Водорастворимые азольные соединения и способ их получения - RU2266909C2
Код документа: RU2266909C2
Описание
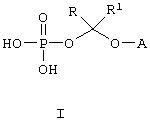
Данное изобретение относится к водорастворимым азольным соединениям, применимым для лечения тяжелых системных грибковых инфекций и пригодных как для перорального, так и, в особенности, для парентерального введения. Более конкретно, изобретение относится к новым водорастворимым пролекарствам общей формулы:
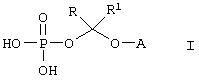
где А обозначает негидроксильный фрагмент триазольного противогрибкового соединения, содержащего вторичную или третичную спиртовую группу, R и R1, каждый независимо обозначает водород или (С1-С6)алкил, или к их фармацевтически приемлемым солям.
ОПИСАНИЕ УРОВНЯ ТЕХНИКИ
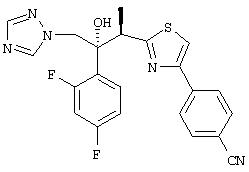
Триазольные противогрибковые соединения хорошо. Из нескольких известных классов один особенно эффективный класс содержит гидроксил при третичном атоме углерода. Например, в Патенте США 5648372 описано, что (2R,3R-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2,4-дифторфенил) -1-(1Н - 1,2,4-триазол-1-ил) -бутан-2-ол проявляет противогрибковую активность.
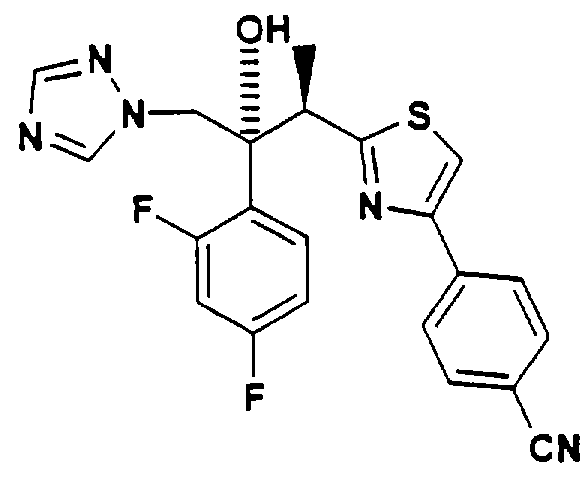
Применение этого класса соединений ограничено их малой растворимостью в воде. Например, растворимость вышеуказанного триазольного соединения в воде при рН 6,8 составляет 0,0006 мг/мл. Это в значительной степени препятствует созданию соответствующих парентеральных лекарственных форм.
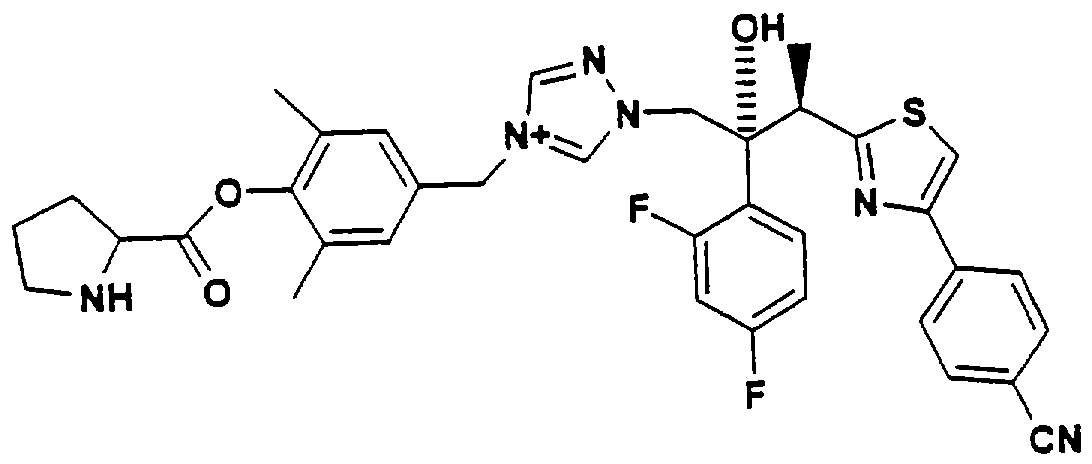
Один способ решения этой проблемы описан в Европейской заявке 829478, где растворимость азольного противогрибкового агента в воде повышали присоединением к азольному фрагменту связанной аминокислоты:
В качестве альтернативы в Международной заявке WO 97/28169 описано, что фосфатный фрагмент может непосредственно присоединяться к гидроксильному фрагменту при третичном атоме углерода противогрибкового соединения, примером является соединение формулы:
В Патенте США 5707977 и в Международной заявке WO 95/19983 описаны водорастворимые пролекарства общей формулы:
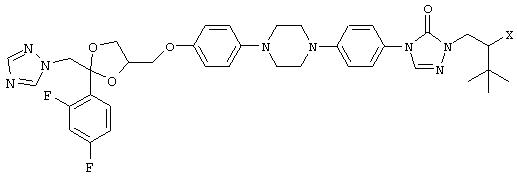
где Х обозначает ОР(O)(ОН)2 или легко гидролизующийся сложный эфир OC(O)RN1R2.
В Международной заявке WO 95/17407 описаны водорастворимые азольные пролекарства общей формулы:
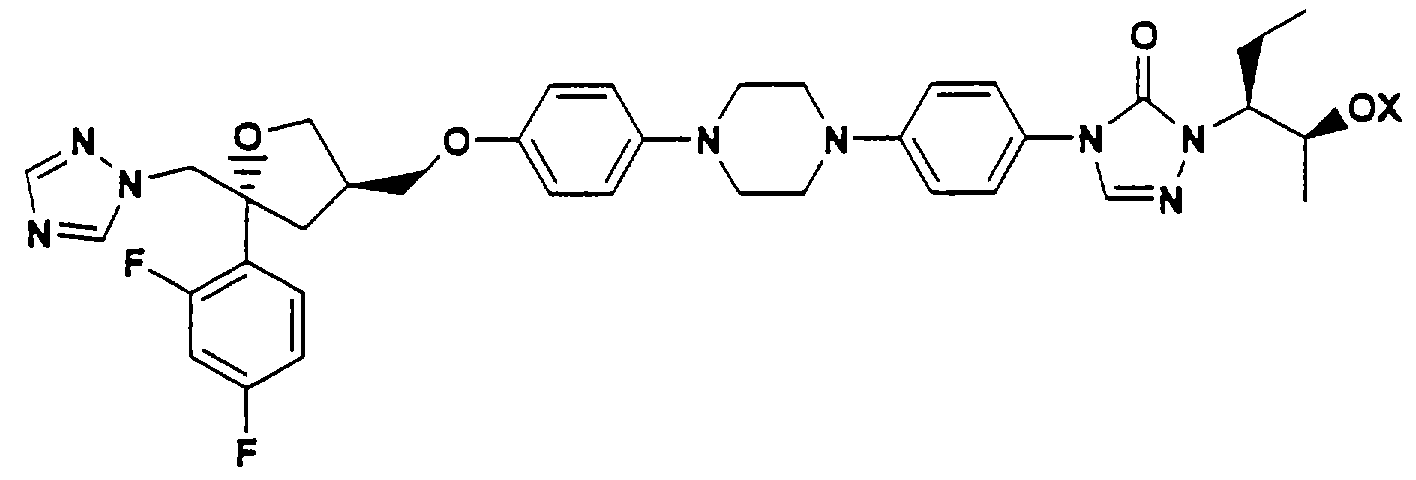
где Х обозначает Р(O)(ОН)2, C(O)-(CHR1)n-OP(O)(OH)2 или С(O)-(CHR1)n-(OCHR1CHR1)mOR2.
В Международной заявке WO 96/38443 описаны водорастворимые азольные пролекарства общей формулы
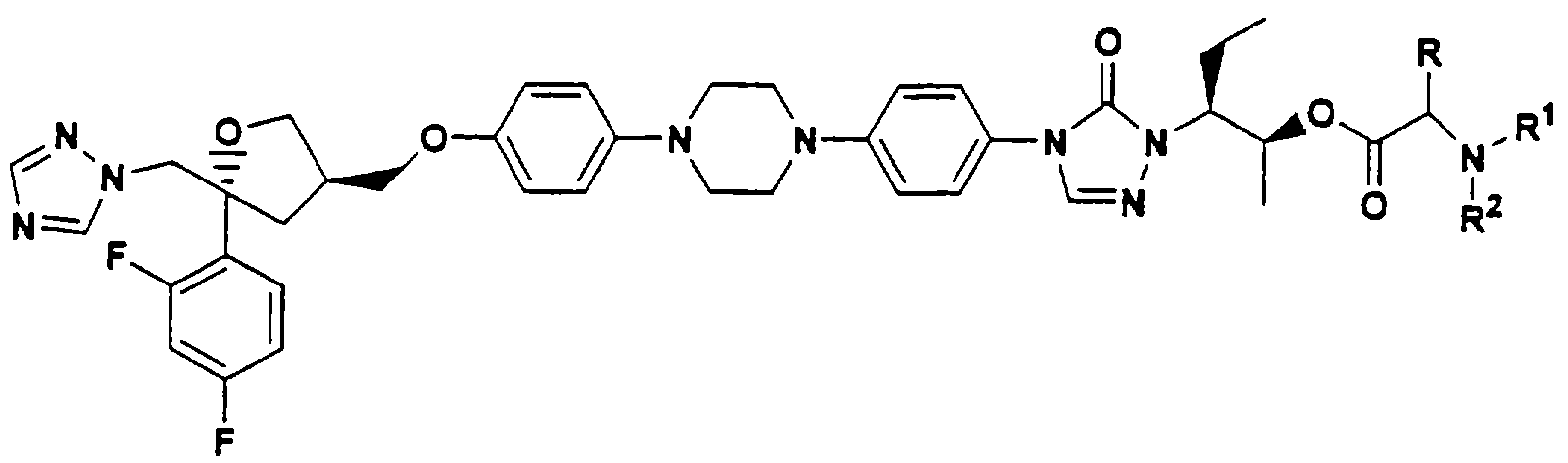
В Патенте США 5883097 описаны водорастворимые аминокислотноазольные пролекарства, такие как сложный эфир глицерина:
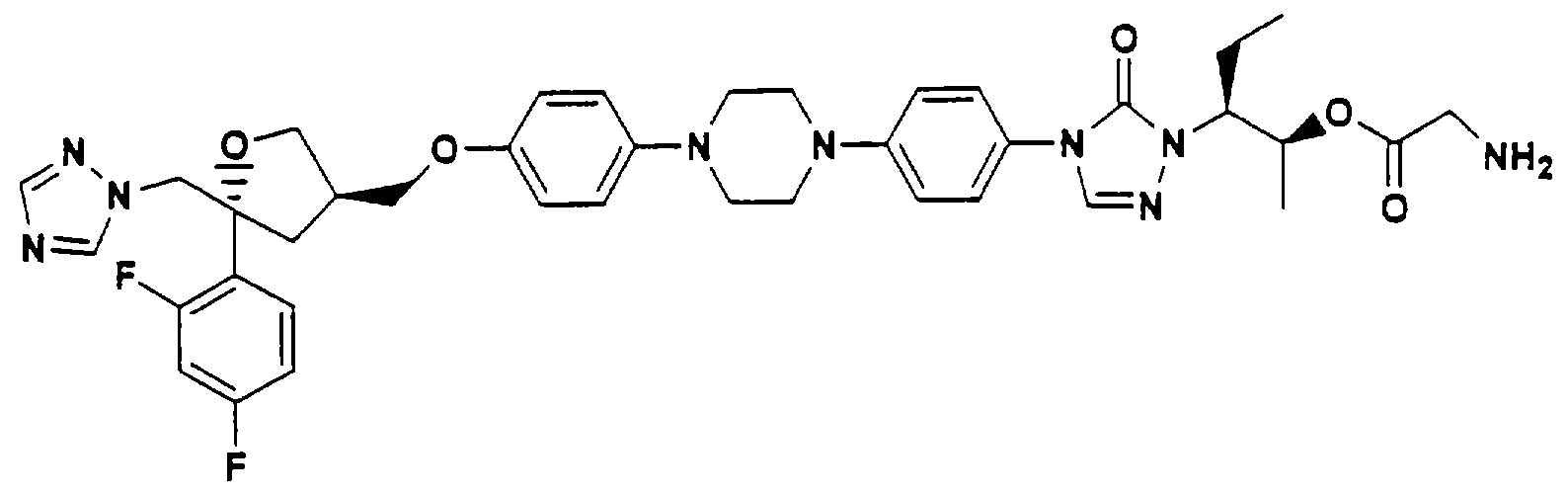
Введение фосфоноксиметильного фрагмента в гидроксилсодержащие лекарственные вещества было описано как способ получения водорастворимых пролекарств гидроксилсодержащих лекарственных веществ.
В Европейской патентной заявке 604910 описаны фосфоноксиметилтаксановые производные общей формулы:
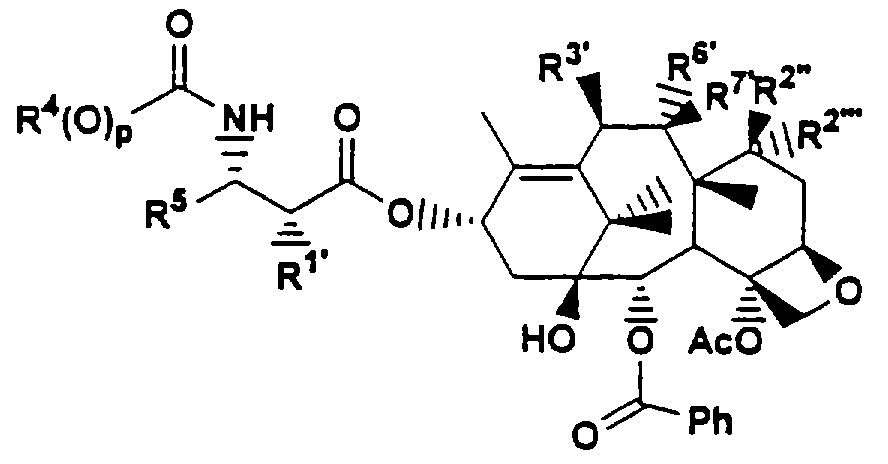
где, по меньшей мере, один из R1), R2) R3), R6) или R7) обозначает ОСН2OP(O)(ОН)2.
В Европейской патентной заявке 639577 описаны производные фосфоноксиметилтаксана формулы Т-[ОСН2(ОСН2)mОР(O)(ОН)2]n, где Т обозначает таксановый фрагмент, где при атоме С13 имеется замещенная 3-амино-2-гидроксипропаноилоксигруппа; n обозначает 1, 2 или 3; m обозначает 0 или целое число от 1 до 6, и их фармацевтически приемлемые соли.
В Международной заявке WO 99/38873 описаны простые O-фосфоноксиметильные эфиры диарил-1,3,4-оксадиазолоновых пролекарств, открывателей калиевых каналов.
Golik, J. et al, Bioorganic and Medicinal Chemistry Letters, 1996 6:1837-1842, описывает новые водорастворимые пролекарства паклитакселя, такие как
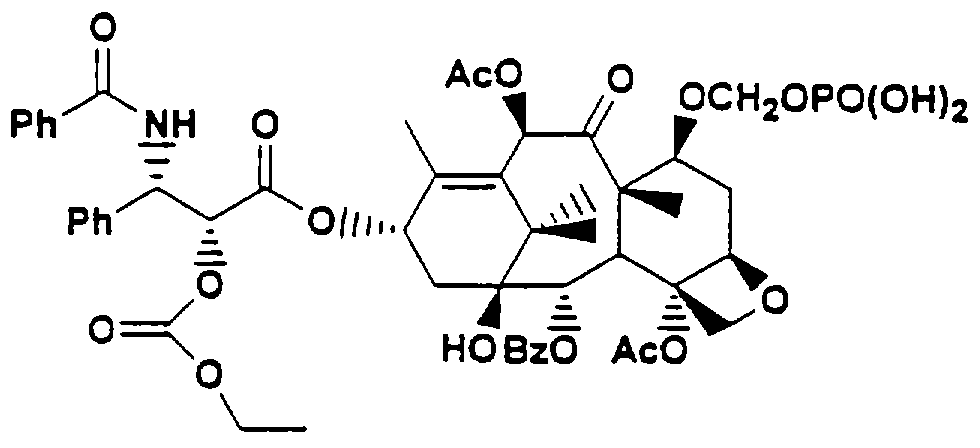
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
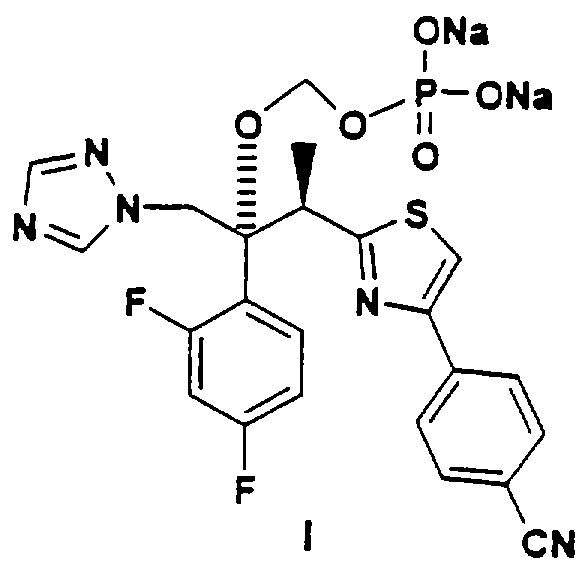
В настоящее время найдено, что триазольные противогрибковые соединения, содержащие вторичную или третичную гидроксильную группу, включая (2R, 3R)-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2,4-дифторфенил)-1-(1Н-1,2,4-триазол-1-ил)-бутан-2-ол, можно превратить в пролекарства со свойствами, превосходящими свойства ранее описанных соединений, путем присоединения фосфатсодержащего фрагмента через связывающую группу. Конкретно, изобретение охватывает соединения формулы:
где А обозначает негидроксильный фрагмент триазольного противогрибкового соединения, содержащего вторичную или третичную спиртовую группу, R и R1, каждый независимо обозначает водород или (С1-С6)алкил, или их фармацевтически приемлемые соли.
Соединения общей формулы I действуют как "пролекарства" при введении in vivo, превращаясь в биологически активный исходный азол в присутствии щелочной фосфатазы.
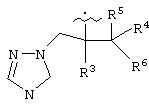
В предпочтительном варианте изобретения А представляет собой негидроксильный фрагмент триазольного противогрибкового соединения, содержащий третичную спиртовую группу:
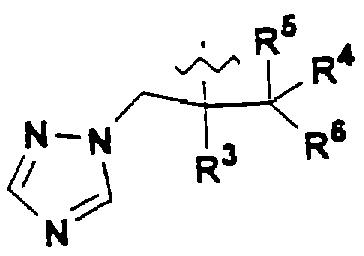
где R3 представляет собой фенил, замещенный одним или более (предпочтительно, 1-3) атомами галогена;
R4 представляет собой Н или СН3;
R5 представляет собой Н или, вместе с R4 может обозначать =СН2;
R6 представляет собой 5- или 6-членный азотсодержащий цикл, который при необходимости может иметь в качестве заместителей одну или более групп, выбранных из галогена, =O, фенила, замещенного одной или более групп, выбранных из CN, (C6H4)-OCH2CF2CHF2 и СН=СН-(С6Н4)-ОСН2CF2CHF2, или фенила, замещенного одной или более групп, выбранных из галогена и метилпиразолила.
Азотсодержащие гетероциклы, которые может обозначать R6, включают тиазолил, пиримидинил или тиазолил.
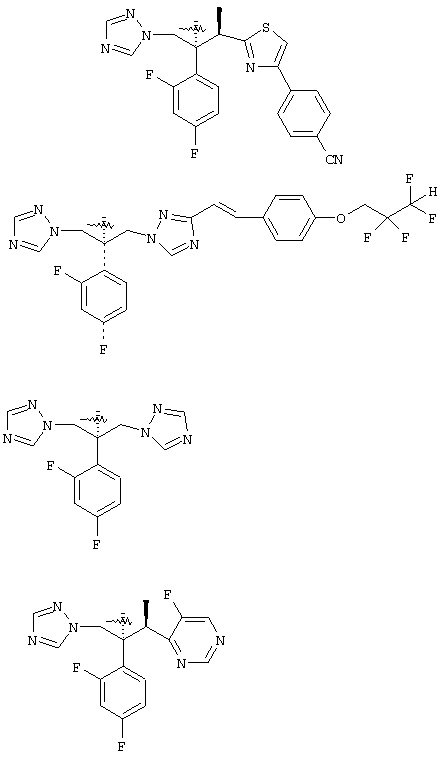
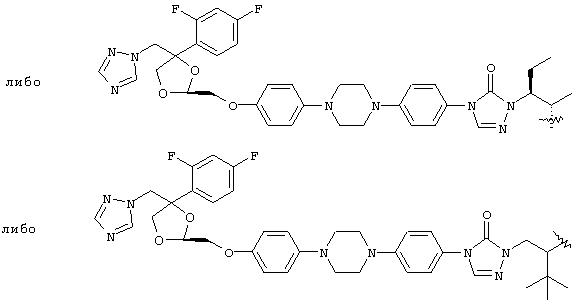
Конкретные примеры А включают, без ограничения, следующие:
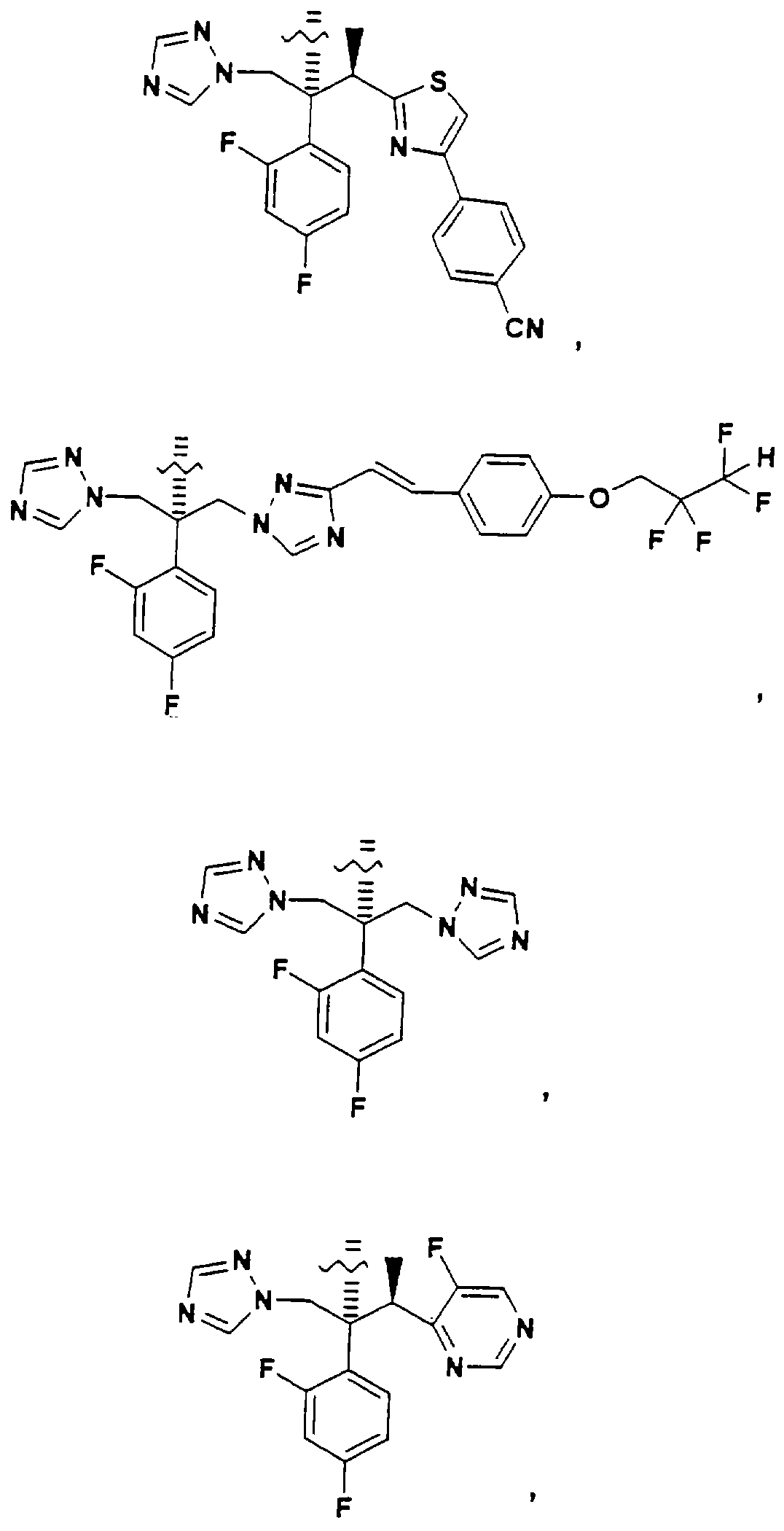
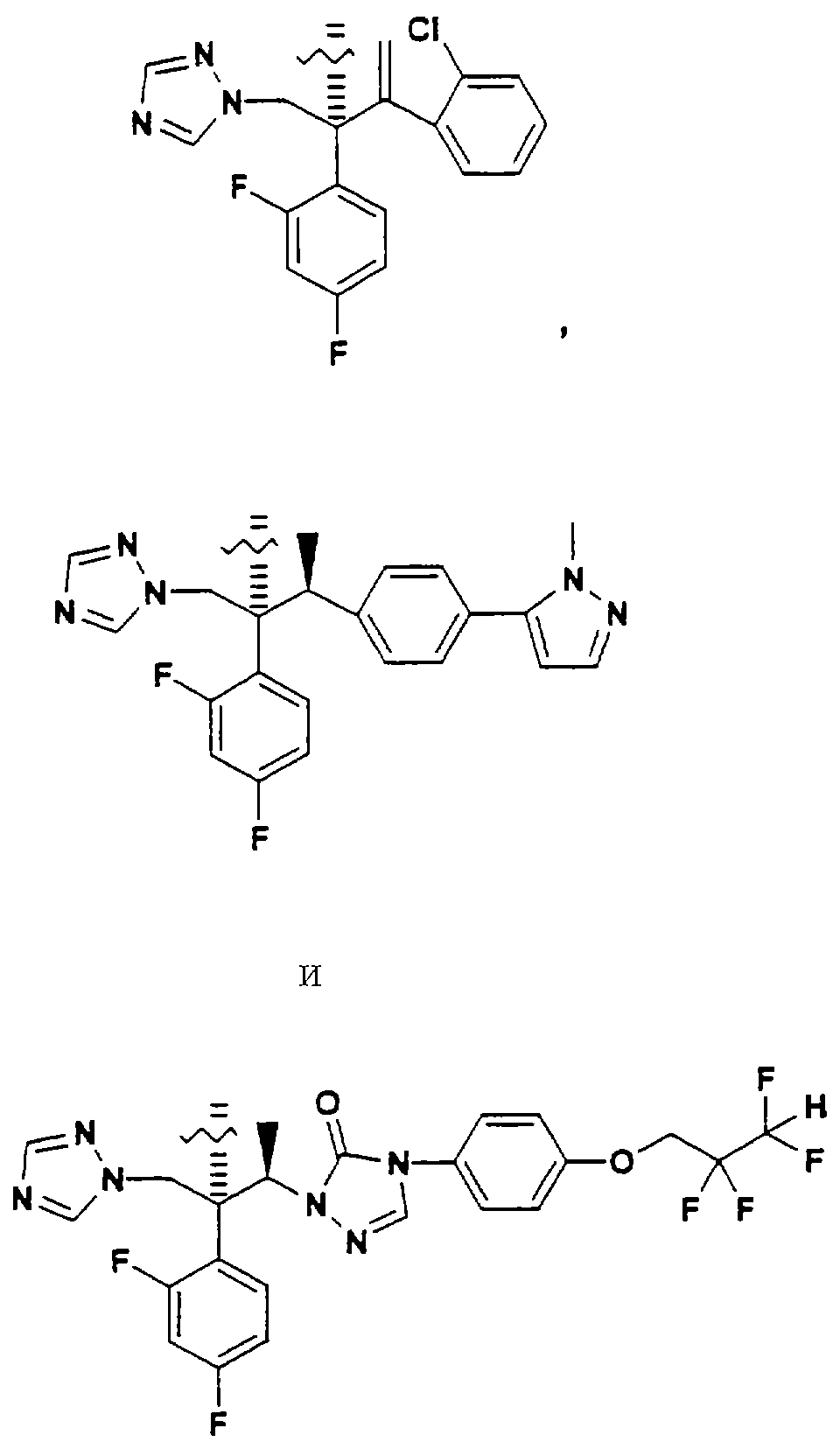
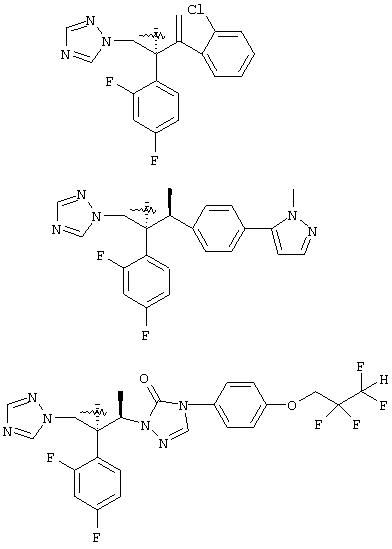
Помимо применения данного изобретения к структурам, содержащим третичную спиртовую группу, его можно применять к противогрибковым агентам, содержащим вторичную спиртовую группу, такую как:
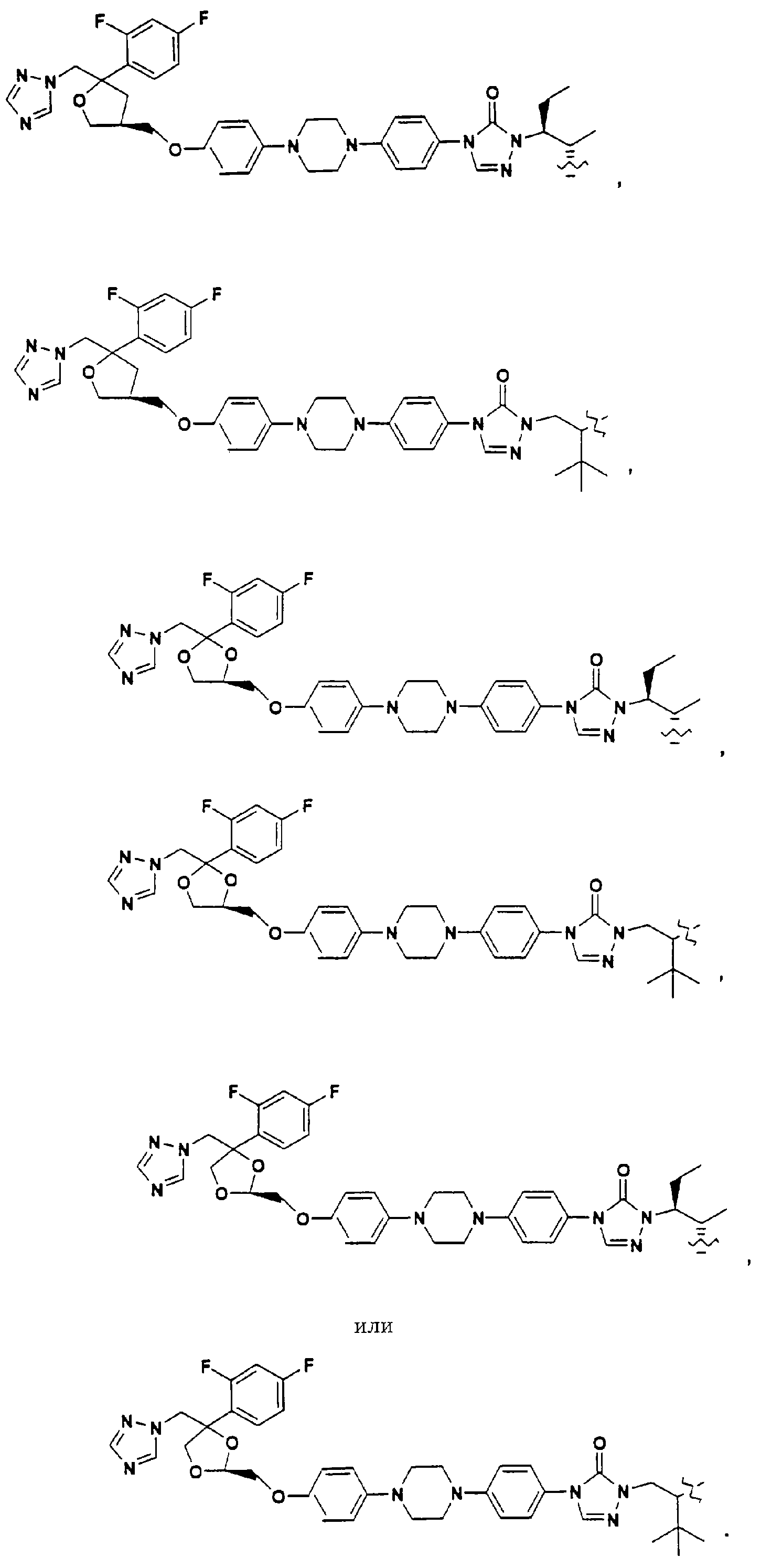
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Употребляемый в данном описании термин "(С1-С6)алкил" относится к насыщенным алифатическим группам с линейной или разветвленной цепью, содержащим 1-6 углеродных атомов, таким как метил, этил, н-пропил, изопропил, н-бутил, втор.-бутил, трет.-бутил, н-пентил, н-гексил и т.д.
Выражение "фармацевтически приемлемая соль", употребляемое в данном описании, охватывает фосфатные соли с такими противоионами, как аммоний, соли металлов, соли с аминокислотами, соли аминов и соли других оснований, таких как пиперидин или морфолин. Предполагается, что как "моно-", так и "бис"-соли охватываются термином "фармацевтически приемлемые соли". Конкретные варианты изобретения включают соли аммония, натрия, кальция, магния, цезия, лития, калия, бария, цинка, алюминия, лизина, аргинина, гистидина, метиламина, этиламина, трет.-бутиламина, циклогексиламина, N-метилглюкомина, этилендиамина, глицина, прокаина, бензатена, диэтаноламина, триэтаноламина, пиперидина и морфолина. Для наиболее предпочтительного варианта изобретения, (2R,3R)-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2,4-дифторфенил)-1-(1Н-1,2, 4-триазол-1-ил)-2-[(дигидроксифосфорилокси)метокси]-бутана, наиболее предпочтительными солями являются соли трет.-бутиламина и лизина, так как их можно получать в виде отдельных полиморфных кристаллических твердых веществ высокой степени чистоты с хорошей растворимостью и стабильностью.
Термин "галоген", применяемый в данном описании, включает хлор, бром, фтор и иод, предпочтительно хлор и фтор, и наиболее предпочтительно фтор.
Соединения по данному изобретению могут быть сольватированными или несольватированными. Предпочтительным сольватом является гидрат.
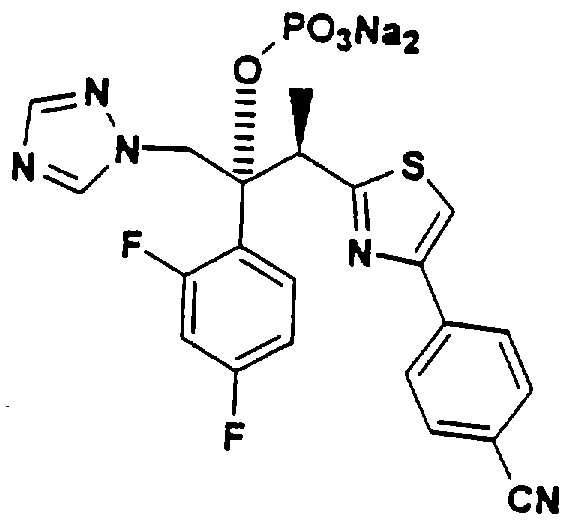
Наиболее предпочтительным вариантом данного изобретения является (2R,3R-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2,4-дифторфенил)-1-(1Н-1,2, 4-триазол-1-ил)-2-[(дигидроксифосфорилокси)метокси]-бутан или его фармацевтически приемлемая соль. Это пролекарство обладает намного более высокой растворимостью (>10мг/мл при рН 7, 5-6 мг/мл при рН 4,3) по сравнению с исходным соединением, что позволяет применять его для парентерального введения, а также для перорального приема. Это соединение также стабильно в растворе, может быть выделено в кристаллическом виде и легко превращается в исходное лекарственное вещество in vivo.
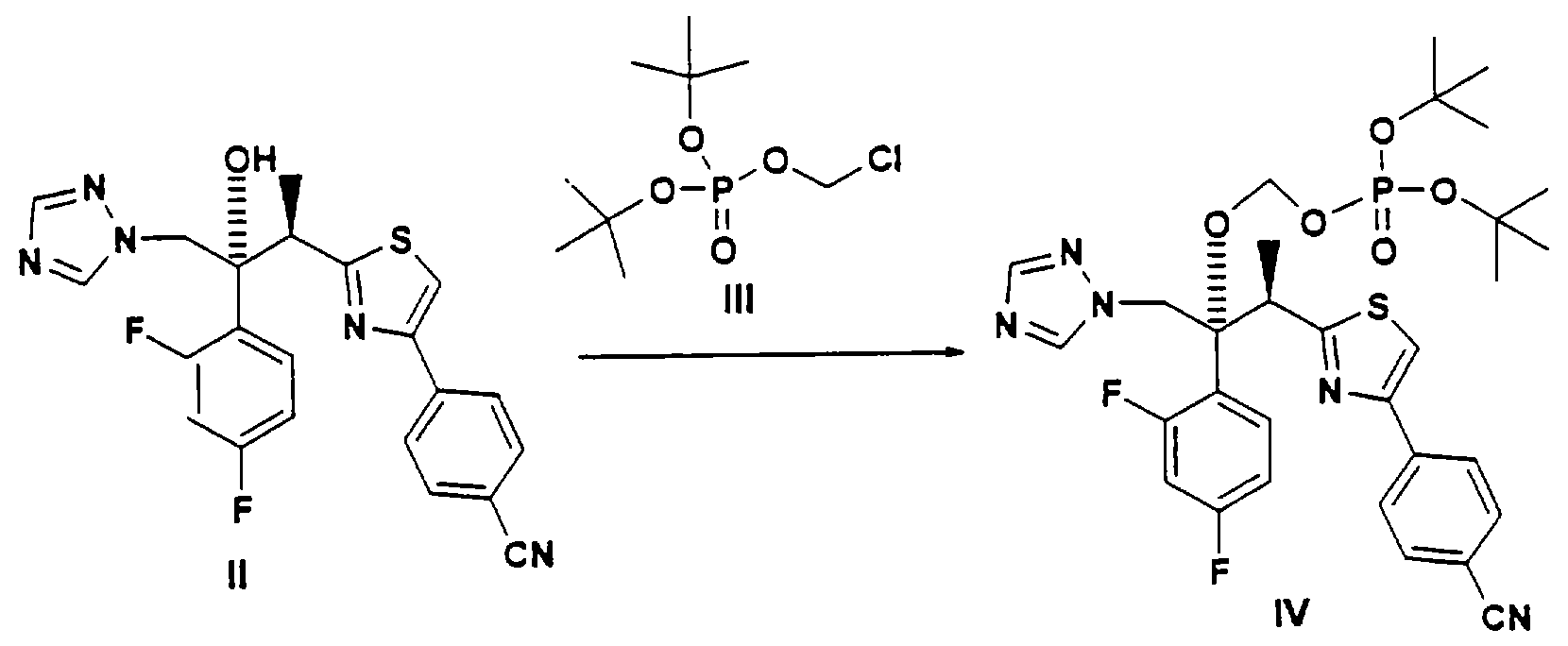
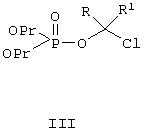
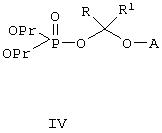
Соединения по данному изобретению можно легко получать по следующей общей схеме. В данном методе А представляет собой негидроксильный фрагмент триазольного противогрибкового соединения, содержащего третичную или вторичную спиртовую группу, Pr обозначает соответствующие защитные группы для гидроксила, такие как трет.-бутил, бензил или аллил, a R и R1, каждый, независимо обозначают водород или (С1-С6)алкил. Наиболее предпочтительно, когда R и R1, оба, обозначают водород.
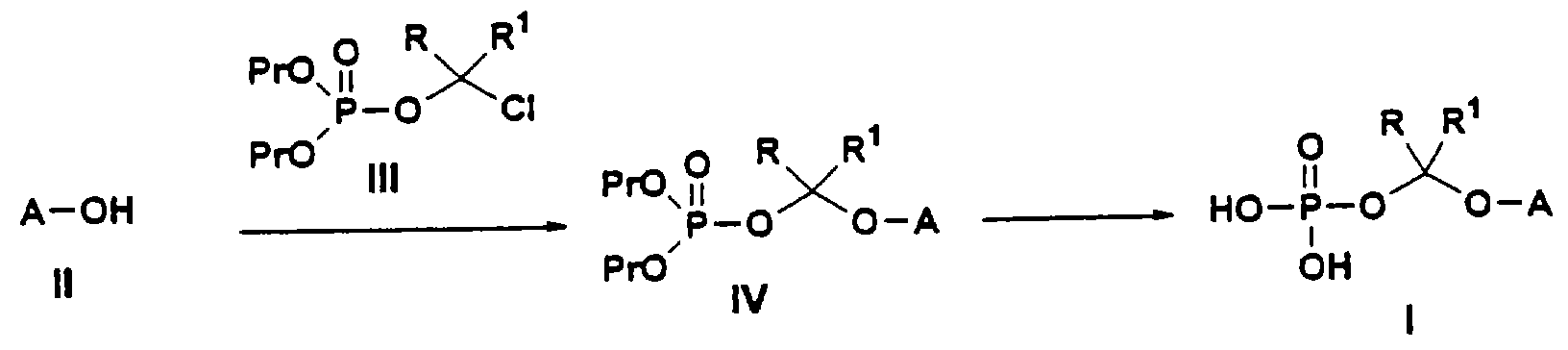
Для осуществления этого метода интересующее нас исходное противогрибковое соединение II превращают в фосфатный интермедиат IV О-алкилированием с хлорсодержащим интермедиатом III в присутствии соответствующего основания, такого как гидрид натрия, гидрид калия, трет.-бутилат натрия, трет.-бутилат калия, натрий-бис(триметилсилил)амид, такие как гидрид натрия плюс натрий-бис(триметилсилил)амид. Эту стадию реакции можно проводить в инертном органическом растворителе, таком как тетрагидрофуран, метилтетрагидрофуран, метил-трет.-бутиловый эфир, диэтиловый эфир или диметилацетамид, при температуре, примерно, 0-50°С, более предпочтительно около 20-40°С, и наиболее предпочтительно около 40°С. Наиболее предпочтительным основанием является гидрид натрия, а наиболее предпочтительным растворителем является тетрагидрофуран. Самыми предпочтительными R и R1 является водород.
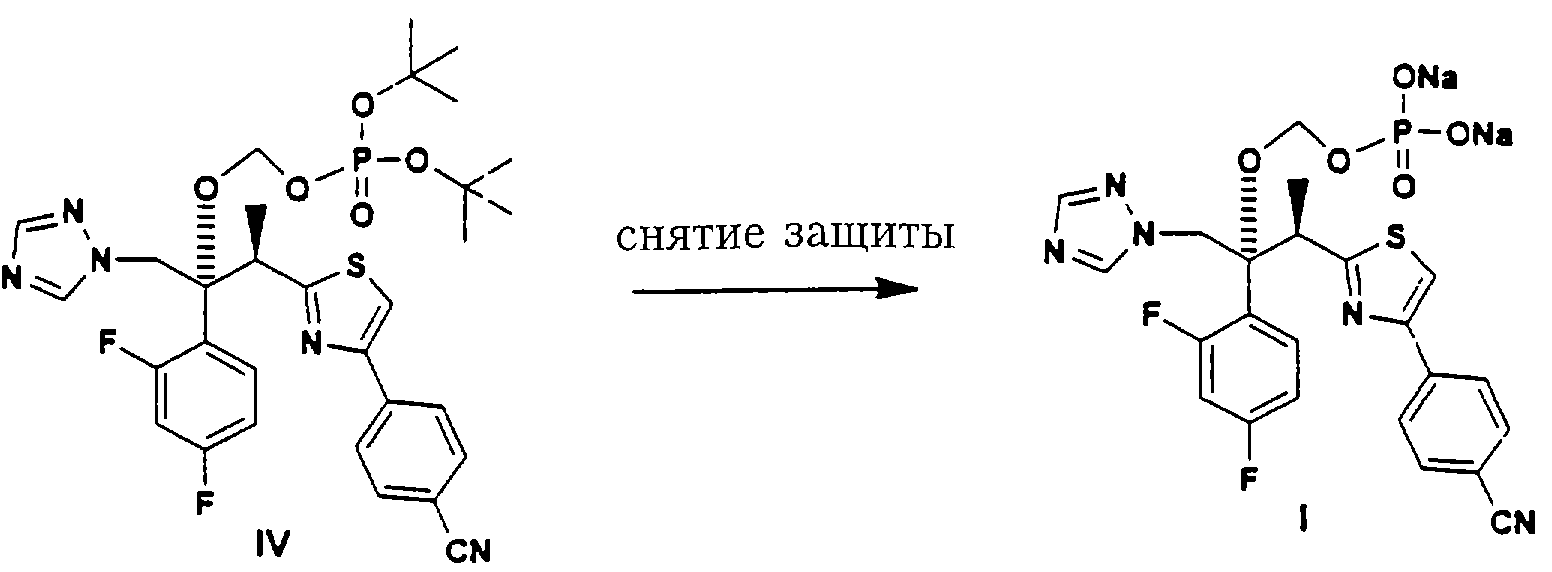
Сложноэфирный интермедиат IV затем подвергают соответствующему депротекционированию с целью снятия гидроксил-защитной группы Pr. Реагенты, применяемые на этой стадии, зависят от конкретной используемой гидроксил-защитной группы, но хорошо известны специалистам в данной области техники. Наиболее предпочтительной гидроксил-защитной группой является трет.-бутильная группа, которую можно снять трифторуксусной кислотой, хлористоводородной кислотой или муравьиной кислотой в подходящем инертном органическом растворителе. Инертным органическим растворителем может быть, например, хлористый метилен, дихлорэтан, метилбензол или трифторметилбензол. В случае предпочтительного депротекционирования ди-трет.-бутилового (сложного) эфира предпочтительно проводить его в трифторуксусной кислоте в хлористом метилене при температуре около 0-40°С, наиболее предпочтительно около 0-5°С.
Конечный продукт I можно затем регенерировать и очистить соответствующими методами, такими как обращенно-фазовая С-18 колоночная хроматография или экстракция растворителем.
Конечный продукт I можно, конечно, превратить соответствующими способами в заданную фармацевтически приемлемую соль.
Позднее было найдено, что применение очищенного реагента III дает довольно низкие выходы интермедиата IV (около 10-35%) в вышеприведенной реакции, что обусловливает низкие общие выходы продукта I. Однако, если на стадии О-алкилирования вышеприведенной реакции добавлять источник иодида, выход, интермедиата IV неожиданно возрастает, примерно, до 90%, тем самым значительно повышается выход конечного продукта I. Полагают, что добавление иодид-иона может привести к in situ образованию соответствующего иодид-интермедиата III' формулы:
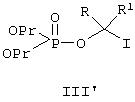
и что использование этого реагента приводит к значительному увеличению выхода фосфатного интермедиата IV. Попытка заместить непосредственно интермедиат III на заранее полученный интермедиат III' на первой стадии вышеописанной реакции, однако, была неудачной вследствие значительно более низкой стабильности йодного реагента III' по сравнению с хлоридным интермедиатом III. Альтернативный способ, который оказался удачным, включает применение йода на стадии O-алкилирования наряду с хлоридным интермедиатом III в присутствии основания, такого как NaH (который также может вести себя по отношению к йоду как восстановитель). Полагают, что иод восстанавливается до иона иода (иодида), который затем превращает хлорид-интермедиат II in situ в иодид-интермедиат III', облегчая эту стадию процесса. Пример, приведенный ниже, иллюстрирует стадию O-алкилирования с помощью элементарного иода, и это является предпочтительным методом проведения этой реакции с целью получения интермедиата IV.
С помощью образования in situ иодидного реагента III' добавлением источника иодид-иона или реакцией иода и реагента III в присутствии сильного основания резко повышающийся выход эфира фосфорной кислоты IV позволяет получать конечный продукт I также со значительно более высоким выходом.
Источником иона иода, предпочтительно, является иодид натрия, но также им может быть иодид лития, иодид цезия, иодид кадмия, иодид кобальта, иодид меди, иодид рубидия, иодид бария, иодид цинка и иодид кальция. На эквивалент исходного соединения А-ОН обычно берут около 2-3 эквивалента соли-иодида.
Когда элементарный иод используют на стадии конденсации, на эквивалент исходного соединения А-ОН берут около 0,1-1,0 эквивалента иода, предпочтительно, 0,5 эквивалента.
Основание и растворители, применяемые при использовании иода или иодид-иона (иона иода), те же самые, что описаны выше, когда реагент III применяется как таковой.
Следует понимать, что если группа заместителей, применяемых в вышеуказанных реакциях, содержит определенные чувствительные функциональные группы, такие как амино- или карбокси-группы, которые могут вызвать нежелательные побочные реакции, такие группы можно защитить подходящими защитными группами, известными специалистам в данной области техники. Соответствующие защитные группы и способы их снятия проиллюстрированы, например, в Protective Groups in Organic Synthesis. Theodora W. Greene (John Wiley and Sons, 1991). Подразумевается, что такие "защищенные" интермедиаты и конечные продукты входят в объем данного открытия и формулы изобретения.
Будет оценено по достоинству, что некоторые продукты, охватываемые Формулой I, могут иметь заместители, которые могут приводить к образованию оптических изомеров. Имеется в виду, что данное изобретение включает в свой объем все такие оптические изомеры, а также их эпимерные смеси, т.е. R- или S- или рацематы.
Фармацевтически активные соединения по данному изобретению можно использовать одни или готовить в виде фармацевтических композиций, содержащих помимо активного триазольного ингредиента фармацевтически приемлемый носитель, адъювант или разбавитель. Соединения можно вводить различными способами, например перорально, местно или парентерально (внутривенная или внутримышечная инъекция). Фармацевтические композиции могут быть в твердом виде, например в виде капсул, таблеток, порошков и т.д., или в жидком виде, например в виде растворов, суспензий или эмульсий. Композиции для инъекций можно готовить в виде стандартной дозы в ампулах или в контейнерах многоразового использования, и они могут содержать добавки, такие как суспендирующие агенты и диспергаторы. Композиции могут быть в форме, готовой к употреблению, или в виде порошка, который готовят к применению в момент доставки с помощью соответствующего носителя, например стерильной воды.
Или же соединения по данному изобретению можно вводить в форме суппозитория или вагинального суппозитория, или их можно применять местно в форме лосьона, раствора или крема. Кроме того, их можно вводить (при концентрации до 10%) в мазь, состоящую из основы - пчелиного воска или мягкого бесцветного парафина, объединенной с нужными стабилизаторами и/или консервантами.
Соединения по изобретению полезны вследствие своей фармакологической активности на животных, включая, в особенности, млекопитающих, и что наиболее важно, человека. Конкретно, соединения по данному изобретению полезны для лечения или предотвращения местных грибковых инфекций, включая инфекции, вызванные видами Candida, Trichophyton, Microsporum или Epidermophyton. Кроме того, они полезны для лечения инфекций слизистой оболочки, вызванных Candida albicans. Их также можно применять при лечении системных грибковых инфекций, вызванных, например, видами Candida albicans, Cryptococcus neoformans, Aspergillus flavus, Aspergillus fumigatus, Coccidioides, Paracoccidioides, Histoplasma или Blastomyces.
Следовательно, согласно другому аспекту изобретения предлагается (создан) способ лечения грибковой инфекции, который заключается во введении терапевтически эффективного количества соединения хозяину, в особенности хозяину-млекопитающему и, что особенно важно, больному человеку. Применение соединений по данному изобретению в качестве фармацевтических препаратов и применение соединений по изобретению в производстве медикаментов для лечения грибковых инфекций также охватывается.
Вводимая доза зависит, в большой степени, от конкретного используемого соединения, конкретной приготовленной композиции, способа введения, природы и состояния хозяина и конкретного обрабатываемого места и организма. Выбор конкретной предпочтительной дозы и способа применения остается в компетенции врача или ветеринара. Однако соединения можно вводить, главным образом, парентерально или перорально хозяину-млекопитающему в количестве, примерно, 5 мг/день - 1 г/день. Эти дозы являются средними, а могут быть индивидуальные случаи, когда требуются более или менее высокие дозы, и такие дозы входят в объем данного изобретения. Кроме того, введение соединений по данному изобретению можно осуществлять в виде разовой или разделенных (многократных) доз.
In vitro оценку противогрибковой активности соединений по изобретению можно осуществлять, определяя минимальную подавляющую концентрацию (MIC, МПК). MIC представляет собой концентрацию испытуемого соединения, которая подавляет рост испытуемых микроорганизмов. На практике серию (ряд) агаровых пластинок, каждая из которых содержит испытуемое соединение, введенное в определенной концентрации, засевают штаммом грибка и затем каждую пластинку инкубируют 48 часов при 37°С. Пластинки исследуют на наличие или отсутствие грибкового роста и отмечают релевантную концентрацию. Микроорганизмы, которые можно использовать в испытании, включают Candida albicans, Aspergillus fumigatus, Trichophyton spp.,, Microsporum spp., Epidermophyton floccosum, Coccidioides immitis и Torulopsos galbrata. Следует признать, что в качестве пролекарств некоторые соединения по изобретению могут быть неактивными in vitro тесте.
In vivo оценку противогрибковой активности соединений по данному изобретению можно осуществлять в серии уровней доз с помощью внутрибрюшинной или внутривенной инъекции или пероральным введением мышам, которым вводят штамм грибка (например, Candida albicans). Активность определяют, сравнивая выживаемость обработанной группы мышей при различных уровнях доз после смерти группы непролеченных (необработанных) мышей. Дозу, при которой испытуемое соединение обеспечивает предупреждение летального исхода вследствие действия инфекции на 50%, отмечают.
Соединения по данному изобретению значительно повышают стабильность исходного триазольного противогрибкового соединения, а также выделяют исходное соединение (т.е. действуют как пролекарства), что демонстрируется в экспериментах S9 на печени человека.
ПОЯСНЯЮЩИЕ ПРИМЕРЫ
Следующие примеры поясняют, иллюстрируют изобретение, но не претендуют на его ограничение. Сокращения, применяемые в примерах, являются обычными сокращениями, хорошо известными специалистам в данной области техники. Некоторые из применяемых сокращений даны ниже:
ч = час(ы)
rt = комнатная температура
ммол = ммол-ь(и)
г = грамм(ы)
ТГФ = тетрагидрофуран
мл = миллилитр(ы)
л = литр(ы)
Et2O = диэтиловый эфир
EtOAc = этилацетат
ТФК = трифторуксусная кислота
СН2Cl2= хлористый метилен
СН3CN = ацетонитрил
В следующих примерах все температуры даны в градусах Цельсия. Температуры плавления определяют на электротермоприборах и не корректируются. Спектры протонного магнитного резонанса (1Н ЯМР) снимают на ЯМР-спектрометрах Bruker 500, Bruker АМ-300 или Varian Gemini 300. Все спектры снимают в CDCl3 или D2O, если не указано иначе. Химические сдвиги даны в единицах (м.д.) относительно тетраметилсилана (TMS) или пика эталона растворителя, а константы спин-спинового взаимодействия даются в герцах (Гц). Мультиплетность сигнала обозначается следующим образом: с, синглет; д, дублет; т, триплет; кв, квартет; м, мультиплет; уш., уширенный сигнал; дд, дублет дублетов; дт, дублет триплетов; и каж. д., кажущийся дублет и т.д. Масс-спектры получены на масс-спектрометрах Kratos MS-50 или Finnegan 4500 методами химической ионизации (DCI, ХИ, изобутен), бомбардировкой быстрыми атомами (FAB, ББА) или ионизацией электрораспылением (ESI).
Аналитическую тонкослойную хроматографию (ТСХ) проводят на пластинах с закрепленным слоем силикагеля (60F-254) и проявляют с помощью УФ-света, паров иода и/или окрашиванием метанольной фосфомолибденовой кислотой. Обращенно-фазовую хроматографию проводят в стеклянной колонке на С 18 силикагеле (Waters Corporation Preparative С 18 125А) под давлением, несколько выше атмосферного.
ПРИМЕР 1
(2R,3R)-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2, 4дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-2-[(дигидроксифосфорилокси)метокси]-бутан, натриевая соль
A. (2R,3R)-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2,4-дифторфенил)-1-(1H-1,2,4-триазол-1-ил)-2-[(ди-трет.бутил-оксифосфорилокси)метокси]-бутан,
В. (2R, 3R)-3-[4-(4-цианофенил)тиазол-2-ил1-2-(2.4-дифторфенил)-1-(1 Н-1,2,4-триазол- 1-ил)-2-[ (дигидроксифосфорилокси] метокси] -бутан, натриевая соль:
Ди-трет.бутилхлорметилфосфат. III:
Ди-трет.бутилхлорметилфосфат, III, можно получать любым из нижеследующих способов.
Способ 1
Ди-трет.бутилфосфат серебра (6,34 г, 20 ммол), который получают, смешивая ди-трет.бутилфосфат (полученный из ди-трет.бутилфосфита методом (Zwierzak and Kluba, Tetrahedron, 1971, 27, 3163) с одним эквивалентом карбоната серебра в 50% водном ацетонитриле и, лиофилизируя досуха, вносят в бензол вместе с хлориодметаном (35 г, 200 ммол) и перемешивают при комнатной температуре в течение 18 часов.
Реакционную смесь фильтруют и фильтрат упаривают в вакууме. Остаток хроматографируют на силикагеле и элюируют: смесь гексанов - этилацетат 2:1. Нужные фракции упаривают досуха, получают вышеназванное соединение III (3,7 г, 71% выход);1ЯМР (CDCl3): 3,63 (д, 2Н, J=17); 1,51 (с, 18Н); Масс-спектр (МН+ = 259).
Способ 2
Тетрабутиламмоний ди-трет.бутилфосфат, готовят, растворяя ди-трет.бутилфосфат [20 г, 94 ммол (получен из ди-трет.бутилфосфита по методу Zwierzak and Kluba, Tetrahedron, 1971, 27, 3163)] в метанольном тетрабутиламмоний гидроксиде (47 мл 1М раствора, 47 ммол). Температура ракционной смеси 23°С и рН 4,33. рН реакционной смеси доводят до рН 6,5-7,0, добавляя метанольный тетрабутиламмоний гидроксид (48 мл 1М раствора, 48 ммол) за 0,2 часа. Реакционную смесь перемешивают 0,5 час, примерно, при 26°С, а затем упаривают в вакууме при температуре бани около 40°С. Сырой остаток трижды чистят азеотропной перегонкой, добавляя толуол (3×100 мл), а затем смесь упаривают в вакууме. Сырой остаток затирают в холодной смеси гексанов (0°С) в течение 1 часа, а затем твердое вещество собирают фильтрованием, промывают минимальным количеством холодной смеси гексанов и сушат, получая первую порцию тетрабутиламмоний ди-трет.бутилфосфата в виде белого твердого вещества (24,0 г). Маточный раствор упаривают в вакууме и затем затирают в холодной смеси гексанов (20 мл) в течение 1 часа. Твердое вещество собирают фильтрованием, промывают минимальным количеством холодной смеси гексанов и сушат, получая вторую порцию тетрабутиламмоний ди-трет.бутилфосфата в виде белого твердого вещества. [(8,5 г, 32,5 г общий выход (77%)]. Раствор тетрабутиламмоний ди-трет.бутилфосфата (218 г, 480 ммол) в бензоле (200 мл) по каплям добавляют при перемешивании к хлориодметану (800 г, 4535 ммол) в течение 1,5 час при комнатной температуре. Реакционную смесь перемешивают еще 1,5 час при комнатной температуре, а затем упаривают в вакууме. Остаток в виде масла растворяют в Et2O и фильтруют, выпавший белый осадок отбрасывают. Органический слой промывают насыщенным раствором NaHCO3 и Н2О/рассол (1:1). Органический слой затем сушат сульфатом магния, фильтруют и упаривают в вакууме, получая коричневокрасное масло (320 г). Это масло хроматографируют на силикагеле, элюируют 20% EtOAc/смесь гексанов, 25% EtOAc/смесь гексанов, затем 30% EtOAc/смесь гексанов. Содержащие продукт фракции упаривают в вакууме, получая золотистое масло. Затем масло разбавляют СН2Cl2 (30 мл), упаривают в вакууме, получая вышеназванное соединение III (61,3 г, 49% выход);1Н ЯМР (бензол-d6): 5,20 (д, 2Н, J=15); 1,22 (с, 18Н).
Способ 3
Иодхлорметан (974 г, 402 мл, 5,35 мол) при 25°С обрабатывают тетрабутиламмоний ди-трет.бутилфосфатом (250 г, 0,553 мол). Фосфат добавляют порциями за 10 минут. Гетерогенная смесь становится прозрачным розовым раствором примерно через 15 минут. Смесь перемешивают три часа и затем на роторном испарителе удаляют иодхлорметан при температуре бани <30°С. Остаток переносят в 1 л трет.бутилметилового эфира и перемешивают 15 минут, при этом выпадает побочный продукт тетрабутиламмоний иодид. Тетрабутиламмоний иодид удаляют фильтрованием в вакууме через воронку со стеклянным фильтром. Фильтрат упаривают на роторном испарителе, получают масло, содержащее 5:1 смеси III и примеси нежелательного димера:

Смесь можно очистить хроматографией на силикагеле, при этом получается III в виде чистого соединения (масло) с выходом ˜60%.
ПРИМЕР 2
(2R, 3R)-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2,4-дифторфенил)-1-(1Н-1,2, 4-триазол-1-ил)-2-[(дигидроксифосфоридокси)метокси]-бутан:
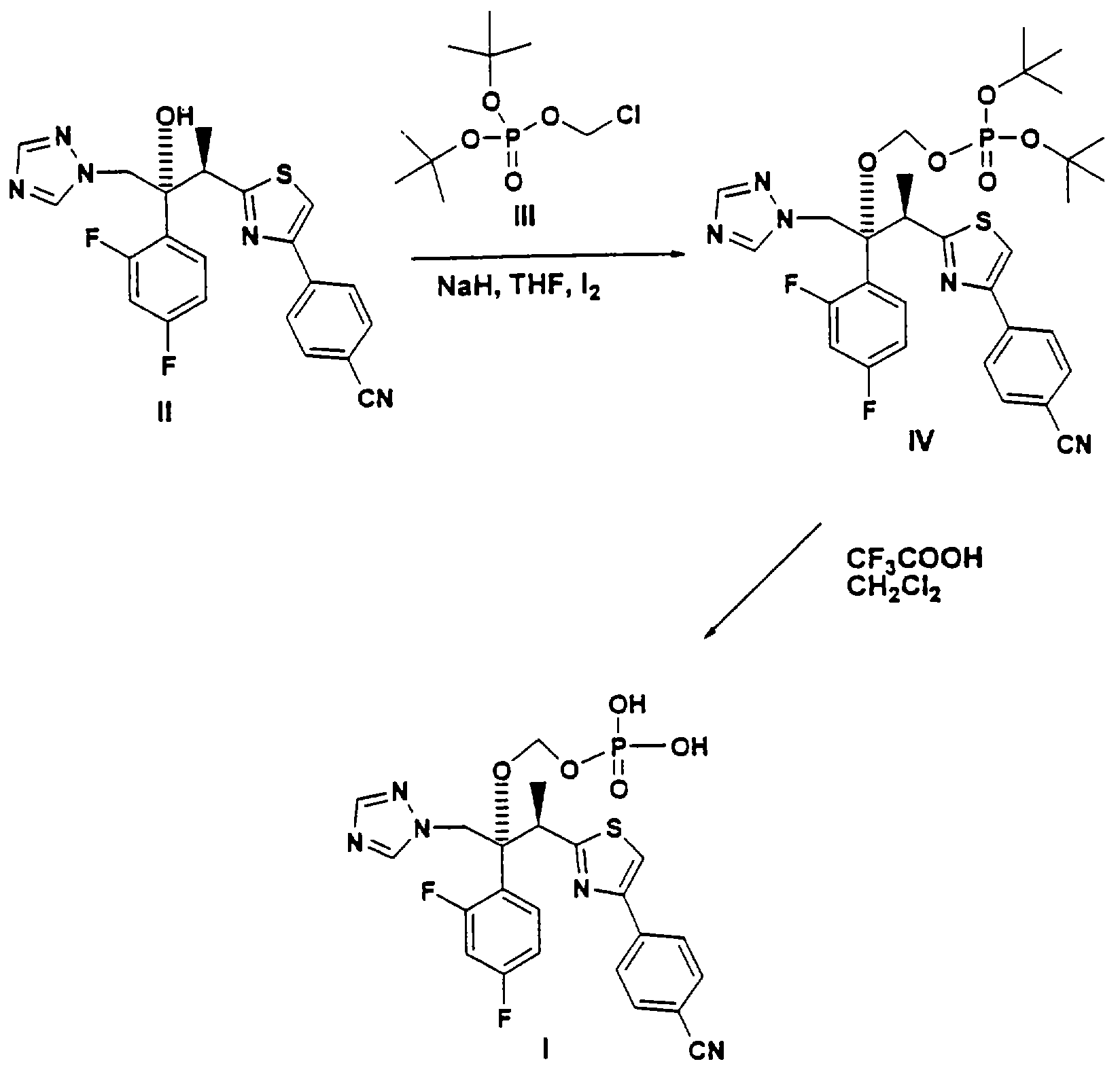
А. В высушенную в сушильном шкафу круглодонную колбу емкостью 1 л, снабженную механической мешалкой, вводом для азота, капельной воронкой с выравниванием давления, резиновой мембраной и датчиком температуры, помещают гидрид натрия (2,89 г, 0,069 мол, 60%) и ТГФ (50 мл). К этой перемешиваемой суспензии по каплям в течение 20 минут при комнатной температуре прибавляют (2R,3R)-3-[4-(4- цианофенил)тиазол-2-ил]-2-(2, 4-дифторфенил)-1-(1Н-1,2,4-триазол-1-ил)бутан-2-ол, II (10 г, 0,023 мол) в 30 мл ТГФ. После перемешивания в течение 45 минут по каплям прибавляют раствор иода (2,99 г, 0,0115 мол) в ТГФ (30 мл) за 10 минут, а затем, за 15 минут, ди-трет.бутилхлорметилфосфат, III (13,29 г, 0,035 мол, ˜68% чистоты). Реакционную смесь перемешивают 4 часа, примерно, при 41°С до завершения реакции. Об окончании реакции судят по ВЭЖХ в процессе реакции. Реакционную смесь выливают в воду со льдом (100 мл). Водную смесь отделяют и экстрагируют этилацетатом (3×50 мл), а объединенные органические вытяжки промывают 10% тиосульфитом натрия (50 мл), водой (50 мл), рассолом (50 мл), сушат сульфатом магния и упаривают в вакууме, получая светложелтое масло (22,8 г, чистота (ВЭЖХ) ˜97%). Сырой продукт непосредственно используют на стадии В.
В. В круглодонную колбу, снабженную магнитной мешалкой, охлаждающей баней, датчиком рН и входом-выходом для азота, помещают продукт стадии А (см. выше) (7,5 г) в СН2Cl2(23 мл) и охлаждают до 0°С. К этому раствору при перемешивании медленно добавляют трифторуксусную кислоту (8,8 мл) и перемешивают в течение 3 час до завершения реакции. Об окончании реакции судят по ВЭЖХ. Реакционную смесь выливают в холодный раствор 2N NaOH (64 мл). Реакционную смесь экстрагируют трет.бутилацетатом (2×65 мл) для удаления всех органических примесей. Водный слой, содержащий вышеназванный продукт в виде натриевой соли, обрабатывают активированным углем (10 г) и фильтруют через слой целита. Прозрачный фильтрат подкисляют 1N HCl до рН 2,5. Свободную кислоту, вышеназванный продукт, экстрагируют этилацетатом (2×50 мл). Объединенные органические вытяжки промывают водой, сушат MgSO4, фильтруют и фильтрат упаривают в вакууме, получая 3,39 г сырого вышеназванного продукта.
ПРИМЕР 3
Бис-лизиновая соль (2R, 3R)-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2, 4-дифторфенил)-1-(1Н-1.2.4-триазол-1-ил)-2-[(дигидроксифосфорилокси)метокси]-бутана
Вышеописанный озаглавленный в Примере 2 продукт растворяют в метаноле (75 мл) и к нему добавляют L-лизин (1,8 г) и нагревают при 60°С в течение 4,5 час. Горячую реакционную смесь фильтруют через слой целита. Фильтрат упаривают, примерно, до 5 мл, смешивают с метанолом (100 мл) и нагревают до 66°С с целью кристаллизовать бис-лизиновую соль. Соль собирают на воронке Бюхнера и сушат в вакууме, получая 3,71 г вышеназванного соединения в виде кристаллического вещества не совсем белого цвета.
ПРИМЕР 4
Соль трет.бутиламина и (2R, 3R)-3-[4-(4-цианофенил)тиазол-2-ил]-2-(2,4дифторфенил)-1-(1Н-1,2,4-триазол-1-ил)-2[(дигидроксифосфорилокси) метокси] -бутана
К раствору озаглавленного в Примере 2 продукта в 50 мл этилацетата добавляют под азотом трет.бутиламин (5,3 мл). Реакционную смесь перемешивают при 40°С примерно в течение 1 час с целью кристаллизовать продукт. Соль бис-трет.бутиламина собирают на воронке Бюхнера и сушат в вакууме, получая 2,21 г вышеназванного соединения в виде кристаллического вещества не совсем белого цвета.
Реферат
Изобретение относится к водорастворимым азольньм соединениям, которые могут найти применение в биологии и медицине. Описывается водорастворимое азольное соединение формулы
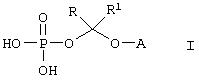
или его фармацевтически приемлемая соль, где R и R1, каждый независимо обозначает водород или (С1-С6)алкил, А обозначает группу формулы
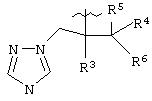
где R3 представляет собой фенильную группу с одним или более атомов галоида в качестве заместителей; R4 представляет собой водород или СН3; R5 представляет собой водород или вместе с R4 может представлять собой =СН3; R6 представляет собой 5- или 6-членный азотсодержащий цикл, который при необходимости может иметь в качестве заместителей одну или более группу, выбранную из галогена, =0, фенила, замещенного одной или более группами, выбранными из CN, (С6H4)-ОСН2CF2CHF2 и СН=СН-(С6Н4)-ОСН2CF2CHF2, или фенила, замещенного одной или более группами, выбранными из галогена и метилпиразолила. Также описывается способ получения водорастворимого азольного соединения. Технический результат - получены новые соединения, которые могут быть полезны в медицине. 6 с. и 9 з.п.ф-лы.
Формула
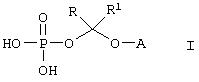
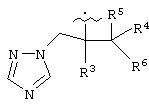
Комментарии