Производные авермектина, фармацевтическая или ветеринарная композиция и способ получения соединений - RU2125059C1
Код документа: RU2125059C1
Чертежи

Описание
Настоящее изобретение относится к новым антипаразитическим средствам, родственным милбемицинам и авермектинам, способам их получения и их препаратам.
Авермектины являются группой антипаразитических средств широкого спектра действия, их ранее именовали соединениями C-076. Их получают ферментацией с участием штамма микроорганизма Streptomyces avermitilis в аэробных условиях в водной питательной среде, содержащей неорганические соли и ассимулируемые источники углерода и азота. Выделение и химическая структура восьми индивидуальных компонентов, которые составляют комплекс C-076, указываются подробно в описании Британского патента N 1573955.
Комплекс С-076 содержит восемь различных, но близко родственных соединений, описанных как С-076 A1a, A1b, A2a, A2b, B1a, B1b, B2a и B2b. Ряд "а" соединений относится к природным авермектинам, у которых 25 - заместитель является (S) - втор-бутилом, и ряд "b" относится к тем соединениям, у которых 25-заместитель является изопропилом. Обозначения "A" и "B" относятся к авермектинам, у которых 5-заместитель является метоксигруппой или гидроксигруппой соответственно, и цифра "1" относится к авермектинам, у которых двойная связь присутствует в положении 22(23), цифра "2" относится к авермектинам, которые не имеют 22 (23)-двойной связи и содержат водород в 22-положении и гидроксигруппу в 23- положении.
В наших заявках на Европейские патенты NN 0214731, 0284176, 0317148, 0308145, 0340832, 0335541 и 0350187 описываются способы получения соединений, родственных авермектинам, но имеющих в 25-положении группу, отличную от изопропила или (S) - втор-бутила, найденного в первоначальных авермектинах, указанных в описании патента Англии N 1573955. Такие соединения можно получить ферментацией с участием определенных штаммов Streptomyces avermitilis в присутствии органических кислот или их производных. Получение таких авермектинов описывается в Journal of Antibiotics (1991), 44, N 3, pp. 357-365.
Милбемицины образуют другую группу родственных макролидов, которые отличаются от авермектинов отсутствием остатка сахара, присоединенного в С-13-положении. Примеры таких соединений описываются в патенте Англии N 1390336 и Европейских патентных публикациях NN 170006, 254583, 334484 и 410615. Кроме этих продуктов ферментации большое число публикаций описывает соединения, полученные полусинтетически из этих продуктов ферментации, многие из которых обладают пригодной антипаразитической активностью. Часть этих способов рассматривается в Maerolide Antibiotics, Omura S., E.d., Academic press, New York (1984) и Davies, H.G., Green, R.H. B Natural product Reports (1986), 3, 87-121 и в Chem. Soc. Rev., 1991, 20, 271-339.
Было показано, что некоторые соединения, которые можно получить синтетически из известных авермектинов и производных авермектинов, обладают неожиданно полезными биологическими свойствами.
В соответствии с одним из аспектов настоящего изобретения предложены соединения формулы 1

в которой пунктирная линия в 22 (23)- положении обозначает возможную дополнительную связь, причем, или эта связь присутствует и R1 отсутствует, или эта связь отсутствует и R1 представляет собой H или OH, R2 представляет собой C1-C8-алкил, или C3-C8-циклоалкил,
R3 представляет собой H,
R4 представляет собой H или группу, способную гидролизоваться in vivo с образованием соединения, у которого R4 представляет собой H, и R5 представляет собой OH.
Если контекст не оговорен особо, все алкильные и алкенильные заместители, имеющие 3 или более атомов углерода, могут иметь цепь нормального или разветвленного строения. Термин "арил" включает фенил, который может быть замещен по меньшей мере одним C1-C6-алкилом, гидроксигруппой, C1-C6-алкоксигруппой, галогеном, нитрогруппой или CF3 . В настоящем изобретении термин "алкил" обозначает алкилы с 1-8 атомами углерода, например метил, этил, пропил, изопропил, бутил, пентил, гексил и подобные алкилы, с цепью нормального или разветвленного строения. Термин "алканоил" обозначает алканоилы с 1-8 атомами углерода, например формил, ацетил, пропионил, бутирил, пентаноил, гексаноил и подобные радикалы.
Термин "карбамоил" обозначает группу -CONR7R8, в которой R7 и R8, одинаковые или разные, представляют собой H, алкил, арил, гетероарил или образуют 4-8-членное ядро, содержащее один или несколько атомов O, N или S.
Группы, гидролизуемые in vivo с образованием соответствующих соединений, у которых эта группа замещена на H, в общем хорошо известны в фармации, целый ряд таких групп пригоден для применения в соединениях настоящего изобретения. Примерами таких групп является C2-C8-алканоил, ароил, карбомоил, C1-C8-алкоксикарбонил и остатки дикарбоновых кислот и аминокислот. Конкретные такие группы указываются в приведенных ниже примерах. Предпочтительны те соединения, у которых R2 - циклогексил, R3 представляет собой H и возможная связь в 22 (23)-положении отсутствует и R1 представляет собой H.
В частности, предпочтительны оксимы моносахаридов, у которых R4 представляет собой H.
Индивидуальные соединения изобретения описаны в примерах, приведенных ниже.
Наиболее предпочтительным соединением является моносахарид-5-оксимино-22,23-дигидро- 25-циклогексилавермектина B1.
В соответствии с другим аспектом изобретения предложен способ получения такого соединения,
который предусматривает стадии: (i) окисления соединения формулы II

для образования соединения формулы III

и (ii) реакции соединения формулы III с соединением формулы
R4 - O - NH2,
где
R4 имеет указанные выше значения, и
R5 представляет собой α - олеандрозилоксигруппу,
и гидролиза полученного соединения в соединение формулы I и (iii), если необходимо, замещения группы R4, когда она представляет собой H, группой, способной гидролизоваться in vivo с образованием соединения, у которого R4 представляет собой H. Если необходимо (iv) (vii) гидрирование соединения для восстановления двойной связи в положении 22 (23) в одинарную связь.
Получение соединений изобретения обсуждается и иллюстрируется ниже.
Соединения настоящего изобретения можно получить из соединений формулы (iv), которые можно получить, как описано в указанных выше патентных публикациях.
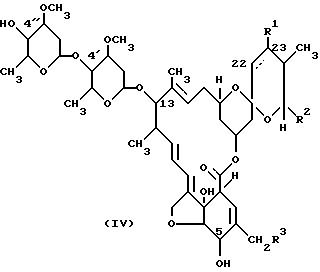
Соединение Па. Двойная связь присутствует, R1 отсутствует.
Соединение Пb. Двойная связь отсутствует, R1 = H.
Соединение Пc. Двойная связь отсутствует, R1 = OH.
Полусинтетические модификации, требуемые для получения соединений формулы I, могут требовать последовательные реакции, точный порядок проведения таких превращений может изменяться. Соединения, имеющие 23-гидроксигруппу (или ее защищенное производное), можно превратить в соответствующее 22, 23-дигидросоединение или соответствующее соединение, имеющее двойную связь в 22(23)- положении, при помощи способов, описанных в патенте США N 4328335. Последние соединения можно также гидрированием превращать в 22, 23-дигидросоединения при применении катализатора Вилкинсона в условиях, описанных в патенте США N 4199569.
Получения соединений изобретения можно достичь прежде всего превращением указанных выше дисахаридов Па, b и c в их соответствующие моносахариды гидролизом. Другой способ получения моносахаридов состоит в прямой ферментации соответствующего агликона, как описано в заявке на Европейский патент N 463677.
По другому способу соединения изобретения можно получить проведением указанных выше синтетических превращений на дисахаридах Па, b или с и последующим гидролизом их в конце в целевые моносахариды.
При желании гидроксигруппы можно ацилировать для получения эфиров с применением таких реагентов, как ангидриды или хлорангидриды и амины кислот в соответствии с общими известными в этой области методиками. Гидроксигруппы можно превратить в оксогруппы окислением диоксидом магния или перрутенатом тетрапропиламмония. Оксосоединение можно обработать гидроксиламином или его О-замещенным аналогом для получения соответствующего оксима.
Соединения изобретения эффективны при лечении различных состояний, вызванных эндопаразитами, включая, в частности, гельминтоз, который наиболее часто вызывается группой паразитических червей, которые описаны как нематоды и которые могут вызвать тяжелые экономические потери при разведении свиней, овец, лошадей и крупного рогатого скота, а также поражает домашних животных и птицу. Эти соединения эффективны также против других нематод, которые поражают различные виды животных, включая, например, Dirofilaria у собак, и различных паразитов, которые могут заражать скот, сопровождающих людей животных, например кошек и собак, а также людей, включая желудочно-кишечных паразитов, например Ancylostoma, Necator, Ascaris, Strongyloides, Trichinella, Capillaria, Toxocara, Toxascaris, Trichuris, Enterobius, и паразитов, которые найдены в крови или других тканях и органах, например филярии и внекишечные стадии Strongyloides и Toxocara, Trichinella.
Эти соединения особенно ценны также при лечении заражения эктопаразитами, включая, в частности, заражения членистоногими эктопаразитами человека, животных и птиц, например, клещами, иксодовыми клещами, вшами, блохами, падальными мухами, жалящими насекомыми и мигрирующими личинками двухкрылых насекомых, которые поражают крупный рогатый скот и лошадей.
Соединения являются также инсектицидами, активными против домашних насекомых - вредителей, например тараканов, моли, кожеедов и комнатных мух, а также пригодными против членистоногих насекомых-вредителей хранимого зерна и сельскохозяйственных культур, например клеща паутинного, тли, гусениц и против мигрирующих ортоптеранов, например саранчи. Мы обнаружили, что соединения в пределах объема настоящего изобретения характеризуются как безопасностью, так и обладают неожиданно очень сильной системной активностью против крылатых насекомых и других важных членистоногих паразитов кошек и собак.
Соединения формулы I можно вводить в виде состава, соответствующего конкретному предусматриваемому способу введения и данным видам животных, которых лечат этими соединениями и которые заражены паразитами или насекомыми. Соединения можно вводить инъекцией подкожно или внутримышечно. Их можно также вводить перорально в форме капсулы, болюса, таблетки, жевательной таблетки или жидкости для вливания лекарственного средства в ротовую полость животного, а также их можно вводить в виде состава для местного введения или имплантата. Для местного введения можно применять состав для окунания, состав для разбрызгивания, порошок, дуст, состав для выливания на животного, состав для нанесения пятен, жидкость для опрыскивания, шампунь, ошейник, ярлык или сбрую. Также составы получают обычным образом в соответствии со стандартной практикой ветеринарии. Так, например, капсулы, болюсы или таблетки модно получить смешиванием активного компонента с пригодным, мелкоизмельченным разбавителем или носителем, дополнительно содержащим дезинтегригующее средство и/или связующее, например, крахмал, лактозу, тальк или стеарат магния. Состав для вливания в ротовую полость животного можно получить диспергированием активного компонента в водном растворе вместе с диспергирующими или смачивающими средствами и инъецируемые препараты можно получить в форме стерильного раствора или эмульсии. Составы для выливания или нанесения пятен можно получить растворением активного компонента в пригодном носителе-наполнителе, например бутилдиголе, жидком парафине или нелетучем сложном эфире с возможным добавлением летучего компонента, например изопропанола. По другому варианту препараты для выливания, нанесения пятен или разбрызгивания на животное можно получить капсулированием для оставления остатка активного компонента на поверхности этого животного. Эти составы будут изменяться в соответствии с массой активного компонента, зависящей от вида обрабатываемого животного-хозяина, тяжести и типа инфицирования и массы тела носителя инфекции. Соединения можно вводить непрерывно, в частности для профилактики, известными методами. Обычная доза для перорального, парентерального введения и введения выливанием на животное от около 0,001 до 10 мг на 1 кг массы тела животного (в виде разовой дозы или в виде разделенной общей дозы на курс лечения от 1 до 5 дней) бывает достаточной, но могут быть случаи, когда есть показания на более высокие или более низкие пределы доз, и это входит в объем настоящего изобретения.
По другому варианту соединения можно вводить вместе с кормом для животного и для этой цели можно получить концентрированную кормовую добавку или премикс для смешивания с обычным кормом для животных.
Для применения в качестве инсектицида и для борьбы с сельскохозяйственными вредителями соединения наносят в виде состава для разбрызгивания, дуста, состава для выливания, эмульсий и подобных форм в соответствии с обычной сельскохозяйственной практикой.
Человеку эти соединения вводят в виде фармацевтически пригодного препарата в соответствии с обычной медицинской практикой.
Получение соединений в соответствии с изобретением иллюстрируется следующими примерами.
Примеры 14-25 относятся к соединениям, в которых R4 представляет собой группу, способную гидролизоваться in vivo.
Пример 1. Моносахарид 22, 23 - дигидроавермектина В1a.
22, 23 -Дигидроавермектин B1a (50 г) растворяли в смеси изопропанола ( 100 мл) и серной кислоты ( 1 мл) и перемешивали при комнатной температуре в атмосфере азота в течение 48 часов. Реакционную смесь выливали на измельченный лед и экстрагировали дихлорметаном ( 2 х 200 мл). Объединенный экстракт промывали водным насыщенным раствором бикарбоната натрия (100 мл), сушили над безводным сульфатом магния и концентрировали в вакууме, получая белые кристаллы (14 г), которые отделяли фильтрованием. Масс-спектр и ЯМР-спектр продукта полностью согласовывались с предполагаемой структурой.
B1b- аналог получали идентичным способом из 22, 23-дигидроавермектина B1b.
Пример 2. Моносахарид 5-оксо-22, 23 -дигидроавермектина В1a.
Моносахарид 22, 23 - дигидроавермектина В1a (14 г) растворяли в диэтиловом эфире (200 мл) и в раствор добавляли активированный диоксид марганца (14 г). Смесь перемешивали при комнатной температуре в течение 4 часов, фильтровали и выпаривали досуха в вакууме, получая заглавный продукт (11,4 г), ЯМР-спектр которого полностью согласовывался с предполагаемой структурой.
В1b - Аналог получали идентичным способом из моносахарида 22, 23 - дигидроавермектина В1b.
Пример 3. Моносахарид 5-оксимино-22,23-дигидроавермектина В1a.
Моносахарид 5-оксо-22,23-дигидроавермектина B1a (1 г) растворяли в сухом пиридине (25 мл) и в раствор добавляли гидрохлорид гидроксиламина (1 г). Реакционную смесь при перемешивании кипятили с обратным холодильником в течение 4 часов и после охлаждения выливали на измельченный лед и экстрагировали дихлорметаном (2 х 50 мл). Объединенный экстракт сушили над сульфатом магния и выпаривали в вакууме, получая неочищенную смолу (1,1 г). Это вещество очищали жидкостной хроматографией при высоком давлении на колонке (41,4 х 250 мм, 8 мкм, ODS- диоксид кремния, Rainin, Dynamax товарный знак), элюируя смесью метанол-вода (83:17) со скоростью 42 мл в минуту. Подходящие фракции объединяли и выпаривали досуха, получая заглавный продукт в виде белого твердого вещества с т.пл. 180 - 190oC. Масс-спектр и ЯМР-спектр его полностью согласовывались с предлагаемой структурой.
5-Оксимино-22, 23 - дигидроавермектин B1b получали аналогичным способом из моносахарида 5-оксо-22,23-дигидроавермектина B1b.
Пример 4.
Моносахарид 22, 23-дигидро-25-циклогексилавермектина B1
25-Циклогексилавермектин B1 (9,9 г) растворяли в толуоле (1 л) и добавляли катализатор Вилкинсона [хлорид три
(трифенил-фосфин)родия(1)] (9,25 г). Раствор гидрировали на большом вибраторе Парра (товарный знак) при комнатной температуре и давлении водорода 3,5 ат. Через 3 часа давление в реакционном сосуде
сбрасывали и выдерживали реакционную смесь 12 часов до добавления следующей порции катализатора (5 г) и гидрировали как и ранее еще 2 часа, после чего в реакционной смеси не осталось исходного
соединения. Раствор фильтровали, выпаривали досуха в вакууме и остаток хроматографировали на диоксиде кремния, элюируя дихлорметаном и затем смесью дихлорметана с метанолом (9: 1). Неочищенный продукт
затем снова хроматографировали на диоксиде кремния (200 г) при элюировании смесью дихлорметана и метанола (19:1), получая после выпаривания растворителя в вакууме нечистый 22,
23-дигидро-25-циклогексилавермектин B1 в виде коричневой пены (10 г). Этот продукт растворяли в смеси изопропанола (200 мл) и серной кислоты (2 мл) и коричневый раствор перемешивали при комнатной
температуре в течение 15 часов и затем выливали в смесь льда и воды (500 мл) и экстрагировали дихлорметаном (3 х 200 мл). Органический слой промывали насыщенным водным раствором бикарбоната калия (100
мл), водой (2 х 50 мл), сушили над безводным сульфатом магния и выпаривали в вакууме, получая неочищенную смолу, которую хроматографировали на диоксиде кремния (100 г) при элюировании дихлорметаном,
затем смесью дихлорметана и этилацетата (2:1), получая заглавное соединение (8,2 г). Масс-спектр и ЯМР-спектр его полностью согласовывается с предполагаемой структурой.
Пример 5.
Моносахарид 5-оксимино-22, 23-дигидро-25-циклогексилавермектина B1
Моносахарид 22,23 - дигидро-25-циклогексилавермектина B1 (8,2 г) окисляли в 5-оксопроизводное при помощи диоксида марганца в
безводном диэтиловом эфире в соответствии со способом примера 2. Неочищенный продукт очищали хроматографией на диоксиде кремния (50 г), получая 5-оксосоединение (3,22 г) в виде желтой пены. Его
растворяли в безводном пиридине (60 мл) и в раствор добавляли гидрохлорид гидроксиламина (3,22 г). После перемешивания в течение 15 часов при комнатной температуре добавляли еще одну порцию
гидрохлорида гидроксиламина (3,22 г) и раствор нагревали при температуре до 50oC до тех пор, пока в реакционной смеси не оставалось исходного соединения. Раствор выливали в воду (50 мл) и
экстрагировали диэтиловым эфиром (3 х 50 мл). Органический слой промывали водой, насыщенным раствором хлористого натрия, сушили над безводным сульфатом натрия и выпаривали досуха в вакууме.
Неочищенный продукт хроматографировали на диоксиде кремния (25 г), элюируя смесью дихлорметана и этилацетата (4:1), и наконец очищали жидкостной хроматографией под высоким давлением при помощи колонки
(41,4 х 250 мм, ODS- диоксид кремния 8 мкм, Rainin) Dynamax (торговая марка), элюируя смесью метанола и воды (9:1) со скоростью 65 мл в минуту. Подходящие фракции объединяли и выпаривали в вакууме,
получая заглавное соединение (1,53 г). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 6. Моносахарид 25-циклогексилавермектина B2
25-Циклогексилавермектин B2 (10 г) суспендировали в изопропаноле (100 мл) и в суспензию добавляли раствор серной кислоты (2 мл) в изопропаноле (100 мл). После перемешивания при комнатной температуре в
течение 24 часов прозрачный раствор выливали на лед (600 г) и экстрагировали дихлорметаном (2 х 100 мл). Органический слой сушили над безводным сульфатом натрия и выпаривали досуха. Остаток растворяли
в тетрахлорметане и раствор выдерживали при 4oC. Кристаллы, которые выделялись медленно, периодически отделяли фильтрованием. Показали, что эти кристаллы представляли собой чистое заглавное
соединение. Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 7. Моносахарид 5-оксимино-25-циклогексилавермектина B2
Способами
примеров 2 и 3 моносахарид 25-циклогексилавермектина B2 превращали в заглавное соединение. Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 8.
Моносахарид 25-циклогексилавермектина B1
25-Циклогексилавермектин B1 (20 г) растворяли в тетрагидрофуране (250 мл) и в раствор добавляли смесь тетрагидрофурана (250 мл), воды (10 мл) и серной
кислоты (10 мл). Смесь перемешивали при комнатной температуре 15 часов и затем выливали в смесь льда (500 г) и воды (1 л) и экстрагировали дихлорметаном (2 х 500 мл). Органический слой промывали
насыщенным раствором хлористого натрия, сушили над безводным сульфатом натрия и выпаривали в вакууме, получая пенообразный продукт. Его хроматографировали на диоксиде кремния (150 г) при элюировании
смесью этилацетата и дихлорметана (1:1), получая неочищенный продукт (13,3 г). Конечную очистку проводили жидкостной хроматографией высокого разрешения (ЖХВР) с обратимой фазой, применяя колонку (41,4
х 250 мм, ODS -диоксид кремния, 8 мкм, Rainin) Dynamax (торговая марка). Элюировали смесью метанола и воды (4:1) со скоростью 70 мл в минуту, получая чистое заглавное соединение. Масс-спектр и
ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 9. Моносахарид 5-оксимино-25-циклогексилавермектина B1
Способами примеров 2 и 3 моносахарид
25-циклогексилавермектина B1 превращали в заглавное соединение. Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 10. Моносахарид
5-O-трет-бутил-диметилсилил-25-циклогексил-22,23-дигидроавермектина B1
Моносахарид 22, 23-дигидро-25-циклогексилавермектина B1 (пример 4) (12,1 г) и имидазол (7,2 г) растворяли в сухом
диметилформамиде (10 мл). В этот раствор при комнатной температуре добавляли хлористый трет-бутилдиметилсилил (7,9 г). Через 18 часов смесь выливали в смесь льда и воды (200 мл), подкисляли до pH 2
при помощи 2N HCl и экстрагировали диэтиловым эфиром (2 х 80 мл). Объединенный экстракт промывали насыщенным водным раствором бикарбоната натрия (50 мл) и водой (50 мл), сушили над безводным сульфатом
натрия и выпаривали в вакууме, получая неочищенный продукт (14,9 г). Продукт далее очищали хроматографией на диоксиде кремния (кизельгель 60, 230 - 240 меш, Merck (300 г), элюируя смесью дихлорметана
и этилацетата (9:1). Подходящие фракции объединяли и выпаривали досуха, получая заглавный продукт (8,35 г). ЯМР-спектр его полностью согласовывался с предполагаемой структурой.
Пример
11. 5-Оксоавермектин B1a
Авермектин B1a (2,4 г) растворяли в диэтиловом эфире (50 мл) и в раствор добавляли активированный диоксид марганца (2,0 г). Смесь перемешивали при комнатной
температуре в течение 18 часов, фильтровали и выпаривали досуха в вакууме, получая заглавный продукт, ЯМР-спектр которого полностью согласовывался с предложенной структурой.
Пример 12.
5-Оксиминоавермектин B1a
5-Оксоавермектин B1a (800 мг) (пример 11) растворяли в пиридине (10 мл) и в раствор добавляли гидрохлорид гидроксиламина (800 мг). После перемешивания при комнатной
температуре в течение 1 часа смесь выливали в смесь льда (50 г) и воды (50 мл), подкисляли до pH концентрированной соляной кислотой и экстрагировали дихлорметаном (3 х 30 мл). Объединенный экстракт
промывали водой (20 мл), сушили над безводным сульфатом натрия и выпаривали досуха при пониженном давлении, получая неочищенный продукт (1 г). Продукт хроматографировали на диоксиде кремния
(кизельгель 60, 230 - 400 меш, Merck)(100 г), элюируя смесью дихлорметана и этилацетата (2:1), и наконец, очищали жидкостной хроматографией при высоком давлении, применяя колонку (41,4 х 250 мм,
ODS-диоксид кремния 8 мкм, Rainin) Dynamax (торговая марка) и элюируя смесью метанола и воды (85:15) со скоростью 70 мл в минуту. Нужные фракции объединяли и выпаривали в вакууме, получая заглавное
соединение (290 мг). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 13. Моносахарид 5-оксимино-авермектина B1a
5-Оксиминоавермектин
B1a (50 мг) (пример 12) растворяли в смеси изопропанола (1 мл) и серной кислоты (10 мкл) и перемешивали при комнатной температуре в атмосфере азота 48 часов. Затем добавляли насыщенный водный раствор
бикарбоната натрия (1 мл) и продукт экстрагировали этилацетатом (2 х 5 мл). Объединенный экстракт сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный неочищенный продукт (25
мг) очищали жидкостной хроматографией под высоким давлением на колонке (24 х 250 мм, ODS -диоксид кремния 5 мкм, Beckman) Ultrasphere (торговая марка), элюируя смесью метанола и воды (85:15) со
скоростью 20 мл в минуту. Подходящие фракции объединяли, получая заглавный продукт. Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 14.
Моносахарид 5-(триметилацетилоксимино-25-циклогексил-22,23-дигидроавермектина B1
В перемешиваемый раствор моносахарида 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) (50
мг) в дихлорметане (2 мл) при комнатной температуре добавляли триэтиламин (72 мл), затем хлористый триметилацетил (80 мл). После выдерживания смеси в течение 18 часов добавляли водный раствор лимонной
кислоты (10 масс./об.% 2 мл). Органический слой отделяли, промывали насыщенным водным раствором хлористого натрия (2 мл), сушили над безводным сульфатом натрия и выпаривали досуха в вакууме, получая
неочищенный продукт, который хроматографировали на диоксиде кремния (кизельгель 60, 230-400 меш, Merck) ( 5 г), элюируя диэтиловым эфиром. Нужные фракции объединяли и выпаривали досуха в вакууме,
получая продукт (53 мг), который далее очищали жидкостной хроматографией под высоким давлением на колонке 21,2 х 250 мм, ODS-диоксид кремния, 5 мкм, Rainin, Dynamax (торговая марка), элюируя со
скоростью 20 мл в минуту смесью метанола и воды (95:5). Подходящие фракции объединяли и выпаривали в вакууме, получая заглавное соединение в виде белого порошка (18 мг). Масс-спектр и ЯМР-спектр его
полностью согласовывались с предполагаемой структурой.
Пример 15. Моносахарид 5-(бензоилоксимино)-25-циклогексил-22, 23-дигидроавермектина B1
Моносахарид
5-осимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) (70 мг) в дихлорметане (30 мл) обрабатывали триэтиламином (50 мл) и хлористым бензилом (100 мл) и целевой продукт экстрагировали
методом, идентичным описанному в примере 42. Очистку проводили жидкостной хроматографией под высоким давлением на колонке (41,4 х 250 мм, ODS- диоксид кремния 8 мкм, Rainin) Dynamax (торговая марка),
элюируя со скоростью 45 мл в минуту смесью метанола и воды (90:10). Подходящие фракции объединяли и выпаривали в вакууме, получая заглавное соединение в виде белого порошка (28 мг). Масс-спектр и
ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 16. Моносахарид 5-(N-метилкарбамоил-оксимино)-25-циклогексил-22,23-дигидроавермектина B1
В
перемешиваемый раствор моносахарида 5-оксимино-25-циклогексил- 22, 23-дигидроавермектина B1 (пример 5) (106 мг) в дихлорметане (10 мл) добавляли метилизоцианат (15 мл) и смесь перемешивали 1 час.
Затем добавляли дополнительное количество метилизоцианата (30 мл) и реакционную смесь перемешивали еще в течение 72 часов до добавления насыщенного водного раствора хлористого натрия (10 мл) и
диэтилового эфира (30 мл). Органический экстракт сушили над безводным сульфатом натрия и выпаривали досуха в вакууме, получая неочищенный продукт (150 мг), который очищали жидкостной хроматографией
под высоким давлением на колонке (41,4 х 250 мм, ODS -диоксид кремния, 8 мкм, Rainin)Dynamax (торговая марка), элюируя со скоростью 45 мл в минуту смесью метанола и воды (91:9). Подходящие фракции
объединяли и выпаривали досуха в вакууме, получая заглавное соединение в виде белого порошка (80 мг). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 17. Моносахарид 5-(N, N-диметилкарбамоилоксимино)-25-циклогексил-22, 23-дигидроавермектина B1
В перемешиваемый раствор моносахарида 5-оксимино-25-циклогексил-22, 23-дигидроавермектина
B1 (пример 5) (50 мг) в дихлорметане (2 мл) при комнатной температуре добавляли триэтиламин (72 мл) и 4 -диметиламинопиридин (1 мл), затем хлористый N,N-диметилкарбамоил (58 мл). Через 3 часа
добавляли дополнительное
количество хлористого N,N-диметилкарбамоила (58 мл) и реакционную смесь выдерживали в течение 18 часов. Затем добавляли водный раствор лимонной кислоты (10 масс./об.%, 2 мл)
и диэтиловый эфир (20 мл). Органический слой отделяли, промывали насыщенным водным раствором хлористого натрия (5 мл), сушили над безводным сульфатом натрия и выпаривали досуха в вакууме, получая
неочищенный продукт, который очищали жидкостной хроматографией под высоким давлением на колонке (21,2 х 250 мм, ODS -диоксид кремния, 5 мкм, Rainin) Dynamax (торговая марка), элюируя со скоростью 10
мл в минуту смесью метанола и воды (90:10). Нужные фракции объединяли и выпаривали в вакууме, получая заглавное соединение в виде белого порошка (18 мг). Масс-спектр и ЯМР-спектр его полностью
согласовывались с предполагаемой структурой.
Пример 18. Моносахарид 5-(4-метилпиперазинил-1-карбонилоксимино)-25-циклогексил-22, 23-дигидроавермектина B1
В перемешиваемый
раствор N-метилпиперазина (0,65 мл) и триэтиламина (1,3 мл) в толуоле (25 мл) при 0oC добавляли по каплям раствор фосгена в толуоле (20%, 5,1 мл) в течение 15 минут. Реакционную смесь
выдерживали для нагревания до комнатной температуры, перемешивали 3 часа, фильтровали и концентрировали приблизительно до 10 мл при пониженном давлении, получая раствор
1-хлоркарбонил-4-метилпиперазина, который обрабатывали моносахаридом 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) (300 мг), триэтиламином (110 мл) и 4-диметиламинопиридином (5 мг)
в дихлорметане (10 мл) при комнатной температуре в соответствии с методом, описанным в примере 45. Очистку продукта проводили хроматографией на диоксиде кремния (кизельгель 60, 230-400 меш, Merck) (35
г), элюируя дихлорметаном. Нужные фракции объединяли и выпаривали в вакууме, получая продукт (53 мг), который далее очищали жидкостной хроматографией под высоким давлением на колонке (21,1 х 250 мм,
ODS -диоксид кремния, 5 мкм, Rainin) Dynamax (торговая марка), элюируя со скоростью 20 мл в минуту смесью метанола и воды (95:5). Нужные фракции объединяли и выпаривали в вакууме, получая заглавное
соединение в виде белого порошка. Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 19. Моносахарид
5-(трет-бутилоксикарбонилоксимино(-25-циклогексил-22, 23-дигидроавермектина B1
В перемешиваемый раствор моносахарида 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) (60 мг)
и триэтиламина (50 мг) в дихлорметане (5 мл) при комнатной температуре добавляли трет-бутиловый эфир угольной кислоты (60 мг). После выдерживания в течение 48 часов реакционную смесь выпаривали досуха
в вакууме, получая остаток, который растворяли в дихлорметане и раствор хроматографировали на диоксиде кремния (кизельгель 60, 230-400 меш, Merck) (5 г), элюируя дихлорметаном. Подходящие фракции
объединяли и выпаривали досуха в вакууме, получая заглавное соединение в виде белого порошка (45 мг). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 20. Моносахарид 5-(N-(4-формилфенил)-карбамоилоксимино)-25-циклогексил-22, 23-дигидроавермектина B1
4-формилфенилизоцианат, полученный в соответствии с методикой, описанной в J. Med.
Chem., 32(10), 2354 (1989), обрабатывали моносахаридом 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) (500 мг) в сухом дихлорметане (50 мл) при комнатной температуре в течение 1
часа в соответствии со способом, описанным в примере 15. Очистку целевого продукта проводили на диоксиде кремния (кизельгель 60, 230 - 400 меш, Merck) (125 г), элюируя смесью гексана и диэтилового
эфира с градиентом от 1:1 до 20:80. Нужные фракции объединяли и выпаривали досуха в вакууме, получая заглавное соединение в виде белого порошка (300 мг). Масс-спектр и ЯМР-спектр его полностью
согласовывались с предполагаемой структурой.
Пример 21. Моносахарид 5-(N-(4-диэтиламинометил)фенил)карбамоилоксимино)-25-циклогексил-22, 23-дигидроавермектина B1
Хлористый
4-диэтиламинометилбензоил, полученный в соответствии с методикой, описанной в публикации патента США US-4623486, обрабатывали моносахаридом 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1
(пример 5) (100 мг) в сухом дихлорметане (50 мл), содержащем триэтиламин (450 мл) и 4-диметиламинопиридин (126 мл), при комнатной температуре в течение 1 часа в соответствии со способом, описанным в
примере 17. Очистку целевого продукта проводили на диоксиде кремния (кизельгель 60, 230-400 меш, Merck) (5 г), элюируя смесью метанола и дихлорметана с градиентом от 0:100 до 10:90. Подходящие фракции
объединяли и выпаривали досуха в вакууме, получая заглавное соединение в виде белого порошка (11 мг). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 22. Моносахарид 5-(N-(4-(4-метил-1-пиперазинилметил)фенил)-карбамоилоксимино) -25-циклогексил-22, 23-дигидроавермектина B1
Хлористый 4-(4-метилпиперазин-1-илметил)бензоил, полученный
в соответствии с методикой, описанной в патентной публикации США US-4623486, обрабатывали моносахаридом 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) способом, идентичным
описанному в примере 20. Заглавное соединение получали в виде белого порошка (18 мг). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 23.
Моносахарид 5-(N-(3-пиридил-карбонил)карбамоилоксимино)-25-циклогексил-22, 23-дигидроавермектина B1
В перемешиваемый раствор никотинамида (4,88 г) в сухом 1,2-дихлорэтане (500 мл) добавляли
по каплям хлористый оксалил (5,24 мл). Смесь кипятили с обратным холодильником в течение 4,5 часов, затем охлаждали и фильтровали. Полученный раствор, содержащий никотиноилизоцианат (50 мл),
обрабатывали моносахаридом 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) (500 мг) в дихлорметане (10 мл) при комнатной температуре. После стояния реакционной смеси в течение 18
часов добавляли дополнительное количество раствора никотиноилизоцианата (25 мл) и смесь выдерживали при комнатной температуре еще 18 часов. Затем смесь выпаривали досуха в вакууме, получая остаток,
который очищали жидкостной хроматографией под высоким давлением на колонке (41,4 х 250 мм, ODS-диоксид кремния, 8 мкм, Rainin) Dynomax (торговая марка), элюируя со скоростью 45 мл в минуту смесью
метанола, ацетонитрила и воды (20:65:15). Нужные фракции объединяли и выпаривали в вакууме, получая заглавное соединение в виде белого порошка. Масс-спектр и ЯМР-спектр его полностью согласовывались с
предполагаемой структурой.
Пример 24. Моносахарид 5-(N-(3-пиридил)-карбамоилоксимино)-25-циклогексил-22, 23-дигидроавермектина B1
В раствор дигидрохлорида гидразида
никотиновой кислоты (2 г) в воде (10 мл) добавляли раствор нитрита натрия (1,6 г) в воде (10 мл), поддерживая температуру ниже 20oC. Затем добавляли диэтиловый эфир (50 мл) и смесь
подщелачивали осторожным добавлением твердого бикарбоната натрия. Органический слой отделяли, промывали водой (20 мл), сушили над безводным сульфатом магния и выпаривали досуха в вакууме, получая
никотинилазид (1,1 г) с т.пл. 54oC. Этот азид (1,1 г) перемешивали в сухом толуоле (10 мл) и нагревали при 100oC в атмосфере азота в течение 8 часов, получая раствор, содержащий
3-пиридилизоцианат. Часть этого раствора (1 мл) обрабатывали моносахаридом 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 ( пример 5) (100 мг) в толуоле (10 мл) при комнатной температуре в
течение 1 часа, затем смесь выливали в смесь диэтилового эфира и воды (1:1, 30 мл). Органический слой отделяли, сушили над безводным сульфатом магния и выпаривали досуха в вакууме, получая остаток
(130 мг), который очищали жидкостной хроматографией под высоким давлением на колонке (41,4 х 250 мм, ОDS -диоксид кремния, 8 мкм, Rainin) Dynamax (торговая марка), элюируя со скоростью 45 мл в минуту
смесью метанола и воды с соотношением 85:15, которое изменяли через 15 минут до соотношения 87:13. Подходящие фракции объединяли и выпаривали в вакууме, получая заглавное соединение в виде белого
порошка (52 мг). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 25. Моносахарид 5-(аллилкарбамоил-оксимино)-25-циклогексил-22,
23-дигидроавермектина B1
Реакцией моносахарида 5-оксимино-25-циклогексил-22, 23-дигидроавермектина B1 (пример 5) (500 мг) с аллилизоцианатом (108 мг) в дихлорметане (50 мл) в соответствии со
способом, описанным в примере 15, получали заглавное соединение в виде белого порошка (352 мг). Масс-спектр и ЯМР-спектр его полностью согласовывались с предполагаемой структурой.
Пример 26. Получение ветеринарной композиции для перорального введения.
Любой из полученных в вышеуказанных примерах продукт смешивают с порошком лактозы и заполняют полученной смесью твердые желатиновые капсулы из расчета 1,0 мг активного соединения на капсулу.
Пример 27. Данные по активности соединений настоящего изобретения в отношении блох.
1. Тест in vitro по определению активности соединений в отношении блох Ctenocephalides felis проводят по следующей общей методике.
1.1. Тестируемые соединения растворяют в ДМСО для получения рабочего раствора с концентрацией 0,2 мг/мл. Аликвоту 25 мл добавляют к 5 мл смеси телячьей крови и цитрата с получением исходной концентрации для тестирования 1 мгк/мл. Делают разведения в ДМСО, и по 25 мкл разбавленных растворов добавляют к 5 мл смеси телячьей крови и цитрата, как описано выше, получая концентрации для тестирования 0,5 мкг/мл, 0,25 мкг/мл и т.д.
1.2. Смесь цитрата и крови теленка готовят добавлением 100 мл раствора цитрата (22 г цитрата натрия, 8,0 г моногидрата лимонной кислоты, 24, 5 г глюкозы в 100 мл воды) к 500 мл крови теленка.
1.3 20-25 взрослых особей блох Ctenocephalides felis (кошачьи блохи) собирают и помещают в камеру для тестирования и выдерживают их перед использованием в течение ночи при 25oC во влажной атмосфере.
1.4. 5 мл смеси телячьей крови и цитрата, содержащей тестируемое соединение, помещают в стеклянную емкость, температуру в которой поддерживают около 37oC с помощью водяной наружной рубашки. Натягивают парафиновую пленку над открытым верхом емкости, получая плотную мембрану, через которую блохи могут питаться. Камеру для тестирования, содержащую блох, осторожно помещают на пленочную мембрану, и затем емкость/камеру для тестирования переворачивают для того, чтобы привести кровь в контакт с мембраной и блохи начали питаться.
1.5. Блохам было позволено питаться в течение 6 часов, после чего камеру для тестирования удаляют и хранят в течение ночи при 25oC во влажной атмосфере.
1.6 Наблюдают блох, которые упали и/или умерли, и фиксируют их процент. Соединения, проявляющие активность при концентрировании 1 мкг/мл, далее тестируют при более низких дозах. В случае, когда соединения тестировали более одного раза (n>1), фиксируют общий процент. Полученные результаты показаны в приложенной таблице.
2. Тестирование in vitro
2. Активность соединения примера 5
настоящего изобретения (ИК-124, 114) была также определена in vivo на мышах.
Тест проводят в соответствии со следующей методикой.
2.1. Блок обычно сохраняют на кошках, получая взрослые особи. 10 голодных взрослых особей блох, которые были изолированы от питания кровью, помещают в стерильные пробирки.
2.2. Тестируемые соединения вводят перорально мышам СД1 весом 20 г, которых затем выдерживают в течение 2-4 часов, анестезируют и помещают в пробирки с блохами.
2.3. Блохи находятся в контакте с мышами в течение 1 часа.
2.4. Затем мышей вынимают из пробирок и наевшихся блох удаляют с мышей и помещают в пробирки, содержащие длинную, узкую полоску бумаги, и оставляют там. Конец пробирки закрывают сеткой с очень мелкими отверстиями.
2.5. Исследуют количество блох, которые были парализованы и/или мертвы через 4, 24 и 48 часов после дозирования, и фиксируют количество умерших или выживших особей.
2.6. Было найдено, что соединение примера 5 (ИК 124, 114) проявляет активность в данном тесте при дозе 3 мг/кг, причем никаких признаков проявления какого-либо токсического действия не наблюдалось.
Реферат
В
настоящем изобретении предложены производные авермектина формулы I, обладающие антипаразитической активностью

Формула
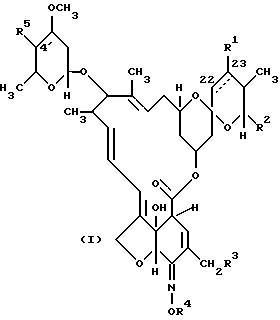
где пунктирная линия в 22(23)-положении обозначает необязательную углерод-углеродную связь, причем эта связь присутствует и R1 отсутствует или эта связь отсутствует и R1 представляет собой Н или ОН;
R2 представляет собой С1-С8-алкил или С3-С8 циклоалкил;
R3 представляет собой Н или ОН;
R4 представляет собой Н или группу, способную гидролизоваться in vivo с образованием соединения, у которого R4 представляет собой Н, и которая представляет собой ацетил, трет-бутилкарбонил, трет-бутоксикарбонил, бензоил, метилпиперазинкарбонил, N-метилкарбамоил, N, N-диметилкарбамоил, формилфенилкарбамоил, N-(4-диэтиламино-метилфенил)карбамоил, N-(4-метил-1-пиперазинметилфенил) карбамоил, N-(3-пиридилкарбонил)карбамоил, N-(3-пиридил)карбамоил или аллилкарбамоил;
R5 представляет собой ОН.
моносахарид 5-оксимино-22, 23-дигидроавермектина B1a,
моносахарид 5-оксимино-22,23-дигидро-25-циклогексилавермектина В1,
моносахарид 5-оксимино-25-циклогексилавермектина В2,
моносахарид 5-оксимино-25-циклогексилавермектина В1,
моносахарид 5-оксимино-авермектина В1а.
моносахарид 5-(триметилацетилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(бензоилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(N-метилкарбамоилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(N, N-диметилкарбамоилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(4-метилпиперазинил-1-карбонилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(трет-бутилоксикарбонилокситмино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(N-(4-формилфенил)карбамоилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(N-(4-диэтиламинометил)фенил)карбамоилоксимино)-25-циклогексил-22, 23-дигидроавермектина В1,
моносахарид 5-(N-(4-метил-1-пиперазинилметил)фенил)карбамоилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(N-3-пиридилкарбонил)карбамоилоксимино-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(N-(3-пиридил)карбамоилоксимино)-25-циклогексил-22,23-дигидроавермектина В1,
моносахарид 5-(N-аллилкарбамоилоксимино)-25-циклогексил-22,23-дигидроаверменктина В1.

где R1, R2, R3, R4 и R5 принимают значения, указанные выше,
который включает стадии:
(i) окисления соединения формулы II
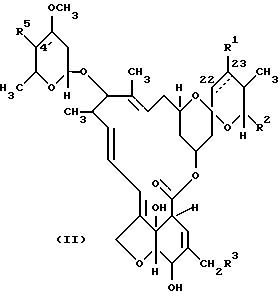
где пунктирная линия, R1, R2 и R3 имеют указанные выше значения;
R5 имеет указанные выше значения или R5 представляет собой α-олеандрозилоксигруппу,
с получением соединения формулы III

(ii) реакцию соединения формулы III с соединением формулы
R4-O - NH2,
где R4 имеет указанные выше значения;
R5 представляет собой α-олеандрозилоксигруппу,
и гидролиза полученного соединения в соединение формулы I,
(iii) если необходимо, замещения группы R4, когда она представляет собой Н, группой, способной гидролизоваться in vivo с образованием соединения, у которого R4 представляет собой Н,
и, если необходимо,
(iv) гидрирование соединения для восстановления двойной связи в положении 22(23) в одинарную связь.
Комментарии