(2s,4r)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановая кислота - RU2726623C2
Код документа: RU2726623C2
Чертежи
Описание
УРОВЕНЬ ТЕХНИКИ
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новому соединению и его кристаллической форме, которое в процессе метаболизма in vivo превращается в соединение, эффективное в качестве ингибитора неприлизина. Изобретение также относится к фармацевтическим композициям, содержащим соединение, способам получения этого соединения и способам применения соединения для лечения таких заболеваний, как гипертония, сердечная недостаточность, легочная гипертензия и почечная болезнь.
УРОВЕНЬ ТЕХНИКИ
Неприлизин (нейтральная эндопептидаза, EC 3.4.24.11) (NEP) представляет собой Zn2+металлопептидазу, связанную с эндотелиальной мембраной, обнаруживаемую во многих органах и тканях, включая мозг, почки, легкие, желудочно-кишечный тракт, сердце и периферическую сосудистую сеть. NEP разрушает и инактивирует ряд эндогенных пептидов, таких как энкефалины, циркулирующий брадикинин, пептиды-ангиотензины и натрийуретические пептиды, последние из которых имеют несколько эффектов, включая, например, вазодилатацию и натрийурез/диурез, а также ингибирование гипертрофии сердца и фиброз желудочков. Таким образом, NEP играет важную роль в гомеостазе кровяного давления и в развитии сердечно-сосудистых заболеваниях.
Ингибиторы NEP, такие как тиофан, кандоксатрил и кандоксатрилат, были изучены в качестве потенциальных терапевтических агентов. Также известны соединения, которые ингибируют как NEP, так и ангиотензин-I превращающий фермент (ACE), такие как омапатрилат, гемпатрилат и сампатрилат. Этот последний класс соединений, известных как ингибиторы вазопептидазы, описан Robl et al. (1999) Exp. Opin. Ther. Patents 9(12): 1665-1677.
Многочисленные ингибиторы NEP описаны в публикации заявки на патент США № 2013/0109639 на имя Hughes et al. Одним из таких соединений является (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановая кислота, которая имеет структуру:
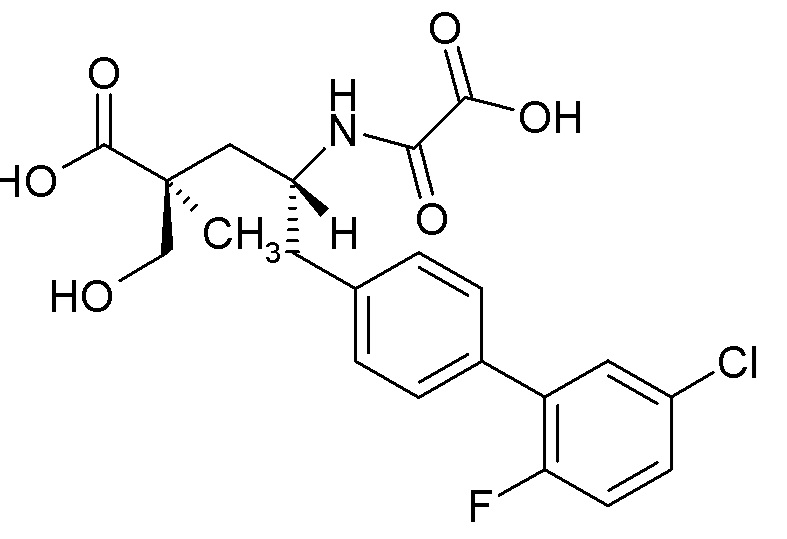
Это соединение демонстрирует мощную ингибирующую активность в отношении NEP (pKi≥9). Однако в доклинических исследованиях было показано, что это соединение имеет очень низкую пероральную биодоступность, что делает его непригодным или нежелательным для перорального введения.
Одним из способов увеличения пероральной биодоступности соединения является получение пролекарства этого соединения. При пероральном введении пролекарство должно иметь приемлемую абсорбцию в ротовой полости и расщепляться in vivo, образуя активное соединение. Что касается ингибиторов NEP, может оказаться предпочтительным, чтобы любое такое пролекарство быстро (например, в течение первого часа после перорального введения) и полностью расщеплялось, образуя начальный болюс активного соединения, способный активировать ответ циклического гуанозинмонофосфата (цГМФ). Также желательно, чтобы пролекарство само по себе имело ингибирующую активность в отношении NEP, в частности, чтобы оно было фармакологически активным до расщепления. Более того, любое такое пролекарство должно быть химически стабильным при хранении в течение длительного периода времени.
Таким образом, существует потребность в пролекарстве (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановой кислоты, которое имеет приемлемую пероральную абсорбции и которое быстро расщепляется in vivo, образуя активное соединение. Пролекарство также может иметь некоторую ингибирующую активность в отношении NEP. Данное изобретение направлено на удовлетворение этой потребности.
Кроме того, для эффективного применения соединения, являющегося ингибитором NEP в качестве терапевтического агента желательно, чтобы оно находилось в форме твердого вещества, которое может быть легко получено и которое имеет приемлемую химическую и физическую стабильность. Например, желательно получить физическую форму, которая является термически стабильной при достаточно высокой температуре, что облегчает обработку и хранение материала. Кристаллические твердые вещества обычно более предпочтительны чем аморфные с точки зрения улучшения чистоты и стабильности готового продукта. Однако образование кристаллических форм органических соединений крайне непредсказуемо. Не существует надежных способов предсказания, какая из форм органического соединения, если таковая имеется, будет кристаллической. Более того, не существует способов предсказания, какая из кристаллических форм, если таковая имеется, будет иметь физические свойства, необходимые для ее применения в качестве фармацевтического агента. Соответственно, имеется потребность в стабильной кристаллической форме, которая имеет достаточно высокую температуру плавления.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому соединению (1), которое превращается in vivo в соединение, обладающее ингибирующей активностью в отношении фермента неприлизина (NEP). Соответственно, ожидается, что это соединение будет полезным и эффективным в качестве терапевтического средства для лечения таких состояний, как гипертония и сердечная недостаточность.
Один из аспектов настоящего изобретения относится к (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановой кислоте (1):
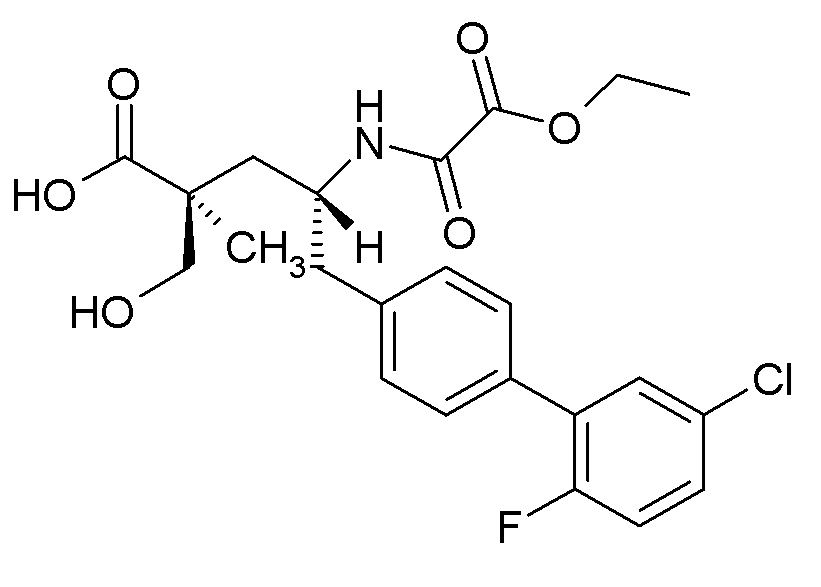
или ее фармцевтически приемлемой соли. Другой аспект настоящего изобретения относится к кристаллической форме соединения 1. В одном из вариантов осуществления кристаллическая форма (1') представляет собой гемикальциевую соль соединения 1.
Другой аспект изобретения относится к фармацевтическим композициям, содержащим один или более фармацевтически приемлемых носителей и соединение 1 или его кристаллическую форму. Такие композиции необязательно могут содержать другие терапевтические агенты, включая, без ограничения, антагонист АТ1-рецепторов, ингибитор ангиотензинпревращающего фермента, ингибитор фосфодиэстеразы (PDE), ингибитор ренина, диуретик или их комбинации.
Соединение 1 обладает ингибирующей активностью в отношении фермента NEP и поэтому предполагается, что оно окажется полезным в качестве терапевтического средства для лечения пациентов, страдающих заболеванием или расстройством, которое лечится путем ингибирования фермента NEP или путем увеличения уровней его пептидных субстратов. Таким образом, один из аспектов изобретения относится к способу лечения пациентов, страдающих заболеванием или расстройством, которое лечится путем ингибирования фермента NEP, включающим введение пациенту терапевтически эффективного количества соединения 1. Другой аспект изобретения относится к способу лечения гипертонии, сердечной недостаточности или заболевания почек, включающему введение субъекту терапевтически эффективного количества соединения 1. Еще один аспект изобретения относится к способу ингибирования фермента NEP у субъекта, включающему введение субъекту, количество соединения 1, ингибирующее NEP.
Поскольку соединение 1 обладает ингибирующей активностью в отношении NEP, оно также полезно в качестве инструмента исследования. Соответственно, один из аспектов изобретения относится к способу применения соединения 1 в качестве инструмента исследования, причем способ включает проведение биологического анализа путем использования соединения 1. Соединение 1 также можно использовать для оценки новых химических соединений. Таким образом, другой аспект изобретения относится к способу оценки испытуемого соединения в биологическом анализе, включающему: (а) проведение биологического анализа с использованием соединения, получая первое аналитическое значение; (b) проведение биологического анализа с использованием соединения по изобретению, получая второе аналитическое значение; где этап (а) выполняют либо до, либо после, либо одновременно с этапом (b); и (c) сравнение первого аналитического значения, полученного на этапе (а), со вторым аналитическим значением, полученным на этапе (b). Примеры биологических анализов включают анализ ингибирования фермента NEP. Еще один аспект изобретения относится к способу изучения биологической системы или образца, содержащего фермент NEP, причем способ включает: (а) взаимодействие биологической системы или образца с соединением по изобретению; и (b) определение эффектов, оказываемых соединением на биологическую систему или образец.
Еще один аспект изобретения относится к способам, применяемым для получения соединения 1 или его кристаллической формы.
Еще один аспект изобретения относится к применению соединения 1 или его кристаллической формы для изготовления лекарственного средства, в частности для изготовления лекарственного средства, которое можно использовать для лечения гипертонии, сердечной недостаточности или заболевания почек. Другой аспект изобретения относится к применению соединения 1 или его кристаллической формы для ингибирования фермента NEP у млекопитающего. В настоящем описании раскрыты и другие аспекты и варианты осуществления изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Различные аспекты настоящего изобретения проиллюстрированы со ссылкой на прилагаемые чертежи.
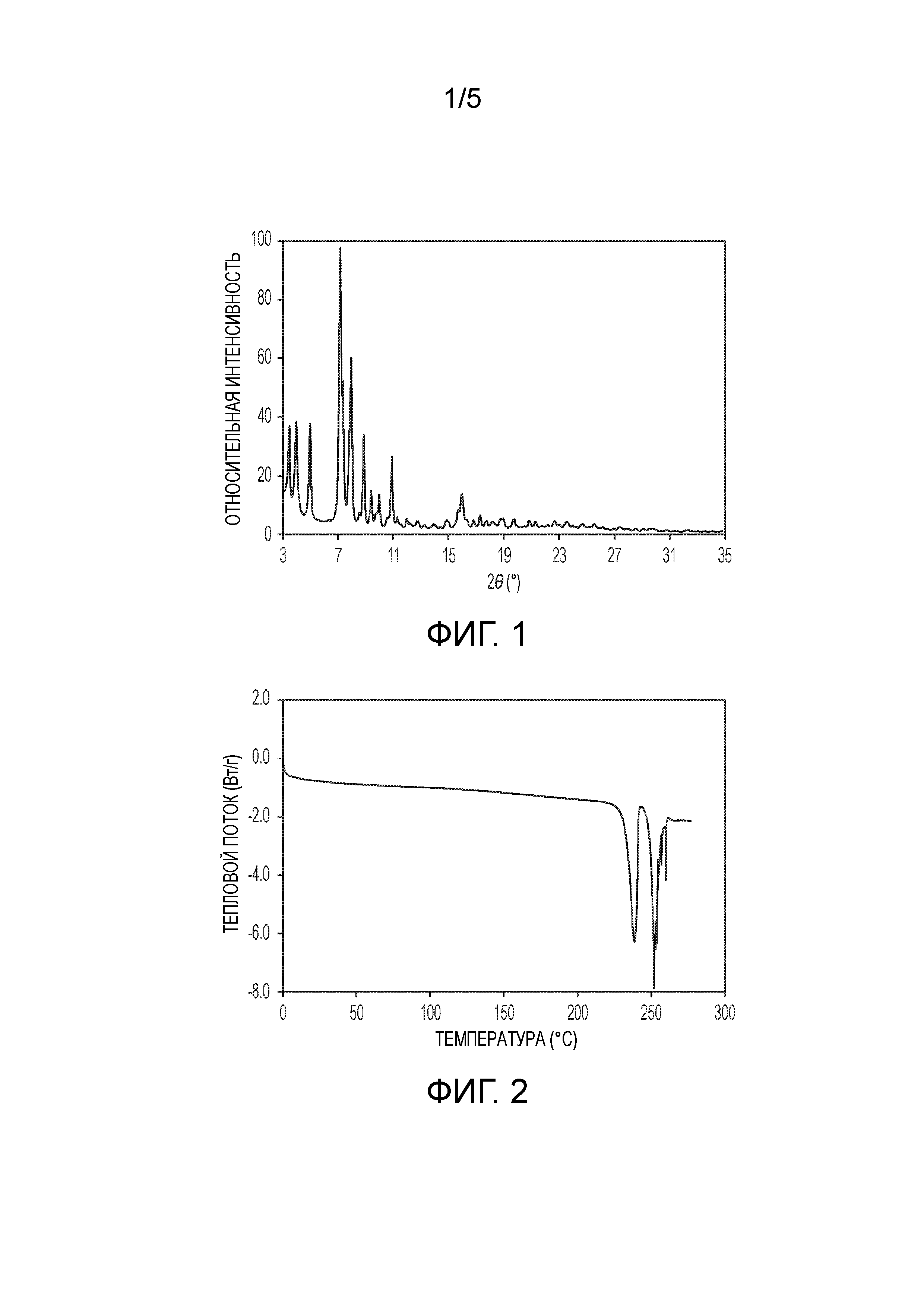
На фиг.1 показана порошковая рентгеновская (PXRD) дифрактограмма кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата кальция (1').
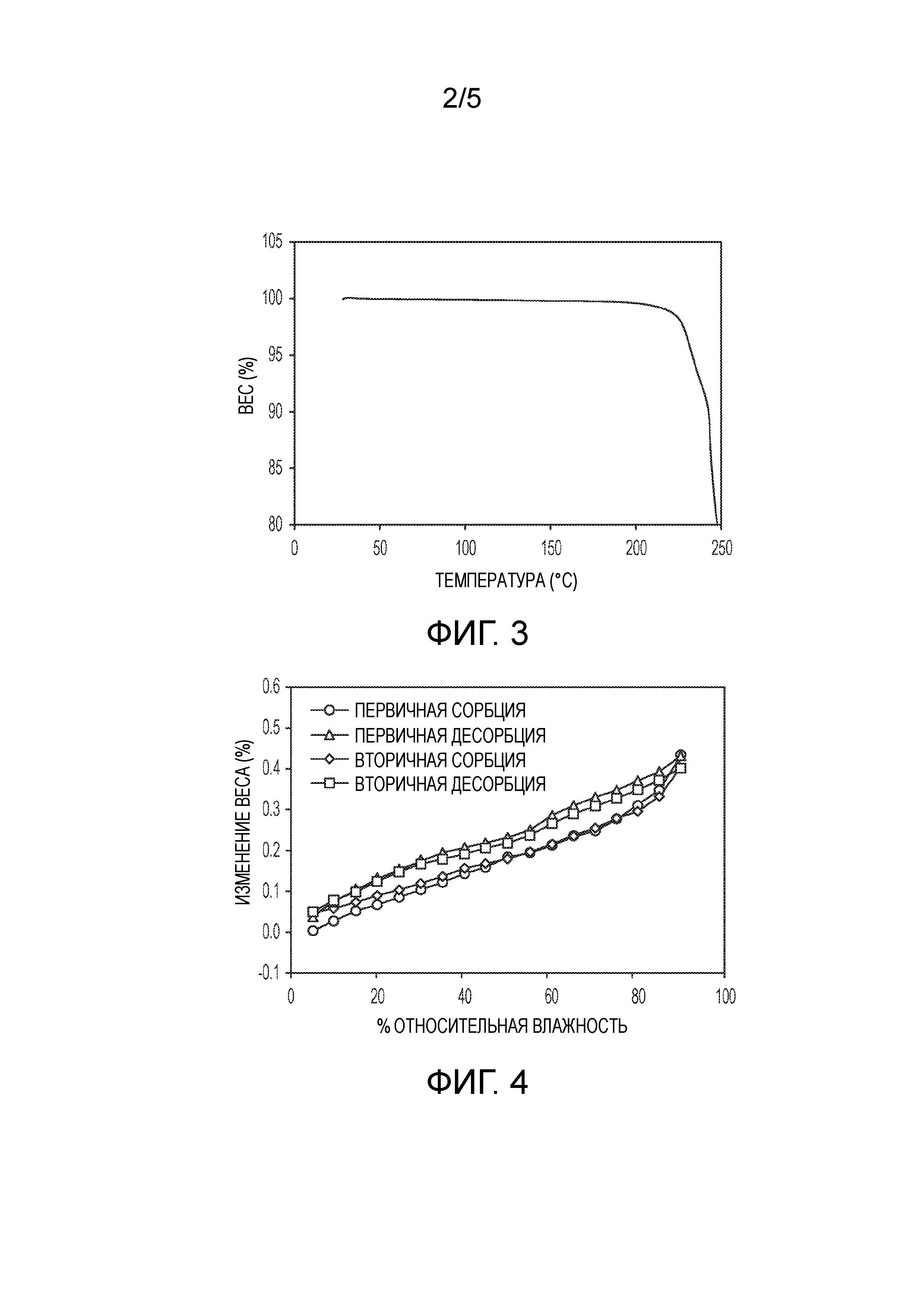
На фиг.2 показана термограмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2- (гидроксиметил)-2-метилпентаноата кальция (1').
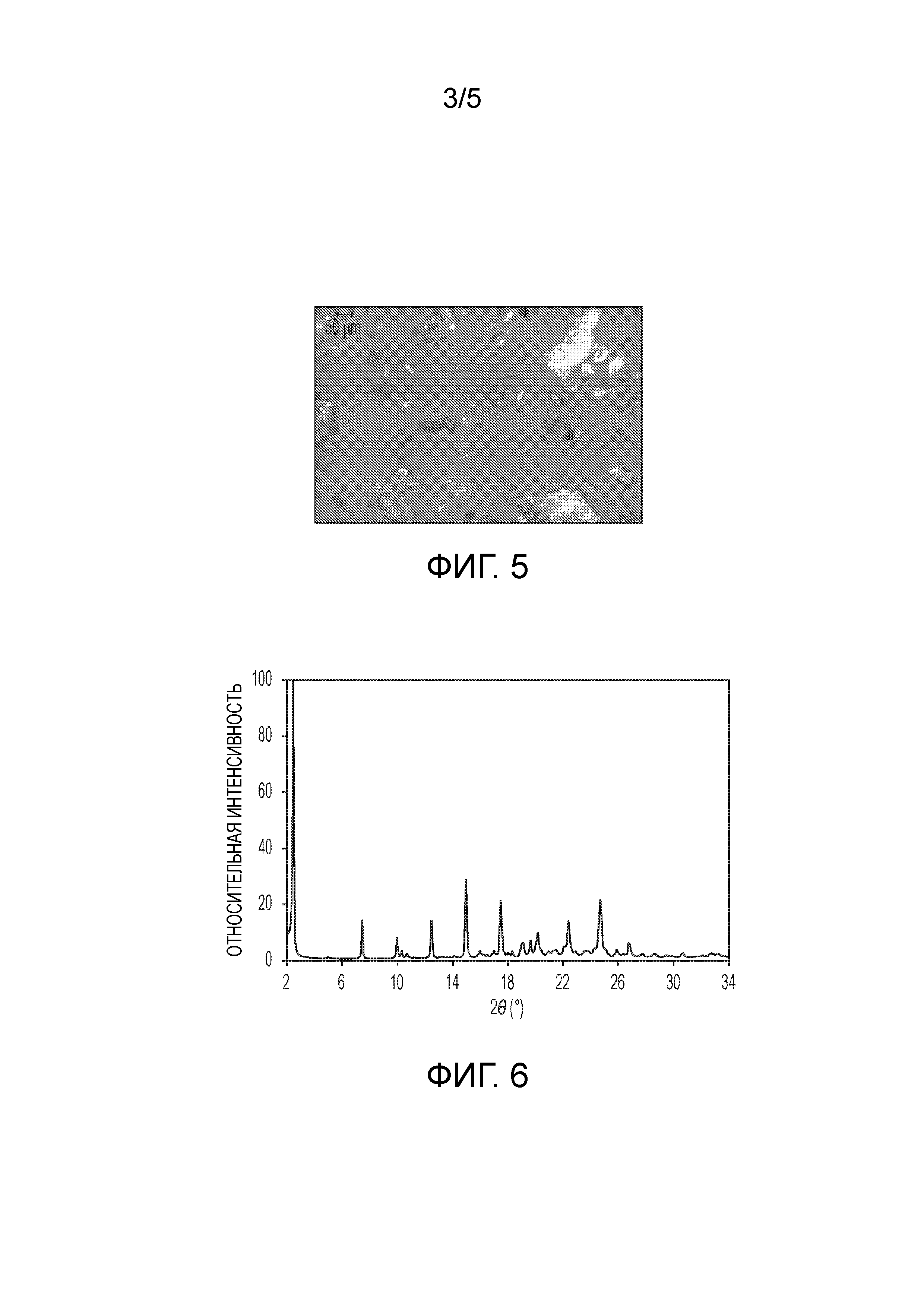
На фиг.3 показан график термического гравиметрического анализа (TGA) кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата кальция (1').
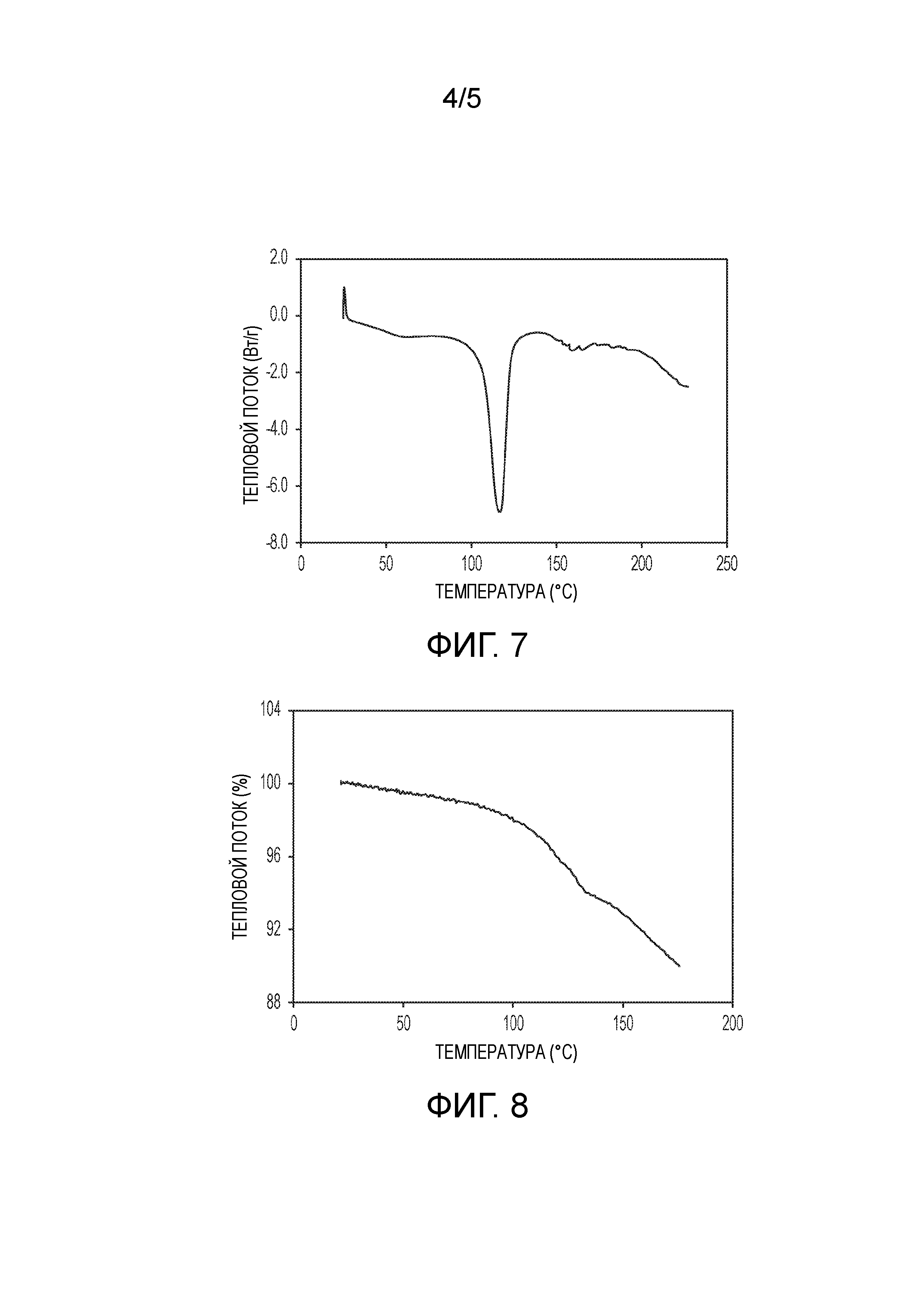
На фиг.4 показана изотерма динамической адсорбции влаги (DMS) кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата кальция (1').
На фиг.5 показано микроскопическое изображение в поляризованном свете (PLM) кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата кальция (1').
На фиг.6 показана порошковая рентгеновская (PXRD) дифрактограмма кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноат аргинина (1").
На фиг.7 показана термограмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата аргинина (1").
На фиг.8 показан график термического гравиметрического анализа (TGA) кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата аргинина (1").
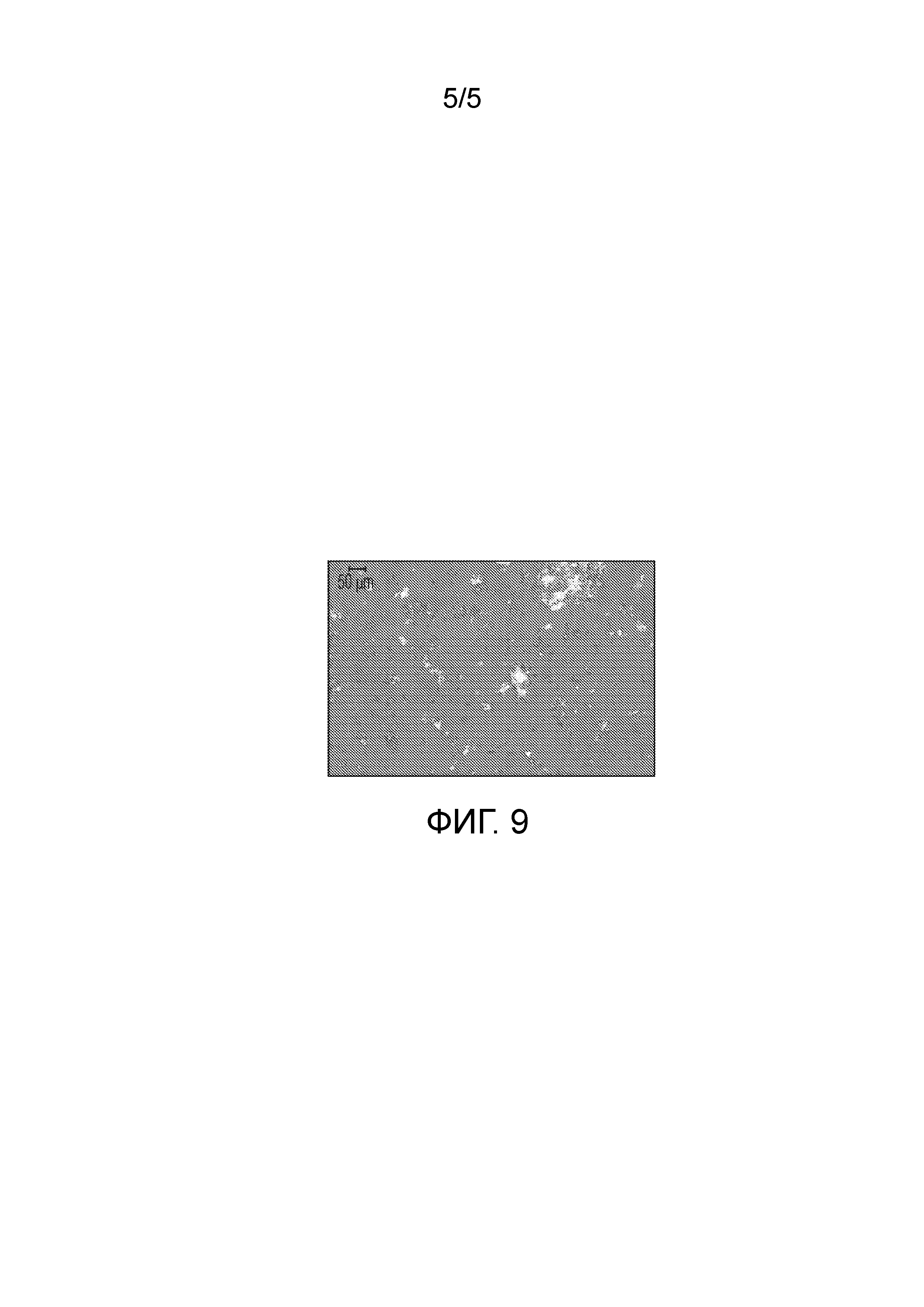
На фиг.9 показано микроскопическое изображение в поляризованном свете (PLM) кристаллической формы (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата аргинина (1").
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В одном из аспектов изобретение относится к (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановой кислоте (1) или ее фармацевтически приемлемой соли.
Соединение 1 по изобретению содержит два хиральных центра, и поэтому это соединение может быть получено и использовано в различных стереоизомерных формах. В частности, атомы углерода имеют конкретную (R,R), (S,S), (S,R) или (R,S) конфигурацию или обогащены стереоизомерной формой, имеющей такую конфигурацию. Показанное и названное соединение 1 находится в конфигурации (S,R). Специалистам в данной области техники будет понятно, что в композициях по изобретению могут присутствовать незначительные количества других стереоизомеров, если не указано иное, при условии, что наличие таких других изомеров не оказывает негативного влияния на эффективность композиции в целом. Отдельные стереоизомеры могут быть получены многочисленными способами, которые хорошо известны в данной области, включая хиральную хроматографию с подходящей хиральной стационарной фазой или носителем или путем их химического превращения в диастереоизомеры, разделения диастереоизомеров обычными способами, такими как хроматография или перекристаллизация, а затем восстановления исходного стереоизомера.
Соединение 1 представляет собой этиловый эфир пролекарства, где этильная часть служит для улучшения липофильности соединения и, следовательно, для улучшения проницаемости активного соединения через пассивную мембрану путем маскирования карбоновой кислоты. Предполагается, что сразу при попадании в организм эта сложноэфирная связь гидролизуется эстеразами, которые содержатся в крови, печени и других органах и тканях.
Было обнаружено, что соединение 1 по изобретению ингибирует фермент неприлизин (NEP). Кроме того, это соединение более эффективно в отношении ингибирования NEP после его превращения in vivo. Таким образом, говоря об активности соединения по изобретению, ясно, что в анализе соединение может проявлять некоторый уровень активности и улучшенный уровень активности после его превращения в активную форму. Одним из показателей способности соединения ингибировать активность NEP является константа ингибирования (pKi). Значение pKi представляет собой отрицательный логарифм по основанию 10 константы диссоциации (Ki), которая обычно выражается в молярных единицах. В процессе метаболизма соединение 1 образует (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановую кислоту ("активный агент или активный метаболит"), который, согласно публикации заявки на патент США № 2013/0109639, имеет в отношении NEP pKi≥9. Другие свойства и применения соединения 1 могут быть продемонстрированы в in vitro и in vivo анализах, которые хорошо известны специалистам в данной области, включая, среди прочего, методы анализа, описанные в публикации заявки на патент США № 2013/0109639.
Соединение 1, а также соединения, которые используются в процессе его синтеза, также могут включать соединения, меченые изотопами, т.е. соединения, у которых один или более типов атомов обогащены атомами, имеющими атомную массу, отличную от атомной массы, преимущественно обнаруживаемой в природе. Примеры изотопов, которые могут быть включены в соединения формулы I, например, включают, без ограничения,2H,3H,13C,14C,15N,18O,17O,35S,36Cl и18F. Особый интерес представляет соединение 1, обогащенное тритием или углеродом-14, которое может быть использовано, например, в исследованиях распределения в тканях; соединение 1, обогащенное дейтерием, особенно на участке метаболизма, где оно превращается, например, в соединение с более высокой метаболической стабильностью; и соединение 1, обогащенное позитрон-излучающим изотопом, таким как11C,18F,15O и13N, которое может быть использовано, например, в исследованиях методом позитронно-эмиссионной томографии (ПЭТ).
В настоящем описании названия химических структур даны согласно перечню ИЮПАК с использованием программного обеспечения ChemDraw (Perkin Elmer, Inc., Cambridge, MA).
ОПРЕДЕЛЕНИЯ
При описании соединения, композиций, методов и способов по изобретению приведенные ниже термины имеют следующие значения, если не указано иное. Кроме того, используемые в настоящем описании формы единственного числа включают соответствующие формы множественного числа, если из контекста в явном виде не следует иное. Термины "содержащий", "включающий" и "имеющий" предназначены для указания включения и означают возможность наличия дополнительных элементов, кроме перечисленных. Все численные значения, выражающие количество ингредиентов, такие свойства, как молекулярная масса, условия реакции и т.д., используемые в настоящем описании, следует понимать как модифицированные во всех случаях термином "примерно", если не указано иное. Соответственно, приведенные в настоящем описании численные значения являются приближениями, которые могут варьировать в зависимости от желаемых свойств, необходимых согласно настоящему изобретению. Не пытаясь ограничить применение доктрины эквивалентов объемом формулы изобретения, каждое численное значение следует трактовать, по меньшей мере, в свете указанных значимых цифр и путем округления при помощи обычных методов округления.
Термин "примерно" или "приблизительно", если используется в контексте поведения соединения 1 при термическом воздействии, означает ±1-3°C. Термин "приблизительно", когда используется в контексте % дозы соединения 1, выделяемого с мочой, определяется пределом погрешности, который обычно примерно в два раза превышает стандартное отклонение или половину ширины 95-процентного доверительного интервала. Термин "приблизительно" в других областях раскрытия может использоваться для обозначения стандартного отклонения или величины вариации или дисперсии набора значений данных.
Используемый в настоящем описании термин "контролируемое высвобождение" является синонимом продолжительного высвобождения и пролонгированного высвобождения и относится к количеству лекарственного средства, доставляемого субъекту в течение длительного периода времени. Как правило, таблетки и капсулы с контролируемым высвобождением высвобождают активное соединение в организме в течение примерно 8, 12, 16 и 24 часов. С другой стороны, термин "немедленное высвобождение" относится к активному соединению, высвобождаемому в организме субъекта в течение непродолжительного периода времени, обычно менее чем примерно 30 минут. Термин "замедленное высвобождение" относится к таблеткам и капсулам, которые высвобождают фармацевтически активную дозу через определенный промежуток времени. Эти лекарственные формы обычно имеют энтеросолюбильное покрытие, предотвращающее высвобождение в желудке, но обеспечивающее высвобождение в кишечнике.
Используемый здесь термин "формула" или "имеющий формулу", или "имеющий структуру" не предназначен для ограничения и используется так же, как обычно используется термин "содержащий". Например, если изображена одна структура, понятно, что включены все стереоизомерные и таутомерные формы, если не указано иное.
Как правило, при описании фармацевтических твердых веществ термин "(2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноат кальция" подразумевает стехиометрическое соотношение карбоксилатного аниона соединения 1 к противоину кальция, равное приблизительно 2:1. Термин гемикальциевая соль соединения 1 эквивалентна кальциевой соли соединения 1. В одном из вариантов осуществления изобретения кристаллическая форма соединения 1' представляет собой негигроскопичное твердое вещество.
Используемый в настоящем описании термин "температура плавления" означает температуру теплового перехода, при которой, используя метод дифференциальной сканирующей калориметрии, наблюдают максимальный эндотермический тепловой поток, который соответствует переходу твердого вещества в жидкую фазу.
Термин "фармацевтически приемлемый" относится к веществу, которое не является биологически или иным образом непригодным для использования в изобретении. Например, термин "фармацевтически приемлемый носитель" относится к веществу, которое может быть включено в композицию и вводиться пациенту, не вызывая нежелательных биологических эффектов и не взаимодействуя неприемлемым образом с другими компонентами композиции. Такие фармацевтически приемлемые вещества обычно удовлетворяют требуемым стандартам токсикологических и производственных испытаний и включают в себя те вещества, которые были определены Управлением США по санитарному надзору за качеством пищевых продуктов и медикаментов как подходящие неактивные ингредиенты.
Термин "фармацевтически приемлемая соль" означает соль, полученную из основания или кислоты, которая является приемлемой для введения пациенту, такому как млекопитающее (например, соли, имеющие приемлемую безопасность для млекопитающих для заданного режима дозирования). При этом очевидно, что не требуется, чтобы соли, охватываемые изобретением, были фармацевтически приемлемыми солями, такими как соли промежуточных соединений, которые не предназначены для введения пациенту. Фармацевтически приемлемые соли могут быть получены из фармацевтически приемлемых неорганических или органических оснований и из фармацевтически приемлемых неорганических или органических кислот. Кроме того, если соединение содержит как группу основания, такую как амин, пиридин или имидазол, так и кислотную группу, такую как карбоновая кислота или тетразол, могут быть образованы цвиттерионы, охватываемые термином "соль", как используется в настоящем описании. Соли, полученные из фармацевтически приемлемых неорганических оснований, включают соли аммония, кальция, меди, железа (III), железа (II), лития, магния, марганца (III), марганца (II), калия, натрия, цинка и т.п. Соли, полученные из фармацевтически приемлемых органических оснований включают соли первичных, вторичных и третичных аминов, включая замещенные амины, циклические амины, амины природного происхождения и т.п., такие как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Соли, полученные из фармацевтически приемлемых неорганических кислот, включают соли борной, углеводородной, галогеноводородной (бромистоводородной, хлористоводородной, фтористоводородной или иодистоводородной), азотной, фосфорной, сульфаминовой и серной кислот. Соли, полученные из фармацевтически приемлемых органических кислот, включают соли алифатических гидроксильных кислот (например, лимонной, глюконовой, гликолевой, молочной, лактобионовой, яблочной и винной кислот), алифатических монокарбоновых кислот (например, уксусной, масляной, муравьиной, пропионовой и трифторуксусной кислот), аминокислот (например, аспарагиновой и глутаминовой кислот), ароматических карбоновых кислот (например, бензойной, п-хлорбензойной, дифенилуксусной, гентизиновой, гиппуровой и трифенилуксусной кислот), ароматических гидроксильных кислот (например, о-гидроксибензойной, п-гидроксибензойной, 1-гидроксинафтален-2-карбоновой и 3-гидроксинафтален-2-карбоновой кислот), аскорбиновых, дикарбоновых кислот (например, фумаровой, малеиновой, щавелевой и янтарной кислот), глюкороновой, миндальной, муциновой, никотиновой, оротовой, памовой, пантотеновой, сульфоновой кислот (например, бензолсульфоновой, камфорсульфоновой, эдизиловой, этансульфоновой, изетионовой, метансульфоновой, нафталинсульфоновой, нафтален-1,5-дисульфоновой, нафтален-2,6-дисульфоновой и п-толуолсульфоновой кислот), ксинафоевой кислоты и т.п.
Термин "терапевтически эффективное количество" означает количество, достаточное для эффективного лечения при введении пациенту, нуждающемуся в этом, т.е. количество лекарственного средства, необходимое для получения желаемого терапевтического эффекта. Например, терапевтически эффективное количество для лечения гипертонии представляет собой количество соединения, необходимое, например, для уменьшения, подавления, устранения или профилактики развития симптомов гипертонии или для лечения основной причины гипертонии. В одном из вариантов осуществления терапевтически эффективное количество представляет собой количество лекарственного средства, необходимое для снижения кровяного давления, или количество лекарственного средства, необходимое для поддержания нормального кровяного давления. С другой стороны, термин "эффективное количество" означает количество, достаточное для получения желаемого результата, который необязательно может быть терапевтическим результатом. Например, при изучении системы, содержащей фермент NEP, "эффективное количество" может быть количеством, необходимым для ингибирования фермента.
Используемый здесь термин "лечащий" или "лечение" означает процесс лечения или лечение заболевания или медицинского состояния (например, гипертонии) у пациента, такого как млекопитающее (в частности человек), которое включает одно или более из следующего: (a) профилактику развития заболевания или состояния, т.е. предупреждение повторения заболевания или состояния, или профилактическое лечение пациента, который предрасположен к развитию этого заболевания или состояния; (b) облегчение тяжести заболевания или состояния, т.е. устранение или регресс заболевания или состояния у пациента; (c) подавление заболевания или состояния, т.е. замедление или остановку развития заболевания или состояния у пациента; или (d) облегчение симптомов заболевания или состояния у пациента. Например, термин "лечение гипертонии" включает профилактику возникновения гипертонии, уменьшение степени тяжести гипертензии, подавление гипертонии и облегчение симптомов гипертонии (например, снижение кровяного давления). Термин "субъект" или "пациент" включает млекопитающих, таких как люди, которые нуждаются в лечении или профилактике заболеваний или которые в настоящее время проходят лечение для предупреждения развития заболевания, или получают лечение конкретного заболевания или состояния, а также испытуемых субъектов, принимающих участие в испытаниях или анализе кристаллического соединения, например, животная модель.
Все другие используемые в настоящем описании термины используются в своих обычных значениях, которые понятны специалистам в области техники, к которой они относятся.
ОСНОВНЫЕ ПРОЦЕДУРЫ СИНТЕЗА
Соединение 1 и его кристаллическая форма в виде соли кальция могут быть синтезированы из легко доступных исходных материалов, описанных ниже и проиллюстрированных в примерах. Понятно, что, в случаях указания типичных или предпочтительных условий проведения процесса (т.е., температуры реакции, времени, молярных соотношений реагентов, растворителей, давления и т.д.) могут применяться и другие условия проведения процесса, если не указано иное. Понятно, что, независимо от конкретных условий проведения процесса (т.е., температуры кристаллизации, времени, молярных соотношений реагентов, растворителей, давления и т.д.), могут быть использованы и другие условия проведения процесса, если не указано иное. В некоторых случаях реакции или кристаллизацию проводили при комнатной температуре, при этом фактическое измерение температуры не проводилось. Понятно, что комнатная температура означает температуру, обычно относящуюся к температуре окружающей среды в лаборатории, которая обычно находится в пределах от примерно 15°С до примерно 30°С, например от примерно 20°С до примерно 25°C. В других случаях, реакции или кристаллизацию проводили при комнатной температуре, которую действительно измеряли и регистрировали.
Любые молярные соотношения, описанные в способах по изобретению, могут быть легко определены различными способами, доступными для специалистов в данной области. Например, такие молярные соотношения могут быть легко определены методом1Н ЯМР. Альтернативно, для определения молярного соотношения можно использовать элементный анализ и ВЭЖХ.
В одном из вариантов осуществления изобретение относится к (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановой кислоте (1) или ее фармацевтически приемлемой соли.
В другом варианте осуществления соединение 1 может быть получено путем смешивания этанола и оксалилхлорида с образованием раствора, взаимодействия бензилового эфира (2S,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановой кислоты с этим раствором и объединения полученной смеси с палладием на углероде в атмосфере водорода.
В другом варианте осуществления соединение 1 может быть получено путем (а) растворения этанола в дихлорметане; (b) добавления оксалилхлорида с образованием раствора, и перемешивания его при комнатной температуре; (с) выпаривания растворителя из раствора; (d) добавления оставшегося раствора к бензиловому эфиру (2S,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановой кислоты, который сначала растворяют в дихлорметане; (e) добавления N,N-диизопропилэтиламина и перемешивания при комнатной температуре; (f) выпаривания растворителя с образованием твердого вещества; (g) объединения твердого вещества с 10 масс.% палладием на углероде в растворителе с образованием смеси; (h) помещения смеси в атмосферу водорода при перемешивания; и (i) отфильтровывания палладия на углероде и вакуумной сушки с образованием соединения 1 в виде твердого вещества. Полученные твердые вещества на этапах (f) и (i) также можно очистить хроматографией.
Получение кристаллической гемикальциевой соли соединения 1 обычно проводят в подходящем инертном разбавителе, примеры которого включают, без ограничения, ацетон, ацетонитрил, этилацетат, метилэтилкетон, метанол, этанол, изопропанол, изобутанол, дихлорметан, метил-трет-бутиловый эфир, циклопентилметиловый эфир, гексаны и т.п., и их смеси, необязательно содержащие воду. Смеси инертных разбавителей (также называемые системами растворителей) включают ацетон с водой, ацетонитрил с водой, этанол и этилацетат, этилацетат и гексан и низшие спирты (C1-6алкил-ОН) с водой, например метанол и вода и изопропанол и вода. Особенно подходящие системы растворителей включают этанол, этанол:вода и этилацетат:этанол, содержащие пропионат кальция или другие соли кальция. По завершении кристаллизации кристаллическое соединение может быть выделено из реакционной смеси любым обычным способом, таким как осаждение, фильтрование, концентрация, центрифугирование, сушка под вакуумом и т.п.
В одном из вариантов осуществления изобретение относится к кристаллической форме (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4- (этоксиоксалиламино)-2-гидроксиметил-2-метилпентановой кислоты. В другом варианте осуществления кристаллическая форма представляет собой (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноат кальция (1').
В другом варианте осуществления кристаллическая форма 1' может быть получена путем (а) растворения (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановой кислоты в этаноле и N,N-диизопропилэтиламине с образованием раствора А; (b) растворения трифторметансульфоната кальция в этаноле с образованием раствора B; (с) добавления по каплям раствора В к раствору А с образованием суспензии; (d) перемешивания при комнатной температуре; и (е) выделения полученных твердых веществ с образованием соединения 1'. Другой вариант осуществления включает в себя вторую стадию кристаллизации. В этом случае, второй процесс кристаллизации дополнительно включает (f) охлаждение соединения 1' до примерно 5°C и добавление холодной смеси этанол:вода при интенсивном перемешивании; и (g) фильтрование и сушку при комнатной температуре с получением соединения 1'.
СВОЙСТВА КРИСТАЛЛИЧЕСКОЙ СТРУКТУРЫ
Как хорошо известно в области анализа порошковой рентгеновской дифракции (PXRD), относительная высота пиков на PXRD дифрактограммах зависит от нескольких факторов, связанных с подготовкой образцов и геометрических параметров инструмента, тогда как положения пиков являются относительно независимыми от деталей эксперимента. PXRD, дифференциальная сканирующая калориметрия (DSC), термогравиметрический анализ (TGA) и оценка динамической адсорбции влаги (DMS) (также известная как анализ сорбции и десорбции влаги) выполняли согласно тому, как описано в настоящей заявке.
В одном из аспектов изобретение относится к кристаллической форме (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановой кислоты. В другом аспекте кристаллическая форма представляет собой (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноат кальция (1'). Эта кристаллическая форма 1' характеризуется PXRD дифрактограммой, в которой положения пиков, по существу, соответствуют пикам, показанным на фиг.1. Пики с относительной интенсивностью выше 0,1% по площади приведены ниже в таблице. На этой дифрактограмме видны острые дифракционные пики в области 3-25° 2θ.
*Значения 2θ представлены как значение ±0,20.
Таким образом, в одном из вариантов осуществления кристаллическая форма 1' характеризуется PXRD дифрактограммой, содержащей дифракционные пики при значениях угла 2θ 7,18±0,2, 7,38±0,2 и 7,97±0,2.
В другом варианте осуществления кристаллическая форма 1' характеризуется PXRD дифрактограммой, содержащей дифракционные пики при значениях угла 2θ 3,98±0,2, 5,00±0,2, 7,18±0,2, 7,38±0,2, 7,97±0,2, 8,87±0,2 и 10,91±0,2.
В другом варианте осуществления кристаллическая форма 1' также характеризуется наличием одного или более дополнительных дифракционных пиков при значениях угла 2θ, выбранных из 3,47±0,2, 9,99±0,2, 15,74±0,2, 15,98±0,2 и 18,98±0,2; а в другом варианте осуществления кристаллическая форма 1' также характеризуется наличием трех или более таких дополнительных дифракционных пиков.
В одном из вариантов осуществления кристаллическая форма 1' характеризуется термограммой DSC, по существу, такой, как показана на фиг.2. Кристаллическая форма 1' характеризуется кривой DSC, регистрируемой при скорости нагревания 10°C в минуту, на которой наблюдается максимум в эндотермическом тепловом потоке при температуре между примерно 237°C и примерно 241°C. На DSC термограмме показана эндотерма плавления с пиком при приблизительно 239°C, началом при 233°C и энтальпией ~67 Дж/г. Вторая эндотерма связана с разложением и другими неизвестными тепловыми эффектами.
В одном из вариантов осуществления кристаллическая форма 1' характеризуется TGA графиком, представленным на фиг.3. На графике TGA показано начало разложения при температуре примерно 225°C. Кристаллическое соединение разлагается после плавления, о чем говорит значительная потеря массы после ~250°C, чему также соответствует вторая эндотерма на кривой DSC.
В одном из вариантов осуществления кристаллическая форма 1' характеризуется изотермой DMS, показанной на фиг.4. Эта форма представляет собой негигроскопичное твердое вещество. Наблюдаемое общее увеличение влажности составляет менее 1% по весу при относительной влажности от 5% до 90%. Между последовательными циклами сорбции-десорбции не наблюдается значительный гистерезис. Твердое вещество, полученное после циклов сорбции-десорбции, имело такую же PXRD дифрактограмму, что и исходное, на что указывает отсутствие изменений в форме после этого эксперимента.
Кристаллическая форма 1' может быть охарактеризована PLM диаграммой, приведенной на фиг.5, на которой показана эта форма в виде тонких двоякопреломляющих кристаллов.
Были получены кристаллы L-аргинин (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата (1"). Однако воздействие окружающей среды на эти кристаллы приводит к их медленному разложению. Кристаллическая форма 1" характеризуется PXRD и кривой DSC, представленными на фиг. 6 и 7, соответственно. Кривую DSC регистрировали при скорости нагревания 10°C в минуту, на которой наблюдали максимум в эндотермическом тепловом потоке при температуре между примерно 113°C и примерно 117°C. На DSC термограмме показана эндотерма плавления с пиком при приблизительно 116,9°C, началом при 106,8°C и энтальпией ~70,3 Дж/г. Кристаллическая форма 1" также характеризуется графиком TGA, представленным на фиг.8, на котором наблюдается потеря массы ~6,5% вплоть до температуры 140°C, при этом постоянная потеря массы также наблюдается после 140°C. Кристаллическая форма 1" может быть охарактеризована PLM диаграммой, представленной на фиг.9, на которой эта форма выглядит в виде тонких двоякопреломляющих кристаллов.
ПРИМЕНЕНИЕ
Экстраполяция in vitro поведения лекарственного средства, наблюдаемое в экспериментах, на его in vivo поведение в организме субъекта постоянно улучшается (см., например, Chiba et al., AAPS J., 2009 June, 11 (2): 262-276). В настоящем изобретении проводили in vitro оценку активности ингибитора неприлизина человека (анализ 1) с целью определения ингибирующей активности соединения 1 в отношении неприлизина. Для этого соединения было получено пороговое значение pKi≥9,0. Однако дополнительно были проведены дополнительные in vivo эксперименты для более точного прогнозирования поведения соединения 1 в организме субъекта.
Критическим параметром при оценке пригодности пролекарства является определение того, насколько быстро это пролекарство превращается в активный агент или активный метаболит. В настоящем изобретении соединение 1, представляющее собой сложный эфир пролекарства, превращается в активный агент или активный метаболит, соединение С2 сравнения, в результате ферментативной реакции, например, гидролиза эстеразой, которая может сильно зависеть от вида. По этой причине предпочтительнее оценивать степень конверсии для нескольких видов в случае экстраполяции на человека. Кроме того, существует несколько свойств, которые полезны при оценке того, будет ли доставлено в плазму количество лекарственного средства, достаточное для достижения необходимого терапевтического эффекта, например, высокая пероральная биодоступность и низкий уровень почечного клиренса у пациентовс нарушенной функцией почек.
В целях настоящего изобретения изучали фармакокинетические свойства при пероральном и внутривенном введениях крысам, собакам и обезьянам для определения пероральной биодоступности соединения 1 по сравнению с его активным метаболитом, соединением С2 сравнения (анализ 2). Кроме того, изучение фармакокинетических свойств проводили при пероральном и внутривенном введении крысам и собакам для сравнения соединения 1 с другими аналогичными с химической точки зрения пролекарствами или соединениями в виде сложных эфиров (анализ 3).
Соединение 1 ингибирует фермент NEP, и поэтому предполагается, что оно будет полезным для лечения и/или профилактики состояний, зависящих от ингибирования NEP. Таким образом, ожидается, что пациенты, страдающие заболеванием или расстройством, которое лечится путем ингибирования фермента NEP или путем увеличения уровней его пептидных субстратов, можно лечить терапевтически эффективным количеством соединения 1. Например, предполагается, что в результате ингибирования NEP соединение 1 будет усиливать биологические эффекты эндогенных пептидов, метаболизм которых происходит с участием NEP, таких как натрийуретические пептиды, бомбезин, брадикинины, кальцитонин, эндотелины, энкефалины, нейротензин, вещество Р и вазоактивный интестинальный пептид. Таким образом, предполагается, что это соединение будет иметь другие физиологические воздействия, например, на почечную, центральную нервную, репродуктивную и желудочно-кишечную системы.
Лекарственные вещества выводятся из организма субъекта различными способами элиминации, которые можно классифицировать как экскреция и биотрансформация. Экскреция относится к удалению интактного нелетучего лекарственного средства в основном почками в мочевой пузырь далее в мочу, в то время как другие пути экскреции включают желчь (печень), пот, слюну, молоко (через лактацию) или другие жидкости тела. Летучие лекарственные вещества, такие как спирт и газообразные анестетики, выделяются легкими с выдыхаемым воздухом. С другой стороны, биотрансформация или метаболизм лекарственного вещества относится к химическому превращению лекарственного вещества в организме в метаболит, и обычно является ферментативным процессом. Исключением является, когда лекарственное вещество претерпевает химические превращения без участия ферментов, например, гидролиз сложного эфира. Ферменты, участвующие в биотрансформации лекарственных веществ, главным образом находятся в печени. Другие ткани, такие как почки, легкие, тонкий кишечник и кожа, также содержат метаболические ферменты.
Фармакокинетические исследования также можно использовать для изучения у субъекта путей элиминации, например почечного клиренса путем экскреции вводимого лекарственного средства с мочой в течение некоторого периода времени. Почечную экскрецию соединения 1 на модели собаки проводили для оценки почечной экскреции как пути элиминации (анализ 4). Этот путь элиминации имеет важное значение для субъектов с нарушенной почечной функцией, которые нуждаются в методах лечения, в минимальной степени включающих очищение посредством почечной экскреции. В одном из вариантов осуществления почечная экскреция у субъекта соединения 1 или его кристаллической формы составляет приблизительно ≤15%, ≤10%, ≤5%, ≤3%, ≤2%, ≤1% или ≤0,5% от введенной дозы в течение 24 часов.
Как описано ниже в разделе анализа, в in vitro анализе фермента NEP и в in vivo определениях биодоступности при пероральном введении и почечной экскреции у разных видов животных наряду с соединением 1 также использовали соединения сравнения с аналогичной химической структурой. Наблюдаемые значимые различия в результатах оказались удивительными. Хотя в одном или более анализах отдельные соединения сравнения демонстрировали свойства, аналогичные свойствам соединения 1, однако только соединение 1 имело высокую ингибирующую активность в отношении неприлизина человека, высокую пероральную биодоступность и низкий уровень почечной экскреции, которые, как ожидается, могут быть использованы для лечения конкретных заболеваний.
Альтернативно, для эффективного использования соединения 1 в качестве терапевтического агента желательно иметь это соединение в твердой форме, которая может быть легко изготовлена и которая имеет приемлемую химическую и физическую стабильность, включая высокую температуру плавления. Кристаллы соединения 1 в виде свободной кислоты не могут быть получены. В конечном итоге были получены кристаллы аргинина и кальция соединения 1, но кристаллы аргинина плавились в условиях окружающей среды, и их дальнейшая разработка была связана с определенными трудностями. С другой стороны, кристаллы кальция были стабильными и плавились при температуре примерно 239°C, и следовательно их можно использовать в дальнейших разработках в качестве терапевтического агента.
Сердечно-сосудистые заболевания
Предполагается, что соединение 1, благодаря усиления им действия вазоактивных пептидов, таких как натрийуретические пептиды и брадикинин, можно использовать для лечения и/или профилактики заболеваний, таких как сердечно-сосудистые заболевания. См., например, Roques et al. (1993) Pharmacol. Rev. 45:87-146 and Dempsey et al. (2009) Amer. J. of Pathology 174(3):782-796. Сердечно-сосудистые заболевания, представляющие особый интерес, включают гипертонию и сердечную недостаточность. Гипертония включает, в качестве иллюстрации, а не ограничения: первичную гипертонию, которая также упоминается как гипертоническая болезнь или идиопатическая гипертензия; вторичную гипертонию; гипертонию с сопутствующей почечной недостаточностью; тяжелую гипертонию с сопутствующей почечной недостаточностью или без нее; легочную гипертензию, включая легочную артериальную гипертензию; и резистентную гипертензию. Сердечная недостаточность включает, в качестве иллюстрации, без ограничения: застойную сердечную недостаточность; острую сердечную недостаточность; хроническую сердечную недостаточность, например, с уменьшенной фракцией выброса левого желудочка (также называемой систолической сердечной недостаточностью) или с сохраненной фракцией выброса левого желудочка (также называемой диастолической сердечной недостаточностью); и острую и хроническую декомпенсированную сердечную недостаточность. Таким образом, в одном из вариантов осуществления изобретение относится к способу лечения гипертонии, особенно первичной гипертонии или легочной артериальной гипертензии, включающей введение пациенту терапевтически эффективного количества соединения 1.
Для лечения первичной гипертонии терапевтически эффективное количество обычно представляет собой количество, достаточное для снижения у пациента кровяного давления. Это относится как к гипертонии от легкой до умеренной, так и тяжелой гипертонии. В случае лечения гипертонии, соединение 1 можно вводить в комбинации с другими терапевтическими агентами, такими как антагонисты альдостерона, ингибиторы альдостеронсинтазы, ингибиторы ангиотензинпревращающего фермента и агент двойного действия, такой как ингибитор ангиотензинпревращающего фермента/ингибитор неприлизина, активаторы и стимуляторы ангиотензинпревращающего фермента 2 (ACE2), вакцины против ангиотензина-II, антидиабетические агенты, антилипидные агенты, антитромботические агенты, антагонисты АТ1-рецепторов и агент двойного действия, такой как антагонист АТ1-рецепторов/ингибитор неприлизина, антагонисты β1-адренергических рецепторов, агент двойного действия, такой как антагонист β-адренергических рецепторов/антагонист α1-рецепторов, блокаторы кальциевых каналов, диуретики, антагонисты рецепторов эндотелина, ингибиторы эндотелин-превращающего фермента, ингибиторы неприлизина, натрийуретические пептиды и их аналоги, антагонисты рецепторов клиренса натрийуретического пептида, доноры оксида азота, нестероидные противовоспалительные средства, ингибиторы фосфодиэстеразы (в частности, ингибиторы PDE-V), агонисты рецепторов простагландина, ингибиторы ренина, стимуляторы и активаторы растворимой гуанилатциклазы и их комбинации. В одном из конкретных вариантов осуществления изобретения соединение по изобретению объединяют с антагонистом АТ1-рецепторов, блокатором кальциевых каналов, мочегонным средством или их комбинацией и используют для лечения первичной гипертонии. В другом конкретном варианте осуществления изобретения соединение по изобретению объединяют с антагонистом АТ1-рецепторов и используют для лечения гипертонии с сопутствующей почечной недостаточностью. В случае лечения резистентной гипертензии соединение можно вводить в комбинации с другими терапевтическими агентами, такими как ингибиторы альдостеронсинтазы.
В случае лечения легочной артериальной гипертензии терапевтически эффективное количество обычно представляет собой количество, достаточное для снижения резистентности легочных сосудов. Другие цели терапии представляют собой улучшение способности пациента переносить физическую нагрузку. Например, в клинических условиях терапевтически эффективное количество может быть количеством, которое улучшает способность пациента ходить, не ощущая дискомфорта, в течение 6 минут (покрывая расстояние приблизительно 20-40 метров). В случае лечения легочной артериальной гипертензии соединение можно вводить в комбинации с другими терапевтическими агентами, такими как антагонисты α-адренергических рецепторов, антагонисты β1-адренергических рецепторов, агонисты β2-адренергических рецепторов, ингибиторы ангиотензинпревращающего фермента, антикоагулянты, блокаторы кальциевых каналов, диуретики, антагонисты рецепторов эндотелина, ингибиторы PDE-V, аналоги простагландина, селективные ингибиторы обратного захвата серотонина и их комбинации. В одном из конкретных вариантов осуществления изобретения соединение 1 объединяют с ингибитором PDE-V или селективным ингибитором обратного захвата серотонина и используют для лечения легочной артериальной гипертензии.
Другой вариант осуществления изобретения относится к способу лечения сердечной недостаточности, в частности застойной сердечной недостаточности (включая систолическую и диастолическую застойную сердечную недостаточность), включающему введение пациенту терапевтически эффективного количества соединения 1. Обычно терапевтически эффективное количество представляет собой количество, достаточное для снижения кровяного давления и/или улучшения функций почек. В клинических условиях терапевтически эффективное количество может представлять собой количество, которое является достаточным для улучшения сердечной гемодинамики, как, например, снижения заклинивающего давления, давления в правом предсердии, давлении наполнения и сосудистого сопротивления. В одном из вариантов осуществления соединение 1 вводят в виде внутривенной лекарственной формы. В случае лечения сердечной недостаточности соединение можно вводить в комбинации с другими терапевтическими агентами, такими как антагонисты аденозиновых рецепторов, усовершенствованные расщепители конечных продуктов гликозилирования, антагонисты альдостерона, антагонисты АТ1-рецепторов, антагонисты β1-адренергических рецепторов, агент двойного действия, такой как антагонист β-адренергических рецепторов/антагонист α1-рецепторов, ингибиторы химазы, дигоксин, диуретики, ингибиторы эндотелин-превращающего фермента (ЕСЕ), антагонисты рецепторов эндотелина, натрийуретические пептиды и их аналоги, антагонисты рецепторов клиренса натрийуретического пептида, доноры оксида азота, аналоги простагландинов, ингибиторы PDE-V, активаторы и стимуляторы растворимой гуанилатциклазы, антагонисты рецепторов вазопрессина. В одном из конкретных вариантов осуществления изобретения соединение 1 объединяют с антагонистом альдостерона, антагонистом β1-адренергических рецепторов, антагонистом АТ1-рецепторов или диуретиком и используют для лечения застойной сердечной недостаточности.
Диарея
В качестве ингибитора NEP, предполагается, что соединение 1 ингибирует деградацию эндогенных энкефалинов, и, таким образом, такие соединения могут также оказаться полезными для лечения диареи, включая инфекционную и секреторную/водянистую диарею. См., например, Baumer et al. (1992) Gut 33:753-758; Farthing (2006) Digestive Diseases 24:47-58; and Marçais-Collado (1987) Eur. J. Pharmacol. 144(2):125-132. В случае лечения диареи соединение 1 можно комбинировать с одним или более дополнительными антидиарейными агентами.
Почечные заболевания
Предполагается, что соединение 1, благодаря усилению им действия вазоактивных пептидов, таких как натрийуретические пептиды и брадикинин, усиливает функцию почек (см. Chen et al. (1999) Circulation 100:2443-2448; Lipkin et al. (1997) Kidney Int. 52:792-801; and Dussaule et al. (1993) Clin. Sci. 84:31-39) и может быть использовано для лечения и/или профилактики заболеваний почек у субъекта с почечной недостаточностью. Заболевания почек, представляющие особый интерес, включают диабетическую нефропатию, хроническое заболевание почек, протеинурию и в частности острое повреждение почек (вызванное, например, сердечно-сосудистой хирургией, химиотерапией или использованием контрастных красителей для медицинской визуализации) или острую почечную недостаточность (см. Sharkovska et al. (2011) Clin. Lab. 57:507-515 and Newaz et al. (2010) Renal Failure 32:384-390).
Почечная недостаточность у субъекта, который страдает хроническим заболеванием почек (CKD), может быть классифицирована в соответствии с Национальным почечным фондом. Инициатива по улучшению качества исходов заболеваний почек (NKF KDOQI). При установлении хронического заболевания почек, т.е., повреждения почек или уровня клубочковой фильтрации (GFR) <60 мл/мин/1,73 м2 в течение≥3 месяцев, стадию заболевания можно определить в соответствии с классификацией по KDOQI CKD. Они включают стадию 1 (повреждение почек с нормальным или повышенным GFR): GFR≥90; стадию 2 (повреждение почек с умеренно уменьшенным GFR): GFR 60-89; стадию 3 (умеренно уменьшенный GFR): GFR 30-59; стадию 4 (серьезное снижение GFR): GFR 15-29; и стадию 5 (почечная недостаточность): GFR<15 (или диализ). GFR определяется в единицах мл/мин/1,73 м2.
Один из вариантов осуществления включает способ лечения субъекта с почечной недостаточностью, включающий введение терапевтически эффективного количества соединения 1 или его кристаллической формы, в частности кристаллической формы 1'. Этот способ дополнительно включает лечение пациента с почечной недостаточностью с гипертонией или сердечной недостаточностью. В случае лечения заболевания почек соединение 1 или его кристаллическую форму, особенно кристаллическую форму 1', можно вводить в комбинации с другими терапевтическими агентами, такими как ингибиторы ангиотензинпревращающего фермента, антагонисты АТ1-рецепторов и диуретики.
Другой вариант осуществления включает способ лечения пациента с почечной недостаточностью, имеющего хроническое заболевание почек с установленным уровнем клубочковой фильтрации (eGFR) между 60 мл/мин/1,73 м2 и 15 мл/мин/1,73 м2, включая введение пациенту терапевтически эффективного количества соединения 1 или его кристаллической формы, в частности кристаллической формы 1'. Другой вариант осуществления включает способ лечения пациента с почечной недостаточностью, имеющего хроническое заболевание почек с установленным уровнем клубочковой фильтрации (eGFR) ≥90 мл/мин/1,73 м2 (этап 1) или eGFR <15 мл/мин/1,73 м2 (этап 5), включающий введение пациенту терапевтически эффективного количества соединения 1 или его кристаллической формы, в частности кристаллической формы 1'. Для целей настоящего изобретения тяжелое заболевание почек можно классифицировать как eGFR <30 мл/мин/1,73 м2. В другом варианте осуществления способ лечения пациента с почечной недостаточностью, имеющего хроническое заболевание почек, классифицированное как стадия 1, стадия 2, стадия 3, стадия 4, стадия 5 или уровни eGFR, охватывающие одну или более из этих стадий с соединением 1 или его кристаллической формой, в частности кристаллической формой 1'.
Профилактическая терапия
Также предполагается, что соединение 1, благодаря усилению им действия натрийуретических пептидов, можно использовать в профилактической терапии в связи с антигипертрофическим и антифибротическим действием натрийуретических пептидов (см. Potter et al. (2009) Handbook of Experimental Pharmacology 191:341-366), например, для профилактики прогрессирования сердечной недостаточности после инфаркта миокарда, профилактики артериального рестеноза после ангиопластики, профилактики утолщения стенок кровеносных сосудов после сосудистых операций, профилактики атеросклероза и профилактики диабетической ангиопатии.
Глаукома
Предполагается, что соединение 1, благодаря усилению им действия натрийуретических пептидов, можно использовать для лечения глаукомы. См., например, Diestelhorst et al. (1989) International Ophthalmology 12:99-101. В случае лечения глаукомы соединение 1 можно комбинировать с одним или более дополнительными антиглаукомными агентами.
Облегчение боли
Предполагается, что соединение 1, в качестве ингибитора NEP, подавляет деградацию эндогенных энкефалинов и, таким образом, такие соединения также можно использовать в качестве анальгетиков. См., например, Roques et al. (1980) Nature 288:286-288 and Thanawala et al. (2008) Current Drug Targets 9:887-894. В случае лечения боли соединение 1 можно комбинировать с одним или более дополнительными антиноцицептивными лекарственными веществами, такими как ингибиторы аминопептидазы N или дипептидилпептидазы III, нестероидными противовоспалительными агентами, ингибиторами обратного захвата моноаминов, миорелаксантов, антагонистов рецепторов NMDA, агонистов опиоидных рецепторов, агонистов серотониновых 5-HT1D рецепторов и трициклических антидепрессантов.
Другие применения
Соединение 1, благодаря его ингибирующим свойствам в отношении NEP, также можно применять в качестве противокашлевого средства, а также для лечения портальной гипертензии, связанной с циррозом печени (см. Sansoe et al. (2005) J. Hepatol. 43:791-798), рака (см. Vesely (2005) J. Investigative Med. 53:360-365), депрессии (см. Noble et al. (2007) Exp. Opin. Ther. Targets 11:145-159), менструальных расстройств, преждевременных родов, преэклампсии, эндометриоза, репродуктивных расстройств (например, мужского и женского бесплодия, синдрома поликистозных яичников, отторжения имплантатов), а также сексуальной дисфункции мужчин и женщин, включая мужскую эректильную дисфункцию и женское половое возбуждение. Более конкретно, предполагается, что соединение 1 можно использовать для лечения женской сексуальной дисфункции (см. Pryde et al. (2006) J. Med. Chem. 49:4409-4424), которая часто определяется как трудность или неспособность пациентки получать сексуальное удовлетворение. Это включает множество разнообразных женских сексуальных расстройств, включая, в качестве иллюстрации, а не ограничения, гипоактивное расстройство сексуального желания, сексуальное возбуждение, оргазмическое расстройство и расстройство сексуальной боли. В случае лечения таких расстройств, особенно женской сексуальной дисфункции, соединение по изобретению может быть объединено с одним или более из следующих вторичных агентов: ингибиторов PDE-V, агонистов допамина, агонистов и/или антагонистов эстрогеновых рецепторов, андрогенов и эстрогенов. Также ожидается, что соединение 1, благодаря свойствам ингибирования NEP, также имеет противовоспалительные свойства и может быть использовано как таковое, в частности, в комбинации со статинами.
Недавние исследования показали, что NEP участвует в регуляции нервной функции при инсулин-дефицитном диабете дефиците инсулина и ожирении, вызванном диетой. Coppey et al. (2011) Neuropharmacology 60:259-266. Следовательно, также предполагается, что соединение 1, благодаря своим свойствам ингибирования NEP, можно использовать для обеспечения защиты от поражения нервов, вызванного диабетом, или ожирения, вызванного диетой.
Количество соединения 1, вводимого за одну дозу, или общее количество, вводимое в сутки, может быть заранее рассчитано или определено для каждого пациента отдельно, учитывая многочисленные факторы, среди которых природа и тяжесть состояния пациента, состояние, на которое направлено лечение, возраст, вес и общее состояние здоровья пациента, переносимость пациентом активного вещества или активного метаболита, путь введения, фармакологические параметры, такие как активность, эффективность, фармакокинетические и токсикологические профили вводимого соединения и любых вторичных агентов и т.п. Лечение пациента, страдающего заболеванием или состоянием (например, гипертонии), может начинаться с предварительно определенной дозы или дозы, определяемой лечащим врачом, и может продолжаться в течение периода времени, необходимого для профилактики, улучшения, подавления или ослабления симптомов заболевания или состояния здоровья. Пациенты, проходящие такое лечение, обычно находятся под наблюдением на регулярной основе для определения эффективности терапии. Например, при лечении гипертонии измерения кровяного давления можно использовать для определения эффективности лечения. Аналогичные показатели для других заболеваний и состояний, указанных в настоящем описании, хорошо известны и легко доступны лечащему врачу. Непрерывное наблюдение врача гарантирует, что оптимальное количество соединения 1 вводится в любой момент времени, а также облегчает определение продолжительности лечения. Это особенно важно, когда вводятся вторичные агенты, поскольку также может возникнуть необходимость в корректировке выбора, дозировки и продолжительности терапии. Таким образом, режим лечения и график дозирования можно регулировать в течение курса терапии таким образом, чтобы вводилось самое минимальное количество активного агента или активного метаболита, которое вызывает желаемую эффективность, и, кроме того, такое введение продолжается только до тех пор, пока это необходимо для успешного лечения заболевания или состояния.
Соединение 1 также можно использовать в качестве промежуточного продукта, пригодного для получения кристаллических форм соединения 1, включая, например, кристаллическую форму 1'.
Инструменты исследования
Поскольку соединение 1 обладает ингибирующей активностью в отношении фермента NEP, оно также может быть использовано в качестве инструмента исследования для анализа или изучения биологических систем или образцов, содержащих фермент NEP, например, для изучения заболеваний, в которых фермент NEP или его пептидные субстраты играют определенную роль. В таких исследованиях, проводимых как in vitro, так и in vivo, может быть использована любая подходящая биологическая система или образец, содержащий фермент NEP. Репрезентативные биологические системы или образцы, подходящие для таких исследований, включают, без ограничения, клетки, клеточные экстракты, плазматические мембраны, образцы тканей, изолированные органы, млекопитающих (таких как мыши, крысы, морские свинки, кролики, собаки, свиньи и т.д.) и т.п., причем млекопитающие представляют особый интерес. В одном конкретном варианте осуществления изобретения активность фермента NEP у млекопитающего ингибируют путем введения соединения 1 в количестве, ингибирующем NEP.
Когда биологическая система или образец, содержащий фермент NEP, используется в качестве инструмента исследования, он обычно контактирует с количеством соединения 1, ингибирующим NEP. После воздействия этого соединения на биологическую систему или образец определяют эффекты ингибирования фермента NEP, используя обычные методы и оборудование, такие как измерение степени связывания рецептора в анализе связывания или измерение лиганд-опосредованных изменений в функциональном анализе. Воздействие включает взаимодействие клеток или ткани с соединением, введение соединения млекопитающему, например, например, i.p., p.o., i.v., s.c. или при помощи ингаляции и т.д. Этот этап определения может включать измерение ответа (количественный анализ) или наблюдение (качественный анализ). Измерение ответа включает, например, определение уровня воздействия соединения на биологическую систему или образец при помощи обычных процедур и оборудования, таких как анализ активности ферментов и измерение изменений, опосредованных субстратом фермента или продуктом, в функциональном анализе. Результаты анализа могут быть использованы для определения уровня активности, а также количества соединения, необходимого для достижения желаемого результата, т.е. количества, ингибирующего фермент NEP. Как правило, этап определения включает определение эффекта ингибирования фермента NEP.
Кроме того, соединение 1 можно использовать в качестве инструмента исследования для оценки других химических соединений и, таким образом, оно также может оказаться полезным в анализе скрининга по обнаружению, например, новых соединений, обладающих ингибирующей активностью в отношении NEP. В этом случае, соединение 1 используют в качестве стандарта в анализе сравнения результатов, полученных для тестируемого соединения и соединения 1 для определения таких тестируемых соединений, которые имеют примерно такую же или более высокую активность, если таковая имеется. Например, данные pKi для тестируемого соединения или группы тестируемых соединений сравнивают с данными pKi для соединения 1 для определения тестируемых соединений, которые имеют необходимые свойства, например, тестируемых соединений, которые имеют примерно такую же или более высокую активность, чем у соединения по изобретению. Этот аспект изобретения включает в качестве отдельных вариантов осуществления как получение данных сравнения (с помощью соответствующих анализов), так и анализ тестовых данных для идентификации представляющих интерес тестируемых соединений. Таким образом, тестируемое соединение может быть оценено в биологическом анализе способом, включающем этапы: (а) проведения биологического анализа с использованием соединения, получая первое аналитическое значение; (b) проведения биологического анализа с соединением 1, получая второе аналитическое значение; где этап (а) выполняют либо до, либо после, либо одновременно с этапом (b); и (c) сравнение первого аналитического значения, полученного на этапе (а), со вторым аналитическим значением, полученным на этапе (b). Примером биологического анализа является анализ ингибирования фермента NEP.
Еще один аспект изобретения относится к способу изучения биологической системы или образца, содержащего фермент NEP, где способ включает: (а) взаимодействие биологической системы или образца с соединением 1; и (b) определение эффектов, оказываемых соединением на биологическую систему или образец.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СОСТАВЫ
Соединение 1 обычно вводят пациенту в форме фармацевтической композиции или состава. Такие фармацевтические композиции могут вводиться пациенту любым приемлемым способом введения, включая, без ограничения, пероральный, ректальный, вагинальный, назальный, ингаляционный, местный (в том числе трансдермальный), глазной и парентеральный способы введения. Кроме того, соединение 1 можно вводить, например, перорально, несколькими дозами в сутки (например, два, три или четыре раза в сутки) в виде одной суточной дозы или одной недельной дозы. Понятно, что в раскрытых в настоящем описании фармацевтических композициях может быть использована любая форма соединения 1 (т.е., свободное основание, свободная кислота, фармацевтически приемлемая соль, сольват и т.д.), которая подходит для конкретного способа введения.
Соответственно, в одном из вариантов осуществления изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение 1. В случае необходимости композиция может содержать другие терапевтические и/или препаратообразующие агенты. При обсуждении композиций «соединение 1» также может упоминаться в настоящем описании как «активный агент», чтобы отличать его от других компонентов состава, таких как носитель. Таким образом, понятно, что термин «активный агент» включает соединения по изобретению, а также его фармацевтически приемлемые соли.
Фармацевтические композиции по изобретению обычно содержат терапевтически эффективное количество соединения 1. Однако специалисты в данной области техники поймут, что его содержание может фармацевтической композиции может быть больше, чем терапевтически эффективное количество, например в нефасованных композициях, или меньше, чем терапевтически эффективное количество, т.е., в отдельных единичных дозах, предназначенных для нескольких введений для достижения терапевтически эффективного количества. Как правило, композиция содержит от 0,01 до 95 масс.% активного агента, в том числе от примерно 0,01-30 масс.%, например примерно от 0,01 до 10 масс.%, при этом фактическое количество зависит от самого состава, способа введения, частоты дозирования и т.д. В одном из вариантов осуществления композиция, подходящая для пероральной лекарственной формы, например, может содержать примерно 5-70 масс.% или примерно 10-60 масс.% активного агента.
В фармацевтических композициях по изобретению может быть использован любой обычный носитель или наполнитель. Выбор конкретного носителя или наполнителя или комбинации носителей или наполнителей зависит от способа введения, используемого для лечения конкретного пациента или состояния здоровья или тяжести заболевания. В связи с этим получение подходящей композиции для конкретного способа введения входит в компетенцию специалиста в области фармацевтики. Кроме того, носители или наполнители, используемые в таких композициях, являются коммерчески доступными. В качестве дополнительной иллюстрации, типичные методы составления лекарственных форм описаны в Remington: The Science and Practice of Pharmacy, 20th Edition, Lippincott Williams & White, Baltimore, Maryland (2000); and H. C. Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems, 7th Edition, Lippincott Williams & White, Baltimore, Maryland (1999).
Типичные примеры материалов, которые могут служить в качестве фармацевтически приемлемых носителей, включают, без ограничения, следующие: сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлозу, такую как микрокристаллическая целлюлоза, и ее производные, такие как карбоксиметилцеллюлоза натрия, этилцеллюлоза и ацетат целлюлозы; соли жирных кислот, такие как стеарат магния; порошкообразный трагакант; солод; желатин; тальк; наполнители, такие как масло какао и суппозиторные воски; масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; полиолы, такие как глицерин, сорбит, маннит и полиэтиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический физиологический раствор; раствор Рингера; этиловый спирт; фосфатные буферные растворы; сжатые пропеллентные газы, такие как хлорфторуглероды и гидрофторуглероды; и другие нетоксичные совместимые вещества, используемые в фармацевтических композициях.
В одном из вариантов осуществления изобретения фармацевтически приемлемым носителем является стеарат магния. Например, фармацевтическая композиция может содержать соединение 1 или кристаллическую форму 1' и стеарат магния в отношении от примерно 3:1 до примерно 10:1 соединения 1 или кристаллической формы 1' к стеарату магния. Другие отношения соединения 1 или кристаллической формы 1' к стеарату магния включают, без ограничения, 1:1, 5:1, 15:1, 20:1, 25:1, 30:1 и 50:1.
Фармацевтические композиции обычно получают путем тщательного перемешивания до получения однородной массы или смешивания активного агента с фармацевтически приемлемым носителем и одним или более необязательными ингредиентами. Полученную равномерно смешанную смесь затем можно формовать или загружать в таблетки, капсулы, пилюли, канистры, картриджи, диспенсеры и т.п. при помощи обычных процедур и оборудования.
В одном из вариантов осуществления фармацевтические композиции пригодны для перорального введения. Подходящие композиции для перорального введения могут находиться в форме капсул, таблеток, пилюль, пастилок, крахмальных капсул, драже, порошков, гранул; растворов или суспензий в водной или неводной жидкости; жидких эмульсий масло-в-воде или вода-в-масле; эликсиров или сиропов; и т.п.; каждая из которых содержит заданное количество активного агента.
Если композиция предназначена для перорального введения в твердой лекарственной форме (капсулы, таблетки, пилюли и т.п.), она обычно содержит активный агент и один или более фармацевтически приемлемых носителей, таких как цитрат натрия, дикальцийфосфат или стеарат магния. Твердые лекарственные формы также могут содержать наполнители или сухие разбавители, такие как крахмалы, микрокристаллическая целлюлоза, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; связующие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или аравийская камедь; увлажнители, такие как глицерин; дезинтегрирующие агенты, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и/или карбонат натрия; замедлители растворения, такие как парафин; ускорители поглощения, такие как соединения четвертичного аммония; смачивающие агенты, такие как цетиловый спирт и/или глицеринмоностеарат; абсорбенты, такие как каолин и/или бентонитовая глина; лубриканты, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и/или их смеси; красители; и буферные агенты. Для целей настоящего изобретения термин "фармацевтически приемлемые носители" включает все термины, такие как носители, наполнители или сухие наполнители, связующие вещества, увлажнители, замедлители растворения, смачивающие агенты, абсорбенты, лубриканты, красители и буферные агенты, описанные выше.
Агенты высвобождения, смачивающие агенты, покрывающие агенты, подсластители, ароматизаторы и отдушки, консерванты и антиоксиданты также могут присутствовать в фармацевтических композициях. Примеры покрывающих агентов для таблеток, капсул, пилюль и т.д. включают вещества, которые используются для энтеросолюбильных покрытий, такие как ацетатфталат целлюлозы, фталат поливинилацетата, фталат гидроксипропилметилцеллюлозы, сополимеры сложного эфира метакриловой кислоты с метакриловой кислотой, ацетат-тримеллитат целлюлозы, карбоксиметилэтилцеллюлоза, гидроксипропил метилцеллюлозы ацетат сукцинат и т.п. Примеры фармацевтически приемлемых антиоксидантов включают: водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, натрия метабисульфат, натрия сульфит и т.п.; маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол, бутилированный гидрокситолуол, лецитин, пропилгаллат, альфа-токоферол и т.п.; и металл-хелатирующие агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота, сорбит, винная кислота, фосфорная кислота и т.п.
Композиции также могут быть составлены для обеспечения медленного или контролируемого высвобождения активного агента с использованием, например, гидроксипропилметилцеллюлозы в различных пропорциях или других полимерных матриц, липосом и/или микросфер. Кроме того, фармацевтические композиции по изобретению могут содержать смолистые агенты и могут быть составлены таким образом, чтобы высвобождение активного агента происходило только или предпочтительно в определенной части желудочно-кишечного тракта, необязательно, с задержкой. Примеры заливочных композиций, которые можно использовать, включают полимерные вещества и воски. Активный агент также может находиться в микроинкапсулированной форме, необязательно с одним или более из вышеописанных наполнителей.
Один из вариантов осуществления изобретения включает пероральную лекарственную форму, содержащую соединение 1 или кристаллическую форму 1' в капсуле, таблетке, жидкости или суспензии. Другой вариант осуществления изобретения относится к пероральной лекарственной форме с немедленным, контролируемым или отсроченным высвобождением соединения 1 или кристаллической формы 1' в организме субъекта. В случае использования капсулы в виде пероральной лекарственной формы, вариант осуществления включает капсулу, состоящую из желатина, полисахаридов или синтетических полимеров. В одном из конкретных вариантов осуществления капсула содержит гидроксипропилметилцеллюлозу.
Подходящие для капсул материалы в соответствии с изобретением выбирают из желатина, производных целлюлозы, крахмала, производных крахмала, хитозана и синтетических пластмасс. Если в качестве материала капсулы используется желатин, его можно использовать в смеси с другими добавками, выбранными из полиэтиленгликоля (ПЭГ), глицерина, сорбита, полипропиленгликоля, блоксополимеров PEO-PPO и других полиспиртов и простых полиэфиров. Если в качестве материала капсулы используют производное целлюлозы, предпочтительными полимерами являются гидроксипропилметилцеллюлоза, гидроксипропилцеллюлоза, метилцеллюлоза, гидроксиметилцеллюлоза и гидроксиэтилцеллюлоза. Если в качестве материала капсулы используются синтетические пластмассы, предпочтительными являются полиэтилен, поликарбонат, полиэфир, полипропилен и полиэтилентерефталат. Особенно предпочтительными являются полиэтилен, поликарбонат или полиэтилентерефталат.
Подходящие жидкие дозированные формы для перорального введения включают, в качестве иллюстрации, фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Жидкие лекарственные формы обычно содержат активный агент и инертный разбавитель, такой как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (например, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот с сорбитом и их смеси. Суспензии могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтиленсорбита и сорбита, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, и их смеси.
Если фармацевтические композиции по изобретению предназначены для перорального введения, они могут быть упакованы в стандартную лекарственную форму. Термин "стандартная лекарственная форма" относится к физически дискретной форме, пригодной для дозирования пациенту, т.е., каждая форма, содержит заданное количество активного агента, рассчитанное для получения желаемого терапевтического эффекта либо отдельно, либо в сочетании с одной или более дополнительными формами. Например, такими стандартными лекарственными формами могут быть капсулы, таблетки, пилюли и т.п.
В другом варианте осуществления композиции по изобретению пригодны для введения при помощи ингаляций и обычно имеют форму аэрозоля или порошка. Такие композиции обычно вводят при помощи известных устройств введения, таких как распылитель, сухой порошок или дозирующий ингалятор. Распылительные устройства создают поток высокоскоростного воздуха, под воздействием которого происходит распыление композиции в виде аэрозоля, который переносится в дыхательные пути пациента. Приведенный в качестве примера состав для распылительного устройства содержит активный агент, растворенный в носителе для образования раствора, или микронизированный и объединенный с носителем для образования суспензии микронизированных частиц вдыхаемого размера. Ингаляторы с сухим порошком предназначены для введения активного вещества в виде свободно текучего порошка, который диспергируется в воздушном потоке пациента во время вдоха. Приведенная в качестве примера композиция сухого порошка включает активный агент, смешанный в сухом виде с наполнителем, таким как лактоза, крахмал, маннит, декстроза, полимолочная кислота, сополимер лактида с гликолидом и их комбинации. Дозирующие ингаляторы выпускают отмеренное количество активного агента при помощи сжатого газа-пропеллента. Приведенный в качестве примера препарат с дозированной количеством включает раствор или суспензию активного агента в сжиженном пропелленте, таком как хлорфторуглерод или гидрофторалкан. Необязательные компоненты таких составов включают сорастворители, такие как этанол или пентан, и поверхностно-активные вещества, такие как триолеат сорбита, олеиновая кислота, лецитин, глицерин и лаурилсульфат натрия. Такие композиции обычно получают путем добавления охлажденного или находящегося под давлением гидрофторалкана в подходящий контейнер, содержащий активный агент, этанол (если присутствует) и поверхностно-активное вещество (если присутствует). Для приготовления суспензии активный агент микронизируют и затем объединяют с пропеллентом. Альтернативно, состав в виде суспензии может быть получен путем распылительной сушки покрытия поверхностно-активного вещества на микронизированные частицы активного агента. Затем состав загружают в аэрозольный контейнер, который образует часть ингалятора.
Соединение 1 и его композиции также можно вводить парентерально, например, путем подкожной, внутривенной, внутримышечной или внутрибрюшинной инъекции. Для такого введения активный агент предоставляется в стерильном растворе, суспензии или эмульсии. Приведенные в качестве примера растворители для приготовления таких составов включают воду, физиологический раствор, электролиты, низкомолекулярные спирты, такие как пропиленгликоль и полиэтиленгликоль, масла, аминокислоты, желатин, сахара, сложные эфиры жирных кислот, такие как этилолеат и т.п. Парентеральные составы также могут содержать один или более антиоксидантов, солюбилизаторов, стабилизаторов, консервантов, смачивающих агентов, эмульгаторов и диспергирующих агентов. Поверхностно-активные вещества, дополнительные стабилизирующие агенты или рН-регулирующие агенты (кислоты, основания или буферы) и антиоксиданты особенно полезны для обеспечения стабильности состава, например, для того, чтобы минимизировать или избежать гидролиза сложноэфирных и амидных связей, которые могут присутствовать в соединении. Эти составы можно сделать стерильными при помощи стерильной среды для инъекций, стерилизирующего агента, фильтрации, облучения или тепла.
Типичные физиологически приемлемые водные носители включают, например, стерильную воду для инъекций, USP; декстрозу для инъекций, USP (например, 2,5, 5,0, 10, 20% декстрозу, включая 5% декстрозу для инъекций (D5/W)); декстрозу и хлорид натрия для инъекций, USP (например, декстрозу с концентрацией от 2,5 до 10% и хлорида натрия с концентрацией от 0,12 (19 мэкв. натрий) до 0,9% (154 мэкв натрий)); маннит для инъекций, USP (например, 5, 10, 15, 20 и 25% маннит); раствор Рингера для инъекций, USP (например, 147 мэкв натрия, 4 мэкв калия, 4,5 мэкв кальция и 156 мэкв хлорида на литр); раствор лактата Рингера для инъекций, USP (например, 2,7 мэкв кальция, 4 мэкв калия, 130 мэкв натрия и 28 мэкв лактата на литр); раствор хлорида натрия для инъекций, USP (например, 0,9% хлорида натрия) и т.п.
При введении пациенту соединение 1 обычно разбавляют в водном носителе в количестве от примерно 0,5 мл до примерно 10 мл водного носителя на 1 мг соединения 1, например от примерно 0,6 до примерно 8 мл носителя на 1 мг соединения 1.
В одном конкретном варианте парентеральная композиция содержит водный раствор циклодекстрина в качестве фармацевтически приемлемого носителя. Подходящие циклодекстрины включают циклические молекулы, содержащие шесть или более единиц α-D-глюкопиранозы, соединенных в положениях 1,4 связями как в амилазе, β-циклодекстрине или циклогептаамилозе. Приведенные в качестве примера циклодекстрины включают производные циклодекстрина, такие как гидроксипропил и сульфобутиловый эфиры циклодекстринов, такие как гидроксипропил-β-циклодекстрин и сульфобутиловый эфир β-циклодекстрина. Приведенные в качестве примера буферы для таких составов включают буферы на основе карбоновой кислоты, такие как цитратный, лактатный и малеатный буферные растворы. В одном из вариантов осуществления изобретения внутривенная лекарственная форма содержит соединение 1 или кристаллическую форму 1' в буферном растворе.
В одном из вариантов осуществления соединение 1 или его фармацевтическая композиция представляет собой лиофилизированный порошок. Как правило, лиофилизированный порошок является стерильным и упакован в герметичный флакон или ампулу или аналогичный контейнер.
Соединение 1 также можно вводить трансдермально при помощи известных трансдермальных систем доставки и наполнителей. Например, соединение 1 можно смешивать с усилителями проницаемости, такими как пропиленгликоль, монолаурат полиэтиленгликоля, азациклоалкан-2-оны и т.п., и вводить в пластырь или аналогичную систему доставки. При необходимости, в таких трансдермальных композициях можно использовать дополнительные наполнители, включающие гелеобразующие агенты, эмульгаторы и буферы.
Вторичные агенты
Для лечения заболевания можно использовать только одно соединение 1 или в комбинации с одним или более дополнительными терапевтическими агентами для получения желаемого терапевтического эффекта. Таким образом, в одном из вариантов осуществления фармацевтические композиции по изобретению содержат другие лекарственные средства, которые вводят вместе с соединением 1. Например, композиция может дополнительно содержать одно или более лекарственных средств (также называемых "вторичными агентами"). Такие терапевтические агенты также хорошо известны в данной области и включают антагонисты аденозининовых рецепторов, антагонисты α-адренергических рецепторов, антагонисты β1-адренергических рецепторов, агонисты β2-адренергических рецепторов, агент двойного действия, такой как антагонист β-адренергических рецепторов/антагонист α1-рецепторов, усовершенствованные расщепители конечных продуктов гликозилирования, антагонисты альдостерона, ингибиторы альдостеронсинтазы, ингибиторы аминопептидазы N, андрогены, ингибиторы ангиотензинпревращающего фермента и агент двойного действия, такой как ингибитор ангиотензинпревращающего фермента/ингибитор неприлизина, активаторы и стимуляторы ангиотензинпревращающего фермента 2, вакцины против ангиотензина II, антикоагулянты, антидиабетические агенты, антидиарейные агенты, средства против глаукомы, антилипидные агенты, антиноцицептивные агенты, антитромботические агенты, антагонисты АТ1-рецепторов и антагонисты АТ1-рецепторов/ингибиторы неприлизина и мультифункциональные блокаторы рецепторов ангиотензина, антагонисты рецепторов брадикинина, блокаторы кальциевых каналов, ингибиторы химазы, дигоксин, диуретики, агонисты допамина, ингибиторы эндотелин-превращающего фермента, антагонисты рецепторов эндотелина, ингибиторы HMG-CoA-редуктазы, эстрогены, агонисты и/или антагонисты рецепторов эстрогена, ингибиторы обратного захвата моноаминов, миорелаксанты, натрийуретические пептиды и их аналоги, антагонисты рецепторов клиренса натрийуретического пептида, ингибиторы неприлизина, доноры оксида азота, нестероидные противовоспалительные агенты, антагонисты рецепторов N-метил d-аспартата, агонисты опиоидных рецепторов, ингибиторы фосфодиэстеразы, аналоги простагландина, агонисты рецепторов простагландина, ингибиторы ренина, селективные ингибиторы обратного захвата серотонина, блокатор натриевых каналов, стимуляторы и активаторы растворимой гуанилатциклазы, трициклические антидепрессанты, антагонисты рецепторов вазопрессина и их комбинации. В настоящем описании приводится подробное описание конкретных примеров этих агентов.
Конкретный вариант осуществления включает фармацевтическую композицию, содержащую соединение 1 или его кристаллическую форму и антагонист АТ1-рецепторов, ингибитор ангиотензинпревращающего фермента, ингибитор фосфодиэстеразы (PDE), ингибитор ренина, диуретик или их комбинации и необязательно один или более фармацевтически приемлемые носителей.
Соответственно, в другом аспекте изобретения фармацевтическая композиция включает соединение 1, второй активный агент и фармацевтически приемлемый носитель. Третий, четвертый и т.д. активные агенты также могут быть включены в композицию. В комбинированной терапии количество вводимого соединения 1, а также количество вторичных агентов может быть меньше количества, обычно вводимого при монотерапии.
Соединение 1 можно физически смешивать со вторым активным агентом с образованием композиции, содержащей оба агента; или каждый агент может присутствовать в отдельных и разных композициях, которые вводят пациенту одновременно или в разные моменты времени. Например, соединение 1 можно объединить со вторым активным агентом при помощи обычных процедур и оборудования для образования комбинации активных агентов, содержащих соединение 1 и второй активный агент. Кроме того, активные агенты могут быть объединены с фармацевтически приемлемым носителем с образованием фармацевтической композиции, содержащей соединение 1, второй активный агент и фармацевтически приемлемый носитель. В этом варианте осуществления компоненты композиции обычно перемешивают или смешивают для образования физической смеси. Затем физическую смесь вводят в терапевтически эффективном количестве, используя любой из описанных здесь способов введения.
Альтернативно, активные агенты могут храниться по отдельности до введения пациенту. В этом варианте осуществления агенты физически не смешивают друг с другом перед введением, но вводят одновременно или в разные моменты времени в виде отдельных композиций. Такие композиции могут быть упакованы по отдельности или могут быть упакованы вместе в наборе. При введении в разные моменты времени вторичный агент обычно вводится менее чем через 24 часа после введения соединения 1, начиная от одновременного введения с соединением по изобретению до примерно 24 часов после введения дозы. Это также называется последовательным введением. Таким образом, соединение 1 можно вводить перорально одновременно или последовательно с другим активным агентом виде двух таблеток, по одной таблетке на каждый активный агент, где последовательный может означать введение сразу после введения соединения 1 или через какой-то определенный промежуток времени (например, спустя один час или через три часа). Также предполагается, что вторичный агент можно вводить более чем через 24 часа после введения соединения 1. Альтернативно, комбинация может вводиться различными способами введения, т.е., первое - перорально, а другое - путем ингаляции.
В одном из вариантов осуществления набор содержит первую лекарственную форму, содержащую соединение 1, и по меньшей мере одну дополнительную лекарственную форму, содержащую один или более вторичных агентов, раскрытых в настоящем описании, в количествах, достаточных для осуществления способов по изобретению. Первая лекарственная форма и вторая (или третья и т.д.) лекарственная форма вместе содержат терапевтически эффективное количество активных агентов для лечения или профилактики заболевания или состояния здоровья у пациента.
Вторичный агент(агенты), если он включен, присутствует в терапевтически эффективном количестве, так что их обычно вводят в количестве, которое обеспечивает терапевтически благоприятный эффект при совместном введении с соединением 1 по изобретению. Вторичный агент может находиться в форме фармацевтически приемлемой соли, сольвата, оптически чистого стереоизомера и т.д. Вторичный агент также может находиться в форме пролекарства, например, соединения, имеющего группу карбоновой кислоты, которая была этерифицирована. Таким образом, вторичные агенты, перечисленные в настоящем описании, предназначены для включения всех таких форм и являются коммерчески доступными или могут быть получены при помощи обычных процедур и реагентов.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антагонистом аденозиновых рецепторов, примеры которого включают наксифиллин, ролофиллин, SLV-320, теофиллин и тонапофиллин.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антагонистом α-адренергических рецепторов, примеры которого включают доксазозин, празозин, тамсулозин и теразозин.
Соединение 1 также можно вводить в комбинации с антагонистом β1-адренергических рецепторов ("β1-блокатор"), примеры которого включают ацебутолол, алпренолол, амосулалол, аротинолол, атенолол, бефунолол, бетаксолол, бевантолол, бисопролол, бопиндолол, буциндолол, букумолол, буфетолол, буфуралол, бунитролол, бупранолол, бубридин, бутофилолол, каразолол, картеолол, карведилол, целипролол, цетамолол, клоранолол, дилевалол, эпанолол, эсмолол, инденолол, лабетолол, левобунолол, мепиндолол, метипранолол, метопролол, такой как метопролола сукцинат и метопролола тартрат, мопролол, надолол, надоксолол, небивалол, нипрадилол, окспренолол, пенбутолол, пербутолол, пиндолол, практолол, пронеталол, пропранолол, соталол, суфиналол, талиндол, тертатолол, тилисолол, тимолол, толипролол, ксибенолол и их комбинации. В одном из конкретных вариантов осуществления β1-антагонист выбран из атенолола, бисопролола, метопролола, пропранолола, соталола и их комбинаций. Обычно, β1-блокатор вводят в количестве, достаточном для обеспечения примерно от 2 до 900 мг на дозу.
В одном из вариантов осуществления соединение 1 вводят в комбинации с агонистом β2-адренергических рецепторов, примеры которого включают альбутерол, битолтерол, фенотерол, формотерол, индакатерол, изоэтарин, левалбутерол, метапротеренол, пирбутерол, сальбутамол, салмефамол, салметерол, тербуталин, вилантерол и т.п. Как правило, агонист β2-адренергических рецепторов вводится в количестве, достаточном для обеспечения примерно 0,05-500 мкг на дозу.
В одном из вариантов осуществления соединение 1 вводят в комбинации с усовершенствованным расщипителем конечных продуктов гликирования (AGE), примеры которого включают алагебриум (или ALT-711) и TRC4149.
В другом варианте осуществления соединение 1 вводят в комбинации с антагонистом альдостерона, примеры которого включают эплеренон, спиронолактон и их комбинации. Как правило, антагонист альдостерона вводится в количестве, достаточном для обеспечения примерно от 5 до 300 мг в день.
В одном из вариантов осуществления соединение 1 вводят в комбинации с ингибитором аминопептидазы N или дипептидилпептидазы III, примеры которого включают бестатин и PC18 (2-амино-4-метилсульфонилбутантиол, метионинтиол).
Соединение 1 также можно вводить в комбинации с ингибитором ангиотензинпревращающего фермента (ACE), примеры которого включают аккуприл, алацеприл, беназеприл, беназеприлат, каптоприл, церанаприл, цилазаприл, делаприл, эналаприл, эналаприлат, фозиноприл, фозиноприлат, имидаприл, лизиноприл, моэксиприл, моноприл, мовелтиприл, пентоприл, периндоприл, квинаприл, квинаприлат, рамиприл, рамиприлат, саралазина ацетат, спираприл, темокаприл, трандолаприл, зофеноприл и их комбинации. В конкретном варианте осуществления ингибитор ACE выбирают из: беназеприла, каптоприла, эналаприла, лизиноприла, рамиприла и их комбинаций. Как правило, ингибитор ACE вводят в количестве, достаточном для обеспечения примерно от 1 до 150 мг в день.
В другом варианте осуществления соединение 1 вводят в комбинации с агентом двойного действия, таким как ингибитор ангиотензинпревращающего фермента/ингибитор неприлизина (ACE/NEP), примеры которого включают: AVE-0848 ((4S,7S,12bR)-7-[3-метил-2(S)-сульфанилбутирамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]-бензазепин-4-карбоновая кислота); AVE-7688 (илепатрил) и его исходное соединение; BMS-182657 (2-[2-оксо-3(S)-[3-фенил-2(S)-сульфанилпропионамидо]-2,3,4,5-тетрагидро-1H-1-бензазепин-1-ил]уксусная кислота); CGS-35601 (N-[1-[4-метил-2(S)-сульфанилпентанамидо]циклопентил-карбонил]-L-триптофан); фазидотрил; фазидотрилат; эналаприлат; ER-32935 ((3R,6S,9aR)-6-[3(S)-метил-2(S)-сульфанилпентанамидо]-5-оксопергидротиазоло[3,2-a]азепин-3-карбоновая кислота); гемпатрилат; MDL-101264 ((4S,7S,12bR)-7-[2(S)-(2-морфолиноацетилтио)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота); MDL-101287 ([4S-[4α,7α(R*),12bβ]]-7-[2-(карбоксиметил)-3-фенилпропионамидо]-6-оксо-1,2,3,4,6,7,8,12b-октагидропиридо[2,1-a][2]бензазепин-4-карбоновая кислота); омапатрилат; RB-105 (N-[2(S)-(меркаптометил)-3(R)-фенилбутил]-L-аланин); сампатрилат; SA-898 ((2R,4R)-N-[2-(2-гидроксифенил)-3-(3-меркаптопропионил)тиазолидин-4-илкарбонил]-L-фенилаланин); Sch-50690 (N-[1(S)-карбокси-2-[N2-(метансульфонил)-L-лизиламино]этил]-L-валил-L-тирозин); и их комбинации. В одном конкретном варианте осуществления ингибитор ACE/NEP выбирают из: AVE-7688, эналаприлата, фазидотрила, фазидотрилата, омапатрилата, сампатрилата и их комбинаций.
В одном из вариантов осуществления соединение 1 вводят в комбинации с активатором или стимулятором ангиотензинпревращающего фермента 2 (ACE2).
В одном из вариантов осуществления соединение 1 вводят в комбинации с вакциной против ангиотензина II, примеры которой включают ATR12181 и CYT006-AngQb.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антикоагулянтом, примеры которого включают: кумарины, такие как варфарин; гепарин; и прямые ингибиторы тромбина, такие как аргатробан, бивалирудин, дабигатран и лепирудин.
В другом варианте осуществления соединение 1 вводят в комбинации с антидиабетическим агентом, примеры которого включают инъецируемые лекарственные средства, а также перорально эффективные лекарственные средства и их комбинации. Примеры инъецируемых лекарственных средств включают инсулин и производные инсулина. Примеры перорально эффективных лекарственных средств включают: бигуаниды, такие как метформин; антагонисты глюкагона; ингибиторы α-глюкозидазы, такие как акарбоза и миглитол; ингибиторы дипептидилпептидазы IV (ингибиторы DPP-IV), такие как алоглиптин, денаглиптин, линаглиптин, саксаглиптин, ситаглиптин и вилдаглиптин; меглитиниды, такие как репаглинид; оксадиазолидиндионы; сульфонилмочевины, такие как хлорпропамид, глимепирид, глипизид, глибурид и толазамид; тиазолидиндионы, такие как пиоглитазон и росиглитазон; и их комбинации.
В другом варианте осуществления соединение 1 вводят в комбинации с антидиарейным лечением. Типичные варианты лечения включают растворы для пероральной регидратации (ORS), лоперамид, дифеноксилат и субсалицилат висмута.
В другом варианте осуществления соединение 1 вводят в комбинации с агентом против глаукомы, примеры которого включают: α-адренергические агонисты, такие как бримонидин; антагонисты β1-адренергических рецепторов; локальные β1-блокаторы, такие как бетаксолол, левобунолол и тимолол; ингибиторы карбоангидразы, такие как ацетазоламид, бринзоламид или дорзоламид; холинергические агонисты, такие как цевимелин и DMXB-анабазин; соединения эпинефрина; миотики, такие как пилокарпин; и аналоги простагландинов.
В другом варианте осуществления соединение 1 вводят в комбинации с антилипидным агентом, примеры которого включают: ингибиторы транспортного белка эфира холестерина (CETP), такие как анацетрапиб, далцетрапиб и торцетрапиб; статины, такие как аторвастатин, флувастатин, ловастатин, правастатин, розувастатин и симвастатин; и их комбинации.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антитромботическим агентом, примеры которого включают: аспирин; антитромбоцитарными агентами, такими как клопидогрел, прасугрел и тиклопидин; гепарином и их комбинациями.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антагонистом АТ1-рецепторов, также известным как блокатор рецепторов ангиотензина II типа 1 (ARB). Типичные ARB включают абитесартан, азилсартан (например, азилсартан медоксомил), бензиллосартан, кандесартан, кандесартан цилексетил, элизартан, эмбусартан, энолтазосартан, эпросартан, EXP3174, фонсартан, форасартан, глициллосартан, ирбесартан, изотеолин, лозартан, медоксомил, милфасартан, олмесартан (например, олмесартан медоксомил), опомисартан, пратосартан, риписартан, саприсартан, саралазин, сармезин, TAK-591, тасосартан, телмисартан, валсартан, золасартан и их комбинации. В конкретном варианте осуществления ARB выбирают из азилсартана медоксомила, кандесартана цилексетила, эпросартана, ирбесартана, лозартана, олмесартана медоксомила, саприсартана, тазосартана, телмисартана, валсартана и их комбинаций. Приведенные в качестве примера соли и/или пролекарства включают кандесартана цилексетил, эпросартана мезилат, калиевая соль лозартана и олмесаратан медоксомил. Как правило, ARB вводят в количестве, достаточном для обеспечения от примерно 4-600 мг на дозу, с приведенными в качестве примера суточными дозами от 20 до 320 мг в день.
Соединение 1 также можно вводить в сочетании с агентом двойного действия, таким как антагонист АТ1-рецепторов/ингибитор неприлизина (ARB/NEP), примеры которого включают соединения, описанные в патентах США №№ 7,879,896 и 8,013,005, оба на имя Allegretti et al., такое как соединение, 4'-{2-этокси-4-этил-5-[((S)-2-меркапто-4-метилпентаноиламино)-метил]имидазол-1-илметил}-3'-фторбифенил-2-карбоновая кислота.
Соединение 1 также можно вводить в комбинации с многофункциональными блокаторами рецепторов ангиотензина, как описано у Kurtz&Klein (2009). Hypertension Research 32:826-834.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антагонистом рецепторов брадикинина, например, икатибантом (HOE-140). Предполагается, что эта комбинированная терапия может оказаться преимущественной для профилактики ангионевротического отека или других нежелательных последствий повышенного уровня брадикинина.
В одном из вариантов осуществления соединение 1 вводят в комбинации с блокатором кальциевых каналов, примеры которого включают амлодипин, анипимил, аранипин, барнидипин, бенциклан, бенидипин, бепридил, клентиазем, цилнидипин, циннаризин, дилтиазем, эфонидипин, элгодипин, этафенон, фелодипин, фендилин, флунаризин, галлопамил, исрадипин, лацидипин, лерканидипин, лидофлазин, ломеризин, манидипин, мибефрадил, никардипин, нифедипин, нигулдипин, нилудипин, нилвадипин, нимодипин, низолдипин, нитрендипин, нивалдипин, пергексилин, прениламин, риозидин, семотиадил, теродилин, тиапамил, верапамил и их комбинации. В конкретном варианте осуществления блокатор кальциевых каналов выбирают из амлодипина, бепридила, дилтиазема, фелодипина, исрадипина, лацидипина, никардипина, нифедипина, нигулдипина, нилудипина, нимодипина, нисолдипина, риозидина, верапамила и их комбинаций. Как правило, блокатор кальциевых каналов вводится в количестве, достаточном для обеспечения от примерно 2-500 мг на дозу.
В одном из вариантов осуществления соединение 1 вводят в комбинации с ингибитором химазы, таким как TPC-806 и 2-(5-формиламино-6-оксо-2-фенил-1,6-дигидропиримидин-1-ил)-N-[{3,4-диоксо-1-фенил-7-(2-пиридилокси)}-2-гептил]ацетамид (NK3201).
В одном из вариантов осуществления соединение 1 вводят в комбинации с диуретиком, примеры которого включают: ингибиторы карбоангидразы, такие как ацетазоламид и дихлорфенамид; петлевые диуретики, которые включают производные сульфонамида, такие как ацетазоламид, амбуцид, азосемид, буметанид, бутазоламид, хлораминофенамид, клофенамид, клопамид, клорексолон, дисульфамид, этоксзоламид, фуросемид, мефрузид, метазоламид, пиретанид, торсемид, трипамид и ксипамид, а также сульфонамидные диуретики, такие как этакриновая кислота и другие соединения феноксиуксусной кислоты, такие как тиениловая кислота, индакринон и квинкарбат; осмотические диуретики, такие как маннит; калийсберегающие диуретики, которые включают антагонисты альдостерона, такие как спиронолактон и ингибиторы Na+ канала, такие как амилорид и триамтерен; тиазидные и тиазидоподобные диуретики, такие как алтиазид, бендронфлуметиазид, бензилгидрохлоротиазид, бензтиазид, бутиазид, хлорталидон, хлортиазид, циклопентиазид, циклотиазид, эпитиазид, этиазид, фенквизон, флюметиазид, гидрохлоротиазид, гидрофлуметиазид, индапамид, метилклотиазид, метикран, метолазон, парафлутизид, политиазид, квинетазон, теклотиазид и трихлорметиазид; и их комбинации. В конкретном варианте осуществления диуретик выбирают из амилорида, буметанида, хлортиазида, хлорталидона, дихлорфенамида, этакриновой кислоты, фуросемида, гидрохлоротиазида, гидрофлуметиазида, индапамида, метилклотиазида, метолазона, торсемида, триамтерена и их комбинаций. Диуретик вводят в количестве, достаточном для обеспечения примерно от 5 до 50 мг в день, более типично 6-25 мг в день, причем общие дозы составляют 6,25 мг, 12,5 мг или 25 мг в день.
Соединение 1 также можно вводить в комбинации с ингибитором эндотелинпревращающего фермента (ЕСЕ), примеры которого включают фосфорамидон, CGS 26303 и их комбинации.
В конкретном варианте осуществления соединение 1 вводят в комбинации с антагонистом рецепторов эндотелина, примеры которого включают: селективные антагонисты рецепторов эндотелина, которые оказывают воздействие на рецепторы эндотелина А, такие как авосентан, амбрисентан, атрасентан, BQ-123, клазосентан, дарусентан, ситаксентан и зиботентан; и антагонисты рецепторов эндотелина с двойным действием, которые влияют на оба рецептора эндотелина А и В, такие как бозентан, мацитентан и тезосентан.
В другом варианте осуществления соединение 1 вводят в комбинации с одним или более ингибиторами HMG-CoA-редуктазы, которые также известны как статины. Типичные статины включают аторвастатин, флувастатин, ловастатин, питавастатин, правастатин, розувастатин и симвастатин.
В одном из вариантов осуществления соединение 1 вводят в комбинации с ингибитором обратного захвата моноаминов, примеры которого включают ингибиторы обратного захвата норэпинефрина, такие как атомоксетин, бупроприон и метаболит бупроприона гидроксибупроприон, мапротилин, ребоксетин и вилоксазин; селективные ингибиторы обратного захвата серотонина (SSRI), такие как циталопрам и метаболит циталопрама десметилциталопрам, дапоксетин, эсциталопрам (например, эсциталопрама оксалат), флуоксетин и метаболит флуоксетина - десметил норфлуоксетина, флувоксамин (например, флувоксамина малеат), пароксетин, сертралин и метаболит сертралина деметилсертралин; агент двойного действия, такой как ингибитор обратного захвата серотонина и норэпинефрина (SNRI), такой как бицифадин, дулоксетин, милнаципран, нефазодон и венлафаксин; и их комбинации.
В другом варианте осуществления соединение 1 вводят в комбинации с миорелаксантом, примеры которого включают: каризопродол, хлорзоксазон, циклобензаприн, дифлунизал, метаксалон, метокарбамол и их комбинации.
В одном из вариантов осуществления соединение 1 вводят в комбинации с натрийуретическим пептидом или аналогом, примеры которого включают: карперитид, CD-NP (Nile Therapeutics), CU-NP, несиритид, PL-3994 (Palatin Technologies, Inc.), уларитид, цендеритид и соединения, описанные Ogawa et al (2004) J.Biol.Chem. 279: 28625-31. Эти соединения также упоминаются как агонисты рецептора А натрийуретического пептида (NPR-А). В другом варианте осуществления соединение 1 вводят в комбинации с антагонистом рецептор С натрийуретического пептида (NPR-C), таким как SC-46542, cANF (4-23) и AP-811 (Veale (2000) Bioorg Med Chem Lett 10: 1949-52). Например, AP-811 продемонстрировал синергизм в сочетании с ингибитором NEP, тиорфаном (Wegner (1995) Clin.Exper.Hypert. 17: 861-876).
В другом варианте осуществления соединение 1 вводят в комбинации с ингибитором неприлизина (NEP), примеры которого включают: AHU-377; кандоксатрил; кандоксатрилат; кандоксатрилат (бензиловый эфир (+)-N-[2(R)-(ацетилтиометил)-3-фенилпропионил]глицина); CGS-24128 (3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-24592 ((S)-3-[3-(бифенил-4-ил)-2-(фосфонометиламино)пропионамидо]пропионовая кислота); CGS-25155 (бензиловый эфир N[9(R)-(ацетилтиометил)-10-оксо-1-азациклодекан-2(S)-илкарбонил]-4(R)-гидрокси-L-пролина); 3-(1-карбамоилциклогексил)пропионовую кислоту, описанные в WO 2006/027680, Hepworth et al. (Pfizer Inc.); JMV-390-1 (2(R)-бензил-3-(N-гидроксикарбамоил)пропионил-L-изолейцил-L-лейцин); экадотрил; фосфорамидон; ретротиорфан; RU-42827 (2-(меркаптометил)-N-(4-пиридинил)бензолпропионамид); RU-44004 (N-(4-морфолинил)-3-фенил-2-(сульфанилметил)пропионамид); SCH-32615 ((S)-N-[N-(1-карбокси-2-фенилэтил)-L-фенилаланил]-β-аланин) и его пролекарство SCH-34826 ((S)-N-[N-[1-[[(2,2-диметил-1,3-диоксолан-4-ил)метокси]карбонил]-2-фенилэтил]-L-фенилаланил]-β-аланин); сиалорфин; SCH-42495 (этиловый эфир N-[2(S)-(ацетилсульфанилметил)-3-(2-метилфенил) пропионил]-L-метионина); спинорфин; SQ-28132 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]лейцин); SQ-28603 (N-[2-(меркаптометил)-1-оксо-3-фенилпропил]-β-аланин); SQ-29072 (7-[[2-(меркаптометил)-1-оксо-3-фенилпропил]амино]гептановая кислота); тиорфан и его пролекарство рацекадотрил; UK-69578 (цис-4-[[[1-[2-карбокси-3- (2-метоксиэтокси)пропил]циклопентил]карбонил]амино]циклогексанкарбоновая кислота); UK-447,841 (2-{1-[3-(4-хлорфенил)пропилкарбомоил]-циклопентилметил}-4-метоксимасляная кислота); UK-505,749 ((R)-2-метил-3-{1-[3-(2-метилбензотиазол-6-ил)пропилкарбомоил]циклопентил}пропионовая кислота); 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановую кислоту и этиловый эфир 5-бифенил-4-ил-4-(3-карбоксипропиониламино)-2-метилпентановой кислоты (WO 2007/056546); даглутарил [(3S,2'R)-3-{1-[2'-(этоксикарбонил)-4'-фенилбутил]-циклопентан-1-карбониламино}-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты], описанной в WO 2007/106708, Khder et al. (Novartis AG); и их комбинации. В конкретном варианте осуществления ингибитор NEP выбирают из AHU-377, кандоксатрила, кандоксатрилата, CGS-24128, фосфорамидона, SCH-32615, SCH-34826, SQ-28603, тиорфана и их комбинаций. В конкретном варианте осуществления ингибитор NEP представляет собой соединение, такое как даглутрил или CGS-26303 ([N-[2-(бифенил-4-ил)-1(S)-(1Н-тетразол-5-ил)этил]амино]метилфосфоновая кислота), которые обладают активностью как ингибитора эндотелинпревращающего фермента (ECE), так и ингибитора NEP. Можно также использовать другие ECE/NEP агенты двойного действия. Ингибитор NEP вводится в количестве, достаточном для обеспечения примерно 20-800 мг в день, с типичными суточными дозами от 50 до 700 мг в день, более предпочтительно 100-600 или 100-300 мг в день.
В одном из вариантов осуществления соединение 1 вводят в комбинации с донором оксида азота, примеры которого включают: никорандил; органические нитраты, такие как пентаэритрит тетранитрат; и сиднонимины, такие как линзидомин и молсидомин.
В другом варианте осуществления соединение 1 вводят в комбинации с нестероидным противовоспалительным агентом (NSAID), примеры которого включают: ацеметацин, ацетилсалициловую кислоту, алклофенак, алминоппрофен, амфенак, амиприлозу, алоксиприн, аниролак, апазон, азапропазон, бенорилат, беноксапрофен, безпиперилон, броперамол, буклоксовую кислоту, капрофен, клиданак, диклофенак, дифлунизал, дифталон, эноликам, этодолак, эторикоксиб, фенбуфен, фенклофенак, фенклозовую кислоту, фенопрофен, фентиазак, фепразон, флуфенамовую кислоту, флуфенизал, флупрофен, флурбипрофен, фурофенак, ибуфенак, ибупрофен, индометацин, индопрофен, изоксепак, изоксикам, кетопрофен, кеторолак, лофемизол, лорноксикам, меклофенамат, меклофенамовую кислоту, мефенаминовую кислоту, мелоксикам, мезаламин, миропрофен, мофебутазон, набуметон, напроксен, нифлумовую кислоту, оксапрозин, оксипинак, оксифенбутазон, фенилбутазон, пироксикам, пирпрофен, пранопрофен, салсалат, судоксикам, сульфасалазин, сулиндак, супрофен, теноксикам, тиопинак, тиапрофеновую кислоту, тиоксапрофен, толфенамовую кислоту, толметин, трифлумидат, зидометацин, зомепирак и их комбинации. В конкретном варианте осуществления NSAID выбирают из этодолака, флурбипрофена, ибупрофена, индометацина, кетопрофена, кеторолака, мелоксикама, напроксена, оксапрозина, пироксикама и их комбинации.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антагонистом рецепторов N-метил-d-аспартата (NMDA), примеры которого включают амантадин, декстрометорфан, декстропропоксифен, кетамин, кетобемидон, мемантин, метадон и т.д.
В другом варианте осуществления соединение 1 вводят в комбинации с агонистом опиоидных рецепторов (также называемым опиоидным анальгетиком). Типичные агонисты опиоидных рецепторов включают: бупренорфин, буторфанол, кодеин, дигидрокодеин, фентанил, гидрокодон, гидроморфон, леваллорфан, леворфанол, меперидин, метадон, морфин, налбуфин, налмефен, налорфин, налоксон, налтрексон, налорфин, оксикодон, оксиморфон, пентазоцин, пропоксифен, трамадол и их комбинации. В некоторых вариантах осуществления агонист опиоидных рецепторов выбирают из кодеина, дигидрокодеина, гидрокодона, гидроморфона, морфина, оксикодона, оксиморфона, трамадола и их комбинаций.
В конкретном варианте осуществления соединение 1 вводят в комбинации с ингибитором фосфодиэстеразы (PDE), в частности ингибитором PDE-V. Типичными ингибиторами PDE-V являются аванафил, лоденафил, мироденафил, силденафил (Ревацио®, Revatio®), тадалафил (Адцирка®, Adcirca®), варденафил (Левитра®, Levitra®) и уденафил.
В другом варианте осуществления соединение 1 вводят в комбинации с аналогом простагландина (также называемым простаноидом или аналогом простациклина). Характерными аналогами простагландинов являются берапрост натрия, биматопрост, эпопростенол, илопрост, латанопрост, тафлупрост, травопрост и трепростинил, причем особый интерес представляют биматопрост, латанопрост и тафлупрост.
В другом варианте осуществления соединение 1 вводят в комбинации с агонистом рецепторов простагландина, примеры которого включают биматопрост, латанопрост, травопрост и т.п.
Соединение 1 также можно вводить в комбинации с ингибитором ренина, примеры которого включают алискирен, эналкирен, ремикирен и их комбинации.
В другом варианте осуществления соединение 1 вводят в комбинации с селективным ингибитором обратного захвата серотонина (SSRI), примеры которого включают: циталопрам и метаболит циталопрама десметилциталопрам, дапоксетин, эсциталопрам (например, оксалат эсциталопрама), флуоксетин и десметиловый метаболит флуоксетина норфлуоксетин, флувоксамин (например, малеат флувоксамина), пароксетин, сертралин и метаболит сертралина деметилсертралин и их комбинации.
В одном из вариантов осуществления соединение 1 вводят в комбинации с агонистом рецепторов серотонина 5-HT1D, примеры которого включают триптаны, такие как алмотриптан, авитриптан, элетриптан, фроватриптан, наратриптан, ризатриптан, суматриптан и золмитриптан.
В одном из вариантов осуществления соединение 1 вводят в комбинации с блокатором натриевых каналов, примеры которых включают карбамазепин, фосфенитоин, ламотриджин, лидокаин, мексилетин, окскарбазепин, фенитоин и их комбинации.
В одном из вариантов осуществления соединение 1 вводят в комбинации со стимулятором или активатором растворимой гуанилатциклазы, примеры которых включают атацигуат, риоцигуат и их комбинации.
В одном из вариантов осуществления соединение 1 вводят в комбинации с трициклическим антидепрессантом (TCA), примеры которого включают амитриптилин, амитриптилиноксид, бутриптилин, кломипрамин, демексиптилин, дезипрамин, дибензепин, диметакрин, досулепин, доксепин, имипрамин, имипраминоксид, лофепрамин, мелитрацен, метапрамин, нитроксазепин, нортриптилин, ноксиптилин, пипофезин, пропизепин, протриптилин, квинупрамин и их комбинации.
В одном из вариантов осуществления соединение 1 вводят в комбинации с антагонистом рецепторов вазопрессина, примеры которого включают кониваптан и толваптан.
Комбинированные вторичные терапевтические агенты также могут быть полезны в дальнейшей комбинированной терапии с соединением по изобретению. Например, соединение по изобретению можно комбинировать с диуретиком и ARB, или блокатором кальциевых каналов и ARB, или с диуретиком и ингибитором ARB, или с блокатором кальциевых каналов и статином. Конкретные примеры включают комбинацию ингибитора ACE эналаприла (в форме малеатной соли) и диуретика гидрохлоротиазида, который продается под торговой маркой Вазеретик® (Vaseretic®) или комбинацию блокатора кальциевых каналов амлодипина (в форме безилатной соли) и ARB олмесартана (в форме пролекарства - медоксомила), или комбинацию блокатора кальциевых каналов и статина, все из которых также могут быть использованы с соединением 1. Другие терапевтические агенты, такие как агонисты α2-адренергических рецепторов и антагонисты рецепторов вазопрессина, также ожно использовать в комбинированной терапии. Приведенные в качестве примера агонисты α2-адренергических рецепторов включают клонидин, дексмедетомидин и гуанфацин.
Приведенные ниже составы иллюстрируют типичные фармацевтические композиции по изобретению.
Иллюстративные твердые желатиновые капсулы для перорального введения
Соединение по изобретению (50 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно перемешивают. Затем полученную композицию загружают в твердые желатиновые капсулы (500 мг композиции на капсулу). Альтернативно, соединение 1 (20 мг) тщательно перемешивают с крахмалом (89 мг), микрокристаллической целлюлозой (89 мг) и стеаратом магния (2 мг). Затем смесь пропускают через сито N 45 меш США и загружают в твердую желатиновую капсулу (200 мг композиции на капсулу).
Альтернативно, соединение 1 (30 г), вторичный агент (20 г), 440 г высушенной распылением лактозы и 10 г стеарата магния тщательно перемешивают и обрабатывают, как описано выше.
Иллюстративная рецептура желатиновой капсулы для перорального введения
Соединение 1 (100 мг) тщательно смешивают с моноолеатом полиоксиэтиленсорбитана (50 мг) и крахмальным порошком (250 мг). Затем смесь загружают в желатиновую капсулу (400 мг композиции на капсулу). Альтернативно, соединение 1 (70 мг) и вторичный агент (30 мг) тщательно смешивают с моноолеатом полиоксиэтиленсорбитана (50 мг) и крахмальным порошком (250 мг), и полученную смесь загружают в желатиновую капсулу (400 мг композиции на капсула).
Альтернативно, соединение 1 (40 мг) тщательно смешивают с микрокристаллической целлюлозой (Avicel PH 103, 259,2 мг) и стеаратом магния (0,8 мг). Затем смесь загружают в желатиновую капсулу (размер №1, белая, непрозрачная) (300 мг композиции на капсулу).
Иллюстративная гидроксипропилметилцеллюлозная (HPMC) капсула для перорального введения
Соединение 1 (50 мг или 100 мг) загружают непосредственно в HPMC капсулу.
Иллюстративный состав таблеток для перорального введения
Соединение 1 (10 мг), крахмал (45 мг) и микрокристаллическую целлюлозу (35 мг) пропускают через сито N 20 меш США и тщательно перемешивают. Полученные таким образом гранулы сушат при 50-60°С и пропускают через сито N 16 меш США. Раствор поливинилпирролидона (4 мг в виде 10%-ного раствора в стерильной воде) смешивают с карбоксиметилкрахмалом натрия (4,5 мг), стеаратом магния (0,5 мг) и тальком (1 мг), и затем эту смесь пропускают через сито N 16 меш США. Затем к гранулам добавляют карбоксиметилкрахмал натрия, стеарат магния и тальк. После смешивания смесь прессуют на таблетирующей машине, получая таблетку массой 100 мг.
Альтернативно, соединение 1 (250 мг) тщательно смешивают с микрокристаллической целлюлозой (400 мг), двуокисью кремния (10 мг) и стеариновой кислотой (5 мг). Затем смесь прессуют с получением таблеток (665 мг композиции на таблетку).
Альтернативно, соединение 1 (400 мг) тщательно смешивают с кукурузным крахмалом (50 мг), кроскармелозой натрия (25 мг), лактозой (120 мг) и стеаратом магния (5 мг). Затем смесь прессуют, получая таблетку с односторонней риской (600 мг композиции на таблетку).
Альтернативно, соединение 1 (100 мг) тщательно смешивают с кукурузным крахмалом (100 мг), водным раствором желатина (20 мг). Смесь сушат и измельчают до мелкодисперсного порошка. Затем микрокристаллическую целлюлозу (50 мг) и стеарат магния (5 мг) смешивают с желатиновой композицией, гранулируют, и полученную смесь прессуют с получением таблеток (100 мг соединения по изобретению на таблетку).
Иллюстративный состав суспензии для перорального введения
Следующие ингредиенты смешивают с образованием суспензии, содержащей 100 мг соединения 1 на 10 мл суспензии:
Иллюстративный жидкий состав для перорального введения
Подходящим жидким составом является состав, содержащий буфер на основе карбоновой кислоты, такой как растворы цитратного, лактатного и малеатного буферов. Например, соединение 1 (которое можно предварительно смешивать с ДМСО) смешивают с 100 мМ аммоний-цитратным буфером и доводят значение рН до 5 или смешивают с 100 мМ раствором лимонной кислоты и доводят значение рН до 2. Такие растворы также могут включать солюбилизирующий наполнитель, такой как циклодекстрин, например раствор может включать 10 мас.% гидроксипропил-β-циклодекстрин.
Другие подходящие составы включают 5% раствор NaHCO3 с циклодекстрином или без него.
Иллюстративный парентеральный IV состав для введения путем инъекций
Соединение 1 (0,2 г) смешивают с раствором 0,4М натрий-ацетатного буфера (2,0 мл). Значение рН полученного раствора доводят до рН 4, используя при необходимости 0,5н. водный раствор соляной кислоты или 0,5н. водный раствор гидроксида натрия, и затем добавляют воду для инъекции в количестве, достаточном для получения конечного объема 20 мл. Затем смесь фильтруют через стерильный фильтр (0,22 мкм), получая стерильный раствор, подходящий для введения путем инъекции.
Следующие составы иллюстрируют типичные фармацевтические композиции по настоящему изобретению.
Примера A состава
Замороженный раствор, подходящий для приготовления инъекционного раствора, получают следующим образом:
Типичная процедура: Наполнители, если они есть, растворяют примерно в 80% воды для инъекций, куда добавляют и растворяют активное соединение 1 или 1'.
Значение рН регулируют 1М гидроксидом натрия до 3-4,5, и затем доводят объем до 95% конечного объема водой для инъекций. При необходимости, проверяют и регулируют величину рН, и доводят объем до конечного объема водой для инъекций. Затем состав фильтруют в стерильных условиях через 0,22 мкм фильтр и помещают в стерильный флакон в асептических условиях. Флакон закрывают, наносят метку и хранят в замороженном виде.
Пример B состава
Лиофилизированный порошок или твердое кристаллическое вещество, подходящее для приготовления инъекционного раствора, получают следующим образом:
Типичная процедура: Наполнители и/или буферные агенты, если имеются, растворяют примерно в 60% воды для инъекций. Активное соединение 1 или 1' добавляют и растворяют, значение рН доводят до 3-4,5 1М гидроксидом натрия, и водой для инъекций доводят объем до 95% от конечного объема. При необходимости, проверяют и регулируют величину рН, и доводят объем до конечного объема водой для инъекций. Затем состав фильтруют в стерильных условиях через 0,22 мкм фильтр и помещают в стерильный флакон в асептических условиях. Затем препарат лиофилизируют с использованием соответствующего цикла лиофилизации. Флакон закрывают (необязательно в условиях неполного вакуума или сухим азотом), помещают и хранят при низких температурах.
Пример C состава
Инъецируемый раствор для внутривенного введения пациенту готовят из приведенного выше примера B состава следующим образом:
Типичная процедура: Лиофилизированный порошок состава примера B (например, содержащий от 10 до 1000 мг активного соединения 1 или 1') восстанавливают в 20 мл стерильной воды, и полученный раствор дополнительно разбавляют 80 мл стерильного физиологического раствора в 100 мл инфузионном пакете. Затем разбавленный раствор вводят пациенту внутривенно в течение 30-120 минут.
Примеры композиций для введения путем ингаляции
Соединение 1 (0,2 мг) микронизируют и затем смешивают с лактозой (25 мг). Затем эту смешанную смесь загружают в желатиновый ингаляционный картридж. Содержимое картриджа вводят, например, при помощи ингалятора для сухого порошка.
Альтернативно, микронизированное соединение 1 (10 г) диспергируют в растворе, полученном путем растворения лецитина (0,2 г) в деминерализованной воде (200 мл). Полученную суспензию сушат распылением и затем микронизируют с образованием микронизированной композиции, содержащей частицы со средним диаметром менее примерно 1,5 мкм. Затем микронизированную композицию загружают в картриджи дозирующего ингалятора, содержащие 1,1,1,2-тетрафторэтан под давлением, в количестве, достаточном для обеспечения от 10 мкг до 500 мкг соединения по изобретению на вводимую ингалятором дозу.
Альтернативно, соединение 1 (25 мг) растворяют в забуференном цитратом (рН 5) изотоническом солевом растворе (125 мл). Смесь перемешивают и обрабатывают ультразвуком до растворения соединения. Значение рН раствора проверяют и при необходимости доводят до 5 путем медленного добавления 1н. водного раствора NaOH. Раствор вводят при распылителя, которое обеспечивает от примерно 10 мкг до примерно 500 мкг соединения 1 на дозу.
ПРИМЕРЫ
Приведенные ниже препараты и примеры используются для иллюстрации конкретных вариантов осуществления изобретения. Однако эти конкретные варианты осуществления не предназначены для ограничения объема изобретения любым способом, если не указано иное.
Следующие сокращения имеют следующие значения, если не указано иное, и любые другие аббревиатуры, используемые и не определенные в настоящем описании, имеют их стандартное общепринятое значение:
AcOH уксусная кислота
BOC т-бутоксикарбонил(-C(O)OC(CH3)3)
(BOC)2O ди-т-бутилдикарбонат
Bn бензил
CPME циклопентилметиловый эфир
DCC 1,3-дициклогексилкарбодиимид
DCM дихлорметан или метилен хлорид
DIPE диизопропиловый эфир
DIPEA N,N-диизопропилэтиламин
DMAP 4-диметиламинопиридин
DMF N,N-диметилформамид
Et3N триэтиламин
EtOH этанол
Et2O диэтиловый эфир ether
EtOAc этилацетат
MeCN ацетонитрил
NaHMDS натрий бис(триметилсилил)амид
Pd(PPh3)4 тетракис(трифенилфосфин)паооадий(0)
PE петролейный эфир
SilicaCat®DPP-Pd катализатор дифенилфосфин палладий (II) на основе оксида кремния
TFA трифторуксусная кислота
THF тетрагидрофуран
Если не указано иное, все материалы, такие как реагенты, исходные вещества и растворители, приобретали у коммерческих поставщиков (таких как Sigma-Aldrich, Fluka Riedel-de Haën и т.п.) и использовали без дальнейшей очистки.
Реакции проводили в атмосфере азота, если не указано иное. Ход реакции контролировали при помощи тонкослойной хроматографии (ТСХ), аналитической высокоэффективной жидкостной хроматографии (аналог ВЭЖХ) и масс-спектрометрии, которые более подробно описаны в конкретных примерах. В аналитической ВЭЖХ, как правило, использовали следующие растворители: растворитель A: 98% H2O/2% MeCN/1.0 mL/L TFA; растворитель B: 90% MeCN/10% H2O/1.0 mL/L TFA.
Реакции проводили, как описано, например, в каждом примере получения; реакционные смеси, как правило, очищали методом экстракции и другими методами очистки, такими как кристаллизация, зависящая от растворителя и температуры, осаждение. Кроме того, реакционные смеси, как правило, очищали методом препаративной ВЭЖХ, как правило, с использованием насадок на колонки Microsorb C18 и Microsorb BDS и обычных элюентов. Наблюдение за ходом реакции, как правило, осуществляли при помощи метода масс-спектрометрии с жидкостной хроматографией (LCMS). Характеристику изомеров определяли методом спектроскопии ядерного эффекта Оверхаузера (NOE). Характеристику продуктов реакции, как правило, получали методом масс-спектроскопии и1H-ЯМР-спектроскопии. В случае измерения методом ЯМР, образцы растворяли в дейтерированном растворителе (CD3OD, CDCl3 или ДМСО-d6), и1H-ЯМР-спектры получали, используя прибор Varian Gemini 2000 (400 МГц), в стандартных условиях наблюдения. Определение соединений методом масс-спектрометрии обычно проводили методом электрораспылительной ионизации (ESMS), используя прибор Applied Biosystems (Foster City, CA) модели API 150 EX или Agilent (Palo Alto, CA) модели 1200 LC/MSD.
Методы измерения
Порошковая рентгеновская дифрактометрия
Анализ методом порошковой рентгеновской дифрактометрии проводили на рентгеновском дифрактометре Bruker D8-Advance. Источником рентгеновского излучения было Cu-Kα излучение с выходным напряжением 40 кВ и током 40 мА. Прибор работал в геометрии Брэгга-Брентано, а для получения параллельного рентгеновского пучка использовали зеркала Гебеля. Любую расходимость пучка ограничивали 0,2° вертикальной щелью расходимости на источнике и щелями Соллера (2,5°) на источнике и детекторе. Для измерения на силиконовый держатель образца с нулевым фоном помещали небольшое количество порошка (5-25 мг) осторожно уплотняя его для образования гладкой поверхности и подвергали воздействию рентгеновского излучения. Образцы сканировали в режиме связанных θ-2θ в диапазоне от 2° до 35° угла 2θ с шагом 0,02° и скоростью сканирования 0,3 секунды на шаг. Сбор данных контролировали с помощью компьютерной программы Bruker DiffracSuite и анализировался с помощью программы Jade (версия 7.5.1). Прибор калибровали, используя стандартный образец корунда в пределах ± 0,02° угла 2θ.
Следует иметь в виду, что геометрия Брэгга-Брентано, используемая при сборе данных, имеет предпочтительную ориентацию. В этих условиях существует вероятность того, что относительные интенсивности дифракционных пиков могут не соответствовать истинным значениям относительной интенсивности, которые были бы получены при идеальном распределении сферических частиц или дифракционной картины, моделируемой по данным, полученным для монокристалла. Также существует вероятность того, что на некоторых дифрактограммах будут не видны некоторые пики из-за выбора предпочтительной ориентации.
Дифференциальная сканирующая калориметрия
Измерения методом DSC проводили, используя прибор TA Instruments модели Q-100 с контроллером Thermal Analyst. Данные собирали и анализировали при помощи компьютерной программы TA Instruments Universal Analysis. Образец аккуратно взвешивали в закрытом алюминиевом контейнере. После 5-минутного периода приведения в изотермическое равновесие при 5°С образец нагревали, линейно повышая температуру нагрева со скоростью 10° С/мин от 0°С до 275°С.
Термогравиметрический анализ
Измерения методом TGA выполняли, используя прибор TA Instruments модели Q-500 высокого разрешения. Данные собирали с помощью контроллера TA Instruments Thermal Analyst и анализировали при помощи компьютерной программы TA Instruments Universal Analysis. Взвешенный образец помещали в платиновый контейнер и сканировали со скоростью нагрева 10°С от температуры окружающей среды до 200°С. Уравнительную и печную камеры во время использования очищены потоком азота.
Микроскопия в поляризованном свете
При изучении методом микроскопии в поляризованном свете (PLM) образцы исследовали под оптическим микроскопом (Olympus BX51) с кросс-поляризованным светофильтром. Изображения собрали с помощью камеры PaxCam, управляемой компьютерной программой PaxIt Imaging (версия 6.4). Образцы готовили на стеклянных слайдах с легким минеральным маслом в качестве иммерсионной среды. В зависимости от размера частиц, для увеличения использовались объектив 4x, 10x или 20x.
Оценка динамической адсорбции влаги
Измерения методом DMS проводили с использованием атмосферного микробаланса VTI, системы SGA-100 (VTI Corp., Hialeah, FL 33016). Использовали взвешенный образец, и в начале анализа устанавливали самую низкую возможную величину (близкую к относительной влажности 0%). Анализ DMS выполняли при скорости сканирования 5% относительной влажности/этап во всем диапазоне влажности от 5 до 90%. Анализ DMS проводили изотермически при 25°C.
Процедуры синтеза и сравнительные примеры
Следующие соединения синтезировали и оценивали их ингибирующую активность в отношении фермента NEP:

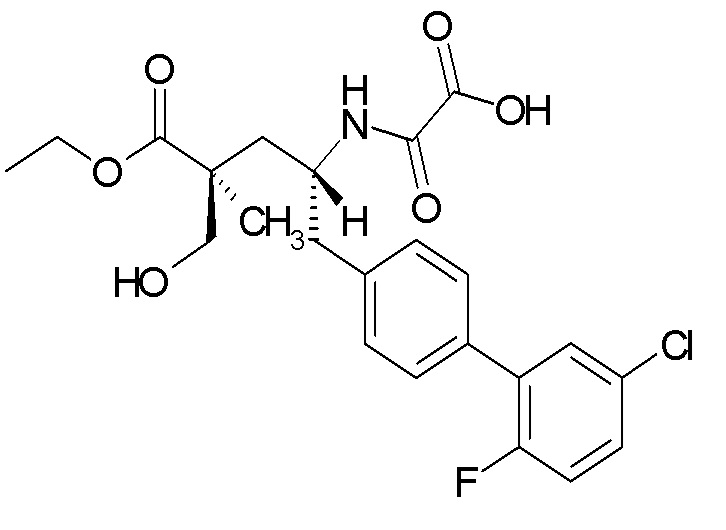
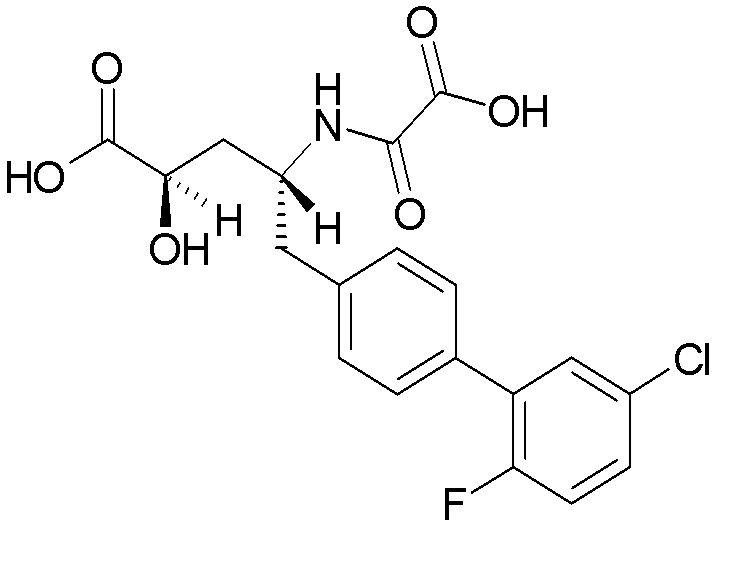
Получение 1: (2S,4R)-4-Амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановая кислота (соединение 7)
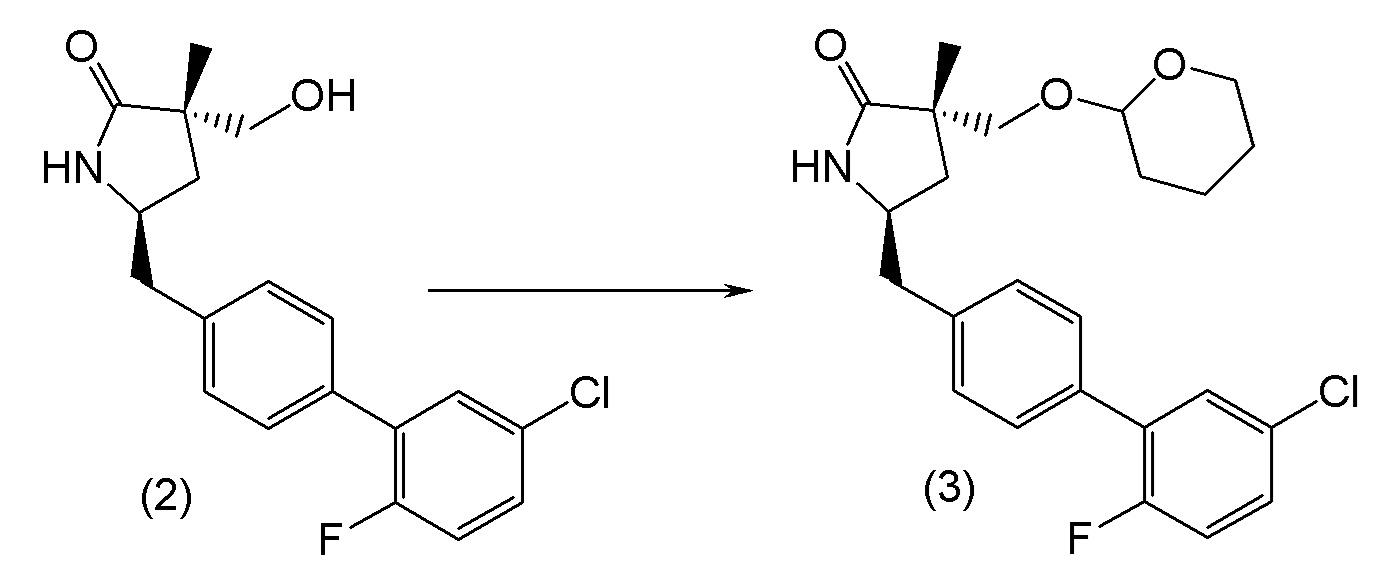
(3S,5R)-5-(5'-Хлор-2'-фторбифенил-4-илметил)-3-гидроксиметил-3-метилпирролидин-2-он (2) (201,0 г, 578 ммоль) объединяли с DCM (4020 мл), получая гомогенный прозрачный коричневый раствор, который затем охлаждали до 0°С при перемешивании. Добавляли 3,4-дигидро-2H-пиран (118 мл, 1,3 моль) и 4-метилбензолсульфоновую кислоту (34,8 г, 202 ммоль), и смесь нагревали до 18,5°С в течение 2 часов, затем перемешивали при 18,5°С в течение ночи (степень конверсии >98%). Реакцию останавливали насыщенным водным раствором NaHCO3 и оставляли для разделения на фазы. Органический слой сушили над Na2SO4, затем фильтровали с последующим удалением растворителя с получением густого темно-коричневого неочищенного продукта (~300 г), который растворяли в DIPE (2 л) и перемешивали при 5°С в течение ночи, получая белую суспензию. Суспензию фильтровали, и твердое вещество сушили в течение 2 дней, получая соединение 3 (113,8 г). Фильтрат сушили, получая густое масло, которое растворяли в DIPE (~100 мл) и перемешивали в течение ночи при 5°С для выделения дополнительного количества соединения 3 (общий выход 225 г).
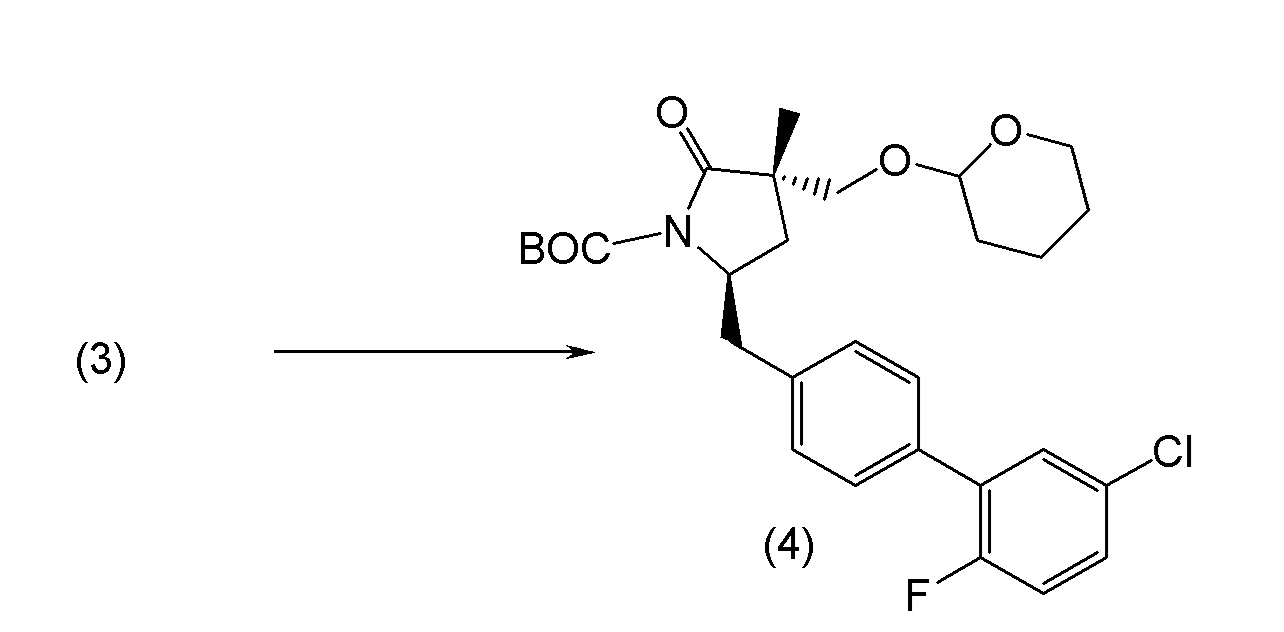
Соединение 3 (208 г, 482 ммоль) растворяли в THF (1912 мл, 23 моль) при перемешивании, получая прозрачный гомогенный раствор. Смесь продували азотом и затем охлаждали до -10°С. Добавляли по каплям 1М NaHMDS в THF (539 мл, 539 ммоль), и смесь перемешивали в течение 30 минут. Ди-трет-бутилдикарбонат (131 г, 602 ммоль) растворяли в THF (393 мл, 4,8 моль), и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакцию останавливали насыщенным водным NH4Cl (5,0 л), и добавляли EtOAc (3,1 л). Фазы разделяли, и органический слой промывали насыщенным водным NaCl (5,0 л). Фазы разделяли, и органический слой сушили над MgSO4. Удаление растворителя дало густое масло, которое при последующей сушке в течение двух дней дало соединение 4 (265 г) в виде вспененного твердого вещества.
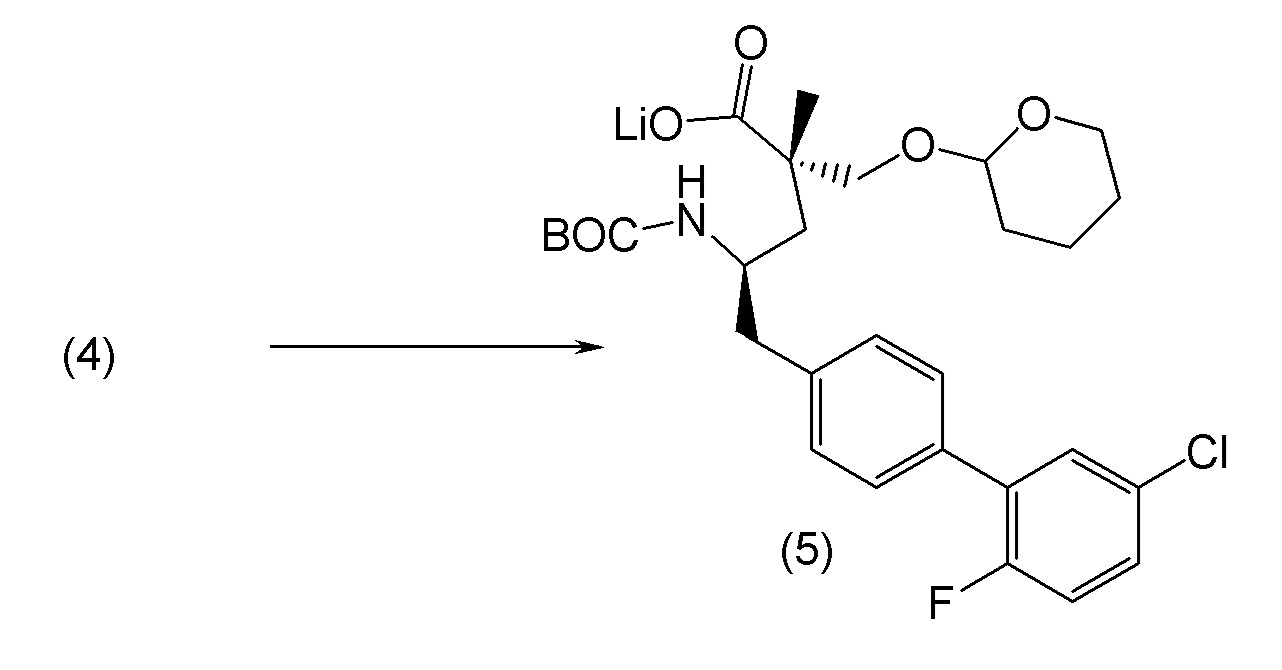
Соединение 4 (265 г, 498 ммоль) растворяли в THF (1,7 л), получая однородный светло-коричневый раствор. Добавляли 1,0М LiOH в воде (1,5 л, 1,5 моль), и полученную смесь перемешивали при комнатной температуре в течение 4 часов. Реакция завершали через 6 часов, но смесь продолжали перемешивать при 15°С в течение ночи. Добавляли EtOAc (1,7 л), и смесь промывали насыщенным водным раствором NH4Cl (1,7 л), и фазы разделяли. Органический слой промывали насыщенным водным NaCl, отделяли, сушили над Na2SO4, затем фильтровали и сушили, получая соединение 5 (300 г) в виде вспененного твердого вещества от не совсем белого до желтого цвета (избыток обусловлен остаточным количеством EtOAc).
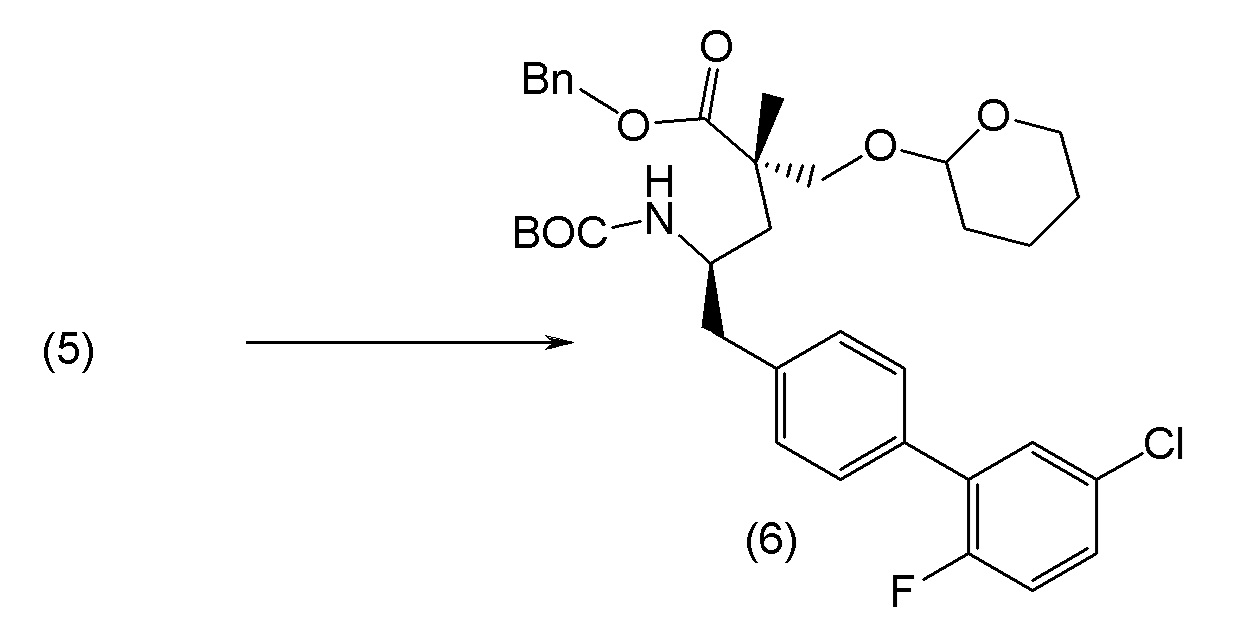
Соединение 5 (277,0 г, 498 ммоль) растворяли в ДМФА (970 мл), получая бесцветный раствор. Добавляли K2CO3 (103 г, 747 ммоль), и полученную смесь перемешивали в течение 15 минут. Одной порцией добавляли бензилбромид (71,1 мл, 598 ммоль), и смесь перемешивали при комнатной температуре в течение ночи; полное превращение происходит через 20 часов. Добавляли NH4Cl (6 л) и EtOAc (1 л), и фазы разделяли. Органический слой промывали насыщенным водным раствором NaCl (6 л) и сушили над Na2SO4 с последующим удалением растворителя с получением неочищенного соединения 6 (335 г), которое использовали непосредственно на следующей стадии.
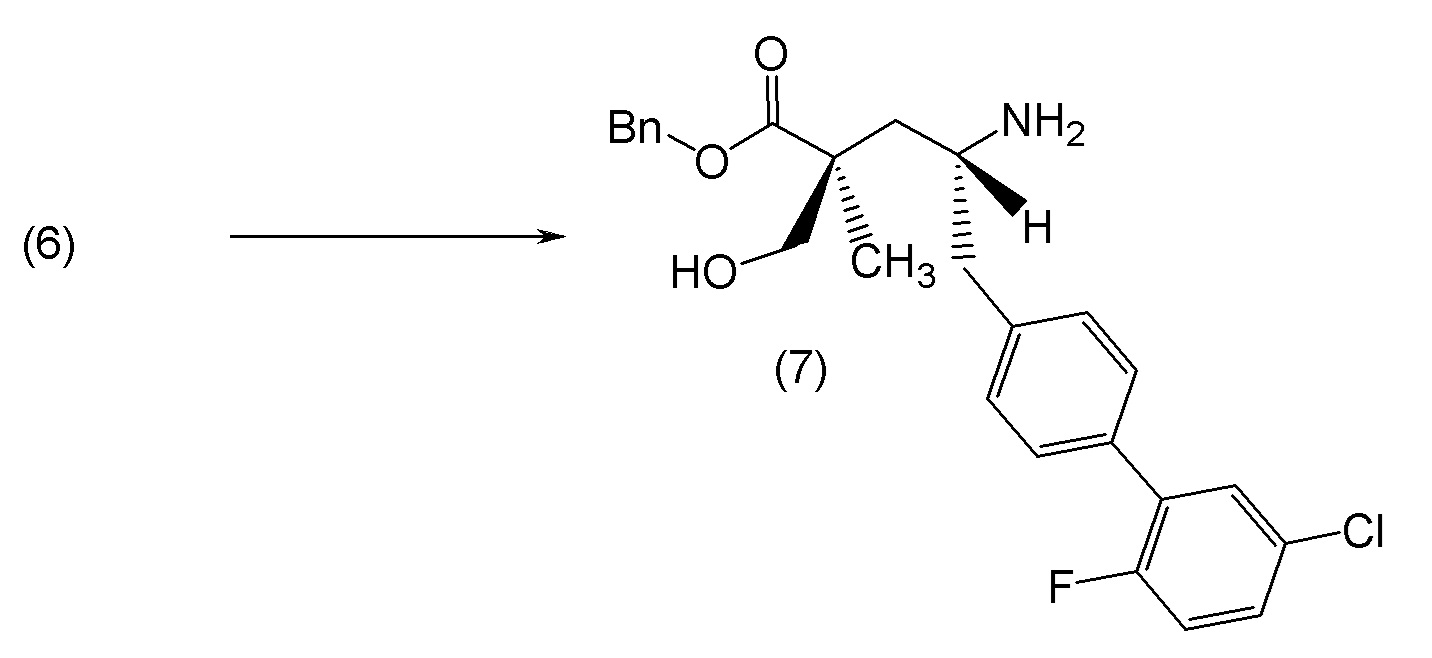
3M HCl (1,7 л, 5,0 моль) в CPME объединяли с соединением 6 (319,0 г, 498 ммоль), и полученную смесь перемешивали при комнатной температуре в течение более 24 часов, получая суспензию (степень конверсии >99%). Добавляли дополнительное количество CPME (1,0 л), и полученную суспензию перемешивали в течение 1 часа. Смесь фильтровали, и влажный осадок промывали CPME (500 мл). После фильтрования и сушки получали соединение 7 (190 г) в виде белой лепешки.
Пример 1. (2S,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-4- (этоксиоксалиламино)-2-гидроксиметил-2-метилпентановая кислота (Соединение 1)
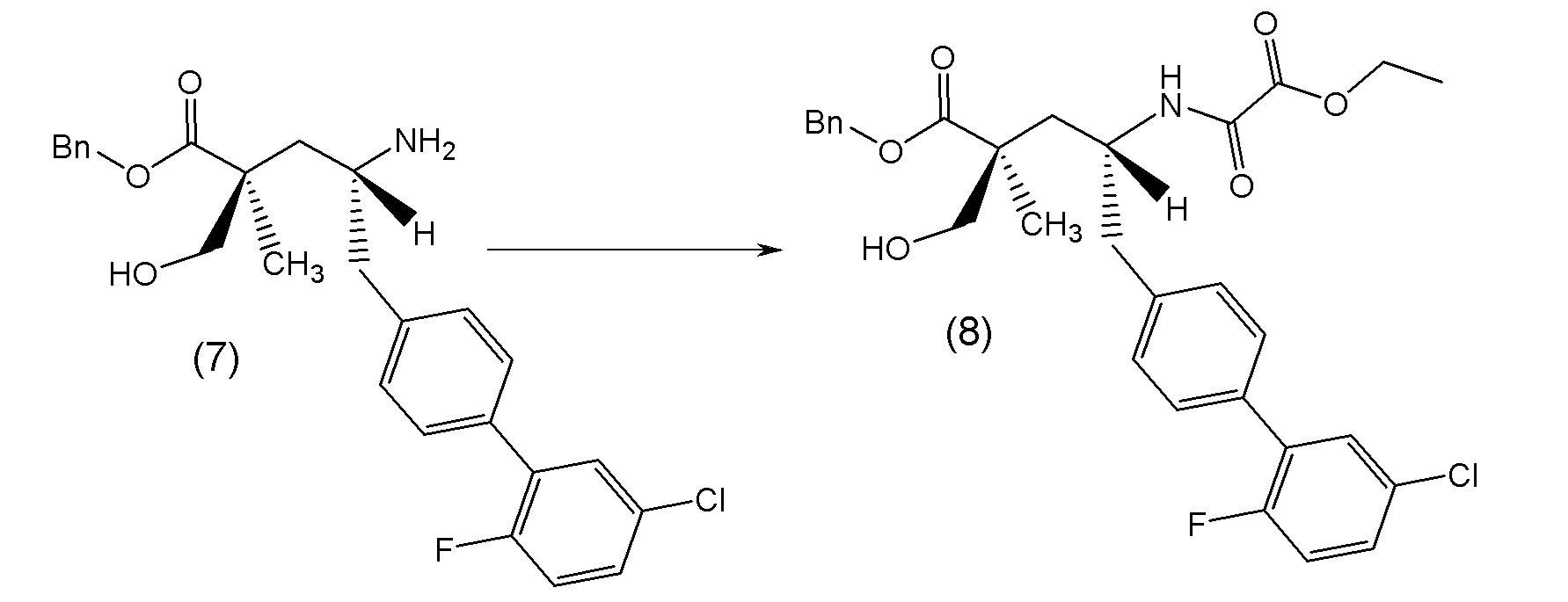
EtOH (576 мкл, 9,9 ммоль) растворяли в DCM (3 мл). Добавляли оксалилхлорид (1,0 мл, 12,1 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 30 минут. Растворитель выпаривали без нагревания, получая раствор, который непосредственно использовали на следующей стадии. Бензиловый эфир (2S,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановой кислоты (7) (1,0 г, 2,2 ммоль) растворяли в DCM (3 мл). Добавляли ранее полученный раствор, затем DIPEA (958 мкл, 5,5 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 15 минут, в этот момент результаты LC/MS показали получение требуемого продукта. Растворитель удаляли под вакуумом, и неочищенный остаток очищали методом нормальной фазовой хроматографии (20-95% EtOAc/гексаны) с получением соединения 8 (1,0 г, 1,9 ммоль), которое сразу использовали на следующей стадии.

Соединение 8 (1,0 г, 1,9 ммоль) объединяли с палладием 10 мас.% на углероде (350 мг, 185 мкмоль), AcOH (5 мл) и EtOAc (5 мл). Смесь помещали в атмосферу водорода и перемешивали при комнатной температуре в течение 2 часов, в этот момент результаты LC/MS показали завершение снятия бензильной защиты. Палладий отфильтровывали, используя 0,2 мкм фильтр PTFE Acrodisc CR, и растворитель удаляли под вакуумом. Неочищенный остаток очищали с помощью обращенно-фазовой хроматографии с получением соединения 1 (400 мг). МС m/z [M+H]+ для C23H25ClFNO6-466,14, вычисленное; 466, найденное.
Кристаллический (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноат кальция (соединение 1')
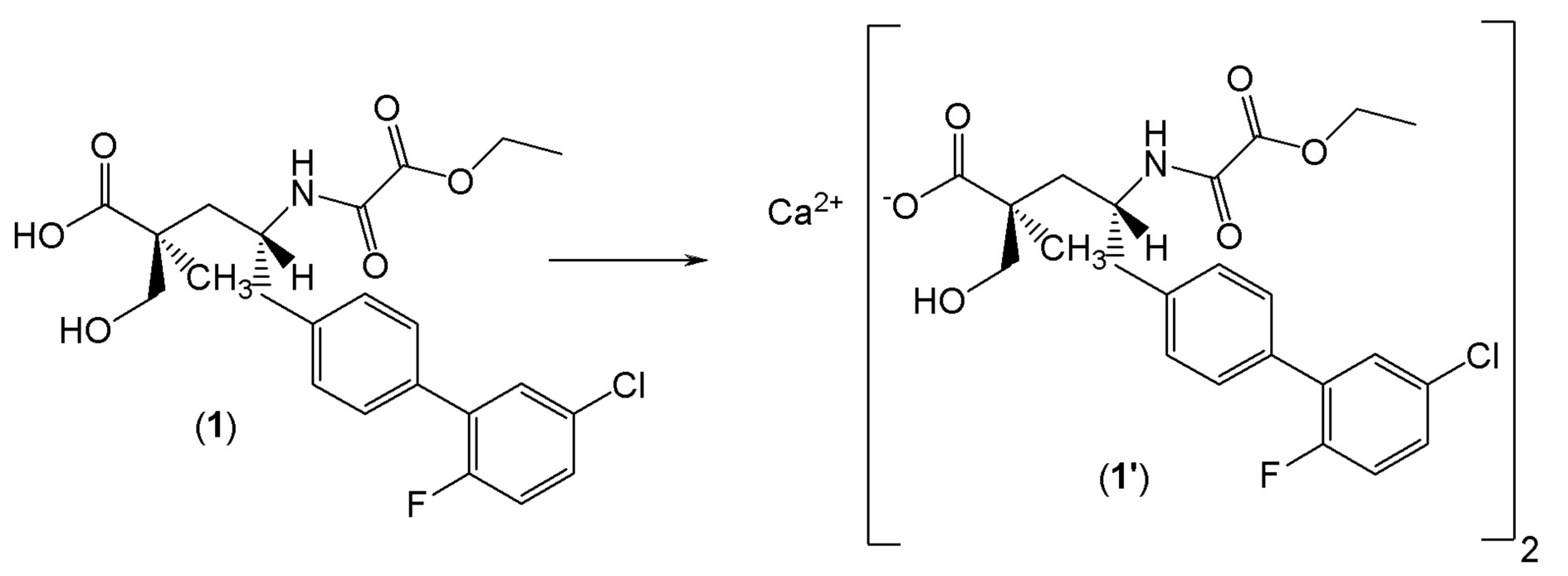
Соединение 1 (30 г, 64,4 ммоль) растворяли в EtOH крепостью 200 (100 мл), и затем к этой смеси добавляли DIPEA (11,25 мл, 64,4 ммоль) при комнатной температуре. Трифторметансульфонат кальция (10,89 г, 32,2 ммоль) растворяли в EtOH (20 мл) и добавляли по каплям к смеси, содержащей соединение 1, с образованием густой суспензии в течение приблизительно 1 часа. Затем густую суспензию перемешивали при комнатной температуре в течение двух дней. Полученную суспензию медленно фильтровали и сушили в течение двух дней, получая 33 г соединения 1' с чистотой >99%. Второй процесс ресуспендирования осуществляли сначала путем охлаждения соединения 1' (33 г, 19,80 ммоль) до 5°С. Затем добавляли холодную смесь EtOH:вода (7:3) (300 мл), и полученную суспензию интенсивно перемешивали в течение 4 дней. Затем суспензию медленно фильтровали и сушили в течение 24 часов непрерывной разбивая образующиеся комки. Затем суспензию сушили на воздухе при комнатной температуре в течение следующих 18 часов, получая 29,5 г Соединения 1' в виде твердого вещества с чистотой >99%. Этот продукт анализировали методами PXRD, DSC и TGA, как описано в настоящем описании; полученные данные представлены на фиг.1-3.
Кристаллический (2S,4R)-5-(5'-хлор-2'-фтор- [1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноат L-аргинина (соединение 1ʺ)
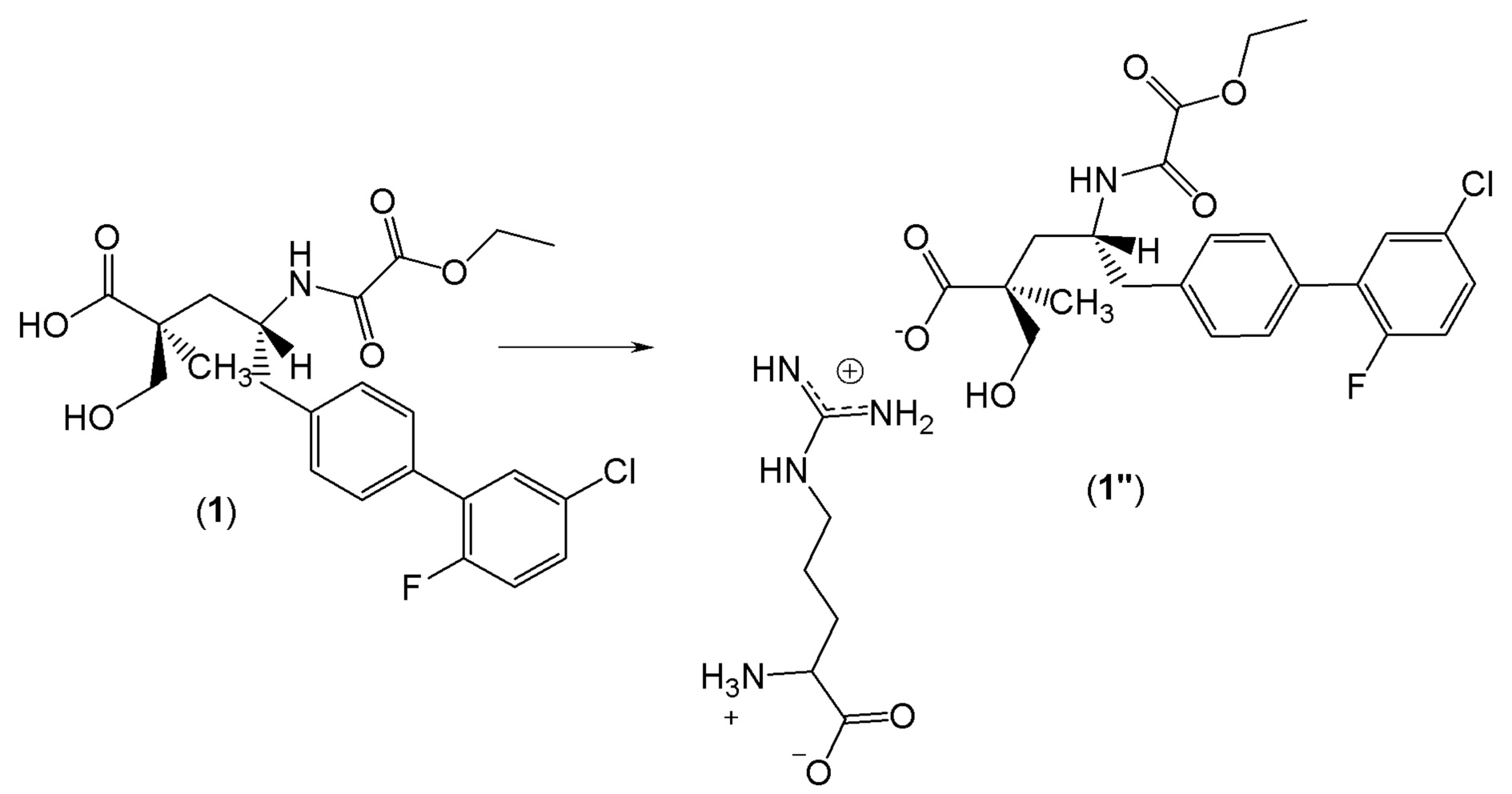
Соединение 1 (181,6 мг) растворяли в EtOH крепостью 200 (0,5 мл) в стеклянном флаконе с получением прозрачного раствора. Раствор охлаждали при -20°C в течение примерно 10 минут. В отдельном стеклянном флаконе растворяли L-аргинин (68 мг) в 0,2 мл воды, и раствор охлаждали при 5°C в течение примерно 10 минут. Прозрачный раствор, содержащий соединение 1, медленно переносили в раствор L-аргинина. Дополнительное количество 0,2 мл EtOH крепостью 200 добавляли во флакон, ранее содержащий соединение 1, и содержимое дополнительно добавляли к флакону, содержащему раствор L-аргинина. Затравочные кристаллы L-аргинина, полученные из более ранней реакции с помощью описанной выше процедуры, добавляли к объединенному раствору, и всю смесь выдерживали при 5°C при осторожном перемешивании, получая кристаллическую суспензию в течение 1-2 дней. Кристаллы соединения 1ʺ фильтровали и анализировали методами PXRD, DSC и TGA, как описано в настоящем описании; полученные данные представлены на фиг.6-8.
Получение 2: (2S,R)-4-трет-бутоксикарбониламино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановая кислота (соединение 10)
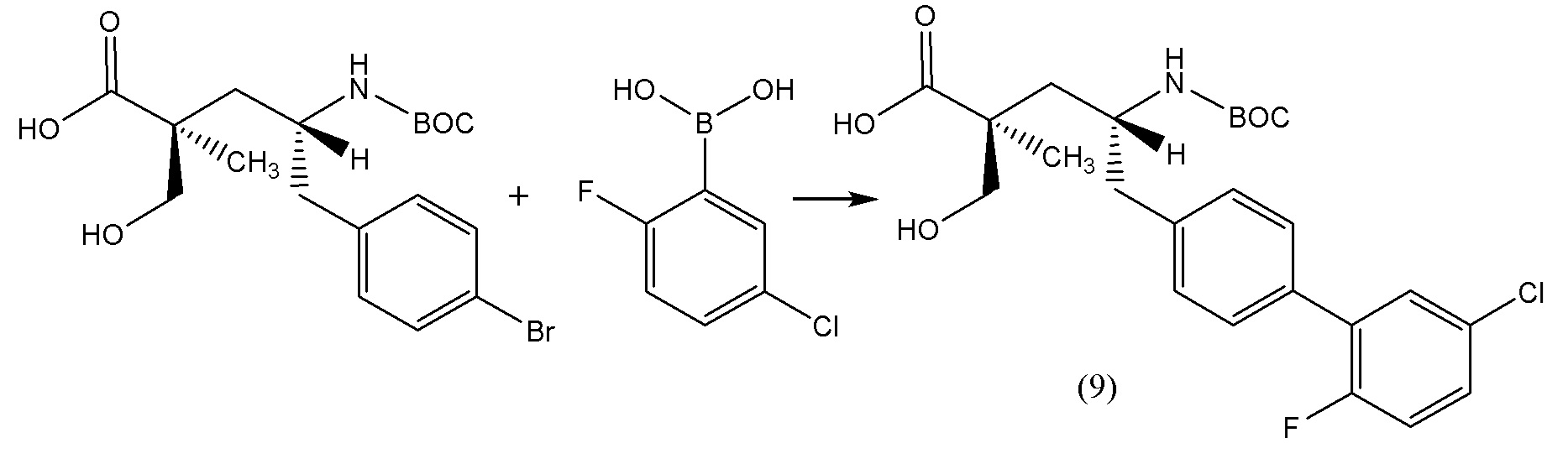
(2S,4R)-5-(4-бромфенил)-4-трет-бутоксикарбониламино-2-гидроксиметил-2-метилпентановую кислоту (1,3 г, 3,1 ммоль) объединяли с Na2CO3 (993 мг, 9,4 ммоль), водой (0,2 Мл) и диоксаном (1,5 мл). Реакционный сосуд закрывали, продували и помещали в атмосферу азота. Быстро добавляли Pd (PPh3)4 (541 мг, 468 мкмоль), и сосуд снова продували. Смесь нагревали в течение 45 минут при 90°С, в этот момент результаты LC/MS показали завершение реакции. Органический слой подкисляли 1н. HCl/водой до рН ~4 и экстрагировали EtOAc. Органический слой отделяли, промывали насыщенным водным раствором NaCl и сушили над MgSO4. Растворитель удаляли под вакуумом, и неочищенный остаток очищали методом обращенно-фазовой хроматографии с получением соединения 9.
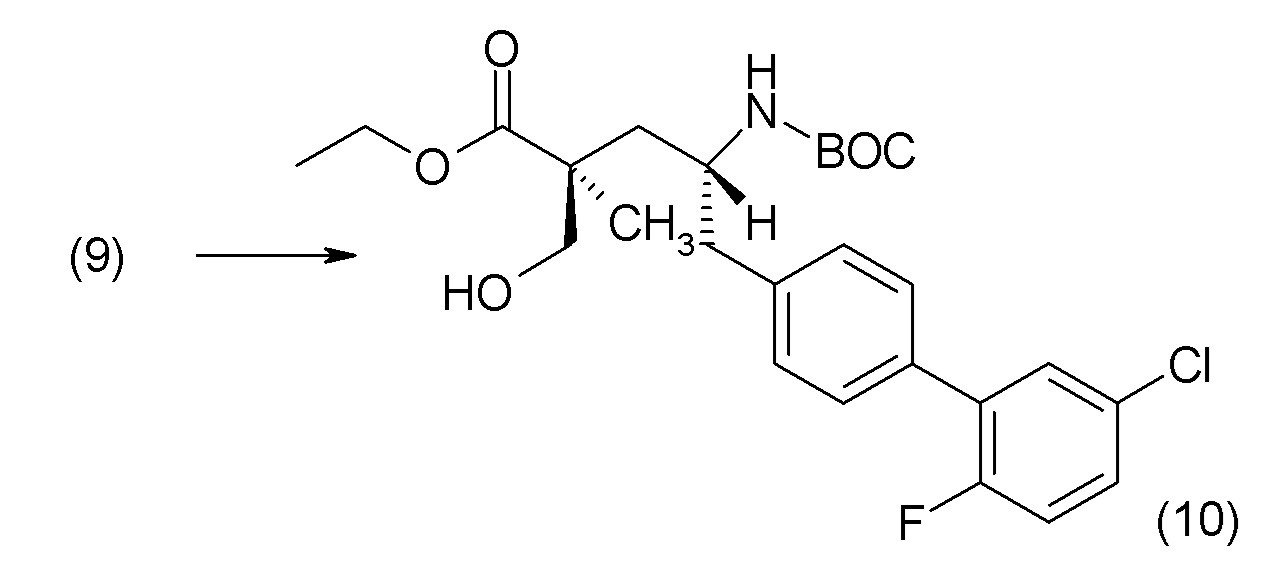
Соединение 9 (1,0 г, 2,1 ммоль) растворяли в EtOH (4 мл) и 4н. HCl в диоксане (4 мл) и перемешивали в течение 3 часов при 60°C. Растворители выпаривали, и неочищенный остаток растворяли в DCM. Добавляли (BOC)2O (472 мкл, 2,031 ммоль) и Et3N (566 мкл, 4,1 ммоль), а затем DMAP (5 мг). Реакционную смесь перемешивали в течение 3 часов. Неочищенный продукт выпаривали и растирали с DCM, и фильтровали без дополнительной очистки с получением соединения 10 (800 мг).
Пример 2: (2S,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановая кислота (соединение С2 сравнения)
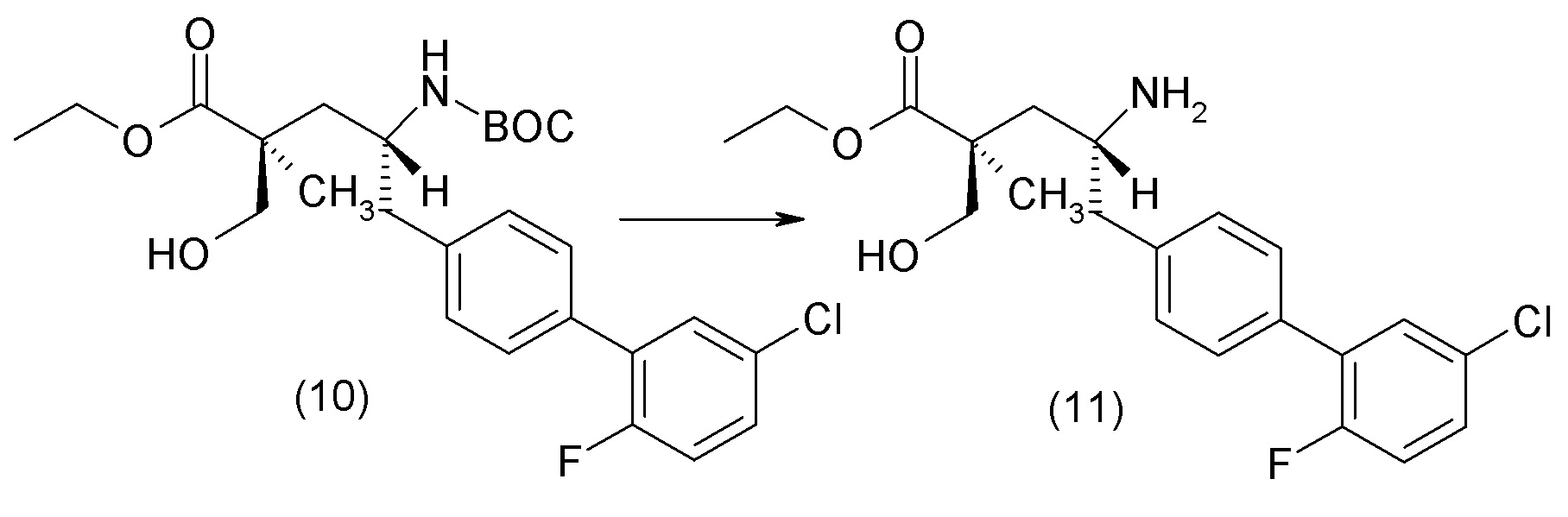
Этиловый эфир (2S,4R)-4-трет-бутоксикарбониламино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановой кислоты (10) (2,6 г, 5,3 ммоль) объединяли с MeCN (5 мл) и 4н. HCl в диоксане (4 мл) и перемешивали в течение 15 минут. Растворитель удаляли центрифужным испарением с получением соединения 11, которое использовали непосредственно на следующей стадии.
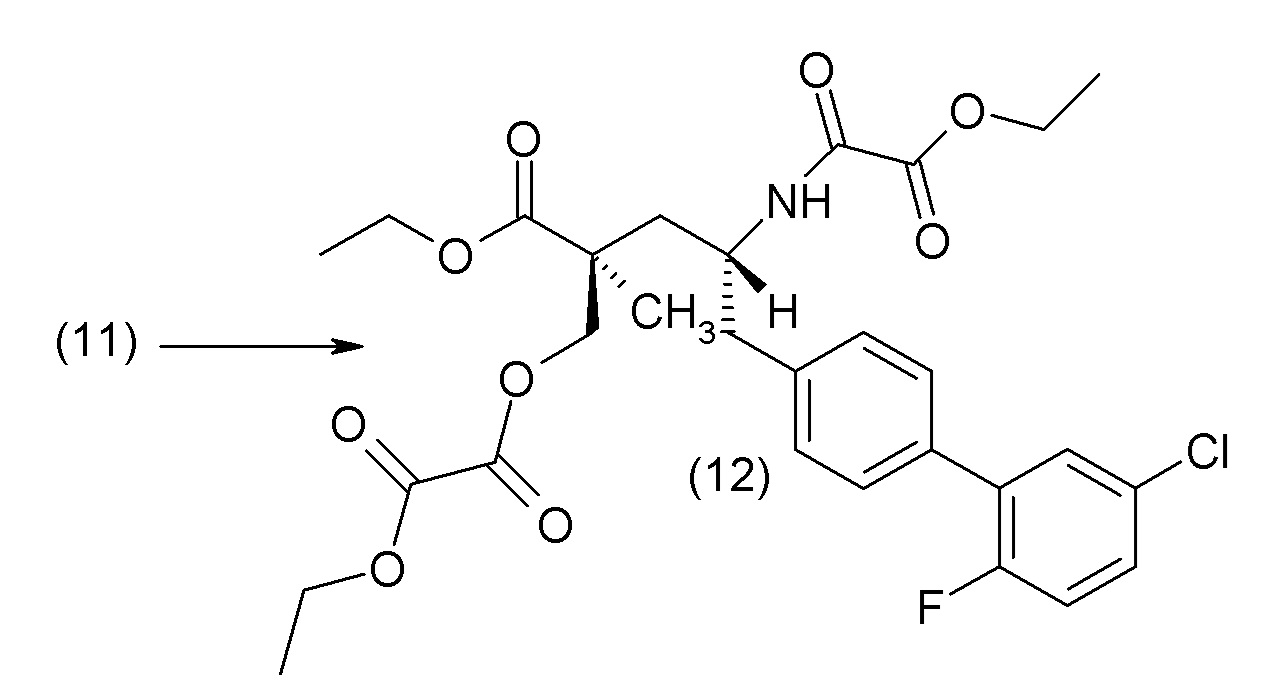
К неочищенному соединению 11 (2,1 г, 5,3 ммоль) в DCM (10 мл) добавляли этил-2-хлор-2-оксоацетат (1,3 мл, 11,7 ммоль) с последующим медленным добавлением Et3N (2,6 мл, 18,7 ммоль). Полученную смесь перемешивали в течение 15 минут, и реакцию контролировали до завершения. Неочищенный продукт очищали флэш-хроматографией (0-100% EtOAc/гексан) с получением соединения 12, которое использовали непосредственно на следующей стадии.
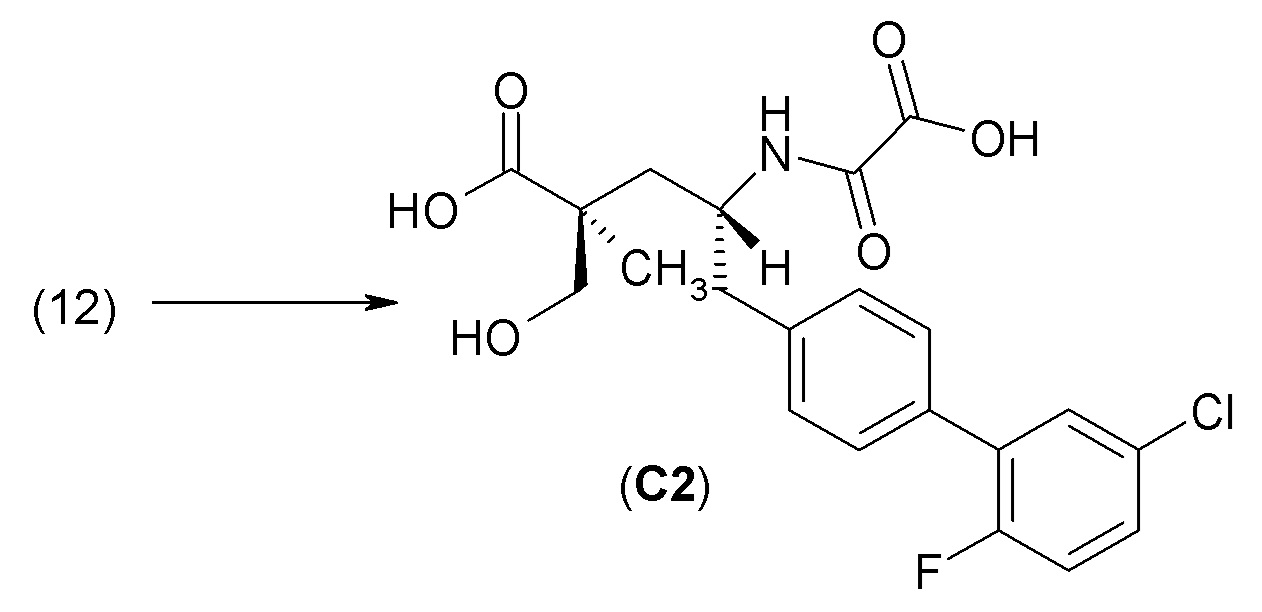
Соединение 12 (2,2 г, 3,7 ммоль) объединяли с THF (5 мл) и NaOH (3,7 мл, 37,0 ммоль) с последующим добавлением воды (10 мл). Полученную смесь перемешивали в течение ночи. Растворитель выпаривали, добавляли AcOH, и продукт очищали методом обращенно-фазовой хроматографии с получением соединения C2 сравнения (540 мг). МС m/z [M+H]+ для C21H21ClFNO6-438,10, вычисленное; 438.2, найденное.
Соединение С2 сравнения описано в примере 11-2 в патенте США № 8,691,868 Hughes et al.
Получение 3: (3S,5R)-5-(5'-Хлор-2'-фторбифенил-4-илметил)-3-гидроксиметил-3-метилпирролидин-2-он (соединение 21)
Раствор (R)-2-амино-3-(4-бромфенил)пропионовой кислоты (3300 г, 13,5 моль, 1,0 экв.) в MeCN (46,2 л) помещали в реакционную колбу, которую продували и поддерживали в атмосфере инертного азота. Раствор NaOH (1081 г, 27,0 моль, 2,0 экв.) в воде (46,2 л) добавляли несколькими порциями при -10°С. К этому добавляли раствор ди-трет-бутилдикарбоната (2948 г, 13,51 моль, 1,0 экв.) в MeCN (6,6 л). Полученный раствор перемешивали в течение ночи при комнатной температуре, затем концентрировали под вакуумом. Полученный раствор разбавляли 45 л воды/льда. Значение рН раствора доводили до 2, используя HCl (1 моль/л). Полученный раствор экстрагировали DCM (50 л ×3), и органические слои объединяли. Полученную смесь промывали насыщенным водным раствором NaCl (50 л), затем сушили над MgSO4 и концентрировали под вакуумом с получением соединения 13 (3720 г) в виде белого твердого вещества.
Раствор соединения 13 (530 г, 1,54 моль, 1,0 экв.) в диоксане (9,54 л) объединяли с (5-хлор-2-фторфенил)борной кислотой (348 г, 2,0 моль, 1,3 экв.), раствором Na2CO3 (228 г, 2,2 моль, 1,4 экв.) в воде (1,1 л) и Pd (PPh3) 4 (8,9 г, 7,7 ммоль, 0,01 экв.) в реакционной колбе, которую продували и поддерживали в атмосфере инертного азота. Полученный раствор кипятили с обратным холодильником в течение 2,5 часов на масляной бане, затем охлаждали до комнатной температуры с помощью воды/ледяной бани. Полученный раствор разбавляли EtOAc (15 л), промывали 1н. HCl (5 л) и насыщенным водным NaCl (5 л ×4). Объединенные органические вещества затем сушили над MgSO4 и концентрировали под вакуумом. Остаток промывали PE (1 л×2) с получением соединения 14 (510 г) в виде коричневого масла.
Раствор соединения 14 (510 г, 1,3 моль, 1,0 экв.) в DCM (5 л) объединяли с 2,2-диметил-1,3-диоксан-4,6-дионом (205 г, 1,4 моль, 1,1 экв.) и 4'-диметиламинопиридином (237 г, 1,9 моль, 1,5 экв.) в реакционной колбе, которую продували и поддерживали в атмосфере инертного азота. Раствор DCC (294 г, 1,4 моль, 1,1 экв.) в DCM (600 мл) добавляли по каплям при перемешивании при -10°С. Полученный раствор перемешивали в течение ночи при комнатной температуре. Твердые вещества фильтровали, и фильтрат промывали 1н. HCl (2 л) и насыщенным водным NaCl (3 л). Объединенные органические вещества сушили над MgSO4. Твердые вещества фильтровали, получая соединение 15 в качестве фильтрата, которое использовали на следующей стадии непосредственно без дальнейшей очистки.
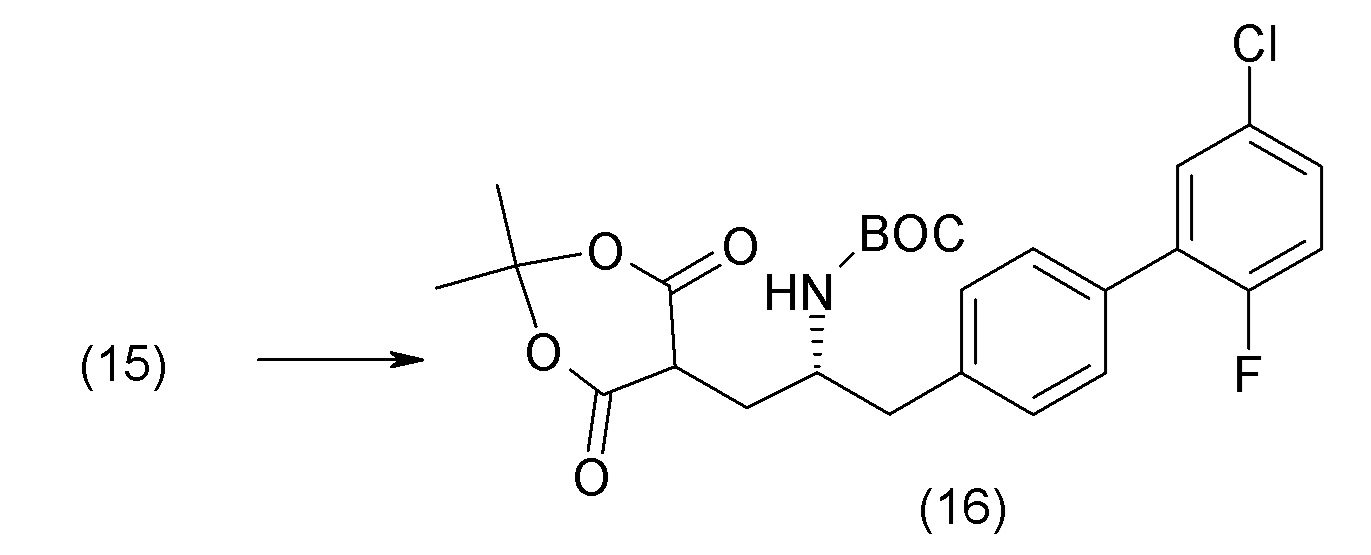
Раствор соединения 15 в DCM (7 л, неочищенного) объединяли с AcOH (600 мл) в реакционной колбе, которую продували и поддерживали в атмосфере инертного азота. NaBH4 (88,8 г, 2,4 моль, 1,8 экв.) добавляли несколькими порциями при -5°С. Полученный раствор перемешивали в течение 3 часов при -5°С. Затем реакцию останавливали путем добавления по каплям насыщенного водного NaCl (1 л). Полученный раствор разбавляли насыщенным водным NaCl (2 л), и полученную смесь промывали водой (2 л ×2), насыщенным водным раствором NaHCO3 (1 л) и насыщенным водным NaCl (2 л). Объединенные органические слои сушили над MgSO4 и концентрировали под вакуумом, получая соединение 16 (520 г) в виде желтого масла.
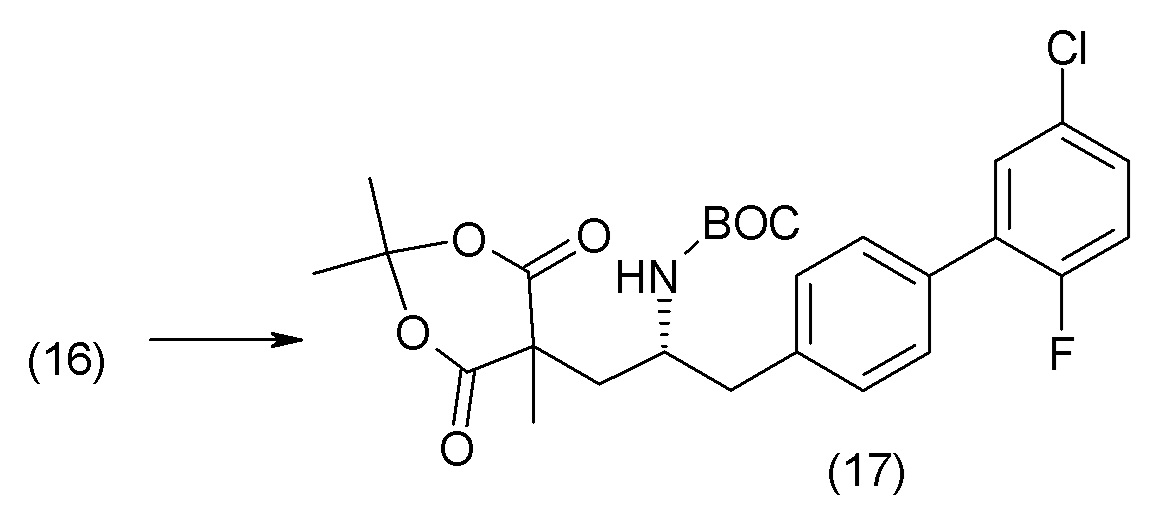
Раствор соединения 16 (520 г, 1,0 моль, 1,0 экв.) в смеси ацетон/ДМФА (1:1) (5,2 л) объединяли с Na2CO3 (163 г, 1,5 моль, 1,5 экв.) и метилиодидом (219 г, 1,5 моль, 1,5 экв.) в реакционной колбе, которую продували и поддерживали в атмосфере инертного азота. Полученный раствор перемешивали в течение ночи при комнатной температуре, затем разбавляли водой (15 л). После перемешивания в течение 1 часа твердые вещества собирали фильтрованием. Остаток растворяли в DCM (5 л). Объединенные органические слои сушили над MgSO4 и концентрировали под вакуумом, получая соединение 17 (520 г) в виде желтого твердого вещества.
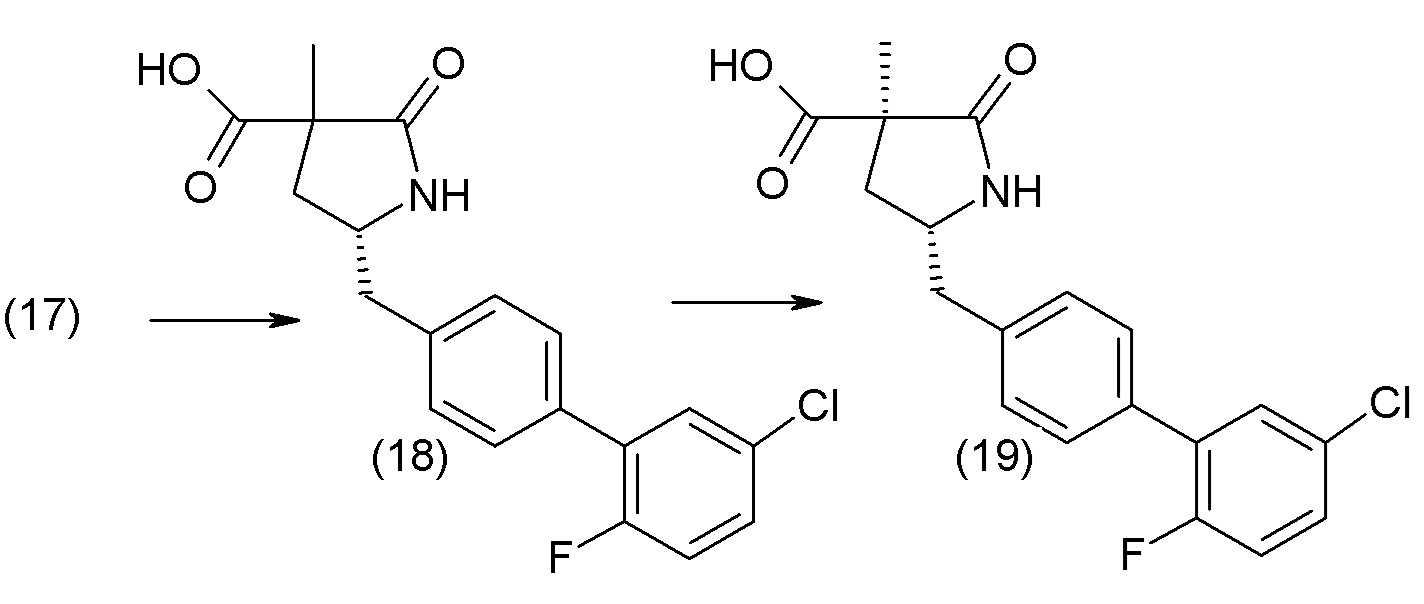
Раствор соединения 17 (520 г, 1,0 моль, 1,0 экв.) в CPME (2,6 л) помещали в реакционную колбу, которую продували и поддерживали в атмосфере инертного азота. Добавляли 4н. раствор HCl в CPME (2,6 л) при -5°С. Полученный раствор перемешивали в течение ночи при комнатной температуре, затем концентрировали до половины объема под вакуумом (получая соединение 18). Твердые вещества собирали фильтрованием, затем промывали смесью EtOAc и DIPE 1:2 с получением соединения 19 (220 г) в виде не совсем белого твердого вещества.
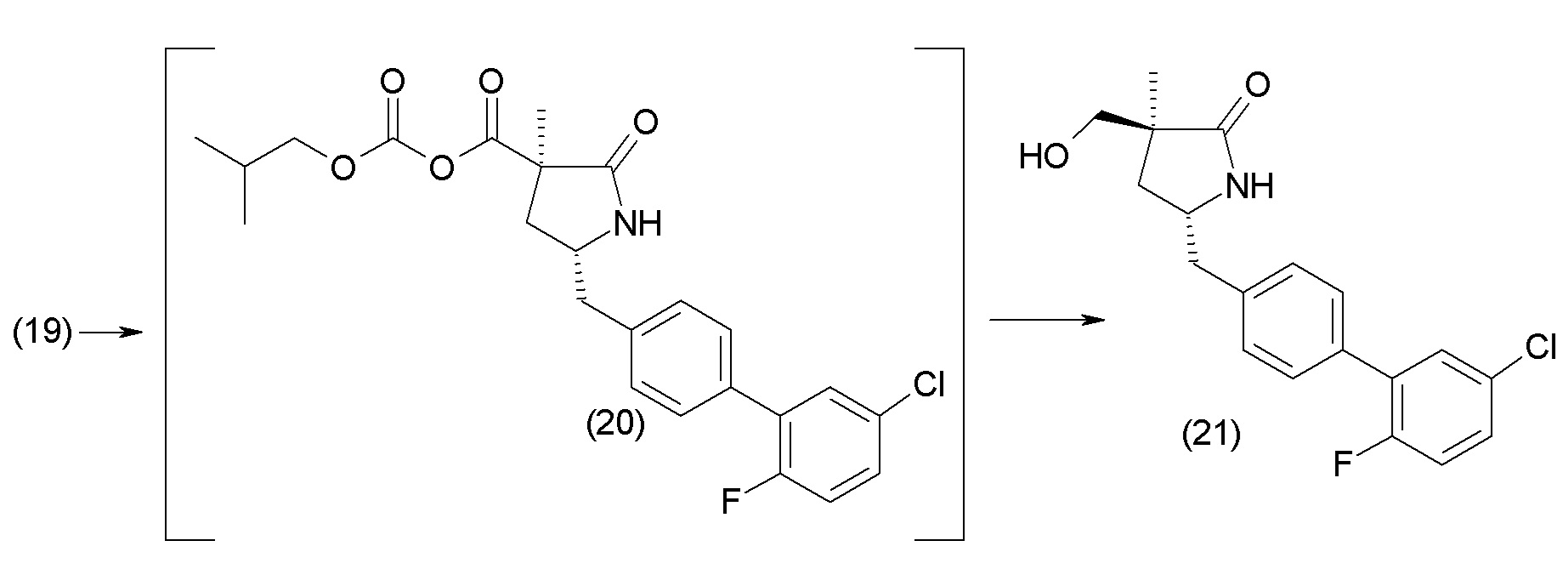
Раствор соединения 19 (218 г, 602,5 ммоль, 1,0 экв.) в THF (4 л) и N-метилморфолин (170 г, 1,7 моль, 2,8 экв.) помещали в реакционную колбу, которую продували и поддерживали в атмосфере инертного азота. Добавляли 2-метилпропилхлорформиат (164,4 г, 1,2 моль, 2,0 экв.) по каплям при перемешивании при -5°С. Полученный раствор перемешивали в течение 20 минут при -5°С. Затем при перемешивании при -5°С добавляли по каплям раствор NaBH4 (91,5 г, 2,4 моль, 4,0 экв.) в воде (400 мл). Полученный раствор перемешивали еще 1 час при комнатной температуре. Затем реакцию останавливали добавлением по каплям 1н. HCl (2,6 л), и полученную смесь перемешивали в течение 1 часа и затем концентрировали под вакуумом. Затем оставшуюся смесь перемешивали еще 1 час, после чего фильтрованием собирали твердые вещества. Твердые вещества промывали водой, растворяли в THF, сушили над Na2SO4 и концентрировали под вакуумом с получением соединения 21 (170 г) в виде белого твердого вещества.
Получение 4: (2S,4R)-4-трет-бутоксикарбониламино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановой кислоты (соединение 23)

(3S,5R)-5-(5'-хлор-2'-фторбифенил-4-илметил)-3-гидроксиметил-3-метилпирролидин-2-он (21) (5,0 г, 14,4 ммоль) растворяли в THF ( 10 мл) и помещали в азот. Раствор помещали на ледяную баню. Добавляли NaHMDS (31,6 мл, 31,6 ммоль), и смесь перемешивали в течение 10 минут от 0°C до комнатной температуры. Затем добавляли (BOC)2O (7,3 мл, 31,6 ммоль), и смесь перемешивали в течение 1 часа при комнатной температуре, в этот момент результаты LC/MS показали завершение реакции. К этому неочищенному раствору добавляли раствор 10н. NaOH (21,6 мл, 216 ммоль) в воде для достижения рН ~12. Добавляли дополнительное количество THF (~10 мл), и раствор перемешивали в течение ночи при комнатной температуре, в этот момент результаты LC/MS показали завершение реакции. Добавляли EtOAc с последующим добавлением 1н. раствора HCl до достижения рН 5. Органический слой экстрагировали, сушили над MgSO4, фильтровали и выпаривали. Неочищенный остаток очищали методом нормально-фазовой хроматографии (50-100% EtOAc/гексан) с получением соединения 22 (2,5 г).
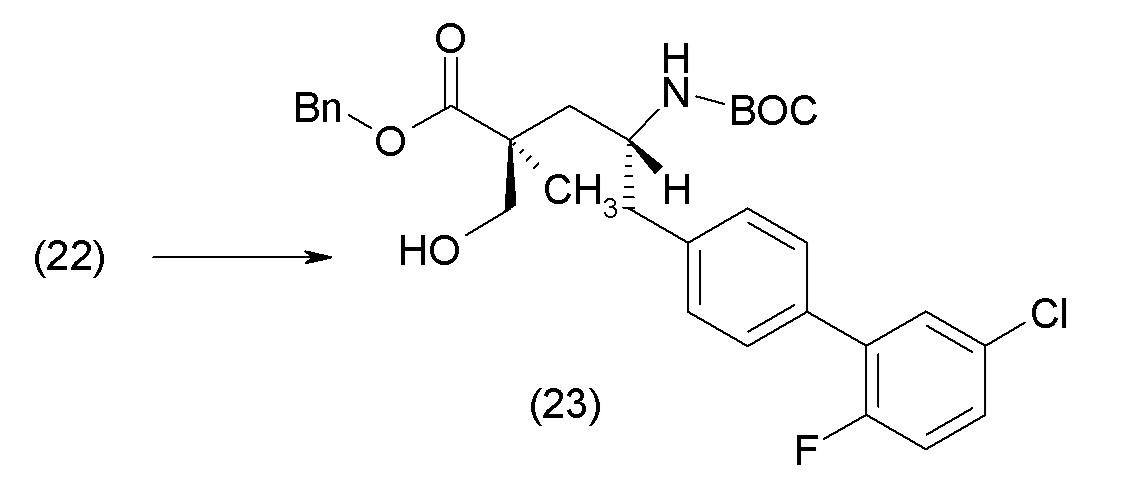
Соединение 22 (550 мг, 1,2 ммоль), K2CO3 (179 мг, 1,3 ммоль) и бензилбромид (154 мкл, 1,3 ммоль) объединяли в DMF (6 мл) и перемешивали в течение 3-4 часов при комнатной температуре, в этот момент результаты LC/MS показали завершение реакции. Растворитель удаляли под вакуумом, и неочищенный остаток очищали методом нормально-фазовой хроматографии с получением соединения 23 (453 мг).
Пример 3: Этиловый эфир (2S,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метил-4-(оксалиламино)пентановой кислоты (соединение С3 сравнения)
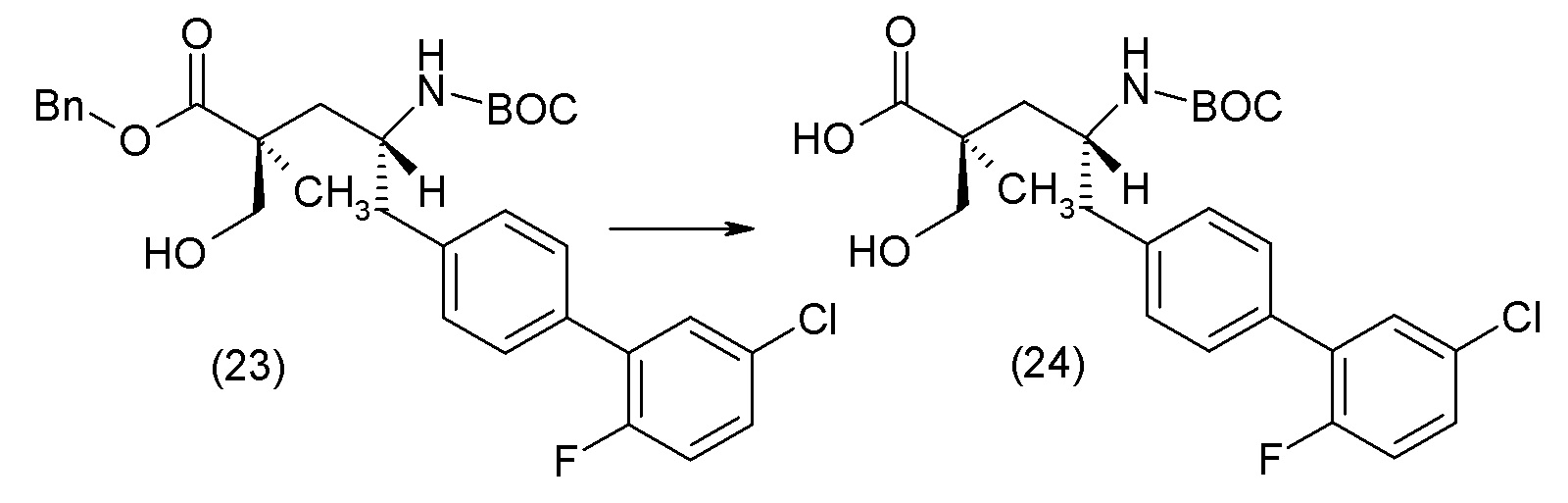
AcOH добавляли к раствору бензилового эфира (2S,4R)-4-трет-бутоксикарбониламино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксиметил-2-метилпентановой кислоты (23) (730 мг, 1,3 ммоль), а затем палладий (140 мг, 131 мкмоль). Полученную смесь перемешивали в атмосфере водорода в течение 3 часов, в этот момент результаты LCMS показали завершение реакции. Смесь фильтровали через 0,2 мкм фильтр PTFE Acrodisc CR и концентрировали с получением соединения 24 (599 мг) в виде прозрачной бесцветной вязкой жидкости.
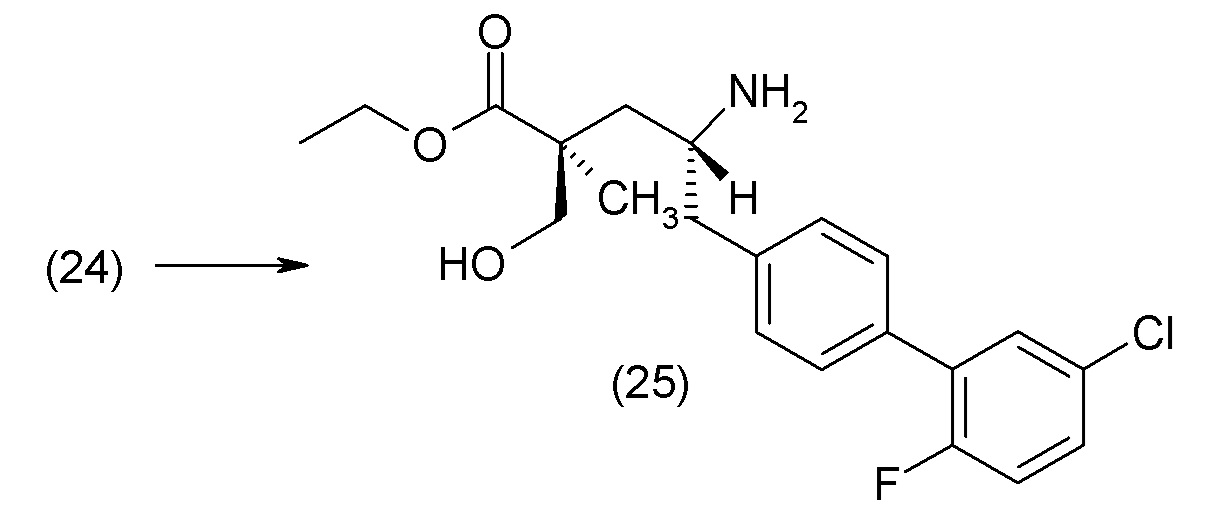
4н. HCl в диоксане (4,8 мл, 19,3 ммоль) добавляли к раствору соединения 24 (599 мг, 1,3 ммоль) в EtOH (5 мл). Полученную смесь перемешивали при 80°С в течение 3 часов, затем концентрировали под вакуумом, получая прозрачную бесцветную жидкость. Неочищенную жидкость очищали методом обращенно-фазовой хроматографии (20-90% MeCN в воде с 0,05% TFA) с получением соединения 25 (455 мг) в виде белой смолистой соли HCl.
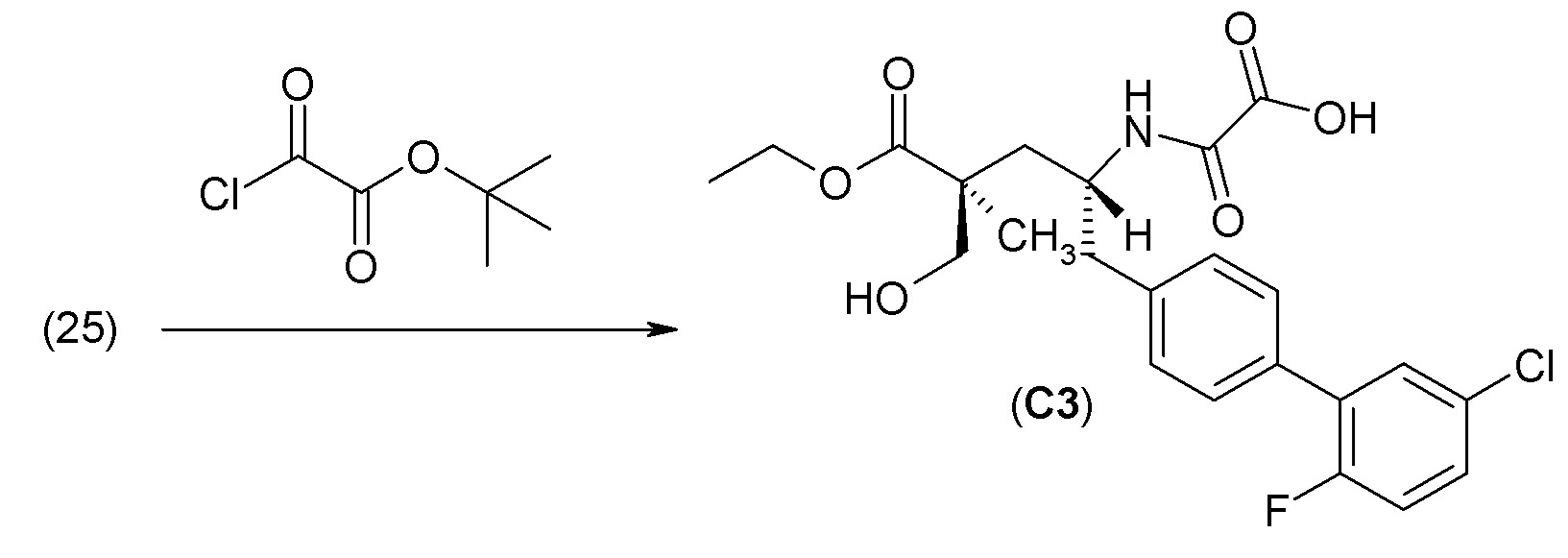
Трет-Бутанол (574 мкл, 6,0 ммоль) добавляли к раствору оксалилхлорида (772 мкл, 9,0 ммоль) в DCM (3 мл) при 0°С, и смесь перемешивали при комнатной температуре в течение 30 минут. Затем реакционную смесь концентрировали под вакуумом с получением трет-бутил-2-хлор-2-оксоацетата (403 мг) в виде прозрачной бесцветной жидкости. Жидкость растворяли в DCM (2,5 мл), получая 1,0М раствор в DCM.
DIPEA (261 мкл, 1,5 ммоль) добавляли к раствору соединения 25 (236 мг, 599 мкмоль) в DCM (3,0 мл) с последующим добавлением трет-бутил-2-хлор-2-оксоацетата (1М раствор в DCM, 659 мкл, 659 мкмоль), и смесь перемешивали при комнатной температуре в течение 15 минут. Затем добавляли TFA (3,0 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. Смесь концентрировали под вакуумом, получая прозрачную бледно-желтую жидкость. Неочищенную жидкость очищали методом обращенно-фазовой хроматографии (20-90% MeCN в воде с 0,05% TFA) с получением соединения C3 сравнения (174 мг) в виде белого твердого вещества. МС m/z [M+H]+ для C23H25ClFNO6-466,14, вычисленное; 466, найденное.
Получение 5: трет-бутиловый эфир (S)-2-(4-Бромбензил)-5-оксопирролидин-1-карбоновой кислоты (соединение 28)

К раствору (R)-2-амино-3-(4-бромфенил)пропионовой кислоты (50 г, 0,2 моль) в MeCN (700 мл) добавляли раствор NaOH (16,4 г, 0,4 моль) в воде (700 мл) при -5°C. После перемешивания в течение 10 минут добавляли раствор (BOC)2O (44,7 г, 0,2 моль) в MeCN (100 мл). Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания MeCN остаток разбавляли DCM (800 мл) и подкисляли 1М HCl до pH 2 при -5°C. Водный раствор экстрагировали DCM (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (500 мл), сушили над Na2SO4 и концентрировали с получением соединения 13 (66,5 г) в виде белого твердого вещества. LC-MC: 366 [M+Na], 709 [2M+Na].
К раствору соединения 13 (66,5 г, 193 мкмоль), кислоты Мельдрума (33,4 г, 232 ммоль) и DMAP (37,7 г, 309 ммоль) в безводном DCM (600 мл) добавляли по каплям раствор DCC (47,9 г, 232 ммоль) в безводном DCM (200 мл) в течение 1 часа при -5°C в атмосфере азота. Смесь перемешивали -5°C в течение 8 часов, затем охлаждали в течение ночи. Наблюдали выпадение кристаллов дициклогексилмочевины. Смесь фильтровали, промывали 5% KHSO4 (5×200 мл) и насыщенным водным NaCl (200 мл), затем сушили над безводным MgSO4 при низкой температуре в течение ночи. После чего раствор выпаривали с получением неочищенного соединения 26 (91 г) в виде светло-желтого твердого вещества. LC-MC: 492 [M+Na], 961 [2M+Na].
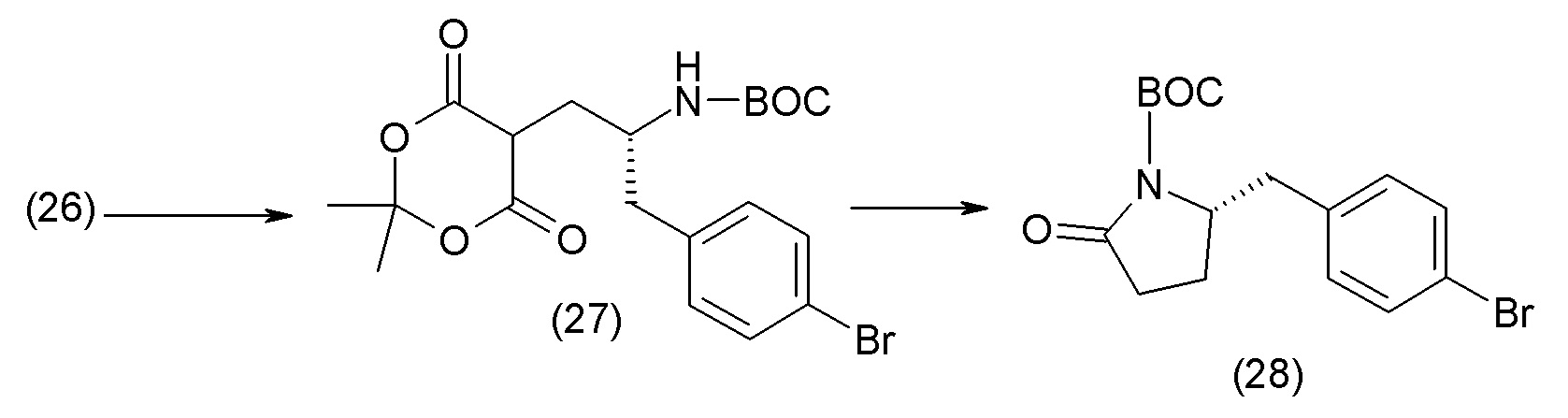
К раствору неочищенного соединения 26 (91 г, 193 ммоль) в безводном DCM (1 л) добавляли AcOH (127,5 г, 2,1 моль) при -5°C в атмосфере азота. Смесь перемешивали при -5°C в течение 30 минут, затем добавляли NaBH4 (18,3 г, 483 ммоль) небольшими порциями в течение 1 часа. После перемешивания в течение еще 1 часа при -5°C добавляли насыщенный водный раствор NaCl (500 мл). Органический слой промывали насыщенным водным раствором NaCl (2×300 мл) и водой (2 раза по 300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который далее очищали промыванием Et2O с получением соединения 27 (68 г) в виде светло-желтого твердого вещества. LC-MC: 478 [M+Na], 933 [2M+Na].
Раствор соединения 27 (68 г, 149 ммоль) в безводном толуоле (500 мл) кипятили с обратным холодильником в атмосфере азота в течение 3 часов. После выпаривания растворителя остаток очищали методом хроматографии (гексан:EtOAc=10:1) с получением соединения 28 (38 г) в виде светло-желтого масла. LC-MC: 376 [M+Na], 729 [2M+Na].
Получение 6: этилового эфира (2R,4R)-4-Амино-5-(4-бромфенил) 2-гидроксипентановой кислоты (соединение 33)
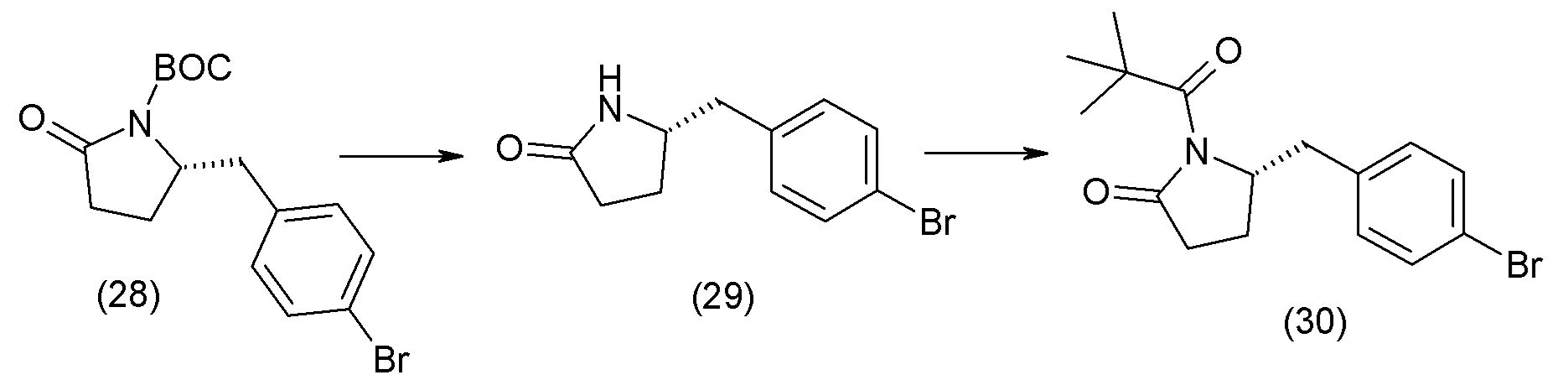
К раствору трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (28) (38 г, 107 ммоль) в безводном DCM (250 мл) добавляли TFA (20 Мл, 0,27 моль) при -5°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. После выпаривания растворителя остаток разбавляли EtOAc (300 мл) и промывали насыщенным водным раствором NaHCO3 (3×200 мл), водой (200 мл), насыщенным водным раствором NaCl (250 мл), сушили над Na2SO4 и концентрировали с получением неочищенного соединения 29 (24 г) в виде светло-желтого твердого вещества. LC-MC: 254 [М+Н].
К раствору NaH (8,6 г, 250 ммоль) в безводном THF (200 мл) по каплям добавляли раствор соединения 29 (24 г, 94 ммоль) в безводном THF (200 мл) в течение 30 минут при 0°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После охлаждения до 0°C, по каплям добавляли пивалоилхлорид (18 г, 150 ммоль) в течение 30 минут. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакцию останавливали насыщенным водным раствором NH4Cl (300 мл) и экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который далее очищали методом хроматографии (гексан:EtOAc=25:1) с получением соединения 30 (18 г ) в виде светло-желтого твердого вещества. LC-MC: 360 (М+Na).
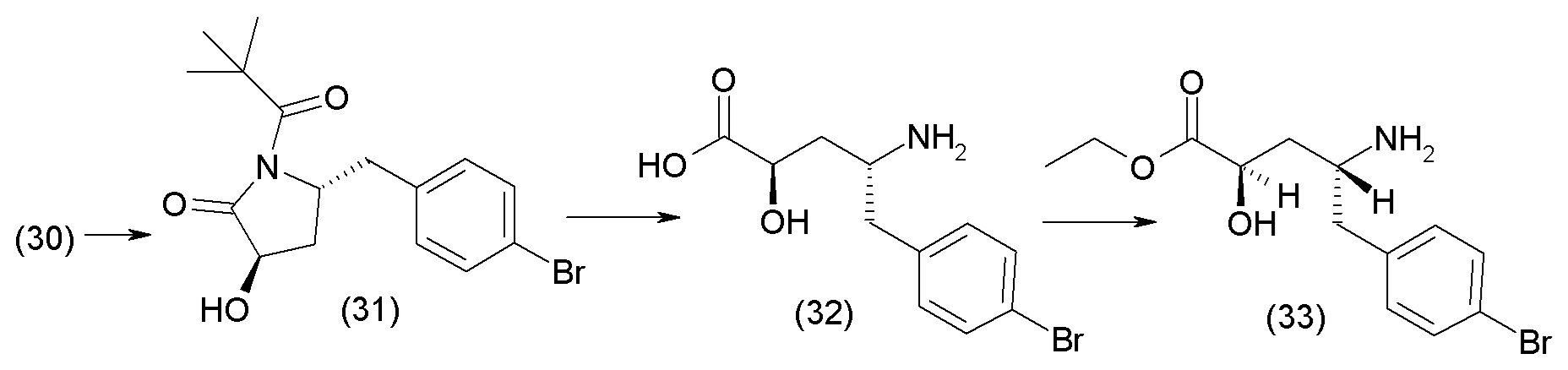
К раствору соединения 30 (18 г, 53 ммоль) в безводном THF (250 мл) добавляли по каплям NaHMDS (47,7 мл, 96 ммоль) в течение 30 минут при -78°C в атмосфере азота. После перемешивания при -78°C в течение 90 минут, добавляли по каплям в раствор (+)-(8,8-дихлоркамфорилсульфонил)-оксазиридина (31,6 г, 106 ммоль) течение 30 минут. После перемешивания при 78°C в течение 2 часов реакцию останавливали насыщенным водным раствором NH4Cl (400 мл) и экстрагировали EtOAc (3×300 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта, который далее очищали методом хроматографии (гексан:EtOAc=15:1) с получением соединения 31 (8,9 г ) в виде светло-желтого твердого вещества. LC-MC: 376 (М+Na).
Раствор соединения 31 (8,9 г, 25 ммоль) в концентрированной HCl (81 мл, 81 ммоль) нагревали при 100°C в течение 16 часов. Затем смесь концентрировали с получением неочищенного продукта, который далее очищали промыванием Et2O с получением соединения 32 (7 г) в виде светло-желтой твердой соли HCl. LC-MC: 323 (М+Н).
Раствор соединения 32 (7 г, 22 ммоль) в EtOH (10 мл) объединяли с 8М HCl в EtOH (120 мл, 960 ммоль) при комнатной температуре. Смесь нагревали при 50°C в течение 16 часов, затем концентрировали. Далее неочищенный продукт очищали промыванием Et2O с получением соединения 33 (6 г) в виде светло-желтой твердой соли HCl. LC-MC: 352 (М+Н).
Препарат 7: трет-бутиловый эфир хлор-оксоуксусной кислоты (соединение 34)
Оксалилхлорид (232 мкл, 2,8 ммоль) и трет-бутиловый спирт (228 мкл) объединяли в эфире (6,7 мл) в атмосфере азота при 0°С. Полученную смесь перемешивали в течение 30 минут при комнатной температуре. Растворитель выпаривали под вакуумом с образованием соединения 34.
Препарат 8: Этиловый эфир (2R,4R)-5-(4-Бромфенил)-4-(трет-бутоксиоксалил-Амино)-2-гидроксипентановой кислоты (соединение 36)
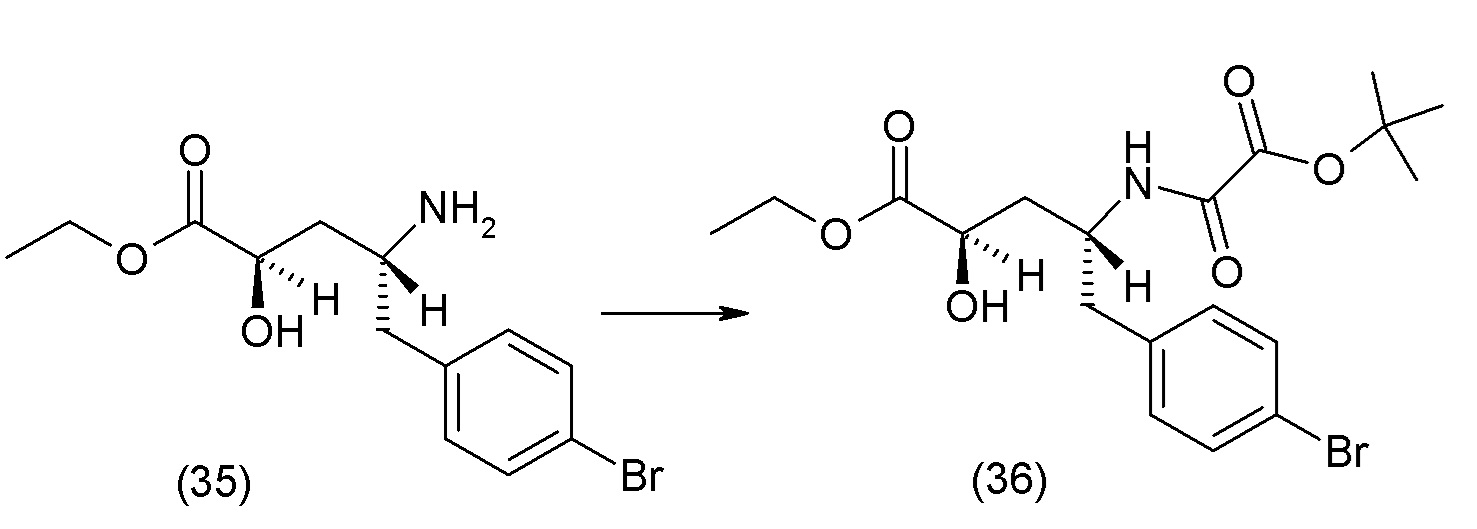
Оксалилхлорид (401 мкл, 4,7 ммоль) в DCM (8 мл) объединяли с трет-бутиловым спиртом (454 мкл, 4,7 ммоль). По каплям добавляли Et3N (198 мкл, 1,4 ммоль), и полученный раствор перемешивали в течение 5 минут. Затем этот раствор добавляли по каплям к раствору этилового эфира (2R,4R)-4-амино-5-(4-бромфенил)2-гидроксипентановой кислоты (35) (150 мг, 474 мкмоль) и Et3N (198 мкл, 1,4 ммоль) в DCM (5 мл) и перемешивали до завершения реакции. Растворители выпаривали и неочищенный продукт очищали методом нормально-фазовой хроматографии (20-100% EtOAc/гексан) с получением соединения 36.
Пример 4: Этиловый эфир (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановой кислоты (соединение С4 сравнения)

(2R,4R)-5-(4-бромфенил)-4-(трет-бутоксиоксалиламино)-2-гидроксипентановую кислоту (36) (48,5 мг, 109 мкмоль) объединяли с 5-хлор-2-фторфенилборной кислотой (22,8 мг, 131 мкмоль) и K2CO3 (45,2 мг, 327 мкмоль) в трет-бутиловом спирте (2 мл) и воде (0,3 мл). Добавляли SilicaCat®DPP-Pd (0,28 ммоль/г лекарственного вещества, 39 мг, 11 мкмоль), и смесь нагревали при 80°С в течение 15 минут; в это время результаты LC/MS показали наличие желаемого продукта. Смесь фильтровали, и фильтрат концентрировали и очищали методом препаративной ВЭЖХ с получением соединения С4 сравнения (20 мг). МС m/z [M+H]+ для C21H21ClFNO6-438,10, вычисленное; 438,0, найденное.
Соединение C4 сравнения описано в примере 5-6 в патенте США № 8,691,868, Hughes et al.
Получение 9: (2R,4R)-4-Амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановая кислота (соединение 42)

К раствору трет-бутилового эфира (S)-2-(4-бромбензил)-5-оксопирролидин-1-карбоновой кислоты (28) (25 г, 70,6 ммоль) в 1,4-диоксане (500 мл) добавляли 5-хлор-2-фторфенилбороновую кислоту (24,6 г, 141 ммоль), Pd (PPh3)4 (4,1 г, 3,5 ммоль) и раствор K2CO3 (17,8 г, 141 ммоль) в воде (90 мл) при комнатной температуре в атмосфере азота. Смесь нагревали до 60°C и перемешивали в течение ночи. Добавляли воду (500 мл), и растворитель выпаривали. Смесь экстрагировали EtOAc (3×200 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (300 мл) и фильтровали. Фильтрат концентрировали с получением неочищенного продукта, который очищали хроматографией с получением соединения 37 (22,7 г) в виде светло-желтого твердого вещества. LC/MS: 829,2 [2M+Na+].
К раствору соединения 37 (4,9 г, 12,1 моль) в DCM (100 мл) добавляли TFA (4,5 мл, 60,7 ммоль) при 0°C в атмосфере азота и перемешивали в течение 1 часа. Смесь нагревали до комнатной температуры в течение 1,5 часов. После выпаривания растворителя остаток разбавляли EtOAc (100 мл), затем промывали насыщенным водным раствором NaHCO3 (3×100 мл), водой (2×100 мл) и насыщенным водным раствором NaCl (100 мл) и затем сушили над Na2SO4. Смесь фильтровали, и фильтрат концентрировали с получением неочищенного соединения 38 (в сочетании с отдельной порцией в общей сложности 16,9 г). LC/MS: 304 [М+Н].
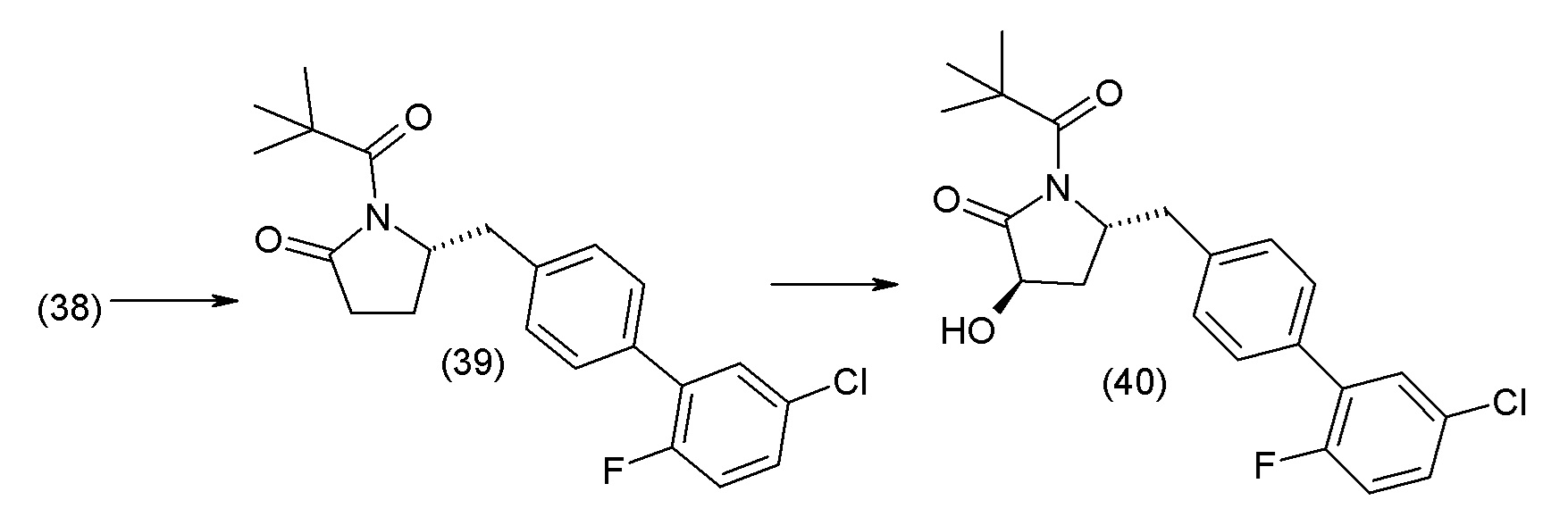
К раствору NaH (2,4 г, 695 ммоль) в THF (200 мл) по каплям добавляли раствор соединения 38 (8,5 г, 278 ммоль) в THF (50 мл) при 0°C в атмосфере азота. Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. После охлаждения до 0°C, по каплям в течение 30 минут добавляли пивалоилхлорид (5 г, 41,7 ммоль). Смесь нагревали до комнатной температуры и перемешивали в течение 9,5 часов. Реакцию останавливали насыщенным водным раствором NH4Cl (250 мл) и экстрагировали EtOAc (3×400 мл). Объединенные органические слои сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали хроматографией с получением соединения 39 (18 г) в виде желтого твердого вещества. LC/MS: 388 [М+Н+].
К раствору соединения 39 (9 г, 23,2 ммоль) в THF (200 мл) добавляли по каплям NaHMDS (20,9 мл, 41,8 ммоль) при -78°C в атмосфере азота. После перемешивания в течение 1 часа при -78°C, по каплям добавляли раствор (+)-(8,8-дихлоркамфорилсульфонил)оксазиридина (10,4 г, 34,8 ммоль) в THF (50 мл). После перемешивания при -78°C в течение 1 часа реакцию останавливали насыщенным водным раствором NH4Cl (50 мл) и экстрагировали EtOAc (3×400 мл). Объединенные органические слои промывали 1М HCl (400 мл), насыщенным водным раствором NaHCO3 (400 мл) и насыщенным водным раствором NaCl (400 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали хроматографией с получением соединения 40 (8,8 г) в виде белого полутвердого вещества. LC-MC: 426,1 [М+Na+].
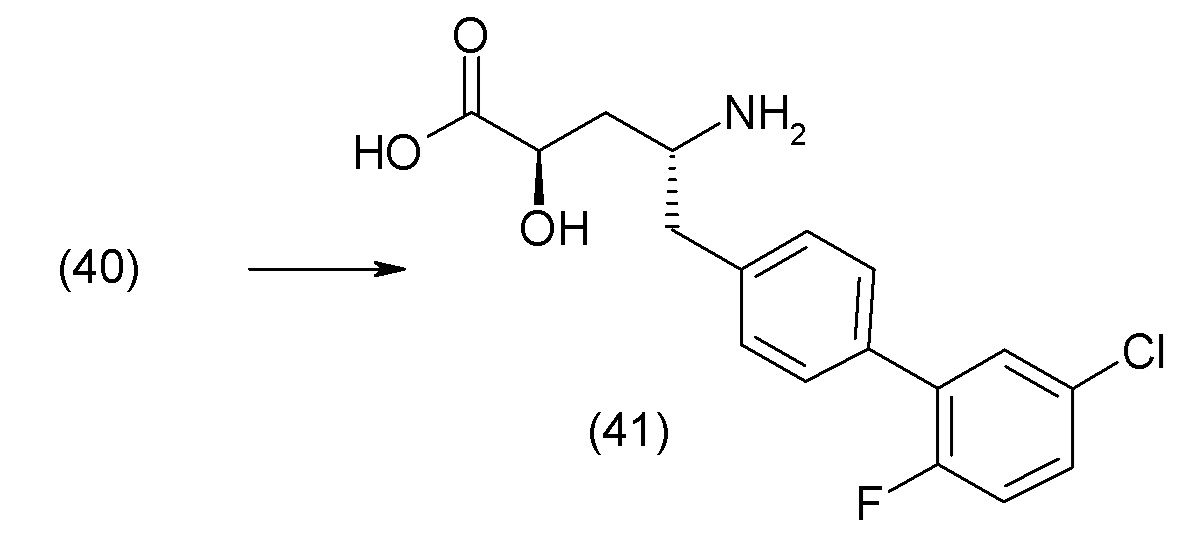
Раствор соединения 40 (8,8 г, 21,8 ммоль) в EtOH (12 мл) добавляли к концентрированной HCl (200 мл), нагревали при 100°C и перемешивали в течение ночи. Затем смесь концентрировали, получая неочищенный продукт, который очищали промыванием Et2O (100 мл) с получением соединения 41 в виде твердой соли HCl (7,5 г). LC-MC: 338 [М+Н+].

Раствор соединения 41 (7,5 г, 20,1 ммоль) в EtOH/HCl (100 мл) нагревали при 50°C в течение ночи. Смесь концентрировали, и неочищенный продукт очищали промыванием Et2O (200 мл) с получением соединения 42 (6,5 г) в виде белой твердой соли HCl. LC-MC: 366,1 [М+Н+].
Пример 5: (2R,4R)-5-(5'-Хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксипентановая кислота (соединение С5 сравнения)
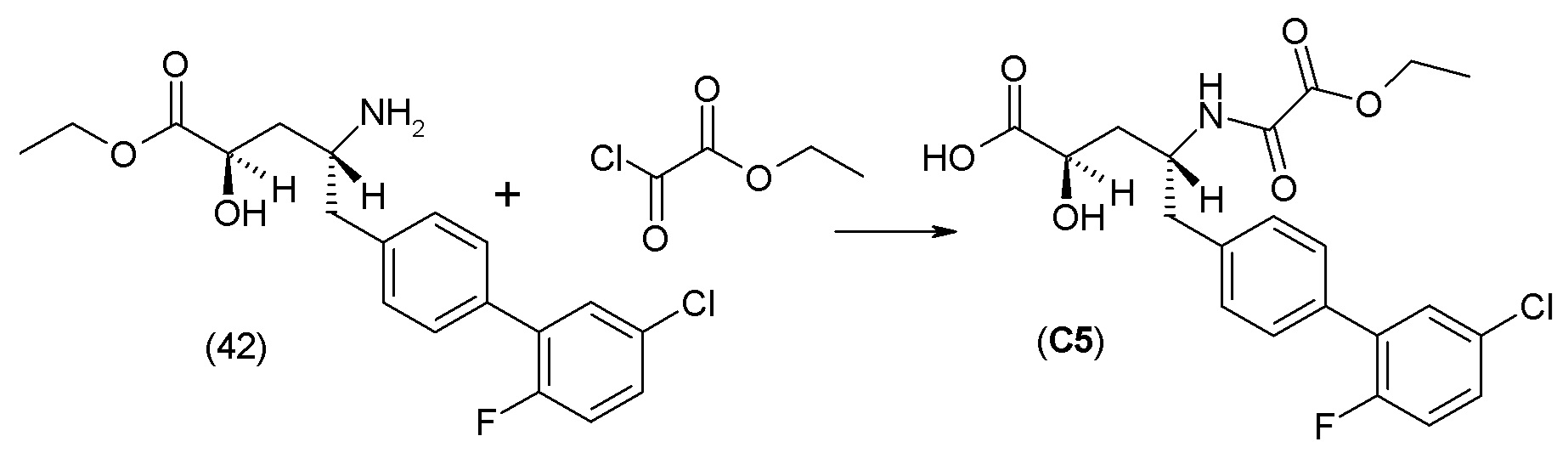
1,0н. HCl (6 мл) добавляли к этиловому эфиру (2R,4R)-4-амино-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидроксипентановой кислоты (42) (114 мг, 313 мкмоль), и смесь перемешивали при 90°С в течение 24 часов, затем концентрировали при пониженном давлении. Продукт в виде цвиттер-иона объединяли с Et3N (157 мкл, 1,1 ммоль) в DCM (6 мл) с последующим добавлением раствора этилоксалилхлорида (34,9 мкл, 313 мкмоль) в DCM (2 мл) при 0°С, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли насыщенный водный раствор NaHCO3 (5 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь экстрагировали DCM (3×3 мл), концентрировали, и остаток очищали методом препаративной ВЭЖХ с получением соединения С5 сравнения (5 мг). МС m/z [M+H]+ для C21H21ClFNO6-438,10, вычисленное; 438.2, найденное.
Соединение C5 сравнения описано в примере 5-14 в патенте США № 8,691,868, Hughes et al.
Пример 6: (2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-2-гидрокси-4-(оксалиламино)пентановая кислота (соединение С6 сравнения)

(2R,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксипентановую кислоту (C5) объединяли с 1М LiOH в воде (2,5 мл, 2,5 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, в этот момент результаты LCMS показали завершение реакции. Растворитель удаляли под вакуумом с получением соединения С6 сравнения (20,8 мг). МС m/z [M+H]+ для C19H17ClFNO6-410,07, вычисленное; 410,0, найденное.
Соединение C6 сравнения описано в примере 5-5 в патенте США № 8,691,868, Hughes et al.
Пример 7. Изучение стабильности кристаллов (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата кальция (1')
Одна из задач при разработке лекарственных препаратов связана с обнаружением стабильной кристаллической формы лекарственного вещества, имеющего достаточно высокую температуру плавления. Проблема, решаемая настоящим изобретением, заключалась в невозможности получения кристаллов свободной кислоты соединения 1. Более того, скрининг большого количества соединений в поисках кристаллической формы не принес успеха, за исключением двух соединений: соль аргинина и кальциевая соль кристаллов (2S,4R)-5-(5'-хлор-2'-фторбифенил-4-ил)-4-(этоксиоксалиламино)-2-гидроксиметил-2-метилпентановой кислоты. Тем не менее, кристаллы аргинина (см. пример 1) плавились в условиях окружающей среды, плавились в условиях окружающей среды, и их дальнейшая разработка была связана с определенными трудностями. С другой стороны, кристаллы кальция оказались стабильными и плавились при температуре около 239°C. По этой причине было проведено ускоренное исследование стабильности соединения 1' при указанных ниже значениях температуры и % относительной влажности (RH).
aОбразец=(масса вещества/общая масса)*100=% (масс./масс.)
bЧистота=(AUC чистого вещества/AUC нечистого вещества)*100=% (a/a)
сnt=не изучалось
Эти данные показывают, что соединение 1' остается относительно стабильным до по меньшей мере трех месяцев при указанных температурах и относительной влажности.
АНАЛИЗЫ
Соединение 1 и соединения С2, С3, С4, С5 и С6 сравнения оценивали в описанных ниже анализах. В приведенной ниже таблице показаны метаболиты, которые могут быть образованы из одного или более соединений, после расщепления в различных участках его структуры.
АНАЛИЗ 1: Анализы in vitro количественного определения ингибиторующей активности в отношении NEP человека и крысы
Ингибирующую активность в отношении неприлизина человека и крысы (EC 3.4.24.11, NEP) определяли, используя описанные ниже анализы in vitro.
Экстракция фермента NEP из почек крысы
NEP крысы получали из почек взрослых крыс Sprague Dawley. Целые почки промывали в холодном забуференном фосфатом физиологическом растворе (PBS) и переносили в охлажденный на льду буфер для лизиса (1% Тритон X-114, 150 мМ NaCl, 50 мМ трис(гидроксиметил)аминометан (Трис), pH 7,5, Bordier (1981) J. Biol. Chem., 256: 1604-1607) в соотношении 5 мл буфера на грамм почки. Образцы гомогенизировали на льду, используя ручной измельчитель тканей Polytron. Гомогенаты центрифугировали при 1000×g в роторе c качающимися стаканами в течение 5 минут при 3°C. Осадок ресуспендировали в 20 мл ледяного буфера для лизиса и инкубировали на льду в течение 30 минут. Затем образцы (15-20 мл) наслаивали на 25 мл ледяного буфера ʺподушкуʺ (6% масс./об. сахарозы, 50 мМ рН 7,5 Трис, 150 мМ NaCl, 0,06%, Тритон X-114), нагревали до 37°C в течение 3-5 минут и центрифугировали при 1000×g в роторе с качающимися стаканами при комнатной температуре в течение 3 минут. Два верхних слоя отсасывали, оставляя вязкий маслянистый осадок, содержащий обогащенную мембраной фракцию. Добавляли глицерин до концентрации 50%, и образцы хранили при -20°С. Проводили количественное определение концентрации белка, используя BCA систему определения с бычьим сывороточным альбумином (BSA) в качестве стандарта.
Анализ ингибирования фермента
Приобретали коммерческий продукт рекомбинантного человеческого NEP (R&D Systems, Minneapolis, MN, номер по каталогу 1182-ZN). Использовали флуорогенный пептидный субстрат Mca-D-Arg-Arg-Leu-Dap-(Dnp)-OH (Medeiros et al., 1997) Braz J. Med. Biol. Res., 30: 1157-62; Anaspec, San Jose, CA).
Анализы выполняли в 384-луночных белых непрозрачных планшетах при 37°С с использованием флюорогенного пептидного субстрата с концентрацией 10 мкМ в буфере для анализа (50 мМ HEPES, pH 7,5, 100 мМ NaCl, 0,01% полиэтиленгликоль сорбитан монолаурат (Твин-20), 10 мкМ ZnSO4). Соответствующие ферменты использовали в концентрациях, которые приводили к количественному протеолизу 1 мкМ субстрата через 20 минут при 37°С.
Тестируемые соединения анализировали в диапазоне концентраций от 10 мкМ до 20 пМ. Тестируемые соединения добавляли к ферменту и инкубировали в течение 30 мин при 37°С перед инициированием реакции путем добавления субстрата. Реакцию останавливали после 20 минутной инкубации при 37°С добавлением ледяной уксусной кислоты до конечной концентрации 3,6% (об./об.).
Пластины считывали на флуорометре с длинами волн возбуждения и излучения, равными, соответственно, 320 нм и 405 нм. Константы ингибирования получали путем построения нелинейной регрессии данных при помощи уравнения (GraphPad Software, Inc., San Diego, CA):
v =v0/[1+(I/K′)],
где v - скорость реакции, v0 - скорость реакции в отсутствие ингибирования, I - концентрация ингибитора, а K' - кажущаяся константа ингибирования.
Эти данные показывают, что активность соединения 1 отношении NEP крысы и человека является одинаковой в случае соединения C2 сравнения, тогда как соединение C3 сравнения имеет очень низкую активность в отношении фермента NEP крысы и человека по сравнению с соединением C2 сравнения. Аналогично, соединение C5 сравнения показало эффективность в отношении NEP крысы и человека, аналогичную для соединения C6 сравнения, тогда как соединение C4 сравнения имеет низкую активность в отношении фермента NEP крысы и человека по сравнению с соединением C6 сравнения.
Соединения 1, C2, C5 и C6 имеют значимую активность в отношении фермента NEP крысы и человека и соответствуют пороговому значению активности pKi ≥ 9,0, необходимому для использования в качестве терапевтического агента, как описано выше.
АНАЛИЗ 2: PO Фармакокинетическое исследование у крыс, собак и обезьян
Каждое фармакокинетическое исследование у крыс, собак или обезьян начинали с приготовления лекарственного состава тестируемого соединения. Соответствующую массу каждого тестируемого соединения добавляли в объем носителя (например, 5% бикарбоната натрия, 5% декстрозы в воде), такой чтобы конечная концентрация каждого соединения была подходящей для дозирования со скоростью 2 мл/кг. Хотя гомогенная суспензия была подходящей для перорального введения, внутривенные дозирующие растворы подвергали стерильной фильтрации (0,2 мкм) перед дозированием, чтобы гарантировать отсутствие каких-либо частиц.
В исследовании на крысах из лаборатории Harlan Laboratories (Indianapolis, IN) получали предварительно канюлированных самцов крыс (по 3 на способ введения) Sprague-Dawley в возрасте от 8 до 10 недель. Крысы получали либо вводимую через желудочный зонд единичную пероральную дозу, либо единичную внутривенную дозу (через боковую хвостовую вену) дозирующего раствора. Конечная доза обычно составляла 0,5-3 мг/кг. Последовательные образцы крови собирали через канюлю, имплантированную в яремную вену, через 3 минуты, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов и 24 часа после введения дозы. Отбор проб выполняли либо вручную, либо с использованием автоматизированных пробоотборников крови. Образцы собирали в микропробирки, содержащие фторид натрия, оксалат калия и параоксон (антикоагулянты и ингибитор эстеразы, соответственно) и обрабатывали для получения плазмы путем центрифугирования в холодильнике.
В исследовании на собаках собакам бигль (по 3 на способ введения) весом от 7 до 12 кг, которые содержались лаборатории Agilux Laboratories (Worcester, MA), через желудочный зонд вводили единичную пероральную дозу дозирующего раствора. Конечная доза обычно составляла 0,1-2 мг/кг. Последовательные образцы крови собирали путем венепункции через 3 минуты, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов и 24 часа после введения дозы. Все образцы собирали вручную в пробирки для микрокапельницы, содержащие фторид натрия, оксалат калия и параоксон, и обрабатывали для получения плазмы путем центрифугирования в холодильнике.
В исследовании с обезьянами самцам яванского макака (по 3 на один способ введения) весом от 4 до 5 кг, содержавшихся в Xenometrics (Stilwell, KS), через желудочный зонд вводили единичную пероральную дозу дозирующего раствора. Конечная доза составляла 2 мг/кг. Серийные образцы крови собирали из головной или подкожной вены перед введением дозы и через 5 минут, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 8 часов, 12 часов и 24 часа после введения дозы. Все образцы собирали в пробирки, содержащие оксалат калия, фторид натрия и параоксон, и обрабатывали для получения плазмы путем центрифугирования в холодильнике.
Образцы плазмы экстрагировали 3 объемами MeCN, содержащим подходящий внутренний стандарт. Экстракты восстанавливали в 3 объемах воды, содержащей 1% муравьиной кислоты, и анализировали методом ВЭЖХ-МС/МС. Данные о концентрации в плазме анализировали при помощи компьютерной программы Phoenix (Pharsight Corp., St. Louis, MO) для расчета фармакокинетических параметров.
Пероральная биодоступность (%F) представляет процент дозы, который достигает системной циркуляции после введения пероральной дозы по сравнению с внутривенной дозой, где вся доза вводится непосредственно в системный кровоток. Он равен отношению площади под кривой концентрация-время после введения пероральной дозы к площади под кривой концентрация-время после введения внутривенной дозы, нормированной для любых различий в уровнях дозы между путями введения.
aAUClast - область под кривой зависимости концентрации в плазме от времени, начиная со времени 0 до момента после дозирования, при котором наблюдалась последняя измеряемая концентрация, оцененная линейным методом трапеций
bНормализована путем деления AUClast на вводимую дозу
Эти данные показывают, что соединение C2 сравнения (активный метаболит соединения 1) имеет низкую пероральную биодоступность, тогда как соединение 1 имеет относительно высокую пероральную биодоступность во всех трех тестируемых моделях животных.
АНАЛИЗ 3: IV/PO фармакокинетическое исследование на крысах и собаках
Каждое фармакокинетическое исследование на крысах или собаках начинали с приготовления лекарственного состава тестируемого соединения. Соответствующую массу каждого тестируемого соединения добавляли в объем носителя (например, 5% бикарбоната натрия, 5% декстрозы в воде), такой чтобы конечная концентрация каждого соединения была подходящей для дозирования со скоростью 2 мл/кг. Хотя гомогенная суспензия была подходящей для перорального введения, внутривенные дозирующие растворы подвергали стерильной фильтрации (0,2 мкм) перед дозированием, чтобы гарантировать отсутствие каких-либо частиц
В исследовании на крысах из лаборатории Harlan Laboratories (Indianapolis, IN) получали предварительно канюлированных самцов крыс (по 3 на способ введения) Sprague-Dawley в возрасте от 8 до 10 недель. Крысы получали либо вводимую через желудочный зонд единичную пероральную дозу, либо единичную внутривенную дозу (через боковую хвостовую вену) дозирующего раствора. Конечная доза обычно составляла 0,5-3 мг/кг. Последовательные образцы крови собирали через канюлю, имплантированную в яремную вену, через 3 минуты, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов и 24 часа после введения дозы. Отбор проб выполняли либо вручную, либо с использованием автоматизированных пробоотборников крови. Образцы собирали в микропробирки, содержащие фторид натрия, оксалат калия и параоксон и обрабатывали для получения плазмы путем центрифугирования в холодильнике.
В исследовании на собаках собакам бигль (по 3 на способ введения) весом от 7 до 12 кг, которые содержались лаборатории Agilux Laboratories (Worcester, MA), получали либо вводимую через желудочный зонд единичную пероральную дозу либо единичную внутривенную дозу (через стационарный катетер) дозирующего раствора. Конечная доза обычно составляла 0,1-2 мг/кг. Последовательные образцы крови собирали путем венепункции через 3 минуты, 15 минут, 30 минут, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов и 24 часа после введения дозы. Все образцы собирали вручную в пробирки для микрокапельницы, содержащие фторид натрия, оксалат калия и параоксон, и обрабатывали для получения плазмы путем центрифугирования в холодильнике.
Образцы плазмы экстрагировали 3 объемами MeCN, содержащим подходящий внутренний стандарт. Экстракты восстанавливали в 3 объемах воды, содержащей 1% муравьиной кислоты, и анализировали методом ВЭЖХ-МС/МС. Данные о концентрации в плазме анализировали при помощи компьютерной программы Phoenix (Pharsight Corp., St. Louis, MO) для расчета фармакокинетических параметров.
Фармакокинетические данные, полученные для крысы
aAUClast - область под кривой зависимости концентрации в плазме от времени, начиная со времени 0 до момента после дозирования, при котором наблюдалась последняя измеряемая концентрация, оцененная линейным методом трапеций
bПероральная биодоступность вычисляется в виде значения AUClast активной молекулы после перорального введения пролекарства, деленного на значение AUClast после внутривенного введения активной молекулы, нормализованное для любых различий в вводимых дозах, выраженных в процентах.
Эти данные, полученные на крысах, показывают, что соединение по изобретению, соединение 1, приводит к значительно большему системному воздействию активного метаболита, чем соединение C3, C5 или C4 (значения биодоступности метаболитов> 100%, 21%, 11% и 9%, соответственно).
Как для соединения 1, так и для соединения С5 сравнения пролекарство не было обнаружено, поэтому можно предположить, что у крысы произошла in vivo полная конверсия соединений-пролекарств в активный метаболит. Оба эти соединения имеют более высокие уровни воздействия метаболита, чем либо соединение C4 (17) сравнения, либо соединение C3 (1,2) сравнения.
Эти пролекарства расщепляются в результате гидролиза эфирных связей, который у крыс происходит быстрее, чем у собак или людей. По этой причине крысиная модель гидролиза сложного эфира не всегда является предсказательной в отношении людей. Таким образом, пролекарства, аналогичные соединению по изобретению, также необходимо изучить у собак, чтобы получить дополнительные данные, позволяющие спрогнозировать скорость расщепления у людей.
Эти данные, полученные на собаках, показывают, что соединение по изобретению, соединение 1, приводит пероральной биодоступности активного метаболита, аналогичной для соединения С4 сравнения, и значительно большей биодоступности, чем соединение С3 или С5 (значения 44%, 46%, 0% и 29%, соответственно).
Эти данные, полученные на собаках, показывают, что пролекарство по изобретению, соединение 1, обеспечивает значительно большее относительное воздействие активной молекулы метаболита (то есть AUClast), чем любое другое изученное соединение или пролекарство. Такой быстрый и глубокий гидролиз пролекарства обеспечивает значительное и неожиданное преимущество соединения по изобретению. У собак соединение 1 расщепляется намного эффективнее, чем соединение C5 сравнения, что приводит к более чем 10-кратному улучшению уровня воздействия (177 против 13). Кроме того, расщепление соединения 1 происходит еще более эффективно по сравнению с соединением С3 сравнения (отношение экспозиции 177 против 0). Величина этой разницы не могла быть предсказана на основе сравнения степени гидролиза соединения С5 сравнения по сравнению с С4 (13 против 1,2).
АНАЛИЗ 4: Почечная экскреция соединения 1 у самцов собак бигль
Важным фактором для обеспечения надлежащего введения лекарственного средства в течение длительного времени и корректной стабильной концентрации лекарственного средства у пациентов является выведение лекарственного средства из организма. В целом, сниженный уровень выведения лекарственного средства приводит к более высокой его концентрации и более сильному воздействию. Для определения почечного клиренса соединения 1 процент вводимой дозы, выделенной с мочой после введения единичной IV дозы, оценивали у самцов собак бигль, как описано ниже.
Самцам собак бигль (N=3) весом 9,58-10,42 кг вводили IV дозу 1,0 мг/кг соединения 1. Соединение 1 вводили в виде лекарственной формы в 5% NaHCO3 в D5W. Собаки голодали в течение ночи, при этом их предварительно обрабатывали пентагастрином (60 мкг/мл, 0,1 мл/кг, IM) примерно за 30 минут до введения дозы для стимуляции секреции желудка. Прием пищи возобновляли примерно через 4 часа после введения. Образцы мочи собирали в пакеты, обложенные льдом во время сбора. Через 24 часа регистрировали массу образца, образец тщательно перемешивали, и получали аликвоты, которые хранили в замороженном виде (-80°C) до проведения биоанализа.
Концентрации соединения 1 в моче собак определяли методом ЖХ/МС/МС. Образцы для исследования мочи (разведенные в плазме) перемешивали на вортексе, и по 50 мкл помещали в 96-луночный планшет. Образцы экстрагировали ацетонитрилом с внутренним стандартом, хризином. Экстракт центрифугировали, а супернатант переносили в новый 96-луночный планшет, и 1 часть образца разбавляли 4 частями воды с 0,2% муравьиной кислотой. Образцы (12 мкл) наносили на колонку Waters Acquity UPLC BEH C18 (50×2,1 мм, 1,7 мкм) со скоростью потока 0,9 мл/мин. Мобильная фаза А состояла из 95:5:0,1 (v:v:v) вода:ацетонитрил:муравьиная кислота, и подвижная фаза В состояла из 50:50:0,1 (v:v:v) метанол:ацетонитрил:муравьиная кислота. Оцененное количество соединения 1 находилось в пределах от 0,001 до 5,00 мкг/мл.
Среднее количество мочи, выделенной в течение 24 часов, и приблизительный % введенной дозы, выведенной с мочой, приведен ниже в таблице.
aСреднее из трех определений
Почечная экскреция соединения 1 у собак составляла примерно <0,5% от вводимой дозы. Эти данные показывают, что соединение 1 имеет низкую почечную экскрецию у самцов собак бигль.
Реферат
Изобретение относится к соединению, имеющему структуру (1), или его фармацевтически приемлемой соли, обладающим свойством ингибитора неприлизина. Изобретение относится также к кристаллической форме (2S,4R)-5-(5'-хлор-2'-фтор-[1,1'-бифенил]-4-ил)-4-(2-этокси-2-оксоацетамидо)-2-(гидроксиметил)-2-метилпентаноата кальция, способам получения указанного соединения и кристаллической формы, содержащей их фармацевтической композиции, пероральной и внутривенной лекарственным формам, применению указанного соединения или кристаллической формы для изготовления лекарственного средства для лечения гипертонии, сердечной недостаточности или почечной недостаточности и способам лечения гипертонии, сердечной недостаточности или почечной недостаточности с их использованием. 12 н. и 13 з.п. ф-лы, 9 ил., 14 табл., 7 пр.(1)
Формула

Документы, цитированные в отчёте о поиске
Ингибиторы неприлизина
Комментарии