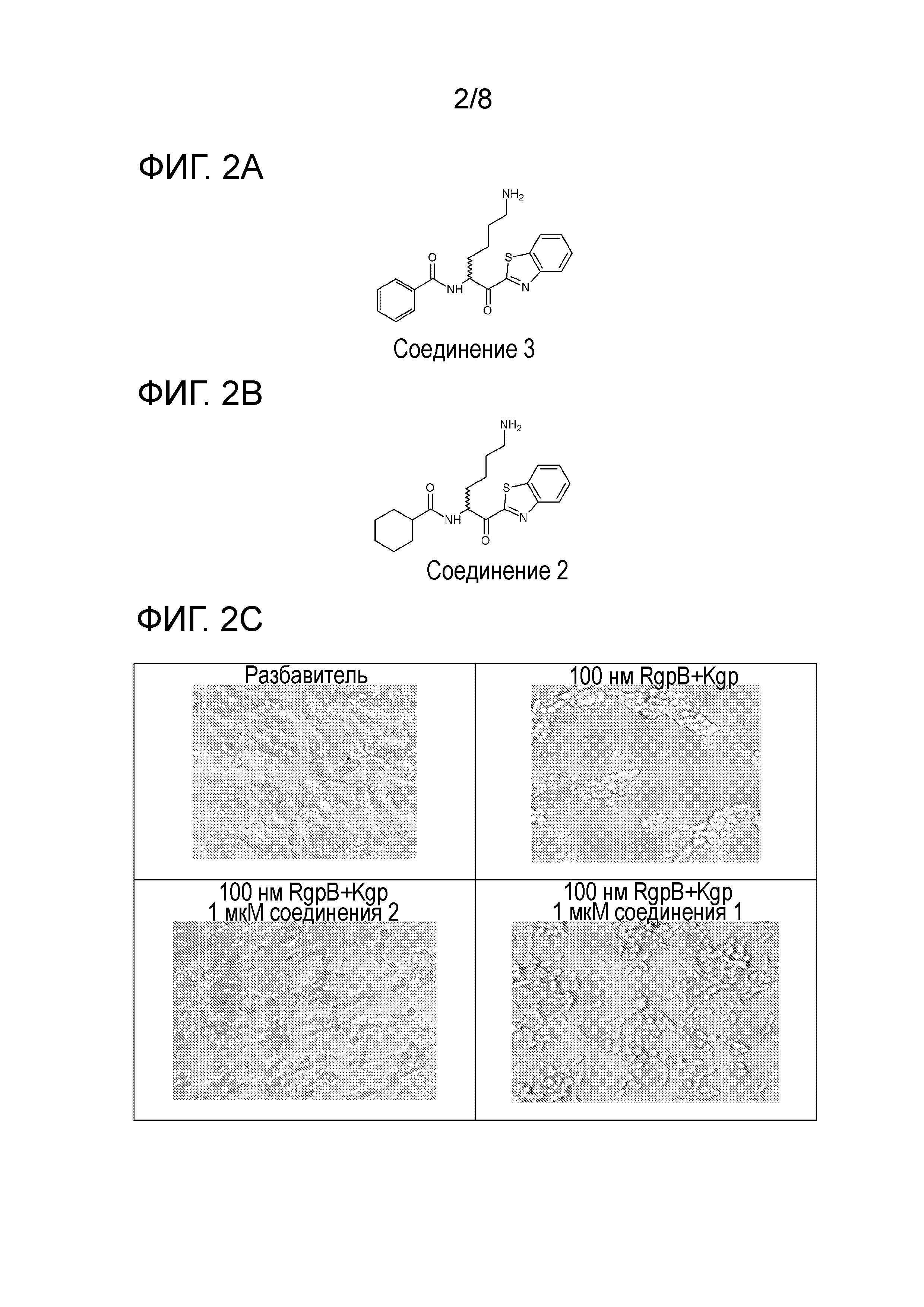
[0010] На фигуре 1А представлена структура 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(R)-лизинил)бензотиазола - соединения 3.
[0011] На фигуре 1В представлена структура 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(R)-лизинил)бензотиазола - соединения 4.
[0012] На фигуре 1C представлены снимки, показывающие, что соединение 4 предотвращает гингипаин-индуцированную гибель дифференцированных SHSY-5Y клеток, а соединение 3 не предотвращает. Значение IC50 Kgp соединения 3 находится в интервале от 10 до 25 нм, а значение IC50 Kgp соединения 4 находится в интервале от 1 до 10 нм.
[0013] На фигуре 2A представлена структура соединения 1.
[0014] На фигуре 2B представлена структура соединения 2.
[0015] На фигуре 2С представлены снимки, показывающие, что соединение 1 и соединение 2 предотвращают гингипаин-индуцированную гибель дифференцированных SHSY-5Y клеток, но соединение 2 является более эффективным. Для соединения 2 значение IC50Kgp находится в интервале от 1 до 10 нМ, значение IC50 трипсина равно 5000 нМ.
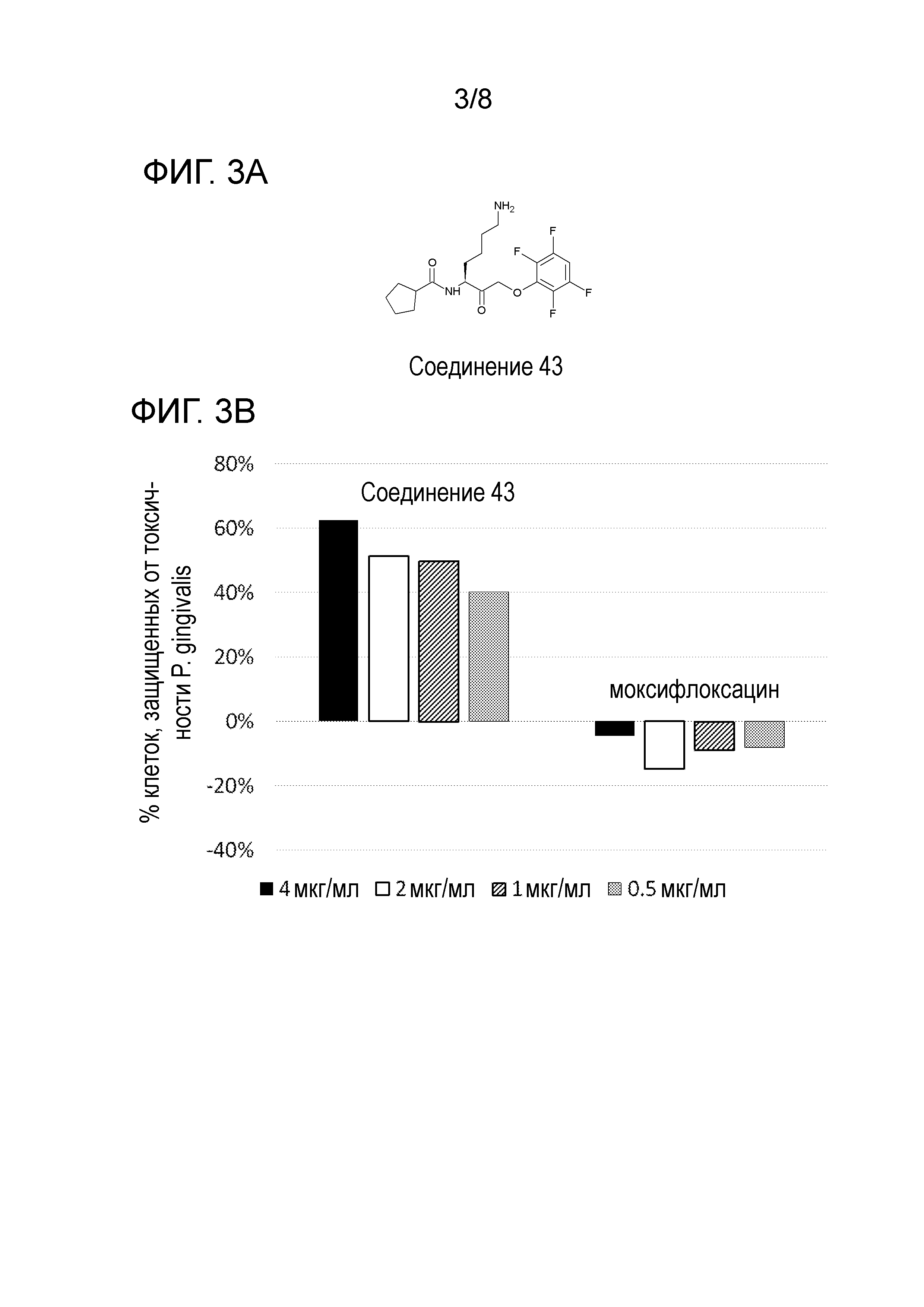
[0016] На фигуре 3А представлена структура соединения 43.
[0017] На фигуре 3B представлена диаграмма, показывающая способность соединения 43, необратимого ингибитора ковалентного лизин-специфичного гингипаина с субнаномольным значением IC50, защищать SHSY5Y клетки от индуцированной Р. gingivali токсичности in vivo в сравнении с антибиотиком моксифлоксацином.
[0018] На фигуре 4 представлены снимки, показывающие, что внутригиппокампальное введение гингипаинов в мозг мыши вызывает нейродегенерацию через 7 суток.
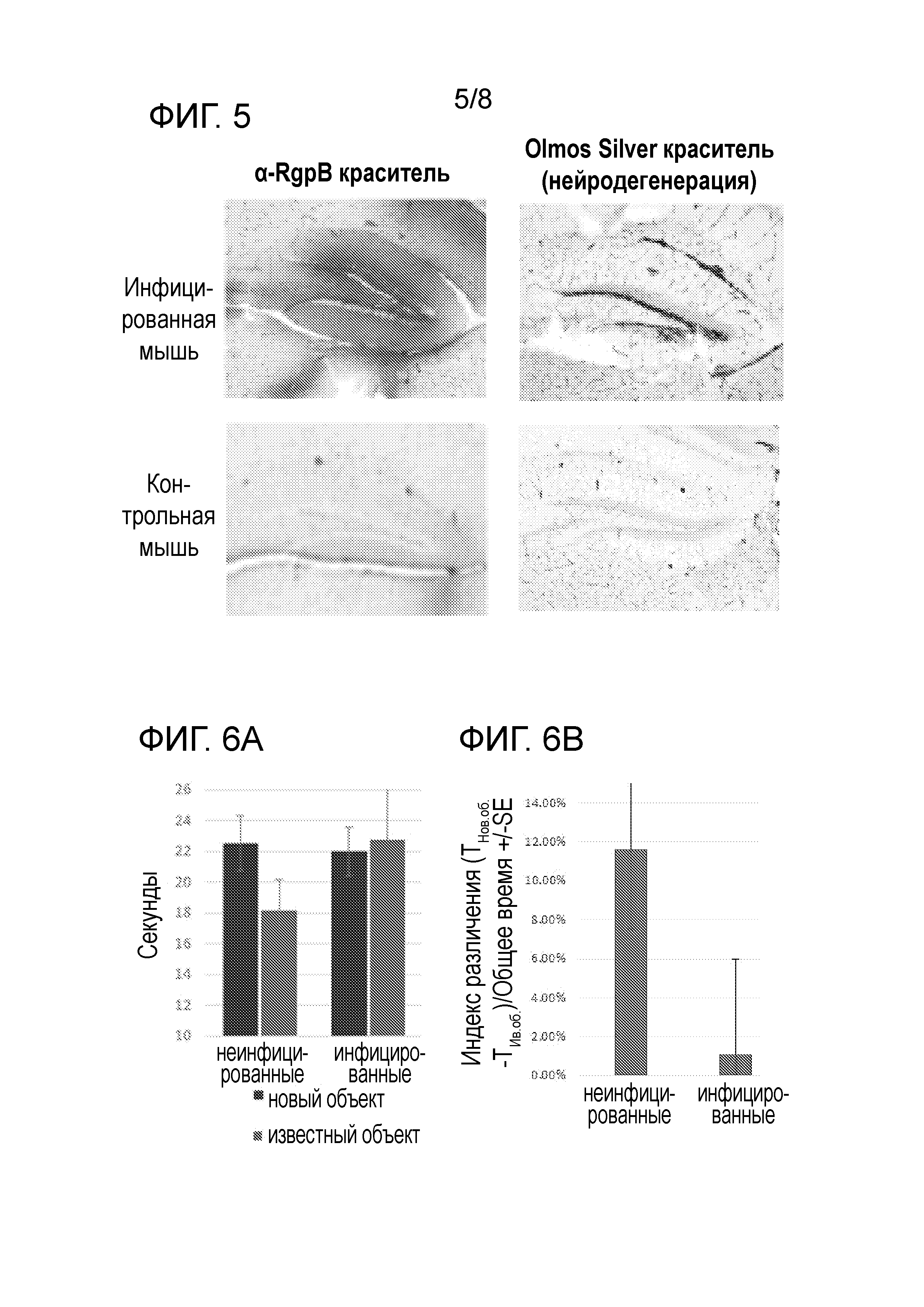
[0019] На фигуре 5 представлены снимки, показывющие, что инфильтрация RgpB в мозг совпадает с нейродегенерацией субгранулярной зоны в гиппокампе BalbC мышей, перорально инфицируемых P. gingivalis в течение 6 недель.
[0020] Фигуре 6A представлена диаграмма, показывающая, нарушение когнитивной функции у мышей дикого типа, инфицированных P. gingivalis, в тесте распознавания нового объекта через 6 недель после инфицирования. Зараженные мыши затрачивают равное количество времени на изучение нового и знакомого объекта, в то время как нормальные мыши затрачивают больше времени на изучение нового объекта.
[0021] На фигуре 6B представлена диаграмма, показывающая значения индекса различения (ТНов.об.-ТИзв.об)/Тобщее для неинфицированных и инфицированных мышей.
[0022] На фигуре 7 представлена диаграмма, показывающая содержание абета 42 («Ab42») в мозге, количественно определенное с помощью иммуноферментного анализа (ELISA), что свидетельствует о способности инфекции вызывать повышение содержания абета в мозге, которое может быть снижено при лечении соединением 2, начиная с середины периода введения инфекции. Кроме того, повышение содержания абета 42 предотвращается, когда бактерии, используемые для инфицирования, не экспрессируют Kgp.
[0023] На фигуре 8 представлены снимки, показывающие, что престарелые собаки с нарушенями когнитивной функции в значительной степени положительны для обнаржения Kgp в мозге.
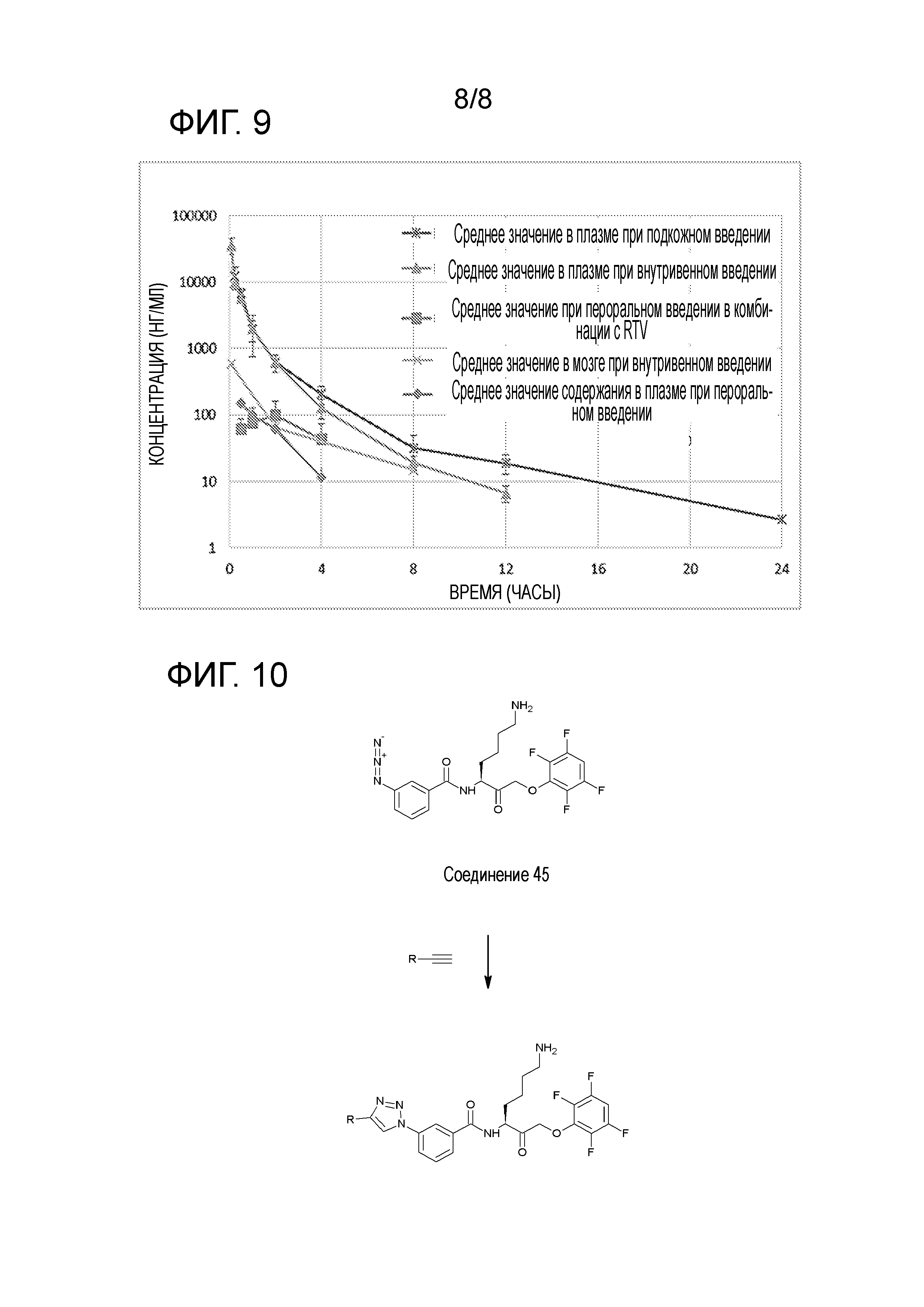
[0024] На фигуре 9 представлены фармакокинетические данные KYT-36, ингибитора Kgp, которые показывают, что ретонавир (RTV) увеличивает период полураспада перорально вводимого KYT-36.
[0025] На фигуре 10 показан пример получения соединения «клик-химии» (click-chemistry), которое может использоваться получения радиоактивно меченых средств визуализации для поэитронно-эмиссионной томографии (ПЭТ)/однофотонной эмиссионной компьютерной томографии (ОФЭКТ) или агентов захвата для in vitro анализов или диагностики. R на фигуре 10 представляет собой радионуклид или фрагмент, содержащий радионуклидный заместитель {например, R=18F-алкилен).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
I. Общие положения
[0026] Было показано, что ингибирование Kgp различными ингибиторами защищает клетки, предотвращает рост бактерий, повышает контроль бактерий иммунной системой и защищает от повторного заражения. Настоящее изобретение предоставляет эффективные и селективные непептидные соединения с улучшенными свойствами по сравнению описанными выше соединениями. Как показано в описании, ингибиторы Kgp по настоящему изобретению могут предотвращать гибель клеток, вызванную гингипаинами или P. gingivalis в модели SH-SY5Y клеток. Соединения могут быть использованы для предотвращения гибели клеток, воспаления и других патологий при различных заболеваниях, связанных с инфекцией P. gingivalis, включая состояния, связанные со старением, такие как болезнь Альцгеймера.
II. Определения
[0027] Термин «алкил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к линейному или разветвленному насыщенному алифатическому радикалу, содержащему указанное число атомов углерода. Алкил может включать любое число атомов углерода, такое как C1-2, C1-3, C1-4, C1-5, C1-6, C1-7, C1-8, C1-9, C1-10, C2--3, C2-4, C2-5, C2-6, C3-4, C3-5, C3-6, C4-5, C4-6 и C5-6. Например, C1-6 алкил включает, но не ограничивается ими, метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, гексил и т.д. Термин «алкил» может также относиться к алкильным группам, содержащим до 20 атомов углерода, таким как, но без ограничения, гептил, октил, нонил, децил и т.д. Алкильные группы могут быть замещенными или незамещенными. «Замещенные алкильные» группы могут быть замещены одной или несколькими группами, выбранными из галогена, гидроксильной группы, амино, алкиламино, амидо, ацила, нитро, циано и алкокси.
[0028] Термин «алкокси», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к группе формулы -OR, в которой R представляет собой алкил. Термин «низший алкоксил» относится к алкоксильному радикалу, содежащему от одного до семи атомов углерода, например метоксильному, этоксильному, пропоксильному, бутоксильному, пентоксильному, гексоксильному или гептоксильному радикалу.
[0029] Термин «циклоалкил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к насыщенному или частично ненасыщенному, моноциклическому, конденсированному бициклическому или мостиковому полициклическому кольцу, содержащему в кольце от 3 до 12 атомов или указанное число атомов. Циклоалкил может включать любое число атомов углерода, такое как C3-6, C4-6, C5-6, C3-8, C4-8, C5-8, C6-8, C3-9, C3-10, C3-11 и C3-12. Насыщенные моноциклические циклоалкильные кольца включают, например, циклопропил, циклобутил, циклопентил, циклогексил и циклооктил. Насыщенные бициклические и полициклические циклоалкильные кольца включают, например, норборнан, [2.2.2]бициклооктан, декагидронафталин и адамантан. Циклоалкильные группы также могут быть частично ненасыщенными, содержащими в кольце одну или несколько двойных или тройных связей. Типичные циклоалкильные группы, которые являются частично ненасыщенными, включают, но без ограничения, циклобутен, циклопентен, циклогексен, циклогексадиен (1,3- и 1, 4-изомеры), циклогептен, циклогептадиен, циклооктен, циклооктадиен (1,3-, 1,4- и 1,5-изомеры), норборнен и норборнадиен. Когда циклоалкил представляет собой насыщенный моноциклический С3-8 циклоалкил, примеры такой группы включают, но без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Когда циклоалкил представляет собой насыщенный моноциклический С3-6 циклоалкил, примеры такой группы включают, но без ограничения, циклопропил, циклобутил, циклопентил и циклогексил. Циклоалкильные группы могут быть замещенными или не замещенными. «Замещенный циклоалкильные» группы могут быть замещены одной или несколькими группами, выбранными из галогена, гидроксильной группы, амино, алкиламино, амидо, ацила, нитро, циано и алкокси. Термин «низший циклоалкил» относится к циклоалкильныому радикалу, содержащему от трех до семи атомов углерода, включая, например, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
[0030] Термин «алкилен», когда используется в настоящем описании, относится к алкильной группе, которая определена выше, связанной по меньшей мере с двумя другими группами (то есть, двухвалентному алкильному радикалу). Эти два фрагмента, связанные с алкиленовой группой, могут присоединяться к одному атому углероду или разным атомам углерода в алкиленовой группе.
[0031] Термин «гетероалкил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к алкильной группе любой подходящей длины, содержащей от 3 до 1 гетероатомов, таких как атомы N, О и S. Например, термин «гетероалкил» может включать простые эфиры, простые тиоэфиры и алкиламины. Могут также использоваться дополнительные гетероатомы, включая, но без ограничения, атомы B, Al, Si и P. Гетероатомы могут быть окисленными с образованием таких групы, как, но без ограничения, -S(O)- и -S(О)2-. Гетероатом гетероалкила может быть замещен водородом алкильной группы с образованием гидроксильной, тио- или аминогруппы. В качестве альтернативы, гетероатом может быть связывающим атомом или может быть вставлен между двумя атомами углерода.
[0032] Термин «гетероалкилен», когда используется в настоящем описании, относится к гетероалкильной группе, которая определена выше, связывающей по меньшей мере две другие группы (то есть, двухвалентному гетероалкильному радикалу). Эти два фрагмента, связанные с гетероалкиленовой группой, могут быть присоединяться к одному атому или различных атомам гетероалкиленовой группы.
[0033] Термин «галоген», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к атому фтора, хлора, брома или йода.
[0034] Термин «галогеналкил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к алкильной группе, в которой некоторые или все атомы водорода замещены атомами галогена. Так же как алкильные группы, галогеналкильные группы могут содержать любое подходящее количество атомов углерода, такое как C1-6. Например, термин «галогеналкил» включает трифторметил, фторметил и т.д. В некоторых случаях термин «перфтор» может быть использован для определения соединения или радикала, в котором все атомы водорода замещены фтором. Например, термин «перфторметил» относится к 1,1,1-трифторметилу.
[0035] Термин «галогеналкокси», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к алкоксильной группе, в которой некоторые или все атомы водорода замещены атомами галогена.
[0036] Термин «галогенциклоалкил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к циклоалкильной группе, в которой некоторые или все атомы водорода замещены атомами галогенов.
[0037] Термин «арил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к ароматической кольцевой системе, содержащей любое подходящее количество атомов в кольце и любое подходящее количество колец. Арильные группы могут содержать в кольце любое подходящее количество атомов, такое как 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или 16 атомов, а также от 6 до 10, от 6 до 12 или от 6 до 14 членов в цикле. Арильные группы могут быть моноциклическими, конденсированными с образованием бициклической {например, бензоциклогексильной) или трициклической группы или могут быть связаны посредством связи с образованием биарильной группы. Типичные арильные группы включают фенил, нафтил и бифенил. Другие арильные группы включают бензил, содержащий метиленовую связывающую группу. Некоторые арильные группы содержат от 6 до 12 членов в кольце, например фенил, нафтил или бифенил. Другие арильные группы включают от 6 до 10 членов в кольце, например фенил или нафтил. Некоторые другие арильные группы содержат 6 членов в кольце, такие как фенил. Арильные группы могут быть замещенными или не замещенными. «Замещенные арильные» группы могут быть замещены одной или несколькими группами, выбранными из галогена, гидроксильной группы, амино, алкиламино, амидо, ацила, нитро, циано и алкокси.
[0038] Термин «гетероарил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к моноциклическому или конденсированному бициклическому или трициклическому ароматическому кольцевому блоку, содержащему от 5 до 16 атомов в кольце, из которых от 1 до 5 атомов являются гетероатомами, такими как атомы N, O или S. Могут также использоваться и дополнительные гетероатомы, в том числе, но без ограничения, атомы B, Al, Si и P. Гетероатомы могут подвергаться окислению с образованием таких групп, как, но без ограничения, -S(O)- и -S(О)2-. Гетероарильные группы могут содержать в кольце любое число атомов, например от 3 до 6, от 4 до 6, от 5 до 6, от 3 до 8, от 4 до 8, от 5 до 8, от 6 до 8, от 3 до 9, от 3 до 10, от 3 до 11 или от 3 до 12 членов в кольце. Гетероарильные группы могут содержать любое подходящее количество гетероатомов, такое как 1, 2, 3, 4 или 5, или от 1 до 2, от 1 до 3, от 1 до 4, от 1 до 5, от 2 до 3, от 2 до 4, от 2 до 5, от 3 до 4 или от 3 до 5. Гетероарильные группы могут содержать в кольце от 5 до 8 членов и от 1 до 4 гетероатомов, или от 5 до 8 членов и от 1 до 3 гетероатомов, от 5 до 6 членов и от 1 до 4 гетероатомов или от 5 до 6 кольцевых членов и от 1 до 3 гетероатомов. Гетероарильная группа может включать такие группы, как пиррол, пиридин, имидазол, пиразол, триазол, тетразол, пиразин, пиримидин, пиридазин, триазин (1,2,3-, 1,2,4- и 1,3,5-изомеры), тиофен, фуран, тиазол, изотиазол, оксазол и изоксазол. Гетероарильные группы также могут конденсированными с ароматическими кольцевыми системами, такими как фенильное кольцо, с образованием таких фрагментов, как, но, без ограничения, бензопирролы, такие как индол и изоиндолы, бензопиридины, такие как хинолин и изохинолин, бензопиразин (хиноксалин), бензопиримидин (хиназолин), бензопиридазины, такие как фталазин и циннолин, бензотиофен и бензофуран. Другие гетероарильные группы включают гетероарильные кольца, соединенные связью, такие как бипиридин. Гетероарильные группы могут быть замещенными или незамещенными. «Замещенные гетероарильные» группы могут быть замещены одной или несколькими группами, выбранными из галогена, гидроксильной группы, амино, алкиламино, амидо, ацил, нитро, циано и алкокси.
[0039] Гетероарильные группы могут присоединяться через атом в любом положении на кольце. Так, например, термин «пиррол» включает 1-, 2- и 3-пиррол, термин «пиридин» включает 2-, 3- и 4-пиридин, термин «имидазол» включает 1-, 2-, 4- и 5-имидазол, термин «пиразол» включает 1-, 3-, 4- и 5-пиразол, термин «триазол» включает 1-, 4- и 5-триазол, термин «тетразол» включает 1- и 5-тетразол, термин «пиримидин» включает 2-, 4-, 5- и 6-пиримидин, термин «пиридазин» включает 3- и 4- пиридазин, термин «1,2,3-триазин» включает 4- и 5-триазин, термин «1,2,4-триазин» включает 3-, 5- и 6-триазин, термин «1,3,5-триазин» включает 2-триазин, термин «тиофен» включает 2- и 3-тиофен, термин «фуран» включает 2- и 3-фуран, термин «тиазол» включает 2-, 4- и 5-тиазол, термин «изотиазол» включает 3-, 4- и 5-изотиазол, термин «оксазол» включает 2-, 4- и 5-оксазол, термин «изоксазол» включает 3-, 4- и 5-изоксазол, термин «индол» включает 1-, 2- и 3-индол, термин «изоиндол» включает 1- и 2-изоиндол, термин «хинолин» включает 2-, 3- и 4-хинолин, термин «изохинолин» включает 1-, 3- и 4-изохинолин, термин «хиназолин» включает в 2- и 4-хиназолин, термин «циннолин» включает в 3- и 4-циннолин, термин «бензотиофен» включает 2- и 3-бензотиофен, и термин «бензофуран» включает в 2- и 3 бензофуран.
[0040] Некоторые гетероарильные группы включают группы, которые содержат в кольце от 5 до 10 членов и от 1 до 3 гетероатомов, включая атомы N, O или S, такие как пиррол, пиридин, имидазол, пиразол, триазол, пиразин, пиримидин, пиридазин, триазин (1,2,3-, 1,2,4- и 1,3,5-изомеры), тиофен, фуран, тиазол, изотиазол, оксазол, изоксазол, индол, изоиндол, хинолин, изохинолин, хиноксалин, хиназолины, фталазин, циннолин, бензотиофен и бензофуран. Другие гетероарильные группы включают группы, которые содержат в кольце от 5 до 8 членов и от 1 до 3 гетероатомов, такие как пиррол, пиридин, имидазол, пиразол, триазол, пиразин, пиримидин, пиридазин, триазин (1,2,3, 1,2,4- и 1,3,5-изомеры), тиофен, фуран, тиазол, изотиазол, оксазол и изоксазол. Некоторые другие гетероарильные группы включают группы, которые содержат в кольце от 9 до 12 членов и от 1 до 3 гетероатомов, такие как индол, изоиндол, хинолин, изохинолин, хиноксалин, хиназолины, фталазин, циннолин, бензотиофны, бензофуран и бипиридин. Кроме того, другие гетероарильные группы включают группы, которые содержат в кольце от 5 до 6 членов и от 1 до 2 гетероатомов, включая атомы N, O или S, такие как пиррол, пиридин, имидазол, пиразол, пиразин, пиримидин, пиридазин, тиофен, фуран, тиазол, изотиазол, оксазол и изоксазол.
[0041] Некоторые гетероарильные группы содержат в кольце от 5 до 10 членов и только гетероатомы азота, такие как пиррол, пиридин, имидазол, пиразол, триазол, пиразин, пиримидин, пиридазин, триазин (1,2,3-, 1,2,4- и 1,3,5-изомеры), индол, изоиндол, хинолин, изохинолин, хиноксалин, хиназолины, фталазин и циннолин. Другие гетероарильные группы содержат в кольце от 5 до 10 членов и только гетероатомы кислорода, такие как фуран и бензофуран. Некоторые другие гетероарильные группы содержат в кольце от 5 до 10 членов и только гетероатомы серы, такие как тиофен и бензотиофен. Кроме того, другие гетероарильные группы содержат в кольце от 5 до 10 атомов и, по меньшей мере, два гетероатома, такие как имидазол, пиразол, триазол, пиразин, пиримидин, пиридазин, триазин (1,2,3-, 1,2,4- и 1,3,5-изомеры), тиазол, изотиазол, оксазол, изоксазол, хиноксалин, хиназолины, фталазин и циннолин.
[0042] Термин «гетероциклил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к насыщенной кольцевой системе, содержащей в кольце от 3 до 12 атомов и от 1 до 4 гетероатомов N, О и S. Могут использоваться дополнительные гетероатомы, в том числе, но без ограничения, атомы B, Al, Si и P. Гетероатомы могут быть окислены с образованием группы, такой как, но без ограничения, -S(O)- и S(O)2. Гетероцикльные группы могут содержать любое количество атомов в кольце, такое как от 3 до 6, от 4 до 6, от 5 до 6, от 3 до 8, от 4 до 8, от 5 до 8, от 6 до 8, от 3 до 9, от 3 до 10, 3 до 11 или от 3 до 12 атомов. В гетероциклильные группы может быть включено любое подходящее количество гетероатомов, такое как 1, 2, 3 или 4 или от 1 до 2, от 1 до 3, от 1 до 4, от 2 до 3, от 2 до 4 или от 3 до 4. Гетероциклильные группы могут включать такие группы, как азиридин, азетидин, пирролидин, пиперидин, азепан, азокан, хинуклидин, пиразолидин, имидазолидин, пиперазин (1,2-, 1,3- и 1,4-изомеры), оксиран, оксетан, тетрагидрофуран, оксан (тетрагидропирано), оксепан, тииран, тиетан, тиолан (тетрагидротиофен), тиан (тетрагидротиопиран), оксазолидин, изоксазолидин, тиазолидин, изотиазолидин, диоксолан, дитиолан, морфолин, тиоморфолин, диоксан или дитиан. Гетероциклильные группы также могут быть конденсированы с ароматическими или неароматическими кольцевыми системами с образованием фрагментов, включая, но без ограничения, индолин. Гетероциклильные группы могут быть незамещенными или замещенными. «Замещенные гетероциклильные» группы могут быть замещены одной или несколькими группами, выбранными из галогена, гидроксильной группы, амино, оксо (=О), алкиламино, амидо, ацил, нитро, циано и алкокси.
[0043] Гетероциклильные группы могут присоединяться через атом в любом положении в кольце. Например, азиридин может представлять собой 1- или 2-азиридин, азетидин может представлять собой 1- или 2-азетидин, пирролидин может представлять собой 1-, 2- или 3 пирролидин, пиперидин может представлять собой 1-, 2-, 3- или 4- пиперидин, пиразолидин может представлять собой 1-, 2-, 3- или 4-пиразолидин, имидазолидин может представлять собой 1-, 2-, 3- или 4-имидазолидин, пиперазин может представлять собой 1-, 2-, 3- или 4-пиперазин, тетрагидрофуран может представлять собой 1- или 2-тетрагидрофуран, оксазолидин может представлять собой 2-, 3-, 4- или 5-оксазолидин, изоксазолидин может представлять собой 2-, 3-, 4- или 5-изоксазолидин, тиазолидин может представлять собой 2-, 3 -, 4- или 5-тиазолидин, изотиазолидин может представлять собой 2-, 3-, 4- или 5-изотиазолидин, и морфолин может представлять собой 2-, 3- или 4-морфолин.
[0044] Когда гетероциклил содержит в кольце от 3 до 8 членов и от 1 до 3 гетероатомов, типичные примеры таких групп включают, но без ограничения, пирролидин, пиперидин, тетрагидрофуран, оксан, тетрагидротиофен, тиан, пиразолидин, имидазолидин, пиперазин, оксазолидин, изоксазолидин, тиазолидин, изотиазолидин, морфолин, тиоморфолин, диоксан и дитиан. Гетероциклил может также образовывать кольцо, содержащее от 5 до 6 членов и от 1 до 2 гетероатомов, типичные примеры которого включают, но без ограничения, пирролидин, пиперидин, тетрагидрофуран, тетрагидротиофен, пиразолидин, имидазолидин, пиперазин, оксазолидин, изоксазолидин, тиазолидин, изотиазолидин и морфолин.
[0045] Термин «тиол-реакционноспособная группа», когда используется в настоящем описании, относится к функциональной группе, способной образовывать обратимую или необратимую ковалентную связь с тиольной группой (т.е. группой структуры «-SH»), например, такой как тиольная группа в α-боковой цепи цистеина. Не ограничивающие примеры тиол-реакционноспособных групп включают тиазол-2-илкарбонил; бензотиазол-2-илкарбонил; оксазол-2-илкарбонил; бензоксазол-2-илкарбонил; пиридин-2-илкарбонил; пиримидин-4-илкарбонил; пиримидин-2-илкарбонил; изоксазол-5-илкарбонил; изоксазол-3-илкарбонил; 1,2,4-оксадиазол-3-илкарбонил; 1,2,4-оксадиазол-5-илкарбонил; малеимидил; пиридинилсульфанил (включая пиридин-2-илдисульфанил); циано; этинил; фторметилкарбонил; ацилоксиметилкарбонил; арилоксиметилкарбонил; алкилсульфонилвинил; и арилсульфонилвинил. Другие тиол-реакционноспособные группы известны специалистам в данной области техники, включая, например, группы, которые описаны Hermanson (Bioconjugate Techniques, 3rd Ed.2013, Academic Press, San Diego).
[0046] Термин «защищенная тиол-реакционноспособная группа» относится к нереакционноспособной части молекулы-предшественника, которая может подвергаться превращению в функциональную группу, способную образовывать обратимую или необратимую ковалентную связь с тиольной группой.
[0047] Термин «защитная группа аминогруппы», когда используется в настоящем описании, относится к химической группе, которая делает аминогруппу инертной, но которая способна удаляться с восстановлением аминогруппы. Примеры защитных групп аминогруппы включают, но без ограничения, бензилоксикарбонил, 9-флуоренилметилоксикарбонил (Fmoc), трет-бутилоксикарбонил (Boc), аллилоксикарбонил (Alloc), ацетамидо, фталимидо и т.п. Другие защитные группы аминогруппы известны специалистам в данной области техники, включая, например, группы, которые описаны Green и Wuts (Protective Groups in Organic Synthesis, 4thEd. 2007, Wiley-Interscience, New York).
[0048] Термин «карбонил», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к фрагменту -C(O)-, то есть к атому углерода, соединенному двойной связью с атомом кислорода и связанному с двумя другими группами с образованием фрагмента, содержащего карбонил.
[0049] Термин «амино», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к фрагменту -NR3, в котором каждая R группа представляет собой Н или алкил. Аминогруппа может быть ионизированной с образованием соответствующего катиона аммония.
[0050] Термин «гидроксильная группа», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к фрагменту -ОН.
[0051] Термин «циано», когда используется в настоящем описании сам по себе или как часть другого заместителя, относится к атому углерода, соединенному тройной связью с атомом азота {т.е. фрагменту -C≡N).
[0052] Термин «карбокси», когда используется в настоящем описании, относится к фрагменту -C(О)ОН. Карбоксильная группа может быть ионизированной с образованием соответствующего карбоксилат-аниона.
[0053] Термин «амидо», когда используется в настоящем описании, относится к фрагменту -NRC(О)R или -C(О)NR2, в котором каждая R группа представляет собой Н или алкил.
[0054] Термин «нитро», когда используется в настоящем описании, относится к фрагменту -NO2.
[0055] Термин «оксо», когда используется в настоящем описании, относится к атому кислорода, который присоединен к соединению двойной связью {т.е. О=).
[0056] Термин «фармацевтически приемлемый эксципиент», когда используется в настоящем описании, относится к веществу, которое способствует введению активного соединения пациенту. Термин «фармацевтически приемлемый» означает, что наполнитель является совместимым с другими ингредиентами препарата и не вреден для его реципиента. Фармацевтические наполнители, используемые в настоящем изобретении, включают, но без ограничения, связующие вещества, инертные добавки, дезинтегрирующие вещества, смазывающие вещества, добавки, способствующие скольжению, покрытия, подсластители, ароматизаторы и красители.
[0057] Термин «соль», когда используется в настоящем описании, относится к солям соединений по изобретению с кислотами или основаниями по изобретению. Иллюстративными примерами фармацевтически приемлемых солей являются соли минеральных кислот (соляной кислоты, бромистоводородной кислоты, фосфорной кислоты и т.п.), органических кислот (уксусной кислоты, пропионовой кислоты, глутаминовой кислоты, лимонной кислоты и т.п.) и четвертичные аммониевые (метилйодид, этилйодид и т.п.) соли. Понятно, что фармацевтически приемлемые соли являются нетоксичными.
[0058] Фармацевтически приемлемые соли кислотных соединений по настоящему изобретению представляют собой соли, образованные с основаниями, а именно катионные соли, такие как соли щелочных и щелочно-земельных металлов, например натрия, лития, калия, кальция, магния, а также аммониевые соли, такие, как соли аммония, триметиламмония, диэтиламмония и трис- (гидроксиметил)метиламмония.
[0059] Аналогично, кислотно-аддитивные соли, такин как соли минеральных кислот, органических карбоновых кислоты и органических сульфоновых кислот рот, например соляной кислоты, метансульфоновой кислоты, малеиновой кислоты, также возможны при условии, что частью структуры является основная группа, такая как пиридил.
[0060] Нейтральные формы соединений могут быть регенерированы контактированием соли с основанием или кислотой и выделением исходного соединения обычным способом. Исходная форма соединения отличается от различных солевых форм по некоторым физическим свойствам, таким как растворимость в полярных растворителях, но в остальном для целей настоящего изобретения соли эквивалентны исходной форме соединения.
[0061] Термины «Porphyromonas gingivalis» и «Р. gingivalis», когда используются в настоящем описании, относятся к грамотрицательной несахаролитической бактерии, которая определена в качестве ключевого патогенного микроорганизма в патогенезе заболевания пародона и родственных состояний. Термин «инфекция Р. gingivalis» относится к вторжению и колонизации P. gingivalis в ткани организма, такие как десны или головной мозг. Инфекция P. gingivalis зачастую характеризуется последующим повреждением тканей и заболеванием.
[0062] Термин «гингипаин», когда используется в настоящем описании, относится к цистеиновым протеазам, экспрессируемым P. gingivalis, обладающим трипсиноподобной специфичностью {т.е. Lys-Хаа и Arg-Xaa). Гингипаины признаны основными факторами вирулентности P. gingivalis и способствуют прикреплению и колонизации бактерий, поглощению питательных веществ, уходу от воздействия защитных сил организма и тканевой инвазии. Термины «лизин-специфичный гингипаин» и «Kgp» используются взаимозаменяемо и относятся P. gingivalis лизин-специфическому гингипаину, известному под ЕС номером EC 3.4.22.47.
[0063] Термины «лечить», «лечение» и «излечение» относятся к любому признаку успеха в лечении или облегчении травмы, патологии, состояния или симптома {например, нарушения когнитивных функций), включая любой объективный или пациентивный параметр, такой как смягчение; ремиссия; уменьшение симптомов или проявления симптомов, травмы, патологии или состояния, которые легче переносятся пациентом; уменьшение частоты или длительности проявления симптомов или состояния; или, в некоторых ситуациях, предотвращение возникновение симптомов. Лечение или облегчение симптомов могут быть основаны на любом объективном или пациентивном параметре; включая, например, результат физического осмотра.
[0064] Термины «эффективное количество» и «терапевтически эффективное количество», когда используются в настоящем описании, относятся к дозе соединения, такого как ингибитор Kgp, которая производит терапевтическое действие, для которого ее вводят. Точная доза будет зависеть от цели лечения и будет определяться специалистом данной области техники с использованием известных способов {см., например, Lieberman, Pharmaceutical Dosage Forms (vols 1 3, 1992); Lloyd, The Art, Science and Technology of Pharmaceutical Compounding (1999); Pickar, Dosage Calculations (1999); Goodman & Gilmanʹs The Pharmacological Basis of Therapeutics, 11th Edition, 2006, Brunton, Ed., McGraw-Hill; Remington: The Science and Practice of Pharmacy, 21st Edition, 2005, Hendrickson, Ed., Lippincott, Williams & Wilkins).
[0065] Термин «болезнь Альцгеймера», когда используется в настоящем описании, относится к прогрессирующему заболеванию центральной нервной системы у человека и других млекопитающих. Оно храктеризуется слабоумием (особенно у пожилых людей); дезориентацией; потерей памяти; затруднением речи, счета или зрительно-пространственной ориентации; и симптомами психиатрических заболеваний. Болезнь Альцгеймера связана с прогрессирующей нейродегенерацией и характерной патологией, точнее, бета-амилоидными бляшками и тау-клубками.
[0066] Термин «пациент», когда используется в настоящем описании, относится к животным, таким как млекопитающие, включая, но без ограничения, приматов {например, людей), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей и т.п.
[0067] Термины «примерно» и «около», когда используются в настоящем описании для корректировки числового значения, указывают на близкий интервал, окружающий это точное числовое значение. Если бы «Х» представлял собой значение, то «примерно X» или «примерно X» указывал бы на значение от 0,9Х до 1,1Х и, более предпочтительно, значение от 0,95X до 1,05X. Любая ссылка на «примерно X» или «около X» конкретно указывает, по меньшей мере, на значения X, 0,95X, 0,96X, 0,97X, 0,98X, 0,99X, 1,01X, 1,02X, 1,03X, 1,04X и 1,05X. Таким образом, «примерно X» и «около X» предназначены показа значения и обеспечения письменной аргументации ограничивающего пункта формулы изобретения, например, «0,98X».
III. Ингибиторы лизин-специфичного гингипаина
[0068] В одном аспекте настоящее изобретение относится к соединению формулы I:

или его фармацевтически приемлемой соли, где
Z представляет собой тиол-реакционноспособную группу или защищенную тиол-реакционноспособную группу;
А выбран из -CH2- и -О-;
В и D независимо выбраны из водорода, галогена, C1-4 галогеналкила и C1-4 галогеналкокси;
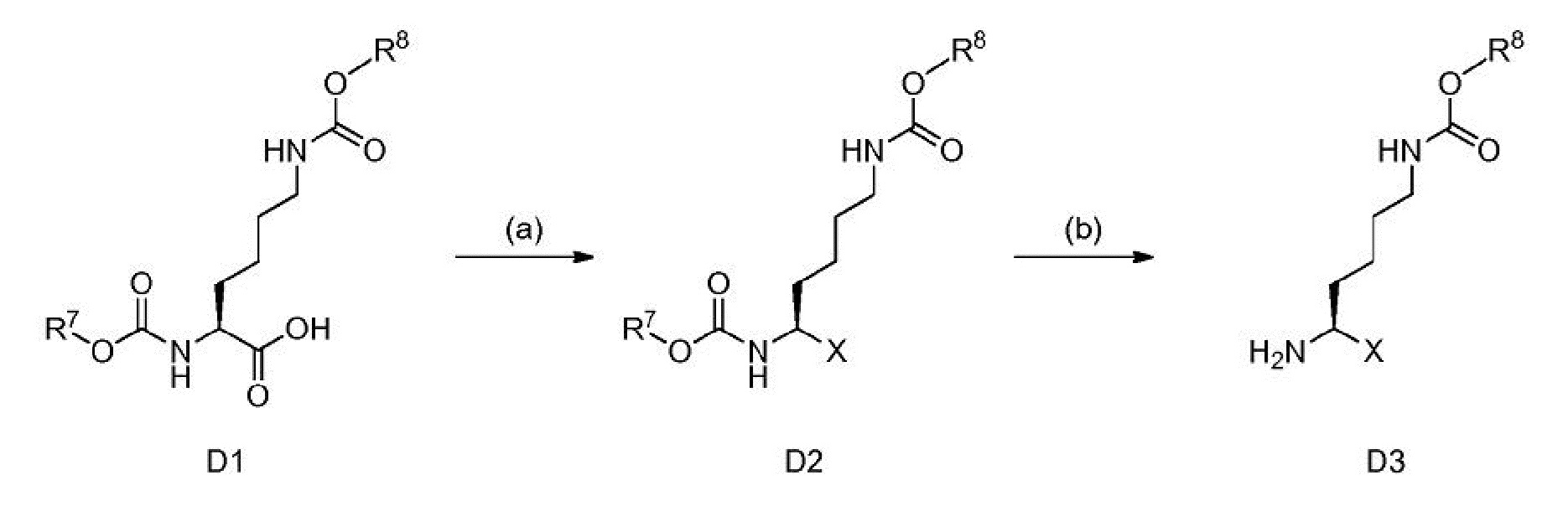
R1 выбран из водорода и защитной группы аминогруппы;
R2 представляет собой водород; и
R3 выбран из C6-10 арила, 5-12-членного гетероарила, C1-8 алкила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила, L R5 и OR6, где
L выбран из -О-, -NR-, C1-4 алкилена и 2-4-членного гетероалкилена, где R выбран из водорода и C1-8 алкила,
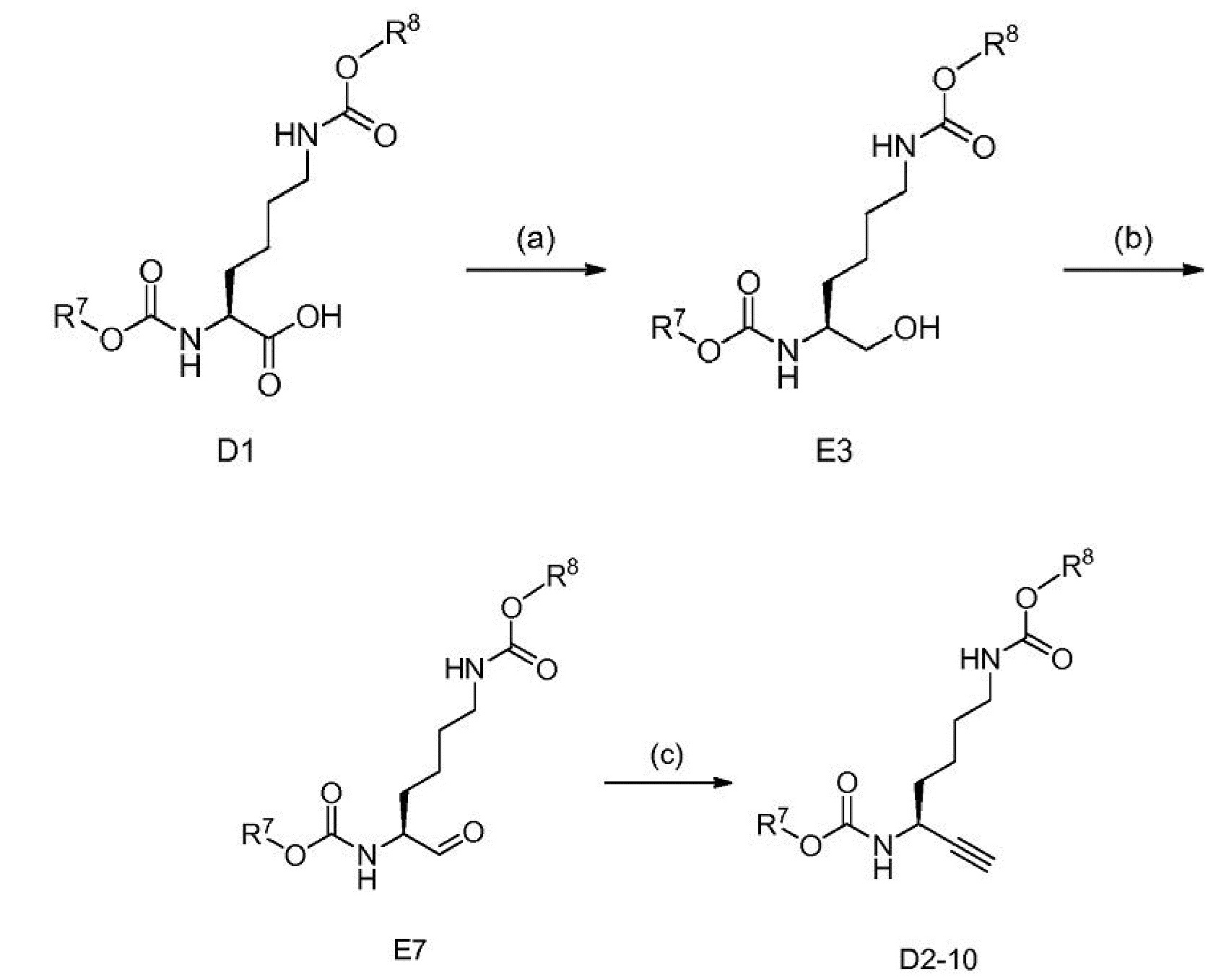
R5 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила и 5-12-членного насыщенного гетероциклила, и
-OR6 и карбонил, к которому он присоединен, образуют защитную группу аминогруппы, и
где R3 является необязательно замещенным одним или несколькокими заместителями, выбранными из галогена, CN, NO2, N3, OH, Ra, Rb, ORa, ORb, (CH2)kC(O)Rc, NRd(CH2)uC(O)Rc, O(CH2)uC(O)Rc, (CH2)kCONRdRd, (CH2)kNRdC(O)Rc, NRd(CH2)uCONRdRd, NRd(CH2)uNRdC(O)Rc, O(CH2)uCONRdRd, O(CH2)uNRdC(O)Rc, (CH2)kS(O)2NRdRd, (CH2)kNRdS(O)2Rc, (CH2)kS(O)2Rc, (CH2)kS(O)Rc, (CH2)kSRd, NRd(CH2)uS(O)2NRdRd, NRd(CH2)uNRdS(O)2Rc, NRd(CH2)uS(O)2Rc, NRd(CH2)uS(O)Rc, NRd(CH2)uSRd, O(CH2)uS(O)2NRdRd, O(CH2)uNRdS(O)2Rc, O(CH2)uS(O)2Rc, O(CH2)uS(O)Rc и O(CH2)uSRc, где:
каждый Raнезависимо выбран из C1-4 алкила и C1-4 галогеналкила,
каждый Rb независимо выбран из C3-6 циклоалкила, C3-6 галогенциклоалкила, C6-10 арила, 5-12-членного гетероарила и 5-12-членного насыщенного гетероциклила,
каждый Rc независимо выбран из -ОН, C1-8 алкила, C1-8 галогеналкила, C3-8 циклоалкила, C3-8 галогенциклоалкила, C6-10 арила, (C6-10 арил)-(C1-8 алкила), 5-12-членного гетероарила и 5-12-членного насыщенного гетероциклила,
каждый Rd независимо выбран из водорода и C1-8 алкила,
каждый подстрочный индекс k независимо выбран из 0, 1, 2, 3, 4, 5 и 6,
каждый подстрочный индекс u независимо выбран из 1, 2, 3, 4, 5 и 6; и
R4 выбран из водорода, галогена, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила и C1-4 галогеналкокси;
при условии, что когда Z представляет собой бензотиазол-2-илкарбонил, А представляет собой -CH2-, а B, D и R1 представляют собой водород, тогда R3 не является бензилоксигруппой, замещенной бензилоксигруппой или 1-(3-фенилпропаноил)пиперидин-3-илом, и
при условии, что когда Z представляет собой феноксиметилкарбонил или замещенный феноксиметилкарбонил, А представляет собой -CH2-, а В и D представляют собой водород, тогда R3 не является (2-фенил)этилом или замещенным (2-фенил)этилом.
[0069] В некоторых вариантах осуществления настоящего изобретения структура соединения формулы I соответствует формуле Iа:

[0070] В некоторых вариантах осуществления настоящего изобретения структура соединения формулы I соответствует структуре соединения формулы Ib:
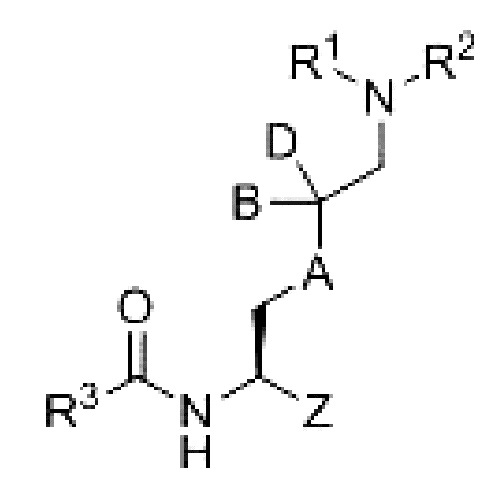
или его фармацевтически приемлемой соли, где
В и D независимо выбраны из водорода, галогена, галогенметила и галогенметокси.
[0071] В некоторых вариантах осуществления изобретение относится к соединениям формулы I, формулы Iа или формулы Ib и их фармацевтически приемлемым солям, где Z выбран из бензотиазол-2-илкарбонила; тиазол-2-илкарбонила; оксазол-2-илкарбонила; бензоксазол-2-илкарбонила; пиридин-2-илкарбонила; пиримидин-4-илкарбонила; пиримидин-2-илкарбонила; изоксазол-5-илкарбонила; изоксазол-3-илкарбонила; 1,2,4-оксадиазол-3-илкарбонила; 1,2,4-оксадиазол-5-илкарбонила; циано; этинила; фторметилкарбонила; ацилоксиметилкарбонила; арилоксиметилкарбонила; алкилсульфонилвинила; и арилсульфонилвинила; каждый из которых является необязательно замещенным одним или несколькими заместителями, выбранными из C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, галогена и -N3.
[0072] В некоторых вариантах осуществления настоящего изобретения Z выбран из бензотиазол-2-илкарбонила, галогенамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила.
[0073] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Iа, которые описаны выше, и их фармацевтически приемлемым солям, где Z выбран из бензотиазол-2-илкарбонила; тиазол-2-илкарбонила; оксазол-2-илкарбонила; бензоксазол-2-илкарбонила; пиридин-2-илкарбонила; пиримидин-4-илкарбонила; пиримидин-2-илкарбонила; изоксазол-5-илкарбонила; изоксазол-3-илкарбонила; 1,2,4-оксадиазол-3-илкарбонила; 1,2,4-оксадиазол-5-илкарбонила; циано; этинила; фторметилкарбонила; ацилоксиметилкарбонила; арилоксиметилкарбонила; алкилсульфонилвинила; и арилсульфонилвинила; где Z является необязательно замещенным одним или несколькими заместителями, выбранными из C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, галогена и N3; и где R3 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и -L-R5. В некоторых таких вариантах осуществления настоящего изобретения R1 и R2 представляют собой Н; В и D независимо выбраны из водорода и фтора; А представляет собой -CH2-; и R4 выбран из водорода и C1-4 алкила.
[0074] В некоторых вариантах осуществления изобретение относится к соединениям формулы Ib, которые описаны выше, и их фармацевтически приемлемым солям, где Z выбран из бензотиазол-2-илкарбонила; тиазол-2-илкарбонила; оксазол-2-илкарбонила; бензоксазол-2-илкарбонила; пиридин-2-илкарбонила; пиримидин-4-илкарбонила; пиримидин-2-илкарбонила; изоксазол-5-илкарбонила; изоксазол-3-илкарбонила; 1,2,4-оксадиазол-3-илкарбонила; 1,2,4-оксадиазол-5-илкарбонила; циано; этинила; фторметилкарбонила; ацилоксиметилкарбонила; арилоксиметилкарбонила; алкилсульфонилвинила; и арилсульфонилвинила; где Z является необязательно замещенным одним или несколькими заместителями, выбранными из C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, галогена и -N3; и где R3 выбран из C6-10 арила, 5 12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и -L-R5. В некоторых таких вариантах осуществления настоящего изобретения R1 и R2 представляют собой Н; В и D независимо выбраны из водорода и фтора; и А представляет собой -CH2-.
[0075] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Iа, которые описаны выше, и их фармацевтически приемлемым солям, где Z выбран из бензотиазол-2-илкарбонила, галогензамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила; и где R3 выбран из C6-10 арила, 5 12-членного гетероарила, C3-8 циклоалкила, 5 12-членного насыщенного гетероциклила и -L-R5. В некоторых таких вариантах осуществления настоящего изобретения R1 и R2 представляют собой Н; В и D независимо выбраны из водорода и фтора; А представляет собой -CH2-; и R4 выбран из водорода и C1-4 алкила.
[0076] В некоторых вариантах осуществления изобретение относится к соединениям формулы Ib, которая описана выше, и их фармацевтически приемлемым солям, где Z выбран из бензотиазол-2-илкарбонила, галогензамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила; и где R3 выбран из C6-10 арила, 5 12-членного гетероарила, C3-8 циклоалкила, 5 12-членного насыщенного гетероциклила и -L-R5. В некоторых таких вариантах осуществления R1 и R2 представляют собой Н; В и D независимо выбраны из водорода и фтора; и А представляет собой CH2.
[0077] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Iа, которые описаны выше, и их фармацевтически приемлемым солям, где Z выбран из бензотиазол-2-илкарбонила; тиазол-2-илкарбонила; оксазол-2-илкарбонила; бензоксазол-2-илкарбонила; пиридин-2-илкарбонила; пиримидин-4-илкарбонила; пиримидин-2-илкарбонила; изоксазол-5-илкарбонила; изоксазол-3-илкарбонил; 1,2,4-оксадиазол-3-илкарбонил; 1,2,4-оксадиазол-5-илкарбонила; циано; этинила; фторметилкарбонила; ацилоксиметилкарбонила; арилоксиметилкарбонила; алкилсульфонилвинила; и арилсульфонилвинила; где Z является необязательно замещенным одним или несколькими заместителями, выбранными из C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, галогена и -N3; и где R3 выбран из C6-10 арила, 5 12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и L R5. В некоторых таких вариантах осуществления R1 и R2 представляют собой Н; В и D независимо выбраны из водорода и фтора; и А представляет собой CH2. В некоторых таких вариантах осуществления R4 выбран из водорода и метила.
[0078] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Iа, которые описаны выше, и их фармацевтически приемлемым солям, где Z выбран из бензотиазол-2-илкарбонила, галогензамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила; и где R3 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и L R5. В некоторых таких вариантах осуществления настоящего изобретения R1 и R2 представляют собой Н; В и D независимо выбраны из водорода и фтора; и А представляет собой -CH2-. В некоторых таких вариантах осуществления настоящего изобретения R4 выбран из водорода и метила.
[0079] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Iа, которые описаны выше, и их фармацевтически приемлемым солям, где:
А представляет собой CH2 ;
В и D представляют собой водород;
Z выбран из бензотиазол-2-илкарбонила, галогензамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила;
R1 и R2 представляют собой Н; и
R3 выбран из C6-10 арила, 5 12-членного гетероарила, C3-8 циклоалкила, 5 12-членного насыщенного гетероциклила и L R5. В некоторых таких вариантах осуществления настоящего изобретения R4 представляет собой водород или метил. В некоторых таких вариантах осуществления R4 представляет собой водород.
[0080] В некоторых вариантах осуществления изобретение относится к соединениям формулы Ib, которая описана выше, и их фармацевтически приемлемым солям, где:
А представляет собой -CH2-;
В и D представляют собой водород;
Z выбран из бензотиазол-2-илкарбонила, галогензамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила;
R1 и R2 представляют собой Н; и
R3 выбран из C6-10 арила, 5 12-членного гетероарила, C3-8 циклоалкила, 5 12-членного насыщенного гетероциклила и L R5.
[0081] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Iа, которые описаны выше, и их фармацевтически приемлемым солям, где:
А представляет собой CH2 ;
В представляет собой водород;
D представляет собой фтор;
Z выбран из бензотиазол-2-илкарбонила, галогензамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила;
R1 и R2 представляют собой Н; и
R3 выбран из C6-10 арила, 5 12-членного гетероарила, C3-8 циклоалкила, 5 12-членного насыщенного гетероциклила и L R5. В некоторых таких вариантах осуществления настоящего изобретения R4 представляет собой водород или метил. В некоторых таких вариантах осуществления R4 представляет собой водород.
[0082] В некоторых вариантах осуществления изобретение относится к соединениям формулы Ib, которая описана выше, и их фармацевтически приемлемым солям, где:
А представляет собой CH2 ;
В представляет собой водород;
D представляет собой фтор;
Z выбран из бензотиазол-2-илкарбонила, галогензамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила;
R1 и R2 представляют собой Н; и
R3 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила, 5 12-членного насыщенного гетероциклила и L R5.
[0083] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Iа, которые описаны выше, и их фармацевтически приемлемым солям, где:
В представляет собой водород;
D представляет собой водород или фтор;
Z представляет бензотиазол-2-илкарбонил или галогензамещенный арилоксиметилкарбонил;
R1 и R2 представляют собой Н;
R3 выбран из циклогексила, циклопентила, морфолино, фенила, пиперидинила, пиридинила, тетрагидрофуранила, тетрагидропиранила, 1,2,3,4-тетрагидронафтила и тиазолила, каждый из которых является необязательно замещенным 1-3 заместителями, выбранными из группы, включающей метил, метокси, трифторметил, ацетил и N3; и
R4 представляет собой водород или метил. В некоторых таких вариантах осуществления настоящего изобретения R4 представляет собой водород. В некоторых таких вариантах осуществления настоящего изобретения D и R4 представляют собой водород. В некоторых таких вариантах осуществления D и R4 представляют собой водород, и Z представляет собой (2,3,5,6-тетрафторфенокси)метилкарбонил.
[0084] В некоторых вариантах осуществления изобретение относится к соединениям формулы Ib, которые описаны выше, и их фармацевтически приемлемым солям, где:
В представляет собой водород;
D представляет собой водород или фтор,
Z представляет бензотиазол-2-илкарбонил или галогензамещенный арилоксиметилкарбонил;
R1 и R2 представляют собой Н; и
R3 выбран из циклогексила, циклопентила, морфолино, фенила, пиперидинила, пиридинила, тетрагидрофуранила, тетрагидропиранила, 1,2,3,4-тетрагидронафтила и тиазолила,
каждый из которых является необязательно замещенным 1-3 заместителями, выбранными из группы, включающей метил, метокси, трифторметил, ацетил и -N3. В некоторых таких вариантах осуществления настоящего изобретения D представляет собой водород. В некоторых таких вариантах осуществления настоящего изобретения D представляет собой водород, и Z представляет собой (2,3,5,6-тетрафторфенокси)метилкарбонил.
[0085] В некоторых вариантах осуществления настоящего изобретения структура соединения формулы I соответствует структуре соединения формулы Ic:
или его фармацевтически приемлемой соли,
где R3 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и L R5,
где L представляет собой C1-4 алкилен.
[0086] В некоторых вариантах осуществления изобретение относится к соединению формулы Ic или его фармацевтически приемлемой соли, где R3 выбран из циклогексила, циклопентила, морфолина, фенила, пиперидинила, пиридинила, тетрагидрофуранила, тетрагидропиранила, 1,2,3,4-тетрагидронафтила и тиазолила, каждый из которых является необязательно замещенным 1-3 заместителями, выбранными из метила, метокси, трифторметила, ацетила и N3.
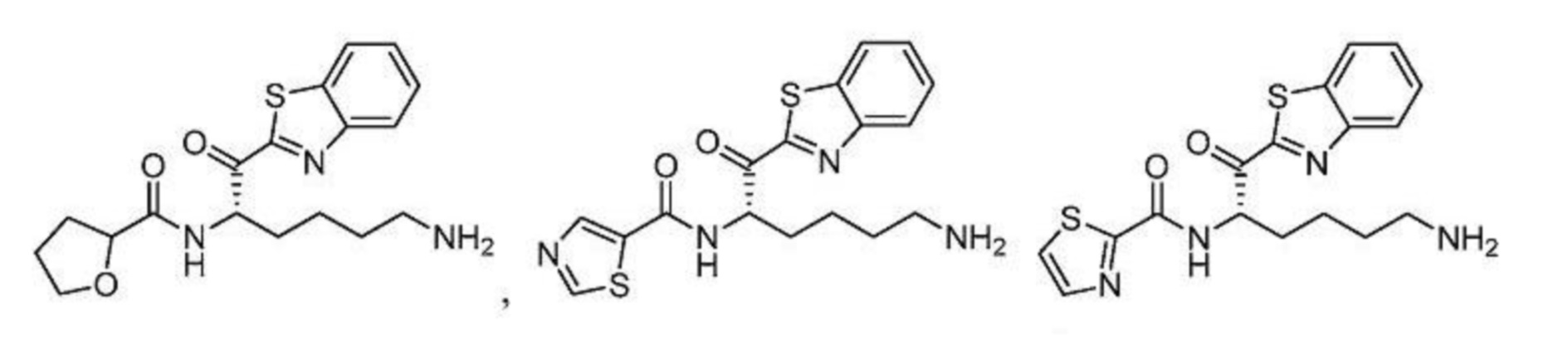
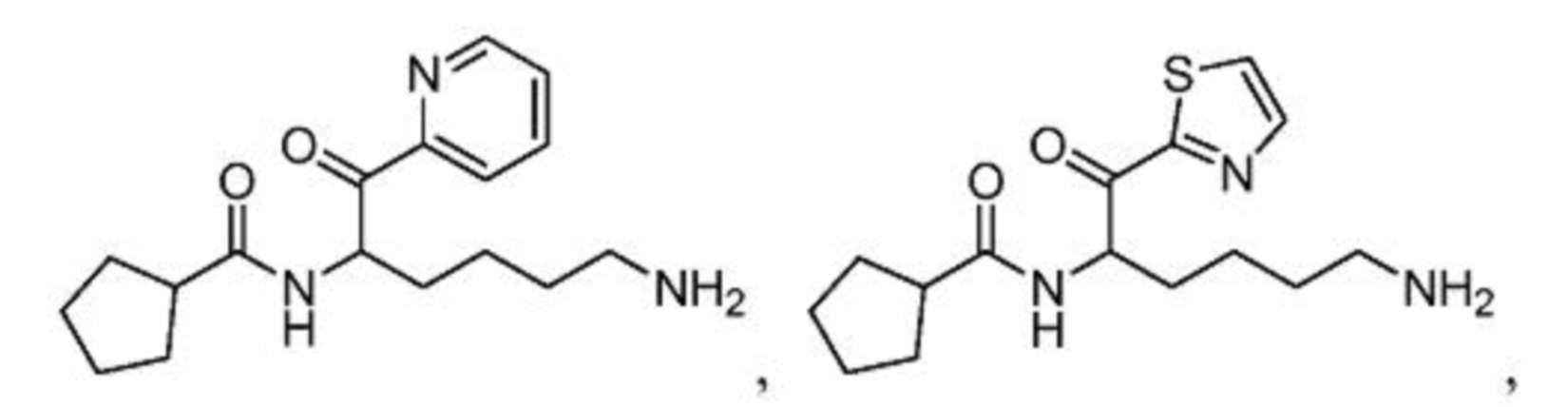
[0087] В некоторых вариантах осуществления настоящего изобретения соединение формулы I, формулы Iа, формулы Ib или формулы Ic выбрано из



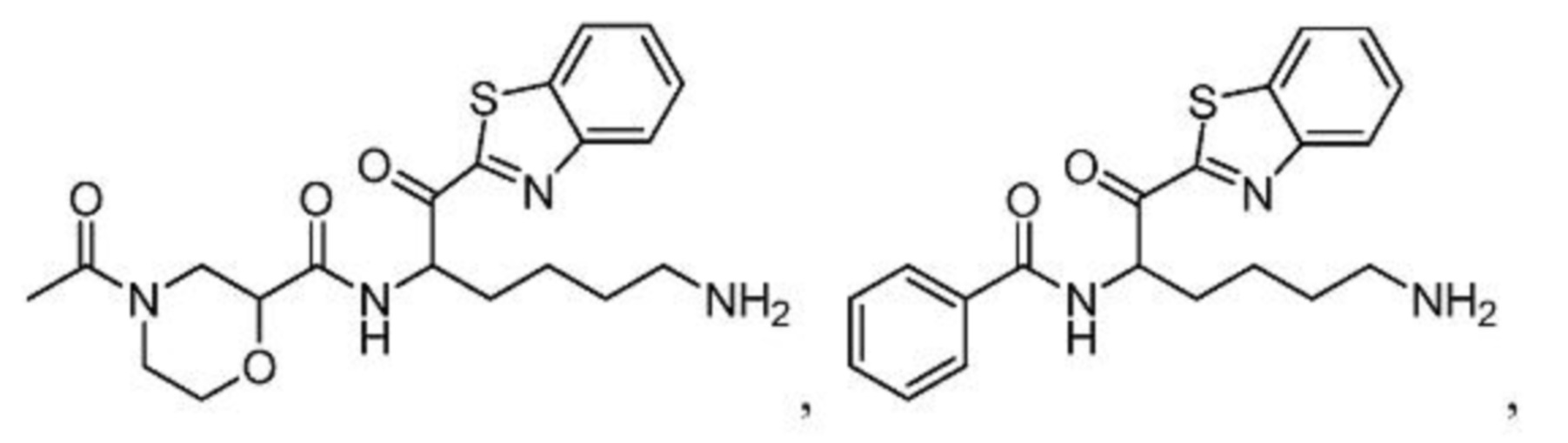
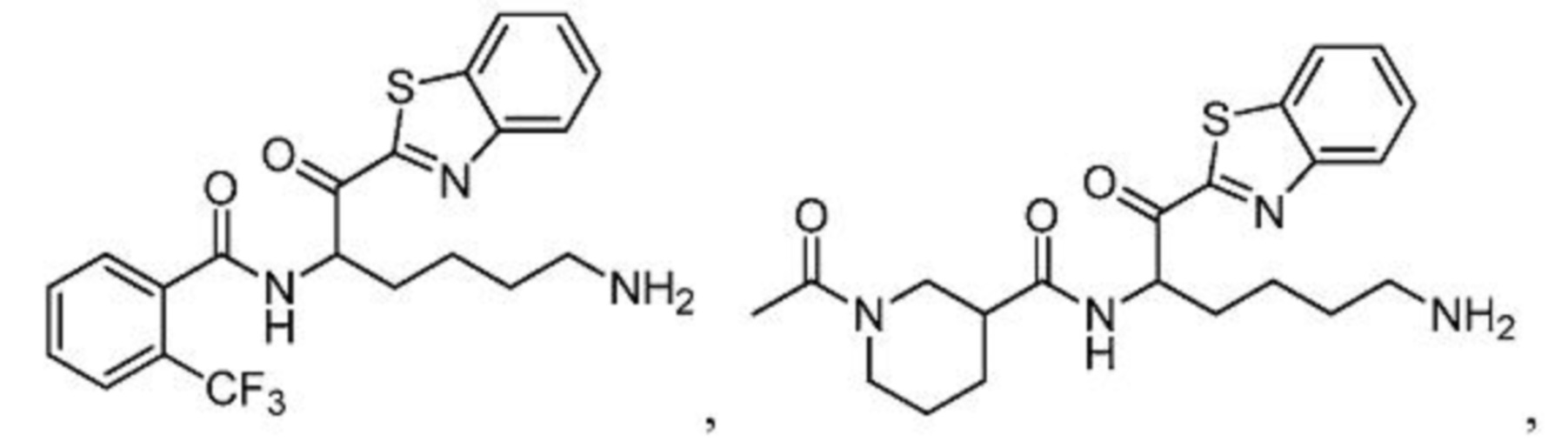


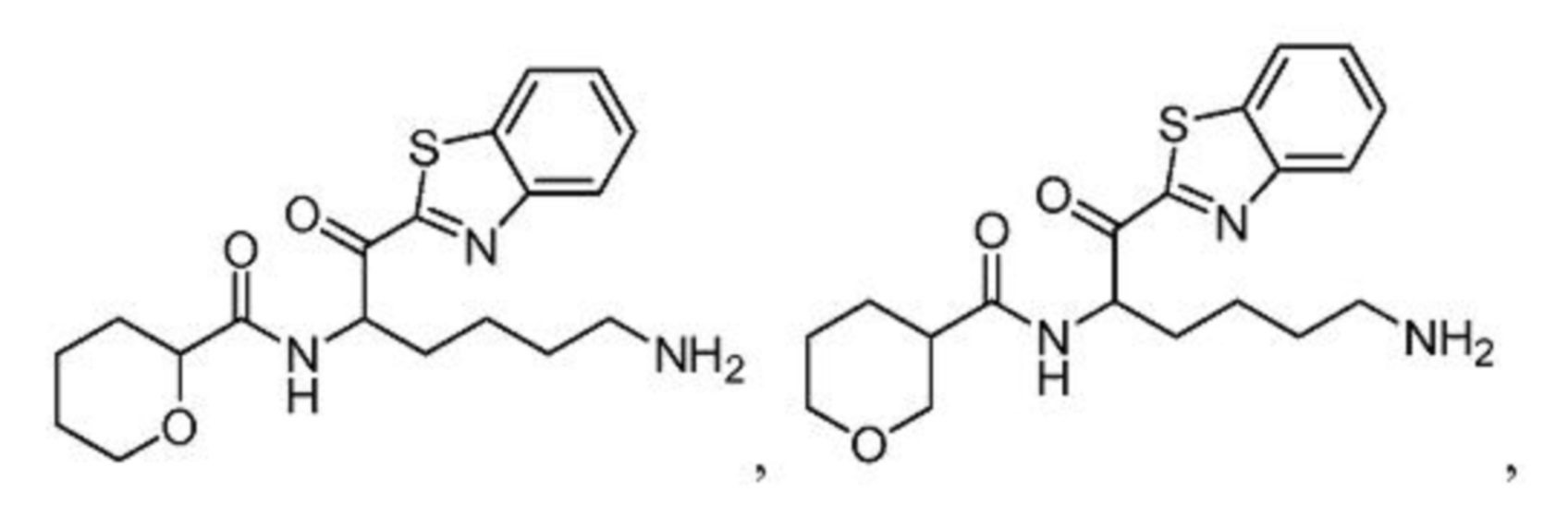

и их фармацевтически приемлемых солей.
[0088] В некоторых вариантах осуществления настоящего изобретения соединение формулы I, формулы Iа, формулы Ib, или формулы Ic выбрано из



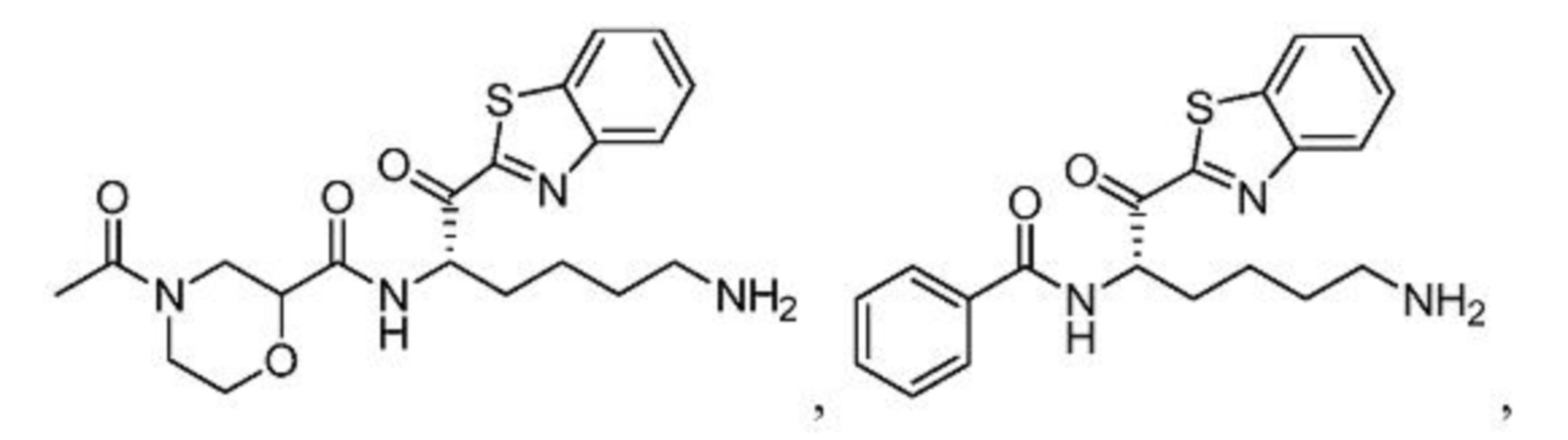


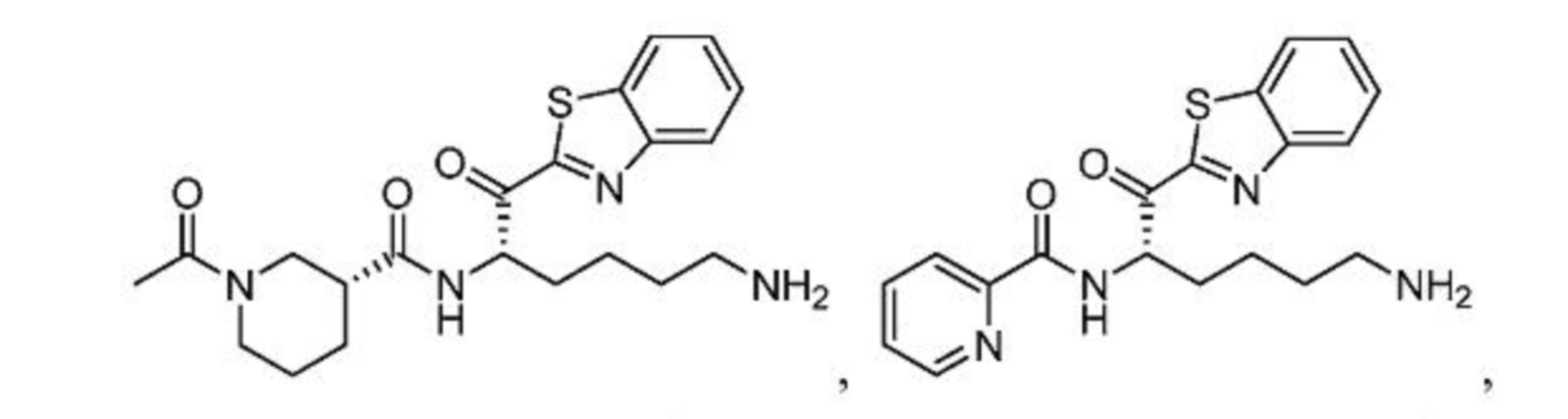

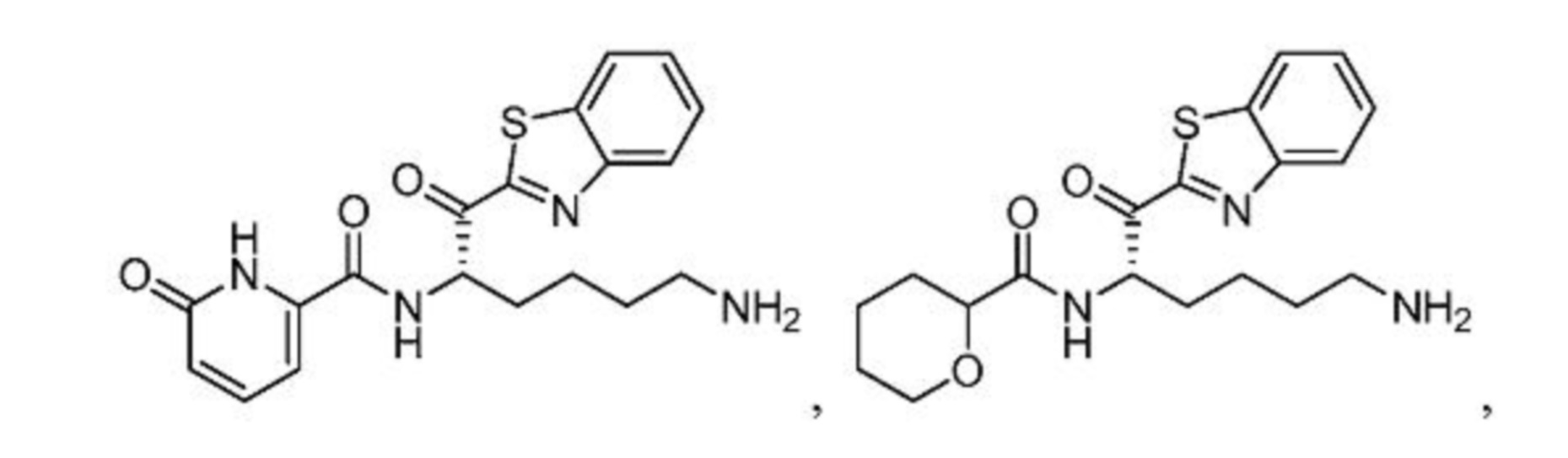

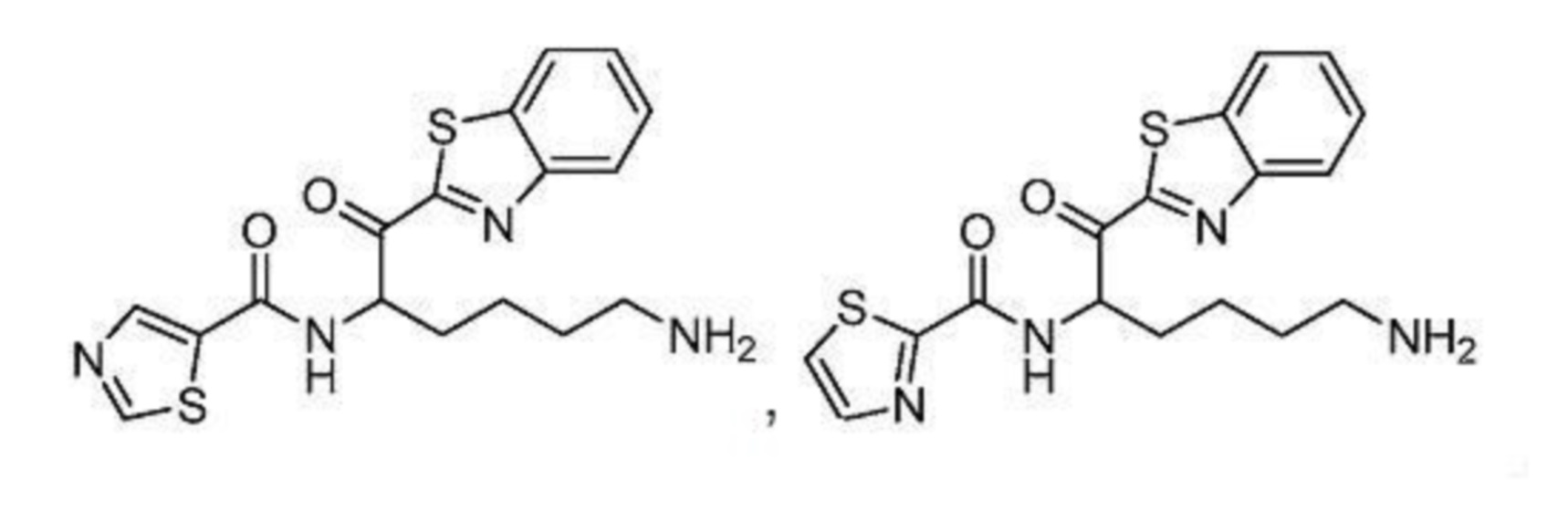
и их фармацевтически приемлемых солей.
[0089] В некоторых вариантах осуществления настоящее изобретение относится к соединениям формулы I, формулы Iа или формулы Ib, где А представляет собой -О-, включая соединения формулы C1:

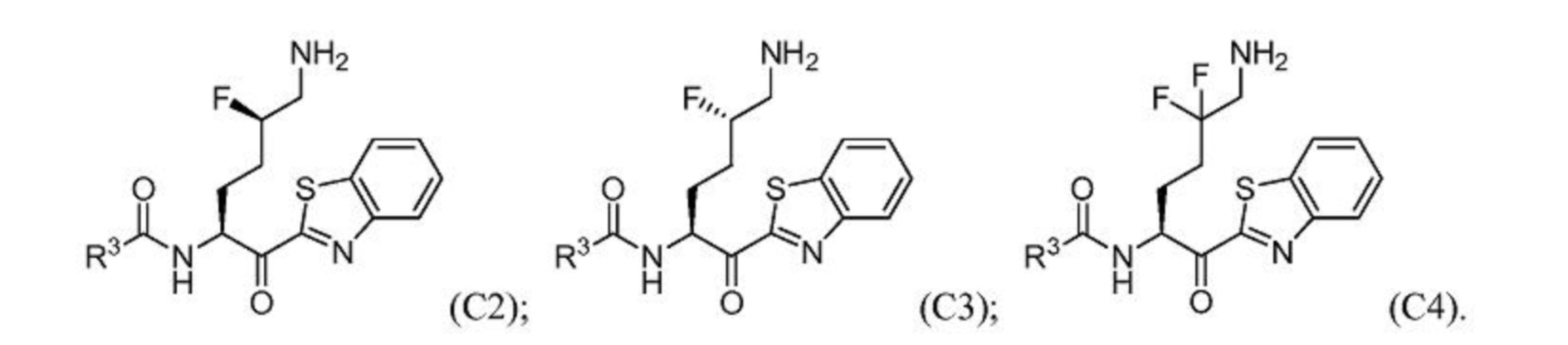
[0090] В некоторых вариантах осуществления изобретение относится к соединениям формулы I, формулы Iа или формулы Ib, где В представляет собой галоген; D представляет собой галоген; или В и D представляют собой галоген; включая соединения формулы С2, формулы С3 и формулы C4:
[0091] В некоторых вариантах осуществления изобретение относится к соединениям формулы I, формулы Iа или формулы 1b, где Z выбран из тиазол-2-илкарбонила; оксазол-2-илкарбонила; бензоксазол-2-илкарбонила; пиридин-2-илкарбонила; пиримидин-4-илкарбонила; пиримидин-2-илкарбонила; изоксазол-5-илкарбонила; изоксазол-3-илкарбонила; 1,2,4-оксадиазол-3-илкарбонила; 1,2,4-оксадиазол-5-илкарбонила; малеимидила; пиридинилдисульфанила (включая пиридин-2-илдисульфанил); циано; этинила; фторметилкарбонила; ацилоксиметилкарбонила; арилоксиметилкарбонила; алкилсульфонилвинила; и арилсульфонил-винила.
[0092] В некоторых вариантах осуществления изобретение относится к соединениям формулы I, формулы Iа или формулы Ib, где Z выбран из тиазол-2-илкарбонила; пиридин-2-илкарбонила; циано; этинила; фторметилкарбонила; и 2,3,5,6-тетрафторфеноксметилкарбонила; включая соединения формулы B1 формулы B2, формулы B3, формулы B4, формулы B5 и формулы B6:


[0093] В некоторых вариантах осуществления изобретение относится к соединению структуры формулы Id:
или его фармацевтически приемлемой соли,
где R3 выбран из С6-10 арила, 5-12-членного гетероарила, циклоалкила, 5 12-членного насыщенного гетероцикла и L R5,
где L представляет собой C1-4 алкилен.
[0094] В некоторых вариантах осуществления изобретение относится к соединению формулы Id или его фармацевтически приемлемой соли, где R3 выбран из циклогексила, циклопентила, морфолина, фенила, пиперидинила, пиридинила, тетрагидрофуранила, тетрагидропиранила, 1,2,3,4-тетрагидронафтила и тиазолил, каждый из которых необязательно замещен 1-3 заместителями, выбранными из метила, метокси, трифторметила, ацетила и -N3. В некоторых таких вариантах осуществления настоящего изобретения R3 представляет собой циклопентил.
[0095] В некоторых вариантах осуществления настоящего изобретения соединение формулы Id выбрано из
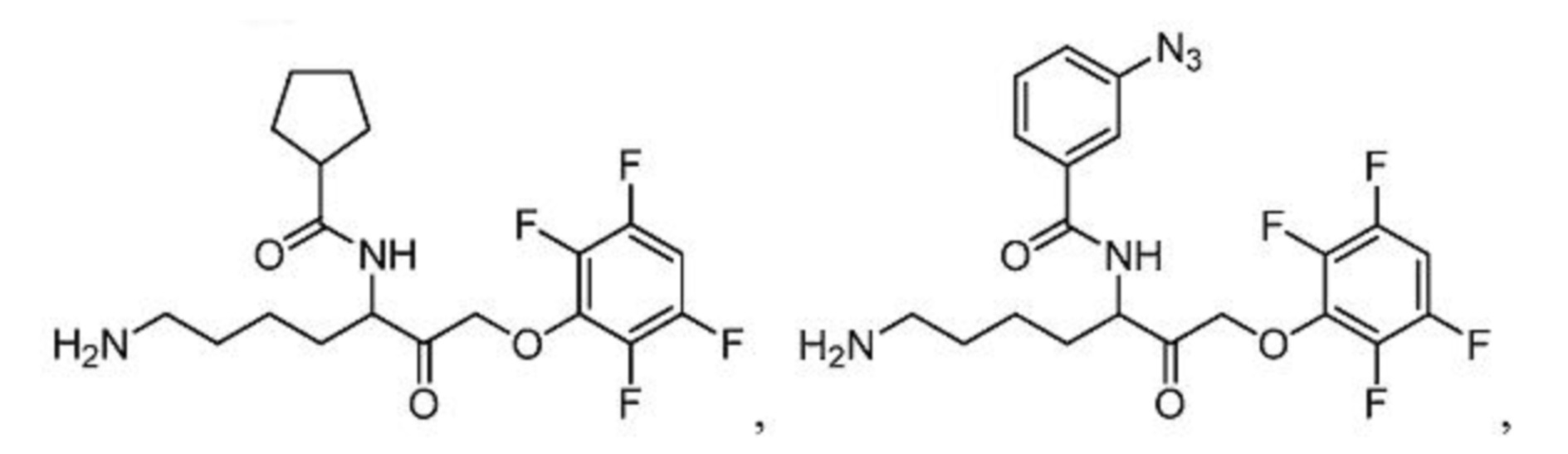
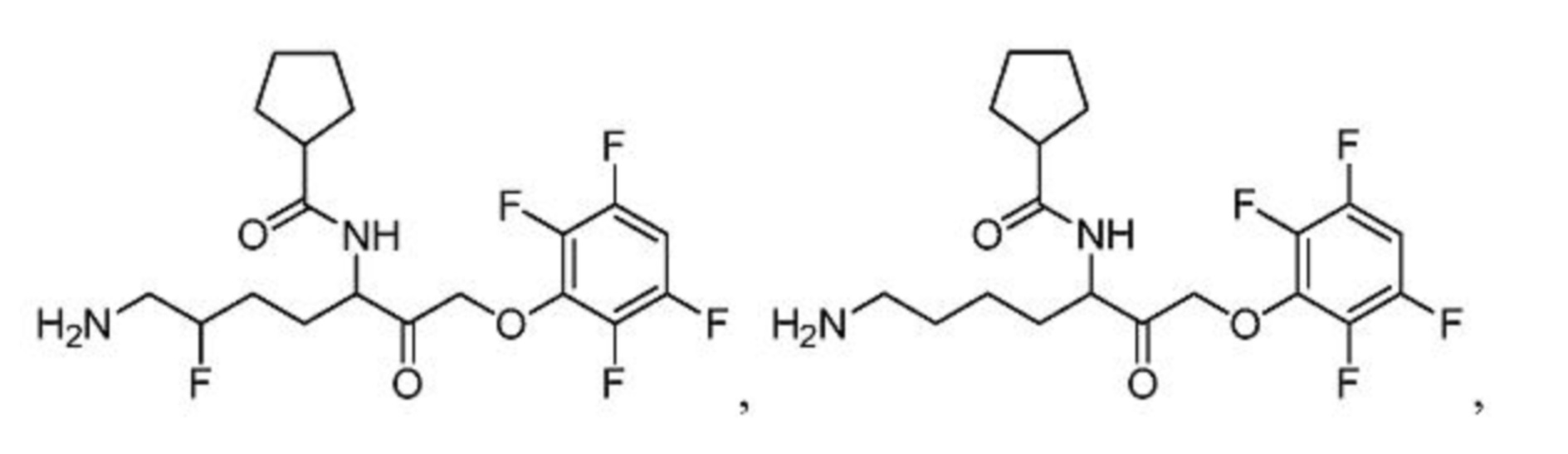
и их фармацевтически приемлемых солей.
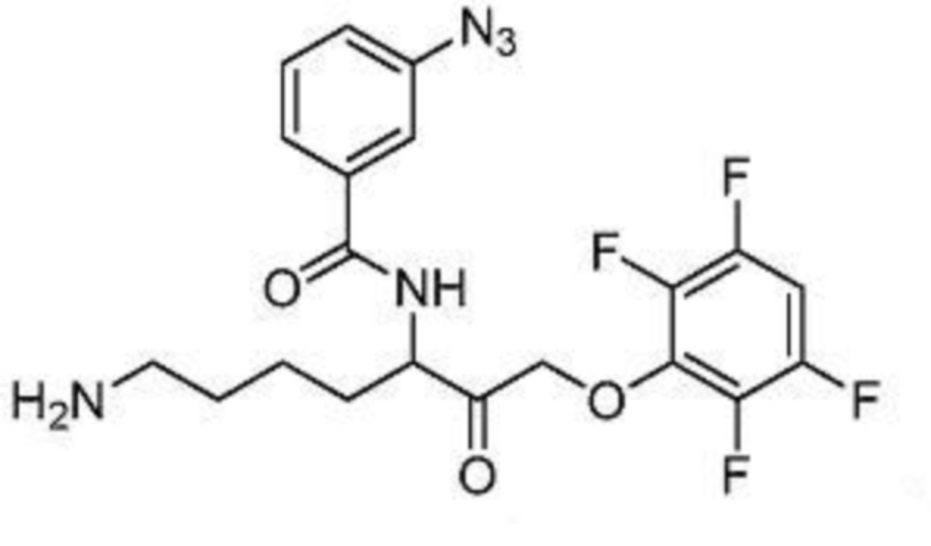
[0096] В некоторых вариантах осуществления настоящего изобретения соединение формулы Id выбрано из

и их фармацевтически приемлемых солей.
[0097] В некоторых вариантах осуществления настоящего изобретения соединение формулы Id выбрано из

и их фармацевтически приемлемых солей.
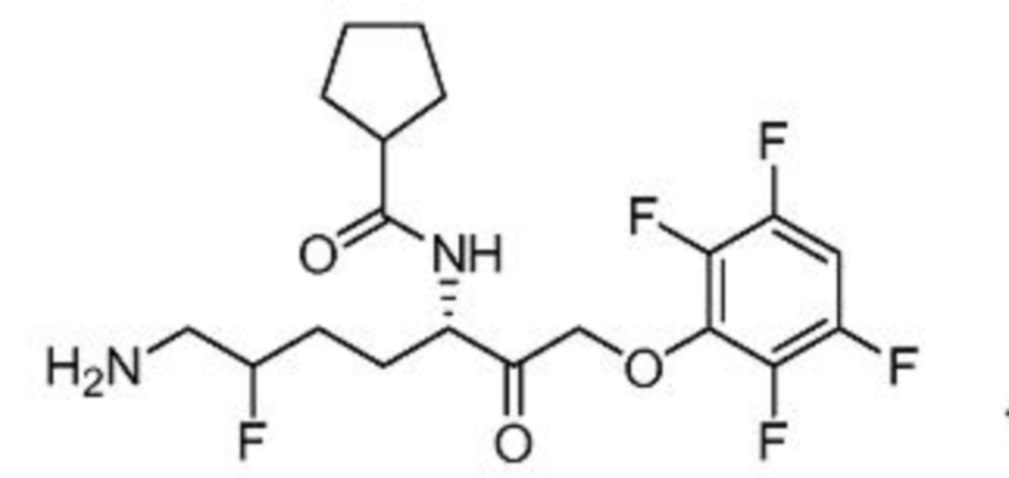
[0098] В некоторых вариантах осуществления настоящего изобретения соединение формулы I, формулы Iа или формулы Ib выбрано из
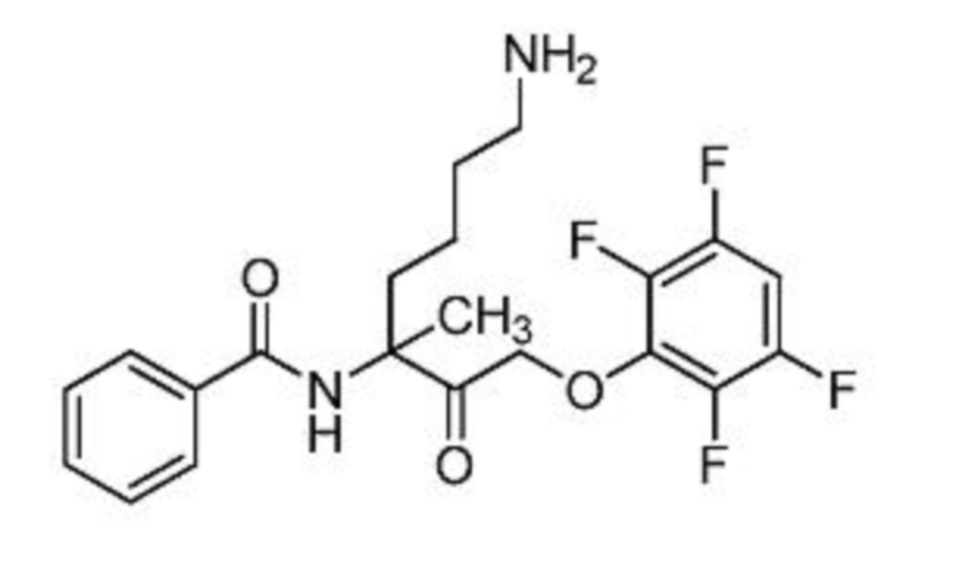
и их фармацевтически приемлемых солей.
[0099] В некоторых вариантах осуществления изобретение относится к соединениям формулы I, формулы Iа или формулы Ib, где Z выбран из пиридин-2-илкарбонила и тиазол-2-илкарбонила, и R3 выбран из C6-10 арила и C3-8 циклоалкила. В некоторых таких вариантах осуществления настоящего изобретения соединение выбрано из

и их фармацевтически приемлемых солей.
[0100] В некоторых вариантах осуществления изобретение относится к соединениям формулы I или формулы Ia, где R4 выбран из C1-4 алкила и C1-4 галгеналкила. В некоторых таких вариантах осуществления изобретения соединение представляет собой

или его фармацевтически приемлемую соль.
[0101] В некоторых вариантах осуществления изобретение относится к соединению формулы I, формулы Ia, формулы Ib, формулы Ic или формулы Id или их фармацевтически приемлемым солям, где R3 выбран из циклогексила; 1-метилциклогексила; 1-метоксициклогексила; циклопентила; морфолин-2-ила; 4-ацетилморфолин-2-ила; фенила; 2-трифторметилфенила; 3-изидофенила; пиперидин-3-ила; 1-ацетилпиперидин-3-ила; пиридин-2-ила; пиридин-3-ила; пиридин-4-ила; 6-оксо-1,6-дигидропиридин-2-ила; тетрагидрофуран-2-ила; тетрагидро-2Н-пиран-2-ила; тетрагидро-2H-пиран-3-ила; тетрагидро-2H-пиран-4-ила; 1,2,3,4-тетрагидронафт-1-ила; 1,2,3,4-тетрагидронафт-2-ила; тиазол-5-ила и тиазол-2-ила.
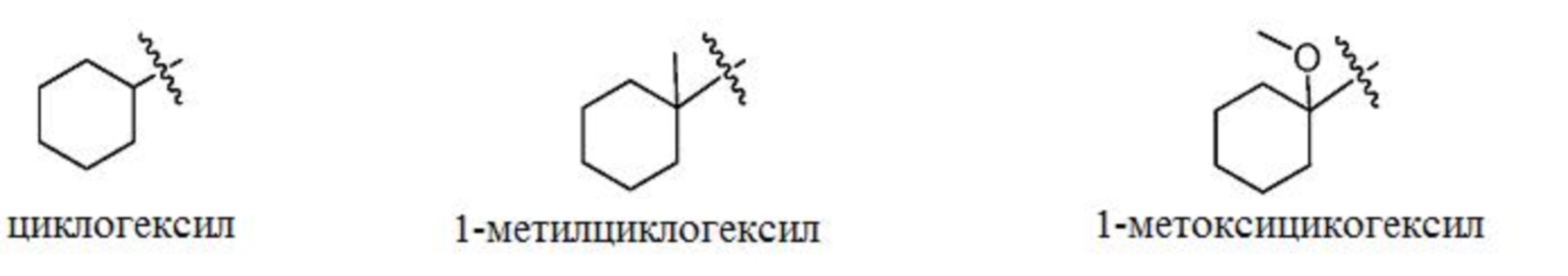
[0102] В некоторых вариантах осуществления изобретение относится к соединению формулы I, формулы Ia, формулы Ib, формулы Ic или формулы Id или их фармацевтически приемлемым солям, где R3 выбран из цикогексила; 1-метилциклогексила; 1-метоксициклогексила; циклопентила; 1,2,3,4-тетрагидронафт-1-ила; и 1,2,3,4-тетрагидронафт-2-ила; радикалы которых представлены ниже. В некоторых таких вариантах осуществления настоящего изобретения R3 обозначает циклопентил.

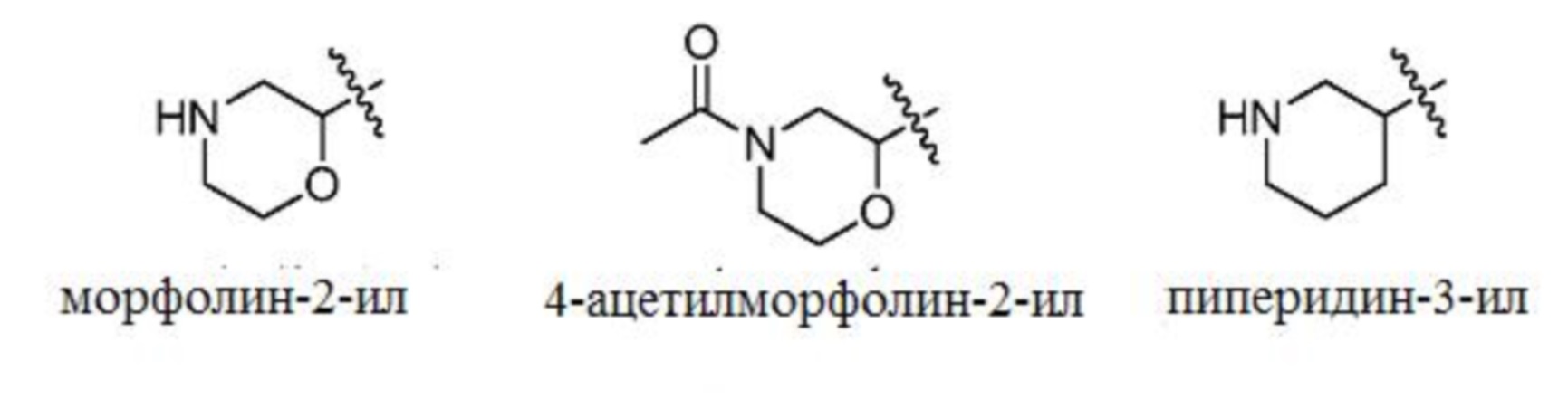
[0103] В некоторых вариантах осуществления изобретение относится к соединению формулы I, формулы Ia, формулы Ib, формулы Ic или формулы Id или их фармацевтически приемлемым солям, где R3 выбран из морфолин-2-ила; 4-ацетилфорфолин-2-ила; пиперидин-3-ила; 1-ацетилпиперидин-3-ила; тетрагидрофуран-2-ила; тетрагидро-2Н-пиран-2-ила; тетрагидро-2H-пиран-3-ила; и тетрагидро-2Н-пиран-4-ила; радикалы которых представлены ниже.


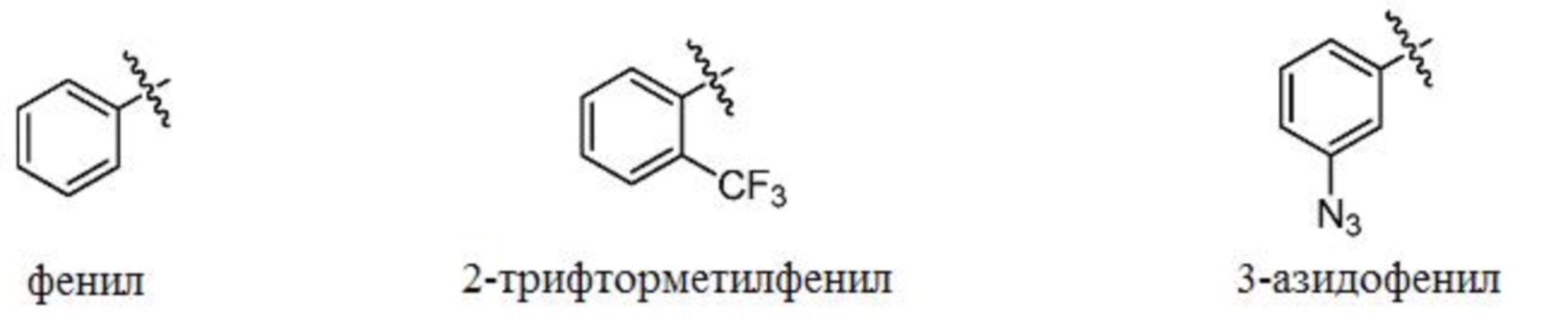
[0104] В некоторых вариантах осуществления изобретение относится к соединению формулы I, формулы Ia, формулы Ib, формулы Ic или формулы Id или их фармацевтически приемлемым солям, где R3 выбран из фенила; 2-трифторметилфенила; 3-азидофенила; пиридин-2-ила; пиридин-3-ила; пиридин-4-ила; 6-оксо-1,6-дигидропиридин-2-ила; тиазол-5-ила; и тиазол-2-ила; радикалы которых представлены ниже.

[0105] Соединения, описанные в данном документе, и способы их применения охватывают получение и применение терапевтически активных энантиомеров или диастереоизомеров описанных соединений. Все такие энантиомеры и диастереоизомеры этих соединений включены в объем настоящего изобретения. Такие соединения могут быть использованы в виде смесей (например, рацемических смесей) или в виде отдельных энантиомеров или диастереоизомеров.
[0106] Соединения по настоящему изобретению могут быть получены таким образом, чтобы они содержали радионуклиды для применения в областях диагностической визуализации, таких как ПЭТ и ОФЭКТ. Например, ингибиторы Kgp, которые описаны в данном изобретении, могут быть получены таким образом, чтобы они содержали один или несколько радионуклидов, выбранных из кислорода-15 (15О), азота-13 (13N), углерода-11 (11С), йода-131 (131I) и фтора-18 (18F). Такие радиоактивно меченые соединения могут быть использованы для ПЭТ визуализации. Соединения по настоящему изобретению также могут быть получены в дейтерированной форме (то есть, в форме, содержащей один или несколько атомов дейтерия,2Н, вместо одного или нескольких атомов водорода), тритированной форме (то есть в форме, содержащей один или несколько атомов трития,3H, вместо одного или нескольких атомов водорода) или14С-меченой форме (то есть, в форме, содержащей один или несколько14С атомов вместо одного или нескольких атомов углерода).
[0107] В других вариантах осуществления изобретение относится к соединениям формулы Ie

и их фармацевтически приемлемым солям, где
Z представляет собой тиол-реакционноспособную группу или защищенную тиол-реакционноспособную группу;
А выбран из CH2 и -О-;
В и D независимо выбраны из водорода, галогена, C1-4 галогеналкила и C1-4 галогеналкокси;
R1 выбран из водорода и защитной группы аминогруппы;
R2 представляет собой водород; и
R3 выбран из C6-10 арила, 5-12-членного гетероарила, C1-8 алкила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила, L R5 и -OR6, где
L выбран из -О-, -NR-, C1-4 алкилена и 2-4-членного гетероалкилена, где R выбран из водорода и C1-8 алкила,
R5 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила и 5-12-членного насыщенного гетероциклила, и
-OR6 и карбонил, к которому он присоединен, образуют защитную группу аминогруппы, и
где R3 является необязательно замещенным одним или несколькими заместителями, выбранными из галогена, CN, NO2, N3, OH, Ra, Rb, ORa, ORb, (CH2)kC(O)Rc, NRd(CH2)uC(O)Rc, O(CH2)uC(O)Rc, (CH2)kCONRdRd, (CH2)kNRdC(O)Rc, NRd(CH2)uCONRdRd, NRd(CH2)uNRdC(O)Rc, O(CH2)uCONRdRd, O(CH2)uNRdC(O)Rc, (CH2)kS(O)2NRdRd, (CH2)kNRdS(O)2Rc, (CH2)kS(O)2Rc, (CH2)kS(O)Rc, (CH2)kSRd, NRd(CH2)uS(O)2NRdRd, NRd(CH2)uNRdS(O)2Rc, NRd(CH2)uS(O)2Rc, NRd(CH2)uS(O)Rc, NRd(CH2)uSRd, O(CH2)uS(O)2NRdRd, O(CH2)uNRdS(O)2Rc, O(CH2)uS(O)2Rc, O(CH2)uS(O)Rc и O(CH2)uSRc, где:
каждый Raнезависимо выбран из C1-4 алкила и C1-4 галогеналкила,
каждый Rb независимо выбран из C3-6 циклоалкила, C3-6 галогенциклоалкила, C6-10 арила, 5-12-членного гетероарила и 5-12-членного насыщенного гетероциклила,
каждый Rc независимо выбран из -ОН, C1-8 алкила, C1-8 галогеналкила, C3-8 циклоалкила, C3-8 галогенциклоалкила, C6-10 арила, (C6-10 арил)-(C1-8 алкила), 5-12-членного гетероарила и 5-12-членного насыщенного гетероциклила,
каждый Rd независимо выбран из водорода и C1-8 алкила,
каждый подстрочный индекс k независимо выбран из 0, 1, 2, 3, 4, 5 и 6, и
каждый подстрочный индекс u независимо выбран из 1, 2, 3, 4, 5 и 6; и
R4 выбран из водорода, галогена, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила и C1-4 галогеналкокси.
[0108] В некоторых вариантах осуществления настоящего изобретения структура соединения формулы Ie соответствует формуле If

[0109] Следует понимать, что соединения по настоящему изобретению не включают [4-амино-1(S)-(бензотиазол-2-карбонилбутил]амид 1-(3-фенилпропионил)пиперидин-3(R,S)-карбоновой кислоты (т.е. A71561).
[0110] Соединения по настоящему изобретению являются высокоэффектвными ингибиторами Kgp, обычно проявляющие значения Ki Kgp и IC50 Kgp менее 1 мкΜ.
[0111] Термин «Ki» относится к константе ингибирования. Значение Ki для конкретного тестируемого соединения может количественно определяться следующим образом. Пятьдесят микролитров (мкл) фермента, такого как Kgp (1 нМ в 50 мМ бис-трис-пропана [рН 8,0], содержащего 1% [об./об.] Troton Х-100 и 5 мМ 2-меркаптоэтанола) добавляют в 1-11 колонки 96-луночного планшета, 100 мкл добавляют к колонку 12. Два мкл раствора тестируемого соединения (100 мкл в 100% ДМСО) добавляют в колонку 12 и перемешивают образец с помощью пипетки три раза. После этого проводят двойное разбавление на планшете с помощью последовательного переноса в соседние лунки. Во все лунки добавляют 50 мкл сукцинил-Ala-Phe-Lys-(7-амидо-4-метилкумарина) ("AMC;" 40 мкΜ в буфере) и перемешивают содержимое. Реакцию контролируют по AMC флуоресценции в течение 15 минут при 25°C, а кривые прогресса автоматически преобразуют в дозы с помощью программного обеспечения Fluoroskan Ascent.
[0112] Способ может использоваться для анализа ферментов, включая Kgp, RgpB, RgpA, трипсин и катепсин В. Для RgpA и RgpB подложкой может быть Z-Arg-AMC. Для трипсина буфер может содержать 10 мМ Tris и 10 мМ CaCl2 (рН 8,0), и подложкой может быть Z-Gly-Gly-Arg-AMC. Для катепсина B буфер может содержать 50 мМ фосфата натрия, 1 мМ EDTA и 10 мМ 2-меркаптоэтанола (рН 6,25), и подожкой может быть Z-Arg-Arg-AMC.
[0113] Константы ингибирования могут быть вычислены с помощью представленного далее уравнения с допущением, что ингибирование является полностью конкурентоспособным:
Vi=(Vmax [S])/([S]+ Km(1+[I]/Ki)
где Vi - наблюдаемая остаточная активность, [S] - концентрация подложки, используемой в анализе, Vmax - максимальная скорость при концентрации ингибитора, равной нулю, Ki - константа диссоциации ингибитора, и [I] - концентрация ингибитора. Кривые могут быть получены с помощью нелинейного регрессионного анализа с использованием фиксированных значений концентраций подложки и значения константы Михаэлиса (Km). Анализ данных может осуществляться с помощью Prism V 2.01 (GraphPad, San Diego, Calif.).
[0114] Термин «IC50» показывает, сколько соединения необходимо для ингибирования данного биологического процесса (или компонента процесса, например фермента, клетки, клеточного рецептора или микроорганизма) на 50 процентов (50%). IC50 соединения может быть определена построением кривой «доза-ответ» и исследованием влияния различных концентраций соединения на реверсирование активности фермента. Исходя из кривой «доза-ответ» могут быть вычислены значения IC50 данного соединения посредством определения концентрации, необходимой для половинного ингибирования максимального биологического ответа фермента.
[0115] Обычно значение Ki Kgp соединений по настоящему изобретению находится в интервале от примерно 0,001 нМ до примерно 500 нМ. Значение Ki Kgp соединения по настоящему изобретению может находиться в интервале, например, от примерно 1 нМ до примерно 20 нМ, от примерно 20 нМ до примерно 40 нМ, от примерно 40 нМ до примерно 60 нМ, от примерно 60 нМ до примерно 80 нМ, от примерно 80 нМ до примерно 100 нМ, от примерно 100 нм до примерно 150 нМ, от примерно 150 нм до примерно 200 нМ, от примерно 200 нм до примерно 250 нМ, от примерно 250 нМ до примерно 300 нМ, от примерно 300 нм до примерно 350 нМ, от примерно 350 нм до примерно 400 нМ, от примерно 400 нм до примерно 450 нМ или от примерно 450 нм до примерно 500 нм. Значение Ki Kgp соединения по настоящему изобрению может находиться в интервале от примерно 0,001 нМ до примерно 0,025 нМ, от примерно 0,025 нМ до примерно 0,050 нМ, от примерно 0,050 нМ до примерно 0,075 нМ, от примерно 0,075 нМ до примерно 0,100 нМ, от примерно 0,100 нМ до примерно 0,250 нМ, от примерно 0,250 нМ до примерно 0,500 нМ, от примерно 0,500 нМ до примерно 0,750 нМ или от примерно 0,750 нМ до примерно 1 нМ.
[0116] Обычо значение IC50 Kgp соединений по настоящему изобретению находится в интервале от примерно 0,001 нМ до примерно 500 нМ. Значение IC50 Kgp соединения по настоящему изобретению может находиться в интервале, например, от примерно 1 нМ до примерно 20 нМ, от примерно 20 нМ до примерно 40 нМ, от примерно 40 нМ до примерно 60 нМ, от примерно 60 нМ до примерно 80 нМ, от примерно 80 нМ до примерно 100 нМ, от примерно 100 нм до примерно 150 нМ, от примерно 150 нм до примерно 200 нМ, от примерно 200 нм до примерно 250 нМ, от примерно 250 нМ до примерно 300 нМ, от примерно 300 нм до примерно 350 нМ, от примерно 350 нм до примерно 400 нМ, от примерно 400 нм до примерно 450 нМ или от примерно 450 нм до примерно 500 нм. Значение IC50 Kgp соединения по настоящему изобретению может находиться в интервале от примерно 0,001 нМ до примерно 0,025 нМ, от примерно 0,025 нМ до примерно 0,050 нМ, от примерно 0,050 нМ до примерно 0,075 нМ, от примерно 0,075 нМ до примерно 0,100 нМ, от примерно 0,100 нМ до примерно 0,250 нМ, от примерно 0,250 нМ до примерно 0,500 нМ, от примерно 0,500 нМ до примерно 0,750 нМ или от примерно 0,750 нМ до примерно 1 нМ.
[0117] В некоторых вариантах осуществления настоящего изобретения значение Ki Kgp ингибитора Kgp по настоящему изобретению составляет 100 нМ или менее. В некоторых вариантах осуществления настоящего изобретения значение Ki Kgp ингибитора Kgp составляет 50 нМ или менее.
[0118] В некоторых вариантах осуществления настоящего изобретения значение IC50 Kgp ингибитора Kgp по настоящему изобретению составляет 50 нМ или менее. В некоторых вариантах осуществления настоящего изобретения значение IC50 Kgp ингибитора Kgp составляет 15 нм или менее.
[0119] Соединения со значением Ki Kgp 15 нм или менее могут быть применимы, в частности, для системного введения. Например, такие соединения могут иметь значения Kgp Ki в интервале от примерно 1 пикомоля (пМ) до примерно 15 наномолей (нМ), от примерно 10 мкМ до примерно 12 нМ, примерно от 100 мкМ до примерно 11 нМ или от примерно 100 пМ до примерно 10 нМ. Значения Ki Kgp таких соединений могут составлять менее 10 нМ (нМ), менее 8 нм, менее 6 нМ или менее 4 нМ.
[0120] Соединения со значениями Ki Kgp, равными или менее 45 нм, могут быть применимы, в частности, для местного введения. Например, значения Ki Kgp таких соединений могут находиться в интервале от примерно 1 пикомолея (пМ) до примерно 40 наномолей (нМ), от примерно 10 мкм до примерно 35 нМ, от примерно 100 мкМ до примерно 30 нМ или от примерно 100 пМ до примерно 25 нМ.
[0121] В некоторых вариантах осуществления настоящего изобретения ингибиторы Kgp по настоящему изобретению являются селективными для Kgp. Термин «селективный» ингибитор Kgp, когда используется в настоящем описании, означает соединение, которое не оказывает существенного влияния на активность протеаз, отличных от Kgp, RgpA и RgpB, при введении в терапевтически эффективной дозе для лечения заболевания или состояния, связанного с инфекцией P. gingivalis. Обычно протеаза, на которую по существу не влияет конкретное соединение, в присутствии соединения проявляет по меньшей мере 90% своей нормальной ферментативной активности в физиологических условиях. Селективные ингибиторы Kgp включают соединения, которые не влияют на активность протеаз, отличных от Kgp, при введении в терапевтически эффективной дозе для лечения нарушения мозговой деятельности, заболевания пародонта, диабета, сердечно-сосудистых заболеваний, артрита, преждевременных родов, пневмонии, рака, заболевая почек, заболевания печени, поражения сетчатки или глаукомы, связанных с инфекцией Р. gingivalis. Предпочтительно, селективные ингибиторы Kgp не оказывают неблагоприятного влияния на каскад коагуляции при введении в терапевтически эффективных количества.
[0122] В некоторых вариантах осуществления изобретение относится ингибитору Kgp со значением Ki Kgp менее 50 нМ. В некоторых таких вариантах осуществления настоящего изобретения значение Ki трипсина составляет более 60 нМ. В некоторых вариантах осуществления настоящего изобретения значение Ki Kgp ингибитора Kgp составляет менее 15 нМ, и соотношение (Ki трипсина)/(Ki Kgp) составляет более 100.
[0123] В некоторых вариантах осуществления изобретение относится к соединениям, которые являются по меньшей мере в 30 раз более селективными в отношении Kgp по сравнению с трипсином или катепсином В. Для некоторых таких соединений значение Ki Kgp равно 0,9 нМ, и значение Ki трипсина и/или катепсиа В равно 30 нМ или более. В некоторых вариантах осуществления настоящего изобретения значение Ki Kgp равно 0,9 нМ, и значение Ki трипсина или /катепсина В равно 115 мкΜ или более. Для некоторых таких соединений IC50 Kgp равно 50 нм или менее, и значение IC50 трипсина равно 100 нм или более. Для некоторых таких соединений значение IC50 Kgp равно 15 нм или менее, и значение IC50 трипсина равно 1 мкΜ или более.
IV. Способы получения соединений
[0124] Некоторые примеры соединений формулы (I) могут быть получены, исходя из некоторых производных лизина D1 и D6, которые описаны ниже, и являются коммерчески доступными или могут быть получены в соответствии с опубликованными в научной литературе методиками.

[0125] В D1 каждый из предпочтительных R7 и R8 может быть удален с помощью химических условий, при которых другой не удаляется. Так, например, R7=бензил может быть удален с использованием водорода и катализатора палладия на угле, но трифторуксусная кислота не воздействует на R7, в то время как R8=трет-бутил может быть удален с помощью трифторуксусной кислоты, но на R7 не влияют водород и катализатор палладий на угле. Другие подходящие благоприятные комбинации R7 и R8 были описаны в научной литературе. Аналогичным образом, в D6, благоприятные комбинации R8 и R9, которые могут удаляться, являются предпочтительными, и некоторые подходящие комбинации были описаны в научной литературе.
[0126] Некоторые D5 могут быть получены с помощью последовательности преобразований D1 - D2 - D3 - D4 - D5. См. схему 1.
Схема 1

[0127] В большинстве случаев превращение D1 в D2 будет включать более одной химической реакции. В соответствии с описанными в литературе методиками, для превращения D1 в D2 могут применяться следующие химические реакции. D1 может подвергаться превращению в Е1 обработкой гидрохлоридом N-метил-О-метилгидроксиламина, органическим основанием (например, Et3N), ингибитором рацемизации (например, HOBt) и дегидратирующии реагентом (например, EDAC) в органическом растворителе (например, ДМФА). См. схему 2, стадию (а). Е1 может подвергаться превращению в D2-5 обработкой литиированным гетероциклом (например, 2-литиобензотиазолом, 2-литиотиазолом или 2-литиопиридином) в органическом растворителе (например, ТГФ) для введения соответствующего R10 (2-бензотиазолила, 2-тиазолила или 2-пиридила). См. схему 2, стадию (b).
Схема 2

[0128] D1 может подвергаться превращению в Е2 обработкой гидрохлоридом аммония, органическим основанием (например, Et3N), ингибитором рацемизации (например, HOBt) и дегидратирующим реагентом (например, EDAC) в органическом растворителе (например, ДМФА). См. схему 2, стадию (с). Е2 может подвергаться превращению в D2-6 обработкой органическим основанием (например, Et3N) и сильным дегидратирующим реагентом (например, комплексом пиридин-триоксид серы) в органическом растворителе (например, CH2Cl2). См. схему 2, стадию (d).
[0129] D1 может подвергаться превращению в D2-8 обработкой фторуксусным ангидридом и Et3N и DMAP в органическом растворителе (например, ДМФА). См. схему 2, стадию (е).
[0130] D1 может подвергаться превращению в Е3 обработкой комплексом боран-диметилсульфид в органическом растворителе (например, ТГФ). См. схему 3, стадию (а). Е3 может подвергаться преобращению в Е7 обработкой органическим основанием (например, Et3N), сильным дегидратирующим реагентом (например, оксалилхлоридом) и диметилсульфоксидом в органическом растворителе (например, CH2Cl2). См. схему 3, стадию (b). Е7 может подвергаться превращению в D2-10 обработкой триметилдиазофосфонацетатом и K2CО3 в спиртовом растворителе (например, метаноле). См. схему 3, стадию (с).
Схема 3
[0131] D1 может подвергаться превращению в Е4 обработкой органическим основанием (например, Et3N), хлорформиатом (например ЕtО2CCl) и диазометаном в органическом растворителе (например, диэтиловом эфире). См. схему 4, стадию (а). Е4 может подвергаться превращению в Е11 обработкой HBr и уксусной кислотой в органическом растворителе (например, ТГФ). См. схему 4, стадию (b). Е11 может подвергаться превращению в D2-9 обработкой спиртом HOR11 (например, 2,3,5,6-тетрафторфенолом) и KF в органическом растворителе (например, ДМФА) для введения соответствующей группы -OR11 (например, 2,3,5,6-тетрафторфенокси). См. схему 4, стадию (с).
Схема 4


[0132] Кроме того, для превращения D1 в другие D2, где Х представляет собой тиол-реакционноспособную группу, которая не показана, может использоваться широкий спектр дополнительных методик, описанных в научной литературе.
[0133] После превращения D1 в D2 (например, в D2-5, D2-6, D2-8, D2-9 или D2-10) R7 может удаляться в подходящих химических условиях с образованием D3 после самопроизвольного декарбоксилирования. D3 или соль D3 (например, гидрохлоридная соль D3) может использоваться в дополнительных стадиях синтеза. D3 может подвергаться обработке карбоновой кислотой R3CО2H, ингибитором рацемизации (например, HOBt) и дегидратирующим рагентом (например, EDAC) в органическом растворителе (например, ДМФА) с получением D4. В качестве альтернативы, D3 может подвергаться обработке R3COX, где Х представляет собой удаляемую группу (например, хлорид), и органическим основанием (например, Et3N) в органическом растворителе (например, CH2Cl2) с получением D4. В качестве альтернативы, D3 может подвергаться обработке изоцианатом в органическом растворителе (например, CH2Cl2) с получением D4. Коммерчески доступен широкий спектр подходящих R3СО2H, R3СОХ и изоцианатов, или они могут быть получены в соответствии с методиками, описанными в научной литературе. R8 может удаляться с использованием соответствующих химических условий с получением D5 после самопроизвольного декарбоксилирования.
[0134] Другие D5 могут быть получены с помощью последовательности превращений D6 - D7 - D4 - D5. См. схему 5.
Схема 5
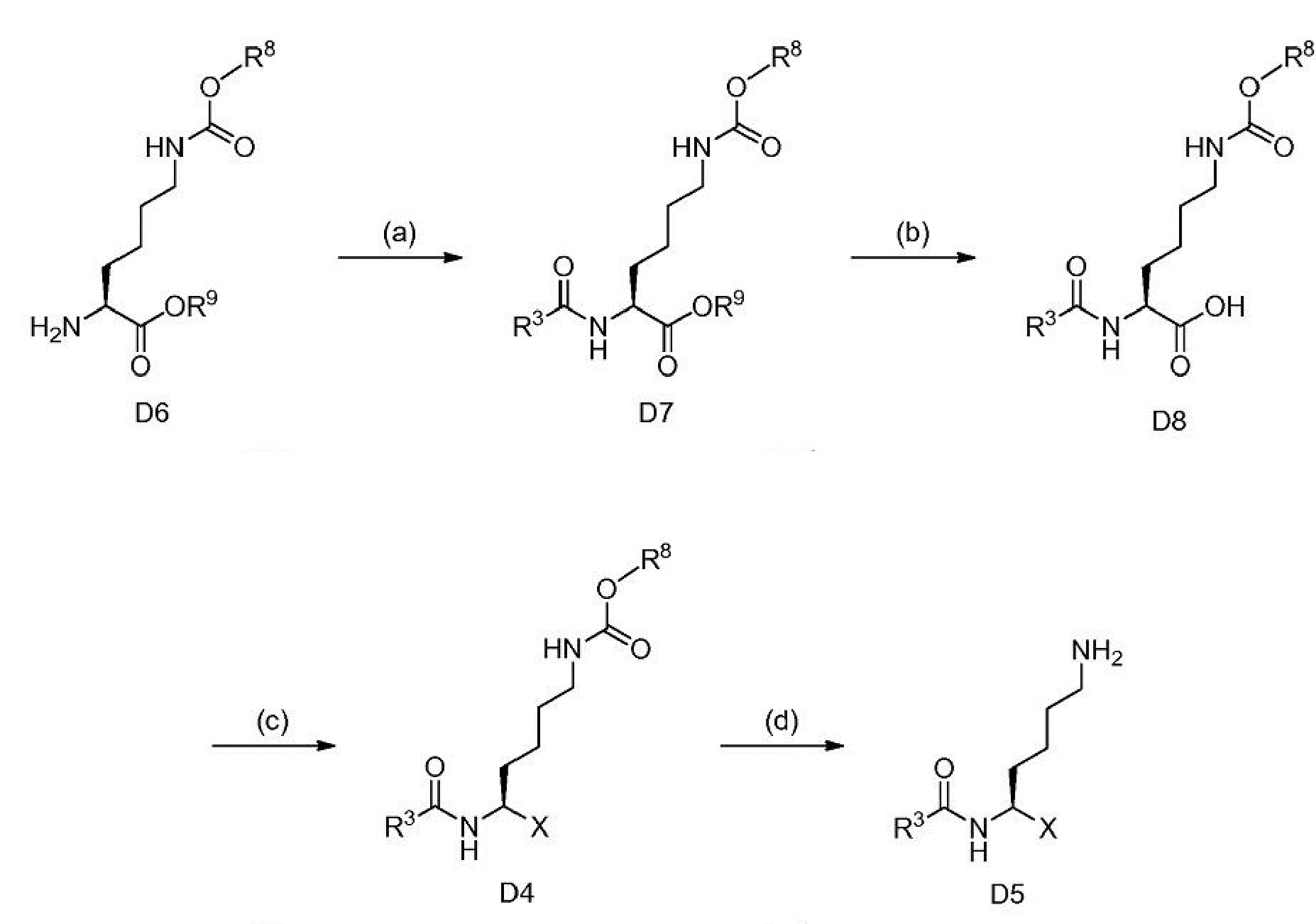
[0135] D6 может подвергаться обработке карбоновой кислотой R3СО2Н, ингибитором рацемизации (например, HOBt) и дегидратирующим реагентом (например, EDAC) в органическом растворителе (например, ДМФА) с получением D7. См. схему 5, стадию (а). В качестве альтернативы, D6 может подвергаться обработке R3COX, где Х представляет собой удаляемую группу (например, хлорид), и органическим основанием (например, Et3N) в органическом растворителе (например, CH2Cl2) с получением D7. В качестве альтернативы, как показано на схеме 1, D3 может подвергаться обработке изоцианатом в органическом растворителе (например, CH2Cl2) с получением D7. Коммерчески доступен широкий спектр подходящих R3СО2H, R3СОХ и изоцианатов, или они могут быть получены в соответствии с методиками, описанными в научной литературе. R9 может быть удален с использованием соответствующих химических условий с получением D8. См. схему 5, стадию (b).
[0136] D8 может подвергаться превращению в D4 с помощью последовательности реакций, аналогичных тем, которые описаны для превращения D1 в D2. См. схему 5, стадию (с). В некоторых вариантах осуществления настоящего изобретения D8 подвргается превращению в D4-9, как показано на схеме 6, стадии (а)-(с). В некоторых вариантах осуществления настоящего изобретения -OR11 в D4-9 представляет собой 2,3,5,6-тетрафторфенокси.
Схема 6

[0137] Кроме того, для превращения D8 в другие D4, где Х представляет собой тиол-реакционноспособную группу, которая не показана, может применяться широкий спектр дополнительных методик, описанных в научной литературе. В соответствии с альтернативной последовательностью получения D4, R8 может быть удален с помощью соответствующих химических условий с получением D5 после самопроизвольного декарбоксилирования. См. схему 5, стадию (d).
[0138] Для получения некоторых примеров соединений формулы (I) необходимо сначала получить аминокислоты с боковыми цепями, которые не присутствуют ни в одной из аминокислот, встречающихся в белках. Опубликован широкий спектр методик получения аминокислот, которые отличаются тем, что содержат боковые цепи, не встречающиеся в природных аминокислотах, включая наиболее применимые и важные методы F1-4:
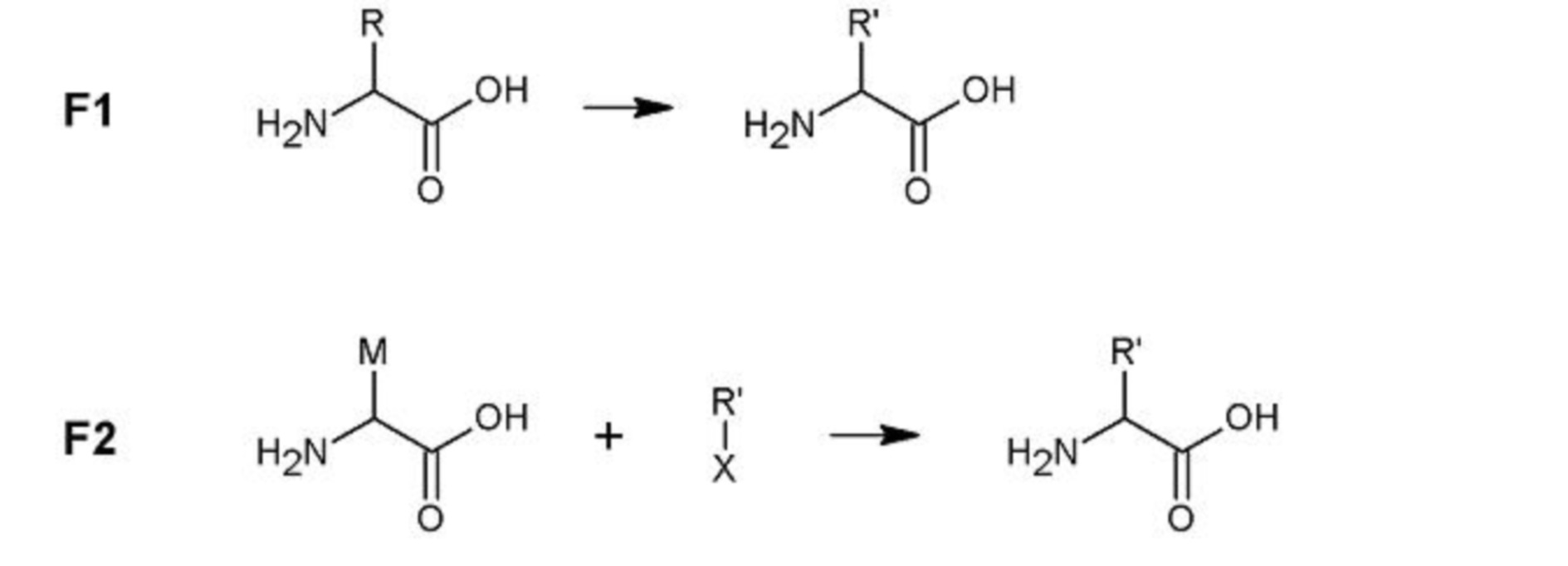

[0139] Хотя это не показано на схемах F1-3, аминнная и карбоксилатная группы обычно защищают перед применением этих способов, и защита удаляется после образования не встречающейся в природе боковой цепи. В F1 природная боковая цепь (R) модифицируется с образованием не встречающейся в природе боковой цепи (R'). Природные аминокислоты серин, глутаминовая кислота и метионин были бы особенно подходящими для получения некоторых примеров соединений формул (I) и (II). В F2 металлированное производное глицина подвергается обработке алкилирующим реагентом (R'X) для введения не существующей в природе боковой цепи (R'). В некоторых случаях металированное производное глицина получают обработкой производного глицина сильным основным металирующим реагентом (например, диизопропиламидом лития или трет-бутоксидом калия). В других случаях исходное производное глицина является достаточно кислым, чтобы подходящим был гораздо менее основный реагент металирования (например, карбонат калия). В последних случаях металированное производное глицина может существовать в виде диссоциированной пары ионов, а не как ковалентно связанное соединение, представленные F2. В F3 металлированное аланин-производное пдвергается обработке алкилирующим реагентом (R'Х) для введения не встречающейся в природе боковой цепи (R'). В большинстве случаев металлированное производное аланина получают обработкой галогенированного производного аланина металлом с низкой валентностью (например, цинковой пылью). Во многих случаях для облечения обработки F3 используют растворимый палладиевый катализатор. В F4 альдегидную группу (R'CHO) подвергают взаимодействию с источником аммониевой группы и источником цианидной группы для получения аминонитрила, который затем подвергают гидролизу для получения аминокислоты с не встречающейся в природе боковой цепью (R'). Такие способы могут использоваться для получения промежуточных C1, С2, С3 и С4, представленных выше.
[0140] После применения подходящих способов для получения аминокислот с не встречающимися в природе боковыми цепями эти аминокислоты могут быть соответствующим образом защищены, как описано для D1 и D6, с последующим применением соответствующих спсобов, как описано для D2-4 и D7-9, для получения аналогов D5, в которых боковая цепь заменена не встречающейся в природе боковой цепью. Таким образом, доступны подходящие способы для обеспечения вариаций R3, Z и СН2АС(В)(D)СН2NH2, которые определены для формул (I) и (II).
V. Композиции/введение
[0141] В связанном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы I и фармацевтически приемлемый эксципиент.
[0142] Фармацевтические композиции могут быть получены любым из способов, хорошо известных в области фармации и доставки лекарственных средств. Обычно, способы получения композиций включают в себя стадию объединения активного ингредиента с носителем, содержащим один или несколько вспомогательных ингредиентов. Фармацевтические композиции обычно получают посредством однородного и тщательного перемешивани активного ингредиента с жидким носителем или тонко измельченным твердым носителем или с обоими и затем, при необходимости, формованием продукта в желаемый препарат. Композиции могут быть получены в лекарственных формах стандартной дозы и/или расфасованы в лекарственные формы стандартной дозы.
[0143] Фармацевтические композиции, содержащие соединения по настоящему изобретению, могут быть получены для перорального введения. Подходящие композиции для перорального введения включают, но без ограничения, таблетки, пастилки, лепешки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, сиропы, эликсиры, растворы, буккальные пластыри, гели для перорального введения, жевательные резинки, жевательные таблетки, шипучие порошки и шипучие таблетки. Композиции для перорального введения могут быть получены в соответствии с любым способом, известным специалистам данной области техники. Такие композиции могут содержать одну или несколько добавок, выбранных из подсластителей, вкусовых добавок, красителей, антиоксидантов и консервантов, для обеспечения фармацевтически изысконных и приятных на вкус препаратов.
[0144] Таблетки обычно содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, включая инертные разбавители, такие как целлюлоза, диоксид кремния, оксид алюминия, карбонат кальция, карбонат натрия, глюкоза, маннит, сорбит, лактоза, фосфат кальция и фосфат натрия; гранулирующие и дезинтегрирующие агенты, такие как кукурузный крахмал и альгиновая кислота; связующие агенты, такие как поливинилпирролидон (PVP), целлюлоза, полиэтиленгликоль (ПЭГ), крахмал, желатин и аравийская камедь; и смазывающие агенты, такие как стеарат магния, стеариновая кислота и тальк. Таблетки могут не иметь покрытия или могут быть покрыты покрытием, энтеральным или другим, с помощью известных методик для замедления дезинтеграции и всасывания в желудочно-кишечном тракте и обеспечения, таким образом, пролонгированного действия в течение более длительного периода времени. Например, для замедления высвобождения действующего вещества может быть использован такой материал, как глицерилмоностеарат или глицерилдистеарат. Таблетки также могут быть покрыты полупроницаемой мембраной и дополнительными полимерными осмогентами в соответствии с известными способами с получением композиций осмотических насосов для контролируемого высвобождения действующего вещества.
[0145] Композиции для перорального введения могут быть получены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем (например, карбонатом кальция, фосфатом кальция или каолином), или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляной средой (такой как арахисовое масло, жидкий парафин или оливковое масло).
[0146] Ингибиторы Kgp также могут вводиться местно в виде раствора, мази, крема, геля или суспензии, а также в форме средств для полоскания рта, глазных капель и т.п. Далее, трансдермальная доставка ингибиторов Kgp может осуществляться с помощью ионтофоретических пластырей и т.п.
[0147] Фармацевтические композиции, содержащие ингибиторы Kgp, могут также быть представлены в форме стерильной водных или масляных растворов и суспензий для инъекций. Стерильные препараты для инъекций могут быть получены с использованием нетоксичных, приемлемых для парентерального введения разбавителей, включая воду, раствор Рингера и изотонический раствор хлорида натрия, и приемлемых растворители, таких как 1,3-бутандиол. Кроме того, в качестве растворителя или суспендирующей среды могут использоваться стерильные нелетучие масла. Для этой цели может использоваться любое мягкое нелетучее масло, включая синтетические моноглицериды, диглицериды или триглицериды.
[0148] Водные суспензии могут содержать один или несколько ингибиторов Kgp в смеси с наполнителями, включая, но без ограничения, суспендирующие агенты, такие как натрийкарбоксиметилцеллюлоза, метилцеллюлоза, маслянистая пропилметилцеллюлоза (oleagino-propylmethylcellulose), альгинат натрия, поливинилпирролидон, трагакантовая камедь и камедь акации; диспергирующие или смачивающие агенты, такие как лецитин, полиоксиэтиленстеарат и полиэтиленсорбитанмоноолеат; и консерванты, такие как этил-, н-пропил- и п-гидроксибензоат. Диспергируемые порошки и гранулы (подходящие для получения водной суспензии путем добавления воды) могут содержать один или несколько ингибиторов Kbp в смеси с диспергирующим агентом, смачивающим агентом, суспендирующим агентом или их комбинациями. Масляные суспензии могут быть получены суспендированием ингибитора Kgp в растительном масле (например, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле) или в минеральном масле (например, жидком парафине). Масляные суспензии могут содержать один или несколько загустителей, например пчелиный воск, твердый парафин или цетиловый спирт. Для хранения композиций в них могут добавляться антиоксидант, такой как аскорбиновая кислота.
[0149] Фармацевтические композиции по настоящему изобретению также могут быть представлены в виде эмульсий типа «масло в воде». Масляная фаза может представлять собой растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например жидкий парафин, или их смеси. Подходящими эмульгирующими агентами могут быть природные камеди, такие как аравийская камедь или трагакантовая камедь; природные фосфолипиды, такие как соевый лецитин; сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и гекситовых ангидридов, такие как сорбитанмоноолеат; и продукты конденсации указанных неполных эфиров с оксидом этилена, например полиоксиэтиленсорбитанмоноолеат.
[0150] Кроме того, настоящее изобретение включает различные способы введения, с помощью которых могут быть доставлены указаннные соединения, для повышения биологической доступности или проникновения через гематоэнцефалический барьер, включая, но без ограничения, внутривенное, интраназальное, подкожное, внутричерепное и пероральное введение. Технология замедленного высвобождения действующего вещества может быть использована для повышения биологической доступности, включая композиции для замедленного высвобождения (sustained-release - SR) действующего вещества, композиции пролонгированного действия (sustained-action - SA), композиции с пролонгированным высвобождением (ER, XR, XL) действующего вещества, композиции медленного выскобождения (timed-release - ТR) действующего вещества, композиции с контролируемым высвобождением (CR) действующего вещества, композиции с модифицированным высвобождением (modified release - MR) действующего вещества, композиции с непрерывным высвобождением действующего вещества, композиции осмотического высвобождения действующего вещества и импланты медленного высвобождения действующего вещества. Указанные альтернативные способы введения ранее не рассматривались для этих соединений, которые, главным образом, предназначались для местной десневой доставки, а не системно.
[0151] Использование гибридных молекул для способствования активному транспорту или использование наночастиц может применяться в некоторых вариантах осуществления настоящего изобретения для повышения транспорта через гематоэнцефалический барьер. Например, липосомы, белки, сконструированные пептидные соединения или антитела, которые связываются с рецепторами и которые транспортируют белки через гематоэнцефалический барьер, включая LPR-1 рецептор, рецептор трансферрина, EGF-подобный фактор роста или переносчик глутатиона, могут использоваться для повышения проникновения в мозг. Могут также использоваться физические методы, включая осмотическое открытие, ультразвук, лазеры, стимуляцию сфенопалантинного ганглия, прямую внутричерепную, интратекальную или внутрижелудочковую доставку с помощью насоса.
[0152] Фармацевтические композиции согласно изобретению могут также включать один или несколько дополнительных активных ингредентов, подходящих для лечения состояний, связанных с инфекцией P. gingivalis. В некоторых вариантах осуществления изобретение относится к фармацевтической композиции, содержащей один или несколько ингибиторов Kgp, которые определены в настоящем описании, в комбинации с одним или несколькими дополнительными активными ингредиентами для лечения болезни Альцгеймера. Некоторые терапевтические средства находятся в стадии разработки и исследования клинического применения для лечения болезни Альцгеймера. Терапевтические стратегии включают в себя снижение уровней циркулирующих бета-амилоида и тау (как описано более подробно ниже), стабилизирующих микротрубочек, удаление атеросклеротических бляшек, модулирование аутофагии, модулирование уровней нейротрансмиттеров и ингибирования GABA(А) α5 рецепторов. Такие терапевтические средства могут поддерживать и/или восстанавливать когнитивные функции у пациентов с болезнью Альцгеймера; замедлять ухудшение когнитивных функций; а также содействовать нейропластичности и восстановлению деятельности головного мозга.
[0153] Активные ингредиенты, которые могут быть объединены с ингибиторами Kgp в фармацевтических композициях, включают, но без ограничения, антибиотики (т.е. бактерицидные соединения и бактериостатические соединения), ингибиторы холинэстеразы, модуляторы альфа-7 никотиновых рецепторов, модуляторы серотонина, модуляторы NMDA, терапевтические средства Αβ-направленного действия, терапевтические средства АpoE-направленного действия, терапевтические средства микроглия-направленного действия, терапевтические средства направленног действия на гематоэнцефалический барьер, терапевтические средства тау-направленного действия, терапевтические средства комплемент-направленного действия, а также противовоспалительные терапевтические средства.
[0154] Любой подходящий антибиотик может быть объединен с одним или несколькими ингибиторами Kgp в фармацевтических композициях по настоящему изобретению. В некоторых вариантах осуществления изобретение относится к фармацевтической композиции, содержащей несколько ингибиторов Kgp и антибиотик со значением MIC50Р. gingivalis менее 25 мкг/мл. Например, MIC50Р. gingivalis антибиотика может составлять менее 20 мкг/мл, менее 15 мкг/мл, менее 10 мкг/мл, менее 8 мкг/мл, менее 6 мкг/мл или менее 5 мкг/мл. В некоторых вариантах осуществления изобретения значение MIC50Р. gingivalis антибиотика составляет менее 1 мкг/мл. В некоторых вариантах осуществления изобретения значение MIC50Р. gingivalis антибиотика составляет менее 0,2 мкг/мл.
[0155] Примеры бактерицидных и бактериостатических соединений включают, но без ограничения, хинолоны (например, моксифлоксацин, гемифлоксацин, ципрофлоксацин, офлаксацин, тровафлоксацин, ситафлоксацин и т.п.), β-лактамы (например, пенициллины, такие как амоксициллин, амоксацилин-клавуланат, пиперациллин-тазобактам, пенициллин G и т.п.; и цефалоспорины, такие как цефтриаксон и т.п.), макролиды (например, эритромицин, азитромицин, кларитромицин и т.п.), карбапенемы (например, дорипенем, имипенем, меропинем, эртапенем и т.п.), тиазолиды (например, тизоксанидин, нитазоксанидин, RM 4807, RM 4809 и т.п.), тетрациклины (например, тетрациклин, миноциклин, доксициклин, эравациклин и т.п.), клиндамицин, метронидазол и сатранидазол. Бактерицидные и бактериостатические соединения также включают соединения, которые ингибируют формирование биопленок или иным образом препятствуют их образованию под действием анаэробных грам-отрицательных бактерий; такие соединения включают оксантел, морантел, тиабендазол и т.п. Композиции по изобретению могут содержать один или несколько ингибиторов Kgp с одним или несколькими (например, двумя, тремя, четырьмя, пятью, шестью или более) бактерицидныма/бактериостатическими соединениями. Композиции, содержащие бактерицидные/бактериостатические соединения, могут дополнительно содержать хлоргексидин (например, хлоргексидина диглюконат) отдельно или в комбинации с соединением цинка (например, ацетатом цинка) и также могут быть использованы в комбинации с вводимыми антибиотиками.
[0156] В некоторых вариантах осуществления настоящего изобретения используется комбинация пенициллина (например, амоксициллина) и метронидазола или комбинация пенициллина (например, амоксициллина, метронидазола) и тетрациклина. В некоторых вариантах осуществления настоящего изобретения антибиотик выбран из миноциклина, доксициклина, метронидазола, амоксициллина, клиндамицина, аугментина, сатранидазола и их комбинаций.
[0157] Примеры подходящих ингибиторов холинэстераз включают, но без ограничения, донепезили, донепезили/мемантин, галантамин, ривастигмин и такрин, а также их фармацевтически приемлемые соли. Примеры подходящих модуляторов серотонина включают, но без ограничения, идадопирдин, RVT-101, циталопрам, эсциталопрам, флуоксетин, флувоксамин, пароксетин и сертралин, а также их фармацевтически приемлемые соли. Примеры подходящих модуляторов альфа-7 никотинового рецептора, включают, но без ограничения, альфа-7 агонисты, такие как энцениклин и APNl 125. Подходящие модуляторы NMDA включают в себя, но без ограничения, антагонисты NMDA рецепторов, такие как мемантин и его производные.
[0158] Фармацевтические композиции по настоящему изобретению могут также содержать активные лекарственные средства, которые направлены на молекулярно-биологические мишени, связанные с неврологическими заболеваниями. Такие мишени включают бета-амилоидные пептиды (также называемые бета-амилоид, абета или Аβ), аполипопротеин Е (называемый также АпоЕ) и ассоциированные с микротрубочками тау (также называемые тау-белками или просто тау).
[0159] Терапевтические средства Αβ-направленного действия включает ингибиторы продуцирования Αβ (такие как ингибиторы бета-секретазы, ингибиторы гаммы-секретазы, активаторы альфа-секретазы), ингибиторы агрегации Αβ, ингибиторы олигомеризации Αβ, стимуляторы функциональной активности Aβ клиренса и т.п. (см., например, Jia, et al.BioMed Research International, 2014. Article ID 837157, doi:10.1155/2014/837157). Примеры терапевтических средств Αβ-направленного действия включают, но без ограничения, антитела, пиоглитазон, бегацестат, аторвастатин, симвастатин, этазолат и трамипросат, а также их фармацевтически приемлемые соли.
[0160] Примеры терапевтических средств АпоЕ-направленного действия включают, но без ограничения, агонисты ретиноидных Х рецепторов (см., Cramer, et al., Science 2012. 335(6075): 1503-1506) и другие терапевтические средства, описанные Liu с соавторами (Nat Rev Neurol. 2013. 9(2): 106-118). Терапевтические средства тау-направленного действия включают, но без ограничения, метилтиониний, лейко-метилтиониний, антитела и терапевтические средства, которые описаны Lee с соавторами (Cold Spring Harb Perspect Med 2011; 1:a006437).
[0161] Фармацевтические композиции по настоящему изобретению могут также содержать терапевтические средства комплемент-целенаправленного действия. Такие терапевтические компоненты средств направленного действия на системы комплемента вовлечены во врожденный иммунный ответ. Терапевтические средства комплемент-направленного действия включают, но без ограничения, терапевтические средства, которые описаны Ricklin и Lambris (Nat. Biotechnology 2007. 25(11): 1265-1275).
[0162] Примеры подходящих противовоспалительных средств включают, но без ограничения, нестероидные противовоспалительные лекартвенные средства (nonsteroidal anti-inflammatory drug - NSAID), такие как апазон, диклофенак, ибупрофен, индометацин, кетопрофен, мелоксикам, напроксен, пироксикам и сулиндак, а также их фармацевтически приемлемые соли.
VI. Способы лечения
[0163] Как описано выше, инфицирование P. gingivalis и гингипаиновая активность были связаны с развитием заболеваний пародонта, болезни Альцгеймера и других нарушений мозговой деятельности, сердечно-сосудистых заболеваний, диабета, рака, заболевания печени, заболевания почек, преждевременными родами, развитием артрита и других расстройств. См: Bostanci, et al. FEMS Microbiol. Lett., 2012. 333(1): 1-9; Ghizoni, et al. J. Appl. Oral. Sci., 2012. 20(1): 104-12; Gatz, et al. Alzheimers Dement., 2006. 2(2): 110-7; Stein, et al. J. Am. Dent. Assoc., 2007. 138(10): 1314-22; quiz 1381-2; Noble, et al. J. Neuro.l Neurosurg. Psychiatry, 2009. 80(11): 1206-11; Sparks Stein, et al. Alzheimers Dement., 2012. 8(3): 196-203; Velsko, et al. PLoS ONE, 2014. 9(5): e97811; Demmer, et al. J. Dent. Res., 2015. 94(9S): 201-S-11S; Atanasova and Yilmaz. Molecular Oral Microbiology, 2014. 29(2): 55-66; Yoneda, et al. BMC Gastroenterol., 2012. 12: 16. Таким образом, ингибиторы Kgp могут использоваться для лечения заболеваний и состояний, таких как нарушения мозговой деятельности, вызванные P. gingivalis или иным образом зависимые от P. gingivalis.
[0164] Соответственно, другой аспект настоящего изобретения относится к способу лечения заболевания или состояния, связанного с инфекцией P. gingivalis. Способ включает введение пациенту эффективного количества соединения формулы Ie

или его фармацевтически приемлемой соли, где
Z представляет собой тиол-реакционноспособную группу или защищенную тиол-реакционноспособную группуа;
А выбран из -CH2- и -О-;
В и D независимо выбраны из водорода, галогена, C1-4 галогеналкила и C1-4 галогеналкокси;
R1 выбран из водорода и защитной группы аминогруппы;
R2 представляет собой водород; и
R3 выбран из C6-10 арила, 5-12-членного гетероарила, C1-8 алкила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила, L R5 и -OR6, где
L выбран из -О-, -NR-, C1-4 алкилена и 2-4-членного гетероалкилена, где R выбран из водорода и C1-8 алкила,
R5 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила и 5-12-членного насыщенного гетероциклила, и
-OR6 и карбонил, к которому он присоединен, образуют защитную группу аминогруппы, и
где R3 является необязательно замещенным одним или несколькими заместителями, выбранными из галогена, CN, NO2, N3, OH, Ra, Rb, ORa, ORb, (CH2)kC(O)Rc, NRd(CH2)uC(O)Rc, O(CH2)uC(O)Rc, (CH2)kCONRdRd, (CH2)kNRdC(O)Rc, NRd(CH2)uCONRdRd, NRd(CH2)uNRdC(O)Rc, O(CH2)uCONRdRd, O(CH2)uNRdC(O)Rc, (CH2)kS(O)2NRdRd, (CH2)kNRdS(O)2Rc, (CH2)kS(O)2Rc, (CH2)kS(O)Rc, (CH2)kSRd, NRd(CH2)uS(O)2NRdRd, NRd(CH2)uNRdS(O)2Rc, NRd(CH2)uS(O)2Rc, NRd(CH2)uS(O)Rc, NRd(CH2)uSRd, O(CH2)uS(O)2NRdRd, O(CH2)uNRdS(O)2Rc, O(CH2)uS(O)2Rc, O(CH2)uS(O)Rcи O(CH2)uSRc, где:
каждый Raнезависимо выбран из C1-4 алкила и C1-4 галогеналкила,
каждый Rb независимо выбран из C3-6 циклоалкила, C3-6 галогенциклоалкила, C6-10 арила, 5-12-членного гетероарила и 5-12-членного насыщенного гетероциклила,
каждый Rc независимо выбран из -ОН, C1-8 алкила, C1-8 галогеналкила, C3-8 циклоалкила, C3-8 галогенциклоалкила, C6-10 арила, (C6-10 арил)-(C1-8 алкила), 5-12-членного гетероарила и 5-12-членного насыщенного гетероциклила,
каждый Rd независимо выбран из водорода и C1-8 алкила,
каждый подстрочный индекс k независимо выбран из 0, 1, 2, 3, 4, 5 и 6, и
каждый подстрочный индекс u независимо выбран из 1, 2, 3, 4, 5 и 6; и
R4 выбран из водорода, галогена, C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила и C1-4 галогеналкокси.
[0165] В некоторых вариантах осуществления настоящего изобретения способ включает введение пациенту одного или нескольких соединений из таблицы 1.
Таблица 1. Соединения для применения в лечении состояний, связанных с инфекцией Р. gingivalis.




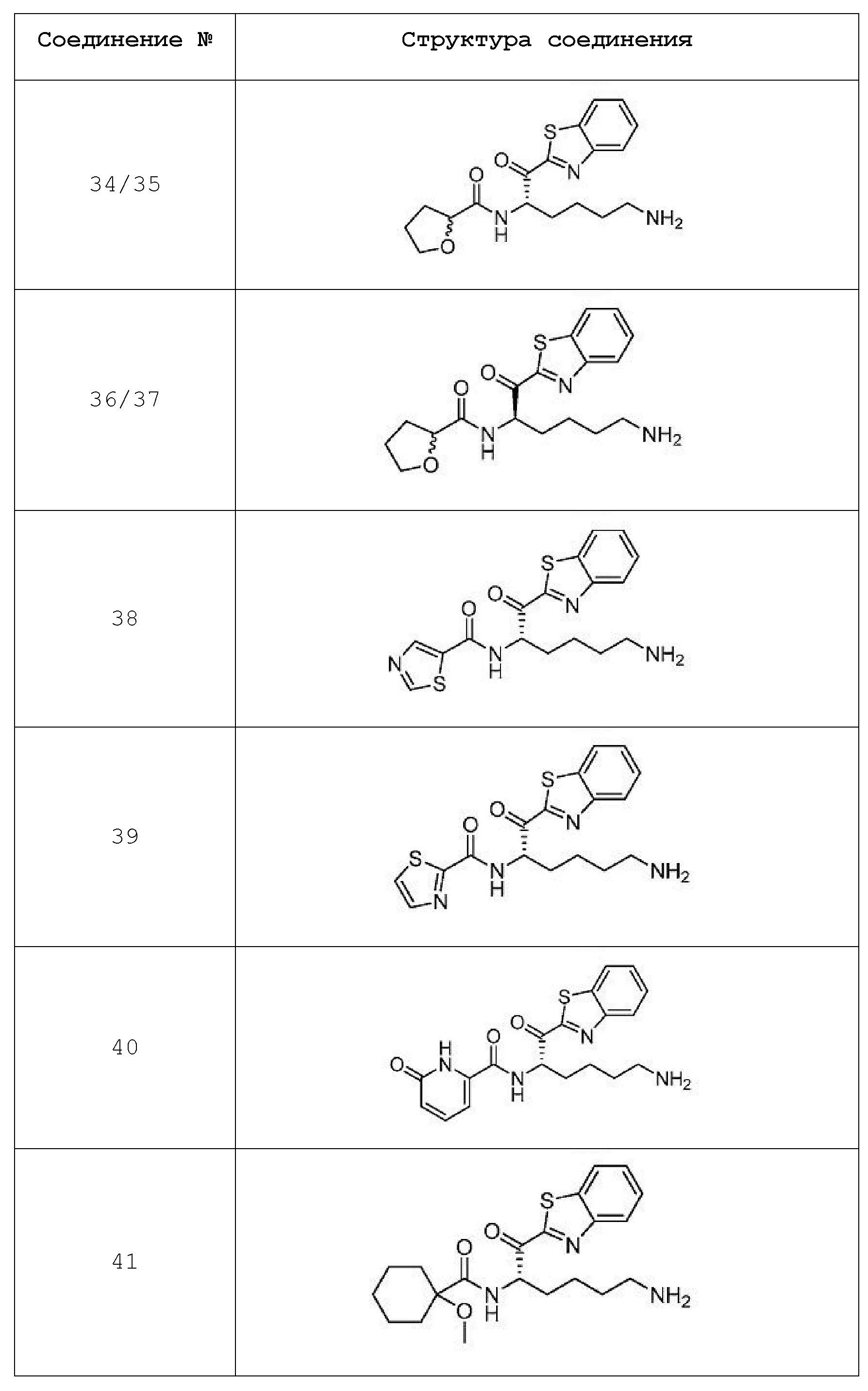
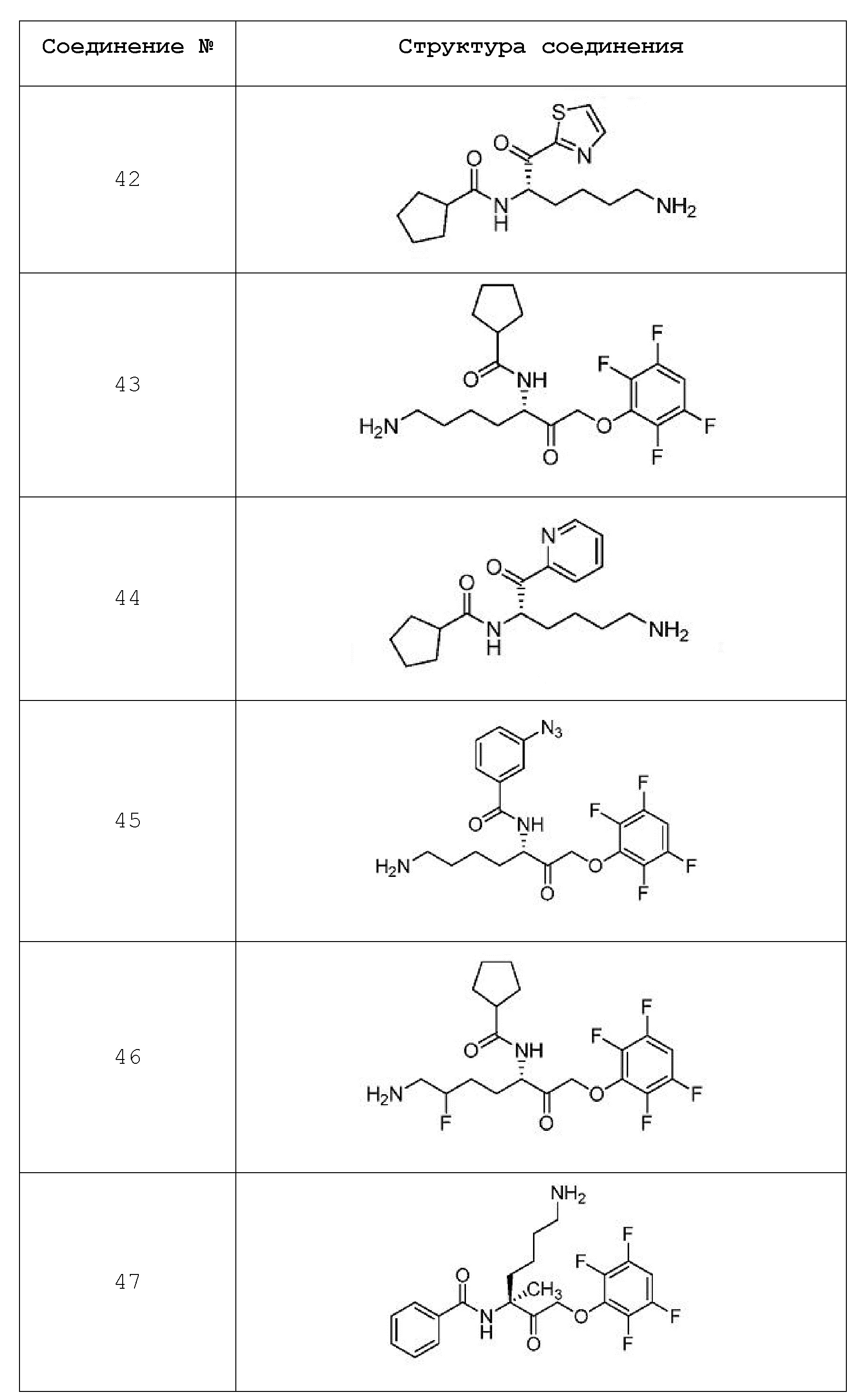

[0166] В некоторых вариантах осуществления изобретение относится к способу лечения заболевания или состояния, связанного с инфекцией P. gingivalis, как описано выше, в котором пациентом является человек или представитель семейства псовых.
[0167] В некоторых вариантах осуществления изобретение относится к способу лечения заболевания или состояния, связанного с инфекцией P. gingivalis, как описано выше, в котором структура соединения формулы Iе соответствует формуле If:

[0168] В некоторых вариантах осуществления изобретение относится к способу лечения заболевания или состояния, связанного с инфекцией P. gingivalis инфекции, как описано выше, где R4 представляет собой водород, а В и D независимо выбраны из водорода, галогена, галогенметила и галогенметокси.
[0169] В некоторых вариантах осуществления настоящего изобретения Z выбран из бензотиазол-2-илкарбонила; тиазол-2-илкарбонила; оксазол-2-илкарбонила; бензоксазол-2-илкарбонила; пиридин-2-илкарбонила; пиримидин-4-илкарбонила; пиримидин-2-илкарбонила; изоксазол-5-илкарбонила; изоксазол-3-илкарбонила; 1,2,4-оксадиазол-3-илкарбонила; 1,2,4-оксадиазол-5-илкарбонила; циано; этинила; фторметилкарбонила; ацилоксиметилкарбонила; арилоксиметилкарбонила; алкилсульфонилвинила; и арилсульфонилвинила; каждый из которых является необязательно замещенным одним или несколькими заместителями, выбранными из C1-4 алкила, C1-4 алкокси, C1-4 галогеналкила, C1-4 галогеналкокси, галогена и -N3.
[0170] В некоторых вариантах осуществления настоящего изобретения Z выбран из бензотиазол-2-илкарбонила, галогенамещенного арилоксиметилкарбонила, пиридин-2-илкарбонила и тиазол-2-илкарбонила.
[0171] В некоторых вариантах осуществления настоящего изобретения способ включает введение соединения структуры формулы Ic:

или его фармацевтически приемлемой соли, где R3 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и -L-R5, где L представляет собой C1-4 алкилен.
[0172] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе, представляет собой соединение формулы Ic или его фармацевтически приемлемую соль, где R3 выбран из циклогексила, циклопентила, морфолино, фенила, пиперидинила, пиридинила, тетрагидрофуранила, тетрагидропиранила, 1,2,3,4-тетрагидронафтила и тиазолила, каждый из которых является необязательно замещенным 1-3 заместителями, выбранными из метила, метокси, трифторметила, ацетила и -N3.
[0173] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе, выбрано из
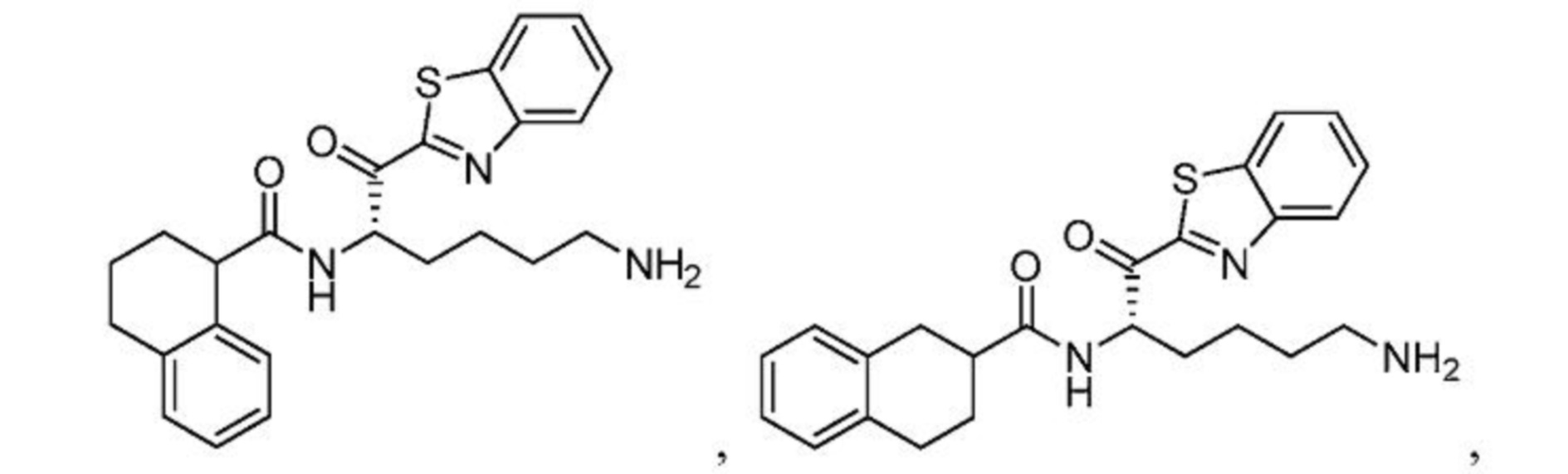

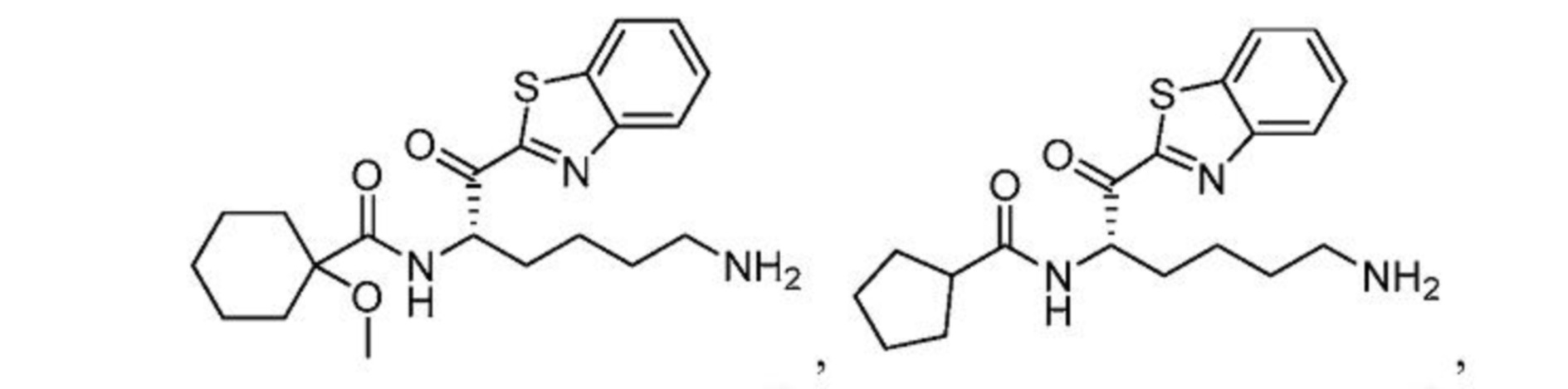

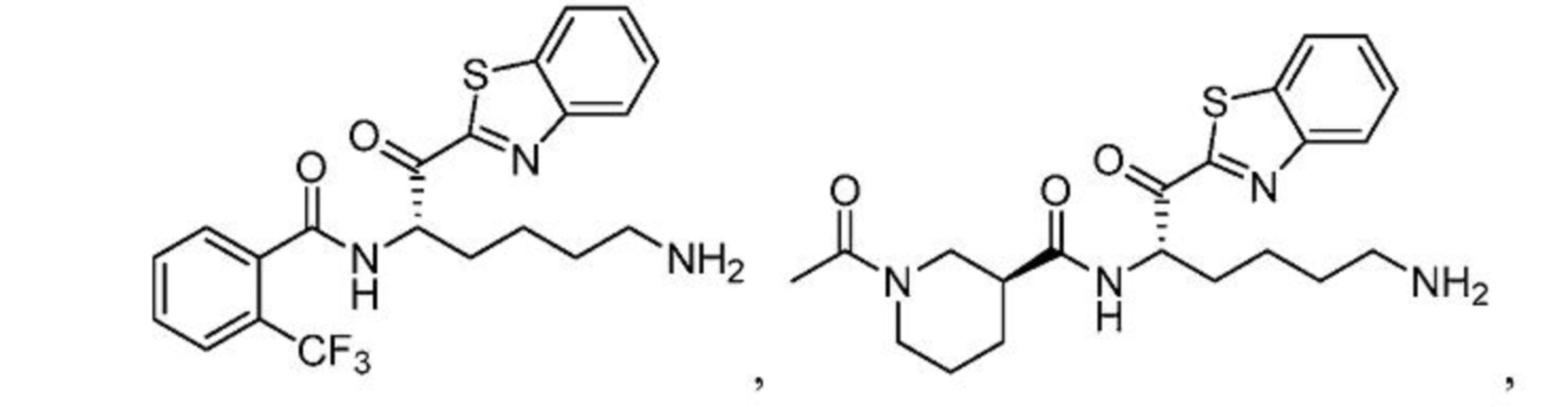
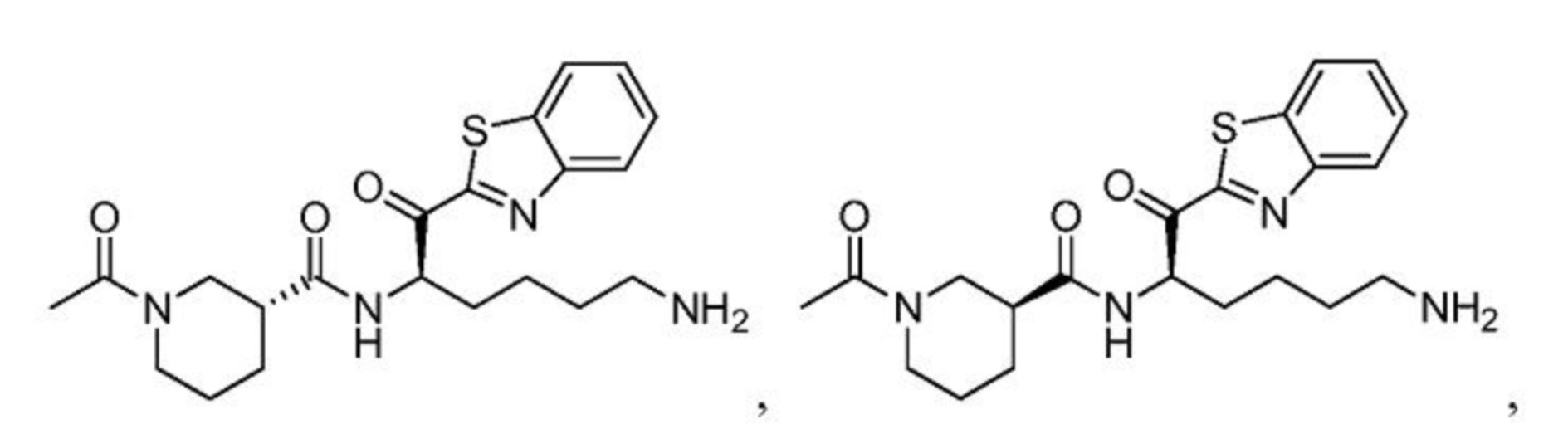




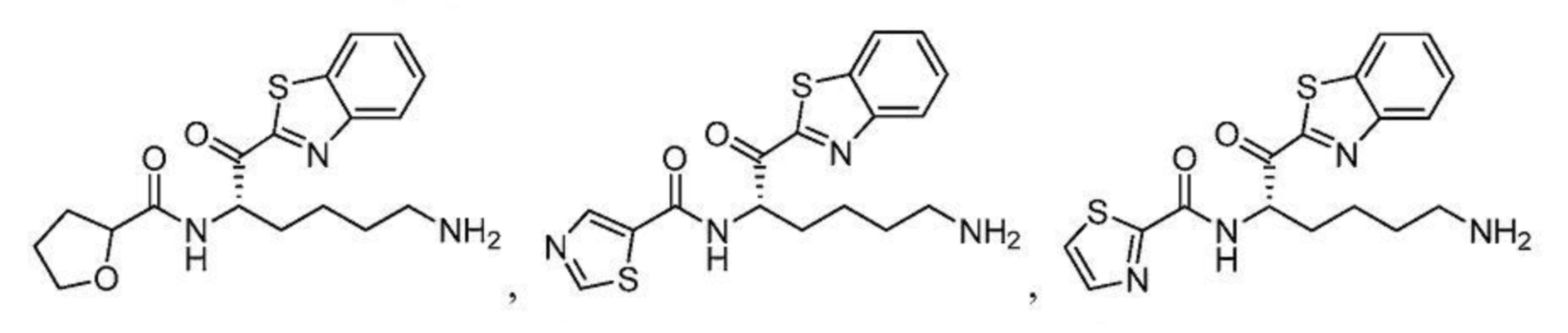
и их фармацевтически приемлемых солей.
[0174] В некоторых вариантах осуществления настоящего изобретения способ включает введение соединения структуры формулы Id
или его фармацевтически приемлемой соли,
где R3 выбран из C6-10 арила, 5-12-членного гетероарила, C3-8 циклоалкила, 5-12-членного насыщенного гетероциклила и L R5,
где L представляет собой C1-4 алкилен.
[0175] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе, представляет собой соединение формулы Id или его фармацевтический приемлемую соль, где R3 выбран из циклогексила, циклопентила, морфолино, фенила, пиперидинила, пиридинила, тетрагидрофуранила, тетрагидропиранила, 1,2,3,4-тетрагидронафтила и тиазолила, каждый из которых является необязательно замещеным 1-3 заместителями, выбранными из метила, метокси, трифторметила, ацетила и -N3.
[0176] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе, выбрано из
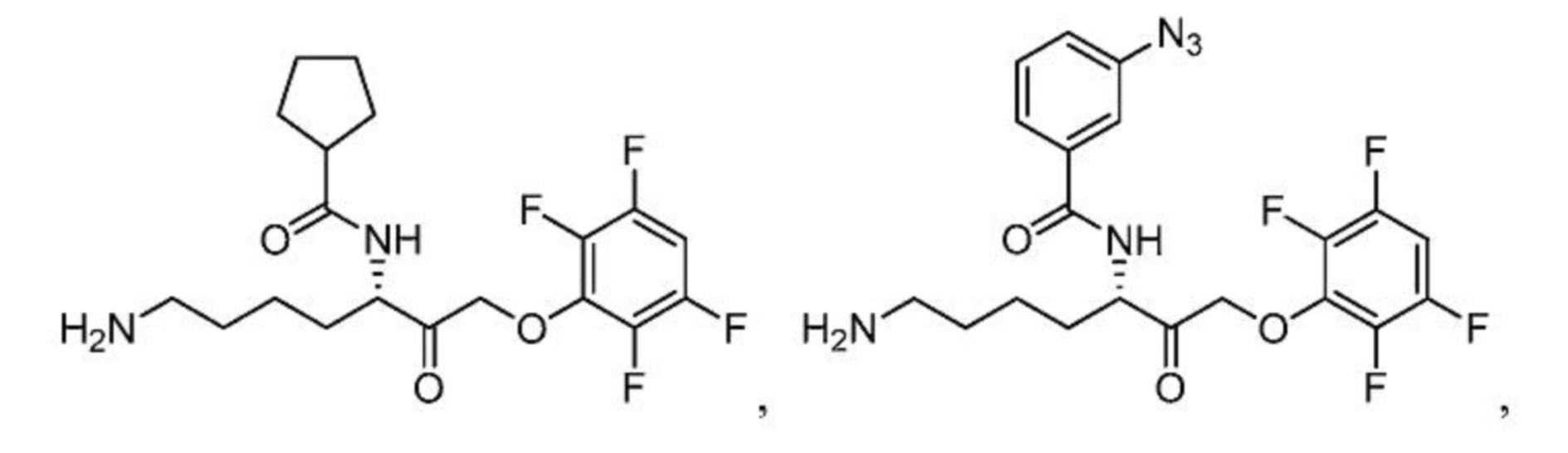

и их фармацевтически приемлемых солей.
[0177] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе, выбрано из

и их фармацевтически приемлемых солей.
[0178] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе по настоящему изобретению, представляет собой соединение формулы I, где Z выбран из пиридин-2-илкарбонила и тиазол-2-илкарбонила, и R3 выбран от С6-10 арила и С3-8 циклоалкила. В некоторых таких вариантах осуществления настоящего изобретения соединение, используемое в способе, выбрано из

и их фармацевтически приемлемых солей.
[0179] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе по настоящему изобретению, представляет собой соединение формулы I, где R4 выбран из C1-4 алкила и C1-4 галогеналкила. В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе, представляет собой

или его фармацевтически приемлемую соль.
[0180] В некоторых вариантах осуществления настоящего изобретения соединение, используемое в способе, выбрано из


и их фармацевтически приемлемых солей.
[0181] В публикации Curtis et al. описано соединение, обозначенное как A71561 (см. Infection and Immunity, 2002. 70(12): 6968-6975). A71561 представляет собой смесь двух диастереомеров: 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(S)-лизинил)бензотиазола и 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(S)-лизинил)бензотиазола. A71561 не является селективным ингибитором Kgp, поскольку его сродство к человеческому трипсину (Ki=30 нМ) слишком близко к сродству к Kgp (Ki=0,9 нМ). Curtis не высказал предположения о том, что A71561 или структурно подобные соединения могут использоваться для лечения нарушений мозговой деятельности (например, болезни Альцгеймера), диабета, сердечно-сосудистых заболеваний, артрита или поражения сетчатки.
[0182] Способ по настоящему изобретению, в частности, охватывает применение 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]лизинил)бензотиазола, 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]лизинил)бензотиазола (соединений формулы (III) и (IV), соответственно), их смесей и фармацевтически приемлемых солей
[0183] В некоторых вариантах осуществления настоящего изобретения соединение формулы (III) не содержит или по существу не содержит (содержит менее 100000 частей на миллион) 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(S)-лизинил)бензотиазол. В некоторых вариантах осуществления настоящего изобретения соединение формулы (III) состоит из 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(S)-лизинил)бензотиазола:

[0184] В некоторых вариантах осуществления настоящего изобретения соединение формулы (III) будет включать (а в некоторых вариантах осуществления изобретения состоит из) смесь(и) стереоизомеров 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(S)-лизинил)бензотиазола и 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(R)-лизинил)бензотиазола:

[0185] Стереоизомеры 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(S)-лизинил)бензотиазол и 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(R)-лизинил)бензотиазол могут присутствовать, например, в соотношении более 20:1. В некоторых вариантах осуществления настоящего изобретение указанное соотношение будет находиться в интервале от 20:1 до 2:1. В некоторых вариантах осуществления настоящего изобретения указанное соотношение будет составлять менее 2:1.
[0186] В некоторых вариантах осуществления настоящего изобретения соединение формулы (IV) будет включать (а в некоторых предпочтительных вариантах осуществления изобретения состоит из) 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(S)-лизинил)бензотиазол(а)

[0187] Таким образом, данное изобретение охватывает применение соединения формулы (IV), которое не содержит или по существу не содержит (содержит менее 100000 частей на миллион) 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(R)-лизинил)бензотиазол(а).
[0188] В некоторых вариантах осуществления настоящего изобретения соединение формулы (IV) содержит смесь 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(S)-лизинил)бензотиазола и 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(R)-лизинил)бензотиазола

[0189] 2-(N-[N-(3-Фенилпропаноил)-(S)-нипекотинил]-(S)-лизинил)бензотиазол и 2-(N-[N-(3-фенилпропаноил)-(S)-нипекотинил]-(R)-лизинил)бензотиазола могут присутствовать, например, в соотношении более 20:1. В некоторых вариантах осуществления настоящего изобретения соотношение находится в интервале от 20:1 до 2:1. В некоторых вариантах осуществления изобретения соотношение составляет менее 2:1.
[0190] В некоторых вариантах осуществления настоящего изобретения заболевание или состояние выбрано из нарушения мозговой деятельности, заболевания пародонта, диабета, сердечно-сосудистого заболевания, артрита, преждевременных родов, пневмонии, рака, заболевания почек, заболевания печени, поражения сетчатки и глаукомы.
[0191] В некоторых вариантах осуществления настоящего изобретения заболевание или состояние представляет собой нарушение мозговой деятельности.
[0192] В некоторых вариантах осуществления настоящего изобретения нарушение мозговой деятельности выбрано из болезни Альцгеймера, синдрома Дауна, эпилепсии, аутизма, болезни Паркинсона, эссенциального тремора, фронто-темпоральной деменции, прогрессирующего надъядерного паралича, бокового амиотрофического склероза, болезни Гантингтона, рассеянного склероза, умеренных когнитивных нарушений, возрастного ухудшения памяти, хронической травматической энцефалопатии, инсульта, болезни телец Леви, множественной системной атрофии, шизофрении и депрессии.
[0193] В некоторых вариантах осуществления нарушение мозговой деятельности представляет собой болезнь Альцгеймера.
[0194] В некоторых вариантах осуществления настоящего изобретения способ дополнительно включает введение пациенту одного или нескольких лекарственных средств, выбранных из ингибиторов холинэстеразы, модуляторов серотонина, модуляторов NMDA, терапевтических средств Αβ-направленного действия, терапевтический средств АпоЕ-направленного действия, терапевтических средств микроглия-направленного действия, терапевтических средств направаленного действия на гематоэнцефалический барьер, терапевтических средств нау-направленного действия, терапевтических средств комплемент-направленного действия и противовоспалительных терапевтических средств.
[0195] В некоторых вариантах осуществления настоящего изобретения заболевание или состояние представляет собой заболевание пародонта. В некоторых вариантах осуществления настоящего изобретения заболевание или состояние представляет собой заболевание печени. В некоторых вариантах осуществления настоящего изобретения заболевание печени представляет собой неалкогольный стеатогепатит. В некоторых вариантах осуществления настоящего изобретения заболевание или состояние представляет собой поражение сетчатки. В некоторых вариантах осуществления поражение сетчатки представляет собой возрастную макулярную дегенерацию.
[0196] В некоторых вариантах осуществления настоящего изобретения заболевание или состояние представляет собой рак. В некоторых вариантах осуществления настоящего изобретения рак представляет собой рак молочной железы, рак ротовой полости, рак поджелудочной железы или мультиформную глиобластому.
[0197] В некоторых вариантах осуществления настоящего изобретения соединения по настоящему изобретению ингибируют активный Kgp в головном мозге млекопитающего, например человека или животного (например, собаки), и являются цитопротективными или нейропротективными. Термин «нейропротективный» означает, что соединения предотвращают аберрантные изменения в нейронах или гибель нейронов. Следовательно, соединения по настоящему изобретению могут применяться, например, при лечении нарушений мозговой деятельности (например, нейродегенеративных заболеваний, таких как болезнь Альцгеймера, синдром Дауна, эпилепсия, аутизм, болезнь Паркинсона, эссенциальный тремор, фронто-темпоральной деменция, прогрессирующий надъядерный паралич, боковой амиотрофический склероз, болезнь Гантингтона, рассеянный склероз, умеренное когнитивное нарушение, возрастное ухудшение памяти, хроническая травматическая энцефалопатия, инсульт, болезнь телец Леви, множественная системная атрофия, шизофрения, депрессия и т.д.), сахарного диабета, сердечно-сосудистого заболевания, артрита, поражения сетчатки (например, возрастной макулярной дегенерации) и глаукомы.
[0198] Как видно из фигуры 1, 2-(N-[N-(3-фенилпропаноил)-(R)-нипекотинил]-(R)-лизинил)бензотиазол эффективен в предотвращении гибели клеток. Как видно из фигуры 2, соединение 1 и соединение 2 эффективны в предотвращении гибели клеток. Кроме того, модификации этих [4-амино-1-(бензотиазол-2-карбонилбутил]амидов 1-(3-фенилпропионил)пиперидин-3-карбоновой кислоты могут улучшить свойства (например, нейропротективные свойства) соединений по изобретению и/или сделать соединения по настоящему изобретению более подходящими для системного введения. Данные in vitro анализа показывает, что соеднения этого типа могут применяться для защиты клеток от гингипаин-индуцированной гибели (фигура 2).
[0199] Игибиторы Kgp, которые описаны в данном изобретении, могут быть введены в любой подходящей дозе способами по настоящему изобретению. Обычно ингибитор Kgp вводится в дозе в интервале от примерно 0,1 мг до примерно 1000 мг на килограмм массы тела пациента (т.е. примерно 0,1-1000 мг/кг). Доза ингибитора Kgp может составлять, например, примерно 0,1-1000 мг/кг, примерно 1-500 мг/кг, примерно 25-250 мг/кг или примерно 50-100 мг/кг. Доза ингибитора Kgp может составлять примерно 1, 2, 3, 4, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 85, 90, 95, 100, 150, 200, 250, 300, 350, 400, 450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950 или 1000 мг/кг. Дозировки могут изменяться в зависимости от потребностей пациента, тяжести заболевания, подлежащего лечению, и конкретного препарата, подлежащего введению. Доза, вводимая пациенту, должна быть достаточной, чтобы привести к благоприятному терапевтическому ответу у пациента. Величина дозы также будет определяться наличием, природой и степенью тяжести каких-либо неблагоприятных побочных эффектов, которые сопровождают введение препарата данному пациенту. Специалист данной области техники способен определить подходящую дозу для конкретной ситуации. Общая дозировка может быть разделена и вводиться частями в течение определенного периода времени, подходящего для лечения судорожного приступа.
[0200] Ингибиторы Kgp могут вводиться в течение периода времени, который будет изменяться в зависимости от природы конкретного расстройства, его тяжести и общего состояния пациента, которому вводится ингибитор Kgp. Введение может осуществляться, например, каждый час, каждые 2 часа, три часа, четыре часа, шесть часов, восемь часов или два раза в день, включая каждые 12 часов или любой промежуточный интервал. Введение может осуществляться один раз в день, один раз каждые 36 часов или 48 часов или один раз в месяц или несколько месяцев. После лечения пациент может отслеживать изменения своего состояния, а также облегчение симптомов расстройства. Дозировка ингибитора KgpА может быть увеличена в случае, если пациент существенно не реагирует на конкретный уровень дозировки, или может быть уменьшена, если наблюдается облегчение симптомов заболевания,и если заболевание было скорректировано или если при конкретной дозировке проявляются неприемлемые побочные эффекты.
[0201] Терапевтически эффективное количество ингибитора Kgp может быть введено пациенту по схеме лечения, включающей интервалы по меньшей мере 1 час, 6 часов, 12 часов, 24 часов, 36 часов или 48 часов между введением доз. Введение может осуществляться с интервалами по меньшей мере 72, 96, 120, 144, 168, 192, 216 или 240 часов (т.е. 3, 4, 5, 6, 7, 8, 9 или 10 дней). В некоторых вариантах осуществления настоящего изобретения введение одного или несколько ингибиторов KgpА проводится постоянно в течение периода от нескольких месяцев до нескольких лет. Соответственно, некоторые варианты осуществления настоящего изобретения относятся к способу лечения заболевания или состояния, связанного с инфекцией P. gingivalis, как описано выше, где соединение вводится пациенту в течение по меньшей мере одного года. В некоторых вариантах осуществления настоящего изобретения соединение вводится пациенту в течение по меньшей мере 10 лет. В некоторых вариантах осуществления соединение вводится пациенту в течение по меньшей мере 60 лет.
[0202] Введение ингибиторов Kgp в соответствии со способами по настоящему изобретению обычно приводит к снижению циркулирующих уровней активного Kgp у пациента и/или снижению уровня активного Kgp в головном мозге. В некоторых вариантах осуществления настоящего изобретения введение ингибитора Kgp в соответствии со способами изобретения приводит, по меньшей мере, к снижению циркулирующих уровней активного Kgp и/или, по меньшей мере, к снижению на 20% содержания активного Kgp в головном мозге. Например, циркулирующие уровни Kgp и/или уровни Kgp в головном мозге предпочтительно снижаются на от примерно 25% до примерно 95%, от примерно 35% до примерно 95%, от примерно 40% до примерно 85% или от примерно 40% до примерно 80% по сравнению с соответствующими уровнями Kgp за 24 часа до первого введения ингибитора Kgp.
[0203] Игибиторы Kgp могут вводиться отдельно или в сочетании с одним или несколькими дополнительными терапевтически активными средствами, как описано выше. Одно или несколько дополнительных терапевтически эффективных средств включают, например, (i) фармацевтически приемлемое средство, которое ингибирует продуцирование аргинин-специфичного гингипаина А (RgpA) и/или аргинин-специфичного гингипаина B, транслокацию RgpB и/или Kgp в большой круг кровообращения или головной мозг и/или патологическое действие (например, нейротоксические эффекты) RgpA, RgpB или KgpА в организме млекопитающего; (ii) противобактериальное средство, которое является бактериостатическим или бактерицидным по отношению к P. gingivalis; (iii) одно или несколько антител, которые связываются с RgpA, RgpB и/или Kgp (например, 18E6, которое связывается с первой половиной иммуноглобулинового домена RgpB; Kgp-специфическое моноклональное антитело, 7B9, которое распознает эпитоп внутри каталитического домена Kgp, антитела 61Bg 1.3 против RgpA, гуманизированные варианты любого из вышеперечисленных антител и т.д.); (iv) эпитопы антител, которые связываются с RgpA, RgpB и/или Kgp или другими белками, экспрессируемыми P. gingivalis; и (v) комбинации любых из вышеперечисленных лекарственных средств.
[0204] Дополнительные терапевтически активные средства включают также средства, снижающие уровень Aβ пептидов, снижающие патогенный уровень тау, стабилизаторы микротрубочек, средства, способные удалять или удаляющие атеросклеротические бляшки, средства, которые снижают уровни циркулирующего β-амилоида и тау, модуляторы аутофагии, регуляторы уровня нейротрансмиттеров, ингибиторы GADA(А) α5 рецепторов, а также дополнительные средства, которые способствуют поддержанию и/или улучшению когнитивных функций и устранению функциональных дефектов при болезни Альцгеймера и/или замедляют нарушение когнитивных функций и развитие функциональных дефектов при болезни Альцгеймера.
[0205] Фармацевтические композиции по настоящему изобретению могут содержать один или несколько ингибиторов Kgp, как описано в настоящем описании, в комбинации с ритонавиром (RTV), который может повышать биологическую доступность и проникновение через гематоэнцефалический барьер. Например, ритонавир обычно комбинируется с пероральными ингибиторами пептидной протеазы ВИЧ для повышения уровней в плазме путем ингибирования фермента Р450 3A4 и, таким образом, снижения пресистемного метаболизма (см. Walmsley, et al., N. Engl. J. Med., 2002. 346(26): 2039-46). Кроме того, RTV связывается с Р-гликопротеином, трансмембранным эффлюксным насосом, который находится во многих тканях, включая гематоэнцефалический барьер, обеспечивая совместно вводимым соединениям лучший доступ к мозгу (см. Marzolini, et al., Mol. Pharm., 2013. 10(6): 2340-9). Таким образом, комбинация RTV и ингибиторов Kgp может использоваться для повышения концентрации в плазме и содержания в мозге ингибиторов гингипаина. Показано, что пероральное введение RTV за 15 минут до введения ингибитора Kgp, KYT-36, увеличивает период его полураспада (фигура 9). Поэтому ожидается, что RTV также будет увеливать период полураспада других ингибиторов Kgp.
[0206] В некоторых вариантах осуществления соединение по настоящему изобретению может вводиться вместе с природными ингибиторами гингипаина, включая мелабарикон C (melabaricone C), выделенный из мускатного ореха, или полифенольные соединения, полученные из растений, таких как клюква, зеленый чай, яблоко и хмель, для лечения или предотвращения нарушений мозговой деятельности. Противомикробные пептиды, существующие и не существующие в природе, включая х-казеиновый пептид (109-137) 34, гистатин 5, CL (14-25), CL (K25A) и CL (R24A, K25A), также могут вводиться в комбинации с ингибиторами Kgp по настоящему изобретению, (см., например, Taniguchi et al., Biopolymers, 2014. 102(5): 379-89).
[0207] Ингибиторы Kgp, которые описаны в данном изобретении, могут вводиться с антителами действия, направленного на гингипаины или другие белков Р. gingivalis. Антитела могут зависеть от повреждения гематоэнцефалического барьера для доступа к мозгу или периферического влияния на гингипаины и распространение P. gingivalis. Антитела могут также способствовать стимулированию эффективности иммунной системы в очистке бактерии. Могут использоваться новые или существующие антитела против RgpA, RgpB или Kgp, включая 18E6 и 7B9. Антитело 61BG 1.3 против RgpA ранее продемонстрировало эффективность при местном применении в предотвращении повторной колонизации P. gingivalis после лечения заболевания пародонта. См. Booth et al., Infect. Immun., 1996. 64(2): 422-7. Антитела предпочтительно необходимо гуманизировать для применения у людей. Могут использоваться методы, известные в области доставки биологических веществ для увеличения периода полураспада и проникновения в мозг, включая, но без ограничения, внутривенное введение, подкожную доставку, интраназальную доставку, интратекальную доставку, векторный перенос и прямую доставку в мозг.
[0208] Способы по изобретению также включают введение ингибиторов Kgp, которые описаны в данном изобретении, с одним или несколькими из следующих дополнительных терапевтически активных средств или их фармацевтически приемлемыми солями: производное аргинина; гистатин 5; бакуловирус р35; модификатор цитокинового ответа точечного мутанта вируса коровьей оспы (CrmA (Asp > Lys)); фенилаланил-уреидо-цитруллинил-валил-циклоаргинал (FA-7°C 1); (ациклокси)метилкетон (Cbz-Phe-Lys-CH2OCO-2,4,6-Me3Ph); пептидилхлорметилкетоны (например, хлорметилкетоновые производные аргинина, хлорметилкетоновые производные лизина и т.п.); фторметилкетоны; бромметилкетоны; кетопептиды; [4-амино-1(S)-(бензотиазол-2-карбонил)бутил]амид 1-(3-фенилпропионил)пиперидин-3(R,S)-карбоновой кислоты (A71561); азапептидный фумарамид; азапептидные акцепторы Михаэля; бензамидиновые соединения; ациклометилкетон; ингибиторы активированного фактора X (например, DX-9065a); недиализуемая клюквенная фракция; клюквенная полифенольная фракция; ингибитор панкреатического трипсина; Cbz-Phe-Lys-CH2O-CO-2,4,6-Me3-Ph;E-64; хлоргексидин; цинк (например, ацетат цинка); или комбинация из двух, трех или более любых из вышеперечисленных терапевтически активных средств.
[0209] В некоторых из этих вариантов осуществления настоящего изобретения Zn может повышать активность и селективность соединений (например, хлоргексидина, бензамидина и т.д.), используемых в способах по настоящему изобретению. Бензамидиновые соединения включают в себя, например, следующие соединения и их производные:
[0210] Ингибитор лизин-специфичного гингипаина по изобретению можно вводить в той же композиции в качестве дополнительного терапевтически активного средства. В качестве альтернативы, дополнительное терапевтически активное средство можно вводить отдельно перед введением, одновременно с введением или после введения ингибитора лизин-специфичного гингипаина.
[0211] Так же как у людей, инфекция P. gingivalis и заболевание пародонта является одним из наиболее распространенных инфекционных заболеваний, поражающих взрослых собак и кошек. Используя взрослых гончих собак, исследователи показали наличие Rgp в образцах тромбоцитов из бляшек гончих собак, получавших специфическую мягкую диету для увеличения образования налета на поверхностях зубов. (См., например, Davis and Head, Front. Pharmacol., 2014. 5: 47; Reichart, et al., Journal of Periodontal Research, 1984. 19(1): 67-75; Kataoka, S., et al., FASEB J., 2014 28(8):3564-78.). Собаки и кошки с P. gingivalis инфекцией и гингипаинами в головном мозге и сердечно-сосудистой системе могут страдать заболеванием пародонта, умренными нарушениями когнитивных функций, возрастным ухудшением памяти, поражением или генерализованным ускоренным старением вследствие гингипаин-индуцированной гибели клеток, которые можно было бы лечить или предотвращать с помощью композиций соединений формулы I, II, III, IV или V.
VII. Способы обнаружения P. gingivalis и диагностика состояний, связанных с инфекцией P. gingivalis
[0212] Настоящее изобретение также относится к диагностическому тесту на гингипаины или P. gingivalis в головном мозге или образцах пациентов с целью диагностики или прогнозирования нарушений мозговой деятельности или выявления наиболее вероятных пациентов для лечения с использований соединений, описанных в данном изобретении. Предшествующий уровень техники показал изменения в профилях сыворотки в присутствии инфекции P. gingivalis. Новый метод анализа для диагностики или прогнозирования риска развития нарушения мозговой деятельности заключается в проведении иммуно-ферментного анализа (enzyme-linked immunosorbent assay - ELISA) а слюне, спинно-мозговой жидкости (CSF) или крови, например, для обнаружения одного или обоих гингипаинов. Предполагается, что уровни содержания гингипаина или других маркеров P. ginigivalis в слюне, крови и CSF должны быть выше у пациентов повышенного риска и пациентов, которые являются наиболее вероятными пациентами для лечения. Разработка ELISA представляет собой довольно простой процесс, известный специалистам в данной области техники, с использованием одного антитела против мишени для захвата мишени и второго меченого антитела против другого эпитопа на мишени для считывания количественных данных. Для этого могут использоваться коммерчески доступные или вновь произведенные антитела. Иммобилизованные или меченые соединения, описанные в настоящем описании (например, с биотином или HRP) могут использоваться для замены одного или обоих антител. Для этой цели могут использоваться соединения, модифицировнные с помощью «клик-химии», как показано на фигуре 10. Диагностика может включать обнаружение одного или нескольких гингипаинов. Для повышения чувствительности может использоваться биотинилирование антитела обнаружения.
[0213] В качестве альтернативы, вместо обнаружения наличия или отсутствия гингипаинов может использоваться анализ их активности в слюне, CSF или крови. Это должно обеспечить преимущество считывания по наиболее биологически релевантному фактору (например, активности), например, с применением лечения или без него. Методы разработки ферментных анализов известны специалистам данной области техники. Тест слюны, известный как тест BANA (тест, позволяющий выявить фермент, гидролизующий бензоиларгининнафтиламид), является коммерчески доступным для применения в стоматологии, чтобы проверить на протеазы из P. gingivalis и 2 других бактерии ротовой полости. Тест BANA представляет собой небольшую пластиковую карту, к которой прикреплены две отдельные матрицы реагентов в виде полосок на карте. Матрица нижнего белого реагента пропитана N-бензоил-DL-аргинин-B-нафтиламидом - BANA). Образцы поддесневых бляшек прикладывают к нижней матрице, а затем на верхнюю матрицу наносится дистиллированная вода. Затем нижняя матрица сгибается для контактирования с верхней матрицей. Матрица верхнего реагента цвета буйволовой кожи содержит хромогенный диазореагент, который вступает в реакцию с одним из продуктов гидролиза, дающую голубое крашивание. Реакция происходит, когда полоска на пластике помещается в инкубатор при 35 градусах C на 5 минут. С помощью субстрата BANA обнаруживаются по меньшей мере 3 различных бактерии полости рта, однако он не является специфическим для P. gingivalis. Тест BANA может быть использован для выявления людей с риском нарушений мозговой деятельности или вероятных пациентов для лечения. В качестве альтернативы, подложка BANA может заменяться в подобных форматах или в жидком тесте специфической подложкой для RgpA, RgpB и/или Kgp.
[0214] Реагенты, которые связываются с активными гингипаинами, включая, но без ограничения, описанные в данном изобретении, могут использоваться для осаждения только активных гингипаинов с последующим обнаружением, например, моноклональных. В качестве альтернативы, антитело или другое вещество с высоким сродством связывания может использоваться для осаждения гингипаина из CSF с последующим протеазным анализом с использованием меченого субстрата, который позволяет увеличить флуоресцентное или колорометрическое считывание по мере того, как меченый субстрат подвергается разложению.
[0215] Настоящее изобретение также относится к диагностической визуализации P. gingivalis или его гингипаинов в человеческом мозге. Любой агент, который связывается с гингипаинами, включая, но без ограничения, соединения по настоящему изобретению и другие соединения, описанные в других литературных источниках, могут быть помечены F18 или другими рентгенографическими маркерами и визуализированы с помощью ПЭТ или ОФЭКТ сканиования. Положительный сигнал будет являться показанием к лечению с помощью соединений, описанных в настоящем изобретении. В предпочтительном, неограничивающем варианте осуществления настоящего изобретения соединение 45, описанное в настоящем изобретении, модифицировано с помощью «клик-химии» для введения радиоактивной метки, которая может визуализироваться с помощью ПЭТ или ОФЭКТ (фигура 10).
VIII. Примеры
Пример 1
[0216] Клетки нейробластомы SH-SY5Y культивируют и дифференцируют в присутствии 5 мкМ ретиноевой кислоты с использованием известных методов [Saberi S., et al. Cell Mol. Neurobiol. 2013. 33:747-751]. Дифференцировку в нейронные клетки контролируют исследованием разрастания нейритов. Дифференцированные клетки подвергают воздействию 100 нМ Kgp и/или RgpB в течение 24 часов в присутствии или в отсутствие ингибиторов Kgp. Результаты записывают с использованием камеры цифрового микроскопа (фигура 1C, фигура 2С). Гингипаины являются токсичными к клеткам, в то время как COR соединения предотвращают цитотоксичность.
Пример 2
[0217] Взрослых самцов мышей (CD-1, примерно 25 г, n=6 на группу) анестезируют, инъекции вводят внутригиппокампально с одной стороны с использованием стандартных стереотаксических методов. Гингипаины RgpB и Kgp, очищенные от P. gingivalis, разбавляют перед инъекцией до 10 мкг/мл. Через семь дней после операции животных анестезируют, перфузируют и гуманно умерщвляют, мозг животных удалялют и рассекают на секции для гистологического анализа. После этого выполняют окрашивание частей гиппокампа с помощью Fluoro-Jade красителя для оценки нейродегенерации (Schmued LC and Hopkins KJ, 2000). Fluoro-Jade-окрашивание идентифицирует клеточные тела, дендриты, аксоны и аксонные терминалы дегенерирующих нейронов, но не окрашивает здоровые нейроны, миелин или элементы сосудов.
[0218] Срезы головного мозга исследуют с помощью эпифлуоресцентного микроскопа (Nikon Microphot FXA), используя систему фильтров, подходящую для визуализации флуоресцеина или флуоресцеинизотиоцианата (FITC). Изображения получают с помощью камеры Leica DC и программного обеспечения Image Analysis (Leica IM50). Нейроны, разлагаюшие Fluoro-Jade C-позитивное окрашивание, проявляются ярко-желто-зеленым окрашиванием на темном фоне и четко идентифицируются в группах животных, обработанных гингипаинами. В группе, обработанной разбавителем, Fluoro-Jade C-позитивные клетки не наблюдаются (фигура 7).
Пример 3
Самок мышей Balb/с получают из Harlan Laboratories (USA) и дают им возможность акклиматизироваться. Мышей возраста 8 недель инфицируют, вводя перорально 109 КОЕ W83 P. gingivalis в 2% Na-КМЦ 2 раза в неделю в течение 6 недель. Контрольным мышам вводят в качестве ложного заражения только 2% Na-КМЦ. Через 6 недель после первичного инфицирования мышей умерщвляют, перфузируют и иссекают головной мозг. Мозг заливают и разрезают на секции. 18E6 иммуногистохимия для RgpB показывает инфильтрацию мозга у 3 из 6 мышей. Краситель De Olmos silver для нейродегенерации показывает окрашивание у 2 из 3 мышей с инфильтрацией (фигура 7).
Пример 4
[0219] Самок мышей Balb/cJ получают от Taconic и дают им возможность акклиматизироваться. Мышей возраста 8 недель заражают, вводя перорально 109 КОЕ W83 P. gingivalis в каждый 3-й день с использованием 4-х введений.
[0220] Тест распознавания нового объекта для исследования когнитивных функций начинают через 6 недель после первого инфицирования. Мышам дают возможность ознакомиться с тестовой клеткой в течение 2 минут за день до ознакомления с объектом. На следующий день мышам представляют для ознакомления в течение 5 минут два голубых деревянных параллелепипеда. Спустя 24 часа мышам представляют в течение 3 минут один голубой параллелепипед (с правой стороны) и одно розовое сердечко (с левой стороны, оба объекта из дерева). Время, в течение которого нос мыши направлен в сторону объекта на расстоянии 2 см от него, записывают. Мыши с ложным заражением тратят, в среднем, больше времени на исследование нового объекта по сравнению с зараженными мышами, которые, в среднем, тратят равное время на оба объекта, показывая нарушение когнитивных функций (фигура 8).
Пример 5
[0221] Самок мышей Balb/Cj получают от Taconic и дают им возможность акклиматизироваться. Мышей возраста 43-44 недель ежедневно в течение 42 дней инфицируют 109 КОЕ W83 P. gingivalis или штамма P. gingivalis с отсутствием экспрессии Kgp. Соединение 2 или носитель (2% ДМСО) вводят подкожно три раза в день в 21-42 дни. На 42 день мышей умерщвляют, мозг удаляют и замораживают. Образец, содержащий ткани гиппокампа, лизируют в буфере RIPA с ингибиторами протеазы и анализируют на экспрессию Abeta42 с использованием набора Amyloid beta 42 ELISA Kit, Mouse (Life Technologies). Инфицированные мыши показывают повышение содержания Аbeta42, что не наблюдается у KgpЕ-нокаутный мышей или у мышей, обработанный соединением 2 (фигура 10).
Пример 6
[0222] Тест оценки нарушений когнитивных функций проводят с использованием престарелых собак, как описано в публикации Araujo et al. [J. Alzheimers Dis, 2011. 26(1): 143-55]. Удаленный мозг заливают, разрезают на секции и оценивают инфильтрацию гингипаина, как описано в примере 1, с 7B9 антителом для Kgp (фигура 11).
Пример 7. Получение бензил-N-((5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогекил]карбамата
Схема 7
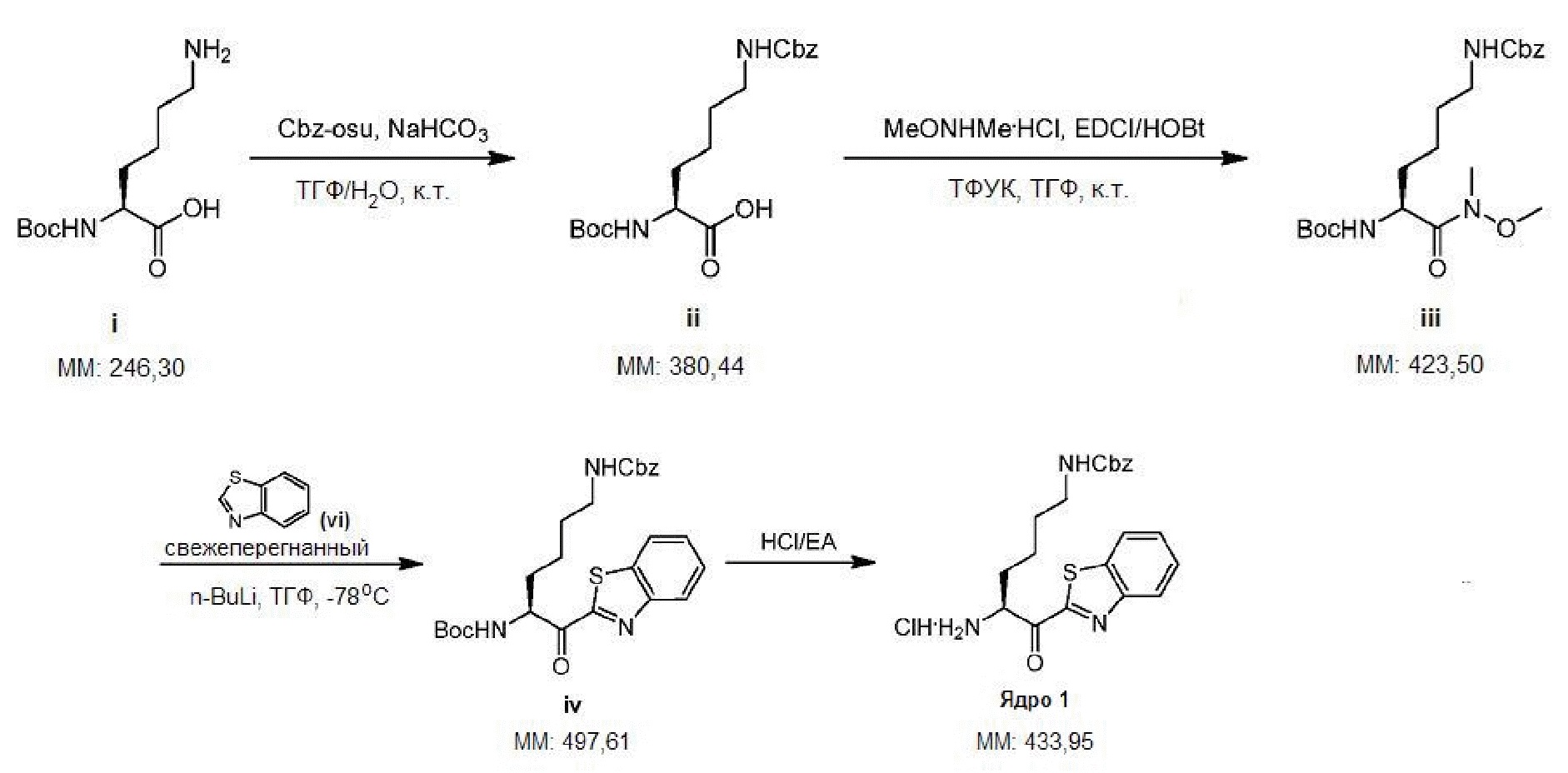
[0223] (2S)-6-(Бензилоксикарбониламино)-2-(трет-бутоксикарбониламин)капроновая кислота (ii). К раствору (2S)-6-амино-2-(трет-бутоксикарбониламино)капроновой кислоты (100,00 г, 406,01 ммоль, 1,00 экв.), NaHCО3 (102,33 г, 1,22 моль, 3,00 экв.) в ТГФ (1000 мл) и Н2О (1000 мл) при 0 °С добавляют CbzOSu (101,19 г, 406,01 ммоль, 1,00 экв.). После этого смесь перемешивают при 30 °С в течение 16 часов. Значение рН смеси доводят до 5-6 с помощью НСl, смесь экстрагируют EA (600 мл × 3), сушат над Na2SО4и концентрируют с получением (2S)-6-(бензилоксикарбониламино)-2-(трет-бутоксикарбониламино)капроновой кислоты (142,00 г, 373,26 ммоль, выход 91,9%) в виде масла желтого цвета.
[0224] трет-Бутил-N-[(1S)-5-(бензилоксикарбониламино)-1- [метокси(метил)карбамоил]пентил]карбамат (iii). К раствору (2S)-6-(бензилоксикарбониламино)-2-(трет-бутоксикарбониламино)капроновой кислоты (142,00 г, 373,26 ммоль, 1,00 экв.), N-метоксиметанамина (54,61 г, 559,89 ммоль, 1,50 экв.), EDCI (93,02 г, 485,24 ммоль, 1,30 экв.), HOBt (65,57 г, 485,24 ммоль, 1,30 экв.) в ДХМ (2 л) при 0 °С по каплям в течение 0,5 часа добавляют TEA (113,31 г, 1,12 моль, 3,00 экв.). После этого смесь перемешивают при 30 °С в течение 16 часов. ТСХ показывает, что реакция завершена. Затем смесь разбавляют водой (1000 мл) и слои разделяют. Органический слой последовательно промывают НСl (водн., 1 М, 500 мл) и насыщенным раствором NaHCО3 (водн., 500 мл), сушат над Na2SО4 и концентрируют с получением сырого продукта, который очищают с помощью колоночной хроматографии (силикагель, элюирование с градиентом: PE:EA от 5:1 до 1:1) с получением трет-бутил-N-[(1S)-5-(бензилоксикарбониламино)-1-[метокси(метил)карбамоил]пентил]карбамата (110,00 г., 259,74 ммоль, выход 69,6%) в виде масла светло-желтого цвета.
[0225] трет-Бутил-N-[(1R)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамат (iv). К смеси 1,3-бензотиазола (9,58 г, 70,83 ммоль, 3,00 экв.) в ТГФ (50 мл) по каплям при -65 °С в атмосфере N2добавляют n-BuLi (2,5 М в ТГФ, 28,3 мл). Смесь перемешивают при -65 °С в атмосфере N2 в течение 1 часа. Затем по каплям при -65 °С в атмосфере N2добавляют трет-бутил-N-[(1R)-5-(бензилоксикарбониламино)-1-[метокси(метил)карбамоил]пентил]карбамат (10,00 г, 23,61 ммоль, 1,00 экв.) в ТГФ (50 мл) и перемешивают реакционную смесь при -65 °С в атмосфере N2 в течение дополнительных 3 часов. ТСХ показывает, что реакция завершена. Смесь гасят насыщенным раствором NH4Cl (водн., 10 мл), после этого смесь экстрагирут EA (100 мл × 3), сушат над Na2SO4 и концентрируют, получая сырой продукт, который очищают с помощью колоночной хроматографии (силикагель, элюирование с градиентом: PE:EA от 5:1 до 3:1) с получением трет-бутил-N-[(1R)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамата (6,20 г, 12,46 ммоль, выход 52,8%) в виде масла светло-желтого цвета.
[0226] Бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамат (ядро 1). К раствору трет-бутил-N-[(1S)-1- (1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамата (10,84 г, 21,78 ммоль, 1,00 экв.) в EA (140 мл) добавляют HCl/EA (4 М, 12 мл). После этого реакционную смесь перемешивают при 30 °С в течение 15 часов. Реакционную смесь фильтруют, осадок на фильтре промывают EA (80 мл) и сушат в вакууме с получением гидрохлорида бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (7,52 г, 17,33 ммоль, выход 79,6%) в виде твердого вещества желтого цвета.
Пример 8. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]бензамида (1).
Схема 8

[0227] Бензил-N-[(5S)-5-бензамидо-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамат (1а). К смеси гидрохлорида бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (400 мг, 921,77 мкмоль, 1,00 экв.), TEA (280 мг, 2,77 ммоль, 3,00 экв.) в ДХМ (10 мл) по каплям при 0 °С в атмосфере N2 добавляют бензоилхлорид (156 мг, 1,11 ммоль, 1,20 экв.). Смесь перемешивают при 0 °С в течение 0,5 часа. ТСХ (РЕ:ЕА=5:1) показывает, что реакция завершена. Смесь промывают водой (5 мл × 3), сушат над Na2SO4и концентрируют с получением бензил-N-[(5S)-5-бензамидо-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 598,09 мкмоль, выход 64,9%) в виде твердого вещества светло-желтого цвета.
[0228] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]бензамид (1). Смесь бензил-N-[(5S)-5-бензамидо-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (250 мг, 498,41 мкмоль, 1,00 экв.) в AcOH (1 мл) добавляют HBr/AcOH (5,5 M, 1 мл). Смесь перемешивают при 30 °С в течение 0,5 часа. ЖХ-МС показывает, что реакция завершена. Добавляют воду (20 мл) и экстрагирут смесь MTBE (5 мл × 2). После этого водный слой лиофилизируют с получением сырого продукта, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]бензамида (20 мг, 41,54 мкмоль, выход 8,33%) в виде твердого вещества светло-желтого цвета. МС m/z=368,1 (МН+).
Пример 9. Получение N-[(1S-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]циклогексанкарбоксамида (2).
Схема 9

[0229]Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(циклогексанкарбониламино)-6-оксогексил]карбамат (2а). К смеси гидрохлорида бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (400 мг, 921,77 мкмоль, 1,00 экв.), циклогексанкарбоновой кислоты (142 мг, 1,11 ммоль, 1,20 экв.), EDCI (230 мг, 1,20 ммоль, 1,30 экв.), HOBt (162 мг, 1,20 ммоль, 1,30 экв.) в ДХМ (10 мл) по каплям при при 0 °С добавляют TEA (280 мг, 2,77 ммоль, 3,00 экв.). Смесь перемешивают при 30 °С в течение 16 часов. ЖХ-МС показывает, что реакция завершена. К смеси добавляют воду (10 мл), экстрагируют смесь с помощью ДХМ (10 мл × 2), промывают водой (10 мл), затем насыщенным раствором NaHCО3 (водн., 10 мл × 2), сушат над Na2SO4и концентрируют с получением сырого продукта, который очищают флэш-хроматографией с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(циклогексанкарбониламино)-6-оксогексил]карбамата (2а; 210 мг, сырой продукт) в виде твердого вещества желтого цвета.
[0230] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]циклогексанкарбоксамид (2). К смеси бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(циклогексанкарбониламино)-6-оксогексил]карбамата (200 мг, 393,98 мкмоль, 1,00 экв.) в АсОН (1 мл) добавляют HBr/AcOH (5,5 М, 1 мл). Смесь перемешивают при 30 °С в течение 0,5 часа. ЖХ-МС показывает, что реакция завершена. К смеси добавляют воду (20 мл) и экстрагирут смесь MTBE (5 мл × 2). Затем водный слой лиофилизируют с получением сырого продукта, который очищают с помощью препаративной ВЭЖХ с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]циклогексанкарбоксамида (2; 10 мг, 20,51 мкмоль, выход 5,21%) в виде твердого вещества светло-желтого цвета. МС m/z=374 (МН+).1H ЯМР (CD3OD, 400 МГц) δ 8,20 (дд, J=7,6, J=0,4, 1H), 8,14 (дд, J=7,6, J=1,2, 1H), 7,66-7,61 (м, 2H), 5,64 (дд, J=4,0, J=10,0, 1H), 3,02-2,93 (м, 2H), 2,38-2,35 (м, 1H), 2,18-2,10 (м, 1H), 1,84-1,55 (м, 9H), 1,48-1,17 (м, 6H).
Пример 10. Получение (3S-N[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамида (3).
Схема 10

[0231] трет-Бутил-(3S)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилат (3c). Смесь (3S)-1-трет-бутоксикарбонилпиперидин-3-карбоновой кислоты (190 мг, 829,59 мкмоль, 1,20 экв.), HATU (315 мг, 829,59 мкмоль, 1,20 экв.), DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) в ДМФА (5 мл) перемешивают при 0 °С в течение 1 часа. После этого у смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают смесь при 30 °С в течение 16 часов. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. К смеси добавляют ЕА (20 мл), промывают полученную смесь водой (10 мл × 3), сушат над Na2SO4 и концентрируют с получением трет-бутил-(3S)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (250 мг, 410,68 мкмоль, выход 59,4%, диастереомерный избыток 55%) в виде твердого вещетсва желтого цвета.
[0232]Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5- [[(3S)пиперидин-3-карбонил]амино]гексил]карбамат (3b). К смеси трет-бутил-(3S)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (250 мг, 410,68 мкмоль, 1,00 экв.) и EA (2 мл) добавляют HCl/EtOAc (4 М, 2 мл). Смесь перемешивают при 30 °С в течение 1,5 часа. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. Реакционную смесь фильтруют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-пиперидин-3-карбонил]амино]гексил]карбамата (3b; 200 мг, сырой продукт, диастереомерный избыток 67%) в виде твердого белого вещества.
[0233] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамат (3а). К смеси бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-пиперидин-3-карбонил]амино]гексил]карбамата (200 мг, 393,21 мкмоль, 1,00 экв.), ТЕА (119 мг, 1,18 ммоль, 3,00 экв.) в ДХМ (5 мл) добавляют 3-фенилпропаноилхлорид (80 мг, 471,85 мкмоль, 1,20 экв.) при 0 °С в атмосфере N2. Смесь перемешивают при 0 °С в течение 0,5 часа. ЖХ-МС показывает, что реакция завершена. Добавляют ДХМ (10 мл), промывают полученную смесь водой (5 мл × 3), сушат над Na2SO4 и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамата (3а; 220 мг, сырой продукт) в виде твердого вещества желтого цвета.
[0234](3S)-N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамид; 2,2,2-трифторуксусная кислота (3). К смеси бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамата (220 мг, 343,33 мкмоль, 1,00 экв.) в AcOH (1 мл) добавляют HBr/AcOH (5,5 М, 1 мл) при 30 °С в атмосфере N2. Смесь перемешивают при 30 °С в атмосфере N2 в течение 0,5 часа. ЖХ-МС показывает, что реакция завершена. Добавляют воду (20 мл) и полученную смесь экстрагирут MTBE (5 мл × 2). Затем водный слой лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (СН3CN/H2О/ТФУК) с получением (3S)-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамида (3; 15 мг, 29,61 мкмоль, выход 8,62%) в виде твердого вещества светло-желтого цвета. МС m/z=507,2 (МН+).
Пример 11. Получение (3R)-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамида (4).
Схема 11

[0235]трет-Бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилат (4c). К раствору (3R)-1-трет-бутоксикарбонилпиперидин-3-карбоновой кислоты (190 мг, 828,72 мкмоль, 1,20 экв.) в ДМФА (10 мл) при 0 °С добавляют HATU (320 мг, 841,60 мкмоль, 1,22 экв.) и DIPEA (267 мг, 2,07 ммоль, 2,99 экв.). После перемешивания в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-(аминохлоранил)-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 30 °С в течение 3 часов. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. Смесь разбавляют H2О (15 мл × 2) и экстрагируют с помощью EA (20 мл × 2). Объединенные органические слои промывают H2О (20 мл × 2), сушат над Na2SO4, фильтруют и концентрируют с получением трет-бутил-(5R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (4с; 600 мг, сырой продукт) в виде масла, которое используют непосредственно в следующей стадии без дополнительной очистки.
[0236] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5- [[(3R)-пиперидин-3-карбонил]амино]гексил]карбамат (4b). К раствору трет-бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (600 мг, 985,63 мкмоль, 1,00 экв.) в EA (10 мл) добавляют HCl/EA (4 М, 4 мл, 16,23 экв.). Реакционную смесь перемешивают при 30 °С в течение 1 часа. Суспензию фильтруют, получая осадок на фильтре, который, как подтверждает ЖХ-МС, представляет собой гидрохлорид бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамата (4b; 700 мг, сырой продукт), который используют непосредственно в следующей стадии без дополнительной очистки.
[0237]Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамат (4а). К раствору гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамата (700 мг, 1,28 ммоль, 1,00 экв.) в ДХМ (10 мл) добавляют TEA (2,19 г, 21,64 ммоль, 16,91 экв.). Смесь охлаждают до 0 °С и по каплям добавляют к смеси 3-фенилпропаноилхлорид (1,14 г, 6,76 ммоль, 5,28 экв.). Полученную смесь перемешивают при 0 °С в течение 30 минут. ЖХ-МС показывает, что реакция завершена. Реакционную смесь гасят H2О (20 мл) и экстрагируют ДХМ (20 мл × 2). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамата (4а; 600 мг, сырой продукт), который используют непосредственно в следующей стадии без дополнительной очистки.
[0238] (3R)-N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамид (4). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамата (600 мг, 936,34 мкмоль, 1,00 экв.) АсОН (10 мл) добавляют HBr/AcOH (936,34 мкмоль, 1,00 экв.) (2 мл, чистота 33%) в атмосфере N2. Реакционную смесь перемешивают при 32 °С в течение 30 минут. ЖХ-МС показывает наличие целевого продукта. Смесь разбавляют H2О (100 мл) и MeOH (20 мл) и промывают МТВЕ (50 мл × 2). Полученный раствор лиофилизируют, получая остаток, который очищают с помощью препаративной ВЭЖХ (подвижная фаза: CH3CN, H2О/ТФУК) с получением (3R)-N-[1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамида; трифторацетат 2,2,2-трифторуксусной кислоты (4; 30 мг, 40,83 мкмоль, выход 4,36%) в виде твердого вещества желтого цвета. МС m/z=507,2 (МН+).
Пример 12. Получение N[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-2-(трифторметил)бензамида (7).
Схема 12

[0239] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[2-(трифторметил)бензоил]амино]гексил]карбамат (7а). К смеси бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (400 мг, 921,77 мкмоль, 1,00 экв.), 2-(трифторметил)бензойной кислоты (210 мг, 1,11 ммоль, 1,20 экв.), HOBt (162 мг, 1,20 ммоль, 1,30 экв.), EDCI (230 мг, 1,20 ммоль, 1,30 экв.) в ДХМ (10 мл) при 0 °С добавляют DIPEA (358 мг, 2,77 ммоль, 3,00 экв.). Полученную смесь перемешивают при 30 °С в течение 16 часов. ЖХ-МС показывает, что реакция завершена. Смесь промывают водой (5 мл × 3), сушат над Na2SO4, концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[2-(трифторметил)бензоил]амино]гексил]карбамата (7а; 500 мг, сырой продукт) в виде твердого вещества желтого цвета.
[0240]N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]-2-(трифторметил)бензамид (7). К смеси бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[2-(трифторметил)бензоил]амино]гексил]карбамата (300 мг, 526,69 мкмоль, 1,00 экв.) в AcOH (1 мл) добавляют HBr/AcOH (5,5 М, 1 мл). Смесь перемешивают при 30 °С в атмосфере N2 в течение 0,5 часа. ЖХ-МС показывает, что реакция завершена. К смеси добавляют воду (20 мл) и экстрагирут полученную смесь MTBE (5 мл × 2). Затем водный слой лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (CH3CN/H2О/ТФУК) с получением трифторацетата N-[1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-2-(трифторметил)бензамида (7; 20 мг, 36,40 мкмоль, выход 6,9%) в виде твердого вещества светло-желтого цвета. МС m/z=437,0 (МН+).
Пример 13. Получение (3S)-1-ацетил-N-(6-амино-1-(бензо[d]тиазол-2-ил)-1-оксогексан-2-ил)пиперидин-3-карбоксамида (8 и 10).
Схема 13

[0241] трет-Бутил-(3S)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбокслат (8c). Смесь (3S)-1-трет-бутоксикарбонилпиперидин-3-карбоновой кислоты (693 мг, 3,02 ммоль, 1,20 экв.), HATU (1,15 г, 3,02 ммоль, 1,20 экв.), DIPEA (977 мг, 7,56 ммоль, 3,00 экв.) в ДМФА (20 мл) перемешивают при 0 °C в течение часа. Затем добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (1,00 г, 2,52 ммоль, 1,00 экв.) и перемешивают полученную смесь при 30 °С в течение 16 часов. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. К смеси добавляют ЕА (100 мл), промывают полученную смесь водой (50 мл × 3), сушат над Na2SO4 и концентрируют с получением трет-бутил-(3S)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (8c; 1,10 г, 1,81 ммоль, выход 71,7%) в виде твердого вещества желтого цвета.
[0242]Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-пиперидин-3-карбонил]амино]гексил]карбамат (8b). К смеси гидрохлорида трет-бутил-(3S)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (1,10 г, 1,81 ммоль, 1,00 экв.) в EA (10 мл) добавляют HCl/EA (4 М, 2 мл) и перемешивают полученную реакционную смесь при 30 °С в течение 2 часов. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. Реакционную смесь фильтруют с получением гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-пиперидин-3-карбонил]амино]гексил]карбамата (8b; 700 мг, сырой продукт) в виде твердого вещества светло-желтого цвета.
[0243] Бензил-N-[(5S)-5-[[(3S)-1-ацетилпиперидин-3-карбонил]амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамат (8а). К смеси гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3S)-пиперидин-3-карбонил]амино]гексил]карбамата (700 мг, 1,28 ммоль, 1,00 экв.), ТЕА (390 мг, 3,85 ммоль, 3,00 экв.) в ДХМ (5 мл) по каплям при 0°C в атмосфере N2добавляют ацетилхлорид (150 мг, 1,93 ммоль, 1,50 экв.) и перемешивают полученную реакционную смесь при 0 °C в атмосфере N2 в течение 1 часа. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. К смеси добавляют ДХМ (20 мл), промывают смесь водой (10 мл × 3), сушат над Na2SO4 и концентрируют с получением бензил-N-[(5S)-5-[[(3S)-1-ацетилпиперидин-3-карбонил]амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (8а; 800 мг, сырой продукт) в виде масла желтого цвета.
[0244] (3S)-1-Ацетил-N-(6-амино-1-(бензо[d]тиазол-2-ил)-1-оксогексан-2-ил)пиперидин-3-карбоксамид (8 и 10). К смеси бензил-N-[(5S)-5-[[(3S)-1-ацетилпиперидин-3-карбонил]амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (800 мг, 1,45 ммоль, 1,00 экв.) и AcOH (5 мл) добавляют HBr/AcOH (5,5 М, 2 мл). После этого реакционную смесь перемешивают при 30 °С в течение 0,5 часа. ЖХ-МС показывает, что реакция завершена. К смеси добавляют воду (30 мл), смесь экстрагирут MTBE (10 мл × 2), водный слой лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (CH3CN/H2О/ТФУК) с получением трифторацетата (3S)-1-ацетил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиперидин-3-карбоксамида (8; 21,20 мг, 39,96 мкмоль, выход 2,8%) и трифторацетата (3S)-1-ацетил-N-[(1R)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиперидин-3-карбоксамида (10; 24,20 мг, 45,61 мкмоль, выход 3,2%) в виде твердых веществ светло-желтого цвета. МС m/z=417,1 (МН+).
Пример 14. Получение (3R)-1-ацетил-N-(6-амино-1-(бензо[d]тиазол-2-ил)-1-оксогексан-2-ил)пиперидин-3-карбоксамида (9 и 11).
Схема 14

[0245]трет-Бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилат (9с). К раствору (3R)-1-трет-бутоксикарбонилпиперидин-3-карбоновой кислоты (633 мг, 2,76 ммоль, 1,20 экв.) в ДМФА (10 мл) при 0 °С добавляют HATU (2,62 г, 6.90 ммоль, 3,00 экв.) и DIPEA (892 мг, 6,90 ммоль, 3,00 экв.). После перемешивания в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-(аминохлоранил)-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (1,00 г, 2,30 ммоль, 1,00 экв.) и перемешивают полученную смесь при 30 °С в течение 6 часов. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. Смесь разбавляют H2О (15 мл × 2) и экстрагируют с помощью ЕА (20 мл × 2). Объединенные органические слои промывают H2О (20 мл × 2), сушат над Na2SO4, фильтруют и концентрируют с получением трет-бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1- карбоксилата (9с; 1,40 г, сырой продукт) в виде масла, которое используют непосредственно в следующей стадии без дополнительной очистки.
[0246] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамат (9b). К раствору трет-бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (821,36 мкмоль, 1,00 экв.) (1,4 г, сырой продукт) в ЕА (20,00 мл) добавляют HCl/EA (4 М, 600,00 мкл, 2,92 экв.). Реакционную смесь перемешивают при 30 °С в течение 4 часов. Суспензию фильтруют и собирают осадок на фильтре с получением гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамата (9b; 2,20 г, сырой продукт), который используют непосредственно в следующей стадии без дополнительной очистки.
[0247] Бензил-N-[(5S)-5-[[(3R)-1-ацетилпиперидин-3-карбонил]амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамат (9а). К раствору гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамата (4,04 ммоль, 1,00 экв.) (2,2 г, сырой продукт) в ДХМ (20 мл) добавляют TEA (1,23 г, 12,11 ммоль, 3,00 экв.). Смесь охлаждают до 0 °С, затем к смеси по каплям добавляют ацетилхлорид (475 мг, 6,05 ммоль, 1,50 экв.). Полученную смесь перемешивают при 0 °С в течение 30 минут. ЖХ-МС показывает, что реакция завершена. Реакционную смесь гасят H2О (30 мл) и экстрагируют ДХМ (20 мл × 2). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-5-[[(3R)-1-ацетилпиперидин-3-карбонил]амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (9а; 1,50 г, сырой продукт), который используют непосредственно в следующей стадии без дополнительной очистки.
[0248](3R)-1-Ацетил-N-(6-амино-1-(бензо[d]тиазол-2-ил)-1-оксогексан-2-ил)пиперидин-3-карбоксамид (9 и 11). К раствору бензил-N-[5-[[(3R)-1-ацетилпиперидин-3-карбонил]амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (2,30 ммоль, 1,00 экв.) (1,5 г, сырой продукт) в АсОН (10 мл) в атмосфере N2добавляют HBr/AcOH (2,30 ммоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 32 °С в течение 30 минут. ЖХ-МС показывает наличие целевого продукта. Смесь разбавляют H2О (100 мл) и MeOH (20 мл) и промывают МТВЕ (50 мл × 2). Полученный раствор лиофилизируют с получением остатка, который очищают с помощью препаративной ВЭЖХ (подвижная фаза: CH3CN/H2О/ТФУК) с получением трифторацетата (3R)-1-ацетил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиперидин-3-карбоксамида (9; 5,30 мг, 9,99 мкмоль, выход 0,4%) (выход для 4 стадий) и трифторацетата (3R)-1-ацетил-N-[(1R)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиперидин-3-карбоксамида (11; 49,70 мг, 119,32 мкмоль, выход 5,2%) (выход для 4 стадий) в виде твердого вещества желтого цвета. МС m/z=417,1 (МН+).
Пример 15. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-2-карбоксамида (12).
Схема 15

[0249]Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-2-карбониламино)гексил]карбамат (12a). К раствору пиридин-2-карбоновой кислоты (102 мг, 829,58 мкмоль, 1,20 экв.) в ТГФ (8 мл) при 0 °С добавляют HATU (315 мг, 829,58 мкмоль, 1,20 экв.) и DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) и перемешивают полученную смесь при 0 °С в течение 15 минут. Затем к смеси при 0 °С добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают реакционную смесь при 30 °С в течение дополнительных 3 часов. ЖХ-МС показывает полное расходование исходного материала. Реакционную смесь гасят H2О (30 мл), экстрагируют ЕА (20 мл × 2), органические фазы объединяют, промывают насыщенным раствором соли (30 мл) и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-2-карбониламино)гексил]карбамата (12а, 698 мг, сырой продукт) в виде твердого вещества желтого цвета.
[0250]N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-2-карбоксамид (12). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-2-карбониламино)гексил]карбамата (682 мг, 1,36 ммоль, 1,00 экв.) в АсОН (3 мл) при 20 °С добавляют HBr/AcOH (1 мл, чистота 33%), реакционную смесь перемешивают при 20 °С в течение 1 часа. ЖХ-МС показывает полное расходование исходного материала и наличие целевого соединения. После перемешивания в течение дополнительных 30 минут реакционную смесь гасят H2О (10 мл) и промывают МТВЕ (20 мл). Органическую фазу экстрагируют H2О (20 мл × 2), водные фазы объединяют и лиофилизируют, с получением сырого продукта, который очищают с помощью препаративной ВЭЖХ (CH3CN/H2O/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-2-карбоксамида (12; 67,44 мг, 139,78 мкмоль, выход 10,3%, энантиомерный избыток 74,4%) в виде твердого вещества желтого цвета. МС m/z=369,1 (МН+).
Пример 16. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-4-карбоксамида (13).
Схема 16
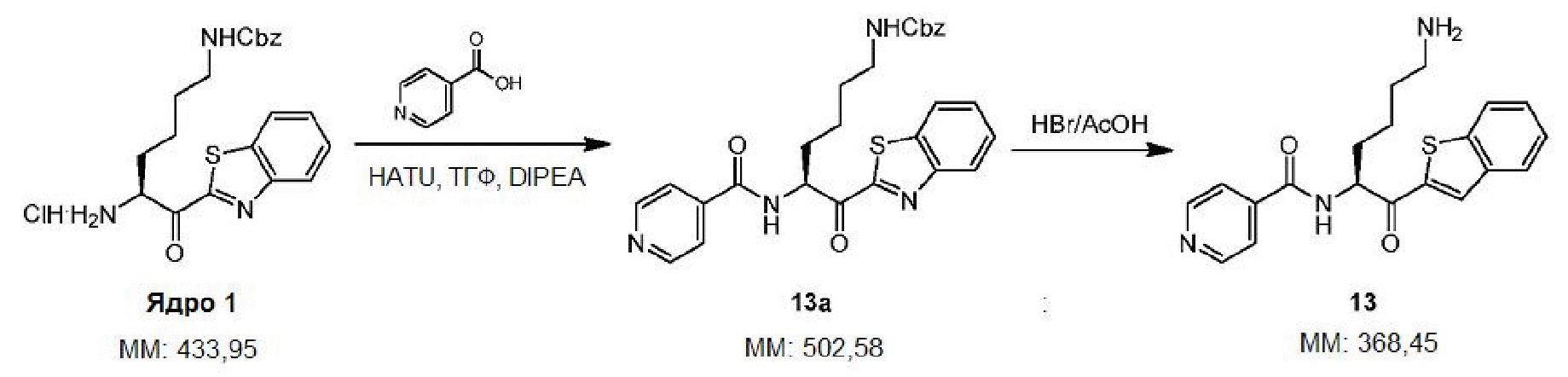
[0251] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-4-карбониламино)гексил]карбамат (13a). К раствору изоникотиновой кислоты (102 мг, 829,58 мкмоль, 1,20 экв.) в ТГФ (8 мл) при 0 °С добавляют DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) и HATU (315,43 мг, 829,58 мкмоль, 1,20 экв.). Реакционную смесь перемешивают при 0 °С в течение 15 минут. После этого при 0 °С добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают реакционную смесь при 30 °С в течение дополнительных 3 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (30 мл), затем экстрагируют EA (20 мл × 2), органические фазы объединяют, промывают насыщенным раствором соли (30 мл) и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-4-карбониламино)гексил]карбамата (13а, 727 мг, сырой продукт) в виде твердого вещества красного цвета, которое используют непосредственно в следующей стадии без дополнительной очистки.
[0252] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-4-карбоксамил (13). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-4- карбониламино)гексил]карбамата (727 мг, 1,45 ммоль, 1,00 экв.) в АсОН (3 мл) добавляют HBr/AcOH (1 мл, чистота 33%). Реакционную смесь перемешивают при 20 °С в течение 3 часов. ЖХ-МС показывает, что исходный материал полностью израсходован. Реакционную смесь гасят H2О (10 мл) и промывают МТВЕ (20 мл). Органическую фазу экстрагируют H2О (20 мл × 2), водные фазы объединяют и лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (СН3CN/H2О/ТФУК) с получением трифторацетата N-[1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-4-карбоксамида (13; 75,78 мг, 157,07 мкмоль, выход 10,8%, энантиомерный избыток 69,3%) в виде твердого вещества желтого цвета. МС m/z=369,0 (МН+).
Пример 17. Получение (3R)-N-[5-амино-1-(1,3-бензотиазол-2-карбонил)пентилl-1-(3-фенилпропаноил)пиперидин-3-карбоксамида (14/15).
Схема 17

[0253]трет-Бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилат (14с). К раствору (3R)-1-трет-бутоксикарбонилпиперидин-3-карбоновой кислоты (3,17 г, 13,83 ммоль, 1,20 экв.) в ДМФА (60 мл) при 0 °С добавляют HATU (5,34 г, 14,06 ммоль, 1,22 экв.) и DIPEA (4,45 г, 34,45 ммоль, 2,99 экв.). После перемешивания при 0 °С в течение 0,5 часа к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (5,00 г, 11,52 ммоль, 1,00 экв.) и перемешивают полученную смесь при 30 °С в течение 3 часов. ТСХ (РЕ:ЕА=3:1) показывает, что реакция завершена. Смесь разбавляют H2О (80 мл × 2), экстрагируют ЕА (100 мл × 2). Органические слои объединяют, промывают H2О (60 мл × 2), сушат над Na2SO4, фильтруют и концентрируют с получением трет-бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1- карбоксилата (9,80 г, сырой продукт) в виде масла, которое используют непосредственно в следующей стадии без дополнительной очистки.
[0254]Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамат (14b). К раствору трет-бутил-(3R)-3-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]пиперидин-1-карбоксилата (16,10 ммоль, 1,00 экв.) (9,8 г, сырой продукт) в EA (100 мл) добавляют HCl/EA (4 М, 20 мл, 4,97 экв.). Реакционную смесь перемешивают при 30 °С в течение 1 часа. Суспензию фильтруют, собирая осадок на фильтре, с получением гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамата (10,90 г, сырой продукт), который используют непосредственно в следующей стадии без дополнительной очистки.
[0255] Бензил-N-[6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамат (14а). К раствору гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-пиперидин-3-карбонил]амино]гексил]карбамата (20 ммоль, 1,00 экв.) (10,9 г, сырой продукт) в ДХМ (100 мл) добавляют TEA (6,07 г, 60,00 ммоль, 3,00 экв.). Смесь охлаждают до 0 °С, а затем к смеси по каплям добавляют 3-фенилпропаноилхлорид (5,06 г, 30,00 ммоль, 1,50 экв.). Полученную смесь перемешивают при 0 °С в течение 30 минут. ЖХ-МС показывает, что реакция завершена. Реакционную смесь гасят H2О (60 мл) и экстрагируют ДХМ (100 × 2 мл). Органическе слои объединяют, сушат над Na2SO4, фильтруют и концентрируют получая сырой продукт, который очищают с помощью колоночной хроматографии (элюирование с градиентом: РЕ:ЕА=от 8:1 до 3:1) с получением бензил-N-[6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамата (2,77 г, 4,32 ммоль, выход 21,6%) в виде твердого вещества желтого цвета.
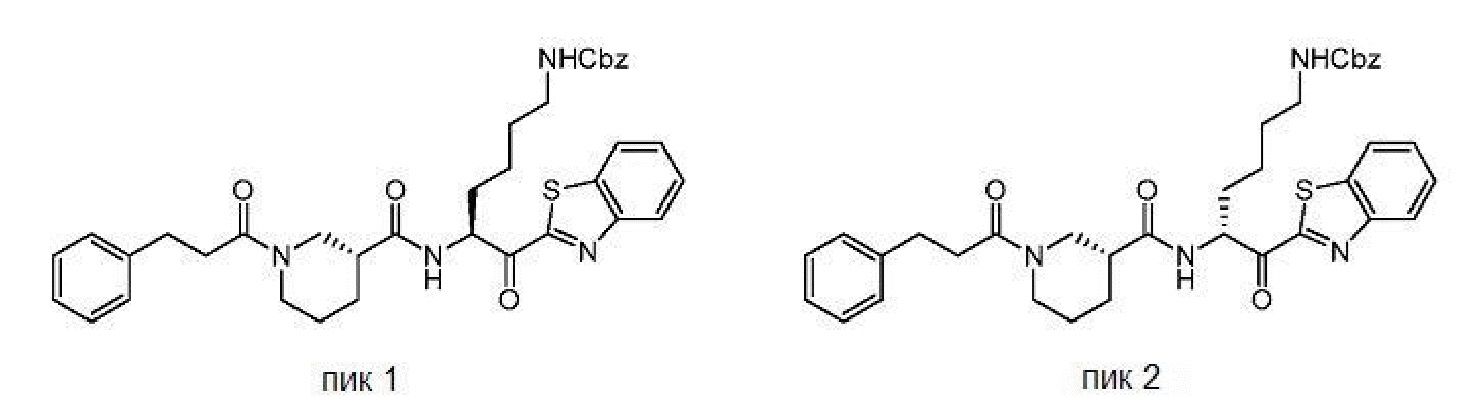
[0256] Бензил-N-[6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамат (1 г) очищают хиральным разделением с помощью SFC (SFC 80, колонка: AD-10 мкм; подвижная фаза: А для СО2 и В для EtOH (0,1% ΝΗ3 Н2O); градиент: D 50%; скорость истечения: 70 мл/мин., обратное давление: 100 бар; температура колонки: 35 °С, длина волны: 220 нм) с получением продуктов (пик 1: 0,4 г) в виде твердого вещества желтого цвета и (пик 2: 0,51 г) в виде масла коричневого цвета.
[0257] (3R)-N-[5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамид; 2,2,2-трифторуксусная кислота (14/15). К раствору бензил-N-[6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамата (400 мг, 624,23 мкмоль, 1,00 экв.) (пик 1) в AcOH (6 мл) в атмосфере N2добавляют HBr/AcOH (624,23 мкмоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 30 °С в течение 1 часа. ЖХ-МС показывает наличие целевого продукта. Смесь разбавляют H2О (100 мл) и MeOH (20 мл) и промывают МТВЕ (50 мл × 2). Полученный раствор лиофилизируют с получением остатка, который очищают с помощью препаративной ВЭЖХ (подвижная фаза: СН3CN/H2О/ТФУК) с получением трифторацетата (3R)-N-[5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамида (14; 39 мг, 76,97 мкмоль, выход 12,3%) в виде твердого вещества светло-желтого цвета. МС m/z=507,2 (МН+).
[0258] К раствору бензил-N-[6-(1,3-бензотиазол-2-ил)-6-оксо-5-[[(3R)-1-(3-фенилпропаноил)пиперидин-3-карбонил]амино]гексил]карбамата (510 мг, 795,89 мкмоль, 1,00 экв.) (пик 2) в АсОН (6 мл) в атмосфере N2добавляют HBr/AcOH (795,89 мкмоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 30 °С в течение 1 часа. ЖХ-МС показывает наличие целевого продукта. Смесь разбавляют H2О (100 мл) и MeOH (20 мл) и промывают МТВЕ (50 мл × 2). Полученный раствор лиофилизируют с получением остатка, который очищают с помощью препаративной ВЭЖХ (подвижная фаза: CH3CN/H2О/ТФУК) с получением трифторацетата (3R)-N-[5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-(3-фенилпропаноил)пиперидин-3-карбоксамида (15; 43 мг, 84,87 мкмоль, выход 10,7%) в виде масла желтого цвета. МС m/z=507,2 (МН+).
Пример 18. Получение N-[(14)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-3-карбоксамида (18/19).
Схема 18

[0259] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-3-карбониламино)гексил]карбамат (18a). К раствору тетрагидропиран-3-карбоновой кислоты (108 мг, 829,58 мкмоль, 1,20 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (390 мг, 1,03 ммоль, 1,48 экв.) и DIPEA (270 мг, 2,09 ммоль, 3,02 экв.). После перемешивания в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 28 °С в течение 4,5 часов. ЖХ-МС показывает, что реакция завершена. Смесь разбавляют H2О (15 мл × 2) и экстрагируют ЕА (20 мл × 2). Органические слои объединяют, промывают H2О (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-3-карбониламино)гексил]карбамата (700 мг, сырой продукт) в виде твердого вещества желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0260]N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-3-карбоксамид (18/19). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-3-карбониламино)гексил]карбамата (1,37 ммоль, 1,00 экв.) (0,7 г, сырой продукт) в АсОН (6 мл) в атмосфере N2добавляют HBr/AcOH (1,37 ммоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 28 °С в течение 1 часа. ЖХ-МС показывает наличие целевого продукта. Смесь разбавляют H2О (10 мл) и MeOH (2 мл) и промывают МТВЕ (10 мл × 2). Полученный раствор разбавляют H2О и затем лиофилизируют с получением остатка, который очищают с помощью препаративной ВЭЖХ (подвижная фаза: CH3CN/H2О/ТФУК) с получением трифторацетатов N-[1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-3-карбоксамида (14 мг - пик 1, соединение 18) и (15 мг - пик 2, соединение 19) в виде твердых веществ желтого цвета. МС m/z=376,1 (МН+).
Пример 19. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-4-карбоксамид (22).
Схема 19

[0261] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-4-карбониламино)гексил]карбамат (22a). К раствору тетрагидропиран-4-карбоновой кислоты (108 мг, 829,58 мкмоль, 1,20 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (390 мг, 1,03 ммоль, 1,48 экв.) и DIPEA (270 мг, 2,09 ммоль, 3,02 экв.). После перемешивания смеси в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 28 °С в течение 4,5 часов. ЖХ-МС показывает, что реакция завершена. Смесь разбавляют H2О (15 мл × 2) и экстрагируют ЕА (20 мл × 2). Органические слои объединяют, промывают H2О (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-4-карбониламино)гексил]карбамата (680 мг, сырой продукт) в виде твердого вещества светло-желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0262] N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-4-карбоксамид (22). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-4-карбониламино)гексил]карбамата (1,33 ммоль, 1,00 экв.) (0,68 г, сырой продукт) в АсОН (5 мл) в атмосфере N2добавляют HBr/AcOH (1,33 ммоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 30 °С в течение 30 минут. ЖХ-МС показывает наличие целевого продукта. Смесь разбавляют H2О (10 мл) и MeOH (2 мл) и промывают МТВЕ (10 мл × 2). Полученный раствор лиофилизируют с получением остатка, который очищают с помощью препаративной ВЭЖХ (подвижная фаза: СН3CN/H2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-4-карбоксамида (45 мг) в виде твердого вещества желтого цвета. МС m/z=376,1 (МН+).
Пример 20. Получение бензил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]карбамата (23).
Схема 20

[0263]трет-Бутил-N-[(5S)-5-(бензилоксикарбониламино)-6-гидроксигексил]карбамат (23d). К раствору (2S)-2-(бензилоксикарбониламино)-6-(трет-бутоксикарбониламино)капроновой кислоты (25,00 г, 65,72 ммоль, 1,00 экв.) в ТГФ (250 мл) добавляют TEA (6,65 г, 65,72 ммоль, 1,00 экв.). Раствор охлаждают до -10 °С и затем к раствору по каплям добавляют изобутилхлорформиат (9,87 г, 72,27 ммоль, 1,10 экв.), в результате чего образуется суспензия. Полученную суспензию перемешивают в течение 2 чаов при 0 °С. Реакционную смесь фильтруют и охлаждают фильтрат до -10 °С. NaBН4 (5,22 г, 138,00 ммоль, 2,10 экв.) растворяют в воде (50 мл) при 0 °С и по каплям добавляют получнный раствор к ТГФ раствору (выделение тяжелого (heavy) СО2). Реакционную смесь оставляют нагреваться до 30 °С и затем перемешивают в течение 3 часов. Реакционную смесь подкисляют 1 М раствором НСl и экстрагируют водную фазу ЕА (400 мл × 2). Органические слои объединяют, промывают водой, насыщенным водным NaHCО3 и раствором соли, сушат над Na2SO4и концентрируют, получая остаток, который очищают с помощью колоночной флэш-хроматографии (элюирование с градиентом: РЕ:ЕА=от 2:1 до 0:1), получая трет-бутил-N-[(5S)-5-(бензилоксикарбониламино)-6-гидроксигексил]карбамат (19,30 г, 52,67 ммоль, выход 80,1%) в виде масла.
[0264] трет-Бутил-N-[(5S)-5-(бензилоксикарбониламино)-6-оксогексил]карбамат (23с). Раствор трет-бутил-N-[(5S)-5-(бензилоксикарбониламино)-6-гидроксигексил]карбамата (19,20 г, 52,39 ммоль, 1,00 экв.) в ДХМ (200 мл) охлаждают до 0 °С. Затем к раствору порциями добавляют реагент Десса-Мартина (22,22 г, 52,39 ммоль, 1,00 экв.). Суспензию оставляют нагреваться до комнатной температуры (30 °С) и перемешивают в течение 14 часов. ТСХ показывает наличие в смеси исходного материала в незначительном количестве. К реакционной смеси добавляют смесь (100 мл: 100 мл) насыщенного водного раствора NaHCО3и 1М раствора Na2S2О3 и энергично перемешивают полученную двухфазную систему в течение 30 мин. Органический слой отделяют и экстрагируют водный слой ДХМ (200 мл). Органические слои объединяют, перегоняют в вакууме, полученное масло переносят в EA (100 мл) и промывают четыре раза смесью NaHCО3/Na2S2О3, водой и насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме с получением сырого трет-бутил-N-[(5S)-5-(бензилоксикарбониламино)-6-оксогексил]карбамата (17,33 г, сырой продукт) в виде масла желтоватого цвета. Подукт используют непосредственно в следующей стадии без дополнительной очистки.
[0265] трет-Бутил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(бензилоксикарбониламино)-6-гидроксигексил]карбамат (23b). Раствор 1,3-бензотиазола (5,56 г, 41,16 ммоль, 3,00 экв.) в ТГФ (100 мл) охлаждают до -78 °С. После этого к раствору по каплям при -78 °С в атмосфере N2добавляют n-BuLi (2,5 М, 16,5 мл, 3,01 экв.) (2,5 М в ТГФ, 16,5 мл). После перемешивания в течение 1 часа при температуре -78 °С к смеси по каплям при -78 °С добавляют трет-бутил-N-[(5S)-5-(бензилоксикарбониламино)-6-оксогексил]карбамат (13,72 ммоль, 1,00 экв.) (5 г, сырой продукт) в ТГФ. Полученную смесь перемешивают в течение дополнительных 3 часов при -78 °С. ЖХ-МС показывает отсутствие исходного материала и наличие целевого продукта. Реакционную смесь гасят NH4Cl (60 мл) и экстрагируют ЕА (100 мл). Органический слой отделяют, промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют, получая сырой продукт, который очищают с помощью колоночной флэш-хроматографии (элюирование с градиентом: PE:ЕА=от 5:1 до 3:1) с получением трет-бутил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(бензилоксикарбониламино)-6-гидроксигексил]карбамата (1,77 г, 3,54 ммоль, выход 25,8%) в виде масла желтого цвета.
[266] Бензил-N-[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(трет-бутоксикарбониламино)пентил]карбамат (23а). Раствор трет-бутил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(бензилоксикарбониламино)-6-гидроксигексил]карбамата (500 мг, 1,00 ммоль, 1,00 экв.) в ДХМ (6 мл) охлаждают до 0 °C. Затем небольшими порциями к раствору добавляют реагент Десса-Мартина (430 мг, 1,01 ммоль, 1,01 экв.). Полученную суспензию оставляют нагреваться до комнатной температуры (28 °С) и перемешивают в течение 14 часов. ТСХ показывает, что исходный материал израсходован полностью. К реакционной смеси добавляют смесь (10 мл: 10 мл) насыщенного водного раствора NaHCО3 и 1 М раствора Na2S2О3 и интенсивно перемешивают полученную двухфазную систему в течение 30 минут. Органический слой отделяют, водный слой экстрагируют ДХМ (20 мл). Органические слои объединяют, отгоняют растворители в вакууме, полученное масло переносят в EA (20 мл), раствор промывают четыре раза смесью NaHCО3/Na2S2О3, водой и насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме, получая сырой продукт, который очищают с помощью флэш-хроматографии с получением бензил-N-[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(трет-бутоксикарбониламино)пентил]карбамата (200 мг, 401,92 мкмоль, выход 40,2%) в виде твердого вещества желтого цвета.
[0267] Бензил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]карбамат (23). К раствору бензил-N-[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(трет-бутоксикарбониламино)пентил]карбамата (200 мг, 401,92 мкмоль, 1,00 экв.) в EA (6 мл) добавляют HCl/EA (4 М, 4 мл, 39,81 экв.). Полученную смесь перемешивают при 28 °С в течение 14 часов. Смесь фильтруют и промывают осадок на фильтре деионизированной водой. Фильтрат лиофилизируют с получением гидрохлорида бензил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]карбамата (43,9 мг) в виде твердого вещества желтого цвета. МС m/z=398,1 (МН+).
Пример 21. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]циклопентанкарбоксамида (24).
Схема 21
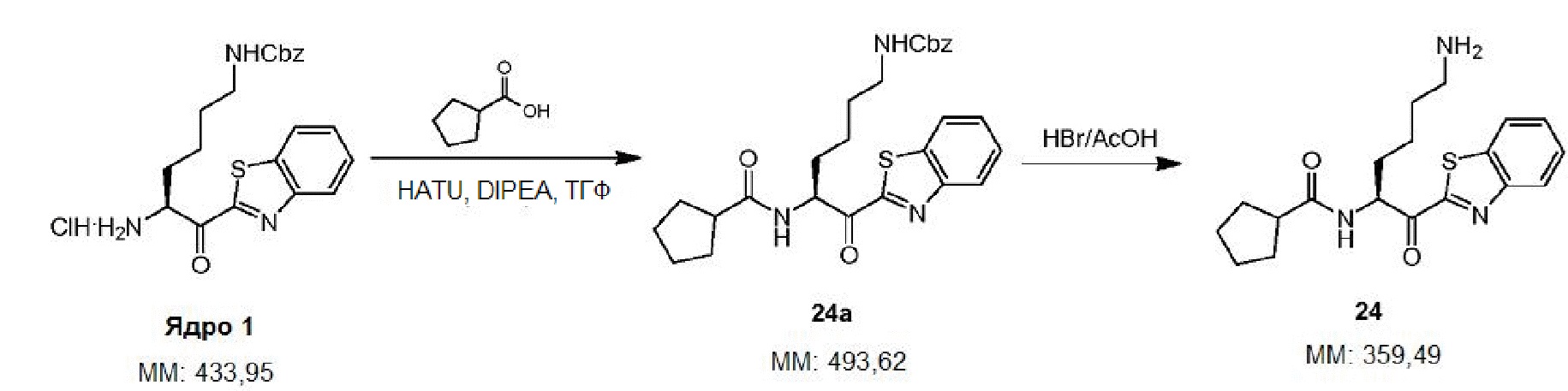
[0268] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(циклопентанкарбониламино)-6-оксогексил]карбамат (24a). К смеси гидрохлорида бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.), циклопентанкарбоновой кислоты (79 мг, 691,32 мкмоль, 1,00 экв.) и DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) в ТГФ (5 мл) добавляют HATU (315 мг, 829,58 мкмоль, 1,20 экв.). Смесь перемешивают при 30 °С в течение 16 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. К смеси добавляют ЕА (50 мл), смесь промывают водой (20 мл × 2), сушат над Na2SO4 и концентрируют, получая сырой продукт, который очищают с помощью колоночной хроматографии (силикагель; элюирование: PE:EA=5:1) с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(циклопентанкарбониламино)-6-оксогексил]карбамата (280 мг, 567,24 мкмоль, выход 82,1%) в виде твердого вещества желтого цвета.
[0269] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]циклоpeнтанкарбоксамид (24). К смеси бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(циклопентанкарбониламино)-6-оксогексил]карбамата (280 мг, 567,24 мкмоль, 1,00 экв.) в АсОН (3 мл) добавляют HBr/AcOH (5,5 М, 1 мл). Полученную смесь перемешивают при 30 °С в течение 1 часа. ЖХ-МС показывает, что исходный материал израсходован полностью. К смеси добавляют воду (50 мл) и полученную смесь экстрагирут MTBE (10 мл × 2). Водный слой лиофилизируют с получением сырого продукта, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]циклопентанкарбоксамида (21,63 мг, 60,17 мкмоль, выход 10,6%) в виде твердого белого вещества. МС m/z=360 (МН+).1H ЯМР (CD3OD, 400 МГц) δ 8,22 (д, J=7,6, 1H), 8,15 (д, J=7,6, 1H), 7,69-7,61 (м, 2H), 5,68 (дд, J=9,6, J=4,0, 1H), 3,07-2,92 (м, 2H), 2,85-2,75 (м, 1H), 2,22-2,12 (м, 1H), 1,95-1,55 (м, 13H).
Пример 22. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-2-карбоксамида (25/26).
Схема 22
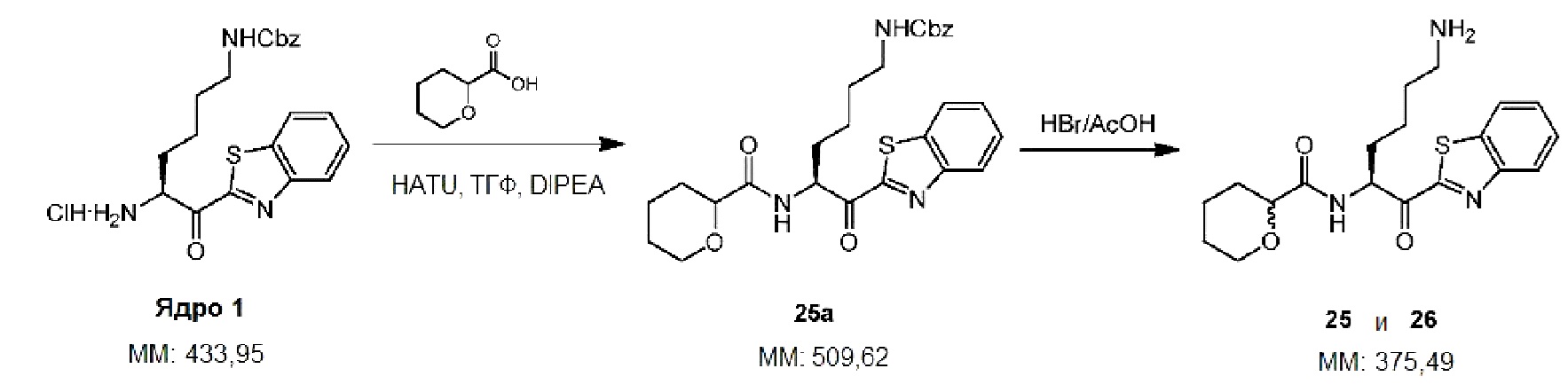
[0270] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-2-карбониламино)гексил]карбамат (2). К раствору тетрагидропиран-2-карбоновой кислоты (108 мг, 829,88 мкмоль, 1,20 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (395 мг, 1,04 ммоль, 1,50 экв.) и DIPEA (270 мг, 2,09 ммоль, 3,02 экв.). После перемешивания при 0 °С в течение 0,5 часа к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 28 °С в течение 4,5 часов. ЖХ-МС показывает, что реакция завершена. Смесь разбавляют H2О (10 мл) и экстрагируют ЕА (20 мл × 2). Органические слои объединяют, промывают H2О (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-2-карбониламино)гексил]карбамата (950 мг, сырой продукт) в виде масла желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0271] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-2-карбоксамид (152/153). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидропиран-2-карбониламино)гексил]карбамата (1,86 ммоль, 1,00 экв.) (0,95 г, сырой продукт) в АсОН (6 мл) в атмосфере N2добавляют HBr/AcOH (1,86 ммоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 30 °С в течение 1 часа. ЖХ-МС показывает наличие в смеси целевого продукта. Смесь разбавляют H2О (10 мл) и MeOH (2 мл) и промывают МТВЕ (10 мл × 2). Полученный раствор лиофилизируют, получая остаток, который очищают с помощью препаративной ВЭЖХ (подвижная фаза: CH3CN/H2О/ТФУК) с получением трифторацетатов N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидропиран-2-карбоксамида (15 мг; пик 1, 25) и (13 мг; пик 2, 26) в виде твердых веществ желтого цвета. МС m/z=376,1 (МН+).
Пример 23. Получение 4-ацетил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]морфолин-2-карбоксамида (29).
Схема 23
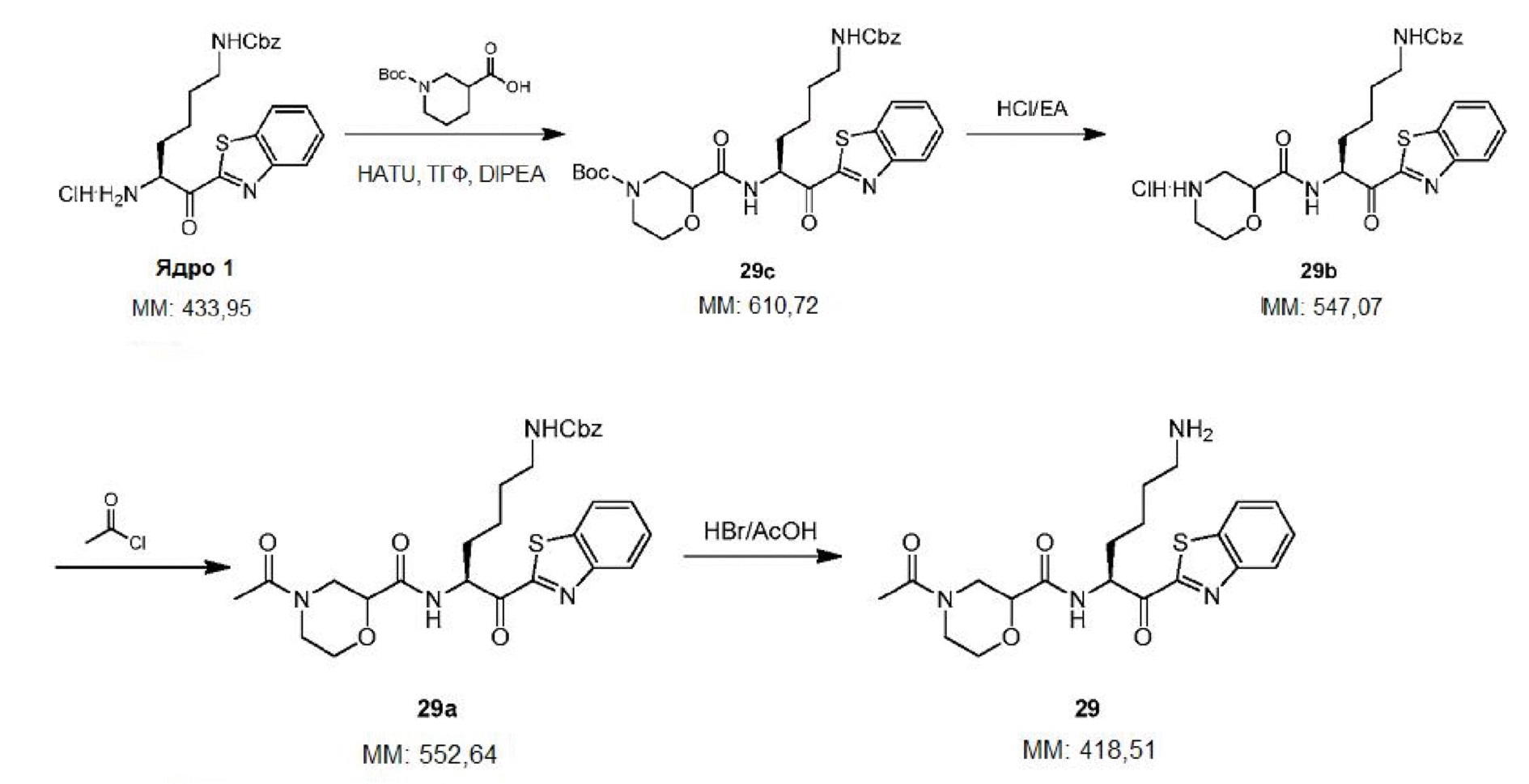
[0272] трет-Бутил-2-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]морфолин-4-карбоксилат (29c). К смеси 4-трет-бутоксикарбонилморфолин-2-карбоновой кислоты (320 мг, 1,38 ммоль, 1,20 экв.), DIPEA (446 мг, 3,45 ммоль, 3,00 экв.) в ТГФ (10 мл) при 0 °С добавляют HATU (525 мг, 1,38 ммоль, 1,20 экв.) и перемешивают полученную смесь при 0 °С в течение 0,5 часа. Затем к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (500 мг, 1,15 ммоль, 1,00 экв.) и перемешивают полученную смесь при 30 °С в течение 3 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. К смеси добавляют ЕА (30 мл), промывают полученную смесь водой (10 мл × 2), сушат над Na2SO4 и концентрируют, получая сырой продукта, который очищают с помощью колоночной хроматографии (силикагель, элюирование с градиентом: PE:EA от 3:1 до 1:1) с получением трет-бутил-2-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламин)пентил]карбамоили]морфолин-4-карбоксилата (600 мг, сырой продукт) в виде твердого вещества желтого цвета.
[0273] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(морфолин-2-карбониламино)-6-оксогексил]карбамат (29b). К смеси трет-бутил-2-[[(1S)-1-(1,3-бензотиазол-2-карбонил)-5-(бензилоксикарбониламино)пентил]карбамоил]морфолин-4-карбоксилата (600 мг, 982,45 мкмоль, 1,00 экв.) и EA (5 мл) добавляют HCl/EA (4 М, 1 мл). Полученную смесь перемешивают при 30 °С в течение 2 часов. ТСХ (РЕ:ЕА=1:1) показывает, что исходный материал израсходован полностью. Смесь фильтруют с получением гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(морфолин-2-карбониламино)-6-оксогексил]карбамата (300 мг, 548,39 мкмоль, выход 55,8%) в виде твердого вещества светло-желтого цвета.
[0274] Бензил-N-[(5S)-5-[(4-ацетилморфолин-2-карбонил)амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамат (29a). К смеси гидрохлорида бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-(морфолин-2-карбониламино)-6-оксогексил]карбамата (300 мг, 548,39 мкмоль, 1,00 экв.) и ТЕА (166 мг, 1,65 ммоль, 3,00 экв.) в ДХМ (5 мл) по каплям при 0 °С в атмосфере N2добавляют ацетилхлорид (86 мг, 1,10 ммоль, 2,00 экв.). Полученную смесь перемешивают при 0 °С в атмосфере N2 в течение 10 минут. ЖХ-МС показывает, что исходный материал израсходован полностью. К смеси добавляют ДХМ (20 мл), смесь промывают водой (10 мл × 3), сушат над Na2SO4 и концентрируют с получением бензил-N-[(5S)-5-[(4-ацетилморфолин-2-карбонил)амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (200 мг, 361,90 мкмоль, выход 67,0%) в виде твердого вещества желтого цвета.
[0275] 4-Ацетил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]морфолин-2-карбоксамид (29). К смеси бензил-N-[(5S)-5-[(4-ацетилморфолин-2-карбонил)амино]-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (200 мг, 361,90 мкмоль, 1,00 экв.) и АсОН (3 мл) добавляют HBr/AcOH (5,5 М, 1 мл). Смесь перемешивают при 30 °С в течение 1 часа. ЖХ-МС показывает, что исходный материал израсходован полностью. К смеси добавляют воду (40 мл) и экстрагируют полученную смесь МТВЕ (10 мл × 2). Водный слой лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата 4-ацетил-N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]морфолин-2-карбоксамида (14,46 мг, 27,15 мкмоль, выход 7,5%) в виде твердого белого вещества. МС m/z=419,1 (МН+).
Пример 24. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-3-карбоксамида (30).
Схема 24

[0276] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-3-карбониламино)гексил]карбамат (30a). К раствору никотиновой кислоты (102 мг, 829,58 мкмоль, 1,20 экв.) в ТГФ (8 мл) при 0 °С добавляют DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) и HATU (315 мг, 829,58 мкмоль, 1,20 экв.) и перемешивают полученную реакционную смесь при 0 °С в течение 15 минут. Затем при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную реакционную смесь при 30 °С в течение дополнительных 3 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (30 мл) и затем экстрагируют ЕА (2×20 мл), органические фазы объединяют, промывают насыщенным раствором соли (30 мл) и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-3-карбониламино)гексил]карбамата (638 мг, сырой продукт) в виде твердого вещества красного цвета, которое используют непосредственно в следующей стадии без дополнительной очистки.
[0277] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-3-карбоксамид (30). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(пиридин-3- карбониламино)гексил]карбамата (638 мг, 1,27 ммоль, 1,00 экв.) в АсОН (3 мл) добавляют HBr/AcOH (1 мл, чистота 33%). Реакционную смесь перемешивают при 30 °С в течение 1 часа. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (10 мл) и промывают МТВЕ (20 мл). Органическую фазу экстрагируют H2О (20 мл × 2), водные фазы объединяют и лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]пиридин-3-карбоксамида (50,96 мг, 105,62 мкмоль, выход 8,3%, энантиомерный избыток 38,2%) в виде твердого вещества желтого цвета. МС m/z=369,1 (МН+).
Пример 25. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетралин-1-карбоксамида (31).
Схема 25
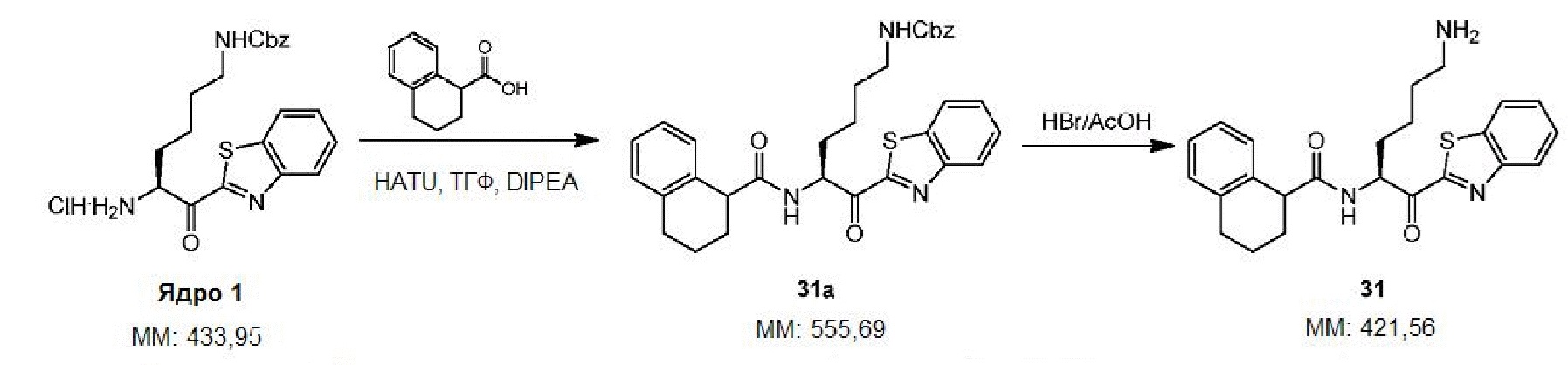
[0278] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетралин-1-карбониламино)гексил]карбамат (31a). К раствору тетралин-1-карбоновой кислоты (150 мг, 851,26 мкмоль, 1,23 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (390 мг, 1,03 ммоль, 1,48 экв.) и DIPEA (270 мг, 2,09 ммоль, 3,02 экв.). После перемешивания в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь перемешивают при 28 °С в течение 5,5 часов. ЖХ-МС показывает, что реакция завершена. Смесь разбавляют H2О (10 мл) и экстрагируют ЕА (20 мл × 2). Органические слои объединяют, промывают H2О (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетралин-1-карбониламино)гексил]карбамата (660,00 мг, сырой продукт) в виде масла желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0279] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетралин-1-карбоксамид (31). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетралин-1- карбониламино)гексил]карбамата (1,19 ммоль, 1,00 экв.) (0,66 г, сырой продукт) в АсОН (6 мл) в атмосфере N2добавляют HBr/AcOH (1,19 ммоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 28 °С в течение 1 часа. ЖХ-МС показывает наличие целевого продукта. Смесь разбавляют H2О (10 мл) и MeOH (2 мл) и промывают МТВЕ (10 мл × 2). Полученный раствор разбавляют H2О и затем лиофилизируют, получая остаток, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетралин-1-карбоксамида (70 мг) в виде твердого вещества желтого цвета. МС m/z=422,1 (МН+).
Пример 26. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетралин-2-карбоксамида (32).
Схема 26
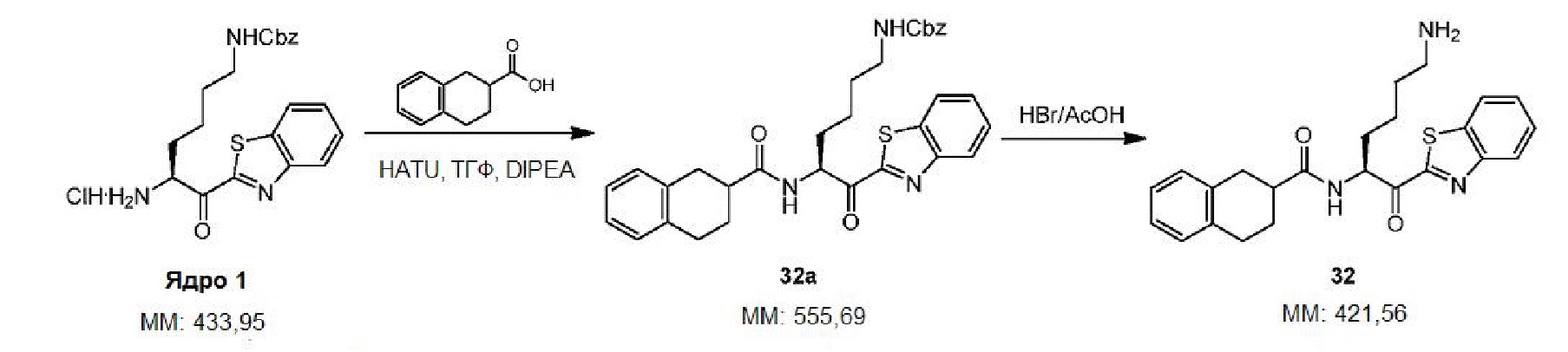
[0280] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетралин-2-карбониламино)гексил]карбамат (32а). К раствору тетралин-2-карбоновой кислоты (150 мг, 851,26 мкмоль, 1,23 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (390 мг, 1,03 ммоль, 1,48 экв.) и DIPEA (270 мг, 2,09 ммоль, 3,02 экв.). После перемешивания смеси в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, добавляют 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 28 °С в течение 5,5 часов. ЖХ-МС показывает, что реакция завершена. Смесь разбавляют H2О (15 мл × 2) и экстрагируют ЕА (20 мл × 2). Органические слои объединяют, промывают H2О (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетралин-2-карбониламино)гексил]карбамата (700 мг, сырой продукт) в виде твердого вещества желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0281] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетралин-2-карбоксамид (32). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетралин-2-карбониламино)гексил]карбамата (1,26 ммоль, 1,00 экв.) (0,7 г, сырой продукт) в АсОН (5 мл) в атмосфере N2добавляют HBr/AcOH (1,26 ммоль, 1,00 экв.) (2 мл). Реакционную смесь перемешивают при 28 °С в течение 0,5 часа. ЖХ-МС показывает, что в реакционной смеси присутствует целевой продукт. Смесь разбавляют H2О (10 мл) и MeOH (2 мл) и промывают МТВЕ (10 мл × 2). Полученный раствор разбавляют H2О и затем лиофилизируют, получая остаток, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением продукта, но ЖХ-МС показывает, что продукт еще не является достаточно чистым. Поэтому продукт дополительно очищают препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением раствора, который лиофилизуют с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетралин-2-карбоксамида (52 мг) в виде твердого вещества желтого цвета. МС m/z=422,2 (МН+).
Пример 27. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-метилциклогексанкарбоксамида (33).
Схема 27
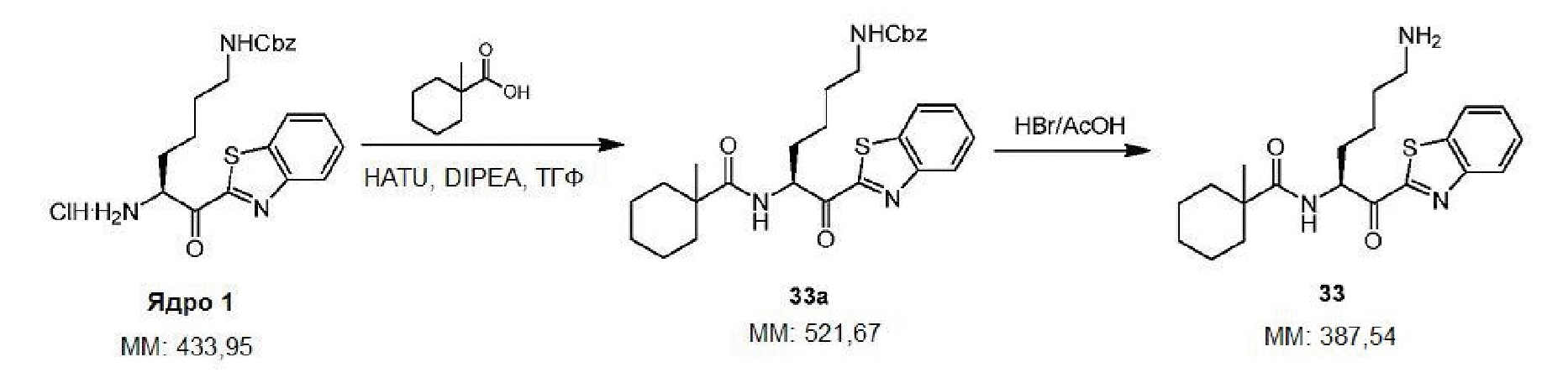
[0282] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-[(1-метилциклогексанкарбонил)амино]-6-оксогексил]карбамат (33a). К смеси 1-метилциклогексанкарбоновой кислоты (118 мг, 829,58 мкмоль, 1,20 экв.) и DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (315 мг, 829,58 мкмоль, 1,20 экв.), полученную реакционную смесь перемешивают при 0 °С в течение 15 минут. После этого к смеси при 0 °С добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают реакционную смесь при 20 °С в течение дополнительных 3 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (10 мл) и экстрагируют ЕА (20 мл × 2). Органические слои объединяют, промывают насыщенным раствором соли (20 мл × 2) и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-[(1-метилциклогексанкарбонил)амино]-6-оксогексил]карбамата (704 мг, сырой продукт) в виде масла желтого цвета, которое используют непосредственно в следующей стадии без дополнительной очистки.
[0283] N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-метилциклогексанкарбоксамид (33). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-[(1-метилциклогексанкарбонил)амино]-6-оксогексил]карбамата (704 мг, 1,26 ммоль, 1,00 экв.) в АсОН (3 мл) добавляют HBr/AcOH (1 мл, 33% чистоты). Реакционную смесь перемешивают при 20 °С в течение 3 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (10 мл) и промывают МТВЕ (20 мл). Органическую фазу экстрагируют H2О (20 мл × 2), водные фазы объединяют и лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-метилциклогексанкарбоксамида (50,68 мг, 101,04 мкмоль, выход 8,0%, 73,1% ее) в виде твердого вещества желтого цвета. МС m/z=388 (МН+).1H ЯМР (CD3OD, 400 МГц) δ 8,19 (д, J=7,6, 1H), 8,12 (д, J=7,6, 1H), 7,66-7,59 (м, 2H), 5,58 (дд, J=10,0, J=4,0, 1H), 3,05-2,94 (м, 2H), 2,23-2,14 (м, 1H), 2,08-2,00 (м, 2H), 1,93-1,71 (м, 3H), 1,62-1,23 (м, 10H), 1,14 (с, 3H).
Пример 28. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидрофуран-2-карбоксамида (34/35).
Схема 28

[0284] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидрофуран-2-карбониламино)гексил]карбамат (2). К смеси тетрагидрофуран-2-карбоновой кислоты (96 мг, 829,58 мкмоль, 1,20 экв.) и DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (315 мг, 829,58 мкмоль, 1,20 экв.). Реакционную смесь перемешивают при 0 °С в течение 15 минут. Затем при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 20 °С в течение дополнительных 3 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (10 мл) и экстрагируют ЕА (20 мл × 2), органические слои объединяют, промывают насыщенным раствором соли (20 мл × 2) и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(2-тетрагидрофуранкарбониламино)гексил]карбамата (517 мг, сырой продукт) в виде масла желтого цвета, которое используют в следующей стадии без дополнительной очистки.
[0285] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидрофуран-2-карбоксамид (164/165). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тетрагидрофуран-2-карбониламино)гексил]карбамата (517 мг, 1,04 ммоль, 1,00 экв.) в AcOH (3 мл) добавляют HBr/AcOH (1 мл, чистота 33%). Реакционную смесь перемешивают при 20 °С в течение 1,5 часа. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (10 мл) и промывают МТВЕ (20 мл). Органическую фазу экстрагируют H2О (20 мл х 2), водные фазы объединяют и лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением двух разделенных диастереомеров трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тетрагидрофуран-2-карбоксамида в виде твердых веществ желтого цвета. Соединение 34 (33,78 мг, 71,04 мкмоль, выход 6,8%), МС m/z=362 (МН+).1H ЯМР (CD3OD, 400 МГц) δ 8,20 (д, J=7,6, 1H), 8,13 (д, J=7,6, 1H), 7,67-7,60 (м, 2H), 5,65 (дд, J=9,2, J=4,0, 1H), 4,37 (дд, J=8,2, J=4,2, 1H), 4,06-4,04 (м, 1H), 3,93-3,90 (м, 1H), 3,00-2,95 (м, 2H), 2,28-2,15 (м, 2H), 1,97-1,81 (м, 4H), 1,77-1,65 (м, 2H), 1,59-1,51 (м, 2H). Соединение 35 (41,00 мг, 86,23 мкмоль, выход 8,3%), МС m/z=362 (МН+).1H ЯМР (CD3OD, 400 МГц) δ 8,21 (дд, J=7,6, 1H), 8,13 (дд, J=7,6, 1H), 7,68-7,60 (м, 2H), 5,71-5,64 (м, 1H), 4,39 (дд, J=8,2, J=5,4, 1H), 4,03 (кв, J=7,0, 1H), 3,89 (д, J=7,4, 1H), 3,01-2,95 (м, 2H), 2,30-2,15 (м, 2H), 2,03-1,82 (м, 4H), 1,77-1,65 (м, 2H), 1,61-1,55 (м, 2H).
Пример 29. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тиазол-5-карбоксамида (38).
Схема 29

[0286] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тиазол-5-карбониламино)гексил]карбамат (38а). К смеси тиазол-5-карбоновой кислоты (107 мг, 829,58 мкмоль, 1,20 экв.) в ТГФ (5 мл) при 0 °С добавляют HATU (315 мг, 829,58 мкмоль, 1,20 экв.) и DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) и перемешивают реакционную смесь при 0 °С в течение 15 минут. Затем при 0 °С добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают реакционную смесь при 20 °С в течение дополнительных 3 часов. ЖХ-МС показывает, что исходный материал израсходован полностью. Реакционную смесь гасят H2О (10 мл) и экстрагируют ЕА (20 мл × 2). Органические слои объединяют, промывают насыщенным раствором соли (20 мл × 2) и упаривают с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тиазол-5-карбониламино)гексил]карбамата (497 мг, сырой продукт) в виде твердого вещества красного цвета.
[0287] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]тиазол-5-карбоксамид (38). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тиазол-5-карбониламино)гексил]карбамата (497 мг, 977,17 мкмоль, 1,00 экв.) в АсОН (3 мл) при 0 °С добавляют HBr/AcOH (1 мл, чистота 33%). Смесь перемешивают при 20 °С в течение 1 часа. ЖХ-МС показывает, что реакция завершена. К смеси добавляют Н2О (10 мл) и промывают смесь MTBE (20 мл). Органическую фазу экстрагируют H2О (20 мл × 2), водные фазы объединяют и лиофилизируют, получая остаток, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тиазол-5-карбоксамида (139,46 мг, 285,49 мкмоль, выход 29,2%, энантиомерный избыток 82,2%) в виде твердого вещества желтого цвета. МС m/z=375,1 (МН+).
Пример 30. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тиазол]-2-карбоксамида (39).
Схема 30

[0288] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тиазол-2-карбониламино)гексил]карбамат (39a). К раствору тиазол-2-карбоновой кислоты (110 мг, 851,79 мкмоль, 1,23 экв.) в ТГФ (6 мл) при 0 °С добавляют HATU (390 мг, 1,03 ммоль, 1,48 экв.) и DIPEA (270 мг, 2,09 ммоль, 3,02 экв.). После перемешивания в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв., НСl) и перемешивают полученную смесь при 28 °С в течение 14 часов. Смесь разбавляют H2О (10 мл) и экстрагируют ЕА (15 мл × 2). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тиазол-2-карбониламино)гексил]карбамата (510 мг, сырой продукт) в виде твердого вещества желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0289] N-[(1S)-5-Амино-1-(1,3-бензотиазол-2-карбонил)пентил]тиазол-2-карбоксамид (39). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-(тиазол-2- карбониламино)гексил]карбамата (1,00 ммоль, 1,00 экв.) (0,51 г, сырой продукт) в АсОН (5 мл) в атмосфере N2добавляют HBr/AcOH (1,00 ммоль, 1,00 экв.) (2 мл, чистота 33%). Реакционную смесь перемешивают при 28 °С в течение 1 часа. ЖХ-МС показывает наличие в реакционной смеси целевого продукта. Смесь разбавляют H2О (10 мл) и MeOH (2 мл) и промывают МТВЕ (10 мл × 2). Полученный раствор разбавляют H2О (80 мл) и затем лиофилизируют, получая остаток, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]тиазол-2-карбоксамида (20 мг) в виде твердого вещества желтого цвета. МС m/z=375,0 (МН+).
Пример 31. Получение N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентилl-6-оксо-1Н-пиридин-2-карбоксамида (40).
Схема 31

[0290] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[(6-оксо-1H-пиридин-2-карбонил)амино]гексил]карбамат (40а). Раствор 6-оксо-1H-пиридин-2-карбоновой кислоты (115 мг, 829,59 мкмоль, 1,20 экв.), DIPEA (268 мг, 2,07 ммоль, 3,00 экв.) и HATU (315 мг, 829,58 мкмоль, 1,20 экв.) в ТГФ (3 мл) перемешивают при 0 °С в течение 15 минут. Затем к смеси при 0 °С добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 25 °С в течение дополнительных 3 часов. ЖХ-МС показывает, что реакция завершена. К смеси добавляют Н2О (10 мл) и экстрагируют смесь ЕА (20 мл × 2). Органические фазы объединяют, промывают насыщенным раствором соли (20 мл) и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[(6-оксо-1Н-пиридин-2-карбонил)амино]гексил]карбамата (612 мг, сырой продукт) в виде твердого вещества желтого цвета.
[0291] N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-6-оксо-1Н-пиридин-2-карбоксамид (40). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-6-оксо-5-[(6-оксо-1Н-пиридин-2-карбонил)амино]гексил]карбамата (612 мг, 1,18 ммоль, 1,00 экв.) в АсОН (3 мл) при 0 °С добавляют HBr/AcOH (1 мл, чистота 33%). Реакционную смесь перемешивают при 20 °С в течение 1 часа. ЖХ-МС показывает наличие в реакционной смеси целевого продукта. Реакционную смесь гасят H2О (10 мл) и промывают МТВЕ (20 мл). Органическую фазу экстрагируют H2О (20 мл × 2), водные фазы объединяют и лиофилизируют, получая сырой продукт, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-6-оксо-1H-пиридин-2-карбоксамида (51,45 мг, 103,22 мкмоль, выход 80,4%, энантиомерный избыток 74,1%) в виде твердого вещества желтого цвета. МС m/z=385,1 (МН+).
Пример 32. Получение (S)-N-(6-амино-1-(бензо[d]тиазол-2-ил)-1-оксогексан-2-ил)-1-метоксициклогексанкарбоксамида (41).
Схема 32

[0292] 1-Метоксициклогексанкарбоновая кислота (41b). К раствору циклогексанона (2,00 г, 20,38 ммоль, 1,00 экв.) в СНСl3 (2,43 г, 20,38 ммоль, 1,00 экв.) (10 мл) при 0-5 °С добавляют раствор КОН (10,00 г, 178,22 ммоль, 8,74 экв.) в MeOH (10 мл). Полученную смесь перемешивают при 23 °С в течение 3 часов. Суспензию фильтруют, фильтрат концентрируют, получая сырый продукт, который разбавляют H2О (20 мл) и экстрагируют ЕА (20 мл х 2). Затем значение рН водного слоя доводят до 5-6 добавлением НСl (1 М). Полученный раствор экстрагируют ДХМ (40 мл × 2). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют с получением 1-метоксициклогексанкарбоновой кислоты (1,21 г, 7,65 ммоль, выход 37,53%) в виде масла желтого цвета.
[0293] Бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-[(1-метоксициклогексанкарбонил)амино]-6-оксогексил]карбамат (41a). К раствору 1-метоксициклогексанкарбоновой кислоты (164 мг, 1,04 ммоль, 1,50 экв.) в ТГФ (6 мл) при 0 °С добавляют HATU (390 мг, 1,03 ммоль, 1,48 экв.) и DIPEA (300 мг, 2,32 ммоль, 3,36 экв.). После перемешивания в течение 0,5 часа при 0 °С к смеси добавляют гидрохлорид бензил-N-[(5S)-5-амино-6-(1,3-бензотиазол-2-ил)-6-оксогексил]карбамата (300 мг, 691,32 мкмоль, 1,00 экв.) и перемешивают полученную смесь при 20 °С в течение 14 часов. Смесь разбавляют H2О (10 мл) и экстрагируют ЕА (15 мл × 2). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют с получением бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-[(1-метоксициклогексанкарбонил)амино]-6-оксогексил]карбамата (830 мг, сырой продукт) в виде твердого вещества желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0294] (S)-N-(6-Амино-1-(бензо[d]тиазол-2-ил)-1-оксогексан-2-ил)-1-метоксициклогексанкарбоксамид (41). К раствору бензил-N-[(5S)-6-(1,3-бензотиазол-2-ил)-5-[(1-метоксициклогексанкарбонил)амино]-6-оксогексил]карбамата (1,00 экв.) (0,83 г, сырой продукт) в АсОН (5 мл) в атмосфере N2добавляют HBr/AcOH (1,00 экв.) (1,5 мл, чистота 33%). Реакционную смесь перемешивают при 18 °С в течение 1 часа. После этого смесь разбавляют H2О (100 мл) и промывают MBTE (40 мл). Водный слой лиофилизируют с получением остатка, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК), получая раствор. Раствор лиофилизируют с получением трифторацетата N-[(1S)-5-амино-1-(1,3-бензотиазол-2-карбонил)пентил]-1-метоксициклогексанкарбоксамида (55 мг, 106,27 мкмоль) в виде твердого вещества светло-желтого цвета. МС m/z=404,2 (МН+).
Пример 33. Получение N[(1S)-5-амино-1-(тиазол-2-карбонил)пентил]циклопентанкарбоксамида (42).
Схема 33
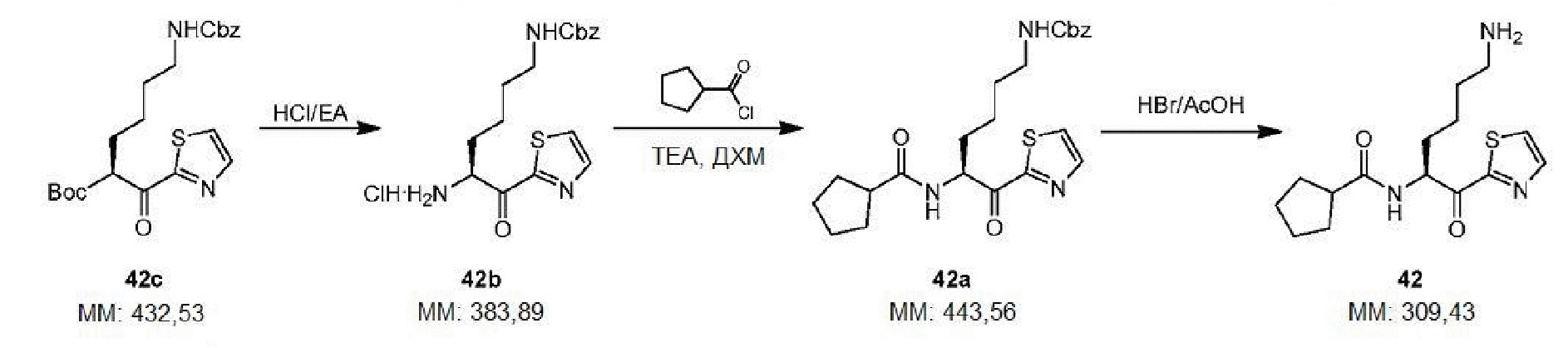
[0295] Бензил-N-[(5S)-5-амино-6-оксо-6-тиазол-2-илгексил]карбамат (42b). К раствору трет-бутил-N-[(1S)-5-(бензилоксикарбониламино)-1-(тиазол-2-карбонил)пентил]карбамата (300 мг, 670,32 мкмоль 1,00 экв.) в EA (10 мл) добавляют HCl/EA (4 М, 2 мл, 11,93 экв.). Реакционную смесь перемешивают при 10 °C в течение 14 часов. ЖХ-МС показывает, что в смеси все еще остается исходный материал и присутствует целевой продукт. К смеси добавляют HCl/ЕА (4 М, 2,00 мл, 11,93 экв.). После этого реакционную смесь перемешивают при 10 °С в течение дополнительных 4 часов. ТСХ (РЕ:ЕА=3:1) показывает, что исходный материал израсходован полностью. Смесь концентрируют с получением гидрохлорида бензил-N-[(5S)-5-амино-6-оксо-6-тиазол-2-илгексил]карбамата (310 мг, сырой продукт, НСl) в виде твердого вещества коричневого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0296] Бензил-N-[(5S)-5-амино-6-оксо-6-тиазол-2-илгексил]карбамат (42a). К раствору гидрохлорида бензил-N-[(5S)-5-амино-6-оксо-6-тиазол-2-илгексил]карбамата (300 мг, 781,47 мкмоль, 1,00 экв., НСl) в ДХМ (10 мл) добавляют TEA (240 мг, 2,37 ммоль, 3,04 экв.) и циклопентанкарбонилхлорид (130 мг, 980,47 мкмоль, 1,25 экв.). Реакционную смесь перемешивают при 10 °С в течение 3 часов. ЖХ-МС показывает, что исходный материал израсходован и основным компонентом смеси является целевой продукт. Раствор промывают H2О (15 мл × 2). Водный слой экстрагируют ДХМ (20 мл × 2). Органические слои объединяют, промывают насыщенным раствором соли (100 мл), сушат над Na2SO4, фильтруют и концентрируют с получением сырого продукта. Сырой продукт очищают с помощью колоночной хроматографии (элюирвание с градиентом: РЕ:ЕА от 4:1 до 2:1) с получением бензил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-6-тиазол-2-илгексил]карбамата (230 мг, 482,23 мкмоль, выход 61,7%, чистота 93%) в виде твердого вещества желтого цвета.
[0297] N-[(1S)-5-Амино-1-(тиазол-2-карбонил)пентил]циклопентанкарбоксамид (42). К раствору бензил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-6-тиазол-2-илгекил]карбамата (210 мг, 473,44 мкмоль, 1,00 экв.) в АсОН (5 мл) в атмосфере N2 добавляют HBr/AcOH (2 мл, 33% чистоты). Реакционную смесь перемешивают при 15 °С в течение 1 часа. ЖХ-МС показывает, что в смеси остается исходный материал и присутствует целевой продукт. Смесь перемешивают при 15 °С в течение дополнительного 1 часа. ЖХ-МС показывает, что в смеси все еще остается исходный материал и присутствует целевой продукт. Смесь промывают деионизированной водой (20 мл), МеОН (2 мл) и МТВЕ (20 мл × 2). Полученный раствор разбавляют деионизированной водой (80 мл) и затем лиофилизируют с получением остатка, значение рН которого доводят до 7-8 добавлением насыщенного водного раствора NaHCО3. Полученный раствор разбавляют деионизированной водой (80 мл) и затем снова лиофилизируют с получением остатка, который очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(тиазол-2-карбонил)пентил]циклопентанкарбоксамида (53 мг) в виде твердого вещества желтого цвета. МС m/z=304,2 (МН+).
Пример 34. Получение N-[(1S)-5-амино-1-[2-(2,3,5,6-тетрафторфенокси)ацетил]пентил]циклопентанкарбоксамида (43).
Схема 34

[0298] (2S)-6-(трет-Бутоксикарбониламино)-2-(циклопентанкарбониламино)капроновая кислота (43d). К раствору (2S)-2-амино-6-(трет-бутоксикарбониламино)капроновой кислоты (5,00 г, 20,30 ммоль, 1,00 экв.) в H2О (40 мл) добавляют NaOH (820 мг, 20,50 ммоль, 1,01 экв.) и Na2СО3 (2,58 г, 24,36 ммоль, 1,20 экв.). После перемешивания в течение 10 минут смесь охлаждают до 0 °С и добавляют при указанной температуре циклопентанкарбонилхлорид (2,96 г, 22,33 ммоль, 1,10 экв.) в EA (20 мл). Реакционную смесь перемешивают при 10 °C в течение 14 часов. ЕА была удаляют, остаток охлаждают до 0 °С, значение рН доводят до 6-7 с помощью твердого KHSO4. Суспензию фильтруют, осадок на фильтре промывают PE (60 мл) и сушат в вакууме с получением (2S)-6-(трет-бутоксикарбониламино)-2-(циклопентанкарбониламино)капроновой кислоты (4,50 г, сырой продукт) в виде твердого белого вещества.
[0299] трет-Бутил-N-[(5S)-5-(циклопентанкарбониламино)-7-диазо-6-оксогептил]карбамат (43с). К раствору (2S)-6-(трет-бутоксикарбониламино)-2-(циклопентанкарбониламино)капроновой кислоты (2,00 г, 5,84 ммоль, 1,00 экв.) в ТГФ (20 мл) добавляют изобутилхлорформиат (798 мг, 5,84 ммоль, 1,00 экв.) и NMM (591 мг, 5,84 ммоль, 1,00 экв.). Смесь перемешивают при -20 °С в течение 2 часов в атмосфере N2. После этого к смеси добавляют диазометан (491 мг, 11,68 ммоль, 2,00 экв.). Смесь перемешивают при 0 °С в течение 10 часов. Смесь разбавляют H2О (20 мл) и экстрагируют ЕА (20 мл × 3). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением остатка, который очищают с помощью хроматографии (силикагель, PE:EA=1:1) с получением трет-бутил-N-[(5S)-5-(циклопентанкарбониламино)-7-диазо-6-оксогептил]карбамата (2,00 г, 5,46 ммоль, выход 93,4%) в виде твердого вещества желтого цвета.
[0300]трет-Бутил-N-[(5S)-7-бром-5-(циклопентанкарбониламино)-6-оксогептил]карбамат (43b). К раствору трет-бутил-N-[(5S)-5-(циклопентанкарбониламино)-7-диазо-6-оксогептил]карбамата (1,00 г, 2,73 ммоль, 1,00 экв.) в ЕА (20 мл) добавляют HBr/АсОН (чистота: 33%, 1 мл). Смесь перемешивают в атмосфере N2при -20 °С в течение 10 минут, затем подщелачивают насыщенным водным раствором NaHCО3 до рН=8 и экстрагируют с помощью ЕА (20 мл × 3). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют с получением трет-бутил-N-[(5S)-7-бром-5-(циклопентанкарбониламино)-6-оксогептил]карбамата (1,10 г, 2,62 ммоль, выход 96,08%) в виде твердого вещества желтого цвета.
[0301] трет-Бутил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-7-(2,3,5,6-тетрафторфенокси)гептил]карбамат (43a). К раствору трет-бутил-N-[(5S)-7-бром-5-(циклопентанкарбониламино)-6-оксогептил]карбамата (1,10 г, 2,62 ммоль, 1,00 экв.) в ДМФА (20 мл) добавляют 2,3,5,6-тетрафторфенол (523 мг, 3,15 ммоль, 1,20 экв.) и KF (457 мг, 7,87 ммоль, 3,00 экв.). Реакционную смесь перемешивают при 20 °С в течение 3 часов. Смесь разбавляют H2О (50 мл) и экстрагируют ЕА (40 мл × 3). Органические слои объединяют, промывают насыщенным раствором соли (50 мл × 5), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают с помощью хроматографии (силикагель; РЕ:ЕА=4:1) с получением трет-бутил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-7-(2,3,5,6-тетрафторфенокси)гептил]карбамата (1,10 г, 2,18 ммоль, выход 83,2%) в виде твердого белого вещества.
[0302] N-[(1S)-5-амино-1-[2-(2,3,5,6-тетрафторфенокси) ацетил]пентил]циклопентанкарбоксамид (43). К раствору трет-бутил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-7-(2,3,5,6-тетрафторфенокси)гептил]карбамата (500 мг, 991,06 мкмоль, 1,00 экв.) в EA (10 мл) добавляют HCl/EA (4 М, 5 мл, 20,18 экв.). Смесь перемешивают при 20 °С в течение 1 часа. После этого смесь разбавляют Н2О (20 мл), водный слой отделяют и лиофилизируют с получением гидрохлорида N-[(1S)-5-амино-1-[2-(2,3,5,6-тетрафторфенокси)ацетил]пентил]циклопентанкарбоксамида (280 мг, 635,12 мкмоль, выход 64,1%) в виде твердого белого вещества. МС m/z=405 (МН+).1H ЯМР ((CD3)2SO, 400 МГц) δ 8,38 (д, J=7,2, 1H), 8,05 (уш.с, 3H), 7,58 (ддд, J=4,5, J=8,3, J=15,5, 1H), 5,25 (д, J=17,6, 1H), 5,18 (д, J=17,6, 1H), 4,29 (ддд, J=2,4, J=4,6, J=10,6, 1H), 2,77-2,64 (м, 3H), 1,78-1,24 (м, 14H).
Пример 35. Получение N-[(1S)-5-амино-1-(пиридин-2-карбонил)пентил]циклопентанкарбоксамида (44).
Схема 35

[0303] Бензил-N-[(5S)-5-амино-6-оксо-6-(2-пиридил)гексил]карбамат (44b). К раствору трет-бутил-N-[(1S)-5-(бензилоксикарбониламино)-1-(пиридин-2-карбонил)пентил]карбамата (480 мг, 1,09 ммоль, 1,00 экв.) в EA (10 мл) добавляют HCl/EA (4 М, 4 мл, 14,68 экв.). Реакционную смесь перемешивают при 10 °С в течение 14 часов. ЖХ-МС показывает, что исходный материал израсходован полностью и в смеси присутствует целевой продукт. Смесь концентрируют с получением гидрохлорида бензил-N-[(5S)-5-амино-6-оксо-6-(2-пиридил)гексил]карбамата (400 мг, сырой продукт) виде твердого вещества желтого цвета. Продукт используют непосредственно в следующей стадии без дополнительной очистки.
[0304] Бензил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-6-(2-пиридил)гексил]карбамат (44a). К раствору гидрохлорида бензил-N-[(5S)-5-амино-6-оксо-6-(2-пиридил)гексил]карбамата (400 мг, 1,06 ммоль, 1,00 экв.) в ДХМ (10 мл) добавляют TEA (320 мг, 3,16 ммоль, 2,98 экв.) и циклопентанкарбонилхлорид (160 мг, 1,21 ммоль, 1,14 экв.). Реакционную смесь перемешивают при 10 °С в течение 3 часов. ЖХ-МС показывает, что исходный материал израсходован, а основным компонентом смеси является целевой продукт. Раствор промывают H2О (15 мл × 2). Водный слой экстрагируют ДХМ (20 мл × 2). Органические слои объединяют, промывают насыщенным водным раствором соли (100 мл), сушат над Na2SO4, фильтруют и концентрируют с получением сырого продукта, который очищают с помощью колоночной хроматографии (элюирование с градиентом: РЕ:ЕА от 4:1 до 2:1) с получением бензил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-6-(2-пиридил)гексил]карбамата (250 мг, 571,39 мкмоль, выход 53,9%) в виде твердого вещества светло-желтого цвета.
[0305] N-[(1S)-5-Амино-1-(пиридин-2-карбонил)пентил]циклопентанкарбоксамид (44). К раствору бензил-N-[(5S)-5-(циклопентанкарбониламино)-6-оксо-6-(2-пиридил)гексил]карбамата (230 мг, 525,68 мкмоль, 1,00 экв.) в АсОН (5 мл) добавляют HBr/AcOH (2 мл, чистота 33%) в атмосфере N2. Реакционную смесь перемешивают при 15 °С в течение 1 часа. ЖХ-МС показывает, что в смеси все еще остается исходный материал и присутствует целевой продукт. Реакционную смесь перемешивают при 15 °С в течение дополнительного 1 часа. ЖХ-МС показывает, что в смеси все еще остается исходный материал и присутствует целевой продукт. Смесь разбавляют деионизированной водой (20 мл) и MeOH (2 мл) и промывают МТВЕ (20 мл × 2). Полученный раствор разбавляют деионизированной водой (80 мл), а затем лиофилизируют с получением остатка, значение рН которого доводят до 7-8 добавлением насыщенного водного раствора NaHCО3. Полученный раствор разбавляют деионизированной водой (80 мл) и затем снова лиофилизируют с получением остатка. Часть сырого продукта очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением целевого продукта, но он содержит циклизованный продукт. Другую часть сырого продукта очищают с помощью препаративной ВЭЖХ (СН3CN/Н2О/ТФУК) с получением трифторацетата N-[(1S)-5-амино-1-(пиридин-2-карбонил)пентил]циклопентанкарбоксамида (56 мг) в виде твердого вещества желтого цвета. МС m/z=304,2 (МН+).
Пример 36. Получение N-[(1S)-5-амино-1-[2-(2,3,5,6-тетрафторфенокси)ацетил]пентил]-3-азидобензамида (45).
Схема 36

[0306] (2S)-2-[(3-Азидобензоил)амино]-6-(трет- бутоксикарбониламино)капроновая кислота (45d). К раствору (2S)-2-амино-6-(трет-бутоксикарбониламино)гексановой кислоты (10,00 г, 40,60 ммоль, 1,00 экв.) в EA/H2О (1/1, 200 мл) добавляют NaOH (1,62 г, 40,60 ммоль, 1,00 экв.), Na2CО3 (4,30 г, 40,60 ммоль, 1,00 экв.) и 3-азидобензоилхлорид (7,37 г, 40,60 ммоль, 1,00 экв.). Смесь перемешивают при 20 °С в течение 4 часов. Смесь подкисляют до рН=4 с помощью KHSO4 и концентрируют при пониженном давлении. Остаток очищают с помощью хроматографии (силикагель, ДХМ/MeOH=10/1) с получением (2S)-2-[(3-азидобензоил)амино]-6-(трет-бутоксикарбониламино)капроновой кислоты (10,00 г, 25,55 ммоль, 62,9% выход) в виде твердого вещества желтого цвета.
[0307]трет-Бутил-N-[(5S)-5-[(3-азидобензоил)амино]-7-диазо-6-оксогептил]карбамат (45c). К раствору (2S)-2-[(3-азидобензоил)амино]-6-(трет-бутоксикарбониламино)капроновой кислоты (3,50 г, 8,94 ммоль, 1,00 экв.) в ТГФ (50 мл) добавляют NMM (904 мг, 8,94 ммоль, 1,00 экв.) и изобутилхлорформиат (1,22 г, 8,94 ммоль, 1,00 экв.). Смесь перемешивают при -20 °С в течение 1 часа в атмосфере N2. К смеси добавляют диазометан (376 мг, 8,94 ммоль, 1,00 экв.). Смесь перемешивают при -20 °С в течение 4 часов в атмосфере N2. Смесь разбавляют H2О (50 мл) и экстрагируют ЕА (50 мл × 3). Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают с помощью хроматографии (силикагель, элюирование: PE/EA=1/1) с получением трет-бутил-N-[(5S)-5-[(3-трихлорэтил)амино]-7-диазо-6-оксогептил]карбамата (2,90 г, 6,98 ммоль, выход 78,1%) в виде твердого вещества желтого цвета.
[0308] трет-Бутил-N-[(5S)-5-[(3-трихлорэтил)амино]-7-бром-6-оксогептил]карбамат (45b). К раствору (3-азидобензоил)-[(1S)-5-(трет-бутоксикарбониламино)-1-(2-диазоацетил)пентил]аммония (2,90 г, 6,96 ммоль, 1,00 экв.) в ЕА (50 мл) добавляют HBr/АсОН (2,56 г, 10,44 ммоль, 1,50 экв., чистота: 33%). Смесь перемешивают при -20 °С в течение 10 минут. Смесь разбавляют H2О (50 мл) и экстрагируют ЕА (50 мл). Органический слой промывают насыщенным раствором соли (50 мл × 3), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением трет-бутил-N-[(5S)-5-[(3-азидобензоил)амино]-7-бром-6-оксогептил]карбамата (2,90 г, сырой продукт, чистота 62,2%) в виде твердого вещества желтого цвета.
[0309] трет-Бутил-N-[(5S)-5-[(3-азидобензоил)амино]-6-оксо-7-(2,3,5,6-тетрафторфенокси)гептил]карбамат (45a). К раствору трет-бутил-N-[(5S)-5-[(3-азидобензоил)амино]-7-бром-6-оксогептил]карбамата (2,90 г, 6,19 ммоль, 1,00 экв.) в ДМФА (20 мл) добавляют 2,3,5,6-тетрафторфенол (1,54 г, 9,29 ммоль, 1,50 экв.) и KF (1,80 г, 30,95 ммоль, 5,00 экв.). Смесь перемешивают при 20 °С в течение 12 часов. Смесь разбавляют H2О (100 мл) и экстрагируют ЕА (100 мл). Органический слой промывают насыщенным раствором соли (100 мл × 5), сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Остаток очищают с помощью хроматографии (силикагель, элюирование: PE:EA=4:1) с получением трет-бутил-N-[(5S)-5-[(3-азидобензоил)амино]-6-оксо-7-(2,3,5,6-тетрафторфенокси)гептил]карбамата (2,80 г, 5,06 ммоль, выход 81,7%) в виде твердого белого вещества.
[0310]N-[(1S)-5-Амино-1-[2-(2,3,5,6-тетрафторфенокси)ацетил]пентил]-3-азидобензамид (45). Раствор трет-бутил-N-[(5S)-5-[(3-азидобензоил)амино]-6-оксо-7-(2,3,5,6-тетрафторфенокси)гептил]карбамата (2,20 г, 3,97 ммоль, 1,00 экв.) в НСl/ЕА (20 мл, 4 М) перемешивают при 20 °С в течение 30 минут. Смесь разбавляют Н2О (20 мл) и лиофилизируют водный слой с получением N-[(1S)-5-амино-1-[2-(2,3,5,6-тетрафторфенокси)ацетил]пентил]-3-азидобензамида в виде твердого белого вещества. МС m/z=454,1 (МН+).
Пример 37. Ингибирование лизин-специфичного гингипаина с помощью соединений по изобретению.
[0311] Способность соединений по настоящее изобретению ингибировать активность лизин-специфичного гингипаина определяют методом флуорогенного анализа, аналогичным описанному в публикации Barret Biochemical Journal. 1980, 187(3), 909. Используют следующие условия анализа. Буфер: рН=7,5, 100 мМ Трис-HCl, 75 мМ NaCl, 2,5 мМ CaCl2, 10 мМ цистеина, 1% ДМСО после всех добавлений. Белок: 0,1 нМ Kgp, выделенного из культуры Porphyromonas gingivalis, как описано в публикациях Pike et al. J. Biol. Chem. 1994, 269(1), 406; Potempa and Nguyen. Current Protocols in Protein Scienc. 2007, 21.20.1-21.20.27. Флуорогенный субстрат: 10 мкМ Z-His-Glu-Lys-MCA. Время=90 минут. Температура=37 °С. Каждое соединение: 10 концентраций, начиная с 100 мкМ или 100 нМ, где более низкие концентрации получают последовательными 3-кратными разбавлениями. Тестированием каждого соединения в указанном диапазоне концентраций определяют концентрацию, необходимую для ингибирования активности лизин-специфичного гингипаина на 50% (далее ʺIC50ʺ). В указанных условиях анализа соотношение «сигнал-шум» является превосходным, и значение Z фактора составляет более 0,6.
[0312] Ингибиторную активность соединений, описанных в данном изобретении, испытывают в отношении Kgp, RgpB и трипсина. Значения IC50Kgp для различных соединений представлены в таблице 2. Значение IC50 RgpB соединения 43 составляет более 475 нМ, а значения IC50RgpB других соединений, представленные в таблице 2, составляют более 2,5 мкΜ. Значения IC50 трипсина всех соединений, представленных в таблице 2, составляют более 1 μΜ.
Таблица 2. Ингибиторная активность соединений по изобретению в отношении лизин-специфичного гингипаина.
[0313] Несмотря на то, что для более доступного понимания изобретение описано с некоторыми подробностями с помощью иллюстраций и примеров, специалисту в данной области техники будет ясно, что некоторые изменения и модификации могут осуществляться в пределах объема прилагаемой формулы изобретения. Кроме того, каждая ссылка, представленная в настоящем описании, введена в качестве ссылки во всей своей полноте в той же степени, как если бы каждая ссылка была отдельно включена в качестве ссылки.
Реферат
Изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, где Z представляет собой галогензамещенный феноксиметилкарбонил; А представляет собой -CH-; В и D представляют собой водород; Rпредставляет собой водород; Rпредставляет собой водород и Rвыбран из группы, состоящей из циклогексила, циклопентила, морфолино, фенила, пиперидинила, пиридинила, тетрагидрофуранила, тетрагидропиранила, 1,2,3,4-тетрагидронафтила и тиазолила, каждый из которых независимо незамещен или независимо замещен 1-3 членами, выбранными из метила, метокси, трифторметила, ацетила и -N; и Rпредставляет собой водород. Соединения формулы I предназначены для ингибирования лизинспецифичного гингипаина. 2 н. и 6 з.п. ф-лы, 10 ил., 2 табл., 37 пр.
Формула



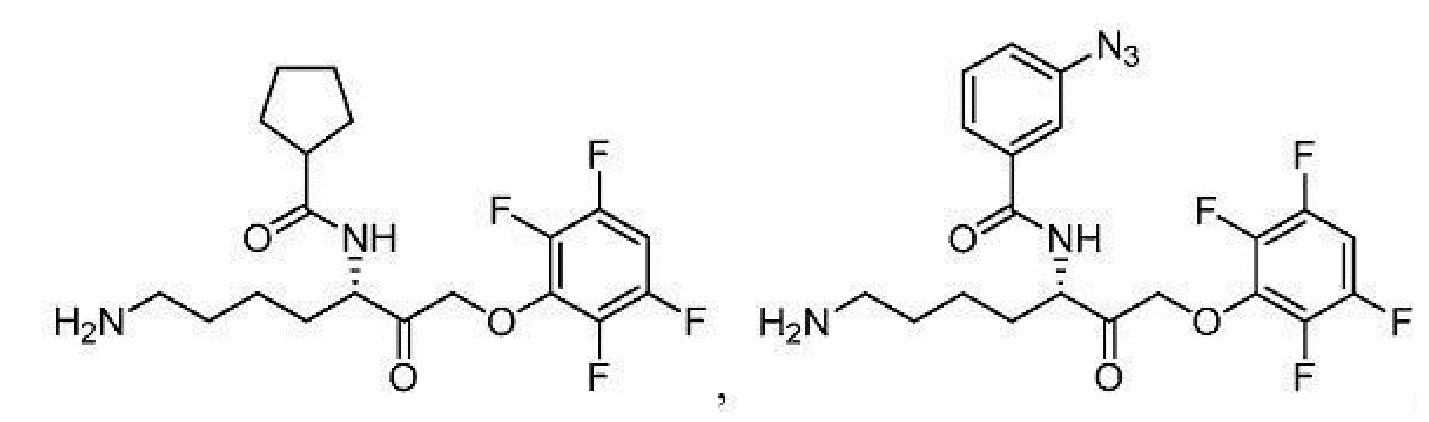
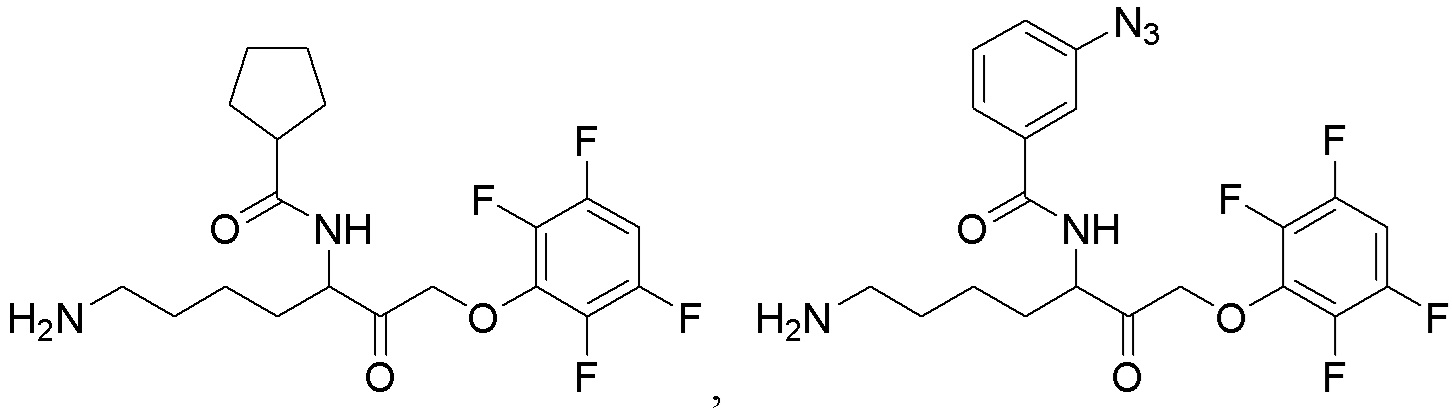
Документы, цитированные в отчёте о поиске
Соединения и композиции в качестве ингибиторов протеазы, активирующей каналы
Комментарии