Использование производных 2-карбоксамид-циклоамино мочевины в лечении egfr-зависимых заболеваний или заболеваний с приобретенной резистентностью к агентам, нацеленным на члены egfr-семейства - RU2589695C2
Код документа: RU2589695C2
Чертежи
Описание
Область изобретения
Настоящее изобретение относится к новому применению конкретных производных 2-карбоксамид-циклоамино-мочевины для лечения заболеваний, зависимых от рецептора эпидермального фактора роста (EGFR) (включающего в себя EGFR1, также называемого HER1 или Erb-B1; EGFR2, также называемого HER2 или Erb-B2; EGFR3, также называемого HER3 или Erb-B3, или EGFR4), или заболеваний с приобретенной резистентностью к агентам, нацеленным на члены семейства EGFR, также настоящее изобретение относится к использованию указанных соединений для производства фармацевтической композиции для лечения указанных заболеваний, к комбинациям указанных соединений с EGFR-модуляторами в целях указанного использования, к способам лечения указанных заболеваний с помощью указанных соединений, и к фармацевтическим композициям для лечения указанных заболеваний, которые содержат указанные соединения в качестве единственных агентов или в комбинации, в частности, с EGFR-модулятором.
Уровень изобретения
Соматические мутации в тирозинкиназовом домене EGFR связывают с клиническим ответом на ингибитор тирозинкиназы EGFR, например, на гефитиниб (Иресса®) или эрлотиниб (Тарцева®) (Paez et al., 2004, EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy, Science, vol.304, 1497-1500). Семейство рецепторов эпидермального фактора роста состоит из четырех подтипов, включающих в себя подтип, являющийся членом семейства ErbB-рецепторов, которые включают в себя EGFR1 (также называемый HER1 или Erb-B1); EGFR2 (также называемый HER2 или Erb-B2); и EGFR3 (также называемый HER3 или Erb-B3); и EGFR4, которые представляют собой трансмембранные белки. Приобретенная резистентность к EGFR-модуляторам возникает у тех пациентов, у которых изначально наблюдался клинический ответ на лечение, но впоследствии выявлено прогрессирование опухолей. Рефракторный ответ на ингибиторы киназы EGFR является примером вторичной мутации резистентности T790M (Kobayashi et al., 2005; EGFR mutation and resistance of non-small cell lung cancer to gefitinib, N. Engl. J. Med., Vol.352, 786-792), что сопоставимо с мутацией (мутациями) резистентности, наблюдаемыми с препаратами Gleevec/Гливек или дазатиниб при хроническом миелолейкозе (ХМЛ) (Gorre et al.; 2002; Bcr-Abl point mutants isolated from patients with imatinib mesylate resistant chronic leukemia remain sensitive to inhibitors of the Bcr-Abl chaperone heat shock protein 90, Blood, vol.100, 3041-3044), или у больных с гастроинтестинальными стромальными опухолями (GIST) (Antonescu et al.; 2005; Acquired resistance to Imatinib in gastrointestinal stromal tumors occurs through secondary gene mutation, Clin. Cancer Res., Vol.11, 4182-4190).
В литературе существуют доказательства наличия активации пути PI3K ниже по отношению к активированному EGFR. Таким образом, генетическая абляция каталитической субъединицы PI3K (p110) в мышиных эмбриональных фибробластах делает клетки устойчивыми к трансформации посредством активированных форм EGFR (Zhao et al.; 2006; The p110 alpha isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation, PNAS, vol.103, 16296-16300). В опухолях, чувствительных к ингибиторам EGFR, часто выявляют сверхэкспрессию партнера HER1 (EGFR1) и HER3 (ErbB-3), одного из четырех членов семейства EGFR, и это коррелирует с конститутивным рекрутингом и активацией PI3K (Engelman et al.; 2005; ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non small cell lung cancer cell lines, PNAS vol.102, 3788-3793; Sergina et al.; 2007; Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER 3; Nature; vol.445, 437-41). Генетические и биохимические характеристики опухолевых биопсий и опухолевых клеточных линий, несущих амплификацию EGFR и резистентность к ингибитору EGFR, выявили состояние конститутивной активации пути PI3K (Engelman et al.; 2006; Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR amplified lung cancer, The Journal of Clinical Investigation, vol.116, 2695-2706).
Было неожиданно обнаружено, что описанные в патенте WO 2010/029082 конкретные производные 2-карбоксамид-циклоамино-мочевины в качестве единственного агента и в комбинации с модуляторами киназы EGFR провоцируют сильное анти-пролиферативное действие и in vivo противоопухолевый ответ клеточных линий раковых клеток молочной железы и желудка с амплифицированными рецепторами EGFR и/или мутантными EGFR1. Таким образом, указанные соединения являются полезными при лечении EGFR-зависимого заболевания.
Сущность изобретения
Настоящее изобретение относится к использованию соединения формулы I (называемого в изобретении "Соединение I",
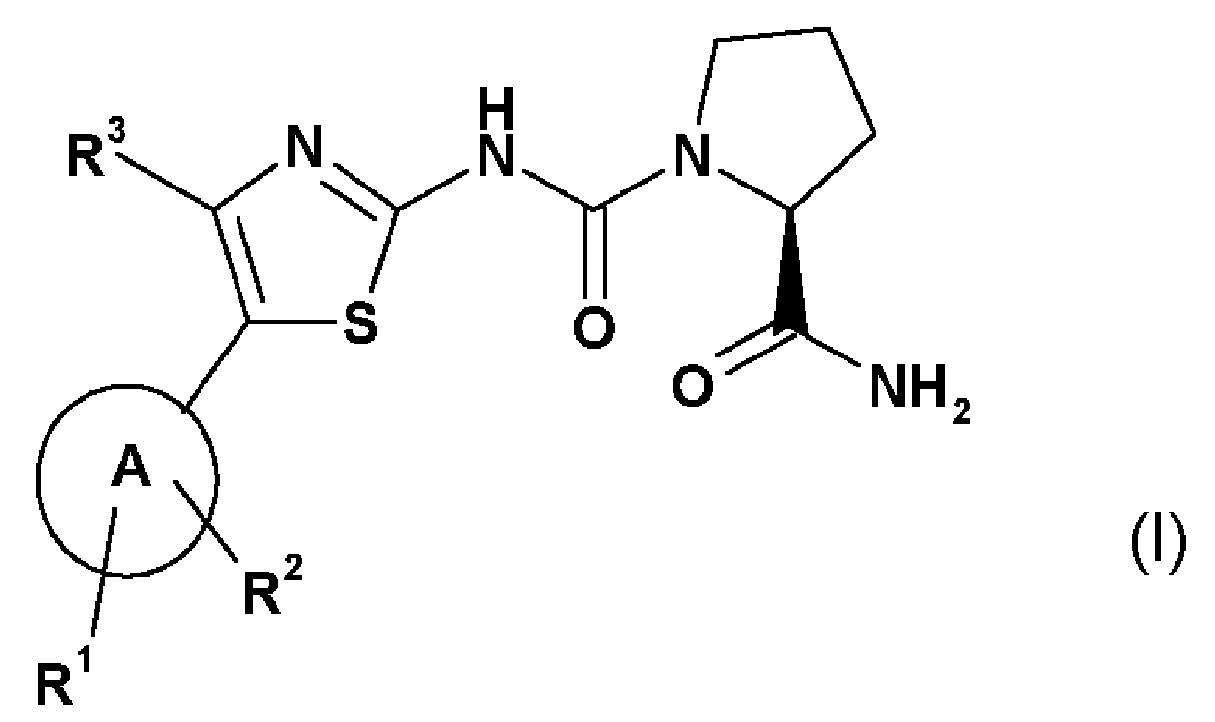
или его соли, где
А обозначает гетероарил, который выбирают из группы, состоящей из:
R1 представляет собой один из следующих заместителей: (1) незамещенный или замещенный, предпочтительно замещенный C1-C7-алкил, при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до девяти: дейтерий, фтор или из следующих остатков от одного до двух C3-C5-циклоалкил; (2) необязательно замещенный C3-C5-циклоалкил, при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до четырех: дейтерий, C1-C4-алкил (предпочтительно метил), фтор, циано, аминокарбонил; (3) необязательно замещенный фенил, при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до двух: дейтерий, галоген, циано, C1-C7-алкил, C1-C7-алкиламино, ди(C1-C7-алкил)амино, C1-C7-алкиламинокарбонил, ди(C1-C7-алкил)аминокарбонил, C1-C7-алкокси; (4) необязательно моно- или ди-замещенные амины, при этом указанные заместители независимо выбирают из следующих функциональных остатков: дейтерий, C1-C7-алкил (который является незамещенным или замещен одним или несколькими заместителями, выбираемыми из группы дейтерия, фтора, хлора, гидрокси), фенилсульфонил (который является незамещенным или замещен одним или несколькими, предпочтительно одним C1-C7-алкилом, C1-C7-алкокси, ди(C1-C7-алкил)амино-C1-C7-алкокси); (5) замещенный сульфонил, при этом указанный заместитель выбирают из следующих функциональных групп: C1-C7-алкил (который является незамещенным или замещен одним или несколькими заместителями, выбираемыми из группы дейтерия, фтора), пирролидино (который является незамещенным или замещен одним или несколькими заместителями, выбираемыми из группы дейтерия, гидрокси, оксо; в частности, замещен одной оксо-группой); (6) фтор, хлор;
R2 представляет собой водород;
R3 представляет собой (1) водород, (2) фтор, хлор, (3) необязательно замещенный метил,
при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до трех: дейтерий, фтор, хлор, диметиламино;
за исключением 2-амид 1-({5-[2-(трет-бутил)пиримидин-4-ил]-4-метил-тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты,
для лечения EGFR-зависимых заболеваний, в особенности злокачественных опухолей, или заболеваний, обусловленных приобретенной резистентностью к EGFR.
Настоящее изобретение также относится к применению соединения формулы I, определение которого приведено выше, или его соли для производства фармацевтической композиции для лечения EGFR-зависимого заболевания, или злокачественного новообразования, или заболевания с приобретенной резистентностью к EGFR-модулятору.
Настоящее изобретение дополнительно относится к применению соединения формулы I для лечения EGFR-зависимых заболеваний, или злокачественных новообразований, или заболеваний с приобретенной резистентностью к EGFR-модулятору, в комбинации с другими активными соединениями, например, с комбинационными партнерами, как описано в WO 2010/029082. Наиболее предпочтительными являются нацеливающие агенты семейства EGFR.
Настоящее изобретение также относится к комбинации, содержащей соединение формулы I и EGFR-модулятор, выбираемый из группы, состоящей из следующих агентов: гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIRW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональное антитело 806, анти-EGFR моноклональное антитело Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, Cdx110, IMC11F8, пертузумаб, трастузумаб, TDM1, Земаб®, Her2 вакцина PX 1041 и HSP90-ингибиторы CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922, и в каждом случае в упомянутой комбинации присутствуют активные ингредиенты в свободной форме или в форме соли, и необязательно присутствует по меньшей мере один фармацевтически приемлемый носитель, и упомянутая комбинация предназначена для одновременного, раздельного или последовательного применения в целях лечения EGFR-зависимых заболеваний, включающих в себя, например, немелкоклеточный рак легкого, рак головы и шеи, колоректальную карциному, рак молочной железы, злокачественные новообразования головного мозга, включающие в себя глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и/или рак яичников.
В другом варианте осуществления настоящее изобретение относится к способу лечения EGFR-зависимых заболеваний или злокачественных опухолей, предпочтительно злокачественных опухолей, которые стали резистентными к модуляторам EGFR-киназы в ходе лечения указанным EGFR-модулятором, и упомянутый способ содержит введение теплокровным животным, нуждающимся в таком введении, терапевтически эффективного количества конкретного производного 2-карбоксамид-циклоамино-мочевины с формулой I, в особенности предпочтительного соединения 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его фармацевтически приемлемой соли, в качестве единственного препарата или в комбинации с EGFR-модулятором.
В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции для лечения EGFR-зависимых заболеваний или заболеваний, которые стали резистентными в ходе лечения EGFR-модулятором, содержащим соединение формулы I, в особенности предпочтительное соединение 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его соль, и по меньшей мере один фармацевтически приемлемый носитель, в качестве единственного препарата или в комбинации с EGFR-модулятором.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы I, в особенности предпочтительного соединения 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его соли для лечения EGFR-зависимых заболеваний или заболеваний с приобретенной резистентностью в ходе лечения EGFR-модулятором.
Описание фигур
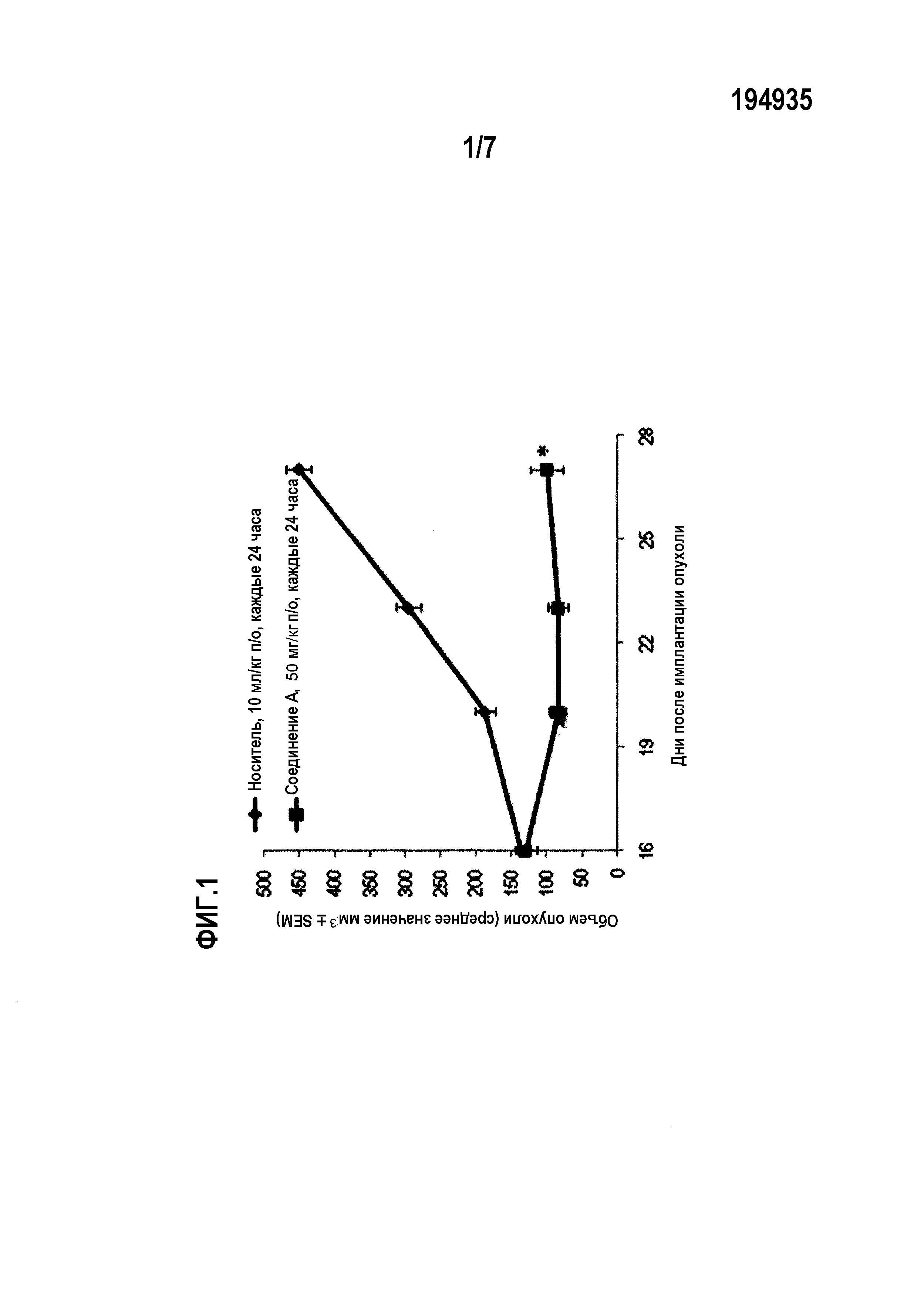
Фиг.1 показывает противоопухолевую активность соединений против мутантных PIK3CA и амплифицированных ErbB2 клеточной линии ВТ474 раковых клеток молочной железы.
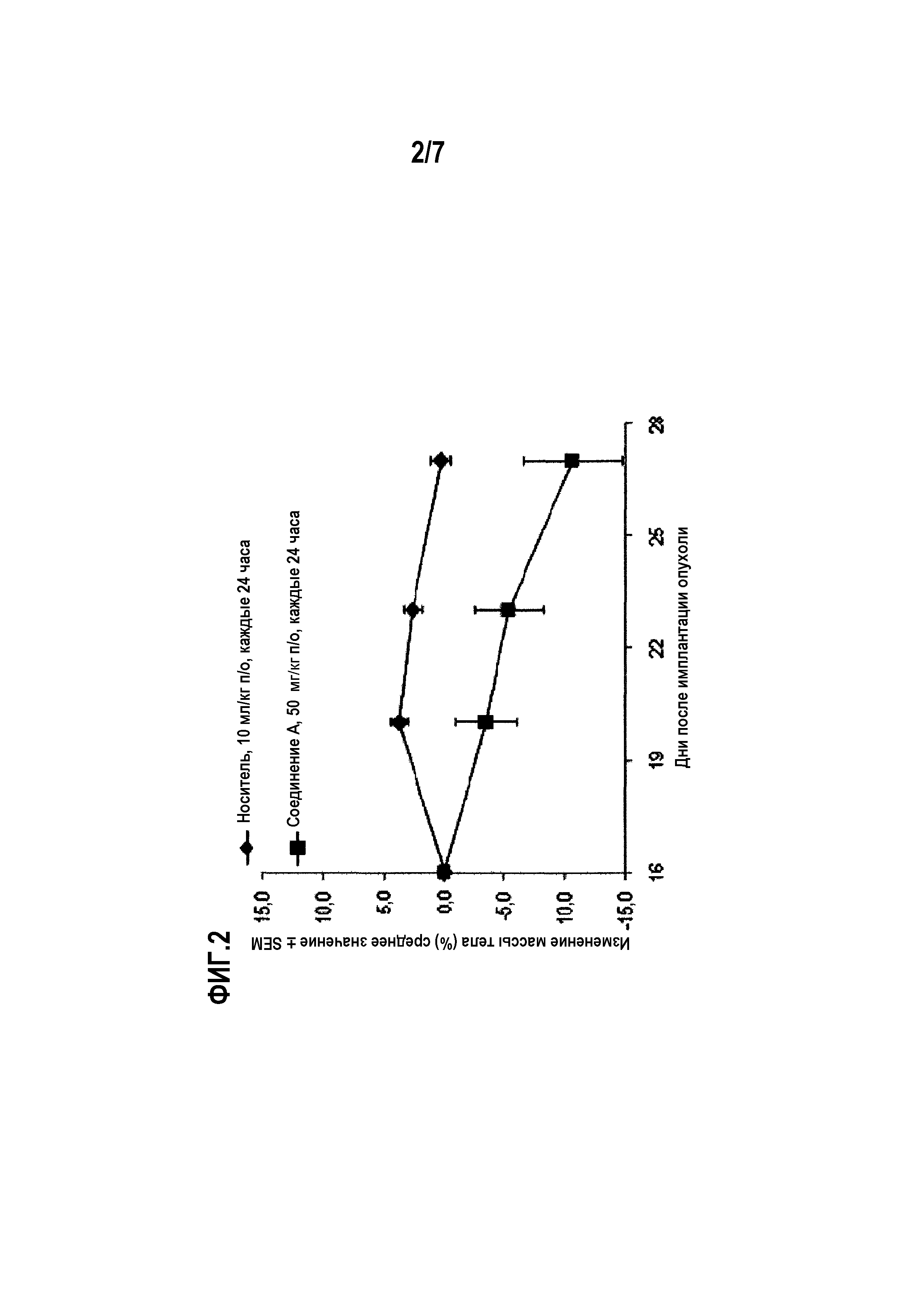
Фиг.2 показывает среднюю массу тела мышей, несущих ортотопические мутантные PIK3CA и амплифицированные ErbB2 клеточной линии ВТ474 раковых клеток молочной железы, в группах мышей, которых лечили носителем и соединением А.
Для тестирования in vivo, показанного на фиг.1 и 2, самкам бестимусных мышей, несущих ортотопические ксенотрансплантаты ВТ474, вводили соединение А или носитель по указанным схемам в указанных дозах. Лечение начинали через 16 дней после имплантации опухолевых клеток и продолжали 11 дней подряд. Статистику по изменению объемов опухолей проводили в одностороннем ANOVA-анализе с апостериорным тестом Даннетта (*р<0,05 по сравнению с контрольным носителем).
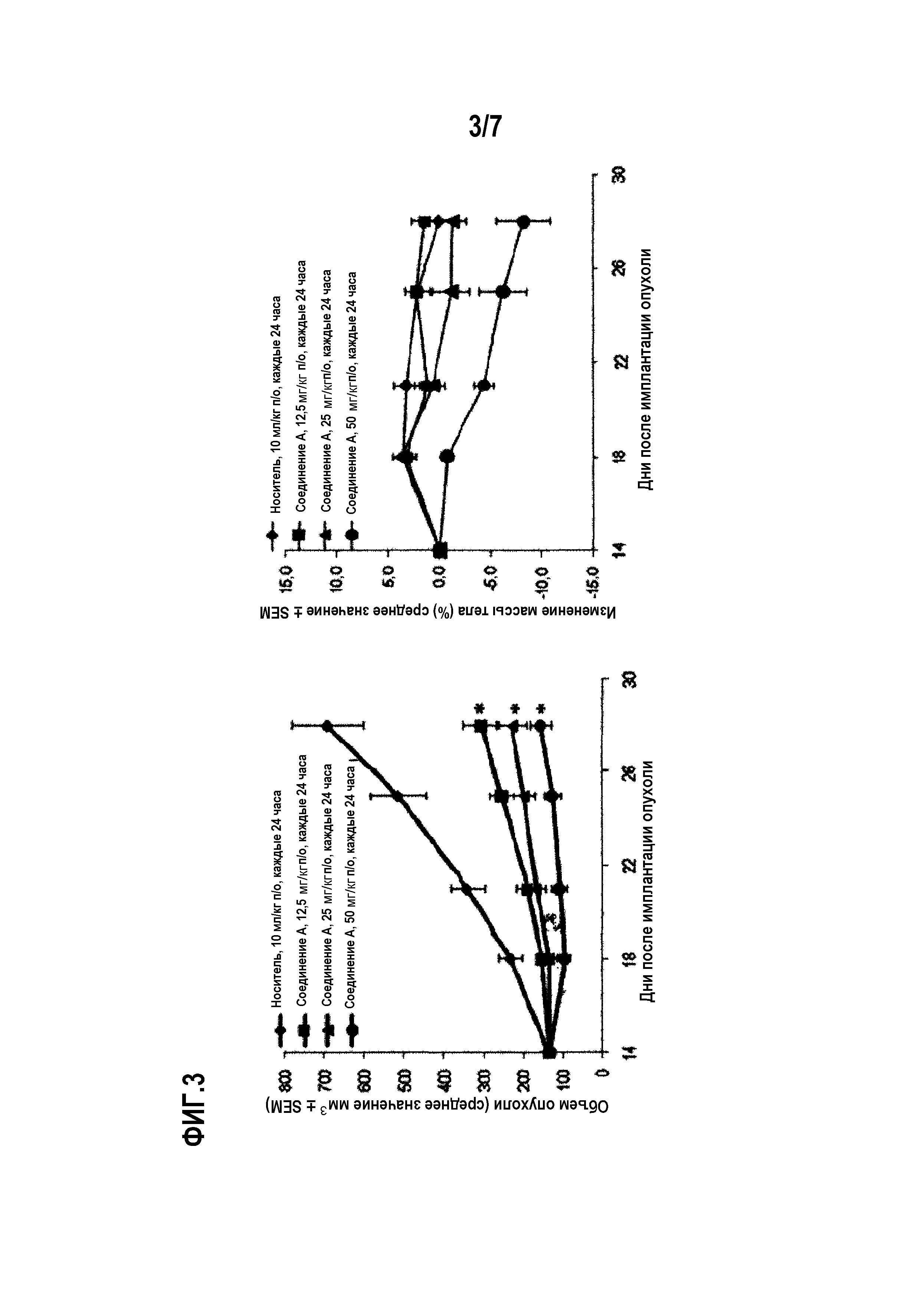
Фиг.3 показывает дозозависимую противоопухолевую активность соединения А в дозе 12,5 мг/кг, 25 мг/кг и 50 мг/кг, которые вводили перорально (п/о) через 24 часа (т.е. каждые 24 часа), по отношению к мутантным PIK3CA и амплифицированным ErbB2 клеточной линии ВТ474 раковых клеток молочной железы.
Для тестирования in vivo, показанного на фиг.3, самкам бестимусных мышей, несущих ортотопические ксенотрансплантаты ВТ474, вводили соединение А или носитель по схеме в дозе 12,5 мг/кг п/о, 25 мг/кг п/о или 50 мг/кг п/о. Лечение начинали через 14 дней после имплантации опухолевых клеток и продолжали 14 дней подряд. Статистику по изменению объемов опухолей проводили в одностороннем ANOVA-анализе с апостериорным тестом Даннетта (*р<0,05 по сравнению с контрольным носителем).
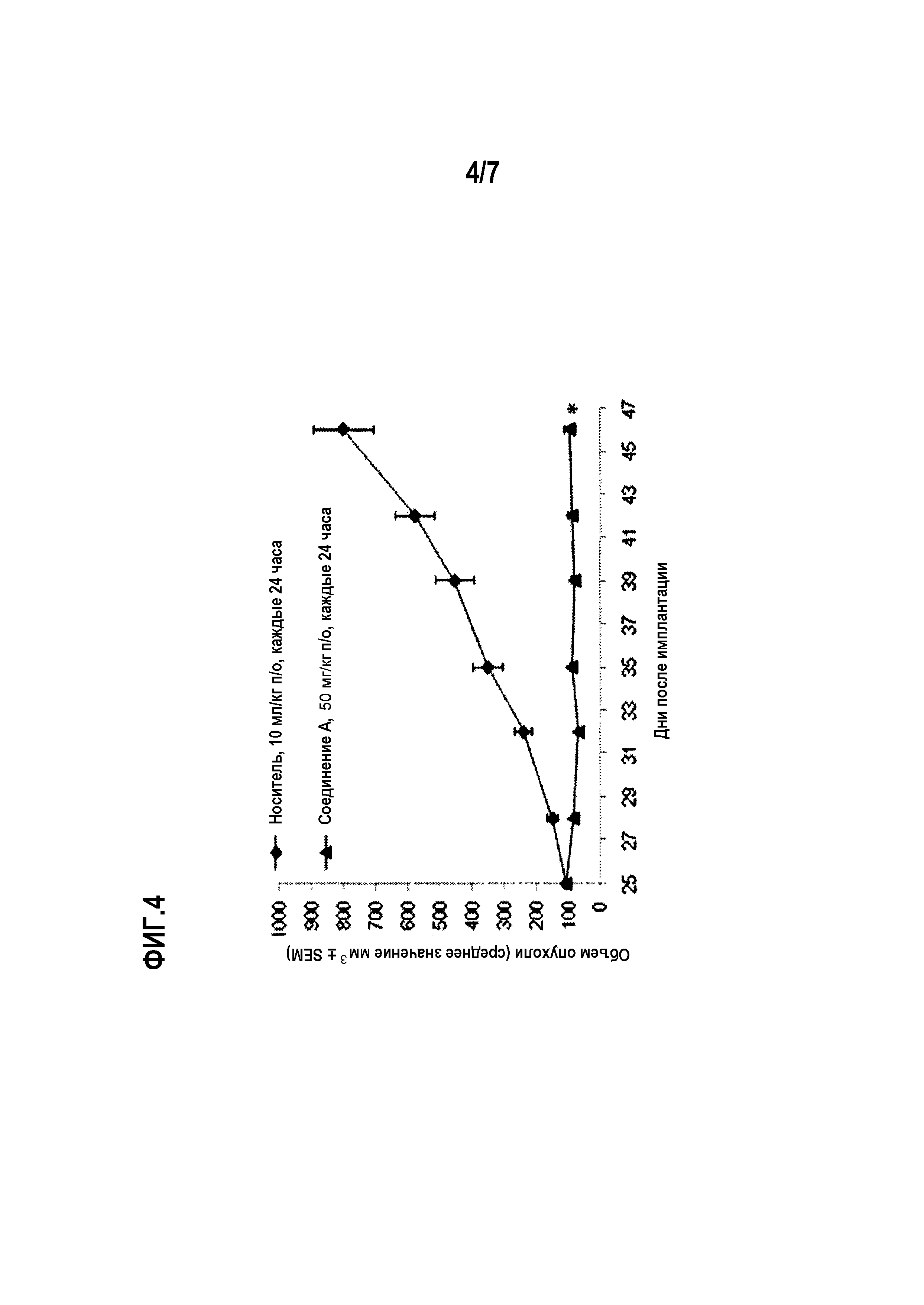
Фиг.4 показывает противоопухолевую активность соединения А по отношению к амплифицированным ErbB2 клеточной линии NCI-N87 клеток рака желудка.
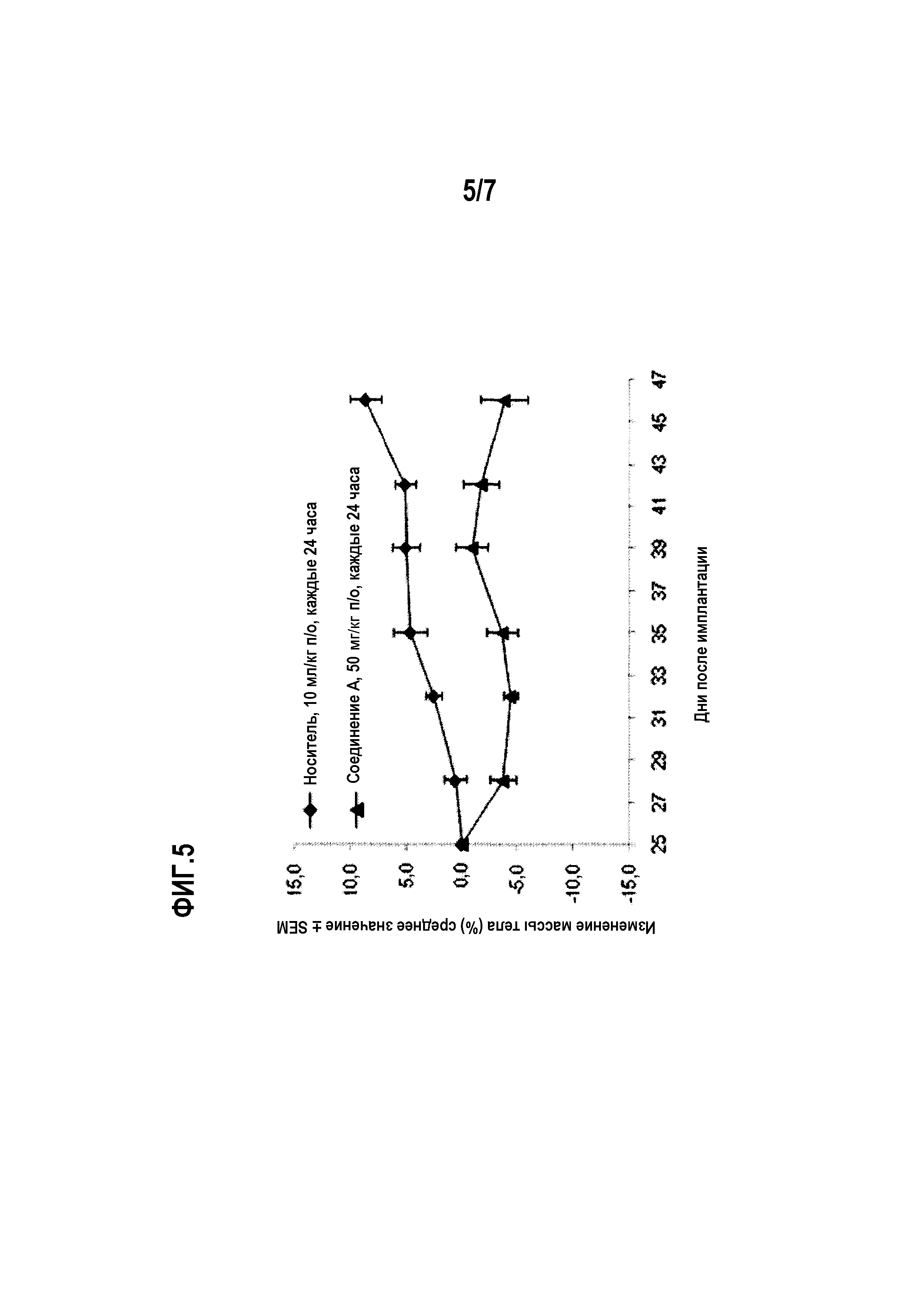
Фиг.5 показывает среднюю массу тела мышей, несущих подкожные амплифицированные ErbB2 клеточной линии NCI-N87 рака желудка, в группах мышей, которых лечили носителем и соединением А.
Для тестирования in vivo, показанного на фиг.3 и 4, самкам бестимусных мышей, несущих подкожные ксенотрансплантаты NCI-N87, вводили соединение А или носитель по указанным схемам в указанных дозах. Лечение начинали через 25 дней после имплантации опухолевых клеток и продолжали 21 день подряд. Статистику по изменению объемов опухолей проводили в одностороннем ANOVA-анализе с апостериорным тестом Даннетта (*р<0,05 по сравнению с контрольным носителем).
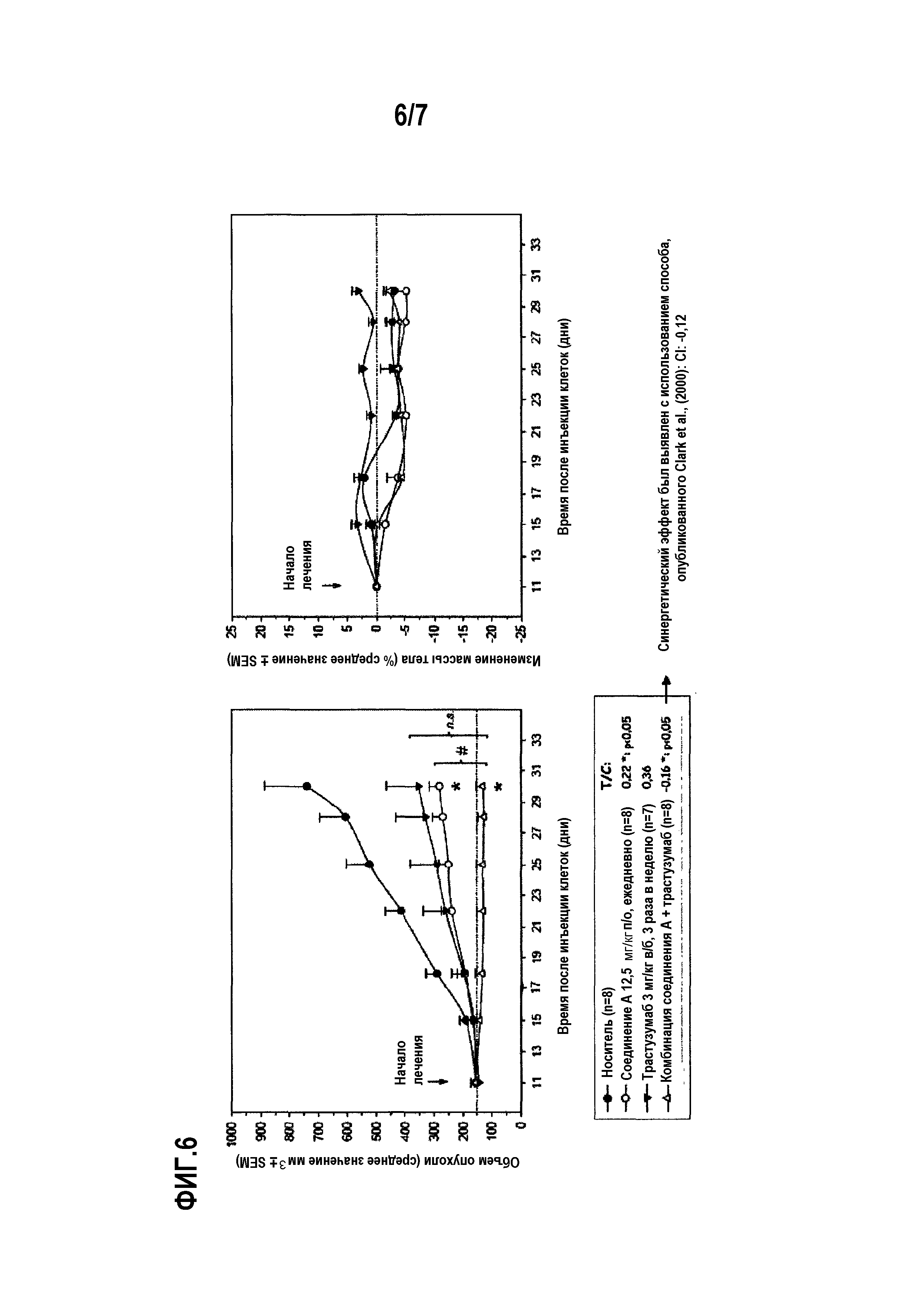
Фиг.6 показывает противоопухолевую активность носителя, соединения А в дозе 12,5 мг/кг п/о один раз в сутки в качестве единственного агента, трастузумаба в дозе 3 мг/кг внутрибрюшинно три раза в неделю в качестве единственного агента и комбинации соединения А и трастузумаба против мутантных PIK3CA и амплифицированных ErbB2 клеточной линии ВТ474 клеток рака молочной железы, и средние корректированные изменения массы тела (представленные как соотношение массы тела в день измерения и исходной массы тела на 11-й день [оба значения корректируются вычитанием начальной массы опухоли], выраженные в процентах для каждого отдельного животного) в группах мышей, получавших носитель, соединение А в качестве единственного агента, трастузумаб в качестве единственного агента и комбинацию соединения А и трастузумаба, при этом мыши имели ортотопические мутантные PIK3CA и амплифицированные ErbB2 клеточных линий ВТ474 клеток рака молочной железы. Значения представляют собой среднее значение ± SEM (стандартная погрешность средней величины); размер выборки (n=7-10 мышей в группе). (*p<0,05, значительное подавление по сравнению с контрольной группой носителя; #: р<0,05, значительное подавление по сравнению с лечением единственным агентом (критерий суммы рангов Манна-Уитни; ns: статистически не значим).
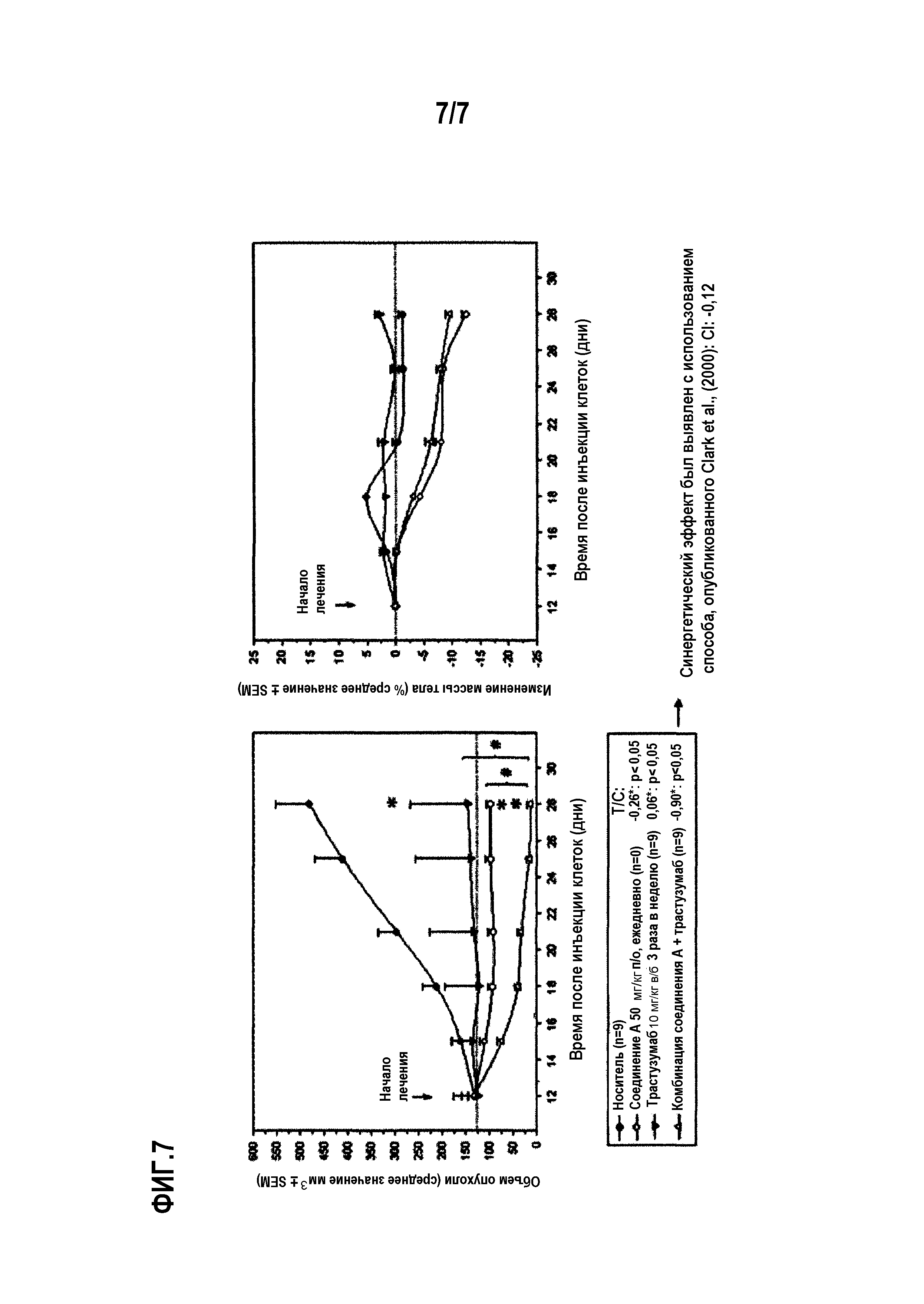
Фиг.7 показывает противоопухолевую активность носителя, соединения А в дозе 50 мг/кг п/о один раз в сутки в качестве единственного агента, трастузумаба в дозе 10 мг/кг внутрибрюшинно три раза в сутки в качестве единственного агента и комбинации соединения А и трастузумаба против мутантных PIK3CA и амплифицированных ErbB2 клеточной линии ВТ474 клеток рака молочной железы, и средние корректированные изменения массы тела (представленные как соотношение массы тела в день измерения и исходной массы тела на 12-й день [оба значения корректируются вычитанием начальной массы опухоли], выраженные в процентах для каждого отдельного животного) в группах мышей, получавших носитель, соединение А в качестве единственного агента, трастузумаб в качестве единственного агента и комбинацию соединения А и трастузумаба, при этом мыши имели ортотопические мутантные PIK3CA и амплифицированные ErbB2 клеточных линий ВТ474 клеток рака молочной железы. Значения представляют собой среднее значение ± SEM; размер выборки (n=9-10 мышей в группе). (*p<0,05, значительное подавление по сравнению с контрольной группой носителя; #: р<0,05, значительное подавление по сравнению с лечением единственным агентом (критерий суммы рангов Манна-Уитни; ns: статистически не значим).
Подробное описание изобретения
В данном описании использованы следующие общепринятые определения, если не указано иное:
"Галоген" (или "гало") обозначает фтор, бром, хлор или йод, в частности, фтор, хлор. Галогензамещенные группы и функциональные остатки, такие как алкил, замещенный галогеном (галогеналкил), могут быть моно-, поли- или пер-галогенированными.
"Гетероатомы" представляют собой атомы, отличные от углерода и водорода, предпочтительно атомы азота (N), кислорода (O) и серы (S), в частности, атом азота.
Углеродсодержащие группы, функциональные остатки или молекулы содержат атомы углерода в количестве от 1 до 7, предпочтительно от 1 до 6, более предпочтительно от 1 до 4, наиболее предпочтительно 1 или 2 атома углерода. Любая нециклическая углеродсодержащая группа или остаток, имеющий более одного атома углерода, представляет собой прямую или разветвленную цепь.
Понятие "низший" или "C1-C7" обозначает радикал, имеющий атомы углерода в количестве включительно до максимум 7, в частности, включительно до максимум 4 атомов углерода, и рассматриваемые радикалы являются или линейными или разветвленными, с одним или несколькими ветвлениями.
"Алкил" относится к алкильной группе с прямой цепью или разветвленной цепью, предпочтительно представляет собой C1-12-алкил с прямой цепью или разветвленной цепью; в частности, предпочтительно представляет собой C1-7-алкил с прямой цепью или разветвленной цепью; например, метил, этил, н- или изо-пропил, н-, изо-, втор- или трет-бутил, н-пентил, н-гексил, н-гептил, н-октил, н-нонил, н-децил, н-ундецил, н-додецил, при этом особо предпочтительным является метил, этил, н-пропил, изо-пропил, и н-бутил и изо-бутил. Алкил может быть незамещенным или замещенным. Примеры заместителей включают в себя без ограничения дейтерий, гидрокси, алкокси, гало и амино. Примером замещенного алкила является трифторметил. Также заместителем алкила может являться циклоалкил. В этом случае примером является функциональная группа (алкил)циклопропил или алкандиил-циклопропил, например, -CH2-циклопропил. C1-C7-алкил предпочтительно представляет собой алкил, имеющий включительно от 1 включительно до 7, предпочтительно включительно от 1 включительно до 4, и является линейным или разветвленным; предпочтительно, низший алкил представляет собой бутил, такой как н-бутил, втор-бутил, изобутил, трет-бутил, пропил, такой как н-пропил или изопропил, этил или, предпочтительно, метил.
Каждая алкильная часть других групп, таких как "алкокси", "алкоксиалкил", "алкоксикарбонил", "алкокси-карбонилалкил", "алкилсульфонил", "алкилсульфоксил", "алкиламино", "галогеналкил", имеют такое же значение, как описано для вышеупомянутого определения "алкила".
"Алкандиил" относится к прямой или разветвленной цепи группы алкандиила, связанной двумя разными атомами углерода с функциональной группой, и предпочтительно представляет собой C1-12 алкандиил с прямой цепью или разветвленной цепью, в частности, предпочтительно представляет собой C1-6 алкандиил с прямой цепью или разветвленной цепью; например, метандиил (-CH2-), 1,2-этандиил (-CH2-CH2-), 1,1-этандиил ((-CH(CH3)-), 1,1-, 1,2-, 1,3-пропандиил и 1,1-, 1,2-, 1,3-, 1,4-бутандиил, при этом особо предпочтительным является метандиил, 1,1-этандиил, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил.
"Алкендиил" относится к группе алкендиила с прямой цепью или разветвленной цепью, связанной с молекулой двумя разными атомами углерода, и предпочтительно представляет собой C2-6 алкендиил с прямой цепью или разветвленной цепью, например, -CH =CH-, -CH=C(CH3)-, -CH=CH-CH2-, -C(CH3)=CH-CH2-, -CH=C(CH3)-CH2-, -CH=CH-C(CH3)H-, -CH=CH-CH=CH-, -C(CH3)=CH-CH=CH-, -CH=C(CH3)-CH=CH-, и особо предпочтительными являются -CH=CH-CH2-, -CH=CH-CH=CH-. Алкендиил может быть замещенным или незамещенным.
"Циклоалкил" относится к насыщенному или частично насыщенному, моноциклическому, конденсированному полициклическому, или спиро-полициклическому углеродному кольцу, имеющему в карбоцикле от 3 до 12 кольцевых атомов. Иллюстративные примеры циклоалкильных групп включают в себя следующие функциональные группы: циклопропил, циклобутил, циклопентил и циклогексил. Циклоалкил может быть незамещенным или замещенным; в определении алкила приведены примеры заместителей, которые также включают в себя непосредственно алкил (например, метил). Такая функциональная группа, как -(CH3)циклопропил, считается замещенным циклоалкилом.
"Арил" относится к ароматической гомоциклической кольцевой системе (т.е. в качестве атомов, образующих кольцо, имеются только атомы углерода) с 6 атомами углерода или больше; арил предпочтительно представляет собой ароматическую функциональную группу, имеющую от 6 до 14 атомов углерода в кольце, более предпочтительно от 6 до 10 атомов углерода в кольце, такую как фенил или нафтил, предпочтительно фенил. Арил может быть незамещенным или замещен одним или несколькими заместителями, предпочтительно до трех, более предпочтительно до двух заместителей, независимо выбираемых из группы, состоящей из незамещенного или замещенного гетероциклила, как описано ниже, в частности, из следующих групп: пирролидинил, например, пирролидино, оксопирролидинил, такой как оксопирролидино, C1-C7-алкил-пирролидинил, 2,5-ди-(C1-C7-алкил)пирролидинил, например, 2,5-ди-(C1-C7-алкил)пирролидино, тетрагидрофуранил, тиофенил, C1-C7-алкилпиразолидинил, пиридинил, C1-C7-алкилпиперидинил, пиперидино, пиперидино, замещенный амино-, или N-моно-, или N,N-ди-[низший алкил, фенил, C1-C7-алканоил и/или фенил-низший алкил)амино, незамещенный или N-низший алкил, замещенный пиперидинолом, связанным посредством кольцевого атома углерода, пиперазино, низший алкилпиперазино, морфолино, тиоморфолино, S-оксотиоморфолино или S,S-диоксотиоморфолино; C1-C7-алкил, амино-C1-C7-алкил, N-C1-C7-алканоиламино-C1-C7-алкил, N-C1-C7-алкансульфонил-амино-C1-C7-алкил, карбамоил-C1-C7-алкил, [N-моно-или N,N-ди-(C1-C7-алкил)карбамоил]-C1-C7-алкил, C1-C7-алкансульфонил-C1-C7-алкил, C1-C7-алкансульфонил-C1-C7-алкил, фенил, нафтил, от моно- до три-[C1-C7-алкил, гало и/или циано]фенил или от моно- до три-[C1-C7-алкил, гало и/или циано]нафтил; C3-C8-циклоалкил, от моно- до три-[C1-C7-алкил и/или гидрокси]-C3-C8-циклоалкил; гало, гидрокси, низший алкокси, низший алкокси-низший алкокси, (низший алкокси)-низший алкокси-низший алкокси, гало-C1-C7-алкокси, фенокси, нафтилокси, фенил- или нафтил-низший алкокси; амино-C1-C7-алкокси, низший-алканоилокси, бензоилокси, нафтоилокси, формил(СНО), амино-, N-моно- или N,N-ди-(C1-C7-алкил)амино-, C1-C7-алканоиламино, C1-C7-алкансульфониламино, карбокси, низший алкоксикарбонил, например, фенил- или нафтил-низший алкоксикарбонил, такой как бензилоксикарбонил; C1-C7-алканоил, такой как ацетил, бензоил, нафтоил, карбамоил, N-моно- или N,N-дизамещенный карбамоил, например, N-моно- или N,N-ди-замещенный карбамоил, в которых заместители выбирают из следующих групп: низший алкил, (низший алкокси)-низший алкил и гидрокси-низший алкил; амидино, гуанидино, уреидо, меркапто, низший алкилтио, фенил- или нафтилтио, фенил- или нафтил-низший алкилтио, низший алкил-фенилтио, низший алкил-нафтилтио, гало-низший алкилмеркапто, сульфо-(-SO3H), низший алкансульфонил, фенил- или нафтил-сульфонил, фенил- или нафтил-низший алкилсульфонил, алкилфенилсульфонил, гало-низший алкилсульфонил, например, трифторметансульфонил; сульфонамидо, бензосульфонамидо, азидо, азидо-C1-C7-алкил, в частности, азидометил, C1-C7-алкансульфонил, сульфамоил, N-моно- или N,N-ди-(C1-C7-алкил)сульфамоил, морфолинсульфонил, тиоморфолинсульфонил, циано и нитро; при этом каждый фенил или нафтил (а также в фенокси или нафтокси), упомянутые выше в качестве заместителя или в качестве части заместителя замещенного алкила (или также замещенного арила, гетероциклила и т.п., упомянутых в изобретении) сами по себе являются незамещенными или замещены одним или несколькими заместителями, например, до трех, предпочтительно 1 или 2 заместителями, независимо выбираемыми из гало, гало-низшего алкила, например, трифторметила, гидрокси, низшего алкокси, азидо, амино, N-моно- или N,N-ди-(низший алкил и/или C1-C7-алканоил)-амино, нитро, карбокси, низшего алкокси-карбонила, карбамоила, циано и/или сульфамоила.
"Гетероциклил" относится к гетероциклическому радикалу, который является ненасыщенным (то есть, имеет максимально возможное число сопряженных двойных связей в кольце (кольцах)), насыщенным или частично насыщенным и, предпочтительно, представляет собой моноциклическое, или, в более широком аспекте изобретения, бициклическое, трициклическое или спироциклическое кольцо, и имеет от 3 до 24, более предпочтительно от 4 до 16, наиболее предпочтительно от 5 до 10, и наиболее предпочтительно 5 или 6 кольцевых атомов; при этом один или несколько, предпочтительно от одного до четырех, в частности, один или два кольцевых атома представляют собой гетероатом (таким образом, остальные кольцевые атомы являются углеродом). Связывающее кольцо (т.е. кольцо, присоединенное к молекуле) предпочтительно имеет от 4 до 12, в частности, от 5 до 7 атомов в кольце. Термин «гетероциклил» также включает в себя гетероарил. Гетероциклический радикал (гетероциклил) может быть незамещенным или замещен одним или несколькими заместителями, предпочтительно имеет от 1 до 3 заместителей, независимо выбираемых из группы, состоящей из заместителей, определенных выше для замещенного алкила, и/или замещен одним или несколькими из следующих заместителей: оксо (=О), тиокарбонил (=S), имино (=NH), имино-низший алкил. Дополнительно, гетероциклил, в частности, представляет собой гетероциклильный радикал, выбираемый из группы, состоящей из следующего: оксиранил, азиринил, азиридинил, 1,2-оксатио-ланил, тиенил (=тиофенил), фуранил, тетрагидрофурил, пиранил, тиопиранил, тиантренил, изобензофуранил, бензофуранил, хроменил, 2H-пирролил, пирролил, пирролинил, пирролидинил, имидазолил, имидазолидинил, бензимидазолил, пиразолил, пиразинил, пиразолидинил, тиазолил, изотиазолил, дитиазолил, оксазолил, изоксазолил, пиридил, пиразинил, пиримидинил, пиперидинил, пиперазинил, пиридазинил, морфолинил, тиоморфолинил, (S-оксо- или S,S-диоксо)тиоморфолинил, индолизинил, азепанил, диазепанил, в частности, 1,4-диазепанил, изоиндолил, 3Н-индолил, индолил, бензимидазолил, кумарил, индазолил, триазолил, тетразолил, пуринил, 4H-хинолизинил, изохинолил, хинолил, тетрагидрохинолил, тетрагидроизохинолил, декагидрохинолил, октагидроизохинолил, бензофуранил, дибензофуранил, бензотиофенил, дибензотиопентил, фталазинил, нафтиридинил, хиноксалил, хиназолинил, циннолинил, птеридинил, карбазолил, бета-карболинил, фенантридинил, акридинил, перимидинил, фенантролинил, фуразанил, феназинил, фенотиазинил, феноксазинил, хроменил, изохроманил, хроманил, бензо[1,3]диоксол-5-ил и 2,3-дигидробензо[1,4]диоксин-6-ил, и каждый из указанных радикалов может быть незамещенным или замещен одним или несколькими заместителями, предпочтительно до трех, выбираемых из вышеупомянутых заместителей для замещенного арила, и/или одним или несколькими из следующих заместителей: оксо (=О), тиокарбонил (=S), имино (=NH), имино-низший алкил.
"Арилалкил" относится к арильной группе, связанной с молекулой через алкильную группу, такую как метильная или этильная группа, предпочтительно фенэтил или бензил, в частности, бензил. Аналогично, циклоалкил-алкил и гетероциклил-алкил представляют собой циклоалкильную группу, связанную с молекулой через алкильную группу, или представляют собой гетероциклическую группу, связанную с молекулой через алкильную группу. В каждом случае арил, гетероциклил, циклоалкил и алкил могут быть замещенными, как указано выше.
«Лечение» включает в себя профилактическое (превентивное) и терапевтическое лечение, а также замедление прогрессирования заболевания или нарушения. Используемый в изобретении термин «замедление прогрессирования» означает введение комбинации пациентам, находящимся на предварительной стадии или на ранней стадии пролиферативного заболевания, подлежащего лечению, и у этих пациентов диагностирована, например, предварительная форма соответствующего заболевания, или эти пациенты находятся, например, в состоянии лечения или в состоянии, возникшем в результате острого случая, на фоне которого существует вероятность развития соответствующего заболевания.
"Заболевания, зависимые от рецептора эпидермального фактора роста" или "EGFR-зависимые заболевания» представляют собой, в частности такие патологии или злокачественные новообразования, при которых наблюдается полезная реакция (например, улучшение одного или нескольких симптомов, отсрочка начала заболевания, вплоть до временного или полного излечения заболевания) на подавление члена семейства рецептора эпидермального фактора роста (при этом заболевания, подлежащие лечению, могут включать в себя пролиферативные заболевания, такие как раковые или опухолевые заболевания). Семейство рецептора эпидермального фактора роста состоит из четырех членов: EGFR1 (также называемый HER1 или Erb-B1); EGFR2 (также называемый HER2 или Erb-B2); EGFR3 (также называемый HER3 или Erb-B3); и EGFR4.
Понятия "фармацевтический препарат" или "фармацевтическая композиция" относятся к смеси или раствору, содержащему по меньшей мере одно терапевтическое соединение для введения млекопитающему, например, человеку, с целью предотвращения, лечения и контроля конкретного заболевания или состояния, поражающего этого млекопитающего.
Понятие "фармацевтически приемлемый" относится к таким соединениям, материалам, композициям и/или лекарственным формам, которые, в рамках обоснованного медицинского заключения, подходят для контакта с тканями млекопитающих, в частности, человека, без чрезмерной токсичности, раздражения, аллергической реакции и других проблем и осложнений, соразмерно с разумным соотношением польза/риск.
"Соли" (которые подразумеваются при упоминании "или их соли" или "или его соли), могут присутствовать в качестве единственного агента или в смеси со свободным соединением формулы (I) и предпочтительно представляют собой фармацевтически приемлемые соли. Такие соли, в частности, фармацевтически приемлемые соли, образованы из соединений формулы (I) с основным атомом азота, например, кислотно-аддитивные соли, предпочтительно с органическими или неорганическими кислотами. Подходящими неорганическими кислотами, например, являются кислоты галогенов, таких как соляная кислота, серная кислота или фосфорная кислота. Подходящими органическими кислотами являются, например, карбоновые кислоты или сульфокислоты, такие как фумаровая кислота или метансульфоновая кислота. В целях выделения или очистки также можно использовать фармацевтически неприемлемые соли, например, пикраты или перхлораты. Для терапевтического применения используют только фармацевтически приемлемые соли или свободные соединения (если они применимы в виде фармацевтических композиций), и поэтому они являются предпочтительными. Учитывая тесную взаимосвязь между новыми соединениями в свободной форме, и новыми соединениями в форме их солей, включающих в себя соли, которые могут быть использованы в качестве промежуточных продуктов, например, при очистке или идентификации новых соединений, любое упоминание о свободных соединениях выше и ниже в настоящем изобретении также следует понимать как относящееся к соответствующим солям, по мере необходимости и целесообразности. Соли соединений формулы (I) предпочтительно являются фармацевтически приемлемыми солями; в данной области техники известны подходящие противоионы, образующие фармацевтически приемлемые соли.
Понятие "комбинация" относится или к фиксированной комбинации в одной лекарственной форме или к нефиксированной комбинации (или набору частей) для совместного введения, при этом соединение формулы (I) и комбинационный партнер (например, другое лекарство, как описано ниже, также называемое "терапевтическим агентом» или «ко-агентом") можно вводить независимо одновременно или раздельно с интервалами времени, в особенности в случаях, когда эти интервалы времени позволяют комбинационным партнерам проявлять совместный эффект, например, синергетический эффект. Подразумевается, что термин "совместное введение" или подобные понятия, используемые в изобретении, охватывают введение выбранного комбинационного партнера одному нуждающемуся в этом введении субъекту (например, пациенту), и подразумевается, что эти понятия включают в себя схемы лечения, согласно которым агенты не обязательно вводятся одним и тем же путем введения или в одно и то же время. Термин "фиксированная комбинация" означает, что активные ингредиенты, например, и соединение формулы (I) и комбинационный партнер, вводятся пациенту одновременно в виде монолитной формы или дозы. Термины "нефиксированная комбинация" или "набор частей" означают, что активные ингредиенты, например, и соединение формулы (I) и комбинационный партнер вводятся пациенту в виде раздельных форм или одновременно, параллельно или последовательно, при отсутствии конкретных ограничений по времени, при этом такое введение обеспечивает терапевтически эффективные уровни двух соединений в организме пациента. Последнее также относится к терапевтической смеси, например, к введению трех или более активных ингредиентов.
"Терапевтически эффективный" предпочтительно относится к количеству, которое является терапевтически эффективным, или в более широком смысле, также профилактически эффективным в отношении прогрессирования пролиферативного заболевания.
Настоящее изобретение относится к использованию конкретных производных 2-карбоксамид-циклоамино-мочевины в качестве единственных агентов или в комбинации, для лечения заболеваний, зависимых от членов семейства рецептора эпидермального фактора роста (EGFR) (включающих в себя EGFR1, также называемый HER1 или Erb-B1; EGFR2 также называемый HER2 или Erb-B2; EGFR3 также называемый HER3 или Erb-B3; и EGFR4).
Конкретные производные 2-карбоксамид-циклоамино мочевины, которые подходят для настоящего изобретения, их получение и подходящие фармацевтические композиции, содержащие указанные производные, описаны в WO 2010/029082 и включают в себя соединения формулы I
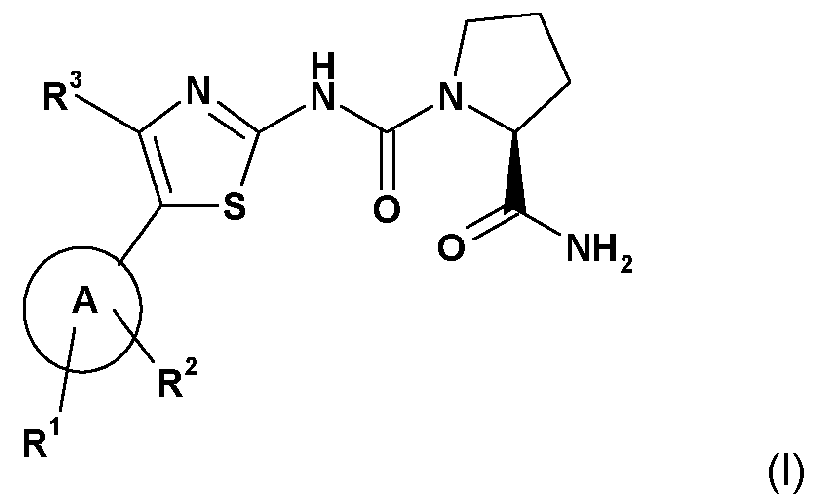
или его соль, где
А обозначает гетероарил, который выбирают из группы, состоящей из:
R1 представляет собой один из следующих заместителей: (1) незамещенный или замещенный, предпочтительно замещенный C1-C7-алкил, при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до девяти: дейтерий, фтор или из следующих остатков от одного до двух C3-C5-циклоалкил; (2) необязательно замещенный C3-C5-циклоалкил, при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до четырех: дейтерий, C1-C4-алкил (предпочтительно метил), фтор, циано, аминокарбонил; (3) необязательно замещенный фенил, при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до двух: дейтерий, галоген, циано, C1-C7-алкил, C1-C7-алкиламино, ди(C1-C7-алкил)амино, C1-C7-алкиламинокарбонил, ди(C1-C7-алкил)аминокарбонил, C1-C7-алкокси; (4) необязательно моно- или ди-замещенные амины, при этом указанные заместители независимо выбирают из следующих функциональных остатков: дейтерий, C1-C7-алкил (который является незамещенным или замещен одним или несколькими заместителями, выбираемыми из группы дейтерия, фтора, хлора, гидрокси), фенилсульфонил (который является незамещенным или замещен одним или несколькими, предпочтительно одним C1-C7-алкилом, C1-C7-алкокси, ди(C1-C7-алкил)амино-C1-C7-алкокси); (5) замещенный сульфонил, при этом указанный заместитель выбирают из следующих функциональных групп: C1-C7-алкил (который является незамещенным или замещен одним или несколькими заместителями, выбираемыми из группы дейтерия, фтора), пирролидино (который является незамещенным или замещен одним или несколькими заместителями, выбираемыми из группы дейтерия, гидрокси, оксо; в частности, замещен одной оксо-группой); (6), фтор, хлор;
R2 представляет собой водород;
R3 представляет собой (1) водород, (2) фтор, хлор, (3) необязательно замещенный метил,
при этом указанные заместители независимо выбирают из одного или более одного функционального остатка, предпочтительно из следующих остатков в количестве от одного до трех: дейтерий, фтор, хлор, диметиламино;
за исключением 2-амид 1-({5-[2-(трет-бутил)пиримидин-4-ил]-4-метил-тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты.
Значения радикалов и символов, используемых в определении соединения формулы I, соответствуют описанию опубликованного патента WO 2010/029082, который включен в настоящую заявку путем ссылки.
Предпочтительным соединением настоящего изобретения является соединение, которое конкретно описано в WO 2010/029082.
В высокой степени предпочтительным соединением настоящего изобретения является 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его фармацевтически приемлемая соль. Синтез 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты описан, например, в патенте WO 2010/029082, как пример 15.
Семейство рецепторов ErbB, включающих в себя EGFR1 (также называемый HER1 или Erb-B1), EGFR2 (также называемый HER2 или Erb-B2), и EGFR3 (также называемый HER3 или Erb-B3) и EGFR4 часто показывает сверхэкспрессию при клеточных пролиферативных нарушениях или в карциномах. Часто сверхэкспрессия ErbB2 (HER2) наблюдается в карциномах молочной железы, яичников и желудка. Несмотря на наличие эффективных терапевтических подходов в отношении ErbB2, у 50% пациентов с амплифицированным/сверхэкспрессируемым HER2 отсутствует реакция на ErbB2-модуляторы, такие как трастузумаб. Клеточные линии ВТ474 и HBCx-5 рака молочной железы клеток и клеточные линии NCI-N87 рака желудка являются полезными моделями при амплификации ErbB2, и обладают высокой онкогенностью in vivo. Противоопухолевую активность ингибиторов PI3K, таких как соединения формулы I, тестировали на обеих моделях ErbB2-индуцированной опухоли.
Неожиданно выявлено, что введение соединения А приводит к подавлению опухолевого роста in vivo в обеих моделях. В группе исследования клеточных линий рака молочной железы, клеток колоректального рака (CRC) и поджелудочной железы, содержащих или не содержащих амплификацию ErbB2, соединение А уменьшает пролиферацию клеток со средней Gl50 при 139±82 нМ в модели рака ВТ474 молочной железы и со средней Gl50 при 687±132 нМ в модели рака HCC1954 молочной железы, и вызывает гибель клеток в обеих клеточных линиях, сверхэкспрессирующих ErbB2.
В модели самок бестимусных голых мышей с ортотопическим ксенотрансплантатом с имплантированным раком ВТ474 молочной железы, соединение А вызывает статистически значимый противоопухолевый эффект в группе, получавшей соединение А в дозах 12,5, 25 и 50 мг/кг, по сравнению с группой, получавшей носитель (р<0,05, ANOVA-анализ с апостериорным тестом Даннетта). Соединение А при пероральном (п/о) введении в дозе 50 мг/кг один раз в сутки вызывает среднее изменение опухоли -29,7±15,6 мм3 в отношении объема опухоли (р<0,01, ANOVA с апостериорным тестом Даннетта) по сравнению с носителем (среднее изменение опухоли 315,1±16,4 мм3), с регрессией 23,0% в первом эксперименте (см. фиг.1). Соединение А при пероральном введении в дозах 12,5, 25 и 50 мг/кг один раз в сутки вызывает, соответственно, среднее изменение объема опухоли 171,8±33,0 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта), 95,5±26,5 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта) и 20,8±15,9 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта) по сравнению с носителем (среднее изменение объема опухоли 557,7±79,0 мм3) во втором эксперименте (см. фиг.3).
Соединение А хорошо переносится в дозах 12,5 и 25 мг/кг, о чем свидетельствуют статистически незначимые изменения средней массы тела в группе, получавшей носитель (0,1±1,2%) и в группе, получавшей соединение А (1,4±1,2% и -1,2+1,2% соответственно). Тем не менее в группе, которой вводили 50 мг/кг соединения А, были выявлены статистически значимые изменения средней массы тела 10,6±4,1% (р<0,05, с использованием парного t-теста) и 8,2±2,6% (р<0,05, с использованием парного t-теста) в двух экспериментах (см. фиг.2).
В модели самок бестимусных голых мышей с подкожным ксенотрансплантатом желудочной линии NCI-N87 соединение А вызывает статистически значимый противоопухолевый эффект в группе, получавшей лечение соединением А по сравнению с носителем (р<0,05, ANOVA), с регрессией 11% в эксперименте (см. фиг.4). В группе, получавшей лечение соединением А перорально (п/о) в дозе 50 мг/кг один раз в сутки, соединение А вызывает среднее изменение объема опухоли от -11,8±17,2 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта) по сравнению с носителем (среднее изменение объема опухоли составляет 694,0±93,5 мм3. Доза соединения А составляет 50 мг/кг. Соединение А хорошо переносится, о чем свидетельствует статистически незначимое среднее изменение массы тела в группе, получавшей в этом эксперименте 50 мг/кг соединения А (3,8±2,1%) (см. фиг.5).
Таким образом, соединение формулы I, в частности, соединение А, является полезным для лечения упомянутых EGFR-зависимых заболеваний, в особенности злокачественных опухолей, или заболеваний, обусловленных приобретенной резистентностью к членам семейства EGFR. Заболевания или злокачественные новообразования, для которых на молекулярном уровне установлена или потенциально существует связь с нарушением регуляции активности EGFR, представляют собой патологии, например, описанные в следующих источниках: "Mendelsohn and Baselga; Status of Epidermal Growth Factor Receptor Antagonists in the Biology and Treatment of Cancer, Journal of Clinical Oncology, 2787-2799"; "Mendelsohn and Baselga; Epidermal Growth Factor Receptor Targeting in Cancer, Seminars in Oncology, Vol.33, 369-385»; Irmer et al., 2007, EGFR kinase domain mutations - functional impact and relevance for lung cancer therapy, Oncogene, 1-9; Roche-Lima et al., EGFR targeting of solid tumors; Cancer Control, 2007, Vol.14 (3), 295-304) все из которых, в том числе приведенные в изобретении ссылки на эти источники, включены в настоящую заявку путем ссылки.
Согласно настоящему изобретению, соединения формулы I, в частности, соединение А предпочтительно применяют для лечения следующих EGFR-зависимых заболеваний, в особенности злокачественных новообразований:
- немелкоклеточный рак легкого
- рак головы и шеи
- колоректальный рак
- рак молочной железы
- злокачественные опухоли мозга, в том числе глиобластома
- рак простаты
- рак мочевого пузыря
- почечно-клеточная карцинома
- рак поджелудочной железы
- рак шейки матки
- рак пищевода
- рак желудка
- рак яичников
или любой комбинации указанных заболеваний.
Дополнительно, настоящее изобретение относится к применению соединения формулы I, описанной выше, или его соли для изготовления фармацевтической композиции для лечения EGFR-зависимого заболевания или злокачественного новообразования.
В одном варианте осуществления настоящее изобретение относится к применению соединения формулы I, в частности, 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его соли для изготовления фармацевтической композиции для лечения EGFR-зависимого заболевания или злокачественного новообразования, или заболевания с приобретенной резистентностью к другим соединениям, нацеленным на члены семейства EGFR.
Соединения согласно настоящему изобретению, нацеленные на члены семейства EGFR, включают в себя модуляторы киназы семейства EGFR, соединения, которые изменяют уровни экспрессии EGFR, или вызывают клеточный иммунный ответ, связанный с экспрессией членов семейства EGFR в опухолевых клетках. Используемый в изобретении термин "EGFR-модулятор" означает соединение или лекарственный препарат, представляющий собой биологическую молекулу или малую молекулу, которая прямо или опосредованно модулирует активность EGFR или пути сигнальной трансдукции EGFR. Прямая или опосредованная модуляция включает в себя активацию, или подавление активности EGFR, или сигнальную трансдукцию пути EGFR. В одном аспекте подавление относится к ингибированию связывания EGFR к EGFR-лиганду, такому как, например, EGF. В другом аспекте ингибирование относится к ингибированию киназной активности EGFR.
Резистентность к лечению EGFR-модулятором может быть приобретена в ходе лечения указанным EGFR-модулятором или может быть обусловлена мутацией или мутациями в белке.
Предпочтительные EGFR-модуляторы проявляют свою активность в качестве ингибиторов функциональной активности EGFR. Соединения, нацеленные на члены семейства EGFR согласно настоящему изобретению, включают в себя без ограничения гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIRW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональное антитело 806, анти-EGFR моноклональное антитело Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, пертузумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, TDM1, Земаб®, Her2 вакцину PX 1041 и HSP90-ингибиторы CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922.
EGFR-модуляторы включают в себя, например, EGFR-специфичные лиганды, ингибиторы EGFR с малыми молекулами и EGFR-моноклональные антитела. В одном аспекте EGFR-модулятор подавляет активность EGFR и/или ингибирует пути сигнальной трансдукции EGFR. В другом аспекте EGFR-модулятор представляет собой EGFR-моноклональное антитело, которое ингибирует активность EGFR и/или ингибирует пути сигнальной трансдукции EGFR.
EGFR-модуляторы включают в себя биологические молекулы или малые молекулы. Биологические молекулы включают в себя все липиды и полимеры моносахаридов, аминокислот и нуклеотидов, имеющие молекулярный вес больше 450. Таким образом, биологические молекулы включают в себя, например, олигосахариды и полисахариды; олигопептиды, полипептиды, пептиды и белки, и олигонуклеотиды и полинуклеотиды. Олигонуклеотиды и полинуклеотиды включают в себя, например, ДНК и РНК.
Биологические молекулы дополнительно включают в себя производные любой из описанных выше молекул. Например, производные биологических молекул включают в себя липиды и производные гликозилирования олигопептидов, полипептидов, пептидов и белков.
Производные биологических молекул дополнительно включают в себя липидные производные олигосахаридов и полисахаридов, например, липополисахариды. Наиболее типичные биологические молекулы представляют собой антитела или функциональные эквиваленты антител. Функциональные эквиваленты антител имеют характеристики связывания, сопоставимые с характеристиками связывания антител, и подавляют рост клеток, экспрессирующих EGFR. Такие функциональные эквиваленты включают в себя, например, химерные, гуманизированные и одноцепочечные антитела, а также их фрагменты.
Функциональные эквиваленты антител также включают в себя полипептиды с аминокислотными последовательностями, по существу идентичными аминокислотным последовательностям вариабельных или гипервариабельных областей антител. Аминокислотная последовательность, которая по существу идентична другой последовательности, но отличается от другой последовательности в отношении одной или нескольких замен, добавлений и/или делеций, считается эквивалентной последовательностью. Предпочтительно, замены, добавления или делеции в белке составляют менее 50%, более предпочтительно менее 25%, и еще более предпочтительно менее 10% по числу аминокислотных остатков в последовательности.
Функциональный эквивалент антитела предпочтительно представляет собой химерное или гуманизированное антитело. Химерное антитело состоит из вариабельной области нечеловеческого антитела и константной области человеческого антитела. Гуманизированное антитело содержит гипервариабельные области (CDR) нечеловеческого антитела. Вариабельная область, отличная от гипервариабельных областей, например, каркасный вариабельный участок, и константная область гуманизированного антитела являются участками человеческого антитела.
Подходящие вариабельные и гипервариабельные области нечеловеческих антител можно получать из антител, вырабатываемых любым млекопитающим, не являющимся человеком, у которого вырабатываются моноклональные антитела. Подходящие примеры млекопитающих, отличных от человека, включают в себя, например, кроликов, крыс, мышей, лошадей, коз или приматов.
Функциональные эквиваленты дополнительно включают в себя фрагменты антител, обладающие характеристиками связывания, которые аналогичны или сопоставимы с характеристиками связывания полного антитела. Подходящие фрагменты антител включают в себя любой фрагмент, который содержит достаточную часть гипервариабельной области (то есть области, определяющей комплементарность) для связывания с тирозинкиназой EGFR со специфичностью и достаточной аффинностью в целях ингибирования роста клеток, экспрессирующих такие рецепторы.
Такие фрагменты, например, могут содержать один или оба Fab-фрагмента или F(ab').sub.2 фрагмент. Предпочтительно, фрагменты антитела содержат все шесть определяющих комплементарность областей из полного антитела, вместе с тем, в объем изобретения также включены функциональные фрагменты, содержащие меньше, чем все такие области, например, фрагменты, содержащие три, четыре, или пять областей CDR.
В одном аспекте фрагменты представляют собой одноцепочечные антитела, или Fv-фрагменты. Одноцепочечные антитела являются полипептидами, которые содержат по меньшей мере вариабельный участок тяжелой цепи антитела, связанный с вариабельным участком легкой цепи посредством соединительного линкера или без линкера. Таким образом, Fv-фрагмент содержит участок связывания полностью. Эти цепи могут продуцироваться в бактериях или в эукариотических клетках.
Антитела и функциональные эквиваленты могут быть членами любого класса иммуноглобулинов, например, IgG, IgM, IgA, IgD или IgE, и их подклассов.
В дополнение к описанным выше биологическим молекулам, полезные для изобретения EGFR-модуляторы также могут представлять собой малые молекулы. Любая молекула, которая не является биологической молекулой, в настоящем изобретении считается малой молекулой. Некоторые примеры малых молекул включают в себя органические соединения, металлоорганические соединения, соли органических и металлоорганических соединений, сахариды, аминокислоты и нуклеотиды. Малые молекулы дополнительно включают в себя молекулы, которые в ином случае могли бы считаться биологическими молекулами, если их молекулярная масса не превышает 450 Да. Таким образом, малые молекулы могут представлять собой липиды, олигосахариды, олигопептиды, олигонуклеотиды и их производные, имеющие молекулярную массу 450 Да или меньше.
В частности, настоящее изобретение относится к лечению заболевания или злокачественного новообразования, которое зависит от членов EGFR-семейства или сопровождается резистентностью, приобретенной в ходе лечения EGFR-модулятором, с помощью соединений формулы I, в частности, соединения А или его соли.
Возможные EGFR-модуляторы, при лечении которыми может возникать резистентность, представляют собой, например, гефитиниб, эрлотиниб, лапатиниб, цетуксимаб, нимотузумаб, панитумумаб, трастузумаб и TDM1.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы (I) для лечения EGFR- зависимого заболевания, или злокачественного новообразования, или заболевания с приобретенной резистентностью к EGFR-модулятору ("заболевания с приобретенной резистентностью к EGFR") в комбинации с другими активными соединениями, например, с комбинационными партнерами, как описано в патенте WO 2010/029082. Наиболее предпочтительными комбинационными партнерами для этого аспекта изобретения являются агенты, нацеленные на EGFR-семейство, такие как, без ограничения, гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIRW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональное антитело 806, анти-EGFR моноклональное антитело-Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, пертузумаб, MDX-214, Cdx110, IMC11F8, пертузумаб, трастузумаб, TDM1, Земаб®, Her2 вакцина PX 1041 и HSP90-ингибиторы CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922.
Еще в одном варианте осуществления настоящее изобретение также относится к комбинации, содержащей соединение формулы I, в частности, 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]-тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) и EGFR-модулятор, выбираемый из группы, состоящей из следующих агентов: гефитиниб, эрлотиниб, лапатиниб, NVP-AEE778, ARRY334543, BIRW2992, BMS690514, пелитиниб, вандетаниб, AV412, анти-EGFR моноклональное антитело 806, анти-EGFR моноклональное антитело-Y90/Re-188, цетуксимаб, панитумумаб, матузумаб, нимотузумаб, залутумумаб, MDX-214, CDX110, IMC11F8, пертузумаб, трастузумаб, TDM1, Земаб®, Her2 вакцина PX 1041 и HSP90-ингибиторы CNF1010, CNF2024, танеспимицин, алвеспимицин, IPI504, SNX5422 и NVP-AUY922, при этом активные ингредиенты во всех случаях присутствуют в свободной форме или в форме соли, и необязательно присутствует по меньшей мере один фармацевтически приемлемый носитель, для одновременного, раздельного или последовательного введения в целях лечения EGFR-зависимых заболеваний, включающих в себя, например, немелкоклеточный рак легкого, рак головы и шеи, колоректальный рак, рак молочной железы, злокачественные опухоли мозга, включающие в себя глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и/или рак яичников.
В частности, настоящее изобретение относится к комбинации соединения формулы I, выбираемой из группы, состоящей из 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты и EGFR-модулятора, выбираемого из группы, состоящей из гефитиниба, эрлотиниба, лапатиниба, цетуксимаба, нимотузумаба, панитумумаба, трастузумаба и TDM1, в котором активные ингредиенты во всех случаях присутствуют в свободной форме или в форме фармацевтически приемлемой соли, и необязательно присутствует по меньшей мере один фармацевтически приемлемый носитель, для одновременного, раздельного или последовательного введения в целях лечения немелкоклеточного рака легкого, рака головы и шеи, колоректального рака, рака молочной железы, злокачественных опухолей мозга, включающих в себя глиобластому, рака предстательной железы, рака мочевого пузыря, почечно-клеточной карциномы, рака поджелудочной железы, рака шейки матки, рака пищевода, рака желудка и рака яичников.
В другом варианте настоящее изобретение относится к способу лечения EGFR-зависимых заболеваний или злокачественных опухолей, предпочтительно злокачественных опухолей с приобретенной резистентность к модуляторам киназы EGFR в ходе лечения указанными EGFR-модулятором, и указанный способ содержит введение теплокровным животным, нуждающимся в таком введении, терапевтически эффективного количества конкретного производного 2-карбоксамид циклоамино-мочевины с формулой I, в особенности, введение предпочтительного соединения 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его фармацевтически приемлемой соли, в качестве единственного агента или в комбинации с EGFR-модулятором.
Заболевания, подлежащие лечению с помощью упомянутого способа, преимущественно представляют собой немелкоклеточный рак легкого рак, рак головы и шеи, колоректальный рак, рак молочной железы, злокачественные опухоли мозга, включающие в себя глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и рак яичников.
В другом варианте настоящее изобретение относится к фармацевтической композиции для лечения EGFR-зависимых заболеваний или заболеваний с приобретенной резистентностью в ходе лечения EGFR-модулятором, и упомянутая фармацевтическая композиция содержит соединение формулы I, в частности, предпочтительное соединение 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его соль, и по меньшей мере один фармацевтически приемлемый носитель, в качестве единственного агента или в комбинации с EGFR-модулятором.
Заболевания, подлежащие лечению упомянутой фармацевтической композицией, преимущественно представляют собой немелкоклеточный рак легкого рак, рак головы и шеи, колоректальный рак, рак молочной железы, злокачественные опухоли мозга, включающие в себя глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и рак яичников.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы I, в частности, предпочтительного соединения 2-амид 1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты (соединение А) или его соли, для лечения EGFR-зависимых заболеваний или заболеваний с приобретенной резистентностью в ходе лечения EGFR-модулятором.
Заболевания, подлежащие лечению упомянутыми соединениями в качестве единственных агентов или в комбинации с EGFR-модулятором, преимущественно представляют собой немелкоклеточный рак легкого, рак головы и шеи, колоректальный рак, рак молочной железы, злокачественные опухоли мозга, включающие в себя глиобластому, рак предстательной железы, рак мочевого пузыря, почечно-клеточную карциному, рак поджелудочной железы, рак шейки матки, рак пищевода, рак желудка и рак яичников.
Соединение формулы (I) также можно применять для достижения полезного эффекта в комбинации с известными способами лечения, например, с введением гормонов или, в особенности, с облучением. В частности, соединение формулы (I) можно использовать в качестве радиосенсибилизатора, в особенности для лечения опухолей, которые проявляют слабую чувствительность к лучевой терапии.
Лечение согласно изобретению может быть симптоматическим или профилактическим.
Соединение формулы I можно вводить в качестве единственного агента или в комбинации с одним или более одного другим терапевтическим соединением; возможная комбинированная терапия может проводиться в форме фиксированных комбинаций, или введение соединения по изобретению и одного или более одного другого терапевтического соединения проводят поочередно или независимо друг от друга, или выполняют комбинированное введение фиксированных комбинаций и одного или более одного другого терапевтического соединения.
Доза активного ингредиента зависит от множества факторов, включающих в себя тип, вид, возраст, вес, пол и состояние здоровья пациента, степень тяжести состояния, подлежащего лечению, пути введения, функции почек и печени пациента и конкретного используемого соединения. Рядовые специалисты в данной области (лечащий врач, клиницист или ветеринар) могут легко определить, и назначить эффективное количество лекарственного средства, необходимого для предотвращения, противодействия или прекращения прогрессирования этого состояния. Для оптимальной точности достижения концентрации препарата в пределах диапазона, в результате которого достигается эффективность, необходима схема введения, основанная на кинетике доступности лекарственных препаратов в целевых зонах. Это подразумевает оценку распределения, равновесного состояния и выведения лекарственного препарата.
Соединения по изобретению можно вводить любым общепринятым путем, в частности, парентерально, например, в форме инъекционных растворов или суспензий, энтерально, например, перорально, например, в форме таблеток или капсул, местно, например, в форме лосьонов, гелей, мазей или кремов, или интраназально, или в форме суппозиториев. Местное применение, например, осуществляют путем нанесения на кожу. Дополнительной формой для местного применения является введение в глаз. Фармацевтические композиции, содержащие соединение по изобретению в комбинации по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем, могут быть изготовлены общепринятым способом путем смешивания с фармацевтически приемлемым носителем или разбавителем.
Фармацевтические композиции содержат соединение формулы I или его соли в количестве, эффективном для лечения одного из вышеупомянутых заболеваний, вместе с фармацевтически приемлемыми носителями, которые подходят для местного, энтерального, например, перорального или ректального, или парентерального введения, и которые могут представлять собой неорганические или органические соединения в твердом или жидком виде. Существуют фармацевтические композиции для перорального введения, в частности, таблетки или желатиновые капсулы, которые содержат активный ингредиент вместе с разбавителями, например, с лактозой, глюкозой, маннитом и/или глицерином, и/или с лубрикантами, и/или с полиэтиленгликолем. Таблетки также могут содержать связующие вещества, например, алюмосиликат магния, крахмалы, например, кукурузный, пшеничный или рисовый крахмал, желатин, метилцеллюлозу, карбоксиметилцеллюлозу натрия и/или поливинилпирролидон, и, если желательно, дезинтегрирующие вещества, например, крахмал, агар, альгиновую кислоту или ее соль, например, альгинат натрия, и/или шипучие смеси, или адсорбенты, красители, ароматизаторы и подсластители. Также можно использовать фармакологически активные соединения настоящего изобретения в виде композиций для парентерального введения или в форме инфузионных растворов. Фармацевтические композиции можно подвергать стерилизации, и/или они могут содержать вспомогательные вещества, например, консерванты, стабилизаторы, увлажняющие соединения и/или эмульгаторы, солюбилизаторы, соли для регуляции осмотического давления и/или буферы. Фармацевтические композиции настоящего изобретения, которые могут, если это желательно, содержать другие фармакологически активные вещества, получают способом, известным per se, например, с помощью общепринятого смешивания, гранулирования, фасовки, растворения или лиофилизации, и содержат активный ингредиент (ингредиенты) в количестве приблизительно от 1% до 99%, в частности, приблизительно от 1% приблизительно до 20%.
Следующие примеры иллюстрируют описанное выше изобретение; вместе с тем, эти примеры не предназначены для какого-либо ограничения объема изобретения. Полезные эффекты фармацевтической комбинации настоящего изобретения также можно продемонстрировать в других тестовых моделях, известных специалистам в данной области.
Пример 1
Эффект соединения А на модели ксенотрансплантатов ВТ474 рака молочной железы
Эксперименты проводили с самками HsdNpa:бестимусных голых-ню мышей в возрасте приблизительно 8-12 недель на начало лечения. Все животные были размещены в оптимальных гигиенических условиях в клетках Makrolon тип III (максимально 10 животных в каждой клетке) со свободным доступом к пище и воде.
Клетки BT-474, представляющие собой человеческие клетки карциномы протоков молочной железы, и амплифицированные ErbB2 с мутацией PIK3CA, K11N1, выращивали в питательной среде DMEM, содержащей 4,5 г/л глюкозы с добавлением 10% инактивированной нагреванием эмбриональной телячьей сыворотки FCS, 2 мМ L-глутамина, 1 мМ пирувата натрия и инкубировали при 37°С в увлажненной атмосфере 5% CO2. Реагенты для клеточных культур были приобретены в компании BioConcept (Allschwil, Switzerland).
Опухоли BT-474 создавали in vivo путем ортотопической инъекции 3×106 клеток в 30 мкл HBSS (Sigma # H8264), содержащем матригель 1 мг/мл (BD # 34234) в третью молочную железу с правой стороны животных. Эксперименты определения эффективности начинали, когда средний размер опухоли достигал примерно 130 мм3 (на 14-16 день после инъекции клеток). Фармакокинетический/фармакодинамический эксперимент (PK/PD) проводили при достижении размеров опухоли 150-250 мм3 (на 22-й день после инъекции клеток). В день инъекции клеток осуществляли подкожную имплантацию пеллета с 0,025 17β эстрадиола в левый бок каждого экспериментального животного под ингаляционным наркозом Forene (R).
Рецептуру соединения А создавали в растворителях N-метилпирролидон/ПЭГ300/Солютол HS15/вода (10:30:20:40% об./об.). Соединение полностью растворялось в NMP(N-метилпирролидон). Затем добавляли полиэтиленгликоль PEG300 и сжиженный Солютол (последовательно с перемешиванием на вортексе после добавления каждого реагента). Воду добавляли непосредственно перед введением животным. Соединение А или носитель вводили перорально в объеме 10 мл/кг.
Объемы опухолей измеряли с помощью калипера и определяли по формуле: длина × диаметр2 × ττ/6. Противоопухолевая активность выражается как % T/C (среднее изменение объема опухоли у леченных животных/среднее изменение объема опухоли у контрольных животных)×100. Показатели регрессии (%) рассчитывали по формуле (средний объем опухоли в конце лечения - средний объем опухоли в начале лечения)/средний объем опухоли в начале лечения)×100. Массу тела и объем опухоли регистрировали два раза в неделю.
Данные представляли в виде среднего значения ± SEM, в случаях, когда это применимо. Во всех тестах уровень значимости устанавливали на уровне р<0,05. Для значений объемов опухолей проводили сравнения между группами лечения и контрольной группой с введением носителя с помощью одностороннего ANOVA-анализа с апостериорным тестом Даннетта. Уровень значимости изменения массы тела внутри группы между началом и окончанием периода лечения определяли с помощью парного t-теста. Сравнения дельта значений массы тела между группами лечения и контроля с введением носителя проводили с помощью одностороннего ANOVA-анализа с апостериорным тестом Даннетта.
В первом эксперименте голым мышам с ортотопической ксенотрансплантацией опухоли BT-474 вводили соединение А перорально ежедневно в дозе 50 мг/кг. Контрольная группа с введением носителя состояла из животных, получавших ежедневно пероральное введение 10 мл/кг смеси NMP/PEG300/Солютол HS15/вода (10:30:20:40% об./об.). Соединение А, вводимое перорально в дозе 50 мг/кг один раз в сутки, вызывало среднее изменение объема опухоли на -29,7±15,6 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта) по сравнению с введением носителя (среднее изменение опухоли 315,1±16,4 мм3), с значением регрессии 23,0% в первом эксперименте.
Во втором эксперименте голым мышам с ортотопической ксенотрансплантацией опухоли BT-474 вводили соединение А перорально ежедневно в дозах 12,5, 25 и 50 мг/кг. Контрольная группа с введением носителя состояла из животных, получавших ежедневно пероральное введение 10 мл/кг смеси NMP/PEG300/Солютол HS15/вода (10:30:20:40% об./об.). Соединение А вызывало статистически значимый противоопухолевый эффект в группе получавшей соединение А в дозах 12,5, 25 и 50 мг/кг по сравнению с группой, получавшей носитель (р<0,05, ANOVA-анализ с апостериорным тестом Даннетта). (См. фиг.1). Соединение А, вводимое перорально в дозах 12,5, 25 и 50 мг/кг один раз в сутки, вызывает среднее изменение объема опухоли на 171,8±33,0 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта), 95,5+26,5 мм3 (р<0,01 ANOVA с апостериорным тестом Даннетта) и на 20,8±15,9 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта) соответственно, по сравнению с введением носителя (среднее изменение объема опухоли на 557,7±79,0 мм3) во втором эксперименте (см. фиг.3).
Соединение А хорошо переносится в дозах 12,5 и 25 мг/кг, о чем свидетельствует статистически не значимое изменение средней массы тела в группе, получавшей носитель (0,1±1,2%) и в группе, получавшей соединение А (1,4±1,2% и -1,2±1,2%, соответственно). Вместе с тем, в группе, получавшей соединение А в дозе 50 мг/кг, было выявлено статистически значимое среднее изменение массы тела 10,6±4,1% (р<0,05, с использованием парного t-теста) и 8,2±2,6% (р<0,05 с использованием парного t-теста) в этих двух экспериментах (см. фиг.2).
Пример 2
Эффект соединения А на модели ксенотрансплантатов рака желудка NCI-N87
Эксперименты проводили с самками HsdNpa:бестимусных голых-ню мышей в возрасте приблизительно 8-12 недель на начало лечения. Все животные были размещены в оптимальных гигиенических условиях в клетках Makrolon тип III (максимально 10 животных в каждой клетке) со свободным доступом к пище и воде.
Клетки NCI-N87 (полученные из Американской коллекции типовых культур), представляющие собой клетки желудочного происхождения и амплифицированные ErbB2, выращивали в питательной среде DMEM, содержащей 4,5 г/л глюкозы с добавлением инактивированной нагреванием 10% эмбриональной телячьей сыворотки FCS, 2 мМ L-глутамина, 1 мМ пирувата натрия. Клетки инкубировали при 37°С в увлажненной атмосфере 5% CO2. Сбор клеток осуществляли с трипсином (0,25% вес./об.) и ЭДТА (0,53 мМ), ресуспендировали в культуральной среде (с добавками) и подсчитывали в системе Casy®. Реагенты для клеточных культур были приобретены в компании BioConcept (Allschwil, Switzerland).
Опухоли NCI-N87 у самок HsdNpa:бестимусных голых-ню мышей прививали путем подкожной трансплантации фрагментов примерно по 40 мг с помощью троакар иглы. Лечение начинали, когда опухоли достигали размера примерно 110 мм3, на 25 день после имплантации.
Рецептуру соединения А создавали в NMP/ПЭГ300/Солютол HS15/вода (10:30:20:40% об./об.). Соединение полностью растворялось в NMP. Затем добавляли ПЭГ300 и сжиженный Солютол (последовательно со встряхиванием после добавления каждого реагента). Воду добавляли непосредственно перед введением животным.
Объемы опухолей измеряли с помощью калипера и определяли по формуле: длина × диаметр2 × ττ/6. Противоопухолевая активность выражается как % T/C (среднее изменение объема опухоли у леченных животных/среднее изменение объема опухоли у контрольных животных)×100. Показатели регрессии (%) рассчитывали по формуле (средний объем опухоли в конце лечения - средний объем опухоли в начале лечения)/средний объем опухоли в начале лечения)×100. Массу тела и объем опухоли регистрировали два раза в неделю.
Данные представляли в виде среднего значения ± SEM, в случаях, когда это применимо. Во всех тестах уровень значимости устанавливали на уровне р<0,05. Для значений объемов опухолей проводили сравнения между группами лечения и контрольной группой с введением носителя с помощью одностороннего ANOVA-анализа с апостериорным тестом Даннетта. Уровень значимости изменения массы тела внутри группы между началом и окончанием периода лечения определяли с помощью парного t-теста. Сравнения дельта значений массы тела между группами лечения и контроля с введением носителя проводили с помощью одностороннего ANOVA-анализа с апостериорным тестом Даннетта.
Животным с опухолью NCI-N87 вводили соединение А перорально ежедневно в дозе 50 мг/кг. Лечение начинали через 18 дней после имплантации опухоли и продолжали в течение 20 дней подряд. Контрольная группа с введением носителя состояла из животных, получавших ежедневно пероральное введение смеси NMP/ПЭГ300/Солютол HS15/вода (10:30:20:40% об./об.). Соединение А вызывало среднее изменение объема опухоли на -11,8±17,2 мм3 (р<0,01, ANOVA с апостериорным тестом Даннетта) по сравнению с введением носителя (среднее изменение опухоли 694,0±93,5 мм3). Соединение А хорошо переносится, о чем свидетельствует статистически не значимое изменение средней массы тела в группе, получавшей в эксперименте соединение А в дозе 50 мг/кг (3,8±2,1%) (см. фиг.4 и 5).
Пример 3
Эффект комбинации соединений и трастузумаба на модели ксенотрансплантатов ВТ474 рака молочной железы
Эксперименты проводили с самками HsdNpa:бестимусных голых-ню мышей в возрасте приблизительно 8-12 недель на начало лечения. Все животные были размещены в оптимальных гигиенических условиях в клетках Makrolon тип III (максимально 10 животных в каждой клетке) со свободным доступом к пище и воде.
Клетки BT-474, представляющие собой человеческие клетки карциномы протоков молочной железы, и амплифицированные ErbB2 с мутацией PIK3CA, K11N1, выращивали в питательной среде DMEM, содержащей 4,5 г/л глюкозы с добавлением инактивированной нагреванием 10% FCS, 2 мМ L-глутамина, 1 мМ пирувата натрия, и инкубировали при 37°С в увлажненной атмосфере 5% CO2. Реагенты для клеточных культур были приобретены в компании BioConcept (Allschwil, Switzerland).
Опухоли BT-474 прививали in vivo путем ортотопической инъекции 3×106 клеток в 30 мкл HBSS (Sigma #H8264), содержащем матригель 1 мг/мл (BD #34234) в третью молочную железу с правой стороны животных. Эксперименты определения эффективности начинали, когда средний размер опухоли достигал примерно 130 мм3 (на 14 день после инъекции клеток). В день инъекции клеток осуществляли подкожную имплантацию пеллета с 0,025 17β эстрадиола в левый бок каждого экспериментального животного под ингаляционным наркозом Forene (R).
Рецептуру соединения А создавали в смеси NMP/ПЭГ300/Солютол HS15/вода (10:30:20:40% об./об.). Соединение полностью растворялось в NMP. Затем добавляли ПЭГ300 и сжиженный Солютол (последовательно с перемешиванием на вортексе после добавления каждого реагента). Воду добавляли непосредственно перед введением животным. Соединение А или носитель вводили перорально в объеме 10 мл/кг. Трастузумаб восстанавливали в фосфатно-буферном растворе (ФБР) и вводили внутрибрюшинно три раза в неделю в концентрации 3 мг/кг.
Объемы опухолей измеряли с помощью калипера и определяли по формуле: длина × диаметр2 × ττ/6. Противоопухолевая активность выражается как % T/C (среднее изменение объема опухоли у леченных животных/среднее изменение объема опухоли у контрольных животных)×100. Показатели регрессии (%) рассчитывали по формуле (средний объем опухоли в конце лечения - средний объем опухоли в начале лечения)/средний объем опухоли в начале лечения)×100. Массу тела и объем опухоли регистрировали два раза в неделю.
Данные представляли в виде среднего значения ± SEM, в случаях, когда это применимо. Во всех тестах уровень значимости устанавливали на уровне р<0,05. Для значений объемов опухолей проводили сравнения между группами лечения и контрольной группой с введением носителя с помощью одностороннего ANOVA-анализа с апостериорным тестом Даннетта. Уровень значимости изменения массы тела в группе между началом и окончанием периода лечения определяли с помощью парного t-теста. Сравнения дельта значений массы тела между группами лечения и контроля с введением носителя проводили с помощью одностороннего ANOVA-анализа и апостериорным тестом Даннетта.
Дополнительно, проводили аппроксимацию лекарственных взаимодействий согласно методике, описанной R. Clarke, Breast Cancer Res. Treat (1997) 46:255-278. Эту методику применяли в отношении изменения объемов опухолей, и она считается полезной для оценки взаимодействия при ограниченных данных. Согласно методике, описанной R. Clarke: для соединения A, B или комбинации AB (с контрольной группой C) предполагается антагонизм, если расчеты демонстрируют AB/C>A/C×B/C, предполагается аддитивный эффект, если AB/C=А/C×B/C, и синергетический эффект, если А×B/C<А/C×B/C.
Соединение А вводили животным с опухолью BT-474 перорально ежедневно в качестве единственного агента в дозе 12,5 мг/кг. Трастузумаб в качестве единственного агента вводили в виде внутрибрюшинных инъекций в дозе 3 мг/кг три раза в неделю. Соединение А вызывало статистически значимый противоопухолевый эффект (р<0,05, ANOVA) с показателем T/C 21,5%. Трастузумаб также вызывал статистически значимый противоопухолевый эффект (р<0,05, ANOVA) с показателем T/C 35,8%. Анализ комбинационного взаимодействия согласно методике, описанной R. Clark., Breast Cancer Res. Treat (1997) 46:255-278, указывал на наличие синергетического противоопухолевого эффекта комбинации в дозах 12,5 мг и 3 мг/кг три раза в неделю, по сравнению с введением единственного агента (см. фиг.6)
Тем не менее указанная комбинация имела только статистические отличия от соединения А в качестве единственного агента (критерий суммы рангов Манна-Уитни), и не отличалась от трастузумаба в качестве единственного агента. Лечение хорошо переносится, поскольку не наблюдалось значительной потери массы тела.
Соединение А вводили животным с опухолью BT-474 перорально ежедневно в качестве единственного агента в дозе 50 мг/кг. Трастузумаб в качестве единственного агента вводили в виде внутрибрюшинных инъекций в дозе 10 мг/кг три раза в неделю. Соединение А вызывало статистически значимый противоопухолевый эффект (р<0,05, критерий суммы рангов Манна-Уитни). Трастузумаб также вызывал статистически значимый противоопухолевый эффект (р<0,05, критерий суммы рангов Манна-Уитни). Анализ комбинационного взаимодействия по методике, описанной R. Clark., Breast Cancer Res. Treat (1997) 46:255-278, указывал на наличие синергетического противоопухолевого эффекта комбинации соединения А в дозе 50 мг в сутки и трастузумаба в дозе 10 мг/кг три раза в неделю, по сравнению с введением единственного агента (см. фиг.7).
Значительная потеря массы тела наблюдалась при введении соединения А в качестве единственного агента или в комбинации с трастузумабом (примерно 12% и примерно 10%, соответственно).
Пример 4
Эффект соединения А в качестве единственного агента и в комбинации с трастузумабом на модели ксенотрансплантатов рака желудка NCI-N87
Эксперименты проводили с самками мышей СВ17 SCID (Fox Chase SCID®, СВ17/ICR-PrkdcSCID, Charles River) в возрасте примерно 7-8 недель и с массой тела (МТ) в диапазоне от 14,7 до 21,2 г на начало лечения. Все животные имели свободный доступ к пище и воде (обратный осмос, 1 ppm CI) с модифицированным и облученным рационом NIH 31 (Modified and Irradiated Lab Diet®), состоящим из 18,0% сырого белка, 5,0% сырого жира и 5,0% сырой клетчатки. Мышей помещали на облученную обогащенным кобальтом подстилку для лабораторных животных (Enrich-o'cobs™ Laboratory Animal Bedding) в статических микроизоляторах с 12-часовым световым циклом при температуре 21-22°С и влажности 40-60%.
Клетки NCI-N87 (полученные из Американской коллекции типовых культур), представляющие собой клетки желудочного происхождения, амплифицированные ErbB2, сохраняли в виде экспоненциально растущих культур в среде RPMI-1640 с добавлением 10% эмбриональной бычьей сыворотки, 2 мМ глутамина, 100 ед/мл пенициллина G натрия и 100 мкг/мл стрептомицина сульфата. Опухолевые клетки культивировали во флаконах для тканевых культур во влажном инкубаторе при температуре 37°C, в атмосфере 5% CO2 и 95% воздуха. Сбор клеток осуществляли с трипсином и суспендировали при концентрации 5×107 клеток/мл в холодном фосфатно-буферном растворе с 50% матригеля. Каждой мыши подкожно в правый бок вводили 1×107 клеток (0,2 мл клеточной суспензии). Объемы опухолей измеряли с помощью калиперов и определяли по формуле: ширина2 × длину/2; вес опухоли предположительно оценивали с учетом, что 1 мг эквивалентен 1 мм3 объема опухоли. Через девять дней после имплантации опухоли (1-й день исследования) мышей с отдельными объемами опухоли от 126 до 320 мм3 разделяли на 12 групп из восьми мышей, при этом средний объем опухоли в группе составлял от 231 до 239 мм3.
Рецептуру соединения А создавали в NMP/ПЭГ300/Солютол HS15/вода (10:30:20:40% об./об.). Соединение полностью растворялось в NMP. Затем добавляли ПЭГ300 и сжиженный Солютол (последовательно с перемешиванием на вортексе после добавления каждого реагента). Воду добавляли непосредственно перед введением животным. Свежие дозы раствора готовили еженедельно? и хранили при 4°C, в защищенном от света месте.
Трастузумаб (Герцептин®, Genentech, 21 мг/мл) каждый раз разводили физиологическим раствором (носитель 2) в каждый день введения.
Лечение начинали через 9 дней после имплантации опухолевых клеток и продолжали в течение 30 последовательных дней (или до достижения объема опухоли = 2000 мм3). Соединение А вводили животным с опухолью NCI-N87 перорально один раз в сутки, и трастузумаб вводили путем внутрибрюшинной инъекции (в/б) два раза в неделю в течение четырех недель. Контроль (группа 1) состояла из животных, получавших смесь NMP/ПЭГ300/Солютол HS15/вода (10:30:20:40% об./об.) (носитель 1) перорально (п/о) один раз в сутки и солевой раствор (носитель 2) путем внутрибрюшинной инъекции (в/б) два раза в неделю в течение четырех недель. При комбинированной терапии вводили трастузумаб в течение 30 минут после введения соединения А. Объем дозы (10 мл/кг подбирали по весу каждого животного, определяемый в день введения, за исключением выходных дней, когда делали перенос предыдущего показателя МТ. Группы 2-4 получали монотерапию соединением А в дозах 12,5, 25 и 50 мг/кг, соответственно. Группы 5 и 6 получали монотерапию трастузумабом в дозах 3 и 10 мг/кг, соответственно. Группы 7-9 получали соединение А в дозах 12,5, 25 и 50 мг/кг, соответственно, и в каждой группе комбинировали это введение с введением 3 мг/кг трастузумаба. Группы 10-12 получали соединение А в дозах 12,5, 25 и 50 мг/кг, соответственно, и в каждой группе это введение комбинировали с введением 10 мг/кг трастузумаба. Введение в группах 4, 9 и 12 было прекращено досрочно. Показатели массы тела регистрировали в дни 1-5, в каждый день введения (кроме выходных), и два раза в неделю в дальнейшем до конца исследования.
Противоопухолевую активность выражали как % T/C (среднее изменение объема опухоли у леченных животных/среднее изменение объема опухоли у контрольных животных)×100. Лечение, при котором значение T/C достигало 40% или меньше, можно классифицировать как потенциально терапевтически активное. Эффективность лечения также можно определять по числу регрессионных ответов, как: (а) частичную регрессию (PR), указывающую, что опухоль составляет 50% или меньше своего объема в первый день исследования в трех последовательных измерениях в исследовании, и эквивалентна или превышает 13,5 мм3 в одном или более чем одном из этих трех измерений, и (б) полную регрессию (CR), указывающую, что объем опухоли меньше чем 13,5 мм3 в течение трех последовательных измерений в ходе исследования.
Данные представлены в виде среднего значения ± SEM, в случаях, когда это применимо. Статистический и графический анализ проводили с помощью программы Prism 3,03 (GraphPad) для Windows. Во всех тестах уровень значимости различий определяли с помощью ANOVA, с помощью теста Бартлетта и апостериорного теста множественного сравнения Даннетта, для сравнения среднего изменения для каждой группы, получавшей лекарственный препарат, со средним значением для группы 1. Если в тесте Бартлетта для равных дисперсий были показаны значительные различия (P=0,0401), группы сравнивали с помощью непараметрического теста Крускала-Уоллиса, в котором были показаны значительные различия между средними изменениями объема (P<0,0001). Достоверность различий между средними изменениями объема в группах с введением лекарственного препарата и в группе 1 анализировали с помощью апостериорного теста множественного сравнения Данна. Двусторонние статистические анализы проводили при Р=0,05. Программа Prism оценивает результаты тестов, как незначительные (н/з) при Р>0,05, значительные (символ «*») при 0,01 Следующие результаты были получены при расчете статистической значимости с помощью теста Крускала-Уоллиса с апостериорным тестом множественного сравнения Данна:
Среди групп 7-9 с введением комбинации соединения А и 3 мг/кг трастузумаба, в группе 7 выявлено значительно более сильное подавление, чем в соответствующих группах 2 и 5 с монотерапией I (P<0,01). Группы 8 и 9 были исключены из статистической оценки из-за небольшой конечной численности группы.
Среди групп 10-12 с введением комбинации соединения А и 10 мг/кг трастузумаба, в группе 10 выявлено значительно более сильнее подавление, чем в группе 2 с монотерапией соединением А, и чем в группе 6 с монотерапией трастузумабом. В группе 11 выявлена значительная активность (P<0,001) и 6 частичных регрессий. Результаты в группе 6 с комбинированным введением были значительно выше, чем результаты группы 3 с монотерапией соединением А и в группе 6 с монотерапией трастузумабом. Группа 12 была исключена из статистических оценок из-за небольшой конечной численности группы.
Пример 5
Эффект соединения А в качестве единственного агента и в комбинации с трастузумабом в модели с ксенотрансплантатом HBCx-5 рака молочной железы
Эксперименты проводили с самками HSD:бестимусных голых Foxn1-ню мышей в возрасте примерно 8-10 недель и весом примерно от 19 до 25 грамм. Мышей оставляли для акклиматизации со свободным доступом к пище и воде в течение 6 дней до начала исследования. Если потеря массы тела достигала 15% по сравнению с первым днем лечения, животным давали рацион DietGel®.
Мышей разделяли на пять групп, в каждой из которых было десять мышей. Лечение начинали через 34 дня после имплантации ксенотрансплантатов опухоли HBCx-5. HBCx-5 представляет собой ксенотрансплантат, полученный из первичной карциномы молочной железы. Она представляет собой протоковую аденокарциному дикого типа TP53 с высокой экспрессией RB и сверхэкспрессией HER2. При имплантации ксенотрансплантата для анестезии мышам вводили 100 мг/кг кетамина гидроксихлорида и 10 мг/кг ксилазина. В межлопаточную область мышам прививали фрагмент опухоли размером 3 мм на 3 мм с применением асептической технологии. Животные были рандомизированы в зависимости от объема опухоли таким образом, чтобы средний объем опухоли и диапазон в группах лечения были статистически сходными. Опухоли измеряли с помощью калиперов три раза в неделю с начала введения. Опухоли рассчитывали по формуле: = ширина2 × длина/2. Размеры опухоли и массу тела измеряли три раза в неделю в течение всего периода лечения. Планируемой конечной точкой эксперимента была фаза лечения в шесть недель и отсутствие последующего этапа.
Соединение А суспендировали в 0,5% метилцеллюлозе (Sigma Aldrich, St. Louis, MO). Суспензию гомогенизировали путем перемешивания на вортексе и обработки ультразвуком. Суспензию сохраняли при 4°C в течение 7 дней в защищенном от света месте. Раствор трастузумаба (Герцептин®, Roche) в концентрации 1 мг/мл делали в каждый день введения. Стоковый раствор в концентрации 21 мг/мл разбавляли стерильной водой для инъекций перед введением в концентрации 1 мг/мл. Капецитабин (Кселода®, Roche) в концентрации 540 мг/мл сохраняли при 4°C в защищенном от света месте. Пеллеты капецитабина измельчали и суспендировали в 0,9% NaCl.
Соединение А вводили ежедневно через желудочный зонд (п/о) в дозе 50 мг/кг в течение 42 дней, в качестве единственного агента или в комбинации с трастузумабом. Трастузумаб вводили в дозе 10 мг/кг внутрибрюшинно один раз в неделю в течение 6 недель. Дозу в соответствии с массой тела подбирали для каждой инъекции.
Капецитабин вводили через желудочный зонд (п/о) в дозе 540 мг/мл пять раз в неделю в течение двух недель (то есть, введение пять дней подряд/в неделю). После одной недели перерыва проводили второй цикл лечения.
Противоопухолевую активность выражали как % T/C (среднее изменение объема опухоли у леченных животных/среднее изменение объема опухоли у контрольных животных)×100. Показатели объема опухоли и относительной массы тела использовали для статистического анализа. Групповое сравнение между группой лечения и контрольной группой проводили с использованием непараметрического теста Манна-Уитни.
В этом исследовании соединение А, вводимое в качестве единственного агента в дозе 50 мг/кг, проявляло значительную противоопухолевую активность на модели с ксенотрансплантатом опухоли HBCx-5 молочной железы (T/C=17,57%, р<0,001). Трастузумаб, вводимый в дозе 10 мг/кг в качестве единственного агента, не показывал какую-либо значительную противоопухолевую активность (T/C - 81,44%). Комбинация соединения А в дозе 50 мг/кг и трастузумаба в дозе 10 мг/кг показывала значительную противоопухолевую активность (T/C - 19,44%, р<0,001). Тем не менее противоопухолевая активность при комбинированном введении не имеет значительного отличия от введения соединения А в качестве единственного агента. Наблюдается отсутствие частичной или полной регрессии.
Соединение А при введении в качестве единственного агента или в комбинации с трастузумабом хорошо переносится. Максимальная потеря массы тела на 9,5% была достигнута через 10 дней после первого введения, и была стабильной до конца исследования. Эта потеря веса была значительной, но приемлемой. Не наблюдалось значительной потери веса при введении трастузумаба в качестве единственного агента. При введении соединения А в комбинации с трастузумабом наблюдалась значительная, но приемлемая потеря веса на 7,9%.
Капецитабин, вводимый в дозе 40 мг/кг в качестве единственного агента, проявлял высокую активность в модели с ксенотрансплантатом опухоли HBCx-5 молочной железы (T/C=9,03%, р<0,001). Капецитабин вызвал значительное и преходящее снижение массы тела на 9,1%.
Пример 6
Эффект соединения А в качестве единственного агента и в комбинации с лапатинибом в модели с ксенотрансплантатом NCI-N87 рака желудка
Изучали эффект соединения А в качестве монотерапии и в комбинации с лапатинибом на модели мышей СВ17 SCID с ксенотрансплантатом NCI-N87 рака желудка с использованием протокола исследования, описанного в примере 4. Самки мышей СВ17 SCID (Fox Chase SCID®, СВ17/ICR-PrkdcSCID, Charles River) были в возрасте 10 недель и имели массу тела (МТ) диапазоне от 15,8 до 21,9 г на 1-й день исследования. Клетки NCI-N87 собирали с применением 5х трипсина. Через восемь дней после имплантации опухоли (1-й день исследования) мышей, имевших отдельные объемы опухоли от 88 до 196 мм3, разделяли на 10 групп из десяти мышей, со средним объемом опухоли в группе от 143 до 149 мм3.
Вместо трастузумаба, используемого в примере 4, для введения применяли лапатиниб (Tykerb®, GlaxoSmithKline, таблетки 250 мг), суспендированный в смеси 0,5% гидроксипропилметилцеллюлоза: 0,1% Tween®80: 99,4% деионизированная вода (носитель 2). Свежую суспензию лапатиниба готовили один раз в неделю и сохраняли при 4°C в защищенном от света месте.
Лечение начинали через восемь дней после имплантации опухолевых клеток и продолжали в течение 59 последовательных дней или до достижения объема опухоли = 1000 мм3. Соединение А вводили перорально один раз в сутки животным с опухолью NCI-N87, и лапатиниб вводили перорально или два раза в сутки (2 р/в сутки) в течение 21 дней, или один раз в сутки в течение 21 дня. При введении 2 р/в сутки первую дозу вводили вечером в 1-й день и последнюю дозу вводили утром на 22-й день. При комбинированной терапии лапатиниб вводили в течение 30 минут после введения соединения А. Вводимый объем (10 мл/кг) подбирали по весу каждого животного, который определяли в день введения, за исключением выходных дней, когда переносили показатели предыдущего измерения МТ. Контроль (группа 1) состоял из животных, получавших смесь NMP/ПЭГ300/Солютол HS15/вода (10:30:20:40% об./об.) (носитель 1) перорально (п/о) один раз в сутки, и смесь 0,5% гидроксипропилметилцеллюлоза: 0,1% Tween®80: 99,4% деионизированная вода (носитель 2) перорально один раз в сутки. Группы 2 и 3 групп получали монотерапию в дозах 12,5 и 25 мг/кг соединения А, соответственно. Группа 4 получала монотерапию лапатинибом в дозе 50 мг/кг 2 р/в сутки. Группы 5 и 6 получали монотерапию в дозах 100 и 150 мг/кг лапатиниба, соответственно, один раз в сутки. Группы 7 и 8 получали соединение А в дозах 12,5 и 25 мг/кг, соответственно, один раз в сутки, и каждую дозу в комбинации с лапатинибом один раз в сутки. Группа 9 получала соединение А в дозе 12,5 мг/кг один раз в сутки в комбинации с 50 мг/кг лапатиниб 2 р/в сутки, при этом соединение А вводили вместе с вечерней дозой лапатиниба. Группа 10 получала 12,5 мг/кг соединения А один раз в сутки в комбинации с 150 мг/кг лапатиниба один раз в сутки. Лечение в группах 7, 8, 9 и 10 было прекращено досрочно. Массу тела регистрировали в дни 1-5, в каждый день лечения (кроме выходных), и два раза в неделю в дальнейшем до конца исследования.
Данные представлены в виде среднего значения ± SEM, в случаях, когда это применимо. Каждое животное подвергали эвтаназии, когда его опухоль достигала конечной точки объема (1000 мм3), или в последний день исследования (день 59). Время до конечной точки (TTE) рассчитывается по формуле: TTE=(log10 (конечная точка объема) - b)/m, где TTE выражается в днях, конечная точка объем в мм3, b обозначает свободный член уравнения регрессии, и m обозначает наклон линии, полученной в линейной регрессии log-трансформированной совокупности данных опухолевого роста. Эффективность лечения определяли по замедлению роста опухоли (TGD), которая была определена как увеличение медианы времени до конечной точки (TTE) в группе лечения по сравнению с контрольной группой (TGD=T-C), выраженное в днях, или в процентах медианы TTE в контрольной группе (% TGD=((Т-С)/C)×100), где T=медиана TTE в группе лечения и C=медиана TTE в обозначенной контрольной группе.
Статистический и графический анализ проводили с помощью программы Prism 3,03 (GraphPad) для Windows. Во всех тестах уровень значимости различий определяли с помощью ANOVA, с помощью теста Бартлетта и апостериорного теста множественного сравнения Даннетта, для сравнения среднего изменения для каждой группы, получавшей лекарственный препарат, со средним значением для группы 1. Выживаемость анализировали по методике Каплана-Мейера. Логранговый критерий использовали для анализа достоверности отличий между общей фактической выживаемостью двух групп, в зависимости от значения времени до конечной точки (TTE). Двусторонние статистические анализы проводили при Р=0,05. Программа Prism оценивает результаты тестов, как незначительные (н/з) при Р>0,05, значительные (символ «*») при 0,01 Следующие результаты были получены при расчете статистической значимости с помощью логрангового критерия:
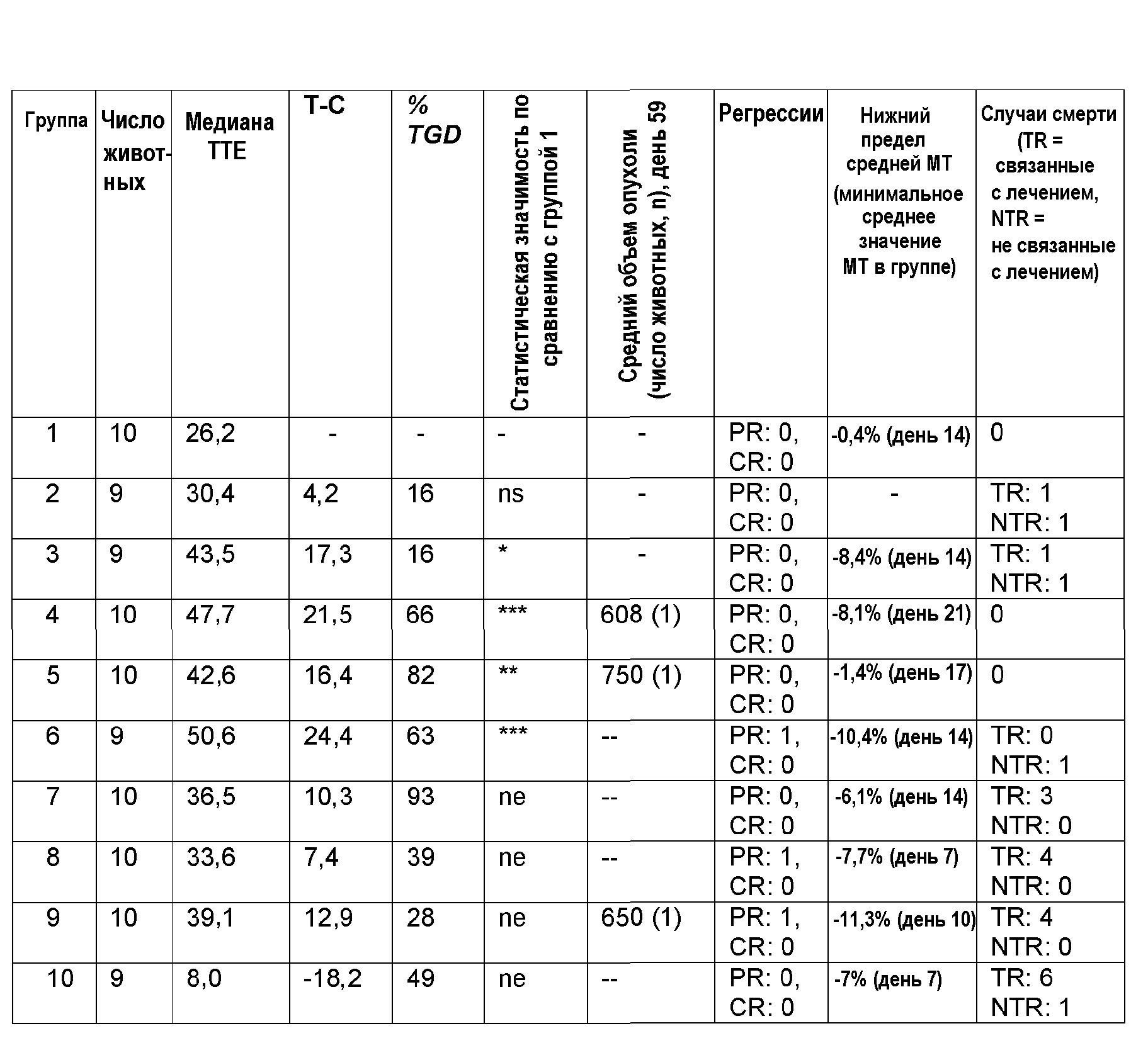
Реферат
Предложена группа изобретений, касающихся лечения злокачественных опухолей. Заявлены: применение 2-амид-1-({4-метил-5-[2-(2,2,2-трифтор-1,1-диметил-этил)пиридин-4-ил]тиазол-2-ил}амид) (S)-пирролидин-1,2-дикарбоновой кислоты или её соли для производства фармацевтических композиций для лечения злокачественной опухоли, комбинация данного соединения с EGFR-модуляторами в целях указанного использования, способ лечения указанного заболевания с помощью этого соединения, и фармацевтические композиции для лечения указанного заболевания. Технический результат: показана регрессия опухоли под действием заявленного соединения, показан синергизм противоопухолевого действия указанного соединения в комбинации с EGFR-модулятором. 4 н. и 6 з.п. ф-лы, 7 ил., 3 табл., 6 пр.
Формула
- немелкоклеточный рак легкого
- рак головы и шеи
- колоректальный рак
- рак молочной железы
- злокачественные опухоли мозга, включающие в себя глиобластому
- рак простаты
- рак мочевого пузыря
- почечно-клеточная карцинома
- рак поджелудочной железы
- рак шейки матки
- рак пищевода
- рак желудка
- рак яичников
или любую их комбинацию.
Комментарии