Материалы для офтальмологических и оториноларингологических устройств - RU2414481C2
Код документа: RU2414481C2
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к улучшенным материалам, применяемым в офтальмологических и оториноларингологических устройствах. В частности, это изобретение относится к мягким, имеющим высокий показатель преломления акриловым материалам, применяемым в устройствах, которые обладают улучшенной прочностью.
Известный уровень техники
В связи с последними достижениями в хирургии катаракты с малыми разрезами особое значение придается разработке мягких складывающихся материалов, пригодных для использования в качестве искусственных хрусталиков (линз). Вообще эти материалы попадают в одну из трех категорий: гидрогели, силиконы и акриловые полимеры.
Вообще материалы на основе гидрогелей имеют относительно низкий показатель преломления, что делает их менее желаемыми, чем другие материалы, потому что для достижения заданной преломляющей способности необходимы более толстые оптические линзы. Силиконовые материалы вообще имеют более высокий показатель преломления, чем гидрогели, но имеют тенденцию разворачиваться взрывообразным способом после того, как их помещают в глаз в свернутом состоянии. Взрывообразное разворачивание может потенциально повредить роговичный эндотелий и/или разорвать природную капсулу хрусталика. Акриловые материалы являются желательными, потому что они обычно имеют высокий показатель преломления и разворачиваются более медленно или контролируемо, чем силиконовые материалы.
Патент США № 5290892 раскрывает акриловые материалы с высоким показателем преломления, пригодные для использования в качестве материала для внутриглазных хрусталиков (“IOL”). Эти акриловые материалы содержат в качестве основных компонентов два арилакриловых мономера. IOL, изготовленные из этих акриловых материалов, можно свернуть или сложить для введения через малые разрезы.
Патент США № 5331073 также раскрывает мягкие акриловые материалы для IOL. Эти материалы содержат в качестве основных компонентов два акриловых мономера, которые определены свойствами их соответствующих гомополимеров. Первый мономер определен как мономер, для которого его гомополимер имеет показатель преломления, по меньшей мере, 1,50. Второй мономер определен как мономер, для которого гомополимер имеет температуру стеклования меньше чем приблизительно 22°С. Эти материалы для IOL также содержат сшивающий компонент. Дополнительно эти материалы могут необязательно содержать четвертый компонент, отличающийся от первых трех компонентов, который получается из гидрофильного мономера. Эти материалы предпочтительно имеют, в общем, менее 15 вес.% гидрофильного компонента.
Патент США № 5693095 раскрывает складываемые, имеющие высокий показатель преломления материалы для офтальмологических хрусталиков, содержащие, по меньшей мере, 90 вес.% только двух основных компонентов: одного арилакрилового гидрофобного мономера и одного гидрофильного мономера. Арилакриловый гидрофобный мономер имеет формулу
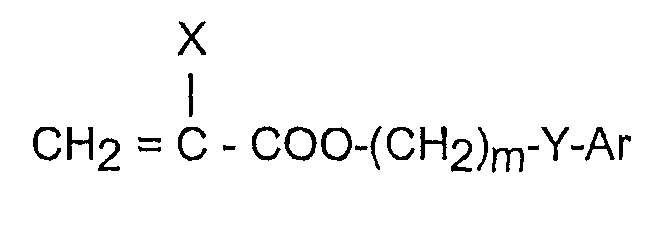
где X представляет собой H или CH3;
m равно от 0 до 6;
Y либо отсутствует, либо представляет собой O, S или NR, где R является H, CH3, CnH2n+1 (n = 1-10), изо-OC3H7, C6H5 или CH2C6H5; и
Ar является любым ароматическим кольцом, которое может быть незамещенным или замещенным группами CH3, C2H5, н-C3H7, изо-C3H7, OCH3, C6H11, Cl, Br, C6H5 или CH2C6H5.
Материалы для хрусталиков, описываемые в '095 патенте США предпочтительно имеют температуру стеклования (“Tg”) приблизительно между -20 и +25°С.
Гибкие внутриглазные хрусталики можно сложить и ввести через маленький разрез. Вообще более мягкий материал можно деформировать в большей степени для того, чтобы его можно было ввести через все более и более маленький разрез. Мягкие акриловые или метакриловые материалы обычно не обладают соответствующей комбинацией прочности, гибкости и неклейких свойств поверхности для того, чтобы дать возможность ввести IOL через такой же малый разрез, как тот, что требуется для силиконовых IOL. Механические свойства силиконовых эластомеров улучшают путем добавления неорганического наполнителя, обычно поверхностно обработанного диоксида кремния. Поверхностно обработанный диоксид кремния также улучшает механические свойства мягких акриловых каучуков, но снижает оптическую прозрачность конечного продукта. Требуются альтернативные наполнители, имеющие показатель преломления, близкий к мягким акриловым каучукам.
Известно, что добавление армирующих наполнителей к мягким полимерам улучшает предел прочности на разрыв и сопротивление разрыву. Армирование придает полимеру жесткость и улучшает прочность посредством ограничения локальной свободы движения полимерных цепей, а также укрепляет структуру посредством введения сетки из слабо связанных точек. Армирующая способность отдельного наполнителя зависит от его характеристик (например, размера частиц и химии поверхности), от типа эластомера, для которого он используется, и от количества присутствующего наполнителя. Традиционные наполнители включают сажу и силикатные наполнители, в которых размер частиц (для максимальной площади поверхности) и смачиваемость (для силы когезии) имеют первостепенное значение. Вообще для эффективного армирования не требуется ковалентное химическое связывание между матрицей и наполнителем. Для обзора см. Boonstra, “Role of particulate fillers in elastomer reinforcement: a review” Polymer 1979, 20, 691 и Gu et al., “Preparation of high strength and optically transparent silicone rubber” Eur.Polym.J. 1998, 34, 1727.
Сущность изобретения
Были изобретены улучшенные мягкие складывающиеся акриловые материалы для устройств, которые, в частности, пригодны для применения в качестве IOL, но которые также полезны в качестве других офтальмологических или оториноларингологических устройств, таких как контактные линзы, кератопротезы, роговичные кольца или вставки, отологические вентиляционные трубки и носовые импланты. Эти полимерные материалы содержат микрофазоворазделенные домены, аналогичные тем, которые можно встретить в обычных блок-сополимерах. Присутствие микрофазоворазделенных доменов улучшает прочность и влияет на поверхностные свойства полимерного материала, не требуя добавления наполнителя. Свойства материалов настоящего изобретения отличаются от статистических сополимеров, полученных с идентичными исходными соотношениями.
Подробное описание изобретения
Если не отмечено иным образом, все количества компонентов представлены на основе весовых % (в/в) («вес.%»).
Материалы для устройств настоящего изобретения являются самоармирующимися полимерными материалами. Материалы представляют собой сополимеры, содержащие а) монофункциональный акрилатный или метакрилатный мономер [1], b) бифункциональный акрилатный или метакрилатный сшивающий агент [2], с) ароматический функциональный метакриловый или акриловый макромономер с концевой акрилатной или метакрилатной группой [3] или [4]. В одном варианте осуществления материалы содержат а) монофункциональный акрилатный или метакрилатный мономер [1], b) бифункциональный акрилатный или метакрилатный сшивающий агент [2], с) ароматический функциональный метакриловый или акриловый макромономер с концевой акрилатной или метакрилатной группой [3] и ароматический функциональный метакриловый или акриловый макромономер с концевой акрилатной или метакрилатной группой [4].
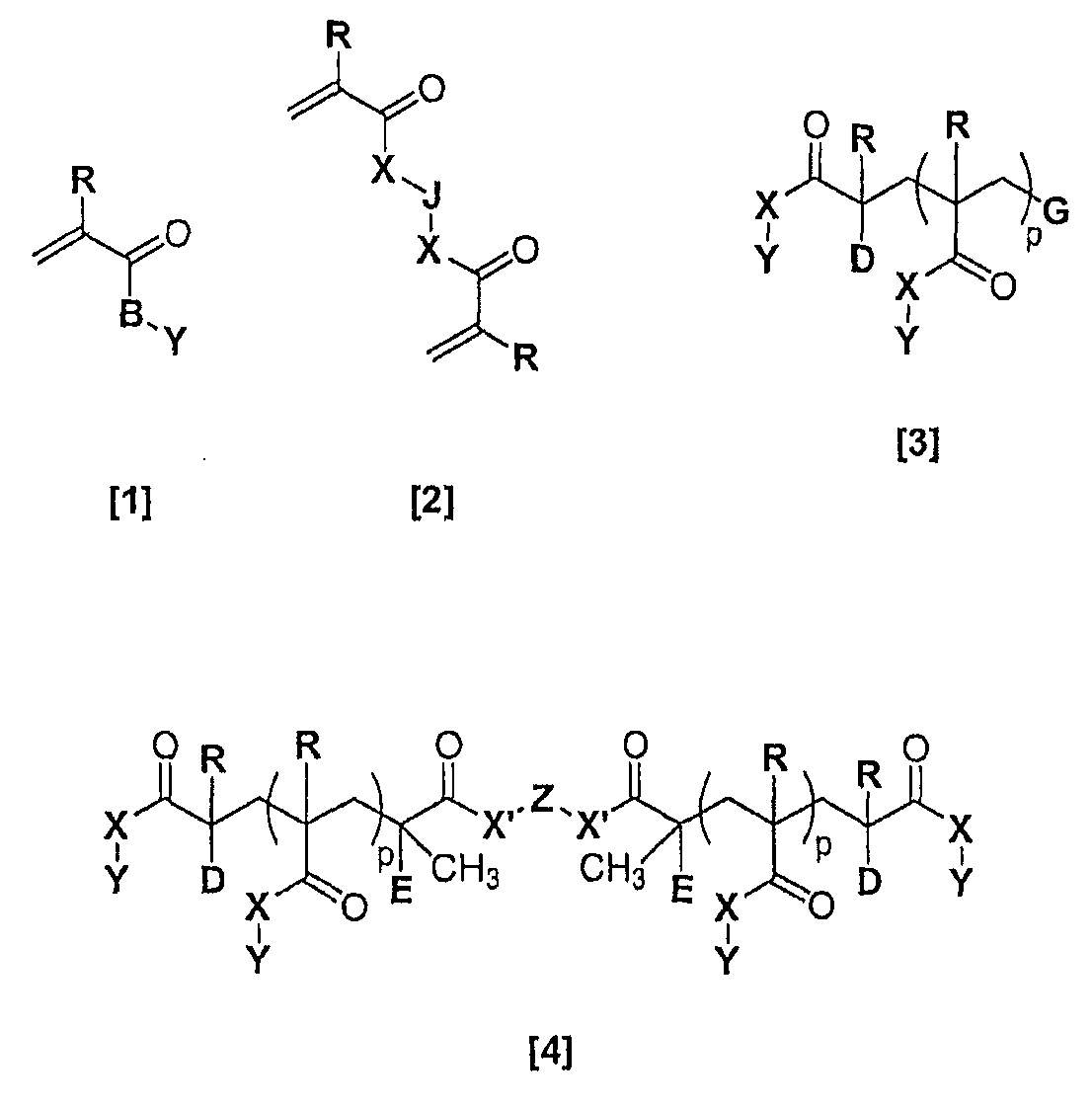
в которых:
B = O(CH2)n, NH(CH2)n или NCH3(CH2)n;
D = Cl, Br, H или -CH2C(=CH2)C(O)XY;
E = H или CH3;
G = H, C(E)(CH3)C(O)X(CH2)nH, C(E)(CH3)C(O)X(CH2CH2O)nCH3 или C(E)(CH3)C(O)X'Z'X'C(O)C(R')=CH2;
R, R' независимо = H, CH3 или CH2CH3;
X, X' независимо = O(CH2)n, NH(CH2)n, NCH3(CH2)n, O(CH2CH2O)nCH2, O(CH2CH2CH2O)nCH2, O(CH2CH2CH2CH2O)nCH2 или отсутствует;
J = (CH2)a, O(CH2CH2O)b, O или отсутствует;
n = 0-6;
Y = C6H5, (CH2)mH, (CH2)mC6H5, OH, CH2CH(OH)CH2OH, (OCH2CH2)mOCH3 или (OCH2CH2)mOCH2CH3;
m = 0-12;
Z, Z' независимо = -(CH2)a-, -(CH2CH2O)a-, -(CH2CH2CH2CH2O)a-, -C6H4-,
-C6H4C(CH3)(CH3)C6H4- или -[(СH(СH2СH3)СH2)(СH2СH2)]q-;
a = 1-12;
b = 1-24;
p = 5-400 и
q = 1-80.
Свободнорадикальная полимеризация этих ингредиентов приводит к сшитой полимерной сетке и, в зависимости от молекулярной массы макромономера, к фазоворазделенным полиакрилатным и полиметакрилатным доменам, аналогичным тем, что обнаруживают в блок-сополимерах. Фазоворазделенные домены влияют на прочность и свойства поверхности получающегося в результате материала и дают сополимер со свойствами материала, отличающимися от свойств, получаемых для статистического сополимера при аналогичном исходном соотношении компонентов.
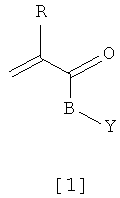
Предпочтительными мономерами формулы [1] являются те, у которых
R = H, B = O(CH2)2, Y = C6H5;
R = H, B = O(CH2)3, Y = C6H5; и
R = СH3, B = O(CH2)4, Y = C6H5.
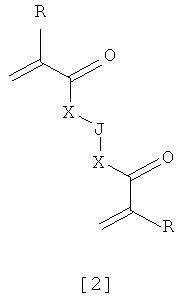
Предпочтительными мономерами формулы [2] являются те, у которых
R = H, X = OCH2, J = (CH2)2;
R = СH3, X = OCH2, J отсутствует; и
R = СH3, X отсутствует, J = O(CH2CH2O)b, где b>10.
Предпочтительными макромерами формулы [3] являются те, у которых
R = H или СН3;
G = C(E)(CH3)C(O)X'Z'X'C(O)C(R')=CH2;
X = O(CH2)n и Х' = О, где n = 1-3;
Y = C6H5;
E = CH3;
Z и Z' = (CH2)2; и
p является таковым, что среднечисловая молекулярная масса (Mn) макромера [3] составляет 5000-50000.
Другими предпочтительными макромерами формулы [3] являются те, у которых
R = H или СН3;
D = -CH2C(=CH2)C(O)XY;
X = O(CH2)n, где n = 1-3;
Y = C6H5;
E = CH3;
Z и Z' = (CH2)2; и
p является таковым, что Mn составляет 5000-50000.
Предпочтительными макромерами формулы [4] являются те, у которых
R = H или СН3;
X = O(CH2)n, где n = 1-3;
Y = C6H5;
E = CH3;
D = -CH2C(=CH2)C(O)XY;
X' = O;
Z' = (CH2)2; и
p является таковым, что Mn составляет 1000-10000.
Мономеры формулы [1] известны, и их можно получить известными способами. См., например, патенты США №№ 5331073 и 5290892. Многие мономеры формулы [1] являются доступными для приобретения у различных производителей.
Мономеры формулы [2] известны, их можно получить известными способами и они являются коммерчески доступными. Предпочтительные мономеры формулы [2] включают этиленгликольдиметакрилат; диэтиленгликольдиметакрилат; 1,6-гександиолдиметакрилат; 1,4-бутандиолдиметакрилат; поли(этиленоксид)диметакрилат (со среднечисловой молекулярной массой 600-1000) и соответствующие им акрилаты.
Макромеры формул [3] и [4] известны. Они являются коммерчески доступными в некоторых случаях и их можно получить известными способами. Макромеры формул [3] и [4] можно получить путем ковалентного присоединения полимеризуемой группы к функциональной концевой группе линейного или разветвленного акрилового или метакрилового полимера. Например, поли(метилметакрилат) с концевой гидроксильной группой можно синтезировать анионной полимеризацией метилметакрилата, затем модифицировать функциональными группами при обрыве цепи этиленоксидом с получением поли(метилметакрилата) с концевой гидроксильной группой. Концевые гидроксильные группы блокированы на одном или обоих концах цепи акрилатными или метакрилатными функциональными группами. Блокирующие группы ковалентно присоединены посредством известных способов, например этерификацией метакрилоилхлоридом или реакцией с изоцианатом с образованием карбаматной связи. См. патенты США №№ 3862077 и 3842059, полное содержание которых введено сюда посредством ссылки.
Макромеры формул [3] и [4] можно также получить в условиях радикальной полимеризации с переносом атома (ATRP). Например, инициатор с концевой гидроксильной группой (гидроксиэтилбромоизобутират) можно смешать с галогенидом меди(I) и солюбилизрующим аминовым лигандом. Это можно использовать для инициирования полимеризации акрилатного или метакрилатного мономера. Получающийся в результате поли(акрилат) или поли(метакрилат) с концевой гидроксильной группой затем может реагировать с метакрилоилхлоридом или изоцианатоэтилметакрилатом. См. патенты США №№ 5852129, 5763548 и 5789487.
Гибкость сополимерного материала настоящего изобретения зависит, главным образом, от температуры стеклования гомополимера, полученного из мономера [1]. Концентрация мономера [1] обычно составляет, по меньшей мере, 35% и предпочтительно 65-85 вес.% от общей концентрации (мономер + макромер + сшивающий агент). Концентрация бифункционального сшивающего агента [2] обычно составляет от 10 до 15 вес.% от общей концентрации, когда R = CH3, X отсутствует, D = O(CH2CH2O)b, где b>5, и предпочтительно менее чем приблизительно 3 вес.% для бифункциональных сшивающих агентов с более низкой молекулярной массой, например, когда R=H, X=OCH2 и D=(CH2)2.
Материалы настоящего изобретения содержат, по меньшей мере, один макромер [3] или [4], но могли бы содержать оба из макромеров [3] и [4]. Общее количество макромеров [3] и [4] зависит от температуры стеклования гомополимера, образуемого из мономера [1], для гарантированного образования гибкого полимерного материала. Сополимерные материалы настоящего изобретения содержат в общем, по меньшей мере, 5 вес.% макромеров [3] и [4]. Для макромеров, у которых R=CH3 или CH2CH3, общая концентрация макромеров [3] и [4], как правило, ниже 50 вес.% и предпочтительно ниже 35 вес.%. Однако для макромеров, у которых R=H, концентрацию макромера можно повысить приблизительно до 50-65 вес.% или более, и, тем не менее, получить сополимерный материал с достаточной гибкостью для того, чтобы позволить ввести IOL, изготовленный из такого материала, через малый хирургический разрез (<2,8 мм). В тех случаях, когда у макромеров [3] или [4] R=H и концентрация выше, чем 50 вес.%, вязкость смеси мономера [1], мономера [2] и макромера [3] и/или [4] становится более важной. Загрузка макромера будет также зависеть от молекулярной массы макромера. Макромеры [3] или [4] склонны к увеличению модуля упругости и уменьшению гибкости получающегося в результате сополимерного материала как функции от его молекулярной массы. При небольших молекулярных массах макромеры [3] или [4], вероятно, будут смешиваемыми, и влияние на Tg будет более сходным с обычным сополимером. При более высоких молекулярных массах или при более высокой концентрации макромера повышенное фазовое разделение может происходить и допускать существование отдельных фаз макромеров и двух Tg. В одном варианте осуществления среднечисловая молекулярная масса макромера [3], имеющего R=CH3 или CH2CH3, составляет 10000-25000.
Сополимерные материалы для устройств настоящего изобретения необязательно содержат один или несколько ингредиентов, выбираемых из группы, состоящей из полимеризуемого поглотителя ультрафиолета и полимеризуемого красителя. Предпочтительно материалы для устройств настоящего изобретения не содержат других ингредиентов, кроме мономеров формулы [1] и [2], макромеров [3] и [4], и полимеризуемых поглотителей ультрафиолета и красителей.
Материалы для устройств настоящего изобретения необязательно содержат химически активные поглотители ультрафиолета или химически активные красители. Известно множество химически активных поглотителей ультрафиолета. Предпочтительным химически активным поглотителем ультрафиолета является 2-(2'-гидрокси-3'-металлил-5'-метилфенил)бензотриазол, коммерчески доступный в виде о-Methallyl Tinuvin P (“oМTP”) на фирме Polysciences, Inc., Warrington, Pennsylvania. Поглотители ультрафиолета обычно присутствуют в количестве приблизительно 0,1-5 вес.%. Подходящие химически активные соединения, поглощающие синий свет, включают соединения, описанные в патенте США № 5470932. Поглотители синего света обычно присутствуют в количестве приблизительно 0,01-0,5 вес.%. Когда их используют для изготовления IOL, материалы для устройств настоящего изобретения предпочтительно содержат как химически активный поглотитель ультрафиолета, так и химически активный краситель.
Для того чтобы сформировать материал для устройств настоящего изобретения, выбранные ингредиенты [1], [2] и [3] и/или [4] смешивают и полимеризуют, используя инициатор радикальной полимеризации, для инициирования полимеризации под действием либо тепла, либо излучения. Материал для устройств предпочтительно полимеризуют в дегазированных полипропиленовых формах в атмосфере азота или в стеклянных формах.
Подходящие инициаторы полимеризации включают термические инициаторы и фотоинициаторы. Предпочтительные термические инициаторы включают свободнорадикальные инициаторы на основе пероксида, такие как трет-бутил(перокси-2-этил)гексаноат и ди-(трет-бутилциклогексил)пероксидикарбонат (коммерчески доступный в качестве Perkadox® 16 на фирме Akzo Chemicals Inc., Chicago, Illinois). Особенно в тех случаях, где материалы настоящего изобретения не содержат хромофора, поглощающего синий свет, предпочтительные инициаторы включают инициаторы на основе бензоилфосфиноксида, такие как 2,4,6-триметилбензоилдифенилфосфиноксид, коммерчески доступный в качестве Lucirin® TPO на фирме BASF Corporation (Charlotte, North Carolina). Инициаторы обычно присутствуют в количестве, равном приблизительно 5% или меньше от общей массы композиции и, более предпочтительно, менее чем 2% от общей массы композиции. Обычно для расчета количеств компонентов массу инициатора не включают в расчеты весовых % композиции.
Отдельные комбинации ингредиентов, описанные выше, и особенность, и количество любых дополнительных компонентов определяются желаемыми свойствами конечного материала для устройства. В предпочтительном варианте осуществления материалы для устройств настоящего изобретения применяют для изготовления IOL, обладающих оптическим диаметром 5,5 или 6 мм, которые сконструированы так, что их можно сжать или растянуть и ввести через хирургический разрез размером 2 мм или меньше.
Материал для устройств предпочтительно имеет показатель преломления в сухом состоянии, по меньшей мере, приблизительно 1,47 и, более предпочтительно, по меньшей мере, приблизительно 1,50, измеренный рефрактометром Аббе при длине волны 589 нм (Na источник света) при 25°С. Оптика, изготовленная из материалов, имеющих показатель преломления, меньший чем 1,47, неизбежно является более толстой, чем оптика той же оптической силы, которая изготовлена из материалов, имеющих более высокий показатель преломления. По существу, IOL оптика, изготовленная из материалов со сравнимыми механическими свойствами и показателем преломления, меньшим чем 1,47, вообще требует сравнительно больших разрезов для имплантации IOL.
Морфология или фазовая структура материала будет зависеть от концентрации макромера, молекулярной массы, его смешиваемости с сеткой сополимера (которая также зависит от молекулярной массы) и способа полимеризации. Поведение микрофазового разделения можно наблюдать посредством дифференциальной сканирующей калориметрии (ДСК). Материалы с микрофазовым разделением будут проявлять две температуры стеклования (“Tg”). Каждая из непрерывной фазы и дискретной фазы будет обладать отдельной Tg. Tg непрерывной фазы будет в основном определять гибкость материала и параметры сворачиваемости-разворачиваемости, и она предпочтительно равна менее чем приблизительно +25°С и более предпочтительно - менее чем приблизительно 0°С. Tg дискретной фазы оказывает меньшее влияние на гибкость материала, чем Tg непрерывной фазы. Tg измеряют дифференциальной сканирующей калориметрией при 10°С/мин и вообще определяют как среднюю точку перехода на кривой теплового потока от температуры.
Материал для устройств предпочтительно имеет удлинение, по меньшей мере, 150%, более предпочтительно - 300% и имеет модуль Юнга менее чем 6,0 МПа, более предпочтительно - менее чем 5,0 МПа. Эти свойства указывают на то, что хрусталики, изготовленные из такого материала, вообще будут легко складываться и не будут трескаться, рваться или расщепляться при складывании. Прочностные свойства образцов полимеров определяли при проведении испытаний на растяжение образцов гантелеобразной формы с общей длиной 20 мм, длиной области захвата 4,88 мм, общей шириной 2,49 мм, шириной в узкой части 0,833 мм с радиусом утолщения 8,83 мм и толщиной 0,9 мм. Тестирование образцов выполняли в стандартных лабораторных условиях при 23±2°С и 50±5% относительной влажности, используя установку для испытаний материалов марки "Инстрон", модели 4400 с 50 Н датчиком нагрузки. Расстояние между захватами составляло 14 мм, скорость ползунка равнялась 500 мм/мин, и образец растягивали до разрыва. Удлинение (деформацию) определяли как долю смещения при разрыве к первоначальному расстоянию между захватами (“удлинение” или “деформация при разрыве %”). Модуль упругости вычисляли как мгновенный наклон кривой напряжение-деформация при 0% деформации (“Модуль Юнга”), 25% деформации (“25% модуль”) и при 100% деформации (“100% модуль”). Сопротивление разрыву измеряли на ненадрезанных под углом 90° образцах (шаблон С) в соответствии с ASTM В624-91 «Стандартный метод тестирования прочности на разрыв обычных вулканизированных каучуков и термопластических эластомеров». Образцы для тестирования имели общую длину 20 мм, длину бороздки 9,0 мм и толщину 0,9 мм. Тестирование образцов выполняли в стандартных лабораторных условиях при 23±2°С, используя установку для испытаний материалов марки "Инстрон", модели 4400 с 50 Н датчиком нагрузки. Расстояние между захватами составляло 9,0 мм, скорость ползунка равнялась 500 мм/мин, и образец растягивали до разрыва. Сопротивление разрыву (“прочность на разрыв”) вычисляли из максимальной силы, получаемой при тестировании, деленной на толщину образца.
IOL, сконструированные из материалов для устройств настоящего изобретения, могут представлять собой любую конструкцию, способную растягиваться или сжиматься до малого размера в поперечном сечении, который может пройти через 2-миллиметровый разрез. Например, IOL могут представлять собой то, что известно как одноблочная или многоблочная конструкция, и содержать оптические и сенсорные компоненты. Оптикой является та часть, которая служит в качестве хрусталика, а сенсорика присоединена к оптике и аналогична рукам, которые держат оптику в соответствующем месте в глазу. Оптика и сенсорика могут быть из одного и того же или из разных материалов. Многоблочные линзы называют так, потому что оптику и сенсорику изготавливают отдельно и затем сенсорику присоединяют к оптике. В одноблочных хрусталиках оптику и сенсорику формируют из одного куска материала. В зависимости от материала сенсорику затем можно срезать или сточить с материала для получения IOL.
В дополнение к IOL материалы настоящего изобретения также пригодны для применения в качестве других офтальмологических или оториноларингологических устройствах, таких как контактные линзы, кератопротезы, роговичные вставки или кольца, отологические вентиляционные трубки и назальные импланты.
Изобретение будет дополнительно проиллюстрировано следующими примерами, которые предназначены для иллюстрации, а не для ограничения объема притязаний.
Пример 1. Синтез поли(2-фенилэтилметакрилата) с концевыми гидроксильными группами.
Синтез выполняли в заполненном N2 боксе с перчатками. В колбу Шленка, содержащую покрытую ПТФЭ магнитную мешалку, загружали 0,3340 г (3,374 ммоль) CuCl, 1,9665 г (11,09 ммоль) N,N,N',N',N''-пентаметилдиэтилентриамина, 30,9268 г (162,57 ммоль) 2-фенилэтилметакрилата (PEMA) и 30 мл анизола. Колбу нагревали на масляной бане до 50°С и добавляли 0,4296 г (2,036 ммоль) 2-гидроксиэтил-2-бромоизобутирата. Реакцию поддерживали при 50°С в течение 3 часов, затем охлаждали до комнатной температуры. Продукт разбавляли этилацетатом и очищали колоночной хроматографией. Продукт дополнительно очищали осаждением из ацетона в большой избыток метанола при 0°С. Продукт отделяли фильтрованием под вакуумом и тщательно промывали охлажденным метанолом, затем сушили под вакуумом при температуре окружающей среды и получали в результате 20,5920 г (67%) твердого вещества белого цвета. Молекулярную массу определяли методом ГПХ в тетрагидрофуране (ТГФ) относительно полистироловых стандартов.
Пример 2. Синтез поли(2-фенилэтилметакрилатного) макромера [3] с концевыми метакрилатными группами
Высушенную в печи колбу Шленка, содержащую покрытую ПТФЭ магнитную мешалку, заполняли N2 и загружали 20,3434 г (0,97 ммоль гидроксильных групп на основании ГПХ Mn) поли(РЕМА) с концевыми гидроксильными группами c Mn 21273. Добавляли безводный дихлорметан (50 мл) и давали полимеру возможность раствориться. Затем добавляли триэтиламин (0,30 мл, 2,15 ммоль). Колбу помещали на ледяную баню, добавляли по каплям при перемешивании метакрилоилхлорид (0,15 мл, 1,55 ммоль). Ледяную баню убирали и реакцию поддерживали при температуре окружающей среды в течение 7 дней в атмосфере азота. Смесь концентрировали на роторном испарителе и неочищенный полимер хроматографировали на колонке с силикагелем с подвижной фазой из дихлорметана. Элюент концентрировали, используя роторный испаритель, и полимер выделяли осаждением из холодного метанола. Продукт выделяли фильтрованием под вакуумом, тщательно промывали метанолом и сушили под вакуумом при температуре окружающей среды, получая 15,8401 г (78%) твердого вещества белого цвета.
Пример 3. Синтез поли(2-фенилэтилакрилата) с концевыми гидроксильными группами.
Синтез выполняли в заполненном N2 боксе с перчатками. В колбу Шленка, содержащую покрытую ПТФЭ магнитную мешалку, загружали 0,2438 г (1,700 ммоль) CuBr, 0,6713 г (3,786 ммоль) N,N,N',N',N''-пентаметилдиэтилентриамина и 7,0941 г (40,26 ммоль) 2-фенилэтилакрилата (PEA) и 30 мл анизола. Колбу нагревали на масляной бане до 50°С и добавляли 0,2840 г (1,346 ммоль) 2-гидроксиэтил-2-бромоизобутирата. Реакцию поддерживали при 50°С в течение 2 часов, затем охлаждали до комнатной температуры. Продукт разбавляли тетрагидрофураном и очищали колоночной хроматографией. Продукт дополнительно очищали осаждением из дихлорметана в большой избыток метанола при -50°С. Продукт отделяли холодным фильтрованием под вакуумом и тщательно промывали охлажденным метанолом, затем сушили под вакуумом при температуре окружающей среды и получали в результате 4,761 г (67%) слегка желтоватой вязкой жидкости. Молекулярную массу определяли методом ГПХ в тетрагидрофуране (ТГФ) относительно полистироловых стандартов.
Пример 4. Синтез поли(2-фенилэтилакрилатного) макромера [4] с концевыми метакрилатными группами
Высушенную в печи 3-горлую круглодонную колбу, содержащую покрытую ПТФЭ магнитную мешалку и снабженную воронкой для добавления, заполняли N2 и загружали 4,7610 г (0,84 ммоль гидроксильных групп на основании ГПХ Mn) поли(РЕА) с концевыми гидроксильными группами c Mn 5676. Добавляли безводный дихлорметан (15 мл) и давали полимеру возможность раствориться. Затем добавляли триэтиламин (0,15 мл, 1,08 ммоль). Колбу помещали на ледяную баню, воронку для добавления заполняли метакрилоилхлоридом (0,14 мл, 1,45 ммоль) и разбавляли его 2 мл дихлорметана. Раствор метакрилоилхлорида добавляли по каплям при перемешивании. Когда добавление заканчивали, ледяную баню убирали и реакцию поддерживали при температуре окружающей среды в течение 4 дней в атмосфере азота. Смесь концентрировали на роторном испарителе и неочищенный полимер хроматографировали на колонке с основным оксидом алюминия с подвижной фазой из дихлорметана. Элюент переносили в делительную воронку и промывали дважды 1 М HCl, дважды деионизированной H2O, затем насыщенным NaCl. Органическую фазу сушили над безводным MgSO4, фильтровали, и растворитель удаляли, используя роторный испаритель, получая 2,9804 г (63%) бесцветной вязкой жидкости.
Пример 5. Сополимеризация 2-фенилэтилметакрилата, 2-фенилэтилакрилата и 1,4-бутандиолдиакрилата
В сцинтилляционный сосуд загружали 0,6204 г (3,261 ммоль, 30,85 вес.%) 2-фенилэтилметакрилата (РЕМА), 1,3261 г (7,526 ммоль, 65,96 вес.%) 2-фенилэтилакрилата (РЕА), 0,0641 г (0,323 ммоль, 3,19 вес.%) 1,4-бутандиолдиакрилата (BDDA) и 0,0264 г (0,161 ммоль) 2-гидрокси-2-метил-1-фенилпропан-1-она (Darocure 1173). Раствор тщательно перемешивали и дегазировали N2. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Получающийся в результате полимер экстрагировали ацетоном в течение 3 часов, промывали свежим ацетоном и оставляли сохнуть на воздухе. Экстрагированный полимер сушили под вакуумом при 60°С в течение, по меньшей мере, 3 часов. Количество экстрагированного вещества определяли гравиметрически. Репрезентативные свойства перечислены в таблице 1.
Пример 6. Сополимеризация поли(2-фенилэтилметакрилатного) макромера (Mn 15460) с концевыми метакрилатными группами с 2-фенилэтилакрилатом и 1,4-бутандиолдиакрилатом
В сцинтилляционный сосуд загружали 0,6114 г (3,214 ммоль мономерных звеньев, 30,45 вес.%) поли(2-фенилэтилметакрилата) с концевыми метакрилатными группами (поли-РЕМА-MA), 1,3336 г (7,568 ммоль, 66,42 вес.%) 2-фенилэтилакрилата (РЕА), 0,0629 г (0,317 ммоль, 3,13 вес.%) 1,4-бутандиолдиакрилата (BDDA) и 0,0247 г (0,150 ммоль) 2-гидрокси-2-метил-1-фенилпропан-1-она (Darocure 1173). Раствор перемешивали до тех пор, пока не растворялся поли-РЕМА-MA, и дегазировали N2. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Получающийся в результате полимер экстрагировали ацетоном в течение 3 часов, промывали свежим ацетоном и оставляли сохнуть на воздухе. Экстрагированный полимер сушили под вакуумом при 60°С в течение, по меньшей мере, 3 часов. Количество экстрагированного вещества определяли гравиметрически. Репрезентативные свойства перечислены в таблице 1.
Пример 7. Сополимеризация поли(2-фенилэтилметакрилатного) макромера (Mn 18470) с концевыми метакрилатными группами с 2-фенилэтилакрилатом и 1,4-бутандиолдиакрилатом
В сцинтилляционный сосуд загружали 0,6114 г (3,214 ммоль мономерных звеньев, 30,47 вес.%) поли-РЕМА-MA, 1,3259 г (7,525 ммоль, 66,09 вес.%) РЕА, 0,0690 г (0,338 ммоль, 3,44 вес.%) BDDA и 0,0227 г (0,138 ммоль) Darocure 1173. Раствор перемешивали до тех пор, пока не растворялся поли-РЕМА-MA, и дегазировали N2. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Получающийся в результате полимер экстрагировали ацетоном в течение 3 часов, промывали свежим ацетоном и оставляли сохнуть на воздухе. Экстрагированный полимер сушили под вакуумом при 60°С в течение, по меньшей мере, 3 часов. Количество экстрагированного вещества определяли гравиметрически. Репрезентативные свойства перечислены в таблице 1.
Пример 8. Сополимеризация поли(2-фенилэтилметакрилатного) макромера (Mn 24624) с концевыми метакрилатными группами с 2-фенилэтилакрилатом и 1,4-бутандиолдиакрилатом
В сцинтилляционный сосуд загружали 1,2218 г (6,422 ммоль мономерных звеньев, 30,55 вес.%) пли-РЕМА-MA, 2,6495 г (15,04 ммоль, 66,26 вес.%) РЕА, 0,1276 г (0,644 ммоль, 3,19 вес.%) BDDA и 0,0407 г (0,248 ммоль, 1,02 вес.%) Darocure 1173. Раствор перемешивали до тех пор, пока не растворялся поли-РЕМА-MA, и фильтровали через стекловолоконный фильтр с размером пор 1 микрон. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Получающийся в результате полимер экстрагировали ацетоном в течение 3 часов, промывали свежим ацетоном и оставляли сохнуть на воздухе. Экстрагированный полимер сушили под вакуумом при 60°С в течение, по меньшей мере, 3 часов. Количество экстрагированного вещества определяли гравиметрически. Репрезентативные свойства перечислены в таблице 1.
Данные таблицы 1 показывают, что добавление макромера [3] улучшает прочностные свойства мягких акриловых полимеров, придавая им повышенную деформируемость без разламывания. Например, в таблице 1 видно, что 2-фенилэтилакрилат-поли(РЕМА)МА привитой сополимер (пример 6) обладает повышенным пределом прочности при разрыве и сопротивлением раздиру по сравнению со статистическим сополимером 2-фенилэтилакрилата и 2-фенилэтилметакрилата с аналогичным исходным соотношением мономеров (пример 5).
Пример 9. Сополимеризация 2-фенилэтилметакрилата, 2-фенилэтилакрилата и 1,4-бутандиолдиакрилата
В сцинтилляционный сосуд загружали 2,5011 г (13,15 ммоль, 25,0 вес.%) РЕМА, 7,4014 г (42,00 ммоль, 74,0 вес.%) РЕА, 0,1007 г (0,508 ммоль, 1,00 вес.%) BDDA и 0,1006 г (0,613 ммоль) Darocure 1173. Раствор тщательно перемешивали и дегазировали N2. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Получающийся в результате полимер экстрагировали ацетоном в течение 3 часов, промывали свежим ацетоном и оставляли сохнуть на воздухе. Экстрагированный полимер сушили под вакуумом при 60°С в течение, по меньшей мере, 3 часов. Количество экстрагированного вещества определяли гравиметрически. Репрезентативные свойства перечислены в таблице 2.
Пример 10. Сополимеризация поли(2-фенилэтилметакрилатного) макромера (Mn 18470) с концевыми метакрилатными группами с 2-фенилэтилакрилатом и 1,4-бутандиолдиакрилатом
В сцинтилляционный сосуд загружали 0,5011 г (2,634 ммоль мономерных звеньев, 24,85 вес.%) поли-РЕМА-MA, 1,4935 г (8,476 ммоль, 74,07 вес.%) РЕА, 0,0216 г (0,109 ммоль, 1,07 вес.%) BDDA и 0,0212 г (0,129 ммоль) Darocure 1173. Раствор перемешивали до тех пор, пока не растворялся поли-РЕМА-MA, и дегазировали N2. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Получающийся в результате полимер экстрагировали ацетоном в течение 3 часов, промывали свежим ацетоном и оставляли сохнуть на воздухе. Экстрагированный полимер сушили под вакуумом при 60°С в течение, по меньшей мере, 3 часов. Количество экстрагированного вещества определяли гравиметрически. Репрезентативные свойства перечислены в таблице 2.
Пример 11. Сополимеризация поли(2-фенилэтилметакрилатного) макромера (Mn 24624) с концевыми метакрилатными группами с 2-фенилэтилакрилатом и 1,4-бутандиолдиакрилатом
В сцинтилляционный сосуд загружали 1,0010 г (5,262 ммоль мономерных звеньев, 24,96 вес.%) поли-РЕМА-MA, 2,9701 г (16,86 ммоль, 74,06 вес.%) РЕА, 0,0393 г (0,198 ммоль, 0,98 вес.%) BDDA и 0,0385 г (0,234 ммоль, 0,96 вес.%) Darocure 1173. Раствор перемешивали до тех пор, пока не растворялся поли-РЕМА-MA, затем дегазировали N2 и фильтровали через стекловолоконный фильтр с размером пор 1 микрон. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Получающийся в результате полимер экстрагировали ацетоном в течение 3 часов, промывали свежим ацетоном и оставляли сохнуть на воздухе. Экстрагированный полимер сушили под вакуумом при 60°С в течение, по меньшей мере, 3 часов. Количество экстрагированного вещества определяли гравиметрически. Репрезентативные свойства перечислены в таблице 2.
Данные таблицы 2 также показывают, что добавление макромера [3] улучшает прочностные свойства мягких акриловых полимеров, придавая им повышенную деформируемость без разламывания. Например, в таблице 2 видно, что 2-фенилэтилакрилат-поли(РЕМА)МА привитой сополимер (пример 10) обладает повышенным пределом прочности при разрыве и сопротивлением раздиру по сравнению со статистическим сополимером 2-фенилэтилакрилата и 2-фенилэтилметакрилата с аналогичным исходным соотношением мономеров (пример 9).
Пример 12. Сополимеризация поли(2-фенилэтилметакрилатного) макромера (Mn 6300) с концевыми метакрилатными группами с 2-фенилэтилакрилатом, 2-(2-метоксиэтокси)этилметакрилатом и 1,4-бутандиолдиакрилатом
В 20-миллилитровый сцинтилляционный сосуд загружали 0,8072 г поли(2-фенилэтилметакрилатного) макромера, 2,5693 г 2-фенилэтилакрилата (РЕА), 0,6131 г 2-(2-метоксиэтокси)этилметакрилата (МЕЕМА) и 0,0410 г 1,4-бутандиолдиакрилата (BDDA). Сосуд закрывали и перемешивали, пока не растворялся макромер. Смесь мономеров фильтровали через стекловолоконный фильтр с размером пор 1 микрон. Композицию дегазировали, барботируя N2 через смесь мономеров. Добавляли ди(4-трет-бутилциклогексил)пероксидикарбонат (Perkadox 16S) (0,0202 г) и раствор тщательно перемешивали. Смесь мономеров дозировали в дегазированные под вакуумом полипропиленовые формы в атмосфере N2. Композицию переносили в полипропиленовые формы и полимеризовали при облучении УФ светом (~3,0 мВт/см2, 365 нм) в течение 30 минут. Заполненные формы помещали на автоматическую конвекцию на 1 час при 70°С, затем на последующую вулканизацию на 2 часа при 110°С. Продукт извлекали из полипропиленовых форм и оставшиеся мономеры удаляли экстракцией ацетона при комнатной температуре. Получившийся полимер сушили под вакуумом при 60°С.
Все эти сополимеры (примеры 5-12) обладали отличной прозрачностью в литом виде.
Привитые сополимеры настоящего изобретения также проявляли пониженную липкость поверхности по сравнению со статистическими сополимерами с идентичным исходным соотношением мономеров, и улучшали технологичность и обращение с IOL.
Изобретение было описано со ссылкой на определенные предпочтительные варианты осуществления; однако следует понимать, что его можно воплотить в других специфических видах или их вариациях, не отступая от его особых или существенных характеристик. Описанные выше варианты осуществления рассматриваются, таким образом, как иллюстративные во всех аспектах и не ограничивающие объем изобретения, который определен в прилагаемой формуле изобретения, а не в предшествующем описании.
Реферат
Изобретение относится к улучшенным материалам, применяемым в офтальмологических и отоларингологических устройствах. Предложен полимерный материал для офтальмологических и отоларингологических устройств, полученный из а) по меньшей мере 35 вес.% от суммы компонентов монофункционального акрилатного или метакрилатного мономера, б) бифункционального акрилатного или метакрилатного сшивающего мономера и в) ароматического функционального метакрилового или акрилового макромономера с концевыми акрилатными или метакрилатными группами. Предложены также офтальмологические или отоларингологические устройства, содержащие заявленный полимерный материал. Технический результат - предложенные материалы имеют высокий показатель преломления и дают возможность получения гибких устройств, обладающих улучшенной прочностью. 3 н. и 16 з.п. ф-лы, 2 табл.
Формула
a) по меньшей мере 35 вес.% от суммы компонентов монофункционального акрилатного или метакрилатного мономера [1];
b) бифункционального акрилатного или метакрилатного сшивающего мономера [2];
c) ароматического функционального метакрилового или акрилового макромономера [3] с концевыми акрилатными или метакрилатными группами и/или ароматического функционального метакрилового или акрилового макромономера [4] с концевыми акрилатными или метакрилатными группами

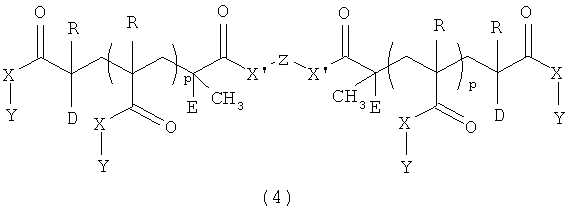
в которых
В=O(CH2)n, NH(CH2)n или NСН3(СН2)n;
D=Cl, Br, H или -CH2C(=CH2)C(O)XY;
Е=Н или СН3;
G=H, С(Е)(СН3)С(O)Х(СН2)nН, С(Е)(СН3)С(O)Х(СН2СН2O)nСН3 или С(Е)(СН3)С(O)Х'Z'Х'С(O)С(R')=СН2;
R, R' независимо =Н, СН3 или СН2СН3;
X, X' независимо = O(CH2)n, NH(CH2)n, NCH3(CH2)n, O(CH2CH2O)nCH2, O(CH2CH2CH2O)nCH2, O(CH2CH2CH2CH2O)nCH2 или отсутствует;
J = (CH2)a, O(CH2CH2O)b, O или отсутствует;
n = 0-6;
Y = C6H5, (CH2)mC6H5;
m=0-12;
Z, Z' независимо = -(CH2)a-, -(CH2CH2O)a-, -(CH2CH2CH2CH2O)a-, -C6H4-, -C6H4C(CH3)(CH3)C6H4- или -[(СH(СH2СH3)СH2)(СH2СH2)]q-;
а=1-12;
b=1-24;
р=5-400 и
q=1-80.
R=Н, В=O(СН2)2, Y=С6Н5;
R=Н, В=O(СН2)3, Y=С6Н5; и
R=СН3, В=O(СН2)4, Y=С6Н5.
R=Н, Х=OCH2, J=(СН2)2;
R=СН3, Х=ОСН2, J отсутствует; и
R=СН3, Х отсутствует, J=O(СН2СН2O)b, где b>10.
R=Н или СН3;
G=С(Е)(СН3)С(O)Х'Z'Х'С(O)С(R')=СН2;
Х=O(СН2)n и X'=О;
Y=С6Н5;
Е=СН3;
Z и Z'=(CH2)2; и
р является таковым, что среднечисловая молекулярная масса (Мn) макромера [3] составляет 5000-50000.
R=Н или СН3;
D=-CH2C(=CH2)C(O)XY;
Х=O(СН2)2;
Y=С6H5;
Е=СН3;
Z и Z'=(CH2)2; и
р является таковым, что Мn составляет 5000-50000.
R=Н или СН3;
Х=O(СН2)2;
Y=С6Н5;
Е=СН3;
D=-CH2C(=CH2)C(O)XY;
X'=О;
Z'=(CH2)2; и
p является таковым, что Мn составляет 1000-10000.
Комментарии