Трициклические дикарбонильные производные и лекарственный препарат на их основе - RU2145606C1
Код документа: RU2145606C1
Чертежи
Описание
Настоящее изобретение относится к трициклическим дикарбонильным производным общих формул
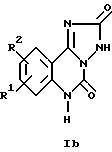
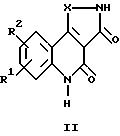
где R1 и R2 каждый независимо друг от друга обозначают водород, (низший)алкил, (низший)алкокси, нитро, трифторметил, амино, галоген, циано или R3R4NS(O)2- и R3 и R4 обозначают (низший)алкил, и R2 может дополнительно обозначать морфолино или триморфолино, 5- или 6-членный гетероцикл с 1-3 атомами азота, необязательно замещенными (низшим)алкилом, гидроксигруппой, аминогруппой или группой -CH2NHCH3, бициклический гетероцикл с 1-3 атомами азота или группу -NR5R6 или -OR5, где R5 и R6 могут быть идентичными или различными и обозначают водород, (низший)алкил, гидрокси(низший)алкил, (низший)алкокси(низший)алкил, амино(низший)алкил или (низший)алкиламино(низший)алкил, и X в формуле II обозначает -CH = CH-, -CH = N-, -NH-, -CO- или -O-, а также к фармацевтически приемлемым солям соединений общих формул Ia, Ib и II.
Указанные соединения и их соли являются новыми, за исключением 2,3,5,6-тетрагидро[1,2,4]триазол[1,5-c]хиназолин-2, 5-диона, и было обнаружено, что они обладают ценными фармакодинамическими свойствами в качестве неконкурентных антагонистов NMDA- и/или AMPA/KA-рецептора, поэтому они могут использоваться в качестве защитных веществ для нервной системы, в частности для лечения или предупреждения ишемической болезни, гипогликемии, гипоксии, мозговых сосудистых спазмов, мышечной спастичности, травм, кровотечений, инфекций (вирусных, бактериальных, амебных, приональных), эпилептических припадков, аутоиммунных заболеваний, симптомов синдрома отмены, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, болезни Гентингтона, интоксикаций, оливопонтоцеребеллярной атрофии, повреждений спинного мозга, шизофрении, депрессий, состояний тревоги, лекарственной зависимости, болей, аутизма и задержки умственного развития.
Предметами настоящего изобретения являются указанные выше соединения и их фармацевтически приемлемые соли как индивидуально, так и в виде терапевтически активных веществ, способ получения этих новых соединений и солей, лекарственных препаратов, содержащих такое соединение или его соль, получение таких лекарственных препаратов, применение указанных выше соединений и солей в качестве защитных веществ для нервной системы, в частности для лечения или предупреждения ишемической болезни, гипогликемии, гипоксии, мозговых сосудистых спазмов, мышечной спастичности, травм, кровотечений, инфекций (вирусных, бактериальных, амебных, приональных), эпилептических припадков, аутоиммунных заболеваний, симптомов синдрома отмены, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, болезни Гентингтона, интоксикаций, оливопонтоцеребеллярной атрофии, повреждений спинного мозга, шизофрении, депрессий, состояний тревоги, лекарственной зависимости, болей, аутизма и задержки умственного развития и применение указанных ранее соединений и солей для получения лекарственных препаратов.
Понятие "низший" включает соединения или группы, содержащие максимально 7, предпочтительно максимально 4 атома углерода.
Понятие "алкил" включает насыщенные углеводородные группы с прямой или разветвленной цепью, такие, как метил, этил, пропил и т.п.
Понятие "алкокси" включает алкильные группы в соответствии с вышеприведенным определением, связанные через атом кислорода, такие, как метокси и т.п.
Понятие "галоген" включает фтор, хлор, бром и йод.
Понятие "5- или 6-членный гетероцикл" включает циклические системы, состоящие из насыщенных или ненасыщенных колец, такие, как, например, пирролил, пирролидинил, имидазолил, пиразолил, пиразолинил, пиразолидинил, триазолил, пиримидинил, пиперазинил, пиперидинил и т. п., причем циклическая система необязательно может быть замещена (низшим)алкилом, гидрокси-, аминогруппой или группой -CH2NHCH3.
Понятие "бициклический гетероцикл" означает циклическую систему, состоящую из двух колец, сконденсированных друг с другом, причем одно из колец представляет собой гетероциклическое кольцо, а второе кольцо обычно представляет собой бензольное кольцо, например, хиноксалинильную группу.
Термин "фармацевтически приемлемые соли" означает соли неорганических и органических кислот, таких, как соляная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфоновая кислота, паратолуолсульфоновая кислота и т.п., а также соли неорганических оснований, таких, как гидроксид натрия или калия. Такие соли могут быть легко получены любым специалистом в данной области техники на основе имеющегося опыта в данной области техники и с учетом природы соединения, которое требуется перевести в соль.
Соединения формул Ia, Ib и II в соответствии с изобретением также могут быть представлены в виде таутомеров, таким образом, объем изобретения 5 включает все их изомеры, а также смеси изомеров.
Предпочтительными
соединениями формулы Ia являются соединения, в которых R1 и R2 каждый обозначают водород, галоген, нитро, метил или метокси, прежде всего следующие соединения:
8,
9-дихлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион;
9-хлор-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин- 3,5-дион;
9-бром-8-нитро-2,3,5,6-тетрагидро-1,2,
4-триазол[4,3-c]хиназолин- 3,5-дион;
8,10-дихлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион;
8,9-диметил-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион;
8-метокси-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион;
8-йод-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион;
8-хлор-9-фтор-2,3,5,6-тетрагидро-1,2,
4-триазол[4,3-c]хиназолин-3,5-дион;
8,9-динитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион; и
8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион.
Кроме того, предпочтительны соединения формулы Ia, в которых R1 обозначает нитро, R2 обозначает пирролидинил или диметиламино. Они, в частности, представляют собой
следующие соединения:
9-диметиламино-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин- 3,5-дион; и
8-нитро-9-пирролидин-1-ил-2,3,5,6-тетрагидро-1,2,4- триазол[4,
3-c]хиназолин-3,5-дион.
Ниже приведены наиболее предпочтительные соединения формулы Ib:
9-хлор-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[1,5-c]хиназолин- 2,5-дион; и
9-имидазол-1-ил-8-нитро-2,3,5,6-тетрагидро-1,2,4- триазол[1,5-c] хиназолин-2,5-дион.
Предпочтительными соединениями общей формулы II являются таковые, в которых R1 и R2 каждый обозначают водород, галоген или нитро, в частности следующие соединения:
7-хлор-2,3,4,5-тетрагидроизоксазол[4,5-c]хинолин-3,4-дион;
7-нитро-2,3,4,
5-тетрагидроизоксазол[4,5-c]хинолин-3,4-дион;
7,8-дихлор-4-гидрокси-2,3-дигидро-1H-пиразол[4,3-c]хинолин-3-он; и
7-хлор-4-гидрокси-2,3-дигидро-1H-пиразол[4,3-c]хинолин-3-он.
Соединения общих формул Ia, Ib и II могут быть получены следующим образом:
а) циклизацией соединения общей формулы
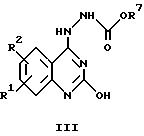
где R1 и R2 имеют указанные выше значения, а R7 обозначает (низший)алкил, с получением соединений общей формулы Ia, или
б) перегруппировкой соединения общей формулы Ia под действием соответствующего основания в протонном растворителе с получением соединений общей формулы Ib, или
в) взаимодействием соединения общей формулы III, в котором R1 обозначает NO2 и R2 обозначает F, с соответствующим гетероциклом с получением соединений формулы Ia, в которых R1 обозначает NO2 и R2 обозначает 5- или 6-членный гетероцикл с 1-3 атомами азота, необязательно замещенный (низшим) алкилом, гидрокси-, аминогруппой или группой -CH2 NHCH3 , или
г) циклизацией соединения общей формулы
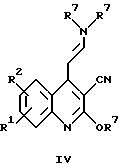
где R1, R2 и R7 имеют указанные выше значения, с получением соединений общей формулы II, в которых X обозначает -CH = CH-, или
д) взаимодействием соединения общей формулы
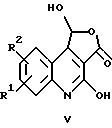
где R1 и R2 имеют указанные выше значения
с гидратом гидразина с получением соединений общей формулы II, в которых X обозначает -CH = N-, или
е) гидрированием соединения общей формулы
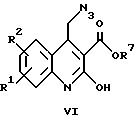
где R1, R2 и R7 имеют указанные выше значения, в присутствии катализатора, циклизацией и спонтанным окислением кислородом с получением соединений общей формулы II, в которых X обозначает -CO-, или
ж) циклизацией соединения общей формулы
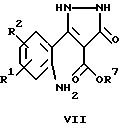
где R1, R2 и R7 имеют указанные выше значения, с получением соединений общей формулы II, в которых X обозначает -NH-, или
з) циклизацией соединения общей формулы
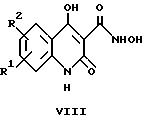
где R1 и R2 имеют указанные выше значения, с получением соединений общей формулы II, в которых X обозначает -O-, и
и) при необходимости превращением полученного соединения формул Ia, Ib или II в фармацевтически приемлемую соль.
Соединения общей формулы Ia могут быть получены в соответствии с вариантом а) указанного выше способа путем циклизации соединения общей формулы III, в котором R1 и R2 имеют приведенные выше значения. Эту реакцию обычно осуществляют путем кипячения при температуре дефлегмации в пригодном растворителе, например, в диметилформамиде.
Соединения общей формулы Ib могут быть получены в соответствии с вариантом б) указанного выше способа путем перегруппировки Димрота соединения общей формулы Ia. Эту реакцию обычно осуществляют в присутствии сильного основания, например, NaOH, и в протонном растворителе, например, в диметилсульфоксиде.
Соединения общей формулы Ia, в которых фенильное кольцо замещено гетероциклом и NO2, обычно получают в соответствии с вариантом в) указанного выше способа. Используя в качестве исходного вещества соединение формулы III, в котором R1 обозначает NO2 и R2 обозначает F, реакцию проводят с соответствующим гетероциклом путем кипячения в течение нескольких часов в растворителе, например, в спирте.
Соединения общей формулы II, в которых X обозначает -CH = CH- и R1 и R2 имеют указанные выше значения, могут быть получены в соответствии с вариантом г) указанного выше способа путем циклизации соединения формулы IV. Эту реакцию обычно проводят в условиях кипячения с обратным холодильником в кислотно-водной реакционной смеси, причем в этом случае наиболее предпочтительна смесь серной кислоты, уксусной кислоты и воды.
Соединения общей формулы II, в которых X обозначает -CH = N- и заместители R1 и R2 имеют указанные выше значения, могут быть получены в соответствии с вариантом д) указанного выше способа. Для этого соединение общей формулы V растворяют в растворителе, например, в диметилсульфоксиде, обычно в атмосфере защитного газа, а затем после добавления гидрата гидразина перемешивают в течение длительного периода времени при комнатной температуре.
Соединения общей формулы II, в которых X обозначает -CO- и R1 и R2 имеют приведенные выше значения, получают в соответствии с вариантом е) указанного выше способа. Эту реакцию обычно проводят путем гидрирования азида формулы VI в присутствии катализатора. Пригодными для этой цели являются катализаторы на основе платины или палладия. Каталитическое гидрирование осуществляют в токе водорода при комнатной температуре после растворения соединения общей формулы VI в смеси, состоящей из спирта, например, метанола, и диметилформамида. Требуемое соединение общей формулы II получают после окисления диметилформамидного раствора в токе кислорода.
В случае, когда X в формуле II обозначает -NH-, такие соединения могут быть получены в соответствии с вариантом ж) указанного выше способа, что может быть осуществлено аналогично тому, как описано для варианта а). Соединение формулы VII обычно подвергают циклизации для получения соединения формулы II путем кипячения при температуре дефлегмации в приемлемом растворителе, например, в диметилформамиде.
Кроме того, соединения общей формулы II, в которых X обозначает -O- и R1 и R2 имеют указанные выше значения, получают путем циклизации соединения общей формулы VIII. Эту реакцию обычно осуществляют в соответствии с вариантом з) указанного выше способа путем суспендирования соответствующего соединения формулы VIII в тетрагидрофуране и последующей обработки тионилхлоридом. Соединения формулы II по изобретению образуются после добавления основания, например, триэтиламина.
Соединения общих формул Ia, Ib и II могут быть переведены в фармацевтически приемлемые соли в соответствии с вариантом и) указанного выше способа. При этом под объем изобретения подпадают не только соли неорганических кислот, но также и соли органических кислот и соли с неорганическими основаниями. Примерами таких солей являются гидрохлориды, гидробромиды, нитраты, сульфаты, фосфаты, цитраты, формиаты, фумараты, малеаты, ацетаты, сукцинаты, тартраты, метансульфонаты, паратолуолсульфонаты, а также натриевые или калиевые соли и т.п. Указанные соли могут быть получены в соответствии с известными способами, которые очевидны для специалистов в данной области техники.
Соединения, используемые в качестве исходных веществ, могут быть получены, например, в соответствии с нижеприведенными реакционными схемами и приведенными далее пояснениями различных реакций.
где R1, R2 и R7 имеют указанные выше значения.
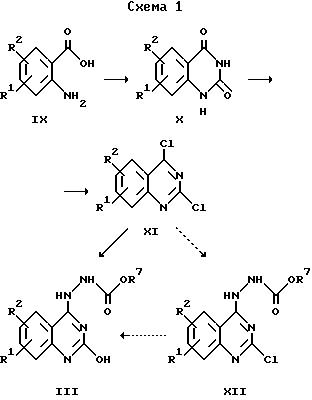
Исходные вещества для получения соединений общей формулы Ia по изобретению могут быть получены в соответствии со схемой I.
Соединение формулы X может быть получено с помощью общеизвестных методов взаимодействием 2-аминобензойной кислоты, содержащей в качестве заместителей R1 и R2, с мочевиной при нагревании в течение нескольких часов. Полученный таким образом хиназолиндион формулы X далее превращают с использованием галогенирующего агента, предпочтительно оксихлорида фосфора, в соединение формулы XI путем перемешивания реакционной смеси при температуре дефлегмации в течение нескольких часов. Соединение формулы IlIa получают взаимодействием соединения формулы XI с алкилкарбазатом в диметилсульфоксиде. В зависимости от природы заместителей R1/R2 это можно осуществлять прямым взаимодействием (например, когда R1 обозначает алкил или алкокси) или с помощью соединений формулы XII (например, когда R1 обозначает галоген). Температура реакции может варьироваться между 70oC и 95oC. Далее полученное таким образом соединение формулы III может быть превращено, следуя варианту а) указанного выше способа, в соединения формулы Ia по изобретению.
где R1, R2 и R7 имеют указанные выше значения.
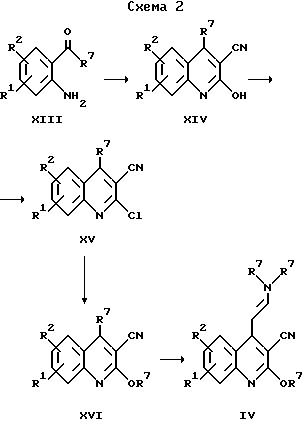
Исходные вещества для получения соединений общей формулы II по изобретению, в которых X обозначает -CH = CH-, могут быть получены в соответствии со схемой 2.
(2-аминофенил)алканон формулы XIII превращают при нагревании и перемешивании в течение нескольких часов с алкилцианацетатом в соединения общей формулы XIV. Полученный таким образом 2-гидроксихинолин-3-карбонитрил общей формулы XIV далее хлорируют в соответствии с общеизвестными методами, например, с помощью оксихлорида фосфора, с получением соединений формулы XV, которые далее суспендируют в спирте, обрабатывают натрием и кипятят с обратным холодильником в течение нескольких часов. Полученное таким образом соединение общей формулы XVI затем обычно подвергают взаимодействию с N,N-диметилформамиддиалкилацеталем в течение нескольких часов при температуре дефлегмации, получая соединение общей формулы IV. Полученные таким образом промежуточные продукты далее могут быть подвергнуты циклизации, следуя варианту г) указанного выше способа, с получением соответствующих соединений общей формулы II по изобретению.
где R1 и R2 имеют указанные выше значения.
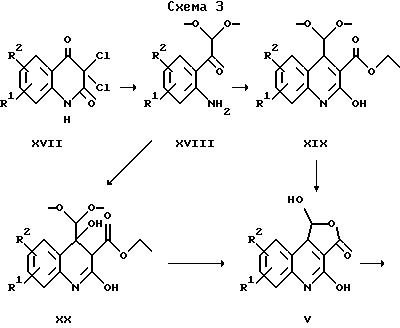
Исходные вещества для получения соединений общей формулы II по изобретению, в которых X обозначает -CH = N-, могут быть получены в соответствии со схемой 3.
Соединения формулы XVIII могут быть получены с помощью общеизвестных методов путем растворения натрия в метаноле, предпочтительно в атмосфере защитного газа, и превращения после добавления метанолового раствора соответствующего производного 3, 3-дихлорхинолиндиона в соединения формулы XVIII. Далее это соединение обычно суспендируют в диэтилмалонате и перемешивают при кипячении с обратным холодильником в течение нескольких часов. Полученный таким образом эфир карбоновой кислоты общей формулы XIX затем обычно обрабатывают соляной кислотой, получая соединения обшей формулы V.
Еще один возможный способ получения соединений общей формулы V включает растворение соединения формулы XVIII в растворителе, предпочтительно в дихлорметане, обработку раствора триэтиламином и этилмалонилхлоридом и перемешивание в течение нескольких минут при охлаждении. Полученное таким образом соединение формулы XX далее обычно перемешивают в смеси, состоящей из уксусной кислоты, воды и серной кислоты, при температуре дефлегмации в течение нескольких часов с получением соединений общей формулы V.
Соединения формулы V служат в качестве исходных веществ для получения соединений общей формулы II, следуя варианту д) указанного выше способа.
где R1, R2 и R7 имеют приведенные выше значения.
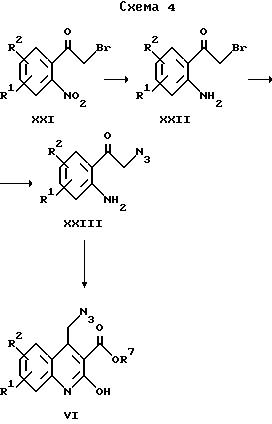
Исходные вещества для получения соединений общей формулы II по изобретению, в которых X обозначает -CO-, могут быть получены в соответствии со схемой 4.
Азотсодержащие соединения формулы XXI, полученные в соответствии с "Helv. Chem. Acta, 1937, 20, 913", восстанавливают до аминосоединений формулы XXII в растворителе, обычно используя серную кислоту, и в присутствии металла, например, медного порошка, при температуре приблизительно 50oC. Затем осуществляют обработку метанольным раствором азида натрия с помощью общеизвестных методов. Полученное соединение формулы XXIII обрабатывают триэтиламином в растворителе, например, в дихлорметане, в атмосфере защитного газа и обрабатывают алкилмалонилхлоридом при 0oC. После перемешивания при комнатной температуре и последующего кипячения с обратным холодильником получают соединения формулы VIa, которые могут быть превращены, следуя варианту е) указанного выше способа, в соответствующие соединения формулы II по изобретению.
где R1, R2 и R7 имеют приведенные выше значения.
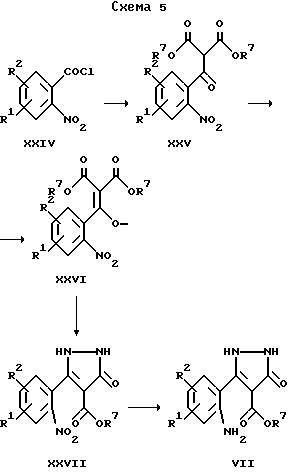
Исходные вещества для получения соединений общей формулы II по изобретению, в которых X обозначает -NH-, могут быть получены в соответствии со схемой 5.
Для получения соединения формулы XXV магниевую стружку обычно обрабатывают в атмосфере защитного газа смесью этанол/четыреххлористый углерод и затем добавляют раствор диалкилмалоната, этанола и простого эфира до тех пор, пока смесь слегка не закипит. После добавления по каплям соответствующего замещенного 2-нитробензоилхлорида в приемлемой смеси растворителей, например, в простом эфире/тетрагидрофуране, получают соединения общей формулы XXV. Далее эти соединения обрабатывают раствором диазометана, получая соединения формулы XXVI. Затем осуществляют гидрирование азотсодержащего соединения до получения аминосоединения формулы VII. Эту реакцию обычно проводят с использованием металлического катализатора, например, с использованием палладиевого катализатора в токе водорода при комнатной температуре.
Соединения формулы VII представляют собой исходные вещества для получения соединений формулы II по изобретению, следуя варианту е) указанного выше способа.
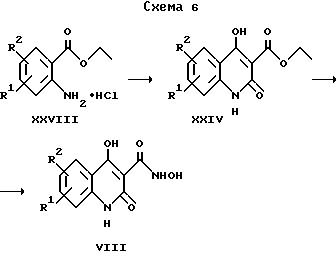
где R1 и R2 имеют приведенные выше значения.
Исходные вещества для получения соединений формулы II по изобретению, в которых X обозначает -O-, могут быть получены в соответствии со схемой 6.
Раствор соединения формулы XXVIII, предпочтительно в ацетоне, обрабатывают триэтиламином, а затем метилмалонилхлоридом. После проведения реакции в течение нескольких часов при комнатной температуре концентрированный раствор обрабатывают этилатом натрия и полученное таким образом хинолиновое соединение формулы XXIX далее превращают с помощью триметилсилилгидроксиламина в соединения формулы VIII.
Образовавшиеся соединения представляют собой исходные вещества для получения соединений формулы II по изобретению, следуя варианту з) указанного выше способа.
Как указано выше, соединения формул Ia, Ib и II по изобретению обладают фармакологической активностью в качестве неконкурентных антагонистов NMDA- и/или AMPA/KA-рецептора. Вследствие этой активности соединения формул Ia, Ib и II и их фармацевтически приемлемые соли могут использоваться в качестве защитных веществ для нервной системы, в частности для лечения или предупреждения ишемической болезни, гипогликемии, гипоксии, мозговых сосудистых спазмов, мышечной спастичности, травм, кровотечений, инфекций (вирусных, бактериальных, амебных, приональных), эпилептических припадков, аутоиммунных заболеваний, симптомов синдрома отмены, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, болезни Гентингтона, интоксикаций, оливопонтоцеребеллярной атрофии, повреждений спинного мозга, шизофрении, депрессий, состояний тревоги, лекарственной зависимости, болей, аутизма и задержки умственного развития.
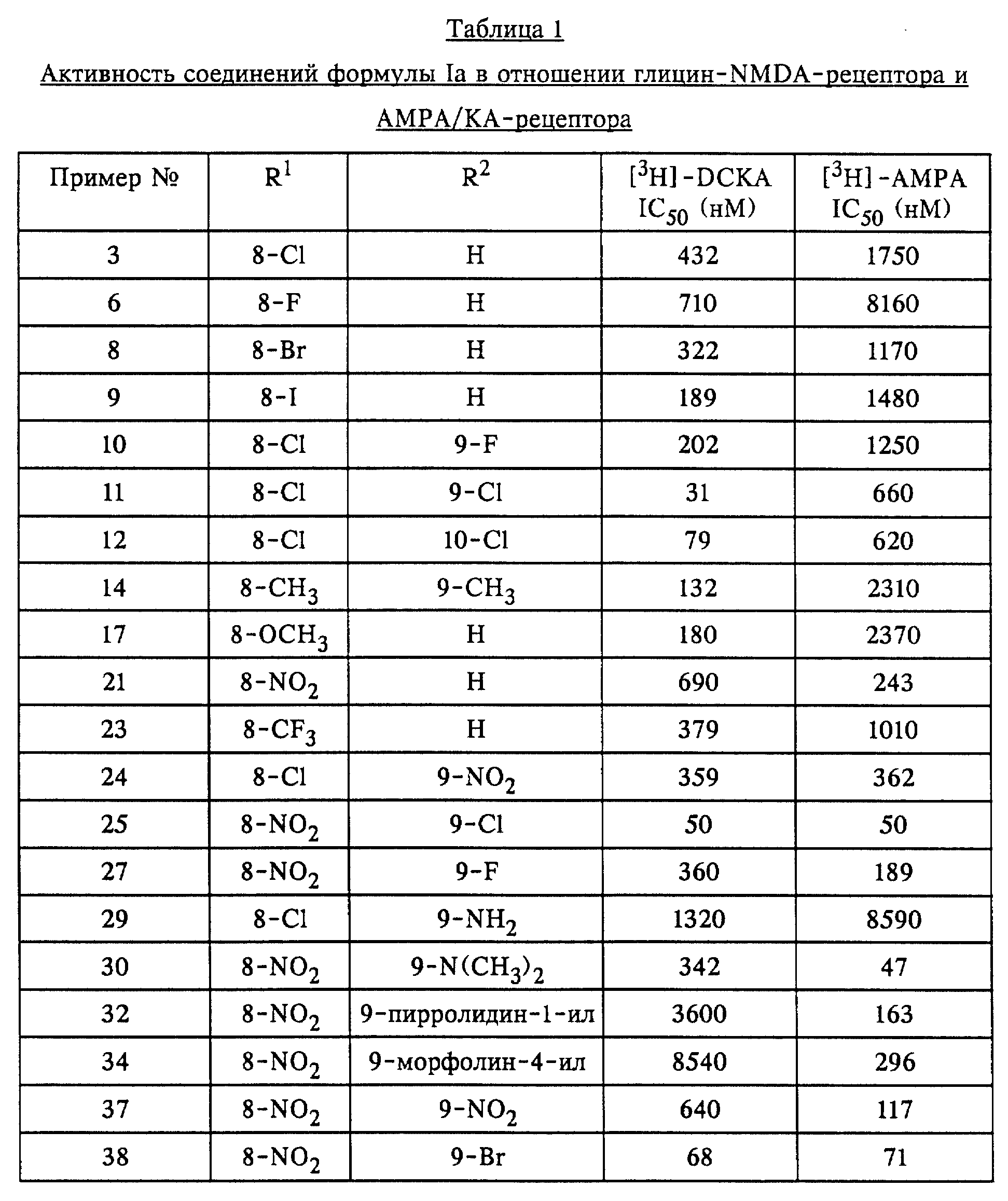
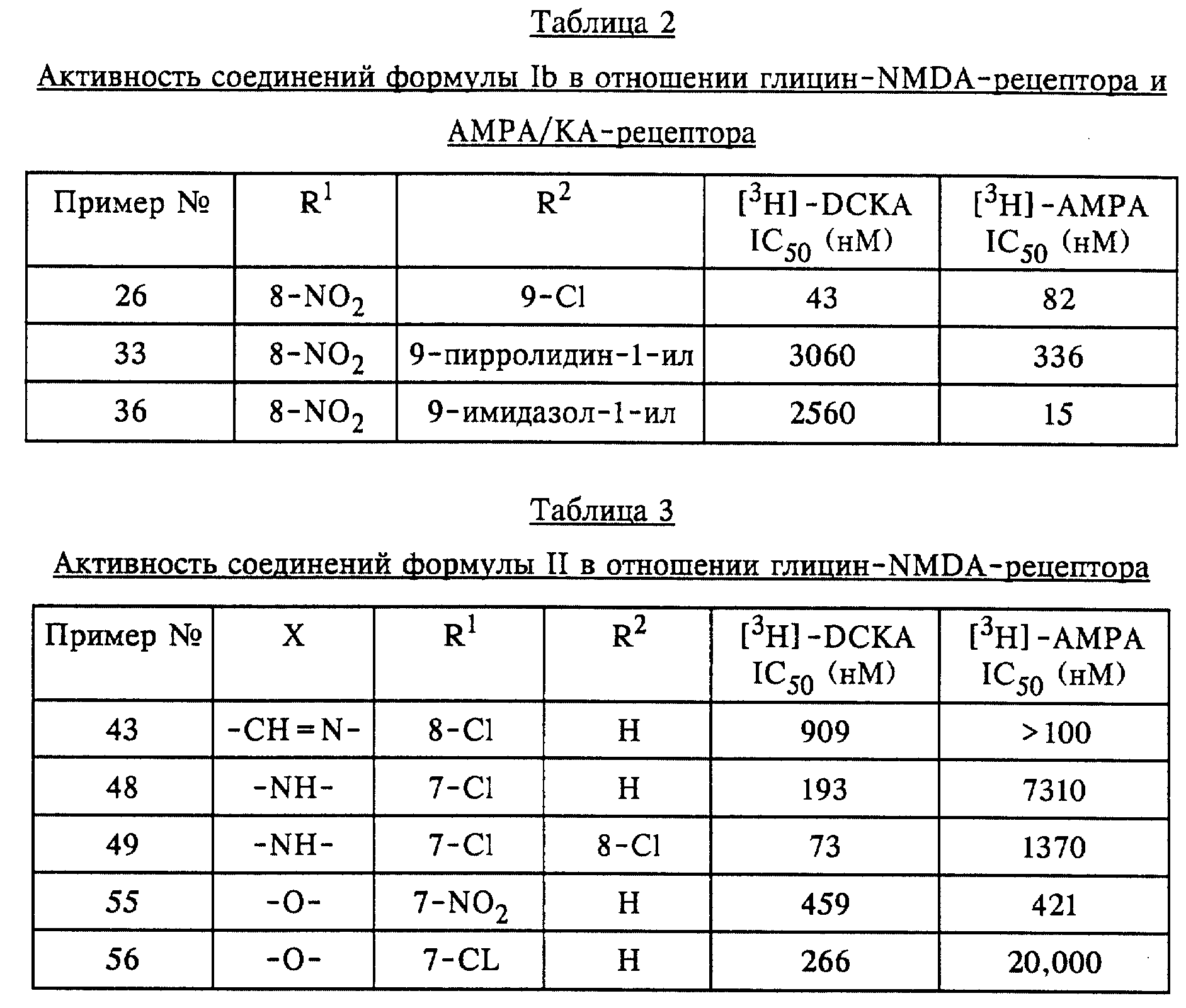
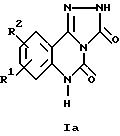
Описанные соединения конкурентно ингибируют связывание3H-DCKA (3H-5,7-дихлоркинуреновой кислоты) с NMDA-рецептором (NMDA = N-метил-D-аспартат) и3H-AMPA (DL-(3H)-амино-3-гидрокси- 5-метилизоксазол-4-пропионовой кислоты) с каинат/AMPA-рецептором.3H-DCKA и3H-AMPA представляют собой специфические лиганды для сайта связывания глицина на NMDA-рецепторе и соответственно сайта связывания глутамата на каинат/AMPA-рецепторе. Эксперименты по связыванию осуществляли аналогично тому, как это описано в следующих работах: связывание3H-DCKA: у B.M. Baron и др., Eur. J. Pharmacol. 206 (1991) 149-154; связывание3H-AMPA: у D. E. Murphy и др., Neurochem. Res. 12 (1987) 775-782.
Для изучения связывания использовали тщательно промытые препараты мембран головного мозга крыс. Использовали следующие концентрации радиолигандов:3H-DCKA - 20 нМ и соответственно3 H-AMPA - 10 нМ.
Для определения неспецифического связывания добавляли соответствующие лиганды с концентрациями 1 мМ глицина и соответственно 1 мМ глутамата. Связанный с мембраной радиолиганд отделяли от несвязанных лигандов путем центрифугирования (15 минут при 40000 кг;3H-DCKA) или фильтрации (комбинация фильтров Whatmann GF/C и GF/B;3H-AMPA).
Описанные соединения применяли в экспериментах по связыванию в различных концентрациях. Концентрации, при которых происходит 50%-ное ингибирование связывания, определяли на основе кривых доза-активность в отношении ингибирования связывания соответствующих радиолигандов. Эти значения (IC50) (в нМ) приведены в таблицах 1-3.
Соединения формул la, lb и II, а также их фармацевтически приемлемые соли могут применяться в качестве лекарственных препаратов, например, в виде фармацевтических препаратов. Фармацевтические препараты могут вводиться энтерально, например, орально, в том числе в форме таблеток, таблеток с оболочкой, драже, желатиновых капсул с твердыми и мягкими оболочками, растворов, эмульсий или суспензий; назально, например, в форме назальных аэрозолей; или ректально, например, в форме суппозиториев. Введение также может осуществляться парентерально, например, подкожно, внутримышечно или внутривенно, например, в виде растворов для инъекций. Для изготовления таблеток, таблеток с покрытием, драже и желатиновых капсул с твердой оболочкой соединения, а также их фармацевтически приемлемые соли могут быть объединены с фармацевтически инертными неорганическими или органическими эксципиентами. В качестве таких эксципиентов, например, для таблеток, драже и желатиновых капсул с твердой оболочкой, могут быть использованы лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли и т.д. Пригодными эксципиентами для желатиновых капсул с мягкой оболочкой являются, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и т.д. Пригодными эксципиентами для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертный сахар, глюкоза и т.д. Пригодными эксципиентами для растворов для инъекций являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.д. Пригодными эксципиентами для суппозиториев являются, например, натуральные или гидрогенизованные масла, воски, жиры, полужидкие или жидкие полиолы и т.д. Фармацевтические препараты, кроме того, могут содержать консерванты, солюбилизаторы, вещества, повышающие вязкость, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, корригенты, соли для изменения осмотического давления, буферы, агенты для образования покрытия или антиокислители. Кроме того, они также могут содержать другие терапевтически полезные вещества.
В соответствии с изобретением соединения общих формул I и II, а также их фармацевтически приемлемые соли могут применяться в качестве защитных веществ для нервной системы, в частности для лечения или предупреждения ишемической болезни, гипогликемии, гипоксии, мозговых сосудистых спазмов, мышечной спастичности, травм, кровотечений, инфекций (вирусных, бактериальных, амебных, приональных), эпилептических припадков, аутоиммунных заболеваний, симптомов синдрома отмены, болезни Альцгеймера, болезни Паркинсона, бокового амиотрофического склероза, болезни Гентингтона, интоксикаций, оливопонтоцеребеллярной атрофии, повреждений спинного мозга, шизофрении, депрессий, состояний тревоги, лекарственной зависимости, болей, аутизма и задержки умственного развития. Дозы могут варьироваться в широких пределах, и их, разумеется, необходимо подбирать в соответствии с индивидуальными требованиями в каждом конкретном случае. При оральном введении суточная доза составляет приблизительно 50-500 мг, хотя верхний предел может быть превышен в том случае, если это показано.
Приведенные ниже примеры служат для более подробной иллюстрации настоящего изобретения. Однако они не ограничивают объем изобретения. Все температуры указаны в градусах Цельсия.
Получение соединений формул Ia и Ib
Пример 1
2,3,5,
6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) 1,2 г (7,4 ммоля) 1,2,3,4-тетрагидрохиназолин-2,4-диона
суспендировали в 10 мл (74 ммоля) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду.
Коричневый осадок сушили и хроматографировали на силикагеле с использованием
метиленхлорида в качестве элюента. Выход: 331 мг (22%) 2,4-дихлорхиназолина в виде белых кристаллов; МС: me/e = 198,200
(M+).
б) К раствору, содержащему 360 мг (1,80 ммоля) 2,4-дихлорхиназолина в 5 мл диметилсульфоксида, добавляли 281 мг (2,7 ммоля) этилкарбазата. Реакционную смесь перемешивали при 80oC в течение 4 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию выдерживали при 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 140 мг (31%) этил-2-гидрокси-4-хиназолинкарбазата в виде белых кристаллов; МС: me/e = 248 (M+).
в) 150 мг (0,6 ммоля) этил-2-гидрокси-4-хиназолинкарбазата в 5 мл диметилформамида выдерживали при температуре дефлегмации в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Таким путем получали 66 мг (54%) 2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде белых кристаллов; МС: me/e = 202 (M+).
Пример 2
7-хлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 5,0 г (0,03 моля) 2-амино-3-хлорбензойной кислоты
и 3,5 г (0,06 моля) мочевины, нагревали до 140oC в течение 2 ч и дополнительно выдерживали при 180oC в течение 24 ч. Полученную коричневую массу перемешивали с 50 мл воды и 50 мл
этилацетата, получая при этом осадок. Его
отфильтровывали, сушили под вакуумом и получали кристаллы бежевого цвета, которые перекристаллизовывали из метанола. Выход: 3,5 г (63%) 8-хлор-1,2,3,
4-тетрагидрохиназолин-2,4-диона в виде белых
кристаллов; tпл 224-225oC.
б) 3,6 г (0,018 моля) 8-хлор-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 16,8 мл (0,18 моля) оксихлорида фосфора и кипятили с обратным холодильником в течение 17 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок экстрагировали дихлорметаном, хроматографировали на силикагеле с использованием дихлорметана в качестве элюента и перекристаллизовывали из диизопропилового эфира. Выход: 2,26 г (53%) 2,4, 8-трихлорхиназолина в виде желтых кристаллов; tпл 155-156oC.
в) К раствору, содержащему 2,26 г (0,009 моля) 2,4,8-трихлорхиназолина в 95 мл диметилсульфоксида, добавляли 1,5 г (0,014 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Желтый осадок отфильтровывали, сушили и перекристаллизовывали из метанола. Выход: 1,56 г (54%) этил-2, 8-дихлорхиназолин-4-илкарбазата в виде белых кристаллов; tпл 213-215oC.
г) Раствор, содержащий 1,56 г (0,0052 моля) этил-2,8-дихлорхиназолин-4-илкарбазата в 47 мл диметилсульфоксида, нагревали до 95oC в течение 6 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Белый осадок отфильтровывали и сушили. Выход: 0,86 г (59%) этил-8-хлор-2-гидроксихиназолин-4-илкарбазата в виде белых кристаллов; tпл 348-350oC.
д) 1,27 г (0,0045 моля) этил-8-хлор-2-гидроксихиназолин-4-илкарбазата в 75 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, сушили под вакуумом и перекристаллизовывали из метанола/тетрагидрофурана. Выход: 0,66 г (62%) 7-хлор-2,3,5,6-тетрагидро-1,2,4-триазол[4, 3-c] хиназолин-3,5-диона в виде желтоватых кристаллов; tпл 348-350oC.
Пример 3
8-хлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 18
г (0,105 моля) 2-амино-4-хлорбензойной кислоты и 12 г (0,20 моля) мочевины, нагревали до 160oC в течение 2 ч и дополнительно выдерживали при 180oC в
течение 2 ч. Полученную
коричневую массу растирали с 200 мл метанола, отфильтровывали и сушили под вакуумом. Выход: 12,2 г (59%) 7-хлор-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде светло-коричневых
кристаллов; МС: me/e =
196 (M+).
б) 10,5 г (0,053 моля) 7-хлор-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 35 мл (0,48 моля) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок отфильтровывали под вакуумом, сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Таким путем получали 7,2 г (58%) 2,4,7-трихлорхиназолина в виде желтых кристаллов; МС: me/e = 232,234 (M+).
в) К раствору, содержащему 0,95 г (4,07 ммоля) 2,4,7-трихлорхиназолина в 40 мл диметилсульфоксида, добавляли 0,845 г (8,13 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 0,45 г (39%) этил-7-хлор-2-гидрокси-4- хиназолинкарбазата в виде белых кристаллов; МС: me/e = 282 (M+).
г) 0,45 г (1,59 ммоля) этил-7-хлор-2-гидрокси-4-хиназолинкарбазата в 10 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 0,18 г (48%) 8-хлор-2,3,5,6-тетрагидро-1,2,4- триазол[4, 3-c]хиназолин-3,5-диона в виде белых кристаллов; МС: me/e = 236 (M+).
Пример 4
9-хлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь,
содержащую 2,0 г (11,6 ммоля) 2-амино-5-хлорбензойной кислоты и 3,48 г (58 ммолей) мочевины, нагревали до 160oC в течение 2 ч и выдерживали дополнительно при 180oC в течение 2
ч.
Полученную коричневую массу растирали с 200 мл метанола, отфильтровывали и сушили под вакуумом. Выход: 1,48 г (65%) 6-хлор-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде белых кристаллов.
б) 1,4 г (7,13 ммоля) 6-хлор-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 7 мл (96 ммолей) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок отфильтровывали, сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Выход: 1 г (60%) 2,4,6-трихлорхиназолина в виде белых кристаллов; МС: me/e = 232,234 (M+).
в) К раствору, содержащему 500 мг (2,1 ммоля) 2,4,6-трихлорхиназолина в 20 мл диметилсульфоксида, добавляли 334 мг (3,2 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 285 мг (47%) этил-6-хлор-2-гидрокси-4-хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 281 (M-H)+.
г) 0,16 г (0,68 ммоля) этил-6-хлор-2-гидрокси-4-хиназолинкарбазата в 15 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 35 мг (26%) 9-хлор-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде белых кристаллов; МС: me/e = 236 (M+).
Пример 5
10-хлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 2,0 г (11 ммолей) 2-амино-6-хлорбензойной кислоты и
1,
4 г (23 ммоля) мочевины, нагревали до 160oC в течение 2 ч и выдерживали дополнительно при 180oC в течение 2 ч. Полученную коричневую массу растирали с 200 мл метанола,
отфильтровывали и сушили под вакуумом. Выход: 1,2 г (53%) 5-хлор-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде белых кристаллов; МС: me/e = 196 (M+);
б) 0,5 г (2,54 ммоля) 5-хлор-1,
2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 7 мл (96 ммолей) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры
и
сливали на ледяную воду. Коричневый осадок отфильтровывали, сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Выход: 260 мг (44%) 2,4,5-трихлорхиназолина
в
виде белых кристаллов; МС: me/e = 232,234 (M+).
в) К раствору, содержащему 200 мг (0,85 ммоля) 2,4,5-трихлорхиназолина в 5 мл диметилсульфоксида, добавляли 134 мг (1,28 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 80 мг (37%) этил-5-хлор-2-гидрокси-4-хиназолинкарбазата в виде белых кристаллов; МС: me/e = 282 (M+).
г) 80 мг (0,28 ммоля) этил-5-хлор-2-гидрокси-4-хиназолинкарбазата в 5 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 38 мг (57%) 10-хлор-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде белых кристаллов; МС: me/e = 236 (M+).
Пример 6
8-Фтор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 0,2 г (1,3 ммоля) 2-амино-4-фторбензойной кислоты и 1,16 г (20 ммолей) мочевины, нагревали до 160oC в течение 2 ч и дополнительно выдерживали при 180oC в течение 2 ч. Полученную коричневую массу растирали с 200 мл метанола, отфильтровывали и сушили под вакуумом. Выход: 167 мг
(70%) 7-фтор-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде желтых кристаллов; МС: me/e = 180 (M+).
б) 164 мг (0,9 ммоля) 7-фтор-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 3,5 мл (48 ммолей) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок отфильтровывали под вакуумом, сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Выход: 130 мг (67%) 2,4-дихлор-7-фторхиназолина в виде кристаллов оранжевого цвета; МС: me/e = 216,218 (M+).
в) К раствору, содержащему 780 мг (3,6 ммоля) 2,4-дихлор-7- фторхиназолина в 10 мл диметилсульфоксида, добавляли 562 мг (5,4 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 0, 58 г (61%) этил-7-фтор-2-гидрокси-4- хиназолинкарбазата в виде кристаллов светло-оранжевого цвета; МС: me/e = 266 (M+).
г) 30 г (0,11 ммоля) этил-7-фтор-2-гидрокси-4-хиназолинкарбазата в 2 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 10 мг (41%) 8-фтор-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде белых кристаллов; МС: me/e = 220 (M+).
Пример 7
9-фтор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 7,2 г (0,046 моля) 2-амино-5-фторбензойной кислоты и
16,7 г (0,28 моля) мочевины, нагревали до 180oC в течение 3 ч. Полученную коричневую массу растирали в ступке, суспендировали в течение ночи в воде, отфильтровывали, промывали водой и
затем
ацетоном и сушили под вакуумом. Выход: 5,1 г (61%) 6-фтор-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде коричневатых кристаллов; tпл 335-340oC (разл.).
б) 5, 1 г (0, 028 моля) 6-фтор-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 39 мл (0,425 моля) оксихлорида фосфора и нагревали до 105oC в течение 18 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок экстрагировали дихлорметаном и хроматографировали на силикагеле с использованием дихлорметана в качестве элюента. Выход: 5,0 г (81%) 2,4-дихлор-6-фторхиназолина в виде белых кристаллов; tпл 133-136oC.
в) К раствору, содержащему 5,5 г (0,025 моля) 2,4-дихлор-6-фторхиназолина в 220 мл диметилсульфоксида, добавляли 3,43 г (0,033 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Осадок отфильтровывали, сушили и суспендировали в 200 мл ацетона. Кристаллы отфильтровывали и сушили под вакуумом. Выход: 3,7 г (55%) этил-6-фтор-2-гидроксихиназолин-4-илкарбазата в виде белых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 266 (C11H11FN4O3+, 10), 220(41), 180(90), 137(100), 109(83), 82(47).
г) 3,7 г (0,014 моля) этил-6-фтор-2-гидроксихиназолин-4-илкарбазата в 75 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали ацетоном и сушили под вакуумом. Выход: 1,7 г (56%) 9-фтор-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3, 5-диона в виде желтоватых кристаллов; tпл > 350oC. МС: me/e = 220 (% от основного пика) = 220 (C9H5FN4O2+, 100), 163(23), 136(26), 135(18), 121(23), 108(21), 43(23).
Пример 8
8-бром-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
63,8 мг (0,92 ммоля) нитрита
натрия добавляли
при 0oC к суспензии, содержащей 210 мг (0,83 ммоля) гидрохлорида 8-амино-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-диона в 5 мл 48%-ной бромистоводородной
кислоты. Смесь
перемешивали при 0oC в течение 1 ч и затем по каплям добавляли 132 мг (0,99 ммоля) бромида меди (I) в 2 мл воды. После перемешивания при комнатной температуре в течение 16 ч
осадок
отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 57 мг (24%) 8-бром-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде желтоватых кристаллов; МС: me/e = 280,
282
(M+).
Пример 9
8-иод-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
63,8 мг (0,92 ммоля) нитрита натрия добавляли при 0oC к
суспензии,
содержащей 210 мг (0,83 ммоля) гидрохлорида 8-амино-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в 10 мл уксусной кислоты и 10 мл серной кислоты. Реакционную смесь
перемешивали при
0oC в течение 1 ч. Далее по каплям добавляли 2,49 г (16,6 ммоля) йодида натрия в 10 мл воды и смесь перемешивали при комнатной температуре в течение еще 1 ч. Осадок
отфильтровывали,
коричневые кристаллы суспендировали в 10 мл 5%-ного раствора тиосульфата натрия, отфильтровывали и сушили под вакуумом. Выход: 145 мг (53%) 8-йод-2,3,5,6-тетрагидро-1,2,4-триазол[4,
3-c] хиназолин-3,
5-диона в виде коричневатых кристаллов; МС: me/e = 328 (M+).
Пример 10
8-хлор-9-фтор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь,
содержащую 8,9 г (0,047 моля) 2-амино-4-хлор-5-фторбензойной кислоты (R.Krishan, S.A. Lang Jr., M.M.Siegel, J. Het. Chem. 1986, 23, 1801) и 17 г (0,28 моля) мочевины, нагревали до
180oC в
течение 3 ч. Полученную коричневую массу растирали в ступке, суспендировали в течение ночи в воде, отфильтровывали, последовательно промывали водой и ацетоном и сушили под
вакуумом. Выход: 7,9 г (78%)
7-хлор-6-фтор-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде коричневатых кристаллов; tпл 320-345oC (разл.). МС: me/e = (% от основного пика) = 214
(C8H4
ClFN2O2+, 76), 171(100), 144(52), 143(30), 116(16), 108(17), 81(35).
б) 1,5 г (0,0070 моля) 7-хлор-6-фтор-1,2,3, 4-тетрагидрохиназолин- 2, 4-диона суспендировали в 9,6 мл (0,10 моля) оксихлорида фосфора и нагревали до 105oC в течение 84 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок экстрагировали дихлорметаном и хроматографировали на силикагеле с использованием дихлорметана в качестве элюента. Выход: 1,45 г (82%) 2,4, 7-трихлор-6-фторхиназолина в виде желтоватых кристаллов; tпл 90-92oC.
в) К раствору, содержащему 4,0 г (0,016 моля) 2,4,7-трихлор-6- фторхиназолина в 160 мл диметилсульфоксида, добавляли 2,16 г (0,021 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Осадок отфильтровывали, промывали этанолом и сушили под вакуумом. Выход: 1,6 г (34%) этил-7-хлор-6-фтор-2-гидроксихиназолин-4-илкарбазата в виде желтых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 300 (C11H10ClFN4O3+, 18), 254(100), 227(14), 197(20), 170(27), 45(22), 31(50), 29(50).
г) 1,6 г (0,0053 моля) этил-7-хлор-6-фтор-2-гидроксихиназолин- 4-илкарбазата в 30 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали ацетоном и сушили под вакуумом. Выход: 0,45 г (33%) 8-хлор-9-фтор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин- 3,5-диона в виде желтоватых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 254 (C9H4ClFN4O2+, 100), 197(12), 170(16), 155(11), 142(8), 58(6).
Пример 11
8,
9-дихлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 0,3 г (1,5 ммоля) 2-амино-4,
5-дихлорбензойной кислоты и 875 мг (15 ммолей) мочевины, нагревали до
160oC в течение 2 ч и дополнительно выдерживали при 180oC в течение 2 ч. Полученную коричневую массу
растирали с 200 мл метанола, отфильтровывали и сушили под вакуумом. Выход:
230 мг (68%) 5,6-дихлор-1,2,3,4-тетрагидрохиназолин- 2,4-диона в виде белых кристаллов; МС: me/e = 230,232 (M+
).
б) 200 мг (0,9 ммоля) 5,6-дихлор-1,2,3, 4-тетрагидрохиназолин-2,4-диона суспендировали в 3,5 мл (48 ммолей) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок отфильтровывали под вакуумом, сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Выход: 110 мг (47%) 2,4,6, 7-тетрахлорхиназолина в виде желтых кристаллов; МС: me/e = 268 (M+).
в) К раствору, содержащему 100 мг (0,4 ммоля) 2,4,6,7-тетрахлорхиназолина в 2 мл диметилсульфоксида, добавляли 53 мг (0,6 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры и кристаллы отфильтровывали и сушили под вакуумом. Выход: 55 мг (47%) этил-6,7-дихлор-2-гидрокси-4-хиназолинкарбазата в виде белых кристаллов; МС: me/e = 316 (M+).
г) 50 мг (0,11 ммоля) этил-6, 7-дихлор-2-гидрокси-4-хиназолинкарбазата в 2 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 20 мг (46%) 8,9-дихлор-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3, 5-диона в виде белых кристаллов; МС: me/e = 270 (M+).
Пример 12
8,10-дихлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую
2 г (10 ммолей) 2-амино-4,6-дихлорбензойной
кислоты и 1,6 г (30 ммолей) мочевины, нагревали до 160oC в течение 2 ч и дополнительно выдерживали при 180oC в течение 2 ч.
Полученную коричневую массу растирали с 200 мл
метанола, отфильтровывали и сушили под вакуумом. Выход: 880 мг (40%) 5,7-дихлор-1,2,3,4-тетрагидрохиназолин- 2,4-диона в виде бежевых кристаллов; МС:
me/e = 230,232 (M+).
б) 0,5 г (2 ммоля) 5,7-дихлор-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 3,5 мл (48 ммолей) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Выход: 276 мг (43%) 2,4,5, 7-тетрахлорхиназолина в виде белых кристаллов; МС: me/e = 268 (M+).
в) К раствору, содержащему 200 мг (0,75 ммоля) 2,4,5,7-тетрахлорхиназолина в 5 мл диметилсульфоксида, добавляли 116 мг (1,1 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 90 мг (38%) этил-5,7-дихлор-2-гидрокси-4-хиназолинкарбазата в виде белых кристаллов; МС: me/e = 316 (M+).
г) 90 мг (0,28 ммоля) этил-5,
7-дихлор-2-гидрокси-4-хиназолинкарбазата в 2 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до
комнатной температуры и сливали на ледяную
воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 15 мг (20%) 8,10-дихлор-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c] хиназолин-3,
5-диона в виде белых кристаллов; МС: me/e =
270,272 (M+)
Пример 13
7,8-диметил-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 5,
96 г (0,036 моля) 2-амино-3,4-диметилбензойной
кислоты и 4,33 г (0,072 ммоля) мочевины, нагревали до 140oC в течение 2 ч и дополнительно выдерживали при 180oC в течение 2 ч.
Полученную коричневую массу перемешивали с 100 мл
воды и 100 мл этилацетата, получая при этом осадок. Осадок отфильтровывали, сушили под вакуумом, получая при
этом светло-коричневатые
кристаллы, которые перекристаллизовывали из
диметилформамида/метанола. Выход: 4,26 г (62%) 7,8-диметил-1,2,3,4- тетрагидрохиназолин-2,4-диона в виде бежевых кристаллов; tпл 305-307oC.
б) 7,96 г (0,042 моля) 7,8-диметил-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 40 мл (0,43 моля) оксихлорида фосфора и выдерживали при температуре кипения с обратным холодильником в течение 16 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок экстрагировали этилацетатом, хроматографировали на силикагеле с использованием дихлорметана в качестве элюента и перекристаллизовывали из этилацетата/н-гексана. Выход: 5,9 г (62%) 2,4-дихлор-7,8-диметилхиназолина в виде белых кристаллов; tпл 145-146oC.
в) К раствору, содержащему 5,83 г (0,025 моля) 2,4-дихлор-7,8-диметилхиназолина в 140 мл диметилсульфоксида, добавляли 5,40 г (0,051 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 5 ч и затем сливали на ледяную воду. Желтый осадок отфильтровывали. Раствор концентрировали и остаток перемешивали в ацетонитриле при комнатной температуре в течение ночи, отделяя при этом белые кристаллы. Кристаллы отфильтровывали под вакуумом и сушили под вакуумом. Выход: 3,21 г (45%) этил-2-гидрокси-7, 8-диметилхиназолин-4-илкарбазата в виде белых кристаллов; tпл 370-372oC.
г) 3,88 г (0,012 моля) этил-2-гидрокси-7,8-диметилхиназолин-4- илкарбазата в 220 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из диметилформамида. Выход: 1,62 г (50%) 7,8-диметил-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин- 3,5-диона в виде желтоватых кристаллов; tпл 380-382oC.
Пример
14
8,9-диметил-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 7,0 г (0,042
моля) 2-амино-4,5-диметилбензойной кислоты и 5,07 г (0,084 моля) мочевины,
нагревали до 140oC в течение 2 ч и дополнительно выдерживали при 180oC в течение 2 ч. Полученную
коричневую массу перемешивали с 50 мл воды и 50 мл этилацетата, получая при этом
осадок. Осадок отфильтровывали, сушили под вакуумом, получая при этом светло-коричневатые кристаллы, которые
перекристаллизовывали из метанола/тетрагидрофурана. Выход: 4,5 г (56%) 6,7-диметил-1,2,3,
4-тетрагидрохиназолин-2,4-диона в виде бежевых кристаллов; tпл 327-329oC.
б) 4,5 г (0,023 моля) 6,7-диметил-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 25 мл (0,27 моля) оксихлорида фосфора и выдерживали при температуре дефлегмации в течение 16 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок экстрагировали дихлорметаном, хроматографировали на силикагеле с использованием дихлорметана в качестве элюента и перекристаллизовывали из этилацетата/н-гексана. Выход: 2,48 г (46%) 2, 4-дихлор-6,7-диметилхиназолина в виде белых кристаллов; tпл 140-142oC.
в) К раствору, содержащему 2,48 г (0,010 моля) 2,4-дихлор-6,7-диметилхиназолина в 60 мл диметилсульфоксида, добавляли 2,33 г (0,020 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 5 ч и затем сливали на ледяную воду. Желтый осадок отфильтровывали, сушили и перекристаллизовывали из диметилформамида/метанола. Выход: 1, 24 г (41%) этил-2-гидрокси-6, 7- диметилхиназолин-4-илкарбазата в виде белых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 276 (C13H16N4O3+, 16), 230(100), 203(20), 174(34), 146(43), 131(22), 31(31).
г) 1,0 г (0, 0030 моля) этил-2-гидрокси-6,7-диметилхиназолин-4- илкарбазата в 57 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из диметилформамида/метанола. Выход: 0,43 г (52%) 8,9-диметил-2, 3,5,6-тетрагидро-1,2,4- триазол[4,3-c]хиназолин-3,5-диона в виде желтоватых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 230 (C11H10 N4O2+, 100), 215(5), 186(3,5), 173(25), 146(32), 131(14), 116(13).
Пример 15
9,10-диметил-2,3,5,6-тетрагидро-1,2,4-триазол[4,
3-c]хиназолин-3,5-дион
а) Смесь, содержащую 11,8 г (0,070 моля) 2-амино-5,
6-диметилбензойной кислоты и 8,52 г (0,14 ммоля) мочевины, нагревали до 140oC в течение 2 ч и
дополнительно выдерживали при 180oC в течение 2 ч. Полученную коричневую массу
перемешивали с 50 мл воды и 50 мл этилацетата, получая при этом осадок. Осадок отфильтровывали и сушили под
вакуумом. Выход: 3,11 г (23%) 5,6-диметил-1,2,3,4-тетрагидрохиназолин- 2,4-диона в виде
коричневых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 190 (C10H10N2O2+, 73), 147(100),
120(20), 118(24), 104(16), 91(19).
6) 3,69 г (0,019 моля) 5,6-диметил-1,2,3, 4-тетрагидрохиназолин-2,4-диона суспендировали в 17,7 мл (0,194 моля) оксихлорида фосфора и выдерживали при температуре кипения с обратным холодильником в течение 16 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок экстрагировали дихлорметаном, хроматографировали на силикагеле с использованием дихлорметана в качестве элюента и перекристаллизовывали из этилацетата/н-гексана. Выход: 2,37 г (54%) 2,4-дихлор-5,6-диметилхиназолина в виде белых кристаллов; tпл 121-123oC.
в) К раствору, содержащему 2,37 г (0,010 моля) 2,4-дихлор-5,6- диметилхиназолина в 60 мл диметилсульфоксида, добавляли 2,2 г (0,020 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 5 ч и затем сливали на ледяную воду. Желтый осадок отфильтровывали и сушили. Выход: 0,71 г (29%) этил-5, 6-диметил-2- гидроксихиназолин-4-илкарбазата в виде желтоватых кристаллов; tпл 358-360oC.
г) 0,67 г (0,0020 моля) этил-5, 6-диметил-2-гидроксихиназолин-4- илкарбазата в 38 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из метанола/диметилформамида. Выход: 0,41 г (74%) 9,10-диметил-2,3,5, 6-тетрагидро-1,2,4- триазол[4,3-c]хиназолин-3,5-диона в виде желтоватых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 230 (C11H10N4O2+, 100), 187(27), 172(42), 145(25).
Пример 16
7-метокси-2,3,5,
6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь,
содержащую 4,7 г (0,028 моля) 2-амино-3-метоксибензойной кислоты и 3,37 г (0,056 моля) мочевины, нагревали до 140oC в
течение 2 ч и дополнительно выдерживали при 180oC в течение
2 ч. Полученную коричневую массу перемешивали с 100 мл воды и 100 мл этилацетата, получая при этом осадок. Осадок отфильтровывали
и сушили под вакуумом. Выход: 3,28 г (61%) 8-метокси-1,2,3,
4-тетрагидрохиназолин- 2,4-диона в виде бежевых кристаллов; tпл 266-268oC.
б) 3,28 г (0,017 моля) 8-метокси-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 15,6 мл (0,17 моля) оксихлорида фосфора и выдерживали при температуре дефлегмации в течение 16 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок экстрагировали дихлорметаном, хроматографировали на силикагеле с использованием дихлорметана в качестве элюента и перекристаллизовывали из диизопропилового эфира. Выход: 2,38 г (61%) 2, 4-дихлор-8-метоксихиназолина в виде белых кристаллов; tпл 161-162oC.
в) К раствору, содержащему 2,38 г (0,010 моля) 2,4-дихлор-8-метоксихиназолина в 60 мл диметилсульфоксида, добавляли 1,59 г (0,015 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 3 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали. Желтый раствор концентрировали и остаток перемешивали в смеси 1:4 ацетонитрил/этанол при комнатной температуре в течение ночи, отделяя при этом белый осадок. Осадок отфильтровывали под вакуумом, сушили под вакуумом и перекристаллизовывали из метанола/простого эфира. Выход: 1,06 г (61%) этил-2-гидрокси-8-метоксихиназолин-4-илкарбазата в виде белых кристаллов; tпл 314-316o C.
г) 1,02 г (0,0036 моля) этил-2-гидрокси-8-метоксихиназолин-4- илкарбазата в 65 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из метанола. Выход: 0,52 г (61%) 7-метокси-2,3,5,6-тетрагидро-1,2, 4-триазол[4,3-c]хиназолин-3,5-диона в виде желтоватых кристаллов; tпл 308-310oC.
Пример 17
8-метокси-2,3,5,
6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,
5-дион
а) Раствор, содержащий 1,0 г (0,015 моля) цианата натрия в 10 мл воды, добавляли по каплям при 70oC в течение 10 минут к
суспензии, содержащей 1,7 г (0,010 моля)
2-амино-4-метоксибензойной кислоты в 17 мл уксусной кислоты. После перемешивания дополнительно в течение 10 минут полученную белую суспензию охлаждали до
комнатной температуры, разбавляли водой и
затем фильтровали под вакуумом. Полученные кристаллы кипятили с обратным холодильником в течение 30 минут в 15 мл 37%-ной водной соляной кислоты, охлаждали,
разбавляли 75 мл воды, фильтровали под
вакуумом, промывали водой и сушили под вакуумом. Выход: 0,73 г (37%) 7-метокси-1,2,3,4-тетрагидрохиназолин- 2,4-диона в виде белых кристаллов; tпл
320-323oC.
б) 10 г (0,052 моля) 7-метокси-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 72 мл (0,78 моля) оксихлорида фосфора и нагревали до 105oC в течение 4 ч. Реакционной смеси давали охладиться до комнатной температуры, обрабатывали толуолом, осторожно сливали на ледяную воду и фильтровали через дикалит (Dicalite). Водную фазу экстрагировали этилацетатом, органические фазы объединяли и концентрировали и остаток хроматографировали на силикагеле с использованием дихлорметана в качестве элюента. Выход: 10,8 г (91%) 2, 4-дихлор-7-метоксихиназолина в виде белых кристаллов; tпл 123-124oC.
в) К раствору, содержащему 4,0 г (0,0175 моля) 2,4-дихлор-7-метоксихиназолина в 160 мл диметилсульфоксида, добавляли 2,72 г (0,026 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Осадок отфильтровывали, раствор концентрировали и остаток суспендировали в метаноле. Суспензию фильтровали под вакуумом, промывали небольшим количеством метанола и сушили под вакуумом. Выход: 1,65 г (34%) этил-2-гидрокси-7-метоксихиназолин-4- илкарбазата в виде белесых кристаллов; tпл 330-332oC.
г) 1,65 г (0,0059 моля) этил-2-гидрокси-7-метоксихиназолин-4- илкарбазата в 30 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из метанола. Выход: 0,58 г (42%) 8-метокси-2,3,5, 6-тетрагидро-1,2,4- триазол[4,3-c]хиназолин-3,5-диона в виде белых кристаллов; tпл 344-346oC.
Пример 18
9-метокси-2,3,5,6-тетрагидро-1,2,4-триазол[4,
3-c]хиназолин-3,5-дион
а) 12,5 г (0,075 моля) 2-амино-5-метоксибензойной кислоты растворяли в 60 мл 2Н соляной
кислоты, при этом через 10 с образовывалась масса, которую нельзя было
перемешать; она превращалась в суспензию после разбавления 120 мл воды. Далее в течение 10 минут при комнатной температуре
добавляли по каплям раствор, содержащий 6,8 г (0,105 моля) цианата натрия в
70 мл воды. После дополнительного перемешивания в течение 16 ч полученную суспензию фильтровали под вакуумом,
последовательно промывали водой и простым эфиром и сушили под вакуумом. Полученные
кристаллы кипятили с обратным холодильником в течение 1 ч в 75 мл 37%-ной водной соляной кислоты, охлаждали,
разбавляли 300 мл воды, фильтровали под вакуумом, промывали водой и сушили под вакуумом.
Выход: 8,6 г (60%) 5-метокси-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде бежевых кристаллов; tпл
330-340oC.
б) 7,3 г (0,038 моля) 5-метокси-1,2,3, 4-тетрагидрохиназолин-2,4-диона суспендировали в 52 мл (0,57 моля) оксихлорида фосфора и нагревали до 105oC в течение 18 ч. Реакционной смеси давали охладиться до комнатной температуры, обрабатывали толуолом, осторожно сливали на ледяную воду и фильтровали через дикалит. Водную фазу экстрагировали этилацетатом, органические фазы объединяли, концентрировали и остаток хроматографировали на силикагеле с использованием дихлорметана в качестве элюента. Выход: 8,0 г (92%) 2, 4-дихлор-6-метоксихиназолина в виде желтоватых кристаллов; tпл 175-177o C.
в) К раствору, содержащему 8,0 г (0,035 моля) 2,4-дихлор-6-метоксихиназолина в 320 мл диметилсульфоксида, добавляли 4,8 г (0,046 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Осадок отфильтровывали под вакуумом, промывали водой и суспендировали в 50 мл метанола. Суспензию фильтровали под вакуумом, промывали небольшим количеством метанола и сушили под вакуумом. Выход: 3,0 г (31%) этил-2-гидрокси-6-метоксихиназолин-4-илкарбазата в виде белых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 278 (C12H14N4O4+, 7,5), 232(100), 217(19), 205(7,5), 191 (9), 176(30), 148(22), 133(26), 45(25), 31(43), 29(38).
г) 3,0 г (0,011 моля) этил-2-гидрокси-6-метоксихиназолин-4-илкарбазата в 70 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из метанола/ацетона. Выход: 1,16 г (46%) 9-метокси-2,3, 5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде бежевых кристаллов; tпл > 350o C. МС: me/e (% от основного пика) = 232 (C10H8N4O3+, 100), 217(26), 190 (6,5), 176(36), 161(14), 148(42), 133(64).
Пример 19
10-метокси-2,3,5,6-тетрагидро-1,2,4-триазол[4,
3-c]хиназолин-3,5-дион
а) Раствор, содержащий 16,6 г (0,255 моля) цианата натрия в 170 мл воды, добавляли по каплям при
комнатной температуре в течение 40 минут к раствору, содержащему 14,2 г
(0,085 моля) 2-амино-6-метоксибензойной кислоты в 150 мл 2Н соляной кислоты. Через 2 ч по каплям добавляли дополнительно 100 мл
2Н соляной кислоты, а затем 8,3 г (0,13 моля) цианата натрия в 85 мл
воды После дополнительного перемешивания в течение 67 ч полученную белую суспензию фильтровали под вакуумом, последовательно
промывали водой и этилацетатом и сушили под вакуумом. Полученные кристаллы
кипятили с обратным холодильником в течение 30 минут в 80 мл 37%-ной водной соляной кислоты, охлаждали, разбавляли 500 мл
воды, фильтровали под вакуумом, промывали водой и затем ацетоном, и сушили под
вакуумом. Выход: 8,35 г (51%) 5-метокси-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде белых кристаллов; tпл
> 350oC. МС: me/e (% от основного пика) = 192 (C9
H8N2O3+, 74), 174(11), 163 (100), 149(27), 146(36), 122(48), 119(57),
107(65).
б) 9,7 г (0,50 моля) 5-метокси-1,2,3, 4-тетрагидрохиназолин-2,4-диона суспендировали в 70 мл (0,76 моля) оксихлорида фосфора и нагревали до 105oC в течение 20 ч. Реакционной смеси давали охладиться до комнатной температуры, обрабатывали толуолом, осторожно сливали на ледяную воду и фильтровали через дикалит. Водную фазу экстрагировали этилацетатом, органические фазы объединяли и концентрировали и остаток хроматографировали на силикагеле с использованием дихлорметана в качестве элюента. Выход: 8,9 г (77%) 2,4-дихлор-5-метоксихиназолина в виде белых кристаллов; tпл 169-170oC.
в) К раствору, содержащему 5,0 г (0,22 моля) 2,4-дихлор-5-метоксихиназолина в 200 мл диметилсульфоксида, добавляли 3,0 г (0,29 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Осадок отфильтровывали, раствор концентрировали и остаток суспендировали в смеси ацетонитрил/этанол 7:3. Остаток отфильтровывали под вакуумом и осадок на фильтре перекристаллизовывали из горячего метанола. Выход: 2,0 г (33%) этил-2-гидрокси-5-метоксихиназолин-4-илкарбазата в виде желтоватых кристаллов; tпл > 350o C. МС: me/e (% от основного пика) = 278 (C12H14N4O4+, 13), 232(100), 203(29), 190(45), 175(26), 161 (40), 145(19), 118(34), 45(40), 31(84).
г) 2,4 г (0,0086 моля) этил-2-гидрокси-5-метоксихиназолин-4-илкарбазата в 50 мл диметилформамида кипятили с обратным холодильником в течение 3 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из диметилформамида. Выход: 0,32 г (16%) 10-метокси-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде белых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 232 (C10H8N4O3+, 100), 203(34), 161 (43), 145(23), 118(29).
Пример 20
7-нитро-2,3,5,6-тетрагидро-1,2,
4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 10,7 г (0,059 моля) 2-амино-3-нитробензойной кислоты и 21,
6 г (0,35 моля) мочевины, нагревали до 180oC в течение 5 ч.
Полученную коричневую массу растирали в ступке, суспендировали в воде в течение ночи, отфильтровывали, последовательно промывали
водой и ацетоном и сушили под вакуумом. Выход: 7,70 г (63%) 8-нитро-1,2,
3,4-тетрагидрохиназолин-2,4-диона в виде коричневатых кристаллов; tпл 272-276oC (разл.).
б) 7,70 г (0,037 моля) 8-нитро-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 51 мл (0,56 моля) оксихлорида фосфора и нагревали до 105oC в течение 40 ч. Суспензии давали охладиться до комнатной температуры, обрабатывали 250 мл толуола, отфильтровывали под вакуумом и осторожно сливали в 0,5 л воды. Водную фазу экстрагировали этилацетатом, органические фазы объединяли и концентрировали и остаток хроматографировали на силикагеле с использованием дихлорметана в качестве элюента. Выход: 4,50 г (50%) 2,4-дихлор-8-нитрохиназолина в виде желтых кристаллов; tпл 155-157oC.
в) К раствору, содержащему 4,40 г (0,018 моля) 2,4-дихлор-8-нитрохиназолина в 160 мл диметилсульфоксида, добавляли 2,44 г (0,023 моля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Осадок отфильтровывали и сушили. Раствор полностью концентрировали; остаток растворяли в метаноле, осадок отфильтровывали под вакуумом и объединяли с первым осадком. Выход: 1,54 г (29%) этил-2-гидрокси-8-нитрохиназолин-4-илкарбазата в виде кристаллов оранжевого цвета; tпл > 350oC. МС: me/e (% от основного пика) = 293 (C11 H11N5O5+, 25), 221 (18), 220(17), 174(30), 146(19), 29(100).
г) 2,20 г (0,0075 моля) этил-2-гидрокси-8-нитрохиназолин-4-илкарбазата в 50 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, суспендировали в течение ночи в метаноле, отфильтровывали под вакуумом и сушили под вакуумом. При этом получали белые кристаллы, которые перекристаллизовывали из метанола. Выход: 1,1 г (59%) 7-нитро-2,3,5, 6-тетрагидро-1,2,4- триазол[4,3-c]хиназолин-3,5-диона в виде желтых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 247 (C9H5N5 O4+, 100), 207(9), 174 (9), 144(12), 130(14), 117(19), 90(14).
Пример 21
8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 5 г (0,027 моля) 2-амино-4-нитробензойной кислоты и 16,5 г (0,27 моля) мочевины, нагревали до 160oC в течение 2 ч и дополнительно выдерживали при 180oC
в течение 2 ч. Полученную коричневую массу растирали с 200 мл метанола, отфильтровывали и сушили под вакуумом. Выход: 4,8 г
(80%) 7-нитро-1,2,3,4-тетрагидрохиназолин-2,4-диона в виде белого твердого
вещества; МС: me/e = 207 (M+).
б) 18,6 мл (0,204 моля) оксихлорида фосфора добавляли к раствору, содержащему 3,5 г (0,017 моля) 7-нитро-1,2,3,4- тетрагидрохиназолин-2, 4-диона и 6,5 мл (0,051 моля) коллидина в 60 мл ацетонитрила, и затем смесь кипятили с обратным холодильником в течение 4 ч. Растворитель удаляли на роторном испарителе и остаток распределяли между 300 мл воды и 300 мл этилацетата. Органическую фазу промывали 200 мл насыщенного раствора NaHCO3 и дважды 200 мл насыщенного раствора хлорида натрия и в завершение сушили над сульфатом натрия. После фильтрации и концентрирования остаток хроматографировали на флорисиле, используя метиленхлорид в качестве элюента. Выход: 2,46 г (60%) 2,4-дихлор-7-нитрохиназолина в виде желтых кристаллов; МС: me/e = 243,245 (M+).
в) К раствору, содержащему 2,45 г (0,01 моля) 2, 4-дихлор-7-нитрохиназолина в 100 мл диметилсульфоксида, добавляли 1,574 г (0,015 моля) этилкарбазата. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 1,77 г (60%) этил-7-нитро-2-гидрокси-4- хиназолинкарбазата в виде желтых кристаллов.
г) 1,7 г (5,4 ммоля) этил-7-нитро-2-гидрокси-4-хиназолинкарбазата в 20 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 1,15 г (77%) 8-нитро-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде желтых кристаллов: МС: me/e = 247 (M+).
Пример 22
9-нитро- 2,3,5,6-тетрагидро-1,2,
4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 2,0 г (10 ммолей)
2-амино-5-нитробензойной кислоты и 6,6 г (110 ммолей) мочевины, нагревали до 160oC в течение 2 ч и
дополнительно выдерживали при 180oC в течение 2 ч. Полученную коричневую массу
растирали с 200 мл метанола, отфильтровывали и сушили под вакуумом. Выход: 2 г (78%) 5-нитро-1,2,3,
4-тетрагидрохиназолин-2,4-диона в виде светло-коричневых кристаллов; МС: me/e = 207 (M+
).
б) 1 г (5 ммолей) 5-нитро-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 7 мл (96 ммолей) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок отфильтровывали, сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Выход: 754 мг (63%) 2,4-дихлор-6-нитрохиназолина в виде белых кристаллов; МС: me/e = 243, 245 (M+).
в) К раствору, содержащему 750 мг (3,1 ммоля) 2, 4-дихлор-6- нитрохиназолина в 15 мл диметилсульфоксида, добавляли 480 мг (4,6 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 640 мг (71%) этил-2-гидрокси-7-нитро-4- хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 293 (M+).
г) 620 мг (2,1 ммоля) этил-2-гидрокси-7-нитро-4-хиназолинкарбазата в 15 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 460 мг (88%) 9-нитро-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3, 5-диона в виде желтых кристаллов; МС: me/e = 247 (M+).
Пример 23
8-трифторметил-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь,
содержащую 1,0 г (0,005 моля) 2-амино-4-трифторметилбензойной кислоты и 2,64 г (0,044 моля) мочевины,
нагревали до 160oC в течение 2 ч и дополнительно выдерживали при 180oC в
течение 2 ч. Полученную коричневую массу растирали с 200 мл метанола, отфильтровывали и сушили под
вакуумом. Выход: 0,7 г (62,5%) 7-трифторметил-1,2,3,4-тетрагидрохиназолин-1,4-диона в виде светлых
коричневатых кристаллов; МС: me/e = 230 (M+).
б) 200 мг (0,87 ммоля) 7-трифторметил-1,2,3,4-тетрагидрохиназолин- 1,4-диона суспендировали в 3,5 мл (48 ммолей) оксихлорида фосфора и нагревали до 120oC в течение 24 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Коричневый осадок отфильтровывали под вакуумом, сушили и хроматографировали на силикагеле с использованием метиленхлорида в качестве элюента. Выход: 142 мг (58%) 2,4-дихлор-7-трифторметилхиназолина в виде кристаллов розового цвета; МС: me/e = 266,286 (M+).
в) К раствору, содержащему 630 мг (2,37 ммоля) 2, 4-дихлор-7- трифторметилхиназолина в 20 мл диметилсульфоксида, добавляли 500 мг (4,8 ммоля) этилкарбазата. Реакционную смесь перемешивали при 70oC в течение 2 ч и затем сливали на ледяную воду. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 0,46 г (61%) этил-2-гидрокси-7-трифторметил-4-хиназолинкарбазата в виде белых кристаллов; МС: me/e = 316 (M+).
г) 0,35 г (1,11 ммоля) этил-2-гидрокси-7-трифторметил-4- хиназолинкарбазата в 10 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 0,11 г (38%) 8-трифторметил-2,3,5,6-тетрагидро-1,2,4- триазол[4, 3-c]хиназолин-3,5-диона в виде белых кристаллов; МС: me/e = 270 (M+).
Пример 24
8-хлор-9-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) 20 г (101,7 ммоля) 7-хлор-1,2,3,
4-тетрагидрохиназолин-2,4-диона растворяли в 100 мл концентрированной серной кислоты и обрабатывали 7 мл концентрированной азотной кислоты. Смесь нагревали до
100oC в течение 10 минут.
После охлаждения реакционную смесь сливали на ледяную воду. Осадок отфильтровывали, сушили в глубоком вакууме и перекристаллизовывали из уксусной кислоты. Выход:
13,3 г (54%) 7-хлор-6-нитро-1,2,3,
4-тетрагидрохиназолин-2,4-диона в виде бежевых кристаллов; МС: me/e = 241 (M+).
б) 2,2 г (9,1 ммоля) 7-хлор-6-нитро-1,2,3, 4-тетрагидрохиназолин- 2,4-диона суспендировали в 15 мл ацетонитрила и кипятили с обратным холодильником в течение 3 ч с 10 мл (110 ммолей) оксихлорида фосфора и 3,6 мл (27,3 ммоля) коллидина. После удаления растворителя остаток суспендировали в метиленхлориде, отфильтровывали через флорисил и сушили под вакуумом. Выход: 1,4 г (55%) 6-нитро-2,4,7-трихлорхиназолина в виде белых кристаллов; МС: me/e = 277,279 (M+).
в) К раствору, содержащему 1,0 г (3,6 ммоля) 6-нитро-2,4,7-трихлорхиназолина в 25 мл диметилсульфоксида, добавляли 0,54 г (5,2 ммоля) этилкарбазата. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем при 70oC в течение 1 ч. Коричневый осадок отфильтровывали и сушили. Коричневые кристаллы суспендировали в 20 мл н-бутанола и суспензию нагревали до 90oC в течение 2 ч. Смеси давали охладиться до комнатной температуры, кристаллы отфильтровывали и сушили под вакуумом. Выход: 660 мг (56%) этил-7-хлор-6-нитро-2-гидрокси-4- хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 327 (M+).
г) 550 мг (1,68 ммоля) этил-7-хлор-6-нитро-2-гидрокси-4- хиназолинкарбазата в 5 мл диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и затем сливали на ледяную воду. Осадок отфильтровывали, промывали метанолом и сушили под вакуумом. Выход: 327 мг (69%) 8-хлор-9-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде кристаллов оранжевого цвета; МС: me/e = 281 (M+).
Пример 25
9-хлор-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 6,0 г (27,7
моля) 2-амино-5-хлор-4-нитробензойной кислоты и 10,0 г (0,166 моля) мочевины,
нагревали до 180oC в течение 2,5 ч. Полученную коричневую массу кипятили с 200 мл воды. Затем ее
отфильтровывали, промывали водой и ацетоном и сушили под вакуумом. Выход: 4,35 г (65%) 2,
4-диоксо-6-хлор-7-нитро-1,2,3,4- тетрагидрохиназолина в виде темно-коричневого порошка; МС: me/e = 243 (M+), 241 (M+).
б) 4,35 г (18,0 ммолей) 2, 4-диоксо-6-хлор-7-нитро-1,2,3,4- тетрагидрохиназолина суспендировали в 40 мл оксихлорида фосфора и нагревали до 140o C в течение 48 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Далее смесь экстрагировали метиленхлоридом и органическую фазу сушили над сульфатом магния, отфильтровывали и концентрировали. Остаток сушили и хроматографировали на силикагеле, используя метиленхлорид в качестве элюента. Выход: 2,535 г (50%) 2,4,6-трихлор-7-нитрохиназолина в виде желтых кристаллов; МС: me/e = 277 (M+), 279 (M+), 281 (M+).
в) К раствору, содержащему 2,535 г (9,1 ммоля) 2,4,6-трихлор-7- нитрохиназолина в 40 мл безводного диметилсульфоксида, добавляли 1,23 г (11,8 ммоля) этилкарбазата. Через 1 ч смесь сливали на ледяную воду. Желтый осадок отфильтровывали, сушили и хроматографировали на силикагеле, используя в качестве элюента смесь (1:1) этилацетат/гексан. Выход: 2, 61 г (83%) этил-2,6-дихлор-7-нитро-4-хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 345 (M+), 347 (M+).
г) Раствор, содержащий 2,6 г (7,5 ммоля) этил-2, 6-дихлор-7-нитро-4- хиназолинкарбазата и 50 мл безводного диметилсульфоксида, нагревали до 100oC в течение 1,5 ч. Затем смесь сливали на лед и желтый осадок отфильтровывали и сушили. Выход: 2,35 г (95%) этил-6-хлор-7-нитро-2-гидрокси-4-хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 327 (M+), 329 (M+).
д) 2,33 г (7,11 ммоля) этил-6-хлор-7-нитро-2-гидрокси-4- хиназолинкарбазата в 100 мл диметилформамида кипятили с обратным холодильником в течение 1,5 ч. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли на роторном испарителе. Остаток растирали с 50 мл ацетона и светло-желтый осадок отфильтровывали, промывали небольшим количеством ацетона и сушили под вакуумом. Выход: 1,284 г (64%) 9-хлор-8-нитро-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде бежевых кристаллов; МС: me/e = 281 (M+), 283 (M+).
Пример 26
Натриевая соль 9-хлор-8-нитро-1-ил-2,3,5,6- тетрагидро[1,2,4] триазол[1,5-c]хиназолин-2,5-диона
5,33 мл 0,1М NaOH добавляли к раствору, содержащему 150 мг (0,
533 ммоля) 9-хлор-8-нитро-1-ил-2,
3,5,6-тетрагидро[1,2,4] триазол[4,3- c]хиназолин-3,5-диона и 10 мл диметилформамида, и смесь перемешивали при комнатной температуре в течение 30 минут. Затем смесь
фильтровали, концентрировали досуха,
трижды обрабатывали деионизированной водой и вновь концентрировали. Остаток сушили в глубоком вакууме. Выход: 142 мг (74%) натриевой соли 9-хлор-8-нитро-1-ил-2,3,5,
6-тетрагидро[1,2,4] триазол[1,5-c]
хиназолин- 2,5-диона в виде красно-коричневого порошка. МС: me/e = 280 (M-Na)-.
Пример 27
9-фтор-8-нитро-2,3,5,6-тетрагидро-1,2,
4-триазол[4,3-c]хиназолин-3,
5-дион
а) 3,8 г (19 ммолей) 2-амино-5-фтор-4-нитробензойной кислоты растворяли в 190 мл этилацетата и этерифицировали при 0oC с помощью диазометана.
Раствор концентрировали и
остаток сушили. Выход: 4,0 г (98%) метил-2-амино-5-фтор-4-нитробензоата в виде желтых кристаллов; МС: me/e = 214 (M+).
б) 4,0 г (18,7 ммоля) метил-2-амино-5-фтор-4-нитробензоата растворяли в 250 мл метанола и насыщали аммонием при -40oC. Далее смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали и остаток растирали с метиленхлоридом. Осадок отфильтровывали под вакуумом и сушили. Выход: 3,17 г (85%) 2-амино-5-фтор-4-нитробензамида в виде желтых кристаллов; МС: me/e = 214 (M+).
в) 3,1 г (15,6 ммоля) 2-амино-5-фтор-4-нитробензамида растворяли в 130 мл тетрагидрофурана и при 0oC обрабатывали 3,1 г (10,4 ммоля) трифосгена. Смеси давали нагреться до комнатной температуры и дополнительно перемешивали в течение 1,5 ч. Затем раствор концентрировали и растирали с водой. Осадок отфильтровывали под вакуумом и сушили. Выход: 2,09 г (60%) 2, 4-диоксо-6-фтор-7-нитро- 1,2,3,4-тетрагидрохиназолина в виде желтоватых кристаллов; МС: me/e = 225 (M+).
г) 2,1 г (9,3 ммоля) 2,4-диоксо-6-фтор-7-нитро-1,2,3, 4- тетрагидрохиназолина суспендировали в 12 мл оксихлорида фосфора и нагревали до 140oC в течение 72 ч. Реакционной смеси давали охладиться до комнатной температуры и сливали на ледяную воду. Затем смесь экстрагировали метиленхлоридом и органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Остаток сушили и хроматографировали на силикагеле, используя метиленхлорид в качестве элюента. Выход: 1,69 г (69%) 2,4-дихлор-6-фтор-7-нитрохиназолина в виде желтых кристаллов; МС: me/e = 261 (M+), 263 (M+), 265 (M+).
д) К раствору, содержащему 2,0 г (7,7 ммоля) 2,4-дихлор-6-фтор-7- нитрохиназолина в 44 мл безводного диметилсульфоксида, добавляли 0,98 г (9,2 ммоля) этилкарбазата. Через 15 минут смесь сливали на ледяную воду. Желтый осадок отфильтровывали и сушили. Выход: 2,13 г (84%) этил-2-хлор-6-фтор-7-нитро-4-хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 329 (M+), 331 (M+).
е) Раствор, содержащий 2,13 г (6,5 ммоля) этил-2-хлор-6-фтор-7- нитро-4-хиназолинкарбазата и 50 мл безводного диметилсульфоксида, нагревали до 100oC в течение 1 ч. Затем смесь сливали на лед и желтый осадок отфильтровывали и сушили. Выход: 1,90 г (94%) этил-6-фтор-7-нитро-2-гидрокси-4-хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 311 (M+).
ж) 404 мг (1,3 ммоля) этил-6-фтор-7-нитро-2-гидрокси-4- хиназолинкарбазата в 20 мл безводного диметилформамида кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли на роторном испарителе. Остаток растирали с небольшим количеством ацетона и осадок оранжевого цвета отфильтровывали и сушили под вакуумом. Выход: 240 мг (70%) 9-фтор-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде кристаллов оранжевого цвета; МС: me/e = 265 (M+).
Пример
28
Гидрохлорид 8-амино-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин- 3,5-диона
1,98 г (8,8 ммоля) дихлорида олова растворяли в 10 мл концентрированной соляной кислоты
при 80oC. К раствору порциями добавляли 494 мг (2 ммоля) 8-нитро-2,3,5,6-тетрагидро-1,2,4- триазол[4,3-c] хиназолин-3,5-диона. После выдержки в течение 2 часов при температуре дефлегмации
осадок
отфильтровывали, промывали водой и сушили под вакуумом. Выход: 315 мг (62%) гидрохлорида 8-амино-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-диона в виде белых кристаллов; МС: me/e =
217
(M-HCl)+.
Пример 29
9-амино-8-хлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин-3,5-дион
672 мг (2,98 ммоля) дихлорида олова растворяли в 3,4 мл
концентрированной соляной кислоты при 80oC. К раствору порциями добавляли 200 мг (0,71 ммоля) 8-хлор-9-нитро-2,3,5,6-тетрагидро-1,2,4- триазол[4,3-c] хиназолин-3,5-диона. После выдержки в
течение 2 ч при температуре дефлегмации осадок
отфильтровывали, промывали водой и сушили под вакуумом. Выход: 137 мг (67%) гидрохлорида 9-амино-8-хлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]
хиназолин-3,5-диона в виде белых кристаллов; МС: me/e = 251 (M-HCl)+.
Пример 30
9-диметиламино-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c]хиназолин- 3,
5-дион
а) Смесь, содержащую 500 мг (1,6 ммоля) этил-6-фтор-7-нитро-2- гидрокси-4-хиназолинкарбазата и 50 мл 33%-ного этанольного раствора диметиламина, перемешивали при комнатной температуре
в течение 24 ч. Затем растворитель сдували и остаток растворяли в 20 мл метиленхлорида. Осадок отфильтровывали и сушили. Выход: 360 мг (67%) этил-(6-диметиламино-7-нитро-2-оксо-1,
2-дигидрохиназолин-4-ил)карбазата в виде порошка оранжевого цвета; МС: me/e = 337 (M+H)+.
б) 350 мг (1,04 ммоля) этил-(6-диметиламино-7-нитро-2-оксо- 1, 2-дигидрохиназолин-4-ил)карбазата в 30 мл безводного диметилформамида выдерживали в течение 3 ч при температуре дефлегмации. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли на роторном испарителе. Остаток растирали с небольшим количеством ацетона, отфильтровывали осадок красного цвета и сушили под вакуумом. Выход: 195 мг (65%) 9-диметиламино-8-нитро- 2,3,5, 6-тетрагидро-1, 2,4-триазол[4,3-c] хиназолин-3,5-диона в виде красного порошка; МС: me/e = 291 (M+H)+.
Пример 31
Натриевая соль 9-диметиламино-8-нитро-1-ил-2,3,5,
6- тетрагидро[1,2,
4] триазол[1,5-c]хиназолин-2,5-диона
К раствору, содержащему 150 мг (0,52 ммоля) 9-диметиламино-8- нитро-1-ил-2,3,5,6-тетрагидро[1,2,4] триазол[4,3-c] хиназолин-3,5-диона и
20 мл
диметилформамида, добавляли 8,05 мл 0,1М NaOH и смесь перемешивали при комнатной температуре в течение 30 минут. Затем смесь концентрировали досуха, обрабатывали трижды деионизированной водой и
вновь
концентрировали. Остаток сушили в глубоком вакууме. Выход: 123 мг (76%) натриевой соли 9-диметиламино-8-нитро-1-ил- 2,3,5,6-тетрагидро[1,2,4]триазол[1,5-c]хиназолин-2,5-диона в виде
красно-коричневого
порошка; МС: me/e = 289 (M-Na)-.
Пример 32
8-нитро-9-пирролидин-1-ил-2,3,5,6-тетрагидро-1,2,4-триазол[4,3- c]хиназолин-3,5-дион
а)
Смесь, содержащую 500
мг (1,6 ммоля) этил-6-фтор-7-нитро-2- гидрокси-4-хиназолинкарбазата и 50 мл пирролидина, перемешивали при комнатной температуре в течение 4 ч. Затем растворитель сдували и
остаток хроматографировали
на силикагеле, используя смесь (95:5) метиленхлорида/метанола в качестве элюента. Выход: 512 мг (88%) этил-(7-нитро-2-оксо-6-пирролидин-1-ил-1,
2-дигидрохиназолин-4-ил)карбазата в виде красного порошка;
МС: me/e = 363 (M+H)+.
б) 500 мг (1,38 ммоля) этил-(7-нитро-2-оксо-6-пирролидин-1-ил- 1, 2-дигидрохиназолин-4-ил)карбазата в 30 мл безводного диметилформамида выдерживали в течение 3 ч при температуре дефлегмации. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли на роторном испарителе. Остаток растирали с небольшим количеством ацетона, отфильтровывали осадок красного цвета и сушили под вакуумом. Выход: 342 мг (78%) 8-нитро-9-пирролидин-1-ил- 2,3,5, 6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде красного порошка; МС: me/e = 317 (M+H)+.
Пример 33
Натриевая соль 8-нитро-9-пирролидин-1-ил-2,3,5,
6-тетрагидро[1,2,4] триазол[1,5-c]хиназолин-2,5-диона
К раствору, содержащему
200 мг (0,63 ммоля) 8-нитро-9-пирролидин- 1-ил-2,3,5,6-тетрагидро[1,2,4] триазол[4,3-c]хиназолин-3,5-диона и 20
мл диметилформамида, добавляли 9,86 мл 0,1М NaOH и смесь перемешивали при комнатной
температуре в течение 30 минут. Затем смесь концентрировали досуха, обрабатывали трижды деионизированной водой и
вновь концентрировали. Остаток сушили в глубоком вакууме. Выход: 193 мг (90%) натриевой
соли 8-нитро-9-пирролидин-1-ил-2,3,5,6- тетрагидро[1,2,4] триазол[1,5-c]хиназолин-2,5-диона в виде
красно-коричневого порошка; МС: me/e = 315 (M-Na)-.
Пример 34
9-морфолин-4-ил-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-дион
а) Смесь,
содержащую 500 мг (1,6 ммоля) этил-6-фтор-7-нитро-2- гидрокси-4-хиназолинкарбазата и 10 мл
морфолина, перемешивали при комнатной температуре в течение 4 ч. Затем растворитель сдували и остаток
растворяли в 20 мл метиленхлорида. Осадок отфильтровывали и сушили. Выход: 360 мг (59%)
этил-(6-морфолин-4-ил-7-нитро-2-оксо- 1,2-дигидрохиназолин-4-ил)карбазата в виде порошка оранжевого цвета; МС:
me/e = 378 (M+).
б) 145 мг (0,38 ммоля) этил-(6-морфолин-4-ил-7-нитро-2-оксо- 1,2-дигидрохиназолин-4-ил)карбазата в 20 мл безводного диметилформамида выдерживали в течение 2 ч при температуре дефлегмации. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли на роторном испарителе. Остаток растирали с небольшим количеством ацетона, отфильтровывали осадок красного цвета и сушили под вакуумом. Выход: 76 мг (60%) 9-морфолин-4-ил-8-нитро- 2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде красно-коричневого порошка; МС: me/e = 333 (M+H)+.
Пример 35
9-(4-метилпиперазин-1-ил)-8-нитро-2,3,5,6-тетрагидро- 1,2,4-триазол[4,3-c]хиназолин-3,5-дион
а) Смесь, содержащую 150 мг (0,48 ммоля)
этил-6-фтор-7-нитро-2- гидрокси-4-хиназолинкарбазата и 5
мл N-метилпиперазина, перемешивали при комнатной температуре в течение 3 ч. Затем растворитель сдували и остаток переносили в 20 мл
метиленхлорида. Осадок отфильтровывали и сушили. Выход: 90 мг (48%)
этил-[6-(4-метилпиперазин-1-ил)-7-нитро-2- оксо-1,2-дигидрохиназолин-4-ил)карбазата в виде порошка оранжевого цвета; МС: me/e = 392
(M+H)+.
б) 73 мг (0,19 ммоля) этил-[6-(4-метилпиперазин-1-ил)-7-нитро-2- оксо-1,2-дигидрохиназолин-4-ил)карбазата в 3 мл безводного диметилсульфоксида нагревали до 15oC в течение 2 ч. Затем растворитель сдували и остаток растирали с небольшим количеством ацетона. Осадок отфильтровывали и сушили под вакуумом. Выход: 49 мг (73%) 9-(4-метилпиперазин-1-ил)-8-нитро-2,3,5,6-тетрагидро-1,2,4- триазол[4, 3-c]хиназолин-3,5-диона в виде порошка оранжево-коричневого цвета; МС: me/e = 346 (M+H)+.
Пример
36
а) Смесь (1:1) соли 9-имидазол-1-ил-8-нитро-2,3,5,
6- тетрагидро[1,2,4] триазол[1,5-c]хиназолин-2,5-диона и имидазола
Смесь, содержащую 500 мг (1,6 ммоля)
этил-6-фтор-7-нитро-2- гидрокси-4-хиназолинкарбазата и 7,5 г (110 ммолей) имидазола,
нагревали до 100oC в течение 3 ч. Затем добавляли 80 мл метанола, отфильтровывали осадок желтого цвета
и сушили. Выход: 547 мг (89%) смеси соли 9-имидазол-1-ил-8-нитро-2,3,5,6-тетрагидро[1,
2,4]триазол[1,5- c]хиназолин-2,5-диона и имидазола; МС: me/e = 314 (M+H)+.
б)
Натриевая соль 9-имидазол-1-ил-8-нитро-2,3,5,6- тетрагидро[1,2,4]триазол[1,5-c]хиназолин-2,
5-диона (1:1)
5,3 мл 0,1М NaOH добавляли к раствору, содержащему смесь (1:1) соли
9-имидазол-1-ил-8-нитро-2,3,5,6-тетрагидро[1,2,4] триазол[1,5- c] хиназолин-2,5-диона с имидазолом и 40 мл
диметилформамида, и смесь перемешивали при комнатной температуре в течение 30 минут. Затем
смесь концентрировали досуха, обрабатывали трижды деионизированной водой и концентрировали. Остаток сушили в
глубоком вакууме. Выход: 134 мг (76%) натриевой соли 9-имидазол-1-ил-8-нитро-2,3,5,
6-тетрагидро[1,2,4] триазол[1,5-c] хиназолин- 2,5-диона в виде оранжевого порошка; МС: me/e = 314 (M-Na+H)+
.
в) 9-имидазол-1-ил-8-нитро-2,3,5,6-тетрагидро[1,2,4] триазол[1,
5- c]хиназолин-2,5-дион
К раствору, содержащему 300 мг (0,84 ммоля) 9-имидазол-1-ил-8- нитро-2,3,5,
6-тетрагидро[1,2,4] триазол[1,5-c] хиназолин-2,5-диона и 20 мл воды, добавляли по каплям 2М
HCl до тех пор, пока значение pH раствора не достигало 1. Осадок отфильтровывали под вакуумом и сушили.
Выход: 242 мг (91%) 9-имидазол-1-ил-8-нитро-2,3,5,6- тетрагидро[1,2,4]триазол[1,5-c]хиназолин-2,
5-диона в виде оранжевого порошка; МС: me/e = 314 (M+H)+.
г)
9-имидазол-1-ил-8-нитро-2,3,5,6-тетрагидро[1,2,4]триазол[4,3- c]хиназолин-3,5-дион
Раствор,
содержащий 100 мг (0,32 ммоля) 9-имидазол-1-ил-8-нитро- 2,3,5,6-тетрагидро[1,2,4] триазол[1,5-c]
хиназолин-2,5-диона и 10 мл ДМФ, нагревали до 80oC в течение 30 минут и затем
концентрировали досуха. После растирания с ацетоном остаток отфильтровывали под вакуумом. Выход: 84 мг (84%)
9-имидазол-1-ил-8-нитро-2,3,5,6- тетрагидро[1,2,4] триазол[4,3-c]хиназолин-3,5-диона; МС:
me/e = 314 (M+H)+.
Пример 37
8,9-динитро-2,3,5,6-тетрагидро-1,2,
4-триазол[4,3-c]хиназолин-3,5-дион
а) 10 г (48,3 ммоля) 7-нитро-1,2,3,
4-тетрагидрохиназолин-2,4-диона добавляли порциями при 0oC к 32 мл дымящей азотной кислоты. Затем смесь
дополнительно перемешивали при комнатной температуре в течение 5 ч. Реакционную
смесь сливали на лед, осадок отфильтровывали под вакуумом и промывали 65%-ной азотной кислотой, разбавленной азотной
кислотой, а затем водой. Выход: 7,15 г (59%) 6,7-динитро-1,2,3,
4- тетрагидрохиназолин-2,4-диона в виде желтого порошка; МС: me/e = 252 (M+).
б) 2 г (7,9 ммоля) 6, 7-динитро-1,2,3,4-тетрагидрохиназолин-2,4-диона суспендировали в 12 мл оксихлорида фосфора и нагревали до 140oC в течение 96 ч. После охлаждения до комнатной температуры реакционную смесь концентрировали досуха. Остаток хроматографировали на силикагеле, используя метиленхлорид в качестве элюента. Выход: 1,11 г (48%) 2,4-дихлор-6,7-динитрохиназолина в виде желтых кристаллов; МС: me/e = 288 (M+), 290(M+), 292 (M+).
в) К раствору, содержащему 1,1 г (3,8 ммоля) 2,4-дихлор-6,7- динитрохиназолина в 25 мл безводного диметилсульфоксида, добавляли 0,475 г (4,57 ммоля) этилкарбазата. Через 15 минут смесь сливали на ледяную воду. Отфильтровывали осадок оранжевого цвета и сушили. Выход: 1,12 г (83%) этил-2-хлор-6, 7-динитро-4-хиназолинкарбазата в виде кристаллов оранжевого цвета; МС: me/e = 356 (M+).
г) Раствор, содержащий 1,1 г (3,1 ммоля) этил-2-хлор-6, 7-динитро- 4-хиназолинкарбазата и 25 мл безводного диметилсульфоксида, нагревали до 100oC в течение 1 часа. Затем смесь сливали на лед, отфильтровывали осадок желтого цвета и сушили. Выход: 0,82 г (79%) этил-6,7-динитро-2-гидрокси-4-хиназолинкарбазата в виде кристаллов желтого цвета; МС: me/e = 338 (M+).
д) 810 мг (2,39 ммоля) этил-6, 7-динитро-2-гидрокси-4-хиназолинкарбазата в 30 мл безводного диметилформамида выдерживали при температуре дефлегмации в течение 3 ч. Реакционной смеси давали охладиться до комнатной температуры и растворитель удаляли на роторном испарителе. Остаток растирали с небольшим количеством ацетона, отфильтровывали осадок оранжевого цвета и сушили под вакуумом. Выход: 560 мг (80%) 8,9-динитро-2,3,5, 6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде оранжевых кристаллов; МС: me/e = 292 (M+).
Пример
38
9-бром-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,
3-c]хиназолин-3,5-дион
а) 25,8 г (93,8 ммоля) метил-2-амино-5-бром-4-нитробензоата растворяли в 800 мл метанола и насыщали
аммиаком при -40oC. Затем смесь перемешивали при
комнатной температуре в течение ночи. Раствор концентрировали и остаток растирали с метиленхлоридом. Осадок отфильтровывали под вакуумом и
сушили. Выход: 18,3 г (75%) 2-амино-5-бром-4-нитробензамида в
виде оранжевых кристаллов; МС: me/e = 259 (M+), 261 (M+).
б) 17,3 г (66,5 ммоля) 2-амино-5-бром-4-нитробензамида растворяли в 500 мл тетрагидрофурана и обрабатывали при 0oC 13 г (10,4 ммоля) трифосгена. Смеси давали нагреться до комнатной температуры и дополнительно перемешивали в течение 1,5 ч. Затем раствор концентрировали и растирали с водой. Осадок отфильтровывали под вакуумом и сушили. Выход: 8,6 г (45%) 2,4-диоксо-6-бром-7-нитро- 1,2,3, 4-тетрагидрохиназолина в виде желтоватых кристаллов; МС: me/e = 285 (M+), 287 (M+).
в) 8,6 г (30,1 ммоля) 2,4-диоксо-6-бром-7-нитро-1,2,3, 4- тетрагидрохиназолина суспендировали в 42 мл оксихлорида фосфора и нагревали до 140oC в течение 96 ч. После охлаждения до комнатной температуры реакционную смесь концентрировали досуха. Остаток хроматографировали на флорисиле, используя метиленхлорид в качестве элюента. Выход: 2,8 г (29%) 2, 4-дихлор-6-бром-7-нитрохиназолина в виде светло-желтых кристаллов; МС: me/e = 321 (M+), 323 (M+), 325 (M+), 327 (M+).
г) 1,0 г (9,6 ммоля) этилкарбазата добавляли к раствору, содержащему 2,6 г (8,1 ммоля) 2, 4-дихлор-6-бром-7-нитрохиназолина в 45 мл безводного диметилсульфоксида. Через 15 минут смесь сливали на ледяную воду. Отфильтровывали осадок желтого цвета и сушили. Выход: 2,88 г (91%) этил-2-хлор-6-бром-7-нитро-4-хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 389 (M+), 391 (M+), 393 (M+).
д) Раствор, содержащий 2,88 г (7, 37 ммоля) этил-6-бром-7-нитро-2-гидрокси-4-хиназолинкарбазата и 55 мл безводного диметилсульфоксида, нагревали до 100oC в течение 1 ч. Затем смесь сливали на лед, отфильтровывали желтый осадок и сушили. Выход: 2,47 г (90%) этил-6-бром-7-нитро-2-гидрокси-4-хиназолинкарбазата в виде желтых кристаллов; МС: me/e = 372 (M+H))+, 374 (M+H)+.
е) 2,45 г (6,6 ммоля) этил-6-бром-7-нитро-2-гидрокси-4- хиназолинкарбазата в 150 мл безводного диметилформамида выдерживали при температуре дефлегмации в течение 2 ч. Реакционной смеси давали охладиться до комнатной температуры и растворитель удаляли на роторном испарителе. Остаток растирали с небольшим количеством ацетона, отфильтровывали осадок оранжевого цвета и сушили под вакуумом. Выход: 1,3 г (61%) 9-бром-8-нитро- 2,3,5,6-тетрагидро-1,2,4-триазол[4,3-c] хиназолин-3,5-диона в виде порошка оранжевого цвета; МС: me/e = 325 (M+), 327 (M+).
Получение
соединений формулы II
Пример 39
5-гидрокси-3,4-дигидропирид[3,4-c]хинолин-4-он
а) Смесь содержащую 4,
34 г (0,022 моля) 2-метокси-4-метилхинолин- 3-карбонитрила и 7,8 г (0,
066 моля) диметилацеталя N,N-диметилформамида, выдерживали в атмосфере аргона при слабом кипячении с обратным холодильником в
течение 65 ч. Смесь охлаждали и хроматографировали на силикагеле,
используя дихлорметан в качестве элюента. Продукт перекристаллизовывали из метанола. С помощью повторной хроматографии маточного
раствора и последующей перекристаллизации получали дополнительную
порцию продукта. Выход: 4,22 г (76%) (E)-4-(2-диметиламиновинил)-2-метоксихинолин-3-карбонитрила в виде желтых кристаллов; tпл 101-103oC.
б) 3,33 г (0,013 моля) (E)-4-(2-диметиламиновинил)-2- метоксихинолин-3-карбонитрила выдерживали при температуре дефлегмации в течение 2 ч в смеси растворителей, состоящей из 4,6 мл серной кислоты, 29 мл уксусной кислоты и 9,6 мл воды. Реакционную смесь охлаждали до комнатной температуры и сливали на 115 мл ледяной воды. Осадок отфильтровывали, промывали водой, сушили под вакуумом и перекристаллизовывали из диметилформамида/метанола. Выход: 1,83 г (65%) 5-гидрокси-3,4-дигидропирид[3,4-c] хинолин-4-она в виде светло-желтых кристаллов; tпл 312-327oC (субл.).
Пример
40
8-хлор-5-гидрокси-3,4-дигидропирид[3,4-c]хинолин-4-он
а) Смесь, содержащую 6,5 г (0,038 моля)
1-(2-амино-4-хлорфенил)этанона и 5,2 г (0,046 моля) этилцианацетата, нагревали до
200oC в течение 4 часов, отгоняя образующуюся смесь воды/этанола. Смесь дополнительно перемешивали при
210oC в течение 2 ч, давали охладиться до комнатной температуры,
образовавшуюся коричневую массу растирали в ступке, суспендировали в этаноле в течение ночи, отфильтровывали, промывали
этанолом и сушили под вакуумом. Выход: 7,28 г (87%)
7-хлор-2-гидрокси-4-метилхинолин-3-карбонитрила в виде светло-коричневых кристаллов; tпл > 350oC; МС: me/e (% от
основного пика) = 218 (C11H7ClN2O2+, 100), 190(53), 189 (35), 163(7,5), 155(13), 140(4), 128(11), 101(6), 77(20).
б) 7, 58 г (0,035 моля) 7-хлор-2-гидрокси-4-метилхинолин-3-карбонитрила суспендировали в 32 мл (0,347 моля) оксихлорида фосфора и перемешивали при 105oC в течение 4 ч. Избыток оксихлорида фосфора отгоняли. Остаток распределяли между 1 л этилацетата и 0,5 л воды и отфильтровывали через дикалит. Затем экстрагировали этилацетатом, сушили над сульфатом магния, фильтровали и концентрировали. Продукт перекристаллизовывали из горячего метанола. Выход: 7,4 г (90%) 2,7-дихлор-4-метилхинолин-3-карбонитрила в виде белых кристаллов; tпл 153-154oC.
в) 7,4 г (0,035 моля) 2,7-дихлор-4-метилхинолин-3-карбонитрила суспендировали в 600 мл метанола. К суспензии добавляли порциями 6,3 г (0,275 моля) натрия при 40oC и смесь кипятили с обратным холодильником в течение 16 ч. Смесь охлаждали до 40oC, к ней медленно добавляли 300 мл воды и перемешивали в ледяной бане в течение 1/2 ч. Полученную суспензию отфильтровывали, промывали водой и сушили под вакуумом. Выход: 6,0 г (83%) 7-хлор-2-метокси-4-метилхинолин- 3-карбонитрила в виде белых кристаллов; tпл 183-185oC.
г) Смесь, содержащую 6,0 г (0,026 моля) 7-хлор-2-метокси-4- метилхинолин-3-карбонитрила и 30,7 г (0,258 моля) диметилацеталя N,N-диметилформамида, выдерживали при слабом кипячении с обратным холодильником в течение 23 ч (по истечении 19 ч к смеси добавляли дополнительную порцию 15,4 г (0,129 моля) диметилацеталя N, N-диметилформамида). Смесь охлаждали и хроматографировали на силикагеле, используя смесь (4:1) дихлорметана/н-гексана в качестве элюента. Продукт перекристаллизовывали из этилацетата. С помощью однократной перекристаллизации маточного раствора получали дополнительную порцию продукта. Выход: 4,0 г (54%) (E)-7-хлор-4-(2-диметиламиновинил)-2-метоксихинолин-3-карбонитрила в виде желтых кристаллов; tпл 174-175oC.
д) 2,5 г (0,0087 моля) (E)-7-хлор-4-(2-диметиламиновинил)-2- метоксихинолин-3-карбонитрила кипятили с обратным холодильником в течение 2 ч в смеси растворителей, состоящей из 3 мл серной кислоты, 19 мл уксусной кислоты и 6, 3 мл воды. Реакционную смесь охлаждали до 0oC и затем сливали на 80 мл ледяной воды. Осадок отфильтровывали, промывали водой, сушили под вакуумом и перекристаллизовывали из диметилформамида/этилацетата. Выход: 1,26 г (59%) 8-хлор-5-гидрокси-3,4-дигидропирид[3,4-c]хинолин-4-она в виде кристаллов бежевого цвета; tпл 340-342oC.
Пример
41
8,9-дихлор-5-гидрокси-3,4-дигидропирид[3,4-c]хинолин-4-он
а) Смесь, содержащую 29,6 г (0,145 моля) 1-(2-амино-4,5- дихлорфенил)этанона и 19,7 г (0,174 моля) этилцианацетата,
нагревали до 210oC в течение 6 часов, отгоняя образующуюся смесь воды/этанола. Смеси давали охладиться до комнатной температуры, образовавшуюся коричневую массу растирали в ступке,
суспендировали в этаноле в течение ночи, отфильтровывали, промывали этанолом и сушили под вакуумом. Выход: 30,2 г (82%) 6,7-дихлор-2-гидрокси-4-метилхинолин- 3-карбонитрила в виде светло-коричневых
кристаллов. Аналитический образец может быть перекристаллизован из этанольной фазы; tпл 310-315oC (разл.).
б) 15 г (0,059 моля) 6, 7-дихлор-2-гидрокси-4-метилхинолин-3-карбонитрила суспендировали в 54 мл (0,59 моля) оксихлорида фосфора и перемешивали при 105oC в течение 3 ч. Избыток оксихлорида фосфора отгоняли. Остаток распределяли между 1 л этилацетата и 0,5 л воды и фильтровали через дикалит. Затем экстрагировали этилацетатом, сушили над сульфатом магния, фильтровали и концентрировали. Продукт перекристаллизовывали из горячего метанола. Выход: 10,7 г (66%) 2,6,7-трихлор-4-метилхинолин-3-карбонитрила в виде белых кристаллов; tпл 195-197oC.
в) 10,7 г (0, 039 моля) 2,6,7-трихлор-4-метилхинолин-3-карбонитрила суспендировали в 1 л метанола. К суспензии добавляли порциями 8,0 г (0,35 моля) натрия при 40oC и смесь выдерживали при температуре кипения с обратным холодильником в течение 16 ч. Смесь охлаждали до 40oC и к ней медленно добавляли 0,5 л воды. Полученную суспензию отфильтровывали, промывали водой и сушили под вакуумом. Продукт с примесями перекристаллизовывали из этилацетата. Выход: 7,42 г (71%) 6,7-дихлор-2-метокси-4-метилхинолин-3-карбонитрила в виде белых кристаллов; tпл 179-181oC.
г) Смесь, содержащую 7,42 г (0,028 моля) 6,7-дихлор-2-метокси-4- метилхинолин-3-карбонитрила и 33,1 г (0,278 моля) диметилацеталя N,N-диметилформамида, выдерживали при слабом кипячении с обратным холодильником в течение 17 ч. Смесь полностью концентрировали и остаток перекристаллизовывали из горячего этилацетата. С помощью перекристаллизации маточного раствора получали дополнительную порцию продукта. Выход: 6,63 г (74%) (E)-6,7-дихлор-4-(2-диметиламиновинил)- 2-метоксихинолин-3-карбонитрила в виде светлых зеленоватых кристаллов; tпл 230-232oC.
д) 2,2 г (0,0069 моля) (E)-6,7-дихлор-4-(2-диметиламиновинил)-2- метоксихинолин-3-карбонитрила кипятили с обратным холодильником в течение 2 ч в смеси растворителей, состоящей из 2,4 мл серной кислоты, 15 мл уксусной кислоты и 5,0 мл воды. Реакционную смесь охлаждали до комнатной температуры и затем сливали на 150 мл ледяной воды. Осадок отфильтровывали, промывали водой, сушили под вакуумом и перекристаллизовывали из диметилформамида. Выход: 1,36 г (70%) 8,9-дихлор-5-гидрокси-3,4-дигидропирид[3,4-c]хинолин-4-она в виде кристаллов светло-желтого цвета; tпл > 350oC; МС: me/e (% от основного пика) = 280 (C12H6Cl2N2O2+, 90), 252(100), 223 (14), 189(31), 162(18), 127(15), 111(16), 97(22), 81(29), 69(43), 55 (44), 41 (52).
Пример 42
5-гидрокси-3,4-дигидропиридазин[4,5-c]хинолин-4-он
а) 6,1 г (0,021 моля)
этил-4-диметоксиметил-2-гидроксихинолин-3- карбоксилата (E. Ziegler, T. Kappe, H.G. Foraita, Monatsh. Chem. 1966, 97, 409) суспендировали в 60 мл 6Н водной соляной кислоты и кипятили с обратным
холодильником в течение 3 минут. Смесь охлаждали до 0oC, обрабатывали 150 мл воды, фильтровали под вакуумом и промывали водой, метанолом и этилацетатом. Выход: 3,5 г (77%) (RS)-1,
4-дигидрокси-1,3-дигидрофур[3,4-c]хинолин-3-она в виде светло-зеленоватых кристаллов; tпл 295-302oC (разл.).
6) 0,54 г (0,0025 моля) (RS)-1,4-дигидрокси-1, 3-дигидрофур[3,4- c]хинолин-3-она растворяли в 5 мл диметилсульфоксида при барботировании аргоном. К раствору добавляли 0,125 г (0,0025 моля) гидрата гидразина и смесь перемешивали при комнатной температуре в течение 4 дней. Полученную суспензию фильтровали под вакуумом и промывали этилацетатом. Выход: 0,28 г (53%) 5-гидрокси-3,4- дигидропиридазин[4,5-c] хинолин-4-она в виде желтых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 213 (C11H7N3O2+, 100), 185(46), 157(18), 129(32), 102(17), 75(10).
Пример 43
8-хлор-5-гидрокси-3,4-дигидропиридазин[4,5-c]хинолин-4-он
а) 16,0 г (0,083 моля) смеси (≈1:2) 7-хлорхинолин-2,4-диола с
5-хлорхинолин-2,
4-диолом (T. Kappe, A. S. Karem, W. Stadlbauer, J. Heterocycl. Chem. 1988, 28, 857) суспендировали в 80 мл диоксана. К суспензии по каплям добавляли 9,4 г (0,17 моля) сульфурилхлорида
при 50oC в течение 10 минут и смесь кипятили с обратным холодильником в течение 20 минут. Смесь охлаждали, сливали на 300 мл ледяной воды, экстрагировали этилацетатом и хроматографировали
на
силикагеле, используя смесь (19:1) толуола/этилацетата в качестве элюента. Оба продукта перекристаллизовывали из простого эфира/н-гексана. Выход: 8,8 г (40%) 3,3,7-трихлор-1,2,3,
4-тетрагидрохинолин-2,
4-диона в виде желто-оранжевых кристаллов; tпл 235-236oC и 10,7 г (49%) 3,3,5-трихлор-1,2,3,4-тетрагидрохинолин-2,4-диона в виде желтых кристаллов; tпл 194-196oC.
б) 3,5 г (0,15 моля) натрия растворяли в 75 мл метанола при барботировании аргоном. В смесь по каплям при 65oC в течение 20 минут добавляли теплый раствор, содержащий 13,4 г (0,051 моля) 3,3,7-трихлор-1,2,3,4-тетрагидрохинолин-2,4-диона в 140 мл метанола. Смесь кипятили с обратным холодильником в течение 15 минут, обрабатывали 150 мл 2Н водного раствора гидроксида натрия и перегоняли до тех пор, пока температура внутри реакционной смеси не достигала 100oC. Смесь охлаждали, экстрагировали этилацетатом, перекристаллизовывали из простого эфира/н-гексана и маточный раствор хроматографировали на силикагеле, используя смесь (1:2) н-гексана/этилацетата в качестве элюента. Выход: 8,9 г (77%) 1-(2-амино-4-хлорфенил)-2,2-диметоксиэтанона в виде желтоватых кристаллов; tпл 81-82oC.
в) 8,9 г (0,039 моля) 1-(2-амино-4-хлорфенил)-2,2-диметоксиэтанона суспендировали в 31 г (0,19 моля) диэтилмалоната и перемешивали при 170oC в течение 18 ч. Смесь охлаждали, разбавляли 50 мл смеси (1:1) этилацетата/н-гексана, фильтровали, концентрировали и хроматографировали на силикагеле, используя смесь (1:1) н-гексана/этилацетата в качестве элюента. Продукт перекристаллизовывали из этилацетата/н-гексана. Выход: 6,63 г (53%) этил-7-хлор-4-диметоксиметил-2-гидроксихинолин-3-карбоксилата в виде белых кристаллов; tпл 134-136oC.
г) 6,63 г (0,020 моля) этил-7-хлор-4-диметоксиметил-2- гидроксихинолин-3-карбоксилата суспендировали в 70 мл 25%-ной водной соляной кислоты и кипятили с обратным холодильником в течение 30 минут. Смесь охлаждали до 0oC, обрабатывали 200 мл воды, фильтровали под вакуумом, промывали водой и этилацетатом и сушили под вакуумом. Продукт перекристаллизовывали из диметилформамида/этилацетата. Выход: 3,85 г (75%) (RS)-7-хлор-l,4-дигидрокси-1,3-дигидрофуро[3,4- c] хинолин-3-она в виде светло-бежевых кристаллов; tпл 270-310oC (разл. ); МС: me/e (% от основного пика) = 251 (C11 H6ClNO4+, 31), 223(45), 205(100), 179(80), 177(64), 151(43), 149(43), 114(44).
д) 2,0 г (0,0079 моля) (RS)-7-хлор-1,4-дигидрокси-1, 3- дигидрофуро[3,4-c] хинолин-3-она растворяли в 20 мл диметилсульфоксида при барботировании аргоном. К раствору добавляли 0,40 г (0,0062 моля) гидрата гидразина и смесь перемешивали при комнатной температуре в течение 90 часов. Полученную суспензию фильтровали под вакуумом и промывали этилацетатом. Выход: 1,21 г (61%) 8-хлор-5-гидрокси-3,4- дигидропиридазин[4,5-c] хинолин-4-она в виде желтых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 247 (C11H6N3O2+, 100), 219(57), 191(15), 163(23), 128(22), 99(13).
Пример 44
8,9-дихлор-5-гидрокси-3,4-дигидропиридазин[4,
5-c]хинолин-4-он
а) 169,8 г (0,738 моля) смеси (≈1:9) 6,7-дихлорхинолин-2,4-диола с 5,
6-дихлорхинолин-2,4-диолом (T. Kappe, A. S. Karem, W. Stadlbauer, J. Heterocycl.
Chem. 1988, 28, 857) суспендировали в 700 мл диоксана. К суспензии по каплям добавляли 274 г (2,03 моля) сульфурилхлорида при 50oC в течение 3/4 ч и смесь перемешивали при 90oC в течение 15 минут. Смесь охлаждали, сливали на 4 л ледяной воды и экстрагировали этилацетатом. Выход: 204 г (93%) смеси с примесями (≈1:9) 3,3,6,7-тетрахлор-1,2,3,4-тетрагидрохинолин-2,4-диона с 3,3,5,6-тетрахлор-1,2,3,4-тетрагидрохинолин-2,4-дионом в виде желтых кристаллов.
б) 47,1 г (2,05 моля) натрия растворяли в 1 л метанола при барботировании аргоном. В смесь по каплям при 65oC в течение 20 минут добавляли теплый раствор, содержащий 204 г (0,68 моля) смеси с примесями (≈1:9) 3,3,6,7-тетрахлор-1,2,3,4-тетрагидрохинолин- 2,4-диона с 3,3,5, 6-тетрахлор-1,2,3,4-тетрагидрохинолин-2,4-дионом в 1,2 л метанола. Смесь кипятили с обратным холодильником в течение 10 минут, обрабатывали 2 л 2 н. водного раствора гидроксида натрия и перегоняли до тех пор, пока температура внутри реакционной смеси не достигала 100oC. Смесь охлаждали, экстрагировали этилацетатом и хроматографировали на силикагеле, используя смесь (4:1) н-гексана/этилацетата в качестве элюента. Оба продукта перекристаллизовывали из простого эфира/н-гексана. Выход: 22,7 г (13%) 1-(2-амино-4,5-дихлорфенил)-2,2-диметоксиэтанона в виде желтоватых кристаллов; tпл 91-92oC и 45,3 г (25%) 1-(2-амино-5,6-дихлорфенил)-2, 2-диметоксиэтанона в виде желтых кристаллов; tпл 81-82oC.
в) 22,7 г (0, 086 моля) 1-(2-амино-4,5-дихлорфенил)-2,2-диметоксиэтанона суспендировали в 69 г (0,43 моля) диэтилмалоната и перемешивали при 170oC в течение 15 ч. Смесь охлаждали, разбавляли 50 мл этилацетата и несколько раз хроматографировали на силикагеле, используя смесь (3:1) н-гексана/этилацетата в качестве элюента. Продукт растворяли в горячем этилацетате и перекристаллизовывали при добавлении н-гексана. Таким путем получали 7,6 г (33%) эдукта в виде масла оранжевого цвета. Выход: 3,1 г (15%) этил-6,7-дихлор-4- диметоксиметил-2-гидроксихинолин-3-карбоксилата в виде беловатых кристаллов; tпл 198-200oC.
г) 4,7 г (0,013 моля) этил-6,7-дихлор-4-диметоксиметил-2- гидроксихинолин-3-карбоксилата суспендировали в 50 мл 25%-ной водной соляной кислоты и перемешивали при 100oC в течение 30 минут. Смесь охлаждали до 0oC, обрабатывали 150 мл воды, фильтровали под вакуумом и сушили под вакуумом. Продукт перекристаллизовывали из диметилформамида/этилацетата. Выход: 2,90 г (78%) (RS)-7,8-дихлор-1, 4-дигидрокси-1,3-дигидрофуро[3,4- c] хинолин-3-она в виде бежево-коричневатых кристаллов; tпл ≈275oC (разл.).
д) 1,76 г (0,0062 моля) (RS)-7,8-дихлор-1, 4-дигидрокси-1,3- дигидрофуро[3,4-c] хинолин-3-она растворяли в 18 мл диметилсульфоксида при барботировании аргоном. К раствору добавляли 0,31 г (0,0062 моля) гидрата гидразина и смесь перемешивали при комнатной температуре в течение 42 ч. Полученную суспензию фильтровали под вакуумом и промывали этилацетатом. Выход: 0,32 г (18%) 8,9-дихлор-5-гидрокси- 3,4-дигидропиридазин[4,5-c]хинолин-4-она в виде желтых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 281 (C11H5Cl2N3O2+, 100), 253(62), 225(16), 197(37), 162(29), 124(13), 99(19).
Пример 45
9,
10-дихлор-5-гидрокси-3,4-дигидропиридазин[4,5-c]хинолин-4-он
а) 13,1 г (0,050 моля) 1-(2-амино-5,
6-дихлорфенил)-2,2-диметоксиэтанона растворяли в 250 мл дихлорметана и обрабатывали 5,5 г (0,
055 моля) триэтиламина. В течение 15 минут при 0oC по каплям добавляли 8,2 г (0,055 моля)
этилмалонилхлорида. Смесь дополнительно перемешивали при 0oC в течение 15 минут,
обрабатывали 150 мл воды, экстрагировали дихлорметаном и хроматографировали на силикагеле, используя
сначала смесь (4:1) дихлорметана/н-гексана, а затем метанол в качестве элюента. Метанольные фракции
концентрировали,
растворяли в горячем этилацетате, фильтровали и концентрировали. Остаток
перекристаллизовывали из простого эфира/н-гексана. Выход: 10,2 г (54%) рацемической смеси этил(цис- и/или
транс-)-5,6-дихлор- 4-диметиоксиметил-2,4-дигидрокси-3,4-дигидрохинолин-3-карбоксилата в виде
белых кристаллов; tпл 265-270oC (разл.).
б) 5,0 г (0,013 моля) рацемической смеси этил(цис- и/или транс-)-5,6-дихлор-4-диметиоксиметил-2,4-дигидрокси-3, 4-дигидрохинолин- 3-карбоксилата суспендировали в смеси, состоящей из 84 мл уксусной кислоты, 28 мл воды и 14 мл серной кислоты, и перемешивали в течение 16 ч при 100oC. Смесь охлаждали, обрабатывали 400 мл воды, фильтровали под вакуумом и сушили под вакуумом. Продукт растворяли в горячем диметилформамиде, фильтровали, неполностью концентрировали и перекристаллизовывали при добавлении этилацетата. Выход: 2,95 г (78%) (RS)-8,9-дихлор-1,4- дигидрокси-1,3-дигидрофур[3,4-c]хинолин-3-она в виде желтоватых кристаллов; tпл 285-290oC (разл.).
в) 2,9 г (0,010 моля) (RS)-8,9-дихлор-1,4-дигидрокси-1,3- дигидрофур[3,4-c] хинолин-3-она растворяли в 60 мл диметилсульфоксида при барботировании аргоном. К раствору добавляли 0,51 г (0, 010 моля) гидрата гидразина и смесь перемешивали при комнатной температуре в течение 50 ч. Получали раствор оранжевого цвета, который обрабатывали 300 мл этилацетата. Осажденный продукт отфильтровывали под вакуумом, промывали этилацетатом и перекристаллизовывали из диметилформамида/этилацетата. Выход: 0,70 г (25%) 9, 10-дихлор-5-гидрокси- 3,4-дигидропиридазин[4,5-c] хинолин-4-она в виде желто-оранжевых кристаллов; tпл ≈270oC; МС: me/e (% от основного пика) = 281 (C11H5Cl2N3O2+, 100), 253(60), 239(19), 213(40), 185(15), 162(23). 148(16), 123(16), 45(58).
Пример 46
8,
10-дихлор-5-гидрокси-3,4-дигидропиридазин[4,5-c]хинолин-4-он
а) 9,1 г (0,
040 моля) 5,7-дихлорхинолин-2,4-диола (полученного аналогично соединениям, описанным у T. Kappe, A. S. Karem, W.
Stadlbauer, J. Heterocycl. Chem. 1988, 28, 857) суспендировали в 60 мл диоксана. К
суспензии по каплям добавляли 14,7 г (0,11 моля) сульфурилхлорида при 50oC в течение 10 минут и смесь
кипятили с обратным холодильником в течение 10 минут. Смесь охлаждали, сливали на 150
мл ледяной воды и экстрагировали этилацетатом. Аналитический образец перекристаллизовывали из метанола. Выход: 12,
3 г (≈100%) 3,3,5,7-тетрахлор-1,2,3,4-тетрагидрохинолин-2,4-диона в виде
желтоватых кристаллов; tпл 248-254oC (разл.).
б) 2,83 г (0,123 моля) натрия растворяли в 60 мл метанола при барботировании аргоном. В этот раствор добавляли по каплям раствор, содержащий 12,3 г (0,040 моля) 3,3,5,7-тетрахлор-1,2,3,4- тетрагидрохинолин-2,4-диона в 300 мл метанола, при температуре кипения с обратным холодильником в течение 10 минут. Смесь кипятили с обратным холодильником в течение 10 минут, обрабатывали 120 мл 2 н. водного раствора гидроксида натрия и перегоняли до тех пор, пока температура внутри реакционной смеси не достигала 100oC. Смесь охлаждали, экстрагировали этилацетатом и хроматографировали на силикагеле, используя смесь (19:1) толуола/этилацетата в качестве элюента. Выход: 4,76 г (43%) 1-(2-амино-4, 6-дихлорфенил)-2,2-диметоксиэтанона в виде красно-оранжевого вязкого масла; МС: me/e (% от основного пика) = 232(1,0), 200(6,4), 188(6,0), 160(2,0), 124(6,5), 75(100).
в) 4,76 г (0, 018 моля) 1-(2-амино-4,6-дихлорфенил)-2,2-диметоксиэтанона растворяли в 50 мл дихлорметана и обрабатывали 2,2 г (0,022 моля) триэтиламина. При 0oC в течение 10 минут по каплям добавляли 3, 3 г (0,022 моля) этилмалонилхлорида. Смесь выдерживали при комнатной температуре в течение 16 ч, обрабатывали 50 мл воды и экстрагировали дихлорметаном. Продукт с примесями растворяли в 50 мл этанола и в течение 15 минут при температуре дефлегмации по каплям добавляли к раствору, содержащему 1,24 г (0,054 моля) натрия в 60 мл этанола. Смесь кипятили с обратным холодильником в течение 1 ч, концентрировали, обрабатывали водой, экстрагировали этилацетатом и хроматографировали на силикагеле, используя смесь (9:1) дихлорметана/н-гексана в качестве элюента. Продукт перекристаллизовывали из простого эфира/н-гексана. Выход: 3,58 г (55%) этил-5,7-дихлор-4-диметоксиметил-2-гидроксихинолин-3-карбоксилата в виде беловатых кристаллов; tпл 167-170oC.
г) 5, 0 г (0,014 моля) этил-5,7-дихлор-4-диметоксиметил-2- гидроксихинолин-3-карбоксилата суспендировали в смеси, состоящей из 84 мл уксусной кислоты, 28 мл воды и 14 мл серной кислоты, и перемешивали при 100oC в течение 16 ч. Смесь охлаждали, обрабатывали 50 мл воды, фильтровали под вакуумом и сушили под вакуумом. Выход: 3,88 г (98%) (RS)-7,9-дихлор-1,4-дигидрокси-1,3-дигидрофуро[3, 4-c]хинолин-3-она в виде белых кристаллов; tпл 280-287oC.
д) 2,05 г (0,0072 моля) (RS)-7,9-дихлор-1,4-дигидрокси-1,3- дигидрофуро[3,4-c] хинолин-3-она растворяли в 20 мл диметилсульфоксида при барботировании аргоном. К раствору добавляли 0,36 г (0,0072 моля) гидрата гидразина и смесь перемешивали при комнатной температуре в течение 89 часов. Полученную суспензию обрабатывали этилацетатом, фильтровали под вакуумом и промывали этилацетатом. Выход: 0,62 г (31%) 8, 10-дихлор-5-гидрокси- 3,4-дигидропиридазин[4,5-c]хинолин-4-она в виде кристаллов бежевого цвета; tпл > 350oC; МС: me/e (% от основного пика) = 281 (C11H5Cl2N3O2+, 100), 253(92), 213(14), 197(17), 162(29), 57(42).
Пример 47
4-гидрокси-2,3-дигидро-1H-пиридазол[4,
3-c]хинолин-3-он
а) 5,35 г (0,22 моля) стружки магния обрабатывали
смесью, состоящей из 5 мл метанола и 0,5 мл четыреххлористого углерода, при барботировании аргоном. После начала реакции к
смеси осторожно по каплям добавляли 75 мл диэтилового эфира. Затем по каплям
добавляли смесь, состоящую из 35,2 г (0,22 моля) диэтилмалоната, 20 мл этанола и 20 мл простого эфира, таким образом, чтобы
поддерживать слабое кипение реакционной смеси. После перемешивания при
температуре кипения с обратным холодильником в течение дополнительных 2,5 ч добавляли по каплям раствор, содержащий 37,1 г (0,20
моля) 2-нитробензоилхлорида в 130 мл диэтилового эфира. Смесь кипятили
с обратным холодильником в течение 1 ч, охлаждали до 0oC, к ней медленно по каплям добавляли 250 мл 2 н. водной серной
кислоты, смесь перемешивали в течение 20 минут, экстрагировали
диэтиловым эфиром, концентрировали и сушили при 120oC в условиях глубокого вакуума. Выход: 53,7 г (86%)
диэтил-2-(2-нитробензоил)малоната в виде жидкости оранжевого цвета. МС: me/e (% от
основного пика) = 309 (C14H15NO7+, 0,3), 264(2,5), 263(2,3),
217(3,7), 205(5,3), 189(8), 159(13), 150(100), 135(21), 104(19), 76(19), 51(22),
44(19), 29(46).
б) Свежеприготовленный раствор диазометана (≈0,038 моля в 150 мл диэтилового эфира) добавляли по каплям при 5oC в течение 2 минут к раствору, содержащему 9,90 г (0,032 моля) диэтил-2-(2-нитробензоил)малоната в 150 мл диэтилового эфира. Раствор желтого цвета выдерживали при комнатной температуре в течение 2 ч, избыток диазометана разлагали с помощью 2 г бензойной кислоты, смесь концентрировали и хроматографировали на силикагеле, используя смесь (9: 1) толуола/этилацетата в качестве элюента. Продукт перегоняли в трубке с шаровым расширением (tкип 210-220oC/при 0,005 торр) и перекристаллизовывали из диэтилового эфира/н-гексана при -40oC. Выход: 9,0 г (87%) диэтил(2-[метокси-(2-нитрофенил)метилен] малоната в виде белых кристаллов; tпл 73-74oC.
в) 3,23 г (0,010 моля) диэтил(2-[метокси-(2-нитрофенил)метилен]малоната растворяли в 70 мл горячего этанола. К охлажденному раствору по каплям добавляли 1,1 г (0,022 моля) гидрата гидразина. Смесь кипятили с обратным холодильником в течение 2 ч, после чего к ней добавляли еще 1,1 г (0,022 моля) гидрата гидразина, смесь кипятили с обратным холодильником в течение 2 ч и концентрировали полностью. Полученное масло переносили в 100 мл этилацетата, кипятили с обратным холодильником и охлаждали. Полученную суспензию фильтровали под вакуумом. Осадок на фильтре переносили в 100 мл воды, доводили до pH 2 с помощью ≈10 мл 1 н. водной соляной кислоты и осажденный продукт отфильтровывали под вакуумом. Продукт перекристаллизовывали из этанола/воды. Выход: 1,93 г (70%) этил-5-(2-нитрофенил)-3-оксо- 2, 3-дигидро-1H-пиразол-4-карбоксилата в виде белых кристаллов; tпл 183-185oC.
г) Раствор, содержащий 0,55 г (0,002 моля) этил-5-(2-нитрофенил)- 3-оксо-2, 3-дигидро-1H-пиразол-4-карбоксилата в 40 мл этанола, обрабатывали 50 мг 10%-ного Pd/C и гидрировали в течение 1,5 ч при комнатной температуре при атмосферном давлении. Катализатор отфильтровывали, фильтрат концентрировали и остаток перекристаллизовывали из этилацетата/гексана. Выход: 0,34 г (69%) этил-5-(2-аминофенил)-3-оксо-2,3-дигидро-1H-пиразол-4-карбоксилата в виде белых кристаллов; tпл > 350oC. МС: me/e (% от основного пика) = 247 (C12H13N3O3+, 18), 201(100), 145(36), 144(33), 117(56), 89(30).
д) 0,25 г (0,001 моля) этил-5-(2-аминофенил)-3-оксо-2,3-дигидро- 1H-пиразол-4-карбоксилата растворяли в 2,5 мл диметилформамида и кипятили с обратным холодильником в течение 2 ч. Смесь охлаждали, обрабатывали 20 мл этилацетата, фильтровали под вакуумом и сушили под вакуумом. Выход: 0,14 г (70%) 4-гидрокси-2,3-дигидро-1H- пиридазол[4,3-c] хинолин-3-она в виде кристаллов бежевого цвета; tпл > 350oC. MC: me/e (% от основного пика) = 201 (C10H7N3O2+, 100), 145(27), 144(32), 127(7), 117(33), 116(24), 89(21), 69(17), 57(21), 43(22).
Пример 48
7-хлор-4-гидрокси-2,3-дигидро-1H-пиридазол[4,3-c]хинолин-3-он
а) 3,52 г (0,145 моля) магниевой
стружки при барботировании аргоном
обрабатывали смесью, состоящей из 3,5 мл этанола и 0,35 мл четыреххлористого углерода. После начала реакции к смеси осторожно по каплям добавляли 50 мл диэтилового
эфира. Затем по каплям добавляли
смесь, состоящую из 23,3 г (0,145 моля) диэтилмалоната, 15 мл этанола и 15 мл диэтилового эфира, таким образом, чтобы поддерживать слабое кипение реакционной смеси.
После перемешивания при
температуре кипения с обратным холодильником в течение 3 ч добавляли по каплям раствор, содержащий 25,8 г (0,12 моля) 4-хлор-2-нитробензоилхлорида в 60 мл простого эфира. Смесь
кипятили с обратным
холодильником в течение 1 ч, охлаждали до 0oC, медленно по каплям добавляли 150 мл 2 н. водной соляной кислоты, смесь перемешивали в течение 10 минут, экстрагировали
диэтиловым эфиром и
концентрировали. Маслянистый остаток перекристаллизовывали из диэтилового эфира/н-гексана при -78oC. Выход: 36,6 г (91%) диэтил-2-(4-хлор-2-нитробензоил)малоната в виде
белых кристаллов;
tпл 43-45oC.
б) Свежеприготовленный раствор диазометана (≈0,034 моля в 150 мл диэтилового эфира) добавляли по каплям при 5oC в течение 2 минут к раствору 10,3 г (0,030 моля) диэтил-2-(4-хлор-2-нитробензоил)малоната в 150 мл диэтилового эфира. Раствор желтого цвета перемешивали при 5oC в течение 1 ч, кипятили с обратным холодильником в течение 10 минут и полностью концентрировали. Выход: 10,8 г (100%) диэтил-2-[(4-хлор-2-нитрофенил)метоксиметилен]малоната в виде желтоватого вязкого масла; MC: me/e (% от основного пика) = 312(17), 297(5), 239(5), 237(13), 225(2), 186(10), 180(11), 154(13), 138(11), 125(19), 112(9), 99(5), 87(6), 75(9), 59(10), 29(100).
в) 10,7 г (0,030 моля) диэтил-2-[(4-хлор-2-нитрофенил) метоксиметилен] малоната растворяли в 200 мл этанола и обрабатывали 6,0 г (0,12 моля) гидрата гидразина. Смесь кипятили с обратным холодильником в течение 1 ч и полностью концентрировали. Полученное масло растворяли в 400 мл воды, pH доводили до 1 с помощью ≈30 мл 25%-ной водной соляной кислоты, обрабатывали 1,5 л этилацетата, отфильтровывали, экстрагировали этилацетатом и концентрировали. Продукт перекристаллизовывали из горячего этилацетата. Выход: 5,2 г (56%) этил-5-(4-хлор-2-нитрофенил)-3-оксо-2,3-дигидро-1H-пиразол-4-карбоксилата в виде белых кристаллов; tпл 230-235oC (разл.).
г) Раствор, содержащий 5,2 г (0,017 моля) этил-5-(4-хлор-2- нитрофенил)-3-оксо-2, 3-дигидро-1H-пиразол-4-карбоксилата в 300 мл тетрагидрофурана, обрабатывали 520 мг 10%-ного Pd/C и гидрировали в течение 21 ч при комнатной температуре и при атмосферном давлении. Катализатор отфильтровывали, фильтрат концентрировали и остаток перекристаллизовывали из этилацетата/н-гексана. Выход: 3,1 г (66%) этил-5-(2-амино-4-хлорфенил)-3-оксо-2,3-дигидро-1H-пиразол-4-карбоксилата в виде белых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 281 (C12H12ClN3O3+, 11), 235(100), 206(2), 178(24), 151(35), 123(12), 114(9), 89(12), 45(14), 31(27).
д) 3,1 г (0,011 моля) этил-5-(2-амино-4-хлорфенил)-3-оксо-2,3- дигидро-1H-пиразол-4-карбоксилата растворяли в 30 мл диметилформамида и кипятили с обратным холодильником в течение 2 ч. Смесь концентрировали, остаток суспендировали в 150 мл метанола при температуре дефлегмации, охлаждали, фильтровали под вакуумом и сушили под вакуумом. Выход: 1,1 г (42%) 7-хлор-4-гидрокси-2,3-дигидро-1H- пиридазол[4,3-c]хинолин-3-она в виде коричневатых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 235 (C10H6ClN3O2+, 100), 201(2), 179(26), 178(34), 151(42), 123(17), 114(14), 89(15).
Пример 49
7,8-дихлор-4-гидрокси-2,3-дигидро-1H-пиридазол[4,3-c]хинолин-3-он
а) 1,95 г (0,080 моля) магниевой стружки обрабатывали смесью, состоящей из 1,95 мл этанола и 0,195 мл четыреххлористого
углерода. После начала реакции к смеси осторожно по каплям добавляли 30 мл диэтилового эфира. Затем по каплям добавляли смесь, состоящую из 1,28 г (0,080 моля) диэтилмалоната, 8 мл этанола и 9 мл
диэтилового эфира, таким образом, чтобы поддерживать слабое кипение реакционной смеси. После перемешивания при температуре кипения с обратным холодильником в течение 2 ч по каплям добавляли раствор,
содержащий 19,5 г (0,072 моля) 4,5-дихлор-2-нитробензоилхлорида в 30 мл диэтилового эфира. Смесь кипятили с обратным холодильником в течение 1 ч, охлаждали до 0oC, медленно по каплям
добавляли 75 мл 2 н. водной соляной кислоты, смесь перемешивали в течение 15 минут, экстрагировали диэтиловым эфиром и концентрировали. Кристаллический остаток перекристаллизовывали из горячего
этанола. Выход: 17,9 г (65%) диэтил-2-(4,5-дихлор-2-нитробензоил)малоната в виде беловатых кристаллов; tпл 93-94oC.
б) Свежеприготовленный раствор диазометана (≈0,052 моля в диэтиловом эфире) добавляли по каплям при 5oC в течение 2 минут к раствору, содержащему 17,9 г (0,047 моля) диэтил-2-(4,5-дихлор-2- нитробензоил)малоната в 350 мл диэтилового эфира. Раствор желтого цвета перемешивали при 5oC в течение 0,5 ч, кипятили с обратным холодильником в течение 10 минут и полностью концентрировали. Выход: 18,6 г (100%) диэтил-2-[(4,5-дихлор-2-нитрофенил)метоксиметилен] малоната в виде желтого вязкого масла; МС: me/e (ISP) = 392,2 (C15H15Cl2NO7+).
в) 18,6 г (0,047 моля) диэтил-2-[(4,5-дихлор-2-нитрофенил) метоксиметилен] малоната растворяли в 400 мл этанола и обрабатывали 9,5 г (0,19 моля) гидрата гидразина. Смесь кипятили с обратным холодильником в течение 1 ч и полностью концентрировали. Остаток суспендировали в 200 мл этилацетата, отфильтровывали под вакуумом и растворяли в 2 л воды. После этого значение pH доводили до 1 с помощью 2 н. водной соляной кислоты и осажденный продукт отфильтровывали под вакуумом, растворяли в горячем этилацетате и перекристаллизовывали при добавлении н-гексана при 0oC. Выход: 11, 8 г (72%) этил-5-(4,5-дихлор-2-нитрофенил)-3-оксо-2,3-дигидро-1H- пиразол-4-карбоксилата в виде светло-бежевых кристаллов; tпл 156-158oC.
г) Раствор, содержащий 5, 0 г (0,014 моля) этил-5-(4,5-дихлор-2- нитрофенил)-3-оксо-2,3-дигидро-1H-пиразол-4-карбоксилата в 200 мл этилацетата, обрабатывали 0,5 г 10%-ного Pd/C и гидрировали в течение 6 ч при комнатной температуре при атмосферном давлении. Катализатор отфильтровывали, фильтрат концентрировали и остаток перекристаллизовывали из этилацетата/н-гексана. Выход: 3,55 г (78%) этил-5-(2-амино-4, 5-дихлорфенил)-3-оксо-2,3-дигидро-1H-пиразол-4- карбоксилата в виде белых кристаллов; tпл 170-175oC (разл.).
д) 0,63 г (0,002 моля) этил-5-(2-амино-4, 5-дихлорфенил)-3-оксо- 2,3-дигидро-1H-пиразол-4-карбоксилата растворяли в 10 мл диметилформамида и кипятили с обратным холодильником в течение 0,5 ч. Смесь охлаждали, суспензию обрабатывали 50 мл этанола, фильтровали под вакуумом и сушили под вакуумом. Выход: 0,18 г (33%) 7,8-дихлор-4-гидрокси-2,3-дигидро-1H-пиридазол[4,3-c] хинолин-3-она в виде коричневатых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 269 (C10H5Cl2N3O2+, 100), 213(31), 185(36), 157(12), 150(15), 125(11), 73(48), 44(68).
Пример 50
4-гидрокси-7,8-диметил-2,3-дигидро-1H-пиридазол[4,3-c]хинолин-3-он
а) 2,96 г (0,122 моля) магниевой стружки при барботировании аргоном
обрабатывали смесью, состоящей из 3 мл этанола и 0,3 мл четыреххлористого углерода. После начала реакции к смеси осторожно по каплям добавляли 90 мл диэтилового эфира. Затем по каплям добавляли смесь,
состоящую из 19,5 г (0,122 моля) диэтилмалоната, 11 мл этанола и 14 мл диэтилового эфира, таким образом, чтобы поддерживать слабое кипение реакционной смеси. После перемешивания при кипячении с
обратным холодильником в течение 2,5 ч по каплям добавляли раствор, содержащий 24,5 г (0,106 моля) 4,5-диметил-2-нитробензоилхлорида, полученного путем обработки 4,5-диметил-2-нитробензойной кислоты
(A. Courtin, H. R. von Tobel, Helv. Chim. Acta 1980, 63, 385) тионилхлоридом, в 300 мл смеси (2:1) диэтилового эфира/тетрагидрофурана. Смесь кипятили с обратным холодильником в течение 1 ч, охлаждали
до 0oC, медленно по каплям добавляли 160 мл 2 н. водной соляной кислоты, смесь перемешивали в течение 10 минут, экстрагировали диэтиловым эфиром и концентрировали. Маслянистый остаток
перекристаллизовывали из диэтилового эфира/н-гексана при -20oC. Выход: 32,7 г (88%) диэтил-2-(4,5-диметил-2-нитробензоил)малоната в виде беловато-бежевых кристаллов; tпл
78-80oC.
б) Свежеприготовленный раствор диазометана (≈0,070 моля в диэтиловом эфире) добавляли по каплям при 5oC в течение 2 минут к раствору, содержащему 20,3 г (0,060 моля) диэтил-2-(4,5-диметил-2- нитробензоил)малоната в 250 мл диэтилового эфира. Раствор желтого цвета перемешивали при 5oC в течение 0,5 ч, выдерживали при комнатной температуре в течение 16 часов и полностью концентрировали. Выход: 21,5 г (100%) диэтил-2-[(4,5-диметил-2-нитрофенил)метоксиметилен] малоната в виде масла оранжевого цвета; МС: me/e (% от основного пика) = 306 (C17H21NO7, 13), 231(38), 189(7), 188(7), 174(14), 158(8), 148(27), 119(47), 106(25), 91(21), 77(20), 29(100).
в) 21,5 г (0,06 моля) диэтил-2-[(4,5-диметил-2-нитрофенил) метоксиметилен] малоната растворяли в 200 мл этанола и обрабатывали 12,3 г (0,245 моля) гидрата гидразина. Смесь кипятили с обратным холодильником в течение 2 ч, охлаждали до 0oC и суспензию фильтровали под вакуумом. Кристаллы растворяли в 1 л воды. После этого значение pH смеси доводили до 1 с помощью 1 н. водной соляной кислоты и осажденный продукт отфильтровывали под вакуумом. Выход: 15,8 г (86%) этил-5-(4,5-диметил-2-нитрофенил)-3-оксо-2,3-дигидро- 1H-пиразол-4-карбоксилата в виде светло-бежевых кристаллов; tпл 166-167oC.
г) Раствор, содержащий 15,8 г (0,051 моля) этил-5-(4,5-диметил-2- нитрофенил)-3-оксо-2,3-дигидро-1H-пиразол-4-карбоксилата в 450 мл этанола, обрабатывали 1,6 г 10%-ного Pd/C и гидрировали в течение 17 ч при комнатной температуре и при атмосферном давлении. Смесь кипятили с обратным холодильником, катализатор отфильтровывали из горячей смеси, фильтрат концентрировали и остаток перекристаллизовывали из этилацетата/н-гексана. Выход: 9,40 г (66%) этил-5-(2-амино-4,5-диметилфенил)-3-оксо-2,3-дигидро-1H-пиразол-4- карбоксилата в виде беловатых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 275 (C14H17N3O3+, 12), 229(100), 214(14), 172(24), 158(16), 144(12), 130(9), 115(11), 69(13), 45(17), 31(32).
д) 4,2 г (0,015 моля) этил-5-(2-амино-4,5-диметилфенил)-3-оксо- 2,3-дигидро-1H-пиразол-4-карбоксилата растворяли в 50 мл диметилформамида и кипятили с обратным холодильником в течение 0,5 ч. Смесь охлаждали, суспензию обрабатывали 50 мл этанола, фильтровали под вакуумом и сушили под вакуумом. Выход: 3,0 г (86%) 4-гидрокси-7,8-диметил-2, 3- дигидро-1H-пиридазол[4,3-c] хинолин-3-она в виде белых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 229 (C12H11N3O2+, 100), 214(14), 172(27), 158(20), 144(14), 130(9), 115(12).
Пример 51
4-гидрокси-7-метокси-2,3-дигидро-1H-пиразол[4,3-c]хинолин-3-он
а)
3,
07 г (0,127 моля) магниевой стружки при барботировании аргоном обрабатывали смесью, состоящей из 3 мл этанола и 0,3 мл четыреххлористого углерода. После начала реакции к смеси осторожно по каплям
добавляли 100 мл диэтилового эфира. Затем по каплям добавляли смесь, состоящую из 20,3 г (0,127 моля) диэтилмалоната, 11,5 мл этанола и 14,5 мл диэтилового эфира, таким образом, чтобы поддерживать
слабое кипение реакционной смеси. После перемешивания при температуре дефлегмации в течение 2,5 ч по каплям добавляли раствор, содержащий 26,1 г (0,12 моля) 4-метокси-2-нитробензоилхлорида в 100 мл
тетрагидрофурана. Смесь кипятили с обратным холодильником в течение 1/4 ч, охлаждали до 0oC, медленно по каплям добавляли 160 мл 2 н. водной соляной кислоты, смесь перемешивали в течение
15
минут, экстрагировали диэтиловым эфиром, концентрировали и хроматографировали на силикагеле, используя дихлорметан в качестве элюента. Выход: 33 г (85%) диэтил-2-(4-метокси-2-нитробензоил)малоната
в
виде темно-оранжевого масла; МС: me/e (% от основного пика) = 293(2), 247(8), 219(12), 180(65), 134(23), 106(43), 63(54), 29(100).
б) Свежеприготовленный раствор диазометана (≈ 0,10 моля в диэтиловом эфире) добавляли по каплям при 5oC в течение 2 минут к раствору, содержащему 33 г (0,097 моля) диэтил-2-(4-метокси-2- нитробензоил)малоната в 200 мл диэтилового эфира. Раствор перемешивали при комнатной температуре в течение 2 часов и полностью концентрировали. Выход: 34 г (≈100%) диэтил-2-[(4-метокси-2- нитрофенил)метоксиметилен]малоната в виде масла оранжевого цвета; МС: me/e (% от основного пика) = 353 (C16H19NO8+, 2), 308(21), 233(29), 189(14), 176(16), 150(24), 121(29), 106(22), 77(12), 63(14), 29(100).
в) 34 г (0,097 моля) диэтил-2-[(4-метокси-2-нитрофенил) метоксиметилен] малоната растворяли в 400 мл этанола и обрабатывали 19,4 г (0,387 моля) гидрата гидразина. Смесь кипятили с обратным холодильником в течение 1,5 ч, охлаждали до 0oC и концентрировали до объема ≈200 мл. Полученные кристаллы затем охлаждали, отфильтровывали под вакуумом и растворяли в 3/4 л воды. После этого значение pH смеси доводили до 2 с помощью 1 н. водной соляной кислоты и осажденный продукт отфильтровывали под вакуумом. Выход: 22,9 г (77%) этил-5-(4-метокси-2-нитрофенил)-3-оксо-2,3-дигидро-1H-пиразол- 4-карбоксилата в виде светло-желтоватых кристаллов; tпл 205-210oC.
г) 22,8 г (0,074 моля) этил-5-(4-метокси-2-нитрофенил)-3-оксо- 2,3-дигидро-1H-пиразол-4-карбоксилата растворяли в 2 л горячего этанола, обрабатывали 2,2 г 10%-ного Pd/C и гидрировали в течение 6 ч при 40oC при атмосферном давлении. Катализатор отфильтровывали и фильтрат полностью концентрировали. Выход: 19 г (91%) этил-5-(2-амино-4-метоксифенил)-3-оксо-2,3-дигидро-1H-пиразол-4-карбоксилата в виде светло-желтоватых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 277 (C13H15N3O4+, 13), 231(100), 188(18), 174(17), 147(10), 132(14).
д) 5,8 г (0,021 моля) этил-5-(2-амино-4-метоксифенил)-3-оксо- 2,3-дигидро-1H-пиразол-4-карбоксилата растворяли в 60 мл диметилформамида и кипятили с обратным холодильником в течение 0,5 ч. Смесь охлаждали, суспензию обрабатывали 120 мл этанола, фильтровали под вакуумом и сушили под вакуумом. Выход: 4,5 г (93%) 4-гидрокси-7-метокси-2, 3-дигидро-1H-пиразол[4,3-c]хинолин-3-она в виде светло-желтоватых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 231 (C11H9N3 O3+, 100), 188(27), 174(20), 146(14), 132(18), 104(8), 77(11), 69(14), 43(16).
Пример 52
4-гидрокси-2,3-дигидро-1H-пиррол[3,4-c]хинолин-3-он
а) Раствор, содержащий 1,03 г (0,0058 моля) 1-(2-аминофенил)-2-азидоэтанона (J. H. Boyer, D. Straw, J. Am. Chem. Soc. 1953, 75, 1642, 2683) в 25 мл дихлорметана, обрабатывали 0,53 г (0,0053
моля) триэтиламина при барботировании аргоном. При 0oC по каплям добавляли 0,76 г (0,005 моля) этилмалонилхлорида и смесь перемешивали в течение 5 минут при 0oC, в течение 1/2 ч
при комнатной температуре и в течение 2,5 ч при температуре дефлегмации. Смесь охлаждали до комнатной температуры, сливали на разбавленную водную соляную кислоту, экстрагировали дихлорметаном и
полностью концентрировали. Остаток растворяли в 30 мл горячего толуола, добавляли 3 капли пиперидина и смесь кипятили с обратным холодильником в течение 2 ч. Смесь охлаждали до 0oC и
полученную суспензию фильтровали под вакуумом. Продукт перекристаллизовывали из горячего этилацетата. Выход: 0,88 г (56%) этил-4-азидометил-2-гидроксихинолин-3-карбоксилата в виде белых кристаллов;
tпл 195-197oC (разл.).
б) 0,27 г (0,0010 моля) этил-4-азидометил-2-гидроксихинолин-3- карбоксилата растворяли в 20 мл горячего диметилформамида и обрабатывали 14 мг 10%-ного Pd/C. В течение 1,5 ч медленно пропускали струю водорода. Суспензию фильтровали в атмосфере аргона и фильтрат частично концентрировали в условиях глубокого вакуума. Остаток обрабатывали 25 мл дегазированного этанола, отфильтровывали, сушили под вакуумом и получали ≈60 мг (30%) 4-гидрокси-2,3-дигидро-1H-пиррол[3,4-c] хинолин-3-она в виде чувствительного к действию воздуха твердого продукта. Маточный раствор полностью концентрировали. Остаток растворяли в 75 мл метанола и медленно подводили струю кислорода в течение 2,5 ч. Смесь фильтровали, концентрировали, хроматографировали на силикагеле, используя смесь (9:1) этилацетата/метанола в качестве элюента, и продукт перекристаллизовывали из горячего этилацетата. Выход: 0,033 г (15%) 4-гидрокси-2, 3-дигидро-1H-пиррол[3, 4-c]хинолин-3-диона в виде желтых кристаллов; tпл 340-350oC (разл. , субл.); МС: me/e (% от основного пика) = 214 (C11H6N2 O3+, 100), 186(9), 171(18), 158(7), 143(33), 115(34), 88(14), 58(16).
Пример 53
7-хлор-4-гидрокси-2,3-дигидро-1H-пиррол[3,4-c]хинолин-1,
3-дион
а) 8,9 г (0,
032 моля) 2-бром-1-(4-хлор-2-нитрофенил)этанона (полученного аналогично 2-бром-1-(2-нитрофенил)этанону, как описано у P. Ruggy, H. Reichwein, Helv. Chem. Acta 1937, 20,
913), растворяли в 66 мл серной
кислоты при 50oC. К раствору добавляли порциями 7,78 г (0,12 грамм-атома) медного порошка таким образом, чтобы поддерживать температуру на уровне
приблизительно 50oC. Смесь
перемешивали при 50oC в течение 15 минут, сливали на лед, добавляли воду до получения объема 510 мл, экстрагировали диэтиловым эфиром и
концентрировали. Остаток перекристаллизовывали из
изопропилового эфира. Выход: 6,26 г (79%) 1-(2-амино-4-хлорфенил)-2-бромэтанона в виде кристаллов бежевого цвета; tпл 112-114o
C.
б) Раствор, содержащий 0,54 г (0, 0084 моля) азида натрия в 50 мл метанола, добавляли при 0oC к раствору 1,98 г (0,008 моля) 1-(2-амино-4-хлорфенил)-2-бромэтанона в 40 мл метанола. Смесь перемешивали при 0oC в течение 1/2 ч, при комнатной температуре в течение 1/2 ч и при температуре кипения с обратным холодильником в течение 1 ч, охлаждали, сливали на воду и экстрагировали диэтиловым эфиром. Продукт перекристаллизовывали из диэтилового эфира/н-гексана. Выход: 1,47 г (87%) 1-(2-амино-4-хлорфенил)-2-азидоэтанона в виде кристаллов бежевого цвета; tпл 107-108oC.
в) Раствор, содержащий 3,92 г (0,0018 моля) 1-(2-амино-4-хлорфенил)- 2-азидоэтанона в 150 мл дихлорметана, обрабатывали 3,76 г (0,0037 моля) триэтиламина при барботировании аргоном. При 0oC по каплям добавляли 4,2 г (0,028 моля) этилмалонилхлорида и смесь перемешивали при 0oC в течение 5 минут, при комнатной температуре в течение 1/2 ч и при температуре кипения с обратным холодильником в течение 24 ч. Смесь полностью концентрировали и хроматографировали на силикагеле, используя смесь (2: 1) циклогексана/этилацетата в качестве элюента. Продукт перекристаллизовывали из диэтилового эфира. Выход: 2,26 г (83%) этил-4-азидометил-7-хлор-2-гидроксихинолин-3-карбоксилата в виде белых кристаллов; tпл 188-190oC.
г) 1,0 г (0,0032 моля) этил-4-азидометил-7-хлор-2-гидроксихинолин- 3-карбоксилата растворяли в смеси, состоящей из 80 мл метанола и 20 мл диметилформамида, и обрабатывали 46 мг 10%-ного Pd/C. В течение 1 ч медленно пропускали струю водорода. Суспензию фильтровали под вакуумом и осадок на фильтре суспендировали в диметилформамиде. Суспензию фильтровали, концентрировали, остаток суспендировали в метаноле и фильтровали под вакуумом. Субстанцию (0,31 г) суспендировали в диметилформамиде и медленно пропускали струю кислорода в течение 4 ч. Смесь концентрировали и продукт перекристаллизовывали из диметилформамида/метанола. Выход: 0,13 г (18%) 7-хлор-4-гидрокси-2,3- дигидро-1H-пиррол[3,4-c] хинолин-3-диона в виде желтых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 248 (C11H5ClN2O3+, 100), 205(23), 177(27), 149(18), 114(20).
Пример 54
7,
8-дихлор-4-гидрокси-2,3-дигидро-1H-пиррол[3,4-c]хинолин-1,3-дион
а) 9,17 г (0,030 моля) 2-бром-1-(4,5-дихлор-2-нитрофенил)этанона
(полученного аналогично 2-бром-1-(2-нитрофенил)этанону, как
описано у P.Ruggy, H.Reichwein, Helv. Chem. Acta 1937, 20, 913), растворяли в 100 мл серной кислоты при 50oC. К раствору
добавляли порциями 7,13 г (0,112 грамм-атома) медного порошка таким
образом, чтобы поддерживать температуру на уровне приблизительно 50oC. Смесь перемешивали при 50oC в течение
15 минут, сливали на лед, добавляли воду до получения объема 470 мл,
смесь экстрагировали диэтиловым эфиром, концентрировали и хроматографировали на силикагеле, используя толуол в качестве элюента.
Продукт перекристаллизовывали из изопропилового эфира. Выход: 6,82 г
(54%) 1-(2-амино-4,5-дихлорфенил)-2-бромэтанона в виде желтых кристаллов; tпл 122-123oC.
б) Раствор, содержащий 1,38 г (0,021 моля) азида натрия в 60 мл метанола, добавляли при 0oC к раствору, содержащему 5,75 г (0,020 моля) 1-(2-амино-4,5-дихлорфенил)-2-бромэтанона в 110 мл метанола. Смесь перемешивали при 0oC в течение 1/2 ч, при комнатной температуре в течение 1/2 ч и при температуре кипения с обратным холодильником в течение 1,5 ч, охлаждали, сливали на воду и экстрагировали диэтиловым эфиром. Продукт перекристаллизовывали из изопропилового эфира. Выход: 4,94 г (100%) 1-(2-амино-4,5-дихлорфенил)- 2-азидоэтанона в виде зеленоватых кристаллов; tпл 136-137oC.
в) Раствор, содержащий 3, 78 г (0,015 моля) 1-(2-амино-4,5- дихлорфенил)-2-азидоэтанона в 150 мл дихлорметана, обрабатывали 3,12 г (0,030 моля) триэтиламина при барботировании аргоном. При 0oC по каплям добавляли 3, 48 г (0,023 моля) этилмалонилхлорида и смесь перемешивали при 0oC в течение 5 минут, при комнатной температуре в течение 1/2 ч и при температуре кипения с обратным холодильником в течение 48 ч. Смесь охлаждали до комнатной температуры, полностью концентрировали и хроматографировали на силикагеле, используя смесь (2:1) циклогексана/этилацетата в качестве элюента. Продукт перекристаллизовывали из этилацетата. Выход: 2,37 г (55%) этил-4-азидометил-6,7-дихлор-2-гидроксихинолин-3-карбоксилата в виде кристаллов бежевого цвета; tпл 197-198oC.
г) 1,0 г (0,003 моля) этил-4-азидометил-6,7-дихлор-2-гидроксихинолин- 3-карбоксилата растворяли в смеси, состоящей из 80 мл метанола и 20 мл диметилформамида, и обрабатывали 50 мг 10%-ного Pd/C. В течение 1,25 ч медленно пропускали струю водорода. Суспензию фильтровали под вакуумом и осадок на фильтре суспендировали в диметилформамиде. Смесь фильтровали, концентрировали, остаток суспендировали в метаноле и фильтровали под вакуумом. Субстанцию (0,31 г) суспендировали в диметилформамиде и медленно пропускали струю кислорода в течение 2 ч. Смесь концентрировали и продукт перекристаллизовывали из диметилформамида/метанола. Выход: 0,27 г (20%) 7,8-дихлор-4-гидрокси- 2,3-дигидро-1H-пиррол[3,4-c] хинолин-3-диона в виде желтых кристаллов; tпл > 350oC; МС: me/e (% от основного пика) = 282 (C11H4Cl2N2O3+, 100), 239(18), 211(29), 183(15), 148(24), 92(10).
Пример 55
7-нитро-2,3,4,5-тетрагидроизоксазол[4,5-c]хинолин-3,4-дион
а) 7,7 мл (55 ммолей)
триэтиламина добавляли по каплям к суспензии, содержащей 10,0 г (4,3 ммоля)
гидрохлорида этил-2-амино-4-нитробензоата в 100 мл ацетона. Затем добавляли 6,55 мл (52 ммоля) метилмалонилхлорида.
Реакционную смесь оставляли для перемешивания при комнатной температуре в течение 48
ч, концентрировали и затем обрабатывали раствором, содержащим 4,2 г (183 ммоля) натрия в 110 мл этанола.
Реакционную смесь кипятили с обратным холодильником в течение 2,5 ч. После удаления
растворителя остаток растворяли в 200 мл воды и нагревали до 60oC в течение 1 ч. Затем смесь фильтровали
через дикалит и после этого значение pH доводили до pH 3,5 с помощью 3 н. HCl.
Выделившийся при этом осадок отфильтровывали, промывали водой и сушили в условиях глубокого вакуума. Выход: 7,2 г (60%)
этил-4-гидрокси-7-нитро-2-оксо-1,2-дигидрохинолин-3-карбоксилата в виде
кристаллов бежевого цвета; МС: me/e = 278 (M+).
б) 4 мл (30 ммолей) триметилсилилгидроксиламина при комнатной температуре добавляли распылением в суспензию, состоящую из 1, 38 г (5 ммолей) этил-4-гидрокси-7-нитро-2-оксо-1,2-дигидрохинолин-3-карбоксилата в 40 мл диоксана. После нагревания до 100oC все вещества медленно переходили в раствор. Реакционную смесь оставляли для перемешивания при 90oC в течение 20 ч, затем ее охлаждали до комнатной температуры и отфильтровывали выделившийся объемный осадок. Этот осадок промывали водой и сушили в условиях глубокого вакуума. Выход: 740 мг (65%) 4,N-дигидрокси-7-нитро-2-оксо-1,2-дигидрохинолин-3-карбоксамида в виде светло-бежевых кристаллов; МС: me/e = 254 (M+).
в) 200 мг (0,75 ммоля) 4,N-дигидрокси-7-нитро-2-оксо-1,2- дигидрохинолин-3-карбоксамида суспендировали в 3 мл тетрагидрофурана и охлаждали до 0oC. Путем распыления добавляли 0,11 мл (1,6 ммоля) тионилхлорида и затем смесь нагревали до комнатной температуры. После перемешивания в течение 16 ч удаляли тетрагидрофуран и остаток обрабатывали 3 мл диоксана. После охлаждения до 0o C путем распыления добавляли 0,32 мл (2,25 ммоля) триэтиламина. Затем смесь нагревали до комнатной температуры и оставляли для перемешивания в течение 2 ч. Образовавшийся коричневый осадок отфильтровывали под вакуумом и перекристаллизовывали из метанола. Выход: 30 мг (16%) 7-нитро-2,3,4, 5-тетрагидроизоксазол[4,5- c] хинолин-3,4-диона в виде светло-коричневых кристаллов; МС: me/e = 247 (M+).
Пример 56
7-хлор-2,3,4,5-тетрагидроизоксазол[4,
5-c]хинолин-3,4-дион
а) 7,7 мл (55 ммолей) триэтиламина добавляли по каплям к суспензии, содержащей
10,0 г (45 ммолей) гидрохлорида этил-2-амино-4-хлорбензоата в 100 мл ацетона. Затем
добавляли 6,8 мл (54 ммоля) метилмалонилхлорида. Реакционную смесь оставляли для перемешивания при комнатной
температуре в течение 48 ч, концентрировали и затем обрабатывали раствором, содержащим 4,2
г (183 ммоля) натрия в 110 мл этанола. Реакционную смесь кипятили с обратным холодильником в течение 2,5 ч.
После удаления растворителя остаток растворяли в 200 мл воды и выдерживали при 60oC в течение 1 ч. Затем смесь фильтровали через дикалит, после чего значение pH доводили до 3,5 с помощью 3
н. HCl. Выделившийся при этом осадок отфильтровывали, промывали водой и сушили в
условиях глубокого вакуума. Выход: 6,94 г (58%) 7-хлор-4-гидрокси-2-оксо-1,2-дигидрохинолин-3-карбоксилата в виде
кристаллов бежевого цвета; МС: me/e = 267 (M+).
б) 2 мл (15 ммолей) триметилсилилгидроксиламина при комнатной температуре путем распыления добавляли в суспензию, состоящую из 2,64 г (10 ммолей) этил-7-хлор-4-гидрокси-2-оксо-1, 2-дигидрохинолин-3-карбоксилата в 20 мл диметилформамида. После нагревания до 100oC все вещества медленно переходили в раствор. Реакционную смесь оставляли для перемешивания при 80oC в течение 2 ч, затем ее охлаждали до комнатной температуры и перемешивали в течение ночи. После добавления 6 мл воды отделяли и отфильтровывали объемный осадок, промывали водой и сушили в условиях глубокого вакуума. Выход: 1,48 г (58%) 7-хлор-4,N-дигидрокси-2-оксо-1,2-дигидрохинолин-3-карбоксамида в виде светло-бежевых кристаллов; МС: me/e = 254 (M+).
в) 1, 48 г (5,8 ммоля) 7-хлор-4,N-дигидрокси-2-оксо-1,2- дигидрохинолин-3-карбоксамида суспендировали в 10 мл тетрагидрофурана и охлаждали до 0oC. Путем распыления добавляли 0,85 мл (11,6 ммоля) тионилхлорида и затем смесь нагревали до комнатной температуры. После перемешивания в течение 1 ч удаляли тетрагидрофуран и остаток обрабатывали 20 мл диоксана. После охлаждения до 0oC путем распыления добавляли 2,5 мл (17,4 ммоля) триэтиламина. Затем смесь нагревали до комнатной температуры и оставляли для перемешивания в течение 2 ч. Образовавшийся коричневый осадок отфильтровывали под вакуумом, далее продукт отделяли от маточного раствора путем подкисления. После перекристаллизации из метанола получали 200 мг (15%) 7-хлор-2,3,4,5-тетрагидроизоксазол[4, 5- c]хинолин-3,4-диона в виде светло-коричневых кристаллов; МС: me/e = 236 (M+).
Пример А
Таблетки,
содержащие следующую композицию, изготавливают обычным
способом: - мг/таблетка
Действующее вещество - 100
Порошкообразная лактоза - 95
Белый кукурузный крахмал - 35
Поливинилпирролидон - 8
Na-карбоксиметилкрахмал - 10
Стеарат магния - 2
Вес таблетки - 250
Пример Б
Таблетки, содержащие следующую композицию,
изготавливают обычным способом:
- мг/таблетка
Действующее вещество - 200
Порошкообразная лактоза - 100
Белый кукурузный крахмал - 64
Поливинилпирролидон - 12
Na-карбоксиметилкрахмал
- 20
Стеарат магния - 4
Вес таблетки - 400
Пример В
Получают капсулы, содержащие следующую композицию: - мг/капсула
Действующее вещество - 50
Кристаллическая лактоза - 60
Микрокристаллическая целлюлоза - 34
Тальк - 5
Стеарат магния - 1
Вес содержимого капсулы - 150
Действующее вещество, имеющее
соответствующий размер частиц, кристаллическую лактозу и микрокристаллическую целлюлозу смешивают друг с другом до однородности, просеивают, а затем примешивают
тальк и стеарат магния. Окончательной
смесью заполняют желатиновые капсулы с твердой оболочкой, имеющие соответствующие размеры.
Реферат
Использование: в медицине в качестве защитных веществ для нервной системы. Раскрыты трициклические дикарбонильные производные формул Ia, Ib, II, где R1 и R2 каждый независимо друг от друга обозначают водород, (низший)алкил, (низший)алкокси, нитро, трифторметил, амино, галоген и R2 может дополнительно обозначать морфолино, 5- или 6-членный гетероцикл с 1 - 3 атомами азота, необязательно замещенный (низшим)алкилом, гидроксигруппой, или группу -NR5R6, где R5 и R6 могут быть идентичными или различными и обозначают водород, (низший)алкил, и Х в формуле II обозначает -CH=CH-, -CH=N-, -NH-, -CO- или -О-, а также фармацевтически приемлемые соли соединений общих формул Ia, Ib и II, за исключением 2,3,5,6-тетрагидро[1,2,4]триазол[1,5-с] хиназолин-2,5-диона, а также лекарственный препарат на их основе. 2 с. и 9 з.п. ф-лы, 3 табл.
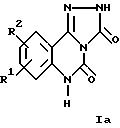
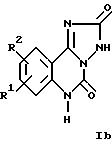
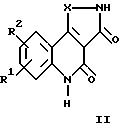
Формула
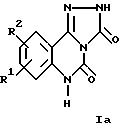
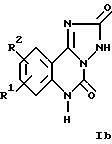
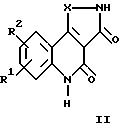
где R1 и R2 каждый независимо друг от друга обозначают водород, (низший)алкил, (низший)алкокси, нитро, трифторметил, амино, галоген;
R2 может дополнительно обозначать морфолино, 5- или 6-членный гетероцикл с 1 - 3 атомами азота, необязательно замещенный (низшим)алкилом, гидроксигруппой, или группу -NR5R6, где R5 и R6 могут быть идентичными или различными и обозначают водород, (низший)алкил;
Х в формуле II обозначает -CH=CH-, -CH=N-, -NH-, -CO- или -О-,
а также фармацевтически приемлемые соли соединений общих формул Ia, Ib и II, за исключением 2, 3,5,6-тетрагидро[1,2,4]триазол[1,5-с]хиназолин-2,5-диона.
9-хлор-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-с] хиназолин-3,5-дион;
9-бром-8-нитро-2,3,5,6-тетрагидро-1,2,4-триазол[4, 3-с] хиназолин-3,5-дион;
8,10-дихлор-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-с]хиназолин-3,5-дион;
8,9-диметил-2,3,5,6-тетрагидро-1,2, 4-триазол[4,3-с]хиназолин-3,5-дион;
8-метокси-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-с]хиназолин-3,5-дион;
8-йод-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-с]хиназолин-3,5-дион;
8-хлор-9-фтор-2,3,5,6-тетрагидро-1,2, 4-триазол[4,3-с]хиназолин-3,5-дион;
8,9-динитро-2,3,5,6-тетрагидро-1,2,4-триазол[4,3-с]хиназолин-3,5-дион;
8-нитро-2,3,5,6-тетрагидро-1,2, 4-триазол[4,3-с]хиназолин-3, 5-дион.
8-нитро-9-пирролидин-1-ил-2,3,5,6-тетрагидро-1,2,4-триазол[4, 3-с]хиназолин-3,5-дион.
9-имидазолил-1-ил-8-нитро-2,3,5,6-тетрагидро[1,2,4]триазол[1,5-с]хиназолил-2,5-дион.
7-нитро-2,3,4, 5-тетрагидроизоксазол[4,5-с]хинолин-3,4-дион;
7,8-дихлор-4-гидрокси-2,3дигидро-1Н-пиразол[4,3-с]хинолин-3-он;
7-хлор-4-гидрокси-2, 3-дигидро-1Н-пиразол[4, 3-с]хинолин-3-он.
24.05.94 - по пп.1 - 11;
17.02.95 - по пп.1 - 11 (разновидности радикалов)
Комментарии