Производные имидазо[1,2-а]пиридина, способы их получения, фармацевтический препарат на их основе, способ ингибирования секреции желудочной кислоты, способ лечения желудочно-кишечных воспалительных заболеваний и способ лечения состояний, в которые вовлеч - RU2193036C2
Код документа: RU2193036C2
Чертежи







Описание
Настоящее изобретение относится к новым соединениям и их терапевтически приемлемым солям, которые ингибируют экзогенно или эндогенно стимулируемую секрецию желудочной кислоты и, следовательно, могут применяться при предупреждении и лечении желудочно-кишечных воспалительных заболеваний. В дальнейших аспектах изобретение относится к соединениям по изобретению для применения в терапии; к способам получения таких новых соединений; к фармацевтическим композициям, содержащим по меньшей мере одно соединение по изобретению или его терапевтически приемлемую соль в качестве активного ингредиента; а также к применению активных соединений в производстве лекарств для медицинского применения, указанного выше.
Замещенные имидазо[1,2-а]пиридины, полезные при лечении пептических язвенных заболеваний, известны в данной области, например, из ЕР-В-0033094 и US 4450164 (Shering Corporation); из ЕР-В-0204285 и US 4725601 (Fujisawa Pharmaceutical Co. ); а также из публикаций J.J.Kaminski ct al. в Journal of Medical Chemistry (vol. 28, 876-892, 1985; vol. 30, 2031-2046, 1987; vol. 30, 2047-2051, 1987; vol. 32, 1686-1700, 1989; и vol. 34,533-541, 1991).
Имидазопиридиновое производное, замещенное в 8-положении 2,4,6-(СН3)3-С6 Н2СН2О, описано в ЕР-В-0033094, а также в J.J.Kaminski et al., J. Med. Chem. , vol. 28, 876-892, 1985 как "Соединение N 49". Однако, согласно последней публикации, указанное соединение не проявляло желательных свойств при тестировании в качестве ингибитора секреции желудочной кислоты.
В качестве обзора фармакологии желудочно-кислотного насоса (Н+, K+-АТФазы) см. Sachs et al. (1995) Annu. Rev. Pharmacol. Toxicol. 35: 277-305.
Неожиданно обнаружено, что соединения формулы I, которые представляют собой замещенные имидазопиридиновые производные, в которых фенильная группировка замещена низшим алкилом в 2- и 6-положении, являются особенно эффективными в качестве ингибиторов желудочно-кишечной Н+, К+-АТФазы и вследствие этого в качестве ингибиторов секреции желудочной кислоты.
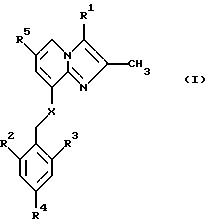
В одном аспекте изобретение таким образом относится к
соединениям общей формулы I:
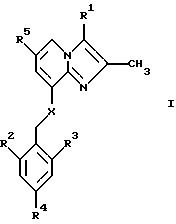
или к их фармацевтически приемлемой соли,
где
R1 представляет собой СН3 или СН2ОН;
R2 представляет собой низший алкил;
R3 представляет собой низший алкил;
R4 представляет собой Н или галоген;
R5 представляет собой Н, галоген или низший алкил;
Х представляет собой NH или О.
Термин "низший алкил", как он используется здесь, обозначает нормальную или разветвленную алкильную группу, имеющую от 1 до 6, предпочтительно от 1 до 4 атомов углерода. Примеры "низшего алкила" включают в себя метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, а также пентил и гексил с нормальной или разветвленной цепью. Предпочтительно "низший алкил" означает метил, этил, н-пропил, изопропил, н-бутил, втор-бутил и трет-бутил.
Термин "галоген" включает в себя фтор-, хлор-, бром- и иод-.
Как чистые энантиомеры, так и рацемические смеси и неравные смеси двух энантиомеров находятся в пределах объема изобретения. Следует понимать, что в пределах объема изобретения находятся все возможные диастереомерные формы (энантиомеры, рацемические смеси и неравные смеси двух энантиомеров). Производные соединений формулы I, которые обладают биологической функцией соединений формулы I, также включены в изобретение.
В зависимости от условий способа конечные продукты формулы I получают либо в нейтральной, либо в форме соли. Как свободное основание, так и соли этих конечных продуктов находятся в пределах объема изобретения.
Соли присоединения кислоты новых соединений могут быть превращены в свободное основание способом, который сам по себе является известным, с использованием основных агентов, таких как щелочи, или с помощью ионного обмена. Полученное свободное основание может также образовывать соли с органическими или неорганическими кислотами.
При получении солей присоединения кислоты предпочтительно используют такие кислоты, которые образуют подходящие терапевтически приемлемые соли. Примерами таких кислот являются галогеноводородные кислоты, такие как соляная кислота, серная кислота, фосфорная кислота, азотная кислота, алифатические, алициклические, ароматические или гетероциклические карбоновые или сульфоновые кислоты, такие как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, п-гидроксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, гидроксиэтансульфоновая кислота, галогенбензолсульфоновая кислота, толуолсульфоновая кислота или нафталинсульфоновая кислота.
Предпочтительные соединения по изобретению представляют собой такие соединения формулы I, где R2 представляет собой СН3 или СН2СН3; R3 представляет собой СН3 или СН2СН3; R4 представляет собой Н, Br, Cl или F; и R5 представляет собой Н, СН3, Вr, Cl или F, более предпочтительно Н, СН3 или F.
Особенно предпочтительными соединениями по изобретению являются:
- 8-(2,6-диметилбензиламино)-2,3,6-триметилимидазо[1,2-а]
пиридин;
- 8-(2,6-диметилбензиламино)-3-гидроксиметил-2-метилимидазо[1,2-а]пиридин;
- 2,3-диметил-8-(2,
6-диметил-4-фторбензиламино)имидазо[1,2-а]пиридин;
- 2,6-диметил-8-(2,
6-диметилбензиламино)-3-гидроксиметилимидазо[1,2-а] пиридин,
- 2,6-диметил-8-(2,
6-диметил-4-фторбензиламино)-3-гидроксиметилимидазо[1,2-а]пиридин;
- 8-(2,
6-диметил-4-фторбензиламино)-2,3,6-триметилимидазо[1,2-а]пиридин;
- 2,3-диметил-8-(2,
6-диметил-4-хлорбензиламино)имидазо[1,2-а]пиридин;
- 2,
6-диметил-8-(2-этил-6-метилбензиламино)-3-гидроксиметилимидазо[1,2-а]пиридин;
- 8-(2,6-диэтилбензиламино)-2,
6-диметил-3-гидроксиметилимидазо[1,2-а] пиридин;
- 8-(2-этил-6-метилбензиламино)-2,3,6-триметилимидазо[1,2-а] пиридин,
- 8-(2,
6-диметил-4-фторбензилокси)-3-гидроксиметил-2-метилимидазо[1,2-а] пиридин;
- 2,6-диметил-8-(2,
6-диметилбензилокси)-3-гидроксиметилимидазо[1,2-а] пиридин;
- 2,
6-диметил-8-(2-этил-4-фтор-6-метилбензиламино)-3-гидроксиметилимидазо[1,2-а]пиридин;
- 8-(2-этил-4-фтор-6-метилбензиламино)-2,3,6-триметилимидазо[1,2-а]пиридин.
Получение
В настоящем изобретении также предлагаются следующие способы А, Б, В, Г и Д производства
соединений с общей формулой 1.
Способ А
Способ А производства соединений с
общей формулой I включает в себя следующие стадии:
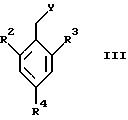
Соединения общей формулы II

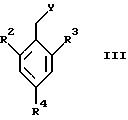
где X1 представляет собой NH2 или ОН, а R1 и R5 являются такими, как определены для формулы I, могут быть подвергнуты взаимодействию с соединениями общей формулы III

где R2, R3 и R4 являются такими, как определены для формулы I, а Y представляет собой отщепляемую группу, такую как галид, тозилокси или мезилокси, с образованием соединений формулы I.
Удобно проводить данную реакцию в инертном растворителе, например в ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле или диметилформамиде, с основанием или без основания. Основание представляет собой, например, гидроксид щелочного металла, такой как гидроксид натрия и гидроксид калия; карбонат щелочного металла, такой как карбонат калия или карбонат натрия; либо органический амин, такой как триэтиламин.
Способ
Б
Способ Б производства соединений с общей формулой I, где Х представляет собой NH, включает в себя
следующие стадии:
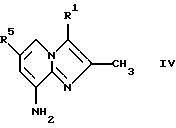
Соединения общей формулы IV

где R1 и R5 являются такими, как определены для формулы I, могут быть подвергнуты взаимодействию с соединениями общей формулы V
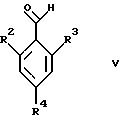
где R2, R3 и R4 являются такими, как определены для формулы I, в присутствии кислоты Льюиса, например хлорида цинка, с образованием соединений формулы VI

где R1, R2, R3, R4 и R5 являются такими, как определены для формулы I, после чего соединения общей формулы VI восстанавливают, например, с использованием борогидрида натрия или цианоборогидрида натрия, с образованием соединений общей формулы I, где Х представляет собой NH. Эти реакции можно проводить в стандартных условиях в инертном растворителе, например в метаноле или этаноле.
Способ В
Способ В производства соединений с общей формулой I, где R1
представляет собой СН2ОН, включает в себя следующие стадии:
Соединения общей формулы VII

где X1 представляет собой NН2 или ОН, а R5 является таким, как определен для формулы I, могут быть подвергнуты взаимодействию с соединениями общей формулы III
где R2, R3 и R4 являются такими, как определены для формулы I, a Y представляет собой отщепляемую группу, такую как галид, тозилокси или мезилокси, с образованием соединений формулы VIII

где R2, R3, R4, R5 и Х являются такими, как определены для формулы I.
Удобно проводить данную реакцию в инертном растворителе, например в ацетоне, ацетонитриле, диметоксиэтане, метаноле, этаноле или диметилформамиде с основанием или без основания. Основание представляет собой, например, гидроксид щелочного металла, такой как гидроксид натрия и гидроксид калия; карбонат щелочного металла, такой как карбонат калия или карбонат натрия; либо органический амин, такой как триэтиламин.
Восстановление соединений общей формулы VIII, например, с использованием алюмогидрида лития в тетрагидрофуране или эфире дает соединения общей формулы I, где R1 представляет собой СН2ОН.
Способ Г
Способ Г производства соединений с общей формулой
I, где R1 представляет собой СН2ОН, а Х представляет
собой NH, включает в себя следующие стадии:
Соединения общей формулы IX

где R5 является таким, как определен для формулы I, могут быть подвергнуты взаимодействию с соединениями общей формулы V

где R2, R3 и R4 являются такими, как определены для формулы I, в присутствии кислоты Льюиса, например хлорида цинка, с образованием соединений формулы Х

где R2, R3, R4 и R5 являются такими, как определены для формулы I, после чего соединения общей формулы Х восстанавливают, например, с использованием борогидрида натрия или цианоборогидрида натрия, с образованием соединений общей формулы XI

где R2, R3, R4 и R5 являются такими, как определены для формулы I. Эти реакции можно проводить в стандартных условиях в инертном растворителе, например в метаноле или этаноле.
Восстановление соединений общей формулы XI, например, с использованием алюмогидрида лития в тетрагидрофуране или эфире, дает соединения общей формулы I, где R1 представляет собой CH2OH, а X представляет собой NH.
Способ Д
Конденсация соединений общей формулы XII

где R2, R3, R4 и R5 являются такими, как определены для формулы I, с использованием α -галокарбонильных промежуточных соединений общей формулы СН3СОСН(Z)СООСН2СН3, где Z представляет собой Вr или Сl, в инертном растворителе, например в ацетонитриле или этаноле, приводит к образованию соединений общей формулы XIII, где R2, R3, R4 и R5 являются такими, как определены для формулы I
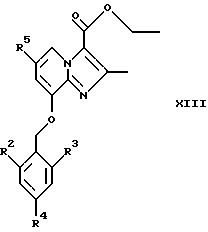
Восстановление соединений общей формулы XIII, например, с помощью использования алюмогидрида лития в тетрагидрофуране или эфире, дает соединения общей формулы I, где R1 представляет собой СН2ОН, а Х представляет собой О.
Медицинское применение
В
дальнейшем аспекте изобретение относится к соединениям формулы I для использования в
терапии, в частности для применения против желудочно-кишечных воспалительных заболеваний. В изобретении также
предлагается применение соединения формулы I в производстве лекарства для ингибирования
секреции желудочной кислоты или для лечения желудочно-кишечных воспалительных заболеваний.
Соединения по изобретению можно таким образом применять для предупреждения и лечения желудочно-кишечных воспалительных заболеваний, а также заболеваний, связанных с желудочной кислотой, таких как гастрит, язва желудка, язва двенадцатиперстной кишки, рефлюкс-эзофагит и синдром Золлингера-Эллисона, у млекопитающих, включая человека. Кроме того, эти соединения можно применять для лечения других желудочно-кишечных расстройств, при которых желудочный антисекреторный эффект является желательным, например, у пациентов с ульцерогенными аденомами поджелудочной железы и у пациентов с острым верхним желудочно-кишечным кровотечением. Их также можно применять у пациентов в ситуациях интенсивной терапии, а также в пред- или послеоперационном периоде для предупреждения аспирации кислоты и стрессового образования язвы.
Типичная суточная доза активного вещества варьирует в широком диапазоне и будет зависеть от различных факторов, таких как, например, индивидуальная потребность каждого пациента, путь введения и заболевание. Как правило, пероральные и парентеральные дозировки должны находиться в диапазоне от 5 до 1000 мг активного вещества в сутки.
Фармацевтические препараты
В еще одном дополнительном аспекте изобретение
относится к фармацевтическим композициям, содержащим в качестве активного ингредиента по меньшей мере одно соединение
по изобретению или его фармацевтически приемлемую соль.
В препараты соединений по изобретению также можно включать другие активные ингредиенты, например антибиотики, такие как амоксициллин (amoxicillin).
Для клинического применения соединения по изобретению включают в фармацевтические препараты для перорального, ректального, парентерального или другого способа введения. В этот фармацевтический препарат включают соединение по изобретению и один или более чем один фармацевтически приемлемый ингредиент. Носитель может находиться в виде твердого, полутвердого или жидкого разбавителя, либо в виде капсулы. Эти фармацевтические препараты составляют следующую задачу изобретения. Обычно количество активных соединений находится между 0,1-95% (мас./мас). препарата, предпочтительно между 0,1-20% (мас. /мас. ), в препаратах для парентерального применения и предпочтительно между 0,1 и 50% (мас./мас.) в препаратах для перорального введения.
При получении фармацевтических препаратов, содержащих соединение по настоящему изобретению, в виде стандартных лекарственных форм для перорального введения, выбранное соединение можно смешивать как с твердыми порошкообразными ингредиентами, такими как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, производные целлюлозы, желатин или другой подходящий ингредиент, так и с разрыхляющими агентами и смазывающими агентами, такими как стеарат магния, стеарат кальция, стеарилфумарат натрия и полиэтиленгликолевые воски. Затем из этой смеси получают гранулы или прессованные таблетки.
Мягкие желатиновые капсулы можно получить с использованием капсул, содержащих смесь активного соединения или соединений по изобретению, растительного масла, жира или другого подходящего наполнителя для мягких желатиновых капсул. В твердые желатиновые капсулы можно включать гранулы активного соединения. Также в твердые желатиновые капсулы, содержащие активное соединение, можно включать твердые порошкообразные ингредиенты, такие как лактоза, сахароза, сорбит, маннит, картофельный крахмал, кукурузный крахмал, амилопектин, производные целлюлозы или желатин.
Стандартные лекарственные формы для ректального введения можно готовить (i) в виде суппозиториев, которые содержат активное вещество, смешанное с нейтральной жировой основой; (ii) в виде желатиновой ректальной капсулы, которая содержит активное вещество в смеси с растительным маслом, парафиновым маслом или другим подходящим наполнителем для желатиновых ректальных капсул; (iii) в виде готовой к употреблению микроклизмы; либо (iv) в виде сухого препарата для микроклизмы, который нужно разводить подходящим растворителем непосредственно перед введением.
Жидкие препараты для перорального введения можно готовить в виде сиропов или суспензий, например растворов или суспензий, содержащих от 0,1 до 20% (мас. /мас. ) активного ингредиента, и остальную часть, состоящую из сахара или сахарных спиртов, а также смеси этанола, воды, глицерина, пропиленгликоля и полиэтиленгликоля. По желанию, в такие жидкие препараты можно включать красящие агенты, корригенты, сахарин и карбоксиметилцеллюлозу либо другой загущающий агент. Жидкие препараты для перорального введения можно также готовить в виде сухого порошка, который нужно разводить подходящим растворителем перед применением.
Растворы для парентерального введения можно готовить в виде раствора соединения по изобретению в фармацевтически приемлемом растворителе, предпочтительно в концентрации от 0,1 до 10% (мас./мас.). Эти растворы могут также содержать стабилизирующие ингредиенты и/или забуферивающие ингредиенты, и их расфасовывают в стандартных дозах в ампулы или флаконы. Растворы для парентерального введения также можно готовить в виде сухого препарата, который нужно разводить подходящим растворителем непосредственно перед применением.
В препараты
соединений по изобретению можно также включать другие активные
ингредиенты, например, для лечения или профилактики состояний, в которые вовлечена инфекция слизистой оболочки желудка человека
Helicobacter pylori. Такие дополнительные активные ингредиенты могут
представлять собой противомикробные агенты, в частности:
- β-лактамные антибиотики, такие как амоксициллин
(amoxycilline), ампициллин (ampicillin), цефалотин (cephalothin), цефаклор
(cefaclor) или цефиксим (cefixime);
- макролиды, такие как эритромицин (Eruthromycin) или кларитромицин
(clarithromycin);
- тетрациклины, такие как тетрациклин (tetracycline) или
доксициклин (doxycycline);
- аминогликозиды, такие как гентамицин (gentamycin), канамицин (kanamycin) или
амикацин (amikacin);
- хинолоны, такие как норфлоксацин (norfloxacin),
ципрофлоксацин (ciprofloxacin) или эноксацин (enoxacin);
- другие, такие как метронидазол (metronidazole),
нитрофурантоин (nitrofurantoin) или хлорамфеникол (chloramphenicol), либо
- препараты, содержащие соли висмута, такие как субцитрат висмута, субсалицилат висмута, субкарбонат висмута,
субнитрат висмута или субгаллат висмута.
ПРИМЕРЫ
Пример 1.1
Синтез 8-(2,6-диметилбензиламино)-2,3,6-триметилимидазо)азо[1,2-а]пиридина гидрохлорида

Перемешанную смесь 8-амино-2.3.6-триметилимидазо[1,2-а)пиридина (0,9 г, 5,1 ммоля), хлорида цинка (II) (0, 84 г, 6,2 ммоля) и 2.6-диметилбензальдегида (0,83 г, 6,2 ммоля) в метаноле (50 мл), обрабатывали цианоборогидридом натрия (0,39 г, 6,2 ммоля) и подвергали дефлегмации в течение 3 ч. Метанол выпаривали при пониженном давлении, и остаток растворяли в метиленхлориде и 2 М гидроксиде натрия (40 мл). Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении.
Остаток очищали дважды с помощью колоночной хроматографии на силикагеле, используя в качестве элюента а) этилацетат: метиленхлорид (1:2) и б) метанол: метиленхлорид (1:20). Маслянистый продукт растворяли в диэтилэфире, обрабатывали смесью диэтилэфир/НCl, и осажденную соль отфильтровывали с получением 0,6 г (36%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 2.33 (s, 3Н), 2.38 (s, 3Н), 2.45 (s, 6H), 2.50 (s, 3Н), 4.40 (d. 211), 6.40 (bs, 1H), 7.95-7.15 (m, 4H).
Пример 1.2
Синтез
2.3-диметил-8-(2,6-диметилбензиламино)-6-фторимидазо[1,2-а] пиридина

Смесь 8-амино-2, 3-диметил-6-фторимидазо[1,2-а] пиридина (0,16 г, 0,89 ммоля), хлорида цинка (II) (0,14 г, 1,04 ммоля) и 2,6-диметилбензальдегида (0,14 г, 1,04 ммоля), перемешанную в метаноле (50 мл), обрабатывали цианоборогидридом натрия (0,065 г, 1,04 ммоля) и подвергали дефлегмации в течение 7 ч. Охлажденную реакционную смесь добавляли к 0,5 М NaOH (20 мл), и осажденные твердые вещества отфильтровывали и очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метанол: метиленхлорид (1: 10). Кристаллизацией из петролейного эфира получали 0,1 г (38%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 2.30 (s, 3Н), 2.34 (s, 3H), 2.40 (s, 6H), 4.35 (d, 2H), 4.95 (bs, 1H), 6.15 (dd, 1H), 7.0-7.20 (m, 4H).
Пример 1.3
Синтез 2,
3-диметпл-8-(2.6-диэтилбензиламино)-имидазо[1.2-a]пиридина
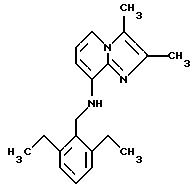
8-амино-2,3-диметил-имидазо[1,2-а] пиридин (0,33 г, 2,0 ммоля) и 2,6-диэтилбензальдегид (0,36 г, 2,2 ммоля) растворяли в метаноле (7 мл). ZnCl2 (0,30 г, 2,2 ммоля) и NaBH3 CN (0,14 г, 2,2 ммоля) последовательно добавляли малыми порциями, и смесь подвергали дефлегмации в атмосфере аргона в течение 3 ч, охлаждали, а затем наливали сверху на водный раствор 1 М NaOH (10 мл). Полученную желтую суспензию экстрагировали дихлорметаном (ДХМ) (3х25 мл), и объединенные органические растворы промывали рассолом, высушивали над Na2SO4, а затем удаляли. Маслянистый остаток (0,4 г) очищали с помощью флэш-хроматографии (ДХМ-ЕtOАс 0%-20% EtOAc) с получением 0,34 г. Обработкой этого маслянистого продукта гексаном (2 мл) получали 0,14 г (23%) в виде очень светлых кристаллов.
1Н-ЯМР (300 МГц, CDCl3): δ 7.2-7.3 (2Н, m), 7.1 (2Н, d), 6.7 (1H, t), 6.2 (1H, d). 4.8 (1H, b), 4.4 (2H, d), 2.7 (4Н, q), 2.3 (6Н, два синглета), 1.2 (6Н, t).
Пример 1.4
Синтез 8-(2.6-диметилбензилокси)-3-гидроксиметил-2-метилимидазо[1,2-а/пиридина

Смесь 8-гидрокси-3-гидроксиметил-2-метилимидазо[1,2-а]пиридина (0,89 г, 5,0 ммолей), карбоната натрия (1,5 г), иодида натрия (0,4 г), 2, 6-диметилбензилхлорида (0,7 г, 4,5 ммоля) и ацетона (60 мл) перемешивали в течение ночи. Добавляли еще карбоната натрия (1,0 г). Реакционную смесь подвергали дефлегмации в течение 2 ч. Реакционную смесь фильтровали, и растворитель удаляли под вакуумом. Остаток ресуспендировали в CH2Cl2/MeOH (100:5) и фильтровали. Вакуумным выпариванием растворителя получали остаток, который очищали с помощью флэш-хроматографии, элюируя СН2Сl2-МеОН (100:4), собирая фракции, и перекристаллизовывали из СН2Сl2/СН3СN с получением 0,37 г соединения, указанного в заголовке.
1Н ЯМР (300 МГц, CDCl3): δ 7,87 (d, J=7.6 Гц, 1H), 7 15-7.08 (m, 1H), 7.0 (d, J=7.6 Гц, 2H), 6.73 (t, J=7.6 Гц,
1H), 6.63 (d, J=7.6
Гц, 1H), 5.23 (s, 2H), 4.83 (s, 2H), 2.4 (s, 6H), 2 28 (s, 3Н)
Пример 1.5
Синтез 2,3-диметил-8-(2,6-диметилбензиламино)имидазо[1,2-a]-пиридина

Смесь 8-амино-2,3-диметилимидазо[1,2-а] пиридина (0,7 г, 4,34 ммоля), карбоната натрия (2,0 г), иодида натрия (0,3 г), 2, 6-диметилбензилхлорида (0,671 г, 4,34 ммоля) и ацетона (30 мл) перемешивали в течение ночи. Реакционную смесь фильтровали, и растворитель удаляли под вакуумом. Остаток растворяли в метиленхлориде и промывали водным NаНСО3. Органический слой отделяли, и растворитель выпаривали. Сырой продукт очищали с помощью флэш-хроматографии, элюируя CH2Cl2 -MeOH, с получением 0,7 г соединения, указанного в заголовке.
1Н ЯМР (300 МГц, CDCl3): δ 7.25 (d, J=7.7 Гц, 1H), 7.14-7.09 (m, 1H), 7.03 (d, J-7.7 Гц, 2Н), 6.73 (t, J=7.7 Гц, 1H), 6,21 (d, J=7.7 Гц, 1H), 4.79 (br "t", 1H), 4.34 (d, J=4.5 Гц, 2Н), 2.38 (s, 6H), 2.34 (s, 6H).
Пример 1.6
Синтез 2,3-диметил-8-(2,
6-диметилбензилокси)имидазо[1,
2-а]пиридина
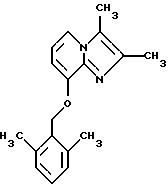
Смесь 8-гидрокси-2,3-диметилимидазо[1,2-а]пиридина (1,2 г, 7,41 ммоля), 2, 6-диметилбензилхлорида (1,145 г, 7,41 ммоля) иодида натрия (0,3 г), карбоната натрия (2,0 г) и ацетона (50 мл) подвергали дефлегмации в течение 3 ч. После добавления метиленхлорида реакционную смесь фильтровали. Растворитель удаляли под вакуумом. Остаток растворяли в CH2Cl2, промывали водным NaHC03, высушивали над Na2 S04 и выпаривали. Остаток подвергали хроматографии на силикагеле, элюируя СН2Сl2-МеОН (100:5) с получением 0,70 г желаемого продукта (из этилацетата-эфира).
1Н ЯМР (300 МГц, CDC13): δ 7.56 (d, J=6.6 Гц, 1Н), 7,1 (t, J=6.6 Гц, 1Н), 6.94-6.85 (m, 3H), 6.73 (d, J=6.6 Гц, 1H), 2.31 (s, 3H), 2.26 (s, 3H), 2.24 (s, 6H).
Пример 1,7
Синтез 2,3-диметил-8-(2-этил-6-метилбензиламино)имидазо [1,2-а] пиридина

8-Амино-2, 3-диметилимидазо[1,2-а] пиридин (0,3 г, 1,86 ммоля) и 2-этил-6-метилбензилхлорид (0,31 г, 1,84 ммоля) растворяли в 5 мл диметоксиэтана. Добавляли иодид калия (0,2 г, 1,2 ммоля) и Na2CO3 (0,3 г, 2,8 ммоля), и смесь подвергали дефлегмации в течение 4 ч. Растворитель выпаривали, и остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь метиленхлорида и этилацетата (60:40). Получали 230 мг (42%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, СDС13): δ
1.22 (t, 3H), 2.35 (s, 6H),
2.39 (s, 3H), 2.70 (q, 2H), 4.35 (d, 2H), 4.81 (t, 1H), 6.21 (d, 1H), 6.73 (t, 1H), 7.01-7.10 (m, 2H), 7.13-7.19 (m, 1H), 7.24 (d, 1H),
Пример 1.8
Синтез
6-бром-2,3-диметил-8-(2,
6-диметилбензиламино) имидазо[1,2-а]пиридина

Смесь 8-амино-6-бром-2, 3-диметилимидазо[1, 2-а] пиридина (1,2 г, 5,0 ммолей), 2,6-диметилбензилхлорида (0,772 г, 5,0 ммолей), карбоната натрия (0,8 г), иодида натрия (0,2 г) и ацетона (45 мл) перемешивали в течение ночи. Добавляли еще 2, 3-диметилбензилхлорида (0,285 г), и реакционную смесь подвергали дефлегмации в течение 5 ч. После добавления ацетона реакционную смесь фильтровали. Растворитель удаляли под вакуумом, и остаток растворяли в СН2Сl2, промывали NaHCO3, высушивали над Na2SO4 и выпаривали. Сырой продукт растворяли в этилацетате и добавляли петролейный эфир. Фильтрованием и выпариванием растворителя получали остаток, который затем перекристаллизовывали из этилацетата с получением 1,45 г соединения, указанного в заголовке.
1 Н-ЯМР (300 МГц, CDCl3): δ 7.37 (d, J=1.5 Гц, 1H), 7.15-7.09 (m, 1H), 7.04 (d. J=7.5 Гц, 2H), 6.28 (d, J=1.5 Гц. 1H), 4.88 ("t", 1H), 4.33 (d, J= 4.13 Гц, 2H), 2.38 (s, 6H), 2.3 (s, 3Н), 2.29 (s, 3Н).
Пример 1.9
Синтез 8-(2,6-диметилбензиламино)-3-гидроксиметил-2-метилимидазо[1,2-а] пиридина

Раствор витрида (бис(2-метоксиэтокси)алюмогидрид натрия, Red-AlTM, Aldrich) (40 мл, 136 ммолей) в толуоле (25 мл) добавляли по каплям к очищенному азотом раствору 3-карбоэтокси-8-(диметилбензиламино)-2-метилимидазо[1,2-а]пиридина (8,0 г, 23,71 ммоля) в толуоле (100 мл). Ледяную баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 105 мин. Реакционную смесь охлаждали до 0oС и гасили добавлением воды (36 мл). Смесь фильтровали, и органический слой промывали водным NaHCO3, высушивали над Na2SO4 и концентрировали. Добавляли ацетонитрил (20 мл), и продукт собирали фильтрованием. Кристаллический продукт дважды промывали ацетонитрилом и высушивали под вакуумом. Выход 5,6 г.
1Н-ЯМР (300 МГц, СDС13): δ 7.58 (d, J=7.1 Гц, 1H), 7.15-7.1 (m, 1H), 7.05 (d, J=7.1 Гц, 2H), 6.74 (t, J=7.1 Гц, 1H), 6.28 (d, J=7.1 Гц, 1H), 4.84 (br t, J=4.5 Гц, 1H), 4.8 (s, 2H), 4.35 (d, J=4.5 Гц, 2H), 2.4 (s, 6H), 2.2 (s, 3Н).
Пример 1.10
Синтез 6-хлор-2,3-диметил-8-(2,6-диметилбензиламино) имидазо[1,
2-а]пиридина

Смесь 8-амино-6-хлор-2,3-диметилимидазо[1,2-а] пиридина (0,894 г, 4,57 ммоля), 2, 6-диметилбензальдегида (0,77 г, 5,7 ммоля), ZnCl2, (1,08 г, 7,92 ммоля), NaB(CN)H3 (0,36 г, 5,7 ммоля) и МеОН (35 мл) подвергали дефлегмации в течение 3,5 ч. Добавляли еще 2, 6-диметилбензальдегида (0,25 г в 4 мл МеОН), ZnCl2, (0,55 г) и NaB(CN)H3 (0,35 г). Реакционную смесь подвергали дефлегмации в течение дополнительных 4 ч. Последовательной обработкой путем добавления 1 M NaOH (150 мл) и воды (50 мл) с последующей экстракцией смеси СН2Cl2, высушиванием и выпариванием растворителя получали твердый остаток. Сырой продукт растворяли в этилацетате и добавляли эфир. Фильтрованием и выпариванием растворителя получали остаток, который затем перекристаллизовывали из этилацетата с получением 0,52 г продукта.
1Н-ЯМР (300 МГц, CDCl3): δ 7.28 (d, J=1.7 Гц, 1H), 7.15-7.1 (m, 1H), 7.04 (d, J=12 Гц, 2H), 6,2 (d, J-1.7 Гц, 1H), 4.89 (br "t", 1H), 4.33 (d, J= 4 Гц, 2H), 2.37 (s, 6H), 2.33 (s, 3H), 2.32 (s, 3H).
Пример 1.11
Синтез 2,3-диметил-8-(2,6-диметил-4-фторбензиламино)имидазо[1,2-а]пиридина
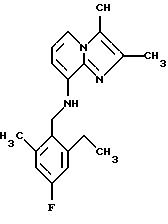
8-Амино-2,3-диметилимидазо[1,2-а] пиридин(0,5 г, 3,1 ммоля) растворяли в ацетонитриле (6 мл). К раствору добавляли 2, 6-диметил-4-фтор-бензилбромид (0,67 г, 3,1 ммоля) и карбонат калия (0,47 г, 3,4 ммоля). Смесь подвергали дефлегмации в течение 16 ч. Добавляли метиленхлорид (12 мл) и раствор хлорида натрия (20 мл). Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Сырой продукт очищали с помощью хроматографии (этилацетат: петролейный эфир 1:1) Получали 400 мг соединения, указанного в заголовке, в виде твердого вещества.
1Н-ЯМР (300 МГц, CDCl3): δ 2.3 (s, 6H), 2.3 (s, 6H), 4,2 (d, 2H), 4.65 (b, 1H), 6.15 (d, 1H), 6.65-6.75 (m, 3H), 7.2 (d, 1H).
Пример 1.12
Синтез 2,6-диметил-8-(2,6-диметилбензиламино)-3-гидроксиметилимидазо[1,2-а]пиридина

Раствор 3-карбоэтокси-2,6-диметил-8-(2,6-диметилбензиламино) имидазо[1,2-а] пиридина (0,4 г, 1,1 ммоля) в 10 мл толуола охлаждали ледяной водой, через 30 мин добавляли Red-AL 65% в толуоле (2.1 г, 6,6 ммоля). Раствор перемешивали в течение 2 ч при комнатной температуре. Добавляли по каплям 10 мл раствора сегнетовой соли (натрия калия тартрата тетрагидрат, 35 г/250 мл воды), добавляли 10 мл толуола, органический слой отделяли и промывали водой, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюент дихлорметан: метанол 9:1, с получением 0,21 г (62%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, СDСl3): δ 1.65 (s, 1H), 2.30 (d. 6H), 2.38 (s, 6H), 4.37 (d, 2H), 4.75 (s. 1H), 4.85 (s, 2H), 6.15 (s, 1H), 7.0-7.15 (m. 3Н), 7.40 (s, 1H).
Пример 1.13
Синтез 2,6-диметил-8-2,6-диметил-4-фторбензиламино)-3-гидроксиметилимидазо[1.2-а] пиридина
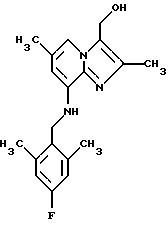
Раствор 0,4 г (1,1 ммоля) 3-карбоэтокси-2,6-диметил-8-(2,6-диметил-4-фторбензиламино)-имидазо[1,2-а] пиридина в 10 мл толуола охлаждали ледяной водой, через 30 мин добавляли Red-AL 65% в толуоле (2,1 г, 6,6 ммоля). Раствор перемешивали в течение 2 ч при комнатной температуре. Добавляли по каплям 10 мл раствора сегнетовой соли (35 г натрия калия тартрата теграгидрата /250 мл воды), добавляли 10 мл толуола, органический слой отделяли и промывали водой, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюент дихлорметан: метанол 95:5, с получением 0,3 г (83%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 2.26 (s, 3Н), 2.33 (s, 3H), 2.37 (s, 6H), 4.28 (d, 2H), 4.70 (s. 1H), 4.82 (s, 2H), 6.14 (s. 1H), 6.75 (d, 2H). 7.42 (s. 1H).
Пример 1.14
Синтез 8-(2,6-диметил-4-фторбензиламино)-2,3,6-триметилимидазо[1,2-пиридина
гидрохлорида

Перемешанную смесь 8-амино-2,3,6-триметилимидазоло[1,2-а]пиридина (0,5 г, 2,85 ммоля), 2,6-диметил-4-фторбеизилбромида (0,7 г, 3,4 ммоля), карбоната калия (0,6 г, 4,6 ммоля), иодида натрия (0,1 г), 15 мл ацетонитрила подвергали дефлегмации в течение ночи. Растворитель выпаривали при пониженном давлении, и остаток растворяли в дихлорметане и промывали водой. Органический слой высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюент гексан: этилацетат 2:1. Маслянистый продукт растворяли в диэтиловом эфире, обрабатывали диэтиловым эфиром/НСl, и осажденную соль отфильтровывали с получением 0,55 г (56%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 2.22 (s, 3H), 2,30 (d, 12H), 4,23 (d, 2H), 4,68 (s, 1H), 6,05 (s, 1H), 6,70 (d, 211), 7,.00 (s, 1Н).
Пример 1.15
Синтез 2.3-димeтил-8-(2,6-димeтил-4-xлopбeнзиламино) имидазо[1,2-a]пиридина
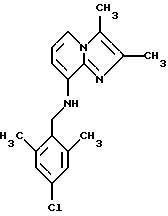
Смесь 4-хлор-2,6-диметилбензилбромида и 2-хлор-4,6-даметилбензилбромида (1,1, 4,68 ммоля) и 8-амино-2,3-диметилимидазо[1,2-а]пиридина (4,65 ммоля) растворяли в 15 мл диметоксиэтана. Добавляли иодид калия (0,5 г, 3,0 ммоля) и Na2CО3 (1 г, 9.4 ммоля). Смесь подвергали дефлегмации в течение 4 ч. Растворитель выпаривали, и остаток очищали с помощью колоночной хроматографии на силикагеле. Продукт элюировали смесью метиленхлорида и этилацетата (70:30). Получали 70 мг соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 2.35 (s, 6H), 4.29 (d, 2H), 4.74 (t, 1H), 6.19 (d, 1H), 6.72 (t, 1H), 7.04 (s, 2H), 7.25 (d. 1H).
Пример 1.16
Синтез 2,
6-диметил-8-(2-этил-6-метилбензиламино)-3-гидроксиметилимидазо[1,2-а]пиридина

3-Карбоэтокси-2, 6-диметил-8-(2-этил-6-метилбензиламино)имидазо[1,2-а] пиридин (1,0 г, 2,8 ммоля) добавляли к тетрагидрофурану (ТГФ) (25 мл) и перемешивали при +5oС. Алюмогидрид лития (0,5 г, 13 ммолей) добавляли порциями в течение 1,5 ч так, чтобы температура оставалась ниже +10oС. После перемешивания этой смеси при комнатной температуре в течение дополнительного 1 ч добавляли по каплям 0, 5 мл воды, затем 0,5 мл 15% гидроксида натрия, а затем 1,5 мл воды. Твердые вещества удаляли с помощью фильтрования и тщательно промывали тетрагидрофураном и метиленхлоридом. Фильтрат и промывки объединяли и высушивали, а растворители удаляли при пониженном давлении. Остаток растворяли в метиленхлориде и промывали водой. Органический слой отделяли, высушивали над сульфатом натрия, выпаривали при пониженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента этилацетат: метиленхлорид (1:1). Кристаллизацией из петролейного эфира: диэтилового эфира (1:1) получали 0,37 г (41%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.25 (t, 3Н), 2.25 (s, 3Н), 2.35 (s, 3H), 2.40 (s, 3Н), 2.70 (q, 2H), 4.35 (d, 2Н), 4.75 (bs, 1H), 4.80 (s, 2H), 6.15 (s, 1H), 7.05-7.25 (m, 3Н), 7.40 (s, 1H).
Пример 1.17
Синтез
8-(2.6-диэтилбензиламино)-2,
6-диметил-3-гидроксиметил имидазо[1,2-а]пиридина

Раствор 3-карбоэтокси-2,6-диметил-8-(2, 6-диэтилбензиламино) имидазо[1,2-а] пиридина (1,75 г, 4,6 ммоля) в 30 мл тетрагидрофурана обрабатывали алюмогидридом лития (0,7 г, 18,5 ммоля) при комнатной температуре в течение 3,5 ч. Реакция была закончена через 4 ч и ее осторожно гасили добавлением по каплям воды (0,7 мл), водного гидроксида натрия (0,7 мл, 15%) и снова воды (2 мл). Смесь экстрагировали хлорформом, и органический слой концентрировали. Остаток перекристаллизовывали в этаноле, и белый кристаллический продукт фильтровали, промывали диэтиловым эфиром и высушивали под вакуумом, что давало выход 1,5 г (96%).
1Н-ЯМР (500 МГц, CDCl3): δ 1.23 (t, 6H), 1.99 (s, 1H), 2.25 (s, 3Н), 2.33 (s, 3H), 2.73 (q, 4Н), 4.34 (d, 2H), 4.80 (s, 3Н), 6.13 (s, 1 Н), 7.09 (d, 2H), 7.22 (t, 1H), 7.40 (s, 1H).
Пример 1.18
Синтез 8-(2,6-диэтилбензиламино) 2,3,6-триметилимидазо[1,2-а]пиридина

Перемешанную смесь 8-амино-2,3,6-триметилимидазо[1,2-а]пиридина (0,5 г, 2,8 ммоля), 2,6-диэтилбензальдегида (0,7 г, 4,3 ммоля) и хлорида цинка (II) (0,44 г, 3 ммоля) в 50 мл метанола обрабатывали цианоборогидридом натрия (0,19 г, 3 ммоля), а затем подвергали дефлегмации в течение 20 ч. Метанол выпаривали при пониженном давлении, и остаток растворяли в дихлорметилене и воде. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток подвергали хроматографии на силикагеле сначала с дихлорметиленом, а затем с дихлорметиленом: этилацетатом (1:1), в результате чего получали 0,42 г соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, СDCl3): δ 1,25 (t, 6Н), 2.28 (s, 3H), 2.30 (s, 3Н), 2.33 (s, 3H), 2.71 (q, 4H), 4.36 (d, 2H), 4.84 (s, 1H),6.10(s, 1H), 7.04-7.23 (m, 4H).
Пример 1.19
Синтез 8-(2-этил-6-метилбензиламино)-2,3,
6-триметилимидазо[1,2-a] пиридина

Перемешанную смесь 8-амино-2,3,6-триметилимидазо[1, 2-а]пиридина (0,5 г, 2,8 ммоля), 2-этил-6-метилбензальдегида (0,45 г, 3 ммоля) и хлорида цинка (II) (0,4 г, 3 ммоля) в 50 мл метанола обрабатывали цианоборогидридом натрия (0,19 г, 3 ммоля) и подвергали дефлегмации в течение 20 ч. Метанол выпаривали при пониженном давлении, и остаток растворяли в дихлорметилене и воде. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток подвергали хроматографии на силикагеле с дихлорметиленом: метанолом (10: 1), что давало выход 0,28 г (33%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.22 (t, 3Н), 2.32 (s, 6Н), 2.34 (s, 3Н), 2.38 (s, 3Н), 2.72 (q, 2H), 4.33 (d, 2Н), 4.77 (s, 1H), 6.08 (s, 1H), 7.03-7.19 (m, 4H).
Пример 1.20
Синтез 8-(2,6-диметил-4-фторбензилокси)-3-гидроксиметил-2-метилимидазо[1,2-а] пиридина

Алюмогидрид лития (0,31 г, 8,4 ммоля) добавляли к тетрагидрофурану (30 мл), и 3-карбоэтокси-8-(2, 6-диметил-4-фторбензилокси)-2-метилимидазо[1,2-а] пиридин (1,5 г, 4,2 ммоля), растворенный в тетрагидрофуране (30 мл), добавляли по каплям в течение 30 мин. Добавляли по каплям 0,31 мл воды, затем 0, 31 мл 15% гидроксида натрия, а затем 0,93 мл воды. Твердые вещества удаляли фильтрованием и тщательно промывали метанолом: метиленхлоридом (1:1). Фильтрат и промывки объединяли, и растворители удаляли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид:метанол (9:1). Обработкой остатка ацетонитрилом получали 0,9 г (69%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, ДМСО-d6): δ 2.25 (s, 3Н), 2.35 (s, 6H), 4.85 (d, 2H), 5.1 (t, 1H), 5.2 (s, 2H), 6.8-7.05 (m, 4H), 7.95 (d, 1H).
Пример 1.21
Синтез 6-бром-8-(2,6-диметил-4-фторбензиламино)-3-гидроксиметил-2-метилимидазо[1,2-а]пиридина

LiBH4 (70 мг) добавляли порциями в течение 4 ч к подвергнутому дефлегмации раствору 6-бром-3-карбоэтокси-8-(2, 6-диметил-4-фторбензиламино)-2-метилимидазо[1,2-а] пиридина (100 мг, 0,23 ммоля) в ТГФ. Реакционную смесь гасили добавлением разбавленной НСl и добавляли метиленхлорид. Органический слой отделяли, высушивали и выпаривали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид: этилацетат (100:10), с получением 40 мг (44%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, СDСl3): δ 7.72 (s, 1h), 6.75 (d, 2h), 6.35 (s, h), 4.9 (t, 1h), 4.8 (s, 2h), 4.3 (d, 2h), 2.35 (s, 6h), 2.25 (s, 3h).
Пример 1.22
Синтез 2,6-диметил-8-(2,
6-диметилбензилокси)-3-гидроксиметилимидазо[1,2-а]пиридина

Смесь витрида (3 мл, 10,2 ммоля) в толуоле (3 мл) добавляли по каплям к очищенному азотом раствору 3-карбоэтокси-2,6-диметил-8-(2, 6-диметилбензилокси)имидазо[1,2-а]пиридина (0,68 г, 1,93 ммоля) в толуоле (15 мл). Ледяную баню удаляли, и реакционную смесь перемешивали при комнатной температуре в течение 2 ч и 15 мин. Реакционную смесь охлаждали до 0oС и гасили добавлением воды (6 мл). Добавляли метиленхлорид/метанол, и реакционную смесь фильтровали. Растворитель удаляли под вакуумом, и остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид: метанол (100:5), с получением 0,35 г (58%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, СDСl3): δ 7.65 (s, 1H), 7.10 (t. 1H), 7.0 (d, 2H), 6.50 (s, 1H), 5.2 (s, 2H), 4.8 (s, 2H), 2.4 (s, 6H), 2.35 (s, 3H), 2.25 (s, 3H).
Пример 1.23
Синтез 8-(2,6-диметил-4-фторбензиламино)-3-гидроксиметил-2-метилимидазо[1,2-а]пиридина

Раствор 0,6 г (1,7 ммоля) 3-карбоэтокси-8-(2,6-диметил-4-фторбензиламино)-2-метилимидазо[1,2-а]пиридина в 30 мл толуола охлаждали ледяной водой. Red-AL 65% 2,1 г (6,6 ммоля) в толуоле добавляли в течение 30 мин. Раствор перемешивали в течение 1 ч при комнатной температуре. Добавляли по каплям 25 мл раствора сегнетовой соли (35 г натрия калия тартрата тетрагидрата /250 мл воды) и отделяли органический слой. Водный слой промывали метиленхлоридом, который отделяли. Объединенные органические растворители высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюент дихлорметан: метанол 95:5, с получением 0,42 г (79%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 2.15 (s, 3Н), 2.35 (s, 6Н), 4.30 (d, 2H), 4.75 (s, 2H), 4.85 (t, 1H), 6,25 (d, 1H), 6.70-6,80 (m, 3Н), 7.55 (d, 1H).
Пример 1.24
Синтез 2,
6-диметил-8-(2-этил-4-фтор-6-метилбензиламино)-2-гидроксиметилимидазо[1,2-а]пиридина

К смеси LiAlH4 (0,08 г, 2,1 ммоля) в тетрагидрофуране (15 мл) добавляли 3-карбоэтокси-2, 6-диметил-8-(2-этил-4-фтор-6-метилбензиламино)имидазо[1,2-а] пиридин (0,4 г, 1,0 ммоль) в тетрагидрофуране (15 мл). После перемешивания этой смеси при комнатной температуре в течение 4 ч добавляли по каплям 0,1 мл воды, затем 0,1 мл 15% гидроксида натрия, а затем 0,3 мл воды. Твердые вещества удаляли фильтрованием и тщательно промывали тетрагидрофураном. Фильтрат и промывки объединяли и высушивали, и растворитель удаляли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид: метанол (9:1). Кристаллизацией из ацетонитрила получали 0,32 г (89%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.2 (t, 3Н), 2.2 (s, 3H), 2.35 (s, 3Н), 2.4 (s, 3H), 2.75 (q, 2Н), 4.3 (d, 2H), 4.75 (bs, 3Н), 6,15 (s, 1H), 6.75-6.85 (m, 2H), 7.45 (s, 1H).
Пример 1.25
Синтез 8-(2-этил-4-фтор-6-метилбензиламино)-2,3,6-триметилимидазо[1,2-а]
пиридина

8-Амино-2,3,6-триметилимидазо[1,2-а] пиридин (0,38 г, 2,16 ммоля) и 2-этил-4-фтор-6-метилбензилбромид (0,50 г, 2,16 ммоля) растворяли в 10 мл диметоксиэтана. Добавляли иодид калия (0,2 г, 1, 2 ммоля) и Na2C03 (0,4 г, 3,8 ммоля), и смесь подвергали дефлегмации в течение 6 ч. Растворитель выпаривали, и остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид и этилацетат (60:40). Получали 203 мг (29%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.21 (t, 3Н), 2.32 (s, 6H), 2.33 (s, 3Н), 2.37 (s, 3H), 2.71 (q, 2H), 4.28 (d, 2H), 4.68 (t, 1H), 6.06 (s, 1H), 6.73-6.80 (m, 2H), 7.05 (s, 1H).
Пример 1.26
Синтез 2,
3-диметил-8-(2,6-диметил-4-фторбензилокси)имидазо[1,2-а] пиридина

2, 3-Диметил-8-гидроксиимидазо[1,2-а] пиридин (1,7 г, 10 ммолей), 2,6-диметил-4-фторбензилбромид (2,3 г, 10 ммолей), иодид натрия (0,5 г, 0,3 ммоля) и карбонат натрия (2,6 г, 28 ммолей) добавляли к ацетону (75 мл) и эту смесь подвергали дефлегмации в течение 6 ч. Добавляли метиленхлорид, смесь фильтровали, и растворители выпаривали при пониженном давлении. Очисткой с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента метиленхлорида: этилацетата (1:2) получали соединение заголовка в виде белого порошка (0,85 г, 28%).
1Н-ЯМР (300 МГц, CDCl3): δ 2.36 (s, 3Н), 2.38 (s, 9Н), 5.15 (s, 2H), 6.57 (d, 1H), 6.68-6.75 (m, 3Н), 7.46 (d, 1H).
Пример 1.27
Синтез 2,
3-диметил-8-(2-этил-6-метилбензилокси)имидазо[1,2-а]пиридинa
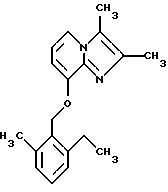
2,3-Диметил-8-гидроксиимидазо[1, 2-а] пиридин (0,8 г, 5 ммолей), 2-этил-6-метилбензилхлорид, иодид натрия (0,25 г, 1,7 ммоля) и карбонат натрия (1,2 г, 11 ммолей) добавляли к ацетону (40 мл), и эту смесь подвергали дефлегмации в течение 5 ч. Ацетон выпаривали, и остаток растворяли в метиленхлориде и промывали водой. Органический растворитель высушивали и выпаривали при пониженном давлении. Остаток очищали дважды с помощью колоночной хроматографии на силикагеле, используя в качестве элюента (а) метиленхлорид: этилацетат (1: 2), (б) метиленхлорид: этилацетат (2:1), с получением соединения, указанного в заголовке (0,02 г, 1,4%).
1Н-ЯМР (300 МГц, CDCl3): δ 1.2 (t, 3H), 2.36 (s, 3Н), 2.38 (s, 3H), 2.40 (s, 3Н), 2.74 (q, 2H), 5.21 (s, 2H), 6.59 (d, lH), 6.7 (t, 1H), 7.04 (m, 2Н), 7.17 (t, lH), 7.45 (d, 1H).
Пример 1.28
Синтез
8-(2-этил-6-метилбензилокси)-3-гидроксиметил-2-метилимидазо[1,2-а]пиридина
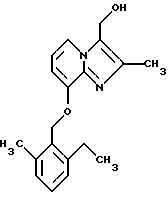
К смеси LiAlH4 (0,08 г, 2,1 ммоля) в тетрагидрофуране (25 мл) добавляли 3-карбоэтокси-8-(2-этил-6-метилбензилокси)-2-метилимидазо[1,2-а] пиридин (1,0 г, 2,8 ммоля) в тетрагидрофуране (25 мл). После перемешивания этой смеси при комнатной температуре в течение 2 ч добавляли по каплям 0,2 мл воды, затем 0,2 мл 15% гидроксида натрия, а затем 0,6 мл воды. Твердые вещества удаляли фильтрованием, и растворитель удаляли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид: метанол (9:1). Кристаллизацией из диэтилового эфира получали 0,52 г (60%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.2 (t, 2Н), 2.25 (s. 3Н), 2.4 (s, 3Н), 2.75 (q, 2H), 4.75 (s, 2H), 5.2 (s, 2H), 6.65-6.75 (m, 2H), 7.0-7.2 (m, 3Н), 7.85 (d, 1H).
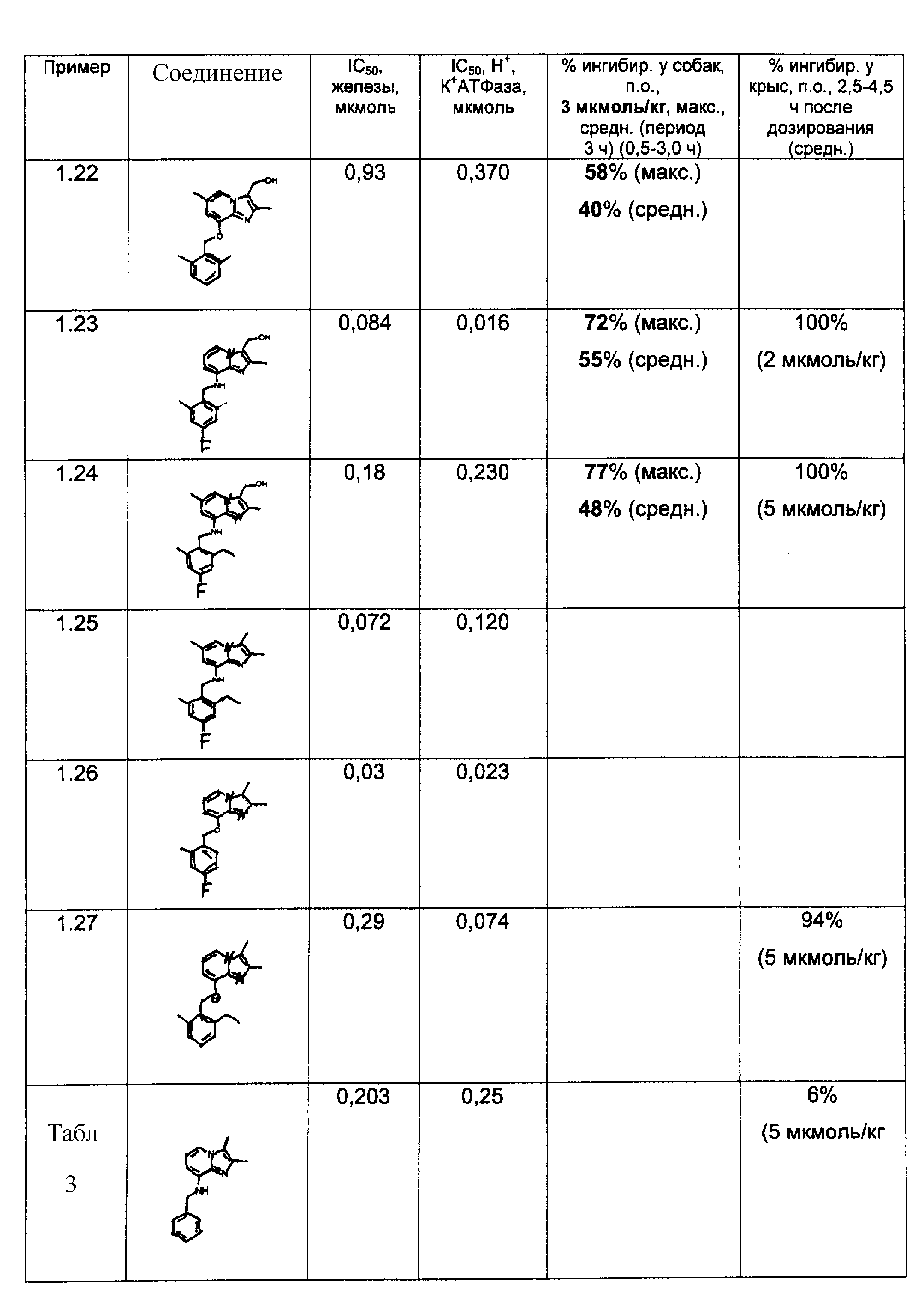
Соединения согласно примерам 1.1- 1.28 представлены в табл.1.
ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНЫХ
СОЕДИНЕНИЙ
Пример 2.1
Синтез 2,
6-диметил-4-фторбензилбромида
Смесь 3,5-диметил-фторбензола (5 г, 0,04 моля), параформальдегида (15 г), бромистоводородной кислоты (70 мл)
(30% в уксусной кислоте) и уксусной кислоты (25 мл)
перемешивали при температуре окружающей среды в течение 4,5 ч. К этой смеси добавляли воду и петролейный эфир, и органический слой отделяли,
высушивали над безводным сульфатом натрия и осторожно
выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с петролейным эфиром в качестве элюента с
получением желаемого продукта (3,7 г, 43%).
1Н-ЯМР (300 МГц, CDCl3): δ 2.5 (s, 6H), 4.55 (s, 2H), 6.75 (d, 2H).
Пример 2.2
Синтез
2-этил-6-метилбензилхлорида
2-этил-6-метилбензиловый спирт (1,0 г, 6,67 ммоля) растворяли в 10 мл метиленхлорида. Добавляли тионилхлорид (1,0 г, 8,5 ммоля). Эту смесь перемешивали в течение
ночи при температуре окружающей
среды. Реакционную смесь выпаривали. Остаток растворяли в метиленхлориде и фильтровали через 5 г силикагеля. Фильтрат выпаривали. Получали 1,0 г (89%) соединения,
указанного в заголовке (масло).
1Н-ЯМР (300 МГц, CDCl3): δ 1.29 (t, 3Н), 2.46 (s. 3H), 2.76 (q, 2H), 4.71 (s, 2H). 7.0-7.2 (m, 3H).
Пример
2.3
Синтез 8-амино-2,3,
6-триметилимидазо[1,2-а] пиридина
К раствору 2,3-диамино-5-метилпиридина (2,0 г, 16 ммолей) в этаноле (100 мл) добавляли 3-бром-2-бутанон (2,4 г, 16 ммолей).
Реакционную смесь подвергали
дефлегмации в течение 16 ч. Добавляли дополнительное количество 3-бром-2-бутанона (1,0 г, 6,7 ммоля) и триэтиламин (1,0 г, 9,9 ммоля), и эту смесь подвергали дефлегмации в
течение 2 ч. Этанол
выпаривали при пониженном давлении, и остаток обрабатывали метиленхлоридом и раствором бикарбоната. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при
пониженном давлении.
Маслянистый остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метанол: метиленхлорид (1:20), с получением желаемого продукта (1,05 г,
37%).
1Н-ЯМР (300 МГц, ДМСО-d6): δ 2.15 (s, 3Н), 2.25 (s, 3Н), 2.3 (s, 3H), 5.45 (bs, 2H),6.05(s, 1H),7.20(s, 1H),
Пример 2.4
Синтез
2-амино-5-фтор-3-нитропиридина
К раствору 2-амино-5-фторпиридина (8,6 г, 77 ммолей) в концентрированной серной кислоте (40 мл) добавляли по каплям (30 мин) дымящуюся азотную кислоту (3,25 мл,
77 ммолей) при температуре +3oС. Эту реакционную смесь перемешивали при комнатной температуре в течение 1 ч и при +55oС в течение 1 ч. Эту смесь наливали на лед, нейтрализовали
10 М гидроксидом натрия и экстрагировали метиленхлоридом. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали дважды с помощью колоночной
хроматографии на силикагеле, используя в качестве элюента (1) метанол: метиленхлорид (1:20) и (2) диэтиловый эфир: петролейный эфир (1:1), с получением соединения, указанного в заголовке (0,44 г, 3,
6%).
1Н-ЯМР (300 МГц, CDCl3): δ 6.65 (bs, 2H), 8.20 (dd, 1H), 8.35 (d, 1H).
Пример 2.5
Синтез 2,3-диамино-5-фторпиридина
К раствору 2-амино-5-фтор-3-нитропиридина (0,42 г, 2,3 ммоля) и железного порошка (1,6 г, 28 ммолей) в этаноле (10 мл) добавляли воду (0,5 мл, 28 ммолей) и соляную кислоту (27 мкл, 0,32 ммоля). Эту
смесь подвергали дефлегмации в течение 1 ч. Добавляли дополнительное количество железного порошка (0,2 г, 3,6 ммоля), и смесь подвергали дефлегмации в течение 30 мин. Реакционную смесь фильтровали
через целит и выпариванием растворителя при пониженном давлении получали 0,3 г (100%) желаемого продукта.
1Н-ЯМР (300 МГц, CDCl3): δ 3.55 (bs, 2H), 4.1 (bs, 2H), 6.7 (dd, 1H), 7.5 (d, 1H).
Пример 2.6
Синтез 8-амино-2,3-диметил-6-фторимидазо[1,2-а] пиридина
Смесь 2,3-диамино-5-фторпиридина (0,3 г, 2,4 ммоля) и
3-бром-2-бутанона (0,36 г, 2,4 ммоля) в этаноле (20 мл) подвергали дефлегмации в течение 10 ч. Растворитель выпаривали при пониженном давлении. Остаток растворяли в метиленхлориде и обрабатывали
раствором бикарбоната. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с метанолом:
метиленхлоридом (1: 20) в качестве элюента с получением 0,16 г (37%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 2,3 (s, 3Н), 2.4 (s, 3H), 46 (bs, 2H), 6.2 (dd, 1H), 7.2 (dd,1H).
Пример 2.7
Синтез 8-амино-6-бром-2,3-диметилимидазо[1,2-а]пиридина
Раствор 2,3-диамино-5-бромпиридина (4,0 г, 21,29
ммоля) и 3-бром-2-бутанона (3,7 г, 24,48 ммоля) в этаноле (40 мл) подвергали дефлегмации в течение ночи. После охлаждения до комнатной температуры кристаллический продукт отфильтровывали и промывали
этанолом и эфиром. Эти кристаллы растворяли в метиленхлориде и нейтрализовали водным NaHCO3. Органический слой отделяли, высушивали над Na2S04 и выпаривали под
вакуумом. Выход 2,3 г.
1Н-ЯМР (300 МГц, CDCl3): δ 7.39 (d, J=1.7 Гц, 1H), 6.36 (d, J=1.7 Гц, 1H), 4.5 (br s,2H), 2.35 (s,3H), 2.3 (s, 3Н).
Пример 2.8
Синтез 3-карбоэтокси-8-(диметилбензиламино)-2-метилимидазо[1,2-а] пиридина
Смесь 8-амино-3-карбоэтокси-2-метилимидазо[1,2-а]пиридина (6,08 г, 27,74 ммоля), 2,
6-диметилбензилхлорида (4,5 г, 29,13 ммоля), карбоната натрия (4,32 г, 43,7 ммоля), иодида натрия (0,7 г) и ацетона (120 мл) перемешивали в течение 30 ч, и кристаллический продукт отфильтровывали.
Этот продукт растворяли в дихлорметане, фильтровали и выпаривали растворитель при пониженном давлении с получением продукта, указанного в заголовке (7,0 г).
1H-ЯMP(300 MГц, CDCl3): δ 8.66 (d, J=11 Гц, 1H), 7,16-7,1 (m, 1H), 7,05 (d, J= 11 Гц, 2H), 6,87 (t, J=11 Гц, 1H), 6,45 (d, J=11 Гц, 1H), 4,86 ("t", 1H), 4,4 (q, J= 7 Гц, 2Н), 4,35 (d, J=3,6 Гц, 2H), 2.65 (s, 3Н), 2,35 (s, 6H), 14 (t, J=7 Гц, 3Н).
Пример 2.9
Синтез 8-амино-6-хлор-2,3-диметилимидазо[1,2-а]пиридина
Смесь 2,3-диамино-5-хлорпиридина (5,26 г, 36,64
ммоля) и 3-бром-2-бутанона (6,2 г, 41,06 ммоля) в этаноле (60 мл) подвергали дефлегмации в течение ночи. После охлаждения до комнатной температуры кристаллический продукт отфильтровывали и промывали
этанолом и эфиром. Эти кристаллы растворяли в метиленхлориде и нейтрализовали водным NaHCO3. Органический слой отделяли, высушивали над Na2S04 и выпаривали под
вакуумом. Выход 3,0 г.
1Н-ЯМР (300 МГц, CDCl3): δ 7.29 (d, J=1.5 Гц, 1Н), 6.26 (d, J=1,5 Гц, 1Н), 4.55 (br s, 2H), 2.4 (s, 3H), 2.3 (s, 3H).
Пример 2.10
Синтез 8-амино-2-карбоэтокси-2,6-диметилимидазо[1,2-а]пиридина
Перемешанную смесь 2,3-диамино-5-метилпиридина (4,0 г, 32,5 ммоля) и (5,9 г, 36,0 ммолей)
этил-хлорацетоацетата в 75 мл абсолютного этанола подвергали дефлегмации в течение ночи. Этанол выпаривали при пониженном давлении. Остаток растворяли в 2 М НС1 и промывали три раза диэтиловым эфиром,
доводили рН до 9 и экстрагировали 3 раза дихлорметаном. Органический слой высушивали над безводным сульфатом натрия и выпаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле с
дихлорметаном: метанолом 95: 5 в качестве элюента с получением продукта, указанного в заголовке 2,0 г (28%).
1Н-ЯМР (300 МГц, CDCl3): δ 1.42 (t, 3Н), 2.28 (s, 3Н), 2.65 (s, 3H), 4.40 (q. 2H), 4.47 (s, 2H), 6.40 (s, 1H), 8.55 (s, 1H).
Пример 2.11
Синтез 3-карбоэтокси-2,6-диметил-8-(2,6-диметилбензиламино) имидазо[1,
2-а]пиридина
Перемешанную смесь 8-амино-2,6-диметилимидазо [1,2-а]пиридина (1,2 г, 5,1 ммоля), хлорида цинка (II) (0,84 г, 6,2 ммоля) и 2,6-диметилбензальдегида (0,84 г, 6,2 ммоля) в 50 мл
метанола обрабатывали цианоборогидридом натрия (0,39 г, 6,2 ммоля) и подвергали дефлегмации в течение 5 ч. Метанол выпаривали при пониженном давлении, и остаток растворяли в дихлорметане и 40 мл 2 М
гидроксида натрия. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюент
петролейный эфир (40-60): изопропиловый эфир 8:2, с выходом 0,8 г (44%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1,44 (t, 3Н), 2.35 (d. 9H), 2.60 (s, 3Н), 4.33 (d, 2H), 4.40 (q, 2H), 4.6 (s, 1H),6,60(s, 1H), 7.10 (d, 2H), 7.25 (m, 1H),8.50(s, 1H).
Пример 2.12
Синтез 3-карбоэтокси-2,6-диметил-8-(2,
6-диметилбензиламино)имидазо[1,2-а]пиридина
Перемешанную смесь 8-амино-2,6-диметилимидазол [1,2-а]пиридина (1,2 г, 5,1 ммоля), хлорида цинка (II) (0,84 г, 6,2 ммоля) и 2,
6-диметилбензальдегида (0,84 г, 6,2 ммоля) в 50 мл метанола обрабатывали цианоборогидридом натрия (0,39 г, 6,2 ммоля) и подвергали дефлегмации в течение 5 ч. Метанол выпаривали при пониженном
давлении,
и остаток растворяли в дихлорметане и 40 мл 2 М гидроксида натрия. Органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с
помощью
колоночной хроматографии на силикагеле, элюент петролейный эфир (40-60): изопропиловый эфир 8:2, с выходом 0,8 г (44%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.44 (t, 3Н), 2.35 (d, 9H), 2.60 (s, 3Н), 4.33 (d, 2H), 4.40 (q, 2H), 4.6 (s, 1H),6.60(s, 1H), 7.10 (d, 2H), 7.25 (m, 1H),8.50(s, 1H).
Пример
2.13
Синтез 3-карбоэтокси-2,6-диметил-8-(2,6-диметил-4-фторбензиламино)-имидазо[1,2-а] пиридина
Перемешанную смесь (1,1 г, 4,7 ммоля) 8-амино-3-карбоэтокси-2,6-диметилимидазо[1,2-а]
пиридина, (1,2 г, 5,7 ммоля) 2,6-диметил-4-фторбензилбромида, (1,0 г, 7,5 ммоля) карбоната калия и (0,1 г) иодида натрия в 15 мл ацетонитрила подвергали дефлегмации в течение ночи. После выпаривания
растворителя при пониженном давлении остаток растворяли в дихлорметане и промывали водой, органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток
очищали с помощью колоночной хроматографии на силикагеле, элюент петролейный эфир (40-60): изопропиловый эфир 7:3, с получением 0,8 г (47%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.42 (t, 3Н), 2.36 (s, 9H), 2.62 (2, 3Н), 4.45 (d, 2H), 4.48 (q, 2H), 4.54 (s, 1H), 6.30 (s, 1H), 6.75 (d, 2H), 8.55 (s, 1H).
Пример
2.14
Cинтез 4-хлор-2,6-димеметилбензилбромида
4-Хлор-3,5-диметилбензол (1,42 г, 0,01 моля) и параформальдегид (0,31 г, 0,01 моля) добавляли к 2 мл гидробромида (33%) в
уксусной
кислоте. Эту смесь перемешивали в течение ночи при +70oС. Реакционную смесь наливали на 25 мл воды и продукт экстрагировали диэтиловым эфиром. Органический слой промывали водой.
Органический слой высушивали (Na2SO4) и выпаривали. Получали 1,1 г продукта (масло). Спектр1Н-ЯМР показывает, что это вещество представляет собой смесь соединения,
указанного в заголовке, и 2-хлор-4,6-диметилбензилбромида. Этот продукт использовали как таковой без какой-либо дальнейшей очистки в следующей стадии синтеза (пример 1.15).
1
Н-ЯМР (300 МГц, CDCl3): δ 2.28 (s, 6H), 4.51 (s, 2H), 7.04 (s, 2H),
Пример 2.15
Синтез 3-карбоэтокси-2,6-диметил-8-(2-этил-6-метилбензиламино)-имидазо[1,
2-а]пиридина
Смесь 8-амино-3-карбоэтокси-2,6-диметилимидазо[1,2-а]пиридина (1,4 г, 6 ммолей), 2-этил-6-метилбензальдегида (0,9 г, 6,5 ммоля), ZnCl2 (1,0 г, 7,4 ммоля),
NaB(CN)H3 (0,41 г, 6,5 ммоля) и МеОН (30 мл) подвергали дефлегмации в течение 5 ч. Добавляли еще ZnCl2 (0,2 г) и NaB(CN)H3 (0,1 г). Реакционную смесь подвергали
дефлегмации в
течение дополнительных 2 ч. Добавляли триэтиламин (2 мл), и смесь перемешивали при комнатной температуре в течение 10 мин. Растворитель выпаривали при пониженном давлении, и остаток
очищали с помощью
колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид. Получали 1,1 г (50%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3 ): δ 1.25 (t, 3H), 1.45 (t, 3Н), 2,30 (s, 6H), 2.6 (s, 3Н), 2.75 (q, 2H), 4.35 (d, 2H), 4.45 (q, 2H), 4.85 (bs, 1H), 6.35 (s, 1H), 7.0-7.25 (m, 3Н), 8.5 (s, 1H).
Пример 2.16
Синтез 3-карбоэтокси-2,6-диметил-8-(2,6-диэтилбеизиламино) имидазо[1,2-а]пиридина
Перемешанную смесь 8-амино-3-карбоэтокси-2,6-диметилимидазо[1,2-а]пиридина (2,
02 г, 8,6 ммоля), хлорида
цинка (II) (1,48 г, 10,8 ммоля) и 2,6-диэтилбензальдегида (2,17 г, 13,4 ммоля) в 50 мл метанола обрабатывали цианоборогидридом натрия (0,65 г, 10,3 ммоля) и подвергали
дефлегмации в течение ночи. Этой
смеси давали охладиться, а затем наливали ее на 80 мл 1 М гидроксида натрия. Образовавшийся осадок отфильтровывали и промывали водой, а затем очищали с помощью
колоночной хроматографии на силикагеле с
дихлорметаном: метанолом (95:5) в качестве элюента. Выход составлял 2,1 г (64%) соединения, указанного в заголовке.
1Н-ЯМР (500 МГц, CDCl3): δ 1.23 (t, 6H), 1.42 (t, 3Н), 2.38 (s, 3Н), 2.61 (s, 3Н), 2.72 (q, 4H), 4.34 (d, 2Н), 4.40 (q, 2H), 4.83 (t, 1 Н), 6.32 (s, 1 Н), 7.11 (d, 2H), 7.24 (t, 1H), 8.51 (s, 1H).
Пример 2.17
Синтез 3-карбоэтокси-8-(2,6-диметил-4-фторбензилокси)-2-метилимидазо[1,2-а]пиридина
Смесь 3-карбоэтокси-8-гидрокси-2-метилимидазо[1,2-а]пиридина (1,
5 г, 6,8 ммоля), 2,
6-диметил-4-фторбензилбромида (1,6 г, 7,5 ммоля), иодида натрия (0,1 г), карбоната калия (1,9 г, 13,6 ммоля) и ацетонитрила (50 мл) подвергали дефлегмации в течение ночи.
Растворитель удаляли под
вакуумом. Остаток растворяли в СН2Сl2, промывали водой, высушивали над Na2SO4 и выпаривали. Остаток подвергали хроматографии на
силикагеле, элюируя
гептаном: изопропиловым эфиром (1:2), с получением 2,0 г (83%) желаемого продукта.
1Н-ЯМР (300 МГц, CDCl3): δ 1.45 (t, 3Н), 2.4 (s, 6H), 2.7 (s, 3Н), 4.45 (q, 2H), 5.2 (s, 2H), 6.7-6.9 (m, 4H), 9.0 (d, 2H).
Пример 2.18
Синтез 8-амино-6-бром-3-карбоэтокси-2-метилимидазо[l,2-a]пиридина
Смесь 2,
3-диамино-5-бромпиридина (2,
5 г, 13,31 ммоля) и этил-2-хлорацетоацетата (2,41 г, 14,64 ммоля) в 35 мл абсолютного этанола подвергали дефлегмации в течение 14 ч. Этанол выпаривали при пониженном
давлении. Остаток растворяли в
метиленхлориде и нейтрализовали водным NaHCO3. Органический слой отделяли, высушивали и выпаривали под вакуумом. Остаток очищали с помощью колоночной
хроматографии на силикагеле с
метиленхлоридом: метанолом (100:3,5) в качестве элюента с получением 1,55 г (39%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 8.9 (s, 1H), 6.65 (s, 1H), 4.6 (bs, 2H), 4.4 (q, 2H), 2.65 (s, 3H), 1.4 (t, 3H).
Пример 2.19
Синтез 6-бром-3-карбоэтокси-8-(2,
6-диметил-4-фторбензиламино)-2-метилимидазо[1,
2-а]пиридина
Смесь 8-амино-6-бром-3-карбоэтокси-2-метилимидазо[1,2-а] пиридина (2,06 г, 691 ммоля), 2,6-диметил-4-фторбензилбромида (1,05 г, 4,
48 ммоля), иодида натрия (0,45 г), карбоната
натрия (2,2 г) и ацетона (40 мл) подвергали дефлегмации в течение 22 ч. Эту реакционную смесь фильтровали Отфильтрованный материал промывали СН2
Сl2. Метиленхлоридный раствор
промывали водой, высушивали и выпаривали под вакуумом. Остаток суспендировали в этаноле/эфире и фильтровали с получением 1,15 г (56%) соединения, указанного в
заголовке.
1
Н-ЯМР (300 МГц, CDCl3): δ 8.85 (s, 1H), 6.8 (d, 2H), 6.55 (s, 1H), 4.9 (t, 1H), 4.4 (q, 2H), 4.3 (d, 2H), 2.6 (s, 3H), 2.4 (s, 6H), 1.45 (t,
3H)
Пример 2.20
Синтез
3-(2,6-диметилбензилокси)-5-метил-2-нитропиридина
К 0,52 г (8,02 ммоля) 87% КОН и 0,15 г q-иодида в 6 мл 95% этанола добавляли раствор
3-гидрокси-5-метил-2-нитропиридина (1,2 г, 7,79 ммоля) в
25 мл этанола. К полученной суспензии калийной соли добавляли по каплям раствор 2,6-диметилбензилхлорида (1,24 г, 8,02 ммоля) в 13 мл этанола.
Эту реакционную смесь подвергали дефлегмации в течение 1
ч. Добавляли еще 87% КОН (0,16 г) и 2,6-диметилбензилхлорида (0,38 г). Эту реакционную смесь подвергали дефлегмации в течение дополнительных
70 мин. Эту смесь фильтровали, и неорганические соли
промывали этанолом и метиленхлоридом. Органический слой выпаривали под вакуумом. Остаток растворяли в метиленхлориде, промывали водным NaHCO3, высушивали и выпаривали под вакуумом. Остаток
суспендировали в эфире/изопропаноле и фильтровали с получением 1,72 г (81%) соединения, указанного в заголовке.
1 Н-ЯМР (500 МГц, CDCl3): δ 7.94 (s, 1H), 7.46 (s, 1H), 7.19 (t, 1H), 7.08 (d, 2H), 5.18 (s, 2H), 2.47 (s, 3H), 2.4 (s, 6H).
Пример 2.21
Синтез 2-амино-3-(2,
6-диметилбензилокси)-5-метилпиридина
Смесь 3-(2,
6-диметилбензилокси)-5-метил-2-нитропиридина (1,9 г, 6.99 ммоля), железного порошка (6,4 г), концентрированной НСl (0,15 мл), воды (1,5 мл) и
95% этанола (35 мл) подвергали дефлегмации в течение 1,0
ч. Эту реакционную смесь фильтровали через целит, и растворитель удаляли при пониженном давлении. Остаток очищали с помощью колоночной
хроматографии на силикагеле, используя в качестве элюента
метиленхлорид: метанол (100:4), с получением 1,56 г (92%) соединения, указанного в заголовке.
1Н-ЯМР (500 МГц, CDCl3): δ 7.57 (s, 1H), 7.2 (t, 1H), 7.09 (d, 2H), 6.95 (s, 1H), 5.02 (s, 2H), 4.45 (bs, 2H), 2,4 (s, 6H), 2.25 (s, 3H).
Пример 2.22
Синтез 2-карбоэтокси-2,
6-диметил-8-(2,6-диметилбензилокси)имидазо[1,2-а] пиридина
Смесь 2-амино-3-(2,6-диметилбензилокси)-5-метилпиридина (1,0 г, 4,13 ммоля) и этил-2-хлорацетоацетата (0,79 г, 4,55 ммоля) в 20 мл
абсолютного этанола подвергали дефлегмации в течение 19 ч.
Добавляли еще этил-2-хлорацетоацетата (0,25 г). Эту реакционную смесь подвергали дефлегмации в течение дополнительных 23 ч. Растворитель
выпаривали под вакуумом, и остаток растворяли в метиленхлориде
и промывали водным NaHCO3. Органический слой высушивали и выпаривали при пониженном давлении. Сырой продукт очищали с помощью
колоночной хроматографии на силикагеле, используя в качестве
элюента метиленхлорид: этилацетат (100:10), с получением 0,68 г (47%) соединения, указанного в заголовке.
1 Н-ЯМР (500 МГц, CDCl3): δ 8.8 (s, 1H), 7,15 (t, 1H), 7.04 (d, 2H), 6.71 (s, 1H), 5.22 (s, 2H), 4.41 (q, 2H), 2.67 (s, 3H), 2.41 (s, 6H), 2.39 (s, 3H), 1.42 (t, 3H).
Пример 2.23
Синтез 2-карбоэтокси-8-(2,
6-диметил-4-фтор-бензиламино)-2-метилимидазо[1,2-a]пиридина
Перемешанную смесь (1,0 г, 4,7 ммоля) 8-амино-3-карбоэтокси-2-метилимидазо[1,2-а]
пиридина, (1,2 г, 5,7 ммоля) 2,
6-диметил-4-фторбензилбромида, (1,0 г, 7,5 ммоля) карбоната калия и (0,1 г) иодида натрия в 15 мл ацетонитрила подвергали дефлегмации в течение ночи. После выпаривания
растворителя при пониженном
давлении остаток растворяли в дихлорметане и промывали водой, органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток
очищали с помощью колоночной
хроматографии на силикагеле, элюент петролейный эфир (40-60): изопропиловый эфир 7: 3, с получением 1,2 г (75%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1,45 (t, 3Н), 2,35 (s, 6H), 2,65 (s, 3Н), 4,40 (q, 2H), 4,40 (q, 2H), 4,85 (t, 1H), 6,40 (d, 1H), 6,75 (d, 2H), 6,85 (t, 1H), 8,70 (d, 1H).
Пример 2.24
Синтез 2-этил-4-фтор-6-метилбензилбромида
Смесь 3-этил-1-фтор-3-метилбензола (1,1 г, 0,008 моля), параформальдегида (1,5 г, 0,05 моля), бромистоводородной
кислоты (4,1 мл, 0,017 моля) (4,1 М
в уксусной кислоте) и уксусной кислоты (2,5 мл) перемешивали при температуре окружающей среды в течение 40 ч. К этой смеси добавляли воду и петролейный эфир
(40-60), и органический слой отделяли,
промывали водой, высушивали над безводным сульфатом натрия и осторожно выпаривали при пониженном давлении. Получали желаемый продукт в виде желтого масла (1,3 г,
72%).
1 Н-ЯМР (300 МГц, CDCl3): δ 1.2 (t, 3Н), 2.35 (s, 3Н), 2.7 (q, 2H), 4.50 (s, 2H), 6.7-6.85 (m, 2H).
Пример 2.25
Синтез
3-карбоэтокси-2,
6-диметил-8-(2-этил-4-фтор-6-метилбензиламино)имидазо[1,2-а]пиридина
Перемешанную смесь (0,7 г, 3,0 ммоля) 8-амино-3-карбоэтокси-2,6-диметилимидазо[1,2-а] пиридина, (0,8 г, 3,
5 ммоля)
2-этил-4-фтор-6-метилбензилбромида, (0,7 г, 4,8 ммоля) карбоната калия и (0,1 г) иодида натрия в 15 мл ацетонитрила подвергали дефлегмации в течение ночи. После выпаривания растворителя при
пониженном
давлении остаток растворяли в дихлорметане и промывали водой, органический слой отделяли, высушивали над сульфатом натрия и выпаривали при пониженном давлении. Остаток очищали с помощью
колоночной
хроматографии на силикагеле, элюент петролейный эфир (40-60): изопропиловый эфир 7:3, с получением 0,4 г (35%) соединения, указанного в заголовке.
1Н-ЯМР (300
МГц, CDCl3): δ 1.25 (t, 3Н), 1.45 (t, 3Н), 2.4 (s, 6H), 2.65 (s, 3Н), 2.75 (q, 2H), 4.3 (d, 2H), 4.4 (q, 2H), 4.75 (bs, 1H), 63 (s, 1H), 6.75-6.85 (m, 2H), 8.5 (s, 1H)
Пример 2.26
Синтез 3-карбоэтокси-8-(2-этил-6-метилбензилокси)-2-метилимидазо[1,2-а] пиридина
Перемешанную смесь 3-карбоэтокси-8-гидрокси-2-метилимидазо[1,2-а]пиридина (0,92 г, 4,2
ммоля), (0,7 г, 4,2
ммоля) 2-этил-6-метилбензилхлорида (0,7 г, 4,2 ммоля), карбоната натрия (1,0 г, 9,4 ммоля) и каталитическое количество иодида калия в ацетонитриле (40 мл) подвергали дефлегмации в
течение 4 ч. После
фильтрования и выпаривания растворителя при пониженном давлении остаток очищали с помощью колоночной хроматографии на силикагеле, используя в качестве элюента метиленхлорид:
этилацетат, с получением 1,
0 г (68%) соединения, указанного в заголовке.
1Н-ЯМР (300 МГц, CDCl3): δ 1.2 (t, 3H), 1.4 (t, 3Н), 2.4 (s, 3H), 2.65 (s, 3H), 2, 75 (q, 2H), 4.4 (q, 2H), 4.40 (q, 2H), 5.25 (s, 2H), 6.85-6.9 (m, 2H), 7.05-7.25 (m, 3H), 8.95 (dd, 1H).
Пример 2.27
Синтез 3-этил-1-фтор-5-метилбензола
Метиллитий
(40 мл, 64 ммоля)
добавляли по каплям при 0oС к суспензии иодида меди (I) (6,42 г, 33,6 ммоля) в диэтиловом эфире (20 мл). После перемешивания при 0oС в течение 30 мин
прозрачный бесцветный
гомогенный раствор купрата, в который добавляли 3-бромметил-1-фтор-5-метилбензол (5,15 г, 25,4 ммоля) в 10 мл диэтилового эфира, охлаждали до -78oС. Температуре
давали медленно повыситься.
Реакцию гасили при -50oС NH4Сl/NН3-буфером (50 мл). Экстракция диэтиловым эфиром (3х50 мл), рассолом (1х100 мл). Органический слой
высушивали над MgSO4,
фильтровали и удаляли растворители с получением 3,3 г (94%) соединения, указанного в заголовке.
1Н-ЯМР (500 МГц, CDCl3): δ 1 22 (t, 3H), 2.32 (s, 3H), 2.60 (q, 2H), 6,69 (d, 2H), 6.78 (s, 1H).
БИОЛОГИЧЕСКИЕ ТЕСТЫ
1. Эксперименты in vitro
Ингибирование секреции кислоты в изолированных
желудочных железах кролика
Ингибирующее воздействие на секрецию кислоты in vitro в изолированных желудочных железах измеряли, как описано Berglindth et al. (1976) Acta Physiol. Scand. 97,
401-414.
Определение
активности Н+, К+-АТФазы
Получение желудочных мембранных везикул: желудочные мембранные везикулы, содержащие Н+, К+-АТФазу, получали из свиных
желудков, как описано ранее Saccomani et al. (1977) Biochim. Biophys. Acta 465, 311-330.
Проницаемые везикулы: мембранную фракцию разбавляли 1 мМ PlPES/Трис, рН 7,4 до получения 1% концентрации сахарозы, гомогенизировали и центрифугировали при 100000xg в течение 2 ч. Полученный осадок суспендировали в воде и дважды лиофилизировали.
Определение активности Н+, K+-АТФазы: проницаемые мембранные везикулы (2,5-5 мкг) инкубировали в течение 15 мин при 37oС в буфере 18 мМ PlPES/Трис, рН 7,4, содержащем 2 мМ MgCl2, 10 мМ КСl и 2 мМ АТФ. Активность АТФазы оценивали по высвобождению неорганического фосфата из АТФ, как описано LeBel et al. (1978) Anal. Biochem. 85, 86-89.
2. Эксперименты in vivo
Ингибирующее воздействие на секрецию кислоты у самок крыс
Используют самок крыс линии Sprague-Dawley. Их снабжают канюлированными фистулами в
желудке (в полости желудка) и в верхней части
двенадцатиперстной кишки для сбора желудочных секретов и введения тестируемых веществ, соответственно. Перед началом тестирования дают восстановительный
период 14 суток после операции.
Перед секреторными тестами животных лишают пищи, но не лишают воды в течение 20 ч. Желудок повторно промывают проточной водой (+37oС) через желудочную канюлю и вводят подкожно 6 мл раствора Рингера глюкозы (Ringer-Glucose). Секрецию кислоты стимулируют инфузией пентагастрина (pentagastrin) и карбахола (carbachol) (20 и 110 нмолей/кг•ч соответственно) в течение 2,5-4 ч (1, 2 мл/ч, подкожно), и в течение этого времени собирают желудочные секреты по фракциям через каждые 30 мин. Тестируемые вещества или растворитель дают либо через 60 мин после начала стимуляции (внутривенное и интрадуоденальное дозирование, 1 мл/кг), либо за 2 ч до начала стимуляции (пероральное дозирование, 5 мл/кг, желудочная канюля закрыта). Временной интервал между дозированием и стимуляцией можно увеличить, чтобы изучить продолжительность действия. Образцы желудочного сока титруют NaOH, 0,1 М до рН 7,0, и выход кислоты вычисляют как произведение объема и концентрации титранта.
Дальнейшие вычисления основаны на усредненных ответах в группе из 4-6 крыс В случае введения в течение стимуляции: выход кислоты в течение периодов после введения тестируемого вещества или носителя выражают в виде фракционных ответов, принимая за 1,0 выход кислоты за 30-минутный период, предшествующий введению. Ингибирование в процентах вычисляют исходя из фракционных ответов, вызванных тестируемым соединением и носителем. В случае введения перед стимуляцией: ингибирование в процентах вычисляют непосредственно по выходу кислоты, регистрируемому после введения тестируемого соединения и носителя.
Биологическая доступность у крысы
Используют взрослых крыс линии Sprague-Dawley. От одних до трех
суток перед экспериментами всех
крыс готовят путем канюлирования левой сонной артерии под анестезией. Крыс, используемых для внутривенных экспериментов, также канюлируют в яремную вену (Popovic
(1960) J. Appl. Physiol. 15, 727-728).
Канюли выводят на поверхность в задней части шеи.
Образцы крови (0,1-0,4 г) берут из сонной артерии в интервалах до 5,5 ч после получения дозы. Образцы замораживают до анализа тестируемого соединения.
Биологическую доступность оценивают путем вычисления соотношения между площадью под кривой концентрации в крови/плазме (ППК), полученной для крысы и собаки, после (1) интрадуоденального или перорального введения и (2) внутривенного введения соответственно.
Площадь под кривой зависимости концентрации от времени, ППК определяют с помощью логарифмического/линейного правила трапеции и экстраполируют до бесконечности путем деления последней определенной концентрации в крови на константу скорости элиминирования в терминальной фазе. Системную биологическую доступность (F%) после интрадуоденального или перорального введения вычисляют как F (%) = (ППК (для перорального или интрадуоденального введения)/ ППК (для внутривенного введения)) х 100.
Ингибирование секреции желудочной кислоты и биологическая доступность у собаки, находящейся в сознании
Используют собак
лабрадор, ретрайвер или гончих
любого пола. Их снабжают дуоденальной фистулой для введения тестируемых соединений или носителя и канюлированной желудочной фистулой или карманом Хейденхейма
(Heidenhaim) для сбора желудочного
секрета.
Перед секреторными тестами животные голодают в течение примерно 18 ч, но имеют свободный доступ к воде. Секрецию желудочной кислоты стимулируют инфузией гистамина дигидрохлорида (12 мл/ч) до 6,5 ч в дозе, дающей примерно 80% индивидуального максимального секреторного ответа, и желудочный сок собирают последовательными фракциями через 30 мин. Тестируемое вещество или носитель вводят перорально, интрадуоденально или внутривенно через 1 или 1,5 ч после начала инфузии гистамина в объеме 0,5 мл/кг массы тела. Следует отметить, что в случае перорального введения собаке с карманом Хейденхейма (Heidenham) тестируемое соединение вводят в главный желудок, секретирующий кислоту.
Кислотность образцов желудочного сока определяют с помощью титрования до рН 7,0 и вычисляют выход кислоты. Выход кислоты за периоды сбора после введения тестируемого вещества или носителя выражают как фракционные ответы, принимая выход кислоты во фракции, предшествующей введению, за 1,0. Ингибирование в процентах вычисляют исходя из фракционных ответов, вызванных тестируемым соединением или носителем.
Образцы крови для анализов концентрации тестируемого соединения в плазме берут в интервалах до 4 ч после дозирования. Плазму отделяют и замораживают в течение 30 мин после сбора, а позже анализируют. Системную биологическую доступность (F%) после перорального или интрадуоденалього введения вычисляют, как описано выше на модели крысы.
Результаты представлены в табл.2-4.
Примеры (А, Б и С)
фармацевтических препаратов, содержащих соединения по изобретению
А 1. Сироп, содержащий 1 мас. % активного соединения
Активное соединение - 1,0 г
Сахар, порошок - 30,0 г
Сахарин - 0,6 г
Глицерин - 5,0 г
Корригент - 0,05 г
Этанол 96%-ный - 5,0 г
Дистиллированная вода, ск. треб. до конечного
объема 100 мл
Сахар и
сахарин растворяют в 60 г теплой воды. После охлаждения этого раствора в нем растворяют активное соединение и затем добавляют раствор глицерина и корригента в этаноле.
Смесь разводят водой до
конечного объема 100 мл.
B 2. Таблетки с энтеросолюбильным покрытием, содержащие 20 мг активного соединения
I. Активное соединение - 200 г
Лактоза - 700 г
Метилцеллюлоза - 6 г
Поливинилпирролидон поперечносшитый - 50 г
Стеарат магния - 15 г
Карбонат натрия - 6 г
Дистиллированная вода, ск.
треб.
II.
Ацетат-фталат целлюлозы - 200 г
Цетиловый спирт - 15 г
изопропанол - 2000 г
Метиленхлорид - 2000 г
I. Активное соединение смешивают с
лактозой и поперечносшитым
поливинилпирролидоном и гранулируют с водным раствором метилцеллюлозы и карбоната натрия. Влажную массу продавливают через сито и сушат гранулят в печи. После сушки
гранулят смешивают со стеаратом
магния. Сухую смесь прессуют с получением таблеточных ядер (10000 таблеток), каждое из которых содержит 20 мг активного соединения, используя таблеточную машину с
пуансонами 6 мм в диаметре.
II. Таблетки, полученные на стадии I, опрыскивают раствором ацетатфталата целлюлозы и цетилового спирта в изопропаноле/метиленхлориде в установке для нанесения покрытия Accela Cota®, Manesty. В результате получают таблетки с массой 110 мг.
С 3. Раствор для внутривенного введения, содержащий 4 мг активного
соединения
активное соединение - 4
г
полиэтиленгликоль 400 для инъекций - 400 г
Динатрия гидрофосфат ск. треб. Стерильная вода до конечного объема 1000 мл
Активное
соединение растворяют в полиэтиленгликоле
400 и добавляют 550 мл воды. рН раствора подводят до 7,4 путем добавления водного раствора динатрия гидрокарбоната, а затем добавляют воду до конечного
объема 1000 мл. Раствор фильтруют через фильтр
22 мкм и немедленно разливают в стерильные ампулы на 10 мл. Ампулы запаивают.
В качестве активного соединения в вышеуказанных препаратах используют соединения по изобретению.
Реферат
Изобретение относится к новым производным имидазо[1,2-а] пиридина ф-лы I или его фармацевтически приемлемым солям, где R1 представляет СН3 или СН2ОН, R2 и R3 - низший алкил, R4 - Н или галоген, R5 - Н, галоген или низший алкил, Х - представляет NH или О. Соединения формулы I ингибируют секрецию желудочной кислоты и могут быть использованы для лечения желудочно-кишечных воспалительных заболеваний. 14 с. и 21 з.п. ф-лы, 4 табл.
Формула

или их фармацевтически приемлемая соль,
где R1 представляет собой СН3 или СН2ОН;
R2 представляет собой низший алкил;
R3 представляет собой низший алкил;
R4 представляет собой Н или галоген;
R5 представляет собой Н, галоген или низший алкил;
Х представляет собой NH или О.

где X1 представляет собой NH2 или ОН, a R1 и R5 являются такими, как определены для формулы I, с соединениями общей формулы III

где R2, R3 и R4 являются такими, как определены для формулы I, a Y представляет собой отщепляемую группу, в инертном растворителе с основанием или без основания, с образованием соединений формулы I.
(а) подвергают взаимодействию соединение формулы IV
где R1 и R5 являются такими, как определены для формулы I, с соединениями формулы V

где R2, R3 и R4 являются такими, как определены для формулы I, предпочтительно в присутствии кислоты Льюиса, в инертном растворителе, с образованием соединений формулы VI

где R1, R2, R3, R4, R5 являются такими, как определены для формулы I;
(б) восстанавливают соединения формулы VI в инертном растворителе при стандартных условиях до соединений общей формулы I, где Х представляет собой NH.
(а) подвергают взаимодействию соединение формулы VII

где X1 представляет собой NH2 или ОН;
R5 является таким, как определен для формулы I,
с соединениями общей формулы III
где R2, R3 и R4 являются такими, как определены для формулы I;
Y представляет собой отщепляемую группу,
в инертном растворителе, с основанием или без основания, с образованием соединений формулы VIII

где R2, R3, R4, R5 и Х являются такими, как определены для формулы I;
(б) восстанавливают соединения формулы VIII в инертном растворителе при стандартных условиях до соединений общей формулы I, где R1 представляет собой СН2 ОН.
(а) подвергают взаимодействию соединение общей формулы IX

где R5 является таким, как определен для формулы I, с соединениями формулы V

где R2, R3 и R4 являются такими, как определены для формулы I,
предпочтительно в присутствии кислоты Льюиса в инертном растворителе с образованием соединений общей формулы Х

где R2, R3, R4 и R5 являются такими, как определены для формулы I;
(б) восстанавливают соединения формулы Х в инертном растворителе при стандартных условиях до соединений формулы XI

где R2, R3, R4 и R5 являются такими, как определены для формулы I;
(в) восстанавливают соединения общей формулы XI в инертном растворителе при стандартных условиях до соединений общей формулы I, где Х представляет собой NH, а R1 представляет собой СН2 ОН.
(а) подвергают взаимодействию соединение общей формулы XII
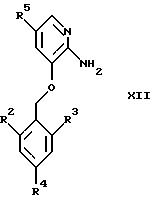
где R2, R3, R4 и R5 являются такими, как определены для формулы I,
с соединениями общей формулы
СН3СОСН(Z)СООСН2СН3,
где Z представляет собой Вr или С1, в инертном растворителе, с образованием соединений общей формулы XIII

где R2, R3, R4 и R5 являются такими, как определены для формулы I;
(б) восстанавливают соединения общей формулы XIII в инертном растворителе при стандартных условиях до соединений общей формулы I, где R1 представляет собой СН2ОН, а Х представляет собой О.
Документы, цитированные в отчёте о поиске
Производные амида 4-оксо-4н-пиридо(1, 2-a)-пиримидин-3-карбоновой кислоты или их кислотно-аддитивные соли
Комментарии