Способ получения макроциклического лактона - RU2234511C2
Код документа: RU2234511C2
Описание
Настоящее изобретение относится к способу получения макроциклического лактона, к способу получения промежуточных соединений и к промежуточным соединениям, используемым при осуществлении этого способа. Изобретение относится, в частности, к способу получения соединения формулы

в которой
R1-R9 независимо друг от друга представляют собой водород или заместитель,
m означает 0, 1 или 2,
n означает 0, 1, 2 или 3 и
А обозначает двойную связь,
В обозначает двойную связь или простую связь,
С обозначает двойную связь,
D обозначает простую связь,
Е и F обозначают двойную связь;
R1 обозначает Н или С1 -С8алкил;
R2 обозначает Н, С1-С8алкил или ОН;
R3 и R4 каждый независимо друг от друга обозначают Н или С1-С8алкил;
R5 обозначает Н или C1-С10алкил;
R6 обозначает Н;
R7 обозначает ОН или ОМе;
R8 и R9 независимо друг от друга обозначают Н или C1-С10алкил,
в свободной форме или в форме соли, заключающемуся в том, что
1) соединение формулы

в которой R1-R7, m, n, А, В, С, D, Е и F имеют те же значения, что и указанные выше для формулы (I), вводят в контакт с биокатализатором, способным избирательно (специфично) окислять спирт в положении 4", с получением соединения формулы

в которой R1, R2, R3, R4, R5, R6, R7, m, n, А, В, С, D, Е и F имеют указанные для формулы (I) значения, и
2) соединение формулы (III) подвергают взаимодействию в присутствии восстановителя с известным амином формулы HN(R8)R9, в которой R8 и R9имеют те же значения, что и указанные для формулы (I).
Методы синтеза соединений формулы (I) описаны в литературе. Однако было установлено, что при осуществлении известных из литературы методов в ходе проведения технологических процессов возникают существенные проблемы, связанные в основном с низким выходом продукта и с необходимостью проведения достаточно трудоемких технологических операций. Так, например, в заявке ЕР-А 0465121 описаны 4’’-кето- и 4’’-амино-4’’-дезоксиавермектиновые соединения и их замещенные аминопроизводные. 4’’-гидроксигруппу таких авермектиновых соединений окисляют до кетоновой группы или заменяют на (замещенную) аминогруппу. Гидроксигруппы в положениях 5 и 23 необходимо защищать во избежание их нежелательного окисления. Для получения соединения, соответствующего приведенной выше формуле (I), 4’’-кето-соединение подвергают аминированию. Таким образом, известные методы в этом отношении не являются удовлетворительными, и поэтому в данной области существует необходимость в разработке доступного усовершенствованного способа получения подобных соединений.
Соединения формул (I), (II) и (III) могут быть представлены в форме их таутомеров. В соответствии с этим выше и в последующем под соединениями формул (I), (II) и (III) понимаются также, когда это целесообразно, и их соответствующие таутомеры, даже если последние и не упоминаются в каждом конкретном случае.
Соединения формул (I), (II) и (III) могут образовывать кислотно-аддитивные соли. Подобные соли образуются, например, с сильными неорганическими кислотами, такими как минеральные кислоты, например с перхлорной, серной, азотной, азотистой, фосфорной или галогенводородной кислотой, с сильными органическими карбоновыми кислотами, такими как незамещенные или замещенные, например галозамещенные, С1-С4алканкарбоновые кислоты, например с уксусной кислотой, с насыщенными или ненасыщенными дикарбоновыми кислотами, например с щавелевой, малоновой, янтарной, малеиновой, фумаровой или фталевой кислотой, с гидроксикарбоновыми кислотами, например с аскорбиновой, молочной, яблочной, винной или лимонной кислотой, либо с бензойной кислотой или с органическими сульфокислотами, такими как незамещенные или замещенные, например галозамещенные, С1-С4алкан- или арилсульфоновые кислоты, например с метан- или n-толуолсульфоновой кислотой. Кроме того, соединения формул (I), (II) и (III), содержащие по меньшей мере одну кислотную группу, могут образовывать соли с основаниями. В качестве примера приемлемых солей с основаниями можно назвать соли металлов, такие как соли щелочных или щелочноземельных металлов, например соли натрия, калия или магния, или соли, образуемые с аммиаком или органическим амином, таким как морфолин, пиперидин, пирролидин, моно-, ди- или три-(низш.)алкиламин, например этил-, диэтил-, триэтил- или диметилпропиламин, или моно-, ди- или тригидрокси-(низш.)алкиламин, например моно-, ди- или триэтаноламин. Помимо этого возможно также образование соответствующих внутренних солей. В соответствии с настоящим изобретением предпочтительны агрохимически приемлемые соли. С учетом тесной взаимосвязи между соединениями формул (I), (II) и (III) в свободной форме и в форме их солей при любом упоминании выше или в последующем описании свободных соединений формул (I), (II) и (III) или их соответствующих солей понимаются также, когда это уместно и целесообразно, соответствующие соли соединений формул (I), (II) и (III) или свободные соединения формул (I), (II) и (III) соответственно. Сказанное относится и к таутомерам соединений формул (I), (II) и (III) и их солям. Следует отметить, что соединения в свободной форме в целом являются предпочтительными в каждом случае.
Предлагаемым в изобретении способом предпочтительно получать соединения формулы (I), в которой
n означает 1,
m означает 1,
А представляет собой двойную связь,
В представляет собой простую связь или двойную связь,
С представляет собой двойную связь,
D представляет собой простую связь,
Е представляет собой двойную связь,
F представляет собой двойную связь либо простую связь и эпоксимостик, либо простую связь и метиленовый мостик,
R1, R2 и R3 означают Н,
R4 означает метил,
R5означает C1-С10алкил, С3-С8циклоалкил или С2-С10алкенил,
R6 означает Н,
R7 означает ОН,
R8 и R9 независимо друг от друга означают Н, С1-С10алкил или С1-С10ацил либо совместно образуют группу -(CH2)q- и
q означает 4, 5 или 6.
Предлагаемым в изобретении способом более предпочтительно получать соединение формулы (I), в которой
n означает 1,
m означает 1,
А, В, С, Е и F представляют собой двойные связи,
D представляет собой простую связь,
R1, R2, и R3 означают Н,
R4 означает метил,
R5 означает втор-бутил или изопропил,
R6 означает Н,
R7 означает ОН,
R8означает метил и
R9 означает H.
Предлагаемым в изобретении способом наиболее предпочтительно получать эмамектин, в частности бензоат эмамектина. Эмамектин представляет собой смесь 4’’-дезокси-4’’-N-метиламиноавермектина B1a/B1b и описан в US 4874749, а также в Journal of Organic Chemistry, т.59 (1994), 7704-7708 под обозначанием МК-244. Соли эмамектина, которые обладают наиболее ценными агрохимическими свойствами, описаны в US 5288710. Соединения формулы (I) являются эффективными пестицидами, прежде всего для борьбы с насекомыми и представителями отряда Acarina (клещей). К указанным вредителям относятся, например, вредители, описанные в заявке ЕР-А 736252. Все такие вредители, упомянутые в указанной заявке, включены в объем настоящего изобретения.
Используемые выше и в последующем описании общие понятия имеют следующие значения, если не указано иное.
Каждая из углеродсодержащих групп и каждое из углеродсодержащих соединений содержат от 1 до 8 включительно, предпочтительно от 1 до 6 включительно, более предпочтительно от 1 до 4 включительно, прежде всего 1 или 2 атома углерода.
Алкил является либо прямоцепочечным, т.е. представляет собой, например, метил, этил, пропил, бутил, пентил или гексил, либо разветвленным, например представляет собой изопропил, изобутил, втор-бутил, трет-бутил, изопентил, неопентил или изогексил.
Алкенил индивидуально либо в качестве структурного элемента других групп и соединений, таких, например, как галоалкенил и арилалкенил, в каждом случае с учетом количества атомов углерода, содержащихся в конкретной группе или конкретном соединении, является либо прямоцепочечным, например представляет собой винил, 1-метилвинил, аллил, 1-бутенил или 2-гексенил, либо разветвленным, например представляет собой изопропенил.
С3-С6Циклоалкил представляет собой циклопропил, циклобутил, циклопентил или циклогексил, прежде всего циклогексил.
Другими объектами изобретения являются следующие:
- указанные выше соединения формулы (III),
- способ получения соединений формулы (III) исходя из соединения формулы (II) в соответствии с описанной выше стадией 1) и
- способ получения соединения формулы (I) исходя из соединения формулы (III) в соответствии с описанной выше стадией 2).
В контексте настоящего изобретения под "биокатализатором" понимаются:
а) живой микроорганизм, например, в виде вегетативных клеток, покоящихся клеток или высушенных вымораживанием клеток,
б) споры указанного микроорганизма,
в) неживой микроорганизм предпочтительно в частично разрушенном виде, т.е. со вскрытой в результате механического или химического воздействия либо в результате распылительной сушки клеточной стенкой/клеточной мембраной,
г) сырые экстракты содержимого клеток указанного микроорганизма и
д) фермент, который обеспечивает (катализирует) превращение соединений формулы (II) в соединения формулы (III).
Наиболее пригодными микроорганизмами для использования в предлагаемом в изобретении способе являются бактерии и грибы. В качестве пригодных для этой цели бактерий особо следует назвать представителей актиномицетов (Actinomycetes), прежде всего из рода Streptomyces. Предпочтительными штаммами рода Streptomyces являются штаммы, выбранные из группы, включающей Streptomyces tubercidicus, Streptomyces chattanoogensis, Streptomyces lydicus, Streptomyces saraceticus и Streptomyces kasugaensis. Наиболее пригодными штаммами для региоселективного окисления гидроксигруппы в положении 4’’ соединений формулы (II) являются, как было установлено, штаммы Streptomyces R-922 (Streptomyces tubercidicus) и прежде всего Streptomyces I-1529 (Streptomyces tubercidicus).
Относящиеся к роду Streptomyces штаммы Streptomyces I-1529 и Streptomyces R-922 в соответствии с Будапештским договором 5 ноября 1999 г. были депонированы в Немецкой коллекции микроорганизмов и клеточных культур (DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Mascheroder Weg 1 b, D-38124 Braunschweig, Germany) под регистрационными номерами DSM-13135 и DSM-13136 соответственно.
К другим штаммам, для которых была выявлена способность осуществлять региоселективное окисление в соответствии с настоящим изобретением, относятся, например, штамм Streptomyces MAAG-7027 (Streptomyces tubercidicus), штамм Streptomyces DSM-40241 (Streptomyces saraceticus, идентифицированный также как Streptomyces chattanoogensis), штамм Streptomyces NRRL-2433 (Streptomyces lydicus ssp. lydicus) и штамм Streptomyces A/96-1208710 (Streptomyces kasugaensis). Все вышеуказанные штаммы обладают высокой степенью родства с относящимися к роду Streptomyces штаммами Streptomyces I-1529 и Streptomyces R-922 соответственно, что подверждается анализом 16s рДНК, выявленная с помощью которого идентичность между такими штаммами составила от 99,4 до 100%.
Соединения формулы (I) являются известными высокоэффективными, соответственно высокоактивными средствами борьбы с вредителями растений. Соединения формулы (II) являются исходными соединениями для получения соединений формулы (I) известными методами, как это описано, например, в ЕР 301806, а также предлагаемым в изобретении способом с использованием микроорганизмов.
В известных методах на первой стадии в соединении формулы (II) защищают атом кислорода в положении 5, а затем это соединение окисляют до 4’’-кетона с последующим превращением в амин и удалением "замаскированной" гидроксигруппы в положении 5, что соответствует обычной технологии использования защитных групп, как это описано у Greene и Wuts, Protective Groups in Organic Synthesis, 1999.
Преимущество предлагаемого в изобретении способа заключается в том, что он является только двухстадийным в отличие от известных методов, которые являются четырехстадийными. Кроме того, при осуществлении этого способа не требуется использовать защитные группы, а сам способ является более безвредным с экологической точки зрения благодаря использованию меньшего количества химикатов. Соединение формулы (III), образующееся на предусмотренной изобретением стадии биокаталитического превращения, известно, например, из ЕР 401029.
В одном из конкретных вариантов предлагаемый в изобретении способ можно осуществлять, в частности, следующим образом.
На первой стадии получают соединения формулы (III). С этой целью соединение формулы (II) можно вводить непосредственно в контакт с биокатализатором, способным селективно окислять спирт в положении 4’’ до кетона формулы (III), и сохранять такой контакт в течение периода времени, достаточного для протекания окислительной реакции.
Наиболее целесообразно при осуществлении способа использовать в качестве биокатализатора микроорганизм, способный обеспечить протекание окислительной реакции согласно изобретению. Подобный микроорганизм предпочтительно культивировать в пригодной для этой цели культуральной среде, стимулирующей быстрое размножение микроогранизма, а также в контролируемых условиях в присутствии соединения формулы (II), продолжая совместную инкубацию этого микроорганизма и его субстрата в течение периода времени, достаточного для протекания окислительной реакции, предпочтительно до достижения степени превращения добавленного в культуральную среду соединения формулы (II) в соединение формулы (III), составляющей от 25 до 99,9%, более предпочтительно от 50 до 99,9%, наиболее предпочтительно от 80 до 99,9%.
Другой, более предпочтительный вариант осуществления предлагаемого в изобретении способа заключается в том, что сначала микроорганизм, способный обеспечить протекание окислительной реакции согласно изобретению, культивируют в контролируемых условиях в пригодной для этой цели культуральной среде, стимулирующей быстрое размножение этого микроогранизма, и затем соответствующими методами, например фильтрацией или центрифугированием, собирают биомассу, образовавшуюся в результате размножения микроорганизма. После этого такую биомассу либо можно сразу же использовать в качестве биокатализатора для превращения соединений формулы (II) в соединения формулы (III), либо ее до использования в реакции можно хранить в охлажденном состоянии как таковую либо после предварительной сушки вымораживанием или распылительной сушки. Затем такой микроорганизм либо в свежесобранном после культивирования виде, либо после его хранения по описанной выше методике и соединение формулы (II) совместно инкубируют в реакционной среде, которая не стимулирует быстрое размножение микроогранизма в течение периода времени, достаточного для протекания окислительной реакции, предпочтительно до достижения степени превращения добавленного в реакционную среду соединения формулы (II) в соединение формулы (III), составляющей от 25 до 99,9%, более предпочтительно от 50 до 99,9%, наиболее предпочтительно от 80 до 99,9%.
Полученный таким путем реакционный продукт формулы (III) можно отделять от исходного материала формулы (II) простыми с технической точки зрения, широко распространенными методами разделения, например фракционированной кристаллизацией или хроматографией. К хроматографическим методам относятся, например, колоночная хроматография, толстослойная хроматография или тонкослойная хроматография на минеральных носителях, таких как силикагель или органические ионообменные смолы (иониты).
В предлагаемом способе вместо клеточных структур в виде вегетативных клеток можно использовать также споры микроорганизмов, для чего сначала собирают споры микрорганизмов, способных избирательно окислять спирт в положении 4’’ до кетона формулы (III), и затем такие споры инкубируют с соединением формулы (II) в течение периода времени, достаточного для протекания соответствующей окислительной реакции. Инкубацию спор и субстрата предпочтительно проводить в отсутствие культуральной среды с целью предотвратить развитие спор.
Соединения формулы (II) используются в качестве субстрата для проведения окислительной реакции в соответствии с настоящим изобретением. Такие соединения известны (см. DE 2717040) или их можно получать из известных соединений известными методами. Эти соединения пригодны для борьбы с паразитами животных и вредителями растений и помимо этого являются ценными исходными соединениями или промежуточными соединениями при получении соединений формулы (I). Соединения формулы (III) можно также получать, используя для окисления соединений формулы (II) не сам микроорганизм, а активные компоненты, источником которых является подобный микроорганизм (согласно определениям, приведенным выше в п.п. б)-д)) и которые способны избирательно окислять спирт в положении 4’’ до кетона формулы (III).
В соответствии с этим еще одним объектом настоящего изобретения является применение способных избирательно окислять спирт в положении 4’’ до кетона формулы (III) иммобилизованных вегетативных клеток микроорганизмов, бесклетоных экстрактов, спор, ферментов и смесей ферментов указанных микроорганизмов.
Для иммобилизации подобных биокатализаторов можно использовать методы, которые известны как таковые. В контексте настоящего изобретения в этом отношении следует назвать прежде всего методы, основанные на адсорбционном связывании подобных биокатализаторов твердыми, обычно водонерастворимыми, носителями либо на ионном или ковалентном их связывании с указанными твердыми носителями, а также на перекрестном сшивании биокатализаторов би- или полифункциональными реагентами, на инкапсулировании (внедрении) в матрицу, на мембранном разделении или на сочетании двух или более вышеуказанных методов.
Адсорбционное связывание водонерастворимыми носителями (адсорбентами) обусловлено главным образом силами межмолекулярного взаимодействия (ван-дер-ваальсовыми силами). В качестве адсорбентов в указанных целях можно использовать большое число неорганических и органических соединений, а также синтетические полимеры.
Методы подобной иммобилизации микроорганизмов описаны у Bickerstaff (под. ред.), 1997 (иммобилизация ферментов и клеток), у van Haecht и др., 1985 (иммобилизация дрожжевых клеток на стекле), у Black и др., 1984 (иммобилизация дрожжевых клеток на высококачественной стали, сложном полиэфире), у Wiegel и Dykstra, 1984 (иммобилизация клостридий на целлюлозе, гемицеллюлозе), у Forberg и Haggstrom, 1984 (иммобилизация клостридий на древесной стружке), а также у Ehrhardt и Rehm, 1985 (иммобилизация псевдомонад на активированном угле). Соответствующую более подробную информацию о применении ферментов, иммобилизованных путем адсорбционного связывания, можно найти, в частности, у Krakowiak и др., 1984 (глюкоамилаза, иммобилизованная на оксиде алюминия), у Cabral и др., 1984 (глюкоамилаза, иммобилизованная на активированном титаном стекле), у Miyawaki и Wingard, 1984 (глюкозооксидаза, иммобилизованная на активированном угле), у Kato и Horikoshi, 1984 (глюкозотрансфераза, иммобилизованная на синтетическом полимере). Ионные связи обусловлены электростатическим притяжением между противоположно заряженными группами носителя (в качестве примера которого можно назвать коммерчески доступные типы ионообменников, например, на основе полисахаридов или синтетических полимеров) и связываемого биокатализатора.
Методы иммобилизации микроорганизмов, основанные на образовании ионных связей, описаны у DiLuccio и Kirwan, 1984 (иммобилизация бактерий из рода азотобактер на Cellex E (целлюлоза)) и у Giard и др., 1977 (иммобилизация животных клеток на ДЭАЭ- ((диэтиламино)этил-) сефадексе). Соответствующие методы иммобилизации ферментов подробно описаны у Angelino и др., 1985 (иммобилизация альдегидоксидазы на октиламино-сефарозе 4В), у Hofstee, 1973 (иммобилизация лактатдегидрогеназы на октиламино-сефадексе), у Kuhn и др., 1980 (иммобилизация глюкозооксидазы на ДЭАЭ-сефадексе, ДЭАЭ-целлюлозе) и в других литературных источниках.
Другой метод иммобилизации основан на силах ковалентной связи, которые обычно возникают в результате образования фиксированной химической связи между биокатализаторами или между биокатализатором и носителем. Пригодными для этой цели носителями являются пористые материалы, такие как различные типы стекол, диоксид кремния или иные нерастворимые неорганические материалы.
Для использования в предлагаемом в изобретении способе микроорганизмы можно иммобилизовать, например, по методам, описанным у Messing и Oppermann, 1979 (иммобилизация Enterobacteria на боросиликатном стекле, иммобилизация дрожжевых клеток на оксиде циркония), у Romanovskaya и др., 1981 (иммобилизация метанообразующих бактерий на силохроме), у Navarro и Durand, 1977 (иммобилизация дрожжевых клеток на пористом кремнеземе).
Для иммобилизации ферментов можно использовать методы, описанные у Weetall и Mason, 1973 (иммобилизация папаина на пористом стекле) и у Monsan и др., 1984 (иммобилизация инвертазы на пористом кремнеземе).
Пригодными для иммобилизации носителями, которые можно использовать при осуществлении предлагаемого в изобретении способа, являются не только уже указанные выше материалы, но и целый ряд природных или синтетических полимеров, таких, например, как целлюлоза, декстран, крахмал, агароза и т.д., или полимеров на основе, например, производных акриловой и метакриловой кислоты, которые обычно используют в производстве реакционноспособных сополимеров. Пригодными реакционноспособными группами, посредством которых образуется связь с биокатализатором, являются реакционноспособные динитрофторфенильные или изотиоцианатные группы или прежде всего оксирановые и кислотноангидридные группы. Другой возможный подход заключается в хлоридной активации несущих карбоксигруппы смол, которые коммерчески доступны, например, под товарными знаками Amberlite® XE-64 и Amberlite® IRC-50.
Иммобилизацию микроорганизмов на природных или синтетических носителях можно осуществлять по методам, описанным у Chipley, 1974 (иммобилизация Bacillus subtilis на агарозе), у Gainer и др., 1980 (иммобилизация бактерий из рода азотобактер на целлюлозе), у Jack и Zajic, 1977 (иммобилизация Micrococcus на карбоксиметилцеллюлозе), у Jirku и др., 1980 (иммобилизация дрожжевых клеток на гидроксиалкилметакрилате), а также у Shimizu и др., 1975 (иммобилизация бактериальных клеток на сополимере этилена и малеинового ангидрида). Для иммобилизации ферментов можно использовать, в частности, методы, описанные у Cannon и др., 1984 (иммобилизация лактатоксидазы на целлюлозе), у Dennis и др., 1984 (иммобилизация химотрипсина на сефарозе), у Ibrahim и др., 1985 (иммобилизация эпоксигидролазы на декстране), у Beddows и др., 1981 (иммобилизация α-галактозидазы на сополимере найлона и акрилата), у Raghunath и др., 1984 (иммобилизация уреазы на метакрилате-акрилате).
В процессе перекрестного сшивания биокатализаторы связываются между собой би- или полифункциональными реагентами, такими, в частности, как глутаровый диальдегид, диизоцианаты, с образованием характерных нерастворимых, обычно гелеобразных, агрегатов с высокой молекулярной массой.
Иммобилизацию микроорганизмов подобными методами можно проводить по технологии, описанной у De Rosa и др., 1981 (иммобилизация бактериальных клеток путем перекрестного сшивания с яичным альбумином с помощью глутарового диальдегида). Методы иммобилизации ферментов, пригодные для использования в соответствии с настоящим изобртением, описаны у Barbaric и др., 1984 (иммобилизация инвертазы путем перекрестного сшивания с дигидразидом адипиновой кислоты), у Talsky и Gianitsopoulos, 1984 (иммобилизация химотрипсина за счет пептидной связи между молекулами фермента без участия агента сшивания), у Workman и Day, 1984 (иммобилизация инулиназы путем перекрестного сшивания содержащих фермент клеток с глутаровым диальдегидом), у Khan и Siddiqi, 1985 (иммобилизация пепсина путем перекрестного сшивания с глутаровым диальдегидом), у Bachmann и др., 1981 (иммобилизация глюкозоизомеразы путем перекрестного сшивания с желатином с помощью глутарового диальдегида), у Kaul и др., 1984 (иммобилизация α-галактозидазы путем перекрестного сшивания с яичным альбумином с помощью глутарового диальдегида).
Инкапсуляция в матрицу заключается во внедрении биокатализаторов в природные или синтетические полимеры, обычно гелеобразной консистенции. Наиболее пригодными материалами матрицы для внедрения клеток, органелл и спор являются природные полимеры, такие как альгинат, карраген, пектин, агар, агароза или желатин, поскольку эти соединения являются не токсичными и защищают клетки при оперировании с ними. Вместе с тем в вышеуказанных целях можно использовать и синтетические полимеры, такие, например, как полиакриламиды, и, в частности, фотосшитые полимеры. Продукты, получаемые инкапсулированием в матрицу, могут иметь самую разнообразную форму, например сферическую, цилиндрическую, волокнистую и пластинчатую (листовую). Иммобилизацию микроорганизмов с помощью природных или синтетических материалов матрицы можно осуществлять по методам, описанным у Mazumder и др., 1985 (иммобилизация бактериальных клеток на фотосшитых полимерах), у Bettmann и Rehm, 1984 (иммобилизация бактериальных клеток на полиакриламидном гидразиде), у Umemura и др., 1984 (иммобилизация бактериальных клеток на каррагене), у Karube и др., 1985 (иммобилизация бактериальных протопластов на агаре-ацетилцеллюлозе), у Cantarella и др., 1984 (иммобилизация дрожжевых клеток на гидроксиэтилметакрилате), у Qureshi и Tamhane, 1985 (иммобилизация дрожжевых клеток на альгинате), у Deo и Gaucher, 1984 (иммобилизация гифомицетов па каррагене), у Eikmeier и Rehm, 1984 (иммобилизация гифомицетов на альгинате), у Bihari и др., 1984 (иммобилизация конидий гифомицетов па полиакриламиде), у Vogel и Brodelius, 1984 (иммобилизация растительных клеток на альгинате, агарозе), у Nakajima и др., 1985 (иммобилизация растительных клеток на агаре, альгинате, каррагене). Для иммобилизации ферментов можно использовать метод, описанный у Mori и др., 1972 (иммобилизация аминоацилазы на полиакриламиде).
Мембранная сепарация заключается в создании специальных определенных зон, в которых протекает реакция. Мембранную сепарацию можно классифицировать на следующие основные типы:
а) микрокапсулирование,
б) технология липосом,
в) использование биокатализатора в мембранных реакторах.
Описанные выше методы иммобилизации можно комбинировать между собой, например комбинировать адсорбцию с перекрестным сшиванием. В этом случае ферменты сначала адсорбируют на носителе, а затем сшивают между собой бифункциональным реагентом.
Инкубацию биокатализаторов, используемых согласно настоящему изобретению, с соединениями формулы (II) для избирательного окисления спирта в положении 4’’ до кетона формулы (III) можно осуществлять по методам, общепринятым в прикладной микробиологии. При этом помимо использования встряхиваемых культур прежде всего следует назвать различные системы ферментации, которые уже достаточно давно используются для микробиологических исследований и в промышленном производстве.
Основное назначение биореакторов состоит в создании оптимальных гидродинамических условий с целью снижения кажущейся константы Михаэлиса и ускорения реакции. Достигается это в основном за счет поддержания необходимой относительной подвижности между биокатализатором и окружающей его средой, что позволяет повысить внешний массообмен до такой степени, при которой он на практике не создает более никаких помех протеканию процесса.
В качестве примера реакторов, пригодных для проведения рассматриваемого процесса, можно назвать реакторы с мешалкой, циркуляционные реакторы, реакторы с неподвижным слоем, реакторы с псевдоожиженным слоем, мембранные реакторы, а также большое число реакторов особых типов, например реакторы с мешалкой с сетчатыми лопастями, ромбоидные реакторы, трубчатые реакторы (W. Hartmeier, Immobilisierte Biokatalysatoren, 1986; W. Crueger и A. Crueger, Biotechnologie-Lehrbuch der angewandten Mikrobiologie, 1984; P. Prave и др., Handbuch der Biotechnologie, 1984). Согласно изобретению предпочтительно использовать реакторы с мешалкой.
Реакторы с мешалкой среди реакторов всех остальных типов нашли наибольшее распространение в биотехнологических процессах ферментации. Реакторы подобного типа обеспечивают быстрое и тщательное смешение субстрата и биокатализатора благодаря высокой эффективности смешения и высокой интенсивности насыщения реакционной смеси килородом.
Преимущества реакторов с мешалкой заключаются в простоте и тем самым экономичности их конструкции, а также в наличии у них хорошо изученных свойств.
При использовании реакторов с мешалкой в принципе возможны два режима проведения технологического процесса, одним из которых является периодический режим, а другим - непрерывный режим.
При работе в периодическом режиме биокатализаторы по завершении процесса удаляют путем сепарации или фильтрации и либо направляют в отходы (вегетативные клетки), либо используют повторно при получении следующей партии продукта (иммобилизованные биокатализаторы).
При работе в непрерывном режиме израсходованный субстрат в реакционном объеме непрерывно, без остановки процесса заменяют на новый субстрат для конечного продукта реакции. При этом биокатализаторы должны постоянно оставаться в реакторе, для чего используют пригодные для этой цели средства (сита, фильтры, устройства рециркуляции).
Культивирование вегетативных клеток микроорганизмов проводят согласно изобретению в соответствии с известными, широко распространенными методами, при этом для упрощения технологического процесса предпочтительно использовать жидкие питательные среды.
Состав питательных сред варьируется в зависимости от используемого микроорганизма. Как правило, предпочтительно использовать комплексные среды с нечетко определенными, легко усвоямыми источниками углерода (С) и азота (N) типа сред, обычно применяемых, например, в том числе и при получении антибиотиков.
Кроме того, культуральная среда должна содержать витамины и требующиеся для роста микроорганизмов ионы металлов, которые, однако, обычно в любом случае присутствуют в соответствующей концентрации в используемой комплексной питательной среде в виде ее компонентов или примесей. При необходимости в питательную среду можно добавлять подобные компоненты, такие, например, как необходимые для роста витамины, а также ионы Na+, К+, Са2+, Mg2+, NH, (SO4)2-, Сl-, (СО3 )2- и микрооэлементы кобальт и марганец, а также, в частности, цинк в виде их солей. Наиболее пригодными источниками азота помимо дрожжевых экстрактов, дрожжевых гидролизатов, дрожжевых автолизатов и дрожжевых клеток являются прежде всего мука соевых бобов, кукурузная мука, овсяная мука, эдамин (продукт, образующийся в результате ферментативного разложения лактальбумина), пептон, гидролизат казеина, жидкость, образующаяся при замачивании зерен кукурузы до набухания, и мясной экстракт.
Предпочтительная концентрация указанных источников азота составляет от 0,1 до 6 г/л. Пригодными источниками углерода являются прежде всего глюкоза, лактоза, сахароза, декстроза, мальтоза, крахмал, церелоза, целлюлоза, маннит, солодовый экстракт и меласса. Предпочтительная коцентрация указанных источников углерода составляет от 1,0 до 25 г/л. В описанном ниже процессе окисления и прежде всего при применении в этом процессе микроорганизмов-представителей рода Streptomyces в качестве источника углерода предпочтительно использовать D-глюкозу, растворимый крахмал или солодовый экстракт, а также церелозу. В соответствии с этим наиболее пригодными культуральными средами для представителей рода Streptomyces являются, например, среды следующих составов:
Среда 1
растворимый крахмал 1,0 г
пептон 0,2 г
дрожжевой экстракт 0,2 г
Объем доводят до 1 л дистиллированной водой, значение рН устанавливают на 7 с помощью NaOH, автоклавируют.
Среда 2
D-глюкоза 4,0 г
солодовый экстракт 10,0 г
дрожжевой экстракт 4,0 г
Объем доводят до 1 л дистиллированной водой, значение рН устанавливают на 7 с помощью NaOH, автоклавируют.
Среда 3
глицерин 10,0 г
декстрин 20,0 г
soytone (Difco Manual, 9-е изд.,
Detroit, Difco Laboratories, 1969) 10,0 г
(NH4)2SO4 2,0 г
СаСО3 2,0 г
Объем доводят до 1 л дистиллированной водой, значение рН устанавливают на 7 с помощью NaOH, автоклавируют.
Среда 4
D-глюкоза 10,0 г
солодовый экстракт 10,0 г
дрожжевой экстракт 3, 0 г
Pharmamedia (Traders Protein,
Southern Cotton Oil Co.,
Мемфис, шт. Теннесси, США) 10,0 г
мясной экстракт 1,0 г
Объем доводят до 1 л дистиллированной водой, значение рН устанавливают на 7 с помощью NaOH, автоклавируют.
Среда 5 (агар ISP-2)
дрожжевой экстракт (Oxoid Ltd.,
Бейсингсток, Гемпшир, Англия) 4 г
D(+)-глюкоза 4 г
бактосолодовый экстракт (Difco
№0186-17-7) 10 г
агар (Difco №0140-01) 20 г
Указанные ингредиенты растворяют в 1 л деминерализованной воды и значение рН устанавливают на 7,0.
Раствор стерилизуют при 121°С в течение 20 мин, охлаждают и выдерживают при 55°С в течение непродолжительного интервала времени, необходимого для непосредственного получения агаровых пластинок.
Среда 6 (среда PHG)
пептон (Sigma 0521) 10 г
дрожжевой экстракт (Difco) 10 г
D-(+)-глюкоза 10 г
NaCl 2 г
MgSO4×7H2O 0,15 г
NaH2PO4×H2O 1,3 г
К2НРО4 4,4 г
Указанные ингредиенты растворяют в 1 л деминерализованной воды и значение рН устанавливают на 7,0.
Вышеописанные среды особо пригодны также для культивирования микроорганизмов-представителей рода Streptomyces и для проведения окислительной реакции. Следует отметить, что приведенные выше данные о составе сред, а также остальных сред, подробно рассмотренных в настоящем описании, приведены только в качестве примеров, иллюстрирующих настоящее изобретение, и не ограничивают его объем.
Помимо собственно состава сред важную роль играет также технология приготовления подобных сред, например последовательность растворения или суспендирования, стерилизация питательного раствора в целом или стерилизация его отдельных компонентов, предотвращение загрязнения питательной среды посторонними примесями, и поэтому подобные параметры для эффективного осуществления предлагаемого в изобретении способа необходимо соответствующим образом оптимизировать.
Следует также отметить, что стерилизация может привести к изменению значения рН питательной среды, а также к выпадению осадка.
Остальные методы культивирования также соответствуют методам, обычно применяемым для культивирования микроорганизмов.
При проведении процесса ферментации в соответствии с настоящим изобретением и небольшом масштабе, включая ферментацию с использованием любых предварительных культур, обычно применяют встряхиваемые культуры, для чего целесообразно использовать стеклянные колбы емкостью от 0,1 до 5 л, предпочтительно от 0,5 до 5 л, заполненные питательной средой в объеме от 0,05 до 2 л, предпочтительно от 0,1 до 2 л. Такие колбы предпочтительно оснащать дефлектором. После автоклавирования и установления величины рН на значение от 4 до 8, прежде всего от 7,0 до 7,5 (для бактерий) или от 6 до 7,5 (для грибов) содержимое колб в стерильных условиях инокулируют культурами соответствующего микроорганизма. Обычно в качестве материала для инокуляции (инокулята) используют предварительную культуру, получаемую из законсервированного инокулята по описанной ниже методике.
Культуры, включая любые предварительные культуры, целесообразно выращивать в аэробных условиях при температуре от примерно 25 до примерно 37°С, предпочтительно от примерно 26 до примерно 30°С, наиболее предпочтительно при примерно 28°С, при непрерывном встряхивании на роторном шюттль-аппарате при скорости вращения ротора от примерно 80 до примерно 300 об/мин, предпочтительно от примерно 100 до 250 об/мин, более предпочтительно при примерно 120 об/мин. В указанных выше условиях оптимальная окислительная активность у штаммов Streptomyces обычно достигается по истечении 1,5-7-дневного культивирования.
По достижении клетками достаточно высокой для протекания необходимой окислительной реакции каталитической активности, преимущественно через 40 ч, добавляют субстрат (соединения формулы (II)), при этом микроорганизмы и подвергаемое окислению соединение можно вводить в контакт между собой различными путями. С практической точки зрения субстрат, т.е. соединение формулы (II), целесообразно добавлять к микроорганизму в питательном растворе.
Подвергаемое окислению соединение можно использовать, например, в порошковом виде или в виде раствора либо в пригодном для этой цели растворителе, таком, например, как диметилформамид, ацетон, диметилсульфоксид, N-метил-2-пирролидон, либо в спиртовом растворителе, таком, например, как метанол, этанол, изопропанол или трет-бутанол, либо в растворителе и виде простого эфира, таком, например, как тетрагидрофуран или 1,4-диоксан (от 0,5 до 15 об.%, предпочтительно 2 об.%), либо в растворителе в виде сложного эфира, таком, например, как этилацетат, либо в углеводородном растворителе, таком, например, как октан, циклогексан, толуол или ксилол, либо в смеси пригодного растворителя и пригодного поверхностно-активного вещества (ПАВ). Под "поверхностно-активным веществом" подразумеваются ионогенные, неионогенные и амфионогенные ПАВ, а также их смеси.
Пригодными анионогенными ПАВ являются водорастворимые мыла и водорастворимые синтетические поверхностно-активные вещества. Пригодными для использования в вышеуказанных целях мылами являются соли щелочных металлов, соли щелочноземельных металлов либо незамещенные или замещенные аммониевые соли высших жирных кислот (С10-С22), например натриевые или калиевые соли олеиновой или стеариновой кислоты, или смесей природных жирных кислот, которые (смеси) можно получать, например, из кокосового или таллового масла. Другими пригодными ПАВ являются также метилтауриновые соли жирных кислот. Более часто, однако, используют так называемые синтетические ПАВ, прежде всего сульфонаты соединений алифатического ряда, сульфаты соединений алифатического ряда, сульфированные бензимидазольные производные или алкиларилсульфонаты. Сульфонаты или сульфаты соединений алифатического ряда, которые обычно применяют в виде солей щелочных металлов, солей щелочноземельных металлов либо незамещенных или замещенных аммониевых солей, содержат С10-С22алкильный радикал, который также включает алкильный фрагмент ацильных радикалов, и представляют собой, например, натриевую или кальциевую соль лигносульфоновой кислоты, додецилсульфата или смеси сульфатов жирных спиртов, получаемых из природных жирных кислот. К подобным соединениям относятся также соли сульфатированных и сульфированных продуктов присоединения жирного спирта к этиленоксиду. Сульфированные бензимидазольные производные предпочтительно содержат 2 группы сульфоновой кислоты и один радикал жирной кислоты, имеющий от 8 до 22 атомов углерода. В качестве примеров алкиларилсульфонатов можно назвать натриевые, кальциевые или триэтаноламиновые соли додецилбензолсульфоновой кислоты, дибутилнафталинсульфоновой кислоты или продукта конденсации нафталинсульфоновой кислоты и формальдегида. Другими пригодными анионогенными ПАВ являются также соли желчных кислот, например натриевые соли холевой кислоты или дезоксихолевой кислоты. Пригодными для применения в вышеуказанных целях являются также соответствующие фосфаты, например соли эфира фосфорной кислоты и аддукта n-нонилфенола с 4-14 молями этиленоксида, или фосфолипиды.
Пригодными катионогенными ПАВ являются тетраалкиламмониевые соли, например бромид цетилтриметиламмония.
Пригодными нейтральными ПАВ являются алкилгликозиды, например алкил-β-D-глюкопиранозиды, алкил-β-D-тиоглюкопиранозиды, алкил-β-D-мальтозиды, содержащие С6-С12алкильный радикал. Другими пригодными нейтральными ПАВ являются глюкамиды, например N,N-бис(3-D-глюконамидопропил)холамид, N,N-бис(3-D-глюконамидопропил)дезоксихоламид, N-метилглюкамиды жирных кислот, содержащие С7-С12ацильный радикал. Кроме того, пригодными для использования в вышеуказанных целях нейтральными ПАВ являются моно- и полидисперсные полиоксиэтилены, например продукты BRIJ®, GENAPOL®, LUBROL® , PLURONIC®, THESIT®, TRITON®, TWEEN®.
Пригодными амфионогенными ПАВ являются N,N,N-триалкилглицины, например N-н-додецил-N,N-диметилглицин. Другими пригодными амфионогенными ПАВ являются ω-N,N,N-триалкиламмонийалкилсульфонаты, например 3-(N-алкил-N,N-диметил)-1-пропансульфонат, содержащий C8-С16алкильный радикал. Помимо этого пригодными амфионогенными ПАВ являются 3-[(3-холамидопропил)диметиламмонио]-1-пропансульфонат и 3-[(3-холамидопропил)диметиламмонио]-2-гидрокси-1-пропансульфонат.
ПАВ, обычно используемые для растворения и в технологии приготовления композиций, описаны, в частности, у Bhairi S.M., "A guide to the Properties and Uses of Detergents in Biology и Biochemistry", Calbiochem-Novabiochem Corp., Сан-Диего, Калифорния (1997), а также в "1999 International McCutcheon’s Emulsifiers and Detergents", изд-во The Manufacturing Confectioner Publishing Co., Глен Рок, шт. Нью-Джерси, США.
Протекание реакции непрерывно контролируют хроматографическими методами, обычно используемыми в области микробиологических исследований.
Настоящее изобретение относится также к культивированию микроорганизмов, способных избирательно окислять спирт в положении 4" до кетона формулы (III), и к инкубации таких микроорганизмов с указанными соединениями в биореакторах, прежде всего в биореакторах типа реактора с мешалкой. С целью обеспечить оптимальную скорость образования целевого продукта в конкретном промышленном ферментере рекомендуется сначала размножать микроорганизмы в виде предварительных культур. Количество помещаемых в ферментер предварительных культур зависит от той концентрации инокулята, которая является оптимальной в каждом конкретном случае. Для стадии ферментации инокулят целесообразно получать в зависимости от используемых микроорганизмов в следующих концентрациях: бактерии - от 0,1 до 3%, грибы - от 5 до 10%, актиномицеты - от 5 до 10%.
Для внесения в небольшие по объему ферментеры (до 20 л) обычно используют предварительные культуры, выращиваемые во встряхиваемых колбах. В этом случае все содержимое колбы вносят в ферментер.
Исходным материалом, используемым для получения предварительных культур, обычно служит законсервированный инокулят, например, в виде лиофилизатов либо в виде замороженного или хранящегося в охлажденном состоянии материала. Законсервированный инокулят, используемый в соответствии с настоящим изобретением, предпочтительно представляет собой материал, хранимый при -80°С.
Инокулят предпочтительно размножать в жидких средах в стеклянных колбах на роторном шюттль-аппарате либо при использовании спор - на твердых питательных субстратах (подложках). Свойства питательного субстрата и условия культивирования, такие, в частности, как температура, величина рН, подача кислорода, необходимо оптимизировать с учетом конкретно используемого микроорганизма или процесса. Продолжительность роста законсервированного инокулята варьируется согласно приведенным ниже данным от нескольких часов до нескольких дней в зависимости от используемого исходного материала:
лиофилизаты 3-10 дней
законсервированные замораживанием культуры:
бактерии 4-18 ч
актиномицеты 1-5
дней
грибы 1-7 дней
хранящиеся в охлажденном состоянии культуры:
бактерии 4-24 ч
актиномицеты 1-3 дня
грибы 15 дней
Если в качестве инокулята используют споры, то такие споры сначала размножают путем выращивания законсервированного инокулята на твердом питательном субстрате в стандартных условиях (стерильная аэрация, камера искусственного климата). При применении питательных субстратов на основе торфа, отрубей, риса или ячменя культуры ежедневно тщательно встряхивают для достижения высокой плотности спор. Другая возможность заключается в культивировании законсервированного инокулята на питательных средах, отвержденных за счет добавления агара либо иных обычно используемых загустителей, при этом предпочтительно использовать питательные среды, которые инициируют индукцию спорообразования.
Продолжительность споруляции в зависимости от используемого микроорганизма и от используемой питательной среды составляет от 7 до 30 дней.
Для внесения в ферментер для выращивания предварительных культур или промышленный ферментер споры либо суспендируют в поверхностно-активных веществах, например в растворе твина 80 (ПАВ, выпускаемое фирмой Sigma-Aldrich Co., Сент-Луис, шт. Миссури, США), и затем совместно с их питательной средой переносят в ферментер, либо при использовании твердой питательной среды - промывают для удаления твердого питательного субстрата, также используя указанные поверхностно-активные вещества. Полученный таким путем содержащий споры раствор используют затем для внесения в ферментеры. Обе описанных выше операции - восстановление спор и внесение в ферментеры - предпочтительно проводить в стерильных условиях.
Для получения соединений формулы (III) согласно настоящему изобретению можно использовать в зависимости от необходимого количества конечного продукта биореакторы различных размеров, вместимость которых составляет от 0,001 до 450 м3.
При использовании биореакторов с мешалкой наиболее важными для оптимального протекания реакции являются следующие параметры ферментации:
1. Температура: Биокаталитическую окислительную реакцию предпочтительно проводить предлагаемым в изобретении способом в пригодном для размножения и развития мезофил интервале температур (от 20 до 45°С).
Оптимальная температура для роста и образования целевого продукта составляет от 20 до 32°С, прежде всего от 24 до 30°С.
2. Аэрация (насыщение кислородом): Скорость аэрации составляет от 0,1 до 2,5 об./об./мин (объемных единиц воздуха на единицу объема жидкости в мин), предпочтительно от 0,3 до 1, 75 об./об./мин. При необходимости скорость аэрации следует согласовывать в процессе ферментации с фактичкской потребностью в кислороде.
3. Давление: Реакторы с мешалкой с целью снизить риск загрязнения их содержимого посторонними примесями обычно работают под небольшим избыточным давлением, составляющим от 0,2 до 1,0 бара, предпочтительно от 0,5 до 0,7 бара.
4. Значение рН: Значение рН может варьироваться в определенных пределах в зависимости от используемого микроорганизма. При применении микроорганизмов из группы актиномицетов начальное значение рН составляет от 6 до 8, предпочтительно от 6,5 до 7,5. При применении грибов начальное значение рН культурального раствора предпочтительно составляет от 4 до 8, более предпочтительно от 6 до 7,5.
5. Перемешивание: Скорость перемешивания зависит от типа используемой мешалки и размера ферментера. Согласно настоящему изобретению предпочтительно использовать мешалки с лопастными колесами дискового типа, скорость вращения которых при объеме реактора с мешалкой, равном 0,002 м3, составляет от 150 до 550 об/мин, прежде всего от 200 до 500 об/мин.
Продолжительность процесса ферментации при проведении предлагаемого в изобретении способа в зависимости от используемого микроорганизма варьируется от 20 ч до 10 дней. Биокаталитическую реакцию прекращают, когда степень превращения субстрата (соединения формулы (II)) в соединение формулы (III) достигает от примерно 25 до примерно 99,9%, более предпочтительно от примерно 50 до примерно 99,9%, наиболее предпочтительно от примерно 80 до примерно 99,9% от его первоначально добавленного количества.
Для определения оптимального момента прекращения окислительной реакции протекание такой реакции контролируют в течение всего процесса ферментации обычными аналитическими методами, прежде всего хроматографическими методами, например с помощью жидкостной хроматографии высокого разрешения (ЖХВР) или тонкослойной хроматографии.
В одном из вариантов осуществления рассмотренного выше способа биореактор можно использовать только для получения биомассы, которую затем собирают фильтрацией или центрифугированием. После этого биомассу, образовавшуюся в результате размножения микроорганизма, либо сразу же используют в качестве биокатализатора для превращения соединений формулы (II) в соединения формулы (III), либо хранят до использования в охлажденном состоянии как таковую либо после предварительной сушки вымораживанием или распылительной сушки. Затем такой микроорганизм либо в свежесобранном после культивирования виде, либо после его хранения по описанной выше методике распределяют далее по другим сосудам, в которых проводят собственно процесс биопревращения, таким как колбы, предпочтительно оборудованные дефлекторами, или по биореакторам с мешалкой. Далее добавляют субстрат (соединение формулы (II)), при этом микроорганизм и подвергаемое окислению соединение можно вводить в контакт между собой различными путями. С практической точки зрения субстрат, т.е. соединение формулы (II), целесообразно добавлять к микроорганизму в забуференном растворе, который не стимулирует быстрое размножение микроорганизма. Подвергаемый окислению субстрат (соединение формулы (II)) можно использовать, например, в порошковом виде или в виде раствора в любом пригодном для этой цели растворителе, например из числа описанных выше.
В одном из предпочтительных вариантов осуществления изобретения субстрат (соединение формулы (II)) сначала растворяют в пригодном растворителе, таком как диметилсульфоксид или твин 40 (ПАВ, выпускаемое фирмой Sigma-Aldrich Co., Сент-Луис, шт. Миссури, США) либо смесь обоих растворителей, и вносят в оборудованные дефлекторами колбы, содержащие буферный раствор, предпочтительно фосфатный буфер, более предпочтительно фосфатный буфер в концентрации 0,07 М с рН 7,0. Полученный раствор затем стерилизуют, после чего добавляют биокатализатор (биомассу, образовавшуюся в результате размножения микроорганизма). Затем эту реакционную смесь инкубируют при комнатной температуре и в зависимости от штамма микроорганизма встряхивают в течение примерно 2-7 дней при скорости вращения ротора от 100 до 150 об/мин, предпочтительно при примерно 120 об/мин.
В другом предпочтительном варианте осуществления изобретения субстрат (соединение формулы (II)) сначала растворяют в пригодном растворителе, таком как диметилсульфоксид или твин 40 (ПАВ, выпускаемое фирмой Sigma-Aldrich Co., Сент-Луис, шт. Миссури, США) либо смесь обоих растворителей, и вносят в оборудованные дефлекторами колбы, содержащие культуральную среду, стимулирующую рост микроорганизма, который предназначен для протекания требуемой окислительной реакции. Полученный раствор затем стерилизуют, после чего добавляют биокатализатор (биомассу, образовавшуюся в результате размножения микроорганизма). Затем эту реакционную смесь инкубируют при комнатной температуре и в зависимости от штамма микроорганизма встряхивают в течение примерно 2-9 дней при скорости вращения ротора от 100 до 150 об/мин, предпочтительно при примерно 120 об/мин.
Согласно еще одному варианту осуществления изобретения получают бесклеточный экстракт с использованием клеток, которые промывают в пригодном для этой цели буферном растворе, ресуспендируют в буфере для разрушения клеток и разрушают, например, с помощью механических средств при температуре от 2 до 15°С, предпочтительно от 3 до 6°С, наиболее предпочтительно при 4°С. Полученную суспензию центрифугириуют и собирают надосадочный бесклеточный экстракт. К полученному таким путем бесклеточному экстракту добавляют растворы, содержащие приемлемое аликвотное количество чужеродной системы-источника электронов, такую как ферредоксин и ферредоксинредуктаза, и субстрат. После добавления субстрата смесь предпочтительно сразу же подвергать тщательному перемешиванию и аэрации. После этого добавляют аликвотное количество NADPH (восстановленный никотинамид-аденин-динуклеотидфосфат) и смесь инкубируют при температуре от 15 до 40°С, предпочтительно от 20 до 35°С, наиболее предпочтительно при 30°С.
Переработку ферментационного бульона для выделения продукта окисления (соединений формулы (III)) можно проводить по методам, обычно применяемым при ферментации (W. Crueger и A. Crueger, 1984; Р. Prave, 1984).
Сначала из реакционного бульона с помощью фильтров, центрифуг или сепараторов удаляют присутствующие в нем в виде частиц компоненты для их экстракции отдельно от фильтрата.
В том случае, если в качестве биокатализатора используют вегетативные или неживые клетки и если часть реакционного продукта (соединения формулы (III)) содержится внутри клеток, такие клетки до экстракции необходимо разрушить. С этой целью можно использовать различные методы разрушения клеток, основанные на механическом, термическом, химическом или ферментативном воздействии.
К механическим методам, пригодным для использования в предлагаемом в изобретении способе, относятся, например, измельчение в шаровых мельницах с мешалкой или в коллоидных мельницах, приложение и сброс давления в гомогенизаторе, а также разрушение клеток под действием ультразвука. К немеханическим методам относятся разрушение клеток путем сушки, лизис клеток под воздействием осмотического шока, химический автолизис и ферментативный лизис клеток.
После удаления присутствующих в виде частиц компонентов реакционный продукт концентрируют путем экстракции культурального раствора и отделенных клеточных компонентов пригодным растворителем или растворителями. Подобную экстракцию можно также проводить с помощью различных, обычно применяемых в процессах ферментации средств, в качестве примера которых можно назвать среди прочего смесители-отстойники, колонны противоточного типа и центрифужные экстракторы.
Существует также возможность концентрировать реакционнй продукт, например, путем мембранной фильтрации, ультрафильтрации, концентрации вымораживанием, с помощью ионообменных методов и т.п.
Последующую переработку сырого реакционного продукта, полученного после экстракции, можно проводить методами, хорошо зарекомендовавшими себя в области микробиологических и химических исследований, а также в промышленности. К подобным методам относятся, например, хроматографические методы, такие как адсорбционная хроматография, ионообменная хроматография, хроматография на молекулярных ситах, аффинная хроматография, гидрофобная хроматография, распределительная хроматография, ковалентная хроматография и т.д., а также различные методы кристаллизации.
Пригодными для экстракции растворителями, которые можно использовать индивидуально либо в смеси между собой, являются ароматические угледоводороды, такие как толуол, смеси ксилолов или замещенные нафталины, фталаты, такие как дибутилфталат или диоктилфталат, алифатические углеводороды, такие как изомеры гексана, гептана, октана или парафинов либо циклогексан, спирты и гликоли, а также их простые и сложные эфиры, такие как метанол, этанол, 2-пропанол, 1-бутанол, трет-бутанол, этиленгликоль, метил-трет-бутиловый эфир, этилацетат, этиленгликольмонометиловый или -моноэтиловый эфир, кетоны, такие как ацетон, 2-бутанон или циклогесанон, хлорированные углеводороды, такие как дихлорметан, хлороформ или четыреххлористый углерод.
Под "поверхностно-активными веществами" подразумеваются также смеси ПАВ. Пригодными анионогенными ПАВ являются водорастворимые мыла и водорастворимые синтетические поверхностно-активные вещества. Пригодными мылами являются соли щелочных металлов, соли щелочноземельных металлов либо незамещенные или замещенные аммониевые соли высших жирных кислот (С10-С22), например натриевые или калиевые соли олеиновой или стеариновой кислоты, или смесей природных жирных кислот, которые (смеси) можно получать, например, из кокосового или таллового масла. Другими пригодными ПАВ являются также метилтауриновые соли жирных кислот. Более часто, однако, используют так называемые синтетические ПАВ, прежде всего сульфонаты соединений алифатического ряда, сульфаты соединений алифатического ряда, сульфированные бензимидазольные производные или алкиларилсульфонаты. Сульфонаты или сульфаты соединений алифатического ряда, которые обычно применяют в виде солей щелочных металлов, солей щелочноземельных металлов либо незамещенных или замещенных аммониевых солей, содержат С10-С22алкильный радикал, который также включает алкильный фрагмент ацильных радикалов, и представляют собой, например, натриевую или кальциевую соль лигносульфоновой кислоты, додецилсульфата или смеси сульфатов жирных спиртов, получаемых из природных жирных кислот. К подобным соединениям относятся также соли сульфатированных и сульфированных продуктов присоединения жирного спирта к этиленоксиду. Сульфированные бензимидазольные производные предпочтительно содержат 2 группы сульфоновой кислоты и один радикал жирной кислоты, имеющий от 8 до 22 атомов углерода. В качестве примеров алкиларилсульфонатов можно назвать натриевые, кальциевые или триэтаноламиновые соли додецилбензолсульфоновой кислоты, дибутилнафталинсульфоновой кислоты или продукта конденсации нафталинсульфоновой кислоты и формальдегида. Пригодными являются также соответствующие фосфаты, например соли эфира фосфорной кислоты и аддукта n-нонилфенола с 4-14 молями этиленоксида, или фосфолипиды. ПАВ, обычно используемые в технологии приготовления композиций, описаны, в частности, в "1986 International McCutcheon’s Emulsifiers and Detergents", изд-во The Manufacturing Confectioner Publishing Co., Глен Рок, шт. Нью-Джерси, США.
Предпочтительный вариант осуществления предлагаемого в изобретении способа заключается в получении 4’’-оксоавермектина путем введения биокатализатора, такого как микроорганизм, способный превращать авермектин в 4’’-оксоавермектин, в контакт с авермектином и выделения полученного 4’’-оксоавермектина из реакционной смеси.
В одном из вариантов предлагаемый в изобретении способ получения соединения формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, заключается в том, что
(1) получают клетки путем инокуляции питательных сред, стимулирующих рост клеток, предварительными культурами микроорганизма, способного превращать соединение формулы (II) в соединение формулы (III), предпочтительно авермектин в 4’’-оксоавермектин,
(2) собирают выращенные клетки,
(3) соединение формулы (II), предпочтительно авермектин, растворяют в пригодном растворителе,
(4) полученный на стадии (3) раствор добавляют к реакционной среде, которая не стимулирует пролиферацию клеток,
(5) собранные на стадии (2) клетки добавляют в реакционную среду, полученную на стадии (4),
(6) реакционную смесь, полученную на стадии (5), встряхивают или перемешивают в присутствии воздуха,
(7) клетки отделяют от реакционной среды,
супернатант и клетки экстрагируют пригодными растворителями,
(9) органические фазы растворителя, полученные на стадии (8), концентрируют,
(10) содержащееся в полученном на стадии (9) экстракте соединение формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, очищают хроматографией или кристаллизацией.
В другом предпочтительном варианте предлагаемый в изобретении способ получения соединения формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, заключается в том, что
(1) получают клетки путем инокуляции питательных сред, стимулирующих рост клеток, предварительными культурами микроорганизма, способного превращать соединение формулы (II) в соединение формулы (III), предпочтительно авермектин в 4’’-оксоавермектин,
(2) собирают выращенные клетки,
(3) соединение формулы (II), предпочтительно авермектин, растворяют в пригодном растворителе,
(4) полученный на стадии (3) раствор добавляют к реакционной среде, которая не стимулирует пролиферацию клеток,
(5) собранные на стадии (2) клетки добавляют в реакционную среду, полученную на стадии (4),
(6) реакционную смесь, полученную на стадии (5), встряхивают или перемешивают в присутствии воздуха,
(7) весь бульон экстрагируют пригодными растворителями,
(8) разделяют фазы,
(9) фазу растворителя, полученную на стадии (8), концентрируют в вакууме,
(10) содержащееся в полученном на стадии (9) экстракте соединение формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, очищают хроматографией или кристаллизацией.
В соответствии еще с одним предпочтительным вариантом предлагаемый в изобретении способ получения соединения формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, заключается в том, что
(1) соединение формулы (II), которое предпочтительно представляет собой авермектин, растворяют в пригодном растворителе,
(2) полученный на стадии (1) раствор добавляют к питательным средам, стимулирующим рост клеток,
(3) питательные среды, полученные на стадии (2), инокулируют предварительными культурами микроорганизма, способного превращать соединение формулы (II) в соединение формулы (III), предпочтительно авермектин в 4’’-оксоавермектин,
(4) культивируют микроорганизм, способный превращать соединение формулы (II) в соединение формулы (III), предпочтительно авермектин в 4’’-оксоавермектин,
(5) клетки отделяют от реакционной среды,
(6) супернатант и клетки экстрагируют пригодными растворителями,
(7) органическую фазу раствора, полученную на стадии (6), концентрируют в вакууме,
(8) содержащееся в полученном на стадии (7) экстракте соединение формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, очищают хроматографией или кристаллизацией.
Согласно следующему предпочтительному варианту предлагаемый в изобретении способ получения соединения формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, заключается в том, что
(1) соединение формулы (II), предпочтительно авермектин, растворяют в пригодном растворителе,
(2) полученный на стадии (1) раствор добавляют к питательным средам, стимулирующим рост клеток,
(3) питательные среды, полученные на стадии (2), инокулируют предварительными культурами микроорганизма, способного превращать авермектин в 4’’-оксоавермектин,
(4) культивируют микроорганизм, способный превращать соединение формулы (II) и соединение формулы (III), предпочтительно авермектин в 4’’-оксоавермектин,
(5) весь бульон экстрагируют приемлемым растворителем,
(6) разделяют фазы,
(7) фазу раствора, полученную на стадии (6), концентрируют в вакууме,
(8) содержащееся в полученном на стадии (7) экстракте соединение формулы (III), которое предпочтительно представляет собой 4’’-оксоавермектин, очищают хроматографией или кристаллизацией.
На второй стадии, которая носит исключительно химический характер, полученное описанным выше путем соединение формулы (III) можно подвергать взаимодействию в присутствии восстановителя с известным амином формулы HN(R8)R9, в которой R8 и R9 имеют те же значения, что и указанные для формулы (I).
Компоненты реакции можно подвергать взаимодействию между собой в отсутствие растворителя, однако предпочтительно их вводить во взаимодействие в присутствии растворителя. Другая возможность состоит в проведении реакции в избыточном количестве одного из компонентов реакции, прежде всего в жидком амине. Однако более предпочтительным обычно является добавление инертного жидкого растворителя или разбавителя. В качестве примеров подобных растворителей или разбавителей можно назвать ароматические, алифатические и алициклические углеводороды и галогенированные углеводороды, такие как бензол, толуол, ксилол, мезителен, тетралин, хлорбензол, дихлорбензол, бромбензол, петролейный эфир, гексан, циклогексан, дихлорметан, трихлорметан, тетрахлорметан, дихлорэтан, трихлорэтен или тетрахлорэтен, сложные эфиры, такие как этиловый эфир уксусной кислоты, простые эфиры, такие как диэтиловый эфир, дипропиловый эфир, диизопропиловый эфир, дибутиловый эфир, трет-бутилметиловый эфир, этиленгликольмонометиловый эфир, этиленгликольмоноэтиловый эфир, этиленгликольдиметиловый эфир, диметоксидиэтиловый эфир, тетрагидрофуран или диоксан, кетоны, такие как ацетон, метилэтилкетон или метилизобутилкетон, спирты, такие как метанол, этанол, пропанол, изопропанол, бутанол, этиленгликоль или глицерин, амиды, такие как N,N-диметилформамид, N, N-диэтилформамид, N,N-диметилацетамид, N-метилпирролидон или триамид гексаметилфосфорной кислоты, нитрилы, такие как ацетонитрил или пропионитрил, и сульфоксиды, такие как диметилсульфоксид, органические кислоты, такие как уксусная кислота, и воду. Предпочтительными растворителями являются простые эфиры, такие как тетрагидрофуран и этиленгликольдиметиловый эфир, прежде всего тетрагидрофуран, спирты, такие как метанол, этанол или изопропанол, галогенированные растворители, такие как дихлорметан или дихлорэтан, ароматические растворители, такие как бензол или толуол, нитрилы, такие как ацетонитрил, амиды, такие как N,N-диметилформамид, карбоновые кислоты, такие как уксусная кислота, и вода, а также их смеси. Наиболее предпочтительными растворителями являются метанол или этанол либо их смеси.
Реакцию предпочтительно проводить в интервале значений рН от 0 до 14, прежде всего от 2 до 10, в некоторых случаях от 6 до 9, преимущественно при рН 9.
Реакцию предпочтительно далее проводить при температуре в пределах от -80 до +140°С, более предпочтительно от -30 до +100°С, в некоторых случаях от -10 до +80°С, прежде всего от 0 до +50°С.
Предпочтительными восстановителями являются гидриды, такие как борогидриды, бораны, муравьиная кислота, формиаты или водород. Более предпочтительны гидриды, такие как борогидрид натрия, борогидрид цинка, борогидрид лития, цианоборогидрид натрия, триацетоксиборогидрид натрия или триацетоксиборогидрид тетраметиламмония. Наиболее предпочтителен борогидрид натрия.
Реакцию можно также проводить, если это возможно, в присутствии некоторых других химических соединений, таких как гомогенные или гетерогенные катализаторы или кислоты. Наиболее пригодны при этом такие кислоты, как соляная кислота, n-толуолсульфоновая кислота, уксусная кислота, пропионовая кислота, винная кислота или фталевая кислота, кислоты Льюиса, такие, например, как тетрахлорид титана, тетраизопропилат титана или хлорид цинка, соли, такие, например, перхлорат магния, ацетат натрия, натрий-калийтартрат, хлорид иттербия или n-толуолсульфонат пиридиния, водоабсорбирующие агенты, такие как сульфат натрия, молекулярные сита или силикагель, либо их смеси. Предпочтительными дополнительными агентами являются кислоты, такие как уксусная кислота, пропионовая кислота или винная кислота, более предпочтительна уксусная кислота. При проведении восстановления с помощью водорода целесообразно добавлять один или несколько пригодных гомогенных или гетерогенных катализаторов. Предпочтительными катализаторами подобного типа являются гетерогенные металлические катализаторы, которые известны в данной области, главным образом Ni-, Pt- или Pd-катализаторы, прежде всего никель Ренея и катализатор Линдлара (Pd-СаСО3-РbО). Пригодными гомогенными катализаторами являются прежде всего комплексные соединения родия, такие как катализаторы Уилкинсона (хлор-трис-трифенил-родий).
Соединения формулы (I), в каждом случае в свободной форме или в форме соли, могут быть представлены в виде одного из возможных изомеров или в виде их смеси, например в зависимости от количества асимметричных атомов углерода в молекуле и их абсолютной и относительной конфигурации и/или в зависимости от конфигурации неароматических двойных связей в молекуле они могут существовать в виде чистых изомеров, таких как антиподы и/или диастереоизомеры, либо в виде смесей изомеров, таких как смеси энантиомеров, например рацематов, смесей диастереоизомеров или смесей рацематов, при этом настоящее изобретение относится и к чистым изомерам, и ко всем возможным смесям изомеров, и поэтому в приведенном выше и в последующем описании под соединениями формулы (I) подразумеваются также их изомеры и их смеси, даже если в каждом отдельном случае и не указаны конкретные подробности касательно стереохимического строения.
Смеси диастереоизомеров и смеси рацематов соединений формулы (I) или их солей, получаемые предлагаемым в изобретении способом или иными методами в зависимости от используемых исходных материалов и выбранной технологии, можно на основе различий физико-химических свойств компонентов таких смесей разделять известными методами, например фракционированной кристаллизацией, перегонкой и/или хроматографией, на чистые диастереоизомеры или рацематы.
Получаемые таким путем смеси энантиомеров, таких как рацематы, можно известными методами разделять на оптические антиподы, например, перекристаллизацией из оптически активного растворителя, хроматографией на хиральных адсорбентах, например жидкостной хроматографией высокого разрешения (ЖХВР) на ацетилцеллюлозе, с помощью пригодных для этой цели микроорганизмов, расщеплением с помощью особых иммобилизованных ферментов, образованием соединений включения, например, с использованием хиральных краун-эфиров, в каковом случае только один энантиомер образует комплекс, или превращением в диастереоизомерные соли, например, взаимодействием основного конечного продукта в виде рацемата с оптически активной кислотой, такой как карбоновая кислота, например камфорная, винная или яблочная кислота, либо сульфоновая кислота, например камфорсульфоновая кислота, и разделением полученной смеси диастереоизомероов, например, на основе различий в их растворимости путем фракционированной кристаллизации, на диастереоизомеры, из которых целевой энантиомер можно выделить воздействием приемлемых, например основных, агентов.
Помимо разделения соответствующих смесей изомеров согласно изобретению существует также возможность получать общеизвестными методами диастереоселективного или энантиоселективного синтеза чистые диастереоизомеры или энантиомеры, например, за счет использования при проведении предлагаемого в изобретении способа исходных материалов с соответствующим стереохимическим строением.
Соединения формул (I) и (III), их кислотно-аддитивные продукты и соли можно также получать в виде их гидратов и/или они могут содержать другие растворители, например растворители, которые могли использоваться для кристаллизации соединений, образующихся в твердом виде.
Настоящее изобретение относится ко всем тем вариантам осуществления способа, в которых соединение, получаемое на любой его стадии в виде исходного соединения или промежуточного продукта, используется в качестве исходного материала с последующим проведением всех или некоторых из оставшихся стадий или в которых исходный материал используется в виде производного либо соли и/или их рацематов или антиподов или прежде всего образуется в реакционных условиях.
Соединения формул (I) и (III), получаемые предлагаемым в изобретении способом или иными методами, можно превращать в другие соединения формул (I) и (III) методами, которые известны как таковые.
В предлагаемом в изобретении способе предпочтительно использовать те исходные материалы и промежуточные продукты, в каждом случае в свободной форме или в форме соли, которые позволяют получать соединения формул (I) и (III) или их соли, которые представлены в начале описания в качестве наиболее предпочтительных.
В том случае, когда R9 представляет собой водород, реакционную стадию 2) можно разбить на две отдельные стадии, на первой из которых взаимодействием соединения формулы (III) с соединением формулы H2N(R8), в которой R8 имеет указанные выше для формулы (I) значения, получают соединение формулы
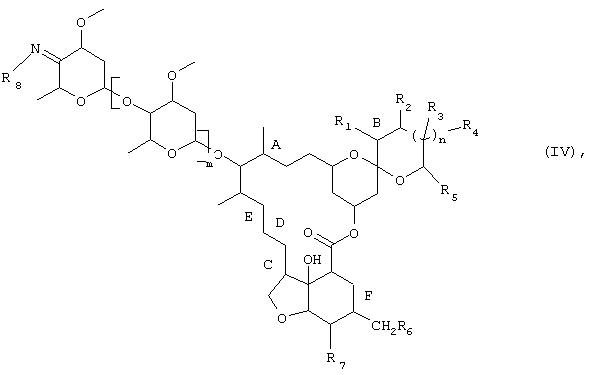
в которой R1, R2, R3, R4, R5, R6, R7, R8, m, n, А, В, С, D, Е и F имеют указанные выше для формулы (I) значения, а на второй стадии это соединение формулы (IV) восстанавливают по методике, описанной выше для стадии 2). Обе указанные отдельные стадии можно осуществлять так называемым методом синтеза в одном реакционном сосуде без выделения соединения формулы (IV), однако в некоторых случаях может оказаться целесообразным выделять соединение формулы (IV) с целью, например, его очистки. Соединения формулы (IV) являются новыми и также составляют один из объектов настоящего изобретения.
ПРИМЕРЫ
Ниже изобретение более подробно рассмотрено на примерах его осуществления. Следует отметить, что эти примеры носят исключительно иллюстративный характер и не ограничивают объем изобретения, если не указано иное. Настоящее изобретение относится прежде всего к способу получения, описанному в приведенных ниже примерах.
Пример 1: Получение клеток
1.1. Штамм Streptomyces tubercidicus I-1529
Предварительные культуры штамма I-1529 (Streptomyces tubercidicus, DSM-13135) выращивали в 20 500-миллилитровых оборудованных дефлектором колбах Эрленмейера, в каждой из которых содержалось 100 мл среды 2, на орбитальном шейкере при 120 об/мин и при 28°С в течение 3 дней.
Эти культуры использовали для инокуляции 40 л среды 4 в 50-литровом ферментере. Клетки выращивали при 28°С при скорости аэрации 0,7 об./об./мин (что соответствует расходу 30 л/мин). Скорость мешалки поддерживали на уровне от 200 до 300 об/мин, контролируя величину рO2 с помощью рО2-датчика во избежание ее уменьшения ниже 25%. После выращивания в течение 2 дней клетки собирали центрифугированием с помощью проточной центрифуги. Таким путем получили 4,2 кг клеток (во влажном состоянии).
1.2. Штамм Streptomyces tubercidicus R-922
Штамм Streptomyces tubercidicus R-922 (DSM-13136) выращивали в чашке Петри на агаре ISP-2 (среда 5). Эту культуру вносили в 4 500-миллилитровые встряхиваемые колбы с дефлектором, каждая из которых содержала 100 мл среды PHG (среда 6). Эти предварительные культуры выращивали на орбитальном шейкере при 120 об/мин и при 28°С в течение 96 ч и затем вносили в 10-литровый ферментер, оборудованный механической мешалкой и содержащий 8 л среды PHG. Такую основную культуру выращивали при 28°С, а также при перемешивании со скоростью 500 об/мин и при скорости аэрации 1,75 об./об./мин (14 л/мин) и давлении 0,7 бара. По завершении логарифмической фазы роста, т.е. примерно через 20 ч, клетки собирали центрифугированием. Выход влажных клеток составил 70-80 г/л культуры. Для последующего использования влажные клетки можно хранить при 4°С предпочтительно не более одной недели.
Пример 2: Проведение реакции
2.1. Покоящаяся культура
2.1.1. Реакционные условия
35,5 г авермектина (технич.) растворяли в 1,05 л смеси диметилсульфоксид/твин 40 в соотношении 1:1. Этот раствор аликвотными количествами по 25 мл распределяли по 42 3-литровым колбам Эрленмейера с дефлектором, каждая из которых содержала 1 л реакционной среды. Далее эти растворы стерилизовали при 121°С в течение 20 мин. После охлаждения до комнатной температуры добавляли 100 г влажных клеток (свежих или после хранения при 4°С не более 4-х дней), полученных согласно примерам 1.1 и 1.2 соответственно. Затем эти реакционные смеси встряхивали при комнатной температуре и при 120 об/мин в течение 4-5 дней.
Состав реакционной среды:
меласса 0,5 г
MgCl 0,5 г
ZnCl2 12,5 мг
MnCl2×4H2O 12,5 мг
CoCl2×6H2O 25 мг
NiCl2 ×6H2O 12,5 мг
CuCl2×2H2O 2,5 мг
NaMoO4 ×2H2O 6,3 мг
1 M HCl 0,15 мл
Объем среды доводят до 1 л добавлением фосфатного буфера в концентрации 70 мМ с рН 6,0, автоклавируют.
2.1.2. Переработка
Реакционные смеси центрифугировали в течение 15 мин при 4°С в 500-миллилитровых полипропиленовых центрифужных пробирках при 13000 g.
Супернатанты из 40 л реакционной смеси объединяли и дважды экстрагировали метил-трет-бутиловым эфиром (0,5 об. экв., 0,4 об. экв.). После этого объединенные метил-трет-бутилэфирные фазы трижды подвергали обратной экстракции 0,185 об. экв. дистиллированной воды. Метил-трет-бутилэфирную фазу концентрировалии в вакууме на роторном испарителе. После сушки остатка получили 10-12 г экстракта S. Водные фазы отбрасывали.
Отделенные центрифугированием клетки из 120-132 центрифужных пробирок экстрагировали следующим образом. Клетки из каждых 24 центрифужных пробирок переносили в одну 2-литровую колбу Эрленмейера. В каждую колбу Эрленмейера добавляли 80 г диатомовой земли (Hyflo Supercell®, очищенная) и 1,2 л ацетона. После ручного перемешивания смесь гомогенизировали с помощью большой магнитной мешалки. Полученную суспензию подвергали вакуум-фильтрации через воронку Бюхнера диаметром 20 см с бумажным фильтром и промывали ацетоном до получения бесцветного элюата. Таким путем получили фильтрат С1 и фильтровальный осадок С1. Фильтрат С1 концентрировали в вакууме на роторном испарителе для удаления ацетона. Затем полученную водную фазу трижды экстрагировали 0,7 л толуола. Объединенные толуольные фазы сушили над безводным сульфатом натрия. В результате фильтрации и упаривания на роторном испарителе в вакууме получили экстракт С1.
Фильтровальный осадок С1 переносили в 2-литровую колбу Эрленмейера и вручную смешивали с 1,5 л толуола. Смесь гомогенизировали с помощью большой магнитной мешалки. Полученную суспензию подвергали вакуум-фильтрации через воронку Бюхнера диаметром 20 см с бумажным фильтром и промывали толуолом до получения бесцветного элюата. Таким путем получили фильтрат С2 и фильтровальный осадок С2. Фильтровальный осадок С2 отбрасывали. Фильтрат С2 концентрировали в вакууме на роторном испарителе с получением экстракта С2, который сушили в глубоком вакууме. Объединеные экстракты С1 и С2 из 40 л реакционной смеси сушили в глубоком вакууме с получением 30-35 г экстракта С.
45 г объединенных экстрактов S и С подвергали экспресс-хроматографии аналогично методу, описанному у Clark-Still и др., на колонке, заполненной 1,5 кг силикагеля (Merck 60, 0,040-0,063 мм), элюируя смесью этилацетат/гексан в соотношении 3:2 при давлении N2 0,5 бара и контролируя протекание процесса с помощью тонкослойной хроматографии. Выход чистого 4"-оксоавермектина составил 5,6 г.
2.2. Пролиферируюшая культура
2.2.1. Реакционные условия
1 г авермектина (технич.) растворяли в 50 мл смеси диметилсульфоксид/твин 40 в соотношении 1:1. Этот раствор аликвотными количествами по 2,5 мл распределяли по 20 500-миллилитровым колбам Эрленмейера с дефлектором, каждая из которых содержала по 100 мл среды 4. Эти растворы стерилизовали при 121°С в течение 20 мин. После охлаждения до комнатной температуры добавляли 5 мл предварительной культуры, полученной согласно примеру 1.1 и 1.2 соответственно. Затем такие инокулированные культуры инкубировали при 28°С в течение 7 дней на орбитальном шейкере при 120 об/мин.
2.2.2. Переработка
Реакционные смеси в течение 15 мин центрифугировали при 4°С в 500-миллилитровых полипропиленовых центрифужных пробирках при 13000 g и обрабатывали аналогично примеру 2.1.2. Таким путем получили 252 мг чистого 4’’-оксоавермектина.
2.3. Бесклеточный биокатализ
2.3.1. Получение бесклеточного экстракта
Исходные растворы:
РР-буфер: 50 мМ К2НРO4/КН2РO4 (рН 7,0).
Буфер для лизиса: 50 мМ К2НРO4/КН2РO4 (рН 7,0), 5 мМ бензамидин, 2 мМ дитиотреитол, 0,5 мМ продукт Pefabloc (фирма Roche Diagnostics).
Субстрат: раствор 10 мг авермектина в 1 мл изопропанола.
6 г влажных клеток после промывки в РР-буфере ресуспендировали в 35 мл буфера для лизиса и разрушали в прессе Френча при 4°С. Полученную суспензию центрифугировали в течение 1 ч при 35000 g. Далее собирали надосадочный бесклеточный экстракт.
2.3.2. Методика проведения анализа по определению ферментативной активности
Исходные растворы:
Ферредоксин: раствор 5 мг ферредоксина (из шпината) вконцентрации 1-3 мг/мл в Трис/НСl-буфере (фирма Fluka) или раствор 5 мг ферредоксина (из Clostridium pasteurianum) в концентрации 1-3 мг/мл в Трис/HCl-буфере (фирма Fluka), или раствор 5 мг ферредоксина (из Porphyra umbilicalis) в концентрации 1-3 мг/мл в Трис/НСl-буфере (фирма Fluka).
Ферредоксин-редуктаза: раствор 1 мг высушенной вымораживанием ферредоксин-редуктазы (из шпината) в концентрации 3, 9 ед. /мг в 1 мл Н2О (фирма Sigma).
NADPH: 100 мМ NADPH в Н2О (фирма Roche Diagnostics).
(Все исходные растворы хранили при -20°С, а при использовании держали на льду.)
Условия ЖХВР:
ЖХВР-хромагограф: Merck-Hitachi.
ЖХВР-колонка: 70×4 мм, Kromasil 100 С18, 3,5 мкм (фирма Macherey-Nagel, Швейцария).
Растворитель А: ацетонитрил, содержащий 0,0075% трифторуксусной кислоты.
Растворитель Б: вода, содержащая 0,01% трифторуксусной кислоты.
Скорость потока: 1,5 мл/мин.
Обнаружение: УФ с длиной волны 243 нм.
Образец: 30 мкл.
Время удерживания: авермектин В1а 3,18 мин; 4’’-оксоавермектин В1а 4,74 мин.
Режим прокачивания: 0,0 мин 75% А, 25% Б.
линейный градиент: до 7, 0 мин 100% А, 0% Б;
до 9,0 мин 100% А, 0% Б.
cтупенчатое изменение: до 9,1 мин 75% А, 25% Б;
до 12,0 мин 75% А, 25% Б.
К 475 мкл бесклеточного экстракта добавляли следующие растворы: 10 мкл ферредоксина, 10 мкл ферредоксин-редуктазы и 1 мкл субстрата. После добавления субстрата смесь сразу же тщательно перемешивали и аэрировали. После этого добавляли 5 мкл NADPH и смесь инкубировали при 30°С в течение 30 мин. Затем к реакционной смеси добавляли 1 мл метил-трет-бутилового эфира и тщательно перемешивали. Далее смесь центрифугировали в течение 2 мин при 14000 об/мин и метил-трет-бутилэфирную фазу переносили в 10-миллилитровую колбу и упаривали в вакууме на роторном испарителе. Остаток растворяли в 200 мкл ацетонитрила и переносили в пробирки для ЖХВР-анализа.
После инжекции образца объемом 30 мкл хроматографический пик появлялся через 4,74 мин, указывая на наличие 4’’-оксоавермектина В1а. Анализ ЖХВР-масс-спектроскопией позволяет соотнести с этим пиком массу в 870 Да, что соответствует молекулярной массе 4’’-оксоавермектина В1а.
При анализе полученного продукта с помощью ЖХВР и ЖХВР-масс-спектроскопии второй пик проявлялся через 2,01 мин, что соответствует кетогидрату 4’’-гидроксиавермектина. Этот факт указывает на то, что бесклеточный экстракт обеспечивает превращение авермектина в результате гидроксилирования в 4’’-гидроксиавермектин, из которого в результате дегидратации образуется 4’’-оксоавермектин.
Вместо ферредоксина из шпината можно использовать, например, ферредоксин из бактерии Clostridium pasteurianum или из красной водоросли Porphyra umbilicalis, что также приводит к биокаталитическому превращению авермектина в 4’’-оксоавермектин.
Пример 3: Штаммы Streptomyces
Штаммы рода Streptomyces, которые можно использовать в предлагаемом в изобретении способе, и данные о степени их родства со штаммами S. tubercidicus I-1529 и R-922, выявленной при анализе 16s рДНК, представлены в таблице.
Пример 4: Получение 4’’-дезокси-4’’-эпи-(метиламино)авермектина В1 формулы
в которой R представляет собой метил или этил.
3 мл уксусной килоты в 30 мл метанола охлаждали до 0-5°С. После этого добавляли газообразный метиламин до тех пор, пока значение рН раствора не установится на 9. К 8,25 мл этого раствора метиламина при 0°С добавляли раствор 1,0 г 4’’-оксоавермектина В1 в 6 мл метанола. Смеси давали нагреться до окружающей температуры и затем перемешивали в течение 50 мин при комнатной температуре. Далее добавляли 90 мг борогидрида натрия в 2,5 мл этанола и полученную смесь перемешивали еще в течение 45 мин. Затем к реакционной смеси добавляли 10 мл этилацетата, органическую фазу трижды экстрагировали насыщенным водным раствором гидрокарбоната натрия, органическую фазу отделяли и сушили над сульфатом натрия. После отгонки растворителя получили 4’’-дезокси-4’’-эпи-(метиламино)авермектин В1. Чистота этого продукта составила более 90%.
Библиография
Angelino S.A.G.F., Muller F., van der Plas H.C., Purification and immobilization of rabbit liver aldehyde oxidase, Biotechnol. Bioeng. 27: 447-455 (1985);
Bachmann S., Gebicka L., Gasyna Z., Immobilization of glucose isomerase on radiation-modified gelatine gel, Starch/Starke 33: 63-66 (1981);
Barbaric S., Kozulic В., Leustek I., Pavlovic В., Cesi V., Mildner P., Crosslinking of glycoenzymes via their carbohydrate chains, 3rd Eur. Congr. Biotechnol., изд-во Verlag Chemie, Weinheim, 1984, т.1, стр.307-312;
Beddows C.G., Guthrie J.T., Abdel-Hay F.I., The use of graft copolymers as enzyme supports immobilization of proteins and enzymes on a hydrolyzed nylon-coacrylonitrile system, Biotechnol. Bioeng. 23: 2885-2889 (1981);
Bettmann H., Rehm H.J., Degradation of phenol by polymer entrapped microorganisms, Appl. Microbiol. Biotechnol. 20: 285-290 (1984);
Bhairi S.M., A guide to the Properties and Uses of Detergents in Biology and Biochemistry, Calbiochem-Novabiochem Corp., San Diego CA (1997);
Bickerstaff G.F. (ред.). Immobilisation of Enzymes and Cells (Methods in Biotechnology Series), изд-во Humana Press, Totowa NJ USA, 1997;
Bihari V., Goswami P.P., Rizvi S.H.M., Kahn A.W., Basu S.K., Vora V.C., Studies on immobilized fungal spores of microbial transformation of steroids: 11a-hydoxylation of progesterone with immobilized spores of Aspergillus ochraceus G8 on polyacrylamide gel and other matrices, Biotechnol. Bioeng. 26: 1403-1408 (1984);
Black G.M., Webb C., Matthews T.M., Atkinson В., Practical reactor systems for yeast cell immobilization using biomass support particles, Biotechnol. Bioeng. 26: 134-141(1984);
Cabral J.M.S., Novais J.M., Cardoso J.P., Coupling of glucoamylase on alkylamine derivative of titanium(IV) activated controlled pore glass with tannic acid, Biotechnol. Bioeng. 26: 386-388 (1984);
Cannon J.J., Chen L.-F., Flickinger M.C., Tsao G.T., The development of an immobilized lactate oxidase system for lactic acid analysis, Biotechnol. Bioeng. 26: 167-173 (1984);
Cantarella M., Migliaresi С., Tafuri M.G., Afani F., Immobilization of yeast cells in hydroxymethacrylate gels, Appl. Microbiol. Biotechnol. 20: 233-237 (1984);
Chipley J.R., Effects of 2,4-dinitrophenol and N,N’-dicyclo-hexylcarbodiimide on cell envelope-associated enzymes of Escherichia coli and Salmonella enteritidis, Microbios. 10: 115-120(1974);
Clark-Still W., Kahn M., Mitra A., Rapid chromatographic technique for preparative separations with moderate resolution, J. Org. Chem. 43: 2923-2925 (1978);
Crueger W., Crueger A., Biotechnologie-Lehrbuch der angewandten Mikrobiologie, 2-е изд., изд-во R. Oldenbourg Verlag Munich, Vienna, 1984;
Dennis K.E., Clark D.S., Bailey J.E., Cho Y.K., Park Y.H., Immobilization of enzymes in porous supports, Biotechnol. Bioeng. 26: 892-900 (1984);
Deo Y.M., Gaucher G.M., Semicontinuous and continuous production of penicillin-G by Penicillium chrysogenum cells immobilized in k-carrageenan beads, Biotechnol. Bioeng. 26: 285-295 (1984);
De Rosa M., Gambacorta A., Lama L., Nicolaus В., Immobilization of thermophilic microbial cells in crude egg white, Biotechnol. Lett 3: 183-188 (1981);
DiLuccio R.C., Kirwan D.J., Effect of dissolved oxygen on nitrogen fixation by A. Vinelandii. II. Ionically adsorbed cells, Biotechnol. Bioeng. 26: 87-91 (1984);
Erhardt H.M., Rehm H.J., Phenol degradation by microorganisms adsorbed on activated carbon, Appl. Microbiol. Biotechnol. 21: 32-36 (1985);
Eikmeier H., Rehm H.J., Production of citric acid with immobilized Aspergillus niger, Appl. Microbiol. Biotechnol. 20: 365-370 (1984);
Forberg С., Haggstrom L., Adsorbed cell systems controlled by the nutrient dosing technique, 3rd Eur. Congr. Biotechnol., изд-во Verlag Chemie, Weinheim, 1984, т.2, стр.115-120;
Gainer J.L., Kirwan D.J., Foster J.A., Seylan E., Use of adsorbed and covalently bound microbes in reactors, Biotechnol. Bioeng. Symp. 10: 35-42 (1980);
Giard D.J., Loeb D.H., Thilly W.G., Wang D.I.C., Levine D.W., Human interferon production with diploid fibroblast cells grown on microcarriers, Biotechnol. Bioeng. 21: 433-442 (1979);
Greene Th.W., Wuts P.G.M., Protective Groups in Organic Synthesis, 3-е изд., изд-во John Wiley & Sons, New York NY, 1999;
Hartmeier W., Immobilisierte Biokatalysatoren, изд-во Springer Verlag, Berlin, Heidelberg, New York, Tokyo, 1986;
Hofstee B.H.J., Immobilization of enzymes through non-covalent binding to substituted agaroses, Biochem. Biophys. Res. Commun. 53: 1137-1144 (1973);
Ibrahim M., Hubert P., Dellacherie E., Magadalou J., Muller J., Siest G., Covalent attachment of epoxide hydrolase to dextran, Enz. Microbiol. Technol. 7: 66-72 (1985);
Jack T.R., Zajic J.E., The enzymatic conversion of L-histidine to urocanic acid by whole cells of Micrococcus luteus immobilized on carbodiimide activated carboxymethylcellulose, Biotechnol. Bioeng. 19: 631 (1977);
Jirku V., Turkova J., Krumphanzl V., Immobilization of yeast with retention of cell division and extracellular production of macromolecules, Biotechnol. Lett. 2: 509-513 (1980);
Karube I., Kawarai M., Matsuoka H., Suzuki S., Production of L-glutamate by immobilized protoplasts, Appl. Microbiol. Biotechnol. 21: 270-272 (1985);
Kato Т., Horikoshi К., Immobilized cyclomaltodextrin glucano-transferase of an alkalophilic Bacillus sp. no 38-2, Biotechnol. Bioeng. 26: 595-598 (1984);
Kaul R., D’Souza S.F., Nadkarni G.B., Hydrolysis of milk lactose by immobilized galactosidase-hen egg white powder, Biotechnol. Bioeng. 26: 901-904 (1984);
Khan S.S., Siddiqi A.M., Studies on chemically aggregated pepsin using glutaraldehyde, Biotechnol. Bioeng. 27: 415-419 (1985);
Krakowiak W., Jach M., Korona J., Sugier H., Immobilization of glucoamylase on activated aluminium oxide, Starch/Starke 36: 396-398 (1984);
Kuhn W., Kirstein D., Mohr P., Darstellung und Eigenschaften tragerfixierter Glukoseoxydase, Acta Biol. med. Germ. 39: 1121-1128 (1980);
Mazumder Т.К., Sonomoto К., Tanaka A., Fukui S., Sequential conversion of cortexolone to prednisolone by immobilized mycelia of Curvularia lunata and immobilized cells of Arthrobacter simplex, App. Microbial. Biotechnol. 21: 154-161 (1985);
Messing R.A., Oppermann R.A., Pore dimensions for accumulating biomass. I. Microbes that reproduce by fission или budding, Biotechnol. Bioeng. 21: 49-58 (1979);
Miyawaki О., Wingardjr L.B., Electrochemical and enzymatic activity of flavin dinucleotide and glucose oxidase immobilized by adsorption on carbon, Biotechnol. Bioeng. 26: 1364-1371 (1984);
Monsan P., Combes D., Alemzadeh I., Invertase covalent grafting onto corn stover, Biotechnol. Bioeng. 26: 658-664 (1984);
Mori Т., Sato Т., Tosa Т., Chibata I., Studies on immobilised enzymes. X. Preparation and properties of aminoacylase entrapped into асrуlамиде gel-lattice, Enzymologia 43: 213-226 (1972);
Nakajima H., Sonomoto К., Usui N., Sato F., Yamada Y., Tanaka A., Fukui S., Entrapment of Lavendula Vera and production of pigments by entrapped cells, J. Biotechnol. 2: 107-117 (1985);
Navarro J.M., Durand G., Modification of yeast metabolism by immobilization onto porous glass, Eur. J. Appl. Microbiol. Biotechnol. 4: 243-254 (1977);
Prave P., Faust U., Sittig W., Sukatsch. D.A., Handbuch Biotechnologie, 2-e изд., изд-во R. Oldenbourg Verlag Munich, Vienna, 1984;
Qureshi N., Tamhane D.V., Production of mead by immobilized whole cells of Saccharomyces cerevisiae, Appl. Microbiol. Biotechnol. 21: 280-281 (1985);
Raghunath К., Rao K.P., Joseph U.T., Preparation and characterization of urease immobilized onto collagen-poly(glycidyl methacrylate) graft copolymer, Biotechnol. Bioeng. 26: 104-109 (1984);
Romanovskaya V.A., Karpenko V.I., Pantskhava E.S., Greenberg T.A., Malashenko Y.R., Catalytic properties of immobilized cells of метан-oxidizing and methanogenic bacteria, под ред. Moo-Young M., Advances in Biotechnology, изд-во Pergamon, Toronto, 1981, т. 3, стр.367-372;
Shimizu S., Morioka H., Tani Y., Ogata K.., Synthesis of coenzyme A by immobilized microbial cells, J. Ferm. Technol. 53: 77-83 (1975);
Talsky G., Gianitsopoulos G., Intermolecular crosslinking of enzymes, 3rd Eur. Congr. Biotechnol., изд-во Verlag Chemie, Weinheim, 1984, т.1, стр.299-305;
Umemura I., Takamatsu S., Sato Т., Tosa Т., Chibata I., Improvement of production of L-aspartic acid using immobilized microbial cells, Appl. Microbiol. Biotechnol. 20: 291-295 (1984);
Van Haecht J.L., Bolipombo M., Rouxhet P.G., Immobilization of Saccaromyces cerevisiae by adhesion: treatment of the cells by AI ions, Biotechnol. Bioeng. 27: 217-224 (1985);
Vogel H.J., Brodelius P., An in vivo 31P NMR comparison of freely suspended and immobilized Catharanthus roseus plant cells, J. Biotechnol. 1: 159-170 (1984);
Weetall H.H., Mason R.D., Studies on immobilized papain, Biotechnol. Bioeng. 15: 455-466 (1973);
Wiegel J., Dykstra M., Clostridium thermocellum: adhesion and sporulation while adhered to cellulose and hemicellulose, Appl. Microbiol. Biotechnol. 20: 59-65 (1984);
Workman W.E., Day D.F., Enzymatic hydrolysis of inulin to fructose by glutaraldehyde fixed yeast cells, Biotechnol. Bioeng. 26: 905-910 (1984).
Указанная в описании патентная литература:
US 5288710,
ЕР-А-0736252,
ЕР 0301806,
ЕР 0401029,
DE 2717040.
Реферат
Изобретение относится к органической химии, в частности к способам получения соединения формулы (I):

в которой m означает 0, 1 или 2; n означает 0, 1, 2 или 3 и А обозначает двойную связь, В обозначает двойную или простую связь, С обозначает двойную связь, D обозначает простую связь, Е и F обозначают двойную связь; r1 обозначает Н или С1-С8алкил; r2 обозначает Н, С1 -С8алкил или ОН; R3 и R4 каждый независимо друг от друга обозначают H или С1-С8алкил; R5 обозначает Н или С1 -С8 алкил; R6 обозначает Н; R7 обозначает ОН; R8 и R9 независимо друг от друга обозначают Н или С1-С10алкил; в свободной форме или в виде соли, заключающийся в том, что соединение формулы (II):
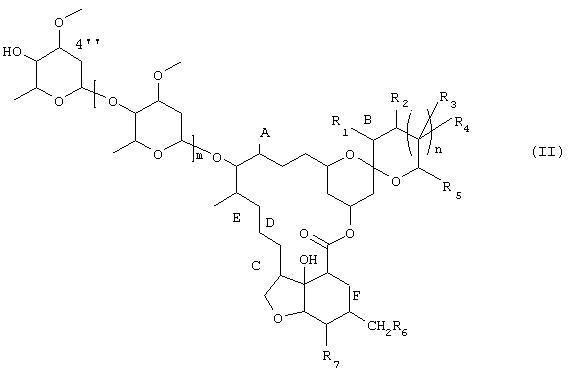
вводят в контакт с биокатализатором, который способен избирательно окислять спирт в положении 4", с получением соединения формулы (III):

в которых R1-R7, m, n, А, В, С, D, Е и F имеют те же значения, что и указанные выше для формулы (I), и соединение формулы (III) подвергают взаимодействию в присутствии восстановителя с известным амином формулы HN(R8)R9, в которой R8 и R9имеют те же значения, что и указанные для формулы (I), с последующим выделением целевого продукта в свободном виде или в виде соли. Предлагаемый способ является двухстадийным, при его осуществлении не требуется использовать защитные группы, а сам способ является более безвредным с экологической точки зрения. 4 с. и 10 з.п. ф-лы, 1 табл.
Формула
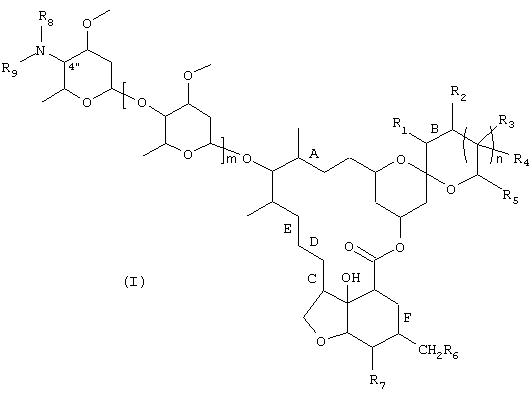
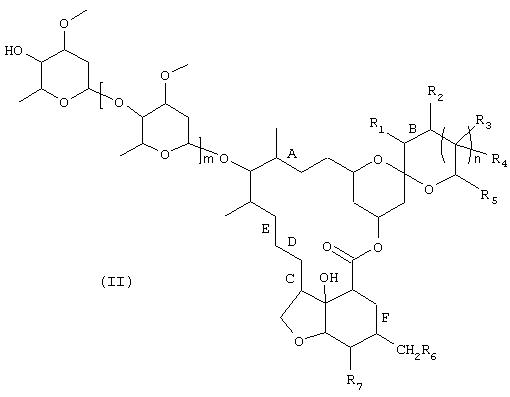

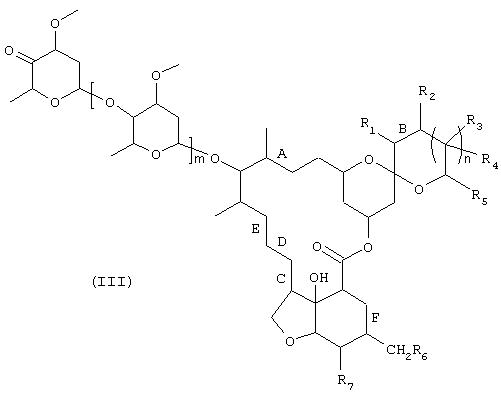

Документы, цитированные в отчёте о поиске
Производные авермектина, фармацевтическая или ветеринарная композиция и способ получения соединений
Комментарии