Новый способ получения метансульфоната 4-аминометил-3-алкоксииминопирролидина (варианты), промежуточные продукты и способ получения хинолоновых антибиотиков - RU2303029C2
Код документа: RU2303029C2
Описание
ОБЛАСТЬ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
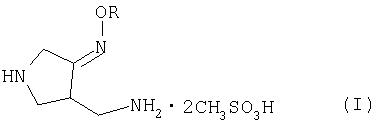
Настоящее изобретение относится к способу получения метансульфоната 4-аминометил-3-алкоксииминопирролидина следующей формулы (I):
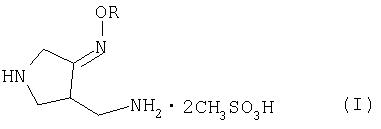
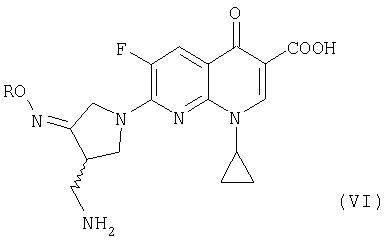
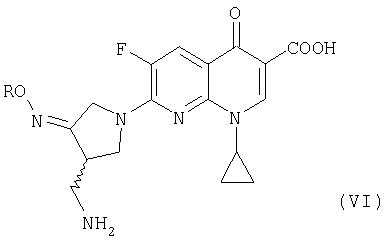
где R представляет собой C1-4 алкил или C1-4 галогеналкил, который применим в качестве промежуточного соединения для получения хинолоновых антибиотиков, особенно (R, S)-7-(3-аминометил-4-син-алкоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты следующей формулы (VI):
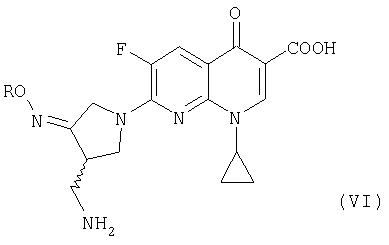
где R является таким, как определено выше, ее соли или гидрата, описанной в патенте США 5 633 262 и ЕР 0 688 772 A1.
Способ по настоящему изобретению реализует новый путь синтеза, имеющий уменьшенное число стадий для получения соединения (I) по сравнению с более ранними способами, посредством чего соединение (I) получают с высоким выходом. Соответственно, настоящее изобретение сокращает стоимость производства и, в конечном счете, вносит вклад в экономичное получение соединения (VI).
УРОВЕНЬ ТЕХНИКИ
WO 99/44991 и WO 01/17961 описывают способ получения соединения (I), изображенный на схеме реакции 1:
Схема реакции 1

где R представляет собой С1-4 алкил или С1-4 галогеналкил, а P и P2 представляют собой одинаковые или отличающиеся защитные группы.
WO 99/44991 конкретно описывает способ получения соединения (3) из соединения (II) в вышеуказанной схеме реакции 1, который состоит из двух стадий восстановления нитрильной группы и одной стадии защиты аминогруппы.
Соединение (1) получают из соединения (II) гидрированием с использованием такого катализатора, как Ra-Ni и т.д., для первого восстановления нитрильной группы. В качестве растворителя используют смесь воды и изопропилового спирта в количестве от 2 до 20 экв. относительно соединения (II).
Соединение (2) получают из соединения (1), защищая аминогруппу. В качестве защитной группы можно использовать формил, ацетил, трифторацетил, бензоил, п-толуолсульфонил, метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил, бензилоксикарбонил, п-метоксибензил, трифенилметил, тетрагидропиранил, пивалоил и т.д. Из данных защитных групп особенно предпочтительным является трет-бутоксикарбонил. Однако (BOC)2O, используемый для введения трет-бутоксикарбонильной группы, является дорогим реагентом, стоимость которого составляет примерно 1/3 от общей стоимости получения соединения (2) из соединения (1). Кроме того, температуру реакции трудно контролировать из-за сильного экзотермического эффекта и высокой скорости реакции. Неспособность контролировать скорость реакции ведет к образованию димера соединения (2). Более того, соединение (2) необходимо отделять экстракционной обработкой и процессом получения твердой фазы. Данные процессы обработки делают указанный способ сложным.
Соединение (3) получают из соединения (2) вторым процессом гидрирования с использованием Pd/C катализатора. Катализатор используют в количестве 0,5-20% (масс.), и для предотвращения восстановления карбонильной группы в 3-положении кольца пирролидина используют амин или буферный раствор.
Способ получения соединения (I) из соединения (4) описан в WO 01/17961, где защитную группу удаляют, используя метансульфоновую кислоту, с получением соли. Однако данный способ требует два процесса перекристаллизации для получения продукта высокого качества. Данные операции снижают производительность и приводят к низкому выходу.
В отличие от приведенной выше схемы реакции 1, EP 0 688 772 A1 описывает способ получения соединения (4) из соединения (II), как изображено на схеме реакции 2.
Схема реакции 2
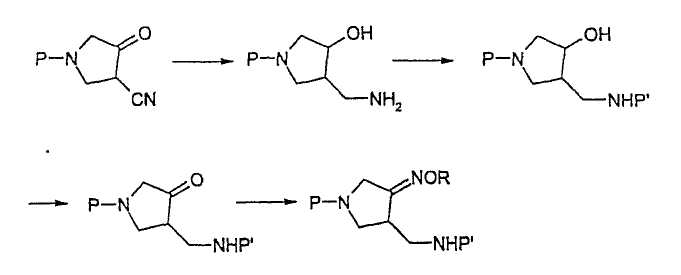
где R представляет собой С1-4 алкил или С1-4 галогеналкил, а P и P2 представляют собой одинаковые или отличающиеся защитные группы.
EP 0 688 772 A1 предлагает способ получения соединения (4) из соединения (II), в котором карбонильные и нитрильные группы восстанавливают одновременно, получая аминоспиртовое промежуточное соединение, и спиртовую группу селективно окисляют, вновь получая карбонильную группу. Данный способ требует реагентов, которые трудно использовать при промышленном производстве, и поэтому он имеет мало достоинств по сравнению со способом WO 99/44991. В частности, данный способ использует гомогенный катализатор для гидрирования нитрильной группы, но получение гомогенного катализатора, его отделение и регенерация после реакции не являются простыми.
ОПИСАНИЕ
Как указано выше, способы получения соединения (I) предшествующего уровня техники имеют такие недостатки, как сложность процесса, высокая стоимость производства, плохая воспроизводимость и т.д., поэтому существует необходимость в разработке нового улучшенного способа.
Обширные исследования привели к настоящему изобретению, в котором двухстадийное гидрирование WO 99/44991 заменяют одностадийным гидрированием, и удается избежать использования дорогих органических реагентов, в частности (BOC)2O, и различных органических растворителей и реагентов.
Поэтому настоящее изобретение предлагает новый и эффективный способ получения соединения (I).
Настоящее изобретение также предлагает способ получения соединения (VI) с использованием полученного вышеуказанным способом соединения (I).
Настоящее изобретение также предлагает новые промежуточные продукты, используемые в способе получения соединения (I).
НАИЛУЧШИЕ ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
В первом аспекте настоящее изобретение предлагает способ получения соединения (I):
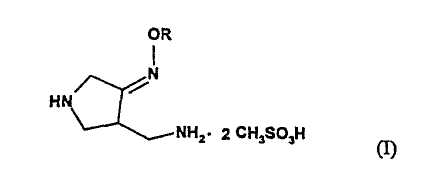
где R представляет собой С1-4 алкил или С1-4 галогеналкил, который включает стадии
(a) взаимодействия соединения (II)

где P представляет собой защитную группу, с алкоксиамином или галогеналкоксиамином или их солью в присутствии основания с получением соединения (III):

где R и P являются такими, как определено выше,
(b) взаимодействия соединения (III) с метансульфоновой кислотой с получением соединения (IV):
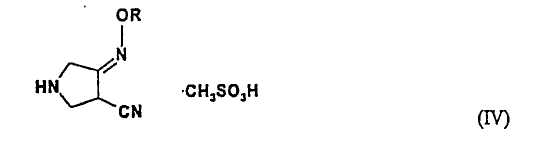
где R является таким, как определено выше, и
(с) добавления метансульфоновой кислоты и катализатора гидрирования к соединению (IV) и осуществления реакции гидрирования указанного соединения с получением соединения (I).
В вышеописанном способе защитная группа P может включать формил, ацетил, трифторацетил, бензоил, п-толуолсульфонил, метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил, бензилоксикарбонил, п-метоксибензил, трифенилметил, тетрагидропиранил, пивалоил и т.д., и наиболее предпочтительной группой является трет-бутоксикарбонил (BOC). Кроме того, R предпочтительно представляет собой метил.
Вышеописанный способ получения соединения (I) можно изобразить следующим образом:
Схема реакции 3
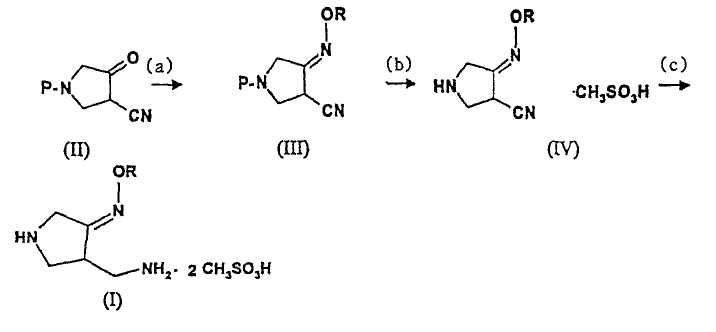
где R представляет собой С1-4 алкил или С1-4 галогеналкил, а P представляет собой защитную группу.
Стадия (a)
Способ превращения соединения (II) в соединение (III) осуществляют в присутствии основания. Предпочтительно используемое основание включает триэтиламин, три-н-бутиламин, диизопропилэтиламин, пиридин, 4-диметиламинопиридин, 4-(4-метилпиперидин-1-ил)пиридин и ацетат натрия. Предпочтительно, основание используют в количестве, составляющем 0,01-10 экв. относительно соединения (II).
Реагент, алкоксиамин или галогеналкоксиамин, предпочтительно, используют в форме аддитивной соли кислоты, в частности в гидрохлоридной форме. Алкоксиамин, галогеналкоксиамин или их соль используют в количестве от 1 до 2 экв. относительно соединения (II).
Растворители, которые можно использовать в данной стадии, включают воду, органический растворитель или их смесь, предпочтительно, С1-6 спирт с неразветвленной или разветвленной цепью, более предпочтительно, MeOH, EtOH или изопропиловый спирт.
Температура реакции и время на стадии (a) могут изменяться в зависимости от используемого основания и растворителя. Температура реакции находится в диапазоне, например, от комнатной температуры до 200°C. Но специалист в данной области может легко определить соответствующую температуру реакции и время в соответствии с используемым основанием и растворителем.
Один наилучший вариант настоящего изобретения состоит в использовании в качестве основания триэтиламина. В данном случае соединение (II) кипятят с обратным холодильником с гидрохлоридом алкоксиамина в присутствии триэтиламина и метанола в течение 22 часов.
Другой наилучший вариант настоящего изобретения состоит в использовании в качестве основания ацетата натрия. В данном случае соединение (II) добавляют к раствору гидрохлорида алкоксиамина и ацетата натрия в этаноле или метаноле и смесь кипятят с обратным холодильником в течение примерно 18 часов.
Другой наилучший вариант настоящего изобретения состоит в использовании в качестве основания пиридина. В данном случае соединение (II) добавляют к раствору гидрохлорида алкоксиамина и пиридина в изопропиловом спирте или метаноле и смесь взаимодействует при перемешивании в течение примерно 5 часов. Данный способ является удобным, поскольку его осуществляют при комнатной температуре.
Чистое соединение (III) можно получить, используя способы проиллюстрированных выше наилучших вариантов, без побочного продукта, что определяется ВЭЖХ.
Стадия (b)
Превращение соединения (III) в соединение (IV) необходимо для удаления защитной группы метансульфоновой кислотой. Метансульфоновую кислоту используют в количестве примерно от 0,5 до 3 экв., предпочтительно, примерно от 1 до 1,2 экв. относительно соединения (III). Защитную группу легко удаляют такой кислотой, как метансульфоновая кислота. Как показано в примерах ниже, методами ЯМР, ВЭЖХ можно показать, что защитная группа легко удаляется кипячением с обратным холодильником в течение 30 минут.
Стадия (c)
Превращение соединения (IV) в желаемое соединение, соединение (I), представляет собой реакцию селективного гидрирования с использованием таких металлических катализаторов Ренея, как никелевый катализатор Ренея, кобальтовый катализатор Ренея и т.д. или металлических катализаторов, нанесенных на носители, такие как активированный уголь, оксид алюминия, диоксид кремния и т.д. Металлы, используемые в качестве активных центров катализатора, включают металлы подобные Ni, Co, Pt, Pd, Ru, Rh, Ir, Cu и т.д. и предшественники палладия, подобные хлориду палладия, нитрату палладия, ацетату палладия и т.д., но палладиевый катализатор является более предпочтительным. Обычно активность катализатора можно изменить с помощью влияния других металлов, добавленных в незначительном количестве в форме со-катализатора, или реакционными условиями, такими как давление, температура и т.д., и таким образом можно контролировать селективность по желаемому продукту. Катализатор гидрирования, особенно предпочтительный для использования по настоящему изобретению, представляет собой Pd катализатор, не только содержащий от 1 до 20% массовых Pd, но также нанесенный на носитель, выбранный из группы, состоящей из угля, диоксида кремния и оксида алюминия. Данный катализатор гидрирования, предпочтительно, используют в количестве от 0,01 до 10% массовых относительно соединения (IV), считая на металлический компонент.
Реакцию гидрирования, предпочтительно, осуществляют в температурном диапазоне от 0 до 50°С и при давлении водорода от 1 до 100 атм.
Метансульфоновую кислоту добавляют к реакционному раствору в количестве от 0,5 до 3 экв., предпочтительно, примерно от 1 до 1,2 экв. относительно соединения (III).
В качестве растворителя, используемого в данной стадии, можно указать один или несколько органических растворителей, выбранных из группы, состоящей из метанола, этанола, н-пропанола, изопропанола, тетрагидрофурана, диметоксиэтана, диоксана, этилацетата и дихлорметана, предпочтительно, метанол.
Реакцию стадии (c) также можно осуществить в присутствии кислоты в качестве вспомогательной добавки. Данная кислота включает хлористоводородную кислоту, азотную кислоту, серную кислоту, уксусную кислоту, метансульфоновую кислоту и т.д., и метансульфоновая кислота является наиболее предпочтительной. Для увеличения выхода предпочтительно добавлять кислоту в таком количестве, чтобы регулировать рН раствора от 1 до 2,5 в течение реакции гидрирования, и кислоту можно добавлять в реакционный раствор во время инициирования или непрерывно в середине реакции.
Во втором аспекте настоящее изобретение предлагает способ получения соединения (I):
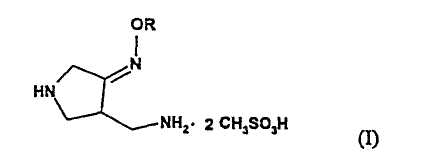
где R представляет собой С1-4 алкил или С1-4 галогеналкил, который включает стадии
(a) взаимодействия соединения (II)

где P представляет собой защитную группу, с алкоксиамином или галогеналкоксиамином или их солью в присутствии основания с получением соединения (III):

где R и P являются такими, как определено выше, и
(b) добавления метансульфоновой кислоты и катализатора гидрирования к соединению (III) и осуществления реакции гидрирования указанного соединения с получением соединения (I).
В вышеуказанном способе R, предпочтительно, представляет собой метил, а P, предпочтительно, представляет собой трет-бутоксикарбонил (BOC).
Вышеуказанный способ получения соединения (I) можно изобразить следующим образом:
Схема реакции 4

где R представляет собой С1-4 алкил или С1-4 галогеналкил, а P представляет собой защитную группу.
Данный второй способ имеет все достоинства, указанные для первого способа, и является более эффективным, поскольку количество стадий снижено с 3 до 2.
Стадия (a)
Стадию (a) второго способа осуществляют в соответствии с тем же методом, как в стадии (a) первого способа.
Стадия (b)
В стадии (b) второго способа одновременно выполняют стадии (b) и (c) первого способа.
Количество метансульфоновой кислоты равно общему количеству метансульфоновой кислоты, используемой в стадиях (b) и (c) первого способа, т.е. примерно от 1 до 6 экв., предпочтительно, примерно от 1,5 до 2,5 экв. относительно соединения (III). Кроме того, катализатор гидрирования вводят в количестве от 0,01 до 10% масс. относительно соединения (III), основываясь на металлическом компоненте.
Растворитель, используемый в стадии (b) второго способа, представляет собой органический растворитель, выбранный из группы, состоящей из метанола, этанола, н-пропанола, изопропанола, тетрагидрофурана, диметоксиэтана, диоксана, этилацетата и дихлорметана, или смесь данного органического растворителя и воды. Более предпочтительной является смесь органического растворителя и воды. При использовании смеси состав смеси представляет собой от 0,2 до 50 объемов органического растворителя к 1 объему воды. Особенно предпочтительным растворителем является смесь метанола и воды.
Кроме того, в данной стадии можно использовать такие же реакционные условия, как в стадии (c) первого способа.
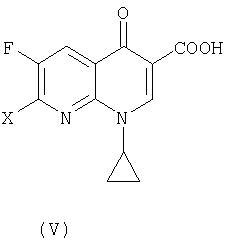
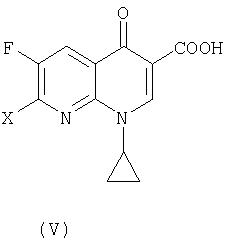
В третьем аспекте настоящее изобретение предлагает способ получения соединения (VI):

где R представляет собой С1-4 алкил или С1-4 галогеналкил, его соли или гидрата, который включает стадию взаимодействия соединения (I):

где R является таким, как определено выше, которое получают в соответствии с первым или вторым способом, как объяснено выше, с соединением (V):

где X представляет собой уходящую группу, предпочтительно галоген.
В данном способе взаимодействие соединения (I) с соединением (V) предпочтительно осуществляют в присутствии основания и в растворителе. Специалист в данной области может легко контролировать конкретные условия реакции, используя для сведения РСТ/GB00/03358.
Предпочтительно, соединение (VI) представляет собой метансульфонат (R,S)-7-(3-аминометил-4-син-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты или его гидрат.
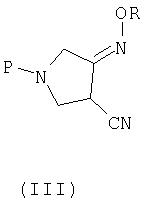
В четвертом аспекте настоящее изобретение предлагает новое соединение (III) следующей формулы:

где R представляет собой С1-4 алкил или С1-4 галогеналкил, а P представляет собой защитную группу.
P, предпочтительно, представляет собой трет-бутоксикарбонил (BOC).
В пятом аспекте настоящее изобретение также предлагает новое соединения (IV) следующей формулы:
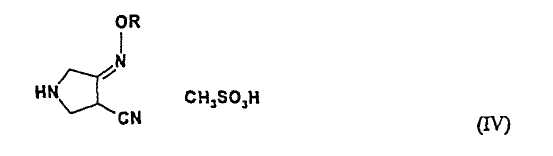
где R представляет собой С1-4 алкил или С1-4 галогеналкил.
Указанные выше соединения (III) и (IV) применимы в качестве промежуточных соединений для получения соединения (I).
Настоящее изобретение будет более конкретно объяснено следующими далее примерами. Однако необходимо понимать, что они не предназначены каким-либо образом ограничивать изобретение.
Пример 1
(1) Синтез соединения (III)

К перемешиваемому раствору соединения (II) (10,5 г, 0,05 моль) в 100 мл метанола в присутствии пиридина (4,84 мл, 1,2 экв.) при комнатной температуре добавляют метоксиламин гидрохлорид (5,0 г, 1,2 экв.). Через 5 часов завершение реакции подтверждают ВЭЖХ при следующих условиях:
Колонка: Capcellpak C18
Растворитель: АН/Н2О/ТФУ=60/40/0,1
Длина волны: 210 нм
Расход: 1 мл/мин.
Температура: комнатная
Летучие компоненты тщательно удаляют под вакуумом и к остатку добавляют этилацетат (50 мл). Органический слой дважды промывают насыщенным водным раствором NaHCO3 (100 мл) и дважды насыщенным раствором соли (100 мл). К органическому слою добавляют безводный сульфат магния для удаления влаги и смесь концентрируют в вакууме, получая соединение (III) (11,09 г, выход 92,8%).
1Н-ЯМР (400 МГц, CDCl3) δ (м.д.): 1,47 (с, 9H), 3,69 (дд, 1H), 3,95(с, 3H), 3,98-4,06 (м, 2H), 4,10˜4,22 (м, 2H).
(2) Синтез соединения (IV)
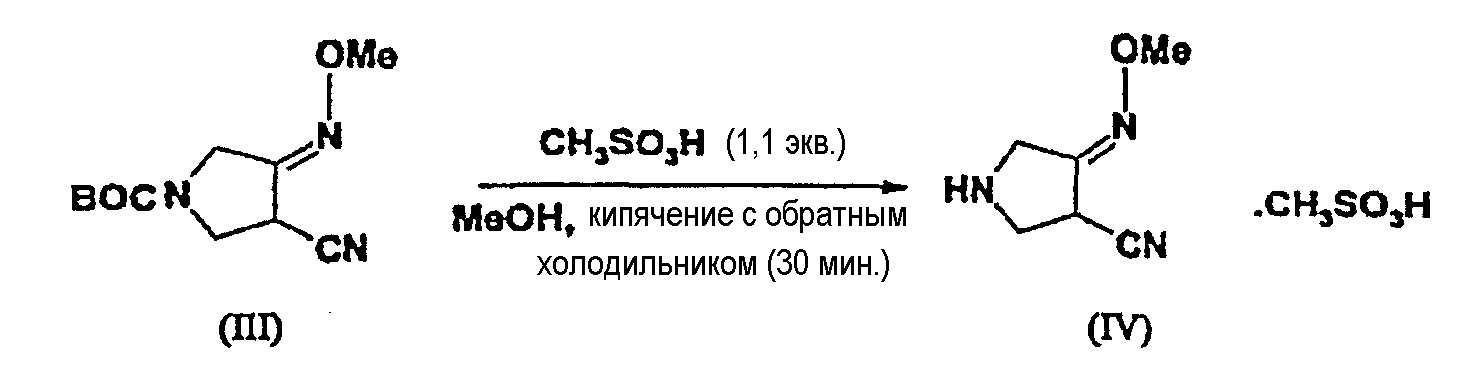
К перемешиваемому раствору соединения (III) (3,0 г, 0,0125 моль) в 25 мл метанола по каплям добавляют метансульфоновую кислоту (0,91 мл, 1,1 экв., 98%) и смесь кипятят с обратным холодильником в течение 30 минут. Реакционную смесь охлаждают до комнатной температуры. Смесь концентрируют при пониженном давлении и перекристаллизовывают, получая соединение (IV) (4,14 г, выход 98,8%).
1Н ЯМР (400 МГц, D2O) δ (м.д.): 2,69 (с, 3H), 3,76-3,82 (дд, 1H), 3,88 (с, 3H), 3,92-3,98 (м, 2H), 4,03-4,21 (дд, 2H).
(3) Синтез соединения (I)

К перемешиваемому раствору соединения (IV) (4,19 г, 0,0125 моль) в 80 мл метанола добавляют Pd/C (0,3 г, влажная основа) в качестве катализатора и метансульфоновую кислоту (1,0 мл, 1,1 экв., 98%) и реакцию гидрирования проводят в течение 24 часов при температуре реакции 25°C и давлении водорода 500 фунт/кв. дюйм (изб). После завершения реакции смесь пропускают через целит для удаления катализатора и фильтрат концентрируют в вакууме. К остатку добавляют метанол (50 мл) и в качестве затравки добавляют соединение (I) (1 мг). Смесь перемешивают при комнатной температуре в течение 1 часа и фильтруют. Полученное в результате твердое вещество растворяют на водяной бане примерно при 50°C, кристаллизуют при -20°C и фильтруют, получая соединение (I) (0,99 г, выход 23,1%).
1Н ЯМР (400 МГц, ДМСО) δ (м.д.): 2,39 (с, 6H), 3,07 (дд, 1H), 3,16 (дд, 1H), 3,24-3,30 (м, 2H), 3,66-3,73 (м, 1H), 3,87 (с, 3H), 3,97 (дд, 2H).
Пример 2
(1) Синтез соединения (III)

К перемешиваемой суспензии соединения (II) (10,5 г, 0,05 моль) в 100 мл метанола добавляют гидрохлорид метоксиламина (5,0 г, 1,2 экв.) и триэтиламин (8,4 г, 1,2 экв.) и смесь кипятят с обратным холодильником в течение 22 часов. Реакционную смесь охлаждают до комнатной температуры. Смесь концентрируют в вакууме и к остатку добавляют этилацетат (50 мл). Органический слой дважды промывают насыщенным водным раствором NaHCO3 (100 мл) и дважды насыщенным раствором соли (100 мл). Добавляют безводный сульфат магния, фильтруют и фильтрат концентрируют в вакууме, получая соединение (III) (11,35 г, выход 95,0%).
(2) Синтез соединения (IV)
Соединение (IV) получают по такой же методике, как в примере 1 (2).
(3) Синтез соединения (I)
Соединение (I) получают по такой же методике, как в примере 1 (3).
Пример 3
(1) Синтез соединения (III)

К перемешиваемой суспензии соединения (II) (10,5 г, 0,05 моль) в 100 мл метанола добавляют гидрохлорид метоксиламина (5,0 г, 1,2 экв.) и ацетат натрия (4,92 г, 1,2 экв.) и смесь кипятят с обратным холодильником в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры. Смесь концентрируют в вакууме и к остатку добавляют этилацетат (50 мл). Органический слой дважды промывают насыщенным водным раствором NaHCO3 (100 мл) и дважды насыщенным раствором соли (100 мл). Добавляют безводный сульфат магния, фильтруют и фильтрат концентрируют в вакууме, получая соединение (III) (11,35 г, выход 95,0%).
(2) Синтез соединения (IV)
Соединение (IV) получают по такой же методике, как в примере 1 (2).
(3) Синтез соединения (I)
Соединение (I) получают по такой же методике, как в примере 1 (3).
Пример 4
(1) Синтез соединения (III)
Соединение (III) получают по такой же методике, как в примере 1 (1).
(2) Синтез соединения (IV)
Соединение (IV) получают по такой же методике, как в примере 1 (2).
(3) Синтез соединения (I)
Соединение (I) (0,72 г, выход 16,8%) получают по такой же методике, как в примере 1 (3), за исключением того, что давление в реакции гидрирования понижают от 500 фунт/кв. дюйм (изб.) до 200 фунт/кв. дюйм (изб.).
Пример 5
(1) Синтез соединения (III)
Соединение (III) получают по такой же методике, как в примере 1 (1).
(2) Синтез соединения (I)
В 100 мл реактор для работы под давлением добавляют соединение (III) (5 г), метанол (40 мл) и воду (10 мл). К данному раствору добавляют 10% Pd/C (0,18 г) и метансульфоновую кислоту (2,2 мл). Смесь встряхивают при 30°C и давлении водорода 100 фунт/кв. дюйм (изб.) в течение 1 часа. Катализатор отфильтровывают и фильтрат полностью концентрируют при пониженном давлении. Остаток растворяют в метаноле (10 мл) и в качестве затравки для образования кристаллов при 5°C добавляют соединение (I) (1 мг). Полученные в результате кристаллы охлаждают до -10°C и отфильтровывают, получая метансульфонат 3-аминометил-4-Z-метилоксииминопирролидина (3,15 г, выход 45%).
Пример 6
Синтез (R, S)-7-(3-аминометил-4-син-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты
К 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1, 8-нафтиридин-3-карбоновой кислоте (3,05 г) в воде (25 мл) при 15-20°C добавляют триэтиламин (5,1 мл) и смесь перемешивают в течение 20 минут. Добавляют соединение (I) (3,86 г), полученное в примере 1, и воду (5 мл) и данную смесь перемешивают при 20-25°C в течение 18 часов. Полученный таким образом продукт отфильтровывают и осадок на фильтре промывают водой (30 мл) и этанолом (30 мл). Сушка при 50°C в вакууме дает указанное в заголовке соединение (4,23 г) в виде твердого белого вещества. Характеристики при идентификации оказываются такими же, как в случае образца установленного строения.
Пример 7
Синтез метансульфоната (R,S)-7-(3-аминометил-4-син-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1, 4-дигидро-1,8-нафтиридин-3-карбоновой кислоты
К суспензии (R,S)-7-(3-аминометил-4-син-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты (1,5 г, содержание 89,9%, 3,36 ммоль) в смеси дихлорметана (23,2 мл) и этанола (2,7 мл) при 30°C добавляют раствор метансульфоновой кислоты (0,33 г, 3,43 ммоль) в дихлорметане (1 мл). Данную смесь перемешивают при 30°C в течение 3 часов, затем охлаждают до 20°C и фильтруют. Осадок на фильтре промывают дихлорметаном (20 мл) и сушат при 50°C в вакууме, получая указанное в заголовке соединение (1,71 г). Характеристики при идентификации оказываются такими же, как в случае образца установленного строения.
Пример 8
Синтез полуторагидрата метансульфоната (R,S)-7-(3-аминометил-4-син-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты
Метансульфонат (R,S)-7-(3-аминометил-4-син-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты (27,5 г, содержание 91%, 51,4 ммоль) перемешивают в смеси изопропанола (150 мл) и воды (75 мл) и затем нагревают, получая прозрачный раствор (52°C). Данный раствор охлаждают до 34°C и в качестве кристаллов затравки к нему добавляют полуторагидрат метансульфоната (R,S)-7-(3-аминометил-4-син-метоксииминопирролидин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидро-1,8-нафтиридин-3-карбоновой кислоты. Полученной таким образом суспензии дают постоять в течение 1 часа при 25°C и затем перемешивают в течение 18 часов. Суспензию охлаждают до 0-4°C, перемешивают в течение 2 часов и фильтруют. Осадок на фильтре промывают изопропанолом (30 мл). Продукт сушат в токе воздуха на вакуум-фильтре в течение 2 часов и далее сушат в вакууме при 50°C. Высушенный продукт увлажняют влажным азотом, получая указанный в заголовке полуторагидрат (22,9 г, 92%). Характеристики при идентификации оказываются такими же, как в случае образца установленного строения.
Эксперимент 1
Для того чтобы определить возможность использования соединения (I), полученного по настоящему изобретению, в качестве вещества для антибиотиков, полученное в примере 1 соединение (I) анализируют ВЭЖХ при следующих условиях:
Колонка: Shodex ODP-50 6E (4,6Ч250 мм, 5 мкм, Asahipak)
Растворитель: АН/Н2О (включающая 5 мМ 1-гексансульфоновой кислоты)/ТФУ=5/95/0,1
Длина волны: 207 нм
Расход: 1 мл/мин.
Температура: 40°C
Содержание примесей и изомеров определяют, основываясь на PAR (отношение площадей пиков), где PAR определяют следующим образом:
PAR (%)=A/B·100
где A обозначает площадь пика каждой примеси, а B обозначает сумму площадей пиков всех примесей за исключением пиков, идентифицированных в чистом растворе (состоящем только из растворителя, не содержащего образец).
Стандарт качества для примесей и изомеров, выраженный в виде PAR, компании заявителя и результаты анализа ВЭЖХ представлены в следующей ниже таблице 1.
Кроме того, в результатах анализа отсутствуют соединения (1), (2), (3), (4) и т.д., которые образуются в случаях более ранних способов получения соединения (I).
Все соединения (I), полученные в соответствии с примерами 2-5, также удовлетворяют стандарту качества.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
При получении метансульфоната 4-аминометил-3-алкоксииминопирролидина, используемого в качестве промежуточного соединения для хинолоновых антибиотиков, настоящее изобретение улучшает пару аспектов предшествующего способа. Общее количество стадий уменьшают до 2-3, приводя к исключению таких операций, как фильтрационная и экстракционная обработка, отдает необходимость использования дорогого реагента, (BOC)2O, различных органических растворителей и реагентов.
Реферат
Изобретение относится к новым улучшенным способам получения метансульфоната 4-аминометил-3-алкоксииминопирролидина формулы (I), которые являются промежуточными продуктами для получения известных хинолоновых соединений формулы VI, обладающих свойствами антибиотиков.

В формулах I и VI R представляет собой С1-4 алкил или С1-4 галогеналкил. Способ получения соединения I включает стадии: (а) взаимодействия соединения (II) с алкоксиамином или галогеналкоксиамином или их солью в присутствии основания с получением соединения (III); (b) взаимодействия соединения (III) с метансульфоновой кислотой с получением соединения (IV); (с) гидрирования соединения (IV) с добавлением достаточных количеств метансульфоновой кислоты и металлсодержащего катализатора гидрирования с получением соединения (I).


При этом можно осуществлять процесс без получения соединения формулы IV (стадии b), добавляя металлсодержащий катализатор гидрирования и соответствующее количество метансульфоновой кислоты непосредственно к соединению формулы (III). Из полученного соединения формулы (I) при взаимодействии с соединением формулы (V) получают формулы (VI). Изобретение также относится к новым соединениям формулы (III) и (IV). 5 н. и 22 з.п. ф-лы, 1 табл.
Формула




Документы, цитированные в отчёте о поиске
Способ получения производных 1,8-нафтиридина или их солей
Комментарии