Новые циклические пептидные соединения - RU2423377C2
Код документа: RU2423377C2
Описание
Область техники
Настоящее изобретение касается нового циклического пептидного соединения или его соли, обладающего ингибирующей активностью в отношении репликации РНК репликона вируса гепатита С (далее в данном описании называемый как HCV). В частном случае настоящее изобретение касается нового циклического пептидного соединения или его соли, способа их получения, фармацевтической композиции, содержащей новое пептидное соединение или его соль, и способа предотвращения и/или терапевтического лечения гепатита С человека или животных.
Уровень техники
Примерное число носителей HCV во всем мире составляет 170 миллионов (приблизительно 3%) и приблизительно 1,5 миллиона в Японии. Даже при комбинированной терапии с применением интерферона (далее в данном описании называемый как INF) и рибавирина (Virazole), доступного в качестве первого назначения при лечении, ее эффективность составляет 40% для всех типов HCV. Более того, ее эффективность составляет только 15-20% для IFN-устойчивого вируса (генотип 1b), в частности, обнаруженного в большом количестве в Японии. С другой стороны, комбинированная терапия часто имеет побочные эффекты. Таким образом, трудно полностью избавиться от вируса, используя доступные в настоящее время способы лечения. В случае когда хронический гепатит не может быть вылечен полностью, он будет легко превращаться в циррозный гепатит (30%) или злокачественную гепатому (25%). В Европе и Соединенных Штатах гепатит С был основным показателем для трансплантации печени. Однако повторное развитие HCV часто встречается даже в трансплантированной печени. Вследствие этих причин общество остро нуждается в новых препаратах, которые улучшают как эффективность, так и безопасность, обладающих более сильным противовирусным действием и способностью к ингибированию гепатита С.
HCV является вирусом, имеющим РНК «плюс-цепь», в качестве генома, и классифицируется в семейства Flaviviridae, в соответствии с анализом основной последовательности генома. В соответствии с четвертым изданием Fields Virology fourth edition, D.Knipe et al ed., Philadelphia, Lippincott Williams & Wilkins 2001, 1127-1161, несмотря на то, что существование HCV прогнозировали в 1970-х, открытие HCV было сложным. Многие годы HCV называли вирусом не-А не-В гепатита. В 1989 году, согласно Choo Q-L et al., Science 244, 359-362 (1989), часть гена данного вируса клонировали из сыворотки инфицированного лабораторного животного и его кДНК последовательность идентифицировали и подтвердили, на основании этого вирус назвали «HCV».
Раскрытие изобретения
Циклоспорин А используется в качестве иммуносупрессора при трансплантации органов. M.Thali et al., Nature 372, 363-365 (1994), описали, что циклоспорин А обладал анти-HIV активностью за счет ингибирования взаимодействия между циклоспорином А и частицей вируса, образующей белок вируса иммунодефицита человека тип 1 (HIV-1) (ВИЧ-1). R.M. Wenger et al. описали в WO 00/01715, что их новый циклоспорин обладает анти-HIV активностью. Более того, K. Inoue et al., на 6-ом международном симпозиуме по гепатиту С и родственным вирусам, 3-6 июня (2000) (6th International Symposium on Hepatitis C и Related Virus, 3-6 June (2000) Bethesda, MD, USA), сделали сообщение, что циклоспорин А обладает анти-HCV активностью, однако до настоящего времени другими группами ученых не была представлена информация, подтверждающая это открытие. HIJIKATA et al. описали в WO 2005/021028, что их модифицированные циклоспорины обладают анти-HCV активностью.
M. Berenguer et al., J. Hepatol 32, 673-684 (2000), описали, что клиническое применение циклоспорина А, служащего в качестве иммуносупрессора, вызывало размножение HCV у пациентов, перенесших трансплантацию.
Следовательно, в силу действия вышеуказанных причин, существует потребность в противогепатитных агентах (против гепатита С), улучшенных в плане активности, переноса в крови, селективности и побочных эффектов, например, по сравнению с циклоспорином А.
Более того, чтобы преобразовать скелет соединений циклоспорина, необходимо соблюдение некоторых жестких условий, таких как высокая температура и высокое давление. С другой стороны, преобразование исходного соединения (FR901459 соединение) в соединения по изобретению с помощью реакции перегруппировки в настоящем изобретении требуется соблюдение мягких кислотных условий вследствие наличия гидроксильной группы в положении 2 исходного соединения.
Целевым циклическим пептидным соединением настоящего изобретения является новое соединение, которое может быть представлено следующей общей формулой (I):

где Х означает


где R1 означает водород или низший алкил;
R2 означает водород, арил или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, цикло(низшего)алкила, низшего алкокси, арила, арил(низший)алкокси, необязательно замещенного карбамоилокси и необязательно замещенного амино;
и

Y означает


где R3 означает цикло(низший)алкил, арил, необязательно замещенную гетероциклическую группу или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, цикло(низшего)алкила, низшего алкокси, арила, арил(низшего)алкокси, низший алкокси(низшего)алкокси, необязательно замещенного амино и -OC(O)NR6R7 (где R6 и R7 означают, каждый независимо, водород или низший алкил или, альтернативно, R6 и R7, вместе с атомом азота, к которому они присоединены, представляют N-содержащую гетероциклическую группу, необязательно замещенную низшим алкилом); и
R4 и R5 означают, каждый независимо, водород или низший алкил; и
------ обозначает одинарную связь или двойную связь;
или его соль,
при условии, что, когда R2 означает водород, R3 означает цикло(низший)алкил, арил, необязательно замещенную гетероциклическую группу, низший алкоксиметил, арил(низший)алкил, трет-бутил, втор-бутил, цикло(низший)алкил(низший)алкил или этил, замещенный подходящим заместителем, выбранным из группы, состоящей из гидрокси, низшего алкокси, арил(низшего)алкокси, низший алкокси(низшего)алкокси, необязательно замещенного амино и -OC(O)NR6R7 (где R6 и R7 являются такими, как определено выше).
Предпочтительные варианты осуществления целевого соединения (I) изложены ниже.
1) Соединение общей формулы (I), где
Х означает

где R1 означает водород или низший алкил; и
R2 означает арил или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, цикло(низшего)алкила, низшего алкокси, арила, арил(низшего)алкокси, ди(низшего)алкилкарбамоилокси и амино, необязательно замещенный одним или двумя подходящими заместителями, выбранными из группы, состоящей из: низшего алкила, бензилоксикарбонила и трет-бутоксикарбонила; и
Y означает

где R3 означает цикло(низший)алкил, арил или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, низшего алкокси и арил(низшего)алкокси,
R4 означает водород; и R5 означает низший алкил;
или его соль.
2) Соединение по 1), где R1 означает низший алкил; и
R2 означает низший алкил,
Y означает

где R3 означает арил или низший алкил, необязательно замещенный гидроксигруппой или низшей алкоксигруппой,
R4 означает водород; и
R5 означает низший алкил; и
-------- фрагмент обозначает двойную связь;
или его соль.
3) Соединение по 2), где R3 означает низший алкил, необязательно замещенный гидроксигруппой или низшей алкоксигруппой;
или его соль.
4) Соединение общей формулы (I), где
Х означает

где R1 означает низший алкил; и
R2 означает водород, и
Y означает


где R3 означает цикло(низший)алкил, арил, гетероциклическую группу, необязательно замещенную низшим алкоксикарбонилом, (низший)алкокси(низшим)алкилом, арил(низшим)алкилом, трет-бутилом, втор-бутилом, цикло(низший)алкил(низшим)алкилом или этилом, замещенным подходящим заместителем, выбранным из группы, состоящей из: гидрокси, низшего алкокси, арил(низшего)алкокси, низший алкокси(низшего)алкокси, -OC(O)NR6R7 (где R6 и R7 означают, каждый независимо, водород или низший алкил или, альтернативно, R6 и R7, вместе с атомом азота, к которому они присоединены, представляют N-содержащую гетероциклическую группу, необязательно замещенную низшим алкилом) и аминогруппы, которая необязательно замещена одним или двумя подходящими заместителями, выбранными из группы, состоящей из: низшего алкила и бензилоксикарбонила; и
R4 и R5 означают, каждый независимо, водород или низший алкил;
или его соль.
5) Соединение по 4), где
Y означает

где R3 означает цикло(низший)алкил, арил, гетероциклическую группу, необязательно замещенную низшим алкоксикарбонилом, трет-бутилом, втор-бутилом или этилом, замещенным подходящим заместителем, выбранным из группы, состоящей из: гидрокси, низшего алкокси, арил(низшего)алкокси, низший алкокси(низшего)алкокси и -OC(O)NR6R7 (где R6 и R7 означают, каждый независимо, водород или низший алкил или, альтернативно, R6 и R7, вместе с атомом азота, к которому они присоединены, представляют N-содержащую гетероциклическую группу, необязательно замещенную низшим алкилом);
R4 означает водород и
R5 означает низший алкил; и
------ фрагмент обозначает двойную связь;
или его соль.
6) Соединение общей формулы (I), где
Х означает

и Y означает

где R3 означает низший алкил; и
R4 и R5 означают, каждый независимо, водород или низший алкил,
или его соль.
7) Соединение по 6), где
Х означает

R4 означает водород;
R5 означает низший алкил; и
------ фрагмент обозначает двойную связь;
или его соль.
Соединение (I) или его соль в настоящем изобретении можно получить способами, проиллюстрированными следующими схемами реакции.
Способ 1

Способ 2



Способ 3



Способ 4

Исходные соединения или их соль в настоящем изобретении можно получить, например, способами, проиллюстрированными на следующих схемах реакции.
Способ А



Способ В


где X и Y являются такими, как определено выше,
L означает уходящую группу,
Р означает аминозащитную группу,
R' означает метоксигруппу или
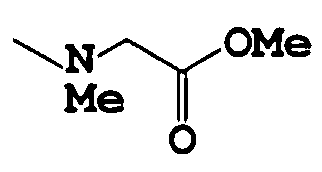
R'' означает H или

n в «n раз» означает 2 или 3.
Способы получения целевых соединений и исходных соединений описаны ниже в данном описании.
Способ 1
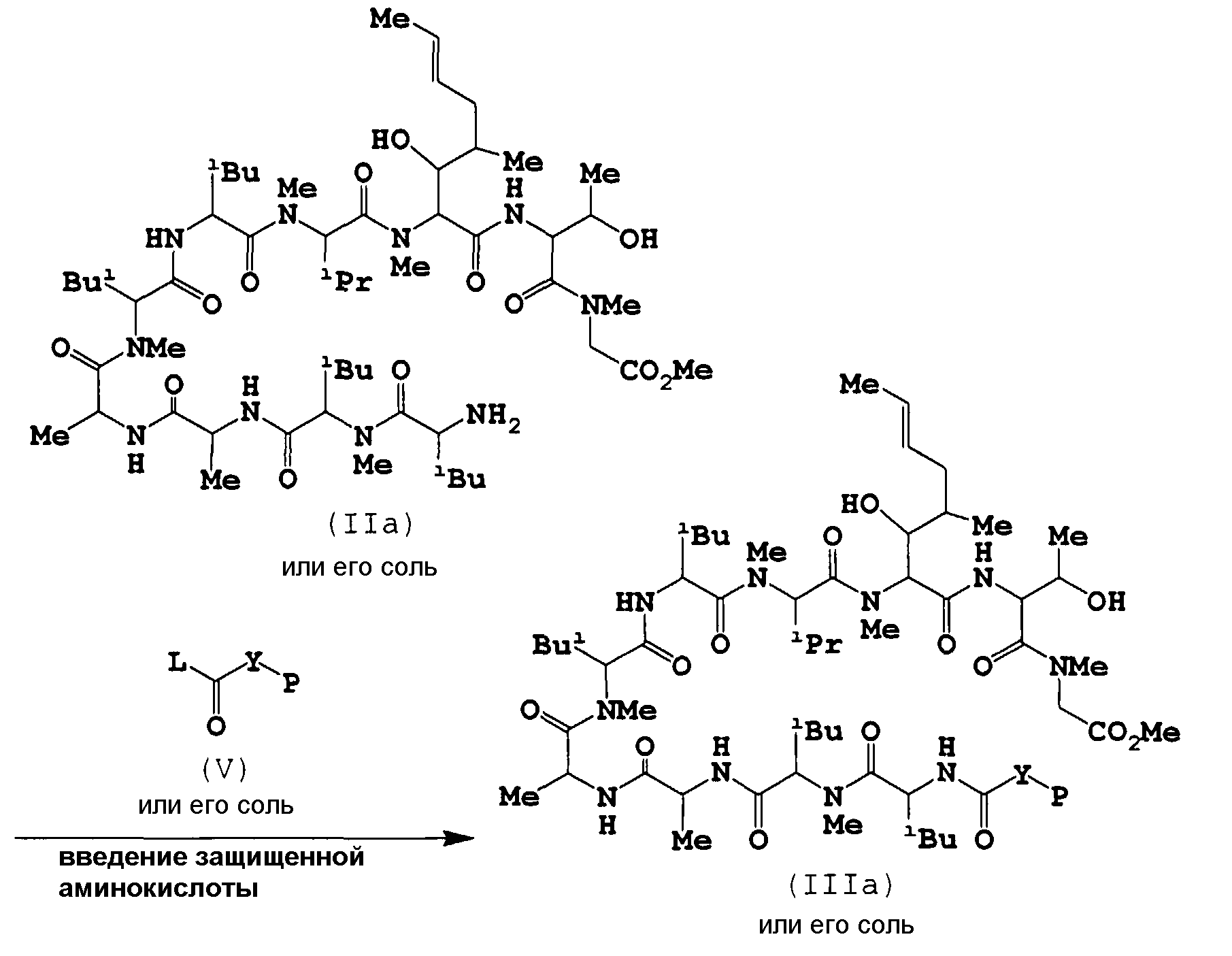
Целевое соединение (Ia) или его соль можно получить из соединения (IIa) или его соли с помощью следующих способов.
а) Введение защищенной аминокислоты
Данная реакция представляет собой амидирование соединения (IIa) с соединением (V).
Обычно соединение (V) является карбоновой кислотой (L означает ОН) или ее реакционно-способным производным (включая ацилгалогенид (например, карбонилхлорид, карбонилбромид и тому подобное), ангидрид кислоты, активированный сложный эфир (например, виниловый эфир, пропаргиловый эфир, 2,4-динитрофениловый эфир, пентафторфениловый эфир, метансульфонилфениловый эфир, диметилиминометиловый эфир, п-нитрофениловый тиоэфир, активированный эфир с N-гидроксисоединением (таким как N-гидроксисукцинимид, N-гидроксибензотриазол и тому подобное) и тому подобное) или тому подобное).
Когда соединение (V) является свободной карбоновой кислотой, реакцию осуществляют предпочтительно в присутствии конденсирующего агента (включая карбодиимид (например, N,N-диизопропилкарбодиимид, N,N'-дициклогексилкарбодиимид, 1-[3-(диметиламино)пропил]-3-этилкарбодиимид и тому подобное), дифенилфосфиновое азидосоединение, дифенилфосфонийхлорид или тому подобное).
Также данную реакцию в настоящем способе обычно осуществляют в присутствии дополнительного компонента, такого как N-гидроксибензотриазол (HOBt), 1-гидрокси-7-азабензотриазол (HOAt), бис(2-оксо-3-оксазолидинил)фосфинийхлорид и тому подобное. Реакция также может быть осуществлена в присутствии органического или неорганического основания, такого как бикарбонат щелочного металла, три(низший)алкиламин, пиридин, N-(низший)алкилморфорин, N,N-ди(низший)алкилбензиламин или тому подобное. Реакцию обычно осуществляют в традиционно использующемся растворителе, таком как вода, ацетон, спирт (например, метанол, этанол, изопропиловый спирт или тому подобное), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид или любые другие органические растворители, которые не оказывают на реакцию неблагоприятное действие, или их смесь.
Температура реакции не ограничивается, и реакцию обычно осуществляют как при охлаждении, так и при нагревании.
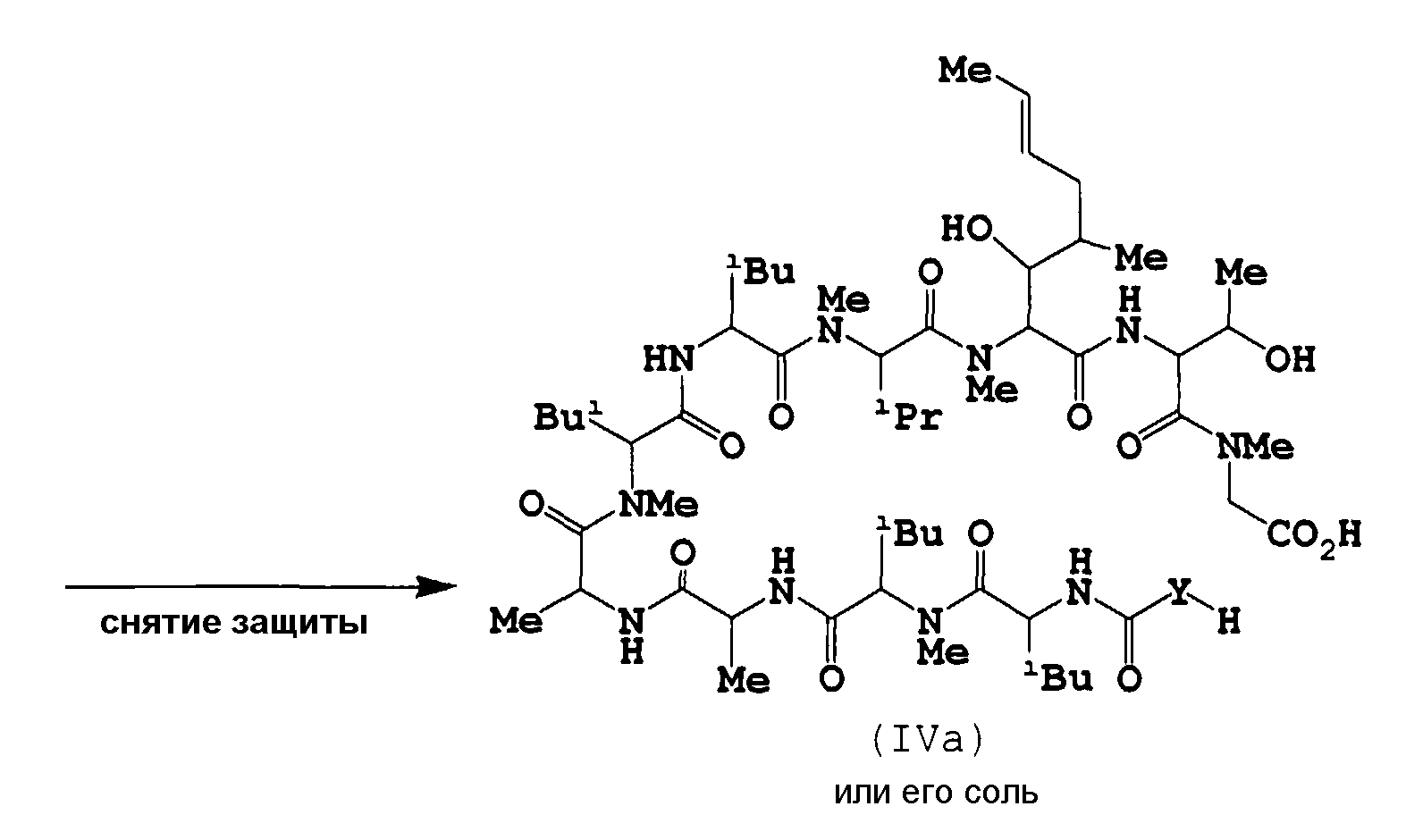
b) Снятие защиты
Данная реакция представляет собой реакцию элиминирования аминозащитной группы соединения (IIIa) или его соли. Данная реакция также является превращением фрагмента метилового эфира соединения (IIIa) или его соли в карбоновую кислоту.
Указанные две реакции осуществляются одновременно (например, что касается примеров получения 68 или 161, описанных ниже) или в двух отдельных реакциях (например, что касается примеров получения 7 и 171 или 153 и 169, описанных ниже) в соответствии с субстратом реакции или условиями реакции.
c) Циклизация
Данную циклизацию осуществляют путем амидирования соединения (IVa), то есть данная реакция может быть осуществлена по аналогичной методике указанного выше способа 1-а), и, следовательно, в качестве используемых реагентов и условий (например, растворитель, температура реакции и т.д.) можно сослаться на используемые в способе 1-а).
Способ 2
Целевое соединение (Ib) или его соль можно получить из соединения (IIb) с использованием последовательности реакций, состоящей из введения защищенной аминокислоты, снятия защиты и циклизации, как проиллюстрировано выше. Каждую реакцию также можно осуществить по аналогичной методике указанного выше способа 1, и, следовательно, в качестве используемых реагентов и условий (например, растворитель, температура реакции и т.д.) можно сослаться на используемые в способе 1.
Способ 3
Целевое соединение (Ic) или его соль можно получить из соединения (IIc) с использованием последовательности реакций, состоящей из введения защищенной аминокислоты и снятия защиты, каждого дважды, и циклизации, как проиллюстрировано выше. Каждую реакцию также можно осуществить по аналогичной методике указанного выше способа 1, и, следовательно, в качестве используемых реагентов и условий (например, растворитель, температура реакции и т.д.) можно сослаться на используемые в способе 1.
Способ 4
Целевое соединение (Id) или его соль можно получить, подвергая соединение (Ic) каталитическому гидрированию.
Подходящие катализаторы, которые используются в каталитическом гидрировании, являются традиционными, такими как платиновые катализаторы (например, платиновая пластина, губчатая платина, черная платина, коллоидная платина, оксид платины, платиновая проволока, т.д.), палладиевые катализаторы (например, губчатый палладий, палладий черный, оксид палладия, палладий на угле, гидроксид палладия на угле, коллоидный палладий, палладий на сульфате бария, палладий на карбонате бария и т.д.) и тому подобное.
Гидрирование обычно осуществляют в традиционном растворителе, таком как вода, ацетон, спирт (например, метанол, этанол, изопропиловый спирт или тому подобное), тетрагидрофуран, диоксан, толуол, метиленхлорид, этилендихлорид, хлороформ, N,N-диметилформамид, N,N-диметилацетамид или любые другие органические растворители, которые не оказывают на реакцию неблагоприятное действие, или их смеси.
Температура реакции не ограничивается, и обычно реакцию осуществляют как при охлаждении, так и при нагревании.
Способ А
Соединение (IX) или его соль можно получить из соединения (VII) или его соли следующими способами.
а) Перегруппировка
Данная реакция представляет собой перегруппировку соединения (VII).
Обычно реакцию осуществляют в присутствии кислоты (такой как трифторуксусная кислота, серная кислота, метансульфоновая кислота или тому подобное). Обычно реакцию осуществляют в традиционных растворителях, таких как вода, ацетон, спирт (например, метанол, этанол, изопропиловый спирт или тому подобное), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид, или других органических растворителях, которые не оказывают на реакцию неблагоприятное действие, или их смеси.
Температура реакции не ограничивается, и обычно реакцию осуществляют как при охлаждении, так и при нагревании.
Реакция настоящего изобретения, вследствие и по причине субстрата, может быть осуществлена в мягких условиях, таких как слабая кислота (п-толуолсульфоновая кислота) и умеренная температура (от температуры окружающей среды до теплого состояния), для того, чтобы сделать возможным проведение перегруппировки соединения селективно.
b) Аминозащита
Данная реакция представляет собой защиту аминофрагмента, который не участвует в перегруппировке. Реакцию обычно осуществляют в традиционных растворителях, таких как вода, спирт (например, метанол, этанол, изопропиловый спирт или тому подобное), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид, или других органических растворителях, которые не оказывают на реакцию неблагоприятное действие, или их смеси.
Температура реакции не ограничивается, и обычно реакцию осуществляют как при охлаждении, так и при нагревании.
Такая последовательность реакций (а) перегруппировка и b) аминозащита) может быть осуществлена способом, описанным в примере получения 156, приведенном ниже, или аналогичными способами.
с) Гидролиз
Соединение (IX) или его соль можно получить из соединения (VIII) или его соли гидролизом.
Гидролиз предпочтительно осуществляют в присутствии основания (включая неорганические основания и органические основания, такие как щелочной металл (например, натрий, калий и т.д.), щелочноземельный металл (например, магний, кальций и т.д.), гидроксид или карбонат, или бикарбонат щелочного металла или щелочноземельного металла, триалкиламин (например, триметиламин и т.д.), гидразин, пиколин, 1,5-диазабицикло[4,3,0]нон-5-ен, 1,8-диазабицикло[5,4,0]ундек-7-ен или тому подобное) или кислоты (включая органическую кислоту (например, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, трифторуксусную кислоту и т.д.), неорганическую кислоту (например, бромистоводородную кислоту, серную кислоту, хлористоводородную кислоту и т.д.) и кислоты Льюиса (например, трибромид бора, хлорид алюминия, трихлорид титана и т.д.)).
Реакцию обычно осуществляют в традиционных растворителях, таких как вода, спирт (например, метанол, этанол, изопропиловый спирт или тому подобное), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, N,N-диметилформамид, или других органических растворителях, которые не оказывают на реакцию неблагоприятное действие, или их смеси.
Жидкое основание или кислота также может использоваться в качестве растворителя.
Температура реакции не ограничивается, и обычно реакцию осуществляют как при охлаждении, так и при нагревании.
Данная реакция может быть осуществлена способом, описанным в примере получения 167, приведенном ниже, или аналогичными способами.
Способ В
Соединение (II) или его соль может быть получено из соединения (IX) или его соли следующими способами.
а) Реакция с соединением (Х)
Данная реакция представляет собой амидирование соединения (IX) соединением (X), то есть данная реакция может быть осуществлена по аналогичной методике указанного выше способа 1-а), и, следовательно, в качестве используемых реагентов и условий реакции (например, растворитель, температура реакции и т.д.) можно сослаться на используемые в способе 1-а).
Данная реакция может быть осуществлена способом, описанным в примере получения 90, приведенном ниже, или аналогичными способами.
b) Снятие защиты
Данная реакция может быть осуществлена по аналогичной методике указанного выше способа 1-b), и, следовательно, в качестве используемых реагентов и условий реакции (например, растворитель, температура реакции и т.д.) можно сослаться на используемые в способе 1-b).
Данная реакция может быть осуществлена способом, описанным в примере получения 152, приведенном ниже, или аналогичными способами.
c) Способ разложения Эдмана (n раз)
Реакцию обычно осуществляют в традиционных растворителях, таких как вода, ацетонитрил, ацетон, спирт (например, метанол, этанол, изопропиловый спирт или тому подобное), тетрагидрофуран, диоксан, толуол, метиленхлорид, хлороформ, этилацетат, N,N-диметилформамид, или других органических растворителях, которые не оказывают на реакцию неблагоприятное действие, или их смеси.
Температура реакции не ограничивается, и обычно реакцию осуществляют как при охлаждении, так и при нагревании.
Реакцию проводят неоднократно до тех пор, пока не будет получено целевое соединение.
Данная реакция может быть осуществлена способом, описанным в примере получения 138, серии 2 и 3, и т.д., приведенном ниже, или аналогичными способами (например, M.K. Eberle et al., J. Org. Chem. 59, 7249-7258 (1994), описавший данный тип способа разложения Эдмана).
Соединение (VII) или его соль (FR901459 соединение) можно получить ферментацией грибка (Stachybotrys chartarum № 19392: регистрационный номер FERM BP-3364), в соответствии со способом, описанным в выложенной заявке на патент Японии Hei 5-271267, например.
Более конкретно, целевое соединение может быть получено способами, описанными в примерах настоящего описания, или аналогичными способами.
Соединения, полученные указанными выше способами 1-4 и А и В, могут быть выделены и очищены традиционными методами, такими как пульверизация, перекристаллизация, колоночная хроматография, высокоэффективная жидкостная хроматография, переосаждение и колоночная хроматография на деминерализованной ионообменной смоле.
Приемлемые соли целевого соединения (I) являются общеизвестными фармацевтически приемлемыми и нетоксичными солями и могут быть солями присоединения основания или кислоты, например соль с неорганическим основанием (таким как соль щелочного металла, например натриевая соль, калиевая соль и т.д., соль щелочноземельного металла, например кальциевая соль, магниевая соль и т.д., соль аммония), соль с органическим основанием (например, органическая аминосоль, например соль триэтиламина, соль диизопропилэтиламина, соль пиридина, соль пиколина, соль этаноламина, соль триэтаноламина, соль дициклогексиламина, соль N'N'-дибензилэтилендиамина и т.д.), соль присоединения неорганической кислоты (такая как гидрохлорид, гидробромид, сульфат, фосфат и т.д.), соль присоединения органической карбоновой кислоты или сульфоновой кислоты (такая как формиат, ацетат, трифторацетат, малеат, тартрат, глюконат, фумарат, метансульфонат, бензолсульфонат, толуолсульфонат и т.д.), соль с основной или кислотной аминокислотой (такой как аргинин, аспаргиновая кислота, глутаминовая кислота и т.д.) и тому подобное.
В вышеуказанном и последующем описании настоящего изобретения подходящие примеры и иллюстрации разнообразных определений, которые включены в объем изобретения, объясняются подробно следующим образом.
Под термином «низший» подразумевается группа, имеющая 1-6, предпочтительно 1-4 атома углерода, если не указано иное.
Подходящие примеры «низшего алкила» и фрагмента «низшего алкила» могут включать прямой или разветвленный радикал, имеющий 1-6 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, трет-пентил, неопентил, гексил, изогексил и тому подобное.
Подходящие примеры «низшего алкилокси» и фрагмента «низшего алкилокси» могут включать прямой или разветвленный радикал, имеющий 1-6 атомов углерода, такой как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентилокси, трет-пентилокси, неопентилокси, гексилокси, изогексилокси и тому подобное.
Подходящие примеры «цикло(низшего)алкила» могут включать циклический алкил, имеющий 3-6 атомов углерода, такой как циклопропил, циклобутил, циклопентил, циклогексил и тому подобное.
Подходящие примеры «арила» и фрагмента «арила» могут включать фенил, который может быть замещен низшим алкилом (например, фенилом, мезитилом, толилом и т.д.), нафтил, антрил, тетрагидронафтил, инденил, тетрагидроинденил и тому подобное.
Подходящие примеры «необязательно замещенной аминогруппы» могут включать аминогруппу, необязательно замещенную одним или двумя подходящими заместителями, такими как низший алкил, амино защитная группа (например, бензилоксикарбонил, трет-бутоксикарбонил (Вос) и тому подобное) и тому подобное.
Подходящие примеры «необязательно замещенной карбамоилоксигруппы» могут включать карбамоилоксигруппу, необязательно замещенную одним или двумя подходящими заместителями, такими как низший алкил, аминозащитная группа (например, бензилоксикарбонил, трет-бутоксикарбонил (Вос) и тому подобное) и тому подобное.
Подходящие примеры «гетероциклической группы» могут включать:
ненасыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1-4 атома азота, например пирролил, пирролинил, имидазолил, пиразолил, пиридил, дигидропиридил, пиримидинил, пиразинил, триазолил (например, 4Н-1,2,4-триазолил, 1Н-1,2,3-триазолил, 2Н-1,2,3-триазолил и т.д.), тетразолил (например, 1Н-тетразолил, 2Н-тетразолил и т.д.), азепинил и т.д.;
насыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1-4 атома азота, например азиридинил, азетинил, пирролидинил, имидазолидинил, пиперидил, пиперазинил, 2,5-метанопиперазинил, гексагидроазепинил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую 1-4 атома азота, например индолил, изоиндолил, индолинил, индолизинил, бензимидазолил, хинолил, изохинолил, индазолил, бензотриазолил, тетрагидрохинолил, тетрагидроизохинолил, тетрагидроиндолил, дигидроиндазолил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, например оксазолил, изоксазолил, оксадиазолил (например, 1,2,4-оксадиазолил, 1,3,4-оксадиазолил, 1,2,5-оксадиазолил и т.д.) и т.д.;
насыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, например морфолинил, сиднонил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, например бензоксазолил, бензоксадиазолил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота, например тиазолил, изотиазолил, тиадиазолил (например, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,3,4-тиадиазолил, 1,2,5-тиадиазолил и т.д.), дигидротиазинил и т.д.;
насыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота, например тиазолидинил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома серы, например тиенил, дигидродитиинил, дигидродитионил и т.д.;
ненасыщенную конденсирванную гетеромоноциклическую группу, содержащую 1 или 2 атома серы и 1-3 атома азота, например бензотиазолил, бензотиадиазолил, имидазотиадиазолил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую один атом кислорода, например фурил и т.д.;
насыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода, например оксиранил, 1,3-диоксоланил, тетрагидрофуранил, тетрагидропиранил и т.д.;
ненасыщенную 3-8-членную (более предпочтительно 5- или 6-членную) гетеромоноциклическую группу, содержащую один атом кислорода и 1 или 2 атома серы, например дигидрооксатиинил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую 1 или 2 атома серы, например бензотиенил, бензодитиинил и т.д.;
ненасыщенную конденсированную гетероциклическую группу, содержащую один атом кислорода и 1 или 2 атома серы, например бензоксадитиинил и т.д.;
насыщенную конденсированную гетеромоноциклическую группу, содержащую 1-3 атома азота, например тетрагидропиридопирролидинил и т.д.; и тому подобное.
В качестве подходящей «N-содержащей гетероциклической группы» можно сослаться на группы, указанные выше, где гетероциклическая группа содержит по меньшей мере один атом азота, такие как пирролидинил, пиперидил, морфолинил, тиазолил, оксазолил и тому подобное.
Подходящая «необязательно замещенная гетероциклическая группа» может включать гетероциклическую группу, как указано выше, которая необязательно замещена подходящим заместителем, таким как низший алкил, низший алкокси, арил, амино, низший алкоксикарбонил и тому подобное.
Термин «галоген» означает фтор, хлор, бром и йод.
V. Lohmann et al., Science 285, 110-113 (1999), сообщили, что они получили клеточные линии гепатомы человека (Huh-7), в которых были введены субгеномные HCV РНК молекулы, и обнаружили, что субгеномная HCV РНК реплицировалась в клеточных линиях с высокой скоростью. Предполагается, что механизм репликации субгеномной HCV РНК в таких клеточных линиях очень похож на репликацию полной длины HCV РНК генома в клетках печени, инфицированных HCV. Следовательно, способ оценки активности соединения (I) при ингибировании РНК репликации в соответствии с настоящим изобретением основан на способе клеточного анализа, который использует Huh-7 клетки, в которые введена субгеномная HCV РНК.
Для того чтобы показать полезность соединения (I) или его соли настоящего изобретения, пример фармакологического теста конкретных соединений в настоящем описании показан следующим образом.
Пример теста
1. Анализ репликона вируса гепатита C с помощью репортера
Ингибирующую активность тестируемых соединений против репликации HCV репликона оценивали с помощью количественного измерения активности люциферазы, продукта репортерного гена, закодированного в системе репликона, описанной Yokota et al., EMBO 4: 602-608 (2003). Ферментативный анализ проводили в соответствии с техническим руководством Steady-Glo(R) системы анализа люциферазы (Promega). Анализ репликона проводили модифицированным способом, предложенным Lohmann et al., Science 285: 110 (1999). Детали описаны более подробно следующим образом.
1) Добавление агента к клеткам
6×103 клеток с HCV репликоном в D-MEM среде, содержащей 5% фетальную телячью сыворотку, высевали в каждую лунку 96-луночного микротитровального планшета (Corning Inc.). После того как клетки инкубировали при 37°C в течение 16 часов в 5% CO2, добавляли тестируемое соединение.
2) Метод анализа люциферазы
После культивирования в течение еще двух дней культуральную среду удаляли и добавляли 25 мкл GIo Lysis буфера в каждую лунку и инкубировали в течение 5 минут. Делая возможным лизис, 25 мкл реагента Steady-Glo(R) анализа добавляли в каждую лунку. После инкубации в течение 5 минут люминесценцию измеряли люминометром, Mithouras LB940 (BERTHOLD TECHNOLOGIES GmbH & Co. KГ), следуя инструкциям изготовителя.
3) Результат теста
Значения активности люциферазы в клетках с репликоном, обработанных при каждой концентрации соединения, использовали для вычисления значения EC50 для каждого соединения, которое давало концентрацию соединения, демонстрирующего 50% уровень ферментативной активности по сравнению с контрольным образцом (не лекарственная группа, содержащая только ДМСО).
Из результатов приведенных выше примеров тестирования понятно, что соединение (I) или его соль настоящего изобретения обладает активностью против вируса гепатита С.
Анти-HCV агент в настоящем изобретении, содержащий соединение (I) или его соль в качестве активного ингредиента, может использоваться в виде фармацевтической композиции, например, в твердой, полутвердой или жидкой форме, в смеси с органическим или неорганическим носителем или эксципиентом, приемлемыми для перорального, сублингвального, буккального, назального, респираторного, парентерального (интрадермального, внутриорганного, подкожного, внутрикожного, внутримышечного, внутрисуставного, введения в центральную вену, в печеночную вену, в периферическую вену, в лимфатическую систему, сердечно-сосудистую систему, артериальную, введения в зрительный орган, включая инъекцию вокруг глаза введение или внутривенного прокапывания вокруг глаза) введения; внутривенного прокапывания в глазное яблоко, аугенную структуру или аугенный слой; в слуховой орган, включая слуховой канал, через папиллярную камеру, наружный и внутренний слуховые каналы, через барабанную перепонку, среднее ухо, внутренний слуховой аппарат, включая спиральный улиточный узел, лабиринт и т.д.; интестинального; ректального; вагинального; через мочеточники и везикального введения. Что касается внутриматочных и перинатальных заболеваний адаптации, то парентеральное введение является предпочтительным, поскольку введение выполняется в сосуды материнской крови или в полые органы, такие как материнские органы, включая матку, шейку матки и влагалище; внутриутробный зародыш, плод, новорожденного и комбинированную ткань; и амниотический мешок, пупочный канатик, пупочную артерию и вену; плаценту и тому подобное. Использование таких способов введения изменяется в зависимости от состояния каждого пациента.
Соединение (I) или его соль может вводиться независимо, в качестве терапевтического агента, или может использоваться как часть назначенных лекарственных препаратов. «Анти-HCV агент» в соответствии с настоящим изобретением может использоваться в виде фармацевтической композиции, например, в твердой, полутвердой или жидкой форме, в смеси по меньшей мере с одним или несколькими подходящими органическими или неорганическими носителями или эксципиентами или другими фармакологическими агентами. Активный ингредиент может быть объединен, например, с обычными фармакологически приемлемыми и нетоксичными носителями в твердой форме, такой как гранулы, таблетки, драже, пастилки, капсулы или суппозитории; кремы; мази; аэрозоли; порошки для инсуффляции; в жидкой форме, такой как растворы, эмульсии или суспензии для инъекции; проглатывания; глазные капли; и любые другие формы, подходящие для применения. При необходимости в вышеуказанные композиции также могут быть включены вспомогательные вещества, такие как стабилизаторы, загустители, увлажнители, затвердители и красители; ароматизаторы или буферы; или любые другие добавки, использующиеся традиционно.
Соединение (I) или его фармацевтически приемлемая соль включена/включены в фармацевтическую композицию в количестве, достаточном для получения желаемого эффекта против гепатита С, влияния на ход или состояние заболевания.
Комбинированное использование IFN и/или рибавирина с соединением (I) или его солью является эффективным против гепатита С.
Для применения данной композиции на людях предпочтительно использовать внутривенное, внутримышечное, легочное, пероральное введение, использование глазных капель или инсуффляцию. Несмотря на то что доза терапевтически эффективного количества соединения (I) варьируется и зависит от возраста и состояния каждого индивидуального пациента, которого подвергают лечению, в случае внутривенного введения, дневная доза 0,001-400 мг соединения (I) на килограмм массы человека, в случае внутримышечного введения, дневная доза 0,1-20 мг соединения (I) на килограмм массы человека, в случае перорального введения, дневная доза 0,5-50 мг соединения (I) на килограмм массы человека в общем случае дается для лечения или предотвращения гепатита С. Однако может потребоваться превышение лимита указанных доз для достижения терапевтических результатов.
Количество липопептидного соединения (I) или его фармацевтически подходящей соли, содержащееся в композиции, для одной единичной дозы настоящего изобретения составляет 0,1-400 мг, более предпочтительно 1-200 мг, еще более предпочтительно 5-100 мг, в частности 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 и 100 мг.
Настоящее изобретение может включать изделие, включающее упаковочные материалы и соединение (I), определенное выше в данном описании, содержащиеся в указанной выше упаковке, где вышеуказанное соединение (I) является терапевтически эффективным для предотвращения и лечения гепатита С и где вышеуказанная упаковка содержит этикетку или письменную информацию, которая указывает, что вышеуказанное соединение (I) может или должно использоваться для предотвращения или лечения гепатита С.
Настоящее изобретение может включать коммерческую упаковку, включающую фармацевтическую композицию, содержащую соединение (I), определенное выше, и письменную информацию, касающуюся него, где утверждается, что соединение (I) может или должно использоваться для предотвращения или лечения гепатита С.
Следует отметить, что соединение (I) или его соль может включать один или несколько стереоизомеров, таких как оптический изомер/изомеры и геометрический изомер/изомеры, вследствие асимметричного атома/атомов углерода и двойной связи/связей, и все такие изомеры и их смесь включены в объем настоящего изобретения.
Соединение (I) или его соль может включать сольватированное соединение (например, гидрат, этилат и т.д.).
Следующие примеры получения и примеры приведены для иллюстрации настоящего изобретения. Однако настоящее изобретение не ограничивается указанными примерами получения и примерами.
Используемые исходные соединения и полученные целевые соединения в следующих примерах 1-83 даны такими, как представлено ниже.
Аббревиатуры, символы и термины, используемые в примерах получения, примерах и формулах приведенного выше и последующего описания настоящего изобретения (включая таблицы), имеют следующие значения.
Пример получения 1
К раствору целевого соединения примера получения 158 ниже (неочищенное вещество 78 г, теоретически 76,9 г) в MeCN (555 мл) добавляли 1 н. HCl (555 мл) при охлаждении на ледяной бане. Смесь нагревали до 30°C и перемешивали при 30°C в течение 3 часов. Полученную смесь нейтрализовали Na2CO3 раствором (29,48 г в H2O 300 мл) и концентрировали в вакууме. Значение pH оставшегося раствора доводили до 8 насыщенным водным раствором NaHCO3 и раствор экстрагировали AcOEt. Органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (3S,6S,9S,12S,15R,18S,21S,24S,27S,30S)-30-[(1R)-1-гидроксиэтил]-27-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-3,6,9,18,21-пентаизобутил-24-изопропил-8,12,15,17,23,26-гексаметил-4,7,10,13,16,19,22,25,28-нонаоксо-2,5,8,11,14,17,20,23,26,29-декаазагентриаконтан-31-оат (неочищенное вещество 70 г, теоретический выход 65,5 г) в виде бледно-коричневого порошка.
Полученный неочищенный продукт использовали в следующей реакции без дальнейшей очистки.
Пример получения 2
К раствору целевого соединения примера получения 1 (неочищенное вещество 70 г, теоретический выход 65,5 г) в AcOEt (660 мл) добавляли изотиоцианатобензол (10 мл) при температуре окружающей среды и значение pH смеси доводили до 7,5 диизопропилэтиламином. Реакционную смесь перемешивали при температуре окружающей среды в течение 1,5 часов. К полученному раствору добавляли N,N-диметилпропандиамин (9,1 г) и перемешивали в течение 5 минут. Реакционную смесь вливали в 0,5 н. HCl (1 л) и экстрагировали AcOEt. Органическую фазу промывали 0,5 н. HCl, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали колоночной хроматографией на силикагеле, при элюировании смесью гексан:AcOEt (2:1-1:1-1:2), получая метил (3S,6S,9S,12S,15R,18S,21S,24S,27S,30S)-1-анилино-30-[(1R)-1-гидроксиэтил]-27-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-3,6,9,18,21-пентаизобутил-24-изопропил-2,8,12,15,17,23,26-гептаметил-4,7,10,13,16,19,22,25,28-нонаоксо-1-тиоксо-2,5,8,11,14,17,20,23,26,29-декаазагентриаконтан-31-оат (44,3 г) в виде бледно-желтого порошка.
Пример получения 3
К раствору целевого соединения примера получения 2 (44,3 г) в MeCN (337 мл) добавляли 1 н. HCl (337 мл) и смесь перемешивали при 30°C в течение 2 часов. Полученную смесь нейтрализовали Na2CO3 раствором (58,8 г в H2O 300 мл) и концентрировали в вакууме. Значение pH оставшегося раствора доводили до 8 насыщенным водным раствором NaHCO3 и раствор экстрагировали AcOEt. Органическую фазу промывали насыщенным NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали колоночной хроматографией на силикагеле, при элюировании смесью CHCl3:MeOH (100:0-97:3), получая метил (2S,5S,8S,11S,14S,17R,20S,23S,26S)-26-амино-2-[(1R)-1-гидроксиэтил]-5-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-11,14,23-триизобутил-8-изопропил-6,9,15,17,20,24,28-гептаметил-4,7,10,13,16,19,22,25-октаоксо-3,6,9,12,15,18,21,24-октаазанонасозан-1-оат (29,1 г) в виде бледно-желтого вещества.
1H-ЯМР (хлороформ-d, δ м.д.): 10,27 (0,5H, д, J=9,0 Гц), 7,38 (0,5H, д, J=8,5 Гц), 7,00 (0,5H, д, J=8,5 Гц), 6,93 (0,5H, д, J=8,5 Гц), 6,89 (1H, д, J=8,5 Гц), 6,84 (0,5H, д, J=8,0 Гц), 6,80 (0,5H, д, J=8,0 Гц), 5,14-5,51 (5H, м), 4,86-5,04 (1H, м), 4,66-4,81 (2H, м), 4,55 (2H, м), 4,31 (1H, м), 4,00 (1H, м), 3,77 (1H, м), 3,76 (1,5H, с), 3,75 (1,5H, с), 3,25 (1,5H, с), 3,14 (1,5H, с), 3,06 (1,5H, с), 3,02 (1,5H, с), 3,01 (1,5H, с), 3,00 (3H, с), 2,71 (1,5H, с), 2,35 (2H, м), 2,03-1,24 (61H, м).
Пример получения 4
К раствору целевого соединения примера получения 59 ниже (3,2 г) в CH2Cl2 (38,5 мл) добавляли ТФУК (9,6 мл) при охлаждении на ледяной бане и смесь перемешивали в течение 2 часов при охлаждении на ледяной бане. К смеси добавляли ТФУК (7 мл) и смесь дополнительно перемешивали в течение 1 часа при охлаждении на ледяной бане. Полученную смесь нейтрализовали Na2CO3 водным раствором (6,6 г в 100 мл H2O) при охлаждении на ледяной бане и концентрировали в вакууме. К оставшемуся раствору добавляли насыщенный водный раствор NaHCO3, для доведения до pH 8, и смесь экстрагировали AcOEt. Органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (3S,6S,9S,12S,15R,18S,21S,24S,27S,30S)-3-втор-бутил-30-[(1R)-1-гидроксиэтил]-27-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,18,21-тетраизобутил-24-изопропил-8,12,15,17,23,26-гексаметил-4,7,10,13,16,19,22,25,28-нонаоксо-2,5,8,11,14,17,20,23,26,29-декаазагентриаконтан-31-оат (3,0 г) в виде бесцветного твердого вещества.
Соединения примеров получения 5-19 получали по методике, аналогичной примеру получения 4.
Пример получения 20
К раствору целевого соединения примера получения 208 ниже (1,20 г) в диоксане (10 мл) добавляли 1 н. LiOH (3,1 мл) при охлаждении на ледяной бане. После перемешивания в течение 3 часов при той же температуре раствор подкисляли 5% лимонной кислотой до pH 5, концентрировали в вакууме, для удаления диоксана, и экстрагировали AcOEt (50 мл). Органический слой промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок растирали с Et2O, получая (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-6-(1-трет-бутоксиэтил)-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24-тетраизобутил-27-изопропил-3,5,11,15,18,20,26,29-октаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оновую кислоту (790 мг) в виде твердого вещества.
Соединения примеров получения 21-24 получали по методике, аналогичной примеру получения 20.
Пример получения 25
К раствору целевого соединения примера получения 4 (81 мг) добавляли (2R)-2-{[(9H-флуорен-9-илметокси)карбонил](метил)амино}пропионовую кислоту (33,5 мг), Bop-Cl (26,2 мг) и диизопропилэтиламин (36 мкл) при охлаждении на ледяной бане. Смесь перемешивали в течение 13 часов при температуре окружающей среды и экстрагировали AcOEt. Органическую фазу промывали 10% водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали препаративной тонкослойной хроматографией (CHCl3:MeOH=90:10), получая метил (5R,8S,11S,14S,17S,20R,23S,26S,29S,32S,35S)-8-втор-бутил-1-(9H-флуорен-9-ил)-35-[(1R)-1-гидроксиэтил]-32-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-11,14,23,26-тетраизобутил-29-изопропил-4,5,7,13,17,20,22,28,31-нонаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-2-окса-4,7,10,13,16,19,22,25,28,31,34-ундекаазагексатриаконтан-36-оат (74 мг) в виде бесцветного твердого вещества.
Соединения примеров получения 26-40 получали по методике, аналогичной примеру получения 25.
Пример получения 41
К раствору целевого соединения примера получения 25 (73 мг) в диоксане (1,9 мл) добавляли 1 н. NaOH (0,49 мл) при температуре окружающей среды и смесь перемешивали в течение 2 часов. К реакционной смеси добавляли 10% водный раствор лимонной кислоты, для доведения pH до 4, и раствор экстрагировали AcOEt. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок растирали с Et2O, получая (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-6-втор-бутил-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24-тетраизобутил-27-изопропил-3,5,11,15,18,20,26,29-октаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оновую кислоту (56 мг) в виде бесцветного порошка.
Соединения примеров получения 42-56 получали по методике, аналогичной примеру получения 41.
Пример получения 57
К раствору целевого соединения примера получения 93 ниже (1,60 г) в N,N-диметилформамиде (16 мл) добавляли пиперидин (1,1 мл) при комнатной температуре. После перемешивания при той же температуре в течение 2 часов реакционную смесь концентрировали в вакууме. Осадок растворяли в AcOEt (60 мл) и раствор промывали 5% водным раствором лимонной кислоты, насыщенным раствором NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Неочищенный продукт очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая метил (2S,5S,8S,11S,14S,17R,20S,23S,26S,29S)-2-[(1R)-1-гидроксиэтил]-5-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-11,14,23,26-тетраизобутил-8-изопропил-6,9,15,17,20,24,30,32,32-нонаметил-29-(метиламино)-4,7,10,13,16,19,22,25,28-нонаоксо-31-окса-3,6,9,12,15,18,21,24,27-нонаазатритриаконтан-1-оат (1,34 г) в виде порошка.
Соединение примера получения 58 получали по методике, аналогичной примеру получения 57.
Пример получения 59
К раствору целевого соединения примера получения 3 (3,0 г) в CH2Cl2 (60 мл) добавляли (2S,3S)-2-[(трет-бутоксикарбонил)(метил)амино]-3-метилпентановую кислоту (839 мг), и HOAt (466 мг), и WSCD (531 мг) при охлаждении на ледяной бане и смесь перемешивали в течение 1,5 часов при охлаждении на ледяной бане. Полученную смесь концентрировали в вакууме и осадок экстрагировали AcOEt. Органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-6-втор-бутил-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24-тетраизобутил-27-изопропил-2,2,5,11,15,18,20,26,29-нонаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-3-окса-5,8,11,14,17,20,23,26,29,32-декаазатетратриаконтан-34-оат (3,22 г) в виде бледно-желтого порошка. Полученный продукт использовали в следующей реакции без дальнейшей очистки.
Соединения примеров получения 60-67 получали по методике, аналогичной примеру получения 59.
Пример получения 68
Метил (6R,9S,12S,15S,18R,21S,24S,27S,30S,33S)-6-втор-бутил-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24-тетраизобутил-27-изопропил-2,2,11,15,18,20,26,29,35-нонаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35-ундекаазагептатриаконтан-37-оат (80 мг) растворяли в смеси 20% ТФУК/CH2Cl2 (2 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 4 часов к раствору добавляли насыщенный водный раствор NaHCO3 до pH 8. Смесь экстрагировали CHCl3 (20 мл) и органический слой промывали насыщенным NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок растворяли в MeOH (4 мл). К раствору добавляли 1 н. LiOH (0,60 мл) при охлаждении на ледяной бане. После перемешивания в течение 1 часа при той же температуре раствор подкисляли 5% водным раствором лимонной кислоты до pH 5, концентрировали в вакууме, для удаления MeOH, и экстрагировали CHCl3 (20 мл). Органический слой промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок растирали с Et2O, получая (5S,8S,11S,14S,17S,20R,23S,26S,29S,32R)-32-амино-5-[(1R)-1-гидроксиэтил]-8-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-14,17,26,29-тетраизобутил-11-изопропил-3,9,12,18,20,23,27,33-октаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-3,6,9,12,15,18,21,24,27,30-декаазапентатриаконтан-1-новую кислоту (45 мг) в виде твердого вещества.
Соединения примеров получения 69-74 получали по методике, аналогичной примеру получения 68.
Пример получения 75
К раствору целевого соединения примера получения 193 ниже (53 мг) в диоксане (0,64 мл) добавляли 1 н. NaOH (0,16 мл) при температуре окружающей среды и смесь перемешивали в течение 2 часов. К реакционной смеси добавляли 10% водный раствор лимонной кислоты, для доведения до pH 4, и раствор экстрагировали AcOEt. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок растирали с Et2O, получая (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-3,6,9,12,21,24-гексаизобутил-27-изопропил-5,11,15,18,20,26,29-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-новую кислоту (18 мг) в виде бесцветного порошка.
Соединения примеров получения 76-89 получали по методике, аналогичной примеру получения 75.
Пример получения 90
К раствору целевого соединения примера получения 167 ниже (5,40 г), гидрохлорида метил{[(2S,3R)-2-амино-3-гидроксибутаноил](метил)амино}ацетата (1,46 г) и HOAt (0,550 г) в CH2Cl2 (80 мл) добавляли раствор WSCD (0,627 г) в CH2Cl2 (4 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 3 часов реакционную смесь концентрировали в вакууме. Осадок растворяли в AcOEt (200 мл) и раствор промывали 0,5 н. HCl, 1 M NaHCO3 и насыщенным раствором соли, сушили над безводным MgSO4 и концентрировали в вакууме, получая метил (6S,12S,15S,18S,21S,24R,27S,30S,33S,36S,39S)-6,39-бис[(1R)-1-гидроксиэтил]-36-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-12,15,18,27,30-пентаизобутил-33-изопропил-2,2,8,11,17,21,24,26,32,35,41-ундекаметил-4,7,10,13,16,19,22,25,28,31,34,37,40-тридекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35,38,41-тридекаазатритетраконтан-43-оат (6,00 г) в виде порошка.
Пример получения 91
К раствору целевого соединения примера получения 3 (120 мг), (2S)-2-[{(2R)-2-[(трет-бутоксикарбонил)(метил)амино]пропаноил}(этил)амино]-3-метилпентановой кислоты (59 мг) и HOAt (19 мг) в CH2Cl2 (6 мл) добавляли раствор WSCD (21 мг) в CH2Cl2 (1 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 1 часа и при комнатной температуре в течение 5 часов реакционный раствор концентрировали в вакууме. Осадок растворяли в AcOEt (20 мл) и раствор промывали 0,5 н. HCl, 1 M NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая метил (6R,9S, 12S,15S,18S,21R,24S,27S,30S,33S,36S)-9-втор-бутил-8-этил-36-[(1R)-1-гидроксиэтил]-33-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-12,15,24,27-тетраизобутил-30-изопропил-2,2,5,6,14,18,21,23,29,32-декаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35-ундекаазагептатриаконтан-37-оат (155 мг) в виде аморфного порошка.
Соединение примера получения 92 получали по методике, аналогичной примеру получения 91.
Пример получения 93
К раствору целевого соединения примера получения 3 (1,20 г), (2S,3R)-3-трет-бутокси-2-{[(9H-флуорен-9-илметокси)карбонил](метил)амино}бутановой кислоты (516 мг) и HOAt (171 мг) в CH2Cl2 (20 мл) добавляли раствор WSCD (195 мг) в CH2Cl2 (1 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 1 часа и при комнатной температуре в течение 4 часов реакционный раствор концентрировали в вакууме. Осадок растворяли в AcOEt (50 мл) и раствор промывали 0,5 н. HCl, 1 M NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая метил (5S,8S,11S,14S,17R,20S,23S,26S,29S,32S)-5-(1-трет-бутоксиэтил)-1-(9H-флуорен-9-ил)-32-[(1R)-1-гидроксиэтил]-29-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-8,11,20,23-тетраизобутил-26-изопропил-4,10,14,17,19,25,28-гептаметил-3,6,9,12,15,18,21,24,27,30-декаоксо-2-окса-4,7,10,13,16,19,22,25,28,31-декаазатритриаконтан-33-оат (1,6 г) в виде аморфного порошка.
Соединение примера получения 94 получали по методике, аналогичной примеру получения 93.
Пример получения 95
К раствору целевого соединения примера получения 166 ниже (1,20 г), (2S,3R)-3-трет-бутокси-2-{[(9H-флуорен-9-илметокси)карбонил](метил)амино}бутановой кислоты (527 мг) и HOAt (174 мг) в CH2Cl2 (20 мл) добавляли раствор WSCD (199 мг) в CH2Cl2 (1 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 1 часа и при комнатной температуре в течение 4 часов реакционный раствор концентрировали в вакууме. Осадок растворяли в AcOEt (50 мл) и раствор промывали 0,5 н. HCl, 1 M NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая метил (5S,8S,11S,14S,17R,20S,23S,26S,29S,32S)-5-(1-трет-бутоксиэтил)-1-(9H-флуорен-9-ил)-32-[(1R)-1-гидроксиэтил]-29-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-8,11,20,23-тетраизобутил-26-изопропил-4,10,14,17,19,25,28,34-октаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-2-окса-4,7,10,13,16,19,22,25,28,31,34-ундекаазагексатриаконтан-36-оат (1,58 г) в виде аморфного порошка.
Соединения примеров получения 96-109 и 111-116 получали по методике, аналогичной примеру получения 95.
Пример получения 110
К раствору целевого соединения примера 25 ниже (190 мг) в пиридине (1,2 мл) добавляли уксусный ангидрид (280 мкл). После перемешивания смеси в течение ночи смесь разбавляли AcOEt, промывали 1 н. водной хлористоводородной кислотой и водным NaHCO3, сушили над MgSO4 и концентрировали. Осадок хроматографировали на силикагеле (гексан/AcOEt=1/4 и затем CH2Cl2/MeOH=9/1), получая (1R)-1-{(2S,5R,8S,11S,14S,17S,20R,23S,26S,29S,32S)-8-(1-трет-бутоксиэтил)-32-[(1R,2R,4E)-1-гидрокси-2-метилгекс-4-ен-1-ил]-11,14,23,26-тетраизобутил-29-изопропил-4,5,7,13,17,20,22,28,31-нонаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2-ил}этил ацетат (193 мг).
Пример получения 117
К раствору целевого соединения примера получения 154 ниже (неочищенное вещество 84 г, теоретический выход 82,5 г) в MeCN (1 л) добавляли 1 н. HCl (1,11 л) при охлаждении на ледяной бане. Смесь нагревали до 30°C и перемешивали при 30°C в течение 4 часов. Полученную смесь нейтрализовали Na2CO3 раствором (58,8 г в H2O 300 мл) и концентрировали в вакууме. Значение pH оставшегося раствора доводили до 8 насыщенным водным раствором NaHCO3 и раствор экстрагировали AcOEt. Органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,12,21,24-пентаизобутил-27-изопропил-5,11,15,18,20,26,29-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оат (неочищенное вещество 78 г, теоретический выход 69,4 г) в виде бледно-коричневого порошка. Полученный неочищенный продукт использовали в следующей реакции без дальнейшей очистки.
Пример получения 118
К раствору целевого соединения примера получения 4 (100 мг) добавляли (2R)-2-[(трет-бутоксикарбонил)(метил)амино]бутановую кислоту (27,6 мг), Bop-Cl (43,2 мг) и диизопропилэтиламин (59 мкл) при охлаждении на ледяной бане. Смесь перемешивали в течение 13 часов при температуре окружающей среды и экстрагировали AcOEt. Органическую фазу промывали 10% водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали препаративной тонкослойной хроматографией (CHCl3:MeOH=90:10), получая метил (6R,9S, 12S,15S,18S,21R,24S,27S,30S,33S,36S)-9-втор-бутил-6-этил-36-(1-гидроксиэтил)-33-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-12,15,24,27-тетраизобутил-30-изопропил-2,2,5,8,14,18,21,23,29,32-декаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35-ундекаазагептатриаконтан-37-оат (64,8 мг) в виде бесцветного твердого вещества.
Соединения примеров получения 119-127 получали по методике, аналогичной примеру получения 118.
Пример получения 128
К раствору целевого соединения примера получения 118 (64,8 мг) в CH2Cl2 (1,2 мл) добавляли ТФУК (0,36 мл) при охлаждении на ледяной бане и смесь перемешивали в течение 2 часов при охлаждении на ледяной бане. К полученному раствору добавляли насыщенный водный раствор NaHCO3, для доведения до pH 8. Смесь экстрагировали AcOEt и органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли. Растворитель удаляли в вакууме, получая метил (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-6-втор-бутил-3-этил-33-(1-гидроксиэтил)-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24-тетраизобутил-27-изопропил-5,11,15,18,20,26,29-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оат (58,6 мг). Полученный продукт использовали в следующей реакции без дальнейшей очистки.
Соединения примеров получения 129-137 получали по методике, аналогичной примеру получения 128.
Пример получения 138
К раствору целевого соединения примера получения 168 ниже (3,70 г) в AcOEt (40 мл) добавляли раствор изотиоцианатобензола (757 мг) в AcOEt (10 мл) при комнатной температуре. После перемешивания в течение 30 минут при той же температуре N,N-диметиламинопропиламин (752 мг) добавляли к раствору. Раствор перемешивали в течение 15 минут, промывали 0,2 н. HCl, NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая порошкообразное вещество. Полученный в результате порошок растворяли в MeCN (100 мл) и 1 н. HCl (60 мл) добавляли при охлаждении на ледяной бане. После перемешивания при комнатной температуре в течение 6 часов раствор нейтрализовали 1 н. Na2CO3 (50 мл), концентрировали в вакууме, для удаления MeCN, и экстрагировали AcOEt (200 мл). Органический слой промывали NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Осадок очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая метил (3S,6S,9S,12S,15R,18S,21S,24S,27S,30S)-30-[(1R)-1-гидроксиэтил]-27-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-3,6,9,18,21-пентаизобутил-24-изопропил-8,12,15,17,23,26,32-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оат (3,19 г) в виде порошка.
Пример получения 139
К раствору целевого соединения примера получения 128 (58,6 мг) в MeOH (1,2 мл) добавляли 1 н. NaOH (0,23 мл) при температуре окружающей среды и смесь перемешивали в течение 2 часов. К реакционной смеси добавляли 10% водный раствор лимонной кислоты, для доведения pH до 4, и раствор экстрагировали AcOEt. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок растирали с Et2O, получая (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-6-втор-бутил-3-этил-33-(1-гидроксиэтил)-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24-тетраизобутил-27-изопропил-5,11,15,18,20,26,29-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-новую кислоту (58,6 мг) в виде бесцветного порошка.
Соединения примеров получения 140-148 получали по методике, аналогичной примеру получения 139.
Пример получения 149
Метил (6R,9S,12S,15S,18S,21R,24S,27S,30S,33S,36S)-9-(1-трет-бутоксиэтил)-6-этил-36-[(1R)-1-гидроксиэтил]-33-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-12,15,24,27-тетраизобутил-30-изопропил-2,2,5,8,14,18,21,23,29,32-декаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35-ундекаазагептатриаконтан-37-оат (110 мг) растворяли в смеси 20% ТФУК/CH2Cl2 (3 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 4 часов к раствору добавляли насыщенный водный раствор NaHCO3, для доведения pH до 8. Смесь экстрагировали CHCl3 (20 мл) и органический слой промывали насыщенным NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок растворяли в MeOH (4 мл). К раствору добавляли LiOH (0,77 мл) при охлаждении на ледяной бане. После перемешивания в течение 1 часа при той же температуре раствор подкисляли 5% водным раствором лимонной кислоты, для доведения pH до 5, концентрировали в вакууме, для удаления MeOH, и экстрагировали AcOEt (20 мл). Органический слой промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок растирали с Et2O, получая (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-3-этил-33-[(1R)-1-гидроксиэтил]-6-(1-гидроксиэтил)-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24-тетраизобутил-27-изопропил-5,11,15,18,20,26,29-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-новую кислоту (70 мг) в виде твердого вещества.
Соединения примеров получения 150 и 151 получали по методике, аналогичной примеру получения 149.
Пример получения 152
Целевое соединение примера получения 90 (6,00 г) растворяли в 20% ТФУК в CH2Cl2 (80 мл) при охлаждении на ледяной бане и смесь перемешивали при той же температуре в течение 4 часов. К раствору добавляли насыщенный водный раствор NaHCO3, для доведения pH до 8, и смесь экстрагировали CHCl3 (200 мл). Органический слой промывали насыщенным NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали, получая метил (5S,8S,11S,14S,17S,20R,23S,26S,29S,32S,38S,39R)-38-амино-39-гидрокси-5-[(1R)-1-гидроксиэтил]-8-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-14,17,26,29,32-пентаизобутил-11-изопропил-3,9,12,18,20,23,27,33,36-нонаметил-4,7,10,13,16,19,22,25,28,31,34,37-додекаоксо-3,6,9,12,15,18,21,24,27,30,33,36-додекаазатетраконтан-1-оат (5,70 г) в виде аморфного порошка.
Соединение примера получения 153 получали по методике, аналогичной примеру получения 152.
Пример получения 154
К раствору целевого соединения примера получения 155 ниже (неочищенное вещество 75,0 г, теоретический выход 71,8 г) в смешанном растворителе (AcOEt 750 мл и пиридин 67,5 мл) добавляли изотиоцианатобензол (19,8 мл) и смесь перемешивали в течение 13 часов. К смеси добавляли пиридин (67,5 мл) и диизопропилэтиламин и значение pH смеси доводили до 8. Смесь перемешивали в течение 3 часов. К полученному раствору добавляли N,N-диметилпропандиамин (19,8 г) и перемешивали в течение 5 минут. Реакционную смесь вливали в 0,5 н. HCl (1 л) и экстрагировали AcOEt.
Органическую фазу промывали 0,5 н. HCl, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (3S,9S,12S,15S,18S,21R,24S,27S,30S,33S,36S)-1-анилино-3,36-бис[(1R)-1-гидроксиэтил]-33-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,15,24,27-пентаизобутил-30-изопропил-5,8,14,18,21,23,29,32-октаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-1-тиоксо-2,5,8,11,14,17,20,23,26,29,32,35-додекаазагептатриаконтан-37-оат (неочищенное вещество 84 г, теоретический выход 82,5 г) в виде коричневого вещества. Полученный неочищенный продукт использовали в следующей реакции без дальнейшей очистки.
Пример получения 155
К раствору целевого соединения примера получения 157 ниже (неочищенное вещество 80,2 г, теоретический выход 77,1 г) в CH2Cl2 добавляли ТФУК (205 мл) при охлаждении на ледяной бане. Смесь перемешивали в течение 2 часов при охлаждении на ледяной бане. Значение pH раствора доводили раствором Na2CO3 (147 г/500 мл H2O) и насыщенным водным раствором NaHCO3 при охлаждении на ледяной бане. Полученный в результате раствор экстрагировали CHCl3. Органическую фазу промывали насыщенным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (2S,5S,8S,11S,14S,17R,20S,23S,26S,29S,35S,36R)-35-амино-36-гидрокси-2-[(1R)-1-гидроксиэтил]-5-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-11,14,23,26,29-пентаизобутил-8-изопропил-6,9,15,17,20,24,30,33-октаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-3,6,9,12,15,18,21,24,27,30,33-ундекаазагептатриаконтан-1-оат (75 г, неочищенное вещество) в виде коричневого порошка. Полученный неочищенный продукт использовали в следующей реакции без дальнейшей очистки.
Пример получения 156
К раствору (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-30-[(1R)-1-гидроксиэтил]-33-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,18,21,24-пентаизобутил-3-изопропил-1,4,10,12,15,19,25,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекона (100 г) в тетрагидрофуране (1 л) добавляли 4-метилбензолсульфоновую кислоту (70,6 г) и смесь перемешивали при 50°C в течение 14 часов. К смеси добавляли 1 н. NaOH, нейтрализовали при охлаждении на ледяной бане и добавляли Boc2O (17,9 г). Значение pH смеси доводили до 8 1 н. NaOH при охлаждении на ледяной бане. Смесь перемешивали при температуре окружающей среды в течение 2,5 часов. Полученную смесь концентрировали в вакууме и экстрагировали AcOEt. Органическую фазу промывали насыщенным водным раствором Na2CO3, 0,1 н. HCl и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая трет-бутил{(3S,6S,9S,12S,15R,18S,21S,24S,27S,33S,34R)-3-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-9,12,21,24,27-пентаизобутил-6-изопропил-4,7,13,15,18,22,28,31,34-нонаметил-2,5,8,11,14,17,20,23,26,29,32-ундекаоксо-1-окса-4,7,10,13,16,19,22,25,28,31-декаазациклотетратриаконтан-33-ил}карбамат (167,2 г) в виде коричневого вещества. Полученный неочищенный продукт использовали в следующей реакции без дальнейшей очистки.
Пример получения 157
К раствору целевого соединения примера получения 167 ниже (71,0 г) в CH2Cl2 (1 л) добавляли гидрохлорид метил (2S,3R)-2-амино-3-гидроксибутаноата (10,8 г) и HOAt (10,8 г) при температуре окружающей среды. К смеси добавляли WSCD (9,9 г) при охлаждении на ледяной бане. Смесь перемешивали в течение 1,5 часов при температуре окружающей среды. Полученную смесь концентрировали в вакууме и экстрагировали AcOEt. Органическую фазу промывали 0,5 н. HCl, насыщенным NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (6S,12S,15S,18S,21S,24R,27S,30S,33S,36S,39S)-6,39-бис[(1R)-1-гидроксиэтил]-36-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-12,15,18,27,30-пентаизобутил-33-изопропил-2,2,8,11,17,21,24,26,32,35-декаметил-4,7,10,13,16,19,22,25,28,31,34,37-додекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35,38-додекаазатетраконтан-40-оат (80,2 г, неочищенное вещество) в виде коричневого вещества. Полученный неочищенный продукт использовали в следующей реакции без дальнейшей очистки.
Пример получения 158
К раствору целевого соединения примера получения 117 (неочищенное вещество 78 г, теоретический выход 69,4 г) в AcOEt (690 мл) добавляли изотиоцианатобензол (11,3 г) при температуре окружающей среды и смесь перемешивали в течение 1 часа. К раствору добавляли диизопропилэтиламин (5 мл) и смесь дополнительно перемешивали в течение 1,5 часов. К полученному раствору добавляли N,N-диметилпропандиамин (9,1 г) и перемешивали в течение 5 минут. Реакционную смесь вливали в 0,5 н. HCl (1 л) и экстрагировали AcOEt. Органическую фазу промывали 0,5 н. HCl, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-1-анилино-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,12,21,24-пентаизобутил-27-изопропил-2,5,11,15,18,20,26,29-октаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-1-тиоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оат (неочищенное вещество 78 г, теоретический выход 76,9 г) в виде коричневого вещества. Полученный неочищенный продукт использовали в следующей реакции без дальнейшей очистки.
Пример получения 159
К раствору целевого соединения примера получения 1 (87 мг) добавляли (2R)-2-{[(9H-флуорен-9-илметокси)карбонил](метил)амино}пропионовую кислоту (36 мг), Bop-Cl (28,2 мг) и диизопропилэтиламин (39 мкл) при охлаждении на ледяной бане. Смесь перемешивали в течение 13 часов при температуре окружающей среды и экстрагировали AcOEt. Органическую фазу промывали 10% водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали препаративной тонкослойной хроматографией (CHCl3:MeOH=90:10), получая метил (5R,8S,11S,14S,17S,20R,23S,26S,29S,32S,35S)-1-(9H-флуорен-9-ил)-35-[(1R)-1-гидроксиэтил]-32-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-8,11,14,23,26-пентаизобутил-29-изопропил-4,5,7,13,17,20,22,28,31-нонаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-2-окса-4,7,10,13,16,19,22,25,28,31,34-ундекаазагексатриаконтан-36-оат (110 мг) в виде бесцветного твердого вещества.
Соединение примера получения 160 получали по методике, аналогичной примеру получения 159.
Пример получения 161
К раствору целевого соединения примера получения 95 (1,55 г) в диоксане (30 мл) добавляли 1 н. LiOH (10 мл) при охлаждении на ледяной бане. После перемешивания в течение 3 часов при той же температуре раствор подкисляли 5% лимонной кислотой, для доведения pH до 5, концентрировали в вакууме, для удаления диоксана, и экстрагировали AcOEt (50 мл). Органический слой промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок растирали с Et2O, получая (5S,8S,11S,14S,17S,20R,23S,26S,29S,32S)-5-[(1R)-1-гидроксиэтил]-8-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-14,17,26,29-тетраизобутил-11-изопропил-3,9,12,18,20,23,27,33,35,35-декаметил-32-(метиламино)-4,7,10,13,16,19,22,25,28,31-декаоксо-34-окса-3,6,9,12,15,18,21,24,27,30-декаазагексатриаконтан-1-новую кислоту в виде твердого вещества.
Соединения примеров получения 162-165 получали по методике, аналогичной примеру получения 161.
Пример получения 166
К раствору целевого соединения примера получения 138 (3,15 г) в AcOEt (40 мл) добавляли раствор изотиоцианатобензола (511 мг) в AcOEt (10 мл) при комнатной температуре. После перемешивания в течение 30 минут при той же температуре N,N-диметиламинопропиламин (527 мг) добавляли к раствору. Раствор перемешивали в течение 15 минут, промывали 0,2 н. HCl, NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая порошок. Полученный в результате порошок растворяли в MeCN (100 мл) и 1 н. HCl (50 мл) добавляли при охлаждении на ледяной бане. После перемешивания при комнатной температуре в течение 6 часов раствор нейтрализовали в Na2CO3 (25 мл), концентрировали в вакууме, для удаления MeCN, и экстрагировали AcOEt (150 мл). Органический слой промывали NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Осадок очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая метил (5S,8S,11S,14S,17S,20R,23S,26S,29S)-29-амино-5-[(1R)-1- гидроксиэтил]-8-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-14,17,26-триизобутил-11-изопропил-3,9,12,18,20,23,27,31- октаметил-4,7,10,13,16,19,22,25,28-нонаоксо-3,6,9,12,15,18,21,24,27-нонаазадотриаконтан-1-оат (2,72 г) в виде порошка.
Пример получения 167
К раствору целевого соединения примера получения 156 (неочищенное вещество 167 г, теоретический выход 108 г) в MeOH (1 л) добавляли 1 н. NaOH (819 мл) при охлаждении на ледяной бане. Смесь перемешивали при температуре окружающей среды в течение 8 часов. К смеси добавляли 1 н. NaOH (82 мл) и смесь перемешивали в течение 2 часов. Полученную смесь нейтрализовали 1 н. HCl и концентрировали в вакууме. Оставшийся раствор подкисляли (pH 3) 1 н. HCl и экстрагировали AcOEt. Органическую фазу промывали 0,1 н. HCl и насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли в вакууме. Полученное в результате масло растирали со смесью AcOEt:гексан=450:1500 мл, получая (6S,12S,15S,18S,21S,24R,27S,30S,33S,36S)-6-[(1R)-1-гидроксиэтил]-36-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-12,15,18,27,30-пентаизобутил-33-изопропил-2,2,8,11,17,21,24,26,32,35-декаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35-ундекаазагептатриаконтан-37-новую кислоту (80,4 г).
Пример получения 168
К раствору целевого соединения примера получения 152 (5,70 г) в AcOEt (100 мл) добавляли раствор изотиоцианатобензола (1,08 г) в AcOEt (10 мл) при комнатной температуре. После перемешивания в течение 2 часов при той же температуре N,N-диметиламинопропиламин (820 мг) добавляли к раствору. Раствор перемешивали в течение 15 минут, промывали 0,2 н. HCl, NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая порошкообразное вещество. Полученный в результате порошок растворяли в MeCN (100 мл) и 1 н. HCl (100 мл) добавляли при охлаждении на ледяной бане. После перемешивания при комнатной температуре в течение 6 часов раствор нейтрализовали в Na2CO3 (50 мл), концентрировали в вакууме, для удаления MeCN, и экстрагировали AcOEt (200 мл). Органический слой промывали NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Осадок очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая метил (6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,12,21,24-пентаизобутил-27-изопропил-5,11,15,18,20,26,29,35-октаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-2,5,8,11,14,17,20,23,26,29,32,35-додекаазагептатриаконтан-37-оат (3,77 г) в виде порошка.
Пример получения 169
К раствору метил (3S,6S,9S,12S,15R,18S,21S,24S,27S,30S)-3-втор-бутил-30-[(1R)-1-гидроксиэтил]-27-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,18,21-тетраизобутил-24-изопропил-8,12,15,17,23,26,32-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оата (230 мг) в MeOH (4 мл) добавляли 1 н. LiOH (1,84 мл) при охлаждении на ледяной бане. После перемешивания в течение 1 часа при той же температуре раствор подкисляли 5% лимонной кислотой, для доведения pH до 5, концентрировали в вакууме, для удаления MeOH, и экстрагировали AcOEt (20 мл). Органический слой промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок растирали с Et2O, получая (3S,6S,9S,12S,15R,18S,21S,24S,27S,30S)-3-втор-бутил-30-[(1R)-1-гидроксиэтил]-27-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,18,21-тетраизобутил-24-изопропил-8,12,15,17,23,26,32-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-новую кислоту (188 мг) в виде твердого вещества.
Соединения примеров получения 170-177 получали по методике, аналогичной примеру получения 169.
Пример получения 178
К раствору целевого соединения примера получения 1 (66 мг) в CH2Cl2 (2,5 мл) добавляли (2R)-2-[(трет-бутоксикарбонил)(метил)амино]-4-метилпентановую кислоту (16,5 мг), и HOAt (9,1 мг), и WSCD (10,4 мг) при охлаждении на ледяной бане и смесь перемешивали в течение 1,5 часов при охлаждении на ледяной бане. Полученную смесь концентрировали в вакууме и осадок экстрагировали AcOEt. Органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (6R,9S,12S,15S,18S,21R,24S,27S,30S,33S,36S)-36-[(1R)-1-гидроксиэтил]-33-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,12,15,24,27-гексаизобутил-30-изопропил-2,2,5,8,14,18,21,23,29,32-декаметил-4,7,10,13,16,19,22,25,28,31,34-ундекаоксо-3-окса-5,8,11,14,17,20,23,26,29,32,35-ундекаазагептатриаконтан-37-оат (57 мг) в виде бледно-желтого порошка. Полученный продукт использовали в следующей реакции без дальнейшей очистки.
Соединения примеров получения 179-192 получали по методике, аналогичной примеру получения 178.
Пример получения 193
К раствору целевого соединения примера получения 178 (57 мг) в CH2Cl2 (1,2 мл) добавляли ТФУК (0,3 мл) при охлаждении на ледяной бане и смесь перемешивали в течение 2 часов при охлаждении на ледяной бане. Полученную смесь нейтрализовали водным раствором NaHCO3 (430 мг в 20 мл H2О) при охлаждении на ледяной бане и концентрировали в вакууме. К оставшемуся раствору добавляли насыщенный водный раствор NaHCO3, для доведения до pH 8, и смесь экстрагировали AcOEt. Органическую фазу промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме, получая метил (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-3,6,9,12,21,24-гексаизобутил-27-изопропил-5,11,15,18,20,26,29-гептаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-оат (53 мг) в виде бесцветного твердого вещества.
Соединения примеров получения 194-207 получали по методике, аналогичной примеру получения 193.
Пример получения 208
К раствору целевого соединения примера получения 57 (1,00 г), (2S)-2-{[(9H-флуорен-9-илметокси)карбонил](метил)амино}кислоты (399 мг) и Bop-Cl (416 мг) в CH2Cl2 (20 мл) добавляли N-этил-N-изопропил-2-пропанамин (422 мг) при охлаждении на ледяной бане. После перемешивания при комнатной температуре в течение ночи реакционный раствор концентрировали в вакууме. Осадок растворяли в AcOEt (50 мл) и раствор промывали 0,5 н. HCl, 1 M NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая метил (5R,8S,11S,14S,17S,20R,23S,26S,29S,32S,35S)-8-(1-трет-бутоксиэтил)-1-(9H-флуорен-9-ил)-35-[(1R)-1-гидроксиэтил]-32-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-11,14,23,26-тетраизобутил-29-изопропил-4,5,7,13,17,20,22,28,31-нонаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-2-окса-4,7,10,13,16,19,22,25,28,31,34-ундекаазагексатриаконтан-36-оат (1,25 г) в виде аморфного порошка.
Соединения примеров получения 209-212 получали по методике, аналогичной примеру получения 208.
Пример получения 213
К раствору целевого соединения примера получения 159 (110 мг) в диоксане (2,4 мл) добавляли 1 н. NaOH (0,6 мл) при температуре окружающей среды и смесь перемешивали в течение 2 часов. К реакционной смеси добавляли 10% водный раствор лимонной кислоты, для доведения pH до 4, и раствор экстрагировали AcOEt. Органическую фазу промывали насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок растирали с Et2O, получая (3R,6S,9S,12S,15S,18R,21S,24S,27S,30S,33S)-33-[(1R)-1-гидроксиэтил]-30-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,12,21,24-пентаизобутил-27-изопропил-3,5,11,15,18,20,26,29-октаметил-4,7,10,13,16,19,22,25,28,31-декаоксо-2,5,8,11,14,17,20,23,26,29,32-ундекаазатетратриаконтан-34-новую кислоту (79 мг) в виде бесцветного порошка.
Соединение примера получения 214 получали по методике, аналогичной примеру получения 213.
Пример получения 215
Раствор целевого соединения примера получения 110 (193 мг) растворяли в 10% ТФУК в CH2Cl2 (6 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 2 часов к реакционному раствору добавляли 1 M водный раствор NaHCO3, для доведения до pH 8. Реакционную смесь экстрагировали CHCl3 (50 мл) и органический слой промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая (1R)-1-{(2S,5R,8S,11S,14S,17S,20R,23S,26S,29S,32S)-8-(1-гидроксиэтил)-32-[(1R,2R,4E)-1-гидрокси-2-метилгекс-4-ен-1-ил]-11,14,23,26-тетраизобутил-29-изопропил-4,5,7,13,17,20,22,28,31-нонаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2-ил}этилацетат (183 мг) в виде аморфного порошка.
Пример получения 216
К раствору целевого соединения примера получения 215 (187 мг) в CH2Cl2 (1,2 мл) добавляли 4-нитрофенилхлорформат (54 мг) и N-метилморфолин (29 мкл). После перемешивания смеси в течение ночи три дополнительные порции 4-нитрофенилхлорформата (54 мг) и N-метилморфолина (29 мкл) добавляли с интервалом 1 час. После полного использования исходного соединения смесь разбавляли AcOEt, промывали 1 н. водной хлористоводородной кислотой и водным NaHCO3, сушили над MgSO4 и концентрировали. Осадок хроматографировали на силикагеле (гексан/AcOEt=1/4 и затем CH2Cl2/MeOH=9/1), получая (1R)-1-[(2S,5R,8S,11S,14S,17S,20R,23S,26S,29S,32S)-32-[(1R,2R,4E)-1-гидрокси-2-метилгекс-4-ен-1-ил]-11,14,23,26-тетраизобутил-29-изопропил-4,5,7,13,17,20,22,28,31-нонаметил-8-(1-{[(4-нитрофенокси)карбонил]окси}этил)-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2-ил]этилацетат (87 мг).
Пример получения 217
К раствору целевого соединения примера получения 216 (27 мг) в тетрагидрофуране (1 мл) добавляли морфолин (4,9 мкл) и смесь перемешивали при комнатной температуре в течение ночи. Осадок хроматографировали на силикагеле (CH2Cl2/MeOH=97/3 до 90/10), получая 1-{(2S,5S,8S,11S,14R,17S,20S,23S,26S,29S,32R)-29-[(1R)-1-ацетоксиэтил]-26-[(1R,2R,4E)-1-гидрокси-2-метилгекс-4-ен-1-ил]-5,8,17,20-тетраизобутил-23-изопропил-1,7,11,14,16,22,25,31,32-нонаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2-ил}этилморфолин-4-карбоксилат (22 мг).
Соединения примеров получения 218-220 получали по методике, аналогичной примеру получения 217, и соединение примера 1 получали по методике, аналогичной примеру получения 4.
Пример 2
К раствору целевого соединения примера получения 139 (42,7 мг) в CH2Cl2 (43 мл) добавляли HOAt (5,5 мг) и WSCD (6,3 мг) при охлаждении на ледяной бане и смесь перемешивали при 5°C в течение 13 часов. Реакционную смесь концентрировали в вакууме и осадок экстрагировали AcOEt. Органическую фазу промывали H2O, 10% водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали препаративной тонкослойной хроматографией (CHCl3:MeOH=95:5), получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-33-втор-бутил-3-этил-6-(1-гидроксиэтил)-9-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-15,18,27,30-тетраизобутил-12-изопропил-1,4,10,13,19,21,24,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (29 мг) в виде бесцветного порошка.
Соединения примеров 3-11 получали по методике, аналогичной примеру 2.
Пример 12
Целевое соединение примера 25 ниже (1,00 г) растворяли в 10% ТФУК в CH2Cl2 (10 мл) при охлаждении на ледяной бане. После перемешивания при той же температуре в течение 2 часов к реакционному раствору добавляли 1 M водный раствор NaHCO3, для доведения pH до 8. Реакционную смесь экстрагировали CHCl3 (50 мл) и органический слой промывали насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме. Полученный осадок очищали на колонке с силикагелем (элюент: 2% MeOH в CHCl3), получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-6-[(1R)-1-гидроксиэтил]-33-(1-гидроксиэтил)-9-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-15,18,27,30-тетраизобутил-12-изопропил-1,3,4,10,13,19,21,24,28-нонаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (625 мг) в виде аморфного порошка.
Соединение примера 13 получали по методике, аналогичной примеру 12.
Пример 14
К раствору целевого соединения примера получения 41 (42,7 мг) в CH2Cl2 (48 мл) добавляли HOAt (7,3 мг) и WSCD (8,3 мг) при охлаждении на ледяной бане и смесь перемешивали при 5°C в течение 13 часов. Реакционную смесь концентрировали в вакууме и осадок экстрагировали AcOEt. Органическую фазу промывали H2O, 10% водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали препаративной тонкослойной хроматографией (CHCl3:MeOH=95:5), получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-33-втор-бутил-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-15,18,27,30-тетраизобутил-12-изопропил-1,3,4,10,13,19,21,24,28-нонаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (29 мг) в виде бесцветного порошка.
Соединения примеров 15-24 получали по методике, аналогичной примеру 14.
Пример 25
К раствору целевого соединения примера получения 20 (785 мг) и HOAt (99 мг) в CH2Cl2 (785 мл) добавляли раствор WSCD (113 мг) в CH2Cl2 (10 мл) в течение 3 часов при охлаждении на ледяной бане. После перемешивания при 5°C в течение ночи раствор концентрировали в вакууме. Осадок растворяли в смеси AcOEt (40 мл)/H2O (40 мл), раствор промывали 0,2 н. HCl, насыщенным NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая порошок. Неочищенный порошок очищали колоночной хроматографией на силикагеле (элюент: гексан:ацетон, 2:1), получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-33-(1-трет-бутоксиэтил)-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-15,18,27,30-тетраизобутил-12-изопропил-1,3,4,10,13,19,21,24,28-нонаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (365 мг) в виде аморфного порошка.
Соединения примеров 26-35 получали по методике, аналогичной примеру 25.
Пример 36
К раствору целевого соединения примера получения 75 (48 мг) в CH2Cl2 (3,7 мл) добавляли HOAt (6,0 мг) и WSCD (6,9 мг) при охлаждении на ледяной бане и смесь перемешивали при 5°C в течение 13 часов. Реакционную смесь концентрировали в вакууме и осадок экстрагировали AcOEt. Органическую фазу промывали H2O, 10% водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали препаративной тонкослойной хроматографией (CHCl3:MeOH=95:5), получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-3,15,18,27,30,33-гексаизобутил-12-изопропил-1,4,10,13,19,21,24,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (34 мг) в виде бесцветного порошка.
Соединения примеров 37-51 получали по методике, аналогичной примеру 36.
Пример 52
К раствору целевого соединения примера получения 161 (920 мг) и HOAt (489 мг) в CH2Cl2 (1000 мл) добавляли раствор WSCD (558 мг) в CH2Cl2 (10 мл) и смесь перемешивали при охлаждении на ледяной бане. После перемешивания при 5°C в течение ночи раствор концентрировали в вакууме. Осадок растворяли в смеси AcOEt (100 мл)/H2O (100 мл), раствор промывали 0,2 н. HCl, насыщенным NaHCO3 и насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме, получая порошок. Неочищенный порошок очищали колоночной хроматографией на силикагеле (элюент: 4% MeOH в CHCl3), получая (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-24-(1-трет-бутоксиэтил)-30-[(1R)-1-гидроксиэтил]-33-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-6,9,18,21-тетраизобутил-3-изопропил-1,4,10,12,15,19,25,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (448 мг) в виде аморфного порошка.
Соединения примеров 53-73 получали по методике, аналогичной примеру 52.
Пример 74
К раствору целевого соединения примера получения 213 (79 мг) в CH2Cl2 (63 мл) добавляли HOAt (10,3 мг) и WSCD (11,7 мг) при охлаждении на ледяной бане и смесь перемешивали при 5°C в течение 13 часов. Реакционную смесь концентрировали в вакууме и осадок экстрагировали AcOEt. Органическую фазу промывали H2O, 10% водным раствором лимонной кислоты, насыщенным водным раствором NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель удаляли в вакууме и осадок очищали препаративной тонкослойной хроматографией (CHCl3:MeOH=95:5), получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-15,18,27,30,33-пентаизобутил-12-изопропил-1,3,4,10,13,19,21,24,28-нонаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (43 мг) в виде бесцветного порошка.
Пример 75
(3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-3-[(Бензилокси)метил]-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R)-1-гидрокси-2-метилгексил]-15,18,27,30,33-пентаизобутил-12-изопропил-1,4,10,13,19,21,24,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (22 мг) растворяли в MeOH (1 мл) и гидрировали при комнатной температуре и атмосферном давлении над 5% Pd на угле. Спустя 2 часа катализатор отфильтровывали и фильтрат концентрировали в вакууме при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (гексан:ацетон, 1:1) с выходом (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-6-[(1R)-1-гидроксиэтил]-3-(гидроксиметил)-9-[(1R,2R)-1-гидрокси-2-метилгексил]-15,18,27,30,33-пентаизобутил-12-изопропил-1,4,10,13,19,21,24,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекона (18 мг).
Пример 76
(3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-3-[(Бензилокси)метил]-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R,4E)-1-гидрокси-2-метил-4-гексен-1-ил]-15,18,27,30-тетраизобутил-12-изопропил-33-(1-метоксиэтил)-1,4,10,13,19,21,24,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (20 мг) растворяли в MeOH (2 мл) и гидрировали при комнатной температуре и атмосферном давлении над Pd на угле. Спустя 40 минут катализатор отфильтровывали и фильтрат концентрировали в вакууме при пониженном давлении. Осадок очищали препаративной тонкослойной хроматографией на силикагеле (CHCl3:MeOH=95:5), получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-6-[(1R)-1-гидроксиэтил]-3-(гидроксиметил)-9-[(1R,2R)-1-гидрокси-2-метилгексил]-15,18,27,30-тетраизобутил-12-изопропил-33-(1-метоксиэтил)-1,4,10,13,19,21,24,28-октаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекан (12 мг) в виде бесцветного порошка.
Пример 77
К раствору целевого соединения примера получения 217 (11,5 мг) в тетрагидрофуране (1 мл) добавляли метилат натрия (25 мкмоль) в MeOH (1 мл) и смесь перемешивали при комнатной температуре в течение ночи. Смесь подвергали очистке ODS, получая 1-{(2S,5S,8S,11S,14R,17S,20S,23S,26S,29S,32R)-29-[(1R)-1-гидроксиэтил]-26-[(1R,2R,4E)-1-гидрокси-2-метилгекс-4-ен-1-ил]-5,8,17,20-тетраизобутил-23-изопропил-1,7,11,14,16,22,25,31,32-нонаметил-3,6,9,12,15,18,21,24,27,30,33-ундекаоксо-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2-ил}этилморфолин-4-карбоксилат (9 мг).
Соединения примеров 78-80 получали по методике, аналогичной примеру 77.
Пример 81
Раствор (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R,4E)-1-гидрокси-2-метилгекс-4-ен-1-ил]-15,18,27,30-тетраизобутил-12-изопропил-33-(1-метоксиэтил)-1,3,4,10,13,19,21,24,28-нонаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекана (60 мг) в MeOH (6,0 мл) гидрировали над 20% Pd/C (50% влажности; 30 мг) при комнатной температуре в течение 2 часов. Смесь фильтровали и фильтрат концентрировали. Осадок выпаривали, получая (3R,6S,9S,12S,15S,18S,21R,24S,27S,30S,33S)-6-[(1R)-1-гидроксиэтил]-9-[(1R,2R)-1-гидрокси-2-метилгексил]-15,18,27,30-тетраизобутил-12-изопропил-33-(1-метоксиэтил)-1,3,4,10,13,19,21,24,28-нонаметил-1,4,7,10,13,16,19,22,25,28,31-ундекаазациклотритриаконтан-2,5,8,11,14,17,20,23,26,29,32-ундекон (56 мг).
Соединения примеров 82 и 83 получали по методике, аналогичной примеру 81.
Структура и данные масс-спектрометрии (ESI-MС [(M+H)+], если не указано иное) для соединений примеров и примеров получения показаны в таблицах 1-42, и пик δ м.д.1H-NMR данных (хлороформ-d, TMS внутренний стандарт) для соединений примеров показан в таблице 43.
Таблицы 1-11
Соединения примеров, представленные следующей формулой

Таблица 1

Таблица 2

Таблица 3

Таблица 4

Таблица 5

Таблица 6

Таблица 7

Таблица 8
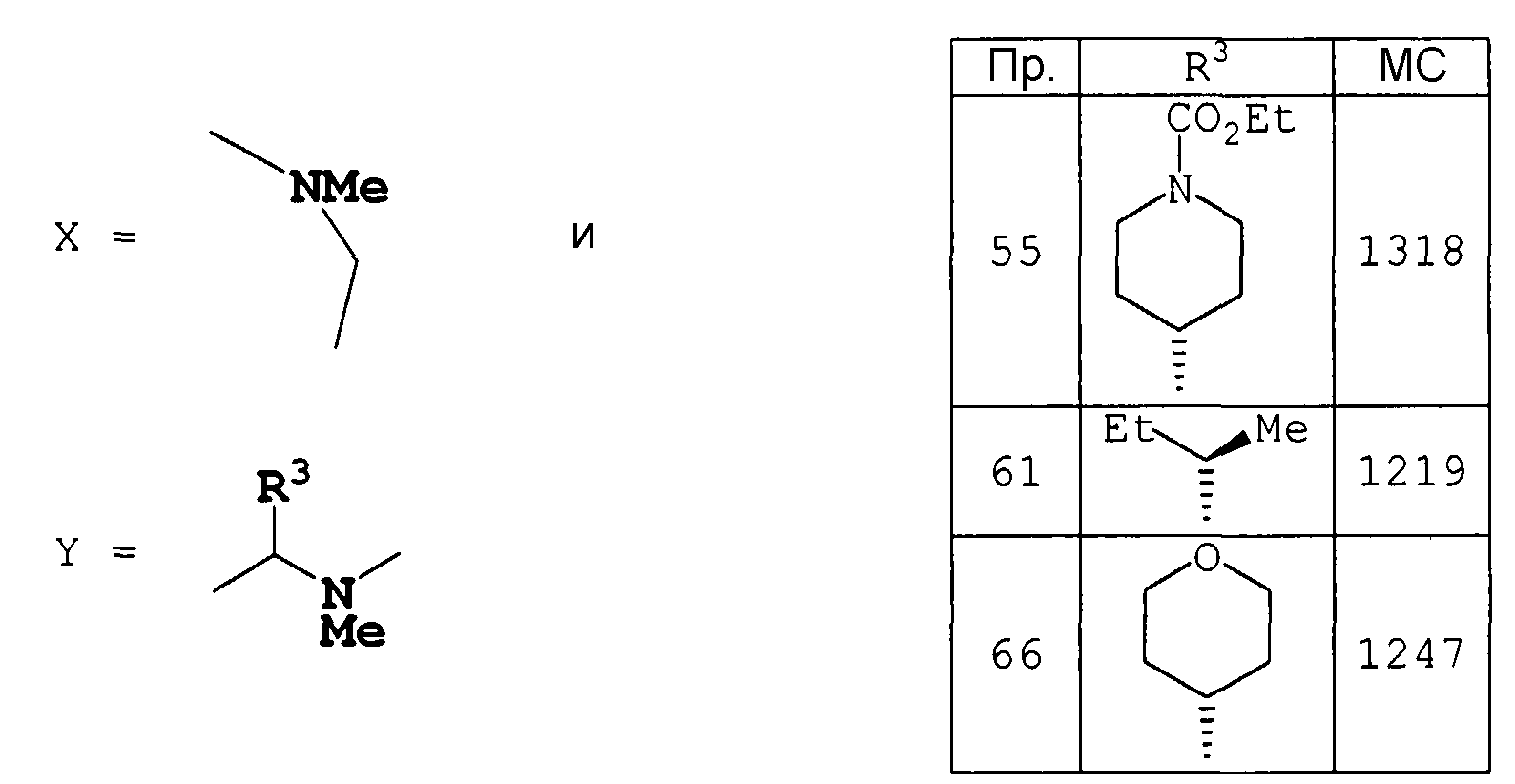
Таблица 9

Таблица 10

Таблица 11

Таблица 12
Соединения примеров, представленные следующей формулой

Таблицы 13-22
Соединения примеров получения, представленные следующей формулой типа 1

Таблица 13 (тип 1-1)

Таблица 14 (тип 1-2)
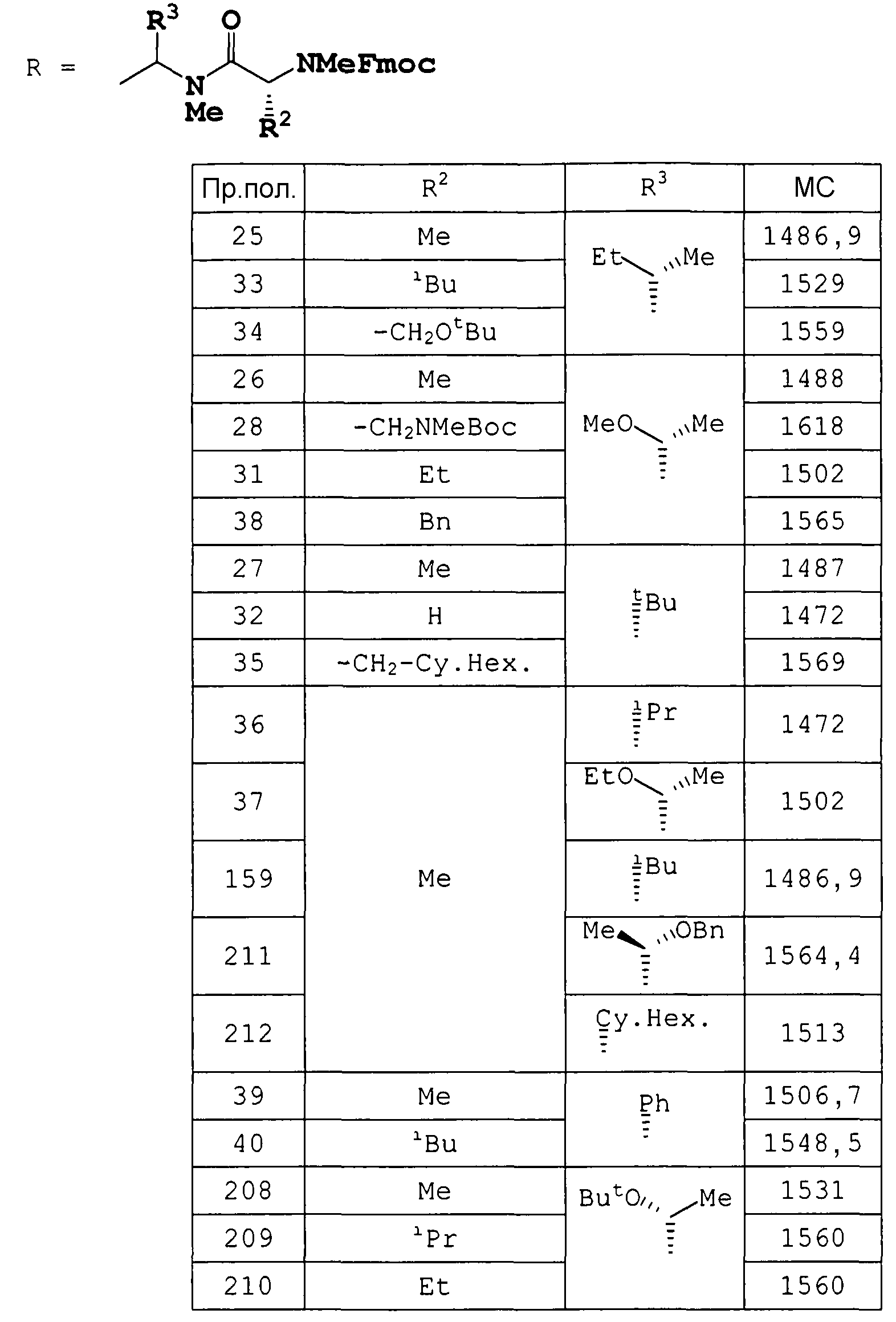
Таблица 15 (тип 1-3)
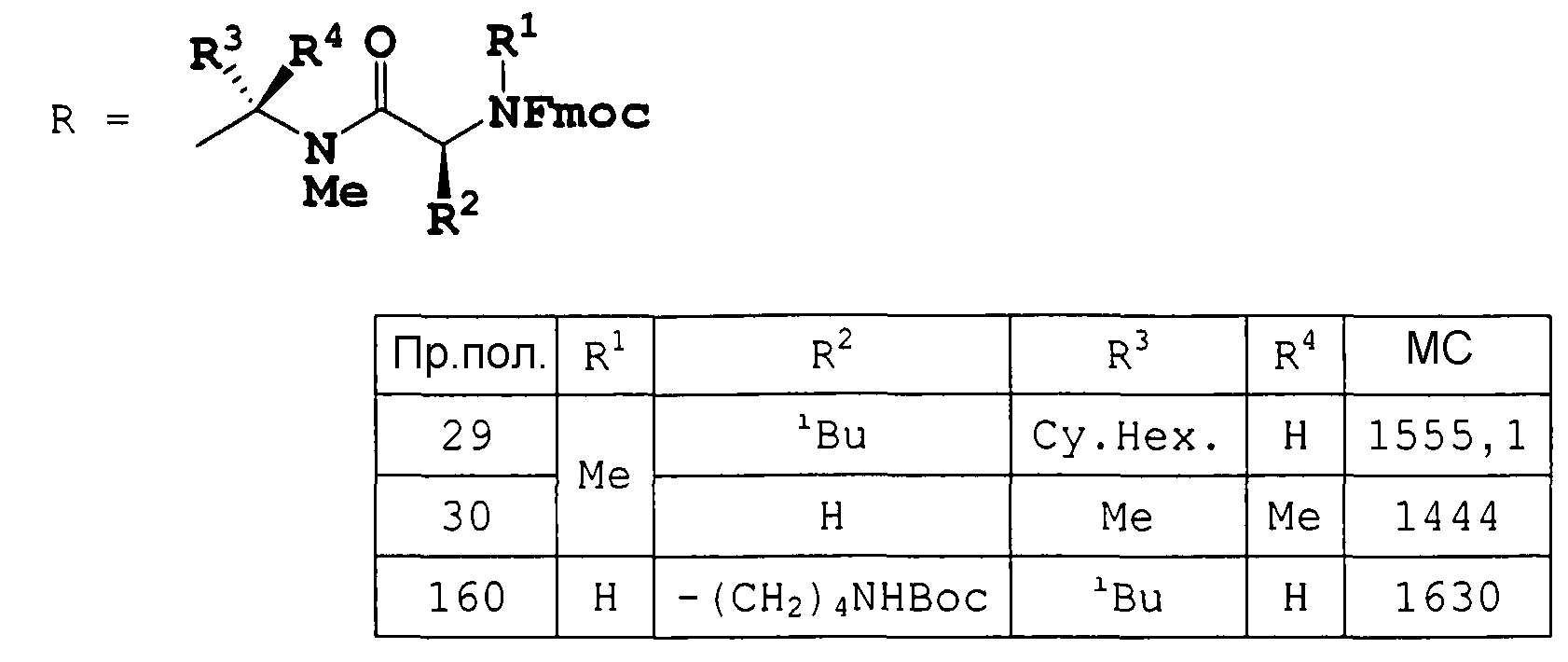
Таблица 16 (тип 1-4)

Таблица 17 (тип 1-5)
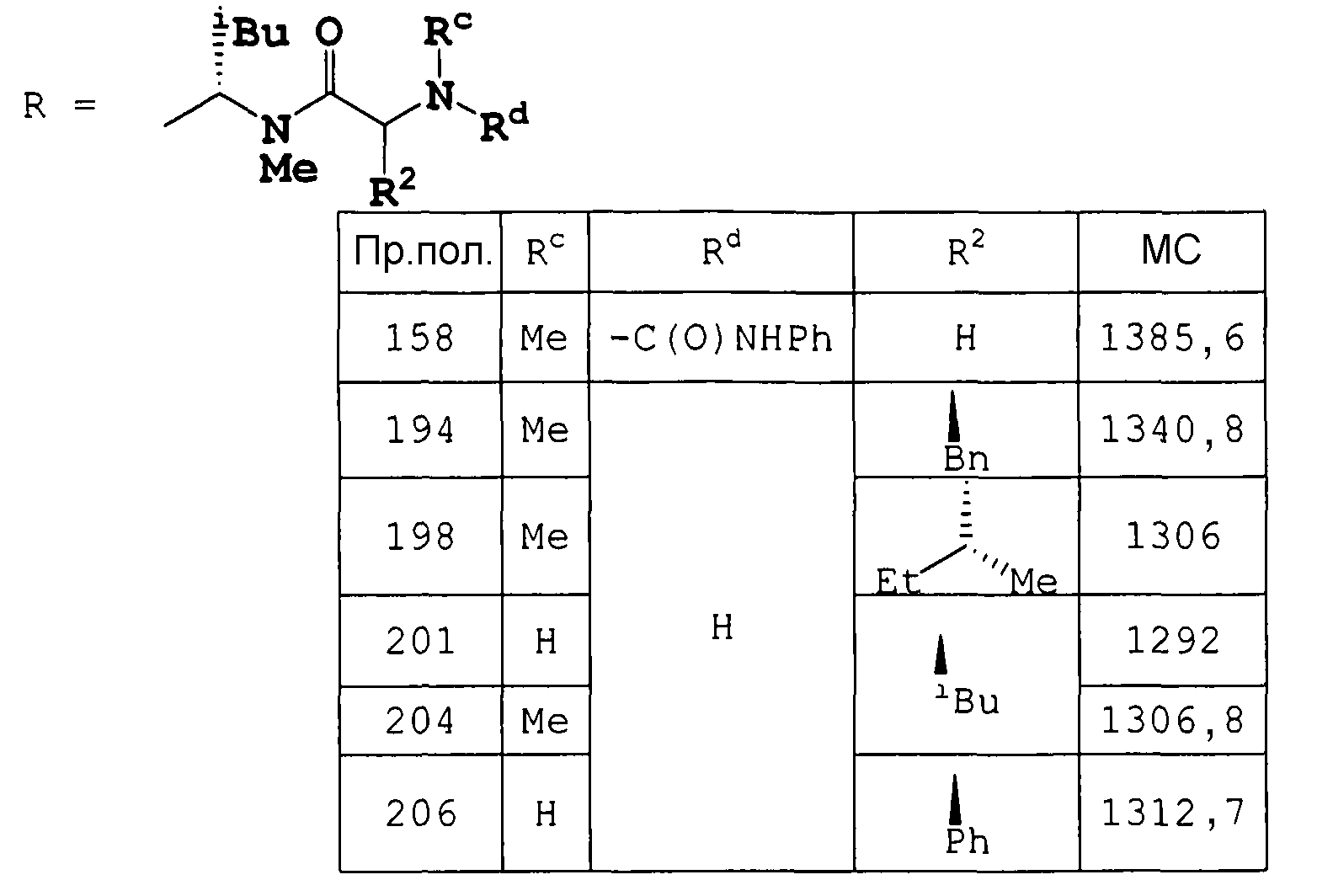
Таблица 18 (тип 1-6)

Таблица 19 (тип 1-7)

Таблица 20 (тип 1-8)

Таблица 21 (тип 1-9)

Таблица 22 (тип 1-10)

Таблицы 23-31
Соединения примеров получения, представленные следующей формулой типа 2
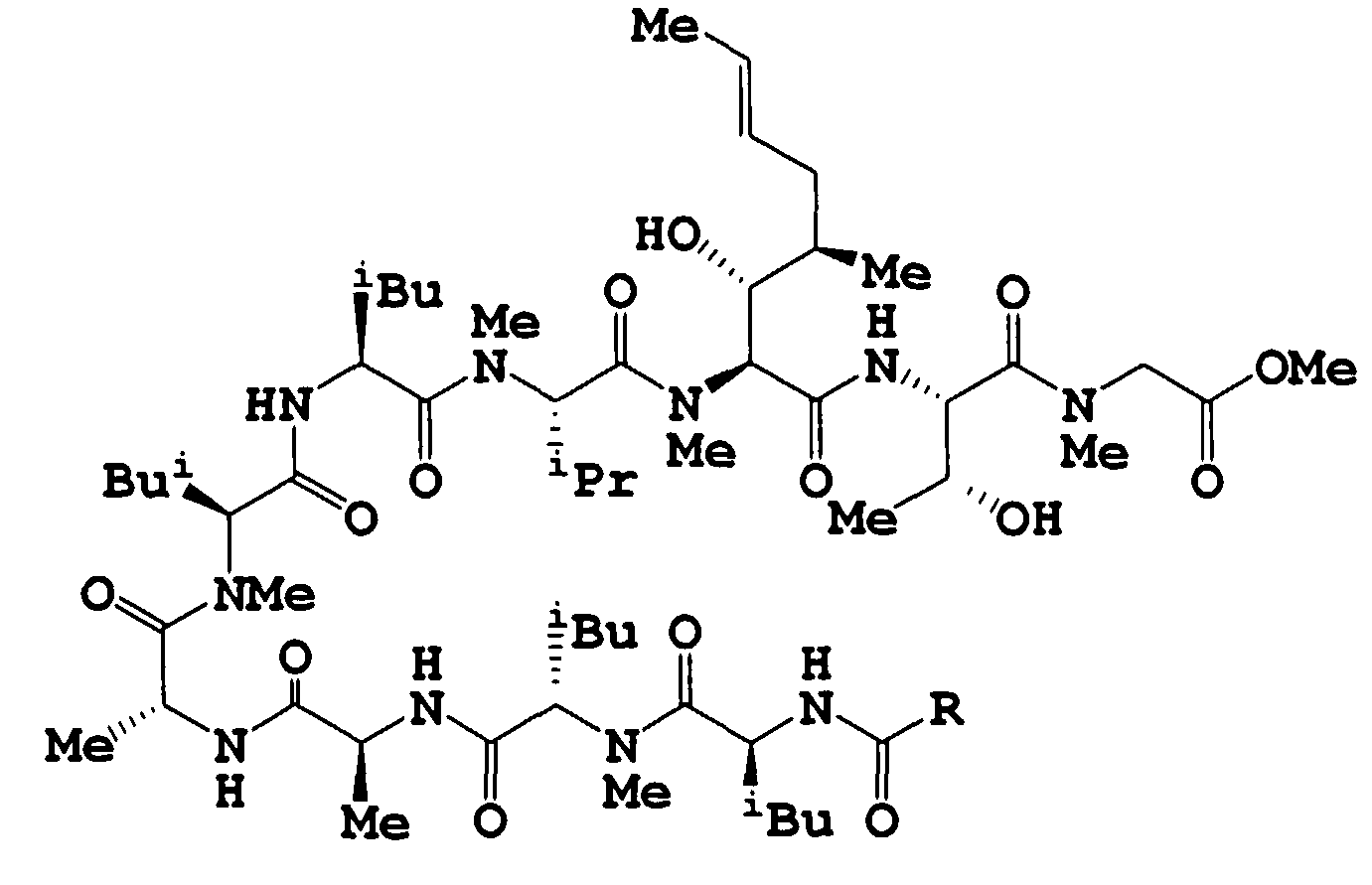
Таблица 23 (тип 2-1)
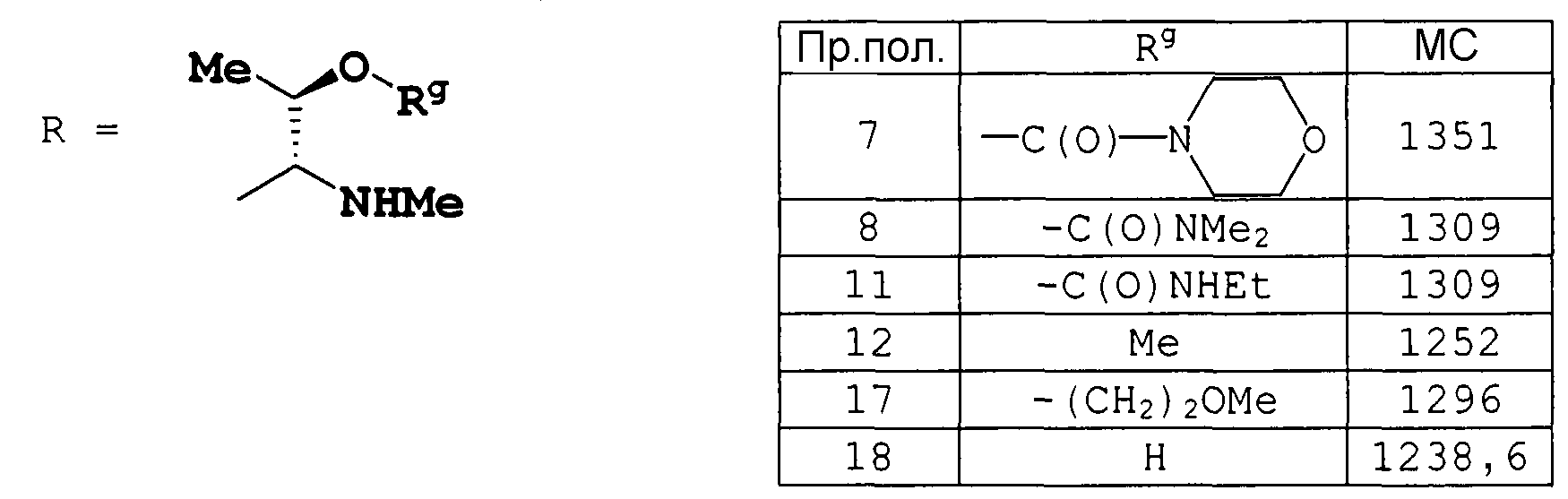
Таблица 24 (тип 2-2)

Таблица 25 (тип 2-3)

Таблица 26 (тип 2-4)

Таблица 27 (тип 2-5)

Таблица 28 (тип 2-6)

Таблица 29 (тип 2-7)

Таблица 30 (тип 2-8)
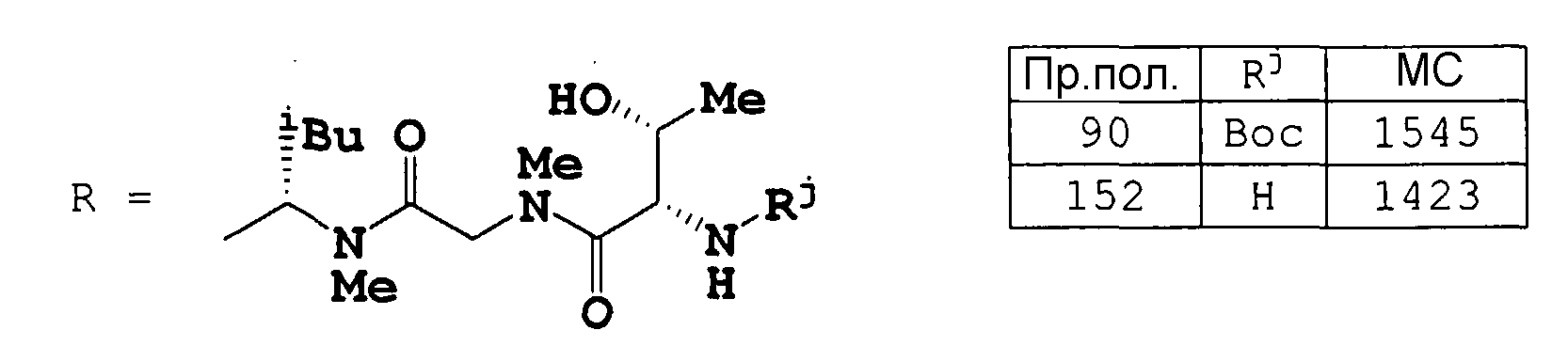
Таблица 31 (тип 2-9)

Таблицы 32-36
Соединения примеров получения, представленные следующей формулой типа 3

Таблица 32 (тип 3-1)

Таблица 33 (тип 3-2)
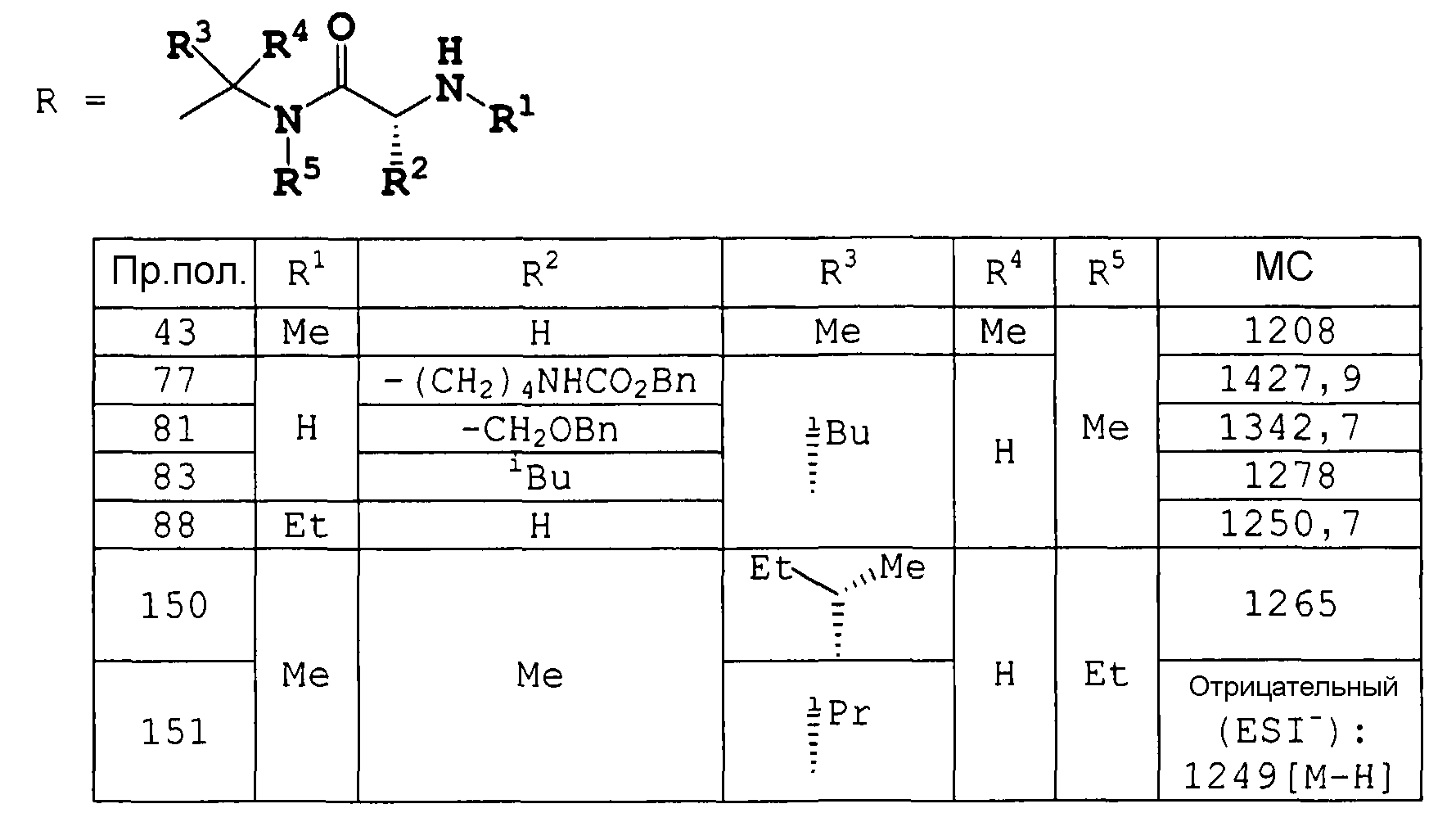
Таблица 34 (тип 3-3)

Таблица 35 (тип 3-4)

Таблица 36 (тип 3-5)

Таблицы 37-40
Соединения примеров получения, представленные следующей формулой типа 4

Таблица 37 (тип 4-1)

Таблица 38 (тип 4-2)
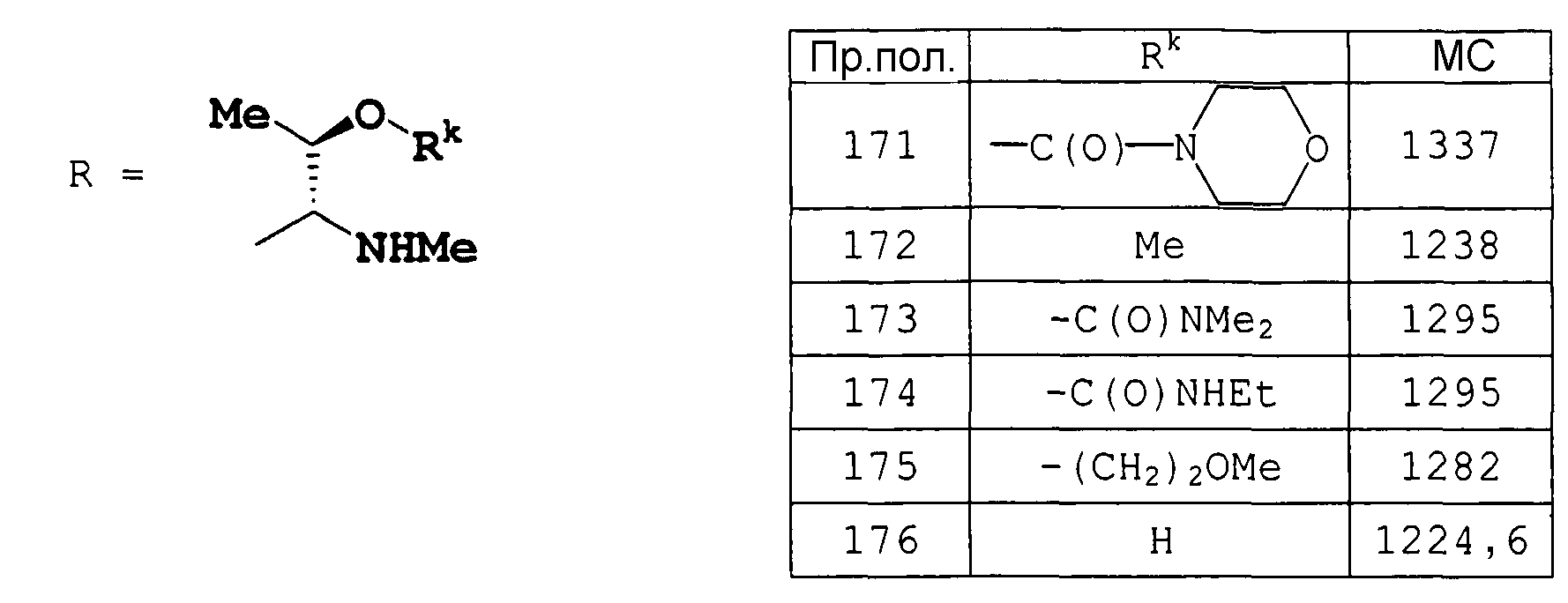
Таблица 39 (тип 4-3)
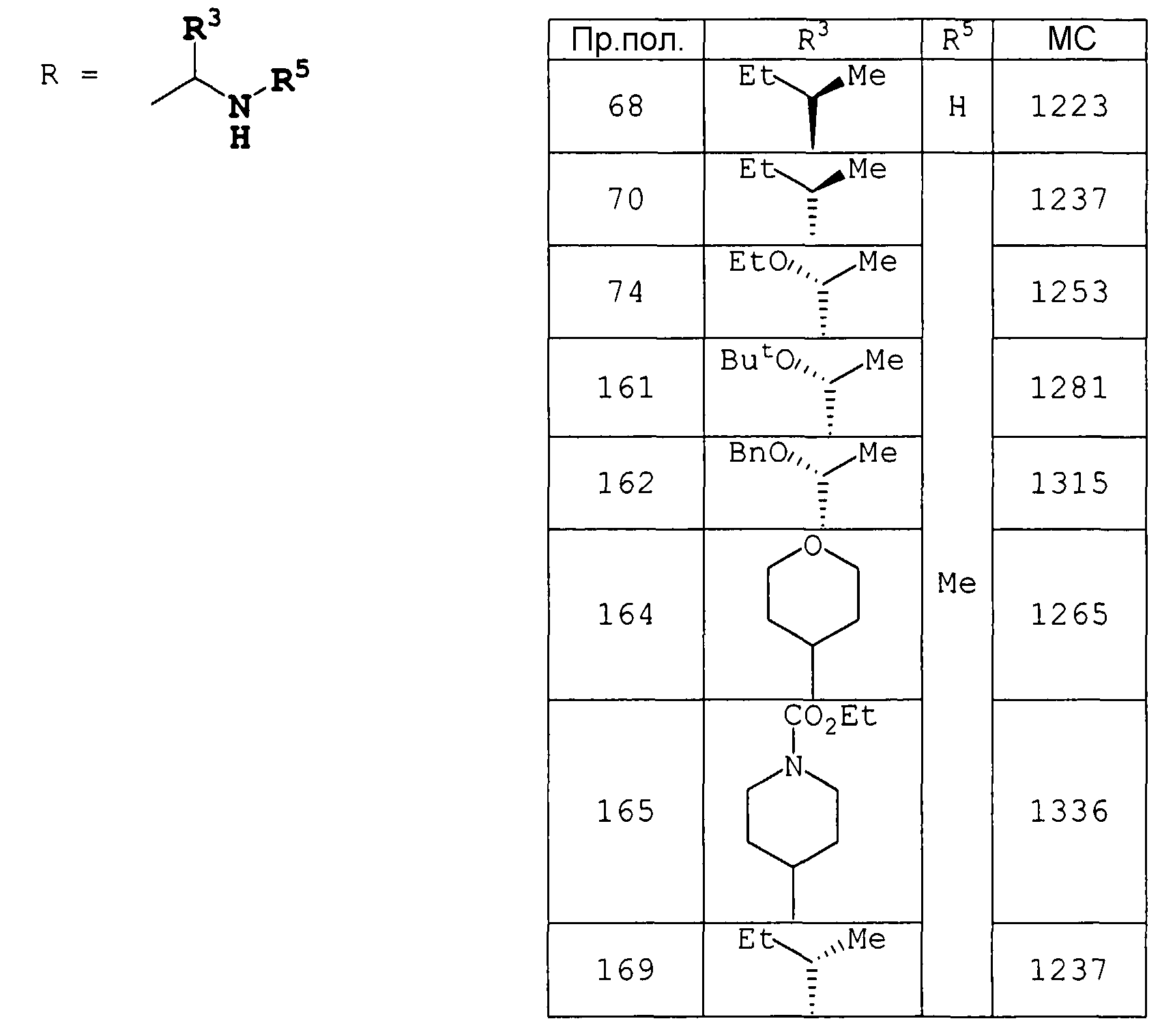
Таблица 40 (тип 4-4)

Таблица 41
Соединения примеров получения другого типа, представленные следующей формулой

Таблица 42
Соединения примеров получения другого типа, представленные следующей формулой
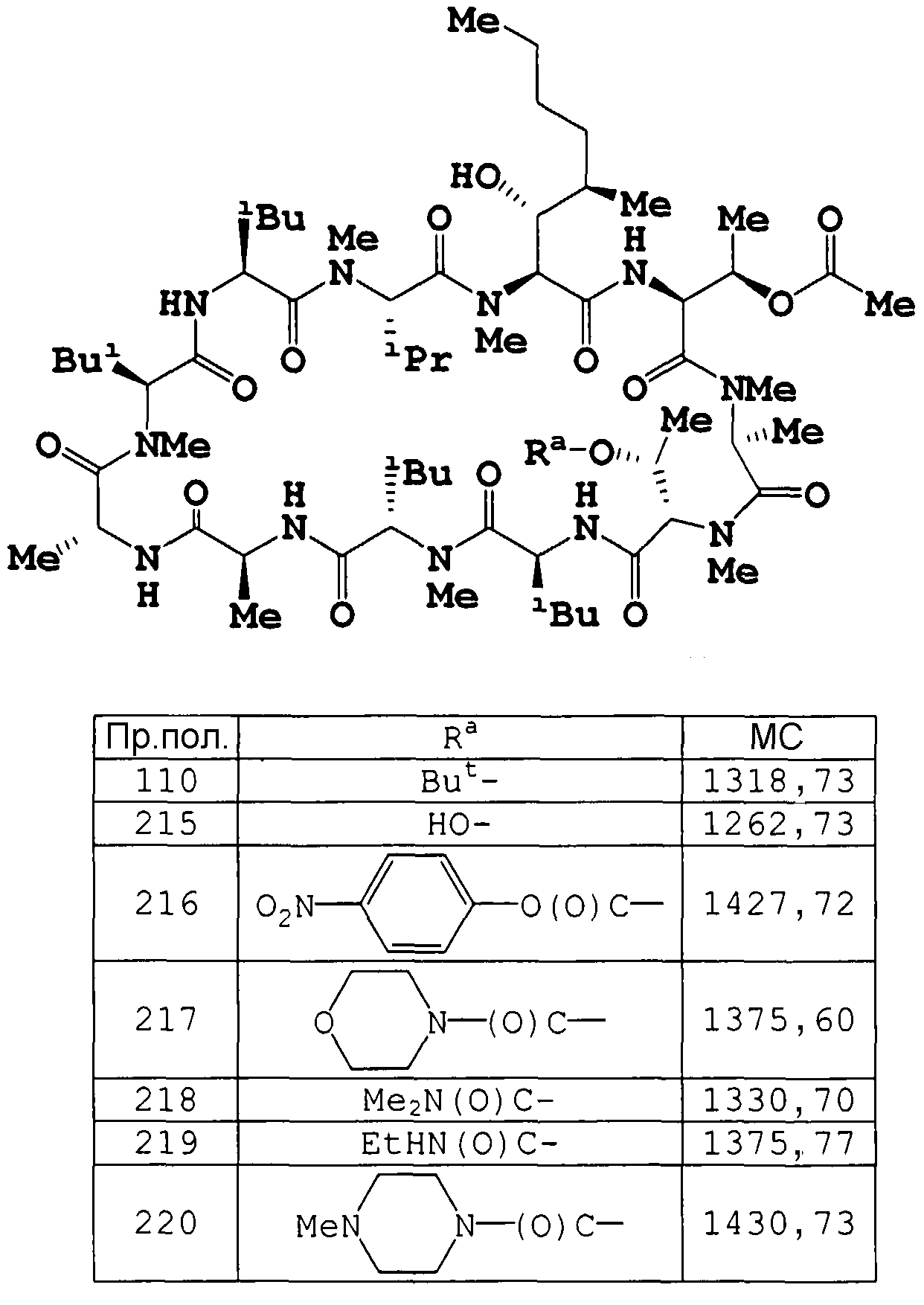
Структура соединений настоящего изобретения показана в следующей таблице 44. Данные соединения могут быть легко получены указанными выше способами получения, способами, описанными в примерах или примерах получения, или способами, хорошо известными специалистам в данной области, или их вариациями.
Таблица 44

Реферат
Настоящее изобретение относится к новому циклическому пептидному соединению или его фармацевтически приемлемой соли, которое обладает активностью против вируса гепатита С, основанной на ингибирующей активности РНК репликации репликона вируса гепатита С, способу его получения, включающему перегруппировку в мягкой кислотной среде и последующие аминокислотные обменные реакции, фармацевтической композиции, содержащей указанное соединение, и применению соединения для получения лекарственного средства, обладающего анти-HCV активностью. 7 н. и 9 з.п. ф-лы, 44 табл.
Формула

где Х означает

где R1 означает водород или низший алкил;
R2 означает водород, фенил или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, цикло(низшего)алкила, низшего алкокси, фенила, фенил(низшего)алкокси, необязательно замещенного низшим алкилом карбамоилокси и необязательно замещенного низшим алкилом, бензилоксикарбонилом или трет-бутоксикарбонилом амино;
и означает насыщенную 5-6-членную N-содержащую гетеромоноциклическую группу, содержащую 1-4 атома азота, или ненасыщенную конденсированную гетероциклическую группу, содержащую 1-4 атома азота;
Y означает


где R3 означает цикло(низший)алкил, фенил, необязательно замещенную низшей алкоксикарбонильной группой гетероциклическую группу, представляющую собой ненасыщенную 5-6-членную гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода или 1-3 атома азота, или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, цикло(низшего)алкила, низшего алкокси, фенила, фенил(низшего)алкокси, низший алкокси(низшего)алкокси, необязательно замещенного низшим алкилом, бензилоксикарбонилом или трет-бутоксикарбонилом амино и -OC(O)NR6R7 (где R6 и R7 означают, каждый независимо, водород или низший алкил, или, альтернативно, R6 и R7, вместе с атомом азота, к которому они присоединены, представляют N-содержащую гетероциклическую группу, представляющую собой ненасыщенную 5-6-членную гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, необязательно замещенную низшим алкилом); и
R4 и R5 означают, каждый независимо, водород или низший алкил; и

или его фармацевтически приемлемая соль,
при условии, что, когда R2 означает водород, R3 означает цикло(низший)алкил, фенил, необязательно замещенную низшей алкоксикарбонильной группой гетероциклическую группу, представляющую собой ненасыщенную 5-6-членную гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода или 1-3 атома азота, низший алкоксиметил, фенил(низший)алкил, трет-бутил, втор-бутил, цикло(низший)алкил(низший)алкил или этил, замещенный подходящим заместителем, выбранным из группы, состоящей из гидрокси, низшего алкокси, фенил(низшего)алкокси, низший алкокси(низшего)алкокси, фенила, необязательно замещенного низшим алкилом, бензилоксикарбонилом или трет-бутоксикарбонилом амино и -ОС(O)NR6R7 (где R6 и R7 являются такими, как определено выше).
Х означает
где R1 означает водород или низший алкил; и
R2 означает фенил или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, цикло(низшего)алкила, низшего алкокси, фенила, фенил(низшего)алкокси, ди(низшего)алкилкарбамоилокси и амино, необязательно замещенного одним или двумя подходящими заместителями, выбранными из группы, состоящей из: низшего алкила, бензилоксикарбонила и трет-бутоксикарбонила; и
Y означает
где R3 означает цикло(низший)алкил, фенил или низший алкил, необязательно замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, низшего алкокси и фенил(низшего)алкокси,
R4 означает водород; и
R5 означает низший алкил;
или его фармацевтически приемлемая соль.
где R1 означает низший алкил; и
R2 означает низший алкил,
Y означает
где R3 означает фенил или низший алкил, необязательно замещенный гидроксигруппой или низшей алкоксигруппой,
R4 означает водород; и
R5 означает низший алкил; и
или его фармацевтически приемлемая соль.
где R3 означает низший алкил, необязательно замещенный гидроксигруппой или низшей алкоксигруппой;
или его фармацевтически приемлемая соль.
Х означает
где R1 означает низший алкил; и
R2 означает водород, и
Y означает
где R3 означает цикло(низший)алкил, фенил, гетероциклическую группу, представляющую собой ненасыщенную 5-6-членную гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода или 1-3 атома азота, необязательно замещенную низшим алкоксикарбонилом, (низший)алкокси(низший)алкил, фенил(низший)алкил, трет-бутил, втор-бутил, цикло(низший)алкил(низший)алкил или этил, замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, низшего алкокси, фенил(низшего)алкокси, низший алкокси(низшего)алкокси, -OC(O)NR6R7 (где R6 и R7 означают, каждый независимо, водород или низший алкил, или, альтернативно, R6 и R7, вместе с атомом азота, к которому они присоединены, представляют N-содержашую гетероциклическую группу, представляющую собой ненасыщенную 5-6-членную гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, необязательно замещенную низшим алкилом) и аминогруппы, которая необязательно замещена одним или двумя подходящими заместителями, выбранными из группы, состоящей из: низшего алкила и бензилоксикарбонила; и
R4 и R5 означают, каждый независимо, водород или низший алкил;
или его фармацевтически приемлемая соль.
Y означает
где R3 означает цикло(низший)алкил, фенил, гетероциклическую группу, представляющую собой ненасыщенную 5-6-членную гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода или 1-3 атома азота, необязательно замещенную низшим алкоксикарбонилом, трет-бутил, втор-бутил или этил, замещенный подходящим заместителем, выбранным из группы, состоящей из: гидрокси, низшего алкокси, фенил(низшего)алкокси, низший алкокси(низшего)алкокси и -OC(O)NR6R7 (где R6 и R7 означают, каждый независимо, водород или низший алкил, или, альтернативно, R6 и R7, вместе с атомом азота, к которому они присоединены, представляют N-содержащую гетероциклическую группу, представляющую собой ненасыщенную 5-6-членную гетеромоноциклическую группу, содержащую 1 или 2 атома кислорода и 1-3 атома азота, необязательно замещенную низшим алкилом);
R4 означает водород и
R5 означает низший алкил; и
или его фармацевтически приемлемая соль.
и Y означает
где R3 означает низший алкил; и
R4 и R5 означают, каждый независимо, водород или низший алкил;
или его фармацевтически приемлемая соль.
Х означает


R4 означает водород и
R5 означает низший алкил; и
или его фармацевтически приемлемая соль.
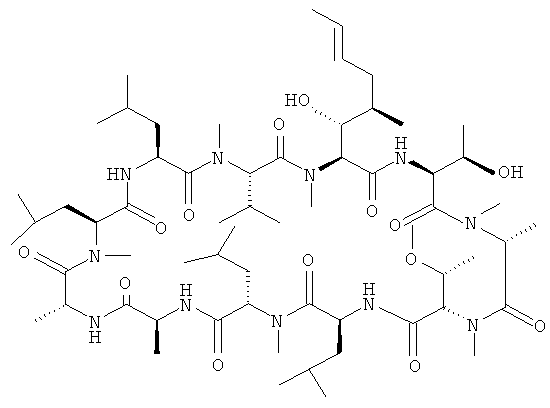
или его фармацевтически приемлемая соль.

или его соли, с соединением формулы (V):

где Y является таким, как определено в п.1,
Р означает аминозащитную группу, и
L означает уходящую группу, или его солью, и непрерывно проведение реакции снятия аминозащитной группы, и далее непрерывно проведение реакции с соединением (VI) формулы:

где Х является таким, как определено в п.1, и
L и Р каждый является таким, как определено выше, или его солью, и проведение реакции снятия аминозащитной группы и реакции циклизации с образованием соединения формулы (Iс):

где Х и Y являются такими, как определено в п.1, или его
фармацевтически приемлемой соли.

где R' означает метоксигруппу или

R" означает водород или

включающий перегруппировку соединения формулы (VII):

или его соли и далее непрерывно проведение реакции аминозащиты, и затем реакции гидролиза, и непрерывно последующее взаимодействие с соединением формулы (X):

где R' является таким, как определено выше, или его солью, и далее непрерывно проведение реакции снятия аминозащитной группы и, наконец, проведение разложения Эдмана 2 или 3 раза.
Документы, цитированные в отчёте о поиске
Смесь аналогов циклоспорина и ее применение в качестве иммуномодулирующего агента
Комментарии