Пиразолхинолины с иммуномодулирующей активностью - RU2328496C2
Код документа: RU2328496C2
Описание
Настоящее изобретение относится к новым гетероциклическим соединениям, к способам их получения, к композициям, содержащим их, и к способам и применению для клинического лечения медицинских состояний, при которых иммуномодуляция может оказать благоприятное действие, включая ревматоидный артрит, рассеянный склероз, сахарный диабет, астму, трансплантацию, системную красную волчанку и псориаз. Более конкретно, настоящее изобретение относится к новым гетероциклическим соединениям, которые являются антагонистами CD80, способными ингибировать взаимодействия между CD80 и CD28.
Иммунная система обладает способностью регулировать гомеостаз между активацией и инактивацией лимфоцитов посредством различных регуляторных механизмов во время и после иммунного ответа. Среди них имеются механизмы, которые специфически ингибируют и/или выключают иммунный ответ. Так, когда антиген представлен молекулами МНС (главного комплекса тканевой совместимости) рецепторам Т-клеток, Т-клетки становятся соответствующим образом активированными только в присутствии дополнительных совместно стимулирующих сигналов. В отсутствие дополнительных сигналов нет активации лимфоцитов и индуцировано или состояние функциональной инактивации, называемое анергией, или толерантности, или Т-клетка специфически уничтожается апоптозом. Один такой совместно стимулирующий сигнал включает взаимодействие CD80 на специализированных, представляющих антиген клетках с CD28 на Т-клетках, которое, как было продемонстрировано, существенно для полной активации Т-клеток (Lenschow et al. (1996) Annu. Rev. Immunol., 14, 233-258).
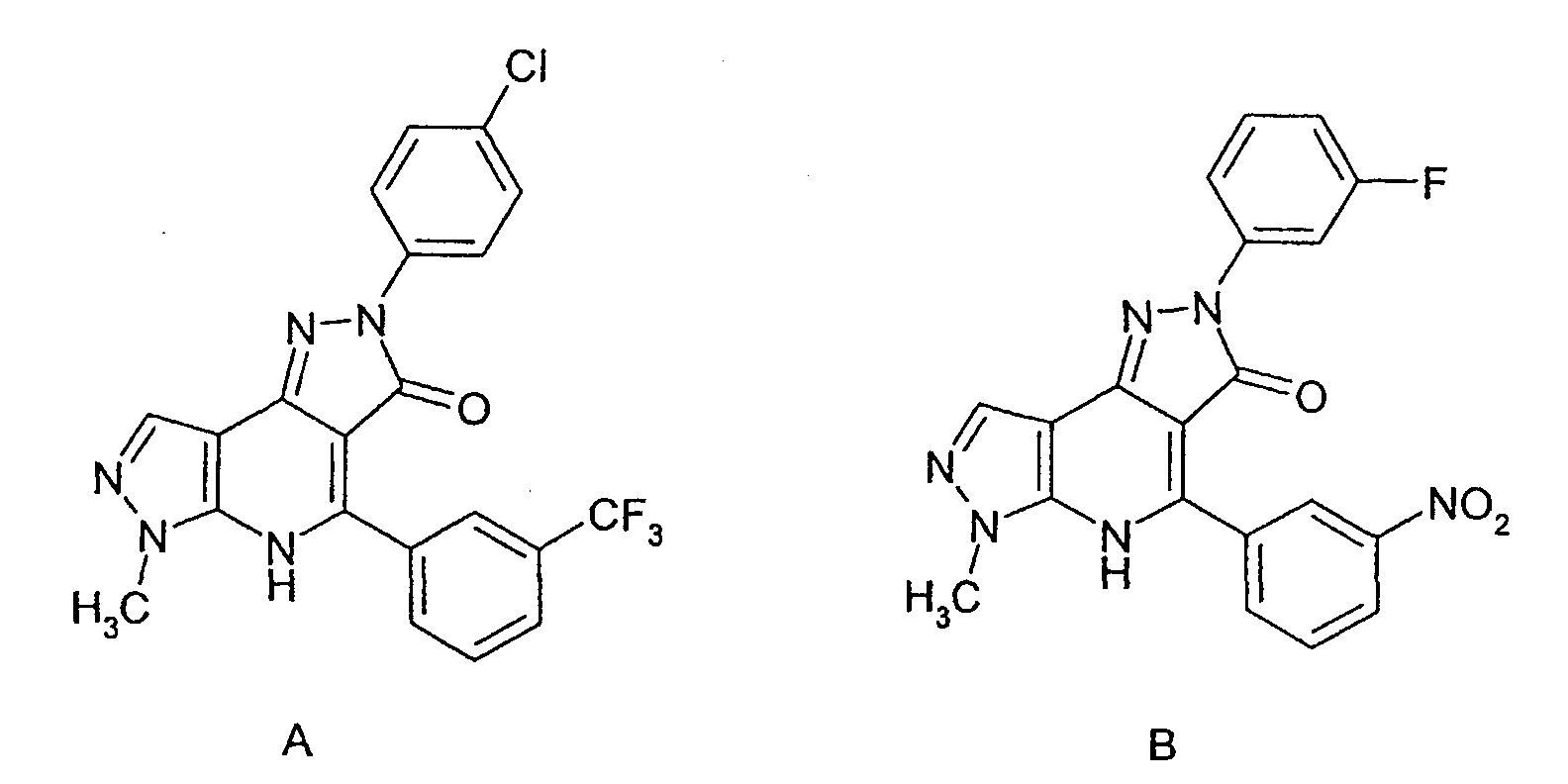
В статье Erbe et al., в J. Biol. Chem. Vol.277, №9, pp.7363-7368 (2002) описываются 3 небольших молекулярных лиганда, которые связываются с CD80 и ингибируют связывание CD80 с CD28 и CTLA4. Два из описанных лигандов представляют собой конденсированные пиразолоны структур А и В:
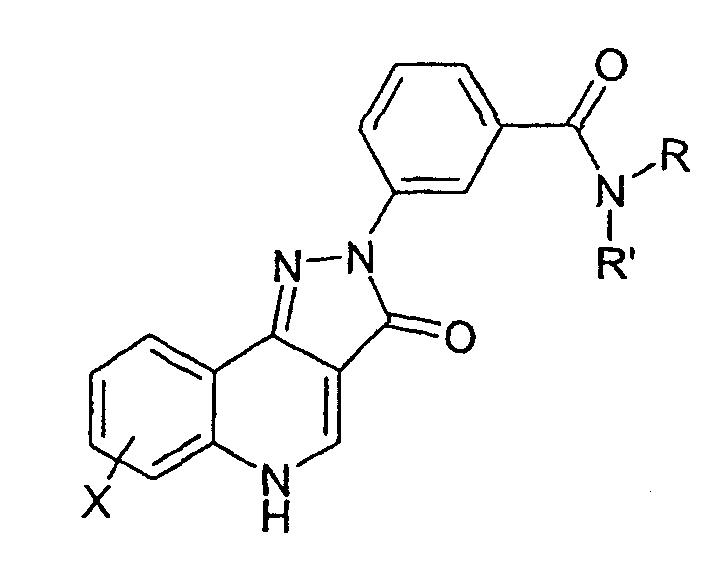
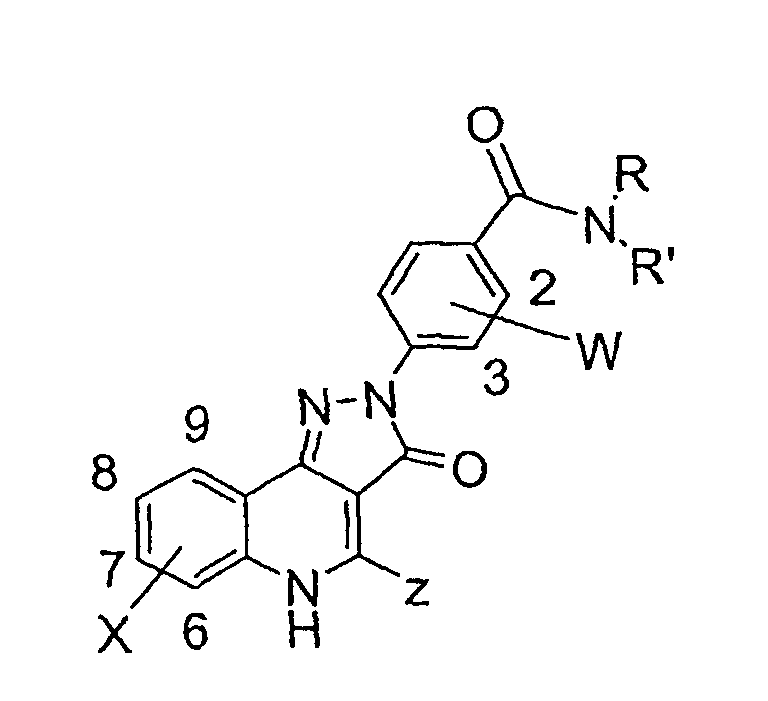
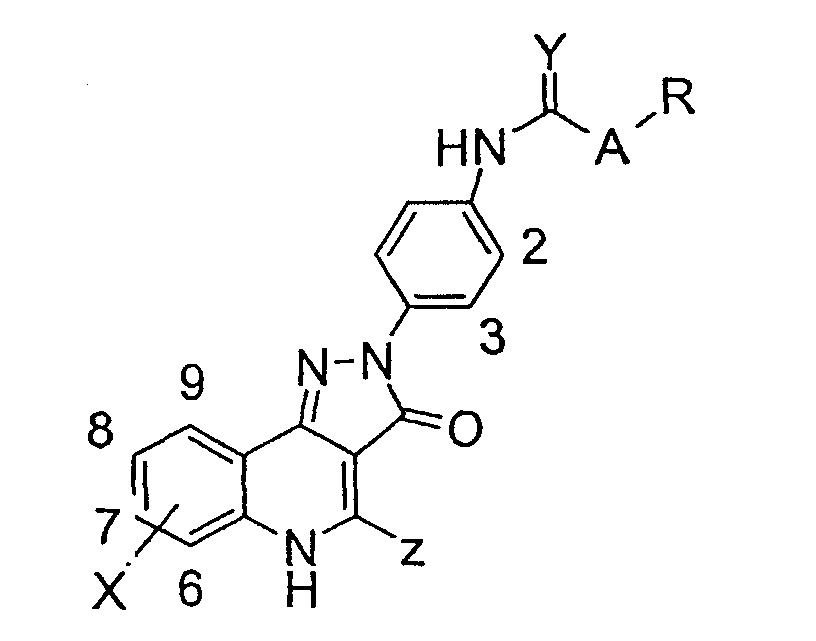
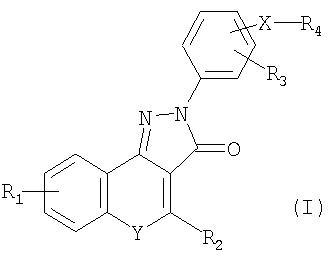
В соответствии с настоящим изобретением предоставляется соединение формулы (I) или его фармацевтически или ветеринарно-приемлемая соль:
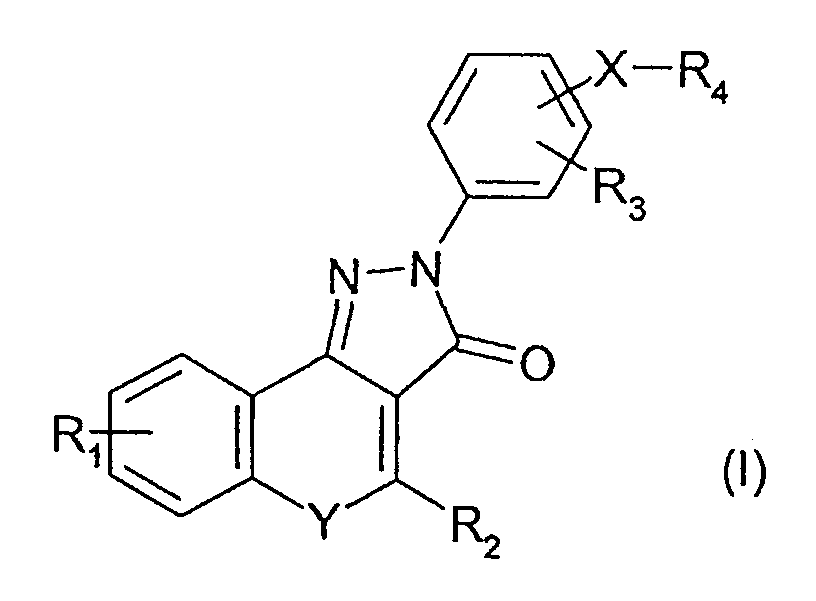
где:
R1 и R3 независимо представляют H; F; Cl; Br; -NO2; -CN; С1-С6алкил, необязательно замещенный F или Cl; или С1-С6алкокси, необязательно замещенный F;
R2 представляет Н или необязательно замещенный С1-С6алкил, С3-С7циклоалкил или необязательно замещенный фенил;
Y представляет -O-, -S-, N-оксид или -N(R5)-, где R5 представляет Н или С1-С6алкил;
Х представляет связь или двухвалентный радикал С1 -С6алкилен;
R4 представляет -C(=O)NR6R7, -NR7C(=O)R6, -NR7C(=O)OR6, -NHC(=O)NHR6 или -NHC(=S)NHR6, где
R6 представляет Н или радикал формулы -(Alk)b-Q, где b равно 0 или 1, и
Alk представляет собой необязательно замещенный двухвалентный С1-С12алкиленовый, С2-С12алкениленовый или С2-С12алкиниленовый радикал с прямой или разветвленной цепью, который может быть прерван одним или несколькими не соседними -O-, -S- или -N(R8)-, где R8 представляет Н или С1-С4алкил, С3-С4алкенил, С3 -С4алкинил или С3-С6циклоалкил, и
Q представляет Н; -CF3; -OH; -SH; -NR8R8, где каждый R8 может быть одинаковым или различным; сложноэфирную группу; или необязательно замещенный фенил, С3-С7циклоалкил, С5-С7циклоалкенил или гетероциклическое кольцо, имеющее от 5 до 8 кольцевых атомов; и
R7 представляет Н или С1-С6алкил; или взятые вместе с атомом или атомами, к которым они присоединены, R6 и R7 образуют необязательно замещенное гетероциклическое кольцо, имеющее от 5 до 8 кольцевых атомов.
Соединения общей формулы (I) являются антагонистами CD80. Они ингибируют взаимодействие между CD80 и CD28 и, таким образом, активацию Т клеток, модулируя посредством этого иммунный ответ.
Соответственно, изобретение также включает:
(i) соединение формулы (I) или его фармацевтически или ветеринарно-приемлемую соль для лечения состояний, при которых иммуномодуляция оказывает благоприятное действие;
(ii) применение соединения формулы (I) или его фармацевтически или ветеринарно-приемлемой соли для изготовления лекарственного средства для лечения состояний, при которых иммуномодуляция оказывает благоприятное действие;
(iii) способ иммуномодуляции у людей и приматов, кроме человека, включающий введение нуждающемуся в таком лечении субъекту иммуномодуляторно эффективной дозы соединения формулы (I) или его фармацевтически или ветеринарно приемлемой соли;
(iv) фармацевтическую или ветеринарную композицию, включающую соединение формулы (I) или его фармацевтически или ветеринарно приемлемую соль вместе с фармацевтически или ветеринарно приемлемым эксципиентом или носителем.
Состояния, при которых иммуномодуляция оказывает благоприятный эффект, включают:
Надпочечниковую недостаточность
Аллергический ангиит и грануломатоз
Амилоидоз
Анкилозирующий спондилит
Астму
Аутоиммунную болезнь Аддисона
Аутоиммунное облысение
Аутоиммунный хронический активный гепатит
Аутоиммунную гемолитическую анемию
Аутоиммунную нейтропению
Аутоиммунную тромбоцитопеническую пурпуру
Аутоиммунные васкулиты
Болезнь Бехчета
Мозжечковую дегенерацию
Хронический активный гепатит
Хроническую воспалительную демиелинирующую полирадикулонейропатию
Герпетиформный дерматит
Сахарный диабет
Миастенический синдром Итона-Лэмберта
Энцефаломиелит
Пузырчатый эпидермолиз
Узловую эритему
Чувствительную к глютену энтеропатию
Сидром Гудпасчура
Болезнь «трансплантат против хозяина»
Синдром Жуллена-Барре
Тироидит Хашимото
Гипертиреоз
Идиопатический гемохроматоз
Идиопатический мембранозный гломерулонефрит
Почечное заболевание с минимальными изменениями
Смешанное заболевание соединительной ткани
Многоочаговую двигательную нейропатию
Рассеянный склероз
Генерализованную миастению
Синдром опсоклонуса-миоклонуса
Псевдопузырчатку
Пузырчатку
Пернициозную анемию
Узелковый полиартериит
Полимиозит/дерматомиозит
Постинфекционные артриты
Первичный склероз желчных протоков
Псориаз
Реактивные артриты
Болезнь Рейтера
Ретинопатию
Ревматоидный артрит
Склерозирующий холангит
Синдром Шегрена
Сидром «ригидного человека»
Подострый тироидит
Системную красную волчанку
Системный склероз (склеродермию)
Височный артериит
Облитерирующий тромбоангиит
Отторжение трансплантата
Аутоиммунный полигландулярный синдром I типа и II типа
Язвенный колит
Увеит
Грануломатоз Вегенера
Используемый в настоящем описании термин «алкилен» относится к прямой или разветвленной алкильной цепи, имеющей две свободные валентности, например -CH2-, -CH2CH2-, -CH2CH2CH2-, -CH(CH3)CH2-, -CH(-CH2CH3)CH2CH2CH3 и -C(CH3)3.
Используемый в настоящем описании термин «гетероарил» относится к 5- или 6-членному ароматическому кольцу, содержащему один или несколько гетероатомов. Такими группами являются, например, тиенил, фурил, пирролил, имидазолил, бензимидазолил, тиазолил, пиразолил, изоксазолил, изотиазолил, триазолил, тиадиазолил, оксадиазолил, пиридинил, пиридазинил, пиримидинил, пиразинил, триазинил.
Используемый в настоящем описании безусловный термин «гетероциклил» или «гетероциклический» включает «гетероарил», как определено выше, и, в частности, означает 5-8-членное ароматическое или неароматическое гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из S, N и O, включая, например, пирролил, фуранил, тиенил, пиперидинил, имидазолил, оксазолил, изоксазолил, тиазолил, тиадиазолил, пиразолил, пиридинил, пирролидинил, пиримидинил, морфолинил, пиперазинил, индолил, морфолинил, бензофуранил, пиранил, изоксазолил, хинуклидинил, азабицикло[3.2.1]октанил, бензимидазолил, метилендиоксифенил, малеимидо и сукцинимидогруппы.
В отсутствие других определений в контексте, где он встречается, термин «замещенный», относящийся к любой указанной в настоящем описании части, означает замещенный одним или несколькими следующими заместителями, а именно (С1-С6)алкилом, трифторметилом, (С1-С6)алкокси (включая особый случай, когда кольцо замещено у атома С соседнего кольца метилендиокси или этилендиокси), трифторметокси, (С1-С6)алкилтио, фенилом, бензилом, фенокси, (С3-С8 )циклоалкилом, гидрокси, меркапто, амино, фтором, бромом, циано, нитро, оксо, -COOH, -SO2OH, -CONH2, -SO2NH2, -CORA, -COORA, -SO2ORA, -NHCORA, -NHSO2RA, -CONHRA, -SO2NHRA, -NHRA, -NRARB, -CONRARB или -SO2NRARB, где RAиRB представляют собой независимо (С1-С6)алкильную группу. В случае, когда «замещенный» означает замещенный (С3-С8)циклоалкилом, фенилом, бензилом или фенокси, его кольцо само по себе может быть замещено любой из указанных выше групп, за исключением (С3-С8)циклоалкила, фенила, бензила или фенокси.
Используемый в настоящем описании безусловный термин «карбоциклил» или «карбоциклический» относится к 5-8-членному кольцу, все кольцевые атомы которого представляют собой углерод.
Некоторые соединения изобретения содержат один или несколько хиральных центров благодаря присутствию асимметричных атомов углерода. Присутствие асимметричных атомов углерода вызывает появление стереоизомеров или дистереоизомеров с R или S стереохимией в каждом хиральном центре. Изобретение включает все такие стереоизомеры и диастереоизомеры и их смеси.
Соли солеобразующих соединений изобретения включают физиологически приемлемые кислотно-аддитивные соли, например гидрохлориды, гидробромиды, сульфаты, метансульфонаты, паратолуолсульфонаты, фосфаты, ацетаты, цитраты, сукцинаты, лактаты, тартраты, фумараты и малеаты; и основно-аддитивные соли, например соли натрия, калия, магния и кальция. Когда соединение содержит аминогруппу, возможны также соли четвертичного амино, и они включены в изобретение.
Соединения изобретения представляют собой следующие примеры структурных вариантов:
R1 может представлять собой, например, H, F, Cl, метил, метокси или метилендиокси. В данном случае предпочтительно, чтобы R1 представлял собой H, Cl или, особенно, F;
R2 может представлять собой, например, H, метил, метокси, циклопропил, фенил или фтор-, хлор-, метил или метокси-замещенный фенил. В данном случае предпочтителен Н или циклопропил;
R3 может представлять собой, например, H, F, Cl, метил, метокси или метилендиокси. В данном случае предпочтительно, чтобы R3 представлял собой F или Cl, и наиболее предпочтительно, чтобы R3 представлял собой Н;
Y может представлять собой, например, -O-, -S- или -N(R5)-, где R5 представляет собой Н или метил. В данном случае предпочтителен -NH- или -S-;
Х может представлять собой, например, связь или радикал -CH2- или -CH2CH2-. В данном случае предпочтительна связь;
R4 представляет -C(=O)NR6R7, -NR7C(=O)R6, -NR7C(=O)OR6, -NHC(=O)NHR6 или -NHC(=S)NHR6. Из них в данном случае предпочтительны -NR7C(=O)R6 и, особенно, -C(=O)NR6R7 и -NHC(=O)NHR6. R7 представляет собой предпочтительно Н, однако широкий диапазон заместителей R6 вызвал появление высоко активных соединений изобретения. Многие иллюстративные заместители R6 показаны в соединениях приведенных ниже примеров;
R6 может представлять собой, например, Н или радикал формулы -Alkb-Q, где b равно 0 или 1, и
Alk может представлять собой, например, радикал -(CH2)n-, -CH((CH2)mCH3)(CH2)n-, -C((CH2)m CH3)((CH2)pCH3)(CH2)n-, -(CH2)n-O-(CH2)m-, -(CH2)n-NH-(CH2 )m- или -(CH2)n-NH-(CH2)m-NH-(CH2)p, где n равно 1, 2, 3 или 4, и m и p независимо равны 0, 1, 2, 3 или 4, и
Q может представлять Н, -ОН, -СООСН3, фенил, циклопропил, циклопентил, циклогексил, пиридил, фурил, тиенил или оксазолил; и
R7 может представлять собой, например, Н, или взятые вместе с атомом или атомами, с которыми они соединены, R6и R7 могут образовывать 5, 6 или 7-членное гетероциклическое кольцо.
Конкретные примеры групп R4 включают группы, присутствующие в соединениях, приведенных в примерах настоящего описания.
Соединения изобретения можно получить синтетическими способами, известными в литературе, из соединений, которые коммерчески доступны или могут быть получены из коммерчески доступных соединений. Например, соединения формулы (I), где R4 представляет собой группу -NR7C(=O)R6, может быть получена ацилированием амина формулы (II) хлорангидридом кислоты формулы (III):
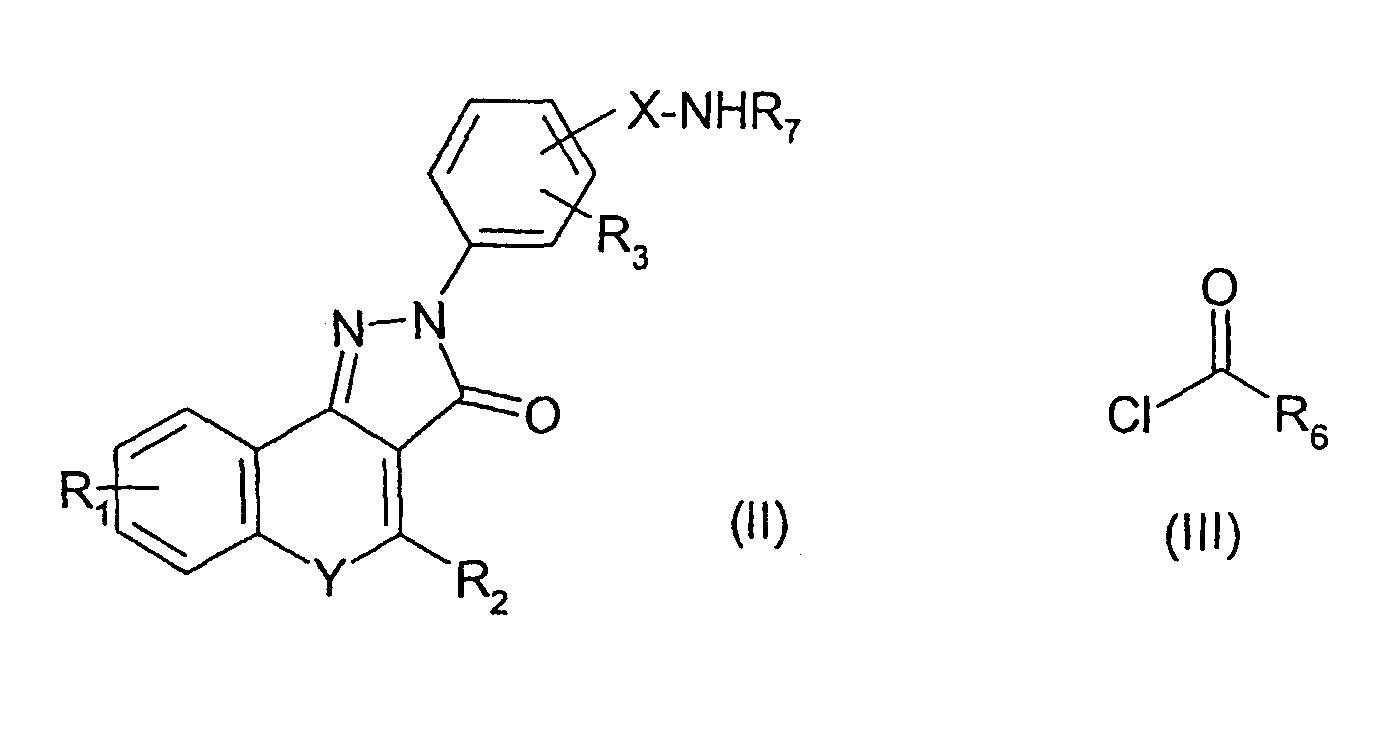
Соединения изобретения, где R4 представляет собой группу -NHC(=O)NHR6, можно получить взаимодействием амина формулы (IIA) с изоцианатом формулы (IIIA)
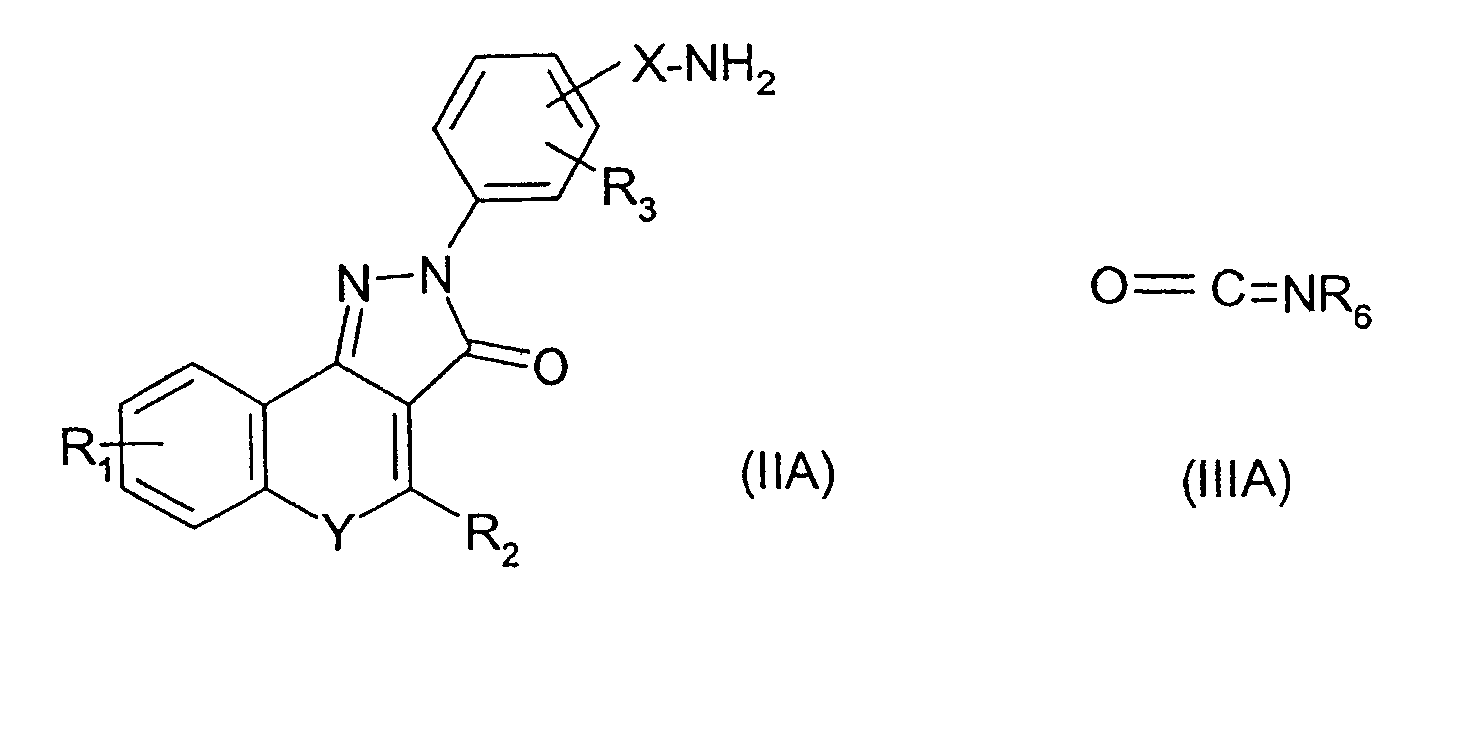
Соединения изобретения, где R4 представляет собой группу -C(=O)NHR6, можно получить взаимодействием хлорангидрида кислоты формулы (IIB) с амином NHR6R7:
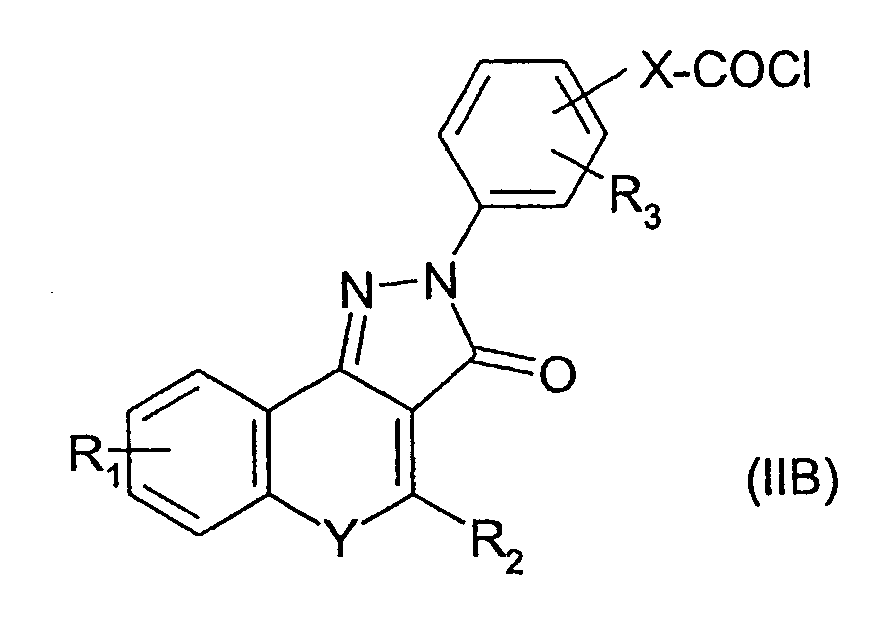
Соединения изобретения, где R4 представляет собой группу -NR7C(=O)OR6, можно получить взаимодействием амина формулы (II) с хлорформиатом ClC(=O)OR6.
Следующие примеры иллюстрируют получение соединений изобретения.
Получение промежуточного соединения 1
2-(4-нитрофенил)-6-фтор-2,5-дигидропиразол[4,3-c]хинолин-3-он
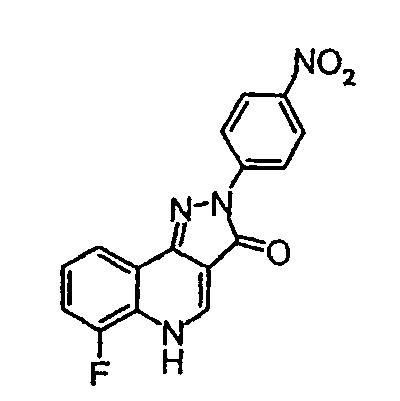
4-нитрофенилгидразин (2,28 г, 0,14 моль) добавляют одной порцией к перемешиваемому раствору этилового эфира 4-хлор-8-фторхинолин-3-карбоновой кислоты (3,58 г, 0,014 моль) в безводном н-бутиловом спирте (50 мл) при комнатной температуре. Смесь кипятят с обратным холодильником в течение 16 ч в атмосфере азота, охлаждают до комнатной температуры и затем фильтруют, чтобы оставить оранжевое твердое вещество. Твердое вещество очищают промыванием последовательно этилацетатом (20 мл) и гептаном (20 мл) и затем окончательно сушат в условиях разрежения с получением пиразолона (3,93 г, 87%) в виде темно-оранжевого твердого вещества, жидкостная хроматография, масс-спектрометрия (ЖХМС) m/z 325,24 [M+H]+при RT 1,47 мин.
Получение промежуточного соединения 2
2-(4-аминофенил)-6-фтор-2,5-дигидропиразол[4,3-c]хинолин-3-он
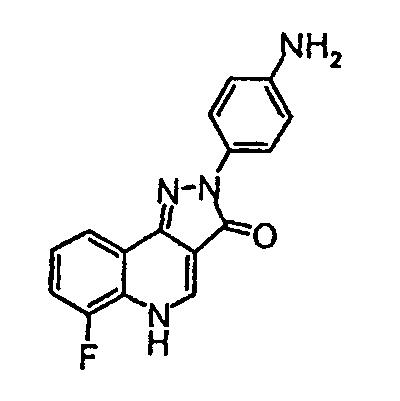
Дигидрохлорид олова (II) (12,5 г, 0,055 моль) добавляют одной порцией к перемешиваемому раствору 2-(4-нитрофенил)-6-фтор-2,5-дигидропиразол[4,3-c]хинолин-3-она (промежуточное соединение 1) (3,59 г, 0,011 моль) в этиловом спирте (110 мл) при комнатной температуре. Смесь затем нагревают до 80°С в течение 8 ч, охлаждают до комнатной температуры и фильтруют с получением желтого твердого вещества. Твердое вещество суспендируют в двухфазном растворе этилацетата (1 л), насыщенного раствора соли Рошеля (500 мл) и насыщенного раствора бикарбоната натрия (500 мл) и перемешивают при комнатной температуре в течение 2 ч. Смесь фильтруют, оставшееся твердое вещество промывают водой и сушат в вакууме с получением указанного в заголовке соединения (3,39 г, 99%) в виде ярко-желтого твердого вещества, ЖХМС m/z 295,30 [M+H]+при RT 0,84 мин.
Пример 1
N-[4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)фенил]-2-метилбутирамид
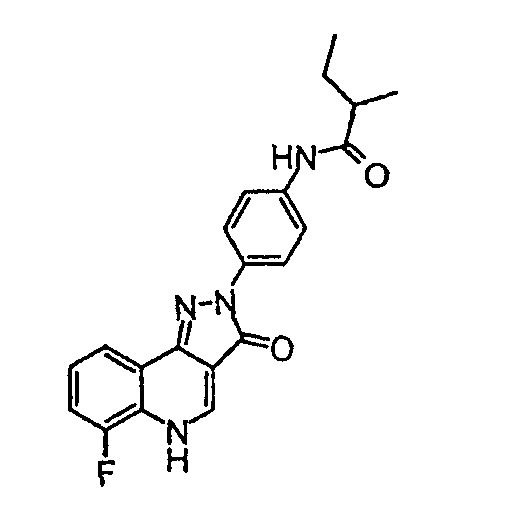
(±)-2-Метилбутирилхлорид (13,6 мкл, 0,11 ммоль) добавляют по каплям в течение 30 с к перемешиваемому раствору 2-(4-аминофенил)-6-фтор-2,5-дигидропиразол[4,3-c]хинолин-3-она (промежуточное соединение 2) (30 мг, 0,10 ммоль), триэтиламина (14 мкл, 0,11 ммоль) и 4-диметиламинопиридина (2,4 мг, 0,02 ммоль) в дихлорметане (1 мл) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение 16 ч. Затем желтое твердое вещество отфильтровывают, очищают промыванием последовательно насыщенным раствором бикарбоната натрия (1 мл), этилацетатом (1 мл) и этиловым спиртом (0,5 мл) и окончательно сушат в условиях разрежения с получением указанного в заголовке соединения (10 мг, 26%) в виде ярко-желтого твердого вещества. ЖХМС m/z 379,36 [M+H]+при RT 1,18 мин. δН (400 МГц, (CD3)2SO) 9,89 (1H, с), 8,52 (1H, с), 8,15 (2H, д, J=9,0 Гц), 8,01 (1H, д, J=1,0 Гц), 7,69 (2H, д J=9,0 Гц), 7,57-7,46 (2H, м), 2,46-2,39 (1H, м), 1,69-1,36 (2H, м), 1,11 (3H, д, J=6,8 Гц), 0,91 (3H, т, J=7,3 Гц).
Указанное в заголовке соединение и соединения последующих примеров испытывали в описанном ниже анализе в разделе «Анализы» для определения их активности в качестве ингибиторов взаимодействия CD80-CD28. Указанное в заголовке настоящее соединение имело оценку активности ***.
Примеры 2-49
Следующие соединения синтезировали, как описано в примере 1, заменяя (±)-2-метилбутирилхлорид соответствующим хлорангидридом кислоты.
Пример 2
[4-(6-фтор-3-оксо-3,5-дигидропиразол[4, 3-c]хинолин-2-ил)фенил]амид 2-метилпентановой кислоты
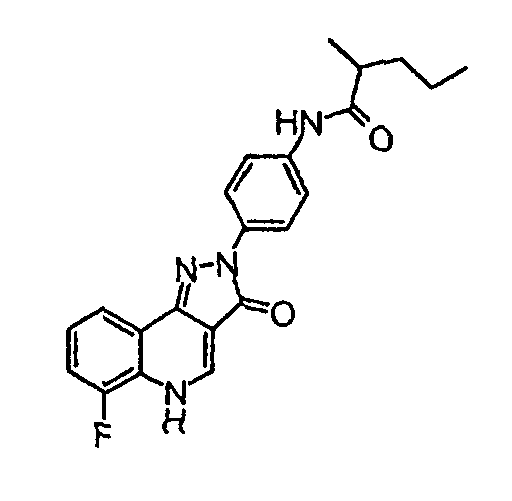
δH (400 МГц, (CD3)2SO) 9,92 (1H, с), 8,53 (1H, с), 8,12 (2H, д, J=9,2 Гц), 8,05 (1H, д, J=7,6 Гц), 7,70 (2H, д, J=9,2 Гц), 7,63-7,53 (2H, м), 1,68-1,58 (1H, м), 1,38-1,28 (3H, м), 1,11 (3H, д, J=6,6 Гц), 0,91 (3H, т, J=7,1 Гц).
Активность ***
Пример 3
[4-(6-фтор-3-оксо-3, 5-дигидропиразол[4,3-c]хинолин-2-ил)фенил]амид 1-метил-1Н-пиррол-2-карбоновой кислоты
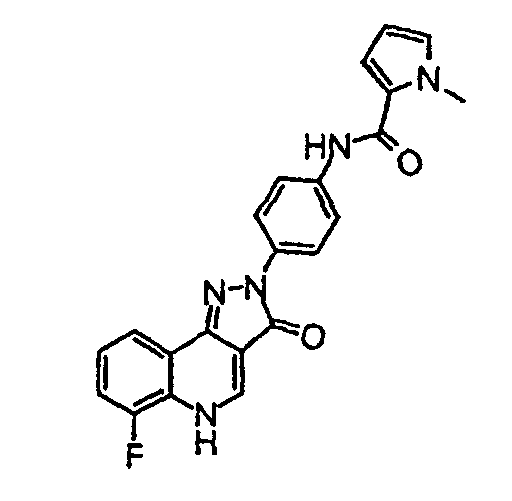
δH (400 МГц, (CD3)2SO) 9,76 (1H, с), 8,50 (1H, с), 8,26 (2H, д, 9,0 Гц), 7,97-7,94 (1H, м), 7,73 (2H, д J=9,0 Гц), 7,39-7,28 (2H, м), 7,07-7, 01 (2H, м), 3,91 (3H, с).
Активность *
Пример 4
N-[4-(6-фтор-3-оксо-3,5-дигидропиразол[4, 3-c]хинолин-2-ил)фенил]-3-метилбутирамид
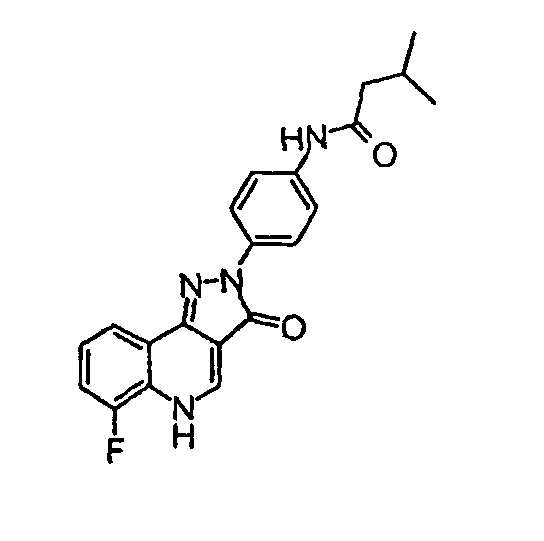
δH (400 МГц, (CD3)2SO) 9,92 (1Н, с), 8,52 (1H, с), 8,14 (2H, д, J=9,2 Гц), 8,01 (1Н, д, J=7,3 Гц), 7,67 (2H, д, J=9,2 Гц), 7,57-7,47 (2H, м), 2,21 (2H, д, J=6,8 Гц), 12, 14-2,07 (1Н, м), 0,96 (6H, д, J=6,6 Гц).
Активность **
Пример 5
[4-(6-фтор-3-оксо-3,5-дигидропиразол[4, 3-c]хинолин-2-ил)фенил]амид 2-пропилпентановой кислоты
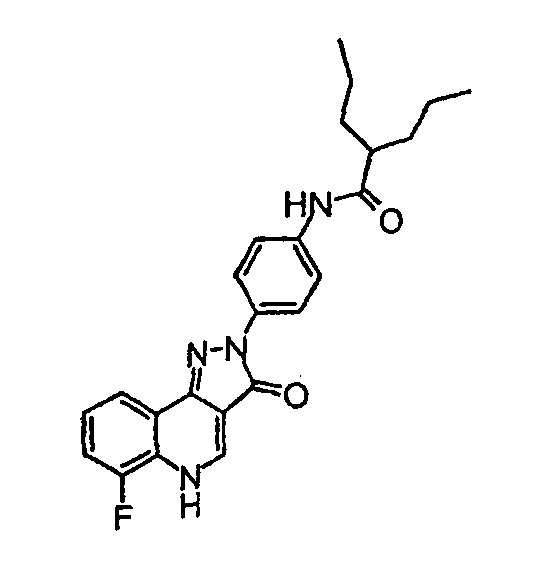
δH (400 МГц, (CD3)2SO) 9,93 (1H, с), 8,53 (1H, с), 8,11 (2H, д, J=9,0 Гц), 8,05 (1H, д, J=7,8 Гц), 7,70 (2H, д, J=9,0 Гц), 7,59-7,46 (2H, м), 2,46-2,35 (1H, м), 1,63-1,27 (4H, м), 0,90 (6H, т, J=7,1 Гц).
Активность *
Пример 6
Метиловый эфир 5-[4-(6-фтор-3-оксо-3, 5-дигидропиразол[4,3-c]хинолин-2-ил)фенилкарбамоил]пентановой кислоты
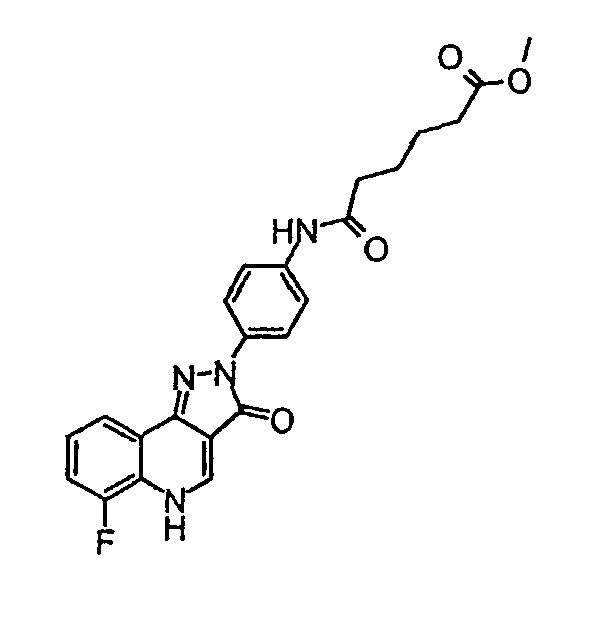
δH (400 МГц, (CD3)2SO) 9,85 (1Н, с), 8,47 (1H, с), 8,25 (2H, д, J=9,0 Гц), 7,91-7,90 (1H, м), 7,59 (2H, д, J=9,0 Гц), 7,29-7,20 (2H, м), 3,61 (3H, с), 2,38-2,28 (4H, м), 1,64-1,50 (4H, м).
Активность ***
Пример 7
N-[4-(6-фтор-3-оксо-3,5-дигидропиразол[4, 3-c]хинолин-2-ил)фенил]-2,2-диметилпропионамид
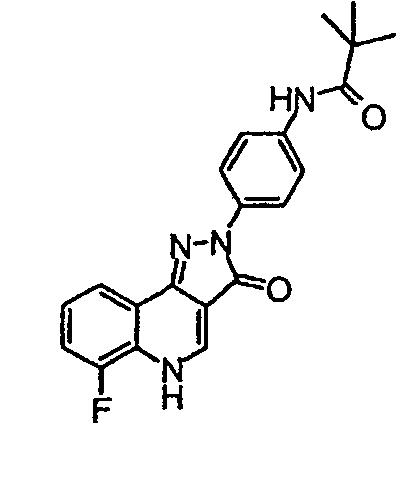
δH (400 МГц, (CD3)2SO) 9,26 (1Н, с), 8,52 (1H, с), 8,15 (2H, д, J=9,2 Гц), 8,03 (1H, д, J=8,8 Гц), 7,71 (2H, д, J=9,2 Гц), 7,56-7,47 (2H, м), 1,26 (9H, с).
Активность **
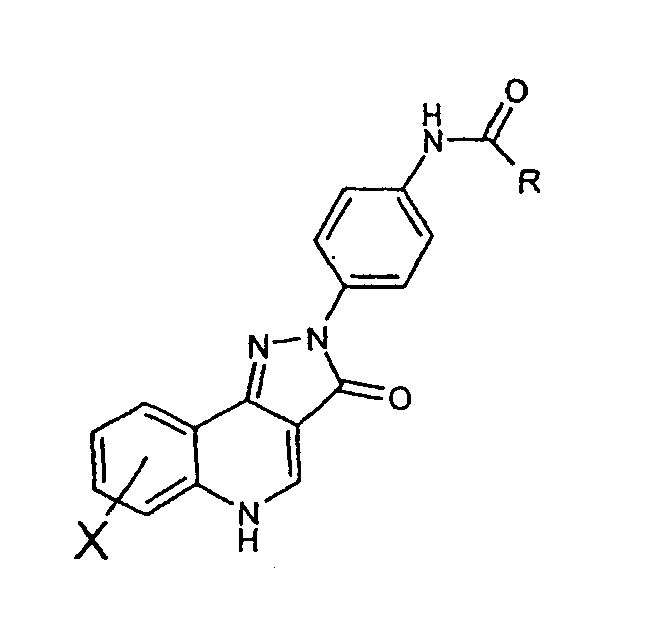
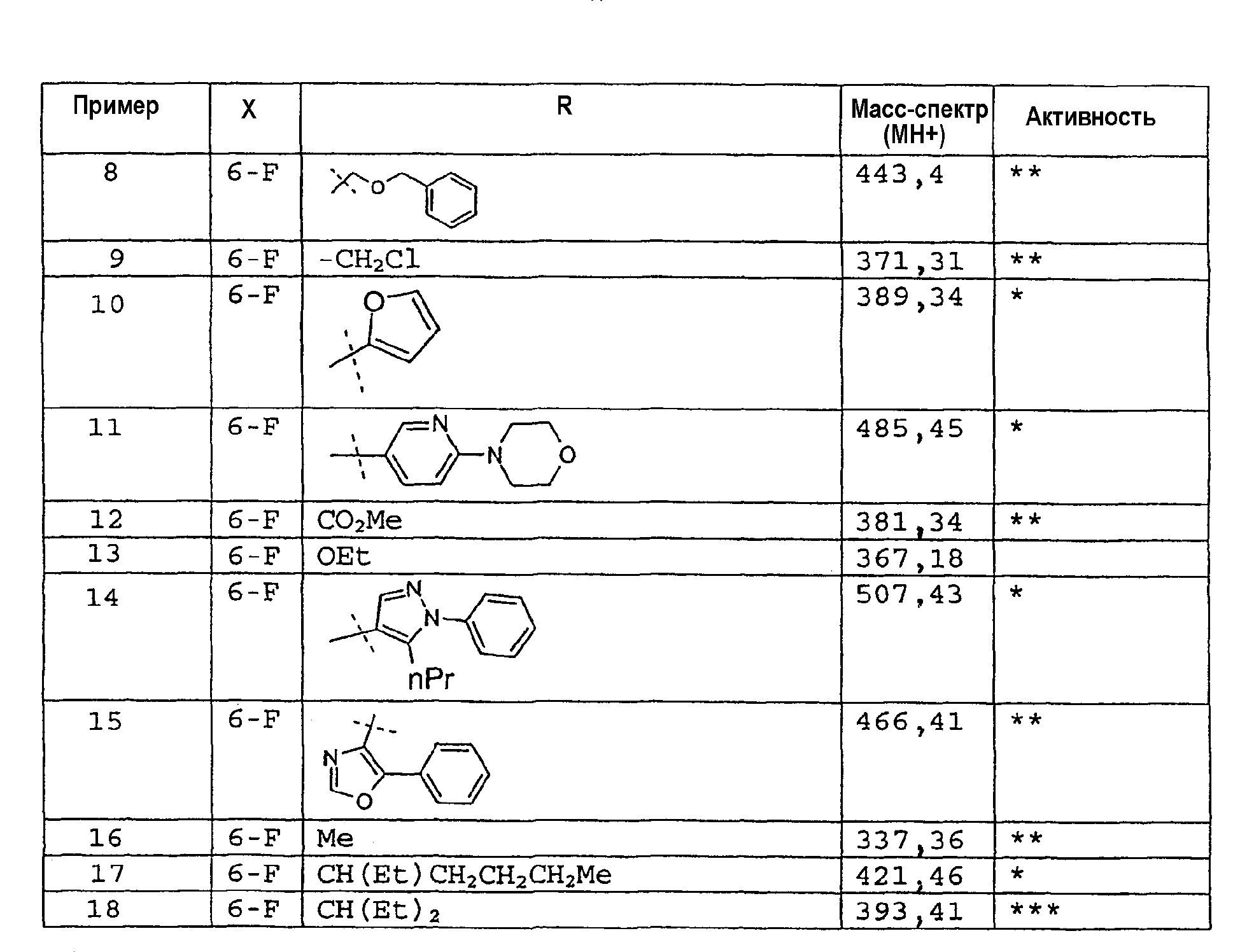
Соединения примеров 8-28 также получали способом примера 1 с использованием соответствующего хлорангидрида кислоты:
Получение промежуточного соединения 3
4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензойная кислота
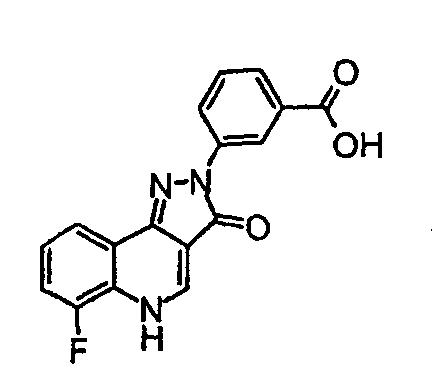
3-гидразинбензойную кислоту (1,91 г, 0,013 моль) добавляют одной порцией в перемешанный раствор этилового эфира 4-хлор-8-фторхинолин-3-карбоновой кислоты (2,93 г, 0,011 моль) в н-бутаноле (60 мл) при комнатной температуре. Раствор кипятят с обратным холодильником в течение 16 ч, охлаждают до комнатной температуры и полученное желтое твердое вещество отфильтровывают, промывают трет-бутилметиловым эфиром и затем сушат. Твердое вещество повторно растворяют в растворе тетрагидрофуран:вода (2:1; 21 мл) и затем добавляют гидроксид лития (1,27 г, 0,031 моль). После перемешивания при комнатной температуре в течение 16 ч концентрированную хлористоводородную кислоту (3 мл) добавляют по каплям к смеси с осаждением желтого твердого вещества, которое фильтруют и сушат в вакууме с получением указанного в заголовке соединения (промежуточное соединение 3) (2,32 г, 63%) в виде ярко-желтого твердого вещества.
Получение промежуточного соединения 4
3-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорид
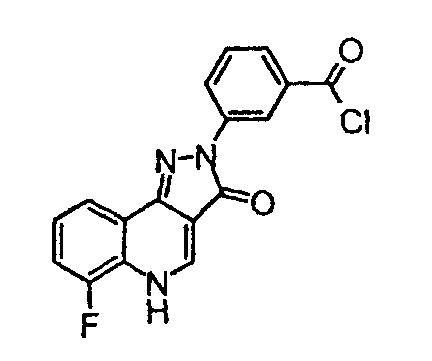
Оксалилхлорид (20 мл, 0,2 моль) добавляют по каплям в течение 2 мин к перемешиваемому раствору 3-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензойной кислоты (промежуточное соединение 3) (2,0 г, 6,1 ммоль) в дихлорметане (10 мл) при комнатной температуре. Затем добавляют N,N-диметилформамид (50 мкл) и полученную смесь нагревают до 50°С в течение 1 ч. Затем раствор охлаждают до комнатной температуры и концентрируют в вакууме с получением указанного в заголовке соединения (промежуточное соединение 4) (2,0 г, 96%) в виде бежевого твердого вещества.
Пример 50
3-(6-фтор-3-оксо-3, 5-дигидропиразол[4,3-c]хинолин-2-ил)-N-(3-метоксипропил)бензамид
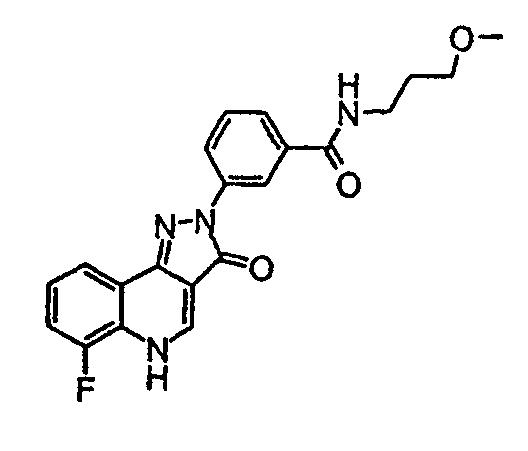
3-метоксипропиламин (0,026 г, 0,29 ммоль) добавляют к перемешиваемому раствору 3-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорида (промежуточное соединение 4) (26 мг, 0,29 ммоль) в тетрагидрофуране (2 мл) и смесь перемешивают при комнатной температуре в течение 15 мин. Затем добавляют триэтиламин (0,2 мл, 1,4 ммоль) и полученную смесь перемешивают в течение ночи. 1М хлористоводородную кислоту (3-4 мл) добавляют по каплям с осаждением желтого твердого вещества, которое фильтруют и сушат в условиях разрежения с получением амида (79 мг, 0,20 ммоль) в виде желтого твердого вещества. ЖХМС m/z 395,25 [M+H]+при RT 1,04 мин; δH (400 МГц, (CD3)2SO) 8,59 (1H, м), 8,57 (1H, с), 8,39 (1H, аппр д, J=9, 3 Гц), 8,08 (1H, аппр д, J=7,3 Гц), 7,66-7,53 (5H, м), 3,37-3,33 (4H, м), 3,27 (3H, с), 1,83-1,77 (2H, м).
Активность **
Пример 51
N-этил-3-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензамид
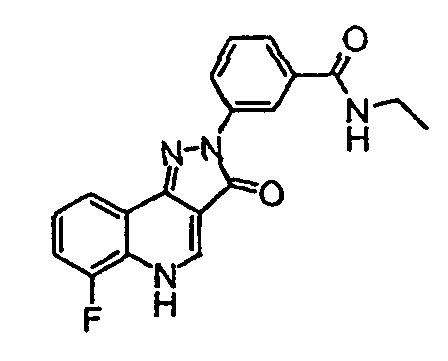
Получают способом примера 50, заменяя 3-метоксипропиламин этиламином.
δH (400 МГц, (CD3)2SO) основной квотированный ротомер; 8,56 (1H, ушир.с), 8,47 (1H, м), 8,21 (2H, д, J=8,5 Гц), 7,94 (2H, д, J=8,5 Гц), 3,96 (3Н, с), 3,31 (2H, кв, J=7,3 Гц), 2,58 (3Н, с), 1,15 (3Н, т, J=7,4 Гц).
Активность **
Пример 52
N-бензил-3-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензамид
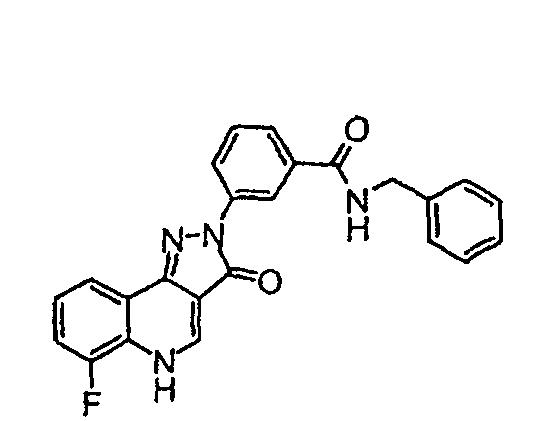
Получают способом примера 50, заменяя 3-метоксипропиламин бензиламином.
ЖХМС m/z 427,16 [M+H]+при RT 1,28 мин.
Активность *
Соединения примеров 53-64 получали способом примера 50 с использованием соответствующего амина.
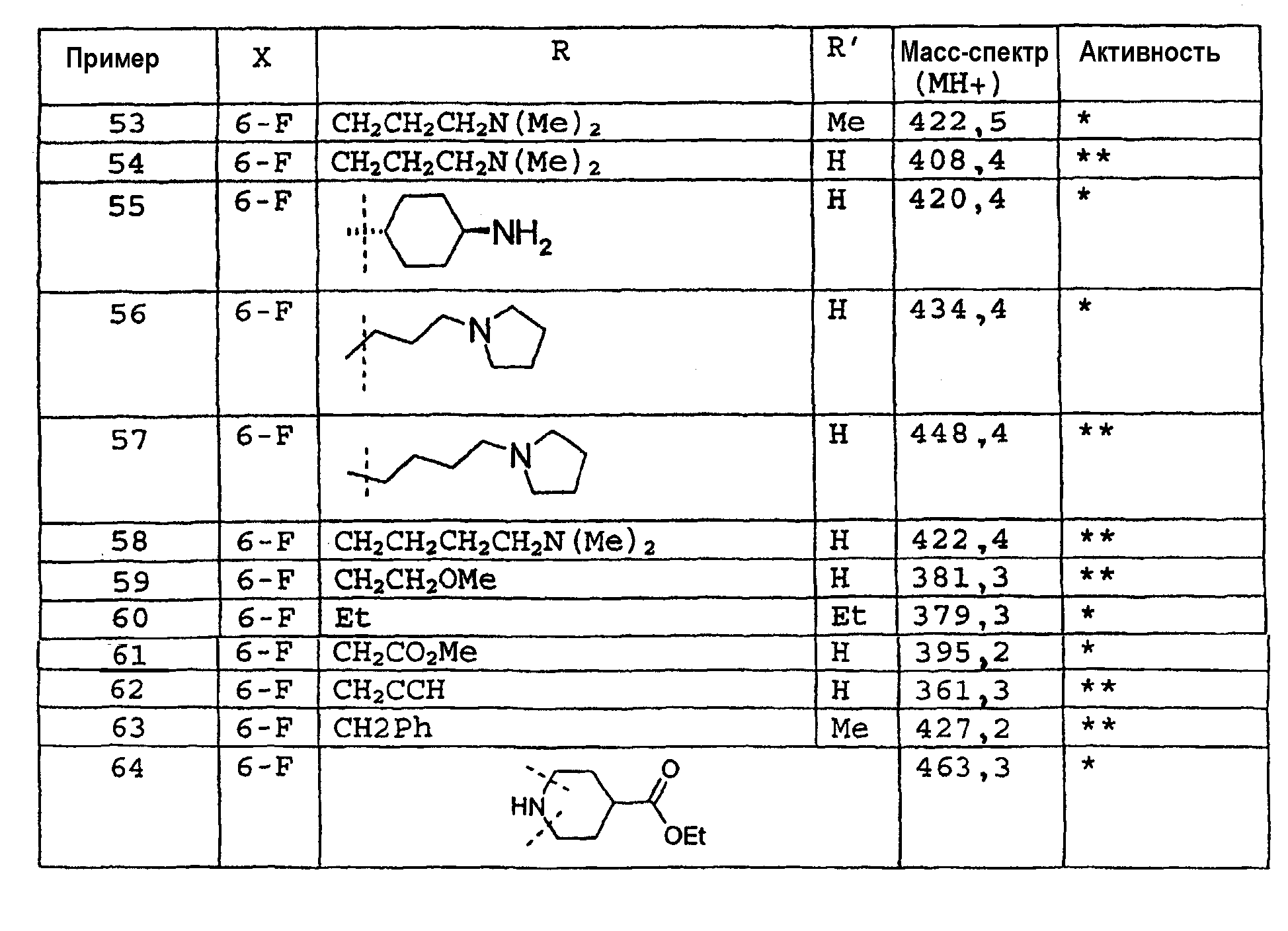
Пример 65
N-(3-диметиламинопропил)-4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил]бензамид
Стадия 1
Этиловый эфир 2-циклопропил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
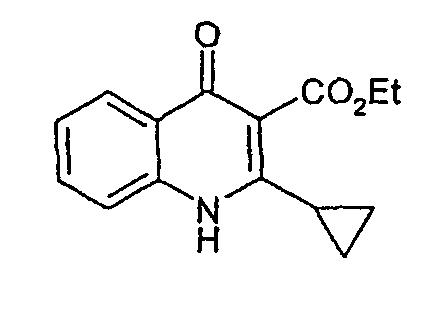
Раствор метилового эфира 3-циклопропил-3-оксопропионовой кислоты (6,2 г, 0,038 моль), 2-этилового эфира 2-аминобезойной кислоты (4,95 г, 0,03 моль) и паратолуолсульфоновой кислоты (0,04 г, 0,2 моль) в толуоле (25 мл) нагревают при 125°С в течение 2 ч; затем 15 мл растворителя отгоняют. К остаточному оранжевому раствору добавляют этоксид натрия (2М, 15 мл) в этаноле (реакционная смесь становится красной). Красную смесь перемешивают при 120°С в течение 2 ч; 15 мл растворителя снова отгоняют. Реакционную смесь оставляют для охлаждения до комнатной температуры, разбавляют этилацетатом (1 л), экстрагируют HCl 0,1М и водой. Объединенный органический экстракт сушат над сульфатом натрия и концентрируют в вакууме с получением оранжевого остатка, который один раз промывают холодным этилацетатом с получением этилового эфира 2-циклопропил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (3,87 г, 53%) в виде не совсем белого твердого вещества. ЖХМС m/z 244,14 [M+H]+при RT 0,78 мин, 89%, m/z 230,11 [Кислота+H]+при RT 1,27 мин, 11%.
δH (400 МГц, (CD3)2SO) 11,04 (1H, с), 8,06 (1H, дд, J1=1,1, J2=8,1), 7,76-7,66 (2H, м), 7,36 (1H, тд, J1=1,1, J2=7,5), 3,89 (3H, с), 2,16 (1H, м), 1,18 (4H, д, J=7,0).
Стадия 2
Этиловый эфир 4-хлор-2-циклопропилхинолин-3-карбоновой кислоты
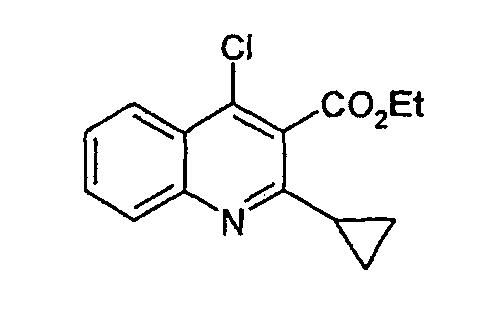
Оксихлорид фосфора (0,77 мл, 0,082 моль) добавляют одной порцией к суспензии этилового эфира 2-циклопропил-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (1,0 г, 0,041 моль) в ацетонитриле и смесь нагревают при 75°С в течение 90 мин (раствор становится прозрачным при температуре выше 65°С). Полученный светло-коричневый раствор выливают в насыщенный бикарбонат натрия (100 мл); суспензию экстрагируют этилацетатом, объединенный органический экстракт сушат и концентрируют в вакууме с получением этилового эфира 4-хлор-2-циклопропилхинолин-3-карбоновой кислоты (1,15 г, 106%) в виде не совсем белого твердого вещества. Rf (AcOEt)=0, 73.
Стадия 3
4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензойная кислота
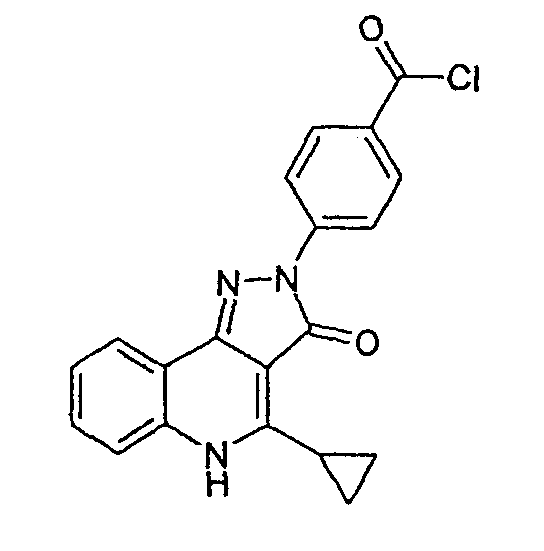
Этиловый эфир 4-хлор-2-циклопропилхинолин-3-карбоновой кислоты (1,15 г, 0,0041 моль) и 4-гидразинбензойной кислоты (1,0 г, 0,0068 моль) перемешивают в этаноле (30 мл) при кипячении с обратным холодильником в течение 16 ч. Светло-желтую суспензию разбавляют гептаном, фильтруют, промывают холодным трет-бутилметиловым эфиром и оставляют сушиться в условиях разрежения с получением неочищенного твердого вещества, содержащего гидразин. Твердое вещество суспендируют в 1М HCl, фильтруют, промывают водой и затем сушат в вакууме с получением 4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензойной кислоты (1,135 г, 80%) в виде желтого твердого вещества. ЖХМС m/z 346,20 [M+H]+при RT 1,05 мин: чистота 96%.
δH (400 МГц, (CD3)2SO) 11,4 (1H, с), 8,43 (2H, д, J=8,1), 8,21 (1H, дд, J=1,2, J=8,1), 8,07 (2H, д, J=8,1), 7,92 (1H, д, J=8,1), 7,67 (1H, т, J=6,6), 7,52 (1H, т, J=6,5), 3,43 (1H, м), 1,59 (2H, м), 1,43 (2H, м).
Стадия 4
4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорид
К суспензии мелко измельченной 4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензойной кислоты (0,19 г, 0,55 ммоль) в дихлорметане (4 мл) добавляют оксалилхлорид (1,6 мл, 0,01 моль) с последующим добавлением капли диметилформамида. Смесь перемешивают в атмосфере азота при 45°С в течение 8 ч. Растворитель затем удаляют в вакууме с получением 4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорида в виде бледно-желтого твердого вещества. ЖХМС m/z 346,20 [M+MeOH-Cl]+при RT 1,46 мин: чистота 95%. Используют без дальнейшей очистки.
Стадия 5
N-(3-диметиламинопропил)-4-(4-циклопропил-3-оксо-3, 5-дигидропиразол[4,3-c]хинолин-2-ил)бензамид
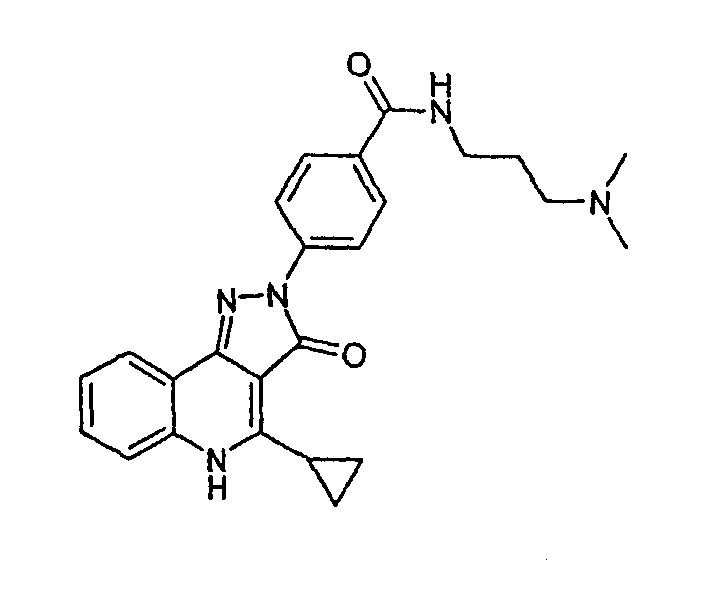
К неполному раствору 4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорида (0,1 г, 0,28 ммоль) в тетрагидрофуране (6 мл) в атмосфере азота добавляют раствор 3-диметиламинопропиламина (0,03 г, 0,3 ммоль) в тетрагидрофуране (3 мл). Смесь перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют при пониженном давлении, желтое твердое вещество промывают небольшим количеством насыщенного бикарбоната натрия, водой и сушат в вакууме с получением N-(3-диметиламинопропил)-4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензамида (57 мг, 47%) в виде желтого твердого вещества. ЖХМС m/z 430,11 [M+H]+при RT 0,99 мин: чистота 100%.
Активность ***
Получение промежуточного соединения 5
4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорид
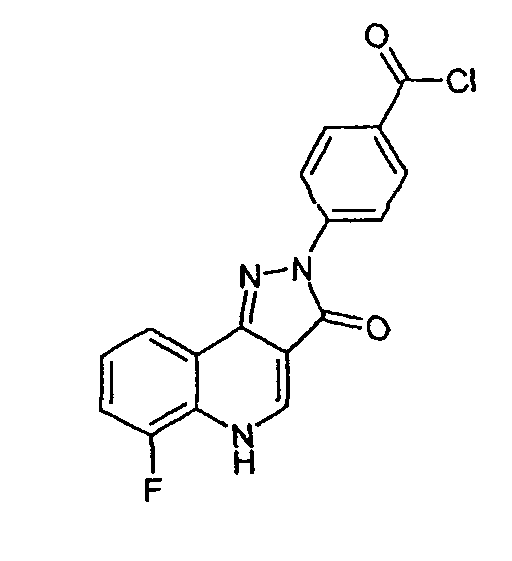
К суспензии мелко измельченной 4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензойной кислоты (1,1 г, 3,4 ммоль) в дихлорметане (6 мл) добавляют оксалилхлорид (2,4 мл, 29 ммоль) с последующим добавлением капли диметилформамида. Смесь перемешивают в атмосфере азота при 45°С в течение 3 ч. Растворитель удаляют в вакууме с получением 4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорида (1,15 г, количественный) в виде бледно-желтого твердого вещества, которое используют без дальнейшей очистки.
Пример 66
Гидрохлорид N-(3-диметиламинопропил)-4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензамида
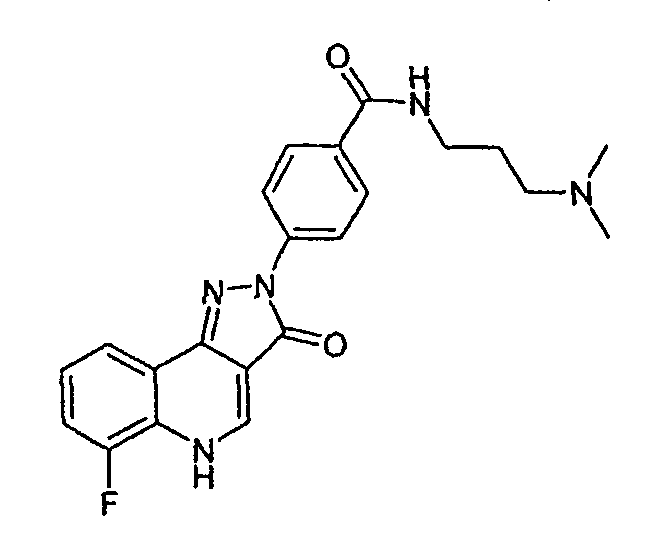
К неполному раствору 4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензоилхлорида (0,1 г, 0,3 ммоль) в тетрагидрофуране (5 мл) в атмосфере азота добавляют раствор 3-диметиламинопропиламина (0,03 г, 0,3 ммоль) в тетрагидрофуране. Смесь перемешивают при комнатной температуре в течение 90 мин. Растворитель удаляют при пониженном давлении и желтое твердое вещество очищают жидкостной хроматографией на силикагеле (градиентное элюирование, MeOH:H2O, Fluka обращенная фаза С18) с получением гидрохлорида N-(3-диметиламинопропил)-4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)бензамида (70 мг, 53%) в виде желтого твердого вещества.
ЖХМС m/z 408,39 [M+H]+при RT 0,89 мин: чистота 90%.
Активность ***
Соединения примеров 67-141 получают аналогичным образом из соответствующего бензоилхлорида и соответствующего амина
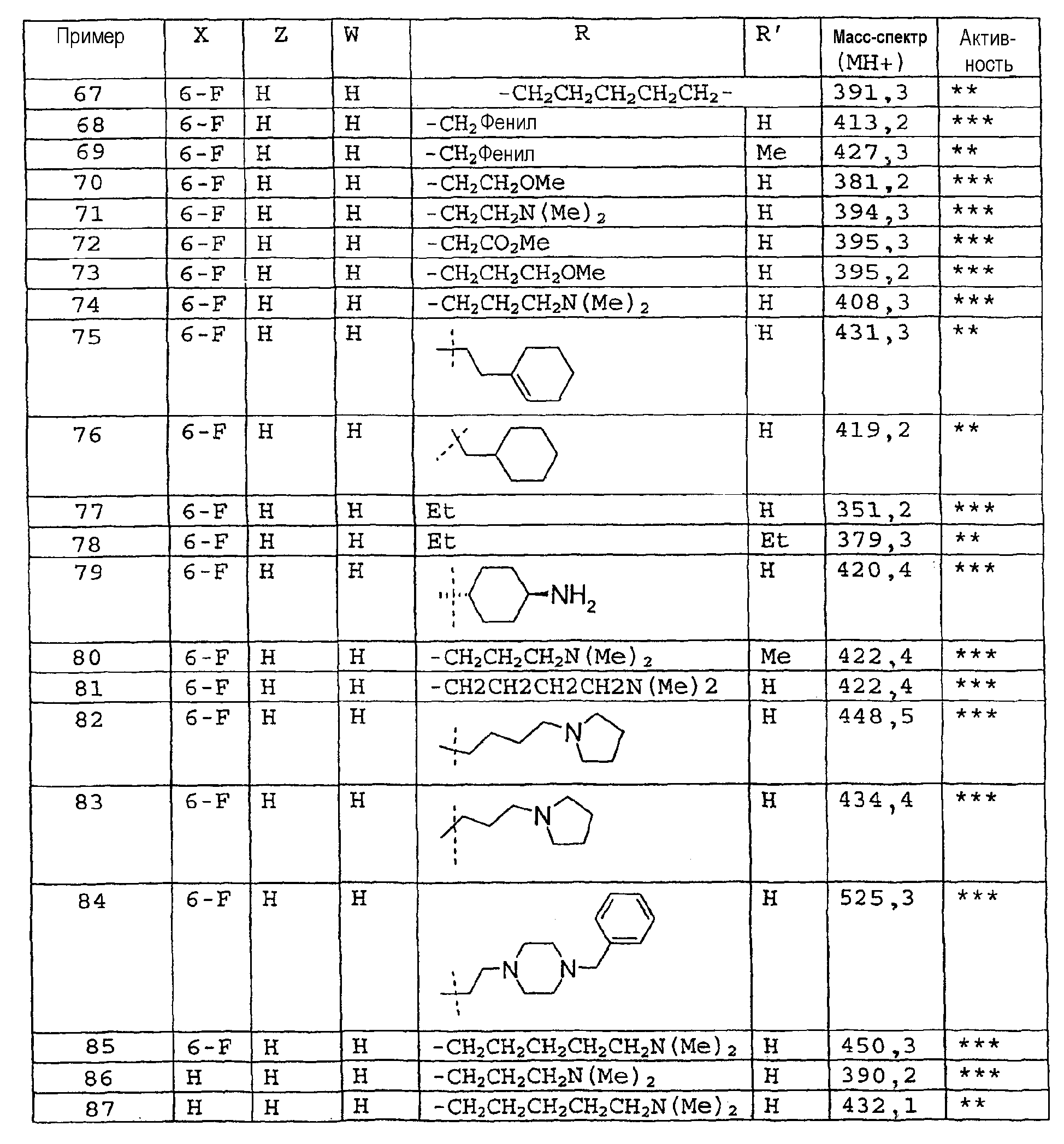
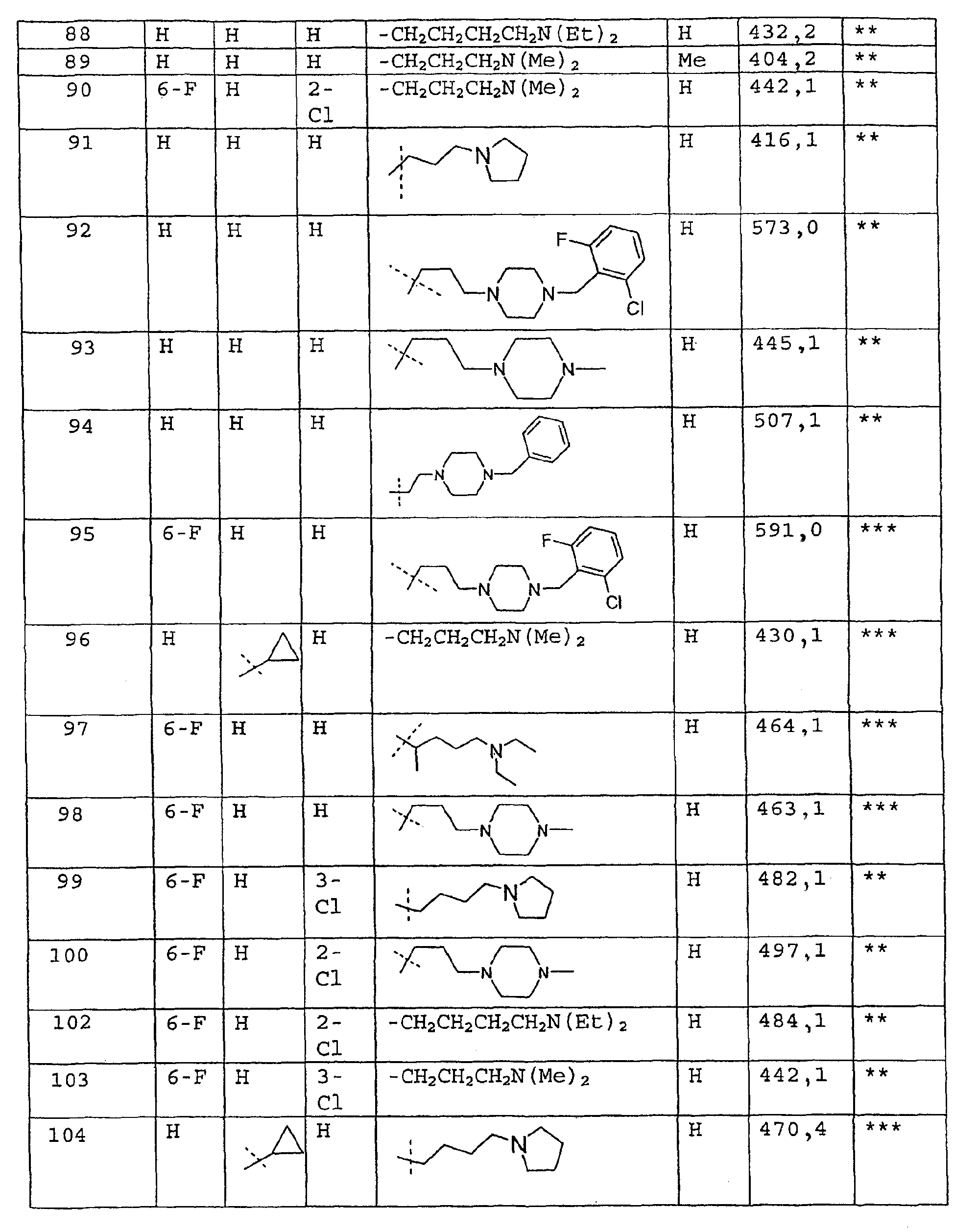
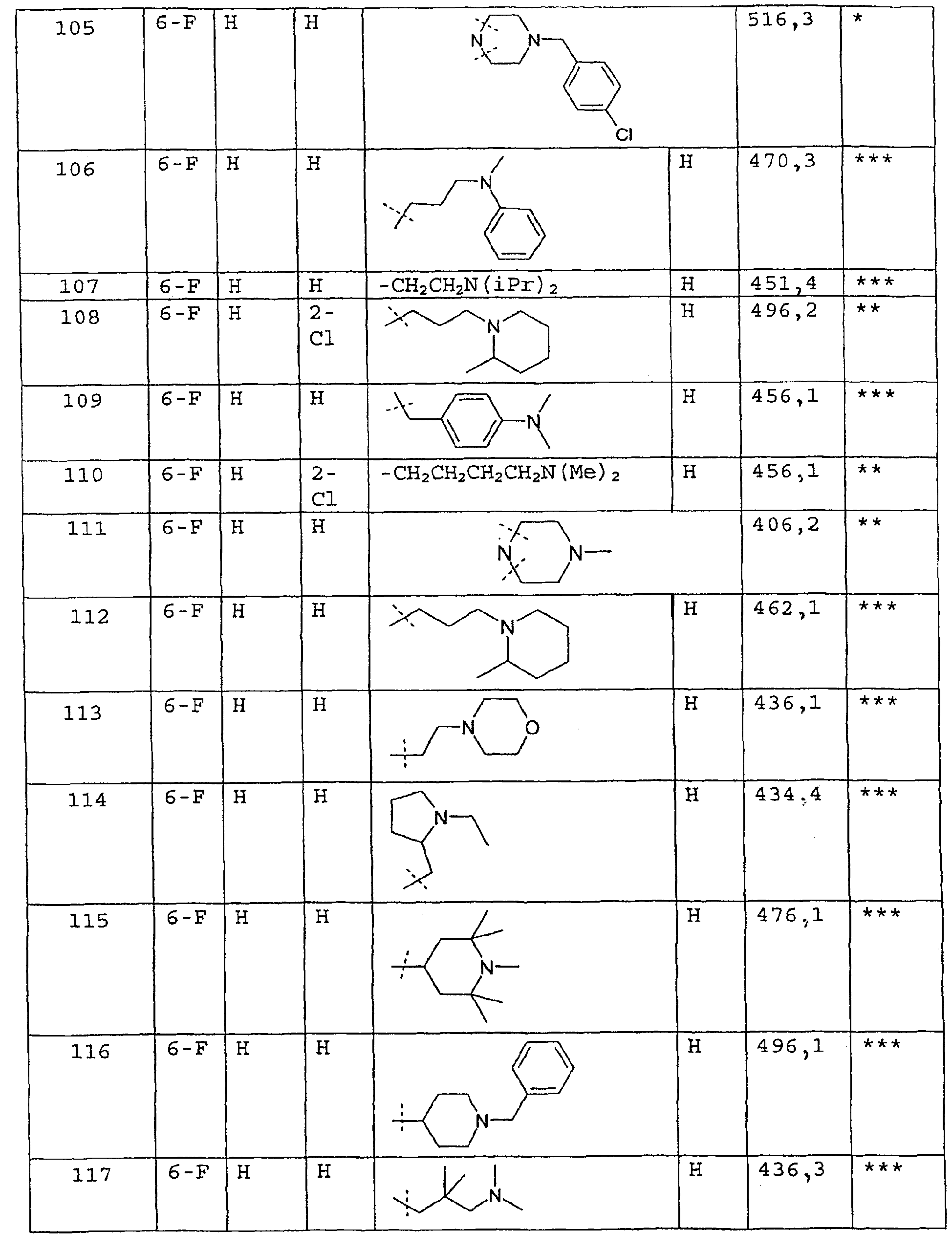
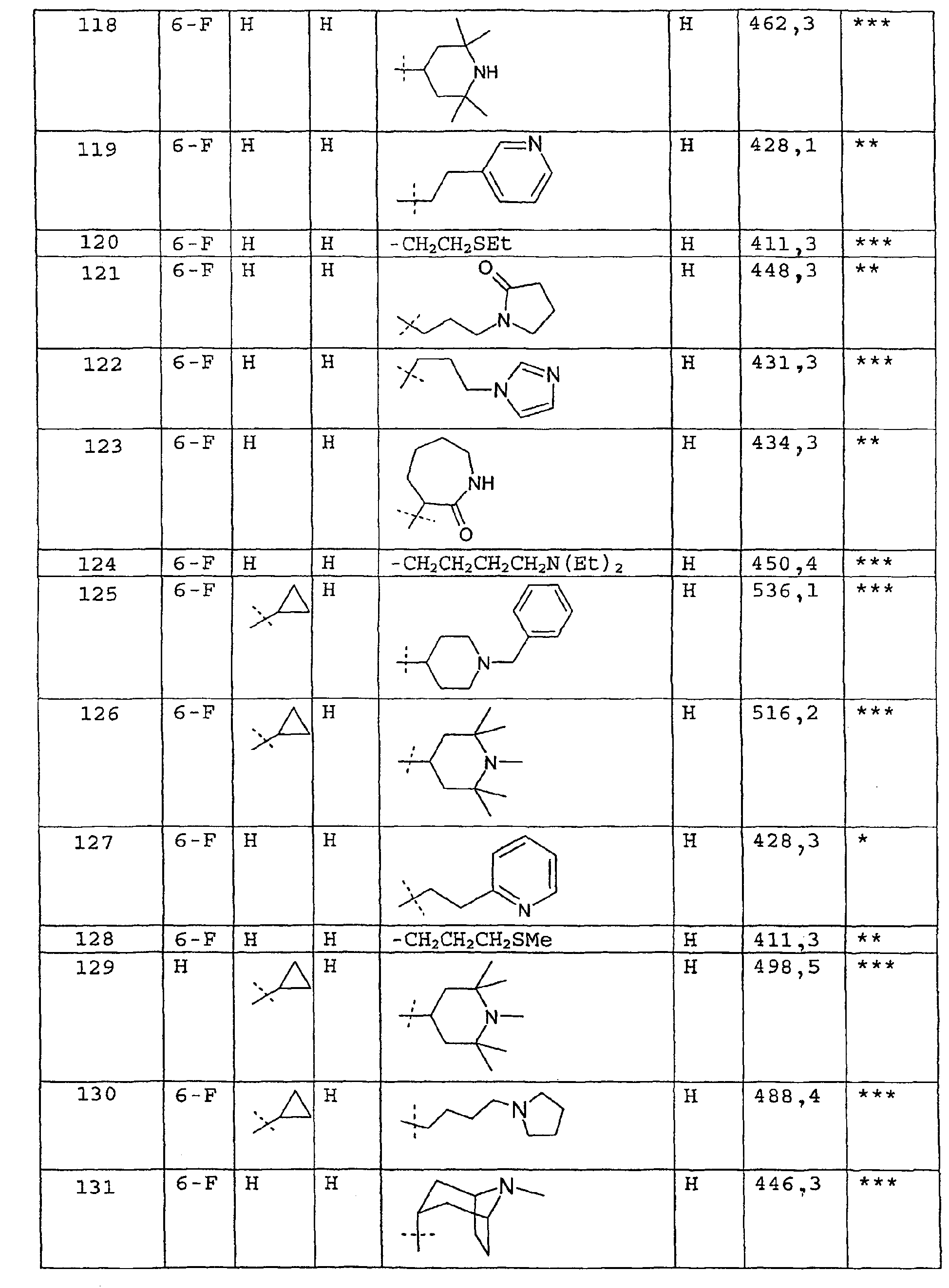
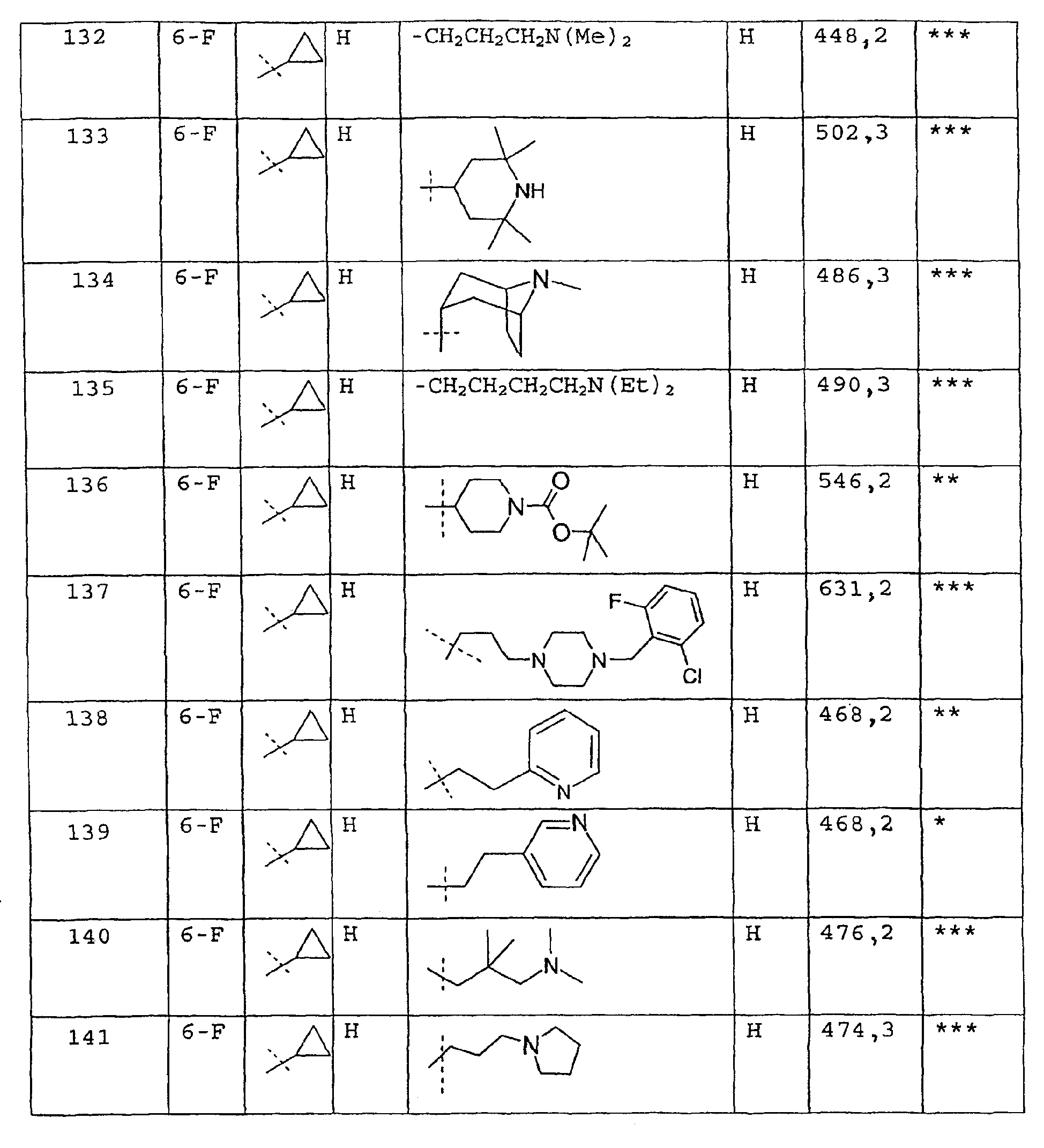
Пример 142
Этиловый эфир {3-[4-(6-фтор-3-оксо-3,5-дигидропиразол[4,3-c]хинолин-2-ил)фенил]уреидо}уксусной кислоты
Этилцианатацетат (31 мг, 0,24 ммоль) добавляют одной порцией к перемешиваемому раствору 2-(4-аминофенил)-6-фтор-2,5-дигидропиразол[4,3-c]хинолин-3-она (промежуточное соединение 2) (50 мг, 0,17 ммоль) в N-диметилформамиде (2 мл) и смесь перемешивают при комнатной температуре в течение 16 ч. Затем к смеси добавляют воду (1 мл) для осаждения твердого вещества, которое отфильтровывают, промывают водой (1 мл) и затем этилацетатом (1 мл) и, наконец, сушат в условиях разрежения с получением мочевины в виде желтого твердого вещества. ЖХМС m/z 424,40 [M+H]+при RT 1, 06 мин.
Активность ***
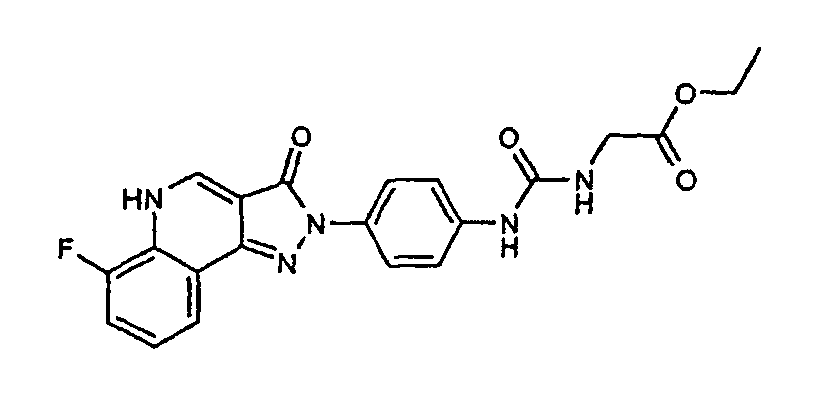
Примеры 143 и 144
Следующие соединения синтезируют способом примера 142, заменяя этилцианатацетат соответствующим изоцианатом, изотиоцианатом или хлорформиатом.
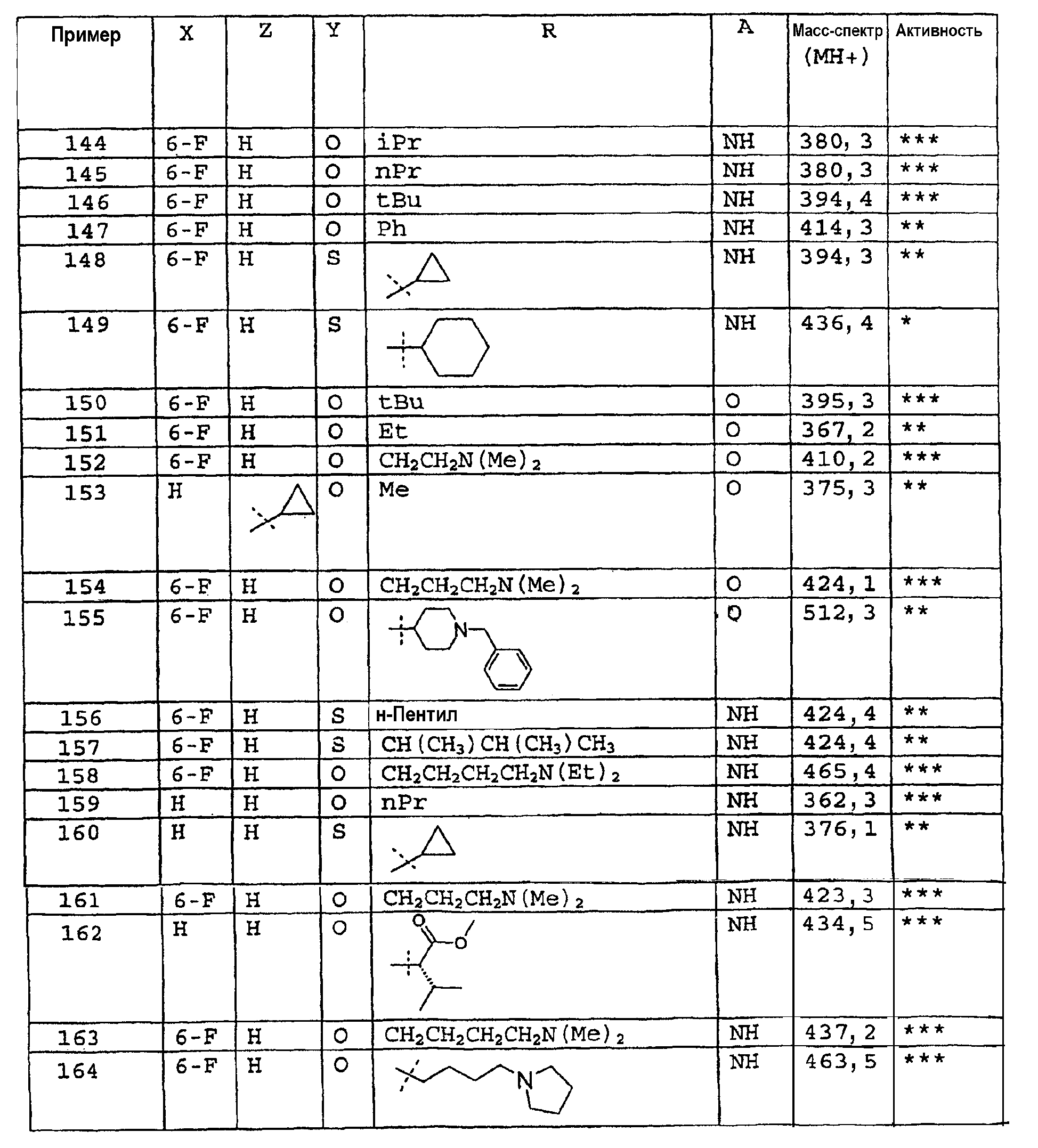
Промежуточное соединение 6: Получение метил-4-оксотиохроман-3-карбоксилата
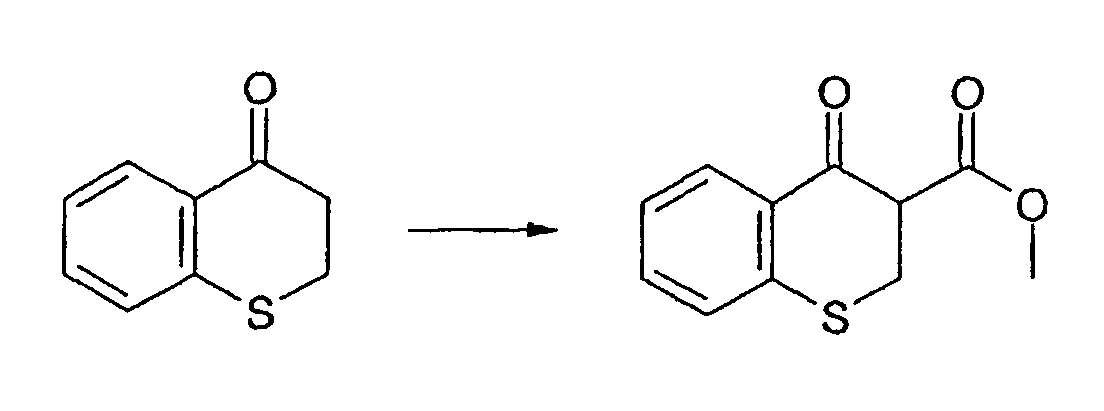
Сухой тетрагидрофуран (60 мл) охлаждают в атмосфере азота до -50-60°С. Добавляют 1М раствор бис(триметилсилил)амида лития в гексане (56 мл, 56 ммоль). Температуру поддерживают при (-50)-(-60)°С и добавляют тиохроман-4-он по каплям в течение 20 мин. Перемешивание продолжают при низкой температуре в течение 60 мин. Метилцианоформиат (4,84 мл, 60,9 ммоль) добавляют по каплям в течение 5 мин к реакционной смеси. Полученную суспензию перемешивают при (-50)-(-60)°С в течение 80 мин и затем дают возможность нагреться до комнатной температуры. Добавляют насыщенный раствор хлорида аммония (100 мл). Фазы разделяют, водную фазу экстрагируют этилацетатом (2×100 мл). Объединенные органические фазы промывают водой (50 мл), сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Получают оранжевое масло и очищают колоночной хроматографией. Указанное в заголовке соединение выделяют в виде желтого твердого вещества (4,70 г, 21,1 ммоль, 42%). ЖХМС: m/z 221 [M-H]*.
Промежуточное соединение 7: Получение 4-(3-оксо-3а,4-дигидро-3Н-тиохромен[4,3-c]пиразол-2-ил)бензойной кислоты
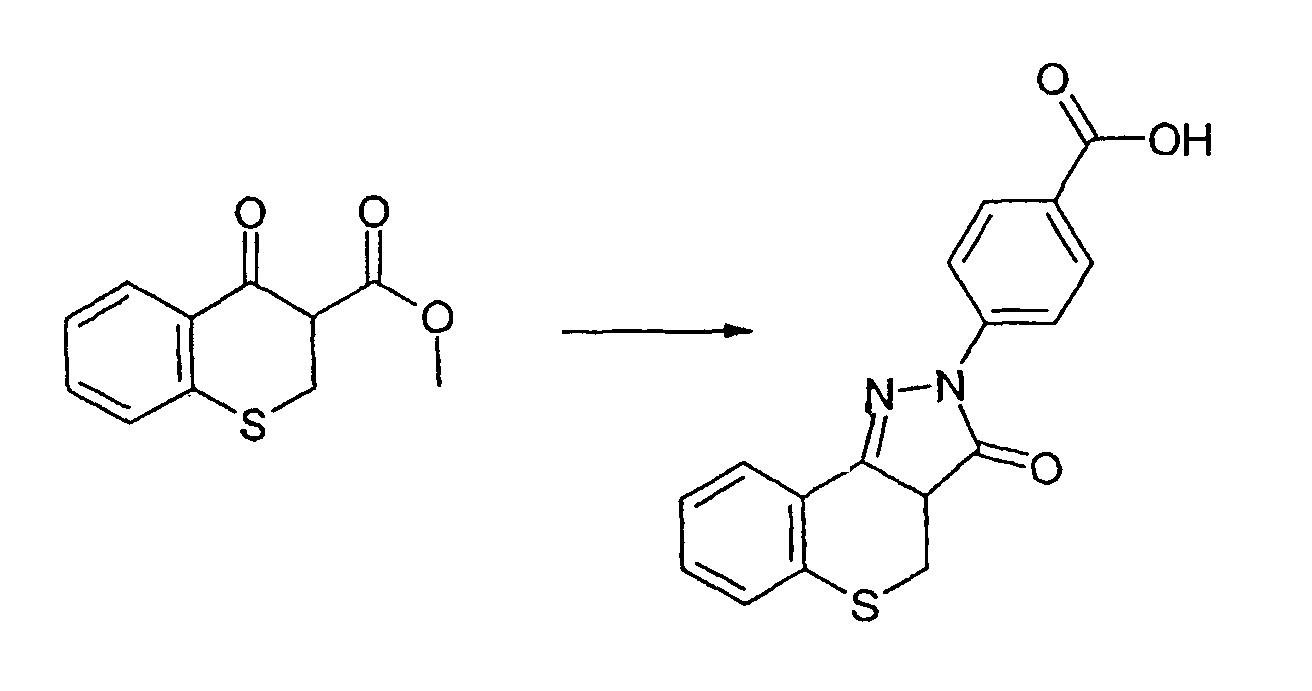
4-оксотиохроман-3-карбоксилат (0,50 г, 2,25 ммоль) и гидразинбензойную кислоту (0,377 г, 2,48 ммоль) перемешивают в уксусной кислоте (6 мл). Смесь кипятят с обратным холодильником в течение 30 мин. Избыточную уксусную кислоту отгоняют с получением коричневого масла. Добавляют диэтиловый эфир, образуется осадок, который отфильтровывают и сушат в вакууме. Неочищенный продукт выделяют в виде красно-коричневого твердого вещества (797 мг). ЖХМС: m/z 325 [M+H]+.Очистку не проводят.
Промежуточное соединение 8: Получение 4-(3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензойной кислоты
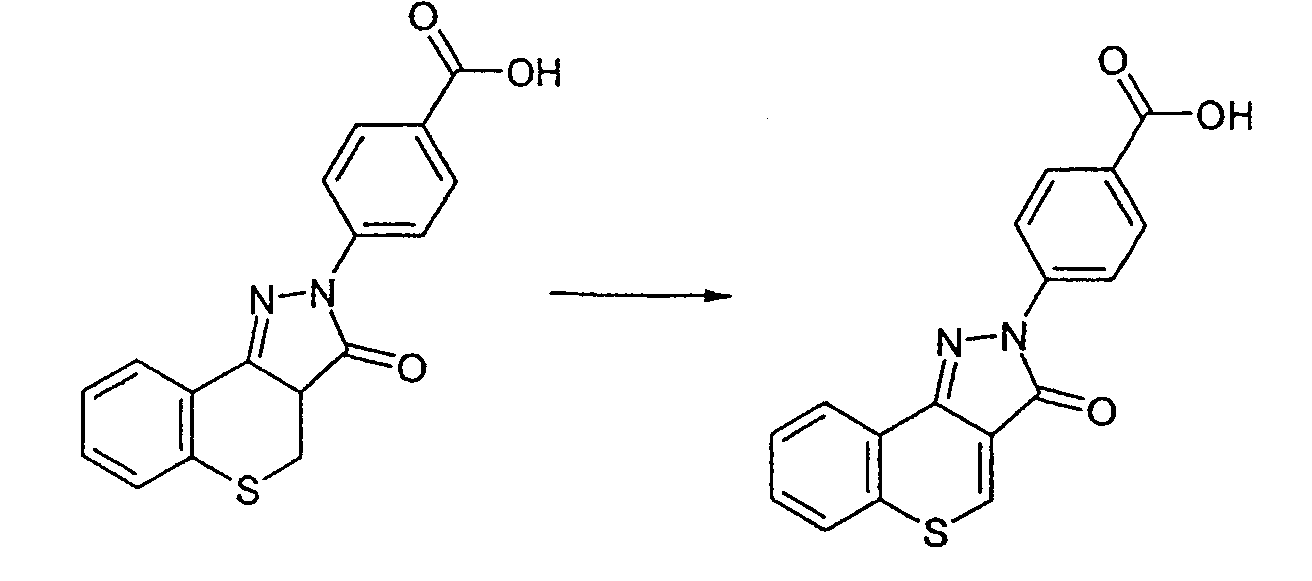
Неочищенную 4-(3-оксо-3а,4-дигидро-3Н-тиохромен[4,3-c]пиразол-2-2-ил)бензойную кислоту (250 мг, 0,77 ммоль) растворяют в диметилсульфоксиде (6 мл). Добавляют О-хлоранил (189 мг, 0,77 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Добавляют воду (20 мл) и твердые вещества отфильтровывают и промывают водой. Осадок на фильтре растирают в толуоле, фильтруют и сушат в вакууме. Указанное в заголовке соединение выделяют в виде темно-коричневого твердого вещества (230 мг, 0,71 ммоль, 92%). ЖХМС: m/z 323 [M+H]+.
Альтернативно, неочищенную 4-(оксо-3а,4-дигидро-3Н-тиохромен[4,3-c]пиразол-2-ил)бензойную кислоту можно перемешивать в диметилсульфоксиде при контакте с воздухом. Было обнаружено, что окисление воздухом обеспечивает чистый продут, однако реакция протекает гораздо медленнее.
Пример 165
Получение N-[3-(диметиламино)пропил]-4-(3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензамида
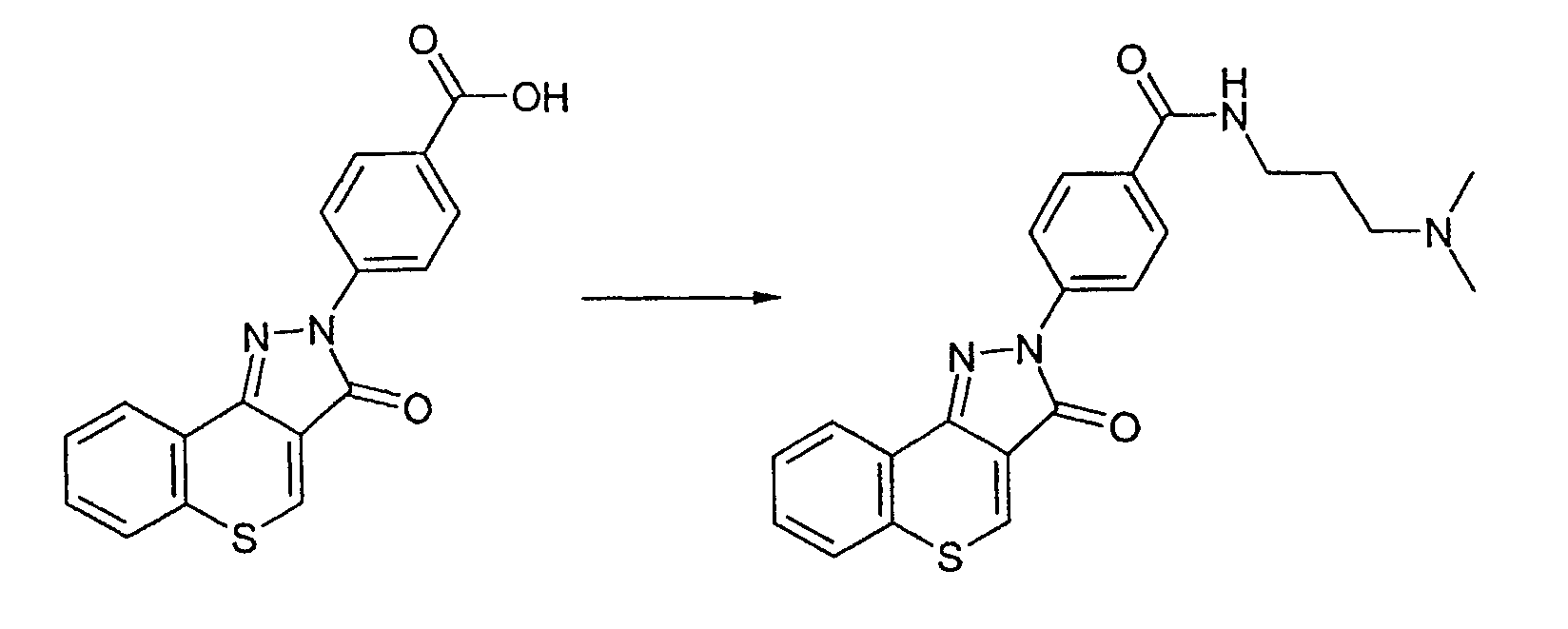
4-(3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензойную кислоту (55 мг, 0,17 ммоль) суспендируют в безводном диметилацетамиде (1 мл). Добавляют диизопропилэтиламин (46,5 мг, 0,36 ммоль, 62 мкл) с последующим добавлением 3-диметиламинопропиламина (17,5 мг, 0,17 ммоль) и гексафторфосфата [(бензотриазол-1-илокси)диметиламинометилен]диметиламмония (65 мг, 0,17 ммоль). Смесь перемешивают при комнатной температуре в течение 4 ч и очищают препаративной ЖХМС. Указанное в заголовке соединение выделяют в виде коричневого твердого вещества. ЖХМС: m/z 407 [M+H]+.
Активность **
Пример 166
Получение N-[(циклогексиламино)пропил]-4-(3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензамида
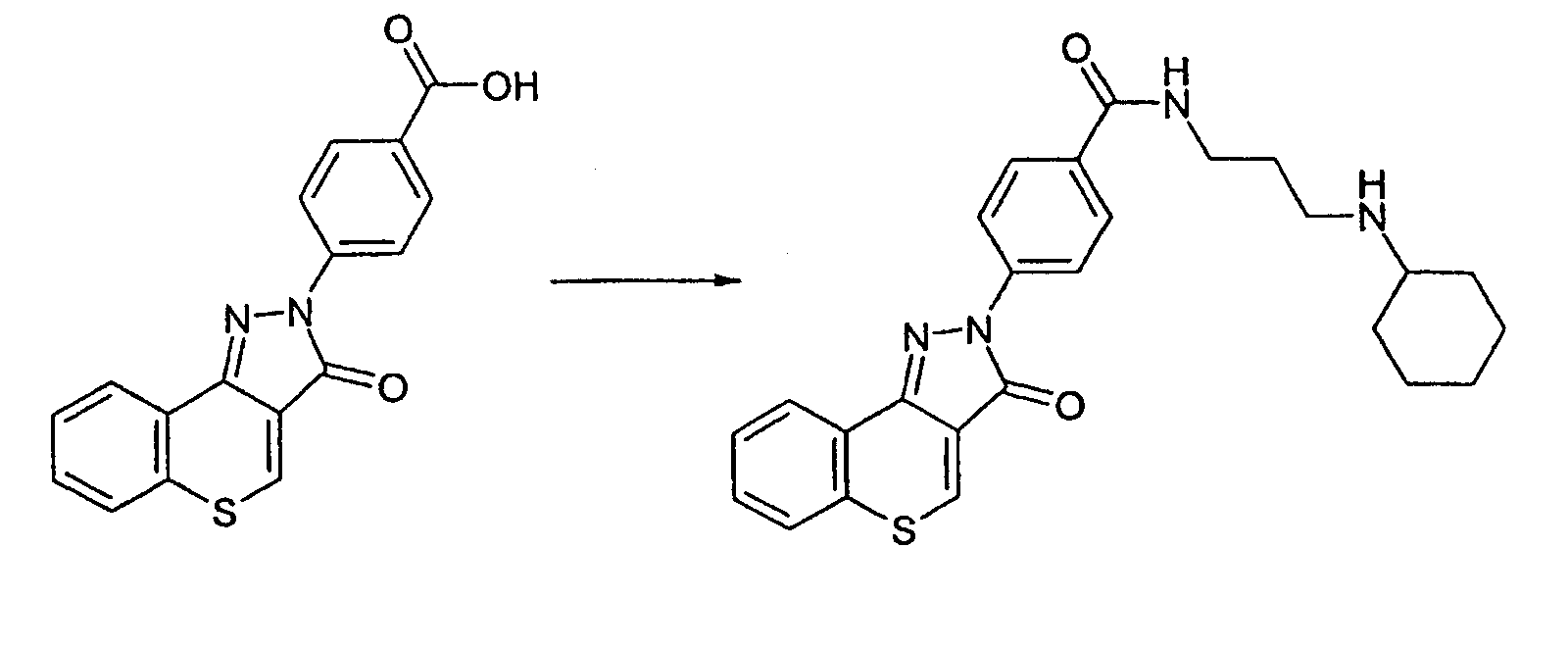
Реакцию проводят, как описано выше. ЖХМС: m/z 461 [M+H]+.
Активность ***
Пример 167
Получение N-(пирролидин-1-илбутил)-4-(3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензамида
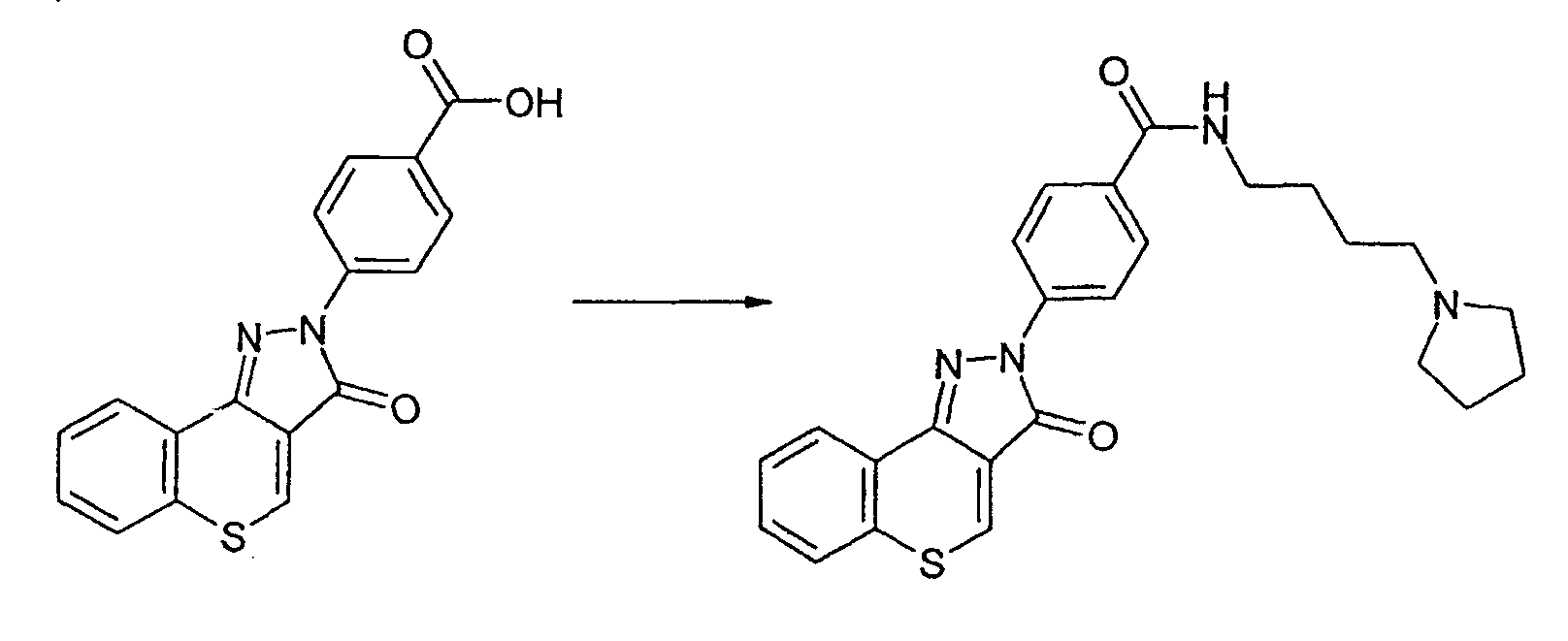
Реакцию проводят, как описано выше. ЖХМС: m/z 447 [M+H]+.
Активность *
Пример 168
Получение 4-(3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)-N-1,2,2,6,6-пентаметилпиперидин-4-илбензамида
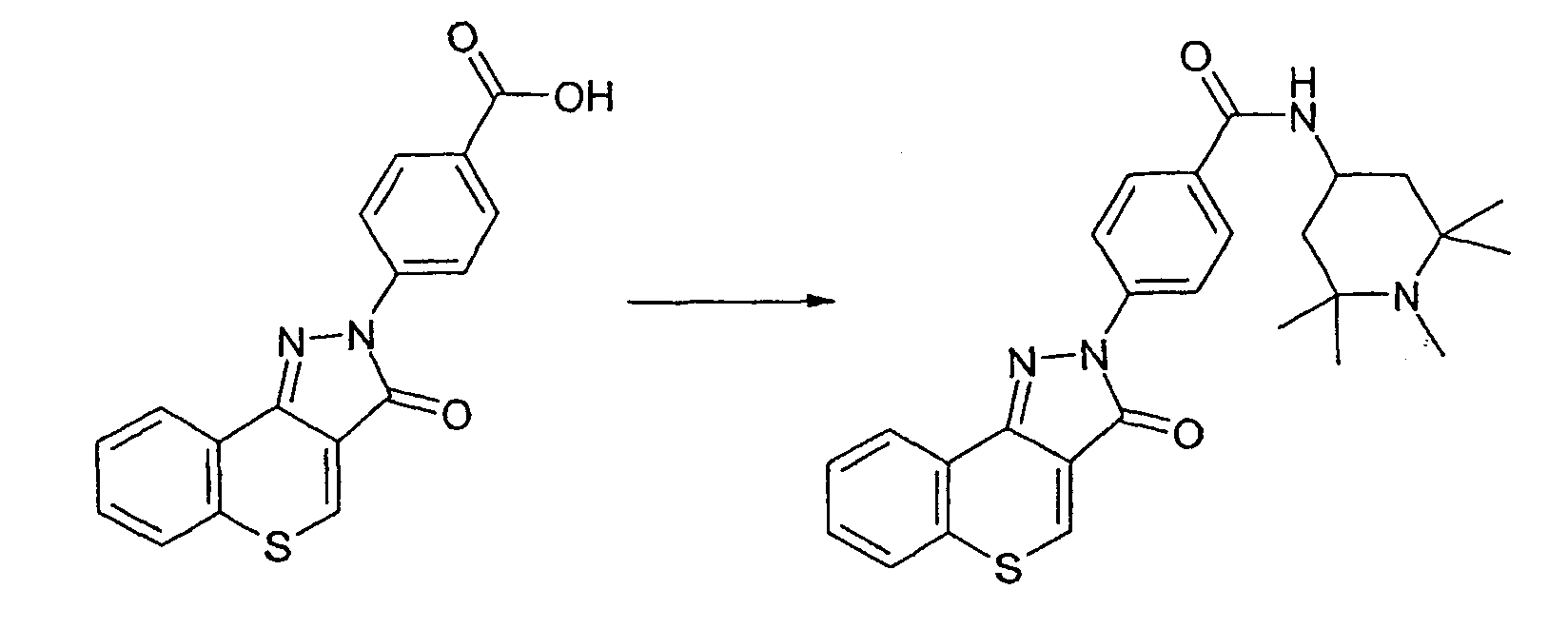
Реакцию проводят, как описано выше. ЖХМС: m/z 475 [M+H]+.
Активность **
Промежуточное соединение 9: Получение 3-[(2-фторфенил)сульфанил]пропановой кислоты
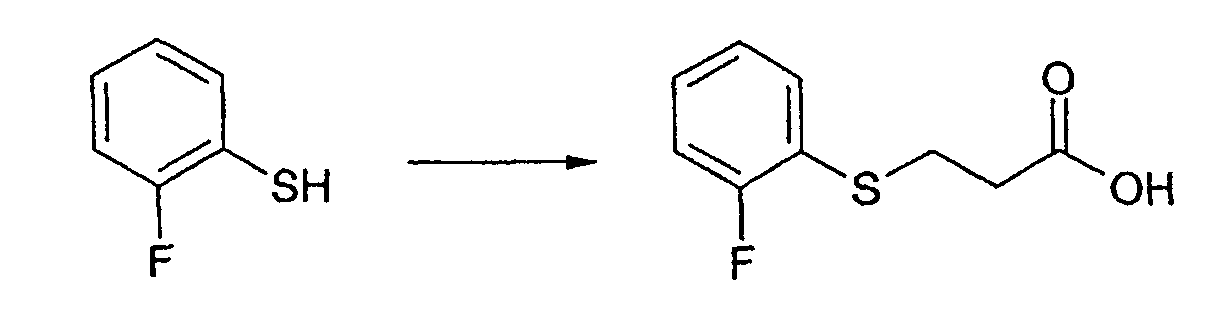
2-фтортиофенол (5,0 г, 39 ммоль) растворяют в тетрагидрофуране (50 мл) в атмосфере азота. Добавляют триэтиламин (3,94 г, 5,33 мл, 85,8 ммоль). Акриловую кислоту (2,81 г, 2,67 мл, 39 ммоль) растворяют в тетрагидрофуране и добавляют по каплям к реакционному раствору в течение 2 ч при комнатной температуре. Смесь перемешивают при комнатной температуре в течение ночи. Добавляют 1М хлористоводородную кислоту (50 мл) и разделяют фазы. Водную фазу промывают этилацетатом (2×50 мл). Комбинированные органические фазы сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Получают желтое масло, которое отверждается при стоянии при комнатной температуре. Твердое вещество растирают в гексане, фильтруют и сушат в вакууме. Указанное в заголовке соединение выделяют в виде не совсем белого твердого вещества (4,19 г, 20,9 ммоль, 54%).
Промежуточное соединение 10: Получение 8-фтор-2, 3-дигидро-4Н-трихромен-4-она
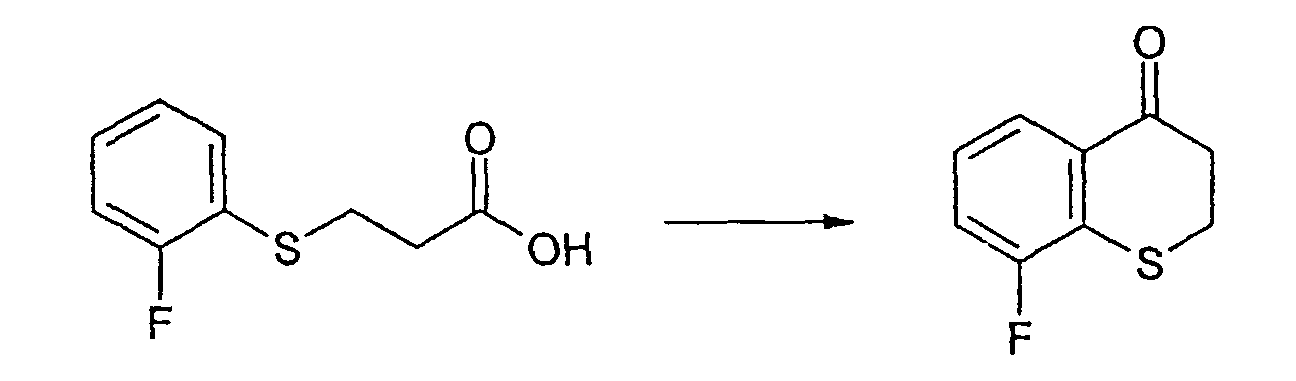
3-[(2-фторфенил)сульфанил]пропановую кислоту (4,0 г, 20 ммоль) смешивают с концентрированной серной кислотой (20 мл) при 0-5°С. Реакционный раствор перемешивают при 0-5°С в течение 3 ч, затем дают возможность нагреться до комнатной температуры в течение ночи. Смесь резко охлаждают внесением по каплям в лед с получением белой суспензии. Водную фазу экстрагируют этилацетатом (1×200 мл, 1×100 мл). Объединенные органические фазы промывают насыщенным раствором бикарбоната натрия (1×50 мл), водой (1×50 мл), 1М хлористоводородной кислотой (50 мл) и водой (2×50 мл). Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Указанное в заголовке соединение выделяют в виде желтого твердого вещества (2,10 г, 11,5 ммоль, 58%).
Промежуточное соединение 11: Получение метил-8-фтор-4-оксотиохроман-3-карбоксилата
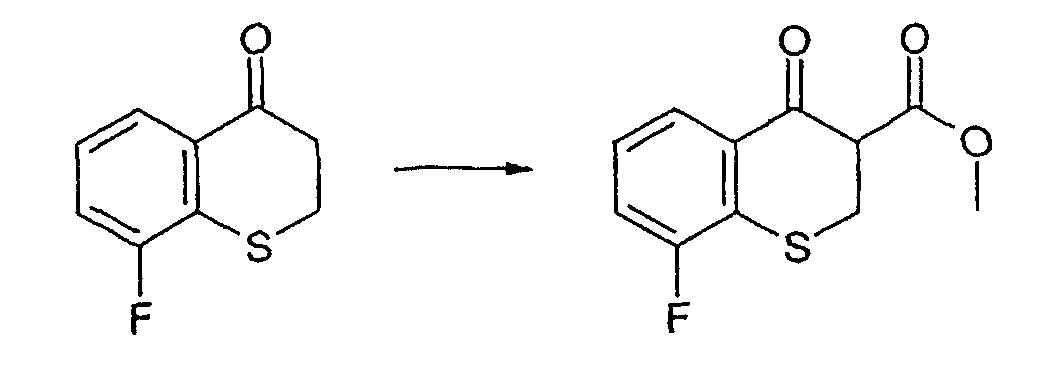
1М раствор гексаметилдисилазида лития в гексане (13,2 мл) растворяют в безводном тетрагидрофуране (20 мл) в атмосфере азота. Раствор охлаждают до -78°С. 8-Фтор-2, 3-дигидро-4Н-тиохромен-4-он (2,00 г, 11 ммоль) растворяют в тетрагидрофуране (40 мл), раствор переносят в капельную воронку и добавляют по каплям в течение 30 мин к реакционной смеси, поддерживая температуру ниже -60°С. Получают оранжевый прозрачный раствор, который перемешивают при температуре от -78°С до -65°С в течение 2 ч. Метилцианоформиат (0,935 г, 0,87 мл) растворяют в тетрагидрофуране (2 мл) и добавляют по каплям к реакционному раствору. Перемешивание продолжают при низкой температуре в течение 1 ч, затем смеси дают возможность нагреться до комнатной температуры. Добавляют насыщенный раствор хлорида аммония (20 мл) и воду (10 мл), фазы смешивают в течение 5 мин и разделяют. Водную фазу промывают этилацетатом (2×100 мл) и объединенные органические фазы сушат над сульфатом магния. Смесь фильтруют и растворитель удаляют в вакууме с получением оранжевого масла. Неочищенное масло очищают колоночной хроматографией; подвижная фаза: гексаны, градиент смеси гексаны/этилацетат [90:10]. Указанное в заголовке соединение выделяют в виде желтого твердого вещества (1,19 г, 4,95 ммоль, 45%).
Промежуточное соединение 12: Получение 4-(6-фтор-3-охсотиохромен[4,3-c]пиразол-2(3Н)-ил)бензойной кислоты
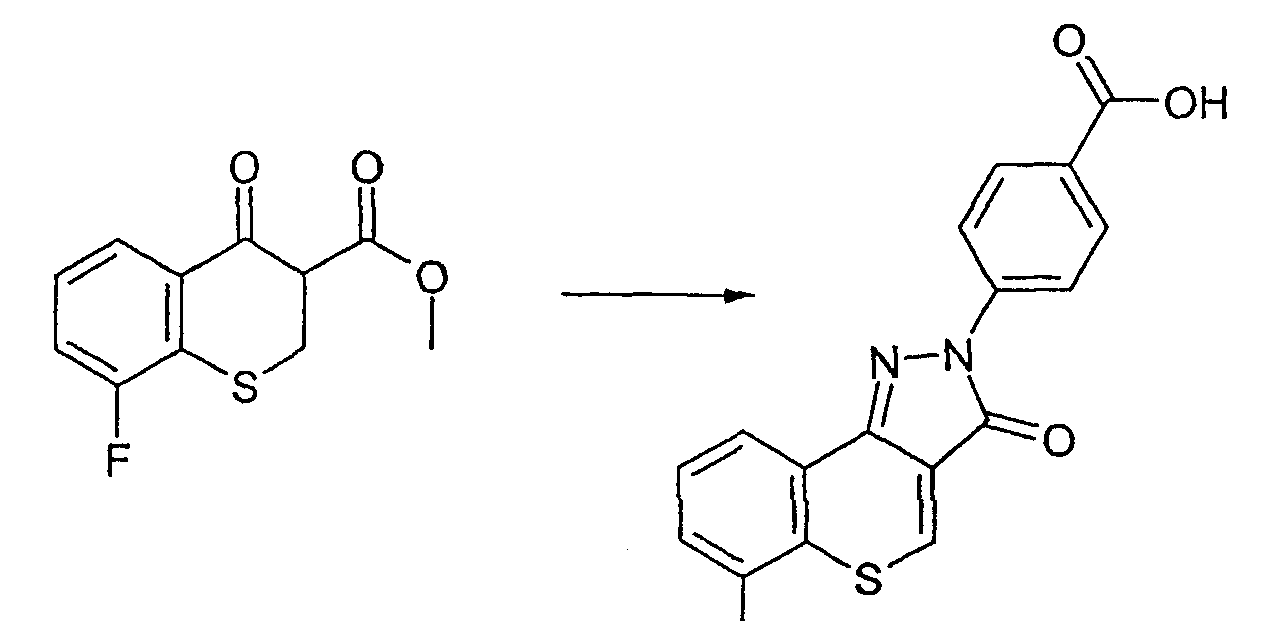
Метил-8-фтор-4-оксотиохроман-3-карбоксилат (1,19 г, 4,95 ммоль) и 4-гидразинбензойную кислоту (755 мг, 4,95 ммоль) смешивают с ледяной уксусной кислотой (10 мл). Смесь кипятят с обратным холодильником в течение 4 ч. Избыточную уксусную кислоту удаляют в вакууме с получением оранжевого масла. Добавляют этилацетат (10 мл) и смесь обрабатывают ультразвуком. Наблюдают осаждение оранжевого твердого вещества. Твердые вещества отфильтровывают и промывают этилацетатом. Осадок на фильтре забирают в диметилсульфоксид (10 мл) и окисляют воздухом при комнатной температуре в течение 1 недели. К реакционной смеси добавляют воду (20 мл), твердые вещества отфильтровывают, суспендируют в этилацетате, фильтруют и сушат в вакууме. Указанное в заголовке соединение выделяют в виде оранжевого порошка (175 мг, 0,51 ммоль, 10%). ЖХМС: m/z 341.
Пример 169
Получение N-[3-(диметиламино)пропил]-4-(6-фтор-3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензамида
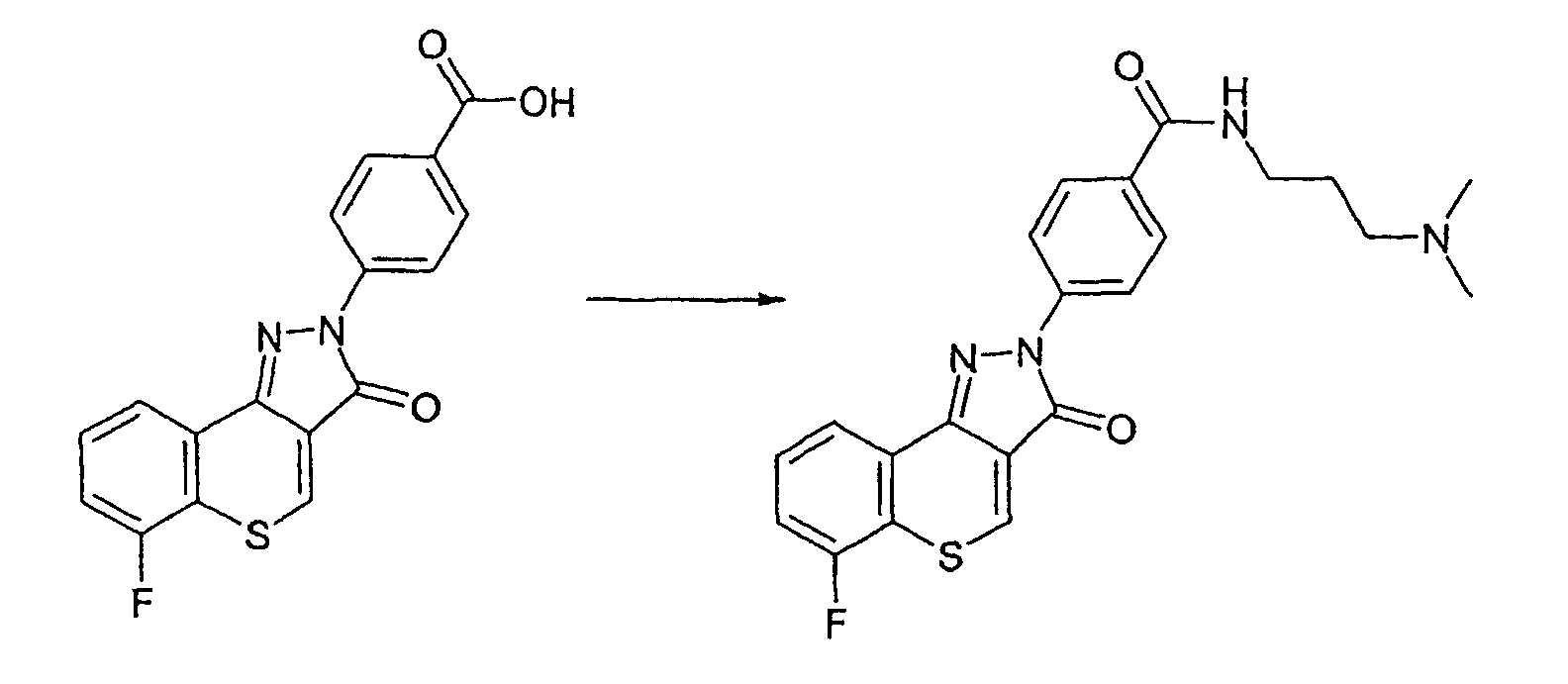
4-(6-фтор-3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензойную кислоту (41 мг, 0,12 ммоль) растворяют в безводном диметилацетамиде (1 мл). Добавляют диизопропилэтиламин (46 мг, 0,36 ммоль, 62 мкл) с последующим добавлением гексафторфосфата [(бензотриазол-1-илокси)диметиламинометилен]диметиламмония (65 мг, 0,17 ммоль) и 3-диметиламинопропиламина (12 мг, 0,12 ммоль). Смесь перемешивают при комнатной температуре в течение ночи и очищают препаративной ВЭЖХ. Указанное в заголовке соединение выделяют в виде коричневого твердого вещества. ЖХМС: m/z 425 [M+H]+.
Активность **
Пример 170
Получение N-[(циклогексиламино)пропил]-4-(6-фтор-3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензамида
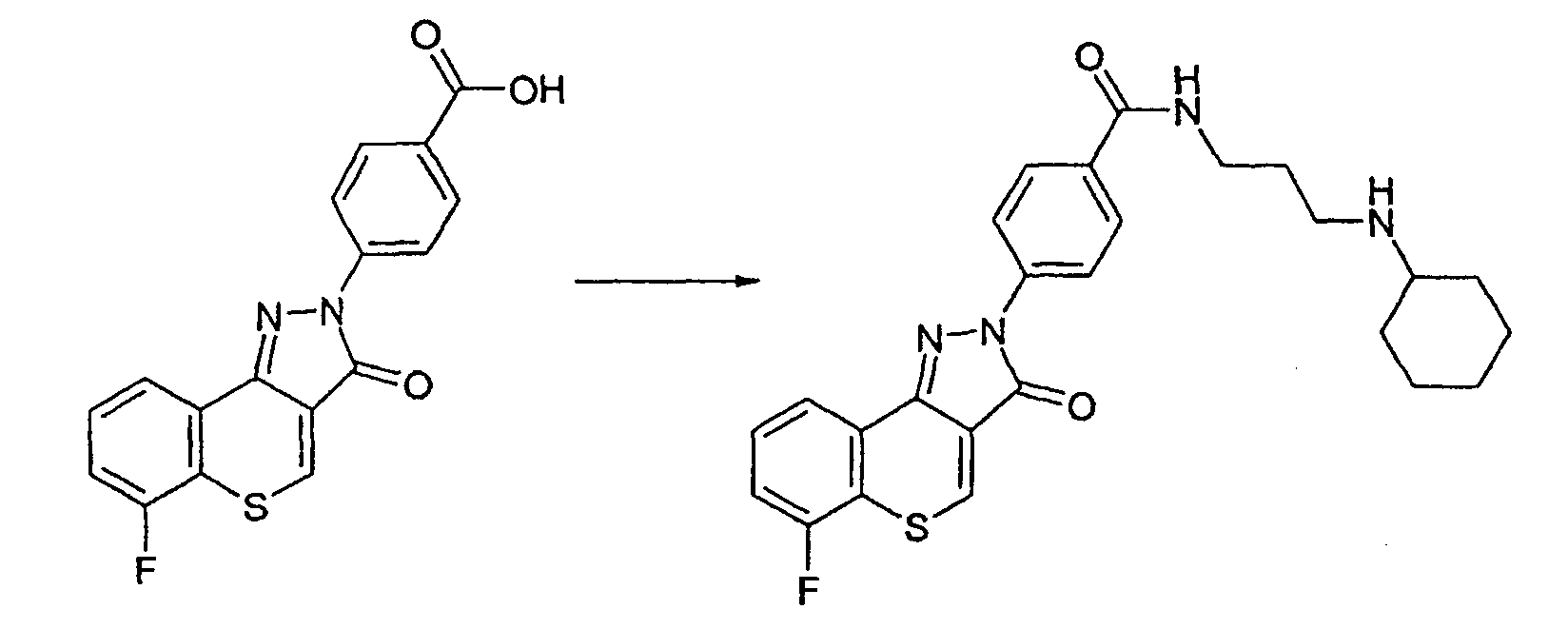
Реакцию проводят, как описано выше. ЖХМС: m/z 479 [M+H]+.
Активность **
Пример 171
Получение N-(пирролидин-1-илбутил)-4-(6-фтор-3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)бензамида
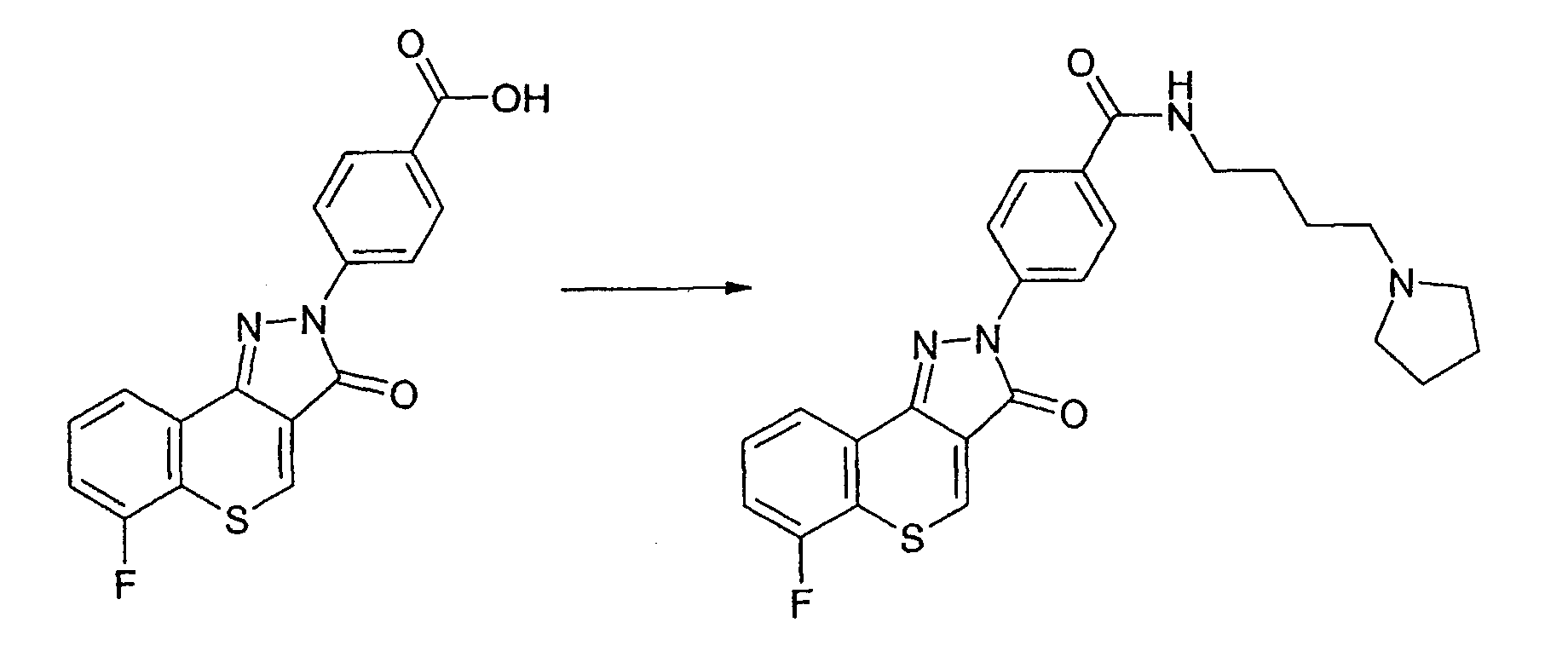
Реакцию проводят, как описано выше. ЖХМС: m/z 465 [M+H]+.
Активность ***
Пример 173
Получение 4-(6-фтор-3-оксотиохромен[4,3-c]пиразол-2(3Н)-ил)-N-1,2,2,6,6-пентаметилпиперидин-4-илбензамида
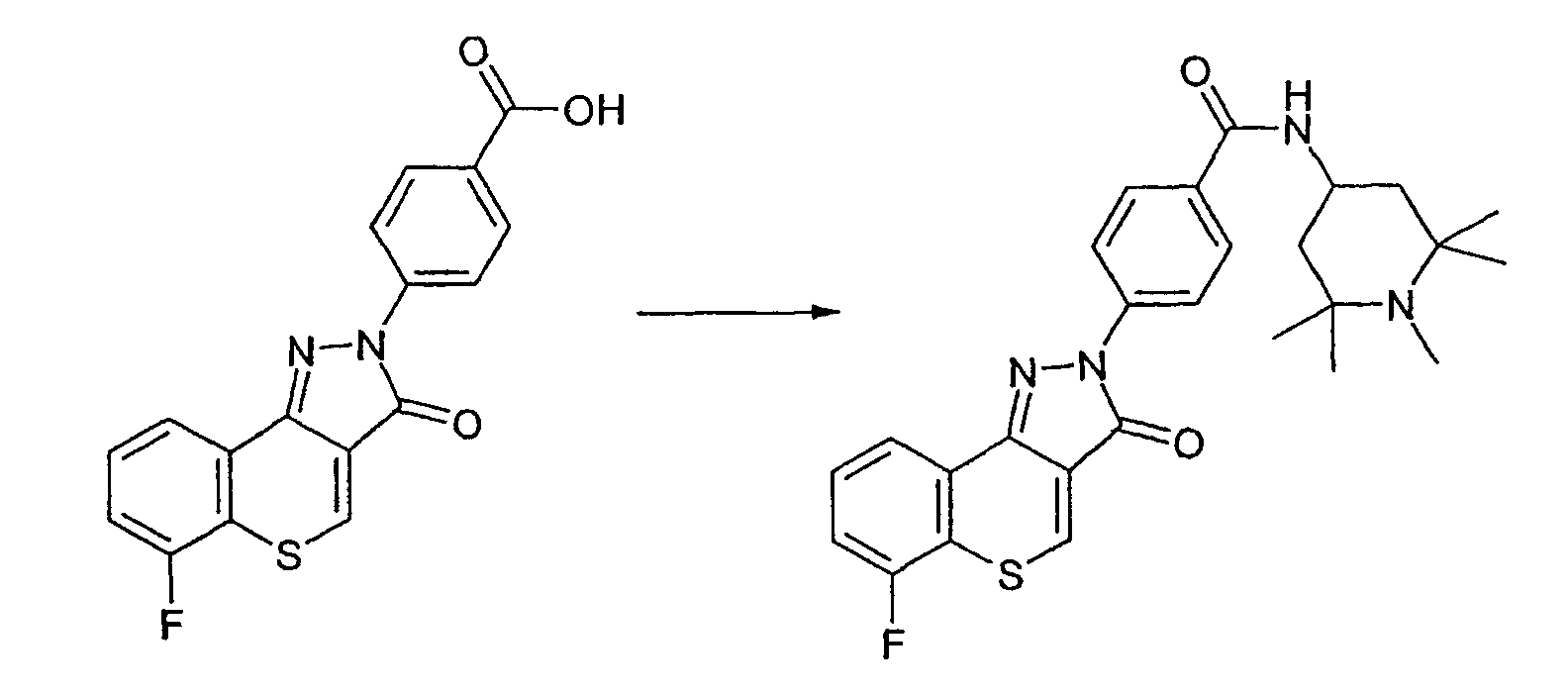
Реакцию проводят, как описано выше. ЖХМС: m/z 493 [M+H]+.
Активность ***
Аналитический раздел
Соединения описанных выше примеров тестировали в бесклеточном анализе однородной разрешаемой во времени флюоресценции (HTRF) для определения их активности в качестве ингибиторов взаимодействия CD80-CD28.
При анализе европий и аллофикоцианин (АРС) опосредованно (через линкеры антител) связывают с CD28 и CD80 с образованием комплекса, который вводит европий и АРС в непосредственную близость для генерирования сигнала. Комплекс включает следующие 6 белков: флюоресцентную метку 1, линкерное антитело 1, слитый белок CD28, слитый белок CD80, линкерное антитело 2 и флюоресцентную метку 2. В таблице ниже более подробно описаны эти реагенты.
При образовании комплекса европий и АРС непосредственно сближены и генерируется сигнал.
Неспецифическое взаимодействие измеряют заменой CD80 слитого белка мышиного фрагмента Fab (C215) (1,9 мкг/мл) мышиным фрагментом Fab (C125). Анализ проводят на черных 384-луночных планшетах в конечном объеме 30 мкл. Аналитический буфер: 50 мМ Tris-HCl, 150 мМ NaCl pH 7,8, содержащий 0,1% BSA (бычий сывороточный альбумин) (мас./об.), добавляют непосредственно перед использованием.
Соединения добавляют к указанным выше реагентам в серии концентраций в диапазоне от 100 мкМ до 1,7 нМ. Реакционную смесь инкубируют в течение 4 ч при комнатной температуре. Проводят двойные измерения с использованием счетчика множества меток Wallac Victor 1420. Первое измерение: возбуждение 340 нм, эмиссия 665 нм, задержка 50 мкс, время окна 200 мкс. Второе измерение: возбуждение 340 нм, эмиссия 615 нм, задержка 50 мкс, время окна 200 мкс. Посчитанные импульсы автоматически корригируются на кроссовер флюоресценции, гашение реакции и фон.
В качестве иллюстрации результаты ЕС50 для соединений примеров 15, 21, 29, 35 и 83 составили соответственно 8 мкМ, 1,9 мкМ, 950 нМ, 148 нМ и 90 нМ. Для удобства величины активности ЕС50 испытанных соединений зарегистрированы выше в итоговой форме в виде:
ЕС50: *=>10 мкМ, **=1-10 мкМ, ***=≤1 мкМ
Реферат
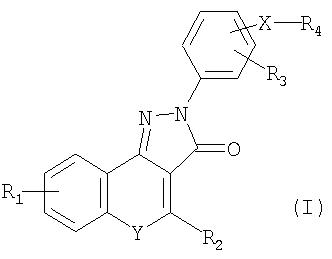
Настоящее изобретение относится к новым соединениям формулы (I) или его фармацевтически или ветеринарно-приемлемым солям:
где: R1 и R3 независимо представляют Н; F; Cl; Br; C1-С6 алкил; R2 представляет Н или С3-С7циклоалкил; Y представляет -S- или -N(R5)-, где R5 представляет Н; Х представляет связь; R4 представляет -С(=O)NR6R7, где R6 представляет Н или радикал формулы -(Alk)b-Q, где b равно 0 или 1, и Alk представляет собой необязательно замещенный C1-С6алкилом, C1-С6алкокси, F, Cl, Br, оксо, СООН, двухвалентный С1-С12алкиленовый, С2-С12алкениленовый с прямой или разветвленной цепью, который может быть прерван одним или несколькими не соседними -О-, -S- или -N(R8)-, где R8 представляет Н или С1-С4алкил, С3 -С4алкенил или С3-С6циклоалкил, и Q представляет Н; -SH; -NR8R8, где каждый R8 может быть одинаковым или различным; группу сложного эфира; или необязательно замещенный C1-С6алкилом, C1-С6алкокси, фенилом, бензилом, фенокси, С3-С8циклоалкилом, амино, фтором, бромом, оксо, -СООН, -CORA, -COORA, NHRA, -NRARB, где RA и RB представляют собой независимо (С1-С6)алкильную группу, фенил, С3-С7циклоалкил, С5-С7циклоалкенил или гетероциклическое кольцо, имеющее от 5 до 8 кольцевых атомов; и R7 представляет Н или C1-С6алкил; или взятые вместе с атомом или атомами, к которым они присоединены, R6 и R7 образуют необязательно замещенное (C1-С6)алкилом, COORA, где RA представляет собой (С1-С6)алкильную группу, фенил, необязательно замещенный F, Cl, Br, гетероциклическое кольцо, имеющее от 5 до 8 кольцевых атомов. Изобретение также относится к N-(3-диметиламинопропил)-4-(4-циклопропил-3-оксо-3,5-дигидропиразол[4,3-с]хинолин-2-ил)бензамиду, к применению соединений по любому из пп.1-10, к способу иммуномодуляции и к фармацевтической или ветеринарной композиции. Технический результат - получение новых биологически активных соединений, которые являются антагонистами CD80, способными ингибировать взаимодействие между CD80 и CD28. 5 н. и 9 з.п. формулы, 1 табл.
Формула
Документы, цитированные в отчёте о поиске
Трициклические дикарбонильные производные и лекарственный препарат на их основе
Комментарии