Производные 4-гидроксипиперидина - RU2178412C2
Код документа: RU2178412C2
Чертежи


Описание
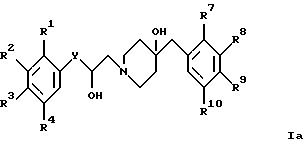
Изобретение относится к соединениям общей формулы
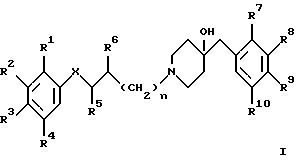
где X обозначает -O-, -NH-, -CH2 -, -CH= , -CO2-, -CONH-, -CON(низший алкил)-, -S- и -SO2-;
R1 - R4, независимо друг от друга, обозначают водород, галоген, гидрокси-, амино-, нитрогруппу, низший алкилсульфониламид, 1- или 2-имидазолил, 1-(1,2,4-триазолил) или ацетамид;
R5, R6, независимо друг от друга, обозначают водород, низший алкил, гидрокси-, низшую алкокси- или оксогруппу;
R7 - R10, независимо друг от друга, обозначают водород, низший алкил, галоген, трифторметил или низшую алкоксигруппу;
n обозначает 0 или 1;
и к их фармацевтически приемлемым кислотно-аддитивным солям.
Соединения формулы I и их соли характеризуются ценными терапевтическими свойствами. Предлагаемые по настоящему изобретению соединения являются селективными блокаторами подтипа N-метил-D-аспартат-рецепторов (NMDA-рецепторов), которые играют ключевую роль в модуляции нейронной активности и пластичности, что делает их основными участниками промежуточных процессов, лежащих в основе развития центральной нервной системы, а также способности к обучению и формированию памяти.
В патологических условиях острой и хронической форм нейродегенерации повышенная активность NMDA-рецепторов является важнейшим фактором инициирования гибели нервных клеток. NMDA-рецепторы состоят из представителей семейств двух подгрупп, а именно: NR-1 (8 различных форм срастания) и NR-2 (от A до D), происходящих от различных генов. Представители семейств этих двух подгрупп проявляют различную локализацию в различных зонах головного мозга. Гетеромерные сочетания представителей NR-1 с различными подгруппами NR-2 обусловливают проявление NMDA-рецепторами различных фармацевтических свойств. Возможные терапевтические показания для применения специфических блокаторов подтипа NMDA-рецепторов включают острые формы нейродегенерации, вызванной, например, внезапным приступом и травмой головного мозга, и хронические формы нейродегенерации, такие, как болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, боковой амиотрофический склероз (БАС), а также нейродегенерацию, связанную с бактериальными или вирусными инфекциями.
Предметом настоящего изобретения являются соединения формулы I и их фармацевтически приемлемые кислотно-аддитивные соли, получение соединений формулы I и их солей, лекарственные препараты, включающие соединения формулы I или их фармацевтически приемлемые кислотно-аддитивные соли, приготовление таких лекарственных препаратов и применение соединений формулы I и их фармацевтически приемлемых солей при лечении и профилактике заболеваний, прежде всего заболеваний и нарушений вышеуказанного типа, а также, следовательно, приготовление соответствующих лекарственных препаратов.
Приведенные ниже определения общих терминов, использованных в данном описании, применимы независимо от того, использованы ли данные конкретные термины индивидуально или в сочетании.
Используемый в данном описании термин "низший алкил" обозначает алкильную группу с прямой или разветвленной цепью, содержащей 1-4 углеродных атома, например, метил, этил, пропил, изопропил, бутил и т. п.
Термин "галоген" обозначает атом хлора, иода, фтора и брома.
Термин "низшая алкоксигруппа" обозначает группу, в которой алкильный остаток определен выше.
Термин "уходящая группа" использован в обычном значении; он относится, например, к галогену, алкилсульфонилокси-, арилсульфонилоксигруппам и т. п. Наиболее предпочтительной уходящей группой в данном случае является атом галогена.
Термин "фармацевтически приемлемые кислотно-аддитивные соли" включает соли с минеральными и органическими кислотами, такими, как соляная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфокислота, п-толуолсульфокислота и т. п.
Соединения формулы I, в которой значения либо R5 или R6, либо их обоих отличны от водорода и каждый обозначает гидроксил или низшую алкильную группу, содержат по меньшей мере один асимметричный углеродный атом. Таким образом, возможно образование двух диастереоизомеров. Настоящее изобретение включает также рацемические смеси и их соответствующие энантиомеры.
Примерами предпочтительных соединений,
у которых X
обозначает O, являются:
1-[2-(4-гидроксифенокси)этил] -4-(4-метилбензил)пиперидин-4-ол;
4-(4-фторбензил)-1-[2-(4-гидроксифенокси)этил] пиперидин-4-ол;
N-(4-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этокси} фенил)метансульфонамид;
N-(4-{ 2-[4-(4-фторбензил)-4-гидроксипиперидин-1- ил] этокси} фенил)метансульфонамид;
N-(4-{
2-[4-(4-хлорбензил)-4-гидроксипиперидин-1- ил] этокси} фенил)метансульфонамид;
N-(4-{ 3-[4-(4-фторбензил)-4-гидроксипиперидин-1- ил] пропокси} фенил)метансульфонамид;
(RS)-1-[2-(4-гидроксифенокси)-1-метилэтил] -4-(4-метилбензил)пиперидин-4-ол.
Примерами предпочтительных соединений, у которых X обозначает NH, являются:
2-(4-бензил-4-гидроксипиперидин-1-ил)-N-(4-гидроксифенил)ацетамид и
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -N-(4-гидроксифенил)ацетамид.
Другими примерами
предпочтительных соединений, у которых X обозначает CH2, являются:
(RS)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенил)пропил] пиперидин-4-ол;
(RS)-1-[2-гидрокси-3-(4-гидроксифенил)пропил] -4-(4-метилбензил)пиперидин-4-ол;
(RS)-4-(4-хлорбензил)-1-[2-гидрокси-3-(4-гидроксифенил)пропил] пиперидин-4-ол.
Предлагаемые
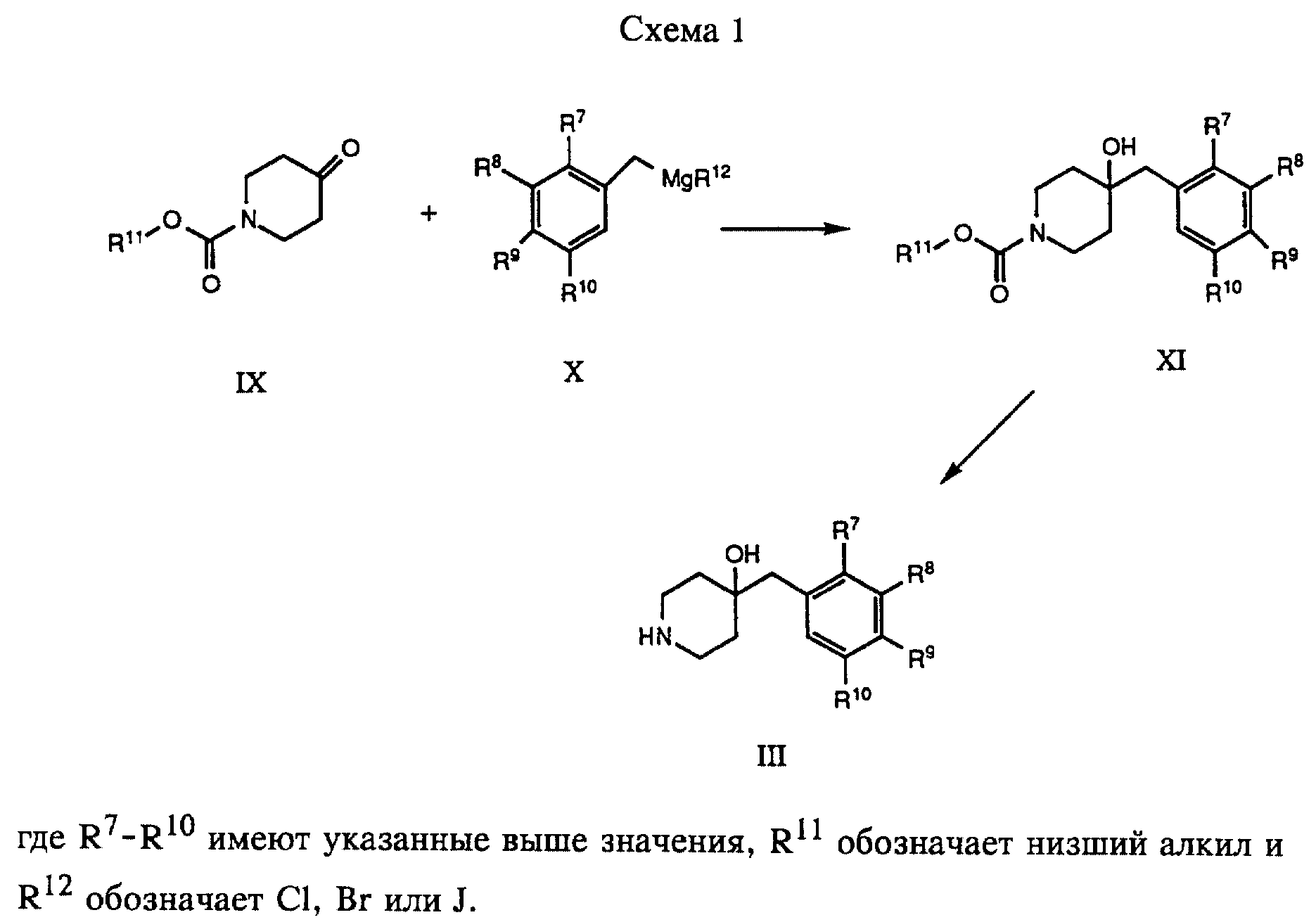
соединения формулы I и их фармацевтически приемлемые соли могут быть получены по методам, известным в данной области техники, например, по описанным ниже способам, включающим
а)
взаимодействие соединения формулы
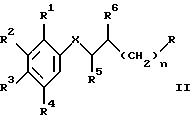
с соединением формулы
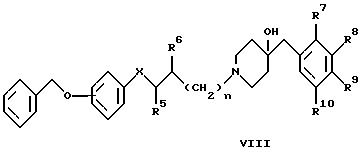
где R1 - R10 и X имеют указанные выше значения, n обозначает 1, R обозначает уходящую группу и R6 обозначает оксо- или гидроксильную группу, или
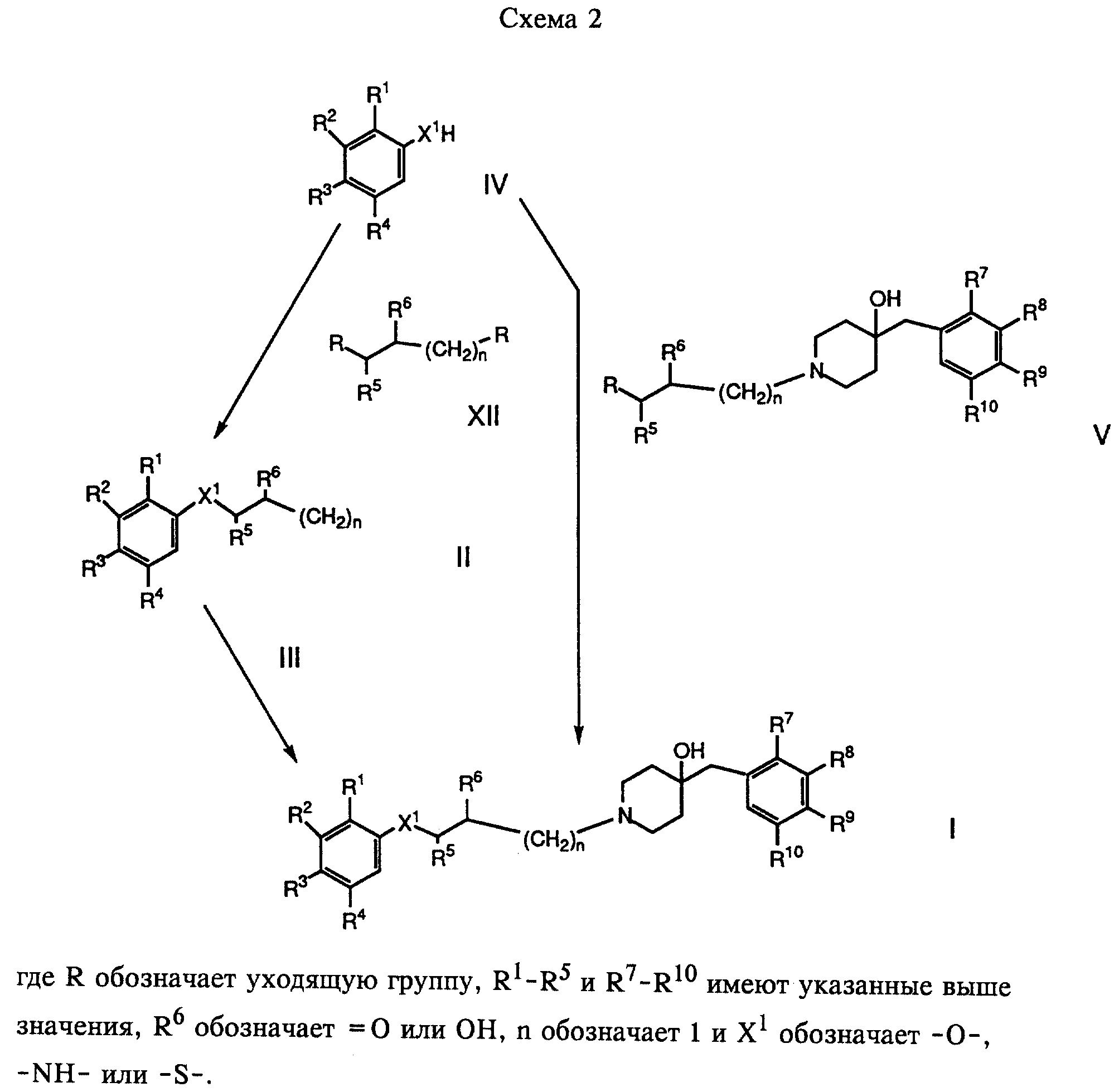
б) взаимодействие соединения формулы
с соединением формулы
где R1 - R10, n и R имеют указанные выше значения, а X обозначает -O-, -NH-, -N-низший алкил- или -S-, или
в) взаимодействие соединения формулы
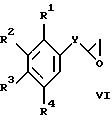
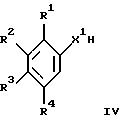
с соединением формулы III с получением соединения формулы
где R1 - R4 и R7 - R10 имеют указанные выше значения, а Y обозначает -XCH2- или -CH2-, или
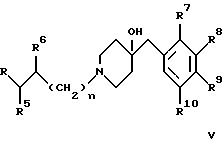
г) взаимодействие соединения формулы IV с соединением формулы
с получением соединения формулы Ia, где R1 - R4 и R7 - R10 имеют указанные выше значения, или
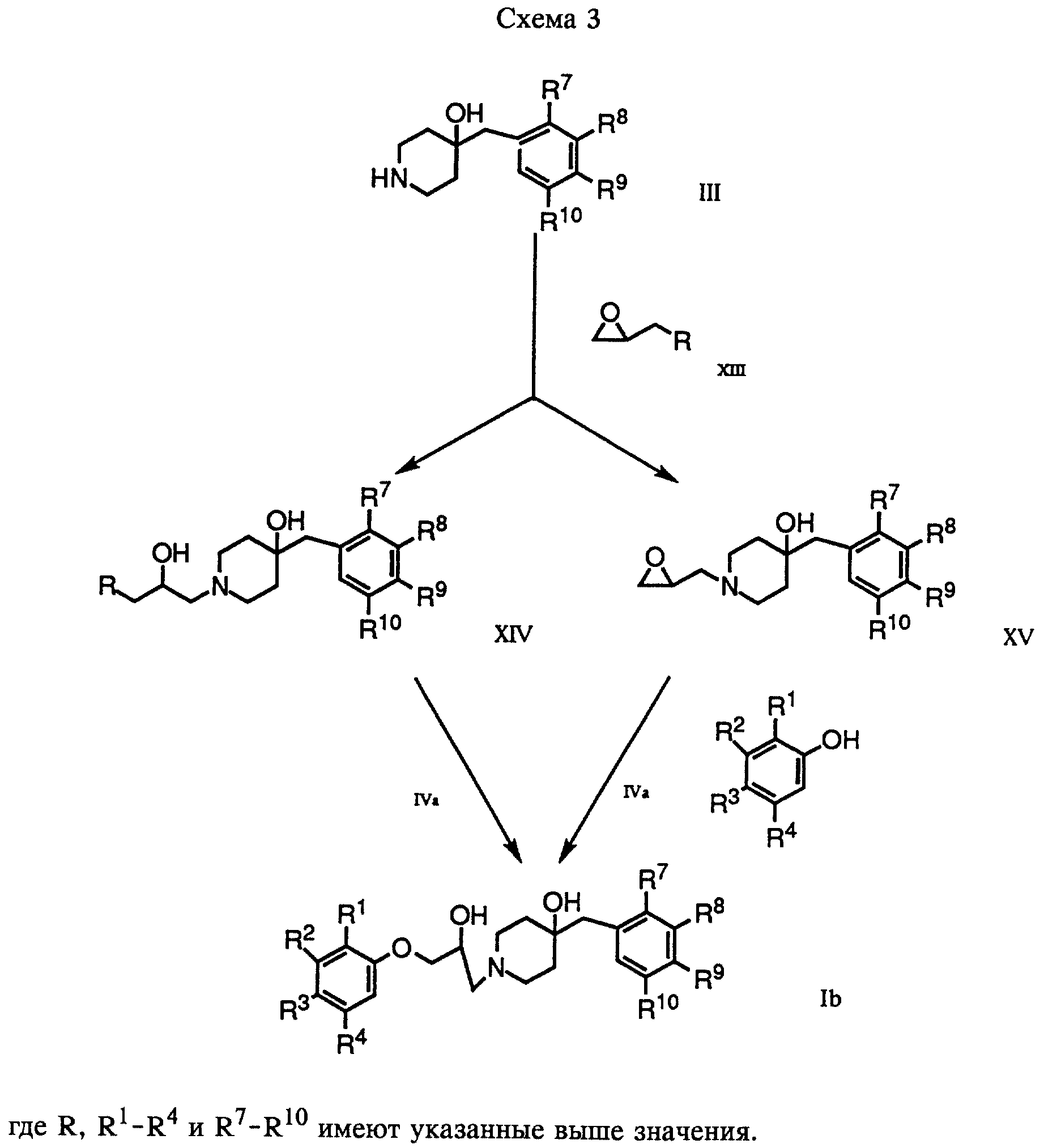
д) дебензилирование соединения формулы
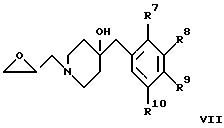
где заместители имеют указанные выше значения, при условии, что ни один из R7 - R10 не обозначает галоген, или
е) взаимодействие соединения формулы I, где один из R1 - R4 обозначает аминогруппу, с (низший)алкилсульфонилгалогенидом с получением соединения формулы I, где один из R1 - R4 обозначает (низший)алкилсульфониламиногруппу, или
ж) восстановление соединения формулы I, где R5 и/или R6 обозначает карбонильную группу, с получением соответствующего гидроксилсодержащего соединения, или
з) окисление соединения формулы I, где X обозначает -S-, с получением соответствующего сульфонилсодержащего (-SO2-) соединения, или
и) отщепление гидроксил- или аминозащитной группы (групп), содержащейся в качестве заместителя(ей) R1 - R4, и
к) при необходимости конверсию полученного соединения формулы I в фармацевтически приемлемую кислотно-аддитивную соль.
В соответствии с вариантом а) смесь соединения формулы III, например, 4-(4-метилбензил)пиперидин-4-ола, и соединения формулы II, например, N-[4-(2-бромэтокси)фенил] метансульфонамида, растворенных в 2-бутаноне, кипятят с обратным холодильником в течение приблизительно 12 часов. Это взаимодействие проводят в присутствии основания, например, карбоната калия. Затем соединение формулы I отделяют обычным путем. Когда в формуле II один из R1 - R4 обозначает гидроксильную группу, эти группы защищают обычно используемыми группами.
Примеры таких групп описаны у Green T. в Protective Groups in Organic Synthesis, глава 7, John Wiley and Sons, Inc. (1981), стр. 218-287. Самыми предпочтительными являются бензилокси-, трет-бутилдиметилсилилоксигруппы или этилоксикарбонильная группа. Эту реакцию можно проводить по известным методам.
В варианте б) описан процесс получения соединений формулы I взаимодействием соединения формулы IV с соединением формулы V.
Предпочтительными соединениями формулы IV являются соответствующие фенолы и амины.
Это взаимодействие проводят в присутствии основания. Предпочтителен карбонат калия. Реакционную смесь кипятят с обратным холодильником в течение примерно 12 часов в приемлемом растворителе, таком, как 2-бутанон, и полученное соединение затем выделяют обычным путем.
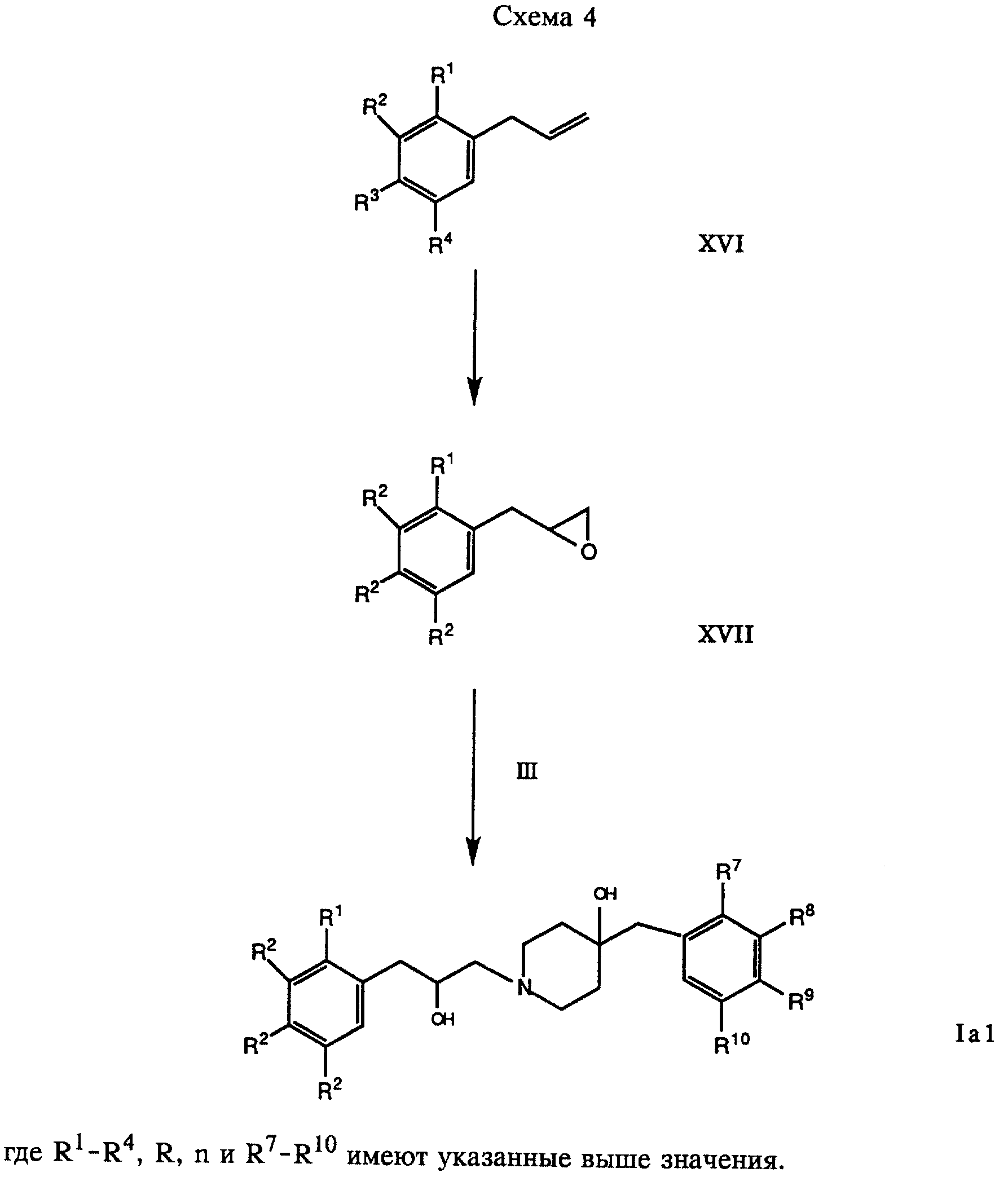
В соответствии с вариантом в) соответствующее оксиранилфенильное производное формулы VI обрабатывают соединением формулы III, получая соответствующее соединение формулы Ia. Это взаимодействие проводят в приемлемом растворителе, таком, как метанол или этанол.
В варианте д) описан процесс получения соединений формулы I, где один из R1 - R4 обозначает гидроксил. Этот процесс проводят дебензилированием соединения формулы VIII при условии, что ни один из R7 - R10 не обозначает галоген. Дебензилирование осуществляют обычным путем. Так, например, соединение формулы VIII растворяют в приемлемом растворителе или смеси растворителей, таких, как этанол и этилацетат, и гидрогенизуют в присутствии Pd на C при комнатной температуре и под атмосферным давлением.
В соответствии с вариантом е) можно получить соединение формулы I, у которого один из R1 - R4 обозначает (низший) алкилсульфониламиногруппу. Такое взаимодействие проводят обработкой соединения формулы I, где один из R1 - R4 обозначает аминогруппу, например, (RS)-1-[3-(4-аминофенокси)-2-гидроксипропил] -4-бензилпиперидин-4-ола, (низший)алкилсульфонилгалогенидом, таким, как метансульфонилхлоридом, в приемлемом растворителе, таком, как хлористый метилен, в присутствии пиридина при комнатной температуре.
В варианте ж) описано восстановление соединения формулы I, где R5 и/или R6 обозначает карбонильную группу, с получением соответствующей гидроксильной группы. Такой процесс проводят обычным путем в присутствии гидрида металла, такого, как LiAlH4 .
В соответствии с вариантом з) соединение формулы I, где X обозначает -S-, окисляют с получением соответствующего сульфонилсодержащего (-SO2-) соединения. Окисление можно проводить в присутствии продукта Oxone® (калиймоноперсульфатной тройной соли) при комнатной температуре.
Приемлемые защитные группы и методы их отщепления известны специалистам в данной области техники, хотя и очевидно, что при этом могут быть использованы только те защитные группы, которые можно удалить по методам, осуществляемым в условиях, которые не влияют на другие структурные элементы соединений.
Кислотно-аддитивные соли соединений формулы I особенно приемлемы для фармацевтического применения.
Исходные материалы для получения соединений формулы I известны или могут быть получены по известным методам, например, в соответствии со следующими реакционными схемами 1 - 5 (см. в конце описания). Эти реакции более подробно описаны в примерах 34-70.
Как указано выше, соединения формулы I и их фармацевтически приемлемые кислотно-аддитивные соли обладают ценными фармакодинамическими свойствами. Они представляют собой селективные блокаторы подтипа NMDA-рецепторов, которые играют ключевую роль в модуляции нейронной активности и пластичности, что делает их ключевыми участниками промежуточных процессов, лежащих в основе развития центральной нервной системы, а также способности к обучению и формированию памяти.
Эти соединения исследовали в соответствии с методами испытаний, описанными ниже.
Метод 1
Связывание 3H-Ro 25-6981 { Ro 25-6981 представляет собой [R-(R*, S*)]
-a-(4-гидроксифенил)-b-метил-4-(фенилметил)-1-пиперидинпропанол}
Использовали самцов белой крысы линии Fullinsdorf весом 150-200 г. Мембраны готовили гомогенизацией цельного головного мозга
без мозжечка и продолговатого мозга с помощью прибора Polytron (10000 об/мин, 30 с) в 25 объемах холодного буфера с pH 7,1, включавшего 50 мМ трис-HCl и 10 мМ ЭДТК. Гомогенат центрифугировали при
48000 g в течение 10 минут при 4oC. Осадок вновь суспендировали с помощью прибора Polytron в таком же объеме буфера и гомогенат инкубировали при 37oC в течение 10 мин. После
центрифугирования осадок гомогенизировали в том же буфере и замораживали при -80oC по меньшей мере на 16 ч, но не более чем на 10 дней. Для испытания на связывание этот гомогенат оттаивали
при 37oC, центрифугировали и осадок трижды промывали по согласно описанному выше в холодном буфере с pH 7,4, включавшем 5 мМ трис-HCl. Конечный осадок вновь суспендировали в том же буфере
и
использовали в конечной концентрации 200 мг белка/мл.
Эксперименты по связыванию 3H-Ro 25-6981 проводили с использованием буфера с pH 7,4, включавшего 50 мМ трис-HCl. Для экспериментов по вытеснению использовали 5 нМ 3H-Ro 25-6981 и неспецифическое связывание определяли с использованием 10 мМ тетрагидроизохинолина; обычно его оценивают как равное 10% от общего. Продолжительность инкубации составляла 2 часа при 4oC, испытание прекращали фильтрацией через фильтры из стекловолокна Whatman GF/B (Unifilter-96, фирма Packard, Цюрих, Швейцария). Фильтры промывали 5 раз холодным буфером. Радиоактивность на фильтре определяли с помощью микросхемного сцинтилляционного счетчика Packard Top-count после добавления 40 мл продукта microscint 40 (фирма Canberra Packard S. A. , Цюрих, Швейцария).
Влияние соединений определяли с использованием минимум 8 концентраций и повторяли по меньшей мере по одному разу. Суммированные нормализованные величины анализировали с помощью программы вычислений с нелинейной регрессией, которая позволяла получать ИК50 и ее относительные верхний и нижний доверительные пределы 95% (RS1, BBN, США).
Метод 2
Связывание 3H-празозина
Использовали самцов белой крысы линии Fullinsdorf весом 150-200 г. Мембраны готовили гомогенизацией цельного
головного мозга без мозжечка и
продолговатого мозга с помощью прибора Polytron (10000 об/мин, 30 с) в 25 объемах холодного буфера с pH 7,1, включавшего 50 мМ трис-HCl и 10 мМ ЭДТК. Гомогенат
центрифугировали при 48000 g в течение
10 минут при 4oC. Осадок вновь суспендировали с помощью прибора Polytron в таком же объеме буфера и гомогенат инкубировали при 37oC в
течение 10 мин. После центрифугирования
осадок гомогенизировали в том же буфере и замораживали при -80oC по меньшей мере на 16 ч, но не более чем на 10 дней. Для испытания на связывание
этот гомогенат оттаивали при 37o
C, центрифугировали и осадок трижды промывали согласно описанному выше в холодном буфере с pH 7,4, включавшем 5 мМ трис-HCl. Конечный осадок вновь
суспендировали в том же буфере и использовали в
конечной концентрации 200 мг белка/мл.
Эксперименты по связыванию 3H-празозина проводили с использованием буфера с pH 7,4, включавшего 50 мМ трис-HCl. Для экспериментов по вытеснению использовали 0,2 нМ 3H-празозина и неспецифическое связывание определяли с использованием 100 мМ хлорпромазина. Продолжительность инкубации составляла 30 минут при комнатной температуре, испытание прекращали фильтрацией через фильтры из стекловолокна Whatman GF/B (Unifilter-96, фирма Canberra Packard S. A. , Цюрих, Швейцария). Фильтры промывали 5 раз холодным буфером. Радиоактивность на фильтре определяли с помощью микросхемного сцинтилляционного счетчика Packard Top-count после добавления 40 мл продукта microscint 40 (фирма Canberra Packard S. A. , Цюрих, Швейцария). Влияние соединений определяли с использованием минимум 8 концентраций и повторяли по меньшей мере по одному разу. Суммированные нормализованные величины анализировали с помощью программы вычислений с нелинейной регрессией, которая позволяла получать ИК50 и ее относительные верхний и нижний доверительные пределы 95% (RS1, BBN, США).
Метод
3
Электрофизиологическое
воздействие на рекомбинантные NMDA-рецепторы
Клоны кДНК, кодирующие подгруппы NMDAR1C и NMDAR2A NMDA-рецептора (номенклатуру подгрупп NMDA-рецептора см.
Hollmann и Heinemann, 1994, Annu. Rev.
Neurosci. 17: 31), выделяли из библиотеки фрагментов кДНК мозга крыс lgtll, как это описано в литературе (см. Sigel и др. , 1994, J. Biol. Chem. 269: 8204).
Клон для подгруппы NMDAR2B NMDA-рецептора
мозга крысы был получен от S. Nakanishi (Киото, Япония). кДНК транскрибировали, присоединяли кэп и хвост поли-(A+) по описанной в литературе
методике (см. Malherbe и др. , 1990, Mol. Brain
Res. 8: 199). Для экспрессии либо сочетания подгрупп NMDAR1C и NMDAR2A, либо подгрупп NMDAR1C и NMDAR2B использовали ооциты южно-африканской шпорцевой
лягушки (Xenopus laevis). В каждый ооцит вводили
приблизительно по 3 фмоля смеси соответствующих разновидностей мРНК в соотношении 1: 1. Через четыре-пять дней по ходу экспериментов с фиксацией
потенциала измеряли ионный ток в каналах NMDA-рецептора
(касательно методов экспрессии ооцитов и фиксации потенциала см. Methfessel и др. , 1986, Pflugers Arch. 407: 577). Мембранный потенциал
фиксировали на уровне -80 мВ и рецепторы активировали с
использованием модифицированного раствора Рингера, содержавшего L-аспартат (Asp) и глицин (Gly) в качестве веществ, обладающих сродством к
NMDA-рецептору. Для любого сочетания подгрупп с целью
объяснить различную чувствительность двух типов рецепторов к агонистам выбирали различные концентрации агонистов (70 мМ Asp плюс 2,5 мМ Gly для
NMDAR1C-NMDAR2A и 15 мМ Asp плюс 0,2 мМ Gly для
NMDAR1C-NMDAR2B). Агонисты применяли в течение 15-секундных интервалов за раз каждые 2,5 мин быстрым переохлаждением ооцита с раствором агониста и
непосредственно перед завершением каждой обработки
измеряли амплитуду индуцируемого этим веществом тока. После серии начальных контрольных обработок как в базисный раствор Рингера, так и в раствор,
содержавший агонист, добавляли испытываемое
вещество-антагонист. Концентрация антагониста, которую применяли для экспрессии ооцитов подгруппы NR2A, составляла 10 ммоля/л, в то время как для
экспрессии ооцитов NR2B применяли 0,1 ммоля/л. Для
каждого соединения и подтипа NMDA-рецептора испытывали по четыре-восемь ооцитов. Воздействию такими соединениями ооциты подвергали в течение 5-30
мин в зависимости от времени, необходимого для
равновесного блокирования тока NMDA-рецептора. Для каждого ооцита уменьшение амплитуды тока выражали в виде процентной доли от контрольного тока,
определенного перед применением соединения. Данные в
таблице 1 являются средними арифметическими значениями этих процентных долей. Определенная таким образом активность некоторых соединений в
соответствии с настоящим изобретением очевидна из данных
таблицы 1 (см. в конце описания).
Приведенные в таблице 1 обозначения соответствуют следующим соединениям:
А
(RS)-1-[2-гидрокси-3-(4-гидроксифенокси)пропил]
-4-(4-метилбензил)пиперидин-4-ол;
Б (S)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенокси)пропил] пиперидин-4-ол;
В
1-[2-(4-гидроксифенокси)этил] -4-(4-метилбензил)пиперидин-4-ол;
Г 4-(4-фторбензил)-1-[2-гидроксифенокси)этил] пиперидин-4-ол;
Д N-(4-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этокси} фенил)метансульфонамид;
Е N-(4-{
2-[4-(4-фторбензил)-4-гидроксипиперидин-1- ил] этокси} фенил)метансульфонамид;
Ж N-(4-{
2-[4-(4-хлорбензил)-4-гидроксипиперидин-1- ил] этокси} фенил)метансульфонамид;
З N-(4-{
3-[4-(4-фторбензил)-4-гидроксипиперидин-1- ил] пропокси} фенил)метансульфонамид;
И
1-[2-(4-гидроксифенокси)-1-метилэтил] -4-(4-метилбензил)пиперидин-4-ол;
К
2-(4-бензил-4-гидроксипиперидин-1-ил)-N-(4-гидроксифенил)ацетамид;
Л
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -N-(4- гидроксифенил)ацетамид;
М
(RS)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенил)пропил] пиперидин-4-ол;
Н
(RS)-1-[2-гидрокси-3-(4-гидроксифенил)пропил] -4-(4- метилбензил)пиперидин-4-ол;
О
(RS)-4-(4-хлорбензил)-1-[2-гидрокси-3-(4- гидроксифенил)пропил] пиперидин-4-ол;
П
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этиловый эфир 4-гидроксибензойной кислоты;
Р
3-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] пропиловый эфир 4-гидроксибензойной кислоты;
С N-[2-(4-бензил-4-гидроксипиперидин-1-ил)этил] -4-гидроксибензамид;
Т 4-гидрокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этил} бензамид;
У
N-[3-(4-бензил-4-гидроксипиперидин-1-ил)пропил] -4-гидроксибензамид;
Ф 4-гидрокси-N-{
3-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] пропил} бензамид;
Ц
(E)-1-[3-(4-гидроксифенил)аллил] -4-(4-метилбензил)пиперидин-4-ол.
Соединения формулы I путем скрининга можно идентифицировать как селективные блокаторы подтипа NMDA-рецепторов, а (для выбранных соединений) преимущество подтипов NMDAR-2R можно подтвердить определением электрофизиологических характеристик с использованием экспрессированных ооцитов клонированных подтипов NMDA-рецептора.
Предлагаемые согласно изобретению соединения формулы I и их соли можно вводить в стандартные фармацевтические дозированные препаративные формы, например, для перорального или парентерального введения совместно с обычными фармацевтическими адъювантами, в частности с органическими или неорганическими инертными носителями, такими, как вода, желатин, лактоза, крахмал, стеарат магния, тальк, растительные масла, камеди, полиалкиленгликоли и т. п. Фармацевтические препараты можно применять в твердом виде, например, в форме таблеток, суппозиториев, капсул, либо в жидком виде, например, в форме растворов, суспензий или эмульсий. К фармацевтическим адъювантам можно добавлять или они могут включать консерванты, стабилизаторы, смачивающие агенты или эмульгаторы, соли для изменения осмотического давления или соли, выполняющие функции буферов. Фармацевтические препараты могут также содержать другие вещества с терапевтическим действием.
Ежедневная доза соединений формулы I, которую необходимо вводить, варьируется в зависимости от конкретно используемого соединения, выбранного пути введения и реципиента. Типичными примерами путей введения соединений формулы I являются пероральный и парентеральный. Ежедневная доза соединения формулы I в композициях для перорального введения в предпочтительном варианте для взрослого пациента составляет от 150 мг до 1,5 г. В составе композиций для парентерального введения ежедневная доза соединения формулы I в предпочтительном варианте для взрослого пациента составляет от 5 до 500 мг.
Более подробно сущность настоящего изобретения проиллюстрирована на приведенных ниже примерах. Во всех случаях температура указана в градусах Цельсия.
Пример
1
Гидрохлорид (RS)-1-[2-гидрокси-3-(4-гидроксифенокси)пропил] -4-(4- метилбензил)пиперидин-4-ола
0,75 г (1,6
ммоля) (RS)-1-[3-(4-бензилоксифенокси)-2-гидроксипропил]
-4-(4- метилбензил)пиперидин-4-ола растворяли в смеси 20 мл этанола и 20 мл этилацетата и при комнатной температуре и под атмосферным давлением
гидрогенизовали в присутствии Pd на C. После
фильтрования и выпаривания растворителя остаток растворяли в 30 мл этанола и 20 мл этилацетата. Добавляли 1,1 эквивалента этанольного раствора HCl с
получением 0,38 г (58%) гидрохлорида
(RS)-1-[2-гидрокси-3-(4-гидроксифенокси)пропил] -4-(4- метилбензил)пиперидин-4-ола в виде бесцветной твердой смеси E/Z-изомеров с tпл 93-96oC
и МС: m/e = 372,5 (M+H+
).
В соответствии общей методикой, описанной в примере 1, получали соединения из примеров 2-5.
Пример 2
Гидрохлорид
(RS)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенокси)пропил] пиперидин-4-ола
Указанное в заголовке соединение с tпл 89-91oC и МС: m/e = 358,4 (M+H+) получали с
использованием (RS)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] пиперидин-4-ола.
Пример 3
Гидрохлорид
(RS)-4-(4-фторбензил)-1-[2-гидрокси-3-(4- гидроксифенокси)пропил)пиперидин-4-ола
Указанное в заголовке соединение с tпл 199-202oC и МС: m/e = 376,4 (M+H+)
получали с использованием (RS)-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] -4-(4-фторбензил)пиперидин-4-ола.
Пример 4
Гидрохлорид
(R)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенокси)пропил] пиперидин-4-ола
Указанное в заголовке соединение с tпл 77-80oC [[α] =]36520 = +48,8o (c = 1,0, метанол) и МС: m/e = 358,5 (M+H+) получали
с использованием (R)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] пиперидин-4-ола.
Пример 5
Гидрохлорид (S)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенокси)пропил]
пиперидин-4-ола
Указанное в заголовке соединение с tпл 122-125oC [[α] =]36520 = -48,0o (c = 1,0, метанол) и МС: m/e = 358,5 (M+H+) получали с использованием
(S)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] пиперидин-4-ола.
Пример 6
Гидрохлорид (RS)-4-бензил-1-[2-гидрокси-3-(4- нитрофенокси)пропил] пиперидин-4-ола
1,9 г (9,7 ммоля) [(4-нитрофенокси)метил] оксирана и 2,0 г (10,7 ммоля) 4-бензил-4-гидроксипиперидина растворяли в 20 мл этанола и кипятили с обратным холодильником в течение 2 ч. После выпаривания
растворителя остаток хроматографировали на силикагеле (CH2Cl2-MeOH, 98: 2). Сырой маслянистый продукт растворяли в 80 мл смеси этилацетата с этанолом (7: 1) и добавляли 1,1
эквивалента этанольного раствора HCl с получением 3,4 г (83%) гидрохлорида (RS)-4-бензил-1-[2-гидрокси-3-(4-нитрофенокси)пропил] пиперидин-4-ола в виде бесцветной твердой смеси E/Z-изомеров с tпл 104-106oC. МС: m/e = 387,4 (M+H+).
Пример 7
Гидрохлорид (RS)-N-{ 4-[3-(4-бензил-4-гидроксипиперидин-1-ил)-2-гидроксипропокси] фенил}
метансульфонамида
0,105 мл (1,4 ммоля) метансульфонилхлорида при комнатной температуре добавляли в суспензию 0,5 г (1,3 ммоля) гидрохлорида (RS)-1-[3-(4-аминофенокси)-2-гидроксипропил]
-4-бензилпиперидин-4-ола в 10 мл CH2Cl2 и 5 мл пиридина. Смесь перемешивали при комнатной температуре в течение ночи, добавляли 15 мл воды и 15 мл рассола и смесь экстрагировали
5 порциями по 25 мл CH2Cl2. Органические фазы объединяли, сушили над Na2SO4 и выпаривали растворитель. Остаток хроматографировали на силикагеле
(этилацетат/MeOH, 96: 4), получая бесцветный продукт в виде масла, который растворяли в 2 мл этанола. Добавляли 1,1 эквивалента этанольного раствора HCl и 50 мл трет-бутилметилового эфира с
получением
0,24 г (38%) гидрохлорида (RS)-N-{ 4-[3-(4-бензил-4-гидроксипиперидин-1-ил)-2- гидроксипропокси] фенил} метансульфонамида в виде бесцветной твердой смеси E/Z-изомеров с tпл
> 230oC (с разложением). МС: m/e = 435,4 (M+H+).
Пример 8
Гидрохлорид 4-бензил-1-[2-(4-гидроксифенокси)этил] пиперидин-4-ола
1,35 г (3,2
ммоля)
4-бензил-1-[2-(4-бензилоксифенокси)этил] пиперидин-4-ола растворяли в смеси 75 мл MeOH и 75 мл этилацетата и при комнатной температуре и под атмосферным давлением гидрогенизовали в присутствии
Pd на
C. После фильтрования и выпаривания растворителя остаток растворяли в 2 мл этанола и 10 мл этилацетата. Добавляли 1,1 эквивалента этанольного раствора HCl с получением 0,85 г (72%) гидрохлорида
4-бензил-1-[2-(4-гидроксифенокси)этил] пиперидин-4-ола в виде бесцветного твердого продукта с tпл 161-163oC и МС: m/e = 328,3 (M+H+).
В соответствии с общей методикой, описанной в примере 8, получали соединения из примеров 9 и 10.
Пример 9
Фумарат 1-[2-(4-гидроксифенокси)этил] -4-(4-метилбензил)пиперидин-4-ола (1: 0.5)
Указанное в заголовке соединение с tпл 216-218oC и МС: m/e = 341 (M+) получали с использованием 1-[2-(4-бензилоксифенокси)этил]
-4-(4-метилбензил)-пиперидин-4-ола.
Пример 10
Гидрохлорид 4-(4-фторбензил)-1-[2-(4-гидроксифенокси)этил] пиперидин-4-ола
Указанное в заголовке соединение с tпл 153-155oC и МС: m/e = 345 (M+) получали с использованием 1-[2-(4-бензилоксифенокси)этил] -4-(4-фторбензил)пиперидин-4-ола.
Пример 11
Гидрохлорид
N-(4-{ 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этокси} фенил)метансульфонамида
Смесь 0,35 г (1,7 ммоля) 4-(4-метилбензил)пиперидин-4-ола, 0,5 г (1,7 ммоля)
N-[4-(2-бромметокси)фенил]
метансульфонамида и 0,25 г (1,8 ммоля) карбоната калия в 20 мл 2-бутанона кипятили с обратным холодильником в течение ночи. Эту смесь охлаждали до комнатной температуры,
добавляли 30 мл H2O
и отделяли органическую фазу. Водную фазу дважды экстрагировали этилацетатом. Затем органические фазы объединяли, сушили над Na2SO4 и выпаривали
растворитель. Остаток
хроматографировали на силикагеле (CH2Cl2/MeOH, 95: 5), получая в виде пены желтоватый продукт, который растворяли в 5 мл этанола и 10 мл этилацетата.
Добавляли 1,1 эквивалента
этанольного раствора HCl с получением 0,32 г (41%) гидрохлорида N-(4-{ 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этокси} фенил)метансульфонамида в виде бесцветного
твердого продукта с tпл > 75-78oC (с разложением) и МС: m/e = 419,5 (M+H+).
В соответствии с общей методикой, описанной в примере 11, получали соединения из примеров 12-14.
Пример 12
Гидрохлорид N-(4-{ 2-[4-(4-фторбензил)-4-гидроксипиперидин-1- ил] этокси} фенил)метансульфонамида
Указанное в заголовке
соединение с tпл
131-134oC и МС: m/e = 423,4 (M+H+) получали с использованием 4-(4-фторбензил)пиперидин-4-ола и N-[4-(2-бромметокси)фенил)метансульфонамида.
Пример 13
Гидрохлорид N-(4-{ 2-[4-(4-хлорбензил)-4-гидроксипиперидин-1- ил] этокси} фенил)метансульфонамида
Указанное в заголовке соединение с tпл 74-77oC и МС: m/e = 439,4
(M+H+) получали с использованием 4-(4-хлорбензил)пиперидин-4-ола и N-[4-(2-бромметокси)фенил)метансульфонамида.
Пример 14
Гидрохлорид
N-(4-{
3-[4-(4-фторбензил)-4-гидроксипиперидин-1- ил] пропокси} фенил)метансульфонамида
Указанное в заголовке соединение с МС: m/e = 437,4 (M+H+) получали с использованием
4-(4-фторбензил)пиперидин-4-ола и N-[4-(2-бромпропокси)фенил] метансульфонамида.
Пример 15
Гидрохлорид (RS)-4-бензил-1-[2-гидрокси-3-(4-[1.2.4] триазол-1-ил-фенокси)пропил]
пиперидин-4-ола
Смесь 0,5 г (1,8 ммоля) 4-бензил-1-(3-хлор-2-гидроксипропил)пиперидин-4-ола, 0,5 г (1,8 ммоля) 4'-(1H-1,2,4-триазол-1-ил)фенола и 0,36 г (2,6 ммоля) карбоната калия в 20 мл
2-бутанона кипятили с обратным холодильником в течение ночи. Эту смесь охлаждали до комнатной температуры, добавляли 50 мл H2O и отделяли органическую фазу. Водную фазу дважды
экстрагировали этилацетатом. Затем органические фазы промывали 2 н. раствором гидроксида натрия и объединяли. Раствор сушили над Na2SO4 и выпаривали растворитель. Остаток
растворяли в 10 мл этанола и 50 мл этилацетата. Добавляли 1,1 эквивалента этанольного раствора HCl, получая 0,69 г (86%) гидрохлорида (RS)-4-бензил-1-[2-гидрокси-3-(4-[1,2,4]
триазол-1-ил-фенокси)пропил] пиперидин-4-ола в виде бесцветной твердой смеси E/Z-изомеров с tпл 198-200oC и МС: m/e = 409,5 (M+H+).
Пример 16
Гидрохлорид (RS)-4-бензил-1-[2-гидрокси-3-(4-имидазол-1-ил-фенокси)пропил] пиперидин-4-ола
Указанное в заголовке соединение с tпл 104-108oC и МС: m/e = 408,6 (М+Н+) получали в соответствии с общей методикой, описанной в примере 15, с использованием 4-бензил-1-(3-хлор-2-гидроксипропил)пиперидин-4-ола и 4-(1-имидазолил)фенола.
Пример 17
Гидрохлорид 4-бензил-1-[3-(4-гидроксифенокси)пропил] пиперидин-4-ола
0,432 г (1 ммоль) 4-бензил-1-[3-(4-бензилоксифенокси)пропил] пиперидин-4-ола растворяли в 50 мл MeOH и при комнатной
температуре и под атмосферным давлением гидрогенизовали в присутствии Pd на C. После фильтрования и выпаривания растворителя остаток растворяли в 3 мл ТГФ и 10 мл диэтилового эфира. Добавляли 1,1
эквивалента эфирного раствора HCl с получением 0,30 г (88%) гидрохлорида 4-бензил-1-[3-(4-гидроксифенокси)пропил] пиперидин-4-ола в виде бесцветного твердого продукта с tпл 64oC
и МС: m/e = 342,3 (M+H+).
В соответствии с общей методикой, описанной в примере 17, получали соединения из примеров 18-22.
Пример 18
Гидрохлорид
4-(4-фторбензил)-1-[3-(4-гидроксифенокси)пропил] пиперидин-4-ола
Указанное в заголовке соединение с МС: m/e = 360,4 (M+H+) получали с использованием
1-[3-(4-бензилоксифенокси)пропил] -4-(4-фторбензил)пиперидин-4-ола.
Пример 19
Гидрохлорид 1-[3-(3-гидроксифенокси)пропил] -4-(4-метилбензил)пиперидин-4-ола
Указанное
в заголовке соединение с МС: m/e = 356,4 (M+H+) получали с использованием 1-[3-(3-бензилоксифенокси)пропил] -4-(4-метилбензил)пиперидин-4-ола.
Пример 20
Гидрохлорид
1-[3-(2-гидроксифенокси)пропил] -4-(4-метилбензил)пиперидин-4-ола
Указанное в заголовке соединение с МС: m/e = 356,4 (M+H+) получали с использованием
1-[3-(2-бензилоксифенокси)пропил] -4-(4-метилбензил)пиперидин-4-ола.
Пример 21
Гидрохлорид 1-[2-(4-гидроксифенокси)этил] -4-(4-метоксибензил)пиперидин-4-ола
Указанное
в заголовке соединение с МС: m/e = 358,3 (M+H+) получали с использованием 1-[2-(4-бензилоксифенокси)этил] -4-(4-метоксибензил)пиперидин-4-ола.
Пример 22
Гидрохлорид
1-[2-(4-гидроксифенокси)-1-метилэтил] -4-(4-метилбензил)пиперидин-4-ола
Указанное в заголовке соединение с МС: m/e = 356,3 (M+H+) получали с использованием
1-[2-(4-бензилоксифенокси)-1-метилэтил] -4-(4-метоксибензил)пиперидин-4-ола.
Пример 23
Гидрохлорид 2-(4-бензил-4-гидроксипиперидин-1-ил)-N-(4-гидроксифенил)ацетамида
0,454 г (1 ммоль) 2-(4-бензил-4-гидроксипиперидин-1-ил)-N-[4-(трет-бутилдиметилсиланилокси)фенил] ацетамида растворяли в 6 мл ТГФ и перемешивали в течение 18 ч при комнатной температуре в присутствии
тетра-н-бутиламмонийфторида/SiO2 (1 г, 1,1 ммоля, 1,1 ммоля/г). Реакцию прекращали добавлением в реакционную смесь 20 мл 20%-ного NH4Cl и водную фазу экстрагировали 3 порциями
по
5 мл этилацетата. Объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат в соотношении 1: 1, а затем
этилацетат), получая в виде пены продукт, который растворяли в MeOH и обрабатывали (0,9 мл) 1 н. HCl. Раствор концентрировали и остаток кипятили с обратным холодильником в присутствии ацетонитрила в
течение 2 ч с получением после охлаждения 0,27 г (72%) гидрохлорида 2-(4-бензил-4-гидроксипиперидин-1-ил)-N-(4-гидроксифенил)ацетамида в виде бесцветной твердой смеси E/Z-изомеров с tпл
222-225oC и МС: m/e = 341,5 (M+H+).
В соответствии с общей методикой, описанной в примере 23, получали соединения из примеров 24-27.
Пример 24
Гидрохлорид 2-[4-гидоокси-4-(4-метилбензил)пиперидин-1-ил] -N-(4-гидроксифенил)ацетамида
Указанное в заголовке соединение с tпл 242-243oC и МС: m/e = 355,4
(M+H+) получали с использованием N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-[4-гидрокси-4-(4- метилбензил)пиперидин-1-ил] ацетамида.
Пример 25
Гидрохлорид
2-[4-(4-хлорбензил)-4-гидроксипиперидин-1-ил] -N-(4-гидроксифенил)ацетамида
Указанное в заголовке соединение с tпл 205-210oC и МС: m/e = 375,3 (M+H+)
получали
с использованием N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-[4-(4-хлорбензил)-4-гидроксипиперидин-1-ил] ацетамида.
Пример 26
Гидрохлорид
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -N-(4- гидроксифенил)пропионамида
Указанное в заголовке соединение с tпл 257oC и МС: m/e = 369,3 (M+H+)
получали с использованием N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-[4-гидрокси-4-(4- метилбензил)пиперидин-1-ил] пропионамида.
Пример 27
Гидрохлорид
3-(4-бензил-4-гидроксипиперидин-1-ил)-N-(4- гидроксифенил)пропионамида
Указанное в заголовке соединение с tпл 140-145oC и МС: m/e = 355,4 (M+H+) получали с
использованием 3-(4-бензил-4-гидроксипиперидин-1-ил)-N-[4- (трет-бутилдиметилсиланилокси)фенил] пропионамида.
Пример 28
Гидрохлорид 4-бензил-1-[2-(4-гидроксифениламино)этил]
пиперидин-4-ола
Раствор 0,97 г (2,13 ммоля) 2-(4-бензил-4-гидроксипиперидин-1-ил)-N-[4- (трет-бутилдиметилсиланилокси)фенил] ацетамида в 5 мл ТГФ по каплям добавляли в суспензию 0,162 г (4,
26
ммоля) LiAlH4 в 5 мл ТГФ при комнатной температуре. По истечении 20 часов выдержки при комнатной температуре реакционную смесь кипятили с обратным холодильником в течение 3 часов. Эту
реакционную смесь охлаждали до 0oC и обрабатывали последовательно 0,2 мл H2O, 0,2 мл 5 н. NaOH и 0,6 мл H2O. После выпаривания ТГФ образовавшийся твердый продукт
отфильтровывали и промывали CH2Cl2. Водную фазу экстрагировали 3 порциями по 10 мл CH2Cl2, объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток растворяли в 5 мл CH2Cl2 и перемешивали в присутствии 0,5 г тетра-н-бутиламмонийфторида/SiO2 (0,55 ммоля, 1,1 ммоля/г). После
выдержки в течение 4 часов при комнатной температуре реакцию прекращали добавлением в реакционную смесь 15 мл 20%-ного NH4Cl и водную фазу экстрагировали 2 порциями по 5 мл CH2
Cl2. Объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле (CH2Cl2/MeOH в соотношении 9:
1,
а затем 4: 1), получая в виде пены продукт, который растворяли в MeOH и обрабатывали 0,6 мл 1 н. HCl. Раствор концентрировали и остаток растворяли в EtOH. Добавлением диэтилового эфира получали 0,
045
г (5,3%) гидрохлорида 4-бензил-1-[2-(4-гидроксифениламино)этил] пиперидин-4-ола в виде бежевого твердого вещества с tпл 130-140oC и МС: m/e = 327,4 (M+H+).
Пример 29
Гидрохлорид 4-бензил-1-[3-(4-гидроксифениламино)пропил] пиперидин-4-ола
Раствор 0,71 г (1,51 ммоля)
3-(4-бензил-4-гидроксипиперидин-1-ил)-N-[4- (трет-бутилдиметилсиланилокси)фенил] пропионамида в 4 мл ТГФ по каплям при 0oC добавляли в суспензию 0,115 г (3,02 ммоля) LiAlH4 в 4
мл ТГФ. Реакционную смесь кипятили с обратным холодильником в течение 30 мин, охлаждали до 0oC и реакцию прекращали осторожным добавлением в реакционную смесь 5 мл H2O. После
разбавления 20 мл H2O реакционную смесь последовательно обрабатывали 2 н. HCl и насыщенным раствором NaHCO3. Водную фазу экстрагировали 3 порциями по 10 мл CH2Cl2, объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле (CH2Cl2/MeOH в соотношении 9: 1, а
затем 4: 1), получая в виде пены продукт, который растворяли в MeOH и обрабатывали избыточным количеством HCl/диэтилового эфира. Раствор концентрировали и остаток растворяли в EtOH. Добавлением
диэтилового эфира получали 0,160 г (26%) гидрохлорида 4-бензил-1-[3-(4-гидроксифениламино)пропил] пиперидин-4-ола в виде бежевого твердого вещества с tпл 213-216oC и МС: m/e =
341,5 (M+H+).
Пример 30
Гидрохлорид (RS)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенил)пропил] пиперидин-4-ола
0,12 г (0,8 ммоля) (RS)-4-оксиранилметилфенола
растворяли в 3 мл MeOH и кипятили с обратным холодильником в течение 3 часов в присутствии 0,19 г (1,0 ммоля) 4-бензил-4-гидроксипиперидина. Реакционную смесь концентрировали и остаток
хроматографировали на силикагеле (CH2Cl2/MeOH, 19: 1, затем 9: 1 и в завершение 4: 1), получая в виде белой пены продукт, который растворяли в 3 мл MeOH и обрабатывали 0,5 мл 1
н. HCl. Раствор концентрировали и остаток растворяли в 2 мл MeOH. Добавлением диэтилового эфира получали 0,112 г (37%) гидрохлорида (RS)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенил)пропил]
пиперидин-4-ола в виде белой твердой смеси E/Z-изомеров с tпл 135-136oC и МС: m/e = 341 (M+).
В соответствии с общей методикой, описанной в примере 30, получали соединения из примеров 31 и 32.
Пример 31
Гидрохлорид (RS)-1-[2-гидрокси-3-(4-гидроксифенил)пропил] -4-(4-метилбензил)пиперидин-4-ола
Указанное в заголовке
соединение с tпл 196-197oC и МС: m/e = 355 (M+) получали с использованием (RS)-4-оксиранилметилфенола и 4-(4-метилбензил)пиперидин-4-ола.
Пример 32
Гидрохлорид (RS)-4-(4-хлорбензил)-1-[2-гидрокси-3-(4-гидроксифенил)пропил] пиперидин-4-ола
Указанное в заголовке соединение с tпл 172-174oC и МС: m/e = 376,4
(M+H+) получали с использованием (RS)-4-оксиранилметилфенола и 4-(4-хлорбензил)пиперидин-4-ола.
Пример 33
Гидрохлорид
1-(4-бензил-4-гидроксипиперидин-1-ил)-3-(4-гидроксифенил)пропан-2-она
0,400 г (0,88 ммоля) 1-(4-бензил-4-гидроксипиперидин-1-ил)-3-[4- (трет-бутилдиметилсиланилокси)фенил] пропан-2-она
растворяли в 4 мл ТГФ и перемешивали в течение 16 часов при комнатной температуре в присутствии 1 мл (1 ммоль) 1 н. тетра-н-бутиламмонийфторида. Реакцию прекращали добавлением в реакционную смесь 15
мл 20%-ного NH4Cl и водную фазу экстрагировали 3 порциями по 20 мл этилацетата. Объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток
хроматографировали на силикагеле (этилацетат), получая в виде желтого масла продукт, который растворяли в 2 мл MeOH и обрабатывали 0,5 мл 1 н. HCl. Раствор концентрировали, остаток растворяли в
изо-PrOH и добавляли в него диэтиловый эфир, получая 0,120 г (36%) гидрохлорида 1-(4-бензил-4-гидроксипиперидин-1-ил)-3-(4-гидроксифенил)пропан-2-она в виде белого твердого вещества с tпл
180-181oC и МС: m/e = 340,3 (M+H+).
Пример 34
Гидрохлорид 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этилового эфира 4-гидроксибензойной кислоты (1:
1)
0,63 г (1,37 ммоля) 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -этилового эфира 4-бензилоксибензойной кислоты растворяли в 12 мл ТГФ и кипятили с обратным холодильником в течение 8
часов в присутствии 31 мг 10%-ного Pd/C под атмосферным давлением водорода. После отфильтровывания катализатора и выпаривания растворителя остаток кристаллизовали в среде 15 мл этилацетата. Белое
твердое вещество растворяли в 10 мл ТГФ и добавляли насыщенный раствор HCl в диэтиловом эфире, получая 0,2 г (37%) гидрохлорида 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этилового эфира
4-гидроксибензойной кислоты в виде бесцветной твердой смеси E/Z-изомеров с tпл 151-152oC и МС: m/e = 370,3 (M+H+).
В соответствии с общей методикой, описанной в примере 34, получали соединение из примера 35.
Пример 35
Гидрохлорид 3-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] пропилового эфира 4-гидроксибензойной кислоты
(1: 1)
Указанное в заголовке соединение с tпл 178-179oC и МС: m/e = 384,3 (M+H+) получали с использованием 3-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил]
пропилового эфира 4-бензилоксибензойной кислоты.
Пример 36
N-[2-(4-бензил-4-гидроксипиперидин-1-ил)этил] -4-гидроксибензамид
150 мг 10%-ного палладия на угле
добавляли в раствор 500 мг (1,12 ммоля) 4-бензилокси-N-[2-(4-гидрокси-4-фенилпиперидин-1-ил)этил] бензамида в 20 мл уксусной кислоты. По истечении 3 ч гидрогенизацию завершали. Катализатор удаляли
фильтрованием через броунмиллерит и выпаривали растворитель. Добавлением 2 мл 10%-ного водного раствора бикарбоната натрия и экстракцией дихлорметаном получали 399 мг (95%)
N-[2-(4-бензил-4-гидроксипиперидин-1-ил)этил] -4-гидроксибензамида в виде светло-желтого твердого вещества.
Пример 37
Гидрохлорид 4-гидрокси-N-{
2-[4-гидрокси-4-(4-метилбензил)-пиперидин-1-ил] этил} бензамида (1: 1)
145 мг 10%-ного палладия на угле добавляли в раствор 780 мг (1,7 ммоля) 4-бензилокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этил} бензамида в 20 мл уксусной кислоты. По истечении 4 ч гидрогенизацию завершали. Катализатор удаляли фильтрованием через броунмиллерит и выпаривали
растворитель. Добавляли 2 мл 10%-ного водного раствора бикарбоната натрия и водный слой экстрагировали дихлорметаном. Органический слой сушили над Na2SO4, фильтровали и
выпаривали. Остаток растворяли в 3 мл дихлорметана и добавляли насыщенный раствор HCl в диэтиловом эфире. Осадок отфильтровывали, получая 460 мг (67%) гидрохлорида 4-бензилокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этил} бензамида (1: 1) в виде белого твердого вещества. МС: me/е = 369 (M+H+).
Пример 38
N-[3-(4-бензил-4-гидроксипиперидин-1-ил)пропил] -4-гидроксибензамид
50 мг 10%-ного палладия на угле добавляли в раствор 185 мг (0,40 ммоля) N-[3-(4-бензил-4-гидроксипиперидин-1-ил)пропил]
-4-бензилоксибензамида в 5 мл уксусной кислоты. По истечении 4 ч гидрогенизацию завершали. Катализатор удаляли фильтрованием через броунмиллерит и выпаривали растворитель. Добавляли 2 мл 10%-ного
водного раствора бикарбоната натрия и водный слой экстрагировали дихлорметаном. Органический слой сушили над Na2SO4, фильтровали и упаривали, получая 130 мг (87%)
N-[3-(4-бензил-4-гидроксипиперидин-1-ил)пропил] -4-гидроксибензамида в виде белого твердого вещества. МС: me/e = 369 (M+H+).
Пример 39
4-гидрокси-N-{
3-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] пропил} бензамида
50 мг 10%-ного палладия на угле добавляли в раствор 220 мг (0,46 ммоля) 4-бензилокси-N-{
3-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] пропил} бензамида в 5 мл уксусной кислоты. По истечении 4 ч гидрогенизацию завершали. Катализатор удаляли фильтрованием через броунмиллерит и выпаривали
растворитель. Добавляли 2 мл 10%-ного водного раствора бикарбоната натрия и водный слой экстрагировали дихлорметаном. Органический слой сушили над Na2SO4, фильтровали и
упаривали,
получая 178 мг (73%) 4-гидрокси-N-{ 3-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] пропил} бензамида в виде белого твердого вещества. МС: me/e = 383 (M+H+).
Пример 40
4-гидрокси-N-{ 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этил} -N-метилбензамида
60 мг 10%-ного палладия на угле добавляли в раствор 226 мг (0,48 ммоля)
4-бензилокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этил} -N-метилбензамида в 6 мл уксусной кислоты. По истечении 4 ч гидрогенизацию завершали. Катализатор удаляли фильтрованием через
броунмиллерит и
выпаривали растворитель. Добавляли 2 мл 10%-ного водного раствора бикарбоната натрия и водный слой экстрагировали дихлорметаном. Органический слой сушили над Na2SO4 и фильтровали,
получая 135 мг (74%) 4-гидрокси-N-{ 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этил} -N-метилбензамида в виде белого твердого вещества. МС: me/e = 383 (M+H+
).
Пример
41
(E)-1-[3-(4-гидроксифенил)аллил] -4-(4-метилбензил)пиперидин-4-ол
В суспензию 324 мг (3,0 экв. ) алюмогидрида лития и 50 мл ТГФ при комнатной
температуре в токе аргона медленно
добавляли раствор 1,0 г (2,85 ммоля) (E)-1-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] -3-(4-гидроксифенил)пропенона в 10 мл ТГФ. После перемешивания в течение 5
ч осторожно добавляли 20 мл 20%-ного
раствора аммонийхлорида и водный слой экстрагировали дихлорметаном. Органический слой сушили над Na2SO4, фильтровали и упаривали.
Маслянистый остаток очищали с помощью
хроматографии (на силикагеле, дихлорметан/метанол/25%-ный водный раствор аммиака в соотношении 140: 10: 1), получая 474 мг (49%) (E)-1-[3-(4-гидроксифенил)аллил]
-4-(4-метилбензил)пиперидин-4-ола в
виде белой пены. МС: me/e = 338 (M+H+).
Синтез полупродуктов
Пример 42
Этиловый эфир
4-гидрокси-4-(4-метилбензил)пиперидин-1-карбоновой кислоты
Раствор 39 мл (0,26 моля) 1-этоксикарбонил-4-пиперидона в 150 мл диэтилового эфира при комнатной температуре по каплям добавляли в
раствор, приготовленный с использованием 237 г (1,28 моля)
4-метилбензилбромида и 31,2 г (1,28 моля) Mg в 300 мл диэтилового эфира. Комнатную температуру поддерживали в течение 45 мин с
перемешиванием и затем смесь кипятили с обратным холодильником в течение
5 ч. После этого смесь охлаждали до 0oC, разбавляли 700 мл диэтилового эфира и гидролизовали 200 мл насыщенного
раствора гидрохлорида аммония и 350 мл воды. Нерастворимый материал удаляли
фильтрованием через броунмиллерит, остаток дважды промывали 2 порциями по 500 мл диэтилового эфира и отделяли органическую
фазу. Водный слой экстрагировали диэтиловым эфиром, органические фазы
объединяли и сушили над MgSO4 и выпаривали растворитель. Остаток хроматографировали на силикагеле (гексан/этилацетат,
2: 1), получая 70,5 г (99%) этилового эфира
4-гидрокси-4-(4-метилбензил)пиперидин-1-карбоновой кислоты в виде желтоватого масла. МС: m/e = 278 (M+H+).
В соответствии с общей методикой, описанной в примере 42, получали соединение из примера 43.
Пример 43
Этиловый эфир 4-(4-фторбензил)-4-гидроксипиперидин-1-карбоновой кислоты
Указанное в заголовке соединение получали с использованием
1-этоксикарбонил-4-пиперидона и 4-фторбензилбромида.
Пример 44
4-(4-метилбензил)пиперидин-4-ол
Смесь 70,5
г (0,25 моля) этилового эфира
4-гидрокси-4-(4-метилбензил)пиперидин-1-карбоновой кислоты с 26 г (0,65 моля) гидроксида натрия в 350 мл этанола и 50 мл воды кипятили с обратным холодильником в течение
2 дней. Добавляли 20 г (0,50
моля) гидроксида натрия и на другой день начинали кипячение с обратным холодильником с последующими охлаждением до комнатной температуры и выпариванием растворителя.
Остаток растворяли в 700 мл CH2Cl2 и 1 л воды, органическую фазу отделяли, а водную фазу экстрагировали CH2Cl2. Органические фазы объединяли, сушили над
MgSO4 и выпаривали
растворитель. Остаток кристаллизовали из н-гексана с получением 34 г (66%) 4-(4-метилбензил)пиперидин-4-ола в виде не совсем белого твердого вещества с tпл
118-121oC и МС: m/e =
206 (M+).
В соответствии с общей методикой, описанной в примере 44, получали соединения из примеров 45 и 46.
Пример 45
4-(4-фторбензил)пиперидин-4-ол
Указанное в заголовке соединение получали с использованием этилового эфира 4-(4-фторбензил)-4-гидроксипиперидин-1-карбоновой кислоты.
Пример
46
4-(4-метоксибензил)пиперидин-4-ол
Указанное в заголовке соединение получали с использованием этилового эфира 4-(4-метоксибензил)-4-гидроксипиперидин-1-карбоновой
кислоты.
Пример 47
(R)-{ [4-(фенилметокси)фенокси] метил} оксиран
Смесь 1,7 г (8,5 ммоля) гидрохинонмонобензилового эфира с 0,19 г (1,7 ммоля)
тетраметиламмонийхлорида в 2,0 мл (25,
5 ммоля) (S)-эпихлоргидрина перемешивали при комнатной температуре в течение 4 дней. Добавляли 30 мл H2O и 50 мл CH2Cl2 и
отделяли органическую фазу. Водную фазу
дважды экстрагировали CH2Cl2. Затем органические фазы объединяли, сушили над Na2SO4 и выпаривали растворитель.
Остаток хроматографировали на силикагеле
(гексан/Et2O, 3: 1) с получением 1,1 г (50%) (R)-{ [4-(фенилметокси)фенокси] метил} оксирана в виде бесцветного твердого вещества с tпл
70-73oC, [[α] =]D20 = -8,1o (c = 1,0,
метанол) и МС: m/e = 256 (M+
).
Пример 48
(RS)-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] -4-(4- метилбензил)пиперидин-4-ол
Смесь 1,0 г (3,4 ммоля)
(RS)-1-хлор-3-[4-(бензилокси)фенокси]
-2-пропанола, 0,70 г (3,4 ммоля) 4-(4-метилбензил)пиперидин-4-ола и 0,50 г (3,6 ммоля) карбоната калия в 2-бутаноне кипятили с обратным холодильником в течение 2
дней. Затем эту смесь охлаждали до
комнатной температуры, добавляли 50 мл H2O и отделяли органическую фазу. Водную фазу дважды экстрагировали этилацетатом. Затем органические фазы
объединяли, сушили над Na2SO4 и выпаривали растворитель. Остаток хроматографировали на силикагеле (CH2Cl2/MeOH, 98: 2) с получением 0,75 г (48%)
(RS)-1-[3-(4-бензилоксифенокси)-2-гидроксипропил]
-4-(4- метилбензил)пиперидин-4-ола в виде бесцветного твердого вещества. МС: m/e = 462,5 (M+H+).
В соответствии с общей методикой, описанной в примере 48, получали полупродукты из примеров 49 и 50.
Пример 49
(RS)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] пиперидин-4-ол
Указанное в заголовке соединение получали с
использованием (RS)-1-хлор-3-[4-(бензилокси)фенокси] -2-пропанола и 4-бензил-4-гидроксипиперидина.
Пример 50
(RS)-1-[3-(4-бензилоксифенокси)-2-гидроксипропил]
-4-(4- фторбензил)пиперидин-4-ол
Указанное в заголовке соединение с МС: m/e = 466,5 (M+H+) получали с использованием
(RS)-1-хлор-3-[4-(бензилокси)фенокси] -2-пропанола и
4-(4-фторбензил)пиперидин-4-ола (из примера 37).
Пример 51
(R)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил]
пиперидин-4-ол
0,55 г (2,2 ммоля) (R)-{
[4-(фенилметокси)фенокси] метил} оксирана и 0,49 г (2,4 ммоля) 4-бензил-4-гидроксипиперидина растворяли в 10 мл этанола и кипятили с обратным
холодильником в течение 2 ч. После выпаривания
растворителя остаток хроматографировали на силикагеле (этилацетат/MeOH, 9: 1) с получением 0,85 г (88%)
(R)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] пиперидин-4-ола в виде бесцветного
масла. МС: m/e = 448,5 (M+H+).
Пример 52
(S)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] пиперидин-4-ол
0,55 г (2,2 ммоля) (S)-{
[4-(фенилметокси)фенокси] метил} оксирана и 0,49 г (2,4 ммоля) 4-бензил-4-гидроксипиперидина
растворяли в 10 мл этанола и кипятили с обратным холодильником в течение 2 ч. После выпаривания
растворителя остаток хроматографировали на силикагеле (этилацетат/MeOH, 9: 1) с получением 0,89 г (92%)
(S)-4-бензил-1-[3-(4-бензилоксифенокси)-2-гидроксипропил] пиперидин-4-ола в виде бесцветного
масла. МС: m/e = 448,5 (M+H+).
Пример 53
Гидрохлорид
(RS)-1-[3-(4-аминофенокси)-2-гидроксипропил] -4-бензилпиперидин-4-ола
3,0 г (7,1 ммоля) гидрохлорида
(RS)-4-бензил-1-[2-гидрокси-3-(4-нитрофенокси)пропил] пиперидин-4-ола растворяли в смеси
250 мл этанола с 80 мл MeOH и при комнатной температуре и под атмосферным давлением гидрогенизовали в
присутствии Pd на C. После фильтрования и выпаривания растворителя остаток растворяли в 20 мл
этанола и 40 мл этилацетата с получением 2,5 г (90%) гидрохлорида
(RS)-1-[3-(4-аминофенокси)-2-гидроксипропил] -4-бензилпиперидин-4-ола в виде бежевой твердой смеси E/Z-изомеров с tпл
93-95oC и МС: m/e = 357,4 (M+H+).
Пример 54
4-бензил-1-(3-хлор-2-гидроксипропил)пиперидин-4-ол
2,5 мл (31 ммоль) (рац. )-эпихлоргидрина,
растворенного в 10 мл диэтилового эфира, при комнатной температуре добавляли в
суспензию 6,0 г (31 ммоля) 4-бензил-4-гидроксипиперидина в 40 мл диэтилового эфира и 40 мл CH2Cl2.
Смесь перемешивали в течение ночи при комнатной температуре, добавляли 50 мл
воды и 50 мл CH2Cl2 и отделяли органическую фазу. Водную фазу экстрагировали CH2Cl2, органические фазы объединяли, сушили над Na2SO4 и
выпаривали растворители. Остаток хроматографировали на силикагеле (этилацетат/MeOH, 9: 1) с получением 1,0 г (13%)
4-бензил-1-(3-хлор-2-гидроксипропил)пиперидин-4-ола в виде бесцветного твердого
вещества с tпл 194-195oC и МС: m/e = (M+H+).
Пример 55
4-бензил-1-[2-(4-бензилоксифенокси)этил] пиперидин-4-ол
Смесь 0,62 г (3,3
ммоля) 4-бензил-4-гидроксипиперидина с 1,0 г (3,3 ммоля) 1-(2-бромэтокси)-4-(фенилметокси)бензола и 0,9 г (6,5
ммоля) карбоната калия в 15 мл 2-бутанона кипятили с обратным холодильником в течение
ночи. Затем смесь охлаждали до комнатной температуры, добавляли 30 мл H2O и отделяли органическую
фазу. Водную фазу дважды экстрагировали этилацетатом. Затем органические фазы объединяли,
сушили над Na2SO4 и выпаривали растворитель с получением 1,35 г (99%)
4-бензил-1-[2-(4-бензилоксифенокси)этил] пиперидин-4-ола в виде желтоватого твердого вещества. МС: m/e = 418,
4 (M+H+).
В соответствии с общей методикой, описанной в примере 55, получали соединения из примеров 56-63.
Пример 56
1-[2-(4-бензилоксифенокси)этил] -4-(4-метилбензил)пиперидин-4-ол
Указанное в заголовке соединение с МС:
m/e = 432,6 (M+H+) получали с использованием
1-(2-бромэтокси)-4-(бензилокси)бензола и 4-(4-метилбензил)пиперидин-4-ола.
Пример 57
1-[2-(4-бензилоксифенокси)этил]
-4-(4-фторбензил)пиперидин-4-ол
Указанное в
заголовке соединение с МС: m/e = 436,5 (M+H+) получали с использованием 1-(2-бромэтокси)-4-(бензилокси)бензола и
4-(4-фторбензил)пиперидин-4-ола.
Пример 58
4-бензил-1-[3-(4-бензилоксифенокси)пропил] пиперидин-4-ол
Указанное в заголовке соединение с МС: m/e = 432,6 (M+H+) получали с использованием
1-(2-бромпропокси)-4-(бензилокси)бензола и 4-бензилпиперидин-4-ола.
Пример 59
1-[3-(4-бензилоксифенокси)пропил]
-4-(4-фторбензил)пиперидин-4-ол
Указанное в заголовке
соединение с МС: m/e = 450,5 (M+H+) получали с использованием 1-(3-бромпропокси)-4-(бензилокси)бензола и
4-(4-фторбензил)пиперидин-4-ола.
Пример 60
1-[3-(3-бензилоксифенокси)пропил] -4-(4-метилбензил)пиперидин-4-ол
Указанное в заголовке соединение с МС: m/e = 446,5
(M+H+) получали с использованием
1-(3-бромпропокси)-3-(фенилметокси)бензола и 4-(4-метилбензил)пиперидин-4-ола.
Пример 61
1-[3-(2-бензилоксифенокси)пропил]
-4-(4-метилбензил)пиперидин-4-ол
Указанное
в заголовке соединение с МС: m/e = 446,5 (M+H+) получали с использованием 1-(3-бромпропокси)-2-(бензилокси)бензола и
4-(4-метилбензил)пиперидин-4-ола.
Пример 62
1-[2-(4-бензилоксифенокси)этил] -4-(4-метоксибензил)пиперидин-4-ол
Указанное в заголовке соединение с МС: m/e = 448,5
(M+H+) получали с использованием
1-(2-бромэтокси)-4-(бензилокси)бензола и 4-(4-метоксибензил)пиперидин-4-ола.
Пример 63
1-[2-(4-бензилоксифенокси)этил]
-4-(4-метоксибензил)пиперидин-4-ол
Указанное в
заголовке соединение с МС: m/e = 446,4 (M+H+) получали с использованием 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил]
-1-метилэтилового эфира метансульфокислоты и 4-бензилоксифенола.
Пример 64
2-(4-бензил-4-гидроксипиперидин-1-ил)-N-[4- (трет-бутилдиметилсиланилокси)фенил] ацетамид
1,14 г (3,8 ммоля) N-[4-(трет-бутилдиметилсиланилокси)фенил]
-2-хлорацетамида растворяли в 12 мл ДМФ и перемешивали в течение 19 ч при комнатной температуре в присутствии 0,79 мл (5,7 ммоля)
триэтиламина и 0,87 г (4,56 ммоля) 4-бензил-4-гидроксипиперидина.
Реакционную смесь концентрировали, растворяли в CH2Cl2 и промывали 2 порциями по 30 мл H2O.
Органическую фазу сушили над Na2SO4 и
концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат в соотношении 1: 1, а затем - этилацетат) с получением 1,32 г
(78%)
2-(4-бензил-4-гидроксипиперидин-1-ил)-N-[4- (трет-бутилдиметилсиланилокси)фенил] ацетамида в виде желтого твердого вещества с tпл 105-108oC и МС: m/e = 455,5 (M+H+
).
В соответствии с общей методикой, описанной в примере 64, получали соединения из примеров 65-68.
Пример 65
N-[4-(трет-бутилдиметилсиланилокси)фенил]
-2-[4-гидрокси-4-(4- метилбензил)пиперидин-1-ил] ацетамид
Указанное в заголовке соединение с tпл 136-138oC и МС: m/e = 468
(M+) получали с использованием
N-[4(трет-бутилдиметилсиланилокси)фенил] -2-хлорацетамида и 4-(4-метилбензил)пиперидин-4-ола.
Пример 66
N-[4-(трет-бутилдиметилсиланилокси)фенил]
-2-[4-(4-хлорбензил)-4- гидроксипиперидин-1-ил] ацетамид
Указанное в заголовке соединение с tпл 135-136oC и МС: m/e = 489,4
(M+) получали с использованием
N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-хлорацетамида и 4-(4-хлорбензил)пиперидин-4-ола.
Пример 67
(RS)-N-[4-(трет-бутилдиметилсиланилокси)фенил]
-2-[4-(4- гидрокси-4-(4-метилбензил)пиперидин-1-ил] пропионамид
Указанное в заголовке соединение с tпл 134-138oC и МС:
m/e = 483,3 (M+H+) получали с
использованием (RS)-N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-хлорпропионамида и 4-(4-метилбензил)пиперидин-4-ола.
Пример 68
3-(4-бензил-4-гидроксипиперидин-1-ил)-N-[4- (трет-бутилдиметилсиланилокси)фенил] пропионамид
Указанное в заголовке соединение с МС: m/e = 468 (M+) получали с использованием
N-[4-(трет-бутилдиметилсиланилокси)фенил] -3-хлорпропионамида и 4-бензил-4-гидроксипиперидина.
Пример 69
N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-хлорацетамид
2,
23 г (10 ммолей) 4-(трет-бутилдиметилсиланилокси)фениламина растворяли в 25 мл ацетона. После добавления 3,2 г (30 ммолей) Na2CO3 по каплям добавляли 0,96 мл (12 ммолей)
хлорацетилхлорида. По истечении 1 часа выдержки при комнатной температуре реакцию прекращали добавлением в реакционную смесь 100 мл H2O и водную фазу экстрагировали 3 порциями по 10 мл
CH2Cl2. Объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат в соотношении 9: 1,
а затем гексан/этилацетат в соотношении 4: 1) с получением 2,55 г (71%) N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-хлорацетамида в виде бесцветного твердого вещества с tпл 107-108oC и МС: m/e = 299 (M+).
В соответствии с общей методикой, описанной в примере 69, получали соединения из примеров 70 и 71.
Пример 70
(RS)-N-[4-(трет-бутилдиметилсиланилокси)фенил] -2-хлорпропионамид
Указанное в заголовке соединение с tпл 74-75oC и МС: m/e = 313 (M+) получали с
использованием 4-(трет-бутилдиметилсиланилокси)фениламина и (RS)-2-хлорпропионилхлорида.
Пример 71
N-[4-(трет-бутилдиметилсиланилокси)фенил] -3-хлорпропионамид
Указанное в заголовке соединение с tпл 126oC и МС: m/e = 313 (M+) получали с использованием 4-(трет-бутилдиметилсиланилокси)фениламина и 3-хлорпропионилхлорида.
Пример 72
4-(трет-бутилдиметилсиланилокси)фениламин
7,3 г (2,9 ммоля) трет-бутилдиметил(4-нитрофенокси)силана растворяли в 75 мл MeOH и при комнатной температуре и под
атмосферным давлением гидрогенизовали в присутствии Pd на C (10%-ный, E 101 N/D) в течение 1 часа. Катализатор отфильтровывали и выпаривали растворитель с получением 6,4 г (99%)
4-(трет-бутилдиметилсиланилокси)фениламина в виде светло-желтого масла с МС: m/e = 223 (M+).
Пример 73
Трет-бутилдиметил(4-нитрофенокси)силан
5,6 г (40
ммолей) 4-нитрофенола растворяли в 200 мл CH2Cl2 и перемешивали при комнатной температуре в присутствии 7,8 г (52 ммоля) трет-бутилдиметилсилилхлорида, 0,1 г (0,8 ммоля)
4-диметиламинопиридина и 7,2 мл (52 ммоля) триэтиламина. После выдержки в течение 30 минут при комнатной температуре реакционную смесь промывали 2 порциями по 200 мл H2O и полученные
водные
фазы экстрагировали 100 мл CH2Cl2. Объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле
(гексан/диэтиловый эфир в соотношении 9: 1) с получением 10 г (100%) трет-бутилдиметил(4-нитрофенокси)силана в виде желтого твердого вещества с tпл 36-38oC и МС: m/e = 253
(M+).
Пример 74
1-(4-бензил-4-гидроксипиперидин-1-ил)-3-[4- (трет-бутилдиметилсиланилокси)фенил] пропан-2-он
В раствор 0,28 мл (3,3 ммоля) оксалилхлорида
в 4 мл
CH2Cl2 при -78oC по каплям добавляли 0,47 мл (6,6 ммоля) ДМСО. По истечении 30 мин добавляли раствор 0,75 г (1,65 ммоля) (RS)-4-бензил-1-{
3-[4-(трет-бутилдиметилсиланилокси)фенил] -2- гидроксипропил} пиперидин-4-ола в 4 мл CH2Cl2. По истечении еще одного часа выдержки при -78oC добавляли 1,8 мл (13,2
ммоля) триэтиламина и реакционной смеси давали медленно нагреться до комнатной температуры. По истечении 1 часа добавляли 15 мл 20%-ного NH4Cl, образовавшуюся водную фазу экстрагировали 3
порциями по 30 мл CH2Cl2. Объединенные органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат в
соотношении 1: 1, а затем этилацетат) с получением 0,4 г (54%) 1-(4-бензил-4-гидроксипиперидин-1-ил)-3-[4- (трет-бутилдиметилсиланилокси)фенил] пропан-2-она в виде желтого масла с МС: m/e = 454,5
(M+H+).
Пример 75
(RS)-4-бензил-1-{ 3-[4-(трет-бутилдиметилсиланилокси)фенил] -2- гидроксипропил)пиперидин-4-ол
0,62 г (2,34 ммоля)
(RS)-трет-бутилдиметил(4-оксиранилметилфенокси)силана растворяли в 8 мл MeOH и перемешивали в течение ночи при комнатной температуре в присутствии 0,9 г (4,68 ммоля) 4-бензил-4-гидроксипиперидина.
Реакционную смесь концентрировали и остаток хроматографировали на силикагеле (гексан/этилацетат в соотношении 1: 1, а затем CH2Cl2/MeOH в соотношении 19: 1) с получением 0,95 г
(90%) (RS)-4-бензил-1-{ 3-[4-(трет-бутилдиметилсиланилокси)фенил] -2- гидроксипропил} пиперидин-4-ола в виде желтого масла с МС: m/e = 456,5 (M+H+).
Пример 76
(RS)-трет-бутилдиметил(4-оксиранилметилфенокси)силан
1 г (6,66 ммоля) (RS)-4-оксиранилметилфенола растворяли в 50 мл CH2Cl2 и перемешивали при комнатной температуре в
присутствии 1,3 г (8,66 ммоля) трет-бутилдиметилсилилхлорида, 0,018 г (0,15 ммоля) 4-диметиламинопиридина и 1,2 мл (8,66 ммоля) триэтиламина. По истечении 22 часов выдержки при комнатной температуре
реакционную смесь промывали 2 порциями по 100 мл H2O и образовавшиеся водные фазы экстрагировали 100 мл CH2Cl2. Объединенные органические фазы сушили над Na2
SO4 и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат в соотношении 9: 1) с получением 1,44 г (82%)
(RS)-трет-бутилдиметил(4-оксиранилметилфенокси)силана в виде
желтого масла с МС: m/e = 264 (M+).
Пример 77
(RS)-4-оксиранилметилфенол
8,9 г (66,3 ммоля)
4-аллилфенола растворяли в 180 мл CH2Cl2
. После добавления 8,4 г (99,5 ммоля) NaHCO3 отдельными порциями добавляли 18 г (73 ммоля) 70%-ной м-хлорнадбензойной кислоты.
После выдержки в течение 6 часов при комнатной температуре
дополнительно добавляли 8,4 г (99,5 ммоля) NaHCO3 и 18 г (73 ммоля) 70%-ной м-хлорнадбензойной кислоты. По истечении 17 часов
реакционную смесь промывали 200 мл насыщенного раствора
NaHCO3 и образовавшиеся водные фазы экстрагировали 3 порциями по 10 мл CH2Cl2. Объединенные органические фазы
промывали 2 порциями по 100 мл насыщенного раствора
Na2S2O3, сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле
(гексан/этилацетат в соотношении 9: 1, а затем 1: 1) с
получением 3,77 г (38%) (RS)-4-оксиранилметилфенола в виде желтого твердого вещества с tпл 54-57oC и МС: m/e = 150 (M+).
Пример 78
4-аллилфенол
В холодный (-78oC) раствор 14,6 мл (95 ммолей) 4-аллиланизола в 300 мл CH2Cl2 по каплям добавляли
100 мл (0,1 моль, 1 моль/л в CH2
Cl2) раствора BBr3. Затем реакционной смеси давали нагреться до комнатной температуры. По истечении 1 часа реакционную смесь охлаждали
до 0oC и реакцию в ней
прекращали медленным добавлением 90 мл H2O. Образовавшуюся водную фазу экстрагировали 2 порциями по 100 мл CH2Cl2. Объединенные
органические фазы сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат в соотношении 9: 1) с получением 11,3 г (89%) 4-аллилфенола в
виде пурпурного масла с МС: m/e = 134
(M+).
Пример 79
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этиловый эфир 4-бензилоксибензойной кислоты
0,685 г
(3 ммоля) 4-бензилоксибензойной кислоты
растворяли в 6 мл ДМФ и отдельными порциями добавляли 0,58 г (3,6 ммоля) 1,1'-карбонилдиимидазола. Реакционную смесь выдерживали при 55-60oC в
течение 20 мин, а затем охлаждали до комнатной
температуры. Добавляли раствор 0,78 г (3,3 ммоля) 1-(2-гидроксиэтил)-4-(4-метилбензил)пиперидин-4-ола в 2 мл ДМФ. Реакционную смесь перемешивали в
течение 23 часов при комнатной температуре и 4 часа
при 60oC. Добавляли 50 мл H2O, а затем CH2Cl2. Органическую фазу промывали насыщенным раствором
NaHCO3, сушили над Na2SO4 и
концентрировали. Остаток хроматографировали на силикагеле (CH2Cl2/MeOH в соотношении 19: 1) с получением 0,65 г
(47%) 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этилового
эфира 4-бензилоксибензойной кислоты в виде бесцветного твердого вещества с tпл 102oC и МС: m/e = 460,3 (M+H+).
В соответствии с общей методикой, описанной в примере 79, получали соединение из примера 80.
Пример 80
3-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] пропиловый эфир 4-бензилоксибензойной кислоты
Указанное в заголовке соединение с МС: m/e = 474,4 (M+H+) получали с использованием
1-(3-гидроксипропил)-4-(4-метилбензил)пиперидин-4-ола.
Пример 81
1-(2-гидроксиэтил)-4-(4-метилбензил)пиперидин-4-ол
Смесь, содержавшую 5,1 г (25 ммолей)
4-(4-метилбензил)пиперидин-4-ола, 1,8 мл (25 ммолей) 2-бромэтанола и 5,2 г (37,5 ммоля) K2
CO3 в 120 мл 2-бутанона, кипятили с обратным холодильником в течение 22 часов.
Добавляли 30 мл H2O и водную фазу экстрагировали этилацетатом. Объединенные органические фазы
сушили над Na2SO4 и концентрировали. Остаток хроматографировали на
силикагеле (CH2Cl2/MeOH в соотношении 9: 1 + 1% NH4OH) с получением 4,3 г 73%
1-(2-гидроксиэтил)-4-(4-метилбензил)пиперидин-4-ола в виде желтого масла с МС: m/e = 235,
3 (M+).
В соответствии с общей методикой, описанной в примере 81, получали соединение из примера 82.
Пример 82
1-(3-гидроксипропил)-4-(4-метилбензил)пиперидин-4-ол
Указанное в заголовке соединение с МС: m/e = 263,3 (M+) получали
с использованием 4-(4-метилбензил)пиперидин-4-ола и
3-бром-1-пропанола.
Пример 83
4-бензилокси-N-(2-гидроксиэтил)бензамид
Раствор 20 г (87,6 ммоля)
4-бензилоксибензойной кислоты, 14,9 г (91,8 ммоля) 1,
1'-карбонилдиимидазола и 80 мл ДМФ перемешивали при 50oC в течение 1 ч. Раствор охлаждали до 0oC и добавляли 81,2 г 25%-ного
водного раствора этаноламина. По истечении 45 мин
осадок отфильтровывали с получением 22,49 г (94,5%) 4-бензилокси-N-(2-гидроксиэтил)бензамида в виде белого твердого вещества. МС: me/e = 271 (M)+.
Пример 84
4-бензилокси-N-(2-хлорэтил)бензамид
Через суспензию 22,49 г (82,8 ммоля) 4-бензилокси-N-(2-гидроксиэтил)бензамида в 130 мл диоксана
барботированием пропускали фосген до растворения всего
нерастворенного материала. Избыток фосгена удаляли током диоксида углерода. Растворитель удаляли под пониженным давлением и остаток сушили в
течение 1 ч при 100oC. Сырой продукт
перекристаллизовывали из этилацетата с получением 16,3 г (68%) 4-бензилокси-N-(2-хлорэтил)бензамида.
Пример 85
4-бензилокси-N-[2-(4-гидрокси-4-фенилпиперидин-1-ил)этил]
бензамид
Смесь 2,1 г (7,25 ммоля) 4-бензилокси-N-(2-хлорэтил)бензамида, 1,386 г (7,25 ммоля) 4-бензил-4-гидроксипиперидина, 2,0 г (14,
5 ммоля) карбоната калия и 40 мл 2-бутанона перемешивали
в течение 15 ч при 60oC. После добавления воды смесь экстрагировали этилацетатом. Органический слой сушили (Na2SO4), фильтровали и упаривали. Остаток очищали
хроматографией (силикагель, хлористый метилен/метанол в соотношении от 95: 5 до 9: 1) с получением 680 мг (21%)
4-бензилокси-N-[2-(4-гидрокси-4-фенилпиперидин-1-ил)этил] бензамида в виде
светло-желтого твердого вещества. МС: me/e = 445 (M+H)+.
Пример 86
4-бензилокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] этил} бензамид
Смесь
2,0 г (6,9 ммоля) 4-бензилокси-N-(2-хлорэтил)бензамида, 1,06 г (5,18 ммоля) 4-гидрокси-4-(метилбензил)пиперидина, 1,43 г
(10,35 ммоля) карбоната калия и 40 мл 2-бутанона перемешивали в течение 15 ч
при 60oC. После добавления воды смесь экстрагировали этилацетатом. Органический слой сушили (Na2
SO4), фильтровали и упаривали. Остаток очищали хроматографией
(силикагель, хлористый метилен/метанол в соотношении от 95: 5 до 9: 1) с получением 445 мг (19%) 4-бензилокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этил} бензамида в виде светло-желтого
твердого вещества. МС: me/e = 459 (M+H)+
Пример 87
4-бензилокси-N-(3-хлорпропил)бензамид
Смесь 5,0 г (21,9 ммоля) 4-бензилоксибензойной кислоты, 3,6 г (1,05 экв.
) 1,1'-карбонилдиимидазола и 50 мл ДМФ перемешивали при 50oC в
течение 1 ч. После охлаждения до комнатной температуры добавляли 3,4 г (26,1 ммоля) 3-хлорпропиламингидрохлорида и 3,5 г (32,
0 ммоля) карбоната натрия и перемешивание продолжали в течение 45 мин.
После добавления воды смесь экстрагировали диэтиловым эфиром. Органический слой сушили (Na2SO4),
фильтровали и упаривали с получением 5,44 г (85%)
4-бензилокси-N-(3-хлорпропил)бензамида в виде белого твердого вещества. МС: me/e = 304 (M+H)+
Пример 88
N-[3-(4-бензил-4-гидроксипиперидин-1-ил)пропил]
-4-бензилоксибензамид
Смесь 0,5 г (1,64 ммоля) 4-бензилокси-N-(3-хлорпропил)бензамида, 0,315 г (1,65 ммоля) 4-бензил-4-гидроксипиперидина, 0,
45 г (3,29 ммоля) карбоната калия и 10 мл
2-бутанона перемешивали при 60oC в течение 48 ч. После добавления воды смесь экстрагировали этилацетатом. Органический слой сушили (Na2
SO4), фильтровали и упаривали.
Остаток очищали хроматографией (силикагель, хлористый метилен/метанол в соотношении от 95: 5 до 9: 1) с получением 185 мг (25%)
N-[3-(4-бензил-4-гидроксипиперидин-1-ил)пропил] -4-бензилоксибензамида в
виде светло-желтого твердого вещества. МС: me/e = 459 (M+H)+.
Пример 89
4-бензилокси-N-{
3-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] пропил} бензамид
Смесь 0,5 г (1,64 ммоля) 4-бензилокси-N-(3-хлорпропил)бензамида, 0,34 г (1,65 ммоля) 4-гидрокси-4-(4-метилбензил)пиперидина,
0,45 г (3,29 ммоля) карбоната калия и 10 мл 2-бутанона перемешивали при
60oC в течение 48 ч. После добавления воды смесь экстрагировали этилацетатом. Органический слой сушили (Na2SO4), фильтровали и упаривали. Остаток очищали
хроматографией (силикагель, хлористый метилен/метанол в соотношении от 95: 5 до 9: 1) с получением 220 мг (28%) 4-бензилокси-N-{
3-[4-гидрокси-4-(4-метилбензил)пиперидин-1- ил] пропил} бензамида в
виде светло-желтого твердого вещества. МС: me/e = 473 (M+H)+.
Пример 90
1-(2-метиламиноэтил)-4-(4-метилбензил)пиперидин-4-ол
Смесь 1,56 г (7,6 ммоля)
4-гидрокси-4-(4-метилбензил)пиперидина, 0,89 г (8,4 ммоля) карбоната натрия, N-метилхлорацетамида и ацетона (14
мл) перемешивали при комнатной температуре в течение 48 ч. После выпаривания
растворителя в суспензию добавляли воду. Водный слой экстрагировали диэтиловым эфиром, органический слой сушили (Na2SO4), фильтровали и упаривали с получением после растирания в
диэтиловом эфире 1,3 г белого кристаллического вещества. Затем его отдельными порциями при 0oC добавляли в
перемешиваемую суспензию 350 мг (9,2 ммоля) алюмогидрида лития в 30 мл ТГФ. После
перемешивания при комнатной температуре в течение ночи и кипячении с обратным холодильником в течение 1 ч реакционную
смесь охлаждали до 0oC и осторожно добавляли в нее (каждый раз по 1 мл)
воду, 15%-ный гидроксид натрия и вновь воду. Фильтрованием, выпариванием растворителя из фильтрата, добавлением воды
и экстракцией хлористым метиленом после сушки (Na2SO4) с
последующим фильтрованием и упариванием получали 480 мг 1-(2-метиламиноэтил)-4-(4-метилбензил)пиперидин-4-ола. МС: me/e
= 263 (M+H)+.
Пример 91
4-бензилокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этил} -N-метилбензамид
Смесь 265 мг (1,16 ммоля)
4-бензилоксибензойной кислоты, 197 мг (1,21 ммоля) 1,1'-карбонилдиимидазола и 7 мл ДМФ
перемешивали при 50oC в течение 1 ч. После охлаждения до комнатной температуры добавляли 335 мг (1,27
ммоля) 1-(2-метиламиноэтил)-4-(4-метилбензил)пиперидин-4-ола и перемешивание продолжали
в течение 1 ч. После добавления воды смесь экстрагировали диэтиловым эфиром. Органический слой сушили (Na2SO4), фильтровали и упаривали. Остаток очищали хроматографией
(силикагель, дихлорметан/метанол в соотношении 95: 5) с получением 226 мг 4-бензилокси-N-{
2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этил} -N-метилбензамида в виде бледно-желтого масла. МС: me/e
= 473 (M+H)+.
Пример 92
(E)-1-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -3-(4- метоксифенил)пропенон
Смесь 3,0 г (16,8 ммоля) 4-метоксикоричной кислоты,
2,78 г (1,05 экв. ) 1,1'-карбонилдиимидазола и 50 мл ДМФ
перемешивали при 50oC в течение 1 ч. После охлаждения до комнатной температуры добавляли 3,63 г (1,05 экв. )
4-(4-метилбензил)-4-гидроксипиперидина и перемешивание продолжали в течение 1
ч. Затем добавляли воду и смесь экстрагировали диэтиловым эфиром. Сушкой (Na2SO4), фильтрованием и
упариванием органического слоя получали 5,87 г (95%)
(E)-1-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -3-(4- метоксифенил)пропенона в виде белой пены. МС: me/e = 365 (M)+.
Пример 93
(E)-1-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -3-(4- гидроксифенил)пропенон
В раствор 1,68 г (4,6 ммоля) (E)-1-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил]
-3-(4- метоксифенил)пропенона
в 50 мл дихлорметана при 0oC добавляли 9,2 мл (2,0 экв. ) 1 М раствора трибромида бора в дихлорметане. После перемешивания при комнатной температуре в течение
4 ч добавляли 50 мл воды и 20
мл водного раствора бикарбоната натрия и водный слой экстрагировали дихлорметаном. Органический слой сушили (Na2SO4), фильтровали и упаривали с
получением 1,423 г (88%)
(E)-1-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] -3-(4- гидроксифенил)пропенона в виде белой пены. МС: me/e = 351 (M)+.
Пример 94.
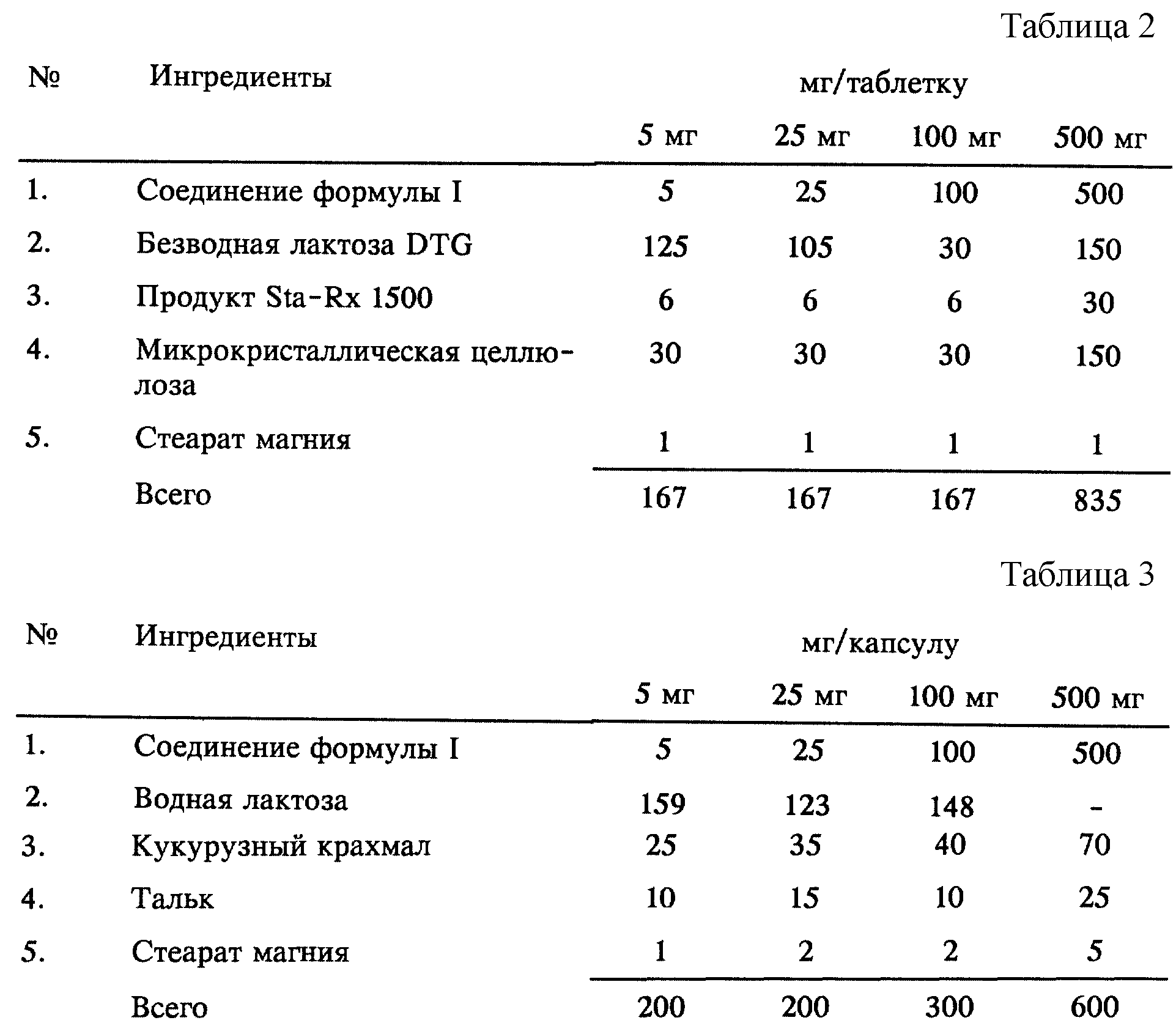
Композиция для таблеток (мокрая грануляция) (см. табл. 2).
Методика приготовления
1. Смешивают ингредиенты 1, 2, 3 и 4 и гранулируют с использованием очищенной
воды.
2. Полученный гранулят сушат при 50oC.
3. Гранулят пропускают через соответствующее измельчительное оборудование.
4. Добавляют ингредиент 5 и перемешивают в течение трех минут, прессуют в соответствующем прессе.
Пример 95
Композиция для капсул (см. табл. 3).
Методика приготовления
1. Ингредиенты 1, 2 и 3
перемешивают в течение 30 минут.
2. Добавляют ингредиенты 4 и 5 и смешивают в течение 3 минут.
3. Наполняют соответствующую капсулу.
Реферат
Изобретение относится к новым производным 4-гидроксипиперидина общей формулы (I), где Х обозначает -О-, -NH-, -CH2-, -СН= , -СО2-, -СОN(низший алкил)- или -CONH-, R1 - R4, независимо друг от друга, - водород, гидрокси, нитрогруппа, низший алкилсульфонамид, 1- или 2-имидазолил, 1-(1,2,4-триазолил), R5 и R6, независимо друг от друга, - водород, низший алкил, гидрокси- или оксогруппа, R7 - R10, независимо друг от друга, - водород, низший алкил, галоген, трифторметил или низшая алкоксигруппа, n = 0 или 1, или к их фармацевтически приемлемым кислотно-аддитивным солям. Изобретение может быть использовано как лекарственное средство в качестве селективного блокатора подтипа NMDA-рецептора. Также раскрыто лекарственное средство на основе 4-гидроксипиперидина. 2 с. и 10 з. п. ф-лы, 3 табл.

Формула
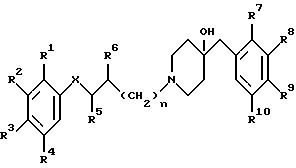
где Х обозначает -О-, -NH-, -CH2-, -CH= , -CO2-, -CONH- или CON(низший алкил)-;
R1 - R4 независимо друг от друга - водород, гидрокси-, нитрогруппа, низший алкилсульфониламид, 1- или 2-имидазолил или 1-(1,2,4-триазолил);
R5 и R6 независимо друг от друга - водород, низший алкил, гидрокси- или оксогруппа;
R7 - R10 независимо друг от друга - водород, низший алкил, галоген, трифторметил или низшая алкоксигруппа;
n = 0 или 1,
и его фармацевтически приемлемые кислотно-аддитивные соли.
1-[2-(4-гидроксифенокси)этил] -4-(4-метилбензил)пиперидин-4-ол;
4-(4-фторбензил)-1-[2-(4-гидроксифенокси)этил] пиперидин-4-ол;
N-(4-{ 2-[4-гидрокси-4-(4-метилбензил)пиперидин-1-ил] этокси} фенил)метансульфонамид;
N-(4-{ 2-[4-(4-фторбензил)-4-гидроксипиперидин-1-ил] этокси} фенил)метансульфонамид;
N-(4-{ 2-[4-(4-хлорбензил)-4-гидроксипиперидин-1-ил] этокси} фенил)метансульфонамид;
N-(4-{ 3-[4-(4-фторбензил)-4-гидроксипиперидин-1-ил] пропокси} фенил)метансульфонамид;
1-[2-(4-гидроксифенокси)-1-метилэтил] -4-(4-метилбензил)пиперидин-4-ол.
(RS)-4-бензил-1-[2-гидрокси-3-(4-гидроксифенил)пропил] пиперидин-4-ол;
(RS)-1-[2-гидрокси-3-(4-гидроксифенил)пропил] -4-(4-метилбензил)пиперидин-4-ол;
(RS)-4-(4-хлорбензил)-1-[2-гидрокси-3-(4-гидроксифенил)пропил] пиперидин-4-ол.
19.07.1996 по пп. 1-12;
01.04.1997 по пп. 1-12 (уточнение признаков).
Комментарии