Композиции, содержащие расщепляемое ферментами пролекарство оксикодона - RU2609412C2
Код документа: RU2609412C2
Чертежи







Описание
Введение
Содержащие кетоны опиоиды, такие как гидрокодон и оксикодон, подвержены неправильному использованию, злоупотреблению и передозировке. Следовательно, необходимо контролировать использование и доступ к этим лекарственным средствам. Контроль доступа к лекарственным средствам является дорогостоящим в осуществлении и может привести к отказу в лечении пациентам, которые не способны явиться лично для введения дозы лекарства. Например, пациентам, страдающим от острой боли, может быть отказано в лечении опиоидом, если они не помещены в стационар. Кроме того, контроль за использованием часто оказывается неэффективен, что приводит к значительным заболеваниям и опасным социальным последствиям.
Раскрытие изобретения
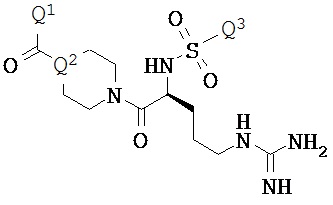
Варианты осуществления изобретения относятся к Соединению КС-8, N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламин]-аргинин-глицин-малоновой кислоте, представленной ниже:

или ее приемлемым солям, сольватам и гидратам. Соединение КС-8 представляет собой пролекарство, которое обеспечивает контролируемое высвобождение оксикодона.
Варианты осуществления настоящего изобретения описывают композицию, которая содержит N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламин]-аргинин-глицин-малоновую кислоту. Соединение КС-8, представленное ниже:

или его фармацевтически приемлемые соли, сольваты и гидраты.
Данное изобретение относится к Соединению КС-8, опиоидному пролекарству, модифицированному кетоном, которое обеспечивает контролируемое высвобождение оксикодона. В модифицированном кетоном опиоидном пролекарстве к оксикодону через енольный атом кислорода оксикодона присоединен фрагмент-предшественник. В модифицированном кетоном опиоидном пролекарстве атом водорода соответствующей енольной группы оксикодона замещен ковалентной связью с фрагментом-предшественником. Фрагмент-предшественник содержит расщепляемый ферментом фрагмент и способную к циклизации спейсерную уходящую группу, так что Соединение КС-8 обеспечивает контролируемое высвобождение оксикодона путем ферментативного расщепления с последующей внутримолекулярной циклизацией. Соединение КС-8 обеспечивает эффективную доставку оксикодона при приеме внутрь.
Настоящее изобретение также описывает фармацевтические композиции и способы их применения, где указанные фармацевтические композиции содержат пролекарство, Соединение КС-8, обеспечивающее контролируемое высвобождение оксикодона путем ферментативного расщепления с последующей внутримолекулярной циклизацией. Такие композиции могут необязательно включать ингибитор, такой как трипсиновый ингибитор, взаимодействующий с ферментом, который является посредником в высвобождении оксикодона из пролекарства, предназначенный для ослабления ферментативного расщепления пролекарства. В данном изобретении предусмотрено, что указанный фермент представляет собой желудочно-кишечный (ЖК) фермент, такой как трипсин.
Краткое описание чертежей
На Фиг. 1 приведен график, иллюстрирующий влияние повышения количества трипсинового ингибитора ("ингибитор", ось X) на фармакокинетический (ФК) параметр (например, максимальную концентрацию, Cmax, лекарственного средства) (ось Y) для фиксированной дозы пролекарства. Влияние ингибитора на ФК параметр пролекарства может меняться от недетектируемого к умеренному и далее к полному ингибированию (т.е. к отсутствию детектируемого высвобождения лекарственного средства).
На Фиг. 2 представлены диаграммы зависимости концентрации лекарственного средства в плазме (ось Y) от времени. Секция А представляет собой фармакокинетическую (ФК) кривую после приема внутрь пролекарства с трипсиновым ингибитором (пунктирная линия), где Cmax лекарственного средства отличается от Cmax пролекарства без ингибитора (сплошная линия). Секция В представляет собой ФК кривую после приема внутрь пролекарства с ингибитором (пунктирная линия), где Cmax и Tmax лекарственного средства отличаются от этих параметров пролекарства без ингибитора (сплошная линия). Секция С представляет собой ФК кривую после приема внутрь пролекарства с ингибитором (пунктирная линия), где Tmax лекарственного средства отличается от Tmax пролекарства без ингибитора (сплошная линия).
На Фиг. 3 представлены характерные ФК кривые зависимости концентрация-доза, которые могут быть быть получены в результате приема нескольких стандартных доз (ось X) по настоящему изобретению. Различные ФК кривые (как показано здесь для типичного ФК параметра - Cmax лекарственного средства (ось Y)) могут быть получены путем корректировки относительных количеств пролекарства и трипсинового ингибитора, содержащихся в одной стандартной дозе, или путем использования в стандартной дозе другого пролекарства или другого ингибитора.
На Фиг. 4 приведено сравнение средних по времени высвобождения концентраций оксикодона в плазме после перорального (ПО) введения крысам увеличивающихся доз пролекарства Соединения КС-8.
На Фиг. 5 приведено сравнение средних по времени высвобождения концентраций оксикодона в плазме после ПО введения собакам пролекарства Соединения КС-8, пролекарства Соединения КС-3, таблеток OxyContin® или гидрохлорида оксикодона.
На Фиг. 6А и Фиг. 6В приведены сравнения средних по времени высвобождения концентраций оксикодона в плазме после ПО введения крысам двух доз пролекарства Соединения КС-8, причем каждая доза использовалась совместно с увеличивающимися количествами трипсинового ингибитора - Соединения 109.
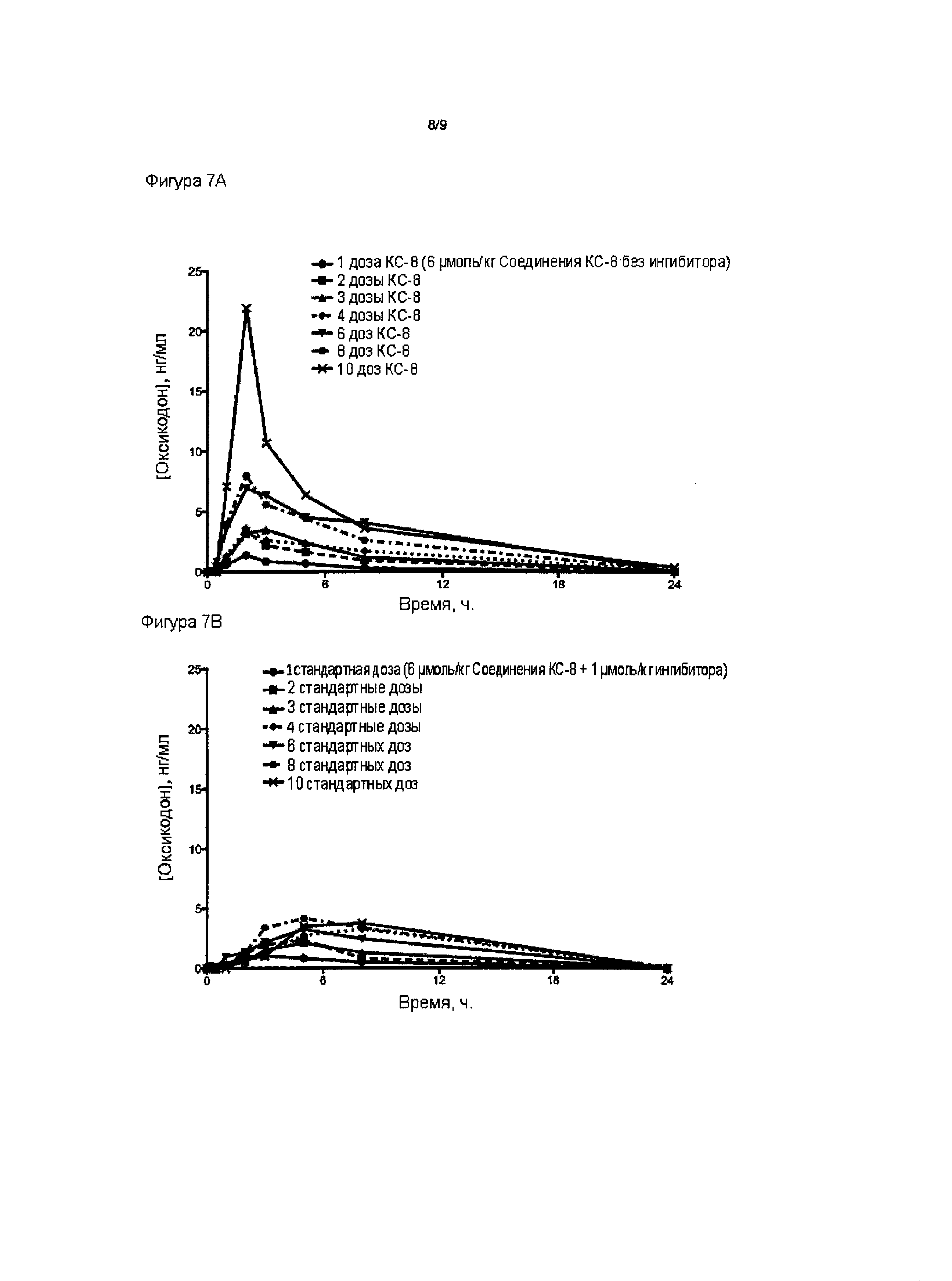
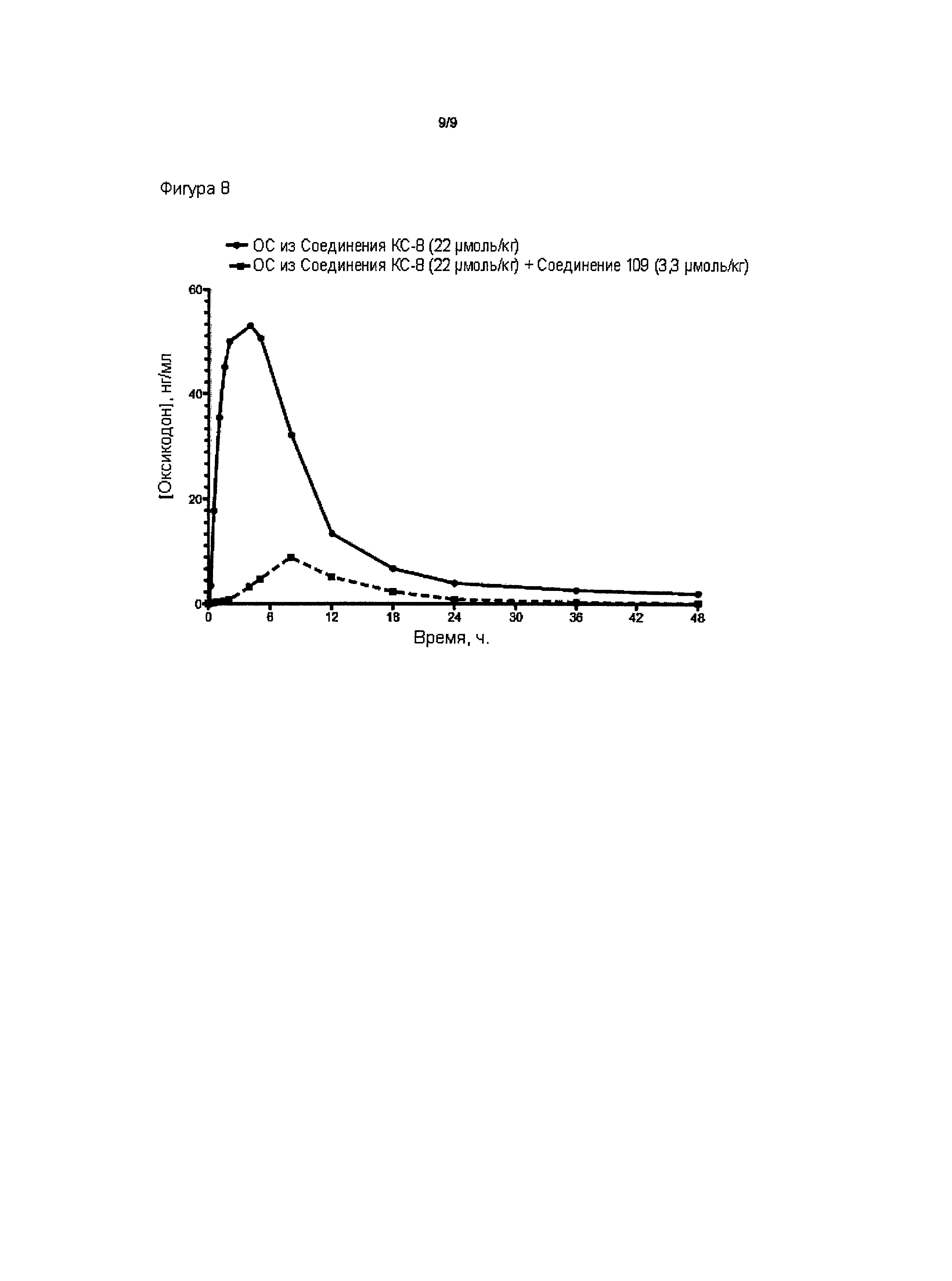
На Фиг. 7А приведено сравнение средних по времени высвобождения концентраций оксикодона в плазме после ПО введения крысам однократной и многократных доз пролекарства Соединения КС-8 в отсутствие трипсинового ингибитора. На Фиг. 7В приведено сравнение средних по времени высвобождения концентраций оксикодона в плазме после ПО введения крысам однократной и многократных стандартных доз, содержащих пролекарство Соединение КС-8 и трипсиновый ингибитор Соединение 109.
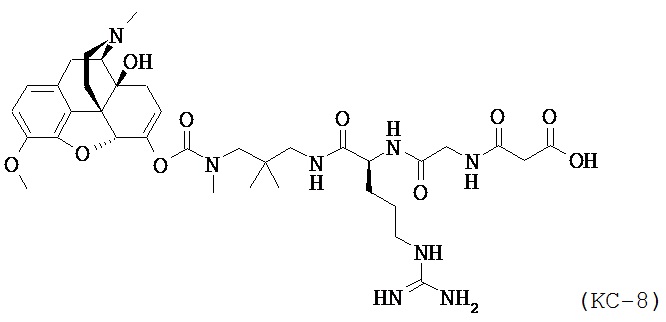
На Фиг. 8 приведено сравнение средних по времени высвобождения концентраций оксикодона в плазме после ПО введения собакам пролекарства Соединения КС-8 в отсутствие или в присутствии трипсинового ингибитора Соединения 109.
Используемые определения
Следующие термины имеют указанные далее значения, если конкретно не будет указано иное. Любые неопределенные здесь выражения имеют значения, общепринятые в той области техники, к которой они относятся.
В настоящем изобретении "стандартная доза" относится к комбинации расщепляемого трипсином пролекарства (например, расщепляемого трипсином пролекарства) и трипсинового ингибитора. "Однократная стандартная доза" представляет собой одну единицу комбинации расщепляемого трипсином пролекарства (например, расщепляемого трипсином пролекарства) и трипсинового ингибитора, при условии, что такая однократная стандартная доза предоставляет терапевтически эффективное количество лекарственного средства (т.е. количество лекарства, достаточное для получения терапевтического эффекта, например доза в пределах соответствующего лекарству терапевтического окна или терапевтического диапазона). Термины "многократная стандартная доза" или "кратная стандартная доза", или "множество стандартных доз" относятся к по меньшей мере двум однократным стандартным дозам.
"Желудочно-кишечный фермент" или "ЖК фермент" относится к ферменту, находящемуся в желудочно-кишечном (ЖК) тракте, который охватывает анатомические местоположения от рта до заднего прохода. Трипсин - это пример ЖК фермента.
"Расщепляемый желудочно-кишечным ферментом фрагмент" или "расщепляемый ЖК ферментом фрагмент" относится к группе, содержащей участок, подверженный расщеплению ЖК ферментом. Например, "расщепляемый трипсином фрагмент" относится к группе, содержащей участок, подверженный расщеплению трипсином.
"Ингибитор желудочно-кишечного фермента" или "ингибитор ЖК фермента" относится к любому средству, способному ингибировать действие желудочно-кишечного фермента на субстрат. Данный термин также включает соли ингибиторов желудочно-кишечных ферментов. Например, "трипсиновый ингибитор" относится к любому средству, способному ингибировать действие трипсина на субстрат.
"Пациент" включает людей, а также других млекопитающих, таких как домашние животные, животные в зоопарке и животные-компаньоны, такие как кошка, собака или лошадь.
"Фармацевтическая композиция" относится к по меньшей мере одному соединению и может дополнительно содержать фармацевтически приемлемый носитель, с которым это соединение вводят пациенту.
"Фармацевтически приемлемый носитель" относится к разбавителю, вспомогательному веществу, наполнителю или носителю, с которым или в котором вводится соединение.
"Фармацевтически приемлемая соль" относится к соли соединения, которая обладает желательной фармакологической активностью соединения. Такие соли включают: (1) кислотные аддитивные соли, образованные неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные; или образованные органическими кислотами, такими как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этан-дисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, третичная бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и подобные; или (2) соли, образованные, когда кислотный протон, присутствующий в соединении, замещен ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; или координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, N-метилглюкамин и подобные.
"Фармакодинамическая (ФД) кривая" относится к кривой эффективности лекарственного средства в пациенте (или субъекте, или потребителе), которая может быть охарактеризована ФД параметрами. "ФД параметры" включают "Emax лекарственного средства" (максимальная эффективность лекарственного средства), "ЕС50 лекарственного средства" (концентрация лекарственного средства при 50% Emax) и побочные эффекты.
"ФК параметр" относится к показателю концентрации лекарственного средства в крови или в плазме, такому как: 1) "Cmax лекарственного средства" - максимальная концентрация лекарственного средства, достигнутая в крови или плазме; 2) "Tmax лекарственного средства" - время, необходимое с момента приема внутрь для достижения Cmax; и 3) "совокупная доза лекарственного средства" - суммарная концентрация лекарственного средства в крови или плазме в течение выбранного периода времени, которая может быть измерена в течение выбранного промежутка времени (t) с помощью площади под кривой (ППК) в ходе высвобождения лекарственного средства. Изменение одного или более ФК параметров обусловливает изменение ФК кривой.
"ФК кривая" относится к кривой концентрации лекарственного средства в крови или в плазме. Такая кривая может быть функцией концентрации лекарства от времени (т.е. "ФК кривая концентрация-время") или функцией концентрации лекарственного средства от количества введенных доз (т.е. "ФК кривая концентрация-доза"). ФК кривая характеризуется ФК параметрами.
"Предотвращение" или "предупреждение", или "профилактика" относится к снижению риска возникновения такого состояния, как боль.
"Пролекарство" относится к производному действующего вещества, которое требует преобразования в организме для высвобождения действующего вещества. В некоторых вариантах осуществления под преобразованием имеют в виду ферментативное превращение. В некоторых вариантах осуществления под преобразованием имеют в виду преобразование, связанное с циклизацией. В некоторых вариантах осуществления под преобразованием имеют в виду сочетание ферментативного превращения и реакции циклизации. Пролекарства зачастую, хотя и необязательно, являются фармакологически неактивными до превращения в действующее вещество.
"Фрагмент-предшественник" относится к форме защитной группы, которая при ее использовании в качестве защиты функциональной группы в действующем веществе превращает действующее вещество в Пролекарство. Обычно, фрагмент-предшественник присоединен к лекарственному средству посредством связи(связей), которые расщепляются in vivo ферментным или неферментным образом.
В настоящем изобретении "сольват" относится к комплексу или агрегату, образованному одной или более молекулами растворенного вещества, например, пролекарством или его фармацевтически приемлемой солью и одной или более молекулами растворителя. Такие сольваты обычно являются кристаллическими твердыми веществами, имеющими в основном постоянное молярное соотношение растворенного вещества и растворителя. Типичные растворители включают, например, воду, метанол, этанол, изопропанол, уксусную кислоту и подобное. Когда растворителем является вода, образованный сольват является гидратом.
"Терапевтически эффективное количество" означает такое количество соединения (например, пролекарства), которое при введении пациенту для предотвращения или лечения такого состояния, как боль, является достаточным для такого лечения. "Терапевтически эффективное количество" будет варьироваться в зависимости от соединения, конкретного состояния и его тяжести, возраста, веса пациента и т.д.
"Терапия" или "лечение" любого состояния, такого как боль, означает, в некоторых вариантах осуществления изобретения, улучшение патологического состояния (т.е., купирование или снижение проявления указанного состояния). В некоторых вариантах осуществления изобретения "терапия" или "лечение" относится к улучшению по меньшей мере одного физического параметра, который может быть незаметен для пациента. В некоторых вариантах осуществления "терапия" или "лечение" относится к замедлению состояния, физически (например, стабилизация заметного симптома), физиологически (например, стабилизация физического параметра), либо и тем, и другим образом. В некоторых вариантах осуществления "терапия" или "лечение" относится к задержке появления патологического состояния.
Подробное описание изобретения
Перед тем, как данное изобретение будет более подробно описано ниже, следует подчеркнуть, что данное изобретение не ограничивается описанными конкретными вариантами осуществления, и, как таковое, может, конечно, в известной степени варьироваться. Также следует понимать, что терминология, применяемая в данном документе, служит только для описания конкретных вариантов осуществления и не предназначена для ограничения объема данного изобретения, который ограничен только приложенной формулой изобретения.
Следует отметить, что используемые в данном документе и в приложенной формуле изобретения термины в единственном числе включают в себя и множественное число, если из контекста явным образом не следует иное. Далее следует отметить, что формула изобретения может быть составлена таким образом, чтобы исключить любой необязательный элемент. В связи с этим, данное предупреждение призвано служить основой для использования такой исчерпывающей терминологии, как "исключительно", "только" и подобное, в связи с перечислением заявленных элементов или применением "отрицательного" ограничения.
Следует понимать, что в настоящем изобретении единственное число какого-либо объекта подразумевает один или несколько таких объектов. Например, соединение означает одно или несколько соединений. По этой причине выражения в единственном числе, "один или более" и "по меньшей мере один" могут использоваться взаимозаменяемо. Аналогичным образом, выражения "содержащий", "включающий" и "имеющий" могут использоваться взаимозаменяемо.
Публикации, обсуждаемые в данном документе, представлены исключительно для раскрытия известного уровня техники, предшествующего дате подачи данной заявки. Изложенное в данном документе не должно толковаться как признание того, что данное изобретение не может предшествовать таким публикациям путем испрашивания приоритета по предшествующему изобретению. Кроме того, указанные даты публикации могут отличаться от фактических дат публикации, и последние может быть необходимо проверить независимо.
Если не указано иное, все технические и научные выражения, применяемые в данном документе, имеют то же значение, которое обычно понимается специалистом в области техники, к которой относится данное изобретение. Хотя любые способы и материалы, подобные или эквивалентные описанным в данном документе, также могут быть применены при осуществлении или исследовании данного изобретения, в данном документе описываются только предпочтительные способы и материалы. Все публикации, упомянутые в данном документе, включены в данный документ посредством ссылки как раскрытие и описание тех способов и/или материалов, в связи с которыми данные публикации цитируются.
Если не указано иное, способы и методики описываемых вариантов осуществления изобретения обычно осуществляются согласно общепринятым способам, хорошо известным в уровне техники, и как описано в различных общих и более конкретных ссылках, которые цитируются и обсуждаются в данном описании. Смотри, например, London, Organic Chemistry, Fourth Edition, New York: Oxford University Press, 2002, pp.360-361, 1084-1085; Smith and March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001.
Номенклатура, используемая в данном документе для именования соединений, проиллюстрирована в Примерах в данном документе. В некоторых случаях эти названия получали с использованием коммерчески доступного программного обеспечения AutoNom (MDL, Сан Леандро, Калифорния).
Следует понимать, что некоторые из свойств настоящего изобретения, которые в целях ясности описаны в отдельных вариантах осуществления, могут также быть представлены в комбинации друг с другом в одном варианте осуществления. И наоборот, различные свойства изобретения, которые для краткости описаны в контексте одного варианта осуществления, могут быть также представлены отдельно или в любой подходящей подкомбинации. Все комбинации вариантов осуществления, имеющие отношение к химическим группам, представленным условными обозначениями, специально и отдельно охвачены настоящим изобретением и раскрыты здесь также, как если бы каждая из этих комбинаций была раскрыта индивидуально и подробно, при том лишь условии, что такие комбинации должны охватывать стабильные соединения (т.е. соединения, которые можно выделить, охарактеризовать и определить их биологическую активность). Кроме того, все подкомбинации химических групп, перечисленные в описывающих такие условные обозначения вариантах осуществления, также специально охвачены данным изобретением и раскрыты здесь так же, как если бы каждая такая подкомбинация химических групп была индивидуально и подробно раскрыта в данном документе.
Общие методики синтезов
Многие общие ссылки, описывающие хорошо известные химические синтетические схемы и условия, используемые для синтеза описываемых соединений, широко доступны (смотри, например, Smith and March, March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Fifth Edition, Wiley-Interscience, 2001; или Vogel, A Textbook of Practical Organic Chemistry, Including Qualitative Organic Analysis, Fourth Edition, New York: Longman, 1978).
Описанные здесь соединения могут быть очищены любыми известными из уровня техники способами, включая хроматографию, такую как, например, высокоэффективная жидкостная хроматография (ВЭЖХ), препаративная тонкослойная хроматография, колоночная флэш-хроматография и ионообменная хроматография. Можно использовать любую подходящую стационарную фазу, включая прямую и обратную фазы, а также ионные смолы. Смотри, например, Introduction to Modern Liquid Chromatography, 2nd Edition, ed. L.R. Snyder and J.J. Kirkland, John Wiley and Sons, 1979; и Thin Layer Chromatography, ed E. Stahl, Springer-Verlag, New York, 1969.
В любом из процессов получения соединений по настоящему изобретению может быть необходимо и/или желательно защитить чувствительные или реакционно-способные группы в любой из молекул, к которой это может иметь отношение. Этого может быть добиться с использованием общепринятых защитных групп как описано в классических трудах, таких как Т.W. Greene and P. G. M. Wuts, "Protective Groups in Organic Synthesis", Fourth edition, Wiley, New York 2006. Защитные группы могут быть удалены на удобной последующей стадии, используя известные из уровня техники способы.
Соединения, описанные в данном документе, могут включать один или более хиральных центров и/или двойных связей и, следовательно, могут существовать в виде различных стереоизомеров, таких как изомеры по двойной связи (т.е. геометрические изомеры), энантиомеры или диастереомеры. Соответственно, все возможные энантиомеры и стереоизомеры соединений, включая стереоизомерно чистые формы (например, геометрически чистую, энантиомерно чистую или диастереомерно чистую), энантиомерные и стереоизомерные смеси, включены в описание соединений в данном документе. Энантиомерные и стереоизомерные смеси могут быть разделены на составляющие их энантиомеры или стереоизомеры с использованием техник разделения или техник хирального синтеза, хорошо известных специалисту в данной области. Соединения также могут существовать в нескольких таутомерных формах, включая енольную форму, кето-форму и их смеси. Соответственно, химические структуры, описанные в данном документе, охватывают все возможные таутомерные формы иллюстрированных соединений.
Описанные соединения также включают изотопно-меченые соединения, в которых один или более атомов имеют атомную массу, отличную от атомной массы, обычно встречающейся в природе. Примеры изотопов, которые могут быть включены в соединения, описываемые в данном документе, включают (без ограничения)2H,3H,11С,13С,14С,15N,18O,17O и т.д. Соединения могут существовать как в несольватированных, так и в сольватированных формах, включая гидратированные формы. В общем, соединения могут быть гидратированными или сольватированными. Некоторые соединения могут существовать в нескольких кристаллических или аморфных формах. В основном, все физические формы являются эквивалентными для применений, описываемых в данном документе, и полагают, что все они включены в объем настоящего изобретения.
Типичные варианты осуществления
Далее будут сделаны ссылки на различные варианты осуществления настоящего изобретения. Следует понимать, что это изобретение не ограничено описываемыми вариантами осуществления. Напротив, предполагается, что оно охватывает все варианты, модификации и эквиваленты, которые входят в объем принятой формулы изобретения.
Варианты осуществления настоящего изобретения описывают Соединение КС-8, N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламин]-аргинин-глицин-малоновую кислоту, представленную ниже:
или ее приемлемые соли, сольваты и гидраты.
Варианты осуществления изобретения описывают также композицию, содержащую N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламин]-аргинин-глицин-малоновую кислоту, Соединение КС-8, представленное ниже:

или его фармацевтически приемлемые соли, сольваты и гидраты.
Данное изобретение описывает Соединение КС-8, модифицированное кетоном опиоидное пролекарство, которое обеспечивает контролируемое высвобождение оксикодона. В модифицированном кетоном опиоидном пролекарстве к оксикодону через енольный атом кислорода оксикодона присоединен фрагмент-предшественник. В модифицированном кетоном опиоидном пролекарстве атом водорода соответствующей енольной группы оксикодона замещен ковалентной связью с фрагментом-предшественником.
В Соединении КС-8 фрагмент-предшественник содержит способную к циклизации спейсерную уходящую группу и расщепляемый фрагмент. В Соединении КС-8 пролекарство модифицированного кетоном оксикодона соответствует соединению, в котором енольный атом кислорода замещен спейсерной уходящей группой, несущей азотный нуклеофил, защищенный расщепляемым ферментом фрагментом, причем конфигурация спейсерной уходящей группы и азотного нуклеофила является такой, чтобы при ферментативном расщеплении расщепляемого фрагмента азотный нуклеофил был способен образовывать циклическую мочевину, освобождая соединение от спейсерной уходящей группы, образуя, таким образом, оксикодон.
Фермент, способный расщеплять расщепляемый ферментом фрагмент, может быть пептидазой, также называемый протеазой, причем фрагмент-предшественник содержит расщепляемый ферментом фрагмент, связанный с нуклеофильным азотом через амидную связь (например, пептидную: -NHC(O)-). В некоторых вариантах осуществления фермент является пищеварительным ферментом белка. Изобретение предусматривает, что фермент представляет собой ЖК фермент, такой как трипсин, а расщепляемый ферментом фрагмент представляет собой расщепляемый ЖК ферментом фрагмент, такой как расщепляемый трипсином фрагмент.
Соответствующее пролекарство позволяет после его приема внутрь контролируемо высвобождать оксикодон. Это пролекарство нуждается в ферментативном расщеплении для инициирования высвобождения оксикодона, и таким образом скорость высвобождения оксикодона зависит как от скорости ферментативного расщепления, так и от скорости циклизации. Соединение КС-8 обеспечивает эффективное контролируемое высвобождение оксикодона благодаря сочетанию высокой скорости ферментативного расщепления и высокой скорости внутримолекулярной циклизации. Пролекарство сконструировано таким образом, что оно не обеспечивает слишком высокое содержание активного препарата в плазме при неправильном приеме и не способно легко разлагаться с выделением активного препарата, кроме как путем ферментативного расщепления с последующей контролируемой циклизацией.
Предпочтительно циклическая группа, образующаяся при высвобождении оксикодона, является фармацевтически приемлемой, в частности фармацевтически приемлемой циклической мочевиной. Следует понимать, что циклические мочевины в основном являются очень стабильными и обладают низкой токсичностью.
Общие методики синтеза соединений
Ниже проиллюстрированы типичные схемы синтеза соединений, раскрытых в данном документе. Соединение КС-8 может быть синтезировано с использованием описанных способов.
Типичные схемы синтеза
Типичный синтез Соединения S-104 показан на Схеме 1. В Схеме 1 для случая Соединения КС-8 n=3; первые и третьи геминальные R1 и R2 представляют собой водород; вторые геминальные R1 и R2 представляют собой метил; R5 представляет собой метил; и PG1 представляет собой защитную группу для амина.
Схема 1

В Схеме 1 Соединение S-100 представляет собой имеющееся в продаже исходное вещество. В альтернативном варианте Соединение S-100 можно синтезировать различными путями, используя находящиеся в продаже исходные вещества и/или исходные вещества, полученные традиционными способами синтеза.
Согласно Схеме 1 аминогруппу Соединения S-100 защищают трифторацетильной группой с получением Соединения S-101. Трифторацетильную группу можно получить путем реакции с использованием таких реагентов, как этилтрифторацетат, трифторацетилхлорид или 1,1,1-трихлор-3,3,3-трифторацетон.
Соединение S-101 защищают по другой аминогруппе с получением Соединения S-102, где PG1 представляет собой защитную группу аминной функции. Защитные группы аминной функции можно найти в T.W. Greene and P. G. М. Wuts, "Protective Groups in Organic Synthesis", Fourth edition, Wiley, New York 2006. Типичные защитные группы для аминогруппы включают (но не ограничиваются только ими) формильные группы; ацильные группы, например алканоильные группы, такие как ацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы, такие как бензил (Bn), тритил (Tr) и 1,1-ди-(4’-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и тому подобные.
В некоторых вариантах осуществления PG1 представляет собой Boc. Условия для образования групп Boc на Соединении S-102 можно найти в монографии Greene and Wuts. Один из способов - это реакция Соединения S-101 с ди-трет-бутилдикарбонатом. Реакция может быть необязательно осуществлена в присутствии активирующего агента, такого как ДМАП. В Схеме 1 в определенных вариантах осуществления трифторацетильная защитная группа и PG1, такая как Boc, представляют собой ортогональные защитные группы.
Согласно Схеме 1 Трифторацетильную группу удаляют из Соединения S-102 с получением Соединения S-103. Условия удаления трифторацетильной группы можно найти в монографии Greene and Wuts. Способы удаления трифторацетильной группы включают гидролиз Соединения S-102. В некоторых случаях условия гидролиза включают реакцию с гидроксидом натрия или гидроксидом лития.
Согласно Схеме 1 группу R5 присоединяют к Соединению S-103 с получением Соединения S-104. Присоединение группы R5 к аминогруппе Соединения S-103 можно осуществить, используя защитные/активирующие группы. В некоторых вариантах осуществления до присоединения группы R5 к аминогруппе Соединения S-103 присоединяют нозильную группу. Нозильную группу можно присоединить, используя нозилхлорид.
В некоторых вариантах осуществления группа R5 представляет собой метил, и ее присоединяют с использованием метилиодида. После присоединения группы R5защитную/активирующую группу можно удалить с получением Соединения S-104. Например, удаление нозильной группы можно провести с помощью тиофенола.
Типичный синтез Соединения S-202 показан на Схеме 2. В Схеме 2 для случая Соединения КС-8 Ra представляет собой гидроксил; n=3; первые и третьи геминальные R1и R2 представляют собой водород; вторые геминальные R1 и R2 представляют собой метил; R5 представляет собой метил; и PG1 представляет собой защитную группу для амина.
Схема 2

В Схеме 2 Соединение S-200 представляет собой имеющееся в продаже исходное вещество. В альтернативном варианте Соединение S-200 можно получить полусинтетическим путем из природных веществ или синтезировать различными путями, используя находящиеся в продаже исходные вещества и/или исходные вещества, полученные традиционными способами синтеза.
Согласно Схеме 2 Соединение S-200 реагирует с Соединением S-104 с образованием Соединения S-201. В этой реакции Соединение S-200 реагирует с аминогруппой Соединения S-104 с образующим карбамат реагентом с получением Соединения S-201. Подходящие образующие карбамат реагенты включают хлорформиаты, такие как 4-нитрофенил-хлорформиат.
Согласно Схеме 2 защитную группу PG1 удаляют из Соединения S-201 с образованием Соединения S-202. Условия для удаления аминогрупп можно найти в монографии Greene and Wuts. Если PG1 представляет собой группу Boc, защитную группу можно удалять в кислых условиях, например обработкой соляной или трифторуксусной кислотой.
Типичный синтез Соединения S-203 показан на Схеме 3. В Схеме 3 для случая Соединения КС-8 Ra представляет собой гидроксил; n=3; первые и третьи геминальные R1и R2 представляют собой водород; вторые геминальные R1 и R2 представляют собой метил; R5 представляет собой метил; R6 представляет собой боковую цепь аргинина; и PG2представляет собой защитную группу аминной функции.
Схема 3

Согласно Схеме 3 Соединение S-202 реагирует с Соединением S-301 с образованием Соединения S-302 в реакции пептидного сочетания. В некоторых вариантах осуществления R6 представляет собой боковую цепь агринина и необязательно защищен. Защитные группы для боковых цепей аргинина известны специалистам в данной области техники и могут быть найдены в монографии Greene and Wuts. В определенных случаях защитная группа для боковой цепи аргинина представляет собой защитную группу сульфонильного типа, такую как 2,2,4,6,7-пентаметилдигидробензофуран (Pbf). Другие защитные группы включают 2,2,5,7,8-пентаметилхроман (Pmc) и 1,2-диметилиндол-3-сульфонил (MIS).
В реакции пептидного сочетания обычно участвует традиционный реагент пептидного сочетания, и ее проводят в традиционных для реакции сочетания условиях, обычно в присутствии триалкиламина, такого как этилдиизопропиламин или диизопропилэтиламин (DIEA). Подходящими реагентами сочетания для использования являются, например, карбодиимиды, такие как этил-3-(3-диметиламино)пропилкарбодиимид (EDC), дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC) и подобные, а также другие хорошо известные реагенты сочетания, такие как N,N’-карбонилдиимидазол, 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (EEDQ), бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат (ВОР), O-(7-азабензотриазол-1-ил)-N,N,N,N’,N’-тетраметилурония гексафторфосфат (HATU) и подобные им. Необязательно, в данной реакции можно применять такие хорошо известные промоторы сочетания, как N-гидроксисукцинимид, 1-гидроксибензотриазол (НОВТ), 1-гидрокси-7-азабензотриазол (HOAT), N,N-диметиламинопиридин (DMAP) и подобные им. Обычно эту реакцию сочетания осуществляют при температуре в интервале от примерно 0°С до примерно 60°С в течение примерно 1-72 часов в инертном растворителе, таком как ТГФ или ДМФА. В некоторых случаях Соединение S-202 реагирует с Соединением S-301 с образованием Соединения S-302 в присутствии HATU.
Согласно Схеме 3 Соединение S-302 преобразуют в Соединение S-303 путем удаления защитной группы аминной функции. Условия для удаления аминогрупп можно найти в монографии Greene and Wuts. Если PG2 представляет собой группу Boc, защитную группу можно удалить в кислых условиях, например обработкой соляной или трифторуксусной кислотой.
Типичный синтез Соединения S-401 показан на Схеме 4. В Схеме 4 для случая Соединения КС-8 Ra представляет собой гидроксил; n=3; первые и третьи геминальные R1 и R2 представляют собой водород; вторые геминальные R1 и R2 представляют собой метил; R5 представляет собой метил; R6 представляет собой боковую цепь аргинина; R7представляет собой боковую цепь глицина; и R8 представляет собой группу малонила.
Схема 4

В Схеме 4 Соединение S-400 представляет собой имеющееся в продаже исходное вещество. В альтернативном варианте Соединение S-400 можно синтезировать путем различных путей синтеза, используя находящиеся в продаже исходные вещества и/или исходные вещества, полученные традиционными способами синтеза.
Согласно Схеме 4 Соединение S-303 реагирует с Соединением S-400 с образованием Соединения S-401 в реакции пептидного сочетания. В реакции пептидного сочетания обычно участвует традиционный реагент пептидного сочетания, и ее проводят в традиционных для реакции сочетания условиях, обычно в присутствии триалкиламина, такого как этилдиизопропиламин или диизопропилэтиламин (DIEA). Подходящими для использования реагентами сочетания являются, например, карбодиимиды, такие как этил-3-(3-диметиламино)пропилкарбодиимид (EDC), дициклогексилкарбодиимид (DCC), диизопропилкарбодиимид (DIC) и подобные, а также другие хорошо известные реагенты сочетания, такие как N,N’-карбонилдиимидазол, 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (EEDQ), бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат (ВОР), O-(7-азабензотриазол-1-ил)-N,N,N,N’,N’-тетраметилурония гексафторфосфат (HATU) и подобные им. Опционально в данной реакции можно использовать такие хорошо известные промотеры сочетания, как N-гидроксисукцинимид, 1-гидроксибензотриазол (НОВТ), 1-гидрокси-7-азабензотриазол (HOAT), N,N-диметиламинопиридин (DMAP) и подобные им. Обычно эту реакцию сочетания проводят при температуре в интервале от примерно 0 до примерно 60°С в течение примерно 1-72 часов в инертном растворителе, таком как ТГФ или ДМФА. В некоторых случаях Соединение S-303 реагирует с Соединением S-400 с образованием Соединения S-401 в присутствии HATU.
В некоторых случаях в Схеме 4 когда Соединение S-400 реагирует с Соединением S-303 с R8, представляющим собой водород, после реакции аминокислотного сочетания присоединяется группа R8, представляющая собой группу малонила. Группа малонила может быть присоединена путем реакции с моно-трет-бутилмалонатом. Реакции с использованием моно-трет-бутилмалоната может способствовать использование активирующих агентов, таких как симметричные ангидриды, O-(бензотриазол-1-ил)-N,N,N’,N’-тетраметилурония гексафторфосфат (HBTU), дициклогексилкарбодиимид (DCC) диизопропилкарбодиимид (DIC)/1-гидроксибезотриазол (HOBt) и бензотриазол-1-ил-окситрис(диметиламино)фосфония гексафторфосфат (ВОР). Группа малонила также может быть присоединена путем реакции с трет-бутиловым эфиром N-карбоксиметилмалоновой кислоты.
Согласно Схеме 4 можно осуществлять удаление и других защитных групп, если другие защитные группы, такие как защитные группы на фрагменте R6, использовались. Условия удаления других защитных групп зависят от природы защитной группы и известны специалистам в данной области. Эти условия можно найти в монографии Greene and Wuts.
Дополнительные типичные схемы синтеза
Типичный синтез Соединения S-503 показан на Схеме 5. В Схеме 5 для случая Соединения КС-8 n=3; первые и третьи геминальные R1 и R2 представляют собой водород; вторые геминальные R1 и R2 представляют собой метил; R5 представляет собой метил; и PG1 и PG2 представляют собой необязательные защитные группы для амина.
Схема 5

В Схеме 5 Соединение S-500 представляет собой имеющееся в продаже исходное вещество. В альтернативном варианте Соединение S-500 можно синтезировать различными путями, используя находящиеся в продаже исходные вещества и/или исходные вещества, полученные традиционными способами синтеза.
Согласно Схеме 5 Соединение S-500 защищают по аминогруппе с получением Соединения S-501, где PG1 и PG2 представляют собой защитные группы для амина. Защитные группы для амина можно найти в монографии Т.W. Greene and P. G. М. Wuts, "Protective Groups in Organic Synthesis", Fourth edition, Wiley, New York 2006. Типичные защитные группы аминной функции включают (но не ограничиваются только ими) формильные группы; ацильные группы, например алканоильные группы, такие как ацетил; алкоксикарбонильные группы, такие как трет-бутоксикарбонил (Boc); арилметоксикарбонильные группы, такие как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметильные группы, такие как бензил (Bn), тритил (Tr) и 1,1-ди-(4’-метоксифенил)метил; силильные группы, такие как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS); и тому подобные.
В некоторых вариантах осуществления PG1 и PG2 представляют собой группы Boc. Условия для получения групп Boc на Соединении S-501 можно найти в монографии Greene and Wuts. Один из способов - это реакция Соединения S-501 с ди-трет-бутилдикарбонатом. Реакция может быть опционально проведена в присутствии активирующего агента, такого как ДМАП.
Согласно Схеме 5 карбоксибензильную группу удаляют из Соединения S-501 с получением Соединения S-502. Условия для удаления карбоксибензильной группы можно найти в монографии Greene and Wuts. Способы удаления карбоксибензильной группы включают гидрогенолиз Соединения S-501 или обработку Соединения S-501 с помощью HBr. Одним из способов удаления карбоксибензильной группы является реакция Соединения S-501 с водородом и палладием.
Согласно Схеме 5 Соединение S-502 подвергают реакции с фосгеном с образованием Соединения S-503. Реакция с фосгеном дает на амино-группе Соединения S-502 ацилхлорид. Фосген может быть заменен другими реагентами, такими как дифосген или трифосген.
Типичный синтез Соединения S-602 показан на Схеме 6. В Схеме 6 для случая Соединения КС-8 Ra представляет собой гидроксил; n=3; первые и третьи геминальные R1и R2 представляют собой водород; вторые геминальные R1 и R2 представляют собой метил; R5 представляет собой метил; и PG1 и PG2 представляют собой необязательные защитные группы аминной функции.
Схема 6

В Схеме 6 Соединение S-600 представляет собой имеющееся в продаже исходное вещество. В альтернативном варианте Соединение S-600 можно получить полусинтетическим путем из природных веществ или синтезировать путем различных путей синтеза, используя находящиеся в продаже исходные вещества и/или исходные вещества, полученные традиционными способами синтеза.
Согласно Схеме 6 Соединение S-600 реагирует с Соединением S-503 с образованием Соединения S-601. В этой реакции енолят Соединения S-600 реагирует с ацетилхлоридом Соединения S-503 с образованием карбамата.
Согласно Схеме 6 необязательные защитные группы PG1 и PG2 удаляют из Соединения S-601 с образованием Соединения S-602. Условия для удаления аминогрупп можно найти в монографии Greene and Wuts. Если PG1 и/или PG2 представляют собой группы Boc, защитные группы можно удалять в кислых условиях, например обработкой соляной или трифторуксусной кислотой.
Соединение S-602 может быть использовано в приведенных выше схемах, таких как Схемы 3 и 4, для получения Соединения КС-8.
Как здесь будет описано более подробно, настоящее изобретение описывает способы и промежуточные соединения, пригодные для получения соединений по настоящему изобретению или его соли, или сольвата, или стереоизомера. Соответственно, настоящее изобретение описывает способ получения соединения по изобретению, включающий:
взаимодействие соединения формулы


Соответственно, как здесь будет описано более подробно, настоящее изобретение описывает способ получения соединения по изобретению, включающий:
взаимодействие соединения формулы


Соответственно, как здесь будет описано более подробно, настоящее изобретение описывает способ получения соединения по изобретению, включающий:
взаимодействие соединения формулы


В одном случае вышеуказанный способ дополнительно включает стадию образования соли соединения по настоящему изобретению. Варианты осуществления также направлены на другие описанные здесь способы; а также на продукт, полученный любым из описанных здесь способов.
Трипсиновые ингибиторы
Как указано в настоящем описании, настоящее изобретение также относится к фармацевтическим композициям и способам их применения, где фармацевтические композиции содержат пролекарство. Соединение КС-8, обеспечивающее контролируемое высвобождение оксикодона путем ферментативного расщепления с последующей внутримолекулярной циклизацией, и ингибитор, такой как трипсиновый ингибитор, взаимодействующий с ферментом, являющимся посредником в ферментативном высвобождении оксикодона из пролекарства, так что ферментативное расщепление пролекарства замедляется. В настоящем описании предполагается, что такой фермент представляет собой трипсин.
В настоящем изобретении термин "трипсиновый ингибитор" относится к любому средству, способному ингибировать действие трипсина на субстрат. Термин "трипсиновый ингибитор" включает также соли трипсинового ингибитора. Способность агента ингибировать трипсин может быть измерена с использованием методов количественного анализа, известных в уровне техники. Например, в типичном количественном анализе одна единица соответствует количеству ингибитора, которое снижает активность трипсина на одну единицу этилового эфира бензоил-L-аргинина (BAEE-U). Одна единица BAEE-U представляет собой количество фермента, которое увеличивает поглощение при 253 нм на 0,001 в минуту при рН 7,6 и 25°С. Смотри, например, K. Ozawa, M. Laskowski, 1966, J. Biol. Chem. 241, 3955 и Y. Birk, 1976, Meth. Enzymol. 45, 700. В некоторых случаях трипсиновый ингибитор может взаимодействовать с активным сайтом трипсина, таким как карман S1 и карман S3/4. Карман S1 содержит остаток аспартата, обладающий сродством к положительно заряженному фрагменту. Карман S3/4 является гидрофобным карманом. Настоящее изобретение относится к специфическим трипсиновым ингибиторам и неспецифическим ингибиторам серин-протеаз.
Из уровня техники известно множество трипсиновых ингибиторов, как специфичных к трипсину, так и таких, которые ингибируют трипсин и другие протеазы, например химотрипсин. Настоящее изобретение описывает трипсиновые ингибиторы, представляющие собой белки, пептиды и малые молекулы. Изобретение описывает трипсиновые ингибиторы, представляющие собой необратимые или обратимые ингибиторы. Изобретение описывает трипсиновые ингибиторы, представляющие собой конкурентные ингибиторы, неконкурентные ингибиторы или неконкурирующие ингибиторы. Изобретение описывает натуральные, синтетические или полусинтетические трипсиновые ингибиторы.
Трипсиновые ингибиторы могут быть получены из различных животных или растительных источников, таких как соя, кукуруза, фасоль лимская и другие бобовые, тыква, подсолнечник, из поджелудочных желез и легких коровы и других животных, белка куриных и индюшиных яиц, детской смеси на основе сои и крови млекопитающих. Трипсиновые ингибиторы также могут иметь микробное происхождение: например, антипаин; смотри, например, Н. Umezawa, 1976, Meth. Enzymol. 45, 678. Трипсиновый ингибитор может также представлять собой имитатор аргинина или имитатор лизина или другое синтетическое соединение, например арилгуанидин, бензамидин, 3,4-дихлороизокумарин, диизопропилфторфосфат, габексата мезилат и фенилметансульфонил фторид или их замещенные варианты или аналоги. В некоторых вариантах осуществления трипсиновые ингибиторы содержат ковалентно модифицируемые группы, как, например, хлоркетоновый фрагмент, альдегидный фрагмент или эпоксидный фрагмент. Другими примерами трипсиновых ингибиторов являются апротинин, камостат и пентамидин.
В настоящем изобретении имитатор аргинина или лизина является соединением, которое способно связываться с Р1 карманом трипсина и/или мешать функционированию активного центра трипсина. Имитатор аргинина или лизина может быть расщепляемым или нерасщепляемым фрагментом.
В одном варианте осуществления трипсиновый ингибитор получают из соевых бобов. Трипсиновые ингибиторы, полученные из соевых бобов (Glycine max), легко доступны и считаются безопасными для потребления человеком. Они включают (но не ограничиваются только ими) SBTI, который ингибирует трипсин, и ингибитор Баумана-Бирка, который ингибирует трипсин и химотрипсин. Такие Трипсиновые ингибиторы можно приобрести, например, у компании Sigma-Aldrich, St. Louis, МО, USA.
Следует иметь в виду, что фармацевтические композиции согласно вариантам осуществления настоящего изобретения могут, кроме того, включать один или несколько других трипсиновых ингибиторов.
Как указано выше, трипсиновый ингибитор может представлять собой имитатор аргинина или лизина или другое синтетическое соединение. В некоторых вариантах осуществления трипсиновый ингибитор представляет собой имитатор аргинина или имитатор лизина, где имитатором аргинина или имитатором лизина является синтетическое соединение.
Некоторые инигибиторы трипсина включают соединения формулы:
в которых:
Q1 выбирают из -O-Q4 или -Q4-СООН, где Q4 является C1-C4 алкилом;
Q2 представляет собой N или СН; и
Q3 представляет собой арил или замещенный арил.
Некоторые инигибиторы трипсина включают соединения формулы:

в которых:
Q5 представляет собой -C(O)-COOH или -NH-Q6-Q7-SO2-C6H5, где
Q6 представляет собой -(CH2)p-COOH;
Q7 представляет собой -(CH2)r-C6H5;
Q8 представляет собой NH;
n является числом от 0 до 2;
o равно нулю или единице;
p является целым числом от одного до трех; и
r является целым числом от одного до трех.
Некоторые инигибиторы трипсина включают соединения формулы:

в которых:
Q5 представляет собой -C(O)-COOH или -NH-Q6-Q7-SO2-C6H5, где
Q6 представляет собой -(CH2)p-COOH;
Q7 представляет собой -(CH2)r-C6H5; и
p является целым числом от одного до трех; и
r является целым числом от одного до трех.
Некоторые трипсиновые ингибиторы включают следующее:









Описание способов получения Соединения 101, Соединения 102, Соединения 103, Соединения 104, Соединения 105, Соединения 107 и Соединения 108 предоставлено в международной публикации РСТ под номером WO 2010/045599 A1, опубликованной 22 апреля 2010, которая включена в данный документ во всей своей полноте посредством ссылки. Соединение 106, Соединение 109 и Соединение 110 можно приобрести, например, у компании Sigma-Aldrich, St. Louis, МО, USA.
В некоторых вариантах осуществления трипсиновым ингибитором является SBTI, BBSI, Соединение 101, Соединение 106, Соединение 108, Соединение 109 или Соединение 110. В некоторых вариантах осуществления трипсиновый ингибитор представляет собой камостат.
В некоторых вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-I:

в котором:
А представляет собой группу следующей формулы:

Rt9 и Rt10 каждый независимо представляет собой атом водорода или группу С1-4 алкила,
Rt8 представляет собой группу, выбранную из следующих формул:



в которых Rt11, Rt12 и Rt13 каждый независимо представляет собой
(1) атом водорода,
(2) фенильную группу,
(3) группу C1-4 алкила, замещеннго фенильной группой,
(4) группу С1-10 алкила,
(5) группу С1-10 алкоксила,
(6) группу C2-10 алкенила, имеющую от 1 до 3 двойных связей,
(7) группу C2-10 алкинила, имеющую от 1 до 2 тройных связей,
(8) группу формулы Rt15-C(O)XRt16,
в которой Rt15 представляет собой одинарную связь или группу C1-8 алкилена,
Х представляет собой атом кислорода или NH-группу, и
Rt16 представляет собой атом водорода, группу C1-4 алкила, фенильную группу или группу C1-4 алкила, замещенного фенильной группой, или
(9) группу С3-7 циклоалкила;
структура

Rt14 представляет собой атом водорода, группу C1-4 алкила, замещенного фенильной группой, или группу формулы COORt17, где Rt17 представляет собой атом водорода, группу C1-4 алкила или группу C1-4 алкила, замещенного фенильной группой;
при условии, что Rt11, Rt12 и Rt13 одновременно не являются атомами водорода;
или его нетоксичные соли, соли присоединения кислот или гидраты.
В некоторых вариантах осуществления трипсиновый ингибитор представляет собой соединение, выбранное из следующего:




В некоторых вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-II:

в которой:
X является NH;
n равно нулю или единице; и
Rt1 выбран из водорода, галогена, нитро-группы, алкила, замещенного алкила, алкокси, карбоксила, алкоксикарбонила, ацила, аминоацила, гуанидина, амидино-группы, карбамида, амино-группы, замещенной амино-группы, гидроксила, циано-группы и -(СН2)m-С(O)-О-(CH2)m-С(O)-N-Rn1Rn2, где каждый m независимо равен числу от 0 до 2; и Rn1 и Rn2 независимо выбраны из водорода и С1-4 алкила.
В некоторых вариантах осуществления в формуле T-II Rt1 представляет собой гуанидино- или амидино-группу.
В некоторых вариантах осуществления в формуле T-II Rt1 представляет собой -(СН2)m-С(O)-О-(CH2)m-С(O)-N-Rn1Rn2, где m равно единице и Rn1 и Rn2 представляют собой метил.
В некоторых вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-III:

в которой:
Х является NH’
n равно нулю или единице;
Lt1 выбирают из -С(O)-O-; -О-С(О)-; -О-(CH2)m-О-; -OCH2-Art2-CH2O-; -С(O)-NRt3- и -NRt3-C(O)-;
Rt3 выбирают из водорода, C1-6 алкила и замещенного C1-6 алкила;
Art1и Art2 независимо представляют собой замещенную или незамещенную арильную группу;
m является числом от 1 до 3; и
Rt2 выбран из водорода, галогена, нитро-группы, алкила, замещенного алкила, алкокси, карбоксила, алкоксикарбонила, ацила, аминоацила, гуанидина, амидино-группы, карбамида, амино-группы, замещенной амино-группы, гидроксила, циано-группы и -(CH2)m-C(O)-O-(CH2)m-C(O)-N-Rn1Rn2, где каждый m независимо равен числу от 0 до 2; и Rn1 и Rn2 независимо выбраны из водорода и C1-4 алкила.
В некоторых вариантах осуществления в формуле T-III Rt2 представляет собой гуанидино- или амидино-группу.
В некоторых вариантах осуществления в формуле T-III Rt2 представляет собой -(CH2)m-C(O)-O-(CH2)m-C(O)-N-Rn1Rn2, где m равно единице и Rn1 и Rn2 представляют собой метил.
В некоторых вариантах осуществления трипсиновый ингибитор представляет собой соединение формулы T-IV:

которой:
каждый Х представляет собой NH;
каждое n независимо равно нулю или единице;
Lt1 выбирают из -С(O)-O-; -О-С(О)-; -O-(СН2)m-O-; -OCH2-Art2-CH2O-; -С(O)-NRt3- и -NRt3-С(O)-;
Rt3 выбирают из водорода, C1-6 алкила и замещенного C1-6 алкила;
Art1и Art2 независимо представляют собой замещенную или незамещенную арильную группу; и
m является числом от 1 до 3.
В некоторых вариантах осуществления в формуле T-IV Art1или Art2 является фенилом.
В некоторых вариантах осуществления в формуле T-IV Art1 или Art2 является нафтилом.
В некоторых вариантах осуществления трипсиновый ингибитор представляет собой Соединение 109.
В некоторых вариантах осуществления трипсиновый ингибитор представляет собой

В некоторых вариантах осуществления трипсиновый ингибитор представляет собой Соединение 110 или его бис-ариламидиновый вариант; см., например, J.D. Geratz, M.C.-F. Cheng и R.R. Tidwell (1976) J Med. Chem. 19, 634-639.
Следует иметь в виду, что данное изобретение также включает ингибиторы других ферментов, вовлеченных в ассимиляцию белка, которые могут быть использованы в комбинации с Соединением КС-8 для ослабления высвобождения оксикодона из пролекарства по изобретению.
Комбинации пролекарства и трипсинового ингибитора
Как уже обсуждалось выше, данное изобретение описывает фармацевтические композиции, содержащие трипсиновый ингибитор и Соединение КС-8 модифицированное кетоном опиоидное пролекарство, которое содержащее расщепляемый трипсином фрагмент-предшественник, который при расщеплении способствует высвобождению оксикодона. Примеры композиций, содержащих Соединение КС-8 и трипсиновый ингибитор, описаны ниже.
Варианты осуществления изобретения описывают фармацевтическую композицию, включающую соединение формул T-I - T-IV и Соединение КС-8 или их фармацевтически приемлемые соли. Варианты осуществления изобретения описывают фармацевтическую композицию, включающую Соединение 109 и Соединение КС-8 или их фармацевтически приемлемые соли.
Некоторые варианты осуществления описывают комбинацию Соединения КС-8 и трипсинового ингибитора, в которой трипсиновый ингибитор проиллюстиррован в следующей таблице.
Комбинации Соединения КС-8 и других лекарственных средств
Настоящее изобретение описывает Соединение КС-8 и дополнительное пролекарство или лекарственное средство, включенные в фармацевтическую композицию. Такие пролекарство или лекарственное средство обеспечивают дополнительное обезболивание или другие преимущества. Их примеры включают опиоиды, парацетамол, нестероидные противовоспалительные средства (НПВС) и другие анальгетики. В одном варианте осуществления Соединение КС-8 может быть объединено с пролекарством опиоидного антагониста или непосредственно с лекарственным. Другие примеры включают лекарственные средства или пролекарства, которые оказывают благоприятное воздействие, отличное от болеутоляющего или дополнительное к нему. Варианты осуществления изобретения описывают фармацевтическую композицию, которая включает Соединение КС-8 и парацетамол или их фармацевтически приемлемые соли.
Такие композиции могут также включать трипсиновый ингибитор. В некоторых вариантах осуществления трипсиновый ингибитор выбирают из SBTI, BBSI, Соединения 101, Соединения 106, Соединения 108, Соединения 109 или Соединения 110. В некоторых вариантах осуществления трипсиновый ингибитор представляет собой Соединение 109. В некоторых вариантах осуществления трипсиновый ингибитор представляет собой камостат.
В некоторых вариантах осуществления фармацевтическая композиция включает Соединение КС-8, неопиоидное лекарственное средство и по меньшей мере один опиоид или опиоидное пролекарство.
Фармацевтические композиции и способы их применения
Как описано в данном документе, варианты осуществления настоящего изобретения относятся также к композиции, содержащей N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламин]-аргинин-глицин-малоновую кислоту, Соединение КС-8. Фармацевтическая композиция по изобретению может дополнительно содержать фармацевтически приемлемый носитель. Композиция обычно изготовлена в удобной форме, подходящей для перорального (включая буккальное и подъязычное) введения, например в виде таблетки, капсулы, тонкой пленки, порошка, суспензии, раствора, сиропа, дисперсии или эмульсии. Композиция может содержать компоненты, общепринятые в фармацевтических препаратах, например один или несколько носителей, связующих веществ, смазочных средств, наполнителей (например, для обеспечения характеристик контролируемого высвобождения), модификаторов рН, подсластителей, объемообразующих средств, красящих веществ или других активных средств.
Пациентами могут быть люди, а также другие млекопитающие, такие как домашние животные, животные в зоопарке и животные-компаньоны, такие как кошка, собака или лошадь.
В другом аспекте варианты осуществления изобретения описывают фармацевтическую композицию, как описано в данном документе выше, предназначенную для применения в лечении боли. Фармацевтическая композиция согласно вариантам осуществления изобретения может быть использована, например, в лечении пациента, страдающего от боли или имеющего риск возникновения боли. Соответственно, настоящее изобретение описывает способы лечения или предупреждения боли у субъектов, включающие введение субъекту описываемой композиции. Настоящее изобретение описывает композицию для использования в терапии или профилактике, или в качестве лекарственного препарата. Настоящее изобретение также описывает использование композиции в производстве лекарственных препаратов, особенно в производстве лекарственных препаратов для лечения или предупреждения боли.
Композиции по настоящему изобретению могут быть использованы в лечении или предупреждении боли, в том числе (но без ограничения) острой боли, хронической боли, невропатической боли, острой травматической боли, боли в суставах, боли при остеоартрите, ревматоидно-артритной боли, мышечно-скелетной боли, боли после стоматологических операций, зубной боли, миофасциальной боли, боли при раке, висцеральной боли, боли при диабете, мышечной боли, невралгической боли после герпеса, хронической боли в области таза, боли при эндометриозе, боли при воспалении органов таза и боли при родах. Острая боль включает без ограничения острую травматическую боль или послеоперационную боль. Хроническая боль включает без ограничения невропатическую боль, артритную боль, боль при остеоартрите, ревматоидно-артритную боль, мышечно-скелетную боль, зубную боль, миофасциальную боль, боль при раке, боль при диабете, висцеральную боль, мышечную боль, невралгическую боль после герпеса, хроническую тазовую боль, боль при эндометриозе, боль при воспалении органов таза и боль в спине.
Данное изобретение описывает применение Соединения КС-8 в лечении боли. Данное изобретение описывает также применение Соединения КС-8 для предотвращения боли.
Данное изобретение описывает применение Соединения КС-8 в изготовлении лекарственного препарата для лечения боли. Данное изобретение описывает также применение Соединения КС-8 в изготовлении лекарственного препарата для предотвращения боли.
В другом аспекте варианты осуществления изобретения касаются способа лечения боли у пациента, нуждающегося в таком лечении, включающего введение эффективного количества фармацевтической композиции, как описано в данном документе выше. В другом аспекте варианты осуществления изобретения касаются способа предотвращения боли у пациента, нуждающегося в таком лечении, включающего введение эффективного количества фармацевтической композиции, как описано в данном документе выше.
Эффективное количество раскрываемой здесь композиции, предназначенной для введения пациенту (т.е. количество, достаточное для обеспечения такого уровня кетон-содержащего опиоида в крови, который достаточен для эффективного лечения или профилактики боли), будет зависеть от биодоступности конкретной композиции, чувствительности конкретной композиции к ферментной активации в кишечнике, а также других факторов, таких как вид, возраст, вес, пол и состояние пациента, путь введения и мнение лечащего врача. Если композиция включает также трипсиновый ингибитор, то количество раскрытой здесь композиции, подлежащее введению пациенту, также будет зависеть и от количества и действенности трипсинового ингибитора, присутствующего в композиции. Обычно доза композиции может быть такой, чтобы содержание Соединения КС-8 находилось в диапазоне от 0,01 миллиграмма пролекарства на килограмм до 20 миллиграммов пролекарства на килограмм (мг/кг) массы тела. Например, композиция, содержащая Соединение КС-8, может вводиться в дозе, эквивалентной дозе свободного оксикодона в диапазоне от 0,02 до 0,5 мг/кг массы тела, или от 0,01 мг/кг до 10 мг/кг массы тела, или от 0,01 до 2 мг/кг массы тела. В одном варианте осуществления композиция может быть введена в такой дозе, чтобы достигаемый в крови уровень оксикодона находился в пределах от 0,5 нг/мл до 10 нг/мл.
Как обсуждалось выше, данное изобретение также описывает фармацевтические композиции, включающие трипсиновый ингибитор и Соединение КС-8 - модифицированное фенолом опиоидное пролекарство, содержащее расщепляемый трипсином фрагмент-предшественник, который при расщеплении способствует высвобождению оксикодона. В таких фармацевтических композициях эффективное количество трипсинового ингибитора, подлежащее введению пациенту (т.е. предназначенное для ослабления высвобождения оксикодона, если введение одного только Соединения КС-8 может привести к передозировке оксикодона), будет зависеть от эффективной дозы Соединения КС-8 и действенности конкретного трипсинового ингибитора, а также других факторов, таких как вид, возраст, вес, пол и состояние пациента, путь введения и мнение лечащего врача. В общем случае доза трипсинового ингибитора может быть в пределах от 0,05 мг до 50 мг на мг Соединения КС-8. В некоторых вариантах осуществления доза трипсинового ингибитора может быть в пределах от 0,001 мг до 50 мг на мг Соединения КС-8. В одном варианте осуществления доза трипсинового ингибитора может быть в пределах от 0,01 наномоль до 100 микромоль на микромоль Соединения КС-8.
Типичные воплощения стандартных доз пролекарства Соединения КС-8 и трипсинового ингибитора, имеющих желаемую фармакокинетическую кривую
Варианты осуществления настоящего изобретения включают композицию, которая содержит (а) пролекарство, содержащее оксикодон, ковалентно связанный через енольный кислород с фрагментом, содержащим расщепляемый трипсином фрагмент, где расщепление трипсином расщепляемого трипсином фрагмента способствует высвобождению оксикодона, где указанное пролекарство представляет собой Соединение КС-8, и (b) трипсиновый ингибитор, взаимодействующий с трипсином, который является посредником в ферментативно контролируемом высвобождении оксикодона из пролекарства после приема такой композиции внутрь.
Варианты осуществления изобретения включают стандартную дозу, содержащую композицию, например фармацевтическую композицию, включающую Соединение КС-8, модифицированное кетоном пролекарство, и трипсиновый ингибитор, где Соединение КС-8 и трипсиновый ингибитор присутствуют в стандартной дозе в количестве, достаточном для обеспечения предварительно выбранной фармакокинетической (ФК) кривой после приема лекарства внутрь. В следующих вариантах осуществления предварительно выбранная ФК кривая содержит по меньшей мере одно значение ФК параметра, которое меньше значения ФК параметра для случая оксикодона, высвобождаемого после приема внутрь эквивалентной дозы Соединения КС-8 в отсутствие ингибитора. В дальнейших вариантах осуществления изобретения это значение ФК параметра выбрано из значения Cmax оксикодона, значения совокупной дозы оксикодона и значения (1/Tmax оксикодона).
В некоторых вариантах осуществления изобретения стандартная доза обеспечивает выбранную заранее ФК кривую после приема внутрь по меньшей мере двух стандартных доз. В родственных вариантах осуществления изобретения выбранная заранее ФК кривая таких стандартных доз отличает от ФК кривой после приема внутрь эквивалентной дозы Соединения КС-8 без ингибитора. В родственных вариантах осуществления изобретения такая стандартная доза обеспечивает линейную ФК кривую при приеме внутрь увеличивающегося числа стандартных доз. В родственных вариантах осуществления изобретения такая стандартная доза обеспечивает нелинейную ФК кривую при приеме внутрь увеличивающегося числа стандартных доз. В родственных вариантах осуществления значение ФК параметра ФК кривой такой стандартной дозы выбирают из значения Cmax оксикодона, значения (1/Tmax оксикодона) и значения совокупной дозы оксикодона.
Варианты осуществления изобретения включают способы лечения пациента, включающие введение любых композиций, например фармацевтических композиций, включающих Соединение КС-8 и трипсиновый ингибитор, или описанных в данном документе стандартных доз нуждающемуся в этом пациенту. Варианты осуществления изобретения включают способы уменьшения побочных эффектов лечения, включающие введение любых описанных в данном документе композиций, например фармацевтических композиций, или стандартных доз нуждающемуся в этом пациенту. Варианты осуществления изобретения включают способы улучшения соблюдения пациентом режима предписанного врачом лечения, включающие управление введением нуждающемуся в этом пациенту любых описанных в данном документе композиций, например фармацевтических композиций, или стандартных доз. Такие варианты осуществления могут обеспечить улучшение соблюдения пациентом предписанного режима лечения пролекарством с ингибитором по сравнению с соблюдением пациентом предписанного режима лечения с использованием лекарственного средства и/или пролекарства без ингибитора.
Варианты осуществления настоящего изобретения включают способы уменьшения риска непреднамеренной передозировки оксикодона, включающие управление введением любых описанных в данном документе композиций, например фармацевтических композиций, или стандартных доз нуждающемуся в этом пациенту.
Варианты осуществления изобретения включают способы получения стандартной дозы, включающие объединение Соединения КС-8 и трипсинового ингибитора в стандартной дозе, где Соединение КС-8 и трипсиновый ингибитор присутствуют в стандартной дозе в количестве, достаточном для уменьшения высвобождения оксикодона из Соединения КС-8.
Варианты осуществления изобретения включают способы предотвращения злоупотребления или неправильного использования множества стандартных доз Соединения КС-8, включающие объединение в стандартной дозе Соединения КС-8 и трипсинового ингибитора, где Соединение КС-8 и трипсиновый ингибитор присутствуют в стандартной дозе в количестве, эффективном для уменьшения высвобождения оксикодона из Соединения КС-8 таким образом, что прием внутрь множества стандартных доз пациентом не приводит к пропорциональному высвобождению оксикодона. В дополнительных вариантах осуществления высвобождение лекарственного препарата уменьшено по сравнению с высвобождением лекарственного препарата из эквивалентной дозы в отсутствие ингибитора.
Один из вариантов осуществления изобретения касается способа определения трипсинового ингибитора и пролекарства Соединения КС-8, пригодных для приготовления стандартной дозы. Такой способ может быть осуществлен, например, как количественный анализ in vitro, количественный анализ in vivo, или количественный анализ ех vivo.
Варианты осуществления данного изобретения включают способы определения трипсинового ингибитора и пролекарства Соединения КС-8, пригодных для приготовления стандартной дозы, включающие объединение Соединения КС-8, трипсинового ингибитора и трипсина в реакционной смеси и определение конверсии пролекарства, где снижение конверсии пролекарства в присутствии трипсинового ингибитора по сравнению с конверсией пролекарства в отсутствие трипсинового ингибитора указывает на то, что пролекарство Соединение КС-8 и трипсиновый ингибитор пригодны для приготовления стандартной дозы.
Варианты осуществления данного изобретения включают способы определения трипсинового ингибитора и пролекарства Соединения КС-8, пригодных для приготовления стандартной дозы, включающие введение животному трипсинового ингибитора и пролекарства Соединения КС-8 и определение конверсии пролекарства, где снижение конверсии в оксикодон в присутствии трипсинового ингибитора по сравнению с конверсией в оксикодон в отсутствие трипсинового ингибитора указывает на то, что трипсиновый ингибитор и пролекарство Соединение КС-8 пригодны для приготовления стандартной дозы. В конкретных вариантах осуществления введение лекарства включает введение животному увеличивающихся доз ингибитора совместно с выбранной фиксированной дозой пролекарства. Определение конверсии пролекарства может способствовать идентификации дозы ингибитора и дозы пролекарства, которые обеспечивают предварительно выбранную фармакокинетическую (ФК) кривую. Такие способы могут быть осуществлены как, например, количественный анализ in vivo или количественный анализ ех vivo.
Варианты осуществления данного изобретения включают способы определения трипсинового ингибитора и пролекарства Соединения КС-8, пригодных для приготовления стандартной дозы, включающие введение в ткани животного трипсинового ингибитора и пролекарства Соединения КС-8 и определение конверсии пролекарства, где снижение конверсии пролекарства в присутствии трипсинового ингибитора по сравнению с конверсией пролекарства в отсутствие трипсинового ингибитора указывает на то, что трипсиновый ингибитор и пролекарство Соединение КС-8 пригодны для приготовления стандартной дозы.
Стандартные дозы пролекарства Соединения КС-8 и трипсинового ингибитора, имеющие желаемую фармакокинетическую кривую
Настоящее изобретение описывает стандартные дозы пролекарства и ингибитора, которые могут обеспечить желаемую фармакокинетическую (ФК) кривую. Стандартные дозы могут обеспечить ФК кривую, отличающуюся от исходной ФК кривой, как описано здесь. Следует понимать, что модифицированная ФК кривая может обеспечить модифицированную фармакодинамическую (ФД) кривую. Многократный прием внутрь такой стандартной дозы также может обеспечить желаемую ФК кривую.
Если специально не указано другое, "стандартная доза", как этот термин употребляется в данном документе, относится к комбинации расщепляемого трипсином пролекарства и трипсинового ингибитора. "Однократная стандартная доза" - это одна единица комбинации расщепляемого трипсином пролекарства и трипсинового ингибитора, где однократная стандартная доза предоставляет терапевтически эффективное количество лекарственного средства (т.е. количество лекарства, достаточное для получения терапевтического эффекта, например доза в пределах соответствующего лекарству терапевтического окна или терапевтического диапазона). "Многократные стандартные дозы" или "кратные стандартные дозы", или "множество стандартных доз" относится к по меньшей мере двум однократным стандартным дозам.
В данном изобретении "ФК кривая" относится к кривой концентрации лекарственного средства в крови или в плазме. Такая кривая может быть функцией концентрации лекарства от времени (т.е. "ФК кривая концентрация-время") или функцией концентрации лекарственного средства от количества введенных доз (т.е. "ФК кривая концентрация-доза"). ФК кривая характеризуется ФК параметрами.
В настоящем изобретении "ФК параметр" относится к уровню концентрации лекарственного средства в крови или в плазме, например: 1) "Cmax лекарственного средства" - максимальная концентрация лекарственного средства, достигаемая в крови или плазме; 2) "Tmax лекарственного средства" - время, необходимое для достижения Cmax с момента приема внутрь; и 3) "совокупная доза лекарственного средства" - сумаррная концентрация лекарственного средства в крови или плазме в течение выбранного периода времени, которая может быть измерена в течение выбранного промежутка времени (t) с использованием площади под кривой (ППК) в ходе высвобождения лекарственного средства. Изменение одного или более ФК параметров обусловливает изменение ФК кривой.
Для описания особенностей стандартных доз в настоящем изобретении "значения ФК параметра", определяющие ФК кривую, включают Cmax лекарственного средства (например, Cmax оксикодона), совокупную дозу лекарственного средства (например, площадь под кривой) (например, совокупная доза оксикодона) и 1/(Tmax лекарственного средства) (уменьшенный параметр 1/Tmax говорит об увеличении Tmax по отношению к сравнительному Tmax) (например, 1/Tmax оксикодона). Так, уменьшение значения ФК параметра относительно сравнительного значения ФК параметра может означать, например, снижение Cmax лекарственного средства, снижение совокупной дозы лекарственного средства и/или увеличение Tmax.
Стандартные дозы по настоящему изобретению могут быть приспособлены для достижения модифицированной ФК кривой, например ФК кривой, которая отличается от кривой, получаемой при введении данной дозы пролекарства в отсутствие ингибитора (т.е. без ингибитора). Например, стандартные дозы могут обеспечить по меньшей мере одно из: сниженного Cmax лекарственного средства, увеличенного Tmax и/или пониженной совокупной дозы лекарственного средства по сравнению с приемом внутрь дозы пролекарства в том же количестве, но в отсутствие ингибитора. Такое изменение происходит благодаря включению в стандартную дозу ингибитора.
В настоящем изобретении "фармакодинамическая (ФД) кривая" относится к кривой эффективности лекарственного средства для данного пациента (или субъекта, или потребителя), которая может быть охарактеризована ФД параметрами. "ФД параметры" включают "Emax лекарственного средства" (максимальная эффективность лекарственного средства), "ЕС50 лекарственного средства" (концентрация лекарственного средства при 50% Emax) и побочные эффекты.
Фиг. 1 представляет схематическую иллюстрацию примера влияния увеличения концентраций ингибитора на ФК параметр Cmax лекарственного средства при фиксированной дозе пролекарства. При низких концентрациях ингибитора заметного влияния на высвобождение лекарственного средства может не наблюдаться, о чем свидетельствует плоский участок графика Cmax лекарственного средства (ось Y) относительно концентрации ингибитора (ось X). По мере увеличения концентрации ингибитора достигается концентрация, при которой высвобождение лекарственного средства из пролекарства замедляется, вызывая снижение или подавление Cmax лекарственного средства. Так, влияние ингибитора на ФК параметр пролекарства для стандартной дозы данного изобретения может меняться от неопределяемого через умеренное и к полному ингибированию (т.е. к отсутствию детектируемого высвобождения лекарственного средства).
Стандартная доза может быть приспособлена для обеспечения желаемой ФК кривой (например, ФК кривой концентрация-время) после приема внутрь однократной дозы. Стандартная доза может быть приспособлена для обеспечения желаемой ФК кривой (например, ФК кривой концентрация-время) после приема внутрь множества стандартных доз (например, по меньшей мере 2, по меньшей мере 3, по меньшей мере 4 или более стандартных доз).
Стандартные дозы, обеспечивающие модифицированную ФК кривую
Комбинация пролекарства и ингибитора в стандартной дозе может обеспечить желаемую (или "заранее выбранную") ФК кривую (например, ФК кривую концентрация-время) после приема однократной дозы внутрь. ФК кривая такой стандартной дозы может быть охарактеризована одним или несколькими параметрами из заранее выбранной Cmax лекарственного средства, заранее выбранного Tmax или заранее выбранной совокупной дозы лекарственного средства. ФК кривая стандартной дозы может отличаться от ФК кривой, полученной с помощью эквивалентной дозы пролекарства в отсутствие ингибитора (т.е. дозы, такой же, как стандартная доза, за исключением того, что в ней нет ингибитора).
Модифицированная ФК кривая может иметь сниженное значение ФК параметра по отношению к сравнительному значению ФК параметра (т.е. значение ФК параметра ФК кривой после приема внутрь дозы пролекарства, которая эквивалентна стандартной дозе за исключением отсутствия ингибитора). Например, стандартная доза может обеспечить сниженную Cmax лекарственного средства, сниженную совокупную дозы лекарственного средства и/или увеличенное Tmax лекарственного средства.
Фиг. 2 представляет схематические графики, показывающие примеры модифицированных ФК кривых концентрация-время однократной стандартной дозы. Секция А представляет собой диаграмму концентрации лекарственного средства в крови или плазме (ось Y) за период времени (ось X) после приема внутрь пролекарства в отсутствие или в присутствии ингибитора. Сплошная верхняя линия Секции А представляет пример концентрации лекарственного средства после приема внутрь пролекарства без ингибитора. Пунктирная нижняя линия Секции А представляет концентрацию лекарственного средства после приема внутрь такой же дозы пролекарства с ингибитором. Прием внутрь ингибитора вместе в пролекарством обеспечивает сниженную Cmax лекарственного средства по сравнению с Cmax лекарственного средства, которая является результатом приема внутрь такого же количества пролекарства в отсутствие ингибитора. Секция А также показывает, что совокупная доза лекарственного средства после приема внутрь пролекарства с ингибитором также снижается по отношению к приему внутрь такого же количества пролекарства без ингибитора.
Секция В Фиг. 2 приводит другой пример стандартной дозы, имеющей модифицированную ФК кривую концентрация-время. Как и в Секции А, сплошная верхняя линия представляет собой концентрацию лекарственного средства от времени в крови или плазме после приема внутрь пролекарства без ингибитора, в то время как пунктирная нижняя линия представляет собой концентрацию лекарственного средства после приема такого же количества пролекарства с ингибитором. В этом примере стандартная доза обеспечивает ФК кривую, имеющую сниженную Cmax лекарственного средства, сниженное воздействие лекарственного средства и замедленное Tmax лекарственного средства (т.е. сниженное (1/Tmax лекарственного средства)) по отношению к введению внутрь такой же дозы пролекарства без ингибитора.
Секция С Фиг. 2 приводит другой пример стандартной дозы, имеющей модифицированную ФК кривую концентрация-время. Как и в Секции А, сплошная линия представляет собой концентрацию лекарственного средства от времени в крови или плазме после приема внутрь пролекарства без ингибитора, в то время как пунктирная линия представляет собой концентрацию лекарственного средства после приема такого же количества пролекарства с ингибитором. В этом примере стандартная доза обеспечивает ФК кривую, имеющую увеличенное Tmax лекарственного средства (т.е. сниженное (1/Tmax лекарственного средства)) по сравнению с введением внутрь такой же дозы пролекарства без ингибитора.
Стандартные дозы, обеспечивающие модифицированную ФК кривую (т.е. сниженную Cmax лекарственного средства и/или увеличенное Tmax лекарственного средства по сравнению с ФК кривой лекарственного средства или ФК кривой пролекарства без ингибитора), находят применение при индивидуальном подборе дозы согласно потребностям пациента (например, путем выбора определенной стандартной дозы и/или выбора режима приема), в уменьшении побочных эффектов и/или для улучшения соблюдения пациентом предписанного режима лечения (по сравнению с побочнымм эффектами или соблюдением пациентом предписанного режима лечения с использованием лекарственного средства или пролекарства без ингибитора). В данном изобретении "соблюдение пациентом предписанного режима лечения" относится к тому, соблюдает ли пациент предписания врача (например, терапевта), включая прием дозы, которая не отличается значительно ни в сторону увеличения, ни в сторону уменьшения от предписанной. Такие стандартные дозы также снижают риск неправильного использования, злоупотребления или передозировки пациентом по сравнению с аналогичным риском(ами), связанным с использованием лекарственного средства или пролекарства без ингибитора. Например, стандартные дозы со сниженной Cmax лекарственного средства обеспечивают меньший «приятный» эффект, чем доза такого же количества лекарственного средства и/или такого же количества пролекарства без ингибитора.
Стандартные дозы, обеспечивающие модифицированные ФК кривые при приеме множества стандартных доз
Стандартная доза по настоящему изобретению может быть приспособлена для обеспечения желаемой ФК кривой (например, ФК кривой концентрация-время или ФК кривой концентрация-доза) после приема внутрь кратной стандартной дозы (например, по меньшей мере 2, по меньшей мере 3, по меньшей мере 4 или более стандартных доз). ФК кривая концентрация-доза означает зависимость между выбранным ФК параметром и числом принятых внутрь однократных стандартных доз. Такая кривая может быть пропорциональной дозе и линейной (линейная ФК кривая) или нелинейной (нелинейная ФК кривая). Модифицированная ФК кривая концентрация-доза может быть обеспечена путем подбора относительных количеств пролекарства и ингибитора, содержащихся в однократной стандартной дозе и/или путем использования другого пролекарства и/или ингибитора.
На Фиг. 3 представлены диаграммы примеров ФК кривых концентрация-доза (проиллюстрированных Cmax лекарственного средства, ось Y), которые могут быть результатом приема множества стандартных доз (ось X) по настоящему изобретению. Каждую кривую можно сравнить с ФК кривой концентрация-доза, полученной путем увеличения доз одного лишь лекарственного средства, где количество лекарственного средства в крови или плазме от одной дозы представляет терапевтически эффективное количество, эквивалентное количеству лекарственного средства, высвобождаемого в кровь или плазму из одной стандартной дозы по настоящему изобретению. Такая ФК кривая "одного лишь лекарственного средства" обычно пропорциональна дозе, при этом она имеет положительный наклон под углом сорок пять градусов. Также следует понимать, что ФК кривую концентрация-доза, полученную при приеме кратной стандартной дозы по настоящему изобретению, также можно сравнивать с другими эталонами, как, например, ФК кривой концентрация-доза, полученной при приеме увеличивающегося числа доз пролекарства без ингибитора, где количество лекарственного средства, высвобождаемого в кровь или плазму из однократной дозы пролекарства в отсутствие ингибитора, представляет собой терапевтически эффективное количество, эквивалентное количеству лекарственного средства, выделенного в кровь или плазму из одной стандартной дозы по настоящему изобретению.
Как проиллюстрировано зависимостью между концентрацией пролекарства и ингибитора на Фиг. 1, стандартная доза может содержать ингибитор в количестве, влияние которого на высвобождение лекарственного средства после приема внутрь невозможно обнаружить. Прием такой кратной стандартной дозы может дать ФК кривую концентрация-доза, в которой соотношение между числом принятых стандартных доз и значением ФК параметра является линейным и имеет положительный наклон, сходный, например, с ФК кривой, пропорциональной дозе увеличивающегося количества одного лишь пролекарства. Секция А Фиг. 3 показывает такую кривую. Стандартные дозы, которые обеспечивают ФК кривую концентрация-доза с таким необнаруживаемым по сравнению с кривой одного только пролекарства изменением в Cmax лекарственного средства in vivo, могут найти применение в пресечении ферментативной конверсии пролекарства из стандартной дозы, содержащей достаточное количество ингибитора для снижения или предотвращения расщепления in vitro расщепляемого ферментом пролекарства соответствующим ферментом.
Секция В Фиг. 3 представляет собой ФК кривую концентрация-доза, в которой соотношение между числом принятых внутрь стандартных доз и значением ФК параметра является линейным и имеет положительный наклон, где кривая демонстрирует меньший наклон по сравнению с Секцией А. Такая стандартная доза обеспечивает кривую, имеющую сниженное значение ФК параметра (например, Cmax лекарственного средства) по отношению к сравнительному значению ФК параметра, демонстрирующего пропорциональность дозе.
ФК кривые концентрация-доза после приема кратной стандартной дозы могут быть нелинейными. Секция С Фиг. 3 представляет пример нелинейной двухфазной ФК кривой концентрация-доза. В этом примере двухфазная ФК кривая концентрация-доза содержит первую фазу, в которой ФК кривая концентрация-доза имеет положительный подъем, а затем вторую фазу, в которой зависимость между числом принятых внутрь стандартных доз и значением ФК параметра (например, Cmax лекарственного средства) остается практически неизмененной (в основном линейной с нулевым наклоном). Для такой стандартной дозы, например, Cmax лекарственного средства может увеличиваться для определенного числа стандартных доз (например, 2, 3, или 4 стандартных доз). Однако, прием дополнительных стандартных доз не приводит к существенному повышению Cmax лекарственного средства.
Секция D Фиг. 3 представляет другой пример нелинейной двухфазной ФК кривой концентрация-доза. В этом примере двухфазная ФК кривая концентрация-доза характеризуется первой фазой, в которой ФК кривая концентрация-доза имеет положительный подъем, и второй фазой, в которой зависимость между числом принятых внутрь стандартных доз и значением ФК параметра (например, Cmax лекарственного средства) является убывающей. Стандартные дозы, обеспечивающие такую ФК кривую концентрация-доза, обеспечивают увеличение Cmax лекарственного средства для определенного числа принятых внутрь стандартных доз (например, 2, 3 или 4 стандартных доз). Однако прием последующих дополнительных стандартных доз не приводит к существенному повышению Cmax лекарственного средства, а, напротив, обеспечивает снижение Cmax лекарственного средства.
Секция Е Фиг. 3 представляет ФК кривую концентрация-доза, в которой зависимость между числом принятых внутрь стандартных доз и ФК параметром (например, Cmax лекарственного средства) является линейной с нулевым наклоном. Такие стандартные дозы не дают значительного увеличения или снижения Cmax лекарственного средства при приеме кратных стандартных доз.
Секция F Фиг. 3 представляет ФК кривую концентрация-доза, в которой зависимость между числом принятых внутрь стандартных доз и значением ФК параметра (например, Cmax лекарственного средства) является линейной с отрицательным наклоном. Так, Cmax лекарственного средства уменьшается при увеличении числа принятых внутрь стандартных доз.
Стандартные дозы, обеспечивающие кривые ФК концентрация-доза в случаях приема внутрь кратной стандартной дозы, находят применение при индивидуальном подборе режима дозирования для обеспечения терапевтического уровня высвобождаемого лекарственного средства при одновременном снижении риска передозировки, неправильного применения и злоупотребления. Такое снижение риска можно сравнивать с эталоном, например с приемом одного лишь лекарственного средства или одного лишь пролекарства. В одном варианте осуществления риск снижен по сравнению с приемом лекарственного средства или пролекарства, который обеспечивает пропорциональную ФК кривую концентрация-доза. Стандартная доза, обеспечивающая ФК кривую концентрация-доза, может снизить риск передозировки пациентом из-за небрежного приема стандартных доз сверх предписанной дозировки. Такая стандартная доза может снизить риск неправильного использования пациентом (например, в результате самолечения). Такая стандартная доза может препятствовать злоупотреблению в результате преднамеренного приема многократных стандартных доз. Например, стандартная доза, обеспечивающая двухфазную ФК кривую концентрация-доза, может позволить увеличить высвобождение лекарственного средства для ограниченного числа принятых внутрь стандартных доз, после чего увеличение высвобождения лекарственного средства с приемом дополнительных стандартных доз не наступает. В другом примере стандартная доза, обеспечивающая ФК кривую концентрация-доза с нулевым наклоном, может позволить сохранять одинаковую кривую высвобождения лекарственного средства независимо от числа принятых стандартных доз.
Прием внутрь кратной стандартной дозы может обеспечить корректировку значения ФК параметра относительно приема многократного количества одной и той же дозы (либо в виде одного лишь лекарственного средства, либо в виде пролекарства) в отсутствие ингибитора таким образом, что, например, прием выбранного числа (например, 2, 3, 4 или более) однократных стандартных доз приведет к уменьшению значения ФК параметра по сравнению с приемом такого же числа доз в отсутствие ингибитора.
Фармацевтические композиции включают такие композиции, которые содержат ингибитор для обеспечения защиты активного вещества от разложения в ЖК тракте. Ингибитор может быть совмещен с лекарственным средством (т.е. не пролекарством) для обеспечения защиты лекарственного средства от разложения в ЖК системе. В данном примере композиция ингибитора и лекарственного средства обеспечивает модифицированную ФК кривую путем увеличения ФК параметра. Ингибитор может также быть совмещен с пролекарством, склонным к разложению ЖК ферментами, и имеющим зону действия вне ЖК тракта. В этой композиции ингибитор защищает принятое внутрь пролекарство в ЖК тракте до его доставки за пределы ЖК тракта и расщепления в предназначенной зоне действия.
Способы, используемые для определения относительных количеств пролекарства и ингибитора в стандартной дозе.
Стандартные дозы, обеспечивающие желаемую ФК кривую, такую как желаемая ФК кривая концентрация-время и/или желаемая ФК кривая концентрация-доза, могут быть получены путем объединения пролекарства и ингибитора в стандартной дозе в относительных количествах, эффективных для обеспечения высвобождения лекарственного средства, которые обеспечивают желаемую ФК кривую лекарственного средства после приема внутрь пациентом.
Пролекарство может быть выбрано в качестве пригодного для использования в стандартной дозе путем определения способности пролекарства высвобождать лекарственное средство под действием ЖК фермента. Это может быть выполнено in vitro, in vivo или ex vivo.
Количественный анализ in vitro может быть проведен путем объединения пролекарства с трипсином в реакционной смеси. Трипсин может присутствовать в реакционной смеси в количестве, достаточном для каталитического расщепления пролекарства. Количественный анализ проводят в подходящих условиях и необязательно в условиях, которые имитируют условия в ЖК тракте субъекта, например, человека. "Конверсия пролекарства" относится к высвобождению лекарственного средства из пролекарства. Конверсия пролекарства может быть оценена путем определения уровня продукта конверсии пролекарства (например, высвобожденного лекарственного средства) и/или путем определения уровня пролекарства, который поддерживается в присутствии трипсина. Конверсия пролекарства может также быть оценена путем определения скорости образования продукта конверсии пролекарства или скорости, с которой расходуется пролекарство. Увеличение содержания высвобожденного лекарственного средства или уменьшение содержания пролекарства свидетельствуют о том, что произошла конверсия пролекарства. Пролекарства, демонстрирующие приемлемый уровень конверсии пролекарства в присутствии трипсина в течение приемлемого периода времени, пригодны для использования в стандартной дозе в сочетании с трипсиновым ингибитором.
Количественный анализ in vivo оценивает пригодность пролекарства для использования в стандартной дозе путем введения пролекарства животному (например, человеку или животному, отличающемуся от человека, например крысе, собаке, свинье и т.д.). Такое введение может быть энтеральным (например, пероральным введением). Конверсия пролекарства может быть определена, например, путем определения продукта конверсии пролекарства (например, высвобождаемого лекарственного средства или метаболита высвобождаемого лекарственного средства) или определения уровня пролекарства в крови или плазме животного в желаемый момент(ы) времени после введения.
Количественный анализ ех vivo, такой как анализ в петле кишки или обратный анализ в петле кишки, может позволить оценить пригодность пролекарства для использования в стандартной дозе путем, например, введения пролекарства в замкнутую петлю кишки животного. Конверсия пролекарства может быть определена путем, например, определения продукта конверсии пролекарства (например, высвобождаемого лекарственного средства или метаболита высвобождаемого лекарственного средства) или определения уровня пролекарства в замкнутой петле кишки животного в желаемый момент(ы) времени после введения.
В целом, ингибиторы выбирают на основании, например, активности взаимодействия с трипсином, который является посредником в высвобождении лекарственного средства из пролекарства, совместно с которым дозируют ингибитор. Такие количественные определения могут быть проведены в присутствии фермента с пролекарством или без него. Ингибиторы также могут быть выбраны согласно их свойствам таким, как период полужизни в ЖК системе, активность, авидность, сродство, молекулярный размер и/или кривая ингибирования фермента (например, крутизна кривой ингибирования в анализе активности фермента, скорость инициирования ингибирования). Ингибиторы для использования в комбинации пролекарство-ингибитор могут быть выбраны путем количественных определений in vitro, in vivo и/или ex vivo.
Один вариант осуществления изобретения относится к способу определения пролекарства и трипсинового ингибитора, подходящих для препарата в стандартной дозе, включающему объединение пролекарства (например, Соединения КС-8), трипсинового ингибитора и трипсина в реакционной смеси и определение конверсии пролекарства. Такую комбинацию исследуют на взаимодействие между пролекарством, ингибитором и ферментом, т.е. исследуют с целью определить, как ингибитор будет взаимодействовать с ферментом, выступающим посредником в контролируемом ферментом высвобождении лекарственного средства из пролекарства. В одном примере осуществления снижение конверсии пролекарства в присутствии трипсинового ингибитора по сравнению с конверсией пролекарства в отсутствие трипсинового ингибитора указывает на то, что пролекарство и трипсиновый ингибитор пригодны для приготовления стандартной дозы. Такой способ может быть количественным анализом in vitro.
Один вариант осуществления изобретения относится к способу определения пролекарства и трипсинового ингибитора, подходящих для препарата в стандартной дозе, включающему введение животному пролекарства (Соединения КС-8) и трипсинового ингибитора и определение конверсии пролекарства. В одном примере осуществления снижение конверсии пролекарства в присутствии трипсинового ингибитора по сравнению с конверсией пролекарства в отсутствие трипсинового ингибитора указывает на то, что пролекарство и трипсиновый ингибитор пригодны для приготовления стандартной дозы. Такой способ может быть количественным анализом in vivo; например, пролекарство и трипсиновый ингибитор могут быть введены перорально. Такой способ может также быть количественным анализом ех vivo; например, пролекарство и трипсиновый ингибитор могут быть введены перорально или в ткани такие, как кишка, которую по меньшей мере временно подвергают их воздействию. Определение может происходить в крови или плазме или в соответствующих тканях. В настоящем изобретении термин «ткани» относится как к самим тканям, так и к содержимому тканей.
Один вариант осуществления изобретения относится к способу определения пролекарства и трипсинового ингибитора, подходящих для препарата в стандартной дозе, включающему введение пролекарства и трипсинового ингибитора в ткани животного, которые были удалены из животного, и определение конверсии пролекарства. В одном примере осуществления снижение конверсии пролекарства в присутствии трипсинового ингибитора по сравнению с конверсией пролекарства в отсутствие трипсинового ингибитора указывает на то, что пролекарство и трипсиновый ингибитор пригодны для приготовления стандартной дозы.
Количественный анализ in vitro может быть проведен путем объединения пролекарства с трипсиновым ингибитором и трипсином в реакционной смеси. Трипсин может присутствовать в реакционной смеси в количестве, достаточном для каталитического расщепления пролекарства, и количественные анализы следует проводить в подходящих условиях, опционально - в условиях, имитирующих условия в ЖК тракте субъекта, например человека. Конверсия пролекарства может быть оценена путем определения уровня продукта конверсии пролекарства (например, высвобожденного лекарственного средства) и/или путем определения уровня пролекарства, поддерживаемого в присутствии трипсина. Конверсия пролекарства может также быть оценена путем определения скорости образования продукта конверсии пролекарства или скорости, с которой расходуется пролекарство. Конверсия пролекарства, модифицированная в результате присутствия ингибитора по сравнению с уровнем конверсии пролекарства в отсутствие ингибитора, указывает на пригодность ингибитора для ослабления конверсии пролекарства и для использования в стандартной дозе по изобретению. Реакционные смеси, имеющие постоянное количество пролекарства и увеличивающиеся количества ингибитора или постоянное количество ингибитора и увеличивающиеся количества пролекарства, могут быть использованы для определения относительных количеств пролекарства и ингибитора, которые обеспечивают желаемую модификацию конверсии пролекарства.
In vivo анализ может определить комбинации пролекарства и ингибитора путем совместного дозирования пролекарства и ингибитора животному. Такое совместное дозирование может быть энтеральным. «Совместное дозирование» относится к введению пролекарства и ингибитора в виде отдельных доз или в виде комбинированных доз (т.е. в одном препарате). Конверсия пролекарства может быть измерена путем, например, определения продукта конверсии пролекарства (например, высвобождаемого лекарственного средства или метаболита лекарственного средства) или определения пролекарства в крови или плазме животного в желаемый момент(ы) времени после введения. Можно определить комбинации пролекарства и ингибитора, которые обеспечивают уровень конверсии пролекарства, дающий желаемую ФК кривую по сравнению, например, с пролекарством без ингибитора.
Комбинации относительных количеств пролекарства и ингибитора, которые обеспечивают желаемую ФК кривую, могут быть идентифицированы путем дозирования животным постоянного количества пролекарства и увеличиваемых количеств ингибитора или постоянного количества ингибитора и увеличиваемых количеств пролекарства. Затем можно определить один или несколько ФК параметров, например Cmax лекарственного средства, Tmax лекарственного средства и совокупную дозу лекарственного средства. Относительные количества пролекарства и ингибитора, которые обеспечивают желаемую ФК кривую, могут быть идентифицированы в виде количеств пролекарства и ингибитора для использования в стандартной дозе. ФК кривая комбинации пролекарства и ингибитора можно, например, характеризоваться сниженным значением ФК параметра по сравнению с пролекарством без ингибитора. Снижение значения ФК параметра комбинации ингибитор-пролекарство (например, снижение Cmax лекарственного средства, снижение 1/(Tmax лекарственного средства), т.е. увеличение Tmax лекарственного средства, или снижение совокупной дозы лекарственного средства) по сравнению с соответствующим значением ФК параметра после введения пролекарства без ингибитора может говорить о том, что комбинация ингибитор-пролекарство может обеспечить желаемую ФК кривую. Количественную оценку можно проводить с различными количествами ингибитора и пролекарства.
Количественный анализ in vivo можно использовать для определения комбинаций пролекарства и ингибитора, которые обеспечивают стандартные дозы, обеспечивающие желаемые ФК кривые концентрация-доза после приема множества стандартных доз (например, по меньшей мере 2, по меньшей мере 3, по меньшей мере 4 или более). Количественный анализ ех vivo можно использовать при прямом введении пролекарства и ингибитора в ткани и/или содержимое тканей животного, как, например, кишечник, включая введение путем инъекции в полость замкнутого кишечника (например, количественный анализ в петле кишки или в петле кишечника или количественный анализ в перевернутой кишке). Ех vivo анализ также можно проводить путем иссечения ткани и/или их содержимого из животного и введения пролекарства и ингибитора в эти ткани и/или их содержимое.
Например, выбирают дозу пролекарства, которая необходима для однократной стандартной дозы (например, количество, обеспечивающее эффективный уровень лекарственного средства в плазме). Затем выбирают множество однократных стандартных доз, для которого необходимо протестировать взаимосвязь между этим множеством и ФК параметром. Например, если необходимо получить ФК кривую концентрация-доза для приема внутрь 2, 3, 4, 5, 6, 7, 8, 9 или 10 стандартных доз, то определяют количество пролекарства, эквивалентное приему внутрь такого же числа стандартных доз (далее - «высокая доза»). Множество стандартных доз можно выбрать на основании числа принятых внутрь таблеток, при котором Cmax лекарственного средства меняется по сравнению с приемом однократной стандартной дозы. Если, например, кривая должна обеспечить предотвращение злоупотребления, то можно выбрать множество равное, например, 10. Можно протестировать разнообразые ингибиторы (например, из перечня ингибиторов), используя различные относительные количества ингибитора и пролекарства. Можно использовать количественные анализы для определения подходящей комбинации(й) ингибитора и пролекарства для того, чтобы получить терапевтически эффективную однократную стандартную дозу, где такая комбинация при приеме в виде множества стандартных доз обеспечивает модифицированный ФК параметр по сравнению с приемом внутрь такого же количества одного лишь лекарственного средства или пролекарства (где однократная доза как одного лекарственного средства, так и пролекарства, высвобождает в кровь или плазму такое же количество лекарственного средства, как и высвобождаемое однократной единицей дозы).
Затем животным одновременно дозируют увеличиваемые количества ингибитора с высокой дозой пролекарства. Определяют уровень дозы ингибитора, который обеспечивает желаемую Cmax лекарственного средства после приема внутрь высокой дозы пролекарства, и устанавливают получающееся в результате отношение ингибитора к пролекарству.
Затем одновременно дозируют пролекарство и ингибитор в количествах, эквивалентных отношению ингибитора к пролекарству, которое обеспечило желаемый результат при высокой дозе пролекарства. Затем оценивают значение интересующего ФК параметра (например, Cmax лекарственного средства). Если результатом является желаемое значение ФК параметра после приема эквивалента однократной стандартной дозы, то однократные стандартные дозы, обеспечивающие желаемую ФК кривую концентрация-доза, считают установленными. Например, в случаях, где нужна нулевая линейная кривая, Cmax лекарственного средства после приема однократной стандартной дозы значительно не увеличивается после приема кратного числа однократных стандартных доз.
Способы получения, составления и упаковки стандартных доз
Стандартные дозы по настоящему изобретению могут быть изготовлены с использованием способов получения, известных из уровня техники, и могут представлять собой различные формы, пригодные для энтерального (включая перороальное, буккальное и подъязычное) введения, например в виде таблетки, капсулы, тонкой пленки, порошка, суспензии, раствора, сиропа, дисперсии или эмульсии. Стандартная доза может содержать компоненты, общепринятые в фармацевтических препаратах, например один или несколько носителей, связующих веществ, смазочных средств, наполнителей (например, для обеспечения характеристик контролируемого высвобождения), модификаторов рН, вкусовых добавок (например, подсластителей), объемообразующих средств, красящих веществ или других активных средств. Стандартные дозы по настоящему изобретению могут включать энтеросолюбильное покрытие или иной компонент(ы) для обеспечения защиты от кислоты в желудке, если это необходимо.
Стандартные дозы могут быть любого размера или формы. Стандартная доза может быть любой формы, пригодной для энтерального введения, например эллипсоидной, линзообразной, круглой, прямоугольной, цилиндрической и тому подобной.
Стандартные дозы, имеющие форму сухих стандартных доз, могут иметь общий вес от примерно 1 микрограмма до примерно 1 грамма, и могут быть от примерно 5 микрограммов до 1,5 грамма, от примерно 50 микрограммов до примерно 1 грамма, от примерно 100 микрограммов до примерно 1 грамма, от примерно 50 микрограммов до примерно 750 миллиграммов и могут быть от примерно 1 микрограмма до 2 граммов.
Стандартные дозы могут включать компоненты в любых относительных количествах. Например, стандартные дозы могут содержать от примерно 0,1% (вес.) до 99% (вес.) активных ингредиентов (например, пролекарства и ингибитора) от общего веса стандартной дозы (0,1% до 99% объединенного веса пролекарства и ингибитора от общего веса однократной стандартной дозы). В некоторых вариантах осуществления стандартные дозы могут содержать от 10% до 50%, от 20% до 40% или примерно 30% (вес.) активных ингредиентов от общего веса стандартной дозы.
Стандартные дозы могут находиться в разнообразных формах и опционально могут иметь пригодную для хранения форму. Например, стандартные дозы могут быть помещены в контейнер, подходящий для содержания фармацевтической композиции. Контейнер может быть, например, бутылкой (например, с закрывающим устройством, таким как крышка), блистерной упаковкой (например, которая может обеспечить одну или более стандартных доз в упаковке), пузырьком, мягкой упаковкой (например, запечатанные мешки из майлара или пластика), ампулой (для однократных стандартных доз в растворе), капельницей, тонкой пленкой, трубкой и тому подобным.
Контейнеры могут включать съемную крышку (например, навинчивающуюся крышку), присоединенную к контейнеру с открытого конца, через который стандартные дозы помещают внутрь контейнера и через который можно получить доступ к ним.
Контейнеры могут включать уплотнение с контролем вскрытия и/или устойчивый к взлому элемент, уплотнение которого нарушается при получении доступа к стандартной дозе, размещенной в контейнере. Такой запечатывающий элемент может быть, например, хрупким элементом, который ломается или изменяется любым другим образом при получении доступа к стандартной дозе, размещенной в контейнере. Примеры такого хрупкого запечатывающего элемента включают запечатывающий элемент, расположенный над открытым концом контейнера таким образом, что доступ к стандартной дозе в контейнере требует нарушения запечатывающего элемента (например, путем отрывания и/или прокалывания запечатывающего элемента). Примеры хрупкого запечатывающего элемента включают хрупкое кольцо, размещенное вокруг открытого конца контейнера и связанное с крышкой таким образом, чтобы это кольцо ломалось при открывании крышки для получения доступа к стандартным дозам в контейнере.
Сухие и жидкие стандартные дозы могут размещаться в контейнере (например, бутылке или упаковке, например в мягком мешке), который своими размером и конфигурацией приспособлен для поддержания стабильности стандартных доз в течение времени, необходимого для отпуска стандартных доз согласно предписанию. Например, контейнеры могут иметь размер и конфигурацию для размещения 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 или более однократных сухих или жидких стандартных доз. Контейнеры могут быть запечатанными или допускать повторное запечатывание. Контейнеры могут быть упакованы в тару (например, для перевозки от производителя в аптеку или другой пункт распределения). Такая тара может быть коробкой, тубусом или иметь другую конфигурацию и может быть изготовлена из любого материала (например, картона, пластика и подобное). Упаковочная система и/или размещенные в ней контейнеры могут иметь одну или более наклеенных этикеток (например, для предоставления информации такой, как номер партии, тип стандартной дозы, производитель и подобное).
Контейнер может включать защищающий от влаги вкладыш и/или светозащитный барьер, например для обеспечения поддержания стабильности активных ингредиентов в стандартных дозах, содержащихся в нем. В случае, если стандартная доза является сухой стандартной дозой, контейнер может включать пакет с осушителем, размещенным внутри контейнера. Контейнер может быть приспособлен для содержания однократной стандартной дозы или кратных стандартных доз. Контейнер может включать контролирующий раздачу механизм такой, как закрывающий механизм, который обеспечивает поддержание режима дозирования.
Стандартные дозы могут быть представлены в твердой или полутвердой форме и могут быть сухой стандартной дозой. «Сухая стандартная доза» относится к стандартной дозе, которая находится в форме, отличающейся от полностью жидкой формы. Примеры сухих стандартных доз включают, например, таблетки, капсулы (например, твердые капсулы, капсулы, содержащие жидкость), тонкую пленку, микрочастицы, гранулы, порошок и подобное. Стандартные дозы могут быть предоставлены как жидкие стандартные дозы, когда стандартные дозы могут быть обеспечены как однократные или многократные дозы препарата, содержащего пролекарство и ингибитор в жидкой форме. Однократные дозы сухой или жидкой стандартной дозы могут быть размещены внутри запечатанного контейнера, а запечатанные контейнеры, опционально, могут находиться в упаковочной системе, например для обеспечения предписанного числа доз, для обеспечения перевозки стандартных доз и тому подобного.
Стандартные дозы могут быть изготовлены таким образом, чтобы пролекарство и ингибитор были представлены в одном носителе, например солюбилизированы или суспендированы в одной и той же матрице. В альтернативном варианте стандартные дозы могут состоять из двух или нескольких частей, где пролекарство и ингибитор могут быть находиться в одной или разных частях, и могут быть находиться в соседних или разделенных частях.
Стандартные дозы могут находиться в контейнере, в котором они размещены, и могут представлять собой часть упаковочной системы (необязательно с инструкцией по пользованию). Например, стандартные дозы, содержащие различные количества пролекарства, могут находиться в различных контейнерах, которые могут быть размещены в более крупных контейнерах (например, для обеспечения защиты стандартных доз во время перевозки). Например, одна или несколько стандартных доз, как описано здесь, могут находиться в разных контейнерах, где стандартные дозы различного состава находятся в разных контейнерах, а разные контейнеры размещены в упаковке для распределения.
В другом примере стандартные дозы могут находиться в двухкамерных диспенсерах, где первая камера содержит препарат пролекарства, а вторая камера содержит препарат ингибитора. Диспенсер можно приспособить для обеспечения перемешивания препарата пролекарства и препарата ингибитора перед приемом внутрь. Например, две камеры диспенсера могут быть разделены удаляемой перегородкой (например, хрупкой перегородкой), которую ломают или удаляют перед вводом, чтобы осуществить смешивание препаратов двух камер. Первая и вторая камеры могут иметь выход в диспенсирующий выход, необязательно через общую камеру. Препараты могут находиться в сухой или жидкой форме или их комбинации. Например, препарат в первой камере может быть жидким, а препарат во второй камере может быть сухим, или оба могут быть сухими или жидкими.
В данном изобретении описаны стандартные дозы, обеспечивающие контролируемое высвобождение пролекарства, ингибитора или как пролекарства, так и ингибитора, где под «контролируемым высвобождением» понимают высвобождение как пролекарства, так и ингибитора из стандартной дозы за выбранный промежуток времени и/или предварительно определенным способом.
Способы применения стандартных доз
Стандартные дозы предпочтительны, поскольку они могут использоваться в способах уменьшения побочных эффектов и/или улучшают переносимость лекарственных средств пациентами, нуждающимися в них, например ограничивая какой-либо ФК параметр, как описано в настоящем документе. Настоящее изобретение относится к способам уменьшения побочных эффектов путем введения стандартной дозы по настоящему изобретению пациенту, нуждающемуся в ней, таким образом, чтобы обеспечить уменьшение побочных эффектов по сравнению с побочными эффектами, вызванными введением лекарственного средства и/или по сравнению с введением пролекарства без ингибитора. Настоящее изобретение также относится к способам улучшения переносимости лекарственного средства путем введения стандартной дозы по настоящему изобретению пациенту, нуждающемуся в нем, таким образом, чтобы обеспечить улучшение переносимости по сравнению с переносимостью самого лекарственного средства и/или по сравнению с введением пролекарства без ингибитора.
Стандартные дозы находят применение в способах улучшения соблюдения пациентом режима лечения, предписанного врачом, включающим управление введением описанной здесь стандартной дозы нуждающемуся в лечении пациенту таким образом, чтобы обеспечить улучшенное соблюдение режима лечения пациентом по сравнению с лечением, включающим в себя введение лекарственного средства и/или по сравнению с введением пролекарства без ингибитора. Такие способы могут помочь увеличить вероятность того, что назначенное доктором лечение проходит согласно предписанию.
Стандартные дозы могут обеспечить повышенное соблюдение пациентом режима лечения и клинический контроль. Например, в результате ограничения ФК параметра (например, такого, как Cmax лекарственного средства или воздействия лекарственного средства) в случае приема многократных (например, двух или более, трех или более или четырех или более) стандартных доз пациент, нуждающийся в повышении дозы лекарственного средства, должен обратиться к врачу. Стандартные дозы могут обеспечить контроль уровня, до которого пациент может легко проводить «самолечение», и могут помочь пациенту регулировать дозу в пределах разрешенного диапазона. Стандартные дозы могут обеспечить уменьшение побочных эффектов путем, например, обеспечения доставки лекарственного препарата в эффективной дозе, но с модифицированной ФК кривой за период лечения, например, как определено сниженными ФК параметрами (например, сниженной Cmax лекарственного средства, сниженной совокупной дозой лекарственного средства).
Стандартные дозы находят применение в способах уменьшения риска непреднамеренной передозировки лекарственного средства после введения многократных доз, которые были приняты одновременно или в течение короткого периода времени. Способы по настоящему изобретению могут уменьшить риск непреднамеренной передозировки по сравнению с риском непреднамеренной передозировки лекарственного средства и/или по сравнению с риском непреднамеренной передозировки пролекарства без ингибитора. Такие способы включают управление введением описанной дозировки пациенту, нуждающемуся в лекарственном средстве, высвобождаемом в результате конверсии пролекарства. Такие способы могут помочь избежать непреднамеренной передозировки из-за преднамеренного или непреднамеренного неправильного использования стандартной дозы.
Настоящее изобретение раскрывает способы снижения неправильного использования и злоупотребления лекарственным средством, а также уменьшения риска передозировки, которая может возникнуть при приеме многократных доз лекарственного средства, например принятых одновременно. В общем, такие способы включают объединение стандартной дозе пролекарства и трипсинового ингибитора, который является влияет на высвобождение лекарственного средства из пролекарства, где ингибитор присутствует в стандартной дозе в количестве, достаточном для ослабления высвобождения лекарственного средства из пролекарства, например после приема внутрь пациентом кратных стандартных доз. Такие способы обеспечивают модифицированную ФК кривую концентрация-доза, обеспечивая при этом терапевтически эффективные уровни однократной стандартной дозы, как это предписано лечащим врачом. Такие способы могут, например, уменьшить риски, сопутствующие неправильному использованию и/или злоупотреблению пролекарством, особенно там, где конверсия пролекарства приводит к высвобождению наркотика или другого наркотического лекарственного средства (например, опиоида). Например, когда пролекарство обеспечивает высвобождение наркотического лекарственного средства, стандартные дозы могут снизить наркотический эффект, который может стать результатом приема кратных стандартных доз наркотического лекарственного средства.
Стандартные дозы могут обеспечить врачам большую свободу в назначении лекарственного средства. Например, врач может предписать режим приема, включающий дозы с различной силой, которые могут включать две или более различные стандартные дозы пролекарства и ингибитора, имеющие различные относительные количества пролекарства, различные количества ингибитора или различные количества как пролекарства, так и ингибитора. Такие стандартные дозы с различной силой могут обеспечить доставку лекарственного средства в соответствии с различными ФК параметрами (например, совокупной дозой лекарственного средства, Cmax лекарственного средства и тому подобным, как описано выше). Например, первая стандартная доза может обеспечить доставку первой дозы лекарственного средства после приема внутрь, а вторая стандартная доза может обеспечить доставку второй дозы после приема внутрь. Первая и вторая дозы пролекарства стандартной дозы могут иметь различную силу, например вторая доза может быть больше, чем первая доза. Доктор может таким образом предписать набор двух или более, трех или более стандартных доз различной силы, что может сопровождаться инструкциями для обеспечения некоторой степени самолечения, например для увеличения доставки опиоидного лекарства согласно потребностям пациента для лечения боли.
Предотвращение несанкционированного использования путем использования опосредованного трипсином высвобождения оксикодона из пролекарства
Данное изобретение описывает композицию, содержащую Соединение КС-8 и трипсиновый ингибитор, которая снижает потенциальное злоупотребление лекарственным средством. Трипсиновый ингибитор может препятствовать возможности потребителя применять трипсин для влияния на высвобождение модифицированного кетоном оксикодона из пролекарства оксикодона in vitro. Например, если злоупотребляющее лицо пытается обработать трипсинон композицию по данном изобретению, включающую Соединение КС-8 и трипсиновый ингибитор, трипсиновый ингибитор может снизить действие добавленного трипсина, препятствуя, тем самым, попыткам высвобождения оксикодона для целей злоупотребления.
Примеры
Следующие далее примеры приведены для того, чтобы предоставить специалистам в данной области полное раскрытие и описание осуществления и применения вариантов осуществления настоящего изобретения, и не предназначены для ограничения объема того решения, которое авторы считают своим изобретением, а также не должны воспринимать как указание на то, что изложенные ниже эксперименты являются всеми возможными или единственными выполненными экспериментами. Были предприняты усилия, чтобы обеспечить точность в отношении используемых количественных характеристик (например, количеств, температур и т.д.), однако следует учитывать и то, что возможны некоторые экспериментальные ошибки и отклонения. Если не указано иное, все части являются весовыми частями, молекулярный вес является средневесовым молекулярным весом, температура измеряется в градусах Цельсия, а давление соответствует атмосферному или приближено к нему. В описании могут применяться стандартные аббревиатуры и сокращения.
Синтез опиоидного пролекарства, модифицированного кетоном
Пример 1: Синтез 6-(N-метил-N-(2-амино)этилкарбамата оксикодона (Соединения КС-19)

Получение соединения А
Бензиловый эфир 2-(аминоэтил)-метил-карбаминовой кислоты (2,0 г, 9,6 ммоля) растворяли в дихлорэтилене (DCE) (20 мл) при комнатной температуре. Добавляли триэтиламин (NEt3) (1,40 мл, 11,5 ммоля), а затем - ди-трет-бутилдикарбонат (BOC2O) (10,5 г, 48 ммоль) и диметиламинопиридин (DMAP) (120 мг). Реакционную смесь перемешивали при комнатной температуре под азотом (N2) в течение 2 ч., а затем нагревали при 60°С в течение 16 часов. Реакционную смесь затем концентрировали. Остаток очищали хроматографией на силикагеле, используя смесь 4/1 гексаны/EtOAc, с получением Соединения А с выходом 86% (3,4 г, 8,3 ммоля). МС: (m/z) рассчит.: 408,2, наблюдаемый пик (M+Na+) 431,9.
Получение соединения В
Соединение А (1,3 г, 3,18 ммоль) растворяли в смеси метанол/EtOAc (10 мл/3 мл соответственно). Смесь дегазировали и насыщали N2. Добавляли палладий на угле (Pd/C) (330 мг, 5% на угле). Смесь встряхивали в колбе аппарата Парра (50 фунтов на квадратный дюйм Н2) в течение 4 ч. Затем смесь фильтровали через подушку целита, и фильтрат концентрировали с получением Соединения В (1,08 г, выход превысил расчетный). Соединение В применяли без дополнительной очистки.
Получение соединения С
Соединение В (500 мг, 1,82 ммоля) и NEt3 (0,4 мл, 2,74 ммоля) смешивали в дихлорметане (4 мл). Смесь добавили к предварительно охлажденному до 0°С раствору фосгена (5,5 мл, 0,5 мол. в толуоле). Реакционную смесь затем перемешивали при 0°С в течение 1 ч. с последующим разбавлением эфиром (20 мл) и фильтровали на фильтровальной бумаге. Фильтрат концентрировали и пропускали через короткую колонку с силикагелем (10 см Х 3 см), элюировали смесью гексаны/EtOAc 3/1. Фракции концентрировали с получением N,N-бис(трет-бутил) N’-2-(хлоркарбонил(метил)амино)этилкарбамата (Соединение С) в виде бесцветного твердого вещества с количественным выходом (615 мг, 1,82 ммоля). МС: (m/z) рассчит.: 336,1, наблюдаемый пик (М+Na+) 359,8.
Синтез 6-(N-метил-N-(2-амино)этилкарбамата оксикодона (Соединения КС-19)

Свободное основание оксикодона (6,5 г, 20,6 ммоля) растворили в сухом дегазированном тетрагидрофуране (120 мл) и смесь охладили до -10°С, используя охлаждающую баню с сухим льдом/ацетоном. Бис(триметилсилил)амид калия (KHMDS) (103,0 мл, 51,6 ммоль, 0,5 мол. в толуоле) был добавлен через полую иглу. Смесь перемешивали в атмосфере N2 при температуре ниже -5°С в течение 30 мин. N,N-бис(трет-бутил) N’-2-(хлоркарбонил(метил)амино)этилкарбамат (8,0 г, 23,7 ммоля) (Соединение С) в ТГФ (30 мл) вводили через полую иглу в течение 15 мин. Смесь перемешивали при -5°С в течение 30 мин. Добавили другую порцию карбамоилхлорида (4,0 г, 11,9 ммоля) в ТГФ (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 2 ч. Добавили бикарбонат натрия (10 мл, насыщ. водн. раствор). Смесь концентрировали в вакууме до половины первоначального объема. Добавили EtOAc (50 мл) и слои разделили. Далее органическую фазу промывали водой (3×20 мл), солевым раствором (40 мл) и затем концентрировали. Остаток очищали хроматографией на силикагеле, используя смесь дихлорметан/метанол (градиент от 100/1 до 100/15) с получением белой пены с выходом 55% (7,0 г, 13,4 ммоля). Этот материал растворили в смеси дихлорметан/трифторуксусная кислота (TFA) 1:1 (20 мл/20 мл) при комнатной температуре и перемешивали в течение 1 ч. Раствор затем концентрировали в вакууме с получением бис-трифторацетата 6-(N-метил-N-(2-амино)этилкарбамата оксикодона (Соединение КС-19) в виде вязкого масла (7,3 г, 11,4 ммоля, с чистотой 99%). МС: (m/z) рассчит.: 415,2, наблюдаемый пик (М+Н+) 416,5.
Пример 2: Синтез N-1-[2-(оксикодон-6-енол-карбонил-метил-амино)-этиламин]-аргинин-малоновой кислоты (Соединение КС-3) [также называемое: N-{(S)-4-гуанидино-1-[2-(метил-[(5R,9R,13S,14S)-4,5а-эпокси-6,7-дидегидро-14-гидрокси-3-метокси-17-метилморфинан-6-окси]карбонил-амино)-этилкарбамоил]-бутил}-малоновая кислота]

Получение соединения D
Раствор N-метилэтилендиамина (27,0 г, 364,0 ммоля) и этилтрифторацетата (96,6 мл, 838,0 ммоль) в смеси ацетонитрила (350 мл) и воды (7,8 мл, 436 ммоль) нагревали с обратным холодильником в течение ночи с перемешиванием. Растворители выпарили в вакууме. Остаток повторно испарили с i-PrOH (3×100 мл) с последующей кристаллизацией путем нагрева и охлаждения из дихлорметана (500 мл). Образовавшиеся кристаллы отфильтровали, промыли дихлорметаном и сушили в вакууме с получением Соединения D (88,3 г, 85%) в виде белого твердого порошка.
Получение соединения Е
Раствор соединения D (88,2 г, 311 ммоль) и ДИПЭА (54,1 мл, 311 ммоль) в ТГФ (350 мл) охладили в ледяной бане с последующим добавлением раствора N-(бензилоксикарбонил)сукцинимида (76,6 г, 307 ммоль) в ТГФ (150 мл) по каплям за 20 минут. Температуру реакционной смеси повысили до комнатной температуры и перемешивание продолжали еще 30 минут. Растворители выпарили и остаток растворили в EtOAc (600 мл). Органический слой экстрагировали 5% води. NaHCO3 (2×150 мл) и солевым раствором (150 мл). Органический слой выпарили с получением Соединения Е в виде желтоватого масла. ЖХ-МС [М+Н] 305,1 (C13H15F3N2O3+H, рассчит.: 305,3). Соединение Е использовали сразу в следующей реакции без очистки в виде раствора в метаноле.
Получение соединения F
К раствору соединения Е (~311 ммоль) в метаноле (1,2 л) добавили раствор LiOH (14,9 г, 622 ммоль) в воде (120 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворители выпарили до 75% начального объема с последующим разбавлением водой (400 мл). Продукт экстрагировали этилацетатом (2×300 мл). Органический слой промыли солевым раствором (200 мл), сушили над MgSO4 и выпарили в вакууме. Остаток растворили в эфире (200 мл) и обработали 2 н раствором HCl в эфире (200 мл). Образовавшийся осадок отфильтровали, промыли эфиром и сушили в вакууме с получением хлористоводородной соли Соединения F (67,8 г, 89%) в виде белого твердого вещества. ЖХ-МС [М+Н] 209,0 (C11H16N2O2+Н, рассчит.: 209,3). Соединение F использовали сразу в следующей реакции без очистки в виде раствора в ДМФА.
Получение соединения G
Раствор Boc-Arg(Pbf)-OH (16,0 г, ~30,4 ммоля), гидрохлорид соединения F (8,2 г, 33,4 ммоля) и ДИПЭА (16,9 мл, 97,2 ммоль) в ДМФА (150 мл) охладили на ледяной бане с последующим добавлением по каплям раствора HATU (13,8 г, 36,4 ммоля) в течение 20 мин. Температуру реакционной смеси повысили до комнатной и продолжали перемешивание еще 1 ч. Реакционную смесь разбавили этилацетатом (1 л) и экстрагировали водой (3×200 мл) и солевым раствором (200 мл). Затем органический слой сушили над MgSO4 и выпарили с получением соединения G (24,4 г, выход превысил количественный) в виде желтоватого масла. ЖХ-МС [М+Н] 717,4 (C35H52N6O8S +Н, рассчит.: 717,9). Соединение G использовали сразу в следующей реакции без очистки в виде раствора в диоксане.
Получение соединения Н
Соединение G (24,4 г, ~30,4 ммоля) растворили в диоксане (150 мл) и обрабатывали 4 н раствором HCl в диоксане (150 мл, 600 ммоль) при комнатной температуре в течение 1 часа. Затем растворитель выпарили. Остаток растворили в i-PrOH (100 мл) и выпарили в вакууме (процедуру повторили дважды). Затем остаток высушили в вакууме с получением Соединения Н (21,1 г, выход превысил количественный) в виде желтоватого твердого вещества. ЖХ-МС [М+Н] 617,5 (C30H44N6O6S +H, рассчит.: 617,8). Соединение Н использовали сразу в следующей реакции без очистки в виде раствора в ДМФА.
Получение соединения I
Раствор соединения Н (21,1 г, ~30,4 ммоля), моно-трет-бутил малоната (5,9 мл, 36,7 ммоля), ВОР (16,2 г, 36,7 ммоля) и ДИПЭА (14,9 мл, 83,5 ммоль) в ДМФА (100 мл) перемешивали при комнатной температуре 1 час. Реакционную смесь разбавили этилацетатом (1 мл) и экстрагировали водой (500 мл), 5% водн. NaHCO3 (500 мл), водой (3×500 мл) и солевым раствором (500 мл). Органический слой сушили над MgSO4, отфильтровали, а затем выпарили с получением соединения I (24,5 г, 97%) в виде желтоватого аморфного твердого вещества. ЖХ-МС [М+Н] 759,6 (C37H54N6O9S +H, рассчит.: 759,9). Соединение I применяли без дополнительной очистки.
Получение соединения J
Соединение I (12,3 г, 16,7 ммоля) растворили в метаноле (100 мл) с последующим добавлением суспензии Pd/C (5% (вес.), 2,0 г) в воде (2 мл). Реакционную смесь подвергли гидрированию (аппарат Парра, 70 фунтов на квадратный дюйм Н2) при комнатной температуре в течение 1 часа. Катализатор затем отфильтровали и промыли метанолом. Фильтрат выпарили в вакууме с получением Соединения J (10,0 г, 99%) в виде бесцветного аморфного твердого вещества. ЖХ-МС [М+Н] 625,5 (C29H48N6O7S +H, рассчит.: 625,8). Соединение J применяли без дополнительной очистки.
Получение свободного основания оксикодона
Гидрохлорид оксикодона (10,0 г, 28,5 ммоля) растворили в хлороформе (150 мл) и промыли 5% водн. NaHCO3 (50 мл). Органический слой сушили над MgSO4 и выпарили. Остаток сушили в вакууме в течение ночи с получением свободного основания оксикодона (8,3 г, 93%) в виде белого твердого вещества.
Получение соединения К
Раствор свободного основания оксикодона (6,6 г, 21,0 ммоль) в ТГФ (400 мл) охладили до -20°С, затем добавили 0,5 мол. раствор KHMDS в толуоле (46,3 мл, 23,1 ммоля). Полученный раствор по каплям добавили к раствору 4-нитро-фенилхлорформиата (4-NPCF) (4,3 г, 21,0 ммоль) в ТГФ (100 мл) в течение 20 мин при -20°С. Реакционную смесь выдерживали при -20°С в течение еще 1 часа с последующей добавкой раствора соединения J (10,0 г, 16,1 ммоля) в ТГФ (200 мл) при -20°С. Реакционной смеси дали нагреться до комнатной температуры и перемешивали в течение ночи. Растворители выпарили в вакууме. Полученный остаток растворили в EtOAc (20 мл) и осадили эфиром (1 л). Образовавшийся осадок отфильтровали, промыли эфиром и высушили в вакууме с получением соединения Н (13,6 г, 87%) в виде грязно-белого твердого вещества. ЖХ-МС [М+Н] 966,9 (C48H67N7O12S +H, рассчит.: 966,2).
Синтез N-1-[2-(оксикодон-6-енол-карбонил-метил-амино)-этиламин]-аргинин-малоновой кислоты (Соединение КС-3)
Соединение К (13,6 мг, 14,1 ммоля) растворили в смеси 5% м-крезола и TFA (100 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 часа с последующим разбавлением этиловым эфиром (1 л). Образовавшийся осадок отфильтровали, промыли эфиром и гексаном и высушили в вакууме с получением трифторацетата Соединения КС-3 (11,4 г, 81%) в виде грязно-белого твердого вещества. ЖХ-МС [М+Н] 658,6 (C31H43N7O9 +H, рассчит.: 658,7).
Неочищенный трифторацетат Соединения КС-3 (11,4 г, 11,4 ммоль) растворили в воде (50 мл). Полученный раствор очистили с использованием ВЭЖХ. [колонка Nanosyn-Pack YMC-GEL-ODS A (100-10) С-18 (75×500 мм); скорость потока: 250 мл/мин; объем впрыска 50 мл; подвижная фаза А: 100% вода, 0,1% TFA; подвижная фаза В: 100% ацетонитрил, 0,1% TFA; изократическое элюирование при 0% B в течение 4 мин, градиентное элюирование от 0% до 10% B в течение 20 мин, изократическое элюирование при 10% B в течение 30 мин, градиентное элюирование от 10% B до 30% B в течение 41 мин; детектировние при 254 нм]. Фракции, содержащие Соединение КС-3, объединяли и концентрировали в вакууме. Противоион TFA в последнем соединении заменили противоионом HCl путем лиофилизации с помощью 0,1 н HCl с получением гидрохлорида Соединения КС-3 (4,2 г, выход 41%) в виде белого твердого вещества. ЖХ-МС [М+Н] 658,6 (C31H43N7O9 +H, рассчит.: 658,7).
Пример 3: Синтез N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламина] (Соединение КС-22) и N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламин]-аргинин-глицин-малоновой кислоты (Соединение КС-8)
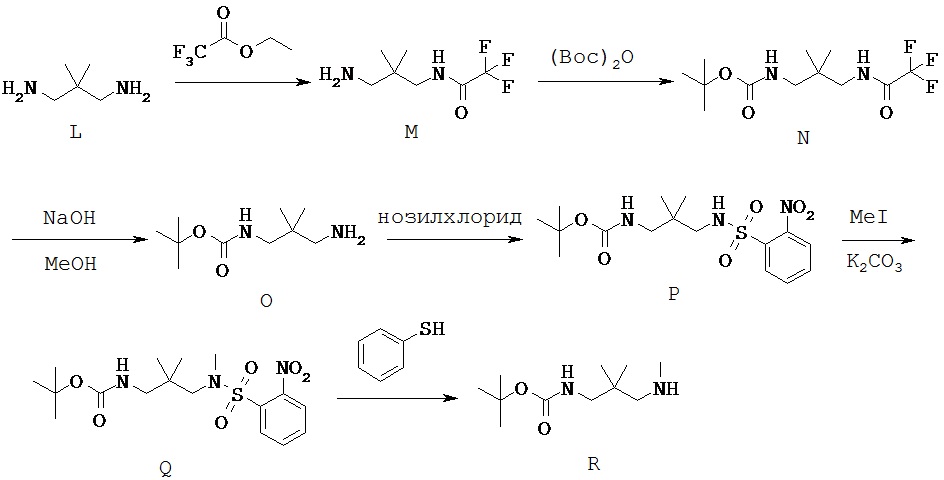
Получение соединения М
Раствор 2,2-диметил-1,3-диаминопропана (Соединение L) (48,0 г, 470,6 ммоля) в ТГФ (1,0 л) охладили на ледяной бане. Этилтрифторацетат (56 мл, 471 ммоль) добавляли в течение 30 мин. с помощью шприца. Смеси дали нагреться до комнатной температуры и продолжали перемешивание в течение 14 часов. Затем смесь концентрировали под вакуумом до половины первоначального объема с образованием неочищенного Соединения М в виде раствора в ТГФ, который затем использовали в следующей реакции без дополнительной очистки. ЖХ-МС [М+Н] 199,6 (C7H13F3N2O3 +H, рассчит.: 199,1).
Получение соединения N
К неочищенному раствору Соединения М (из предыдущей стадии) в ТГФ (500 мл), захоложенному на ледяной бане, малыми порциями в течение 15 мин. добавили (Вос)2O. Смесь перемешивали при комнатной температуре 15 часов. Продукт реакции затем концентрировали под вакуумом с образованием промежуточного Соединения N с выходом 84% (в две стадии) (120,0 г, 402,4 ммоля) в виде липкого масла. ЖХ-МС [М+Н] 299,2 (C12H21F3N2O3 +H, рассчит.: 299,2). Соединение N сразу использовали в следующей реакции без дальнейшей очистки.
Получение соединения О
Соединение N (120 г, 403 ммоля) растворили в метаноле (500 мл) и перемешивали при комнатной температуре. NaOH (100 мл, 10 н водн.) добавили по каплям. Смесь затем перемешивали в заранее нагретой масляной бане при 50°С в течение 3 ч. Смесь охладили до комнатной температуры и разбавили водой (500 мл). Растворители затем выпарили в вакууме. Продукт экстрагировали хлороформом (3×100 мл). Объединенный раствор в хлороформе сушили над Na2SO4, отфильтровали и концентрировали под вакуумом с получением неочищенного Соединения О с выходом 95% (77,0 г, 381 ммоль). ЖХ-МС [М+Н] 203,8 (C10H22N2O2 +H, рассчит.: 203,2). Соединение О сразу использовали в следующей реакции без дальнейшей очистки.
Получение соединения Р
Соединение О (97,0 г, 480 ммоль) растворили в метиленхлориде (750 мл). К раствору добавили К2СО3 (75,0 г, 542,6 ммоля) одной порцией, а затем постепенно добавили 2-нозилхлорид (108,0 г, 487,3 ммоля). Реакционную смесь перемешивали при комнатной температуре 15 часов. Затем добавили воду (200 мл) и разделили полученные слои. Водный слой снова экстрагировали метиленхлоридом. Объединенный раствор в метиленхлориде сушили над Na2SO4, фильтровали и концентрировали под вакуумом. Остаток очищали хроматографией на силикагеле, используя смесь 3/1 гексаны/EtOAc, с получением промежуточного Соединения Р с выходом 83% (155,0 г, 400,5 ммоля) в виде белого твердого вещества. ЖХ-МС [М+Н] 388,8 (C16H25N3O6S +H, рассчит.: 388,1).
Получение соединения R
Соединение Р (155,0 г, 400,5 ммоля) растворили в ДМФА (500 мл) при комнатной температуре. Добавили K2CO3 (83,0 г, 600 ммоль) одной порцией. Затем смесь охладили на водно-ледяной бане. MeI (37,0 мл, 593 ммоля) добавили малыми порциями через шприц в течение 10 мин. Затем смесь нагрели до комнатной температуры и продолжали перемешивание, при этой температуре в течение еще 2 часов. Смесь концентрировали в вакууме до получения остатка объемом ~50 мл. Оставшаяся смесь, содержащую промежуточное Соединение Q, охладили на водно-ледяной бане. Продолжая перемешивание, с помощью шприца добавили тиофенол (100 мл, 978 ммоль). Полученную смесь перемешивали при комнатной температуре 6 часов. Затем добавили воду (500 мл). Смесь экстрагировали этилацетатом (100 мл, затем 2×500 мл). Объединенные экстракты в этилацетате экстрагировали с помощью 2 н HCl (400 мл, затем 2×200 мл). Экстракты в HCl собрали и промыли дихлорметаном (500 мл). Кислый раствор затем охлаждали на водно-ледяной бане и защелачивали путем добавления Юн NaOH до рН ~13. Затем для экстракции водного раствора использовали хлороформ (400 мл, затем 2×200 мл). Объединенные растворы в хлороформе сушили над Na2SO4 и фильтровали. После выпаривания растворителей под вакуумом получали Соединение R с выходом 67% (58,0 г, 268,5 ммоля) в виде слегка желтого масла. ЖХ-МС [М+Н] 217,6 (C11H24N2O2+H, рассчит.: 217,2).

Получение соединения S
Свободное основание оксикодона (10,0 г, 31,75 ммоля) растворили в сухом тетрагидрофуране (150 мл) и смесь охладили до -70°С, используя охлаждающую баню с сухим льдом/ацетоном. KHMDS (64,0 мл, 128,0 ммоль, 0,5 мол. в толуоле) добавляли с помощью шприца в течение 15 мин. Смесь перемешивали под N2 дополнительно в течение 30 мин. (температура бани -70°С). В отдельную колбу добавили 4-нитрофенил-хлорформиат (6,4 г, 31,75 ммоля) и ТГФ (10 мл). Эту смесь также охладили до -70°С используя охлаждающую баню с сухим льдом/ацетоном. Смесь из первой колбы (содержащую депротонированный оксикодон) затем перенесли с помощью полой иглы во вторую колбу (содержащую 4-нитрофенил-хлорформиат). Перенос осуществляли за ~30 минут, причем в обеих колбах в процессе переноса поддерживали температуру -70°С. Полученную реакционную смесь дополнительно перемешивали при -70°С в течение 30 мин. Затем с помощью шприца добавили раствор Соединения R (6,9 г, 31,94 ммоля). Смесь выдерживали 30 мин. при -70°С при перемешивании, а затем концентрировали под вакуумом с получением гелеобразного остатка (удалив ~90% растворителя). Остаток выдерживали при комнатной температуре в течение 15 часов. Затем его растворили в этилацетате (200 мл) и промыли насыщ. водн. NaHCO3 (5×50 мл), водой (3×40 мл) и солевым раствором (50 мл). Затем остаток из сконцентрированного слоя этилацетата очищали хроматографией на силикагеле, используя 10/1 CH3Cl/МеОН с получением Соединения S с выходом 62% (11,0 г, 19,7 ммоля). ЖХ-МС [М+Н] 559,1 (C30H43N3O7 +Н, рассчит.: 558,3).
Получение N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламина] (Соединение КС-22)
Раствор Соединения S (11,0 г, 19,7 ммоля) обрабатывали смесью TFA и метиленхлорида (30 мл/30 мл) в течение 2 ч. при комнатной температуре. Растворители затем удаляли в вакууме до получения остатка объемом ~5 мл. Затем добавляли этиловый эфир (250 мл) для осаждения продукта. Полученный осадок отфильтровывали, промывали этиловым эфиром (50 мл) и сушили с получением неочищенного Соединения КС-22 с выходом 97% (11,0 г, 19,2 ммоля, с чистотой 90%) в виде белого твердого вещества. ЖХ-МС [М+Н] 458,9 (C25H35N3O5+Н, рассчит.: 458,3). Соединение КС-22 сразу использовали в следующей реакции без дальнейшей очистки.
Получение соединения U
Раствор Boc-Arg(Pbf)-OH (9,4 г, 17,8 ммоля). Соединения КС-22 (11,2 г, 19,7 ммоля) и NEt3 (10,0 мл, 71,7 ммоль) в ДМФА (80 мл) охладили в ледяной бане с последующим добавлением HATU (6,8 г, 17,9 ммоля) порциями в течение 10 мин. Ледяную баню убрали, и реакционную смесь перемешивали при комнатной температуре дополнительно в течение 1 часа. Смесь разбавили этилацетатом (150 мл) и экстрагировали водой (3×50 мл) и солевым раствором (50 мл). Затем органический слой сушили над Na2SO4 и отфильтровали; после выпаривания растворителей под вакуумом получили неочищенное Соединение U. Соединение U очищали с помощью флэш-хроматографии, используя метиленхлорид и метанол с получением Соединения U с выходом 79% (13,7 г, 14,2 ммоля) в виде твердой пены. ЖХ-МС [М+Н] 967,5 (C49H71N7O11S +H, рассчит.: 966,5).
Получение соединения V
Раствор Соединения U (13,7 г, 14,2 ммоля) обрабатывали соляной кислотой (4,0 мол. раствором в 1,4-диоксане, 40 мл) при комнатной температуре в течение 90 мин. Растворители удаляли в вакууме и остаток обрабатывали этиловым эфиром (100 мл). Полученный осадок отфильтровывали, промывали этиловым эфиром (2×25 мл) и сушили с получением неочищенного Соединения V с выходом 91% (12,1 г, 12,9 ммоля) в виде белого твердого вещества. ЖХ-МС [М+Н] 867,8 (C44H63N7O9S +H, рассчит.: 866,4). Соединение V сразу использовали в следующей реакции без дальнейшей очистки.
Получение соединения Х
К раствору Соединения V (73,3 г, 78,14 ммоля, в форме гидрохлорида), трет-бутилового эфира N-карбоксиметил-малоновой кислоты (Соединение W) (17,0 г, 78,34 ммоля) и NEt3 (33,0 г, 236,7 ммоля) в ДМФА (500 мл) при 0°С порциями за 10 мин. добавили HATU (30,6 г, 80,47 ммоля). Реакционную смесь перемешивали при комнатной температуре 1 ч. Добавили воду (500 мл) и смесь экстрагировали этилацетатом (750 мл). Экстракты этилацетата промывали водой (2×250 мл), NaHCO3 (2×200 мл) и солевым раствором (250 мл). Органический слой сушили над Na2SO4 и фильтровали. Раствор концентрировали и остаток очищали в колонке с силикагелем, используя градиент 1-10% метанола в метиленхлориде, с получением Соединения Х с выходом 43% (36,0 г, 33,8 ммоля) в виде белого твердого вещества. ЖХ-МС [М+Н] 1067,2 (C53H76N8O13S +H, рассчит.: 1065,5).
Получение N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламина]-аргинин-глицин-малоновой кислоты (Соединение КС-8)
Соединение Х (36,0 г, 33,8 ммоля) обрабатывали смесью TFA (60 мл) и м-крезола (2,0 мл) при комнатной температуре. Протекание реакции контролировали с помощью ЖХ/МС. После 4 часов смесь концентрировали в вакууме, удаляя большую часть летучих (удалено ~90% растворителя). Остаток обрабатывали этиловым эфиром (1 л) с образованием белого осадка. Прозрачный супернатант удаляли, и остаток промывали этиловым эфиром (1 л). Твердое вещество затем концентрировали и подвергали ВЭЖХ. [колонка Nanosyn-Pack Microsorb (100-10) С-18 (50×300 мм); скорость потока: 100, мл/мин; объем впрыска 15 мл; подвижная фаза А: 100% вода, 0,1% TFA; подвижная фаза В: 100% ацетонитрил, 0,1% TFA; градиентное элюирование от 0% до 20% B в течение 30 мин, изократическое элюирование при 20% B в течение 30 мин, градиентное элюирование от 20% B до 45% B в течение 35 мин; детектирование при 254 нм]. Фракции, содержащие целевое соединение, объединили и концентрировали в вакууме. Остаток растворяли в ацетонитриле (60 мл) и 0,1 н соляной кислоте (200 мл) и лиофилизировали с получением Соединения КС-8 с выходом 69,6% (19,5 г, 23,5 ммоля, чистота 99,4%) в виде белой пены. ЖХ-МС [М+Н] 758,5 (C36H52N8O10 +H, рассчит.: 757,4).
Биологические данные
Пример 4: Фармакокинетика Соединения КС-8 после введения крысам перорально (ПО)
Этот Пример иллюстрирует высвобождение оксикодона в плазму после перорального (ПО) введения Соединения КС-8 крысам.
Раствор Соединения КС-8 в физиологическом растворе (который может быть приготовлен, как описано в примерах в данном документе) вводили, как показано в Таблице 1, путем принудительного кормления через желудочный зонд самцам крысы Спраг-Доули с введенным в яремную вену катетером (четыре крысы на группу), которых не кормили 16-18 часов до перорального введения. Через указанные промежутки времени брали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин., и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили при -80°С до проведения анализа с помощью высокоэффективной жидкостной хроматографии/масс спектрометрии (ВЭЖХ/МС).
В Таблице 1 и на Фиг. 4 представлены суммарные полученные дозы оксикодона для крыс, которым вводили разные дозы Соединения КС-8. Результаты в Таблице 1 приведены для каждой группы крыс как (а) максимальная концентрация оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) время после введения Соединения КС-8, после которого концентрация оксикодона в плазме максимальна (Tmax) (среднее ± стандартное отклонение), и площадь под кривой (AUC) от 0 до 24 часов (среднее ± стандартное отклонение (СО)).
На Фиг. 4 приведено сравнение усредненных концентраций в плазме относительно времени высвобождения оксикодона после ПО введения крысам увеличивающихся доз Соединения КС-8.
Результаты в Таблице 1 и Фиг. 4 свидетельствуют о том, что концентрации оксикодона в плазме крыс повышаются пропорционально дозе Соединения КС-8.
Пример 5: Фармакокинетика Соединения КС-8 после введения собакам ПО
Этот Пример иллюстрирует высвобождение оксикодона в плазму после перорального (ПО) введения Соединения КС-8 собакам. Этот Пример также сопоставляет такое высвобождение с высвобождением оксикодона из Соединения КС-3 - пролекарства оксикодона, котором, в отличие от Соединения КС-8, не имеет геминальных диметиловых групп в циклизуемой спейсерной уходящей группе, а также не имеет глицина в расщепляемом трипсином фрагменте. Также приведено сравнение уровней оксикодона в плазме у собак, которым введен оксикодон или OxyContin® в таблетках.
Исследование А
Чистокровных молодых взрослых/взрослых самцов гончих не кормили в течение ночи. Увеличивающиеся дозы Соединения КС-8 (как показано в Таблице 2А), 4,15 мг/кг (5,7 мкмоль/кг) Соединения КС-3 (каждое из которых можно получить, как описано в примерах данного изобретения), или 2 мг/кг (5,7 мкмоль/кг) гидрохлорида оксикодона (Johnson Matthey Pharmaceutical Materials, West Deptford, NJ, USA) вводили в воде через желудочный зонд (в Таблице 2А указано число собак в группе). Дополнительно одной группе из 4-х собак вводили по одной таблетке OxyContin® (гидрохлорид оксикодона с контролируемым высвобождением) С-II на каждую собаку (NDC 59011-420-10, Purdue Pharma, Stamford, CT, USA). После таблеток вводили примерно 5 мл воды для облегчения проглатывания. Дозы оксикодона и таблеток OxyContin® выбирали так, чтобы обеспечить примерно эквимолярные количества. Образцы крови брали у каждого животного из яремной вены через различные промежутки времени в течение периода 24 ч, центрифугировали и 0,8 мл плазмы переносили из каждого образца в чистую пробирку, содержащую 8 мкл муравьиной кислоты; образцы встряхивали на шейкере типа Vortex и затем немедленно помещали на сухой лед, а затем хранили в морозильной камере при -80°С до проведения анализа ВЭЖХ/МС.
В Таблице 2А и на Фиг. 5 представлены суммарные полученные дозы оксикодона у собак, которым вводили указанные соединения. Результаты в Таблице 2А приведены для каждой группы собак как (а) максимальная концентрация оксикодона (ОС) в плазме (Cmax) (среднее ± стандартное отклонение), (b) время после введения соединения, после которого концентрация оксикодона в плазме максимальна (Tmax) (среднее ± стандартное отклонение), и площадь под кривой (AUC) от 0 до 24 часов (среднее ± стандартное отклонение (СО)).
На Фиг. 5 приведено сравнение усредненных по времени после высвобождения концентраций в плазме оксикодона после ПО введения собакам Соединения КС-8, Соединения КС-3, таблеток OxyContin® или гидрохлорида оксикодона.
Результаты в Таблице 2А и на Фиг. 5 говорят о том, что пероральное введение Соединения КС-8 собакам приводит к подавлению Cmax оксикодона, увеличению Tmax оксикодона и более продолжительному времени воздействия оксикодона (AUC) по сравнению в введением самого оксикодона. Соединение КС-8 также приводит к значительно повышенному высвобождению оксикодона в плазму собак (более высокие Cmax и AUC) по сравнению с Соединением КС-3. ФК кривая оксикодона в плазме, высвобождаемого в результате перорального введения собакам Соединения КС-8, похожа на ФК кривую таблеток OxyContin® больше, чем на ФК кривую оксикодона; продолжительность воздействия лекарственного средства у Соединения КС-8 по меньшей мере такая же, как и у таблеток OxyContin®.
Исследование В
Также, в отдельном эксперименте, собакам вводили Соединение КС-8 в дозах, указанных в Таблице 2В, и в различные периоды времени в течение периода 48 ч брали образцы. В остальном процедуры были такими же, как описано в Исследовании А.
В Таблице 2В представлены суммарные полученные дозы оксикодона для собак, которым вводили увеличивающиеся дозы Соединения КС-8. Результаты приведены таким же образом, как и в Таблице 2А, за исключением AUC, которое вычисляли за период от 0 до 48 ч.
Результаты в Таблице 2В показывают, что Соединение КС-8 дает воспроизводимые пропорциональные дозе ФК кривые у собак.
Пример 6: Расщепление пролекарства in vitro под действием трипсина, а также скорость циклизации уходящей спейсерной группы Соединения КС-8
Этот пример позволяет оценить способность трипсина расщеплять пролекарство оксикодона Соединение КС-8. Этот пример также позволяет оценить скорость циклизации и высвобождения оксикодона из Соединения КС-22, которое является идентичным Соединению КС-8 за исключением того, что Соединение КС-22 не содержит расщепляемого трипсином фрагмента.
Соединение КС-8 инкубировали с трипсином из бычьей поджелудочной железы (Кат. № Т8003, тип I, ~10000 ВАЕЕ единиц/мг белка, Sigma-Aldrich, St. Louis, МО, USA). В частности, реакционные смеси включали 0,761 ммоля Соединения КС-8⋅2HCl, 22,5 ммоля хлорида кальция, от 40 до 172 ммоля буфера Tris pH 8 и 0,25% ДМСО с различными активностями трипсина. Реакции проводили при 37°С в течение 24 часов. Образцы отбирали в определенные моменты времени, переносили в 0,5% муравьиную кислоту в ацетонитриле для прекращения активности трипсина и хранили при температуре ниже -70°С до анализа с помощью ЖХ-МС/МС.
Скорости высвобождения в результате циклизации измеряли, отслеживая скорость расходования Соединения КС-22 (при начальной концентрации 2,18 ммоля) в 50 ммолях фосфатного буфера с рН 7,4 при 20°С.
В Таблице 3 показаны результаты воздействия трипсина на Соединение КС-8. Результаты представлены как время полужизни пролекарства, подвергаемого воздействию трипсина (т.е. время полужизни пролекарства с трипсином), в часах и скорость образования оксикодона в мкмолях в час на ВАЕЕ единицу (мкмоль/ч/ВАЕЕ U) трипсина. Таблица 3 показывает скорость циклизации способной к циклизации уходящей спейсерной группы Соединения КС-22. Результаты выражены как время полужизни, получаемое при расходовании соединения.
Результаты в Таблице 3 говорят о том, что Соединение КС-8 может расщепляться трипсином, и что спейсерная уходящая группа Соединения КС-8 может циклизоваться, причем последний результат иллюстрируется непосредственно скоростью циклизации Соединения КС-22.
Пример 7: Пероральное введение крысам Соединения КС-8 совместно с трипсиновым ингибитором - Соединением 109
Этот пример демонстрирует способность трипсинового ингибитора влиять на способность Соединения КС-8 высвобождать оксикодон в плазму при пероральном введении Соединения КС-8 крысам.
Растворы Соединения КС-8 в физиологическом растворе (которые могут быть приготовлены, как описано в примерах данного изобретения) дозировали, как указано в Таблице 4. Одновременно с соединением RC-8 крысам вводили увеличивающиеся концентрации трипсинового ингибитора Соединения 109 (Кат. №3081, Tocris Bioscience, Ellisville, МО, USA, или Кат. № WS 38665, Waterstone Technology, Carmel, IN, USA) путем принудительного кормления через желудочный зонд самцов крысы Спраг-Доули с введенным в яремную вену катетером (по четыре крысы в группе), которых не кормили 16-18 часов до перорального дозирования. Через указанные промежутки времени отбирали пробы крови, выделяли плазму путем центрифугирования при 5400 об/мин при 4°С в течение 5 мин., и 100 микролитров (мкл) плазмы переносили из каждого образца в чистую пробирку, содержащую 2 мкл 50%-ной муравьиной кислоты. Пробирки встряхивали на шейкере типа Vortex в течение 5-10 секунд, немедленно помещали на сухой лед, а затем хранили в морозильной камере при -80°С до проведения анализа с помощью ВЭЖХ/МС.
Фиг.6А и Фиг. 6В показывают суммарные полученные дозы оксикодона для крыс, которым вводили различные дозы Соединения КС-8 в присутствии или в отсутствие Соединения 109.
На Фиг. 6А сравниваются средние по времени высвобождения концентрации оксикодона в плазме после перорального введения крысам 5 мг/кг (6 мкммоль/кг) пролекарства Соединения КС-8 совместно с увеличивающимися количествами трипсинового ингибитора Соединения 109.
На Фиг. 6В сравниваются средние по времени высвобождения концентрации оксикодона в плазме после перорального введения крысам 50 мг/кг (60 мкмоль/кг) пролекарства Соединения КС-8 совместно с увеличивающимися количествами трипсинового ингибитора Соединения 109.
Результаты на Фиг. 6А и Фиг. 6В указывают на способность Соединения 109 ослаблять способность Соединения КС-8 высвобождать оксикодон у крыс дозозависимым образом, о чем свидетельствуют сниженная Cmax и/или увеличенное Tmax.
Пример 8: Пероральное введение крысам однократной стандартной дозы и многократных стандартных доз композиции, включающей пролекарство Соединение КС-8 и трипсиновый ингибитор Соединение 109
Этот Пример иллюстрирует результаты перорального введения крысам однократной и многократных стандартных доз, содержащих пролекарство Соединение КС-8 и трипсиновый ингибитор Соединение 109.
Растворы Соединения КС-8 в физиологическом растворе (которые могут быть приготовлены, как описано в примерах данного изобретения) перорально вводили крысам (по 4 крысы в группе) при увеличивающихся концентрациях, изменяющихся от 5 до 50 мг/кг (от 6 до 60 мкмоль/кг), где стандартная доза была представлена 5 мг/кг (6 мкмоль/кг) Соединения КС-8 в отсутствие трипсинового ингибитора.
Второму комплекту крыс (по 4 крысы в группе) перорально дозировали пролекарство Соединение КС-8 и трипсиновый ингибитор Соединение 109 (Кат. №3081, Tocris Bioscience, или Кат. № WS38665, Waterstone Technology), как описано ниже и указано в Таблице 5. В частности, раствор композиции, содержащей 5 мг/кг (6 мкмоль/кг) Соединения КС-8 и 0,5 мг/кг (1 мкмоль/кг) Соединения 109, в физиологическом растворе, представляющий собой однократную стандартную дозу, вводили через желудочный зонд группе из 4-х крыс. Следует отметить, что отношение моль:моль трипсинового ингибитора к пролекарству (109: КС-8) составляет 0,17:1; с учетом этого, такая стандартная доза здесь называется стандартной дозой 109:КС-8 (0,17:1). Схожим образом дополнительным группам из 4-х крыс вводили растворы, представляющие собой 2 стандартные дозы, 3 стандартные дозы, 4 стандартные дозы, 6 стандартных доз, 8 стандартных доз, 10 стандартных доз (т.е. как указано в Таблице 5) 109:КС-8 (0,17:1) в физиологическом растворе.
Все крысы представляли собой самцов крысы Спраг-Доули с введенным в яремную вену катетером, которых не кормили 16-18 часов до перорального дозирования. Дозирование, сбор образцов и процедуры анализа были сходны с таковыми, описанными в Примере 4.
Таблица 5 (верхняя половина) и Фиг. 7А показывают суммарные полученные дозы оксикодона в плазме крыс, которым ввели 1, 2, 3, 4, 6, 8 и 10 доз Соединения КС-8 в отсутствие трипсинового ингибитора. Таблица 5 (нижняя половина) и Фиг. 7В показывают суммарные полученные дозы оксикодона в плазме крыс, которым ввели 1, 2, 3, 4, 6, 8 и 10 стандартных доз 109:КС-8 (0,17:1) стандартной дозы. Значения Cmax, Tmax и AUC оксикодона представлены, как описано в Примере 4.
На Фиг. 7А сравниваются средние по времени высвобождения концентрации оксикодона в плазме после ПО введения однократной дозы и многократных доз Соединения КС-8, введенных в отсутствие трипсинового ингибитора.
На Фиг. 7В сравниваются средние по времени высвобождения концентрации оксикодона в плазме после ПО введения однократной стандартной дозы и многократных стандартных доз композиции, включающей пролекарство Соединение КС-8 и трипсиновый ингибитор Соединение 109.
Результаты в Таблице 5, на Фиг. 7А и Фиг. 7В свидетельствуют о том, что введение множества стандартных доз (на примере 1, 2, 3, 4, 6, 8 и 10 стандартных доз 109:КС-8 (0.17:1)) приводит к ФК кривой концентрации оксикодона в плазме от времени, которая не пропорциональна ФК кривой зависимости концентрации оксикодона в плазме от времени для однократной стандартной дозы. Кроме того, ФК кривая многократных стандартный доз (например, Фиг. 7В) отличается от ФК кривой эквивалентной дозы пролекарства в отсутствие трипсинового ингибитора (например, Фиг. 7А).
Пример 9: Пероральное введение собакам Соединения КС-8 совместно с трипсиновым ингибитором Соединением 109
Этот Пример иллюстрирует способность трипсинового ингибитора влиять на способность Соединения КС-8 высвобождать оксикодон в плазму при пероральном (ПО) введении Соединения КС-8 собакам.
Чистокровных молодых взрослых/взрослых самцов гончих не кормили в течение ночи. Соединение КС-8 (которое может быть приготовлено, как описано в примерах данного изобретения) перорально через желудочный зонд дозировали в воде в количестве 18,2 мг/кг (22 мкмоль/кг) одновременно с 1,8 мг/кг (3,3 мкмоль/кг) Соединения 109 (Кат. №3081, Tocris Bioscience, Ellisville или Кат. № WS38665, Waterstone Technology) или без него, как описано в Таблице 6. Образцы крови собирали, обрабатывали и анализировали, как в Примере 5.
Таблица 6 и Фиг. 8 представляют результирующую совокупную дозу оксикодона у собак, которым ввели Соединение КС-8 в присутствии или в отсутствие Соединения 109. Результаты в Таблице 6 представлены для каждой группы из четырех собак, как описано в Примере 5.
На Фиг. 8 сравниваются средние по времени высвобождения концентрации оксикодона в плазме после ПО введения собакам Соединения КС-8 совместно с трипсиновым ингибитором Соединением 109 или без него.
Результаты в Таблице 6 и на Фиг. 8 свидетельствуют о способности Соединения 109 ослаблять высвобождение оксикодона из Соединения КС-8, как путем снижения Cmax и AUC, так и путем увеличения Tmax.
Хотя данное изобретение описано со ссылками на его конкретные варианты осуществления, специалисту в данной области техники должно быть понятно, что в изобретение могут быть внесены различные изменения, а его признаки могут быть заменены эквивалентными без отхождения от истинной сущности и объема данного изобретения. К тому же, для приспособления изобретения к конкретной ситуации, материалу, композиции материала, процессу, этапу или этапам процесса, в цели, сущность и объем данного изобретения могут быть внесены многочисленные модификации. Предполагается, что все эти модификации охвачены объемом прилагаемой формулы изобретения.
Реферат
Изобретение относится к соединению KC-8, представляющему собой N-1-[3-(оксикодон-6-енол-карбонил-метил-амино)-2,2-диметил-пропиламин]-аргинин-глицин-малоновую кислоту, или его фармацевтически приемлемым солям, сольватам или гидратам, которые обеспечивают контролируемое высвобождение оксикодона путем ферментативного расщепления с последующей внутримолекулярной циклизацией. Изобретение относится также к фармацевтическим композициям для применения в лечении или предупреждении боли, в том числе к композициям, дополнительно содержащим трипсиновый ингибитор, к способу уменьшения потенциальной возможности злоупотребления указанной композицией, стандартной дозе, содержащей эту композицию, способу ее изготовления и способам выбора соединения и трипсинового ингибитора, пригодных для изготовления стандартной дозы. 8 н. и 9 з.п. ф-лы, 6 табл., 8 ил., 9 пр.
Формула
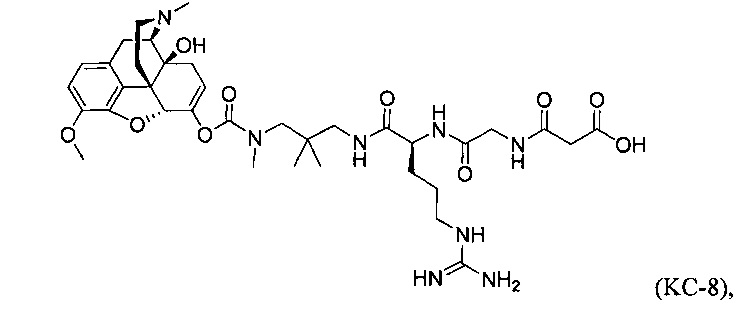

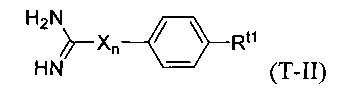


Документы, цитированные в отчёте о поиске
Препаративные формы гидрокодона с контролируемым высвобождением
Комментарии