2-карбоксамид циклоамино производные мочевины в комбинации с ингибиторами hsp90 для лечения пролиферативных заболеваний - RU2624493C2
Код документа: RU2624493C2
Чертежи








Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к фармацевтической комбинации, содержащей 2-карбоксамид циклоамино производное мочевины формулы (I) и ингибиторы белка теплового шока 90, а также к применению таких комбинаций при лечении пролиферативных заболеваний, более конкретно, PI3K-зависимых заболеваний, более конкретно, PI3K-альфа-зависимых заболеваний.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Метаболический путь PI3K/Akt/mTOR является важным, четко регулируемым путем выживания для нормальной клетки. Фосфатидилинозитол 3-киназы (PI3K) являются широко представленными липидными киназами, которые катализируют перенос фосфата в положение D-3' инозитольных липидов с образованием фосфоинозитол-3-фосфата (PIP), фосфоинозитол-3,4-дифосфата (PIP2) и фосфоинозитол-3,4,5-трифосфата (PIP3). Эти продукты PI3K-катализируемых реакций выступают в качестве вторичных мессенджеров и играют центральные роли в ключевых клеточных процессах, включая рост клеток, дифференциацию, подвижность, пролиферацию и выживание.
Из двух классов 1 PI3K, класс 1A PI3K представляет собой гетеродимеры, состоящие из каталитической p110 субъединицы (α, β, δ изоформы), конструктивно связанной с регуляторной субъединицей, которая может представлять собой p85α, p55α, p50α, p85β или р55γ. Подкласс класса 1B содержит один член семейства, гетеродимер, состоящий из каталитической p110γ субъединицы, связанной с одной из двух регуляторных субъединиц, P101 или P84 (Fruman et al., Annu Rev. Biochem. 67:481 (1998); Suire et al., Curr. Biol. 15:566 (2005)).
Во многих случаях PIP2 и PIP3 рекрутируют AKT к плазматической мембране, где она выступает в качестве узловой точки для многих внутриклеточных сигнальных путей, важных для роста и выживания (Fantl et al., Cell 69:413-423(1992); Bader et al., Nature Rev. Cancer 5:921 (2005); Vivanco и Sawyer, Nature Rev. Cancer 2:489 (2002)). Нарушенная регуляция PI3K, которая часто повышает выживаемость через активацию AKT, является одним из наиболее распространенных событий при злокачественных новообразованиях у человека и, как было показано, происходит на нескольких уровнях. Подавляющий опухоль ген PTEN, который дефосфорилирует фосфоинозитиды в положении 3' инозитольного кольца и тем самым противодействует активности PI3K, функционально исключен в различных видах опухолях. В других опухолях гены для изоформы p110α, PIK3CA и для AKT являются амплифицированными и повышающими экспрессию белка из их генных продуктов, что было продемонстрировано на нескольких злокачественных новообразованиях человека.
Кроме того, соматические миссенс-мутации в PIK3CA, которые активируют дополнительные сигнальные пути, описывались достаточно часто для широкого разнообразия злокачественных новообразований человека (Kang at el., Proc. Natl. Acad. Sci. USA 102:802 (2005); Samuels et al., Science 304:554 (2004); Samuels et al., Cancer Cell 7:561-573 (2005)). Таким образом, известно, что ингибиторы PI3K альфа имеют особое значение при лечении пролиферативного заболевания и других расстройств.
Кроме того, белок теплового шока 90 (Hsp90) признан в качестве мишени для противораковых средств. Hsp90 является весьма распространенным и важным белком, который действует как молекулярный шаперон для обеспечения устойчивости конформации, формы и функции белков-клиентов. Семейство Hsp90 шаперонов включает четыре члена: Hsp90α и Hsp90β, оба локализованы в цитозоле, GRP94 в эндоплазматическом ретикулуме и TRAP1 в митохондриях. Hsp90 является распространенным клеточным шапероном, составляющим примерно 1%-2% общего белка.
Среди белков стресса Hsp90 является уникальным, т.к. он не требуется для биосинтеза большинства полипептидов. Hsp90 образует комплексы с онкогенными белками, называемыми ʺбелки-клиентыʺ, которые являются конформационно лабильными передатчиками сигналов, играющими важную роль в контроле роста, в выживании клеток и в развитии тканей.
Такое связывание предотвращает деградацию этих белков-клиентов. Подсемейство белков-клиентов Hsp90, таких как Raf, AKT, phospho-AKT, CDK4 и семейство EGFR, включающее ErbB2, представляет собой группу онкогенных сигнальных молекул, играющих основную роль в росте, дифференциации и апоптозе клеток, всех процессов, которые играют важную роль для раковых клеток. Ингибирование присущей ему АТФазной активности белка Hsp90 нарушает взаимодействие с белками-клиентами Hsp90, что приводит к их деградации по убиквитин-протеосомному пути.
Шапероны Hsp90, которые характеризуются консервативным АТФ-связывающим сайтом в N-концевом домене, относятся к небольшому подсемейству АТФаз и известны как ДНК-гираза, Hsp90, гистидинкиназа и подсемейство MutL (GHKL). Шапероновая (образование вторичной структуры белков) активность Hsp90 зависит от его АТФазной активности, которая снижается в очищенном ферменте. Однако было установлено, что АТФазная активность Hsp90 повышается при его связывании с белками, так называемыми ко-шаперонами. Таким образом, белки Hsp90 функционируют in vivo в качестве субъединиц крупных, динамичных белковых комплексов. Hsp90 важен для выживания эукариотических клеток и сверхэкспрессирован во многих опухолях.
Несмотря на многочисленные варианты лечения пациентов с пролиферативными заболеваниями, остаются потребность в эффективных и безопасных терапевтических средствах и потребность в их преимущественном применении в комбинационной терапии. Неожиданно было обнаружено, что определенные соединения 2-карбоксамидциклоамино производные мочевины формулы (I), которые были описаны в WO 2010/029082, в комбинации с ингибиторами Hsp90 вызывают сильную анти-пролиферативную активность и in vivo противоопухолевый ответ.
Совместная обработка раковых клеток ингибитором Hsp90 и ингибитором PI3K, в частности, высоко специфическим ингибитором PI3K альфа соединения формулы (I), является особенно эффективной, так как она сочетает в себе ингибирование компонентов проксимальных путей, таких как рецепторные тирозинкиназы (в основном, мишеневые посредством ингибирования Hsp90), с другим ингибитором (PI3K ингибитором), который также действует близко к верхней части сигнального каскада. Дополнительное преимущество ингибирования Hsp90 может возникнуть в результате его действия на другие сигнальные компоненты в метаболическом пути PI3K/Akt/mTOR, как например, на AKT и pAKT, и его широкого действия на многие белки-клиенты.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтической комбинации, содержащей (a) соединение формулы (I)

где
A представляет собой гетероарил, выбранный из группы, состоящей из:

R1 представляет собой один из следующих заместителей: (1) незамещенный или замещенный, предпочтительно, замещенный C1-C7-алкил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, из от одной до девяти, следующих групп: дейтерий, фтор, или из от одной до двух следующих групп C3-C5-циклоалкил; (2) необязательно замещенный C3-C5-циклоалкил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, из от одной до четырех следующих групп: дейтерий, C1-C4-алкил (предпочтительно, метил), фтор, циано, аминокарбонил; (3) необязательно замещенный фенил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, из от одной до двух, следующих групп: дейтерий, галоген, циано, C1-C7-алкил, C1-C7-алкиламино, ди(C1-C7-алкил)амино, C1-C7-алкиламинокарбонил, ди(C1-C7-алкил)аминокарбонил, C1-C7-алкокси; (4) необязательно моно- или дизамещенный амин; где указанные заместители независимо выбраны из следующих групп: дейтерий, C1-C7-алкил (который является незамещенным или замещенным одним или несколькими заместителями, выбранными из группы, включающей дейтерий, фтор, хлор, гидрокси), фенилсульфонил (который является незамещенным или замещенным одним или несколькими, предпочтительно, одним, C1-C7-алкилом, C1-C7-алкокси, ди(C1-C7-алкил)амино-C1-C7-алкокси); (5) замещенный сульфонил; где указанный заместитель выбран из следующих групп: C1-C7-алкил (который является незамещенным или замещенным одним или несколькими заместителями, выбранными из группы из дейтерия, фтора), пирролидино (который является незамещенным или замещенным одним или несколькими заместителями, выбранными из группы из дейтерия, гидрокси, оксо; в частности один оксо); (6) фтор, хлор;
R2 представляет собой водород;
R3 представляет собой (1) водород, (2) фтор, хлор, (3) необязательно замещенный метил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, из от одной до трех, следующих групп: дейтерий, фтор, хлор, диметиламино;
при исключении 2-амида 1-({5-[2-(трет-бутил)пиримидин-4-ил]-4-метилтиазол-2-ил}амида) (S)пирролидин-1,2-дикарбоновой кислоты,
или его фармацевтически приемлемую соль; и (b) по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль. Такая комбинация может быть предназначена для одновременного, раздельного или последовательного использования при лечении пролиферативного заболевания.
В предпочтительном варианте осуществления изобретения фармацевтическая комбинация по настоящему изобретению включает соединение формулы (I), выбранное из 2-амида 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)пирролидин-1,2-дикарбоновой кислоты (“соединение A”) или его фармацевтически приемлемой соли.
Фармацевтическая комбинация по настоящему изобретению включает, по меньшей мере, одно мишеневое соединение, снижающее или ингибирующее присущую Hsp90 АТФазную активность и/или деградадирующее, нацеливающее, снижающее или ингибирующее белки-клиенты Hsp90 посредством убиквитин протеосомного метаболизма. Такие соединения далее указываются как ʺингибиторы белка теплового шока 90ʺ или ʺингибиторы Hsp90ʺ. Примеры ингибиторов Hsp90, подходящих для использования в настоящем изобретении, включают, но этим не ограничиваются, производное гелданамицина, танеспимицин (17-аллиламино-17-деметоксигелданамицин) (известный также как KOS-953 и 17-AAG); радицикол; метансульфонат 6-хлор-9-(4-метокси-3,5-диметилпиридин-2-илметил)-9H-пурин-2-амина (известный также как CNF2024); IPI504; SNX5422; этиламид 5-(2,4-дигидрокси-5-изопропилфенил)-4-(4-морфолин-4-илметилфенил)изоксазол-3-карбоновой кислоты (AUY922); и (R)-2-амино-7-[4-фтор-2-(6-метоксипиридин-2-ил)фенил]-4-метил-7,8-дигидро-6H-пиридо[4,3-d]пиримидин-5-он (HSP990).
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль. В одном варианте осуществления изобретения указанная фармацевтическая композиция по настоящему изобретению предназначена для использования при лечении пролиферативного заболевания.
Кроме того, настоящее изобретение относится к применению фармацевтической комбинации, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль, при получении лекарственного средства для лечения пролиферативного заболевания.
Кроме того, настоящее изобретение относится к способу лечения пролиферативного заболевания у субъекта, при необходимости этого, включающему введение указанному субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, и, по меньшей мере, одного ингибитора Hsp90 или его фармацевтически приемлемой соли. В соответствии с настоящим изобретением соединение формулы (I) и ингибитор Hsp90 могут быть введены либо в виде одной фармацевтической композиции, в виде отдельных композиций, либо последовательно.
Кроме того, настоящее изобретение относится к набору, содержащему соединение формулы (I) по п. 1 или его фармацевтически приемлемую соль, и, по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль.
ОПИСАНИЕ ФИГУР
На фигуре 1 показана противоопухолевая активность соединения A против PIK3CA мутантной линии клеток рака желудка HGC-27.
На фигуре 2 показана масса тела у групп мышей, несущих HGC-27, обработанных растворителем и соединением A.
Для in vivo исследований, представленных на фигурах 1 и 2, бестимусных самок мышей, несущих подкожные ксенотрансплантаты HGC-27, обрабатывали соединением A (соед. A) или растворителем в указанных дозах и режимах. Обработку начинали через 12 дней после прививки опухолевых клеток и продолжали в течение 12 последующих дней. Статистику по изменению объемов опухолей проводили с использованием однофакторной ANOVA с апостериорным критерием Данетта (*p<0,05 против контролей растворителей).
На фигуре 3 показана противоопухолевая активность растворителя, 12,5 мг/кг перорально, один раз в день (qd) отдельного агента соединения A, 50 мг/кг, внутривенно, два раза в неделю (2qw) отдельного агента AUY922 и комбинации соединения A с AUY922 против PIK3CA мутантной линии клеток рака желудка HGC-27. Значения являются средним ± SEM; размером образца (n=10 мышей в группе). (*p<0,05, значительное ингибирование по сравнению с контрольными группами, обработанными растворителем и обработанными отдельным агентом (ранговый критерий Манна-Уитни с апостериорным критерием Стьюдента-Ньюмана-Келса).
На фигуре 4 показаны усредненные изменения массы тела (представленные как отношение массы тела в день измерения и начальной массы тела в день 12 [оба откорректированные путем вычитания первичной массы опухоли], выраженные в процентах для каждого отдельного животного) для групп мышей с PIK3CA мутантной линией клеток рака желудка HGC-27, обработанных растворителем, 12,5 мг/кг соединения A, 50 мг/кг AUY922 и комбинацией соединения A в дозе 25 мг/кг и AUY922 в дозе 50 мг/кг.
На фигуре 5 показана противоопухолевая активность растворителя, 25 мг/кг перорально qd отдельного агента соединения A, 50 мг/кг, внутривенно, 2qw отдельного агента AUY922 и комбинации соединения A с AUY922 против PIK3CA мутантной линии клеток рака желудка HGC-27. Значения являются средним ± SEM; размером образца (n=10 мышей в группе). (*p<0,05, значительное ингибирование по сравнению с контрольными группами, обработанными растворителем и обработанными отдельным агентом (ранговый критерий Манна-Уитни с апостериорным критерием Данна).
На фигуре 6 показаны усредненные изменения массы тела (представленные как отношение массы тела в день измерения и начальной массы тела в день 12 [оба откорректированные путем вычитания первичной массы опухоли], выраженные в процентах для каждого отдельного животного) для групп мышей с PIK3CA мутантной линией клеток рака желудка HGC-27, обработанных растворителем, 25 мг/кг соединения A, 50 мг/кг AUY922 и комбинацией соединения A в дозе 25 мг/кг и AUY922 в дозе 50 мг/кг.
На фигуре 7 показана противоопухолевая активность растворителя, 50 мг/кг перорально qd отдельного агента соединения A, 50 мг/кг, внутривенно, 2qw отдельного агента AUY922 и комбинации соединения A с AUY922 против PIK3CA мутантной линии клеток рака желудка HGC-27. Значения являются средним ± SEM; размером образца (n=10 мышей в группе). (*p<0,05, значительное ингибирование по сравнению с контрольными группами, обработанными растворителем и обработанными отдельным агентом (ранговый критерий Манна-Уитни с апостериорным критерием Стьюдента-Ньюмана-Келса).
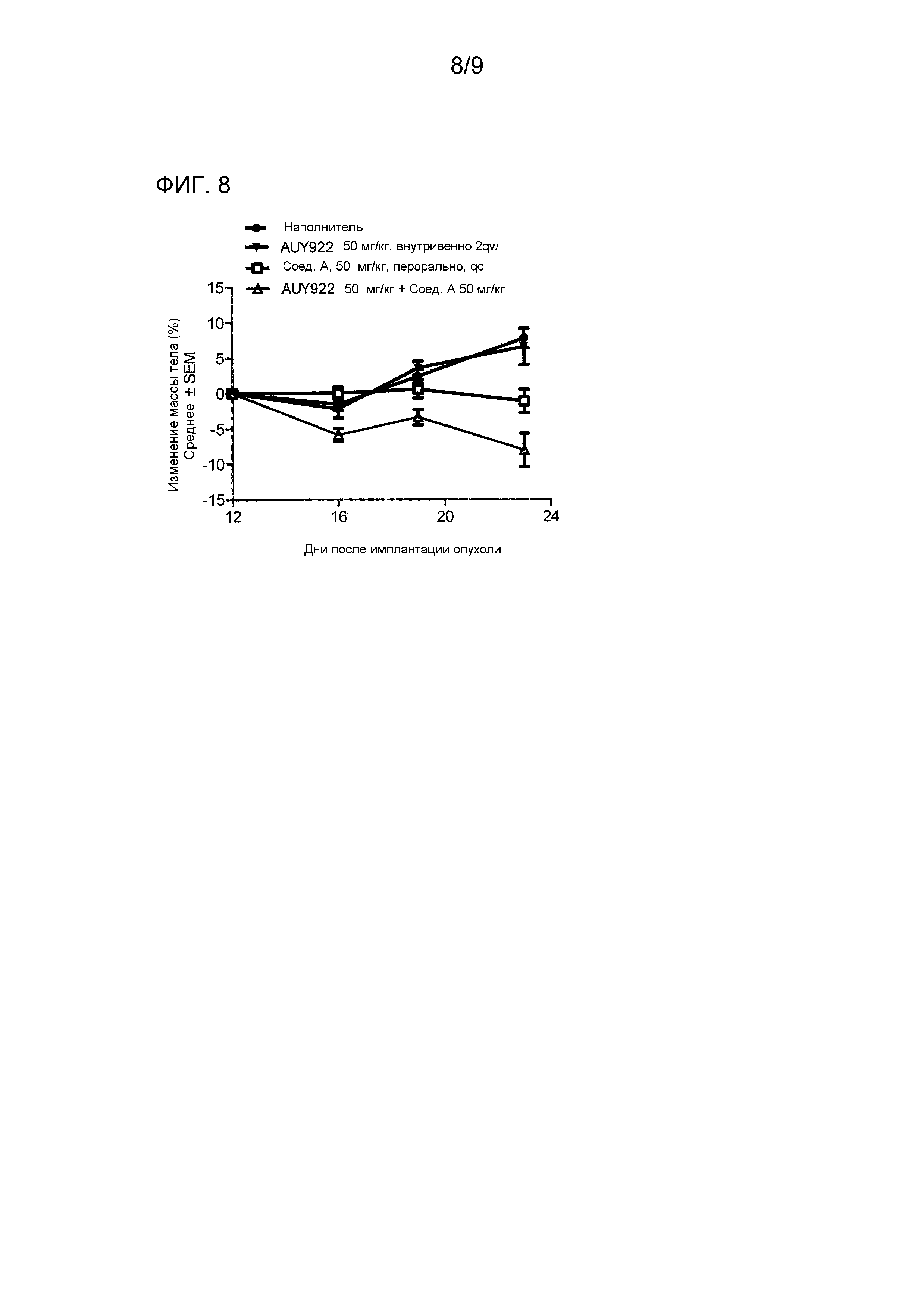
На фигуре 8 показаны усредненные изменения массы тела (представленные как отношение массы тела в день измерения и начальной массы тела в день 12 [оба откорректированные путем вычитания первичной массы опухоли], выраженные в процентах для каждого отдельного животного) для групп мышей с мутантной PIK3CA линией клеток рака желудка HGC-27, обработанных растворителем, 50 мг/кг соединения A, 50 мг/кг AUY922 и комбинацией соединения A в дозе 50 мг/кг и AUY922 в дозе 50 мг/кг.
На фигуре 9 показаны (a) частичный рост опухоли и (b) средние изменения массы тела при обработке растворителем/плацебо (n=5), 40 мг/кг перорально qd отдельным агентом соединением A (n=7), 50 мг/кг, внутривенно, 2qw отдельным агентом AUY922 (n=8) и комбинацией 40 мг/кг перорально qd соединения A и 50 мг/кг AUY922 (n=5) против клеточной линии меланомы A375.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Следующие общие определения предоставлены для лучшего понимания изобретения:
“Галоген” (или “гало”) обозначает фтор, бром, хлор или йод, в частности, фтор, хлор. Галогензамещенные группы и фрагменты, такие как алкил, замещенный галогеном (галогеналкил), могут быть моно-, поли- или пергалогенированными.
“Гетероатомы” представляют собой атомы, иные, чем углерод и водород, предпочтительно, азот (N), кислород (O) или серу (S), в частности, азот.
“Углеродсодержащие группы”, фрагменты или молекулы содержат от 1 до 7, предпочтительно, от 1 до 6, более предпочтительно, от 1 до 4, наиболее предпочтительно, 1 или 2, атомов углерода. Любая нециклическая углеродсодержащая группа или фрагмент с более чем 1 атомом углерода является прямой или разветвленной.
Префикс “низший” или “C1-C7” обозначает радикал, содержащий вплоть до и включая максимум 7, особенно вплоть до и включая максимум 4, атома углерода, и эти радикалы являются либо линейными, либо разветвленными с одним или множеством разветвлений.
“Алкил” относится к алкильной группе с прямой цепью или разветвленной цепью, предпочтительно, представляет собой прямой или разветвленный C1-12-алкил, особенно предпочтительно, представляет собой прямой или разветвленный C1-7-алкил; например, метил, этил, н- или изопропил, н-, изо-, втор- или трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил, н-ундецил, н-додецил, при особом предпочтении, отдаваемом метилу, этилу, н-пропилу, изо-пропилу и н-бутилу и изобутилу. Алкил может быть незамещенным или замещенным. Примеры заместителей включают, но этим не ограничиваются, дейтерий, гидрокси, алкокси, галоген и амино. Примером замещенного алкила является трифторметил. Циклоалкил также может быть заместителем для алкила. Примером такого случая является фрагмент (алкил)-циклопропил или алкандиил-циклопропил, например -CH2-циклопропил. C1-C7-алкил, предпочтительно, представляет собой алкил, содержащий от и включая 1 вплоть до и включая 7, предпочтительно, от и включая 1 до и включая 4, атомов углерода, и является линейным или разветвленным; предпочтительно, низший алкил представляет собой бутил, такой как н-бутил, втор-бутил, изобутил, трет-бутил, пропил, такой как н-пропил или изопропил, этил или, предпочтительно, метил.
Каждая алкильная часть других групп, таких как “алкокси”, “алкоксиалкил”, “алкоксикарбонил”, “алкоксикарбонилалкил”, “алкилсульфонил”, “алкилсульфоксил”, “алкиламино”, “галогеналкил”, будет иметь те же значения, что описаны выше для определения “алкил”.
“Алкандиил” относится алкандиильной группе с прямой цепью или разветвленной цепью, присоединенной двумя разными атомами углерода к фрагменту, он, предпочтительно, представляет собой прямой или разветвленный C1-12 алкандиил, особенно предпочтительно, представляет собой прямой или разветвленный C1-6 алкандиил; например, метандиил (-CH2-), 1,2-этандиил (-CH2-CH2-), 1,1-этандиил ((-CH(CH3)-), 1,1-, 1,2-, 1,3-пропандиил и 1,1-, 1,2-, 1,3-, 1,4-бутандиил, при особом предпочтении, отдаваемом метандиилу, 1,1-этандиилу, 1,2-этандиилу, 1,3-пропандиилу, 1,4-бутандиилу.
“Алкендиил” относится к алкендиильной группе с прямой цепью или разветвленной цепью, присоединенной двумя разными атомами углерода к молекуле, он, предпочтительно, представляет собой прямой или разветвленный C2-6-алкандиил; например, -CH=CH-, -CH=C(CH3)-, -CH=CH-CH2-, -C(CH3)=CH-CH2-, -CH=C(CH3)-CH2-, -CH=CH-C(CH3)H-, -CH=CH-CH=CH-, -C(CH3)=CH-CH=CH-, -CH=C(CH3)-CH=CH-, при особом предпочтении, отдаваемом -CH=CH-CH2-, -CH=CH-CH=CH-. Алкендиил может быть замещенным или незамещенным.
“Циклоалкил” относится к насыщенному или частично насыщенному моноциклическому конденсированному полициклическому или спиро-полициклическому карбоциклу, содержащему от 3 до 12 кольцевых атомов в карбоцикле. Иллюстративные примеры циклоалкильных групп включают следующие фрагменты: циклопропил, циклобутил, циклопентил и циклогексил. Циклоалкил может быть незамещенным или замещенным; примеры заместителей представлены при определении алкила и также включают сам алкил (например, метил). Фрагмент, подобный -(CH3)-циклопропилу, рассматривается как замещенный циклоалкил.
“Арил” относится к ароматической гомоциклической кольцевой системе (то есть, только углерод в качестве образующих кольцо атомов) с 6 или более атомами углерода; арил предпочтительно представляет собой ароматический фрагмент, содержащий от 6 до 14 кольцевых атомов углерода, более предпочтительно, от 6 до 10 кольцевых атомов углерода, такой как фенил или нафтил, предпочтительно, фенил. Арил может быть незамещенным или замещенным одним или несколькими, предпочтительно, вплоть до трех, более предпочтительно, вплоть до двух, заместителями, независимо выбранными из группы, включающей незамещенный или замещенный гетероциклил, как описано далее, особенно пирролидинил, такой как пирролидино, оксопирролидинил, такой как оксопирролидино, C1-C7-алкилпирролидинил, 2,5-ди-(C1-C7-алкил)пирролидинил, такой как 2,5-ди-(C1-C7-алкил)пирролидино, тетрагидрофуранил, тиофенил, C1-C7-алкилпиразолидинил, пиридинил, C1-C7-алкилпиперидинил, пиперидино, пиперидино, замещенный амино, или н-моно-, или N,N-ди-(низший алкил, фенил, C1-C7-алканоил и/или фенил-низший алкил)амино, незамещенный или замещенный н-низшим алкилом пиперидинил, присоединенный через кольцевой атом углерода, пиперазино, низший алкилпиперазино, морфолино, тиоморфолино, S-оксотиоморфолино или S,S-диоксотиоморфолино; C1-C7-алкил, амино-C1-C7-алкил, н-C1-C7-алканоиламино-C1-C7-алкил, н-C1-C7-алкансульфониламино-C1-C7-алкил, карбамоил-C1-C7-алкил, [N-моно- или N,N-ди-(C1-C7-алкил)карбамоил]-C1-C7-алкил, C1-C7-алкансульфинил-C1-C7-алкил, C1-C7-алкансульфонил-C1-C7-алкил, фенил, нафтил, от моно- до три-[C1-C7-алкил, галоген и/или циано]фенил или от моно- до три-[C1-C7-алкил, галоген и/или циано]нафтил; C3-C8-циклоалкил, от моно- до три-[C1-C7-алкил и/или гидрокси]-C3-C8-циклоалкил; галоген, гидрокси, низший алкокси, низший алкокси-низший алкокси, (низший алкокси)-низший алкокси-низший алкокси, галоген-C1-C7-алкокси, фенокси, нафтилокси, фенил- или нафтил-низший алкокси; амино-C1-C7-алкокси, низший-алканоилокси, бензоилокси, нафтоилокси, формил (CHO), амино, н-моно- или N,N-ди-(C1-C7-алкил)амино, C1-C7-алканоиламино, C1-C7-алкансульфониламино, карбокси, низший алкоксикарбонил, например; фенил- или нафтил-низший алкоксикарбонил, такой как бензилоксикарбонил; C1-C7-алканоил, такой как ацетил, бензоил, нафтоил, карбамоил, н-моно- или N,N-дизамещенный карбамоил, такой как н-моно- или N,N-ди-замещенный карбамоил, где заместители выбраны из низшего алкила, (низший алкокси)-низшего алкила и гидрокси-низшего алкила; амидино, гуанидино, уреидо, меркапто, низший алкилтио, фенил- или нафтилтио, фенил- или нафтил-низший алкилтио, низший алкилфенилтио, низший алкилнафтилтио, галоген-низший алкилмеркапто, сульфо (-SO3H), низший алкансульфонил, фенил- или нафтилсульфонил, фенил- или нафтил-низший алкилсульфонил, алкилфенилсульфонил, галоген-низший алкилсульфонил, такой как трифторметансульфонил; сульфонамидо, бензосульфонамидо, азидо, азидо-C1-C7-алкил, особенно азидометил, C1-C7-алкансульфонил, сульфамоил, н-моно- или N,N-ди-(C1-C7-алкил)сульфамоил, морфолиносульфонил, тиоморфолиносульфонил, циано и нитро; где каждый фенил или нафтил (также в фенокси или нафтокси), указанные выше в качестве заместителя или части заместителя замещенного алкила (или также замещенного арила, гетероциклила и так далее, указанных в данном документе), сами являются незамещенными или замещенными одним или несколькими, например, вплоть до трех, предпочтительно, 1 или 2, заместителями, независимо выбранными из галогена, галоген-низшего алкила, такого как трифторметил, гидрокси, низший алкокси, азидо, амино, н-моно- или N,N-ди-(низший алкил и/или C1-C7-алканоил)амино, нитро, карбокси, низший-алкоксикарбонил, карбамоил, циано и/или сульфамоил.
“Гетероциклил” относится к гетероциклическому радикалу, который является ненасыщенным (= несущим максимально возможное количество сопряженных двойных связей в кольце(ах)), насыщенным или частично насыщенным и, предпочтительно, представляет собой моноциклическое или, в более широком аспекте данного изобретения, бициклическое, трициклическое или спироциклическое кольцо; и содержит от 3 до 24, более предпочтительно, от 4 до 16, наиболее предпочтительно, от 5 до 10, и, наиболее предпочтительно, 5 или 6 кольцевых атомов; где один или несколько, предпочтительно, от одного до четырех, особенно, одно или два кольцевых атома являются гетероатомом (остальные кольцевые атомы, следовательно, являются углеродами). Связывающее кольцо (то есть кольцо, присоединенное к молекуле), предпочтительно, содержит от 4 до 12, особенно от 5 до 7 кольцевых атомов. Термин гетероциклил также включает гетероарил. Гетероциклический радикал (гетероциклил) может быть незамещенным или замещенным одним или несколькими, особенно от 1 до 3, заместителями, независимо выбранными из группы, включающей заместители, определенные выше для замещенного алкила, и/или из одного или нескольких следующих заместителей: оксо (=O), тиокарбонил (=S), имино(=NH), имино-низший алкил. Кроме того, гетероциклил, особенно, представляет собой гетероциклильный радикал, выбранный из группы, состоящей из оксиранила, азиринила, азиридинила, 1,2-оксатиоланила, тиенила (= тиофенил), фуранила, тетрагидрофурила, пиранила, тиопиранила, тиантренила, изобензофуранила, бензофуранила, хроменила, 2Н-пирролила, пирролила, пирролинила, пирролидинила, имидазолила, имидазолидинила, бензимидазолила, пиразолила, пиразинила, пиразолидинила, тиазолила, изотиазолила, дитиазолила, оксазолила, изоксазолила, пиридила, пиразинила, пиримидинила, пиперидинила, пиперазинила, пиридазинила, морфолинила, тиоморфолинила, (S-оксо или S,S-диоксо)тиоморфолинила, индолизинила, азепанила, диазепанила, особенно 1,4-диазепанила, изоиндолила, 3H-индолила, индолила, бензимидазолила, кумарила, индазолила, триазолила, тетразолила, пуринила, 4Н-хинолизинила, изохинолила, хинолила, тетрагидрохинолила, тетрагидроизохинолила, декагидрохинолила, октагидроизохинолила, бензофуранила, дибензофуранила, бензотиофенила, дибензотиофенила, фталазинила, нафтиридинила, хиноксалила, хиназолинила, хиназолинила, циннолинила, птеридинила, карбазолила, бета-карболинила, фенантридинила, акридинила, перимидинила, фенантролинила, фуразанила, феназинила, фенотиазинила, феноксазинила, хроменила, изохроманила, хроманила, бензо[1,3]диоксол-5-ила и 2,3-дигидробензо[1,4]диоксин-6-ила, где каждый из этих радикалов является незамещенным или замещенным одним или несколькими, предпочтительно, вплоть до трех, заместителями, выбранными из тех, которые указаны выше для замещенного арила, и/или из одного или нескольких следующих заместителей: оксо (=O), тиокарбонила (=S), имино(=NH), имино-низшего алкила.
“Арилалкил” относится к арильной группе, присоединенной к молекуле через алкильную группу, такую как метильная или этильная группа, предпочтительно, фенэтилу или бензилу, в частности, бензилу. Подобным же образом, циклоалкил-алкил и гетероциклил-алкил представляют собой циклоалкильную группу, присоединенную к молекуле через алкильную группу, или гетероциклильную группу, присоединенную к молекуле через алкильную группу. В каждом случае арил, гетероциклил, циклоалкил и алкил могут быть замещены, как определено выше.
“Соли” (которые подразумеваются в термине “или его соли” или “или его соль”), могут быть представлены самостоятельно или в смеси со свободным соединением, например, соединением формулы (I), и являются, предпочтительно, фармацевтически приемлемыми солями. Такие соли соединений формулы (I) получают, например, в виде кислотно-аддитивных солей, предпочтительно, с органическими и неорганическими кислотами, из соединений формулы (I), обладающих основным атомом азота. Подходящими неорганическими кислотами являются, например, галогенсодержащие кислоты, такие как хлористоводородная кислота, серная кислота или фосфорная кислота. Подходящими органическими кислотами являются, например, карбоновые кислоты или сульфоновые кислоты, такие как фумаровая кислота или метансульфоновая кислота. Для целей выделения или очистки возможно также использовать фармацевтически неприемлемые соли, например, пикраты или перхлораты. Для терапевтического применения используются только фармацевтически приемлемые соли или свободные соединения (где это применимо в форме фармацевтических препаратов), и они иногда являются предпочтительными. Ввиду тесной взаимосвязи между новыми соединениями в свободной форме и теми, которые находятся в форме своих солей, включая те соли, которые могут быть использованы в качестве промежуточных, например, при очистке или характеризации новых соединений, любую ссылку на свободные соединения выше и далее следует понимать как ссылку также на соответствующие соли, в зависимости от обстоятельств и целесообразности. Соли соединений формулы (I) являются, предпочтительно, фармацевтически приемлемыми солями; подходящие образующие противоионы фармацевтически приемлемые соли известны в данной области.
“Комбинация” относится либо к комбинации постоянного состава в виде одной стандартной лекарственной формы, либо к комбинации непостоянного состава (или набора компонентов) для объединенного введения, где соединение формулы (I) и компонент комбинации (например, другое лекарственное средство, как описано далее, указываемое также как “терапевтический агент” или “со-агент”) могут быть введены независимо в одно и то же время или раздельно в промежутках времени, особенно, когда эти промежутки времени позволяют компонентам комбинации проявлять совместное, например, синергетическое действие. Термин “объединенное введение” или тому подобное, как используется в данном документе, означает введение выбранного компонента комбинации одному субъекту, при необходимости этого (например, пациенту), и предназначен для охвата лечебных режимов, при которых агенты не обязательно вводятся одним и одним и тем же путем введения или в одно и то же время. Термин “комбинация определенного состава” означает, что активные ингредиенты, например, соединение формулы (I) и компонент комбинации, оба водятся пациенту одновременно в виде единого целого или единой дозировки. Термины “комбинация неопределенного состава” или “набор компонентов” означает, что активные ингредиенты, например, соединение формулы (I) и компонент комбинации, оба водятся пациенту в виде раздельных компонентов либо одновременно, параллельно, либо последовательно, без конкретных ограничений во времени, где такое введение обеспечивает терапевтически эффективный уровень двух соединений в организме пациента. Последнее также относится к смешанной терапии, например, введение трех или более активных ингредиентов.
“Лечение” включает профилактическое (превентивное) и терапевтическое лечение, а также задержку прогрессирования расстройства или заболевания. Термин “профилактическое” означает предупреждение появления или рецидива заболеваний, сопровождающих пролиферативные заболевания. Термин “задержка прогрессирования”, как используется в настоящем документе, означает введение комбинации пациентам, находящимся в предварительной стадии или на ранней стадии пролиферативного заболевания, подлежащего лечению, например, пациентам, у которых диагностирована предварительная стадии соответствующего заболевания, или пациентам, находящимся в состоянии, например, процесса медицинского лечения, или состоянии в результате несчастного случая, при которых имеется вероятность развития соответствующей болезни.
Термин “субъект” предназначен для включения животных. Примерами субъектов являются млекопитающие, например, люди, собаки, коровы, лошади, свиньи, овцы, козы, кошки, мыши, кролики, крысы и трансгенные животные. В некоторых вариантах осуществления изобретения субъектом является человек, например, человек, страдающий от, рискующий пострадать от или потенциально способный страдать от опухолевого заболевания мозга. Особенно предпочтительно, субъектом является человек.
“Фармацевтический препарат” или “фармацевтическая композиция” относятся к смеси или раствору, содержащим, по меньшей мере, одно терапевтическое соединение для введения млекопитающему, например, человеку, чтобы предотвратить, лечить или контролировать конкретное заболевание или состояние, влияющее на млекопитающее.
“Со-назначение”, “со-введение”, или “объединенное введение”, или тому подобное означают введение выбранных терапевтических средств одному пациенту, и предназначены для включения в лечебные режимы, согласно которым средства не обязательно вводятся одним и тем же путем введения или в одно и то же время.
“Фармацевтически приемлемый” относится к таким соединениям, веществам, композициям и/или дозированным формам, которые, в рамках обоснованного медицинского заключения, подходят для контакта с тканями млекопитающих, особенно людей, без чрезмерной токсичности, раздражения, аллергической реакции и других проблемных осложнений, соизмеримых с разумным отношением польза/риск.
“Терапевтически эффективный”, предпочтительно, относится к количеству, которое терапевтически или в более широком смысле также профилактически эффективны против прогрессирования пролиферативного заболевания.
“Стандартная фармацевтическая композиция” относится к стандартному носителю или основе, разработанных для обеспечения пациента эффективным количеством обоих терапевтических агентов. Стандартная основа предназначена для доставки эффективного количества каждого из агентов, а также любых фармацевтически приемлемых носителей или инертных наполнителей. В некоторых вариантах осуществления изобретения основой является таблетка, капсула, пилюля или пластырь. В других вариантах осуществления основой является раствор или суспензия.
“Интервал доз” относится к высшему или низшему пределу приемлемого варианта количества конкретного агента. Обычно пациенту, проходящему лечение, может вводиться доза агента в любом количестве в пределах указанного диапазона.
Термин “примерно” или “приблизительно” обычно обозначает в пределах 20%, более предпочтительно, в пределах 10%, и наиболее предпочтительно, даже в пределах 5% данного значения или интервала. Альтернативно, особенно в биологических системах, термин “примерно” означает в пределах примерно log (то есть, порядка величины) предпочтительно, в пределах в два раза от данного значения.
Настоящее изобретение относится к фармацевтической комбинации, содержащей (a) соединение формулы (I), как определено далее, или его фармацевтически приемлемую соль; и (b) по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль. Такая комбинация может быть для одновременного, раздельного или последовательного использования при лечении пролиферативного заболевания.
Конкретные 2-карбоксамидциклоамино производные мочевины, которые является подходящими по настоящему изобретению, их получение и подходящие, содержащие их фармацевтические препараты описаны в WO 2010/029082 и включают соединения формулы (I)

где
A представляет собой гетероарил, выбранный из группы, состоящей из:

R1 представляет собой один из следующих заместителей: (1) незамещенный или замещенный, предпочтительно, замещенный C1-C7-алкил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, из от одной до девяти следующих групп: дейтерий, фтор, или из от одной до двух следующих групп C3-C5-циклоалкил; (2) необязательно замещенный C3-C5-циклоалкил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, из от одной до четырех следующих групп: дейтерий, C1-C4-алкил (предпочтительно, метил), фтор, циано, аминокарбонил; (3) необязательно замещенный фенил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, из от одной до двух следующих групп: дейтерий, галоген, циано, C1-C7-алкил, C1-C7-алкиламино, ди(C1-C7-алкил)амино, C1-C7-алкиламинокарбонил, ди(C1-C7-алкил)аминокарбонил, C1-C7-алкокси; (4) необязательно моно- или ди- замещенный амин; где указанные заместители независимо выбраны из следующих групп: дейтерий, C1-C7-алкил (который является незамещенным или замещенным одним или несколькими заместителями, выбранными из группы из дейтерия, фтора, хлора, гидрокси), фенилсульфонил (который является незамещенным или замещенным одним или несколькими, предпочтительно, одним, C1-C7-алкил, C1-C7-алкокси, ди(C1-C7-алкил)амино-C1-C7-алкокси); (5) замещенный сульфонил; где указанный заместитель выбран из следующих групп: C1-C7-алкил (который является незамещенным или замещенным одним или несколькими заместителями, выбранными из группы из дейтерия, фтора), пирролидино (который является незамещенным или замещенным одним или несколькими заместителями, выбранными из группы из дейтерия, гидрокси, оксо; в частности, один оксо); (6) фтор, хлор;
R2представляет собой водород;
R3 представляет собой (1) водород, (2) фтор, хлор, (3) необязательно замещенный метил, где указанные заместители независимо выбраны из одной или нескольких, предпочтительно, от одного до трех из следующих групп: дейтерий, фтор, хлор, диметиламино;
при исключении 2-амида 1-({5-[2-(трет-бутил)пиримидин-4-ил]-4-метилтиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты.
Как используется в данном описании, радикалы и символы соединения формулы (I) имеют значения, описанные в WO 2010/029082, который приведен здесь в качестве ссылки в полном объеме.
Как описано в WO2010/029082, было обнаружено, что указанные соединения 2-карбоксамидциклоамино производных мочевины формулы (I) обладают значительной ингибирующей активностью в отношении фосфатидилинозитол 3-киназы (или PI3K). Указанные соединения формулы (I) обладают выгодными фармакологическими свойствами в качестве ингибитора PI3K и проявляют высокую избирательность в отношении подтипа PI3-киназы альфа по сравнению с подтипами бета и/или дельта и/или гамма.
Предпочтительным соединением формулы (I) по настоящему изобретению является соединение, которое конкретно описано в WO2010/029082. Весьма предпочтительным соединением по настоящему изобретению является 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение A) или его фармацевтически приемлемая соль. Синтез 2-амида 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты описан в WO2010/029082 в виде примера 15.
Фармацевтические комбинации по настоящему изобретению включают, по меньшей мере, одно мишеневое соединение, снижающее или ингибирующее присущую Hsp90 АТФазную активность и/или деградадирующее, нацеливающее, снижающее или ингибирующее белки-клиенты Hsp90 посредством убиквитин протеосомного метаболизма. Такие соединения далее указываются как ʺингибиторы белка теплового шока 90ʺ или ʺингибиторы Hsp90ʺ.
Подходящие ингибиторы Hsp90 включают, но этим не ограничиваются,
(a) производное гелданамицина, танеспимицин (17-аллиламино-17-деметоксигелданамицин) (известный также как KOS-953 и 17-AAG), который доступен от фирмы Sigma-Aldrich Co, LLC (St. Louis, Missouri) и описан в патенте США № 4261989, датированным 14 апреля, 1981, который включен в настоящую заявку посредством ссылки, и другие гелданамицин-родственные соединения;
(b) радицикол, который доступен от фирмы Sigma-Aldrich Co, LLC (St. Louis, Missouri);
(c) метансульфонат 6-хлор-9-(4-метокси-3,5-диметилпиридин-2-илметил)-9H-пурин-2-амина (известный также как CNF2024) (Conforma Therapeuticals Corp.);
(d) IPI504;
(e) SNX5422;
(f) этиламид 5-(2,4-дигидрокси-5-изопропилфенил)-4-(4-морфолин-4-илметилфенил)изоксазол-3-карбоновой кислоты (AUY922), структура которого и способ получения которого описаны в заявке PCT № WO04/072051, опубликованной 26 августа, 2004, который включен в настоящую заявку посредством ссылки; и
(g) (R)-2-амино-7-[4-фтор-2-(6-метоксипиридин-2-ил)фенил]-4-метил-7,8-дигидро-6H-пиридо[4,3-d]пиримидин-5-он (HSP990), структура которого и способ получения которого описаны в патенте США № 2007-0123546, опубликованном 31 мая, 2007, который включен в настоящую заявку посредством ссылки;
и их фармацевтически приемлемые соли.
Предпочтительными ингибиторами Hsp90 по настоящему изобретению являются этиламид 5-(2,4-дигидрокси-5-изопропилфенил)-4-(4-морфолин-4-илметилфенил)изоксазол-3-карбоновой кислоты (AUY922) и (R)-2-амино-7-[4-фтор-2-(6-метоксипиридин-2-ил)фенил]-4-метил-7,8-дигидро-6H-пиридо[4,3-d]пиримидин-5-он (HSP990) или их фармацевтически приемлемые соли.
Также включенными являются их фармацевтически приемлемые соли, соответствующие рацематы, диастереоизомеры, энантиомеры, таутомеры, а также соответствующие кристаллические модификации описанных выше соединений, когда они имеются, например, сольваты, гидраты и полиморфы, которые описаны в настоящем документе. Соединения, используемые в качестве активных ингредиентов в комбинациях по настоящему изобретению, могут быть получены и введены, как описано в цитированных документах, соответственно. Также в пределах объема данного изобретения предложена комбинация из более чем двух отдельных активных ингредиентов, как указано выше, то есть, фармацевтическая комбинация в пределах объема данного изобретения может включать три активных ингредиента или больше.
В одном варианте настоящего изобретения фармацевтическая комбинация содержит соединение формулы (I) которое представляет собой 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты или его фармацевтически приемлемую соль, и, по меньшей мере, один ингибитор Hsp90, выбранный из этиламида 5-(2,4-дигидрокси-5-изопропилфенил)-4-(4-морфолин-4-илметилфенил)изоксазол-3-карбоновой кислоты (AUY922), (R)-2-амино-7-[4-фтор-2-(6-метоксипиридин-2-ил)фенил]-4-метил-7,8-дигидро-6H-пиридо[4,3-d]пиримидин-5-она (HSP990) или их фармацевтически приемлемых солей.
В одном варианте настоящего изобретения фармацевтическая комбинация содержит соединение формулы (I) которое представляет собой 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты или их фармацевтически приемлемые соли и, по меньшей мере, один ингибитор Hsp90 этиламид 5-(2,4-дигидрокси-5-изопропилфенил)-4-(4-морфолин-4-илметилфенил)изоксазол-3-карбоновой кислоты (AUY922) или его фармацевтически приемлемую соль.
В настоящее время было неожиданно обнаружено, что комбинация соединения формулы (I), которое представляет собой альфа-специфический ингибитор PI3K и, по меньшей мере, один ингибитор Hsp90 обладает полезными терапевтическими свойствами, которые делают его особенно полезным для лечения пролиферативных заболеваний, в частности, злокачественного новообразования.
В одном аспекте настоящее изобретение касается фармацевтической комбинации, содержащей (a) соединение формулы (I), в частности, соединение 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты или его фармацевтически приемлемую соль, и (b), по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль, для применения при лечении пролиферативного заболевания, в частности, злокачественного новообразования.
В одном аспекте настоящее изобретение касается применения фармацевтической комбинации, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль, при получении лекарственного средства для лечения пролиферативного заболевания.
В одном аспекте, кроме того, настоящее изобретение относится к способу лечения пролиферативного заболевания у субъекта, при необходимости этого, включающее введение указанному субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, и, по меньшей мере, одного ингибитора Hsp90 или его фармацевтически приемлемой соли. В соответствии с настоящим изобретением соединение формулы (I) и ингибитор Hsp90 могут быть введены либо в виде одной фармацевтической композиции, в виде отдельных композиций, либо последовательно.
Предпочтительно, настоящее изобретение может быть использовано при лечении млекопитающего, особенно людей, страдающих от пролиферативного заболевания, такого как злокачественное новообразование.
Чтобы продемонстрировать, что комбинация соединения формулы (I) и, по меньшей мере, одного ингибитора Hsp90 являются особенно подходящими для эффективного лечения пролиферативных заболеваний с хорошим запасом терапевтических и других преимуществ, могут быть проведены клинические испытания способами, известными специалистам в данной области.
Подходящими клиническими исследованиями являются, например, немаскированные исследования, исследования по эскалации дозы у пациентов с пролиферативными заболеваниями. Такие исследования доказывают, в частности, синергизм активных ингредиентов комбинаций по изобретению. Положительные эффекты могут быть определены непосредственно по результатам этих исследований, которые известны специалисту в данной области. Такие исследования, в частности, подходят для сравнения действия монотерапии с использованием активных ингредиентов и комбинации по изобретению. Предпочтительно, дозу агента (a) повышают до тех пор, пока не будет достигнута максимально переносимая доза, а агент (b) вводят в фиксированной дозе. Альтернативно, агент (a) вводят в фиксированной дозе, а дозу агента (b) повышают. Каждый пациент получает дозы агента (a) либо ежедневно, либо периодически. Эффективность лечения может быть определена в таких исследованиях, например, после 12, 18 или 24 недель путем оценки симптомов баллами каждые 6 недель.
Введение фармацевтической комбинации по изобретению приводит не только к благоприятному эффекту, например, синергетическому терапевтическому эффекту, например, с точки зрения ослабления, задержки прогрессирования или ингибирования симптомов, но также к другим неожиданным благоприятным эффектам, например, ослаблению побочных эффектов, улучшению качества жизни или снижению заболеваемости по сравнению с монотерапией с применением только одного из агентов (a) или агентов (b), которые используются в комбинации по изобретению.
Еще одним преимуществом является то, что в комбинации по изобретению могут быть использованы более низкие дозы активных ингредиентов, например, необходимые дозы не только часто являются меньшими, но также и применяются реже, что может уменьшить тяжесть протекания болезни или снизить побочные эффекты. Это находится в соответствии с пожеланиями и требованиями самих пациентов, проходящих лечение.
Одной из целей настоящего изобретения является разработка фармацевтической композиции, содержащей такое количество каждого из компонентов комбинации, агента (a) и (b) по изобретению, которое вместе является терапевтически эффективным для лечения или предотвращения пролиферативных заболеваний. В одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и, по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль. В одном варианте осуществления изобретения такая фармацевтическая композиция по настоящему изобретению предназначена для использования при лечении пролиферативного заболевания. В соответствии с настоящим изобретением агент (a) и агент (b) могут быть введены вместе в единой фармацевтической композиции, раздельно в одной объединенной стандартной лекарственной форме или в двух отдельных стандартных лекарственных формах, либо последовательно. Стандартная лекарственная форма может также представлять собой комбинацию определенного состава.
Фармацевтические композиции для раздельного введения агента (a) и агента (b) или для введения в виде комбинации определенного состава (то есть, одной галеновой композиции, содержащей, по меньшей мере, два компонента комбинации (a) и (b)) в соответствии с изобретением, могут быть получены известным способом, и являются подходящими для энтерального, такого как пероральное или ректальное, местное, и парентеральное введение субъектам, включая млекопитающих (теплокровные животные), таких как люди, содержащими терапевтически эффективное количество, по меньшей мере, одного фармакологически активного компонента комбинации отдельно, например, как указано выше, или в комбинации с одним или несколькими фармацевтически приемлемыми носителями или разбавителями, особенно подходящими для энтерального или парентерального применения. Подходящие фармацевтические композиции содержат, например, от примерно 0,1% до примерно 99,9%, предпочтительно, от примерно 1% до примерно 60%, активного(ых) ингредиента(ов).
Фармацевтическими композициями при комбинационной терапии для энтерального или парентерального введения представлены, например, в виде стандартных лекарственных форм, таких как покрытые сахаром таблетки, таблетки, капсулы или суппозитории, ампулы, растворы для инъекций или суспензии для инъекций. Местное введение осуществляется, например, в кожу или в глаз, например, в виде лосьонов, гелей, мазей или кремов, или в назальной форме, или в виде суппозитория. Если не указано иного, их получают известными способами, например, путем обычных методов смешивания, гранулирования, нанесения сахарного покрытия, растворения или процессов лиофилизации. Следует иметь в виду, что единица содержания агента (a) или агента (b), входящая в состав отдельной дозы каждой лекарственной формы не должна быть представлена в эффективном количестве, поскольку необходимое эффективное количество может быть достигнуто путем введения нескольких дозированных единиц.
Фармацевтические композиции могут содержать один или несколько фармацевтических приемлемых носителей или разбавителей и могут быть получены обычными способами путем смешивания одного или обоих компонентов комбинаций с фармацевтически приемлемым носителем или разбавителем. Примеры фармацевтически приемлемых разбавителей включают, но этим не ограничиваются, лактозу, декстрозу, манит, и/или глицерин, и/или лубриканты, и/или полиэтиленгликоль. Примеры фармацевтически приемлемых связующих включают, но этим не ограничиваются, алюмосиликат магния, крахмалы, такие как кукурузный, пшеничный или рисовый крахмал, желатин, метилцеллюлозу, натрий карбоксиметилцеллюлозу и/или поливинилпирролидон, и, если желательно, фармацевтически приемлемые разрыхлители, включая, но этим не ограничиваясь, крахмалы, агар, альгиновую кислоту или ее соль, такую как альгинат натрия, и/или шипучие смеси, или адсорбенты, красители, ароматизаторы и подсластители. Также можно использовать соединения по настоящему изобретению в виде парентерально вводимых композиций или в виде инфузионных растворов. Фармацевтические композиции могут быть стерилизованы и/или могут содержать инертные наполнители, например, консерванты, стабилизаторы, смачивающие соединения и/или эмульгаторы, солюбилизаторы, соли для регулирования осмотического давления и/или буферы.
В частности, терапевтически эффективное количество каждого компонента в комбинации по изобретению может быть введено одновременно или последовательно и в любом порядке, и компоненты могут быть введены раздельно или в виде комбинации установленного состава. Например, способ профилактики или лечения пролиферативных заболеваний в соответствии с изобретением может включать: (i) введение первого агента (a) в свободном виде или в виде или фармацевтически приемлемой соли; и (ii) введение агента (b) в свободном виде или в виде фармацевтически приемлемой соли, одновременно или последовательно в любом порядке, в объединенных терапевтически эффективных количествах, предпочтительно, в синергетически эффективных количествах, например, в суточных или дробных дозах, соответствующих описанным, в данном документе количествах. Отдельные компоненты комбинации по изобретению могут быть введены раздельно в различное время при проведении терапии или параллельно в виде раздельных или единых форм комбинации. Кроме того, термин введение также охватывает применение пролекарства компонента комбинации, которое преобразуется in vivo в непосредственно компонент комбинации. Настоящее изобретение, таким образом, следует рассматривать как охватывающее все такие режимы одновременного или чередующегося лечения, и термин ʺвведениеʺ следует интерпретировать соответствующим образом.
Эффективная дозировка каждого агента (a) или агента (b) комбинации компонентов, используемых в комбинации по изобретению, может меняться в зависимости от используемого конкретного соединения или фармацевтической композиции, способа введения, состояния, подвергаемого лечению, тяжести состояния, подвергаемого лечению. Таким образом, режим дозирования комбинации по изобретению выбирают в соответствии с целым рядом факторов, включая тип, вид, возраст, вес, пол и состояние здоровья пациента; тяжесть состояния, подлежащего лечению; путь введения; функционирование почек и печени у пациента; и конкретное используемое соединение. Лечащий врач, клиницист или ветеринар могут легко определить и прописать эффективное количество лекарственного средства, необходимое для предотвращения и пресечения или остановки прогрессирования заболевания. Оптимальная точность в достижении концентрации лекарственного средства в пределах диапазона, обеспечивающего эффективность требований режима, основывается на кинетике доступности лекарственного средства к мишеневым участкам. Это включает в себя рассмотрение вопроса о распределении, равновесии и вывода лекарственного средства.
Для целей настоящего изобретения терапевтически эффективная доза обычно будет представлять собой общую суточную дозу, вводимую хозяину в виде одной или разделенных доз. Соединение формулы (I) может быть введено хозяину в суточной дозе в интервале, например, от примерно 0,05 до примерно 50 мг/кг массы тела реципиента, предпочтительно, примерно 0,1-25 мг/кг массы тела реципиента, более предпочтительно, от примерно 0,5 до 10 мг/кг массы тела реципиента. При введении человеку весом 70 кг интервал дозирования соединения формулы (I) был бы, наиболее предпочтительно, примерно 35-700 мг в сутки. Агент (b) может быть введен хозяину в суточной дозе в интервале, например, от примерно 0,001 до 1000 мг/кг массы тела реципиента и, более предпочтительно, от 1,0 до 30 мг/кг массы тела реципиента. Композиции единиц дозировок могут содержать такие количества их дольных единиц, чтобы восполнить суточную дозу.
Следующим преимуществом является то, что могут быть использованы уменьшенные дозы активных ингредиентов комбинации по изобретению, например, необходимые дозы являются часто меньше, но также и применяются реже, или могут быть использованы для того, чтобы уменьшить частоту проявлений побочных эффектов. Это соответствует желаниям и требованиям пациентов, проходящих лечение.
Комбинация соединения формулы (I) и ингибитора Hsp90 может быть отдельной или объединенной, по меньшей мере, с одним другим фармацевтически активным соединением для использования при этих патологиях. Указанные активные соединения могут быть объединены в том же самом фармацевтическом препарате или представлены в виде ʺнабора компонентовʺ объединенных препаратов, в том смысле, что компоненты комбинации могут быть дозированы независимо или с использованием других комбинаций определенных составов с отличающимся количеством компонентов комбинации, то есть, одновременно или в различные моменты времени. Части набора компонентов могут быть введены, например, одновременно или в хронологическом порядке, то есть в различные моменты времени с равными и или различными интервалами времени для любой части набора компонентов. Неограничивающие примеры соединений, которые могут быть указаны для использования в комбинации сочетания соединения формулы (I) и, по меньшей мере, одного ингибитора Hsp90, представляют собой химиотерапевтические лекарственные средства, такие как анастрозол, гидрохлорид доксорубицина, флутамид, дексаметазон, доцетаксел, цисплатин, паклитаксел и т.д. Кроме того, комбинация соединения пиримидиламинобензамида и ингибитора Hsp90 может быть объединена с другими ингибиторами передачи сигнала или другими онкоген-мишеневыми лекарственными средствами с ожиданием, что это приведет к значительный синергии.
Комбинация по настоящему изобретению является особенно полезной при лечении пролиферативных заболеваний. Термин ʺпролиферативное заболеваниеʺ включает, но этим не ограничивается, злокачественное новообразование, опухоль, гиперплазию, рестеноз, гипертрофию сердца, иммунное расстройство и воспаление.
Примеры пролиферативного заболевания, которое можно лечить комбинацией по настоящему изобретению, представляют собой, например, злокачественные новообразования, включая, например, саркому; рак легких; бронхов; простату; молочной железы (включая спорадические злокачественные новообразования молочной железы и страдающих болезнью Каудена); поджелудочной железы; двенадцатиперстной кишки или желудка; толстой кишки; прямой кишки; аденому ободочной и прямой кишки; рак щитовидной железы; печени; внутрипеченочных желчных протоков; гепатоцеллюлярный; надпочечников; желудка; глиому; глиобластому; рак эндометрия; почки; почечной лоханки; мочевого пузыря; тела матки; шейки матки; влагалища; яичников; множественную миелому; рак пищевода; лейкоз; острый миелобластный лейкоз; хронический миелолейкоз; лимфолейкоз; миелоидный лейкоз; рак мозга; полости рта и глотки; гортани; тонкого кишечника; неходжкинскую лимфому; меланому; ворсинчатую аденому толстой кишки; неоплазию; неоплазию эпителиального характера; лимфомы; карциному молочной железы; базально-клеточную карциному; плоскоклеточный рак; актинический кератоз; опухоли шеи или головы; истинную полицитемию; эссенциальную тромбоцитемию; миелофиброз с миелоидной метаплазией; и болезнь Вальденстрема.
Далее примеры включают истинную полицитемию, эссенциальную тромбоцитемию, миелофиброз с миелоидной метаплазией, астму, COPD, ARDS, синдром Леффлера, эозинофильную пневмонию, паразитическое (в частности, метазоа) заражение (включая тропическую эозинофилию), бронхолегочный аспергиллез, узелковый полиартериит (включая синдром Чардж-Стросса), эозинофильную гранулему, связанные с эозинофилами расстройства, влияющие на дыхательные пути, вызванные реакцией на лекарственное средство, псориаз, контактный дерматит, атопический дерматит, очаговую алопецию, мультиформную эритему, герпетиформный дерматит, склеродермию, витилиго, аллергический ангиит, крапивницу, буллезный пемфигоид, красную волчанку, пемфизус, приобретенный буллезный эпидермолиз, аутоиммунные гематологические расстройства (например, гемолитическую анемию, апластическую анемию, анемию при недостаточности красных кровяных телец и идиопатическую тромбоцитопению), системную красную волчанку, полихондрит, склеродермию, гранулематоз Вегенера, дерматомиозит, хронический активный гепатит, миастению, синдром Стивена-Джонсона, идиопатическую спру, аутоиммунное воспалительное заболевание кишечника (например неспецифический язвенный колит и болезнь Крона), эндокринную офтальмопатию, болезнь Грейвса, саркоидоз, альвеолит, хронический аллергический пневмонит, рассеянный склероз, первичный билиарный цирроз, увеит (передний и задний), интерстициальный фиброз легких, псориатический артрит, гломерулонефрит, сердечно-сосудистые заболевания, атеросклероз, гипертонию, тромбоз глубоких вен, инсульт, инфаркт миокарда, нестабильную стенокардию, тромбоэмболию, эмболию легочной артерии, тромболитические заболевания, острую артериальную ишемию, периферийные тромботические окклюзии, ишемическую болезнь сердца, реперфузионные травмы, ретинопатию, такую как диабетическая ретинопатия или гипербарическая кислород-индуцированная ретинопатия, и состояния, характеризующиеся повышенным внутриглазным давлением или секрецией глазной водянистой влаги, такие как глаукома.
В одном варианте осуществления изобретения пролиферативное заболевание, которое можно лечить комбинацией по настоящему изобретению, представляет собой злокачественное новообразование, которое можно успешно лечить путем ингибирования HSP90 и/или PI3K, включая, например, рак желудка, легких и бронхов; простаты; груди; поджелудочной железы; толстой кишки; прямой кишки; щитовидной железы; печени и внутрипеченочных желчных протоков; почек и почечной лоханки; мочевого пузыря; тела матки; шейки матки; яичников; множественную миелому; рак пищевода; острый миелобластный лейкоз; хронический миелолейкоз; лимфолейкоз; миелоидный лейкоз; рак мозга; полости рта и глотки; гортани; тонкого кишечника; неходжкинскую лимфому; меланомы; и ворсинчатую аденому толстой кишки.
В одном варианте осуществления изобретения пролиферативное заболевание, которое можно лечить комбинацией по настоящему изобретению, представляет собой рак пищевода, двенадцатиперстной кишки или желудка.
Когда упоминаются опухоль, опухолевое заболевание, саркома, карцинома или злокачественное новообразование, также альтернативно подразумеваются метастазы в основном органе или ткани и/или в любом другом месте, независимо от расположения опухоли и/или метастазов.
Комбинация по настоящему изобретению особенно полезна при лечении пролиферативных заболеваний, в частности, злокачественных опухолей и других злокачественных новообразований, опосредованных фосфатидилинозит 3-киназой (PI3K), в частности, альфа-субъединицей PI3K, и/или Hsp90 (или зависящие от PI3K или Hsp90). Пролиферативные заболевания могут включать такие, которые показывают сверхэкспрессию или амплификацию PI3K альфа, соматическую мутацию PIK3CA или мутации зародышевой линии, или соматическую мутацию PTEN или мутации и транслокации p85α, которые служат для до-регулирования комплекса p85-P110.
В одном варианте осуществления изобретения настоящее изобретение относится к способу лечения пролиферативного расстройства, включающему введение указанному субъекту терапевтически эффективного количества соединения формулы (I), выбранного из 2-амида 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение A) или его фармацевтически приемлемой соли и, по меньшей мере, одного ингибитора Hsp90, выбранного из производного гелданамицина, танеспимицина (17-аллиламино-17-деметоксигелданамицин) (известного также как KOS-953 и 17-AAG); радицикола; метансульфоната 6-хлор-9-(4-метокси-3,5-диметилпиридин-2-илметил)-9H-пурин-2-амина (известного также как CNF2024); IPI504; SNX5422; этиламида 5-(2,4-дигидрокси-5-изопропилфенил)-4-(4-морфолин-4-илметилфенил)изоксазол-3-карбоновой кислоты (AUY922); и (R)-2-амино-7-[4-фтор-2-(6-метоксипиридин-2-ил)фенил]-4-метил-7,8-дигидро-6H-пиридо[4,3-d]пиримидин-5-она (HSP990) или его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к набору, содержащему соединение формулы (I), в частности, 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты, или его фармацевтически приемлемую соль, и, по меньшей мере, один ингибитор Hsp90 или его фармацевтически приемлемую соль, и упаковочный вкладыш или этикетку для обеспечения инструкцией по лечению пролиферативного заболевания.
Кроме того, настоящее изобретение относится к набору, содержащему соединение формулы (I), в частности, 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты, или его фармацевтически приемлемую соль, и упаковочный вкладыш или этикетку для обеспечения инструкцией по лечению пролиферативного заболевания путем одновременного введения, по меньшей мере, одного ингибитора Hsp90 или его фармацевтически приемлемой соли.
Следующие примеры иллюстрируют изобретение, описанное выше; они, однако, не предназначены для ограничения объема изобретения каким-либо образом. Положительные эффекты фармацевтической комбинации по настоящему изобретению могут также быть определены с помощью других исследовательских моделей, известных специалисту в данной области техники.
Пример 1 - Эффект соединения на ксенотрансплантатной модели рака желудка HGC-27 на самках бестимусных голых мышей
Эксперименты проводились на самках бестимусных голых мышей HSD:Athymic Nude-FoxN1nu приблизительно 8-12 недельного возраста в начале лечения. Все животные были получены от фирмы Harlan (South Easton, MA) и размещены при оптимальных гигиенических условиях в клетках microisolator с фильтруемым воздухозаборником (максимум 5 животных на клетку) со свободным доступом к продуктам питания и воде.
Клетки HGC-27, которые являлись клетками карциномы желудка человека с мутацией PIK3CA (c1624G>А, p.E542K) и PTEN null, выращивали в культуральной среде MEM, содержащей 1% заменимой аминокислоты с 10% инактивированной нагреванием FCS, и инкубировали при 37°С в 5% СО2 в увлажненной атмосфере. Реагенты клеточных культур получали от фирмы Invitrogen (Carlsbad, CA).
Опухоли HGC-27 прививали in vivo путем инъекции 5×106 клеток в 200 мкл (100 мкл PBS+100 мкл Matrigel) (Cat #354234, BD Bioscience, Bedford, MA) подкожно в правый бок животных. Проведение экспериментов начинали, когда опухоли достигали среднего размера около 230 мм3 (12-ый день после инъекции клеток).
Соединение A готовили в 0,5%-ной метилцеллюлозе (MC). 80 мг соединения A добавляли к 16 мл 0,5% MC, затем перемешивали на вортексе и воздействовали ультразвуком на водяной бане сонификатором в течение 1 ч с получением гомогенной суспензии 5 мг/мл. Для разбавления 5 мг/мл раствора до 2,5 мг/мл и 1,25 мг/мл для дозирования использовали 0,5% MC. Соединение A или растворитель вводили перорально объемом 10 мл/кг. Эта суспензия стабильна в течение одной недели при комнатной температуре.
AUY922 мезилат готовили в D5 воде. Поправочный коэффициент для свободного основания соединения был 1,21. Для получения 50 мг/кг свободного основания AUY922, 60,5 мг AUY922 мезилата добавляли к 5,0 мл D5 воды, и воздействовали ультразвуком на водяной бане сонификатором до тех пор, пока раствор не становился прозрачным. AUY922 вводили внутривенно (i.v.) объемом 5 мл/кг два раза в неделю. AUY922 каждый раз готовили свежим.
Объемы опухолей измеряли с помощью штангенциркуля и определяли по формуле: длина × диаметр2 × π/6. Противоопухолевую активность выражали как T/C %, которое определяли по формуле: (среднее изменение объема опухоли обработанных животных/среднее изменение объема опухоли контрольных животных)×100. Регрессии (%) рассчитывали по формуле ((средний объем опухоли в конце обработки-средний объем опухоли в начале обработки)/средний объем опухоли в начале обработки)×100. Массу тела и объемы опухолей регистрировали два раза в неделю.
В случае необходимости данные представляли в виде средних ± SEM. Для всех исследований уровень статистической значимости устанавливали при p<0,05. Для объемов опухолей сравнения между обработанными группами и контрольными группами, обработанными растворителем, выполняли с использованием однофакторного ANOVA с последующим тестом Даннетта. Сравнения объемов опухолей между обработанными группами выполняли с использованием дисперсионного анализа Краскела-Уоллеса однофакторного ANOVA с апостериорным множественным сравнением Стьюдента-Ньюмана-Келса или критерия Данна. В первом эксперименте соединение A вводили перорально ежедневно в подкожные ксенотрансплантаты HGC-27 мышей с опухолями в дозах 12,5 мг/кг, 25 мг/кг и 50 мг/кг. Контроли растворителем заключались в ежедневном введении животным 10 мл/кг 0,5% MC перорально и внутривенном введении 5 мл/кг D5W два раза в неделю.
Соединение A, вводимое перорально в количестве 12,5 мг/кг, 25 мг/кг и 50 мг/кг один раз в день, давало T/C % как 39,4%, 35,5% и 7,1%, соответственно (фиг. 1). AUY922, вводимое в количестве 50 мг/кг свободного основания два раза в неделю, давало T/C (%) как 60,5% (фиг. 3). Комбинация соединения A в количестве 12,5 мг/кг с AUY922 в количестве 50 мг/кг свободного основания приводила к T/C (%) как 16,6% (фиг. 3). Комбинация соединения A в количестве 25 мг/кг с AUY922 в количестве 50 мг/кг свободного основания приводила к 29,48%-ной регрессии опухоли (фиг. 5); и комбинация соединения A в количестве 50 мг/кг с AUY922 в количестве 50 мг/кг свободного основания приводила к 85,1%-ной регрессии опухоли (фиг. 7). 23-ый день являлся последним днем измерения опухоли.
Соединение A производило статистически значимый противоопухолевый эффект при дозах 50 мг/кг по сравнению с группой, обработанной растворителем (p<0,05, ANOVA, апостериорное множественное сравнение критерия Даннета). (Смотри фигуру 1). Соединение A, водимое перорально в дозах 12,5, 25 и 50 мг/кг один раз в день, давало среднее изменение объема опухоли в 515±85 мм3, 465±111 мм3 и 93±77 мм3(p<0,05, ANOVA и апостериорное множественное сравнение критерия Даннета), соответственно, по сравнению с группой, обработанной растворителем (среднее изменение объема опухоли составляло 1309±169 мм3) (Смотри фигуру 1). AUY922 вызывало среднее изменение объема опухоли в 792,2±159 мм3.
Соединение A, вводимое перорально в дозах 12,5, 25 и 50 мг/кг один раз в день в комбинации с AUY922 в дозе 50 мг/кг два раза в неделю, давало среднее изменение объема опухоли в 217±68 мм3 (p<0,05, в сравнении с растворителем и обоими отдельными агентами в анализе Краскела-Уоллеса ANOVA с апостериорным множественным сравнением Стьюдента-Ньюмана-Келса), -68±36 мм3(p<0,05, в сравнении с растворителем и обработанной AUY922 группой в анализе Краскела-Уоллеса однофакторной ANOVA с апостериорным критерием Даннета), и -196±21 мм3(p<0,05, в сравнении с растворителем и обоими отдельными агентами в анализе Краскела-Уоллеса однофакторной ANOVA с апостериорным множественным сравнением Стьюдента-Ньюмана-Келса), соответственно (смотри фигуры 3, 5 и 7).
Соединение A хорошо переносилось в дозах 12,5, 25 мг/кг и 50 мг/кг, как показывало отличие массы тела у группы, обработанной растворителем (7,8±1,4%), и группы, обработанной соединением A (5,3±1,4%, 2,2±1,1% и -1,1±1,6%, соответственно). В группе, обработанной AUY922, было показано изменение массы тела как 6,6±2,6%.
Соединение A, вводимое перорально в дозах 12,5, 25 и 50 мг/кг один раз в день в комбинации с AUY922 в дозе 50 мг/кг два раза в неделю, переносилось при всех дозах (0,9±1,5%, -3,0±2,4%, 8,06±2,4%) (смотри фигуры 4, 6 и 8).
Пример 2 - Эффект соединения на ксенотрансплантатной модели рака желудка HGC-27 у самок бестимусных голых мышей
Процедуру, описанную в примере 1, проводили со следующими модификациями:
Обработку начинали на 20-ый день после имплантации опухолевых клеток из 5 миллионов клеток HCG-27, когда средний объем опухоли составлял 316 мм3 (164-485 мм3). Животным вводили либо: (a) контроли растворителей, состоящие из суточной дозы введения животным 10 мл/кг 0,5% MC перорально и внутривенного введения 5 мл/кг D5W два раза в неделю, (b) 50 мг/кг AUY922, 2 q.w., внутривенно, (c) соединение A, либо 25 мг/кг, либо 50 мг/кг, q.d., перорально, (d) комбинацию AUY922, 50 мг/кг, 2 q.w., внутривенно, и соединение A, 25 мг/кг, q.d., перорально, либо (e) комбинацию AUY922, 50 мг/кг, 2 q.w., внутривенно, и соединение A, 50 мг/кг, q.d., перорально. Обработка продолжалась в течение 14 дней.
В данном эксперименте соединение A в дозах 25 и 50 мг/кг приводило к значительному ингибированию роста опухоли с 11% T/C (p<0,05 s. растворитель) и 10% T/C (p<0,05 vs. растворитель), соответственно. AUY922 в дозе 50 мг/кг приводил к 57% T/C, что не являлось значительным по сравнению с группой, обработанной растворителем. Соединение A в дозах 25 и 50 мг/кг в комбинации с AUY922 при дозе 50 мг/кг приводило к -11% T/T0 (p<0,05 vs. групп, обработанной растворителем или AUY922) и -57% T/T0 (p<0,05 vs. групп, обработанных растворителем, AUY922 или соединением A, соответственно.
Пример 3 - Эффект соединения на ксенотрансплантатной модели рака желудка NCI-N87 у самок бестимусных голых мышей
Эксперименты проводили на самках мышей Hsd:Athymic Nude-nu CPB приблизительно 10-12 недельного возраста в начале обработки. Все животные были получены от фирмы Harlan (Winkelmann, Germany) и содержались в оптимальных гигиенических условиях в клетках типа Makrolon типа III (максимум 5 животных на клетку) со свободным доступом к пище и воде.
Клетки NCI-N87, которые являются клетками карциномы желудка человека, выращивали в культуральной среде DMEM, содержащей 4,5 г/л глюкозы, дополненной 10% инактивированной нагреванием FCS, 2 мМ L-лютамина, 1 мМ пирувата натрия. Клетки инкубировали при температуре 37°C в увлажненной атмосфере 5%-ного CO2. Клетки выделяли с трипсином (0,25% масс./объем)-EDTA (0,53 мМ), вновь суспендировали в культуральной среде (с добавками) и подсчитывали с помощью системы Casy®. Реагенты клеточных культур получали от фирмы BioConcept (Allschwil, Switzerland).
Опухоли NCI-N87 прививали путем инъекции от 8×106 до 1×107 клеток (в HBSS, содержащей 50% объем/объем Matrigel) подкожно иглой калибра 23. Когда опухоли были привиты и достигали размеров между 180 и 210 мм3, животных распределяли на обрабатываемые группы и начинали обработку.
Соединение A готовили в NMP/PEG300/Solutol HS15/воде (10:30:20:40% объем/объем). Соединение сначала полностью растворяли в NMP, и воду добавляли непосредственно перед введением животным. Соединение A или растворитель вводили перорально объемом 10 мл/кг. Данная суспензия являлась стабильной в течение одной недели при комнатной температуре.
AUY922 мезилат готовили в D5 воде (5%-ая глюкоза в воде). Все дозы AUY922 относились к эквиваленту свободного основания. AUY922 вводили объемом 10 мл/кг два раза в неделю.
В случае необходимости данные представлялись в виде средних ±SEM. Для всех исследований, уровень статистической значимости устанавливали при p<0,05. Для объемов опухолей сравнения между обработанными группами и контрольными группами, обработанными растворителем, выполняли с использованием ANOVA с последующим тестом Даннетта. Парные сравнения проводили с использованием однофакторного ANOVA с последующим критерием Тьюки. Уровень значимости изменения массы тела в пределах группы между началом и концом периода лечения определяли с использованием парного критерия Стьюдента. Сравнение массы тела между обрабатываемой группой и группой с контролем растворителем осуществляли с использованием однофакторного ANOVA с апостериорным критерием Даннета. Расчеты выполняли с использованием программы GraphPad Prism 4 для Windows (GraphPad Software Inc.).
Кроме того, приближение взаимодействий лекарственного средства осуществляли с использованием способа, описанного Clarke R., “Issues in experimental design и endpoint analysis in the study of experimental cytotoxic агенты in vivo in breast cancer и other models”, Breast Cancer Res. Treat., 46, 255-78 (1997). Это применимо к ΔTV (объем опухоли).
Сравнения объемов опухолей между обработанными группами выполняли с использованием дисперсионного анализа Краскела-Уоллеса однофакторного ANOVA с апостериорным множественным сравнением Стьюдента-Ньюмана-Келса или критерия Данна.
Первый эксперимент:
Самок бестимусных голых мышей обрабатывали перорально один раз в день 50 мг/кг соединения A, отдельно или в комбинации с 50 мг/кг AUY922, вводимого внутривенно два раза в неделю. Контроли растворителей включали получение животными суточного перорального введения смеси NMP/PEG300/Solutol HS15/вода (10:30:20:40% объем/объем), в добавление к внутривенному введению 10 мл/кг раствора 5%-ой глюкозы в воде.
В виде единственного агента соединение A давало статистически значительный противоопухолевый эффект с T/C как 4,2% (p<0,05, однофакторный ANOVA, апостериорный критерий Данетта) и среднее изменение объема опухоли (мм3 ± SEM) составляло -15,1±21,4. AUY922 (50 мг/кг), используемый в виде единственного агента, приводил к 7% регрессии опухоли, а когда был объединен с соединением A, давал 72,3% регрессии. Оба эффекта значительно отличались от контролей растворителей (p<0,05, ANOVA).
Контроли растворителей приводили к среднему изменению объема опухоли (мм3 ± SEM) как 248,9±20,4. AUY922 (50 мг/кг), используемый в виде единственного агента, давал среднее изменение объема опухоли равное -15,1±21,4, и соединение A, используемое в виде единственного агента, давало среднее изменение объема опухоли (мм3 ± SEM) равное 1,4±18,8. Комбинация AUY922 и соединения A давала среднее изменение объема опухоли (мм3 ± SEM) равное -155,8±14,7. Кроме того, группа, обработанная комбинацией, существенно отличалась от их обоих, соединения A и AUY922, вводимых в виде единственных агентов (p<0,05, однофакторный ANOVA, апостериорный критерий Тьюки).
Кроме того, анализ возможных взаимодействий соединений по способу, описанному Clarke R. (1997), указывал на синергетический противоопухолевый эффект комбинации AUY922 и соединения A:

Пример 5 - Эффект соединения A на ксенотрансплантатной модели меланомы A375 у самок бестимусных голых мышей
Эксперименты проводили на бестимусных голых самках мышей Harlan Hsd:Npa весом приблизительно 20-25 г. A375 опухоли прививали путем инъекции 4×106 клеток A375, которые представляли собой клетки меланомы, подкожно в спину мышей. Через (10) дней после прививки образовывалась опухоль. Приблизительно через 30 дней после прививки отбирали 32 животных и разделяли на 4 обрабатываемые группы (n=8).
Контроль плацебо готовили в 1% карбоксиметилцеллюлозе (CMC) для введения один раз в день перорально (плацебо 1) и в виде 10 мл/кг 2,5 мл глюкозы для введения внутривенно или интраперитонеально два раза в неделю (плацебо 2).
Соединение A готовили в дозе 40 мг/кг соединения A путем растворения в 1% (масс./объем) карбоксиметилцеллюлозе (CMC), содержащей 5% (объем/объем) Tween-80. Соединение A или растворитель вводили перорально в дозе объемом 10 мг/мл один раз в день.
AUY922 готовили в D5 воде (5%-ая глюкоза в воде). Все дозы AUY922 относятся к эквиваленту свободного основания. AUY922 вводили внутривенно объемом 10 мг/мл и в дозе 50 мг/кг, два раза в неделю.
Каждую группу мышей обрабатывали в течение 11 дней одним из следующих: (a) плацебо 1 и плацебо 2 (группа 1), (b) 40 мг/кг соединения A перорально один раз в день (группа 2), (c) 50 мг/кг AUY922 внутривенно два раза в неделю (группа 3), (e) комбинация 40 мг/кг соединения A перорально один раз в день и 50 мг/кг AUY922 внутривенно два раза в неделю (группа 4).
В случае необходимости данные представляли в виде средних ±SEM. Для всех исследований уровень статистической значимости устанавливали при p<0,05. Сравнения объемов опухолей между обработанными группами осуществляли с использованием одностороннего дисперсионного анализа Краскела-Уоллиса с ранговым дисперсионным анализом или критерием Тьюки.
Полученные в соответствии с вышеописанным методом эксперимента данные частичного роста опухоли и изменения массы тела обработанных мышей показаны на фигуре 9.
Пример 6 - Эффект соединения A на ксенотрансплантатной модели меланомы A375 у самок бестимусных голых мышей
Эксперименты проводили на самках бестимусных голых мышей Harlan Hsd:Npa весом приблизительно 20-25 г. Опухоли A375 прививали путем инъекции 4×106 клеток A375, которые представляли собой клетки меланомы, подкожно в спину мышей. Через (10) дней после прививки образовывалась опухоль. Приблизительно через 30 дней после прививки отбирали 32 животных и разделяли на 4 обрабатываемые группы (n=8).
Контроль плацебо готовили в 1% карбоксиметилцеллюлозе (CMC) для введения один раз в день перорально (плацебо 1) и в виде 10 мл/кг 2,5 мл глюкозы для введения внутривенно или интраперитонеально два раза в неделю (плацебо 2).
Соединение A готовили в дозе 40 мг/кг соединения A путем растворения в 1% (масс./объем) карбоксиметилцеллюлозе (CMC), содержащей 5% (объем/объем) Tween-80. Соединение A или растворитель вводили перорально в дозе объемом 10 мг/мл один раз в день.
AUY922 готовили в D5 воде (5%-ая глюкоза в воде). Все дозы AUY922 относились к эквиваленту свободного основания. AUY922 вводили внутривенно объемом 10 мг/мл и в дозе 50 мг/кг, два раза в неделю.
Каждую группу мышей обрабатывали в течение 11 дней одним из следующих: (a) плацебо 1 и плацебо 2 (группа 1), (b) 40 мг/кг соединения A перорально один раз в день (группа 2), (c) 50 мг/кг AUY922 внутривенно два раза в неделю (группа 3), (d) комбинация 40 мг/кг соединения A перорально один раз в день и 50 мг/кг AUY922 внутривенно два раза в неделю (группа 4).
В случае необходимости данные представлялись в виде средних ± SEM. Для всех исследований уровень статистической значимости устанавливали при p<0,05. Сравнения объемов опухолей между обработанными группами проводили с использованием одностороннего дисперсионного анализа Краскела-Уоллиса с ранговым дисперсионным анализом или критерием Тьюки.
Полученные в соответствии с вышеописанным методом эксперимента данные среднего частичного роста опухоли и изменения массы тела обработанных мышей показаны на фигуре 9.
Реферат
Группа изобретений относится к фармации и онкотерапии. Предложено: фармацевтическая комбинация для лечения меланомы, содержащая: (a) 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметилэтил)пиридин-4-ил]тиазол-2-ил}амида) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его фармацевтически приемлемую соль; и (b) по меньшей мере один ингибитор Hsp90, представляющий собой этиламид 5-(2,4-дигидрокси-5-изопропилфенил)-4-(4-морфолин-4-илметил-фенил)изоксазол-3-карбоновой кислоты (AUY922), или его фармацевтически приемлемую соль; применение указанной комбинации для получения лекарственного средства для лечения меланомы и соответствующий способ лечения меланомы. Технический результат: синергизм действия соединений комбинации в снижении объёма опухоли. 3 н. и 1 з.п. ф-лы, 9 ил., 2 табл., 6 пр.
Комментарии