Получение вакцин, содержащих поверхностный антиген вируса гепатита в и поверхностно-активное вещество - RU2444374C2
Код документа: RU2444374C2
Чертежи







Описание
Все упомянутые здесь документы включены сюда в полном объеме в виде ссылки.
Область техники
Настоящее изобретение относится к способам получения комбинированных вакцин, где вакцины содержат смешанные иммуногены из более чем одного патогена, так что введение вакцины приводит к одновременной иммунизации субъекта против более чем одного патогена. В частности, изобретение касается применения поверхностно-активных веществ при получении комбинированных вакцин.
Уровень техники
Вакцины, содержащие антигены из более чем одного патогенного организма в пределах одной дозировки, известны как "поливалентные", или "комбинированные" вакцины. В Европе и США были одобрены для использования человеком различные комбинированные вакцины, включая в себя трехвалентные вакцины против дифтерии, столбняка и коклюша (вакцины "DTP") и трехвалентные вакцины против кори, свинки и краснухи (вакцины "MMR").
Преимущество комбинированных вакцин заключается в уменьшении количества инъекций пациентам, что приводит к клиническому преимуществу в связи с повешенной восприимчивостью (например, см. главу 29 документа 1), особенно в случае вакцинации детей. Однако при этом существуют трудности в изготовлении, возникающие из-за ряда факторов, включая физическую и биохимическую несовместимость антигенов и других компонентов; иммунологическое взаимодействие и стабильность.
Включение неантигенных компонентов в вакцины необходимо, но может вызвать трудности. В комбинированных вакцинах поверхностно-активные вещества представляют собой определенную проблему, потому что для оптимальной активности одного антигена требуется поверхностно-активное вещество, тогда как для другого наличие поверхностно-активного вещества может сказываться отрицательно. Кроме того, включение поверхностно-активных веществ в вакцины для детей вызывает сложности для некоторых групп пациентов, даже при том, что поверхностно-активное вещество считается безопасным.
Особенный интерес в области вакцин представляют поверхностно-активные вещества сложных эфиров полиоксиэтиленсорбитана, особенно такие как полисорбат 20 (также известный как 'Tween 20', или полиоксиэтиленсорбитанмонолаурат) и полисорбат 80 (также известный как 'Tween 80', или полиоксиэтиленсорбитанмоноолеат). Полисорбат 20 включен в моновалентную вакцину гепатита А HAVRIX™, а полисорбат 80 включен в комбинированные вакцины, такие как вакцины серии TRIPEDIA™ и INFANRIX™. Эти два поверхностно-активных вещества также используют для стабилизации жидких ротавирусных вакцин [2].
Полисорбаты используют также при получении комбинированных вакцин, которые содержат поверхностный антиген гепатита B ('HBsAg'); например, в документах 3 и 4 раскрыт способ получения четырехвалентной вакцины D-T-P-HBsAg, в которой отсутствует взаимодействие с фосфолипидным компонентом HBsAg за счет добавления неионогенного поверхностно-активного вещества, такого как Tween 20, Tween 80 или Triton X-100. По данным на фигуре 2 документов 3 и 4 (здесь фигура 1) видно, что поверхностно-активное вещество требуется для поддержания антигенности HBsAg, но является менее важным для других компонентов. Самая высокая измеренная концентрация поверхностно-активного вещества составляла 10 мкг/мл при 20 мкг/мл HBsAg, и это обеспечивало наилучшую антигенность.
Согласно способу, описанному в документах 3 и 4, неионогенный детергент добавляли после очистки HBsAg. Однако добавление детергентов после очистки HBsAg не является оптимальным, поскольку это требует отдельной стадии во время получения, которая увеличивает время процесса и также увеличивает риск контаминации HBsAg. При использовании контаминированного компонента при получении комбинированной вакцины возможные потери оказываются большими, чем при получении моновалентных вакцин, например, если контаминированный компонент HBsAg смешивают с чистым D-T-P-компонентом, то вся смесь D-T-P-HBsAg должна быть признана негодной, а не только HBsAg.
Для комбинированных вакцин, содержащих неионогенные поверхностно-активные вещества, существует потребность в способах получения, в которых поверхностно-активное вещество не добавляют в виде отдельного компонента во время выполнения способа.
Краткое описание изобретения
Вместо того, чтобы добавлять неионогенные поверхностно-активные вещества к антигенам после того, как они были очищены [3, 4], в настоящем изобретении их используют во время очистки антигена. Таким образом, поверхностно-активное вещество может выполнять свою функцию в конечной комбинированной вакцине, но риск контаминации снижается (а значит, и риск потери всей комбинированной вакцины после получения).
Таким образом, изобретение обеспечивает способ получения комбинированной вакцины, где вакцина включает в себя (i) неионогенное поверхностно-активное вещество, (ii) поверхностный антиген вируса гепатита B (HBV) и (iii) антиген по меньшей мере из одного не-HBV-патогена, и где способ включает в себя (i) очистку поверхностного антигена HBV от рекомбинантных дрожжевых клеток, где очистка включает в себя стадию, в которой дрожжевые клетки разрушаются в присутствии неионогенного поверхностно-активного вещества, с получением очищенного компонента HBsAg; и (ii) объединение очищенного компонента HBsAg по меньшей мере с одним дополнительным антигеном из не-HBV-патогена, для получения комбинированной вакцины.
Для предотвращения описанных выше проблем, связанных с контаминацией, способ не включает в себя стадию добавления неионогенного поверхностно-активного вещества как отдельного компонента после очистки HBsAg. Возможно присутствие поверхностно-активного вещества (или других поверхностно-активных веществ, ионных или неионных) в других компонентах антигена, с которыми объединяют HBsAg, для получения комбинированной вакцины, но не должно происходить добавление поверхностно-активного вещества как самостоятельного отдельного компонента. Таким образом, поверхностно-активное вещество не добавляется в виде отдельного компонента к очищенному компоненту HBsAg, и оно не добавляется во время объединения антигенов.
Изобретение также обеспечивает способ получения комбинированной вакцины, где вакцина включает в себя (i) неионогенное поверхностно-активное вещество, (ii) поверхностный антиген вируса гепатита B (HBV) и (iii) антиген по меньшей мере из одного не-HBV-патогена, и где способ включает в себя стадию объединения очищенного поверхностного антигена HBV по меньшей мере с одним дополнительным антигеном из не-HBV-патогена, с получением комбинированной вакцины, где очищенный поверхностный антиген HBV был получен способом, при котором рекомбинантные HBsAg-экспрессирующие дрожжевые клетки разрушают в присутствии неионогенного поверхностно-активного вещества. И снова, поверхностно-активное вещество отдельно не добавляют.
Изобретение также обеспечивает иммуногенную композицию, включающую в себя (i) неионогенное поверхностно-активное вещество, (ii) поверхностный антиген вируса гепатита B (HBV) и (iii) антиген по меньшей мере из одного не-HBV-патогена, где поверхностный антиген HBV был получен способом, при котором рекомбинантные HBsAg-экспрессирующие дрожжевые клетки разрушают в присутствии неионогенного поверхностно-активного вещества. И снова, HBsAg был получен без отдельного добавления поверхностно-активного вещества. Этот продукт можно отличить от продуктов, где HBsAg был получен другими способами, поскольку неионогенное поверхностно-активное вещество, использованное при очистке, сохраняется в частице HBsAg.
Неионогенное поверхностно-активное вещество
Согласно изобретению, можно использовать различные неионогенные поверхностно-активные вещества [5], и, в частности, те, которые обычно используются в вакцинных препаратах. Предпочтительными являются органические поверхностно-активные вещества. Обычно они представляют собой продукт реакции алкиленоксида (например, этиленоксида) со спиртом жирного ряда, жирной кислотой, алкилфенолом, алкиламином или другим соответствующим соединением, имеющим по меньшей мере один активный водородный атом. Для большинства поверхностно-активных веществ обычно длина углеродной цепи спирта, амина и кислоты составляет С8-С18. Обычные алкилфенолы представляют собой нонилфенол и октилфенол. Особенно предпочтительными являются поверхностно-активные вещества, содержащие поли(оксиэтен)ые остатки.
Например, согласно изобретению, могут быть использованы без ограничения следующие поверхностно-активные вещества: поверхностно-активные вещества сложных эфиров полиоксиэтиленсорбитана (обычно называемые Tween), в частности, полисорбат 20 и полисорбат 80; сополимеры этиленоксида (EO), пропиленоксида (PO), и/или бутиленоксида (BO), продаваемые под товарным знаком DOWFAX™, такие как линейные блок-сополимеры EO/PO; октоксинолы, с варьируемым числом повторов этокси(окси-1,2-этанедил)ьных групп, с октоксинолом-9 (Triton X-100 или t-октилфеноксиполиэтоксиэтанол) является особенно интересным; (октилфенокси)полиэтоксиэтанол (IGEPAL CA-630/NP-40); полиоксиэтилированные жирные эфиры, производные лаурилового, цетилового, стеарилового и олеилового спиртов (известные как поверхностно-активные вещества Brij), такие как монолауриловый простой эфир триэтиленгликоля (Brij 30); и сложные эфиры сорбитана (обычно называемые SPAN), такие как триолеат сорбитана (Span 85) и сорбитанмонолаурат.
Согласно изобретению, в частности, может быть использован полисорбат 20. Это поверхностно-активное вещество имеет установленный профиль безопасности при введении человеку, включая профиль безопасности в случае вакцин.
Поверхностно-активные вещества могут быть классифицированы по их ГЛБ (гидрофильно-липофильный баланс). Предпочтительные поверхностно-активные вещества согласно изобретению имеют ГЛБ по меньшей мере 10, предпочтительно, по меньшей мере 15, и более предпочтительно, по меньшей мере, 16.
Неионогенное поверхностно-активное вещество является компонентом композиций согласно изобретению. Чтобы избежать введения больших доз поверхностно-активного вещества пациенту, предпочтительные концентрации поверхностно-активного вещества в композиции должны составлять не более 30 мкг/мл, например, ≤25 мкг/мл, ≤20 мкг/мл, ≤15 мкг/мл, ≤10 мкг/мл, ≤5 мкг/мл, и т.д. Предпочтительная концентрация составляет ≤10 мкг/мл.
В качестве альтернативного варианта определения концентрации поверхностно-активного вещества, предпочтительное количество поверхностно-активного вещества в композиции должно составлять меньше чем 50 мкг (например, ≤40 мкг, ≤30 мкг, ≤25 мкг, ≤20 мкг, ≤15 мкг, ≤10 мкг, и т.д.) на каждые 100 мкг HBsAg. Аналогично, в документах 3 и 4 предлагается, что массовое соотношение поверхностно-активное вещество:HBsAg составляет меньше чем 50%. Предпочтительным является меньше чем 25 мкг поверхностно-активного вещества на 100 мкг HBsAg.
Как будет подробно описано ниже, в предпочтительном способе согласно изобретению способов используют предварительно смешенный компонент, включающий в себя анатоксины столбняка и дифтерии. Предпочтительно, этот D-T-компонент по существу не содержит неионогенные поверхностно-активные вещества, и, в частности, не содержит полисорбаты 20 и 80. Аналогично, предварительно смешанный D-T-Pw-компонент не содержит неионогенные поверхностно-активные вещества, например, полисорбаты 20 и 80.
Поверхностные антигены вируса гепатита В
Вирус гепатита В (HBV) является одним из известных агентов, вызывающих вирусный гепатит. Вирион HBV состоит из внутреннего ядра, окруженного внешней белковой оболочкой или капсидом, а вирусное ядро содержит вирусный геном ДНК. Основным компонентом капсида является белок, известный как поверхностный антиген HBV или, более часто, 'HBsAg', который типично представляет собой полипептид с 226 аминокислотами с молекулярной массой ~24 кДа. Все существующие вакцины от гепатита B содержат HBsAg, и при введении этого антигена нормальному вакцинируемому пациенту, он стимулирует выработку анти-HBsAg антител, которые предохраняют против HBV инфекции.
Для получения вакцины HBsAg может быть получен двумя способами. Первый способ включает в себя выделение антигена в форме частиц из плазмы носителя хронического гепатита B, поскольку при HBV инфекции большие количества HBsAg синтезируются в печени и высвобождаются в кровоток. Второй путь включает в себя экспрессию белка методами рекомбинантных ДНК. Для применения в способе согласно изобретению HBsAg представляет собой рекомбинантно экспрессированный в дрожжевых клетках. Подходящие дрожжи включают в себя Saccharomyces (такие как S.cerevisiae) или Hanensula (такие как H.polymorpha) клетки-хозяева.
В отличие от нативного HBsAg (т.е. выделенного из плазмы продукта), экспрессированный дрожжами HBsAg в общем случае является негликозилированным, и он представляет собой наиболее предпочтительный вид HBsAg для использования согласно изобретению. Экспрессированный дрожжами HBsAg является высоко иммуногенным и может быть получен без риска контаминации продукта крови.
В общем случае HBsAg представлен в виде по существу сферических частиц (средний диаметр приблизительно 20 нм), включая в себя липидный матрикс, включающий в себя фосфолипиды. Экспрессированные дрожжами частицы HBsAg могут включать в себя фосфатидилинозитол, который не обнаружен в природных вирионах HBV. Также частицы могут включать в себя нетоксичное количество ЛПС для стимуляции иммунной системы [6].
Предпочтительный HBsAg - из HBV подтипа adw2.
В данной области известно множество способов очистки HBsAg (например, см. документы 7-33). Эти способы раскрывают получение моновалентных препаратов HBsAg, но, в отличие от способа, раскрытого в документах 3 и 4, ни один из них не относится к очистке HBsAg, особенно для использования в комбинированных вакцинах. Любой из этих и других способов может быть использован, при условии, что способ является подходящим для очистки антигена после экспрессии в рекомбинантных дрожжевых клетках, где очистка включает в себя стадию, в которой дрожжевые клетки разрушаются в присутствии неионогенного поверхностно-активного вещества.
Предпочтительный способ очистки HBsAg после разрушения клетки включает в себя: ультрафильтрацию; эксклюзионную хроматографию; анионообменную хроматографию; ультрацентрифугирование; обессоливание; и стерильную фильтрацию. Лизаты могут осаждаться после разрушения клетки (например, используя полиэтиленгликоль), оставляя HBsAg в растворе, готовом для ультрафильтрации.
После очистки HBsAg может быть подвергнут диализу (например, с цистеином), который может быть использован для удаления ртутных консервантов, таких как тимеросал, который может быть использован во время получения HBsAg [30, 34].
Количества HBsAg обычно выражены в микрограммах, и обычное количество HBsAg на вакцинную дозу составляет 10 мкг.
Кроме 'S' последовательности, поверхностный антиген может включать в себя всю или часть pre-S последовательности, такую как всю или часть pre-S1 и/или pre-S2 последовательности.
Антигены не-HBV
Иммуногенные композиции согласно изобретению включают в себя по меньшей мере один защитный антиген по меньшей мере из одного не-HBV-патогена. Не-HBV-патоген(ы) могут быть вирусными и/или бактериальными.
Типичные вирусные патогены без ограничения включают в себя: полиовирус; вирус гепатита А; вирус гриппа; вирус кори; вирус свинки; вирус краснухи; и вирус ветряной оспы.
Типичные бактериальные патогены без ограничения включают в себя: Corynebacterium diphtheriae, Clostridium tetani, Bordetella pertussis; Haemophilus influenzae, включая тип b и нетипируемые штаммы; Neisseria meningitidis, включая серотипы A, B, C, W135 и/или Y; Streptococcus pneumoniae, включая серотипы 4, 6B, 9V, 14, 18C, 19F и 23F; и Moraxella catarrhalis.
Corynebacterium diphtheriae вызывает дифтерию. Дифтерийный токсин может быть обработан (например, формалином или формальдегидом) для удаления токсичности, при этом сохраняется способность вызывать выработку специфических антител против токсина после инъекции. Такие дифтерийные анатоксины используют в противодифтерийных вакцинах, и они более подробно раскрыты в главе 13 документа 1. Предпочтительными дифтерийными анатоксинами являются таковые, полученные посредством обработки формальдегидом. Дифтерийный анатоксин может быть получен культивированием C.diphtheriae в питательной среде (например, среда Фентона или среда Линггода и Фентона), в которую может быть добавлен бычий экстракт, с последующей обработкой формальдегидом, ультрафильтрацией и осаждением. Затем токсоидный материал может быть обработан методами, включающими в себя стерильную фильтрацию и/или диализ.
Clostridium tetani вызывает столбняк. Столбнячный токсин может быть обработан для получения защитного анатоксина. Такие анатоксины используют в вакцинах против столбняка и более подробно они раскрыты в главе 27 документа 1. Предпочтительными столбнячными анатоксинами являются таковые, полученные посредством обработки формальдегидом. Столбнячный анатоксин может быть получен культивированием C.tetani в питательной (например, среда Латама, полученная из коровьевого казеина) с последующей обработкой формальдегидом, ультрафильтрацией и осаждением. Затем материал может быть обработан методами, включающими в себя стерильную фильтрацию и/или диализ.
Bordetella pertussis вызывает коклюш. Коклюшные антигены в вакцинах являются либо клеточными (целая клетка, в виде инактивированной клетки B.pertussis), либо бесклеточными ('aP'). Препараты клеточных коклюшных антигенов широко описаны в литературе [например, см., глава 21 документ 1], например, они могут быть получены инактивацией нагреванием фазы I культуры B. pertussis. В случае, если используют бесклеточные антигены, то включают один, два или (предпочтительно) три из следующих антигенов: (1) детоксифицированный коклюшный токсин (коклюшный анатоксин или 'PT'); (2) филаментный гемагглютинин ('FHA'); (3) пертактин (также называемый как 'белок наружной мембраны 69 килоДальтон'). Эти три антигена, предпочтительно, готовят выделением из культуры B.pertussis, выращенную в модифицированной жидкой среде Stainer-Scholte. PT и FHA могут быть выделены из ферментационного бульона (например. адсорбцией на гидроксиапатитном геле), тогда как пертактин может быть экстагирован из клеток посредством термической обработки и флокуляции (например, используя хлорид бария). Антигены могут быть очищены при использовании поочередных стадий хроматографии и/или осаждения. PT и FHA могут быть очищены гидрофобной хроматографией, афинной хроматографией и эксклюзионной хроматографией. Пертактин может быть очищен ионообменной хроматографией, гидрофобной хроматографией и эксклюзионной хроматографией. FHA и пертактин могут быть обработаны формальдегидом перед использованием согласно изобретению. PT, предпочтительно, детоксифицирован обработкой формальдегидом и/или глютаральдегидом. В качестве альтернативы химической детоксикации PT может представлять собой мутантный PT, в котором ферментативная активность уменьшена за счет мутагенеза [35], но предпочтительным является химическая детоксикация.
Haemophilus influenzae типа ('Ηib') вызывает бактериальный менингит. Обычно, вакцины Hib в основе содержат капсульный сахаридный антиген [например, глава 14 документа 1], такие препараты широко описаны в литературе [например, документы 36-45]. Сахарид конъюгирован Hib с белком-носителем для усиления иммуногенности, особенно для детей. Обычные белки-носители представляют собой столбнячный анатоксин, дифтерийный анатоксин, производное дифтерийного анатоксина CRM197, белок D Н.influenzae и комплекс наружного белка мембраны менингококков от серологической группы B. Столбнячный анатоксин является предпочтительным носителем, и используется в продукте, обычно называемом 'PRP-T'. PRP-T может быть получен активацией бромцианом капсульного полисахарида Hib, соединяя активированный сахарид с линкером адипиновой кислоты (такие как (1-этил-3-(3-диметиламинопропил) карбодиимид), обычно гидрохлоридная соль), и затем проводят реакцию линкер-сахаридного комплекса с белком-носителем столбнячного анатоксина. Сахаридная часть конъюгата может включать в себя непроцессированный полирибозилрибит фосфат (PRP), полученные из бактерий Hib, и/или фрагменты непроцессированного PRP. Могут быть использованы конъюгаты с соотношением (вес./вес.) сахарид:белок составляющим между 1:5 (т.е. избыток белка) и 5:1 (т.е. избыток сахарида), например, соотношение, составляющее между 1:2 и 5:1, и соотношение, составляющее между 1:1,25 и 1:2,5. Однако в предпочтительных вакцинах соотношение сахарида к белку-носителю по весу составляет между 1:2,5 и 1:3,5. В вакцинах, где столбнячный анатоксин присутствует к качестве как антигена, так и как белка-носителя соотношение сахарида к белку-носителю по весу в конъюгате может составлять между 1:0,3 и 1:2 [46]. Введение конъюгата Hib, предпочтительно, приводит к концентрации против-PRP антитела составляющей ≥0,15 мкг/мл, и более, предпочтительно, ≥1 мкг/мл, и это является стандартными пороговыми реакциями.
Neisseria meningitidis вызывает бактериальный менингит. На основе капсульного полисахарида организма были идентифицированы различные серологические группы N.meningitidis, включая в себя A, B, C, Н, I, K, L, 29E, W135, X, Y и Z. Серологическими группами, наиболее ассоциируемыми с заболеванием, являются A, B, C, W135 и Y. Существующие вакцины против серологических групп A, C, W135 и Y имеют в основе капсульные сахаридные антигены, но этот принцип не подходит для серологической группы B, вместо них используют белковые антигены и носители наружной мембраны. Капсульные сахариды конъюгированы с белками-носителями для усиления иммуногенности. Типичными белками-носителями являются столбнячный анатоксин, дифтерийный анатоксин, производное дифтерийного анатоксина CRM197 и белкок D H.influenzae. Сахаридная часть конъюгата может включать в себя непроцессированный сахарид, полученный от менингококков, и/или его фрагменты. Сахариды серогруппы C могут быть получены как от OAc+, так и от OAc-штаммов. Для сахаридов серогруппы А, предпочтительно, по меньшей мере 50% (например, по меньшей мере 60%, 70%, 80%, 90%, 95% или более) маннозаминных остатков O-ацетилированы по положению C-3. Менингококковые конъюгаты с сахаридом:белком с соотношением (вес./вес.) между 1:10 (то есть избыток белка), и 10:1 (т.e. избыток сахарида) могут быть использованы например, соотношения между 1:5 и 5:1, между 1:2,5 и 2,5:1, или между 1:1,25 и 1,25:1. Введение конъюгата, предпочтительно, приводит к увеличению титра в серологическом бактерицидном анализе (SBA) для релевантной серогруппы по меньшей в 4 раза, и, предпочтительно, по меньшей мере в 8 раз. Титры SBA можно измерять, используя комплемент детеныша кролика или человеческий комплемент [47].
Streptococcus pneumoniae вызывает бактериальный менингит. Что касается Hib и менингококков, то существующие вакцины имеют в основе капсульные сахариды. Предпочтительно, включать сахариды от более чем одного серотипа С.pneumoniae и особенно от по меньшей мере серотипов 6B, 14, 19F и 23F. Другие серотипы, предпочтительно, выбирают из: 1, 3, 4, 5, 7F, 9V и 18C. Например, широко используются смеси полисахаридов от 23 различных серотипов, а также конъюгированные вакцины с полисахаридами из от 5 до 11 различных серотипов [48]. Например, PrevNar™ [49] содержит конъюгированные антигены от семи серотипов (4, 6B, 9V, 14, 18C, 19F, и 23F). Сахариды, предпочтительно, конъюгированы с белками-носителями [например, документы 50-52]. Типичными белками-носителями являются столбнячный анатоксин, дифтерийный анатоксин, производное дифтерийного анатоксина CRM197, и белок D H.influenzae.
Сахариды в продукте PrevNar™ отдельно конъюгированы с CRM197 восстановительным аминированием по 2 мкг каждого сахарида на 0,5 мл дозу (4 мкг серотипа 6B). В качестве альтернативы использования сахаридных антигенов от пневмококка, композиция может включать в себя один или более полипептидных антигенов. Доступны геномные последовательности для нескольких штаммов пневмококка [53, 54] и могут быть подвергнуты обратному вакцинологическому анализу [55-58] для идентификации подходящих полипептидных антигенов [59, 60]. Например, композиция может включать в себя один или более из следующих антигенов: PhtA, PhtD, PhtB, PhtE, SpsA, LytB, LytC, LytA, Sp125, Sp101, Sp128, Sp130 и Sp130, как указано в документе 61. Композиция может включать в себя более чем один (т.е. 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 или 14) указанных антигенов. В некоторых вариантах выполнения изобретения, композиция может включать в себя как сахарид, так и полипептидные антигены от пневмококка. Они могут быть использованы как простая смесь, или пневмококковый сахаридный антиген может быть конъюгирован с пневмококковым белком. Соответствующие белки-носители для таких вариантов выполнения изобретения включают в себя пневмококковые белковые антигены, упомянутые выше [61].
Moraxella catarrhalis вызывает средний отит и синусит, и является нетипичной причиной ларингита. В настоящее время вакцины исследуются, как сообщается в документе 62.
Как и HBV, HAV вызывает гепатит. Вакцины HAV раскрыты в главе 15 документа 1. Предпочтительный компонент HAV имеет в основе инактивированный вирус, и инактивация может быть проведена обработкой формалином. Вирус может быть выращен на эмбриональных диплоидных фибробластах человека, таких как MRC-5 клетки. Предпочтительным штаммом HAV является HM175, хотя также может быть использован CR326F. Клетки выращивают в условиях, обеспечивающих рост вируса. Клетки лизируют, и полученную суспензию можно очищать ультрафильтрацией и гель-проникающей хроматографией.
Полиовирус вызывает полиомиелит. Вместо того, чтобы использовать пероральную вакцину полиовируса, в изобретении используется инактивированная вакцина вируса полиомиелита (IPV), как раскрыто более подробно в главе 24 документа 1. Полиовирусы могут быть выращены в клеточной культуре, и предпочтительная культура из клеточной линии Vero, полученной из почки обезьяны. Для удобства клетки Vero могут быть культивированы на микроносителях. После выращивания вирионы могут быть очищены, используя методы ультрафильтрации, диафильтрации и хроматографии. Перед введением пациентам полиовирусы должны быть инактивированы, это осуществляют обработкой формальдегидом. Полиомиелит может быть вызван одним из трех типов полиовируса. Эти три типа похожи и вызывают идентичные симптомы, но они значительно различаются антигенами, и заражение одним типом не предохраняет против заражения другими. Поэтому согласно изобретению, предпочтительно, использовать три полиовирусных антигена: полиовирус типа 1 (например, штамм Mahoney), полиовирус типа 2 (например, штамм MEF-I), и полиовирус типа 3 (например, штамм Saukett). Вирусы, предпочтительно, выращивают, очищают и инактивируют по отдельности, и затем объединяют, получая трехвалентную смесь для использования согласно изобретению. Количество IPV обычно выражают в 'DU' единицах ("единица D-антигена" [63]).
Антигены, предохраняющие против вирусов кори, свинки и краснухи, обычно являются живыми вирусами, которые содержатся в известных моновалентных и трехвалентных вакцинах ('MMR'). Вакцины вируса кори описаны более подробно в главе 19 документа 1. Вакцины вируса свинки описаны более подробно в главе 20 документа 1. Вакцины вируса краснухи описаны более подробно в главе 26 документа 1. Типичсный штамм вируса кори включает в себя: Moraten; Connaught; Schwarz; Edmonston-Zagreb; CAM-70; AIK-C; TD97; Leningrad-16; Shanghai-191; и т.д. Штаммы Schwarz и Moraten наиболее часто используемые в США и Европе. Типичный штамм вируса свинки включает в себя: Jeryl Lynn; RIT 4385; Urabe; Hoshino; Rubini; Leningrad-3; Leningrad-Zagreb; Miyahara; Torii; NK M-46; S-12 и т.д. Штаммы Jeryl Lynn, RIT 4385, Urabe и Leningrad-Zagreb наиболее часто повсеместно используемые. Типичный штамм вируса краснухи включает в себя: RA27/3; Matsuba; TCRB 19; Takahashi; Matsuura; TP-336 и т.д. Штамм RA27/3 наиболее часто используемый на западе.
Антигены VZV, предохраняющие против ветрянки, обычно являются живыми вирусами, основанными на штаммах вируса Oka. Вакцины VZV описаны более подробно в главе 28 документа 1.
Антигены вируса гриппа описаны более подробно в главах 17 и 18 документа 1. В широком понимании вакцины вируса гриппа могут быть основаны на живом вирусе или инактивированном вирусе, и инактивированные вакцины могут быть основаны на целом вирусе, 'расщепленном' вирусе или на очищенных поверхностных антигенах (включая гемагглютинин и нейраминидазы). Вирусы, использованные для получения вакцины, могут быть выращены как на яйцах, так и на культуре клеток. Вакцинные штаммы вируса гриппа изменяются от сезона к сезону. В настоящий межпандемический период вакцины обычно включают в себя два штамма гриппа (H1N1 и H3N2) и один штамм гриппа B, и обычными являются трехвалентные вакцины. Также могут быть использованы согласно изобретению вирусы от пандемичных штаммов (то есть штамм, к которому вакцинный реципиент и большая часть человеческой популяции являются иммунологически наивными), такие как штаммы подтипов H2, H5, H7 или H9 (в частности, вирус гриппа А), и вакцины от гриппа для пандемичных штаммов могут быть моновалентные или могут быть основаны на обычной трехвалентной вакцине, дополненной пандемичным штаммом. Вирус гриппа может быть химерным штаммом, и может быть получен методами "обратной генетики". Вирус может быть аттенюирован. Вирус может быть чувствительным к изменению температуры. Вирус может быть адаптирован к холоду.
Антигенные компоненты от этих патогенов для использования в вакцинах обычно обозначают упрощенными названиями: 'D' для дифтерийного анатоксина; 'T' для столбнячного анатоксина; 'P' для коклюшного антигена, где 'Pa' для ацеллюлярного и 'Pw' для клеточного; 'Hib' для капсульного сахарида b H.influenzae; 'MenA', 'MenB', 'MenC', MenW и 'MenY' для соответствующих менингококковых серогрупп; 'IPV' для инактивированного полиовируса и 'Spn' для пневмококка.
При объединении антигенных компонентов с HBsAg для получения поливалентной композиции, антигены могут быть добавлены по отдельности, или они могут быть заранее смешаны, перед объединением с HBsAg. В случае если используют D и T антигены, предпочтительно, использование заранее смешанного D-T-компонента. Этот двухвалентный компонент может быть использован в способе согласно изобретению, например, он может быть объединен HBsAg для получения трехвалентного D-T-HBV-компонента. Как вариант, D-T-компонент может быть объединен с дополнительными антигенами не-HBV (например, с ацеллюлярными коклюшными антигенами), и такой компонент затем может быть объединен с HBsAg, и т.д. В случае если используют антигены D, T и Pw, предпочтительно, использовать заранее смешанный компонент D-T-Pw, и затем использовать этот компонент при выполнении способа согласно изобретению.
В случае включения адъюванта в композицию согласно изобретению, он также может быть добавлен на различных стадиях. Как правило, антигены объединяют с адъювантами до использования в способе согласно изобретению (например, двухвалентная D-T смесь адсорбируют на адъюванте(ах) соли алюминия до использования в способе согласно изобретению, который может быть обеспечен отдельным получением анатоксинов, адсорбируя каждый из них на отдельном адъюванте из гидроксида алюминия, и затем смешивая два адсорбированных анатоксина (необязательно, с дополнительным адъювантом) для получения материала для использования в способе согласно изобретению), но также возможно добавлять адъювант после смешивания антигенов, или добавление антигенов к адъюванту (например, начиная с водного адъюванта, затем добавить антигены, или по отдельности, или заранее смешанные). Как описано ниже, HbsAg-компонент, предпочтительно, адсорбируют на адъюванте из фосфата алюминия до объединения с антигенными компонентами не-HBV.
Предпочтительные композиции согласно изобретению включают в себя по меньшей мере антигены D, T и P (сравни с документами 3 и 4) в дополнение к HBsAg. Особенно предпочтительными являются композиции со следующими комбинациями:
- HBsAg, D, T.
- HBsAg, D, T, Pw.
- HBsAg, D, T, Pw, Hib.
- HBsAg, D, T, Pw, Hib, MenA, MenC.
- HBsAg, D, T, Pw, Hib, MenA, MenC, MenW135.
- HBsAg, D, T, Pw, Hib, MenA, MenC, MenY.
- HBsAg, D, T, Pw, Hib, MenA, MenC, MenW135, MenY.
- HBsAg, D, T, Pa.
- HBsAg, D, T, Pa, Hib.
- HBsAg, D, T, Pa, полиовирус.
- HBsAg, D, T, Pa, полиовирус, Hib.
- HbsAg, D, T, Pa, полиовирус, Hib, MenC.
- HBsAg, D, T, Pa, полиовирус, Hib, MenC, MenA.
- HBsAg, D, T, Pa, полиовирус, Hib, MenC, MenY.
- HbsAg, D, T, Pa, полиовирус, Hib, MenC, MenW135.
- HBsAg, D, T, Pa, полиовирус, Hib, MenC, MenA, MenW135, MenY.
- HBsAg, Hib.
- HBsAg, вирус гепатита А.
Эти композиции могут состоять из вышеперечисленных антигенов или дополнительно могут включать в себя антигены от других патогенов. Таким образом, они могут быть использованы отдельно или как компоненты дополнительных вакцин.
В некоторых вариантах выполнения изобретения композиция не является 5-валентной D-T-Pa-HBV-IPV [30]. Таким образом, композиция может включать в себя компонент Pw и/или по меньшей мере один конъюгат.
Адъюванты
Предпочтительные иммуногенные композиции согласно изобретению включают в себя адъювант, и этот адъювант, предпочтительно, включает в себя одну или более солей алюминия, и, в частности, адъювант из фосфата алюминия и/или адъювант из гидроксида алюминия.
Антигенные компоненты, используемые в способе согласно изобретению, предпочтительно, должны включать в себя алюминиевые адъюванты до использования в способе, т.е. они 'предварительно смешиваются' или 'предварительно адсорбируются' с адъювантом(ами).
В композициях, включающих в себя HBsAg и дифтерийный анатоксин, дифтерийный анатоксин может быть адсорбирован на адъюванте из гидроксида алюминия.
В композициях, включающих в себя HBsAg и столбнячный анатоксин, столбнячный анатоксин может быть адсорбирован на адъюванта гидроксида алюминия, но это не является обязательным (например, может быть использован с адсорбцией между 0-10% от общего количества столбнячного анатоксина).
В композициях, включающих в себя HBsAg и цельноклеточный коклюшный антиген, антиген wP, предпочтительно, объединен с адъювантом из гидроксида алюминия и/или адъювантом из фосфата алюминия.
В композициях, включающих в себя HBsAg и ацеллюлярный коклюшный антиген(ы), коклюшный антиген(ы) может быть адсорбирован на один или более адъювантов из солей алюминия, или может быть добавлен в неадсорбированном состоянии.
В случае, если пертактин присутствует в композиции, то, предпочтительно, он должен быть адсорбирован на адъюванте из гидроксида алюминия до использования в способе согласно изобретению. PT и FHA могут быть адсорбированы на адъюванте из гидроксида алюминия или фосфата алюминия до использования в способе согласно изобретению. В предпочтительных вариантах выполнения изобретения, FHA PT5 и пертактин отдельно предварительно адсорбируются на гидроксиде алюминия до использования в способе согласно изобретению.
В композициях, включающих в себя HBsAg и антигены Hib, конъюгат Hib может быть неадсорбированным, но, предпочтительно, он адсорбирован на адъюванте из фосфата алюминия [64]. Такая адсорбция часто используется в вакцинах, включающих в себя антигены D-T-Pw-Hib-HBsAg. Другие конъюгированные антигены (т.е. менингококки, пневмококки), аналогично могут быть адсорбированы на соли алюминия (например, фосфате) или могут быть неадсорбированы [65].
Антигены IPV обычно не адсорбируют на адъюванты до использования в способе согласно изобретению, но они могут адсорбироваться на алюминиевых адъюванте(ах) других компонентов.
В композиции HBsAg может быть адсорбирован на фосфате алюминия методами, описанными в документе 66. Адсорбция на фосфате алюминия не используется в известном продукте ENGERIX-B™ (в котором HBsAg адсорбирован на гидроксиде алюминия), но присутствует в продуктах HEPACCINE™ и RECOMBIVAX™. Как указано в документе 61, в качестве адъюванта для HBsAg, фосфат алюминия может быть лучше, чем гидроксид алюминия. Хотя в конечной вакцине HBsAg может быть адсорбирован на адъюванте из гидроксида алюминия (как в известном продукте ENGERIX-B™), или может быть неадсорбированным, он, как правило, будет адсорбирован на адъюванте из фосфата алюминия. Кроме того, предпочтительно, его предварительно адсорбируют на фосфате алюминия до использования в способе согласно изобретению.
В случае, если в способе согласно изобретению используется компонент, в котором дифтерийные и столбнячные анатоксины смешаны до объединения с HBsAg, то эта D-T смесь, предпочтительно, содержит адъювант из гидроксида алюминия, на который адсорбированы антигены D и T.
В случае, если в способе согласно изобретению используется компонент, в котором дифтерийный анатоксин, столбнячный анатоксин и цельноклеточный коклюшный антиген смешаны до объединения с HBsAg, то эта D-T-Pw смесь, предпочтительно, содержит как адъювант из гидроксида алюминия, на который адсорбированы антигены D и T, так и адъювант из фосфата алюминия.
Используемые в настоящее время адъюванты алюминия обычно называют адъюванты из "гидроксида алюминия" или как адъюванты из "фосфата алюминия". Эти названия, используемые для удобства, однако они не отражают точное описание фактически присутствующего в них химического вещества (например, см. главу 9 документа 68). Согласно изобретению можно использовать любые из "гидроксидов" или "фосфатных" солей, которые обычно используются как адъюванты.
Типичные адъюванты, известные как "гидроксид алюминия", представляют собой соли оксигидроксида алюминия, которые обычно являются по меньшей мере частично кристаллическими. Оксигидроксид алюминия, который может быть представлен формулой AlO(OH), можно отличить от других соединений алюминия, таких как гидроксид алюминия Al(OH)3, инфракрасной (ИК) спектроскопией, в частности, по наличию адсорбционной линии при 1070 см-1 и ярковыраженного плеча при 3090-3100 см-1 (глава 9 документа 68).
Типичные адъюванты, известные как "фосфат алюминия", представляют собой гидроксифосфаты алюминия, часто также содержащие малое количество сульфата. Они могут быть получены осаждением, при этом условия реакции, а также концентрации во время осаждения влияют на степень замещения в соли фосфата гироксилом. Гидроксифосфаты в общем случае имеют молярное соотношение PO4/Al, составляющее между 0,3 и 0,99. Гидроксифосфаты можно отличить от AlPO4 наличием гидроксильных групп. Например, ИК спектральная полоса при 3164 см-1 (например, при нагревании до 200°C) показывает наличие гидроксилов в структуре (глава 9 документа 68).
Мольное соотношение PO4/Al3+ адъюванта фосфата алюминия в общем случае составляет между 0,3 и 1,2, предпочтительно, между 0,8 и 1,2, и более предпочтительно, 0,95+0,1. Фосфат алюминия в общем случае аморфный, особенно в случае гидроксифосфатной соли. Типичным адъювантом является аморфный гидроксифосфат алюминия с мольным соотношением PO4/Al, составляющим между 0,84 и 0,92, включая 0,6 мг Al3+/мл. Фосфат алюминия в общем случае представлен в виде частиц. Диаметр частиц обычно находится в диапазоне 0,5-20 мкм (например, около 5-10 мкм) после адсорбции любого антигена.
PZC фосфата алюминия обратно пропорционально степени замещения фосфата гидроксилом, и эта степень замещения может изменяться в зависимости от условий реакции и концентрации реагентов, используемых для получения соли осаждением. PZC также можно варьировать, изменяя концентрацию свободных фосфатных ионов в растворе (больше фосфата = более кислое PZC) или добавляя буфер, такой как гистидиновый буфер (делает PZC более основной). PZC фосфатов алюминия, используемых согласно изобретению, в общем случае составляет между 4,0 и 7,0, более предпочтительно, между 5,0 и 6,5 например, приблизительно 5,7.
Раствор фосфата алюминия, используемый для получения композиции согласно изобретению, может содержать буфер (например, фосфатный, или гистидиновый, или Tris буфер), но это не всегда необходимо. Предпочтительно, раствор фосфата алюминия стерильный и апирогенный. Раствор фосфата алюминия может включать в себя свободные водные фосфорнокислые ионы, например, содержащиеся в концентрации между 1,0 и 20 мМ, предпочтительно, между 5 и 15 мМ, и более предпочтительно, приблизительно 10 мМ. Раствор фосфата алюминия также может включать в себя хлорид натрия. Концентрация хлорида натрия находится, предпочтительно, в диапазоне от 0,1 до 100 мг/мл (т.е. 0,5-50 мг/мл, 1-20 мг/мл, 2-10 мг/мл), и более, предпочтительно, приблизительно 3±1 мг/мл. Наличие NaCl способствует правильному измерению pH перед адсорбцией антигенов.
В некоторых вариантах выполнения изобретения изобретение исключает композиции, которые включают в себя эмульсию типа "масло-в-воде", содержащую смесь полисорбата 80, Span 85 и сквалена [69]. В некоторых вариантах выполнения изобретения изобретение может исключать композиции, которые включают в себя смесь масла, α-токоферола и полисорбата 80 [70]. В некоторых вариантах выполнения изобретения изобретение может исключать композиции, которые включают в себя в себя сапониновый адъювант (такие как QS21) и неионогенное поверхностно-активное вещество (такое как полисорбат 40, 60 или 80). В некоторых вариантах выполнения изобретения изобретение может исключать композиции, которые включают в себя сапониновый адъювант, метаболизируемое масло и неионогенное поверхностно-активное вещество [71]. В некоторых вариантах выполнения изобретения изобретение может исключать композиции, которые включают в себя сапониновый адъювант, эмульсии "масло-в-воде” эмульсии и стерин [72]. В некоторых вариантах выполнения изобретения изобретение может исключать композиции, которые включают в себя 3d-MPL, QS21, триглицерид и эмульсию типа "масло-в-воде" [73].
Объединение очищенного HBsAg с дополнительными антигеном(ами)
Способ согласно изобретению включает в себя стадию, в которой очищенный HBsAg, объединяют по меньшей мере с одним антигеном по меньшей мере из одного не-HBV-патогена.
Антигены могут быть объединены индивидуально последовательно, или они могут быть заранее перемешаны и добавлены вместе. Например, 4-валентная DTP-HBsAg вакцина может быть получена способом, включающим в себя последовательное добавление антигенов HBsAg, D, T и P в сосуд, или предварительным смешиванием антигенов D, T и P и затем объединения HBsAg и смеси DTP.
Антигенные компоненты могут быть объединены в любом требуемом порядке.
Антиген(ы) от не-HBV-патогена(ов) может включать в себя поверхностно-активное вещество, и оно может быть таким же или отличным от неионогенного поверхностно-активного вещества, применяемого при очистке HBsAg. Способ согласно изобретению особенно эффективен, в случае, если дополнительный антигенный(е) компонент(ы) включает в себя поверхностно-активное вещество, поскольку позволяет избежать множества отдельных стадий добавления поверхностно-активного вещества. Предпочтительными являеются способы, в которых один или более не-HBV компонентов включают в себя полисорбат 80.
В случае, если дифтерийный и столбнячный анатоксины включены в композицию согласно изобретению, предпочтительно, их предварительно смешивают до объединения с HBsAg. Таким образом, способ согласно изобретению включает в себя объединение первого компонента, включающего в себя HBsAg со вторым компонентом, включающим в себя как антигены D, так и T. Аналогично, в случае, если дифтерийный анатоксин, столбнячный анатоксин и клеточный коклюшный антиген включены в композицию, предпочтительно, их предварительно смешивают, и таким образом, способ согласно изобретению включает в себя объединение первого компонента, включающего в себя HBsAg, со вторым компонентом, включающим в себя антигены D, T и Pw.
В случае, если используют D-T смесь, соотношение дифтерийного анатоксина к столбнячному анатоксину в вакцинах согласно изобретению обычно составляет между 2:1 и 3:1 (измеренное в Lf единицах), предпочтительно, между 2,4:1 и 2,6:1, и более предпочтительно, 2,5:1.
В случае, если в композицию согласно изобретению включают адъювант, он также может быть добавлен на различных стадиях. Как правило, антигены объединяют с адъювантами до использования в способе согласно изобретению (например, двухвалентную D-T смесь адсорбируют на адъюванте(ах) соли алюминия до использования в способе согласно изобретению), но также возможно добавлять адъювант после смешивания антигенов, или добавлять антигены к адъюванту (т.е. начиная с водного адъюванта, затем добавить антигены, или по отдельности или предварительно смешанные). Как описано выше, компонент HBsAg может быть адсорбирован на адъюванте из фосфата алюминия до объединения с антигенными компонентами не-HBV.
Комбинированная вакцина
Композиции согласно изобретению могут включать в себя: (a) антигенный компонент; и (b) неантигенный компонент. Антигенный компонент может включать в себя или состоять из антигенов, раскрытых выше. Неантигенный компонент может включать в себя носители, адъюванты, инертные наполнители, буфер и т.д., как описано более подробно ниже. Эти неантигенные компоненты могут иметь различные источники. Например, они могут присутствовать в одном из антигенных или адъювантных компонентах, которые используют при получении, или могут быть добавлены отдельно от этих компонентов.
Предпочтительные композиции согласно изобретению включают в себя один или более фармацевтический носитель(ей) и/или инертный наполнитель(ей).
Для регулировки тоничности, предпочтительно, включать физиологическую соль, такую как натриевая соль. Предпочтительным является хлорид натрия (NaCl), который может присутствовать между 1 и 20 мг/мл.
В общем случае, осмоляльность композиции составляет между 200 мОсм/кг и 400 мОсм/кг, предпочтительно, между 240-360 мОсм/кг, и более предпочтительно, попадает в диапазон 280-320 мОсм/кг. Как ранее сообщалось, осмоляльность не влияет на появление боли при вакцинации [74], но тем не менее соблюдение осмоляльности в этом диапазоне предпочтительно.
Композиции согласно изобретению могут включать в себя один или более буферы. Обычные буферы включают в себя: фосфорнокислый буфер; трис-буфер; борнокислый буфер; сукцинатный буфер; гистидиновый буфер или цитратный буфер. Количество буфера обычно будет составлять в диапазоне 5-2 мМ.
pH композиции согласно изобретению в общем случае составляет между 5,0 и 7,5, и обычно между 5,0 и 6,0 для оптимальной стабильности или, в случае использования дифтерийного анатоксина и/или столбнячного анатоксина, между 6,0 и 7,0. Поэтому способ согласно изобретению может включать в себя стадию доведения pH полученной вакцины перед упаковкой.
Композиции согласно изобретению, предпочтительно, стерильны.
Композиции согласно изобретению, предпочтительно, непирогенны, например, содержание <1 EU (эндотоксиновая единица, стандартная мера) на дозу, и, предпочтительно, <0,1 EU на дозу.
Композиции согласно изобретению, предпочтительно, не содержат глютена.
Из-за того что HBsAg адсорбирован, конечный вакцинный продукт может быть получен в виде мутной суспензии. Такой внешний вид означает, что микробная контаминация совсем не наблюдается, и таким образом вакцина, предпочтительно, содержит противомикробный компонент. Это особенно важно, если вакцину упаковывают в мультидозовые контейнеры. Предпочтительные используемые антимикробные компоненты представляют собой 2-феноксиэтанол и тимеросал. Однако предпочтительно, не использовать ртутные консерванты (например, тимеросал) при осуществлении способа согласно изобретению. Таким образом, от 1 до всех используемых в способе компонентов могут по существу не содержать ртутный консервант (особенно: двухвалентный D-T-компонент; IPV компонент; конъюгатный компонент). Однако присутствие следовых количеств неизбежно, если компонент (особенно HBsAg) был обработан таким консервантом до использования согласно изобретению. Однако для безопасности предпочтительным является конечная композиция, которая содержит менее чем приблизительно 25 нг/мл ртути. Более предпочтительно, конечный вакцинный продукт не содержит обнаруживаемых количеств тимеросала. В общем случае это достигается путем удаления ртутного консерванта из антигенного препарата перед его добавлением при выполнении способа согласно изобретению или избегая применения тимеросала при получении компонентов, используемых для получения композиции.
В случае использования двухвалентной смеси D-T при осуществлении способа согласно изобретению, она не должна содержать тимеросал. В некоторых вариантах выполнения изобретения, D-T смесь может включать в себя 2-феноксиэтанол, но в других она не содержит ни тимеросал, ни 2-феноксиэтанол. В случае использования трехвалентной D-T-Pw смеси при осуществлении способа согласно изобретению, она не должна содержать 2-феноксиэтанол, но может включать в себя тимеросал.
При получении разбавления компонентов до требуемых конечных концентраций обычно выполняют WFI (вода для инъекции).
Концентрация фосфата алюминия в композиции согласно изобретению в пересчете на Al3+, предпочтительно, составляет менее чем 5 мг/мл, например, ≤4 мг/мл, ≤3 мг/мл, ≤2 мг/мл, ≤1 мг/мл и т.д.
Концентрация HBsAg в композиции согласно изобретению, предпочтительно, составляет менее чем 60 мкг/мл, например ≤55 мкг/мл, ≤50 мкг/мл, ≤45 мкг/мл, ≤40 мкг/мл и т.д. Обычная концентрация составляет приблизительно 20 мкг/мл.
Концентрация дифтерийного анатоксина в композиции согласно изобретению обычно составляет по меньшей мере 50 МЕ/мл. Концентрация столбнячного анатоксина в композиции согласно изобретению обычно составлет по меньшей мере 100 МЕ/мл.
Соотношение дифтерийного анатоксина к столбнячному анатоксину в композициях согласно изобретению обычно составляет между 2:1 и 3:1 (измеренное в Lf единицах), предпочтительно, между 2,4:1 и 2,6:1, и наиболее предпочтительно, 2,5:1.
Количество антигена wP в композициях согласно изобретению обычно составляет по меньшей мере 8 МЕ/мл.
Количество конъюгата Hib, измеренного как сахарид, в композициях согласно изобретению обычно составляет между 10 и 30 мкг/мл.
Количество HAV антигена, измеренного в EU (единицы Elisa), обычно составляет по меньшей мере 600 EU/мл.
Количество IPV антигена зависит от серотипа штамма. Для вируса типа 1 композиция обычно содержит приблизительно 80 DU/мл. Для вируса типа 2 композиция обычно содержит приблизительно 16 DU/мл. Для вируса типа 3 композиция обычно содержит приблизительно 65 DU/мл.
Количество менингококкового конъюгата, измеренного как сахарид, в композициях согласно изобретению обычно составляет между 5 и 25 мкг/мл для каждой серогруппы.
Количество пневмококкового конъюгата, измеренного как сахарид, в композициях согласно изобретению обычно составляет между 2 и 20 мкг/мл для каждого серотипа.
Композиции согласно изобретению, предпочтительно, вводятся пациентам в дозе 0,5 мл. Указание дозы в 0,5 мл подразумевает, включение нормального отклонения, например, 0,5 мл±0,05 мл.
Изобретение может предоставлять продукт, который можно упаковывать на отдельные дозы для доставки для введения пациентам. Упомянутые выше концентрации представляют собой обычные концентрации в полученных упакованных дозах, и таким образом концентрации в полученной вакцине может быть выше (например, для уменьшения до конечных концентраций разбавлением).
В общем случае композиции согласно изобретению будут в водном виде.
Остаточные вещества от отдельных антигенных компонентов также могут присутствовать в следовых количествах в конечной вакцине, полученной способом согласно изобретению. Например, если для получения дифтерийного, столбнячного и коклюшного анатоксинов используют формальдегид, то конечный вакцинный продукт может содержать следовые количества формальдегида (например, меньше чем 10 мкг/мл, предпочтительно, <5 мкг/мл). При получении препаратов полиовируса возможно использование сред или стабилизаторов (например, среда 199), и они могут попасть в конечную вакцину. Аналогично, свободные аминокислоты (например, аланин, аргинин, аспартат, цистеин и/или цистин, глутамат, глютамин, глицин, гистидин, пролин и/или гидроксипролин, изолейцин, лейцин, лизин, метионин, фенилаланин, серин, треонин, триптофан, тирозин и/или валин), витамины (например, холин, аскорбат и т.д.), динатрий фосфат, первичный кислый фосфат калия, кальций, глюкоза, сульфат аденина, феноловый красный, натрия ацетат, калия хлорид и т.д. могут присутствовать в конечной вакцине в количестве ≤100 мкг/мл, предпочтительно, <10 мкг/мл, каждая. Другие компоненты антигенных препаратов, такие как неомицин (например, сульфат неомицина, особенно от IPV компонента), полимиксин B (например, полимиксин B сульфат, особенно от IPV компонента), и т.д. могут также присутствовать в субнанограммовых количествах на дозу. Возможные дополнительные компоненты конечной вакцины, которые содержатся в антигенных препаратах, являются результатом в количестве меньшем, чем имеет место при обычной очистке антигенов. Могут присутствовать малые количества белков и/или геномных ДНК B.pertussis, C.diphtheriae, C.tetani и S.cerevisiae. Чтобы минимизировать количества этих остаточных компонентов, антигенные препараты, предпочтительно, обрабатывают для их удаления, перед использованием антигенов в способе согласно изобретению.
В случае, если используют IPV компонент, его обычно выращивают на Vero клетках. Конечная вакцина, предпочтительно, содержит меньше чем 10 нг/мл, предпочтительно, ≤1 нг/мл, например, ≤500 пг/мл или ≤50 пг/мл ДНК Vero клетки, например, меньше чем 10 нг/мл ДНК Vero клетки, длина которой составляет ≥50 пар оснований.
Упаковка композиций согласно изобретению
После объединения HBsAg и адъювантов, способов согласно изобретению может включать в себя стадию извлечения и упаковки 0,5 мл образца смеси в контейнер. В случае мультидозового контейнера, количества многократных доз извлекают и упаковывают вместе в одном контейнере.
Способ согласно изобретению может включать в себя дополнительную стадию упаковки вакцины в контейнер для использования. Подходящие контейнеры включают в себя ампулы и шприцы одноразового применения (предпочтительно, стерильные).
В случае, если композиции согласно изобретению упакованы в ампулы, они, предпочтительно, выполнены из стекла или пластикового материала. Стерилизацию ампул, предпочтительно, проводят до заполнения их композицией. Для того чтобы избежать проблем с чувствительными к латексу пациентами, ампулы, предпочтительно, закупоривают пробкой, не содержащей латекса. Ампула может включать в себя одну дозу вакцины или она может включать в себя более чем одну дозу ('мультидозовая' ампула), например, на 10 доз. В случае использования мультидозовой ампулы, каждую дозу извлекают стерильной иглой и шприцом в строгих асептических условиях, стараясь избежать контаминации содержимого ампулы. Предпочтительно, ампулы выполнены из бесцветного стекла.
Ампула может иметь колпачок (например, наконечник Люэра), адаптированный таким образом, что предварительно набранный шприц может быть вставлен в колпачок, и содержимое шприца может быть введено в пробирку (например, для восстановления лиофилизированного материала), и содержимое ампулы может быть введено обратно в шприц. После удаления шприца из ампулы, можно присоединить иглу и композиция может быть введена пациенту. Колпачок, предпочтительно, расположен внутри наконечника или крышки, так что наконечник или крышка должны быть удалены для получения доступа к колпачку.
В случае, если композиция упакована в шприц, шприц обычно не содержит присоединенную к нему иглу, хотя отдельная игла может поставляться со шприцом как набор и для применения. Предпочтительными являются безопасные иглы. Обычными являются 1-дюймовые номер 25, 1-дюймовые номер 23 и 5/8-дюймовые номер 25 иглы. На шприцы могут быть нанесены удаляемые метки, на которых напечатаны номер партии и срок годности содержимого, для облегчения ведения протокола. Поршень в шприце, предпочтительно, имеет ограничитель для предовращения случайного удаления поршня во время применения. В шприцах может быть латексный резиновый колпачок и/или поршень. Одноразовые шприцы содержат одну дозу вакцины. В общем случае на шприце будет колпачок для герметичного крепления иглы и колпачок, предпочтительно, выполнен из бутилкаучука. Если шприц и игла упакованы отдельно, то, предпочтительно, игла оснащена бутилкаучуковой крышкой. Предпочтительным является серый бутилкаучук. Предпочтительными шприцами являются таковые, продаваемые под торговой маркой "Tip-Lok"™.
В случае использования стеклянных емкостей (например, шприца или ампулы), предпочтительно, использовать емкости, выполненные из боросиликатного стекла, а не из известково-натриевого стекла.
После упаковки композиции в емкость, емкость может быть помещена в коробки для сбыта, например в картонные коробки, и затем коробки маркируют характеристиками вакцины, например торговое название, список антигенов в вакцине (например, 'рекомбинантный гепатит B' и т.д.), назначение содержимого (например, 'Одноразовые предварительно наполненные шприцы Tip-Lok' или '10×0,5 мл ампул с однократной дозой), дозу (например, 'каждая содержит одну дозу 0,5 мл'), предупреждения (например, 'только для взрослых' или 'только для педиатрического применения), срок годности, показания (например, 'активная иммунизация против инфекции вируса гепатита B (HBV), вызванной всеми известными подтипами для пациентов с почечной недостаточностью (включая предгемодиализные и гемодиализные) для пациентов в возрасте от 15 лет и старше' и т.д.), номер патента и т.д. Каждая коробка может содержать более одной упакованной вакцины, например пять или десять упакованных вакцин (особенно в случае ампул). Если вакцина содержится в шприце, то на упаковке может быть изображение шприца.
Вакцина может быть упакована вместе (например, в одной коробке) с листком, указывающим характеристики вакцины, например, инструкцию для введения, характеристики антигенов, находящихся в вакцине и т.д. Инструкции могут также содержать предупреждения, например, иметь быстро доступный раствор адреналина в случае анафилактической реакции после вакцинации и т.д. Упакованную вакцину, предпочтительно, хранят при между 2°C и 8°C. Она не должна быть заморожена.
Вакцины могут быть обеспечены в полностью жидкой форме (например, когда все антигенные компоненты находятся в водном растворе или суспензии) во время получения, или они могут быть получены в форме, когда некоторые компоненты находятся в жидкой форме, а другие находятся в лиофилизированной форме. Таким образом, конечная вакцина может быть получена для немедленного приема путем смешивания вместе двух компонентов: (a) первого компонента, включающего в себя водные антигены; и (b) второго компонента, включающего в себя лиофилизированные антигены. Эти два компонента, предпочтительно, находятся в отдельных емкостях (например, ампулах и/или шприцах), и изобретение обеспечивает набор, включающий в себя компоненты (a) и (b). Этот формат особенно полезен в случае вакцин, включающих в себя конъюгатный компонент, особенно Hib и/или менингококковый и/или пневмококковый конъюгат, поскольку они более стабильны в лиофилизированном виде. Таким образом, конъюгаты могут быть лиофилизированы до их применения согласно изобретению. Дополнительные компоненты также могут быть добавлены до сушки сублимацией, например, как стабилизаторы. Предпочтительные используемые стабилизаторы представляют собой лактозу, сахарозу и маннит, а также их смеси, например смеси лактозы/сахарозы, смеси сахарозы/маннита и т.д. Таким образом, конечная вакцина может содержать лактозу и/или сахарозу. Использование смеси сахароза/маннит может ускорить сушку.
Таким образом, изобретение обеспечивает способ получения двухконтейнерной комбинированной вакцины, включающей в себя следующие стадии:
- получение водной комбинированной вакцины как описано выше, но где упомянутый один или более антигенов не включают в себя антиген конъюгированного капсульного сахарида;
- упаковка указанной комбинированной вакцины в первый контейнер (например, шприц);
- получение антигена конъюгированного капсульного сахарида в лиофилизированной форме;
- упаковка указанного лиофилизированного антигена во второй контейнер (например, ампулу); и
- упаковка первого контейнера и второго контейнера в набор.
Затем набор может быть поставлен врачам.
Компоненты D, T, P и HbsAg, предпочтительно, находятся в жидкой форме.
Способы лечения и введения вакцины
Композиции согласно изобретению являются подходящими для введения человеку, и изобретение обеспечивает способ усиления иммунного ответа у пациента, включающий в себя стадию введения пациенту композиции согласно изобретению.
Также изобретение обеспечивает композиции согласно изобретению для применения в медицине.
Также изобретение обеспечивает применение (i) очищенного HBsAg от рекомбинантных дрожжевых клеток, где способ очистки включает в себя разрушение дрожжевых клеток в присутствии неионогенного поверхностно-активного вещества, и (ii) одного или более антигенов не-HBV, при получении лекарственного средства для введения пациенту.
Иммуногенные композиции согласно изобретению, предпочтительно, представляют собой вакцины, используемые для профилактики и/или лечения по меньшей мере вирусной инфекции гепатита B. У пациентов, получавших композиции согласно изобретению, предпочтительно титр, измеренный спустя 6 недель после первой иммунизации, анти-HBsAg GM в сыворотке составляет ≥500 мМЕ/мл. Более предпочтительно, титр составляет ≥500 мМЕ/мл, при измерении спустя 12 месяцев.
Для того чтобы получить наиболее полную эффективность, обычный график первичной вакцинации для ребенка может включать в себя введение более чем одной доза. Например, дозы могут быть введены в: 0 и 6 месяцев (время 0 соответствует введению первой дозы); в 0, 1, 2 и 6 месяцев; в 0 день, 21 день и затем третья доза между 6 и 12 месяцами; в 2, 4 и 6 месяцев; в 3, 4 и 5 месяцев; в 6, 10 и 14 недель или в 0, 1, 2, 6 и 12 месяцев.
Также композиции могут быть использованы в качестве ревакцинирующих доз, например, для детей двух лет.
Композиции согласно изобретению могут быть введены внутримышечной инъекцией, например, в руку или ногу.
Вакцины, полученные в соответствии с изобретением, могут быть введены пациентам одновременно с пневмококковой конъюгированной вакциной, такой как Prevnar™.
В случае, если композиции согласно изобретению включают в себя адъювант на основе алюминия, может происходить осаждение компонентов во время хранения. Композиции следует взбалтывать до введения пациенту. Такая композиция будет представлять собой мутную белую суспензию.
Предпочтительные вакцины
Конкретные варианты поливалентных иммуногенных композиций согласно изобретению включают в себя:
• пятивалентная композиция, включающая в себя HBsAg, D, T, Ра и IPV. Вакцина представлена в водной фазе. Она включает в себя в качестве адъювантов как фосфат алюминия, так и гидроксид алюминия. HBsAg адсорбирован на фосфате алюминия. D, T и Pa адсорбированы на гидроксиде алюминия. Количества на мл: приблизительно 50 Lf дифтерийного анатоксина; приблизительно 20 Lf столбнячного анатоксина; приблизительно 50 мкг PT; приблизительно 50 мкг FHA; приблизительно 16 мкг пертактина; приблизительно 20 мкг HBsAg; приблизительно 80 DU полиовируса типа 1; приблизительно 16 DU полиовируса типа 2; приблизительно 64 DU полиовируса типа 3. Доза: приблизительно 0,5 мл. Может присутствовать в предзаполненном шприце.
• пятивалентная композиция, включающая в себя HBsAg, D, T, Ра и IPV. Вакцина представлена в водной фазе. Она включает в себя в качестве адъювантов как фосфат алюминия, так и гидроксид алюминия. HBsAg адсорбирован на фосфате алюминия. D, T и Pa адсорбированы на гидроксиде алюминия. Количества на мл: по меньшей мере 60 МЕ дифтерийного анатоксина; по меньшей мере 80 МЕ столбнячного анатоксина; приблизительно 50 мкг PT; приблизительно 50 мкг FHA; приблизительно 16 мкг пертактина; приблизительно 20 мкг HBsAg; приблизительно 80 DU полиовируса типа 1; приблизительно 16 DU полиовируса типа 2; приблизительно 64 DU полиовируса типа 3. Доза: приблизительно 0,5 мл. Может находиться в предварительно заполненном шприце.
• четырехвалентная композиция, включающая в себя HBsAg, D, T и Pw. Компоненты представлены в водной фазе. Она включает в себя в качестве адъювантов как фосфат алюминия, так и гидроксид алюминия. HBsAg адсорбирован на фосфате алюминия. D и T адсорбированы на гидроксиде алюминия. Композиция включает тимеросал, но, предпочтительно, не содержит 2-феноксиэтанол. Количества на мл: по меньшей мере 60 МЕ дифтерийного анатоксина; по меньшей мере 120 МЕ столбнячного анатоксина; по меньшей мере 8 МЕ Pw; приблизительно 20 мкг HBsAg. Доза: приблизительно 0,5 мл.
• пятивалентная композиция, включающая в себя HBsAg, D, T, Pw и конъюгат Hib-T. Компоненты HBsAg, D, T и Pw представлены в водной фазе; Hib-T лиофилизирован. Она включает в себя в качестве адъювантов как фосфат алюминия, так и гидроксид алюминия. D и T адсорбированы на гидроксиде алюминия. HBsAg и Hib-T адсорбированы на фосфате алюминия. Лиофилизированный Hib-T включает в себя лактозу. Водный компонент может включать в себя тимеросал. Количества на мл: по меньшей мере 60 МЕ дифтерийного анатоксина; по меньшей мере 120 МЕ столбнячного анатоксина (плюс между 5-25 мкг столбнячного анатоксина в качестве носителя Hib-T); по меньшей мере 8 МЕ Pw; приблизительно 20 мкг HBsAg; приблизительно 5 мкг Hib-T, измеренного как сахарид. Доза: приблизительно 0,5 мл.
• семивалентная композиция, включающая в себя HBsAg, D, T, Pw и три конъюгата: конъюгат конъюгат Hib-T, конъюгат MenA и конъюгат MenC. Компоненты HbsAg, D, T и Pw представлены в водной фазе; три конъюгата лиофилизированы. Она включает в себя в качестве адъювантов как фосфат алюминия, так и гидроксид алюминия. D и T адсорбированы на гидроксиде алюминия. HBsAg адсорбирован на фосфате алюминия. Лиофилизированный компонент может включать в себя лактозу и/или сахарозу. Водный компонент может включать в себя тимеросал. Потенциальные количества на мл: по меньшей мере 60 МЕ дифтерийного анатоксина; по меньшей мере 120 МЕ столбнячного анатоксина (плюс между 5-25 мкг столбнячного анатоксина как носителя Hib-T); по меньшей мере 8 МЕ Pw; приблизительно 20 мкг HBsAg; приблизительно 5 мкг каждого конъюгата, измеренного как сахарид. Доза: приблизительно 0,5 мл.
Эти композиции могут быть использованы самостоятельно в качестве вакцин или в качестве компонентов для других вакцин. Например, изобретение обеспечивает шестивалентную композицию, включающую в себя пятивалентную композицию HBsAg-D-T-Ра-IPV, описанную выше, плюс лиофилизированный конъюгат Hib-T. Лиофилизированный Hib-T, предпочтительно, не адсорбирован на соли алюминия. Изобретение также обеспечивает семивалентную композицию, включающую в себя пятивалентную композицию HBsAg-D-T-Pa-IPV, описанную выше, плюс лиофилизированные конъюгаты Hib-T и MenC. Изобретение также обеспечивает восьмивалентную композицию, включающую в себя пятивалентную композицию HBsAg-D-T-Pa-IPV, описанную выше, плюс лиофилизированные конъюгаты Hib-T, MenC и MenY. Изобретение также обеспечивает пятивалентную композицию, включающую в себя четырехвалентную композицию HBsAg-D-T-Pw, описанную выше, плюс лиофилизированный конъюгат Hib-T. Изобретение также обеспечивает семивалентную композицию, включающую в себя четырехвалентную композицию HBsAg-D-T-Pw, описанную выше, плюс лиофилизированная смесь конъюгата Hib-T, конъюгата MenA и конъюгата MenC. Конечные вакцины могут быть получены реконструированием лиофилизированного материала водными HBsAg-содержащими материалами при применении, как лиофилизированные, так и водные компоненты, предпочтительно, упакованы вместе в наборе, как описано выше.
Конкретные способы согласно изобретению включают в себя таковые, которые включают в себя следующие стадии:
• Очистка HBsAg в соответствии с изобретением; адсорбция HBsAg на адъюванте из фосфата алюминия; получение не содержащей тимеросал двухвалентной D-T смеси с адъювантом из гидроксида алюминия; получение PT, FHA и пертактин для Ра компонента; получение IPV антигенов, как смешанных типов 1, 2 и 3, предпочтительно, без адъюванта соли алюминия; объединение D-T, Pa IPV и HBsAg в любом порядке, в итоге получая пятивалентную комбинацию; при необходимости, упаковка в шприц.
• Очистка HBsAg в соответствии с изобретением; адсорбция HBsAg на адъюванте из фосфата алюминия; получение не содержащей 2-феноксиэтанол и содержащей тимеросал трехвалентной D-T-Pw смеси с адъювантами фосфата алюминия и гидроксидом алюминия; объединение D-T-Pw и HBsAg для получения конечной четырехвалентной комбинации; если требуется, упаковка в шприц; если требуется, упаковка в комбинации с лиофилизированным конъюгированным компонентом(ами), например Hib-T, MenA, MenC.
• Очистка HBsAg в соответствии с изобретением; адсорбция HBsAg на адъюванте из фосфата алюминия; получение не содержащей 2-феноксиэтанол и содержащей тимеросал трехвалентной D-T-Pw смеси с адъювантами фосфата алюминия и гидроксидом алюминия; получение лиофилизированного конъюгата Hib-T; объединение D-T-Pw и HBsAg для получения водного четырехвалентного компонента; упаковка водного четырехвалентного компонента в стеклянную ампулу; упаковка лиофилизированного Hib-T и/или MenC и/или MenY в стеклянную ампулу; объединение этих двух ампул в одном наборе для восстановления с получением пятивалентной комбинированной вакцины. Стеклянные ампулы могут быть выполнены из стекла типа 1 и иметь резиновые бутилкаучуковые пробки.
• Очистка HBsAg в соответствии с изобретением; адсорбция HBsAg на адъюванте из фосфата алюминия; получение не содержащей 2-феноксиэтанол и содержащей тимеросал трехвалентной D-T-Pw смеси с адъювантами фосфата алюминия и гидроксидом алюминия; получение антигенов IPV, смешанных типов 1, 2 и 3, предпочтительно, без адъюванта соли алюминия; объединение D-T-Pw, IPV и HBsAg в любом порядке для получения конечной пятивалентной комбинации; если требуется, упаковка в шприц; если требуется, упаковка в комбинации с лиофилизированным конъюгированным компонентом(ами), например Hib-T, MenA, MenC.
Дополнительные компоненты могут быть добавлены на любой стадии, например хлорид натрия, адъюванты, консерванты и т.д. Такие способы могут быть использованы, например, для получения вакцин, описанных выше.
Различные стадии способа могут быть выполнены по существу одновременно или по отдельности. Они могут быть выполнены в одном месте или в различных местах, даже, например, в различных странах. Очистку HBsAg и лиофилизацию Hib-T можно проводить в разных местах.
Белки-носители для конъюгатов
Конъюгированные сахаридные антигены включают в себя белок-носитель, к которому ковалентно присоединен сахарид, или напрямую, или посредством линкера. Общие сведения о способах конъюгирования могут быть найдены в документе 45.
Для применения в качестве носителей известны различные белки, и предпочтительными белками-носителями являются бактериальные токсины или анатоксины, такие как дифтерийный анатоксин или столбнячный анатоксин. Другие подходящие белки-носители без ограничения включают в себя мутантный дифтерийный токсин CRM197 [75-77], белок наружной мембраны N.meningitidis [78], синтетические пептиды [79,80], белки теплового шока [81, 82], коклюшные белки [83, 84], цитокины [85], лимфокины [85], гормоны [85], факторы роста [85], искусственные белки, включающие в себя множественные эпитопы CD4+ Т-клеток человека от различных патогенных антигенов [86], таких как N19 [87], D белок от H.influenzae [88, 89], пневмококковый поверхностный белок PspA [90], пневмолизин [91], белки захвата железа [92], токсин A или B от C.difficile [93], белки S.agalactiae [94] и т.д.
Сахарид к носителю, предпочтительно, присоединен посредством -NH2 группы, например, в боковой цепи остатка лизина в белке-носителе или остатка аргинина. Также возможно присоединение посредством -SH групп (например, в боковой цепи цистеина).
Предпочтительными являются конъюгаты с соотношением сахарид:белок (вес./вес.), составляющим между 1:5 (т.е. избыток белка) и 5:1 (т.е. избыток сахарида).
Композиции могут включать в себя небольшое количество свободного носителя. Не включая носитель, добавленный как отдельный антиген, количество несвязанного носителя, предпочтительно, составляет не больше чем 5% от общего количества белка-носителя в композиции, и более, предпочтительно, составляет меньше чем 2% по массе.
Возможно включать более одного типа белка-носителя в композицию, например, для уменьшения риска супрессии носителем.
Белок-носитель для N.meningitidis конъюгатов может представлять собой белок D от Н.influenzae. Этот белок более подробно описан в документах 95 и 96, а его применение в качестве белка-носителя в конъюгатах описано в документе 97. Термин "белок D" включает в себя фрагменты нативного непроцессированного белка, как раскрыто в документе 97, а также слитые белки, включающие в себя или непроцессированный белок D или его фрагменты (например, слияние фрагмента белка вируса гриппа NS1 и фрагмента белка D). При конъюгировании фрагменты сохраняют способность конвертировать T-независимые сахаридные антигены в T-зависимые антигены. Обычные фрагменты включают в себя по меньшей мере 1/3 N-конца белка D. Белок может быть экспрессирован в E.coli [96], и для применения в изобретении такой рекомбинантный материал предпочтителен [97].
Общие сведения
Термин "включающий" ("comprising") охватывает как "включающий" ("including"), так и "состоящий" ("consisting"), например, композиция, "включающая в себя" X может состоять исключительно из X или может включать в себя что-либо дополнительно, например, X+Y.
Слово "по существу" не исключает "полностью", например, композиция, которая "по существу не содержит" Y может быть вообще не содержать Y. Если требуется, слово "по существу" может быть опущено при определении изобретения.
Термин "приблизительно" при числовом обозначении x означает, например, x±10%.
Если конкретно не определено, способ, включающий в себя стадию смешивания двух или больше компонентов, не требует указания специфического порядка смешивания. Таким образом, компоненты могут быть смешаны в любом порядке. В случае, если используют три компонента, тогда два компонента могут быть объединены друг с другом, а затем комбинация может быть объединена с третьим компонентом, и т.д.
Когда антиген описан как "адсорбированный" на адъюванте, предпочтительно, что по меньшей мере 50% (по массе) этого антигена адсорбированы, например, 50%, 60%, 70%, 80%, 90%, 95%, 98% или больше. Предпочтительно, чтобы дифтерийный анатоксин и столбнячный анатоксин были оба полностью адсорбированы, т.е. ни один не обнаруживается в супернатанте. Может быть использована полная адсорбция HBsAg.
Количества дифтерийного анатоксина могут быть выражены в международных единицах (МЕ). Например, NIBSC поставляет 'Diphtheria Toxoid Adsorbed Third International Standard 1999' [98,99], который содержит 160 МЕ на ампулу. В качестве альтернативы системе МЕ, используют единицы Lf ("единицы флокуляции" или "известковую флокуляционную дозу"), определяемые как количество анатоксина, которое при смешивании с одной международной единицей антитоксина, образует оптимальную флокуляционную смесь [100]. Например, NIBSC поставляет 'Diphtheria Toxoid, Plain' [101], который содержит 300 LF на ампулу, а также 'The 1st International Reference Reagent For Diphtheria Toxoid For Flocculation Test' [102], который содержит 900 LF на ампулу.
Количества столбнячного анатоксина могут быть выражены в международных единицах (МЕ). Например, NIBSC поставляет 'Tetanus Toxoid Adsorbed Third International Standard 2000' [103,104], который содержит 469 МЕ на ампулу. В качестве альтернативы системе МЕ, используют единицы Lf ("единицы флокуляции" или "limes flocculating dose"), определяемые как количество анатоксина, которое при смешивании с одной международной единицей антитоксина, образует оптимальную флокуляционную смесь [100]. NIBSC поставляет 'The 1st International Reference Reagent for Tetanus Toxoid For Flocculation Test' [105], который содержит 1000 LF на ампулу.
Количества антигенов wP могут быть выражены в международных единицах (МЕ). Например, NIBSC поставляет 'Third International Standard For Pertussis Vaccine' [106], который содержит 46 МЕ на ампулу. Каждая ампула содержит высушенный замораживанием остаток 2 мл аликвоты водного раствора, который содержал 10 литров бактерийной суспензии (эквивалентно 180 единицам непрозрачности согласно стандарту непрозрачности США) разбавленной восьмью литрами M/15 буфера Соренсена с pH 7,0. В качестве альтернативы системе МЕ, также используют единицы 'OU' ("единицы непрозрачности") (например, 4 OU составляют приблизительно 1 МЕ).
Количества конъюгата в общем случае указываются в пересчете на массу сахарида (т.е. доза конъюгата (носитель+сахарид) в целом выше заявленной дозы) для того, чтобы избежать варьирования при выборе носителя.
В случае использования в культуре клеток животного (и, в частности, коровьего) материала, он должен быть получен из источника, не содержащего передающихся спонгиозных энцефалопатий (TSE), и, в частности, не содержащего бычей спонгиозной энцефалопатии (BSE).
КРАТКОЕ ОПИСАНИЕ ФИГУР
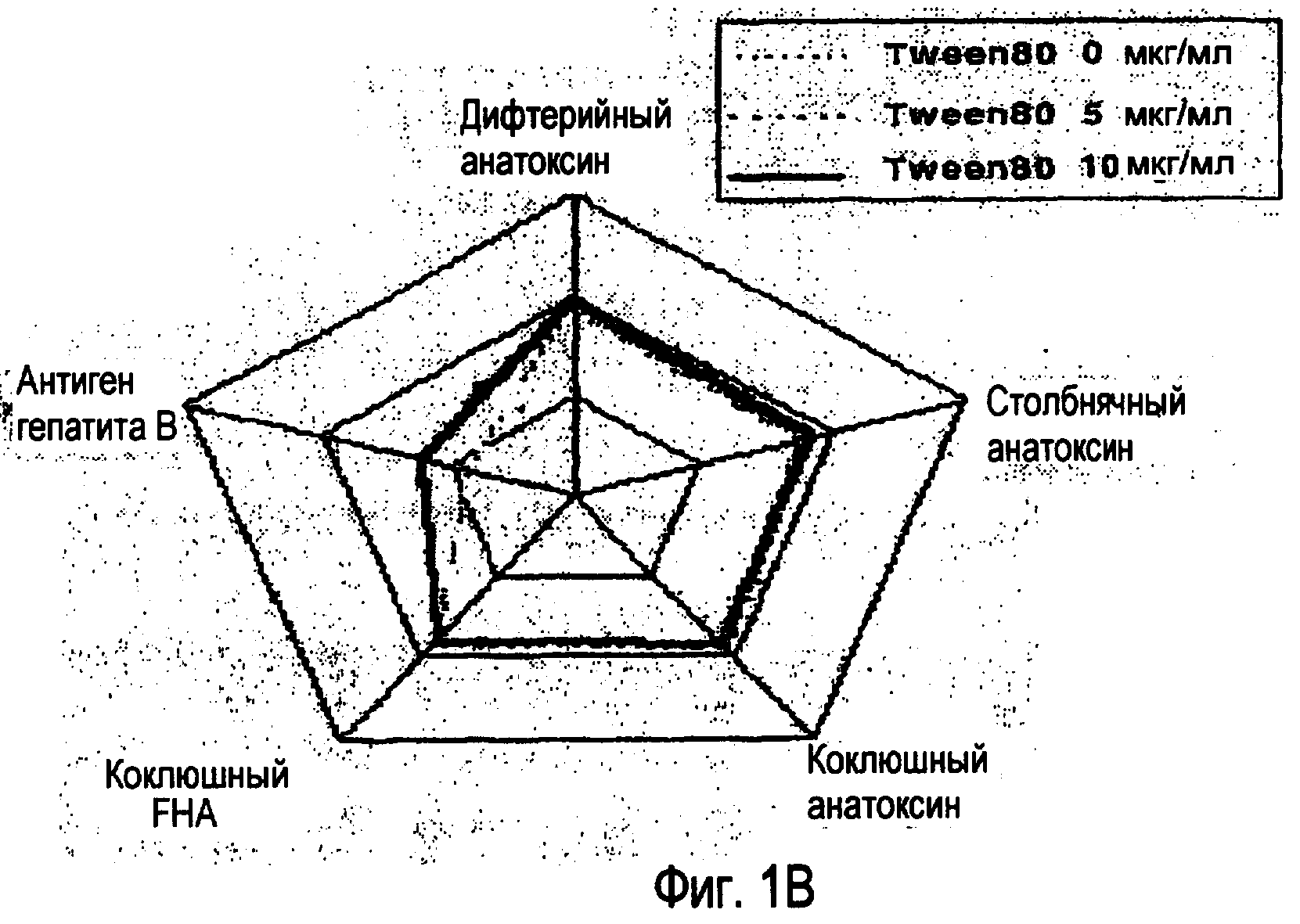
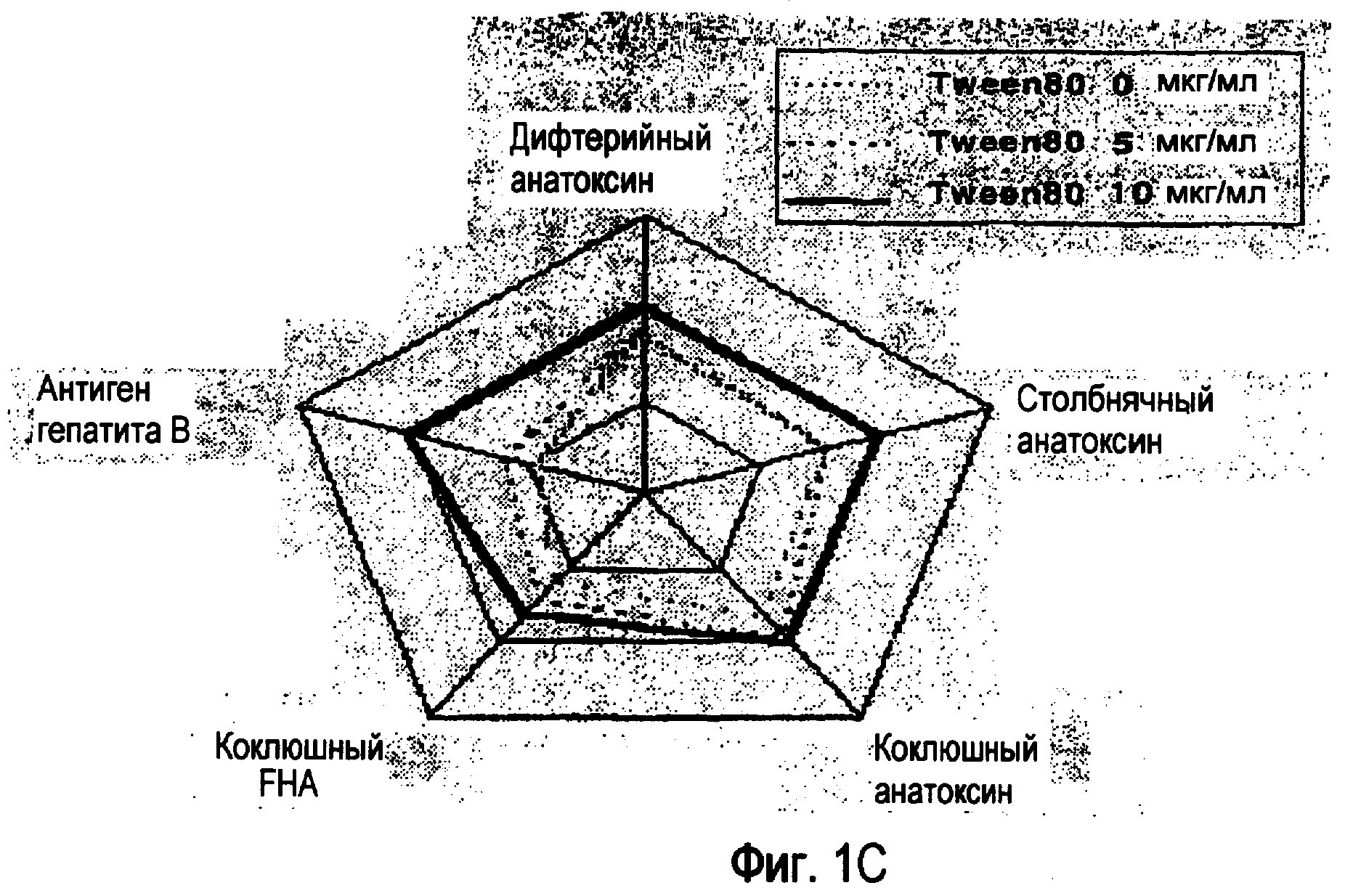
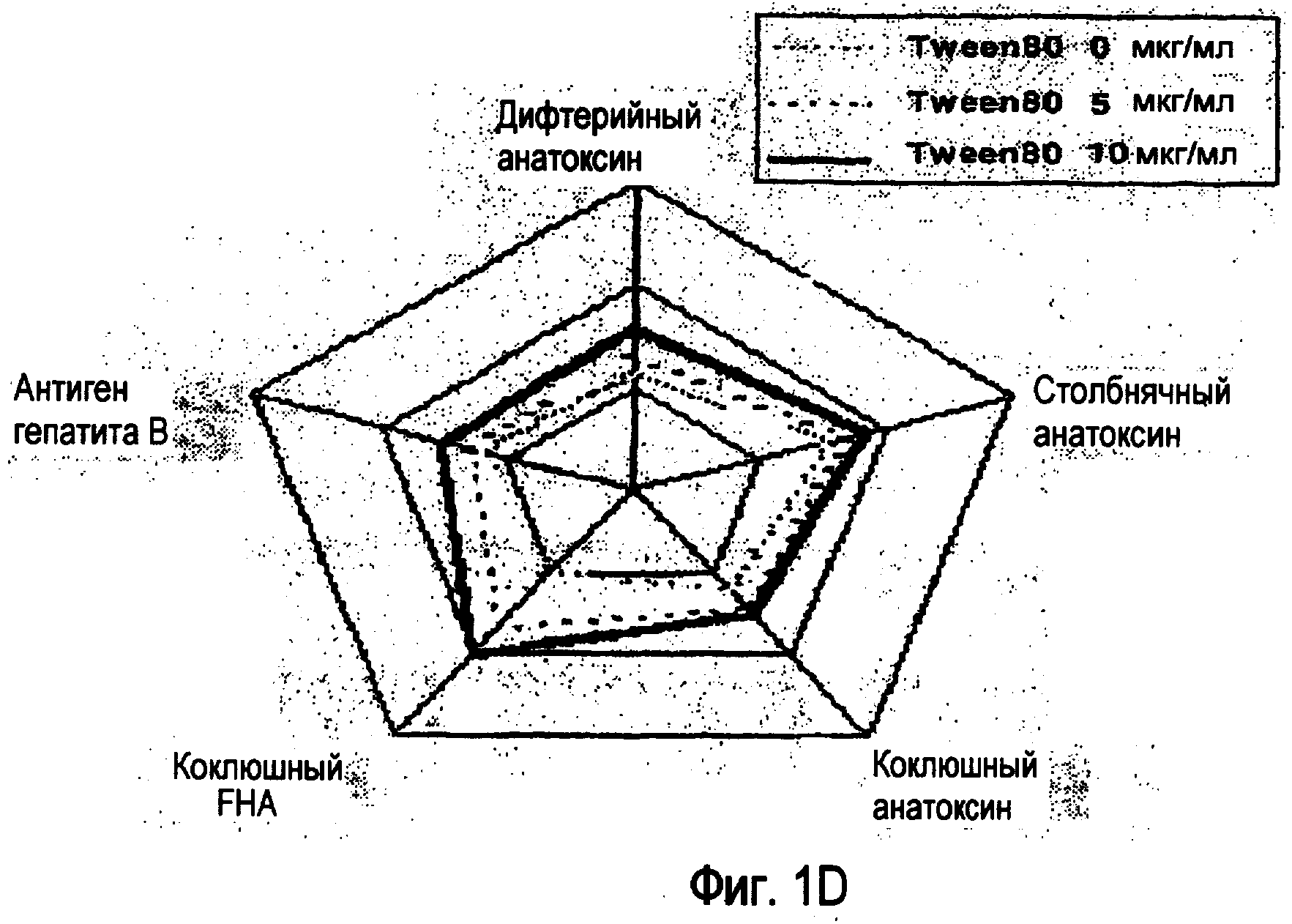
Фигура 1 является копией фигуры 2 из документов 3 и 4, в которых раскрывается, что фигура поясняет "антигенность и иммунитет, на основе адсорбционного способа, где образец 1 представляет собой образец, в котором добавлен избыток геля оксида алюминия после объединения составляющих вакцины, и образец 2 представляет собой образец, в котором упомянутый адсорбент добавлен той же концентрации до объединения". Фигуры 1A и 1B иллюстрируют относительную антигенность образцов 1 и 2 соответственно. Фигуры 1C и 1D иллюстрируют относительный уровень образования антител против каждого антигена образцов 1 и 2 соответственно.
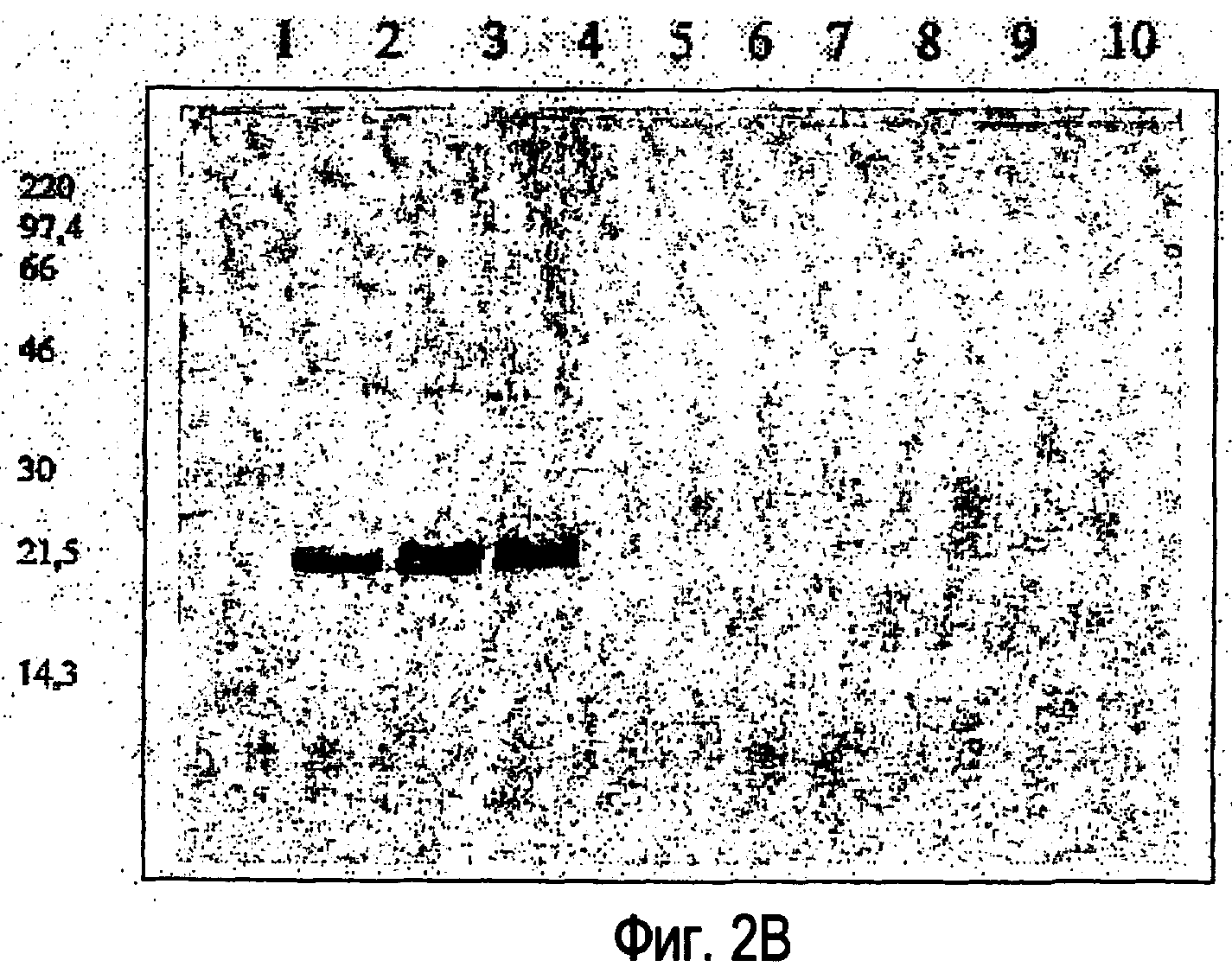
Фигура 2 показывает результаты вестерн-блоттинга по стабильности HBsAg в композиции. На фигуре 2A полосы означают: (1) образец буфера Лэммли (LSB); (2) MW маркеры; (3-5) 1 мкг трех отдельных контрольных препаратов HBsAg; (6-8) супернатант трех отдельных пятивалентных партий; (9-10) LSB. На фигурах 2B и 2C полосы означают: (1) MW маркеры; (2-4) 1 мкг трех отдельных контрольных препаратов HBsAg; (5-7) супернатант трех отдельных пятивалентных партий, хранившихся в течение 2 недель при 2-8°C; (8-10) супернатант трех отдельных пятивалентных партий, хранившихся в течение 2 недель при 36-38°C. На фигуре 2D полосы означают: (1) MW маркеры; (2) контрольный HBsAg; (3-5) супернатант трех отдельных пятивалентных партий.
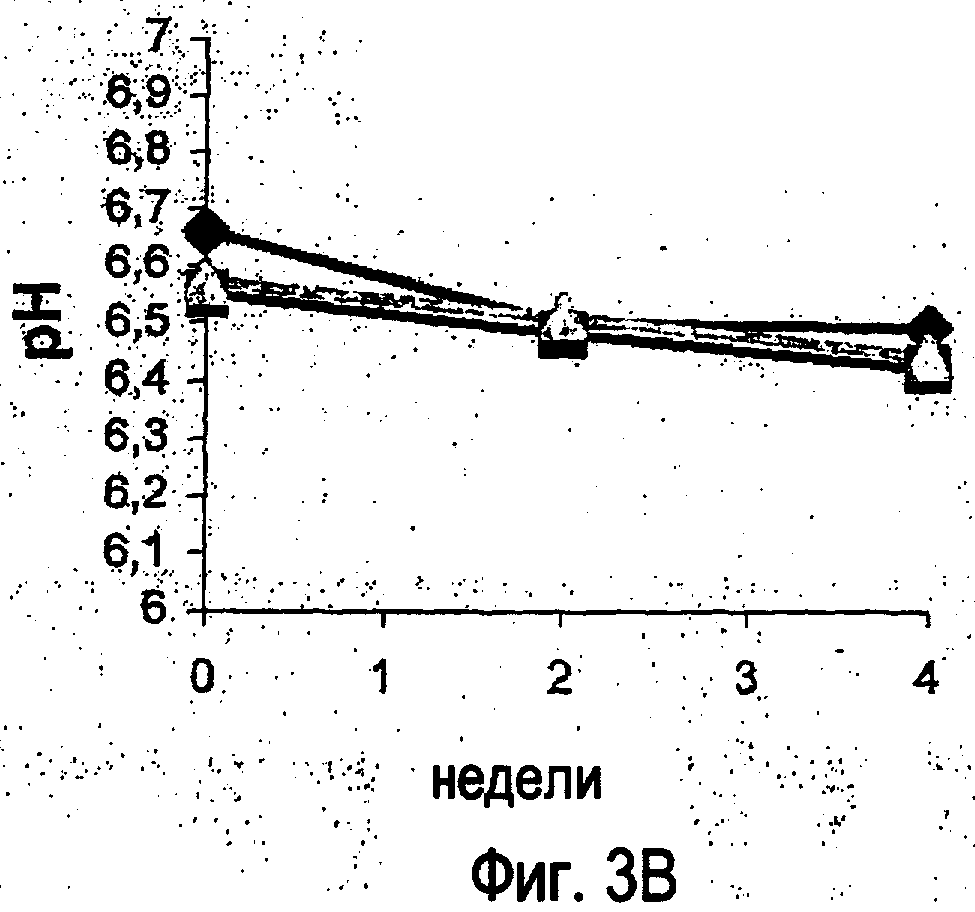
На фигуре 3 показано изменение pH пятивалентной композиции с течением времени.
На фигуре 4 показаны результаты вестерн-блоттинга стабильности HBsAg в восьмивалентной композиции. На фигурах 4A и 4B: полоса 1 содержит MW маркеры; полоса 2 содержит контрольный HBsAg 1 мкг/мл; полоса 3 содержит супернатант восьмивалентной композиции. На фигуре 4B: полоса 4 содержит LSB; полоса 5 содержит такой же контроль как в полосе 2; полоса 6 содержит экстракт DOC/TCA.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Экспрессия HBsAg и очистка
Были получены дрожжевые хозяева H.polymorpha, кодирующие HBsAg [107,108]. Готовили 100 литров среды в ферментере на 300 литров и инокулировали с дрожжами. Ферментацию продолжали до тех пор, пока содержание клеток составляло 100 грамм/литр. На этой стадии, поскольку хозяин является метилотрофом, добавляли метанол и прекращали ферментацию. Объем конечной культуры составлял 160-170 литров. Клетки отделяли от питательной среды центрифугированием.
Вместо добавления неионогенных поверхностно-активных веществ к HBsAg после очистки, как описано в документах 3 и 4, поверхностно-активное вещество использовали во время очистки белка. В частности, собранные дрожжевые клетки суспендировали в фосфатном буфере, содержащем 0,1-0,2% полисорбата 20 и 10 мМ EDTA. Суспензию охлаждали и разрушали, используя гомогенизатор высокого давления для жидкостей.
Затем гомогенизированную клеточную суспензию очищали центрифугированием. После добавления NaCl к супернатанту, медленно добавляли полиэтиленгликоль до концентрации 3-5% для осаждения. Раствор размешивали в течение 5 минут, оставляли в течение 30 минут и центрифугировали в течение 10 минут. Медленно к супернатанту добавляли еще PEG до 8-10%, размешивали и оставляли в течение 2 часов. Этот раствор центрифугировали в течение 10 минут.
Коллоидный диоксид кремния добавляли к супернатанту до концентрации 1-2%. Раствор размешивали в течение 12 часов, центрифугировали и осадок полностью растворяли в борном буфере, содержащим натрия дезоксихолат. После встряхивания в течение 100-120 минут в водяной бане, раствор центрифугировали и собирали супернатант. Супернатант загружали в DEAE колонку, уравновешенную Tris/Cl буфером при скорости потока 200 мл/мин, и элюировали Tris/Cl буфером, содержащим раствор NaCl. Хлорид цезия добавляли к DEAE элюату до концентрации 1,2 г/мл и ультрацентрифугировали раствор при 40000 об/мин. Обессоленный CsCl пул разбавляли PBS до концентрации белка 400 мкг/мл, добавляли формалин до концентрации 0,025%, и смесь оставляли на 72 часа. В конце удаляли остаточный формалин, используя PBS.
Таким образом, HBsAg был очищен включением полисорбата 20, но при этом поверхностно-активное вещество было добавлено до разрушения рекомбинантных клеток, вместо добавления после очищения антигена.
Полностью жидкие поливалентные вакцины
Экспрессированные дрожжами HBsAg, дифтерийный анатоксин, столбнячный анатоксин и клеточные коклюшные антигены добавляли к суспензии адъюванта соли алюминия. Доводили pH смеси и затем добавляли конъюгат Hib-CRM197, таким образом, чтобы он не адсорбировался на алюминиевом адъюванте. Этим способом получали пятивалентную вакцину со следующим составом:
В другой работе отдельные менингококковые-CRM197 конъюгаты от каждой из серогрупп C, W135 и Y добавляли после CRM-Hib компонента для получения восьмивалентной вакцины:
Важным показателем стабильности и эффективности вакцины является процент гидролиза конъюгата Hib, клинический предел составляет 25% свободного сахарида (в документе 109 сообщается, что 20% не вызывали клинической иммуногенности). Этот параметр измеряли в пятивалентной вакцине посредством HPAEC-PAD, который позволяет проводить прямой количественный анализ неконъюгированных углеводов в пикомолярных концентрациях с минимальным разделением и поглощением. Анализ фокусируется на количестве свободного сахарида.
Количество свободного сахарида измеряли и выражали как процент от общего количества. Следующие результаты были получены:
Максимальное значение в 25% свободного сахарида клинически допустимо. Все значения были ниже этого порога, и составляли менее 6,5% в течение 4 недель при 2-8°C. В условиях тепловой нагрузки (4 недели при 36-38°С) отмечали более высокий уровень, но все же значительно ниже значения в 25%, при этом максимум составлял 11,6% для партии 1. В более ранней работе с поливалентными Hib вакцинами было показано, что гидролиз в случае одного месяца хранения при 36-38°C больше гидролиза CRM-Hib в случае двух лет хранения при 2-8°C. Таким образом, допустимый гидролиз может ожидаться по меньшей мере в течение 2 лет при нормальных условия хранения.
HPAEC-PAD анализ свободного сахарида выполняли через 6 месяцев при 2-8°C. Были получены следующие данные:
Таким образом, наблюдали только небольшое процентное увеличение свободного сахарида через 6 месяцев по сравнению с 4 неделями, при этом значения оставались значительно ниже значения в 25%. Таким образом, CRM197-Hib очень стабилен в этих трех препаратах.
На фигуре 3A показано изменение pH в пятивалентной вакцине за 6 месяцев для трех партий, хранившихся при 2-8°C. На фигуре 3B показано изменение pH за 4 недели для трех партий, хранившихся при 36-38°C. При 2-8°C pH был стабильным более чем 6 месяцев, тогда как в условиях тепловой нагрузки наблюдали небольшое понижение pH на 0,1 единицу после 2 недель и еще небольшое понижение после 4 недель. Тем не менее, все значения pH оставались в пределах допустимого диапазона 6,0-7,0.
Осмолярность всех трех пятивалентных партий составляла между 312 и 315 мОсм/кг, с центральным целевым диапазоном, составляющим 240-360 мОсм/кг для инъекционных вакцин.
Определение активности и иммуногенности антигенов важно для оценки эффективности комбинированной вакцины. Определяли активность дифтерийного, столбнячного и коклюшного антигенов в пятивалентной вакцине и исследовали иммуногенность как CRM-Hib, так и HBsAg. Выполняли анализ ELISA для определения концентрации специфичных антител после иммунизации. Иммуногенность HBsAg представляли на мышиной модели и различных графиках иммунизации в соответствии с таковыми, используемыми для активности HBV.
Получали следующие значения активности DTP:
Для каждого из этих трех антигенов результаты тестов на активность значительно превышают допустимые нижние пределы и эти результаты являются показателем хорошей эффективности для этих трех антигенов.
Для оценки иммуногенности HBsAg, группам из 10 CD1 мышей в дни 0 и 14 вводили пятивалентную вакцину подкожной инъекцией (0,5 мл, разведенную солевым раствором 1:4). Забор крови у мышей проводили на 21 день, и посредством ELISA проводили оценку HBsAg-специфичных антител, используя либо (a) тест "Enzygnost Anti-HBs II" (Dade Behring) или (b) тест "Ausab EIA" (Abbott). Эти тесты ELISA имеют различные форматы и различную чувствительность к HBsAg. Т.е. средние геометрические значения титра не сопоставимы между этими двумя тестами. Однако в рамках каждого теста значения GMT для сыворотки были оптимальными.
Были получены следующие данные:
Все значения GMT, полученные в этом исследовании иммуногенности мышей, выше, чем значения опубликованные в литературе. Процент респондеров соответственно высок для обоих антигенов при оптимальном уровне ~100%.
Для оценки иммуногенности Hib, группам из 8 CD1 мышей в дни 0, 10 и 20 вводили пятивалентную вакцину подкожной инъекцией (0,5 мл, разведенную солевым раствором 1:4). Забор крови у мышей проводили на день 34 и посредством ELISA проводили оценку Hib-специфичных антител. Были получены следующие данные:
Адсорбция HBsAg на адъюванте из алюминия является важным фактором для иммуногенности вакцины, и этот параметр оценивали посредством иммуноблота. Иммуноблот получали следующим образом: вакцинный супернатант объемом 1 мл осаждали DOC/TCA и денатурировали LSB и затем загружали в 12% акриламидную SDS-PAGE; 1 мкг каждой партии HBsAg загружали в качестве контроля; анти-HBsAg препарат антитела козла использовали в качестве первичного антитела (разведение 1:1000), и POD конъюгат против-козла (разведение 1:2500) использовали в качестве вторичного антитела.
Результаты для пятивалентной вакцины показаны на фигуре 2. Полосы 6-8 на фигуре 2A показывают, что в композиции отсутствует обнаружимый растворимый HBsAg в начале отсчета времени, и полосы 5-10 на фигурах 2B и 2C, подтверждают что это так и остается после 2 недель и 4 недель хранения при 2-8°C или 36-38°C. В трех различных партиях ~99% HBsAg остаются адсорбированными на адъюванте при этих различных условиях.
Дополнительное исследование стабильности проводили после 6 месяцев хранения при 2-8°C (фигура 2D). Значение адсорбции сохранялось ~99% для каждой из этих трех партий.
Положительные контроли, использованные на фигуре 2, содержали 1 мкг HBsAg. Была замечена одна полоса, соответствующая S пептиду (24 кДа) плюс полоса, характеризующая агрегаты (~45кДа). Pre-S2, не был замечен.
Также исследовали адсорбцию HBsAg в восьмивалентной вакцине. Образец 1 мл центрифугировали при 3500 об/мин в течение 10 минут. В чистую пробирку отделяли супернатант и преципитировали с DOC/TCA. Пеллету ресуспендировали в 200 мкл экстракционного буфера, кипятили в течение 5 минут и центрифугировали при 1300 об/мин 10 минут. 20 мкл этого экстракта и супернатанта, преципитированого с DOC/TCA, загружали в 12% SDS-PAGE для вестерн-блоттинга.
На фигуре 4A показана восьмивалентная вакцина в начале отсчета времени, а на фигуре 4B показана вакцина после 8 месяцев хранения при 2-8°C. Отсутствие какого-либо значительного окрашивания в линиях 3 и 6 на фигуре 4B (конечно меньшее окрашивание по сравнению с таковым при 1 мкг HBsAg в контрольных линиях 2 и 5) показывает, что адсорбция HBsAg сохраняется стабильной в этот период хранения.
Жидкая/лиофилизированная поливалентная вакцина
Были получены три следующих антигенных компонента:
- Трехвалентный компонент D-T-Pw: готовили компонент D-T-Pw, включающий в себя дифтерийный анатоксин, адсорбированный на адъюванте из гидроксида алюминия, столбнячный анатоксин, также адсорбированный на адъюванте из гидроксида алюминия, и клеточный коклюшный антиген. Компонент D-T-Pw также включает в себя адъювант из фосфата алюминия. Этот компонент содержит тимеросал, но не содержит 2-феноксиэтанол.
- HbsAg-компонент: экспрессировали HBsAg и очищали от рекомбинантных дрожжей и адсорбировали на антиген из фосфата алюминия [110].
- конъюгатный компонент Hib: Hib полисахарид готовили из Hib, штамм 20752, и после активации бромцианом и дериватизации адипингидразидом, спейсер ковалентно присоединяли к столбнячному анатоксину посредством карбодиимидной конденсации, при весовом соотношении сахарид:носитель, составляющим приблизительно 1:3. Конъюгат адсорбировали на адъюванте из фосфата алюминия [64] и затем лиофилизировали с лактозой.
D-T-Pw-компонент смешивали с HbsAg-компонентом. Концентрация HBsAg составляла 20 мкг/мл. Концентрация D составляла ≥60 IU. Концентрация T составляла ≥120 IU. Концентрация Pw составляла ≥8 IU. Четырехвалентную смесь, в виде мутной белой суспензии, упаковывали в водной фазе в стеклянные ампулы с бутилкаучуковой пробкой, с содержанием 1 дозы (0,5 мл), 2 доз (1 мл) или 10 доз (5 мл). Лиофилизированный Hib компонент также помещали в ампулы. Количество порошка на пробирку отмеряли таким образом, чтобы получить 5 мкг/мл (измеренного по сахариду) после восстановления, с 1 дозой, 2 дозами или 10 дозами на пробирку.
Эти две ампулы упаковывали вместе в коробки. Коробки содержат либо по 1 пробирке каждого компонента (или однодозовых или многодозовых ампул), или по 100 ампул каждого компонента.
Для введения пациентам водный D-T-Pw-HBsAg материал извлекают в шприц, и вводят в пробирку с лиофилизированным конъюгатом. После реактивации лиофилизированного материала, его извлекают обратно в шприц через свежую иглу, получая пятивалентную вакцину, готовую для введения пациентам. Могут быть использованы различные 3-дозовые графики первичной иммунизации: 2, 4 и 6 месяцев; 3, 4, и 5 месяцев; или 6, 10 и 14 недель. Вакцина также может быть использована в качестве бустера на втором году жизни.
10 дозовые ампулы дают достаточно материала для иммунизации 10 пациентов из одной ампулы. Для восстановления используют первый шприц, а затем отдельные дозы извлекаются с новыми шприцами, одна доза на шприц. В качестве альтернативного (менее предпочтительного) варианта выполнения, 10 доз извлекают в один шприц, и заменяют иглы при введении пациентами.
Должно быть понятно, что изобретение было описано посредством только примеров, и могут быть выполнены модификации изобретения, которые также будут входить в притязания согласно изобретению.
ДОКУМЕНТЫ (включены в полном объеме посредством указания ссылки на них)
[1] Vaccines. (eds. Plotkin & Orenstein). 4th edition, 2004, ISBN: 0-7216-9688-0.
[2] US patent 6616931.
[3] WO 02/055105.
[4] US 2004/0048336.
[5] Nonionic Surfactants: Organic Chemistry. Nico M. van Os, ed. ISBN: 0 824 79997 6.
[6] Vanlandschoot et at. (2005) J Gen Virol 86:323-31.
[7] Hilleman (1993) pages 17-40 of Hepatitis B vaccines in clinical practice ISBN 0-8247-8780-3.
[8] Gerin et al. (1969) J Virol 4:763.
[9] Gerin et al. (1969) J Virol 7:569.
[10] Siebke et al. (1972) Acta Pathol Microbiol Scan sect B 80:935.
[11] Pillot et al. (1976) J Clin Microbiol 4:205.
[12] Duimel & Krijnen (1972) Vox Sang 23:249.
[13] Neurath et al. (1974) PNAS USA 71:2663.
[14] Charm & Wong (1975) Biotechnol Bioeng 16:593.
[15] US patent 4,857,317.
[16] US patent 4,683,294.
[17] Houwen et al. (1975) J Immunol Methods 8:185.
[18] Sitrin et al. (1993) pages 83-101 of Hepatitis B vaccines in clinical practice ISBN 0-8247-8780-3.
[19] Wampler et al. (1985) PNAS USA 82:6830-4.
[20] Chi et al. (1994) Ann N Y Acad Sci 721:365-73.
[21] Agraz et al. (1994) J Chromatogr A 672:25-33.
[22] Mason et al. (1992) PNAS USA 89:11745-9.
[23] Deml et al. (1999) J Virol Methods 79:205-17.
[24] Ibarra et al. (1999) J Chromatogr B Biomed Sci Appl 735:271-7.
[25] WO 90/10058.
[26] Japanese patent application JP 63239234.
[27] US patent 4,694,074.
[28] EP-0341733.
[29] US patent 5,242,812.
[30] WO 02/12287.
[31] Russian patent application RU-2128707.
[32] Hardy et al. (2000) J Biotechnol 77:157-67.
[33] Dogan et al. (2000) Biotechnol Prog 16:435-41.
[34] WO 03/066094.
[35] Rappuoli et al. (1991) TIBTECH 9:232-238.
[36] Ramsay et al. (2001) Lancet 357(9251):195-196.
[37] Lindberg (1999) Vaccine 17 Suppl 2:S28-36.
[38] Buttery & Moxon (2000) JR Coll Physicians Lond 34:163-168.
[39] Ahmad & Chapnick (1999) Infect Dis Clin North Am 13:113-133, vii.
[40] Goldblatt (1998) J. Med. Microbiol. 47:563-567.
[41] European patent 0477508.
[42] US patent 5,306,492.
[43] WO 98/42721.
[44] Conjugate Vaccines (eds. Cruse et al.) ISBN 3805549326, particularly vol. 10:48-114.
[45] Hermanson (1996) Bioconjugate Techniques ISBN: 0123423368 or 012342335X.
[46] WO 96/40242.
[47] W.H.O. Tech Rep Ser. 594:51, 1976.
[48] Zielen et al. (2000) Infect. Immun 68:1435-1440.
[49] Darkes & Plosker (2002) Paediatr Drugs 4:609-630.
[50] Watson (2000) Pediatr Infect Dis J 19:331-332.
[51] Rubin (2000) Pediatr Clin North Am 47:269-285, v.
[52] Jedrzejas (2001) Microbiol Mol Biol Rev 65:187-207.
[53] Tettelin et al. (2001) Science 293:498-506.
[54] Hoskins et al. (2001) J Bacteriol 183:5709-5717.
[55] Rappuoli (2000) Curr Opin Microbiol 3:445-450.
[56] Rappuoli (2001) Vaccine 19:2688-2691.
[57] Masignani et al. (2002) Expert Opin Biol Ther 2:895-905.
[58] Mora et al. (2003) Drug Discov Today 8:459-464.
[59] Wizemann et al. (2001) Infect Immun 69:1593-1598.
[60] Rigden et al. (2003) Crit Rev Biochem Mol Biol 38:143-168.
[61] WO 02/22167.
[62] McMichael & Green (2003) Curr Opin Investig Drugs 4:953-8.
[63] Module 6 of WHO's The immunological basis for immunization series (Robertson).
[64] WO 97/00697.
[65] WO 02/00249.
[66] US patent 6013264.
[67] US patent 4,624,918.
[68] Vaccine Design: The Subunit and Adjuvant Approach (eds. Powell & Newman) Plenum Press 1995 (ISBN 0-306-44867-X).
[69] WO 90/14837.
[70] US patent 6146632.
[71] WO 99/11241.
[72] WO 99/12565.
[73] WO 98/56414.
[74] Nony et al. (2001) Vaccine 27:3645-51.
[75] Anonymous (Jan 2002) Research Disclosure, 453077.
[76] Anderson (1983) Infect Immun 39(1):233-238.
[77] Anderson et al. (1985) J Clin Invest 76(1):52-59.
[78] EP-A-0372501.
[79] EP-A-0378881.
[80] EP-A-0427347.
[81] WO 93/17712.
[82] WO 94/03208.
[83] WO 98/58668.
[84] EP-A-0471177.
[85] WO 91/01146.
[86] Falugi et al. (2001) Eur J Immunol 31:3816-24.
[87] Baraldo et al. (2004) Infect Immun 72:4884-87.
[88] EP-A-0594610.
[89] WO 00/56360.
[90] WO 02/091998.
[91] Kuo et al. (1995) Infect Immun 63:2706-13.
[92] WO 01/72337.
[93] WO 00/61761.
[94] WO 2004/041157.
[95] WO 91/18926 and US patents 5858677, 5888517, 5989828, 6025484 & 6139846.
[96] Janson et al. (1991) Infect Immun 59:119-25.
[97] WO 00/56360.
[98] Sesardic et al. (2001) Biologicals 29:107-22.
[99] NIBSC code: 98/560.
[100] Module 1 of WHO's The immunological basis for immunization series (Galazka).
[101] NIBSC code: 69/017.
[102] NIBSC code: DIFT.
[103] Sesardic et al. (2002) Biologicals 30:49-68.
[104] NIBSC code: 98/552.
[105] NIBSC code: TEFT.
[106] NTBSC code: 66/303.
[107] Diminsky et al. (1997) Vaccine 15:637-47.
[108] Heijtink et al (2002) Vaccine 20:2191-6.
[109] Sturgess et al. (1999) Vaccine 17:1169-1178.
[110] WO 93/24148.
Реферат
Изобретение относится к области медицины и фармакологии и представляет собой способ получения комбинированной вакцины, где вакцина содержит: (i) неионогенное поверхностно-активное вещество, которое включает поли(оксиэтен)овые остатки, где неионогенное поверхностно-активное вещество присутствует в вакцине в количестве ≤30 мкг/мл; (ii) поверхностный антиген (HBsAg) вируса гепатита В (HBV), где HBsAg присутствует в вакцине в количестве <60 мкг/мл; и (iii) антиген по меньшей мере из одного не-HBV-патогена, где по меньшей мере один не-HBV-патоген включает C.diphtheriae и C.tetani; и антигены из этих двух патогенов представляют собой дифтерийный анатоксин и столбнячный анатоксин; и где способ включает: (i) очистку поверхностного антигена HBV от рекомбинантных дрожжевых клеток, где очистка включает в себя стадию, на которой дрожжевые клетки разрушают в присутствии неионогенного поверхностно-активного вещества, с образованием очищенного HbsAg-компонента; и (ii) объединения очищенного HBsAg-компонента по меньшей мере с одним дополнительным антигеном из не-HBV-патогена, с получением комбинированной вакцины. Изобретение обеспечивает снижение риска контаминации и времени, затрачиваемого на изготовление вакцины. 6 н. и 39 з.п. ф-лы, 7 табл., 12 ил.
Формула
(i) неионогенное поверхностно-активное вещество, которое включает поли(оксиэтен)овые остатки, где неионогенное поверхностно-активное вещество присутствует в вакцине в количестве ≤30 мкг/мл;
(ii) поверхностный антиген (HBsAg) вируса гепатита В (HBV), где HBsAg присутствует в вакцине в количестве <60 мкг/мл; и
(iii) антиген по меньшей мере из одного не-HBV-патогена, где по меньшей мере один не-HBV-патоген включает C.diphtheriae и C.tetani; и антигены из этих двух патогенов представляют собой дифтерийный анатоксин и столбнячный анатоксин; и где способ включает:
(i) очистку поверхностного антигена HBV от рекомбинантных дрожжевых клеток, где очистка включает в себя стадию, на которой дрожжевые клетки разрушают в присутствии неионогенного поверхностно-активного вещества с образованием очищенного HbsAg-компонента; и
(ii) объединение очищенного HBsAg-компонента по меньшей мере с одним дополнительным антигеном из не-HBV-патогена с получением комбинированной вакцины.
(i) неионогенное поверхностно-активное вещество, которое включает поли(оксиэтен)овые остатки, где неионогенное поверхностно-активное вещество присутствует в вакцине в количестве ≤30 мкг/мл;
(ii) поверхностный антиген (HBsAg) вируса гепатита В (HBV), где HBsAg присутствует в вакцине в количестве <60 мкг/мл; и
(iii) антиген по меньшей мере из одного не-HBV-патогена, где по меньшей мере один не-HBV-патоген включает C.diphtheriae и C.tetani; и антигены из этих двух патогенов представляют собой дифтерийный анатоксин и столбнячный анатоксин; и где способ включает стадию объединения очищенного HBsAg-компонента по меньшей мере с одним дополнительным антигеном из не-HBV-патогена с образованием комбинированной вакцины, где HBsAg получен способом, в котором рекомбинантные HBsAg-экспрессирующие дрожжевые клетки разрушают в присутствии неионогенного поверхностно-активного вещества.
(i) неионогенное поверхностно-активное вещество, которое включает поли(оксиэтен)овые остатки, где неионогенное поверхностно-активное вещество присутствует в вакцине в количестве ≤30 мкг/мл;
(ii) поверхностный антиген (HBsAg) вируса гепатита В (HBV), где HBsAg присутствует в вакцине в количестве <60 мкг/мл; и
(iii) антиген по меньшей мере из одного не-HBV-патогена, где по меньшей мере один не-HBV-патоген включает C.diphtheriae и C.tetani; и антигены из этих двух патогенов представляют собой дифтерийный анатоксин и столбнячный анатоксин.
Документы, цитированные в отчёте о поиске
Способ очистки поверхностного антигена вируса гепатита b, содержащего пре s2 пептид, из рекомбинантных клеток дрожжей, вакцина для иммунизации против гепатита b
Комментарии