Устойчивые лекарственные формы с регулируемым выделением, имеющие акриловые полимерные покрытия, (варианты) и способ их получения - RU2127587C1
Код документа: RU2127587C1
Чертежи





Описание
Важным аспектом всех видов небольших дозированных форм является их стабильность. Стабильность фармацевтической дозированной формы касается сохранения ее физических, химических, микробиологических, лечебных и токсикологических свойств, в частности содержимого и оболочки при хранении. Требования по исследованию стабильности рассматриваются, например, в Good Manyfacturing Practices (GMPs), the USP, а также в новых лекарственных заявках (NDAs) и в новых исследованиях лекарственных заявках (INDS).
Подбор ингредиентов, используемых для поддержания выделения дозированных лекарственных форм, часто представляет особую проблему, которая относится к их физической стабильности при хранении. Например, известно, что воски, используемые в таких лекарственных формах, претерпевают физические изменения при длительном хранении и поэтому предусматривается их стабилизация как в процессе производства, так и для предотвращения возможных изменений. Известно, что вещества, представляющие собой жиры и воски, при их использовании в очищенном состоянии могут кристаллизоваться в неустойчивые формы, вызывая непредсказуемые изменения в их пригодности при испытании стабильности в процессе производства и при дальнейшем хранении.
Известно, что во многих случаях требуется проявить определенное искусство для получения лекарственных форм с регулируемым выделением лекарства, таких как обеспечение стабильной формы индивидуальных ингредиентов до их введения в продукт, требование того, чтобы обработка не меняла эти условия, замедление нестабильности путем введения дополнительных добавок и создания условий для достижения индивидуальными ингредиентами дозированных форм стабильного состояния до окончательного получения продукта.
Признано также, что содержание в продукте влаги также может оказывать влияние на его стабильность. Изменения в в степени гидратации полимерной пленки, такой как этилцеллюлоза, может изменять степень водопроницаемости и пригодность лекарства. Кроме того, известно, что связующие, такие как акация, становятся менее растворимыми при действии влаги и тепла. Однако содержание влаги в продукте может быть подвергнуто достаточно успешному регулированию при определенной обработке и непосредственно при упаковке продукта.
Гидрофобные полимеры, такие как определенные производные целлюлозы, зеин, акриловые смолы, воски, высшие алифатические спирты и полимеры молочной и гликолевой кислот были использованы в предшествующих работах для создания регулируемых дозировочных форм. Способ использования таких полимеров для создания регулируемых дозировочных форм, таких как таблетки, капсулы, суппозитории, шарики, гранулы или микрошарики, состоит в покрытии индивидуальных дозированных единиц такими гидрофобными полимерами. Из предшествующих работ известно, что такие гидрофобные покрытия могут быть получены из растворов, суспензий или в сухом состоянии. Так как большая часть таких полимеров обладает низкой растворимостью в воде, они используются обычно растворением полимера в органическом растворителе, напылением раствора на индивидуальную лекарственную форму, (такую, как гранула или таблетка) и удалением растворителя.
В патенте США N 4990341 описана твердая дозированная лекарственная форма, включающая эффективное количество гидроморфона или его соли в матрице, имеющей требуемую скорость растворения in vitro. В качестве матрицы могут использоваться любые матрицы, которые обеспечивают скорость растворения гидроморфона, не зависящую от pH, например гидрофильные полимеры, включая акриловые смолы, длинноцепочечные (C8-C20) замещенные или незамещенные углеводороды и полиалкиленгликоли.
Водные дисперсии гидрофобных полимеров были использованы в работах, описанных в предшествующем уровне, для покрытия фармацевтических дозированных форм для эстетических целей, таких как покрытие пленкой таблеток или гранул, или для изменения вкуса. Однако такие дозированные формы используют для непосредственного выделения активного лекарственного средства, содержащегося в дозированной форме, при приеме лекарства.
Попытки получения устойчивых лекарственных форм с использованием водных дисперсий гидрофобных полимеров не были успешными из-за проблем, связанных со стабильностью.
Таким образом, желательным является получение лекарственной формы с регулируемым выделением из водных дисперсий гидрофобных полимеров. Однако на сегодняшний день, попытки получения устойчивых фармацевтических лекарственных форм с регулируемым выделением при использовании водных дисперсий гидрофобных полимеров не были успешными из-за проблем со стабильностью.
В частности, при покрытии таких фармацевтических форм с использованием водных дисперсий полимеров для получения активного лекарства (лекарств) с характеристикой выделения до нескольких часов или более из работ предшествующего уровня известно, что характеристика выделения при растворении изменяется при старении. Это было показано недавно Munday и др., Drug Devel and Indus Phar 17 (15) 2135 - 2143 (1991), который отметил влияние хранения на степень выделения лекарства для мини-таблеток теофилина, покрытых пленкой этилцеллюлозы с ПЭГ (при отношении 2:1; величина покрытия = 3% по весу), этилцеллюлозы с Eudragit®L (при отношении 2:1; величина покрытия = 3% по весу) и Eudragit®L (величина покрытия = 1,5% по весу) при изменении температур и относительных влажностей. Образцы были подвергнуты изотермическому старению при 28oC, 35oC и 45oC при относительной влажности (RH), поддерживаемой в интервале 55 - 60%, при циклических условиях при 45oC при 55% RH в течение 24 часов, затем при 28oC и 20% RH в течение 24 часов, затем при 5oC и 10% RH, поддерживаемых в течение 24 часов, после чего цикл повторялся с изменением условий каждые 24 часа между 45oC и 55% RH и 28oC и 0% RH. Процесс старения, вызванный хранением при вышеперечисленных стрессовых условиях, задерживал растворение, независимо от природы полимерной пленки. Было установлено, что наибольшее снижение в скорости выделения происходит в первые 21 день (изотермическое хранение) после покрытия.
Известно, что такой проблемы, связанной с нестабильностью, не существует при применении полимеров из органического растворителя. Использование органических растворителей при приготовлении полимерных покрытий рассматривается как сомнительное, так как лекарственные формы характеризуются обычно присущими им проблемами в отношении способности к воспламенению, канцерогенности, загрязнения окружающей среды, общей безопасности.
Кроме того, попытки получения фармацевтических лекарственных форм с регулируемым выделением с использованием органических покрытий оказались в значительной степени безуспешности из-за проблем, связанных со стабильностью и изменением скорости выделения лекарства при хранении.
Например, в данной области техники желательным является получение лекарственной формы с регулируемым выделением, которое используют ингибирующее покрытие, полученное из водной дисперсии акрилового полимера, такого как Eudragit®, коммерчески доступный из фирмы Rohm Pharma. Однако до настоящего времени невозможно было получить лекарственную форму с регулируемым выделением, которая является стабильной в различных условиях хранения.
Более детально, известно, что покрытие с регулируемым выделением, содержащее Eudragit®, нестабильно при условиях отверждения, рекомендуемых изготовителем, - при 45oC в течение двух часов.
Таким образом, целью данного изобретения является создание лекарственной дозированной формы с регулируемым выделением для орального приема, которая имела бы регулирующее выделение покрытие, полученное из водной дисперсии акрилового полимера, так, чтобы в основном характеристика стабильности растворения медикамента сохранялась бы при различных условиях хранения.
Далее, целью данного изобретения является создание дозированной формы с регулируемым выделением, получаемой с использованием покрытия, наносимого из водной дисперсии акриловой смолы, которая является стабильной в стрессовых условиях, включая длительные периоды воздействия высокой температуры и высокой влажности.
Эти и другие цели достигаются данным изобретением, которое относится к твердым дозированным формам, которые имеют регулирующее выделение покрытие, получаемое из водной дисперсии акриловой смолы, которая обеспечивает в основном стабильное выделение образцом содержащегося в нем терапевтически активного агента(ов).
Далее, представленное изобретение относится к удивительному открытию, состоящему в том, что когда покрытая лекарственная форма подвергается действию определенных повышенных или "стрессовых" условий - температур и влажности - в течение определенного отрезка времени, может быть достигнута желаемая граничная точка, после которой скорость выделения терапевтически активного агента существенно не меняется при старении в условиях широкого интервала температур и/или влажности. Это удивительное открытие делает возможным регулирование выделения покрытий данного изобретения для многих видов фармацевтических дозированных форм для создания устойчивых фармацевтических дозированных продуктов с регулируемым выделением.
Данное изобретение относится также к твердой дозированной форме, включающей сердцевину, содержащую терапевтически активный агент, и покрытие, полученное из водной дисперсии акриловой смолы в количестве, достаточном для обеспечения регулируемого выделения терапевтически активного агента, когда дозированная форма подвергается действию водных растворов, например желудочной жидкости. Твердая дозированная форма подвергается отверждению после нанесения покрытия таким образом, чтобы выделение терапевтически активного агента в основном не изменялось при воздействии повышенной температуры и/или влажности.
Данное изобретение относится также к твердой устойчивой дозированной форме с регулируемым выделением - для орального приема, включающей множество инертных фармацевтически приемлемых гранул, покрытых терапевтически активным агентом, и покрытых сверху акриловой смолой, имеющей толщину, достаточную для достижения регулируемого выделения названного терапевтически активного агента при помещении твердой дозированной формы в водные растворы. Покрытые гранулы подвергаются отверждению в течение определенного периода времени при температуре, выше переходной температуры стеклования (Tg) пластифицированного акрилового полимера с получением конечного продукта, который имеет характеристику растворения, на которую, в основном, не влияют воздействия длительного хранения при повышенной температуре и/или влажности.
Данное изобретение относится, далее, к устойчивой твердой лекарственной дозированной форме, содержащей терапевтически активный агент, покрытый сверху пластифицированным акриловым полимером, при этом покрытая дозированная форма подвергается отверждению при эффективной температуре, выше Tg пластифицированного акрилового полимера, в течение такого периода времени, при котором характеристика растворения устойчивого лекарственного средства существенно не меняется под действием хранения в условиях повышенной температуры и/или влажности.
Данное изобретение касается также способа получения устойчивой лекарственной формы с регулируемым выделением, включающей субстрат, покрытый пластифицированным акриловым полимером. Способ включает этапы: получение водной дисперсии акрилового полимера и предпочтительно пластифицированного, получение субстрата, включающего терапевтически активный агент, покрытие сверху субстрата достаточным количеством дисперсии акрилового полимера для получения предварительно определенного регулируемого выделения активного агента при помещении покрытых частиц в водные растворы и отверждение покрытого субстрата при эффективной температуре, выше Tg пластифицированного акрилового полимера в течение такого периода времени, при котором достигается граничная точка, при которой характеристика растворения лекарственного препарата в основном не меняется под действием хранения в условиях повышенных или изменяющихся температуры и/или влажности.
При дальнейшем осуществлении способ, кроме того, включает этап определения граничной точки для конкретной лекарственной формы выдержкой ее при различных стадиях приведенного выше отверждения и получения характеристик растворения для лекарственной формы до тех пор, пока не будут в основном стабилизированы характеристики растворения. Затем, если это необходимо, лекарственная форма модифицируется для получения нужной характеристики растворения терапевтически активного агента, основанной на граничной точке.
В конкретных предложенных осуществлениях данного изобретения акриловый полимер, включающий регулирующее выделение покрытие, состоит из одного или большего числа аммонийметакрилатных сополимеров. Аммонийметакрилатные сополимеры являются хорошо известными в данной области техники и описаны в NF XVII как полностью полимеризованные сополимеры акриловых и метакриловых кислых эфиров с низким содержанием четвертичных аммониевых групп.
Для того чтобы получить лекарственные формы с регулируемым выделением, обычно бывает необходимо покрыть субстрат, содержащий терапевтически активный агент, достаточным количеством водной дисперсии акрилового полимера с тем, чтобы получить степень привеса приблизительно от 5 до 15 процентов, хотя наружное покрытие может быть меньше или больше в зависимости от физических свойств терапевтически активного агента и желаемой скорости выделения, включения пластификатора в водную дисперсию акрилового полимера, например, способа его введения.
Примером подходящей лекарственной формы с регулируемым выделением согласно данному изобретению является скорость растворения дозированной формы in vitro, когда эта величина, измеренная методом USP Paddle при 100 м.д. в 900 мл водного буферного раствора (pH между 1.6 и 7.2) при 37oC лежит в интервале между 12.5 и 42.5% (по весу) терапевтически активного агента, выделенного спустя 1 час, между 25 и 55% (по весу) выделенного спустя 2 часа, между 45 и 75% (по весу), выделяемого спустя 4 часа и более, чем 55% (по весу) выделяемого спустя 6 часов. Этот пример, конечно, не следует рассматривать как в какой-либо степени ограничивающий изобретение.
Для того чтобы получить необходимую характеристику растворения для данного, терапевтически активного агента, такого как детально описанный выше, необходимо соединять два или больше аммонийметакрилатных сополимеров, имеющих различные физические свойства. Например, известно, что изменением молярного отношения четвертичных аммониевых групп к нейтральным (мет)акриловым эфирам можно изменить свойства проницаемости образующегося покрытия.
В предложенном осуществлении данного изобретения акриловое покрытие получается из двух акриловых смоляных лаков, используемых в форме водных дисперсий, коммерчески доступных из фирмы Rohm Pharma под торговой маркой Eudragit®RL30D и Eudragit®RS30D соответственно. Eudragit® RL30D и Eudragit®RS30D представляют собой сополимеры акриловых и метакриловых эфиров с низким содержанием четвертичных аммониевых групп, причем молярное отношение аммониевых групп к остальным нейтральным (мет)акриловым эфирам в Eudragit®RL30D составляет 1:20, а в Eudragit®RS30D - 1: 40. Величина молекулярной массы около 150.000. Кодовые обозначения RL (высокая проницаемость) и RS (низкая проницаемость) относятся к свойствам проницаемости этих агентов. Смеси Eudragit®RL/RS являются нерастворимыми в воде и в пищеварительных жидкостях. Однако образованные покрытия являются набухающими и проницаемыми в водных растворах и пищеварительных жидкостях.
Дисперсии Eudragit®RL/RS данного изобретения могут быть смешаны в любом нужном отношении с тем, чтобы в конечном счете получить лекарственную форму с регулируемым выделением, имеющую нужную характеристику растворения. Нужные лекарственные формы с регулируемым выделением могут быть получены, например, из замедляющего покрытия, получаемого из

Кроме того, для модифицирования и характеристики растворения изменением относительных количеств различных акриловых смоляных лаков характеристика растворения конечного продукта может быть модифицирована также, например, увеличением или снижением толщины замедляющего покрытия.
Водные дисперсии акриловых полимеров, примененные в качестве покрытий в данном изобретении могут быть использованы в таблетках, шариках (или гранулах), микрошариках, зернах, пилюлях, гранулах ионообменной смолы и других системах мультичастиц для получения необходимого регулируемого выделения терапевтически активного агента. Гранулы, шарики или пилюли и т.д., полученные в соответствии с данным изобретением, могут существовать в виде капсул или любых других дозированных лекарственных форм.
Лекарственные покрытия данного изобретения должны быть способны к образованию прочной целостной пленки, которая является гладкой и первоклассной, способной удерживать пигменты и другие добавки к покрытию, нетоксичной, инертной и нелипкой.
Предполагается, что акриловые покрытия, использованные в данном изобретении, включают эффективное количество подходящего пластифицирующего агента, так как было установлено, что использование пластификатора будет способствовать улучшению физических свойств пленки. Например, использование пластификатора может повысить эластичность пленки и снизить температуру пленкообразования дисперсии. Пластифицирование акриловой смолы может выполняться либо посредством так называемой "внутренней пластификации", либо "внешней пластификацией".
Внутренняя пластификация обычно относится непосредственно к молекулярным модификациям полимера в процессе его изготовления, например, сополимеризацией, такой как изменение и/или замещение функциональных групп, регулирование длины боковых цепей или регулирование длины полимера. Такие способы обычно не используются при формировании покрытия из раствора.
Внешняя пластификация предусматривает введение вещества в пленочный раствор с тем, чтобы достичь необходимых изменений в пленочных свойствах сухой пленки.
Пригодность пластификатора зависит от его средства или сольватирующей способности по отношению к полимеру и от его эффективности при смешении с полимер-полимерными композициями. Такая активность придает желаемую гибкость, снижая молекулярную жесткость.
Обычно количество пластификатора, включенного в раствор, предназначенный для покрытия, основывается на концентрации пленкообразующего, например составляет в большинстве случаев приблизительно от 1 до 50 процентов по весу от веса пленкообразующего. Однако концентрация пластификатора конкретно может быть определена только после внимательного эксперимента с конкретным раствором, применяемым для покрытия, и способом его применения.
Наиболее предпочтительным является введение в водную дисперсию акрилового полимера около 20% пластификатора.
Важным показателем при определении пригодности пластификатора для полимера является переходная температура стеклования полимера (Tg). Переходная температура стеклования относится к температуре или температурному интервалу, в котором наблюдается существенное изменение физических свойств полимера. Это изменение не отражает изменения в состоянии, но скорее относится к макромолекулярной подвижности полимера. Ниже Tg подвижность полимерной цепи сильно ограничена. Таким образом, для данного полимера, если величина его Tg выше комнатной температуры, полимер ведет себя подобно стеклу, являясь твердым, непластичным и скорее хрупким, и эти свойства могут быть до некоторой степени ограничивающими в процессе покрытия пленкой, так как покрытые дозированные формы могут подвергаться определенному воздействию внешних напряжений.
Введение подходящего пластификатора в полимерную матрицу эффективно снижает Tg, так что при обычных условиях пленка является более мягкой, более пластичной и часто более прочной, и, таким образом, она в большей степени способна выдерживать механическое напряжение.
Другими аспектами пригодности пластификаторов являются способность пластификатора действовать как хороший "набухающий агент" для этилцеллюлозы и нерастворимость пластификатора в воде.
Примерами подходящих пластификаторов для акриловых полимеров, соответствующими данному изобретению, но не ограничивающими его, могут служить эфиры лимонной кислоты, такие как триэтилцитрат NF XVI, трибутилцитрат, дибутилфталат и, возможно, 1,2-пропиленгликоль. Другие пластификаторы, которые оказываются подходящими для придания эластичности пленкам, образующимся из акриловых пленок, являются такие, как Eudragit®RL/RS, лаковые растворы, включая полиэтиленгликоли, пропиленгликоль, диэтилфталат, касторовое масло и триацетин. Особенно предпочтительным пластификатором для водных дисперсий этилцеллозы является в соответствии с данным изобретением триэтилцитрат.
Далее, было установлено, что добавление небольших количеств талька снижает тенденцию водной дисперсии к склеиванию в процессе обработки и действует как полирующая присадка.
Устойчивые лекарственные формы с регулируемым выделением в соответствии с данным изобретением медленно выделяют терапевтически активный компонент, например, при проглатывании и воздействии желудочных соков. Характеристика регулируемого выделения лекарственных форм данного изобретения может изменяться, например, изменением количества наносимого сверху покрытия, изменением способа введения пластификатора, изменением количества пластификатора по отношению к акриловой смоле, введением дополнительных ингредиентов или наполнителей, изменением способа производства и т.д.
В соответствии с данным изобретением может быть использовано большое число различных терапевтически активных агентов. Терапевтически активные агенты (например, фармацевтические агенты, которые могут быть использованы в композициях данного изобретения), включают как водорастворимые, так и нерастворимые в воде лекарственные препараты. Примерами таких терапевтически активных агентов являются антигистамины (например, дименгидринат, дифенгидринат, хлорфенирамин и дихлорфенирамин малеат), анальгетики (например, аспирин, кодеин, морфин, дигидроморфин, оксикодон и т.д.), противовоспалительные агенты (например, напроксин, дихлофенак, индометацин, ибупрофен, ацетаминофен, аспирин, сулиндак), желудочно-кишечные и противорвотные (например, метоклопрамид), антиэпилептические (например, фенитоин, мепробамат и нитрезепам), сосудорасширяющие (например, нифедипин, папаверин, дильтиазем и никардирин), противокашлевые агенты и отхаркивающие средства (например, кодеин фосфат), антиастматики (например, теофиллин), антиспазмолитические (например, атропин, скополамин), гормоны (например, инсулин, лепарин), диуретики (например, элтакримовая кислота, бендрофлюазид), антигипотензивные (например, пропранолол, клонидин), бронхолитические (например, альбутерол), противовоспалительные стероиды (например, гидрокортизон, триамцинолон, преднизон), антибиотики (например, тетрациклин), антигемморроидальные, гипнотические, психотропные, противопоносные, муколитические, седативные, противоотечные, слабительные, антацидные, витамины, стимулирующие (включая средства, сдерживающие аппетит, такие, как фенилпропаноламин). Приведенное выше перечисление не является исключительным.
В некоторых предлагаемых осуществлениях терапевтически активный агент включает гидроморфин, оксикодон, дигидрокодеин, кодеин, гидроморфон, дигидроморфин, морфин, бупренорфин, соли любых из перечисленных соединений и их смеси и т.п. В других предложенных осуществлениях терапевтически активный агент включает теофиллин.
При использовании дисперсии акриловой смолы для покрытия фармацевтически инертных гранул, таких как гранулы nu pariel 18/20, множество образующихся таким образом устойчивых твердых гранул с регулируемым выделением может быть помещено в желатиновую капсулу в количестве, достаточном для обеспечения эффективного регулирования выделяемой дозы при проглатывании и контактировании с желудочным соком. В этом воплощении гранулы, покрытые терапевтически активным агентом получаются, например, растворением терапевтически активного агента в воде и последующим напылением раствора на субстрат, например, гранулы nu pariel 18/20 с использованием вкладыша Wurster. Необязательно, до осуществления покрытия гранул, могут быть добавлены также дополнительные ингредиенты для того, чтобы содействовать соединению гидроморфона с гранулами, и/или для окрашивания раствора и т.д. Например, продукт, который содержит гидроксипропилметилцеллюлозу и т.д. с окрашивающим веществом или без него может добавляться к раствору и смешиваться с раствором (например, в течение 1 часа) до нанесения его на гранулы. Образующийся покрытый субстрат, в данном случае гранулы, может быть необязательно покрыт сверху защитным агентом с тем, чтобы отделить терапевтически активный агент от акрилового покрытия. Примером подходящего защитного агента является гидроксипропилметилцеллюлоза. Однако может быть использовано любое известное в данной области техники пленкообразующее. Предпочтительным является, чтобы защитный агент не оказывал влияния на скорость растворения конечного продукта.
Гидроморфоновые, защищенные гидроксипропилметилцеллюлозой HPMC (необязательно) гранулы затем могут быть покрыты сверху акриловым полимером. Предпочтительно далее, чтобы дисперсия акрилового полимера включала эффективное количество пластификатора, например триэтилцитрата. Предварительно готовятся дисперсии акриловых смол, таких как различные коммерчески доступные формы

Покрывающие растворы данного изобретения предпочтительно содержат, помимо пленкообразующего, пластификатор и растворяющую систему (например, воду), краситель для придания продукту внешней привлекательности и отличительности. Краситель может быть добавлен к раствору терапевтически активного агента вместе или дополнительно к верхнему покрытию. Подходящими компонентами для придания цвета лекарственной форме являются двуокись титана и окрашенные пигменты, такие как пигменты на основе окиси железа. Однако введение пигментов может увеличивать замедляющее воздействие покрытия. Или же может быть использован любой подходящий способ окрашивания лекарственных форм, относящихся к данному изобретению.
Пластифицированное покрытие акрилового полимера может наноситься на субстрат, содержащий терапевтически активный агент, методом напыления с использованием любого подходящего оборудования для напыления, известного в данной области техники. В предложенном методе используется система псевдоожиженного кипящего слоя Wurster, в которой воздушная струя, впрыснутая снизу, флюидизирует материал сердцевины и осуществляет высушивание в то время, как производится напыление акрилового полимерного покрытия. Для того чтобы получить достаточное количество покрытия, предварительно контролировалось выделение терапевтически активного агента при помещении названного покрытого субстрата в водные растворы, предпочтительно применялся, например, желудочный сок, при этом принимались во внимание физические характеристики терапевтически активного агента, способ введения пластификатора и т.д.
После покрытия гранул акриловой смолой необязательно далее применялось верхнее покрытие пленкообразующего, такое как Opadry®. Это верхнее покрытие, если оно используется вообще, имеет целью существенно снизить агломерацию гранул.
Затем, покрытые гранулы отверждаются для того, чтобы получить стабильную скорость выделения терапевтически активного агента.
По традиции отверждение покрытых лекарственных форм проводится для Eudragit®, если оно вообще проводится, посредством псевдоожиженного воздействия при 45oC в течение 2 часов после нанесения. Такое стандартное нанесение рекомендуется Rohm Pharma, так как эта температура находится выше переходной температуры стеклования (Tg) Eudragit®RS30, пластифицированного триэтилцитратом при содержании 20% твердого вещества. Такое рекомендованное отверждение не стабилизует характеристику растворения лекарственной формы при старении, как это будет продемонстрировано приводимыми здесь далее примерами.
Этап отверждения согласно данному изобретению выполняется так, что покрытый субстрат, например гранулы, подвергается действию температуры, более высокой, чем Tg покрытой лекарственной формы, при непрерывном отверждении до достижения граничной точки, при которой покрытая лекарственная форма достигает характеристики растворения, существенно не меняющейся под воздействием хранения в условиях повышенной температуры и/или влажности. Обычно, время отверждения составляет от 24 часов или более, а температура отверждения может быть, например, около 45oC. Далее, в данном изобретении было установлено, что необходимо подвергнуть покрытый субстрат действию уровней влажности выше комнатных условий в течение этапа отверждения для того, чтобы получить устойчивый конечный продукт.
Один из возможных механизмов для изменения характеристики растворения продуктов предшествующего уровня, отверждаемых стандартными методами, состоит в том, что эти продукты непрерывно отверждаются при старении и могут никогда не достигнуть стабилизованной конечной точки, при которой продукт приобретает в основном постоянную характеристику растворения. В противоположность этому, отвержденные продукты данного изобретения дают скорость выделения терапевтически активного агента, которая существенно не меняется в процессе старения повышением температуры и влажности.
В предложенных воплощениях данного изобретения стабилизованный продукт получается воздействием на покрытый субстрат теплового отверждения при температуре, выше Tg пластифицированного акрилового полимера, в течение требуемого периода времени при оптимальных значениях температуры и времени для конкретной лекарственной формы, которые определяются экспериментально.
В конкретных воплощениях данного изобретения устойчивый продукт получается посредством теплового отверждения, проводимого при температуре около 45oC в течение периода времени приблизительно от 24 до 48 часов. Таким образом, в некоторых воплощениях предпочтительным может быть отверждение продукта, например, в течение 36 часов. В некоторых предлагаемых воплощениях продукт отверждается в течение приблизительно 48 часов. Здесь рассматривается также, что некоторые продукты, имеющие покрытия с регулируемым выделением в соответствии с данным изобретением, могут требовать большего, чем 48 часов времени отверждения, например 60 часов или более.
Когда покрытия с регулируемым выделением, соответствующие данному изобретению, применяются к таблеткам, сердцевина таблетки (то есть субстрат) может содержать активный агент вместе с любым фармацевтически допустимым инертным фармацевтическим наполнителем (разбавителем), включающим, но не ограниченным сахарозой, декстрозой, лактозой, микрокристаллической целлюлозой, ксилитом, фруктозой, сорбитом, их смесями и т.п. Перед прессованием ингредиентов сердцевины таблеток к вышеперечисленным ингредиентам наполнителей может добавляться также эффективное количество любого принятого фармацевтического смазывающего вещества, включающего кальциевые или магниевые мыла. Наиболее предпочтительным является стеарат магния в количестве приблизительно 0.5 - 3% по весу от веса твердой дозированной формы.
Таблетки, покрытые сверху достаточным количеством покрытия из акриловой смолы для обеспечения регулируемого выделения лекарственной формы согласно данному изобретению, могут быть получены и отверждены аналогично тому, как это описано для приготовления гранул. Специалисту понятно, что необходимые условия отверждения, касающиеся конкретной повышенной температуры, повышенной влажности и промежутка времени, необходимых для получения устойчивого продукта, будут зависеть от конкретной лекарственной формы.
Следующие примеры иллюстрируют различные аспекты представленного изобретения. Их не следует рассматривать как ограничивающие каким-либо образом притязания изобретения.
Пример 1. Получение гранул гидроморфона.
Гранулы гидроморфона были получены растворением гидроморфона HCl в воде с добавлением Opadry®Y-5-1442, светлого розового (продукт коммерчески доступный от West Point, Pennsylvania, который содержит гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, двуокись титана, полиэтиленгликоль и DC Red NO 30 Алюминиевую композицию), 20% по весу и смешением в течение часа и последующим напылением на nupariel 18/20 гранулы с использованием вкладыша Wurster. Образующаяся композиция имеет состав, приведенный в табл. 1.
Пример 2. Ингибирующее покрытие - нет этапа отверждения.
В примере 2 гидроморфоновые гранулы, полученные в соответствии с примером 1, были покрыты сверху Eudragit® 30D до 5% привеса, как показано в табл. 2. Конечное высушивание не проводилось.
Гидроморфоновые гранулы испытывались на исходное растворение и затем хранились в течение одного месяца в "суровых" условиях - при 37oC / 80% RH (RH = относительная влажность). Спустя один месяц было установлено, что гранулы подвергались агломерированию.
Испытания растворимости проводились в соответствии с методом USP Basket при 37oC, 100 RPM, первый час 700 мл желудочного сока при pH 1.2, затем изменялось до 900 мл при 7.5. Растворимость осуществлялась помещением открытой капсулы, содержащей соответствующий вес гранул, в сосуд. Результаты приводятся в табл. 3.
Приведенные в табл. 3 результаты указывают на то, что произошло существенное замедление в растворимости гидроморфона HCl из гранул с покрытием под действием старения в "ускоренных" условиях.
Пример 3. Защищающее ингибирующее покрытие.
Для того чтобы определить, связано ли замедление растворения гидроморфоновых гранул в примере 2 с проблемой стабильности между гидроморфоном и ингибитором, в примере 3 были получены Nu pariel гидроморфоновые гранулы в соответствии с примером 1, затем они были покрыты сверху 5% НРМС (ГПМЦ) и испытывались без ингибирующего слоя. Испытания растворимости проводились сразу же и после хранения в "ускоренных" условиях: 37oC в сухой атмосфере и 37oC /80% RH. Результаты по испытаниям растворимости для примера 3 приводятся в табл. 4.
Результаты, приведенные в примере 3, показывают, что покрытые гранулы, не включающие ингибирующего покрытия, были устойчивы.
Для того чтобы определить относительную влажность в "сухих условиях" в печи, определялась относительная влажность в наполненном водой эксикаторе в печи при 60oC следующим образом. Сначала, в пластмассовый эксикатор наливалось около 500 грамм очищенной воды и вставлялся металлический аппарат. В верхнюю часть аппарата помещался индикатор ареометр/температура, а эксикатор закрывался и помещался на 24 часа в печь с температурой 60oC. Через 24 часа относительная влажность в эксикаторе была 85%, в то время как температура была еще 60oC. При помещении одного ареометра на 24 часа в печь с температурой 60oC относительная влажность была 9% при 60oC.
Пример 4. Отверждение в образцах предшествующего уровня техники (в соответствии с литературными рекомендациями).
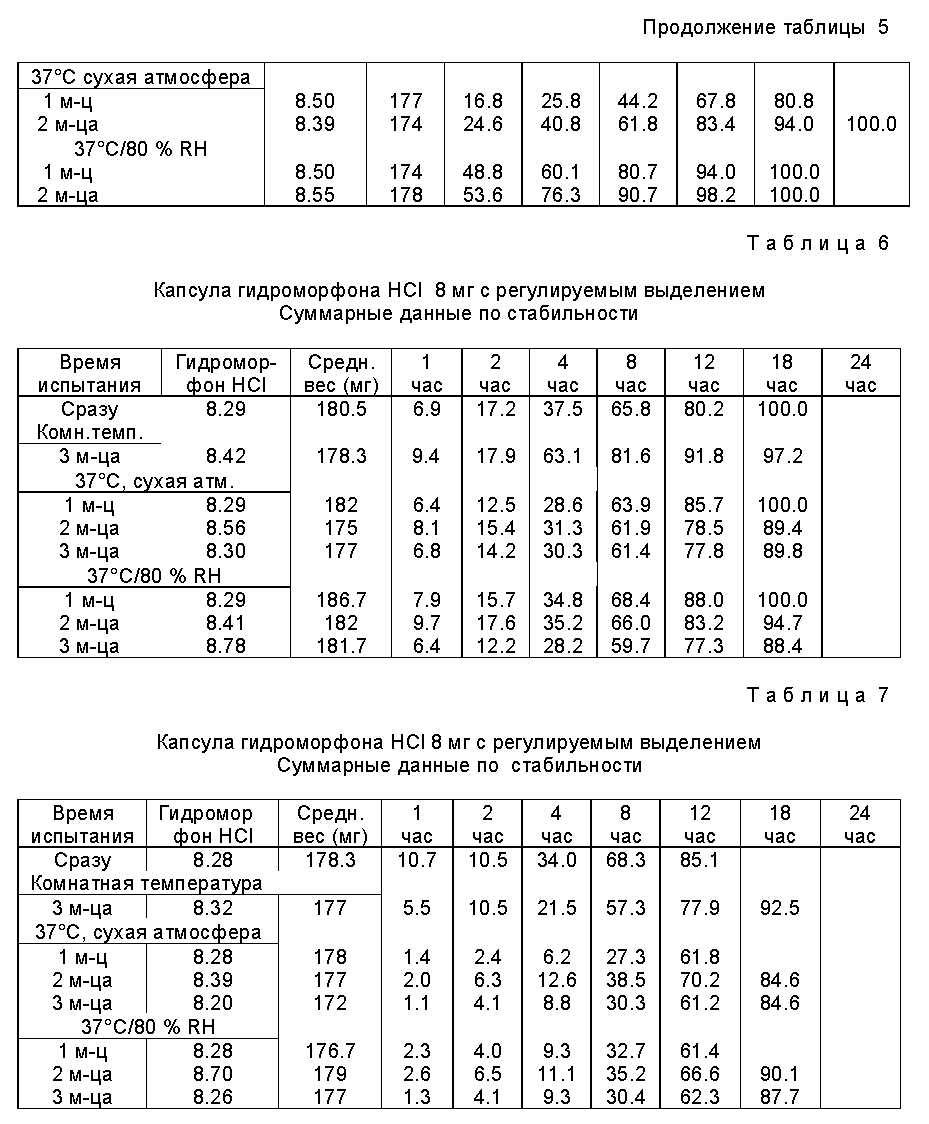
В примере 4, гидроморфоновые гранулы, полученные в соответствии с примером 3, были покрыты Eudragit®RS до 5% привеса. После нанесения покрытия гранулы были высушены (отверждены) при 45oC в псевдоожиженном сушителе в течение 2 часов. Температура поддерживается выше Tg Eudragit®RS 30D, пластифицированного триэтилцитратом в количестве 20% от содержания твердых продуктов. Испытания по растворению проводились сразу же и после хранения при 37oC в сухой атмосфере и в условиях 37oC/80% RH. Результаты представлены в табл. 5.
Из полученных в табл. 5 результатов можно видеть, что растворение гидроморфона из гранул подвергается значительному изменению под действием хранения и что короткий этап отверждения, рекомендованный в литературе и использованный в примере 4, не помогает в решении проблем стабильность/отверждение.
Примеры 5 - 6. Защитное ингибирующее покрытие.
В примере 5 Eudragit®-ом покрытые гранулы, полученные в соответствии с примером 4, покрывались сверху 5% НРМС (ГПМЦ) для защиты ингибирующего покрытия от окружающей среды. Испытания на растворение проводилось сразу же, после хранения при комнатной температуре (RT) при 37oC в течение 3-х месяцев и после хранения при 37oC в сухой атмосфере и при 37oC/80% RH. Результаты приводятся в табл. 6.
В примере 6 покрытые Eudragit®-ом гранулы, приготовленные в соответствии с примером 4, которые не были отверждены, были покрыты сверху 5% НРМС для защиты ингибирующего покрытия от окружающей среды. Испытания по растворению проводились сразу же и после хранения при 37oC в сухой атмосфере и при 37oC /80% RH. Результаты приводятся в табл. 7.
Как можно видеть из результатов, приведенных в табл. 6 и 7, помимо предотвращения агрегирования шариков (в частности, шариков, покрытых Eudragit®-ом) в "ускоренных" условиях, исходное покрытие сверху НРМС не стабилизирует продукты примеров 5 и 6. На основании этих результатов, однако, можно предположить, что несмотря на изменение растворения в "ускоренных" условиях конечная точка отверждения не может быть достигнута ни в сухой атмосфере, ни во влажных условиях при 37oC.
Примеры 7 - 9. Оптимизирующее отверждение и ингредиенты ингибирующего покрытия.
Результаты, полученные в примерах 2 - 7, показали, что растворение гранул, покрытых сверху ингибирующим покрытием, по существу, замедляется и не более. Однако достижение конечных точек растворения было слишком медленным.
Так как гидроморфин в лекарственных формах был защищен от окружающего воздействия, можно было предположить, что воздействие "ускоренных" условий (например, 37oC /80% RH), оказывало действие на дальнейшее "отверждение" ингибирующего слоя. Таким образом, были проведены дополнительные испытания для определения условий обработки, требуемых в процессе производства, для отверждения продукта до его граничной точки растворения.
Для того чтобы получить лекарственную форму, имеющую более подходящую кривую растворения, и, вместо того чтобы снизить покрытие до привеса менее чем 5%, был введен более растворимый Eudragit®RL (метакриловый эфир 1:20 четвертичных аммониевых групп) в ингибирующее покрытие.
В примерах 7 - 9 гидроморфиновые гранулы готовились согласно примеру 5. В примере 7 ингибирующее покрытие состояло из 100% Eudragit®RL. В примере 8 ингибирующее покрытие состояло из 50% Eudragit®RL и 50%Eudragit®RS. Наконец, в примере 9 ингибирующее покрытие состояло из 10% Eudragit®RL:90%Eudragit®RS. Каждый из примеров 7 - 9 были покрыты до общего привеса 5%.
Каждый из покрытий примеров 7 - 9, защищенные покрытием НРМС, отверждались до 1, 2, 6, 8, 12 и 21 дня при 45oC в сухой атмосфере, и при этом проводилось изучение растворения, осуществляемое как это описано в примере 2.
Только пример 9 показал нужную характеристику выделения и кривая была завершена всего за один день. Изучение растворения продуктов примеров 7 и 8 показало такое же немедленное освобождение продуктов, при этом количество/тип использованного ингибитора являются недостаточными для предотвращения немедленного выделения лекарственного средства (то есть около 100% выделяется спустя один час) даже после того, как лекарственные формы были отверждены. пример 9 далее был испытан при хранении в "ускоренных" условиях следующим образом. После отверждения в течение 21 дня образцы примера 9 помещались в печь при 37oC/80% RH, и испытания растворимости, проводимые как показано в примере 2, проводились спустя 7 и 30 дней. Типичные характеристики растворения для примера 9 (средние результаты для трех образцов) представлены ниже в табл. 8.
Результаты, приведенные в табл. 8, демонстрируют, что характеристика растворения через 1 месяц не дает замедления по сравнению с образцом сразу после отверждения, даже для образца, испытанного в "ускоренных" условиях. Таким образом, после отверждения в течение 24 часов при 45oC, метакрилатное покрытие пленкой с регулируемым выделением было в основном стабилизовано.
Примеры 10 - 12. Оптимизирующая толщина ингибирующего покрытия.
В примерах 10 - 12 был проведен дополнительный эксперимент по определению оптимального веса метакрилатного полимера, используемого для получения необходимой характеристики выделения и для определения воспроизводимости и эффективности этапа отверждения в 48 часов при 45oC в сухой атмосфере. Были изготовлены три партии с различным весом метакрилата и отверждены при 45oC в сухой печи.
В примере 10 гидроморфоновые гранулы готовились в соответствии с примером 3 с рецептурой, приведенной в табл. 9.
Затем гранулы гидроморфона были подвергнуты дальнейшей обработки в соответствии с примером 5. В примере 10 ингибирующим покрытием было покрытие из Eudragit®RS и Eudragit®RL в соотношении 90 : 10 (5% покрытия по весу). Рецептура для примера 10 приведена в табл. 10.
Примеры 11 и 12 готовились аналогично примеру 10. В примере 11 ингибирующее покрытие состояло из Eudragit®RS и Eudragit®RL в соотношении 90:10 (8% по весу от веса покрытия). Рецептуры для примеров 11 и 12 приведены соответственно в табл. 11 и 12.
Каждый из образцов примеров 10 - 12 отверждался на бумажном линейном поддоне в печи при 45oC в течение двух дней после нанесения покрытия с регулируемым выделением из Eudragit®-a с покрытием сверху 5% ГПМЦ.
Изучение растворения проводилось в соответствии с процедурой, описанной в примерах 10 - 12.
Исходная характеристика растворения (после отверждения) примера 10 показала, что она сходна с примером 9 (оба продукта были покрыты сверху покрытием в 5% по весу Eudragit®.). После отверждения в течение 2 дней образцы примера 10 были подвергнуты дальнейшим испытаниям при комнатной температуре и в "ускоренных" условиях - 37oC /80% RH, 37oC в сухой атмосфере и 50oC в сухой атмосфере. Полученные характеристики растворения для примера 10 (усредненные результаты для трех образцов) представлены в табл. 13.
Как можно видеть из результатов растворения, относящихся к примеру 10, хотя и не была взята характеристика растворения спустя 1 день после отверждения, соответствующий результат, спустя 2 дня после отверждения, в основном близок к результатам, полученным для образцов примера 9 через 1 и 2 суток после отверждения. Таким образом, можно предположить, что продукт примера 10 был также устойчив спустя 1 сутки после отверждения.
После отверждения в течение 2 дней образцы примера 11 испытывались на растворение, а затем образцы примера 11 подвергались воздействию "ускоренных" условий - 37oC/80% RH в течение одного месяца.
В табл. 14 представлены характеристики исходного растворения (средние результаты для трех образцов).
Как можно видеть из результатов растворения, представленных для примера 11, данные, полученные через два дня отверждения, в основном аналогичны соответствующим данным, полученным при хранении в "ускоренных" условиях - при 37oC/80% RH, и, следовательно, образцы примера 11 выявляют стабильность после двух дней отверждения. Далее, полученные в примере 11 результаты растворения показывают более медленную скорость выделения гидроморфона, как это можно ожидать из данных для более толстого ингибирующего покрытия.
Через 2 дня отверждения образцы примера 12 подвергались испытаниям на растворение, а затем образцы примера 12 подвергались дальнейшим испытаниям после хранения в течение одного месяца при комнатной температуре и при "ускоренных" условиях - 37oC/80% RH, 37oC в сухой атмосфере и 50oC в сухой атмосфере. Характерные данные по растворению (средние данные для трех образцов) для примера 12 приведены в табл. 15.
Как можно видеть из представленных выше результатов растворения для образцов примера 12, полученные для образцов примера 12 результаты выявляют более медленную скорость выделения гидроморфона по сравнению с более тонкими ингибирующими покрытиями примеров 10 и 11, как это ожидалось.
Общие результаты, полученные через 2 дня после отверждения в основном аналогичны результатам, полученным при хранении в "ускоренных" условиях при 37oC /80% RH за исключением процента лекарственного препарата, относящегося к растворенному в точках при 8 и при 12 часах. Эти результаты можно объяснить тем, что при большей величине привеса ингибирующего покрытия может быть необходим более длительный период отверждения покрытия для достижения устойчивости лекарственной формы.
Пример 13
Гранулы, покрытые сульфатом морфина
В примере 13 этап отверждения данного изобретения применялся
к лекарственной форме, в которой в качестве лекарственного средства был использован сульфат морфина.
Суспензия сульфата морфина и ГПМН (Opadry®clear У-5-7095) была нанесена на гранулы 18/20 меш nupariel в псевдоожиженном слое сушителя и аппарате Wurster при исходной температуре 60oC. Затем в качестве защищающего покрытия была нанесена суспензия основного пленкообразователя Opadry lavender ys - 1-4729 ГПМЦ в количестве 5% привеса.
После окончания нанесения верхнего покрытия морфинсульфатные гранулы были затем покрыты сверху ингибирующим покрытием из смеси Eudragit®RS30D и Eudragit®RL30D при соотношении RS к RL 90:10 и уровне привеса 5%. Нанесение этой смеси Eudragit® RS30D и Eudragit®RL30D наряду с тальком (введенным в качестве присадки против липкости) и триэтилцитратом (пластификатор) осуществлялось при начальной температуре 35oC с использованием аппарата Wurster-а.
Как только было завершено нанесение верхнего ингибирующего покрытия, морфинсульфатные гранулы подвергались покрытию конечным покрытием из Opadry® lavender ys-1-4729 при привесе 5%.
После окончания обработки последним пленкообразующим морфиносульфатные гранулы отверждались в течение 2 дней на бумажном линейном поддоне в сухой печи при 45oC. После отверждения гранулы помещались в желатиновые капсулы с эффективностью 30 мг морфин сульфата. Окончательная рецептура дана в табл. 16.
Затем было проведено изучение устойчивости растворения на продукте примера 13 после названного выше этапа отверждения при хранении в условиях комнатной температуры, 37oC/80% RH, 37oC в сухой атмосфере и 50oC с сухой атмосфере после одного и двух месяцев. Результаты приведены в табл. 17.
Результаты, приведенные в табл. 17, показывают, что обработка отверждением стабилизует характеристику растворения морфинсульфата до граничной точки скорости растворения, которая остается в основном постоянной даже для образцов, хранившихся в "ускоренных" условиях.
Образцы, приведенные выше, не являются исключительными. Много других видов осуществления данного изобретения являются очевидными для специалиста и рассматриваются как подпадающие под действие приложенных притязаний.
Реферат
Способ получения твердой дозированной лекарственной формы для орального применения включает приготовление твердого субстрата, содержащего активный ингредиент, приготовление водной дисперсии акрилового сополимера, покрытие субстрата водной дисперсией с получением полимерного покрытия. Покрытие обеспечивает регулируемое выделение активного ингредиента. В качестве акрилового сополимера используют пластифицированную смесь из аммонийметакрилатных сополимеров, которые являются сополимерами акриловых и метакриловых эфиров с низким содержанием четвертичных аммониевых групп. Нанесенное на субстрат покрытие отверждают при температуре выше температуры стеклования пластифицированной смеси акриловых сополимеров в течение по меньшей мере 24 ч. Полученные твердые дозированные формы имеют устойчивые характеристики растворения, практически не меняющиеся в процессе хранения при повышенной температуре и/или повышенной относительной влажности. 6 с. и 28 з.п.ф-лы, 17 табл.
Комментарии