Производные 1,2,4-триазола или их фармацевтически приемлемые соли, фармацевтическая композиция и промежуточные соединения - RU2095358C1
Код документа: RU2095358C1
Чертежи

Описание
Изобретение относится к новым производным триазола, которые обладают антифунгицидным действием и могут быть использованы для лечения грибковых заболеваний у животных, включая людей.
Изобретение относится к
соединениям формулы
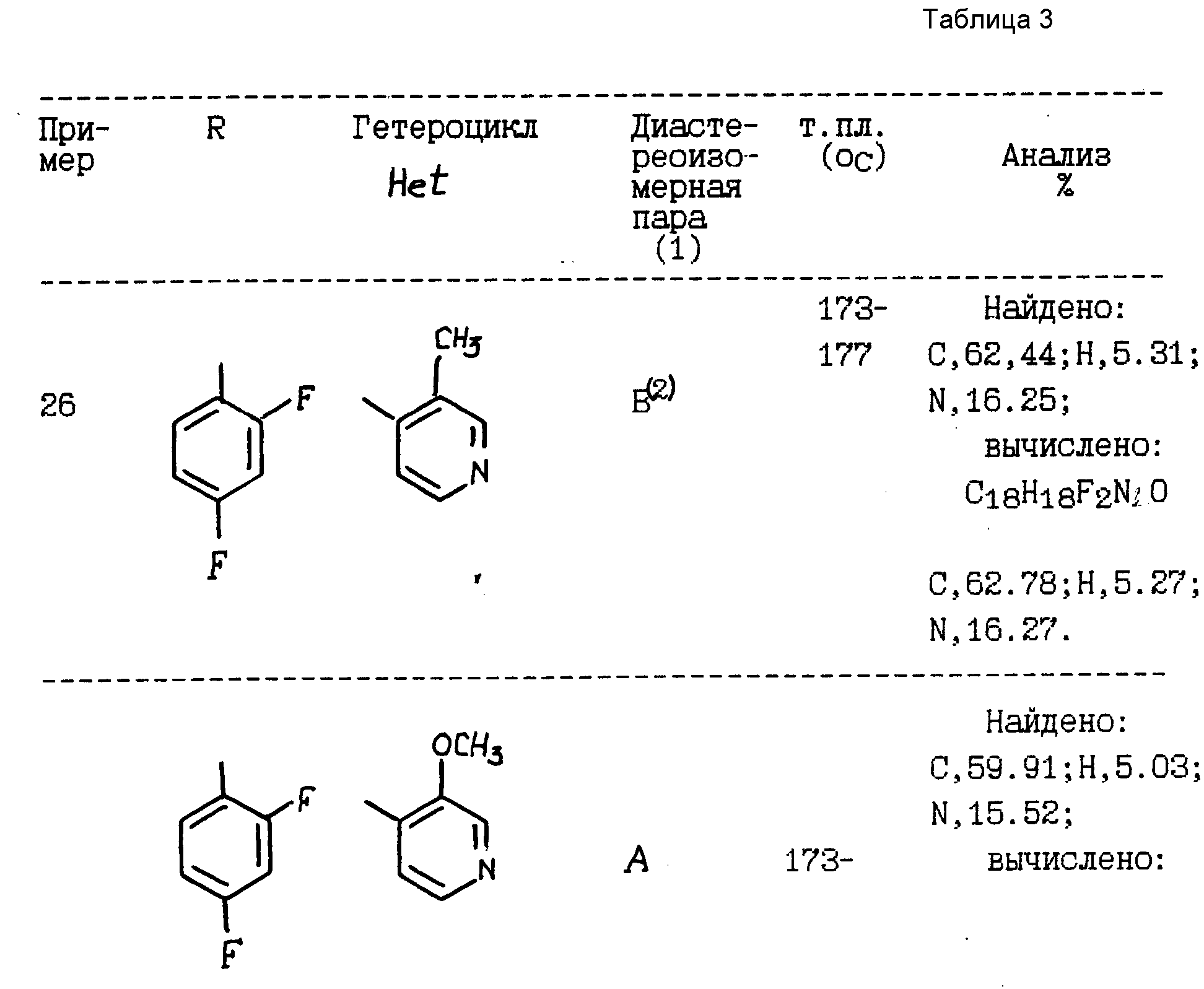
и их фармацевтически приемлемым солям, в котором:
R обозначает фенил, замещенный 1-2 заместителями, каждый из которых независимо друг от друга выбирают из галоида;
R1 обозначает C1-4 алкил;
R2 обозначает H или C1-4 алкил; и
"Het", который прикреплен к смежному атому углерода кольцевым атомом углерода, выбирают из пиридинила, пиридазинила, пиримидинила или пиразинила, при этом "Het" необязательно замещается C1-4 алкилом, C1-4 алкокси, галоидом, CN, NH2, или -NHCO2(C1-C4) алкилом.
В одном из аспектов, изобретение обеспечивает получение соединений формулы (l) и их фармацевтически приемлемых солей, где "Het" выбирают из числа таких радикалов, как 2- и 4-пиридинил, пиридазинил, 2- и 4-пиримидинил и пиразинил, "Het" необязательно замещен C1-C4 алкилом, C1-C4 алкокси, галоидом, CN, NH2, или -NHCO2(C1-C4 алкилом); и радикалы R, R1 и R2 имеют указанные выше значения для соединений формулы (l).
В другом аспекте "Het", представляющий собой пиридинил, пиридазинил, пиримидинил или пиразинил "Het", необязательно содержит в качестве заместителей C1-C4 алкил, С1-С4 алкокси, гало или NH2.
Когда радикал "Het" содержит заместители, то предпочтительно 1 или 2, наиболее предпочтительно 1 заместитель.
Галоид означает F, Cl, Br или I.
C3 и C4 алкильные и алкоксильные группы могут быть прямыми или разветвленными.
Когда радикал является замещенной фенильной группой, то это включает например, 2-фторфенил, 2-хлорфенил, 2-бромфенил, 2-иодфенил, 2-трифторметилфенил, 2,4-дихлорфенил, 2,4-дифторфенил, 2-хлор-4-фторфенил, 2-фтор-4-хлорфенил и 2,5-дифторфенил.
Предпочтительно радикал R представляет собой фенильную группу, замещенную 1-2 галоидами (предпочтительно F или Cl).
Также более предпочтительно, чтобы радикал R представлял собой 2,4-дифторфенил, 2,4-дихлорфенил, 2-фторфенил или 2-хлорфенил.
Наиболее предпочтительно, чтобы R представлял собой 2, 4-дифторфенил.
Предпочтительно радикал R1 означает метил и радикал R2 H или метил.
Наиболее предпочтительно радикал R1 означает метил и радикал R2 H.
Предпочтительно радикал "Het" выбирать из таких групп, как пиридинил, пиридазинил, пиримидинил или пиразинил, необязательно замещенных 1 или 2 заместителями, каждый из которых независимо выбирают из C1-C4 алкила, C1-C4 алкокси, гало, CN, NH2 и -NHCO2(C1-C4 алкила).
Также более предпочтительно "Het" выбирают из числа таких групп, как пиридинил, пиридазинил, пиримидинил и пиразинил; все необязательно замещены одним из заместителей CN, NH2 или -NHCO2(C1-C4 алкил).
Предпочтительными пиридинильными и пиримидинильными группами являются 2- и 4-пиридинил и 2- и 4-пиримидинил, все необязательно замещенные как указано выше.
Но все же более предпочтительно "Het" выбирают из числа таких групп, как пиридинил (предпочтительно 2- и 4-пиридинил), пиридазинил, 2- и 4-пиримидинил и пиразинил, все необязательно замещенные одним из заместителей CN, NH2 или -NHCO2(C1-C4 алкил).
Наиболее предпочтительно "Het" представляет собой 2-пиридинил, 4-пиридинил или 4-пиримидинил.
Фармацевтически приемлемые соли соединений формулы (l) включают соли, полученные путем добавления кислот, которые обеспечивают получение нетоксичных солей, таких как гидрохлориды, гидробромиды, гидроиодиды, сульфаты или бисульфаты, фосфаты или кислые фосфаты, ацетаты, малеаты, фумараты, лактаты, тартраты, питраты, глюконаты, бензоаты, метансульфонаты, бензосульфонаты и пара-толуиленсульфонаты.
Особенно предпочтительными
отдельными соединениями являются следующие:
2-/2,
4-дифторфенил/-3-/пиридин-2-ил/-1-/1Н-1,2,4-триазол-1-ил/бутан-2-ол,
2-/2,4-дифторфенил/-3-/пиридин-4-ил/-1-/1Н-1,2,
4-триазол-1-ил/бутан-2-ол и
2-/2,
4-дифторфенил/-3-/пиримидин-4-ил/-1-/1Н-1,2,4-триазол-1-ил/бутан-2-ол; и фармацевтически приемлемые соли.
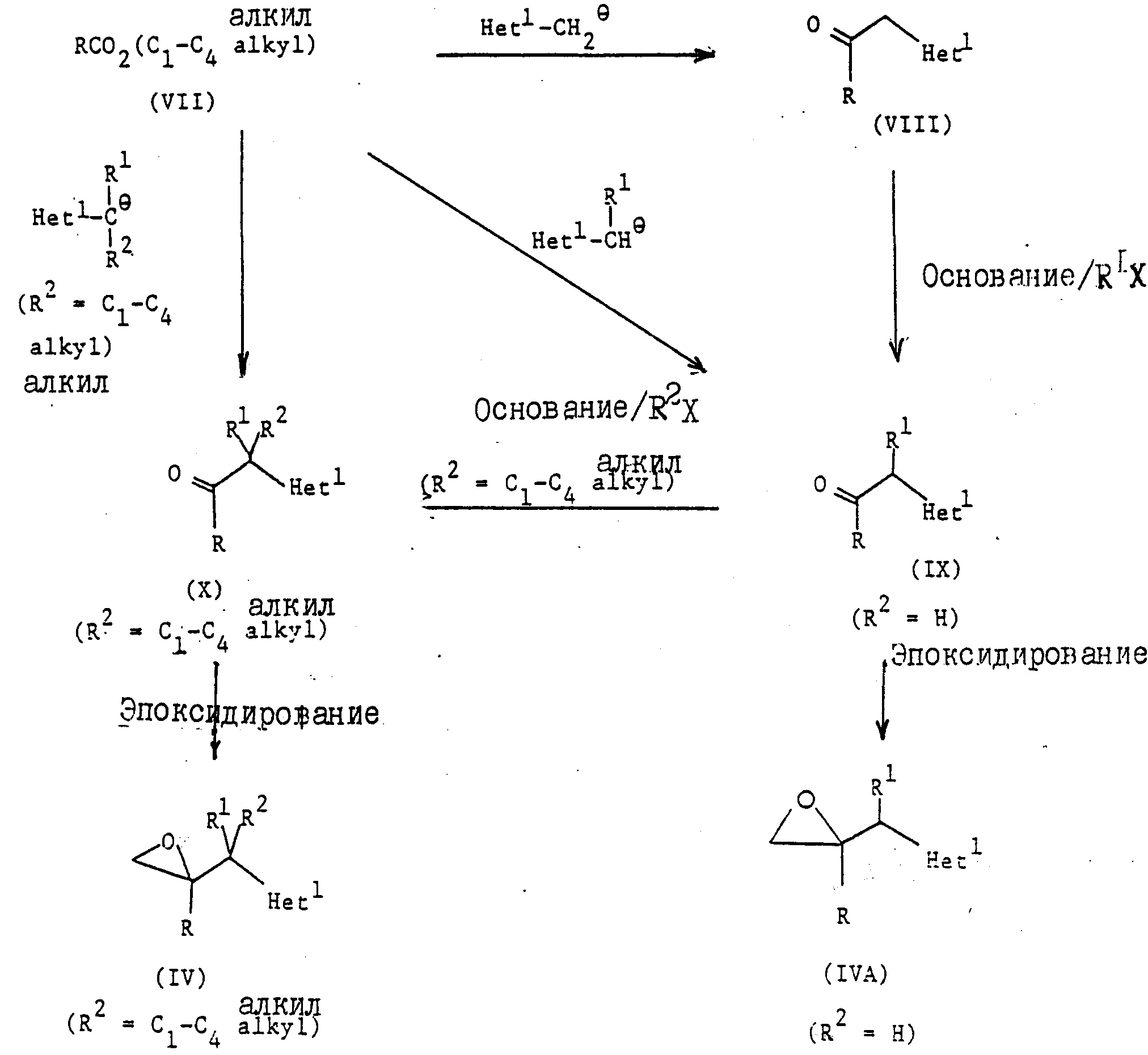
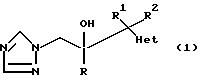
Соединения формулы (l) изобретения получают следующими способами.
1. Соединения формулы
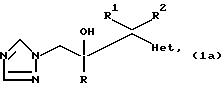
где радикалы R, R1, и R2 имеют те же значения, что указаны для формулы (l), и "Het" представляет собой пиридинил, пиридазинил, пиримидинил или пиразинил, причем "Het" необязательно замещен C1-C4 алкилом, C1-C4 алкокси, гало; или CN могут быть получены следующим образом:

где радикалы R, R1, R2 и "Het" имеют те же значения, что указаны для формулы (lА).
Согласно типичной методике, соединения формулы (II) подвергаются депротонированию путем добавления приблизительно одного эквивалента соответствующего сильного основания, например, диизопропиламида лития, осуществляют взаимодействие полученной соли (предпочтительно литиевой, натриевой или калиевой соли). in situ с кетоном формулы (III). Реакцию обычно осуществляют от -80o до -50oC, предпочтительно при -70oC, в среде соответствующего органического растворителя, например, тетрагидрофурана или диэтилового эфира, в инертной атмосфере, например, атмосфере азота или аргона.
Исходные соединения формулы (II) известны или
могут быть получены обычными методами (см. раздел "Примеры"). Исходные материалы (III) являются известными соединениями (смотри, например, EP-A-44605,
EP-A-69442 или патент Великобритании 1 464 224)
или могут быть получены аналогичными способами; или
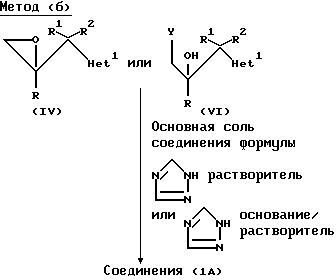
где радикалы R, R1, R2 и "Het" имеют те же значения, что указаны для (lА), и Y представляет собой отщепляющуюся группу, например, хлор, бром или C1-C4 алкансульфонилокси группа (такая как метансульфонилокси). Примерами приемлемых солей 1Н-1,2,4-триазола являются соли щелочных металлов (предпочтительно натрия) и тетраалкиламмония (предпочтительно тетра-н-бутиламмония (см. патент США 4 259505).
Реакцию предпочтительно осуществляют, используя в качестве исходного материала эпоксид (lY). Если в этом процессе используют соединение формулы (Yl), то механизм реакции диктует, чтобы по крайней мере частично, эпоксид формулы (lY) образовывался in situ в условиях реакции. Поэтому такой процесс в этом отношении аналогичен тому, что предусматривает использование эпоксида (lY) в качестве исходного материала.
Когда используют соль 1Н-1,2,4-триазол, то реакцию обычно осуществляют при температуре от комнатной до 100oC, предпочтительно примерно при 60oC, если применяют натриевую соль 1Н-1,2,4-триазол, и предпочтительно при комнатной температуре, когда применяют соответствующую тетра-н-бутиламмониевую соль, в среде соответствующего органического растворителя, например, N,N-диметилформамида или тетрагидрофурана.
В другом варианте, реакцию можно осуществить, используя 1Н-1,2,4-триазол в присутствии основания, например, Na2CO3 или K2CO3, предпочтительно от 50oC до 100oC в среде соответствующего органического растворителя, например, N, N-диметилформамида или метанола.
Промежуточные соединения формулы (lY) и (Yl) могут быть получены обычными методами, например,
как описано в разделе
"Примеры", и как показано на схемах A и B:
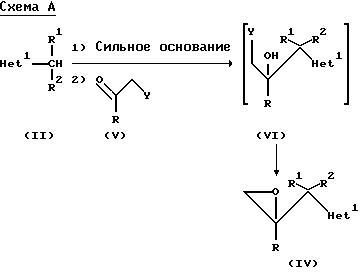
где радикалы R, R1 , R2 и "Het" имеют значения, указанные для формулы (lА), а Y представляет собой отщепляющуюся группу, предпочтительно C1 или Br.
В обычной методике соединения формулы (II) депротонируют путем добавления приблизительно одного эквивалента соответствующего сильного основания, например, диизопропиламида лития, и образующееся металлоорганическое промежуточное соединение взаимодействует in situ с соединением формулы (Y). Реакцию обычно осуществляют при температуре от -80o до -50oC, предпочтительно примерно при -70oC, в среде соответствующего органического растворителя, например, тетрагидрофурана или диэтилового эфира, в инертной атмосфере, например, атмосфере азота или аргона. Промежуточное соединение (Yl) не надо выделять, и обычно оно претерпевает циклизацию in situ после перемешивания при более высокой температуре (например, при комнатной температуре), и образует оксиран формулы (lY).
Соединения формулы (Yl), когда Y представляет собой хлор или бром, также могут быть получены путем взаимодействия эпоксида (lY) с соответствующим галогенводородом в безводных условиях; или
Схема B представлена в конце описания,
где радикалы R, R1, R2 и "Het" имеют значения, указанные для формулы (lА), и X соответствующая отщепляемая группа, например, Cl,
Br, l или метансульфонилокси.
Согласно стандартной методике, соединения формул (YIII), (lХ) и (Х) получают непосредственно из сложного эфира формулы (YII) по реакции с металлоорганическим промежуточным соединением, полученным как правило путем депротонирования соединения формулы Het1-CH3 или Het1-CHR1R2 (соединение II), где Het1, R1 и R2 имеют те же значения, что указаны для формулы (lA), примерно с одним эквивалентом соответствующего сильного основания, например, диизопропиламидом лития. Реакция обычно осуществляют при температуре от -80o до -50oC, предпочтительно примерно при -70oC, в среде соответствующего органического растворителя, например, тетрагидрофурана или диэтилового эфира, в инертной атмосфере, например, атмосфере азота или аргона.
Хотя на схеме B и не показано, но соединения формулы (VIII)
или (IX), когда "Het" представляет собой 3-пиридинил или 5-пиримидинил и радикалы R и R1 имеют те же значения, что указаны для формулы (1A), можно легко получить из сложного эфира формулы
(VII) по реакции с металлоорганическим промежуточным соединением, полученным депротонированием соединения формулы
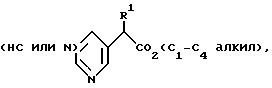
где радикалы R1 представляет собой водород или C1-C4 алкил, in situ по той же методике, что описана в предыдущем параграфе. Промежуточный β-кетоэфир, полученный после переработки, подвергаются затем гидролизу/декарбоксилированию путем обработки соответствующей сильной минеральной кислотой, например, концентрированной соляной кислотой, предпочтительно при кипячении, с целью получения соединения формулы (VIII) или (IX).
В другом варианте, соединения формулы (IX) и (X) можно получить по реакции соответственно соединения формулы (VIII) или (IX) приблизительно с одним эквивалентом соответствующего основания, например, гидрида натрия, с последующим алкилированием образовавшегося карбаниона in situ с помощью соответствующего агента алкирования. Реакция обычно осуществляется при температуре от 0oC до комнатной температуры в среде соответствующего органического растворителя, например, N,N-диметилформамида.
Предпочтительно алкилирование соединения формулы (VIII) или (IX) осуществлять в условиях переноса фаз, например, с
использование
NaOH[CH3(CH2)3]4N⊕⊖HSO4/H2O/CHCl3//C1-C4 алкил/X
где X представляет
собой предпочтительно иод, при температуре от 0oC до комнатной температуры, как правило, при комнатной температуре.
Эпоксидирование кетонов формулы (IX) или (X) осуществляют с помощью обычных методов, например, с использованием метилида диметилоксосульфония (см. например, j A. C. S. (1965), 87, 13 53) или хлорметиллития (смотри, например, Tel. Lett. (1986), 795).
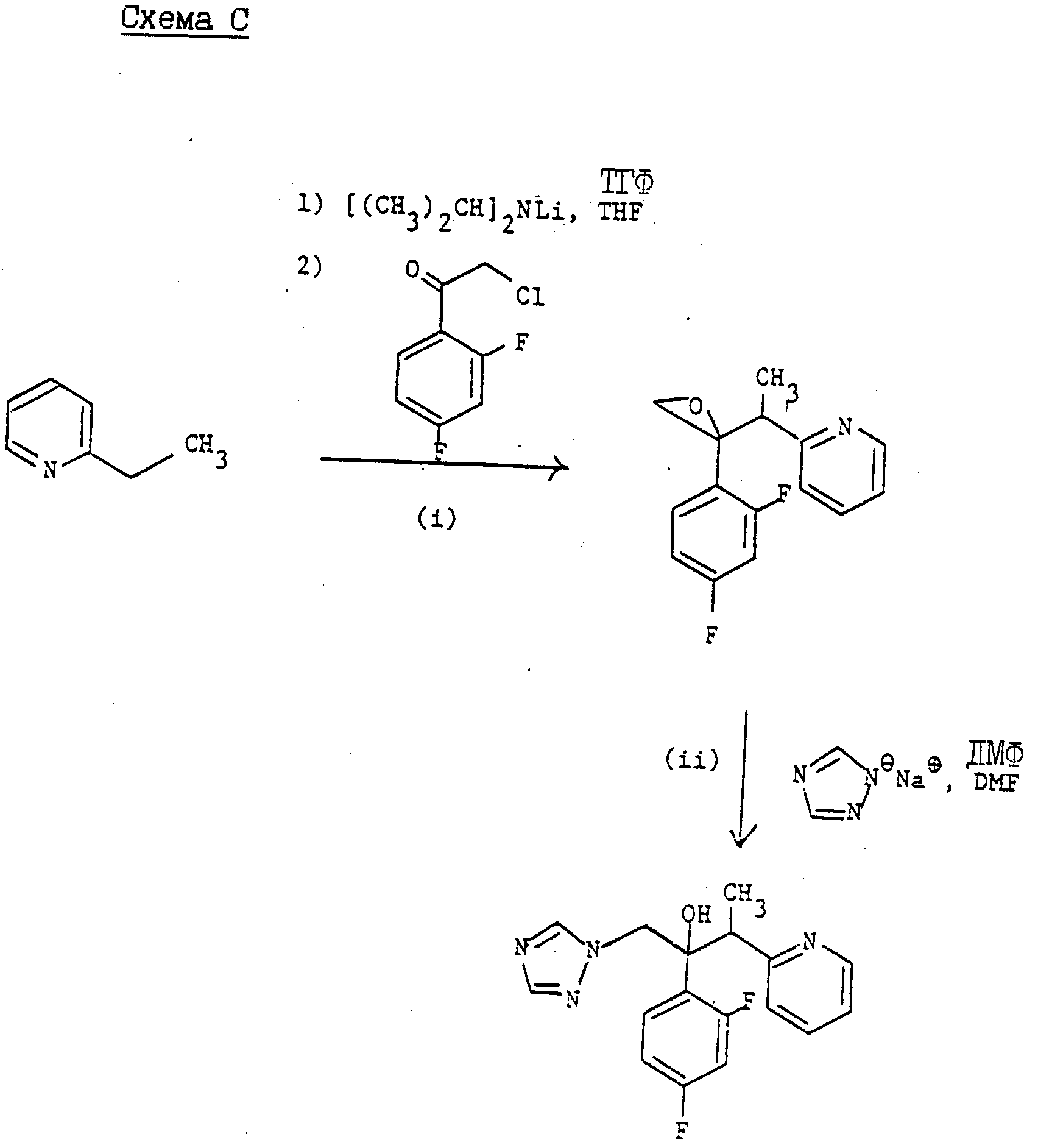
(2) Соединение формулы (l), в которой "Het" является монозамещенным цианогруппой по атому углерода ядра, расположенному рядом с атомом азота ядра, где "Het" представляет собой пиридинил, пиридазинил, пиримидинил, пиразинил или триазинил, и радикалы R, R1 и R2 и имеют те же значения, что указаны для формулы (l), наиболее удобно получать из незамещенных предшественников "Het" способом, показанным на схеме C, представленной в конце описания.
Методика проиллюстрирована для соединений формулы (l); где "Het" представляет собой пиридинил, хотя аналогичная ментодология применима во всех случаях, где "Het" имеет значения, указанные выше при описании этого метода, при условии, что "Het" должен иметь по крайней мере один незамещенный атом углерода в ядре, который расположен рядом с атомом азота ядра, который является N-окисленным.
"Het" предпочтительно представляет собой пиридинил или пиримидинил в этом методе.
В зависимости от конкретной группы "Het" и/или положения ее присоединения существует вероятность образования двух региоизомеров в этом процессе. Такие региоизомеры, если они образовались, можно разделить с помощью обычных методов, например, методом колончатой хроматографии.
Как правило соединение формулы (lB) окисляют с целью получения N-оксида формулы lC. Реакцию предпочтительную осуществляют, используя 3-хлорпероксибензойную кислоту, в среде соответствующего растворителя, например, дихлорметана, при температуре от 0oC до температуре его кипения, предпочтительно при комнатной температуре. В другом варианте окисления можно осуществлять, используя пероксид водорода в соответствующей C1-C4 алкановой кислоте, например, уксусной кислоте.
В результате обработки N-оксида (lC), N,N-диметилкарбамоилхлоридом, с последующей обработкой либо триметилсилицианидом или цианидом калия по методу W.K. Fife (j.Org. Chem. 48 1375 (1983) и др. Hiterocycles 22, 1121 (1984) циано-замещенное соединение (lE). Реакцию предпочтительно осуществляют, используя N, N-диметилкарбамоил хлорид и триметилсилил цианид в дихлорметане при комнатной температуре, а можно ее проводить ступенчато, например, добавляя сначала N,N-диметилкарбамоил хлорид к N-оксиду и перемешивая смесь в течение некоторого времени до добавления триметилсилил цианида.
(3) Некоторые из соединений формулы (l) можно получить из других соединений формулы (l) путем "интерконверсии функциональных групп", осуществляемой следующим образом.
(а) Цианогруппа в "Het" может быть превращена в -NHCO2(C1-C4 алкил) в результате осуществления следующей ступенчатой процедуры.
(1) Цианосоединение первоначально обрабатывают C1-C4 алканом, например, метанолом, в кислотных условиях и обычно при кипячении для превращения цианогруппы в -CO2(C1-C4 алкильную) группу.
В другом варианте, в результате гидролиза цианосоединения обычно в щелочных или кислотных условиях образуется соответствующая карбоновая кислота, которую затем можно этерифицировать с помощью C1-C4 алканола в кислотных условиях.
(2) Сложнооэфирную группу превращают в -CONHNH2 группу путем обработки сложного эфира гидразином (предпочтительно гидратом гидразина) в среде соответствующего органического растворителя, например, C1-C4 алканола, такого как изопропанол, при температуре от комнатной до, и предпочтительно, температуры его кипения.
(3) И в конце концов -CONHNH2 группу превышают в целевую -NHCO2 (C1-C4) группу в условиях реакции перегруппировки Куртиуса, например, путем обработки гидразином карбоновой кислоты с
азотистой кислотой, предпочтительно при температуре примерно 0oC, с последующей переработкой полученного промежутка азида и его обработкой C1-C4 алканолом,
предпочтительно при кипячении;
(в) -NHCO2(C1
-C4) заместитель в радикале "Het" можно превратить в аминный заместитель путем гидролиза в щелочных условиях,
например, при использовании водного раствора гидроксида, натрия или калия в
C1 C4 алканоле (например, этаноле или изопропаноле) при кипячении;
(c) Аминозаместитель в
радикале "Het" можно превратить в заместитель формулы -NH(C2-C4 алканоил) путем ацилирования либо C2-C4 алканоил галогенидом либо ангидридом кислоты
формулы (C1-C4 алканоил)2О. Когда используют
алканоил галогенид, то реакцию обычно проводят при температуре от 0oC до комнатной температуры в среде
соответствующего органического растворителя, например, хлористого метилена, и в
присутствии соответствующего акцептора кислоты, например, триэтиламина или пиридина. Реакцию можно также проводить,
используя пиридин и как растворитель, и как акцептор кислоты. Когда используют
ангидрид, то реакцию обычно проводят при температуре до температуре кипения, предпочтительно при 100oC, в
среде приемлемого органического растворителя, например, C2-C4
алкановой кислоты;
(d) Аминозаместитель на радикале "Het" можно превратить в заместитель формулы -NHCHO с
помощью обычных методов, например, формилированием в присутствии уксусно-муравьиного
ангидрида; или
(e) Аминозаместитель на радикале "Het" можно превратить в галозаместитель сначала по
реакции с нитритом натрия в приемлемой водной минеральной кислоте, например, водном
растворе соляной кислоты или серной кислоты, предпочтительно при температуре порядка 0oC, с целью
получения промежуточной диазониевой соли. В результате последующей обработки:
(I)
хлоридом или бромидом меди (l) в радикал "Het" вводятся в качестве заместителей атомы хлорида или брома;
(II) иодидом калия происходит введение в "Het" в качестве заместителя атома иода;
или
(III) фторборная кислота вызывает осаждение фторбората диазония, который можно отфильтровать,
высушить и подвергнуть термическому разложению с целью введения в "Het" фтора в качестве
заместителя.
Все из вышеприведенных реакций являются традиционными, а соответствующие реагенты и условия реакций для их превращений и методы выделения целевых продуктов хорошо известны специалистам по литературе и представленных ниже примерах.
В том случае, когда R1
идентичен радикалу R2, соединения формулы (l) содержит по крайней мере один
хиральный центр и поэтому существуют в виде пары энантиомеров или диастереоизомерных пар энантиомеров. Когда
радикалы R1 и R2 имеют различные значения, соединения формулы (l)
содержат по крайней мере два хиральных центра (*), и поэтому существуют по крайней мере две диастереоизомерные
пары энантиомеров, например
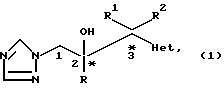
Изобретение охватывает оба отдельных стереоизомера соединений формулы (l), а также их смеси. Разделение можно осуществить с помощью обычных методов, например, методом фракционной кристаллизации, хроматографически или ГПЖХ стереоизомерной смеси основного соединения или соответствующей соли или его производного. Наиболее предпочтительно диастереоизомеры или разделенные диастереоизомерные пары энантиомеров соединений формулы (l), содержащих два хиральных центра, получать из разделенных промежуточных, как проиллюстрировано ниже в разделе "Примеры".
Предпочтительные соединения формулы (l), где R2 представляет собой H, имеют (2R, 3S) конфигурацию, т.е.
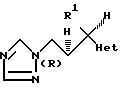
Особенно предпочтительными
отдельными диастереоизомерами являются следующие:
/2R,
3S/-2-/2,4-дифторфенил/-3-/пиридин-2-ил/-1-/1Н- 1,2,4-триазол-1-ил/бутан-2-ол,
/2R, 3S/-2-/2,
4-дифторфенил/-3-/пиридин-4-ил/-1-/1H- 1,2,4-триазол-1 -ил/бутан-2-ол и
/2R, 3S/-2-/2,
4-дифторфенил/-3/пиримидин-4-ил/-1-/1H- 1,2,4-триазол-1-ил/бутан-2-ол; и их фармацевтически приемлемые
соли.
Фармацевтически приемлемые соли легко получать путем смещения растворов, содержащих эквимолярные количества свободного основания и выбранной кислоты. Соль обычно выпадает из раствора в осадок и ее можно отфильтровать, или ее можно извлечь путем испарения растворителя.
Соединения формулы (l) и их соли являются антифунгицидными агентами, пригодными для лечения или профилактики грибковых заболеваний у животных, а также людей. Например, они могут быть использованы для лечения топических фунгицидных инфекций у человека, вызванных помимо других организмов такими классами как Candida, Trichophyton, Microsporcem или Epidermophyton или микозных инфекций, вызванных Candida albicans (например, ). Их можно также использовать для лечения хронических грибковых инфекций, вызванных, например, Candida albicans, Criptococcus neoformans, Aspergillus flavus, Aspergillus furmigatus, Coocidioides, Paracoccidioides, Histoplasma или Blastomyces.
Соединения изобретения, как было установлено, обладают неожиданно высокой эффективностью против клинически важных грибков Aspergillus sp.
Оценку противогрибкового действия in vitro соединений изобретения можно провести методом определения минимальной ингибирующей концентрации (м.и.к.), которая представляет собой концентрацию испытуемых соединений в соответствующей среде, при которой прекращается рост конкретных микроорганизмов. На практике, ряд агаровых пластинок, каждая из которых содержала испытуемое соединение в определенной концентрации, инокулировали стандартной культурой, например, Candida albicans и затем каждую пластинку выдержали в течение 48 ч при 37oC. Затем пластинки изучали на наличие или отсутствие роста грибков и фиксировали соответствующее значение м.и.к. Другие микроорганизмы, используемые в таких опытах, могут включать Aspergillus fumigatus, Trichophyton spp, Microsporum spp, Epidermophuton flaccosum, Coccidioides immitis и Torulopsis glabrata.
Оценку соединений данного изобретения in vivo можно осуществлять, вводя внутрижелудочно или внутривенно или орально последовательные дозировки вещества мышам, инокулированных, например, штампом Candida albicans или Aspergillus fumigatus. Активность основана на выживании обратной группы мышей после смерти необработанной группы мышек. Уровень дозы, при которой соединение обеспечивает 50% защиты от летального эффекта инфекции (РД50), фиксируется.
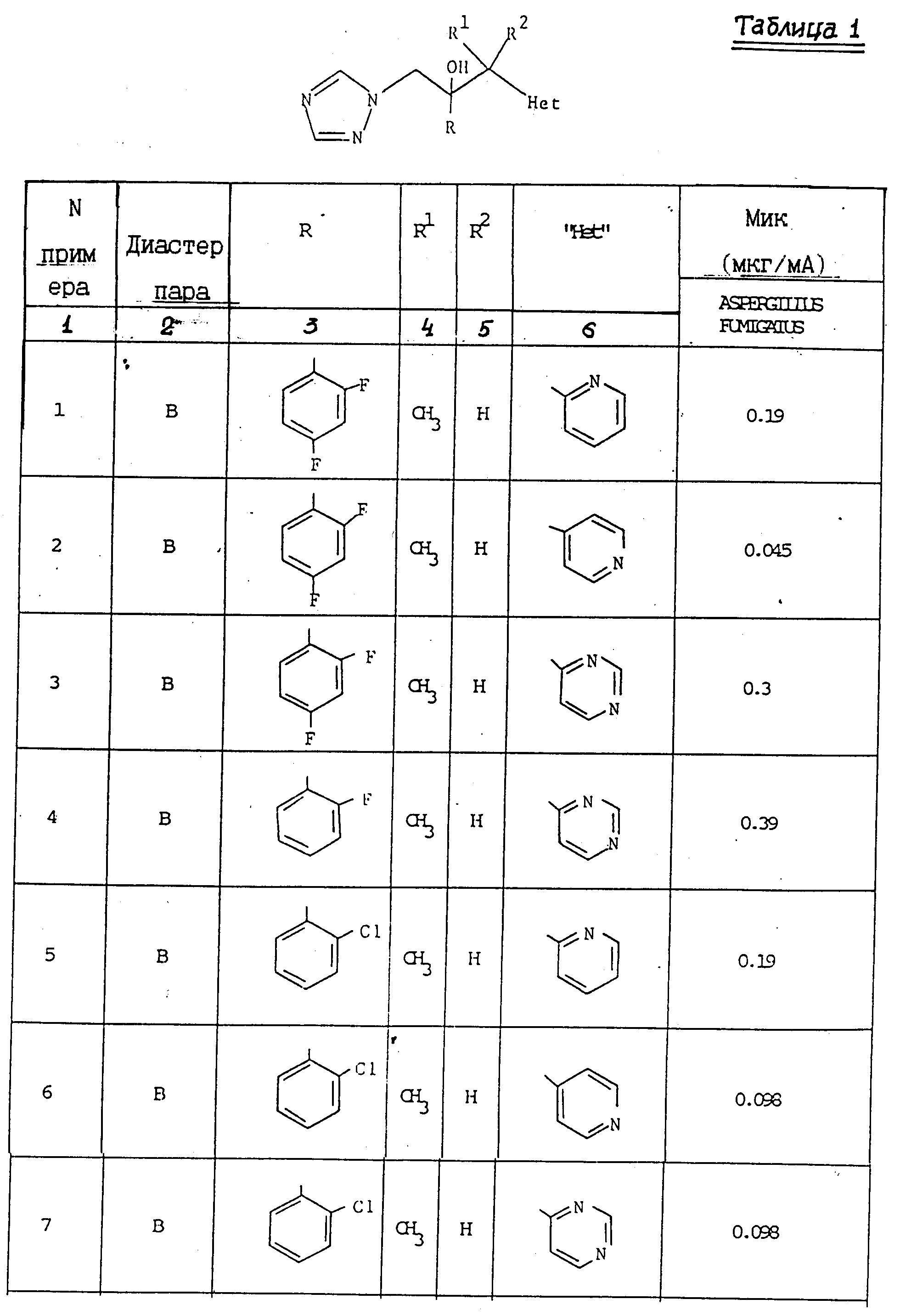
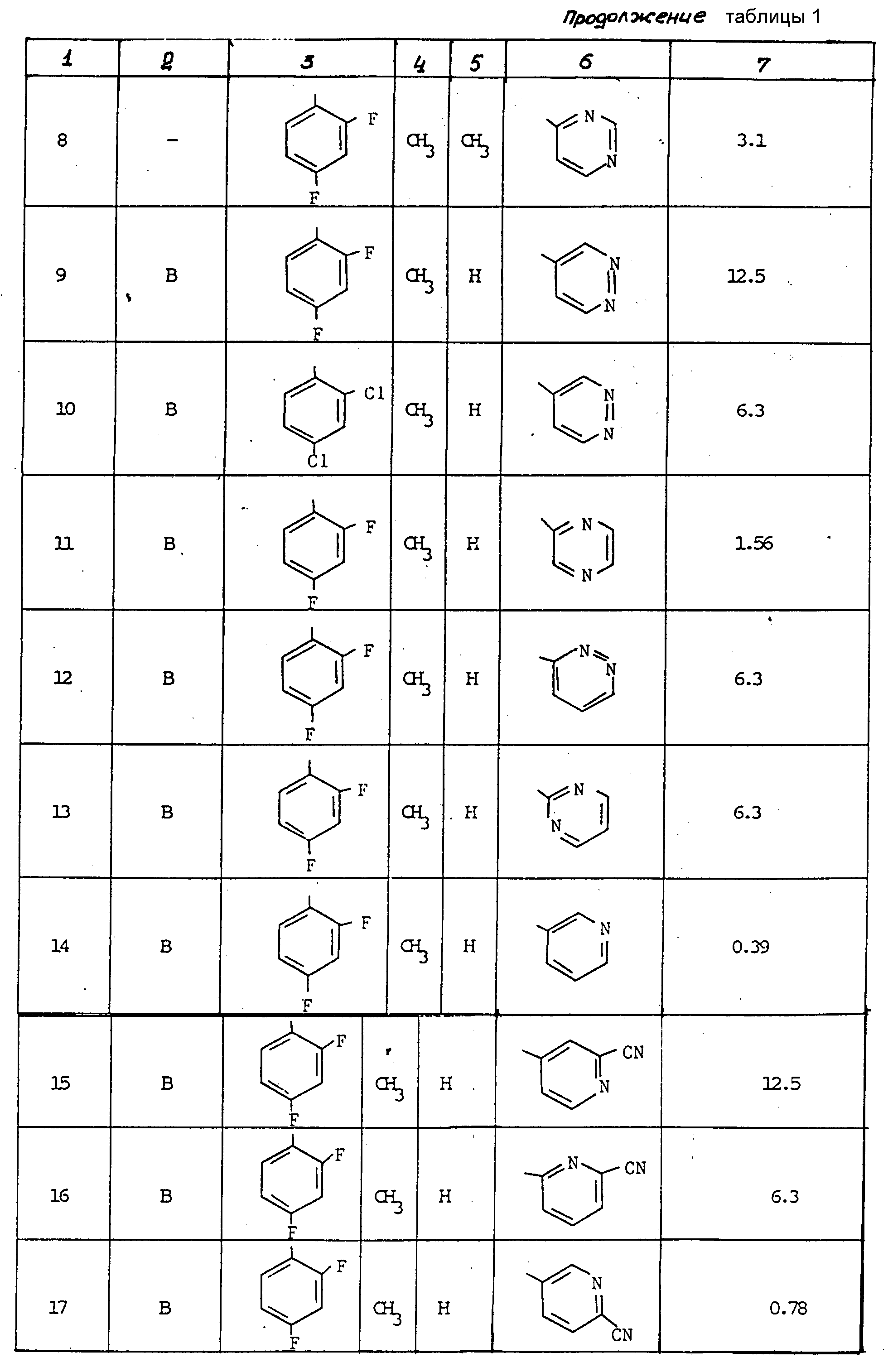
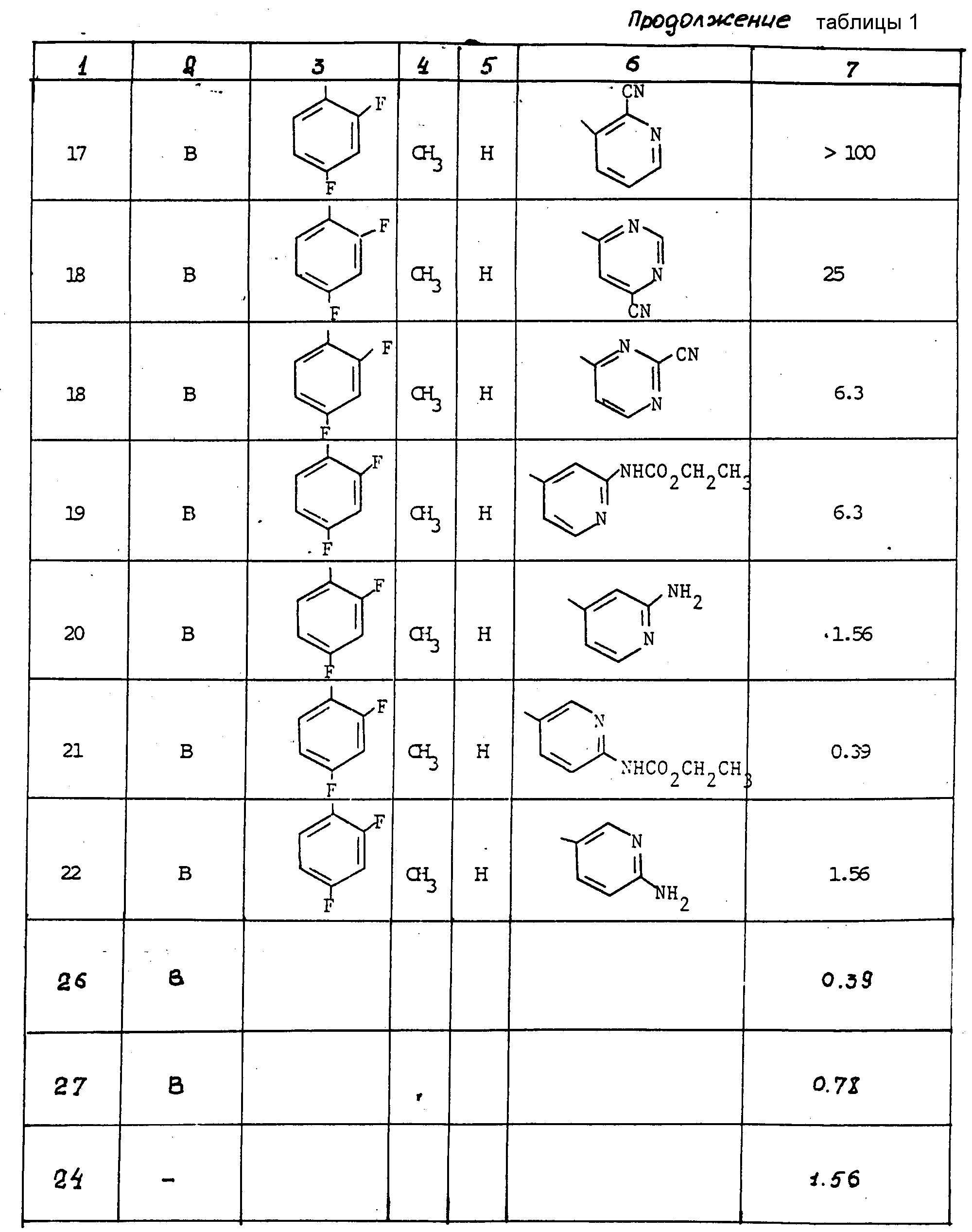
Антифунгицидная активность соединений в отношении Aspergillus fumigatus была измерена in vitro по методу, изложенному выше. Результаты представлены в табл. 1.
Для лечения людей антифунгицидные соединения формулы (l) и их соли можно вводить самостоятельно, но как правило их надо вводить в смеси с фармацевтическим носителем, выбранным в зависимости от выбранного способа введения и принятой фармацевтической практики. Например, их можно вводить орально в форме таблеток, содержащих такие ингредиенты, как крахмал или лактоза, или в форме капсул либо самостоятельно либо в смеси с носителями, или в форме элексиров или суспензий, содержащих отдушки или окрашивающие агенты. Их можно вводить парэнтерально, например, внутривенно, внутримышечно или подкожно. Для парэнтерального введения их лучше всего использовать в форме стерильного водного раствора, который может содержать другие вещества, например, достаточно солей или глюкозы, чтобы сделать раствор изотопным крови.
Для орального парэнтального введения в человеческий организм дневной уровень дозы антифунгицидных соединений формулы (l) и их солей будет составлять от 0,01 до 20 кг/мг (единоразово или в дробных дозах) при введении либо орально либо парэнтерально. Таким образом, таблетки или капсулы соединений формулы (l) будет содержать от 5 мг до 0,5 г активнодействующего соединения для введения единоразово или дважды или большее число раз в день. В любом случае врач определит необходимую дозировку, которая будет наиболее подходящим для отдельного пациента и которая будет зависеть от возраста, веса и чувствительности конкретного пациента. Вышеприведенные дозы даны как усредненный пример; безусловно, могут быть отдельные случаи, когда оптимальными будут более высокие или низкие дозировки, они также входят в объем притязаний данного изобретения.
В другом варианте, антифунгицидные соединения формулы (l) можно вводить в форме суппозиториев или свечей, или их можно наносить наружно в виде лосьенов, растворов, кремов, мазей или пудры. Например, они могут быть включены в состав крема, состоящего из водной эмульсии полиэтиленгликолей или жидкого парафина; или их можно вводить, в концентрации от 1 до 10% в состав мази, содержащей белый воск или белый мягкий парафин в качестве основы вместе с такими стабилизаторами, какие могут быть необходимы.
Кроме того, было установлено, что соединение формулы (l), где R1 и R2 представляют собой H, а радикалы R или "Het" имеют те же значения, что указаны выше для формулы (l), обладают антифунгицидным действием в животных, и что они особенно эффективны против грибка Aspergillus sp.
Таким образом, данное изобретение относится также к фармацевтическим композициям, содержащим соединение формулы (l) или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем.
Далее изобретение относится к соединениям формулы (l) или их фармацевтически приемлемым солям для использования в качестве медикаментов, в частности, в качестве антифунгицидного агента.
Изобретение также относится к использованию соединений формулы (l) или их фармацевтически приемлемым солям или их композициям для получения антифунгицидных агентов.
Кроме того, изобретение включает способ лечения животных (включая людей) с целью излечения или предотвращения грибковой инфекции, который включает обработку указанного живого организма эффективным количеством соединения формулы (l) или фармацевтически приемлемой солью или композицией на его основе.
Данное изобретение также включает любые новые промежуточные соединения, раскрытые в нем, такие как соединения формулы (IV), (VI), (IX) и (X).
Данное изобретение проиллюстрировано с помощью следующих примеров, в которых все значения температуры даны вoC.
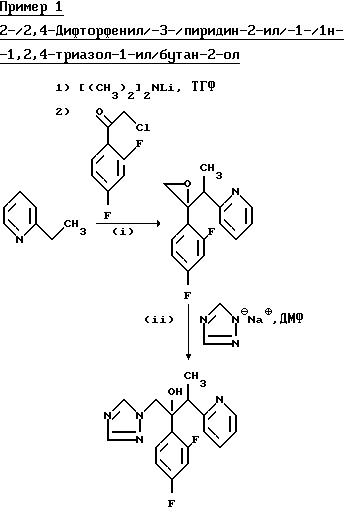
(l) 2-/2,4-Дифторфенил/-2-/1-пиридин-2-ил/этил/оксиран.
К раствору диизопропиламина (3, 18 г) в сухом тетрагидрофуране (50 мл) при перемешивании добавили H-бутиллитий (19,7 мл 1,6 M раствора в гексане) при 70oC под атмосферой сухого азота. Раствор перемешивали при температуре -70oC в течение 0,17 ч, затем 0,17 ч при 0oC, а затем его повторно охлаждали до -70oC. 2-Этилпиридин (3,37 г) добавляли в течение 0,08 ч и образовавшийся красный раствор перемешивали при -70oC в течение 0,33 ч, а затем с помощью шприца добавляли его при перемешивании к раствору 2-хлор-2',4' дифторацетофенона (5,00 г) в сухом тетрагидрофуране (50 мл) при -70oC. Раствор перемешивали при -70o в течение 3 ч, а затем при комнатной температуре в течение 18 ч. Добавили воду (4 мл) и раствор выпаривали. Оставшееся масло обработали водой (80 мл) и дихлорметаном (100 мл). Органический слой отделили, промыли водой (80 мл), а затем проэкстрагировали 2H соляной кислотой (2 х 80 мл). Объединенные кислотные экстракты подщелочили до pH 12 с помощью 2H раствора гидроксида натрия и проэкстрагировали дихлорметаном (3 х 75 мл). Объединенные органические фракции сушили (Na2SO4), выпаривали и остаток подвергали хроматографии на силикагеле. После элюирования этилацетатом, соединения и выпаривания соответствующих фракций получили указанное соединение (2,25 г) в виде желтого масла, которое использовали непосредственно на следующей стадии.
(II) 2-/2,4-Дифторфенил/-3-/пиридин-2-ил/-1-/1H-1,2,4-триазол- 1-ил/бутан-2-ол.
Смесь продукта, полученного в части (1), (2,20 г) и натриевой соли 1H-1,2,4-триазола (1,53 г) в N,N-диметилформамиде (15 мл) нагревали при 60o при перемешивании в течение 18 ч, а затем выпаривали. Добавляли воду (50 мл) и смесь проэкстрагировали этилацетатом (3 х 50 мл). Объединенные экстракты осушили (Na2SO4), выпаривали и остаток подвергли хроматографической очистке на силикагеле. После элюирования этилацетатом, соединения и выпаривали соответствующих фракций получили указанное соединение, диастереозиомерную пару A (0,93 г), Tпл146-148o (из простого эфира).
Результаты анализа,
Найдено:
C 61,69
H 4,73
N 16,88
По расчету для C17H16F2N4
O:
C 61,
81
H 4,88
N 16,96
После дополнительного элюирования, соединения и выпаривания соответствующих фракций получили указанное соединение, диастереоизомерную пару
B, (0,63 г.),
Tпл151-152o (из простого эфира).
Результат анализа,
Найдено:
C 61,68
H 4,79
N 17,01
По расчету для
C17
H16F2N4O:
C 61,81
H 4,88
N 16,96
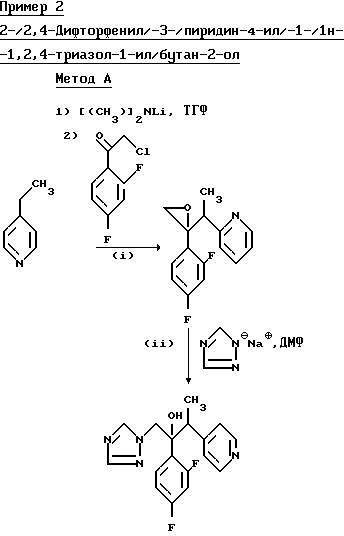
(l) 2-/2,4-Дифторфенил/-2-/1- пиридин-4-ил-этил/оксиран.
Диизопропиламид лития получили путем добавления н-бутил-лития (19,7 мл 1,6 М раствора в гексане) к раствору диизопропиламина (3,18 г) в сухом тетрагидрофуране (50 мл), и полученный раствор последовательно обработали 4-этилпиридином (3,37 г) и раствором 2-хлор-2',4'-дифторацетофенона (5,00 г) в сухом тетрагидрофуране (50 мл) согласно методике, описанной в примере 1 (l). Обработка реакционной смеси как указано выше позволила получить указанное соединение (1,05 г) в виде желтого масла, которое непосредственно использовали на следующей стадии.
(II) 2-/2,4-Дифторфенил/3--/пиридин-4-ил/-1-/1H-1,2,4-триазол-1-ил/ бутан -2-ол.
В результате обработки продукта,
полученного в части
(1) (1,02 г), натриевой солью 1H-1,2,4-триазола (0,71 г) в N,N-диметилформамиде (10 мл) по методике примера 1 (II) с последующей хроматографической очисткой сырого продукта на
силикагеле с
использованием дихлорметана/метанола (93:3) в качестве элюента после соединения и выпаривания соответствующих фракций получили указанное соединение, диастереоизомерную пару А (0,22 г),
Тпл.
161-163o (из простого эфира)
Результаты анализа,
Найдено:
C 61,87
H 4,89
N 16,96
По расчету для C17H16F2N4O:
C 61,81
H 4,88
N 16,96
После дополнительного элюирования смесью дихлорметан/ метанол (97:3), соединения и выпаривания
соответствующих
фракций получили указанное соединение, диастереоизомерную пару B, (0,35 г), Тпл. 156-158o (из простого эфира).
Результаты анализа,
Найдено:
C
61,79
H 4,86
N 17,32
По расчету для C17H16F2N4O:
C 61,81
H 4,88
N 16,96
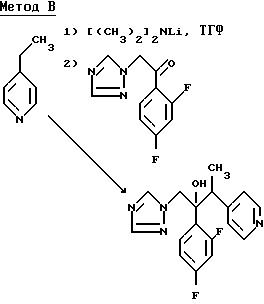
2-/2,4-Дифторфенил/-3-/пиридин/4/ил/-1-/1H-1,2,4-триазол-1- ил/бутан-2-ол.
Раствор диизопропиламида лития приготовили так, как описано в примере 1(l) ЕРА-0357241 из диизопропиламина (40,4 г) и н-бутиллития (160 мл 2.5 М раствора в гексане) в сухом тетрагидрофуране (800 мл) под атмосферой сухого азота. К этому раствору при -70o добавили 4-этилпиридин (42,8 г), по каплям, при перемешивании, в течение 0,17 ч. Раствор перемешивали при -70o в течение 0,33 ч, а затем добавили в течение 0,33 ч раствор 1-/2,4-дифторфенил/2-/1H-1,2,4-триазол-1-ил/этанола (89,2 г) в сухом тетрагидрофуране (350 мл). Раствор перемешивали при -70oС еще в течение 0, 75 ч а затем по каплям добавили уксусную кислоту (40 мл). Раствору дали нагреться до комнатной температуры и разбавили водой. Смесь проэкстрагировали трижды простым эфиром и объединенные экстракты промыли водой. Водные фракции проэкстрагировали один раз этилацетатом, органические экстракты объединили, осушили (Na2SO4) и выпарили. Остаток растворили в кипящем дихлорметане, добавили равный объем простого эфира, а затем раствору дали охладиться. Выпавший осадок отфильтровали и получили исходный кетон (17,5 г). Фильтрат выпарили и остаток подвергли хроматографической очистке на силикагеле. После первичного элюирования смесью этилацетат/гексан (1: 1) получили дополнительное количество исходного кетона. В результате дальнейшего элюирования этилацетатом получили фракции, содержащие указанное соединение, диастереоизомерную пару А (без дальнейшей обработки). Затем растворитель заменили на этилацетат/метанол (19:1) и элюирование продолжали до получения чистых фракций, содержащих указанное соединение, диастереоизомерную пару B. Эти фракции соединили, выпарили и остаток перекристаллизовали из смеси дихлорметана, простого эфира, в результате чего получили указанное соединение, диастереоизомерную пару B, (20,5 г), Тпл. 155-157oС (ЯРМ/300 МГц) - спектр идентичен ЯМР-спектру, полученному для образца диастереоизомерной пары B, полученной как описано в ЕРА-0357241.
После перекристаллизации из ацетонитрила получили полиморф, Тпл. 165-166,5oС.
Результаты анализа,
Найдено:
C 61,69
H 4,85
N 16,85
По расчету для C17H16F2N4O:
C 61,81
H 4,88
N 16,96
Рентгеноскопическая кристаллография подтвердила стереохимию диастереоизомерной пары B, как рацемической смеси 2R, 3S) и (2S, 3R) диастереоизомеров.
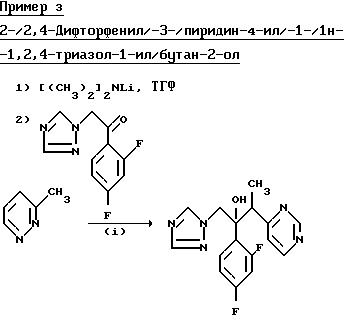
К раствору диизопропиламина (1,01 г) в сухом тетрагидрофуране (30 мл) при -70oС, под атмосферой сухого азота, при перемешивании добавили н-бутиллитий (4,0 мл 2,5 М раствора в гексане). Раствор перемешивали при -70oС в течение 0,17 ч, затем 0,17 ч при 0oС, после чего повторно охладили до -70oС. Добавили 4-этилпиримидин (1,08 г), раствор перемешивали при -70oС в течение 0,75 ч. В течение 0,17 ч добавили раствор 1-/2,4-дифторфенил/-2-/1H-1,2,4-триазол-1-ил/этинона (2.23 г) в сухом тетрагидрофуране (30 мл). Раствор перемешивали при -70oС в течение 1 ч, а затем добавили уксусную кислоту (1 мл). Раствору дали нагреться до комнатной температуры, а затем разбавили водой. Смесь трижды проэкстрагировали этилацетатом, и объединенные экстракты промыли водой и осушивали (Na2SO4). Растворитель выпарили и остаток подвергли хроматографической очистке на силикагеле. После первичного элюирования смесью этилацетат/гексан (3:2) получили восстановленный исходный кетон. После дальнейшего элюирования этилацетатом, соединения и выпаривания соответствующих фракций получили указанное соединение, диастереоизомерную пару А, (0,305 г), Тпл. 114-115,5oС (из простого эфира/гексана).
Результаты
анализа,
Найдено:
C 57,76
H 4,45
N 21,26
По расчету для C16H15F2N5O:
C 58,00
H 4,56
N 21,14
В результате дальнейшего элюирования этилацетатом/метанолом (19:1) соединения и выпаривания соответвтующих фракций получили указанное соединение, диастереоизомерную пару B, (0,215),
Тпл. 104-105oС (из простого эфира/гексана).
Результаты анализа,
Найдено:
C 57,63
H 4,44
N 21,36
По расчету для C16H15F2N5O:
C 58,00
H 4,56
N 21,14
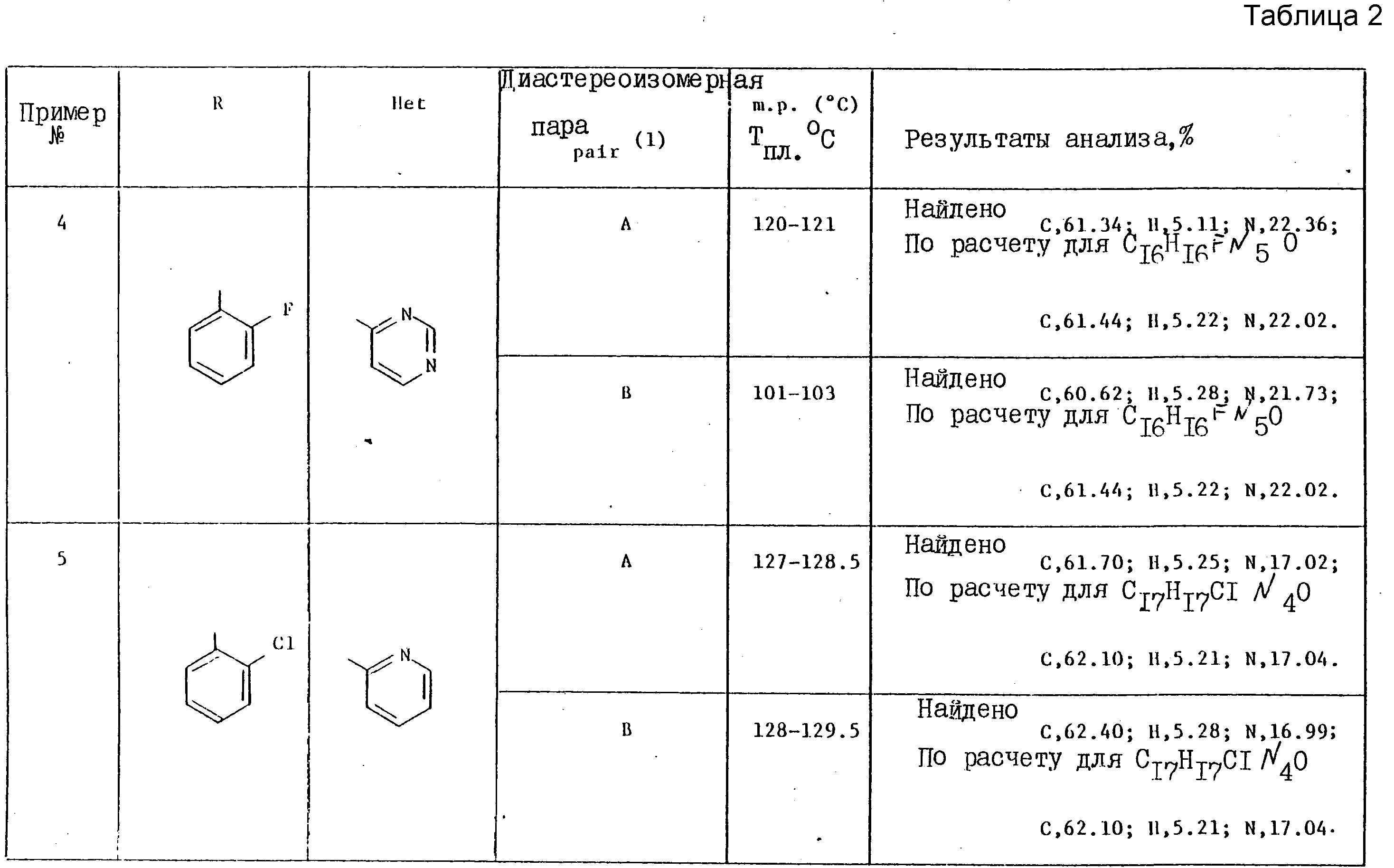
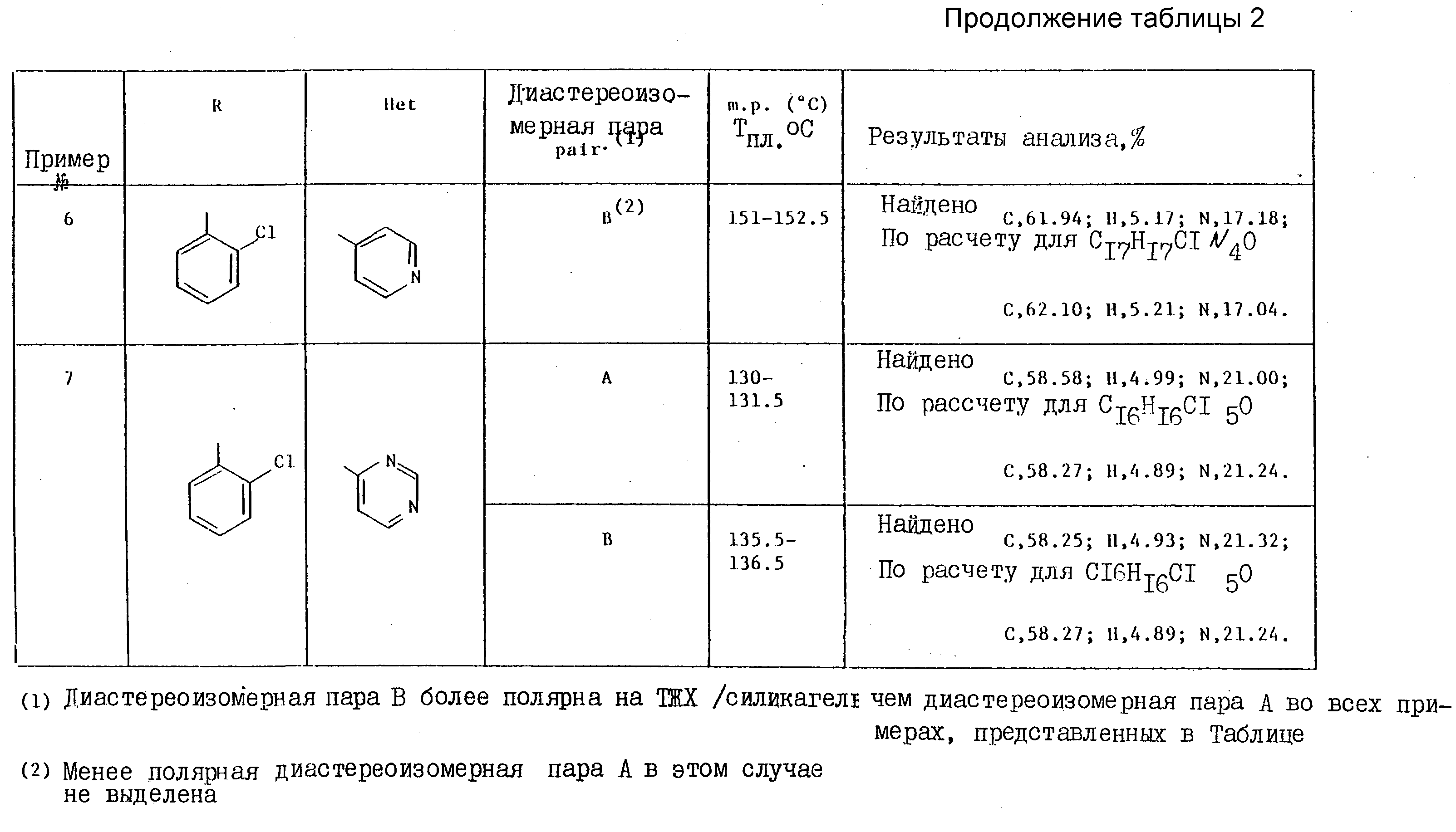
Примеры 4-7. Представленные ниже в таблице примеры соединений общей формулы
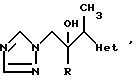
были получены методами, аналогичными тем, что использованы в примере 2, путем обработки соответствующего этилгетероцикла диизопропиламидом лития, с последующей реакцией полученного карбаниона in situ с соответствующим производным 1-арил-2-/1Н- 1,2,4-триазол-1-ил/этанона (табл. 2).
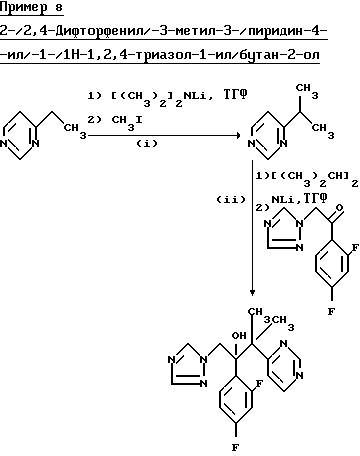
(l) 4-/1-Метилэтил/пиримидин.
Раствор диизопропиламида лития приготовили так, как описано в примере l(l) ЕРА-0357241 из диизопропиламина (6,88 г) и н-бутиллития (27,0 мл 2,5 М раствора в гексане) в сухом тетрагидрофуране (180 мл) под атмосферой сухого азота. К этому раствору добавили по каплям, при -70oC, в течение 0,17 ч раствор 4-этилпиримидана (7,35 г) в сухом тетрагидрофуране (20 мл). Раствор перемешивали при -70oC в течение 0,75 ч, а затем добавляли иодометан (11,60 г). Смесь перемешивали еще в течение 3 ч, а затем нагрели до комнатной температуры. Добавили воду и раствор выпарили до малого объема, затем обработали этилацетатом и водой. Органический слой отделили, водный слой трижды проэкстрагировали этилацетатом, и органические фракции обхекдинили и осушили (Na2SO4). После выпаривания растворителя получили масло, которое подвергли очистке на хроматографической колонке с силикагелем, используя в качестве элюента дихлорметан/простой эфир (9:1). Фракции, содержащие продукт, объединили и выпарили, оставшееся масло перегнали и получили указанное соединение, (3,14 г), Тпл. 52-56oC при 15 мм.
(II) 2-/2,4-Дифторфенил/-3-метил-3-/пиримидин-4-ил/-1- /1Н-1,2, 4-триазол-1-ил/бутан-2-ол.
В результате обработки продукта, полученного в части (1) (2,46 г) диизопропиламидом лития (0,02 моля) в сухом тетрагидрофуране, с последующей обработкой 1-/2, 4-дифторфенил/-2-/1Н-1,2,4-триазол-1-ил/этанолом (4,49 г) по методике Примера 3 получили указанное соединение, (0,185 г), Тпл. 126-127oС (из простого эфира).
Результаты анализа,
Найдено:
C 59,15
H 4,87
N 20,41
По расчету для C17H17F2N5O:
C 59,12
H 4,
96
N 20,28
Пример 9. 2-/2,4-Дифторфенил/-3-/пиридазин- 4-ил/-1-/1Н-1,2,4- триазол-1-ил/бутан- 2-ол.
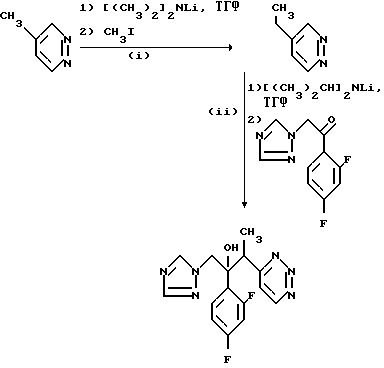
(l) 4-Этилпиридазин.
Раствор диизопропиламида лития приготовили как описано в примере 1 (1) ЕРА-057241 из диизопропиламина (17,9 г) и н-бутиллития (70,4 мл 2,5 М раствора в гексане) в сухом тетрагидрофуране (300 мл) под атмосферой сухого азота. К этому раствору добавили при -70oС 4-метилпиридазин, по каплям, при перемешивании, обеспечивая температурный режим не выше -60oС. Медленно, при перемешивании добавили иодометал (27,25 г), раствор перемешивали при 70oС в течение 1 ч, а затем дали ему нагреться до комнатной температуры. Добавили воду и раствор выпаривали до малого объема. Раствор трижды проэкстрагировали дихлорметаном и объединенные экстракты осушили (Na2SO4) и выпаривали. Остаток подвергли хроматографической очистке на силикагеле, используя в качестве элюента этилацетат. Фракции, содержащие продукт, объединили и выпарили, и оставшееся масло перегнали, в результате чего получили указанное соединение, (10,4 г) Ткип. 65-66oС при 0,1 мм.
ЯМР (300 МГц).
δ (CDCl3) 1.21 (T, 3H, j= 7,6 Гц, CH2CH3), 2,61 (кв, 2H, j=7,6 Гц, CH2CH3), 7,24 (м, 1H, Наром) 8.97 (м, 2H, Наром) млн. долей.
(ll) 2-/2, 4-Дифторфенил/-3-/пиридазин-4-ил/-1-/1Н-1,2,4- триазол-1-ил/бутан-2-ол.
Раствор диизопропиламида лития приготовили как описано в примере 1(l) ЕРА-0357241 из диизопропиламина (2, 02 г) и н-бутиллития (8,0 мл 2,5 М раствора в гексане) в сухом тетрагидрофуране (60 мл).
К этому раствору добавили по каплям, при перемешивании, при -70oС 4-этилпиридазин (2,16 г). Желтый раствор перемешивали в течение 0,4 ч при -70oС; а затем добавили раствор 1-/2,4-дифторфенил/-2-/1Н-1,2,4-триазол-1-ил/этанона (4,46 г) в сухом тетрагидрофуране (20 мл), поддерживая температуру ниже -65oС. Раствор перемешивали еще в течение 1 ч при этой температуре, а затем добавили уксусную кислоту (1 мл). Раствору дали нагреться до комнатной температуры и разбавили его водой. Смесь трижды проэкстрагировали этилацетатом и объединенные органические эекстракты промыли водой и осушили (Na2SO4). После выпаривания растворителя получили сырой продукт. Далее сырой продукт получили после экстракции объединенных водных фракций дихлорметаном, полученные таким образом обе части сырого продукта объединили и подвергли фроматографической очистке на силикагеле. После элюирования смесью дихлорметан/метанол (50: 1) сначала получили исходный кетон. Дальнейшее элюирование тем же растворителем позволило получить после соединения и выпаривания соответствующих фракций указанное соединение, диастереоизомерную пару А, (0,98 г), Тпл. 172-174oС (из дихлорметан/простой эфир).
Результаты анализа:
Найдено:
C 57,80
H 4,57
N 21,08
По расчету для C16H15F2N5O:
C 58,00
H 4,
56
N 21,14
После дальнейшего элюирования смесью дихлорметан/метанол (50:1), соединения и выпаривания соответствующих фракций получили указанное соединение, диастереоизомерную пару B,
(1.58 г/, Тпл.
187-188oС (из ацетонитрила).
Результаты анализа,
Найдено,
С 58,00
Н 4,54
N 21,14
По расчету для
C16H15
F2N5O:
C 58,00
Н 4,56
N 21,14
Пример 10. 2-/2,4- Дихлорфенил/-3-/пиридазин-4-ил/-1-/1Н-1,2,
4- триазол-1-ил/бутан-2-ол.
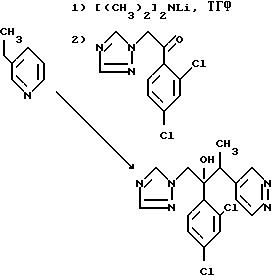
В результате обработки 4-этилпиридддазина (2,16 г) диизопропиламидом лития (0,02 моля) в сухом тетрагидрофуране, а затем 1-/2,4- дихлорфенил/-2-/1Н-1,2,4-триазол-1-ил/этаноном (5,12 г) по методу, описанному в примере 9(ll), получили указанное соединение, диастереоизомерную пару А, (1,24 г), Тпл 174-177oC.
Результаты анализа,
Найдено:
C 52,22
H 4,12
N 19,05
По расчету для C16H15CL2N5O
C 52,75
H 4,15
N 19,23
и указанное соединение, диастереоизомерную пару B, (1.45 г), Тпл. 173-176oC.
Результаты анализа,
Найдено:
C 52,41
H 4,08
N 18,85
По расчету для C16H15Cl2N5O
C 52,75
H 4,
15
N 19,23
Пример 11. 2-/2,4-Дифторфенил/- 3-/пиразин-2-ил/-1-/1H-1,2,4- триазол-1-ил/бутан-2-ол.
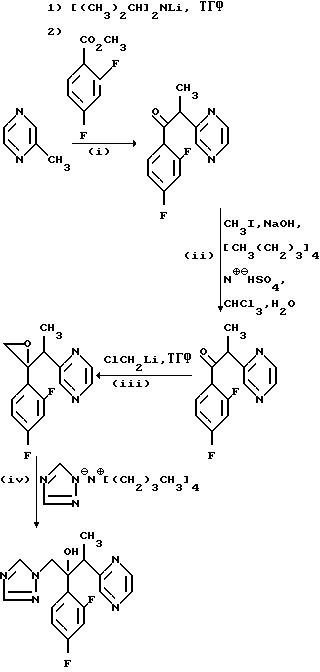
(l)1-/2,4-Дифторфенил/-2-/пиразин-2- ил/этанон.
Раствор диизопропиламида лития приготовили, используя н-бутиллитий (20 мл, 2.5 М раствора в гексане) и диизопропиламин (5,06 г) в сухом тетрагидрофуране (100 мл) под атмосферой сухого азота, как описано в примере 1(l). К этому раствору при -70oС добавили 2-метилпиразин (4,70 г) и образовавшийся раствор пурпурного цвета перемешивали при -70oС в течение 0,5 ч. Раствор метил 2,4-дифторбензоата (8,60 г) в сухом тетрагидрофуране (75 мл) добавляли в течение 0,5 ч и перемешивание продолжали при -70oС еще в течение 0,5 ч. Добавили уксусную кислоту (10 мл) и температуре дали возможность повыситься до комнатной. Раствор разбавили водой и pH довели до 7 с помощью бикарбоната натрия. Смесь трижды проэкстрагировали этилацетатом и объединенные органические экстракты промыли водой и осушили (Na2SO4). Растворитель выпарили и остаток подвергли хроматографической очистке на силикагеле. В результате элюирования смесью этилацетат/гексан (3: 7), соединения и выпаривания соответствующих фракций получили твердое вещество, которое перекристаллизовали из гексана и получили указанное целевое соединение, (5,90 г), Тпл. 107-108oС.
Результаты анализа,
Найдено:
C 61,50
H 3,32
N 12,02
По расчету для C12H8F2N2O:
C 61,54
H 3,44
N 11,96
(ll) 1-(2,
4-Дифторфенил/-2-/пиразин-2-ил/пропанол-1-он.
Раствор гидроксида натрия (1,98 г) в воде (40 мл) по каплям добавили к перемешиваемому, охлажденному на льду раствору продукта, полученного в части (1) (5,80 г), иодометану (8,79 г) и кислому сульфату тетра-н-бутиламмония (8,40 г) в хлороформе (40 мл). Смесь интенсивно перемешивали при комнатной температуре в течение 3 ч, а затем разбавили водой и дихлорметаном. Добавили уксусную кислоту (3 мл) и pH водного слоя довели до 7 с помощью бикарбоната натрия. Органический слой отделили, дважды промыли водой и осушили (Na2SO4). После выпаривания растворителя получили сырой продукт в виде масла (5,57 г), использовали без дополнительной очистки (наличие 10% исходного материала (продукта части (1) идентифицировано методом ЯМР-спектроскопии).
(lll) 2-/2,4-Дифторфенил/-2-/1-/пиразин-2-ил/этил-оксиран.
н-Бутиллитий (9.3 мл 2,5 М раствора в гексане) (при перемешивании добавили к раствору продукта, полученного в части (ll) (5,50 г), и бромхлорметана (3Ю16 г) в сухом тетрагидрофуране (125 мл), охлажденному до -70oС, под атмосферой сухого азота с такой скоростью, что температура смеси не повышалась свыше -65oС. Раствор перемешивали при -70o в течение 6 ч, а затем при комнатной температуре в течение 18 ч. Раствор разбавили водой и трижды проэкстрагировали этилацетатом. Объединенные органические экстракты промыли водой и осушили (Na2SO4). После выпаривания растворителя получили масло, которое пропустили через хроматографическую колонку с силикагелем. После элюирования смесью этилацетат/гексан (1:5) получили масло (4,80 г), которое по данным ЯМР-спектроскопии содержало са. 70% указанного соединения вместе с примесями. Этот продукт использовался непосредственно без дополнительной очистки.
(IV) 2/2, 4-Дифторфенил/-3-/пиразин-2-ил/-1-/1H-1,2, 4-триазол-1-ил/бутан--2-ол.
Тетра-н-бутиламмониевую соль 1H-1,2,4-триазол (см. патент США 4 259 505) (5,45 г) при перемешивании добавили к раствору продукта, полученного в части (III) (2,30 г) в сухом тетрагидрофуране (25 мл) при комнатной температуре, и перемешивание продолжали с течение 4 дней. Затем растворитель выпаривали и остаток обработали водой и этилацетатом. Добавили уксусную кислоту (1 мл) и смесь профильтровали через Avicel (торговая марка для фильтра на основе целлюлозы). Органический слой отделили, трижды промыли водой и осушили (Na2SO4). Растворитель выпарили и остаток пропустили через хроматографическую колонку с силикагелем. Колонку сначала элюировали смесь этилацетат/гексан (3: 2) для удаления примесей. В результате последующего элюирования смесью этилацетат/гексан (9:1), соединения и выпаривания соответствующих фракций получили указанное соединение, диастереоизомерную пару А. (0,85 г), Тпл. 107-109oС (из дихлорметан/гексана).
Результаты анализа,
Найдено:
C 57,76
H 4,44
N 21,31
По расчету
для C16H15F2
N5O
C 58,00
H 4,56
N 21,14
После дополнительного элюирования смесью этилацетат/метанол (19:1), соединения и
выпаривания соответствующих фракций получали
указанное соединение, диастереоизомерную пару B, (0.29 г), Тпл 133-135oС (из смеси дихлорметан/гексан).
Результаты
анализа,
Найдено:
C 57,82
H 4,53
N 21,00
По расчету для C16H15F2N5O
C 58,00
H 4,56
N
21,14
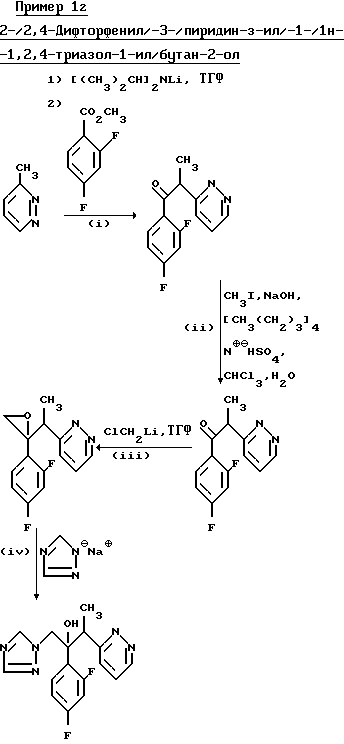
(l) 1-/2,4-Дифторфенил/-2-/пиридазин-3-ил/этанон.
В результате обработки 3-метилпирадазин (4,70 г) диизопропиламидом лития (0,05 моля) в сухом тетрагидрофуране, а затем метил 2,4-дифторбензоатом (8,80 г) согласно методу, описанному в примере 11(l), получили указанное соединение, (3,40 г), Тпл 115,5-117,5oС (из простого эфира).
Результаты анализа,
Найдено:
C 61,69
H 3,40
N 11,
77
По расчету для C12
H8F2N2O
C 61,54
H 3,44
N 11,96
(II) 1-/2,4-Дифторфенил/-2-/пиридазин-3-ил/пропан-1-он.
Метилированием продукта, полученного в части (1) (3,30 г), иодометаном (5,0 г) по методике, описанной в примере 11 (II) получили указанное соединение в виде смолы (2,25 г), которую использовали непосредственно на следующей стадии.
(III) 2-/2,4-Дифторфенил/-2-/1-/пиридазин-3-ил/этил/оксиран.
В результате обработки продукта, полученного в части (II), (2,0 г) бромхлорметаном (1,15 г) и н-бутиллитием (5,28 мл 1,6 М раствора в гексане) по методу, описанному в примере 11(III), получили указанное соединение в виде смелы (1,20 г), которую использовали непосредственно на следующей стадии.
(IV) 2-/2,4-Дифторфенил/-3-/пиридазин-3-/-1-/1Н-1,2,4-триазол-1-ил/бутан-2-ол.
В результате обработки продукта, полученного в части (III), (1.15 г) натриевой солью 1Н-1,2,4-триазол (0.80 г) в N,N-диметилформамиде (15 мл) по методу, описанному в примере 1(ll), с последующей хроматографией сырого продукта на силикагеле с использованием в качестве элюента смеси дихлорметан/метанол (50: 1), после соединения и выпаривания соответствующих фракций сначала получили указанное соединение, диастереоизомерную пару А, (0.35 г), Tпл. 134-135o С (тз простого эфира).
Результаты анализа,
Найдено:
C 58,04
H 4,57
N 20,87
По расчету для
C16H15F2N5O
C 58,00
H 4,56
N 21,14
В результате дальнейшего элюирования тем же растворителем, после соединения и
выпаривания соответствующих фракций получили указанное
соединение, диастереоизомерную пару В, в виде аморфной пены (84 мг).
ЯМР (300 МГц).
(CDCl3) 1,20 (д, 2H, j 7,2 Гц, CH3), 3,95 (кв, 1H, j 7,2 Гц, CHCH3), 4,04 и 4,91 (g, 1H, j 14,2 Гц, CH2); 6,18 (c, 1H, OH), 6,82 (м, 2H, Hаром), 7,67 (м, 1H, Hаром), 7,56 (м, 2H, Hаром), 7,64 (с, 1H, Hаром), 7,94 (с, 1H, Hаром), 9,18 (м, 1H, Hаром) млн. долей.
Пример 13. 2-/2, 4- Дифторфенил/-3- /пиридазин-2-ил/-1-/1Н-1,2,4-триазол-1-ил/бутан-2-ол.
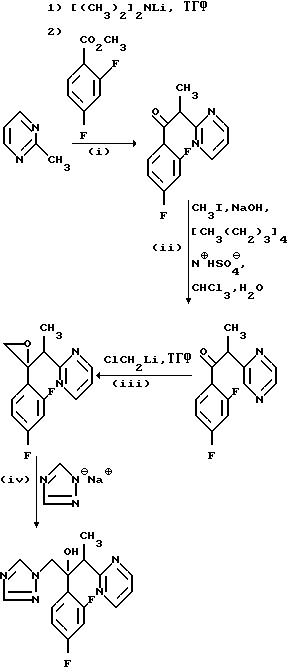
(l) 1-/2, 4-Дифторфенил/-2-/пиридазин-2-ил/этанол
В результате обработки 2-метилпиримидина (8.50 г) диизопропиламидом лития (0.09 моля) в сухом тетрагидрофуране, а затем метил 2,4-дифторбензоатом (15.5 г) по методу, описанному в примере 11/l, получили указанное соединение (3,65 г), Tпл. 86-88oС (из гексана).
Результаты анализа,
Найдено:
C 61,67
H 3,41
N 12,01
По расчету для C12H8F2N2O
C 61,54
H 3,44
N 11,96
(II) 1-/2,
4-Дифторфенил/-2-/пиридизан-2-ил/пропан-1-он.
Путем метилирования продукта, полученного в части (1), (3.50 г) иодометаном (5.32 г) по методу, описанному в примере 11(II), получили указанное соединение (3,30 г), Tпл. 118-119oС.
Результаты анализа,
Найдено:
C 63,17
H 4,18
N 11,02
По расчету для
C13H10F2N2O
C 62,9
H 4,
06
N 11,29
(III) 2-/2,4-Дифторфенил/-2-/1-/пиримидин-2-ил/этил оксиран.
В результате обработки продукта, полученного в части (II), (3,10 г) хлорметиллитием (полученным из бромхлорметана (1,78 г) и 1,6 М раствора н-бутиллития в гексане (8,20 мл) по методу, описанному в примере 11 (III), получили указанное соединение в виде смолы (2,25 г), которую использовали непосредственно на следующей стадии.
(IV) 2-/2,4-Дифторфенил/-3-/пиримидин-2-ил/-1-/1H-1,2, 4-триазол-1-ил/бутан-2-ол.
В результате продукта, полученного в части (III), (0,80 г) натриевой солью 1H-1,2,4-триазол (0,82 г) в N,N-диметилформамиде по методу, описанному в примере 12 (IV), с последующей хроматографией сырого продукта на силикагеле при использовании в качестве элюента этилацетата, после соединения и выпаривания соответствующих фракций получили сначала указанное соединение, диастереоизомерную пару А, (0,26 г), Tпл. 193-195oС (из дихлорметана простого эфира).
Результаты анализа,
Найдено:
C 57,50
H 4,57
N 21,03
По расчету для C16
H15F2N5O
C 58,00
H 4,56
N 21,14
После дальнейшего элюирования
смесью этилацетат/метанол (20:1), соединения и выпаривания
соответствующих фракций получили указанное соединение, диастереоизомерную пару В, (0,055 г), Tпл. 104-106oС (из
простого эфира).
Результаты анализа,
Найдено:
C 57,27
H 4,37
N 20,55
По расчету для C16H15F2N5
O
C 58,00
H 4,56
N 21,14
Пример 14. 2-/2,4-Дифторфенил/-3-/пиридин-3-ил/-1-/1H-1,2,4-триазол-1-ил/бутан-2-ол.
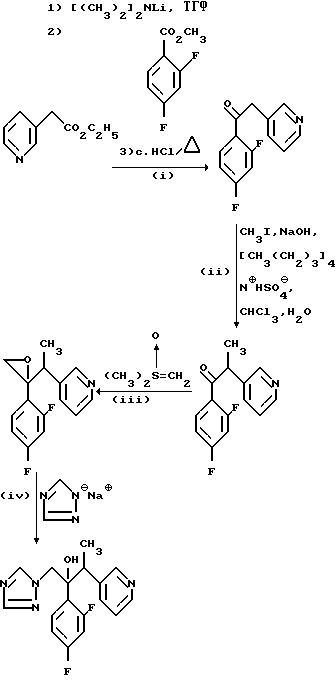
(I) 1-/2,4-Дифторфенил/- 2-/пиридин-3-ил/этанол.
Раствор диизопропиламида лития приготовили, используя н-бутиллитий (66 мл 1,6 М раствора в гексане) и диизопропиламин (10,8 г) в сухом тетрагидрофуране (200 мл), в атмосфере сухого азота, как описано в примере 1 (I). К этому раствору при -79oС, по каплям добавили этил 3-пиридилацетат. Густую смесь перемешивали при -70oС в течение 0,25 ч, а затем добавили в течение 0,05 ч раствор метил 2,4-дифторбензоата (18,36 г) в сухом тетрагидрофуране (100 мл). Охлаждающую баню удалили и смесь перемешивали при комнатной температуре в течение 5 ч. Добавили уксусную кислоту (12 мл) и смесь разбавили водой и этилацетатом. Органический слой отделили, осушили (Na2SO4) и выпаривали, в результате чего получили масло, которое прокипятили с концентрированной соляной кислотой (40 мл) в течение 5 ч. Раствор выпарили, остаток растворили в воде и добавили концентрированный раствор аммиака до са pH 7. Смесь дважды проэкстрагировали атилацетатом и объединенные экстракты промыли рассолом и осушили (Na2SO4 ). После выпаривания растворителя получили масло, которое пропустили через хроматографическую колонку с силикагелем. После элюирования смесью дихлорметан/этилацетат (70:30) получили указанное соединение в виде масла (6,98 г), которое использовали непосредственно на следующей стадии.
(II) 1-/2,4-Дифторфенил/-2-/пиридин--3-ил/пропан-1-он.
Путем металирования продукта, полученного на стадии (I), (5,0 г) иодометаном (7.60 г) по методу, описанному в примере 11 (II), получили указанное соединение в виде масла (3,90 г), которое использовали непосредственно на следующей стадии.
(III) 2-/2, 4-Дифторфенил/-2-1-/пиридин-3-ил/этил оксиран.
Раствор метилида диметилсульфоксония (36,5 мл 0,6 М раствора в тетрагидрофуране) по каплям, при перемешивании добавили к раствору продукта, полученного в части (II), (4,36 г) в тетрагидрофуране (35 мл) при -29oС. Раствору дали нагреться до комнатной температуры и перемешивание продолжали в течение 18 ч, а затем разбавили водой. Смесь проэкстрагировали этилацетатом, и объединенные экстракты осушили (Na2SO4).После выпаривания растворителя получили указанное соединение в виде масла (4,50 г), которое использовали непосредственного на следующей стадии.
(IV) 2-/2,4-Дифторфенил/-3-/пиридин-3-ил/-1-/1H-1,2, 4-триазол-1- ил/бутан-2-ол.
Путем обработки продукта, полученного в части (III), (4,30 г) натриевой солью 1H-1,2,4-триазола (3,0 г) в N,N-диметилформамиде (50 мл) по методу, описанному в примере 1(II), с последующей хроматографией сырого продукта на силикагеле с использованием этилацетата в качестве элюента, соединения и выпаривания соответствующих фракций сначала указанное соединение, диастереоизомерную пару А, (1,13 г), Тпл. 113-114oС (из простого эфира).
Результаты анализа,
Найдено:
C 62,10
H
4,90
N 16,96
По расчету для C17H16F2N4O:
C 61,81
H 4,88
N 16,96
В результате дальнейшего элюирования
смесью этилацетат/метанол ж (20:1), после соединения и выпаривания
соответствующих фракций получили указанное соединение, диастереоизомерную пару В, (1,25 г), Тпл. 115-116oС (из
простого эфира).
Результаты анализа,
Найдено:
C 61,92
H 4,95
N 16,87
По расчету для C17H16F2N4
O:
C 61,81
H 4,88
N 16,96
Пример 15. 2/2,4-Дифторфенил/-3- /2-цианопиридин-4-ил)-1-/1H-1,2,4-триазол- 1-ил/бутан-2-ол.
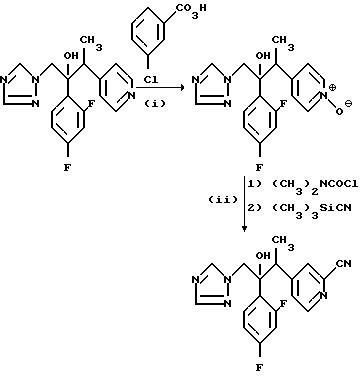
(I) 2/2,4-Дифторфенил/-3-/1-оксидопиридин-4-ил/-1-/1H-1,2,4-триазол- 1-ил/бутан-2-ол.
Раствор 2/2, 4-дифторфенил/-3- /пиридин-4-ил/-1-/1H-1,2, 4-триазол- 1-ил/бутан-2-ол/ диастереоизомерная пара В из примера 2/ (20,0 г) и 85% м/м 3-хлорнадбензойной кислоты (12.3 г) в дихлорметане (250 мл) перемешивали при комнатной температуре в течение 18 ч. Затем добавили еще 3-хлорнадбензойную кислоту (2,50 г) и перемешивание осуществляли в течение 24 ч. Раствор выпарили и остаток растворили в простом эфире. Осадок образовавшийся при стоянии, отфильтровали, и пропустили через хроматографическую колонку с силикагелем. После элюирования смесью дихлорметан/метанол/ 0,88 аммиачный раствор (100: 4: 0.5) получили указанное соединение в виде твердого вещества, (20,00 г), Тпл. 195-198oС.
(II) 2/2,4- Дифторфенил/-3-/2-цианопиридин- 4-ил/-1-/1H-1,2, 4-триазол- 1-ил/бутан-2-ол.
Смесь продукта, полученного в части (I), (20,0 г) и N,N-диметилкарбамоил хлорида (6,80 г) в дихлорметане (250 мл) перемешивали при комнатной температуре в течение 2,5 дней, в результате чего получили прозрачный раствор. Добавили триметилсилил цианид (6,35 г) и перемешивание продолжали еще в течение 48 ч. Затем добавили дополнительные количества N, N-диметилкарбамоил хлорида (1,30 г) и триметилсилил цианида (1,30 г) и раствор перемешивали еще в течение 36 ч. После этого реакционную смесь последовательно промыли 10%-ным раствором карбоната калия, рассолом и осушили (MgSO4). Поле выпаривания растворителя получили твердое вещество, которое перемешали с простым серным эфиром, отфильтровали и получили указанное соединение, (19,2 г), Тпл. 188-189oС.
Результаты анализа,
Найдено:
C 60,89
H 4.24
N 19,44
По расчету для C18H15F2N5O:
C 60,84
H 4,25
N 19,71
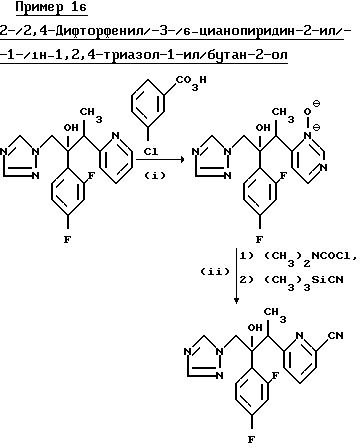
(I) 2-/2, 4-Дифторфенил/-3-/1-оксидопиридин-2-ил/-1- -/1H-1,2, 4-триазол-1-ил/бутан-2-ол.
Раствор 2-/2,4-дифторфенил/-3-/1-пиридин-2-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан- 2-ола (диастереоизомерная пара B из примера 1) (1,60 г) и 85% -ной м/м 3-хлорнадбензойной кислоты (1,60 г) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 36 ч, а затем переработали как описано в примере 15(l), в результате чего получили указанное соединение, (0.92 г), Тпл.159-160oC.
Результаты анализа,
Найдено:
C 59,27
H 4,96
N 16,58
По расчету для C17H16F2N4O2:
C 58,96
H 4,45
N 15,47
(II) 2-/2,
4-Дифторфенил/-3-/6-цианопиридин-2-ил/-1- -/1H-1,2,
4-триазол-1-ил/бутан-2-ол.
Смесь продукта, полученного в части (1), (0,90 г), N,N-диметилкарбамоил хлорида (0,80 г) и триметилсилил цианида (0,80 г) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 7 дней и полученный раствор выпарили. Остаток обработали 5H соляной кислотой (10 мл) и смесь перемешивали на ультразвуковой бане в течение 0,05 ч, в результате чего получили прозрачный раствор. Осадок, образовавшийся при стоянии, отфильтровали, промыли ацетоном, а затем простым эфиром и высушили, в результате получили указанное соединение в виде хлористоводородной соли, (0.28 г), Тпл. 219oС (с разложением).
Результаты анализа,
Найдено:
C
55,19
H 4,10
N 18,00
По расчету для C18H15F2N5O•HCl:
C 55,18
H 4,12
N 17,87
Кислотный
фильтр подщелочили (ca. pH 8) с помощью 0,88
раствора аммиака и раствор проэкстрагировали дихлорметаном. Органический экстракт осушили (MgSO4) и выпарили. Остаток растерли с простым эфиром
и профильтровали, в результате чего получили
указанное соединение в виде свободного основания, (0,13 г), Тпл.144-146oС.
Результаты анализа,
Найдено:
C 60,85
H 4,25
N 19,
71
По расчету для C18H15F2N5O:
C 60,48
H 4,17
N 19,90
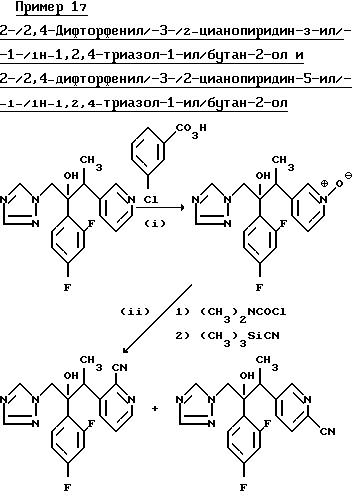
(I) 2-/2,4-Дифторфенил/-3-/1-оксидопиридин-3-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол.
Раствор 2-/2, 4-дифторфенил/-3-/пиридин-3-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ола (диастереоизомерная пара B из примера 14) (1,00 г) и 85% -ной м/м 3-хлорнадбензойной кислоты (1,30 г) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 18 ч, а затем выпарили. Остаток перемешали с простым серным эфиром, твердое вещество отфильтровали и высушили в результате чего получили указанное соединение (0793 г) Тпл.-190-193oС.
(II) 2-/2,4-дифторфенил/-3-/2- цианопиридин-3-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол и 2-/2, 4-дифторфенил/-3-/2-цианопиридин-5-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол.
Смесь продукта, полученного в части (l), (0,93 г), N,N-диметилкарбамоил хлорида (0,40 г) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение ночи. Добавили триметилсилилцианид (0,40 г) и перемешивание продолжили еще в течение 60 ч. Раствор промыли 10%-ным раствором карбоната натрия, водный слой отделили и промыли дихлорметаном. Органические слои соединили, осушили (MgSO4) и выпарили. Остаток пропустили через хроматографическую колонку с силикагелем, используя смесь гексан/изопропанол (4:1) в качестве элюента, в результате чего получили 2-/2,4-дифторфенил/-3-/2- цианопиридин-5-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол, (0,18 г), Т.пл.136-141o С.
Результаты анализа,
Найдено:
C 60,89
H 4,59
N 19,47
По расчету для C18H15F2N5O:
C
60,84
H 4,25
N 19,71
ЯМР (300 МГц).
δ (CDCl3)= 1,17 (d, 3H, j=7,1 Гц, CH3), 3,47 (q, 1H, j=7,1 Гц, CHCH3), 3,81 и 4, 85 (d, 1H, j=13,8 Гц, CH2), 5,19 (S,1H, OH), 6,81 (m, 2H, Hаром), 7,47 (m, 1H, Hаром), 7,75 (d,1H, j=8 Гц, пиридин H-3), 7,76 и 7,79 (S, 1H, триазол H), 8,10 (м, 1H, пиридин H-4), 8,80 (d, 1H, j=1,8 Гц, пиридин H-6) млн. долей.
После дальнейшего элюирования той же смесью растворителей, соединения и выпаривания соответствующих фракций получили 2-/2, 4-дифторфенил/-3-/2-цианопиридин-3-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол, (0,23 г), Т.пл.180-182oС.
Результаты анализа,
Найдено:
C 60,85
H 4,33
N 19,51
По расчету для C18H15F2N5O:
C 60,84
H 4,25
N 19,71
ЯМР (300
МГц).
d (CDCl3)= 1,17 (g, 3H, j=7,0 Гц, CH), 3.82 и 5.17 (g, 1H, j=13,8 Гц, CH2), 4.05 (кв. 1H, j=7,0 Гц, CHCH3), 5,21 (c. 1H, OH), 6,82 (м. 2H, Hаром), 7,46 (м. 1H, Hаром), 7,60 (м. 1H, пиридин H-5), 7,76 и 7,83 (c. 1H, триазол H), 8,32 (м, 1H, пиридин H-4), 8,68 (м. 1H, пиридин H-6) млн. долей.
Пример 18. 2-/2, 4-Дифторфенил/-3-/2-цианопиридин-4-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол и 2-/2,4-Дифторфенил/-3-/6-цианопиридин-4-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол.
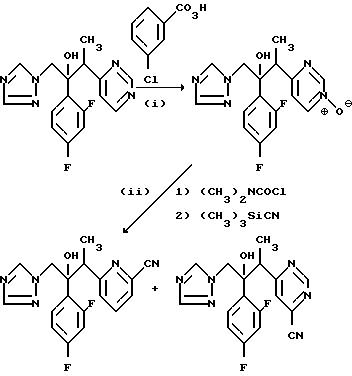
(I) 2-/2,4-Дифторфенил/-3-/1-оксидопиримидин-2-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол.
Раствор 2-/2,4-дифторфенил/-3-/1-пиримидин-4-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ола (диастереоизомерная пара B из примера 3) (3,31 г) и 85% -ной м/м 3-хлорнадбензойной кислоты (2,03 г) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 48 ч. Добавили дополнительно 2,03 г 85%-ной м/м 3-хлорнадбензойной кислоты и перемешивание продолжали еще в течение 18 ч. После осуществления всех операций, описанных в примере 15(l), получили указанное соединение, (0,80 г), Тпл.157-160oС.
(II) 2-/2, 4-Дифторфенил/-3-/2- цианопиридин-4-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол и 2-/2,4-дифторфенил/-3-/6-цианопиридин-4-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол.
Смесь продукта, полученного в части (l), (0,80 г), N,N-диметилкарбамои- хлорида (0,50 г) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 2 ч. Добавили триметилсилил цианид (0,50 г) и перемешивание продолжали еще в течение 6 дней. Раствор выпарили и остаток пропустили через хроматографическую колонку с силикагелем. После элюирования смесью дихлорметан/метанол (100: 1) получили продукт, который повторно пропустили через хроматографическую колонку с силикагелем. Элюирование начинали простым эфиром, и полярность элюента постепенно увеличивали путем добавления до 6% (по объему) метанола. После объединения и выпаривания исходных фракций содержащих продукт, получили 2-/2,4-дифторфенил/-3-/6-цианопиримидин-4-ил/-1- -/1H-1,2,4-триазол-1-ил/бутан-2-ол, (30 мг), Тпл. 148-149oC.
ЯМР (300 МГц).
δ (CDCl3)=1,16 (д, 3H, j=7,17 Гц, CH3), 3,77 (кв. 1H, j=7,17 Гц, CHCH3), 4,09 и 4,88 (g, 1H, j=14,15 Гц, CH2), 5,74 (с. 1H, OH), 6,85 (м, 2H, Hаром), 7,55 (м. 1H, Hаром), 7,69 и 7.87 (c, 1H, триазол H), 7,89 (g, 1H, j=1 Гц, пипимидин H-5), 9.24 (g, 1H, j=1 Гц, пиримидин H-2) млн. долей.
После дальнейшего элюирования, соединения и выпаривания соответствующих фракций получили 2-/2, 4-дифторфенил/-3-/2-цианопиримидин-4-ил/-1- -/1H-1,2, 4-триазол-1-ил/бутан-2-ол, (203 мг), Т.пл.155-157oС.
Результаты анализа,
Найдено:
C 57,
30
H 3,96
N 23,69
По расчету
для C17H14F2N6O:
C 57,36
H 3.97
N 23.36
ЯМР (300 МГц).
d (CDCl3)=1,17 (g, 3H, j=7,16 Гц, CH3), 3,73 (q, 1H, j=7,16 Гц, CHCH3), 3.99 и 4.99 (g, 1H, j=14,2 Гц, CH2), 5.39 (c, 1H, OH), 6,82 (м. 2H, Hаром), 7,51 (м. 1H, Hаром), 7,71 и 7, 88 (c, 1H, триазол H), 7,77 (g, 1H, j=5,3 Гц, пиримидин H-5), 8,84 (g, 1H, j=5,3 Гц, пиримидин H-6) млн. долей.
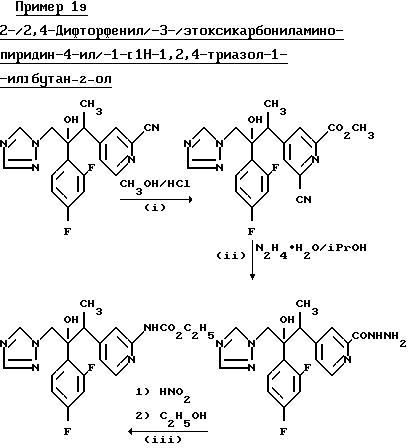
(I) 2-/2,4-Дифторфенил/-3-/2-метоксикарбонилпиридин-4-ил/ -1-/1H-1,2,4-триазол-1-ил/бутан-2-ол.
Суспензию 2-/2, 4-дифторфенил/-3-/2-цианопиридин-4-ил/1-1H- 1,2,4,-триазол-1-ил/бутан-2-ола (см. пример 15) (5,0 г) в метаноле (50 мл) насытили газообразным хлористым водородом, кипятили в течение 2 ч, а затем оставили стоять при комнатной температуре в течение 18 ч. Раствор выпарили и остаток подщелочили с помощью разбавленного раствора бикарбоната натрия. Смесь проэкстрагировали несколько раз дихлорметаном и объединенные экстракты осушили (MgSO4) и выпарили. После перекристаллизации остатки из металацетата получили указанное соединение, (4,90 г), Тпл. 182-183oС.
(II) 4-3-/2,4-Дифторфенил/-3-окси-4-/1H-1,2,4-триазол-1-ил/бут-2- ил пирин-2-карбоновой кислоты гидразид.
Раствор продукта, полученного в части (I), (3,80 г) и гидрата гидразина (6.9 мл) в изопропаноле (20 мл) кипятили в течение 2,5 ч, а затем выпарили. К остатку добавили воду и смесь проэкстрагировали несколько раз дихлорметаном. Объединенные экстракты промыли рассолом и осушили (MgSO4). После выпаривания растворителя получили указанное соединение (3,30 г) в виде аморфной пены, которое использовали непосредственно на следующей стадии.
(III) 2/2,4-дифторфенил/-3-/2-этоксикарбониламинопиридин-4-ил)-1- /1H-1,2,4-триазол-1-ил/бутан-2-ол.
Продукт, полученный в части (II), (1,40 г) растворили в 6Н соляной кислоты и раствор охладили до 0oС. По каплям, при перемешивании добавили раствор нитрита натрия (0,276 г) в воде (2 мл) и перемешивание продолжали в течение 1 ч. Затем раствор подщелочили раствором бикарбоната натрия и полученную смесь несколько раз проэкстрагировали дихлорметаном. Объединенные органические экстракты осушили (MgSO4) и выпарили. Остаток растворили в этаноле (50 мл) и раствор кипятили в течение 2,5 ч, а затем выпарили. Остаток перекристаллизовали из простого эфира и получили указанное соединение, (1,12 г), Тпл. 177-179Сo.
Результаты анализа,
Найдено:
C 57,90
H 5,25
N 16,81
По расчету для C20H21F2N5O3:
C 57,55
H 5,07
N 16,78
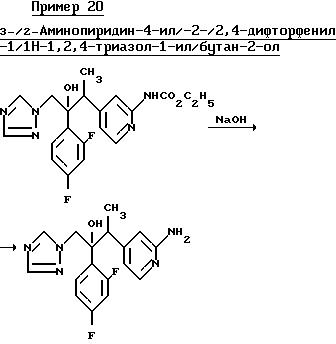
Раствор продукта, полученного в примере 19, (0,80 г), в этаноле (30 мл), содержащий 40%-ный раствор гидроксида натрия (2,0 мл), кипятили в течение 2 ч, а затем выпаривали. К остатку добавили воду и смесь несколько раз проэкстрагировали этилацетатом. Объединенные органические экстракты осушили (MgSO4) и выпарили, в результате чего получили смолу. Смолу растворили в простом эфире и после кристаллизации при стоянии этого раствора получили указанное соединение, (0,45 г), Тпл. 182-185oС.
Результаты анализа,
Найдено:
C 59,34
H 5,03
N 19,92
По расчету для
C17H17F2N5O:
C 59,13
H 4,96
N 20,28
Пример 21. 2/2,
4-Дифторфенил/-3-/2-этоксикарбониламинопиридин-5-ил)-1- /1H-1,2,
4-триазол-1-ил/бутан-2-ол.
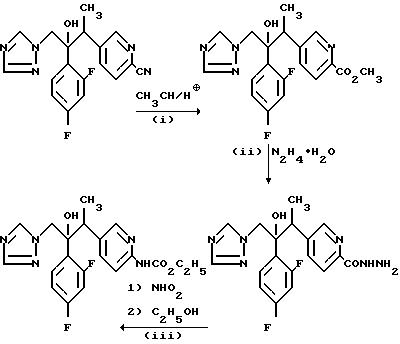
(l) 2/2, 4-Дифторфенил/-3-/2-метоксикарбонилпиридин-5-ил)-1- /1H-1,2,4-триазол-1-ил/бутан-2-ол.
В результате обработки 2-/2,4-дифторфенил/-3-/2-цианопиридин-5- ил/-1-/1H-1,2, 4-триазол-1-ил/бутан-2-ола (см. пример 17) (1,0 г) метанолом (20 мл) в присутствии хлористого водорода по методу, описанному в примере 19 (I) получили указанное соединение в виде смолы, (0,75 г), которое использовали непосредственно на следующей стадии.
(II) 5-[/3-/2,4-Дифторфенил/-3-окси-4-/1Н-1,2,4-триа- зол-1-ил/]бут-2-ил/пиридин-2-карбоновой кислоты гидразид.
В результате обработки продукта, полученного в части (1), (0,75 г), гидратом гидразина (2,0 мл) в изопропаноле (10 мл) по методу, описанному в примере 19 (II), получили указанное соединение, (0,36 г), в виде аморфной пены, которое использовали непосредственно на следующей стадии.
(III) 2-/2,4-Дифторфенил/-3-/2-этоксикарбониламинопиридин-5- ил/-1-/1Н-1,2, 4-триазол-1-ил/бутан-2-ол.
В результате обработки продукта, полученного в части (II), (0,36 г) азотистой кислотой с последующим нагреванием образовавшегося азидного промежуточного
соединения в этаноле по методу, описанному в примере 19(III) получили сырой продукт, который пропустили через хроматографическую колонку с силикагелем. В результате элюирования этилацетатом,
соединения и выпаривания соответствующих фракций получили твердое вещество, которое кристаллизовали из смеси этилацетата/простой эфир и получили указанное соединение, (0,12 г), Тпл.
167-168oC
Результаты анализа,
Найдено:
C 57,81
H 5,00
N 16,46
По расчету для C20H21F2N5
O3:
C 57,55
H 5,07
N 16,78
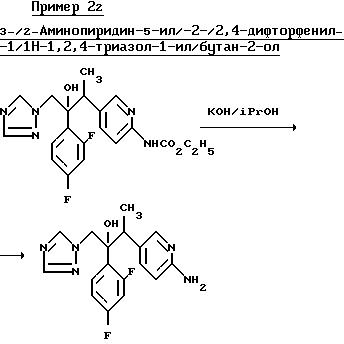
Раствор продукта, полученного в примере 21, (70 мл) в изопропаноле (4 мл), содержащей 50% водного гидроксила калия (4-капли), кипятили в течении 4 ч, а затем выпарили. К остатку добавили воду и смесь проэкстрагировали несколько раз этилацетатом. Объединенные органические экстракты промыли водой и осушили (MgSO4). После выпаривания растворителя получили указанное соединение в виде аморфной пены (49 мг).
ЯМР (300 МГц).
δ (СДСI3)1,06, (g, 3H, j=7,12 Гц, CH3), 3.23 (кв. 1H, j=7,12 Гц, СНСН3), 3,93 и 4,77 (g, 1H, j=14,2 Гц, СН2), 4,63 (уширенные с. 2H, N H2), 6,54 (g, 1H, j= 8,5 Гц, пиридин H-3), 6,75 (м, 2H, Hаром), 7,45 (м, 1Н, Наром), са. 7,70 (м, 1Н, пиридин Н-4), 7,71 и 7, 76 (С, триазол Н), 8,04 (S, 1H, пиридин Н-6) млн. долей.
Пример 23. /-/-/2R, 3S/-2-/2,4-Дифторфенил/-3-/пиридин-4-ил/-1- /1Н-1,2,4-триазол-1-ил/бутан-2-ол.
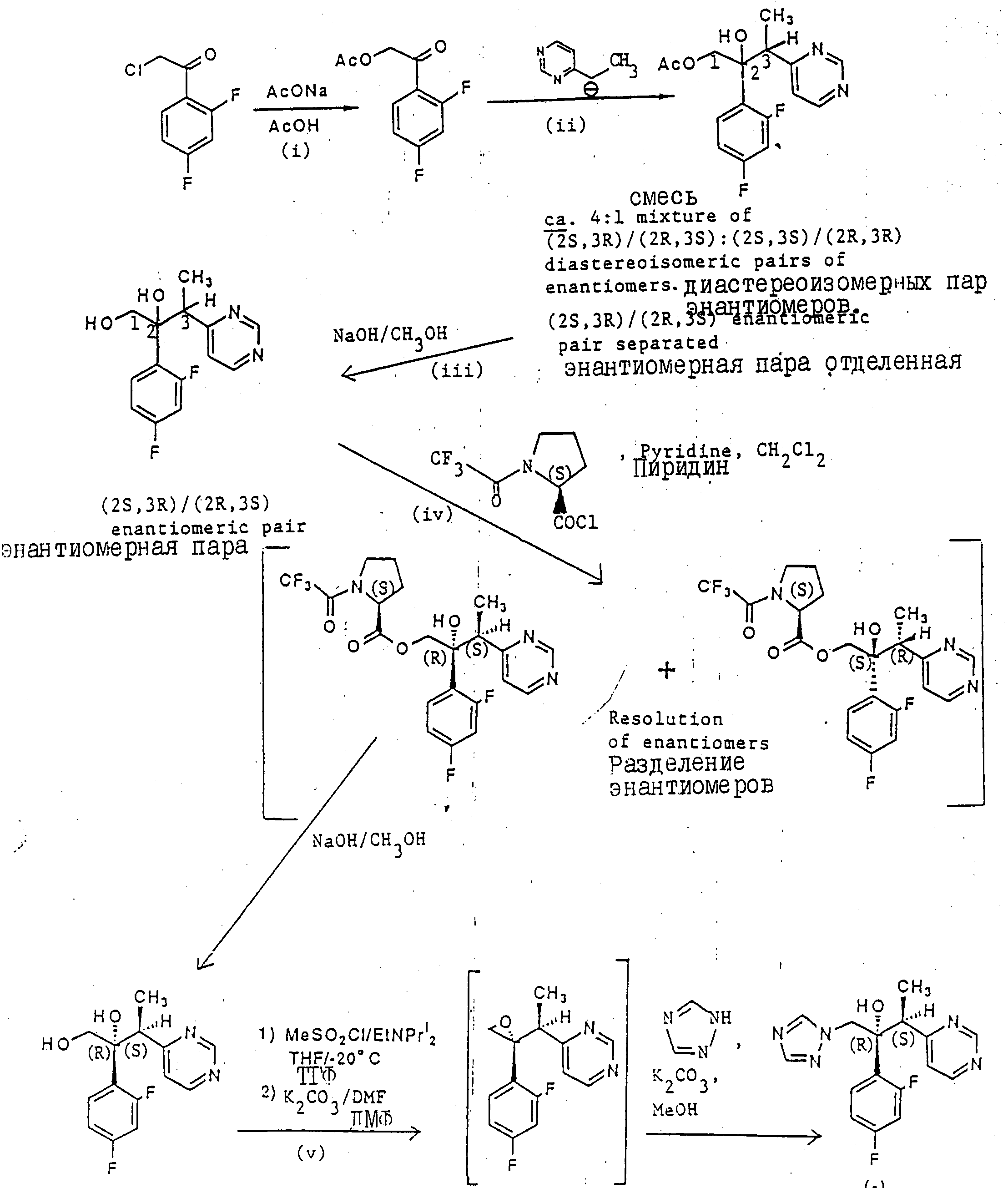
(l) 2-Ацетокси-2,,4, дифторацетофенон.
Раствор 2-хлор-2', 4'- дифторацетофенона (19,0 г) и безводного ацетата натрия (16,4 г) в уксусной кислоте (50 мл) кипятили в течении 4 ч, а затем выпарили. Остаток обработали этилацетатом и водой. Органический слой отделили, промыли раствором бикарбоната натрия и осушили (Na2SO4). После выпаривания растворителя получили масло, которое растерли с гексаном. Образовавшееся вещество, отфильтровали, промыли гексаном и осушили, в результате чего получили указанное соединение (16,2 г), Тпл. 54-56oС (см. конец описания).
(II) /±/-/2R, 3S/ и /2S, 3R/ -1-Ацетокси-2-/2,4-дифторфенил -/3-пиримидин-4-ил/бутан-2-ол.
Раствор диизопропиламина (30,3 г) в сухом тетрагидрофуране (400 мл) последовательно обработали н-бутиллитием (188 мл 1,6 М раствора в гексане) и 4-этилпиримидном (32,4 г) по методу, описанному в примере 3. Раствор продукта, полученного в части (1), (64,0 г) в сухом тетрагидрофуране (400 мл) добавили при перемешивании в течение 0,58 ч при температуре от -40 до -50oС. Затем добавили уксусную кислоту (30 мл), и раствору дали нагреться до комнатной температуры. Добавили простой эфир (1000 мл) и воду (1000 мл) и смесь встряхнули. Органический слой отделили, промыли рассолом и осушили (MgSO4). Растворители выпарили и остаток пропустили через хроматографическую колонку с селикагелем. В результате элюирования смесью простой эфир/гексан (1:4) получили сначала исходный кетон. После дальнейшего элюирования смесью простой эфир/гексан (1:1), при постепенном снижении доли гексана вплоть до чистого простого эфира, получили полукристаллическое вещество, состоящее из /±/-энантиометрической смеси указанных соединений вместе с -/2R,3R/- и /2S, 3S/- диастереоизомерной парой энантиомеров. Простой эфир добавляли до получения прозрачного раствора, а затем добавили гексан (20% по объему). Смесь охладили и образовавшийся осадок отфильтровали, промыли гексаном и осушили, в результате чего получили /±/- энантиомерную смесь указанных соединений, (23,3 г), Тпл.102-103, 5oС.
Результаты анализа,
Найдено:
C 59,68
H 5,09
N 8,55
По расчету для C16H16F2N2
O3:
C 59,62
H 5,00
N 8,69
(III) /±/-/2R, 3S/ и /2S, 3R/-2-/2,4-Дифторфенил/-3-/пиримидин-4-ил/бутан-1,2-диол.
2H раствор гидроксид натрия при перемешивании, в течение 0,25 ч добавили к раствору продукта, полученного в части (ll), (23.3 г) в метаноле (80 мл) и перемешивание осуществляли еще в течение 0,25 ч. Добавили воду (150 мл) и смесь охладили. Осадок отфильтровали, промыли водой и осушили, в результате чего получили указанные соединения, (17,4 г), Тпл 148-150,5oС.
Результаты анализа,
Найдено:
C 59,80
H 5,09
N 10,12
По расчету для C14H14F2N2O2:
C 60,
00
H 5,04
N 10,00
(IV) /-/-/2R, 3S/-2-/2,4-Дифторфенил/-3-/пиримидин-4-ил/ бутан-1,2-диол.
/S/ -N-/Трифторацетил/пропил хлорид (72 мл 1.0 М раствора в дихлорметане) по каплям, в течение 0,5 ч добавляли к охлажденному льду раствору продукта, полученного в части (lll) (16,7 г), и пиридину (8,7 мл) в сухом дихлорментане (50 мл). Раствор перемешивали в течение 0,5 ч, а затем выпаривали дихлорметан. Добавили этилацетат и воду и смесь подкислили до pH 3 с помощью 2H соляной кислоты. Органический слой отделили, последовательно промыли 0,1H соляной кислоты и водой, а затем осушили (Na2SО4). Растворитель выпаривали и остаток пропускали через хроматографическую колонку с силикагелем. Колонку элюировали смесью гексан (простой эфир) диэтиламин (65:30:5), и фракции, содержащие исходный продукт, соединили и выпарили. Остаток перекристаллизовали из диизопропилового эфира и получили /S/-N-/трифторацетил/пропиловый сложный эфир указанного (2R, 3S/-энантиомера, /4,78 г/, Тпл. 91-92,5oС.
В результате последующего элюирования колонки получили фракции, содержащие смесь обоих (2R, 3S)-энантиомеров в виде их (S) -N-/трифторацетил/пропиловых сложных эфиров. Соответствующие фракции соединили и выпарили, а этот остаток соединили с остатком, полученным после выпаривания маточных растворов при кристаллизации. Смесь растворили в небольшом количестве диизопропилового простого эфира, в раствор поместили зародышеобразующие кристаллы чистого (2R, 3S)-продукта и охлаждали в течение 4 ч. После фильтрования получили еще 1,9 г чистого (2R, 3S)-энантиомера в виде (S)-N-/трифторацетил/пропилового сложного эфира.
Абсолютная стереохимия этого продукта была подтверждена методом рентгеноструктурной кристаллографии.
Вышеназванный сложный эфир указанного соединения (6,0 г) растворили в метаноле (28 мл) и добавили 2H раствор гидроксида натрия (14 мл). Через 0,25 ч добавили воду (100 мл) и смесь охладили на льду в течение 1 ч. Осадок отфильтровали, промывали водой и осушили, в результате чего получили указанное соединение, (2,50 г), Тпл 147-148,5oС.
Результаты анализа,
Найдено:
C 59,94
H 5,16
N 9,97
По расчету для C14H14
F2N2O2:
C 60,00
H 5,04
N 17,00
(IV) /-/-/2R, 3S/-2-/2,4-Дифторфенил/-3-/пиримидин-4-ил/-1-/1H-1,2,4-триазол-1-ил/бутан-2-ол.
Метансульфонилхлорид (1,15 г) добавили при перемешивании к раствору продукта, полученного в части (lУ), (2,35 г) и диизопропилэтиамина (2,38 г) в сухом тетрагидрофуране (30 мл) при температуре от -10 до -20oС под атмосферой сухого азота. Раствор перемешивали при той же температуре в течение 1 ч, а затем добавили безводный карбонат калия (7,0 г) и сухой N, N-диметилформамид (25 мл). Смесь перемешивали при комнатной температуре в течение 1,5 ч, а затем обработали водой и простым эфиром. Органический слой отделили, промыли водой и осушили (Na2 SO4). После выпаривания растворителя получили масло, которое немедленно растворили в метаноле (50 мл). Добавили 1H-1,2,4-триазол (6,0 г) и безводный карбонат калия (6,0 г) и смесь нагревали при 60oС и перемешивали в течение 40 ч, а затем выпаривали. Остаток обработали смесью этилацетат (простой эфир) 1:1 и водой, органический слой отделили и промыли водой, затем осушили (Na2SO4). Растворитель выпарили и остаток пропустили через хроматографическую колонку с силикагелем. После первичного элюирования этилацетатом получили фракции, содержащие примеси, последующее элюирование смесью этилацетат/метанол (20:1), соединение и выпаривание соответствующих фракций позволило получить указанное соединение, (0,87 г), Тпл. 55-58oС, -65,1oС (0,55 в метаноле).
ЯМР (300 МГц).
d (СДCI3) 1,13 (g, 3H, j 7,12 Гц, CH3), 3.68 (кв. 1H, j 7,12 Гц, CHCH3), 4,16 и 4,78 (g, 1H, j 14,1 Гц, CH2), 6.60 (c, 1H, OH), 6,82 (м, 2H, Hаром), 7,44 (g, 1H, j 5,0 Гц, пиримидин H-5), 7,57 (м, 1H, Hаром), 7,61 и 7,96 (c, 1H, триазол H), 8,77 (g, 1H, j 5.0 Гц, пиримидин H-6), 9,17 (c, 1H, пиримидин H-2) млн. долей.
Считается, что если в вышеприведенных примерах выделяют диастереоизомерную пару В, то она представляет собой смесь (2R, 3S) (2S, 3R) дистереоизомеров.
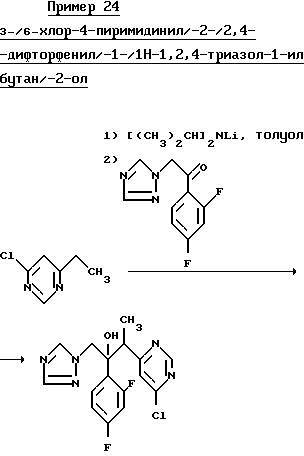
N-бутиллитий (16.0 мл 2.5 М раствор в гексане) добавили в перемешиваемый раствор диизопропиламина (5,60 г) в сухом толуоле (80 мл) при -20oC в атмосфере сухого азота. Раствор перемешивали при -20oC в течение 0,5 ч, затем перемешивали при 0oC еще 0,5 ч, а затем вновь охладили уже до -70oC. В течение 0,08 ч добавили раствор 4-хлор-6 этилпиримидина (5.70 г) в толуоле (20 мл), образовавшийся раствор желтого цвета перемешивали при -70oC в течение 1 ч, а затем добавили раствор 1-/2,4-дифторфенил/-2-/1H-1,2,4-триазол-1-ил/этанол (8.92 г) в сухом тетрагидрофуране (40 мл) в течение 0,25 ч. Раствор перемешивали при -70oC еще 0,7 ч, после чего по каплям добавили 5 мл уксусной кислоты. Раствор оставили охлаждаться до комнатной температуре и разбавили водой. Смесь проэкстрагировали дважды этилацетатом и объединенные органические экстракты промыли рассолом, высушили над MgSO4 и после упаривания растворителя получили твердый остаток коричневого цвета. Растерли твердый остаток с эфиром и получили озаглавленное соединение в виде бледно-желтого остатка твердого (6,45 г), Тпл. 164-165o C.
Анализ
Найдено:
C 52,
89
H 4,01
N 19,06
Согласно расчету для формулы C16H14ClF2N5O:
C 52,23
H 3,83
N 19,15
Пример
25. /2R, 3R/-2-/2,4-дифторфенил/-3-/пиримидин-4-ил/-1-/1H-1,2,4- -триазол-1-ил/бутан-2-ол-димезилат.
К перемешиваемому раствору 2-/2,4-дифторфенил/-3-/пиримидин-4-мл/-1-/1H1,2, 4-триазол-1-ил/бутан- -2-ола (0.70 г) в ацетоне (7 мл) добавили по каплям метансульфокислоту (0.40 г). Смесь охладили во льду в течение 1 ч, добавили 7 мл ацетона, выпавший желтый осадок отфильтровали. Осадок перемешали с ацетоном (приготовили взвесь (5 мл) и смесь нагревали с обратным холодильником с добавлением воды (0,5 мл) до получения прозрачного раствора. Раствор отфильтровали через складчатый целлюлозно-бумажный фильтр, фильтрат охладили во льду и добавили три аликвозы 5 мл ацетона через десятиминутный интервал. После этого смесь перемешивали в течение 1,5 ч после фильтрования получили кристаллический остаток бледно-желтого цвета (0,51 г), Тпл. 168-169oC.
Анализ,
Рассчитано на C18H23N5O7F2S2:
C 41,30
H 4,33
N 13,38
S 12,25
F 7,26
Найдено:
C 41,12
H 4,41
N 13,
14
S 12,35
F 7,06
Примеры 26 и 27. По примерам, приведенным ниже в таблице, были получены соединения общей формулы:-
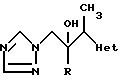
по способу, аналогичным способу, используемому в примере 3, обработкой соответствующего этилгетероцикла диизопропиламидом лития с последующей реакцией полученного карбаниона in situ с соответствующим производным 1-арил-2-(1H-1,2,4-триазол-1-ил)-этанона (табл.3).
Получение фармацевтических композиций для внутреннего введения, содержащих соединение примера 23.
Получено четыре композиции ((I) (IV)). Композиции (I) (III) получены методы А и композиции (IV) методом В.
Композиция (1): 20,64 мг/мл раствора соединения Примера 23 в смеси 30: 25:5:40, объемных, глицерин:пропиленгликоль:этанол:вода.
Композиция (II): 23,31 мг/мл раствора соединения Примера 23 в смеси 30: 10:60, объемных, пропиленгликоль:этанол:вода.
Композиция (III): 22,07 мг/мл раствора соединения примера 23 в смеси 40: 30:30, объемных, глицерин:пропиленгликоль:вода.
Композиция (IV): 20,7 мг/мл раствора соединения примера 23 в 0,9 мас. солевом растворе, содержащем 100 мг/мл гидроксипропил- β циклодекстрина.
Метод А.
1. Пригодное количество основы из сорастворителей получают смешением вместе всех компонентов в соотношении, описанном выше.
2. В подходящий сосуд помещают упомянутый выше раствор в объеме, равном 80% от требуемого объема конечного продукта.
3. К раствору прибавляют требуемое количество соединения примера 23 и перемешивают до растворения.
4. Раствор доводят до объема (100%) добавлением предварительно полученной основы пункта 1. Результирующий раствор перемешивают до гомогенного состояния.
5. Раствор фильтруют через стерильный 0,2 микронный стерилизованный фильтр и асептически заполняют пригодные стерильные контейнеры.
Методы В.
1. К воде, составляющей по объему 80% от конечного объема прибавляют при температуре 40oC требуемое количество гидроксипропил- b циклодекстрина и хлористого натрия.
2. Раствор перемешивают до растворения.
3. После растворения гидроксипропил- b -циклодекcтрина и хлористого натрия раствор оставляют охлаждаться до комнатной температуры.
4. К раствору прибавляют требуемое количество соединения примера 23 и перемешивают до растворения.
5. После растворения всего соединения раствора доводят до объема (100%) добавлением воды и перемешивают до обеспечения тщательного смешения.
6. Раствор фильтруют через стерильный 0,2 микронный стерилизованный фильтр и асептически наполняют пригодные стерильные контейнеры.
Реферат
Использование: в
качестве антифунгицидных препаратов, для лечения грибковых заболеваний у
животных, включая людей. Сущность изобретения: производные 1,2,4-триазола формулы
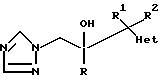
где R - фенил, замещенный 1-2 галоидами; R1 - C1-4 алкил; R2 - водород или C1-4 алкил; Het - пиридинил, пиридазинил, пиримидинил или пиразинил, незамещенные или замещенные C1-4 алкилом, C1-4 алкокси, галоидом, цианом, амином или -NHCO2 (C1-4) алкилом; или их фармацевтически приемлемые соли; фармацевтическая композиция на их основе и промежуточные соединения для получения производных 1,2,4-триазола общих формул
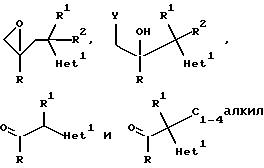
где R1, R2 и R имеют указанные выше значения; Het1 - пиридинил, пиридазинил, пирамидинил или пиразинил, возможно замещенный C1-4 алкилом, C1-4 алкокси, галоидом, циано или нитрогруппой, Y - удаляемая группа. 6 с. и 13 з.п.ф-лы, 3 табл.
Формула
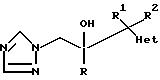
или их фармацевтически приемлемые соли,
где R фенил, замещенный 1 2 заместителями, каждый из которых независимо друг от друга выбирают из галоида;
R1 С1 C4 алкил;
R2 Н или С1 C4алкил,
Het, который присоединен к смежному атому углерода кольцевым атомом углерода, выбирают из пиридинила, пиридазинила, пиримидинила или пиразинила, при этом Het необязательно замещен С1 C4алкилом, С1 - C4алкокси, галоидом, цианом, аминогруппой или -NHCO2(С1 - C4алкилом.
или их фармацевтически приемлемая соль.
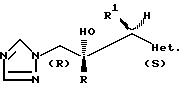
13. Фармацевтическая композиция, обладающая противогрибковой активностью, содержащая активный ингредиент и фармацевтически приемлемый носитель или разбавитель, отличающийся тем, что в качестве активного ингредиента она содержит эффективное количество соединения формулы I по п.1 или его фармацевтически приемлемую соль.
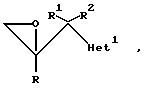
где R, R1, R2 имеют значения, указанные в п.1, Het1 представляет собой пиридинил, пиридазинил, пиримидинил или пиразинил, необязательно замещенные С1 C4алкилом, С1-4алкокси, галоидом, циано- или нитрогруппой.
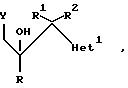
где R, R1, R2 и Het имеют значения, указанные выше, У означает удаляемую группу.
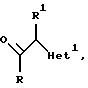
где R, R1 и Het1 имеют значения, указанные выше.
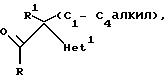
где R, R1 и Het1 имеют значения, указанные выше.
Комментарии