Новые композиции для предупреждения и/или лечения лизосомных болезней накопления - RU2608520C2
Код документа: RU2608520C2
Описание
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[0001] Настоящая заявка испрашивает приоритет по предварительной заявке США №61/252806, поданной 19 октября 2009 г., содержимое которой включено в настоящем документе ссылкой.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[0002] Настоящее изобретение относится к новым соединениям, известным как фармакологические шапероны, а также способам использования таковых для предупреждения и/или лечения лизосомных болезней накопления. В частности, настоящее изобретение обеспечивает способы для предупреждения и/или лечения болезни Гоше.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
[0003] Лизосомные болезни накопления вызываются дефектом лизосомной функции, который приводит к накоплению веществ внутри лизосомы клеток. Этот дефект обычно является следствием недостаточности отдельного фермента, который требуется для метаболизма липида, гликогена, гликопротеина или мукополисахарида. Болезнь Гоше, самая распространенная лизосомная болезнь накопления, характеризуется накоплением гликолипида глюкоцереброзида (также известного как гликозилцерамид). Симптомы болезни Гоше включают увеличенную селезенку и печень, нарушение функции печени, заболевания скелета и поражения костей, которые могут причинять боль, сильные неврологические осложнения, увеличение лимфатических узлов и (изредка) прилежащих суставов, вздутый живот, коричневатый оттенок кожи, анемию, низкое содержание тромбоцитов и желтые жировые отложения на склере. К тому же, люди, пораженные болезнью Гоше, могут также быть более восприимчивыми к инфекции.
[0004] Существует потребность в способах предупреждения и/или лечения лизосомных болезней накопления, которые дают пациентам более высокое качество жизни и достигают лучшего клинического результата. В частности, существует потребность в способах предупреждения и/или лечения болезни Гоше, которые дают пациентам более высокое качество жизни и достигают лучшего клинического результата.
КРАТКОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0005] Настоящее изобретение относится к новым соединениям, а также композициям и способам их использования для предупреждения и/или лечения лизосомной болезни накопления у пациента с риском развития или диагнозом этого заболевания, которые включают введение пациенту, нуждающемуся в лечении, эффективного количества соединения, описанного в настоящем документе.
[0006] В одном аспекте изобретение относится к соединению, а также композициям и способам их использования для предупреждения и/или лечения лизосомной болезни накопления у пациента с риском развития или диагнозом этого заболевания, которые включают введение пациенту, нуждающемуся в лечении, эффективного количества соединения, определенного формулой I
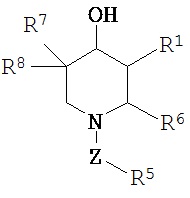
где
R1 представляет собой C(R2)(R3)(R4);
R2 представляет собой водород, -ОН или галоген;
R3 представляет собой водород, -ОН, галоген или C1-8 алкил;
R4 представляет собой галоген, C1-8 алкил, замещенный C1-8 алкил, арил, замещенный арил, алкилциклоалкил или замещенный алкилциклоалкил;
R3 и R4 могут быть соединены с атомом углерода, к которому они присоединены, с образованием циклоалкильного кольца, которое может быть необязательно замещено, предпочтительно галогеном и более предпочтительно одним или несколькими атомами фтора;
R6 представляет собой водород, C1-8 алкил, замещенный C1-8 алкил, арилалкил, замещенный арилалкил, алкиларил или замещенный алкиларил;
Z является необязательным, если Z присутствует, то представляет собой -(CH2)1-8-, -С(=О) -, -S(=O)2NH-, -S(=O)2-, -C(=S)NH-, -S(=O)2-CH3, C(=O)-NH-, -S(=O)2-NR9R10, -C(=O)C1-8 алкил или -С(=O)СН(NH2)СН3;
R9 представляет собой водород, C1-8 алкил или замещенный C1-8 алкил;
R10 представляет собой водород, C1-8 алкил или замещенный C1-8 алкил;
R5 представляет собой водород, C1-8 алкил, замещенный C1-8 алкил, арил, замещенный арил, C1-8 алкенил, замещенный C1-8 алкенил, арилалкил, замещенный арилалкил, алкиларил, замещенный алкиларил, аминоарилалкил или замещенный аминоарилалкил;
R7 представляет собой -ОН или галоген; и R8 представляет собой водород, галоген или C1-8 алкил,
при условии, что R2 и R3 оба не могут быть атомами водорода, если R4 представляет собой галоген, Z не присутствует, R7 представляет собой -ОН, R5, R6 и R8представляют собой атомы водорода.
[0007] Специалист в настоящем уровне техники понимает, что R2, R3 и R4 в вышеупомянутых формулах I, II, и III не будут выбраны так, чтобы привести к нестабильной молекуле.
[0008] В другом аспекте настоящее изобретение относится к соединению, а также композициям и способам их использования для предупреждения и/или лечения лизосомной болезни накопления у пациента с риском развития или диагнозом этого заболевания, которые включают введение пациенту, нуждающемуся в лечении, эффективного количества соединения, определенного формулой II
где R1 представляет собой C(R2)(R3)(R4);
R2 представляет собой водород, -ОН или галоген;
R3 представляет собой водород, -ОН, галоген или -СН3;
R4 представляет собой галоген, -СН3, фенил, фторфенил, метилфенил, циклогексилметил, где, если R4 представляет собой галоген, оба R2 и R3 не могут быть атомами водорода;
R3 и R4 могут быть соединены с атомом углерода, к которому они присоединены, с образованием циклоалкильного кольца, которое может быть необязательно замещено одним или несколькими атомами галогена;
R6 представляет собой водород, фенилалкил или замещенный фенилалкил;
Z является необязательным, если Z присутствует, то представляет собой -(СН3)-, -С(=O) -, -S(=O)2NH-, -S(=O)2-, -S(=O)2-СН3, C(=O)-NH-, -S(=O)2NR9R10, -C(=S)-NH- или -С(=O)2-СН3,
R9 представляет собой водород или СН3;
R10 представляет собой водород или СН3;
R5 представляет собой водород или аминофенилалкил;
R7 представляет собой -ОН или галоген; и
R8 представляет собой водород, галоген или -СН3,
при условии, что R2 и R3 оба не могут быть атомами водорода, если R4 представляет собой галоген, Z не присутствует, R7 представляет собой -ОН, R5, R6 и R8 представляют собой атомы водорода.
[0009] В другом аспекте изобретение относится к соединению, а также композициям и способам их использования для предупреждения и/или лечения лизосомной болезни накопления у пациента с риском развития или диагнозом этого заболевания, которые включают введение пациенту, нуждающемуся в лечении, эффективного количества соединения, определенного формулой III

где R1 представляет собой C(R2)(R3)(R4);
R2 представляет собой водород, -ОН или галоген;
R3 представляет собой водород, -ОН, галоген или -СН3;
R4 представляет собой галоген, -СН3, фенил, фторфенил, метилфенил, циклогексилметил, где, если R4 представляет собой галоген, R2 и R3 оба не могут быть атомами водорода;
R3 и R4 могут быть соединены с атомом углерода, к которому они присоединены, с образованием циклоалкильного кольца, которое может быть необязательно замещено одним или несколькими атомами галогена;
R7 представляет собой -ОН или галоген; и
R8 представляет собой водород, галоген или -СН3,
при условии, что R2 и R3 оба не могут быть атомами водорода, если R4 представляет собой галоген, R7 представляет собой -ОН и R6 и R8 представляют собой атомы водорода.
[0010] В другом аспекте изобретение относится к соединению, а также композициям и способам их использования для предупреждения и/или лечения лизосомной болезни накопления у пациента с риском развития или диагнозом этого заболевания, которые включают введение пациенту, нуждающемуся в лечении, эффективного количества соединения, выбранного из следующего
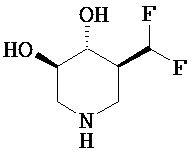
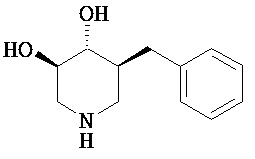
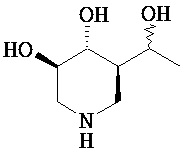
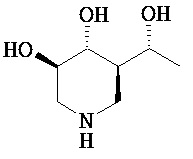
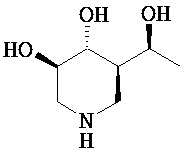

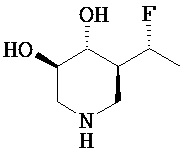

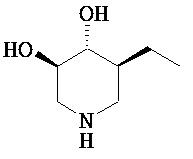
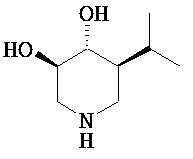
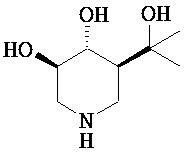
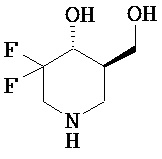

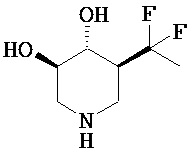
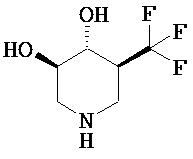
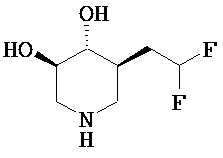

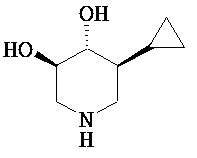
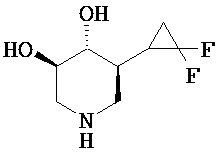
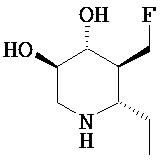
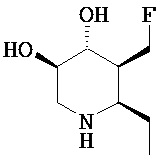
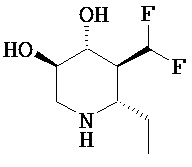
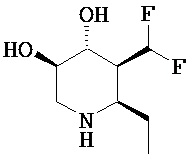
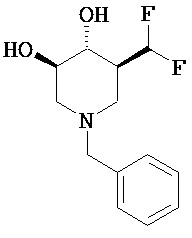
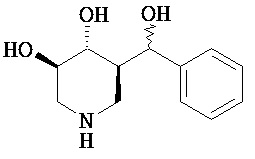
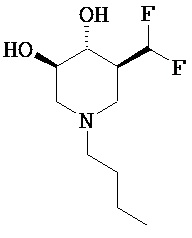
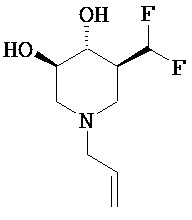
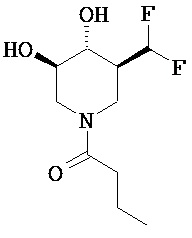
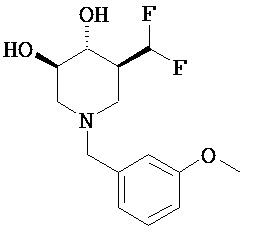

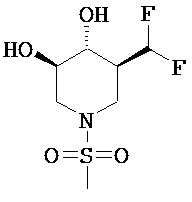
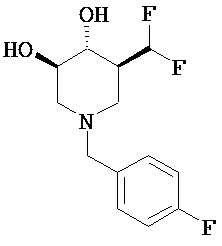


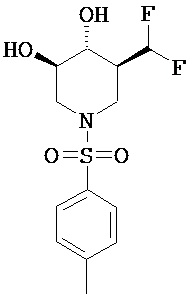
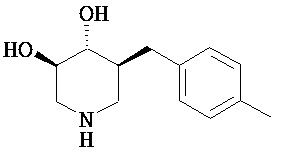


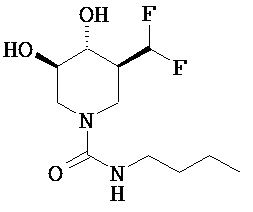
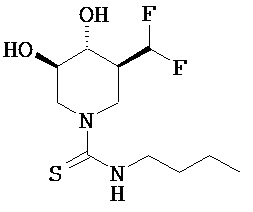
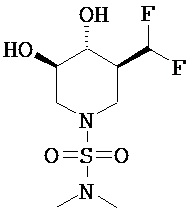
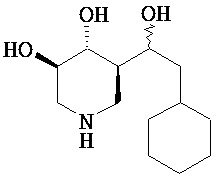
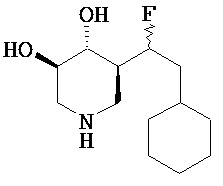
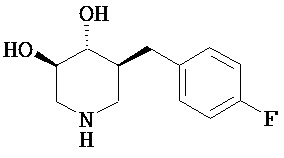
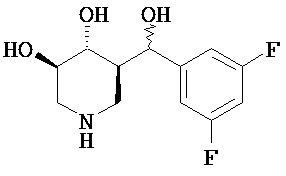
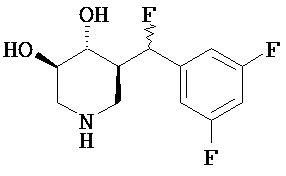
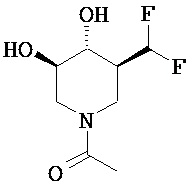
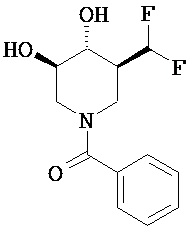
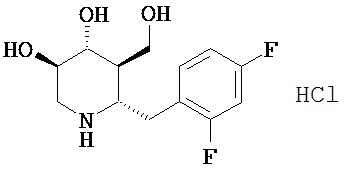

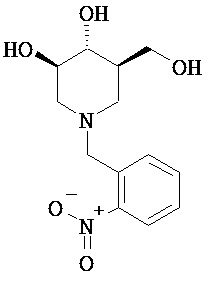

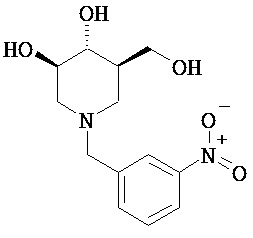
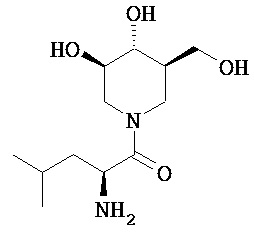
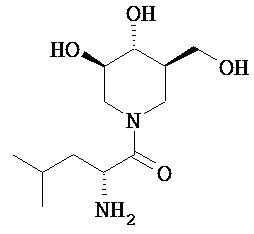
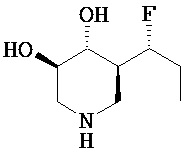
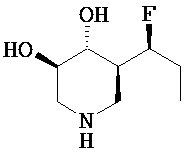
или его фармацевтически приемлемая соль, сольват или пролекарство.
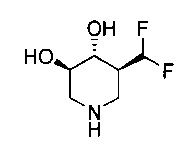
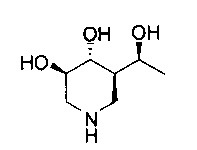
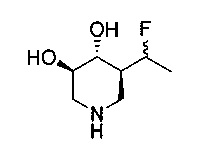
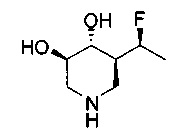
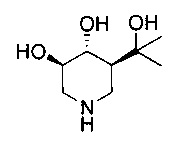
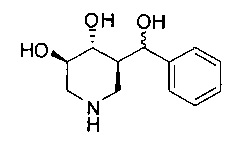
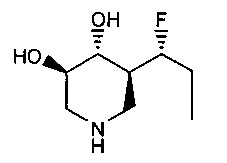
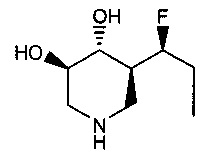
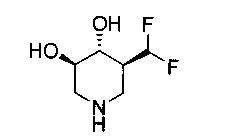
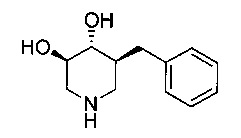
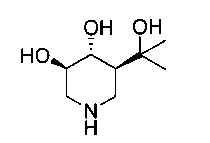
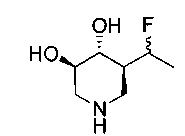
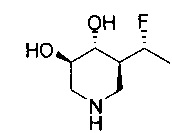
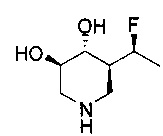
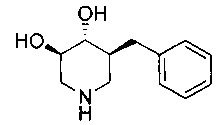
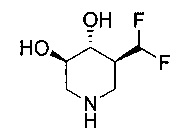
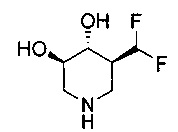
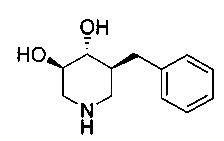
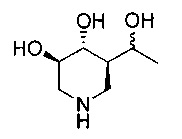
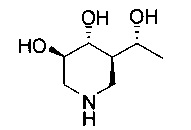
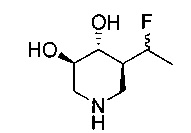
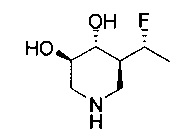
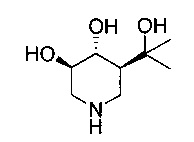
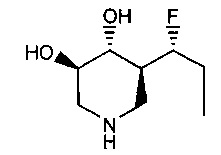
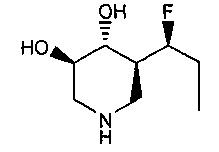
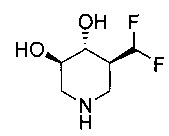
[0011] В одном варианте осуществления соединение представляет собой (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, (3R,4R,5S)-5-бензилпиперидин-3,4-диол или его фармацевтически приемлемую соль, сольват или пролекарство. В одном варианте осуществления соединение представляет собой (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол или его фармацевтически приемлемую соль, сольват или пролекарство. В одном варианте осуществления соединение представляет собой (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол или его фармацевтически приемлемую соль, сольват или пролекарство. В одном варианте осуществления соединение представляет собой (3R,4R,5S)-5-бензилпиперидин-3,4-диол или его фармацевтически приемлемую соль, сольват или пролекарство.
[0012] В одном варианте осуществления лизосомная болезнь накопления связана с накоплением по меньшей мере одного гликолипида. В одном варианте осуществления лизосомная болезнь накопления связана с накоплением по меньшей мере одного гликосфинголипида. В одном варианте осуществления лизосомная болезнь накопления связана с накоплением глюкоцереброзида. В одном варианте осуществления лизосомная болезнь накопления связана с недостаточностью в глюкоцереброзидазе. В одном варианте осуществления лизосомная болезнь накопления связана с мутацией в глюкоцереброзидазе. В одном варианте осуществления лизосомная болезнь накопления представляет собой болезнь Ниманна-Пика. В одном варианте осуществления лизосомная болезнь накопления представляет собой болезнь Гоше. В одном варианте осуществления способ дополнительно включает введение эффективного количества по меньшей мере одного другого терапевтического средства. В одном варианте осуществления способ содержит по меньшей мере одно другое терапевтическое средство, которое представляет собой имиглюцеразу или 1,5-(бутилимино)-1,5-дидеокси-D-глюцитол.
[0013] Настоящее изобретение также относится к способам предупреждения и/или лечения болезни Гоше у пациента с риском развития или диагнозом этого заболевания, который включает введение пациенту, нуждающемуся в лечении, эффективного количества композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль, сольват или пролекарство.
[0014] В одном варианте осуществления способ включает введение соединения (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диола, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диола, (3R,4R,5S)-5-бензилпиперидин-3,4-диола или его фармацевтически приемлемой соли, сольвата или пролекарства. В одном варианте осуществления способ включает введение соединения (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диола или его фармацевтически приемлемой соли, сольвата или пролекарства. В одном варианте осуществления способ включает введение соединения (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диола или его фармацевтически приемлемой соли, сольвата или пролекарства. В одном варианте осуществления способ включает введение соединения (3R,4R,5S)-5-бензилпиперидин-3,4-диола или его фармацевтически приемлемой соли, сольвата или пролекарства.
[0015] В одном варианте осуществления способ дополнительно включает введение эффективного количества по меньшей мере одного другого терапевтического средства. В одном варианте осуществления по меньшей мере одним другим терапевтическим средством является имиглюцеразу или 1,5-(бутилимино)-1,5-дидеокси-D-глюцитол.
[0016] Настоящее изобретение также относится к набору, содержащему:
- контейнер, содержащий эффективное количество любого из соединений настоящего изобретения отдельно или в комбинации; и
- инструкции по его применению для предупреждения и/или лечения лизосомной болезни накопления.
[0017] В одном варианте осуществления лизосомная болезнь накопления представляет собой болезнь Гоше.
[0018] Настоящее изобретение также относится к способам для усиления активности глюкоцереброзидазы в клетке ex vivo с применением 5-(фторметил)пиперидин-3,4-диола, 5-(хлорметил)пиперидин-3,4-диола или их фармацевтически приемлемой соли, сольвата или пролекарства, или любой комбинации двух или нескольких из этого.
[0019] К тому же, настоящее изобретение относится к способам диагностирования пациентов, подлежащим лечению, включащим контакт ех vivo клетки у пациента с риском развития или с диагнозом лизосомной болезни накопления терапевтическим средством, которое представляет собой 5-(фторметил)пиперидин-3,4-диол, 5-(хлорметил)пиперидин-3,4-диол, или его фармацевтически приемлемую соль, сольват или пролекарство, или любую комбинацию двух или нескольких из этого и испытания лизата клетки для активности лизосомный глюкоцереброзидазы, где увеличение активности в лизосомной глюкоцереброзидазе относительно другой клетки, которая не обработана терапевтическим средством, указывает на то, что пациент подлежит лечению. В одном варианте осуществления лизосомная болезнь накопления представляет собой болезнь Гоше.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0020] Используемые в настоящем документе выражения должны характеризоваться изложенными ниже значениями.
[0021] Используемое в настоящем документе выражение "лечение" означает улучшение одного или нескольких симптомов, связанных с упомянутым расстройством.
[0022] Используемое в настоящем документе выражение "предупреждение" означает ослабление симптома упомянутого расстройства.
[0023] Используемая в настоящем документе фраза "эффективное количество" означает количество, эффективное для предупреждения и/или лечения пациента с риском развития или с диагнозом упомянутого расстройства, и, таким образом, образующее желаемый терапевтический эффект.
[0024] Используемое в настоящем документе выражение "пациент" означает млекопитающее (например, человек).
[0025] Используемая в настоящем документе фраза "лизосомная болезнь накопления" относится к любой из группы болезней, возникающей вследствие анормального метаболизма, приводящего к накоплению субстрата в лизосоме. Таблица 1 содержит неограничивающий список типичных лизосомных болезней накопления и дефектных ферментов, связанных с ними.
[0026] Наиболее распространенная лизосомная болезнь накопления, Болезнь Гоше, характеризуется накоплением гликолипида глюкоцереброзида (также известного как гликозилцерамид). Было описано три фенотипа для болезни Гоше, которые отмечаются отсутствием (тип 1) или присутствием нейрологических поражений в период детства (тип 2) или юности (тип 3). Например, см. Grabowski, Gaucher's disease. Adv Hum Genet 1993; 21:377-441.
[0027] Эти три типа болезни Гоше наследуются аутомосомно-рецессивным образом. Оба родителя должны быть носителями для того, чтобы ребенок был поражен. Если оба родители являются носителями, существует для каждой беременности вероятность того, что ребенок будет поражен, составляет один из четырех, или 25%. Генетическое консультирование и генетическое тестирование рекомендуется семьям, которые могут быть носителями мутаций. Каждый тип связан с определенной мутацией. В общей сложности, известно около 80 мутаций, приводящих к болезни Гоше (см., например, McKusick, V.A.: Mendelian Inheritance in Man. A Catalog of Human Genes and Genetic Disorders. Baltimore: Johns Hopkins University Press, 1998 (12th edition)).
[0028] Болезнь Гоше типа 1 является общеэтнической, но особенно распространена среди лиц потомков евреев-ашкенази, с частотой носительства 1 из 17 евреев-ашкенази. Мутации N370S и 84GG представляют собой наиболее частые мутации гена глюкоцереброзидазы среди евреев-ашкенази, с частотами 1 из 17,5 для N370S и 1 из 400 для 84GG в обычной здоровой популяции ашкенази, и связываются с умеренной и сильной болезнью Гоше соответственно. Мутация 84GG встречается почти исключительно среди евреев-ашкенази. Другие редкие варианты гена глюкоцереброзидазы, установленные у пациентов потомков ашкенази с болезнью Гоше, включают L444P, IVS2+1G→A, V394L, и R496H. В отличие от картины болезни Гоше типа 1 у евреев-ашкенази болезнь Гоше типа 1 у японских пациентов обычно бывает суровой и нарастающей (см., Ida et al., Type 1 Gaucher Disease Patients: Phenotypic Expression and Natural History in Japanese Patients, Blood Cells, Molecules and Diseases, 1984, 24(5): 73-81). К тому же, болезнь Гоше типа 3, связанная с одной или двумя копиями генного варианта глюкоцереброзидазы L444P, преобладает у шведских пациентов из провинции Норрботтен.
[0029] Окончательный диагноз болезни Гоше ставят после генетического тестирования. Поскольку существуют многочисленные различные мутации, секвенирование гена глюкоцереброзидазы иногда является необходимым для подтверждения диагноза. Пренатальная диагностика является доступной, и она полезна при известном факторе генетического риска. Однако диагноз болезни Гоше может также вытекать из биохимических нарушений, таких как высокий уровень щелочной фосфатазы, ангиотензин-превращающего фермента (АСЕ) и иммуноглобулинов, или при анализе клеток, обнаруживающем цитоплазму типа "гофрированная бумага" и перегруженные гликолипидами макрофаги. В частности, болезнь Ниманна-Пика является подобной в том, что она характеризуется накоплением GM2-ганглиозидов и GM1-ганглиозидов в дополнение к глюкоцереброзиду (Vanier et al., Brain Pathology. 1998; 8: 163-74).
[0030] Симптомы болезни Гоше включают следующее:
- Безболезненные увеличение печени и селезенки (размер селезенки может составлять 1500-3000 мл, по сравнению с нормальным размером 50-200 мл)
- Гиперспленизм: быстрое и преждевременное разрушение клеток крови, приводящее к анемии, нейтропении и тромбоцитопении (с повышенным риском инфекции и кровотечения)
- Цирроз печени, хотя редко
- Нейрологические симптомы наблюдаются только в некоторых типах болезни Гоше (см. ниже):
- Тип II: тяжелые судороги, гипертония, умственная отсталость, апноэ
- Тип III: мышечные судороги известные как миоклонус, судороги, слабоумие, апраксия глазной мышцы
- Остеопороз: у 75% развиваются заметные аномалии костей вследствие накопленного гликозилцерамида. Часто описывается деформация дистальной части бедра в форме колбы Эрленмейера
- Желтовато-коричневая пигментация кожи
Соединения
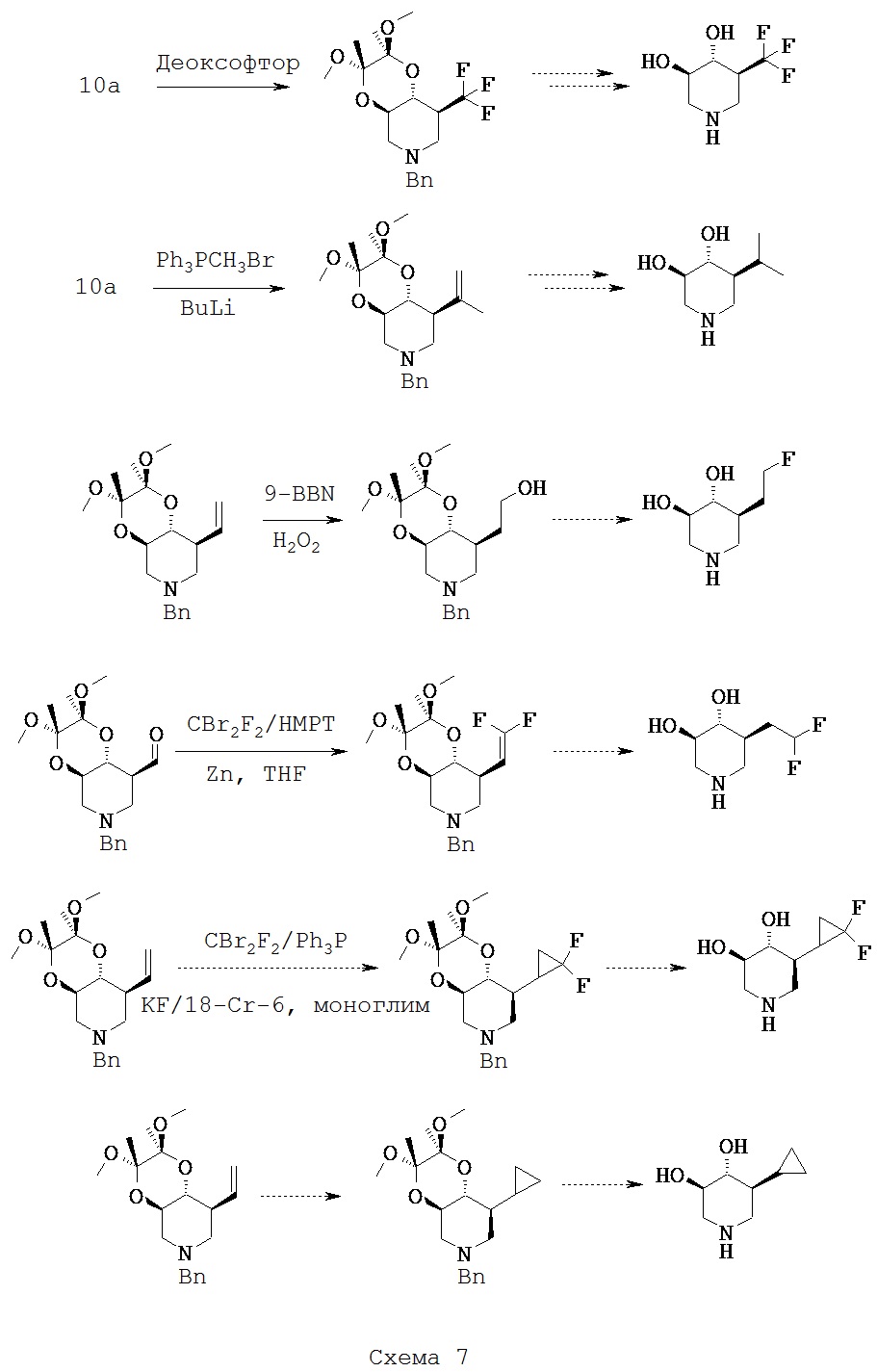
[0031] Ниже представлены новые соединения по настоящему изобретению:
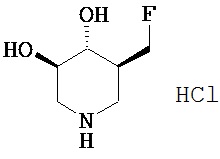
Химический процесс
[0032] Композиции по настоящему изобретению могут быть получены согласно одной или нескольким следующим схемам.
Схема процесса 1
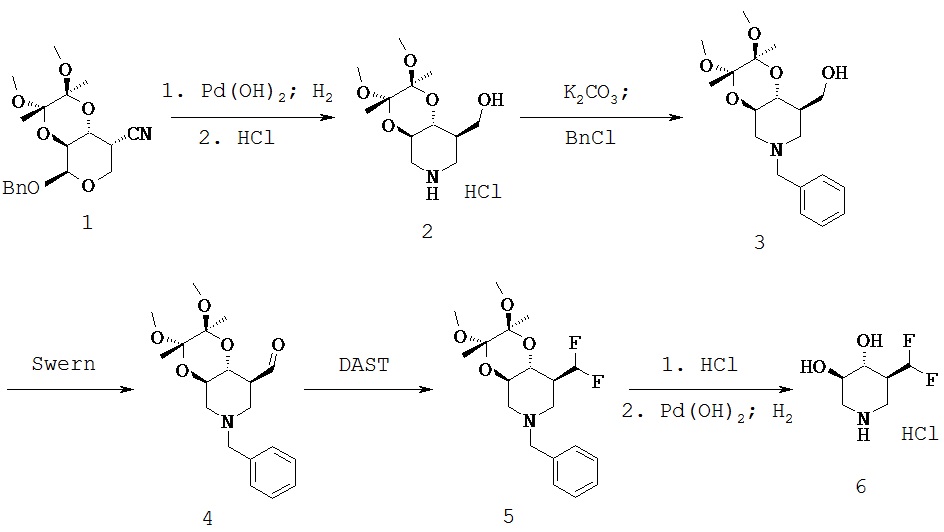
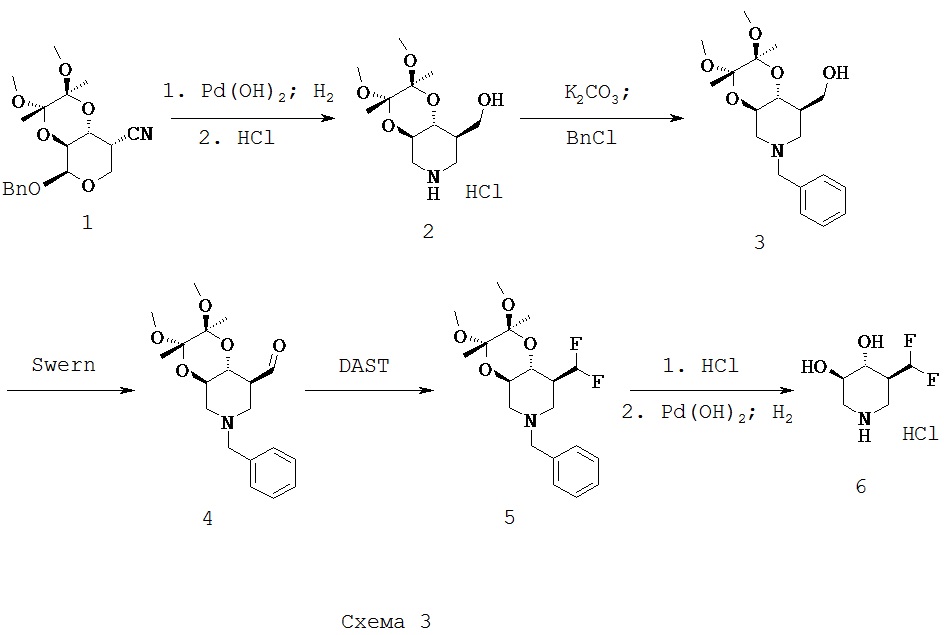
[0033] ((2S,3S,4аR,8R,8аR)-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин-8-ил)метанола гидрохлорид(2). Раствор 1 (20,0 г, 55,0 ммоль) в МеОН (500 мл) объединяли с Pd(OH)2 (4-6 г) и формиатом аммония (14 г, 220 ммоль) и смесь нагревали при 50-55°С. Дополнительные количества (3×100,0 ммоль) формиата аммония добавляли в течение следующих 8 часов. После заключительного добавления реакционную смесь дополнительно перемешивали и нагревали дополнительно 16 часов при 50-55°С. Катализатор удаляли путем фильтрации и фильтрат выпаривали в вакууме. Неочищенный продукт растворяли в ацетоне (150 мл), отфильтровывали и добавляли HCl в 2-РrОН. После затравки и затем охлаждения в ледяной бане продукт собирали в виде белого кристаллического твердого вещества (11,0 г, 71%).1Н ЯМР (DMSO-d6) 9,45 (s, 2H), 4,80 (t, 1H, ex), 3,85 (m, 1H), 3,0-3,75 (m, 11H), 2,8 (q, 2H), 1,95 (m, 1H), 1,2 (2, 6H).
[0034] ((2S,3S,4аR,8R,8аR)-6-бензил-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин-8-ил)метанол (3). В раствор 2 (14,85 г, 50,0 ммоль) в DMF (200 мл) добавляли К2СО3 (17,25 г, 125 ммоль) и смесь перемешивали при 40°С в течение приблизительно 4 часов. На этой стадии добавляли одну часть BnCl (5,7 мл, 50,0 ммоль) и реакционную смесь перемешивали при 40°С в течение ночи. Растворитель выпаривали в вакууме и остаток суспендировали в воде (600 мл) и добавляли НСl для растворения остатка. Раствор промывали Et2O и затем превращали в основание при помощи Na2CO3. Раствор экстрагировали EtOAc (2×) и объединенные экстракты промывали водой и затем рассолом и затем сушили MgSO4. Раствор фильтровали и фильтрат выпаривали в вакууме с получением названного соединения (17,2 г, >95%) в виде бесцветного до бледно-желтого вязкого масла, которое использовали без дополнительной очистки.1H ЯМР (CDCl3) 7,3 (m, 5Н), 3,6-3,8 (m, 2H), 3,5 (s, 3H), 3,4 (t, 1H), 3,26 (s, 3H), 3,268 (s, 3H), 2,9 (m, 2H), 2,2 (br s, 1H), 2,05 (m, 1H), 1,85 (t, 1H), 1,28 (s, 3H), 1,26 (s, 3H).
[0035] ((2S,3S,4аR,8R,8аR)-6-бензил-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин-8-ил)карбоксальдегид (Общая методика А) (4). В раствор DMSO (7,3 г, 96,9 ммоль) в CH2Cl2 (150 мл), охлажденный до -78°С, добавляли раствор оксалилхлорида (6,1 мл, 72,8 ммоль) по каплям в CH2Cl2. После завершения добавления реакционную смесь перемешивали дополнительно 30 минут, при этом по каплям добавляли раствор 3 (17,0 г, 48,4 ммоль) в CH2Cl2. После завершения добавления реакционную смесь перемешивали в течение 1 часа при -78°С и затем добавляли по каплям диизопропилэтиламин (34,4 мл, 193 ммоль). После завершения этого добавления охлаждающую баню удаляли и реакционную смесь оставляли нагреваться до 0°С, когда добавляли насыщенный NaHCO3. Смесь разбавляли некоторым количеством дополнительного CH2Cl2 и затем органический слой отделяли и сушили над MgSO4. После фильтрации растворитель выпаривали в вакууме и неочищенный продукт очищали хроматографией на силикагеле (Hex/EtOAc) с получением названного соединения (12,7 г, 75%) в виде вязкого масла.1Н ЯМР (CDCl3) 9,73 (s, 1Н), 7,2 (m, 5H), 3,75 (m, 2H), 3,5 (q, 2H), 3,2 (2s, 6H), 2,7-3,0 (m, 3H), 2,05 (m, 2H), 1,25 (2s, 6H).
[0036] ((2S,3S,4аR,8S,8аR)-6-бензил-8,8-дифторметил-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридина гидрохлорид (Общая методика В) (5). В раствор DAST (1,4 мл, 10,3 ммоль) в СН2Сl2 (50 мл), охлажденный до -15°С, по каплям добавляли раствор 4 (2,4 г, 6,9 ммоль). Через 10 минут ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение ночи. На этой стадии реакционную смесь снова охлаждали в ледяной бане и реакцию гасили путем добавления насыщенного NaHCO3 (сначала по каплям, поскольку это дает в результате легкий экзотермический эффект). Органический слой разделяли и сушили над Nа2SO4, отфильтровывали и растворитель выпаривали в вакууме с получением желтого масла. Остаток очищали хроматографией на силикагеле (Hex/EtOAc) с получением названного соединения (1,6 г, 62%) в виде бесцветного масла.1Н ЯМР (CDCl3) 7,2 (m, 5H), 6,0 (dt, 1Н), 3,75 (m, 1Н), 3,55 (m, 3H), 3,2 (2s, 6H), 2,95 (m, 1Н), 2,85 (m, 1Н), 2,3 (m, 2H), 1,5 (br s, 1Н), 1,2 (2s, 6H).
[0037] (3R,4R,5S)-5-(дифторметил)пиперидин 3,4-диол гидрохлорид (Общая методика С) (6). Соединение 5 (1,6 г, 4,3 ммоль) нагревали при возгонке в смеси ЕtOН/Н2O/НСl (40 мл/40 мл/5 мл) и реакцию наблюдали при помощи HPLC, пока исходный материал не будет больше определяться. Растворитель выпаривали в вакууме и затем совместно выпаривали 2× с EtOH. Остаток растворяли в МеОН и гидрировали над Pd(OH)2. При завершении катализатор удаляли путем фильтрации и фильтрат выпаривали в вакууме. Остаток перекристаллизовывали из EtOH (50 мл) до названного соединения (0,55 г, 66%) в виде белого твердого вещества (т.пл. 168-170°С).1H ЯМР (D2O) 6,15 (dt, 1Н), 4,3-4,8 (m, 2H), 3,0 (t, 1Н), 2,85 (t, 1Н), 2,3 (m, 1Н).
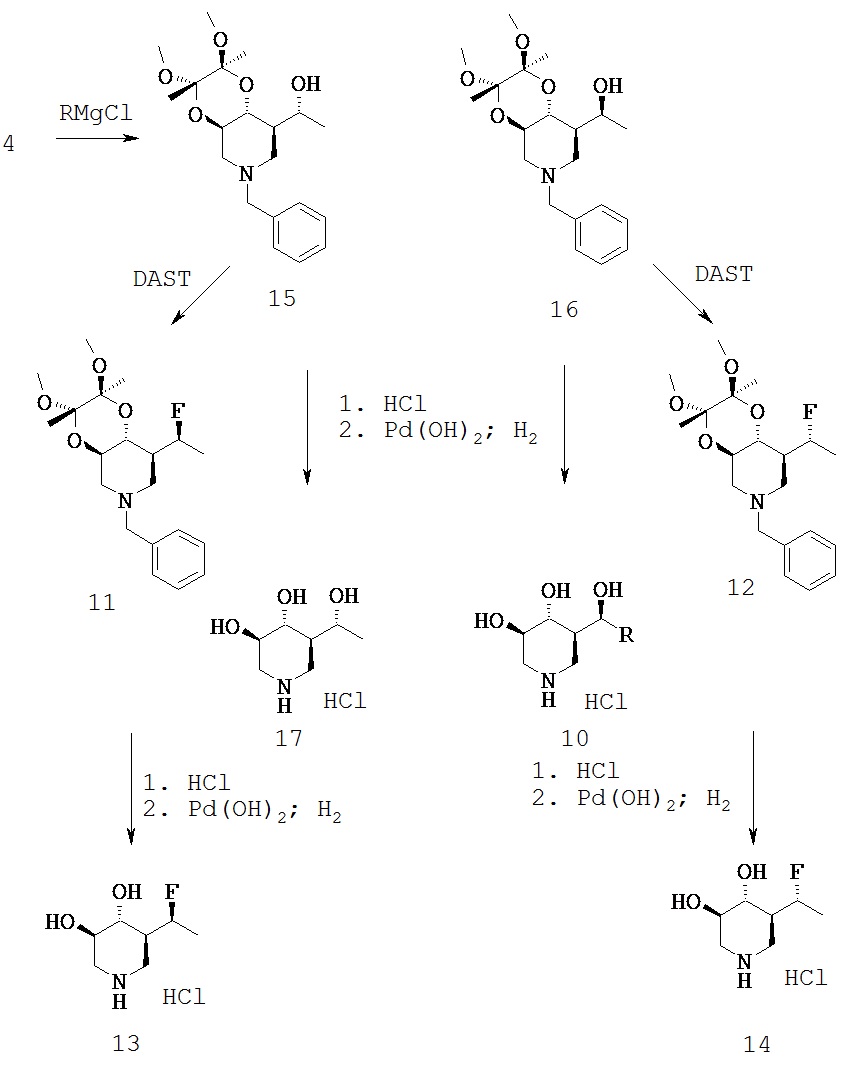
[0038] (R) и (S)-1-((2S,3S,4аR,8R,8аR)-6-бензил-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин-8-ил)этанол (Общая методика D) (15/16). В раствор 4 (7,0 г, 20,0 ммоль) в сухом THF (100 мл) добавляли MeMgBr (20,0 мл, 1,4 М в 3:1 THF/толуол) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакцию гасили насыщенным NH4Cl и смесь экстрагировали EtOAc (2×). Объединенные экстракты промывали рассолом, сушили над Na2SO4 и фильтрат выпаривали в вакууме. Остаток очищали хроматографией на силикагеле (гексан/2-РrOН) с получением основного изомера (15) (1,6 г, 24,6%).1H ЯМР (CDCl3) 7,3 (m, 5Н), 4,15 (m, 1Н), 3,5-3,9 (m, 3Н), 3,3 (2s, 6Н), 2,85 (m, 2Н), 2,0 (2m, 4Н), 1,3 (2s, 6Н), 1,2 (d, 3Н). Побочный изомер (16) был также выделен (0,55 г, 7,5%) 7,3 (m, 5H), 3,75 (m, 2H), 3,5 (m, 2H), 3,2 (2s, 6H), 2,8 (m, 2H), 2,0 (t, 1H), 1,75 (m, 2H), 1,2 (2s, 6H), 1,0 (d, 3H).
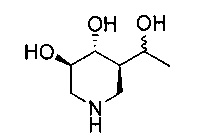
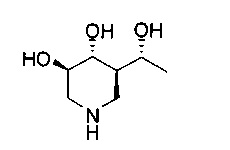
[0039] (3R,4R,5R)-5((R)-1-гидроксиэтил)пиперидин 3,4-диол (17). Соединение 15 (0,55 г, 1,5 ммоль) перемешивали в смеси 9/1 TFA:H2O (20 мл) пока исходный материал больше не будет определяться при помощи HPLC. Летучие соединения удаляли и остаток совместно выпаривали 2-3х с ЕtOН и затем растворяли в ЕtOН и обрабатывали твердым К2СО3. После фильтрации твердого вещества фильтрат выпаривали в вакууме и остаток превращали в соль НСl и гидрировали над Pd(OH)2. Катализатор отфильтровывали и фильтрат выпаривали в вакууме. Неочищенный продукт очищали с применением ионобменной смолы (Dowex 50WX8-200) с элюированием 0,1 н NH4OH. Соответствующие фракции объединяли и лиофилизировали с получением названного соединения (0,12 г, 50%).1Н ЯМР (D2O) 4,2 (q, 1H), 3,65 (m, 1H), 3,45 (m, 3Н), 2,8 (m, 2H), 1,65 (m, 1H), 1,15 (d, 3Н).
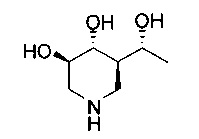
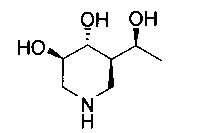
[0040] (3R,4R,5R)-5((S)-1-гидроксиэтил)пиперидин 3,4-диол (10). С соединения 16 (0,34 г, 0,93 ммоль) снимали защитные группы, как описано выше, с получением названного соединения (0,11 г, 75%).1Н ЯМР (D2O) 4,15 (m, 2H), 3,5 (m, 1H), 3,35 (t, 1H), 3,15 (m, 2H), 1,8 (m, 1H), 1,1 (d, 3Н).
[0041] ((2S,3S,4аR,8R,8аR)-6-бензил-8(S)-(1фторэтил)-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин (11). Соединение 15 (1,8 г, 5,0 ммоль) фторировали с применением общей методики В. Хроматографией на силикагеле (Нех/ЕtOАс) получали названное соединение (0,42 г, 23%).1Н ЯМР (CDCl3) 7,25 (m, 5H), 4,7-4,9 (dq, 1H), 3,75 (m, 2H), 3,4 (m, 2H), 3,2 (2s, 6H), 2,8 (m, 2H), 2,0 (m, 3Н), 1,35 (dd, 3Н), 1,2 (2s, 6H).
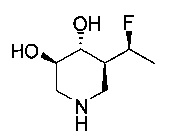
[0042] (3R,4R,5R)-5((8)-1-фторэтил)пиперидин 3,4-диол гидрохлорид (13). С соединения 11 (0,42 г, 1,14 ммоль) снимали защиту, как описано в основной методике С. После удаления катализатора фильтрат выпаривали в вакууме и затем совместно выпаривали с EtOH (2×). Полученный остаток перетирали с ацетоном с получением названного соединения (0,20 г, 88%) в виде белого твердого вещества.1Н ЯМР (DMSO-d6) 9,0 (br s, 2H), 5,6 (d, 1H, ex), 5,4 (d, 1H, ex), 5,0-5,2 (dq, 1H), 3,55 (m, 1H), 3,2 (m, 2H), 2,9 (t, 1H), 2,7 (t, 1H), 2,2 (m, 1H), 1,3 (dd, 3H).
[0043] ((2S,3S,4аR,8R,8аR)-6-бензил-8(R)-(1фторэтил)-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин (12). Соединение 16 (0,55 г, 1,5 ммоль) фторировали с применением общей методики В с получением названного соединения (0,22 г, 40%).1Н ЯМР (CDCl3) 7,3 (m, 5H), 5,0 (dq, 1H), 3,8 (m, 1H), 3,5-3,75 (m, 3H), 3,3 (2s, 6H), 3,0 (d, 1H), 2,9 (m, 1H), 2.1 (m, 2H), 1,85 (m, 1H), 1,3 (2s, 6H).
[0044] (3R,4R,5R)-5((R)-(1-фторэтил)пиперидин 3,4-диол гидрохлорид (14). С соединения 12 (0,22 г, 0,6 ммоль) снимали защитные группы, как описано в основной методике С. После удаления катализатора фильтрат выпаривали в вакууме и затем совместно выпаривали с EtOH (2×). Полученный остаток перетирали с ацетоном с получением названного соединения (0,08 г, 67%) в виде белого твердого вещества.1Н ЯМР (D2О) 5,1 (dq, 1H), 3,5 (m, 4H), 2,8 (m, 2H), 1,8 (m, 1H), 1,3 (dd, 3H).
Схема процесса 2
[0045] ((2S,3S,4аR,8R,8аR)-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин-8-ил)метанола гидрохлорид(2). Раствор 1 (20,0 г, 55,0 ммоль) в МеОН (500 мл) объединяли с Pd(OH)2 (4-6 г) и формиатом аммония (14 г, 220 ммоль) и смесь нагревали при 50-55°С. Дополнительные количества (3×100,0 ммоль) формиата аммония добавляли в течение следующих 8 часов. После заключительного добавления реакционную смесь дополнительно перемешивали и нагревали дополнительно 16 часов при 50-55°С. Катализатор удаляли путем фильтрации и фильтрат выпаривали в вакууме. Неочищенный продукт растворяли в ацетоне (150 мл), отфильтровывали и добавляли HCl в 2-РrОН. После затравки и затем охлаждения в ледяной бане продукт собирали в виде белого кристаллического твердого вещества (11,0 г, 71%).1Н ЯМР (DMSO-d6) 9,45 (s, 2H), 4,80 (t, 1H, ex), 3,85 (m, 1H), 3,0-3,75 (m, 11H), 2,8 (q, 2H), 1,95 (m, 1H), 1,2 (2, 6H).
[0046] ((2S,3S,4аR,8R,8аR)-6-бензил-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин-8-ил)метанол (3). В раствор 2 (14,85 г, 50,0 ммоль) в DMF (200 мл) добавляли К2СО3 (17,25 г, 125 ммоль) и смесь перемешивали при 40°С в течение приблизительно 4 часов. На этой стадии добавляли одну часть BnCl (5,7 мл, 50,0 ммоль) и реакционную смесь перемешивали при 40°С в течение ночи. Растворитель выпаривали в вакууме и остаток суспендировали в воде (600 мл) и НСl добавляли для растворения остатка. Раствор промывали Et2O и затем превращали в основание при помощи Nа2СО3. Раствор экстрагировали EtOAc (2×) и объединенные экстракты промывали водой и затем рассолом и затем сушили MgSO4. Раствор фильтровали и фильтрат выпаривали в вакууме с получением названного соединения (17,2 г, >95%) в виде бесцветного до бледно-желтого вязкого масла, которое использовали без дополнительной очистки.1H ЯМР (CDCl3) 7,3 (m, 5Н), 3,6-3,8 (m, 2H), 3,5 (s, 3Н), 3,4 (t, 1H), 3,26 (s, 3Н), 3,268 (s, 3Н), 2,9 (m, 2H), 2,2 (br s, 1H), 2,05 (m, 1H), 1,85 (t, 1H), 1,28 (s, 3Н), 1,26 (s, 3Н).
[0047] ((2S,3S,4аR,8R,8аR)-6-бензил-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридин-8-ил)карбоксальдегид (Общая методика А) (4). В раствор DMSO (7,3 г, 96,9 ммоль) в СН2Сl2 (150 мл) охлажденный до -78°С добавляли раствор оксалилхлорида (6,1 мл, 72,8 ммоль) по каплям в CH2Cl2. После завершения добавления реакционную смесь перемешивали дополнительно 30 минут, при этом раствор 3 (17,0 г, 48,4 ммоль) в СН2Сl2 добавляли по каплям. После завершения добавления реакционную смесь перемешивали в течение 1 часа при -78°С и затем по каплям добавляли диизопропилэтиламин (34,4 мл, 193 ммоль). После завершения этого добавления охлаждающую баню удаляли и реакционную смесь оставляли нагреваться до 0°С, пока добавляли насыщенный NаНСО3. Смесь разбавляли некоторым количеством дополнительного СН2Сl2 и затем органический слой отделяли и сушили над MgSO4. После фильтрации растворитель выпаривали в вакууме и неочищенный продукт очищали хроматографией на силикагеле (Нех/ЕtOАс) с получением названного соединения (12,7 г, 75%) в виде вязкого масла.1H ЯМР (CDCl3) 9,73 (s, 1Н), 7,2 (m, 5H), 3,75 (m, 2H), 3,5 (q, 2H), 3,2 (2s, 6H), 2,7-3,0 (m, 3Н), 2,05 (m, 2H), 1,25 (2s, 6H).
[0048] ((2S,3S,4аR,8S,8аR)-6-бензил-8,8-дифторметил-2,3-диметокси-2,3-диметилоктагидро-[1,4]диоксино[2,3-с]пиридина гидрохлорид (Общая методика В) (5). В раствор DAST (1,4 мл, 10,3 ммоль) в CH2Cl2 (50 мл), охлажденный до -15°С, добавляли по каплям раствор 4 (2,4 г, 6,9 ммоль). Через 10 минут ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение ночи. На этой стадии реакционную смесь снова охлаждали в ледяной бане и реакцию гасили путем добавления насыщенного NaHCO3 (сначала по каплям, поскольку это дает в результате легкий экзотермический эффект). Органический слой разделяли и сушили над Nа2SO4, отфильтровывали и растворитель выпаривали в вакууме с получением желтого масла. Остаток очищали хроматографией на силикагеле (Нех/ЕtOАс) с получением названного соединения (1,6 г, 62%) в виде бесцветного масла.1Н ЯМР (CDCl3) 7,2 (m, 5H), 6,0 (dt, 1Н), 3,75 (m, 1Н), 3,55 (m, 3Н), 3,2 (2s, 6H), 2,95 (m, 1Н), 2,85 (m, 1Н), 2,3 (m, 2H), 1,5 (br s, 1Н), 1,2 (2s, 6H).
[0049] (3R,4R,5S)-5-(дифторметил)пиперидин 3,4-диола гидрохлорид (Общая методика С) (6). Соединение 5 (1,6 г, 4,3 ммоль) нагревали при возгонке в смеси EtOH/H2O/HCl (40 мл/40 мл/5 мл) и реакцию наблюдали при помощи HPLC, пока исходный материал не будет больше определяться. Растворитель выпаривали в вакууме и затем совместно выпаривали 2× с EtOH. Остаток растворяли в МеОН и гидрировали над Pd(OH)2. При завершении катализатор удаляли путем фильтрации и фильтрат выпаривали в вакууме. Остаток перекристаллизовывали из EtOH (50 мл) до названного соединения (0,55 г, 66%) в виде белого твердого вещества (т.пл. 168-170°С).1Н ЯМР (D20) 6,15 (dt, 1H), 4,3-4,8 (m, 2H), 3,0 (t, 1H), 2,85 (t, 1H), 2,3 (m, 1H).

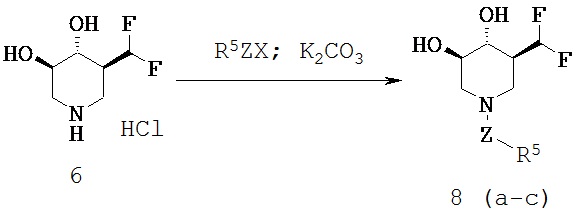
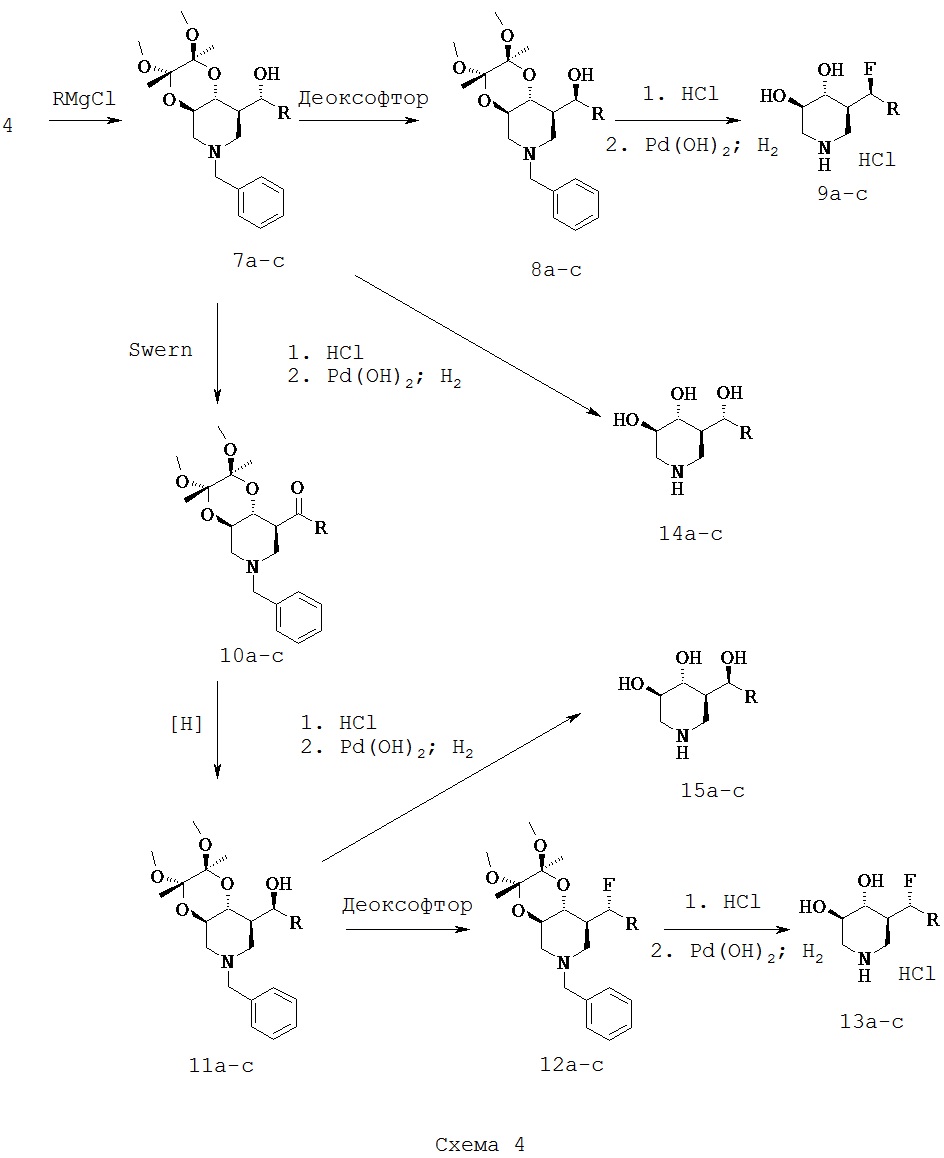
[0050] (3R,4R,5S)-1.бутил-5-(дифторметил)пиперидин 3,4-диол (Общая методика D) (7a; R5=Bu). Смесь 6 (0,30 г, 1,4 ммоль), К2СО3 (0,48 г, 3,5 ммоль) и BuBr (0,20 г, 1,4 ммоль) объединяли в DMF (10 мл) и нагревали в течение ночи при 60°С. Растворитель выпаривали в вакууме и остаток растворяли в EtOAc, промывали водой и затем рассолом и сушили над Na2SO4. После фильтрации фильтрат выпаривали в вакууме с получением неочищенного продукта, который очищали хроматографией (CH2Cl2/(9:1) MeOH/NH4OH) с получением названного соединения (0,25 г, 80%) в виде бесцветного сиропа. MH+=224.1Н ЯМР (DMSO-d6) 6,2 (t, 1H, J=57 Гц), 5,13 (d, 1H, ex), 4,91 (d, 1H, ex), 3,3 (m, 1H), 3,1 (m, 1H), 2,9 (m, 2H), 2,3 (m, 2H), 1,95 (m, 2H), 1,75 (t, 1H), 1,2-1,5 (2m, 4H), 0,9 (t, 3H).
[0051] (3R,4R,5S)-1.Аллил-5-(дифторметил)пиперидин 3,4-диол (7b; R5=аллил). Последующей общей методикой D с применением аллилбромида (0,17 г, 1,4 ммоль) получали названное соединение в виде белого твердого вещества (0,22 г, 76%). MH+=208.1Н ЯМР (DMSO-d6) 6,2 (t, 1H, J=57 Гц), 5,8 (m, 1H), 5,2 (m, 3H), 4,92 (d, 1H), 3,3 (m, 1H), 3,1 (1H), 2,95 (d, 2H), 2,85 (d, 2H), 1,9 (br m, 2H), 1,75 (t, 1H).
[0052] (3R,4R,5S)-5-(дифторметил)-1-(4-фторбензил)пиперидин 3,4-диол (7с; R5=4-фторбензил). Последующей общей методикой D, за исключением, что реакцию проводили при комнатной температуре и с применением 4-фторбензилбромида (0,26 г, 1,4 ммоль), получали названное соединение в виде белого твердого вещества (0,22 г, 56%). МН+=276.1Н ЯМР (DMSO-d6) 7,4 (m, 2H), 7,15 (m, 2H), 6,2 (t, 1H, J=57 Гц), 5,2 (d, 1H, ex), 4,9 (d, 1H, ex), 3,5 (q, 2H), 3,3 (m, 1H), 3,1 (m, 1H), 2,8 (m, 2H), 2,0 (m, 2H), 1,8 (t, 1H).
[0053] (3R,4R,5S)-5-(дифторметил)-1-(4-метилбензил)пиперидин 3,4-диол (7d; R5=4-метилбензил). Последующей общей методикой D за исключением, что реакцию проводили при комнатной температуре и с применением 4-метилбензилбромида (0,26 г, 1,4 ммоль) получали названное соединение в виде белого твердого вещества (0,30, 81%). MH+=272.1H ЯМР (DMSO-d6) 7,2 (m, 4H), 6,2 (t, 1H, J=57 Гц), 5,2 (d, 1H, ex), 4,9 (d, 1H, ex), 3,5 (q, 2H), 3,3 (1H), 3,05 (m, 1H), 2,8 (m, 2H), 2,5 (s, 3Н), 1,95 (m, 2H), 1,8 (t, 1H).
[0054] (3R,4R,5S)-5-(дифторметил)-1-(4-метоксилбензил)пиперидин 3,4-диол (7е; R5=4-метоксилбензил). Последующей общей методикой D за исключением, что реакцию проводили при комнатной температуре и с применением 4-метоксилбензилхлорида (0,26 г, 1,4 ммоль), получали названное соединение в виде бесцветного сиропа (0,19 г, 49%). МН+=288.1Н ЯМР (DMSO-d6) 7,3 (m, 1H), 6,85 (m, 3Н), 6,2 (t, 1H, J=57 Гц), 5,2 (d, 1H, ex), 4,9 (d, 1H, ex), 3,75 (s, 3Н), 3,5 (q, 2H), 3,4 (m, 1H), 3,1 (m, 1H), 2,85(m, 2H), 1,95 (m, 2H), 1,8 (t, 1H).
[0055] 1-((3S,4R,5R)-3-(дифторметил)-4,5-дигидроксипиперидин-1-ил)пентан-1-он (8a; Z=CO; R5=бутил). Последующей общей методикой D, за исключением того, что реакция проходила при комнатной температуре и с применением пентаноилхлорида (0,17 г, 1,4 ммоль), получали названное соединение в виде белого твердого вещества (0,26 г, 71%). MH+=252.1Н ЯМР (DMSO-d6) 5,9-6,5 (dt, 1H), 5,35 (m, 1H, ex), 5,25 (m, 1H), ex), 4,2 (dd, 1H), 3,75 (dd, 1H), 3,35 (m, 2H), 3,1 (m, 1H), 2,85 (m, 1H), 2,3 (t, 2H), 1,9 br m, 1H), 1,4 (m, 2H), 1,25 (m, 2H), 0,85 (t, 3H).
[0056] (3R,4R,5S)-5-(дифторметил)-1-(метансульфонил)пиперидин 3,4-диол (8b; Z=SO2; R5=Me). Последующей общей методикой D за исключением того, что реакция проходила при комнатной температуре и с применением метан сульфонилхлорида (0,16 г, 1,4 ммоль), получали названное соединение в виде белого твердого вещества (0,17 г, 51%).1Н ЯМР (DMSO-d6) 6,2 (t, 1H, J=53 Гц), 5,43 (d, 1H, ex), 5,38 (d, 1H, ex), 3,2-3,7 (m, 4H), 2,95 (s, 3H), 2,85 (m, 1H), 2,7 (t, 1H), 2,1 (br s, 1H). (3R,4R,5S)-5-(дифторметил)-1-тозилпиперидин 3,4-диол (8b; Z=SO2; R5=Ph). Последующей общей методикой D за исключением того, что реакция проходила при комнатной температуре и с применением толуолсульфонилхлорида (0,26, 1,4 ммоль), получали названное соединение в виде белого твердого вещества (0,35 г, 67%).1Н ЯМР (DMSO-d6) 7,6 (d, 2H), 7,45 (d, 2H), 6,25 (t, 1H, J=53 Гц), 5,4 (2d, 2H, ex), 3,3-3,55 (m, 4H), 3,2 (m, 1H), 2,5 (m, 3H), 2,4 (t, 1H), 2,1 (m, 1H).
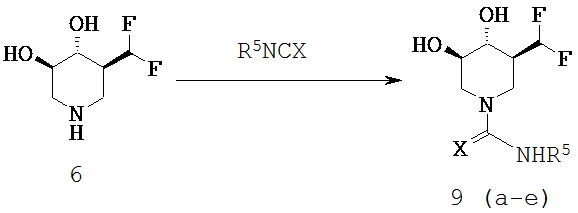
[0057] (3S,4R,5R)-3-(дифторметил)-4,5-дигидрокси-N-пропилпиперидин-1-карбоксамид (Общая методика Е) (9а; Х=О; R5=пропил). В раствор 6 (свободное основание) (0,29 г, 1,2 ммоль) в сухом DMF (5 мл) добавляли пропилизоцианат (0,10 г, 1,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали в вакууме и остаток очищали хроматографией (CH2Cl2/MeOH) с получением названного соединения в виде белого твердого вещества (0,14 г, 48%). MH+=253.1Н ЯМР (DMSO-d6) 6,7 (t, 1H), 6,22 (t, 1H, J=53 Гц), 5,25 (d, 1H, ex), 5,15 (d, 1H, ex), 4,05 (d, 1H), 3,9 (d, 1H), 3,3 (m, 2H), 3,0 (q, 2H), 2,5 (m, 1H), 1,8 (br d, 1H), 1,4 (m, 2H), 0,85 (t, 3H).
[0058] (3S,4R,5R)-3-(дифторметил)-4,5-дигидрокси-N-фенилпиперидин-1-карбоксамид (9b; X=O; R5=фенил). Последующей общей методикой Е и с применением фенилизоцианата (0,14 г, 1,2 ммоль) получали названное соединение в виде белого твердого вещества (0,21 г, 62%). МН+=287,1Н ЯМР (DMSO-d6) 8,7 (s, 1H), 7,45 (d, 2H), 7,3 (t, 2H), 6,95 (t, 1H), 6,3 (t, 1H, J=53 Гц), 5,35 (d, 1H), 5,25 (d, 1H), 4,1 (t, 2H), 3,3 (m, 2H), 2,85 (t, 1H), 2,75 (t, 1H), 1,95 (br d, 1H).
[0059] (3S,4R,5R)-3-(дифторметил)-4,5-дигидрокси-N-бутилпиперидин-1-карбоксамид (9с; X=О; R5=бутил). Последующей общей методикой Е и с применением бутилизоцианата (0,12 г, 1,2 ммоль) получали названное соединение в виде белого твердого вещества (0,24 г, 76%). МН+=267.1H ЯМР (DMSO-d6) 6,6 (t, 1H), 6,2 (t, 1H, J=53 Гц), 5,25 (d, 1H), 5,1 (d, 1H), 4,05 (d, 1H), 3,9 (d, 1H), 3,35 (m, 2H), 3,05 (q, 2H), 2,65 (t, 1H), 2,45 (m, 1H), 1,8 (br d, 1H), 1,2-1,4 (2m, 4H), 0,85 (t, 3H).
[0060] (3S,4R,5R)-3-(дифтopмeтил)-4,5-дигидpoкcи-N-бyтилпипepидин-1-карботиоамид (9d; X=S; R5=бутил). Последующей общей методикой Е и с применением бутилизотиоцианата (0,14 г, 1,2 ммоль) получали названное соединение в виде бесцветного сиропа (0,21 г, 63%). МН+=283.1Н ЯМР (DMSO-d6) 7,85 (t, 1H), 6,25 (t, 1H), 5,35 (2d, 2H), 4,8 (d, 1H), 4,45 (d, 1H), 3,45 (m, 2H), 3,25 (m, 1H), 3,05 (t, 1H), 2,8 (t, 1H), 1,85 (br d, 1H), 1,4 (m, 2H), 1,35 (m, 2H), 1,1 (m, 1H), 0,95 (t, 3H).
[0061] (3S,4R,5R)-3-(дифторметил)-4,5-дигидрокси-N-фенилпиперидин-1-карботиоамид (9е; Х=S; R5=фенил). Последующей общей методикой Е и с применением фенилизотиоцианата (0,16 г, 1,2 ммоль) получали названное соединение в виде белого твердого вещества (0,31 г, 86%). MH+=303.1Н ЯМР (DMSO-d6) 9,5 (s, 1Н), 7,3 (m, 4H), 7,1 (t, 1H), 6,35 (t, 1H), 5,35 (2d, 2H), 4,85 (d, 1H), 4,55 (d, 1H), 3,45 (m, 2H), 3,2 (t, 1H), 3,0 (t, 1H), 2,05 (br d, 1H).
[0062] Соединения по настоящему изобретению также могут быть получены специалистом настоящей области с применением следующих общих схем:
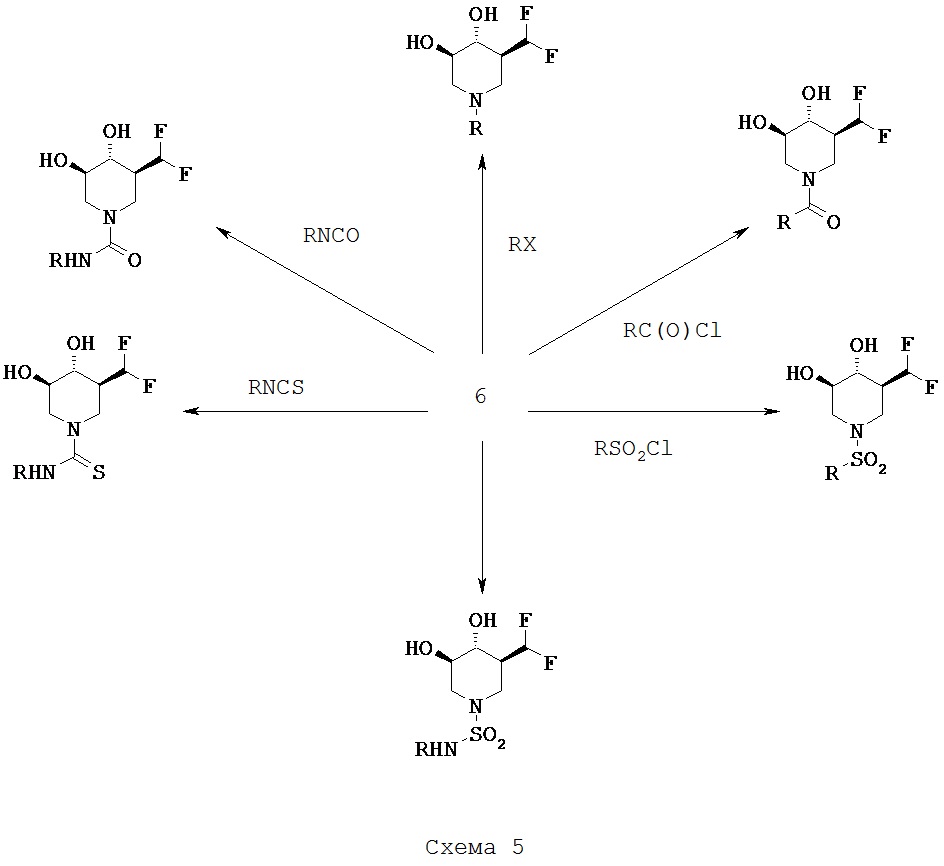
Соли, сольваты и пролекарства
[0063] Соединения настоящего изобретения включают фармацевтически приемлемые соли, сольваты и пролекарственные средства раскрытых здесь соединений. Фармацевтически приемлемые соли включают соли, полученные из неорганических оснований, таких как Li, Na, К, Са, Mr, Fe, Cu, Zn, Mn; соли органических оснований, таких как N,N'-диацетилэтилендиамин, глюкамин, триэтиламин, холин, гидроксид, дициклогексиламин, метформин, бензиламин, триалкиламин, тиамин; хиральные основания, такие как алкилфениламин, глицинол, фенилглицинол, соли природных аминокислот, таких как глицин, аланин, валин, лейцин, изолейцин, норлейцин, тирозин, цистин, цистеин, метионин, пролин, гидроксипролин, гистидин, омитин, лизин, аргинин, серии; неприродные аминокислоты, такие как D-изомеры или замещенные аминокислоты; гуанидин, замещенный гуанидин, где заместители выбраны из нитро, амино, алкила, алкенила, алкинила, аммониевых или замещенных аммониевых солей и солей алюминия. Соли могут включать кислотно-аддитивные соли, приемлемыми из которых являются гидрохлориды, сульфаты, нитраты, фосфаты, перхлораты, бораты, гидрогалиды, ацетаты, тартаты, малеаты, цитраты, сукцинаты, пальмоаты, метансульфонаты, бензоаты, салицилаты, бензолсульфонаты, аскорбаты, глицерофосфаты, кетогутараты. В одном варианте осуществления фармацевтически приемлемая соль соединений, раскрытых в настоящем изобретении является гидрохлоридной солью.
[0064] «Сольват» обозначает физическую ассоциацию соединения с одной или несколькими молекулами растворителя. Эта физическая ассоциация включает различные степени ионной и ковалентной связи, включая водородную связь. В некоторых случаях сольват может быть способен к изоляции, например, когда одна или несколько молекул растворителя включаются в кристаллическую решетку кристаллического твердого вещества. «Сольват» охватывает как фазу раствора, так и выделяемые сольваты. «Гидрат» представляет собой сольват, в котором растворителем является молекула H2O. Другие неограничивающие примеры подходящих сольватов включают спирты (например, этанолаты, метанолаты и т.п.).
[0065] Пролекарства представляют собой соединения, которые превращаются in vivo в активную форму (см., например, R.В.Silverman, 1992, «The Organic Chemistry of Drug Design and Drug Action», Academic Press, Глава 8, включена в настоящий документ посредством ссылки). Дополнительно, обсуждение пролекарств представлено в Higuchi and V.Stella, Pro-drugs as Novel Delivery Systems, Volume 14 of the A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, Edward В. Roche, ed., American Pharmaceutical Association and Pergamon Press, 1987, обе из которых включены в настоящий документ посредством сслыки на них. Пролекарства могут использоваться для изменения биораспределения (например, допускать соединениям, которые обычно не могут попасть в реакционноспособный участок протеазы) или фармакокинетики для отдельного соединения. Например, карбоксильная кислотная группа может быть подвергнута эстерификации, например, метильной группой или этильной группой, давая сложный эфир. Когда сложный эфир вводится субъекту, сложный эфир расщепляется, ферментативно или неферментативно, восстановительно, окислительно или путем гидролиза, открывая анионную группу. Анионная группа может быть эстерифицирована компонентами (например, ацилоксиметиловые сложные эфиры), которые расщепляются, высвобождая промежуточное соединение, которое затем разлагается, давая активное соединение.
[0066] Примеры пролекарств и их использование хорошо известны в настоящем уровне техники (см., например, Berge et al. (1977) «Pharmaceutical Salts», J. Pharm. Sci. 66:1-19). Пролекарства могут готовиться in situ во время финального выделения и очистки соединений, или отдельно реакцией очищенного соединения с подходящим средством для получения производных. Например, гидроксильные группы могут превращаться в сложные эфиры путем воздействия карбоновой кислоты в присутствии катализатора. Примеры расщепляемых спиртовых пролекарственных фрагментов включают замещенные и незамещенные, разветвленные или неразветвленные низшие алкильные сложноэфирные фрагменты (например, этиловые сложные эфиры), низшие алкениловые сложные эфиры, динизшие алкил-аминовые низшие-алкиловые сложные эфиры (например, диметиламиноэтиловый сложный эфир), ациламиновые низшие алкиловые сложные эфиры, ацилокси низшие алкиловые сложные эфиры (например, пивалоилоксиметиловый сложный эфир), ариловые сложные эфиры (фениловые сложные эфиры), арил-низшие алкиловые сложные эфиры (например, бензиловый сложный эфир), замещенные (например, метил, галоген или метокси заместители) ариловые и арил-низшие алкиловые сложные эфиры, амиды, низшие-алкиловые амиды, динизшие алкиловые амиды и гидрокси амиды.
[0067] Все стереоизомеры (например, геометрические изомеры, оптические изомеры и т.п.) соединений, раскрытых в настоящем документе (включая те соли, сольваты и пролекарства этих соединений, а также соли и сольваты пролекарств), такие как те, которые могут существовать благодаря асимметричным углеродам в различных заместителях, включая энантиомерные формы (которые могут существовать даже в отсутствие асимметричных углеродов), ротамерные формы, атропизомеры и диастереомерные формы, предусматриваются внутри объема настоящего изобретения. Отдельные стереоизомеры этих соединений могут, например, быть в основном свободными от других изомеров, или могут быть смешанными, например, как рацематы или со всеми другими, или другими выбранными, стереоизомерами. Хиральные центры вышеупомянутых соединений могут иметь S или R конфигурацию, как определяется в Рекомендации ИЮПАК 1974. Применение выражений «соль», «сольват», «пролекарство» и т.п., подразумевает равное применение к соли, сольвату и пролекарству энантиомеров, стереоизомеров, ротамеров, таутомеров, рацематов или пролекарств соединения настоящего изобретения, раскрытых в настоящем документе.
Составы
[0068] Терапевтическое средство(средства) может составляться, чтобы подходить для любого способа введения, включая, например, пероральный в форме таблеток или капсул, или жидкости, или в стрерильном водном растворе для инъекций. Когда терапевтическое средство(средства) составляется для перорального введения, таблетки или капсулы могут готовиться обычным способом с фармацевтически приемлемым наполнителем, таким как вяжущие вещества (например, желатинированный маисовый крахмал, поливинилпирролидон или гидроксипропил метилцеллюлоза); наполнители (например, лактоза, мироккристаллическая целлюлоза или кальция водород фосфат); скользящие вещества (например, стеарат магния, тальк или кремнезем); разрыхлители (например, картофельный крахмал или натрия крахмал гликолят) или смачивающие средства (например, натрия лаурил сульфат). Таблетки могут покрываться способами, хорошо известными в настоящем уровне техники. Жидкие препараты для перорального введения могут принимать форму, например, растворов, сиропов или суспензий, или они могут быть в виде сухого продукта для разбавления водой или другим подходящим растворителем перед применением. Такие жидкие препараты могут быть приготовлены обычным способом с фармацевтически приемлемыми добавками, такими как суспендирующие средства (например, сироп сорбита, производные целлюлозы или гидрогенизированные пищевые жиры); эмульгирующие средства (например, лецитин или гуммиарабик); неводные растворители (например, миндальное масло, жирные сложные эфиры, этиловый спирт или фракционированные растительные масла); или консерванты (например, метил, или пропил-р-гидроксибензоаты, или сорбиновая кислота). Жидкие препараты также могут содержать приемлемые буферные соли, ароматизаторы, красящие средства или подсластители. Препараты для перорального введения могут составляться соответственно, чтобы давать контролируемое или длительное высвобождения терапевтического средства(средств).
[0069] В определенных вариантах осуществления настоящего изобретения терапевтическое средство(средства) вводится в лекарственной форме, предусматривающей системное поглощение, так, чтобы терапевтическое средство(средства) могли пересекать гематэнцефалический барьер, так, чтобы оказывать влияние на нервные клетки. Например, фармацевтические составы терапевтического средства(средств), подходящие для парэнтерального/инъекционного применения, обычно включают стерильные водные растворы (если растворимы в воде) или дисперсии и стерильные порошки для немедленного приготовления стерильных инъекционных растворов или дисперсий. Во всех случаях форма должна быть стерильной и должна быть жидкой в пределах существования легкой возможности введения через шприц. Она должна быть стабильной в условиях производства и хранения и должна предохраняться от загрязняющего воздействия микроорганизмов, таких как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, полиэтиленгликоль и т.п.), их подходящей смесью или растительными маслами. Соответствующая текучесть может поддерживаться, например, применением оболочки, такой как лецитин, поддержанием требуемого размера частиц в случае дисперсий и применением поверхностно-активных веществ. Предотвращение действия микроорганизмов может обуславливаться различными антибактериальными и противогрибковыми средствами, например парабенами, хлорбутанолом, фенолом, бензиловым спиртом, сорбиновой кислотой и т.п. Во многих случаях будет иметь смысл включение изотонических средств, например сахаров или хлорида натрия. Продленная адсобция инъекционных композиций может обуславливаться применением в композиции средств, задерживающих адсорбцию, например моностеарата алюминия или желатина.
[0070] Стерильные инъекционные растворы готовят включением терапевтического средства(средств) в требуемом количестве в соответствующий растворитель с различными другими ингредиентами, перечисленными выше, при необходимости, с последующей фильтрацией или терминальной стерилизацией. Обычно дисперсии готовят введением различных простерилизованных активных ингредиентов в стерильный растворитель, содержащий основную дисперсионную среду и требуемые другие ингредиенты из перечисленных выше. В случае стерильных порошков для приготовления стерильных инъекционных растворов, предпочтительными способами приготовления являются вакуумная сушка и лиофилизация, дающие порошок активного ингредиента с добавлением любых дополнительных желательных ингредиентов из их раствора, предварительно стерилизованного фильтрованием.
[0071] Состав может содержать наполнитель. Фармацевтически приемлемыми наполнителями, которые могут включаться в состав, являются буферы, такие как цитратный буфер, фосфатный буфер, ацетатный буфер и бикарбонатный буфер, аминокислоты, мочевина, спирты, аскорбиновая кислота, фосфолипиды; белки, такие как сывороточный альбумин, коллаген и желатин; соли, такие как ЭДТА или ЭГТА и хлорид натрия; липосомы; поливинилпироллидон; сахара, такие как декстран, маннитол, сорбит и глицерин; пропиленгликоль и полиэтиленгликоль (например, PEG-4000, PEG-6000); глицерин; глицин или другие аминокислоты; и липиды. Буферные системы для применения с составами включают цитратный; ацетатный; бикарбонатный и фосфатный буферы. Фосфатные буфер является предпочтительным вариантом осуществления.
[0072] Состав может также содержать неионный детергепт. Предпочтительные неионные детергенты включают Полисорбат 20, Полисорбат 80, Тритон Х-100, Тритон Х-114, Нонидет Р-40, октил α-глюкозид, октил Р-глюкозид, Бридж 35, Плюроник и Твин 20.
Пути введения
[0073] Терапевтическое средство(средства) может вводиться перорально или парентерально, включая внутривенно, подкожно, внутриартериально, внутрибрюшинно, глазное введение, внутримышечно, буккально, ректально, вагинально, интраорбитально, интрацеребрально, внутрикожно, интракраниально, интраспинально, внутрижелудочково, интратекально, интрацистернально, внутрикапсулярно, внутрилегочно, интраназально, трансмукозально, трансдермально или путем ингаляции. В одном предпочтительном варианте осуществления терапевтическое средство(средства) вводится перорально.
[0074] Введение терапевтического средства(средств) может осуществляться путем периодических инъекций болюса состава или может вводиться внутривенным или внутрибрюшинным введением из резервуара, внешнего (например, контейнер для внутривенных вливаний) или внутреннего (например, биоразрушаемый имплант). См., например, патенты США №4407957 и №5798113, каждый включен в настоящем документе ссылкой. Способы внутрилегочной доставки и аппаратура описываются, например, в патентах США №5654007, №5780014 и №5814607, каждый включен в настоящем документе ссылкой. Другие пригодные системы парентеральной доставки включают этилен-винилацетатные сополимерные частицы, осмотические насосы, имплантируемые инфузионные системы, насосную доставку, инкапсулированную клеточную доставку, липосомальную доставку, игольную инъекцию, безыгольную инъекцию, ингалятор, аэрозоль, электропорацию и трансдермальный пластырь. Безыгольный инъекторные устройства описываются в патентах США №5879327; №5520639; №5846233 и №5704911, спецификации которых включены в настоящем документе ссылкой. Любой из составов, описанных выше, может вводиться с использованием этих способов.
[0075] Подкожные инъекции имеют преимущества, предусматривая самовведение, кроме того, также приводя к удлиненному периоду полужизни в плазме, по сравнению с внутривенным введением. Кроме того, большое разнообразие устройств, разработанных для удобства пациента, таких как перезаполняемые шприцы-ручки и безыгольные инъекционные устройства, может использоваться с составами настоящего изобретения, как обсуждается в настоящем документе.
Дозировка
[0076] Подходящий фармацевтический препарат находится в стандартной лекарственной форме. В такой форме препарат разделяют на стандартные дозы, подходящего размера, содержащие соответствующие количества активного компонента, например эффективное количество для достижения желаемой цели. В определенных вариантах осуществления терапевтическое средство(средства) вводится в одной или нескольких ежедневных дозах (например, раз в день, два раза в день, три раза в день). В определенных вариантах осуществления терапевтическое средство(средства) вводится с перерывами.
[0077] Примерные режимы дозирования описываются в Международной патентной заявке PCT/US08/61764, опубликованной как WO 2008/134628 11 июня 2008, и предварительной патентной заявке США 61/108,192, зарегистрированной 24 октября 2008, обе из которых включены ссылкой в настоящий документ во всей своей полноте. В одном варианте осуществления терапевтическое средство(средства) вводится в прерывистом режиме дозирования, включающем начальную «ударную дозу», даваемую ежедневно, с последующим периодом неежедневного интервального дозирования.
[0078] Количество эффективного терапевтического средства(средств) для предупреждения или лечения упомянутого расстройства может определяться в каждом конкретном случае специалистами в настоящем уровне техники. Количество и частота введения терапевтического средства(средств) будут регулироваться в соответствии с решением лечащего клинициста (врача), с учетом таких факторов, как возраст, состояние и размеры тела пациента, а также риска развития расстройства или тяжести симптомов упомянутого расстройства, которое лечат.
Комбинированная лекарственная терапия
[0079] Терапевтическое средство(средства) настоящего изобретения могут вводиться в комбинации с по меньшей мере одним другим терапевтическим средством. Введение терапевтического средства(средств) настоящего изобретения с по меньшей мере одним другим терапевтическим средством понимается, как включающее введение, которое является последовательным или совместным. В одном варианте осуществления терапевтические средства вводятся в отдельных лекарственных формах. В другом варианте осуществления два или более терапевтических средства вводятся одновременно в одной и той же лекарственной форме.
[0080] В определенных вариантах осуществления терапевтическое средство(средства) настоящего изобретения вводятся в комбинации с по меньшей мере одним другим терапевтическим средством, которое представляет собой антидискинетическое средство (например, карбидопа, леводопа), антибактериальное средство (например, миглустат), противоопухолевое средство (например, бусульфан, циклофосфамид), желудочно-кишечное средство (например, метилпреднизолон), витамин (например, кальцитриол, холекальциферол, эргокальциферолы, витамин D), сосудосуживающее средство (например, кальцитриол). В одном предпочтительном варианте осуществления вышеупомянутые другие терапевтические средства вводятся, если расстройство представляет собой болезнь Гоше.
[0081] В определенных вариантах осуществления терапевтическое средство(средства) настоящего изобретения вводятся в комбинации с аллопрегналоном, диетой с низким содержание холестерина или средствами, снижающими холестерин, такими как статины (например, Lipitor®); фибраты, такие как фенофибрат (Lipidil®); ниацин; и/или связывающие смолы, такие как холестирамин (Questran®).
[0082] В одном варианте осуществления терапевтическое средство(средства) настоящего изобретения вводится в комбинации с генной терапией. Генная терапия предусматривает как замещение генов, таких как ген глюкоцереброзидазы, так и введение ингибирующих РНК (siRNA) для гена SNCA. Генная терапия описывается более детально в Патенте США №7446098, заявка на который была подана 17 февраля 2004.
[0083] В одном варианте осуществления терапевтическое средство(средства) настоящего изобретения вводится в комбинации с по меньшей мере одним другим терапевтическим средством, которое представляет собой противовоспалительное средство (например, ибупрофен или другое NSAID).
[0084] В одном варианте осуществления терапевтическое средство(средства) настоящего изобретения вводится в комбинации с субстратным ингибитором глюкоцереброзидазы, таким как N-бутил-деоксинойиримицин (Zavesca®; миглустат можно получить в Actelion Pharmaceuticals, US, Inc., South San Francisco, CA, USA).
[0085] Комбинации терапевтического средства(средств) настоящего изобретения по меньшей мере с одним другим терапевтическим средством, которое представляет собой терапевтическое средство для одного или нескольких других лизосомных ферментов, также предусматриваются. Таблица 2 содержит неограничивающий список терапевтических средств для лизосомных ферментов.
[0086] В определенных вариантах осуществления терапевтическое средство(средства) настоящего изобретения вводятся в комбинации по меньшей мере с одним терапевтическим средством, которое представляет собой антидискинетическое средство (например, карбидопа, леводопа), антибактериальное средство (например, циклоспорин, миглустат, пириметамин), противоопухолевое средство (например, алемтузумаб, азатиоприн, бусульфан, клофарабин, циклофосфамид, мелфалан, метотрексат, ритуксимаб), противоревматическое средство (например, ритуксимаб), желудочно-кишечное средство (например, метилпреднизолон), витамин (например, кальцитриол, холекальциферол, эргокальциферолы, фолиевая кислота, витамин D), средство контроля размножения (например, метотрексат), средство для дыхательной системы (например, тетрагидрозолин), сосудосуживающее средство (например, кальцитриол, тетрагидрозолин).
[0087] В определенных вариантах осуществления терапевтическое средство(средства) настоящего изобретения вводятся в комбинации по меньшей мере с одним терапевтическим средством, которое представляет собой терапевтическое средство для β-гексозаминидазы А и/или терапевтическое средство для кислой β-галактозидазы. В определенных вариантах осуществления терапевтическое средство(средства) настоящего изобретения вводятся в комбинации по меньшей мере с одним терапевтическим средством, которое представляет собой антибактериальное средство (например, миглустат), противоопухолевое средство (например, алемтузумаб, бусульфан, циклофосфамид), желудочно-кишечное средство (например, метилпреднизолон). В одном варианте осуществления вышеупомянутая комбинация вводится людям, подверженным риску или с диагностированной болезнью Ниманна-Пика (например, болезнь Ниманна-Пика типа С).
ПРИМЕРЫ
[0088] Настоящее изобретение далее описывается посредством примеров, представленных ниже. Применение таких примеров является только иллюстративным и ни в коей мере не ограничивает объем и содержание настоящего изобретения или любого приведенного в примере выражения. Подобным образом, настоящее изобретение не ограничивается никаким особенным предпочтительным вариантом осуществления, описанным в настоящем документе. В действительности, множество модификаций и разновидностей настоящего изобретения будут очевидны для специалистов в настоящей области техники при прочтении этого описания. Настоящее изобретение, следовательно, ограничивается только выражениями приложенной формулы изобретения наряду с полным объемом эквивалентов, для которых формула является правомочной.
ПРИМЕР 1: Определение констант ингибирования
[0089] Связывающую способность (определяемая здесь К, константой связывания) глюкоцереброзидазы к новому соединению настоящего изобретения определяли эмпирически, используя ферментные ингибиторные анализы. Кратко, ферментные ингибиторные анализы контролируют способность тестируемого соединения связываться и предотвращать гидролиз флуорогенного субстрата в зависимости от концентрации. В частности, ферментативная активность рекомбинантной человеческой глюкоцереброзидазы (rhGCase; Cerezyme®, Genzyme Corp.) измеряли, используя 4-метилумбеллиферил-β-D-глюкопиранозидный (4-MU-β-D-Glc) флуорогенный субстрат в отсутствии или в присутствии переменных количеств каждого тестируемого соединения. Полученные данные анализировали сравнением всех тестируемых образцов с неингибированным контрольным образцом (без соединения; соответствующий 100% ферментативной активности) для определения остаточной ферментативной активности в присутствии тестируемого соединения. Нормализованные данные остаточной активности затем нанесли на график (на y - оси) относительно концентрации тестируемого соединения (на x - оси) для экстраполяции концентрации тестируемого соединения, приводящей к 50% ингибированию ферментативной активности (обозначается как IC50). Значение IC50 для каждого тестируемого соединения затем вставили в уравнение Ченга-Прусоффа (подробно рассмотрено ниже) для получения константы абсолютного ингибирования Ki, которая точно отражает связывающую способность глюкоцереброзидазы к тестируемому соединению. Ферментные ингибиторные анализы проводили как при рН 7,0 (рН эндоплазматического ретикулума), так и при рН 5,2 (рН лизосом), чтобы разобраться в связывающей способности (т.е. эффективности) соединения к глюкоцереброзидазе в эндоплазматическом ретикулуме и лизосоме.
In vitro анализ
[0090] Различные концентрации тестируемого соединения приготовили в буфере «М», содержащем 50 мМ натрий-фосфатного буфера с 0,25% таурохлората натрия при рН 7,0 и рН 5,2. Фермент (Cerezyme®, рекомбинантная форма человеческого фермента β-глюкоцереброзидазы) также разводили в том же самом буфере «М» при рН 7,0 и рН 5,2. Раствор субстрата состоял из 3 мМ 4-метилумбеллиферон-β-D-глюкопиранозида в буфере «М» с 0,15% Тритона Х-100 при обоих рН. Пять микролитров разведенного фермента добавили к 15 мкл различных концентраций ингибитора или одного лишь буфера «М» и инкубировали при 37°С в течение 1 часа с 50 мкл приготовленного субстрата для оценки β-глюкозидазной активности при рН 7,0 и рН 5,2. Реакции останавливали добавлением равного объема 0,4 М глицина, рН 10,6. Флуоресцению измеряли на сканирующем спектрофотометре для прочтения планшетов со скоростью 1 сек/лунка, с использованием 355 нм возбуждения и 460 нм излучения. Инкубации без добавленных ферментов или без добавленных ингибиторов использовали для определения отсутствия ферментативной активности и максимальной активности, соответственно, и нормализовали % ингибирования для данного анализа. Результаты таких анализов ингибирования in vitro для контрольного соединения IFG-тартрата и нескольких тестируемых соединений обобщаются ниже в таблице 2А.
In situ анализ
[0091] Эффект новых соединений настоящего изобретения на активность лизосомной глюкоцереброзидазы анализировали in situ, используя фибробласты, полученные от нормального человека. Клетки, высеянные в 48-луночные планшеты, инкубировали с указанными концентрациями соединения в течение 16-24 ч. Для анализов эффекта дозы, клетки инкубировали с in situ субстратом 5-(пентафторбензоиламино)флуоресцин ди-β-D-глюкопиранозидом (PFBFDβGlu) в течение 1 часа и затем лизировали для определения объема гидролиза субстрата в присутствии соединения. В анализе использовали диапазон из 12 концентраций, охватывающих 5 порядков величины, центрированных на IC50. В частности, использовали следующие диапазоны концентрации: (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, (3R,4R,5R)-5-(1-гидроксиэтил)-пиперидин-3,4-диол, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорид и (3R,4R,5S)-5-бензилпиперидин-3,4-диол: от 1,0×10-3 до 3,0×10-9 М; (3R,4R,5R)-5-(1-гидроксиэтил)-пиперидин-3,4-диол: от 1,0×10-4 до 3,0×10-10 М; и (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол: от 1,0×10-3 до 3,0×10-11 М; где соединение последовательно разводили 1:3 от самой высокой концентрации в указанных диапазонах. Ингибирование определяли как соотношение активности в присутствии соединения к активности в отсутствии соединения. Для анализов отмывки клетки обрабатывали соединением в течение 16-24 часов при концентрации, равной IC90. Клетки тщательно промыли и инкубировали в среде без лекарственного средства, чтобы обеспечить вытекание чистого соединения из клеток. В клетках затем проверяли активность лизосомной глюкоцереброзидазы с 2-часовыми интервалами в течение общего периода 8 часов после удаления соединения. Увеличение активности с течением времени аппроксимировали с единой экспоненциальной функцией для определения времени отмывки соединения. Результаты этих in situ анализов ингибирования для контрольного соединения IFG-тартрата и нескольких тестируемых соединений обещаются ниже в таблице 2В.
[0092] По сравнению с контрольным соединением IFG-тартратом отмечено следующее: (i) обнаружено, что тестируемые соединения (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, (3R,4R,5R)-5-(1-гидроксиэтил)-пиперидин-3,4-диол, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорид и (3R,4R,5S)-5-бензилпиперидин-3,4-диол вызывают концентрация-зависимое увеличение активности глюкоцереброзидазы и улучшенную ферментативную активность до того же максимального уровня, что и контрольное соединение IFG-тартрат, при намного более низкой концентрации; (ii) тестируемые соединения (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол и (3R,4R,5S)-5-бензилпиперидин-3,4-диол отмывались из лизосомного компартмента (in situ отмывка) намного быстрее, чем контрольное соединение IFG-тартрат; и (iii) тестируемые соединения (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, (3R,4R,5R)-5-(1-гидроксиэтил)-пиперидин-3,4-диол, (3R,4R,5R)-5-(1-гидроксиэтил)-пиперидин-3,4-диол, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорид и (3R,4R,5S)-5-бензилпиперидин-3,4-диол ингибировали активность глюкоцереброзидазы.
ПРИМЕР 2: Проникновение через гематоэнцефалический барьер
[0093] Проникновение через гематоэнцефалический барьер (ВВВ) контрольного соединения IFG-тартрата и нескольких соединений настоящего изобретения (а именно, (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диола, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диола, (3R,4R,5R)-5-(1-гидроксиэтил)-пиперидин-3,4-диола, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорида и (3R,4R,5S)-5-бензилпиперидин-3,4-диола) анализировали после орального введения мышам. Для этой цели, 8-недельным самцам мыши дикого типа (C57BL/6) вводили однократную 30 мг/кг (эквивалент свободного основания) п/о дозу контрольного или тестируемого соединения через желудочный зонд (n=3 мыши на момент времени). Растворы дозировок готовили на воде. После введения доз, мышей умерщвляли СО2 в следующие моменты времени: 0-, 0,5-, 1-, и 4-часа после введения дозы. После умерщвления, собрали цельную кровь из нижней полой вены в пробирки с литий-гепарином. Аналогично, собрали мозг от каждой мыши. Плазму получили центрифугированием цельной крови при 2700×g в течение 10 минут при 4°С с последующим хранением на сухом льду. Мозг целиком промыли в холодном PBS для удаления загрязняющей его крови, промокнули до сухости, быстро заморозили на сухом льду и, наконец, хранили при -80°С до проведения анализа. Для приготовления образцов мозга для анализа, 50-100 мг ткани гомогенизировали в 400 мкл воды/мг ткани. Образцы затем очищали центрифугированием. Затем, 25 мкл супернатанта мозгового гомогената или 25 мкл плазмы смешали с 25 мкл ацетонитрил:водой (95/5). К этому добавили 25 мкл ацетонитрила и 50 мкл внутреннего стандарта (100 нг/мл IFG-тартрата 13C2-15N в 0,5% муравьиной кислоте в (70:30) ацетонитрил:метаноле). Образцы снова очистили центрифугированием и 75 мкл супернатанта смешали с 75 мкл ацетонитрила. Затем образцы проанализировали на уровни соединения с помощью LC-MS/MS в PPD Inc. (3230 Deming Way, Middleton, WI 53562). Вкратце, использовали колонку Thermo Betasil, Silica-100, 50×3 мм, 5 мк, уравновешенную смесью подвижной фазы, состоящей из 5 мМ формиата аммония и 0,05% муравьиной кислоты в (А) 95:5 ацетонитрил:воде или (В) 70:20:10 метанол:вода:ацетонитриле. Для анализа впрыскивали между 20 и 30 мкл образца. Для вычисления концентраций лекарственного средства, исходные данные для плазмы (нг/мл) и мозга (нг/г) преобразовали в нм, используя молекулярный вес соответствующего соединения и принимая, что 1 г ткани эквивалентен 1 мл объема. Концентрация как функция времени нанесена на график в GraphPad Prism версия 4.02.
[0094] Уровни в плазме и мозге, определенные у мышей, которым была введена однократная 30 мг/кг (эквивалент в чистом основании) п/о доза контрольного соединения (т.е. IFG-тартрата) или тестируемого соединения (т.е. (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диола, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диола, (3R,4R,5R)-5-(1-гидроксиэтил)-пиперидин-3,4-диола, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорида или (3R,4R,5S)-5-бензилпиперидин-3,4-диола), отражают, что (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорид и (3R,4R,5S)-5-бензилпиперидин-3,4-диол переходят через гематоэнцефалический барьер более легко по сравнению с IFG-тартратом. Дополнительно, более высокие уровни (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диола, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диола, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорида и (3R,4R,5S)-5-бензилпиперидин-3,4-диола обнаружены в мозге, чем наблюдаемые после введения IFG-тартрата.
ПРИМЕР 3: Усиление глюкоцереброзидазы
[0095] Способность перорально введенного тестируемого соединения ((3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорид или (3R,4R,5S)-5-бензилпиперидин-3,4-диол) повышать уровни глюкоцереброзидазы оценивали на мышах. Для этой цели, 8-недельным самцам мыши дикого типа (C57BL/6) вводили однократную п/о (через желудочный зонд) дозу соединения настоящего изобретения (т.е. (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, (3R,4R,5S)-5-((R)-1-фторпропил)пиперидин-3,4-диол гидрохлорид или (3R,4R,5S)-5-бензилпиперидин-3,4-диол). Детали вводимой дозы для каждого соединения даны в таблицах 3А и 3В. Растворы дозировок готовили на воде. Соединения вводили в течение 2 недель следующим образом: неделя 1, Пн-Пт (включены), Сб-Вс (исключены); неделя 2, Пн-Чт (включены); вскрытие в пятницу. Таким образом, всего 9 доз (растворы дозировок приготавливали свежими ежедневно) давали каждой мыши, с 24-часовой отмывкой между последней дозой и вскрытием.
[0096] После завершения введения доз, мышей умертвили с СО; и цельную кровь брали из нижней полой вены в пробирки с литий-гепарином. Плазму собирали центрифугированием крови при 2700g в течение 10 минут при 4°С. Ткани печени, селезенки, легкого и мозга ткани отделили, промыли в холодном PBS, промокнули до сухости, быстро заморозили на сухом льду и хранили при -80°С до проведения анализа. Уровни глюкоцереброзидазы измеряли, гомогенизируя приблизительно 50 мг ткани в 500 мкл буфера Мак-Илвейна (Ml) (100 мМ цитрата натрия, 200 мМ двузамещенного фосфата натрия, 0,25% таухлората натрия и 0,1% Тритона Х-100, рН 5,2) при рН 5,2 в течение 3-5 секунд на льду с помощью микрогомогенизатора. Затем гомогенаты инкубировали при комнатной температуре без и с 2,5 мМ кондуритол-В-эпоксидом (СВЕ) в течение 30 мин. Наконец, добавили 3,7 мМ 4-метилумбериферрил-β-глюкозидного (4-MUG) субстрата и инкубировали при 37°C в течение 60 мин. Реакции останавливали добавлением 0,4 М глицина, рН 10,6. Флуоресценцию измеряли на сканирующем спектрофотометре для прочтения планшетов со скоростью 1 сек/лунка, используя 355 нм возбуждение и 460 нм излучение. Общий белок определяли в лизатах, используя набор MicroBCA в соответствии с инструкциями производителя. Стандартную кривую 4-метилумбеллиферона (4-MU), в диапазоне от 1,0 нМ до 50 мкМ, пустили параллельно для перевода исходных данных флуоресценции в абсолютную активность глюкоцереброзидазы (в присутствии и отсутствии СВЕ) и выразили как наномоли высвобожденного 4-MU на миллиграмм белка в час (нмоль/мг белка/ч). Уровни глюкоцереброзидазы и белка рассчитали, используя Microsoft Excel (Редмонд, Вашингтон) и GraphPad Prism версия 4.02.
[0097] Таблицы 3А и 3В обобщают введенные дозы для каждого соединения, изученные на мышах, как описано выше, а также полученный уровень усиления глюкоцереброзидазы в мозге и селезенке, соответственно, концентрацию соединения в ткани, концентрацию соединения в анализе глюкоцереброзидазы и константу ингибирования (Ki).
[0098] Как отражено в таблицах 3А и 3В, у мышей, которым вводили (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол, или (3R,4R,5S)-5-бензилпиперидин-3,4-диол, показано значительное усиление глюкоцереброзидазы в мозге и селезенке.
ПРИМЕР 4: Фармакокинетика у крыс
[0099] Данные фармакокинетики (РК) получали на крысах для оценки биодоступности тестируемого соединения. В частности, рассчитывали следующие параметры РК: биодоступность, измеренная как площадь под кривой концентрация/время (AUC), доступную долю дозы (%F; дополнительно определена ниже), клиренс (CL), объем распределения (Vd), и период полужизни (t½). Для этой цели, 8-недельным самцам крысы линии Спраг Доули давали или однократную внутривенную (в/в) дозу, эквивалентную 3 мг/кг свободного основания, или однократные, вводящиеся п/о (через зонд), дозы тестируемого соединения, эквивалентные 10, 30, и 100 мг/кг свободного основания. На каждую группу дозировки использовали по три крысы. Кровь собирали в течение 24-часового периода. Моменты времени для забора крови после внутривенного введения были: 0, 2,5, 5, 10, 15, 30, 45 мин, 1, 2, 4, 8, 12, и 24 часа; моменты времени для сбора крови после п/о введений были: 0, 5, 15, 30, 45 мин, 1, 2, 3, 4, 8, 12, и 24 часа. Образцы плазмы исследовали на уровни соединения с помощью LC-MS/MS в PPD. Исходные данные анализировали некомпартменым методом в Win-nonLin для вычисления VD, %F, CL и t½.
[00100] Различные фармакокинетические параметры для (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диола, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диола и (3R,4R,5S)-5-бензилпиперидин-3,4-диола на основе вышеупомянутого исследования раскрываются ниже в таблицах 4A-D.
[00101] Как отражено в таблицах 4A-D, (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол и (3R,4R,5S)-5-бензилпиперидин-3,4-диол имеют подходящие фармакокинетические профили для разработки лекарственного средства. В частности, (3R,4R,5S)-5-(дифторметил)пиперидин-3,4-диол, (3R,4R,5S)-5-(1-фторэтил)пиперидин-3,4-диол и (3R,4R,5S)-5-бензилпиперидин-3,4-диол демонстрируют прекрасную оральную биодоступность (приблизительно 50-100%) и пропорциональность доле, период полужизни 1,0-4,0 ч и объем распределения, означающий достаточное проникновение в периферические ткани.
Реферат
Изобретение относится к способу лечения пациента с диагнозом болезни Гоше, который включает введение пациенту, нуждающемуся в лечении, эффективного количества по меньшей мере одного соединения, выбранного из группы, состоящей из:,,,,,,,,,,,.Изобретение также относится к варианту способа лечения пациента с диагнозом болезни Гоше и к набору, обладающему активностью в отношении глюкоцереброзидазы, содержащему контейнер с эффективным количеством по меньшей мере одного соединения изобретения. Технический результат: предложен способ лечения пациента с диагнозом болезни Гоше новыми производными пиперидин-3,4-диола. 3 н. и 8 з.п. ф-лы, 10 табл., 4 пр.
Формула








Документы, цитированные в отчёте о поиске
Производные 4-гидроксипиперидина
Комментарии