Средство для ингибирования фермента тирозил-днк-фосфодиэстеразы 1 человека - RU2612875C1
Код документа: RU2612875C1
Описание
Изобретение относится к молекулярной биологии, биохимии и биотехнологии, конкретно к соединениям, представляющим собой производные хроменона общей формулы I:

где, n=1 или 2,
или II:

у которых выявлена биологическая активность, заключающаяся в способности ингибировать действие фермента тирозил-ДНК-фосфодиэстеразы 1 человека (Tdp1).
В последние годы ведутся активные поиски ингибиторов фермента тирозил-ДНК-фосфодиэстеразы 1 (Tdp1), который рассматривается как перспективный фермент-мишень при создании лекарственных препаратов для лечения онкологических и нейродегенеративных заболеваний [Cortes Ledesma, 2009].
Tdp1 относится к классу фосфодиэстераз - ферментов, расщепляющих фосфодиэфирные связи [Interthal, 2001]. Tdp1 играет важную роль в удалении повреждений ДНК, создаваемых топоизомеразой 1 (Top1) в присутствии ее ингибитора камптотецина. Таким образом, Tdp1 противостоит ингибиторам Top1, которые являются достаточно эффективными противоопухолевыми препаратами [см. обзоры Pommier, 2010; Pommier, 2006]. Предполагается, что именно Tdp1 ответственна за лекарственную устойчивость некоторых видов рака [Dexheimer, 2008; Beretta, 2010]. Эта гипотеза подтверждается рядом исследований: мыши, нокаутные по Tdp1, и человеческие клеточные линии, имеющие мутацию SCAN1, гиперчувствительны к камптотецину [El-Khamisy, 2009; Das, 2009; Katyal, 2007; Hirano, 2007]. И, наоборот, в клетках с повышенным уровнем экспрессии Tdp1 камптотецин и этопозид вызывают меньше повреждений ДНК [Barthelmes, 2004; Nivens, 2004]. Таким образом, сочетание препаратов, воздействующих на Top1 и Tdp1, может существенно повысить эффективность химиотерапии. Терапевтическим эффектом ингибиторов Tdp1 может быть селективное увеличение активности ингибиторов Top1 в опухолях с нарушениями в процессах репарации ДНК и контроля клеточного цикла.
Кроме того, показано, что подавление активности Tdp1 делает опухолевые клетки гиперчувствительными к противораковом препаратам с другими механизмами действия: темозоломиду (метилирование пуринов) [Alagoz, 2013], метилметансульфонату (образование апуриновых/апиримидиновых сайтов), блеомицину (одноцепочечные/двухцепочечные разрывы с 3'-фосфогликолятами), перекиси водорода и ионизирующему излучению (разрывы и др. виды повреждений) [Murai, 2012].
В литературе описано немного ингибиторов Tdp1, и, как правило, они обладают умеренным ингибирующим действием (в диапазоне 100-1 мкМ) [Antony, 2007; Nguyen, 2012; Dexheimer, 2008; Zakharenko, 2015].
Наиболее близким к заявляемому средству – прототипом - является фурамидин, представляющий собой гетероциклический диамидин [Antony, 2007] общей формулы III:

Недостатком известного средства являются неудовлетворительные ингибиторные характеристики (IС50 для одноцепочечной ДНК порядка 100 мкМ).
Задачей изобретения является создание более эффективного ингибитора Tdp1.
Поставленная техническая задача решается применением соединений, представляющих собой производные хроменона общей формулы I или II, у которых выявлена биологическая активность, заключающаяся в подавлении активности Tdp1.
Соединения I и II могут быть синтезированы взаимодействием бромида IV с замещенными кумаринами V и VI в соответствии со следующей схемой:

Соединение IV может быть синтезировано, например, из монотерпеноида миртеналя VII в соответствии со следующей схемой:
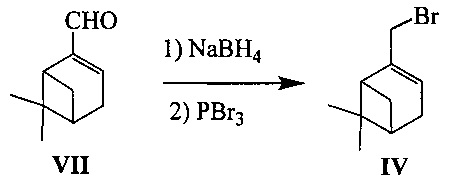
Для получения обоих энантиомеров соединения IV в качестве исходных соединений использовались (+)- и (-)-миртеналь (VII).
Соединения Va, b и VI могут быть синтезированы, например, взаимодействием резорцина VIII с эфирами соответствующих β-кетокислот по следующей схеме:

Структура полученных соединений I и II подтверждена данными ЯМР-спектроскопии и масс-спектрометрии.
Результаты тестирования соединений Ia (n=1), Ib (n=2) и II приведены в таблице.
Технический результат: получен класс эффективных ингибиторов Tdp1 с хорошими ингибирующими характеристиками (IС50 для одноцепочечной ДНК от 0.13 до 1.6 мкМ).
В случае соединений Ia и II производные (+)-миртеналя оказались более активны, чем соответствующие (-)-энантиомеры, в то же время оба энантиомера соединения Ib проявили одинаковую активность.
Соединения общей формулы I и II, после проведения углубленных фармакологических исследований, могут использоваться для дальнейшей разработки новых высокоэффективных противораковых средств.

Ниже приводятся конкретные примеры реализации заявляемого технического решения.
Пример 1. Синтез 7-(((1R,5S)-6,6-диметилбицикло[3.1.1]гепт-2-ен-2-ил)метокси)-2,3-дигидроциклопента[с]хромен-4(1H)-она ((-)-Iа)

К 0.101 г (0.5 ммоль) соединения Va в 5 мл этанола прибавили 0.104 г (0.75 ммоль) K2СО3 и 0.161 г (0.75 ммоль) бромида (-)-IV при 25°С и перемешивании. Перемешивали при комнатной температуре 15 минут, затем нагревали при 80°С в течение 5 часов. Горячий раствор отфильтровали, фильтрат выдерживали при -10°С в течение 48 часов. Продукт (-)-Iа выделили перекристаллизацией из этанола, выход составил 0.067 г (40%).
Тпл=145°С,
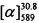
Пример 2. Синтез 7-(((1S,5R)-6,6-диметилбицикло[3.1.1]гепт-2-ен-2-ил)метокси)-2,3-дигидроциклопента[с]хромен-4(1H)-она ((+)-Ia)

К 0.101 г (0.5 ммоль) соединения Va в 5 мл этанола прибавили 0.104 г (0.75 ммоль) K2CO3 и 0.161 г (0.75 ммоль) бромида (+)-IV при 25°С и перемешивании. Перемешивали при комнатной температуре 15 минут, затем нагревали при 80°С в течение 5 часов. Горячий раствор отфильтровали, фильтрат выдерживали при -10°С в течение 48 часов. Продукт (+)-Ia выделили перекристаллизацией из этанола, выход составил 0.093 г (55%).
Тпл=140°С.

Пример 3. Синтез 3-(((1R,5S)-6,6-диметилбицикло[3.1.1]гепт-2-ен-2-ил)метокси)-7,8,9,10-тетрагидро-6H-бензо[с]хромен-6-она ((-)-Ib)

К 0.108 г (0.5 ммоль) соединения Vb в 5 мл этанола прибавили 0.104 г (0.75 ммоль) K2СО3 и 0.161 г (0.75 ммоль) бромида (-)-IV при 25°С и перемешивании. Перемешивали при комнатной температуре 15 минут, затем нагревали при 80°С в течение 5 часов. Горячий раствор отфильтровали, фильтрат выдерживали при -10°С в течение 48 часов. Продукт (-)-Ib выделили перекристаллизацией из этанола, выход составил 0.067 г (38%).
Тпл=110°С.

Пример 4. Синтез 3-(((1S,5R)-6,6-диметилбицикло[3.1.1]гепт-2-ен-2-ил)метокси)-7,8,9,10-тетрагидро-6H-бензо[с]хромен-6-она ((+)-Ib)

К 0.108 г (0.5 ммоль) соединения Vb в 5 мл этанола прибавили 0.104 г (0.75 ммоль) K2СO3 и 0.161 г (0.75 ммоль) бромида (+)-IV при 25°С и перемешивании. Перемешивали при комнатной температуре 15 минут, затем нагревали при 80°С в течение 5 часов. Горячий раствор отфильтровали, фильтрат выдерживали при -10°С в течение 48 часов. Продукт (+)-Ib выделили перекристаллизацией из этанола, выход составил 0.067 г (38%).
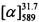
Пример 5. Синтез 7-(((1R,5S)-6,6-диметилбицикло[3.1.1]гепт-2-ен-2-ил)метокси)-4-метил-2H-хромен-2-она ((-)-II)

К 0.108 г (0.5 ммоль) соединения VI в 5 мл этанола прибавили 0.104 г (0.75 ммоль) K2СO3 и 0.132 г (0.75 ммоль) бромида (-)-IV при 25°С и перемешивании. Перемешивали при комнатной температуре 15 минут, затем нагревали при 80°С в течение 5 часов. Горячий раствор отфильтровали, фильтрат выдерживали при -10°С в течение 48 часов, упарили. Продукт (-)-II выделили колоночной хроматографией на силикагеле, элюент - раствор, содержащий от 25 до 100% хлороформа в гексане. Выход составил 0.084 г (54%).
Тпл=70°С.

Пример 6. Синтез 7-(((1S,5R)-6,6-диметилбицикло[3.1.1]гепт-2-ен-2-ил)метокси)-4-метил-2H-хромен-2-она ((+)-II)

К 0.108 г (0.5 ммоль) соединения VI в 5 мл этанола прибавили 0.104 г (0.75 ммоль) K2СO3 и 0.132 г (0.75 ммоль) бромида (+)-IV при 25°С и перемешивании. Перемешивали при комнатной температуре 15 минут, затем нагревали при 80°С в течение 5 часов. Горячий раствор отфильтровали, фильтрат выдерживали при -10°С в течение 48 часов, упарили. Продукт (+)-II выделили колоночной хроматографией на силикагеле, элюент - раствор, содержащий от 25 до 100% хлороформа в гексане. Выход составил 0.047 г (30%).

Пример 7. Исследование влияния предлагаемых соединений на активность Tdp1
Рекомбинантная тирозил-ДНК-фосфодиэстераза 1 человека (КФ 3.1.4.) была экспрессирована в системе Escherichia coli (плазмида рЕТ 16B-Tdp1 любезно предоставлена доктором Кальдекотт К.У., Университет Сассекса, Великобритания) и выделена, как описано [Interthal, 2001].
В качестве тест-системы для определения ингибирующих свойств предлагаемых соединений использована реакция удаления тушителя флуоресценции Black Hole Quencher 1 (BHQ1) с 3'-конца олигонуклеотида, катализируемая Tdp1. На 5'-конце олигонуклеотида находится (5,6)-FAM - флуорофор, интенсивность флуоресценции которого возрастает при удалении тушителя. Для измерения флуоресценции использовался флуориметр POLARstar OPTIMA производства BMG LABTECH.
Реакционные смеси объемом 200 мкл содержали буфер (50 мМ Tris-HCl, рН 8.0; 50 мМ NaCl; 7 мМ меркаптоэтанол), 50 нМ олигонуклеотид и различные концентрации ингибиторов. Реакция запускалась добавлением Tdp1 до конечной концентрации 1.3 нМ. Измерения проводились в линейном диапазоне зависимости скорости реакции от времени (до 8 минут) через каждые 55 секунд. Влияние предлагаемых соединений оценивали по величине IC50 (концентрация ингибитора, при которой активность фермента снижена наполовину). Обсчет значений IC50 проводился с помощью программы MARS Data Analisys 2.0 (BMG LABTECH).
Величины IC50 для изученных соединений приведены в таблице.
Использованная литература
- Alagoz М, et al., Nucleic Acids Res., 2013, [Epub ahead of print].
- Antony, S et al., Nucleic Acids Res. 2007, 35, 4474-4484.
- Barthelmes HU, et al., J Biol Chem. 2004, 279, 55618-25565.
- Beretta GL, et al., Curr. Med. Chem. 2010, 17, 1500-1508.
- Cortes Ledesma F, et al., Nature, 2009, 461, 674-678.
- Das BB, et al., The EMBO Journal., 2009, 28, 3667-3680.
- Dexheimer TS, et al., Anticancer Agents Med Chem. 2008, 8, 381-389.
- El-Khamisy SF, et al., DNA Repair (Amst)., 2009, 8 760-766.
- Hirano R, et al., EMBO J., 2007, 26, 4732-4743.
- Interthal H, et al., Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 12009-12014.
- Katyal S, et al, EMBO J., 2007, 26, 4720-4731.
- Khomenko TM, et al., Letters in Drug Design & Discovery, 2009, 6, 464-467.
- Kutuzov MM, et al., Biopolym. and Cell, 2012, 28, 239-241.
- Nguyen TX, et al., J. Med. Chem., 2012, 55, 4457-4478.
- Nivens MC, et al., Cancer Chemother Pharmacol., 2004, 53, 107-115.
- Murai J, et al., J Biol Chem. 2012, 287,12848-12857.
- Pommier Y. Nat. Rev. Cancer, 2006, 6, 789-802.
- Pommier Y, et al. Chem Biol., 2010, 17, 421-433.
- Zakharenko A, et al., Bioorg. Med. Chem., 2015, 23, 2044-2052.
Реферат
Изобретение относится к молекулярной биологии, биохимии и биотехнологии. Предложено средство, представляющее собой производное хроменона:, где n=1 или 2,илипроявляющее способность ингибировать действие фермента тирозил-ДНК-фосфодиэстеразы 1 человека. Предлагаемое средство расширяет арсенал ингибиторов фермента тирозил-ДНК-фосфодиэстеразы 1 человека и может быть использовано для разработки противораковых лекарственных препаратов. 1 табл., 7 пр.
Формула


Комментарии