Бензопираны и бензоксепины, содержащие их фармацевтические композиции, способ их получения - RU2228333C2
Код документа: RU2228333C2
Описание
Настоящее изобретение относится к бензопиранам и бензоксепинам, которые можно применять для лечения дислипидемии, атеросклероза и диабета, к содержащим эти вещества фармацевтическим препаратам и к способу изготовления названных соединений.
Кроме того, изобретение относится к применению этих соединений для изготовления лекарственных препаратов для лечения дислипидемии, атеросклероза и диабета.
В большинстве стран одними из самых распространенных заболеваний являются сердечно-сосудистые, они составляют основную причину смертности населения. От этих заболеваний страдают примерно треть мужчин, не достигших 60 лет; женщины этой возрастной категории подвержены подобным заболеваниям примерно в 10 меньше, но с возрастом риск возрастает, и после 65 лет женщины становятся уязвимыми для сердечно-сосудистых болезней в той же мере, что и мужчины. Такие сердечно-сосудистые заболевания, как коронарная болезнь, инсульты, рестеноз и заболевания периферической сосудистой системы, остаются основной причиной смертности и инвалидности по всему миру.
В то время как образ жизни и неправильный режим питания могут ускорить развитие сердечно-сосудистых заболеваний, генетическая предрасположенность, которая обуславливает дислипидемию, является важным фактором в развитии инсультов и смертей.
По всей вероятности, развитие атеросклероза связано, главным образом, с дислипидемией, что означает аномальные уровни липопротеинов в плазме крови. Эта дисфункция с особой очевидностью проявляется при коронарной болезни, диабетах и ожирении.
Традиционная концепция, объясняющая развитие атеросклероза, основывалась, главным образом, на метаболизме холестерина и метаболизме триглицеридов.
Тем не менее, после опубликования исследований Randle с сотрудниками (Lancet, 1963, 785-789), была предложена новая концепция; цикл глюкоза-жирная кислота, или цикл Randle, который описывает правила установления равновесия между метаболизмом липидов в терминах триглицеридов и холестерина, и окисления глюкозы. Изобретатели разработали новую программу, имея целью
обнаружить новые соединения, которые могли бы одновременно воздействовать как на метаболизм липидов, так и на метаболизм глюкозы.
Фибраты - хорошо известные терапевтические агенты с механизмом действия через "активированные рецепторы размножающейся пероксисомы". Именно эти рецепторы ответственны за метаболизм липидов в организме (изоформ PPARα). В последние десять лет тиазолидиндионы описывались как мощные гипогликемические агенты в организме животных и человека. В литературе имеются сообщения, что тиазолидиндионы - мощные селективные активаторы другого изоформа PPAR: различных PPARγ (Lehmann et al., J. Biol. Chem., 1995, 270, 12953-12956).
Изобретатели открыли новый класс соединений, которые являются мощными активаторами изоформов PPARα и PPARγ. Благодаря своей активности эти соединения проявляют значительное гиполипидемическое и гипогликемическое действие.
По изобретению соединения описываются представленной ниже формулой (I)

в которой:
Х представляет собой О или S;
А есть либо двухвалентный радикал -(СН2)s -CO-(CH2)t-, либо двухвалентный радикал -(CH2)s-CR3R4-(CH2)t-, в которых
s=t=0 или одно из s и t имеет значение 0, а остальные равны 1;
R4 представляет собой атом водорода или алкильную группу (C1-C15);
R1 и R2 независимо представляют собой определенные ниже цепочки Z; атом водорода; алкильную группу (C1-C18); алкенильную группу (C2-C18); алкинильную группу (C2-C18); арильную группу (С6-С10), которая может быть замещенной атомом галогена, алкильной группой (C1-C5), которая может быть галогенизированной, или алкоксигруппой (C1-C5), которая может быть галогенизированной; или моно- либо бициклическую гетероарильную группу (C4-C12),
включающую один или более гетероатомов, выбранных из О, N и S, которая может быть замещенной атомом галогена, алкильной группой (C1-C5), которая может быть галогенизированной или алкоксигруппой (C1-C5), которая может быть галогенизированной;
R3 может обозначать любое из соединений, описанных выше как R1 и R2, за исключением цепочки Z; кроме того,
R3 и R4 в сочетании образуют алкиленовую цепочку (C2-C6), которая может быть замещенной атомом галогена или алкоксигруппой (C1-C5), которая может быть галогенизированной;
R выбрано из атомов галогенов; цианогруппы, нитрогруппы; карбоксильной группы, алкоксикарбонильной группы (C1-C18), которая может быть галогенизированной; группы Ra-CO-NH- или RaRbN-CO- [в которой Ra и Rb независимо представляют собой алкильную группу (C1-C18), которая может быть галогенизированной; атом водорода; (С6-С10) арил, или (С6-С10)арил(C1-C5)алкил (где арильные группы могут быть замещены атомами галогенов, алкильной группой (C1-C5), которая может быть галогенизированной, или алкоксигруппой (C1-C5), которая может быть галогенизированной); (С3-С12)циклоалкила, который может быть замещенным атомом галогена, алкильной группой (C1-C5), которая может быть галогенизированной, или алкоксигруппой (C1-C5), которая может быть галогенизированной)]; алкильной группы (C1-C18), которая может быть галогенизированной; алкоксигруппы (C1-C18), которая может быть галогенизированной и (С6-С10) арила, (С6-С10) арил (C1-С5)алкила, (С6-С10) алкоксила, (C3-C12) циклоалкила, (С3-C12) циклоалкенила, циклоалкоксигруппы (С3-C12), циклоалкенилоксигруппы (С3-С12), или (С6-С10)арилоксикарбонила, в которых арильные, циклоалкильные и циклоалкенильные группы могут быть замещены атомом галогена, алкильной группой (C1-C5), которая может быть галогенизированной, или алкоксигруппы (C1 -C5), которая может быть галогенизированной;
р может принимать значения 0, 1, 2, 3 или 4; Z представляет собой радикал

где n принимает значения 1 или 2;
группы R’ независимо представляют атом водорода; алкильную группу (C1-C5); арильную группу (С6-С10), которая может быть замещена атомом галогена, алкильной группой (C1-C5), которая может быть галогенизированной, или алкоксигруппой (C1-C5), которая может быть галогенизированной; или моно- либо бициклическую гетероарильную группу (C4-C12), включающую один или более гетероатомов из набора О, N и S, которая может быть замещена атомом галогена, алкильной группой (C1-C5), которая может быть галогенизированной, или алкоксигруппой (C1 -C5), которая может быть галогенизированной;
Y может представлять -ОН; алкоксигруппу (C1-C5); или группу -NRcRd (в которой Rc и Rd независимо представляют атом водорода; алкил (C1-C5); циклоалкил (С3-С8), который может быть замещен атомом галогена, алкильной группой (C1-C5), которая может быть галогенизированной, или алкоксигруппой (C1-C5), которая может быть галогенизированной; арильную группу (С6 -С10), которая может быть замещенной атомом галогена, алкилом (C1-C5), который может быть галогенизированным, или алкоксигруппой (C1-C5), которая может быть галогенизированной); понятно, что по отдельности каждый из радикалов R1 и R2представляет собой цепочку Z.
Предметом изобретения являются также соли названных соединений с кислотами или основаниями - в зависимости от присутствующих в молекуле функциональных групп, - пригодные для использования в фармацевтике.
Если соединение, которое описывается формулой (I), включает кислотную функциональную группу, например - функциональную карбоксигруппу, то последняя может образовывать соль с неорганическим или органическим основанием.
Следует отметить, как пример солей с органическими или неорганическими основаниями, соли, образованные с металлами, в особенности - щелочными, щелочноземельными и переходными металлами (такими, как натрий, калий, кальций, магний или алюминий), или с основаниями, такими как аммиак, или вторичные либо четвертичные амины (например, диэтиламин, триэтиламин, пиперидин, пиперазин или морфолин), или с основными аминокислотами, либо с озаминами (например, меглумин), либо с аминоспиртами (такими, как 3-аминобутанол и 2-аминоэтанол).
Если соединение с формулой (I) включает основную функциональную группу, например - атом азота, то названная группа может образовывать соль с органической или неорганической кислотой.
В качестве примеров солей с органическими или неорганическими кислотами, следует указать гидрохлоридные, гидробромидные, сульфатные, гидросульфатные, дигидрофосфатные, малеатные, фумаратные, 2-нафталенсульфонатные и пара-толуинсульфонатные.
Предметом изобретения являются также соли, которые дают возможность надлежащим образом производить разделение или кристаллизацию соединений с формулой (I), такие, как соли пикриновой кислоты, щавелевой кислоты, или оптически активной кислоты, например винной, дибензоилвинной, манделиновой или камфоросульфонильной кислоты.
Формула (I) охватывает все типы геометрических изомеров и стереоизомеров соединений с формулой (I).
В настоящем изобретении термин "алкил" определяет линейный или разветвленный углеводородзамещенный радикал, такой как метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, рентил, гексил, гептил, октил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил или октадецил.
Если алкильная группа является замещенной одним или более атомов галогенов, то она представляет собой преимущественно перфторалкил, и в частности - пентафторэтил или трифторметил.
Термин "алкоксигруппа" подразумевает определенную выше алкильную группу, связанную с атомом кислорода, например метокси-, этокси-, изопропилокси-, бутокси- и гексилоксирадикалы.
Термин "алкиленовая группа" подразумевает линейные или разветвленные алкиленовые группы, или двухвалентные радикалы, которые представляют собой линейные или разветвленные двухвалентные алкильные цепочки.
Термин "циклоалкил" определяет насыщенные углеводородзамещенные группы, которые могут быть моно- или
полициклическими и содержать от 3 до 12 атомов углерода, преимущественно - от 3 до 8. Особо предпочтительными являются моноциклические циклоалкильные группы, такие, как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил и циклододецил.
По данному изобретению, термин "циклоалкенил" подразумевает циклоалкильную группу, в которой имеется не менее одной двойной связи.
Термин "галоген" подразумевает атом фтора, хлора, брома или иода.
Термин "арил" подразумевает моно- или бициклическую ароматическую углеводородсодержащую группу, включающую от 6 до 10 атомов углерода, например - фенил или нафтил.
Термин "моно- или бициклический гетероарил" определяет моноциклические или бициклические ароматические группы, которые содержат один или более эндомостиковые гетероатомы, например - фурил, тиенил, пирролил, оксазолил, изоксазолил, тиазолил, изотиазолил, имидазолил, пиразолил, оксадиазолил, триазолил, тиадиазолил, пиридинил, пиридазинил, пиразинил, триазинил, индолизинил, индолил, изоиндолил, бензофурил, бензотиенил, индазолил, бензимидазолил, бензотиазолил, пуринил, хинолил, хинолизинил, изохинолил, циннолинил, фталазинил, хиназолинил, хиноксалинил, птеридинил бензоксепинил.
Предпочтительно гетероарилы должны включать от 4 до 10 углеродных атомов и 1 или 2 гетероатома.
Алкенильные и алкинильные группы могут иметь более чем одну ненасыщенную связь. Алкенильные группы содержат ненасыщенные связи этиленового типа, а алкинильные - ацетиленового типа.
Группы (С6-С10) арил, (С3-С8) циклоалкил, гетероарил и циклоалкенил могут быть замещенными. Выражение "могут быть замещенными атомом галогена, группой (C1-C5), которая может быть галогенизированной, или (C1-C5)алкоксигруппой, которая может быть галогенизированной" означает, что названные арильные, циклоалкильные, гетероарильные и циклоалкенильные
группы могут быть замещенными одним или более заместителей из нижеприведенных:
атомы галогенов;
алкильные группы, которые могут быть замещенными одним или более атомов галогенов, и
алкоксигруппы, которые могут быть замещенными одним или более атомов галогенов.
Таким же образом, если замещенными являются алкиленовые цепочки, то они включают один или более одинаковых или различных заместителей из следующего набора: атомы галогенов и алкоксигруппы, которые могут быть галогенизированными.
В контексте изобретения выражение "могут быть галогенизированными" означает: могут быть замещенными одним или более атомов галогенов.
В контексте настоящего изобретения термин "бензоксепин" используется для обозначения бензо[b]оксепиновой структуры с формулой

По данному изобретению, предпочтительными являются соединения, в которых А представляет собой радикал
-(СН2)s-СR3R4-(CH2)t-
где s, t, r3 и R4 определены выше для формулы (I).
Другая предпочтительная группа соединений, соответствующих формуле (I), состоит из:
- соединений, в которых:
Х представляет собой О;
А представляет собой -СR3R4- или -CH2-CR3R4-, в которых незамещенная метиленовая группа образует связь с X;
R1 и R2 независимо представляют собой Z; H; (C1-C15)алкил; (C1-C15) алкенил или фенил, произвольно замещенный (C1-C5)алкилом, алкоксигруппой (C1-C5), атомом галогена или -СF3;
R3 принимает одно из значений, представленных выше для R1 и R2, за исключением Z;
R выбрано из следующего набора: (C1-С9)алкил; (C1-C5)алкоксигруппа; фенил или фенилкарбонил, которые могут быть замещенными атомом галогена, (C1-С5) алкилом, (C1-C5)алкоксигруппой, -СF3 или -ОСF3; атомом галогена; -СF3и -ОСF3;
Z представляет собой радикал со следующей формулой:

где n равно 1;
R’ представляет собой (C1-C5) алкил.
Из этих соединений предпочтительными являются те, в которых:
Х представляет собой О;
А представляет собой -СR3R4-;
Z представляет собой

- или альтернативный вариант, в котором:
Х есть О;
А представляет собой -СН2-СR3R4-, в котором незамещенная метиленовая группа образует связь с X;
R1 и R2 независимо представляют собой Z, атом водорода, или (C1-C5) алкил;
R3 принимает одно из значений, определенных выше для R1 и R2, за исключением Z;
Z представляет собой

R’ есть метил или фенил. Предпочтительно, чтобы Y представлял собой:
-ОН
-(C1-C5)алкоксигруппу и
-NRcRd, где Rc и Rd определены выше для формулы (I).
Особо предпочтительно, чтобы Y представлял собой -ОН или -(C1-C5)-алкоксигруппу.
Аналогично, предпочтительно, чтобы p принимало значения 0, 1 или 2.
По особенно предпочтительному варианту осуществления данного изобретения, в соединениях из групп, определенных выше как предпочтительные, p и Y принимают одно из указанных значений.
В качестве примера преимущественных соединений можно указать следующие:
- (2Е, 4Е)-5-(2-пентил-2Н-1-бензопиран-3-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Z, 4Е)-5-(2-пентил-2Н-1-бензопиран-3-ил)-3-метил-пента-2, 4-диеновая кислота;
- (2Е, 4Е)-5-(2,2-диметил-6-метокси-2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(2,2-диметил-2Н-1-бензопиран-3-ил)-3-метилпента-2, 4-диеновая кислота;
- (2Z, 4Е)-5-(2,2-диметил-2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е,4Е)-5-[2-(нон-6-енил)-2Н-1-бензопиран-3-ил]-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(4-фенил-2Н-1-бензопиран-3-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(6-нонил-2Н-1-бензопиран-3-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(6-фенил-2Н-1-бензопиран-3-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(2-нонил-2Н-1-бензопиран-3-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(4-метил-2Н-1-бензопиран-3-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Z, 4Е)-5-(2Н-1-бензопиран-3-ил)-3-3-метил-пента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(2-ундеканил-2Н-1-бензопиран-3-ил)-3-метилпента-2, 4-диеновая кислота;
- (2Е, 4Е)-5-(2-фенил-2Н-1-бензопиран-3-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(5-метил-2,3-дигидробензоксепин-4-ил)-3-метил-пента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-метокси-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота; и [sic]
- (2Е, 4Е)-5-(2,3-дигидробензоксепин-4-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-метокси-2,3-дигидробензоксепин-5-ил)-3-фенилпента-2,4-диеновая кислота;
- (2Z, 4Е)-5-(3,3-диметил-7-метокси-2, 3-дигидробензоксепин-5-ил)-3-фенилпента-2,4-диеновая кислота;
- (2Z, 4Е)-5-(3,3-диметил-7-метокси-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7,8-диметокси-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-2,3-дигидро-7-(пара-хлоробензоил) бензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-хлоро-2, 3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7,8-дихлоро-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-бромо-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-фторо-8-хлоро-2,3-дигидробензоксепин-5-ил)-3-метилпента-2, 4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-фторо-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-трифторметил-2, 3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-7-фенил-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3,7-триметил-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(3,3-диметил-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
- (2Е, 4Е)-5-(9-метокси-3,3-диметил-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота;
их эфиры, допустимые для применения в фармацевтике, например этиловые эфиры.
В патенте FR 2698873 заявлены соединения с формулой

которые являются сильными активаторами калиевых каналов клеточных мембран.
По названному документу ни один из заместителей от R1 до R7 не представляет собой цепочку Z, как это определено по настоящему изобретению.
В патенте US 5391569 бензопираны с формулой

предложено применять для лечения остеопороза и воспалений. Эти соединения отличаются от заявленных в настоящем изобретении тем, что алкенильные цепочки имеют три двойных связи, в то время как по настоящему изобретению цепочки Z имеют либо две, либо четыре двойных связи.
Необходимо отметить, что гиполипидемическая и гипогликемическая активность соединений, заявленных в настоящем изобретении, не имеет отношения к активности соединений, заявленных в патентах US 5391569 и FR 2698273.
Соединения с формулой (I) могут быть получены при использовании одного из двух описанных ниже способов А или В, которые составляют второй предмет настоящего изобретения.
Способ А позволяет синтезировать соединения с формулой (I), в которой n равно 1.
Этот способ включает стадии, состоящие из: (al) приготовление илида
- либо в реакции основания с фосфонатом, имеющим формулу
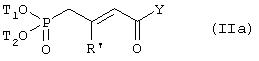
в которой R’ определено в пункте 1 формулы изобретения;
T1 и Т2 независимо представляют собой (C1-C5)алкил и Y есть (C1-C5)алкоксигруппа,
- или в реакции основания с фосфониевой солью, которая описывается формулой (IIb)

в которой R’ определено в п.1 формулы изобретения;
Т3, Т4 и Т5 независимо представляют собой (C1-C5)алкил или (С6-С10)арил, который может быть замещенным (C1-C5)алкилом;
Y представляет (C1-C5)алкоксигруппу и
hal есть атом галогена;
(bl) проведения реакции илида, полученного на стадии (al), с альдегидом, который описывается формулой

в которой
R, p, Х и А имеют значения, определенные в п.1 формулы изобретения;
каждый из радикалов R’1 и R’2 представляет собой -СНО, и остальные принимают одно из значений, указанных в п.1 формулы изобретения для R1 и R2, за исключением цепочки Z, такие, чтобы в результате реакции получалось соединение с формулой (I), для случая, когда n равно 1 и Y есть алкоксигруппа (C1-C5);
(cl) если это целесообразно, то эфир, полученный на описанной выше стадии (bl), в кислотной или основной среде превращают в соответствующую карбоновую кислоту, которая описывается формулой (I), если Y представляет собой ОН;
(dl) при необходимости, осуществляют реакцию функциональной группы карбоновой кислоты, соответствующей формуле (1), которая получена на стадии с амином, имеющим формулу HNRcRd, в которой Rc и Rd определены в п.1 формулы
изобретения, необязательно после активирования карбоксильной функциональной группы, так, чтобы приготовить соответствующее соединение с формулой (I), в которой Y представляет собой -NRcRd.
На стадии (b1) используется либо реакция Wittig, либо Horner-Emmons или Wadsworth-Emmons.
В результате получается реактив илид.
Если илид получают из фосфониевой соли (соединение IIb), следует использовать реакцию Wittig.
Если илид получают из фосфоната (соединение IIа), надо использовать реакцию Horner-Emmons или Wadsworth-Emmons.
На стадии a1 илид получают в реакции основания либо с соединением IIа, либо с соединением IIb. Применяемое основание должно быть очень сильным, чтобы переместить протон с позиции α к фосфонату.
Основание обычно выбирают из гидридов щелочных металлов, карбонатов щелочных металлов, амидов щелочных металлов, (C1-С10)алкила лития и металлоалкоголятов щелочных металлов.
Следует отметить, что в представленном ниже примере, описано применение гидрида натрия, карбоната калия, н-бутила лития, трет-бутоксида калия, амида лития или амида натрия.
В контексте данного изобретения предпочтительными основаниями являются гидрид натрия и трет-бутоксид калия.
Реакцию основания с соединениями (IIа) или (IIb) проводят в растворе, предпочтительно в апротонном растворителе, и особо предпочтительно - в растворителе, в котором растворяются фосфонат (IIа) или фосфониевая соль (IIb) соответственно.
Подходящими являются апротонные растворители; в качестве примеров, которыми круг возможных растворителей не ограничивается, можно указать ароматические углеводороды (например - бензол и толуол), эфиры (например - диэтиловый эфир, диоксан и тетрагидрофуран) и их смеси.
Выбор растворителя зависит, главным образом, от типа илида (соединения IIа или IIb).
По главному предмету изобретения, стадия (b2) реакции альдегида с илидом проводится при добавлении альдегида (III) к исходной реакционной смеси, полученной на стадии (аl), без выделения промежуточного илида.
Таким образом, желательно, чтобы применяемый на стадии (аl) растворитель был способен растворять также и альдегид (III). Тем не менее, другой предмет изобретения состоит в добавлении раствора альдегида (III) в растворителе к исходной реакционной смеси, полученной на стадии (al). При этих условиях нет необходимости выбирать при проведении стадии (al) растворитель, в котором будет растворяться также и соединение (III).
Температура, при которой осуществляют стадию (al), зависит от кислотности соединений (IIа) или (IIb) соответственно, это зависит от того, какая энергия необходима для переноса α-протона к фосфорану, не говоря уже о том, что на выбор температуры проведения реакции непосредственно влияет тип применяемого основания. Таким образом, чем сильнее основание, тем ниже температура проведения реакции. Если в качестве основания используется н-бутил лития, то желательно, чтобы температура находилась в диапазоне от -80 до -40°С, предпочтительно - между -80 и -70°С. В этом случае растворитель следует выбирать таким образом, чтобы он позволял удовлетворить подобным жестким условиям проведения реакции: этим требованиям очень хорошо удовлетворяют эфиры.
Если основание представляет собой металлоалкоголят щелочного металла, то реакцию можно проводить при температурах в диапазоне от 10 до 100°С. Более того, если это основание вступает в реакцию с фосфонатом (IIа), то оптимально поддерживать температуру в диапазоне от 15 до 70°С.
Если применяемое основание представляет собой гидрид щелочного металла, то диапазон допустимых температур находится между -10 и 50° С. Кроме того, если это соединение вступает в реакцию с фосфонатом (IIа), то оптимальными будут температуры от -5 до 30°С.
На стадии (аl) необходимо использовать стехиометрические количества основания, чтобы реализовать перенос α-протона к фосфору соединений (IIа) или (IIb) соответственно. Тем не менее, можно применять очень небольшой избыток основания для того, чтобы реакция образования илида проходила до конца. Таким образом, соотношение молярностей основания и соединений (IIа) или (IIb) соответственно должно быть между 1 и 1,2, предпочтительно - между 1 и 1,1, еще лучше - между 1 и 1,05.
По данному изобретению на концентрацию в реакционной смеси соединений (IIа) или (IIb) соответственно строгие ограничения не накладываются. В общем случае концентрация может находиться в диапазоне между 0,01 и 10 моль/л, предпочтительно - между 0,1 и 1 моль/л.
На стадии (bl) альдегид (III) реагирует с илидом, полученным на стадии (al).
Предпочтительнее добавлять альдегид (III) к исходной реакционной смеси, полученной в результате осуществления стадии (al).
Альдегид (III) можно добавлять к реакционной смеси либо непосредственно, либо предварительно растворяя его в растворителе, предпочтительно - в апротонном.
В качестве предпочтительных, следует назвать растворители типа ароматических углеводородов, эфира, N-диметилформамида, диметилсульфоксида, N-метилпирролидона или Р[N(СН3)2]3 и их смеси, которые указаны выше.
Какой бы метод действий не был выбран, предпочтительно концентрация альдегида (III) в реакционной смеси должна находиться в диапазоне между 6×10-3 и 0,6 моль/л, главным образом - между 0,01 и 0,7 моль/л.
Температура, при которой илид вступает в реакцию с альдегидом (III), зависит от относительной реакционноспособности этих двух реагентов.
Следует отметить, что илид, приготовленный из фосфоната (IIа), более реакционноспособен, чем илид, полученный из соединения (IIb). Следовательно, способ А, включающий проведение реакции типа Horner-Emmons с использованием
фосфоната (IIа), является особо предпочтительным.
При проведении реакции илида с альдегидом(III) температура должна находиться в диапазоне между -10 и 50°С, предпочтительный интервал - от -5 до 30°С.
В большинстве случаев реакции на стадиях (аl) и (bl) осуществляют при различных температурных условиях, поэтому смесь, полученную на стадии (аl), перед использованием ее в качестве реагента для проведения стадии (bl), необходимо довести до соответствующей температуры.
В еще большем числе случаев, при получения соединений с формулой (I), в которой n равно 1, а Y представляет собой алкоксигруппу (C1-С5), полученную в результате осуществления стадий (аl) и (bl), квалифицированный персонал может использовать в качестве пособия представленные в литературе сведения об условиях проведения реакций Wittig и Homer-Emmons.
Стадия (сl) позволяет произвести гидролиз эфира с формулой (I), полученного на стадии (bl). Эту стадию предпочтительно осуществляют в основной среде. Для реализации стадии (сl) могут быть использованы основания, которые обычно применяют для омыления эфиров. Предпочтительно, это должны быть неорганические основания щелочных металлов гидроксидного типа (NaOH, KОН) или карбонатного типа (K2СО3, Na2СО3).
В общем случае гидролиз функциональной группы эфира производят в растворителе протонного типа. Особенно хорошо подходят для этих целей (C1-C5) алканол [sic], вода и их смеси.
При проведении гидролиза, температура предпочтительно должна находиться в диапазоне от 0 до 100°С, например - в интервале от 20 до 80°С при проведении реакции с гидроксидом натрия в смеси воды и метанола.
Обычно по отношению к эфиру с формулой (1) используют от 1 до 5 эквивалентов основания, предпочтительно - от 1 до 2 эквивалентов.
Хотя это не относится к основному предмету изобретения, гидролиз функциональной группы эфира может быть
произведен в кислой среде.
Чтобы определить оптимальные условия для гидролиза функциональной группы эфира, квалифицированный персонал может обратиться, например, к следующей литературе: Protective Groups in Organic Synthesis, Greene T.W. и Wuts P.G.M., издательства John Wiley & Sons, 1991, а также Protecting Groups, Kocienski P.J., 1994, издательства Georg Thieme Verlag.
Стадию (dl) осуществляют для приготовления соединений с формулой (I), в которой Y представляет собой -NRcRd.
Эта стадия включает обычную реакцию карбоновой кислоты, полученной на стадии (сl), с амином, который описывается формулой NHRcRd. Особо предпочтительно в этой реакции с амином NHRcRd использовать активированную форму карбоновой кислоты с формулой (1). Такими активированными формами являются, например, ангидрид карбоновой кислоты, кислый хлорид или смешанный ангидрид. Амидирование карбоксильной функциональной группы может быть произведено любым квалифицированным работником.
Способ В позволяет приготовить соединение с формулой (I), в которой n=2. Этот способ включает проведение следующих стадий:
(а2) приготовление илида
- либо путем проведения реакции основания с фосфонатом, имеющим формулу (IVa)

в которой
R’j и R’k независимо представляют собой группу R’, как определено в п.1 формулы изобретения;
T6 и Т7 независимо представляют собой (C1-C5)алкил; Y представляет алкоксигруппу (C1-C5);
- либо в реакции основания с фосфониевой солью, которая описывается формулой (IVb)

в которой
R’j и R’k независимо представляют группу R’, как определено в п.1 формулы изобретения;
Т8, Т9 и Т10 независимо представляют собой (С1-С5)алкил или (С6-С10)арил, который может быть замещен (C1-C5)алкилом;
Y представляет алкоксигруппу (C1-С5) и hal представляет атом галогена;
(b2) проведение реакции илида, полученного на стадии (а2), с альдегидом, который описывается следующей формулой:

в которой
R, р, Х и А определены в п.1 формулы изобретения [sic];
каждый в отдельности из радикалов R’1 и R’2 представляет собой -СНО, а остальные принимают одно из значений, названных в п.1 формулы изобретения [sic] для R1 и R2, за исключением цепочки Z, чтобы получить соединение с формулой I [sic], в которой n равно 2, а Y есть алкоксигруппа (C1-C5);
(с2) если требуется эфир, полученный на описанной выше стадии (b2), в кислой или основной среде, превратить в соответствующую карбоновую кислоту с формулой (I), в которой Y представляет группу ОН;
(d2) если требуется приготовить соответствующее соединение с формулой (I), в которой Y представляет собой -NRcRd, то проводят реакцию функциональной группы карбоновой кислоты соединения с формулой (I), полученного на стадии (с2), с амином, который описывается формулой HNRcRd, где Rc и Rd определены в п.1 формулы изобретения [sic], причем карбоксильная функциональная группа может быть активированной.
Общие условия, при которых реализуют стадии от (аl) до (dl) применимы также и при осуществлении описанных выше стадий от (а2) до (d2).
Фосфонат с формулой (IIа) получают с использованием обычных методик, например, в реакции Арбузова. В частном случае, когда T1 и Т2 одинаковы, проводят реакцию фосфита с формулой (IX)

в которой T1 имеет значение, представленное выше при описании формулы (IIа), и галида (X)

где R’ и Y имеют такие же значения, что и определенные выше для формулы (IIа), а hal представляет собой атом галогена.
Такого же типа реакция позволяет получить фосфонат с формулой (IVa).
Фосфониевые соли с формулой (IIb) легко можно получить в известной реакции фосфина (V)

где Т3, Т4 и Т5 имеют такие же значения, что и в формуле (IIb), и галида (VI)

где R’, Y и hal принимают такие же значения, что и в формуле (IIb).
Аналогично фосфониевую соль с формулой (IVb) можно легко получить в реакции фосфина (VII)
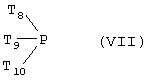
где Т8, Т9 и Т10 соответствуют определенным выше для формулы (IVb), с галидом (VIII)

где hal, R’j, R’k и Y соответствуют определенным выше для формулы (IVb). Условия этого эксперимента описаны в статье A. Zumbrunn с соавторами в журнале Helv. Chim. Acta, 1985, 68, 1519.
Альдегиды, которые описываются формулой (III), имеются в продаже, кроме того, их можно легко приготовить из имеющихся в продаже реактивов путем применения одного из следующих способов.
Способ С получения альдегида с формулой (III)

где R’1 представляет собой -СНО.
Последовательность проведения реакций при реализации способа С показана на схеме 1.
На стадии (i) осуществляется восстановительное алкилирование кетона (XI). Эта стадия включает реакцию кетона (XI) с металлорганическим соединением
СН3-М
в котором М представляет собой -Mg-hal (где hal -атом галогена), или Li.
Схема 1

На стадии (i) условия проведения реакции определяются в зависимости от природы металлорганического соединения.
Если металлорганическое соединение [sic] представляет собой реагент Grignard, то предпочтительно использовать растворители типа диэтилового эфира, тетрагидрофурана, диоксана, бензола, толуола и аналогичных. Реакцию следует проводить при температуре от -78 до 100°С, предпочтительно - от -10 до 70°С. На стадии (ii) производят дегидратацию соединения с формулой (XII), которую можно осуществить, подвергая это соединение воздействию органической или неорганической кислоты.
В качестве примеров наиболее подходящих [sic] кислот, можно указать соляную, серную, азотную, трифторметансульфоновую, уксусную, трифторуксусную, пара-толуолсульфоновую или пара-нитробензойную.
Наиболее подходящими растворителями для реагентов в этих реакциях являются те, которые обычно применяются в этой сфере при условии, что они способны растворять соединения с формулой (XII). В этом отношении предпочтительны ароматические углеводороды, такие как бензол и толуол.
На стадии (iii) осуществляют бромирование радикалов соединения с формулой (XIII). Эту реакцию можно провести традиционным способом - путем воздействия бромирующим агентом либо при облучении, либо при нагревании; можно также применить инициатор типа пероксида или азосоединений.
В качестве бромирующих агентов можно использовать, например бром или N-сукцинимид.
Для инициирования радикалов можно использовать, например, α,α’-азобисизобутиронитрил или трет-бутилпероксид.
Стадия (iv) включает, на первом этапе, проведение реакции гексаметилентетрамина с соединением (XIV), а на втором - обработку продукта смесью уксусной и соляной кислот.
Отношение молярностей гексаметилентетрамина и соединения (XIV) предпочтительно должно находиться в интервале от 1 до 3, более предпочтительно - от 1 до 2.
Температура проведении реакции гексаметилентетрамина и соединения (XIV) предпочтительно должна находиться в диапазоне от 20 до 120°С, более предпочтительно - между 30 и 80°С. Преимущественно, реакцию проводят в растворителе. В качестве примеров подходящих растворителей, следует указать алифатические, ароматические или циклоалифатические углеводороды, которые могут быть галогенизированными, например - хлороформ, четыреххлористый углерод, тетрахлорэтилен и хлорбензол.
Последующая обработка кислотами включает, в качестве первого этапа, обработку полученной смеси раствором уксусной кислоты при температуре от 20 до 120°С, предпочтительно - от 30 до 80°С. На втором этапе к реакционной смеси добавляют
концентрированную соляную кислоту, поддерживая в реакторе ту же температуру - между 20 и 120°С, предпочтительно - между 30 и 80°С.
Способ D для приготовления соединений с формулой (III)

в которой R’2 есть -СНО, А обозначает -СR3R4, как определено выше, а Х представляет собой О.
Согласно этому способу альдегид с формулой (XV)

в которой R и p принимают те же значения, которые определены выше для формулы (III), реагирует с соединением, которое описывается формулой (XVI)

в которой R3 и R4 определены выше для формулы (III), в присутствии сильного основания.
Эта реакция - стехиометрическая. Тем не менее, предпочтительно проводить ее при небольшом избытке соединения (XV), чтобы соотношение молярностей реагентов (XV) и (XVI) находилось в интервале от 1 до 1,5, лучше - от 1 до 1,3.
В этой реакции преимущественно следует использовать неорганические основания, например - NaOH, КОН, NaHCO3, Na2CO3, KНСО3 или K2СО3. По главному предмету изобретения используется основание K2СО3.
Необходимое количество основания определяется количеством участвующего в реакции соединения (XV).
Таким образом, соотношение молярностей основания и соединения (XV) предпочтительно должно находиться в интервале между 1 и 1,2.
Эту реакцию можно провести в растворителе. Тип растворителя зависит от природы применяемого основания и присутствующих реагентов. Если в качестве основания используется K2СО3, предпочтительными растворителями являются эфиры, в особенности - диоксан, тетрагидрофуран, или диэтиловый эфир. Если реакцию проводят в присутствии растворителя, то концентрации реагентов в реакционной смеси предпочтительно должны находиться в диапазоне от 0,05 до 5 моль/л, лучше - между 0,08 и 1,2 моль/л. Предпочтительно, чтобы при проведении реакции температура находилась в диапазоне от 30 до 150°С, лучше - между 50 и 120°С, например - между 90 и 100°С.
Способ Е получения альдегида (III) с формулой

в которой R’2 представляет собой группу -СОН [sic]. Последовательность проведения реакций по способу Е показана на схеме 2.
Схема 2

Способ Е включает использование обычных реакций органической химии. На стадии (i) карбоновую кислоту (XVII)
этерифицируют спиртом с формулой RнОН, в которой Rнпредставляет собой группу (C1-C6)алкил.
В общем случае, этерификацию производят в кислой среде. Особенно целесообразно применять каталитические количества кислот типа серной или пара-толуолсульфоновой. Тем не менее, реакцию можно проводить и при избытке кислоты.
Желательно проводить реакцию при значительном избытке спирта RH-OH. Аналогично предпочтительно вводить в реакционную смесь дегидратирующий агент, например - типа молекулярных сит. Температура проведения реакции должна находиться в диапазоне от 20 до 120°С, предпочтительно - между 50 и 100°С, что является оптимальным.
Во многих случаях спирт Rh-OH можно использовать в качестве растворителя.
По данному изобретению, природа группы RH, которая вводится на стадии (i), не имеет никакого значения.
На следующей стадии эфир (XVIII) восстанавливают до спирта (XIX). Восстановление может быть проведено по любой из известных методик.
В качестве восстанавливающего агента может быть использован, например, литий-алюминиевый гидрид, а также боргидрид лития, диизобутилалюминиевый гидрид, триэтилборгидрид лития, ВН3-SMe2 при протоке тетрагидрофурана, НSi(ОЕt)3 или даже боргидрид натрия.
На стадии (iii) спирт (XIX) окисляют до альдегида таким образом, чтобы получить альдегид (XX). Окисление производят любым из известных способов. Целесообразно избегать последующего окисления альдегида до кислоты. По этой причине подходящий окислитель следует выбирать из ряда MnO2, диметилсульфоксид, реагент Collins, реагент Corey, пиридиндихромат, Ag2СО3 на целите, горячая HNO3 в жидком глиме, Pb(ОАс)4-пиридин, цериевоаммониевый нитрат или N-метилморфолин-N-оксид.
Способ F получения соединений с формулой III [sic]

в которой R’2 представляет группу -СНО.
По данному способу соединения с формулой (III) получают в реакции смеси оксихлорида фосфора с диметилформамидом и соединения с формулой (XXI)

в которой R, p, X, А и R’1 соответствуют представленным выше определениям для формулы (III).
Предпочтительно использовать отношение молярности оксихлорида фосфора к молярности соединения (XXI), а также молярности диметилформамида к молярности соединения (XXI) в диапазоне от 1 до 3, лучше - от 1 до 2, например между 1 и 1,5.
Диметилформамид и оксихлорид фосфора целесообразно использовать в равных количествах.
Один из способов проведения реакции включает приготовление раствора реагентов оксихлорида фосфора и диметилформамида в растворителе, к которому затем добавляют раствор соединения (XXI).
Раствор реагентов обычно приготавливают путем добавления оксихлорида фосфора к раствору диметилформамида в растворителе. В качестве подходящего растворителя можно использовать галогенизированные алифатические углеводороды (например, дихлорметан) или ацетонитрил.
Добавление POCl3 к раствору ДМФ предпочтительно производить при низких температурах, конкретно - при температурах между -40 и 15°С, предпочтительно - между -10 и 10°С, еще лучше - между -5 и +5°С.
К этому раствору добавляют соединение с формулой (XXI), предпочтительно - в виде раствора в растворителе.
По главному предмету изобретения, следует применять тот же растворитель, который использовался для приготовления раствора реагентов.
Реакция соединения (XXI) с системой реагентов DMF/POCl3 производится при температуре, которая находится между 15 и 100°С, предпочтительно - между 18 и 70°С.
Соединение с формулой (XXI) можно легко получить из соответствующего кетона с формулой (XI)

в которой R, p, Х и А соответствуют определенным выше для формулы (XXI). Можно, например, приготовить это соединение путем проведения реакций, аналогичных описанным выше в способе С (Схема 1: стадии (i) и (ii)). Короче говоря, можно провести реакцию кетона (XI) с металлорганическим соединением, соответствующим формуле R’1-М, где М представляет собой атом лития или радикал -Mg-hal, hal - атом галогена. Полученное соединение, соответствующее формуле
в которой R, р, Х и А соответствуют введенным выше определениям, подвергают обработке в кислой среде.
Кетоны с формулой (XI), альдегиды с формулой (XV) и кислоты с формулой (XVII) имеются в продаже, кроме того, их можно легко приготовить из имеющихся в продаже реактивов по традиционным методикам органической химии.
Другой предмет изобретения составляют новые соединения, которые описываются формулой

в которой:
А представляет собой двухвалентный радикал - (СН2)3-CR3R4-(СН2)t-, где одно из s и t равно 0, а второе 1;
R3, R4, R, р и Х соответствуют определенным выше для формулы (I) и
одно из R’1 и R’2 обозначает -СНО, а другое принимает одно из значений, приведенных выше для R1 и R2применительно к формуле (I), за исключением цепочки Z.
Среди этих соединений предпочтительными являются те, в которых R’1 представляет собой -СНО.
Другая группа предпочтительных соединений состоит из соединений, которые описываются приведенной выше формулой (IIIa), в которой
Х обозначает О;
А обозначает -СН2-СR3R4-, в котором незамещенная метиленовая группа образует связь с X;
R’1 или R’2 обозначают Н; (C1-C15)алкил; (C1-С15)алкенил; или фенил, который может быть замещенным (C1-C5)алкилом, (C1-C5)алкоксигруппой, атомом галогена или -CF3;
R3 принимает одно из значений, определенных выше для R’1 или R’2, но не может представлять собой -СНО;
R4 обозначает атом водорода или (C1-C15)алкил;
R обозначает одну из групп ряда (C1-C9)алкил; (C1-С3)алкоксигруппа; фенил; или фенилкарбонил, который может быть замещенным атомом галогена, (C1-C5)алкилом, (C1-C5 )алкоксигруппой; -CF3, или -ОСF3; атом галогена; -СF3; и -ОСF3; и
p равно 0, 1 или 2.
Еще более предпочтительны соединения, в которых:
Х обознаяает О;
А обозначает –СН2-СR3R4-, в котором незамещенная метиленовая группа образует связь с X;
R’1 или R’2 представляет собой атом водорода;
R3 представляет собой атом водорода или группу (C1-C5)алкил, например - метил;
R4 обозначает (C1-C15)алкил, предпочтительно (C1-C5)алкил, например - метил;
R может обозначать атом галогена, CF3, (C1-C5)алкоксигруппу, фенил и пара-хлорбензоил;
p равно 0, 1 или 2.
В качестве примеров таких соединений следует указать:
3,3-диметил-5-формил-7-бром-2,3-дигидробензоксепин,
3,3-диметил-5-формил-9-метокси-2,3-дигидробензоксепин,
3,3-диметил-5-формил-7,8-дихлор-2, 3-дигидробензоксепин,
3,3-диметил-5-формил-7-фтор-8-хлор-2,3-дигидробензоксепин,
3,3-диметил-5-формил-7-(пара-хлорбензоил)-2,3-дигидробензоксепин,
3, 3-диметил-5-формил-7-трифторметил-2,3-дигидробензоксепин,
3,3-диметил-5-формил-7-фтор-2,3-дигидробензоксепин,
3,3-диметил-5-формил-7-хлор-2,3-дигидробензоксепин,
3,3-диметил-5-формил-7,8-диметокси-2,3-дигидробензоксепин,
3,3-диметил-5-формил-7-фенил-2,3-дигидробензоксепин,
3,3-диметил-5-формил-2,3-дигидробензоксепин,
3,3-диметил-5-формил-7-метокси-2,3-дигидробензоксепин.
В другом аспекте изобретение относится к промежуточным соединениям, которые описываются формулой
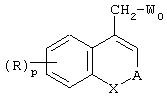
в которой:
А обозначает двухвалентный радикал -(СН2)s -CR3R4-(СН2)t-, где одно из s и t равно 0, а другое 1;
R3, R4, R, p и X соответствуют определенным выше для формулы (I) и
W0 обозначает -СН3- или -CH2Br.
Среди этих соединений предпочтительными являются те, в которых:
R3 представляет собой Н, (C1-C15)алкил, (C1-C15)алкенил или фенил, который может быть замещенным (C1-C5)алкилом, (C1-C5)алкоксигруппой, атомом галогена или -CF3;
R4 обозначает атом водорода или (C1-C15)алкил;
R может обозначать одну из групп ряда: (C1-C9)алкил; (C1-C5)алкоксигруппа; фенил; или фенилкарбонил, который может быть замещенным атомом галогена, (C1-C5 )алкилом, (C1-C5)алкоксигруппой, -CF3 или -ОСF3; атом галогена; -СF3; и -ОСF3; и
p равно 0, 1 или 2.
Еще лучше, если R3, R4, R и р принимают значения, представленные в следующих группах:
R3 представляет собой атом водорода или группу (C1 -C5)алкил, такую, как метил;
R4 представляет собой (C1-C15)алкил, предпочтительно (C1-C5)алкил, например - метил;
R может обозначать атом галогена, СF3, (C1-C5)алкоксигруппу, фенил и пара-хлорбензоил;
p есть 0, 1 или 2.
Примеры соединений, в которых W0=-СН3, приведены в Таблице 4, которая представлена после примеров.
Следует также отметить 3,3,5-триметил-7-метокси-2, 3-дигидробензоксепин.
Примеры соединений, в которых Wo=-CH2Br сведены в приведенной после примеров Таблице 5.
Также можно упомянуть о 3, 3-диметил-5-бромометил-7-метокси-2,3-дигидробензоксепине.
Кроме того, настоящее изобретение относится к промежуточным соединениям формулы (IIIb)

в которой:
R’1 представляет собой атом водорода, (C1-C5)алкильную группу или фенил;
R3 и R4 независимо друг от друга выбирают из атома водорода, (C1-C18)алкильной группы или (C2 -C18)алкенильной группы.
Среди этих соединений предпочтение отдается таковым, где R’1 обозначает атом водорода.
В качестве примеров можно упомянуть о
-2,2-диметил-3-формил-2Н-1-бензопиране;
-2-[нон-3-енил]-3-формил-2Н-1-бензопиране;
-2-ундецил-3-формил-2Н-1-бензопиране,
-2-пентил-3-формил-2Н-1-бензопиране;
-2-нонил-3-формил-2Н-1-бензопиране;
-4-метил-3-формил-2Н-1-бензопиране и
-4-фенил-3-формил-2Н-1-бензопиране.
Далее, данное изобретение относится к фармацевтическим композициям, содержащим фармацевтически эффективное количество вышеприведенного соединения формулы (I) в комбинации с одним или несколькими фармацевтически приемлемыми носителями.
Эти соединения можно применять орально в форме гранул с немедленным или контролируемым высвобождением, твердых желатиновых капсул или таблеток, внутривенно в форме раствора для инъекций, чрезкожно в форме адгезивного чрезкожного
компонента либо местно в форме раствора, крема или геля.
Твордую композицию для орального применения изготавливают путем добавления к активному соединению наполнителя и, если это целесообразно, связующего компонента, разделительного агента, смазочного материала, красителя или ароматизирующего вещества с последующим формованием смеси в таблетки, таблетки с покрытием, гранулы, порошок либо капсулы.
Примерами наполнителей могут служить лактоза, кукурузный крахмал, сахароза, глюкоза, сорбит, кристаллическая целлюлоза и диоксид кремния, а в качестве примеров связующих компонентов можно привести поли(виниловый спирт), поли(виниловый эфир), этилцеллюлозу, метилцеллюлозу, аравийскую камедь, трагакант, желатин, шеллак, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, лимоннокислый кальций, декстрин и пектин. Примерами смазочных материалов могут служить стеарат магния, тальк, полиэтиленгликоль, диоксид кремния и гидрогенизированные растительные масла. Краситель может быть любым, при условии, что он может применяться в лекарствах. В качестве примеров ароматизирующих веществ можно привести какаовый порошок, мяту в травяной форме, ароматический порошок, мяту в форме масла, борнеол и коричный порошок. Не стоит и упоминать, что таблетка или гранула может быть порыта сахарозой, желатином и т.д.
Форму для инъекций, содержащую в качестве активного компонента соединение по настоящему изобретению, изготавливают, если это целесообразно, путем смешивания указанного соединения с регулятором кислотности, буфером, суспендирующим агентом, солюбилизатором, стабилизатором, агентом тоничности и/или противостарителем с последующим преобразованием смеси в форму, пригодную для внутривенных, подкожных или внутримышечных инъекций, что осуществляется одним из обычных методов.
Примерами суспендирующих агентов могут служить метилцеллюлоза, полисорбат 80, гидроксиэтилцеллюлоза, аравийская камедь, порошковый трагакант, натрийкарбоксиметилцеллюлоза и полиэтоксилированный монолаурат сорбита.
В качестве примеров солюбилизаторов можно привести касторовое масло, укрепленное полиоксиэтиленом, полисорбат 80, никотинамид, полиэтоксилированный монолаурат сорбита и этиловый эфир жирной кислоты касторового масла.
Кроме того, в число стабилизаторов входят сульфит натрия, метасульфит натрия и простой эфир, а в качестве противостарителей можно применять метил-пара-гидроксибензоат, этил-пара-гидроксибензоат, сорбиновую кислоту, фенил, крезол и хлорокрезол.
Далее, целью данного изобретения является применение активного соединения, которое выбирают из соединений приведенной выше формулы (I), в приготовлении лекарства, предназначенного для предупреждения или лечения дислипидемий, атеросклероза или диабета. Гиполипидемическая и гипогликемическая активность соединений по настоящему изобретению была продемонстрирована in vitro и in vivo в ходе следующих испытаний.
1) Доказательство активности in vitro.
Гиполипидемическое и гипогликемическое действие соединений по данному изобретению обуславливается их способностью активации изоформов PPARα и PPARγ .
Анализ активации PPARα и PPARγ основывается на трансфекции ДНК, дающей возможность экспрессировать репортерный ген (ген люциферазы) под управлением PPARs, либо эндогенного в случае PPARγ, либо экзогенного в случае PPARα. В состав репортерной плазмиды J3TkLuc входят три копии сигнальных элементов для PPARs человеческого гена аро A-II (Staels В. и др. (1995), J. Clin. Invest., 95, 705-712), клонируемые выше промотора гена тимидинкиназы симплексного вируса герпеса в плазмиде pGL3. Этот репортерный ген был получен путем субклонирования, в плазмиде pGL3, плазмиде J3TkCAT, описанной выше (Fajas L. и др. (1997), J. Biol. Chem., 272, 18779-18789). Используемыми клетками являются таковые CV1 зеленой макаки, трансформированные вирусом SV40, которые экспрессируют PPARγ (Forman В. и др. (1995), Cell, 83, 803-812), и человеческие клетки SK-Hep1, не экспрессирующие PPARs. Эти клетки высевали с плотностью
20000 клеток на ячейку (96-ячейковые планшеты) и заражали 150 нг репортерной ДНК в комплексе со смесью липидов. В случае клеток SK-Hep1, совместно трансфектируется вектор экспрессии PPARα, описанный у Sher Т. и др. (1993), Biochemistry, 32, 5598-5604. Через 5 часов клетки дважды промывают и инкубируют в течение 36 часов в присутствии испытуемого соединения в свежей культуральной среде, содержащей 10% эмбриональной телячьей сыворотки. В завершение инкубирования клетки лизируют и измеряют степень активности люциферазы. Эту активность выражают относительно контрольного значения.
В качестве примера, соединение из Примера 16b, описанное ниже, ((2Е,4Е)-5-(3,3-диметил-7-метокси-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота) усиливает, в этих условиях, сигнал люциферазы на 300% в клетках CV1 и на 250% - в клетках SK-Hep1. В присутствии репортерного вектора pGL3 без сигнального элемента PPAR соединение из Примера 16b не проявляет активности ни в одном, ни в другом типе клеток.
2) Доказательство активности in vivo.
Антидиабетическая и гиполипидемическая активность соединений формулы I [sic] определялась орально на мышах db/db.
Каждую особь из двухмесячных мышей db/db обрабатывают в течение 15 дней соединением из Примера 16 (100 мг/кг/день). В каждую исследуемую группу входят семь животных. Через три дня (D3) и пятнадцать дней (D15) обработки после слабой анестезии с прекращением кормления в течение 4 часов берут заглазничные пробы.
Были произведены следующие измерения.
Количественное определение гликемии (глюкозная оксидаза) на D3 и D15 и липидных характеристик относительно сывороток на D15 (COBAS): триглицериды, общий холестерин (CHOL), HDL-холестерин (HDL-C) и свободные жирные кислоты (FFA) (комплект для количественного определения фирм

Полученные результаты сведены в таблицу 1a. Значения, приведенные в этой таблице, являются средними ± стандартная погрешность.
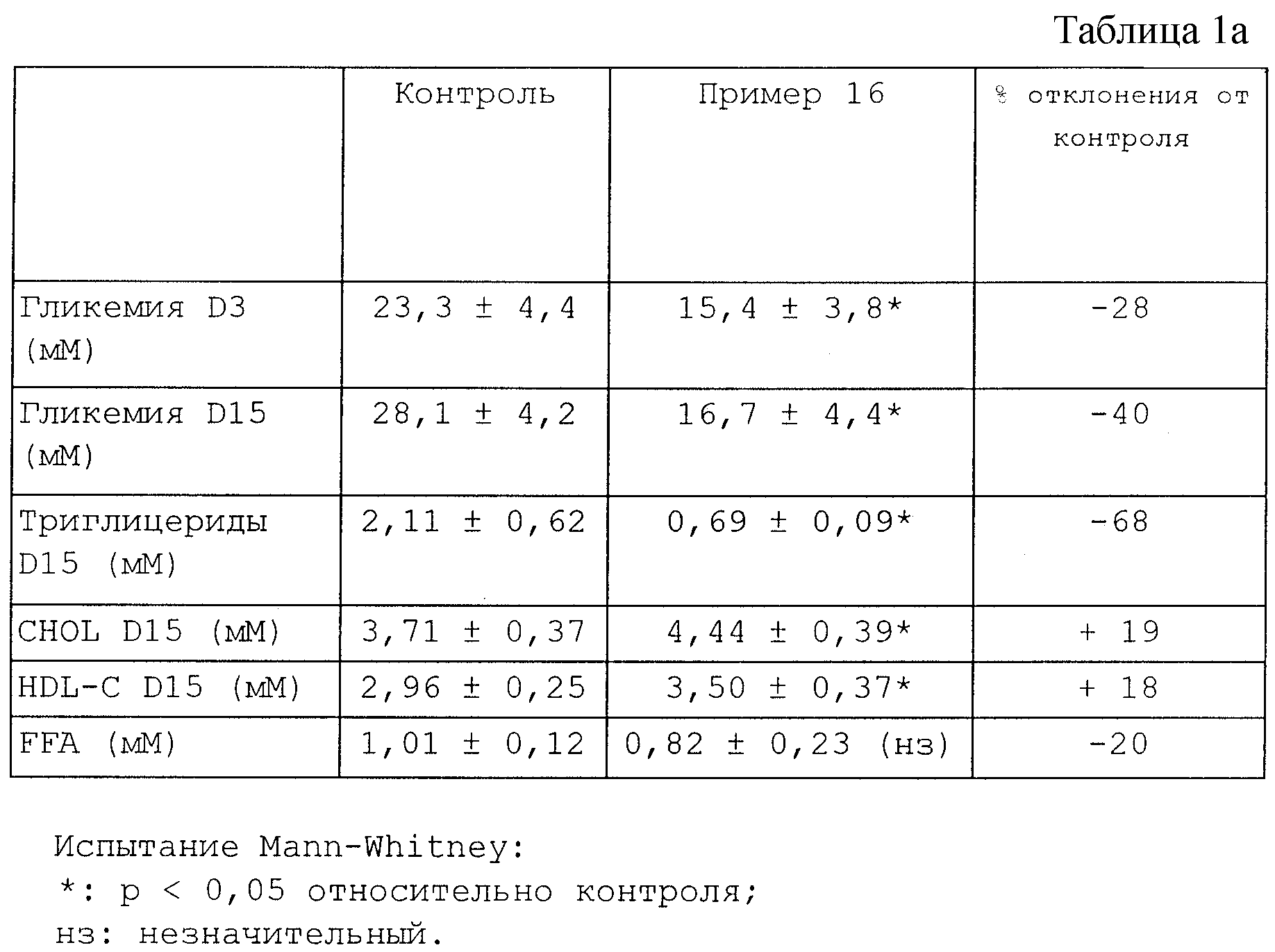
Эти результаты однозначно свидетельствуют о гиполипидемической и антидиабетической активности соединений по настоящему изобретению.
Следующие примеры позволяют проиллюстрировать данное изобретение без ограничения последнего.
Данные протонного ядерного магнитного резонанса (ЯМР) приводятся с использованием следующих сокращений: s обозначает синглет, d - дублет, t - триплет, q - квартет, о - октет и m - мультиплет. Химические сдвиги d выражают в промилях; Т.пл. обозначает точку плавления, а Т.кип. - точку кипения.
ПРИМЕР 1
(2Е,4Е)-5-(2-Пентил-2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновая кислота и сложный этиловый эфир (I: р=0; R1=Н; Х=О; А=-СR3R4-; R3=Н; R4=н-С5Н11; R2=Z; n=1; R’=-СН3;
соединение 1a [sic]: Y=-ОСН2СН3;
соединение 1b [sic]: Y=-ОН).
а) 2-н-Пентил-3-формил-2Н-1-бензопиран.
Смесь из 22,0 г (0,18 моля) салицилальдегида, 25,0 г (0,198 моля) 2-октенала и 24,8 г (0,18 моля) карбоната калия в 200 мл диоксана в инертной атмосфере нагревают с обратным холодильником в течение 2,5 ч. Далее реакционную смесь доводят до комнатной температуры (20-25°С), а затем разбавляют, добавляя 1,5 литра воды. Наблюдается образование масла, которое экстрагируют метиленхлоридом. Органическую фазу сушат над безводным сульфатом натрия, а затем концентрируют при пониженном давлении. Таким образом получают указанное в заголовке соединение в форме оранжевого масла, которое дистиллируют при пониженном давлении (Т.кип. (0,44 мм рт.ст.) 132-140°С). Путем дистилляции получают 20 г указанного в заголовке продукта (выход 48%).
б) Сложный этиловый эфир (2Е,4Е)-5-(2-пентил-2Н-1-бензопиран-3-ил)-3-метилпента-2, 4-диеновой кислоты (соединение 1а).
3 г (75,5 ммоля) гидрида натрия в форме 60%-ной суспензии в масле добавляют при 0°С к раствору (в инертной атмосфере) из 19,9 г (75,5 ммоля) сложного этилового эфира диэтил(2-метил-3-карбоксипроп-2-енил)фосфоната в 200 мл тетрагидрофурана.
Смеси дают прореагировать в течение 20 минут при 0°С, а затем в течение 15 минут при комнатной температуре (от 20 до 25°С). После этого к реакционной смеси добавляют 2 мл DMPU (1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон). Затем реакционную смесь охлаждают до 0°С и при такой температуре добавляют раствор из 14,5 г (62,9 ммоля) 2-н-пентил-3-формил-2Н-1-бензопирана в 145 мл тетрагидрофурана.
Смеси дают прореагировать в течение 1 часа при 0°С, а затем избыточный гидрид натрия удаляют, добавляя холодную воду (приблизительно 0°С). После этого с использованием этилацетата проводят экстракцию. Органическую фазу промывают водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. Получают указанное в заголовке соединение в форме масла, которое очищают быстрым хроматографическим методом с использованием в качестве элюента смеси 98/2 циклогексан/этилацетат. Таким образом получают 2,4 г изомера 2Z кислоты 1a [sic] и 7,5 г изомера 2Е кислоты 1а [sic].
Соединение 2Z:
1H ЯМР (CDCl3, 300 МГц) δ (промили): 0,8 (3Н, m), 1,2-1,8 (11Н, m), 2,0 (3Н, s), 4,1 (2Н, q, J=7 Гц), 5,2 (1Н, m), 5,6 (1H, s), 6,4 (1H, s), 6,5 (1H, d, J=16 Гц), 6,8-7,1 (4Н, m), 7,7 (1H, d, J=16 Гц).
Соединение 2Е:
1H ЯМР (CDCl3, 300 МГц) δ (промили): 0,8 (3Н, t, J=7 Гц), 1,2-1,8 (11H, m), 2, 3 (3Н, s), 4,1 (2Н, q, J=7 Гц), 5,0 (1H, m), 5,8 (1H, s), 6,1 (1H, d, J=16 Гц), 6,4 (1H, s), 6, 8-7, 1 (4Н, m).
в) (2Е,4Е)-5-(2-Пентил-2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновая кислота (соединение 1b).
5,0 г (30,8 ммоля) гидроокиси натрия в форме раствора в 100 мл воды добавляют к раствору из 10,5 г (30,8 ммоля) полученного на предыдущей стадии б) сложного этилового эфира в растворе [sic] со 105 мл метанола.
Реакционную смесь нагревают с обратным холодильником в течение 3 часов. Она становится прозрачной.
Затем реакционной смеси дают остыть до комнатной температуры (20-25°С), после чего при пониженном давлении выпаривают метанол. Осадок покрывают 600 мл воды, а затем смесь дважды промывают диэтиловым эфиром. После этого водную фазу подкисляют 5N водным раствором соляной кислоты. Выпадает пастообразный осадок, который экстрагируют метиленхлоридом. Органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Таким образом получают пастообразное сухое вещество, которое рекристаллизируют из 50 мл метанола. Указанное в заголовке соединение выделяют в форме белого сухого вещества (выход 86%).
Соединение 2Е: Т.пл. 118-120°С.
1H ЯМР (DMSO [sic], 300 МГц) δ (промили): 0,88 (3Н, t, J=7 Гц), 1, 7-1,3 (8Н, m), 2,3 (3Н, s), 5,3 (1Н, d, J=2,3 Гц), 6,0 (1Н, s), 6,6 (1Н, d, J=16 Гц), 7,2-6,8 (6Н, m).
Соединение 2Z:
Т.пл. 161-163°С.
1H ЯМР (d6-DMSO, 300 МГц) δ (промили): 0,8 (3Н, t, J=3 Гц), 1,8-1,2 (8Н, m), 2,0 (3Н, s), 5,1 (1Н, d, J=10 Гц), 5,7 (1H, s), 6,5 (1H, s), 6,6 (1H, d, J=16 Гц), 7,1-6,8 (4Н, m), 7,7 (1H, d, J=16 Гц).
ПРИМЕР 2
(2Е,4Е)-5-(2,2-Диметил-6-метокси-2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновая кислота и сложный этиловый эфир (I: р=1; R=6-ОСН3; Х=О; А=-СR3R4-; R3=R4=-СН3; R1=Z; R2=Н; n=1; R’=-СН3; соединение 2а [sic]: Y=ОСН2СН3; соединение 2b [sic]: Y=-ОН).
а) 6-Метокси-2,2-диметилхроман-4-он.
19,8 мл (1,6 эквивалента) пирролидина по каплям добавляют к охлажденному ниже 25°С раствору из 25 г (0,15 моля) 2’-гидрокси-5’-метоксиацетофенона, 12,1 мл (1,1 эквивалента) ацетона и 140 мл толуола. Реакционную смесь оставляют перемешиваться в течение 16 часов при 25°С.
Затем добавляют 290 мл ацетона и реакционную смесь нагревают с обратным холодильником в течение 4 часов. Далее ее концентрируют при пониженном давлении и экстрагируют этилацетатом. Органическую фазу сначала промывают насыщенным водным раствором хлорида натрия, затем раствором 1N гидроокиси натрия и наконец раствором 1N соляной кислоты, после чего промывают водой. Затем органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Осадок (соединение оранжевого цвета) растворяют в диизопропиловом эфире. Нерастворимое вещество отфильтровывают и органическую фазу концентрируют при пониженном давлении. Получают коричневую пасту, которую очищают быстрым хроматографическим методом с использованием в качестве элюента дихлорометана. Получают 9,4 г желтого масла.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 7,2 (1Н, d, J=3,15 Гц), 7 (1Н, d, J=3,17 Гц), 6,75 (1Н, d, J=8,99 Гц), 3,7 (3Н, s), 2,6 (2Н, s), 1,35 (6Н, s).
ИК (см-1): 1638,4.
б) 6-Метокси-2,2,4-триметилхроман-4-ол.
9,4 г (45,6 ммоля) 6-метокси-2,2-диметилхроман-4-она, растворенных в 150 мл тетрагидрофурана, по каплям добавляют к раствору из 33,5 мл (0,1 моля) 3М метилхлорида магния в тетрагидрофуране в инертной атмосфере с температурой 50°С. Смесь нагревают с обратным холодильником в течение 4 часов. Далее реакционную смесь охлаждают в ванне со льдом и гидролизуют, по каплям добавляя воду. После этого реакционную смесь выливают на раствор хлористого аммония. После экстрагирования этилацетатом органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Получают 10 г указанного в заголовке соединения.
ИК (см-1): 3400-3500.
в) 6-Метокси-2,2,4-триметил-2Н-1-бензопиран.
Смесь из 10 г (0,045 моля) 6-метокси-2,2,4-триметилхроман-4-ола, 0,25 г (1,45 ммоля) пара-толуолсульфокислоты и 150 мл толуола нагревают с обратным холодильником в течение 4 часов в 500-миллилитровой колбе с четырьмя горлами, оснащенной прибором Дина и Старка.
После охлаждения раствора до комнатной температуры (25°С) органическую фазу промывают сначала раствором кислого углекислого натрия, а затем водой. Органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Таким образом получают темное масло (9,4 г).
г) 4-Бромометил-6-метокси-2,2-диметил-2Н-1-бензопиран.
Раствор из 9,1 г (0, 045 моля) 6-метокси-2,2, 4-триметил-2Н-1-бензопирана, 8 г (0,045 моля) N-
бромосукцинимида и 0,25 г α,α’-азобисизобутиронитрила в 100 мл тетрахлорида углерода нагревают с обратным холодильником в течение 4 часов. Нерастворимое вещество отфильтровывают и органическую фазу промывают теплой водой (30°С), после чего сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Таким образом получают 13 г темного масла.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 6,6-6,85 (3Н, m), 5,75 (1Н, s), 4,15 (2Н, s), 3,7 (3Н, s), 1,3 (6Н, s).
д) 6-Метокси-4-формил-2,2-диметил-2Н-1-бензопиран.
Смесь из 12,7 г (0,045 моля) 4-бромометил-6-метокси-2, 2-диметил-2Н-1-бензопирана, 125 мл хлороформа и 8,8 г (0,059 моля) гексаметилентетрамина нагревают с обратным холодильником в течение 2 часов. Далее реакционную смесь концентрируют при пониженном давлении. Получают оранжевый осадок, который покрывают 75%-ным водным раствором уксусной кислоты (133 мл). Этот раствор нагревают с обратным холодильником в течение 90 минут, затем добавляют 20 мл концентрированной соляной кислоты и полученный раствор вновь нагревают с обратным холодильником в течение 30 минут. В горячий раствор добавляют 80 мл воды. Смесь оставляют перемешиваться при 25° С в течение 30 минут, затем ее экстрагируют диэтиловым эфиром и эфирную фазу сушат над безводным сульфатом натрия. Далее органическую фазу концентрируют при пониженном давлении. Таким образом получают 8,6 г темного масла, которое очищают быстрым хроматографическим методом с использованием в качестве элюента дихлорометана. Выделяют 2,7 г желтого масла.
ИК (см-1): 1696,3, 1487, 1261.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 9,6 (1Н, s), 7,75 (1Н, s), 6,7 (2Н, s), 6,4 (1Н, s), 3,75 (3Н, s), 1,4 (6Н, s).
е) Сложный этиловый эфир (2Е,4Е)-5-(2,2-диметил-6-метокси-2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновой кислоты (соединение 2а).
Раствор из 3 мл (12,3 ммоля) сложного этилового эфира диэтил(2-метил-3-карбоксипроп-2-енил)-фосфоната (41% транс) в 20 мл добавляют к раствору из 1,38 г (12,3 ммоля) трет-бутокиси калия и 20 мл тетрагидрофурана в инертной атмосфере (экзотермическая реакция). Реакционную смесь оставляют перемешиваться в течение 1 часа, а затем охлаждают до 10°С. К раствору добавляют 2,7 г (12,3 ммоля) 6-метокси-4-формил-2,2-диметил-2Н-1-бензопирана в 20 мл тетрагидрофурана. Смесь оставляют перемешиваться в течение 16 часов при 25°С, а затем охлаждают и добавляют воду. Реакционную смесь экстрагируют диэтиловым эфиром. После этого органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Получают 4 г оранжевого масла, которое очищают быстрым хроматографическим методом с использованием в качестве элюента смеси циклогексана и диизопропилового эфира. Получают 1,9 г желтого масла.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 6,8-6,5 (5Н, m), 5,8 (2Н, s), 4,1 (2Н, q), 3,7 (3Н, s), 2,3 (3Н, s), 1,3 (6Н, s), 1,2 (3Н, m).
ж) (2Е,4Е)-5-(2,2-Диметил-6-метокси-2Н-1-бензопиран-3-ил)-3-метилпента-2,4-диеновая кислота (соединение 2b).
Раствор из 1,9 г (5,7 ммоля) сложного этилового эфира, полученного на стадии е), 30 мл метанола, 0,3 г (1,3 эквивалента) гидроокиси натрия и 10 мл воды нагревают с обратным холодильником в течение 2 ч. Затем реакционную смесь концентрируют при пониженном давлении и осадок покрывают водой. Смесь подкисляют водным раствором 1N соляной кислоты. Выпавший осадок (желтого цвета) сперва отфильтровывают, а затем промывают водой и сушат при пониженном давлении. В сухой форме выделяют 1,3 г указанного в заголовке соединения. Т.пл.=140°С.
ИК (см-1): 1685, 1602, 1487, 1266.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 6,55-6,8 (5Н, m), 5,8 (2Н, 2s), 3,7 (3Н, s), 2,35 (3Н, s), 1,4 (6Н, s).
Микроанализ:
Теоретический: С=72%, Н=6,66%, О=21,33%.
Рассчитанный: С=71,74%, Н=6,81%, О=20,76%.
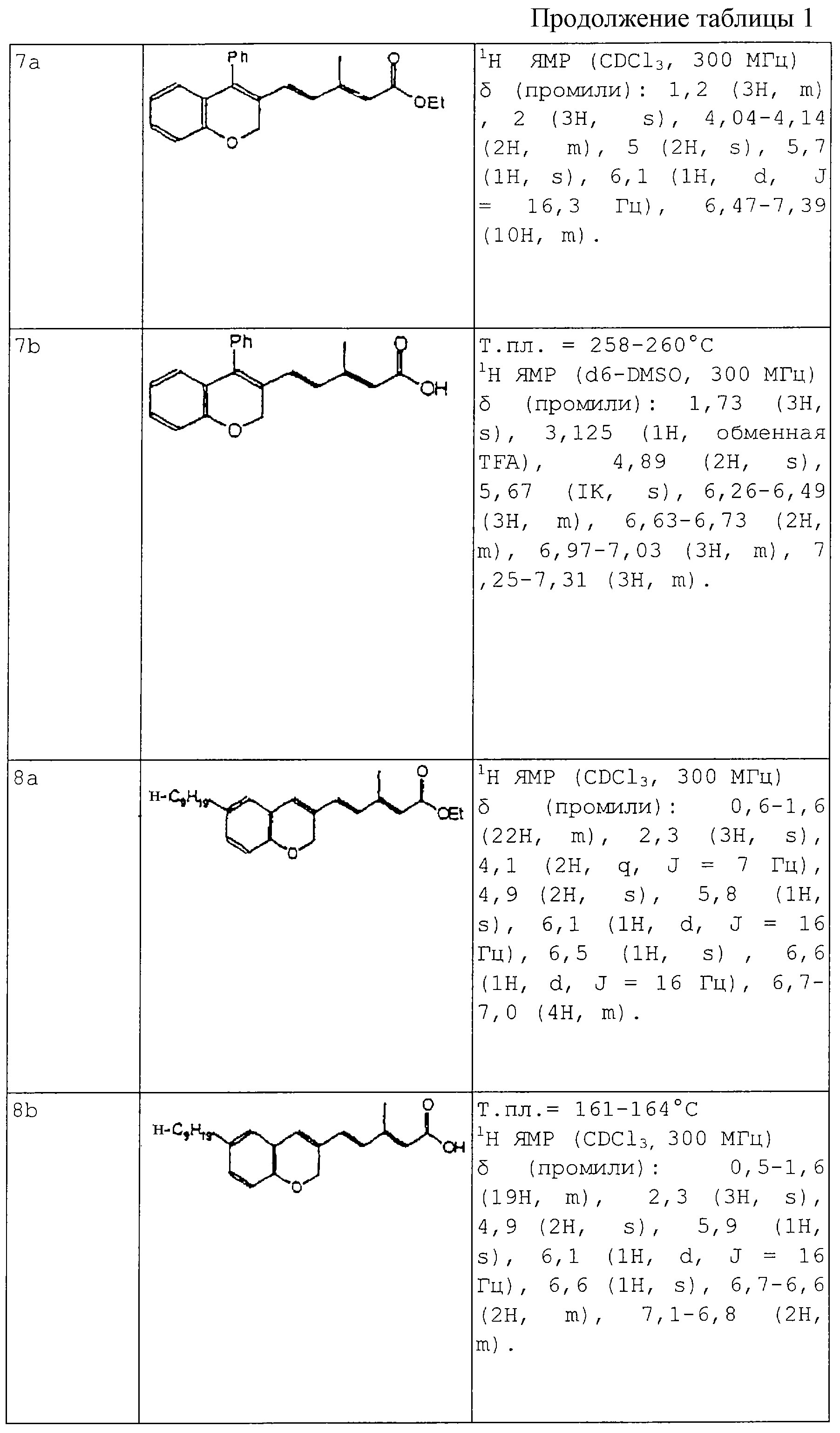
ПРИМЕРЫ 3-14
Применяя способы, проиллюстрированные в предыдущих Примерах 1 и 2, синтезируют следующие соединения из Примеров 3-14 (где Et обозначает этил, Ph представляет собой фенил и TFA обозначает трифтороуксусную кислоту).





ПРИМЕР 15
(2Е,4Е)-5-(5-Метил-2,3-дигидробензоксепин-4-ил)-3-метилпента-2,4 -диеновая кислота (I: р=0; R1=-СН3; X=0; А=-CH2-CH2-; R2=Z; n=1; R’[пропуск]-СН3;
соединение 15а [sic]: Y=-О-СН2-СН3;
соеинение 15b [sic]: Y=-ОН).
а) 5-Метил-2,3,4,5-тетрагидробензоксепин-5-ол.
Раствор [sic] из 33,5 мл (0,1 моля) 3М метилхлорида магния с тетрагидрофураном в инертной атмосфере
нагревают до 50°С. Довольно быстро добавляют 11,3 г (0,069 моля) 3,4-дигидро-2Н-бензоксепин-5-она, растворенных в 150 мл тетрагидрофурана. Этот раствор нагревают с обратным холодильником в течение 4 часов и оставляют перемешиваться в течение ночи при комнатной температуре. Далее реакционую смесь слабо гидролизуют водой при пониженной температуре. Затем в реакционную смесь заливают раствор хлористого аммония (120 г на литр). Последний экстрагируют этилацетатом, органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Получают 14,7 г желтого масла, которое очищают методом кристаллизации из изоооктана. Таким образом получают 11,9 г указанного в заголовке соединения с точкой плавления 92° С.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 7,5 (1Н, d, J=1,73 Гц), 7,2-7 (3Н, m), 4,15-3,9 (2Н, m), 2,5 (1Н, s), 2,1 (2Н, m), 1,95 (2Н, m), 1,6 (3Н, s).
б) 5-Метил-2,3-дигидробензоксепин.
14,9 г (0,1 моля) 5-метил-2,3,4,5-тетрагидробензоксепин-5-ола, 300 мл толуола и пара-толуолсульфокислоту на кончике шпателя загружают в 1-литровый реактор, оснащенный прибором Дина и Старка. Раствор нагревают с обратным холодильником в течение 2 часов с удалением воды по мере ее образования. Затем реакционную смесь нейтрализуют с использованием 5% водного раствора кислого углекислого натрия и органический раствор промывают водой. Далее органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Получают 16 г указанного в заголовке соединения, которые очищают методом дистилляции, т.кип. (2,5 мм рт.ст.) 80-90°С. Получают 10 г бесцветной жидкости.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 7,3-6,9 (4Н, m), 5,9 (1Н, m), 4,15 (2Н, t), 2,4 (2Н, m), 2,05 (3Н, s).
в) 5-Метил-4-формил-2, 3-дигидробензоксепин.
24 мл диметилформамида выливают на 150 мл ацетонитрила в инертной атмосфере в 500-миллилитровой колбе с четырьмя горлами. Раствор охлаждают до 0°С и затем добавляют 28,8 мл хлорангидрида фосфорной кислоты. Далее реакционную смесь оставляют перемешиваться при 5°С в течение 20 минут. После этого к реакционной смеси добавляют раствор из 8,2 г (0,051 моля) 5-метил-2,3-дигидробензоксепина в 24 мл диметилформамида и 20 мл ацетонитрила. Температуре дают медленно подняться до комнатной, после чего реакционную смесь нагревают до 60°С. Далее, приостановив нагревание, ее оставляют перемешиваться в течение 16 часов. После этого реакционную смесь нагревают с обратным холодильником и обрабатывают ледяной водой. Смесь нейтрализуют гидроокисью натрия и экстрагируют этилацетатом. Органическую фазу трижды промывают водным раствором [пропуск], сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Полученный продукт очищают быстрым хроматографическим методом с использованием в качестве элюента дихлорометана. Получают 4 г указанного в заголовке соединения.
1H ЯМР (CDCl3 , 300 МГц) δ (промили): 10,45 (1Н, s), 7,25-7,5 (4Н, m), 4,6 (2Н, m), 2,7 (5Н, s+m).
г) Сложный этиловый эфир (2Е,4Е)-5-(5-метил-2,3-дигидробензоксепин-4-ил)-3-метилпента-2, 4-диеновой кислоты (соединение 15а).
Раствор из 2,3 г (0,02 моля) трет-бутокиси калия в 40 мл тетрагидрофурана в 250-миллилитровой колбе с четырьмя горлами доводят до 50°С в инертной атмосфере. Далее к раствору добавляют раствор 6,1 мл (0,02 моля) сложного этилового эфира диэтил(2-метил-3-карбоксипроп-2-енил)фосфоната в 20 мл тетрагидрофурана в инертной атмосфере. Реакционную смесь оставляют перемешиваться при 50°С в течение 20 минут. Затем смесь охлаждают до 0°С и по каплям добавляют раствор из 4 г (0,02 моля) 5-метил-4-формил-2, 3-дигидробензоксепина в 20 мл тетрагидрофурана.
Реакционную смесь оставляют перемешиваться при комнатной температуре (20-25°С) в течение 16 часов. После охлаждения в реакционную смесь заливают воду и экстрагируют этилацетатом. Органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Получают 7,2 г оранжевого масла, которое очищают быстрым хроматографическим методом с использованием в качестве элюента смеси 80/20 циклогексан/диизопропиловый эфир. Таким образом получают 3,4 г указанного в заголовке соединения.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 7,25-7 (5Н, m), 6,3 (1Н, d, J=15,78 Гц), 5,8 (1Н, s), 4,4 (2Н, m), 4,1 (2Н, m), 2,35 (5Н, m +s), 2,2 (3Н, s), 1,2 (3Н, t).
д) (2Е,4Е)-5-(5-Метил-2,3-дигидробензоксепин-4-ил)-3-метилпента-2,4-диеновая кислота (соединение 15b).
0,7 г (0,0175 моля) гидроокиси натрия, растворенных в 60 мл воды, добавляют к раствору из 3,4 г (0,011 моля) полученного на предыдущей стадии г) сложного этилового эфира в 60 мл метанола. Реакционную смесь нагревают с обратным холодильником в течение 2 часов. Затем ее концентрируют при пониженном давлении. Осадок покрывают водой (нерастворимый). Смесь подкисляют раствором 5N соляной кислоты. Выпавший осадок отфильтровывают, промывают водой и сушат. В результате рекристаллизации из этанола получают 1,5 г указанного в заголовке соединения с точкой плавления 199-202°С.
ИК (см-1): кислотный пик при 2500-3000, 1674.
1H ЯМР (DMSO [sic], 300 МГц): 7,3-6,9 (5Н, m), 6,5-6,6 (1J [sic], d, J=15,81 Гц), 5,8 (1Н, s), 4,3 (2Н, t), 2,4 (2Н, t), 2,25 (3Н, s), 2,1 (3Н, s).
ПРИМЕР 16
(2Е,4Е)-5-(3,3-Диметил-7-метокси-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота (I: р=1; R=7-O-СН3; Х=О; А=-CH2-CR3R4; R3=R4=-СН3; R2=H; R1=Z; n=1; R’=-СН3;
соединение 16а [sic]: Y=-О-СН2СН3;
соединение 16b [sic]: Y=-ОН).
a) 3-(4-пара-метоксифенокси)-2,2-диметилпропан-1-ол.
Раствор из 391 г (1,19 моля) [3-(4-пара-метоксифенокси)-2,2-диметилпропан]сульфоната калия в 1,69 литра воды нагревают при 50°С в течение 15 минут до полного растворения сульфоната.
К этому раствору по каплям добавляют 156 мл концентрированной соляной кислоты (1, 5 эквивалента) и нагревают с обратным холодильником в течение 2,5 часов. В результате охлаждения реакционной смеси в ванне со льдом выпадает осадок, который растворяют, добавляя диэтиловый эфир. Органическую фазу промывают водой и сушат над безводным сульфатом натрия. После фильтрации органическую фазу концентрируют при пониженном давлении. Таким образом получают 241,2 г указанного в заголовке соединения (выход 96%) с точкой плавления 68°С.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 7,05 (4Н, s), 4 (3Н, s), 3,97 (2Н, s), 3,8 (2Н, s), 2,35 (1Н, s), 1,25 (6Н, s).
ИК (см-1): 3350-3250, 2995, 1520, 1470.
б) 3-(4-пара-метоксифенокси)-2,2-диметилпропилметансульфонат.
191,5 мл (1,38 моля) триэтиламина добавляют к раствору из 241 г (1,15 моля) полученного на вышеприведенной стадии а) спирта в 700 мл толуола при -10-0°С. При температуре ниже 10°С к этому раствору добавляют 100 мл (1,265 моля) метансульфонилхлорида.
После 2-часовой реакции добавляют 550 мл соляной кислоты в нормальной концентрации и экстрагируют этилацетатом. Органическую фазу промывают насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом натрия. Далее органическую фазу фильтруют и концентрируют при пониженном давлении. Получают 321 г оранжевого масла,
которое кристаллизуется при комнатной температуре и имеет точку плавления 78°С (выход 96%).
1H ЯМР (СDCl3, 300 МГц) δ (промили): 6,75 (4Н, s), 4,1 (2Н, s), 3,7 (3Н, s), 3,6 (2Н, s), 2,85 (3Н, s), 1,05 (6Н, s).
в) 4-(4-пара-метоксифенокси)-3,3-диметилбутиронитрил.
40 г (1,5 эквивалента) цианистого натрия добавляют к раствору из 160,8 г (0,557 моля) полученного на вышеприведенной стадии б) метансульфоната, растворенного в 600 мл диметилсульфоксида. Раствор доводят до 150°С и выдерживают при такой температуре в течение 3 часов, после чего реакционной смеси в течение 16 часов дают охладиться до комнатной температуры. Затем реакционную смесь охлаждают в ванне со льдом и добавляют воду. Выпадает осадок, который экстрагируют этилацетатом. Органическую фазу дважды промывают 400 мл водного раствора гидроокиси натрия (5 г на литр) и сушат над безводным сульфатом натрия. Далее органическую фазу концентрируют при пониженном давлении и получают 122 г оранжевого масла, которое кристаллизуется (выход 100%) и имеет точку плавления 64°С.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 6,75 (4Н, s), 3,7 (3Н, s), 3,6 (2Н, s), 2,4 (2Н, s), 1,15 (6Н, s).
г) 4-(4-пара-метоксифенокси)-3, 3-диметилмасляная кислота.
Раствор из 47 г (0,215 моля) полученного на предыдущей стадии в) соединения, 41,3 г (4 эквивалента) гидроокиси калия и 280 мл этиленгликоля доводят до 150°С в течение 3,75 часов. После охлаждения в реакционную смесь заливают воду и экстрагируют диэтиловым эфиром. Водную фазу подкисляют 5N соляной кислотой до рН 1, параллельно перемешивая. Осадок отфильтровывают, промывают Н2O и экстрагируют метиленхлоридом. Органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Получают 37 г оранжевого масла, которое
кристаллизуется при комнатной температуре и имеет точку плавления 66°С (выход 73%).
ИК (см-1): кислотный пик 1695.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 6,7 (4Н, s), 3,6 (3Н, s), 3,55 (2Н, s), 2,3 (2Н, s), 1 (6Н, s).
д) 7-Метокси-3,3-диметил-3, 4-дигидро-2Н-бензоксепин-5-он.
Раствор из 900 г полифосфорной кислоты в 1,1 литра толуола доводят до 90°С. К этому раствору добавляют 200 г (0,839 моля) 4-(4-пара-метоксифенокси)-3,3-диметилмасляной кислоты, растворенной в 550 мл толуола. Смесь выдерживают с перемешиванием при 90°С в течение 4 часов. Затем реакционную смесь охлаждают до комнатной температуры (20-25°С). От толуола отделяют густое масло. После охлаждения масло покрывают 500 мл ледяной воды и экстрагируют этилацетатом. Органическую фазу сперва промывают 500 мл (1N) водного раствора гидроокиси натрия, а затем 500 мл воды. Органическую фазу сушат над безводным сульфатом натрия. Этилацетат и толуол выпаривают при пониженном давлении. Получают 165 г желтого масла с общим выходом 89%.
ИК (см-1): 2960, 1680, 1489, 1462, 1419.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 7,1 (1H, m), 6, 95 (2H, s), 3,7 (2Н, s), 3,65 (3Н, s), 2,6 (2Н, s), 1,05 (6Н, s).
e) 7-Метокси-3,3,5-триметил-2,3,4,5-тетрагидробензоксепин-5-ол.
190 мл (0,57 моля) 3М метилхлорида магния в растворе с тетрагидрофураном загружают в 2-литровый реактор в инертной атмосфере. Раствор доводят до 50°С и быстро добавляют 82 г (0,372 моля) полученного на предыдущей стадии д) кетона, растворенного в 1,1 литра тетрагидрофурана. Полученный раствор нагревают с обратным холодильником в течение 4 часов. Далее реакционную смесь охлаждают в ванне со льдом и добавляют воду. Затем смесь выливают на насыщенный водный раствор хлористого аммония.
После этого проводят экстракцию этилацетатом, а органическую фазу отделяют осаждением и сушат над безводным сульфатом натрия. Эту органическую фазу концентрируют при пониженном давлении и получают 85 г оранжевого масла, которое кристаллизуется при комнатной температуре (выход 96%).
1 H ЯМР (СDCl3, 300 МГц) [пропуск]: 7,05-6,6 (3Н, m), 3,7 (5Н, s), 2,15 (1Н, s), 1,8 (2Н, d), 1,55 (3Н, s), 1,05 (3Н, s), 1 (3Н, s).
ж) 7-Метокси-3,3,5-триметил-2, 3-дигидробензоксепин.
169 г (0,716 моля) полученного на предыдущей стадии е) соединения в 2,25 литра толуола и 5 г пара-толуолсульфокислоты вводят в 4-литровый реактор, оснащенный прибором Дина и Старка. Смесь нагревают с обратным холодильником в течение 4 часов. По мере образования воды ее удаляют. Таким образом удаляется 11,5 мл воды. Реакционной смеси дают охладиться до комнатной температуры. Затем ее выливают на 5% водный раствор кислого углекислого натрия. Затем происходит отделение осаждением, после чего органическую фазу промывают водой и сушат над безводным сульфатом натрия. Далее органическую фазу концентрируют при пониженном давлении. Получают 155 г темного масла. Выход 99%.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 6,9-6,6 (3Н, m), 5,6 (1Н, s), 3,7 (5Н, 2s), 2,05 (3Н, s), 1,05 (6Н, s).
з) 5-Бромометил-7-метокси-3,3-диметил-2,3-дигидробензоксепин.
155 г (0,71 моля) полученного на предыдущей стадии соединения, 130 г (0,73 моля) N-бромосукцинимида и 4 г α,α’-азобисизобутиронитрила вводят в 1,6 литра тетрахлорида углерода. Раствор нагревают с обратным холодильником в течение 4 часов и затем охлаждают до 25°С. Образовавшееся сухое вещество отфильтровывают и органическую фазу несколько раз промывают теплой водой (30-40°С). Отделение осуществляется
осаждением, после чего органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Таким образом получают 210 г коричневого масла, соответствующего указанному в заголовке продукту.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 7,1-6,7 (3Н, m), 6 (1Н, s), 4,3 (2Н, s), 3,75 (5Н, s), 1,1 (6Н, s).
и) 7-Метокси-5-формил-3,3-диметил-2,3-дигидробензоксепин.
210 г (0,71 моля) полученного на предыдущей стадии з) соединения и 150 г (1,5 эквивалента) гексаметилентетрамина вводят в 2 литра хлороформа. Раствор нагревают с обратным холодильником в течение 2 часов. Далее реакционную смесь концентрируют при пониженном давлении. Выпавший коричневый осадок покрывают 2,55 литра [sic] 75%-ного водного раствора уксусной кислоты. Смесь нагревают с обратным холодильником в течение 90 минут, затем добавляют 385 мл концентрированной соляной кислоты, после чего смесь вновь нагревают с обратным холодильником в течение 30 минут. К реакционной смеси при слегка повышенной температуре добавляют воду. Смесь вновь [sic] оставляют на 30 минут при комнатной температуре (20-25°С), а затем экстрагируют этилацетатом. Органическую фазу отделяют осаждением, затем промывают водным раствором кислого углекислого натрия, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Таким образом получают 140 г темно-коричневого масла, которое очищают быстрым хроматографическим методом с использованием в качестве элюента дихлорометана. После очищения выделяют 98 г указанного в заголовке соединения. Выход 60%.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 9,5 (1Н, s), 7,6 (1Н, s), 6,9 (1Н, d), 6,7 (1Н, d, J=8,85 Гц), 6,43 (1Н, s), 3,82 (2Н, s), 3,73 (3Н, s), 1,18 (6Н, s).
к) Сложный этиловый эфир (2Е,4Е)-5-(3,3-диметил-7-метокси-2, 3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновой кислоты (соединение 16а).
64,5 мл (0,263 моля) сложного этилового эфира диэтил(2-метил-3-карбоксипроп-2-енил)фосфоната (41% транс) в 250 мл тетрагидрофурана по каплям добавляют к раствору из 29,6 г (0,263 моля) трет-бутокиси калия в 500 мл тетрагидрофурана. Реакционную смесь оставляют перемешиваться в течение 1 часа, а затем охлаждают в ванне со льдом и по каплям добавляют 43 г (0,185 моля) полученного на предыдущей стадии и) соединения, растворенного в 250 мл тетрагидрофурана. Реакционную смесь вновь оставляют перемешиваться при 25°С в течение 16 часов, после чего ее охлаждают в ванне со льдом и гидролизуют, добавляя воду. Затем проводят экстракцию этилацетатом, а органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Таким образом получают 88 г темно-оранжевого масла, которое очищают быстрым хроматографическим методом с использованием в качестве элюента смеси циклогексан/диизопропиловый эфир. Таким образом выделяют 46,8 г желтого масла.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 6,9 (1Н, d, J=2, 95 Гц), 6,7-6,6 (3Н, m), 6,35 (1H, d, J=15,42 Гц), 6 (1Н, s), 5,8 (1H, s), 4,1 (2Н, m), 3,8 (2Н, s), 3,7 (3Н, s), 2,3 (3Н, s), 1,25 (3Н, m), 1,08 (6Н, s).
л) (2E,4E)-5-(3, 3-Диметил-7-метокси-2,3-дигидробензоксепин-5-ил)-3-метилпента-2,4-диеновая кислота (соединение 16b).
7,2 г (0,18 моля) гидроокиси натрия, растворенной в 500 мл воды, добавляют к раствору из 46 г (0,134 моля) полученного на предыдущей стадии к) соединения в 500 мл метанола. Эту смесь нагревают с обратным холодильником в течение 4 часов и концентрируют при пониженном давлении. Выпавший пастообразный осадок растворяют в диэтиловом эфире и промывают водой. Водную фазу подкисляют 5N соляной кислотой до рН 1. Выпавший желтый осадок растворяют в метиленхлориде. Органическую фазу сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Получают 44 г масла, которое рекристаллизируется из этанола. Таким образом выделяют 18 г указанного в заголовке соединения.
ИК (см-1): 3000, 2900, 1680, 1599, 1271, кислотный пик 2400-2800.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 7,1 (1Н, d, J=8,64 Гц), 7-6,85 (3Н, m), 6,65 (1H, d, J=15,36 Гц), 6,2 (1Н, s), 6 (1H, s), 4,05 (2Н, s), 3,95 (3Н, s), 2,55 (3Н, s), 1,3 (6Н, s).
ПРИМЕР 17
(2Е,4Е)-5-(2, 3-Дигидробензоксепин-4-ил)-3-метилпента-2,4-диеновая кислота (I: p=0; R1=H; X=О; А=-СН2-СН2-; R2=Z; n=1; R’=СН3;
соединение 17а [sic]: Y=О-СН2-СН3;
соединение 17b [sic]: Y=-ОН).
а) 2,3-Дигидро-4-(этоксикарбонил)-бензоксепин.
Смесь из 8,2 г (0,043 моля) 2, 3-дигидро-4-карбоксибензоксепина в 150 мл этанола и 5 мл концентрированной серной кислоты нагревают с обратным холодильником в течение 6 часов. Далее реакционную смесь концентрируют при пониженном давлении и осадок покрывают водой. Смесь экстрагируют этилацетатом. Органическую фазу сперва промывают кислым углекислым натрием, а затем водой и сушат над безводным сульфатом натрия, после чего коцентрируют при пониженном давлении. Таким образом получают 8,5 г оранжевого масла.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 7,6 (1H, s), 7,35-7 (4Н, m), 4,2 (4Н, m), 3 (2Н, m), 1,3 (3Н, m).
ИК (см-1): 1705.
б) 2,3-Дигидро-4-(гидроксиметил)-бензоксепин. Приготавливают раствор из 1,4 г (0,036 моля) алюмогидрида лития в 80 мл диэтилового эфира в инертной атмосфере. Этот раствор охлаждают в ванне со льдом и солью до 0°С и затем добавляют 8 г (0,036 моля) полученного на предыдущей стадии а) соединения, растворенного в 80 мл диэтилового эфира. Реакционную смесь оставляют при 0°С на 1 час, после чего, поддерживая ее температуру в диапазоне 0-10°С, добавляют воду.
Затем экстрагируют диэтиловым эфиром и водные исходные растворы насыщают хлоридом натрия. Далее органические фазы сушат над безводным сульфатом натрия и концентрируют при пониженном давлении, получая 7 г бесцветного масла. Результаты инфракрасной спектроскопии свидетельствуют о присутствии ОН-спиртового пика.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 7,15-6,9 (4Н, m), 6,3 (1Н, s), 4,2 (4Н, m), 2,6 (2Н, m), 1,8 (1Н, s).
в) 2,3-Дигидро-4-формилбензоксепин.
Смесь из 6,3 г (0,036 моля) полученного на предыдущей стадии б) соединения, 250 мл хлороформа и 50 г (0,575 моля) двуокиси марганца выдерживают с перемешиванием в течение 16 часов при комнатной температуре (25°С).
После фильтрации через диоксид кремния реакционную смесь концентрируют при пониженном давлении, получая 5 г желтого масла.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 9,5 (1Н, s), 7,3-7 (5Н, m), 4,2 (2Н, m), 2,8 (2Н, m).
г) Сложный этиловый эфир (2Е,4Е)-5-(2,3-дигидробензоксепин-4-ил)-3-метилпента-2,4-диеновой кислоты (соединение 17а).
0,72 г (0,03 моля) гидрида натрия и 20 мл тетрагидрофурана загружают в 250-миллилитровую колбу с четырьмя горлами в инертной атмосфере. К этому раствору добавляют раствор из 7,5 мл (0,03 моля) сложного этилового эфира диэтил(2-метил-3-карбоксипроп-2-енил)фосфонатной кислоты [sic] в 20 мл тетрагидрофурана. Смесь оставляют перемешиваться в течение 1 часа при комнатной температуре (25°С), а затем добавляют 5 г (0,0287 моля) полученного на предыдущей стадии в) соединения в 40 мл тетрагидрофурана. Смесь вновь оставляют перемешиваться в течение 16 часов при 25°С. Далее реакционную смесь гидролизуют водой и выливают на насыщенный водный раствор хлорида натрия. После этого проводят экстракцию этилацетатом и органическую фазу сушат над безводным сульфатом натрия. После этого органическую фазу концентрируют при пониженном давлении. Таким образом получают 9,3 г масла, которое очищают быстрым хроматографическим методом с использованием в качестве элюента смеси циклогексана и этилацетата. Таким образом выделяют 5,8 г масла, которое кристаллизуется при комнатной температуре. Этот продукт рекристаллизуют из диизопропилового эфира и получают 2,7 г указанного в заголовке соединения с точкой плавления 100°С.
ИК (см-1): пик 1700.
1H ЯМР (СDCl3, 300 МГц) [пропуск]: 7,4-7 (4Н, m), 6,9 (1Н, d, J=15,82 Гц), 6,65 (1Н, s), 6,3 (1H, d, J=15,8 Гц), 5,95 (1H, s), 4,4 (2Н, m), 4,3 (2Н, m), 2,95 (2Н, m), 2,5 (3Н, s), 1,4 (3Н, m).
д) (2Е,4Е)-5-(2,3-Дигидробензоксепин-4-ил)-3-метилпента-2, 4-диеновая кислота (соединение 17b).
Раствор из 0,4 г (0,01 моля) гидроокиси натрия в 40 мл воды добавляют к раствору из 2,7 г полученного на предыдущей стадии г) соединения в 40 мл метанола. Этот раствор нагревают с обратным холодильником в течение 3 часов. К этой смеси добавляют новые 50 мл метанола и нагревание с обратным холодильником продолжают еще в течение 3 часов. Далее реакционную смесь концентрируют при пониженном давлении и осадок покрывают водой. Полученный раствор подкисляют до рН 1 с использованием 1N соляной кислоты. Выпавший осадок отфильтровывают, промывают водой и сушат. Получают 2,3 г указанного в заголовке соединения, которое рекристаллизуют из метанола. Выделяют 1,1 г очищенного продукта с точкой плавления 220-222°С.
ИК (см-1): 1669, 1588, 1262, 2400-3200.
1H ЯМР (CDCl3, 300 МГц) δ (промили): 7,3-6,9 (5Н, m), 6,7 (1H, s), 6,5-6,4 (1H, d, J=15,87 Гц), 5,8 (1H, s), 4,2 (2Н, m), 2,8 (2Н, m), 2,3 (3Н, s).
ПРИМЕРЫ 18-30
Соединения из Примеров 18-30, сведенные в Таблицу 2, получают с помощью методик, проиллюстрированных в предыдущих примерах.

















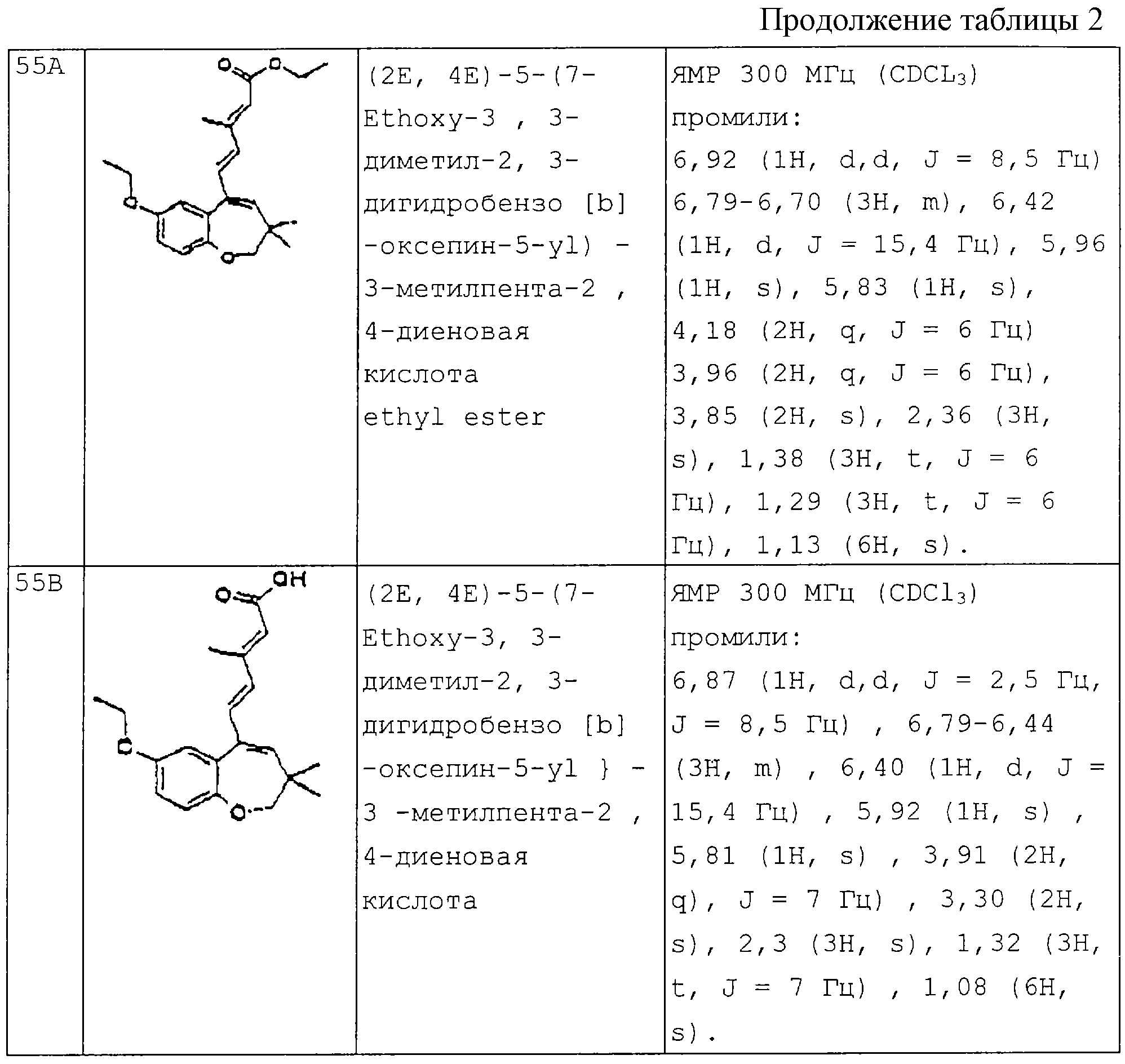
К тому же следующие способы получения иллюстрируют синтез промежуточных соединений для изготовления соединений из вышеприведенных Примеров 1-31.
Получение 1
3,3-Диметил-7-фенил-3,4-дигидро-2Н-бензоксепин-5-он
8,5 г 7-бромо-3,3-диметил-3,4-дигидро-2Н-бензоксепин-5-она (0,0316 моля), 95 мл толуола, 34,7 мл 2М раствора карбоната натрия, 46 мл этанола, 4,19 г фениборной [sic] кислоты (0,0344 моля, 1,09 эквивалента) и 0,677 г тетракис(трифенилфосфин)палладия загружают в 250-миллилитровый реактор.
Реакционную смесь нагревают с обратным холодильником в течение 8 ч и дают охладиться до комнатной температуры. Реакционную массу выливают в смесь из 70 мл воды, 56,8 мл 30%-ного водного раствора гидроксида аммония и 78 мл 2М водного раствора карбоната натрия. Катализатор отфильтровывают через диоксид кремния. Затем трижды 100 мл этилацетата проводят экстракцию и органические фазы триждый промывают 100 мл воды, сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Выделяют 14 г масла, которое очищают методом колоночной хроматографии с использованием смеси 4/1 гексан/этилацетат. Таким образом получают 8,5 г указанного в заголовке соединения в форме жидкости.
1H ЯМР (СDCl3, 300 МГц) δ (промили): 7,88-7,25 (8Н, m), 3,98 (2Н, s), 2,74 (2Н, s), 1,15 (6Н, s).
Получение 2
(4-пара-Хлорофенил)(3,3,5-триметил-2,3-дигидробензоксепин-7-ил)кетон [sic]
0, 627 г стружки магния [sic] (0,0258 моля), 20 мл безводного тетрагидрофурана и кристаллический йод вводят в 250-миллилитровый реактор. К этому раствору медленно добавляют смесь из 8,3 г 7-бромо-3,3, 5-триметил-2,3-дигидробензоксепина в 30 мл безводного тетрагидрофурана. Смесь нагревают с обратным холодильником в течение 1,5 ч и дают охладиться до комнатной температуры (раствор А).
5 г пара-хлоробензоилхлорида и 20 мл безводного тетрагидрофурана вводят во второй 250-миллилитровый реактор. Этот раствор охлаждают до 0°С и медленно добавляют к нему приготовленный (см. выше) раствор А, поддерживая температуру реакционной смеси ниже 5°С. Смеси дают возвратиться к комнатной температуре и перемешивают в течение 20 ч. После охлаждения до 5°С проводят гидролиз, медленно добавляя 3,2 мл водного раствора 1N соляной кислоты, смесь разбавляют водой и трижды экстрагируют 100 мл диэтилового эфира. Органическую фазу промывают 1N водным раствором гидроокиси натрия и дважды - 100 мл воды, после чего сушат над безводным сульфатом натрия и концентрируют при пониженном давлении. Таким образом получают 10,5 г масла, которое очищают методом колоночной хроматографии с использованием в качестве элюента смеси 10/1 гексан/этилацетат. После очистки выделяют 3,6 г указанного в заголовке соединения в форме жидкости. Выход 38,7%.
1H ЯМР (DMSO [sic], 300 МГц) δ (промили): 8,04-6,96 (7Н, m), 5,73 (1Н, s), 3,83 (2Н, s), 2,06 (3Н, s), 1,05 (6Н, s).
Получение 3
4-Метил-3-формил-2Н-1-бензопиран
6,3 г (40,63 ммоля) РОСl3 выливают, при 0°С, в 50-миллилитровую колбу с четырьмя горлами в инертной атмосфере, содержащую 3,0 г (40,63 ммоля) диметилформамида. Реакционную смесь выдерживают при 5°С в течение 15 минут и при такой температуре вливают раствор из 4,8 г (32,5 ммоля) 4-метил-2Н-1-бензопирана в 25 мл метиленхлорида.
Реакционной смеси дают возвратиться к комнатной температуре и перемешивают в течение 2 часов. Затем реакционную смесь быстро выливают в смесь воды и льда и экстрагируют этилацетатом.
Органическую фазу промывают 5%-ным водным раствором кислого углекислого натрия и затем водой. После сушки над безводным сульфатом натрия органическую фазу концентрируют при пониженном давлении. Получают масло, которое медленно кристаллизуется. Его рекристаллизуют из 25 мл изопропилового эфира. Таким образом получают 2,0 г указанного в заголовке соединения в форме сухого вещества с точкой плавления 68-70°С (выход=35%).
1H ЯМР (СDCl3, 300 МГц) δ (промили): 2,4 (3Н, s), 4,8 (2Н, s), 6,8-7,0 (2Н, m), 7,2-7,4 (2Н, m), 10,1 (1Н, s).
Т.пл.=68-70°С.
Получение 4
4-Фенил-3-формил-2Н-1-бензопиран
71 мл (0,773 моля) хлорангидрида фосфорной кислоты по каплям добавляют к 300 мл диметилформамида при 0°С. Реакционную смесь оставляют перемешиваться в течение 20 минут при 0-5°С. К реакционной смеси добавляют раствор из 16,1 г (77,3 ммоля) 4-фенил-2Н-1-бензопирана в 22,5 мл диметилформамида. Реакционной смеси дают остыть до комнатной температуры и в течение 7 часов доводят до 60°С. Затем реакционную смесь выливают в смесь льда с водой, нейтрализованную NaOH. Органическую фазу экстрагируют этилацетатом и трижды промывают водой. После сушки над безводным сульфатом натрия органическую фазу концентрируют при пониженном давлении. Таким образом получают пастообразное сухое вещество. Используют метод колоночной хроматографии со смесью 1/1 метиленхлорид/циклогексан в качестве элюента, получая 15,3 г указанного в заголовке соединения в форме желтого сухого вещества с точкой плавления 82-83°С (выход=83%).
1H ЯМР (CDCl3, 300 МГц) δ (промили): 5 (2Н, s), 6,79-6,89 (3Н, m), 7,21-7,26 (3Н, m), 7,39-7,42 (3Н, m), 9,38 (1Н, s).
Характеристические физико-химические данные в отношении некоторых промежуточных продуктов приведены в Таблице 3.



Характеристические физико-химические данные в отношении некоторых промежуточных соединений типа 5-метил-2,3-дигидробензоксепина сведены в Таблицу 4.



Характеристические физико-химические данные в отношении некоторых производных 5-бромометил-2,3-дигидробензоксепина сведены в Таблицу 5.


Реферат
Изобретения относятся к новым производным бензопирана и бензоксепина формулы (I)

в которой X, A, r1 и (R)p имеют указанные в п.1 значения, способам их получения, промежуточным соединениям, а также к фармацевтической композиции, содержащей соединения формулы I, которая обладает гиполипидемической и гипогликемической активностью. Это делает возможным ее использование для лечения дислипидемии, атеросклероза и диабета. 7 с. и.,13 з.п. ф-лы., 5 табл.
Формула
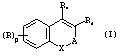

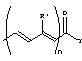











Комментарии