Препаративные формы гидрокодона с контролируемым высвобождением - RU2230556C2
Код документа: RU2230556C2
Чертежи

Описание
Вследствие трудностей, имеющих место при фармакотерапии боли, в частности хронической боли, идеальными лекарственными средствами являются опиоидные анальгетики, подлежащие введению в качестве препаративных форм с контролируемым высвобождением.
Целью всех препаративных форм с контролируемым высвобождением является обеспечение более длительного периода фармакологического действия после введения, чем после введения дозированных лекарственных форм с немедленным высвобождением. Такие более длительные периоды ответной реакции имеют много терапевтических преимуществ, которые не достигаются с помощью соответствующих короткодействующих препаратов с немедленным высвобождением. Таким образом, терапия может продолжаться без прерывания сна пациента, что особенно важно, например, при лечении пациента с целью регуляции сильной боли (например, пациент после хирургического вмешательства, пациент со злокачественной опухолью и т.д.) или тех пациентов, которые испытывают головные боли типа мигрени при пробуждении, также как ослабленные пациенты, для которых необходим сон.
Несмотря на то, что общепринятая терапия быстродействующими лекарственными средствами сопровождается тщательным введением с небольшими интервалами для поддержания эффективных устойчивых концентраций лекарственного средства в плазме, имеют место пики и провалы уровня активного лекарственного средства в плазме по причине быстрого всасывания, системной экскреции соединения и из-за метаболической инактивации, вызывая таким образом проблемы при поддерживающей терапии пациента. Другим важным преимуществом лекарственных препаратов длительного действия является улучшение соблюдения пациентом терапии вследствие исключения пропущенных из-за забывчивости пациента доз.
В области фармацевтики известен способ получения композиций, которые обеспечивают контролируемое высвобождение фармакологически активных веществ, содержащихся в композициях, после перорального введения людям и животным. Такие композиции с медленным высвобождением используются для задержки всасывания лекарственного средства до достижения его определенного количества в пищеварительном тракте. Такое контролируемое высвобождение лекарственного средства в пищеварительном тракте поддерживает в дальнейшем требуемую концентрацию указанного лекарственного средства в кровотоке в течение более длительного времени, чем то, что имеет место при введении общепринятых лекарственных форм с быстрым высвобождением.
Известные из предшествующего уровня способы получения и применения композиций, обеспечивающих контролируемое высвобождение активного соединения из носителя, в основном относится к высвобождению активного вещества в физиологическую жидкость пищеварительного тракта. Однако признано, что простое присутствие активного вещества в жидкостях желудочно-кишечного тракта само по себе не гарантирует биодоступности.
Для его всасывания активное лекарственное средство должно быть в растворе. Время, требуемое для данного распределения активного вещества из отдельной дозы, определяется как отношение количества активного лекарственного вещества, высвобождаемого из стандартной лекарственной формы за определенное время, что основано на способе тестирования, проводимого в стандартизированных условиях. Физиологические жидкости желудочно-кишечного тракта являются средами для определения времени растворения. Из предшествующего уровня известно много удовлетворительных тестовых процедур для измерения времени растворения фармацевтических композиций, и эти тестовые процедуры описаны во всем мире в официальных руководствах.
Хотя имеется много разных факторов, которые влияют на время растворения лекарственного вещества из носителя, время растворения, определенное для фармакологически активного вещества их конкретной композиции является относительно постоянным и воспроизводимым. К различным факторам, которые могут влиять на время растворения, относятся площадь поверхности лекарственного вещества, контактирующей со средой растворителя, рН раствора, растворимость вещества в конкретной среде растворителя и движущие силы концентрации насыщения растворенного вещества в среде растворителя. Таким образом, концентрация растворения активного лекарственного вещества динамически модифицируется до ее стабильного состояния, поскольку компоненты удаляются из среды растворителя путем всасывания через участок ткани. В физиологических условиях концентрация насыщения растворенных веществ восполняется из резерва лекарственной формы для поддержания универсальной и постоянной концентрации растворения в среде растворителя, что обеспечивает стабильное всасывание.
Транспорт через тканевые участки всасывания желудочно-кишечного тракта подчиняется силам осмотического уравнения Доннана (Donnan) на обеих сторонах мембраны, поскольку направление движущей силы представляет собой разницу между концентрациями активного вещества по каждую сторону мембраны, т.е. между количеством, растворенным в жидкостях желудочно-кишечного тракта, и количеством, находящимся в крови. Поскольку концентрация в крови постоянно меняется за счет растворения, циркуляторных изменений, накопления в тканях, метаболических превращений и системной экскреции, поток активного вещества направляется из желудочно-кишечного тракта в кровоток.
Для получения лекарственных форм с контролируемым высвобождением использовали различные способы. В данной области известны специальным образом покрытые пилюли, таблетки и капсулы, где медленное высвобождение активного лекарственного средства осуществляется за счет селективного разрушения покрытия препарата или за счет смешивания со специальным матриксом для воздействия на высвобождение лекарственного вещества. Известные препаративные формы с контролируемым высвобождением обеспечивают последовательное высвобождение разовой дозы активного соединения за предварительно определенные периоды после введения.
Конкретные примеры опиоидных препаративных форм с контролируемым высвобождением, о которых сообщается в патентной литературе, включают, например, описанные в патентах США №4990341 и 4844909 (Goldie et al.), переданных правопреемнику настоящего изобретения и включенных в описание в качестве ссылки, и они описывают композиции гидроморфона, где скорость растворения in vitro лекарственной формы при измерении способами “лопасти” (Paddle) или “корзины” (Basket) согласно USP при 100 об/мин в 900 мл водного буфера (рН между 1,6 и 7,2) при 37°С находится между 12,5 и 42,5% (по массе) гидроморфона, высвобождаемого через 1 час, между 25 и 55% (по массе) высвобождения через 2 часа, между 45 и 75% (по массе) высвобождения через 4 часа и между 55 и 85% (по массе) высвобождения через 6 часов, причем скорость высвобождения in vitro не зависела от рН в интервале между 1,6 и 7,2 и была выбрана таким образом, что пик концентрации гидроморфона в плазме, полученный in vivo, имел место в интервале между 2 и 4 часами после введения лекарственной формы. При помощи данных препаративных форм гидроморфина достигалось по крайней мере 12 часов обезболивания.
Предоставление дозированных препаратов с контролируемым высвобождением для других опиоидных анальгезирующих лекарственных средств, которые могут применяться для снижения боли, представляется крайне желательным. Кроме того, крайне желательным представляется предоставление таких препаратов с контролируемым высвобождением с фармакокинетическими свойствами, которые обеспечивают наиболее эффективное управление болью у пациентов, которые нуждаются в терапии боли.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является существенное повышение эффективности и качества управления болью у пациентов, испытывающих умеренную боль.
Целью настоящего изобретения является предоставление биодоступных препаративных форм гидрокодона, которые существенно повышают эффективность и качество управления болью у пациентов, испытывающих умеренную боль.
Еще одной целью настоящего изобретения является предоставление биодоступных препаративных форм гидрокодона с контролируемым высвобождением, которые обеспечивают существенное увеличение длительности действия по сравнению с препаративными формами гидрокодона с немедленным высвобождением, но которые обеспечивают раннее начало анальгезии.
Дальнейшей целью изобретения является предоставление вводимых перорально препаративных форм гидрокодона с контролируемым высвобождением, подходящих для введения два раза в день, которые обеспечивают раннее начало терапевтического действия и которые после достижения максимальной концентрации в течение интервала дозировки обеспечивают относительно ровный профиль в плазме крови, означающий, что уровень опиоида в плазме обеспечивает соотношение C12/Cmax от 0,55 до 0,85, и которые обеспечивают эффективное обезболивание пациента. В альтернативных вариантах лекарственная форма обеспечивает соотношение C12/Cmax от 0, 65 до 0,75.
Вышеуказанные и другие цели достигают благодаря настоящему изобретению, которое в некоторых вариантах относится к твердым пероральным дозированным формам с контролируемым высвобождением, включающим эффективное для анальгезии количество гидрокодона или его фармацевтически приемлемой соли и достаточное количество вещества для контролируемого высвобождения для того, чтобы сделать дозированную форму подходящей для введения два раза в день, причем лекарственная форма после одного введения пациенту или группе пациентов обеспечивает время для достижения пика концентрации гидрокодона в плазме in vivo предпочтительно в интервале примерно от 2 до 8 часов (Тmах) и после достижения максимальной концентрации обеспечивает соотношение C12/Cmax от 0,55 до 0,85.
В некоторых предпочтительных вариантах лекарственная форма с контролируемым высвобождением обеспечивает высвобождение in vitro примерно от 18 до 42,5% по массе гидрокодона или его соли из лекарственной формы за один час, при измерении способом “корзины” (Basket) согласно USP при 100 об/мин в 700 мл искусственного желудочного сока (SGF) в течение 55 мин при 37°С, и затем переключая на 900 мл искусственного кишечного сока (SIF) при 37°С.
В некоторых предпочтительных вариантах скорость растворения in vitro лекарственной формы гидрокодона при измерении способом “корзины” (Basket) согласно USP при 100 об/мин в 900 мл водного буфера при рН 1,2 и 7,5 при 37°С находится примерно от 25 до 65% по массе гидрокодона или его соли, высвобождаемого через 2 часа, примерно от 45 до 85% по массе гидрокодона или его соли, высвобождаемого через 4 часа, и примерно более 60% по массе гидрокодона или его соли, высвобождаемого через 8 часов. Хотя скорость высвобождения in vitro может быть по требованию рН-независимой или рН-зависимой, в предпочтительных вариантах изобретения высвобождение гидрокодона является рН-независимым.
Некоторые предпочтительные варианты относятся к лекарственным формам для контролируемого высвобождения, включающим терапевтически эффективное количество гидрокодона, где лекарственная форма обеспечивает концентрацию гидрокодона в плазме, равную по крайней мере 5 или 6 нг/мл, через 12 часов после введения и обеспечивает концентрацию гидрокодона в плазме, равную по крайней мере около 8 нг/мл, примерно через 2-8 часов после введения.
Другие предпочтительные варианты изобретения относятся к пероральной лекарственной форме гидрокодона с контролируемым высвобождением для введения два раза в сутки, которая обеспечивает Сmax гидрокодона, составляющую менее чем 50% от Сmах при введении эквивалентной дозы контрольной препаративной формы гидрокодона с немедленным высвобождением (например, Lortab®), и которая обеспечивает эффективную анальгезию в течение 12-часового интервала между дозами.
Другие предпочтительные варианты изобретения относятся к пероральной лекарственной форме гидрокодона с контролируемым высвобождением для введения два раза в сутки, где лекарственная форма обеспечивает время достижения 80% Сmах, составляющее примерно от 90 до 150%, предпочтительно от 90 до 110% от времени достижения 80% Сmах при введении эквивалентной дозы контрольной гидрокодоновой препаративной формы сравнения с немедленным высвобождением (например, Lortab). Предпочтительно время достижения 80% Сmах гидрокодона для лекарственной формы с контролируемым высвобождением составляет примерно от 0,5 до 1,5 часов, наиболее предпочтительно примерно от 0,8 до 1,2 часов. В альтернативных вариантах время достижения 80% Сmах гидрокодона для лекарственной формы с контролируемым высвобождением составляет примерно от 0,75 до 2 часов, наиболее предпочтительно примерно от 0,9 до 1,5 часов.
Другие предпочтительные варианты изобретения относятся к пероральной лекарственной форме гидрокодона с контролируемым высвобождением для введения два раза в сутки, где лекарственная форма обеспечивает время достижения 90% Сmах, составляющее примерно от 150 до 400%, предпочтительно от 150 до 250%, от времени достижения 90% Cmax при введении эквивалентной дозы контрольной гидрокодоновой препаративной формы сравнения с немедленным высвобождением. Предпочтительно время достижения 90% Cmax гидрокодона для лекарственной формы с контролируемым высвобождением составляет примерно от 1,5 до 2,5 часов, наиболее предпочтительно примерно от 1,8 до 2,2 часов. В альтернативных вариантах время достижения 90% Cmaxгидрокодона для лекарственной формы с контролируемым высвобождением составляет примерно от 1,5 до 4 часов, наиболее предпочтительно примерно от 1,8 до 2,5 часов.
Другие предпочтительные варианты изобретения относятся к пероральной лекарственной форме гидрокодона с контролируемым высвобождением для введения два раза в сутки, где лекарственная форма поддерживает концентрацию в плазме в пределах 80% Cmax примерно от 0,5 до 10 часов, предпочтительно примерно от 1 до 9 часов или примерно от 4 до 8 часов.
Другие предпочтительные варианты изобретения относятся к пероральной лекарственной форме гидрокодона с контролируемым высвобождением для введения два раза в сутки, где лекарственная форма поддерживает концентрацию гидрокодона в плазме в пределах 90% Cmax примерно от 1 до 6,5 часов, предпочтительно примерно от 2 до 5 часов или примерно от 2 до 6,5 часов.
Другие предпочтительные варианты изобретения относятся к пероральной лекарственной форме гидрокодона с контролируемым высвобождением для введения два раза в сутки, которая обеспечивает среднюю скорость всасывания in vivo от введения до Тmах, составляющую примерно от 1,5 до 5 мг/час, и обеспечивает среднюю скорость всасывания in vivo от Тmах до конца интервала между дозами, составляющую менее чем примерно 0,5 мг/час, основываясь на пероральном введении лекарственной формы, содержащей 15 мг гидрокодона битартрата. Предпочтительно лекарственная форма обеспечивает среднюю скорость всасывания in vivo от введения до Тmах, составляющую примерно от 2 до 4 мг/час, и обеспечивает среднюю скорость всасывания in vivo от Тmах до конца 12-часового интервала между дозами, составляющую примерно от 0,08 до 0,4 мг/час, основываясь на пероральном введении лекарственной формы, содержащей 15 мг гидрокодона битартрата.
Другие предпочтительные варианты изобретения относятся к пероральной лекарственной форме гидрокодона с контролируемым высвобождением для введения два раза в сутки, которая обеспечивает скорость всасывания в течение периода времени от Тmах примерно до 12 часов после перорального введения лекарственной формы, которая составляет примерно от 55 до 85% от скорости элиминации (eliminatia) в течение того же периода времени.
Вышеуказанные варианты изобретения, также как и другие варианты, предпочтительно обеспечивают время до Тmах, в 3-4 раза большее, чем Тmах, эквивалентной дозы ссылочного гидрокодонового препарата с немедленным высвобождением. Предпочтительно Тmах препаративных форм с длительным высвобождением находится в интервале примерно от 2 до 8 часов, от 3 до 7 часов или от 4 до 6 часов после перорального введения.
Настоящее изобретение также относится к препаративным формам гидрокодона, которые обеспечивают Сmах гидрокодона, составляющую менее чем примерно 50%, предпочтительно менее чем 40% от Сmах, которая обеспечивается эквивалентной дозой ссылочного продукта с немедленным высвобождением.
Например, неожиданно обнаружено, что когда получали препарат гидрокодона с системой доставки, как описано в патентах США №4861598 и 4970075, Сmах гидрокодона, которая обеспечивалась системой доставки, выраженная как процент от Сmах ссылочного продукта с немедленным высвобождением, была существенно ниже, чем то же значение, рассчитанное для препарата оксикодона, полученного с той же системой доставки. Данный феномен был очевидным, несмотря на тот факт, что препаративные формы оксикодона и гидрокодона с контролируемым высвобождением проявляли сходные параметры растворения in vitro.
Когда препаративные формы по настоящему изобретению получают с использованием систем доставки, описанных в патентах США №4861598 и 4970075, Сmах системы доставки, выраженная как процент от Сmах ссылочного продукта с немедленным высвобождением, составляет менее чем примерно 50% и менее чем примерно 40% в предпочтительных вариантах, тогда как оксикодон характеризуется рассчитанным значением, составляющим более чем 50%.
Термин “гидрокодон” определяется для целей изобретения как охватывающий свободное основание гидрокодон, также как фармацевтически приемлемые соли и комплексы гидрокодона.
Термин “способ лопасти (Paddle) и корзины (Basket) согласно USP” представляет способ лопасти (Paddle) и корзины (Basket), описанный, например, в US Pharmacopoeia XXII (1990), включенной в описание в качестве ссылки.
Термин “рН-зависимый” для целей настоящего изобретения определяется как обладающий характеристиками (например, растворением), которые изменяются в зависимости от рН окружающей среды.
Термин “рН-независимый” для целей настоящего изобретения определяется как обладающий характеристиками (например, растворением), на которые рН окружающей среды существенно не влияет.
Термин “биодоступность” определяется для целей настоящего изобретения как степень, в которой лекарственное средство (например, гидрокодон) всасывается из стандартных лекарственных форм.
Термин “контролируемое высвобождение” определяется для целей настоящего изобретения как высвобождение лекарственного средства (например, гидрокодона) с такой скоростью, что концентрации в крови (например, в плазме) поддерживаются в терапевтических пределах, но ниже токсических концентраций, в течение периода времени, примерно равного 12 часам или большего.
Термин “Сmax” означает максимальную концентрацию в плазме, которая достигается в течение интервала между дозами.
Термин “Тmax” означает время достижения максимальной концентрации в плазме (Cmax).
Термин T1/2(abs) означает время, необходимое для переноса в плазму половины всасываемой дозы опиоида.
Термин “устойчивое состояние” означает, что концентрация в плазме данного лекарственного средства достигнута и поддерживается последовательными дозами лекарственного средства на уровне или выше минимальной эффективной терапевтической концентрации и ниже минимальной токсической концентрации в плазме данного лекарственного средства. Для опиоидных анальгетиков минимальная эффективная терапевтическая концентрация частично определяется степенью обезболивания, достигнутой у данного пациента. Специалистам в данной области будет хорошо понятно, что измерение боли в высокой степени субъективно, и среди пациентов могут иметь место большие индивидуальные различия.
Термины “поддерживающая терапия” и “хроническая терапия” определяются для целей настоящего изобретения как лекарственная терапия, назначенная пациенту после того, как пациента подвергали титрованию опиоидными анальгетиками до определенного выше устойчивого состояния.
Термин “минимальная эффективная анальгетическая концентрация” или “МЭАК” по отношению к концентрациям опиоидов, таких как гидрокодон, очень трудно определить количественно. Однако в общем это означает минимально эффективную анальгетическую концентрацию гидрокодона в плазме, при концентрациях ниже которой аналгезия не обеспечивается. Хотя имеет место непрямая взаимосвязь, например, между концентрациями гидрокодона в плазме и аналгезией, более высокие и пролонгированные концентрации гидрокодона в плазме в общем ассоциируются с наилучшим обезболиванием. Между временем пика концентрации гидрокодона в плазме и временем пика эффекта лекарственного средства имеется время отставания или гистерезис. Это остается верным для лечения опиоидными анальгетиками в общем.
Термин “среднее резонансное время” (СРВ) определяется как среднее время, в течение которого молекула лекарственного средства остается в организме. Эта расчетная величина, которая является функцией всасывания, распределения и выделения, частично зависит от лекарственной формы, содержащей активный ингредиент.
Для целей изобретения, кроме специально указанных далее случаев, термин “пациент” означает, что описание (или пункт формулы изобретения) относится к фармакокинетическим параметрам индивидуального пациента или субъекта.
Термин “группа пациентов” означает, что описание (или пункт формулы изобретения) относится к среднему значению фармакокинетических параметров по крайней мере двух пациентов или субъектов.
Термин “прорыв боли” (breakthrough pain) означает боль, которую испытывает пациент, несмотря на тот факт, что пациенту вводили эффективные в общем смысле количества твердых лекарственных форм по изобретению с длительным высвобождением, содержащих гидроморфон.
Термин “избавление” (rescue) относится к дозе анальгетика, которую вводят пациенту, испытывающему прорыв боли.
Термин “эффективное управление болью” означает объективную оценку, которую дает врач ответной реакции пациента (испытываемая боль против побочных эффектов) на анальгезирующее лечение, также как субъективную оценку терапевтического лечения пациентом, подвергающимся этому лечению. Специалисту будет ясно, что эффективная анальгезия будет изменяться в зависимости от многих факторов, включая индивидуальную вариабельность пациентов.
Термин “контрольная гидрокодоновая препаративная форма с немедленным высвобождением” для целей настоящего изобретения представляет собой эквивалентное количество гидрокодона из Lortab®, коммерчески доступного от USB Pharma, Inc, или фармацевтического продукта, который обеспечивает немедленное высвобождение гидрокодона или его соли.
Для целей изобретения описанные препаративные формы с контролируемым высвобождением и контрольные препаративные формы с немедленным высвобождением соответствуют друг другу в плане дозы. Фармакокинетические параметры таких препаративных форм (например, AUC и Сmax) линейно возрастают от одной дозировки к другой. Поэтому фармакокинетические параметры конкретной дозы могут выводиться из параметров другой дозы той же препаративной формы.
Для целей изобретения, если не указано по-другому, описанные фармакокинетические параметры основаны на введении разовой дозы препаративной формы гидрокодона индивидуальному пациенту. Фармакокинетические параметры, основанные на исследовании группы пациентов, будут обозначены как “средние” данные.
Термин “первое введение” означает разовую дозу по настоящему изобретению в начале терапии, введенную индивидуальному пациенту или группе пациентов.
Пероральные твердые лекарственные формы с контролируемым высвобождением по настоящему изобретению могут содержать неожиданно мало опиоида. Возможно, что пероральные твердые лекарственные формы с контролируемым высвобождением по настоящему изобретению могут иметь существенно меньшую суточную дозировку по сравнению с общепринятыми продуктами с немедленным высвобождением без различия по эффективности анальгезии. При сравнимых суточных дозировках с использованием пероральных твердых лекарственных форм с контролируемым высвобождением по настоящему изобретению в результате можно получить большую эффективность по сравнению с общепринятыми продуктами с немедленным высвобождением.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Приложенные фигуры иллюстрируют варианты изобретения и не подразумевают ограничений объема изобретения, охваченного формулой изобретения.
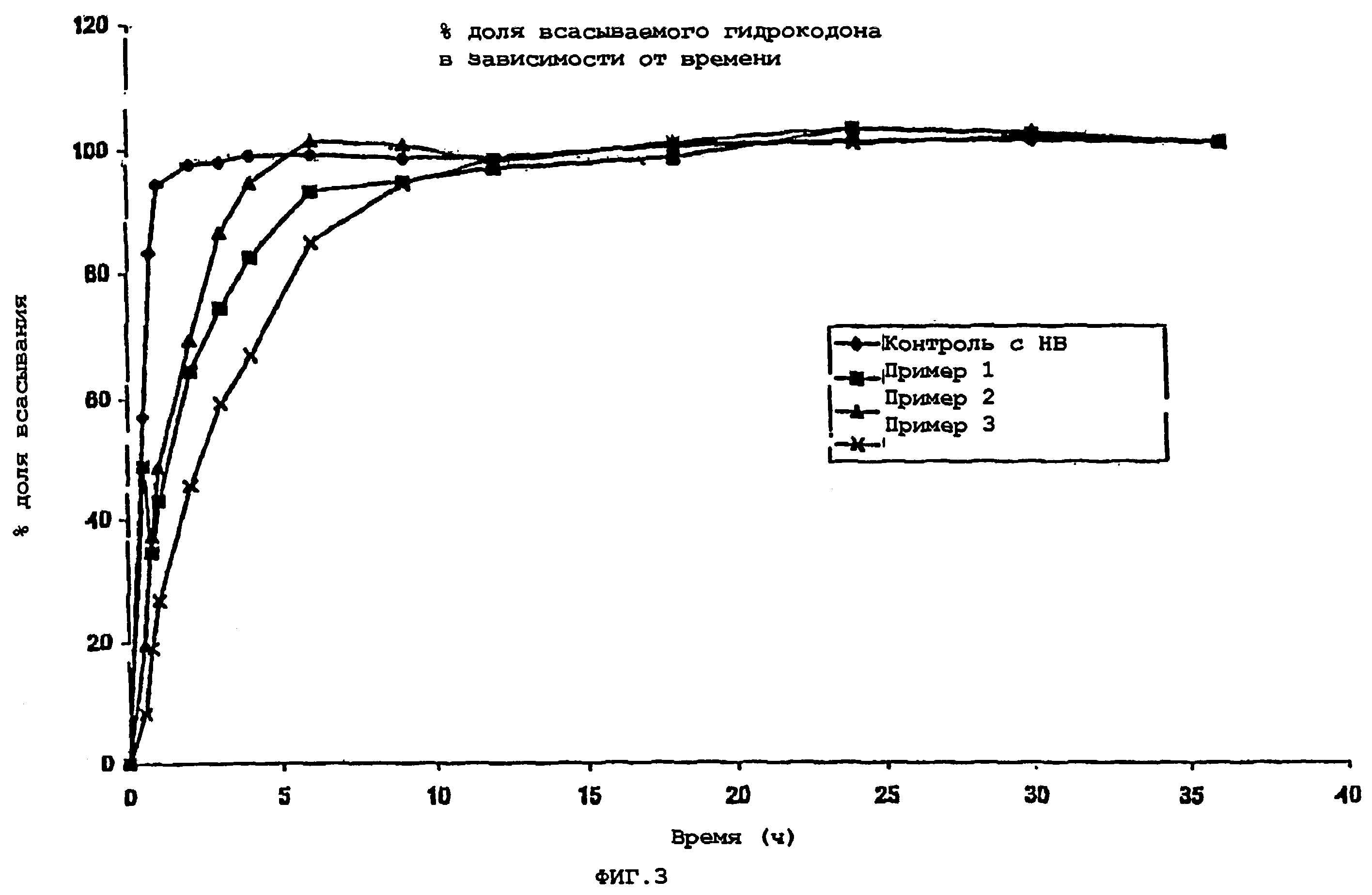
Фигура 1 является графическим представлением средней концентрации гидрокодона в плазме по примеру 1, примеру 2, примеру 3 и эквивалентной дозы гидрокодона с немедленным высвобождением.
Фигура 2 является графическим представлением средней концентрации в плазме по примеру 1, примеру 2 и примеру 3 против разных образцов оксикодона с контролируемым высвобождением, полученных согласно процедурам по примеру 4, и разных образцов морфина с контролируемым высвобождением, полученных согласно процедурам по примеру 5.
Фигура 3 является графическим представлением % доли гидрокодона, всасываемого за период времени по примеру 1, примеру 2, примеру 3, и эквивалентной дозы гидрокодона с немедленным высвобождением.
ПОДРОБНОЕ ОПИСАНИЕ
Вышеуказанные варианты изобретения могут обеспечиваться широким разнообразием препаративных форм с контролируемым высвобождением, известным специалистам в данной области. Например, подходящие лекарственные формы с контролируемым высвобождением описаны в патенmax США №4861598 и 4970075, включенных в описание в качестве ссылки.
В некоторых вариантах настоящего изобретения эффективное количество опиоида в форме с немедленным высвобождением включено в препаративную форму. Форма опиоида с немедленным высвобождением включается в количестве, которое является эффективным для уменьшения времени достижения максимальной концентрации опиоида в крови (например, в плазме), так что Тmaх уменьшается, например, до времени примерно от 2 до 5 часов или примерно от 2 до 4 часов. Обнаружено, что путем включения такого эффективного количества опиоида с немедленным высвобождением в стандартную дозу значительно снижаются относительно сильные боли, которые испытывает пациент. В таких вариантах эффективное количество опиоида в форме с немедленным высвобождением может покрывать субстраты по настоящему изобретению. Например, там, где длительное высвобождение опиоида из препаративной формы является следствием наличия покрытия для контролируемого высвобождения, слой для немедленного высвобождения может наноситься поверх покрытия для контролируемого высвобождения. С другой стороны, слой для немедленного высвобождения может покрывать поверхность субстратов, в которых опиоид включен в матрикс для контролируемого высвобождения. Там, где множество субстратов для длительного высвобождения, включающих эффективную стандартную дозу опиоида (например, системы мультимикрочастиц, включающие гранулы, сферы, шарики и тому подобное), входит в состав твердой желатиновой капсулы, часть дозы опиоида с немедленным высвобождением может помещаться в состав желатиновой капсулы путем включения в капсулу достаточного количества опиоида с немедленным высвобождением в виде порошка или гранул. Альтернативно желатиновая капсула сама может покрываться слоем опиоида с немедленным высвобождением. Специалисту в данной области известны и другие, альтернативные способы включения части опиоида с немедленным высвобождением в состав стандартной дозы. Такие альтернативы считаются охваченными прилагаемой формулой изобретения.
Одним из преимуществ опиоидных лекарственных форм по настоящему изобретению является то, что терапевтические концентрации в общем достигаются по существу без значительного повышения интенсивности и/или степени сопутствующих побочных эффектов, таких как тошнота, рвота или сонливость, которые часто ассоциируются с высокими концентрациями опиоидов в крови. Также очевидно предполагать, что применение настоящих лекарственных форм приводит к снижению риска развития лекарственной зависимости.
АКТИВНЫЙ АГЕНТ
Пероральные твердые лекарственные формы с контролируемым высвобождением по настоящему изобретению предпочтительно включают примерно от 0,5 до 1250 мг гидрокодона или эквивалентное количество его фармацевтически приемлемой соли. В более предпочтительных изобретениях лекарственная форма может включать примерно от 5 до 60 мг, например 15 мг. Подходящие фармацевтически приемлемые соли гидрокодона включают гидрокодона битартрат, гидрокодона битартрата гидрат, гидрокодона гидрохлорид, гидрокодона п-толуолсульфонат, гидрокодона фосфат, гидрокодона тиосемикарбазон, гидрокодона сульфат, гидрокодона трифторацетат, гидрокодона гемипентагидрат, гидрокодона пентафторпропионат, гидрокодона п-нитрофенилгидразон, гидрокодона о-метилоксим, гидрокодона семикарбазон, гидрокодона гидробромид, гидрокодона мукат, гидрокодона олеат, гидрокодона двухосновный фосфат, гидрокодона одноосновный фосфат, неорганическую соль гидрокодона, органическую соль гидрокодона, гидрокодона ацетата тригидрат, гидрокодона бис(гептафторбутират), гидрокодона бис(метилкарбамат), гидрокодона бис(пентафторпропионат), гидрокодона бис(пиридинкарбоксилат), гидрокодона бис(трифторацетат), гидрокодона хлоргидрат и гидрокодона сульфата пентагидрат. Предпочтительно гидрокодон представлен как соль битартрат.
Лекарственные формы по настоящему изобретению могут также включать одно или несколько дополнительных лекарственных средств, которые могут действовать синергически с гидрокодоновыми анальгетиками по настоящему изобретению или не действуют таким образом. Примеры таких дополнительных лекарственных средств включают нестероидные противовоспалительные средства, включая ибупрофен, диклофенак, напроксен, беноксапрофен, флурбипрофен, фенопрофен, флубифен, кетопрофен, индопрофен, пиропрофен, карпрофен, оксапрозин, прамопрофен, муропрофен, триоксапрофен, супрофен, аминопрофен, тиапрофеновую кислоту, флупрофен, буклоксовую кислоту, индометацин, сулиндак, толметин, зомепирак, тиопинак, зидометацин, ацеметацин, фентиазак, клинадак, окспинак, мефенамовую кислоту, меклофенамовую кислоту, флуфенамовую кислоту, нифлумовую кислоту, толфенамовую кислоту, дифлуризал, флуфенизал, пироксикам, судоксикам или изоксикам и тому подобное. Такие нестероидные противовоспалительные средства также включают ингибиторы циклоксигеназы, такие как целекоксиб (SC-58635), DUP-697, флозулид (GCP-28238), мелоксикам, 6-метокси-2-нафтилуксусная кислота (6-MNA), Vioxx (MK-966), набуметон (пролекарственное вещество для 6-MNA), нимесулид, NS-398, SC-5766, SC-58215 и Т-614, такие как амантадин (1-аминоамантадин), мемантин (3,5-диметиламиноадамантон), их смеси и их фармацевтически приемлемые соли.
Другие дополнительные лекарственные средства включают нетоксичные антагонисты рецептора NMDA, такие как декстрорфан, декстрометорфан,
3-(1-нафталенил)-5-(фосфонометил)-L-фенилаланин;
3-(1-нафталенил)-5-(фосфонометил)-DL-фенилаланин;
1-(3, 5-диметилфенил)нафтален и 2-(3,5-диметилфенил)нафтален;
2SR,4RS-4-(((1Н-тетразол-5-ил)метил)окси)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-((((1H-тетразол-5-ил)метил)окси)метил)пиперидин-2-карбоновая кислота;
Е и Z 2SR-4-(О-(1Н-тетразол-5-ил)метил)кетоксимино)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-((1Н-тетразол-5-ил)тио)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-((1Н-тетразол-5-ил)тио)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-(5-меркапто-1Н-тетразол-1-ил)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-(5-меркапто-2Н-тетразол-2-ил)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-(5-меркапто-1Н-тетразол-1-ил)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-(5-меркапто-2Н-тетразол-2-ил)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-(((1Н-тетразол-5-ил)тио)метил)пиперидин-2-карбоновая кислота;
2SR, 4RS-4-((5-меркапто-1Н-тетразол-1-ил)метил)пиперидин-2-карбоновая кислота или
2SR, 4RS-4-((5-меркапто-2Н-тетразол-2-ил)метил)пиперидин-2-карбоновая кислота,
их смеси и их фармацевтически приемлемые соли.
Другие подходящие дополнительные лекарственные средства, которые могут входить в состав лекарственных форм по настоящему изобретению, включают ацетаминофен, аспирин, нейроактивные стероиды (как те, что описаны в U.S. Serial No. 09/026520 от 20 февраля 1998, включенном в описание в качестве ссылки) и другие неопиоидные анальгетики.
Например, если второе (неопиоидное) лекарственное средство включено в лекарственную форму, такое лекарственное средство может включаться в форму с контролируемым высвобождением или в форму с немедленным высвобождением.Дополнительное лекарственное средство может включаться в состав матрикса для контролируемого высвобождения вместе с опиоидом, включаться в состав покрытия для контролируемого высвобождения, включаться в виде отдельного слоя для контролируемого высвобождения или слоя для немедленного высвобождения, или может включаться в виде порошка, гранул и т.д., в состав желатиновой капсулы с субстратами по настоящему изобретению.
В некоторых предпочтительных вариантax настоящего изобретения эффективное количество гидрокодона в форме с немедленным высвобождением включено в стандартную лекарственную форму гидрокодона с контролируемым высвобождением, подлежащую введению. Препаративная форма гидрокодона с немедленным высвобождением включается в количестве, которое является эффективным для уменьшения времени достижения Сmax гидрокодона в крови (например, в плазме). В таких вариантах эффективное количество гидрокодона в форме с немедленным высвобождением может покрывать субстраты по настоящему изобретению. Например, там, где длительное высвобождение гидрокодона из препаративной формы является следствием наличия покрытия для контролируемого высвобождения, слой для немедленного высвобождения может наноситься поверх покрытия для контролируемого высвобождения. С другой стороны, слой для немедленного высвобождения может наноситься на поверхность субстратов, в которых опиоид включен в матрикс для контролируемого высвобождения. Там, где множество субстратов для длительного высвобождения, включающих эффективную стандартную дозу гидрокодона (например, системы множества микрочастиц, включающие шарики, сферы, гранулы и тому подобное), входит в состав твердой желатиновой капсулы, часть дозы опиоида для немедленного высвобождения может помещаться в состав желатиновой капсулы путем включения в капсулу достаточного количества гидрокодона с немедленным высвобождением в виде порошка или гранул. Альтернативно желатиновая капсула сама может быть покрыта слоем гидрокодона с немедленным высвобождением. Специалисту в данной области известны и другие, альтернативные способы включения части гидроморфона с немедленным высвобождением в состав стандартной дозы. Такие альтернативы считаются охваченными прилагаемой формулой изобретения. Обнаружено, что путем включения такого эффективного количества гидрокодона с немедленным высвобождением в отдельную дозу значительно снижаются относительно сильные боли, которые испытывает пациент.
ЛЕКАРСТВЕННЫЕ ФОРМЫ
Лекарственные формы с контролируемым высвобождением могут необязательно включать вещество для контролируемого высвобождения, которое входит в состав матрикса вместе с гидрокодоном или наносится в виде покрытия для длительного высвобождения на субстрате, включающем лекарственное вещество (термин “субстрат” охватывает шарики, гранулы, сфероиды, таблетки, ядра таблеток и т.д.). Вещество для контролируемого высвобождения может быть гидрофобным или гидрофильным. Пероральные лекарственные формы по изобретению могут предоставляться, например, в виде гранул, сфероидов, шариков (здесь и далее обобщенно обозначаемых как “мультимикрочастицы”; multiparticulates). Количество мультимикрочастиц, эффективное для получения требуемой дозы опиоида за период времени, может помещаться в капсулу или может входить в состав любой другой подходящей твердой лекарственной формы, например спрессовано в таблетку. С другой стороны, пероральная лекарственная форма по настоящему изобретению может быть получена в виде ядра таблетки, окруженной покрытием для контролируемого высвобождения, или в виде таблетки, включающей матрикс лекарственного вещества, вещество для контролируемого высвобождения и необязательно другие требуемые с точки зрения фармацевтики ингредиенты (например, растворители, связующие вещества, красители, смазки и т.д.).
ПРЕПАРАТИВНЫЕ ФОРМЫ С МАТРИКСОМ ДЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ
В некоторых предпочтительных вариантax настоящего изобретения препаративные формы с контролируемым высвобождением получаются путем применения матрикса (например, таблеток матрикса), который включает вещество для контролируемого высвобождения, как описано выше. Лекарственная форма, включающая матрикс, обеспечивающий скорости растворения опиоида in vitro, лежащие в предпочтительных пределах, и она высвобождает опиоид по рН-зависимому или рН-независимому типу. Вещества, подходящие для включения в состав матрикса для контролируемого высвобождения, зависят от способа, используемого для получения матрикса. Пероральная лекарственная форма может содержать от 1 до 80% (по массе) по крайней мере одного гидрофильного или гидрофобного вещества для контролируемого высвобождения.
Неограничивающий список подходящих веществ для контролируемого высвобождения, которые могут входить в состав матрикса для контролируемого высвобождения по изобретению, включает гидрофильные и/или гидрофобные вещества, такие как резины, простые эфиры целлюлозы, акриловые смолы, вещества белкового происхождения, воски, шеллак и масла, такие как гидрированное касторовое масло, гидрированное растительное масло. Однако по настоящему изобретению может использоваться любое фармацевтически приемлемое гидрофобное или гидрофильное вещество, способное обеспечивать контролируемое высвобождение опиоида. Предпочтительные полимеры для контролируемого высвобождения включают алкилцеллюлозы, такие как этилцеллюлоза, полимеры и сополимеры акриловой и метакриловой кислот, и простые эфиры целлюлозы, особенно гидроксиалкилцеллюлозы (особенно гидроксипропилметилцеллюлоза) и карбоксиалкилцеллюлозы. Предпочтительные полимеры и сополимеры акриловой и метакриловой кислот включают метилметакрилат, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, сополимер аминоалкилметакрилата, поли(акриловую кислоту), поли(метакриловую кислоту), сополимер метакриловой кислоты и алкиламина, полиметилметакрилат, поли(метакриловая кислота) (ангидрид), полиметакрилат, полиакриламид, поли(ангидрид метакриловой кислоты) и сополимеры глицидилметакрилата. В некоторых предпочтительных вариантах в матриксах по изобретению применяются смеси любых из вышеуказанных веществ для контролируемого высвобождения.
Матрикс также может включать связующее вещество. В таких вариантax связующее вещество способствует контролируемому высвобождению гидрокодона из матрикса для контролируемого высвобождения.
Предпочтительные гидрофобные связующие вещества являются нерастворимыми в воде с более или менее выраженными тенденциями к гидрофильности и/или гидрофобности. Предпочтительно гидрофобные связующие вещества, которые могут применяться по изобретению, характеризуются температурой плавления примерно от 30 до 200°С, предпочтительно примерно от 45 до 90°С. Когда гидрофобное вещество представляет углеводород, углеводород предпочтительно характеризуется температурой плавления от 25 до 90°С. Из длинноцепочечных углеводородных (С8-С50) продуктов предпочтительными являются жирные (алифатические) спирты. Пероральная лекарственная форма может содержать свыше 80% (по массе) по крайней мере одного перевариваемого длинноцепочечного углеводорода.
Предпочтительно пероральная лекарственная форма содержит до 80% (по массе) по крайней мере одного полиалкиленгликоля. Конкретно гидрофобное связующее вещество может включать натуральные или синтетические воски, жирные спирты (такие как лауриловый, миристиловый, стеариловый, цетиловый или предпочтительно цетостеариловый спирт), жирные кислоты, без ограничения включая сложные эфиры жирных кислот, глицериды жирных кислот (моно-, ди- и триглицериды), гидрированные жиры, углеводороды, нормальные воски, стеариновую кислоту, стеариловый спирт, и гидрофобные и гидрофильные вещества, обладающие углеводородным скелетом. Подходящие воски включают, например, пчелиный воск, гликовоск, касторовый воск и воск карнаубы. Для целей настоящего изобретения воскоподобное вещество определяется как любое вещество, которое при комнатной температуре обычно является твердым и обладает температурой плавления примерно от 30 до 100°С.
Предпочтительные гидрофобные связующие вещества, которые могут использоваться по настоящему изобретению, включают перевариваемые длинноцепочечные (C8-C50, предпочтительно С12-С40) замещенные или незамещенные углеводороды, такие как жирные кислоты, жирные спирты, глицериловые сложные эфиры жирных кислот, минеральные и растительные масла, натуральные и синтетические воски и полиалкиленгликоли. Предпочтительными являются углеводороды, характеризующиеся температурой плавления между 25 и 90°С. Из длинноцепочечных углеводородных связующих веществ предпочтительными в некоторых вариантax являются жирные (алифатические) спирты. Пероральная форма может содержать до 80% (по массе) по крайней мере одного перевариваемого длинноцепочечного углеводорода.
В некоторых предпочтительных изобретениях в матриксную композицию включают комбинацию двух или более гидрофобных связующих веществ. Если включают дополнительное связующее вещество, его предпочтительно выбирают из натуральных и синтетических восков, жирных кислот, жирных спиртов и их смесей. Примеры включают пчелиный воск, воск карнаубы, стеариновую кислоту и стеариловый спирт. Этот список не следует считать исключающим.
Один конкретный подходящий матрикс для контролируемого высвобождения включает по крайней мере одну водорастворимую гидроалкилцеллюлозу, по крайней мере один C12-С36-, предпочтительно С14-С22-алифатический спирт и необязательно по крайней мере один полиалкиленгликоль. Гидроксиалкилцеллюлоза предпочтительно представляет собой гидроксиалкилцеллюлозу с алкилом от C1 до С6, такую как гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза и особенно гидроксиэтилцеллюлоза. Количество по крайней мере одной гидроксиалкилцеллюлозы в настоящей пероральной лекарственной форме будет определяться, кроме прочего, требуемой точной скоростью высвобождения опиоида. Алифатический спирт может быть, например, лауриловым спиртом, миристиловым спиртом или стеариловым спиртом. Однако в особенно предпочтительных вариантах настоящей пероральной лекарственной формы по крайней мере один алифатический спирт представляет собой цетиловый спирт или цетостеариловый спирт. Количество по крайней мере одного алифатического спирта в настоящей пероральной лекарственной форме будет определяться, как указано выше, требуемой точной скоростью высвобождения опиоида. Оно также будет зависеть от того, присутствует или отсутствует в пероральной лекарственной форме по крайней мере один полиалкиленгликоль. В отсутствие по крайней мере одного полиалкиленгликоля пероральная лекарственная форма предпочтительно содержит от 20 до 50% (по маcсе.) алифатического спирта. Когда полиалкиленгликоль присутствует в пероральной лекарственной форме, совместная масса алифатического спирта и полиалкиленгликоля предпочтительно составляет от 20 до 50% (по маcсе.) от общей дозировки.
В одном предпочтительном осуществлении соотношение, например, по крайней мере одной гидроалкилцеллюлозы или акриловой смолы и по крайней мере одного алифатического спирта/полиалкиленгликоля в значительной степени определяет скорость высвобождения опиоида из препаративной формы. Предпочтительным является соотношение по крайней мере одной гидроалкилцеллюлозы и по крайней мере одного алифатического спирта/полиалкиленгликоля, лежащее между 1:2 и 1:4, причем особенно предпочтительным является соотношение между 1:3 и 1:4.
Полиалкиленгликоль может, например, представлять собой полипропиленгликоль или полиэтиленгликоль, который является предпочтительным. Предпочтительным является средняя молекулярная масса полиэтиленгликоля, лежащая между 1000 и 15000, особенно между 1500 и 12000.
Другой подходящий матрикс для контролируемого высвобождения включает алкилцеллюлозу (особенно, этилцеллюлозу), С12-С36-алифатический спирт и необязательно полиалкиленгликоль.
В дополнение к вышеуказанным ингредиентам матрикс для контролируемого высвобождения может также содержать подходящие количества других веществ, например растворителей, смазывающих веществ, связующих веществ, средств, способствующих гранулированию, красителей, вкусовых добавок и скользящих веществ, которые являются общепринятыми в области фармацевтики.
Для того чтобы облегчить получение твердой пероральной лекарственной формы с контролируемым высвобождением по настоящему изобретению, в дальнейшем аспекте настоящего изобретения описан способ получения твердой пероральной лекарственной формы с контролируемым высвобождением по настоящему изобретению, включающий введение опиоидов или их солей в матрикс для контролируемого высвобождения.
Введение в матрикс может выполняться, например, путем
(a) формирования гранул, включающих по крайней мере одно гидрофобное или гидрофильное вещество, как описано выше (например, водорастворимую гидроксиалкилцеллюлозу), вместе с гидрокодоном;
(b) смешивания гранул, содержащих по крайней мере одно гидрофобное и/или гидрофильное вещество, по крайней мере с одним C12-С36 алифатическим спиртом и
(c) необязательно прессования и придания формы гранулам.
Гранулы могут формироваться любым из способов, хорошо известных специалистам в области получения фармацевтических препаратов. Например, по одному из предпочтительных способов гранулы могут формироваться путем влажного гранулирования водой гидроалкилцеллюлозы/опиоида. В наиболее предпочтительном варианте данного способа количество воды, добавляемой во время стадии влажного гранулирования, предпочтительно в 1,5-5 раз больше, особенно в 1,75-3,5 раз больше сухой массы опиоида.
Матриксы по настоящему изобретению могут также быть получены способом гранулирования расплава. В таком случае опиоид в хорошо измельченной форме объединяют со связующим веществом (также в форме частиц) и другими необязательными инертными ингредиентами и после этого смесь измельчают, например, путем механической обработки в интенсивно смешивающем устройстве для образования частиц (гранул, сфер). После этого частицы (гранулы, сферы) могут просеиваться через сито для получения частиц нужного размера. Связующее вещество предпочтительно находится в форме частиц и обладает температурой плавления, большей чем примерно 40°С. Подходящие связующие вещества включают, например, гидрированное касторовое масло, гидрированное растительное масло, другие гидрированные жиры, жирные спирты, сложные эфиры жирных кислот, глицериды жирных кислот и тому подобное.
Матриксы для контролируемого высвобождения могут также быть получены, например, способами гранулирования расплава или экструзии расплава. В общем, способы гранулирования расплава включают плавление твердого в обычных условиях гидрофобного связующего вещества, например воска, и введение в него порошкообразного лекарственного средства. Для получения лекарственной формы с контролируемым высвобождением может быть необходимым введение гидрофобного вещества для контролируемого высвобождения, например этилцеллюлозы или нерастворимого в воде акрилового полимера, в расплавленное воскообразное гидрофобное связующее вещество. Примеры препаративных форм с контролируемым высвобождением, полученных способами гранулирования расплавлением, описаны, например, в патенте США №4861598, переданном правопреемнику настоящего изобретения и включенном в описание в качестве ссылки.
Дополнительное гидрофобное связующее вещество может включать одно или несколько нерастворимых в воде воскоподобных термопластических веществ, возможно, смешанных с одним или несколькими воскоподобными термопластическими веществами, менее гидрофобными, чем указанные воскоподобные термопластические вещества. Для достижения для контролируемого высвобождения индивидуальные воскоподобные вещества в композиции не должны по существу разрушаться и растворяться в желудочно-кишечных жидкостях во время начальных фаз высвобождения. Применимые нерастворимые в воде воскоподобные связующие вещества могут обладать водорастворимостью, меньшей чем примерно 1:5000 (маc./маc.).
В дополнение к вышеуказанным ингредиентам матрикс для контролируемого высвобождения может также содержать подходящие количествa других материалов, например растворителей, смазывающих веществ, связующих веществ, средств, способствующих гранулированию, красителей, корригентов и скользящих веществ, которые являются общепринятыми в области фармацевтики, в количествах примерно до 50% по массе от массы частицы, если требуется. Количества этих добавочных материалов будут достаточными для обеспечения требуемого эффекта требуемой фармацевтической формой.
Конкретные примеры фармацевтически приемлемых носителей и наполнителей, которые могут использоваться для составления пероральных лекарственных форм, описаны в Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986), включенной сюда в качестве ссылки.
Получение подходящих матриксов экструзии расплава по настоящему изобретению могут, например, включать стадии смешивания опиоидного анальгетика вместе с веществом для контролируемого высвобождения и предпочтительно со связующим веществом до получения гомогенной смеси. Затем гомогенную смесь нагревают до температуры, достаточной для того, чтобы по крайней мере размягчить смесь до степени, достаточной для ее экструзии. Затем полученную гомогенную смесь подвергают экструзии с использованием, например, экструдера с двумя шнеками, с образованием нитей. Продукт экструзии предпочтительно охлаждают и нарезают на мультимикрочастицы любыми способами, известными в данной области. Нити охлаждают и нарезают на мультимикрочастицы. Затем мультимикрочастицы разделяют на отдельные дозы. Продукт экструзии предпочтительно имеет диаметр примерно от 0,1 до 5 мм и обеспечивает контролируемое высвобождение терапевтически активного агента в течение периода времени примерно от 8 до 24 часов.
Необязательный способ для получения препаративных форм методом экструзии расплава по настоящему изобретению включает непосредственное отмеривание в экструдер гидрофобного вещества для контролируемого высвобождения, терапевтически активного средства и необязательного связующего вещества; нагревание гомогенной смеси; экструзию гомогенной смеси с формированием таким образом нитей; охлаждение нитей, содержащих гомогенную смесь; нарезание нитей на частицы размером примерно от 0,1 до 12 мм и разделение указанных частиц на отдельные дозы. В данном аспекте изобретения выполняется относительно продолжительная производственная процедура.
Пластификаторы, такие как те, что описаны выше, могут включаться в матриксы, полученные экструзией расплава. Пластификатор предпочтительно включается в количестве примерно от 0,1 до 30% по массе от матрикса. Другие фармацевтические наполнители, например тальк, моно- или полисахариды, красители, корригенты, смазки и тому подобные, могут включаться по требованию в матриксы для контролируемого высвобождения по настоящему изобретению. Включаемые количества зависят от того, какая требуемая характеристика должна быть достигнута.
Диаметр отверстия экструдера или выходного порта может настраиваться для изменения толщины нитей экструзии. Более того, выходная часть экструдера не должна быть круглой; она может быть продолговатой, прямоугольной и т.д. Система мультимикрочастиц экструзии расплава может, например, находиться в виде гранул, сфероидов и шариков в зависимости от отверстия экструдера. Для целей настоящего изобретения термины “мультимикрочастица(ы) экструзии расплава” и “система(ы) мультимикрочастиц экструзии расплава” и “частицы экструзии расплава” будут относиться к множеству единиц, предпочтительно в пределах сходных размеров и/или формы, и содержащих одно или нескольких активных средств и один или несколько наполнителей, предпочтительно включающих гидрофобное вещество для контролируемого высвобождения. Предпочтительно мультимикрочастицы экструзии расплава имеют длину в пределах примерно от 0,1 до 12 мм в длину и имеют диаметр примерно от 0,1 до 5 мм. В дополнение, следует понимать, что мультимикрочастицы экструзии расплава могут быть любой геометрической формы в пределах размеров, такой как, например, шарики, семена, гранулы и т.д. Альтернативно продукты экструзии могут просто нарезаться на требуемую длину и разделяться на отдельные дозы терапевтически активного вещества без необходимости этапа сферообразования.
В одном предпочтительном осуществлении получают пероральные лекарственные формы, которые включают эффективное количество мультимикрочастиц, полученных способом экструзии расплава внутри капсулы. Например, множество мультимикрочастиц экструзии расплава может помещаться в желатиновую капсулу в количестве, достаточном для обеспечения дозы для контролируемого высвобождения при приеме внутрь и контакте с желудочным соком.
В другом предпочтительном осуществлении подходящее количество мультимикрочастиц продукта экструзии спрессовано в пероральные таблетки при помощи общепринятого оборудования для таблетирования с использованием стандартных способов. Способы и композиции для изготовления таблеток (прессованных и штампованных), капсул (твердых и мягких желатиновых капсул) и пилюль также описаны в Remington’s Pharmaceutical Sciences (Arthur Osol, editor), 1553-1593 (1980), включенном в описание в качестве ссылки.
Еще в одном предпочтительном варианте из продукта экструзии могут формироваться таблетки, как указано в патенте США №4957681 (Klimesch et al.), описанном в дополнительных деталях выше и включенном в описание в качестве ссылки.
Необязательно матриксные системы мультимикрочастиц с контролируемым высвобождением могут покрываться покрытием для контролируемого высвобождения, таким как покрытия для контролируемого высвобождения, описанные выше, или ими может далее покрываться желатиновая капсула. Такие покрытия предпочтительно включают достаточное количество гидрофобного и/или гидрофильного вещества для контролируемого высвобождения с получением уровня прироста массы примерно от 2 до 25%, хотя покрытие может сильно зависеть, кроме других факторов, например, от физических свойств применяемого конкретного опиоидного анальгетика и требуемой скорости высвобождения.
Лекарственные формы по настоящему изобретению могут далее включать комбинации мультимикрочастиц, полученных способом экструзии расплава, содержащих один или более опиоидных анальгетиков. Более того, лекарственные формы могут также включать некоторое количество терапевтически активного средства немедленного высвобождения для быстрого терапевтического действия. Терапевтически активное средство немедленного высвобождения может включаться, например, в виде отдельных шариков в желатиновую капсулу или может, например, покрывать поверхность гранул или мультимикрочастиц экструзии расплава. Отдельные лекарственные формы по настоящему изобретению могут также содержать, например, комбинации гранул для контролируемого высвобождения и матриксных мультимикрочастиц для достижения требуемого эффекта.
Препаративные формы с контролируемым высвобождением по настоящему изобретению предпочтительно медленно высвобождают терапевтически активное средство при приеме внутрь и воздействии желудочного сока, а затем кишечного сока. Профиль для контролируемого высвобождения композиций экструзии расплава по изобретению может изменяться, например, путем вариации количества вещества для контролируемого высвобождения, путем вариации количества пластификатора по отношению к другим составляющим матрикса, гидрофобному материалу, путем включения дополнительных ингредиентов или наполнителей, путем изменения способа производства и т.д.
В других вариантax изобретения препаративные формы экструзии расплава получают без включения терапевтически активного средства, который добавляют после в продукт экструзии. Такие препаративные формы обычно содержат терапевтически активное средство, смешанное вместе с матриксным продуктом экструзии, и затем смесь подлежит таблетированию для получения препаративной формы медленного высвобождения. Такие препаративные формы могут быть удобны, например, в случаях, когда терапевтически активный агент, включенный в препаративную форму, чувствителен к температуре, требуемой для размягчения гидрофобного вещества и/или задерживающего вещества.
Типичные системы продукции для получения путем экструзии расплава, подходящие для применения по настоящему изобретению, включают подходящий двигатель экструдера, имеющий переменную скорость и контроль постоянства вращения, контроли “старт-стоп” и амперметр. В дополнение система продукции включает консоль контроля температуры, устройство охлаждения и температурные индикаторы по длине экструдера. В дополнение система продукции включает экструдер, такой как экструдер с двумя шнеками, который состоит из двух противовращающих сцепленных между собой шнеков, находящихся внутри цилиндра или барабана, имеющего на своем конце отверстие или матрицу. Загружаемые материалы вносятся через входную воронку и перемещаются через барабан с помощью шнеков, и с силой проталкиваются через матрицу с образованием нитей, которые затем передаются по типу длинной ленты конвейера для того, чтобы дать им охладиться и направить их в измельчитель или другое подходящее устройство для преобразования нитей экструзии в систему мультимикрочастиц. Измельчитель может состоять из валиков, фиксированного ножа, вращающегося резака и тому подобного. Подходящие инструменты и системы доступны от распространителей, таких как C.W.Brabender Instruments, Inc., South Hackensack, Нью-Джерси. Другие подходящие устройства известны обычным специалистам в данной области.
Дальнейший аспект изобретения относится к описанному выше получению мультимикрочастиц способом экструзии расплава, контролирующим количество воздуха, включаемое в продукт экструзии. При контроле количества воздуха, включаемого в продукт экструзии, было неожиданно обнаружено, что скорость высвобождения терапевтически активного средства, например, из мультимикрочастиц продукта экструзии может значительно изменяться. В некоторых вариантax было неожиданно обнаружено, что может также изменяться рН-зависимость продукта экструзии.
Таким образом, в дальнейшем аспекте изобретения продукт экструзии расплава получают способом, который по существу исключает воздух во время экструзионной фазы процесса. Это может осуществляться, например, с использованием экструдера Лейстрица (Leistritz), имеющего вакуумную приставку. Неожиданно было обнаружено, что мультимикрочастицы, полученные по изобретению с использованием экструдера Лейстрица в вакууме, представляют продукт экструзии расплава, обладающий другими физическими характеристиками. В частности, продукт экструзии, является по существу непористым, что видно при увеличении, например, с использованием сканирующего электронного микроскопа, который предоставляет ЭМС (электронные микрограммы сканирования). Вопреки общепринятому мнению, было обнаружено, что такие по существу непористые препаративные формы обеспечивают скорейшее высвобождение терапевтически активного средства по отношению к той же композиции, полученной без вакуума. ЭМС мультимикрочастиц, полученных с использованием экструдера в вакууме, оказываются очень однородными, и мультимикрочастицы имеют тенденцию быть более прочными, чем мультимикрочастицы, полученные без вакуума. Имеется наблюдение, что, по крайней мере в некоторых препаративных формах, применение экструзии в вакууме обеспечивает продукт экструзии в виде мультимикрочастиц, который в большей степени рН-зависим, чем соответствующая ему препаратквная форма, полученная без вакуума.
СПОСОБЫ ПОЛУЧЕНИЯ МАТРИКСНЫХ ГРАНУЛ
Лекарственные формы с контролируемым высвобождением по настоящему изобретению могут также быть получены в виде матриксных шариков. Матриксные шарики включают сферообразующее средство и гидрокодон.
Гидрокодон предпочтительно составляет примерно от 0,1 до 99% по массе от матриксного шарика. Предпочтительно гидрокодсн включен в количестве примерно от 0,1 до 50% по массе от матриксного шарика.
Сферообразующие средства, которые могут использоваться для получения матриксных шариков по настоящему изобретению, включают любое известное в данной области сферообразующее средство. Предпочтительными являются производные целлюлозы, а особенно предпочтительной является микрокристаллическая целлюлоза. Подходящая микрокристаллическая целлюлоза представляет собой, например, вещество, которое продается как Avicel РН 101 (торговая марка, FMC Corporation). Сферообразующее средство предпочтительно включается в количестве примерно от 1 до 99% по массе от матриксного шарика.
В дополнение к активному ингредиенту и сферообразующему средству сфероиды также могут содержать связующее вещество. Подходящие связующие вещества, такие как водорастворимые полимеры с низкой вязкостью, хорошо известны специалистам в области фармацевтики. Однако предпочтительными являются водорастворимые низкогидроксилированные алкилцеллюлозы, такие как гидроксипропилцеллюлоза.
В дополнение к опиоидному анальгетику и сферообразующему средству препаративные формы матриксных шариков по настоящему изобретению могут включать вещество для контролируемого высвобождения, такое как те, что описаны здесь выше. Предпочтительными веществами для контролируемого высвобождения для включения в препаративные формы матриксных шариков включают полимеры или сополимеры акриловой и метакриловой кислот и этилцеллюлозу. При его наличии в препаративной форме вещество для контролируемого высвобождения включается в количестве примерно от 1 до 80% по массе от массы матриксного шарика. Вещество для контролируемого высвобождения предпочтительно включается в композицию матриксных шариков в количестве, эффективном для обеспечения для контролируемого высвобождения опиоидного анальгетика из шарика.
В препаративные формы матриксных шариков могут быть включены средства, облегчающие фармацевтическую обработку, такие как связующие вещества, растворители и тому подобные. Количества этих средств, включенных в препаративную форму, изменяются в зависимости от требуемого действия, которое должна оказывать препаративная форма.
На матриксные шарики может быть нанесено покрытие для контролируемого высвобождения, включающее вещество для контролируемого высвобождения, такое как те, что описаны выше. Покрытие для контролируемого высвобождения применяется в количестве, составляющем прирост по массе примерно от 5 до 30%. Количество покрытия для контролируемого высвобождения, подлежащего применению, варьирует в зависимости от разных факторов, например композиции матриксных шариков и химических и/или физических свойств опиоидного анальгетика (т.е. гидрокодона).
Матриксные гранулы в основном получают гранулированием сферообразующего средства вместе с опиоидным анальгетиком, например, путем влажного гранулирования. Затем продукт гранулирования подвергается сферообразованию с получением матриксных шариков. Затем матриксные шарики необязательно покрывают покрытием для контролируемого высвобождения способами, такими как те, что описаны выше.
Другой способ получения матриксных гранул осуществляется, например, путем (а) формирования гранул, включающих по крайней мере одну гидроксиалкилцеллюлозу и опиоид или соль опиоида; (b) смешивания гранул, содержащих гидроксиалкилцеллюлозу, с по крайней мере одним С12-С36 -алифатическим спиртом и (с) необязательно компрессии гранул и придания им формы. Предпочтительно гранулы образуют путем влажного гранулирования гидроксиалкилцеллюлозы/опиоида водой. По особо предпочтительному осуществлению данного способа количество воды, добавляемой во время стадии влажного гранулирования, предпочтительно в 1,5-5 раз больше, особенно в 1,75-3,5 раз больше, сухой массы опиоида.
В других альтернативных вариантax средство для сферообразования вместе с активным ингредиентом может подвергаться сферообразованию с формированием сфероидов. Предпочтительной является микрокристаллическая целлюлоза. Подходящая микрокристаллическая целлюлозы представляет собой, например, вещество, которое продается как Avicel PH 101 (торговая марка, FMC Corporation). В таких вариантax, в дополнение к активному ингредиенту и сферообразующему средству, сфероиды также могут содержать связующее вещество. Подходящие связующие вещества, такие как водорастворимые полимеры с низкой вязкостью, хорошо известны специалистам в области фармацевтики. Однако предпочтительными являются водорастворимые низкогидроксилированные алкилцеллюлозы, такие как гидроксипропилцеллюлоза. Дополнительно (или альтернативно) сфероиды могут содержать нерастворимый в воде полимер, особенно акриловый полимер, акриловый сополимер, такой как сополимер метакриловая кислота-этилакрилат, или этилцеллюлозу. В таких вариантax покрытие длительного высвобождения будет в основном включать нерастворимое в воде вещество, такое как (а) воск, один или в смеси с жирным спиртом, или (b) шеллак или зеин.
ПРЕПАРАТИВНЫЕ ФОРМЫ ШАРИКОВ С КОНТРОЛИРУЕМЫМ ВЫСВОБОЖДЕНИЕМ
В одном особенно предпочтительном осуществлении пероральная лекарственная форма включает эффективное количество сфероидов для контролируемого высвобождения, содержащееся внутри желатиновой капсулы.
В другом предпочтительном осуществлении настоящего изобретения лекарственная форма с контролируемым высвобождением включает активный ингредиент, покрытый покрытием для контролируемого высвобождения, включающим вещество для контролируемого высвобождения. Термин “сфероид” известен в области фармацевтики и означает, например, сферическую гранулу, имеющую диаметр между 0,1 и 2,5 мм, особенно между 0, 5 и 2 мм.
Сфероиды предпочтительно покрыты пленкой с веществом для контролируемого высвобождения, которая обеспечивает высвобождение опиоида (или соли) с контролируемой скоростью в водную среду. Пленочное покрытие выбрано так, что, при прочих постоянных свойствах, достигается скорость высвобождения in vitro, указанная выше (например, по крайней мере около 12,5% высвобождения за 1 час). Препаративные формы с контролируемым высвобождением по настоящему изобретению предпочтительно продуцируют прочную сплошную пленку, которая является гладкой и элегантной, способной удерживать пигменты и другие добавки покрытия, нетоксичной, инертной и неклейкой.
ПОКРЫТИЯ
Лекарственные формы по настоящему изобретению могут быть необязательно покрыты одним или несколькими покрытиями, подходящими для регуляции высвобождения и для защиты препарата. В одном варианте имеются покрытия, обеспечивающие или рН-зависимое, или рН-независимое высвобождение, например, под воздействием желудочно-кишечной жидкости. Когда требуется рН-независимое покрытие, наносится покрытие для достижения оптимального высвобождения независимо от изменений рН в окружающей жидкости, т.е. в ЖК тракте. Другие предпочтительные варианты включают рН-зависимое покрытие, которое высвобождает опиоид в требуемых областях желудочно-кишечного (ЖК) тракта, например в желудке или тонком кишечнике, так что имеет место профиль всасывания, способный обеспечить у пациента по крайней мере около двенадцати часов и предпочтительно до двадцати четырех часов анальгезии. Также возможно составить препаративные формы, которые высвобождают часть дозы в требуемой области ЖК тракта, например в желудке, а оставшуюся часть дозы высвобождают в другой области ЖК тракта, например в тонком кишечнике.
Препаративные формы по изобретению, в которых нанесено рН-зависимое покрытие, могут также иметь эффект повторного действия, когда незащищенное лекарственное средство наносится поверх внутреннего покрытия и высвобождается в желудке, тогда как остаток, будучи защищенным внутренним покрытием, высвобождается в желудочно-кишечном тракте ниже. рН-зависимые покрытия, которые могут использоваться по настоящему изобретению, включают вещество для контролируемого высвобождения, такое как, например, шеллак, фталат ацетата целлюлозы (CAP), фталат поливинилацетата (PVAP), фталат гидроксипропилметилцеллюлозы и сложноэфирные сополимеры акриловой кислоты, зеин и тому подобное.
В другом предпочтительном осуществлении настоящее изобретение относится к стабилизированным твердым контролируемым дозированным формам, включающим опиоид, покрытый гидрофобным веществом для контролируемого высвобождения, выбранным из (i) алкилцеллюлозы; (ii) акрилового полимера или (iii) их смесей. Покрытие может применяться в форме органического или водного раствора или дисперсии.
В некоторых предпочтительных вариантax покрытие для контролируемого высвобождения производится из водной дисперсии гидрофобного вещества для контролируемого высвобождения. Покрытый субстрат, содержащий опиоид(ы) (например, ядро таблетки или инертные фармацевтические гранулы или сфероиды), затем отверждают до достижения конечной точки, при которой субстрат характеризуется стабильным растворением. Точку конца отверждения можно определить путем сравнения профиля (кривой) растворения лекарственной формы немедленно после отверждения с профилем (кривой) растворения лекарственной формы после воздействия условий ускоренного хранения, например, по крайней мере в течение одного месяца при температуре 40°С и относительной влажности 75%. Данные композиции в деталях описаны в патентax США №5273760 и 5286493, переданных правопреемнику настоящего изобретения и включенных в описание в качестве ссылки. Другие примеры препаративных форм с контролируемым высвобождением и покрытий, которые могут использоваться по настоящему изобретению, включают патенты США №5324351, 5356467 и 5472712, принадлежащие правопреемнику настоящего изобретения, включенные в описание в качестве ссылки в своей целостности.
В предпочтительных вариантax покрытия для контролируемого высвобождения включают пластификатор, такой как те, что описаны здесь ниже.
В некоторых вариантax имеется необходимость покрывать субстрат, включающий опиоидный анальгетик, достаточным количеством водной дисперсии, например, алкилцеллюлозы или акрилового полимера, для получения уровня прироста массы примерно от 2 до 50%, например примерно от 2 до 25%, чтобы получить композицию с контролируемым высвобождением. Покрытие может в большей или меньшей степени зависеть от физических свойств терапевтически активного средства и требуемой скорости высвобождения, включения в водную дисперсию пластификатора и, например, способа его включения.
ПОЛИМЕРЫ АЛКИЛЦЕЛЛЮЛОЗЫ
Целлюлозные материалы и полимеры, включая алкилцеллюлозу, представляют собой вещества для контролируемого высвобождения, хорошо подходящие для покрытия субстратов, например шариков, таблеток и т.д., по изобретению. Например, один из предпочтительных алкилцеллюлозных полимеров представляет этилцеллютюзу, хотя специалист в данной области поймет, что другие полимеры целлюлозы и/или алкилцеллюлозы могут быть использованы, сами по себе или в любой комбинации, в качестве всего или части гидрофобного покрытия по изобретению.
Одной коммерчески доступной водной дисперсией этилцеллюлозы является Aquacoat® (FMC Corp., Филадельфия, Пенсильвания, США). Aquacoat® получают путем растворения этилцеллюлозы в несмешивающемся с водой органическом растворителе и последующего эмульгирования его в воде в присутствии поверхностно-активного вещества и стабилизатора. После гомогенизации с получением субмикронных капель органический растворитель выпаривают в вакууме с образованием псевдолатекса. Во время производственной фазы в псевдолатекс не добавляют пластификатор. Таким образом, перед его использованием в качестве покрытия, Aquacoat® необходимо смешать в своей лаборатории с подходящим пластификатором.
Другая водная дисперсия этилцеллюлозы коммерчески доступна как Surelease® (Colorcon, Inc., Уэст-Пойнт, Пенсильвания, США). Продукт получают путем введения пластификатора в дисперсию во время производственного процесса. Горячий расплав полимера, пластификатор (дибутилсебакат) и стабилизатор (олеиновая кислота) получают в виде гомогенной смеси, которую затем разбавляют раствором щелочи для получения водной дисперсии, которая может непосредственно применяться в отношении субстратов.
АКРИЛОВЫЕ ПОЛИМЕРЫ
В других предпочтительных вариантах настоящего изобретения вещество для контролируемого высвобождения, включающее покрытие для контролируемого высвобождения, представляет собой фармацевтически приемлемый акриловый полимер, включая в качестве неограничивающих примеров сополимеры акриловой кислоты и метакриловой кислоты, сополимеры метилметакрилата, этоксиэтилметакрилаты, цианоэтилметакрилат, поли(акриловую кислоту), поли(метакриловую кислоту), сополимер метакриловой кислоты и алкиламина, полиметилметакрилат, полиметакрилат, сополимер полиметилметакрилата, полиакриламид, сополимер аминоалкилметакрилата, поли(ангидрид метакриловой кислоты) и сополимеры глицидилметакрилата.
В некоторых предпочтительных вариантax акриловый полимер включает один или несколько сополимеров аминометакрилата. Сополимеры аминометакрилата хорошо известны в данной области и описаны в NF XVII как полностью полимеризованные сополимеры сложных эфиров акриловой и метакриловой кислот с низким содержанием четвертичных аминогрупп.
Для получения требуемого профиля растворения может быть необходимым включение двух или более сополимеров аминометакрилата, обладающих разными физическими свойствами, такими как разные молярные соотношения четвертичных аминогрупп и нейтральных (мет)акриловых сложных эфиров.
Некоторые полимеры типа сложных эфиров метакриловой кислоты могут использоваться для получения рН-зависимых покрытий, применимых по настоящему изобретению. Например, имеется семейство сополимеров, синтезированных из диэтиламиноэтилметакрилата и других нейтральных метакриловых сложных эфиров, также известных как сополимеры метакриловой кислоты или полимерные метакрилаты, коммерчески доступных как Eudragit® от

В некоторых предпочтительных вариантах акриловое покрытие включает смесь двух лаков на основе акриловых смол, коммерчески доступных от Rohm Pharma под торговыми названиями Eudragit RL30D и Eudragit RS30D соответственно. Eudragit RL30D и Eudragit RS30D представляют собой сополимеры акриловых и метакриловых сложных эфиров с низким содержанием четвертичных аминогрупп, причем молярное соотношение аминогрупп и оставшихся нейтральных (мет)акриловых сложных эфиров составляет 1:20 в Eudragit RL30D и 1:40 в Eudragit RS30D. Средняя молекулярная масса равна примерно 150000. Кодовые обозначения RL (высокая проницаемость) и RS (низкая проницаемость) относятся к свойствам проницаемости данных средств. Смеси Eudragit® RL/RS нерастворимы в воде и пищеварительных соках.
Дисперсии Eudragit® RL/RS по настоящему изобретению могут смешиваться вместе в любом требуемом соотношении для получения в конечном счете препаративной формы с контролируемым высвобождением, характеризующейся требуемым профилем растворения. Требуемые композиции с контролируемым высвобождением могут быть получены, в частности, из задерживающего покрытия, полученного из 100% Eudragit® RL, 50% Eudragit® RL : 50% Eudragit® RS и 10% Eudragit® RL: 90% Eudragit® RS. Специалист в данной области, конечно, поймет, что также могут использоваться другие акриловые полимеры, такие как, например, Eudragit® L.
ПЛАСТИФИКАТОРЫ
В вариантax настоящего изобретения, где покрытие включает водную дисперсию гидрофобного вещества для контролируемого высвобождения, включение эффективного количества пластификатора в водной дисперсии еще больше улучшит физические свойства покрытия для контролируемого высвобождения. Поскольку, например, этилцеллюлоза характеризуется относительно высокой температурой перехода в стекловидное состояние и не образует гибких пленок при нормальных условиях покрытия, является предпочтительным включать пластификатор в этилцеллюлозные покрытия, содержащие покрытия для контролируемого высвобождения, перед их применением в качестве покрывающего материала. Как правило, количество пластификатора, включаемое в раствор покрытия, основывается на концентрации пленкообразователя, например, чаще всего примерно от 1 до 50% по массе от массы пленкообразователя. Однако правильно определить концентрацию пластификатора можно только после тщательного экспериментирования с конкретным раствором покрытия и способом применения.
Примеры подходящих пластификаторов для этилцеллюлозы включают нерастворимые в воде пластификаторы, такие как дибутилсебакат, диэтилфталат, триэтилцитрат, трибутилцитрат и триацетин, хотя возможно, что могут применяться другие нерастворимые в воде пластификаторы (такие как ацетилированные моноглицериды, сложные эфиры фталата, касторовое масло и т.д.). Особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы по настоящему изобретению является триэтилцитрат.
Примеры подходящих пластификаторов для акриловых полимеров по настоящему изобретению включают в качестве неограничивающих примеров сложные эфиры лимонной кислоты, такие как метилцитрат NF XVI, трибутилцитрат, дибутилфталат и, возможно, 1,2-пропиленгликоль. Другие пластификаторы, которые оказались подходящими для повышения эластичности пленок, образованных акриловыми пленками, такими как лаковые растворы Eudragit® RL/RS, включают полиэтиленгликоли, пропиленгликоль, диэтилфталат, касторовое масло и триацетин. Особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы по настоящему изобретению является триэтилцитрат.
Далее было обнаружено, что добавление к покрытию для контролируемого высвобождения малого количества талька снижает склонность водной дисперсии к прилипанию во время обработки, и тальк действует в качестве полирующего средства.
ПОЛУЧЕНИЕ ПРЕПАРАТИВНЫХ ФОРМ В ВИДЕ ШАРИКОВ С ПОКРЫТИЕМ
Когда водная дисперсия гидрофобного вещества применяется для покрытия субстратов, например, инертных фармацевтических гранул, таких как шарики nu pariel 18/20, множество полученных стабилизированных твердых шариков для контролируемого высвобождения может после этого помещаться в желатиновую капсулу в количестве, достаточном для обеспечения эффективной дозы с контролируемым высвобождением при приеме внутрь и контакте с окружающей жидкостью, например с желудочным соком в качестве среды растворения.
Стабилизированные композиции шариков для контролируемого высвобождения по настоящему изобретению медленно высвобождают опиоидный анальгетик, например, при приеме внутрь и воздействии желудочных соков и затем кишечных соков. Профиль для контролируемого высвобождения композиций по изобретению может изменяться, например, путем вариации количества покрытия водной дисперсией гидрофобного вещества для контролируемого высвобождения, путем изменения способа, по которому пластификатор добавляют к водной дисперсии гидрофобного вещества для контролируемого высвобождения, путем вариации количества пластификатора по отношению к количеству гидрофобного вещества для контролируемого высвобождения, путем включения дополнительных ингредиентов или наполнителей, путем изменения способа производства и т.д. Профиль растворения конечного продукта может также изменяться, например, путем увеличения или уменьшения толщины покрытия для контролируемого высвобождения.
Субстраты, покрытые терапевтически активным средством, получают, например, путем растворения терапевтически активного средства в воде и последующего распыления раствора на субстрат, например на шарики nu pariel 18/20, с использованием вставки Вустера (Wuster insert). Необязательно дополнительные ингредиенты также добавляют перед покрытием шариков для того, чтобы способствовать связыванию опиоида с шариками и/или для окрашивания раствора и т.д. Например, продукт, который включает гидроксипропилметилцеллюлозу и т.д., с красителем (например, Opadry®, коммерчески доступный от Colorcon, Inc.) или без него, может добавляться к раствору и смешиваться с раствором (например, примерно в течение 1 часа) перед его применением по отношению к субстрату. Полученный субстрат с покрытием может необязательно покрываться сверху барьерным средством для отделения терапевтически активного средства от гидрофобного покрытия для контролируемого высвобождения.
Примером подходящего барьерного средства является средство, которое включает гидроксипропилметилцеллюлозу. Однако может применяться любой пленкообразователь, известный в данной области. Предпочтительно, чтобы барьерное средство не влияло на скорость растворения конечного продукта.
Затем субстраты могут быть покрыты сверху гидрофобным веществом для контролируемого высвобождения. Водная дисперсия гидрофобного вещества для контролируемого высвобождения, кроме того, предпочтительно включает эффективное количество пластификатора, например триэтилцитрата. Могут применяться предварительно полученные водные дисперсии этилцеллюлозы, такие как Aquacoat® и Shurelease®. При использовании Shurelease® нет необходимости отдельно добавлять пластификатор. Альтернативно могут применяться водные дисперсии акриловых полимеров, такие как Euragit®.
Покрывающие растворы по настоящему изобретению предпочтительно содержат, в дополнение к пленкообразователю, пластификатор и систему растворителя (т.е. воду), краситель для обеспечения элегантного вида и возможности отличить продукт. Краситель может добавляться к раствору терапевтически активного средства вместо или в дополнение к его добавлению в водную дисперсию гидрофобного вещества. Например, краситель может добавляться в Aquacoat® путем применения основанных на спирте или пропиленгликоле красящих дисперсий, молотых алюминиевых красящих лаков и белил, таких как диоксид титана, с добавлением красителя к раствору водорастворимого полимера с усилием сдвига и с применением затем малого усилия сдвига по отношению к пластифицированному Aquacoat®. Альтернативно может использоваться любой приемлемый способ окрашивания композиций по настоящему изобретению. Подходящие ингредиенты для окрашивания композиции при использовании водной дисперсии акрилового полимера включают диоксид титана и цветные пигменты, такие как пигменты оксидов железа. Включение пигментов может, однако, увеличить задерживающий эффект покрытия.
Пластифицированные водные дисперсии гидрофобного вещества для контролируемого высвобождения могут применяться по отношению к субстрату, включающему терапевтическое средство, путем распыления посредством любого подходящего оборудования для распыления, известного в данной области. По предпочтительному способу применяется система псевдоожиженного слоя Вурстера (Wurster fluidized-bed system), в которой струя воздуха, выбрасываемая снизу, псевдоожижает материал ядра и оказывает высушивающее действие во время распыления акрилового полимерного покрытия. С учетом физических характеристик терапевтически активного средства, способа включения пластификатора и т.д. предпочтительно применяется количество водной дисперсии гидрофобного вещества, достаточное для получения предварительно установленного для контролируемого высвобождения указанного терапевтически активного средства при воздействии на указанный покрытый субстрат водных растворов, например желудочного сока. После покрытия гидрофобным веществом для контролируемого высвобождения шарики необязательно подвергаются дальнейшему покрытию пленкообразователем, таким как Opadry®. Если данное покрытие и проводится, то для того чтобы по существу снизить агломерацию шариков.
Высвобождение терапевтически активного средства из препаративной формы с контролируемым высвобождением по настоящему изобретению может, кроме того, подвергаться воздействию, т.е. доводиться до требуемой скорости, путем добавления одного или нескольких средств, модулирующих высвобождение, или предусматривая наличие одного или нескольких проходов в покрытии. Соотношение гидрофобного вещества для контролируемого высвобождения и водорастворимого вещества определяется, помимо других факторов, требуемой скоростью высвобождения и характеристиками растворимости выбранных веществ.
Средства, модулирующие высвобождение, которые действуют как порообразователи, могут быть органическими и неорганическими, и включают вещества, которые могут растворяться, экстрагироваться или выщелачиваться из покрытия в среде применения. Порообразователи могут включать одно или более гидрофильных веществ, таких как гидроксипропилметилцеллюлоза.
Покрытия для контролируемого высвобождения по настоящему изобретению также могут включать средства, способствующие эрозии, такие как крахмал или смолы.
Покрытия для контролируемого высвобождения по настоящему изобретению также могут включать вещества, применимые для получения тонкого микропористого слоя в среде использования, такие как поликарбонаты, состоящие из линейных полиэфиров угольной кислоты, в которых карбонатные группы повторяются в виде полимерной цепи.
Средство, модулирующее высвобождение, также может включать полупроницаемый полимер. В некоторых предпочтительных вариантax средство, модулирующее высвобождение, выбрано из гидроксипропилметилцеллюлозы, лактозы, стеаратов металлов и смесей любых из вышеуказанных.
Покрытия для контролируемого высвобождения по настоящему изобретению также могут включать средства выхода, включающие по крайней мере один проход, отверстие или тому подобное. Проход может быть образован такими способами, описанными в патентax США №3845770; 3916889; 4063064 и 4088864, которые все включены в описание в качестве ссылки. Проход может иметь любую форму, как круглую, так и треугольную, квадратную, эллиптическую, неправильную и т.д.
Другой способ получения шариков с контролируемым высвобождением, подходящих примерно для 24-часового введения, состоит в наслаивании порошка. Патент США №5411745, переданный правопреемнику настоящего изобретения и включенный в описание в качестве ссылки в своей целостности, содержит руководство по приготовлению 24-часовой препаративной формы морфина, полученной способами наслаивания порошка, привлекающими средства обработки, состоящие по существу из мельчайших частиц гидрата лактозы. Шарики порошкового наслаивания получают путем распыления водного связывающего раствора на инертные шарики для создания липкой поверхности и последующего распыления на липкие шарики порошка, представляющего собой гомогенную смесь сульфата морфина и мельчайших частиц гидрата лактозы. Затем шарики сушат и покрывают гидрофобным веществом, таким как те, что описаны выше, для получения требуемого высвобождения лекарственного средства при воздействии на конечную композицию окружающих жидкостей. Подходящее количество шариков с контролируемым высвобождением затем, например, инкапсулируют для получения конечной лекарственной формы, которая обеспечивает эффективные концентрации морфина в плазме в течение 12 часов.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ
Последующие примеры иллюстрируют различные аспекты настоящего изобретения. Они никоим образом не подразумевают истолкования, ограничивающего формулу изобретения.
Пример 1
Таблетки с замедленным высвобождением гидрокодона получали по рецепту, указанному ниже в таблице I.

Таблетки получали по следующей процедуре.
1. Замедляющая дисперсия: смешивают Eudragit RS30D и триацетин с использованием миксера мгновенного смешивания.
2. Расплавляют стеариловый спирт.
3. Распыляют замедляющую высвобождение дисперсию на гидрокодона битартрат, высушенную распылением лактозу и повидон, используя гранулятор с псевдоожиженным слоем.
4. Сушат партию на противне из нержавеющей стали в течение 15 минут, или до постоянного веса.
5. Вносят в партию расплавленный стеариловый спирт, используя миксер Хобарта (Hobart).
6. Сушат вощеную гранулированную массу на противне из нержавеющей стали в течение 30 минут, или до достижения температуры массы, равной или менее 35°С.
7. Размалывают охлажденную гранулированную массу посредством CoMil.
8. Обрабатывают гранулированную массу в качестве смазки тальком и стеаратом магния, используя миксер Хобарта (Hobart).
9. Прессуют гранулированную массу в таблетки при помощи пресса для таблетирования.
Затем таблетки тестировали на предмет растворения, используя следующую процедуру.
1. Устройство: способ 1 по USP (корзина), 100 об/мин.
2. Среда: 700 мл ИЖС (SGF) в течение 55 мин, после чего 900 мл ИКС (SIF) без ферментов.
3. Время забора образцов: 1, 2, 4, 8 и 12 часов.
4. Анализ: высокоэффективная жидкостная хроматография. Параметры растворения приведены ниже в таблице II.

Затем определяли Cmaxи Tmax для примера 1 и для контрольного стандарта с немедленным высвобождением в исследовании биодоступности, сравнивающем действие на здоровых людей 15 мг гидрокодона, введенного в виде препаративной формы с немедленным высвобождением (Lortab 7,5 мг × 2), и вышеуказанной композиции с KB (контролируемое высвобождение), что приведено ниже в таблице III.

Пример 2
Таблетки с замедленным высвобождением гидрокодона получали по рецепту, указанному ниже в таблице IV, следуя процедуре примера 1.

Затем определяют параметры растворения, используя процедуру по примеру 1. Результаты приведены ниже в таблице V.

Пример 3
Капсулы с замедленным высвобождением гидрокодона получают по рецепту, указанному ниже в таблице VI.

Капсулы получают по следующей процедуре.
1. Смешивают молотые стеариловый спирт, Eudragit RLPO, гидрокодона битартрат и Eudragit RSPO с использованием миксера Хобарта (Hobart).
2. Гранулируют массу экструзии с использованием устройства подачи порошка, экструдера плавления (оборудованного матрицей 6×1 мм), конвейера, лазерного микрометра (lasermike) и измельчителя при следующих условиях:

Скорость подачи порошка 40 г/мин, скорость шнека 185 об/мин, вакуум 980 мбар.
Конвейер - соответствующий диаметру продукта экструзии, равному 1 мм.
Измельчитель - обеспечивающий нарезание частиц 1 мм длиной.
3. Просеивают частицы через сито №16 и №20. Собирают материал, прошедший через сито №16 и задержавшийся на сите №20.
4. Заполняют частицами пустые желатиновые капсулы №2. Разброс: NLT 114 мг и NMT 126 мг.
Параметры растворения определяли затем, используя процедуру по примеру 1. Результаты приведены ниже в таблице VII.

Пример 4
Таблетки с замедленным высвобождением оксикодона получали по рецепту, указанному ниже в таблице VIII.

Таблетки получают по следующей процедуре.
1. Гранулирование: распыляют дисперсию Eudragit/триацетин на оксикодон-HCl, высушенную распылением лактозу и повидон, используя гранулятор с подвижным дном.
2. Перемалывание: отбирают гранулированную массу и пропускают через мельницу.
3. Вощение: расплавляют стеариловый спирт и добавляют в молотую гранулированную массу с использованием миксера. Дают охладиться.
4. Перемалывание: пропускают охлажденную гранулированную массу через мельницу.
5. Смазка: обрабатывают гранулированную массу в качестве смазки тальком и стеаратом магния, используя миксер.
6. Прессовка: прессуют гранулированную массу в таблетки при помощи пресса таблетирования.
7. Покрытие пленкой: наносят водное пленочное покрытие на таблетки.
Затем таблетки тестируют на предмет растворения с использованием следующей процедуры.
1. Устройство: способ 2 по USP (лопасть), 150 об/мин.
2. Среда: 700 мл ИЖС (SGF) в течение первого часа, после чего давали 900 мл фосфатного буфера до рН 7,5.
3. Время забора образцов: 1, 2, 4, 8, 12, 18 и 24 часа.
4. Анализ: высокоэффективная жидкостная хроматография. Параметры растворения приведены ниже в таблице IX.

Затем определяют Cmax и Tmax для примера 4 и для контрольного стандарта с немедленным высвобождением в исследовании биодоступности, что приведено ниже в таблице X.

Пример 5
Таблетки с замедленным высвобождением морфина получали по рецепту, указанному ниже в таблице XI.

Таблетки получали по следующей процедуре.
1. Гранулирование: добавляют воду к сульфату морфина, высушенной распылением лактозе и гидроксиэтилцеллюлозе в миксере и высушивают, используя гранулятор с псевдоожиженным слоем.
2. Просеивание: отбирают гранулированную массу и пропускают через сито.
3. Вощение: расплавляют цетостеариловый спирт и добавляют в молотую гранулированную массу с использованием миксера. Дают охладиться.
4. Просеивание: отбирают гранулированную массу и пропускают через сито.
5. Смазка: обрабатывают гранулированную массу в качестве смазки тальком и стеаратом магния, используя миксер.
6. Прессовка: прессуют гранулированную массу в таблетки при помощи пресса таблетирования.
7. Покрытие пленкой: обрабатывают таблетки водным пленочным покрытием.
Затем таблетки тестируют на предмет растворения с использованием следующей процедуры.
1. Устройство: способ 1 по USP (корзина), 50 об/мин.
2. Среда: 900 мл чистой воды, 37°С.
3. Время забора образцов: 1, 2, 3, 4, и 6 часов.
4. Анализ: УФ-детекция, 285 нм и 305 нм, способ 2 точек с использованием 5-см ячейки.
Параметры растворения приведены ниже в таблице XII.

Затем определяют Cmax и Tmax для примера 5 и для контрольного стандарта с немедленным высвобождением в исследовании биодоступности, что приведено ниже в таблице XIII.

Пример 6
Фармакокинетические параметры по примеру 1, примеру 4 и примеру 5 сравнивали друг с другом. Неожиданно было обнаружено, что даже несмотря на то, что растворение таблеток с контролируемым высвобождением гидрокодона-НСl по примеру 1 было очень сходным с растворением таблеток с контролируемым высвобождением оксикодона по примеру 4 и таблеток с контролируемым высвобождением морфина по примеру 5, соотношение Cmax KB к таковой НВ для композиции гидрокодона составляет 38%, тогда как таблеткам оксикодона и таблеткам морфина соответствуют значения выше 50%. Результаты сравнения приведены в таблице XIV ниже.

Пример 7
Проводили фармакокинетическое сравнение действия разовой дозы препаративной формы гидрокодона с немедленным высвобождением по примеру 1, примеру 2, примеру 3 и двух таблеток гидрокодона битартрата (7,5 мг)/ацетаминофена (500 мг) с немедленным высвобождением (пример НВ) на здоровых добровольцах, натощак, при четырехкратном лечении с открытой этикеткой. Концентрации в плазме, соответствующие данным композициям, приведены в таблицах XV-XVIII ниже.


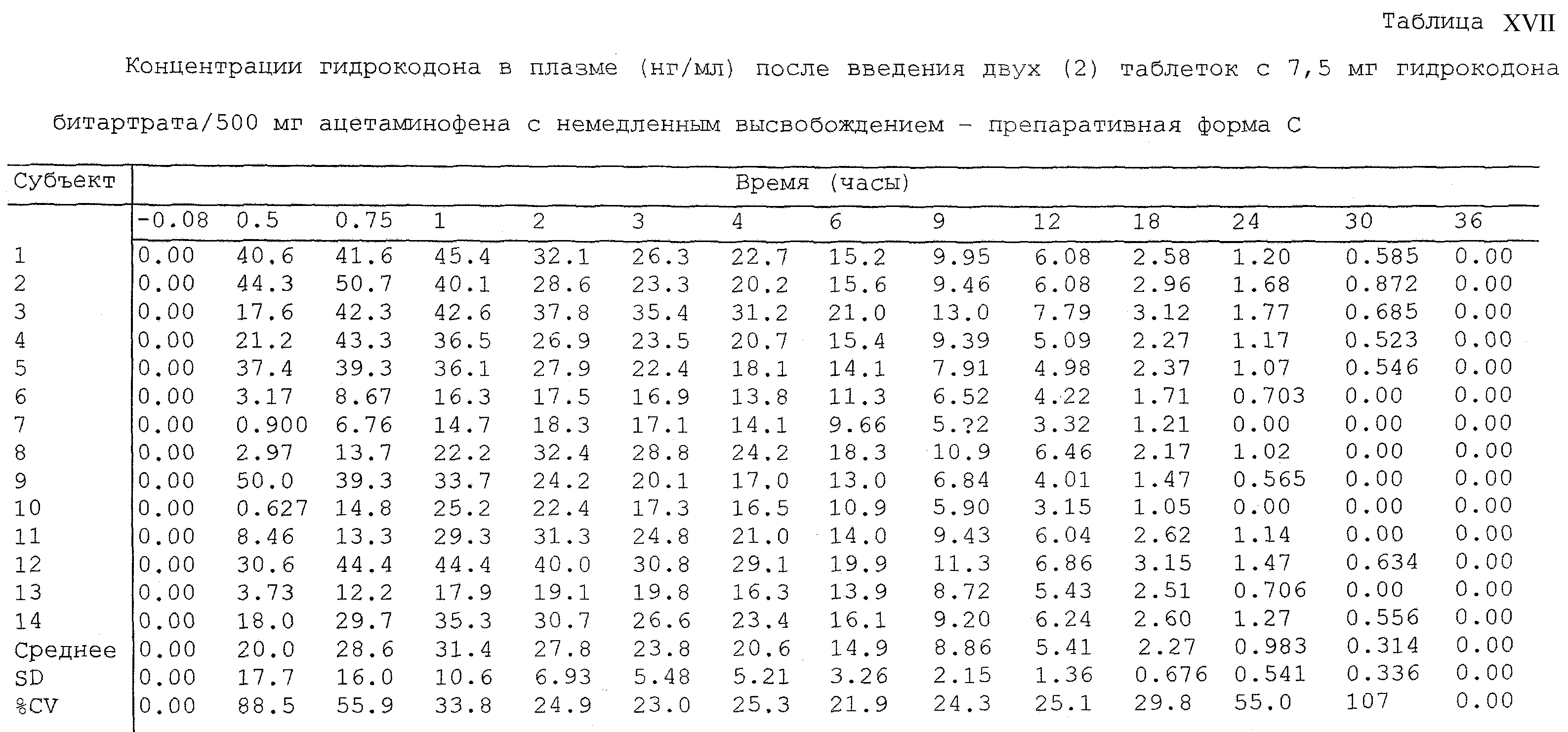

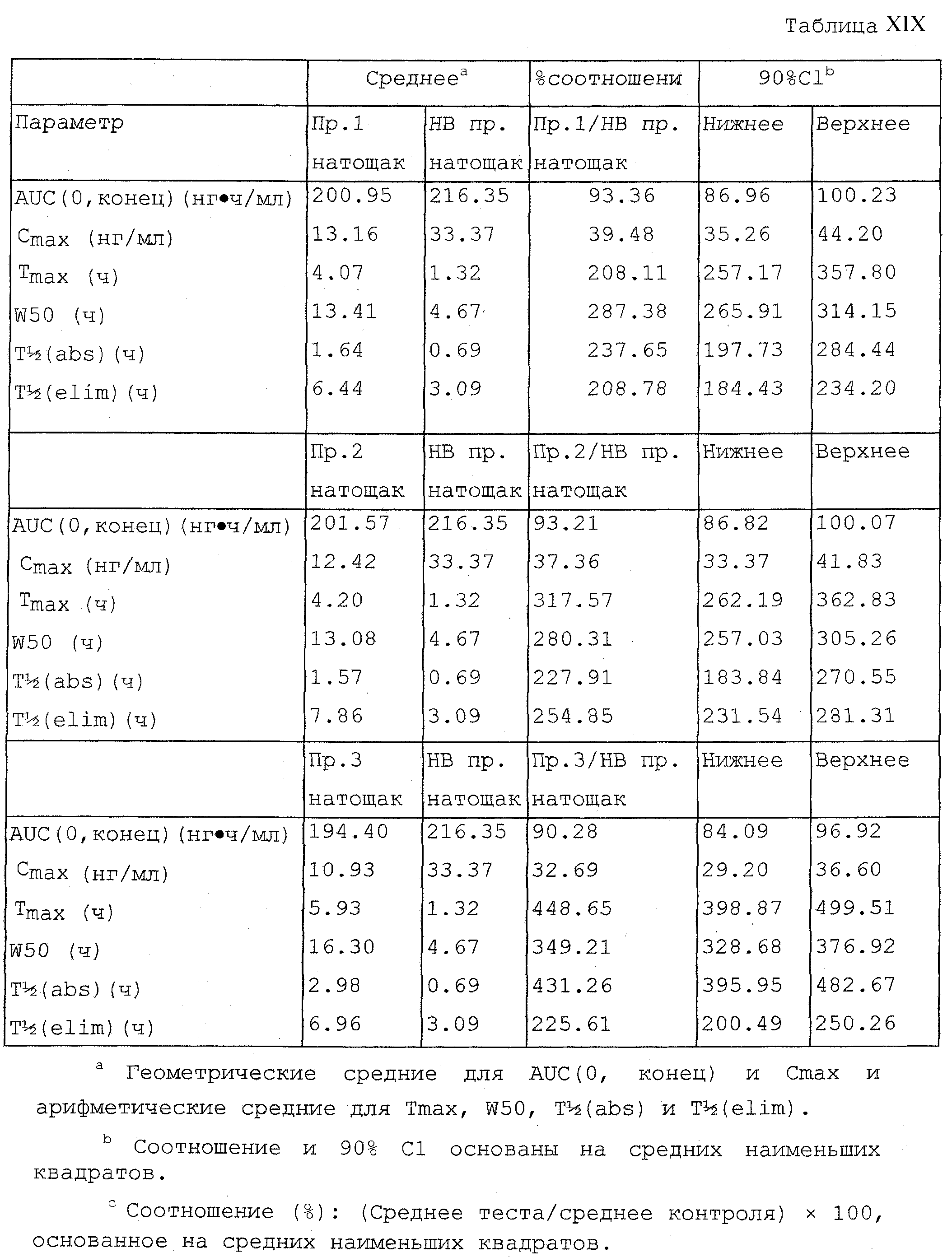
Пример 8
Таблетки с замедленным высвобождением гидрокодона получали по рецепту, указанному ниже в таблице XX.

Таблетки получали по следующей процедуре.
1. Перемалывание: пропускают хлопья стеарилового спирта через мельницу.
2. Смешивание: смешивают гидрокодона битаратрат, вторичный кислый фосфат кальция, глицерилбегенат и микрокристаллическую целлюлозу в подходящем смесителе.
3. Экструзия: постепенно загружают перемешанный материал в экструдер с двумя шнеками при повышенной температуре для размягчения и образования продукта экструзии.
4. Охлаждение: дают продукту экструзии охладиться на конвейере.
5. Перемалывание: пропускают охлажденный экструдат через мельницу с получением гранулированной массы с подходящим размером частиц.
6. Смешивание: смешивают молотый продукт экструзии со стеаратом магния.
7. Прессовка: смешивают полученную в результате гранулированную массу с использованием пресса для таблеток.
8. Покрытие: получают раствор, покрывающей пленки, путем диспергирования Opadry в очищенной воде и нанесение его на ядра таблеток.
Затем таблетки тестировали на предмет растворения с использованием следующей процедуры.
1. Устройство: способ 2 по USP (корзина), 100 об/мин.
2. Среда: 700 мл ИЖС (SGF, без ферментов) в течение 55 мин, после чего давали 900 мл фосфатного буфера до рН 7,5.
3. Время забора образцов: 1, 2, 4, 8 и 12 часов.
4. Анализ: высокоэффективная жидкостная хроматография. Параметры растворения приведены ниже в таблице XXI.

Пример 9
Фармакокинетическое исследование 3 способами, представляющее сравнение действия разовой дозы таблеток с контролируемым высвобождением 15 мг гидрокодона (пример 8) после еды и натощак и 15 мг гидрокодона с немедленным высвобождением (2×7,5 мг, таблетки), выполнялось за две дозы Q6H на здоровых добровольцах, натощак.
Затем определяли Сmах и Тmах для примера 8 и для контрольного стандарта с немедленным высвобождением в исследовании биодоступности, что приведено ниже в таблицах XXII и XXIII.


Реферат
Описана твердая пероральная лекарственная форма гидрокодона с контролируемым высвобождением (варианты). Лекарственная форма включает эффективное для аналгезии количество гидрокодона или его фармацевтически приемлемой соли, составляющее от 0,5 до 1250 мг, и вещество для контролируемого высвобождения, достаточное для приведения лекарственной формы в состояние, подходящее для введения два раза в сутки пациенту. Лекарственная форма обеспечивает среднее отношение С12/Сmax, составляющее от 0,55 до 0,85, причем указанная лекарственная форма обеспечивает терапевтический эффект по крайней мере в течение 12 часов. Изобретение обеспечивает повышение эффективности и качества управления болью у пациентов, испытывающих умеренную боль. 12 c. и 30 з.п. ф-лы, 3 ил., 24 табл.

Формула
Документы, цитированные в отчёте о поиске
Способ уменьшения диапазона суточных доз препаратов, содержащих оксикодон, композиция с контролируемым высвобождением, твердая лекарственная форма и таблетка, содержащие оксикодон
Устойчивые лекарственные формы с регулируемым выделением, имеющие акриловые полимерные покрытия, (варианты) и способ их получения
Комментарии